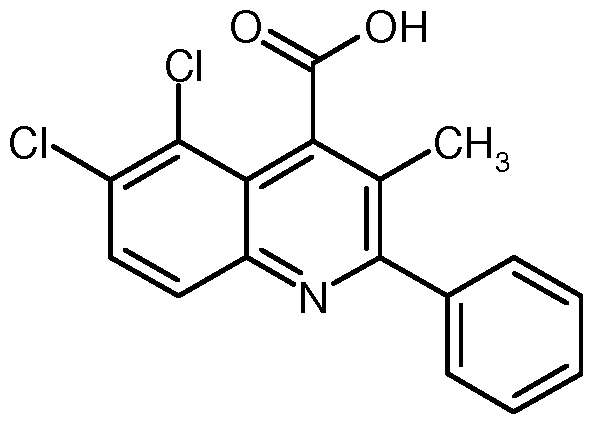
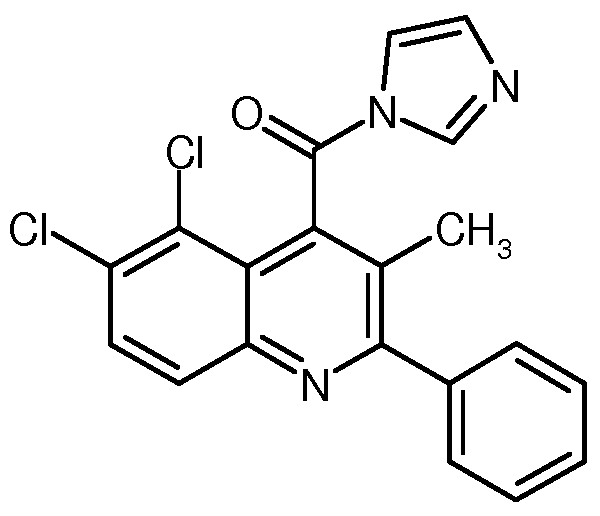
SUBSTITUIERTE N.2-D I ARYLCHI NOLI N-4-CARBOXAM IDE UND IHRE
ANTI-INFLAM MATORISCHE VERWENDUNG
Die vorliegende Anmeldung betrifft neue substituierte /V,2-Diarylchinolin-4-carboxamid-Derivate, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Behandlung und/oder Prävention von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prävention von Krankheiten, insbesondere zur Behandlung und/oder Prävention von fibrotischen und inflammatorischen Erkrankungen.
Prostaglandin F2alpha (PGF2a) gehört zur Familie der bioaktiven Prostaglandine, die Derivate der Arachidonsäure darstellen. Nach Freisetzung aus Membranphospholipiden durch A2-Phospho- lipasen wird die Arachidonsäure durch Cyclooxygenasen zu dem Prostaglandin H2 (PGH2) oxi- diert, welches durch die PGF-Synthase weiter zu PGF2a umgewandelt wird. Zu einem wesentlich geringeren Anteil kann PGF2a auch aus anderen Prostaglandinen wie PGE2 oder PGD2 enzyma- tisch gebildet werden [Watanabe et al., /. Biol. Chem. 1985, 260:7035-7041] . PGF2a wird nicht gespeichert, sondern nach Synthese sofort freigesetzt, wodurch es lokal seine Wirkungen entfaltet. PGF2a ist ein instabiles Molekül (tm < 1 Minute), welches schnell enzymatisch in Lunge, Leber und Niere zu einem inaktiven Metaboliten, 15-Ketodihydro-PGF2a, umgelagert wird [Basu et al., Acta Chem. Scand. 1992, 46: 108-110]. 15-Ketodihydro-PGF2a ist in größeren Mengen im Plasma und später auch im Urin sowohl unter physiologischen als auch pathophysiologischen Bedingungen detektierbar.
Die biologischen Effekte von PGF2a kommen durch die Bindung und Aktivierung eines membran- ständigen Rezeptors, des PGF2a-Rezeptors oder auch so genannten FP-Rezeptors, zustande. Der FP-Rezeptor gehört zu den G-Protein-gekoppelten Rezeptoren, die durch sieben Transmembrandomänen charakterisiert sind. Neben dem humanen FP-Rezeptor konnten auch die FP-Rezeptoren von Maus und Ratte Moniert werden [Abramovitz et al., /. Biol. Chem. 1994, 269:2632-2636; Sugimoto et al., /. Biol. Chem. 1994, 269: 1356-1360; Kitanaka et al., Prostaglandins 1994, 48:31- 41]. Im Menschen existieren zwei Isoformen des FP-Rezeptors, FPA und FPB. Von den Prosta- noid-Rezeptoren ist der FP-Rezeptor am wenigsten selektiv, da an ihn neben PGF2a noch PGD2 und PGE2 mit nanomolaren Affinitäten binden [Woodward et al., Pharmacol. Rev. 2011, 63:471- 538]. Stimulation des FP-Rezeptors führt primär zur Gq-abhängigen Aktivierung der Phospho- lipase C, was in Freisetzung von Calcium und einer Aktivierung der Diacylglycerol-abhängigen Proteinkinase C (PKC) resultiert. Der erhöhte intrazelluläre Calciumspiegel führt zur Calmodulin- vermittelten Stimulation der Myosin-leichte Ketten-Kinase (MLCK). Neben der Kopplung an das G-Protein Gq kann der FP-Rezeptor über G12/G13 auch die Rho/Rhokinase-Signaltransduktions- kaskade aktivieren sowie über Gi-Kopplung alternativ den Raf/MEK/MAP-Signalweg stimulieren [Woodward et al., Pharmacol. Rev. 2011, 63:471-538].
PGF2a ist an der Regulation zahlreicher physiologischer Funktionen, wie z.B. Ovarialfunktionen, Embryonalentwicklung, Veränderungen in der Gebärmutterschleimhaut, Gebärmutterkontraktion, Luteolyse und in der Induktion von Geburtswehen und der Entbindung, beteiligt. PGF2a wird auch im Endometrium in Epithelialzellen synthetisiert, wo es die zelluläre Proliferation stimuliert [Woodward et al., Pharmacol. Rev. 2011, 63:471-538]. Außerdem ist PGF2a ein potenter Stimu- lator der Glattmuskel-, Gefäß- und Bronchokonstriktion und ist in akuten und chronischen inflammatorischen Prozessen involviert [Basu, Mol. Cells 2010, 30:383-391] . In der Niere ist PGF2a an der Wasserabsorption, Natriurese und Diurese beteiligt. In den Augen reguliert PGF2a den intraokularen Druck. PGF2a spielt auch eine wichtige Rolle im Knochenmetabolismus: Das Prosta- glandin stimuliert den Natrium-abhängigen Transport von anorganischem Phosphat in Osteoblasten und es steigert die Freisetzung von Interleukin-6 und des vaskulären endothelialen Wachstumsfaktors (VEGF) in Osteoblasten; außerdem ist PGF2a ein starkes Mitogen und ein Überlebensfaktor für Osteoblasten [Agas et al., /. Cell Physiol. 2013, 228:25-29]. Darüber hinaus konnte gezeigt werden, dass die PGF2a-FP-Rezeptoraktivierung in verschiedenen kardiovaskulären Dys- funktionen, wie z.B. Myokardinfarkt und Hypertension, involviert ist [Zhang et al., Frontiers in Pharmacol. 2010, 1 : 1-7]. Stabilere Analoga von PGF2a wurden zur Ostrussynchronisierung und zur Beeinflussung humaner Reproduktionsfunktionen entwickelt sowie zur Reduktion des intraokularen Drucks für die Behandlung des Glaukoms eingesetzt [Basu, Mol. Cells 2010, 30:383- 391]. In Patienten mit idiopathischer pulmonaler Fibrose (IPF) konnte gezeigt werden, dass der stabile PGF2a-Metabolit 15-Ketodihydro-PGF2a im Plasma signifikant erhöht ist und dass die Spiegel von 15-Ketodihydro-PGF2a mit funktionalen Parametern, wie z.B. der forcierten Vitalkapazität (FVC), der Diffusionsstrecke von Kohlenmonoxid in der Lunge (DLCO) und dem 6-Minuten- Gehtest, korrelieren. Außerdem konnte ein Zusammenhang zwischen erhöhtem Plasma-15-Keto- dihydro-PGF2a und der Mortalität der Patienten festgestellt werden [Aihara et al., PLoS One 2013, 8: 1-6] . In Übereinstimmung damit wurde auch gezeigt, dass die Stimulation von humanen Lungenfibroblasten mit natürlich vorkommenden Silikastäuben, die im Menschen bei chronischer Inhalation zu Silikose und als Folge Lungenfibrose führen können, eine starke Hochregulation der PGF2a-Synthese bewirkt [O'Reilly et al., Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288: L1010-L1016]. In der Bleomycin-induzierten Lungenfibrose in Mäusen führte das Ausschalten des FP-Rezeptors durch Knock-down (FP-/-) zu deutlich reduzierter pulmonaler Fibrose im Vergleich zu Wildtyp-Mäusen [Oga et al., Nat. Med. 2009, 15: 1426-1430] . In FP-/- Mäusen war nach Bleomycin-Gabe eine signifikante Reduktion des Hydroxyprolingehalts sowie ein verminderte Induktion von profibrotischen Genen im Lungengewebe zu sehen. Außerdem war die Funktion der Lunge in FP-/- Mäusen im Vergleich zu den Wildtyp-Mäusen deutlich verbessert. In humanen
Lungenfibroblasten stimuliert PGF2a die Proliferation und die Kollagenproduktion über den FP- Rezeptor. Da dies unabhängig von dem profibrotischen Mediator TGFß erfolgt, stellt die PGF2 / FP-Rezeptor-Signalkaskade einen eigenständigen Weg bei der Entstehung der Lungenfibrose dar [Oga et al., Nat. Med. 2009, 15: 1426-1430] . Diese Befunde zeigen, dass der FP-Rezeptor ein thera- peutisches Zielprotein zur Behandlung von IPF ist [Olman, Nat. Med. 2009, 15: 1360-1361]. Die Beteiligung von PGF2a bei der Induktion von fibrotischen Veränderungen konnte auch an kardialen Maus-Fibroblasten [Ding et al., Int. J. Biochem. & Cell Biol. 2012, 44: 1031-1039], in einem Tiermodell der Sklerodermie [Kanno et al., Arthritis Rheum. 2013, 65: 492-502] sowie an Syn- oviozyten aus Patienten mit Kniegelenksarthrose [Bastiaansen et al. Arthritis Rheum. 2013, 65: 2070-2080] gezeigt werden.
Es wird daher angenommen, dass der FP-Rezeptor bei vielen Erkrankungen, Verletzungen und pathologischen Veränderungen, deren Entstehung und/oder Progression mit einem entzündlichen Geschehen und/oder einem proliferativen und fibroproliferativen Gewebe- und Gefäßumbau in Zusammenhang steht, eine wichtige Rolle spielt. Dies können insbesondere Erkrankungen und/ oder Schädigungen der Lunge, des Herz-Kreislauf-Systems oder der Niere sein, oder es kann sich hierbei um eine Erkrankung des Blutes, eine Krebs-Erkrankung oder um andere entzündliche Erkrankungen handeln.
In diesem Zusammenhang zu nennende Erkrankungen und Schädigungen der Lunge sind insbesondere die idiopathische Lungenfibrose, die pulmonale Hypertonie, das Bronchiolitis obliterans-Syn- drom (BOS), die chronisch-obstruktive Lungenerkrankung (COPD), Asthma und zystische Fibrose. Erkrankungen und Schädigungen des Herz-Kreislauf-Systems, in denen der FP-Rezeptor involviert ist, sind zum Beispiel Gewebeveränderungen nach einem Myokardinfarkt und bei der Herzinsuffizienz. Erkrankungen der Niere sind zum Beispiel Niereninsuffizienz und Nierenversagen. Eine Erkrankung des Blutes ist zum Beispiel die Sichelzellanämie. Beispiele für einen Gewebeab- und -umbau bei Krebsprozessen sind das Einwandern von Krebszellen in das gesunde Gewebe (Metastasenbildung) und die Neuausbildung von versorgenden Blutgefäßen (Neo-Angiogenese). Andere entzündliche Krankheiten, bei denen der FP-Rezeptor eine Rolle spielt, sind zum Beispiel Arthrose und Multiple Sklerose.
Die idiopathische Lungenfibrose oder idiopathische pulmonale Fibrose (IPF) ist eine progrediente Lungenerkrankung, die unbehandelt durchschnittlich innerhalb von 2.5 bis 3.5 Jahren nach Diagnosestellung zum Tode führt. Die Patienten sind zum Zeitpunkt der Diagnosestellung meist älter als 60 Jahre, und Männer sind etwas häufiger betroffen als Frauen. Der Krankheitsbeginn der IPF ist schleichend und durch eine zunehmende Atemnot und trockenen Reizhusten gekennzeichnet. IPF gehört zur Gruppe der idiopathischen interstitiellen Pneumonien (IIP), einer heterogenen
Gruppe von Lungenerkrankungen, die durch Fibrose und Inflammation unterschiedlichen Grades charakterisiert sind und die mit Hilfe klinischer, bildgebender und feingeweblicher Kriterien unterschieden werden. Innerhalb dieser Gruppe hat die idiopathische pulmonale Fibrose aufgrund ihrer Häufigkeit und des aggressiven Verlaufs eine besondere Bedeutung [Ley et al., Am. J. Respir. Crit. Care Med. 183, 431-440 (2011)]. Die IPF kann entweder sporadisch oder familiär gehäuft auftreten. Die Ursachen sind derzeit nicht geklärt. In den letzten Jahren wurden jedoch zahlreiche Hinweise dafür gefunden, dass eine chronische Schädigung des Alveolarepithels zur Freisetzung von profibrotischen Zytokinen/Mediatoren führt, gefolgt von einer gesteigerten Fibroblastenpro- liferation und einer vermehrten Kollagenfaserbildung, wodurch es zu einer fleckenförmigen Fibro- se und der typischen honigwabenartigen Struktur der Lunge kommt [Strieter et al., Chest 136, 1364-1370 (2009)]. Die klinischen Folgen der Fibrosierung sind eine Abnahme der Elastizität des Lungengewebes, eine Verminderung der Diffusionskapazität sowie die Entwicklung einer schweren Hypoxie. Lungenfunktionell kann entsprechend eine Verschlechterung der forcierten Vitalkapazität (FVC) und der Diffusionskapazität (DLCO) festgestellt werden. Wesentliche und pro- gnostisch bedeutende Komorbiditäten der IPF sind die akute Exazerbation und die pulmonale Hypertonie [Beck et al., Pneumologe 10, 105-111 (2013)]. Die Prävalenz der pulmonalen Hypertonie bei interstitiellen Lungenerkrankungen liegt bei 10-40% [Lettieri et al, Chest 129, 746-752 (2006); Behr et al, Eur. Respir. J. 31, 1357-1367 (2008)]. Es gibt gegenwärtig keine kurative Behandlung für die IPF - mit Ausnahme der Lungentransplantation. Die Pulmonale Hypertonie (PH) ist eine progrediente Lungenerkrankung, die unbehandelt durchschnittlich innerhalb von 2.8 Jahren nach Diagnosestellung zum Tode führt. Definitionsgemäß liegt bei einer chronischen pulmonalen Hypertonie ein pulmonal-arterieller Mitteldruck (mPAP) von > 25 mmHg in Ruhe oder > 30 mmHg unter Belastung vor (Normal wert < 20 mmHg). Die Pathophysiologie der pulmonalen Hypertonie ist gekennzeichnet durch Vasokonstriktion und ein Remodeling der Pulmonalgefäße. Bei der chronischen PH kommt es zu einer Neomuskularisierung primär nicht muskularisierter Lungengefäße, und die Gefäßmuskulatur der bereits muskularisierten Gefäße nimmt an Umfang zu. Durch diese zunehmende Obliteration der Lungenstrombahn kommt es zu einer progredienten Belastung des rechten Herzens, die zu einer verminderten Auswurfleistung des rechten Herzens führt und letztlich in einem Rechtsherzversagen endet [M. Humbert et al., /. Am. Coli. Cardiol. 2004, 43, 13S-24S] . Wenn auch die idiopathische (oder primäre) pulmonal-arterielle Hypertonie (ΓΡΑΗ) eine sehr seltene Erkrankung ist, so ist die sekundäre pulmonale Hypertonie (non-PAH PH, NPAHPH) weit verbreitet, und es wird zur Zeit angenommen, dass PH die dritthäufigste kardiovaskuläre Krankheitsgruppe nach koronarer Herzkrankheit und systemischem Bluthochdruck ist [Naeije, in: A. J. Peacock et al. (Eds.), Pulmonary Circulation. Diseases and their treatment, 3rd edition, Hodder Arnold Publ., 2011, S. 3] . Die Einteilung der pulmonalen Hypertonie in verschiedene Gruppen gemäß der jeweiligen Ätiologie erfolgt seit 2008
nach der Dana Point-Klassifikation [D. Montana und G. Simonneau, in: A. J. Peacock et al. (Eds.), Pulmonary Circulation. Diseases and their treatment, 3rd edition, Hodder Arnold Publ., 2011, S. 197-206].
Trotz aller Fortschritte in der Therapie der PH gibt es bisher keine Aussicht auf Heilung dieser schwerwiegenden Erkrankung. Auf dem Markt befindliche Therapien (z.B. Prostacyclin-Analoga, Endothelinrezeptor-Antagonisten, Phosphodiesterase-Inhibitoren) sind in der Lage, die Lebensqualität, die körperliche Belastbarkeit und die Prognose der Patienten zu verbessern. Hierbei handelt es sich um systemisch applizierte, primär hämodynamisch wirkende Therapieprinzipien, die den Gefäßtonus beeinflussen. Die Anwendbarkeit dieser Medikamente ist durch die z. T. gravie- renden Nebenwirkungen und/oder aufwendigen Applikationsformen eingeschränkt. Der Zeitraum, über den unter einer spezifischen Monotherapie die klinische Situation der Patienten stabilisiert oder verbessert werden kann, ist begrenzt (z.B. aufgrund einer Toleranzentwicklung). Es erfolgt schließlich eine Therapieeskalation und somit eine Kombinationstherapie, bei der mehrere Medikamente gleichzeitig gegeben werden müssen. Zur Zeit sind diese Standardtherapeutika nur zur Behandlung der pulmonal-arteriellen Hypertonie (PAH) zugelassen. Bei sekundären Formen der PH, wie z.B. PH-COPD, scheiterten diese Therapieprinzipien (z.B. Sildenafil, Bosentan) in klinischen Studien, da sie infolge einer unselektiven Vasodilatation zu einer Absenkung (Entsättigung) des arteriellen Sauerstoffgehalts bei den Patienten führten. Ursache hierfür ist wahrscheinlich eine ungünstige Beeinflussung der Ventilations-Perfusions-Anpassung innerhalb der Lunge bei hetero- genen Lungenerkrankungen aufgrund der systemischen Gabe unselektiver Vasodilatatoren [I. Blanco et al, Am. J. Respir. Grit. Care Med. 2010, 181, 270-278; D. Stolz et al, Eur. Respir. J. 2008, 32, 619-628].
Neue Kombinationstherapien sind eine der aussichtsreichsten zukünftigen Therapieoptionen zur Behandlung der pulmonalen Hypertonie. In diesem Zusammenhang ist die Erkundung neuer pharmakologischer Mechanismen zur Behandlung der PH von besonderem Interesse [Ghofrani et al, Herz 2005, 30, 296-302; E. B. Rosenzweig, Expert Opin. Emerging Drugs 2006, 11, 609-619; T. Ito et al, Curr. Med. Chem. 2007, 14, 719-733]. Vor allem solche neuen Therapieansätze, die mit den bereits auf dem Markt befindlichen Therapiekonzepten kombinierbar sind, könnten Grundlage einer effizienteren Behandlung sein und somit einen großen Vorteil für die Patienten bringen. Im Sinne der vorliegenden Erfindung schließt der Begriff pulmonale Hypertonie sowohl primäre als auch sekundäre Unterformen (NPAHPH) ein, wie sie nach der Dana Point-Klassifikation gemäß ihrer jeweiligen Ätiologie definiert worden sind [D. Montana und G. Simonneau, in: A. J. Peacock et al. (Eds.), Pulmonary Circulation. Diseases and their treatment, 3rd edition, Hodder Arnold Publ., 2011, S. 197-206; Hoeper et al, J. Am. Coli. Cardiol, 2009, 54 (1), Suppl. S, S85-
S96]. Hierzu gehört insbesondere in Gruppe 1 die pulmonal-arterielle Hypertonie (PAH), zu der unter anderem die idiopathischen und die familiären Formen zählen (IPAH bzw. FPAH). Des weiteren umfasst PAH auch die persistierende pulmonale Hypertonie bei Neugeborenen sowie die assoziierte pulmonal-arterielle Hypertonie (AP AH), welche assoziiert ist mit Kollagenosen, kon- genitalen systemisch-pulmonalen Shuntvitien, portaler Hypertension, HIV -Infektionen, der Einnahme bestimmter Drogen und Medikamente (z.B. von Appetitzüglern), mit Erkrankungen mit einer signifikanten venösen/kapillären Beteiligung wie der pulmonal-venookklusiven Erkrankung und der pulmonal-kapillären Hämangiomatose, oder mit anderen Erkrankungen wie Schilddrüsenerkrankungen, Glykogenspeicherkrankheiten, Morbus Gaucher, hereditärer Teleangiektasie, Hämoglobinopathien, myeloproliferativen Erkrankungen und Splenektomie. In Gruppe 2 der Dana Point-Klassifikation werden PH-Patienten mit einer ursächlichen Linksherzerkrankung, wie ventrikulären, atrialen oder valvulären Erkrankungen, zusammengefasst. Gruppe 3 umfasst Formen der pulmonalen Hypertonie, die mit einer Lungenerkrankung, wie z.B. chronisch-obstruktiver Lungenerkrankung (COPD), interstitieller Lungenkrankheit (ILD), Lungenfibrose (IPF), und/oder einer Hypoxämie (z.B. Schlafapnoe-Syndrom, alveoläre Hypoventilation, chronische Höhenkrankheit, anlagebedingte Fehlbildungen) assoziiert sind. Zur Gruppe 4 zählen PH-Patienten mit chronisch- thrombotischen und/oder embolischen Erkrankungen, z.B. bei thromboembolischer Obstruktion von proximalen und distalen Lungenarterien (CTEPH) oder bei nicht-thrombotischen Embolisie- rungen (z.B. infolge von Tumorerkrankungen, Parasiten, Fremdkörpern). Seltenere Formen der pulmonalen Hypertonie, wie z.B. bei Patienten mit Sarkoidose, Histiozytose X oder Lymphangio- matose, sind in der Gruppe 5 zusammengefasst.
Beim Bronchiolitis obliterans-Syndrom (BOS) handelt es sich um eine chronische Abstoßungsreaktion nach erfolgter Lungentransplantation. Innerhalb der ersten fünf Jahre nach Lungentransplantation sind ca. 50-60% aller Patienten, innerhalb der ersten neun Jahre über 90% der Patienten betroffen [Estenne et al, Am. J. Respir. Grit. Care Med. 166, 440-444 (2003)]. Die Ursache der Erkrankung ist nicht geklärt. Trotz vieler Fortschritte bei der Behandlung von Transplantationspatienten haben sich die BOS-Fallzahlen in den vergangenen Jahren kaum verändert. Das BOS ist die wichtigste langfristige Komplikation bei Lungentransplantationen und gilt als Hauptgrund dafür, dass die Überlebensraten nach wie vor deutlich unter denen anderer Organtransplantationen liegen. Beim BOS handelt es sich um ein entzündliches Geschehen, das mit Veränderungen des Lungengewebes einhergeht, die vor allem die kleinen Atemwege betreffen. Die Schädigung und entzündlichen Veränderungen der Epithelzellen sowie der subepithelialen Strukturen der kleineren Atemwege führen aufgrund einer nicht effektiven Regeneration des Epithels und einer aberrierenden Gewebereparation zu einer exzessiven Fibroproliferation. Es kommt zur Vernarbung und schließlich Zerstörung der Bronchiolen sowie zu Pfropfen von Granulationsgewebe in den kleinen Atemwegen und Alveolen, gelegentlich auch mit vaskulärer Beteiligung. Die Diagnose wird auf-
grund der Lungenfunktion gestellt. Beim BOS kommt es zu einer Verschlechterung des FEV1 im Vergleich zum Durchschnitt der zwei besten postoperativ gemessenen Werte. Gegenwärtig gibt es keine kurative Behandlung für BOS. Ein Teil der Patienten verbessert sich unter intensivierter Immunsuppression, bei den nicht darauf ansprechenden Patienten kommt es zu einer anhaltenden Verschlechterung, so dass eine erneute Transplantation (Retransplantation) indiziert ist.
Die chronisch-obstruktive Lungenerkrankung (COPD) ist eine langsam fortschreitende Lungenerkrankung, die durch eine Behinderung der Atemströmung charakterisiert ist, welche durch ein Lungenemphysem und/oder eine chronische Bronchitis hervorgerufen wird. Die ersten Symptome der Erkrankung zeigen sich in der Regel ab dem vierten bis fünften Lebensjahrzehnt. In den darauffolgenden Lebensjahren verschlimmert sich häufig die Kurzatmigkeit und es manifestiert sich Husten, verbunden mit einem ausgiebigen und stellenweise eitrigen Auswurf und einer Stenose-Atmung bis hin zu einer Atemnot (Dyspnoe). COPD ist in erster Linie eine Krankheit von Rauchern: Rauchen ist verantwortlich für 90% aller COPD-Fälle und 80-90% aller COPD-Todes- fälle. COPD ist ein großes medizinisches Problem und stellt weltweit die sechsthäufigste Todes- Ursache dar. Von den über 45-jährigen Menschen sind ca. 4-6% betroffen. Obwohl die Behinderung der Atemströmung nur partiell und zeitlich befristet sein kann, ist COPD nicht heilbar. Behandlungsziel ist folglich eine Verbesserung der Lebensqualität, die Linderung der Symptome, die Verhinderung akuter Verschlechterungen und die Verlangsamung der fortschreitenden Beeinträchtigung der Lungenfunktion. Bestehende Pharmakotherapien, die sich seit den letzten zwei bis drei Jahrzehnten kaum geändert haben, sind das Verwenden von Bronchodilatoren, um blockierte Atemwege zu öffnen, und in bestimmten Situationen Kortikosteroide, um die Entzündung der Lunge einzudämmen [P. J. Barnes, N. Engl. J. Med. 343, 269-280 (2000)] . Die chronische Entzündung der Lunge, hervorgerufen durch Zigarettenrauch oder andere Reizstoffe, ist die treibende Kraft der Krankheitsentwicklung. Der zugrunde liegende Mechanismus beinhaltet Immunzellen, die im Zuge der inflammatorischen Reaktion der Lunge Proteasen und verschiedene Zytokine ausschütten, die zu einem Lungenemphysem und Remodeling der Bronchien führen.
Die Aufgabe der vorliegenden Erfindung liegt in der Identifizierung und Bereitstellung neuer Substanzen, die potente, chemisch und metabolisch stabile, nicht-prostanoide Antagonisten des FP- Rezeptors darstellen und sich als solche zur Behandlung und/oder Prävention insbesondere von fibrotischen und inflammatorischen Erkrankungen eignen.
Unter anderem aus WO 95/32948-A1, WO 96/02509-A1 und WO 97/19926-A1 sind 2-Arylchino- lin-4-carboxamide als NK3- oder duale NK/NKs-Antagonisten bekannt, welche sich zur Behandlung von Erkrankungen der Lunge und des Zentralnervensystems eignen. In WO 2004/045614-A1 werden bestimmte Chinolincarboxamide als Glucokinase-Liganden für die Behandlung von Dia-
betes beschrieben. In WO 2006/094237-A2 werden Chinolin-Derivate als Sirtuin-Modulatoren offenbart, die zur Behandlung verschiedenartiger Erkrankungen eingesetzt werden können. In WO 2011/009540- A2 werden bicyclische Carboxamide mit pestizider Wirkung beschrieben. In WO 2011/153553-A2 werden verschiedene bicyclische Heteroaryl-Verbindungen als Kinase-Inhibito- ren für die Behandlung insbesondere von Krebserkrankungen beansprucht. In EP 2 415 755-A1 sind unter anderem Chinolin-Derivate beschrieben, die sich zur Behandlung von Erkrankungen eignen, welche mit der Aktivität des Plasminogen- Aktivator-Inhibitor- 1 (PAI-1) assoziiert sind. In WO 2013/074059-A2 werden verschiedene Chinolin-4-carboxamid-Derivate aufgeführt, die als Inhibitoren von Cytosin-Deaminasen zur Verstärkung einer DNA-Transfektion von Zellen dienen können. In WO 2013/164326- AI werden /V,3-Diphenylnaphthalin-1 -carboxamide als Agonisten des EP2-Prostaglandin-Rezeptors zur Behandlung von Atemwegserkrankungen offenbart. In WO 2014/117090-A1 werden verschiedene 2-Arylchinolin-Derivate als Inhibitoren von Metalloenzymen beschrieben. Zwischenzeitlich wurden in WO 2015/094912-A1 unter anderem substituierte /V,2-Diphenylchinolin-4-carboxamid-Derivate offenbart, die sich als Antagonisten des Prostaglan- din EP4-Rezeptors zur Behandlung von Arthritis und damit verbundenen Schmerzzuständen eignen.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)
in welcher R
A für Wasserstoff, Halogen, Pentafluorsulfanyl, Cyano, Nitro, (Ci-C -Alkyl, Hydroxy, (Ci-C
4)-Alkoxy, Amino oder eine Gruppe der Formel -NH-C(=0)-R
6, -NH-C(=0)-NH-R
6 oder -S(=0)„-R
7 steht, wobei (Ci-C -Alkyl und (Ci-C -Alkoxy bis zu dreifach mit Fluor substituiert sein können,
und worin
R6 Wasserstoff oder (Ci-C4)-Alkyl, das bis zu dreifach mit Fluor substituiert sein kann, bedeutet,
R7 (Ci-C- -Alkyl, das mit Hydro xy, Methoxy oder Ethoxy oder bis zu dreifach mit Fluor substituiert sein kann, bedeutet und n die Zahl 0, 1 oder 2 bedeutet, D für C-RD oder N steht, E für C-RE oder N steht, G für C-RG oder N steht, wobei maximal zwei der Ringglieder D, E und G zugleich für N stehen, und worin
RD und RE unabhängig voneinander Wasserstoff, Fluor, Chlor, Methyl, Trifluormethyl, Methoxy oder Trifluormethoxy bedeuten und
RG Wasserstoff, Fluor, Chlor, Brom, Methyl oder Trifluormethyl bedeutet,
Z für OH oder eine Gruppe der Formel -NH-R8, -NH-SO2-R9 oder -NH-SO2-NR10AR10B steht, worin
R8 Wasserstoff oder (Ci-C -Alkyl, das bis zu dreifach mit Fluor substituiert sein kann, bedeutet,
R9 (Ci-C -Alkyl, das bis zu dreifach mit Fluor substituiert sein kann, oder Phenyl bedeutet und
R10A und R10B unabhängig voneinander Wasserstoff oder (Ci-C4)-Alkyl, das bis zu dreifach mit Fluor substituiert sein kann, bedeuten,
R1 für Halogen, Trifluormethoxy, (Trifluormethyl)sulfanyl, Pentafluorsulfanyl, (Ci-C4)-Alkyl, Trimethylsilyl, Cyclopropyl oder Cyclobutyl steht, wobei (Ci-C -Alkyl bis zu dreifach mit Fluor und Cyclopropyl und Cyclobutyl bis zu zweifach mit Fluor substituiert sein können,
R2, R3 und R4 unabhängig voneinander für Wasserstoff, Fluor, Chlor, Methyl oder Trifluormethyl stehen,
R5 für (Ci-C -Alkyl, das bis zu dreifach mit Fluor substituiert sein kann, oder für Fluor, Chlor, Methoxy oder Cyclopropyl steht, und
Ar für Phenyl, das ein- oder zweifach, gleich oder verschieden, mit Fluor und Chlor substituiert sein kann, oder für Pyridyl oder Thienyl steht, sowie ihre -Oxide, Salze, Solvate, Salze der -Oxide und Solvate der -Oxide und Salze.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt. Erfindungsgemäße Verbindungen sind ebenso -Oxide der Verbindungen der Formel (I) sowie deren Salze, Solvate und Solvate der Salze.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung, Reinigung oder Lagerung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen insbesondere die von üblichen Basen abgeleiteten Salze, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze), Zinksalze sowie Ammoniumsalze abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-
Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, /V,/V-Diisopro- pylethylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dimethylaminoethanol, Diethyl- aminoethanol, Tris(hydroxymethyl)aminomethan, Cholin, Procain, Dicyclohexylamin, Dibenzyl- amin, /V-Mefhylmorpholin, /V-Mefhylpiperidin, Arginin, Lysin und 1,2-Ethylendiamin. Darüber hinaus umfassen physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen auch Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethansulfonsäure, Benzolsulfonsäure, Toluolsulfonsäure, Naphthalindisulfonsäure, Ameisensäure, Essigsäure, Trifluoressigsäure, Propionsäure, Bernsteinsäure, Fumarsäure, Maleinsäure, Milch- säure, Weinsäure, Äpfelsäure, Zitronensäure, Gluconsäure, Benzoesäure und Embonsäure.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in unterschiedlichen stereoisomeren Formen existieren, d.h. in Gestalt von Konfigurationsisomeren oder gegebenenfalls auch als Konformationsisomere (Enantiomere und/oder Diastereomere, einschließlich solcher bei Atropisomeren). Die vorliegende Erfindung umfasst deshalb die Enantiomere und Dia- stereomere sowie ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/ oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren. Vorzugsweise werden hierfür chromatographische Verfahren angewandt, insbesondere die HPLC -Chromatographie an achiralen bzw. chiralen Trennphasen. Im Falle von Carbonsäuren als Zwischen- oder Endprodukten kann alternativ auch eine Trennung über diastereomere Salze mit Hilfe chiraler Amin-Basen erfolgen.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Die vorliegende Erfindung umfasst auch alle geeigneten isotopischen Varianten der erfindungsgemäßen Verbindungen. Unter einer isotopischen Variante einer erfindungsgemäßen Verbindung wird hierbei eine Verbindung verstanden, in welcher mindestens ein Atom innerhalb der erfindungsgemäßen Verbindung gegen ein anderes Atom der gleichen Ordnungszahl, jedoch mit einer anderen Atommasse als der gewöhnlich oder überwiegend in der Natur vorkommenden Atommasse ausgetauscht ist. Beispiele für Isotope, die in eine erfindungsgemäße Verbindung inkor-
poriert werden können, sind solche von Wasserstoff, Kohlenstoff, Stickstoff, Sauerstoff, Phosphor, Schwefel, Fluor, Chlor, Brom und Iod, wie Ή (Deuterium), Ή (Tritium), 13C, 14C, 15N, 170, 180, 32P, 33P, 33S, 34S, 35S, 36S, 18F, 36C1, 82Br, 123I, 124I, 129I und 131L Bestimmte isotopische Varianten einer erfindungsgemäßen Verbindung, wie insbesondere solche, bei denen ein oder mehrere radio- aktive Isotope inkorporiert sind, können von Nutzen sein beispielsweise für die Untersuchung des Wirkmechanismus oder der Wirkstoff -Verteilung im Körper; aufgrund der vergleichsweise leichten Herstell- und Detektierbarkeit sind hierfür insbesondere mit 3H- oder 14C -Isotopen markierte Verbindungen geeignet. Darüber hinaus kann der Einbau von Isotopen, wie beispielsweise von Deuterium, zu bestimmten therapeutischen Vorteilen als Folge einer größeren metabolischen Sta- bilität der Verbindung führen, wie beispielsweise zu einer Verlängerung der Halbwertszeit im Körper oder zu einer Reduktion der erforderlichen Wirkdosis; solche Modifikationen der erfindungsgemäßen Verbindungen können daher gegebenenfalls auch eine bevorzugte Ausführungsform der vorliegenden Erfindung darstellen. Isotopische Varianten der erfindungsgemäßen Verbindungen können nach allgemein gebräuchlichen, dem Fachmann bekannten Verfahren hergestellt werden, so beispielsweise nach den weiter unten beschriebenen Methoden und den bei den Ausführungsbeispielen wiedergegebenen Vorschriften, indem hierbei entsprechende isotopische Modifikationen der jeweiligen Reagentien und/oder Ausgangsverbindungen eingesetzt werden.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" bezeichnet hierbei Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper auf beispielsweise metabolischem oder hydrolytischem Wege zu erfindungsgemäßen Verbindungen umgesetzt werden.
Insbesondere umfasst die vorliegende Erfindung als Prodrugs hydrolysierbare Ester-Derivate der erfindungsgemäßen Carbonsäuren der Formel (I) [mit Z = OH] . Hierunter werden Ester verstanden, die in physiologischen Medien, unter den Bedingungen der im weiteren beschriebenen biolo- gischen Tests und insbesondere in vivo auf enzymatischem oder chemischem Wege zu den freien Carbonsäuren, als den biologisch hauptsächlich aktiven Verbindungen, hydrolysiert werden können. Als solche Ester werden (Ci-C -Alkylester, in welchen die Alkylgruppe geradkettig oder verzweigt sein kann, bevorzugt. Besonders bevorzugt sind Methyl-, Ethyl- oder ieri.-Butylester.
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
(Ci -C Q-Alkyl steht im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, Isobutyl, sec.-Butyl und ieri.-Butyl.
(Ci-C4)-Alkoxy steht im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkoxy- rest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy, Isobutoxy, sec.-Butoxy und ieri.-Butoxy.
Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und lod ein. Bevorzugt sind Chlor, Fluor oder Brom, besonders bevorzugt Fluor oder Chlor.
Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Eine Substitution mit einem oder mit zwei gleichen oder verschiedenen Substituenten ist bevorzugt. Besonders bevorzugt ist die Substitution mit einem Substituenten.
Eine besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
RA für Wasserstoff, Fluor, Chlor, Brom, Cyano, Nitro, Methyl, Trifluormethyl, Hydroxy, Methoxy, Trifluormethoxy oder eine Gruppe der Formel -S(=0)n-R7 steht, worin
R7 Methyl oder Trifluormethyl bedeutet und n die Zahl 0 oder 2 bedeutet, D für C-RD oder N steht, worin
RD Wasserstoff oder Fluor bedeutet, E für C-H steht und
G für C-RG oder N steht, worin
RG Wasserstoff, Fluor oder Chlor bedeutet, sowie ihre Salze, Solvate und Solvate der Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindung Formel (I), in welcher
RA für Fluor, Chlor, Brom, Cyano, Methyl, Trifluormethyl, Methoxy, Trifluormethoxy oder eine Gruppe der Formel -S(=0)n-R7 steht, worin
R7 Methyl oder Trifluormethyl bedeutet und n die Zahl 0, 1 oder 2 bedeutet,
D für C-H steht, E für C-H steht und
G für C-RG oder N steht, worin RG Wasserstoff, Fluor oder Chlor bedeutet, sowie ihre Salze, Solvate und Solvate der Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Z für OH steht, sowie ihre Salze, Solvate und Solvate der Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R1 für Chlor, Brom, Iod, Methyl, Ethyl, Isopropyl, Trifluormethyl, Trifluormethoxy, (Tri- fluormethyl)sulfanyl, Pentafluorsulfanyl oder Trimethylsilyl steht, R2 und R3 jeweils für Wasserstoff stehen und
R4 für Wasserstoff, Fluor oder Chlor steht, sowie ihre Salze, Solvate und Solvate der Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R1 für Brom steht und
R2, R3 und R4 jeweils für Wasserstoff stehen, sowie ihre Salze, Solvate und Solvate der Salze. Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R1 und R4 jeweils für Chlor stehen und
R2 und R3 jeweils für Wasserstoff stehen, sowie ihre Salze, Solvate und Solvate der Salze.
Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
R5 für Methyl steht, sowie ihre Salze, Solvate und Solvate der Salze. Eine weitere besondere Ausführungsform der vorliegenden Erfindung umfasst Verbindungen der Formel (I), in welcher
Ar für Phenyl, das einfach mit Fluor substituiert sein kann, oder für Pyridyl steht, sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher RA für Wasserstoff, Fluor, Chlor, Brom, Cyano, Methyl, Trifluormethyl, Methoxy, Trifluor- methoxy oder eine Gruppe der Formel -S(=0)n-R7 steht, worin
R7 Methyl oder Trifluormethyl bedeutet und n die Zahl 0 oder 2 bedeutet,
D für C-RD oder N steht, worin
RD Wasserstoff oder Fluor bedeutet, E für C-H steht, G für C-RG oder N steht, worin RG Wasserstoff, Fluor oder Chlor bedeutet,
Z für OH steht,
R1 für Chlor, Brom, lod, Methyl, Ethyl, Isopropyl, Trifluormethyl, Trifluormethoxy, (Tri- fluormethyl)sulfanyl, Pentafluorsulfanyl oder Trimethylsilyl steht,
R2 und R3 jeweils für Wasserstoff stehen, R4 für Wasserstoff, Fluor oder Chlor steht,
R5 für Methyl, Chlor oder Cyclopropyl steht, und
Ar für Phenyl, das einfach mit Fluor substituiert sein kann, oder für Pyridyl steht, sowie ihre Salze, Solvate und Solvate der Salze. Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
RA für Fluor, Chlor, Cyano, Methyl, Trifluormethyl, Methoxy, Trifluormethoxy oder eine Gruppe der Formel -S(=0)n-R7 steht, worin
R7 Methyl oder Trifluormethyl bedeutet und n die Zahl 0 oder 2 bedeutet, D für C-H steht, E für C-H steht, G für C-RG oder N steht, worin
RG Wasserstoff, Fluor oder Chlor bedeutet,
Z für OH steht, für Chlor, Brom, Methyl, Trifluormethyl oder Trimethylsilyl steht,
R2 und R3 jeweils für Wasserstoff stehen, R4 für Wasserstoff oder Chlor steht, R5 für Methyl steht, und
Ar für Phenyl, das einfach mit Fluor substituiert sein kann, oder für 4-Pyridyl steht, sowie ihre Salze, Solvate und Solvate der Salze. Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche. Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Verbindungen, dadurch gekennzeichnet, dass man eine Verbindung der Formel (II)
in welcher R
1, R
2, R
3, R
4, R
5 und Ar die oben angegebenen Bedeutungen haben, unter Aktivierung der Carbonsäure-Funktion mit einer Amin- Verbindung der Formel (III)
in welcher RA, D, E und G die oben angegebenen Bedeutungen haben
und
T für (Ci-C4)-Alkyl oder Benzyl steht,
zu einer Verbindung der Formel (IV)
in welcher RA, D, E, G, R1, R2, R3, R4, R5, Ar und T die oben angegebenen Bedeutungen haben, kuppelt und anschließend den Ester-Rest T zur erfindungsgemäßen Carbonsäure der Formel (I-A)
in welcher R
A, D, E, G, R
1, R
2, R
3, R
4, R
5 und Ar die oben angegebenen Bedeutungen haben, abspaltet, sowie gegebenenfalls die Carbonsäure (I-A) in das entsprechende Säurechlorid der Formel (V)
in welcher R
A, D, E, G, R
1, R
2, R
3, R
4, R
5 und Ar die oben angegebenen Bedeutungen haben, überführt und dieses nachfolgend mit einer Verbindung der Formel (VI)
H
2N— R
8 (VI), in welcher R
8 die oben angegebene Bedeutung hat, zum erfindungsgemäßen Carbonsäureamid der Formel (I-B)
in welcher R
A, D, E, G, R
1, R
2, R
3, R
4, R
5, R
8 und Ar die oben angegebenen Bedeutungen haben, umsetzt, und die so erhaltenen Verbindungen der Formeln (I-A) und (I-B) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Basen oder Säuren in ihre Solvate, Salze und/oder Solvate der Salze überführt.
