WO2012004258A1 - Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen - Google Patents
Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen Download PDFInfo
- Publication number
- WO2012004258A1 WO2012004258A1 PCT/EP2011/061305 EP2011061305W WO2012004258A1 WO 2012004258 A1 WO2012004258 A1 WO 2012004258A1 EP 2011061305 W EP2011061305 W EP 2011061305W WO 2012004258 A1 WO2012004258 A1 WO 2012004258A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fluorine
- compound
- hydrogen
- formula
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *c(cc12)cnc1[n](*)nc2-c(nn1)nc(N)c1IC(O)=O Chemical compound *c(cc12)cnc1[n](*)nc2-c(nn1)nc(N)c1IC(O)=O 0.000 description 5
- MZDSTWAEWHPRKG-UHFFFAOYSA-N CC(C)(C(OC)=O)c(nnc(-c(c1c2)n[n](Cc(cccc3F)c3F)c1ncc2F)n1)c1O Chemical compound CC(C)(C(OC)=O)c(nnc(-c(c1c2)n[n](Cc(cccc3F)c3F)c1ncc2F)n1)c1O MZDSTWAEWHPRKG-UHFFFAOYSA-N 0.000 description 1
- KDQAUUHNKJTNIM-UHFFFAOYSA-N CC(CC(N1)=O)c2c1nc(-c1n[n](Cc(cccc3)c3F)c3ncccc13)nc2N Chemical compound CC(CC(N1)=O)c2c1nc(-c1n[n](Cc(cccc3)c3F)c3ncccc13)nc2N KDQAUUHNKJTNIM-UHFFFAOYSA-N 0.000 description 1
- DXFPGNDROFMFEK-UHFFFAOYSA-N CCOC(C1(CC1)c(cnc(-c(c1c2)n[n](Cc(cccc3)c3F)c1ncc2F)n1)c1Cl)=O Chemical compound CCOC(C1(CC1)c(cnc(-c(c1c2)n[n](Cc(cccc3)c3F)c1ncc2F)n1)c1Cl)=O DXFPGNDROFMFEK-UHFFFAOYSA-N 0.000 description 1
- PQJXNBBEXOBJMB-UHFFFAOYSA-N CCOC(C1(CC1)c(cnc(-c1n[n](Cc(cccc2)c2F)c2ncccc12)n1)c1Cl)=O Chemical compound CCOC(C1(CC1)c(cnc(-c1n[n](Cc(cccc2)c2F)c2ncccc12)n1)c1Cl)=O PQJXNBBEXOBJMB-UHFFFAOYSA-N 0.000 description 1
- KTABMVBHLSIDES-UHFFFAOYSA-N CCOC(Cc(c(C(F)F)nc(-c1n[n](Cc(cccc2)c2F)c2ncccc12)n1)c1Cl)=O Chemical compound CCOC(Cc(c(C(F)F)nc(-c1n[n](Cc(cccc2)c2F)c2ncccc12)n1)c1Cl)=O KTABMVBHLSIDES-UHFFFAOYSA-N 0.000 description 1
- GTVALKQTBJETMJ-UHFFFAOYSA-N Fc1cc(F)c(C[n](c2c3cccn2)nc3I)cc1 Chemical compound Fc1cc(F)c(C[n](c2c3cccn2)nc3I)cc1 GTVALKQTBJETMJ-UHFFFAOYSA-N 0.000 description 1
- GRVMXAPAKZZGOV-UHFFFAOYSA-N NC(c1n[n](Cc(ccc(F)c2)c2F)c2c1cccn2)=N Chemical compound NC(c1n[n](Cc(ccc(F)c2)c2F)c2c1cccn2)=N GRVMXAPAKZZGOV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/527—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim spiro-condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
Definitions
- the present application relates to novel fused pyrimidines and triazines, processes for their preparation, their use alone or in combinations for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of diseases, in particular for treatment and / or prophylaxis of cardiovascular disease.
- cyclic guanosine monophosphate cGMP
- NO nitric oxide
- the guanylate cyclases catalyze the biosynthesis of cGMP from guanosine triphosphate (GTP).
- GTP guanosine triphosphate
- the previously known members of this family can be divided into two groups according to both structural features and the nature of the ligands: the particulate guanylate cyclases stimulable by natriuretic peptides and the soluble guanylate cyclases stimulable by NO.
- the soluble guanylate cyclases consist of two subunits and most likely contain one heme per heterodimer that is part of the regulatory center. This is central to the activation mechanism. NO can bind to the iron atom of the heme and thus significantly increase the activity of the enzyme. On the other hand, heme-free preparations can not be stimulated by NO. Also, carbon monoxide (CO) is able to bind to the central iron atom of the heme, with stimulation by CO being significantly less than by NO.
- CO carbon monoxide
- guanylate cyclase plays a crucial role in various physiological processes, in particular in the relaxation and proliferation of smooth muscle cells, platelet aggregation and adhesion, neuronal signaling and diseases based on a disturbance of the above operations.
- the NO / cGMP system may be suppressed, which may, for example, lead to hypertension, platelet activation, increased cell proliferation, endothelial dysfunction, arteriosclerosis, angina pectoris, heart failure, myocardial infarction, thrombosis, stroke and sexual dysfunction.
- a NO-independent treatment option for such diseases which is aimed at influencing the cGMP pathway in organisms, is a promising approach on account of the expected high efficiency and low side effects.
- WO 00/06569 discloses fused pyrazole derivatives and, in WO 03/095451, carbamate-substituted 3-pyrimidinyl-pyrazolopyridines as stimulators of soluble guanylate cyclase.
- WO 2010/065275 discloses substituted pyrrolo and dihydropyridopyrimidines as sGC activators.
- the object of the present invention was to provide new substances which act as stimulators of soluble guanylate cyclase and have a similar or improved therapeutic profile compared to the compounds known from the prior art, for example with respect to their in vivo properties and / or their pharmacokinetic behavior.
- the present invention relates to compounds of the general formula (I)
- A is nitrogen or CR, where R 3 is hydrogen, deuterium, fluorine, difluoromethyl, trifluoromethyl, (C 1 -C 4 ) -alkyl, cyclopropyl or cyclobutyl,
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- # is the point of attachment to the pyrimidine or triazine ring
- p is a number 0, 1 or 2
- R 4A represents hydrogen, fluorine, (C 1 -C 4 ) -alkyl or hydroxyl
- R 4B is hydrogen, fluorine, (C 1 -C 4 ) -alkyl, trifluoromethyl, (C 1 -C 4 ) -alkoxycarbonyl-amino or phenyl, in which (C 1 -C 4 -alkyl having 1 or 2 substituents independently of one another selected from the group fluorine , Trifluoromethyl, hydroxy, hydroxycarbonyl and (C 1 -C 4 ) -alkoxycarbonyl, or
- R 4A and R 4B together with the carbon atom to which they are attached form an oxo group, a 3- to 6-membered carbocycle or a 4- to 6-membered heterocycle, wherein the 3- to 6-membered carbocycle and the 4- to 6-membered heterocycle having 1 or 2 substituents independently selected from the group fluorine and (Ci-C 4 ) alkyl may be substituted, or
- R 4A and R 4B together with the carbon atom to which they are attached, a
- R 5A is hydrogen, fluorine, (C 1 -C 4) -alkyl or hydroxyl
- R is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or trifluoromethyl
- R 1 is hydrogen or fluorine
- R 2 is benzyl, wherein benzyl is substituted by 1 to 3 substituents fluorine, and their N-oxides, salts, solvates, salts of N-oxides and solvates of N-oxides and salts.
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts comprising the compounds of the formulas below and their salts, solvates and solvates of the salts and of the formula (I) encompassed by formula (I), hereinafter referred to as exemplary compounds and their salts, solvates and solvates of the salts, as far as the compounds of formula (I), the compounds mentioned below are not already salts, solvates and solvates of the salts.
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. Also included are salts which are themselves unsuitable for pharmaceutical applications but can be used, for example, for the isolation or purification of the compounds of the invention.
- Physiologically acceptable salts of the compounds of the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric, hydrobromic, sulfuric, phosphoric, methanesulfonic, ethanesulfonic, toluenesulfonic, benzenesulfonic, naphthalenedisulfonic, formic, acetic, trifluoroacetic, propionic, lactic, tartaric, malic, citric, fumaric, maleic and benzoic acids.
- Salts of hydrochloric, hydrobromic, sulfuric, phosphoric, methanesulfonic, ethanesulfonic, toluenesulfonic, benzenesulfonic, naphthalenedisulfonic formic, acetic, trifluoroacetic, propionic, lactic, tartaric, malic, citric, fumaric, maleic and benzoic
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms, such as, by way of example and by way of illustration, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts
- Solvates in the context of the invention are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water. As solvates, hydrates are preferred in the context of the present invention.
- the compounds according to the invention may exist in different stereoisomeric forms, ie in the form of configurational isomers or optionally also as conformational isomers (enantiomers and / or diastereomers, including those in the case of atropisomers). The present invention therefore encompasses the enantiomers and diastereomers and their respective mixtures.
- the stereoisomerically uniform components can be isolated in a known manner; Preferably, chromatographic methods are used for this, in particular HPLC chromatography on achiral or chiral phase.
- the present invention encompasses all tautomeric forms.
- the present invention also includes all suitable isotopic variants of the compounds of the invention.
- An isotopic variant of a compound according to the invention is understood to mean a compound in which at least one atom within the compound according to the invention is exchanged for another atom of the same atomic number but with a different atomic mass than the atomic mass that usually or predominantly occurs in nature.
- isotopes which can be incorporated into a compound of the invention are those of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2 H (deuterium), 3 H (tritium), 13 C, 14 C, 15 N, 17 0, 18 0, 32 P, 33 P, 33 S, 34 S, 35 S, 36 S, 18 F, 36 Cl, 82 Br, 123 I, 124 I, 129 I and 131 I.
- isotopic variants of a compound of the invention such as, in particular, those in which one or more radioactive isotopes are incorporated, may be useful, for example, for the study of the mechanism of action or drug distribution in the body; Due to the comparatively easy production and detectability, compounds labeled with 3 H or 14 C isotopes in particular are suitable for this purpose.
- isotopes such as deuterium may result in certain therapeutic benefits as a result of greater metabolic stability of the compound, such as prolonging the body's half-life or reducing the required effective dose;
- Such modifications of the compounds of the invention may therefore optionally also constitute a preferred embodiment of the present invention.
- Isotopic variants of the compounds according to the invention can be prepared by the processes known to the person skilled in the art, for example by the methods described below and the rules given in the exemplary embodiments, by using appropriate isotopic modifications of the respective reagents and / or starting compounds.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs refers to compounds which themselves may be biologically active or inactive, but are converted during their residence time in the body to compounds of the invention (for example metabolically or hydrolytically). Unless otherwise specified, in the context of the present invention, the substituents have the following meaning:
- alkyl is a linear or branched alkyl radical having 1 to 4 carbon atoms. Examples which may be mentioned are: methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, 1-methylpropyl, tert-butyl.
- Carbocycle in the context of the invention is a monocyclic, saturated carbocycle having 3 to 6 ring carbon atoms. By way of example and preferably mention may be made of: cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- Alkenyl in the context of the invention represents a linear or branched alkenyl radical having 2 to 4 carbon atoms and one double bond.
- alkenyl radical having 2 to 4 carbon atoms and one double bond.
- Alkoxycarbonyl in the context of the invention is a linear or branched alkoxy radical having 1 to 4 carbon atoms and an oxygen-bonded carbonyl group. Examples which may be mentioned by way of example include methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl and tert-butoxycarbonyl.
- Alkoxycarbonylamino in the context of the invention represents an amino group having a linear or branched alkoxycarbonyl substituent which has 1 to 4 carbon atoms in the alkyl chain and is linked via the carbonyl group to the nitrogen atom.
- Heterocycle in the context of the invention is a saturated heterocycle having a total of 4 to 6 ring atoms, which contains one or two ring heteroatoms from the series N, O, S, SO and / or SO 2 and is linked via a ring carbon atom.
- Preferred are azetidinyl, oxetanyl, pyrrolidinyl, tetrahydrofuranyl, piperidinyl and tetrahydropyranyl.
- Halogen is in the context of the invention for fluorine, chlorine, bromine and iodine. Preference is given to bromine and iodine.
- radicals are substituted in the compounds according to the invention, the radicals can, unless otherwise specified, be monosubstituted or polysubstituted. In the context of the present invention, the meaning is independent of each other for all radicals which occur repeatedly. Substitution with one, two or three identical or different substituents is preferred.
- A is nitrogen or CR 3 , where
- R 3 is hydrogen, deuterium, fluorine, difluoromethyl, trifluoromethyl, (C 1 -C 4 -alkyl, cyclopropyl or cyclobutyl,
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- # is the point of attachment to the pyrimidine or triazine ring
- p is a number 0, 1 or 2
- R 4A represents hydrogen, fluorine, (C r C4) alkyl, hydroxy,
- R 4B is hydrogen, fluorine, (Ci-C alkyl or trifluoromethyl, or
- R 4A and R 4B together with the carbon atom to which they are attached form an oxo group, a 3- to 6-membered carbocycle or a 4- to 6-membered heterocycle, for hydrogen, fluorine, (Ci-C - Alkyl or hydroxy, R 5B is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or trifluoromethyl, R 1 is hydrogen or fluorine, R 2 is benzyl, where benzyl is substituted by 1 to 3 substituents fluorine, and their N-oxides, Salts, solvates, salts of N-oxides and solvates of N-oxides and salts.
- A is nitrogen or CR 3 , where
- R 3 is hydrogen, fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl or cyclopropyl,
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- # represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number 0 or 1
- R 4A is hydrogen, fluorine, methyl, ethyl or hydroxy
- R 4B is hydrogen, fluorine, methyl, ethyl, trifluoromethyl, methoxycarbonylammo or phenyl, wherein methyl and ethyl may be substituted with 1 or 2 substituents independently selected from the group of fluorine, trifluoromethyl and hydroxy, or
- R 4A and R 4B together with the carbon atom to which they are attached, a
- R 5A is hydrogen, fluorine, methyl, ethyl or hydroxy
- R 5B is hydrogen, fluorine, methyl, ethyl or trifluoromethyl
- R 1 is hydrogen or fluorine
- R 2 is benzyl, wherein benzyl is substituted by 1 to 3 substituents fluorine, and their salts, solvates and solvates of the salts.
- A is nitrogen or CR 3 , where
- R 3 is hydrogen, difluoromethyl, trifluoromethyl, methyl, ethyl or cyclopropyl,
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- # is the point of attachment to the pyrimidine or triazine ring
- p is a number 0, 1 or 2
- R 4A is hydrogen, fluorine, methyl, ethyl or hydroxy
- R 4B is hydrogen, fluorine, methyl, ethyl or trifluoromethyl, or R 4A and R 4B together with the carbon atom to which they are attached form a cyclopropyl, cyclobutyl, cyclopentyl, azetidinyl, pyrrolidinyl, tetrahydrofuranyl, piperidinyl or tetrahydropyranyl ring,
- R 5A is hydrogen, fluorine, methyl, ethyl or hydroxy
- R 5B is hydrogen, fluorine, methyl, ethyl or trifluoromethyl
- R 1 is hydrogen or fluorine
- R 2 is benzyl, wherein benzyl is substituted by 1 or 2 substituents fluorine, and their salts, solvates and solvates of the salts.
- A is CR 3 , where
- R 3 L represents hydrogen, a group -CR * 4A R 4B - (CR 5A R 5B) p - #, where
- # represents the point of attachment to the pyrimidine ring
- p stands for a number
- R 4A is hydrogen, fluorine, methyl or hydroxy
- R 4B is hydrogen, fluorine, methyl or trifluoromethyl, or
- R 4A and R 4B together with the carbon atom to which they are attached, a
- R 1 is hydrogen or fluorine
- R 2 is benzyl, where benzyl is substituted by 1 or 2 substituents fluorine, and their salts, solvates and solvates of the salts.
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- # represents the point of attachment to the triazine ring
- p stands for a number
- R 4A is hydrogen, fluorine or methyl
- R 4B is hydrogen, fluorine, methyl or trifluoromethyl, or
- R 4A and R 4B together with the carbon atom to which they are attached, a
- R 1 is hydrogen or fluorine
- R 2 is benzyl, wherein benzyl is substituted by 1 or 2 substituents fluorine, and their salts, solvates and solvates of the salts.
- A is nitrogen or CR 3 , where
- R 3 L represents hydrogen, a group -CR * 4A R 4B - (CR 5A R 5B) p - #, where
- # represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 4A is hydrogen, fluorine, methyl or hydroxy
- R 4B is hydrogen, fluorine, methyl or trifluoromethyl, or
- R 4A and R 4B together with the carbon atom to which they are attached, a
- R 1 is hydrogen or fluorine
- R 2 is a group of the formula
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #
- # represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 4A and R 4B together with the carbon atom to which they are attached, a
- * represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 4A is hydrogen, fluorine, methyl or hydroxy
- R 4B is hydrogen, fluorine, methyl or trifluoromethyl, and their salts, solvates and solvates of the salts.
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- * represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 4A is methyl
- R 4B is methyl
- their salts, solvates and solvates of the salts are methyl
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- * represents the point of attachment to the carbonyl group
- # represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 4A and R 4B together with the carbon atom to which they are attached, a
- R 2 is benzyl, wherein benzyl is substituted by 1 or 2 substituents fluorine, and their salts, solvates and solvates of the salts.
- R 2 is a group of the formula
- Another object of the present invention are compounds of formula (XV),
- # is the point of attachment to the triazine ring
- p is a number 0, 1 or 2
- R 4A represents hydrogen, fluorine, (C 1 -C 4 ) -alkyl or hydroxyl
- R 4B is hydrogen, fluorine, (C 1 -C 4 ) -alkyl, trifluoromethyl, (C 1 -C 4 ) -alkoxycarbonyl-amino or phenyl, in which (C 1 -C 4 ) -alkyl having 1 or 2 substituents independently of one another selected from Group fluorine, trifluoromethyl, hydroxy, hydroxycarbonyl and (C 1 -C 4) - alkoxycarbonyl may be substituted, or
- R 4A and R 4B together with the carbon atom to which they are attached form a 3- to
- R 5B is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or trifluoromethyl,
- R 1 is hydrogen or fluorine
- R 2 is benzyl, where benzyl is substituted by 1 to 3 fluorine substituents,
- T 4 is (C r C 4 ) -alkyl, and their salts, solvates and solvates of the salts.
- Another object of the present invention are compounds of formula (XIII),
- # is the point of attachment to the triazine ring
- p is a number 0, 1 or 2
- R 4A represents hydrogen, fluorine, (C 1 -C 4 ) -alkyl or hydroxyl
- R 4B is hydrogen, fluorine, (Ci-C) alkyl, trifluoromethyl, (C r C4) alkoxycarbonyl amino or phenyl, wherein (C 1 -C 4) -alkyl having 1 or 2 substituents independently of one another can be substituted from the group of fluorine, trifluoromethyl, hydroxy, hydroxycarbonyl and (C 1 -C 4 ) -alkoxycarbonyl, or
- R 4A and R 4B together with the carbon atom to which they are attached form a 3- to
- 6-membered carbocycle or a 4- to 6-membered heterocycle wherein the 3- to 6-membered carbocycle and the 4- to 6-membered heterocycle having 1 or 2 substituents independently selected from the group fluorine and (Ci-C4 ) Alkyl,
- R 5A is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or hydroxyl
- R 5B is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or trifluoromethyl,
- R 1 is hydrogen or fluorine
- R 2 is benzyl, where benzyl is substituted by 1 to 3 fluorine substituents,
- T 4 is (C r C 4 ) -alkyl, and their salts, solvates and solvates of the salts.
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- # is the point of attachment to the triazine ring
- p is a number 0, 1 or 2
- R 4A is hydrogen, fluorine, methyl, ethyl or hydroxy
- R 4B is hydrogen, fluorine, methyl, ethyl or trifluoromethyl, or
- R 4A and R 4B together with the carbon atom to which they are attached, a
- R 5A represents hydrogen, fluorine, methyl, ethyl or hydroxy
- R 5B is hydrogen, fluorine, methyl, ethyl or trifluoromethyl
- R 1 is hydrogen or fluorine
- R 2 is benzyl, where benzyl is substituted by 1 or 2 substituents fluorine, T 4 is methyl or ethyl, and their salts, solvates and solvates of the salts.
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, where
- # represents the point of attachment to the triazine ring
- p stands for a number
- R 4A is methyl
- R 4B is methyl
- their salts, solvates and solvates of the salts are methyl
- L represents a group * -CR 4A R 4B - (CR 5A R 5B ) p - #, in which
- # represents the point of attachment to the triazine ring
- p is a number 0, R 4A and R 4B together with the carbon atom to which they are attached, one
- the invention further provides a process for the preparation of the compounds of the formula (I) according to the invention which comprises reacting a compound of the formula (II)
- T 1 is (C r C 4) -alkyl, to a compound of formula (IV)
- # is the point of attachment to the pyrimidine or triazine ring
- p is a number 1 or 2
- R 4A represents hydrogen, fluorine, (C 1 -C 4 ) -alkyl or hydroxyl
- R 4B is hydrogen, fluorine, (Ci-C) -alkyl or trifluoromethyl, or
- R 4A and R 4B together with the carbon atom to which they are attached, a
- Oxo group form a 3- to 6-membered carbocycle or a 4- to 6-membered heterocycle
- R 5A is hydrogen, fluorine, (C r C4) alkyl or hydroxy
- R 5B is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or trifluoromethyl
- T 2 is (C r C 4 ) -alkyl, to give a compound of formula (IV-B)
- T 3 represents (C r C 4) -alkyl, to a compound of formula (VIII)
- T is (C r C4) alkyl
- L, R 1 and R 2 are each as defined above, cyclized, and optionally the resulting compounds of the formulas (IA), (IB), (IC) and (ID) optionally with the appropriate (i) solvents and / or (ii) converting acids or bases into their solvates, salts and / or solvates of the salts.
- Inert solvents for process step (II) + (III) or (VI) - > (IV) are, for example, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers such as diethyl ether, Dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N- Methylpyrrolidone (NMP), pyridine, acetonitrile, sulfolane or water. It is likewise possible to use mixtures of the solvents
- Suitable bases for process step (II) + (III) or (VI) -> (IV) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali metal bicarbonates such as sodium or potassium hydrogen carbonate, alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 5-diazabicyclo [4.3.0] non-5-ene (DBN).
