WO2009135808A2 - Verfahren zur herstellung von 1,3,4-substituierten pyrazolverbindungen - Google Patents
Verfahren zur herstellung von 1,3,4-substituierten pyrazolverbindungen Download PDFInfo
- Publication number
- WO2009135808A2 WO2009135808A2 PCT/EP2009/055328 EP2009055328W WO2009135808A2 WO 2009135808 A2 WO2009135808 A2 WO 2009135808A2 EP 2009055328 W EP2009055328 W EP 2009055328W WO 2009135808 A2 WO2009135808 A2 WO 2009135808A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- alkyl
- compound
- optionally substituted
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(N(*)*)(F)F Chemical compound *C(N(*)*)(F)F 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/72—Hydrazones
- C07C251/74—Hydrazones having doubly-bound carbon atoms of hydrazone groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C251/76—Hydrazones having doubly-bound carbon atoms of hydrazone groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/72—Hydrazones
- C07C251/84—Hydrazones having doubly-bound carbon atoms of hydrazone groups being part of rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/72—Hydrazones
- C07C251/86—Hydrazones having doubly-bound carbon atoms of hydrazone groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
Definitions
- the present invention relates to a process for the preparation of 1, 3,4-substituted pyrazole compounds of the formula I,
- X is a group CX 1 X 2 X 3 , wherein
- X 1 , X 2 and X 3 independently of one another are hydrogen, fluorine or chlorine, where X 1 can also be C 1 -C 6 -alkyl or C 1 -C 4 -haloalkyl and where at least one of the radicals X 1 , X 2 is different from hydrogen , R 1 stands for Ci-C4-alkyl or cyclopropyl, and R 2 is CN or a group CÜ2R 2a, wherein
- R 2a is C 5 -C 6 -cycloalkyl, optionally substituted phenyl or C 1 -C 6 -alkyl, which may optionally be substituted by C 1 -C 4 -alkoxy, phenyl or C 1 -C 6 -cycloalkyl.
- Pyrazoles of general formula I are important starting materials for a number of pharmaceutical active ingredients and crop protection active ingredients, in particular of 1,3-substituted pyrazol-4-ylcarboxylic anilides, as described, for example, in US Pat. No. 5,498,624, EP 545099 A1, EP 589301 A1, WO 92/12970, US Pat. WO 03/066610, WO 2006/024389, WO 2007/003603, WO 2007/006806.
- 1, 3,4-substituted pyrazole compounds of the formula I is typically carried out by cyclization of suitable 1,3-difunctional compounds with substituted hydrazine compounds or by reaction of 1, 3-difunctional compounds with hydrazine, followed by alkylation to introduce the substituent on the nitrogen ( 1 position).
- a principal disadvantage of this approach is the lack of regioselectivity of the cyclization of 1, 3-difunctional compounds with substituted hydrazine compounds as well as the lack of regioselectivity of N-alkylation of pyrazoles, so that in both cases in addition to the desired 1, 3,4-substituted pyrazole compound of Formula I (1, 3-isomer) is also the 1, 4,5-substituted isomer of the formula I '(1, 5-isomer) is formed.
- WO 2003/051820 and WO 2005/042468 describe the cyclization of 2-haloacyl-3-aminoacrylic acid esters with alkylhydrazines to give 1-alkyl-3-haloalkylpyrazole-4-carboxylic acid esters.
- the selectivity to the desired isomer is unsatisfactory.
- WO 2008/022777 describes a process for preparing 1-substituted-3- (dihalomethyl) pyrazole-4-carboxylic acid esters in which vinylogous amidinium salts which are prepared by reacting ⁇ - (halomethyl) -difluoromethylamines with acrylates in the presence of a Lewis Acid, are reacted with substituted hydrazines.
- the selectivity to the desired isomer is unsatisfactory.
- the invention is therefore based on the object to provide a process for the preparation of 1, 3,4-substituted pyrazole compounds of the formula I cited above, which provides the desired 1, 3-isomer of the formula I with high yields and good selectivity. It has surprisingly been found that 1, 3,4-substituted pyrazole compounds of the formula I defined in the introduction can be prepared in a simple manner with high yields and high regioselectivity with respect to the desired 1,3-isomer, if suitable 1,3-difunctional compounds are used Compounds of formula II described below first with a hydrazone of the formula III described below and the resulting intermediate is treated with an acid in the presence of water.