Die Kupplungsreaktion (II) + (III)— (IV) [Amid-Bildung] kann entweder auf direktem Weg mit Hilfe eines Kondensations- oder Aktivierungsmittels oder über die Zwischenstufe eines aus (II) erhältlichen Carbonsäurechlorids oder Carbonsäureimidazolids erfolgen.
Als solche Kondensations- oder Aktivierungsmittel eignen sich beispielsweise Carbodiimide wie N,N'-Diefhyl-, N,N'-Dipropyl-, N,N'-Diisopropyl-, N,N'-Dicyclohexylcarbodiimid (DCC) oder ^-(S-Dimethylaminopropy^-ZV-ethylcarbodiimid-Hydrochlorid (EDC), Phosgen-Derivate wie '-Carbonyldiimidazol (CDI) oder Isobutylchlorformiat, 1,2-Oxazolium- Verbindungen wie
2- Ethyl-5-phenyl-l,2-oxazolium-3-sulfat oder 2-ieri.-Butyl-5-methylisoxazolium-perchlorat, Acyl- amino- Verbindungen wie 2-Ethoxy-l-ethoxycarbonyl-l,2-dihydrochinolin, α-Chlorenamine wie l-Chlor-/V,/V,2-trimethylprop-l-en-l-amin, 1,3,5-Triazin-Derivate wie 4-(4,6-Dimefhoxy-l,3,5-tri- azin-2-yl)-4-methylmorpholiniumchlorid, Phosphor- Verbindungen wie n-Propanphosphonsäure- anhydrid (PPA), Cyanophosphonsäurediethylester, Diphenylphosphorylazid (DPPA), Bis-(2-oxo-
3- oxazolidinyl)-phosphorylchlorid, Benzotriazol- 1 -yloxy-tris(dimethylamino)phosphonium-hexa- fluorophosphat oder Benzotriazol- l-yloxy-tris(pyrrolidino)phosphonium-hexafluorophosphat (Py- BOP), oder Uronium- Verbindungen wie (^-(Benzotriazol-l-y^- A'./V'./V'-tetramethyluronium- tetrafluoroborat (TBTU), 0-(Benzotriazol- 1 -y^- A'./V'./V'-tetramethyluronium-hexafluorophosphat (HBTU), 0-( lii-6-Chlorbenzotriazol-l -yl)- 1 , 1,3,3-tetramethyluronium-tetrafluoroborat (TCTU), (^-(T-Azabenzotriazol-l-y^- A'./V'./V'-tetramethyluronium-hexafluorophosphat (HATU) oder 2-(2- Oxo-l-(2 /)-pyridyl)-l,l,3,3-tetramethyluronium-tetrafluoroborat (TPTU), gegebenenfalls in Kom-
bination mit weiteren Hilfsstoffen wie 1-Hydroxybenzotriazol (HOBt) oder -Hydroxysuccinimid (HOSu), sowie als Basen Alkalicarbonate, z.B. Natrium- oder Kaliumcarbonat, oder tertiäre Amin- basen wie Triethylamin, N-Mefhylmorpholin (NMM), N-Mefhylpiperidin (NMP), N, N-Diisopro- pylethylamin, Pyridin oder 4-/V,/V-Dimethylaminopyridin (DMAP). Als Kondensations- oder Akti- vierungsmittel bevorzugt eingesetzt wird l-Chlor-AyV,2-trimethylprop-l-en-l-amin in Verbindung mit Pyridin oder (^-(T-Azabenzotriazol-l-y^-A'. /V'./V'-tetramethyluronium-hexafluorophosphat (HATU) in Kombination mit -Diisopropylefhylamin.
Bei zweistufiger Reaktionsführung über die aus (Π) erhältlichen Carbonsäurechloride oder Carbon- säureimidazolide wird die Kupplung mit der Amin-Komponente (ΠΙ) in Gegenwart einer üblichen Base durchgeführt, wie beispielsweise Natrium- oder Kaliumcarbonat, Triethylamin, N,N-Oiiso- propylethylamin, N-Mefhylmorpholin (NMM), N-Mefhylpiperidin (NMP), Pyridin, 2,6-Dimefhyl- pyridin, 4-N, N-Dimethylaminopyridin (DMAP), l,8-Diazabicyclo[5.4.0]undec-7-en (DBU), 1,5- Diazabicyclo[4.3.0]non-5-en (DBN), Natrium- oder Kaliummethanolat, Natrium- oder Kalium- ethanolat, Natrium- oder Kalium-ieri.-butylat oder Natrium- oder Kaliumhydrid. Im Falle der Car- bonsäurechloride wird für die Kupplung bevorzugt Pyridin und bei Carbonsäureimidazoliden bevorzugt Kalium-ieri.-butylat oder Natriumhydrid als Base verwendet.
Inerte Lösungsmittel für die genannten Kupplungsreaktionen sind - je nach eingesetztem Verfahren - beispielsweise Ether wie Diethylether, Diisopropylether, Methyl-ieri.-butylether, Tetrahydro- furan, 1,4-Dioxan, 1,2-Dimethoxyethan oder Bis(2-methoxyethyl)ether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Pentan, Hexan oder Cyclohexan, Halogenkohlenwasserstoffe wie Dichlor - methan, Trichlormethan, Tetrachlormethan, 1,2-Dichlorethan, Trichlorethylen oder Chlorbenzol, oder polar-aprotische Lösungsmittel wie Aceton, Methylethylketon, Ethylacetat, Acetonitril, Butyronitril, Pyridin, Dimethylsulfoxid (DMSO), Ν,Ν-Dimethylformamid (DMF), W-Dimefhyl- propylenharnstoff (DMPU) oder /V-Methylpyrrolidinon (NMP). Auch können Gemische solcher Lösungsmittel eingesetzt werden. Bevorzugt werden Dichlormethan, Tetrahydrofuran, N,N-Oi- methylformamid oder Gemische hiervon verwendet. Die Kupplungen werden im Allgemeinen in einem Temperaturbereich von -20°C bis +60°C, bevorzugt bei 0°C bis +40°C durchgeführt.
Bevorzugte Kupplungsmethode ist die Umsetzung eines aus (II) abgeleiteten Carbonsäureimidazo- lids mit der Amin- Verbindung (III). Die Carbonsäureimidazolide selbst sind nach bekanntem Verfahren durch Umsetzung von (II) mit '-Carbonyldiimidazol (CDI) bei erhöhter Temperatur (+60°C bis +150°C) in einem entsprechend höhersiedenden Lösungsmittel wie -Dimefhylformamid (DMF) erhältlich. Die Herstellung der Carbonsäurechloride geschieht auf übliche Weise durch Behandlung von (II) mit Thionyl- chlorid oder Oxalylchlorid in einem inerten Lösungsmittel wie Dichlormethan.
Die Abspaltung der Ester-Gruppe T im Verfahrensschritt (IV)— (I-A) wird nach üblichen Methoden durchgeführt, indem man den Ester in einem inerten Lösungsmittel mit einer Säure oder Base behandelt, wobei bei letzterer Variante das zunächst entstehende Salz der Carbonsäure durch nachfolgende Behandlung mit Säure in die freie Carbonsäure überführt wird. Im Falle der tert.- Butylester erfolgt die Esterspaltung vorzugsweise mit einer Säure. Methyl- und Ethylester werden bevorzugt mittels einer Base gespalten. Benzylester können alternativ auch durch Hydrierung (Hydrogenolyse) in Gegenwart eines geeigneten Katalysators, wie beispielsweise Palladium auf Aktivkohle, abgespalten werden.
Als inerte Lösungsmittel eignen sich für diese Reaktionen Wasser und die für eine Esterspaltung üblichen organischen Lösungsmittel. Hierzu zählen insbesondere Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder ieri.-Butanol, Ether wie Diethylether, Tetrahydrofuran, 1,4-Dioxan oder 1,2-Dimethoxyethan, oder andere Lösungsmittel wie Dichlormethan, Acetonitril, -Dimefhylformamid oder Dimethylsulfoxid. Ebenso ist es möglich, Gemische dieser Lösungsmittel einzusetzen. Im Falle einer basischen Ester-Hydrolyse werden bevorzugt Gemische von Wasser mit Tetrahydrofuran, 1,4-Dioxan, Methanol und/oder Ethanol verwendet. Im Falle der Umsetzung mit Trifluoressigsäure wird bevorzugt Dichlormethan und im Falle der Umsetzung mit Chlorwasserstoff bevorzugt 1,4-Dioxan, jeweils unter wasserfreien Bedingungen, verwendet.
Als Basen sind die für eine Hydrolyse-Reaktion üblichen anorganischen Basen geeignet. Hierzu gehören insbesondere Alkali- oder Erdalkalihydroxide wie beispielsweise Lithium-, Natrium-, Kalium- oder Bariumhydroxid, oder Alkali- oder Erdalkalicarbonate wie Natrium-, Kalium- oder Calciumcarbonat. Bevorzugt wird Lithium- oder Natriumhydroxid eingesetzt.
Als Säuren eignen sich für die Esterspaltung im Allgemeinen Schwefelsäure, Chlorwasserstoff/ Salzsäure, Bromwasserstoff/Bromwasserstoffsäure, Phosphorsäure, Essigsäure, Trifluoressigsäure, Toluolsulfonsäure, Methansulfonsäure oder Trifluormethansulfonsäure oder deren Gemische gege- benenfalls unter Zusatz von Wasser. Bevorzugt werden Chlorwasserstoff oder Trifluoressigsäure verwendet.
Die Esterspaltung wird in der Regel in einem Temperaturbereich von -20°C bis +100°C, bevorzugt bei 0°C bis +80°C durchgeführt.
Die Herstellung des Säurechlorids (V) erfolgt auf übliche Weise durch Behandlung der Carbon- säure (I-A) mit Oxalylchlorid oder Thionylchlorid in einem inerten Lösungsmittel wie Dichlormethan, Trichlormethan oder 1,2-Dichlorethan, gegebenenfalls unter Verwendung einer kleinen Menge an /V,/V-Dimethylformamid als Katalysator. Die Reaktion wird im Allgemeinen bei einer Temperatur von 0°C bis +30°C durchgeführt.
Die nachfolgende Amid-Bildung im Verfahrensschritt (V) + (VI)— > (I-B) erfolgt gewöhnlich in Gegenwart eines größeren Überschusses an der Amin-Komponente (VI). Alternativ kann auch eine übliche tertiäre Aminbase als Hilfsbase eingesetzt werden, wie beispielsweise Triethylamin, /V,/V-Diisopropylethylamin, N-Methylmorpholin (NMM), N-Mefhylpiperidin (NMP), Pyridin, 2,6-Dimethylpyridin oder 4-/V,/V-Dimethylaminopyridin (DMAP).
Inerte Lösungsmittel für diese Umsetzung sind beispielsweise Ether wie Diethylether, Diisopropyl- ether, Methyl- tert. -b ty lether, Tetrahydrofuran, 1,4-Dioxan, 1,2-Dimethoxyethan oder Bis(2-meth- oxyethyl)ether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Pentan, Hexan oder Cyclohexan, Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, 1,2-Dichlor- ethan, Trichlorethylen oder Chlorbenzol, polar-aprotische Lösungsmittel wie Aceton, Methylethyl- keton, Ethylacetat, Acetonitril, Butyronitril, Pyridin, Dimethylsulfoxid (DMSO), NN-Dimethyl- formamid (DMF), N, N'-Dimefhylpropylenharnstoff (DMPU) oder N-Mefhylpyrrolidinon (NMP), oder auch Wasser. Ebenso können Gemische solcher Lösungsmittel eingesetzt werden. Bevorzugt wird Wasser oder ein Gemisch von Wasser mit Tetrahydrofuran, 1,4-Dioxan, 1,2-Dimethoxyethan oder Aceton verwendet. Die Reaktion wird in der Regel bei einer Temperatur von 0°C bis +40°C durchgeführt.
Erfindungsgemäße Verbindungen der Formel (I), in welcher Z für eine Gruppe der Formel -NH-SO2-R9 oder -NH-SO2-NR10AR10B steht, können in Analogie zur oben beschriebenen Amid- Bildung (V) + (VI)— (I-B) durch basenvermittelte Umsetzung des Säurechlorids (V) mit einer Verbindung der Formel (VI-A) bzw. (VI-B)
(VI-A) (VI-B), in welchen R9, R10A und R10B die oben angegebenen Bedeutungen haben, erhalten werden. Die Reaktion erfolgt vorzugsweise unter Verwendung von Natriumhydrid als Base in Tetrahydrofuran oder -Dimefhylformamid als inertem Lösungsmittel bei einer Tem- peratur von 0°C bis +50°C.
Weitere erfindungsgemäße Verbindungen der Formel (I) können, falls zweckmäßig, auch durch Umwandlungen von funktionellen Gruppen einzelner Reste und Substituenten, insbesondere den unter RA, R1 und R5 aufgeführten, hergestellt werden, wobei von anderen, nach obigen Verfahren erhaltenen Verbindungen der Formel (I) oder deren Vorstufen ausgegangen wird. Diese Umwand-
lungen werden nach üblichen, dem Fachmann geläufigen Methoden durchgeführt und umfassen beispielsweise Reaktionen wie nukleophile oder elektrophile Substitutionsreaktionen, Übergangsmetall -vermittelte Kupplungsreaktionen, Herstellungs- und Additionsreaktionen von Metallorga- nylen (z.B. Grignard- Verbindungen oder Lithiumorganylen), Oxidations- und Reduktionsreak- tionen, Hydrierung, Halogenierung (z.B. Fluorierung, Bromierung), Dehalogenierung, Aminierung, Alkylierung und Acylierung, die Bildung von Carbonsäureestern, Carbonsäureamiden und Sulfonamiden, die Esterspaltung und -hydrolyse sowie die Einführung und Entfernung temporärer Schutzgruppen.
Die Verbindungen der Formel (II) können in Abhängigkeit vom jeweiligen Substitutionsmuster da- durch hergestellt werden, dass man in Analogie zu literaturbekannten Verfahren entweder
[A] ein Isatin-Derivat der Formel (VII)
in welcher R
1, R
2, R
3 und R
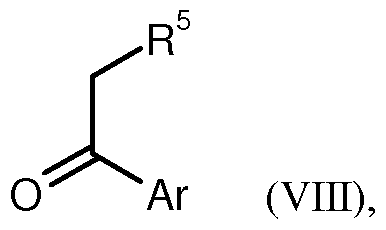
4 die oben angegebenen Bedeutungen haben, in einer Säure- oder Base- vermittelten Kondensationsreaktion mit einer Ketomethylen- Verbindung der Formel (VIII)
in welcher R
5 und Ar die oben angegebenen Bedeutungen haben, zur Verbindung der Formel (Π)
in welcher R , R , R , R , R und Ar die oben angegebenen Bedeutungen haben, umsetzt oder einen oriÄo-Aminophenylessigsäureester der Formel (ΓΧ)
in welcher R
1 die oben angegebene Bedeutung hat, in einer Säure-induzierten Kondensationsreaktion mit einer Diketo-Verbindung der Formel
(X)
in welcher R
5 und Ar die oben angegebenen Bedeutungen haben, zu einer Verbindung der Formel (II-A)
in welcher R
1, R
5 und Ar die oben angegebenen Bedeutungen haben, umsetzt. Die Kondensation des Isatin-Derivats (VII) mit der Ketomethylen- Verbindung (VIII) zur Chinolin- 4-carbonsäure (II) in Variante [A] kann durch Erhitzen der Reaktanden in Gegenwart einer wäss- rigen Säure, wie Schwefelsäure oder konzentrierte Salzsäure, oder in Gegenwart einer wässrigen Base, wie Natron- oder Kalilauge, erzielt werden. Bei Verwendung einer Säure wird für die Umsetzung vorzugsweise Essigsäure als Lösungsmittel eingesetzt; bei basischer Reaktionsführung
wird bevorzugt ein alkoholisches Lösungsmittel wie Methanol oder Ethanol verwendet. Die Kondensation wird im Allgemeinen in einem Temperaturbereich von +70°C bis +120°C durchgeführt [vgl. z.B. K. Lackey und D. D. Sternbach, Synthesis, 993-997 (1993); A. N. Boa et al., Bioorg. Med. Chem. 13 (6), 1945-1967 (2005)]. Die Kondensationsreaktion nach Variante [B] zur Chinolin-4-carbonsäure (Π-Α) erfolgt auf analoge Weise durch Erhitzen des ori^o-Aminophenylessigsäureesters (ΓΧ) und des Diketons (X) mit wässriger Säure, insbesondere konzentrierter Salzsäure. Als inertes Lösungsmittel für die Umsetzung wird auch hier bevorzugt Essigsäure eingesetzt.
Der oriÄo-Aminophenylessigsäureester (IX) seinerseits kann in Anlehnung an ein in der Literatur beschriebenes Verfahren durch basenvermittelte Reaktion des α-Chloressigsäureesters (XI)
mit dem Nitrophenyl-Derivat (ΧΠ)
in welcher R
1 die oben angegebene Bedeutung hat, zum oriÄo-Nitrophenylessigsäureester (ΧΠΙ)
in welcher R
1 die oben angegebene Bedeutung hat, und nachfolgende Reduktion der Nitro-Gruppe, beispielsweise durch katalytische Hydrierung, erhalten werden [vgl. P. Beier et al., /. Org. Chem. 76, 4781-4786 (2011)] . Die Verbindungen der Formel (ΠΙ) sind kommerziell erhältlich oder ihre Herstellung ist in der Literatur beschrieben, oder sie können, ausgehend von anderen kommerziell erhältlichen Verbin-
dungen, nach dem Fachmann geläufigen, literaturbekannten Methoden hergestellt werden. Beispiele hierzu sind in den nachfolgenden Reaktionsschemata wiedergegeben. Detaillierte Vorschriften und weitere Literaturangaben befinden sich auch im Experimentellen Teil im Abschnitt zur Herstellung der Ausgangsverbindungen und Intermediate. Die Verbindungen der Formeln (VI), (VI-A), (VI-B), (VII), (VIII), (X), (XI) und (ΧΠ) sind gleichfalls kommerziell erhältlich oder als solche in der Literatur beschrieben, oder sie können, ausgehend von anderen kommerziell erhältlichen Verbindungen, auf einfache Weise in Analogie zu literaturbekannten Verfahren hergestellt werden.
Die Herstellung der erfindungsgemäßen Verbindungen kann durch die folgenden Reaktions- Schemata beispielhaft veranschaulicht werden:
Schema 1
Die erfindungsgemäßen Verbindungen besitzen wertvolle pharmakologische Eigenschaften und können zur Vorbeugung und Behandlung von Erkrankungen bei Menschen und Tieren verwendet werden.
Die erfindungsgemäßen Verbindungen stellen potente, chemisch und metabolisch stabile Antagonisten des FP-Rezeptors dar und eignen sich daher zur Behandlung und/oder Prävention von Erkrankungen und pathologischen Prozessen, insbesondere solcher, bei denen im Zuge eines Entzündungsgeschehens und/oder eines Gewebe- oder Gefäßumbaus der FP-Rezeptor involviert ist. Dazu zählen im Sinne der vorliegenden Erfindung insbesondere Erkrankungen wie die Gruppe der interstitiellen idiopathischen Pneumonien, zu denen die idiopathische pulmonale Fibrose (IPF), die akute interstitielle Pneumonie, nicht-spezifische interstitielle Pneumonien, lymphoide interstitielle Pneumonien, respiratorische Bronchiolitis mit interstitieller Lungenerkrankung, kryptogene organisierende Pneumonien, desquamative interstitielle Pneumonien und nicht-klassifizierbare idiopa- thische interstitielle Pneumonien gehören, ferner granulomatöse interstitielle Lungenerkrankungen, interstitielle Lungenerkrankungen bekannter Ursache und andere interstitielle Lungenerkrankungen unbekannter Ursache, die pulmonale arterielle Hypertonie (PAH) und andere Formen der pulmonalen Hypertonie (PH), das Bronchiolitis obliterans-Syndrom (BOS), die chronisch-obstruktive Lungenerkrankung (COPD), Lungensarkoidose, das akute Atemwegssyndrom (ARDS), akute Lungenschädigung (ALI), alpha- 1-Antitrypsin-Defizienz (AATD), Lungenemphysem (z.B. durch Zigarettenrauch induziertes Lungenemphysem), zystische Fibrose (CF), entzündliche und fibro- tische Erkrankungen der Niere, chronische Darmentzündungen (IBD, Morbus Crohn, Colitis ulcerosa), Peritonitis, Peritonealfibrose, rheumatoide Erkrankungen, multiple Sklerose, entzündliche und fibrotische Hauterkrankungen, Sichelzellanämie sowie entzündliche und fibrotische Augen- erkrankungen.
Die erfindungsgemäßen Verbindungen können weiterhin verwendet werden zur Behandlung und/ oder Prävention von asthmatischen Erkrankungen unterschiedlicher Schweregrade mit intermittierendem oder persistierendem Verlauf (refraktäres Asthma, bronchiales Asthma, allergisches Asthma, intrinsisches Asthma, extrinsisches Asthma, durch Medikamente oder durch Staub indu- ziertes Asthma), von verschiedenen Formen der Bronchitis (chronische Bronchitis, infektiöse Bronchitis, eosinophile Bronchitis), von Bronchiektasien, Pneumonie, Farmerlunge und verwandten Krankheiten, Husten- und Erkältungskrankheiten (chronischer entzündlicher Husten, iatrogener Husten), Nasenschleimhautentzündungen (einschließlich medikamentöse Rhinitis, vasomotorische Rhinitis und jahreszeitabhängige, allergische Rhinitis, z.B. Heuschnupfen) und von Polypen. Die erfindungsgemäßen Verbindungen können darüber hinaus zur Behandlung und/oder Prävention von kardiovaskulären Erkrankungen eingesetzt werden, wie beispielsweise Bluthochdruck (Hypertonie), Herzinsuffizienz, koronare Herzerkrankung, stabile und instabile Angina pectoris, renale Hypertonie, periphere und kardiale Gefäßerkrankungen, Arrhythmien, Rhythmusstörungen der Vorhöfe und der Kammern sowie Überleitungsstörungen wie beispielsweise atrio-ventrikuläre Blockaden des Grades I-III, supraventrikuläre Tachyarrhythmie, Vorhofflimmern, Vorhofflattern, Kammerflimmern, Kammerflattern, ventrikuläre Tachyarrhythmie, Torsade de pointes-Tachykar- die, Extrasystolen des Vorhofs und des Ventrikels, AV-junktionale Extrasystolen, Sick-Sinus- Syndrom, Synkopen, AV-Knoten-Reentry-Tachykardie, Wolff-Parkinson-White-Syndrom, akutes Koronarsyndrom (ACS), autoimmune Herzerkrankungen (Perikarditis, Endokarditis, Valvolitis, Aortitis, Kardiomyopathien), Boxerkardiomyopathie, Aneurysmen, Schock wie kardiogener Schock, septischer Schock und anaphylaktischer Schock, ferner zur Behandlung und/oder Prävention von thromboembolischen Erkrankungen und Ischämien, wie myokardiale Ischämie, Myokardinfarkt, Hirnschlag, Herzhypertrophie, transistorische und ischämische Attacken, Präeklampsie, entzündliche kardiovaskuläre Erkrankungen, Spasmen der Koronararterien und peripherer Arterien, Ödembildung wie beispielsweise pulmonales Ödem, Hirnödem, renales Ödem oder Herzinsuffizienz-bedingtes Ödem, periphere Durchblutungsstörungen, Reperfusionsschäden, arterielle und venöse Thrombosen, Mikroalbuminurie, Herzmuskelschwäche, endotheliale Dysfunktion, mikro- und makrovaskuläre Schädigungen (Vaskulitis), sowie zur Verhinderung von Restenosen beispielsweise nach Thrombolyse-Therapien, percutan-transluminalen Angioplastien (PTA), per- cutan-transluminalen Koronarangioplastien (PTCA), Herztransplantationen und Bypass-Operati- onen.
Im Sinne der vorliegenden Erfindung umfasst der Begriff Herzinsuffizienz sowohl akute als auch chronische Erscheinungsformen der Herzinsuffizienz wie auch spezifische oder verwandte Krankheitsformen hiervon, wie akute dekompensierte Herzinsuffizienz, Rechtsherzinsuffizienz, Links- herzinsuffizienz, Globalinsuffizienz, ischämische Kardiomyopathie, dilatative Kardiomyopathie,
hypertrophe Kardiomyopathie, idiopathische Kardiomyopathie, diabetische Kardiomyopathie, angeborene Herzfehler, Herzklappenfehler, Herzinsuffizienz bei Herzklappenfehlern, Mitralklappen- stenose, Mitralklappeninsuffizienz, Aortenklappenstenose, Aortenklappeninsuffizienz, Trikuspi- dalstenose, Trikuspidalinsuffizienz, Pulmonalklappenstenose, Pulmonalklappeninsuffizienz, kom- binierte Herzklappenfehler, Herzmuskelentzündung (Myokarditis), chronische Myokarditis, akute Myokarditis, virale Myokarditis, diabetische Herzinsuffizienz, alkoholtoxische Kardiomyopathie, kardiale Speichererkrankungen sowie diastolische und systolische Herzinsuffizienz.
Die erfindungsgemäßen Verbindungen eignen sich außerdem zur Behandlung und/oder Prävention von Nierenerkrankungen, insbesondere von Niereninsuffizienz und Nierenversagen. Im Sinne der vorliegenden Erfindung umfassen die Begriffe Niereninsuffizienz und Nierenversagen sowohl akute als auch chronische Erscheinungsformen hiervon wie auch diesen zugrundeliegende oder verwandte Nierenerkrankungen, wie renale Hypoperfusion, intradialytische Hypotonie, obstruktive Uropathie, Glomerulopathien, Glomerulonephritis, akute Glomerulonephritis, Glomerulosklerose, tubulointerstitielle Erkrankungen, nephropathische Erkrankungen wie primäre und angeborene Nierenerkrankung, Nierenentzündung, immunologische Nierenerkrankungen wie Nierentransplantat-Abstoßung und Immunkomplex-induzierte Nierenerkrankungen, durch toxische Substanzen induzierte Nephropathie, Kontrastmittel-induzierte Nephropathie, diabetische und nicht-diabetische Nephropathie, Pyelonephritis, Nierenzysten, Nephrosklerose, hypertensive Nephrosklerose und nephrotisches Syndrom, welche diagnostisch beispielsweise durch abnorm verminderte Krea- tinin- und/oder Wasser-Ausscheidung, abnorm erhöhte Blutkonzentrationen von Harnstoff, Stickstoff, Kalium und/oder Kreatinin, veränderte Aktivität von Nierenenzymen wie z.B. Glutamylsyn- thetase, veränderte Urinosmolarität oder Urinmenge, erhöhte Mikroalbuminurie, Makroalbumin- urie, Läsionen an Glomerula und Arteriolen, tubuläre Dilatation, Hyperphosphatämie und/oder die Notwendigkeit zur Dialyse charakterisiert werden können. Die vorliegende Erfindung umfasst auch die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Folgeerscheinungen einer Niereninsuffizienz, wie beispielsweise Hypertonie, Lungenödem, Herzinsuffizienz, Urämie, Anämie, Elektrolytstörungen (z.B. Hyperkalämie, Hyponaträmie) und Störungen im Knochen- und Kohlenhydrat-Metabolismus.
Darüber hinaus sind die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen des Urogenitalsystems geeignet, wie beispielsweise benignes Prostata-Syndrom (BPS), benigne Prostatahyperplasie (BPH), benigne Prostatavergrößerung (BPE), Blasenentleerungsstörungen (BOO), untere Harnwegssyndrome (LUTS), neurogene überaktive Blase (OAB), Inkontinenz wie beispielsweise Misch-, Drang-, Stress- oder Überlauf-Inkontinenz (MUI, UUI, SUI, OUI), Beckenschmerzen sowie erektile Dysfunktion und weibliche sexuelle Dysfunktion.
Die erfindungsgemäßen Verbindungen können auch zur Behandlung von Erkrankungen des weiblichen Reproduktionssystems, wie Uterusmyome, Endometriose, Dysmenorrhöe und vorzeitige Geburtswehen, verwendet werden. Weiterhin eignen sie sich zur Prophylaxe oder Behandlung von Hirsutismus und Hypertrichose. Zudem besitzen die erfindungsgemäßen Verbindungen anti-inflammatorische Wirkung und können daher als entzündungshemmende Mittel zur Behandlung und/oder Prävention von Sepsis (SIRS), multiplem Organversagen (MODS, MOF), entzündlichen Erkrankungen der Niere, chronischen Darmentzündungen (IBD, Morbus Crohn, Colitis ulcerosa), Pankreatitis, Peritonitis, Cystitis, Urethritis, Prostatitis, Epidimytitis, Oophoritis, Salpingitis, Vulvovaginitis, rheumatoiden Erkrankun- gen, Arthrose, entzündlichen Erkrankungen des Zentralnervensystems, multipler Sklerose, entzündlichen Hauterkrankungen und entzündlichen Augenerkrankungen eingesetzt werden.
Die erfindungsgemäßen Verbindungen sind ferner zur Behandlung und/oder Prävention von fibro- tischen Erkrankungen der inneren Organe, wie beispielsweise der Lunge, des Herzens, der Niere, des Knochenmarks und insbesondere der Leber, sowie von dermatologischen Fibrosen und fibro- tischen Erkrankungen des Auges geeignet. Im Sinne der vorliegenden Erfindung umfasst der Begriff fibrotische Erkrankungen insbesondere solche Erkrankungen wie Leberfibrose, Leberzirrhose, Lungenfibrose, Endomyokardfibrose, Nephropathie, Glomerulonephritis, interstitielle Nieren- fibrose, fibrotische Schäden in Folge von Diabetes, Knochenmarksfibrose, Peritonealfibrose und ähnliche fibrotische Erkrankungen, Sklerodermie, Morphaea, Keloide, hypertrophe Narbenbil- dung, Naevi, diabetische Retinopathie, proliferative Vitroretinopathie und Erkrankungen des Bindegewebes (z.B. Sarkoidose). Die erfindungsgemäßen Verbindungen können ebenso verwendet werden zur Förderung der Wundheilung, zur Bekämpfung postoperativer Narbenbildung, z.B. nach Glaukom-Operationen, und zu kosmetischen Zwecken bei alternder oder verhornender Haut.
Auch können die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Anämien verwendet werden, wie hämolytischen Anämien, insbesondere Hämoglobinopathien wie Sichelzell anämie und Thalassämien, megaloblastären Anämien, Eisenmangel-Anämien, Anämien durch akuten Blutverlust, Verdrängungsanämien und aplastischen Anämien.
Die erfindungsgemäßen Verbindungen sind zudem zur Behandlung von Krebserkrankungen geeignet, wie beispielsweise von Hautkrebs, Hirntumoren, Brustkrebs, Knochenmarktumoren, Leuk- ämien, Liposarcomen, Karzinomen des Magen-Darm- Traktes, der Leber, Bauchspeicheldrüse, Lunge, Niere, Harnleiter, Prostata und des Genitaltraktes sowie von bösartigen Tumoren des lymphoproliferativen Systems, wie z.B. Hodgkin's und Non-Hodgkin's Lymphom.
Darüber hinaus können die erfindungsgemäßen Verbindungen eingesetzt werden zur Behandlung und/oder Prävention von Arteriosklerose, Lipidstoffwechselstörungen und Dyslipidämien (Hypo- lipoproteinämie, Hypertriglyceridämie, Hyperlipidämie, kombinierte Hyperlipidämien, Hyper- cholesterolämie, Abetalipoproteinämie, Sitosterolämie), Xanthomatose, Tangier-Krankheit, Fett- sucht (Adipositas), Fettleibigkeit (Obesitas), metabolischen Erkrankungen (Metabolisches Syndrom, Hyperglykämie, Insulin-abhängiger Diabetes, nicht-Insulin-abhängiger Diabetes, Gestationsdiabetes, Hyperinsulinämie, Insulinresistenz, Glukose-Intoleranz und diabetische Spätfolgen wie Retinopathie, Nephropathie und Neuropathie), von Erkrankungen des Gastrointestinaltrakts und des Abdomen (Glossitis, Gingivitis, Periodontitis, Oesophagitis, eosinophile Gastroenteritis, Mastocytose, Morbus Crohn, Colitis, Proctitis, Pruritis ani, Diarrhöe, Zöliakie, Hepatitis, Leber - fibrose, Leberzirrhose, Pankreatitis und Cholecystitis), von Erkrankungen des Zentralen Nervensystems und von neurodegenerativen Störungen (Schlaganfall, Alzheimer'sche Krankheit, Parkin- son'sche Krankheit, Demenz, Epilepsie, Depressionen, Multiple Sklerose), Immunerkrankungen, Schilddrüsenerkrankungen (Hyperthyreose), Hauterkrankungen (Psoriasis, Akne, Ekzeme, Neuro- dermitis, vielfältige Formen der Dermatitis wie z.B. Dermatitis abacribus, Dermatitis actinica, Dermatitis allergica, Dermatitis ammoniacalis, Dermatitis artefacta, Dermatitis autogenica, Dermatitis atrophicans, Dermatitis calorica, Dermatitis combustionis, Dermatitis congelationis, Dermatitis cosmetica, Dermatitis escharotica, Dermatitis exfoliativa, Dermatitis gangraenose, Dermatitis haemostatica, Dermatitis herpetiformis, Dermatitis lichenoides, Dermatitis linearis, Dermatitis maligna, Dermatitis medimencatosa, Dermatitis palmaris et plantaris, Dermatitis parasitaria, Dermatitis photoallergica, Dermatitis phototoxica, Dermatitis pustularis, Dermatitis seborrhoica, Dermatitis solaris, Dermatitis toxica, Dermatitis ulcerosa, Dermatitis veneata, infektiöse Dermatitis, pyogene Dermatitis und Rosazea-artige Dermatitis, sowie Keratitis, Bullosis, Vasculitis, Cellulitis, Panniculitis, Lupus erythematodes, Erythema, Lymphome, Hautkrebs, Sweet-Syndrom, Weber- Christian-Syndrom, Narbenbildung, Warzenbildung, Frostbeulen), von entzündlichen Augenerkrankungen (Saccoidosis, Blepharitis, Conjunctivitis, Iritis, Uveitis, Chorioiditis, Ophthalmitis), viralen Erkrankungen (durch Influenza-, Adeno- und Coronaviren, wie z.B. HPV, HCMV, HIV, SARS), von Erkrankungen des Skelettknochens und der Gelenke sowie der Skelettmuskel (vielfältige Formen der Arthritis wie z.B. Arthritis alcaptonurica, Arthritis ankylosans, Arthritis dys- enterica, Arthritis exsudativa, Arthritis fungosa, Arthritis gonorrhoica, Arthritis mutilans, Arthritis psoriatica, Arthritis purulenta, Arthritis rheumatica, Arthritis serosa, Arthritis syphilitica, Arthritis tuberculosa, Arthritis urica, Arthritis villonodularis pigmentosa, atypische Arthritis, hämophile Arthritis, juvenile chronische Arthritis, rheumatoide Arthritis und metastatische Arthritis, des weiteren das Still-Syndrom, Felty-Syndrom, Sjörgen-Syndrom, Clutton-Syndrom, Poncet-Syndrom, Pott-Syndrom und Reiter- Syndrom, vielfältige Formen der Arthropathien wie z.B. Arthropathie deformans, Arthropathie neuropathica, Arthropathie ovaripriva, Arthropathie psoriatica und
Arthropathie tabica, systemische Sklerosen, vielfältige Formen der entzündlichen Myopathien wie z.B. Myopathie epidemica, Myopathie fibrosa, Myopathie myoglobinurica, Myopathie ossificans, Myopathie ossificans neurotica, Myopathie ossificans progressiva multiplex, Myopathie purulenta, Myopathie rheumatica, Myopathie trichinosa, Myopathie tropica und Myopathie typhosa, sowie das Günther-Syndrom und das Münchmeyer-Syndrom), von entzündlichen Arterienveränderungen (vielfältige Formen der Arteritis wie z.B. Endarteritis, Mesarteritis, Periarteritis, Panarteritis, Arteritis rheumatica, Arteritis deformans, Arteritis temporalis, Arteritis cranialis, Arteritis giganto- cellularis und Arteritis granulomatosa, sowie das Horton-Syndrom, Churg-Strauss-Syndrom und die Takayasu-Arteritis), des Muckle-Well-Syndroms, der Kikuchi-Krankheit, von Polychondritis, Sklerodermia sowie von weiteren Erkrankungen mit einer entzündlichen oder immunologischen Komponente, wie beispielsweise Katarakt, Kachexie, Osteoporose, Gicht, Inkontinenz, Lepra, Sezary-Syndrom und paraneoplastisches Syndrom, bei Abstossungsreaktionen nach Organtransplantationen und zur Wundheilung und Angiogenese insbesondere bei chronischen Wunden.
Aufgrund ihres biochemischen und pharmakologischen Eigenschaftsprofils eignen sich die erfin- dungsgemäßen Verbindungen insbesondere zur Behandlung und/oder Prävention von interstitiellen Lungenerkrankungen, vor allem der idiopathischen Lungenfibrose (IPF), sowie von pulmonaler Hypertonie (PH), Bronchiolitis obliterans-Syndrom (BOS), entzündlichen und fibrotischen Haut- und Augenerkrankungen und fibrotischen Erkrankungen der inneren Organe.
Die zuvor genannten, gut charakterisierten Krankheiten des Menschen können mit vergleichbarer Ätiologie auch in anderen Säugetieren vorkommen und dort ebenfalls mit den Verbindungen der vorliegenden Erfindung behandelt werden.
Im Sinne der vorliegenden Erfindung umfasst der Begriff "Behandlung" oder "behandeln" ein Hemmen, Verzögern, Aufhalten, Lindern, Abschwächen, Einschränken, Verringern, Unterdrücken, Zurückdrängen oder Heilen einer Krankheit, eines Leidens, einer Erkrankung, einer Verletzung oder einer gesundheitlichen Störung, der Entfaltung, des Verlaufs oder des Fortschreitens solcher Zustände und/oder der Symptome solcher Zustände. Der Begriff "Therapie" wird hierbei als synonym mit dem Begriff "Behandlung" verstanden.
Die Begriffe "Prävention", "Prophylaxe" oder "Vorbeugung" werden im Rahmen der vorliegenden Erfindung synonym verwendet und bezeichnen das Vermeiden oder Vermindern des Risikos, eine Krankheit, ein Leiden, eine Erkrankung, eine Verletzung oder eine gesundheitliche Störung, eine Entfaltung oder ein Fortschreiten solcher Zustände und/oder die Symptome solcher Zustände zu bekommen, zu erfahren, zu erleiden oder zu haben.
Die Behandlung oder die Prävention einer Krankheit, eines Leidens, einer Erkrankung, einer Verletzung oder einer gesundheitlichen Störung können teilweise oder vollständig erfolgen.
Weiterer Gegenstand der vorliegenden Erfindung ist somit die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen, zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen in einem Verfahren zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen. Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit einer oder mehreren anderen pharmakologisch wirksamen Substanzen eingesetzt werden, solange diese Kombination nicht zu unerwünschten und inakzeptablen Nebenwirkungen führt. Weiterer Gegenstand der vorliegenden Erfindung sind daher Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prävention der zuvor genannten Erkrankungen. Als hierfür geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt: · organische Nitrate und NO-Donatoren, wie beispielsweise Natriumnitroprussid, Nitroglycerin, Isosorbidmononitrat, Isosorbiddinitrat, Molsidomin oder SIN-1, sowie inhalatives NO;
• Verbindungen, die den Abbau von cyclischem Guanosinmonophosphat (cGMP) und/oder cyclischem Adenosinmonophosphat (cAMP) inhibieren, wie beispielsweise Inhibitoren der Phosphodiesterasen (PDE) 1, 2, 3, 4 und/oder 5, insbesondere PDE 5-Inhibitoren wie Sildenafil, Vardenafil, Tadalafil, Udenafil, Dasantafil, Avanafil, Mirodenafil oder Lodenafil;
• NO- und Häm-unabhängige Aktivatoren der löslichen Guanylatcyclase (sGC), wie insbesondere die in WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 und WO 02/070510 beschriebenen Verbindungen;
• NO-unabhängige, jedoch Häm-abhängige Stimulatoren der löslichen Guanylatcyclase (sGC), wie insbesondere Riociguat, Nelociguat und Vericiguat sowie die in WO 00/06568, WO 00/
06569, WO 02/42301, WO 03/095451, WO 2011/147809, WO 2012/004258, WO 2012/028647 und WO 2012/059549 beschriebenen Verbindungen;
• Prostacyclin-Analoga und IP-Rezeptor-Agonisten, wie beispielhaft und vorzugsweise Iloprost, Beraprost, Treprostinil, Epoprostenol oder Selexipag; · Endothelin-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan;
• Verbindungen, die die humane neutrophile Elastase (HNE) inhibieren, wie beispielhaft und vorzugsweise Sivelestat oder DX-890 (Reltran);
• die Signaltransduktionskaskade inhibierende Verbindungen, beispielhaft und vorzugsweise aus der Gruppe der Kinase-Inhibitoren, insbesondere aus der Gruppe der Tyrosinkinase- und/oder
Serin/Threoninkinase-Inhibitoren, wie beispielhaft und vorzugsweise Nintedanib, Dasatinib, Nilotinib, Bosutinib, Regorafenib, Sorafenib, Sunitinib, Cediranib, Axitinib, Telatinib, Imati- nib, Brivanib, Pazopanib, Vatalanib, Gefitinib, Erlotinib, Lapatinib, Canertinib, Lestaurtinib, Pelitinib, Semaxanib oder Tandutinib; · Verbindungen, die den Ab- und Umbau der Extrazellulärmatrix inhibieren, beispielhaft und vorzugsweise Inhibitoren der Matrix-Metalloproteasen (MMPs), insbesondere Inhibitoren von Stromelysin, Kollagenasen, Gelatinasen und Aggrecanasen (hierbei vor allem von MMP-1, MMP-3, MMP-8, MMP-9, MMP-10, MMP-11 und MMP-13) sowie der Metallo-Elastase (MMP-12); · Verbindungen, die die Bindung von Serotonin an dessen Rezeptor blockieren, beispielhaft und vorzugsweise Antagonisten des 5 -HT2B -Rezeptors wie PRX -08066;
• Antagonisten von Wachstumsfaktoren, Zytokinen und Chemokinen, beispielhaft und vorzugsweise Antagonisten von TGF-ß, CTGF, IL-1, IL-4, IL-5, IL-6, IL-8, IL-13 und Integrinen;
• die Rho-Kinase inhibierende Verbindungen, wie beispielhaft und vorzugsweise Fasudil, Y-27632, SLx-2119, BF-66851, BF-66852, BF-66853, KI-23095 oder BA-1049;
• Verbindungen, die die lösliche Epoxidhydrolase (sEH) inhibieren, wie beispielsweise N,N'-Oi- cyclohexylharnstoff, 12-(3-Adamantan-l-yl-ureido)-dodecansäure oder l-Adamantan-l-yl-3-{5- [2-(2-ethoxyethoxy)ethoxy]pentyl } -harnstoff ;
• den Energiestoffwechsel des Herzens beeinflussende Verbindungen, wie beispielhaft und vor- zugsweise Etomoxir, Dichloracetat, Ranolazin oder Trimetazidin;
• anti-obstruktiv wirkende Mittel, wie sie z.B. zur Therapie der chronisch-obstruktiven Lungenerkrankung (COPD) oder eines Asthma bronchiale eingesetzt werden, beispielhaft und vorzugsweise aus der Gruppe der inhalativ oder systemisch angewendeten ß-adrenergen Rezeptor- Agonisten (ß-Mimetika) und der inhalativ angewendeten anti-muscarinergen Substanzen; · entzündungshemmende, immunmodulierende, immunsuppressive und/oder zytotoxische Mittel, beispielhaft und vorzugsweise aus der Gruppe der systemisch oder inhalativ angewendeten Corticosteroide sowie Acetylcystein, Montelukast, Azathioprin, Cyclophosphamid, Hydroxy- carbamid, Azithromycin, Pirfenidon oder Etanercept;
• antifibrotisch wirkende Mittel, wie beispielhaft und vorzugsweise Adenosin-A2b-Rezeptor- Antagonisten, Sphingosin-l-phosphat-Rezeptor 3 (S1P3) -Antagonisten, Autotaxin-Inhibitoren,
Lysophosphatidsäure-Rezeptor 1 (LPA-1)- und Lysophosphatidsäure-Rezeptor 2 (LPA-2)-Ant- agonisten, Lysyloxidase (LOX) -Inhibitoren, Lysyloxidase-like-2-Inhibitoren, CTGF-Inhibito- ren, IL-4-Antagonisten, IL- 13- Antagonisten, avß6-Integrin-Antagonisten, TGF-ß -Antagonisten, Inhibitoren des Wnt-Signalwegs oder CCR2-Antagonisten; · antithrombotisch wirkende Mittel, beispielhaft und vorzugsweise aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien und der profibrinolytischen Substanzen;
• den Blutdruck senkende Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, Vasopeptidase-lnhibitoren, Endothelin-Antagonisten, Renin-Inhibitoren, α-Rezeptoren-Blocker, ß-Rezeptoren-Blocker, Mineralocorticoid-Rezeptor-Antagonisten sowie der Diuretika;
• den Fettstoffwechsel verändernde Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie beispielhaft und vorzugsweise HMG-CoA-Reduktase- oder Squalensynthese -Inhibitoren, der ACAT-Inhibitoren, CETP- Inhibitoren, MTP-Inhibitoren, PPAR-a-, PPAR-γ- und/oder PPAR-8-Agonisten, Cholesterin- Absorptionshemmer, Lipase-Inhibitoren, polymeren Gallensäureadsorber, Gallensäure -Reab- sorptionshemmer und Lipoprotein(a)-Antagonisten; und/oder
• Chemotherapeutika, wie sie z.B. zur Therapie von Neubildungen (Neoplasien) der Lunge oder anderer Organe eingesetzt werden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ß-adrenergen Rezeptor-Agonisten, wie beispielhaft und vor- zugsweise Albuterol, Isoproterenol, Metaproterenol, Terbutalin, Fenoterol, Formoterol, Repro- terol, Salbutamol oder Salmeterol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einer anti-muscarinergen Substanz, wie beispielhaft und vorzugsweise Ipratropiumbromid, Tiotropiumbromid oder Oxitropiumbromid, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Corticosteroid, wie beispielhaft und vorzugsweise Prednison, Prednisolon, Methylprednisolon, Triamcinolon, Dexamethason, Beclomethason, Betamethason, Flunisolid, Budesonid oder Fluticason, verabreicht.