- alkali metal hydroxides such as lithium, sodium or potassium hydroxide
- alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate
- alkali metal bicarbonates such as sodium or potassium hydrogen carbonate
- alkali metal alcoholates such as
- the reaction (II) + (III) or (VI) -> (IV) is generally in a temperature range from + 20 ° C to + 150 ° C, preferably at + 75 ° C to + 100 ° C, optionally in a microwave oven.
- the reaction can be carried out at normal, elevated or at reduced pressure (for example from 0.5 to 5 bar). Generally, one works at normal pressure.
- the process step (IV) -> (V) is carried out with or without solvent.
- Suitable solvents are all organic solvents which are inert under the reaction conditions. Preferred solvent is dimethoxyethane.
- the reaction (IV) -> (V) is generally carried out in a temperature range of + 20 ° C to + 100 ° C, preferably in the range of + 50 ° C to + 100 ° C, optionally in a microwave.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the process step (IV) -> (V) is generally carried out with a molar ratio of 10 to 30 moles of isopentyl nitrite and 10 to 30 moles of the iodine equivalent based on 1 mole of the compound of formula (IV).
- iodine source in implementing (IV) - » ⁇ (V) are suitable, for example, diiodomethane or a mixture of cesium, iodine and copper (I) iodide.
- Inert solvents for process step (V) -> (IA) are alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol or 1,2-ethanediol, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or even water. It is likewise possible to use mixtures of the solvents mentioned. Preferred is DMF.
- the reduction (V) -> (I-A) is carried out with hydrogen in conjunction with transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- the reaction (V) -> (I-A) is generally carried out in a temperature range of + 20 ° C to + 50 ° C.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (II) + (VII) - > (VIII) are, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers, such as diethyl ether, dioxane, dimethoxyethane , Tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ethers, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (NMP), pyridine or acetonitrile. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to methanol or
- Suitable bases for process step (II) + (VII) -> (VIII) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali metal bicarbonates such as sodium or potassium bicarbonate , Alkali metal such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, l, 8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l , 5-diazabicyclo [4.3.0] non-5-ene (DBN). Preference is given to sodium methoxide or sodium ethoxide.
- alkali metal hydroxides such as lithium, sodium or potassium hydroxide
- alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate
- alkali metal bicarbonates
- the reaction (II) + (VII) -> (VIII) is generally carried out in a temperature range from + 50 ° C to + 120 ° C, preferably from + 50 ° C to + 100 ° C, optionally in a microwave.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the reactions (VIII) -> (IX) and (XIII) -> (XIV) can be carried out in a solvent which is inert under the reaction conditions or without solvent.
- Preferred solvent is sulfolane.
- the reactions (VIII) - > (IX) and (XIII) - > (XIV) are generally carried out in a temperature range from + 70 ° C to + 150 ° C, preferably from + 80 ° C to + 130 ° C, if necessary in a microwave.
- the reaction can be carried out at normal or elevated pressure (for example in the range from 0.5 to 5 bar). Generally, one works at normal pressure.
- reaction (XIII) - > (XIV) without solvent in a temperature range of 0 ° C to + 50 ° C at atmospheric pressure.
- the process step (IX) -> (X) is carried out by reaction with sodium azide with intermediate formation of the azide derivatives, which are further reduced directly to the corresponding amines.
- Inert solvents for the azide formation are, for example, ethers, such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons, such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO).
- DMF dimethylformamide
- DMSO dimethyl sulfoxide
- N, N'-dimethylpropylene urea (DMPU), N-methylpyrrolidone (NMP), pyridine, acetonitrile or sulfolane. It is likewise possible to use mixtures of the solvents mentioned. Preferred is DMF.
- the azide is generally formed in a temperature range from + 50 ° C to + 100 ° C, preferably from + 60 ° C to + 80 ° C, at atmospheric pressure.
- the reduction is carried out in an inert solvent such as, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol or 1,2-ethanediol, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether.
- alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol or 1,2-ethanediol
- ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol
- DMF dimethylformamide
- DMSO dimethyl sulfoxide
- DMPU NN'-dimethyl propyleneurea
- ⁇ N-methylpyrrolidone
- pyridine acetonitrile or even water.
- DMF dimethylformamide
- DMSO dimethyl sulfoxide
- DMPU NN'-dimethyl propyleneurea
- ⁇ N-methylpyrrolidone
- pyridine acetonitrile
- acetonitrile even water.
- Preferred is DMF.
- the reduction takes place at + 10 ° C to + 30 ° C with hydrogen in conjunction with transition metal catalysts such as palladium (10% on activated carbon), platinum dioxide or palladium hydroxide, or without hydrogen with stannous chloride and hydrochloric acid.
- transition metal catalysts such as palladium (10% on activated carbon), platinum dioxide or palladium hydroxide, or without hydrogen with stannous chloride and hydrochloric acid.
- reaction (IX) -> (X) can also be carried out in a stage analogous to process step (XIV) -> (XV).
- the process step (XIV) -> (XV) takes place in a solvent which is inert under the reaction conditions.
- Suitable solvents are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or else Water. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to acetonitrile.
- the reaction (XIV) -> (XV) is generally carried out in a temperature range from + 20 ° C to + 100 ° C, preferably from + 40 ° C to + 70 ° C, optionally in a microwave.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the cyclizations (X) -> (IC) and (XV) -> (ID) are carried out in a solvent which is inert under the reaction conditions, for example alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert.
- a solvent which is inert under the reaction conditions, for example alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert.
- Suitable bases for the process steps (X) - » ⁇ (IC) and (XV) -» ⁇ (ID) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali metal bicarbonates such as sodium or potassium bicarbonate, alkali metal such as sodium or potassium, sodium or potassium or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, l, 8-diazabicyclo [5.4.0] undec-7 -en (DBU) or l, 5-diazabicyclo [4.3.0] non-5-ene (DBN).
- alkali metal hydroxides such as lithium, sodium or potassium hydroxide
- alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate
- alkali metal bicarbonates such as sodium or potassium bicarbonate
- alkali metal such as sodium or potassium
- the reactions (X) -> (IC) and (XV) -> (ID) are generally carried out in a temperature range from 0 ° C to + 50 ° C, preferably from + 10 ° C to + 30 ° C, optionally in one Microwave.
- the reaction can be carried out at normal or elevated pressure (for example in the range from 0.5 to 5 bar). Generally, one works at normal pressure.
- the reactions (XIII) - (XIV) - (XV) - (I-D) are preferably carried out simultaneously in a one-pot reaction without isolation of the intermediates.
- Inert solvents for the process step (XI) + (XII) - > (XIII) are, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers, such as diethyl ether, dioxane, Dimethoxyethane, tetrahydrofuran, glycol dimethacrylate or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N -Methylpyrrolidone ( ⁇ ), pyridine or acetonitrile. It is likewise possible to use mixtures of the solvents mentioned. Preference is given
- the reaction (XI) + (XII) -> (XIII) is generally carried out in a temperature range from + 50 ° C to + 120 ° C, preferably from + 50 ° C to + 100 ° C, optionally in a microwave.
- the implementation can be carried out at normal or elevated pressure (eg in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (II) -> (XI) are, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers, such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, Hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (NMP), pyridine or acetonitrile.
- alcohols such as methanol, ethanol, n-propanol, isopropanol, n-but
- Suitable bases for process step (II) -> ⁇ (XI) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali metal hydrogen carbonates such as sodium or potassium hydrogen carbonate, alkali metal alkoxides such as Sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) or 1.5 Diazabicyclo [4.3.0] non-5-ene (DBN).
- DBU 1,8-diazabicyclo [5.4.0] undec-7-ene
- DBN Diazabicyclo [4.3.0] non-5-ene
- triethylamine Preferably triethylamine.
- the reaction (II) -> (XI) is generally carried out in a temperature range from 0 ° C to + 60 ° C, preferably from + 10 ° C to + 30 ° C.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). In general, one works at normal pressure.
- Other compounds of the invention may optionally also be prepared by conversions of functional groups of individual substituents, in particular those listed under L and R 3 , starting from the compounds of the formula (I), (IX) or (XIV) obtained by the above method.
- transformations are carried out by conventional methods known to those skilled in the art and include, for example, reactions such as nucleophilic and electrophilic substitutions, oxidations, reductions, hydrogenations, transition metal-catalyzed coupling reactions, elimination, alkylation, amination, esterification, ester cleavage, etherification, ether cleavage, formation of carbonamides, and introduction and removal of temporary protection groups.
- the following synthetic schemes (Schemes 5, 6, 11, and 12) exemplify preferred transformations:
- the compounds of the formula (II) are known from the literature (see, for example, WO 03/095451, Example 6A) or can be prepared by reacting a compound of the formula (XVI)
- X 1 represents a suitable leaving group, such as, for example, halogen, tosylate or mesylate, in a compound of the formula (XX)
- R 1 and R 2 each have the meanings given above, reacting and finally reacting these under acidic conditions with an ammonia equivalent.
- Inert solvents for process step (XVI) -> (XVII) are alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol or 1,2-ethanediol, ethers, such as diethyl ether, dioxane, tetrahydrofuran , Glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (NMP), Pyridine, acetonitrile or water. It is likewise possible to use mixtures of the solvents mentioned. Preference is 1, 2-ethanediol.
- the reaction (XVI) -> (XVII) is generally carried out in a temperature range of + 60 ° C to + 200 ° C, preferably at + 120 ° C to + 180 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for the reaction (XVII) - [ (XVIII) are, for example, halogenated hydrocarbons, such as dichloromethane, trichloromethane, tetrachloromethane, trichlorethylene or chlorobenzene, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol.
- Dimethyl ether, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine or acetonitrile.
- DMF dimethylformamide
- DMSO dimethyl sulfoxide
- DMPU NN'-dimethylpropyleneurea
- ⁇ N-methylpyrrolidone
- pyridine acetonitrile.
- DMF dimethylformamide
- Suitable Lewis acids for process step (XVII) -> (XVIII) are boron trifluoride-diethyl ether complex, cerium (IV) ammonium nitrate (CAN), tin (II) chloride, lithium perchlorate, zinc (II) chloride, indium (III) chloride or indium (III) bromide. Boron trifluoride diethyl ether complex is preferred.
- the reaction (XVII) -> (XVIII) is generally carried out in a temperature range of -78 ° C to + 40 ° C, preferably at 0 ° C to + 20 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for the reaction (XVIII) + (XIX) - > (XX) are, for example, halogenated hydrocarbons such as dichloromethane, trichloromethane, tetrachloromethane, trichlorethylene or chlorobenzene, ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents such as Dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile. Preferred is DMF.
- halogenated hydrocarbons such as dichloromethane, trichloromethane, tetrachloromethane, trichlorethylene or chlorobenzene
- ethers such as diethyl ether, dioxane
- Suitable bases for the process step (XVIII) + (XIX) -> (XX) are alkali metal hydrides such as potassium hydride or sodium hydride, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali metal bicarbonates such as sodium or potassium bicarbonate, alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, organometallic compounds such as butyl lithium or phenyllithium, or organic amines such as triethylamine, Diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 5-diazabicyclo [4.3.0] non-5-ene (DBN). Cesium carbonate is preferred.
- the reaction (XVIII) + (XIX) -> (XX) is generally carried out in a temperature range from 0 ° C to + 60 ° C, preferably at + 10 ° C to + 25 ° C.
- the reaction can be carried out at normal, elevated or at reduced pressure (for example from 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (XX) - > (XXI) are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons, such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or others Solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine or acetonitrile. It is likewise possible to use mixtures of the solvents mentioned. Preferred is DMSO.
- the reaction (XX) -> (XXI) is generally carried out in a temperature range from + 20 ° C to + 180 ° C, preferably at + 100 ° C to + 160 ° C, optionally in a microwave.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- reaction (XXI) -> (II) is carried out according to the methods known in the art in a two-step process initially to form the iminoester with sodium methoxide in methanol at 0 ° C to + 40 ° C and subsequent nucleophilic addition of an ammonia equivalent such as ammonia or ammonium chloride in a suitable acid to form the amidine (III) at +50 to + 150 ° C.
- an ammonia equivalent such as ammonia or ammonium chloride
- Suitable acids for the formation of the amidine (II) are inorganic acids such as hydrochloric acid / hydrochloric acid, sulfuric acid, polyphosphoric acid or phosphoric acid or organic acids such as acetic acid, trifluoroacetic acid or formic acid.
- inorganic acids such as hydrochloric acid / hydrochloric acid, sulfuric acid, polyphosphoric acid or phosphoric acid
- organic acids such as acetic acid, trifluoroacetic acid or formic acid.
- hydrochloric acid or acetic acid are used.
- the compounds of the invention are potent stimulators of soluble guanylate cyclase, have valuable pharmacological properties, and have an improved therapeutic profile, such as in their in vivo properties and / or their pharmacokinetic behavior. They are therefore suitable for the treatment and / or prophylaxis of diseases in humans and animals.
- the compounds of the invention cause vasorelaxation and inhibition of platelet aggregation and lead to a reduction in blood pressure and to an increase in coronary blood flow. These effects are mediated by a direct stimulation of soluble guanylate cyclase and an intracellular cGMP increase.
- the compounds according to the invention enhance the action of substances which increase the cGMP level, such as, for example, EDHF (endothelium-derived relaxing factor), NO donors, protoporphyrin IX, arachidonic acid or phenylhydrazine derivatives.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of cardiovascular, pulmonary, thromboembolic and fibrotic disorders.
- the compounds according to the invention can therefore be used in medicaments for the treatment and / or prophylaxis of cardiovascular diseases such as hypertension, acute and chronic heart failure, coronary heart disease, stable and unstable angina pectoris, peripheral and cardiac vascular diseases, arrhythmias, arrhythmia of the atria and the chambers and conduction disorders such for example atrio-ventricular blockades grade I-III (AB block I-III), supraventricular tachyarrhythmia, atrial fibrillation, atrial flutter, ventricular fibrillation, ventricular tachyarrhythmia, torsade de pointes tachycardia, atrial and ventricular extrasystoles, atrioventricular extrasystoles, Sick sinus syndrome, syncope, AV nodal reentrant tachycardia, Wolff-
- cardiac failure also includes more specific or related forms of disease such as acutely decompensated heart failure, right heart failure, left ventricular failure, global insufficiency, ischemic cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, idiopathic cardiomyopathy, congenital heart defects, valvular heart failure, cardiac valvulopathy, mitral valve stenosis, mitral valves - insufficiency, aortic valve stenosis, aortic valve insufficiency, tricuspid stenosis, tricuspid insufficiency, pulmonary valve stenosis, pulmonary valvular insufficiency, combined valvular heart failure, myocarditis, chronic myocarditis, acute myocarditis, viral myocarditis, diabetic heart failure, alcoholic cardiomyopathy, cardiac disease, diastolic heart failure and systolic heart failure.
- ischemic cardiomyopathy dilated cardiomyopathy
- the compounds according to the invention may also be used for the treatment and / or prophylaxis of arteriosclerosis, lipid metabolism disorders, hypolipoproteinemias, dyslipidemias, hypertriglyceridemias, hyperlipidemias, hypercholesterolemias, abetelipoproteinaemia, sitosterolemia, xanthomatosis, Tangier's disease, obesity (obesity) and combined hyperlipidemias and the metabolic syndrome.
- the compounds of the invention may be used for the treatment and / or prophylaxis of primary and secondary Raynaud's phenomenon, microcirculatory disorders, claudication, peripheral and autonomic neuropathies, diabetic microangiopathies, diabetic retinopathy, diabetic ulcers on the extremities, gangrenous, CREST syndrome, erythematosis, onychomycosis , rheumatic diseases and to promote wound healing.
- the compounds according to the invention are suitable for the treatment of urological diseases such as benign prostatic syndrome (BPS), benign prostatic hyperplasia (BPH), benign prostate enlargement (BPE), bladder emptying disorder (BOO), lower urinary tract syndromes (LUTS, including Feiine's urological syndrome ( FUS)), diseases of the urogenital system including neurogenic overactive bladder (OAB) and (IC), incontinence (UI) such as mixed, urge, stress, or overflow incontinence (MUI, UUI, SUI, OUI), Pelvic pain, benign and malignant diseases of the organs of the male and female urogenital system.
- BPS benign prostatic syndrome
- BPH benign prostatic hyperplasia
- BPE benign prostate enlargement
- BOO bladder emptying disorder
- LUTS lower urinary tract syndromes
- FUS lower urinary tract syndromes
- UI incontinence
- MUI mixed, urge, stress, or overflow incontinence
- UUI UUI
- SUI S
- kidney diseases in particular of acute and chronic renal insufficiency, as well as of acute and chronic renal failure.
- renal insufficiency includes both acute and chronic manifestations of renal insufficiency, as well as underlying or related renal diseases such as renal hypoperfusion, intradialytic hypotension, obstructive uropathy, glomerulopathies, glomerulonephritis, acute glomerulonephritis, glomerulosclerosis, tubulo-interstitial disorders, nephropathic disorders such as primary and congenital kidney disease, nephritis, immunological kidney disease such as renal transplant rejection, immune complex-induced kidney disease diseases, nephropathy induced by toxic substances, contrast agent-induced nephropathy, diabetic and nondiabetic nephropathy, pyelonephritis, renal cysts, nephrosclerosis, hyperten
- the present invention also encompasses the use of the compounds of the invention for the treatment and / or prophylaxis of sequelae of renal insufficiency, such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte imbalances (eg, hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- sequelae of renal insufficiency such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte imbalances (eg, hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of asthmatic diseases, pulmonary arterial hypertension (PAH) and other forms of pulmonary hypertension (PH), including left heart disease, HIV, sickle cell anemia, thromboembolism (CTEPH), sarcoidosis, COPD or Pulmonary fibrosis-associated pulmonary hypertension, chronic obstructive pulmonary disease (COPD), acute respiratory tract syndrome (ARDS), acute lung injury (ALI), alpha-1-antitrypsin deficiency (AATD), pulmonary fibrosis, pulmonary emphysema (eg, cigarette smoke-induced Pulmonary emphysema) and cystic fibrosis (CF).
- PAH pulmonary arterial hypertension
- PH pulmonary hypertension
- COPD chronic obstructive pulmonary disease
- ARDS acute respiratory tract syndrome
- ALI acute lung injury
- AATD alpha-1-antitrypsin deficiency
- CF
- the compounds described in the present invention are also agents for controlling diseases in the central nervous system, which are characterized by disorders of the NO / cGMP system.
- they are suitable for improving the perception, concentration, learning performance or memory performance after cognitive disorders, such as those found in situations / diseases / syndromes such as mild cognitive impairment, age-associated learning and memory disorders, age-associated memory loss, vascular dementia, cranial Brain trauma, stroke, post-stroke dementia, post-traumatic traumatic brain injury, general attention deficit disorder, impaired concentration in children with learning and memory problems, Alzheimer's disease, dementia with Lewy's disease.
- Corpuscles dementia with degeneration of the frontal lobes including Pick's syndrome, Parkinson's disease, progressive nuclear palsy, dementia with corticobasal degeneration, amyolateral sclerosis (ALS), Huntington's disease, demyelinization, multiple sclerosis, thalamic degeneration, Creutzfeld-Jacob dementia , HIV Dementia, schizophrenia with dementia or Korsakoff's psychosis. They are also suitable for the treatment and / or prophylaxis of diseases of the central nervous system such as states of anxiety, tension and depression, central nervous conditional sexual dysfunctions and sleep disorders as well as for the regulation of pathological disorders of food, consumption and addiction.
- diseases of the central nervous system such as states of anxiety, tension and depression, central nervous conditional sexual dysfunctions and sleep disorders as well as for the regulation of pathological disorders of food, consumption and addiction.
- the compounds according to the invention are also suitable for regulating cerebral blood flow and are effective agents for combating migraine. They are also suitable for the prophylaxis and control of the consequences of cerebral infarct events (Apoplexia cerebri) such as stroke, cerebral ischaemias and craniocerebral trauma , Likewise, the compounds of the invention can be used to combat pain and tinnitus.
- the compounds of the invention have anti-inflammatory action and can therefore be used as anti-inflammatory agents for the treatment and / or prophylaxis of sepsis (SIRS), multiple organ failure (MODS, MOF), inflammatory diseases of the kidney, chronic inflammatory bowel disease (IBD, Crohn's Disease, UC), pancreatitis , Peritonitis, rheumatoid diseases, inflammatory skin diseases and inflammatory eye diseases.
- SIRS sepsis
- MODS multiple organ failure
- IBD chronic inflammatory bowel disease
- UC chronic inflammatory bowel disease
- pancreatitis atitis
- Peritonitis rheumatoid diseases
- inflammatory skin diseases and inflammatory eye diseases.
- the compounds of the invention can also be used for the treatment and / or prophylaxis of autoimmune diseases.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of fibrotic disorders of the internal organs such as, for example, the lung, the heart, the kidney, the bone marrow and in particular the liver, as well as dermatological fibroses and fibrotic disorders of the eye.
- fibrotic disorders includes in particular the following terms: liver fibrosis, cirrhosis, pulmonary fibrosis, endomyocardial fibrosis, nephropathy, glomerulonephritis, interstitial renal fibrosis, fibrotic damage as a result of diabetes, bone marrow fibrosis and similar fibrotic disorders, scleroderma, morphea, keloids, hypertrophic scarring (also after surgical interventions), nevi, diabetic retinopathy, proliferative vitroretinopathy and connective tissue disorders (eg sarcoidosis).
- the compounds according to the invention are suitable for combating postoperative scar formation, for example as a consequence of glaucoma operations.
- the compounds according to the invention can likewise be used cosmetically for aging and keratinizing skin.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of hepatitis, neoplasm, osteoporosis, glaucoma and gastroparesis.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of heart failure, angina pectoris, hypertension, pulmonary hypertension, ischaemia, vascular disease, renal insufficiency, thromboembolic disorders, fibrotic diseases and arteriosclerosis.
- the present invention furthermore relates to the compounds according to the invention for use in a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular disorders, renal insufficiency, thromboembolic disorders, fibrotic disorders and atherosclerosis.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of heart failure, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular diseases, renal insufficiency, thromboembolic disorders, fibrotic diseases and arteriosclerosis.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an effective amount of at least one of the compounds of the invention.
