TWI723572B - 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 - Google Patents
具有四氫吡喃基甲基之吡啶酮衍生物及其用途 Download PDFInfo
- Publication number
- TWI723572B TWI723572B TW108136526A TW108136526A TWI723572B TW I723572 B TWI723572 B TW I723572B TW 108136526 A TW108136526 A TW 108136526A TW 108136526 A TW108136526 A TW 108136526A TW I723572 B TWI723572 B TW I723572B
- Authority
- TW
- Taiwan
- Prior art keywords
- compound
- pyran
- tetrahydro
- ylmethyl
- cancer
- Prior art date
Links
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical class OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 title abstract 2
- 125000006173 tetrahydropyranylmethyl group Chemical group 0.000 title abstract 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 449
- 150000003839 salts Chemical class 0.000 claims abstract description 66
- 201000010099 disease Diseases 0.000 claims abstract description 43
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 43
- 206010028980 Neoplasm Diseases 0.000 claims description 99
- -1 oxygen-1-(tetrahydro-2H-pyran-4-ylmethyl)-1,4-dihydropyridine-3-carboxamide Chemical compound 0.000 claims description 82
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 70
- 238000004519 manufacturing process Methods 0.000 claims description 66
- 201000011510 cancer Diseases 0.000 claims description 50
- 239000003814 drug Substances 0.000 claims description 43
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 35
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 31
- 239000005551 L01XE03 - Erlotinib Substances 0.000 claims description 30
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 claims description 30
- 229960001433 erlotinib Drugs 0.000 claims description 30
- 108091000080 Phosphotransferase Proteins 0.000 claims description 28
- 102000020233 phosphotransferase Human genes 0.000 claims description 28
- ZHNUHDYFZUAESO-UHFFFAOYSA-N formamide Substances NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 claims description 27
- 125000005843 halogen group Chemical group 0.000 claims description 25
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 23
- 229910052757 nitrogen Inorganic materials 0.000 claims description 21
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 21
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 17
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 17
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 14
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 13
- 229910019142 PO4 Inorganic materials 0.000 claims description 13
- 239000010452 phosphate Substances 0.000 claims description 13
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 claims description 13
- 239000005483 tyrosine kinase inhibitor Substances 0.000 claims description 13
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 12
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 12
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 claims description 12
- 206010027476 Metastases Diseases 0.000 claims description 10
- 229910052805 deuterium Inorganic materials 0.000 claims description 10
- 230000009401 metastasis Effects 0.000 claims description 10
- 206010059866 Drug resistance Diseases 0.000 claims description 9
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical group Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims description 9
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 claims description 9
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 9
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 8
- 201000005202 lung cancer Diseases 0.000 claims description 8
- 208000020816 lung neoplasm Diseases 0.000 claims description 8
- 206010006187 Breast cancer Diseases 0.000 claims description 7
- 208000026310 Breast neoplasm Diseases 0.000 claims description 7
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 7
- 229910002651 NO3 Inorganic materials 0.000 claims description 7
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 claims description 7
- 206010033128 Ovarian cancer Diseases 0.000 claims description 7
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 7
- 229940077388 benzenesulfonate Drugs 0.000 claims description 7
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 claims description 7
- 229940079593 drug Drugs 0.000 claims description 7
- 125000006414 CCl Chemical group ClC* 0.000 claims description 6
- 206010009944 Colon cancer Diseases 0.000 claims description 6
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 6
- 206010017758 gastric cancer Diseases 0.000 claims description 6
- 230000003463 hyperproliferative effect Effects 0.000 claims description 6
- 201000001441 melanoma Diseases 0.000 claims description 6
- 201000011549 stomach cancer Diseases 0.000 claims description 6
- 206010014733 Endometrial cancer Diseases 0.000 claims description 5
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 5
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 5
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 5
- 206010060862 Prostate cancer Diseases 0.000 claims description 5
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 5
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 5
- 125000004429 atom Chemical group 0.000 claims description 5
- 208000029742 colonic neoplasm Diseases 0.000 claims description 5
- 201000004101 esophageal cancer Diseases 0.000 claims description 5
- 208000032839 leukemia Diseases 0.000 claims description 5
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 5
- 201000002510 thyroid cancer Diseases 0.000 claims description 5
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 4
- 206010029260 Neuroblastoma Diseases 0.000 claims description 4
- 206010038389 Renal cancer Diseases 0.000 claims description 4
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 4
- 201000010982 kidney cancer Diseases 0.000 claims description 4
- XTEGVFVZDVNBPF-UHFFFAOYSA-L naphthalene-1,5-disulfonate(2-) Chemical compound C1=CC=C2C(S(=O)(=O)[O-])=CC=CC2=C1S([O-])(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-L 0.000 claims description 4
- 201000008968 osteosarcoma Diseases 0.000 claims description 4
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- YQGUQZWOHCDPTK-UHFFFAOYSA-N N-[4-[2-amino-5-[1-(oxan-4-yl)pyrazol-4-yl]pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)C=1C=NN(C=1)C1CCOCC1 YQGUQZWOHCDPTK-UHFFFAOYSA-N 0.000 claims description 3
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 3
- 150000001975 deuterium Chemical group 0.000 claims description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 3
- 229910052739 hydrogen Inorganic materials 0.000 claims description 3
- 239000001257 hydrogen Substances 0.000 claims description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 125000004434 sulfur atom Chemical group 0.000 claims description 3
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 3
- 206010046766 uterine cancer Diseases 0.000 claims description 3
- 239000005411 L01XE02 - Gefitinib Substances 0.000 claims description 2
- 229960002584 gefitinib Drugs 0.000 claims description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical group C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 claims description 2
- 208000002409 gliosarcoma Diseases 0.000 claims description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical group CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 claims 2
- RQAATFYANFZYNJ-UHFFFAOYSA-N N-[4-[2-amino-5-(1-ethylpyrazol-4-yl)pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)C=1C=NN(C=1)CC RQAATFYANFZYNJ-UHFFFAOYSA-N 0.000 claims 1
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 claims 1
- 239000013078 crystal Substances 0.000 abstract description 104
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 223
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 150
- 238000006243 chemical reaction Methods 0.000 description 129
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 114
- 239000007787 solid Substances 0.000 description 93
- 238000000634 powder X-ray diffraction Methods 0.000 description 86
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 81
- 239000000243 solution Substances 0.000 description 74
- 238000000034 method Methods 0.000 description 70
- 239000002904 solvent Substances 0.000 description 66
- 230000002829 reductive effect Effects 0.000 description 65
- 238000005160 1H NMR spectroscopy Methods 0.000 description 64
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 59
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 54
- 229910052802 copper Inorganic materials 0.000 description 54
- 239000010949 copper Substances 0.000 description 54
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 48
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 45
- 239000000543 intermediate Substances 0.000 description 45
- 238000001914 filtration Methods 0.000 description 41
- 238000010898 silica gel chromatography Methods 0.000 description 40
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 38
- 239000012044 organic layer Substances 0.000 description 37
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- 230000015572 biosynthetic process Effects 0.000 description 34
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 32
- 239000007858 starting material Substances 0.000 description 32
- 238000003786 synthesis reaction Methods 0.000 description 32
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical class CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 31
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 31
- 150000001335 aliphatic alkanes Chemical class 0.000 description 30
- 230000002401 inhibitory effect Effects 0.000 description 28
- 238000012360 testing method Methods 0.000 description 28
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 27
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 26
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 25
- 238000009835 boiling Methods 0.000 description 25
- 210000004027 cell Anatomy 0.000 description 25
- 239000000203 mixture Substances 0.000 description 25
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 24
- 238000003756 stirring Methods 0.000 description 24
- 230000001678 irradiating effect Effects 0.000 description 23
- 239000007864 aqueous solution Substances 0.000 description 22
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 22
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical class C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 19
- 229910000027 potassium carbonate Inorganic materials 0.000 description 19
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 18
- 239000002585 base Substances 0.000 description 18
- 238000000921 elemental analysis Methods 0.000 description 18
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 17
- 229910000024 caesium carbonate Inorganic materials 0.000 description 17
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 17
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 16
- 239000012046 mixed solvent Substances 0.000 description 16
- FWFGVMYFCODZRD-UHFFFAOYSA-N oxidanium;hydrogen sulfate Chemical compound O.OS(O)(=O)=O FWFGVMYFCODZRD-UHFFFAOYSA-N 0.000 description 16
- 239000000047 product Substances 0.000 description 16
- 239000003112 inhibitor Substances 0.000 description 15
- 230000005764 inhibitory process Effects 0.000 description 15
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- 239000011259 mixed solution Substances 0.000 description 14
- 239000002253 acid Substances 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 241000699670 Mus sp. Species 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 239000004327 boric acid Substances 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- 230000000259 anti-tumor effect Effects 0.000 description 11
- 239000012467 final product Substances 0.000 description 11
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 11
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 11
- 230000035755 proliferation Effects 0.000 description 11
- XHFLOLLMZOTPSM-UHFFFAOYSA-M sodium;hydrogen carbonate;hydrate Chemical class [OH-].[Na+].OC(O)=O XHFLOLLMZOTPSM-UHFFFAOYSA-M 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 239000012299 nitrogen atmosphere Substances 0.000 description 10
- 108090000765 processed proteins & peptides Proteins 0.000 description 10
- 238000005406 washing Methods 0.000 description 10
- 150000007529 inorganic bases Chemical class 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 238000002054 transplantation Methods 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 239000004480 active ingredient Substances 0.000 description 8
- 125000000217 alkyl group Chemical group 0.000 description 8
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 8
- 239000003054 catalyst Substances 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 8
- 229940098779 methanesulfonic acid Drugs 0.000 description 8
- 150000007530 organic bases Chemical class 0.000 description 8
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 8
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 8
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 8
- 108700020796 Oncogene Proteins 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 7
- 239000002246 antineoplastic agent Substances 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 238000010586 diagram Methods 0.000 description 7
- 239000003480 eluent Substances 0.000 description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- 241000208340 Araliaceae Species 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- 235000005035 Panax pseudoginseng ssp. pseudoginseng Nutrition 0.000 description 6
- 235000003140 Panax quinquefolius Nutrition 0.000 description 6
- GPDHNZNLPKYHCN-DZOOLQPHSA-N [[(z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium;hexafluorophosphate Chemical compound F[P-](F)(F)(F)(F)F.CCOC(=O)C(\C#N)=N/OC(=[N+](C)C)N1CCOCC1 GPDHNZNLPKYHCN-DZOOLQPHSA-N 0.000 description 6
- 235000011054 acetic acid Nutrition 0.000 description 6
- 125000001246 bromo group Chemical group Br* 0.000 description 6
- 230000008859 change Effects 0.000 description 6
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 6
- 239000012156 elution solvent Substances 0.000 description 6
- 235000008434 ginseng Nutrition 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000011159 matrix material Substances 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 6
- 239000002953 phosphate buffered saline Substances 0.000 description 6
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 6
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 6
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 5
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 5
- 241000009298 Trigla lyra Species 0.000 description 5
- 102100037236 Tyrosine-protein kinase receptor UFO Human genes 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 229940100198 alkylating agent Drugs 0.000 description 5
- 239000002168 alkylating agent Substances 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 5
- 235000019253 formic acid Nutrition 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 230000009545 invasion Effects 0.000 description 5
- 208000017169 kidney disease Diseases 0.000 description 5
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 125000006239 protecting group Chemical group 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 229910000029 sodium carbonate Inorganic materials 0.000 description 5
- 239000012453 solvate Substances 0.000 description 5
- 230000002194 synthesizing effect Effects 0.000 description 5
- 229910052723 transition metal Inorganic materials 0.000 description 5
- 150000003624 transition metals Chemical class 0.000 description 5
- 125000005500 uronium group Chemical group 0.000 description 5
- 239000003981 vehicle Substances 0.000 description 5
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-bis(diphenylphosphino)propane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 4
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 4
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 4
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- VSXLNJTYRFDLNH-UHFFFAOYSA-N 5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound CC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O VSXLNJTYRFDLNH-UHFFFAOYSA-N 0.000 description 4
- 229940126062 Compound A Drugs 0.000 description 4
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 101150022345 GAS6 gene Proteins 0.000 description 4
- 239000007995 HEPES buffer Substances 0.000 description 4
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 4
- 238000002441 X-ray diffraction Methods 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 238000004364 calculation method Methods 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 229910002091 carbon monoxide Inorganic materials 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000012292 cell migration Effects 0.000 description 4
- 230000005754 cellular signaling Effects 0.000 description 4
- 239000012295 chemical reaction liquid Substances 0.000 description 4
- 208000033679 diabetic kidney disease Diseases 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 230000032050 esterification Effects 0.000 description 4
- 238000005886 esterification reaction Methods 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 150000004677 hydrates Chemical class 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 238000004020 luminiscence type Methods 0.000 description 4
- 230000005012 migration Effects 0.000 description 4
- 238000013508 migration Methods 0.000 description 4
- 230000035772 mutation Effects 0.000 description 4
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 235000011056 potassium acetate Nutrition 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- 238000001308 synthesis method Methods 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical group OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 4
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 4
- 229960003862 vemurafenib Drugs 0.000 description 4
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 4
- 239000011534 wash buffer Substances 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 3
- IFCSWGQBKVTSFE-UHFFFAOYSA-N 3-(4-aminophenyl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(C=2C=CC(N)=CC=2)=C1 IFCSWGQBKVTSFE-UHFFFAOYSA-N 0.000 description 3
- PYZATJISPIITJV-UHFFFAOYSA-N 5-bromo-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound BrC=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O PYZATJISPIITJV-UHFFFAOYSA-N 0.000 description 3
- 101150018445 Axl gene Proteins 0.000 description 3
- JPVNIXANASYDQI-UHFFFAOYSA-N COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)B1OC(C(O1)(C)C)(C)C Chemical compound COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)B1OC(C(O1)(C)C)(C)C JPVNIXANASYDQI-UHFFFAOYSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 3
- 206010018367 Glomerulonephritis chronic Diseases 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 208000007766 Kaposi sarcoma Diseases 0.000 description 3
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 3
- XNRVGTHNYCNCFF-UHFFFAOYSA-N Lapatinib ditosylate monohydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.CC1=CC=C(S(O)(=O)=O)C=C1.O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 XNRVGTHNYCNCFF-UHFFFAOYSA-N 0.000 description 3
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 3
- MEEFJMIHPGPRHH-UHFFFAOYSA-N NC(C(C1)=CNC=C1C1=NC=CC=C1)=O.[O] Chemical compound NC(C(C1)=CNC=C1C1=NC=CC=C1)=O.[O] MEEFJMIHPGPRHH-UHFFFAOYSA-N 0.000 description 3
- BSXPETOIHMTHPK-UHFFFAOYSA-N NC(C1=CC=CN(CC2CCOCC2)C1)=O.[O] Chemical compound NC(C1=CC=CN(CC2CCOCC2)C1)=O.[O] BSXPETOIHMTHPK-UHFFFAOYSA-N 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 206010048955 Retinal toxicity Diseases 0.000 description 3
- 230000001154 acute effect Effects 0.000 description 3
- 230000033115 angiogenesis Effects 0.000 description 3
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 229960004562 carboplatin Drugs 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000007850 degeneration Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 229940071111 erlotinib 25 mg Drugs 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- 239000000367 immunologic factor Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 229960004891 lapatinib Drugs 0.000 description 3
- 238000011580 nude mouse model Methods 0.000 description 3
- 210000002747 omentum Anatomy 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 238000010791 quenching Methods 0.000 description 3
- 230000000171 quenching effect Effects 0.000 description 3
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 3
- 230000004043 responsiveness Effects 0.000 description 3
- 231100000385 retinal toxicity Toxicity 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000004055 small Interfering RNA Substances 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 238000010189 synthetic method Methods 0.000 description 3
- 238000010998 test method Methods 0.000 description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 3
- 229960004528 vincristine Drugs 0.000 description 3
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 3
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 3
- KMSKQZKKOZQFFG-YXRRJAAWSA-N (7S,9S)-7-[[(2R,4S,5S,6S)-4-amino-6-methyl-5-[[(2R)-2-oxanyl]oxy]-2-oxanyl]oxy]-6,9,11-trihydroxy-9-(2-hydroxy-1-oxoethyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@@H]1CCCCO1 KMSKQZKKOZQFFG-YXRRJAAWSA-N 0.000 description 2
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 description 2
- 239000001211 (E)-4-phenylbut-3-en-2-one Substances 0.000 description 2
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- MYBLAOJMRYYKMS-RTRLPJTCSA-N 1-(2-chloroethyl)-1-nitroso-3-[(3r,4r,5s,6r)-2,4,5-trihydroxy-6-(hydroxymethyl)oxan-3-yl]urea Chemical compound OC[C@H]1OC(O)[C@H](NC(=O)N(CCCl)N=O)[C@@H](O)[C@@H]1O MYBLAOJMRYYKMS-RTRLPJTCSA-N 0.000 description 2
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 2
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 2
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 2
- AIXGNRNTXUKZLC-UHFFFAOYSA-N 2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C(F)=C1 AIXGNRNTXUKZLC-UHFFFAOYSA-N 0.000 description 2
- SDTMFDGELKWGFT-UHFFFAOYSA-N 2-methylpropan-2-olate Chemical compound CC(C)(C)[O-] SDTMFDGELKWGFT-UHFFFAOYSA-N 0.000 description 2
- REZBRRWYFOIXJN-UHFFFAOYSA-N 3-(4-aminophenyl)-5-bromopyridin-2-amine Chemical compound C1=CC(N)=CC=C1C1=CC(Br)=CN=C1N REZBRRWYFOIXJN-UHFFFAOYSA-N 0.000 description 2
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 2
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 2
- HUTRLPYFPQSQBA-UHFFFAOYSA-N 3-bromo-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(Br)=C1 HUTRLPYFPQSQBA-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- KCHQXPGUJBVNTN-UHFFFAOYSA-N 4,4-diphenylbut-3-en-2-one Chemical compound C=1C=CC=CC=1C(=CC(=O)C)C1=CC=CC=C1 KCHQXPGUJBVNTN-UHFFFAOYSA-N 0.000 description 2
- ZBWDZCYDOMKEQA-UHFFFAOYSA-N 4-(4-methylphenyl)-3-oxobutanoic acid Chemical compound CC1=CC=C(CC(=O)CC(O)=O)C=C1 ZBWDZCYDOMKEQA-UHFFFAOYSA-N 0.000 description 2
- WHSIIJQOEGXWSN-RLTMCGQMSA-N 4-bromo-2,3,5-trideuterio-6-(trideuteriomethoxy)phenol Chemical compound BrC1=C(C(=C(C(=C1[2H])OC([2H])([2H])[2H])O)[2H])[2H] WHSIIJQOEGXWSN-RLTMCGQMSA-N 0.000 description 2
- NRTSMZFIDWZGFJ-UHFFFAOYSA-N 5-(4-cyclopropyl-2-fluorophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound C1(CC1)C1=CC(=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O)F NRTSMZFIDWZGFJ-UHFFFAOYSA-N 0.000 description 2
- ZYDXJCGHPMGSOG-UHFFFAOYSA-N 5-bromo-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC(Br)=CN=C1N ZYDXJCGHPMGSOG-UHFFFAOYSA-N 0.000 description 2
- WOVKYSAHUYNSMH-RRKCRQDMSA-N 5-bromodeoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-RRKCRQDMSA-N 0.000 description 2
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- RURLVUZRUFHCJO-UHFFFAOYSA-N Chromomycin A3 Natural products COC(C1Cc2cc3cc(OC4CC(OC(=O)C)C(OC5CC(O)C(OC)C(C)O5)C(C)O4)c(C)c(O)c3c(O)c2C(=O)C1OC6CC(OC7CC(C)(O)C(OC(=O)C)C(C)O7)C(O)C(C)O6)C(=O)C(O)C(C)O RURLVUZRUFHCJO-UHFFFAOYSA-N 0.000 description 2
- MPONAPFARZGDTH-UHFFFAOYSA-N Cl.OS(O)=O Chemical compound Cl.OS(O)=O MPONAPFARZGDTH-UHFFFAOYSA-N 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 101150039808 Egfr gene Proteins 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 2
- 208000032612 Glial tumor Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 102000005720 Glutathione transferase Human genes 0.000 description 2
- 108010070675 Glutathione transferase Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 2
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 2
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 2
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- SBQHEYGRLCNAMO-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-bromo-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)Br)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC SBQHEYGRLCNAMO-UHFFFAOYSA-N 0.000 description 2
- MVHPGOCNSYOLJS-UHFFFAOYSA-N N-[6-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]pyridin-3-yl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=NC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC MVHPGOCNSYOLJS-UHFFFAOYSA-N 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- 150000001204 N-oxides Chemical class 0.000 description 2
- XKDGOMGVZTUHGZ-UHFFFAOYSA-N NC(C(C1)=CN(CC2CCOCC2)C=C1C1=NC=CC=C1)=O.[O] Chemical compound NC(C(C1)=CN(CC2CCOCC2)C=C1C1=NC=CC=C1)=O.[O] XKDGOMGVZTUHGZ-UHFFFAOYSA-N 0.000 description 2
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 108010057150 Peplomycin Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 206010038910 Retinitis Diseases 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 108091027967 Small hairpin RNA Proteins 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 208000007536 Thrombosis Diseases 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- 208000005475 Vascular calcification Diseases 0.000 description 2
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 2
- 229960004176 aclarubicin Drugs 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 238000005576 amination reaction Methods 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 125000002344 aminooxy group Chemical group [H]N([H])O[*] 0.000 description 2
- 229940045799 anthracyclines and related substance Drugs 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 230000004900 autophagic degradation Effects 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 229930008407 benzylideneacetone Natural products 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000004709 cell invasion Effects 0.000 description 2
- 239000013592 cell lysate Substances 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229960005395 cetuximab Drugs 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 229960001480 chlorozotocin Drugs 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- ZYVSOIYQKUDENJ-WKSBCEQHSA-N chromomycin A3 Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@@H]1OC(C)=O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@@H](O)[C@H](O[C@@H]3O[C@@H](C)[C@H](OC(C)=O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@@H]1C[C@@H](O)[C@@H](OC)[C@@H](C)O1 ZYVSOIYQKUDENJ-WKSBCEQHSA-N 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 201000006754 cone-rod dystrophy Diseases 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 2
- YKGMKSIHIVVYKY-UHFFFAOYSA-N dabrafenib mesylate Chemical compound CS(O)(=O)=O.S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 YKGMKSIHIVVYKY-UHFFFAOYSA-N 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 229960002448 dasatinib Drugs 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 239000000032 diagnostic agent Substances 0.000 description 2
- 229940039227 diagnostic agent Drugs 0.000 description 2
- BSHICDXRSZQYBP-UHFFFAOYSA-N dichloromethane;palladium(2+) Chemical compound [Pd+2].ClCCl BSHICDXRSZQYBP-UHFFFAOYSA-N 0.000 description 2
- 125000006222 dimethylaminomethyl group Chemical group [H]C([H])([H])N(C([H])([H])[H])C([H])([H])* 0.000 description 2
- GPAYUJZHTULNBE-UHFFFAOYSA-N diphenylphosphine Chemical compound C=1C=CC=CC=1PC1=CC=CC=C1 GPAYUJZHTULNBE-UHFFFAOYSA-N 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 229940121647 egfr inhibitor Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- YJGVMLPVUAXIQN-UHFFFAOYSA-N epipodophyllotoxin Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YJGVMLPVUAXIQN-UHFFFAOYSA-N 0.000 description 2
- 108700021358 erbB-1 Genes Proteins 0.000 description 2
- 229960002568 ethinylestradiol Drugs 0.000 description 2
- PYUFYLOTFMNRSN-UHFFFAOYSA-N ethyl 4-(5-methylpyridin-2-yl)-3-oxobutanoate Chemical compound CC=1C=CC(=NC=1)CC(CC(=O)OCC)=O PYUFYLOTFMNRSN-UHFFFAOYSA-N 0.000 description 2
- UIKUIBBGEJWAIP-UHFFFAOYSA-N ethyl 5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound CC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OCC)=O UIKUIBBGEJWAIP-UHFFFAOYSA-N 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 210000004211 gastric acid Anatomy 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 201000011066 hemangioma Diseases 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 230000036210 malignancy Effects 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- IZXGZAJMDLJLMF-UHFFFAOYSA-N methylaminomethanol Chemical class CNCO IZXGZAJMDLJLMF-UHFFFAOYSA-N 0.000 description 2
- 230000011987 methylation Effects 0.000 description 2
- 238000007069 methylation reaction Methods 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 2
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 2
- 229960001346 nilotinib Drugs 0.000 description 2
- 229950010203 nimotuzumab Drugs 0.000 description 2
- 229960001420 nimustine Drugs 0.000 description 2
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- QJPQVXSHYBGQGM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QJPQVXSHYBGQGM-UHFFFAOYSA-N 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- DPBLXKKOBLCELK-UHFFFAOYSA-N pentan-1-amine Chemical compound CCCCCN DPBLXKKOBLCELK-UHFFFAOYSA-N 0.000 description 2
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 2
- 229950003180 peplomycin Drugs 0.000 description 2
- TWHXWYVOWJCXSI-UHFFFAOYSA-N phosphoric acid;hydrate Chemical compound O.OP(O)(O)=O TWHXWYVOWJCXSI-UHFFFAOYSA-N 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 238000011533 pre-incubation Methods 0.000 description 2
- 238000012746 preparative thin layer chromatography Methods 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 239000001294 propane Substances 0.000 description 2
- 239000012264 purified product Substances 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- 229960003440 semustine Drugs 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 229960003787 sorafenib Drugs 0.000 description 2
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 2
- 229960001796 sunitinib Drugs 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 229960001674 tegafur Drugs 0.000 description 2
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 2
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 2