Unter antithrombotisch wirkenden Mittel werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien und der profibrinolytischen Substanzen verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximela- gatran, Melagatran, Dabigatran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem GPITb/nia-Antagonisten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Riva- roxaban, Apixaban, Fidexaban, Razaxaban, Fondaparinux, Idraparinux, DU-176b, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR- 126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten, Renin-Inhibitoren, α-Rezeptoren-Blocker, ß-Rezeptoren-Blocker, Mineralocorticoid-Rezeptor- Antagonisten sowie der Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Calcium- Antagonisten, wie beispielhaft und vorzugsweise Nifedipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem cti-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ß -Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Metipra- nolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol, Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucindolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin AII-Antagonisten, wie beispielhaft und vorzugs- weise Losartan, Candesartan, Valsartan, Telmisartan oder Embursatan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Lisinopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Endothelin-Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Renin-Inhibitor, wie beispielhaft und vorzugsweise Aliskiren, SPP-600 oder SPP-800, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Mineralocorticoid-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Spironolacton, Eplerenon oder Finerenon, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise Furosemid, Bumetanid, Torsemid, Bendroflumethiazid, Chlorthiazid, Hydrochlorthiazid, Hydroflumethiazid, Methyclothiazid, Polythiazid, Trichlormethiazid, Chlorthalidon, Indapamid, Metolazon, Quineth- azon, Acetazolamid, Dichlorphenamid, Methazolamid, Glycerin, Isosorbid, Mannitol, Amilorid oder Triamteren, verabreicht.
Unter den Fettstoffwechsel verändernden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der CETP-Inhibitoren, Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, MTP-Inhibi- toren, PPAR-a-, PPAR-γ- und/oder PPAR-8-Agonisten, Cholesterin-Absorptionshemmer, polyme- ren Gallensäureadsorber, Gallensäure -Reabsorptionshemmer, Lipase-Inhibitoren sowie der Lipoproteine-Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP-Inhibitor, wie beispielhaft und vorzugsweise Torcetrapib (CP-529 414), JJT-705 oder CETP-vaccine (Avant), verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidrezeptor-Agonisten, wie beispielhaft und vorzugsweise D-Thyroxin, 3,5,3'-Triiodothyronin (T3), CGS 23425 oder Axitirome (CGS 26214), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem HMG-CoA-Reduktase-Inhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK-475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS-201038, R-103757 oder JTT-130, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-y-Agonisten, wie beispielhaft und vorzugsweise Pioglita- zone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-8-Agonisten, wie beispielhaft und vorzugsweise GW 501516 oder BAY 68-5042, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterin- Absorptionshemmer, wie beispielhaft und vorzugsweise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimid, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Gallensäure -Reabsorptionshemmer, wie beispielhaft und vorzugsweise ASBT (= IBAT)-Inhibitoren wie z.B. AZD-7806, S-8921, AK-105, BARI- 1741, SC-435 oder SC-635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Lipoprotein(a)-Antagonisten, wie beispielhaft und vorzugsweise Gemcabene calcium (CI-1027) oder Nicotinsäure, verabreicht.
Besonders bevorzugt sind Kombinationen der erfindungsgemäßen Verbindungen mit einem oder mehreren weiteren Wirkstoffen ausgewählt aus der Gruppe bestehend aus PDE 5 -Inhibitoren, sGC- Aktivatoren, sGC-Stimulatoren, Prostacyclin-Analoga, IP-Rezeptor-Agonisten, Endothelin-Antagonisten, die Signaltransduktionskaskade inhibierenden Verbindungen und Pirfenidon. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otisch oder als Implantat oder Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden. Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Frei- setzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophilisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. inhalativ, intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern. Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer, Dosieraerosole), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augenpräparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipo-
phile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale und die parenterale Applikation, insbesondere die orale, die intravenöse und die intrapulmonale (inhalative) Applikation. Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige Poly- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg kg, vorzugsweise etwa 0.01 bis 0.5 mg kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht. Bei intrapulmonaler Applikation beträgt die Menge im Allgemeinen etwa 0.1 bis 50 mg je Inhalation. Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
A. Beispiele
Abkürzungen und Akronyme: abs. absolut
Ac Acetyl
AIBN 2,2'-Azobis(2-methylpropionitril), Azoisobuttersäurenitril aq. wässrig, wässrige Lösung
br. breit (bei NMR-Signal)
Bsp. Beispiel
Bu Butyl
c Konzentration
ca. circa, ungefähr
cat. katalytisch
CDI N,N'-Carbonyldiimidazol
CI chemische Ionisation (bei MS)
d Dublett (bei NMR)
d Tag(e)
DAST -Diethylaminoschwefeltrifluorid
DC Dünnschichtchromatographie
DCI direkte chemische Ionisation (bei MS)
dd Dublett von Dublett (bei NMR)
DMF N, N-Dimefhylformamid
DMSO Dimethylsulfoxid
dt Dublett von Triplett (bei NMR)
AT Erwärmung, Temperaturerhöhung (in Reaktionsschemata) d. Th. der Theorie (bei chemischer Ausbeute)
EI Elektronenstoß-Ionisation (bei MS)
eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl
h Stunde(n)
HATU 0-(7-Azabenzotriazol-l-yl)-/V,/V,/V',/V'-tetramethyluronium- hexafluorophosphat
HOAc Essigsäure
HPLC Hochdruck-, Hochleistungsflüssigchromatographie iPr Isopropyl
konz. konzentriert (bei Lösung)
LC Flüssigchromatographie
LC/MS Flüssigchromatographie-gekoppelte Massenspektrometrie
Lit. Literatur(stelle)
m Multiplett (bei NMR)
MCPBA meta-Chlorperbenzoesäure, 3-Chlorperbenzoesäure
Me Methyl
min Minute(n)
MS Massenspektrometrie
NBS /V-Bromsuccinimid
NMR Kernresonanzspektrometrie
Pd/C Palladium auf Aktivkohle
Pr Propyl
q (oder quart) Quartett (bei NMR)
qd Quartett von Dublett (bei NMR)
quant. quantitativ (bei chemischer Ausbeute)
quint Quintett (bei NMR)
Rf Retentionsindex (bei DC)
RP reverse phase (Umkehrphase, bei HPLC)
RT Raumtemperatur
Rt Retentionszeit (bei HPLC, LC/MS)
s Singulett (bei NMR)
sept Septett (bei NMR)
t Triplett (bei NMR)
tBu teri.-Butyl
td Triplett von Dublett (bei NMR)
TFA Trifluoressigsäure
THF Tetrahydrofuran
UV Ultraviolett-Spektrometrie
v/v Volumen zu Volumen- Verhältnis (einer Lösung) zus. zusammen
HPLC- und LC/MS-Methoden:
Methode 1 (LC/MS):
Instrument: Waters Acquity SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1.8 μ, 50 x 1 mm; Eluent A: 1 1 Wasser + 0.25 ml 99%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%-ige Ameisensäure; Gradient: 0.0 min 90% A -> 1.2 min 5% A -> 2.0 min 5% A; Ofen: 50°C; Fluss: 0.40 ml/min; UV-Detektion: 208-400 nm.
Methode 2 (LC/MS):
Instrument MS: Waters Micromass QM; Instrument HPLC: Agilent 1100 Serie; Säule: Agilent ZORBAX Extend-C18, 3.0 x 50 mm, 3.5 μ; Eluent A: 1 1 Wasser + 0.01 mol Ammoniumcarbonat, Eluent B: 1 1 Acetonitril; Gradient: 0.0 min 98% A -> 0.2 min 98% A -> 3.0 min 5% A -> 4.5 min 5% A; Ofen: 40°C; Fluss: 1.75 ml/min; UV-Detektion: 210 nm.
Methode 3 (LC/MS):
Instrument MS: Agilent MS Quad 6150; Instrument HPLC: Agilent 1290; Säule: Waters Acquity UPLC HSS T3 1.8 μ, 50 x 2.1 mm; Eluent A: 1 1 Wasser + 0.25 ml 99%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%-ige Ameisensäure; Gradient: 0.0 min 90% A -> 0.3 min 90% A -> 1.7 min 5% A -> 3.0 min 5% A; Ofen: 50°C; Fluss: 1.20 ml/min; UV-Detektion: 205-305 nm.
Methode 4 (LC/MS):
Instrument MS: Agilent 6130; Instrument HPLC: Agilent 1200; UV DAD; Säule: Waters XBridge BEH XP 2.5 μιη, 2.1 x 50 mm; Eluent A: Ammoniumacetat (10 mM) + Wasser/Methanol/ Acetonitril (9.0:0.6:0.4), Eluent B: Ammoniumacetat (10 mM) + Wasser/Methanol/Acetonitril (1.0:5.4:3.6); Gradient A/B: 80/20 (0.0 min) -> 80/20 (1.5 min) -> 0/100 (2.5 min); Fluss: 0.6 ml/ min; Temperatur: 35°C; UV-Detektion: 215 und 238 nm.
Methode 5 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 125 x 30 mm; Eluent: Acetonitril/Wasser mit 0.1% TFA; Gradient: 0- 5.00 min 10:90, Probeninjektion bei 3.00 min, 5.00-23.00 min bis 95:5, 23.00-30.00 min 95:5, 30.00-30.50 min bis 10:90, 30.50-31.20 min 10:90.
Methode 6 (präparative HPLC):
Säule: Chromatorex C18, 125 x 40 mm; Eluent A: Wasser + 0.05% TFA, Eluent B: Acetonitril; Gradient: 0.0 min 20% B -> 4.0 min 20% B -> 30 min 95% B -> 35 min 95% B -> 36 min 20% B; Fluss: 50 ml/min.
Methode 7 (präparative HPLC):
Säule: Chromatorex C18, 250 x 30 mm; Eluent A: Wasser, Eluent B: Acetonitril; Gradient: 0.0 min 60% B -> 4.5 min 80% B -> 11.5 min 100% B -> 12 min 100% B -> 14.75 min 60% B; Fluss: 50 ml/min. Methode 8 (präparative HPLC):
Säule: Chromatorex C18, 250 x 30 mm; Eluent A : Wasser, Eluent B : Acetonitril; Gradient: 0.0 min 40% B -> 4.5 min 60% B -> 11.5 min 80% B - -> 12 min 100% B - -> 14.75 min 40% B; Fluss: 50 ml/min.
Methode 9 (präparative HPLC):
Säule: Chromatorex C18, 250 x 30 mm; Eluent A: Wasser + 0.1% TFA, Eluent B : Acetonitril; Gradient: 0.0 min 40% B -> 4.5 min 60% B -> 11.5 min 80% B -> 12 min 100% B -> 14.75 min 40% B; Fluss: 50 ml/min.
Methode 10 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 250 x 30 mm; Fluss: 50 ml/min; Laufzeit: 18 min; Detektion: 210 nm; Eluent: Wasser + 0.1% Ameisensäure/Methanol; Gradient: 20% Methanol (4.25 min)— > 40% Methanol (4.5 min) -> 60% Methanol (11.5 min) -> 100% Methanol (12 min) -> 100% Methanol (14.5 min) -> 20% Methanol (14.75 min) -> 20% Methanol (18 min).
Methode 11 (präparative HPLC):
Säule: GromSil C18, 250 x 30 mm, 10 μιη; Fluss: 50 ml/min; Eluent A: Wasser, Eluent B: Aceto- nitril; Gradient: 0 min 30% B -> 4.25 min 30% B -> 4.5 min 50% B -> 11.5 min 70% B -> 12 min 100% B -> 14.5 min 100% B -> 14.75 min 30% B -> 18 min 30% B; Detektion: 210 nm.
Methode 12 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 125 x 30 mm; Eluent: AcetonitrilAVasser mit 0.1% TFA; Gradient: 0- 5.00 min 10:90, Probeninjektion bei 3.00 min, 5.00-23.00 min bis 95:5, 23.00-30.00 min 95:5, 30.00-30.50 min bis 10:90, 30.50-31.20 min 10:90.
Methode 13 (präparative HPLC):
Säule: Chromatorex C18, 250 x 30 mm; Eluent A: Wasser, Eluent B: Acetonitril; Gradient: 0.0 min 30% B -> 4.5 min 50% B -> 11.5 min 70% B -> 12 min 100% B -> 14.75 min 30% B; Fluss: 50 ml/min.
Methode 14 (präparative HPLC):
Säule: Kinetex 5 μιη C18 100 A, 150 x 21.2 mm; Eluent A: Wasser + 0.2% Ameisensäure, Eluent B: Acetonitril; Gradient: 70% A, 30% B, isokratisch; Fluss: 25 ml/min.
Methode 15 (präparative HPLC):
Säule: Chromatorex C18, 125 x 30 mm; Eluent A: Wasser + 0.05% TFA, Eluent B: Acetonitril; Gradient: 0.0 min 20% B -> 3.2 min 20% B -> 18 min 95% B -> 23 min 95% B -> 24 min 20% B; Fluss: 50 ml/min.
Methode 16 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 125 x 30 mm; Eluent: AcetonitrilAVasser mit 0.1% TFA; Gradient: 0- 6.00 min 5:95, Probeninjektion bei 3.00 min, 6.00-27.00 min bis 35:65, 27.00-30.00 min 95:5, 30.00-33.00 min bis 5:95.
Methode 17 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 250 x 30 mm; Fluss: 50 ml/min; Laufzeit: 18 min; Detektion: 210 nm; Eluent: Wasser + 0.1% Ameisensäure/Methanol; Gradient: 20% Methanol (4.25 min)— > 40% Methanol (4.5 min) -> 60% Methanol (11.5 min) -> 100% Methanol (12 min) -> 100% Methanol (14.5 min) -> 20% Methanol (14.75 min) -> 20% Methanol (18 min).
Methode 18 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 125 x 30 mm; Eluent: AcetonitrilAVasser mit 0.1% TFA; Fluss: 75 ml/min; Gradient: 0-5.50 min 10:90, Probeninjektion bei 3.00 min, 5.50-17.65 min bis 95:5, 17.65- 19.48 min 95:5, 19.48-19.66 min bis 10:90, 19.66-20.72 min 10:90.
Methode 19 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 125 x 30 mm; Eluent: AcetonitrilAVasser mit 0.1% TFA; Gradient: 0- 6.00 min 35:65, Probeninjektion bei 3.00 min, 6.00-27.00 min bis 80:20, 27.00-30.00 min 95:5, 30.00-33.00 min bis 35:65. Methode 20 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 125 x 30 mm; Eluent: AcetonitrilAVasser mit 0.1% TFA; Gradient: 0- 6.00 min 35:65, Probeninjektion bei 3.00 min, 6.00-27.00 min bis 80:20, 27.00-51.00 min 95:5, 51.00-53.00 min bis 35:65.
Methode 21 (LC/MS):
Instrument: Agilent MS Quad 6150 mit HPLC Agilent 1290; Säule: Waters Acquity UPLC HSS T3 1.8 μιη, 50 mm x 2.1 mm; Eluent A: 1 1 Wasser + 0.25 ml 99%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%-ige Ameisensäure; Gradient: 0.0 min 90% A— > 0.3 min 90% A— > 1.7 min 5% A -> 3.0 min 5% A; Fluss: 1.20 ml/min; Ofen: 50°C; UV-Detektion: 205-305 nm.
Methode 22 (LC/MS):
Instrument: Micromass Quattro Premier mit Waters UPLC Acquity; Säule: Thermo Hypersil GOLD 1.9 μιη, 50 mm x 1 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 97% A— > 0.5 min 97% A— > 3.2 min 5% A -> 4.0 min 5% A; Ofen: 50°C; Fluss: 0.30 ml/min; UV-Detektion: 210 nm.
Methode 23 (LC/MS):
Instrument: Waters Acquity SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1.8 μιη, 50 mm x 1 mm; Eluent A: 1 1 Wasser + 0.25 ml 99%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.25 ml 99%-ige Ameisensäure; Gradient: 0.0 min 95% A -> 6.0 min 5% A -> 7.5 min 5% A; Ofen: 50°C; Fluss: 0.35 ml/min; UV-Detektion: 210-400 nm.
Methode 24 (präparative HPLC):
Säule: Reprosil C18, 10 μιη, 250 x 40 mm; Eluent: Acetonitril/Wasser mit 0.1% TFA; Gradient: 0- 6.00 min 10:90, Probeninjektion bei 3.00 min, 6.00-27.00 min bis 95:5, 27.00-38.00 min 95:5, 38.00-39.00 min bis 10:90, 39.00-40.20 min 10:90. Weitere Angaben:
Die Prozentangaben in den folgenden Beispiel- und Testbeschreibungen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen. Reinheitsangaben beziehen sich in der Regel auf entsprechende Peak-Integrationen im LC/MS- Chromatogramm, können aber zusätzlich auch unter Zuhilfenahme des ^-NMR-Spektrums ermittelt worden sein. Wenn keine Reinheit angegeben ist, handelt es sich in der Regel um eine 100%-Reinheit laut automatischer Peak-Integration im LC/MS-Chromatogramm oder die Reinheit wurde nicht explizit ermittelt. Angaben zu Ausbeuten in % d. Th. sind in der Regel reinheitskorrigiert, sofern eine Reinheit <100% angegeben ist. Bei lösungsmittelhaltigen oder verunreinigten Chargen kann die Ausbeute
formal ">100%" betragen; in diesen Fällen ist die Ausbeute nicht lösungsmittel- bzw. reinheits- korrigiert.
Die nachfolgenden Beschreibungen der Kopplungsmuster von ^-NMR-Signalen wurden teilweise direkt den Vorschlägen des ACD SpecManagers (ACD/Labs Release 12.00, Product version 12.5) entnommen und nicht notwendigerweise streng hinterfragt. Teilweise wurden die Vorschläge des SpecManagers manuell angepasst. Manuell angepasste bzw. zugewiesene Beschreibungen orientieren sich in der Regel an dem optischen Erscheinungsbild der betreffenden Signale und entsprechen nicht notwendigerweise einer strengen, physikalisch korrekten Interpretation. In der Regel bezieht sich die Angabe zur chemischen Verschiebung auf das Zentrum des betreffenden Signals. Bei brei- ten Multipletts erfolgt die Angabe eines Intervalls. Durch Lösungsmittel oder Wasser verdeckte Signale wurden entweder tentativ zugeordnet oder sind nicht aufgeführt. Stark verbreiterte Signale - z.B. verursacht durch schnelle Rotation von Molekülteilen oder aufgrund von austauschenden Protonen - wurden ebenfalls tentativ zugeordnet (oft als breites Multiplett oder breites Singulett bezeichnet) oder sind nicht aufgeführt. Schmelzpunkte und Schmelzbereiche, soweit angegeben, sind nicht korrigiert.
Für alle Reaktanden oder Reagenzien, deren Herstellung im Folgenden nicht explizit beschrieben ist, gilt, dass sie von allgemein zugänglichen Quellen kommerziell bezogen wurden. Für alle übrigen Reaktanden oder Reagenzien, deren Herstellung im Folgenden ebenfalls nicht beschrieben ist und die nicht kommerziell erhältlich waren oder von Quellen bezogen wurden, die nicht allge- mein zugänglich sind, ist ein Verweis auf die veröffentlichte Literatur angegeben, in der ihre Herstellung beschrieben ist.
Wenn bei den im Folgenden beschriebenen Synthese-Intermediaten und Ausführungsbeispielen der Erfindung eine Verbindung in der Form eines Salzes der korrespondierenden Base bzw. Säure aufgeführt ist, so ist die exakte stöchiometrische Zusammensetzung eines solchen Salzes, wie es nach dem jeweiligen Herstell- und/oder Reinigungsverfahren erhalten wurde, in der Regel nicht bekannt. Sofern nicht genauer spezifiziert, sind daher Namens- und Strukturformel-Zusätze wie beispielsweise "Hydrochlorid", "Formiat", "Acetat", "Trifluoracetat", "Natrium-Salz" bzw. "x HCl", "x HCOOH", "x CH3COOH", "x CF3COOH", "x Na+" bei solchen Salzen nicht stöchiometrisch zu verstehen, sondern haben allein deskriptiven Charakter bezüglich der enthaltenen salzbildenden Komponenten.
Ausgangsverbindungen und Intermediate: Beispiel 1A
6-Brom-3-methyl-2-phenylchinolin-4-carbonsäure
100.0 g (398.16 mmol, Reinheit 90%) 5-Brom-l#-indol-2,3-dion und 59.4 g (442.41 mmol) 1-Phenylpropan-l-on wurden mit 1.2 Liter Essigsäure versetzt und 20 min bei 75°C gerührt. Danach wurden zur Reaktionsmischung 400 ml konz. Salzsäure gegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Die Reaktionslösung wurde dann unter Rühren in ein Gemisch aus 10 Liter 1 N Salzsäure, 9.2 Liter Wasser und 840 ml konz. Salzsäure gegeben. Die Mischung wurde mit 1 Liter Eiswasser versetzt, und der Niederschlag wurde mit Hilfe einer Fritte abfiltriert. Der Filterrückstand wurde zweimal mit 500 ml Wasser gewaschen, danach zweimal mit jeweils 150 ml eines 3: 1-Gemisches aus ieri.-Butylmethy lether und Aceton ausgerührt und erneut abfiltriert. Der Rückstand wurde noch dreimal mit jeweils 100 ml feri.-Butylmethylether ausgerührt, erneut abfiltriert und schließlich im Vakuum getrocknet. Es wurden 117.96 g (78% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.39 (br. s, 1H), 8.01 (d, 1H), 7.94-7.90 (m, 2H), 7.63-7.61 (m, 2H), 7.56-7.49 (m, 3H), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.76 min, m/z = 343 [M+H]+.
Beispiel 2A (6-Brom-3-methyl-2-phenylchinolin-4-yl)( 1 ii-imidazol- 1 -yl)methanon
100.0 g (292.23 mmol) der Verbindung aus Beispiel 1A wurden in 1.5 Liter DMF suspendiert und bei RT mit 95.0 g (584.45 mmol) /V,/V'-Carbonyldiimidazol versetzt. Das Reaktionsgemisch wurde zunächst 4 h bei 60°C und dann über Nacht bei RT gerührt. Unter Eisbadkühlung wurden 1.5 Liter Eiswasser langsam zugegeben und die Mischung dann für drei Tage in den Kühlschrank gestellt. Der ausgefallene Feststoff wurde über eine Fritte abfiltriert, dreimal mit 250 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 103.41 g (85% d. Th., Reinheit 94%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 8.6-8.0 (br. m, 1H), 8.09 (d, 1H), 8.0-7.5 (br. m, 1H), 7.97 (dd, 1H), 7.82 (s, 1H), 7.72-7.66 (m, 2H), 7.59-7.48 (m, 3H), 7.22 (br. s, 1H), 2.25 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.16 min, m/z = 392/394 [M+H]+.
Beispiel 3A
6-Fluor-3-methyl-2-phenylchinolin-4-carbonsäure
OO g (6.06 mmol) 5-Fluor-li7-indol-2,3-dion wurden in 16.5 ml Essigsäure vorgelegt und mit 813 mg (6.06 mmol) 1-Phenylpropan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 5.5 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde 3 h bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abgesaugt. Der Feststoff wurde mit Wasser ge- waschen und im Vakuum getrocknet. Es wurden 670 mg (Reinheit 35%, 14% d. Th.) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): R
t = 0.64 min, m/z = 282 [M+H]
+. Beispiel 4A
(6-Fluor-3-methyl-2-phenylchinolin-4-yl)(lii-imidazol-l-yl)methanon
Zu einer Lösung von 1.50 g (5.33 mmol) der Verbindung aus Beispiel 3A in 23 ml DMF wurden bei RT 951 mg (5.87 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 3 h bei 60°C gerührt. Anschließend wurden weitere 300 mg (1.07 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde weitere 15 h bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch unter Rühren in 100 ml Wasser eingetragen und mit etwas Eis versetzt. Der entstandene Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 1.57 g (89% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-8.0 (br. m, 1H), 8.22 (dd, 1H), 8.0-7.5 (br. m, 1H), 7.76 (td, 1H), 7.69 (dd, 2H), 7.59-7.47 (m, 3H), 7.41 (dd, 1H), 7.21 (br. s, 1H), 2.25 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.05 min, m/z = 332 [M+H]+. Beispiel 5A
6-Iod-3-methyl-2-phenylchinolin-4-carbonsäure
20.0 g (73.25 mmol) 5-Iod-li7-indol-2,3-dion wurden in 200 ml Essigsäure vorgelegt und mit 9.83 g (73.25 mmol) 1-Phenylpropan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C
gerührt. Danach wurden 66 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Anschließend wurde die Reaktionslösung vorsichtig unter Rühren in Wasser eingetragen. Der entstandene Niederschlag wurde abfiltriert und zweimal mit Wasser und zweimal mit wenig ieri.-Butylmethylether gewaschen. Nach Trocknen über Nacht im Vakuum wurden 11.10 g (32% d. Th., Reinheit 82%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.36 (br. s, 1H), 8.13 (d, 1H), 8.05 (dd, 1H), 7.84 (d, 1H), 7.66-7.57 (m, 2H), 7.57-7.41 (m, 3H), 2.39 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.78 min, m/z = 390 [M+H]+.
Beispiel 6A (l f-Imidazol-l-yl)(6-iod-3-methyl-2-phenylchinolin-4-yl)methanon
600 mg (1.23 mmol, Reinheit 80%) der Verbindung aus Beispiel 5A wurden in 5.5 ml DMF gelöst und bei RT mit 400 mg (2.47 mmol) /V,/V'-Carbonyldiimidazol versetzt. Das Reaktionsgemisch wurde über Nacht bei 60°C gerührt und anschließend nach Abkühlen auf RT mit Wasser und Essigsäureethylester versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (80 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 5: 1, Biotage). Nach Entfernen des Lösungsmittels im Vakuum wurde der Rückstand über Nacht im Vakuum getrocknet. Es wurden 568 mg (98% d. Th., Reinheit 94%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 9.50-6.00 (br. m, 2H), 8.10 (dd, 1H), 7.95 (s, 1H), 7.91 (d, 1H), 7.72-7.65 (m, 2H), 7.60-7.42 (m, 3H), 7.22 (br. s, 1H), 2.24 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.13 min, m/z = 440 [M+H]+.
Beispiel 7A
3,6-Dimethyl-2-phenylchinolin-4-carbonsäure
2.00 g (12.41 mmol) 5-Mefhyl-li7-indol-2,3-dion wurden in 33.7 ml Essigsäure vorgelegt und mit 1.66 g (12.41 mmol) 1-Phenylpropan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 11.3 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 400 ml 1 M Salzsäure gegeben und dieses Gemisch dann drei Tage bei RT stehen gelassen. Der ausgefallene Feststoff wurde abfiltriert, mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 700 mg (20% d. Th., Reinheit 100%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.53 min, m/z = 278 [M+H]+.
Beispiel 8A
(3,6-Dimethyl-2-phenylchinolin-4-yl)( 1 ii-imidazol- 1 -yl)methanon
Zu einer Lösung von 464 mg (1.68 mmol) der Verbindung aus Beispiel 7A in 7 ml DMF wurden bei RT 299 mg (1.84 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 4 h bei 60°C gerührt. Anschließend wurden weitere 30 mg (0.18 mmol) /V,/V'-Carbonyldiimidazol hinzugefügt und das Gemisch eine weitere Stunde bei 60°C gerührt. Nach Abkühlen auf RT wurde mit Wasser versetzt und dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wur- den einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, fil-
triert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen, erneut eingeengt und im Vakuum getrocknet. Es wurden 505 mg (92% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-8.0 (br. m, 1H), 8.04 (d, 1H), 8.0-7.5 (br. m, 1H), 7.98-7.59 (m, 3H), 7.57-7.45 (m, 3H), 7.34 (s, 1H), 7.26-7.09 (m, 1H), 2.46 (s, 3H), 2.23 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.05 min, m/z = 328 [M+H]+.
Beispiel 9A
6-Ethyl-3-methyl-2-phenylchinolin-4-carbonsäure
1.00 g (5.71 mmol) 5-Efhyl-li7-indol-2,3-dion wurden in 15.5 ml Essigsäure vorgelegt und mit 766 mg (5.71 mmol) 1-Phenylpropan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 5.2 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abfiltriert. Der Feststoff wurde mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 780 mg (46% d. Th., Reinheit 99%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.63 min, m/z = 292 [M+H]+.
Beispiel 10A
(6-Ethy 1-3 -methyl-2-phenylchinolin-4-yl) ( 1 ii-imidazol- 1 -yl)methanon
Zu einer Lösung von 640 mg (2.20 mmol) der Verbindung aus Beispiel 9A in 10 ml DMF wurden bei RT 392 mg (2.42 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 3.5 h bei 60°C gerührt. Anschließend wurde das Gemisch auf RT abgekühlt, mit Wasser versetzt und drei- mal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt und der Rückstand im Vakuum getrocknet. Der Rückstand wurde mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 7:3, Biotage). Es wurden 362 mg (46% d. Th., Reinheit 95%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 8.5-8.0 (br. m, 1H), 8.07 (d, 1H), 8.0-7.5 (br. m, 1H), 7.73 (dd, 1H), 7.70-7.65 (m, 2H), 7.57-7.47 (m, 3H), 7.32 (br. s, 1H), 7.22 (br. s, 1H), 2.76 (q, 2H), 2.24 (s, 3H), 1.19 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.18 min, m/z = 342 [M+H]+. Beispiel IIA 6-Isopropyl-3-methyl-2-phenylchinolin-4-carbonsäure
2.50 g (13.21 mmol) 5-rsopropyl-li7-indol-2,3-dion wurden mit 36 ml Essigsäure und 1.77 g (13.21 mmol) 1-Phenylpropan-l-on versetzt, und das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Dann wurden 12 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 500 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abfiltriert. Der Feststoff wurde mit Wasser gewa-
schen und im Vakuum getrocknet. Es wurden 2.04 g (31% d. Th., Reinheit 62%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.66 min, m/z = 306 [M+H]+. Beispiel 12A 3-Methyl-2-phenyl-6-(trifluormethyl)chinolin-4-carbonsäure
1.00 g (4.65 mmol) 5-(Trifluormethyl)-li7-indol-2,3-dion wurden mit 12.6 ml Essigsäure und 624 mg (4.65 mmol) 1-Phenylpropan-l-on versetzt, und das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Dann wurden 4.2 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abfiltriert. Der Feststoff wurde mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 1.17 g (75% d. Th., Reinheit 100%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.91 min, m/z = 332 [M+H]+. Beispiel 13A
3-Methyl-2-phenyl-6-(trifluormethoxy)chinolin-4-carbonsäure
Zu einem Gemisch von 2.5 g (10.82 mmol) 5-(Trifluormefhoxy)-li7-indol-2,3-dion in 25 ml Essigsäure wurden 1.45 g (10.82 mmol) 1-Phenylpropan-l-on gegeben. Nach 5 min Rühren bei 75°C wurde mit 8 ml konz. Salzsäure versetzt, und das Gemisch wurde 5 h bei 110°C weiter gerührt.
Nach Abkühlen auf RT und Stehenlassen über Nacht wurde das Gemisch unter Rühren in 500 ml 1 M Salzsäure eingetragen. Nach einigen Minuten wurde der entstandene Feststoff abfiltriert, zweimal mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 3.16 g (77% d. Th., Reinheit 92%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.41 (br. s, 1H), 8.21 (d, 1H), 7.80 (dd, 1H), 7.69 (d, 1H), 7.66-7.58 (m, 2H), 7.57-7.47 (m, 3H), 2.41 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.97 min, m/z = 348 [M+H]+.
Beispiel 14A
(l i-Imidazol-l-yl)[3-methyl-2-phenyl-6-(trifluormethoxy)chinolin-4-yl]methanon
Zu einer Lösung von 1.50 g (3.97 mmol, Reinheit 92%) der Verbindung aus Beispiel 13A in 15 ml DMF wurden bei RT 709 mg (4.37 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 3 h bei 60°C gerührt. Anschließend wurden weitere 709 mg (4.37 mmol) NN'-Carbonyl- diimidazol hinzugefügt, und das Gemisch wurde weitere 4 h bei 60°C gerührt. Nach Stehenlassen über Nacht bei RT wurde zunächst eine weitere Stunde bei 80°C gerührt. Danach wurden nochmals 709 mg (4.37 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde erneut für 4 h bei 100°C gerührt. Nach Stehenlassen über Nacht bei RT wurde das Gemisch unter Rühren in Eiswasser eingetragen und mittels 10%-iger wässriger Zitronensäure -Lösung auf pH 4 gestellt. Anschließend wurde zweimal mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie vorgereinigt (100 g Kieselgel, Lauf mittel Cyclohexan/Ethylacetat 7:3, Bio tage). Das so erhaltene Produkt wurde in Pentan verrührt, und der vorhandene Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 1.28 g (81 % d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 8.5-8.0 (br. m, 1H), 8.30 (d, 1H), 8.0-7.5 (br. m, 1H), 7.84 (dd, 1H), 7.72-7.66 (m, 2H), 7.59-7.50 (m, 4H), 7.21 (br. s, 1H), 2.26 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 398 [M+H]+. Beispiel 15A
6-Brom-3-ethyl-2-phenylchinolin-4-carbonsäure
1.00 g (4.42 mmol) 5-Brom-li7-indol-2,3-dion wurden in 12.0 ml Essigsäure vorgelegt und mit 656 mg (4.42 mmol) 1-Phenylbutan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 4.0 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abgesaugt. Der Feststoff wurde mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 1.20 g (55% d. Th., Reinheit 72%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.88 min, m/z = 357 [M+H]+. Beispiel 16A
(6-Brom-3-ethyl-2-phenylchinolin-4-yl)(lii-imidazol-l-yl)methanon
Zu einer Lösung von 500 mg (1.40 mmol) der Verbindung aus Beispiel 15 A in 6 ml DMF wurden bei RT 250 mg (1.54 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 5 h bei 60°C Badtemperatur gerührt. Anschließend wurde das Gemisch mit jeweils 100 ml Wasser und Ethylacetat versetzt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extra-
hiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlor - methan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 7:3, Biotage). Es wurden 259 mg (45% d. Th., Reinheit 100%) der Titel- Verbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-7.5 (br. m, 2H), 8.08 (d, 1H), 7.97 (dd, 1H), 7.77 (br. s, 1H), 7.69-7.60 (m, 2H), 7.59-7.47 (m, 3H), 7.22 (br. s, 1H), 2.82-2.69 (m, 1H), 2.53-2.44 (m, 1H, teilweise verdeckt), 0.81 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.16 min, m/z = 406/408 [M+H]+. Beispiel 17A
6-Brom-2-phenyl-3-propylchinolin-4-carbonsäure
300 mg (1.33 mmol) 5-Brom-li7-indol-2,3-dion wurden in 3.6 ml Essigsäure vorgelegt und mit 237 mg (1.46 mmol) 1-Phenylpentan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 1.2 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abgesaugt. Der Feststoff wurde mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 246 mg (35% d. Th., Reinheit 70%) der Titelverbindung erhalten. LC/MS (Methode 1, ESIpos): Rt = 0.97 min, m/z = 371 [M+H]+.
Beispiel 18A
(6-Brom-2-phenyl-3-propylchinolin-4-yl)(lii-imidazol-l-yl)methanon
Zu einer Lösung von 500 mg (1.35 mmol) der Verbindung aus Beispiel 17A in 6 ml DMF wurden bei RT 241 mg (1.49 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 5 h bei 60°C Badtemperatur gerührt. Anschließend wurde das Gemisch mit jeweils 100 ml Wasser und Ethylacetat versetzt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlor - methan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 7:3, Biotage). Es wurden 355 mg (62% d. Th., Reinheit 100%) der Titel- Verbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-7.5 (br. m, 2H), 8.08 (d, 1H), 7.97 (dd, 1H), 7.77 (br. s, 1H), 7.68-7.62 (m, 2H), 7.58-7.48 (m, 3H), 7.22 (br. s, 1H), 2.81-2.70 (m, 1H), 2.47-2.36 (m, 1H), 1.30-1.08 (m, 2H), 0.55 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.25 min, m/z = 420/422 [M+H]+. Beispiel 19A
6-Brom-7-chlor-3-methyl-2-phenylchinolin-4-carbonsäure
Zu einem Gemisch von 521 mg (2.00 mmol) 5-Brom-6-chlor-li7-indol-2,3-dion in 4.5 ml Essigsäure wurden 268 mg (2.00 mmol) Propiophenon gegeben. Nach 5 min Rühren bei 75°C wurde mit 1.5 ml konz. Salzsäure versetzt, und das Gemisch wurde 6 h bei 110°C weiter gerührt. Nach Abkühlen auf RT und Stehenlassen über Nacht wurde das Gemisch weitere 6 h bei 110°C gerührt.
Danach wurde das Gemisch unter Rühren in 100 ml 1 N Salzsäure eingetragen. Nach einigen Minuten wurde der entstandene Feststoff abfiltriert, zweimal mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 612 mg (58% d. Th., Reinheit 71%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.45 (br. s, 1H), 8.35 (s, 1H), 8.15 (s, 1H), 7.64-7.58 (m, 2H), 7.57-7.48 (m, 3H), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 376/378 [M+H]+.
Beispiel 20A
(6-Brom-7-chlor-3-methyl-2-phenylchinolin-4-yl)(lii-imidazol-l-yl)methanon
Zu einer Lösung von 608 mg (1.15 mmol, Reinheit 71%) der Verbindung aus Beispiel 19A in 3 ml DMF wurden bei RT 204 mg (1.26 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 8 h bei 60°C gerührt. Anschließend wurde das Gemisch mit jeweils 100 ml Wasser und Ethylacetat versetzt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung ge- waschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlor - methan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 7:3, Biotage). Es wurden 446 mg (86% d. Th., Reinheit 94%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-7.7 (br. m, 2H), 8.44 (s, 1H), 8.09 (s, 1H), 7.72- 7.65 (m, 2H), 7.60-7.50 (m, 3H), 7.21 (br. s, 1H), 2.24 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.21 min, m/z = 426/428 [M+H]+.
Beispiel 21 A
6,7-Dichlor-3-methyl-2-phenylchinolin-4-carbonsäure
10.0 g (46.29 mmol) eines Regioisomerengemisches aus 4,5-Dichlor-li7-indol-2,3-dion und 5,6- Dichlor-l#-indol-2,3-dion [ca. 1: 1, Darstellung beschrieben in /. Med. Chem. 2004, 47 (4), 935- 946] wurden in 136 ml Essigsäure vorgelegt und mit 6.21 g (46.29 mmol) 1-Phenylpropan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Dann wurden 42 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Anschließend wurde die Reaktionslösung vorsichtig unter Rühren in Wasser eingetragen. Der entstandene Niederschlag wurde abfiltriert und mittels Säulenchromatographie vorgereinigt (Kieselgel, Laufmittel Ethylacetat/Methanol 10: 1). Das so erhaltene Produktgemisch wurde in 120 ml eines Acetonitril/ Methanol/W asser/Trifluoressigsäure-Gemisches in der Wärme gelöst und mittels präparativer HPLC in die Regioisomere aufgetrennt [Säule: Kinetix C18, 5 μιη, 100 x 21.2 mm; Fluss: 25 ml/ min; Detektion: 210 nm; Injektionsvolumen: 1.0 ml; Temperatur: 35°C; Eluent: 45% Wasser / 50% Acetonitril / 5% Ameisensäure (1% in Wasser), isokratisch; Laufzeit: 4.3 min]. Es wurden 380 mg (2.2% d. Th., Reinheit 90%) der Titelverbindung sowie 300 mg (1.9% d. Th., Reinheit 100%) der regioisomeren Verbindung aus Beispiel 23A erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.54 (br. s, 1H), 8.37 (s, 1H), 8.00 (s, 1H), 7.68-7.58 (m, 2H), 7.58-7.47 (m, 3H), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.99 min, m/z = 332 [M+H]+. Beispiel 22A (6,7-Dichlor-3-methyl-2-phenylchinolin-4-yl)(lii-imidazol-l-yl)methanon
Zu einer Lösung von 328 mg (0.99 mmol) der Verbindung aus Beispiel 21 A in 4.4 ml DMF wurden bei RT 320 mg (1.98 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Anschließend wurde das Gemisch mit Wasser und Ethylacetat versetzt. Nach Phasentrennung wurde die wässrige Phase dreimal mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (80 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 2: 1, Biotage). Es wurden so 295 mg (70% d. Th., Reinheit 90%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 9.25-6.25 (br. m, 2H), 8.46 (s, 1H), 7.97 (s, 1H), 7.75- 7.63 (m, 2H), 7.63-7.38 (m, 3H), 7.21 (br. s, 1H), 2.24 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.19 min, m/z = 382 [M+H]+.
