WO2013161958A1 - 新規な発現ベクター - Google Patents
新規な発現ベクター Download PDFInfo
- Publication number
- WO2013161958A1 WO2013161958A1 PCT/JP2013/062251 JP2013062251W WO2013161958A1 WO 2013161958 A1 WO2013161958 A1 WO 2013161958A1 JP 2013062251 W JP2013062251 W JP 2013062251W WO 2013161958 A1 WO2013161958 A1 WO 2013161958A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gene
- expression vector
- ribosome binding
- internal ribosome
- binding site
- Prior art date
Links
- 239000013604 expression vector Substances 0.000 title claims abstract description 138
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 230
- 230000014509 gene expression Effects 0.000 claims abstract description 108
- 108010022394 Threonine synthase Proteins 0.000 claims abstract description 59
- 210000004962 mammalian cell Anatomy 0.000 claims abstract description 52
- 102000004169 proteins and genes Human genes 0.000 claims abstract description 46
- 108020002326 glutamine synthetase Proteins 0.000 claims abstract description 29
- 102000005396 glutamine synthetase Human genes 0.000 claims abstract description 28
- 238000004519 manufacturing process Methods 0.000 claims abstract description 17
- 210000004027 cell Anatomy 0.000 claims description 150
- 239000003112 inhibitor Substances 0.000 claims description 28
- 206010059866 Drug resistance Diseases 0.000 claims description 25
- 239000003814 drug Substances 0.000 claims description 23
- 229940079593 drug Drugs 0.000 claims description 22
- 239000002773 nucleotide Substances 0.000 claims description 21
- 125000003729 nucleotide group Chemical group 0.000 claims description 21
- 241000710188 Encephalomyocarditis virus Species 0.000 claims description 20
- 102000004190 Enzymes Human genes 0.000 claims description 20
- 108090000790 Enzymes Proteins 0.000 claims description 20
- 229940088598 enzyme Drugs 0.000 claims description 20
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 claims description 20
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 claims description 19
- 108010061174 Thyrotropin Proteins 0.000 claims description 17
- 102000011923 Thyrotropin Human genes 0.000 claims description 17
- 230000035772 mutation Effects 0.000 claims description 17
- 108091026898 Leader sequence (mRNA) Proteins 0.000 claims description 15
- 241000700605 Viruses Species 0.000 claims description 15
- 108091081024 Start codon Proteins 0.000 claims description 13
- 102000010292 Peptide Elongation Factor 1 Human genes 0.000 claims description 12
- 108010077524 Peptide Elongation Factor 1 Proteins 0.000 claims description 12
- 210000004978 chinese hamster ovary cell Anatomy 0.000 claims description 12
- 238000012258 culturing Methods 0.000 claims description 12
- 229930193140 Neomycin Natural products 0.000 claims description 11
- 229960004927 neomycin Drugs 0.000 claims description 11
- 229950010131 puromycin Drugs 0.000 claims description 10
- 241001529936 Murinae Species 0.000 claims description 9
- 241000701022 Cytomegalovirus Species 0.000 claims description 8
- 241000709664 Picornaviridae Species 0.000 claims description 8
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 claims description 8
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 claims description 8
- 230000002132 lysosomal effect Effects 0.000 claims description 8
- 229960000187 tissue plasminogen activator Drugs 0.000 claims description 8
- 108090000394 Erythropoietin Proteins 0.000 claims description 7
- 102000003951 Erythropoietin Human genes 0.000 claims description 7
- 239000003166 dihydrofolate reductase inhibitor Substances 0.000 claims description 7
- 229940105423 erythropoietin Drugs 0.000 claims description 7
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 claims description 7
- 229940117937 Dihydrofolate reductase inhibitor Drugs 0.000 claims description 6
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 claims description 6
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 claims description 6
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 claims description 5
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 claims description 5
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 claims description 5
- 229940028334 follicle stimulating hormone Drugs 0.000 claims description 5
- 102000015081 Blood Coagulation Factors Human genes 0.000 claims description 4
- 108010039209 Blood Coagulation Factors Proteins 0.000 claims description 4
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 claims description 4
- 102000004547 Glucosylceramidase Human genes 0.000 claims description 4
- 108010017544 Glucosylceramidase Proteins 0.000 claims description 4
- 102000004627 Iduronidase Human genes 0.000 claims description 4
- 108010003381 Iduronidase Proteins 0.000 claims description 4
- 102000014150 Interferons Human genes 0.000 claims description 4
- 108010050904 Interferons Proteins 0.000 claims description 4
- AEMOLEFTQBMNLQ-HNFCZKTMSA-N L-idopyranuronic acid Chemical compound OC1O[C@@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-HNFCZKTMSA-N 0.000 claims description 4
- 108010079274 Thrombomodulin Proteins 0.000 claims description 4
- 239000002253 acid Substances 0.000 claims description 4
- 239000003114 blood coagulation factor Substances 0.000 claims description 4
- 229940019700 blood coagulation factors Drugs 0.000 claims description 4
- 230000002950 deficient Effects 0.000 claims description 4
- 108010089296 galsulfase Proteins 0.000 claims description 4
- 229960005390 galsulfase Drugs 0.000 claims description 4
- 229940079322 interferon Drugs 0.000 claims description 4
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims description 3
- 241000283690 Bos taurus Species 0.000 claims description 3
- 241000711573 Coronaviridae Species 0.000 claims description 3
- 108700006830 Drosophila Antp Proteins 0.000 claims description 3
- 241000710198 Foot-and-mouth disease virus Species 0.000 claims description 3
- 241000709721 Hepatovirus A Species 0.000 claims description 3
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 claims description 3
- 102100024295 Maltase-glucoamylase Human genes 0.000 claims description 3
- 102000005840 alpha-Galactosidase Human genes 0.000 claims description 3
- 108010030291 alpha-Galactosidase Proteins 0.000 claims description 3
- 108010028144 alpha-Glucosidases Proteins 0.000 claims description 3
- 201000002491 encephalomyelitis Diseases 0.000 claims description 3
- 230000000968 intestinal effect Effects 0.000 claims description 3
- 102000006395 Globulins Human genes 0.000 claims description 2
- 108010044091 Globulins Proteins 0.000 claims description 2
- 241000711549 Hepacivirus C Species 0.000 claims description 2
- 241001180873 Saposhnikovia divaricata Species 0.000 claims description 2
- 102000040945 Transcription factor Human genes 0.000 claims description 2
- 230000036039 immunity Effects 0.000 claims description 2
- 102000012607 Thrombomodulin Human genes 0.000 claims 1
- 239000013598 vector Substances 0.000 abstract description 26
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 abstract description 24
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 abstract description 24
- 239000003550 marker Substances 0.000 description 92
- 108020004414 DNA Proteins 0.000 description 73
- 101150074355 GS gene Proteins 0.000 description 68
- 239000012634 fragment Substances 0.000 description 56
- 102000004419 dihydrofolate reductase Human genes 0.000 description 52
- 238000010276 construction Methods 0.000 description 50
- SXTAYKAGBXMACB-UHFFFAOYSA-N methionine sulfoximine Chemical compound CS(=N)(=O)CCC(N)C(O)=O SXTAYKAGBXMACB-UHFFFAOYSA-N 0.000 description 38
- 235000018102 proteins Nutrition 0.000 description 38
- 239000002609 medium Substances 0.000 description 35
- 108091008146 restriction endonucleases Proteins 0.000 description 30
- 101150074155 DHFR gene Proteins 0.000 description 28
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 23
- 239000000243 solution Substances 0.000 description 23
- 229960000485 methotrexate Drugs 0.000 description 22
- 241000699666 Mus <mouse, genus> Species 0.000 description 21
- 229960004641 rituximab Drugs 0.000 description 19
- 108010030074 endodeoxyribonuclease MluI Proteins 0.000 description 16
- QAPSNMNOIOSXSQ-YNEHKIRRSA-N 1-[(2r,4s,5r)-4-[tert-butyl(dimethyl)silyl]oxy-5-(hydroxymethyl)oxolan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O[Si](C)(C)C(C)(C)C)C1 QAPSNMNOIOSXSQ-YNEHKIRRSA-N 0.000 description 14
- 102100023795 Elafin Human genes 0.000 description 12
- 101001048718 Homo sapiens Elafin Proteins 0.000 description 12
- 239000006152 selective media Substances 0.000 description 11
- 239000000126 substance Substances 0.000 description 11
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 description 10
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 10
- 238000000034 method Methods 0.000 description 10
- 238000011144 upstream manufacturing Methods 0.000 description 8
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 7
- 239000012228 culture supernatant Substances 0.000 description 7
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 230000008488 polyadenylation Effects 0.000 description 6
- 241000699802 Cricetulus griseus Species 0.000 description 5
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 5
- 239000003623 enhancer Substances 0.000 description 5
- 230000004545 gene duplication Effects 0.000 description 5
- 238000013518 transcription Methods 0.000 description 5
- 230000035897 transcription Effects 0.000 description 5
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- 150000001413 amino acids Chemical group 0.000 description 4
- 229960003896 aminopterin Drugs 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000013612 plasmid Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 239000012086 standard solution Substances 0.000 description 4
- MSTNYGQPCMXVAQ-RYUDHWBXSA-N (6S)-5,6,7,8-tetrahydrofolic acid Chemical compound C([C@H]1CNC=2N=C(NC(=O)C=2N1)N)NC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 MSTNYGQPCMXVAQ-RYUDHWBXSA-N 0.000 description 3
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 3
- 241000255789 Bombyx mori Species 0.000 description 3
- 101001099217 Thermotoga maritima (strain ATCC 43589 / DSM 3109 / JCM 10099 / NBRC 100826 / MSB8) Triosephosphate isomerase Proteins 0.000 description 3
- 102100026966 Thrombomodulin Human genes 0.000 description 3
- 229940125644 antibody drug Drugs 0.000 description 3
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 3
- 229930189065 blasticidin Natural products 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 210000001672 ovary Anatomy 0.000 description 3
- 239000005460 tetrahydrofolate Substances 0.000 description 3
- 229940104230 thymidine Drugs 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 241000186361 Actinobacteria <class> Species 0.000 description 2
- 241000271566 Aves Species 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 241000221198 Basidiomycota Species 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 241000938605 Crocodylia Species 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- 241000255925 Diptera Species 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 241000991587 Enterovirus C Species 0.000 description 2
- 108010076282 Factor IX Proteins 0.000 description 2
- 108010023321 Factor VII Proteins 0.000 description 2
- 108010054218 Factor VIII Proteins 0.000 description 2
- 102000001690 Factor VIII Human genes 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 2
- 241001634830 Geometridae Species 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000633601 Homo sapiens Thyrotropin subunit beta Proteins 0.000 description 2
- 108020005350 Initiator Codon Proteins 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 241000255777 Lepidoptera Species 0.000 description 2
- 241000244206 Nematoda Species 0.000 description 2
- 102000011755 Phosphoglycerate Kinase Human genes 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 241000256251 Spodoptera frugiperda Species 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 101710087237 Whey acidic protein Proteins 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 230000002238 attenuated effect Effects 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- OZRNSSUDZOLUSN-LBPRGKRZSA-N dihydrofolic acid Chemical compound N=1C=2C(=O)NC(N)=NC=2NCC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OZRNSSUDZOLUSN-LBPRGKRZSA-N 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- 239000000833 heterodimer Substances 0.000 description 2
- 244000144972 livestock Species 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N phenylalanine group Chemical group N[C@@H](CC1=CC=CC=C1)C(=O)O COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 125000000430 tryptophan group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 2
- 108700026220 vif Genes Proteins 0.000 description 2
- 241000235349 Ascomycota Species 0.000 description 1
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 102000002464 Galactosidases Human genes 0.000 description 1
- 108010093031 Galactosidases Proteins 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 102000004366 Glucosidases Human genes 0.000 description 1
- 108010056771 Glucosidases Proteins 0.000 description 1
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 1
- 101000899240 Homo sapiens Endoplasmic reticulum chaperone BiP Proteins 0.000 description 1
- 101001038874 Homo sapiens Glycoprotein hormones alpha chain Proteins 0.000 description 1
- 241000702626 Infectious bursal disease virus Species 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 101100278853 Mus musculus Dhfr gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- ACFIXJIJDZMPPO-NNYOXOHSSA-N NADPH Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](OP(O)(O)=O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-NNYOXOHSSA-N 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 241001575928 Siler Species 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 108091036066 Three prime untranslated region Proteins 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000023555 blood coagulation Effects 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 208000031513 cyst Diseases 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229960000301 factor viii Drugs 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 238000011577 humanized mouse model Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- 210000000944 nerve tissue Anatomy 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images












Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
- C07K14/505—Erythropoietin [EPO]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/575—Hormones
- C07K14/59—Follicle-stimulating hormone [FSH]; Chorionic gonadotropins, e.g.hCG [human chorionic gonadotropin]; Luteinising hormone [LH]; Thyroid-stimulating hormone [TSH]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2887—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against CD20
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0012—Oxidoreductases (1.) acting on nitrogen containing compounds as donors (1.4, 1.5, 1.6, 1.7)
- C12N9/0026—Oxidoreductases (1.) acting on nitrogen containing compounds as donors (1.4, 1.5, 1.6, 1.7) acting on CH-NH groups of donors (1.5)
- C12N9/0028—Oxidoreductases (1.) acting on nitrogen containing compounds as donors (1.4, 1.5, 1.6, 1.7) acting on CH-NH groups of donors (1.5) with NAD or NADP as acceptor (1.5.1)
- C12N9/003—Dihydrofolate reductase [DHFR] (1.5.1.3)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/93—Ligases (6)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/10—Immunoglobulins specific features characterized by their source of isolation or production
- C07K2317/14—Specific host cells or culture conditions, e.g. components, pH or temperature
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/10—Plasmid DNA
- C12N2800/106—Plasmid DNA for vertebrates
- C12N2800/107—Plasmid DNA for vertebrates for mammalian
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/42—Vector systems having a special element relevant for transcription being an intron or intervening sequence for splicing and/or stability of RNA
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/50—Vector systems having a special element relevant for transcription regulating RNA stability, not being an intron, e.g. poly A signal
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2830/00—Vector systems having a special element relevant for transcription
- C12N2830/60—Vector systems having a special element relevant for transcription from viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2840/00—Vectors comprising a special translation-regulating system
- C12N2840/60—Vectors comprising a special translation-regulating system from viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y105/00—Oxidoreductases acting on the CH-NH group of donors (1.5)
- C12Y105/01—Oxidoreductases acting on the CH-NH group of donors (1.5) with NAD+ or NADP+ as acceptor (1.5.1)
- C12Y105/01003—Dihydrofolate reductase (1.5.1.3)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y603/00—Ligases forming carbon-nitrogen bonds (6.3)
- C12Y603/01—Acid-ammonia (or amine)ligases (amide synthases)(6.3.1)
- C12Y603/01002—Glutamate-ammonia ligase (6.3.1.2)
Definitions
- the present invention relates to a novel expression vector for efficiently expressing a recombinant protein in mammalian cells. More specifically, the present invention relates to a gene expression control site, a gene encoding a desired protein downstream thereof, and an internal ribosome downstream thereof.
- the gene further comprises a gene encoding glutamine synthetase downstream of the binding site, and further comprises a dihydrofolate reductase gene downstream of the gene expression control site or a gene expression control site different from the gene expression control site , Relating to expression vectors.