- the present invention further provides a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular diseases, renal insufficiency, thromboembolic disorders, fibrotic diseases and atherosclerosis, using an effective amount of at least one of the compounds according to the invention ,
- the compounds of the invention may be used alone or as needed in combination with other agents.
- Another object of the present invention are pharmaceuticals, containing at least one of the compounds according to the invention and one or more further active compounds, in particular for the treatment and / or prophylaxis of the abovementioned disorders.
- suitable combination active ingredients may be mentioned by way of example and preferably:
- organic nitrates and NO donors such as sodium nitroprusside, nitroglycerin, isosorbide mononitrate, isosorbide dinitrate, molsidomine or SIN-1, and inhaled NO;
- cGMP cyclic guanosine monophosphate
- PDE phosphodiesterases
- Antithrombotic agents by way of example and preferably from the group of thrombocyte aggregation inhibitors, anticoagulants or profibrinolytic substances;
- Antihypertensive agents by way of example and preferably from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor B-relaxer, beta-receptor blocker, mineralocorticoid receptor - antagonists and diuretics; and / or ⁇ fat metabolism-altering agents, by way of example and preferably from the group of thyroid receptor agonists, cholesterol synthesis inhibitors such as by way of example and preferably HMG-CoA reductase or squalene synthesis inhibitors, ACAT inhibitors, CETP inhibitors, MTP inhibitors , PPAR alpha, PPAR gamma and / or PPAR delta agonists, cholesterol absorption inhibitors, lipase inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, and lipoprotein (a) antagonists.
- angiotensin AII antagonists by way of
- Antithrombotic agents are preferably understood as meaning compounds from the group of platelet aggregation inhibitors, anticoagulants or profibrinolytic substances.
- the compounds according to the invention are administered in combination with a platelet aggregation inhibitor, such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- a platelet aggregation inhibitor such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- the compounds according to the invention are administered in combination with a thrombin inhibitor, such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- a thrombin inhibitor such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- the compounds according to the invention are administered in combination with a GPIIb / IIIa antagonist, such as, by way of example and by way of preference, tirofiban or abciximab.
- the compounds according to the invention are used in combination with a factor Xa inhibitor, such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD 31 12, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 or SSR-128428.
- a factor Xa inhibitor such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD 31 12, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-10
- the compounds according to the invention are administered in combination with heparin or a low molecular weight (LMW) heparin derivative.
- LMW low molecular weight
- the compounds according to the invention are administered in combination with a vitamin K antagonist, such as by way of example and preferably coumarin.
- antihypertensive agents are preferably compounds from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor B-relaxer, beta-receptor blocker, mineralocorticoid receptor Tor antagonists and diuretics understood.
- the compounds according to the invention are administered in combination with a calcium antagonist, such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- a calcium antagonist such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- the compounds according to the invention are administered in combination with an alpha-1-receptor blocker, such as by way of example and preferably prazosin.
- the compounds according to the invention are used in combination with a beta-receptor blocker, such as by way of example and preferably propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipranolol, nadolol, mepindolol, carazalol, sotalol, Metoprolol, betaxolol, celiprolol, bisoprolol, carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol or bucindolol.
- a beta-receptor blocker such as by way of example and preferably propranolol, atenolol, timolol
- the compounds according to the invention are administered in combination with an angiotensin AII antagonist, such as by way of example and preferably losartan, candesartan, valsartan, telmisartan or embursatan.
- an ACE inhibitor such as, by way of example and by way of preference, enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- the compounds according to the invention are administered in combination with an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- the compounds of the invention are administered in combination with a renin inhibitor, such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- a mineralocorticoid receptor antagonist such as by way of example and preferably spironolactone or eplerenone.
- the compounds of the present invention are used in combination with a loop diuretic such as furosemide, torasemide, bumetanide and piretanide with potassium sparing diuretics such as amiloride and triamterene with aldosterone antagonists such as spironolactone, potassium canrenoate and eplerenone and thiazide diuretics such as Hydrochlorothiazide, chlorthalidone, xipamide, and indapamide.
- a loop diuretic such as furosemide, torasemide, bumetanide and piretanide
- potassium sparing diuretics such as amiloride and triamterene with aldosterone antagonists such as spironolactone, potassium canrenoate and eplerenone and thiazide diuretics
- Hydrochlorothiazide chlorthalidone
- xipamide xipamide
- indapamide indapamide
- lipid metabolizing agents are preferably compounds from the group of CETP inhibitors, thyroid receptor agonists, cholesterol synthesis inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors, the ACAT inhibitors, MTP inhibitors, PPAR-alpha, PPAR gamma and / or PPAR delta agonists, cholesterol absorption inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, lipase inhibitors and the lipoprotein (a) antagonists understood.
- CETP inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors
- ACAT inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors
- MTP inhibitors MTP inhibitors
- PPAR-alpha PPAR-alpha
- PPAR gamma and / or PPAR delta agonists cholesterol absorption inhibitors
- polymeric bile acid adsorbers
- the compounds according to the invention are administered in combination with a CETP inhibitor, such as, for example and preferably, dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- a CETP inhibitor such as, for example and preferably, dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- the compounds of the invention are administered in combination with a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- the compounds according to the invention are administered in combination with an HMG-CoA reductase inhibitor from the class of statins, such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin or pitavastatin.
- the compounds according to the invention are administered in combination with a squalene synthesis inhibitor, such as by way of example and preferably BMS
- the compounds according to the invention are administered in combination with an ACAT inhibitor, such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- an ACAT inhibitor such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- the compounds according to the invention are administered in combination with an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- the compounds of the invention are administered in combination with a PPAR-gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- a PPAR-gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- the compounds according to the invention are administered in combination with a PPAR delta agonist, such as by way of example and preferably GW 501516 or BAY 68-5042.
- a PPAR delta agonist such as by way of example and preferably GW 501516 or BAY 68-5042.
- the compounds according to the invention are administered in combination with a cholesterol absorption inhibitor, such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- the compounds according to the invention are administered in combination with a lipase inhibitor, such as, for example and preferably, orlistat.
- a lipase inhibitor such as, for example and preferably, orlistat.
- the compounds of the invention are administered in combination with a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- ASBT IBAT
- the compounds of the invention are administered in combination with a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- compositions containing at least one compound of the invention are pharmaceutical compositions containing at least one compound of the invention, usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the compounds according to the invention can act systemically and / or locally. For this purpose, they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds of the invention rapidly and / or modified donating application forms containing the compounds of the invention in crystalline and / or amorphized and / or dissolved form, such.
- Tablets uncoated or coated tablets, for example with enteric or delayed-release or insoluble coatings which control the release of the compound of the invention
- tablets or films / wafers rapidly breaking down in the oral cavity, films / lyophilisates
- capsules e.g. Soft gelatin capsules
- dragees granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally).
- a resorption step e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar
- absorption e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally.
- parenteral administration are suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- inhalant medicaments including powder inhalers, nebulizers
- nasal drops solutions or sprays
- lingual, sublingual or buccal are suitable applying tablets, films / wafers or capsules, suppositories, ear or eye preparations, vaginal capsules, aqueous suspensions (lotions, shaking mixtures), lipophilic suspensions, ointments, creams, transdermal therapeutic systems (eg patches), milk, pastes, foams, powdered litter , Implants or stents.
- Preference is given to oral or parenteral administration, in particular oral administration.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitol oleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- stabilizers for example, antioxidants such as ascorbic acid
- dyes eg, inorganic pigments such as iron oxides
- flavor and / or odoriferous for example, antioxidants such ascorbic acid
- dyes eg, inorganic pigments such as iron oxides
- the dosage is about 0.01 to 100 mg / kg, preferably about 0.01 to 20 mg / kg and most preferably 0.1 to 10 mg / kg of body weight.
- Instrament Waters ACQUITY SQD UPLC System; Column: Waters Acquity UPLC HSS T3 1,8 ⁇ 50 x 1mm; Eluent A: 1 l of water + 0.25 ml of 99% formic acid, eluent B: 1 l of acetonitrile + 0.25 ml of 99% formic acid; Gradient: 0.0 min 90% A -> 1.2 min 5% A -> 2.0 min 5% A; Oven: 50 ° C; Flow: 0.40 ml / min; UV detection: 210 - 400 nm.
- Device Type MS Waters ZQ; Device type HPLC: Agilent 1 100 Series; UV DAD; Column: Thermo Hypersil GOLD 3 ⁇ 20 mm x 4 mm; Eluent A: 1 l of water + 0.5 ml of 50% formic acid, eluent B: 1 l of acetonitrile + 0.5 ml of 50% formic acid; Gradient: 0.0 min 100% A -> 3.0 min 10% A-> 4.0 min 10% A, oven: 55 ° C; Flow 2ml / min; UV detection: 210 nm.
- the diazonium salt thus prepared was added in portions to a 0 ° C cold solution of 12.81 g (85.45 mmol) of sodium iodide in acetone (329 ml) and the mixture stirred for 30 min at RT.
- the reaction mixture was added to ice water (1.8 L) and extracted twice with ethyl acetate (487 mL each).
- the collected organic phases were washed with saturated aqueous sodium chloride solution (244 ml), dried, filtered and concentrated. 12.1 g (86% purity, 60% of theory) of the title compound were obtained as a solid.
- the crude product was reacted without further purification.
- Example 13A 440 mg (11.00 mmol) of sodium hydride (60% strength in mineral oil) were initially charged in 30 ml of THF and 726 mg (11.00 mmol) of malononitrile were added in portions. Thereafter, 2.3 g (11.00 mmol) of the compound obtained in Example 69A in THF (50 ml) was added. It was stirred for 6 h at RT and then heated to 50 ° C overnight. After cooling, the batch was washed with saturated aqueous sodium bicarbonate solution and extracted with water Extracted ethyl acetate. The combined organic phases were washed with saturated aqueous sodium chloride solution and then dried with sodium sulfate, filtered and concentrated. The residue (2.66 g) was dried under high vacuum for 1 h and then reacted without further purification.
- Example 13A 440 mg (11.00 mmol) of sodium hydride (60% strength in mineral oil) were initially charged in 30 ml of THF and 726 mg (11
- Example 1A 5,887 g (19,256 mmol) of Example 1A were initially charged in tert-butanol (50 ml) and treated with 2,593 g (23.107 mmol) of potassium tert-butoxide. Subsequently, 3.2 g (19.256 mmol) of Example 10A in tert-butanol (25 ml) were added dropwise and the mixture was heated to reflux overnight. The next day another 0.64 g (3.851 mmol) of Example 10A was added and the mixture was heated to reflux for a further day. After cooling, a precipitate was filtered off, which was washed with diethyl ether. It was then slurried in water and filtered once more from and washed with diethyl ether. After drying under high vacuum, it was possible to obtain 6.65 g of the title compound (85% of theory).
- Example 9A In analogy to the preparation of Example 2, 4.18 g (12.035 mmol) of Example 9A were reacted with 2.20 g (13.239 mmol) of Example 10A. 3.72 g of the title compound were obtained (73% of theory).
- Example 13A In analogy to the preparation of Example 13A, 2,257 g (7,382 mmol) of Example 1A were reacted with 1,434 g (7,382 mmol) of Example 12A. 566 mg of the title compound were obtained (17% of theory).
- Example 16A Ethyl ⁇ 2- [1- (2-fluorobenzyl) -1 H -pyrazolo [3,4-b] pyridin-3-yl] -4-hydroxypyrimidin-5-yl ⁇ acetate
- Example 16A 55.00 g (135 mmol) of Example 16A were initially charged in sulfolane (220 ml) and admixed with 41.40 g (270 mmol) of phosphoryl chloride. Thereafter, it was heated to 120 ° C for 1 h. After cooling, it was added to warm water (1 500 ml) and then neutralized with solid sodium bicarbonate. The precipitate which formed was filtered off with suction and washed with water. Thereafter, it was further purified by chromatography on silica gel (mobile phase: cyclohexane / ethyl acetate 3: 2). After drying under high vacuum, 43.0 g of the title compound were obtained (73% of theory).
- Example 17A 10.00 g (23.482 mmol) of Example 17A were initially charged in DMF (200 ml) and treated with 2.290 g (35.223 mmol) of sodium azide. Thereafter, it was heated to 60 ° C for 1 h. After cooling, the reaction mixture was added to water and extracted three times with ethyl acetate. The organic phases were combined and washed once with saturated aqueous sodium chloride solution, then dried over sodium sulfate, filtered and concentrated. The residue was used without further purification in the next step.
- Example 17A 1.00 g (2.348 mmol) of Example 17A were initially charged in THF (15 ml) and DMF (15 ml) and treated with 469 mg (11.741 mmol) of sodium hydride (60%) and stirred for 15 min at RT. Thereafter, 0.607 ml (7.054 mmol) of 1,2-dibromoethane were added and the mixture was stirred at RT for a further 30 min. The mixture was combined with water and ethyl acetate, the phases were separated and the organic Phase extracted twice with ethyl acetate. The combined organic phases were washed once with saturated aqueous sodium chloride solution and dried with sodium sulfate, filtered and concentrated.
- Example 20A 550 mg (about 0.913 mmol) of Example 20A were reacted in analogy to the procedure of Example 18A. The title compound thus obtained was used in the next step without further purification.
- Example 21A 418 mg (0.913 mmol) of Example 21 A were hydrogenated in analogy to the procedure of Example 19A. The title binding thus obtained was used without further purification in the next step.
- Example 25 A 600 mg (1312 mmol) of Example 25 A were reacted. There were obtained 1.7 g of the title compound (about 36% purity, contaminated with sulfolane).
- Example 26A In analogy to 20A, 1.7 g (about 1.31 mmol) of Example 26A were reacted with methyl iodide. There were obtained 1.24 g of the title compound (about 36% purity, contaminated with sulfolane).
- Example 27A In analogy to Example 18A, 1.24 g (about 1312 mmol) of Example 27A were reacted. There was obtained the title compound (contaminated with sulfolane), which was further reacted without determination of the yield, since the azide-containing solution was not further concentrated.
- Example 29A In analogy to Example 8A, 76.00 g (204.78 mmol) of Example 29A were reacted. 57.00 g (94% pure, 97% of theory) of the title compound were obtained.
- Example 30A In analogy to Example 9A, 56.00 g (207,221 mmol) of Example 30A were reacted. 19.00 g of the title compound were obtained (26% of theory).
- Example 31A 15 g (43187 mmol) of Example 31A were reacted analogously to the procedure of Example 16A. 14.30 g of the title compound (75% of theory) were obtained.
- Example 32A 14.20 g (33.380 mmol) of Example 32A were reacted analogously to the procedure of Example 17A. 13.20 g of the title compound (88% of theory) were obtained.
- Example 7A In analogy to the synthesis of Example 7A, 54.00 g (215.98 mmol) of 3-iodo-1H-pyrazolo [3,4-b] pyridine (synthesis described in WO 2006/130673 Example 4) were mixed with 50.18 g (237,578) 2,3- Implemented difluorobenzyl bromide. The crude product was mixed with ice-water, filtered off with suction and the precipitate was washed with isopropanol and pentane and then dried under high vacuum. There were obtained 73.00 g of the title compound (85% of theory).
- Example 34A In analogy to the synthesis of Example 8A, 72.00 g (194,002 mmol) of Example 34A were reacted. There were obtained 50.00 g (93% pure, 88% of theory) of the title compound.
- Example 35A In analogy to the synthesis of Example 9A, 49.00 g (181.318 mmol) of Example 35A were reacted. 29.00 g of the title compound were obtained (46% of theory).
- Example 36A 15 g (43187 mmol) of Example 36A were reacted analogously to the procedure of Example 16A. 13.20 g of the title compound (69% of theory) were obtained.
- Example 37A 13.10 g (30.795 mmol) of Example 37A were reacted analogously to the procedure described in Example 17A. 12.10 g of the title compound (88% of theory) were obtained.
- Example 38A 800 mg (1.802 mmol) of Example 38A were reacted with methyl iodide. There were obtained 1.00 g (purity 84%) of the title compound, which were reacted further without further purification.
- Example 40A The solution obtained in Example 40A was hydrogenated in analogy to Example 19A.
- the crude compound thus obtained (0.863 g, about 94% purity) was used without further purification in the next stage.
- Example 16A In analogy to Example 16A, 3.74 g (12,234 mmol) of Example 1A and 3.21 1 g of Example 42A were reacted. After working up, 763 mg (14% of theory) of the title compound were obtained.
- Example 43A 762 mg (1.817 mmol) of Example 43A were initially charged in methanol (20 ml) and treated with 3 drops of conc. Sulfuric acid added. A slurry was obtained which could be stirred again by further addition of methanol (15 ml). It was heated to reflux for 1 h. After cooling, the product was filtered off with suction and washed with methanol and dried under high vacuum. 685 mg (87% of theory) of the title compound were obtained.
- Example 44A 683 mg (1.576 mmol) of Example 44A were initially charged in phosphoryl chloride (2.423 ml), treated with 470 mg (3.151 mmol) of diethylaniline and heated to 90 ° C for 2 days. After cooling, the mixture was added to warm water, the resulting precipitate was filtered off with suction and washed with water and dried under high vacuum. 724 mg (100% of theory) of the title compound were obtained.
- Example 45A 712 mg (1576 mmol) of Example 45A were reacted with methyl iodide. There were 867 mg (about 75% purity) of the title compound, which were used without further purification in the next step.
- Example 46A 867 mg (about 1.355 mmol) of Example 46A were reacted in analogy to the procedure of Example 18A.
- the title compound thus obtained was used in the next step without further purification. A yield was not determined because the azide-containing solution was not concentrated to dryness.
- Example 33A In analogy to Example 20A, 800 mg (1.802 mmol) of Example 33A were reacted with methyl iodide. There were obtained 1.05 g (78% purity) of the title compound, which were used without further purification in the next step.
- Example 48A 1.05 g (about 1.736 mmol) of Example 48A were reacted in analogy to the procedure of Example 18A. The title compound thus obtained was used in the next step without further purification. A yield was not determined because the azide-containing solution was not concentrated to dryness.
- Example 49A The solution obtained under Example 49A was hydrogenated in analogy to the procedure of Example 19A.
- the title compound thus obtained (0.755 g, about 76% purity) was used without further purification in the next step.
- Example 51A 2,174 g (7,112 mmol) of Example 1A and 1.3 g (7.823 mmol) of Example 51A were initially charged in 20 ml of methanol and then 423 mg (7.823 mmol) of sodium methoxide were added in portions at RT. The mixture was stirred for 10 min at RT and then heated overnight to reflux. After cooling, the reaction was treated with acetic acid (0.5 ml) and water (20 ml) and ice bath cooled. The precipitate was filtered off, washed with water and methanol and then dried under high vacuum. 2.51 g of the title compound were obtained (87% of theory).
- Example 1A 694 mg (2.271 mmol) of Example 1A and 500 mg (2.271 mmol) of Example 14A were initially charged in 10 ml of tert-butanol and then treated at RT with 305 mg (2.725 mmol) of potassium tert-butoxide in portions. The mixture was stirred for 10 min at RT and then heated for 2 days to reflux. After cooling, the reaction mixture was treated with water and ethyl acetate. The precipitate was sucked off. The filtrate was concentrated, treated with a little ethyl acetate and diethyl ether and the resulting precipitate was filtered off with suction. The combined solids fractions were then dried under high vacuum. 588 mg of the title compound were obtained (53% of theory).
- Example 13A 5.00 g (12.394 mmol) of Example 13A were initially charged in iso-pentyl nitrite (35.87 ml) and diiodomethane (1.16 mol, 93.71 ml) and heated to 85 ° C. for 12 h. After cooling, it was filtered off from solids, concentrated and then purified by chromatography on silica gel (mobile phase: first cyclohexane-dichloromethane gradient, then dichloromethane-methanol gradient). There were obtained 5.50 g of the title compound (67% of theory).
- Example 14A 3,325 g (7,890 mmol) of Example 14A were reacted in analogy to Example 55A. There was obtained 3.65 g of the title compound (87% of theory, 61% purity).
- Example 53A 530 mg (1314 mmol) of Example 53A were reacted analogously to the procedure of Example 57A. 171 mg of the title compound were obtained (25% of theory).
- Example 54A 556 mg (1.216 mmol) of Example 54A were reacted in analogy to Example 57A. There were obtained 605 mg of the title compound (87% of theory).
- Example 9A 23,000 g (66.22 mmol) of Example 9A were dissolved in 322 ml of ethanol and treated at 0 ° C with 26,804 g (264.88 mmol) of triethylamine and 6.027 g (66.22 mmol) of hydrazine hydrate (55% solution in water). The mixture was stirred overnight at rt and then added to 1.715 l of a 10% aqueous sodium chloride solution and extracted twice with ethyl acetate. The combined organic phases were washed with 10% aqueous sodium chloride solution, dried over sodium sulfate and concentrated on a rotary evaporator. The residue was purified on silica gel (eluent: dichloromethane / methanol, 95: 5). There were obtained 15,000 g (75% of theory) of the title compound.
- Example 60A 1,780 g (38.97 mmol) of Example 60A were dissolved in 353 ml of ethanol and treated with 14,667 g (77.94 mmol) of dimethyl 2,2-dimethyl-3-oxobutanedioate (described in J. Am. Chem. Soc. 124 (14), 3680-3691; 2002). The mixture was heated to reflux overnight. After cooling, the solid was filtered off with suction, washed with a little ethanol and the filtrate was concentrated. The residue was purified on silica gel (eluent: dichloromethane / acetone, 95: 5). There was obtained 10,000 g (58% of theory) of the title compound.
- Example 6A 10,000 g (38.02 mmol) of Example 6A were dissolved in 270 ml of dimethylformamide, and 8,858 g (41.82 mmol) of 1- (bromomethyl) -2,3-difluorobenzene and 13,627 g (41.82 mmol) of cesium carbonate were added. It was stirred for 2 h at room temperature and then ethyl acetate and water were added. The organic phase was separated and the aqueous phase extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated on a rotary evaporator. There were obtained 8,460 g (56% of theory) of the target compound. The residue was used without further purification in the next step.
- Example 65A (2,3-Difluorobenzyl) -5-fluoro-1H-pyrazolo [3,4-b] pyridine-3-carboximidohydrazide
- Example 6A 10,000 g (38.02 mmol) of Example 6A were dissolved in 270 ml of dimethylformamide and 8658 g (41.82 mmol) of 1- (bromomethyl) -2,4-difluorobenzene and 13,627 g (41.82 mmol) of cesium carbonate added. It was stirred for 2 h at room temperature and then ethyl acetate and water were added. The organic phase was separated and the aqueous phase extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated on a rotary evaporator. 8,340 g (53% of theory, purity about 93%) of the target compound were obtained. The residue was used without further purification in the next step.