Beispiel 23A
5,6-Dichlor-3-methyl-2-phenylchinolin-4-carbonsäure
Wie unter Beispiel 21A beschrieben, wurden aus 10.0 g (46.29 mmol) eines Regioisomeren- gemisches aus 4,5-Dichlor-li7-indol-2,3-dion und 5,6-Dichlor-li7-indol-2,3-dion (ca. 1: 1) 300 mg (1.9% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.92 (d, 1H), 7.82 (d, 1H), 7.58-7.43 (m, 5H), 2.29 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 0.95 min, m/z = 332 [M+H]+. Beispiel 24A
(5,6-Dichlor-3-methyl-2-phenylchinolin-4-yl)(lii-imidazol-l-yl)methanon
Zu einer Lösung von 274 mg (0.83 mmol) der Verbindung aus Beispiel 23A in 3.7 ml DMF wurden bei RT 268 mg (1.65 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde über Nacht bei 60°C gerührt. Anschließend wurde das Gemisch 3 h in einer Mikrowellenapparatur bei 150°C gerührt. Nach Zugabe von weiteren 268 mg (1.65 mmol) /V,/V'-Carbonyldiimidazol wurde das Gemisch nochmals 1 h in der Mikrowelle bei 150°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Wasser und Ethylacetat versetzt, und nach Phasentrennung wurde die wässrige Phase dreimal mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und einge- engt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (80 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 2: 1 -> Ethylacetat/Methanol 10: 1, Biotage). Es wurden 108 mg (32% d. Th., Reinheit 94%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 9.0-7.0 (br. m, 2H), 8.19 (d, 1H), 8.07 (d, 1H), 7.72- 7.65 (m, 2H), 7.59-7.50 (m, 3H), 7.28 (br. s, 1H), 2.25 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.17 min, m/z = 382 [M+H]+.
Beispiel 25A
6-Brom-2-(4-fluorphenyl)-3-methylchinolin-4-carbonsäure
1.00 g (4.42 mmol) 5-Brom-li7-indol-2,3-dion wurden in 12.0 ml Essigsäure vorgelegt und mit 673 mg (4.42 mmol) l-(4-Fluorphenyl)propan-l-on versetzt. Das Reaktionsgemisch wurde 5 min
bei 75°C gerührt. Anschließend wurden 4.0 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abfiltriert. Der Feststoff wurde mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 1.29 g (73% d. Th., Reinheit 90%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.88 min, m/z = 360 [M+H]+.
Beispiel 26A
[6-Brom-2-(4-fluorphenyl)-3-methylchinolin-4-yl] ( 1 ii-imidazol- 1 -yl)methanon
Zu einer Lösung von 500 mg (1.39 mmol) der Verbindung aus Beispiel 25 A in 6 ml DMF wurden bei RT 248 mg (1.53 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde zunächst 5 h bei 60°C Badtemperatur und dann 14 h bei RT gerührt. Anschließend wurden weitere 124 mg (0.76 mmol) /V,/V'-Carbonyldiimidazol hinzugefügt, und das Gemisch wurde nochmals 7 h bei 60°C Badtemperatur gerührt. Danach wurde das Gemisch mit jeweils 100 ml Wasser und Ethyl- acetat versetzt, und nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Di- chlormethan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 7:3, Biotage). Es wurden 402 mg (71% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-7.5 (br. m, 2H), 8.09 (d, 1H), 7.97 (dd, 1H), 7.82 (br. s, 1H), 7.79-7.72 (m, 2H), 7.37 (t, 2H), 7.21 (br. s, 1H), 2.25 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.13 min, m/z = 410/412 [M+H]+.
Beispiel 27A
6-Brom-2-(3-fluorphenyl)-3-methylchinolin-4-carbonsäure
1.00 g (4.42 mmol) 5-Brom-li7-indol-2,3-dion wurden in 12.0 ml Essigsäure vorgelegt und mit 673 mg (4.42 mmol) l-(3-Fluorphenyl)propan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 4.0 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abfiltriert. Der Feststoff wurde mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 1.20 g (63% d. Th., Reinheit 83%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.94 min, m/z = 360 [M+H]+.
Beispiel 28A
[6-Brom-2-(3-fluorphenyl)-3-methylchinolin-4-yl](lii-imidazol-l-yl)methanon
Zu einer Lösung von 500 mg (1.39 mmol) der Verbindung aus Beispiel 27 A in 6 ml DMF wurden bei RT 248 mg (1.53 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 7 h bei 60°C Badtemperatur gerührt. Anschließend wurde das Gemisch mit jeweils 100 ml Wasser und Ethylacetat versetzt, und nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid- Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in
Dichlormethan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 7:3, Biotage). Es wurden 344 mg (60% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-8.0 (br. m, 1H), 8.10 (d, 1H), 8.0-7.5 (br. m, 1H), 7.98 (dd, 1H), 7.84 (br. s, 1H), 7.64-7.49 (m, 3H), 7.41-7.32 (m, 1H), 7.22 (br. s, 1H), 2.27-2.22 (m, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.16 min, m/z = 410/412 [M+H]+. Beispiel 29A
6-Brom-2-(2-fluorphenyl)-3-methylchinolin-4-carbonsäure
1.00 g (4.42 mmol) 5-Brom-li7-indol-2,3-dion wurden in 12.0 ml Essigsäure vorgelegt und mit 673 mg (4.42 mmol) l-(2-Fluorphenyl)propan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 4.0 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben und der ausgefallene Feststoff abfiltriert. Der Feststoff wurde mit Wasser gewaschen, im Vakuum getrocknet und dann mit Dichlormethan verrührt. Das Lösungsmittel wurde abgesaugt und der Rückstand im Vakuum getrocknet. Es wurden 649 mg (37% d. Th., Reinheit 90%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.90 min, m/z = 360 [M+H]+. Beispiel 30A
[6-Brom-2-(2-fluorphenyl)-3-methylchinolin-4-yl] ( 1 ii-imidazol- 1 -yl)methanon
Zu einer Lösung von 500 mg (1.39 mmol) der Verbindung aus Beispiel 29A in 6 ml DMF wurden bei RT 248 mg (1.53 mmol) /V,/V'-Carbonyldiimidazol gegeben. Das Gemisch wurde 5 h bei 60°C Badtemperatur gerührt und anschließend 14 h bei RT stehen gelassen. Danach wurden weitere 124 mg (0.77 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde 8 h bei 80°C Badtemperatur gerührt und anschließend 14 h bei RT stehen gelassen. Danach wurden nochmals 162 mg (1.00 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde weitere 7 h bei 100°C Badtemperatur gerührt und anschließend 14 h bei RT stehen gelassen. Danach wurden wieder 162 mg (1.00 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde wei- tere 7 h bei 145°C Badtemperatur gerührt und anschließend 14 h bei RT stehen gelassen. Danach wurden erneut 162 mg (1.00 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde weitere 7 h bei 145°C Badtemperatur gerührt und anschließend 14 h bei RT stehen gelassen. Danach wurden abermals 162 mg (1.00 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde weitere 7 h bei 145°C Badtemperatur gerührt und anschließend 14 h bei RT stehen gelassen. Danach schließlich wurde das Gemisch mit jeweils 25 ml Ethylacetat und gesättigter wässriger Natriumchlorid-Lösung versetzt, und nach Phasentrennung wurde die wässrige Phase zweimal mit jeweils 50 ml Ethylacetat extrahiert. Die vereinten organischen Phasen wurden über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Säulenchromatographie gereinigt (100 g Kieselgel, Laufmittel Cyclohexan/ Ethylacetat 7:3, Biotage). Es wurden 405 mg (71% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-7.5 (br. m, 2H), 8.11 (d, 1H), 8.00 (dd, 1H), 7.87 (br. s, 1H), 7.67-7.56 (m, 2H), 7.45-7.36 (m, 2H), 7.21 (br. s, 1H), 2.14 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.10 min, m/z = 410/412 [M+H]+. Beispiel 31 A
6-Brom-3-methyl-2-(pyridin-4-yl)chinolin-4-carbonsäure
Methode A:
1.00 g (4.42 mmol) 5-Brom-li7-indol-2,3-dion wurden in 12.0 ml Essigsäure vorgelegt und mit 598 mg (4.42 mmol) l-(Pyridin-4-yl)propan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 4.0 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde 16 h bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml Wasser gegeben und der ausgefallene Feststoff abfiltriert. Das Filtrat wurde im Vakuum eingeengt und der Rückstand mittels präparativer HPLC (Methode 17) gereinigt. Es wurden 200 mg (13% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.56 (br. s, 1H), 8.79-8.70 (m, 2H), 8.08-8.01 (m, 1H), 7.98-7.92 (m, 2H), 7.66 (d, 2H), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.45 min, m/z = 343/345 [M+H]+.
Methode B (allgemeine Versuchs Vorschrift):
Zu einer Lösung des entsprechenden Isatins (li7-Indol-2,3-dions) in Ethanol (Konzentration zwischen 0.5 und 0.75 M) wurden tropfenweise sechs Äquivalente einer 8.5 M wässrigen Kaliumhydroxid-Lösung gegeben. Anschließend wurde ein Äquivalent l-(Pyridin-4-yl)propan-l-on hinzugegeben, und das Gemisch wurde über Nacht unter Rückfluss gerührt. Nach Abkühlen auf RT und Entfernen des Lösungsmittels im Vakuum wurde der Rückstand in Wasser aufgenommen und die wässrige Phase mit Ethylacetat extrahiert. Anschließend wurde die wässrige Phase durch Zusatz von Salzsäure auf pH 4-5 eingestellt. Der entstandene Feststoff wurde abfiltriert, mit wenig Wasser und Ethylacetat gewaschen und im Vakuum getrocknet. Die betreffende Zielverbindung wurde in einer Ausbeute von 35-70% d. Th. erhalten.
Beispiel 32A
Ethyl-6-amino-2-chlor-5-fluornicotinat
Ein Gemisch von 5.00 g (21.00 mmol) Efhyl-2,6-dichlor-5-fluornicotinat [beschrieben in US 2008/ 0171732, Example 44 (b)] und 105 ml (210 mmol) einer 2 M Lösung von Ammoniak in Ethanol wurde auf 5 Mikrowellengefäße verteilt und 1.5 h in einer Mikrowellenapparatur auf 120°C er- hitzt. Nach Abkühlen auf RT wurde das Lösungsmittel entfernt und der Rückstand mit Ethylacetat versetzt. Es wurde einmal mit Wasser gewaschen und die wässrige Phase danach einmal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Das so erhaltene Rohprodukt wurde mit dem Rohprodukt eines analog durchgeführten Experiments [einge- setzte Menge an Ethyl-2,6-dichlor-5-fluornicotinat 1.00 g (4.20 mmol)] vereinigt. Dieses Material wurde dann mittels präparativer HPLC gereinigt [Säule: Daicel C18 Bio Spring Column, 10 μιη, 300 x 100 mm; Fluss: 250 ml/min; Detektion: 210 nm; Injektionsvolumen: 20 ml; Temperatur: 22°C; Eluent: Acetonitril/W asser-Gradient] . Es wurden 2.60 g der Titelverbindung erhalten (Reinheit 100%, 47% d. Th. bezogen auf insgesamt 6.0 g Ethyl-2,6-dichlor-5-fluornicotinat). Außerdem wurden 290 mg der isomeren Verbindung Ethyl-2-amino-6-chlor-5-fluornicotinat erhalten (Reinheit 100%, 5% d. Th. bezogen auf insgesamt 6.0 g Ethyl-2,6-dichlor-5-fluornicotinat).
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 7.80 (d, 1H), 7.49 (br. s, 2H), 4.23 (q, 2H), 1.28 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.82 min, m/z = 219 [M+H]+. Beispiel 33A
Ethyl-6-amino-5-fluornicotinat-Trifluoressigsäuresalz
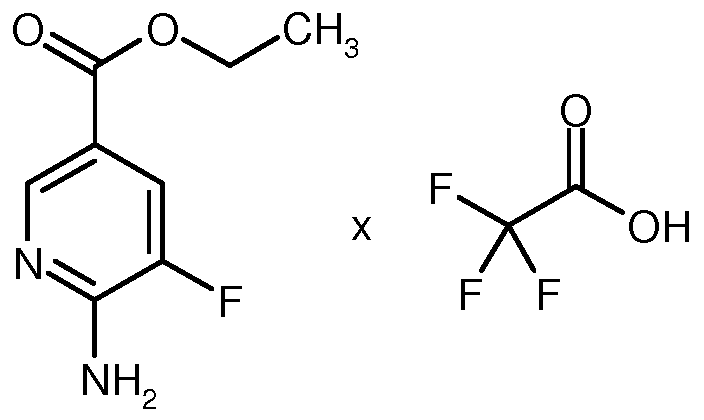
Ein unter Argon angesetztes Gemisch von 500 mg (2.29 mmol) der Verbindung aus Beispiel 32A, 100 mg (94 mmol) Palladium auf Aktivkohle (10% Pd) und 324 mg (3.20 mmol) Triethylamin in 20 ml Ethanol wurde 6 h bei RT unter Normaldruck hydriert. Anschließend wurden weitere 100 mg (94 mmol) Palladium auf Aktivkohle (10% Pd) und 324 mg (3.20 mmol) Triethylamin hin- zugegeben, und es wurde weitere 16 h bei RT unter Normaldruck hydriert. Danach wurden nochmals 100 mg (94 mmol) Palladium auf Aktivkohle (10% Pd) und 324 mg (3.20 mmol) Triethylamin hinzugefügt, und es wurde erneut 6 h bei RT unter Normaldruck hydriert. Danach wurde das Gemisch über Kieselgur filtriert und der Filterrückstand mit Ethanol und Ethylacetat gewaschen. Das Filtrat wurde eingeengt und der Rückstand mit Wasser verrührt. Der vorhandene Feststoff wurde abfiltriert, mit Wasser gewaschen und im Vakuum getrocknet. Das so erhaltene Rohprodukt wurde mittels präparativer HPLC (Methode 16) gereinigt. Die vereinigten Produktfraktionen wurden eingeengt, der Rückstand in Dichlormethan aufgenommen, erneut eingeengt und schließlich im Vakuum getrocknet. Es wurden 555 mg (81% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d
6): δ [ppm] = 8.37-8.35 (m, 1H), 7.69 (dd, 1H), 7.4-5.5 (br. m, ~2H), 4.25 (q, 2H), 1.29 (t, 3H).
LC/MS (Methode 2, ESIpos): Rt = 1.83 min, m/z = 185 [M+H]+. Beispiel 34A
Ethyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}benzoat
300 mg (0.88 mmol) der Verbindung aus Beispiel 1A wurden mit 16 ml Dichlormethan und dann mit 0.19 ml (1.40 mmol) l-Chlor-/V,/V,2-trimethylprop-l-en-l-amin versetzt. Das Gemisch wurde 30 min bei RT gerührt und dann mit 0.21 ml (2.63 mmol) Pyridin und 145 mg (0.88 mmol) Ethyl-
4-aminobenzoat, gelöst in 4 ml Dichlormethan, versetzt. Das Reaktionsgemisch wurde anschließend 30 min bei 50°C und danach 90 min bei 70°C gerührt. Nach Abkühlen auf RT wurde das Lösungsmittel im Vakuum entfernt, und der Rückstand wurde ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 7) gereinigt. Es wurden 177 mg (41% d. Th., Reinheit 100%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.31 min, m/z = 489/491 [M+H]+. Beispiel 35A
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}benzoylchlorid
Zu einer Suspension von 200 mg (0.43 mmol) der Verbindung aus Beispiel 1 in 5 ml Dichlormethan wurden nacheinander ein Tropfen DMF und langsam 110 mg (0.87 mmol) Oxalylchlorid gegeben. Nach 1 h Rühren bei RT wurden weitere 59 mg (0.46 mmol) Oxalylchlorid hinzugefügt, und das Gemisch wurde nochmals 30 min bei RT gerührt. Anschließend wurde das Gemisch eingeengt und der Rückstand im Vakuum getrocknet. Die Titelverbindung wurde in einer Reinheit von 98% laut LC/MS erhalten (die analytische Probe wurde mit Methanol gequencht).
LC/MS (Methode 3, ESIpos): Rt = 1.52 min, m/z = 475/477 [M-Cl+OCH3+H]+. Beispiel 36A
Methyl-5-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}pyrimidin-2-carboxylat
150 mg (0.38 mmol) der Verbindung aus Beispiel 2A und 59 mg (0.38 mmol) Mefhyl-5-amino- pyrimidin-2-carboxylat wurden in 3 ml DMF gelöst. Das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden 64 mg (0.57 mmol) Kalium-ieri.-butylat hinzugegeben, und das Reaktions- gemisch wurde über Nacht bei RT weiter gerührt. Danach wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Nach Entfernen des Lösungsmittel- Wasser-Gemisches wurde der Rückstand über Nacht im Vakuum getrocknet. Es wurden 23 mg (12% d. Th., Reinheit 95%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.63 (s, 1H), 9.32 (s, 2H), 8.11 (d, 1H), 8.05 (d, 1H), 7.95 (dd, 1H), 7.70-7.59 (m, 2H), 7.59-7.49 (m, 3H), 3.92 (s, 3H), 2.42 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.05 min, m/z = 477/479 [M+H]+.
Beispiel 37A
Ethyl-6-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-fluornicotinat
Zu einer Lösung von 200 mg (0.51 mmol) der Verbindung aus Beispiel 2A und 152 mg (0.51 mmol) der Verbindung aus Beispiel 33A in 2 ml DMF wurden bei RT 114 mg (1.02 mmol) Kalium-ieri. -butylat in kleinen Portionen hinzugegeben. Das Gemisch wurde 15 min bei RT ge- rührt. Anschließend wurden weitere 29 mg (0.26 mmol) Kalium-ieri. -butylat in kleinen Portionen hinzugegeben und es wurde nochmals 15 min bei RT gerührt. Danach wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 5) gereinigt. Es wurden 195 mg (74% d. Th., Reinheit 99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.66 (s, 1H), 8.89 (br. s, 1H), 8.35 (d, 1H), 8.09-7.91 (m, 3H), 7.67-7.49 (m, 5H), 4.39 (q, 2H), 2.47 (s, 3H), 1.36 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.18 min, m/z = 508/510 [M+H]+.
Beispiel 38A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
1.00 g (2.55 mmol) der Verbindung aus Beispiel 2A wurden in 10 ml DMF gelöst. Zu der Lösung wurden 474 mg (2.80 mmol) Methyl-4-amino-3-fluorbenzoat und 429 mg (3.82 mmol) Kalium- ieri.-butylat gegeben. Das Reaktionsgemisch wurde 1 h bei RT gerührt und dann in ein Gemisch aus 50 ml Eiswasser, 50 ml gesättigter Ammoniumchlorid-Lösung und 100 ml Ethylacetat eingerührt. Die organische Phase wurde abgetrennt, zweimal mit Wasser und einmal mit gesättigter Natriumchlorid-Lösung gewaschen und über Magnesiumsulfat getrocknet. Das Lösungsmittel wurde im Vakuum entfernt und der Rückstand mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel 5% Ethylace tat/95% Cyclohexan -> 15% Ethylacetat/85% Cyclohexan -> 45% Ethyl- acetat/55% Cyclohexan, Biotage). Die produkthaltigen Fraktionen wurden eingeengt und der Rückstand in 10 ml ieri.-Butylmethylether verrührt. Der Feststoff wurde abfiltriert, zweimal mit 5 ml ieri.-Butylmethylether gewaschen und anschließend drei Tage in 4 ml Ethylacetat verrührt. Der Feststoff wurde danach wieder abfiltriert und im Vakuum getrocknet. Es wurden 810 mg (64% d. Th., Reinheit 99%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.10 (s, 1H), 8.27 (t, 1H), 8.05 (d, 1H), 7.99 (d, 1H), 7.95-7.90 (m, 2H), 7.85 (dd, 1H), 7.64-7.62 (m, 2H), 7.58-7.51 (m, 3H), 3.89 (s, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.27 min, m/z = 493/495 [M+H]+.
Beispiel 39A tert. -Butyl-4- { [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoat
Zu einem Gemisch von 1.00 g (2.09 mmol) der Verbindung aus Beispiel 7 in 50 ml THF wurden 912 mg (4.17 mmol) ieri.-Butyl-trichloracetimidat und 59 mg (0.42 mmol) Bortrifluorid-Diefhyl- ether-Komplex gegeben und das Gemisch 1 h bei RT gerührt. Anschließend wurden weitere 912 mg (4.17 mmol) ieri.-Butyl-trichloracetimidat hinzugefügt und das Gemisch nochmals 1 h bei RT gerührt. Nach Zugabe von Dichlormethan wurde das Gemisch mit Wasser gewaschen und die wässrige Phase einmal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel Cyclohexan/Ethylacetat 85: 15, Biotage). Es wurden 939 mg (84% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.06 (s, 1H), 8.22 (t, 1H), 8.05 (d, 1H), 7.98 (d, 1H), 7.94 (dd, 1H), 7.85 (dd, 1H), 7.78 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.49 (m, 3H), 2.43 (s, 3H), 1.57 (s, 9H). LC/MS (Methode 1, ESIpos): Rt = 1.41 min, m/z = 535/537 [M+H]+. Beispiel 40A
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoylchlorid
Zu einer Suspension von 250 mg (0.52 mmol) der Verbindung aus Beispiel 7 in 2.5 ml Dichlormethan wurden nacheinander ein Tropfen DMF und langsam 110 mg (0.87 mmol) Oxalylchlorid gegeben. Nach Verdünnen mit weiteren 2.5 ml Dichlormethan und 1 h Rühren bei RT wurde das Gemisch eingeengt und der Rückstand im Vakuum getrocknet. Die Titelverbindung wurde in einer Reinheit von 94% laut LC/MS erhalten (die analytische Probe wurde mit Methanol gequencht).
LC/MS (Methode 3, ESIpos): Rt = 1.55 min, m/z = 493/495 [M-Cl+OCH3+H]+.
Beispiel 41 A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-fluorbenzoat
300 mg (0.88 mmol) der Verbindung aus Beispiel 1A wurden mit 3 ml Dichlormethan und dann mit 0.19 ml (1.40 mmol) l-Chlor-/V,/V,2-trimethylprop-l-en-l-amin versetzt. Das Gemisch wurde 30 min bei RT gerührt und dann mit 0.21 ml (2.63 mmol) Pyridin und 148 mg (0.88 mmol)
Methyl-4-amino-2-fluorbenzoat versetzt. Das Reaktionsgemisch wurde anschließend 3 h bei 70°C gerührt. Nach Abkühlen auf RT wurde das Lösungsmittel im Vakuum entfernt und der Rückstand ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 7) gereinigt. Es wurden 124 mg (23% d. Th., Reinheit 81%) der Titelverbindung erhalten. LC/MS (Methode 1, ESIpos): Rt = 1.27 min, m/z = 493/495 [M+H]+.
Beispiel 42A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-chlorbenzoat
Zu einer Lösung von 1.50 g (3.82 mmol) der Verbindung aus Beispiel 2A und 710 mg (3.82 mmol) Methyl-4-amino-3-chlorbenzoat in 15 ml DMF wurden bei RT 430 mg (3.82 mmol) Kalium-ieri.- butylat in kleinen Portionen hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden nochmals 215 mg (1.91 mmol) Kalium-feri.-butylat in kleinen Portionen hinzugegeben und es wurde weitere 2 h bei RT gerührt. Danach wurde das Gemisch unter Rühren in 40 ml einer 10%-igen wässrigen Zitronensäure-Lösung eingetragen, woraufhin ein Feststoff ausfiel. Nach Verdünnen mit Wasser wurde der Feststoff abfiltriert, mit Wasser gewaschen und im Vakuum getrocknet. Das so erhaltene Rohprodukt wurde mittels Säulenchromatographie gereinigt (100 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 85: 15, Biotage). Es wurden 1.60 g (80% d. Th., Reinheit 98%) der Titel Verbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 10.96 (s, 1H), 8.14-8.08 (m, 3H), 8.07-8.02 (m, 2H), 7.94 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.50 (m, 3H), 3.90 (s, 3H), 2.48 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.32 min, m/z = 509/511 [M+H]+.
Beispiel 43A
Methyl-3-brom-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}benzoat
Zu einer Lösung von 200 mg (0.51 mmol) der Verbindung aus Beispiel 2A und 129 mg (0.56 mmol) Methyl-4-amino-3-brombenzoat in 2 ml DMF wurden bei RT 57 mg (0.51 mmol) Kalium- ieri.-butylat in kleinen Portionen hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden nochmals 29 mg (0.26 mmol) Kalium-feri.-butylat in kleinen Portionen hinzugegeben und es wurde weitere 15 min bei RT gerührt. Danach wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure -Lösung versetzt, mit Wasser verdünnt und zweimal mit Efhyl- acetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie vorgereinigt (25 g Kieselgel, Laufmittel Cyclohexan/ Ethylacetat 85: 15, Biotage) und anschließend mittels präparativer HPLC (Methode 5) nachgereinigt. Es wurden 145 mg (51% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.91 (s, 1H), 8.25 (d, 1H), 8.14 (d, 1H), 8.10-8.07 (m, 1H), 8.07-8.02 (m, 2H), 7.94 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.50 (m, 3H), 3.90 (s, 3H), 2.50 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.33 min, m/z = 553/555/557 [M+H]+. Beispiel 44A Ethyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3,5-dichlorbenzoat
Zu einer Lösung von 200 mg (0.51 mmol) der Verbindung aus Beispiel 2A und 119 mg (0.51 mmol) Ethyl-4-amino-3,5-dichlorbenzoat in 2 ml DMF wurden bei RT 57 mg (0.51 mmol) Kalium- ieri.-butylat in kleinen Portionen hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. An- schließend wurden nochmals 29 mg (0.26 mmol) Kalium-feri.-butylat in kleinen Portionen hinzugegeben und es wurde weitere 15 min bei RT gerührt. Danach wurde das Gemisch direkt mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden mit gesättigter Natriumhydrogencarbonat-Lösung neutralisiert und bis auf ein Restvolumen an wäss- riger Phase eingeengt. Der entstandene Feststoff wurde abfiltriert, zweimal mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 76 mg (24% d. Th., Reinheit 90%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.26 (s, 1H), 8.43-8.37 (m, 1H), 8.13 (s, 1H), 8.09- 8.02 (m, 1H), 7.99-7.92 (m, 1H), 7.73 (s, 1H), 7.68-7.48 (m, 5H), 4.38 (q, 2H), 2.58 (s, 3H, teilweise verdeckt), 1.36 (t, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.41 min, m/z = 557/559/561 [M+H]+.
Beispiel 45A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino }-3-methylbenzoat
300 mg (0.88 mmol) der Verbindung aus Beispiel 1A wurden mit 3 ml Dichlormethan und dann mit 0.19 ml (1.40 mmol) l-Chlor-/V,/V,2-trimethylprop-l-en-l-amin versetzt. Das Gemisch wurde 30 min bei RT gerührt und dann mit 0.21 ml (2.63 mmol) Pyridin und 145 mg (0.88 mmol) Methyl-4-amino-3-methylbenzoat versetzt. Das Reaktionsgemisch wurde anschließend 3 h bei 70°C gerührt. Nach Abkühlen auf RT wurde das Lösungsmittel im Vakuum entfernt und der Rückstand ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 7) gereinigt. Es wurden 128 mg (25% d. Th., Reinheit 84%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.27 min, m/z = 489/491 [M+H]+. Beispiel 46A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-methoxybenzoat
Zu einer Lösung von 820 mg (2.09 mmol) der Verbindung aus Beispiel 2A und 379 mg (2.09 mmol) Methyl-4-amino-3-methoxybenzoat in 8 ml DMF wurden bei RT 235 mg (2.09 mmol) Kalium-ieri. -butylat in kleinen Portionen hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden nochmals 117 mg (1.05 mmol) Kalium-ieri. -butylat in kleinen Por- tionen hinzugegeben und es wurde weitere 2 h bei RT gerührt. Danach wurde das Gemisch unter Rühren in 20 ml einer 10%-igen wässrigen Zitronensäure-Lösung eingetragen, woraufhin ein Feststoff ausfiel. Nach Verdünnen mit Wasser wurde der Feststoff abfiltriert, mit Wasser gewaschen und im Vakuum getrocknet. Das so erhaltene Rohprodukt wurde mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 85: 15, Biotage). Es wurden 709 mg (67% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.53 (s, 1H), 8.17 (d, 1H), 8.06-8.00 (m, 2H), 7.92 (dd, 1H), 7.69 (dd, 1H), 7.65-7.60 (m, 3H), 7.59-7.47 (m, 3H), 3.94 (s, 3H), 3.89 (s, 3H), 2.42 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.25 min, m/z = 505/507 [M+H]+. Beispiel 47A
Ethyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(trifluormethoxy)benzoat
150 mg (0.38 mmol) der Verbindung aus Beispiel 2A und 95 mg (0.38 mmol) Ethyl-4-amino-3- (trifluormethoxy)benzoat wurden in 3 ml DMF gelöst. Es wurden 86 mg (0.76 mmol) Kalium-ieri.- butylat hinzugegeben, und das Gemisch wurde 15 min bei RT gerührt. Danach wurden weitere 22 mg (0.19 mmol) Kalium-ieri. -butylat langsam hinzugegeben. Das Reaktionsgemisch wurde über Nacht bei RT weiter gerührt und dann ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Nach Entfernen des Lösungsmittel-Wasser-Gemisches wurde der Rückstand
über Nacht im Vakuum getrocknet. Es wurden 75 mg (34% d. Th., Reinheit 100%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.38 min, m/z = 573/575 [M+H]+. Beispiel 48A Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino }-3-(methylsulfanyl)benzoat
Zu einer Lösung von 500 mg (1.28 mmol) der Verbindung aus Beispiel 2A und 267 mg (1.28 mmol, Reinheit 94%) Methyl-4-amino-3-(methylsulfanyl)benzoat [beschrieben in Org. Prep. Proced. Int. 2003, 35 (5), 520-524] in 5 ml DMF wurden bei RT 144 mg (1.28 mmol) Kalium- ieri.-butylat in kleinen Portionen hinzugegeben. Das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden weitere 72 mg (0.64 mmol) Kalium-feri.-butylat in kleinen Portionen hinzugegeben und es wurde weitere 15 min bei RT gerührt. Danach wurde das Gemisch mit 10 ml einer 10%-igen wässrigen Zitronensäure-Lösung versetzt, mit Wasser verdünnt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natrium- chlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/ Ethylacetat 85: 15, Biotage). Es wurden 418 mg (61 % d. Th., Reinheit 96%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 10.73 (s, IH), 8.26 (d, IH), 8.04 (d, IH), 7.96-7.91 (m, 2H), 7.88 (dd, IH), 7.74 (d, IH), 7.65-7.61 (m, 2H), 7.60-7.49 (m, 3H), 3.90 (s, 3H), 2.57 (s, 3H), 2.52-2.50 (m, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.27 min, m/z = 521/523 [M+H]
Beispiel 49A
Methyl-4-{ [(6-brom-3-methyl-2^henylchinolin-4-yl)carbonyl]amino}-3-(methylsulfi
Zu einer Lösung von 150 mg (0.29 mmol) der Verbindung aus Beispiel 48A in 3 ml Dichlormethan wurden 71 mg (0.29 mmol, Reinheit 70%) 3-Chlorperbenzoesäure gegeben und das Gemisch 5 min bei RT gerührt. Anschließend wurden weitere 20 mg (0.08 mmol) 3-Chlorperbenzoesäure hinzugegeben, und das Gemisch wurde weitere 5 min bei RT gerührt. Danach wurden erneut 20 mg (0.08 mmol) 3-Chlorperbenzoesäure hinzugefügt, und das Gemisch wurde nochmals 5 min bei RT gerührt. Anschließend wurde das Lösungsmittel entfernt. Der Rückstand wurde in Acetonitril aufgenommen und mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden 150 mg (97% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.24 (s, 1H), 8.54 (d, 1H), 8.22 (dd, 1H), 8.09-8.04 (m, 2H), 8.01-7.93 (m, 1H), 7.86 (d, 1H), 7.67-7.62 (m, 2H), 7.61-7.49 (m, 3H), 3.93 (s, 3H), 2.92 (s, 3H), 2.50 (s, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.13 min, m/z = 537/539 [M+H]
Beispiel 50A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(methylsulfonyl)benzoat
Methode A:
Zu einer Lösung von 150 mg (0.29 mmol) der Verbindung aus Beispiel 48A in 3 ml Dichlormethan wurden 141 mg (0.58 mmol, Reinheit 70%) 3-Chlorperbenzoesäure gegeben. Das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde das Gemisch mit 5 ml 1 M Natronlauge versetzt, mit Wasser verdünnt und dreimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 106 mg (61% d. Th., Reinheit 92%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.76 (s, 1H), 8.55 (d, 1H), 8.42-8.32 (m, 2H), 8.26 (d, 1H), 8.04 (d, 1H), 7.94 (dd, 1H), 7.66-7.61 (m, 2H), 7.59-7.49 (m, 3H), 3.94 (s, 3H), 3.42 (s, 3H), 2.50-2.48 (m, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.23 min, m/z = 553/555 [M+H]+. Methode B:
Zu einer Lösung von 1.00 g (1.86 mmol) der Verbindung aus Beispiel 49 A in 20 ml Dichlormethan wurden 917 mg (3.72 mmol, Reinheit 70%) 3-Chlorperbenzoesäure gegeben. Das Gemisch wurde 2 h bei RT gerührt. Anschließend wurde das Gemisch mit 35 ml 1 M Natronlauge versetzt, mit Wasser verdünnt und dreimal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrock- net, filtriert und eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 881 mg (67% d. Th., Reinheit 78%) der Titel Verbindung erhalten.
Beispiel 51 A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(ethylsulfony oxybenzoat
Zu einer Lösung von 200 mg (0.51 mmol) der Verbindung aus Beispiel 2A und 139 mg (0.51 mmol) Methyl-4-amino-5-(ethylsulfonyl)-2-methoxybenzoat in 2 ml DMF wurden bei RT 57 mg (0.51 mmol) Kalium-ieri. -butylat in kleinen Portionen hinzugegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden weitere 29 mg (0.26 mmol) Kalium-ieri. -butylat in kleinen Portionen hinzugegeben und es wurde nochmals 15 min bei RT gerührt. Danach wurde das Gemisch mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden mit gesättigter Natriumhydrogencarbonat-Lösung neutralisiert und bis auf ein Restvolumen an wässriger Phase eingeengt. Der entstandene Feststoff wurde abfiltriert, zweimal mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 235 mg (77% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d
6): δ [ppm] = 10.61 (s, 1H), 8.25 (br. s, 2H), 8.18 (br. s, 1H), 8.08- 8.01 (m, 1H), 7.98-7.90 (m, 1H), 7.68-7.60 (m, 2H), 7.54 (s, 3H), 4.03 (s, 3H), 3.85 (s, 3H), 3.44 (q, 2H), 2.50 (s, 3H, verdeckt), 1.14 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.32 min, m/z = 597/599 [M+H]+.
Beispiel 52A Dimethyl-3,3'-disulfandiylbis(4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}- benzoat)
Zu einem Gemisch von 2.24 g (4.17 mmol) der Verbindung aus Beispiel 49A und 1.38 g (12.92 mmol) 2,6-Dimethylpyridin in 18.5 ml Acetonitril wurden bei -20°C 2.63 g (12.50 mmol) Trifluor- essigsäureanhydrid tropfenweise hinzugegeben. Das Gemisch wurde 1 h bei -10°C bis 0°C gerührt. Danach wurde das Gemisch im Vakuum eingeengt, und der Rückstand wurde bei 0°C mit 14 ml eines auf 0°C gekühlten und entgasten Gemisches aus Methanol und Triethylamin (1: 1) versetzt. Anschließend wurde 30 min bei RT gerührt und dann die flüchtigen Bestandteile im Vakuum entfernt. Der Rückstand wurde mit 4 ml eines Gemisches aus Methanol und 6 M Salzsäure versetzt und 20 min bei 50°C gerührt. Anschließend wurde eingeengt, und der Rückstand wurde in Efhyl- acetat aufgenommen und das Gemisch einmal mit Wasser gewaschen. Die wässrige Phase wurde einmal mit Ethylacetat rückextrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen, auf 10 g neutrales Aluminiumoxid aufgezogen und mittels Säulenchromatographie gereinigt (Kieselgel, Cyclohexan/Ethylacetat 7:3, Biotage, mit Vorsäule). Es wurden Fraktionen erhalten, die die Titel Verbindung enthielten, sowie weitere Fraktionen, die die als Beispiel 72A aufgeführte Verbindung als Nebenprodukt der Reaktion enthielten. Die Fraktionen, die die Titelverbindung enthielten, wurden eingeengt und der Rückstand in Acetonitril verrührt. Der vorhandene Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden so 608 mg (13% d. Th., Reinheit ca. 90%) der Titelverbindung erhalten. LC/MS (Methode 1, ESIpos): Rt = 1.59 min, m/z = 1011/1013/1015 [M+H]+.
Die Fraktionen, die die als Beispiel 72A aufgeführte Verbindung enthielten, wurden eingeengt und mittels präparativer HPLC nachgereinigt (Säule: Sunfire C18, 5 μιη, 100 mm x 30 mm; Eluent: Wasser/ Acetonitril/2% Ameisensäure in Wasser; Gradient: 50:30:20— > 5:90:5, 10 min; Injektionsvolumen: 1.0 ml; Fluss: 75 ml/min; Temperatur: 40°C; Detektion: 210 nm). Es wurden 210 mg (9% d. Th., Reinheit 100%) des Nebenprodukts erhalten (Analytik siehe unter Beispiel 72A).
Beispiel 53A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(trifluorm
benzoat
Zu einem Gemisch von 300 mg (0.30 mmol, Reinheit 90%) der Verbindung aus Beispiel 52A in 3 ml DMF wurden 105 mg (0.89 mmol) Natriumhydroxymethansulfinat (Rongalit
™) gegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden 196 mg (0.59 mmol) 3,3- Dimethyl-l-(trifluormethyl)-l,2-benziodoxol hinzugefügt, und das Gemisch wurde weitere 30 min bei RT gerührt. Danach wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 5) gereinigt. Es wurden 140 mg (91% d. Th., Reinheit 100%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.40 min, m/z = 575/577 [M+H]+.
Aus einem analog durchgeführten Experiment wurde das folgende H-NMR der Titelverbindung erhalten [eingesetzte Menge an Verbindung aus Beispiel 52A: 100 mg (0.10 mmol)]: Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.37 (s, 1H), 8.36 (s, 1H), 8.28 (dd, 1H), 8.16 (d, 1H), 8.06 (d, 1H), 8.00-7.92 (m, 2H), 7.67-7.61 (m, 2H), 7.60-7.49 (m, 3H), 3.92 (s, 3H), 2.47 (s, 3H).
Beispiel 54A
Methyl-3-fluor-4-{ [(6-fluor-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}benzoat
Zu einer Lösung von 200 mg (0.60 mmol) der Verbindung aus Beispiel 4A und 102 mg (0.60 mmol) Methyl-4-amino-3-fluorbenzoat in 2.5 ml DMF wurden bei RT 68 mg (0.60 mmol) Kalium- ieri.-butylat in kleinen Portionen hinzugegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden weitere 34 mg (0.31 mmol) Kalium-ieri.-butylat in kleinen Portionen hinzugefügt und es wurde weitere 15 min bei RT gerührt. Danach wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure -Lösung versetzt, mit Wasser verdünnt und zweimal mit Efhyl- acetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wur- de mittels Säulenchromatographie gereinigt (25 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 85: 15, Biotage). Es wurden 198 mg (76% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.07 (s, 1H), 8.28 (t, 1H), 8.17 (dd, 1H), 7.91 (d, 1H), 7.84 (dd, 1H), 7.73 (td, 1H), 7.65-7.59 (m, 2H), 7.59-7.47 (m, 4H), 3.89 (s, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.16 min, m/z = 433 [M+H]+. Beispiel 55A
Methyl-4-{ [(3,6-dimethyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 200 mg (0.61 mmol) der Verbindung aus Beispiel 8A und 103 mg (0.61 mmol) Methyl-4-amino-3-fluorbenzoat in 2.4 ml DMF wurden bei RT 69 mg (0.61 mmol) Kalium- ieri.-butylat in kleinen Portionen hinzugegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden weitere 34 mg (0.30 mmol) Kalium-feri.-butylat in kleinen Portionen hinzugefügt und es wurde weitere 15 min bei RT gerührt. Danach wurden 21 mg (0.12 mmol) Methyl- 4-amino-3-fluorbenzoat sowie 14 mg (0.12 mmol) Kalium-ieri. -butylat hinzugegeben, und das Gemisch wurde nochmals 30 min bei RT gerührt. Anschließend wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure -Lösung versetzt, mit Wasser verdünnt und zweimal mit Efhyl- acetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden 134 mg (47% d. Th., Reinheit 92%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.06 (s, 1H), 8.31 (t, 1H), 8.00 (d, 1H), 7.91 (dd, 1H), 7.84 (dd, 1H), 7.67 (dd, 1H), 7.65-7.60 (m, 3H), 7.59-7.48 (m, 3H), 3.89 (s, 3H), 2.41 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.16 min, m/z = 429 [M+H]+.
Beispiel 56A Methyl-4-{ [(6-ethyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 200 mg (0.59 mmol) der Verbindung aus Beispiel 10A und 99 mg (0.59 mmol) Methyl-4-amino-3-fluorbenzoat in 2 ml DMF wurden bei RT 66 mg (0.59 mmol) Kalium- ieri.-butylat in kleinen Portionen hinzugegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden weitere 29 mg (0.26 mmol) Kalium-ieri.-butylat in kleinen Portionen hinzugefügt und es wurde weitere 15 min bei RT gerührt. Danach wurden 40 mg (0.12 mmol) der Verbindung aus Beispiel 10A sowie 20 mg (0.12 mmol) Methyl-4-amino-3-fluorbenzoat hinzugegeben, und das Gemisch wurde nochmals 30 min bei RT gerührt. Anschließend wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure-Lösung versetzt, mit Wasser verdünnt und zwei- mal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkt- haltigen Fraktionen wurden eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden 195 mg (69% d. Th., Reinheit 91%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.06 (s, IH), 8.26 (t, IH), 8.03 (d, IH), 7.92 (dd, IH), 7.85 (dd, IH), 7.72 (dd, IH), 7.66-7.60 (m, 3H), 7.59-7.48 (m, 3H), 3.92-3.87 (m, 3H), 2.84 (q, 2H), 2.41 (s, 3H), 1.26 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.25 min, m/z = 443 [M+H]+. Beispiel 57A
Methyl-3-fluor-4-{ [(6-isopropyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}benzoat
670 mg (2.19 mmol) der Verbindung aus Beispiel I IA wurden mit 3 ml Dichlormethan und dann mit 0.46 ml (3.51 mmol) l-Chlor-/V,/V,2-trimethylprop-l-en-l-amin versetzt. Das Gemisch wurde 30 min bei RT gerührt und danach mit 0.53 ml (6.55 mmol) Pyridin und 371 mg (2.19 mmol) Methyl-4-amino-3-fluorbenzoat versetzt. Das Reaktionsgemisch wurde 10 h bei 60°C gerührt. Nach Abkühlen auf RT wurde das Lösungsmittel im Vakuum entfernt, und der Rückstand wurde ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 8) gereinigt. Es wurden 108 mg (10% d. Th., Reinheit 96%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.32 min, m/z = 457 [M+H]+. Beispiel 58A
Methyl-3-fluor-4-({ [3-methyl-2-phenyl-6-(trifluormethyl)chinolin-4-yl]carbonyl}amino)benzoat
300 mg (0.91 mmol) der Verbindung aus Beispiel 12A wurden mit 2 ml Dichlormethan und dann mit 0.19 ml (1.45 mmol) l-Chlor-AyV,2-trimethylprop-l-en-l-amin versetzt. Das Gemisch wurde 30 min bei RT gerührt und danach mit 0.22 ml (2.72 mmol) Pyridin und 153 mg (0.91 mmol) Methyl-4-amino-3-fluorbenzoat versetzt. Das Reaktionsgemisch wurde 4 h bei 60°C gerührt. Nach Abkühlen auf RT wurde das Lösungsmittel im Vakuum entfernt, und der Rückstand wurde ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 7) gereinigt. Es wurden 105 mg (20% d. Th., Reinheit 83%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 0.91 min, m/z = 483 [M+H]+.