- a method for producing a recombinant protein using a mammalian cell transformed with an expression vector incorporating a gene encoding a desired protein is a widely used technique in industrial fields such as pharmaceutical production.
- Galactosidase A iduronic acid 2-sulfatase, glucocerebrosidase, galsulfase, ⁇ -L-iduronidase, lysosomal enzymes such as acid ⁇ -glucosidase, tissue plasminogen activator (t-PA), blood coagulation factor VII, blood coagulation Blood coagulation factors such as factor VIII and blood coagulation factor IX, erythropoietin, interferon, thrombomodulin, follicle stimulating hormone, granulocyte colony stimulating factor (G-CSF), various antibody drugs, etc. are manufactured using this technology. Is commercially available.
- a gene control site that induces strong gene expression such as a promoter derived from cytomegalovirus (CMV), an SV40 early promoter (SV40 enhancer / promoter), an elongation factor 1 ⁇ (EF-1) promoter, etc. It is common to use a product into which a gene encoding a desired protein is incorporated downstream. Mammalian cells introduced with such an expression vector express the desired protein incorporated in the expression vector, but the expression level varies depending on the individual cells and is not uniform. Therefore, in order to efficiently produce a recombinant protein, it is necessary to select a cell having a high expression level of a desired protein from mammalian cells into which an expression vector has been introduced. In order to perform this selection step, a gene that functions as a selection marker is incorporated into the expression vector.
- CMV cytomegalovirus
- SV40 enhancer / promoter SV40 enhancer / promoter
- EF-1 elongation factor 1 ⁇
- the most common selection marker is an enzyme (drug resistance marker) that degrades drugs such as puromycin and neomycin.
- Mammalian cells die in the presence of the drug above a certain concentration.
- mammalian cells into which an expression vector has been introduced can be decomposed by the drug selection marker incorporated in the expression vector, and can be detoxified or attenuated, so that it can survive even in the presence of the drug. Become. Therefore, when cells into which an expression vector has been introduced are cultured in a medium containing the drug at a certain concentration, only cells that express the drug selection marker at a high level proliferate and are selected.
- Cells expressing a drug selectable marker at a high level tend to express a gene encoding a desired protein simultaneously incorporated into an expression vector at a high level. As a result, a mammal expressing a desired protein at a high level Cells are obtained.
- Dihydrofolate reductase is an enzyme that reduces dihydrofolate to tetrahydrofolate.
- MTX methotrexate
- Glutamine synthase is an enzyme that synthesizes glutamine from glutamic acid and ammonia.
- MSX methionine sulfoximine
- the cells die.
- an expression vector incorporating glutamine synthetase as a selectable marker is introduced into a mammalian cell, the expression level of glutamine synthetase increases in the cell, so that it can grow even in the presence of a higher concentration of MSX. It becomes.
- Patent Document 1 describes that the use of the GS gene and methionine sulphoximine (MSX) can increase the copy number of vector DNA as compared with the case of using the DHFR gene and methotrexate (MTX). ing.
- Patent Document 2 also discloses that by using the GS gene and MSX, the copy number of a different foreign gene is also increased in the host cell DNA along with the increase in the copy number of the GS gene. And thus increase the level of production of the desired polypeptide.
- an expression vector containing a selection marker is suitable for the production of an efficient recombinant protein and is widely used.
- a gene encoding a desired protein and a gene encoding a selection marker are generally incorporated under different gene control sites (Patent Document 3).
- a method is also known in which a gene encoding a desired protein and a selection marker is serially incorporated under one gene control site (Patent Documents 4 to 7).
- the desired protein and the selection marker are known.
- An internal ribosome binding site IRS: internal ribosome entry site
- Various internal ribosome binding sites are known, such as those derived from picornavirus, poliovirus, encephalomyocarditis virus, and chicken infectious bursal disease virus (Patent Documents 8 to 10).
- an expression vector incorporating herpes simplex virus thymidine kinase as a selectable marker downstream of the internal ribosome binding site (Patent Document 11), 3 or more using two or more internal ribosome binding sites
- An expression vector (Patent Document 12) in which these genes are bound is known.
- JP-T 63-502955 Japanese Patent Publication No. 5-504050 JP 2009-273427 A JP 59-173096
- An object of the present invention is to provide a novel expression vector for efficiently expressing a recombinant protein in mammalian cells, a mammalian cell transformed with the vector, and a method for producing the mammalian cell.
- the present inventors have found that a gene expression control site, a gene encoding a desired protein such as human thyroid stimulating hormone (hTSH) downstream, an internal ribosome binding site downstream, and Using an expression vector further containing a gene encoding glutamine synthetase downstream and further containing a gene encoding dihydrofolate reductase downstream of the gene expression control site or another gene expression control site, The present inventors have found that a gene encoding the protein can be expressed at a high level by transforming and. That is, the present invention provides the following. 1.
- hTSH human thyroid stimulating hormone
- An expression vector for expressing a protein which encodes a gene expression control site (A), a gene encoding the protein downstream thereof, an internal ribosome binding site downstream, and a glutamine synthetase further downstream
- An expression vector comprising a gene and further comprising a dihydrofolate reductase gene downstream of the gene expression control site (A) or a gene expression control site (B) different from the gene expression control site (A) .
- the gene expression control site (A) and / or the gene expression control site (B) is selected from the group consisting of a cytomegalovirus-derived promoter, an SV40 early promoter, and an elongation factor 1 promoter. Expression vector. 3.
- the internal ribosome binding site is a picornaviridae virus, foot-and-mouth disease virus, hepatitis A virus, hepatitis C virus, coronavirus, bovine intestinal virus, siler murine encephalomyelitis virus, coxsackie type B virus, human immunity
- the expression vector according to 1 or 2 above which is derived from a 5 'untranslated region of a virus or gene selected from the group consisting of a globulin heavy chain binding protein gene, a Drosophila antennapedia gene, and a Drosophila ultravitracs gene. 4).
- the drug resistance gene is a puromycin or neomycin resistance gene.
- the gene encoding the protein is a human-derived gene. 18.
- the gene derived from human is lysosomal enzyme, tissue plasminogen activator (t-PA), blood coagulation factor, erythropoietin, interferon, thrombomodulin, thyroid stimulating hormone (TSH), follicle stimulating hormone, granulocyte colony stimulating factor (G -CSF), and the expression vector according to 17 above, which is selected from the group consisting of genes encoding antibodies. 19. 18. The expression vector according to 17 above, wherein the human-derived gene is a gene encoding a lysosomal enzyme. 20. 19.
- 25 (A) introducing the expression vector of any one of 1 to 21 into mammalian cells; (B) a step of selectively culturing mammalian cells into which the expression vector has been introduced in the presence of a dihydrofolate reductase inhibitor, and (c) the presence of a glutamine synthetase inhibitor in cells selected by the selective culture.
- a method for producing a transformed cell expressing a gene encoding the protein further comprising a step of selective culturing below.
- A introducing the expression vector of any one of 14 to 16 into a mammalian cell;
- B a step of selectively culturing mammalian cells into which the expression vector has been introduced in the presence of a dihydrofolate reductase inhibitor; and
- c the presence of a glutamine synthetase inhibitor in cells selected by the selective culture.
- a method for producing a transformed cell that expresses a gene encoding the protein further comprising a step of selective culturing below, between the step (a) and the step (b), or the step (b)
- a production method further comprising the step of selectively culturing mammalian cells into which the expression vector has been introduced during the step (c) in the presence of a drug corresponding to the drug resistance gene.
- an expression vector for efficiently expressing a desired recombinant protein in a mammalian cell can be obtained.
- a transformed cell that efficiently produces the recombinant protein can be obtained. If the transformed cells thus obtained are used, the production cost of the recombinant protein can be greatly reduced.
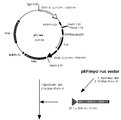
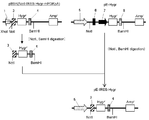
- the flowchart which shows the construction method of a pE-neo vector The flowchart which shows the construction method of a pE-neo vector.
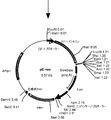
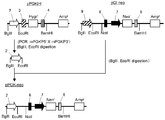
- the flowchart which shows the construction method of a pE-hygr vector The flowchart which shows the construction method of a pE-hygr vector.
- the flowchart which shows the construction method of a pE-hygr vector The flowchart which shows the construction method of a pE-hygr vector.
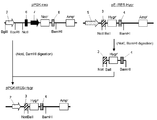
- the flowchart which shows the construction method of pE-IRES-GS-puro The flowchart which shows the construction method of pE-IRES-GS-puro.
- the flowchart which shows the construction method of pE-IRES-GS-puro The flowchart which shows the construction method of pE-IRES-GS-puro.
- the flowchart which shows the construction method of pE-IRES-GS-puro The flowchart which shows the construction method of pE-IRES-GS-puro.
- the flowchart which shows the construction method of pE-IRES-GS-puro The flowchart which shows the construction method of pE-IRES-GS-puro.
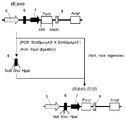
- the flowchart which shows the construction method of pE-mIRES-GS-puro The flowchart which shows the construction method of pE-mIRES-GS.
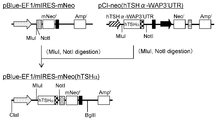
- the flowchart which shows the construction method of pE-mIRES-GS-mNeo The flowchart which shows the construction method of pBlue-EF1 / mIRES-mNeo.
- the flowchart which shows the construction method of pBlue-EF1 / SVpA The flowchart which shows the construction method of pE-mDHFR31.
- the flowchart which shows the construction method of pCI-neo TH ⁇ -WAP3′UTR).
- the flowchart which shows the construction method of pE-mIRES-GS-mNeo hTSH ⁇ ).
- the flowchart which shows the construction method of pBlue-EF1 / mIRES-mNeo (hTSH ⁇ ).
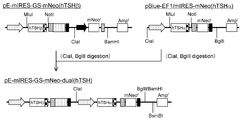
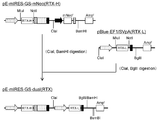
- the flowchart which shows the construction method of pE-mIRES-GS-mNeo-dual (hTSH).
- the flowchart which shows the construction method of pE-mIRES-GS-mNeo-dual + mDHFR31 (hTSH).
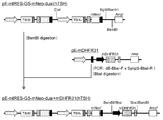
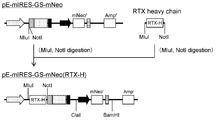
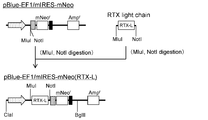
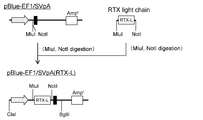
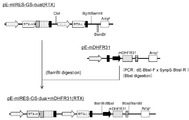
- the flowchart which shows the construction method of pE-mIRES-GS-mNeo-dual (RTX).
- the flowchart which shows the construction method of pE-mIRES-GS-dual (RTX).
- the term “gene” means a structural gene.
- a “gene expression control site” is a region on DNA that can control (regulate) the frequency of transcription of a gene present downstream thereof, and is generally referred to as a promoter or promoter gene. is there. Gene expression control sites exist on almost all upstream sides of genes expressed in vivo, and control (regulate) the frequency of transcription, and their nucleotide sequences are diverse.
- the gene expression control site that can be used in the present invention is not particularly limited as long as it can strongly induce transcription of a gene incorporated downstream thereof in mammalian cells, but is preferably cytomegalovirus. (CMV) -derived promoters, virus-derived promoters such as the SV40 early promoter, and elongation factor 1 ⁇ (EF-1) promoter.
- CMV cytomegalovirus
- an “internal ribosome binding site” refers to a region (structure) within a mRNA chain, to which a ribosome can directly bind and initiate translation independent of a cap structure, or by being transcribed.
- the “gene encoding an internal ribosome binding site” is a region (structure) of a DNA chain that is transcribed to generate the region.
- the internal ribosome binding site is generally referred to as IRES (internal ribosome entry site), and Picornaviridae virus (poliovirus, rhinovirus, mouse encephalomyocarditis virus, etc.), foot-and-mouth disease virus, hepatitis A virus, type C Hepatitis virus, coronavirus, bovine intestinal virus, Siler's murine encephalomyelitis virus, Coxsackie B virus 5 'untranslated region, human immunoglobulin heavy chain binding protein, Drosophila antennapedia, Drosophila ultrabitrax, etc. It is found in the 5 'untranslated region of the gene.
- the IRES is a region consisting of about 450 bp present in the 5 ′ untranslated region of mRNA.
- the “virus 5 ′ untranslated region” is a 5 ′ untranslated region of viral mRNA, or a region (structure) of a DNA strand that is transcribed to produce this region.
- the internal ribosome binding site is not particularly limited as long as it functions as an internal ribosome binding site in mammalian cells, particularly cells derived from the ovary of Chinese hamsters (CHO cells). Can also be used. Among them, preferably an internal ribosome binding site derived from the 5 ′ untranslated region of the virus, more preferably an internal ribosome binding site derived from the 5 ′ untranslated region of the Picornaviridae virus, more preferably mouse brain myocardium An internal ribosome binding site derived from the 5 'untranslated region of the flame virus.
- the internal ribosome binding site having a wild-type base sequence can be used as it is.
- a mutant internal ribosome binding site obtained by adding one or more mutations (for example, substitution, deletion, and / or insertion) to the base sequence of these wild type internal ribosome binding sites is also a mammal. Any substance can be used as long as it functions as an internal ribosome binding site in animal cells (particularly in CHO cells).
- a chimeric internal ribosome binding site in which two or more internal ribosome binding sites are fused can also be used.
- the expression level of the GS gene is controlled by placing a gene encoding a glutamine synthase (GS gene) under the control of the internal ribosome binding site.
- GS gene glutamine synthase
- the culture for selecting not only the cell into which the GS gene has been introduced but also the cell into which the selection marker has been introduced is referred to as “selective culture” and is used for selecting the introduced cells.
- the medium is referred to as “selective medium”.
- a suitable one is appropriately selected from various internal ribosome binding sites.
- a wild-type internal ribosome binding site with mutations can also be used.
- start codons ATG
- ATG start codons
- an internal ribosome binding site in which a part of these start codons is destroyed can be used.
- disruption means that a certain gene sequence is mutated so that the function inherent to the gene sequence is not exhibited.
- the start codons to be destroyed by mutation are preferably the second and third start codons from the 5 ′ side, more preferably Second start codon.
- SEQ ID NO: 2 5′-ATGataatnnngccacaaccnnn-3 ′: n is an arbitrary base, but three n are initiation codons ATG The same shall apply hereinafter.
- SEQ ID NO: 7 5′-ATGataannnngccacaaccnnn-3 ′
- SEQ ID NO: 3 5′-ATGataatnnngccacaaccATG-3 ′
- SEQ ID NO: 8 5 And the base sequence of '-ATGataannnngccacaaccATG-3').
- the 3 ′ end is an internal ribosome binding site that is the base sequence of SEQ ID NO: 4 (5′-ATGataagcttgccacaaccATG-3 ′), and the second start codon from the 5 ′ side was destroyed by mutation.
- the internal ribosome binding site of wild-type mouse encephalomyocarditis virus comprises the nucleotide sequence of SEQ ID NO: 5.
- the base sequence of sequence number 6 is mentioned.
- the expression level of the GS gene arranged downstream of the wild-type and / or mutant-type internal ribosome binding site can be further controlled by another method.
- the expression level of the GS gene can be decreased.