- 229960001278 teniposide Drugs 0.000 description 2
- 229960004072 thrombin Drugs 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- BWHOZHOGCMHOBV-BQYQJAHWSA-N trans-benzylideneacetone Chemical compound CC(=O)\C=C\C1=CC=CC=C1 BWHOZHOGCMHOBV-BQYQJAHWSA-N 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- RCVDPBFUMYUKPB-UHFFFAOYSA-N (3,4-dimethoxyphenyl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=C1OC RCVDPBFUMYUKPB-UHFFFAOYSA-N 0.000 description 1
- WPVBHUUZDFUIJA-UHFFFAOYSA-N (3-fluoro-4-methylphenyl)boronic acid Chemical compound CC1=CC=C(B(O)O)C=C1F WPVBHUUZDFUIJA-UHFFFAOYSA-N 0.000 description 1
- BMQDAIUNAGXSKR-UHFFFAOYSA-N (3-hydroxy-2,3-dimethylbutan-2-yl)oxyboronic acid Chemical compound CC(C)(O)C(C)(C)OB(O)O BMQDAIUNAGXSKR-UHFFFAOYSA-N 0.000 description 1
- YJGVMLPVUAXIQN-LGWHJFRWSA-N (5s,5ar,8ar,9r)-5-hydroxy-9-(3,4,5-trimethoxyphenyl)-5a,6,8a,9-tetrahydro-5h-[2]benzofuro[5,6-f][1,3]benzodioxol-8-one Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-LGWHJFRWSA-N 0.000 description 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- HKBJSSIHFXNWFN-UHFFFAOYSA-N 1,3-diphenylbut-3-en-2-one Chemical compound C=1C=CC=CC=1C(=C)C(=O)CC1=CC=CC=C1 HKBJSSIHFXNWFN-UHFFFAOYSA-N 0.000 description 1
- OMRFOTIFDZORHU-UHFFFAOYSA-N 1-(oxan-4-ylmethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CN(CC2CCOCC2)N=C1 OMRFOTIFDZORHU-UHFFFAOYSA-N 0.000 description 1
- GFTXALYVNPNJLF-UHFFFAOYSA-N 1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound O=C1C(=CN(C=C1)CC1CCOCC1)C(=O)N GFTXALYVNPNJLF-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- MEXZZLMLPPPMCH-UHFFFAOYSA-N 1-phenyl-n-(2-phenylethoxy)methanamine Chemical class C=1C=CC=CC=1CNOCCC1=CC=CC=C1 MEXZZLMLPPPMCH-UHFFFAOYSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 1
- PNBIYFPZODYMOO-UHFFFAOYSA-N 2-(4-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=C(Br)C=C1F PNBIYFPZODYMOO-UHFFFAOYSA-N 0.000 description 1
- QVKJPKIMVKZJGM-UHFFFAOYSA-N 2-(4-cyclopropyl-2-fluorophenyl)acetic acid Chemical compound C1(CC1)C1=CC(=C(C=C1)CC(=O)O)F QVKJPKIMVKZJGM-UHFFFAOYSA-N 0.000 description 1
- GXXXUZIRGXYDFP-UHFFFAOYSA-N 2-(4-methylphenyl)acetic acid Chemical compound CC1=CC=C(CC(O)=O)C=C1 GXXXUZIRGXYDFP-UHFFFAOYSA-N 0.000 description 1
- TYHTUJMBDHMENY-UHFFFAOYSA-N 2-(5-methylpyridin-2-yl)acetic acid Chemical compound CC1=CC=C(CC(O)=O)N=C1 TYHTUJMBDHMENY-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- WFSJROCEOJANPD-UHFFFAOYSA-N 2-methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound C1=C(O)C(OC)=CC(B2OC(C)(C)C(C)(C)O2)=C1 WFSJROCEOJANPD-UHFFFAOYSA-N 0.000 description 1
- JEECGGXGHJUYMN-UHFFFAOYSA-N 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound C1=NC(C)=CC=C1B1OC(C)(C)C(C)(C)O1 JEECGGXGHJUYMN-UHFFFAOYSA-N 0.000 description 1
- XQQBUAPQHNYYRS-UHFFFAOYSA-N 2-methylthiophene Chemical compound CC1=CC=CS1 XQQBUAPQHNYYRS-UHFFFAOYSA-N 0.000 description 1
- DWYHDSLIWMUSOO-UHFFFAOYSA-N 2-phenyl-1h-benzimidazole Chemical group C1=CC=CC=C1C1=NC2=CC=CC=C2N1 DWYHDSLIWMUSOO-UHFFFAOYSA-N 0.000 description 1
- YCIMNLLNPGFGHC-RHQRLBAQSA-N 3,4,5,6-tetradeuteriobenzene-1,2-diol Chemical compound [2H]C1=C([2H])C([2H])=C(O)C(O)=C1[2H] YCIMNLLNPGFGHC-RHQRLBAQSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- RUDFGVCQAZXNAJ-UHFFFAOYSA-N 3-(4-amino-2-fluorophenyl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound C1=C(OC)C(OC)=CC=C1C1=CN=C(N)C(C=2C(=CC(N)=CC=2)F)=C1 RUDFGVCQAZXNAJ-UHFFFAOYSA-N 0.000 description 1
- WOUBEGBESRQENP-UHFFFAOYSA-N 3-(4-amino-3-fluorophenyl)-4-methoxypyridin-2-amine Chemical compound NC1=C(C=C(C=C1)C=1C(=NC=CC=1OC)N)F WOUBEGBESRQENP-UHFFFAOYSA-N 0.000 description 1
- SHIZYMRFBUULOJ-UHFFFAOYSA-N 3-(4-amino-3-fluorophenyl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound NC1=C(C=C(C=C1)C=1C(=NC=C(C=1)C1=CC(=C(C=C1)OC)OC)N)F SHIZYMRFBUULOJ-UHFFFAOYSA-N 0.000 description 1
- QAOPVULVILQLPY-UHFFFAOYSA-N 3-(5-amino-3-fluoropyridin-2-yl)-5-(3,4-dimethoxyphenyl)pyridin-2-amine Chemical compound COC=1C=C(C=CC=1OC)C=1C=C(C(=NC=1)N)C1=NC=C(C=C1F)N QAOPVULVILQLPY-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OYZOHRSCEASBER-UHFFFAOYSA-N 3-bromo-5-iodopyridin-2-amine Chemical compound NC1=NC=C(I)C=C1Br OYZOHRSCEASBER-UHFFFAOYSA-N 0.000 description 1
- JZMJGLPDACZKBC-UHFFFAOYSA-N 3-chloro-5-iodopyridine Chemical compound ClC1=CN=CC(I)=C1 JZMJGLPDACZKBC-UHFFFAOYSA-N 0.000 description 1
- FLMNWVXAEGUVNY-UHFFFAOYSA-N 3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C=C1F FLMNWVXAEGUVNY-UHFFFAOYSA-N 0.000 description 1
- QBPYHICVLNIUPW-UHFFFAOYSA-N 3-iodo-4-methoxypyridin-2-amine Chemical compound COC1=CC=NC(N)=C1I QBPYHICVLNIUPW-UHFFFAOYSA-N 0.000 description 1
- FPHXYKLKNOEKTQ-UHFFFAOYSA-N 4,4-diphenylbutan-2-one Chemical compound C=1C=CC=CC=1C(CC(=O)C)C1=CC=CC=C1 FPHXYKLKNOEKTQ-UHFFFAOYSA-N 0.000 description 1
- TVOJIBGZFYMWDT-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1h-pyrazole Chemical compound O1C(C)(C)C(C)(C)OB1C1=CNN=C1 TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 1
- ZANPJXNYBVVNSD-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(N)C=C1 ZANPJXNYBVVNSD-UHFFFAOYSA-N 0.000 description 1
- XHLXYECYFZTASB-UHFFFAOYSA-N 4-(4-bromophenyl)-3-oxobutanoic acid Chemical compound OC(=O)CC(=O)CC1=CC=C(Br)C=C1 XHLXYECYFZTASB-UHFFFAOYSA-N 0.000 description 1
- LMOOYAKLEOGKJR-UHFFFAOYSA-N 4-(bromomethyl)oxane Chemical compound BrCC1CCOCC1 LMOOYAKLEOGKJR-UHFFFAOYSA-N 0.000 description 1
- QOWSWEBLNVACCL-UHFFFAOYSA-N 4-Bromophenyl acetate Chemical compound OC(=O)CC1=CC=C(Br)C=C1 QOWSWEBLNVACCL-UHFFFAOYSA-N 0.000 description 1
- XWNSFEAWWGGSKJ-UHFFFAOYSA-N 4-acetyl-4-methylheptanedinitrile Chemical compound N#CCCC(C)(C(=O)C)CCC#N XWNSFEAWWGGSKJ-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- 125000000590 4-methylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- LLPFPLXIDMRGBA-UHFFFAOYSA-N 5-(4-bromophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)O)=O LLPFPLXIDMRGBA-UHFFFAOYSA-N 0.000 description 1
- NGUDGIXCTSONFQ-UHFFFAOYSA-N 5-(5-methylpyridin-2-yl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylic acid Chemical group CC=1C=CC(=NC=1)C1=CN(C=C(C1=O)C(=O)O)CC1CCOCC1 NGUDGIXCTSONFQ-UHFFFAOYSA-N 0.000 description 1
- IVILGUFRMDBUEQ-UHFFFAOYSA-N 5-iodopyridin-2-amine Chemical compound NC1=CC=C(I)C=N1 IVILGUFRMDBUEQ-UHFFFAOYSA-N 0.000 description 1
- GMMWNTHAIOJBSD-UHFFFAOYSA-N 6-chloro-4-n-(2,3-dimethylphenyl)-2-n,2-n-dimethylpyrimidine-2,4-diamine Chemical group CN(C)C1=NC(Cl)=CC(NC=2C(=C(C)C=CC=2)C)=N1 GMMWNTHAIOJBSD-UHFFFAOYSA-N 0.000 description 1
- HAZMBQGWYYGRES-UHFFFAOYSA-N 6-chloro-5-fluoropyridin-3-amine Chemical compound NC1=CN=C(Cl)C(F)=C1 HAZMBQGWYYGRES-UHFFFAOYSA-N 0.000 description 1
- NKGPJODWTZCHGF-KQYNXXCUSA-N 6-thioinosinic acid Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(S)=C2N=C1 NKGPJODWTZCHGF-KQYNXXCUSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- WOVKYSAHUYNSMH-UHFFFAOYSA-N BROMODEOXYURIDINE Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-UHFFFAOYSA-N 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- 208000031648 Body Weight Changes Diseases 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- JHOGQJHPYMBQKC-UHFFFAOYSA-N C1(=CC=CC=C1)P(CCCP(C1=CC=CC=C1)C1=CC=CC=C1)C1=CC=CC=C1.C1(=CC=CC=C1)P(CCCP(C1=CC=CC=C1)C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound C1(=CC=CC=C1)P(CCCP(C1=CC=CC=C1)C1=CC=CC=C1)C1=CC=CC=C1.C1(=CC=CC=C1)P(CCCP(C1=CC=CC=C1)C1=CC=CC=C1)C1=CC=CC=C1 JHOGQJHPYMBQKC-UHFFFAOYSA-N 0.000 description 1
- DFVYOEWFOJYXQS-UHFFFAOYSA-N CC[K].OC(=O)CC(O)=O Chemical compound CC[K].OC(=O)CC(O)=O DFVYOEWFOJYXQS-UHFFFAOYSA-N 0.000 description 1
- JCTVKBUHQNZYPU-UHFFFAOYSA-N COC1=C(C=C(C=C1)C2=CC(=CN=C2)Cl)OC Chemical compound COC1=C(C=C(C=C1)C2=CC(=CN=C2)Cl)OC JCTVKBUHQNZYPU-UHFFFAOYSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- YCGFHHACUCVQPI-UMSFTDKQSA-N Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc(cc2)ccc2-c2cc(-c(cc3OC)ccc3OC[C@H]3OCCOC3)cnc2N)=O)C1=O Chemical compound Cc(cc1)ccc1C1=CN(CC2CCOCC2)C=C(C(Nc(cc2)ccc2-c2cc(-c(cc3OC)ccc3OC[C@H]3OCCOC3)cnc2N)=O)C1=O YCGFHHACUCVQPI-UMSFTDKQSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical class C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283074 Equus asinus Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical class NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 206010015548 Euthanasia Diseases 0.000 description 1
- 206010015719 Exsanguination Diseases 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 102100031487 Growth arrest-specific protein 6 Human genes 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 208000017891 HER2 positive breast carcinoma Diseases 0.000 description 1
- 108010068250 Herpes Simplex Virus Protein Vmw65 Proteins 0.000 description 1
- 101000923005 Homo sapiens Growth arrest-specific protein 6 Proteins 0.000 description 1
- 101000973901 Homo sapiens Tyrosine-protein kinase Mer Proteins 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- UCEQXRCJXIVODC-PMACEKPBSA-N LSM-1131 Chemical compound C1CCC2=CC=CC3=C2N1C=C3[C@@H]1C(=O)NC(=O)[C@H]1C1=CNC2=CC=CC=C12 UCEQXRCJXIVODC-PMACEKPBSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- FQISKWAFAHGMGT-SGJOWKDISA-M Methylprednisolone sodium succinate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCC([O-])=O)CC[C@H]21 FQISKWAFAHGMGT-SGJOWKDISA-M 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- YYGLURGKDYINIT-UHFFFAOYSA-N N-(5-iodopyridin-2-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound IC=1C=CC(=NC=1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 YYGLURGKDYINIT-UHFFFAOYSA-N 0.000 description 1
- RJAXTNPUCKJOER-UHFFFAOYSA-N N-(6-iodopyridin-3-yl)-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound IC1=CC=C(C=N1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1 RJAXTNPUCKJOER-UHFFFAOYSA-N 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- NDHJRDQILUOWLR-UHFFFAOYSA-N N-[4-(2-amino-5-bromopyridin-3-yl)-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=C(C=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)F)Br NDHJRDQILUOWLR-UHFFFAOYSA-N 0.000 description 1
- NATXQLRQRJHHRS-UHFFFAOYSA-N N-[4-(2-amino-5-bromopyridin-3-yl)phenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)Br NATXQLRQRJHHRS-UHFFFAOYSA-N 0.000 description 1
- VNBRGSXVFBYQNN-UHFFFAOYSA-N N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-4-ethoxy-1-(4-fluorophenyl)-2-oxo-3-pyridinecarboxamide Chemical compound O=C1C(C(=O)NC=2C=C(F)C(OC=3C(=C(N)N=CC=3)Cl)=CC=2)=C(OCC)C=CN1C1=CC=C(F)C=C1 VNBRGSXVFBYQNN-UHFFFAOYSA-N 0.000 description 1
- GEAJYTNNHUHUPD-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]-3-fluorophenyl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound C1(C=2C=C(C3=CC=C(OC)C(OC)=C3)C=NC=2N)=CC=C(NC(=O)C=2C(=O)C(C3=CC=C(C=C3)C)=CN(C=2)CC2CCOCC2)C=C1F GEAJYTNNHUHUPD-UHFFFAOYSA-N 0.000 description 1
- HVZYRAXHTSGBOE-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(4-cyclopropylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C1CC1)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC HVZYRAXHTSGBOE-UHFFFAOYSA-N 0.000 description 1
- JXIOZYRODOOPLU-UHFFFAOYSA-N N-[4-[2-amino-5-(3,4-dimethoxyphenyl)pyridin-3-yl]phenyl]-5-(5-methylthiophen-2-yl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=CC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C=1SC(=CC=1)C)CC1CCOCC1)C1=CC(=C(C=C1)OC)OC JXIOZYRODOOPLU-UHFFFAOYSA-N 0.000 description 1
- WSNUZOLBHLCQOI-UHFFFAOYSA-N N-[6-(2-amino-5-bromopyridin-3-yl)pyridin-3-yl]-5-(4-methylphenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxamide Chemical compound NC1=NC=C(C=C1C1=NC=C(C=C1)NC(=O)C1=CN(C=C(C1=O)C1=CC=C(C=C1)C)CC1CCOCC1)Br WSNUZOLBHLCQOI-UHFFFAOYSA-N 0.000 description 1
- KCTZOTUQSGYWLV-UHFFFAOYSA-N N1C=NC=C2N=CC=C21 Chemical group N1C=NC=C2N=CC=C21 KCTZOTUQSGYWLV-UHFFFAOYSA-N 0.000 description 1
- 101710204212 Neocarzinostatin Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- AFLXUQUGROGEFA-UHFFFAOYSA-N Nitrogen mustard N-oxide Chemical compound ClCC[N+]([O-])(C)CCCl AFLXUQUGROGEFA-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- BZSGJAREDHFGPI-UHFFFAOYSA-N O1CCCC1.CB1OB(OB(O1)C)C Chemical compound O1CCCC1.CB1OB(OB(O1)C)C BZSGJAREDHFGPI-UHFFFAOYSA-N 0.000 description 1
- YRXHPYYZMCPPGK-UHFFFAOYSA-N O=C1C(C(C=C2)=CC=C2Br)=CNC=C1 Chemical compound O=C1C(C(C=C2)=CC=C2Br)=CNC=C1 YRXHPYYZMCPPGK-UHFFFAOYSA-N 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 229940122907 Phosphatase inhibitor Drugs 0.000 description 1
- 239000004153 Potassium bromate Substances 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 229920000519 Sizofiran Polymers 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 208000000277 Splenic Neoplasms Diseases 0.000 description 1
- 108091005729 TAM receptors Proteins 0.000 description 1
- 108091077436 Tam family Proteins 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 1
- 208000000728 Thymus Neoplasms Diseases 0.000 description 1
- MSCCTZZBYHQMQJ-AZAGJHQNSA-N Tocopheryl nicotinate Chemical compound C([C@@](OC1=C(C)C=2C)(C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)CC1=C(C)C=2OC(=O)C1=CC=CN=C1 MSCCTZZBYHQMQJ-AZAGJHQNSA-N 0.000 description 1
- CDJJKTLOZJAGIZ-UHFFFAOYSA-N Tolylacetate Chemical compound CC(=O)OC1=CC=C(C)C=C1 CDJJKTLOZJAGIZ-UHFFFAOYSA-N 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 1
- 229930003268 Vitamin C Natural products 0.000 description 1
- 229950002684 aceglatone Drugs 0.000 description 1
- ZOZKYEHVNDEUCO-XUTVFYLZSA-N aceglatone Chemical compound O1C(=O)[C@H](OC(C)=O)[C@@H]2OC(=O)[C@@H](OC(=O)C)[C@@H]21 ZOZKYEHVNDEUCO-XUTVFYLZSA-N 0.000 description 1
- MNZMECMQTYGSOI-UHFFFAOYSA-N acetic acid;hydron;bromide Chemical compound Br.CC(O)=O MNZMECMQTYGSOI-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 108010023337 axl receptor tyrosine kinase Proteins 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical class C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- MVIOINXPSFUJEN-UHFFFAOYSA-N benzenesulfonic acid;hydrate Chemical compound O.OS(=O)(=O)C1=CC=CC=C1 MVIOINXPSFUJEN-UHFFFAOYSA-N 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- 230000004579 body weight change Effects 0.000 description 1
- PLJIUMGEXZJCGD-UHFFFAOYSA-N boric acid;1,2-dimethoxybenzene Chemical compound OB(O)O.COC1=CC=CC=C1OC PLJIUMGEXZJCGD-UHFFFAOYSA-N 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229950004398 broxuridine Drugs 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 210000005252 bulbus oculi Anatomy 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 230000006208 butylation Effects 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 239000003560 cancer drug Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000005587 carbonate group Chemical group 0.000 description 1
- JYYOBHFYCIDXHH-UHFFFAOYSA-N carbonic acid;hydrate Chemical compound O.OC(O)=O JYYOBHFYCIDXHH-UHFFFAOYSA-N 0.000 description 1
- 230000006315 carbonylation Effects 0.000 description 1
- 238000005810 carbonylation reaction Methods 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 239000012829 chemotherapy agent Substances 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- VUHJZBBCZGVNDZ-TTYLFXKOSA-N chlormadinone Chemical compound C1=C(Cl)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 VUHJZBBCZGVNDZ-TTYLFXKOSA-N 0.000 description 1
- 229960003996 chlormadinone Drugs 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 150000003946 cyclohexylamines Chemical class 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- FMGSKLZLMKYGDP-USOAJAOKSA-N dehydroepiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC=C21 FMGSKLZLMKYGDP-USOAJAOKSA-N 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 1
- YZDNEKBXKIZKRK-UHFFFAOYSA-N dicyclohexyl-[2-[2,4-di(propan-2-yl)phenyl]-1-propan-2-ylcyclohexa-2,4-dien-1-yl]phosphane Chemical group CC(C)C1=CC(C(C)C)=CC=C1C1=CC=CCC1(C(C)C)P(C1CCCCC1)C1CCCCC1 YZDNEKBXKIZKRK-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical class OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 150000005332 diethylamines Chemical class 0.000 description 1
- NLORYLAYLIXTID-ISLYRVAYSA-N diethylstilbestrol diphosphate Chemical compound C=1C=C(OP(O)(O)=O)C=CC=1C(/CC)=C(\CC)C1=CC=C(OP(O)(O)=O)C=C1 NLORYLAYLIXTID-ISLYRVAYSA-N 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 201000006828 endometrial hyperplasia Diseases 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- HNGDOSBFYRVIEY-UHFFFAOYSA-N ethanesulfonic acid;hydrate Chemical compound O.CCS(O)(=O)=O HNGDOSBFYRVIEY-UHFFFAOYSA-N 0.000 description 1
- FXPHJTKVWZVEGA-UHFFFAOYSA-N ethenyl hydrogen carbonate Chemical group OC(=O)OC=C FXPHJTKVWZVEGA-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- BRYNITBKFYVFBV-UHFFFAOYSA-N ethyl 4-(4-bromophenyl)-3-oxobutanoate Chemical compound CCOC(=O)CC(=O)CC1=CC=C(Br)C=C1 BRYNITBKFYVFBV-UHFFFAOYSA-N 0.000 description 1
- KKAFIMYLMXUFKH-UHFFFAOYSA-N ethyl 5-(4-bromophenyl)-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound BrC1=CC=C(C=C1)C=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OCC)=O KKAFIMYLMXUFKH-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000001508 eye Anatomy 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000834 fixative Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 229960000297 fosfestrol Drugs 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 150000002301 glucosamine derivatives Chemical class 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 229960002743 glutamine Drugs 0.000 description 1
- 230000009422 growth inhibiting effect Effects 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000003505 heat denaturation Methods 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 1
- 230000036732 histological change Effects 0.000 description 1
- 102000045684 human MERTK Human genes 0.000 description 1
- GVLGAFRNYJVHBC-UHFFFAOYSA-N hydrate;hydrobromide Chemical compound O.Br GVLGAFRNYJVHBC-UHFFFAOYSA-N 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- 238000011503 in vivo imaging Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical group C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- ZCYVEMRRCGMTRW-YPZZEJLDSA-N iodine-125 Chemical compound [125I] ZCYVEMRRCGMTRW-YPZZEJLDSA-N 0.000 description 1
- 229940044173 iodine-125 Drugs 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- FRQMUZJSZHZSGN-HBNHAYAOSA-N medroxyprogesterone Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FRQMUZJSZHZSGN-HBNHAYAOSA-N 0.000 description 1
- 229960004616 medroxyprogesterone Drugs 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229960003578 metenolone Drugs 0.000 description 1
- OIXMUQLVDNPHNS-UHFFFAOYSA-N methanesulfonic acid;hydrate Chemical compound O.CS(O)(=O)=O OIXMUQLVDNPHNS-UHFFFAOYSA-N 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- ANJQEDFWRSLVBR-VHUDCFPWSA-N methenolone Chemical compound C1C[C@@H]2[C@@]3(C)C(C)=CC(=O)C[C@@H]3CC[C@H]2[C@@H]2CC[C@H](O)[C@]21C ANJQEDFWRSLVBR-VHUDCFPWSA-N 0.000 description 1
- 238000006063 methoxycarbonylation reaction Methods 0.000 description 1
- CYRBTLNOTDUNLV-UHFFFAOYSA-N methyl 1,2-dihydropyridine-3-carboxylate Chemical compound COC(=O)C1=CC=CNC1 CYRBTLNOTDUNLV-UHFFFAOYSA-N 0.000 description 1
- LSEMVPNOOZDQLJ-UHFFFAOYSA-N methyl 5-bromo-1-(oxan-4-ylmethyl)-4-oxopyridine-3-carboxylate Chemical compound BrC=1C(C(=CN(C=1)CC1CCOCC1)C(=O)OC)=O LSEMVPNOOZDQLJ-UHFFFAOYSA-N 0.000 description 1
- MOSJHTAFHHUNRB-UHFFFAOYSA-N methyl 5-bromo-4-oxo-1h-pyridine-3-carboxylate Chemical compound COC(=O)C1=CNC=C(Br)C1=O MOSJHTAFHHUNRB-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical group [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 229960004719 nandrolone Drugs 0.000 description 1
- NPAGDVCDWIYMMC-IZPLOLCNSA-N nandrolone Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 NPAGDVCDWIYMMC-IZPLOLCNSA-N 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 208000025189 neoplasm of testis Diseases 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- FGHSTPNOXKDLKU-UHFFFAOYSA-N nitric acid;hydrate Chemical compound O.O[N+]([O-])=O FGHSTPNOXKDLKU-UHFFFAOYSA-N 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229960005244 oxymetholone Drugs 0.000 description 1
- ICMWWNHDUZJFDW-DHODBPELSA-N oxymetholone Chemical compound C([C@@H]1CC2)C(=O)\C(=C/O)C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@](C)(O)[C@@]2(C)CC1 ICMWWNHDUZJFDW-DHODBPELSA-N 0.000 description 1
- ICMWWNHDUZJFDW-UHFFFAOYSA-N oxymetholone Natural products C1CC2CC(=O)C(=CO)CC2(C)C2C1C1CCC(C)(O)C1(C)CC2 ICMWWNHDUZJFDW-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 229940100684 pentylamine Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- 125000005328 phosphinyl group Chemical group [PH2](=O)* 0.000 description 1
- 108091008695 photoreceptors Proteins 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- 229960001237 podophyllotoxin Drugs 0.000 description 1
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 description 1
- YVCVYCSAAZQOJI-UHFFFAOYSA-N podophyllotoxin Natural products COC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YVCVYCSAAZQOJI-UHFFFAOYSA-N 0.000 description 1
- 108010001062 polysaccharide-K Proteins 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229940094037 potassium bromate Drugs 0.000 description 1
- 235000019396 potassium bromate Nutrition 0.000 description 1
- WVUCPRGADMCTBN-UHFFFAOYSA-M potassium;3-ethoxy-3-oxopropanoate Chemical compound [K+].CCOC(=O)CC([O-])=O WVUCPRGADMCTBN-UHFFFAOYSA-M 0.000 description 1
- 229960002847 prasterone Drugs 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 230000004144 purine metabolism Effects 0.000 description 1
- 230000004147 pyrimidine metabolism Effects 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- JSTXDUPGNXVYPZ-UHFFFAOYSA-N ramosine Natural products CC1C2C(CC(=C)C3CC(O)C(O)(CO)C3C2OC1=O)OC(=O)C(=C)CO JSTXDUPGNXVYPZ-UHFFFAOYSA-N 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 239000013557 residual solvent Substances 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 229950001403 sizofiran Drugs 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 201000002471 spleen cancer Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 150000003431 steroids Chemical group 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 125000000565 sulfonamide group Chemical group 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 150000003527 tetrahydropyrans Chemical group 0.000 description 1
- 125000005942 tetrahydropyridyl group Chemical group 0.000 description 1
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical class C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 229950005976 tivantinib Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 150000003852 triazoles Chemical group 0.000 description 1
- OVCXRBARSPBVMC-UHFFFAOYSA-N triazolopyridine Chemical compound C=1N2C(C(C)C)=NN=C2C=CC=1C=1OC=NC=1C1=CC=C(F)C=C1 OVCXRBARSPBVMC-UHFFFAOYSA-N 0.000 description 1
- YWBFPKPWMSWWEA-UHFFFAOYSA-O triazolopyrimidine Chemical group BrC1=CC=CC(C=2N=C3N=CN[N+]3=C(NCC=3C=CN=CC=3)C=2)=C1 YWBFPKPWMSWWEA-UHFFFAOYSA-O 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- 239000003656 tris buffered saline Substances 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N urea group Chemical group NC(=O)N XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 235000019154 vitamin C Nutrition 0.000 description 1
- 239000011718 vitamin C Substances 0.000 description 1
- 229940045997 vitamin a Drugs 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
Images






















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/02—Sulfonic acids having sulfo groups bound to acyclic carbon atoms
- C07C309/03—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C309/04—Sulfonic acids having sulfo groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton containing only one sulfo group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/33—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems
- C07C309/34—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton of six-membered aromatic rings being part of condensed ring systems formed by two rings
- C07C309/35—Naphthalene sulfonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Urology & Nephrology (AREA)
- Physical Education & Sports Medicine (AREA)
- Endocrinology (AREA)
- Gastroenterology & Hepatology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Reproductive Health (AREA)
- Pulmonology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Dermatology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本發明之課題係提供一種新穎化合物或其鹽、或彼等之結晶,其可用於抑制Axl,而治療由Axl之功能亢進所致的疾病、治療與Axl之功能亢進相關的疾病、及/或治療伴隨Axl之功能亢進的疾病。
本發明之解決手段係提供具有各種取代基之以下述式(I)所示的具有四氫吡喃基甲基之吡啶酮衍生物或其鹽、或彼等之結晶(其中,式(I)中之R1
、R2
、R3
、R4
、R5
、W、X、Y、Z各自如說明書中所定義同義。)
Description
本發明係關於具有Axl抑制活性的化合物或其鹽。
Axl係一種受體型酪胺酸激酶,其屬於以成長停止特異性基因6(Gas6)蛋白質作為配位體的Tyro3-Axl-Mer(TAM)受體酪胺酸激酶家族(receptor tyrosine kinase family),當初係被認定為慢性骨髓性白血病中的轉形基因(transforming gene) (非專利文獻1)。
已有報告指出Gas6/Axl訊息傳導系統會調節細胞存活(cell survival)、細胞分裂、自噬作用(autophagy)、細胞移動(migration)、血管新生、血小板凝集、NK細胞分化等各式各樣的細胞性反應(非專利文獻2),亦已有多數報告指出於原發性結腸癌(非專利文獻3)、胃癌(非專利文獻4)、食道癌(非專利文獻5)、黑色素瘤(非專利文獻6)、卵巢癌(非專利文獻7)、腎臓癌(非專利文獻8)、子宮內膜癌(非專利文獻9)、甲狀腺癌(非專利文獻10)等之癌組織中會過度表現。又,亦已呈示Axl之存在與肺癌中之淋巴結狀態與分期以及乳癌中的ER表現有很大關係(非專利文獻11)。
此外,Axl亦已於免疫(非專利文獻12)、血小板功能(非專利文獻13)、精子形成(非專利文獻14)、血管鈣化(非專利文獻15)、凝血酶誘導血管平滑肌細胞(VSMC)增殖(非專利文獻16)、及各種之腎臓疾病,例如急性及慢性絲球體腎炎、糖尿病性腎病變及慢性同種移植排斥反應(非專利文獻17)方面顯示具有作用;Axl抑制劑不僅被期待有益於癌症(包含癌瘤(carcinoma)、肉瘤等之固態腫瘤及白血病及淋巴性惡性疾病)之治療,而且亦被期待有益於血管疾病(包含血栓症、動脈粥狀硬化症及再狹窄,但未限於此等)、腎臓疾病(包含急性及慢性絲球體腎炎、糖尿病性腎病變及移植排斥反應,但未限於此等)、及任意的血管生成為嚴重的疾病(包含糖尿病性視網膜病變、網膜症、乾癬、類風溼性關節炎、粉瘤(atheroma)、卡波西肉瘤(Kaposi’s sarcoma)及血管瘤,但未限於此等)等之多數疾病的治療。
另一方面,有就Axl所屬之TAM受體酪胺酸激酶家族之一員的Mer而言,由於其同型組合(homozygous)之突變而引起體染色體隱性色素性視網膜炎(autosomal recessive pigmentous retinitis)之報告(非專利文獻24);此外,也有Mer的某種突變係與孩童時期發病之錐狀細胞-桿狀細胞失養症(Rod-cone dystrophy)有關之報告(非專利文獻25)。
就抑制Axl的化合物而言,已有報告指出具有磺醯胺結構的化合物(專利文獻3)、具有吡咯并嘧啶結構的化合物(專利文獻4、專利文獻5)、具有吡啶及吡 結構的化合物(專利文獻6)、具有吡結構的化合物(專利文獻7)、具有吡基苯并咪唑結構的化合物(專利文獻8)、具有吲哚酮(indolinone)結構的化合物(專利文獻9)、具有三唑并吡啶及三唑并嘧啶結構的化合物(專利文獻10)、具有咪唑結構的化合物(專利文獻11)、具有三唑結構的化合物(專利文獻12、專利文獻13、專利文獻14、專利文獻15、專利文獻16、專利文獻17、專利文獻20、專利文獻24、專利文獻25、專利文獻26、專利文獻27、專利文獻28)、具有嘧啶二胺結構的化合物(專利文獻18)、具有嘧啶結構的化合物(專利文獻19、非專利文獻18、非專利文獻22)、具有喹啉基氧基苯基磺醯胺結構的化合物(專利文獻21)、具有喹啉結構的化合物(專利文獻22、專利文獻30、非專利文獻21)、具有吡啶結構的化合物(專利文獻23、專利文獻33、非專利文獻19)、具有脲結構的化合物(專利文獻29)、具有2,4-二取代芳基醯胺結構的化合物(非專利文獻20)、具有開環類固醇結構的化合物(非專利文獻23)、具有2環性嘧啶結構的化合物(專利文獻31、專利文獻32、專利文獻34)等。
[先前技術文獻]
[專利文獻]
結構的化合物(專利文獻6)、具有吡結構的化合物(專利文獻7)、具有吡基苯并咪唑結構的化合物(專利文獻8)、具有吲哚酮(indolinone)結構的化合物(專利文獻9)、具有三唑并吡啶及三唑并嘧啶結構的化合物(專利文獻10)、具有咪唑結構的化合物(專利文獻11)、具有三唑結構的化合物(專利文獻12、專利文獻13、專利文獻14、專利文獻15、專利文獻16、專利文獻17、專利文獻20、專利文獻24、專利文獻25、專利文獻26、專利文獻27、專利文獻28)、具有嘧啶二胺結構的化合物(專利文獻18)、具有嘧啶結構的化合物(專利文獻19、非專利文獻18、非專利文獻22)、具有喹啉基氧基苯基磺醯胺結構的化合物(專利文獻21)、具有喹啉結構的化合物(專利文獻22、專利文獻30、非專利文獻21)、具有吡啶結構的化合物(專利文獻23、專利文獻33、非專利文獻19)、具有脲結構的化合物(專利文獻29)、具有2,4-二取代芳基醯胺結構的化合物(非專利文獻20)、具有開環類固醇結構的化合物(非專利文獻23)、具有2環性嘧啶結構的化合物(專利文獻31、專利文獻32、專利文獻34)等。
[先前技術文獻]
[專利文獻]
[專利文獻1]國際公開公報第2008/025820號
[專利文獻2]國際公開公報第2008/074997號
[專利文獻3]國際公開公報第2008/128072號
[專利文獻4]US專利申請公開公報第20100204221號
[專利文獻5]國際公開公報第2010/090764號
[專利文獻6]國際公開公報第2009/053737號
[專利文獻7]國際公開公報第2009/007390號
[專利文獻8]國際公開公報第2009/024825號
[專利文獻9]國際公開公報第2007/057399號
[專利文獻10]國際公開公報第2009/047514號
[專利文獻11]國際公開公報第2009/058801號
[專利文獻12]國際公開公報第2008/083367號
[專利文獻13]國際公開公報第2008/083353號
[專利文獻14]國際公開公報第2010/005879號
[專利文獻15]國際公開公報第2008/083357號
[專利文獻16]國際公開公報第2008/083356號
[專利文獻17]國際公開公報第2008/083354號
[專利文獻18]國際公開公報第2008/045978號
[專利文獻19]國際公開公報第2007/070872號
[專利文獻20]國際公開公報第2007/030680號
[專利文獻21]國際公開公報第2011/045084號
[專利文獻22]國際公開公報第2009/127417號
[專利文獻23]國際公開公報第2007/066187號
[專利文獻24]國際公開公報第2009/054864號
[專利文獻25]國際公開公報第2010/005876號
[專利文獻26]國際公開公報第2009/054864號
[專利文獻27]US專利申請公開公報第20090111816號
[專利文獻28]US專利申請公開公報第20100168416號
[專利文獻29]國際公開公報第2009/138799號
[專利文獻30]US專利申請公開公報第20090274693號
[專利文獻31]US專利申請公開公報第20100069369號
[專利文獻32]US專利申請公開公報第20070142402號
[專利文獻33]國際公開公報第2013/115280號
[專利文獻34]國際公開公報第2013/162061號
[非專利文獻]
[非專利文獻1]O'Bryan et al., Mol. Cell. Biol., 11, 5031 (1991)
[非專利文獻2]Rachel MA Linger et al., Expert Opin. Ther. Targets, 14, 1073 (2010)
[非專利文獻3]Craven et al., Int. J. Cancer., 60, 791 (1995)
[非專利文獻4]Sawabu et al., Mol. Carcinog., 46, 155 (2007)
[非專利文獻5]Nemoto et al., Pathobiology, 65, 195 (1997)
[非專利文獻6]Quong et al., Melanoma Res., 4, 313 (1994)
[非專利文獻7]Sun et al., Oncology, 66, 450 (2004)
[非專利文獻8]Chung et al., DNA Cell Biol., 22, 533 (2003)
[非專利文獻9]Sun et al., Ann. Oncol., 14, 898 (2003)
[非專利文獻10]Ito et al., Thyroid, 9, 563 (1999)
[非專利文獻11]Berclaz et al., Ann. Oncol., 12, 819 (2001)
[非專利文獻12]Lu et al., Science, 293, 306 (2001)
[非專利文獻13]Angelillo-Scherrer et al., Nat. Med., 7, 215 (2001)
[非專利文獻14]Lu et al., Nature, 398, 723 (1999)
[非專利文獻15]Son et al., Eur. J. Pharmacol., 556, 1 (2007)
[非專利文獻16]Nakano et al., J. Biol. Chem., 270, 5702 (1995)
[非專利文獻17]Yanagita et al., J. Clin. Invest., 110, 239 (2002)
[非專利文獻18]AlexisMollard et al., Med. Chem. Lett., 2, 907 (2011)
[非專利文獻19]Gretchen M. Schroeder et al., J. Med. Chem., 52, 1251 (2009)
[非專利文獻20]Carl R. Illig et al., Bioorg. Med. Chem. Lett., 18, 1642 (2008)
[非專利文獻21]Yi-Xiang Zhang et al., Cancer Res., 68, 1905 (2008)
[非專利文獻22]D Mahadevan et al., Oncogene, 26, 3909 (2007)
[非專利文獻23]Daowan Lai et al., Bioorg. Med. Chem., 19, 6873 (2011)
[非專利文獻24]Ksantini M.,Eur J Ophthalmol., 22, 647 (2012)
[非專利文獻25]Mackay et al., Molecular Vision., 16, 369 (2010)
[發明欲解決之課題]
本發明係提供對Axl之抑制特異性高,安全性更高的Axl抑制化合物。又,本發明係提供含有Axl抑制化合物之以Axl之功能亢進作為原因的疾病之治療劑、與Axl之功能亢進有關的疾病之治療劑、及/或伴隨Axl之功能亢進的疾病之治療劑,例如抗癌劑。
[解決課題之手段]
上述專利文獻33之實施例9的化合物(以下為Compound A)被確認到雖具有高Axl抑制活性,但在小鼠長期間投與試驗中,卻引起不可逆的網膜光受體變性。對與Axl同為TAM家族之一員的Mer而言,鑑於有報告指出其不活化與網膜細胞變性的關連性、及專利文獻33之實施例9的化合物對Mer亦具有抑制活性,本發明者認為該化合物的Mer抑制係涉及視網膜毒性。
本發明者專心研討的結果,發現具有以下述通式(I)所表示結構的化合物或其鹽係Axl抑制特異性高、對Mer的抑制活性低,又發現在小鼠長期間投與試驗中亦不會引起視網膜毒性。
亦即,本發明係關於如下之[1]至[53]。
[1]一種以通式(I)所表示之化合物或其藥學上容許鹽, [式(I)中,
W、X、Y係各自獨立地表示氮原子、C-H、C-F或C-Cl;
Z係表示氮原子、C-H、C-F、C-Cl或C-C1
~C6
烷基;
R1
係表示下述式(II-1)或(II-2),
[式(I)中,
W、X、Y係各自獨立地表示氮原子、C-H、C-F或C-Cl;
Z係表示氮原子、C-H、C-F、C-Cl或C-C1
~C6
烷基;
R1
係表示下述式(II-1)或(II-2), (式(II-1)中,
Q係表示氮原子、C-H或C-F;
T係表示氮原子或C-H;
U係表示氮原子或C-H;
R6
係表示鹵素原子、C1
~C6
烷基、C3
~C6
環烷基、氰基或三氟甲氧基;
式(II-2)中,
V係表示硫原子或氧原子;
R7
係表示C1
~C6
烷基。)
R2
係表示氫原子、鹵素原子、C1
~C6
烷基、C1
~C6
烷氧基或氰基;
R3
係表示氫原子或C1
~C6
烷基;
R4
係表示氫原子、鹵素原子或C1
~C6
烷基;
R5
係表示氫原子或下述式(III-1)、(III-2)、(III-3)或(III-4),
(式(II-1)中,
Q係表示氮原子、C-H或C-F;
T係表示氮原子或C-H;
U係表示氮原子或C-H;
R6
係表示鹵素原子、C1
~C6
烷基、C3
~C6
環烷基、氰基或三氟甲氧基;
式(II-2)中,
V係表示硫原子或氧原子;
R7
係表示C1
~C6
烷基。)
R2
係表示氫原子、鹵素原子、C1
~C6
烷基、C1
~C6
烷氧基或氰基;
R3
係表示氫原子或C1
~C6
烷基;
R4
係表示氫原子、鹵素原子或C1
~C6
烷基;
R5
係表示氫原子或下述式(III-1)、(III-2)、(III-3)或(III-4), (式(III-1)中,
R8
及R12
係各自獨立地表示氫原子或氘原子;
R9
係表示氫原子、鹵素原子或C1
~C6
烷氧基;
R10
係表示氫原子、可經雜環烷基取代的C1
~C6
烷基或C1
~C6
可經可被烷基取代之雜環烷基取代的C1
~C6
烷氧基;
R11
係表示氫原子或C1
~C6
烷氧基或經氘取代的C1
~C6
烷氧基;
式(III-2)中,
R13
係表示可經雜環烷基取代的C1
~C6
烷基或雜環烷基;
式(III-3)中,
R14
係表示氫原子或C1
~C6
烷基;
R15
係表示氫原子、C1
~C6
烷基或C1
~C6
烷氧基;
R16
係表示氫原子或鹵素原子;
式(III-4)中,
R17
係表示C1
~C6
烷氧基;
R18
係表示C1
~C6
烷氧基)]。
[2]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二
(式(III-1)中,
R8
及R12
係各自獨立地表示氫原子或氘原子;
R9
係表示氫原子、鹵素原子或C1
~C6
烷氧基;
R10
係表示氫原子、可經雜環烷基取代的C1
~C6
烷基或C1
~C6
可經可被烷基取代之雜環烷基取代的C1
~C6
烷氧基;
R11
係表示氫原子或C1
~C6
烷氧基或經氘取代的C1
~C6
烷氧基;
式(III-2)中,
R13
係表示可經雜環烷基取代的C1
~C6
烷基或雜環烷基;
式(III-3)中,
R14
係表示氫原子或C1
~C6
烷基;
R15
係表示氫原子、C1
~C6
烷基或C1
~C6
烷氧基;
R16
係表示氫原子或鹵素原子;
式(III-4)中,
R17
係表示C1
~C6
烷氧基;
R18
係表示C1
~C6
烷氧基)]。
[2]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二 烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽,
烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽, 。
[3]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽,
。
[3]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽, 。
[4]一種以下述式所表示之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽,
。
[4]一種以下述式所表示之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽, 。
[5]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽,
。
[5]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽, 。
[6]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽,
。
[6]一種以下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽, 。
[7]一種以下述式所表示之N-{4-[2-胺基-5-(1-乙基-1H-吡唑-4-基)吡啶-3-基]-3-氟苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽,
。
[7]一種以下述式所表示之N-{4-[2-胺基-5-(1-乙基-1H-吡唑-4-基)吡啶-3-基]-3-氟苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽, 。
[8]一種以下述式所表示之N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽,
。
[8]一種以下述式所表示之N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽, 。
[9]一種以下述式所表示之N-[6-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)嗒-3-基]-5-(5-甲基噻吩-2-基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽,
。
[9]一種以下述式所表示之N-[6-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)嗒-3-基]-5-(5-甲基噻吩-2-基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽, 。
[10]一種以下述式所表示之N-[6-(2-胺基-5-{3-甲氧基-4-[2-(
。
[10]一種以下述式所表示之N-[6-(2-胺基-5-{3-甲氧基-4-[2-( 啉-4-基)乙氧基]苯基}吡啶-3-基)嗒-3-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽,
啉-4-基)乙氧基]苯基}吡啶-3-基)嗒-3-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽, 。
[11]一種如[2]所記載之化合物的氫溴酸鹽、硝酸鹽、硫酸鹽、磷酸鹽、甲磺酸鹽、乙磺酸鹽、苯磺酸鹽或p-甲苯磺酸鹽。
[12]一種如[4]所記載之化合物的甲磺酸鹽。
[13]一種如[5]所記載之化合物的甲磺酸鹽、磷酸鹽、萘-1,5-二磺酸鹽或硫酸鹽。
[14]一種如[11]所記載的甲磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74、7.56、8.96、11.38、12.36、14.78、15.60、16.16、18.70及24.10呈現特徵波峰。
[15]一種如[11]所記載之氫溴酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.84、7.72、9.40、11.62、14.92、15.48、16.70、18.88、19.32及24.40呈現特徵波峰。
[16]一種如[11]所記載之硝酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.82、7.66、9.28、9.52、11.54、15.26、15.54、16.62、19.24及24.56呈現特徵波峰。
[17]一種如[11]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74、7.56、8.92、9.58、11.36、12.38、14.68,15.64、16.06及24.38呈現特徵波峰。
[18]一種如[11]所記載之磷酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74、7.56、8.80、9.56、11.34、14.56、15.74、23.68、24.34及24.68呈現特徵波峰。
[19]一種如[11]所記載之乙磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=6.72、7.90、12.02、13.40、16.90、17.88、19.00、19.80、21.26及24.18呈現特徵波峰。
[20]一種如[11]所記載之苯磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=9.22、10.60、10.82、11.10、13.40、15.78、17.50、18.66、21.02及26.10呈現特徵波峰。
[21]一種如[11]所記載之p-甲苯磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=4.18、5.12、13.44、14.98、16.96、17.44、18.92、19.72、20.16及23.04呈現特徵波峰。
[22]一種如[13]所記載之磷酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=4.28、8.42、8.64、10.54、12.72、13.48、15.90、17.00、17.46及21.26呈現特徵波峰。
[23]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.66、6.42、7.32、9.76、11.00、12.88、18.42、19.62、20.54及24.22呈現特徵波峰。
[24]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.40、7.32、9.76、17.38、18.42、19.64、20.56、22.90及24.20呈現特徵波峰。
[25]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.40、7.30、9.76、17.34、18.38、19.34、20.56、21.52及22.94呈現特徵波峰。
[26]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.62、6.38、7.28、9.74、17.30、18.36、19.54、20.52、22.86及24.14呈現特徵波峰。
[27]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.40、7.30、9.76、12.86、18.40、19.62、20.54、22.92及24.20呈現特徵波峰。
[28]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.36、7.30、18.36、19.04、19.42、19.70、20.12、20.42及21.32呈現特徵波峰。
[29]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=5.62、7.18、9.22、10.36、15.56、16.40及20.86呈現特徵波峰。
[30]一種如[13]所記載之萘-1,5-二磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=6.14、6.98、11.24、14.84、17.48、19.54、20.94、22.38、23.20及24.70呈現特徵波峰。
[31]一種如[13]所記載之萘-1,5-二磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=9.24、9.58、14.00、14.46、16.70、17.02、18.22、20.24、21.64及25.52呈現特徵波峰。
[32]一種Axl抑制劑,其係包含如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[33]一種醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[34]一種用以治療由Axl激酶功能亢進所致的疾病、與Axl激酶功能亢進相關的疾病、及/或伴隨Axl激酶功能亢進的疾病之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[35]一種用以治療過度增殖性疾病(hyperproliferative diseases)之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[36]一種用以治療癌症之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[37]一種用以預防癌症的轉移之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[38]一種用以解除癌症的耐藥性之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[39]一種用以使癌症的耐藥性獲得抑制之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[40]如[36]至[39]中任一項所記載的醫藥,其中癌症係選自乳癌、結腸癌、前列腺癌、肺癌、胃癌、卵巢癌、子宮內膜癌、腎癌、肝細胞癌、甲狀腺癌、子宮癌、食道癌、鱗狀上皮細胞癌(squamous cell carcinoma)、白血病、骨肉瘤(osteosarcoma)、黑色素瘤、神經膠肉瘤(gliosarcoma)、神經母細胞瘤(neuroblastoma)及胰臓癌。
[41]一種醫藥組成物,其係含有如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽及藥學上容許的載體。
[42]一種治療由Axl激酶功能亢進所致的疾病、與Axl激酶功能亢進相關的疾病、及/或伴隨Axl激酶功能亢進的疾病之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[43]一種治療過度增殖性疾病之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[44]一種治療癌症之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[45]一種預防癌症轉移之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[46]一種解除癌症的耐藥性方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[47]一種使癌症的耐藥性獲得抑制之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[48]如[44]至[49]中任一項所記載之方法,其中癌症係選自乳癌、結腸癌、前列腺癌、肺癌、胃癌、卵巢癌、子宮內膜癌、腎癌、肝細胞癌、甲狀腺癌、子宮癌、食道癌、鱗狀上皮細胞癌、白血病、骨肉瘤、黑色素瘤、神經膠肉瘤及神經母細胞瘤。
[49]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於醫藥製造之用途。
[50]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於治療癌症之用途。
[51]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於預防癌症的轉移之用途。
[52]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於解除癌症的耐藥性之用途。
[53]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於使癌症的耐藥性獲得抑制之用途。
[發明之效果]
。
[11]一種如[2]所記載之化合物的氫溴酸鹽、硝酸鹽、硫酸鹽、磷酸鹽、甲磺酸鹽、乙磺酸鹽、苯磺酸鹽或p-甲苯磺酸鹽。
[12]一種如[4]所記載之化合物的甲磺酸鹽。
[13]一種如[5]所記載之化合物的甲磺酸鹽、磷酸鹽、萘-1,5-二磺酸鹽或硫酸鹽。
[14]一種如[11]所記載的甲磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74、7.56、8.96、11.38、12.36、14.78、15.60、16.16、18.70及24.10呈現特徵波峰。
[15]一種如[11]所記載之氫溴酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.84、7.72、9.40、11.62、14.92、15.48、16.70、18.88、19.32及24.40呈現特徵波峰。
[16]一種如[11]所記載之硝酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.82、7.66、9.28、9.52、11.54、15.26、15.54、16.62、19.24及24.56呈現特徵波峰。
[17]一種如[11]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74、7.56、8.92、9.58、11.36、12.38、14.68,15.64、16.06及24.38呈現特徵波峰。
[18]一種如[11]所記載之磷酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74、7.56、8.80、9.56、11.34、14.56、15.74、23.68、24.34及24.68呈現特徵波峰。
[19]一種如[11]所記載之乙磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=6.72、7.90、12.02、13.40、16.90、17.88、19.00、19.80、21.26及24.18呈現特徵波峰。
[20]一種如[11]所記載之苯磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=9.22、10.60、10.82、11.10、13.40、15.78、17.50、18.66、21.02及26.10呈現特徵波峰。
[21]一種如[11]所記載之p-甲苯磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=4.18、5.12、13.44、14.98、16.96、17.44、18.92、19.72、20.16及23.04呈現特徵波峰。
[22]一種如[13]所記載之磷酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=4.28、8.42、8.64、10.54、12.72、13.48、15.90、17.00、17.46及21.26呈現特徵波峰。
[23]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.66、6.42、7.32、9.76、11.00、12.88、18.42、19.62、20.54及24.22呈現特徵波峰。
[24]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.40、7.32、9.76、17.38、18.42、19.64、20.56、22.90及24.20呈現特徵波峰。
[25]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.40、7.30、9.76、17.34、18.38、19.34、20.56、21.52及22.94呈現特徵波峰。
[26]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.62、6.38、7.28、9.74、17.30、18.36、19.54、20.52、22.86及24.14呈現特徵波峰。
[27]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.40、7.30、9.76、12.86、18.40、19.62、20.54、22.92及24.20呈現特徵波峰。
[28]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.36、7.30、18.36、19.04、19.42、19.70、20.12、20.42及21.32呈現特徵波峰。
[29]一種如[13]所記載之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=5.62、7.18、9.22、10.36、15.56、16.40及20.86呈現特徵波峰。
[30]一種如[13]所記載之萘-1,5-二磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=6.14、6.98、11.24、14.84、17.48、19.54、20.94、22.38、23.20及24.70呈現特徵波峰。
[31]一種如[13]所記載之萘-1,5-二磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=9.24、9.58、14.00、14.46、16.70、17.02、18.22、20.24、21.64及25.52呈現特徵波峰。
[32]一種Axl抑制劑,其係包含如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[33]一種醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[34]一種用以治療由Axl激酶功能亢進所致的疾病、與Axl激酶功能亢進相關的疾病、及/或伴隨Axl激酶功能亢進的疾病之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[35]一種用以治療過度增殖性疾病(hyperproliferative diseases)之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[36]一種用以治療癌症之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[37]一種用以預防癌症的轉移之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[38]一種用以解除癌症的耐藥性之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[39]一種用以使癌症的耐藥性獲得抑制之醫藥,其係以如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽作為有效成分。
[40]如[36]至[39]中任一項所記載的醫藥,其中癌症係選自乳癌、結腸癌、前列腺癌、肺癌、胃癌、卵巢癌、子宮內膜癌、腎癌、肝細胞癌、甲狀腺癌、子宮癌、食道癌、鱗狀上皮細胞癌(squamous cell carcinoma)、白血病、骨肉瘤(osteosarcoma)、黑色素瘤、神經膠肉瘤(gliosarcoma)、神經母細胞瘤(neuroblastoma)及胰臓癌。
[41]一種醫藥組成物,其係含有如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽及藥學上容許的載體。
[42]一種治療由Axl激酶功能亢進所致的疾病、與Axl激酶功能亢進相關的疾病、及/或伴隨Axl激酶功能亢進的疾病之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[43]一種治療過度增殖性疾病之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[44]一種治療癌症之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[45]一種預防癌症轉移之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[46]一種解除癌症的耐藥性方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[47]一種使癌症的耐藥性獲得抑制之方法,其特徵在於使用如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽。
[48]如[44]至[49]中任一項所記載之方法,其中癌症係選自乳癌、結腸癌、前列腺癌、肺癌、胃癌、卵巢癌、子宮內膜癌、腎癌、肝細胞癌、甲狀腺癌、子宮癌、食道癌、鱗狀上皮細胞癌、白血病、骨肉瘤、黑色素瘤、神經膠肉瘤及神經母細胞瘤。
[49]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於醫藥製造之用途。
[50]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於治療癌症之用途。
[51]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於預防癌症的轉移之用途。
[52]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於解除癌症的耐藥性之用途。
[53]一種如[1]至[10]中任一項所記載的化合物或其藥學上容許鹽用於使癌症的耐藥性獲得抑制之用途。
[發明之效果]
依據本發明,提供具有Axl抑制活性之以上述式(I)所表示之化合物。此等化合物可用於作為由Axl功能亢進所致的疾病、與Axl功能亢進有關的疾病、及/或伴隨Axl功能亢進的疾病之治療劑,例如抗癌劑。
[用以實施發明的形態]
本發明中所謂的Axl係指由Axl基因所編碼的蛋白質。「Axl」包含完全長度之由Axl基因所編碼的Axl蛋白質或由Axl基因突變體(包含缺失突變體、取代突變體或加成突變體)所編碼的Axl蛋白質等。本發明中所謂的「Axl」亦包含來自各種動物種的同系物。
本發明中所謂的「Axl抑制劑」係指抑制Axl作為酪胺酸激酶之功能的試劑。
本發明中用語「腫瘤」(tumor)及「癌(症)」(cancer)可交換使用。又,本發明中有將腫瘤(tumor)、惡性腫瘤(malignant tumor)、癌(症)(cancer)、惡性新生物(malignant neoplasm)、癌瘤(carcinoma)、肉瘤(sarcoma)等統稱為「腫瘤」或「癌(症)」的情形。
本發明中,
所謂的「C1
~C6
烷基」係指碳數1~6之直鏈、支鏈之烷基。作為「C1
~C6
烷基」,可列舉例如:甲基、乙基、丙基、異丙基、丁基、三級丁基等。
所謂的「C1
~C6
烷氧基」係指具有碳數1~6之直鏈、支鏈之烷基的烷氧基。作為「C1
~C6
烷氧基」,可列舉例如:甲氧基、乙氧基、丙氧基、異丙氧基、丁氧基等。
作為「鹵素原子」,可列舉氟原子、氯原子、溴原子、碘原子等。
所謂的「環烷基」只要未特別提及,係指碳數3~8之環狀的烷基,其可列舉環丙基、環丁基、環戊基、環己基等。
所謂的「雜環烷基」係指一價的飽和雜環基,可列舉環上有氮原子的飽和雜環基、環上有氧原子的飽和雜環基等,可列舉由例如:吡咯啶、咪唑啉、哌啶、哌、吖呾(azetidine)、啉、二烷、氧環丁烷、四氫吡喃、 啶(quinuclidine)衍生的一價基等。
所謂的「環烯基」可列舉於上述之「環烷基」上具有1個以上的雙鍵等之不飽和鍵者,可列舉例如:環戊烯基、環己烯基等。
所謂的「雜環烯基」可列舉於上述之「雜環烷基」上具有1個以上的雙鍵等之不飽和鍵者,可列舉例如:四氫吡啶基、二氫吡喃基。以下說明關於式(I)中之各取代基。
下述通式(I)中,
啶(quinuclidine)衍生的一價基等。
所謂的「環烯基」可列舉於上述之「環烷基」上具有1個以上的雙鍵等之不飽和鍵者,可列舉例如:環戊烯基、環己烯基等。
所謂的「雜環烯基」可列舉於上述之「雜環烷基」上具有1個以上的雙鍵等之不飽和鍵者,可列舉例如:四氫吡啶基、二氫吡喃基。以下說明關於式(I)中之各取代基。
下述通式(I)中, W、X、Y係各自獨立地表示氮原子、C-H、C-F或C-Cl。
Z係表示氮原子、C-H、C-C1
~C6
烷氧基或C-Cl。
R1
係表示下述式(II-1)或(II-2),
W、X、Y係各自獨立地表示氮原子、C-H、C-F或C-Cl。
Z係表示氮原子、C-H、C-C1
~C6
烷氧基或C-Cl。
R1
係表示下述式(II-1)或(II-2), (式(II-1)中,Q係表示氮原子、C-H或C-F;T係表示氮原子或C-H;U係表示氮原子或C-H;R6
係表示鹵素原子、C1
~C6
烷基、C3
~C6
環烷基、氰基或三氟甲氧基;式(II-2)中,V係表示硫原子或氧原子;R7
係表示C1
~C6
烷基。)。
R2
係表示氫原子、鹵素原子、C1
~C6
烷基、C1
~C6
烷氧基或氰基。其中作為C1
~C6
烷基,較佳為甲基、乙基或丙基;作為C1
~C6
烷氧基,較佳為甲氧基、乙氧基。
R3
係表示氫原子或C1
~C6
烷基,作為C1
~C6
烷基,較佳為甲基、乙基或丙基。
R4
係表示氫原子、鹵素原子或C1
~C6
烷基,作為R4
,較佳為氫原子、氟原子、氯原子、甲基或乙基。
R5
係表示氫原子或下述式(III-1)、(III-2)、(III-3)或(III-4),
(式(II-1)中,Q係表示氮原子、C-H或C-F;T係表示氮原子或C-H;U係表示氮原子或C-H;R6
係表示鹵素原子、C1
~C6
烷基、C3
~C6
環烷基、氰基或三氟甲氧基;式(II-2)中,V係表示硫原子或氧原子;R7
係表示C1
~C6
烷基。)。
R2
係表示氫原子、鹵素原子、C1
~C6
烷基、C1
~C6
烷氧基或氰基。其中作為C1
~C6
烷基,較佳為甲基、乙基或丙基;作為C1
~C6
烷氧基,較佳為甲氧基、乙氧基。
R3
係表示氫原子或C1
~C6
烷基,作為C1
~C6
烷基,較佳為甲基、乙基或丙基。
R4
係表示氫原子、鹵素原子或C1
~C6
烷基,作為R4
,較佳為氫原子、氟原子、氯原子、甲基或乙基。
R5
係表示氫原子或下述式(III-1)、(III-2)、(III-3)或(III-4), (式(III-1)中,
R8
及R12
係各自獨立地表示氫原子或氘原子;
R9
係表示氫原子、鹵素原子或C1
~C6
烷氧基;
R10
係表示氫原子、可經雜環烷基取代的C1
~C6
烷基或可經可被C1
~C6
烷基取代之雜環烷基取代的C1
~C6
烷氧基;
R11
係表示氫原子或C1
~C6
烷氧基或經氘取代的C1
~C6
烷氧基;
式(III-2)中,
R13
係表示可經雜環烷基取代的C1
~C6
烷基或雜環烷基;
式(III-3)中,
R14
係表示氫原子或C1
~C6
烷基;
R15
係表示氫原子、C1
~C6
烷基或C1
~C6
烷氧基;
R16
係表示氫原子或鹵素原子;
式(III-4)中,
R17
係表示C1
~C6
烷氧基;
R18
係表示C1
~C6
烷氧基。)。
R5
為以式(III-1)所表示之基的情形,R10
中的雜環烷基較佳為二烷基、啉基、哌基或四氫吡喃基;C1
~C6
烷基較佳為甲氧基或乙氧基。R11
中的C1
~C6
烷氧基較佳為甲氧基或乙氧基。
R5
為以式(III-2)所表示之基的情形,R13
中的雜環烷基較佳為二烷基、啉基、哌基或四氫吡喃基,R13
中的C1
~C6
烷基較佳為甲基或乙基。
又,以通式(I)所表示之化合物或其藥學上容許鹽係以下述所示化合物中之任一者或其藥學上容許鹽為進一步較佳。
下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,
(式(III-1)中,
R8
及R12
係各自獨立地表示氫原子或氘原子;
R9
係表示氫原子、鹵素原子或C1
~C6
烷氧基;
R10
係表示氫原子、可經雜環烷基取代的C1
~C6
烷基或可經可被C1
~C6
烷基取代之雜環烷基取代的C1
~C6
烷氧基;
R11
係表示氫原子或C1
~C6
烷氧基或經氘取代的C1
~C6
烷氧基;
式(III-2)中,
R13
係表示可經雜環烷基取代的C1
~C6
烷基或雜環烷基;
式(III-3)中,
R14
係表示氫原子或C1
~C6
烷基;
R15
係表示氫原子、C1
~C6
烷基或C1
~C6
烷氧基;
R16
係表示氫原子或鹵素原子;
式(III-4)中,
R17
係表示C1
~C6
烷氧基;
R18
係表示C1
~C6
烷氧基。)。
R5
為以式(III-1)所表示之基的情形,R10
中的雜環烷基較佳為二烷基、啉基、哌基或四氫吡喃基;C1
~C6
烷基較佳為甲氧基或乙氧基。R11
中的C1
~C6
烷氧基較佳為甲氧基或乙氧基。
R5
為以式(III-2)所表示之基的情形,R13
中的雜環烷基較佳為二烷基、啉基、哌基或四氫吡喃基,R13
中的C1
~C6
烷基較佳為甲基或乙基。
又,以通式(I)所表示之化合物或其藥學上容許鹽係以下述所示化合物中之任一者或其藥學上容許鹽為進一步較佳。
下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, ;
下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,
;
下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, ;
下述式所表示之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,
;
下述式所表示之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,  ;
下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺,
;
下述式所表示之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺, ;
下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺,
;
下述式所表示之N-[4-(2-胺基-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺, ;
下述式所表示之N-{4-[2-胺基-5-(1-乙基-1H-吡唑-4-基)吡啶-3-基]-3-氟苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,
;
下述式所表示之N-{4-[2-胺基-5-(1-乙基-1H-吡唑-4-基)吡啶-3-基]-3-氟苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, ;
下述式所表示之N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,
;
下述式所表示之N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, ;
下述式所表示之N-[6-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)嗒-3-基]-5-(5-甲基噻吩-2-基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,
;
下述式所表示之N-[6-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)嗒-3-基]-5-(5-甲基噻吩-2-基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, ;所表示之N-[6-(2-胺基-5-{3-甲氧基-4-[2-(啉-4-基)乙氧基]苯基}吡啶-3-基)嗒-3-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺,
;所表示之N-[6-(2-胺基-5-{3-甲氧基-4-[2-(啉-4-基)乙氧基]苯基}吡啶-3-基)嗒-3-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, 。
。
本發明之以式(I)所表示之化合物存有立體異構物或源自不對稱碳原子的光學異構物,但此等立體異構物、光學異構物及此等之混合物之任一者亦包含於本發明。
以本發明之通式(I)所表示之化合物具有胺基等之鹼性基的情形,依所欲可作成醫藥上容許鹽。作為此種鹽,可列舉例如鹽酸鹽、氫溴酸鹽、氫碘酸鹽等之氫鹵酸鹽;硝酸鹽、過氯酸鹽、硫酸鹽、磷酸鹽等之無機酸鹽;甲磺酸鹽、三氟甲磺酸鹽、乙磺酸鹽等之低級烷烴磺酸鹽;苯磺酸鹽、p-甲苯磺酸鹽等之芳基磺酸鹽;甲酸、乙酸、蘋果酸、反丁烯二酸鹽、琥珀酸鹽、檸檬酸鹽、酒石酸鹽、乙二酸鹽、馬來酸鹽等之有機酸鹽;及鳥胺酸酸鹽、麩胺酸鹽、天冬胺酸鹽等之胺基酸鹽;較佳為氫鹵酸鹽及有機酸鹽。
以本發明之通式(I)所表示之化合物具有羧基等之酸性基的情形,一般而言可形成鹼加成鹽。作為醫藥上容許鹽可列舉例如:鈉鹽、鉀鹽、鋰鹽等之鹼金屬鹽;鈣鹽、鎂鹽等之鹼土類金屬鹽;銨鹽等之無機鹽;二苄基胺鹽、啉鹽、苯基甘胺酸烷基酯鹽、伸乙二胺鹽、N-甲基還原葡糖胺鹽、二乙胺鹽、三乙胺鹽、環己基胺鹽、二環己基胺鹽、N,N’-二苄基伸乙二胺鹽、二乙醇胺鹽、N-苄基-N-(2-苯基乙氧基)胺鹽、哌鹽、四甲基銨鹽、參(羥基甲基)胺基甲烷鹽等之有機胺鹽等。
以本發明之通式(I)所表示之化合物或其鹽亦有以游離體(free form)或溶劑合物存在。亦有藉由吸收空氣中之水分等而以水合物存在。就溶劑合物而言,只要是醫藥上可容許者則無特別限定,但具體而言,較佳為水合物、乙醇合物等。又,以通式(I)所表示的本發明化合物中存在氮原子的情形,亦可形成N-氧化物,此等溶劑合物及N-氧化物亦包含於本發明之範圍。再者,本化合物或其鹽中亦包含彼等之結晶。
更具體而言,作為鹽,可列舉N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之氫溴酸鹽、硝酸鹽、硫酸鹽、磷酸鹽、乙磺酸鹽、苯磺酸鹽或p-甲苯磺酸鹽。
又,可列舉N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之甲磺酸鹽。
此外,可列舉N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之甲磺酸鹽、磷酸鹽或硫酸鹽。
本發明的別的態樣係關於本發明的化合物或鹽之結晶。其中所謂的結晶係指其內部結構為構成原子(或其集團)三維地有規律地重複之固體,其係有別於不具有此種規律的內部結構的非晶固體。
即使是相同化合物的結晶,依結晶化之條件,亦會生成具有複數不同之內部結構及物理化學之性質的結晶(結晶多形),但本發明之結晶可為此等結晶多形之任一者,亦可為2種以上的結晶多形之混合物。
本發明之結晶有經由放置於大氣中吸收水分,而附著附著水的情形、或藉由在一般大氣條件下加熱至25乃至150℃等而形成水合物的情形。此外,本發明之結晶亦有於附著殘留溶劑或於溶劑合物中,含有結晶化時之溶劑的情形。
本說明書中,有基於粉末X射線繞射之數據表示本發明之結晶,而粉末X射線繞射係可藉由一般該領域中所使用之手法來進行測定・解析,例如:可根據實施例所記載的方法來進行。又,一般而言,水合物或脫水物有時會因結晶水之附著、脫離,而改變其晶格常數,致使粉末X射線繞射中之繞射角(2θ)改變。又,波峰之強度亦有因結晶之成長面等的差異(結晶習慣)等而變化。因此,基於粉末X射線繞射之數據表示本發明之結晶時,除了包含粉末X射線繞射中波峰之繞射角及X射線繞射圖為一致的結晶外,由彼等所獲得的水合物及脫水物亦包含於本發明的範圍。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之甲磺酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之甲磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74, 7.56, 8.96, 11.38, 12.36, 14.78, 15.60, 16.16, 18.70及24.10呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖1之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之氫溴酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之氫溴酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.84, 7.72, 9.40, 11.62, 14.92, 15.48, 16.70, 18.88, 19.32及24.40呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖2之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之硝酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之硝酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.82, 7.66, 9.28, 9.52, 11.54, 15.26, 15.54, 16.62, 19.24及24.56呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖3之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之硫酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74, 7.56, 8.92, 9.58, 11.36, 12.38, 14.68, 15.64, 16.06及24.38呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖4之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之磷酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之磷酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.74, 7.56, 8.80, 9.56, 11.34, 14.56, 15.74, 23.68, 24.34及24.68呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖5之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之乙磺酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之乙磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=6.72, 7.90, 12.02, 13.40, 16.90, 17.88, 19.00, 19.80, 21.26及24.18呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖6之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之苯磺酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之苯磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=9.22, 10.60, 10.82, 11.10, 13.40, 15.78, 17.50, 18.66, 21.02及26.10呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖7之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之p-甲苯磺酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之p-甲苯磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=4.18, 5.12, 13.44, 14.98, 16.96, 17.44, 18.92, 19.72, 20.16及23.04呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖8之粉末X射線繞射圖。
更具體之本發明的一態樣為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之磷酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之磷酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=4.28, 8.42, 8.64, 10.54, 12.72, 13.48, 15.90, 17.00, 17.46及21.26呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖9之粉末X射線繞射圖。
更具體之本發明的一態樣為N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.66, 6.42, 7.32, 9.76, 11.00, 12.88, 18.42, 19.62, 20.54及24.22呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖10之粉末X射線繞射圖。其他較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64, 6.40, 7.32, 9.76, 17.38, 18.42, 19.64, 20.56, 22.90及24.20呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖11之粉末X射線繞射圖。其他較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64, 6.40, 7.30, 9.76, 17.34, 18.38, 19.34, 20.56, 21.52及22.94呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖12之粉末X射線繞射圖。其他較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.62, 6.38, 7.28, 9.74, 17.30, 18.36, 19.54, 20.52, 22.86及24.14呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖13之粉末X射線繞射圖。其他較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.40、7.30、9.76、12.86、18.40、19.62、20.54、22.92及24.20呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖14之粉末X射線繞射圖。其他較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=3.64、6.36、7.30、18.36、19.04、19.42、19.70、20.12、20.42及21.32呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖15之粉末X射線繞射圖。其他較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之硫酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=5.62、7.18、9.22、10.36、15.56、16.40及20.86呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖16之粉末X射線繞射圖。
更具體之本發明的一態樣為N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之萘-1,5-二磺酸鹽的結晶。進一步較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之萘-1,5-二磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=6.14、6.98、11.24、14.84、17.48、19.54、20.94、22.38、23.20及24.70呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖17之粉末X射線繞射圖。其他較佳為關於N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺之萘-1,5-二磺酸鹽的結晶,其於以銅之Kα線(波長λ=1.54Å)之照射所得之粉末X射線繞射圖中,於繞射角度2θ=9.24、9.58、14.00、14.46、16.70、17.02、18.22、20.24、21.64及25.52呈現特徵波峰。該結晶於銅之Kα線(波長λ=1.54Å)之照射中呈現圖18之粉末X射線繞射圖。
以本發明之通式(I)所表示之化合物依取代基之種類或組合,可存在有順式體、反式體等之幾何異構物、互變異構物或d體、l體等之光學異構物等之各種異構物,但本發明之化合物在沒有特別限定的情形下,亦包含彼等全部之異構物、立體異構物及任一比率之此等異構物及立體異構物混合物。
以本發明之通式(I)所表示之化合物,亦可於一個或複數個所構成的原子上包含非天然比率之原子同位素。作為原子同位素可列舉例如:氘(2
H)(本說明書中亦有寫成D者。)、氚(3
H)、碘-125(125
I)或碳-14(14
C)等。此等化合物可用於作為治療或預防劑、研究試劑例如檢定試劑,及診斷劑,例如:作為體內影像診斷劑。以通式(I)所表示之化合物之所有同位素變體,不論是否具有放射性,均包含於本發明之範圍內。
又,本發明係藉由在活體內之生理條件下,經酵素、胃酸等反應,而轉換成本發明之醫藥組成物之有效成分的以通式(I)所表示之化合物的化合物,亦即,因酵素引起的氧化、還原、水解等而轉變成以通式(I)所表示之化合物的化合物、或因胃酸等引起的水解等而轉變成以通式(I)所表示之化合物的「醫藥上容許之前藥化合物」亦包含於本發明。
就上述前藥而言,以通式(I)所表示之化合物上有胺基存在的情形,可列舉其胺基係經醯基化、烷基化、磷酸化之化合物(例如:其胺基係經二十醯基化、丙胺醯基化、戊胺羰基化、(5-甲基-2-側氧-1,3-二氧戊二烯-4-基)甲氧基羰基化、四氫呋喃基化、吡咯啶基甲基化、三甲基乙醯基氧基甲基化、三級丁基化之化合物等)等;以通式(I)所表示之化合物上有羥基存在的情形,可列舉其羥基係經醯基化、烷基化、磷酸化、硼酸化之化合物(例如:其羥基係經乙醯基化、棕櫚醯基化、丙醯基化、三甲基乙醯基化、琥珀醯基化、延胡索醯基化、丙胺醯基化、二甲基胺基甲基羰基化之化合物等。)等。又,以通式(I)所表示之化合物上有羧基存在的情形,可列舉其羧基係經酯化、醯胺化之化合物(例如:其羧基係經乙基酯化、苯基酯化、羧基甲基酯化、二甲基胺基甲基酯化、三甲基乙醯基氧基甲基酯化、乙氧基羰基氧基乙基酯化、醯胺化或甲基醯胺化之化合物等。)等。
本發明之化合物的前藥可以周知之方法由以通式(I)所表示之化合物製造。又,本發明之化合物的前藥亦包含如廣川書店1990年刊「醫藥品之開發」第7卷分子設計163頁~198頁中所記載之以生理的條件下轉變成以通式(I)所表示之化合物者。
接著,說明關於以通式(I)所表示之化合物之代表性的製造法。本發明之化合物可依照各種製造法製造,以下所示製造法係為一例,本發明不應被解釋成限定於此等。此外,反應時,因應需要可將取代基以適當之保護基保護來進行,保護基之種類沒有特別限定。 通式(I)係如上述般,可分割成部位A、部位B、部位C來考量。作為主要的合成流程可列舉:首先使化合物(B)與化合物(C-I)鍵結,再使其複合物與化合物(A-I)鍵結之方法;或首先使化合物(A-I)與化合物(B)鍵結,再使其複合物與化合物(C-I)鍵結之方法;或首先使化合物(B)與化合物(C-I)鍵結,再使其複合物與化合物(A-II)鍵結後,將溴基轉換成R1
之方法;或將化合物(BC)之溴基轉換成R5
後,使化合物(A-I)或化合物(A-II)與鍵結之方法;或首先使化合物(A-I)與化合物(BC)鍵結後,將溴基轉換成R5
之方法;或使化合物(A-I)化合物(B)複合物與化合物(C-II)鍵結後,將溴基轉換成R5
之方法等,但並未特別限定。
通式(I)係如上述般,可分割成部位A、部位B、部位C來考量。作為主要的合成流程可列舉:首先使化合物(B)與化合物(C-I)鍵結,再使其複合物與化合物(A-I)鍵結之方法;或首先使化合物(A-I)與化合物(B)鍵結,再使其複合物與化合物(C-I)鍵結之方法;或首先使化合物(B)與化合物(C-I)鍵結,再使其複合物與化合物(A-II)鍵結後,將溴基轉換成R1
之方法;或將化合物(BC)之溴基轉換成R5
後,使化合物(A-I)或化合物(A-II)與鍵結之方法;或首先使化合物(A-I)與化合物(BC)鍵結後,將溴基轉換成R5
之方法;或使化合物(A-I)化合物(B)複合物與化合物(C-II)鍵結後,將溴基轉換成R5
之方法等,但並未特別限定。 [式中,R1
~R5
、W、X、Y、Z係與前述同義。R31
係為了使化合物(B)與化合物(C-I)或化合物(C-II)鍵結所必要的取代基,表示鹵素基、三氟甲磺酸基、硼酸、硼酸酯。R32
係為了使化合物(B)與化合物(C-I)鍵結所必要的取代基,表示鹵素基、三氟甲磺酸基、硼酸、硼酸酯。R33
係為了使化合物(B)與化合物(C-II)鍵結所必要的取代基,表示硼酸、硼酸酯。]
[式中,R1
~R5
、W、X、Y、Z係與前述同義。R31
係為了使化合物(B)與化合物(C-I)或化合物(C-II)鍵結所必要的取代基,表示鹵素基、三氟甲磺酸基、硼酸、硼酸酯。R32
係為了使化合物(B)與化合物(C-I)鍵結所必要的取代基,表示鹵素基、三氟甲磺酸基、硼酸、硼酸酯。R33
係為了使化合物(B)與化合物(C-II)鍵結所必要的取代基,表示硼酸、硼酸酯。]
首先說明關於化合物(A-I)之合成法。
[製造法1]
化合物(A-I)可以如下述反應式1的方法合成,但並未特別限定。
反應式1 [式中,R1
~R3
、Q、T及U係與前述同義。R41
係表示鹵素基。R42
係羧酸之保護基,其表示C1
-C6
烷基。R43
表示硼酸、硼酸酯。R44
表示C1
~C6
烷基及環C1
-C6
烷基。化合物(1d)係被化合物(1e)包含在內。]
化合物(1c)之合成
將市售或經適當合成的化合物(1a),在碳酸鉀、碳酸鈉、碳酸銫、乙酸鉀、磷酸鉀、三級丁氧化物或三乙基胺等之有機或無機鹼、參(二苯亞甲基丙酮)-二鈀(0)、肆(三苯基膦)鈀(0)、雙(三苯基膦)氯化鈀(II)、[1,1’-雙(二苯基膦基)-二茂鐵]氯化鈀(II)二氯甲烷錯合物、乙酸鈀(II)、碘化銅(I)氯化銅(I)等之過渡金屬觸媒存在下,與市售或以於製造法4、製造法5、製造法6所示之方法合成的化合物(1b)進行耦合反應,藉此可得到化合物(1c)。又,上述反應亦可在三苯基膦、三環己基膦、二苯亞甲基丙酮、1,3-雙(二苯基膦基)丙烷(1,3-bis(diphenyl phosphino)propane)、2-二環己基膦基-2’,4’,6’-三異丙基聯苯、4,5-雙(二苯基膦基)-9,9-二甲基二苯并吡喃等之配位子存在下實施。又,上述反應可併用2種以上之上述過渡金屬觸媒來進行。溶劑係可列舉1,4-二烷、1,2-二甲氧基乙烷、甲醇、甲苯、水或此等之混合溶劑,但未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。又,上述處理亦可藉由在密封管中、在微波下來進行。相對於化合物(1a)而言,有機或無機鹼係可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。相對於化合物(1c)而言,過渡金屬觸媒、配位子係可使用0.001至1莫耳當量。較佳為可使用0.05至1莫耳當量。相對於化合物(1c)而言,硼酸酯係可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(1d)之合成
可將化合物(1c)以硫酸、鹽酸等之酸、或氫氧化鈉、碳酸鉀等之鹼進行水解處理而得到。相對於化合物(1c)而言,鹼或酸可使用1至過剩莫耳當量。反應中所使用之溶劑可列舉甲醇、乙醇、丙醇、水等、或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。
化合物(1f)之合成
可將市售或經適當合成的化合物(1e)活性化,而在丙二酸單酯或其鹽與氯化鎂存在下,使之反應而得到。作為羧酸之活性化的方法,可列舉使用1,1’-羰基二咪唑的方法、經由醯氯(acid chloride)的方法。反應可因應需要添加鹼,作為鹼可列舉例如三乙基胺等。相對於化合物(1e)而言,鹼可使用1至過剩莫耳當量。較佳為使用2至10莫耳當量。反應中所使用之溶劑可列舉四氫呋喃、二烷、乙腈、N,N-二甲基甲醯胺、甲苯等、或此等混合溶劑,但並未特別限定。反應溫度通常在-78℃至100℃或至溶劑之沸點的範圍,但較佳為室溫附近至100℃的範圍。
化合物(1g)之合成
可將化合物(1f)以N,N-二甲基甲醯胺二甲基縮醛處理而得到。相對於化合物(1f)而言,N,N-二甲基甲醯胺二甲基縮醛可使用2至過剩莫耳當量。反應中所使用之溶劑可列舉甲苯、二甲苯、二氯甲烷、乙醇、N,N-二甲基甲醯胺、乙酸乙酯等、或此等混合溶劑,但並未特別限定。或即使在無溶劑下亦可進行。反應溫度通常在0℃至300℃的範圍,但較佳為室溫附近至130℃的範圍。
化合物(1i)之合成
可將化合物(1g)以市售或經適當合成的化合物(1h)處理而得到。反應中所使用之溶劑可列舉甲苯、乙酸乙酯、乙醇、1,4-二烷等、或此等混合溶劑,但並未特別限定。反應溫度通常在-78℃至130℃或至溶劑之沸點的範圍,但較佳為室溫附近至溶劑之沸點的範圍。又,上述反應可因應需要添加碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼、或乙酸、氯化氫、溴化氫、硫酸等之酸來進行。相對於化合物(1g)而言,化合物(1h)、鹼或酸可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(A-I)之合成
可藉由與化合物(1d)之合成同樣的操作而得到。
[式中,R1
~R3
、Q、T及U係與前述同義。R41
係表示鹵素基。R42
係羧酸之保護基,其表示C1
-C6
烷基。R43
表示硼酸、硼酸酯。R44
表示C1
~C6
烷基及環C1
-C6
烷基。化合物(1d)係被化合物(1e)包含在內。]
化合物(1c)之合成
將市售或經適當合成的化合物(1a),在碳酸鉀、碳酸鈉、碳酸銫、乙酸鉀、磷酸鉀、三級丁氧化物或三乙基胺等之有機或無機鹼、參(二苯亞甲基丙酮)-二鈀(0)、肆(三苯基膦)鈀(0)、雙(三苯基膦)氯化鈀(II)、[1,1’-雙(二苯基膦基)-二茂鐵]氯化鈀(II)二氯甲烷錯合物、乙酸鈀(II)、碘化銅(I)氯化銅(I)等之過渡金屬觸媒存在下,與市售或以於製造法4、製造法5、製造法6所示之方法合成的化合物(1b)進行耦合反應,藉此可得到化合物(1c)。又,上述反應亦可在三苯基膦、三環己基膦、二苯亞甲基丙酮、1,3-雙(二苯基膦基)丙烷(1,3-bis(diphenyl phosphino)propane)、2-二環己基膦基-2’,4’,6’-三異丙基聯苯、4,5-雙(二苯基膦基)-9,9-二甲基二苯并吡喃等之配位子存在下實施。又,上述反應可併用2種以上之上述過渡金屬觸媒來進行。溶劑係可列舉1,4-二烷、1,2-二甲氧基乙烷、甲醇、甲苯、水或此等之混合溶劑,但未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。又,上述處理亦可藉由在密封管中、在微波下來進行。相對於化合物(1a)而言,有機或無機鹼係可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。相對於化合物(1c)而言,過渡金屬觸媒、配位子係可使用0.001至1莫耳當量。較佳為可使用0.05至1莫耳當量。相對於化合物(1c)而言,硼酸酯係可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(1d)之合成
可將化合物(1c)以硫酸、鹽酸等之酸、或氫氧化鈉、碳酸鉀等之鹼進行水解處理而得到。相對於化合物(1c)而言,鹼或酸可使用1至過剩莫耳當量。反應中所使用之溶劑可列舉甲醇、乙醇、丙醇、水等、或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。
化合物(1f)之合成
可將市售或經適當合成的化合物(1e)活性化,而在丙二酸單酯或其鹽與氯化鎂存在下,使之反應而得到。作為羧酸之活性化的方法,可列舉使用1,1’-羰基二咪唑的方法、經由醯氯(acid chloride)的方法。反應可因應需要添加鹼,作為鹼可列舉例如三乙基胺等。相對於化合物(1e)而言,鹼可使用1至過剩莫耳當量。較佳為使用2至10莫耳當量。反應中所使用之溶劑可列舉四氫呋喃、二烷、乙腈、N,N-二甲基甲醯胺、甲苯等、或此等混合溶劑,但並未特別限定。反應溫度通常在-78℃至100℃或至溶劑之沸點的範圍,但較佳為室溫附近至100℃的範圍。
化合物(1g)之合成
可將化合物(1f)以N,N-二甲基甲醯胺二甲基縮醛處理而得到。相對於化合物(1f)而言,N,N-二甲基甲醯胺二甲基縮醛可使用2至過剩莫耳當量。反應中所使用之溶劑可列舉甲苯、二甲苯、二氯甲烷、乙醇、N,N-二甲基甲醯胺、乙酸乙酯等、或此等混合溶劑,但並未特別限定。或即使在無溶劑下亦可進行。反應溫度通常在0℃至300℃的範圍,但較佳為室溫附近至130℃的範圍。
化合物(1i)之合成
可將化合物(1g)以市售或經適當合成的化合物(1h)處理而得到。反應中所使用之溶劑可列舉甲苯、乙酸乙酯、乙醇、1,4-二烷等、或此等混合溶劑,但並未特別限定。反應溫度通常在-78℃至130℃或至溶劑之沸點的範圍,但較佳為室溫附近至溶劑之沸點的範圍。又,上述反應可因應需要添加碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼、或乙酸、氯化氫、溴化氫、硫酸等之酸來進行。相對於化合物(1g)而言,化合物(1h)、鹼或酸可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(A-I)之合成
可藉由與化合物(1d)之合成同樣的操作而得到。
[製造法2]
又,化合物(1i)亦可藉由如下述反應式2之方法合成,但並未特別限定。
反應式2 [式中,R1
、R42
係與前述同義。R45
係表示鹵素基、甲磺醯氧基、三氟甲磺醯氧基、p-甲苯磺醯基氧基。化合物(2c)係被化合物(1i)包含在內。]
化合物(2a)之合成
化合物(2a)係可為市售、或參考J.Heterocyclic Chem.17,359(1980)、Chem.Pharm.Bull.43,450(1995)等之已經被報告的報告例而由化合物(1f)合成。
化合物(2c)之合成
可將化合物(2a)在碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(2b)處理而得到。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(2a)而言,化合物(2b)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
接著說明關於化合物(A-II)之合成法。
[式中,R1
、R42
係與前述同義。R45
係表示鹵素基、甲磺醯氧基、三氟甲磺醯氧基、p-甲苯磺醯基氧基。化合物(2c)係被化合物(1i)包含在內。]
化合物(2a)之合成
化合物(2a)係可為市售、或參考J.Heterocyclic Chem.17,359(1980)、Chem.Pharm.Bull.43,450(1995)等之已經被報告的報告例而由化合物(1f)合成。
化合物(2c)之合成
可將化合物(2a)在碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(2b)處理而得到。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(2a)而言,化合物(2b)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
接著說明關於化合物(A-II)之合成法。
[製造法3]
化合物(A-II)可以如下述反應式3之方法合成,但並未特別限定。
反應式3 [式中,R42
、R45
係與前述同義。]
化合物(3b)之合成
可藉由將市售、或參考J.Med.Chem.51,5330(2008)等之已經被報告的報告例而容易地合成之化合物(3a),在碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(2b)處理而得到。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(3a)而言,化合物(2b)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(A-II)之合成
可藉由與[製造法1]中之化合物(A-I)之合成同樣的操作而得到。
接著說明關於化合物(C-I)之合成法。
R5
為氫原子的情形,化合物(C-I)可為市售品或可由市售品以文獻已知之方法容易地合成。
R5
為氫原子以外的情形,化合物(C-I)可以如下述製造法4至製造法7之方法合成。亦即,選擇如於製造法4至製造法6所示之方法中任一者,合成構成R5
的化合物(4c)、化合物(5h)或化合物(6c)後,使用此等化合物或市售化合物,而可以製造法7合成化合物(C-I)。但,並未特別限定於此等。
[式中,R42
、R45
係與前述同義。]
化合物(3b)之合成
可藉由將市售、或參考J.Med.Chem.51,5330(2008)等之已經被報告的報告例而容易地合成之化合物(3a),在碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(2b)處理而得到。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(3a)而言,化合物(2b)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(A-II)之合成
可藉由與[製造法1]中之化合物(A-I)之合成同樣的操作而得到。
接著說明關於化合物(C-I)之合成法。
R5
為氫原子的情形,化合物(C-I)可為市售品或可由市售品以文獻已知之方法容易地合成。
R5
為氫原子以外的情形,化合物(C-I)可以如下述製造法4至製造法7之方法合成。亦即,選擇如於製造法4至製造法6所示之方法中任一者,合成構成R5
的化合物(4c)、化合物(5h)或化合物(6c)後,使用此等化合物或市售化合物,而可以製造法7合成化合物(C-I)。但,並未特別限定於此等。
[製造法4]
反應式4 [式中,R8
、R9
、R11
、R12
、R43
、R45
係與前述同義。R51
係表示可經雜環烷基取代的C1
-C6
烷基。]
化合物(4c)之合成
可藉由將市售的、或經適當合成的化合物(4a),於碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(4b)處理而得到。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(4a)而言,化合物(4b)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
[式中,R8
、R9
、R11
、R12
、R43
、R45
係與前述同義。R51
係表示可經雜環烷基取代的C1
-C6
烷基。]
化合物(4c)之合成
可藉由將市售的、或經適當合成的化合物(4a),於碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(4b)處理而得到。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈或此等混合溶劑,但並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(4a)而言,化合物(4b)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
[製造法5]
反應式5 [式中,R8
、R9
、R12
、R43
、R45
、R51
係與前述同義。R61
係表示氘或氫原子。R62
係表示羥基、及經氘取代的羥基之保護基。作為保護基,可列舉以四氫吡喃基所代表的醚基、以三級丁基二苯基矽烷基所代表的矽烷基、以乙醯基所代表的酯基、以乙烯基碳酸酯基所代表的碳酸酯基等。R63
係表示經氘取代的C1
-C6
烷基。]
化合物(5b)之合成
可藉由僅保護市售的、或經適當合成的化合物(5a)的2個羥基中之一個而得到化合物(5b)。例如:藉由將化合物(5a)在p-甲苯磺酸吡啶鹽等之酸存在下,以3,4-二氫-2H-吡喃處理,可得到僅將一個羥基以四氫吡喃基保護之化合物(5b)。但,羥基保護之方法並未特別限定於此等。反應中所使用之溶劑可列舉二氯甲烷、氯仿、甲苯或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(5a)而言,化合物(4b)、酸可使用0.01至過剩莫耳當量。較佳為可使用0.01至1莫耳當量。
化合物(5d)之合成
利用將化合物(5b)在碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(5c)處理,而可得到化合物(5d)。可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈、丙酮或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(5b)而言,化合物(5c)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(5e)之合成
可藉由將化合物(5d)以鹽酸、乙酸、p-甲苯磺酸吡啶鹽等之酸處理,得到化合物(5e)。反應中所使用之溶劑可列舉乙醇、甲醇、異丙醇、水或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(5d)而言,化合物(4b)、酸可使用0.01至過剩莫耳當量。較佳為可使用0.01至1莫耳當量。
化合物(5f)之合成
利用將化合物(5e)以N-溴琥珀酸醯亞胺(N-Bromosuccinimide)、溴酸鉀等之溴化試劑處理,而可得到化合物(5f)。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、30%氫溴酸-乙酸溶液、乙酸或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於溴化試劑而言,化合物(5e)可使用0.1至過剩莫耳當量。較佳為可使用0.1至5莫耳當量。
化合物(5g)之合成
可藉由與[製造法4]同樣的操作而得到。
化合物(5h)之合成
藉由將化合物(5g)在碳酸鉀、碳酸鈉、碳酸銫、乙酸鉀、磷酸鉀、三級丁氧化物或三乙基胺等之有機或無機鹼、參(二苯亞甲基丙酮)-二鈀(0)、雙(三苯基膦)氯化鈀(II)、[1,1’-雙(二苯基膦基)-二茂鐵]氯化鈀(II)二氯甲烷錯合物、[1,1’-雙(二苯基膦基)-二茂鐵]氯化鈀(II)二氯甲烷錯合物、1,3-雙(二苯基膦基)丙烷]氯化鎳(II)、等之過渡金屬觸媒、三苯基膦、三環己基膦、二苯亞甲基丙酮、1,3-雙(二苯基膦基)丙烷、2-二環己基膦基-2’,4’,6’-三異丙基聯苯、4,5-雙(二苯基膦基)-9,9-二甲基二苯并吡喃等之配位子存在下,以雙聯頻哪醇基二硼烷Bis(pinacolato)diboron、頻哪醇硼酸酯(pinacol boride)等之硼酸酯處理,而可得到。溶劑可列舉1,4-二烷、1,2-二甲氧基乙烷、甲醇、甲苯或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。又,上述處理亦可藉由在密封管中、在微波下來進行。相對於化合物(5g)而言,有機或無機鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。相對於化合物(5g)而言,過渡金屬觸媒、配位子可使用0.001至1莫耳當量。較佳為可使用0.05至1莫耳當量。相對於化合物(5g)而言,硼酸酯可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
[式中,R8
、R9
、R12
、R43
、R45
、R51
係與前述同義。R61
係表示氘或氫原子。R62
係表示羥基、及經氘取代的羥基之保護基。作為保護基,可列舉以四氫吡喃基所代表的醚基、以三級丁基二苯基矽烷基所代表的矽烷基、以乙醯基所代表的酯基、以乙烯基碳酸酯基所代表的碳酸酯基等。R63
係表示經氘取代的C1
-C6
烷基。]
化合物(5b)之合成
可藉由僅保護市售的、或經適當合成的化合物(5a)的2個羥基中之一個而得到化合物(5b)。例如:藉由將化合物(5a)在p-甲苯磺酸吡啶鹽等之酸存在下,以3,4-二氫-2H-吡喃處理,可得到僅將一個羥基以四氫吡喃基保護之化合物(5b)。但,羥基保護之方法並未特別限定於此等。反應中所使用之溶劑可列舉二氯甲烷、氯仿、甲苯或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(5a)而言,化合物(4b)、酸可使用0.01至過剩莫耳當量。較佳為可使用0.01至1莫耳當量。