Beispiel 59A Methyl-3-fluor-4-({ [3-methyl-2-phenyl-6-(trifluormethoxy)chinolin-4-yl]carbonyl}amino)benzoat
Zu einer Lösung von 200 mg (0.50 mmol) der Verbindung aus Beispiel 14A und 85 mg (0.50 mmol) Methyl-4-amino-3-fluorbenzoat in 2 ml DMF wurden bei RT 56 mg (0.50 mmol) Kalium- ieri.-butylat in kleinen Portionen hinzugegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden weitere 28 mg (0.26 mmol) Kalium-ieri.-butylat in kleinen Portionen hinzugefügt und es wurde weitere 15 min bei RT gerührt. Danach wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure -Lösung versetzt, mit Wasser verdünnt und zweimal mit Ethyl- acetat extrahiert. Die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wur- de mittels Säulenchromatographie gereinigt (25 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 85: 15, Biotage). Die vereinigten produkthaltigen Fraktionen wurden eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden 166 mg (60% d. Th., Reinheit 91%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.11 (s, 1H), 8.27-8.17 (m, 2H), 7.92 (dd, 1H), 7.86 (dd, 1H), 7.82 (dd, 1H), 7.75-7.73 (m, 1H), 7.66-7.61 (m, 2H), 7.60-7.44 (m, 4H), 6.77 (t, 1H), 3.89 (s, 3H), 2.45 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.26 min, m/z = 499 [M+H]+. Beispiel 60A ieri.-Butyl-3-fluor-4-({ [3-methyl-2-phenyl-6-(trimethylsilyl)chinolin-4-yl]carbonyl}amino)benzoat
Zu einem Gemisch von 214 mg (0.40 mmol) der Verbindung aus Beispiel 39A und 64 mg (0.44 mmol) Hexamethyldisilan in einem Gemisch aus 1 ml Toluol und 1 ml Wasser wurden 7 mg (0.02 mmol) Allylpalladiumchlorid-Dimer, 11 mg (0.04 mmol) (2-Hydroxyphenyl)diphenylphosphan, 19 mg (0.48 mmol) Natriumhydroxid sowie 14 mg (0.044 mmol) Tetrabutylammoniumbromid gegeben. Das Gemisch wurde 5 h bei 100°C gerührt und danach 14 h bei RT stehen gelassen. Anschließend wurden weitere 64 mg (0.44 mmol) Hexamethyldisilan, 7 mg (0.02 mmol) Allylpalladiumchlorid-Dimer sowie 11 mg (0.04 mmol) (2-Hydroxyphenyl)diphenylphosphan hinzuge- fügt, und das Gemisch wurde nochmals 7 h bei 100°C gerührt. Nach anschließendem dreitägigen Stehenlassen bei RT wurde das Gemisch mit Ethylacetat verdünnt und mit Wasser gewaschen. Die wässrige Phase wurde einmal mit Ethylacetat rückextrahiert, und die vereinten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 5) gerei- nigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, und der Rückstand wurde in Dichlormethan aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden so 39 mg (18% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.03 (s, 1H), 8.10-7.99 (m, 3H), 7.94 (dd, 1H), 7.85 (dd, 1H), 7.80 (dd, 1H), 7.66-7.60 (m, 2H), 7.59-7.48 (m, 3H), 2.43 (s, 3H), 1.57 (s, 9H), 0.32 (s, 9H).
LC/MS (Methode 1, ESIpos): Rt = 1.51 min, m/z = 529 [M+H]+. Beispiel 61 A
Methyl-4-( { [6-brom-3-(brommethyl)-2-phenylchinolin-4-yl] carbonyl } amino)-3-fluorbenzoat
Zu einer Lösung von 1.07 g (2.17 mmol) der Verbindung aus Beispiel 38A in 10 ml Acetonitril unter Argon wurden bei RT 463 mg (2.60 mmol) /V-Bromsuccinimid (NBS) und 36 mg (0.22 mmol) 2,2'-Azobis(2-methylpropionitril) (AIBN) gegeben, und das Gemisch wurde auf 80°C Badtemperatur erhitzt. Sodann wurden 10 ml Tetrachlorkohlenstoff hinzugegeben, und das Gemisch wurde 1 h bei 80°C Badtemperatur gerührt. Anschließend wurden weitere 232 mg (1.30 mmol) /V-Bromsuccinimid (NBS) sowie 21 mg (0.13 mmol) 2,2'-Azobis(2-methylpropionitril) (ΑΓΒΝ) hinzugefügt, und das Gemisch wurde weitere 8 h bei 80°C Badtemperatur gerührt. Nach Abkühlen auf RT wurde der vorhandene Feststoff abfiltriert und zweimal mit 2 ml Acetonitril gewaschen. Nach Trocknen des Feststoffs im Vakuum wurden 656 mg (44% d. Th., Reinheit 84%) einer ersten Charge der Titelverbindung erhalten. Das Filtrat wurde eingeengt und der Rückstand zusammen mit einem analog erhaltenen Rückstand aus einem vorhergehenden Experiment [eingesetzte Menge an Verbindung aus Beispiel 38A: 100 mg (0.20 mmol)] mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel Cyclohexan/Ethylacetat 9: 1). Es wurden so 260 mg einer zweiten Charge der Titelverbindung erhalten (Reinheit 90%, 17% d. Th. bezogen auf insgesamt 1.17 g (2.37 mmol) eingesetzter Menge an Verbindung aus Beispiel 38A). Darüber hinaus wurden als Nebenprodukt der Reaktion 194 mg (10% d. Th, Reinheit 70%) der Verbindung aus Beispiel 62A erhalten (siehe dort).
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.23 (s, IH), 8.25 (t, IH), 8.10-8.01 (m, 3H), 7.93 (dd, IH), 7.87 (dd, IH), 7.72-7.66 (m, 2H), 7.64-7.56 (m, 3H), 4.73 (dd, 2H), 3.90 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.27 min, m/z = 571/573/575 [M+H]+.
Beispiel 62A Methyl-4-({ [6-brom-3-(dibrommethyl)-2-phenylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Die Titelverbindung (194 mg, Reinheit 70%) wurde als Nebenprodukt der in Beispiel 61A beschriebenen Umsetzung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.26 (s, IH), 8.38 (t, IH), 8.14-8.07 (m, 3H), 7.94 (dd, IH), 7.87 (dd, IH), 7.65-7.59 (m, 5H), 7.03 (s, IH), 3.90 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.32 min, m/z = 649/651/653/655 [M+H]+.
Beispiel 63A
Methyl-4-({ [6-brom-3-(fluormethyl)-2-phenylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Zu einem Gemisch von 232 mg (0.37 mmol, Reinheit 90%) der Verbindung aus Beispiel 61 A in 18 ml Acetonitril unter Argon wurden 130 mg (1.02 mmol) Silberfluorid gegeben, und das Gemisch wurde 1.5 h bei 80°C Badtemperatur erhitzt. Nach Abkühlen auf RT wurde das Gemisch mit jeweils 100 ml Ethylacetat und gesättigter wässriger Natriumhydrogencarbonat-Lösung versetzt. Nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Säulenchromatographie gereinigt (25 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 9: 1, Biotage). Es wurden so 77 mg (41% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.19 (s, 1H), 8.28 (t, 1H), 8.15-8.10 (m, 2H), 8.07 (dd, 1H), 7.93 (dd, 1H), 7.86 (dd, 1H), 7.70-7.64 (m, 2H), 7.62-7.54 (m, 3H), 5.63 (s, 1H), 5.51 (s, 1H), 3.89 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.22 min, m/z = 511/513 [M+H]+. Beispiel 64A
Methyl-4-({ [6-brom-3-(difluormethyl)-2-phenylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Zu einem Gemisch von 170 mg (0.18 mmol, Reinheit 70%) der Verbindung aus Beispiel 62A in 6 ml Acetonitril unter Argon wurden 93 mg (0.73 mmol) Silberfluorid gegeben, und das Gemisch wurde 1 h bei 80°C Badtemperatur erhitzt. Nach Abkühlen auf RT wurde das Gemisch mit jeweils 50 ml Ethylacetat und gesättigter wässriger Natriumhydrogencarbonat-Lösung versetzt. Nach Phasentrennung wurde die wässrige Phase einmal mit 50 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Säulenchromatographie gereinigt (25 g Kieselgel, Lauf mittel Cyclohexan/Ethylacetat 9: 1, Biotage). Es wurden so 28 mg (27% d. Th., Reinheit 95%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.16 (s, 1H), 8.34-8.28 (m, 1H), 8.17-8.11 (m, 3H), 7.92 (dd, 1H), 7.85 (dd, 1H), 7.65-7.55 (m, 5H), 7.08 (t, 1H), 3.89 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.22 min, m/z = 529/531 [M+H]+.
Beispiel 65A Methyl-4-{ [(6-brom-3-ethyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 238 mg (0.59 mmol) der Verbindung aus Beispiel 16A und 99 mg (0.59 mmol) Methyl-4-amino-3-fluorbenzoat in 2.3 ml DMF unter Argon wurden 0.88 ml (0.88 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure- Lösung versetzt, mit Wasser verdünnt und zweimal mit jeweils 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit 40 ml gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlor - methan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 4: 1, Biotage). Es wurden 206 mg (69% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.13 (s, 1H), 8.22 (t, 1H), 8.06-7.89 (m, 4H), 7.85 (dd, 1H), 7.60-7.49 (m, 5H), 3.89 (s, 3H), 2.91-2.76 (m, 2H), 0.98 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.27 min, m/z = 507/509 [M+H]+. Beispiel 66A
Methyl-4-{ [(6-brom-2-phenyl-3-propylchinolin-4-yl)carbonyl]amino }-3-fluorbenzoat
Zu einer Lösung von 170 mg (0.40 mmol) der Verbindung aus Beispiel 18A und 68 mg (0.40 mmol) Methyl-4-amino-3-fluorbenzoat in 2 ml DMF unter Argon wurden 0.61 ml (0.61 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure- Lösung versetzt, mit Wasser verdünnt und zweimal mit jeweils 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit 40 ml gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlor - methan aufgenommen und mittels Säulenchromatographie gereinigt (25 g Kieselgel, Lauf mittel Cyclohexan/Ethylacetat 9: 1, Biotage). Es wurden 151 mg (72% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.11 (s, 1H), 8.18 (t, 1H), 8.03 (d, 1H), 7.99 (d, 1H), 7.97-7.90 (m, 2H), 7.86 (dd, 1H), 7.59-7.50 (m, 5H), 3.89 (s, 3H), 2.79 (t, 2H), 1.47-1.30 (m, 2H), 0.67 (t, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.31 min, m/z = 521/523 [M+H]+.
Beispiel 67A
Methyl-4-{ [(6-brom-7-chlor-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino }-3-fluorbenzoat
Zu einer Lösung von 200 mg (0.47 mmol) der Verbindung aus Beispiel 20A und 87 mg (0.52 mmol) Methyl-4-amino-3-fluorbenzoat in 1 ml DMF unter Argon wurden bei RT 53 mg (0.47 mmol) Kalium-ieri.-butylat in kleinen Portionen hinzugegeben, und das Gemisch wurde 15 min bei RT gerührt. Anschließend wurden weitere 26 mg (0.24 mmol) Kalium-ieri.-butylat in kleinen Portionen hinzugefügt und es wurde nochmals 15 min bei RT gerührt. Danach wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure -Lösung versetzt und mit 6 ml Wasser verdünnt. Der entstandene Feststoff wurde abfiltriert, zweimal mit Wasser gewaschen und im Vakuum getrocknet. Der Feststoff wurde dann in Dichlormethan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 9: 1, Biotage). Es wurden 74 mg (30% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.11 (s, 1H), 8.39 (s, 1H), 8.28 (t, 1H), 8.20 (s, 1H), 7.92 (dd, 1H), 7.85 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.49 (m, 3H), 3.89 (s, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.32 min, m/z = 527/529 [M+H]+. Beispiel 68A
Methyl-4-({ [6-brom-2-(4-fluorphenyl)-3-methylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Zu einer Lösung von 200 mg (0.49 mmol) der Verbindung aus Beispiel 26A und 82 mg (0.49 mmol) Methyl-4-amino-3-fluorbenzoat in 2 ml DMF unter Argon wurden 0.73 ml (0.73 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure- Lösung versetzt, mit Wasser verdünnt und zweimal mit jeweils 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit 40 ml gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlor - methan aufgenommen und mittels Säulenchromatographie gereinigt (50 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 4: 1, Biotage). Es wurden 193 mg (77% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.08 (s, 1H), 8.27 (t, 1H), 8.05 (d, 1H), 8.00-7.97 (m, 1H), 7.96-7.88 (m, 2H), 7.85 (dd, 1H), 7.73-7.65 (m, 2H), 7.38 (t, 2H), 3.89 (s, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.25 min, m/z = 511/513 [M+H]+. Beispiel 69A
Methyl-4-({ [6-brom-2-(3-fluorphenyl)-3-methylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Zu einer Lösung von 170 mg (0.41 mmol) der Verbindung aus Beispiel 28A und 70 mg (0.41 mmol) Methyl-4-amino-3-fluorbenzoat in 2 ml DMF unter Argon wurden 0.62 ml (0.62 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure- Lösung versetzt, mit Wasser verdünnt und zweimal mit jeweils 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Säulenchromatographie gereinigt (25 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 9: 1, Biotage). Es wurden 108 mg (51% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.09 (s, 1H), 8.27 (t, 1H), 8.06 (d, 1H), 8.01-7.90 (m 3H), 7.85 (dd, 1H), 7.66-7.55 (m, 1H), 7.51-7.44 (m, 2H), 7.41-7.33 (m, 1H), 3.89 (s, 3H), 2.44 (s 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.25 min, m/z = 511/513 [M+H]+.
Beispiel 70A
Methyl-4-({ [6-brom-2-(2-fluorphenyl)-3-methylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Zu einer Lösung von 200 mg (0.49 mmol) der Verbindung aus Beispiel 30A und 82 mg (0.49 mmol) Methyl-4-amino-3-fluorbenzoat in 2.4 ml DMF unter Argon wurden 0.73 ml (0.73 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch mit 4 ml einer 10%-igen wässrigen Zitronensäure- Lösung versetzt, mit Wasser verdünnt und zweimal mit jeweils 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mit 4 ml Acetonitril versetzt und das Gemisch im Ultraschallbad behandelt, woraufhin ein Feststoff ausfiel. Der Feststoff wurde abfiltriert, mit 4 ml Acetonitril gewaschen und im Vakuum getrocknet. Es wurden 164 mg (63% d. Th., Reinheit 96%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.18 (s, 1H), 8.25 (t, 1H), 8.09-8.04 (m, 1H), 8.02 (d, 1H), 7.99-7.94 (m, 1H), 7.91 (dd, 1H), 7.85 (dd, 1H), 7.66-7.57 (m, 1H), 7.57-7.50 (m, 1H), 7.45- 7.37 (m, 2H), 3.89 (s, 3H), 2.32 (s, 3H). LC/MS (Methode 3, ESIpos): Rt = 1.54 min, m/z = 511/513 [M+H]+.
Beispiel 71 A
Ethyl-4-({ [6-brom-3-methyl-2-(pyridin-4-yl)chinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Zu einer Suspension von 500 mg (1.46 mmol) der Verbindung aus Beispiel 31 A in 50 ml Dichlormethan wurden einige Tropfen DMF und dann 0.57 ml (6.56 mmol) Oxalylchlorid gegeben. Nach 15 min Rühren bei RT wurde das Lösungsmittel entfernt, und der Rückstand wurde in 15 ml Pyri- din aufgenommen. Anschließend wurden 321 mg (1.75mmol) Ethyl-4-amino-3-fluorbenzoat hinzugegeben, und das Gemisch wurde über Nacht bei 80°C gerührt. Nach Abkühlen auf RT wurde das Lösungsmittel im Vakuum entfernt und der Rückstand in 25 ml Dichlormethan aufgenommen. Die Lösung wurde mit Wasser gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel Dichlormethan/ Methanol 97:3). Es wurden 90 mg (12% d. Th.) der Titelverbindung erhalten, welche direkt in der Folgestufe eingesetzt wurden.
Beispiel 72A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(methoxymethyl)- sulfanyl]benzoat
Die Titelverbindung wurde als Nebenprodukt bei der Darstellung der Verbindung aus Beispiel 52A erhalten (Beschreibung siehe dort).
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.78 (s, 1H), 8.25 (dd, 2H), 8.04 (d, 1H), 7.95 (td, 2H), 7.83 (d, 1H), 7.66-7.61 (m, 2H), 7.60-7.51 (m, 3H), 5.10 (s, 2H), 3.88 (s, 3H), 3.36 (s, 3H), 2.50 (s, 3H, verdeckt).
LC/MS (Methode 21, ESIpos): Rt = 1.40 min, m/z = 551/553 [M+H]+.
Beispiel 73A
Methyl-4-amino-3-[(trifluormethyl)sulfanyl]benzoat
Zu einem Gemisch von 400 mg (1.10 mmol) Dimethyl 3,3'-disulfandiylbis(4-aminobenzoat) [beschrieben in Org. Prep. Proc. Int. 2003, 35 (5), 520-524] in 10 ml DMF wurden 389 mg (3.29 mmol) Natriumhydroxymethansulfinat (Rongalit™) gegeben, und das Reaktionsgemisch wurde 15 min bei RT gerührt. Anschließend wurden 725 mg (2.20 mmol) 3,3-Dimethyl-l-(trifluor- methyl)-l,2-benziodoxol hinzugefügt. Nach 30 min Rühren bei RT wurde das Gemisch direkt mittels präparativer HPLC (Methode 5) aufgereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt und im Vakuum getrocknet. Der Rückstand wurde in Dichlormethan aufgenommen, die Lösung wurde erneut eingeengt und der Rückstand wurde abermals im Vakuum getrocknet. Es wurden 185 mg (67% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.94 (d, 1H), 7.77 (dd, 1H), 6.83 (d, 1H), 6.71 (s, 2H), 3.77 (s, 3H).
LC/MS (Methode 22, ESIpos): Rt = 2.16 min, m/z = 252 [M+H]+.
Beispiel 74A
Methyl-4-amino-3-[(trifluormethyl)sulfonyl]benzoat
Zu einem Gemisch von 200 mg (0.80 mmol) der Verbindung aus Beispiel 73A in 5 ml Acetonitril wurden bei 0°C 3.3 mg (0.016 mmol) Ruthenium(III)chlorid und 511 mg (2.39 mmol) Natrium- periodat gegeben. Anschließend wurden 5 ml auf 0°C vorgekühltes Wasser hinzugegeben, und das Gemisch wurde 15 min bei 0°C und dann über Nacht bei RT gerührt. Danach wurden weitere 3.3 mg (0.016 mmol) Ruthenium(III)chlorid und 511 mg (2.39 mmol) Natriumperiodat hinzugegeben, und das Gemisch wurde nochmals 2 h bei RT gerührt. Anschließend wurde das Gemisch mit Ethylacetat versetzt und einmal mit Wasser gewaschen. Die wässrige Phase wurde einmal mit Ethylacetat rückextrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 18) gereinigt. Die vereinigten produkt- haltigen Fraktionen wurden eingeengt und im Vakuum getrocknet. Der Rückstand wurde in Di- chlormethan aufgenommen, die Lösung wurde erneut eingeengt und der Rückstand wurde abermals im Vakuum getrocknet. Es wurden 50 mg (21 % d. Th., Reinheit 96%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.11 (d, 1H), 7.98 (dd, 1H), 7.34 (br. s, 2H), 7.03 (d, 1H), 3.81 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.94 min, m/z = 284 [M+H]+. Beispiel 75A Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino }-3-[(trifluormethyl)- sulfonyl]benzoat
Zu einer Lösung von 62 mg (0.16 mmol) der Verbindung aus Beispiel 2A und 45 mg (0.16 mmol) der Verbindung aus Beispiel 74A in 0.65 ml DMF unter Argon wurden 0.24 ml (0.24 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF gegeben. Das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde das Gemisch mit 0.018 ml (0.24 mmol) TFA versetzt und direkt mittels prä- parativer HPLC (Methode 18) gereinigt. Es wurden 45 mg (46% d. Th., Reinheit 96%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.03 (s, 1H), 8.62 (dd, 1H), 8.57 (s, 1H), 8.41 (d, 1H), 8.09-8.02 (m, 2H), 7.95 (dd, 1H), 7.67-7.60 (m, 2H), 7.60-7.49 (m, 3H), 3.96 (s, 3H), 2.48 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.42 min, m/z = 607/609 [M+H]+.
Beispiel 76A
Methyl-4- { [(6-brom-3-methyl- 1 -oxido-2-phenylchinolin-4-yl)carbonyl] amino } -3- [(trifluormethyl)- sulfanyl]benzoat
Zu einer Lösung von 90 mg (0.16 mmol) der Verbindung aus Beispiel 53A in 5 ml Dichlormethan wurden 58 mg (0.24 mmol, Reinheit 70%) 3-Chlorperbenzoesäure gegeben, und das Gemisch wurde 3 h bei RT gerührt. Anschließend wurden weitere 39 mg (0.16 mmol, Reinheit 70%) 3-Chlor- perbenzoesäure hinzugegeben, und das Gemisch wurde erneut 2.5 h bei RT gerührt. Danach wurden weitere 20 mg (0.08 mmol, Reinheit 70%) 3-Chlorperbenzoesäure hinzugegeben, und das Gemisch wurde nochmals 1 h bei RT gerührt. Danach wurde das Gemisch mit 5 ml gesättigter wäss- riger Natriumhydrogencarbonat-Lösung versetzt, einige Minuten verrührt und dann mit 1 ml 10%- iger Natriumthiosulfat-Lösung versetzt. Nach ca. 80 h Stehenlassen bei RT wurde mit Dichlor - methan und Wasser verdünnt, und nach Phasentrennung wurde die wässrige Phase einmal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 5) gereinigt. Nach Trocknen im Vakuum wurden 48 mg (52% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.28 (s, IH), 8.52 (d, IH), 8.34 (s, IH), 8.26 (dd, IH), 8.22 (d, IH), 8.01 (dd, IH), 7.98 (d, IH), 7.62-7.56 (m, 2H), 7.56-7.50 (m, IH), 7.44 (d, 2H), 3.92 (s, 3H), 2.19 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.21 min, m/z = 591/593 [M+H]+.
Beispiel 77A 6-Brom-5-chlor-3-methyl-2-phenylchinolin-4-carbonsäure
2.50 g (9.60 mmol) 5-Brom-4-chlor-li7-indol-2,3-dion wurden in 21.6 ml Essigsäure vorgelegt und mit 1.29 g (9.60 mmol) 1-Phenylpropan-l-on versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 7.2 ml konzentrierte Salzsäure hinzugegeben, und das Ge- misch wurde 28 h bei 105°C gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 100 ml 1 M Salzsäure gegeben, und der ausgefallene Feststoff wurde abfiltriert. Der Feststoff wurde zweimal mit Wasser gewaschen, in Ethylacetat suspendiert und die Suspension dreimal mit 75 ml 1 M Natronlauge extrahiert. Die vereinigten Natronlauge-Phasen wurden dann mit konzentrierter Salzsäure auf pH 4 gestellt. Der ausgefallene Feststoff wurde abfiltriert, zweimal mit Was- ser gewaschen und im Vakuum unter Erhalt einer ersten Zwischencharge der Zielverbindung getrocknet. Die zuvor erhaltene Ethylacetat-Phase wurde zweimal mit 100 ml Wasser extrahiert, die beiden wässrigen Phasen wurden mit konzentrierter Salzsäure auf pH 1 gestellt und zweimal mit 100 ml Ethylacetat extrahiert. Die vereinigten Ethylacetat-Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde unter Erhalt einer zweiten Zwischencharge der Zielverbindung getrocknet. Beide Zwischenchargen wurden sodann vereinigt, erneut in 100 ml 1 M Natronlauge aufgenommen und zweimal mit 50 ml Ethylacetat extrahiert. Die wässrige Phase wurde mit konzentrierter Salzsäure auf pH 4 gestellt, und der entstandene Feststoff wurde abfiltriert, zweimal mit 10 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden so 2.23 g (53% d. Th., Reinheit 86%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.17 (br. s, 1H), 8.11 (d, 1H), 7.98 (d, 1H), 7.66-7.59 (m, 2H), 7.58-7.50 (m, 3H), 2.39 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.97 min, m/z = 376/378 [M+H]+.
Beispiel 78A
(6-Brom-5-chlor-3-methyl-2-phenylchinolin-4-yl)(lii-imidazol-l-yl)methanon
Zu einer Lösung von 2.20 g (5.02 mmol, Reinheit 86%) der Verbindung aus Beispiel 77A in 12 ml DMF wurden bei RT 1.42 g (8.74 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 6 h bei 120°C Badtemperatur gerührt. Anschließend wurden weitere 552 mg (3.40 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde erneut 4 h bei 120°C Badtemperatur gerührt. Danach wurden nochmals 552 mg (3.40 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde weitere 5 h bei 140°C Badtemperatur gerührt. Danach wurde das Gemisch mit jeweils 100 ml Wasser und Ethylacetat versetzt, und nach Phasentrennung wurde die wässrige Phase einmal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden ein- mal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in Dichlormethan aufgenommen und mittels Säulenchromatographie gereinigt (100 g Kieselgel, Laufmittel Cyclohexan/Ethylacetat 7:3, Biotage). Es wurden 362 mg (17% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.5-7.5 (br. m, 2H), 8.18 (d, 1H), 8.09 (d, 1H), 7.71- 7.66 (m, 2H), 7.59-7.49 (m, 3H), 7.27 (br. s, 1H), 2.25 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.25 min, m/z = 426/428 [M+H]+.
Beispiel 79A
Methyl-4-{ [(6-brom-5-chlor-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 357 mg (0.84 mmol) der Verbindung aus Beispiel 78A und 156 mg (0.92 mmol) Methyl-4-amino-3-fluorbenzoat in 1.0 ml DMF unter Argon wurden 0.84 ml (0.84 mmol) einer 1 M Lösung von Kalium-feri. -butylat in THF gegeben. Das Gemisch wurde 2.5 h bei RT gerührt. Anschließend wurden erneut 0.84 ml (0.84 mmol) einer 1 M Lösung von Kalium-ieri.- butylat in THF hinzugegeben, und das Gemisch wurde über Nacht bei RT weiter gerührt. Danach wurde das Gemisch mit 30 ml 10%-iger wässriger Zitronensäure-Lösung und 30 ml Wasser versetzt. Der gebildete Niederschlag wurde abfiltriert, zweimal mit 10 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 289 mg einer Produktcharge erhalten, in der die Titelverbindung laut LC/MS-Analytik zu 4% Reinheit (3% d. Th.) enthalten war. Dieses Material wurde ohne weitere Aufreinigung in Folgereaktionen eingesetzt.
LC/MS (Methode 23, ESIpos): Rt = 4.16 min, m/z = 527/529 [M+H]+.
Beispiel 80A
Methyl-4-[( { 6-brom-3- [brom(difluor)methyl] -2-phenylchinolin-4-yl } carbonyl)amino] -3-fluor- benzoat
Zu einem Gemisch von 133 mg (0.25 mmol) der Verbindung aus Beispiel 64A in 4 ml Tetrachlormethan wurden 103 mg (0.58 mmol) /V-Bromsuccinimid und 2.1 mg (0.013 mmol) 2,2'-Azobis-2- methylpropannitril gegeben. Das Gemisch wurde 24 h mit einer UV -Lampe bei 300 W bestrahlt, wobei die Innentemperatur auf 80°-90°C anstieg. Nach dieser Zeit wurden erneut 103 mg (0.58 mmol) /V-Bromsuccinimid und 2.1 mg (0.013 mmol) 2,2'-Azobis-2-methylpropannitril hinzugegeben, und das Gemisch wurde weitere 48 h mit einer UV-Lampe bei 300 W und einer Innentemperatur von 80°-90°C bestrahlt. Anschließend wurden nochmals 103 mg (0.58 mmol) /V-Brom- succinimid und 2.1 mg (0.013 mmol) 2,2'-Azobis-2-methylpropannitril hinzugefügt, und das Ge- misch wurde erneut 24 h mit einer UV-Lampe bei 300 W und einer Innentemperatur von 80°-90°C bestrahlt. Danach wurde das Lösungsmittel entfernt und der Rückstand mittels präparativer HPLC (Methode 20) gereinigt. Es wurden 35 mg (18% d. Th., Reinheit 76%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.31 min, m/z = 607/609/611 [M+H]+. Beispiel 81 A
Methyl-4-({ [6-brom-2-phenyl-3-(trifluormethyl)chinolin-4-yl]carbonyl}amino)-3-fluorbenzoat
Eine Lösung von 32 mg (0.04 mmol, Reinheit 76%) der Verbindung aus Beispiel 80A in 1.6 ml Acetonitril unter Argon wurde mit 27 mg (0.21 mmol) Silber(I)fluorid versetzt und 30 min bei 80°C gerührt. Nach Abkühlen auf RT wurden die Festbestandteile abfiltriert, und das Filtrat wurde mittels präparativer HPLC (Methode 19) gereinigt. Es wurden 4.5 mg (18% d. Th., 90% Reinheit) der Titelverbindung erhalten.
LC/MS (Methode 23, ESIpos): Rt = 4.22 min, m/z = 547/549 [M+H]+. Beispiel 82A
Methyl-4-{ [(6-chlor-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 210 mg (0.71 mmol) 6-Chlor-3-methyl-2-phenylchinolin-4-carbonsäure in 5 ml DMF wurden bei RT 239 mg (1.41 mmol) Methyl-4-amino-3-fluorbenzoat, 402 mg (1.06 mmol) HATU und 182 mg (1.41 mmol) /V,/V-Diisopropylethylamin gegeben. Das Gemisch wurde
1 h bei 60°C gerührt. Nach dem Abkühlen auf RT wurde das Gemisch in 10%-ige wässrige Zitronensäure-Lösung eingetragen, und der gebildete Niederschlag wurde abfiltriert, dreimal mit Wasser gewaschen und im Vakuum getrocknet. Das so erhaltene Material wurde in 4.5 ml DMF suspendiert, bei RT mit 117 mg (0.69 mmol) Methyl-4-amino-3-fluorbenzoat und 0.94 ml (0.94 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF versetzt und 30 min bei RT gerührt. Danach wurde das Gemisch in 30 ml 10%-ige Zitronensäure-Lösung eingetragen, und es wurde zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einem Gemisch aus DMSO, Wasser und Acetonitril aufgenommen und nach Abfiltration einiger Schwebstoffe mittels präparativer HPLC (Methode 19) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden bis auf ein Restvolumen an wässriger Phase eingeengt, und der wässrige Rückstand wurde zweimal mit Ethylacetat extrahiert. Die vereinigten Ethylacetat-Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 78 mg (25% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.10 (s, 1H), 8.28 (t, 1H), 8.15-8.07 (m, 1H), 7.91 (d, 1H), 7.88-7.79 (m, 3H), 7.66-7.60 (m, 2H), 7.60-7.49 (m, 3H), 3.89 (s, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.19 min, m/z = 449 [M+H]+.
Beispiel 83A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(methoxy:
sulfonyl]benzoat
Zu einer Lösung von 134 mg (0.24 mmol) der Verbindung aus Beispiel 72A in 3.5 ml Dichlor - methan wurden 120 mg (0.49 mmol, Reinheit 70%) 3-Chlorperbenzoesäure gegeben. Das Gemisch
wurde 15 min bei RT gerührt. Anschließend wurde das Gemisch mit Dichlormethan verdünnt und mit gesättigter Natriumhydrogencarbonat-Lösung gewaschen. Die wässrige Phase wurde einmal mit Dichlormethan rückextrahiert, und die vereinigten organischen Phasen wurden über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 18) gereinigt. Nach Trocknen im Vakuum wurden 77 mg (54% d. Th., Reinheit 100%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.24 min, m/z = 583/585 [M+H]+.
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 10.63 (s, 1H), 8.51-8.46 (m, 2H), 8.43 (dd, 1H), 8.20 (d, 1H), 8.05 (d, 1H), 7.95 (dd, 1H), 7.67-7.62 (m, 2H), 7.59-7.49 (m, 3H), 5.03 (s, 2H), 3.94 (s, 3H), 3.41 (s, 3H).
Beispiel 84A
6-ieri. -Butyl-3-methyl-2-phenylchinolin-4-carbonsäure
5.00 g (24.60 mmol) 5-ieri. -Butyl-li7-indol-2,3-dion wurden in 50 ml Essigsäure vorgelegt und mit 3.30 g (24.60 mmol) 1-Phenyl-l-propanon versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 18 ml konzentrierte Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C gerührt. Nach dem Abkühlen auf RT wurde das Reaktionsgemisch auf 1 Liter 1 M Salzsäure gegeben und der ausgefallene Feststoff abfiltriert. Der Feststoff wurde mit Wasser gewaschen, an der Luft getrocknet und dann mit 50 ml Acetonitril verrührt. Der Feststoff wurde erneut abfiltriert und an der Luft und zuletzt im Vakuum getrocknet. Es wurden 4.85 g (61% d. Th., Reinheit 99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.09 (br. s, 1H), 7.99 (d, 1H), 7.92 (dd, 1H), 7.66 (d, 1H), 7.62-7.57 (m, 2H), 7.55-7.45 (m, 3H), 2.37 (s, 3H), 1.39 (s, 9H).
LC/MS (Methode 1, ESIpos): Rt = 0.69 min, m/z = 320 [M+H]+.
Beispiel 85A
Methyl-4-{ [(6-ter -butyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino }-3-fluorbenzoat
Zu einer Lösung von 200 mg (0.63 mmol) der Verbindung aus Beispiel 84A und 127 mg (0.75 mmol) Methyl-4-amino-3-fluorbenzoat in 5.0 ml DMF unter Argon wurden 357 mg (0.94 mmol) HATU und 162 mg (1.25 mmol) /V,/V-Diisopropylefhylamin gegeben. Das Gemisch wurde über Nacht bei 60°C gerührt. Anschließend wurden erneut 127 mg (0.51 mmol) Methyl-4-amino-3- fluorbenzoat hinzugegeben, und das Gemisch wurde weitere 7.5 h bei 60°C gerührt und anschließend ca. 80 h bei RT stehen gelassen. Danach wurden 0.94 ml (0.94 mmol) einer 1 M Lösung von Kalium-ieri. -butylat in THF hinzugegeben, und das Gemisch wurde nach einigem Rühren eine weitere Nacht bei RT stehen gelassen. Es wurden erneut 0.94 ml (0.94 mmol) einer 1 M Lösung von Kalium-ieri. -butylat in THF hinzugegeben, und das Gemisch wurde zunächst 8 h bei RT gerührt und dann eine weitere Nacht bei RT stehen gelassen. Danach wurde das Gemisch in ca. 40 ml 5%-ige wässrige Zitronensäure -Lösung eingetragen, und es wurde zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 18) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden 86 mg (28% d. Th., Reinheit 98%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.07 (s, 1H), 8.10 (t, 1H), 8.05 (d, 1H), 7.96 (dd, 1H), 7.92 (dd, 1H), 7.87 (dd, 1H), 7.77 (d, 1H), 7.65-7.60 (m, 2H), 7.59-7.49 (m, 3H), 3.89 (s, 3H), 2.42 (s, 3H), 1.38 (s, 9H).
LC/MS (Methode 1, ESIpos): Rt = 1.28 min, m/z = 471 [M+H]
Beispiel 86A ieri.-Butyl-[2-nitro-5-(pentafluor- 6-sulfanyl)phenyl]acetat
24.90 g (99.94 mmol) l-Nitro-4-(pentafluor- 6-sulfanyl)benzol wurden in 200 ml wasserfreiem DMF unter Argon vorgelegt. Die Lösung wurde auf -30°C gekühlt und mit 15.05 g (14.30 ml, 99.94 mmol) ieri.-Butyl-chloracetat versetzt. Dann wurde eine Lösung von 44.86 g (339.74 mmol) Kalium-ieri. -butylat in 400 ml wasserfreiem DMF langsam hinzugetropft. Es bildete sich eine tiefblaue Lösung. Das Reaktionsgemisch wurde über Nacht gerührt, wobei es sich langsam auf Raumtemperatur erwärmte. Danach wurde die Reaktionslösung unter Rühren vorsichtig auf ein ieri.-Butylmethy lether/W asser-Gemisch gegeben. Die Phasen wurden getrennt, und die organische Phase wurde einmal mit Wasser gewaschen und über Natriumsulfat getrocknet. Das Lösungsmittel wurde entfernt, und der Rückstand wurde mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel Cyclohexan/Ethylacetat 20: 1). Nach Entfernen des Lösungsmittels wurde der Rückstand in Pentan verrührt. Der Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 13.67 g (38% d. Th., Reinheit >99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 8.28 (d, 1H), 8.26 (d, 1H), 8.14 (dd, 1H), 4.12 (s, 2H), 1.39 (s, 9H).
LC/MS (Methode 22, ESIneg): Rt = 2.64 min, m/z = 362 [M-H]".
Beispiel 87A ieri.-Butyl-[2-amino-5-(pentafluor-
6-sulfanyl)phenyl]acetat
13.67 g (37.63 mmol) der Verbindung aus Beispiel 86A wurden zusammen mit 1.98 g (1.88 mmol) Palladium auf Aktivkohle (10%) in 500 ml Ethanol vorgelegt. Das Reaktionsgemisch wurde 3 h bei RT unter Normaldruck hydriert. Nach Beendigung der Reaktion wurde der Palladium-Kata- lysator über Celite abfiltriert und das Filtrat eingeengt. Der Rückstand wurde mit einem Pentan/ ieri.-Butylmethy lether-Gemisch verrührt, und der Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 10 g (80% d. Th., Reinheit >99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 7.49-7.36 (m, 2H), 6.68 (d, 1H), 5.75 (s, 2H), 3.50 (s, 2H), 1.40 (s, 9H). LC/MS (Methode 22, ESIpos): Rt = 2.53 min, m/z = 278 [M-C4H8+H]+. Beispiel 88A
3-Methyl-6-(pentafluor- 6-sulfanyl)-2-phenylchinolin-4-carbonsäure
4.79 g (14.38 mmol) der Verbindung aus Beispiel 87 A wurden in 71.4 ml Essigsäure vorgelegt. 2.13 g (1.94 ml, 14.38 mmol) l-Phenylpropan-l,2-dion wurden hinzugegeben, und das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Dann wurden 23.8 ml konz. Salzsäure hinzugegeben, und es wurde über Nacht bei 105°C weiter gerührt. Das Reaktionsgemisch wurde danach unter Rühren vorsichtig auf ein Ethylacetat/W asser-Gemisch gegeben. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wur- den über Natriumsulfat getrocknet und das Lösungsmittel entfernt. Der Rückstand wurde mit einem Pentan/ieri. -Butylmethy lether-Gemisch verrührt. Der Feststoff wurde abfiltriert und dann
mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel Ethylacetat/Methanol 10: 1). Nach Entfernen des Lösungsmittels wurde der Feststoff im Vakuum getrocknet. Es wurden 980 mg (17% d. Th., Reinheit 99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 15.02 (br. s, 1H), 8.31 (d, 1H), 8.26-8.14 (m, 2H), 7.67-7.58 (m, 2H), 7.58-7.48 (m, 3H), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.00 min, m/z = 390 [M+H]+.
Beispiel 89A l f-Imidazol-l-yl[3-methyl-6-(pentafluor- 6-sulfanyl)-2-phenylchinolin-4-yl]methanon
838 mg (2.15 mmol) der Verbindung aus Beispiel 88A wurden in 5 ml DMF gelöst und bei RT mit 698 mg (4.30 mmol) /V,/V'-Carbonyldiimidazol versetzt. Das Reaktionsgemisch wurde über Nacht bei 60°C gerührt. Danach wurden weitere 174 mg (1.08 mmol) /V,/V'-Carbonyldiimidazol hinzugegeben, und das Gemisch wurde eine weitere Stunde bei 60°C gerührt. Es wurden nochmals 349 mg (2.15 mmol) /V,/V'-Carbonyldiimidazol hinzugefügt und erneut 3 h bei 60°C gerührt. Das Reak- tionsgemisch wurde dann mit Wasser und ieri.-Butylmethy lether versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit feri.-Butylmethylether extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet. Nach Entfernen des Lösungsmittels wurde der Rückstand über Nacht im Vakuum getrocknet. Es wurden 520 mg (52% d. Th., Reinheit 95%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d
6): δ [ppm] = 8.90-7.37 (br. m, 2H), 8.36 (d, 1H), 8.28 (dd, 1H), 8.04 (br. s, 1H), 7.78-7.67 (m, 2H), 7.63-7.49 (m, 3H), 7.23 (br. s, 1H), 2.28 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.18 min, m/z = 440 [M+H]+.
Beispiel 90A
Methyl-3-fluor-4-({ [3-methyl-6-(pentafluor- 6-sulfanyl)-2-phenylchinolin-4-yl]carbonyl} benzoat
100 mg (0.23 mmol) der Verbindung aus Beispiel 89A und 39 mg (0.23 mmol) Mefhyl-4-amino-3- fluorbenzoat wurden in 2 ml DMF gelöst. 0.57 ml (0.57 mmol) einer 1 M Lösung von Kalium- ieri.-butylat in THF wurden hinzugegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde mit Ethylacetat und Wasser versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit Wasser gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einem Pentan/ieri.-Butylmethy lether-Gemisch verrührt. Der Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 117 mg (87% d. Th., Reinheit 91%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.17 (s, 1H), 8.35-8.22 (m, 3H), 8.15 (t, 1H), 7.96- 7.84 (m, 2H), 7.71-7.63 (m, 2H), 7.63-7.44 (m, 3H), 3.89 (s, 3H), 2.48 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.33 min, m/z = 541 [M+H]+.
Beispiel 91 A
Methyl-6-iod-3-methyl-2-phenylchinolin-4-carboxylat
22.4 g (57.5 mmol) der Verbindung aus Beispiel 5A wurden zusammen mit 28.1 g (86.23 mmol) Cäsiumcarbonat in 224 ml Acetonitril unter Argon vorgelegt. Es wurden 3.6 ml (57.5 mmol) Iod- methan bei RT hinzugegeben. Das Reaktionsgemisch wurde auf 40°C erwärmt und 1 h gerührt. Anschließend wurden weitere 3.6 ml (57.5 mmol) Iodmethan hinzugegeben, und es wurde nochmals 2 h bei 40°C gerührt. Das Reaktionsgemisch wurde dann auf RT abgekühlt und mit Ethyl- acetat und Wasser versetzt. Die Phasen wurden getrennt, und die organische Phase wurde einmal mit gesättigter Natriumcarbonat-Lösung gewaschen. Es bildete sich ein Niederschlag, welcher über Kieselgur abfiltriert wurde. Das Filtrat wurde über Natriumsulfat getrocknet, filtriert und einge- engt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel Cyclo- hexan/Ethylacetat 10: 1). Nach Trocknen im Vakuum wurden 12.7 g (55% d. Th., Reinheit 96%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.12 (d, 1H), 8.06 (dd, 1H), 7.84 (d, 1H), 7.64-7.59 (m, 2H), 7.57-7.47 (m, 3H), 4.07 (s, 3H), 2.35 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.26 min, m/z = 404 [M+H]+.