- the base sequence that inhibits transcription is not particularly limited, and examples thereof include a polymerase addition signal (5′-aataa-3 ′). Examples of base sequences that inhibit translation include a stop codon that induces reading through.
- glucose synthase is not particularly limited as long as it can synthesize glutamine from glutamic acid and ammonia, and belongs to mammals, reptiles, birds, amphibians, and Lepidoptera. Bombyx mori, Spodoptera frugiperda, Geometridae, and other insects such as Drosophila belonging to Diptera, prokaryotes, nematodes, yeasts, actinomycetes, filamentous fungi, offspring Although it may be derived from any organism including cysts, basidiomycetes and plants, it is preferably a mammal, and a human or Chinese hamster (especially a CHO cell) can be suitably used.
- the term “endogenous GS gene” refers to a glutamine synthase gene that is inherently present on the genome of the cell into which the expression vector is introduced, and the term “exogenous GS gene”. , refers to a glutamine synthase gene introduced into cells by an expression vector.
- glucose synthase inhibitor is not particularly limited as long as it can inhibit the activity of the above glutamine synthase, and any one can be used, but methionine sulfoximine is preferred. (MSX).
- GS inhibitor glutamine synthetase inhibitor
- cells with a low expression level of glutamine synthase die without being able to synthesize glutamine.
- cells with a relatively high expression level can be selectively obtained.
- the increase in the expression level of glutamine synthetase is mainly due to an increase in the copy number of the GS gene integrated on the chromosome due to gene duplication.
- the gene encoding the desired protein incorporated into the expression vector together with the glutamine synthetase also increases the copy number due to gene duplication, and as a result, cells that highly express the desired protein are obtained.
- CHO cells have an endogenous GS gene
- this endogenous GS gene may increase the copy number due to duplication of the endogenous GS gene by a glutamine synthetase inhibitor.
- the cells proliferate in the presence of the glutamine synthetase inhibitor, and cells that highly express the desired protein cannot be obtained.
- This problem can be solved by further incorporating a second selectable marker gene different from the GS gene into the expression vector, and selecting cells using this second selectable marker.
- an expression vector containing a GS gene and a second selection marker gene different from the GS gene is introduced into the cell.
- the cells into which the expression vector has been introduced are selectively cultured with the second selection marker before being selectively cultured with the GS gene.
- the gene encoding the desired protein and the GS gene can be increased by duplication. As a result, cells expressing the desired protein at a certain high level can be obtained.
- the copy number of the exogenous GS gene increases due to duplication, but the endogenous GS gene does not increase due to duplication.
- the selection medium (second selection medium) used in the selection by the second selection marker is an agent that inhibits the activity of the gene product encoded by the second selection marker, has cytotoxicity, and is used for the second selection.
- a substance for selecting cells by a second selection marker such as a substance detoxified or attenuated by the gene product encoded by the marker, or a second selection marker introduced
- a substance in which later cells do not exhibit auxotrophy that is, a substance in which only cells before introduction of the second selection marker exhibit auxotrophy
- examples of selective substances added to the medium include methotrexate (MTX) and aminopterin.
- the selection substances removed from the medium are hypoxanthine and thymidine.
- cells with a further increased expression level of the second selection marker are obtained by increasing the concentration of the selection substance stepwise. be able to.
- the increase in the expression level of the second selectable marker is mainly due to the increase in the copy number of the second selectable marker due to gene duplication. Therefore, by this method, other genes incorporated into the expression vector, that is, the GS gene And the gene encoding the desired protein can be increased more efficiently by duplication.
- the selection marker is switched to the GS gene to select cells.
- the copy number of the exogenous GS gene is increased compared to that of the endogenous GS gene. Therefore, at this point, by selecting the cell by switching the selection marker to the GS gene, the exogenous GS gene that is dominant in number may be amplified by duplication as compared with the endogenous GS gene. Is probabilistically higher.
- the duplication of the endogenous GS gene can be suppressed and the duplication of the exogenous GS gene can be promoted during the selection by the GS gene. Can do. As a result, cells that highly express the desired protein can be efficiently obtained.
- the second selectable marker is provided with a gene expression control site (second gene expression control site) separate from the gene expression control site for controlling the expression of the recombinant protein, and downstream thereof. May be incorporated.
- the second selectable marker is provided with a second internal ribosome binding site upstream thereof, through which a gene encoding the desired recombinant protein and an internal ribosome binding site downstream of the gene are encoded. May be incorporated in the region between or in the region downstream of the GS gene. Thereby, the expression level of the second selection marker can be controlled by the second internal ribosome binding site.
- the second internal ribosome binding site may have the same base sequence as the internal ribosome binding site upstream of the GS gene, or may have a different base sequence. Further, the second internal ribosome binding site can be arbitrarily selected from the various internal ribosome binding sites described above.
- the second selectable marker is not particularly limited as long as it is a selectable marker different from the GS gene, but in principle, the gene conferring drug resistance to mammalian cells (drug resistant gene) is the second selectable marker. Not included in selection marker. However, when a third selectable marker gene described later is incorporated into an expression vector and mammalian cells are further selected using this gene, a drug resistance gene can be used as the second selectable marker.
- the dihydrofolate reductase (DHFR) gene can be suitably used as the second selection marker.
- DHFR dihydrofolate reductase
- Lepidoptera It may be derived from any organism including insects such as Drosophila, prokaryotes, nematodes, yeasts, actinomycetes, filamentous fungi, ascomycetes, basidiomycetes and plants, but preferably from mammals Yes, those derived from humans, mice or Chinese hamsters (particularly those derived from CHO cells) can be preferably used.
- insects such as Drosophila, prokaryotes, nematodes, yeasts, actinomycetes, filamentous fungi, ascomycetes, basidiomycetes and plants
- mammals Yes those derived from humans, mice or Chinese hamsters (particularly those derived from CHO cells)
- mutant enzymes in which mutations are introduced into these wild-type enzymes can be used as long as they have enzyme activity, and these are also included in “dihydrofolate reductase”.
- Such mutant enzymes include, for example, those having different enzyme activities and those having different sensitivities to dihydrofolate reductase inhibitors compared to wild-type enzymes.
- dihydrofolate reductase that can be preferably used include mouse wild-type DHFR and mutant DHFR in which phenylalanine at position 31 from the N-terminal of mouse wild-type DHFR is substituted with tryptophan (Mclvor RS et al., Nucleic Acids). Research, 18 (23), 7025-32 (1990)).
- nucleotide sequence and amino acid sequence of mouse wild-type DHFR are shown in SEQ ID NO: 9 and SEQ ID NO: 10
- nucleotide sequence and amino acid sequence of the mutant DHFR are shown in SEQ ID NO: 11 and SEQ ID NO: 12.
- endogenous DHFR gene refers to a DHFR gene inherently present on the genome of a cell into which an expression vector is introduced
- exogenous DHFR gene refers to expression. This refers to the DHFR gene introduced into cells by a vector.
- dihydrofolate reductase inhibitor is not particularly limited as long as it can inhibit the activity of the dihydrofolate reductase (DHFR), and any of them can be used.
- a folic acid antagonist more preferably methotrexate (MTX) and aminopterin.
- DHFR inhibitor dihydrofolate reductase inhibitor
- tetrahydrofolate is synthesized in cells with low DHFR expression. Since they die without being able to do so, cells with a relatively high expression level of DHFR will be selectively obtained.
- concentration of the DHFR inhibitor stepwise, cells with a higher expression level of DHFR can be obtained.
- the increase in the expression level of DHFR is mainly due to the fact that the DHFR gene integrated on the chromosome increases the copy number due to gene duplication.
- the copy number of other genes incorporated into the expression vector that is, the GS gene and the gene encoding the desired protein
- the copy number of other genes incorporated into the expression vector that is, the GS gene and the gene encoding the desired protein
- a cell can be obtained in which the desired protein is highly expressed to some extent and the copy number of the exogenous GS gene is increased compared to that of the endogenous GS gene.
- the exogenous GS gene that is predominant in number may be amplified by duplication compared to the endogenous GS gene. Become expensive.
- cells that highly express the desired protein can be efficiently obtained.
- the maximum concentration is preferably 0.25 to 5 ⁇ M, more preferably 0.5 to 1.5 ⁇ M when the DHFR inhibitor is methotrexate, Preferably it is about 1.0 ⁇ M.
- a third selection marker may be further introduced into the expression vector in addition to the GS gene and the second selection marker.
- the drug resistance gene can be suitably used as the third selection marker gene.
- the drug resistance gene that can be used as the third selectable marker is not particularly limited as long as it is a gene that can confer drug resistance to mammalian cells.
- the cell is purified with puromycin, hygromycin, It is a gene that can confer resistance to drugs such as blasticidin and neomycin.
- drugs such as puromycin, hygromycin, blasticidin, and neomycin are “drugs corresponding to drug resistance genes”, respectively.
- These drug resistance genes include puromycin resistance gene, hygromycin resistance gene, blasticidin resistance gene, and neomycin resistance gene.
- the third selectable marker is provided by providing a gene expression control site (third gene expression control site) separate from the gene expression control site where the recombinant protein is controlled and incorporating it downstream of the gene expression control site. , The amount of expression can be controlled.
- the third selectable marker has a second internal ribosome binding site upstream thereof (if a second internal ribosome binding site is provided upstream of the second selectable marker, It may be incorporated into a region between the gene encoding the recombinant protein and the internal ribosome binding site, a region downstream of the GS gene, or the like via an internal ribosome binding site. Thereby, the expression level of the third selection marker can be controlled by the second internal ribosome binding site.
- the second internal ribosome binding site may be the same as the internal ribosome binding site upstream of the GS gene and the internal ribosome binding site upstream (if provided) of the second selectable marker, Another thing may be used.
- the second internal ribosome binding site upstream of the third selectable marker can be arbitrarily selected from the above various internal ribosome binding sites. Also, the second internal ribosome binding site may be optimized by adding mutation as appropriate, as described above.
- the drug resistance gene used as the third selection marker gene here is selected from drug resistance genes other than the second selection marker.
- the third selection marker can amplify the GS gene without duplicating the endogenous GS gene. Accordingly, the exogenous GS gene is duplicated without duplicating the endogenous GS gene by selecting cells with the third selectable marker prior to or after selection with the second selectable marker. be able to. As a result, cells that highly express the desired protein can be obtained more efficiently.
- the animal species of the gene encoding the recombinant protein incorporated into the expression vector is not particularly limited, including those derived from mammals including humans.
- the gene when the vector of the present invention is used for the production of a medical drug, it is generally derived from a human, and when it is used for the production of a drug for livestock, it is generally Is a gene derived from the same species of livestock as the subject of treatment.
- the type of gene encoding the recombinant protein is not particularly limited, but preferably ⁇ -galactosidase A, iduronic acid 2-sulfatase, glucocerebrosidase, galsulfase, ⁇ -L-iduronidase, acid ⁇ - Lysosomal enzymes such as glucosidase, tissue plasminogen activator (t-PA), blood coagulation factor VII, blood coagulation factor VIII, blood coagulation factor IX and other blood coagulation factors, erythropoietin, interferon, thrombomodulin, follicle stimulating hormone , Granulocyte colony stimulating factor (G-CSF), DNase I, thyroid stimulating hormone (TSH) or a gene encoding various antibody drugs.
- antibody drugs include mouse antibodies, humanized mouse antibodies, human / mouse chimeric antibodies, human antibodies, and the like, for example, rituximab which is a human / mouse chimeric anti
- the gene encoding either the antibody heavy chain or the antibody light chain is incorporated into a vector so that its expression is controlled by a gene expression control site that controls the expression of the GS gene.
- the gene encoding the other strand is incorporated so that its expression is controlled by a gene expression control site arranged in the vector separately.
- the gene expression site used at this time is preferably the same type of gene expression site so that the antibody heavy chain and the antibody light chain are expressed at the same level.
- the expression of a gene encoding any subunit that forms the heterodimer is controlled by a gene expression control site that controls the expression of the GS gene.
- the gene encoding the other subunit is incorporated so that its expression is controlled by a gene expression control site arranged in the vector separately.
- the gene expression site used at this time is preferably the same type of gene expression site so that the two types of subunits are expressed at the same level. Proteins that form such heterodimers include follicle-stimulating hormone and thyroid-stimulating hormone (TSH).
- introduction of an expression vector into a mammalian cell is performed for the purpose of expressing a gene encoding the recombinant protein in the mammalian cell, and any method can be used as long as this object can be achieved. May be used.
- Expression vectors are usually circular plasmids, which can be introduced into cells even if they remain circular or after being cleaved with a restriction enzyme to form a straight chain.
- the mammalian cells into which the expression vector is introduced are not particularly limited as long as the target recombinant protein can be expressed, but organs, muscle tissues, skin tissues, connective tissues, nerves removed from the living body. It may be any of primary culture cells, subculture cells, and cells established so that the traits are stable even when subcultured, from cells collected from tissues, blood, bone marrow, and the like. Further, the cells may be normal cells or cancerous cells. Cells that can be used particularly preferably are CHO cells derived from Chinese hamster ovary, human fibroblasts, and COS cells derived from African green monkey kidney fibroblasts.
- the mammalian cell into which the expression vector is introduced may be a cell having a mutation in the endogenous gene corresponding to the second selection marker.
- the mutation of the endogenous gene corresponding to the second selectable marker is one that reduces or eliminates the expression of the gene, or reduces or loses the function of the protein encoded by the endogenous gene.
- the selection pressure by the selection substance is strongly applied to the exogenous second selection marker, and the duplication of the second selection marker is further promoted.
- the second selectable marker is a DHFR gene
- the mutation of the endogenous DHFR gene decreases the expression level of dihydrofolate reductase or deletes the dihydrofolate reductase
- the DHFR gene is selected at the time of selection with the DHFR gene. Selection pressure by the inhibitor is exclusively applied to the exogenous DHFR gene, and duplication of the exogenous DHFR gene is further promoted.
- Mammalian cells into which the expression vector has been introduced are cultured in a selection medium for selecting cells with the second selection marker prior to selection with the GS gene.
- the medium at this time is a medium to which a DHFR inhibitor is added when the second selection marker is DHFR.
- aminopterin and methotrexate (MTX) are preferably used as DHFR inhibitors.
- the second selection marker is a drug resistance gene, it is a drug corresponding to the drug resistance gene.
- a mammalian cell into which an expression vector is introduced can have a mutation in the endogenous DHFR gene that reduces or deletes the expression level of DHFR.
- An example of such cells is the DG44 strain.
- the DG44 strain does not express the endogenous DHFR gene. Therefore, when the cell line is used, the selective pressure in selective culture is applied only to the exogenous DHFR gene, so that the exogenous DHFR gene is efficiently increased by duplication. Can be made.
- the selective culture can be performed in a selective medium containing a DHFR inhibitor such as aminopterin and methotrexate (MTX).
- MTX methotrexate
- the maximum concentration is preferably 0.25 to 5 ⁇ M, more preferably 0.5 to 1.5 ⁇ M when the DHFR inhibitor is methotrexate, Preferably it is about 1.0 ⁇ M.
- the culture for maintaining or growing the cells is carried out in a medium to which these are added.
- cells introduced with the exogenous DHFR gene do not have to be auxotrophic for hypoxanthine and thymidine, so it is not necessary to add them to the medium. Therefore, when an expression vector containing a DHFR gene as a second selection marker is introduced into a cell having no or almost no DHFR activity derived from the endogenous DHFR gene due to deletion of the endogenous DHFR gene, etc.