化合物(5d)之合成
利用將化合物(5b)在碳酸鉀、碳酸銫、三級丁醇鉀或三乙基胺等之有機或無機鹼存在下,以化合物(5c)處理,而可得到化合物(5d)。可列舉N,N-二甲基甲醯胺、二甲亞碸、乙腈、丙酮或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(5b)而言,化合物(5c)、鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
化合物(5e)之合成
可藉由將化合物(5d)以鹽酸、乙酸、p-甲苯磺酸吡啶鹽等之酸處理,得到化合物(5e)。反應中所使用之溶劑可列舉乙醇、甲醇、異丙醇、水或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於化合物(5d)而言,化合物(4b)、酸可使用0.01至過剩莫耳當量。較佳為可使用0.01至1莫耳當量。
化合物(5f)之合成
利用將化合物(5e)以N-溴琥珀酸醯亞胺(N-Bromosuccinimide)、溴酸鉀等之溴化試劑處理,而可得到化合物(5f)。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、30%氫溴酸-乙酸溶液、乙酸或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。相對於溴化試劑而言,化合物(5e)可使用0.1至過剩莫耳當量。較佳為可使用0.1至5莫耳當量。
化合物(5g)之合成
可藉由與[製造法4]同樣的操作而得到。
化合物(5h)之合成
藉由將化合物(5g)在碳酸鉀、碳酸鈉、碳酸銫、乙酸鉀、磷酸鉀、三級丁氧化物或三乙基胺等之有機或無機鹼、參(二苯亞甲基丙酮)-二鈀(0)、雙(三苯基膦)氯化鈀(II)、[1,1’-雙(二苯基膦基)-二茂鐵]氯化鈀(II)二氯甲烷錯合物、[1,1’-雙(二苯基膦基)-二茂鐵]氯化鈀(II)二氯甲烷錯合物、1,3-雙(二苯基膦基)丙烷]氯化鎳(II)、等之過渡金屬觸媒、三苯基膦、三環己基膦、二苯亞甲基丙酮、1,3-雙(二苯基膦基)丙烷、2-二環己基膦基-2’,4’,6’-三異丙基聯苯、4,5-雙(二苯基膦基)-9,9-二甲基二苯并吡喃等之配位子存在下,以雙聯頻哪醇基二硼烷Bis(pinacolato)diboron、頻哪醇硼酸酯(pinacol boride)等之硼酸酯處理,而可得到。溶劑可列舉1,4-二烷、1,2-二甲氧基乙烷、甲醇、甲苯或此等混合溶劑,並未特別限定。反應溫度通常在0℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至溶劑之沸點的範圍。又,上述處理亦可藉由在密封管中、在微波下來進行。相對於化合物(5g)而言,有機或無機鹼可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。相對於化合物(5g)而言,過渡金屬觸媒、配位子可使用0.001至1莫耳當量。較佳為可使用0.05至1莫耳當量。相對於化合物(5g)而言,硼酸酯可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
[製造法6]
反應式6 [式中,R13
、R43
、R45
係與前述同義。]
化合物(6c)之合成
可使用市售的、或經適當合成的化合物(6a)及(6b),藉由與[製造法4]同樣的操作,而得到。
[式中,R13
、R43
、R45
係與前述同義。]
化合物(6c)之合成
可使用市售的、或經適當合成的化合物(6a)及(6b),藉由與[製造法4]同樣的操作,而得到。
[製造法7]
反應式7 [式中,R5
、R43
係與前述同義。R81
係表示鹵素基。化合物(7a)係包含化合物(4c)、化合物(5h)或化合物(6c)之硼酸、硼酸酯化合物。化合物(7c)及化合物(7d)係被化合物(C-I)包含在內。]
化合物(7c)之合成
可與[製造法1]中之化合物(1c)之合成同樣地,將化合物(7b)與市售的、或以製造法4、製造法5、製造法6所示之方法合成的化合物(7a)進行耦合反應,而得到化合物(7c)。
化合物(7d)之合成
可使用市售的、或經適當合成的化合物(7c),藉由與[製造法5]之化合物(5h)之合成同樣的操作,而得到。
接著說明關於化合物(B)化合物(C-I)複合物之合成法。
化合物(B)化合物(C-I)複合物可藉由如[製造法8]所示,將化合物(BC)之溴基轉換成R5
之方法合成,但並未特別限定。
[式中,R5
、R43
係與前述同義。R81
係表示鹵素基。化合物(7a)係包含化合物(4c)、化合物(5h)或化合物(6c)之硼酸、硼酸酯化合物。化合物(7c)及化合物(7d)係被化合物(C-I)包含在內。]
化合物(7c)之合成
可與[製造法1]中之化合物(1c)之合成同樣地,將化合物(7b)與市售的、或以製造法4、製造法5、製造法6所示之方法合成的化合物(7a)進行耦合反應,而得到化合物(7c)。
化合物(7d)之合成
可使用市售的、或經適當合成的化合物(7c),藉由與[製造法5]之化合物(5h)之合成同樣的操作,而得到。
接著說明關於化合物(B)化合物(C-I)複合物之合成法。
化合物(B)化合物(C-I)複合物可藉由如[製造法8]所示,將化合物(BC)之溴基轉換成R5
之方法合成,但並未特別限定。
[製造法8]
反應式8 [式中,R4
、R5
、W、X、Y、Z,R43
係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使用市售的、或經適當合成的化合物(BC)或化合物(8),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
又,化合物(8)亦可藉由於[製造法9]、[製造法10]所示之方法合成,但並未特別限定。
[式中,R4
、R5
、W、X、Y、Z,R43
係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使用市售的、或經適當合成的化合物(BC)或化合物(8),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
又,化合物(8)亦可藉由於[製造法9]、[製造法10]所示之方法合成,但並未特別限定。
[製造法9]
反應式9 [式中,R4
、R5
、W、X、Y、Z、R81
係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使用市售的、或經適當合成的化合物(9a)及(9b),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
[式中,R4
、R5
、W、X、Y、Z、R81
係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使用市售的、或經適當合成的化合物(9a)及(9b),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
[製造法10]
反應式10 [式中,R4
、R5
、W、X、Y、Z、R81
係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使用市售的、或經適當合成的化合物(7d)及化合物(10),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於使化合物(B)化合物(C-I)複合物與化合物(A-I)結合之合成法。
使化合物(B)化合物(C-I)複合物與化合物(A-I)結合,係可藉由以下之製造法11或製造法12進行。
[式中,R4
、R5
、W、X、Y、Z、R81
係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使用市售的、或經適當合成的化合物(7d)及化合物(10),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於使化合物(B)化合物(C-I)複合物與化合物(A-I)結合之合成法。
使化合物(B)化合物(C-I)複合物與化合物(A-I)結合,係可藉由以下之製造法11或製造法12進行。
[製造法11]
可以如下述反應式11之方法合成,但並未特別限定。
反應式11 [式中,R1
~R5
、W、X、Y、Z係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使化合物(A-I)與化合物(8)在縮合劑存在下反應而得到。作為縮合劑可列舉N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(N-[1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholino]-uronium hexafluorophosphate)(COMU)等,但並未限定於此。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二氯甲烷或此等混合溶劑,並未特別限定。反應溫度通常在-78℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至100℃的範圍。相對於化合物(A-I)而言,縮合劑可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
[式中,R1
~R5
、W、X、Y、Z係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
可使化合物(A-I)與化合物(8)在縮合劑存在下反應而得到。作為縮合劑可列舉N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(N-[1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholino]-uronium hexafluorophosphate)(COMU)等,但並未限定於此。反應中所使用之溶劑可列舉N,N-二甲基甲醯胺、二氯甲烷或此等混合溶劑,並未特別限定。反應溫度通常在-78℃至200℃或至溶劑之沸點的範圍,但較佳為0℃至100℃的範圍。相對於化合物(A-I)而言,縮合劑可使用1至過剩莫耳當量。較佳為可使用1至5莫耳當量。
[製造法12]
可以如下述反應式12之方法合成,但並未特別限定。
反應式12 [式中,R1
~R5
、W、X、Y、Z係與前述同義。化合物(8)係被化合物(B)化合物(C-I)之複合物包含在內。]
可將化合物(A-I)以亞硫醯氯、草醯氯等之酸鹵化試劑(acid halide agent)處理得到化合物(12)後,以化合物(8)處理,而得到。反應中所使用之溶劑可列舉二氯甲烷、二氯乙烷或此等混合溶劑,並未特別限定。相對於化合物(A-I)而言,酸鹵化試劑可使用0.9至過剩莫耳當量。較佳為可使用0.9至2莫耳當量。
接著說明關於化合物(A-I)化合物(B)複合物之合成法。
[式中,R1
~R5
、W、X、Y、Z係與前述同義。化合物(8)係被化合物(B)化合物(C-I)之複合物包含在內。]
可將化合物(A-I)以亞硫醯氯、草醯氯等之酸鹵化試劑(acid halide agent)處理得到化合物(12)後,以化合物(8)處理,而得到。反應中所使用之溶劑可列舉二氯甲烷、二氯乙烷或此等混合溶劑,並未特別限定。相對於化合物(A-I)而言,酸鹵化試劑可使用0.9至過剩莫耳當量。較佳為可使用0.9至2莫耳當量。
接著說明關於化合物(A-I)化合物(B)複合物之合成法。
[製造法13]
可以如下述反應式13之方法合成,但並未特別限定。
反應式13 [式中,R1
~R4
、W、X、Y、Z、R31
係與前述同義。化合物(13)係被化合物(A-I)化合物(B)複合物包含在內。]
化合物(13)之合成
可化合物(B)為市售品、或由市售品以文獻已知之方法可容易地合成。使用化合物(A-I)與市售的、或經適當合成的化合物(B),藉由與[製造法11]同樣的操作,而得到。
接著說明關於使化合物(A-I)化合物(B)複合物與化合物(C-I)結合之合成法。
[式中,R1
~R4
、W、X、Y、Z、R31
係與前述同義。化合物(13)係被化合物(A-I)化合物(B)複合物包含在內。]
化合物(13)之合成
可化合物(B)為市售品、或由市售品以文獻已知之方法可容易地合成。使用化合物(A-I)與市售的、或經適當合成的化合物(B),藉由與[製造法11]同樣的操作,而得到。
接著說明關於使化合物(A-I)化合物(B)複合物與化合物(C-I)結合之合成法。
[製造法14]
可以如下述反應式14之方法合成,但並未特別限定。
反應式14 [式中,R1
~R5
、W,X,Y,Z係與前述同義。R31
為鹵素基、三氟甲磺酸基等之情形,R32
係表示硼酸、硼酸酯。R31
為硼酸、硼酸酯的情形,R32
係表示鹵素基、三氟甲磺酸基等。化合物(13)係被化合物(A-I)化合物(B)複合物包含在內。]
化合物(11)之合成
可以使用化合物(13)與市售的、或經適當合成的化合物(C-I),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於使化合物(B)化合物(C-I)複合物與化合物(A-II)結合之合成法。
[式中,R1
~R5
、W,X,Y,Z係與前述同義。R31
為鹵素基、三氟甲磺酸基等之情形,R32
係表示硼酸、硼酸酯。R31
為硼酸、硼酸酯的情形,R32
係表示鹵素基、三氟甲磺酸基等。化合物(13)係被化合物(A-I)化合物(B)複合物包含在內。]
化合物(11)之合成
可以使用化合物(13)與市售的、或經適當合成的化合物(C-I),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於使化合物(B)化合物(C-I)複合物與化合物(A-II)結合之合成法。
[製造法15]
可以如下述反應式15之方法合成,但並未特別限定。
反應式15 [式中,R4
~R5
、W,X,Y,Z係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
化合物(15)之合成
可以使用化合物(A-II)與化合物(8),藉由與[製造法11]同樣的操作,而得到。
接著說明關於將化合物(A-II)化合物(B)化合物(C-I)複合物之溴基轉換成R1
之合成法。
[式中,R4
~R5
、W,X,Y,Z係與前述同義。化合物(8)係被化合物(B)化合物(C-I)複合物包含在內。]
化合物(15)之合成
可以使用化合物(A-II)與化合物(8),藉由與[製造法11]同樣的操作,而得到。
接著說明關於將化合物(A-II)化合物(B)化合物(C-I)複合物之溴基轉換成R1
之合成法。
[製造法16]
可以如下述反應式16之方法合成,但並未特別限定。
反應式16 [式中,R1
、R4
~R5
、W、X、Y、Z、R43
係與前述同義。化合物(15)係被化合物(A-I)化合物(B)化合物(C-I)複合物包含在內。]
化合物(16b)之合成
可以使用化合物(15)與市售的、或經適當合成的化合物(16a),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於將化合物(A-I)化合物(B)化合物(C-I)複合物之鹵素基轉換成烷基之合成法。
[式中,R1
、R4
~R5
、W、X、Y、Z、R43
係與前述同義。化合物(15)係被化合物(A-I)化合物(B)化合物(C-I)複合物包含在內。]
化合物(16b)之合成
可以使用化合物(15)與市售的、或經適當合成的化合物(16a),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於將化合物(A-I)化合物(B)化合物(C-I)複合物之鹵素基轉換成烷基之合成法。
[製造法17]
可以如下述反應式17之方法合成,但並未特別限定。
反應式17 [式中,R2
~R5
、Q、T、U、W、X、Y、Z、R43
、R44
係與前述同義。化合物(17a)、化合物(17b)係被化合物(A-I)化合物(B)化合物(C-I)複合物包含在內。]
化合物(17b)之合成
可以使用化合物(17a)與市售的、或經適當合成的化合物(1b),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於使化合物(A-I)與化合物(BC)結合後,將溴基轉換成R5
的合成法。
[式中,R2
~R5
、Q、T、U、W、X、Y、Z、R43
、R44
係與前述同義。化合物(17a)、化合物(17b)係被化合物(A-I)化合物(B)化合物(C-I)複合物包含在內。]
化合物(17b)之合成
可以使用化合物(17a)與市售的、或經適當合成的化合物(1b),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
接著說明關於使化合物(A-I)與化合物(BC)結合後,將溴基轉換成R5
的合成法。
[製造法18]
可以如下述反應式18之方法合成,但並未特別限定。
反應式18 [式中,R1
~R5
、W、X、Y、Z、R43
係與前述同義。]
化合物(18)之合成
可使用化合物(A-I)與化合物(BC),藉由與[製造法11]同樣的操作,而得到。
化合物(11)之合成
可使用化合物(18)與市售的、或經適當合成的化合物(7a),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
又,化合物(18)亦可藉由如[製造法19]之方法合成。
[式中,R1
~R5
、W、X、Y、Z、R43
係與前述同義。]
化合物(18)之合成
可使用化合物(A-I)與化合物(BC),藉由與[製造法11]同樣的操作,而得到。
化合物(11)之合成
可使用化合物(18)與市售的、或經適當合成的化合物(7a),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
又,化合物(18)亦可藉由如[製造法19]之方法合成。
[製造法19]
反應式19 [式中,R1
~R4
、R33
、R81
、W、X、Y、Z係與前述同義。化合物(19)係被化合物(13)包含在內。]
化合物(C-II)為市售品、或可由市售品以文獻已知之方法容易地合成。可使用化合物(19)與市售的、或經適當合成的化合物(C-II),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
又,化合物(11)亦可藉由製造法20製造。
[式中,R1
~R4
、R33
、R81
、W、X、Y、Z係與前述同義。化合物(19)係被化合物(13)包含在內。]
化合物(C-II)為市售品、或可由市售品以文獻已知之方法容易地合成。可使用化合物(19)與市售的、或經適當合成的化合物(C-II),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
又,化合物(11)亦可藉由製造法20製造。
[製造法20]
反應式20 [式中,R1
~R4
、R43
、W、X、Y、Z係與前述同義。R201
係表示氫原子或鹵素基。R202
係表示亦可形成環之C1
~C6
烷基胺基或亦可形成環之氧基C1
~C6
烷基胺基。化合物(20d)係被化合物(11)包含在內。]
化合物(20b)之合成
可使用化合物(18)與市售的、或經適當合成的化合物(20a),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
化合物(20d)之合成
可使化合物(20b)在還原劑存在下,與市售的、或經適當合成的化合物(20c)反應而得到。作為所使用的還原劑,可列舉三乙醯氧基硼氫鈉(Sodium Tri(acetoxy)borohydride)、氰基硼氫化鈉、硼氫化鈉等。作為所使用的溶劑,可列舉甲醇、乙醇、水、四氫呋喃、二烷、N,N-二甲基甲醯胺、二氯甲烷、乙酸等、或此等混合溶劑,並未特別限定。反應溫度通常在-78℃至100℃或至溶劑之沸點的範圍,但較佳為0℃至100℃的範圍。
[式中,R1
~R4
、R43
、W、X、Y、Z係與前述同義。R201
係表示氫原子或鹵素基。R202
係表示亦可形成環之C1
~C6
烷基胺基或亦可形成環之氧基C1
~C6
烷基胺基。化合物(20d)係被化合物(11)包含在內。]
化合物(20b)之合成
可使用化合物(18)與市售的、或經適當合成的化合物(20a),藉由與[製造法1]之化合物(1c)之合成同樣的操作,而得到。
化合物(20d)之合成
可使化合物(20b)在還原劑存在下,與市售的、或經適當合成的化合物(20c)反應而得到。作為所使用的還原劑,可列舉三乙醯氧基硼氫鈉(Sodium Tri(acetoxy)borohydride)、氰基硼氫化鈉、硼氫化鈉等。作為所使用的溶劑,可列舉甲醇、乙醇、水、四氫呋喃、二烷、N,N-二甲基甲醯胺、二氯甲烷、乙酸等、或此等混合溶劑,並未特別限定。反應溫度通常在-78℃至100℃或至溶劑之沸點的範圍,但較佳為0℃至100℃的範圍。
如前述,Gas6/Axl訊息傳導系統,由已報告有調節細胞存活(cell survival)、細胞分裂、自噬作用(autophagy)、細胞移動(migration)、血管新生、血小板凝集、NK細胞分化等各式各樣的細胞性反應(非專利文獻2),Axl抑制劑係可用於用以治療由Axl激酶功能亢進所致的疾病、與Axl激酶功能亢進相關的疾病、及/或伴隨Axl激酶功能亢進的疾病。
特別是,本發明之化合物係Axl抑制特異性高,由於亦不會引起視網膜毒性,故可更安全地使用。
就由Axl激酶功能亢進所致的疾病、與Axl激酶功能亢進相關的疾病、伴隨Axl激酶功能亢進的疾病而言,可列舉具有可見到Axl之基因及/或蛋白質的過剩表現的組織的疾病、具有可見到Axl之磷酸化活性上升的組織的疾病等。
作為上述疾病之例,可列舉例如:過度增殖性疾病、作為過度增殖性疾病之例,可列舉子宮內膜增殖症、凝血酶誘導血管平滑肌細胞(VSMC)增殖良性腫瘤、惡性腫瘤(癌)、急性及慢性絲球體腎炎、糖尿病性腎病變等,但並未限定於此等。
再者,Axl於免疫(非專利文獻12)、血小板功能(非專利文獻13)、精子形成(非專利文獻14)、血管鈣化(非專利文獻15)、(非專利文獻16)及各種腎臓疾病、慢性同種移植排斥反應(非專利文獻17)中亦呈現具有作用,Axl抑制劑係可用於治療血管疾病(包含血栓症、動脈粥狀硬化症及再狹窄,但未限於此等)及任意的血管生成嚴重的疾病(包含糖尿病性視網膜病變、網膜症、乾癬、類風溼性關節炎、粉瘤、卡波西肉瘤及血管瘤,但未限於此等)等多數疾病。
本發明之化合物由於會抑制Axl,故可用於治療上述記載之疾病。
更佳為,本發明之化合物係可用於治療各種癌症。就癌症而言,可列舉例如:乳癌、結腸癌、前列腺癌、肺癌、胃癌、卵巢癌、子宮頸癌、子宮內膜癌、子宮體癌、腎癌、肝細胞癌、甲狀腺癌、食道癌、鱗狀上皮細胞癌、白血病、骨肉瘤、黑色素瘤、神經膠肉瘤、神經母細胞瘤、卵巢癌、頭頸部癌、睾丸腫瘤、大腸癌、血液癌(hematologic cancer)、視網膜母細胞瘤、胰臓癌等,但未限定於此等之癌症。
關於癌症與Axl之關係,由增殖抑制、轉移・移動(migration)・侵入(invasion)抑制、耐藥性解除之觀點而言,已有各種報告。
有報告指出Axl之顯性負性突變體(dominant negative mutant)抑制腦腫瘤之增殖(Vajkoczy et al., PNAS 2006, 103, 5799)。有報告指出源自神經膠母細胞瘤(glioblastoma)患者的組織中,可見到Axl表現、Axl/Gas6共表現的情形,腫瘤增殖明顯地快速發展,患者生存期間縮短(Hutterer et al., Clin Cancer Res 14, 130 (2008))。有報告指出Axl之shRNA會抑制乳癌細胞之增殖(Yi-Xiang et al., Cancer Res 68, 1905 (2008))。由此等報告,可清楚知道Axl抑制劑可用於抑制癌症之細胞增殖。
另一方面,有報告指出Axl之顯性負性突變體可抑制細胞之移動(migration)及侵入(invasion)(Zhang et al., Cancer Res 68, 1905 (2008), Vajkoczy et al., PNAS 103, 5799 (2006), Holland et al., Cancer Res 65, 9294 (2005))。有報告指出Axl-shRNA於活體中可抑制轉移(Li et al., oncogene 28, 3442(2009))。有報告指出抗Axl抗體、siRNA於小鼠模式可抑制腫瘤增殖與轉移(Li et al., Oncogene 28, 3442(2009), Ye et al., Oncogene 29, 5254(2010))。有報告指出Axl促進細胞之侵入(invasion)(Tai et al., Oncogene 27, 4044(2008))。有報告指出為Axl抑制劑的R-428可抑制轉移性乳癌之擴散模式(Holland et al., Cancer Res 70, 1544(2010))。有報告指出Axl抗體、Axl之shRNA及Axl抑制劑NA80x1可抑制乳癌細胞之移動、侵入(invasion)(Yi-Xiang et al., Cancer Res 68, 1905(2008))。此外,亦有複數報告指出Axl係參與前列腺癌、脾臓癌、轉移性卵巢癌、胸腺癌等之轉移、惡性化。由此等報告可清楚明白Axl抑制劑可用於癌症之轉移、細胞之移動(migration)、侵入(invasion)的抑制、治療、預防等。
又,報告指出Axl抑制劑解除胃癌中的伊馬替尼(imatinib)耐受性(Mahadevan et al., Oncogene 26, 3909 (2007))。於阿黴素(doxorubicin)、VP16、順鉑(cisplatin)等之化學療法劑耐受性中,於急性骨髓性白血病中呈現Axl被誘導(Hong et al., Cancer Letters 268, 314 (2008))。報告指出HER-2陽性乳癌細胞中的拉帕替尼(lapatinib)耐受性中可見到Axl的活性化(Liu et al., Cancer Res 69, 6871(2009))。報告指出Axl參與PLX4032(vemurafenib)耐受性機制(Johannessen et al., Nature 468, 968(2010))。此外,報告指出Axl參與替莫唑胺(temozolomode)、卡鉑(carboplatin)、長春新鹼(vincristine)耐受性(AK Keeating et al., Mol Cancer Ther 9(5), 1298(2010))。由此等報告,Axl抑制劑係可清楚明白可用於耐藥性之解除,例如:對於各種抗癌劑的耐受性之解除。
本案的Axl抑制劑,如本案試驗例所示,藉由與埃羅替尼併用投與,可抑制腫瘤對埃羅替尼的耐受性化。又,由藉由投與埃羅替尼可觀察到Axl蛋白質之有意的誘導,認為Axl參與癌症的耐藥性之獲得。
又,對於對埃羅替尼產生耐受性的腫瘤,藉由併用本案之Axl抑制劑,由於可以恢復對埃羅替尼的反應性,因此認為Axl抑制劑對癌症的耐藥性之解除為有效。
再者,報告指出Axl參與腎臓之纖維化、糖尿病性腎病變等之腎疾病(特表2005-517412),可清楚知道Axl抑制劑可用於上述腎疾病,此外可用於特發性肺纖維症等之纖維化疾病之治療。
化合物之Axl抑制活性可藉由例如:本案之試驗例記載的方法來測定,但未限定於此。
細胞之增殖抑制活性係可使用本技術領域者通常使用的增殖抑制試驗法來檢查。細胞之增殖抑制活性係可藉由比較於試驗化合物之存在下或不存在下的細胞(例如:腫瘤細胞)之增殖程度來實施。增殖程度例如:可使用測定活細胞的試驗系統來檢查。作為活細胞之測定方法,可列舉例如:[3
H]-胸苷之攝取試驗、BrdU法或MTT分析等。
又,活體內之抗腫瘤活性可使用本技術領域者通常使用的抗腫瘤試驗法來檢查。例如,將各種腫瘤細胞移植至小鼠、大鼠等,確認移植細胞之存活附著後,將本發明之化合物經口投與、靜脈內投與等,數日~數週後,藉由比較無藥劑投與組中的腫瘤增殖與化合物投與組中的腫瘤增殖,可確認本發明之活體內的抗腫瘤活性。
其他,有報告指出關於轉移抑制活性、侵入(invasion)抑制活性、移動(migration)抑制活性、耐藥性解除活性,與Axl之各自之活性的關連性,而可以上述所列舉的文獻中記載之試驗方法來測定。
本發明之醫藥組成物包含本發明之化合物與藥學上容許的載體,其可作為靜脈內注射、肌肉內注射、皮下注射等之各種注射劑,或可藉由經口投與或經皮投與等之各式各樣的方法投與。所謂的藥學上容許的載體係指參與將本發明之化合物或含有本發明之化合物的組成物從某器官或臓器輸送至其他器官或臓器的藥學容許材料(例如:賦形劑、稀釋劑、添加劑、溶劑等)。
就製劑之調製方法而言,係因應投與而選擇適當的製劑(例如:口服劑或注射劑),其可以通常所使用之各種製劑的調製法調製。作為口服劑可例示例如:錠劑、散劑、顆粒劑、膠囊劑、丸劑、片劑(troche)、溶液劑、糖漿劑、酏劑、乳劑(emulsion)、或油性或水性之懸浮液等。經口投與之情形,無論是直接以游離體或鹽型皆可。水性製劑可以利用與藥學容許酸形成酸加成物、或做成鈉等之鹼金屬鹽來調製。注射劑的情形,可於製劑中使用安定劑、防腐劑或溶解輔助劑等。亦可將可能包含此等輔助劑等的溶液收納於容器後,藉由冷凍乾燥等做成固形製劑而作為使用時調製之製劑。又,可將一次投與量收納於一個容器中,亦可將複數次投與量收納於一個容器中。
作為固形製劑,可列舉例如:錠劑、散劑、顆粒劑、膠囊劑、丸劑、或片劑。此等固形製劑含有本發明之化合物的同時,亦可含有藥學容許的添加物。作為添加物可列舉例如:填充劑類、增量劑類、結合劑類、崩解劑類、溶解促進劑類、濕潤劑類或潤滑劑類,因應需要選擇並混合此等,而可加以製劑化。
作為液體製劑,可列舉例如:溶液劑、糖漿劑、酏劑、乳劑、或懸浮劑。此等液體製劑含有本發明之化合物的同時亦可含有藥學容許的添加物。作為添加物,可列舉例如:懸浮化劑或乳化劑,因應需要選擇而混合此等,而可製劑化。
本發明之化合物可用於哺乳類、尤其是用於人類的癌症治療。投與量及投與間隔係依疾病的部位、患者的身高、體重、性別或病歷,根據醫師的判斷而可適當地選擇。將本發明之化合物對人類投與的情形,投與量範圍係平均1日約0.01mg/kg體重~約500mg/kg體重,較佳為約0.1mg/kg體重~約100mg/kg體重。對人類投與的情形較佳為每1日1回,或分2至4次投與,較佳為以適當的間隔重複。又,1日量係由醫師判斷,按需要亦可超過上述之量。
本發明之化合物亦可與其他抗腫瘤劑併用而來使用。除了如上述所示的埃羅替尼的酪胺酸激酶抑制劑(TKI)之外,可列舉例如:抗腫瘤抗生素、抗腫瘤性植物成分、BRM(生物反應調節劑(biological response modifiers))、賀爾蒙、維他命、抗腫瘤性抗體、其他分子標的藥、其他抗腫瘤劑等。
更具體而言,作為烷基化劑,可列舉例如:氮芥(nitrogen mustard)、氮芥N-氧化物或氯芥苯丁酸(chlorambucil)等之烷基化劑、卡波醌(carboquone)或塞替派(thiotepa)等之氮丙啶系烷基化劑、二溴甘露糖醇(dibromomannitol)或二溴半乳糖醇(dibromodulcitol)等之環氧化物系烷基化劑、卡莫司汀(carmustine)、洛莫司丁(lomustinum)、司莫司汀(semustine)、尼莫司汀(nimustine)鹽酸鹽、鏈脲黴素(streptozocin)、氯脲黴素(chlorozotocin)或雷莫司汀(ranimustine)等之亞硝基脲系烷基化劑、白消安(busulfan)、英丙舒凡甲苯磺酸鹽(improsulfan tosilate)或達卡巴嗪(dacarbazine)等。
作為各種代謝拮抗劑,可列舉例如:6-巰基嘌呤、6-硫鳥嘌呤或硫肌苷(thioinosine)等之嘌呤代謝拮抗劑、氟尿嘧啶、替加氟(tegafur)、替加氟・尿嘧啶、卡莫氟(carmofur)、去氧氟尿苷(doxifluridine)、溴尿苷(broxuridine)、阿糖胞苷(cytarabine)或是伊洛胞苷(enocitabin)等之嘧啶代謝拮抗劑、胺甲喋呤(methotrexate)或三甲蝶呤(trimetrexate)等之葉酸代謝拮抗劑等。
作為抗腫瘤性抗生素可列舉例如:絲裂黴素C(mitomycin C)、博來黴素(bleomycin)、培洛黴素(peplomycin)、道諾黴素(daunorubicin)、阿克拉黴素(aclarubicin)、阿黴素(doxorubicin)、吡柔比星(pirarubicin)、THP-亞德利亞黴素(THP-adriamycin)、4’-表阿黴素(4’-epidoxorubicin)或表柔比星(epirubicin)等之蒽環(anthracycline)系抗生素抗腫瘤劑、色黴素A 3(chromomycin A 3)或放線菌素D(Actinomycin D)等。
作為抗腫瘤性植物成分,可列舉例如:長春地辛(vindesine)、長春新鹼或是長春鹼(vinblastine)等之長春花生物鹼(vinca alkaloid)類、太平洋紫杉醇(paclitaxel)、多烯紫杉醇(docetaxel)等之紫杉烷(taxane)類、或依托泊(etoposide)或替尼泊甙(teniposide)等之鬼臼毒素(epipodophyllotoxin)類。
作為BRM,可列舉例如:腫瘤壞死因子或吲哚美辛(indomethacin)等。
作為賀爾蒙,可列舉例如:氫化可體松(hydrocortisone)、地塞米松(dexamethasone)、甲基去氫皮質醇(methylprednisolone)、去氫皮質醇(prednisolone)、去氫異雄甾酮(prasterone)、貝皮質醇(betamethasone)、曲安西隆(triamcinolone)、羥甲睪丸素(oxymetholone)、睾甾酮(nandrolone)、美替諾隆(metenolone)、磷雌酚(fosfestrol)、乙炔雌二醇(ethinyl estradiol)、氯地孕酮(chlormadinone)或甲羥助孕酮(medroxyprogesterone)等。
作為維他命,可列舉例如:維他命C或維他命A等。
作為抗腫瘤性抗體、分子標的藥,可列舉曲妥珠單抗(trastuzumab)、利妥昔單抗(rituximab)、西妥昔單抗(cetuximab)、尼妥單珠單抗(nimotuzumab)、地諾單抗(denosumab)、貝伐單抗(bevacizumab)、英夫利昔單抗(infliximab)、甲磺酸伊馬替尼(imatinib mesilate)、吉非替尼(gefitinib)、埃羅替尼(erlotinib)、舒尼替尼(sunitinib)、拉帕替尼(lapatinib)、索拉非尼(sorafenib)、達沙替尼(dasatinib)、尼羅替尼(nilotinib)、維姆非尼(vemurafenib)、替凡替尼(tivantinib)等。
作為其他抗腫瘤劑,可列舉例如:順鉑、卡鉑、奧沙利鉑(oxaliplatin)、它莫西芬(tamoxifen)、喜樹鹼(camptothecin)、依弗醯胺(ifosfamide)、環磷醯胺(cyclophosphamide)、黴法蘭(melphalan)、L-天冬醯胺酸酶(L-asparaginase)、醋葡醛內酯(aceglatone)、西佐喃(sizofiran)、必醫你舒(Picibanil)、丙卡巴肼(procarbazine)、哌泊溴烷(pipobroman)、新制癌菌素(neocarzinostatin)、羥基脲、烏苯美司(ubenimex)或雲芝多糖(krestin)等。
本發明亦包含以投與本發明化合物或其鹽為特徵之癌症預防方法及/或治療方法。
再者,本發明亦包含用以製造前述醫藥之本發明之化合物、其鹽或彼等之溶劑合物之用途。
藉由以下所示之實施例具體說明本發明,但本發明並未限定於此等,此等之任何意義不被作限定地解釋。又,本說明書中,未特別記載的試劑、溶劑及起始材料可由市售之供給源容易獲得。
[實施例]
簡稱
DMF: N,N-二甲基甲醯胺
THF:四氫呋喃
HATU: N,N,N',N'-四甲基-O-(7-吖苯并三唑-1-基)脲鎓六氟磷酸鹽
HOAt: 1-羥基-7-吖苯并三唑
DIPEA: N,N-二異丙基乙基胺
COMU:(1-氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基-啉基-碳正離子六氟磷酸鹽(1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate
TFA:三氟乙酸
EDC・HCl: 1-乙基-3-(3-二甲基胺基丙基)碳二亞胺(carbodiimide)鹽酸鹽
HOBt: 1-羥基苯并三唑
DMAP: 4-二甲基胺基吡啶
PLC:製備用薄層層析(preparative thin-layer chromatography)
HPLC:高效液相層析
(中間體1a)5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3甲酸 (步驟1)4-(4-甲基苯基)-
3-側氧基丁酸乙酯
將75.0g(499mmol)之(4-甲基苯基)乙酸溶解於1.00L之四氫呋喃中,添加105g(649mmol)之羰基二咪唑,於室溫攪拌40分鐘。於反應液中添加111g(649mmol)之丙二酸單乙基酯鉀鹽、57.1g(599mmol)之無水氯化鎂,於60℃加熱攪拌6小時。將反應液冷卻至室溫,添加2.00L之乙酸乙酯,以1.00L之1當量鹽酸洗淨。將有機層以飽和碳酸氫鈉水、飽和食鹽水依序洗淨。將有機層以無水硫酸鈉乾燥後,將溶劑於減壓下餾去。將所得到之油狀物質於減壓下與甲苯共沸,得到86.8g(產率78.8%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) g(47.18-7.07 (4H, m), 4.17 (2H, q, J = 7.5 Hz), 3.78 (2H, s), 3.43 (2H, s), 2.33 (3H, s), 1.26 (3H, t, J = 7.5 Hz).
MS (ESI) m/z : 221 [(M+H)+
].
(步驟2)5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸乙酯
將43.0g(195mmol)之4-(4-甲基苯基)-
3-側氧基丁酸乙酯溶解於390ml之甲苯,添加130ml(976mmol)之N,N-二甲基甲醯胺二甲基縮醛,藉由Dean-Stark裝置邊捕集生成的甲醇,邊於100℃加熱攪拌5小時。將反應液冷卻至室溫,於減壓下濃縮。將殘渣溶解於390ml之乙醇,添加26.0ml(215mmol)之四氫-2H-吡喃-4-基甲胺,於60℃加熱攪拌9小時。將反應液冷卻至室溫,於減壓下濃縮。將殘渣使用矽膠管柱層析(SHOKO scientific公司,洗提溶劑:二氯甲烷/乙酸乙酯=75/25~20/80)純化,得到36.9g(產率53.2%)之呈焦糖狀物質的標題化合物。1
H-NMR(CDCl3
) δ: 8.10 (1H, d, J = 2.4 Hz), 7.48 (2H, d, J = 8.0 Hz), 7.32 (1H, d, J = 2.4 Hz), 7.20 (2H, d, J = 8.0 Hz), 4.38 (2H, q, J = 7.0 Hz), 4.02 (2H, dd, J = 11.5, 4.0 Hz), 3.74 (2H, d, J = 7.3 Hz), 3.38 (2H, td, J = 11.5, 2.0 Hz), 2.08-2.00 (1H, m), 1.63-1.56 (2H, m), 1.35-1.42 (5H, m).
MS (APCI) m/z : 356 [(M+H)+
].
(步驟3)5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
將36.9g(104mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸乙酯溶解於519ml之甲醇,添加311ml(311mmol)之1當量氫氧化鈉水溶液,於室溫攪拌1小時。將反應液於減壓下濃縮,於殘渣中添加400ml之1當量鹽酸,以二氯甲烷萃取。將有機層以無水硫酸鈉乾燥後,將溶劑於減壓下餾去。將殘渣以甲醇進行漿體洗淨,得到31.6g(產率92.9%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 8.46 (1H, d, J = 2.4 Hz), 7.56 (1H, d, J = 2.4 Hz), 7.48 (2H, d, J = 8.5 Hz), 7.28 (2H, d, J = 8.5 Hz), 4.03 (2H, dd, J = 11.3, 4.0 Hz), 3.89 (2H, d, J = 7.5 Hz), 3.38 (2H, t, J = 11.3 Hz), 2.40 (3H, s), 2.16-2.04 (1H, m), 1.62-1.53 (3H, m), 1.49-1.37 (2H, m).
MS (APCI) m/z : 328 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表1之中間體。
[表1-1]
(步驟1)4-(4-甲基苯基)-
3-側氧基丁酸乙酯
將75.0g(499mmol)之(4-甲基苯基)乙酸溶解於1.00L之四氫呋喃中,添加105g(649mmol)之羰基二咪唑,於室溫攪拌40分鐘。於反應液中添加111g(649mmol)之丙二酸單乙基酯鉀鹽、57.1g(599mmol)之無水氯化鎂,於60℃加熱攪拌6小時。將反應液冷卻至室溫,添加2.00L之乙酸乙酯,以1.00L之1當量鹽酸洗淨。將有機層以飽和碳酸氫鈉水、飽和食鹽水依序洗淨。將有機層以無水硫酸鈉乾燥後,將溶劑於減壓下餾去。將所得到之油狀物質於減壓下與甲苯共沸,得到86.8g(產率78.8%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) g(47.18-7.07 (4H, m), 4.17 (2H, q, J = 7.5 Hz), 3.78 (2H, s), 3.43 (2H, s), 2.33 (3H, s), 1.26 (3H, t, J = 7.5 Hz).
MS (ESI) m/z : 221 [(M+H)+
].
(步驟2)5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸乙酯
將43.0g(195mmol)之4-(4-甲基苯基)-
3-側氧基丁酸乙酯溶解於390ml之甲苯,添加130ml(976mmol)之N,N-二甲基甲醯胺二甲基縮醛,藉由Dean-Stark裝置邊捕集生成的甲醇,邊於100℃加熱攪拌5小時。將反應液冷卻至室溫,於減壓下濃縮。將殘渣溶解於390ml之乙醇,添加26.0ml(215mmol)之四氫-2H-吡喃-4-基甲胺,於60℃加熱攪拌9小時。將反應液冷卻至室溫,於減壓下濃縮。將殘渣使用矽膠管柱層析(SHOKO scientific公司,洗提溶劑:二氯甲烷/乙酸乙酯=75/25~20/80)純化,得到36.9g(產率53.2%)之呈焦糖狀物質的標題化合物。1
H-NMR(CDCl3
) δ: 8.10 (1H, d, J = 2.4 Hz), 7.48 (2H, d, J = 8.0 Hz), 7.32 (1H, d, J = 2.4 Hz), 7.20 (2H, d, J = 8.0 Hz), 4.38 (2H, q, J = 7.0 Hz), 4.02 (2H, dd, J = 11.5, 4.0 Hz), 3.74 (2H, d, J = 7.3 Hz), 3.38 (2H, td, J = 11.5, 2.0 Hz), 2.08-2.00 (1H, m), 1.63-1.56 (2H, m), 1.35-1.42 (5H, m).
MS (APCI) m/z : 356 [(M+H)+
].
(步驟3)5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
將36.9g(104mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸乙酯溶解於519ml之甲醇,添加311ml(311mmol)之1當量氫氧化鈉水溶液,於室溫攪拌1小時。將反應液於減壓下濃縮,於殘渣中添加400ml之1當量鹽酸,以二氯甲烷萃取。將有機層以無水硫酸鈉乾燥後,將溶劑於減壓下餾去。將殘渣以甲醇進行漿體洗淨,得到31.6g(產率92.9%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 8.46 (1H, d, J = 2.4 Hz), 7.56 (1H, d, J = 2.4 Hz), 7.48 (2H, d, J = 8.5 Hz), 7.28 (2H, d, J = 8.5 Hz), 4.03 (2H, dd, J = 11.3, 4.0 Hz), 3.89 (2H, d, J = 7.5 Hz), 3.38 (2H, t, J = 11.3 Hz), 2.40 (3H, s), 2.16-2.04 (1H, m), 1.62-1.53 (3H, m), 1.49-1.37 (2H, m).
MS (APCI) m/z : 328 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表1之中間體。
[表1-1] 
[表1-2] 
[表1-3] 
[表1-4] 
[表1-5] 
(中間體2)5-(4-環丙基-2-氟苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 (步驟1)(4-溴-2-氟苯基)乙酸乙酯
於2.00g(8.58mmol)之(4-溴-2-氟苯基)乙酸的40.0mL二氯甲烷溶液中,添加0.969mL(0.791g,17.2mmol)之乙醇、0.105g(0.858mmol)之4-二甲基胺基吡啶、1.97g(10.3mmol)之1-(3-二甲基胺基丙基)-3-乙基碳二亞胺鹽酸鹽(EDAC・HCl),於室溫攪拌整夜。於反應液添加1當量鹽酸,以二氯甲烷萃取3次,將有機層以飽和碳酸氫鈉水、飽和食鹽水之順序洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=(100/0~87/13))純化,得到1.96g(產率87.5%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 7.27-7.24 (2H, m), 7.18-7.14 (1H, m), 4.17 (2H, q, J = 7.0 Hz), 3.62 (2H, s), 1.26 (3H, t, J = 7.0 Hz).
(步驟2)(4-環丙基-2-氟苯基)乙酸乙酯
於1.95g(7.47mmol)之於步驟1合成之(4-溴-2-氟苯基)乙酸乙酯之30.0mL甲苯懸浮液中,添加0.962g(11.2mmol)之環丙基硼酸、0.419g(1.49mmol)之三環己基膦、5.55g(26.1mmol)之磷酸三鉀、6.00mL之水,經氮取代後,添加0.168g(0.747mmol)之乙酸鈀(II),於100℃攪拌2小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=100/0~87/13)純化,進一步以胺基矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=100/0~87/13)純化,得到1.59g(產率95.8%)之呈油狀物的標題化合物。1
H-NMR (CDCl3
) δ: 7.12 (1H, t, J = 7.9 Hz), 6.84-6.82 (1H, m), 6.76-6.72 (1H, m), 4.16 (2H, q, J = 7.0 Hz), 3.61 (2H, s), 1.90-1.83 (1H, m), 1.25 (3H, t, J = 7.0 Hz), 1.00-0.95 (2H, m), 0.70-0.66 (2H, m).
MS (APCI) m/z : 223 [(M+H)+
].
(步驟3)(4-環丙基-2-氟苯基)乙酸
於1.59g(7.15mmol)之於步驟2合成之(4-環丙基-2-氟苯基)乙酸乙酯之30.0mL四氫呋喃溶液中,添加15.0mL之甲醇與14.3mL(14.3mmol)之1mol/L氫氧化鈉水溶液,於室溫攪拌整夜。將有機溶劑減壓餾去後,於殘渣中添加水,以二乙基醚洗淨。於水層添加1當量鹽酸而呈酸性後,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。藉由過濾、減壓濃縮,得到1.36g(7.00mmol)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 7.11 (1H, t, J = 7.9 Hz), 6.83 (1H, dd, J = 7.9, 1.8 Hz), 6.75 (1H, dd, J = 11.2, 1.8 Hz), 3.65 (2H, s), 1.90-1.83 (1H, m), 1.00-0.95 (2H, m), 0.70-0.66 (2H, m).
(步驟4)5-(4-環丙基-2-氟苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
使用(4-環丙基-2-氟苯基)乙酸代替中間體1a之步驟1之(4-甲基苯基)乙酸,其後,與中間體1a之步驟1至步驟3同樣地進行反應,得到呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.80 (1H, d, J = 2.4 Hz), 8.31 (1H, d, J = 2.4 Hz), 7.34 (1H, t, J = 8.2 Hz), 7.03-6.99 (2H, m), 4.12 (2H, d, J = 7.3 Hz), 3.85 (2H, dd, J = 11.2, 3.3 Hz), 3.24 (2H, t, J = 11.2 Hz), 2.12-1.97 (2H, m), 1.43 (2H, br d, J = 10.9 Hz), 1.33-1.22 (2H, m), 1.04-0.99 (2H, m), 0.77-0.73 (2H, m).
MS (APCI) m/z : 372 [(M+H)+
].
(步驟1)(4-溴-2-氟苯基)乙酸乙酯
於2.00g(8.58mmol)之(4-溴-2-氟苯基)乙酸的40.0mL二氯甲烷溶液中,添加0.969mL(0.791g,17.2mmol)之乙醇、0.105g(0.858mmol)之4-二甲基胺基吡啶、1.97g(10.3mmol)之1-(3-二甲基胺基丙基)-3-乙基碳二亞胺鹽酸鹽(EDAC・HCl),於室溫攪拌整夜。於反應液添加1當量鹽酸,以二氯甲烷萃取3次,將有機層以飽和碳酸氫鈉水、飽和食鹽水之順序洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=(100/0~87/13))純化,得到1.96g(產率87.5%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 7.27-7.24 (2H, m), 7.18-7.14 (1H, m), 4.17 (2H, q, J = 7.0 Hz), 3.62 (2H, s), 1.26 (3H, t, J = 7.0 Hz).
(步驟2)(4-環丙基-2-氟苯基)乙酸乙酯
於1.95g(7.47mmol)之於步驟1合成之(4-溴-2-氟苯基)乙酸乙酯之30.0mL甲苯懸浮液中,添加0.962g(11.2mmol)之環丙基硼酸、0.419g(1.49mmol)之三環己基膦、5.55g(26.1mmol)之磷酸三鉀、6.00mL之水,經氮取代後,添加0.168g(0.747mmol)之乙酸鈀(II),於100℃攪拌2小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=100/0~87/13)純化,進一步以胺基矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=100/0~87/13)純化,得到1.59g(產率95.8%)之呈油狀物的標題化合物。1
H-NMR (CDCl3
) δ: 7.12 (1H, t, J = 7.9 Hz), 6.84-6.82 (1H, m), 6.76-6.72 (1H, m), 4.16 (2H, q, J = 7.0 Hz), 3.61 (2H, s), 1.90-1.83 (1H, m), 1.25 (3H, t, J = 7.0 Hz), 1.00-0.95 (2H, m), 0.70-0.66 (2H, m).
MS (APCI) m/z : 223 [(M+H)+
].
(步驟3)(4-環丙基-2-氟苯基)乙酸
於1.59g(7.15mmol)之於步驟2合成之(4-環丙基-2-氟苯基)乙酸乙酯之30.0mL四氫呋喃溶液中,添加15.0mL之甲醇與14.3mL(14.3mmol)之1mol/L氫氧化鈉水溶液,於室溫攪拌整夜。將有機溶劑減壓餾去後,於殘渣中添加水,以二乙基醚洗淨。於水層添加1當量鹽酸而呈酸性後,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。藉由過濾、減壓濃縮,得到1.36g(7.00mmol)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 7.11 (1H, t, J = 7.9 Hz), 6.83 (1H, dd, J = 7.9, 1.8 Hz), 6.75 (1H, dd, J = 11.2, 1.8 Hz), 3.65 (2H, s), 1.90-1.83 (1H, m), 1.00-0.95 (2H, m), 0.70-0.66 (2H, m).
(步驟4)5-(4-環丙基-2-氟苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
使用(4-環丙基-2-氟苯基)乙酸代替中間體1a之步驟1之(4-甲基苯基)乙酸,其後,與中間體1a之步驟1至步驟3同樣地進行反應,得到呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.80 (1H, d, J = 2.4 Hz), 8.31 (1H, d, J = 2.4 Hz), 7.34 (1H, t, J = 8.2 Hz), 7.03-6.99 (2H, m), 4.12 (2H, d, J = 7.3 Hz), 3.85 (2H, dd, J = 11.2, 3.3 Hz), 3.24 (2H, t, J = 11.2 Hz), 2.12-1.97 (2H, m), 1.43 (2H, br d, J = 10.9 Hz), 1.33-1.22 (2H, m), 1.04-0.99 (2H, m), 0.77-0.73 (2H, m).
MS (APCI) m/z : 372 [(M+H)+
].
(中間體3)5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲酸 (步驟1)4-(5-甲基吡啶-2-基)-3-側氧基丁酸乙酯
於0.500g(3.31mmol)之(5-甲基吡啶-2-基)乙酸之15.0mL四氫呋喃溶液中,添加0.805g(4.96mmol)之1,1’-羰基雙-1H-咪唑,於室溫攪拌2小時半。於別的反應容器中,將1.69g(9.92mmol)之丙二酸單乙酯鉀鹽與0.945g(9.92mmol)之氯化鎂懸浮於25.0ml之四氫呋喃中,於冰冷下,添加2.38mL(1.74g, 17.2mmol)之三乙基胺,於室溫攪拌2小時半後,再次冰冷,添加於上述調整之活性酯的溶液,於室溫攪拌整夜。於反應液中添加10%檸檬酸水溶液,以乙酸乙酯萃取5次,將有機層以飽和碳酸氫鈉水、飽和食鹽水之順序洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;n-己烷/乙酸乙酯=57/43~36/64)純化,得到0.634g(產率86.6%)之呈油狀物的標題化合物。
MS (APCI) m/z : 222 [(M+H)+
].
(步驟2、3)5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲酸
使用4-(5-甲基吡啶-2-基)-3-側氧基丁酸 乙酯取代中間體1a之步驟2之4-(4-甲基苯基)-
3-側氧基丁酸 乙酯,與中間體1a之步驟2至步驟3同樣地進行反應,得到呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.86 (1H, d, J = 2.4 Hz), 8.80 (1H, d, J = 2.4 Hz), 8.53 (1H, s), 8.38 (1H, d, J = 8.5 Hz), 7.72 (1H, dd, J = 8.5, 2.1 Hz), 4.24 (2H, d, J = 7.3 Hz), 3.84 (2H, dd, J = 11.2, 3.3 Hz), 3.24 (2H, t, J = 11.2 Hz), 2.35 (3H, s), 2.11-2.01 (1H, m), 1.44 (2H, br d, J = 10.9 Hz), 1.35-1.24 (2H, m).
MS (APCI) m/z : 329 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表2之中間體。
[表2]
(步驟1)4-(5-甲基吡啶-2-基)-3-側氧基丁酸乙酯
於0.500g(3.31mmol)之(5-甲基吡啶-2-基)乙酸之15.0mL四氫呋喃溶液中,添加0.805g(4.96mmol)之1,1’-羰基雙-1H-咪唑,於室溫攪拌2小時半。於別的反應容器中,將1.69g(9.92mmol)之丙二酸單乙酯鉀鹽與0.945g(9.92mmol)之氯化鎂懸浮於25.0ml之四氫呋喃中,於冰冷下,添加2.38mL(1.74g, 17.2mmol)之三乙基胺,於室溫攪拌2小時半後,再次冰冷,添加於上述調整之活性酯的溶液,於室溫攪拌整夜。於反應液中添加10%檸檬酸水溶液,以乙酸乙酯萃取5次,將有機層以飽和碳酸氫鈉水、飽和食鹽水之順序洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;n-己烷/乙酸乙酯=57/43~36/64)純化,得到0.634g(產率86.6%)之呈油狀物的標題化合物。
MS (APCI) m/z : 222 [(M+H)+
].
(步驟2、3)5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲酸
使用4-(5-甲基吡啶-2-基)-3-側氧基丁酸 乙酯取代中間體1a之步驟2之4-(4-甲基苯基)-
3-側氧基丁酸 乙酯,與中間體1a之步驟2至步驟3同樣地進行反應,得到呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.86 (1H, d, J = 2.4 Hz), 8.80 (1H, d, J = 2.4 Hz), 8.53 (1H, s), 8.38 (1H, d, J = 8.5 Hz), 7.72 (1H, dd, J = 8.5, 2.1 Hz), 4.24 (2H, d, J = 7.3 Hz), 3.84 (2H, dd, J = 11.2, 3.3 Hz), 3.24 (2H, t, J = 11.2 Hz), 2.35 (3H, s), 2.11-2.01 (1H, m), 1.44 (2H, br d, J = 10.9 Hz), 1.35-1.24 (2H, m).
MS (APCI) m/z : 329 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表2之中間體。
[表2] 
(中間體4a)5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 (步驟1)4-(4-溴苯基)-3-側氧基丁酸 乙酯
使用2-(4-溴苯基)乙酸代替中間體1a之步驟1之(4-甲基苯基)乙酸,以下以同樣之操作進行反應,得到呈油狀物的標題化合物。1
H-NMR (CDCl3
) δ: 7.47 (2H, d, J = 8.5 Hz), 7.09 (2H, d, J = 8.5 Hz), 4.18 (2H, q, J = 7.1 Hz), 3.81 (2H, s), 3.46 (2H, s), 1.27 (3H, t, J = 7.1 Hz).
(步驟2)5-(4-溴苯基)-4-側氧-1,4-二氫吡啶-3-甲酸乙酯
於5.00g(17.5mmol)之於步驟1合成之4-(4-溴苯基)-3-側氧基丁酸 乙酯之50.0mL乙醇溶液中,添加1.56g(19.3mmol)之1,3,5-三與1.43g(21.0mmol)之乙醇鈉,於85℃攪拌4小時。放冷至室溫後,減壓濃縮,於所得到之殘渣中添加1當量鹽酸而呈酸性後,濾取析出之固體,將固體以丙酮洗淨、乾燥,得到3.73g之呈固體的標題化合物之粗純化物。
MS (APCI) m/z : 322 [(M+H)+
].
(步驟3)5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 乙酯
於0.322g之於步驟2合成之5-(4-溴苯基)-4-側氧-1,4-二氫吡啶-3-甲酸 乙酯之粗純化物之45.0mL N,N-二甲基甲醯胺懸浮液中,添加0.977g(3.00mmol)之碳酸銫,於80℃攪拌15分鐘後,添加0.583g(3.00mmol)之甲磺酸四氫-2H-吡喃-4-基甲酯,於80℃攪拌4小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;n-己烷/乙酸乙酯=57/43~36/64)純化,得到0.234g(2步驟產率36.7%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.30 (1H, d, J = 2.4 Hz), 8.02 (1H, d, J = 2.4 Hz), 7.59 (4H, s), 4.20 (2H, q, J = 7.0 Hz), 3.92 (2H, d, J = 7.3 Hz), 3.85 (2H, dd, J = 10.9, 3.0 Hz), 3.25 (2H, t, J = 10.9 Hz), 2.09-1.99 (1H, m), 1.43 (2H, br d, J = 10.9 Hz), 1.32-1.24 (5H, m).
MS (APCI) m/z : 420 [(M+H)+
].
(步驟4)5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
使用5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 乙酯代替中間體1a之步驟3之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 乙酯,以下以同樣之操作進行反應,得到呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.79 (1H, d, J = 1.8 Hz), 8.45 (1H, d, J = 1.8 Hz), 7.70-7.65 (4H, m), 4.14 (2H, d, J = 7.3 Hz), 3.85 (2H, dd, J = 11.2, 3.3 Hz), 3.24 (2H, t, J = 11.2 Hz), 2.16-2.06 (1H, m), 1.43 (2H, br d, J = 10.9 Hz), 1.34-1.24 (2H, m).
MS (APCI) m/z : 392 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表3之中間體。
[表3]
(步驟1)4-(4-溴苯基)-3-側氧基丁酸 乙酯
使用2-(4-溴苯基)乙酸代替中間體1a之步驟1之(4-甲基苯基)乙酸,以下以同樣之操作進行反應,得到呈油狀物的標題化合物。1
H-NMR (CDCl3
) δ: 7.47 (2H, d, J = 8.5 Hz), 7.09 (2H, d, J = 8.5 Hz), 4.18 (2H, q, J = 7.1 Hz), 3.81 (2H, s), 3.46 (2H, s), 1.27 (3H, t, J = 7.1 Hz).
(步驟2)5-(4-溴苯基)-4-側氧-1,4-二氫吡啶-3-甲酸乙酯
於5.00g(17.5mmol)之於步驟1合成之4-(4-溴苯基)-3-側氧基丁酸 乙酯之50.0mL乙醇溶液中,添加1.56g(19.3mmol)之1,3,5-三與1.43g(21.0mmol)之乙醇鈉,於85℃攪拌4小時。放冷至室溫後,減壓濃縮,於所得到之殘渣中添加1當量鹽酸而呈酸性後,濾取析出之固體,將固體以丙酮洗淨、乾燥,得到3.73g之呈固體的標題化合物之粗純化物。
MS (APCI) m/z : 322 [(M+H)+
].
(步驟3)5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 乙酯
於0.322g之於步驟2合成之5-(4-溴苯基)-4-側氧-1,4-二氫吡啶-3-甲酸 乙酯之粗純化物之45.0mL N,N-二甲基甲醯胺懸浮液中,添加0.977g(3.00mmol)之碳酸銫,於80℃攪拌15分鐘後,添加0.583g(3.00mmol)之甲磺酸四氫-2H-吡喃-4-基甲酯,於80℃攪拌4小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;n-己烷/乙酸乙酯=57/43~36/64)純化,得到0.234g(2步驟產率36.7%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.30 (1H, d, J = 2.4 Hz), 8.02 (1H, d, J = 2.4 Hz), 7.59 (4H, s), 4.20 (2H, q, J = 7.0 Hz), 3.92 (2H, d, J = 7.3 Hz), 3.85 (2H, dd, J = 10.9, 3.0 Hz), 3.25 (2H, t, J = 10.9 Hz), 2.09-1.99 (1H, m), 1.43 (2H, br d, J = 10.9 Hz), 1.32-1.24 (5H, m).
MS (APCI) m/z : 420 [(M+H)+
].
(步驟4)5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
使用5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 乙酯代替中間體1a之步驟3之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 乙酯,以下以同樣之操作進行反應,得到呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.79 (1H, d, J = 1.8 Hz), 8.45 (1H, d, J = 1.8 Hz), 7.70-7.65 (4H, m), 4.14 (2H, d, J = 7.3 Hz), 3.85 (2H, dd, J = 11.2, 3.3 Hz), 3.24 (2H, t, J = 11.2 Hz), 2.16-2.06 (1H, m), 1.43 (2H, br d, J = 10.9 Hz), 1.34-1.24 (2H, m).
MS (APCI) m/z : 392 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表3之中間體。
[表3] 
(中間體5a)
(2S)-2-{[2-甲氧基-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯氧基]甲基}-1,4-二烷 將0.500g(1.99mmol)之2-甲氧基-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)酚溶解於N,N-二甲基甲醯胺(10.0ml),添加0.401g(2.04mmol)之甲磺酸 (2S)-1,4-二烷-2-基甲酯,於90℃攪拌10小時。將反應液冷卻至室溫,添加乙酸乙酯與水,進行分液操作。將有機層以飽和食鹽水洗淨後,以無水硫酸鎂乾燥。於減壓下餾去溶劑,將殘渣藉由矽膠管柱層析(Biotage 公司、洗提溶劑;己烷/乙酸乙酯=100/0~50/50)純化,得到0.451g(產率64.4%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 7.39 (1H, d, J = 7.9 Hz), 7.28 (1H, s), 6.89 (1H, d, J = 7.9 Hz), 4.09-4.03 (2H, m), 4.00-3.93 (2H, m), 3.89 (3H, s), 3.86-3.63 (4H, m), 3.57-3.50 (1H, m), 1.34 (12H, s).
同樣地進行,由所對應的起始原料合成表4之中間體。
[表4]
將0.500g(1.99mmol)之2-甲氧基-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)酚溶解於N,N-二甲基甲醯胺(10.0ml),添加0.401g(2.04mmol)之甲磺酸 (2S)-1,4-二烷-2-基甲酯,於90℃攪拌10小時。將反應液冷卻至室溫,添加乙酸乙酯與水,進行分液操作。將有機層以飽和食鹽水洗淨後,以無水硫酸鎂乾燥。於減壓下餾去溶劑,將殘渣藉由矽膠管柱層析(Biotage 公司、洗提溶劑;己烷/乙酸乙酯=100/0~50/50)純化,得到0.451g(產率64.4%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 7.39 (1H, d, J = 7.9 Hz), 7.28 (1H, s), 6.89 (1H, d, J = 7.9 Hz), 4.09-4.03 (2H, m), 4.00-3.93 (2H, m), 3.89 (3H, s), 3.86-3.63 (4H, m), 3.57-3.50 (1H, m), 1.34 (12H, s).
同樣地進行,由所對應的起始原料合成表4之中間體。
[表4] 
(中間體6)1-(四氫-2H-吡喃-4-基甲基)-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)-1H-吡唑 使用4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)-1H-吡唑、及4-(溴甲基)四氫吡喃,作為起始原料,與中間體5a同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 7.79 (1H, s), 7.65 (1H, s), 4.00 (2H, d, J = 7.5 Hz), 3.96 (2H, dd, J = 11.5, 4.5 Hz), 3.35 (2H, td, J = 11.5, 2.0 Hz), 2.23-2.11 (1H, m), 1.48 (2H, d, J = 11.5 Hz), 1.40-1.28 (14H, m).
MS (APCI) m/z : 293 [(M+H)+
].