Beispiel 92A
Methyl-6-formyl-3-methyl-2-phenylchinolin-4-carboxylat
5.0 g (12.4 mmol) der Verbindung aus Beispiel 91A wurden in 98 ml wasserfreiem THF unter Ar- gon gelöst und auf -50°C abgekühlt. Anschließend wurden langsam nacheinander 35.4 ml (37.2 mmol) einer 1.05 M Lösung von Isopropylmagnesiumchlorid/Lithiumchlorid-Komplex in THF und 3.2 ml (37.2 mmol) 1,4-Dioxan zugetropft. Das Reaktionsgemisch wurde 1 h bei -50°C gerührt
und anschließend auf -78°C abgekühlt. Es wurden dann 9.5 ml (124 mmol) absolutes DMF zugetropft. Das Reaktionsgemisch wurde über Nacht unter Rühren auf RT kommen gelassen und dann mit Ethylacetat und Wasser versetzt. Die Phasen wurden getrennt, und die organische Phase wurde einmal mit Wasser gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Beim Ver- 5 such, den Rückstand mittels Säulenchromatographie zu reinigen (Kieselgel, Laufmittel Cyclohexan/Ethylacetat 6: 1), fiel das Produkt auf der Säule aus. Die chromatographische Reinigung wurde daraufhin abgebrochen und das Silicagel mit Ethylacetat verrührt. Nach Filtration wurde das Filtrat eingeengt. Der Rückstand wurde in Methanol verrührt und der Feststoff abfiltriert und im Vakuum getrocknet. Es wurden 2.29 g (59% d. Th., Reinheit 98%) der Titelverbindung erhallt) ten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.24 (s, 1H), 8.44 (d, 1H), 8.24-8.12 (m, 2H), 7.71- 7.60 (m, 2H), 7.59-7.47 (m, 3H), 4.12 (s, 3H), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.06 min, m/z = 306 [M+H]+.
Beispiel 93A
15 Methyl-6-(difluormethyl)-3-methyl-2-phenylchinolin-4-carboxylat
1.0 g (3.2 mmol) der Verbindung aus Beispiel 92A wurden in 40 ml Dichlormethan gelöst. Das Gemisch wurde auf -78°C abgekühlt und langsam mit 1.4 g (7.86 mmol, Reinheit 90%) N-Ethyl-N- (trifluor- 4-sulfanyl)ethanamin (DAST) versetzt. Das Reaktionsgemisch wurde über Nacht gerührt,
20 wobei es sich auf RT erwärmte, und dann mit gesättigter wässriger Natriumhydrogencarbonat- Lösung versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (Kieselgel, Laufmittel Cyclohexan/Ethylacetat 5: 1). Nach Entfernen des Lösungsmittels wurde der Rückstand
25 im Vakuum getrocknet. Es wurden 737 mg (69% d. Th., Reinheit >99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 8.21 (d, 1H), 8.02 (br. s, 1H), 7.95 (d, 1H), 7.69-7.61 (m, 2H), 7.59-7.48 (m, 3H), 7.28 (t, 1H), 4.09 (s, 3H), 2.38 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 328 [M+H]+.
Beispiel 94A 6-(Difluormethyl)-3-methyl-2-phenylchinolin-4-carbonsäure
100 mg (0.31 mmol) der Verbindung aus Beispiel 93A wurden in 5 ml eines THF/Methanol- Gemisches (5: 1) gelöst und mit 1.53 ml (1.53 mmol) einer 1 M Lösung von Lithiumhydroxid in Wasser versetzt. Das Reaktionsgemisch wurde 7 h bei 50°C gerührt und dann auf RT abgekühlt und mit Ethylacetat und Wasser versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde mit 1 M Salzsäure auf pH 1-2 gestellt und dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Extrakte wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einem Pentan/ieri.-Butylmethy lether-Gemisch verrührt, und der Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 61 mg (96% d. Th., Reinheit 99%) der Titelverbin- dung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.37 (br. s, 1H), 8.20 (d, 1H), 8.02 (d, 1H), 7.92 (dd, 1H), 7.67-7.61 (m, 2H), 7.59-7.49 (m, 3H), 7.33 (t, 1H), 2.42 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.72 min, m/z = 314 [M+H]+.
Beispiel 95A Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2,3-difluorbenzoat
150 mg (0.38 mmol) der Verbindung aus Beispiel 2A und 72 mg (0.38 mmol) Methyl-4-amino-2,3- difluorbenzoat wurden in 3.4 ml DMF gelöst. 0.96 ml (0.96 mmol) einer 1 M Lösung von Kalium- ieri.-butylat in THF wurden hinzugegeben, und das Gemisch wurde 1 h bei RT gerührt. Danach wurde mit Ethylacetat und Wasser versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit Wasser gewaschen, über Natriumsulfat getrocknet und das Lösungsmittel wurde am Rotationsverdampfer entfernt. Der Rückstand wurde im Vakuum getrocknet. Es wurden 110 mg (50% d. Th., Reinheit 89%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.29 (s, 1H), 8.12-8.00 (m, 2H), 7.98 (d, 1H), 7.93 (dd, 1H), 7.87-7.77 (m, 1H), 7.66-7.60 (m, 2H), 7.60-7.48 (m, 3H), 3.90 (s, 3H), 2.43 (s, 3H).
LC/MS (Methode 23, ESIpos): Rt = 4.00 min, m/z = 511/513 [M+H]+.
Beispiel 96A
Methyl-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2,5-difluorbenzoat
150 mg (0.38 mmol) der Verbindung aus Beispiel 2A und 72 mg (0.38 mmol) Mefhyl-4-amino-2,5- difluorbenzoat wurden in 3.4 ml DMF gelöst. 0.96 ml (0.96 mmol) einer 1 M Lösung von Kalium- ieri.-butylat in THF wurden hinzugegeben, und das Gemisch wurde 1 h bei RT gerührt. Danach wurde mit Ethylacetat und Wasser versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden mit Wasser gewaschen, über Natriumsulfat getrocknet und das Lösungsmittel wurde entfernt. Der Rückstand wurde mittels Säulenchromatographie gereinigt (25 g Kieselgel, Laufmittel Cyclohexan/Ethyl- acetat 4: 1, Biotage Isolera™ One). Nach Entfernen des Lösungsmittels wurde der Rückstand im Vakuum getrocknet. Es wurden 102 mg (44% d. Th., Reinheit 85%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.27 (s, 1H), 8.31 (dd, 1H), 8.04 (d, 1H), 8.01 (d, 1H), 7.93 (dd, 1H), 7.81 (dd, 1H), 7.65-7.59 (m, 2H), 7.59-7.49 (m, 3H), 3.88 (s, 3H), 2.41 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.23 min, m/z = 511/513 [M+H]+.
Beispiel 97A 6-Brom-3-fluor-2-phenylchinolin-4-carbonsäure
O^OH
1.75 g (6.97 mmol, Reinheit 90%) 5-Brom-li7-indol-2,3-dion wurden in 15 ml Essigsäure vorgelegt und mit 0.96 g (6.97 mmol) 2-Fluor-l-phenylethanon versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 5 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 115°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktions- gemisch auf 100 ml 1 M Salzsäure gegeben. Der ausgefallene Feststoff wurde abfiltriert, zweimal mit 10 ml Wasser gewaschen und im Vakuum getrocknet. Der Rückstand wurde mittels präparativer HPLC (Methode 18) gereinigt. Es wurden 501 mg (20% d. Th., Reinheit 98%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 14.66 (br. s, 1H), 8.21 (d, 1H), 8.11 (d, 1H), 8.04-7.96 (m, 3H), 7.62-7.56 (m, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.98 min, m/z = 346/348 [M+H]+.
Beispiel 98A
Methyl-4-{ [(6-brom-3-fluor-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 200 mg (0.58 mmol) der Verbindung aus Beispiel 97A in 4.1 ml DMF wurden bei RT 195 mg (1.16 mmol) Methyl-4-amino-3-fluorbenzoat, 330 mg (0.87 mmol) HATU und 149 mg (1.16 mmol) /V,/V-Diisopropylethylamin gegeben. Das Gemisch wurde zunächst 4.5 h bei 60°C gerührt und dann zwei Tage bei RT stehen gelassen. Danach wurde das Gemisch in 10%-ige wässrige Zitronensäure-Lösung eingetragen und zweimal mit 30 ml Ethylacetat extrahiert. Die ver- einigten organischen Phasen wurden einmal mit 60 ml gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Der Rückstand wurde in 3 ml DMSO und 3 ml Acetonitril aufgenommen und mittels präparativer HPLC (Methode 18) gereinigt. Es wurden 51 mg (16% d. Th., Reinheit 91%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d
6): δ [ppm] = 11.25 (s, 1H), 8.39 (t, 1H), 8.17-8.12 (m, 2H), 8.09- 8.03 (m, 2H), 8.00 (dd, 1H), 7.92 (dd, 1H), 7.85 (dd, 1H), 7.65-7.57 (m, 3H), 3.89 (s, 3H).
LC/MS (Methode 23, ESIpos): Rt = 4.30 min, m/z = 497/499 [M+H]+.
Beispiel 99A 3-Chlor-6-iod-2-phenylchinolin-4-carbonsäure
10.00 g (36.63 mmol) 5-Iod-li7-indol-2,3-dion wurden in 100 ml Essigsäure vorgelegt und mit 5.66 g (36.63 mmol) 2-Chlor-l-phenylethanon versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 5 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben. Der ausgefallene Feststoff wurde abfiltriert, zweimal mit Wasser gewaschen und im Vakuum getrocknet. Der Feststoff wurde danach in 50 ml Acetonitril verrührt, erneut abfiltriert und wieder im Vakuum getrocknet. Es wurden 4.45 g (16% d. Th., Reinheit 54%) der Titelverbindung erhalten. LC/MS (Methode 1, ESIpos): Rt = 0.94 min, m/z = 409 [M+H]+.
Beispiel 100A
(3-Chlor-6-iod-2-phenylchinolin-4-yl)( 1 ii-imidazol- 1 -yl)methanon
Zu einer Lösung von 700 mg (0.92 mmol, Reinheit 54%) der Verbindung aus Beispiel 99A in 6 ml DMF wurden bei RT 165 mg (1.02 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 2 h bei 60°C gerührt. Anschließend wurden weitere 165 mg (1.02 mmol) /V,/V'-Carbonyl- diimidazol hinzugegeben, und das Gemisch wurde nochmals 4 h bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch unter Rühren in 50 ml Wasser eingetragen. Der entstandene Feststoff wurde abfiltriert, zweimal mit jeweils 2 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 699 mg (">100%" d. Th., noch Lösungsmittel-haltig, Reinheit 81% laut LC/MS) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.17 min, m/z = 460 [M+H]+. Beispiel 101A
Methyl-4-{ [(3-chlor-6-iod-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 110 mg (0.23 mmol) der Verbindung aus Beispiel 100A und 61 mg (0.36 mmol) Methyl-4-amino-3-fluorbenzoat in 2 ml DMF wurden 0.36 ml (0.36 mmol) einer 1 M Lösung von Kalium-ieri. -butylat in THF gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch direkt mittels präparativer HPLC (Methode 24) gereinigt. Es wurden 46 mg (31% d. Th., Reinheit 90%) der Titelverbindung erhalten.
LC/MS (Methode 23, ESIpos): Rt = 4.26 min, m/z = 561 [M+H]+.
Beispiel 102A 6-Brom-3-chlor-2-phenylchinolin-4-carbonsäure
10.00 g (44.24 mmol) 5-Brom-li7-indol-2,3-dion wurden in 120 ml Essigsäure vorgelegt und mit 6.84 g (44.24 mmol) 2-Chlor-l-phenylethanon versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 5 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 105°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben. Der ausgefallene Feststoff wurde abfiltriert, zweimal mit Wasser gewaschen und im Vakuum getrocknet. Der Feststoff wurde danach in 50 ml Acetonitril verrührt, erneut abfiltriert und wieder im Vakuum getrocknet. Es wurden 5.60 g (29% d. Th., Reinheit 82%) der Titelverbindung erhalten. LC/MS (Methode 1, ESIpos): Rt = 0.88 min, m/z = 362/364 [M+H]+.
Beispiel 103A
(6-Brom-3-chlor-2-phenylchinolin-4-yl)( 1 ii-imidazol- 1 -yl)methanon
Zu einer Lösung von 500 mg (1.13 mmol, Reinheit 82%) der Verbindung aus Beispiel 102A in 5 ml DMF wurden bei RT 202 mg (1.24 mmol) /V,/V'-Carbonyldiimidazol gegeben, und das Gemisch wurde 2 h bei 60°C gerührt. Anschließend wurden weitere 202 mg (1.24 mmol) Ν,Ν'- Carbonyldiimidazol hinzugegeben, und das Gemisch wurde nochmals 4 h bei 60°C gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 50 ml wässriger Zitronensäure-Lösung versetzt. Der entstandene Feststoff wurde abfiltriert, zweimal mit jeweils 2 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 553 mg (95% d. Th., Reinheit 80%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.14 min, m/z = 412/414 [M+H]+.
Beispiel 104A
Methyl-4-{ [(6-brom-3-chlor-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einem Gemisch von 80 mg (0.19 mmol) der Verbindung aus Beispiel 103A und 49 mg (0.29 mmol) Methyl-4-amino-3-fluorbenzoat in 2 ml DMF wurden 0.29 ml (0.29 mmol) einer 1 M Lösung von Kalium-ieri. -butylat in THF gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch direkt mittels präparativer HPLC (Methode 24) gereinigt. Es wurden 45 mg (38% d. Th., Reinheit 85%) der Titelverbindung erhalten.
LC/MS (Methode 23, ESIpos): Rt = 4.19 min, m/z = 513/515 [M+H]+. Beispiel 105A
6-Brom-3-cyclopropyl-2-phenylchinolin-4-carbonsäure
Methode A:
1.75 g (6.97 mmol, Reinheit 90%) 5-Brom-li7-indol-2,3-dion wurden in 15 ml Essigsäure vorge- legt und mit 1.12 g (6.97 mmol) 2-Cyclopropyl-l-phenylethanon [Darstellung beschrieben in WO 2009/143049-Al, S. 182, Compound 99 A] versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C
gerührt. Anschließend wurden 5 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde 2.5 h bei 110°C und danach über Nacht bei RT weiter gerührt. Das Reaktionsgemisch wurde dann auf 100 ml 1 M Salzsäure gegeben. Der ausgefallene Feststoff wurde abfiltriert, zweimal mit 10 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 2.23 g eines Rohprodukts erhalten. 200 mg dieses Rohprodukts wurden mittels präparativer HPLC (Methode 4) gereinigt. Daraus wurden 43 mg (1.5% d. Th. bezogen auf 6.97 mmol Edukt, Reinheit 93%) der Titelverbindung erhalten.
Methode B:
Zu einer Lösung von 2.03 g (8.12 mmol, Reinheit 90%) 5-Brom-li7-indol-2,3-dion in 20 ml Ethanol wurden bei RT 1.95 g (12.18 mmol) 2-Cyclopropyl-l-phenylethanon [Darstellung beschrieben in WO 2009/143049-A1, S. 182, Compound 99 A] und 2.05 g (36.56 mmol) Kaliumhydroxid gegeben. Das Reaktionsgemisch wurde 1 h bei 100°C Badtemperatur gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 300 ml Wasser versetzt und mit konz. Salzsäure auf pH 2 eingestellt. Es wurde zweimal mit je 20 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einem Gemisch aus 30 ml DMSO und 10 ml Acetonitril suspendiert, und der verbliebene Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden so 107 mg (4% d. Th., Reinheit 100%) einer ersten Charge der Titelverbindung gewonnen. Das Filtrat wurde eingeengt und der Rückstand mittels präparativer HPLC (Methode 3) gereinigt, wodurch 750 mg (25% d. Th., Reinheit 100%) einer zweiten Charge der Titelverbindung erhalten wurden.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 14.22 (br. s, 1H), 8.01 (d, 1H), 7.97 (d, 1H), 7.93 (dd, 1H), 7.76-7.71 (m, 2H), 7.54-7.46 (m, 3H), 2.38-2.28 (m, 1H), 0.76-0.66 (m, 2H), 0.33-0.25 (m, 2H).
LC/MS (Methode 1, ESIpos): Rt = 0.91 min, m/z = 368/370 [M+H]+. Beispiel 106A
Methyl-4-{ [(6-brom-3-cyclopropyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 400 mg (1.09 mmol) der Verbindung aus Beispiel 105 A in 6 ml DMF wurden bei RT 367 mg (2.17 mmol) Methyl-4-amino-3-fluorbenzoat, 620 mg (1.63 mmol) HATU und 281 mg (2.17 mmol) A^ -Diisopropylethylamin gegeben. Das Gemisch wurde 5 h bei 60°C ge- rührt. Danach wurden weitere 140 mg (1.09 mmol) /VyV-Diisopropylefhylamin hinzugegeben, und das Gemisch wurde nochmals 2 h bei 60°C gerührt. Nach dem Abkühlen auf RT wurden 2.17 ml (2.17 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF hinzugefügt, und das Gemisch wurde 18 h bei RT gerührt. Anschließend wurden erneut 2.17 ml (2.17 mmol) einer 1 M Lösung von Kalium-ieri. -butylat in THF hinzugegeben, und das Gemisch wurde nochmals 2 h bei RT ge- rührt. Danach wurde das Gemisch in 80 ml einer 10%-igen wässrigen Zitronensäure -Lösung eingetragen, und es wurde zweimal mit je 50 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einem Gemisch aus DMSO und Wasser aufgenommen und mittels präparativer HPLC (Methode 18) gereinigt. Es wurden 3.4 mg (0.6% d. Th., Reinheit 100%) einer ersten Charge der Titelverbindung sowie 33 mg (5% d. Th., Reinheit 77%) einer zweiten Charge der Titelverbindung erhalten.
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 11.04 (s, 1H), 8.31 (t, 1H), 8.07-8.03 (m, 2H), 7.96- 7.91 (m, 2H), 7.86 (dd, 1H), 7.76 (dd, 2H), 7.56-7.48 (m, 3H), 3.90 (s, 3H), 2.43-2.32 (m, 1H), 0.68 (d, 2H), 0.32 (d, 2H).
LC/MS (Methode 1, ESIpos): Rt = 1.31 min, m/z = 519/521 [M+H]+. Beispiel 107 A
6-Brom-3,8-dimethyl-2-phenylchinolin-4-carbonsäure
3.00 g (12.50 mmol) 5-Brom-7-methyl-li7-indol-2,3-dion wurden in 34 ml Essigsäure vorgelegt und mit 1.68 g (12.50 mmol) 1-Phenyl-l-propanon versetzt. Das Reaktionsgemisch wurde 5 min bei 75°C gerührt. Anschließend wurden 11 ml konz. Salzsäure hinzugegeben, und das Gemisch wurde über Nacht bei 115°C weiter gerührt. Nach Abkühlen auf RT wurde das Reaktionsgemisch auf 200 ml 1 M Salzsäure gegeben. Der ausgefallene Feststoff wurde abfiltriert, zweimal mit 10 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 3.02 g (64% d. Th., Reinheit 94%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 14.31 (br. s, 1H), 7.83 (s, 1H), 7.77-7.63 (m, 3H), 7.57- 7.49 (m, 3H), 2.70 (s, 3H), 2.41 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 356/358 [M+H]+.
Beispiel 108A
Methyl-4-{ [(6-brom-3,8-dimethyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Zu einer Lösung von 500 mg (1.32 mmol, Reinheit 94%) der Verbindung aus Beispiel 107A in 9 ml DMF wurden bei RT 448 mg (2.65 mmol) Methyl-4-amino-3-fluorbenzoat, 755 mg (1.98 mmol) HATU und 342 mg (2.65 mmol) /VyV-Diisopropylefhylamin gegeben. Das Gemisch wurde
2 h bei 60°C gerührt. Danach wurden weitere 171 mg (1.32 mmol) /V,/V-Diisopropylefhylamin hinzugegeben, und das Gemisch wurde nochmals 7 h bei 60°C gerührt. Nach dem Abkühlen auf RT wurden 2.65 ml (2.65 mmol) einer 1 M Lösung von Kalium-ieri.-butylat in THF hinzugefügt, und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurden erneut 2.65 ml (2.65 mmol) einer I M Lösung von Kalium-ieri.-butylat in THF hinzugegeben, und das Gemisch wurde für zwei Tage bei RT weiter gerührt. Danach wurde das Gemisch in 200 ml einer 10%-igen wässrigen Zitronensäure-Lösung eingetragen. Der entstandene Niederschlag wurde abfiltriert und im Vakuum getrocknet. Es wurden 651 mg (78% d. Th., Reinheit 80%) der Titelverbindung erhalten.
LC/MS (Methode 1, ESIpos): Rt = 1.35 min, m/z = 507/509 [M+H]+.
Ausführungsbeispiele : Beispiel 1
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}benzoesäure
177 mg (0.36 mmol) der Verbindung aus Beispiel 34A wurden zunächst mit 4.5 ml THF und dann mit einer Lösung von 13 mg (0.54 mmol) Lithiumhydroxid in 1.5 ml Wasser versetzt. Das Reaktionsgemisch wurde acht Tage bei RT gerührt und anschließend mit 2 M Salzsäure auf pH 1-2 eingestellt. Der ausgefallene Feststoff wurde abfiltriert, mit Wasser gewaschen und über Nacht im Vakuum bei 60°C getrocknet. Es wurden 109 mg (65% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.25 (s, 1H), 8.06 (d, 1H), 8.01-7.99 (m, 2H), 7.95 (dd, 1H), 7.92-7.87 (m, 3H), 7.65-7.63 (m, 2H), 7.58-7.51 (m, 3H), 2.41 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 461/463 [M+H]+.
Beispiel 2
6-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}nicotinsäure-Imidazolsalz
100 mg (0.26 mmol) der Verbindung aus Beispiel 2A wurden nacheinander mit 2 ml THF, 47 mg (0.31 mmol) Methyl-6-aminonicotinat und 22 mg (0.56 mmol) Natriumhydrid (60% in Mineralöl) versetzt. Das Reaktionsgemisch wurde über Nacht bei RT gerührt und danach ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 8) gereinigt. Es wurden 15 mg (11% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d
6): δ [ppm] = 12.54 (br. s, IH), 11.65 (s, IH), 8.85 (d, IH), 8.43 (d, IH), 8.36 (dd, IH), 8.03 (d, IH), 7.93-7.90 (m, 2H), 7.64-7.61 (m, 4H), 7.58-7.49 (m, 4H), 7.01 (s, IH), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.07 min, m/z = 462/464 [M+H]
Beispiel 3 5-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}pyrazin-2-carbonsäure
100 mg (0.26 mmol) der Verbindung aus Beispiel 2A und 43 mg (0.26 mmol) Efhyl-5-amino- pyrazin-2-carboxylat wurden in 2 ml DMF gelöst. Zu der Lösung wurden 72 mg (0.64 mmol) Kalium-ieri. -butylat portionsweise hinzugegeben. Das Reaktionsgemisch wurde über Nacht bei RT gerührt und danach mit 1.3 ml (1.3 mmol) 1 M Natronlauge versetzt. Nach 1 h Rühren bei 80°C und Abkühlen auf RT wurde mit 1 M Salzsäure auf pH 1-2 eingestellt. Das Gemisch wurde mit Ethylacetat extrahiert, und die organische Phase wurde abgetrennt und mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat und Entfernen des Lösungsmittels am Rotationsverdampfer wurde der Rückstand in wenig DMF aufgenom- men und mittels präparativer HPLC (Methode 6) gereinigt. Nach Entfernen des Lösungsmittel- Wasser-Gemisches wurde der Rückstand in wenig Acetonitril aufgenommen, mit Wasser versetzt und dann lyophilisiert. Da noch Lösungsmittelrückstände vorhanden waren, wurde das Lyophilisat in Dichlormethan und Ethylacetat umgelöst, wieder mit Wasser versetzt und erneut lyophilisiert. Das resultierende Lyophilisat wurde in Dichlormethan und ieri.-Butanol nochmals umgelöst und nach Versetzen mit Wasser wieder lyophilisiert. Das so erhaltene Lyophilisat wurde insgesamt 9 h im Hochvakuum bei 100°C getrocknet. Es wurden auf diese Weise 39 mg (29% d. Th., Reinheit 90%) der Titelverbindung erhalten.
Ή-NMR (600 MHz, DMSO-de): δ [ppm] = 13.54 (br. s, 1H), 12.03 (s, 1H), 9.71 (s, 1H), 9.01 (s, 1H), 8.10 (d, 1H), 8.04 (d, 1H), 7.93 (dd, 1H), 7.66-7.60 (m, 2H), 7.58-7.50 (m, 3H), 2.41 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 0.96 min, m/z = 463/465 [M+H]+.
Beispiel 4
5-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino }pyrimidin-2-carbonsäure
Eine Lösung von 23 mg (0.05 mmol) der Verbindung aus Beispiel 36A in 0.7 ml eines THF/ Methanol-Gemisches (5: 1) wurde mit 0.24 ml (0.24 mmol) 1 M Natronlauge versetzt und 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 15) gereinigt. Das Acetonitril-Wasser-Gemisch wurde am Rotationsverdampfer entfernt und der Rückstand über Nacht im Vakuum getrocknet. Es wurden 10 mg (22% d. Th., Reinheit 98%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, CD3OD): δ [ppm] = 9.41 (br. s, 2H), 8.12 (s, 1H), 8.05 (d, 1H), 7.95 (d, 1H), 7.71-7.51 (m, 5H), 2.50 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 0.90 min, m/z = 463/465 [M+H]+.
Beispiel 5
5-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-6-methylpyridin-2-carbonsäure
250 mg (0.64 mmol) der Verbindung aus Beispiel 2A wurden mit 10 ml THF, 127 mg (0.77 mmol) Methyl-5-amino-6-methylpyridin-2-carboxylat und 56 mg (1.40 mmol) Natriumhydrid (60% in Mineralöl) versetzt. Das Reaktionsgemisch wurde zwei Tage bei RT gerührt, dann mit Wasser versetzt, mit Salzsäure angesäuert und mit Ethylacetat extrahiert. Die organische Phase wurde abge- trennt und das Lösungsmittel am Rotationsverdampfer entfernt. Der Rückstand wurde mittels prä- parativer HPLC (Methode 11) gereinigt. Es wurden 41 mg (13% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.76 (s, 1H), 8.43 (d, 1H), 8.07-8.02 (m, 3H), 7.97-7.94 (m, 1H), 7.66-7.64 (m, 2H), 7.59-7.49 (m, 3H), 2.58 (s, 3H), 2.48 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 0.96 min, m/z = 476/478 [M+H]+.
Beispiel 6
6-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-fluornicotinsäure
Zu einer Lösung von 156 mg (0.31 mmol) der Verbindung aus Beispiel 37A in 3.9 ml THF und 0.8 ml Methanol wurden bei RT 1.54 ml (1.54 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel entfernt. Der verbliebene Rückstand wurde mit Wasser verdünnt, und das Gemisch wurde unter Rühren mit 1 M Salzsäure sauer gestellt. Der entstandene Feststoff wurde abfiltriert und zweimal mit Wasser gewaschen. Nach Trocknen im Vakuum wurden 131 mg (89% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.67 (br. s, 1H), 11.60 (s, 1H), 8.86 (br. s, 1H), 8.30 (d, 1H), 8.10-7.89 (m, 3H), 7.67-7.60 (m, 2H), 7.58-7.49 (m, 3H), 2.47 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.99 min, m/z = 480/482 [M+H]+.
Beispiel 7
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
100 mg (0.20 mmol) der Verbindung aus Beispiel 38A wurden in 2.5 ml THF und 0.5 ml Methanol gelöst. Zu der Lösung wurden 0.61 ml (0.61 mmol) 1 M Natronlauge gegeben. Das Reaktionsgemisch wurde 1 h unter Rückfluss gerührt. Das Volumen der Lösung wurde dann am Rotationsverdampfer reduziert, und die aufkonzentrierte Lösung wurde mit 0.6 ml 1 M Salzsäure auf pH 3 eingestellt. Es wurde mit 5 ml Wasser verdünnt und der ausgefallene Feststoff abfiltriert. Der Fest- stoff wurde mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 94 mg (97% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.27 (br. s, IH), 11.04 (s, IH), 8.17 (t, IH), 8.05 (d, IH), 7.99 (d, IH), 7.94 (dd, IH), 7.88 (dd, IH), 7.80 (dd, IH), 7.65-7.61 (m, 2H), 7.58-7.50 (m, 3H), 2.44 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 479/481 [M+H]+.
Beispiel 8
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2-fluorbenzoesäure
95 mg (0.19 mmol) der Verbindung aus Beispiel 41A wurden mit 2 ml THF und 1.9 ml 4 M Natronlauge versetzt. Das Reaktionsgemisch wurde über Nacht bei 80°C gerührt. Danach wurde die organische Phase abgetrennt und ohne weitere Aufarbeitung mittels präparativer HPLC (Me- thode 9) gereinigt. Es wurden 16 mg (17% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.38 (s, 1H), 8.07-8.04 (m, 1H), 7.96-7.92 (m, 3H), 7.85 (dd, 1H), 7.65-7.62 (m, 2H), 7.58-7.50 (m, 4H), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.11 min, m/z = 479/481 [M+H]+.
Beispiel 9 4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-chlorbenzoesäure
Zu einer Lösung von 1.53 g (3.00 mmol) der Verbindung aus Beispiel 42A in 38 ml THF und 8 ml Methanol wurden bei RT 15 ml (15 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h
unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel entfernt. Der verbliebene Rückstand wurde mit Wasser verdünnt, und das Gemisch wurde unter Rühren mit 1 M Salzsäure sauer gestellt. Der vorhandene Feststoff wurde abfiltriert und zweimal mit Wasser und einmal mit tert. -Butylmethylether gewaschen. Nach Trocknen im Vakuum wurden 1.40 g (94% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.34 (br. s, 1H), 10.93 (s, 1H), 8.13-7.99 (m, 5H), 7.94 (dd, 1H), 7.68-7.60 (m, 2H), 7.59-7.49 (m, 3H), 2.48 (s, 3H, teilweise verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.11 min, m/z = 495/497 [M+H]+.
Beispiel 10 3-Brom-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino }benzoesäure
Zu einer Lösung von 120 mg (0.22 mmol) der Verbindung aus Beispiel 43A in 2.5 ml THF und 0.5 ml Methanol wurde bei RT 1.0 ml (1.0 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch in 30 ml Wasser eingetragen und anschließend unter Rühren mit 1 M Salzsäure sauer gestellt. Der vorhandene Feststoff wurde abfiltriert und zweimal mit Wasser und einmal mit tert. -Butylmethylether gewaschen. Nach Trocknen im Vakuum wurden 113 mg (95% d. Th., Reinheit 98%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.35 (s, 1H), 10.88 (s, 1H), 8.23 (d, 1H), 8.14 (d, 1H), 8.09-8.02 (m, 2H), 8.01-7.97 (m, 1H), 7.94 (dd, 1H), 7.66-7.61 (m, 2H), 7.59-7.49 (m, 3H), 2.50 (s, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.13 min, m/z = 539/541/543 [M+H]
Beispiel 11
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-iodbenzo
100 mg (0.26 mmol) der Verbindung aus Beispiel 2A und 71 mg (0.26 mmol) Mefhyl-4-amino-3- iodbenzoat wurden in 2 ml DMF gelöst. Anschließend wurden 72 mg (0.64 mmol) Kalium-ieri.- butylat portionsweise hinzugegeben, und das Reaktionsgemisch wurde über Nacht bei RT gerührt. Danach wurde das Gemisch mit 1.3 ml (1.3 mmol) 1 M Natronlauge versetzt und 1 h bei 80°C gerührt. Nach Abkühlen auf RT wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Nach Entfernen des Lösungsmittel-Wasser-Gemisches wurde der Rückstand über Nacht im Vakuum getrocknet. Es wurden 81 mg (53% d. Th., Reinheit 99%) der Titelverbindung erhalten.
Ή-NMR (500 MHz, DMSO-de): δ [ppm] = 13.30 (br. s, 1H), 10.77 (s, 1H), 8.45 (d, 1H), 8.17 (d, 1H), 8.10-8.01 (m, 2H), 7.94 (dd, 1H), 7.86 (d, 1H), 7.68-7.60 (m, 2H), 7.60-7.49 (m, 3H), 2.53 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.11 min, m/z = 586/588 [M+H]+. Beispiel 12
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3,5-difluorbenzoesäure
500 mg (1.28 mmol) der Verbindung aus Beispiel 2A und 286 mg (1.53 mmol) Mefhyl-4-amino- 3,5-difluorbenzoat wurden in 10 ml THF gelöst und bei RT mit 112 mg (2.80 mmol) Natriumhydrid (60% in Mineralöl) versetzt. Nach Rühren über Nacht wurde das Lösungsmittel am Rota- tionsverdampfer entfernt. Der Rückstand wurde ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 13) gereinigt. Das so erhaltene Produkt wurde mit Acetonitril verrührt, und der resultierende Feststoff wurde abfiltriert und mit Acetonitril gewaschen. Nach Trocknen im Vakuum wurden 124 mg (18% d. Th., Reinheit 94%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.69 (s, 1H), 8.08 (d, 1H), 8.06-8.03 (d, 1H), 7.96- 7.94 (dd, 1H), 7.65-7.63 (m, 2H), 7.58-7.50 (m, 5H), 2.48 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 497/499 [M+H]+.
Beispiel 13
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3,5-dichlorbenzoesäure
Zu einer Lösung von 62 mg (0.11 mmol) der Verbindung aus Beispiel 44A in 1.4 ml THF und 0.3 ml Methanol wurden bei RT 0.56 ml (0.56 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel entfernt. Der verbliebene Rückstand wurde mit Wasser verdünnt, und das Gemisch wurde unter Rühren mit 1 M Salzsäure sauer gestellt. Der entstandene Feststoff wurde abfiltriert und zweimal mit Wasser gewaschen. Nach Trocknen an der Luft wurde der Feststoff in DMSO aufgenommen und mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan/Methanol aufgenommen und das Ge- misch wurde erneut eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 20 mg (34% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.68 (br. s, 1H), 11.22 (s, 1H), 8.42 (d, 1H), 8.16-8.01 (m, 3H), 7.95 (dd, 1H), 7.67-7.61 (m, 2H), 7.59-7.48 (m, 3H), 2.58 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.10 min, m/z = 529/531/533 [M+H]+. Beispiel 14
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-methylbenzoesäure
128 mg (0.26 mmol) der Verbindung aus Beispiel 45A wurden mit 3.3 ml THF und dann mit einer Lösung von 9 mg (0.39 mmol) Lithiumhydroxid in 1.1 ml Wasser versetzt. Das Reaktionsgemisch wurde zunächst zwei Tage bei RT gerührt, anschließend mit 2.6 ml 4 M Natronlauge versetzt und weitere 24 h bei RT gerührt. Danach wurde das Reaktionsgemisch 3 h auf 100°C erhitzt. Nach Abkühlen auf RT wurde mit Salzsäure auf pH 1-2 eingestellt. Der ausgefallene Feststoff wurde abgesaugt, mit Wasser gewaschen und im Vakuum getrocknet. Es wurden 70 mg (56% d. Th., Reinheit 99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.57 (s, 1H), 8.06 (d, 1H), 8.02 (d, 1H), 7.95 (dd, 1H), 7.92-7.87 (m, 3H), 7.66-7.64 (m, 2H), 7.59-7.51 (m, 3H), 2.54 (s, 3H), 2.38 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.09 min, m/z = 475/477 [M+H]+.
Beispiel 15
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(trifluormethyl)benzoesäure
Zu einer Lösung von 300 mg (0.87 mmol) der Verbindung aus Beispiel 1A in 3 ml DMF wurden bei RT 267 mg (0.96 mmol) 4-(4,6-Dimethoxy-l,3,5-triazin-2-yl)-4-methylmorpholiniumchlorid und 211 mg (0.96 mmol) Methyl-4-amino-3-(trifluormethyl)benzoat gegeben. Das Gemisch wurde 20 h bei RT gerührt. Anschließend wurden langsam ca. 100 mg (ca. 2.63 mmol) Natriumhydrid (60%-ige Dispersion in Mineralöl) portionsweise hinzugegeben, und das Gemisch wurde zunächst 1 h bei RT gerührt und dann 2 Tage bei RT stehen gelassen. Danach wurde mit Ethylacetat verdünnt und mit Wasser gewaschen. Die wässrige Phase wurde einmal mit Ethylacetat extrahiert, und die vereinigten organischen Phasen wurden über Magnesiumsulfat getrocknet, filtriert und ein- geengt. Der Rückstand wurde mittels präparativer HPLC (Methode 5) gereinigt. Die vereinten pro- dukthaltigen Fraktionen wurden bis auf ein Restvolumen an wässriger Phase eingeengt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden wieder über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde in einem Methanol/Dichlor- methan-Gemisch aufgenommen, erneut eingeengt und schließlich im Vakuum getrocknet. Es wur- den 153 mg (33% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.54 (br. s, 1H), 10.95 (s, 1H), 8.35 (dd, 1H), 8.31- 8.29 (m, 1H), 8.08-8.00 (m, 3H), 7.95 (dd, 1H), 7.67-7.61 (m, 2H), 7.60-7.50 (m, 3H), 2.50 (s, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.17 min, m/z = 529/531 [M+H]+. Beispiel 16
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-cyanobenzoesäure
250 mg (0.64 mmol) der Verbindung aus Beispiel 2A und 133 mg (0.70 mmol) Efhyl-4-amino-3- cyanobenzoat wurden mit 2 ml THF und 56 mg (1.40 mmol) Natriumhydrid (60% in Mineralöl) versetzt. Das Reaktionsgemisch wurde über Nacht bei RT gerührt. Anschließend wurde das Ge- misch mit Wasser versetzt, mit 2 M Salzsäure angesäuert und mit Ethylacetat extrahiert. Die organische Phase wurde abgetrennt und mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat und Entfernen des Lösungsmittels wurde der Rückstand mit Acetonitril verrührt. Der Feststoff wurde abfiltriert, mit Acetonitril gewaschen und dann mittels präparativer HPLC (Methode 8) gereinigt. Es wurden 102 mg (31% d. Th., Reinheit 96%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.54 (br. s, 1H), 11.54 (s, 1H), 8.39 (d, 1H), 8.32 (dd, 1H), 8.17 (d, 1H), 8.06 (d, 1H), 7.96 (dd, 1H), 7.91 (d, 1H), 7.65-7.63 (m, 2H), 7.59-7.51 (m, 3H), 2.49 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.07 min, m/z = 486/488 [M+H]+. Beispiel 17
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-nitrobenzoesäure
100 mg (0.26 mmol) der Verbindung aus Beispiel 2A und 60 mg (0.31 mmol) Mefhyl-4-amino-3- nitrobenzoat wurden mit 2 ml THF versetzt. Dann wurden bei RT 22 mg (0.56 mmol) Natriumhydrid (60% in Mineralöl) hinzugegeben. Nach Rühren über Nacht bei RT wurde das Reaktions- gemisch mit wenigen Tropfen Wasser versetzt und ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 8) gereinigt. Es wurden 110 mg (85% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.62 (br. s, IH), 11.65 (s, IH), 8.45 (d, IH), 8.32-8.29 (dd, IH), 8.12 (d, IH), 8.06 (d, IH), 7.96 (dd, IH), 7.81 (d, IH), 7.65-7.61 (m, 2H), 7.59-7.51 (m, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 507 [M+H]+.
Beispiel 18
3-Amino-4- { [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino Jbenzoesäure
282 mg (0.56 mmol) der Verbindung aus Beispiel 17 wurden mit 1.6 ml Essigsäure, 2.5 ml Ethanol, 2.5 ml Wasser und 628 mg (2.79 mmol) Zinn(II)chlorid versetzt. Das Reaktionsgemisch wurde 3 h bei 80°C gerührt und dann ohne weitere Aufarbeitung mittels präparativer HPLC (Me- thode 13) gereinigt. Es wurden 211 mg (80% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.69 (br. s, IH), 10.30 (s, IH), 8.05 (d, IH), 8.01 (d, IH), 7.94 (dd, IH), 7.74 (d, IH), 7.65-7.63 (m, 2H), 7.58-7.50 (m, 3H), 7.47 (d, IH), 7.28 (dd, IH), 5.25 (br. s, 2H), 2.46 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.97 min, m/z = 476/478 [M+H]+. Beispiel 19
3-Acetamido-4- { [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino Jbenzoesäure
100 mg (0.21 mmol) der Verbindung aus Beispiel 18 wurden mit 1 ml DMF und 0.12 ml (0.84 mmol) Triethylamin versetzt. Bei 0°C wurden 0.02 ml (0.25 mmol) Acetylchlorid zugetropft. Nach 5 min wurde das Eisbad entfernt, und das Reaktionsgemisch wurde über Nacht bei RT gerührt. Das Gemisch wurde dann ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 8) gereinigt. Das Acetonitril-Wasser-Gemisch wurde am Rotationsverdampfer entfernt, und der resultierende Rückstand wurde durch nochmalige präparative HPLC (Methode 13) nachgereinigt. Es wurden 14 mg (13% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.07 (br. s, 1H), 10.60 (s, 1H), 9.64 (s, 1H), 8.04-7.99 (m, 4H), 7.93 (dd, 1H), 7.82 (d, 1H), 7.63-7.61 (m, 2H), 7.57-7.50 (m, 3H), 2.45 (s, 3H), 2.04 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.99 min, m/z = 518/520 [M+H]+. Beispiel 20
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(ethylcarbamoyl)amino]benzoe-
100 mg (0.21 mmol) der Verbindung aus Beispiel 18 wurden mit 1 ml DMF und 0.12 ml (0.84 mmol) Triethylamin versetzt. Bei 0°C wurden 0.02 ml (0.25 mmol) Ethylisocyanat zugetropft. Nach 5 min wurde das Eisbad entfernt, und das Reaktionsgemisch wurde 3 h bei RT gerührt. Der ausgefallene Feststoff wurde danach abfiltriert, mit Acetonitril gewaschen, im Vakuum getrocknet und dann mittels präparativer HPLC (Methode 14) gereinigt. Es wurden 38 mg (33% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.72 (s, 1H), 8.12 (s, 1H), 8.06-8.01 (m, 3H), 7.93 (dd, 1H), 7.83 (d, 1H), 7.70 (d, 1H), 7.63-7.61 (m, 2H), 7.58-7.45 (m, 3H), 6.76 (m, 1H), 3.06 (m, 2H), 2.46 (s, 3H), 0.98 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.03 min, m/z = 547/549 [M+H]+. Beispiel 21
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-methoxybenzoesäure
Zu einer Lösung von 660 mg (1.31 mmol) der Verbindung aus Beispiel 46A in 16.5 ml THF und 3.5 ml Methanol wurden bei RT 6.6 ml (6.6 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel entfernt. Der verbliebene Rückstand wurde mit Wasser verdünnt, und das Gemisch wurde unter Rühren mit 1 M Salzsäure sauer gestellt. Der vorhandene Feststoff wurde abfiltriert und zweimal mit Wasser und einmal mit ieri.-Butylmethylether gewaschen. Nach Trocknen im Vakuum wurden 599 mg (93% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.02 (br. s, 1H), 10.50 (br. s, 1H), 8.17-7.84 (m, 4H), 7.75-7.43 (m, 7H), 3.93 (s, 3H), 2.43 (s, 3H, teilweise verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.09 min, m/z = 491/493 [M+H]+.