- Hypoxanthine and thymidine-free medium can also be used to selectively culture cells into which an exogenous DHFR gene has been introduced.
- the culture medium which added the DHFR inhibitor to the hypoxanthine and the thymidine-free culture medium can also be used as a selective culture medium.
- the expression vector-introduced cells are cultured in a medium for selecting cells into which the GS gene has been introduced.
- the medium at this time is a glutamine-free or low glutamine medium supplemented with a glutamine synthetase inhibitor (for example, MSX).
- the concentration of the GS inhibitor added to the selective medium By gradually increasing the concentration of the GS inhibitor added to the selective medium, it is possible to select an expression vector-introduced cell having a higher expression level of the GS gene. This is because only the expression vector-introduced cells in which the number of GS genes incorporated into the genome of the expression vector-introduced cell increased due to duplication and the expression level of the GS gene increased relatively during the selective culture process. Caused by proliferation. At this time, since the number of copies of the gene encoding the recombinant protein incorporated in the expression vector also increases, the expression level of the gene also increases.
- the expression vector-introduced cell thus selected is referred to as a transformed cell.
- the maximum concentration is preferably 100 to 1000 ⁇ M when the GS inhibitor is methionine sulfoximine (MSX). More preferably 200 to 500 ⁇ M, and still more preferably about 300 ⁇ M.
- the cells into which the expression vector has been introduced are added to the selective culture by the second selective culture, and the selective medium for selecting with the third selective marker. It can also be selectively cultured.
- the selective culture using the third selection marker may be performed either before or after the second selective culture.
- the third selectable marker is a drug resistance gene. Therefore, a drug corresponding to the drug resistance gene is added to the selective medium. At this time, cells with a higher expression level of the drug resistance gene can be obtained by increasing the concentration of the drug in the selective medium stepwise.
- the maximum concentration is preferably 3 to 30 ⁇ M, more preferably 5 to 20 ⁇ M, more preferably about 10 ⁇ M.
- the maximum concentration is preferably 0.4 to 1.5 mg / mL, more preferably 0.8 to 1.2 mg / mL, and further preferably about 1 mg / mL.
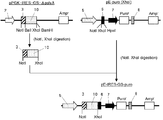
- the pE-neo vector was digested with SfiI and BstXI, and an approximately 1 kbp region containing a neomycin resistance gene was excised (FIG. 2-1).
- the hygromycin gene was amplified by PCR reaction using pcDNA3.1 / Hygro (+) (Invitrogen) as a template and using the primer Hyg-Sfi5 ′ (SEQ ID NO: 13) and primer Hyg-BstX3 ′ (SEQ ID NO: 14). ( Figure 2-2).
- the amplified hygromycin gene was digested with SfiI and BstXI and inserted into the above pE-neo vector to construct a pE-hygr vector (FIG. 2-3).
- Base from 5 ′ end including binding site (IRES), hygromycin resistance gene (Hygr gene), and polyadenylation region (mPGKpA) of mouse phosphoglycerate kinase (mPGK)
- the region consisting of 1 to 6 is the “XhoI site”
- the bases 120 to 715 followed by the bases 716 to 718 (atg) are the “internal ribosome binding site derived from the 5 ′ untranslated region of mouse encephalomyocarditis virus
- a region consisting of bases 716 to 1741 containing bases 716 to 718 (atg) is a "base sequence encoding a hygromycin resistance gene”
- a region consisting of bases 1747 to 2210 is "mouse phosphoglycerate kinase" (mPGK) polyadeni Bases sequence containing a region ", and 3 'regions of six bases (bases 2211-2216) of the terminal is" BamHI site
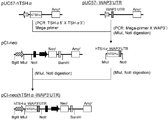
- a DNA fragment containing a part of EMCV IRES was amplified by PCR using pBSK (IRES-Hygr-mPGKpA) as a template and using primer IRES5 ′ (SEQ ID NO: 17) and primer IRES3 ′ (SEQ ID NO: 18).
- This DNA fragment is digested with restriction enzymes (XhoI and HindIII) and pBSK This was inserted between the XhoI and HindIII sites of (IRES-Hygr-mPGKpA) and designated pBSK (NotI-IRES-Hygr-mPGKpA) (FIG. 3-2).
- pBSK NotI-IRES-Hygr-mPGKpA
- restriction enzymes NotI and BamHI
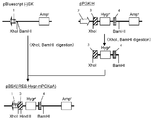
- This DNA fragment was digested with restriction enzymes (BglII and EcoRI) and inserted between the BglII and EcoRI sites of pCI-neo (Promega) to obtain pPGK-neo (FIGS. 3-4).
- pE-IRES-Hygr was digested with restriction enzymes (NotI and BamHI) to cut out a DNA fragment (IRES-Hygr) and inserted between NotI and BamHI sites of pPGK-neo, which was designated as pPGK-IRES-Hygr ( Fig. 3-5).
- CDNA was prepared from CHO-K1 cells, and using this as a template, a DNA fragment containing the GS gene was amplified by PCR using primer GS5 ′ (SEQ ID NO: 22) and primer GS3 ′ (SEQ ID NO: 23). This DNA fragment was digested with restriction enzymes (BalI and BamHI) and inserted between the BalI and BamHI sites of pPGK-IRES-Hygr to obtain pPGK-IRES-GS- ⁇ polyA (FIGS. 3-6).
- restriction enzymes BalI and BamHI
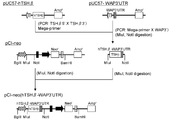
- pCAGIPuro (Miyahara Puromycin resistance by PCR using M. et.al., J. Biol. Chem. 275,613-618 (2000)) as template and primer puro5 ′ (SEQ ID NO: 24) and primer puro3 ′ (SEQ ID NO: 25).
- puro r gene of the base sequence (SEQ ID NO: 26.5 'end including a base region consisting of 2-7' AflII site "region consisting of bases 8-607 followed this" puromycin resistance gene ( puro r gene) a nucleotide sequence encoding a ", and, the next region consisting of bases 608 to 619 is" BstXI site ".) was from consisting DNA fragment was amplified (the amino acid corresponding to puro r gene The sequence is that shown in SEQ ID NO: 27).
- This DNA fragment was digested with restriction enzymes (AflII and BstXI) and inserted between the AflII and BstXI sites of the expression vector pE-neo to obtain pE-puro (FIGS. 3-7).
- DNA fragment containing SV40 late polyadenylation region was amplified by PCR using pE-puro as a template and primer SV40polyA5 ′ (SEQ ID NO: 28) and primer SV40polyA3 ′ (SEQ ID NO: 29).
- This DNA fragment was digested with restriction enzymes (NotI and HpaI) and inserted between NotI and HpaI sites of the expression vector pE-puro, which was designated as pE-puro (XhoI) (FIGS. 3-8).
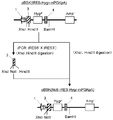
- pPGK-IRES-GS- ⁇ polyA is digested with restriction enzymes (NotI and XhoI), a DNA fragment containing the IRES-GS region is excised, and inserted between NotI and XhoI sites of the expression vector pE-puro (XhoI). This was designated as pE-IRES-GS-puro ( Figure 3-9).
- This DNA fragment is digested with restriction enzymes (NotI and PstI), and the excised DNA fragment is inserted between NotI and PstI sites of the expression vector pE-IRES-GS-puro, which is inserted into pE-mIRES-GS-puro. (FIG. 4).
- a DNA fragment containing the SV40 late polyA region was amplified by PCR using the expression vector pE-neo as a template and the primer SV40polyA5'-2 (SEQ ID NO: 32) and primer SV40polyA3'-2 (SEQ ID NO: 33). This DNA fragment was digested with restriction enzymes (XhoI and BamHI) and inserted between the XhoI and BamHI sites of pE-mIRES-GS-puro to obtain pE-mIRES-GS (FIG. 5).
- restriction enzymes XhoI and BamHI
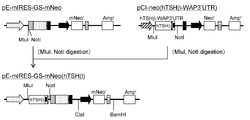
- a DNA fragment containing the mNeo r gene was amplified by PCR using the primer mNeoB5 ′ (SEQ ID NO: 36) and primer mNeoB3 ′ (SEQ ID NO: 37) using the pCI-mNeo vector as a template.
- This DNA fragment was digested with restriction enzyme (EagI) and pUC57-IE1 (Gene It was inserted into NotI of Script Corp. and called pUC57-IE1-mNeo.
- the pCI-neo as a template a region using the primers MNeoC5 '(SEQ ID NO: 38) and primer MNeoC3' (SEQ ID NO: 39), containing the 3 'region and synthetic poly A signal of the neomycin-resistant gene by PCR (neo r gene) This was used as a mega primer (neo-polyA).
- pUC57-IE1-mNeo as a template and using primer mNeoD5 '(SEQ ID NO: 40) and megaprimer (neo-polyA)
- a region containing the mNeo r gene and synthetic poly A signal was amplified by PCR. .
- the obtained DNA fragment was digested with restriction enzymes (AflII and BamHI) and inserted between the AflII and BamHI sites of pE-mIRES-GS-puro to obtain pE-mIRES-GS-mNeo (FIG. 6).
- pE-mIRES-GS-mNeo was digested with a restriction enzyme (PvuI), and a DNA fragment containing the mutant neomycin resistance gene (mNeo r gene) and a DNA fragment containing the SV40 late polyA region were excised.
- mNeo r gene mutant neomycin resistance gene
- mNeo r gene mutant neomycin resistance gene
- DNA fragment containing the SV40 late polyA region was excised.
- the DNA fragment containing the SV40 late polyA region as a template, using the primer SVpA-Mega-F (SEQ ID NO: 41) and primer SVpA-BstXI-R (SEQ ID NO: 42), the DNA fragment containing the SV40 late polyA region by PCR was amplified.
- This gene fragment and the DNA fragment containing the mNeo r gene were joined together by PCR to produce a DNA fragment containing the SV40 late polyA region downstream of the mNeo r gene.
- This DNA fragment was digested with a restriction enzyme (BstXI) and inserted between the BstXI sites of pBlue-EF1 / mIRES to obtain pBlue-EF1 / mIRES-mNeo (FIG. 7).
- pGEM mDHFR31
- F31W restriction enzymes
- the region consisting of bases 1 to 6 is the “MluI site”, and the region consisting of bases 14 to 364 is “coding the hTSH ⁇ chain And a region comprising bases 365 to 372 following this is a “NotI site”), and a DNA containing the human thyroid stimulating hormone ⁇ chain (hTSH ⁇ ) gene (SEQ ID NO: 47, 5 ′ end)
- the region consisting of bases 1 to 6 is the “MluI site”
- the region consisting of bases 14 to 433 is the “base sequence encoding the TSH ⁇ chain”
- the subsequent region consisting of bases 434 to 441 is the “NotI site” And incorporated into pUC57, respectively, to form pUC57-hTSH ⁇ and pUC57-hTSH ⁇ (Shanghai ShineGene Molecular Biotech).
- a DNA (SEQ ID NO: 48) containing the 3 'untranslated region (WAP3'UTR) of rabbit whey acidic protein (WAP) was synthesized and incorporated into pUC57 to obtain pUC57-WAP3'UTR (Shanghai ShineGene Molecular Biotech).
- PUC57- including WAP3'UTR A DNA fragment having the TSH ⁇ gene and WAP3′UTR downstream thereof was amplified by PCR using WAP3′UTR as a template and megaprimer TSH ⁇ and primer WAP3 ′ (SEQ ID NO: 53) (hTSH ⁇ -WAP3′UTR).
- a DNA fragment having TSH ⁇ gene and WAP3'UTR downstream was amplified by PCR using pUC57-WAP3'UTR containing WAP3'UTR as a template and megaprimer TSH ⁇ and primer WAP3 '(hTSH ⁇ -WAP3 'UTR).
- Each obtained DNA fragment was digested with restriction enzymes (MluI and NotI) and inserted between the MluI and NotI sites of pCI-neo, and pCI-neo (hTSH ⁇ -WAP3'UTR) and pCI-neo (hTSH ⁇ -WAP3, respectively) 'UTR) (FIGS. 10-1 and 10-2).
- restriction enzymes MluI and NotI
- pCI-neo (hTSH ⁇ )
- pCI-neo (hTSH ⁇ -WAP3′UTR)
- pCI-neo (hTSH ⁇ -WAP3′UTR)
- MluI and NotI restriction enzymes
- a DNA fragment containing the hTSH ⁇ gene and WAP3′UTR was excised. This DNA fragment was inserted between the MluI and NotI sites of pE-mIRES-GS-mNeo to obtain pE-mIRES-GS-mNeo (hTSH ⁇ ) (FIG. 11).
- pCI-neo (hTSH ⁇ )
- pCI-neo (hTSH ⁇ -WAP3′UTR)
- MluI and NotI restriction enzymes
- a DNA fragment containing the hTSH ⁇ gene and WAP3′UTR was excised. This DNA fragment was inserted between the MluI and NotI sites of pBlue-EF1 / mIRES-mNeo to obtain pBlue-EF1 / mIRES-mNeo (hTSH ⁇ ) (FIG. 12).
- This DNA fragment was inserted between the ClaI and BamHI sites of pE-mIRES-GS-mNeo (hTSH ⁇ ), and this was inserted into the human thyroid stimulating hormone (hTSH) expression vector pE-mIRES-GS-mNeo-dual (hTSH). (FIG. 13).
- the obtained DNA fragment was digested with a restriction enzyme (BbsI) and inserted between the BsmBI sites of pE-mIRES-GS-mNeo-dual (hTSH), which was then inserted into the human thyroid stimulating hormone (hTSH) expression vector pE- It was set as mIRES-GS-mNeo-dual + mDHFR31 (hTSH) (FIG. 14).
- BbsI restriction enzyme
- hTSH human thyroid stimulating hormone
- This DNA is digested with restriction enzymes (MluI and NotI), and the resulting DNA fragment is inserted between the MluI and NotI sites of pE-mIRES-GS-mNeo, which is inserted into pE-mIRES-GS-mNeo (RTX-H (FIG. 15).
- This DNA is digested with restriction enzymes (MluI and NotI), and the resulting DNA fragment is inserted between the MluI and NotI sites of pBlue-EF1 / mIRES-mNeo, which is inserted into pBlue-EF1 / mIRES-mNeo (RTX-L ) (FIG. 16).
- pE-mIRES-GS-mNeo-dual (RTX) pBlue-EF1 / mIRES-mNeo (RTX-L) is digested with restriction enzymes (ClaI and BglII), and the resulting DNA fragment is inserted between the ClaI and BamHI sites of pE-mIRES-GS-mNeo (RTX-H) This was designated pE-mIRES-GS-mNeo-dual (RTX) as a rituximab expression vector (FIG. 17).
- pBlue-EF1 / SVpA (RTX-L) is digested with restriction enzymes (ClaI and BglII), and the resulting DNA fragment is inserted between the ClaI and BamHI sites of pE-mIRES-GS-mNeo (RTX-H). This was designated as pE-mIRES-GS-dual (RTX) (FIG. 18-2).
- SV40 early promoter (SV40 enhancer / promoter, mutant DHFR (F31W) by PCR using pE-mDHFR31 as a template and primer dE-BbsI-F (SEQ ID NO: 58) and primer SynpA-BbsI-R (SEQ ID NO: 59) And a DNA fragment containing a synthetic polyadenylation signal, which was digested with a restriction enzyme (BbsI) and inserted between the BsmBI sites of pE-mIRES-GS-dual (RTX), which was expressed by rituximab
- the vector for use was pE-mIRES-GS-dual + mDHFR31 (RTX) (FIG. 18-3).