使用4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)-1H-吡唑、及4-(溴甲基)四氫吡喃,作為起始原料,與中間體5a同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 7.79 (1H, s), 7.65 (1H, s), 4.00 (2H, d, J = 7.5 Hz), 3.96 (2H, dd, J = 11.5, 4.5 Hz), 3.35 (2H, td, J = 11.5, 2.0 Hz), 2.23-2.11 (1H, m), 1.48 (2H, d, J = 11.5 Hz), 1.40-1.28 (14H, m).
MS (APCI) m/z : 293 [(M+H)+
].
(中間體7)(2R)-2-[({2-[(2
H3
)甲基氧基]-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)(2
H3
)苯基}氧基)甲基}-1,4-二烷 (步驟1)4-溴-2,3,5-三氘-6-(三氘甲氧基)酚
4-溴-2-[(2
H3
)甲基氧基](2
H3
)酚
將12.0g(105mmol)之(2
H4
)苯-1,2-二醇懸浮於105ml之二氯甲烷中,添加9.53ml(105mmol)之3,4-二氫-2H-吡喃、396mg(1.58mmol)之p-甲苯磺酸吡啶鹽,於室溫攪拌3小時。於反應液中添加飽和碳酸氫鈉水,攪拌10分鐘。將有機層以飽和食鹽水洗淨,藉由無水硫酸鈉乾燥,於減壓下餾去溶劑。將所得到的油狀物質溶解於207ml之丙酮中,添加45.8g(330mmol)之碳酸鉀、14.4ml(228mmol)之三氘碘化甲烷(tri-deuterated iodomethane),於氮氣流下,於40℃加熱攪拌24小時。將反應液冷卻至室溫後,以KIRIYAMA過濾,將濾液於減壓下濃縮。於殘渣中添加己烷,再度以KIRIYAMA過濾後,將濾液於減壓下濃縮。將所得到之油狀物質溶解於181ml之乙醇,添加341mg(1.36mmol)之p-甲苯磺酸吡啶鹽,於65℃加熱攪拌3小時。冷卻至室溫後,於減壓下濃縮。將殘渣溶解於90.0ml之N,N-二甲基甲醯胺,將16.1g(90.6mmol)之N-溴琥珀醯亞胺(N-Bromosuccinimide)於冰冷下每次少量添加,於冰冷下攪拌1小時。於反應液中添加500ml之二乙基醚後,添加50ml之8.85mol/L硫代硫酸鈉水溶液,使反應停止。將有機層以無水硫酸鎂乾燥後,於減壓下濃縮。將殘渣使用胺基矽膠管柱層析(SHOKO scientific 公司,洗提溶劑;己烷/乙酸乙酯=90/10~55/45)純化,得到6.97g(產率28.3%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 5.57 (1H, s).
MS (CI) m/z: 207 [(M-H)+
].
(步驟2)(2R)-2-[({4-溴-2-[(2
H3
)甲基氧基](2
H3
)苯基}氧基)甲基]-1,4-二烷
將2.00g(9.57mmol)之4-溴-2-[(2
H3
)甲基氧基](2
H3
)酚溶解於48.0ml之N,N-二甲基甲醯胺,添加2.64g(19.1mmol)之碳酸鉀、1.88g(9.57mmol)之甲磺酸 (2R)-1,4-二烷-2-基甲酯,於90℃加熱攪拌4小時。將反應液冷卻至室溫,添加乙酸乙酯,濾去不溶物。將濾液於減壓下濃縮,將殘渣使用矽膠管柱層析(SHOKO scientific 公司,洗提溶劑;己烷/乙酸乙酯=100/0~70/30)純化,得到1.75g(產率59.2%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 4.06-3.48 (9H, m).
MS (APCI) m/z : 309 [(M+H)+
].
(步驟3)(2R)-2-[({2-[(2
H3
)甲基氧基]-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)(2
H3
)苯基}氧基)甲基}-1,4-二烷
將800mg(2.59mmol)之(2R)-2-[({4-溴-2-[(2
H3
)甲基氧基](2
H3
)苯基}氧基)甲基]-1,4-二烷懸浮於5.20ml之甲苯中,添加140mg(259μmol)之1,3-雙(二苯基膦基)丙烷]氯化鎳(II)、107mg(0.259mmol)之1,3-雙(二苯基膦基)丙烷、0.744ml(5.17mmol)之頻哪醇硼酸酯、1.10ml(7.76mmol)之三乙基胺,於氮氣流下,於100℃加熱攪拌16小時。將反應液冷卻至室溫,以乙酸乙酯稀釋後,將不溶物以Celite濾去。將濾液於減壓下濃縮,將殘渣使用矽膠管柱層析(SHOKO scientific 公司,洗提溶劑;己烷/乙酸乙酯=100/0~85/15)純化,得到840mg(91.1%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 4.15-4.02 (2H, m), 4.00-3.93 (2H, m), 3.87-3.62 (4H, m), 3.57-3.49 (1H, m), 1.34 (12H, s).
MS (ESI) m/z : 357 [(M+H)+
].
(步驟1)4-溴-2,3,5-三氘-6-(三氘甲氧基)酚
4-溴-2-[(2
H3
)甲基氧基](2
H3
)酚
將12.0g(105mmol)之(2
H4
)苯-1,2-二醇懸浮於105ml之二氯甲烷中,添加9.53ml(105mmol)之3,4-二氫-2H-吡喃、396mg(1.58mmol)之p-甲苯磺酸吡啶鹽,於室溫攪拌3小時。於反應液中添加飽和碳酸氫鈉水,攪拌10分鐘。將有機層以飽和食鹽水洗淨,藉由無水硫酸鈉乾燥,於減壓下餾去溶劑。將所得到的油狀物質溶解於207ml之丙酮中,添加45.8g(330mmol)之碳酸鉀、14.4ml(228mmol)之三氘碘化甲烷(tri-deuterated iodomethane),於氮氣流下,於40℃加熱攪拌24小時。將反應液冷卻至室溫後,以KIRIYAMA過濾,將濾液於減壓下濃縮。於殘渣中添加己烷,再度以KIRIYAMA過濾後,將濾液於減壓下濃縮。將所得到之油狀物質溶解於181ml之乙醇,添加341mg(1.36mmol)之p-甲苯磺酸吡啶鹽,於65℃加熱攪拌3小時。冷卻至室溫後,於減壓下濃縮。將殘渣溶解於90.0ml之N,N-二甲基甲醯胺,將16.1g(90.6mmol)之N-溴琥珀醯亞胺(N-Bromosuccinimide)於冰冷下每次少量添加,於冰冷下攪拌1小時。於反應液中添加500ml之二乙基醚後,添加50ml之8.85mol/L硫代硫酸鈉水溶液,使反應停止。將有機層以無水硫酸鎂乾燥後,於減壓下濃縮。將殘渣使用胺基矽膠管柱層析(SHOKO scientific 公司,洗提溶劑;己烷/乙酸乙酯=90/10~55/45)純化,得到6.97g(產率28.3%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 5.57 (1H, s).
MS (CI) m/z: 207 [(M-H)+
].
(步驟2)(2R)-2-[({4-溴-2-[(2
H3
)甲基氧基](2
H3
)苯基}氧基)甲基]-1,4-二烷
將2.00g(9.57mmol)之4-溴-2-[(2
H3
)甲基氧基](2
H3
)酚溶解於48.0ml之N,N-二甲基甲醯胺,添加2.64g(19.1mmol)之碳酸鉀、1.88g(9.57mmol)之甲磺酸 (2R)-1,4-二烷-2-基甲酯,於90℃加熱攪拌4小時。將反應液冷卻至室溫,添加乙酸乙酯,濾去不溶物。將濾液於減壓下濃縮,將殘渣使用矽膠管柱層析(SHOKO scientific 公司,洗提溶劑;己烷/乙酸乙酯=100/0~70/30)純化,得到1.75g(產率59.2%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 4.06-3.48 (9H, m).
MS (APCI) m/z : 309 [(M+H)+
].
(步驟3)(2R)-2-[({2-[(2
H3
)甲基氧基]-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)(2
H3
)苯基}氧基)甲基}-1,4-二烷
將800mg(2.59mmol)之(2R)-2-[({4-溴-2-[(2
H3
)甲基氧基](2
H3
)苯基}氧基)甲基]-1,4-二烷懸浮於5.20ml之甲苯中,添加140mg(259μmol)之1,3-雙(二苯基膦基)丙烷]氯化鎳(II)、107mg(0.259mmol)之1,3-雙(二苯基膦基)丙烷、0.744ml(5.17mmol)之頻哪醇硼酸酯、1.10ml(7.76mmol)之三乙基胺,於氮氣流下,於100℃加熱攪拌16小時。將反應液冷卻至室溫,以乙酸乙酯稀釋後,將不溶物以Celite濾去。將濾液於減壓下濃縮,將殘渣使用矽膠管柱層析(SHOKO scientific 公司,洗提溶劑;己烷/乙酸乙酯=100/0~85/15)純化,得到840mg(91.1%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 4.15-4.02 (2H, m), 4.00-3.93 (2H, m), 3.87-3.62 (4H, m), 3.57-3.49 (1H, m), 1.34 (12H, s).
MS (ESI) m/z : 357 [(M+H)+
].
(中間體8)
3-(4-胺基苯基)-5-{4-[(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-2-胺 將0.510g(1.93mmol)之以專利(WO2013/115280 A1)記載方法合成之3-(4-胺基苯基)-5-溴吡啶-2-胺溶解於1,4-二烷(5.00ml),添加水(1.00ml)、0.451g(1.29mmol)之(2S)-2-{[2-甲氧基-4- (4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯氧基]甲基}-1,4-二烷、0.0744g(0.0644mmol)之肆(三苯基膦)鈀(0)、及0.356g(2.58mmol)之碳酸鉀,於氮氣環境下,於100℃攪拌5小時。於反應液中添加二氯甲烷與水,進行分液操作。將有機層以無水硫酸鈉乾燥,將溶劑於減壓下餾去。將殘渣使用矽膠管柱層析(Biotage 公司,洗提溶劑;乙酸乙酯/甲醇=99/1~90/10)純化,得到0.139g(產率26.5%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 8.24-8.22 (1H, m), 7.53-7.51 (1H, m), 7.32-7.27 (2H, m), 7.07-6.99 (2H, m), 6.98-6.93 (1H, m), 6.81-6.76 (2H, m), 4.62-4.58 (2H, m), 4.14-4.01 (3H, m), 4.01-3.93 (3H, m), 3.92-3.63 (7H, m), 3.59-3.50 (1H, m).
MS (ESI) m/z : 408 [(M+H)+
]
將0.510g(1.93mmol)之以專利(WO2013/115280 A1)記載方法合成之3-(4-胺基苯基)-5-溴吡啶-2-胺溶解於1,4-二烷(5.00ml),添加水(1.00ml)、0.451g(1.29mmol)之(2S)-2-{[2-甲氧基-4- (4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯氧基]甲基}-1,4-二烷、0.0744g(0.0644mmol)之肆(三苯基膦)鈀(0)、及0.356g(2.58mmol)之碳酸鉀,於氮氣環境下,於100℃攪拌5小時。於反應液中添加二氯甲烷與水,進行分液操作。將有機層以無水硫酸鈉乾燥,將溶劑於減壓下餾去。將殘渣使用矽膠管柱層析(Biotage 公司,洗提溶劑;乙酸乙酯/甲醇=99/1~90/10)純化,得到0.139g(產率26.5%)之呈油狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 8.24-8.22 (1H, m), 7.53-7.51 (1H, m), 7.32-7.27 (2H, m), 7.07-6.99 (2H, m), 6.98-6.93 (1H, m), 6.81-6.76 (2H, m), 4.62-4.58 (2H, m), 4.14-4.01 (3H, m), 4.01-3.93 (3H, m), 3.92-3.63 (7H, m), 3.59-3.50 (1H, m).
MS (ESI) m/z : 408 [(M+H)+
]
(中間體9a)3-溴-5-{4- [(2S)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-2-胺 於0.533g(1.79mmol)之3-溴-5-碘吡啶-2-胺、0.625g(1.79mmol)之(2S)-2-{[2-甲氧基-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯氧基]甲基 }-1,4-二烷、0.378g(3.57mmol)之碳酸鈉、0.103g(0.0892mmol)之肆(三苯基膦)鈀(0)中,添加9.00mL之1,4-二烷與1.80mL之水,於氮氣環境下,於90℃攪拌7小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;氯仿/乙酸乙酯=59/41~38/62)純化,得到0.388g(產率55.0%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.28 (1H, d, J = 1.8 Hz), 8.05 (1H, d, J = 2.4 Hz), 7.17 (1H, d, J = 2.4 Hz), 7.10 (1H, dd, J = 8.5, 2.4 Hz), 6.99 (1H, d, J = 8.5 Hz), 6.28 (2H, s), 3.98-3.74 (8H, m), 3.69-3.60 (2H, m), 3.52-3.46 (1H, m), 3.42-3.36 (1H, m).
MS (APCI) m/z : 395 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表5之中間體。
[表5-1]
於0.533g(1.79mmol)之3-溴-5-碘吡啶-2-胺、0.625g(1.79mmol)之(2S)-2-{[2-甲氧基-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯氧基]甲基 }-1,4-二烷、0.378g(3.57mmol)之碳酸鈉、0.103g(0.0892mmol)之肆(三苯基膦)鈀(0)中,添加9.00mL之1,4-二烷與1.80mL之水,於氮氣環境下,於90℃攪拌7小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;氯仿/乙酸乙酯=59/41~38/62)純化,得到0.388g(產率55.0%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.28 (1H, d, J = 1.8 Hz), 8.05 (1H, d, J = 2.4 Hz), 7.17 (1H, d, J = 2.4 Hz), 7.10 (1H, dd, J = 8.5, 2.4 Hz), 6.99 (1H, d, J = 8.5 Hz), 6.28 (2H, s), 3.98-3.74 (8H, m), 3.69-3.60 (2H, m), 3.52-3.46 (1H, m), 3.42-3.36 (1H, m).
MS (APCI) m/z : 395 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表5之中間體。
[表5-1] 
[表5-2]
| 中間體 9e |  |  |
| 中間體 9f |  |  |
| 中間體 9g |  |  |
(中間體10a)
3-(4-胺基-2-氟苯基)-5-{4- [(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-2-胺 使用3-氟-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、以專利(WO2013/115280 A1)記載之方法合成之3-溴-5-{4- [(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-2-胺作為起始原料,使用1,4-二烷及水作為溶劑,使用肆(三苯基膦)鈀(0) 作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌5小時,與中間體8同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 8.27 (1H, d, J = 2.4 Hz), 7.54 (1H, d, J = 2.4 Hz), 7.17 (1H, t, J = 8.0 Hz), 7.06-7.01 (2H, m), 6.95 (1H, d, J = 8.0 Hz), 6.55 (1H, dd, J = 8.0, 2.4 Hz), 6.51 (1H, dd, J = 11.5, 2.4 Hz), 4.52 (2H, br s), 4.13-3.52 (12H, m).
MS (APCI) m/z : 426 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表6之中間體。
[表6]
使用3-氟-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、以專利(WO2013/115280 A1)記載之方法合成之3-溴-5-{4- [(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-2-胺作為起始原料,使用1,4-二烷及水作為溶劑,使用肆(三苯基膦)鈀(0) 作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌5小時,與中間體8同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 8.27 (1H, d, J = 2.4 Hz), 7.54 (1H, d, J = 2.4 Hz), 7.17 (1H, t, J = 8.0 Hz), 7.06-7.01 (2H, m), 6.95 (1H, d, J = 8.0 Hz), 6.55 (1H, dd, J = 8.0, 2.4 Hz), 6.51 (1H, dd, J = 11.5, 2.4 Hz), 4.52 (2H, br s), 4.13-3.52 (12H, m).
MS (APCI) m/z : 426 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表6之中間體。
[表6] 
(中間體11a)3-(4-胺基-3-氟苯基)-5-(3,4-二甲氧基苯基)吡啶-2-胺 於1.30g(4.20mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-溴-5-(3,4-二甲氧基苯基)吡啶-2-胺、1.20g(5.06mmol)之2-氟-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、0.340g(0.416mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、4.10g(12.6mmol)之碳酸銫中,添加25.0mL之1,4-二烷與5.00mL之水,於80℃攪拌5小時。放冷至室溫後,於反應液中添加水與二氯甲烷,將不溶物以Celite過濾濾去,將濾液以二氯甲烷萃取3次後,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,氯仿/乙酸乙酯=50/50~0/100)純化後,以胺基矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=25/75~0/100→乙酸乙酯/甲醇=100/0~90/10)純化。減壓濃縮後,將所得到之固體懸浮於二乙基醚中,藉由進行濾取、乾燥,得到0.928g(產率65.1%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.20 (1H, d, J = 2.4 Hz), 7.52 (1H, d, J = 2.4 Hz), 7.18-7.11 (3H, m), 7.06 (1H, dd, J = 7.9, 2.4 Hz), 6.98 (1H, d, J = 8.5 Hz), 6.87-6.83 (1H, m), 5.61 (2H, s), 5.30 (2H, s), 3.82 (3H, s), 3.77 (3H, s).
MS (APCI) m/z : 340 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表7之中間體。
[表7]
於1.30g(4.20mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-溴-5-(3,4-二甲氧基苯基)吡啶-2-胺、1.20g(5.06mmol)之2-氟-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、0.340g(0.416mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、4.10g(12.6mmol)之碳酸銫中,添加25.0mL之1,4-二烷與5.00mL之水,於80℃攪拌5小時。放冷至室溫後,於反應液中添加水與二氯甲烷,將不溶物以Celite過濾濾去,將濾液以二氯甲烷萃取3次後,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,氯仿/乙酸乙酯=50/50~0/100)純化後,以胺基矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=25/75~0/100→乙酸乙酯/甲醇=100/0~90/10)純化。減壓濃縮後,將所得到之固體懸浮於二乙基醚中,藉由進行濾取、乾燥,得到0.928g(產率65.1%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.20 (1H, d, J = 2.4 Hz), 7.52 (1H, d, J = 2.4 Hz), 7.18-7.11 (3H, m), 7.06 (1H, dd, J = 7.9, 2.4 Hz), 6.98 (1H, d, J = 8.5 Hz), 6.87-6.83 (1H, m), 5.61 (2H, s), 5.30 (2H, s), 3.82 (3H, s), 3.77 (3H, s).
MS (APCI) m/z : 340 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表7之中間體。
[表7] 
(中間體12a)3-(4-胺基-3-氟苯基)-4-甲氧基吡啶-2-胺 於0.500g(2.00mmol)之3-碘-4-甲氧基吡啶-2-胺、0.498g(2.10mmol)之2-氟-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、0.231g(0.200mmol)之肆(三苯基膦)鈀(0)、0.636g(6.00mmol)之碳酸鈉中,添加10.0mL之1,4-二烷與2.00mL之水,於氮氣環境下,於80℃攪拌12小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~90/10)純化後,以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~90/10)純化,得到0.177g(產率38.0%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 7.83 (1H, d, J = 5.5 Hz), 7.65-7.53 (1H, m), 6.83-6.72 (3H, m), 6.39 (1H, d, J = 6.1 Hz), 5.18 (2H, s), 5.10 (2H, s), 3.65 (3H, s).
MS (APCI) m/z : 234 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表8之中間體。
[表8]
於0.500g(2.00mmol)之3-碘-4-甲氧基吡啶-2-胺、0.498g(2.10mmol)之2-氟-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、0.231g(0.200mmol)之肆(三苯基膦)鈀(0)、0.636g(6.00mmol)之碳酸鈉中,添加10.0mL之1,4-二烷與2.00mL之水,於氮氣環境下,於80℃攪拌12小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~90/10)純化後,以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~90/10)純化,得到0.177g(產率38.0%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 7.83 (1H, d, J = 5.5 Hz), 7.65-7.53 (1H, m), 6.83-6.72 (3H, m), 6.39 (1H, d, J = 6.1 Hz), 5.18 (2H, s), 5.10 (2H, s), 3.65 (3H, s).
MS (APCI) m/z : 234 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表8之中間體。
[表8] 
(中間體13)3-(4-胺基苯基)-5-(3,4-二甲氧基苯基)吡-2-胺 (步驟1)3-氯-5-(3,4-二甲氧基苯基)吡-2-胺
於0.510g(2.00mmol)之按照專利(WO2011/110545 A1)記載之方法合成之3-氯-5-碘吡-2-胺、0.363g(2.00mmol)之(3,4-二甲氧基苯基)硼酸、0.163g(0.200mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、1.30g(4.00mmol)之碳酸銫中,添加7.50mL之1,4-二烷與2.50mL之水,於80℃攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,氯仿/乙酸乙酯=100/0~90/10)純化,進一步以胺基矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=25/75~0/100)純化,得到0.412g(產率77.7%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.57 (1H, s), 7.47-7.45 (2H, m), 7.01 (1H, d, J = 7.9 Hz), 6.85 (2H, s), 3.83 (3H, s), 3.79 (3H, s).
MS (APCI) m/z : 266 [(M+H)+
].
(步驟2)3-(4-胺基苯基)-5-(3,4-二甲氧基苯基)吡-2-胺
於0.0800g(0.301mmol)之於步驟1合成之3-氯-5-(3,4-二甲氧基苯基)吡-2-胺、0.0990g (0.452mmol)之4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、0.0246g(0.030mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、0.294g(0.903mmol)之碳酸銫中,添加2.00mL之1,4-二烷與0.200mL之水,於100℃攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,僅乙酸乙酯)純化,得到0.0940g(產率96.8%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.39 (1H, s), 7.54-7.50 (4H, m), 7.01 (1H, d, J = 8.5 Hz), 6.67 (2H, d, J = 8.5 Hz), 6.00 (2H, s), 5.44 (2H, s), 3.83 (3H, s), 3.79 (3H, s).
MS (APCI) m/z : 323 [(M+H)+
].
(步驟1)3-氯-5-(3,4-二甲氧基苯基)吡-2-胺
於0.510g(2.00mmol)之按照專利(WO2011/110545 A1)記載之方法合成之3-氯-5-碘吡-2-胺、0.363g(2.00mmol)之(3,4-二甲氧基苯基)硼酸、0.163g(0.200mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、1.30g(4.00mmol)之碳酸銫中,添加7.50mL之1,4-二烷與2.50mL之水,於80℃攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,氯仿/乙酸乙酯=100/0~90/10)純化,進一步以胺基矽膠管柱層析(山善公司,n-己烷/乙酸乙酯=25/75~0/100)純化,得到0.412g(產率77.7%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.57 (1H, s), 7.47-7.45 (2H, m), 7.01 (1H, d, J = 7.9 Hz), 6.85 (2H, s), 3.83 (3H, s), 3.79 (3H, s).
MS (APCI) m/z : 266 [(M+H)+
].
(步驟2)3-(4-胺基苯基)-5-(3,4-二甲氧基苯基)吡-2-胺
於0.0800g(0.301mmol)之於步驟1合成之3-氯-5-(3,4-二甲氧基苯基)吡-2-胺、0.0990g (0.452mmol)之4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯胺、0.0246g(0.030mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、0.294g(0.903mmol)之碳酸銫中,添加2.00mL之1,4-二烷與0.200mL之水,於100℃攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,僅乙酸乙酯)純化,得到0.0940g(產率96.8%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.39 (1H, s), 7.54-7.50 (4H, m), 7.01 (1H, d, J = 8.5 Hz), 6.67 (2H, d, J = 8.5 Hz), 6.00 (2H, s), 5.44 (2H, s), 3.83 (3H, s), 3.79 (3H, s).
MS (APCI) m/z : 323 [(M+H)+
].
(中間體14a)N-(6-氯嗒-3-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 將3.00g(9.16mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸溶解於30.0ml之N,N-二甲基甲醯胺,添加4.32g(10.1mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU),於室溫攪拌1小時。其後,添加1.31g(10.1mmol)之3-胺基-6-氯嗒,於室溫攪拌3日。將反應液以乙酸乙酯稀釋後,1當量鹽酸,將有機層以飽和碳酸氫鈉水、飽和食鹽水依序洗淨,將有機層以無水硫酸鈉乾燥。將溶劑於減壓下濃縮,將殘渣以矽膠管柱層析 (SHOKO scientific 公司,乙酸乙酯/甲醇=100/0~96/4)純化,得到非晶質固體。將其以甲醇進行漿體洗淨,得到2.10g(產率52.2%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.68 (1H, s), 8.59 (1H, d, J = 9.0 Hz), 8.48 (1H, d, J = 2.0 Hz), 7.51-7.46 (4H, m), 7.26 (2H, d, J = 8.0 Hz), 4.03 (2H, dd, J = 11.5, 4.0 Hz), 3.87 (2H, d, J = 7.5 Hz), 3.39 (2H, t, J = 11.5 Hz), 2.40 (3H, s), 2.16-2.06 (1H, m), 1.64-1.55 (2H, m), 1.50-1.38 (2H, m).
MS (APCI) m/z : 439 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表9之中間體。
[表9-1]
將3.00g(9.16mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸溶解於30.0ml之N,N-二甲基甲醯胺,添加4.32g(10.1mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU),於室溫攪拌1小時。其後,添加1.31g(10.1mmol)之3-胺基-6-氯嗒,於室溫攪拌3日。將反應液以乙酸乙酯稀釋後,1當量鹽酸,將有機層以飽和碳酸氫鈉水、飽和食鹽水依序洗淨,將有機層以無水硫酸鈉乾燥。將溶劑於減壓下濃縮,將殘渣以矽膠管柱層析 (SHOKO scientific 公司,乙酸乙酯/甲醇=100/0~96/4)純化,得到非晶質固體。將其以甲醇進行漿體洗淨,得到2.10g(產率52.2%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.68 (1H, s), 8.59 (1H, d, J = 9.0 Hz), 8.48 (1H, d, J = 2.0 Hz), 7.51-7.46 (4H, m), 7.26 (2H, d, J = 8.0 Hz), 4.03 (2H, dd, J = 11.5, 4.0 Hz), 3.87 (2H, d, J = 7.5 Hz), 3.39 (2H, t, J = 11.5 Hz), 2.40 (3H, s), 2.16-2.06 (1H, m), 1.64-1.55 (2H, m), 1.50-1.38 (2H, m).
MS (APCI) m/z : 439 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表9之中間體。
[表9-1] 
[表9-2] 
(中間體16a)N-(5-碘吡啶-2-基)-5-(4-甲基苯基) -4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 於300mg(0.916mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸之1ml二甲基乙醯胺溶液中,冰水浴下,添加0.087ml(1.19mmol)之亞硫醯氯。攪拌0.5小時後,冰水浴下,添加201mg(0.916mmol)之5-碘吡啶-2-胺、0.23ml(1.37mmol)之二異丙基乙基胺,緩緩升溫至室溫。於室溫攪拌5小時後,於反應液中添加水,濾取不溶物。以己烷洗淨後,於減壓下,於50度乾燥1小時。以矽膠管柱層析(氯仿/甲醇=99/1~95/5)純化,得到302mg(產率62.3%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.19 (1H, s), 8.53 (1H, d, J = 2.4 Hz), 8.49 (1H, d, J = 2.4 Hz), 8.15 (1H, d, J = 8.5 Hz), 7.95 (1H, dd, J = 8.5, 2.4 Hz), 7.51 (2H, d, J = 7.9 Hz), 7.46 (1H, d, J = 2.4 Hz), 7.28-7.23 (2H, m), 4.03 (2H, dd, J = 11.0, 3.7 Hz), 3.85 (2H, d, J = 7.3 Hz), 3.42-3.34 (2H, m), 2.40 (3H, s), 2.15-2.03 (1H, m), 1.63-1.51 (2H, m), 1.49-1.37 (2H, m).
MS (ESI/APCI) m/z : 530 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表10之中間體。
[表10]
於300mg(0.916mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸之1ml二甲基乙醯胺溶液中,冰水浴下,添加0.087ml(1.19mmol)之亞硫醯氯。攪拌0.5小時後,冰水浴下,添加201mg(0.916mmol)之5-碘吡啶-2-胺、0.23ml(1.37mmol)之二異丙基乙基胺,緩緩升溫至室溫。於室溫攪拌5小時後,於反應液中添加水,濾取不溶物。以己烷洗淨後,於減壓下,於50度乾燥1小時。以矽膠管柱層析(氯仿/甲醇=99/1~95/5)純化,得到302mg(產率62.3%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.19 (1H, s), 8.53 (1H, d, J = 2.4 Hz), 8.49 (1H, d, J = 2.4 Hz), 8.15 (1H, d, J = 8.5 Hz), 7.95 (1H, dd, J = 8.5, 2.4 Hz), 7.51 (2H, d, J = 7.9 Hz), 7.46 (1H, d, J = 2.4 Hz), 7.28-7.23 (2H, m), 4.03 (2H, dd, J = 11.0, 3.7 Hz), 3.85 (2H, d, J = 7.3 Hz), 3.42-3.34 (2H, m), 2.40 (3H, s), 2.15-2.03 (1H, m), 1.63-1.51 (2H, m), 1.49-1.37 (2H, m).
MS (ESI/APCI) m/z : 530 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表10之中間體。
[表10] 
(中間體17a)5-(3,4-二甲氧基苯基)-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺 於2.48g(8.02mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-溴-5-(3,4-二甲氧基苯基)吡啶-2-胺、3.05g(12.0mmol)之雙(頻哪醇)二硼烷、0.441g(0.481mmol)之參(二苯亞甲基丙酮)鈀(0)、0.315g(1.12mmol)之三環己基膦、1.18g(12.0mmol)之乙酸鉀中,添加40.0mL之1,4-二烷,於氮氣環境下,於80℃攪拌6小時半。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨。過濾、減壓濃縮後,將所得到之固體懸浮於二乙基醚中,藉由進行濾取、乾燥,得到2.20g(產率77.0%)之呈黃色固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.36 (1H, d, J = 3.0 Hz), 7.86 (1H, d, J = 3.0 Hz), 7.09-6.97 (3H, m), 6.18 (2H, s), 3.83 (3H, s), 3.77 (3H, s), 1.32 (12H, s).
同樣地進行,由所對應的起始原料合成表11之中間體。
[表11-1]
於2.48g(8.02mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-溴-5-(3,4-二甲氧基苯基)吡啶-2-胺、3.05g(12.0mmol)之雙(頻哪醇)二硼烷、0.441g(0.481mmol)之參(二苯亞甲基丙酮)鈀(0)、0.315g(1.12mmol)之三環己基膦、1.18g(12.0mmol)之乙酸鉀中,添加40.0mL之1,4-二烷,於氮氣環境下,於80℃攪拌6小時半。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨。過濾、減壓濃縮後,將所得到之固體懸浮於二乙基醚中,藉由進行濾取、乾燥,得到2.20g(產率77.0%)之呈黃色固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.36 (1H, d, J = 3.0 Hz), 7.86 (1H, d, J = 3.0 Hz), 7.09-6.97 (3H, m), 6.18 (2H, s), 3.83 (3H, s), 3.77 (3H, s), 1.32 (12H, s).
同樣地進行,由所對應的起始原料合成表11之中間體。
[表11-1] 
[表11-2] 
(中間體18)5’-(3,4-二甲氧基苯基)-3-氟-2,3’-聯吡啶-2’,5-二胺 於0.0300g(0.205mmol)之6-氯-5-氟吡啶-3-胺、0.109g(0.307mmol)之5-(3,4-二甲氧基苯基)-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺、0.0167g(0.0205mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、0.200g(0.614mmol)之碳酸銫中,添加2.00mL之1,4-二烷與0.200mL之水,於100℃攪拌3小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(SHOKO scientific 公司,乙酸乙酯/甲醇=100/0~85/15)純化,得到0.0690g(產率99.0%)之呈油狀物的標題化合物。1
H-NMR (CDCl3
) δ: 8.27 (1H, d, J = 2.4 Hz), 7.98-7.95 (2H, m), 7.10-7.04 (2H, m), 6.94 (1H, d, J = 7.9 Hz), 6.85 (1H, dd, J = 12.1, 2.4 Hz), 5.99 (2H, s), 3.99-3.92 (8H, m).
MS (APCI) m/z : 341 [(M+H)+
].
於0.0300g(0.205mmol)之6-氯-5-氟吡啶-3-胺、0.109g(0.307mmol)之5-(3,4-二甲氧基苯基)-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺、0.0167g(0.0205mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、0.200g(0.614mmol)之碳酸銫中,添加2.00mL之1,4-二烷與0.200mL之水,於100℃攪拌3小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(SHOKO scientific 公司,乙酸乙酯/甲醇=100/0~85/15)純化,得到0.0690g(產率99.0%)之呈油狀物的標題化合物。1
H-NMR (CDCl3
) δ: 8.27 (1H, d, J = 2.4 Hz), 7.98-7.95 (2H, m), 7.10-7.04 (2H, m), 6.94 (1H, d, J = 7.9 Hz), 6.85 (1H, dd, J = 12.1, 2.4 Hz), 5.99 (2H, s), 3.99-3.92 (8H, m).
MS (APCI) m/z : 341 [(M+H)+
].
(中間體19)5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸 (步驟1)5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸甲酯
於2.36g(10.2mmol)之按照文獻(J.Med.Chem.2008,51,5330-5341)記載之方法合成之5-溴-4-羥基吡啶-3-甲酸甲酯中,添加9.94g(30.5mmol)之碳酸銫與30.0mL之N,N-二甲基甲醯胺,於80℃攪拌15分鐘後,添加5.46g(30.5mmol)之4-(溴甲基)四氫-2H-吡喃,於80℃攪拌11小時後,於室溫放置整夜。於反應液中添加水與飽和食鹽水,以乙酸乙酯萃取3次後,以二氯甲烷萃取5次,合併有機層,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以胺基矽膠管柱層析(山善公司,洗提溶劑;乙酸乙酯/甲醇=100/0~85/15)純化,得到2.83g之呈油狀物的標題化合物之粗純化物。
MS (APCI) m/z : 330 [(M+H)+
].
(步驟2)5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
於2.83g之於步驟1合成之5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸甲酯之粗純化物之50.0mL四氫呋喃溶液中,添加25.0mL之甲醇與25.7mL(25.7mmol)之1當量氫氧化鈉水溶液,於室溫攪拌整夜。將有機溶劑減壓餾去後,於殘渣中添加水,以乙酸乙酯洗淨2次。於水層中添加1當量鹽酸而呈酸性後,添加乙酸乙酯。濾取析出之固體後,將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之固體懸浮於二乙基醚中,濾取,合併先前所得到之固體,藉由乾燥,得到1.93g(2步驟產率60.0%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.79 (1H, d, J = 1.8 Hz), 8.75 (1H, d, J = 1.8 Hz), 4.08 (2H, d, J = 7.9 Hz), 3.84 (2H, dd, J = 11.6, 3.4 Hz), 3.23 (2H, t, J = 11.6 Hz), 2.10-2.00 (1H, m), 1.40 (2H, br d, J = 10.4 Hz), 1.30-1.20 (2H, m).
MS (APCI) m/z : 316 [(M+H)+
].
(步驟1)5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸甲酯
於2.36g(10.2mmol)之按照文獻(J.Med.Chem.2008,51,5330-5341)記載之方法合成之5-溴-4-羥基吡啶-3-甲酸甲酯中,添加9.94g(30.5mmol)之碳酸銫與30.0mL之N,N-二甲基甲醯胺,於80℃攪拌15分鐘後,添加5.46g(30.5mmol)之4-(溴甲基)四氫-2H-吡喃,於80℃攪拌11小時後,於室溫放置整夜。於反應液中添加水與飽和食鹽水,以乙酸乙酯萃取3次後,以二氯甲烷萃取5次,合併有機層,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以胺基矽膠管柱層析(山善公司,洗提溶劑;乙酸乙酯/甲醇=100/0~85/15)純化,得到2.83g之呈油狀物的標題化合物之粗純化物。
MS (APCI) m/z : 330 [(M+H)+
].
(步驟2)5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸
於2.83g之於步驟1合成之5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸甲酯之粗純化物之50.0mL四氫呋喃溶液中,添加25.0mL之甲醇與25.7mL(25.7mmol)之1當量氫氧化鈉水溶液,於室溫攪拌整夜。將有機溶劑減壓餾去後,於殘渣中添加水,以乙酸乙酯洗淨2次。於水層中添加1當量鹽酸而呈酸性後,添加乙酸乙酯。濾取析出之固體後,將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之固體懸浮於二乙基醚中,濾取,合併先前所得到之固體,藉由乾燥,得到1.93g(2步驟產率60.0%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 8.79 (1H, d, J = 1.8 Hz), 8.75 (1H, d, J = 1.8 Hz), 4.08 (2H, d, J = 7.9 Hz), 3.84 (2H, dd, J = 11.6, 3.4 Hz), 3.23 (2H, t, J = 11.6 Hz), 2.10-2.00 (1H, m), 1.40 (2H, br d, J = 10.4 Hz), 1.30-1.20 (2H, m).
MS (APCI) m/z : 316 [(M+H)+
].
(中間體20)N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 於0.754g(2.39mmol)之5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸、0.730g(2.27mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-(4-胺基苯基)-5-(3,4-二甲氧基苯基)吡啶-2-胺與1.27g(2.95mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU)中,添加12.0mL之N,N-二甲基甲醯胺後,添加0.593mL(0.440g,3.41mmol)之N,N-二異丙基乙基胺,於室溫攪拌整夜。將反應液減壓濃縮後,於殘渣中添加飽和碳酸氫鈉水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;乙酸乙酯/甲醇=100/0~85/15)純化。減壓濃縮後,於殘渣中添加二乙基醚而固化,藉由將固體進行濾取、乾燥,得到0.724g(產率51.5%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.61 (1H, s), 8.74 (1H, d, J = 2.4 Hz), 8.63 (1H, d, J = 2.4 Hz), 8.26 (1H, d, J = 2.4 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.64 (1H, d, J = 2.4 Hz), 7.55 (2H, d, J = 8.5 Hz), 7.20-7.14 (2H, m), 6.99 (1H, d, J = 8.5 Hz), 5.77 (2H, s), 4.07 (2H, d, J = 7.3 Hz), 3.87-3.83 (5H, m), 3.77 (3H, s), 3.26 (2H, t, J = 10.7 Hz), 2.11-2.02 (1H, m), 1.42 (2H, br d, J = 10.9 Hz), 1.33-1.23 (2H, m).
MS (APCI) m/z : 619 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表12之中間體。
[表12]
於0.754g(2.39mmol)之5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸、0.730g(2.27mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-(4-胺基苯基)-5-(3,4-二甲氧基苯基)吡啶-2-胺與1.27g(2.95mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU)中,添加12.0mL之N,N-二甲基甲醯胺後,添加0.593mL(0.440g,3.41mmol)之N,N-二異丙基乙基胺,於室溫攪拌整夜。將反應液減壓濃縮後,於殘渣中添加飽和碳酸氫鈉水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,洗提溶劑;乙酸乙酯/甲醇=100/0~85/15)純化。減壓濃縮後,於殘渣中添加二乙基醚而固化,藉由將固體進行濾取、乾燥,得到0.724g(產率51.5%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.61 (1H, s), 8.74 (1H, d, J = 2.4 Hz), 8.63 (1H, d, J = 2.4 Hz), 8.26 (1H, d, J = 2.4 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.64 (1H, d, J = 2.4 Hz), 7.55 (2H, d, J = 8.5 Hz), 7.20-7.14 (2H, m), 6.99 (1H, d, J = 8.5 Hz), 5.77 (2H, s), 4.07 (2H, d, J = 7.3 Hz), 3.87-3.83 (5H, m), 3.77 (3H, s), 3.26 (2H, t, J = 10.7 Hz), 2.11-2.02 (1H, m), 1.42 (2H, br d, J = 10.9 Hz), 1.33-1.23 (2H, m).
MS (APCI) m/z : 619 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表12之中間體。
[表12] 
(中間體21)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基) -3-氟苯基]-5-溴-1’-[(4-氰基四氫-2H-吡喃-4-基) 甲基]-4’-側氧-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺 於0.168g(0.401mmol)之5-溴-1’-[(4-氰基四氫-2H-吡喃-4-基) 甲基]-4’-側氧-1’,4’-二氫-2,3’-聯吡啶-5’-甲酸之5.00mL N,N-二甲基甲醯胺懸浮液中,添加0.172g(0.401mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU)與0.0825mL(0.0612g,0.474mmol)之N,N-二異丙基乙基胺,於室溫攪拌1小時。此時反應液變得均一。添加0.155g(0.364mmol)之3-(4-胺基-2-氟苯基)-5-{4- [(2R)-1,4-二烷-2-基甲氧基] -3-甲氧基苯基}吡啶-2-胺,於室溫攪拌整夜。將反應液減壓濃縮後,於殘渣中添加飽和碳酸氫鈉水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~85/15)純化後,以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~93/7)純化,得到0.238g(產率79.1%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.95 (1H, s), 8.89-8.80 (3H, m), 8.59 (1H, d, J = 8.5 Hz), 8.31 (1H, d, J = 2.4 Hz), 8.18 (1H, dd, J = 8.5, 2.4 Hz), 7.96-7.92 (1H, m), 7.63 (1H, d, J = 2.4 Hz), 7.51-7.42 (2H, m), 7.19 (1H, d, J = 2.4 Hz), 7.11 (1H, dd, J = 8.5, 2.4 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.71 (2H, s), 4.71 (2H, s), 3.97-3.75 (10H, m), 3.69-3.59 (2H, m), 3.53-3.30 (4H, m), 1.88-1.78 (4H, m).
MS (APCI) m/z : 825 [(M+H)+
].
於0.168g(0.401mmol)之5-溴-1’-[(4-氰基四氫-2H-吡喃-4-基) 甲基]-4’-側氧-1’,4’-二氫-2,3’-聯吡啶-5’-甲酸之5.00mL N,N-二甲基甲醯胺懸浮液中,添加0.172g(0.401mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU)與0.0825mL(0.0612g,0.474mmol)之N,N-二異丙基乙基胺,於室溫攪拌1小時。此時反應液變得均一。添加0.155g(0.364mmol)之3-(4-胺基-2-氟苯基)-5-{4- [(2R)-1,4-二烷-2-基甲氧基] -3-甲氧基苯基}吡啶-2-胺,於室溫攪拌整夜。將反應液減壓濃縮後,於殘渣中添加飽和碳酸氫鈉水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~85/15)純化後,以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~93/7)純化,得到0.238g(產率79.1%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.95 (1H, s), 8.89-8.80 (3H, m), 8.59 (1H, d, J = 8.5 Hz), 8.31 (1H, d, J = 2.4 Hz), 8.18 (1H, dd, J = 8.5, 2.4 Hz), 7.96-7.92 (1H, m), 7.63 (1H, d, J = 2.4 Hz), 7.51-7.42 (2H, m), 7.19 (1H, d, J = 2.4 Hz), 7.11 (1H, dd, J = 8.5, 2.4 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.71 (2H, s), 4.71 (2H, s), 3.97-3.75 (10H, m), 3.69-3.59 (2H, m), 3.53-3.30 (4H, m), 1.88-1.78 (4H, m).
MS (APCI) m/z : 825 [(M+H)+
].
(實施例1)
N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 將0.121g(0.367mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸溶解於3.00ml之N,N-二甲基甲醯胺,添加0.236g(0.367mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU),於室溫攪拌30分鐘。其後,添加以0.149g(0.367mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-(4-胺基苯基)-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-2-胺,於室溫攪拌4小時。於反應液中添加水,濾取析出之固體。將其使用矽膠管柱層析(Biotage 公司、洗提溶劑;二氯甲烷/甲醇=99/1~95/5)純化,得到0.18g(產率69.8%)之呈泡狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 12.85 (1H, s), 8.57 (1H, d, J = 2.4 Hz), 8.25 (1H, d, J = 1.8 Hz), 7.87 (2H, d, J = 8.5 Hz), 7.57 (1H, d, J = 1.8 Hz), 7.47 (5H, dd, J = 8.2, 5.8 Hz), 7.29 (2H, d, J = 8.5 Hz), 7.08-7.02 (2H, m), 6.98-6.94 (1H, m), 4.71 (2H, s), 4.11-3.94 (6H, m), 3.93-3.79 (8H, m), 3.79-3.63 (1H, m), 3.59-3.51 (1H, m), 3.44-3.35 (2H, m), 2.41 (3H, s), 2.18-2.04 (1H, m), 1.64-1.58 (2H, m), 1.51-1.35 (2H, m).
MS (ESI) m/z : 717 [(M+H)+
]
同樣地進行,由所對應的起始原料A及B合成表13之最終物。
[表13-1]
將0.121g(0.367mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸溶解於3.00ml之N,N-二甲基甲醯胺,添加0.236g(0.367mmol)之N-[1-(氰基-2-乙氧基-2-側氧基亞乙基胺基氧基)二甲基胺基(啉基)]脲鎓六氟磷酸鹽(COMU),於室溫攪拌30分鐘。其後,添加以0.149g(0.367mmol)之以專利(WO2013/115280 A1)記載之方法合成之3-(4-胺基苯基)-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-2-胺,於室溫攪拌4小時。於反應液中添加水,濾取析出之固體。將其使用矽膠管柱層析(Biotage 公司、洗提溶劑;二氯甲烷/甲醇=99/1~95/5)純化,得到0.18g(產率69.8%)之呈泡狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 12.85 (1H, s), 8.57 (1H, d, J = 2.4 Hz), 8.25 (1H, d, J = 1.8 Hz), 7.87 (2H, d, J = 8.5 Hz), 7.57 (1H, d, J = 1.8 Hz), 7.47 (5H, dd, J = 8.2, 5.8 Hz), 7.29 (2H, d, J = 8.5 Hz), 7.08-7.02 (2H, m), 6.98-6.94 (1H, m), 4.71 (2H, s), 4.11-3.94 (6H, m), 3.93-3.79 (8H, m), 3.79-3.63 (1H, m), 3.59-3.51 (1H, m), 3.44-3.35 (2H, m), 2.41 (3H, s), 2.18-2.04 (1H, m), 1.64-1.58 (2H, m), 1.51-1.35 (2H, m).