Beispiel 22
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-hydroxybenzoesäure
Zu einer Suspension von 90 mg (0.18 mmol) der Verbindung aus Beispiel 21 in 2 ml Dichlor - methan wurden bei 0°C 0.55 ml (0.55 mmol) einer 1 M Lösung von Bortribromid in Dichlor - methan gegeben. Das Gemisch wurde 2 h bei RT gerührt. Das so erhaltene Reaktionsgemisch wurde dann mit einem analog erhaltenen Reaktionsgemisch aus einem vorhergehenden Experiment [eingesetzte Menge an Verbindung aus Beispiel 21: 10 mg (0.02 mmol)] vereinigt. Nach Entfernen des Lösungsmittels wurde der Rückstand in DMSO aufgenommen und mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen im Vakuum wurden 86 mg (Reinheit 100%, 89% d. Th., bezogen auf insgesamt 100 mg (0.20 mmol) der Verbindung aus Beispiel 21) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 12.79 (br. s, 1H), 10.37 (s, 1H), 10.35 (s, 1H), 8.09- 7.99 (m, 3H), 7.91 (dd, 1H), 7.66-7.59 (m, 2H), 7.59-7.46 (m, 5H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.99 min, m/z = 477/479 [M+H]+. Beispiel 23
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(trifluormethoxy)benzoesäure
Eine Lösung von 75 mg (0.13 mmol) der Verbindung aus Beispiel 47 A in 2 ml eines THF/ Methanol-Gemisches (5: 1) wurde mit 0.65 ml (0.65 mmol) 1 M Natronlauge versetzt und 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Das Acetonitril-W asser-Gemisch wurde am Rotationsverdampfer entfernt und der Rückstand über Nacht im Vakuum getrocknet. Es wurden 49 mg (69% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.42 (br. s, 1H), 11.14 (s, 1H), 8.29 (d, 1H), 8.12-8.01 (m, 2H), 7.98-7.91 (m, 3H), 7.68-7.60 (m, 2H), 7.60-7.49 (m, 3H), 2.43 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 545/547 [M+H]
Beispiel 24
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(methylsulfanyl)benzoesäure
Zu einer Lösung von 100 mg (0.19 mmol) der Verbindung aus Beispiel 48 A in 2.5 ml THF und 0.5 ml Methanol wurden bei RT 0.50 ml (0.50 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel entfernt. Der verbliebene Rückstand wurde mit Wasser verdünnt, und das Gemisch wurde unter Rühren mit 1 M Salzsäure sauer gestellt. Nach einigen Minuten Rühren wurde der vorhandene Feststoff abfiltriert und zweimal mit Wasser gewaschen. Nach Trocknen im Vakuum wurden 88 mg (86% d. Th., Reinheit 95%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.17 (br. s, 1H), 10.70 (s, 1H), 8.27 (d, 1H), 8.04 (d, 1H), 7.96-7.90 (m, 2H), 7.86 (dd, 1H), 7.69 (d, 1H), 7.66-7.60 (m, 2H), 7.59-7.49 (m, 3H), 2.56 (s, 3H, teilweise verdeckt), 2.50 (s, 3H, teilweise verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.09 min, m/z = 507/509 [M+H]+.
Beispiel 25
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(methylsulfinyl)benzoesäure
Zu einer Lösung von 140 mg (0.26 mmol) der Verbindung aus Beispiel 49A in 3.5 ml THF und 0.7 ml Methanol wurden bei RT 1.31 ml (1.31 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.10 ml (1.31 mmol) Trifluor- essigsäure zugesetzt. Anschließend wurde das Gemisch mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 126 mg (93% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.39 (br. s, 1H), 11.22 (s, 1H), 8.53 (d, 1H), 8.20 (dd, 1H), 8.11-8.03 (m, 2H), 7.97 (dd, 1H), 7.81 (d, 1H), 7.67-7.61 (m, 2H), 7.61-7.50 (m, 3H), 2.92 (s, 3H), 2.50 (s, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 0.97 min, m/z = 523/525 [M+H]+. Beispiel 26
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-(methylsulfonyl)benzoesäure
Zu einer Lösung von 100 mg (0.18 mmol) der Verbindung aus Beispiel 50A in 2.5 ml THF und 0.5 ml Methanol wurden bei RT 0.91 ml (0.91 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.07 ml (0.91 mmol) Trifluor- essigsäure zugesetzt. Anschließend wurde das Gemisch mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 83 mg (85% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.55 (br. s, 1H), 10.75 (s, 1H), 8.54 (d, 1H), 8.37 (dd, 1H), 8.32-8.24 (m, 2H), 8.04 (d, 1H), 7.94 (dd, 1H), 7.67-7.61 (m, 2H), 7.60-7.49 (m, 3H), 3.41 (s, 3H), 2.50 (s, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 539/541 [M+H]+. Beispiel 27 4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-5-(ethylsulfonyl)-2-methoxy- benzoe säure
Zu einer Lösung von 221 mg (0.37 mmol) der Verbindung aus Beispiel 51A in 4.7 ml THF und 1.0 ml Methanol wurden bei RT 1.86 ml (1.86 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel entfernt. Der verbliebene Rückstand wurde mit Wasser verdünnt, und das Gemisch wurde unter Rühren mit 1 M Salzsäure sauer gestellt. Der entstandene Feststoff wurde abfiltriert und zweimal mit Wasser gewaschen. Nach Trocknen im Vakuum wurden 180 mg (84% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.03 (br. s, 1H), 10.61 (s, 1H), 8.27-8.22 (m, 2H), 8.12 (s, 1H), 8.08-8.01 (m, 1H), 7.95 (dd, 1H), 7.68-7.61 (m, 2H), 7.60-7.49 (m, 3H), 4.02 (s, 3H), 3.47-3.36 (m, 2H, teilweise verdeckt), 2.49 (s, 3H, verdeckt), 1.14 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.12 min, m/z = 583/585 [M+H]+.
Beispiel 28
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(trifluormethyl)sulfanyl]benzoe- säure
Eine Gemisch von 50 mg (0.09 mmol) der Verbindung aus Beispiel 53A in 1.6 ml THF und 0.3 ml Methanol wurde mit 0.32 ml (0.32 mmol) 1 M Natronlauge versetzt und anschließend 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit ca. 0.03 ml Trifluoressigsäure auf pH 4 eingestellt und ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 5) gereinigt. Nach Entfernen des Lösungsmittel-Wasser-Gemisches wurde der Rückstand in Dichlor- methan aufgenommen, das Gemisch wurde erneut eingeengt und der Rückstand wurde im Vakuum getrocknet. Es wurden 35 mg (72% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.46 (br. s, 1H), 11.34 (br. s, 1H), 8.50-7.80 (m, 6H), 7.73-7.33 (m, 5H), 2.47 (s, 3H, teilweise verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.19 min, m/z = 561/563 [M+H]+.
Beispiel 29
3-Fluor-4- { [(6-fluor-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino Jbenzoesäure
Zu einer Lösung von 184 mg (0.43 mmol) der Verbindung aus Beispiel 54A in 5.5 ml THF und 1.1 ml Methanol wurden bei RT 2.14 ml (2.14 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel entfernt. Die verbliebene wässrige Lösung wurde unter Rühren in 20 ml I M Salzsäure eingetragen. Nach einigen Minuten Rühren wurde der entstandene Feststoff abfiltriert und zweimal mit Wasser und einmal mit Pentan gewaschen. Nach Trocknen im Vakuum wurden 148 mg (83% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.24 (br. s, 1H), 11.03 (s, 1H), 8.22 (t, 1H), 8.17 (dd, 1H), 7.88 (dd, 1H), 7.80 (dd, 1H), 7.73 (td, 1H), 7.64-7.59 (m, 2H), 7.59-7.49 (m, 4H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.99 min, m/z = 419 [M+H]+. Beispiel 30
3-Fluor-4- { [(6-iod-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino Jbenzoesäure
100 mg (0.23 mmol) der Verbindung aus Beispiel 6A und 39 mg (0.23 mmol) Mefhyl-4-amino-3- fluorbenzoat wurden in 1.7 ml DMF gelöst. Zu der Lösung wurden 64 mg (0.57 mmol) Kalium- ieri.-butylat portionsweise hinzugegeben. Das Reaktionsgemisch wurde 1 h bei RT gerührt und dann mit 1.3 ml (1.3 mmol) 1 M Natronlauge versetzt. Das Gemisch wurde anschließend 1 h bei 80°C gerührt. Nach Abkühlen auf RT wurde das Gemisch ohne weitere Aufarbeitung mittels prä- parativer HPLC (Methode 6) gereinigt. Nach Entfernen des Lösungsmittel-Wasser-Gemisches und Trocknen des Rückstands im Vakuum wurden 76 mg (62% d. Th., Reinheit 98%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.25 (br. s, 1H), 11.04 (s, 1H), 8.19 (t, 1H), 8.18 (d, 1H), 8.06 (dd, 1H), 7.93-7.84 (m, 2H), 7.82 (dd, 1H), 7.66-7.59 (m, 2H), 7.59-7.47 (m, 3H), 2.42 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.07 min, m/z = 527 [M+H]+.
Beispiel 31 4-{ [(3,6-Dimethyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
Zu einer Lösung von 124 mg (0.27 mmol, Reinheit 93%) der Verbindung aus Beispiel 55A in 3.5 ml THF und 0.7 ml Methanol wurden bei RT 1.35 ml (1.35 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.10 ml (1.35 mmol) Trifluoressigsäure hinzugesetzt. Anschließend wurde das Gemisch mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen im Vakuum wurden 83 mg (75% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.17 (br. s, 1H), 10.99 (s, 1H), 8.24 (t, 1H), 7.98 (d, 1H), 7.88 (dd, 1H), 7.80 (dd, 1H), 7.68-7.58 (m, 4H), 7.58-7.47 (m, 3H), 2.53 (s, 3H, teilweise verdeckt), 2.40 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.01 min, m/z = 415 [M+H]+.
Beispiel 32
4-{ [(6-Ethyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
Zu einer Lösung von 186 mg (0.38 mmol, Reinheit 91%) der Verbindung aus Beispiel 56A in 5.0 ml THF und 1.0 ml Methanol wurden bei RT 1.92 ml (1.92 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.15 ml (2.00 mmol) Trifluoressigsäure hinzugesetzt. Nach Stehenlassen über Nacht wurde das Gemisch mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen im Vakuum wurden 139 mg (85% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.30 (br. s, 1H), 11.25-10.78 (m, 1H), 8.20 (t, 1H), 8.03 (d, 1H), 7.89 (dd, 1H), 7.81 (dd, 1H), 7.72 (dd, 1H), 7.66-7.60 (m, 3H), 7.59-7.49 (m, 3H), 2.84 (q, 2H), 2.41 (s, 3H), 1.27 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.06 min, m/z = 429 [M+H]+.
Beispiel 33 3-Fluor-4- { [(6-isopropyl-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino Jbenzoesäure
108 mg (0.24 mmol) der Verbindung aus Beispiel 57A wurden mit 2.4 ml THF und 2.4 ml 4 M Natronlauge versetzt. Das Reaktionsgemisch wurde 10 h bei 60°C gerührt. Danach wurde die organische Phase abgetrennt und ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 9) gereinigt. Das Acetonitril-Wasser-Gemisch wurde am Rotationsverdampfer entfernt und der Rückstand mit Acetonitril verrührt. Der Feststoff wurde abfiltriert und im Vakuum getrocknet. Es wurden 45 mg (43% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.26 (br. s, 1H), 11.01 (s, 1H), 8.14 (t, 1H), 8.02 (d, 1H), 7.92-7.71 (m, 3H), 7.67-7.44 (m, 6H), 3.20-3.04 (m, 1H), 2.41 (s, 3H), 1.29 (d, 6H). LC/MS (Methode 1, ESIpos): Rt = 1.10 min, m/z = 443 [M+H]+.
Beispiel 34
3-Fluor-4-({ [3-methyl-2-phenyl-6-(trifluormethyl)chinolin-4-yl]carbonyl}amino)benzoesäure
105 mg (0.22 mol) der Verbindung aus Beispiel 58A wurden mit 2.2 ml THF und 2.2 ml 4 M Natronlauge versetzt. Das Reaktionsgemisch wurde über Nacht bei 60°C gerührt. Danach wurde die organische Phase abgetrennt und ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 9) gereinigt. Nach Entfernen des Lösungsmittel-Wasser-Gemisches wurde der Rückstand über Nacht im Vakuum bei 60°C getrocknet. Es wurden 97 mg (95% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.30 (br. s, 1H), 11.13 (s, 1H), 8.32 (d, 1H), 8.18-8.14 (m, 2H), 8.08 (dd, 1H), 7.90 (dd, 1H), 7.83 (dd, 1H), 7.68-7.66 (m, 2H), 7.61-7.53 (m, 3H), 2.48 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.13 min, m/z = 469 [M+H]+. Beispiel 35
3-Fluor-4-({ [3-methyl-2-phenyl-6-(trifluormethoxy)chinolin-4-yl]carbonyl}amino)benzoesäure
Zu einer Lösung von 150 mg (0.27 mmol, Reinheit 91%) der Verbindung aus Beispiel 59A in 3.5 ml THF und 0.7 ml Methanol wurden bei RT 1.38 ml (1.38 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.11 ml (1.38 mmol) Trifluoressigsäure hinzugesetzt. Anschließend wurde das Gemisch mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen im Vakuum wurden 115 mg (87% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.25 (br. s, 1H), 11.07 (s, 1H), 8.24 (d, 1H), 8.16 (t, 1H), 7.89 (dd, 1H), 7.82 (dd, 2H), 7.74 (s, 1H), 7.68-7.61 (m, 2H), 7.60-7.48 (m, 3H), 2.45 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 485 [M+H]+. Beispiel 36
3-Fluor-4-({ [3-methyl-2-phenyl-6-(trimethylsilyl)chinolin-4-yl]carbonyl}amino)benzoesäure
Zu einem Gemisch von 34 mg (0.06 mmol) der Verbindung aus Beispiel 60A in 1 ml Dichlor - methan wurden 0.5 ml (6.49 mmol) Trifluoressigsäure gegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch eingeengt. Der Rückstand wurde in DMSO aufgenommen und mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Ge- misch wurde erneut eingeengt. Nach Trocknen im Vakuum wurden 25 mg (81% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.19 (br. s, 1H), 11.03 (s, 1H), 8.11-7.99 (m, 3H), 7.97-7.87 (m, 2H), 7.83 (d, 1H), 7.67-7.61 (m, 2H), 7.60-7.47 (m, 3H), 2.44 (s, 3H), 0.32 (s, 9H).
LC/MS (Methode 1, ESIpos): Rt = 1.19 min, m/z = 473 [M+H]+. Beispiel 37
4-({ [6-Brom-3-(fluormethyl)-2-phenylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
Zu einer Lösung von 60 mg (0.12 mmol) der Verbindung aus Beispiel 63A in 1.5 ml THF und 0.3 ml Methanol wurden bei RT 0.59 ml (0.59 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 30 min unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.05 ml (0.60 mmol) Tri- fluoressigsäure hinzugesetzt. Anschließend wurde das Gemisch mittels präparativer HPLC (Methode 12) gereinigt. Es wurden 39 mg (66% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 13.23 (br. s, IH), 11.16 (s, IH), 8.23 (t, IH), 8.15-8.10 (m, 2H), 8.07 (dd, IH), 7.90 (dd, IH), 7.82 (dd, IH), 7.70-7.64 (m, 2H), 7.63-7.54 (m, 3H), 5.57
(dd, 2H). LC/MS (Methode 1, ESIpos): Rt = 1.05 min, m/z = 497/499 [M+H]+. Beispiel 38
4-({ [6-Brom-3-(difluormethyl)-2-phenylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
Zu einer Lösung von 20 mg (0.04 mmol) der Verbindung aus Beispiel 64A in 0.6 ml THF und 0.13 ml Methanol wurden bei RT 0.19 ml (0.19 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 30 min unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.015 ml (0.19 mmol) Trifluoressigsäure hinzugesetzt. Anschließend wurde das Gemisch mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in Dichlormethan aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen im Vakuum wurden 11 mg (54% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.24 (br. s, 1H), 11.13 (s, 1H), 8.26 (t, 1H), 8.18-8.09 (m, 3H), 7.90 (dd, 1H), 7.81 (dd, 1H), 7.65-7.54 (m, 5H), 7.09 (t, 1H). LC/MS (Methode 1, ESIpos): Rt = 1.05 min, m/z = 515/517 [M+H]+.
Beispiel 39
4- { [(6-Brom-3-ethyl-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoesäure
Zu einer Lösung von 187 mg (0.37 mmol) der Verbindung aus Beispiel 65A in 5.8 ml THF und 1.2 ml Methanol wurden bei RT 1.86 ml (1.86 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 30 min unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel destillativ entfernt, und die verbliebene wässrige Phase wurde mit 0.14 ml (1.86 mmol) Trifluoressigsäure versetzt. Der entstandene Feststoff wurde abfiltriert, zweimal mit 1 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 178 mg (98% d. Th., Reinheit 100%) der Titelver- bindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 10.99 (s, 1H), 8.05-7.97 (m, 3H), 7.94 (dd, 1H), 7.84 (dd, 1H), 7.76 (dd, 1H), 7.60-7.48 (m, 5H), 2.83 (d, 2H), 0.99 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.07 min, m/z = 493/495 [M+H]+.
Beispiel 40
4- { [(6-Brom-2-phenyl-3-propylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoesäure
Zu einer Lösung von 139 mg (0.27 mmol) der Verbindung aus Beispiel 66A in 4.2 ml THF und 0.9 ml Methanol wurden bei RT 1.34 ml (1.34 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 3 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel destillativ entfernt, und die verbliebene wässrige Phase wurde mit 0.10 ml (1.34 mmol) Trifluor- essigsäure versetzt. Der entstandene Feststoff wurde abfiltriert, zweimal mit 1 ml Wasser gewa- sehen und im Vakuum getrocknet. Es wurden 86 mg (64% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 10.74 (s, 1H), 8.04-7.99 (m, 2H), 7.94 (dd, 1H), 7.74 (dd, 1H), 7.70-7.62 (m, 2H), 7.59-7.48 (m, 5H), 2.88-2.74 (m, 2H), 1.40 (br. s, 2H), 0.69 (t, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.14 min, m/z = 507/509 [M+H]+. Beispiel 41
4- { [(6-Brom-7-chlor-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino} -3-fluorbenzoesäure
Zu einer Lösung von 52 mg (0.10 mmol) der Verbindung aus Beispiel 67A in 1.3 ml THF und 0.25 ml Methanol wurden bei RT 0.49 ml (0.49 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 30 min unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.04 ml (0.50 mmol) Trifluoressigsäure hinzugesetzt. Der entstandene Feststoff wurde abfiltriert und zweimal mit 1 ml THF gewaschen. Nach Trocknen im Vakuum wurden 13 mg (26% d. Th., Reinheit 100%) einer ersten Charge der Titelverbindung erhalten. Das Filtrat wurde eingeengt, und der Rückstand wurde in einem Gemisch aus DMSO, THF und Wasser aufgenommen und mittels präparativer HPLC (Methode 12) gereinigt. Es wurden so 30 mg (60% d. Th., Reinheit 100%) einer zweiten Charge der Titelverbindung erhalten.
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 13.25 (br. s, 1H), 11.08 (s, 1H), 8.39 (s, 1H), 8.23 (t, 1H), 8.20 (s, 1H), 7.89 (dd, 1H), 7.82 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.50 (m, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.16 min, m/z = 513/515 [M+H]+.
Beispiel 42 4- { [(6,7-Dichlor-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoesäure
100 mg (0.26 mmol) der Verbindung aus Beispiel 22A und 44.4 mg (0.26 mmol) Mefhyl-4-amino- 3-fluorbenzoat wurden in 2 ml DMF gelöst. Zu der Lösung wurden 73 mg (0.65 mmol) Kalium- ieri.-butylat portionsweise hinzugegeben. Das Reaktionsgemisch wurde 1 h bei RT gerührt und dann mit 1.3 ml (1.3 mmol) 1 M Natronlauge versetzt. Das Gemisch wurde anschließend 1 h bei 80°C gerührt. Nach Abkühlen auf RT wurde das Gemisch ohne weitere Aufarbeitung mittels prä- parativer HPLC (Methode 6) gereinigt. Nach Entfernen des Lösungsmittel-Wasser-Gemisches und Trocknen des Rückstands im Vakuum wurden 78 mg (59% d. Th., Reinheit 93%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.25 (br. s, 1H), 11.07 (s, 1H), 8.41 (s, 1H), 8.24 (t, 1H), 8.06 (s, 1H), 7.89 (dd, 1H), 7.81 (dd, 1H), 7.69-7.60 (m, 2H), 7.60-7.48 (m, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 469/471 [M+H]+.
Beispiel 43
4-({ [6-Brom-2-(4-fluorphenyl)-3-methylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
Zu einer Lösung von 173 mg (0.34 mmol) der Verbindung aus Beispiel 68A in 5.4 ml THF und 1.1 ml Methanol wurden bei RT 1.71 ml (1.71 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 30 min unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungs- mittel destillativ entfernt, und die verbliebene wässrige Phase wurde mit 0.13 ml (1.71 mmol) Tri- fluoressigsäure versetzt. Der entstandene Feststoff wurde abfiltriert, zweimal mit 1 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 166 mg (99% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.20 (br. s, IH), 11.04 (s, IH), 8.19 (t, IH), 8.05 (d, IH), 7.99 (d, IH), 7.94 (dd, IH), 7.88 (dd, IH), 7.81 (dd, IH), 7.73-7.65 (m, 2H), 7.42-7.33 (m, 2H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.06 min, m/z = 497/499 [M+H]+.
Beispiel 44
4-({ [6-Brom-2-(3-fluorphenyl)-3-methylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
Zu einer Lösung von 96 mg (0.19 mmol) der Verbindung aus Beispiel 69A in 3.0 ml THF und 0.6 ml Methanol wurden bei RT 0.95 ml (0.95 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 3 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel destillativ entfernt, und die verbliebene wässrige Phase wurde mit 0.07 ml (0.95 mmol) Trifluor- essigsäure versetzt. Der entstandene Feststoff wurde abfiltriert, zweimal mit 1 ml Wasser gewaschen und im Vakuum getrocknet. Anschließend wurde der Feststoff in DMSO aufgenommen und mittels präparativer HPLC (Methode 12) gereinigt. Es wurden 68 mg (72% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.25 (s, 1H), 11.05 (s, 1H), 8.21 (t, 1H), 8.06 (d, 1H), 8.00 (d, 1H), 7.95 (dd, 1H), 7.89 (dd, 1H), 7.82 (dd, 1H), 7.64-7.57 (m, 1H), 7.51-7.44 (m, 2H), 7.42-7.33 (m, 1H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.09 min, m/z = 497/499 [M+H]
Beispiel 45 4-({ [6-Brom-2-(2-fluorphenyl)-3-methylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
Zu einer Lösung von 95 mg (0.19 mmol) der Verbindung aus Beispiel 70A in 3 ml THF und 0.6 ml Methanol wurden bei RT 0.94 ml (0.94 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 3 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das organische Lösungsmittel destilla- tiv entfernt, und die verbliebene wässrige Phase wurde mit 0.07 ml (0.94 mmol) Trifluoressigsäure versetzt. Der entstandene Feststoff wurde abfiltriert, zweimal mit 1 ml Wasser gewaschen und im Vakuum getrocknet. Es wurden 83 mg (86% d. Th., Reinheit 96%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.25 (br. s, 1H), 11.14 (s, 1H), 8.19 (t, 1H), 8.06 (d, 1H), 8.02 (d, 1H), 7.96 (dd, 1H), 7.89 (dd, 1H), 7.82 (dd, 1H), 7.65-7.50 (m, 2H), 7.45-7.37 (m, 2H), 2.32 (s, 3H).
LC/MS (Methode 3, ESIpos): Rt = 1.34 min, m/z = 497/499 [M+H]+.
Beispiel 46
4-({ [6-Brom-3-methyl-2-(pyridin-4-yl)chinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
Ein Gemisch von 90 mg (0.18 mmol) der Verbindung aus Beispiel 71 A in 1 ml (1 mmol) 1 M Natronlauge, 1 ml Methanol und 1 ml THF wurde über Nacht bei RT gerührt. Anschließend wurde das Gemisch durch Zugabe einiger Tropfen 1 M Salzsäure auf pH 7 gestellt und das Lösungsmittel im Vakuum entfernt. Der Rückstand wurde mittels präparativer RP-HPLC gereinigt (Eluent: Gradient Acetonitril/10 mM aq. Ammoniumcarbonat). Es wurden 55 mg (63% d. Th.) der Titelverbindung erhalten.
Ή-NMR (300 MHz, DMSO-d6): δ [ppm] = 8.79 (d, 2H), 8.14 (m, 2H), 8.05 (d, 1H), 7.92 (m, 2H), 7.82 (m, 1H), 7.72 (m, 2H), 2.55 (s, 3H). LC/MS (Methode 4, ESIpos): Rt = 1.36 min, m/z = 479/481 [M+H]+.
Beispiel 47
6-Brom-/V-(4-carbamoylphenyl)-3-methyl-2-phenylchinolin-4-carboxamid
Zu 208 mg (0.43 mmol) der Verbindung aus Beispiel 35A wurden 3.5 ml (30 mmol) Ammoniak- Lösung (33% in Wasser) langsam hinzugegeben, und das Gemisch wurde 1 h bei RT gerührt. Anschließend wurde das Gemisch mit Wasser verdünnt, und der entstandene Feststoff wurde abfiltriert, zweimal mit Wasser gewaschen und an der Luft getrocknet. Das Rohprodukt wurde mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in einem Gemisch aus Dichlormethan und Methanol aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 113 mg (57% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.10 (s, 1H), 8.05 (d, 1H), 7.97-7.89 (m, 5H), 7.84 (d, 2H), 7.66-7.60 (m, 2H), 7.59-7.47 (m, 3H), 7.31 (br. s, 1H), 2.41 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.92 min, m/z = 460/462 [M+H]+.
Beispiel 48
6-Brom-/V-(4-carbamoyl-2-fluorphenyl)-3-methyl-2-phenylchinolin-4-carboxamid
Zu 260 mg (0.52 mmol) der Verbindung aus Beispiel 40 A wurden 4.0 ml (33.9 mmol) Ammoniak- Lösung (33% in Wasser) langsam hinzugegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde der entstandene Feststoff abfiltriert, zweimal mit Wasser gewaschen und an der Luft getrocknet. Das Rohprodukt wurde mittels präparativer HPLC (Methode 5) gereinigt. Die vereinigten produkthaltigen Fraktionen wurden eingeengt, der Rückstand wurde in einem Gemisch aus Dichlormethan und Methanol aufgenommen und das Gemisch wurde erneut eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 181 mg (73% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d
6): δ [ppm] = 10.96 (s, 1H), 8.12-8.02 (m, 3H), 8.00 (d, 1H), 7.94 (dd, 1H), 7.87-7.80 (m, 2H), 7.66-7.61 (m, 2H), 7.60-7.48 (m, 4H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.98 min, m/z = 478/480 [M+H]+.
Beispiel 49 4- { [(6-Brom-3-methyl- 1 -oxido-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoesäure
Zu einer Suspension von 100 mg (0.21 mmol) der Verbindung aus Beispiel 7 in 3 ml Dichlormethan wurden bei RT 113 mg (0.46 mmol, Gehalt 70%, Rest Wasser) 3-Chlorperbenzoesäure, suspendiert in 1 ml Dichlormethan, gegeben. Das Gemisch wurde 2 h bei RT gerührt. Anschlie- Bend wurden 0.5 ml THF hinzugegeben, und das Gemisch wurde 3 Tage bei RT stehen gelassen. Danach wurden nochmals 113 mg (0.46 mmol, Gehalt 70%) 3-Chlorperbenzoesäure, suspendiert in 1 ml Dichlormethan, hinzugefügt, und das Gemisch wurde einen weiteren Tag bei RT gerührt. Anschließend wurden weitere 57 mg (0.23 mmol, Gehalt 70%) 3-Chlorperbenzoesäure, suspendiert in 0.5 ml Dichlormethan, hinzufügt, und das Gemisch wurde nochmals einen Tag bei RT gerührt. Das Gemisch wurde dann mit jeweils 20 ml gesättigter wässriger Natriumhydrogencarbonat-Lösung und Dichlormethan versetzt, und nach Phasentrennung wurde die wässrige Phase einmal mit 20 ml Dichlormethan extrahiert. Die wässrige Phase wurde durch Zusatz von konzentrierter Salzsäure auf pH 1 gestellt und noch zweimal mit jeweils 30 ml Dichlormethan extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wur- de in DMSO aufgenommen und mittels präparativer HPLC (Methode 12) gereinigt. Es wurden so 67 mg (65% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.24 (br. s, 1H), 10.98 (s, 1H), 8.51 (d, 1H), 8.23 (t, 1H), 8.07 (d, 1H), 7.99 (dd, 1H), 7.88 (dd, 1H), 7.80 (dd, 1H), 7.63-7.48 (m, 3H), 7.43 (d, 2H), 2.16 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 0.87 min, m/z = 495/497 [M+H]+. Beispiel 50
Natrium-4-{ [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoat
Methode A:
50 mg (0.10 mmol) der Verbindung aus Beispiel 7 wurden in 1.5 ml THF gelöst und tropfenweise mit 1 M Natronlauge versetzt, bis pH 8 erreicht war. Das Gemisch wurde dann am Rotationsverdampfer bis zur Trockene eingeengt. Der Rückstand wurde in wenig Acetonitril und Methanol gelöst und über Nacht lyophilisiert. Es wurden 50 mg (95% d. Th., Reinheit 99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 10.73 (s, 1H), 8.03-8.01 (m, 2H), 7.93-7.91 (d, 1H), 7.71 (s, 2H), 7.66-7.62 (m, 3H), 7.57-7.50 (m, 3H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.07 min, m/z = 479/481 [M+H]+.
Methode B:
2.0 g (4.18 mmol) der Verbindung aus Beispiel 7 wurden mit 100 ml 1,4-Dioxan versetzt und das Gemisch kurz zum Sieden erhitzt. Zu der heißen Lösung wurde eine Lösung von 167 mg (4.18 mmol) Natriumhydroxid in 20 ml Wasser gegeben. Nach Abkühlen auf RT wurde das Gemisch an
der Luft bei RT bis zum Verdunsten des Lösungsmittels stehengelassen. Nach Umfüllen wurden 2.3 g der Titelverbindung erhalten (quant., Reinheit 100%, noch wasserhaltig).
Beispiel 51
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure-L-Argininsalz
1.1 g (2.30 mmol) der Verbindung aus Beispiel 7 wurden mit 200 ml Methanol und 5 ml DMF versetzt, und das Gemisch wurde kurz zum Sieden erhitzt. Zu der heißen Lösung wurde eine Lösung von 401 mg (2.30 mmol) L-(+)-Arginin in 30 ml Wasser gegeben. Nach Abkühlen auf RT wurde das Gemisch bei RT an der Luft bis zum Verdunsten des Lösungsmittels stehengelassen. Der verbliebene Rückstand wurde in 40 ml Wasser suspendiert und einen Tag bei RT gerührt. Danach wurde die Suspension filtriert und der Feststoff bei RT an der Luft getrocknet. Es wurden 1.0 g (73% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 10.76 (br. s, 1H), 8.04 (d, 1H), 8.00 (d, 1H), 8.0-7.7 (br. m, ~2H), 7.93 (dd, 1H), 7.89-7.82 (m, 1H), 7.77 (dd, 1H), 7.69 (dd, 1H), 7.65-7.60 (m, 2H), 7.55 (d, 3H), 3.30-3.03 (m, 2H, teilweise verdeckt), 2.44 (s, 3H), 1.81-1.53 (m, 4H).
LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 479/481 [M+H]+.
Beispiel 52
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(methoxymethyl)sulfanyl]- benzoe säure
Zu einer Lösung von 50 mg (0.09 mmol) der Verbindung aus Beispiel 72A in 1.5 ml THF und 0.4 ml Methanol wurden bei RT 0.46 ml (0.46 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.035 ml (0.46 mmol) Trifluoressigsäure versetzt und mittels präparativer HPLC (Methode 18) gereinigt. Nach Entfernen des Lösungsmittels wurde der Rückstand in einem Dichlormethan/Methanol-Gemisch aufgenommen und die Lösung wieder eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 42 mg (87% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.14 (br. s, 1H), 10.75 (s, 1H), 8.25 (dd, 2H), 8.04 (d, 1H), 7.97-7.89 (m, 2H), 7.78 (d, 1H), 7.66-7.61 (m, 2H), 7.60-7.49 (m, 3H), 5.09 (s, 2H), 3.34 (s, 3H, verdeckt), 2.50 (s, 3H, verdeckt).
LC/MS (Methode 1, ESIpos): Rt = 1.12 min, m/z = 537/539 [M+H]+.
Beispiel 53
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(trifluormethyl)sulfonyl]- benzoe säure
Zu einer Lösung von 45 mg (0.07 mmol) der Verbindung aus Beispiel 75A in 1.2 ml THF und 0.3 ml Methanol wurden bei RT 0.37 ml (0.37 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 2 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.029 ml (0.37 mmol) Trifluoressigsäure versetzt und mittels präparativer HPLC (Methode 18) gereinigt. Nach Entfernen des Lösungsmittels wurde der Rückstand in einem Dichlormethan/Methanol-Gemisch aufgenommen und die Lösung wieder eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 41 mg (94% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.91 (br. s, 1H), 11.01 (s, 1H), 8.60 (dd, 1H), 8.58- 8.55 (m, 1H), 8.37 (d, 1H), 8.07-8.04 (m, 2H), 7.95 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.50 (m, 3H), 2.48 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.25 min, m/z = 593/595 [M+H]+.
Beispiel 54
4-{ [(6-Brom-3-methyl-l-oxido-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(trifluormethyl)- sulfanyl]benzoesäure
Zu einer Lösung von 43 mg (0.07 mmol) der Verbindung aus Beispiel 76A in 1.5 ml THF und 0.3 ml Methanol wurden bei RT 0.37 ml (0.37 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.028 ml (0.37 mmol) Trifluoressigsäure versetzt und mittels präparativer HPLC (Methode 5) gereinigt. Nach Entfernen des Lösungsmittels wurde der Rückstand in Dichlormethan aufgenommen und die Lösung wieder eingeengt. Nach Trocknen des Rückstands im Vakuum wurden 33 mg (78% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 13.45 (br. s, 1H), 11.27 (s, 1H), 8.53 (d, 1H), 8.33 (s, 1H), 8.25-8.21 (m, 2H), 8.01 (dd, 1H), 7.93 (d, 1H), 7.62-7.57 (m, 2H), 7.56-7.50 (m, 1H), 7.45 (d, 2H), 2.19 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.00 min, m/z = 577/579 [M+H]+.
Beispiel 55
4-{ [(6-Brom-5-chlor-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
Zu einer Lösung von 285 mg (0.02 mmol, Reinheit 4%) der Verbindung aus Beispiel 79A in 7 ml THF und 1.5 ml Methanol wurden bei RT 0.09 ml (0.09 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 2 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.008 ml (0.11 mmol) Trifluoressigsäure versetzt und ca. 80 h bei RT stehen gelassen. Danach wurde der entstandene Niederschlag abfiltriert und zweimal mit 0.5 ml THF nachgewaschen. Der im Filtrat erneut entstandene Niederschlag wurde ebenso abfiltriert. Das Filtrat wurde eingeengt, und der erhaltene Rückstand wurde mittels präparativer HPLC (Methode 19) vorgereinigt. Das so gewonnene Produkt wurde ein weiteres Mal mittels präparativer HPLC gereinigt [Säule: Sunfire C18, 5 μιη, 100 mm x 30 mm; Fluss: 75 ml/min; Detektion: 210 nm; Injektionsvolumen: 2.0 ml; Temperatur: 40°C; Eluent: Wasser/Acetonitril/(Acetonitril/2% Ameisensäure in Wasser 80:20); Gradient: 70: 10:20 -> 5:90:5, 10 min]. Es wurden auf diese Weise 6.1 mg (55% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.16 (br. s, 1H), 10.99 (s, 1H), 8.33 (t, 1H), 8.13 (d, 1H), 8.02 (d, 1H), 7.88 (dd, 1H), 7.77 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.53 (m, 3H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.10 min, m/z = 513/515 [M+H]+.
Beispiel 56
6-Brom-/V-[2-fluor-4-(methylcarbamoyl)phenyl]-3-methyl-2-phenylchinolin-4-carboxamid
Zu 298 mg (0.60 mmol) der Verbindung aus Beispiel 40A in 3 ml THF wurden bei 0°C langsam 1.90 ml (3.80 mmol) einer 2 M Lösung von Methylamin in THF gegeben, und das Gemisch wurde 2 h bei RT gerührt. Anschließend wurden weitere 3 ml THF und 1 ml (1.90 mmol) der Methyl- amin-Lösung hinzugegeben, und das Gemisch wurde in einer Mikrowellenapparatur 30 min auf 70°C erhitzt. Nach Abkühlen auf RT wurde erneut 1 ml (1.90 mmol) der Methylamin-Lösung hinzugefügt, und das Gemisch wurde weitere 15 min bei 100°C in der Mikrowelle erhitzt. Nach Abkühlen auf RT wurde das Gemisch mit gesättigter Natriumchlorid-Lösung, Wasser und 1 M Salzsäure versetzt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden einmal mit 1 M Natronlauge gewaschen, über Magnesiumsulfat getrocknet und eingeengt. Der Rückstand wurde mittels präparativer HPLC (Methode 12) gereinigt. Es wurden 43 mg (15% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.96 (s, 1H), 8.55 (q, 1H), 8.09 (t, 1H), 8.04 (d, 1H), 8.00 (d, 1H), 7.94 (dd, 1H), 7.82-7.76 (m, 2H), 7.65-7.61 (m, 2H), 7.59-7.50 (m, 3H), 2.81 (d, 3H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.00 min, m/z = 492/494 [M+H]+.
Beispiel 57
4-({ [6-Brom-2-phenyl-3-(trifluormethyl)chinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
Zu einer Lösung von 4.5 mg (0.008 mmol) der Verbindung aus Beispiel 81A in 1 ml THF und 0.5 ml Methanol wurden bei RT 0.03 ml (0.03 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 14 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.003 ml (0.035 mmol) Trifluoressigsäure versetzt und eingeengt. Der Rückstand wurde mittels präparativer HPLC gereinigt (Säule: Phenomenex Luna C18, 5 μιη, 100 mm x 21.2 mm; Fluss: 25 ml/min; Eluent: Wasser mit 1% Ameisensäure/ Acetonitril; Gradient: 95:5— 5:95, 16 min). Es wurden so 4.5 mg (92% d. Th., Reinheit 90%) der Titelverbindung erhalten. Außerdem wurden als Nebenkomponente 0.5 mg (9% d. Th., Reinheit 92%) der als Beispiel 62 aufgeführten Verbindung iso- liert (Analytik siehe dort).
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 11.27 (s, 1H), 8.28-8.10 (m, 4H), 7.91 (dd, 1H), 7.83 (dd, 1H), 7.56-7.54 (m, 5H).
LC/MS (Methode 23, ESIpos): Rt = 3.54 min, m/z = 533/535 [M+H]+.
Beispiel 58 4-{ [(6-Brom-3,8-dimethyl-2-phenylchinolin-4-yl)carbonyl]amino}benzoesäure
Zu einer Lösung von 42 mg (0.08 mmol, Reinheit 96%) Methyl-4-{ [(6-brom-3,8-dimethyl-2- phenylchinolin-4-yl)carbonyl]amino}benzoat (kommerziell erhältlich) in 1.5 ml THF und 0.3 ml Methanol wurden bei RT 0.4 ml (0.4 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.03 ml (0.4 mmol) Trifluoressigsäure versetzt und mittels präparativer HPLC (Methode 5) gereinigt. Es wurden so 30 mg (78% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 12.83 (br. s, 1H), 11.16 (s, 1H), 8.00 (d, 2H), 7.89 (d, 2H), 7.86-7.84 (m, 1H), 7.73 (d, 1H), 7.70-7.64 (m, 2H), 7.60-7.49 (m, 3H), 2.73 (s, 3H), 2.42 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.23 min, m/z = 475/477 [M+H]+.
Beispiel 59
4-{ [(6-Chlor-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
Zu einer Lösung von 60 mg (0.13 mmol) der Verbindung aus Beispiel 82A in 2.7 ml THF und 0.5 ml Methanol wurden bei RT 0.39 ml (0.39 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.045 ml (0.59 mmol) Trifluoressigsäure versetzt, mit 2 ml DMSO verdünnt und dann mittels präparativer HPLC (Methode 18) gereinigt. Es wurden 51 mg (88% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.22 (br. s, 1H), 11.06 (s, 1H), 8.22 (t, 1H), 8.15-8.09 (m, 1H), 7.89 (dd, 1H), 7.86-7.78 (m, 3H), 7.66-7.60 (m, 2H), 7.59-7.49 (m, 3H), 2.43 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.06 min, m/z = 435 [M+H]+.
Beispiel 60
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-[(methoxymethyl)sulfonyl]- benzoe säure
Zu einer Lösung von 70 mg (0.12 mmol) der Verbindung aus Beispiel 83A in 2.6 ml THF und 0.5 ml Methanol wurden bei RT 0.60 ml (0.60 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.047 ml (0.61 mmol) Trifluoressigsäure versetzt und mittels präparativer HPLC (Methode 18) gereinigt. Es wurden 64 mg (91% d. Th., Reinheit 98%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.50 (br. s, 1H), 10.62 (s, 1H), 8.49 (d, 1H), 8.46-8.37 (m, 2H), 8.21 (d, 1H), 8.05 (d, 1H), 7.95 (dd, 1H), 7.67-7.61 (m, 2H), 7.59-7.50 (m, 3H), 5.02 (s, 2H), 3.42 (s, 3H), 2.49 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 569/571 [M+H]+.
Beispiel 61
4-{ [(6-ieri.-Butyl-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
Zu einer Lösung von 80 mg (0.17 mmol) der Verbindung aus Beispiel 85A in 3.5 ml THF und 0.7 ml Methanol wurden bei RT 0.86 ml (0.86 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit 0.066 ml (0.86 mmol) Trifluoressigsäure versetzt und mittels präparativer HPLC (Methode 5) gereinigt. Es wur- den 61 mg (77% d. Th., Reinheit 98%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.24 (br. s, 1H), 11.01 (s, 1H), 8.07-8.00 (m, 2H), 7.94 (dd, 1H), 7.89 (d, 1H), 7.83 (dd, 1H), 7.77 (d, 1H), 7.64-7.59 (m, 2H), 7.59-7.48 (m, 3H), 2.42 (s, 3H), 1.38 (s, 9H).
LC/MS (Methode 1, ESIpos): Rt = 1.09 min, m/z = 457 [M+H]+. Beispiel 62
3-Brom-4-({ [6-brom-2-phenyl-3-(trifluormethyl)chinolin-4-yl]carbonyl}amino)-5-fluorbenzoe- säure
Die Titelverbindung wurde als Nebenkomponente bei der Herstellung und Aufreinigung der Ver- bindung aus Beispiel 57 erhalten (Beschreibung siehe dort).
Ή-NMR (500 MHz, DMSO-d6): δ [ppm] = 11.28 (s, 1H), 8.49 (d, 1H), 8.23 (dd, 1H), 8.18 (d, 1H), 8.13-8.10 (m, 1H), 7.94 (dd, 1H), 7.57-7.52 (m, 5H).
LC/MS (Methode 1, ESIpos): Rt = 1.14 min, m/z = 611/613/615 [M+H]+.