- CHO-K1 cells derived from Chinese hamster ovary were electroporated (Invitrogen), pE-mIRES-GS-mNeo-dual (hTSH), pE-mIRES-GS-mNeo-dual + mDHFR31 (hTSH) , PE-mIRES-GS-mNeo-dual (RTX) and pE-mIRES-GS-dual + mDHFR31 (RTX), respectively.
- the transformed cells were subjected to selective culture using a selective medium to obtain hTSH-expressing cells and RTX-expressing cells.
- pE-mIRES-GS-mNeo-dual + mDHFR31 TSH
- pE-mIRES-GS-dual + mDHFR31 RTX
- CD Opti CHO medium Invitrogen
- WAKO methotrexate
- WAKO methotrexate
- the medium was replaced with CD Opti CHO medium (Invitrogen) containing methionine sulphoximine (SIGMA), and cultured while gradually increasing the concentration of methionine sulphoximine contained in the medium.
- the cells were cultured in a medium containing methionine sulfoximine at a concentration of 300 ⁇ M, and cells resistant to methionine sulfoximine were selectively grown.
- the cells obtained here were used as bulk cells.
- the bulk cells obtained by the above selective culture were seeded in a 96-well plate at 1 to 3 cells / well, and cultured for about 2 weeks until colonies were formed in CD-Opti-CHO medium (Invitrogen).
- the concentration of hTSH and RTX in the culture supernatant was measured to obtain highly expressed clonal cells.
- methionine sulfoximine was added to the medium at a concentration of 300 ⁇ M.
- the clonal cells with high expression thus obtained were subjected to the following experiment as hTSH-expressing cells and RTX-expressing cells.
- mouse anti-hTSH-beta monoclonal antibody (Leinco Technologies) solution was added to a 96-well plate and allowed to stand at room temperature for 1 hour to immobilize the antibody on the plate. Discard the solution and wash the plate 3 times with TBS-T solution (Tris: 0.05M, NaCl: 0.138 M, KCl: 0.0027M, 0.05% Tween20, pH 8.0), then plate with 1% BSA / TBS-T solution. 200 ⁇ L each was added to the plate and allowed to stand at room temperature for 1 hour to block the plate.
- TBS-T solution Tris: 0.05M, NaCl: 0.138 M, KCl: 0.0027M, 0.05% Tween20, pH 8.0
- the plate was washed 3 times with TBS-T solution, and 100 ⁇ L of the sampled culture supernatant was added to the plate as a specimen and allowed to stand at room temperature for 1 hour. At this time, the culture supernatant was diluted with TBS-T solution as necessary. Also, hTSH (NIBSC) with TBS-T solution 0.244-62.5 A solution diluted to a concentration of ng / mL was added as a standard solution to the plate in the same manner as the sample, and allowed to stand.
- TBS-T solution 100 ⁇ L of the sampled culture supernatant was added to the plate as a specimen and allowed to stand at room temperature for 1 hour. At this time, the culture supernatant was diluted with TBS-T solution as necessary.
- hTSH (NIBSC) with TBS-T solution 0.244-62.5 A solution diluted to a concentration of ng / mL was added as a standard solution to the plate in the same manner as the sample, and allowed to stand.
- the plate was washed 3 times with TBS-T solution, and 100 ⁇ L of HRP-labeled mouse anti-hTSH-alpha monoclonal antibody (Leinco Technologies) was added to the plate as a secondary antibody and allowed to stand at room temperature for 1 hour. .
- the plate was washed 3 times with TBS-T solution, HRP substrate (WAKO) was added, and the plate was allowed to stand at room temperature for 15 minutes, and then the reaction was stopped by adding sulfuric acid.
- the absorbance at 490 nm was measured with a microwell plate reader, and the measured value of the sample was interpolated into the calibration curve obtained from the measured value of the absorbance of the standard solution to obtain the concentration of hTSH contained in the sample.
- Rituximab expression was measured using the Human IgG ELISA Quantitation Set (Bethyl Laboratories). 100 ⁇ L each of goat anti-hIgG antibody (Bethyl Laboratories) solution was added to a 96-well plate and allowed to stand at room temperature for 1 hour to immobilize the antibody on the plate. After discarding the solution and washing the plate 3 times with TBS-T solution, 200 ⁇ L of 1% BSA / TBS-T solution was added to the plate and allowed to stand at room temperature for 1 hour to block the plate.
- the plate was washed 3 times with TBS-T solution, and 100 ⁇ L of the sampled culture supernatant was added to the plate as a specimen and allowed to stand at room temperature for 1 hour. At this time, the culture supernatant was diluted with TBS-T solution as necessary.
- rituximab (Chugai Pharmaceutical Co., Ltd.) diluted with TBS-T solution to a concentration of 3.9 to 500 ng / mL was added as a standard solution to the plate and allowed to stand.
- the plate was washed three times with TBS-T solution, 100 ⁇ L of HRP-labeled goat anti-hIgG antibody (Bethyl Laboratories) was added to the plate as a secondary antibody, and allowed to stand at room temperature for 1 hour.
- the plate was washed 3 times with TBS-T solution, HRP substrate (WAKO) was added, and the plate was allowed to stand at room temperature for 15 minutes, and then the reaction was stopped by adding sulfuric acid. 490 with a microwell plate reader The absorbance at nm was measured, and the measured value of the sample was interpolated into a calibration curve obtained from the measured value of the absorbance of the standard solution, thereby obtaining the concentration of rituximab contained in the sample.
- pE-mIRES-GS which is an expression vector for hTSH containing the elongation factor 1 promoter (EF-1p), hTSH gene, mutant internal ribosome binding site (mIRES), and GS gene as a first selection marker in this order.
- hTSH pE-mIRES-GS-mNeo-dual + mDHFR31
- hTSH pE-mIRES-GS-mNeo-dual + mDHFR31
- RTX -mNeo-dual
- RTX pE-mIRES-GS-dual + mDHFR31
- the present invention can express a recombinant protein at a high level using mammalian cells, it can be used for reducing the production cost of a product containing the recombinant protein, for example, a pharmaceutical product containing the recombinant protein. can do.
- LacZ promoter 2 mPGK promoter 3 Internal ribosome binding site derived from wild-type mouse encephalomyocarditis virus containing the nucleotide sequence shown in SEQ ID NO: 1 (EMCV-IRES) 3a Internal ribosome binding site derived from a mutant mouse encephalomyocarditis virus containing the nucleotide sequence shown in SEQ ID NO: 4 (EMCV-mIRES) 4 Polyadenylation region of mPGK (mPGKpA) 5 Base sequence containing EF-1p and first intron 6 SV40 late polypolyadenylation region 7 Region containing SV40 early promoter (SV40 enhancer / promoter) 8 Synthetic polypolyadenylation region 9 Region containing cytomegalovirus promoter 10 Glutamine synthase gene
- Sequence number 2 Modified murine encephalomyocarditis virus
- n is a, c, g, or t
- Sequence number 3 Modified murine encephalomyocarditis virus
- n is a, c, g, or t
- Sequence number 4 Modified murine encephalomyocarditis virus
- Sequence number 6 Modified murine encephalomyocarditis virus
- Sequence number 7 Modified murine encephalomyocarditis virus
- n is a, c, g, or t
- Sequence number 8 Modified murine encephalomyocarditis virus
- n is a, c, g, or t
- Sequence number 11 Modified Mus musculus DHFR
- Sequence number 12 Synthetic Construct Sequence number 13: Primer Hyg-Sfi5 ', synthetic sequence Sequence number 14: Primer Hyg-BstX3 ', synthetic sequence Sequence number
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Gastroenterology & Hepatology (AREA)
- Toxicology (AREA)
- Immunology (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
1.蛋白質を発現させるための発現ベクターであって,遺伝子発現制御部位(A),並びに,その下流に該蛋白質をコードする遺伝子,更に下流に内部リボソーム結合部位,及び更に下流にグルタミン合成酵素をコードする遺伝子を含み,且つ,該遺伝子発現制御部位(A)の又は該遺伝子発現制御部位(A)とは別の遺伝子発現制御部位(B)の下流にジヒドロ葉酸レダクターゼ遺伝子を更に含んでなる,発現ベクター。
2.該遺伝子発現制御部位(A)及び/又は該遺伝子発現制御部位(B)が,サイトメガロウイルス由来のプロモーター,SV40初期プロモーター,伸長因子1プロモーターからなる群から選択されるものである,上記1の発現ベクター。
3.該内部リボソーム結合部位が,ピコルナウイルス科のウイルス,口蹄疫ウイルス,A型肝炎ウイルス,C型肝炎ウイルス,コロナウイルス,ウシ腸内ウイルス,サイラーのネズミ脳脊髄炎ウイルス,コクサッキーB型ウイルス,ヒト免疫グロブリン重鎖結合蛋白質遺伝子,ショウジョウバエアンテナペディア遺伝子,ショウジョウバエウルトラビトラックス遺伝子からなる群から選択されるウイルス又は遺伝子の5'非翻訳領域に由来するものである,上記1又は2の発現ベクター。
4.該内部リボソーム結合部位が,ピコルナウイルス科のウイルスの5'非翻訳領域に由来するものである,上記1又は2の発現ベクター,
5.該内部リボソーム結合部位が,マウス脳心筋炎ウイルスの5'非翻訳領域に由来するものである,上記1又は2の発現ベクター。
6.該内部リボソーム結合部位が,野生型の内部リボソーム結合部位の塩基配列に,1又は2以上の変異を加えたものである,上記1ないし5の何れかの発現ベクター,
7.該野生型の内部リボソーム結合部位の塩基配列が複数の開始コドンを含み,該変異により,それら複数の開始コドンのうち一部が破壊されたものである,上記6の発現ベクター。
8.該内部リボソーム結合部位が,配列番号1の塩基配列を含むものである,上記5の発現ベクター。
9.該内部リボソーム結合部位が,配列番号2の塩基配列を含むものである,上記5の発現ベクター。
10.該内部リボソーム結合部位が,配列番号3の塩基配列を含むものである,上記5の発現ベクター。
11.該内部リボソーム結合部位が,配列番号4の塩基配列を含むものである,上記5の発現ベクター。
12.該内部リボソーム結合部位が,配列番号5の塩基配列を含むものである,上記5の発現ベクター。
13.該内部リボソーム結合部位が,配列番号6の塩基配列を含むものである,上記5の発現ベクター。
14.該蛋白質をコードする該遺伝子と該内部リボソーム結合部位との間の領域又はグルタミン合成酵素をコードする該遺伝子の下流の領域において,該内部リボソーム結合部位とは別の内部リボソーム結合部位及びその下流に薬剤耐性遺伝子を更に含んでなる,上記1ないし13の何れかの発現ベクター。
15.該遺伝子発現制御部位(A)及び遺伝子発現制御部位(B)とは別に,更なる遺伝子発現制御部位(C)及びその下流に薬剤耐性遺伝子を更に含んでなる,上記1ないし13の何れかの発現ベクター。
16.該薬剤耐性遺伝子が,ピューロマイシン又はネオマイシン耐性遺伝子である,上記14又は15の発現ベクター。