- Example 68A 0.383 g (16.68 mmol) of sodium were stirred in portions into 118 ml of methanol. After completion of gas evolution, 5,270 g (about 16.68 mmol) of Example 68A were added in portions and the mixture was stirred for 2 h at room temperature. 1.070 g (20.01 mmol) of ammonium chloride and 3.895 g (64.86 mmol) of acetic acid were added and the mixture was refluxed overnight. After cooling, the batch was concentrated on a rotary evaporator and the residue was combined with ethyl acetate and 1N sodium hydroxide solution, forming a solid. The solid was filtered off with suction, washed with ethyl acetate and dried under high vacuum. 5,640 g (93% of theory) of the target compound were obtained.
- Example 69A 5,640 g (15.44 mmol) of Example 69A were dissolved in 76 ml of ethanol and treated at 0 ° C with 6,249 g (61.76 mmol) of triethylamine and 0.966 g (15.44 mmol) of hydrazine hydrate (80% solution in water). The mixture was stirred at RT overnight and then concentrated by rotary evaporation. There were obtained 5.30 g (100% of theory, purity 93%) of the title compound. The product was used without further purification in the next step.
- Example 69A 5,300 g (about 15.42 mmol) of Example 69A were dissolved in 140 ml of ethanol, containing 5,805 g (30.85 mmol) of dimethyl 2,2-dimethyl-3-oxobutanedioate (described in J. Am. Chem. Soc., 124 (14). , 3680-3691, 2002) and stirred overnight. The resulting solid was filtered off with suction, washed with a little ethanol and the filtrate was concentrated. The residue was stirred with diethyl ether, filtered off, washed with a little diethyl ether and dried under high vacuum. There were obtained 2,290 g (32% of theory) of the title compound.
- Example 4 700 mg (1,943 mmol) of Example 4 were suspended in acetic acid (14 ml) and treated with 281 mg (4.079 mmol) of sodium nitrite and a few drops of water. After 30 min at RT, water was added and a precipitate was filtered off, washed with water and then dried under high vacuum. 756 mg of the title compound (99% of theory) were obtained.
- Example 72A 124 mg (0.319 mmol) of Example 72A were initially charged in trifluoroacetic acid (2.5 ml) and admixed with 41 mg (0.637 mmol) of zinc dust and stirred at RT overnight. It was then concentrated and the raw material (about 150 mg) used without further purification in the next stage.
- Example 36A In analogy to Example 70A, 10.00 g (28.791 mmol) of Example 36A were reacted. 11.74 g of the title compound were obtained in about 60% purity (81% of theory). The compound was used without further purification in the next step.
- Example 74A was dissolved in 300 ml of ethanol containing 8,037 g (42,710 mmol) of dimethyl 2,2-dimethyl-3-oxobutanedioate (described in J. Am. Chem. Soc. 124 (14), 10.67 g (about 21.355 mmol). , 3680-3691, 2002) and stirred at 50 ° C overnight. After cooling, it was filtered from a precipitate and washed with ethanol. The residue was concentrated and then taken up in methanol (50 ml) and acetonitrile (50 ml) and purified by preparative HPLC (acetonitrile-water gradient). There was obtained 0.75 g (8% of theory) of the title compound.
- Example 31A In analogy to Example 70A, 10.00 g (28.791 mmol) of Example 31A were reacted. This gave 9.31 g of the title compound in about 82% purity (87% of theory). The compound was used without further purification in the next step.
- Example 76A 6.80 g (about 18,446 mmol) of Example 76A were dissolved in 272 ml of ethanol, with 6,942 g (36,892 mmol) of dimethyl 2,2-dimethyl-3-oxobutanedioate (described in J. Am. Chem. Soc., 124 (14). , 3680-3691, 2002) and stirred at 50 ° C overnight. After cooling, it was filtered from a precipitate and washed with ethanol. The residue was taken up in a lot of ethanol, whereupon a solid formed again. This was filtered off and washed with ethanol and diethyl ether. The solid was dried to give the title compound 2.21 g (26% of theory).
- Example 78A In analogy to Example 8A, 12.1 g (29.72 mmol) of Example 78A were reacted. There were obtained 9.35 g of the title compound in 79% purity (81% of theory).
- Example 9A In analogy to Example 9A, 9.1 g of the crude compound of Example 79A were reacted. This gave 7.77 g of the title compound (86% of theory).
- Example 80A In analogy to the synthesis of Example 70A, 7.77 g (20.271 mmol) of Example 80A were reacted. There were obtained 6.85 g of the title compound (99% of theory). The compound was used without further purification in the next step.
- Example 9A 2.00 g (5.73 mmol) of Example 9A and 1.42 g (6.58 mmol) of diethyl 2-formylbutandioate (synthesis described in WO 2005/73234, page 43) were initially charged in 50 ml of ethanol. Subsequently, 4.27 ml of sodium ethylate solution (21% strength in ethanol, 11.5 mmol) were added dropwise. Then it was heated to reflux for 9 h. After cooling, the reaction mixture was treated with 25 ml of water and then with 1N hydrochloric acid. The resulting precipitate was filtered off with suction and washed successively with methanol (20 ml) and with diethyl ether (20 ml). After drying under high vacuum, 1.53 g of the title compound were obtained (63% of theory).
- Example 83A 1.53 g (3.60 mmol) of Example 83A were initially charged in 3.35 ml (36.0 mmol) of phosphoryl chloride. Then, 0.684 ml (5.40 mmol) of N.N-dimethylaniline was added. Subsequently, lh was reacted in an oil bath at 150 ° C. The volatile components were separated on a rotary evaporator and then the residue was carefully stirred into 50 ml of 2M aqueous sodium carbonate solution. The mixture was stirred for 15 min and the solid filtered off. The solid was taken up in ethyl acetate, washed with saturated aqueous sodium chloride solution and the organic phase was dried with magnesium sulfate. The solvent was removed by distillation under reduced pressure and the residue was dried overnight under high vacuum. There was thus obtained 1.44 g of the title compound (90% of theory).
- Example 86A 1.15 g (2.41 mmol) of Example 86A were initially charged in 30 ml of DMF. Then, 500 mg (10%) of palladium on charcoal was added. Hydrogen pressure was then hydrogenated at 1 atmosphere for 3 h. The reaction mixture was filtered through Celite. It was washed with DMF and the volatile components were separated on a rotary evaporator. The crude material thus obtained was dried in a high vacuum overnight. Thus, 1.28 g of crude material was obtained, which contained the title compound in about 90% purity (LC-MS). The material thus obtained was used without further purification.
- Example 1 Exemplary embodiments: Example 1
- Example 55A 1.00 g (1944 mmol) of Example 55A were added in 425 mg of palladium on carbon (10%) in DMF (50 ml) and hydrogenated for 4 hours at normal hydrogen pressure. It was then filtered through Celite and concentrated to dryness. The residue was purified by preparative HPLC (acetonitrile-water (+0.05% formic acid) gradient). 500 mg of the title compound were obtained (66% of theory).
- Example 56A 1.74 g (about 1,944 mmol) of Example 56A were reacted in analogy to Example 1. 644 mg of the title compound were obtained (78% of theory).
- Example 57A 96 mg (0.177 mmol) of Example 57A were reacted in analogy to Example 1. There was obtained 51 mg of the title compound (68% of theory).
- Example 22A 394 mg (0.913 mmol) of Example 22A were reacted in analogy to the procedure under Example 4. There were obtained 186 mg of the title compound (53% of theory).
- Example 28A The crude product from Example 28A (1.24 g, about 1312 mmol) was hydrogenated analogously to the procedure of Example 19A. It was then filtered through Celite and concentrated. The residue was purified by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid). 101 mg of the title compound were obtained (17% of theory).
- Example 8 2 '- [1- (2,4-Difluorobenzyl) -1H-pyrazolo [3,4-b] pyridin-3-yl] spiro [cyclopropane-l, 5'-pyrrolo [2,3-d] pyrimidine ] -6 '(7'H) -on
- Example 33A 0.80 g (1.802 mmol) of Example 33A were dissolved in 15 ml of DMF under argon, combined with 360 mg (9,012 mmol) of sodium hydride (60% suspension in mineral oil) and stirred at RT for 15 min. Subsequently, 1.015 g (5.407 mmol) of 1,2-dibromoethane were added and stirred at RT for 2 h. Water and saturated aqueous sodium chloride solution were added, and the mixture was extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated on a rotary evaporator. 848 mg (purity 54%, 54% of theory) of the intermediate product were obtained
- the intermediate from step a) was dissolved in 13 ml of DMF and admixed with 95 mg (1.461 mmol) of sodium azide.
- the reaction mixture was stirred for 7 h at 60 ° C and added after cooling to water. It was extracted three times with ethyl acetate.
- the combined organic phases were washed with saturated aqueous sodium chloride solution, dried over sodium sulfate, treated with 10 ml of DMF and the ethyl acetate removed at 80 mbar on a rotary evaporator.
- the product-containing DMF solution was mixed with 20 ml of DMF and treated with 250 mg of palladium (10% on charcoal) and hydrogenated at atmospheric pressure overnight.
- the reaction solution was filtered through Celite and the filter cake was washed with DMF.
- the combined organic phases were concentrated on a rotary evaporator.
- the intermediate from stage b) was taken up in 20 ml of THF, combined under an argon atmosphere with 109 mg (0.974 mmol) of potassium tert-butoxide and stirred for 2 h at RT.
- the intermediate from step a) was admixed with 35 ml of phosphoryl chloride and stirred overnight.
- the reaction mixture was dissolved in 500 ml of acetonitrile and stirred with ice cooling in 300 ml of concentrated ammonia solution. The mixture was stirred for 2 h at RT and the reaction mixture was concentrated on a rotary evaporator.
- the intermediate from step a) was dissolved in 13 ml of DMF and mixed with 131 mg (2.020 mmol) of sodium azide.
- the reaction mixture was stirred for 7 h at 60 ° C and added after cooling to water. It was extracted three times with ethyl acetate.
- the combined organic phases were washed with saturated aqueous sodium chloride solution, dried over sodium sulfate, treated with 10 ml of DMF and the ethyl acetate removed at 80 mbar on a rotary evaporator.
- the product-containing DMF solution was mixed with 20 ml of DMF and treated with 250 mg of palladium (10% on carbon) and hydrogenated at atmospheric pressure for 2 h.
- the reaction solution was filtered through Celite and the filter cake was washed with DMF.
- the combined organic phases were concentrated on a rotary evaporator.
- Example 47A The solution obtained in Example 47A was hydrogenated in analogy to Example 19A. After purification by preparative HPLC (eluent: acetonitrile / water with 0.1% formic acid), 48 mg of the title compound were obtained (7% of theory).
- Example 58A 168 mg (0.327 mmol) of Example 58A were reacted in analogy to Example 1. There were obtained 71 mg of the title compound (56% of theory).
- Example 59A 603 mg (1.061 mmol) of the compound obtained in Example 59A were reacted in analogy to Example 1. 174 mg of the title compound were obtained (37% of theory).
- Example 4 2.00 g (5.550 mmol) of Example 4 were initially charged in dioxane (200 ml) and treated with 3.079 g (27.751 mmol) of selenium dioxide and heated to reflux for 2 h. After cooling, it was filtered and the Filtrate was concentrated and purified by chromatography on silica gel (mobile phase: cyclohexane / ethyl acetate 1: 1). 890 mg of the title compound were obtained (42% of theory).
- Example 16 200 mg (0.534 mmol) of Example 16 were initially charged in THF (10 ml) at 0 ° C. and admixed with 0.356 ml (1.069 mmol) of methylmagnesium bromide (3M solution in diethyl ether). After 15 min at 0 ° C was warmed to RT for 1 h. Subsequently, the batch was added to saturated aqueous ammonium chloride solution and extracted three times with ethyl acetate. The organic phases were combined, washed once with water and once with saturated aqueous sodium chloride solution. It was then dried over sodium sulfate, filtered and concentrated. The residue was purified by preparative HPLC (acetonitrile: water (+0.05% formic acid) gradient). 55 mg of the title compound were obtained (53% of theory).
- Example 16 150 mg (0.401 mmol) of Example 16 were initially charged in 1,2-dimethoxyethane (3 ml) at RT and admixed with 6 mg (0.04 mmol) of cesium fluoride. Subsequently, 177 ⁇ (1 .202 mmol) (trifluoromethyl) trimethylsilane were added dropwise and the mixture was stirred overnight at RT. Thereafter, the following reagents were again added: 1,2-dimethoxyethane (2 ml), cesium fluoride (20 mg) and (trifluoromethyl) trimethylsilane (177 ⁇ ).
- Example 16 75 mg (0.200 mmol) of Example 16 were initially charged in THF (5 ml) at 0 ° C. and admixed with 0.134 ml of a 3M solution of phenylmagnesium bromide in diethyl ether (0.401 mmol). After 15 min at 0 ° C was warmed to RT for 1 h. Subsequently, the batch was added to saturated aqueous ammonium chloride solution and extracted three times with ethyl acetate. The organic phases were combined, washed once with water and once with saturated aqueous sodium chloride solution. It was then dried over sodium sulfate, filtered and concentrated.
- Example 17 100 mg (0.256 mmol) of Example 17 were initially charged in dichloromethane (4,795 ml) at -78 ° C., and 40.61 ⁇ l (0.307 mmol) of diethylaminosulfur trifluoride were added. The mixture was then warmed slowly to RT overnight. Thereafter, another 40.61 ⁇ (0.307 mmol) of diethylaminosulfur trifluoride was added and the mixture was stirred at RT for a further night. The reaction was then diluted with dichloromethane and extracted with saturated aqueous sodium bicarbonate solution. The organic phase was dried over sodium sulfate, filtered and concentrated.
- Example 24 60 mg (0.122 mmol) of Example 24 were stirred in dichloromethane (1 ml) and trifluoroacetic acid (1 ml) for 30 min at RT. It was then concentrated and the residue taken up in acetonitrile and treated with water. A precipitate was filtered off, which was washed with acetonitrile and dried under high vacuum. This gave 42 mg of the title compound (80% of theory).
- Example 73A 150 mg (about 0.319 mmol) of Example 73A were initially charged in pyridine (2 ml) and then methyl chloroformate (24 ⁇ in 1 ml of dichloromethane) was added until complete conversion was achieved. It was then concentrated and the residue was purified by preparative HPLC (acetonitrile-water (+0.05% formic acid) gradient). There were obtained 1 1 mg of the title compound in 90% purity (7% of theory).
- Example 87A 1.28 g (2.84 mmol) of Example 87A were reacted in analogy to the procedure of Example 4 with 478 mg (4.26 mmol) of potassium tert-butoxide in 60 ml of THF. There was obtained 426 mg of the title compound (36% of theory).
- Example 4 500 mg (1.388 mmol) of Example 4 were initially charged in acetone (50 ml), treated with 0.274 ml (2.775 mmol) of piperidine and heated to reflux for 1 h. After cooling, it was filtered from a precipitate, washed with acetone and the filtrate was concentrated in vacuo. This residue was then purified by chromatography on silica gel (mobile phase: cyclohexane / ethyl acetate gradient). The material thus obtained was reslurried in ethyl acetate, filtered off, washed with ethyl acetate and dried under high vacuum. 101 mg of the title compound were obtained as a solid (18% of theory).
- the force of contraction is detected with Statham UC2 cells, amplified and digitized via A / D converters (DAS-1802 HC, Keithley Instruments Munich) and registered in parallel on chart recorders.
- DAS-1802 HC A / D converters
- phenylephrine is added cumulatively to the bath in increasing concentration.
- the substance to be examined is added in each subsequent course in increasing dosages and the height of the contraction is compared with the height of the contraction achieved in the last predistortion. This is used to calculate the concentration required to reduce the level of the control value by 50% (IC 5 o value).
- the standard application volume is 5 ⁇ , the DMSO content in the bath solution corresponds to 0.1%.
- the cellular activity of the compounds of the invention is measured on a recombinant guanylate cyclase reporter cell line as described in F. Wunder et al., Anal. Biochem. 339, 104-112 (2005).
- a commercially available telemetry system from DATA SCIENCES INTERNATIONAL DSI, USA is used for the blood pressure measurement on awake rats described below.
- the system consists of 3 main components:
- Physiotel® receivers connected to a data acquisition computer via a multiplexer (DSI Data Exchange Matrix).
- DSI Data Exchange Matrix DSI Data Exchange Matrix
- the experimental animals are kept individually in macroion cages type 3 after transmitter implantation. You have free access to standard food and water.
- the day - night rhythm in the experimental laboratory is changed by room lighting at 6:00 in the morning and at 19:00 in the evening.
- the TAI 1 PA - C40 telemetry transmitters are surgically implanted into the experimental animals under aseptic conditions at least 14 days before the first trial.