Beispiel 63
3-Fluor-4-({ [3-methyl-6-(pentafluor- 6-sulfanyl)-2-phenylchinolin-4-yl]carbonyl}amino)ben säure
110 mg (0.20 mmol) der Verbindung aus Beispiel 90A wurden in 4 ml eines THF/Methanol- Gemisches (5: 1) gelöst und mit 1.02 ml (1.02 mmol) einer 1 M Lösung von Lithiumhydroxid in Wasser versetzt. Das Reaktionsgemisch wurde 3 h bei 50°C gerührt. Anschließend wurde das Gemisch auf RT abgekühlt, mit 4 M Salzsäure auf pH 1-2 gestellt und ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Die Produktfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 85 mg (79% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.28 (br. s, 1H), 11.14 (s, 1H), 8.35-8.22 (m, 3H), 8.09 (t, 1H), 7.90 (dd, 1H), 7.84 (dd, 1H), 7.73-7.63 (m, 2H), 7.63-7.50 (m, 3H), 2.49 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.14 min, m/z = 527 [M+H]+. Beispiel 64
4-({ [6-(Difluormethyl)-3-methyl-2-phenylchinolin-4-yl]carbonyl}amino)-3-fluorbenzoesäure
55 mg (0.18 mmol) der Verbindung aus Beispiel 94A wurden in 2 ml DMF unter Argon gelöst und bei RT mit 57 mg (0.35 mmol) /V,/V'-Carbonyldiimidazol versetzt. Das Reaktionsgemisch wurde bei 60°C über Nacht gerührt und anschließend mit Wasser und Ethylacetat versetzt. Die Phasen wurden getrennt, und die wässrige Phase wurde dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet und eingeengt. Der Rückstand wurde in 1.55 ml DMF gelöst und mit 29.8 mg (0.26 mmol) Methyl-4-amino-3-fluorbenzoat versetzt. Anschließend wurden 0.44 ml (0.44 mmol) einer 1 M Lösung von Kalium-ieri. -butylat in THF hinzugegeben. Das Reaktionsgemisch wurde über Nacht bei RT gerührt und im Anschluss mit 0.88 ml (0.88 mmol) einer 1 M Lösung von Lithiumhydroxid in Wasser versetzt. Das Reaktionsgemisch wurde dann 3 h bei 50°C gerührt. Das Gemisch wurde danach auf RT abgekühlt, mit 4 M Salzsäure auf pH 1-2 gestellt und ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Die Produktfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden so 20 mg (23% d. Th., Reinheit 90%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.24 (br. s, 1H), 11.08 (s, 1H), 8.35-8.17 (m, 2H), 8.09 (s, 1H), 7.98-7.87 (m, 2H), 7.82 (dd, 1H), 7.67-7.62 (m, 2H), 7.60-7.51 (m, 3H), 7.31 (t, 1H), 2.45 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.03 min, m/z = 451 [M+H]+. Beispiel 65 4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2,3-difluorbenzoesäure
105 mg (0.21 mmol) der Verbindung aus Beispiel 95 A wurden in 5 ml eines THF/Methanol- Gemisches (5: 1) gelöst und mit 1.03 ml (1.03 mmol) einer 1 M Lösung von Lithiumhydroxid in Wasser versetzt. Das Reaktionsgemisch wurde 3 h bei 50°C gerührt, danach auf RT abgekühlt und mit 4 M Salzsäure auf pH 1-2 gestellt. Anschließend wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Die Produktfraktionen wurden eingeengt, der Rückstand wurde lyophilisiert und danach aus Wasser umkristallisiert. Nach Trocknen im Vakuum wurden 69 mg (68% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.51 (br. s, 1H), 11.24 (s, 1H), 8.08-8.03 (m, 1H), 8.03-7.96 (m, 2H), 7.93 (dd, 1H), 7.85-7.74 (m, 1H), 7.66-7.60 (m, 2H), 7.60-7.48 (m, 3H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.03 min, m/z = 497/499 [M+H]+. Beispiel 66
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-2,5-difluorbenzoesäure
99 mg (0.17 mmol, Reinheit 85%) der Verbindung aus Beispiel 96A wurden in 5 ml eines THF/ Methanol-Gemisches (5: 1) gelöst und mit 0.82 ml (0.82 mmol) einer 1 M Lösung von Lithiumhydroxid in Wasser versetzt. Das Reaktionsgemisch wurde 3 h bei 50°C gerührt, danach auf RT abgekühlt und mit 4 M Salzsäure auf pH 1-2 gestellt. Anschließend wurde das Gemisch ohne weitere Aufarbeitung mittels präparativer HPLC (Methode 6) gereinigt. Die Produktfraktionen wurden eingeengt und der Rückstand im Vakuum getrocknet. Es wurden 81 mg (99% d. Th., Reinheit >99%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.47 (br. s, 1H), 11.23 (s, 1H), 8.25 (dd, 1H), 8.04 (d, 1H), 8.01 (d, 1H), 7.93 (dd, 1H), 7.77 (dd, 1H), 7.66-7.60 (m, 2H), 7.60-7.48 (m, 3H), 2.41 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.07 min, m/z = 497/499 [M+H]+.
Beispiel 67
6-Brom-/V-(2-fluor-4-{ [(trifluormethyl)sulfonyl]carbamoyl}phenyl)-3-methyl-2-phenylchinolin-4- carboxamid
Zu einer Suspension von 20 mg (0.50 mmol) Natriumhydrid (60% in Mineralöl) in 2.5 ml THF wurden 75 mg (0.50 mmol) Trifluormethansulfonsäureamid gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurden 124 mg (0.25 mmol) der Verbindung aus Beispiel 40A hinzugegeben, und das Gemisch wurde weitere 30 min bei RT gerührt. Danach wurde das Gemisch eingeengt. Der Rückstand wurde in DMSO aufgenommen und mittels präparativer HPLC (Methode 18) gereinigt. Die produkthaltigen Fraktionen wurden eingeengt, und der Rückstand wurde in einem Gemisch aus Dichlormethan und Methanol aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden 77 mg (50% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.90 (s, 1H), 8.07-7.98 (m, 3H), 7.95 (dd, 1H), 7.85 (dd, 1H), 7.75 (dd, 1H), 7.66-7.61 (m, 2H), 7.60-7.50 (m, 3H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.10 min, m/z = 610/612 [M+H]+.
Beispiel 68 6-Brom-/V- { 4-[(dimethylsulf amoyl)carbamoyl] -2-fluorphenyl } -3 -methyl-2-phenylchinolin-4- carboxamid
Zu einer Suspension von 20 mg (0.50 mmol) Natriumhydrid (60% in Mineralöl) in 2.5 ml THF wurden 62 mg (0.50 mmol) A^ -Dimefhylsulfonsäurediamid gegeben, und das Gemisch wurde 30 min bei RT gerührt. Anschließend wurde 1 ml DMF zugesetzt und das Gemisch 2 h bei 50°C gerührt. Nach Abkühlen auf RT wurden 124 mg (0.25 mmol) der Verbindung aus Beispiel 40 A hinzugegeben, und das Gemisch wurde weitere 30 min bei RT gerührt. Danach wurde das Gemisch eingeengt. Der Rückstand wurde in DMSO aufgenommen und mittels präparativer HPLC (Methode 18) gereinigt. Die produkthaltigen Fraktionen wurden eingeengt, und der Rückstand wurde in einem Gemisch aus Dichlormethan und Methanol aufgenommen, erneut eingeengt und dann im Vakuum getrocknet. Es wurden 40 mg (27% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 11.92 (s, 1H), 11.08 (s, 1H), 8.22 (t, 1H), 8.05 (d, 1H), 7.99 (d, 1H), 7.95 (d, 1H), 7.94-7.87 (m, 2H), 7.66-7.61 (m, 2H), 7.59-7.48 (m, 3H), 2.91 (s, 6H), 2.43 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.11 min, m/z = 585/587 [M+H]+. Beispiel 69
Kalium-4- { [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoat
1.10 g (2.30 mmol) der Verbindung aus Beispiel 7 wurden mit 200 ml Methanol und 5 ml DMF versetzt und das Gemisch kurz zum Sieden erhitzt. In die heiße Lösung wurde eine Lösung von 129 mg (2.30 mmol) Kaliumhydroxid in 30 ml Wasser gegeben. Nach Abkühlen auf RT wurde das Gemisch bei RT an der Luft bis zum Verdunsten des Lösungsmittels stehengelassen. Ein Teil des Rückstandes wurde 1-1.5 h bei 200°C getempert. Nach Abkühlen auf RT und Umfüllen wurden daraus 0.50 g (42% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 10.72 (br. s, 1H), 8.05-8.00 (m, 2H), 7.95-7.90 (m, 1H), 7.76-7.68 (m, 2H), 7.67-7.60 (m, 3H), 7.59-7.49 (m, 3H), 2.45 (s, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.05 min, m/z = 479/481 [M+H]+.
Beispiel 70
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure-L-Lysinsalz
2.0 g (4.18 mmol) der Verbindung aus Beispiel 7 wurden mit 100 ml 1,4-Dioxan versetzt und das Gemisch kurz zum Sieden erhitzt. In die heiße Lösung wurde eine Lösung von 610 mg (4.18 mmol) L-Lysin in 20 ml Wasser gegeben. Nach Abkühlen auf RT wurde das Gemisch bei RT an der Luft bis zum Verdunsten des Lösungsmittels stehengelassen. Der Rückstand wurde in 20 ml eines EthanolA asser-Gemisches (1: 1) suspendiert und eine Woche bei RT gerührt. Anschließend wurde der Feststoff abfiltriert und bei RT an der Luft getrocknet. Es wurden 2.2 g der Titelverbindung erhalten (84% d. Th., Reinheit 100%, noch Lösungsmittel-haltig).
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.70 (br. s, 1H), 8.03 (d, 1H), 8.00 (d, 1H), 7.93 (dd, 1H), 7.82 (t, 1H), 7.75 (dd, 1H), 7.68 (dd, 1H), 7.65-7.60 (m, 2H), 7.59-7.49 (m, 3H), 3.16 (t, 1H), 2.75 (t, 2H), 2.44 (s, 3H), 1.79-1.29 (m, 6H).
LC/MS (Methode 1, ESIpos): Rt = 1.05 min, m/z = 479/481 [M+H]+.
Beispiel 71
2-Hydroxy-/V,/V,/V-trimethylethanaminium-4- { [(6-brom-3-methyl-2-phenylchinolin-4-yl)carbonyl] - amino } -3-fluorbenzoat
2.0 g (4.18 mmol) der Verbindung aus Beispiel 7 wurden mit 100 ml 1,4-Dioxan versetzt und das Gemisch kurz zum Sieden erhitzt. In die heiße Lösung wurde ein Gemisch von 1.23 g (5.09 mmol) einer 50%-gewichtsprozentigen wässrigen Lösung von 2-Hydroxy-/V,/V,/V-trimethylethanaminium- hydroxid (Cholin-Hydroxid) und 20 ml Wasser gegeben. Nach Abkühlen auf RT wurde das Ge- misch bei RT an der Luft bis zum Verdunsten des Lösungsmittels stehengelassen. Es wurden 2.5 g der Titelverbindung erhalten (quant., Reinheit 100%, noch wasserhaltig).
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.62 (br. s, 1H), 8.07-7.98 (m, 2H), 7.96-7.87 (m, 1H), 7.74-7.65 (m, 2H), 7.65-7.59 (m, 3H), 7.59-7.47 (m, 3H), 3.87-3.82 (m, 2H), 3.43-3.38 (m, 2H), 3.11 (s, 9H), 2.44 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.06 min, m/z = 479/481 [M+H]+. Beispiel 72
4-{ [(6-Brom-3-methyl-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure-2-Amino-2- (hydroxymethyl)propan-l,3-diol-Salz
2.0 g (4.18 mmol) der Verbindung aus Beispiel 7 wurden mit 100 ml 1,4-Dioxan versetzt und das Gemisch kurz zum Sieden erhitzt. In die heiße Lösung wurde eine Lösung von 507 mg (4.18 mmol) 2-Amino-2-(hydroxymethyl)propan-l,3-diol (TRIS) in 20 ml Wasser gegeben. Nach Abkühlen auf RT wurde das Gemisch bei RT an der Luft bis zum Verdunsten des Lösungsmittels stehengelassen. Nach Umfüllen wurden 2.5 g der Titelverbindung erhalten (quant., Reinheit 100%, noch wasserhaltig). Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 10.89 (br. s, 1H), 8.04 (d, 1H), 8.01 (d, 1H), 7.97-7.90 (m, 2H), 7.80 (dd, 1H), 7.72 (dd, 1H), 7.66-7.60 (m, 2H), 7.60-7.49 (m, 3H), 3.44 (s, 6H), 2.45 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.07 min, m/z = 479/481 [M+H]+.
Beispiel 73 4- { [(6-Brom-3-fluor-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoesäure
Zu einer Lösung von 45 mg (0.06 mmol, Reinheit 69%) der Verbindung aus Beispiel 98A in 1.0 ml THF und 0.2 ml Methanol wurden bei RT 0.31 ml (0.31 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1.5 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurden 0.03 ml (0.37 mmol) Trifluoressigsäure hinzugegeben. Anschließend wurde das Gemisch direkt mittels präpara- tiver HPLC (Methode 18) gereinigt. Es wurden 5 mg (16% d. Th., Reinheit 94%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.22 (br. s, 1H), 11.21 (s, 1H), 8.33 (t, 1H), 8.17-8.12 (m, 2H), 8.09-8.04 (m, 2H), 8.00 (dd, 1H), 7.89 (dd, 1H), 7.81 (dd, 1H), 7.65-7.57 (m, 3H). LC/MS (Methode 1, ESIpos): Rt = 1.14 min, m/z = 483/485 [M+H]+.
Beispiel 74
4-{ [(6-Brom-3-methoxy-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
Die Titelverbindung wurde als weiteres Produkt der in Beispiel 73 beschriebenen Umsetzung erhalten. Bei der Aufreinigung des Reaktionsgemisches durch präparative HPLC nach Methode 18 (siehe Beispiel 73) war eine weitere Produktfraktion gewonnen worden, welche durch erneute präparative HPLC nachgereinigt wurde [Säule: Kinetix C18, 5 μιη, 100 x 21.2 mm; Fluss: 60 ml/min; Detektion: 210 nm; Injektionsvolumen: 1.0 ml; Temperatur: 40°C; Eluent: Gradient 95% Wasser / 0% Acetonitril / 5% (Acetonitril/Wasser 80:20 + 2% Ameisensäure) -> 30% Wasser / 65% Aceto- nitril / 5% (Acetonitril/Wasser 80:20 + 2% Ameisensäure); Laufzeit: 10.8 min]. Es wurden auf diese Weise 9 mg (29% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 11.03 (s, 1H), 8.23 (t, 1H), 8.07 (d, 1H), 8.02-7.98 (m, 3H), 7.91 (dd, 1H), 7.88 (d, 1H), 7.80 (d, 1H), 7.61-7.53 (m, 3H), 3.65 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.08 min, m/z = 495/497 [M+H]+.
Beispiel 75
4-{ [(3-Chlor-6-iod-2-phenylchinolin-4-yl)carbonyl]amino}-3-fluorbenzoesäure
Zu einer Lösung von 46 mg (0.07 mmol, Reinheit 90%) der Verbindung aus Beispiel 101A in 2.3 ml THF und 0.5 ml Methanol wurden bei RT 0.44 ml (0.44 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 20 h bei RT gerührt. Anschließend wurde mit Trifluoressigsäure auf pH 3 angesäuert und das Gemisch mittels präparativer HPLC (Methode 18) gereinigt. Es wurden 32 mg (77% d. Th., Reinheit 98%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.24 (br. s, 1H), 11.20 (s, 1H), 8.31 (t, 1H), 8.21 (d, 1H), 8.17 (dd, 1H), 7.94 (d, 1H), 7.90 (dd, 1H), 7.82 (dd, 1H), 7.78-7.73 (m, 2H), 7.61-7.52 (m, 3H).
LC/MS (Methode 1, ESIpos): R
t = 1.11 min, m/z = 547 [M+H]
+.
Beispiel 76
4- { [(6-Brom-3-chlor-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoesäure
Zu einer Lösung von 45 mg (0.07 mmol, Reinheit 85%) der Verbindung aus Beispiel 104A in 2.3 ml THF und 0.5 ml Methanol wurden bei RT 0.44 ml (0.44 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 20 h bei RT gerührt. Anschließend wurde mit Trifluoressigsäure auf pH 3 angesäuert und das Gemisch mittels präparativer HPLC (Methode 18) vorgereinigt. Das so erhaltene Produkt wurde in 10 ml eines Acetonitril/Methanol/DMSO/Ameisensäure -Gemisches in der Wärme gelöst und durch erneute präparative HPLC nachgereinigt [Säule: Kinetix C18, 5 μιη, 100 x 21.2 mm; Fluss: 25 ml/min; Detektion: 210 nm; Injektionsvolumen: 2.75 ml; Temperatur: 35°C; Eluent: Gradient 50% Wasser / 45% Acetonitril / 5% Ameisensäure (1% in Wasser) -> 20% Wasser / 75% Acetonitril / 5% Ameisensäure (1% in Wasser); Laufzeit: 6.0 min]. Es wurden so 22 mg (56% d. Th., Reinheit 95%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d
6): δ [ppm] = 13.24 (br. s, 1H), 11.21 (s, 1H), 8.32 (t, 1H), 8.14-8.09 (m, 1H), 8.07-8.02 (m, 2H), 7.92-7.87 (m, 1H), 7.81 (dd, 1H), 7.78-7.73 (m, 2H), 7.61-7.54 (m, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.09 min, m/z = 499/501 [M+H]+.
Beispiel 77 4- { [(6-Brom-3-cyclopropyl-2-phenylchinolin-4-yl)carbonyl]amino} -3-fluorbenzoesäure
Zu einer Lösung von 33 mg (0.05 mmol, Reinheit 77%) der Verbindung aus Beispiel 106A in 1.5 ml THF und 0.3 ml Methanol wurden bei RT 0.073 ml (0.073 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 2 h bei RT stehen gelassen. Anschließend wurde das Gemisch 5 h bei 60°C gerührt und dann wieder 2 Tage bei RT stehen gelassen. Nach Zugabe von weiteren 0.15 ml (0.15 mmol) 1 M Natronlauge wurde das Gemisch nochmals einen Tag bei 60°C gerührt. Danach wurde mit Trifluoressigsäure auf pH 3 angesäuert und das Gemisch mittels präparativer HPLC (Methode 18) gereinigt. Es wurden 19 mg (75% d. Th., Reinheit 100%) der Titelverbindung erhalten. Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 13.24 (br. s, 1H), 11.00 (s, 1H), 8.25 (t, 1H), 8.07-8.02 (m, 2H), 7.94 (dd, 1H), 7.90 (dd, 1H), 7.82 (dd, 1H), 7.76 (dd, 2H), 7.57-7.46 (m, 3H), 2.46-2.30 (m, 1H), 0.68 (d, 2H), 0.33 (d, 2H).
LC/MS (Methode 1, ESIpos): Rt = 1.15 min, m/z = 505/507 [M+H]+. Beispiel 78 4- { [(6-Brom-3,8-dimethyl-2-phenylchinolin-4-yl)carbonyl] amino } -3-fluorbenzoesäure
Zu einer Lösung von 649 mg (1.02 mmol, Reinheit 80%) der Verbindung aus Beispiel 108A in 13 ml THF und 2.6 ml Methanol wurden bei RT 4.3 ml (4.3 mmol) 1 M Natronlauge gegeben, und das Gemisch wurde 1.5 h unter Rückfluss gerührt. Nach Abkühlen auf RT wurde das Gemisch mit Trifluoressigsäure auf pH 3 angesäuert und dann mittels präparativer HPLC gereinigt [Säule: Kinetix C18, 5 μιη, 100 x 21.2 mm; Fluss: 60 ml/min; Detektion: 210 nm; Injektionsvolumen: 1.0 ml; Temperatur: 40°C; Eluent: Gradient 50% Wasser / 30% Acetonitril / 20% (Acetonitril/ Wasser 80:20 + 2% Ameisensäure)— > 30% Wasser / 65% Acetonitril / 5% (Acetonitril/Wasser 80:20 + 2% Ameisensäure); Laufzeit: 5.7 min]. Es wurden 419 mg (83% d. Th., Reinheit 100%) der Titelverbindung erhalten.
Ή-NMR (400 MHz, DMSO-de): δ [ppm] = 13.25 (br. s, 1H), 11.02 (s, 1H), 8.19 (t, 1H), 7.89 (dd, 1H), 7.86-7.83 (m, 1H), 7.81 (td, 2H), 7.70-7.64 (m, 2H), 7.60-7.49 (m, 3H), 2.73 (s, 3H), 2.45 (s, 3H).
LC/MS (Methode 1, ESIpos): Rt = 1.19 min, m/z = 493/495 [M+H]+.
B. Bewertung der pharmakologischen Wirksamkeit
Die pharmakologische Aktivität der erfindungsgemäßen Verbindungen kann durch in vitro- und in vzvo-Untersuchungen, wie sie dem Fachmann bekannt sind, nachgewiesen werden. Die nachfolgenden Anwendungsbeispiele beschreiben die biologische Wirkung der erfindungsgemäßen Verbindungen, ohne die Erfindung auf diese Beispiele zu beschränken.
Abkürzungen und Akronyme:
BSA bovines Serumalbumin
CRTH2 Chemoattractant Receptor-homologous molecule expressed on
T-Helper type 2 cells
DMEM Dulbecco's modified Eagle's medium
DMSO Dimethylsulfoxid
DP PGD2-Rezeptor
EC50 halbmaximale effektive Konzentration
Em Emission
EP PGE2-Rezeptor
Ex Exzitation
Fa. Firma (Bezugsquelle)
FCS fötales Kälberserum
FP PGF2a-Rezeptor
HEPES 2-[4-(2-Hydroxyethyl)piperazin-l-yl]ethansulfonsäure
IC50 halbmaximale inhibitorische Konzentration
IP PGI2-rezeptor
MES 2-(/V-Morpholino)efhansulfonsäure
Pen/Strep Penicillin/Streptomycin
PGD2 Prostaglandin D2
PGE2 Prostaglandin E2
PGF2a Prostaglandin F2a
PGI2 Prostaglandin Γ2
TC tissue culture
TP Thromboxan A2-Rezeptor
Tris Tris(hydroxymethyl)aminomethan
v/v Volumen zu Volumen- Verhältnis (einer Lösung)
w/w Gewicht zu Gewicht- Verhältnis (einer Lösung)
B-l. In νί Γο-Test auf Hemmung der humanen FP-Rezeptor- Aktivität Variante B-1A:
Für die Charakterisierung von Testsubstanzen auf FP-Antagonismus wurde der PGF2a-induzierte Calcium-Flux in FP-exprimierenden CHEM1 -Zellen (Millipore, HTS093C) verwendet. 3000 Zellen in 25 μΐ Vollmedium [DMEM F12, 10% FCS, 1.35 mM Natriumpyruvat, 20 mM HEPES, 4 mM GlutaMAX™, 2% Natriumbicarbonat, 1 % Pen/Strep, 1% lOOx nicht-essentielle Aminosäuren] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC -Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Fluo-8 AM-Beladungspuffer [Calcium-freie Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 4.8 mM NaHC03, pH 7.4), 2 mM CaCl2, lx Smart- Block (Fa. CANDOR Bioscience GmbH), 4.5 mM Probenecid, 5 μΜ Fluo-8 AM, 0.016% Plu- ronic®, 0.04% Brilliant black] ersetzt und bei 37°C / 5% C02 für 30 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit Calcium-freier Tyrode / 2 mM CaCl2 vorverdünnt. 10 μΐ der vorverdünnten Substanzlösung werden zu den Fluo-8-beladenen Zellen gegeben und bei 37°C / 5% C02 für 10 Minuten inkubiert. Der FP-Rezeptor wird durch Zugabe von 20 μΐ 3 nM (finale Konzentration) PGF2a in Calcium-freier Tyrode / 2 mM CaCl2 / 0.04% Brilliant black aktiviert, und der Calcium-Flux wird durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) bestimmt.
In der folgenden Tabelle 1A sind für individuelle Ausführungsbeispiele der Erfindung die IC50- Werte aus diesem Assay aufgeführt (zum Teil als Mittelwerte aus mehreren unabhängigen Einzelbestimmungen):
Tabelle 1A
Beispiel Nr. FP-Rezeptoraktivität Beispiel Nr. FP-Rezeptoraktivität
IC50 [μηιοΙ/L] IC50 [μηιοΙ/L]
1 0.166 6 0.079
2 0.148 7 0.020
3 0.170 8 0.795
4 0.880 9 0.065
5 0.905 10 0.041
Beispiel Nr. FP-Rezeptoraktivität Beispiel Nr. FP-Rezeptoraktivität ICso [μηιοΙ/L] ICso [μηιοΙ/L]
11 0.053 40 0.560
12 0.022 41 0.025
13 0.150 42 0.033
14 0.617 43 0.052
15 0.045 44 0.033
16 0.278 45 0.205
17 0.060 46 0.140
18 0.335 47 0.197
19 0.650 48 0.102
20 0.530 49 0.120
21 0.204 50 0.037
22 0.290 51 0.091
23 0.057 52 0.017
24 0.124 53 0.031
25 0.720 54 0.102
26 0.086 55 0.049
27 0.097 56 0.285
28 0.017 57 0.437
29 0.890 58 0.557
30 0.006 59 0.203
31 0.165 60 0.027
32 0.245 61 0.131
33 0.272 62 0.301
34 0.093 63 0.246
35 0.290 64 0.362
36 0.078 65 0.105
37 0.039 66 0.282
38 0.265 67 0.239
39 0.190 68 0.318
Beispiel Nr. FP-Rezeptoraktivität Beispiel Nr. FP-Rezeptoraktivität ICso [μηιοΙ/L] ICso [μηιοΙ/L]
69 0.104 75 0.009
70 0.089 76 0.017
71 0.090 77 0.027
72 0.079 78 0.154
74 0.220
Variante B-1B:
Für die Charakterisierung von Testsubstanzen auf FP-Antagonismus wurde der PGF2a-induzierte Calcium-Flux in rekombinant FP-exprimierenden CHO-Zellen, die zusätzlich das Ca2+-Sensorpro- tein GCaMP6 exprimieren, verwendet.
3000 Zellen in 25 μΐ Vollmedium [DMEM F12, 10% FCS, 1.35 mM Natriumpyruvat, 20 mM HEPES, 4 mM GlutaMAX™, 2% Natriumbicarbonat, 1% Pen/Strep, 1% lOOx nicht-essentielle Aminosäuren] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC -Platte, schwarz mit klarem Boden) ausgesät und bei 37°C, 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Puffer [Calcium-freie Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 4.8 mM NaHC03, pH 7.4), 2 mM CaCl2, 0.01% BSA] ersetzt und bei 37°C, 5% CO2 für 30 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1:50 mit Calcium-freier Tyrode / 2 mM CaCh / 0.01% BSA vorverdünnt. 10 μΐ der vorverdünnten Substanzlösung werden zu den Zellen gegeben und bei 37°C, 5% CO2 für 10 Minuten inkubiert. Der FP-Rezeptor wird durch Zugabe von 20 μΐ 3 nM (finale Konzentration) PGF2a in Calcium-freier Tyrode / 2 mM CaCh aktiviert, und der Calcium-Flux wird durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden in einem Fluoreszenzmessgerät (FLIPR Tetra®, MolecularDevices) bestimmt.
In der folgenden Tabelle 1B sind für individuelle Ausführungsbeispiele der Erfindung die IC50- Werte aus diesem Assay aufgeführt (zum Teil als Mittelwerte aus mehreren unabhängigen Einzelbestimmungen):
Tabelle 1B
B-2. In vitro FP-Rezeptorbindung-Inhibitionstest Für den FP-Rezeptorbindungstest werden humane rekombinante Prostanoid-FP-Rezeptoren, expri- miert in HEK293-Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268510). 80 μg Membran werden mit 1 nM [3H]-PGF2a für 60 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in An- Wesenheit von 1 μΜ Cloprostenol bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGF2a zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Abramovitz et al., /. Biol. Chem. 1994, 269 (4): 2632] .
B-3. In vitro CRTH2-Rezeptorbindung-Inhibitionstest Für diesen Test werden humane rekombinante Prostanoid-CRTH2-Rezeptoren, exprimiert in CHO- Kl-Zellen, in modifiziertem Tris-HCl-Puffer, pH 7.4, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268030). 4 μg Membran werden mit 1 nM [3H]-PGD2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 1 μΜ PGD2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGD2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Sugimoto et al., /. Pharmacol. Exp. Ther. 2003, 305 (1): 347] .
B-4. In vitro DP-Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-DP-Rezeptoren, exprimiert in Chem-1- Zellen, in modifiziertem HEPES-Puffer, pH 7.4, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268060). 10 μg Membran werden mit 2 nM [3H]-PGD2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 1 μΜ PGD2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGD2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Wright et al., Br. J. Pharmacol. 1998, 123 (7): 1317; Sharif et al., Br. J. Pharmacol. 2000, 131 (6): 1025] .
B-5. In vitro EP 1 -Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-EP 1 -Rezeptoren, exprimiert in HEK293- Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268110). 14 μg Membran werden mit 1 nM [3H]-PGE2 für 60 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Abra- movitz et al., Biochim. Biophys. Acta 2000, 1483 (2): 285; Funk et al., /. Biol. Chem. 1993, 268 (35): 26767] .
B-6. In vitro EP2-Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-EP2-Rezeptoren, exprimiert in HEK293- Zellen, in modifiziertem MES/KOH-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268200). 25 mg/ml Membran werden mit 4 nM [3H]-PGE2 für 120 Minuten bei 25 °C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermes- sen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Bastien et al., /. Biol. Chem. 1994, 269 (16): 11873; Boie et al., Eur. J. Pharmacol. 1997, 340 (2-3): 227] .
B-7. In vitro EP3-Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-EP3-Rezeptoren, exprimiert in HEK293- Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268310). 3 μg Membran werden mit 0.5 nM [3H]-PGE2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Schmidt et al., Eur. J. Biochem. 1995, 228 (1): 23] .
B-8. In vitro EP4-Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-EP4-Rezeptoren, exprimiert in Chem-1- Zellen, in modifiziertem MES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268420). 3 μg Membran werden mit 1 nM [3H]-PGE2 für 120 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ PGE2 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-PGE2 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Davis et al., Br. J. Pharmacol. 2000, 130 (8): 1919] .
B-9. In vitro IP-Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-IP-Rezeptoren, exprimiert in HEK293- Zellen, in modifiziertem HEPES-Puffer, pH 6.0, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #268600). 15 μg Membran werden mit 5 nM [3H]-Hoprost für 60 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 10 μΜ Iloprost bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H] -Iloprost zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosiswirkungskurve getestet [Lit.: Armstrong et al., Br. J. Pharmacol. 1989, 97 (3): 657; Boie et al., /. Biol. Chem. 1994, 269 (16): 12173] .
B-10. In vitro TP-Rezeptorbindung-Inhibitionstest
Für diesen Test werden humane rekombinante Prostanoid-TP-Rezeptoren, exprimiert in HEK-293 EBNA-Zellen, in modifiziertem Tris/HCl-Puffer, pH 7.4, verwendet. Die Durchführung dieses Tests wird kommerziell angeboten (Fa. Eurofins Panlabs, Katalog #285510). 18.4 μg Membran werden mit 5 nM [3H] -SQ-29,548 für 30 Minuten bei 25°C inkubiert. Die Menge an Membranprotein kann von Lot zu Lot variieren und wird bei Bedarf angepasst. Unspezifische Bindung wird in Anwesenheit von 1 μΜ SQ-29,548 bestimmt. Die Membranen werden gefiltert, gewaschen und dann vermessen, um die spezifische Bindung von [3H]-SQ-29,548 zu bestimmen. Substanzen werden auf inhibitorische Aktivität bei einer Konzentration von 10 μΜ oder in Form einer Dosis- wirkungskurve getestet [Lit.: Saussy Jr. et al., /. Biol. Chem. 1986, 261 : 3025; Hedberg et al., /. Pharmacol. Exp. Ther. 1988, 245: 786] .
B-l l . In νί Γο-Test auf DP-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf DP-Agonismus und -Antagonismus wurde der PGD2-induzierte Calcium-Flux in DP-exprimierenden CHEM 1 -Zellen (Millipore, HTS091C) ver- wendet: 3000 Zellen in 25 μΐ Vollmedium [DMEM, 4.5 g/1 Glucose, 10% Hitze-inaktiviertes FCS, 1% lOOx nicht-essentielle Aminosäuren, 10 mM HEPES, 0.25 mg/ml Geneticin (G418), 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC- Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 4.8 mM NaHC03, pH 7.4) / 2 mM CaC vorverdünnt. Für die DP-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΐ der vorverdünnten Substanzlösung zu den mit Calcium- Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die DP-Antagonismus-Messung wird der DP-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΐ -76 nM (2 x EC50, finale Konzentration) PGD2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: T. Matsuoka et al. (2000) Science 287: 2013-2017; S. Narumiya und G. A. Fitzgerald (2001) /. Clin. Invest. 108: 25-30] .
B-12. In νί Γο-Test auf EPl-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EPl-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EPl-exprimierenden CHEM1 -Zellen (Millipore, HTS099C) verwendet: 3000 Zellen in 25 μΐ Vollmedium [DMEM, 4.5 g/1 Glucose, 10% Hitze-inaktiviertes FCS, 1% lOOx nicht-essentielle Aminosäuren, 10 mM HEPES, 0.25 mg/ml Geneticin (G418), 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC- Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsub- stanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 4.8 mM NaHC03, pH 7.4) / 2 mM CaC vorverdünnt. Für die EPl-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΐ der vorverdünnten Substanzlösung zu den mit Calcium- Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP 1 -Antagonismus-Messung wird der EPl-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΐ ~6 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaCL aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: Y. Matsuoka et al. (2005) Proc. Natl. Acad. Sei. USA 102: 16066-16071; S. Narumiya und G. A. Fitzgerald (2001) /. Clin. Invest. 108: 25-30; K. Watanabe et al. (1999) Cancer Res. 59: 5093-5096].
B-13. In vitro-Test auf EP2-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EP2-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EP2-exprimierenden CHEM9-Zellen (Millipore, HTS 185C) verwendet: 3000 Zellen in 25 μΐ Plattiermedium [DMEM, 4.5 g/1 Glucose, 4 mM Glutamin, 10% Hitze-inaktiviertes FCS, 1% lOOx nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCh, 4.8 mM NaHCOs, pH 7.4) / 2 mM
CaCh vorverdünnt. Für die EP2-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΐ der vorverdünnten Substanzlösung zu den mit Calcium- Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP2- Antagonismus-Messung wird der EP2-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΐ -22 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaCh aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: C. R. Kennedy et al. (1999) Nat. Med. 5: 217-220; S. Narumiya und G. A. Fitzgerald (2001) /. Clin, luvest. 108: 25-30; N. Yang et al. (2003) /. Clin, luvest. 111 : 727-735] .
B-14. In νί Γο-Test auf EP3-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EP3-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EP3 (splice- Variante 6)-exprimierenden CHEM1 -Zellen (Milli- pore, HTS092C) verwendet: 3000 Zellen in 25 μΐ Plattiermedium [DMEM, 4.5 g/1 Glucose, 4 mM Glutamin, 10% Hitze-inaktiviertes FCS, 1% lOOx nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multi titerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C 1 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 4.8 mM NaHC03, pH 7.4) / 2 mM CaCh vorverdünnt. Für die EP3-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΐ der vorverdünnten Substanzlösung zu den mit Cal- cium-Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP3-Antagonismus-Messung wird der EP3-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΐ ~2 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaCh aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: M. Kotani et al. (1995) Mol. Pharmacol. 48: 869-879; M. Kotani et al. (1997) Genomics 40: 425-434; T. Kunikata et al. (2005) Nat. Immunol. 6: 524-531 ; S. Narumiya und G. A. Fitzgerald (2001) /. Clin. Invest. 108: 25-30; F. Ushikubi et al. (1998) Nature 395: 281-284].
B-15. In νί Γο-Test auf EP4-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf EP4-Agonismus und -Antagonismus wurde der PGE2-induzierte Calcium-Flux in EP4-exprimierenden CHEM1 -Zellen (Millipore, HTS 142C) verwendet: 3000 Zellen in 25 μΐ Plattiermedium [DMEM, 4.5 g/1 Glucose, 4 mM Glutamin, 10% Hitze-inaktiviertes FCS, 1% lOOx nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C 1 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsub- stanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 4.8 mM NaHC03, pH 7.4) / 2 mM CaC vorverdünnt. Für die EP4-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΐ der vorverdünnten Substanzlösung zu den mit Calcium- Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die EP4- Antagonismus-Messung wird der EP4-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΐ -26 nM (2 x EC50, finale Konzentration) PGE2 in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluores- zenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: S. Narumiya und G. A. Fitzgerald (2001) /. Clin. Invest. 108: 25-30; M. Nguyen et al. (1997) Nature 390: 78-81; K. Yoshida et al. (2002) Proc. Natl. Acad. Sei. USA 99: 4580-4585] .
B-16. In vitro-Test auf IP-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf ΓΡ-Agonismus und -Antagonismus wurde der Iloprost-induzierte Calcium-Flux in ΓΡ-exprimierenden CHEM1 -Zellen (Millipore, HTS 131C) verwendet: 3000 Zellen in 25 μΐ Plattiermedium [DMEM, 4.5 g/1 Glucose, 4 mM Glutamin, 10% Hitze-inaktiviertes FCS, 1% lOOx nicht-essentielle Aminosäuren, 10 mM HEPES, 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCh, 4.8 mM NaHCOs, pH 7.4) / 2 mM
CaCh vorverdünnt. Für die IP-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΐ der vorverdünnten Substanzlösung zu den mit Calcium- Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die IP-Antagonismus-Messung wird der IP-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΐ -106 nM (2 x EC50, finale Konzentration) Iloprost in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: S. Narumiya et al. (1999) Physiol. Rev. 79: 1193-1226; T. Murata et al. (1997) Nature 388: 678-682; Y. Cheng et al. (2002) Science 296: 539-541 ; C. H. Xiao et al. (2001) Circulation 104: 2210-2215; G. A. Fitzgerald (2004) N. Engl. J. Med. 351 : 1709-1711].
B-17. In vitro-Test auf TP-Agonismus und -Antagonismus
Für die Charakterisierung von Testsubstanzen auf TP-Agonismus und -Antagonismus wurde der U46619-induzierte Calcium-Flux in TP-exprimierenden CHEMl-Zellen (Millipore, HTS081C) verwendet: 3000 Zellen in 25 μΐ Plattiermedium [DMEM, 10% Hitze-inaktiviertes FCS, 1 % lOOx nicht-essentielle Aminosäuren, 10 mM HEPES, 0.25 mg/ml Geneticin (G418), 100 U/ml Penicillin und Streptomycin] werden je Vertiefung einer 384-Multititerplatte (Fa. Greiner, TC-Platte, schwarz mit klarem Boden) ausgesät und bei 37°C / 5% CO2 für 24 Stunden inkubiert. Vor der Messung wird das Medium durch 30 μΐ Calcium-Farbstoff-Beladungspuffer (FLIPR Calcium Assay, Molecular Devices) ersetzt und bei 37°C / 5% CO2 für 60 Minuten inkubiert. Die Testsubstanz wird in DMSO in verschiedenen Konzentrationen als Dosiswirkungskurve vorbereitet (Startkonzentration 10 mM, Verdünnungsfaktor 3.16) und 1 :50 mit zum Beispiel Calcium-freier Tyrode (130 mM NaCl, 5 mM KCl, 20 mM HEPES, 1 mM MgCl2, 4.8 mM NaHC03, pH 7.4) / 2 mM CaC vorverdünnt. Für die TP-Agonismus-Messung werden in einem Fluoreszenzmessgerät (FLIPR Tetra®, Molecular Devices) 10 μΐ der vorverdünnten Substanzlösung zu den mit Calcium- Farbstoff beladenen Zellen gegeben und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt. Danach werden die Zellen bei 37°C / 5% CO2 für 10 Minuten inkubiert. Für die TP- Antagonismus-Messung wird der TP-Rezeptor im FLIPR Tetra® durch Zugabe von 20 μΐ -88 nM (2 x EC50, finale Konzentration) U46619 in zum Beispiel Calcium-freier Tyrode / 2 mM CaC aktiviert und der Calcium-Flux durch Messung der Fluoreszenz bei Ex. 470 nm / Em. 525 nm für 120 Sekunden bestimmt [Lit.: S. Ali et al. (1993) /. Biol. Chem. 268: 17397-17403; K. Hanasaki et al. (1989) Biochem. Pharmacol. 38: 2967-2976; M. Hirata et al. (1991) Nature 349: 617-620] .
B-18. Tiermodell der Bleomycin-iriduzierten pulmonalen Fibrose
Die Bleomycin-induzierte Lungenfibrose bei der Maus oder Ratte ist ein weit verbreitetes Tiermodell für die Lungenfibrose. Bleomycin ist ein Glykopeptid-Antibiotikum, das in der Onkologie zur Therapie von Hodentumoren, Hodgkin- und Non-Hodgkin-Tumoren eingesetzt wird. Es wird renal eliminiert, besitzt eine Halbwertszeit von ca. 3 Stunden und beeinflusst als Zytostatikum verschiedene Phasen des Teilungszyklus [Lazo et ah, Cancer Chemother. Biol. Response Modif. 15, 44-50 (1994)]. Sein anti-neoplastischer Effekt beruht auf einer oxidativ-schädigenden Wirkung auf DNA [Hay et ah, Arch. Toxicol. 65, 81-94 (1991)] . Das Lungengewebe ist gegenüber Bleomycin in besonderer Weise gefährdet, da hier sog. Cysteinhydrolasen, welche in anderen Geweben zu einer Inaktivierung von Bleomycin führen, nur in geringer Anzahl vorhanden sind. Nach Gabe von Bleomycin kommt es bei den Tieren zu einem "acute respiratory distress Syndrome" (ARDS) mit anschliessender Entwicklung einer Lungenfibrose.
Die Verabreichung des Bleomycins kann in einfacher oder mehrfacher Gabe intratracheal, inhalativ, intravenös oder intraperitoneal erfolgen. Die Behandlung der Tiere mit der Testsubstanz (per gavage, durch Zusatz im Futter oder Trinkwasser, per osmotischer Minipumpe, per subkutaner oder intraperitonealer Injektion oder per Inhalation) beginnt am Tag der ersten Applikation des Bleomycins oder therapeutisch 3-14 Tage später und erstreckt sich über einen Zeitraum von 2-6 Wochen. Am Studienende werden eine bronchio-alveoläre Lavage zur Bestimmung des Zellgehaltes und der pro-inflammatorischen und pro-fibrotischen Marker sowie eine histologische Beurtei- lung der Lungenfibrose durchgeführt.
B-19. Tiermodell der D012-Quarz-induzierten pulmonalen Fibrose
DQ12-Quarz-induzierte Lungenfibrose an Maus und Ratte ist ein weit verbreitetes Tiermodell für Lungenfibrose [Shimbori et ah, Exp. Lung Res. 36, 292-301 (2010)] . DQ12-Quarz ist ein durch Brechen beziehungsweise Mahlen hochaktiver Quarz. Die intratracheale oder inhalative Applika- tion von DQ12-Quarz führt bei Mäusen und Ratten zu einer Alveolarproteinose gefolgt von einer interstitiellen Lungenfibrose. Die Tiere erhalten eine einfache oder mehrfache intratracheale oder inhalative Instillation von DQ12-Quarz. Die Behandlung der Tiere mit der Testsubstanz (per gavage, durch Zusatz im Futter oder Trinkwasser, per osmotischer Minipumpe, per subkutaner oder intraperitonealer Injektion oder per Inhalation) beginnt am Tag der ersten Instillation des Sili- kats oder therapeutisch 3-14 Tage später und erstreckt sich über einen Zeitraum von 3-12 Wochen. Am Studienende werden eine bronchio-alveoläre Lavage zur Bestimmung des Zellgehalts und der pro-inflammatorischen und pro-fibrotischen Marker sowie eine histologische Beurteilung der Lungenfibrose durchgeführt.
B-20. Tiermodell der DQ12-Quarz- oder FITC-induzierten pulmonalen Inflammation
Eine intratracheale Gabe von DQ12-Quarz oder Fluoresceinisothiocyanat (FITC) bei Maus und Ratte führt zu einer Inflammation in der Lunge [Shimbori et ah , Exp. Lung Res. 36, 292-301 (2010)] . Die Tiere werden am Tag der Instillation von DQ12-Quarz oder FITC oder einen Tag später für eine Dauer von 24 h bis zu 7 Tagen mit der Testsubstanz behandelt (per gavage, durch Zusatz im Futter oder Trinkwasser, per osmotischer Minipumpe, per subkutaner oder intraperitonealer Injektion oder per Inhalation). Am Versuchsende wird eine bronchio-alveoläre Lavage zur Bestimmung des Zellgehaltes und der pro-inflammatorischen und pro-fibrotischen Marker durchgeführt.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Tablette: Zusammensetzung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm. Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt. i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.