17.該蛋白質をコードする該遺伝子が,ヒト由来の遺伝子である上記1ないし16の何れかの発現ベクター。
18.ヒト由来の該遺伝子が,リソソーム酵素,組織プラスミノーゲンアクチベーター(t-PA),血液凝固因子,エリスロポエチン,インターフェロン,トロンボモジュリン,甲状腺刺激ホルモン(TSH),卵胞刺激ホルモン,顆粒球コロニー刺激因子(G-CSF),及び抗体をコードする遺伝子からなる群から選択されるものである,上記17の発現ベクター。
19.ヒト由来の該遺伝子が,リソソーム酵素をコードする遺伝子である,上記17の発現ベクター。
20.該リソソーム酵素が,α-ガラクトシダーゼA,イズロン酸2-スルファターゼ,グルコセレブロシダーゼ,ガルスルファーゼ,α-L-イズロニダーゼ,及び酸性α-グルコシダーゼからなる群から選択されるものである,上記19の発現ベクター。
21.ヒト由来の該遺伝子が,エリスロポエチンをコードする遺伝子である上記17の発現ベクター。
22.上記1ないし21の何れかの発現ベクターで形質転換された哺乳動物細胞。
23.該哺乳動物細胞が,内因性のジヒドロ葉酸レダクターゼ遺伝子を欠損したものである,上記22の細胞。
24.該哺乳動物細胞が,CHO細胞である,上記22又は23の細胞。
25.(a)上記1ないし21の何れかの発現ベクターを,哺乳動物細胞に導入するステップ,
(b)該発現ベクターが導入された哺乳動物細胞を,ジヒドロ葉酸レダクターゼ阻害剤の存在下で選択培養するステップ,及び
(c)該選択培養により選択された細胞を,グルタミン合成酵素阻害剤の存在下で更に選択培養するステップ
を含む,該蛋白質をコードする遺伝子を発現する形質転換細胞の製造方法。
26.(a)上記14ないし16の何れかの発現ベクターを,哺乳動物細胞に導入するステップ,
(b)該発現ベクターが導入された哺乳動物細胞を,ジヒドロ葉酸レダクターゼ阻害剤の存在下で選択培養するステップ,及び
(c)該選択培養により選択された細胞を,グルタミン合成酵素阻害剤の存在下で更に選択培養するステップ
を含む,該蛋白質をコードする遺伝子を発現する形質転換細胞の製造方法であって,該ステップ(a)と該ステップ(b)の間,又は該ステップ(b)と該ステップ(c)の間に,該発現ベクターが導入された哺乳動物細胞を,該薬剤耐性遺伝子に対応する薬剤の存在下で選択培養するステップを更に含む,製造方法。
pEF/myc/nucベクター(インビトロジェン社)を,KpnIとNcoIで消化し,EF-1プロモーター及びその第一イントロンを含む領域を切り出し,これをT4DNAポリメラーゼで平滑末端化処理した。別に,pCI-neo(インビトロジェン社)を,BglII及びEcoRIで消化して,CMVのエンハンサー/プロモーター及びイントロンを含む領域を切除した後に,T4 DNA ポリメラーゼで平滑末端化処理した。これに,上記のEF-1αプロモーター及びその第一イントロンを含む領域(平滑末端化処理後のもの)を挿入して,pE-neoベクターを構築した(図1-1及び図1-2)。
発現ベクターpPGKIH(Miyahara M. et.al., J. Biol. Chem. 275,613-618(2000) )を制限酵素(XhoI及びBamHI)で消化し,マウス脳心筋炎ウイルス(EMCV)に由来する内部リボソーム結合部位(IRES),ハイグロマイシン耐性遺伝子(Hygr遺伝子)及びマウスホスホグリセリン酸キナーゼ(mPGK)のポリアデニル化領域(mPGKpA)を含む塩基配列IRES-Hygr-mPGKpA(配列番号15。5'末端から,塩基1~6からなる領域が「XhoIサイト」,塩基120~715及びこれに続く塩基716~718(atg)からなる領域が「マウス脳心筋炎ウイルスの5'非翻訳領域に由来する内部リボソーム結合部位を含む塩基配列」,該塩基716~718(atg)を含む塩基716~1741からなる領域が「ハイグロマイシン耐性遺伝子をコードする塩基配列」,塩基1747~2210からなる領域が「マウスホスホグリセリン酸キナーゼ(mPGK)のポリアデニル化領域を含む塩基配列」,及び3'末端の6個の塩基(塩基2211~2216)からなる領域が「BamHIサイト」である。)よりなるDNA断片を切り出した(なお,Hygrr遺伝子に対応するアミノ酸配列は,配列番号16で示されたものである。)。このDNA断片をpBluescript SK(-) (Stratagene社)のXhoI及びBamHIサイト間に挿入し,これをpBSK(IRES-Hygr-mPGKpA)とした(図3-1)。
(IRES-Hygr-mPGKpA) のXhoI及びHindIIIサイト間に挿入し,これをpBSK (NotI-IRES-Hygr-mPGKpA) とした(図3-2)。pBSK(NotI-IRES-Hygr-mPGKpA)を制限酵素(NotI及びBamHI)で消化し,pE-hygrベクターのNotI及びBamHIサイト間に挿入し,これをプラスミドpE-IRES-Hygrとした(図3-3)。
M. et.al., J. Biol. Chem. 275,613-618(2000))を鋳型とし,プライマーpuro5'(配列番号24)及びプライマーpuro3'(配列番号25)を用いて,PCRによりピューロマイシン耐性遺伝子(puror遺伝子)を含む塩基配列(配列番号26。5'末端から,塩基2~7からなる領域が「AflIIサイト」,これに続く塩基8~607からなる領域が「ピューロマイシン耐性遺伝子(puror遺伝子)をコードする塩基配列」,及び,その次の塩基608~619からなる領域が「BstXIサイト」である。)よりなるDNA断片を増幅させた(なお,puror遺伝子に対応するアミノ酸配列は,配列番号27で示されたものである。)。このDNA断片を制限酵素(AflII及びBstXI)で消化し,発現ベクターpE-neoのAflII及びBstXIサイト間に挿入し,これをpE-puroとした(図3-7)。
発現ベクターpE-IRES-GS-puroを鋳型とし,プライマーmIRES-GS5'(配列番号30)及びプライマーmIRES-GS3'(配列番号31)を用いて,PCRにより,EMCVのIRESからGSにかけての領域を増幅させ,EMCVのIRESの5'側から2番目に位置する開始コドン(ATG)に変異を加えて破壊したDNA断片を増幅させた。発現ベクターpE-IRES-GS-puroを鋳型として,このDNA断片と上記のプライマーIRES5'を用いて,PCRによりIRESからGSにかけての上記領域を含むDNA断片を増幅させた。このDNA断片を制限酵素(NotI及びPstI)で消化し,切り出されたDNA断片を,発現ベクターpE-IRES-GS-puroのNotI及びPstIサイト間に挿入し,これをpE-mIRES-GS-puroとした(図4)。
発現ベクターpE-neoを鋳型とし,プライマーSV40polyA5'-2(配列番号32)及びプライマーSV40polyA3'-2(配列番号33)を用いて,PCRによりSV40 後期polyA領域を含むDNA断片を増幅させた。このDNA断片を制限酵素(XhoI及びBamHI)で消化し,pE-mIRES-GS-puroのXhoI及びBamHIサイト間に挿入し,これをpE-mIRES-GSとした(図5)。
〔pE-mIRES-GS-mNeoの構築〕
pCI-neo(インビトロジェン社)を鋳型とし,プライマーmNeoA5'(配列番号34)及びプライマーmNeoA3'(配列番号35)を用いて,PCRによりこのプラスミド全長を増幅させ,セルフライゲーションによりE182D変異型ネオマイシン耐性遺伝子(mNeor遺伝子)を含むプラスミドpCI-mNeoを構築した。pCI-mNeoベクターを鋳型としてプライマーmNeoB5'(配列番号36)及びプライマーmNeoB3'(配列番号37)を用いてPCRによりmNeor遺伝子を含むDNA断片を増幅させた。このDNA断片を制限酵素(EagI)で消化し,pUC57-IE1(Gene
Script社)のNotIに挿入し,これをpUC57-IE1-mNeoとした。pCI-neoを鋳型とし,プライマーmNeoC5'(配列番号38)及びプライマーmNeoC3'(配列番号39)を用いて,PCRによりネオマイシン耐性遺伝子(neor遺伝子)の3'領域及び合成ポリAシグナルを含む領域を増幅させ,これをメガプライマー(neo-polyA)とした。次に,pUC57-IE1-mNeoを鋳型とし,,プライマーmNeoD5'(配列番号40)及びメガプライマー(neo-polyA)を用いて,PCRによりmNeor遺伝子及び合成ポリAシグナルを含む領域を増幅させた。得られたDNA断片を制限酵素(AflII及びBamHI)で消化し,pE-mIRES-GS-puroのAflII及びBamHIサイト間に挿入し,これをpE-mIRES-GS-mNeoとした(図6)。
pE-mIRES-GS-mNeoを制限酵素(EcoRI)で消化し,伸長因子1プロモーター(EF-1p),変異型内部リボソーム結合部位(mIRES)及びグルタミン合成酵素(GS)の一部を含むDNA断片を切り出した。このDNA断片をpBluescript
SK(+) (Stratagene社)のEcoRIサイトに挿入し,これをpBlue-EF1/mIRESとした(図7)。
発現ベクターpE-mIRES-GS-mNeoを鋳型とし,プライマーSVpA-Not-F(配列番号43)及びプライマーSVpA-BstXI-R(配列番号44)を用いて,PCRによりSV40 後期polyA領域を含むDNA断片を増幅させた。このDNA断片を制限酵素(NotI及びBstXI)で消化し,pBlue-EF1/mIRESのNotI及びBstXIサイト間に挿入し,これをpBlue-EF1/SVpAとした(図8)。
マウスの野生型DHFRのN末端から31位のフェニルアラニンがトリプトファンに置換されたものである配列番号11の変異型DHFR(F31W)遺伝子(mDHFR31)を含むDNA(配列番号45)を合成した。このDNAをpGEM-T easy vector(Promega社)の3'-T overhangs箇所に挿入し,これをpGEM(mDHFR31)とした。pGEM(mDHFR31)を制限酵素(AflII及びBstXI)で消化し,変異型DHFR(F31W)を含むDNA断片を切り出し,このDNA断片をpE-neoのAflII及びBstXIサイト間に挿入し,これをpE-mDHFR31とした(図9)。
ヒト甲状腺刺激ホルモンのα鎖(hTSHα)遺伝子を含むDNA(配列番号46。5'末端から,塩基1~6からなる領域が「MluIサイト」,塩基14~364からなる領域が「hTSHα鎖をコードする塩基配列」,及び,これに続く塩基365~372からなる領域が「NotIサイト」である。),及びヒト甲状腺刺激ホルモンのβ鎖(hTSHβ)遺伝子を含むDNA(配列番号47。5'末端から,塩基1~6からなる領域が「MluIサイト」,塩基14~433からなる領域が「TSHβ鎖をコードする塩基配列」,及び,これに続く塩基434~441からなる領域が「NotIサイト」である。)を合成し,それぞれpUC57に組み込んで,pUC57-hTSHα,pUC57-hTSHβとした(Shanghai ShineGene Molecular
Biotech社)。また,ウサギ乳清酸性タンパク質(WAP)の3'側非翻訳領域(WAP3'UTR)を含むDNA(配列番号48)を合成し,pUC57に組み込んで,pUC57-WAP3'UTRとした(Shanghai ShineGene Molecular Biotech社)。
WAP3'UTRを鋳型とし,メガプライマーTSHαとプライマーWAP3'(配列番号53)を用いて,PCRによりTSHα遺伝子とその下流にWAP3'UTRを有するDNA断片を増幅させた(hTSHα-WAP3'UTR)。また,WAP3'UTRを含むpUC57- WAP3'UTRを鋳型とし,メガプライマーTSHβとプライマーWAP3'を用いて,PCRによりTSHβ遺伝子とその下流にWAP3'UTRを有するDNA断片を増幅させた(hTSHβ-WAP3'UTR)。得られた各DNA断片を制限酵素(MluI及びNotI)で消化し,pCI-neoのMluI及びNotIサイト間に挿入し,それぞれpCI-neo(hTSHα-WAP3'UTR)及びpCI-neo(hTSHβ-WAP3'UTR)とした(図10-1及び図10-2)。
pCI-neo(hTSHβ-WAP3'UTR)を制限酵素(MluI及びNotI)で消化し,hTSHβ遺伝子及びWAP3'UTRを含むDNA断片を切り出した。このDNA断片をpE-mIRES-GS-mNeoのMluI及びNotIサイト間に挿入し,これをpE-mIRES-GS-mNeo(hTSHβ)とした(図11)。
pCI-neo(hTSHα-WAP3'UTR)を制限酵素(MluI及びNotI)で消化し,hTSHα遺伝子及びWAP3'UTRを含むDNA断片を切り出した。このDNA断片をpBlue-EF1/mIRES-mNeoのMluI及びNotIサイト間に挿入し,これをpBlue-EF1/mIRES-mNeo(hTSHα)とした(図12)。
pBlue-EF1/mIRES-mNeo(hTSHα)を制限酵素(ClaI及びBglII)で消化し,伸長因子1プロモーター(EF-1p),hTSHα遺伝子,mIRES及び変異型ネオマイシン耐性遺伝子(mNeor遺伝子)を含むDNA断片を切り出した。このDNA断片をpE-mIRES-GS-mNeo(hTSHβ)のClaI及びBamHIサイト間に挿入し,これをヒト甲状腺刺激ホルモン(hTSH)発現用ベクターのpE-mIRES-GS-mNeo-dual(hTSH)とした(図13)。
pE-mDHFR31を鋳型として,プライマーdE-BbsI-F(配列番号54)及びプライマーSynpA-BbsI-R(配列番号55)を用いてPCRによりSV40初期プロモーター(SV40エンハンサー/プロモーター),変異型DHFR(F31W)及び合成ポリアデニレーションシグナル(合成ポリアデニル化領域)を含むDNA断片を増幅させた。得られたDNA断片を制限酵素(BbsI)で消化し,pE-mIRES-GS-mNeo-dual(hTSH)のBsmBIサイト間に挿入し,これをヒト甲状腺刺激ホルモン(hTSH)発現用ベクターのpE-mIRES-GS-mNeo-dual+mDHFR31(hTSH)とした(図14)。
リツキシマブ重鎖遺伝子を含むDNA(配列番号56。5'末端から,塩基1~6からなる領域が「MluIサイト」,塩基18~1430からなる領域が「リツキシマブ重鎖をコードする塩基配列」,及び,これに続く塩基1431~1438からなる領域が「NotIサイト」である。)を合成した。このDNAを制限酵素(MluI及びNotI)で消化し,得られたDNA断片をpE-mIRES-GS-mNeoのMluI及びNotIサイト間に挿入し,これをpE-mIRES-GS-mNeo(RTX-H)とした(図15)。
リツキシマブ軽鎖遺伝子を含むDNA(配列番号57。5'末端から,塩基1~6からなる領域が「MluIサイト」,塩基18~725からなる領域が「リツキシマブ軽鎖をコードする塩基配列」,及び,これに続く塩基726~733からなる領域が「NotIサイト」である。)を合成した。このDNAを制限酵素(MluI及びNotI)で消化し,得られたDNA断片をpBlue-EF1/mIRES-mNeoのMluI及びNotIサイト間に挿入し,これをpBlue-EF1/mIRES-mNeo(RTX-L)とした(図16)。
pBlue-EF1/mIRES-mNeo(RTX-L)を制限酵素(ClaI及びBglII)で消化し,得られたDNA断片をpE-mIRES-GS-mNeo(RTX-H)のClaI及びBamHIサイト間に挿入し,これをリツキシマブ発現用ベクターのpE-mIRES-GS-mNeo-dual(RTX)とした(図17)。
配列番号57のリツキシマブ軽鎖遺伝子を含むDNAを含む合成DNAを,制限酵素(MluI及びNotI)で消化し,得られたDNA断片をpBlue-EF1/SVpAのMluI及びNotIサイト間に挿入し,これをpBlue-EF1/SVpA(RTX-L)とした(図18-1)。
pE-mDHFR31を鋳型として,プライマーdE-BbsI-F(配列番号58)及びプライマーSynpA-BbsI-R(配列番号59)を用いてPCRによりSV40初期プロモーター(SV40エンハンサー/プロモーター,変異型DHFR(F31W)及び合成ポリアデニレーションシグナルを含むDNA断片を増幅させた。このDNA断片を制限酵素(BbsI)で消化し,pE-mIRES-GS-dual(RTX)のBsmBIサイト間に挿入し,これをリツキシマブ発現用ベクターのpE-mIRES-GS-dual+mDHFR31(RTX)とした(図18-3)。
チャイニーズハムスターの卵巣に由来する細胞のCHO-K1細胞をエレクトロポレーション(Invitrogen社)により,pE-mIRES-GS-mNeo-dual(hTSH),pE-mIRES-GS-mNeo-dual+mDHFR31(hTSH),pE-mIRES-GS-mNeo-dual(RTX)及びpE-mIRES-GS-dual+mDHFR31(RTX)でそれぞれ形質転換した。形質転換後の細胞を,選択培地を用いて選択培養を行い,hTSH発現細胞及びRTX発現細胞を得た。
で形質転換させた細胞の選択培養においては,まず細胞を,メトトレキセート(WAKO社)を含むCD Opti CHO培地(Invitrogen社)で,培地中に含まれるメトトレキセートの濃度を段階的に上昇させながら培養し,最終的にメトトレキセートを0.5μMで含む培地で培養し,メトトレキセートに耐性を示す細胞を選択的に増殖させた。次いで,培地をメチオニンスルホキシミン(SIGMA社)を含むCD Opti CHO培地(Invitrogen社)に置き換えて,培地中に含まれるメチオニンスルホキシミンの濃度を段階的に上昇させながら培養し,最終的にメチオニンスルホキシミンを300μMの濃度で含む培地で培養し,メチオニンスルホキシミンに耐性を示す細胞を選択的に増殖させた。ここで得られた細胞をバルク細胞とした。
選択培養して得たhTSH発現細胞を2×105
個/mLの細胞密度で300μMのメチオニンスルホキシミンを含有する5mLのCD Opti CHO培地で,37℃,5%
CO2存在下で10日間培養した。培養,3,7及び10日目に培養上清をサンプリングした。
ng/mLの濃度に希釈したものを標準溶液として,検体と同様にプレートに加えて静置した。液を捨て,プレートをTBS-T液で3回洗浄後,HRP標識したマウス抗hTSH-alphaモノクロナール抗体(Leinco Technologies社)を2次抗体としてプレートに100μLずつ加え,室温で1時間静置した。プレートをTBS-T液で3回洗浄後,HRP基質(WAKO社)を加え,室温で15分間静置した後,硫酸を加えて反応を停止した。マイクロウエルプレートリーダーで490nmにおける吸光度を測定して,標準溶液の吸光度の測定値から得られた検量線に,検体の測定値を内挿し,検体に含まれるhTSHの濃度を求めた。
選択培養して得たRTX発現細胞を2×105
個/mLの細胞密度で300μMのメチオニンスルホキシミンを含有する5mLのCD Opti CHO培地で,37℃,5%
CO2存在下で10日間培養した。培養,3,7及び10日目に培養上清をサンプリングした。
Laboratories社)を用いて行った。ヤギ抗hIgG抗体(Bethyl Laboratories社)溶液を96ウェルプレートに100μLずつ加え,室温で1時間静置して抗体をプレートに固定させた。液を捨て,プレートをTBS-T液で3回洗浄した後,1%BSA/TBS-T液をプレートに200μLずつ加え,室温で1時間静置してプレートをブロッキングした。液を捨て,プレートをTBS-T液で3回洗浄後,サンプリングした培養上清を検体としてプレートに100μLずつ加え,室温で1時間静置した。このとき,必要に応じて,培養上清をTBS-T液で希釈した。また,リツキシマブ(中外製薬社)をTBS-T液で3.9~500 ng/mLの濃度に希釈したものを標準溶液として,検体と同様にプレートに加えて静置した。液を捨て,プレートをTBS-T液で3回洗浄後,HRP標識したヤギ抗hIgG抗体(Bethyl Laboratories社)を2次抗体としてプレートに100μLずつ加え,室温で1時間静置した。プレートをTBS-T液で3回洗浄後,HRP基質(WAKO社)を加え,室温で15分間静置した後,硫酸を加えて反応を停止した。マイクロウエルプレートリーダーで490