- the animals so instrumented are repeatedly used after healing of the wound and ingrowth of the implant.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Hospice & Palliative Care (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description







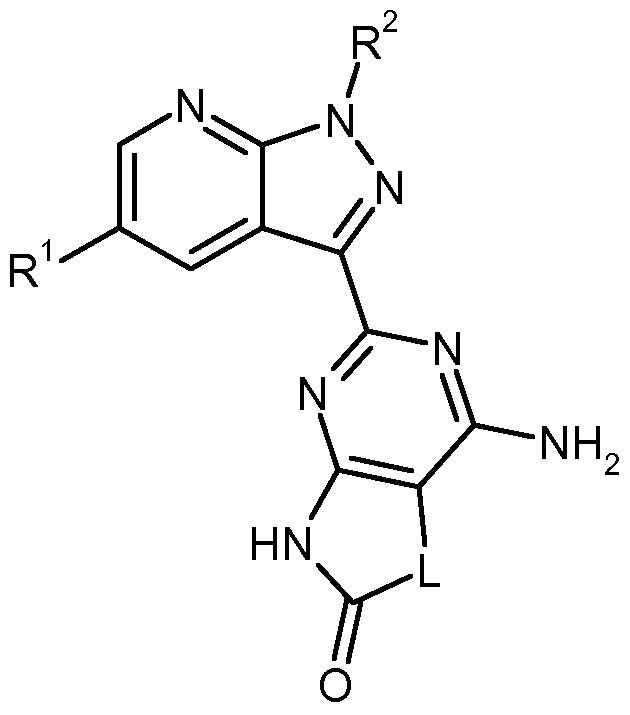
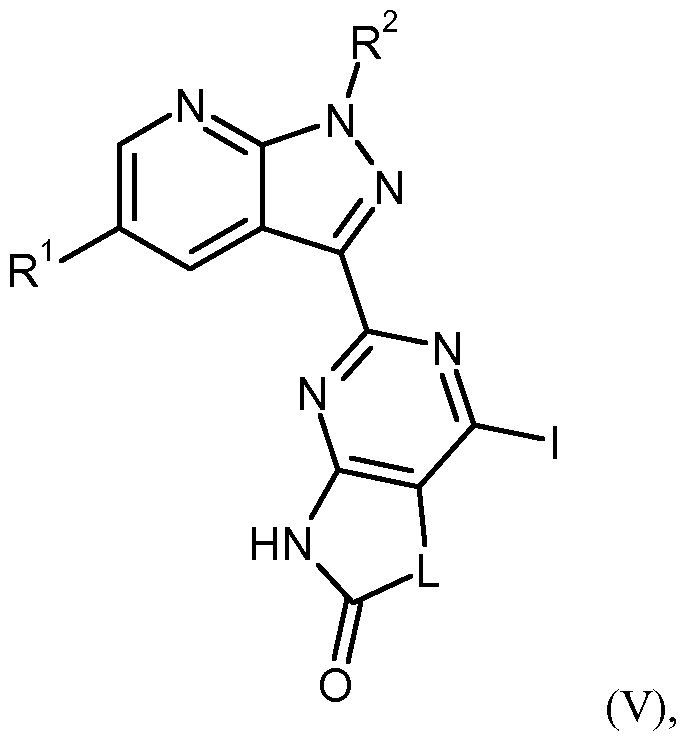


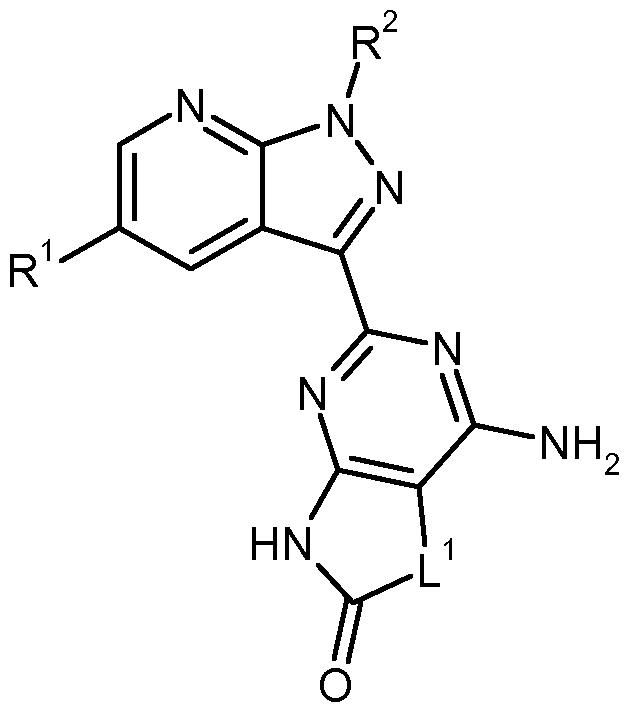

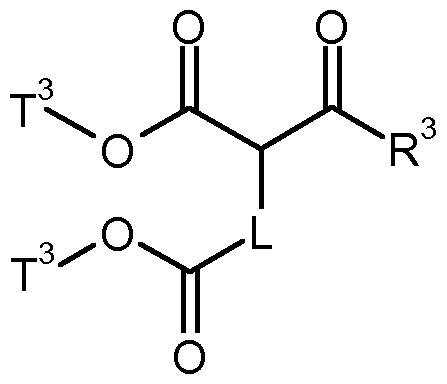
















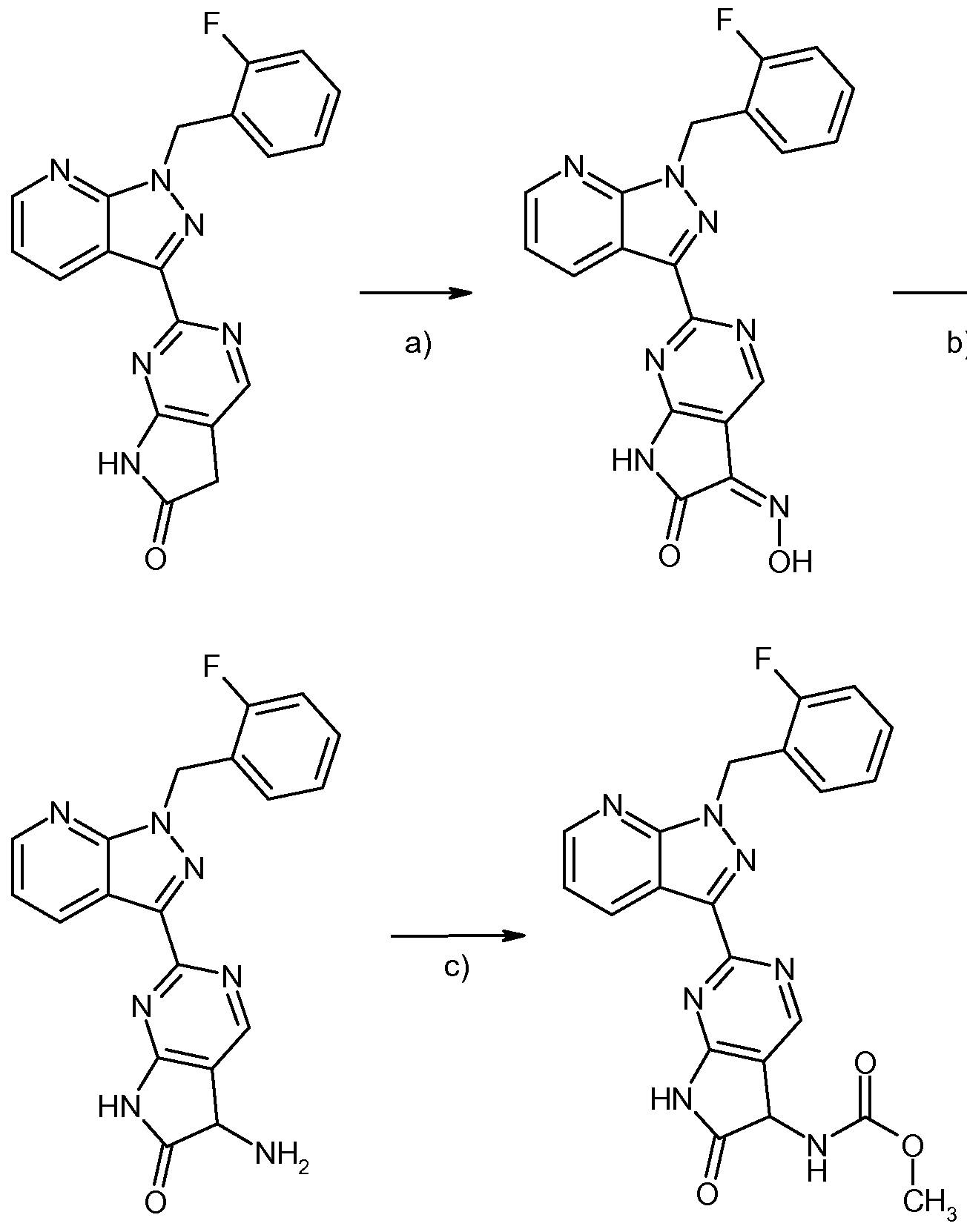







































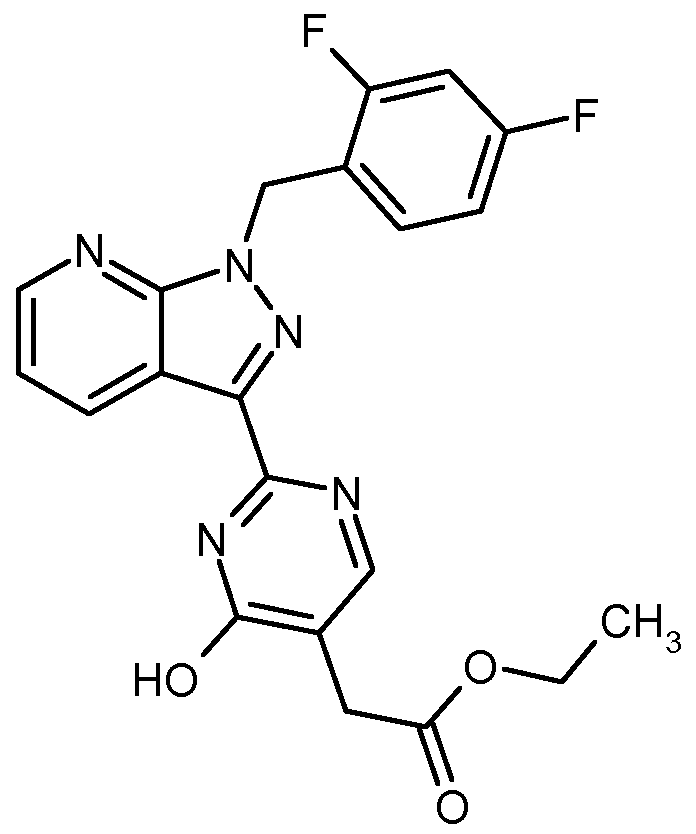

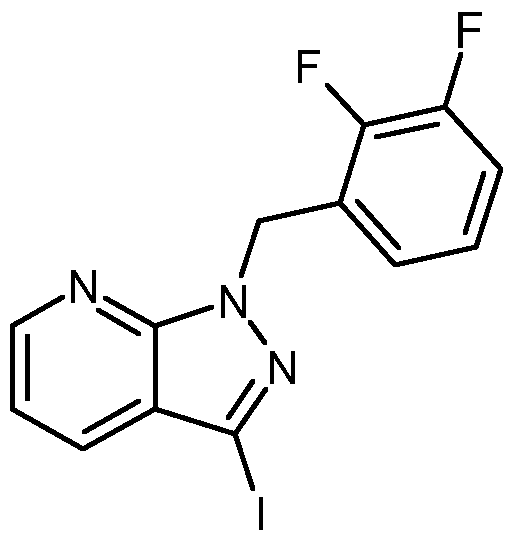

























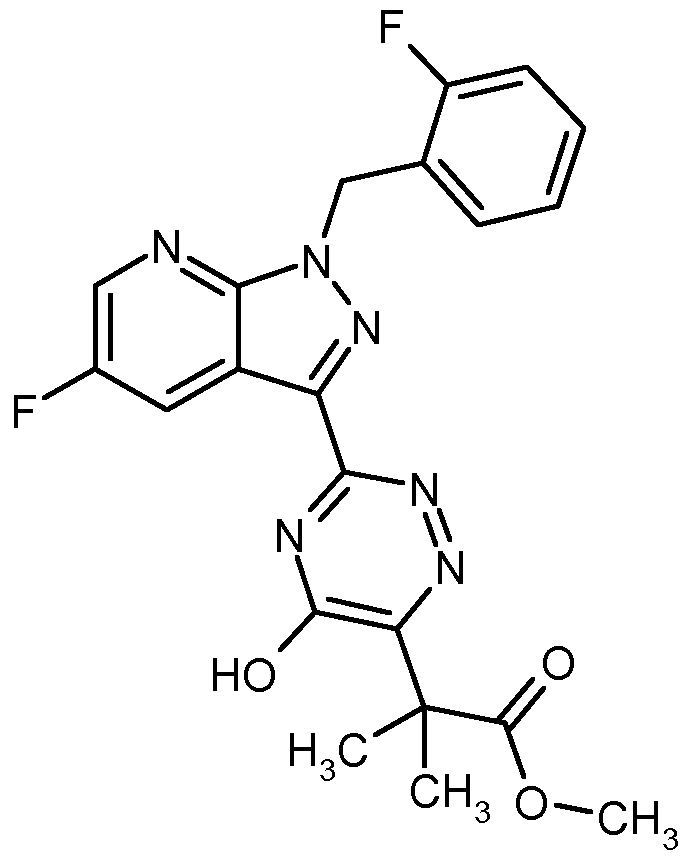





























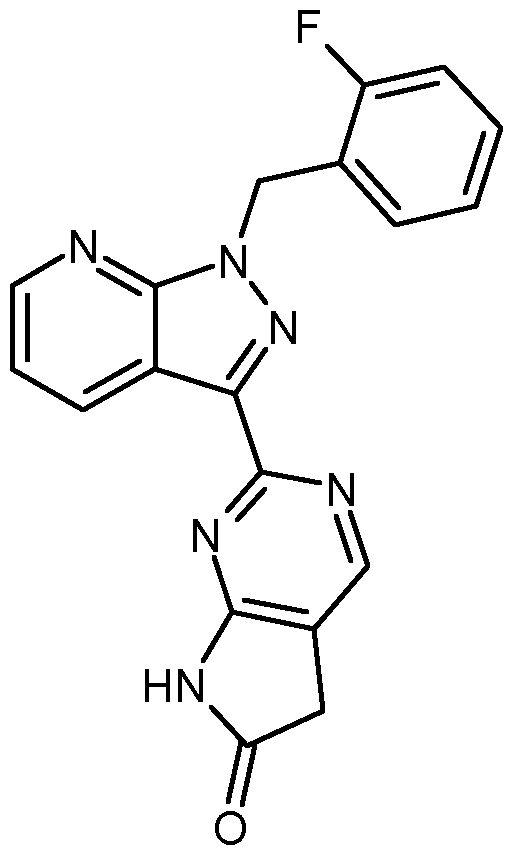





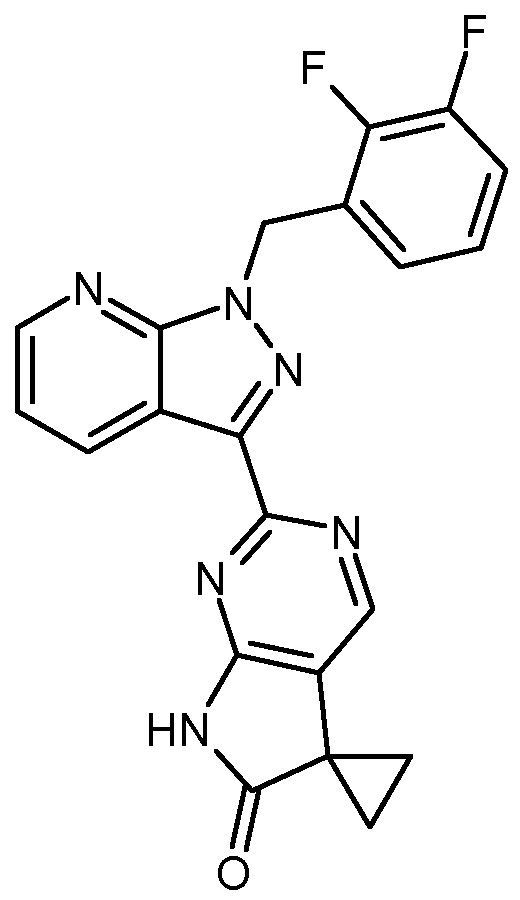













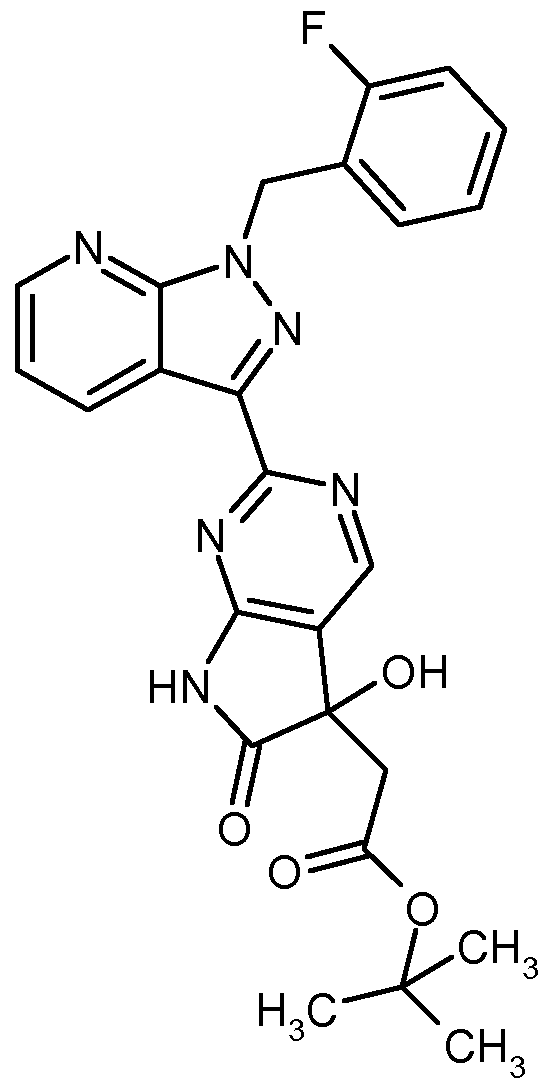
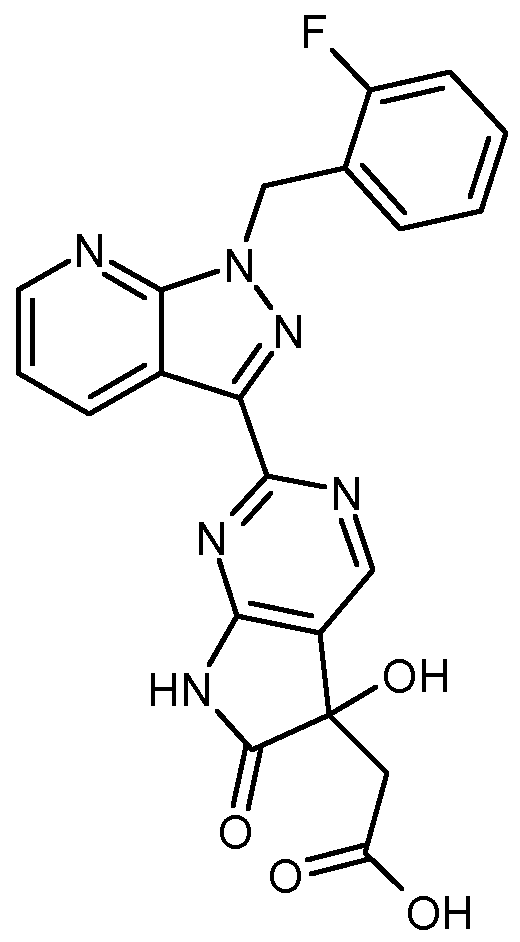







Claims
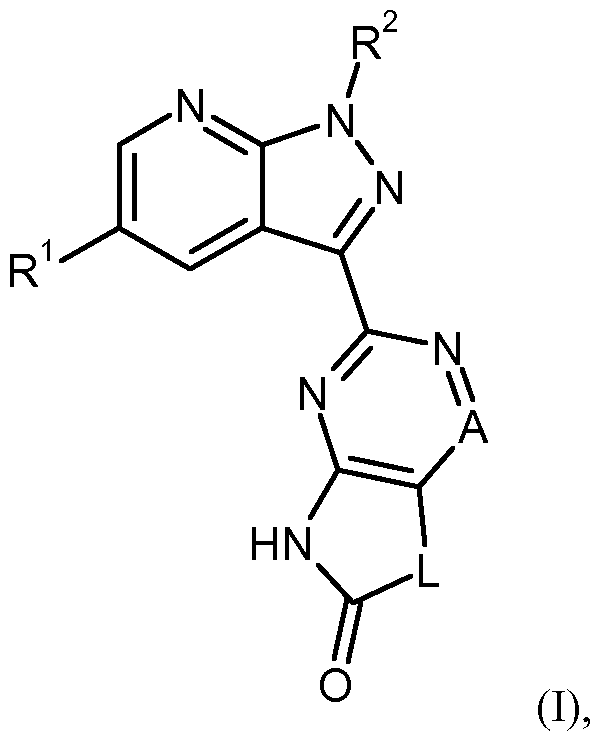


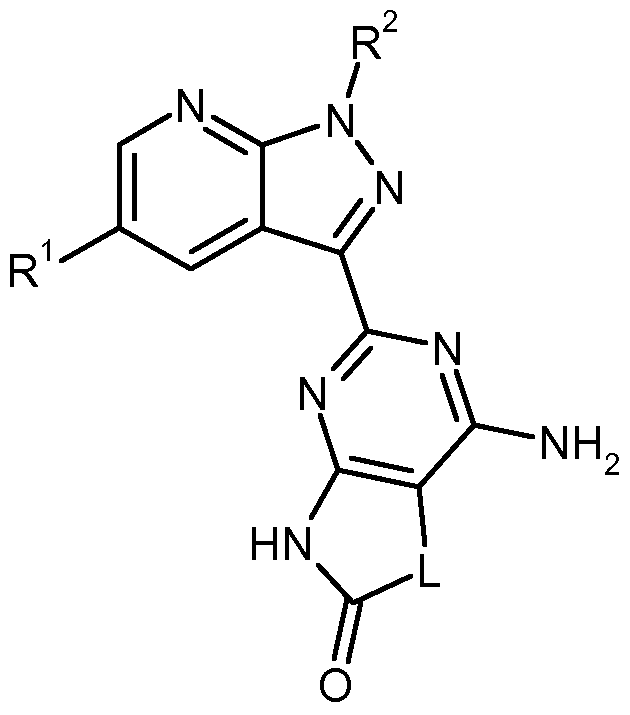





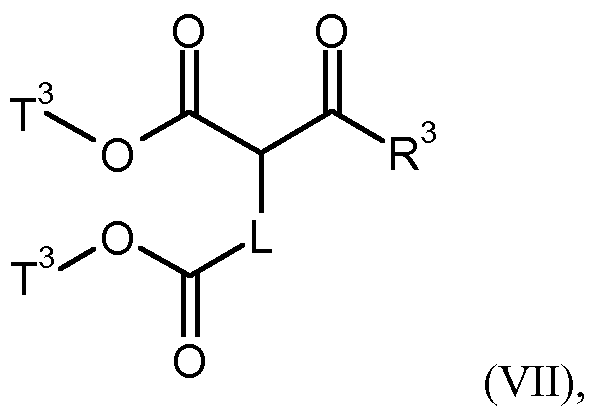










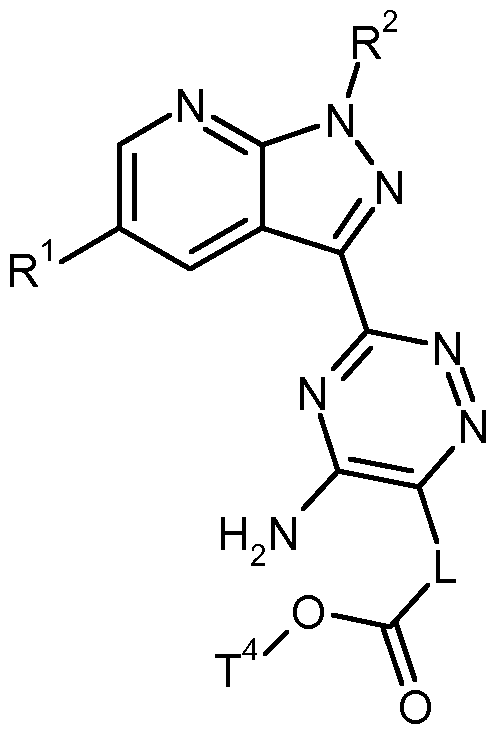
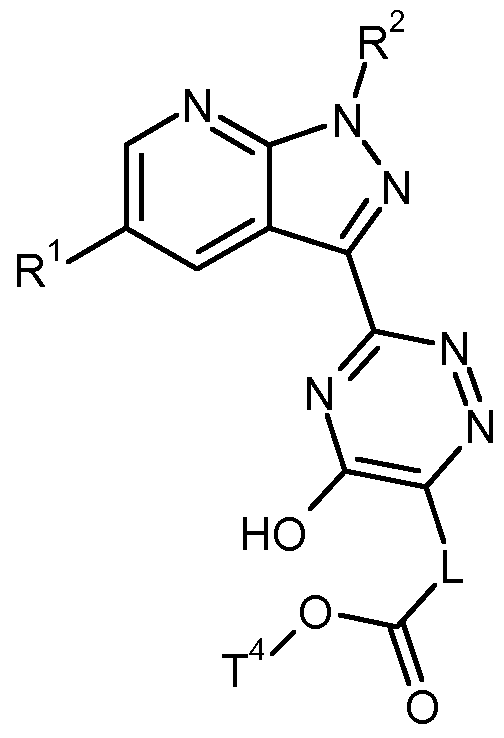
Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013517357A JP6114189B2 (ja) | 2010-07-09 | 2011-07-05 | 縮合ピリミジンおよびトリアジンならびに心血管障害の処置および/または予防のためのその使用 |
| EA201390060A EA201390060A1 (ru) | 2010-07-09 | 2011-07-05 | Аннелированные пиримидины и триазины и их применение для лечения и/или профилактики сердечно-сосудистых заболеваний |
| EP11732409.5A EP2590979A1 (de) | 2010-07-09 | 2011-07-05 | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen |
| AU2011275825A AU2011275825A1 (en) | 2010-07-09 | 2011-07-05 | Ring-fused pyrimidines and triazines and use thereof for the treatment and/or prophylaxis of cardiovascular diseases |
| PH1/2013/500055A PH12013500055A1 (en) | 2010-07-09 | 2011-07-05 | Ring-fused pyrimidines and triazines and use thereof for the treatment and/or prophylaxis of cardiovascular diseases |
| US13/806,425 US9216978B2 (en) | 2010-07-09 | 2011-07-05 | Ring-fused pyrimidines and triazines and use thereof for the treatment and/or prophylaxis of cardiovascular diseases |
| MX2013000198A MX2013000198A (es) | 2010-07-09 | 2011-07-05 | Pirimidinadas y triazinas condensadas y su uso. |
| CN201180043624.6A CN103097387B (zh) | 2010-07-09 | 2011-07-05 | 环稠合的嘧啶和三嗪以及其用于治疗和/或预防心血管疾病的用途 |
| SG2012093043A SG186749A1 (en) | 2010-07-09 | 2011-07-05 | Ring-fused pyrimidines and triazines and use thereof for the treatment and/or prophylaxis of cardiovascular diseases |
| KR1020137003410A KR20130132393A (ko) | 2010-07-09 | 2011-07-05 | 고리-융합된 피리미딘 및 트리아진, 및 심혈관 질환의 치료 및/또는 예방을 위한 그의 용도 |
| CA2804470A CA2804470A1 (en) | 2010-07-09 | 2011-07-05 | Ring-fused pyrimidines and triazines and use thereof for the treatment and/or prophylaxis of cardiovascular diseases |
| BR112013000596A BR112013000596A2 (pt) | 2010-07-09 | 2011-07-05 | piridinas e tiazinas fusionadas e seus usos. |
| ZA2012/09434A ZA201209434B (en) | 2010-07-09 | 2012-12-12 | Fused pyrimidines and triazines and their use |
| CU2013000007A CU20130007A7 (es) | 2010-07-09 | 2013-01-08 | Pirimidinas y triazinas condensadas y su uso |
| CR20130008A CR20130008A (es) | 2010-07-09 | 2013-01-08 | Anillos condensados de pirimidinas y triazinas y su uso para el tratamiento y/o profilaxis de enfermedades cardiovasculares |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102010031149A DE102010031149A1 (de) | 2010-07-09 | 2010-07-09 | Annellierte Pyrimidine und Triazine und ihre Verwendung |
| DE102010031149.9 | 2010-07-09 | ||
| DE102011003315A DE102011003315A1 (de) | 2011-01-28 | 2011-01-28 | Annellierte Pyrimindine und Triazine und ihre Verwendung |
| DE102011003315.7 | 2011-01-28 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2012004258A1 true WO2012004258A1 (de) | 2012-01-12 |
| WO2012004258A9 WO2012004258A9 (de) | 2012-06-07 |
Family
ID=44513289
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/061305 Ceased WO2012004258A1 (de) | 2010-07-09 | 2011-07-05 | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US9216978B2 (de) |
| EP (3) | EP2708539A1 (de) |
| JP (1) | JP6114189B2 (de) |
| KR (1) | KR20130132393A (de) |
| CN (2) | CN106977530A (de) |
| AU (1) | AU2011275825A1 (de) |
| BR (1) | BR112013000596A2 (de) |
| CA (1) | CA2804470A1 (de) |
| CL (1) | CL2013000083A1 (de) |
| CR (1) | CR20130008A (de) |
| CU (1) | CU20130007A7 (de) |
| DO (1) | DOP2013000007A (de) |
| EA (1) | EA201390060A1 (de) |
| EC (1) | ECSP13012379A (de) |
| MX (1) | MX2013000198A (de) |
| PE (1) | PE20130779A1 (de) |
| PH (1) | PH12013500055A1 (de) |
| SG (1) | SG186749A1 (de) |
| WO (1) | WO2012004258A1 (de) |
| ZA (1) | ZA201209434B (de) |
Cited By (117)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013030288A1 (de) | 2011-09-02 | 2013-03-07 | Bayer Intellectual Property Gmbh | Substituierte annellierte pyrimidine und ihre verwendung |
| JP2013527196A (ja) * | 2010-05-27 | 2013-06-27 | メルク・シャープ・エンド・ドーム・コーポレイション | 可溶性グアニル酸シクラーゼアクチベーター |
| WO2013104598A2 (de) | 2012-01-11 | 2013-07-18 | Bayer Intellectual Property Gmbh | Substituierte, annellierte imidazole und pyrazole und ihre verwendung |
| WO2013104703A1 (de) | 2012-01-11 | 2013-07-18 | Bayer Pharma Aktiengesellschaft | Substituierte annellierte pyrimidine und triazine und ihre verwendung |
| WO2013131923A1 (de) * | 2012-03-06 | 2013-09-12 | Bayer Intellectual Property Gmbh | Substituierte azabicyclen und ihre verwendung |
| WO2014012935A1 (de) | 2012-07-20 | 2014-01-23 | Bayer Pharma Aktiengesellschaft | Substituierte aminoindan- und aminotetralin-carbonsäuren und ihre verwendung |
| WO2014012934A1 (de) | 2012-07-20 | 2014-01-23 | Bayer Pharma Aktiengesellschaft | Neue 5-aminotetrahydrochinolin-2-carbonsäuren und ihre verwendung |
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| WO2014131760A1 (de) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Trifluormethyl-substituierte annellierte pyrimidine und ihre verwendung |
| WO2014131741A1 (de) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Benzyl-substituierte pyrazolopyridine und ihre verwendung |
| WO2015004105A1 (de) | 2013-07-10 | 2015-01-15 | Bayer Pharma Aktiengesellschaft | Benzyl-1h-pyrazolo[3,4-b]pyridine und ihre verwendung |
| WO2015036563A1 (de) | 2013-09-16 | 2015-03-19 | Bayer Pharma Aktiengesellschaft | Disubstituierte trifluormethylpyrimidinone und ihre verwendung als ccr2 antagonisten |
| WO2015052065A1 (de) | 2013-10-07 | 2015-04-16 | Bayer Pharma Aktiengesellschaft | Cyclische thienouracil-carboxamide und ihre verwendung |
| WO2015088886A1 (en) * | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2015088885A1 (en) * | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| WO2015150363A1 (de) | 2014-04-03 | 2015-10-08 | Bayer Pharma Aktiengesellschaft | 2,5-disubstituierte cyclopentancarbonsäuren und ihre verwendung |
| WO2015150350A1 (de) | 2014-04-03 | 2015-10-08 | Bayer Pharma Aktiengesellschaft | 2,5-disubstituierte cyclopentancarbonsäuren zur behandlung von atemwegserkrankungen |
| WO2015150362A2 (de) | 2014-04-03 | 2015-10-08 | Bayer Pharma Aktiengesellschaft | Chirale 2,5-disubstituierte cyclopentancarbonsäure-derivate und ihre verwendung |
| WO2016030354A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Amino-substituierte annellierte pyrimidine und ihre verwendung |
| WO2016030362A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Substituierte annellierte pyrimidine und ihre verwendung |
| WO2016037954A1 (de) | 2014-09-09 | 2016-03-17 | Bayer Pharma Aktiengesellschaft | Substituierte n,2-diarylchinolin-4-carboxamide und ihre anti-inflammatorische verwendung |
| WO2016113205A1 (de) | 2015-01-13 | 2016-07-21 | Bayer Pharma Aktiengesellschaft | Substituierte pentafluorethylpyrimidinone und ihre verwendung |
| WO2016150901A1 (de) | 2015-03-26 | 2016-09-29 | Bayer Pharma Aktiengesellschaft | Heterocyclylmethyl-thienouracile als antagonisten des adenosin-a2b-rezeptors |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| WO2017021011A1 (fr) | 2014-08-07 | 2017-02-09 | C.E.R.M.E.X. Constructions Etudes Et Recherches De Materiels Pour L'emballage D'expedition | Groupement de produits |
| US9637550B2 (en) | 2008-12-18 | 2017-05-02 | Oregon Health & Science University | Anti-fXI antibodies and methods of use |
| US9636399B2 (en) | 2007-11-21 | 2017-05-02 | Oregon Health & Science University | Anti-factor XI monoclonal antibodies and methods of use thereof |
| WO2017097671A1 (de) | 2015-12-10 | 2017-06-15 | Bayer Pharma Aktiengesellschaft | Substituierte perhydropyrrolo[3,4-c]pyrrol-derivate und ihre verwendung |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| US9688699B2 (en) | 2014-02-19 | 2017-06-27 | Bayer Pharma Aktiengesellschaft | 3-(pyrimidine-2-yl)imidazo[1,2-a]pyridines |
| WO2017121700A1 (de) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | 1,3-disubstituierte 1h-pyrazolo[3,4-b]pyridin- derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
| WO2017121693A1 (de) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituierte thiazol- und thiadiazolamide und ihre verwendung |
| WO2017121692A1 (de) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituierte sulfamide und ihre verwendung |
| WO2017153235A1 (de) | 2016-03-09 | 2017-09-14 | Bayer Pharma Aktiengesellschaft | Substituierte n-cyclo-3-aryl-1-naphthamide und ihre verwendung |
| WO2017153231A1 (de) | 2016-03-09 | 2017-09-14 | Bayer Pharma Aktiengesellschaft | Substituierte n-cyclo-2-arylisochinolinon-4-carboxamide und ihre verwendung |
| WO2017153234A1 (de) | 2016-03-09 | 2017-09-14 | Bayer Pharma Aktiengesellschaft | Substituierte n-cyclo-2-arylchinolin-4-carboxamide und ihre verwendung |
| US9771352B2 (en) | 2014-11-03 | 2017-09-26 | Bayer Pharma Aktiengesellschaft | Hydroxyalkyl-substituted phenyltriazole derivatives and uses thereof |
| US9776997B2 (en) | 2013-06-04 | 2017-10-03 | Bayer Pharma Aktiengesellschaft | 3-aryl-substituted imidazo[1,2-A]pyridines and their use |
| US9783614B2 (en) | 2012-05-10 | 2017-10-10 | Bayer Pharma Aktiengesellschaft | Antibodies capable of binding to the coagulation Factor XI and/or its activated form factor Xia and uses thereof |
| WO2017191107A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted phenyltriazole derivatives and uses thereof |
| WO2017191105A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted aryltriazole derivatives and uses thereof |
| WO2017191115A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Oxoalkyl-substituted phenyltriazole derivatives and uses thereof |
| WO2017191102A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted pyridinyltriazole derivatives and uses thereof |
| WO2017191117A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | V1a receptor antagonists for use in the treatment of renal diseases |
| WO2017191114A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Aktiengesellschaft | Hydroxyalkyl-substituted heteroaryltriazole derivatives and uses thereof |
| US9822130B2 (en) | 2014-11-21 | 2017-11-21 | Merck Sharp & Dohme Corp. | Triazolo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| WO2018011017A1 (de) | 2016-07-11 | 2018-01-18 | Bayer Pharma Aktiengesellschaft | 7-substituierte 1-pyridyl-naphthyridin-3-carbonsäureamide und ihre verwendung |
| WO2018015196A1 (de) | 2016-07-20 | 2018-01-25 | Bayer Aktiengesellschaft | Substituierte diazaheterobicyclische verbindungen und ihre verwendung |
| WO2018041771A1 (de) | 2016-09-02 | 2018-03-08 | Bayer Pharma Aktiengesellschaft | (1-methylcyclopropyl)methyl-substituierte thienouracile und ihre verwendung |
| EP3296298A1 (de) | 2016-09-14 | 2018-03-21 | Bayer Pharma Aktiengesellschaft | 7-substituierte 1-aryl-naphthyridin-3-carbonsäureamide und ihre verwendung |
| WO2018050510A1 (de) | 2016-09-14 | 2018-03-22 | Bayer Aktiengesellschaft | 7-substituierte 1-aryl-naphthyridin-3-carbonsäureamide und ihre verwendung |
| WO2018054846A1 (de) | 2016-09-23 | 2018-03-29 | Bayer Aktiengesellschaft | N3-cyclisch substituierte thienouracile und ihre verwendung |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| WO2018069222A1 (en) | 2016-10-14 | 2018-04-19 | Bayer Aktiengesellschaft | Substituted 6-(1h-pyrazol-1-yl)pyrimidin-4-amine derivatives and uses thereof |
| WO2018073144A1 (en) | 2016-10-20 | 2018-04-26 | Bayer Pharma Aktiengesellschaft | Hydroxyalkyl-substituted triazole derivatives and uses thereof |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| EP3338764A1 (de) | 2016-12-21 | 2018-06-27 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| EP3338803A1 (de) | 2016-12-21 | 2018-06-27 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| WO2018114503A1 (de) | 2016-12-21 | 2018-06-28 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| WO2018114501A1 (de) | 2016-12-21 | 2018-06-28 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| WO2018189012A1 (de) | 2017-04-10 | 2018-10-18 | Bayer Aktiengesellschaft | Substituierte n-arylethyl-2-aminochinolin-4-carboxamide und ihre verwendung |
| WO2018189011A1 (de) | 2017-04-10 | 2018-10-18 | Bayer Aktiengesellschaft | Substituierte n-arylethyl-2-arylchinolin-4-carboxamide und ihre verwendung |
| US10138236B2 (en) | 2014-09-24 | 2018-11-27 | Bayer Pharma Aktiengesellschaft | Factor xia-inhibiting pyridobenzazepine and pyridobenzazocine derivatives |
| WO2018228909A1 (en) | 2017-06-14 | 2018-12-20 | Bayer Pharma Aktiengesellschaft | Substituted bridged diazepane derivatives and use thereof as task-1 and task-3 inhibitors |
| WO2018228907A1 (de) | 2017-06-14 | 2018-12-20 | Bayer Aktiengesellschaft | Diazabicyclisch substituierte imidazopyrimidine und ihre verwendung zur behandlung von atemstörungen |
| WO2019081303A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081291A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | PRODRUGS OF SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081302A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081307A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081292A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | PRODRUGS OF SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081299A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | AMINE SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081306A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019091847A1 (de) | 2017-11-07 | 2019-05-16 | Bayer Aktiengesellschaft | Substituierte 2,4-dihydro-3h-1,2,4-triazol-3-one und ihre verwendung |
| US10292970B2 (en) | 2014-12-02 | 2019-05-21 | Bayer Pharma Aktiengesellschaft | Heteroaryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2019105881A1 (de) | 2017-12-01 | 2019-06-06 | Bayer Pharma Aktiengesellschaft | Verfahren zur herstellung von (3s)-3-(4-chlor-3-{[(2s,3r)-2-(4-chlorphenyl)-4,4,4-trifluor-3-methylbutanoyl]amino}phenyl)-3-cyclopropylpropansäure und dessen kristalline form für die verwendung als pharmazeutischer wirkstoff |
| US10364229B2 (en) | 2011-11-25 | 2019-07-30 | Adverio Pharma Gmbh | Crystalline substituted 5-fluoro-1H-pyrazolopyridines and process for preparing |
| EP3553079A1 (de) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Mit natriuretischem peptid vom c-typ gepfropfte antikörper |
| EP3553082A1 (de) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Natriuretische peptidgepfropfte antikörper im gehirn |