MS (ESI) m/z : 717 [(M+H)+
]
同樣地進行,由所對應的起始原料A及B合成表13之最終物。
[表13-1] 
[表13-2] 
[表13-3] 
[表13-4] 
[表13-5] 
[表13-6] 
[表13-7] 
[表13-8] 
[表13-9] 
[表13-10] 
[表13-11] 
[表13-12] 
[表13-13] 
[表13-14] 
[表13-15] 
[表13-16] 
[表13-17] 
[表13-18] 
[表13-19] 
[表13-20] 
[表13-21] 
[表13-22] 
[表13-23] 
[表13-24] 
[表13-25] 
[表13-26] 
(實施例68)
N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]-3-氟苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽 將0.127g(0.389mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸溶解於N-甲基‐2‐吡咯啶酮(3.00ml),添加0.0277ml(0.382mmol)之亞硫醯氯,於室溫攪拌30分鐘。其後,於冰冷下,添加0.121g(0.354mmol)之3-(4-胺基-2-氟苯基)-5-(3,4-二甲氧基苯基)吡啶-2-胺,於室溫攪拌5小時。再度冰冷,以飽和碳酸氫鈉中和後,以乙酸乙酯萃取。將有機層以飽和食鹽水洗淨後,以無水硫酸鎂乾燥,於減壓下餾去溶劑。將殘渣使用矽膠管柱層析(Biotage 公司,洗提溶劑;二氯甲烷/甲醇=99/1~96/4)純化,得到0.106g(產率46.2%)之呈泡狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 12.98 (1H, s), 8.56 (1H, d, J = 2.4 Hz), 8.31 (1H, d, J = 2.4 Hz), 7.92 (1H, dd, J = 12.1, 1.8 Hz), 7.59 (1H, d, J = 2.4 Hz), 7.49-7.43 (4H, m), 7.36 (1H, t, J = 8.2 Hz), 7.29 (2H, d, J = 7.9 Hz), 7.08 (1H, dd, J = 8.2, 2.1 Hz), 7.03 (1H, d, J = 1.8 Hz), 6.93 (1H, d, J = 7.9 Hz), 4.57-4.52 (2H, m), 4.07-4.00 (2H, m), 3.94 (3H, s), 3.92 (3H, s), 3.88 (2H, d, J = 7.3 Hz), 3.44-3.35 (2H, m), 2.41 (3H, s), 2.19-2.04 (1H, m), 1.65-1.57 (2H, m), 1.51-1.38 (2H, m).
MS (ESI) m/z : 649 [(M+H)+
]
同樣地進行,由所對應的起始原料合成表14之最終物。
[表14]
將0.127g(0.389mmol)之5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲酸溶解於N-甲基‐2‐吡咯啶酮(3.00ml),添加0.0277ml(0.382mmol)之亞硫醯氯,於室溫攪拌30分鐘。其後,於冰冷下,添加0.121g(0.354mmol)之3-(4-胺基-2-氟苯基)-5-(3,4-二甲氧基苯基)吡啶-2-胺,於室溫攪拌5小時。再度冰冷,以飽和碳酸氫鈉中和後,以乙酸乙酯萃取。將有機層以飽和食鹽水洗淨後,以無水硫酸鎂乾燥,於減壓下餾去溶劑。將殘渣使用矽膠管柱層析(Biotage 公司,洗提溶劑;二氯甲烷/甲醇=99/1~96/4)純化,得到0.106g(產率46.2%)之呈泡狀物質的標題化合物。1
H-NMR (CDCl3
) δ: 12.98 (1H, s), 8.56 (1H, d, J = 2.4 Hz), 8.31 (1H, d, J = 2.4 Hz), 7.92 (1H, dd, J = 12.1, 1.8 Hz), 7.59 (1H, d, J = 2.4 Hz), 7.49-7.43 (4H, m), 7.36 (1H, t, J = 8.2 Hz), 7.29 (2H, d, J = 7.9 Hz), 7.08 (1H, dd, J = 8.2, 2.1 Hz), 7.03 (1H, d, J = 1.8 Hz), 6.93 (1H, d, J = 7.9 Hz), 4.57-4.52 (2H, m), 4.07-4.00 (2H, m), 3.94 (3H, s), 3.92 (3H, s), 3.88 (2H, d, J = 7.3 Hz), 3.44-3.35 (2H, m), 2.41 (3H, s), 2.19-2.04 (1H, m), 1.65-1.57 (2H, m), 1.51-1.38 (2H, m).
MS (ESI) m/z : 649 [(M+H)+
]
同樣地進行,由所對應的起始原料合成表14之最終物。
[表14] 
(實施例71)N-[4-(2-胺基-5-{4-[(4-甲基哌-1-基) 甲基] 苯基 }吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽 (步驟1)N-[4-(2-胺基-5-溴吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用3-(4-胺基苯基)-5-溴吡啶-2-胺作為起始原料,與實施例1同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 12.88-12.85 (1H, m), 8.56 (1H, d, J = 2.4 Hz), 8.08 (1H, d, J = 2.4 Hz), 7.87-7.84 (2H, m), 7.49-7.45 (4H, m), 7.43-7.39 (2H, m), 7.31-7.27 (2H, m), 4.63-4.58 (2H, m), 4.06-4.00 (2H, m), 3.89-3.86 (2H, m), 3.43-3.35 (2H, m), 2.41 (3H, s), 2.17-2.06 (1H, m), 1.65-1.38 (4H, m).
MS (ES+APCI) m/z : 573 [(M+H)+
].
(步驟2)N-[4-(2-胺基-5-{4-[(4-甲基哌-1-基) 甲基] 苯基 }吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用N-[4-(2-胺基-5-溴吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺與1-甲基-4-[[4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯基]甲基]哌作為起始原料,使用1,4-二烷及水作為溶劑,使用肆(三苯基膦)鈀(0) 作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌2小時,與中間體8同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 12.87-12.85 (1H, m), 8.57 (1H, d, J = 2.4 Hz), 8.31 (1H, d, J = 2.4 Hz), 7.87 (2H, d, J = 7.9 Hz), 7.61 (1H, d, J = 2.4 Hz), 7.52-7.45 (7H, m), 7.37 (2H, d, J = 7.9 Hz), 7.29 (2H, d, J = 7.9 Hz), 4.69-4.64 (2H, m), 4.06-4.00 (2H, m), 3.87 (2H, d, J = 7.3 Hz), 3.54 (2H, s), 3.43-3.35 (2H, m), 2.63-2.34 (8H, m), 2.41 (3H, s), 2.29 (3H, s), 2.15-2.07 (1H, m), 1.64-1.57 (2H, m), 1.50-1.38 (2H, m).
MS (ES+APCI) m/z : 683 [(M+H)+
].
(步驟3)N-[4-(2-胺基-5-{4-[(4-甲基哌-1-基) 甲基] 苯基 }吡啶-3-基)苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
將34.0mg(49.8μmol)N-[4-(2-胺基-5-{4-[(4-甲基哌-1-基) 甲基] 苯基 }吡啶-3-基)苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺溶解於3.00 ml之氯仿,於室溫添加49.8μL(49.8μmol)之1mol/L甲磺酸/乙醇溶液後,將溶劑於減壓下蒸餾去除。於所得到之殘渣中,添加二異丙基醚,濾取析出之固體,得到25.7mg(產率66.3%)之標題化合物。1
H-NMR (CDCl3
) δ: 12.92-12.90 (1H, m), 8.57 (1H, d, J = 2.4 Hz), 8.20 (1H, d, J = 2.4 Hz), 7.91-7.87 (2H, m), 7.69 (1H, d, J = 2.4 Hz), 7.52-7.44 (7H, m), 7.39-7.35 (2H, m), 7.31-7.27 (2H, m), 4.07-4.00 (2H, m), 3.88 (2H, d, J = 7.3 Hz), 3.69-3.63 (2H, m), 3.44-3.35 (2H, m), 3.08-2.61 (8H, m), 2.86 (3H, s), 2.41 (3H, s), 2.18 (3H, s), 2.16-2.06 (1H, m), 1.65-1.58 (2H, m), 1.51-1.39 (2H, m).
MS (ES+APCI) m/z : 683 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表15之最終物。
[表15-1]
(步驟1)N-[4-(2-胺基-5-溴吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用3-(4-胺基苯基)-5-溴吡啶-2-胺作為起始原料,與實施例1同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 12.88-12.85 (1H, m), 8.56 (1H, d, J = 2.4 Hz), 8.08 (1H, d, J = 2.4 Hz), 7.87-7.84 (2H, m), 7.49-7.45 (4H, m), 7.43-7.39 (2H, m), 7.31-7.27 (2H, m), 4.63-4.58 (2H, m), 4.06-4.00 (2H, m), 3.89-3.86 (2H, m), 3.43-3.35 (2H, m), 2.41 (3H, s), 2.17-2.06 (1H, m), 1.65-1.38 (4H, m).
MS (ES+APCI) m/z : 573 [(M+H)+
].
(步驟2)N-[4-(2-胺基-5-{4-[(4-甲基哌-1-基) 甲基] 苯基 }吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用N-[4-(2-胺基-5-溴吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺與1-甲基-4-[[4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)苯基]甲基]哌作為起始原料,使用1,4-二烷及水作為溶劑,使用肆(三苯基膦)鈀(0) 作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌2小時,與中間體8同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 12.87-12.85 (1H, m), 8.57 (1H, d, J = 2.4 Hz), 8.31 (1H, d, J = 2.4 Hz), 7.87 (2H, d, J = 7.9 Hz), 7.61 (1H, d, J = 2.4 Hz), 7.52-7.45 (7H, m), 7.37 (2H, d, J = 7.9 Hz), 7.29 (2H, d, J = 7.9 Hz), 4.69-4.64 (2H, m), 4.06-4.00 (2H, m), 3.87 (2H, d, J = 7.3 Hz), 3.54 (2H, s), 3.43-3.35 (2H, m), 2.63-2.34 (8H, m), 2.41 (3H, s), 2.29 (3H, s), 2.15-2.07 (1H, m), 1.64-1.57 (2H, m), 1.50-1.38 (2H, m).
MS (ES+APCI) m/z : 683 [(M+H)+
].
(步驟3)N-[4-(2-胺基-5-{4-[(4-甲基哌-1-基) 甲基] 苯基 }吡啶-3-基)苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
將34.0mg(49.8μmol)N-[4-(2-胺基-5-{4-[(4-甲基哌-1-基) 甲基] 苯基 }吡啶-3-基)苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺溶解於3.00 ml之氯仿,於室溫添加49.8μL(49.8μmol)之1mol/L甲磺酸/乙醇溶液後,將溶劑於減壓下蒸餾去除。於所得到之殘渣中,添加二異丙基醚,濾取析出之固體,得到25.7mg(產率66.3%)之標題化合物。1
H-NMR (CDCl3
) δ: 12.92-12.90 (1H, m), 8.57 (1H, d, J = 2.4 Hz), 8.20 (1H, d, J = 2.4 Hz), 7.91-7.87 (2H, m), 7.69 (1H, d, J = 2.4 Hz), 7.52-7.44 (7H, m), 7.39-7.35 (2H, m), 7.31-7.27 (2H, m), 4.07-4.00 (2H, m), 3.88 (2H, d, J = 7.3 Hz), 3.69-3.63 (2H, m), 3.44-3.35 (2H, m), 3.08-2.61 (8H, m), 2.86 (3H, s), 2.41 (3H, s), 2.18 (3H, s), 2.16-2.06 (1H, m), 1.65-1.58 (2H, m), 1.51-1.39 (2H, m).
MS (ES+APCI) m/z : 683 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表15之最終物。
[表15-1] 
[表15-2] 
[表15-3] 
[表15-4] 
(實施例83)N-(4-{2-胺基-5-[3-氟-4-(啉-4-基甲基)苯基]吡啶-3-基}苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽 (步驟1)N-{4-[2-胺基-5-(3-氟-4-甲醯基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用於實施例71之步驟1得到之N-[4-(2-胺基-5-溴吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺及3-氟-4-甲醯基苯基硼酸,使用1,4-二烷及水(比率10:1)作為溶劑,使用肆(三苯基膦)鈀(0)作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌7小時,與中間體8同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 12.89 (1H, s), 10.37-10.35 (1H, m), 8.58-8.57 (1H, m), 8.37 (1H, d, J = 2.4 Hz), 7.94-7.87 (3H, m), 7.64 (1H, d, J = 2.4 Hz), 7.49-7.45 (6H, m), 7.36 (1H, dd, J = 12.1, 1.8 Hz), 7.31-7.28 (2H, m), 4.86-4.81 (2H, m), 4.07-4.01 (2H, m), 3.88 (2H, d, J = 7.3 Hz), 3.43-3.35 (2H, m), 2.41 (3H, s), 2.17-2.07 (1H, m), 1.64-1.53 (2H, m), 1.51-1.39 (2H, m).
MS (ES+APCI) m/z : 617 [(M+H)+
].
(步驟2)N-(4-{2-胺基-5-[3-氟-4-(啉-4-基甲基)苯基]吡啶-3-基}苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
將70.0mg(0.114mmol)之N-{4-[2-胺基-5-(3-氟-4-甲醯基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺懸浮於2.27ml之二氯乙烷中,添加19.8μl(0.227mmol)之啉,於室溫攪拌5分鐘。於反應液中添加48.1mg(0.227mmol)之三乙醯氧基硼氫化鈉,於室溫攪拌3小時。於反應液添加水及碳酸氫鈉,以氯仿萃取,以無水硫酸鈉乾燥。將溶劑於減壓下餾去,將所得到之殘渣以矽膠管柱層析(Biotage 公司,二氯甲烷/乙醇=100/1~50/1)純化,得到固體。將其溶解於8.00ml之氯仿,添加91.2μl(91.2μmol)之1M甲磺酸/乙醇溶液,於減壓下濃縮。於殘渣中添加二乙基醚,濾取析出之固體,得到30.5mg(34.7%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.21-13.18 (1H, m), 10.01-9.83 (1H, m), 8.72 (1H, d, J = 2.4 Hz), 8.42 (1H, d, J = 2.4 Hz), 8.19 (1H, d, J = 2.4 Hz), 8.10-8.03 (1H, m), 7.91-7.81 (3H, m), 7.78-7.73 (1H, m), 7.69-7.63 (1H, m), 7.61-7.55 (4H, m), 7.27 (2H, d, J = 7.9 Hz), 4.49-4.36 (2H, m), 4.12 (2H, d, J = 7.3 Hz), 4.03-3.92 (2H, m), 3.90-3.84 (2H, m), 3.72-3.59 (2H, m), 3.47-3.10 (8H, m), 2.36 (3H, s), 2.32 (3H, s), 2.17-2.05 (1H, m), 1.50-1.42 (2H, m), 1.38-1.25 (2H, m).
MS (ES+APCI) m/z : 688 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表16之最終物。
[表16]
(步驟1)N-{4-[2-胺基-5-(3-氟-4-甲醯基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用於實施例71之步驟1得到之N-[4-(2-胺基-5-溴吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺及3-氟-4-甲醯基苯基硼酸,使用1,4-二烷及水(比率10:1)作為溶劑,使用肆(三苯基膦)鈀(0)作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌7小時,與中間體8同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 12.89 (1H, s), 10.37-10.35 (1H, m), 8.58-8.57 (1H, m), 8.37 (1H, d, J = 2.4 Hz), 7.94-7.87 (3H, m), 7.64 (1H, d, J = 2.4 Hz), 7.49-7.45 (6H, m), 7.36 (1H, dd, J = 12.1, 1.8 Hz), 7.31-7.28 (2H, m), 4.86-4.81 (2H, m), 4.07-4.01 (2H, m), 3.88 (2H, d, J = 7.3 Hz), 3.43-3.35 (2H, m), 2.41 (3H, s), 2.17-2.07 (1H, m), 1.64-1.53 (2H, m), 1.51-1.39 (2H, m).
MS (ES+APCI) m/z : 617 [(M+H)+
].
(步驟2)N-(4-{2-胺基-5-[3-氟-4-(啉-4-基甲基)苯基]吡啶-3-基}苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
將70.0mg(0.114mmol)之N-{4-[2-胺基-5-(3-氟-4-甲醯基苯基)吡啶-3-基]苯基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺懸浮於2.27ml之二氯乙烷中,添加19.8μl(0.227mmol)之啉,於室溫攪拌5分鐘。於反應液中添加48.1mg(0.227mmol)之三乙醯氧基硼氫化鈉,於室溫攪拌3小時。於反應液添加水及碳酸氫鈉,以氯仿萃取,以無水硫酸鈉乾燥。將溶劑於減壓下餾去,將所得到之殘渣以矽膠管柱層析(Biotage 公司,二氯甲烷/乙醇=100/1~50/1)純化,得到固體。將其溶解於8.00ml之氯仿,添加91.2μl(91.2μmol)之1M甲磺酸/乙醇溶液,於減壓下濃縮。於殘渣中添加二乙基醚,濾取析出之固體,得到30.5mg(34.7%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.21-13.18 (1H, m), 10.01-9.83 (1H, m), 8.72 (1H, d, J = 2.4 Hz), 8.42 (1H, d, J = 2.4 Hz), 8.19 (1H, d, J = 2.4 Hz), 8.10-8.03 (1H, m), 7.91-7.81 (3H, m), 7.78-7.73 (1H, m), 7.69-7.63 (1H, m), 7.61-7.55 (4H, m), 7.27 (2H, d, J = 7.9 Hz), 4.49-4.36 (2H, m), 4.12 (2H, d, J = 7.3 Hz), 4.03-3.92 (2H, m), 3.90-3.84 (2H, m), 3.72-3.59 (2H, m), 3.47-3.10 (8H, m), 2.36 (3H, s), 2.32 (3H, s), 2.17-2.05 (1H, m), 1.50-1.42 (2H, m), 1.38-1.25 (2H, m).
MS (ES+APCI) m/z : 688 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表16之最終物。
[表16] 
(實施例85)N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽 (步驟1)N-[4-(2-胺基-5-溴吡啶-3-基)-3-氟苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用以專利(WO2013/115280 A1)記載之方法合成之3-(4-胺基-2-氟苯基)-5-溴-吡啶-2-胺作為起始原料,與實施例1同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 13.01-12.98 (1H, m), 8.55 (1H, d, J = 2.4 Hz), 8.13 (1H, d, J = 2.4 Hz), 7.91 (1H, dd, J = 12.1, 2.4 Hz), 7.51-7.41 (5H, m), 7.32-7.25 (3H, m), 4.54-4.50 (2H, m), 4.07-4.01 (2H, m), 3.88 (2H, d, J = 7.3 Hz), 3.43-3.35 (2H, m), 2.41 (3H, s), 2.16-2.05 (1H, m), 1.64-1.54 (2H, m), 1.51-1.38 (2H, m).
MS (ES+APCI) m/z : 591 [(M+H)+].
(步驟2)N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
使用於步驟1所得到之N-[4-(2-胺基-5-溴吡啶-3-基)-3-氟苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺及1-(四氫-2H-吡喃-4-基)-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)-1H-吡唑作為起始原料,使用1,4-二烷及水(比率10:1)作為溶劑,使用肆(三苯基膦)鈀(0)作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌7小時,與中間體8同樣地進行,得到N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺。使用其,藉由與實施例71步驟3同樣之操作得到標題化合物。1
H-NMR (DMSO-D6
) δ: 13.38-13.36 (1H, m), 8.73 (1H, d, J = 2.4 Hz), 8.42-8.40 (1H, m), 8.26-8.19 (3H, m), 8.03-7.98 (2H, m), 7.61-7.44 (6H, m), 7.27 (2H, d, J = 7.9 Hz), 4.44-4.35 (1H, m), 4.12 (2H, d, J = 6.7 Hz), 3.99-3.93 (2H, m), 3.90-3.84 (2H, m), 3.52-3.43 (2H, m), 3.31-3.23 (2H, m), 2.36 (3H, s), 2.31 (3H, s), 2.17-2.06 (1H, m), 2.05-1.98 (2H, m), 1.97-1.84 (2H, m), 1.49-1.42 (2H, m), 1.37-1.25 (2H, m).
MS (ES+APCI) m/z : 663 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表17之最終物。
[表17]
(步驟1)N-[4-(2-胺基-5-溴吡啶-3-基)-3-氟苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
使用以專利(WO2013/115280 A1)記載之方法合成之3-(4-胺基-2-氟苯基)-5-溴-吡啶-2-胺作為起始原料,與實施例1同樣地得到標題化合物。1
H-NMR (CDCl3
) δ: 13.01-12.98 (1H, m), 8.55 (1H, d, J = 2.4 Hz), 8.13 (1H, d, J = 2.4 Hz), 7.91 (1H, dd, J = 12.1, 2.4 Hz), 7.51-7.41 (5H, m), 7.32-7.25 (3H, m), 4.54-4.50 (2H, m), 4.07-4.01 (2H, m), 3.88 (2H, d, J = 7.3 Hz), 3.43-3.35 (2H, m), 2.41 (3H, s), 2.16-2.05 (1H, m), 1.64-1.54 (2H, m), 1.51-1.38 (2H, m).
MS (ES+APCI) m/z : 591 [(M+H)+].
(步驟2)N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
使用於步驟1所得到之N-[4-(2-胺基-5-溴吡啶-3-基)-3-氟苯基] -5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺及1-(四氫-2H-吡喃-4-基)-4-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)-1H-吡唑作為起始原料,使用1,4-二烷及水(比率10:1)作為溶劑,使用肆(三苯基膦)鈀(0)作為觸媒,使用碳酸鉀作為鹼,於反應溫度100℃加熱攪拌7小時,與中間體8同樣地進行,得到N-(4-{2-胺基-5-[1-(四氫-2H-吡喃-4-基)-1H-吡唑-4-基]吡啶-3-基}-3-氟苯基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺。使用其,藉由與實施例71步驟3同樣之操作得到標題化合物。1
H-NMR (DMSO-D6
) δ: 13.38-13.36 (1H, m), 8.73 (1H, d, J = 2.4 Hz), 8.42-8.40 (1H, m), 8.26-8.19 (3H, m), 8.03-7.98 (2H, m), 7.61-7.44 (6H, m), 7.27 (2H, d, J = 7.9 Hz), 4.44-4.35 (1H, m), 4.12 (2H, d, J = 6.7 Hz), 3.99-3.93 (2H, m), 3.90-3.84 (2H, m), 3.52-3.43 (2H, m), 3.31-3.23 (2H, m), 2.36 (3H, s), 2.31 (3H, s), 2.17-2.06 (1H, m), 2.05-1.98 (2H, m), 1.97-1.84 (2H, m), 1.49-1.42 (2H, m), 1.37-1.25 (2H, m).
MS (ES+APCI) m/z : 663 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表17之最終物。
[表17] 
(實施例88 )N-[2’-胺基-5’-(3,4-二甲氧基苯基)-2,3’-聯吡啶-5-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽 (步驟1)N-(2’-胺基-5’-溴-2,3’-聯吡啶-5-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
於200mg(0.669mmol)之5-溴-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺之10.0ml二烷、1.00ml水溶液中,添加354mg(0.669mmol)之N-(6-碘吡啶-3-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、277mg(2.01mmol)之碳酸鉀、38.0mg(0.0344mmol)之肆(三苯基膦)鈀(0),於氮氣環境下,於90℃攪拌3小時。進一步添加200mg(0.669mmol)之5-溴-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺,於90℃攪拌5小時。
放冷後,添加水、二氯甲烷、甲醇,進行分液操作。將有機層以無水硫酸鈉乾燥後,過濾,將溶液濃縮。以乙酸乙酯洗淨殘渣,得到352mg(91.6%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.05 (1H, s), 8.94 (1H, d, J = 2.4 Hz), 8.56 (1H, d, J = 2.4 Hz), 8.32 (1H, dd, J = 8.5, 2.4 Hz), 8.10 (1H, d, J = 1.8 Hz), 7.92 (1H, d, J = 2.4 Hz), 7.68 (1H, d, J = 9.2 Hz), 7.50-7.44 (3H, m), 7.29 (2H, d, J = 7.9 Hz), 6.76 (2H, br s), 4.04 (2H, dd, J = 11.9, 3.4 Hz), 3.89 (2H, d, J = 7.3 Hz), 3.39 (2H, t, J = 11.0 Hz), 2.42 (3H, s), 2.16-2.06 (1H, m), 1.65-1.57 (2H, m), 1.51-1.38 (2H, m).
MS (APCI) m/z : 574 [(M+H)+
].
(步驟2)N-[2’-胺基-5’-(3,4-二甲氧基苯基)-2,3’-聯吡啶-5-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
於130mg(0.226mmol)之N-(2’-胺基-5’-溴-2,3’-聯吡啶-5-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, 50mg(0.272mmol)之3,4-二甲氧基苯基硼酸之10ml二烷、1ml水懸浮液中,添加94mg(0.679mmol)之碳酸鉀、13mg(0.0113mmol)之肆(三苯基膦)鈀(0),於氮氣環境下,於100℃攪拌2小時。將反應液以二氯甲烷稀釋後,水洗。將有機層以無水硫酸鈉乾燥,過濾後,餾去溶劑。將殘渣以矽膠管柱層析(二氯甲烷/甲醇=19/1,使用胺基矽膠管柱作為前置管柱)純化,得到69.0mg(48.3%)之呈固體的N-[2’-胺基-5’-(3,4-二甲氧基苯基)-2,3’-聯吡啶-5-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺。於其之1.00ml二氯甲烷、1.00ml乙醇溶液中,添加10.5mg(0.192mmol)之甲磺酸,餾去溶劑。將殘渣懸浮於二氯甲烷/乙酸乙酯/異丙基醚中,濾取不溶物,得到65.0mg(81.8%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 14.93 (1H, s), 13.27 (1H, s), 9.03 (1H, d, J = 2.4 Hz), 8.57 (1H, s), 8.41-8.36 (2H, m), 7.91-7.85 (2H, m), 7.52 (1H, d, J = 2.4 Hz), 7.46 (2H, d, J = 8.5 Hz), 7.30 (2H, d, J = 7.9 Hz), 7.05 (1H, dd, J = 8.2, 2.1 Hz), 7.00-6.94 (2H, m), 4.04 (2H, dd, J = 11.6, 3.7 Hz), 3.96 (3H, s), 3.95 (3H, s), 3.91 (2H, d, J = 7.3 Hz), 3.40 (2H, t, J = 11.0 Hz), 2.95 (3H, s), 2.42 (3H, s), 2.18-2.07 (1H, m), 1.65-1.55 (2H, m), 1.52-1.39 (2H, m).
MS (ESI) m/z : 632 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表18之最終物。
[表18-1]
(步驟1)N-(2’-胺基-5’-溴-2,3’-聯吡啶-5-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺
於200mg(0.669mmol)之5-溴-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺之10.0ml二烷、1.00ml水溶液中,添加354mg(0.669mmol)之N-(6-碘吡啶-3-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、277mg(2.01mmol)之碳酸鉀、38.0mg(0.0344mmol)之肆(三苯基膦)鈀(0),於氮氣環境下,於90℃攪拌3小時。進一步添加200mg(0.669mmol)之5-溴-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺,於90℃攪拌5小時。
放冷後,添加水、二氯甲烷、甲醇,進行分液操作。將有機層以無水硫酸鈉乾燥後,過濾,將溶液濃縮。以乙酸乙酯洗淨殘渣,得到352mg(91.6%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.05 (1H, s), 8.94 (1H, d, J = 2.4 Hz), 8.56 (1H, d, J = 2.4 Hz), 8.32 (1H, dd, J = 8.5, 2.4 Hz), 8.10 (1H, d, J = 1.8 Hz), 7.92 (1H, d, J = 2.4 Hz), 7.68 (1H, d, J = 9.2 Hz), 7.50-7.44 (3H, m), 7.29 (2H, d, J = 7.9 Hz), 6.76 (2H, br s), 4.04 (2H, dd, J = 11.9, 3.4 Hz), 3.89 (2H, d, J = 7.3 Hz), 3.39 (2H, t, J = 11.0 Hz), 2.42 (3H, s), 2.16-2.06 (1H, m), 1.65-1.57 (2H, m), 1.51-1.38 (2H, m).
MS (APCI) m/z : 574 [(M+H)+
].
(步驟2)N-[2’-胺基-5’-(3,4-二甲氧基苯基)-2,3’-聯吡啶-5-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽
於130mg(0.226mmol)之N-(2’-胺基-5’-溴-2,3’-聯吡啶-5-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺, 50mg(0.272mmol)之3,4-二甲氧基苯基硼酸之10ml二烷、1ml水懸浮液中,添加94mg(0.679mmol)之碳酸鉀、13mg(0.0113mmol)之肆(三苯基膦)鈀(0),於氮氣環境下,於100℃攪拌2小時。將反應液以二氯甲烷稀釋後,水洗。將有機層以無水硫酸鈉乾燥,過濾後,餾去溶劑。將殘渣以矽膠管柱層析(二氯甲烷/甲醇=19/1,使用胺基矽膠管柱作為前置管柱)純化,得到69.0mg(48.3%)之呈固體的N-[2’-胺基-5’-(3,4-二甲氧基苯基)-2,3’-聯吡啶-5-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺。於其之1.00ml二氯甲烷、1.00ml乙醇溶液中,添加10.5mg(0.192mmol)之甲磺酸,餾去溶劑。將殘渣懸浮於二氯甲烷/乙酸乙酯/異丙基醚中,濾取不溶物,得到65.0mg(81.8%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 14.93 (1H, s), 13.27 (1H, s), 9.03 (1H, d, J = 2.4 Hz), 8.57 (1H, s), 8.41-8.36 (2H, m), 7.91-7.85 (2H, m), 7.52 (1H, d, J = 2.4 Hz), 7.46 (2H, d, J = 8.5 Hz), 7.30 (2H, d, J = 7.9 Hz), 7.05 (1H, dd, J = 8.2, 2.1 Hz), 7.00-6.94 (2H, m), 4.04 (2H, dd, J = 11.6, 3.7 Hz), 3.96 (3H, s), 3.95 (3H, s), 3.91 (2H, d, J = 7.3 Hz), 3.40 (2H, t, J = 11.0 Hz), 2.95 (3H, s), 2.42 (3H, s), 2.18-2.07 (1H, m), 1.65-1.55 (2H, m), 1.52-1.39 (2H, m).
MS (ESI) m/z : 632 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表18之最終物。
[表18-1] 
[表18-2] 
[表18-3] 
[表18-4] 
[表18-5] 
(實施例100)N-{6-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基] 嗒-3-基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 於0.0800g(0.182mmol)之5-(3,4-二甲氧基苯基)-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺、0.0974g(0.273mmol)之N-(6-氯嗒-3-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.0167g(0.0182mmol)之參(二苯亞甲基丙酮)鈀(0)、0.0348g(0.0729mmol)之2-二環己基膦基-2,4’,6’-三異丙基聯苯(XPhos)、0.0229g(0.547mmol)之碳酸鈉中,添加2.00mL之1,4-二烷與0.200mL之水,於100℃攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~80/20)純化。減壓濃縮後,將所得到之固體懸浮於乙醇中,藉由進行濾取、乾燥,得到0.067g(產率58.1%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.94 (1H, s), 8.78 (1H, d, J = 2.4 Hz), 8.64 (1H, d, J = 9.7 Hz), 8.52 (1H, d, J = 9.7 Hz), 8.42 (1H, d, J = 2.4 Hz), 8.25-8.21 (2H, m), 7.61 (2H, d, J = 7.9 Hz), 7.48 (2H, s), 7.28-7.23 (4H, m), 7.02 (1H, d, J = 8.5 Hz), 4.12 (2H, d, J = 7.3 Hz), 3.91-3.83 (5H, m), 3.79 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.37 (3H, s), 2.16-2.07 (1H, m), 1.47 (2H, br d, J = 10.9 Hz), 1.37-1.26 (2H, m).
MS (APCI) m/z : 633 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表19之最終物。
[表19-1]
於0.0800g(0.182mmol)之5-(3,4-二甲氧基苯基)-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺、0.0974g(0.273mmol)之N-(6-氯嗒-3-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.0167g(0.0182mmol)之參(二苯亞甲基丙酮)鈀(0)、0.0348g(0.0729mmol)之2-二環己基膦基-2,4’,6’-三異丙基聯苯(XPhos)、0.0229g(0.547mmol)之碳酸鈉中,添加2.00mL之1,4-二烷與0.200mL之水,於100℃攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~80/20)純化。減壓濃縮後,將所得到之固體懸浮於乙醇中,藉由進行濾取、乾燥,得到0.067g(產率58.1%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.94 (1H, s), 8.78 (1H, d, J = 2.4 Hz), 8.64 (1H, d, J = 9.7 Hz), 8.52 (1H, d, J = 9.7 Hz), 8.42 (1H, d, J = 2.4 Hz), 8.25-8.21 (2H, m), 7.61 (2H, d, J = 7.9 Hz), 7.48 (2H, s), 7.28-7.23 (4H, m), 7.02 (1H, d, J = 8.5 Hz), 4.12 (2H, d, J = 7.3 Hz), 3.91-3.83 (5H, m), 3.79 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.37 (3H, s), 2.16-2.07 (1H, m), 1.47 (2H, br d, J = 10.9 Hz), 1.37-1.26 (2H, m).
MS (APCI) m/z : 633 [(M+H)+
].
同樣地進行,由所對應的起始原料A及B合成表19之最終物。
[表19-1] 
[表19-2] 
[表19-3] 
[表19-4] 
(實施例110)N-[6-(2-胺基-5-{3-甲氧基-4-[2-(啉-4-基)乙氧基]苯基}吡啶-3-基)嗒-3-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 於0.245g(0.600mmol)之3-溴-5-{3-甲氧基-4-[2-(啉-4-基)乙氧基]苯基}吡啶-2-胺、0.229g(0.900mmol)之雙(頻哪醇)二硼烷、0.0330g(0.0360mmol)之參(二苯亞甲基丙酮)二鈀(0)、0.0236g(0.0840mmol)之三環己基膦、0.0883g(0.900mmol)之乙酸鉀中,添加3.00mL之1,4-二烷,於氮氣環境下,於80℃攪拌6小時。放冷至室溫後,過濾反應液,將濾液減壓濃縮。於所得到之殘渣中,添加0.175g(0.399mmol)之N-(6-氯嗒-3-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.0326g(0.0399mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II)二氯甲烷錯合物(1:1)、0.390g(1.20mmol)之碳酸銫、4.00mL之1,4-二烷、0.400mL之水,於氮氣環境下,於100℃攪拌4小時。放冷至室溫後,於反應液中添加飽和碳酸氫鈉水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~80/20)純化。減壓濃縮後,將所得到之固體懸浮於乙酸乙酯中,藉由進行濾取、乾燥,得到0.153g(產率52.4%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.77 (1H, s), 8.67 (1H, d, J = 9.7 Hz), 8.51 (1H, d, J = 2.4 Hz), 8.35 (1H, d, J = 2.4 Hz), 7.96-7.94 (2H, m), 7.53-7.48 (3H, m), 7.27 (2H, d, J = 7.9 Hz), 7.08-7.05 (2H, m), 6.98 (1H, d, J = 7.9 Hz), 6.89 (2H, s), 4.21 (2H, t, J = 6.1 Hz), 4.03 (2H, dd, J = 11.2, 3.3 Hz), 3.94 (3H, s), 3.87 (2H, d, J = 7.3 Hz), 3.76 (4H, t, J = 4.9 Hz), 3.39 (2H, t, J = 11.2 Hz), 2.88 (2H, t, J = 6.1 Hz), 2.63-2.61 (4H, m), 2.41 (3H, s), 2.15-2.07 (1H, m), 1.62-1.60 (2H, br d、J = 11.2 Hz), 1.49-1.39 (2H, m).
MS (APCI) m/z : 732 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表20之最終物。
[表20]
於0.245g(0.600mmol)之3-溴-5-{3-甲氧基-4-[2-(啉-4-基)乙氧基]苯基}吡啶-2-胺、0.229g(0.900mmol)之雙(頻哪醇)二硼烷、0.0330g(0.0360mmol)之參(二苯亞甲基丙酮)二鈀(0)、0.0236g(0.0840mmol)之三環己基膦、0.0883g(0.900mmol)之乙酸鉀中,添加3.00mL之1,4-二烷,於氮氣環境下,於80℃攪拌6小時。放冷至室溫後,過濾反應液,將濾液減壓濃縮。於所得到之殘渣中,添加0.175g(0.399mmol)之N-(6-氯嗒-3-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.0326g(0.0399mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II)二氯甲烷錯合物(1:1)、0.390g(1.20mmol)之碳酸銫、4.00mL之1,4-二烷、0.400mL之水,於氮氣環境下,於100℃攪拌4小時。放冷至室溫後,於反應液中添加飽和碳酸氫鈉水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~80/20)純化。減壓濃縮後,將所得到之固體懸浮於乙酸乙酯中,藉由進行濾取、乾燥,得到0.153g(產率52.4%)之呈固體的標題化合物。1
H-NMR (CDCl3
) δ: 13.77 (1H, s), 8.67 (1H, d, J = 9.7 Hz), 8.51 (1H, d, J = 2.4 Hz), 8.35 (1H, d, J = 2.4 Hz), 7.96-7.94 (2H, m), 7.53-7.48 (3H, m), 7.27 (2H, d, J = 7.9 Hz), 7.08-7.05 (2H, m), 6.98 (1H, d, J = 7.9 Hz), 6.89 (2H, s), 4.21 (2H, t, J = 6.1 Hz), 4.03 (2H, dd, J = 11.2, 3.3 Hz), 3.94 (3H, s), 3.87 (2H, d, J = 7.3 Hz), 3.76 (4H, t, J = 4.9 Hz), 3.39 (2H, t, J = 11.2 Hz), 2.88 (2H, t, J = 6.1 Hz), 2.63-2.61 (4H, m), 2.41 (3H, s), 2.15-2.07 (1H, m), 1.62-1.60 (2H, br d、J = 11.2 Hz), 1.49-1.39 (2H, m).
MS (APCI) m/z : 732 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表20之最終物。
[表20] 
(實施例112)N-{5-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]吡-2-基}-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 於0.111g(0.310mmol)之5-(3,4-二甲氧基苯基)-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺、0.100g(0.207mmol)之N-(5-溴吡-2-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.0169g(0.0207mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II)二氯甲烷體(1:1)、0.202g(0.621mmol)之碳酸銫中,添加2.00mL之1,4-二烷與0.400mL之水,於80℃攪拌1小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~90/10)純化。減壓濃縮後,將殘渣使用乙酸乙酯與二乙基醚固化,藉由將固體進行濾取、乾燥,得到0.111g(產率84.8%)之呈固體的標題化合物。
1H-NMR (DMSO-D6) δ: 13.61 (1H, s), 9.58 (1H, d, J = 1.2 Hz), 9.17 (1H, d, J = 2.4 Hz), 8.79 (1H, d, J = 2.4 Hz), 8.39 (1H, d, J = 2.4 Hz), 8.32 (1H, d, J = 2.4 Hz), 8.20 (1H, d, J = 2.4 Hz), 7.59 (2H, d, J = 8.5 Hz), 7.29-7.23 (6H, m), 7.02 (1H, d, J = 8.5 Hz), 4.13 (2H, d, J = 6.7 Hz), 3.89-3.85 (5H, m), 3.79 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.36 (3H, s), 2.16-2.07 (1H, m), 1.49-1.44 (2H, m), 1.37-1.27 (2H, m).
MS (APCI) m/z : 633 [(M+H)+
].
於0.111g(0.310mmol)之5-(3,4-二甲氧基苯基)-3-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶-2-胺、0.100g(0.207mmol)之N-(5-溴吡-2-基)-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.0169g(0.0207mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II)二氯甲烷體(1:1)、0.202g(0.621mmol)之碳酸銫中,添加2.00mL之1,4-二烷與0.400mL之水,於80℃攪拌1小時。放冷至室溫後,於反應液中添加水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~90/10)純化。減壓濃縮後,將殘渣使用乙酸乙酯與二乙基醚固化,藉由將固體進行濾取、乾燥,得到0.111g(產率84.8%)之呈固體的標題化合物。
1H-NMR (DMSO-D6) δ: 13.61 (1H, s), 9.58 (1H, d, J = 1.2 Hz), 9.17 (1H, d, J = 2.4 Hz), 8.79 (1H, d, J = 2.4 Hz), 8.39 (1H, d, J = 2.4 Hz), 8.32 (1H, d, J = 2.4 Hz), 8.20 (1H, d, J = 2.4 Hz), 7.59 (2H, d, J = 8.5 Hz), 7.29-7.23 (6H, m), 7.02 (1H, d, J = 8.5 Hz), 4.13 (2H, d, J = 6.7 Hz), 3.89-3.85 (5H, m), 3.79 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.36 (3H, s), 2.16-2.07 (1H, m), 1.49-1.44 (2H, m), 1.37-1.27 (2H, m).
MS (APCI) m/z : 633 [(M+H)+
].
(實施例113)N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-環丙基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 於0.350g(0.503mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之5.00mL甲苯懸浮液中,添加0.0648g(0.755mmol)之環丙基硼酸、0.0282g(0.101mmol)之三環己基膦、0.374g(1.76mmol)之磷酸三鉀、1.00mL之水,氮取代後,添加0.0113g(0.0503mmol)之乙酸鈀(II),加溫至100℃後,添加2.00mL之1,4-二烷,於同溫度攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~85/15)純化,進一步以高效液相層析(NOMURA Developsil Combi,乙腈/水/0.1%甲酸)純化。將有機溶劑減壓餾去後,於殘渣中添加飽和碳酸氫鈉水,藉由將析出之固體進行濾取、乾燥,得到0.200g(產率60.5%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.11 (1H, s), 8.71 (1H, d, J = 2.4 Hz), 8.25 (1H, d, J = 2.4 Hz), 8.16 (1H, d, J = 2.4 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.63 (1H, d, J = 2.4 Hz), 7.58-7.52 (4H, m), 7.20-7.14 (4H, m), 6.99 (1H, d, J = 8.5 Hz), 5.76 (2H, s), 4.10 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.16-2.04 (1H, m), 2.00-1.93 (1H, m), 1.46 (2H, br d, J = 10.9 Hz), 1.35-1.26 (2H, m), 1.02-0.97 (2H, m), 0.73-0.69 (2H, m).
MS (APCI) m/z : 657[(M+H)+
].
於0.350g(0.503mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(4-溴苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺之5.00mL甲苯懸浮液中,添加0.0648g(0.755mmol)之環丙基硼酸、0.0282g(0.101mmol)之三環己基膦、0.374g(1.76mmol)之磷酸三鉀、1.00mL之水,氮取代後,添加0.0113g(0.0503mmol)之乙酸鈀(II),加溫至100℃後,添加2.00mL之1,4-二烷,於同溫度攪拌4小時。放冷至室溫後,於反應液中添加水,以二氯甲烷萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=100/0~85/15)純化,進一步以高效液相層析(NOMURA Developsil Combi,乙腈/水/0.1%甲酸)純化。將有機溶劑減壓餾去後,於殘渣中添加飽和碳酸氫鈉水,藉由將析出之固體進行濾取、乾燥,得到0.200g(產率60.5%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.11 (1H, s), 8.71 (1H, d, J = 2.4 Hz), 8.25 (1H, d, J = 2.4 Hz), 8.16 (1H, d, J = 2.4 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.63 (1H, d, J = 2.4 Hz), 7.58-7.52 (4H, m), 7.20-7.14 (4H, m), 6.99 (1H, d, J = 8.5 Hz), 5.76 (2H, s), 4.10 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.16-2.04 (1H, m), 2.00-1.93 (1H, m), 1.46 (2H, br d, J = 10.9 Hz), 1.35-1.26 (2H, m), 1.02-0.97 (2H, m), 0.73-0.69 (2H, m).
MS (APCI) m/z : 657[(M+H)+
].
(實施例114)N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺 於0.620g(1.00mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.480g(1.70mmol)之5-甲基吡啶-2-硼酸N-苯基二乙醇胺酯、0.0762g(0.400mmol)之碘化銅(I)中,添加6.00mL之甲苯與1.85mL之甲醇、1.84g(13.3mmol)之碳酸鉀之1.20mL水溶液,氮取代後,添加0.116g(0.100mmol)之肆(三苯基膦)鈀(0),於100℃攪拌4小時。放冷至室溫後,於反應液中添加乙酸乙酯與水,將不溶物濾去。將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~85/15)純化。減壓濃縮後,於殘渣中添加乙酸乙酯固化,藉由將固體進行濾取、乾燥,得到0.229g(產率36.2%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.98 (1H, s), 8.75 (1H, d, J = 2.4 Hz), 8.71 (1H, d, J = 2.4 Hz), 8.30 (1H, d, J = 2.4 Hz), 8.25 (1H, d, J = 2.4 Hz), 8.02 (1H, dd, J = 8.5, 2.4 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.61 (1H, d, J = 2.4 Hz), 7.53 (2H, d, J = 8.5 Hz), 7.34 (1H, d, J = 8.5 Hz), 7.19-7.13 (2H, m), 6.98 (1H, d, J = 8.5 Hz), 5.69 (2H, s), 4.12 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 11.0 Hz), 2.52 (3H, s), 2.17-2.07 (1H, m), 1.47 (2H, br d, J = 11.0 Hz), 1.37-1.26 (2H, m).
MS (APCI) m/z: 632 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表21之最終物。
[表21]
於0.620g(1.00mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.480g(1.70mmol)之5-甲基吡啶-2-硼酸N-苯基二乙醇胺酯、0.0762g(0.400mmol)之碘化銅(I)中,添加6.00mL之甲苯與1.85mL之甲醇、1.84g(13.3mmol)之碳酸鉀之1.20mL水溶液,氮取代後,添加0.116g(0.100mmol)之肆(三苯基膦)鈀(0),於100℃攪拌4小時。放冷至室溫後,於反應液中添加乙酸乙酯與水,將不溶物濾去。將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~85/15)純化。減壓濃縮後,於殘渣中添加乙酸乙酯固化,藉由將固體進行濾取、乾燥,得到0.229g(產率36.2%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.98 (1H, s), 8.75 (1H, d, J = 2.4 Hz), 8.71 (1H, d, J = 2.4 Hz), 8.30 (1H, d, J = 2.4 Hz), 8.25 (1H, d, J = 2.4 Hz), 8.02 (1H, dd, J = 8.5, 2.4 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.61 (1H, d, J = 2.4 Hz), 7.53 (2H, d, J = 8.5 Hz), 7.34 (1H, d, J = 8.5 Hz), 7.19-7.13 (2H, m), 6.98 (1H, d, J = 8.5 Hz), 5.69 (2H, s), 4.12 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 11.0 Hz), 2.52 (3H, s), 2.17-2.07 (1H, m), 1.47 (2H, br d, J = 11.0 Hz), 1.37-1.26 (2H, m).