nmにおける吸光度を測定して,標準溶液の吸光度の測定値から得られた検量線に,検体の測定値を内挿し,検体に含まれるリツキシマブの濃度を求めた。
ヒト甲状腺刺激ホルモン(hTSH)発現用ベクターのpE-mIRES-GS-mNeo-dual(hTSH)で形質転換させたCHO細胞では,培養開始後10日目の培地中のhTSHの濃度は,バルク細胞では22.0μg/mL,クローン細胞では29.2μg/mLであった。一方,第2の選択マーカーとしてDHFR遺伝子を組み込んだhTSH発現用ベクターであるpE-mIRES-GS-mNeo-dual+mDHFR31(hTSH)で形質転換させたCHO細胞では,培養開始後10日目の培地中のhTSHの濃度は,バルク細胞では35.3μg/mL,クローン細胞では85.2μg/mLであった(表1)。
すなわち,伸長因子1プロモーター(EF-1p),hTSH遺伝子,変異型内部リボソーム結合部位(mIRES),及び第1の選択マーカーとしてGS遺伝子をこの順で含むhTSH発現用ベクターであるpE-mIRES-GS-mNeo-dual(hTSH)と比較して,更に第2の選択マーカーとしてDHFR遺伝子を組み込んだhTSH発現用ベクターであるpE-mIRES-GS-mNeo-dual+mDHFR31(hTSH)で形質転換させたCHO細胞では,hTSHの発現量が,バルク細胞間で約1.6倍に,クローン細胞間で約2.9倍と,飛躍的に増加した。

RTX発現用ベクターのpE-mIRES-GS-mNeo-dual(RTX)で形質転換させたCHO細胞では,培養開始後10日目の培地中のリツキシマブの濃度は,バルク細胞では188.6μg/mL,クローン細胞では318.9μg/mLであった。一方,第2の選択マーカーとしてDHFR遺伝子を組み込んだRTX発現用ベクターであるpE-mIRES-GS-dual+mDHFR31(RTX)で形質転換させたCHO細胞では,培養開始後10日目の培地中のリツキシマブの濃度は,バルク細胞では174.1μg/mL,クローン細胞では401.5μg/mLであった(表2)。
すなわち,伸長因子1プロモーター(EF-1p),リツキシマブ遺伝子,変異型内部リボソーム結合部位(mIRES),及び第1の選択マーカーとしてGS遺伝子をこの順で含むRTX発現用ベクターであるpE-mIRES-GS-mNeo-dual(RTX)と比較して,更に第2の選択マーカーとしてDHFR遺伝子を組み込んだRTX発現用ベクターであるpE-mIRES-GS-dual+mDHFR31(RTX)で形質転換させた細胞では,RTXの発現量が,バルク細胞間ではほとんど同レベルであるものの,クローン細胞間では約1.26倍に増加した。

これらの結果は,遺伝子発現制御部位の下流に,外来遺伝子,変異型内部リボソーム結合部位,及び第1の選択マーカーとしてグルタミン合成酵素をこの順で組み込み,更に,第2の選択マーカーとしてジヒドロ葉酸レダクターゼ遺伝子を組み込んだ発現ベクターが,外来遺伝子を高レベルで発現させることができる発現ベクターとして機能することを示すものである。
2 mPGKプロモーター
3 配列番号1に示す塩基配列を含む野生型マウス脳心筋炎ウイルス由来の内部リボソーム結合部位(EMCV-IRES)
3a 配列番号4に示す塩基配列を含む変異型マウス脳心筋炎ウイルス由来の内部リボソーム結合部位(EMCV-mIRES)
4 mPGKのポリアデニル化領域(mPGKpA)
5 EF-1p及び第一イントロンを含む塩基配列
6 SV40後期ポリポリアデニル化領域
7 SV40初期プロモーター(SV40エンハンサー/プロモーター)を含む領域
8 合成ポリポリアデニル化領域
9 サイトメガロウイルスプロモーターを含む領域
10 グルタミン合成酵素遺伝子
encephalomyocarditis virus; n is a, c, g, or t
配列番号3:Modified murine
encephalomyocarditis virus; n is a, c, g, or t
配列番号4:Modified murine
encephalomyocarditis virus
配列番号6:Modified murine
encephalomyocarditis virus
配列番号7:Modified murine encephalomyocarditis
virus; n is a, c, g, or t
配列番号8:Modified murine
encephalomyocarditis virus; n is a, c, g, or t
配列番号11:Modified Mus musculus DHFR
配列番号12:Synthetic Construct
配列番号13:Primer Hyg-Sfi5', synthetic
sequence
配列番号14:Primer Hyg-BstX3', synthetic sequence
配列番号15:IRES-Hygr-mPGKpA, synthetic
sequence
配列番号16:Synthetic Construct
配列番号17:Primer IRES5', synthetic
sequence
配列番号18:Primer IRES3', synthetic
sequence
配列番号19:Primer mPGKP5', synthetic
sequence
配列番号20:Primer mPGKP3', synthetic
sequence
配列番号21:mPGKp, synthetic sequence
配列番号22:Primer GS5', synthetic
sequence
配列番号23:Primer GS3', synthetic
sequence
配列番号24:Primer puro5', synthetic
sequence
配列番号25:Primer puro3', synthetic
sequence
配列番号26:Synthetic sequence containing
puromycin resistance gene
配列番号27:Synthetic Construct
配列番号28:Primer SV40polyA5', synthetic
sequence
配列番号29:Primer SV40polyA3', synthetic
sequence
配列番号30:Primer mIRES-GS5', synthetic
sequence
配列番号31:Primer mIRES-GS3', synthetic
sequence
配列番号32:Primer SV40polyA5'-2,
synthetic sequence
配列番号33:Primer SV40polyA3'-2,
synthetic sequence
配列番号34:Primer mNeoA5', synthetic
sequence
配列番号35:Primer mNeoA3', synthetic
sequence
配列番号36:Primer mNeoB5', synthetic
sequence
配列番号37:Primer mNeoB3', synthetic
sequence
配列番号38:Primer mNeoC5', synthetic
sequence
配列番号39:Primer mNeoC3', synthetic
sequence
配列番号40:Primer mNeoD5', synthetic
sequence
配列番号41:Primer SVpA-Mega-F, synthetic
sequence
配列番号42:Primer SVpA-BstXI-R, synthetic
sequence
配列番号43:Primer SVpA-Not-F, synthetic
sequence
配列番号44:Primer SVpA-BstXI-R, synthetic
sequence
配列番号45:Synthetic sequence containing
Modified DHFR(F31W)
配列番号46:Synthetic sequence containing
human TSH alpha
配列番号47:Synthetic sequence containing
human TSH beta
配列番号48:Synthetic sequence containing
WAP3'UTR
配列番号49:Primer TSHalpha5', synthetic
sequence
配列番号50:Primer TSHalpha3', synthetic
sequence
配列番号51:Primer TSHbeta5', synthetic
sequence
配列番号52:Primer TSHbeta3', synthetic
sequence
配列番号53:Primer WAP3', synthetic
sequence
配列番号54:Primer dE-BbsI-F, synthetic
sequence
配列番号55:Primer SynpA-BbsI-R, synthetic
sequence
配列番号56:Rituximab heavy chain,
synthetic sequence
配列番号57:Rituximab light chain,
synthetic sequence
配列番号58:Primer dE-BbsI-F, synthetic
sequence
配列番号59:Primer SynpA-BbsI-R, synthetic
sequence
Claims (26)
- 蛋白質を発現させるための発現ベクターであって,遺伝子発現制御部位(A),並びに,その下流に該蛋白質をコードする遺伝子,更に下流に内部リボソーム結合部位,及び更に下流にグルタミン合成酵素をコードする遺伝子を含み,且つ,該遺伝子発現制御部位(A)の又は該遺伝子発現制御部位(A)とは別の遺伝子発現制御部位(B)の下流にジヒドロ葉酸レダクターゼ遺伝子を更に含んでなる,発現ベクター。
- 該遺伝子発現制御部位(A)及び/又は該遺伝子発現制御部位(B)が,サイトメガロウイルス由来のプロモーター,SV40初期プロモーター,伸長因子1プロモーターからなる群から選択されるものである,請求項1の発現ベクター。
- 該内部リボソーム結合部位が,ピコルナウイルス科のウイルス,口蹄疫ウイルス,A型肝炎ウイルス,C型肝炎ウイルス,コロナウイルス,ウシ腸内ウイルス,サイラーのネズミ脳脊髄炎ウイルス,コクサッキーB型ウイルス,ヒト免疫グロブリン重鎖結合蛋白質遺伝子,ショウジョウバエアンテナペディア遺伝子,ショウジョウバエウルトラビトラックス遺伝子からなる群から選択されるウイルス又は遺伝子の5'非翻訳領域に由来するものである,請求項1又は2の発現ベクター。
- 該内部リボソーム結合部位が,ピコルナウイルス科のウイルスの5'非翻訳領域に由来するものである,請求項1又は2の発現ベクター,
- 該内部リボソーム結合部位が,マウス脳心筋炎ウイルスの5'非翻訳領域に由来するものである,請求項1又は2の発現ベクター。
- 該内部リボソーム結合部位が,野生型の内部リボソーム結合部位の塩基配列に,1又は2以上の変異を加えたものである,請求項1ないし5の何れかの発現ベクター,
- 該野生型の内部リボソーム結合部位の塩基配列が複数の開始コドンを含み,該変異により,それら複数の開始コドンのうち一部が破壊されたものである,請求項6の発現ベクター。
- 該内部リボソーム結合部位が,配列番号1の塩基配列を含むものである,請求項5の発現ベクター。
- 該内部リボソーム結合部位が,配列番号2の塩基配列を含むものである,請求項5の発現ベクター。
- 該内部リボソーム結合部位が,配列番号3の塩基配列を含むものである,請求項5の発現ベクター。
- 該内部リボソーム結合部位が,配列番号4の塩基配列を含むものである,請求項5の発現ベクター。
- 該内部リボソーム結合部位が,配列番号5の塩基配列を含むものである,請求項5の発現ベクター。
- 該内部リボソーム結合部位が,配列番号6の塩基配列を含むものである,請求項5の発現ベクター。
- 該蛋白質をコードする該遺伝子と該内部リボソーム結合部位との間の領域又はグルタミン合成酵素をコードする該遺伝子の下流の領域において,該内部リボソーム結合部位とは別の内部リボソーム結合部位及びその下流に薬剤耐性遺伝子を更に含んでなる,請求項1ないし13の何れかの発現ベクター。
- 該遺伝子発現制御部位(A)及び遺伝子発現制御部位(B)とは別に,更なる遺伝子発現制御部位(C)及びその下流に薬剤耐性遺伝子を更に含んでなる,請求項1ないし13の何れかの発現ベクター。
- 該薬剤耐性遺伝子が,ピューロマイシン又はネオマイシン耐性遺伝子である,請求項14又は15の発現ベクター。
- 該蛋白質をコードする該遺伝子が,ヒト由来の遺伝子である請求項1ないし16の何れかの発現ベクター。
- ヒト由来の該遺伝子が,リソソーム酵素,組織プラスミノーゲンアクチベーター(t-PA),血液凝固因子,エリスロポエチン,インターフェロン,トロンボモジュリン,甲状腺刺激ホルモン(TSH),卵胞刺激ホルモン,顆粒球コロニー刺激因子(G-CSF),及び抗体をコードする遺伝子からなる群から選択されるものである,請求項17の発現ベクター。
- ヒト由来の該遺伝子が,リソソーム酵素をコードする遺伝子である,請求項17の発現ベクター。
- 該リソソーム酵素が,α-ガラクトシダーゼA,イズロン酸2-スルファターゼ,グルコセレブロシダーゼ,ガルスルファーゼ,α-L-イズロニダーゼ,及び酸性α-グルコシダーゼからなる群から選択されるものである,請求項19の発現ベクター。
- ヒト由来の該遺伝子が,エリスロポエチンをコードする遺伝子である請求項17の発現ベクター。
- 請求項1ないし21の何れかの発現ベクターで形質転換された哺乳動物細胞。
- 該哺乳動物細胞が,内因性のジヒドロ葉酸レダクターゼ遺伝子を欠損したものである,請求項22の細胞。
- 該哺乳動物細胞が,CHO細胞である,請求項22又は23の細胞。
- (a)請求項1ないし21の何れかの発現ベクターを,哺乳動物細胞に導入するステップ,
(b)該発現ベクターが導入された哺乳動物細胞を,ジヒドロ葉酸レダクターゼ阻害剤の存在下で選択培養するステップ,及び
(c)該選択培養により選択された細胞を,グルタミン合成酵素阻害剤の存在下で更に選択培養するステップ
を含む,該蛋白質をコードする遺伝子を発現する形質転換細胞の製造方法。 - (a)請求項14ないし16の何れかの発現ベクターを,哺乳動物細胞に導入するステップ,
(b)該発現ベクターが導入された哺乳動物細胞を,ジヒドロ葉酸レダクターゼ阻害剤の存在下で選択培養するステップ,及び
(c)該選択培養により選択された細胞を,グルタミン合成酵素阻害剤の存在下で更に選択培養するステップ
を含む,該蛋白質をコードする遺伝子を発現する形質転換細胞の製造方法であって,該ステップ(a)と該ステップ(b)の間,又は該ステップ(b)と該ステップ(c)の間に,該発現ベクターが導入された哺乳動物細胞を,該薬剤耐性遺伝子に対応する薬剤の存在下で選択培養するステップを更に含む,製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/396,929 US9657310B2 (en) | 2012-04-27 | 2013-04-25 | Expression vector |
| JP2014512690A JP6279466B2 (ja) | 2012-04-27 | 2013-04-25 | 新規な発現ベクター |
| EP13782136.9A EP2848687B1 (en) | 2012-04-27 | 2013-04-25 | Novel expression vector |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-102796 | 2012-04-27 | ||
| JP2012102796 | 2012-04-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013161958A1 true WO2013161958A1 (ja) | 2013-10-31 |
Family
ID=49483257
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/062251 WO2013161958A1 (ja) | 2012-04-27 | 2013-04-25 | 新規な発現ベクター |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9657310B2 (ja) |
| EP (1) | EP2848687B1 (ja) |
| JP (1) | JP6279466B2 (ja) |
| WO (1) | WO2013161958A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017523780A (ja) * | 2014-07-25 | 2017-08-24 | デルフィ ジェネティクス | タンパク質を産生させるために改善した宿主細胞 |
| WO2021075526A1 (ja) | 2019-10-17 | 2021-04-22 | Jcrファーマ株式会社 | 血清アルブミンと成長ホルモンの融合蛋白質の製造方法 |
| WO2021085518A1 (ja) | 2019-10-30 | 2021-05-06 | Jcrファーマ株式会社 | 血清アルブミンと成長ホルモンの融合蛋白質を含有する水性医薬組成物 |
| EP3845644A1 (en) | 2015-09-08 | 2021-07-07 | JCR Pharmaceuticals Co., Ltd. | Novel human serum albumin mutant |
| WO2022045273A1 (ja) | 2020-08-28 | 2022-03-03 | Jcrファーマ株式会社 | α-N-アセチルグルコサミニダーゼの変異体 |
| WO2022191158A1 (ja) | 2021-03-09 | 2022-09-15 | Jcrファーマ株式会社 | 抗体-リソソーム酵素融合蛋白質の製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9567577B2 (en) * | 2012-03-12 | 2017-02-14 | Ares Trading S.A. | Expression vector comprising a polynucleotide encoding a modified glutamine synthetase and a method for preparing a target protein employing the same |
| CA3076369A1 (en) | 2017-10-02 | 2019-04-11 | Denali Therapeutics Inc. | Fusion proteins comprising enzyme replacement therapy enzymes |
| WO2020227206A1 (en) * | 2019-05-07 | 2020-11-12 | Amgen Inc. | Vectors and expression systems for producing recombinant proteins |
| AU2022245592A1 (en) | 2021-03-24 | 2023-10-19 | Jcr Pharmaceuticals Co., Ltd. | Stable aqueous pharmaceutical composition or freeze-dried pharmaceutical composition |
| US20240269244A1 (en) | 2021-05-12 | 2024-08-15 | Jcr Pharmaceuticals Co., Ltd. | Pharmaceutical Composition for Treatment of Mucopolysaccharidosis Type 1 |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59173096A (ja) | 1983-01-19 | 1984-09-29 | ジエネンテツク・インコーポレイテツド | ポリシストロン性発現ベクターおよびそれを用いた方法 |