| EP3553081A1 (de) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Natriuretische peptidgepfropfte antikörper im vorhof |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| WO2019219517A1 (en) | 2018-05-17 | 2019-11-21 | Bayer Aktiengesellschaft | Substituted dihydropyrazolo pyrazine carboxamide derivatives |
| US10525041B2 (en) | 2016-05-03 | 2020-01-07 | Bayer Pharma Aktiengesellschaft | Fluoroalkyl-substituted aryltriazole derivatives and uses thereof |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020020789A1 (de) | 2018-07-24 | 2020-01-30 | Bayer Aktiengesellschaft | Oral applizierbare pharmazeutische darreichungsform mit modifizierter freisetzung |
| WO2020020790A1 (de) | 2018-07-24 | 2020-01-30 | Bayer Aktiengesellschaft | Oral applizierbare pharmazeutische darreichungsform mit modifizierter freisetzung |
| WO2020109109A1 (de) | 2018-11-27 | 2020-06-04 | Bayer Aktiengesellschaft | Verfahren zur herstellung von pharmazeutischen darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| US10722501B2 (en) | 2016-05-09 | 2020-07-28 | Bayer Aktiengesellschaft | Substituted 5,6,7,8-tetrahydro[1,2,4]triazolo[4,3-A]pyridine-3(2H)-ones and 2,5,6,7-tetrahydro-3H-pyrrolo[2,1-C][1,2,4]triazol-3-ones, and use thereof |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| US10759794B2 (en) | 2015-12-10 | 2020-09-01 | Bayer Pharma Aktiengesellschaft | 2-phenyl-3-(piperazinomethyl)imidazo[1,2-A]pyridine derivatives as blockers of task-1 and task-2 channels, for the treatment of sleep-related breathing disorders |
| WO2020225095A1 (en) | 2019-05-07 | 2020-11-12 | Bayer Aktiengesellschaft | Masp inhibitory compounds and uses thereof |
| WO2020245342A1 (en) | 2019-06-07 | 2020-12-10 | Bayer Aktiengesellschaft | The use of sgc activators for the treatment of ophthalmologic diseases |
| WO2021089683A1 (en) | 2019-11-06 | 2021-05-14 | Bayer Aktiengesellschaft | Inhibitors of adrenoreceptor adrac2 |
| EP3822268A1 (de) | 2019-11-15 | 2021-05-19 | Bayer Aktiengesellschaft | Substituierte hydantoinamide als adamts7 antagonisten |
| EP3822265A1 (de) | 2019-11-15 | 2021-05-19 | Bayer AG | Substituierte hydantoinamide als adamts7 antagonisten |
| WO2021094210A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted pyrazine carboxamide derivatives as prostaglandin ep3 receptor antagonists |
| WO2021094208A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted imidazo pyrimidine ep3 antagonists |
| WO2021094209A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted pyrrolo triazine carboxamide derivatives as prostaglandin ep3 receptor antagonists |
| WO2021167458A1 (en) | 2020-02-21 | 2021-08-26 | Universiteit Maastricht | Use of a soluble guanylate cyclase (sgc) stimulator or of a combination of a sgc stimulator and an sgc activator for conditions wherein the heme group of sgc is oxidized or wherein sgc is deficient in heme |
| WO2021172982A1 (en) | 2020-02-26 | 2021-09-02 | Universiteit Maastricht | Therapeutic combination for the treatment of brain ischemia and said therapeutic combination for use in the treatment of brain ischemia |
| WO2022096394A1 (en) | 2020-11-04 | 2022-05-12 | Bayer Aktiengesellschaft | Masp inhibitory compounds and uses thereof |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
| WO2022112213A1 (en) | 2020-11-30 | 2022-06-02 | Bayer Aktiengesellschaft | Crystalline forms of 3-[[3-(4-chlorophenyl)-5-oxo-4-((2s)-3,3,3-trifluoro- 2-hydroxypropyl)-4,5-dihydro-1h-1,2,4-triazol-1-yl]methyl]-1-[3- (trifluoromethyl)pyridin-2-yl]-1h-1,2,4-triazole-5-carboxamide |
| EP4011904A1 (de) | 2020-12-14 | 2022-06-15 | Bayer Aktiengesellschaft | Masp-hemmende verbindungen und deren verwendungen |
| EP4011873A1 (de) | 2020-12-10 | 2022-06-15 | Bayer Aktiengesellschaft | Substituierte pyrazolo-piperidin-carbonsäuren |
| EP4011874A1 (de) | 2020-12-10 | 2022-06-15 | Bayer Aktiengesellschaft | Substituierte pyrazolo-piperidin-carbonsäuren |
| WO2022122914A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| WO2022122917A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | The use of sgc activators for the treatment of ophthalmologic diseases |
| WO2022122916A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolyl piperidine carboxylic acids |
| US11377417B2 (en) | 2011-04-13 | 2022-07-05 | Bayer Intellectual Property Gmbh | Branched 3-phenylpropionic acid derivatives and their use |
| WO2023126437A1 (en) | 2021-12-29 | 2023-07-06 | Bayer Aktiengesellschaft | Treatment of cardiopulmonary disorders |
| WO2023126438A1 (en) | 2021-12-29 | 2023-07-06 | Bayer Aktiengesellschaft | Pharmaceutical dry powder inhalation formulation |
| WO2023126436A1 (en) | 2021-12-29 | 2023-07-06 | Bayer Aktiengesellschaft | Process for preparing (5s)-{[2-(4-carboxyphenyl)ethyl] |2-(2-{|3-chloro-4'-(trifluoromethyl)biphenyl-4- yl]methoxy}phenyl)ethyl]aminol-5,6,7,8-tetrahydroquinoline-2-carboxylic acid and its crystalline forms for use as pharmaceutically active compound |
| WO2023237577A1 (en) | 2022-06-09 | 2023-12-14 | Bayer Aktiengesellschaft | Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in women |
| EP4144741A4 (de) * | 2020-04-30 | 2024-05-22 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Tricyclische verbindung auf pyrimidinbasis und verwendung davon |
| WO2025068514A1 (en) | 2023-09-28 | 2025-04-03 | Bayer Aktiengesellschaft | Substituted heterocyclic carboxamindes and use thereof |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL2604608T3 (pl) | 2009-11-27 | 2016-02-29 | Adverio Pharma Gmbh | Sposób wytwarzania {4,6-diamino-2-[1-(2-fluorobenzylo)-1H-pirazolo[3,4-b]pirydyn-3-ylo]pirymidyn-5-ylo}metylokarbaminianu metylu |
| US9284301B2 (en) | 2010-03-25 | 2016-03-15 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| DE102010021637A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung |
| CN103180327B (zh) | 2010-07-09 | 2016-08-10 | 拜耳知识产权有限责任公司 | 稠环的4-氨基嘧啶及其作为可溶性鸟甘酸环化酶的刺激物的用途 |
| DE102010040233A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclische Aza-Heterocyclen und ihre Verwendung |
| JP6005134B2 (ja) | 2011-04-21 | 2016-10-12 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | フルオロアルキル置換ピラゾロピリジンおよびその使用 |
| DE102012200360A1 (de) * | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte Triazine und ihre Verwendung |
| MX2015010725A (es) | 2013-02-21 | 2016-05-31 | Adverio Pharma Gmbh | Formas de metil {4,6-diamino-2-[1-(2-fluorobencil)-1h-pirazolo [3,4-b] piridino-3-il] pirimidino-5-il} metil carbamato. |
| US9796733B2 (en) | 2014-06-04 | 2017-10-24 | Merck Sharp & Dohme Corp. | Imidazo-pyrazine derivatives useful as soluble guanylate cyclase activators |
| WO2016191334A1 (en) | 2015-05-27 | 2016-12-01 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| WO2016191335A1 (en) | 2015-05-28 | 2016-12-01 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| US10438785B2 (en) * | 2015-08-31 | 2019-10-08 | Shimadzu Corporation | Method for quantitative analysis of high-molecular compound and data-processing device for the quantitative analysis |
| WO2017107052A1 (en) | 2015-12-22 | 2017-06-29 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase stimulators |
| WO2017197555A1 (en) | 2016-05-16 | 2017-11-23 | Merck Sharp & Dohme Corp. | Fused pyrazine derivatives useful as soluble guanylate cyclase stimulators |
| EP3458063A4 (de) | 2016-05-18 | 2020-02-26 | Merck Sharp & Dohme Corp. | Verfahren zur verwendung von triazolo-pyrazinyl-löslichen guanylatcyclaseaktivatoren in fibrotischen erkrankungen |
| CN107964018A (zh) * | 2016-10-19 | 2018-04-27 | 中国人民解放军军事医学科学院毒物药物研究所 | 取代嘌呤酮类衍生物及其医药用途 |
| ES2924359T3 (es) | 2017-04-11 | 2022-10-06 | Sunshine Lake Pharma Co Ltd | Compuestos de indazol sustituidos con flúor y usos de los mismos |
| US10905667B2 (en) | 2018-07-24 | 2021-02-02 | Bayer Pharma Aktiengesellschaft | Orally administrable modified-release pharmaceutical dosage form |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0634413A1 (de) | 1993-07-13 | 1995-01-18 | Rhone Poulenc Agriculture Ltd. | Herbizide |
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| US5976523A (en) * | 1995-05-16 | 1999-11-02 | Mitsui Pharmaceuticals, Inc. | Method for healing compromised tissues using pyrimidine derivatives |
| WO2000006569A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Mit sechsgliedrigen heterocyclischen ringen kondensierte substituierte pyrazolderivate |
| WO2002059083A2 (en) * | 2000-10-23 | 2002-08-01 | Smithkline Beecham Corporation | Novel compounds |
| WO2003095451A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Carbamat-substituierte pyrazolopyridine |
| CN1613849A (zh) | 2003-11-03 | 2005-05-11 | 上海药明康德新药开发有限公司 | 2-氯-5-氟-烟酸酯及酸的制备方法 |
| WO2005073234A2 (en) | 2004-01-31 | 2005-08-11 | Actimis Pharmaceuticals, Inc. | Imidazo[1,2-c]pyrimidinylacetic acid derivatives |
| EP1626045A1 (de) | 2003-05-09 | 2006-02-15 | Asahi Glass Company, Limited | Verfahren zur herstellung von 3-substituiertem 2-chlor-5-fluorpyridin bzw. einem salz davon |
| WO2006130673A1 (en) | 2005-05-31 | 2006-12-07 | Janssen Pharmaceutica, N.V. | 3-benzoimidazolyl-pyrazolopyridines useful in treating kinase disorders |
| WO2009018415A1 (en) | 2007-07-31 | 2009-02-05 | Vertex Pharmaceuticals Incorporated | Process for preparing 5-fluoro-1h-pyrazolo [3, 4-b] pyridin-3-amine and derivatives thereof |
| WO2010065275A1 (en) | 2008-11-25 | 2010-06-10 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63139949A (ja) | 1986-12-02 | 1988-06-11 | Fuji Photo Film Co Ltd | 新規ピラゾロン染料 |
| JPH09295946A (ja) * | 1995-05-16 | 1997-11-18 | Mitsui Pharmaceut Inc | 医薬組成物 |
| US6451805B1 (en) | 1997-11-14 | 2002-09-17 | Bayer Aktiengesellschaft | Substituted pyrazole derivatives for the treatment of cardiocirculatory diseases |
| DE19834047A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Substituierte Pyrazolderivate |
| DE10021069A1 (de) | 2000-04-28 | 2001-10-31 | Bayer Ag | Substituiertes Pyrazolderivat |
| ES2231581T3 (es) | 2000-11-22 | 2005-05-16 | Bayer Healthcare Ag | Nuevos derivados de pirazolopiridina sustituidos con lactama. |
| DE10132416A1 (de) | 2001-07-04 | 2003-01-16 | Bayer Ag | Neue Morpholin-überbrückte Pyrazolopyridinderivate |
| DE10232571A1 (de) | 2002-07-18 | 2004-02-05 | Bayer Ag | 4-Aminosubstituierte Pyrimidinderivate |
| ATE451363T1 (de) | 2005-01-26 | 2009-12-15 | Schering Corp | 3-(indazol-5-yl)-(1,2,4)triazinderivate und verwandte verbindungen als proteinkinaseinhibitoren zur behandlung von krebs |
| DE102006043443A1 (de) | 2006-09-15 | 2008-03-27 | Bayer Healthcare Ag | Neue aza-bicyclische Verbindungen und ihre Verwendung |
| WO2011115804A1 (en) | 2010-03-17 | 2011-09-22 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| DE102010021637A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung |
| KR20140019004A (ko) | 2010-05-27 | 2014-02-13 | 머크 샤프 앤드 돔 코포레이션 | 가용성 구아닐레이트 시클라제 활성화제 |
| EP2687210A1 (de) | 2010-06-25 | 2014-01-22 | Bayer Intellectual Property GmbH | Verwendung von Stimulatoren und Aktivatoren der löslichen Guanylatzyklase zur Behandlung von Sichelzellanämie und Konservierung von Blutersatzstoffen |
| CN103180327B (zh) | 2010-07-09 | 2016-08-10 | 拜耳知识产权有限责任公司 | 稠环的4-氨基嘧啶及其作为可溶性鸟甘酸环化酶的刺激物的用途 |
| DE102010040233A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclische Aza-Heterocyclen und ihre Verwendung |
| DE102010043380A1 (de) | 2010-11-04 | 2012-05-10 | Bayer Schering Pharma Aktiengesellschaft | Benzyl-substituierte Carbamate und ihre Verwendung |
| JP6005134B2 (ja) | 2011-04-21 | 2016-10-12 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | フルオロアルキル置換ピラゾロピリジンおよびその使用 |
| CA2834901A1 (en) | 2011-05-06 | 2012-11-15 | Bayer Intellectual Property Gmbh | Substituted imidazopyridines and imidazopyridazines and the use thereof |
| CN103906752B (zh) | 2011-07-06 | 2016-08-24 | 拜耳知识产权有限责任公司 | 杂芳基取代的吡唑并吡啶及其用作可溶性鸟苷酸环化酶刺激剂的用途 |
| JP6096778B2 (ja) | 2011-09-01 | 2017-03-15 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | ピロロピラジンキナーゼ阻害剤 |
| CN104039784B (zh) | 2011-09-02 | 2017-08-22 | 拜耳知识产权有限责任公司 | 取代的增环嘧啶及其用途 |
| DE102012200352A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte, annellierte Imidazole und Pyrazole und ihre Verwendung |
| DE102012200349A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung |
| WO2013131923A1 (de) | 2012-03-06 | 2013-09-12 | Bayer Intellectual Property Gmbh | Substituierte azabicyclen und ihre verwendung |
| MX2015010966A (es) | 2013-03-01 | 2016-04-04 | Bayer Pharma AG | Pirimidinas fusionadas sustituidas con trifluorometilo y sus usos. |
-
2011
- 2011-07-05 EA EA201390060A patent/EA201390060A1/ru unknown
- 2011-07-05 CA CA2804470A patent/CA2804470A1/en not_active Abandoned
- 2011-07-05 EP EP13180879.2A patent/EP2708539A1/de not_active Withdrawn
- 2011-07-05 WO PCT/EP2011/061305 patent/WO2012004258A1/de not_active Ceased
- 2011-07-05 PE PE2013000009A patent/PE20130779A1/es not_active Application Discontinuation
- 2011-07-05 JP JP2013517357A patent/JP6114189B2/ja not_active Expired - Fee Related
- 2011-07-05 EP EP13180880.0A patent/EP2682394A1/de not_active Withdrawn
- 2011-07-05 SG SG2012093043A patent/SG186749A1/en unknown
- 2011-07-05 CN CN201610884283.XA patent/CN106977530A/zh active Pending
- 2011-07-05 CN CN201180043624.6A patent/CN103097387B/zh not_active Expired - Fee Related
- 2011-07-05 AU AU2011275825A patent/AU2011275825A1/en not_active Abandoned
- 2011-07-05 PH PH1/2013/500055A patent/PH12013500055A1/en unknown
- 2011-07-05 MX MX2013000198A patent/MX2013000198A/es not_active Application Discontinuation
- 2011-07-05 US US13/806,425 patent/US9216978B2/en not_active Expired - Fee Related
- 2011-07-05 EP EP11732409.5A patent/EP2590979A1/de not_active Withdrawn
- 2011-07-05 KR KR1020137003410A patent/KR20130132393A/ko not_active Withdrawn
- 2011-07-05 BR BR112013000596A patent/BR112013000596A2/pt not_active IP Right Cessation
-
2012
- 2012-12-12 ZA ZA2012/09434A patent/ZA201209434B/en unknown
-
2013
- 2013-01-07 DO DO2013000007A patent/DOP2013000007A/es unknown
- 2013-01-08 CR CR20130008A patent/CR20130008A/es unknown
- 2013-01-08 CU CU2013000007A patent/CU20130007A7/es unknown
- 2013-01-09 EC ECSP13012379 patent/ECSP13012379A/es unknown
- 2013-01-09 CL CL2013000083A patent/CL2013000083A1/es unknown
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0634413A1 (de) | 1993-07-13 | 1995-01-18 | Rhone Poulenc Agriculture Ltd. | Herbizide |
| US5976523A (en) * | 1995-05-16 | 1999-11-02 | Mitsui Pharmaceuticals, Inc. | Method for healing compromised tissues using pyrimidine derivatives |
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| WO2000006569A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Mit sechsgliedrigen heterocyclischen ringen kondensierte substituierte pyrazolderivate |
| WO2002059083A2 (en) * | 2000-10-23 | 2002-08-01 | Smithkline Beecham Corporation | Novel compounds |
| WO2003095451A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Carbamat-substituierte pyrazolopyridine |
| EP1626045A1 (de) | 2003-05-09 | 2006-02-15 | Asahi Glass Company, Limited | Verfahren zur herstellung von 3-substituiertem 2-chlor-5-fluorpyridin bzw. einem salz davon |
| CN1613849A (zh) | 2003-11-03 | 2005-05-11 | 上海药明康德新药开发有限公司 | 2-氯-5-氟-烟酸酯及酸的制备方法 |
| WO2005073234A2 (en) | 2004-01-31 | 2005-08-11 | Actimis Pharmaceuticals, Inc. | Imidazo[1,2-c]pyrimidinylacetic acid derivatives |