MS (APCI) m/z: 632 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表21之最終物。
[表21] 
(實施例116) N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-6’-甲基-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫-3,3’-聯吡啶-5-甲醯胺 於氮氣環境下,於0.400g(0.646mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.212g(0.969mmol)之2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶、0.631g(1.94mmol)之碳酸銫、0.0527g(0.0646mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)中,添加5.00mL之1,4-二烷與1.00mL之水,於80℃攪拌4小時。放冷至室溫後,於反應液中添加水與乙酸乙酯,將不溶物濾去。將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~90/10)純化,進一步以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~5/95)純化。減壓濃縮後,將殘渣以高效液相層析(NOMURA Developsil Combi,乙腈/水/0.1%甲酸)純化。將有機溶劑減壓餾去後,於殘渣中添加飽和碳酸氫鈉水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。藉由過濾、減壓濃縮,得到0.129g(產率31.6%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.08 (1H, s), 8.76 (1H, d, J = 2.4 Hz), 8.69 (1H, d, J = 2.4 Hz), 8.51-8.47 (2H, m), 8.26 (1H, d, J = 2.4 Hz), 7.84 (2H, d, J = 8.5 Hz), 7.71 (1H, dd, J = 8.5, 2.4 Hz), 7.62 (1H, d, J = 2.4 Hz), 7.55 (2H, d, J = 8.5 Hz), 7.20-7.14 (2H, m), 6.99 (1H, d, J = 8.5 Hz), 5.69 (2H, s), 4.20 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 11.3 Hz), 2.36 (3H, s), 2.11-2.03 (1H, m), 1.47 (2H, br d, J = 11.0 Hz), 1.37-1.26 (2H, m).
MS (APCI) m/z: 632 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表22之最終物。
[表22]
於氮氣環境下,於0.400g(0.646mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.212g(0.969mmol)之2-甲基-5-(4,4,5,5-四甲基-1,3,2-二氧環戊硼烷-2-基)吡啶、0.631g(1.94mmol)之碳酸銫、0.0527g(0.0646mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)中,添加5.00mL之1,4-二烷與1.00mL之水,於80℃攪拌4小時。放冷至室溫後,於反應液中添加水與乙酸乙酯,將不溶物濾去。將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~90/10)純化,進一步以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~5/95)純化。減壓濃縮後,將殘渣以高效液相層析(NOMURA Developsil Combi,乙腈/水/0.1%甲酸)純化。將有機溶劑減壓餾去後,於殘渣中添加飽和碳酸氫鈉水,以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。藉由過濾、減壓濃縮,得到0.129g(產率31.6%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.08 (1H, s), 8.76 (1H, d, J = 2.4 Hz), 8.69 (1H, d, J = 2.4 Hz), 8.51-8.47 (2H, m), 8.26 (1H, d, J = 2.4 Hz), 7.84 (2H, d, J = 8.5 Hz), 7.71 (1H, dd, J = 8.5, 2.4 Hz), 7.62 (1H, d, J = 2.4 Hz), 7.55 (2H, d, J = 8.5 Hz), 7.20-7.14 (2H, m), 6.99 (1H, d, J = 8.5 Hz), 5.69 (2H, s), 4.20 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 11.3 Hz), 2.36 (3H, s), 2.11-2.03 (1H, m), 1.47 (2H, br d, J = 11.0 Hz), 1.37-1.26 (2H, m).
MS (APCI) m/z: 632 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表22之最終物。
[表22] 
(實施例119) N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-(5-甲基噻吩-2-基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺 於0.200g(0.323mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.055g(0.387mmol)之(5-甲基-2-噻吩基)硼酸、0.134g(0.969mmol)之碳酸鉀、0.0373g(0.0323mmol)之肆(三苯基膦)鈀(0)中,添加5.00mL之1,2-二甲氧基乙烷與0.500mL之水,於90℃攪拌12小時後,放冷。於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~80/20)純化,以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~93/7)純化。進一步藉由以高效液相層析(NOMURA Developsil Combi,乙腈/水/0.1%甲酸)純化,得到0.0860g(產率41.8%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.90 (1H, s), 8.69-8.66 (2H, m), 8.26 (1H, d, J = 2.4 Hz), 7.84 (2H, d, J = 8.5 Hz), 7.62 (1H, d, J = 2.4 Hz), 7.57-7.54 (3H, m), 7.20-7.14 (2H, m), 6.99 (1H, d, J = 8.5 Hz), 6.87-6.85 (1H, m), 5.70 (2H, s), 4.14 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.49 (3H, s), 2.16-2.10 (1H, m), 1.45 (2H, br d, J = 10.9 Hz), 1.38-1.27 (2H, m).
MS (APCI) m/z : 637 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表23之最終物。
[表23]
於0.200g(0.323mmol)之N-{4-[2-胺基-5-(3,4-二甲氧基苯基)吡啶-3-基]苯基}-5-溴-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺、0.055g(0.387mmol)之(5-甲基-2-噻吩基)硼酸、0.134g(0.969mmol)之碳酸鉀、0.0373g(0.0323mmol)之肆(三苯基膦)鈀(0)中,添加5.00mL之1,2-二甲氧基乙烷與0.500mL之水,於90℃攪拌12小時後,放冷。於反應液中添加水,以二氯甲烷萃取3次,將有機層以無水硫酸鈉乾燥。過濾、減壓濃縮後,將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~80/20)純化,以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~93/7)純化。進一步藉由以高效液相層析(NOMURA Developsil Combi,乙腈/水/0.1%甲酸)純化,得到0.0860g(產率41.8%)之呈固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 12.90 (1H, s), 8.69-8.66 (2H, m), 8.26 (1H, d, J = 2.4 Hz), 7.84 (2H, d, J = 8.5 Hz), 7.62 (1H, d, J = 2.4 Hz), 7.57-7.54 (3H, m), 7.20-7.14 (2H, m), 6.99 (1H, d, J = 8.5 Hz), 6.87-6.85 (1H, m), 5.70 (2H, s), 4.14 (2H, d, J = 7.3 Hz), 3.88-3.83 (5H, m), 3.77 (3H, s), 3.27 (2H, t, J = 10.9 Hz), 2.49 (3H, s), 2.16-2.10 (1H, m), 1.45 (2H, br d, J = 10.9 Hz), 1.38-1.27 (2H, m).
MS (APCI) m/z : 637 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表23之最終物。
[表23] 
(實施例121)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基] -1’-[(4-氰基四氫-2H-吡喃-4-基) 甲基]-5-甲基-4’-側氧-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺 於氮氣環境下,於0.230g(0.279mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基] -3-甲氧基苯基}吡啶-3-基) -3-氟苯基]-5-溴-1’-[(4-氰基四氫-2H-吡喃-4-基) 甲基]-4’-側氧-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺、0.0228g(0.0279mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、0.272g(0.836mmol)之碳酸銫中,添加5.00mL之1,4-二烷、0.500mL之水、0.0960mL(0.334mmol)之50%三甲基硼環氧烷四氫呋喃溶液(3.5mol/L),於100℃攪拌3小時半。放冷至室溫後,於反應液中添加飽和碳酸氫鈉水與乙酸乙酯,將不溶物濾去。將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~80/20)純化後,進一步以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~93/7)純化,得到0.145g(產率68.4%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.07 (1H, s), 8.86 (1H, d, J = 2.4 Hz), 8.80 (1H, d, J = 2.4 Hz), 8.52-8.48 (2H, m), 8.31 (1H, d, J = 2.4 Hz), 7.94 (1H, dd, J = 12.1, 2.4 Hz), 7.72 (1H, dd, J = 8.2, 2.4 Hz), 7.63 (1H, d, J = 2.4 Hz), 7.50-7.42 (2H, m), 7.18 (1H, d, J = 2.4 Hz), 7.11 (1H, dd, J = 8.5, 2.4 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.71 (2H, s), 4.70 (2H, s), 3.97-3.74 (10H, m), 3.69-3.60 (2H, m), 3.52-3.32 (4H, m), 2.36 (3H, s), 1.88-1.77 (4H, m).
MS (APCI) m/z : 761 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表24之最終物。
[表24]
於氮氣環境下,於0.230g(0.279mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基] -3-甲氧基苯基}吡啶-3-基) -3-氟苯基]-5-溴-1’-[(4-氰基四氫-2H-吡喃-4-基) 甲基]-4’-側氧-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺、0.0228g(0.0279mmol)之[1,1’-雙(二苯基膦基)二茂鐵]二氯化鈀(II) 二氯甲烷錯合物(1:1)、0.272g(0.836mmol)之碳酸銫中,添加5.00mL之1,4-二烷、0.500mL之水、0.0960mL(0.334mmol)之50%三甲基硼環氧烷四氫呋喃溶液(3.5mol/L),於100℃攪拌3小時半。放冷至室溫後,於反應液中添加飽和碳酸氫鈉水與乙酸乙酯,將不溶物濾去。將濾液以乙酸乙酯萃取3次,將有機層以飽和食鹽水洗淨後,以無水硫酸鈉乾燥。過濾、減壓濃縮後,藉由將所得到之殘渣以矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~80/20)純化後,進一步以胺基矽膠管柱層析(山善公司,乙酸乙酯/甲醇=99/1~93/7)純化,得到0.145g(產率68.4%)之呈非晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.07 (1H, s), 8.86 (1H, d, J = 2.4 Hz), 8.80 (1H, d, J = 2.4 Hz), 8.52-8.48 (2H, m), 8.31 (1H, d, J = 2.4 Hz), 7.94 (1H, dd, J = 12.1, 2.4 Hz), 7.72 (1H, dd, J = 8.2, 2.4 Hz), 7.63 (1H, d, J = 2.4 Hz), 7.50-7.42 (2H, m), 7.18 (1H, d, J = 2.4 Hz), 7.11 (1H, dd, J = 8.5, 2.4 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.71 (2H, s), 4.70 (2H, s), 3.97-3.74 (10H, m), 3.69-3.60 (2H, m), 3.52-3.32 (4H, m), 2.36 (3H, s), 1.88-1.77 (4H, m).
MS (APCI) m/z : 761 [(M+H)+
].
同樣地進行,由所對應的起始原料合成表24之最終物。
[表24] 
(實施例124)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’- (四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺甲磺酸鹽(非晶性固體) 於23.9g(32.5mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’- 二氫-2,3’-聯吡啶-5’-甲醯胺之110mL二氯甲烷溶液中,添加2.11mL(3.12g,32.5mmol)之甲磺酸之36.0mL乙醇溶液,於室溫攪拌15分鐘。將反應液減壓濃縮後,將所得到之非晶性固體懸浮於乙酸乙酯中,藉由進行濾取、乾燥,得到25.7g(產率95.1%)之呈非晶性固體的標題化合物。
1H-NMR (DMSO-D6) δ: 13.27 (1H, s), 8.79 (1H, d, J = 2.4 Hz), 8.72 (1H, d, J = 2.4 Hz), 8.54 (1H, d, J = 2.4 Hz), 8.46 (1H, d, J = 8.5 Hz), 8.34 (1H, d, J = 2.4 Hz), 8.29 (1H, d, J = 2.4 Hz), 8.03-7.99 (1H, m), 7.78 (1H, d, J = 8.5 Hz), 7.59-7.51 (4H, m), 7.31 (1H, d, J = 2.4 Hz), 7.25 (1H, dd, J = 8.5, 2.4 Hz), 7.06 (1H, d, J = 8.5 Hz), 4.21 (2H, d, J = 7.3 Hz), 4.01-3.93 (2H, m), 3.90-3.74 (8H, m), 3.69-3.24 (6H, m), 2.37 (3H, s), 2.34 (3H, s), 2.13-2.02 (1H, m), 1.47 (2H, br d, J = 10.9 Hz), 1.37-1.28 (2H, m).
MS (APCI) m/z : 736[(M+H)+
].
作為元素分析値C41
H42
FN5
O7
・1.0CH3
SO3
H・1.0H2
O
計算値:C, 59.35; H, 5.69; N, 8.24; F, 2.24; S, 3.77.
實測値:C, 59.38; H, 5.80; N, 8.10; F, 2.23; S, 3.71.
同樣地進行,由所對應的起始原料得到表25之呈非晶性固體的各甲磺酸鹽。
[表25-1]
於23.9g(32.5mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’- 二氫-2,3’-聯吡啶-5’-甲醯胺之110mL二氯甲烷溶液中,添加2.11mL(3.12g,32.5mmol)之甲磺酸之36.0mL乙醇溶液,於室溫攪拌15分鐘。將反應液減壓濃縮後,將所得到之非晶性固體懸浮於乙酸乙酯中,藉由進行濾取、乾燥,得到25.7g(產率95.1%)之呈非晶性固體的標題化合物。
1H-NMR (DMSO-D6) δ: 13.27 (1H, s), 8.79 (1H, d, J = 2.4 Hz), 8.72 (1H, d, J = 2.4 Hz), 8.54 (1H, d, J = 2.4 Hz), 8.46 (1H, d, J = 8.5 Hz), 8.34 (1H, d, J = 2.4 Hz), 8.29 (1H, d, J = 2.4 Hz), 8.03-7.99 (1H, m), 7.78 (1H, d, J = 8.5 Hz), 7.59-7.51 (4H, m), 7.31 (1H, d, J = 2.4 Hz), 7.25 (1H, dd, J = 8.5, 2.4 Hz), 7.06 (1H, d, J = 8.5 Hz), 4.21 (2H, d, J = 7.3 Hz), 4.01-3.93 (2H, m), 3.90-3.74 (8H, m), 3.69-3.24 (6H, m), 2.37 (3H, s), 2.34 (3H, s), 2.13-2.02 (1H, m), 1.47 (2H, br d, J = 10.9 Hz), 1.37-1.28 (2H, m).
MS (APCI) m/z : 736[(M+H)+
].
作為元素分析値C41
H42
FN5
O7
・1.0CH3
SO3
H・1.0H2
O
計算値:C, 59.35; H, 5.69; N, 8.24; F, 2.24; S, 3.77.
實測値:C, 59.38; H, 5.80; N, 8.10; F, 2.23; S, 3.71.
同樣地進行,由所對應的起始原料得到表25之呈非晶性固體的各甲磺酸鹽。
[表25-1] 
[表25-2] 
[表25-3] 
以下,N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺係實施例1之化合物,N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’- 二氫-2,3’-聯吡啶-5’-甲醯胺係實施例34之化合物。
(實施例130)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺甲磺酸鹽水合物 將46.3g(62.6mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺懸浮於244ml之丙酮中,添加27.0ml水。於混液中,每次少量添加4.27ml(65.8mmol)之甲磺酸。將反應液於室溫攪拌30小時後,藉由布赫納漏斗(Büchner funnel)抽吸過濾,將所濾取之固體以水/丙酮混合液(水/丙酮=1/9)洗淨,得到47.5g(產率87.5%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.0 Hz), 8.28 (1H, d, J = 2.0 Hz), 8.21 (1H, d, J = 2.0 Hz), 8.19 (1H, d, J = 2.0 Hz), 7.90 (2H, d, J = 8.5 Hz), 7.60-7.52 (6H, m), 7.32 (1H, d, J = 2.0 Hz), 7.27 (3H, d, J = 8.5 Hz), 7.06 (1H, d, J = 8.5 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.89-3.22 (14H, m), 2.36 (3H, s), 2.32 (3H, s), 2.17-2.04 (1H, m), 1.46 (2H, d, J = 11.0 Hz), 1.37-1.25 (2H, m).
MS (APCI) m/z : 717 [(M+H)+
].
作為元素分析値C42
H44
N4
O7
・1.0CH3
SO3
H・3.0H2
O
計算値:C, 59.57; H, 6.28; N, 6.46; S, 3.70.
實測値:C, 59.75; H, 6.29; N, 6.48; S, 3.76.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖1中之最大波峰強度設為100的情形下之相對強度22以上的波峰顯示於表26。
[表26]
將46.3g(62.6mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺懸浮於244ml之丙酮中,添加27.0ml水。於混液中,每次少量添加4.27ml(65.8mmol)之甲磺酸。將反應液於室溫攪拌30小時後,藉由布赫納漏斗(Büchner funnel)抽吸過濾,將所濾取之固體以水/丙酮混合液(水/丙酮=1/9)洗淨,得到47.5g(產率87.5%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.0 Hz), 8.28 (1H, d, J = 2.0 Hz), 8.21 (1H, d, J = 2.0 Hz), 8.19 (1H, d, J = 2.0 Hz), 7.90 (2H, d, J = 8.5 Hz), 7.60-7.52 (6H, m), 7.32 (1H, d, J = 2.0 Hz), 7.27 (3H, d, J = 8.5 Hz), 7.06 (1H, d, J = 8.5 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.89-3.22 (14H, m), 2.36 (3H, s), 2.32 (3H, s), 2.17-2.04 (1H, m), 1.46 (2H, d, J = 11.0 Hz), 1.37-1.25 (2H, m).
MS (APCI) m/z : 717 [(M+H)+
].
作為元素分析値C42
H44
N4
O7
・1.0CH3
SO3
H・3.0H2
O
計算値:C, 59.57; H, 6.28; N, 6.46; S, 3.70.
實測値:C, 59.75; H, 6.29; N, 6.48; S, 3.76.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖1中之最大波峰強度設為100的情形下之相對強度22以上的波峰顯示於表26。
[表26] 
(實施例131)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺氫溴酸鹽水合物 於250mg(349μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加3.99ml之甲醇、633μl之水。其後,添加365μl(365μmol)之1.00mol/L氫溴酸水溶液。將混合液於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到243mg(產率81.4%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.5 Hz), 8.28 (1H, d, J = 2.5 Hz), 8.21 (1H, d, J = 2.5 Hz), 8.19 (1H, d, J = 2.5 Hz), 7.90 (2H, d, J = 8.8 Hz), 7.60-7.55 (4H, m), 7.50 (2H, br s), 7.31 (1H, d, J = 2.5 Hz), 7.29-7.23 (3H, m), 7.06 (1H, d, J = 8.8 Hz), 4.12 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.89-3.24 (14H, m), 2.36 (3H, s), 2.16-2.05 (1H, m), 1.46 (2H, d, J = 12.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・1HBr・3.3H2
O
計算値:C,58.85; H,6.07; N,6.53;Br, 9.32.
實測値:C,58.91; H,5.98; N,6.58;Br, 9.42.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖2中之最大波峰強度設為100的情形下之相對強度19以上的波峰顯示於表27。
[表27]
於250mg(349μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加3.99ml之甲醇、633μl之水。其後,添加365μl(365μmol)之1.00mol/L氫溴酸水溶液。將混合液於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到243mg(產率81.4%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.5 Hz), 8.28 (1H, d, J = 2.5 Hz), 8.21 (1H, d, J = 2.5 Hz), 8.19 (1H, d, J = 2.5 Hz), 7.90 (2H, d, J = 8.8 Hz), 7.60-7.55 (4H, m), 7.50 (2H, br s), 7.31 (1H, d, J = 2.5 Hz), 7.29-7.23 (3H, m), 7.06 (1H, d, J = 8.8 Hz), 4.12 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.89-3.24 (14H, m), 2.36 (3H, s), 2.16-2.05 (1H, m), 1.46 (2H, d, J = 12.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・1HBr・3.3H2
O
計算値:C,58.85; H,6.07; N,6.53;Br, 9.32.
實測値:C,58.91; H,5.98; N,6.58;Br, 9.42.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖2中之最大波峰強度設為100的情形下之相對強度19以上的波峰顯示於表27。
[表27] 
(實施例132)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺硝酸鹽水合物 於249mg(347μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加1.49ml之甲醇、9.10μl之水。其後,添加364μl(364μmol)之1.00mol/L硝酸水溶液。將混合液於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到252mg(產率87.0%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.5 Hz), 8.27 (1H, d, J = 2.5 Hz), 8.21 (1H, d, J = 2.5 Hz), 8.19 (1H, d, J = 2.5 Hz), 7.90 (2H, d, J = 8.8 Hz), 7.60-7.55 (4H, m), 7.49 (2H, br s), 7.31 (1H, d, J = 2.4 Hz), 7.29-7.24 (3H, m), 7.06 (1H, d, J = 8.8 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.90-3.22 (14H, m), 2.36 (3H, s), 2.17-2.04 (1H, m), 1.46 (2H, d, J = 12.5 Hz), 1.38-1.26 (2H, m).
作為元素分析値C42
H44
N4
O7
・1HNO3
・3H2
O
計算値:C,60.49; H,6.16; N,8.40.
實測値:C,60.58; H,6.15; N,8.43.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖3中之最大波峰強度設為100的情形下之相對強度34以上的波峰顯示於表28。
[表28]
於249mg(347μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加1.49ml之甲醇、9.10μl之水。其後,添加364μl(364μmol)之1.00mol/L硝酸水溶液。將混合液於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到252mg(產率87.0%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.5 Hz), 8.27 (1H, d, J = 2.5 Hz), 8.21 (1H, d, J = 2.5 Hz), 8.19 (1H, d, J = 2.5 Hz), 7.90 (2H, d, J = 8.8 Hz), 7.60-7.55 (4H, m), 7.49 (2H, br s), 7.31 (1H, d, J = 2.4 Hz), 7.29-7.24 (3H, m), 7.06 (1H, d, J = 8.8 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.90-3.22 (14H, m), 2.36 (3H, s), 2.17-2.04 (1H, m), 1.46 (2H, d, J = 12.5 Hz), 1.38-1.26 (2H, m).
作為元素分析値C42
H44
N4
O7
・1HNO3
・3H2
O
計算値:C,60.49; H,6.16; N,8.40.
實測値:C,60.58; H,6.15; N,8.43.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖3中之最大波峰強度設為100的情形下之相對強度34以上的波峰顯示於表28。
[表28] 
(實施例133)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺硫酸鹽水合物 於249mg(347μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加3.98ml之甲醇、632μl之水。其後,添加363μl(363μmol)之1.00mol/L 硫酸水溶液。將混合液於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到271mg(產率90.4%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.20 (1H, s), 8.72 (1H, d, J = 2.4 Hz), 8.27 (1H, d, J = 2.4 Hz), 8.18 (1H, d, J = 2.4 Hz), 8.08 (1H, s), 7.88 (2H, d, J = 8.5 Hz), 7.57 (4H, t, J = 8.5 Hz), 7.30-7.03 (7H, m), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.92 (2H, m), 3.91-3.21 (14H, m), 2.36 (3H, s), 2.16-2.05 (1H, m), 1.46 (2H, d, J = 12.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・0.75H2
SO4
・4H2
O
計算値:C,58.49; H,6.25; N,6.50; S, 2.79.
實測値:C,58.53; H,6.13; N,6.52; S, 2.80..
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖4中之最大波峰強度設為100的情形下之相對強度13以上的波峰顯示於表29。
[表29]
於249mg(347μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加3.98ml之甲醇、632μl之水。其後,添加363μl(363μmol)之1.00mol/L 硫酸水溶液。將混合液於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到271mg(產率90.4%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.20 (1H, s), 8.72 (1H, d, J = 2.4 Hz), 8.27 (1H, d, J = 2.4 Hz), 8.18 (1H, d, J = 2.4 Hz), 8.08 (1H, s), 7.88 (2H, d, J = 8.5 Hz), 7.57 (4H, t, J = 8.5 Hz), 7.30-7.03 (7H, m), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.92 (2H, m), 3.91-3.21 (14H, m), 2.36 (3H, s), 2.16-2.05 (1H, m), 1.46 (2H, d, J = 12.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・0.75H2
SO4
・4H2
O
計算値:C,58.49; H,6.25; N,6.50; S, 2.79.
實測値:C,58.53; H,6.13; N,6.52; S, 2.80..
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖4中之最大波峰強度設為100的情形下之相對強度13以上的波峰顯示於表29。
[表29] 
(實施例134)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺磷酸鹽水合物 於251mg(350μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加501μL之甲醇、1.28ml之水。其後,添加729μl(367μmol)之0.504mol/L磷酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到254mg(產率82.8%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.11 (1H, s), 8.72 (1H, d, J = 2.5 Hz), 8.25 (1H, d, J = 2.5 Hz), 8.17 (1H, d, J = 2.5 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.62 (1H, d, J = 2.5 Hz), 7.58 (2H, d, J = 8.5 Hz), 7.53 (2H, d, J = 8.5 Hz), 7.26 (2H, d, J = 8.5 Hz), 7.20 (1H, d, J = 2.0 Hz), 7.13 (1H, dd, J = 8.5, 2.0 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.73 (2H, br s), 4.11 (2H, d, J = 7.5 Hz), 3.98-3.90 (2H, m), 3.89-3.22 (14H, m), 2.36 (3H, s), 2.16-2.04 (1H, m), 1.46 (2H, d, J = 11.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・1.0H3
PO4
・3.5H2
O
計算値:C,57.46; H,6.20; N,6.38; P, 3.53.
實測値:C,57.41; H,6.20; N,6.41; P, 3.41.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖5中之最大波峰強度設為100的情形下之相對強度20以上的波峰顯示於表30。
[表30]
於251mg(350μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加501μL之甲醇、1.28ml之水。其後,添加729μl(367μmol)之0.504mol/L磷酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到254mg(產率82.8%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.11 (1H, s), 8.72 (1H, d, J = 2.5 Hz), 8.25 (1H, d, J = 2.5 Hz), 8.17 (1H, d, J = 2.5 Hz), 7.82 (2H, d, J = 8.5 Hz), 7.62 (1H, d, J = 2.5 Hz), 7.58 (2H, d, J = 8.5 Hz), 7.53 (2H, d, J = 8.5 Hz), 7.26 (2H, d, J = 8.5 Hz), 7.20 (1H, d, J = 2.0 Hz), 7.13 (1H, dd, J = 8.5, 2.0 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.73 (2H, br s), 4.11 (2H, d, J = 7.5 Hz), 3.98-3.90 (2H, m), 3.89-3.22 (14H, m), 2.36 (3H, s), 2.16-2.04 (1H, m), 1.46 (2H, d, J = 11.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・1.0H3
PO4
・3.5H2
O
計算値:C,57.46; H,6.20; N,6.38; P, 3.53.
實測値:C,57.41; H,6.20; N,6.41; P, 3.41.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖5中之最大波峰強度設為100的情形下之相對強度20以上的波峰顯示於表30。
[表30] 
(實施例135)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺乙磺酸鹽水合物 於251mg(350μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加1.00ml之甲醇、3.65ml之水。其後,添加368μl(368μmol)之1.00mol/L乙磺酸水溶液。於混合液中少量添加以下述所示之方法取得的種晶,於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到265mg(產率81.8%)之呈結晶性固體的標題化合物。
種晶之取得方法
於10.4mg(14.5μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加41.6μL之甲醇、151μl之水。其後,添加15.2μl(15.2μmol)之1.00mol/L乙磺酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到種晶10.9mg。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.0 Hz), 8.28 (1H, d, J = 2.0 Hz), 8.20 (2H, dd, J = 8.5, 2.0 Hz), 7.90 (2H, d, J = 8.5 Hz), 7.61-7.49 (6H, m), 7.32 (1H, d, J = 2.0 Hz), 7.29-7.24 (3H, m), 7.06 (1H, d, J = 8.5 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.87-3.24 (14H, m), 2.41-2.31 (5H, m), 2.16-2.04 (1H, m), 1.46 (2H, d, J = 11.5 Hz), 1.37-1.25 (2H, m), 1.06 (3H, t, J = 7.5 Hz).
作為元素分析値C42
H44
N4
O7
・1.0C2
H5
SO3
H・5.5H2
O
計算値:C,57.07; H,6.64; N,6.05; S, 3.46.
實測値:C,57.13; H,6.78; N,6.08; S, 3.60.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖6中之最大波峰強度設為100的情形下之相對強度5以上的波峰顯示於表31。
[表31]
於251mg(350μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加1.00ml之甲醇、3.65ml之水。其後,添加368μl(368μmol)之1.00mol/L乙磺酸水溶液。於混合液中少量添加以下述所示之方法取得的種晶,於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到265mg(產率81.8%)之呈結晶性固體的標題化合物。
種晶之取得方法
於10.4mg(14.5μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加41.6μL之甲醇、151μl之水。其後,添加15.2μl(15.2μmol)之1.00mol/L乙磺酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到種晶10.9mg。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.0 Hz), 8.28 (1H, d, J = 2.0 Hz), 8.20 (2H, dd, J = 8.5, 2.0 Hz), 7.90 (2H, d, J = 8.5 Hz), 7.61-7.49 (6H, m), 7.32 (1H, d, J = 2.0 Hz), 7.29-7.24 (3H, m), 7.06 (1H, d, J = 8.5 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.87-3.24 (14H, m), 2.41-2.31 (5H, m), 2.16-2.04 (1H, m), 1.46 (2H, d, J = 11.5 Hz), 1.37-1.25 (2H, m), 1.06 (3H, t, J = 7.5 Hz).
作為元素分析値C42
H44
N4
O7
・1.0C2
H5
SO3
H・5.5H2
O
計算値:C,57.07; H,6.64; N,6.05; S, 3.46.
實測値:C,57.13; H,6.78; N,6.08; S, 3.60.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖6中之最大波峰強度設為100的情形下之相對強度5以上的波峰顯示於表31。
[表31] 
(實施例136)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺苯磺酸鹽水合物 於250mg(349μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加1.00ml之甲醇、3.64ml之水。其後,添加366μl(366μmol)之1.00mol/L苯磺酸水溶液。使用超音波洗淨機製成懸浮液後,於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到287mg(產率88.3%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.0 Hz), 8.27 (1H, d, J = 2.0 Hz), 8.19 (2H, d, J = 2.0 Hz), 7.90 (2H, d, J = 8.5 Hz), 7.61-7.55 (6H, m), 7.44 (2H, br s), 7.35-7.24 (7H, m), 7.06 (1H, d, J = 8.5 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.90-3.22 (14H, m), 2.36 (3H, s), 2.17-2.04 (1H, m), 1.46 (2H, d, J = 11.6 Hz), 1.31 (2H, td, J = 12.2, 8.5 Hz).
作為元素分析値C42
H44
N4
O7
・1.0C6
H5
SO3
H・3H2
O
計算値:C,62.05; H,6.08; N,6.03; S, 3.45.
實測値:C,62.18; H,6.07; N,6.08; S, 3.46.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖7中之最大波峰強度設為100的情形下之相對強度29以上的波峰顯示於表32。
[表32]
於250mg(349μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加1.00ml之甲醇、3.64ml之水。其後,添加366μl(366μmol)之1.00mol/L苯磺酸水溶液。使用超音波洗淨機製成懸浮液後,於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到287mg(產率88.3%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.72 (1H, d, J = 2.0 Hz), 8.27 (1H, d, J = 2.0 Hz), 8.19 (2H, d, J = 2.0 Hz), 7.90 (2H, d, J = 8.5 Hz), 7.61-7.55 (6H, m), 7.44 (2H, br s), 7.35-7.24 (7H, m), 7.06 (1H, d, J = 8.5 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.90-3.22 (14H, m), 2.36 (3H, s), 2.17-2.04 (1H, m), 1.46 (2H, d, J = 11.6 Hz), 1.31 (2H, td, J = 12.2, 8.5 Hz).
作為元素分析値C42
H44
N4
O7
・1.0C6
H5
SO3
H・3H2
O
計算値:C,62.05; H,6.08; N,6.03; S, 3.45.
實測値:C,62.18; H,6.07; N,6.08; S, 3.46.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖7中之最大波峰強度設為100的情形下之相對強度29以上的波峰顯示於表32。
[表32] 
(實施例137)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺p-甲苯磺酸鹽 於268mg(373μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加2.14ml之甲醇、145μl之水。其後,添加390μl(390μmol)之1.00mol/Lp-甲苯磺酸水溶液。於混合液中少量添加以下述所示之方法取得的種晶,於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到255mg(產率77.0%)之呈結晶性固體的標題化合物。
種晶之取得方法
於10.3mg(14.4μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加165μL之甲醇、26.2μl之水。其後,添加15.0μl(15.0μmol)之1.00mol/Lp-甲苯磺酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到種晶10.7mg。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.73 (1H, d, J = 2.0 Hz), 8.28 (1H, d, J = 2.0 Hz), 8.21 (1H, d, J = 2.0 Hz), 8.19 (1H, d, J = 2.0 Hz), 7.90 (2H, d, J = 8.0 Hz), 7.60-7.45 (8H, m), 7.31 (1H, d, J = 2.0 Hz), 7.29-7.24 (3H, m), 7.11 (2H, d, J = 8.0 Hz), 7.06 (1H, d, J = 8.0 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.02-3.93 (2H, m), 3.90-3.22 (14H, m), 2.36 (3H, s), 2.29 (3H, s), 2.16-2.04 (1H, m), 1.46 (2H, d, J = 11.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・1.0C7
H7
SO3
H
計算値:C,66.19; H,5.89; N,6.30; S, 3.61.
實測値:C,65.91; H,5.97; N,6.26; S, 3.59.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖8中之最大波峰強度設為100的情形下之相對強度23以上的波峰顯示於表33。
[表33]
於268mg(373μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加2.14ml之甲醇、145μl之水。其後,添加390μl(390μmol)之1.00mol/Lp-甲苯磺酸水溶液。於混合液中少量添加以下述所示之方法取得的種晶,於40℃攪拌約21小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到255mg(產率77.0%)之呈結晶性固體的標題化合物。
種晶之取得方法
於10.3mg(14.4μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺中,添加165μL之甲醇、26.2μl之水。其後,添加15.0μl(15.0μmol)之1.00mol/Lp-甲苯磺酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到種晶10.7mg。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.73 (1H, d, J = 2.0 Hz), 8.28 (1H, d, J = 2.0 Hz), 8.21 (1H, d, J = 2.0 Hz), 8.19 (1H, d, J = 2.0 Hz), 7.90 (2H, d, J = 8.0 Hz), 7.60-7.45 (8H, m), 7.31 (1H, d, J = 2.0 Hz), 7.29-7.24 (3H, m), 7.11 (2H, d, J = 8.0 Hz), 7.06 (1H, d, J = 8.0 Hz), 4.11 (2H, d, J = 7.5 Hz), 4.02-3.93 (2H, m), 3.90-3.22 (14H, m), 2.36 (3H, s), 2.29 (3H, s), 2.16-2.04 (1H, m), 1.46 (2H, d, J = 11.5 Hz), 1.37-1.25 (2H, m).
作為元素分析値C42
H44
N4
O7
・1.0C7
H7
SO3
H
計算値:C,66.19; H,5.89; N,6.30; S, 3.61.
實測値:C,65.91; H,5.97; N,6.26; S, 3.59.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖8中之最大波峰強度設為100的情形下之相對強度23以上的波峰顯示於表33。
[表33] 
(實施例138)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺磷酸鹽水合物 將50.0mg(67.9μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺懸浮於400μl之丙酮中,添加64.4μl之水。其後,添加35.7μl(143μmol)之4.00mol/L磷酸水溶液,於40℃攪拌3小時後,添加500μl之20%含水丙酮,進一步攪拌約21小時。然後,於室溫攪拌約30分鐘後,藉由桐山漏斗抽吸過濾,得到42.5mg(產率69.7%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.77 (1H, d, J = 2.0 Hz), 8.69 (1H, d, J = 2.0 Hz), 8.51 (1H, d, J = 2.0 Hz), 8.47 (1H, d, J = 8.5 Hz), 8.30 (1H, d, J = 2.0 Hz), 7.93 (1H, dd, J = 12.5, 2.0 Hz), 7.71 (1H, dd, J = 8.5, 2.0 Hz), 7.64 (1H, d, J = 2.0 Hz), 7.49-7.41 (2H, m), 7.19 (1H, d, J = 2.0 Hz), 7.12 (1H, dd, J = 8.5, 2.0 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.77 (2H, br s), 4.21 (2H, d, J = 7.5 Hz), 3.98-3.24 (16H, m), 2.36 (3H, s), 2.13-2.01 (1H, m), 1.46 (2H, d, J = 12.0 Hz), 1.38-1.26 (2H, m).
MS (APCI) m/z: 736[(M+H)+
].
作為元素分析値C41
H42
FN5
O7
・1.0H3
PO4
・3.5H2
O
計算値:C,54.91; H,5.84; N,7.81; F,2.12; P, 3.45.
實測値:C,54.95; H,5.54; N,7.86; F,2.20; P, 3.19.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖9中之最大波峰強度設為100的情形下之相對強度18以上的波峰顯示於表34。
[表34]
將50.0mg(67.9μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺懸浮於400μl之丙酮中,添加64.4μl之水。其後,添加35.7μl(143μmol)之4.00mol/L磷酸水溶液,於40℃攪拌3小時後,添加500μl之20%含水丙酮,進一步攪拌約21小時。然後,於室溫攪拌約30分鐘後,藉由桐山漏斗抽吸過濾,得到42.5mg(產率69.7%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.22 (1H, s), 8.77 (1H, d, J = 2.0 Hz), 8.69 (1H, d, J = 2.0 Hz), 8.51 (1H, d, J = 2.0 Hz), 8.47 (1H, d, J = 8.5 Hz), 8.30 (1H, d, J = 2.0 Hz), 7.93 (1H, dd, J = 12.5, 2.0 Hz), 7.71 (1H, dd, J = 8.5, 2.0 Hz), 7.64 (1H, d, J = 2.0 Hz), 7.49-7.41 (2H, m), 7.19 (1H, d, J = 2.0 Hz), 7.12 (1H, dd, J = 8.5, 2.0 Hz), 6.99 (1H, d, J = 8.5 Hz), 5.77 (2H, br s), 4.21 (2H, d, J = 7.5 Hz), 3.98-3.24 (16H, m), 2.36 (3H, s), 2.13-2.01 (1H, m), 1.46 (2H, d, J = 12.0 Hz), 1.38-1.26 (2H, m).
MS (APCI) m/z: 736[(M+H)+
].
作為元素分析値C41
H42
FN5
O7
・1.0H3
PO4
・3.5H2
O
計算値:C,54.91; H,5.84; N,7.81; F,2.12; P, 3.45.
實測値:C,54.95; H,5.54; N,7.86; F,2.20; P, 3.19.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖9中之最大波峰強度設為100的情形下之相對強度18以上的波峰顯示於表34。
[表34] 
(實施例139)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺硫酸鹽水合物 將80.0g(109mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺懸浮於1.28L之丙酮中,添加247ml之水。於40℃滴下71.7ml(430mmol)之6.00mol/L硫酸。將反應液於40℃攪拌3日後,藉由布赫納漏斗抽吸過濾,濾取所析出之固體。將固體以水/丙酮混合液(水/丙酮=1/9)洗淨,得到92.3g(產率88.3%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.04 (1H, br s), 8.83 (2H, d, J = 9.5 Hz), 8.63 (1H, s), 8.44 (1H, d, J = 8.0 Hz), 8.34 (2H, s), 8.05-7.98 (2H, m), 7.72 (2H, br s), 7.59-7.52 (2H, m), 7.32 (1H, d, J = 2.0 Hz), 7.26 (1H, dd, J = 8.0, 2.0 Hz), 7.07 (1H, d, J = 8.0 Hz), 4.22 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.90-3.74 (8H, m), 3.70-3.59 (2H, m), 3.53-3.45 (1H, m), 3.40 (1H, dd, J = 11.0, 10.0 Hz), 3.27 (2H, t, J = 11.0 Hz), 2.43 (3H, s), 2.16-2.05 (1H, m), 1.51-1.43 (2H, m), 1.39-1.26 (2H, m).
MS (APCI) m/z : 736[(M+H)+
].
作為元素分析値C41
H42
FN5
O7
・1.8H2
SO4
・3.0H2
O
計算値:C, 50.96; H, 5.38; N, 7.25; F, 1.97; S, 5.97.
實測値:C, 50.98; H, 5.36; N, 7.23; F, 1.97; S, 6.19.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖10中之最大波峰強度設為100的情形下之相對強度6以上的波峰顯示於表35。
[表35]
將80.0g(109mmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺懸浮於1.28L之丙酮中,添加247ml之水。於40℃滴下71.7ml(430mmol)之6.00mol/L硫酸。將反應液於40℃攪拌3日後,藉由布赫納漏斗抽吸過濾,濾取所析出之固體。將固體以水/丙酮混合液(水/丙酮=1/9)洗淨,得到92.3g(產率88.3%)之呈結晶性固體的標題化合物。1
H-NMR (DMSO-D6
) δ: 13.04 (1H, br s), 8.83 (2H, d, J = 9.5 Hz), 8.63 (1H, s), 8.44 (1H, d, J = 8.0 Hz), 8.34 (2H, s), 8.05-7.98 (2H, m), 7.72 (2H, br s), 7.59-7.52 (2H, m), 7.32 (1H, d, J = 2.0 Hz), 7.26 (1H, dd, J = 8.0, 2.0 Hz), 7.07 (1H, d, J = 8.0 Hz), 4.22 (2H, d, J = 7.5 Hz), 4.01-3.93 (2H, m), 3.90-3.74 (8H, m), 3.70-3.59 (2H, m), 3.53-3.45 (1H, m), 3.40 (1H, dd, J = 11.0, 10.0 Hz), 3.27 (2H, t, J = 11.0 Hz), 2.43 (3H, s), 2.16-2.05 (1H, m), 1.51-1.43 (2H, m), 1.39-1.26 (2H, m).
MS (APCI) m/z : 736[(M+H)+
].
作為元素分析値C41
H42
FN5
O7
・1.8H2
SO4
・3.0H2
O
計算値:C, 50.96; H, 5.38; N, 7.25; F, 1.97; S, 5.97.
實測値:C, 50.98; H, 5.36; N, 7.23; F, 1.97; S, 6.19.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖10中之最大波峰強度設為100的情形下之相對強度6以上的波峰顯示於表35。
[表35] 
(實施例140)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺硫酸鹽水合物
於39.8mg(54.1μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺中,添加637μL之丙酮、78.3μl之水。其後,添加80.9μl(80.9μmol)之1.00mol/L硫酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到36.7mg(產率71.6%)之呈結晶性固體的標題化合物。
作為元素分析値C41
H42
FN5
O7
・1.60H2
SO4
・3.0H2
O
計算値:C,52.01; H,5.45; N,7.40; F,2.01; S,5.42.
實測値:C,52.07; H,5.24; N,7.25; F,2.09; S,5.50.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖11中之最大波峰強度設為100的情形下之相對強度12以上的波峰顯示於表36。
[表36] 
(實施例141)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺硫酸鹽水合物
於40.5mg(55.0μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺中,添加648μL之丙酮、24.8μl之水。其後,添加137μl(137μmol)之1.00mol/L硫酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到47.2mg(產率88.8%)之呈結晶性固體的標題化合物。
作為元素分析値C41
H42
FN5
O7
・1.80H2
SO4
・3.0H2
O
計算値:C,50.96; H,5.38; N,7.25; F,1.97; S,5.97.
實測値:C,50.85; H,5.20; N,7.06; F,2.09; S,5.99.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖12中之最大波峰強度設為100的情形下之相對強度7以上的波峰顯示於表37。
[表37] 
(實施例142)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺硫酸鹽水合物
於39.8mg(54.0μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺中,添加636μL之丙酮、112μl之水。其後,添加46.7μl(270μmol)之5.79mol/L硫酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到47.5mg(產率89.2%)之呈結晶性固體的標題化合物。
作為元素分析値C41
H42
FN5
O7
・2.00H2
SO4
・3.0H2
O
計算値:C,49.94; H,5.32; N,7.10; F,1.93; S,6.50.
實測値:C,49.75; H,5.08; N,6.91; F,2.20; S,6.69.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖13中之最大波峰強度設為100的情形下之相對強度11以上的波峰顯示於表38。
[表38] 
(實施例143)N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺硫酸鹽水合物
於40.2mg(54.6μmol)之N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺中,添加643μL之丙酮、51.8μl之水。其後,添加109μl(109μmol)之1.00mol/L硫酸水溶液。將混合液於40℃攪拌約24小時,接著,於室溫攪拌約30分鐘,藉由桐山漏斗抽吸過濾,濾取所析出之固體。其後,風乾,得到44.4mg(產率84.6%)之呈結晶性固體的標題化合物。
作為元素分析値C41
H42
FN5
O7
・1.75H2
SO4
・3.0H2
O
計算値:C, 51.22; H, 5.40; N, 7.28; F, 1.98; S, 5.84.
實測値:C, 50.92; H, 5.19; N, 7.11; F, 2.25; S, 5.81.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖14中之最大波峰強度設為100的情形下之相對強度11以上的波峰顯示於表39。
[表39] 
(實施例144)N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺硫酸鹽水合物 於2.11g(2.88mmol)之N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺中,添加12.7ml之甲醇、2.72ml之水,滴下5.72ml(5.72mmol)之1.00mol/L硫酸。將反應液於25℃攪拌約21小時後,濾取所析出之固體。於室溫減壓乾燥約18小時,得到2.39g(產率85.8%)之標題化合物。
作為元素分析値C41
H42
N5
O7
F·1.75H2
SO4
·3.5H2
O
計算値: C, 50.74; H, 5.45; N, 7.22; F, 1.96; S, 5.78.
實測値: C, 50.70; H, 5.33; N, 7.13; F, 2.01; S, 5.82.
將粉末X射線繞射(CuKα、λ=1.54Å、掃描速度=20º/min)之繞射圖案於圖15中之最大波峰強度設為100的情形下之相對強度18以上的波峰顯示於表40。
[表40]
於2.11g(2.88mmol)之N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺中,添加12.7ml之甲醇、2.72ml之水,滴下5.72ml(5.72mmol)之1.00mol/L硫酸。將反應液於25℃攪拌約21小時後,濾取所析出之固體。於室溫減壓乾燥約18小時,得到2.39g(產率85.8%)之標題化合物。
作為元素分析値C41
H42
N5
O7
F·1.75H2
SO4
·3.5H2
O
計算値: C, 50.74; H, 5.45; N, 7.22; F, 1.96; S, 5.78.