| JPS60199387A (ja) | 1984-02-06 | 1985-10-08 | インタ−フエロン・サイエンシズ・インコ−ポレ−テツド | 縦列配列した遺伝子をもつハイブリツド核酸配列 |
| JPS63502955A (ja) | 1986-01-23 | 1988-11-02 | セルテク・リミテツド | 組換えdna配列類、それらを含有するベクタ−類およびそれらの使用方法 |
| JPH04500004A (ja) | 1988-07-29 | 1992-01-09 | ザイモジェネティクス,インコーポレイティド | 真核細胞におけるポリシストロン性メッセージの高率翻訳 |
| JPH05504050A (ja) | 1989-10-25 | 1993-07-01 | アルスイス ホールディングス アクチエンゲゼルシャフト | 組換えdna法及びそこに使用するベクター |
| JPH06509713A (ja) | 1991-08-07 | 1994-11-02 | アンダースン,ダブリュー.,フレンチ | 内部リボソームエントリー部位を含むレトロウイルスベクター |
| JPH08502644A (ja) | 1992-08-27 | 1996-03-26 | バイアースドルフ アーゲー | 多シストロン発現ユニットおよびそれらの使用 |
| JPH08256776A (ja) | 1995-03-24 | 1996-10-08 | Morinaga Milk Ind Co Ltd | 高効率発現ベクター |
| JPH10327871A (ja) | 1997-05-26 | 1998-12-15 | Chemo Sero Therapeut Res Inst | 多分子発現カセット及びその利用 |
| JP2004520016A (ja) | 2000-10-06 | 2004-07-08 | オックスフォード バイオメディカ(ユーケイ)リミテッド | ベクターシステム |
| WO2005040364A1 (ja) * | 2003-10-24 | 2005-05-06 | Juridical Foundation The Chemo-Sero-Therapeutic Research Institute | 新規なタンパク質高産生組換え動物細胞、その作製方法及びそれを用いたタンパク質を大量産生する方法 |
| JP2008539785A (ja) | 2005-05-16 | 2008-11-20 | モルフォテック、インク. | 所望の表現型を示す細胞を選択するために制御されたベクター |
| JP2009273427A (ja) | 2008-05-16 | 2009-11-26 | Jcr Pharmaceuticals Co Ltd | 組換え体ヒトfshの製造方法 |
| CN102409060A (zh) * | 2010-09-26 | 2012-04-11 | 北京华安科创生物技术有限公司 | Gs-dhfr双基因筛选表达载体及其构建方法与应用 |
| WO2012063799A1 (ja) * | 2010-11-08 | 2012-05-18 | 日本ケミカルリサーチ株式会社 | 新規発現ベクター |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002542834A (ja) * | 1999-04-29 | 2002-12-17 | アールフス・ユニバーシティー | レトロウイルスベクターでのires転写カセットからの異種遺伝子群の発現 |
| IL149820A0 (en) * | 2002-05-23 | 2002-11-10 | Curetech Ltd | Humanized immunomodulatory monoclonal antibodies for the treatment of neoplastic disease or immunodeficiency |
| WO2006004988A2 (en) * | 2004-06-30 | 2006-01-12 | Mayo Foundation For Medical Education And Research | B7-dc binding antibody |
| EP2468881A3 (en) * | 2005-07-21 | 2012-08-15 | Abbott Laboratories | Multiple gene expression including sorf contructs and methods with polyproteins, pro-proteins, and proteolysis |
| EP2089424B1 (en) * | 2006-12-06 | 2015-07-01 | JCR PHARMACEUTICALS Co., LTD. | Method for production of human erythropoietin |
| JP5570980B2 (ja) * | 2007-06-28 | 2014-08-13 | ラティオファーム ゲーエムベーハー | Fsh生産細胞クローン |
| TWI488640B (zh) * | 2008-04-16 | 2015-06-21 | Ferring Int Ct Sa | 藥學製劑 |
| AU2012203048A1 (en) * | 2011-05-24 | 2012-12-13 | Agency For Science, Technology And Research | IRES mediated multicistronic vectors |
| CN102321668A (zh) * | 2011-07-06 | 2012-01-18 | 中国人民解放军军事医学科学院野战输血研究所 | 一种表达重组人凝血因子ⅶ的方法及其专用载体 |
-
2013
- 2013-04-25 US US14/396,929 patent/US9657310B2/en active Active
- 2013-04-25 WO PCT/JP2013/062251 patent/WO2013161958A1/ja active Application Filing
- 2013-04-25 JP JP2014512690A patent/JP6279466B2/ja active Active
- 2013-04-25 EP EP13782136.9A patent/EP2848687B1/en active Active
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59173096A (ja) | 1983-01-19 | 1984-09-29 | ジエネンテツク・インコーポレイテツド | ポリシストロン性発現ベクターおよびそれを用いた方法 |
| JPS60199387A (ja) | 1984-02-06 | 1985-10-08 | インタ−フエロン・サイエンシズ・インコ−ポレ−テツド | 縦列配列した遺伝子をもつハイブリツド核酸配列 |
| JPS63502955A (ja) | 1986-01-23 | 1988-11-02 | セルテク・リミテツド | 組換えdna配列類、それらを含有するベクタ−類およびそれらの使用方法 |
| JPH04500004A (ja) | 1988-07-29 | 1992-01-09 | ザイモジェネティクス,インコーポレイティド | 真核細胞におけるポリシストロン性メッセージの高率翻訳 |
| JPH05504050A (ja) | 1989-10-25 | 1993-07-01 | アルスイス ホールディングス アクチエンゲゼルシャフト | 組換えdna法及びそこに使用するベクター |
| JPH06509713A (ja) | 1991-08-07 | 1994-11-02 | アンダースン,ダブリュー.,フレンチ | 内部リボソームエントリー部位を含むレトロウイルスベクター |
| JPH08502644A (ja) | 1992-08-27 | 1996-03-26 | バイアースドルフ アーゲー | 多シストロン発現ユニットおよびそれらの使用 |
| JPH08256776A (ja) | 1995-03-24 | 1996-10-08 | Morinaga Milk Ind Co Ltd | 高効率発現ベクター |
| JPH10327871A (ja) | 1997-05-26 | 1998-12-15 | Chemo Sero Therapeut Res Inst | 多分子発現カセット及びその利用 |
| JP2004520016A (ja) | 2000-10-06 | 2004-07-08 | オックスフォード バイオメディカ(ユーケイ)リミテッド | ベクターシステム |
| WO2005040364A1 (ja) * | 2003-10-24 | 2005-05-06 | Juridical Foundation The Chemo-Sero-Therapeutic Research Institute | 新規なタンパク質高産生組換え動物細胞、その作製方法及びそれを用いたタンパク質を大量産生する方法 |
| JP2008539785A (ja) | 2005-05-16 | 2008-11-20 | モルフォテック、インク. | 所望の表現型を示す細胞を選択するために制御されたベクター |
| JP2009273427A (ja) | 2008-05-16 | 2009-11-26 | Jcr Pharmaceuticals Co Ltd | 組換え体ヒトfshの製造方法 |
| CN102409060A (zh) * | 2010-09-26 | 2012-04-11 | 北京华安科创生物技术有限公司 | Gs-dhfr双基因筛选表达载体及其构建方法与应用 |
| WO2012063799A1 (ja) * | 2010-11-08 | 2012-05-18 | 日本ケミカルリサーチ株式会社 | 新規発現ベクター |
Non-Patent Citations (6)
| Title |
|---|
| CACCIATOREA, J.J ET AL.: "Gene amplification and vector engineering to achieve rapid and high-level therapeutic protein production using the Dhfr-based CHO cell selection system", BIOTECHNOL. ADV, vol. 28, no. 6, 2010, pages 673 - 681, XP027331807 * |
| KAUFMAN RJ. ET AL., MOL CEL BIOL., vol. 2, 1982, pages 1304 - 19 |
| MARTIN, P ET AL.: "Development of a new bicistronic retroviral vector with strong IRES activity", BMC BIOTECHNOL, vol. 6, no. 4, 2006, pages 1 - 9, XP021017066 * |
| MCLVOR RS ET AL., NUCLEIC ACIDS RESEARCH, vol. 18, no. 23, 1990, pages 7025 - 32 |
| MIYAHARA M., J. BIOL. CHEM., vol. 275, 2000, pages 613 - 618 |
| WANG, Z. ET AL.: "Dual gene amplification and selection system with dihydrofolate reductase and glutamine synthetase genes effectively increase the foreign gene expression", J. EXP. CLIN. VIROL, vol. 16, no. 1, 2002, pages 59 - 61, XP008175472 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017523780A (ja) * | 2014-07-25 | 2017-08-24 | デルフィ ジェネティクス | タンパク質を産生させるために改善した宿主細胞 |
| US10718001B2 (en) | 2014-07-25 | 2020-07-21 | Delphi Genetics | Host cell for producing proteins |
| JP2020202828A (ja) * | 2014-07-25 | 2020-12-24 | デルフィ ジェネティクス | タンパク質を産生させるために改善した宿主細胞 |
| JP7126824B2 (ja) | 2014-07-25 | 2022-08-29 | アール.ピー.シェーラー テクノロジーズ,エルエルシー | タンパク質を産生させるために改善した宿主細胞 |
| EP3845644A1 (en) | 2015-09-08 | 2021-07-07 | JCR Pharmaceuticals Co., Ltd. | Novel human serum albumin mutant |
| EP4374913A2 (en) | 2015-09-08 | 2024-05-29 | JCR Pharmaceuticals Co., Ltd. | Novel human serum albumin mutant |
| WO2021075526A1 (ja) | 2019-10-17 | 2021-04-22 | Jcrファーマ株式会社 | 血清アルブミンと成長ホルモンの融合蛋白質の製造方法 |
| WO2021085518A1 (ja) | 2019-10-30 | 2021-05-06 | Jcrファーマ株式会社 | 血清アルブミンと成長ホルモンの融合蛋白質を含有する水性医薬組成物 |
| WO2022045273A1 (ja) | 2020-08-28 | 2022-03-03 | Jcrファーマ株式会社 | α-N-アセチルグルコサミニダーゼの変異体 |
| WO2022191158A1 (ja) | 2021-03-09 | 2022-09-15 | Jcrファーマ株式会社 | 抗体-リソソーム酵素融合蛋白質の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2848687B1 (en) | 2017-09-13 |
| JPWO2013161958A1 (ja) | 2015-12-24 |
| US20150337331A1 (en) | 2015-11-26 |
| EP2848687A4 (en) | 2015-12-02 |
| EP2848687A1 (en) | 2015-03-18 |
| JP6279466B2 (ja) | 2018-02-14 |
| US9657310B2 (en) | 2017-05-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6279466B2 (ja) | 新規な発現ベクター | |
| JP5913122B2 (ja) | 新規発現ベクター | |
| JP6152402B2 (ja) | タンパク質の生産方法 | |
| JP5432117B2 (ja) | 宿主細胞中の組換えタンパク質の発現を増進するための組換え発現ベクター要素(rEVE) | |
| JP4554370B2 (ja) | 多重遺伝子プラスミドおよび組み換えポリペプチドの発現増加法 | |
| ES2840725T3 (es) | Procedimiento de producción | |
| RU2494147C2 (ru) | Вектор экспрессии млекопитающих | |
| JP4817514B2 (ja) | 新規動物細胞用ベクターおよびその使用 | |
| US9567577B2 (en) | Expression vector comprising a polynucleotide encoding a modified glutamine synthetase and a method for preparing a target protein employing the same | |
| JP5835636B2 (ja) | 抗体高生産性細胞の樹立のための発現ベクター及び抗体高生産性細胞 | |
| JP2010512776A (ja) | 治療への応用性が高いことで知られている蛋白質を生産するための高転写性活性細胞株 | |
| Picanço-Castro et al. | Patents in therapeutic recombinant protein production using mammalian cells | |
| RU2656142C1 (ru) | РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pBiPr-ABIgA1FI6-ht ДЛЯ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ИММУНОГЛОБУЛИНА А ИЗОТИПА IGA1 | |
| WO2012081629A1 (ja) | タンパク質の生産方法 | |
| JP6150143B2 (ja) | 薬剤選択融合遺伝子を含む高生産性細胞の樹立のための発現ベクター | |
| RU2822889C1 (ru) | Способ получения димерной формы мутантного иммуноглобулина IgA2m1-изотипа в клетках млекопитающих | |
| RU2822890C1 (ru) | Способ получения димерной формы иммуноглобулина IgA1-изотипа в клетках млекопитающих | |
| RU2664184C2 (ru) | РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pBiPr-ABIgA1FI6-Intht ДЛЯ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ИММУНОГЛОБУЛИНА А ИЗОТИПА IgA1 | |
| RU2671477C2 (ru) | РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pBiPr-ABIgA2m1F16-ht ДЛЯ ПОЛУЧЕНИЯ РЕКОМБИНАНТНОГО ИММУНОГЛОБУЛИНА А ИЗОТИПА IGA2m1 | |
| WO2019225372A1 (ja) | 新規ベクターおよびその利用 | |
| JP2012055303A (ja) | 遺伝子増幅を含む高生産性細胞の樹立のための発現ベクター | |
| JP2004248509A (ja) | 真核細胞内における配列特異的dna組換え |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13782136 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014512690 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013782136 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013782136 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14396929 Country of ref document: US |