| WO2006130673A1 (en) | 2005-05-31 | 2006-12-07 | Janssen Pharmaceutica, N.V. | 3-benzoimidazolyl-pyrazolopyridines useful in treating kinase disorders |
| WO2009018415A1 (en) | 2007-07-31 | 2009-02-05 | Vertex Pharmaceuticals Incorporated | Process for preparing 5-fluoro-1h-pyrazolo [3, 4-b] pyridin-3-amine and derivatives thereof |
| WO2010065275A1 (en) | 2008-11-25 | 2010-06-10 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
Non-Patent Citations (14)
| Title |
|---|
| F. WUNDER ET AL., ANAL. BIOCHEM., vol. 339, 2005, pages 104 - 112 |
| GOLDBERG ET AL., J. BIOL. CHEM., vol. 252, 1977, pages 1279 |
| HETEROCYCLES, 1985, pages 1135 - 1141 |
| J. AM. CHEM. SOC., vol. 124, no. 14, 2002, pages 3680 - 3691 |
| JOURNAL OFORGANIC CHEMISTRY, 1996, pages 2690 |
| KLAUS WITTE, KAI HU, JOHANNA SWIATEK, CLAUDIA MÜSSIG, GEORG ERTL, BJÖRN LEMMER: "Experimental heart failure in rats: effects on cardiovascular circadian rhythms and on myocardial ?-adrenergic signaling", CARDIOVASC RES, vol. 47, no. 2, 2000, pages 203 - 405 |
| KOZO OKAMOTO: "Spontaneous hypertension in rats", INT REV EXP PATHOL, vol. 7, 1969, pages 227 - 270 |
| MAARTEN VAN DEN BUUSE: "Circadian Rhythms of Blood Pressure, Heart Rate, and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio-Telemetry", PHYSIOLOGY & BEHAVIOR, vol. 55, no. 4, 1994, pages 783 - 787 |
| MÜLSCH ET AL., BRIT. J. PHARMACOL., vol. 120, 1997, pages 681 |
| PETTIBONE ET AL., EUR. J. PHARMACOL., vol. 116, 1985, pages 307 |
| SYNTH. COMMUN., vol. 9, no. 7, 1979, pages 603 - 7 |
| WINN M., J. MED. CHEM., vol. 36, 1993, pages 2676 - 7688 |
| WU ET AL., BLOOD, vol. 84, 1994, pages 4226 |
| YU ET AL., BRIT. J. PHARMACOL., vol. 114, 1995, pages 1587 |
Cited By (172)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9636399B2 (en) | 2007-11-21 | 2017-05-02 | Oregon Health & Science University | Anti-factor XI monoclonal antibodies and methods of use thereof |
| US10577428B2 (en) | 2007-11-21 | 2020-03-03 | Oregon Health & Science University | Anti-factor XI monoclonal antibodies and methods of use thereof |
| US9637550B2 (en) | 2008-12-18 | 2017-05-02 | Oregon Health & Science University | Anti-fXI antibodies and methods of use |
| JP2013527196A (ja) * | 2010-05-27 | 2013-06-27 | メルク・シャープ・エンド・ドーム・コーポレイション | 可溶性グアニル酸シクラーゼアクチベーター |
| US11377417B2 (en) | 2011-04-13 | 2022-07-05 | Bayer Intellectual Property Gmbh | Branched 3-phenylpropionic acid derivatives and their use |
| WO2013030288A1 (de) | 2011-09-02 | 2013-03-07 | Bayer Intellectual Property Gmbh | Substituierte annellierte pyrimidine und ihre verwendung |
| US10633356B2 (en) | 2011-11-25 | 2020-04-28 | Adverio Pharma Gmbh | Hydrates of substituted 5-fluoro-1H-pyrazolopyridines |
| US10633357B2 (en) | 2011-11-25 | 2020-04-28 | Adverio Pharma Gmbh | Intermediates and process for preparing intermediates in the production of substituted pyrazolopyridines |
| US10364229B2 (en) | 2011-11-25 | 2019-07-30 | Adverio Pharma Gmbh | Crystalline substituted 5-fluoro-1H-pyrazolopyridines and process for preparing |
| US9505786B2 (en) | 2012-01-11 | 2016-11-29 | Bayer Pharma Aktiengesellschaft | Substituted annulated triazines and use thereof |
| EA025837B1 (ru) * | 2012-01-11 | 2017-02-28 | Байер Интеллектуэль Проперти Гмбх | Замещенные аннеллированные пиримидины и триазины |
| CN104321324A (zh) * | 2012-01-11 | 2015-01-28 | 拜耳知识产权有限责任公司 | 取代的稠合咪唑类和吡唑类化合物及其用途 |
| WO2013104598A3 (de) * | 2012-01-11 | 2013-10-10 | Bayer Intellectual Property Gmbh | Substituierte, annellierte imidazole und pyrazole und ihre verwendung |
| WO2013104703A1 (de) | 2012-01-11 | 2013-07-18 | Bayer Pharma Aktiengesellschaft | Substituierte annellierte pyrimidine und triazine und ihre verwendung |
| WO2013104598A2 (de) | 2012-01-11 | 2013-07-18 | Bayer Intellectual Property Gmbh | Substituierte, annellierte imidazole und pyrazole und ihre verwendung |
| US9498480B2 (en) | 2012-03-06 | 2016-11-22 | Bayer Intellectual Property Gmbh | Substituted azabicycles and use thereof |
| CN104395313A (zh) * | 2012-03-06 | 2015-03-04 | 拜耳知识产权有限责任公司 | 取代的氮杂双环及其用途 |
| CN104395313B (zh) * | 2012-03-06 | 2017-03-08 | 拜耳知识产权有限责任公司 | 取代的氮杂双环及其用途 |
| JP2015509515A (ja) * | 2012-03-06 | 2015-03-30 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | 置換アザ二環およびその使用 |
| WO2013131923A1 (de) * | 2012-03-06 | 2013-09-12 | Bayer Intellectual Property Gmbh | Substituierte azabicyclen und ihre verwendung |
| US9783614B2 (en) | 2012-05-10 | 2017-10-10 | Bayer Pharma Aktiengesellschaft | Antibodies capable of binding to the coagulation Factor XI and/or its activated form factor Xia and uses thereof |
| US10221247B2 (en) | 2012-05-10 | 2019-03-05 | Bayer Pharma Aktiengesellschaft | Antibodies capable of binding to the coagulation factor XIa and uses thereof |
| US10040866B2 (en) | 2012-05-10 | 2018-08-07 | Bayer Pharma Aktiengesellschaft | Nucleic acids and host cells expressing antibodies capable of binding to the coagulation factor XIa and uses thereof |
| US9688636B2 (en) | 2012-07-20 | 2017-06-27 | Bayer Pharma Aktiengesellschaft | 5-aminotetrahydroquinoline-2-carboxylic acids and their use |
| WO2014012935A1 (de) | 2012-07-20 | 2014-01-23 | Bayer Pharma Aktiengesellschaft | Substituierte aminoindan- und aminotetralin-carbonsäuren und ihre verwendung |
| US10053428B2 (en) | 2012-07-20 | 2018-08-21 | Bayer Pharma Aktiengesellschaft | 5-aminotetrahydroquinoline-2-carboxylic acids and their use |
| US9387203B2 (en) | 2012-07-20 | 2016-07-12 | Bayer Pharma Aktiengesellschaft | Substituted aminoindane- and aminotetralinecarboxylic acids and the use thereof |
| WO2014012934A1 (de) | 2012-07-20 | 2014-01-23 | Bayer Pharma Aktiengesellschaft | Neue 5-aminotetrahydrochinolin-2-carbonsäuren und ihre verwendung |
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| WO2014131760A1 (de) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Trifluormethyl-substituierte annellierte pyrimidine und ihre verwendung |
| WO2014131741A1 (de) | 2013-03-01 | 2014-09-04 | Bayer Pharma Aktiengesellschaft | Benzyl-substituierte pyrazolopyridine und ihre verwendung |
| US9776997B2 (en) | 2013-06-04 | 2017-10-03 | Bayer Pharma Aktiengesellschaft | 3-aryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2015004105A1 (de) | 2013-07-10 | 2015-01-15 | Bayer Pharma Aktiengesellschaft | Benzyl-1h-pyrazolo[3,4-b]pyridine und ihre verwendung |
| JP2016523944A (ja) * | 2013-07-10 | 2016-08-12 | バイエル・ファルマ・アクティエンゲゼルシャフト | ベンジル−1H−ピラゾロ[3,4−b]ピリジンおよびその使用 |
| US9605008B2 (en) | 2013-07-10 | 2017-03-28 | Bayer Pharma Aktiengesellschaft | Benzyl-1H-pyrazolo[3,4-b]pyridines and use thereof |
| WO2015036563A1 (de) | 2013-09-16 | 2015-03-19 | Bayer Pharma Aktiengesellschaft | Disubstituierte trifluormethylpyrimidinone und ihre verwendung als ccr2 antagonisten |
| WO2015052065A1 (de) | 2013-10-07 | 2015-04-16 | Bayer Pharma Aktiengesellschaft | Cyclische thienouracil-carboxamide und ihre verwendung |
| US9783552B2 (en) | 2013-12-11 | 2017-10-10 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2015088886A1 (en) * | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| US9611278B2 (en) | 2013-12-11 | 2017-04-04 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2015088885A1 (en) * | 2013-12-11 | 2015-06-18 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| US9688699B2 (en) | 2014-02-19 | 2017-06-27 | Bayer Pharma Aktiengesellschaft | 3-(pyrimidine-2-yl)imidazo[1,2-a]pyridines |
| WO2015150350A1 (de) | 2014-04-03 | 2015-10-08 | Bayer Pharma Aktiengesellschaft | 2,5-disubstituierte cyclopentancarbonsäuren zur behandlung von atemwegserkrankungen |
| WO2015150362A2 (de) | 2014-04-03 | 2015-10-08 | Bayer Pharma Aktiengesellschaft | Chirale 2,5-disubstituierte cyclopentancarbonsäure-derivate und ihre verwendung |
| WO2015150363A1 (de) | 2014-04-03 | 2015-10-08 | Bayer Pharma Aktiengesellschaft | 2,5-disubstituierte cyclopentancarbonsäuren und ihre verwendung |
| WO2017021011A1 (fr) | 2014-08-07 | 2017-02-09 | C.E.R.M.E.X. Constructions Etudes Et Recherches De Materiels Pour L'emballage D'expedition | Groupement de produits |
| WO2016030362A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Substituierte annellierte pyrimidine und ihre verwendung |
| WO2016030354A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Amino-substituierte annellierte pyrimidine und ihre verwendung |
| US10189788B2 (en) | 2014-09-09 | 2019-01-29 | Bayer Pharma Aktiengesellschaft | Substituted N,2-diarylquinoline-4-carboxamides and the use thereof as anti-inflammatory agents |
| WO2016037954A1 (de) | 2014-09-09 | 2016-03-17 | Bayer Pharma Aktiengesellschaft | Substituierte n,2-diarylchinolin-4-carboxamide und ihre anti-inflammatorische verwendung |
| US10479765B2 (en) | 2014-09-09 | 2019-11-19 | Bayer Pharma Aktiengesellschaft | Substituted N,2-diarylquinoline-4-carboxamides and the use thereof as anti-inflammatory agents |
| US10138236B2 (en) | 2014-09-24 | 2018-11-27 | Bayer Pharma Aktiengesellschaft | Factor xia-inhibiting pyridobenzazepine and pyridobenzazocine derivatives |
| US9771352B2 (en) | 2014-11-03 | 2017-09-26 | Bayer Pharma Aktiengesellschaft | Hydroxyalkyl-substituted phenyltriazole derivatives and uses thereof |
| US9822130B2 (en) | 2014-11-21 | 2017-11-21 | Merck Sharp & Dohme Corp. | Triazolo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| US10292970B2 (en) | 2014-12-02 | 2019-05-21 | Bayer Pharma Aktiengesellschaft | Heteroaryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2016113205A1 (de) | 2015-01-13 | 2016-07-21 | Bayer Pharma Aktiengesellschaft | Substituierte pentafluorethylpyrimidinone und ihre verwendung |
| WO2016150901A1 (de) | 2015-03-26 | 2016-09-29 | Bayer Pharma Aktiengesellschaft | Heterocyclylmethyl-thienouracile als antagonisten des adenosin-a2b-rezeptors |
| US10428083B2 (en) | 2015-03-26 | 2019-10-01 | Bayer Pharma Aktiengesellschaft | Heterocyclylmethyl-thienouracile as antagonists of the adenosine-A2B-receptor |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| US12427131B2 (en) | 2015-07-23 | 2025-09-30 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sGC) in combination with an inhibitor of neutral endopeptidase (NEP inhibitor) and/or an angiotensin AII antagonist and the use thereof |
| US11166932B2 (en) | 2015-07-23 | 2021-11-09 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sGC) in combination with an inhibitor of neutral endopeptidase (NEP inhibitor) and/or an angiotensin AII antagonist and the use thereof |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| WO2017097671A1 (de) | 2015-12-10 | 2017-06-15 | Bayer Pharma Aktiengesellschaft | Substituierte perhydropyrrolo[3,4-c]pyrrol-derivate und ihre verwendung |
| US10759794B2 (en) | 2015-12-10 | 2020-09-01 | Bayer Pharma Aktiengesellschaft | 2-phenyl-3-(piperazinomethyl)imidazo[1,2-A]pyridine derivatives as blockers of task-1 and task-2 channels, for the treatment of sleep-related breathing disorders |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| WO2017121700A1 (de) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | 1,3-disubstituierte 1h-pyrazolo[3,4-b]pyridin- derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
| WO2017121693A1 (de) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituierte thiazol- und thiadiazolamide und ihre verwendung |
| WO2017121692A1 (de) | 2016-01-15 | 2017-07-20 | Bayer Pharma Aktiengesellschaft | Substituierte sulfamide und ihre verwendung |
| WO2017153235A1 (de) | 2016-03-09 | 2017-09-14 | Bayer Pharma Aktiengesellschaft | Substituierte n-cyclo-3-aryl-1-naphthamide und ihre verwendung |
| WO2017153231A1 (de) | 2016-03-09 | 2017-09-14 | Bayer Pharma Aktiengesellschaft | Substituierte n-cyclo-2-arylisochinolinon-4-carboxamide und ihre verwendung |
| WO2017153234A1 (de) | 2016-03-09 | 2017-09-14 | Bayer Pharma Aktiengesellschaft | Substituierte n-cyclo-2-arylchinolin-4-carboxamide und ihre verwendung |
| US11091463B2 (en) | 2016-05-03 | 2021-08-17 | Bayer Pharma Aktiengesellschaft | Amide-substituted pyridinyltriazole derivatives and uses thereof |
| WO2017191105A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted aryltriazole derivatives and uses thereof |
| WO2017191102A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted pyridinyltriazole derivatives and uses thereof |
| WO2017191117A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | V1a receptor antagonists for use in the treatment of renal diseases |
| US10472348B2 (en) | 2016-05-03 | 2019-11-12 | Bayer Pharma Aktiengesellschaft | Amide-substituted pyridinyltriazole derivatives and uses thereof |
| US9988367B2 (en) | 2016-05-03 | 2018-06-05 | Bayer Pharma Aktiengesellschaft | Amide-substituted pyridinyltriazole derivatives and uses thereof |
| WO2017191107A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted phenyltriazole derivatives and uses thereof |
| US10800746B2 (en) | 2016-05-03 | 2020-10-13 | Bayer Pharma Aktiengesellschaft | Oxoalkyl-substituted phenyltriazole derivatives and uses thereof |
| US10815205B2 (en) | 2016-05-03 | 2020-10-27 | Bayer Pharma Aktiengesellschaft | Amide-substituted phenyltriazole derivatives and uses thereof |
| US10525041B2 (en) | 2016-05-03 | 2020-01-07 | Bayer Pharma Aktiengesellschaft | Fluoroalkyl-substituted aryltriazole derivatives and uses thereof |
| US10526314B2 (en) | 2016-05-03 | 2020-01-07 | Bayer Aktiengesellschaft | Hydroxyalkyl-substituted heteroaryltriazole derivatives and uses thereof |
| WO2017191114A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Aktiengesellschaft | Hydroxyalkyl-substituted heteroaryltriazole derivatives and uses thereof |
| WO2017191115A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Oxoalkyl-substituted phenyltriazole derivatives and uses thereof |
| US10722501B2 (en) | 2016-05-09 | 2020-07-28 | Bayer Aktiengesellschaft | Substituted 5,6,7,8-tetrahydro[1,2,4]triazolo[4,3-A]pyridine-3(2H)-ones and 2,5,6,7-tetrahydro-3H-pyrrolo[2,1-C][1,2,4]triazol-3-ones, and use thereof |
| WO2018011017A1 (de) | 2016-07-11 | 2018-01-18 | Bayer Pharma Aktiengesellschaft | 7-substituierte 1-pyridyl-naphthyridin-3-carbonsäureamide und ihre verwendung |
| WO2018015196A1 (de) | 2016-07-20 | 2018-01-25 | Bayer Aktiengesellschaft | Substituierte diazaheterobicyclische verbindungen und ihre verwendung |
| WO2018041771A1 (de) | 2016-09-02 | 2018-03-08 | Bayer Pharma Aktiengesellschaft | (1-methylcyclopropyl)methyl-substituierte thienouracile und ihre verwendung |
| EP3296298A1 (de) | 2016-09-14 | 2018-03-21 | Bayer Pharma Aktiengesellschaft | 7-substituierte 1-aryl-naphthyridin-3-carbonsäureamide und ihre verwendung |
| WO2018050510A1 (de) | 2016-09-14 | 2018-03-22 | Bayer Aktiengesellschaft | 7-substituierte 1-aryl-naphthyridin-3-carbonsäureamide und ihre verwendung |
| WO2018054846A1 (de) | 2016-09-23 | 2018-03-29 | Bayer Aktiengesellschaft | N3-cyclisch substituierte thienouracile und ihre verwendung |
| US11684621B2 (en) | 2016-10-11 | 2023-06-27 | Bayer Pharma Aktiengesellschaft | Combination containing sGC stimulators and mineralocorticoid receptor antagonists |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
| US10918639B2 (en) | 2016-10-11 | 2021-02-16 | Bayer Pharma Aktiengesellschaft | Combination containing SGC stimulators and mineralocorticoid receptor antagonists |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| WO2018069222A1 (en) | 2016-10-14 | 2018-04-19 | Bayer Aktiengesellschaft | Substituted 6-(1h-pyrazol-1-yl)pyrimidin-4-amine derivatives and uses thereof |