實測値: C, 50.70; H, 5.33; N, 7.13; F, 2.01; S, 5.82.
將粉末X射線繞射(CuKα、λ=1.54Å、掃描速度=20º/min)之繞射圖案於圖15中之最大波峰強度設為100的情形下之相對強度18以上的波峰顯示於表40。
[表40] 
(實施例145)N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺硫酸鹽水合物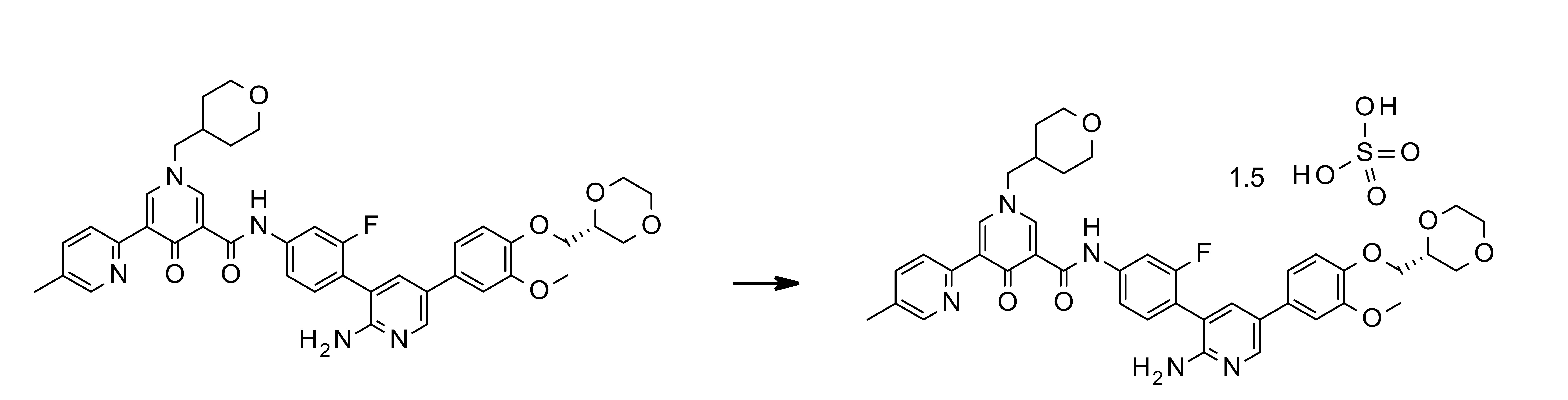 於2.44g之實施例139所記載的硫酸鹽水合物中,添加24.4ml之20%含水甲醇,於5℃攪拌約27小時。濾取固體後,於室溫減壓乾燥4.5小時,得到2.30g(產率93.5%)之標題化合物。
作為元素分析値C41
H42
N5
O7
F·1.5H2
SO4
·5.0H2
O
計算値: C, 50.61; H, 5.70; N, 7.20; F, 1.95; S, 4.94.
實測値: C, 50.59; H, 5.55; N, 7.24; F, 2.04; S, 5.09.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖16中之最大波峰強度設為100的情形下之相對強度13以上的波峰顯示於表41。
[表41]
於2.44g之實施例139所記載的硫酸鹽水合物中,添加24.4ml之20%含水甲醇,於5℃攪拌約27小時。濾取固體後,於室溫減壓乾燥4.5小時,得到2.30g(產率93.5%)之標題化合物。
作為元素分析値C41
H42
N5
O7
F·1.5H2
SO4
·5.0H2
O
計算値: C, 50.61; H, 5.70; N, 7.20; F, 1.95; S, 4.94.
實測値: C, 50.59; H, 5.55; N, 7.24; F, 2.04; S, 5.09.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖16中之最大波峰強度設為100的情形下之相對強度13以上的波峰顯示於表41。
[表41] 
(實施例146)N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺萘-1,5-二磺酸鹽水合物 於199.34mg(271μmol)之N-{4-[ 2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺中,添加3.19ml之甲醇、285μl(284μmol)之1mol/L萘-1,5-二磺酸水溶液、513μl之水。將反應液於40℃攪拌約26小時,於室溫攪拌約0.5小時。濾取固體後,風乾一晩,得到285.7mg(產率94.6%)之標題化合物。1
H-NMR (DMSO-D6
) δ: 12.97 (1H, br s), 8.90-8.80 (4H, m), 8.64 (1H, s), 8.42 (1H, d, J = 8.5 Hz), 8.33 (2H, s), 8.11-7.97 (2H, m), 7.92 (2H, d, J = 7.0 Hz), 7.71 (2H, br s), 7.59-7.52 (2H, m), 7.40 (2H, dd, J = 8.5, 7.0 Hz), 7.31 (1H, d, J = 2.5 Hz), 7.26 (1H, dd, J = 8.5, 2.5 Hz), 7.06 (1H, d, J = 8.5 Hz), 4.22 (2H, d, J = 7.0 Hz), 3.99-3.24 (16H, m), 2.43 (3H, s), 2.18-2.05 (1H, m), 1.47 (2H, d, J = 10.5 Hz), 1.39-1.26 (2H, m).
MS (APCI) m/z : 736 [(M+H)+
].
作為元素分析値C41
H42
N5
O7
F·1.0C10
H8
O6
S2
·5.0H2
O
計算値: C, 54.98; H, 5.43; N, 6.29; F, 1.71; S, 5.76.
實測値: C, 54.74; H, 5.37; N, 6.24; F, 1.92; S, 5.82.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖17中之最大波峰強度設為100的情形下之相對強度45以上的波峰顯示於表42。
[表42]
於199.34mg(271μmol)之N-{4-[ 2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺中,添加3.19ml之甲醇、285μl(284μmol)之1mol/L萘-1,5-二磺酸水溶液、513μl之水。將反應液於40℃攪拌約26小時,於室溫攪拌約0.5小時。濾取固體後,風乾一晩,得到285.7mg(產率94.6%)之標題化合物。1
H-NMR (DMSO-D6
) δ: 12.97 (1H, br s), 8.90-8.80 (4H, m), 8.64 (1H, s), 8.42 (1H, d, J = 8.5 Hz), 8.33 (2H, s), 8.11-7.97 (2H, m), 7.92 (2H, d, J = 7.0 Hz), 7.71 (2H, br s), 7.59-7.52 (2H, m), 7.40 (2H, dd, J = 8.5, 7.0 Hz), 7.31 (1H, d, J = 2.5 Hz), 7.26 (1H, dd, J = 8.5, 2.5 Hz), 7.06 (1H, d, J = 8.5 Hz), 4.22 (2H, d, J = 7.0 Hz), 3.99-3.24 (16H, m), 2.43 (3H, s), 2.18-2.05 (1H, m), 1.47 (2H, d, J = 10.5 Hz), 1.39-1.26 (2H, m).
MS (APCI) m/z : 736 [(M+H)+
].
作為元素分析値C41
H42
N5
O7
F·1.0C10
H8
O6
S2
·5.0H2
O
計算値: C, 54.98; H, 5.43; N, 6.29; F, 1.71; S, 5.76.
實測値: C, 54.74; H, 5.37; N, 6.24; F, 1.92; S, 5.82.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖17中之最大波峰強度設為100的情形下之相對強度45以上的波峰顯示於表42。
[表42] 
(實施例147)N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺萘-1,5-二磺酸鹽水合物 於199.50mg(271μmol)之N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺中,添加3.19ml之丙酮、285μl(284μmol)之1mol/L萘-1,5-二磺酸水溶液、513μl之水。將反應液於40℃攪拌約26小時、於室溫攪拌約0.5小時。濾取固體後,風乾一晩,得到290.9mg(產率97.9%)之標題化合物。
作為元素分析値C41
H42
N5
O7
F·1.0C10
H8
O6
S2
·4.0H2
O
計算値: C, 55.88; H, 5.33; N, 6.39; F, 1.73; S, 5.85.
實測値: C, 55.71; H, 5.45; N, 6.18; F, 1.82; S, 5.62.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖18中之最大波峰強度設為100的情形下之相對強度42以上的波峰顯示於表43。
[表43]
於199.50mg(271μmol)之N-{4-[2-胺基-5-(4-{[(2R)-1,4-二烷-2-基]甲氧基}-3-甲氧基苯基)-3-吡啶基]-3-氟苯基}-5-(5-甲基-2-吡啶基)-4-側氧-1-(四氫吡喃-4-基甲基)吡啶-3-甲醯胺中,添加3.19ml之丙酮、285μl(284μmol)之1mol/L萘-1,5-二磺酸水溶液、513μl之水。將反應液於40℃攪拌約26小時、於室溫攪拌約0.5小時。濾取固體後,風乾一晩,得到290.9mg(產率97.9%)之標題化合物。
作為元素分析値C41
H42
N5
O7
F·1.0C10
H8
O6
S2
·4.0H2
O
計算値: C, 55.88; H, 5.33; N, 6.39; F, 1.73; S, 5.85.
實測値: C, 55.71; H, 5.45; N, 6.18; F, 1.82; S, 5.62.
將粉末X射線繞射(CuKα,λ=1.54Å,掃描速度=20º/min)之繞射圖案於圖18中之最大波峰強度設為100的情形下之相對強度42以上的波峰顯示於表43。
[表43] 
(試驗例1無細胞的Axl激酶抑制活性)
使用激酶反應緩衝液(100mM HEPES(pH 7.4)、0.003% Brij-35、0.004% Tween-20、1mM DTT、10mM MgCl2
),作成含170ng/ml之AXL(人類 AXL之細胞內區域第464~885號之胺基酸與麩胱甘肽轉移酶之融合蛋白質於桿狀病毒表現系統中表現,以麩胱甘肽瓊脂糖凝膠(Glutathione Sepharose)層析純化。CARNABIO股份有限公司,商品目錄編號08-107)的激酶稀釋溶液,於384孔盤之各孔中各添加19μl。
接著,使用DMSO稀釋試驗化合物,將此稀釋液於各孔中各添加1μl。
於室溫進行30分鐘之預培養後,作成分別含有1.5μM、10μM之基質胜肽(FL-Peptide 30(5FAM-KKKKEEIYFFF-CONH2
),Caliper Life Sciences,商品目錄編號760430)及ATP 的溶液,藉由將其於各孔中各添加5μl,使激酶反應開始進行。將培養盤於28℃培養1.5小時,於各孔中添加40μl之終止緩衝液(Termination Buffer solution)(100mM HEPES(pH7.4)、0.015% Brij-35、40mM EDTA、0.1% Coating Reagent3) ,使反應停止。
反應溶液中之基質胜肽與磷酸化胜肽係藉由EZ Reader II(Caliper Life Sciences)分離、定量。
激酶反應係以由基質胜肽波峰高度(S)與磷酸化胜肽波峰高度(P)所計算之生成物比(P/(P+S))來進行評價。
抑制率(Inhibition)係依下式求得(由EZ Reader II系統之軟體自動算出)。
抑制率(%)=100×(1–Ci
/C0
)
其中,Ci
表示添加試驗化合物的情形之生成物比,C0
表示添加DMSO代替試驗化合物的情形之生成物比。
由對於12種試驗化合物濃度的抑制率之數據,藉由使用下式之非線性迴歸(4參數-邏輯式迴歸)求得IC50
。
抑制率(%)=底部+(頂部–底部)/(1+([化合物]/IC50
)斜 率
)
(試驗例2無細胞的Mer激酶抑制活性)
使用激酶反應緩衝液(100mM HEPES(pH 7.4)、0.003% Brij-35、0.004% Tween-20、1mM DTT、10mM MgCl2
),作成含20ng/ml之Mer(人類 MER之細胞內區域第528~999號之胺基酸與麩胱甘肽轉移酶之融合蛋白質於桿狀病毒表現系統中表現,以麩胱甘肽瓊脂糖凝膠層析與離子交換層析純化。CARNABIO股份有限公司,商品目錄編號08-108)的激酶稀釋溶液,於384孔盤之各孔中各添加19μl。
接著,使用DMSO稀釋試驗化合物,將此稀釋液於各孔中各添加1μl。
於室溫進行20分鐘之預培養後,做成含5mM之基質胜肽(FL-Peptide 27(5FAM-EFPIYDFLPAKKK-CONH2
),Caliper Life Sciences,商品目錄編號760424)及ATP之溶液,藉由將其於各孔中各添加5μl,使激酶反應開始進行。將培養盤於28℃培養45分鐘,於各孔中添加40μl之終止緩衝液(100mM HEPES(pH7.4)、0.015% Brij-35、40mM EDTA、0.1% Coating Reagent3),使反應停止。
反應溶液中之基質胜肽與磷酸化胜肽係藉由EZ Reader II(Caliper Life Sciences)分離、定量。
激酶反應係以由基質胜肽波峰高度(S)與磷酸化胜肽波峰高度(P)所計算之生成物比(P/(P+S)) 來進行評價。
抑制率(Inhibition)係依下式求得(由EZ Reader II系統之軟體自動算出)。
抑制率(%)=100×(1–Ci/C0
)
其中,Ci表示添加試驗化合物的情形之生成物比,C0
表示添加DMSO代替試驗化合物的情形之生成物比。
由對於12種試驗化合物濃度的抑制率之數據,藉由使用下式之非線性迴歸(4參數-邏輯式迴歸)求得IC50
。
抑制率(%)=底部+(頂部–底部)/(1+([化合物]/IC50
)斜率
)
表44中,顯示ATP濃度為1mM之條件下之Axl激酶抑制活性作為IC50
(nM)値,Mer激酶抑制作為IC50
(nM)値,進一步顯示對Mer激酶之Axl激酶選擇性(倍)。
[表44-1] 
[表44-2] 
[表44-3] 
[表44-4] 
[表44-5] 
[表44-6] 
[表44-7] 
(試驗例3細胞內Axl磷酸化抑制活性)
使用源自人類非小細胞肺癌之細胞株NCI-H1299,實施磷酸化Axl(以下pAxl)抑制試驗。
將NCI-H1299細胞懸浮於培養基(含10%胎牛血清之RPMI1640培養基),於96孔之多孔盤中各自以15000細胞/100μl/孔接種,於37℃、5% CO2
存在下培養1日。隔日去除培養基,添加100μl之培養基,於37℃、5% CO2
下培養1日。將試驗化合物溶解於DMSO中,以未添加FBS之培養基稀釋,作為檢體溶液(DMSO濃度2%)。將培養基或添加檢體之培養基於孔中添加25μl (DMSO濃度0.4%,於37℃、5% CO2
存在下培養1小時。
使用使GAS6(R&D,商品編號:885-GS)成為6μg/ml之未添加FBS之培養基稀釋,對各孔添加25μl,攪拌後,於37℃、5% CO2
存在下培養10分鐘。
捨棄上清液,於孔中添加0.1ml之以磷酸緩衝液(PBS)將37%福馬林液稀釋為4%之溶液(以下,4%福馬林液),於室溫靜置10分鐘。接著捨棄4%福馬林液,添加0.2ml之以PBS將TritonX-100稀釋為0.1%之溶液(以下,洗滌緩衝液),以傾析器捨棄,於紙巾上去除多餘水分。
接著,於10.7mL之洗滌緩衝液中添加110μl之10%NaN3
與H2
O2
(以下,淬息緩衝液(quenching buffer)),對孔添加0.1ml,於室溫靜置15分鐘。
捨棄淬息緩衝液,添加0.2 ml之洗滌緩衝液,以傾析器捨棄,於紙巾上去除多餘水分。於洗滌緩衝液中,添加最終濃度5%之脫脂乳(WAKO #198-10605) (阻斷緩衝液(blocking buffer)),對孔添加0.25ml,於室溫靜置1小時。
捨棄阻斷緩衝液,使Anti-phospho-Axl(Y702)(D12B2)兔單株抗體(Cell Signaling,商品目錄編號5724) 以1/1000之濃度反應,於4℃靜置一晩。以洗滌緩衝液重複5次洗淨操作,使Peroxidase AffiniPure Donkey Anti-Rabbit IgG(H+L)(Jackson ImmunoResearch,商品目錄編號711-035-152)以1/2000之濃度,於室溫反應1小時。同樣地進行洗淨操作,添加0.05ml之Super Signal ELISA pico chemi luminescent substrate(Thermo Scientific,商品目錄編號37069),輕輕攪拌後培養20分鐘。其後,以ARVO sx (Perkin ElMer)測定發光,測定pAxl (Y702)程度。
pAxl抑制活性係以以下之式求得。
抑制率%=100-(A-B)×100/(T-B)
A:被檢化合物之測定値
B:添加抑制將近100%磷酸化之濃度的陽性對照化合物的反應液之發光値(例如添加BMS-777607 1μM的反應液之發光値)
T:未添加化合物的反應液之發光値
由複數濃度之pAxl抑制活性的數據,藉由GraphPad Prism4求得50%抑制濃度(IC50
)。
於表45中,將細胞內Axl磷酸化抑制活性顯示作為IC50
(nM)値。
[表45-1] 
[表45-2] 
(試驗例4眼之病理組織學的檢査)
1.動物:NOG雌性小鼠
2.藥物:
Compound A 1鹽酸鹽2水合物
實施例124
實施例128
實施例129
3.投與液調製:
將Compound A、實施例124實施例128及實施例129懸浮於0.5%甲基纖維素溶液(0.5% MC,和光純藥工業)。
4.投與方法:
4連續投與、1日停藥、5連續投與、2日停藥、5連續投與、2日停藥、5連續投與、2日停藥、4連續投與。
5.病理學的檢査
於投與期間結束日之隔日,於異氟醚(isoflurane)麻醉下,放血安樂死後,採取眼球,以戴維森固定劑(Davidson's fixative)固定後,石蠟包埋及製作蘇木素-伊紅染色標本,實施病理組織學的檢査。
Compound A1鹽酸鹽2水合物:於100mg/kg投與組全例(8例)中,看見輕度~中度之網膜的外顆粒層及桿狀錘狀體層之變性及薄化。
實施例124、實施例128、實施例129:於100及200mg/kg投與組全例(8例)中,於網膜上看見有組織學的變化。
(試驗例5:抗腫瘤試驗)
將NIH-3T3-Axl#7(將經插入全長Axl cDNA之pLXSN反轉錄病毒轉染至NIH3T3細胞而製作之細胞)Block移植至NOG雌性小鼠,於推定腫瘤體積達到約500 mm3
的時間點,將Compound A1鹽酸鹽2水合物(25、4.2、0.7 mg/kg)、實施例124(50、12.5、3.1、0.8 mg/kg)及實施例128(50、12.5、3.1、0.8 mg/kg)以1日2次(bid)5連續投與(經口投與)。Compound A在25及4.2 mg/kg明確地顯示腫瘤縮退效果,實施例124及實施例128在3.1 mg/kg投與期間中,幾乎完全地抑制腫瘤增殖,在50及12.5mg/kg明確地顯示腫瘤縮退效果。
(試驗例6:與埃羅替尼之活體內併用效果之研討)
將於EGFR基因exon 19具有缺失突變且對EGFR抑制藥顯示高反應性之HCC827肺癌細胞,使用磷酸緩衝生理食鹽水懸浮成5×107
cells/ml,將0.1 mL之所調製的細胞懸浮液移植至裸鼠(雌性,5 週齡)之皮下。於大部分的小鼠之推定腫瘤體積達到約450 mm3
的時間點(腫瘤移植後第52日),進行根據腫瘤體積値之分組,強制經口投與25 mg/kg埃羅替尼(LC Laboratories)(1日1回: qd)或50 mg/kg N-[4-(2-胺基-5-{4-[(2R)-1,4-二烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧 -1’- (四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺(Compound B)硫酸鹽水合物(1日2回: bid)(單獨投與或兩劑之併用投與)。投與係自分組隔日(Day 1)以一週5日之比例(星期六日停藥)進行,媒介物及Compound B硫酸鹽水合物單獨投與組係進行至Day 46,埃羅替尼單獨投與組係進行至Day 86,併用投與組係進行至Day 106。隨著時間的經過以電子數位游標尺測量腫瘤之長徑(mm)及短徑(mm),根據以下所示計算式(1)算出推定腫瘤體積而作成圖。又,隨著時間的經過使用小動物用自動天秤測定體重,根據以下所示之計算式(2)算出體重變化率(Body weight change %),研討藥劑投與對體重之影響,同時使用最近的體重測定結果算出投與量。
推定腫瘤體積(mm3
)=各個體之推定腫瘤體積之平均値…(1)
各個體之推定腫瘤體積=An×Bn2
/2
An:第n日之腫瘤長徑
Bn:第n日之腫瘤短徑
體重變化(%)=各個體之體重變化率之平均値…(2)
各個體之體重變化率=(1-BWn/BWs)×100
BWn:第n日之體重
BWs:投與開始日之體重
就埃羅替尼單劑而言,投與後可立即確認腫瘤退縮效果,但其後獲得耐性,於Day 77超過投與開始時之大小。然而,就與Compound B硫酸鹽水合物之併用組而言,即使於Day 107也沒達到投與開始時之大小(圖19-1)。在埃羅替尼與Compound B硫酸鹽水合物之併用組中,沒有觀察到特別顯著的體重減少之降低(圖19-2)。
(試驗例7:因埃羅替尼投與所致之AXL表現上升之研討)
將HCC827肺癌細胞使用磷酸緩衝生理食鹽水懸浮使成為5×107
cells/ml,將0.1 mL之所調製的細胞懸浮液移植至裸鼠(雌性,5 週齡)之皮下。於大部分的小鼠之推定腫瘤體積達到約450 mm3
的時間點(腫瘤移植後第52日),進行根據腫瘤體積値之分組(Day 0),以一週5日之比例(星期六日停藥)進行強制經口投與25 mg/kg、12.5 mg/kg、或者6.25 mg/kg之埃羅替尼(LC Laboratories)。媒介物投與組係進行至Day 46、埃羅替尼投與組係進行至Day 86。隨著時間的經過以電子數位游標尺測量腫瘤長徑(mm)及短徑(mm),根據以下所示之計算式(1)算出推定腫瘤體積而作成圖(圖20-1)。由各組2-6隻之腫瘤塊調製細胞溶解產物(Cell lysate),根據西方墨點轉漬法(Western blot)確認AXL之表現,將各個帶(band)藉由ImageQuant LAS4000 (GE Health Care)定量化(圖20-2)。關於細胞溶解產物之調製,及西方墨點轉漬法係如下進行。將100 mg左右之腫瘤塊於包含磷酸酶抑制劑(Roche-diagnostics,04 906 837 001)與蛋白酶抑制劑(Roche-diagnostics,11 836 153 001)之溶解緩衝液(Lysis Buffer)(Cell signaling technology,9803)中均質化,將上清液作為溶解產物試樣。將此試樣之蛋白質以緩衝液(Life technologies,NP0008及NP0009)與熱變性固定而供給西方墨點轉漬法使用。將15 μg之試樣進行電泳,轉印至硝基纖維素膜,經1小時的阻斷後與兔抗AXL抗體(Cell signaling technology,9803、1/1000)或兔抗Actin抗體(Santa Cruz Biotechnology,SC-1616,1/2000)於冷藏下反應一晩。以三羥甲基胺基甲烷緩衝生理食鹽水(Tris-buffered saline)洗淨後,與HRP標識抗兔IgG抗體(Cell signaling technology,7074,1/2000)於室溫下反應1小時,以三羥甲基胺基甲烷緩衝生理食鹽水洗淨後,以HRP基質(Merck Millipore,WBLUF0500)使其發光。
藉由西方墨點轉漬法確認AXL之表現,藉由Welch test確認各個帶藉由ImageQuant LAS4000定量化之結果、埃羅替尼投與組之AXL之表現有意義地提升。
(試驗例8:與埃羅替尼之活體內併用效果之研討2)
將HCC827肺癌細胞使用磷酸緩衝生理食鹽水懸浮成4×107
cells/mL,將0.1 mL之所調製之細胞懸浮液移植至裸鼠(雌性,5 週齡)之皮下。於腫瘤細胞移植後第52日,對推定腫瘤體積為200 mm3
以上小於700 mm3
之小鼠(86隻/129隻),強制經口投與25 mg/kg之埃羅替尼(LC Laboratories)(1日1回: qd)。投與係自分組隔日(移植後第53日)以一週5日之比例(星期六日停藥)進行,使用腫瘤暫時退縮而可確認明確地再增殖之帶有癌之小鼠而實施分組(6隻/組,移植後第115日),分成群組1:埃羅替尼+媒介物投與組(平均推定腫瘤體積275 mm3
)、群組2:埃羅替尼+Compound B硫酸鹽水合物 50 mg/kg (平均推定腫瘤體積 284 mm3
)、群組3:埃羅替尼+Compound B硫酸鹽水合物 25 mg/kg(平均推定腫瘤體積 268 mm3
)之3組,研討對於埃羅替尼耐性化之腫瘤,可否藉由Compound B硫酸鹽水合物之併用,而使埃羅替尼之效果再現。Compound B硫酸鹽水合物之投與係自移植後第116日,繼續以一週5日之比例(星期六日停藥)1日2次投與(bid)。就群組1而言,腫瘤增殖至與當初移植時之同程度的推定腫瘤體積,但就群組2及群組3而言,可確認因埃羅替尼所致之明確地增殖抑制效果。就群組2而言,即使於移植後第140日,仍可維持開始併用投與之移植後第116日之推定腫瘤體積。
無。
圖1於實施例130所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖2於實施例131所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖3於實施例132所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖4於實施例133所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖5於實施例134所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖6於實施例135所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖7於實施例136所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖8於實施例137所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖9於實施例138所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖10於實施例139所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖11於實施例140所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖12於實施例141所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖13於實施例142所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖14於實施例143所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖15於實施例144所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖16於實施例145所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖17於實施例146所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖18於實施例147所得到之結晶的粉末X射線繞射圖。圖之縱軸係以計數/秒(cps)之單位表示繞射強度(Intensity);橫軸係表示繞射角度2θ之値。
圖19-1係顯示埃羅替尼(erlotinib)與本案之Axl抑制化合物對於在EGFR基因exon 19有缺失突變(deletion mutation)而顯示對EGFR抑制藥有高反應性之HCC827小鼠皮下移植腫瘤之併用效果的圖。符號×表示媒介物(Vehicle)投與組(一日3回投與(Tid))、符號白圈表示Compound B硫酸鹽水合物50mg/kg(一日2回投與(bid))、符號黑圈表示埃羅替尼25mg/kg(一日1回投與(qd))、符號白三角表示Compound B硫酸鹽水合物50mg/kg(bid)+埃羅替尼25mg/kg(qd)。橫軸表示投與開始後的日數。縱軸表示根據腫瘤直徑所算出的推定腫瘤體積。
圖19-2係埃羅替尼與本案之Axl抑制化合物之併用試驗中,顯示各個體之體重變化率的平均値之圖(圖式符號與圖19-1相同)。
圖20-1係顯示埃羅替尼對HCC827小鼠皮下移植腫瘤之抗腫瘤效果圖。符號×表示媒介物投與組、符號白圈表示埃羅替尼25mg/kg、符號黑圈表示埃羅替尼12.5mg/kg、符號白三角表示埃羅替尼6.25mg/kg。橫軸表示投與開始後的日數。縱軸表示根據腫瘤直徑所算出的推定腫瘤體積。
圖20-2係顯示在HCC827小鼠皮下移植腫瘤中之埃羅替尼投與時的Axl蛋白質表現量變化之圖。

無。
Claims (22)
- 一種醫藥,其特徵為以通式(I)所表示之化合物或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與,



- 一種醫藥,其特徵為以下述式
 烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
- 一種醫藥,其特徵為以下述式
 烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)苯基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
- 一種醫藥,其特徵為以下述式

- 一種醫藥,其特徵為以下述式
 烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
- 一種醫藥,其特徵為以下述式
 烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)-3-氟苯基]-5-甲基-4’-側氧-1’-(四氫-2H-吡喃-4-基甲基)-1’,4’-二氫-2,3’-聯吡啶-5’-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
- 一種醫藥,其特徵為以下述式

- 一種醫藥,其特徵為以下述式

- 一種醫藥,其特徵為以下述式
 烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)嗒
烷-2-基甲氧基]-3-甲氧基苯基}吡啶-3-基)嗒 -3-基]-5-(5-甲基噻吩-2-基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
-3-基]-5-(5-甲基噻吩-2-基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
- 一種醫藥,其特徵為以下述式
 啉-4-基)乙氧基]苯基}吡啶-3-基)嗒
啉-4-基)乙氧基]苯基}吡啶-3-基)嗒 -3-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
-3-基]-5-(4-甲基苯基)-4-側氧-1-(四氫-2H-吡喃-4-基甲基)-1,4-二氫吡啶-3-甲醯胺或其藥學上容許鹽、與酪胺酸激酶抑制劑係組合而投與。
- 如請求項2之醫藥,其中藥學上容許鹽為氫溴酸鹽、硝酸鹽、硫酸鹽、磷酸鹽、甲磺酸鹽、乙磺酸鹽、苯磺酸鹽或p-甲苯磺酸鹽。
- 如請求項4之醫藥,其中藥學上容許鹽為甲磺酸鹽。
- 如請求項5之醫藥,其中藥學上容許鹽為甲磺酸鹽、磷酸鹽、萘-1,5-二磺酸鹽或硫酸鹽。
- 如請求項5之醫藥,其中藥學上容許鹽為硫酸鹽。
- 如請求項1至14中任一項之醫藥,其中酪胺酸激酶抑制劑為吉非替尼(gefitinib)或埃羅替尼(erlotinib)。
- 一種如請求項1至15中任一項之醫藥的用途,其係用以製造用於由Axl激酶功能亢進所致的疾病、與Axl激酶功能亢進相關的疾病、及/或伴隨Axl激酶功 能亢進的疾病的治療之藥劑。
- 一種如請求項1至15中任一項之醫藥的用途,其係用以製造用於過度增殖性疾病(hyperproliferative diseases)的治療之藥劑。
- 一種如請求項1至15中任一項之醫藥的用途,其係用以製造用於使癌症的耐藥性獲得抑制之藥劑。
- 一種如請求項1至15中任一項之醫藥的用途,其係用以製造用於預防癌症的轉移之藥劑。
- 一種如請求項1至15中任一項之醫藥的用途,其係用以製造用於解除癌症的耐藥性之藥劑。
- 一種如請求項1至15中任一項之醫藥的用途,其係用以製造用於癌症的治療之藥劑。
- 如請求項18至21中任一項之醫藥的用途,其中癌症係選自乳癌、結腸癌、前列腺癌、肺癌、胃癌、卵巢癌、子宮內膜癌、腎癌、肝細胞癌、甲狀腺癌、子宮癌、食道癌、鱗狀上皮細胞癌(squamous cell carcinoma)、白血病、骨肉瘤(osteosarcoma)、黑色素瘤、神經膠肉瘤(gliosarcoma)、神經母細胞瘤(neuroblastoma)及胰臓癌。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014139628 | 2014-07-07 | ||
| JP2014-139628 | 2014-07-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW202003507A TW202003507A (zh) | 2020-01-16 |
| TWI723572B true TWI723572B (zh) | 2021-04-01 |
Family
ID=55064330
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW108136526A TWI723572B (zh) | 2014-07-07 | 2015-07-03 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
| TW104121592A TWI690525B (zh) | 2014-07-07 | 2015-07-03 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW104121592A TWI690525B (zh) | 2014-07-07 | 2015-07-03 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
Country Status (27)
| Country | Link |
|---|---|
| US (3) | US10442797B2 (zh) |
| EP (2) | EP3868757A1 (zh) |
| JP (2) | JP6086516B2 (zh) |
| KR (1) | KR102348190B1 (zh) |
| CN (2) | CN106604920B (zh) |
| AU (1) | AU2015288648B2 (zh) |
| CA (1) | CA2954488C (zh) |
| CO (1) | CO2017001178A2 (zh) |
| CY (1) | CY1124537T1 (zh) |
| DK (1) | DK3168219T3 (zh) |
| ES (1) | ES2875360T3 (zh) |
| HR (1) | HRP20210899T1 (zh) |
| HU (1) | HUE054534T2 (zh) |
| IL (2) | IL249948B (zh) |
| LT (1) | LT3168219T (zh) |
| MX (1) | MX2017000330A (zh) |
| MY (1) | MY194307A (zh) |
| PH (1) | PH12017500048A1 (zh) |
| PL (1) | PL3168219T3 (zh) |
| PT (1) | PT3168219T (zh) |
| RS (1) | RS61941B1 (zh) |
| RU (1) | RU2707953C2 (zh) |
| SG (2) | SG11201700063RA (zh) |
| SI (1) | SI3168219T1 (zh) |
| TW (2) | TWI723572B (zh) |
| WO (1) | WO2016006706A1 (zh) |
| ZA (1) | ZA201700139B (zh) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI723572B (zh) | 2014-07-07 | 2021-04-01 | 日商第一三共股份有限公司 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
| JP6885390B2 (ja) | 2016-02-26 | 2021-06-16 | 小野薬品工業株式会社 | Axl阻害剤と免疫チェックポイント阻害剤とを組み合わせて投与することを特徴とする癌治療のための医薬 |
| CN108250200A (zh) * | 2016-12-28 | 2018-07-06 | 中国科学院上海药物研究所 | 一种具有Axl抑制活性的化合物及其制备和应用 |
| TWI796326B (zh) * | 2017-03-24 | 2023-03-21 | 日商第一三共股份有限公司 | 含有axl抑制劑與egfr酪胺酸激酶抑制藥的醫藥品及其用途 |
| JP7156287B2 (ja) | 2017-08-23 | 2022-10-19 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含むがん治療剤 |
| WO2019074116A1 (ja) | 2017-10-13 | 2019-04-18 | 小野薬品工業株式会社 | Axl阻害剤を有効成分として含む固形がん治療剤 |
| JP7446236B2 (ja) * | 2017-12-22 | 2024-03-08 | ラヴェンナ ファーマシューティカルズ,インコーポレイテッド | ホスファチジルイノシトールリン酸キナーゼ阻害剤としてのアリール-ビピリジンアミン誘導体 |
| CN110041316B (zh) * | 2018-01-17 | 2022-04-19 | 药捷安康(南京)科技股份有限公司 | Tam家族激酶/和csf1r激酶抑制剂及其用途 |
| AU2019212959A1 (en) * | 2018-01-29 | 2020-09-17 | Capulus Therapeutics, Llc | SREBP inhibitors comprising a 6-membered central ring |
| TWI840378B (zh) * | 2018-07-04 | 2024-05-01 | 日商第一三共股份有限公司 | 二芳基吡啶衍生物之製造方法 |
| CN114364798A (zh) | 2019-03-21 | 2022-04-15 | 欧恩科斯欧公司 | 用于治疗癌症的Dbait分子与激酶抑制剂的组合 |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| JPWO2022220227A1 (zh) * | 2021-04-14 | 2022-10-20 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130281428A1 (en) * | 2012-01-31 | 2013-10-24 | Daiichi Sankyo Company, Limited | Pyridone Derivatives |
Family Cites Families (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1136485A1 (en) * | 2000-03-23 | 2001-09-26 | Sanofi-Synthelabo | Aminophenyl pyrimidone derivatives |
| GB0319241D0 (en) | 2003-08-15 | 2003-09-17 | Hotpods Ltd | Oven and a food delivery vehicle comprising said oven |
| CA2621503C (en) | 2005-09-07 | 2014-05-20 | Rigel Pharmaceuticals, Inc. | Triazole derivatives useful as axl inhibitors |
| WO2007057399A2 (en) | 2005-11-15 | 2007-05-24 | Boehringer Ingelheim International Gmbh | Treatment of cancer with indole derivatives |
| SI1959955T1 (sl) | 2005-12-05 | 2011-02-28 | Pfizer Prod Inc | Postopek zdravljenja abnormalne celične rasti |
| ES2562428T3 (es) | 2005-12-15 | 2016-03-04 | Rigel Pharmaceuticals, Inc. | Inhibidores de cinasa y sus usos |
| EP1900727A1 (en) | 2006-08-30 | 2008-03-19 | Cellzome Ag | Aminopyridine derivatives as kinase inhibitors |
| WO2008045978A1 (en) | 2006-10-10 | 2008-04-17 | Rigel Pharmaceuticals, Inc. | Pinane-substituted pyrimidinediamine derivatives useful as axl inhibitors |
| GB0625659D0 (en) | 2006-12-21 | 2007-01-31 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| WO2008083356A1 (en) | 2006-12-29 | 2008-07-10 | Rigel Pharmaceuticals, Inc. | Substituted triazoles useful as axl inhibitors |
| ES2404668T3 (es) | 2006-12-29 | 2013-05-28 | Rigel Pharmaceuticals, Inc. | Triazoles sustituidos con arilo bicíclico puenteado y heteroarilo bicíclico puenteado, útiles como agentes inhibidores del axl |
| ES2672172T3 (es) | 2006-12-29 | 2018-06-12 | Rigel Pharmaceuticals, Inc. | Triazoles N3-heteroarilsustituidos y triazoles N5-heteroarilsustituidos útiles como inhibidores de Axl |
| CA2710043C (en) | 2006-12-29 | 2016-02-09 | Rigel Pharmaceuticals, Inc. | Bicyclic aryl and bicyclic heteroaryl substituted triazoles useful as axl inhibitors |
| ES2460894T3 (es) | 2006-12-29 | 2014-05-14 | Rigel Pharmaceuticals, Inc. | Triazoles sustituidos con heteroarilo policíclico útiles como inhibidores de Axl |
| CA2682733A1 (en) | 2007-04-13 | 2008-10-23 | Supergen, Inc. | Axl kinase inhibitors |
| GB0713259D0 (en) | 2007-07-09 | 2007-08-15 | Astrazeneca Ab | Pyrazine derivatives 954 |
| JP2010533159A (ja) | 2007-07-09 | 2010-10-21 | アストラゼネカ アクチボラグ | 化合物947 |
| WO2009024825A1 (en) | 2007-08-21 | 2009-02-26 | Astrazeneca Ab | 2-pyrazinylbenzimidazole derivatives as receptor tyrosine kinase inhibitors |
| GB0719803D0 (en) * | 2007-10-10 | 2007-11-21 | Cancer Rec Tech Ltd | Therapeutic compounds and their use |
| RU2010120536A (ru) * | 2007-10-25 | 2011-11-27 | Астразенека Аб (Se) | Производные пиридина и пиразина, полезные для лечения клеточных проферативных расстройств |
| AU2008315746A1 (en) | 2007-10-25 | 2009-04-30 | Astrazeneca Ab | Pyridine and pyrazine derivatives useful in the treatment of cell proliferative disorders |
| SI2205592T1 (sl) | 2007-10-26 | 2013-09-30 | Rigel Pharmaceuticals, Inc. | Triazoli substituirani s policikličnim arilom in triazoli substituirani s policikličnim heteroarilom uporabni kot Axl inhibitorji |
| CN101917994A (zh) | 2007-11-02 | 2010-12-15 | 詹森药业有限公司 | Cfms抑制剂用于治疗或预防骨癌以及与骨癌相关的骨丢失和骨痛的用途 |
| KR101596527B1 (ko) | 2008-01-23 | 2016-02-22 | 브리스톨-마이어스 스큅 컴퍼니 | 4-피리디논 화합물 및 암에 대한 그의 용도 |
| WO2009094427A1 (en) | 2008-01-23 | 2009-07-30 | Bristol-Myers Squibb Company | 4-pyridinone compounds and their use for cancer |
| CA2718538A1 (en) | 2008-04-16 | 2009-10-22 | Max-Planck-Gesellschaft Zur Forderung Der Wissenschaften E.V. | Quinoline derivatives as axl kinase inhibitors |
| UY31800A (es) | 2008-05-05 | 2009-11-10 | Smithkline Beckman Corp | Metodo de tratamiento de cancer usando un inhibidor de cmet y axl y un inhibidor de erbb |
| WO2009138799A1 (en) | 2008-05-14 | 2009-11-19 | Astex Therapeutics Limited | Therapeutic uses of 1-cycl0pr0pyl-3 - [3- ( 5 -morpholin- 4 -ylmethyl- 1h-benz0imidaz0l- 2 -yl) -lh-pyrazol-4-yl] -urea |
| WO2010005876A2 (en) | 2008-07-09 | 2010-01-14 | Rigel Pharmaceuticals, Inc. | Polycyclic heteroaryl substituted triazoles useful as axl inhibitors |
| PT2328888E (pt) | 2008-07-09 | 2013-01-29 | Rigel Pharmaceuticals Inc | Triazóis bicíclicos em ponte substituídos com heteroarilos úteis como inibidores axl |
| US20100204221A1 (en) | 2009-02-09 | 2010-08-12 | Hariprasad Vankayalapati | Pyrrolopyrimidinyl axl kinase inhibitors |
| MX2011011326A (es) * | 2009-04-27 | 2012-02-13 | Elan Pharm Inc | Antagonistas de piridinona de las integrinas alfa-4. |
| EP2311809A1 (en) | 2009-10-16 | 2011-04-20 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Quinolinyloxyphenylsulfonamides |
| AR080754A1 (es) | 2010-03-09 | 2012-05-09 | Janssen Pharmaceutica Nv | Derivados de imidazo (1,2-a) pirazina y su uso como inhibidores de pde10 |
| JP5970003B2 (ja) | 2011-03-04 | 2016-08-17 | ローカス ファーマシューティカルズ インコーポレイテッド | アミノピラジン化合物 |
| WO2013162061A1 (ja) | 2012-04-26 | 2013-10-31 | 第一三共株式会社 | 二環性ピリミジン化合物 |
| TWI723572B (zh) | 2014-07-07 | 2021-04-01 | 日商第一三共股份有限公司 | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 |
-
2015
- 2015-07-03 TW TW108136526A patent/TWI723572B/zh active
- 2015-07-03 TW TW104121592A patent/TWI690525B/zh active
- 2015-07-06 MY MYPI2017000012A patent/MY194307A/en unknown
- 2015-07-06 JP JP2016532991A patent/JP6086516B2/ja active Active
- 2015-07-06 CN CN201580047744.1A patent/CN106604920B/zh active Active
- 2015-07-06 CA CA2954488A patent/CA2954488C/en active Active
- 2015-07-06 HU HUE15818440A patent/HUE054534T2/hu unknown
- 2015-07-06 MX MX2017000330A patent/MX2017000330A/es unknown
- 2015-07-06 RS RS20210707A patent/RS61941B1/sr unknown
- 2015-07-06 RU RU2017103753A patent/RU2707953C2/ru active
- 2015-07-06 WO PCT/JP2015/069976 patent/WO2016006706A1/ja active Application Filing
- 2015-07-06 US US15/036,981 patent/US10442797B2/en active Active
- 2015-07-06 EP EP21166082.4A patent/EP3868757A1/en active Pending
- 2015-07-06 SI SI201531589T patent/SI3168219T1/sl unknown
- 2015-07-06 AU AU2015288648A patent/AU2015288648B2/en active Active
- 2015-07-06 CN CN202010427596.9A patent/CN111467346B/zh active Active
- 2015-07-06 SG SG11201700063RA patent/SG11201700063RA/en unknown
- 2015-07-06 LT LTEP15818440.8T patent/LT3168219T/lt unknown
- 2015-07-06 DK DK15818440.8T patent/DK3168219T3/da active
- 2015-07-06 ES ES15818440T patent/ES2875360T3/es active Active
- 2015-07-06 SG SG10201900144TA patent/SG10201900144TA/en unknown
- 2015-07-06 PT PT158184408T patent/PT3168219T/pt unknown
- 2015-07-06 KR KR1020177000534A patent/KR102348190B1/ko active IP Right Grant
- 2015-07-06 EP EP15818440.8A patent/EP3168219B1/en active Active
- 2015-07-06 PL PL15818440T patent/PL3168219T3/pl unknown
-
2017
- 2017-01-05 IL IL249948A patent/IL249948B/en active IP Right Grant
- 2017-01-06 ZA ZA2017/00139A patent/ZA201700139B/en unknown
- 2017-01-06 PH PH12017500048A patent/PH12017500048A1/en unknown
- 2017-02-01 JP JP2017016682A patent/JP6203439B2/ja active Active
- 2017-02-07 CO CONC2017/0001178A patent/CO2017001178A2/es unknown
-
2019
- 2019-09-18 US US16/575,177 patent/US11208403B2/en active Active
-
2020
- 2020-10-18 IL IL278124A patent/IL278124B/en unknown
-
2021
- 2021-06-07 HR HRP20210899TT patent/HRP20210899T1/hr unknown
- 2021-06-30 CY CY20211100586T patent/CY1124537T1/el unknown
- 2021-08-12 US US17/400,795 patent/US20220002277A1/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130281428A1 (en) * | 2012-01-31 | 2013-10-24 | Daiichi Sankyo Company, Limited | Pyridone Derivatives |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI723572B (zh) | 具有四氫吡喃基甲基之吡啶酮衍生物及其用途 | |
| JP6703075B2 (ja) | Rhoキナーゼ阻害剤 | |
| KR102078530B1 (ko) | 피리돈 유도체들 | |
| KR102189529B1 (ko) | 키누레닌 경로의 억제제 | |
| JP5960688B2 (ja) | プロテインキナーゼ調節物質としての新規3,5−二置換−3h−[1,2,3]トリアゾロ[4,5−b]ピリジン化合物 | |
| TW201904964A (zh) | 作為hpk1之抑制劑的啶 | |
| KR20210137422A (ko) | 티로신 키나아제 억제제로서의 퀴나졸린 유도체, 조성물, 이들의 제조 방법 및 이들의 용도 | |
| CN113195471B (zh) | 多取代吡啶酮类衍生物及其在医药上的应用 | |
| AU2019390863A2 (en) | Heterocyclic compound | |
| JP2023550591A (ja) | EGFR阻害薬としての置換1H-ピラゾロ[4,3-c]及び誘導体 | |
| BR112017000242B1 (pt) | Composto, cristal, inibidor de axl, medicamento, composição farmacêutica, e, uso de um composto | |
| CN115433210A (zh) | 一类具有免疫调节功能的化合物的制备和应用 | |
| CN103748095A (zh) | 二环杂芳族化合物 |