| WO2018073144A1 (en) | 2016-10-20 | 2018-04-26 | Bayer Pharma Aktiengesellschaft | Hydroxyalkyl-substituted triazole derivatives and uses thereof |
| US10927098B2 (en) | 2016-10-20 | 2021-02-23 | Bayer Pharma Aktiengesellschaft | Hydroxyalkyl-substituted triazole derivatives and uses thereof |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| WO2018114501A1 (de) | 2016-12-21 | 2018-06-28 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| WO2018114503A1 (de) | 2016-12-21 | 2018-06-28 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| EP3338803A1 (de) | 2016-12-21 | 2018-06-27 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| EP3338764A1 (de) | 2016-12-21 | 2018-06-27 | Bayer Pharma Aktiengesellschaft | Pharmazeutische darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| WO2018189012A1 (de) | 2017-04-10 | 2018-10-18 | Bayer Aktiengesellschaft | Substituierte n-arylethyl-2-aminochinolin-4-carboxamide und ihre verwendung |
| WO2018189011A1 (de) | 2017-04-10 | 2018-10-18 | Bayer Aktiengesellschaft | Substituierte n-arylethyl-2-arylchinolin-4-carboxamide und ihre verwendung |
| WO2018228909A1 (en) | 2017-06-14 | 2018-12-20 | Bayer Pharma Aktiengesellschaft | Substituted bridged diazepane derivatives and use thereof as task-1 and task-3 inhibitors |
| WO2018228907A1 (de) | 2017-06-14 | 2018-12-20 | Bayer Aktiengesellschaft | Diazabicyclisch substituierte imidazopyrimidine und ihre verwendung zur behandlung von atemstörungen |
| WO2019081303A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081291A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | PRODRUGS OF SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081302A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081307A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019081292A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | PRODRUGS OF SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| US11331314B2 (en) | 2017-10-24 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Amine substituted triazole derivatives and uses thereof |
| US11298367B2 (en) | 2017-10-24 | 2022-04-12 | Bayer Aktiengesellschaft | Prodrugs of substituted triazole derivatives and uses thereof |
| US11230540B2 (en) | 2017-10-24 | 2022-01-25 | Bayer Pharma Aktiengesellschaft | Substituted triazole derivatives and uses thereof |
| US11173151B2 (en) | 2017-10-24 | 2021-11-16 | Bayer Aktiengesellschaft | Substituted triazole derivatives and uses thereof |
| WO2019081299A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | AMINE SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| US11149023B2 (en) | 2017-10-24 | 2021-10-19 | Bayer Pharma Aktiengesellschaft | Substituted triazole derivatives and uses thereof |
| WO2019081306A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Pharma Aktiengesellschaft | SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
| WO2019091847A1 (de) | 2017-11-07 | 2019-05-16 | Bayer Aktiengesellschaft | Substituierte 2,4-dihydro-3h-1,2,4-triazol-3-one und ihre verwendung |
| WO2019105881A1 (de) | 2017-12-01 | 2019-06-06 | Bayer Pharma Aktiengesellschaft | Verfahren zur herstellung von (3s)-3-(4-chlor-3-{[(2s,3r)-2-(4-chlorphenyl)-4,4,4-trifluor-3-methylbutanoyl]amino}phenyl)-3-cyclopropylpropansäure und dessen kristalline form für die verwendung als pharmazeutischer wirkstoff |
| US11332435B2 (en) | 2017-12-01 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Method for producing (3S)-3-(4-chloro-3-{[(2S,3R)-2-(4- chlorophenyl)-4,4,4-trifluoro-3-methylbutanoyl]amino}phenyl)-3-cyclo-propylpropanoic acid and the crystalline form thereof for use as a pharmaceutical ingredient |
| EP3553082A1 (de) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Natriuretische peptidgepfropfte antikörper im gehirn |
| WO2019197477A1 (en) | 2018-04-12 | 2019-10-17 | Bayer Aktiengesellschaft | C-type natriuretic peptide engrafted antibodies |
| EP3553079A1 (de) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Mit natriuretischem peptid vom c-typ gepfropfte antikörper |
| US12331094B2 (en) | 2018-04-12 | 2025-06-17 | Bayer Aktiengesellschaft | Atrial natriuretic peptide engrafted antibodies |
| US12390508B2 (en) | 2018-04-12 | 2025-08-19 | Boehringer Ingelheim International Gmbh | Brain natriuretic peptide engrafted antibodies |
| EP3553081A1 (de) | 2018-04-12 | 2019-10-16 | Bayer Aktiengesellschaft | Natriuretische peptidgepfropfte antikörper im vorhof |
| WO2019197470A1 (en) | 2018-04-12 | 2019-10-17 | Bayer Aktiengesellschaft | Atrial natriuretic peptide engrafted antibodies |
| WO2019197475A1 (en) | 2018-04-12 | 2019-10-17 | Bayer Aktiengesellschaft | Brain natriuretic peptide engrafted antibodies |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| WO2019219517A1 (en) | 2018-05-17 | 2019-11-21 | Bayer Aktiengesellschaft | Substituted dihydropyrazolo pyrazine carboxamide derivatives |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020020790A1 (de) | 2018-07-24 | 2020-01-30 | Bayer Aktiengesellschaft | Oral applizierbare pharmazeutische darreichungsform mit modifizierter freisetzung |
| WO2020020789A1 (de) | 2018-07-24 | 2020-01-30 | Bayer Aktiengesellschaft | Oral applizierbare pharmazeutische darreichungsform mit modifizierter freisetzung |
| WO2020109109A1 (de) | 2018-11-27 | 2020-06-04 | Bayer Aktiengesellschaft | Verfahren zur herstellung von pharmazeutischen darreichungsformen enthaltend inhibitoren von task-1 und task-3 kanälen und deren verwendung für die therapie von atemstörungen |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| WO2020225095A1 (en) | 2019-05-07 | 2020-11-12 | Bayer Aktiengesellschaft | Masp inhibitory compounds and uses thereof |
| US12503491B2 (en) | 2019-05-07 | 2025-12-23 | Bayer Aktiengesellschaft | MASP inhibitory compounds and uses thereof |
| WO2020245342A1 (en) | 2019-06-07 | 2020-12-10 | Bayer Aktiengesellschaft | The use of sgc activators for the treatment of ophthalmologic diseases |
| WO2021089683A1 (en) | 2019-11-06 | 2021-05-14 | Bayer Aktiengesellschaft | Inhibitors of adrenoreceptor adrac2 |
| WO2021094210A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted pyrazine carboxamide derivatives as prostaglandin ep3 receptor antagonists |
| WO2021094209A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted pyrrolo triazine carboxamide derivatives as prostaglandin ep3 receptor antagonists |
| WO2021094208A1 (en) | 2019-11-12 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted imidazo pyrimidine ep3 antagonists |
| EP3822265A1 (de) | 2019-11-15 | 2021-05-19 | Bayer AG | Substituierte hydantoinamide als adamts7 antagonisten |
| WO2021094434A1 (en) | 2019-11-15 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted hydantoinamides as adamts7 antagonists |
| WO2021094436A1 (en) | 2019-11-15 | 2021-05-20 | Bayer Aktiengesellschaft | Substituted hydantoinamides as adamts7 antagonists |
| US12398120B2 (en) | 2019-11-15 | 2025-08-26 | Bayer Aktiengesellschaft | Substituted hydantoinamides as ADAMTS7 antagonists |
| EP3822268A1 (de) | 2019-11-15 | 2021-05-19 | Bayer Aktiengesellschaft | Substituierte hydantoinamide als adamts7 antagonisten |
| WO2021167458A1 (en) | 2020-02-21 | 2021-08-26 | Universiteit Maastricht | Use of a soluble guanylate cyclase (sgc) stimulator or of a combination of a sgc stimulator and an sgc activator for conditions wherein the heme group of sgc is oxidized or wherein sgc is deficient in heme |
| WO2021172982A1 (en) | 2020-02-26 | 2021-09-02 | Universiteit Maastricht | Therapeutic combination for the treatment of brain ischemia and said therapeutic combination for use in the treatment of brain ischemia |
| EP4144741A4 (de) * | 2020-04-30 | 2024-05-22 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Tricyclische verbindung auf pyrimidinbasis und verwendung davon |
| WO2022096394A1 (en) | 2020-11-04 | 2022-05-12 | Bayer Aktiengesellschaft | Masp inhibitory compounds and uses thereof |
| WO2022112213A1 (en) | 2020-11-30 | 2022-06-02 | Bayer Aktiengesellschaft | Crystalline forms of 3-[[3-(4-chlorophenyl)-5-oxo-4-((2s)-3,3,3-trifluoro- 2-hydroxypropyl)-4,5-dihydro-1h-1,2,4-triazol-1-yl]methyl]-1-[3- (trifluoromethyl)pyridin-2-yl]-1h-1,2,4-triazole-5-carboxamide |
| WO2022122914A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| US12195448B2 (en) | 2020-12-10 | 2025-01-14 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| US12595247B2 (en) | 2020-12-10 | 2026-04-07 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| EP4011873A1 (de) | 2020-12-10 | 2022-06-15 | Bayer Aktiengesellschaft | Substituierte pyrazolo-piperidin-carbonsäuren |
| EP4011874A1 (de) | 2020-12-10 | 2022-06-15 | Bayer Aktiengesellschaft | Substituierte pyrazolo-piperidin-carbonsäuren |
| WO2022122913A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| WO2022122916A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolyl piperidine carboxylic acids |
| WO2022122917A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | The use of sgc activators for the treatment of ophthalmologic diseases |
| WO2022122910A1 (en) | 2020-12-10 | 2022-06-16 | Bayer Aktiengesellschaft | Substituted pyrazolo piperidine carboxylic acids |
| EP4011904A1 (de) | 2020-12-14 | 2022-06-15 | Bayer Aktiengesellschaft | Masp-hemmende verbindungen und deren verwendungen |
| US12202805B2 (en) | 2021-12-29 | 2025-01-21 | Bayer Aktiengesellschaft | Process for preparing (5S)-{[2-(4-carboxyphenyl)ethyl] |2-(2-{|3-chloro-4'-(trifluoromethyl)biphenyl-4-yl]methoxy}phenyl)ethyl]aminol-5,6,7,8-tetrahydroquinoline-2-carboxylic acid and its crystalline forms for use as pharmaceutically active compound |
| US12138256B2 (en) | 2021-12-29 | 2024-11-12 | Bayer Aktiengesellschaft | Treatment of cardiopulmonary disorders |
| WO2023126436A1 (en) | 2021-12-29 | 2023-07-06 | Bayer Aktiengesellschaft | Process for preparing (5s)-{[2-(4-carboxyphenyl)ethyl] |2-(2-{|3-chloro-4'-(trifluoromethyl)biphenyl-4- yl]methoxy}phenyl)ethyl]aminol-5,6,7,8-tetrahydroquinoline-2-carboxylic acid and its crystalline forms for use as pharmaceutically active compound |
| WO2023126438A1 (en) | 2021-12-29 | 2023-07-06 | Bayer Aktiengesellschaft | Pharmaceutical dry powder inhalation formulation |
| WO2023126437A1 (en) | 2021-12-29 | 2023-07-06 | Bayer Aktiengesellschaft | Treatment of cardiopulmonary disorders |
| WO2023237577A1 (en) | 2022-06-09 | 2023-12-14 | Bayer Aktiengesellschaft | Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in women |
| WO2025068514A1 (en) | 2023-09-28 | 2025-04-03 | Bayer Aktiengesellschaft | Substituted heterocyclic carboxamindes and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| CL2013000083A1 (es) | 2013-04-12 |
| EP2590979A1 (de) | 2013-05-15 |
| JP6114189B2 (ja) | 2017-04-12 |
| CA2804470A1 (en) | 2012-01-12 |
| WO2012004258A9 (de) | 2012-06-07 |
| CN106977530A (zh) | 2017-07-25 |
| ZA201209434B (en) | 2014-02-26 |
| CR20130008A (es) | 2013-07-18 |
| CN103097387B (zh) | 2016-11-02 |
| AU2011275825A1 (en) | 2013-02-07 |
| EP2708539A1 (de) | 2014-03-19 |
| DOP2013000007A (es) | 2013-04-30 |
| KR20130132393A (ko) | 2013-12-04 |
| EA201390060A1 (ru) | 2013-07-30 |
| MX2013000198A (es) | 2013-01-28 |
| EP2682394A1 (de) | 2014-01-08 |
| US9216978B2 (en) | 2015-12-22 |
| PE20130779A1 (es) | 2013-06-21 |
| BR112013000596A2 (pt) | 2019-09-24 |
| JP2013532162A (ja) | 2013-08-15 |
| CN103097387A (zh) | 2013-05-08 |
| US20140350020A1 (en) | 2014-11-27 |
| SG186749A1 (en) | 2013-02-28 |
| ECSP13012379A (es) | 2013-02-28 |
| CU20130007A7 (es) | 2013-06-28 |
| PH12013500055A1 (en) | 2017-08-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2802587B1 (de) | Substituierte, annellierte imidazole und pyrazole und ihre verwendung | |
| WO2012004258A1 (de) | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen | |
| EP2590987B1 (de) | Annellierte 4-aminopyrimidine und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2699578B1 (de) | Fluoralkyl-substituierte pyrazolopyridine und ihre verwendung | |
| EP2729476B1 (de) | Heteroaryl-substituierte pyrazolopyridine und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2751106B1 (de) | Substituierte annellierte pyrimidine und ihre verwendung | |
| EP2822951B1 (de) | Substituierte azabicyclen und ihre verwendung | |
| DE102012200349A1 (de) | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung | |
| EP2576547A1 (de) | Substituierte 5-fluor-1h-pyrazolopyridine und ihre verwendung | |
| EP2635577A1 (de) | Substituierte 6-fluor-1h-pyrazolo[4,3-b]pyridine und ihre verwendung | |
| EP2705037A1 (de) | Substituierte imidazopyridine und imidazopyridazine und ihre verwendung | |
| EP2635576A1 (de) | Benzyl-substituierte carbamate und ihre verwendung | |
| EP3019506A1 (de) | Benzyl-1h-pyrazolo[3,4-b]pyridine und ihre verwendung | |
| WO2017121700A1 (de) | 1,3-disubstituierte 1h-pyrazolo[3,4-b]pyridin- derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| WO2012010576A1 (de) | Carbamat-substituierte diaminopyrimidine und ihre verwendung | |
| DE102011075399A1 (de) | Substituierte Imidazopyridine und Imidazopyridazine und ihre Verwendung | |
| DE102011007891A1 (de) | Annellierte 4-Aminopyrimidine und ihre Verwendung | |
| DE102011007890A1 (de) | Fluoralkyl-substituierte Pyrazolopyridine und ihre Verwendung | |
| WO2017121692A1 (de) | Substituierte sulfamide und ihre verwendung | |
| DE102011078715A1 (de) | Heteroaryl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102011003315A1 (de) | Annellierte Pyrimindine und Triazine und ihre Verwendung | |
| DE102010031149A1 (de) | Annellierte Pyrimidine und Triazine und ihre Verwendung | |
| DE102012200356A1 (de) | Substituierte Imidazopyridine und Imidazopyridazine und ihre Verwendung | |
| DE102012200354A1 (de) | Heteroaryl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102012200357A1 (de) | Fluoralkyl-substituierte Pyrazolopyridine und ihre Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180043624.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11732409 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011732409 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 000009-2013 Country of ref document: PE |
|
| ENP | Entry into the national phase |
Ref document number: 2804470 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2013/000198 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2013517357 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2013-000008 Country of ref document: CR Ref document number: 12013500055 Country of ref document: PH |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013000083 Country of ref document: CL Ref document number: 13003185 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201390060 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2011275825 Country of ref document: AU Date of ref document: 20110705 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20137003410 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201301525 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13806425 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013000596 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013000596 Country of ref document: BR Kind code of ref document: A2 Effective date: 20130109 |