- the present invention relates to a process for preparing 1, 3-substituted pyrazole compounds of the formula I defined at the outset, which comprises the following steps:
- Y is oxygen, a group NR ⁇ 1 or a group [NR y2 R y3 J + Z ", where R y1 , R y2 and R y3 independently of one another are C 1 -C 6 -alkyl, C 5 -C 6 -cycloalkyl, optionally substituted phenyl or optionally substituted phenyl-Ci-C4-alkyl, or
- R y2 and R y3 together with the nitrogen atom to which they are attached represent an N-bonded 5- to 8-membered, saturated, optionally substituted heterocycle which, in addition to the nitrogen atom, has 1 or 2 further heteroatoms selected from N , O and S may be ring atoms, and Z "is an anion;
- R 3 is OR 3a or a group NR 3b R 3c wherein R 3a , R 3b and R 3c independently of one another are C 1 -C 6 -alkyl, C 5 -C 6 -cycloalkyl, optionally substituted phenyl or optionally substituted phenyl-Ci -C4-alkyl, stand, or
- R 3b and R 3c together with the nitrogen atom to which they are attached, represent an N-linked 5- to 8-membered, saturated, optionally substituted heterocycle which, in addition to the nitrogen atom 1 or 2 further heteroatoms selected from among N, O and S as ring atoms,
- variable R 1 has the meanings given for formula I,
- R 4 and R 5 are independently hydrogen, Ci-C ⁇ -alkyl, which may be optionally substituted by Ci-C 4 -alkoxy, phenyl or Cs-C ⁇ -cycloalkyl, Cs-C ⁇ -cycloalkyl or optionally substituted phenyl, wherein at least one of R 4 or R 5 is other than hydrogen, and wherein
- R 4 and R 5 together with the carbon atom to which they are attached may also stand for a 5- to 10-membered saturated carbocycle which is optionally mono- or polysubstituted by C 1 -C 4 -alkyl groups and / or optionally substituted phenyl and / or one or two fused phenyl rings;
- the process according to the invention has a number of advantages.
- low temperatures are not required to achieve the desired selectivity and step i) and step ii ) can be carried out at moderate temperatures, for example in the range from 10 to 180 ° C., in particular in the range from 20 to 150 ° C.
- the reaction in steps i) and ii) may be carried out at temperatures up to -20 0 C and at lower temperatures, for example, which is not required to achieve the desired regioselectivity.
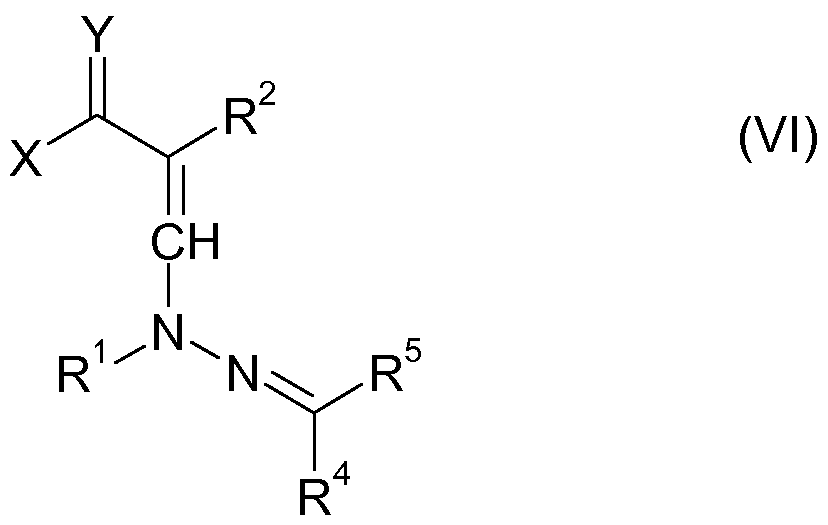
- step i) of the process according to the invention the compound of the formula VI shown below is formed, which can usually be isolated:
- halogen refers to each of fluorine, bromine, chlorine or iodine, especially fluorine, chlorine or bromine.
- C 1 -C 6 -alkyl denotes a saturated, straight-chain or branched hydrocarbon group comprising 1 to 6 carbon atoms, especially 1 to 4 carbon atoms, for example methyl, ethyl, propyl, 1-methylethyl, butyl tyl, 1-methylpropyl, 2-methylpropyl, 1, 1-dimethylethyl, pentyl, 1-methylbutyl, 2-methylbutyl, 3-methylbutyl, 2,2-dimethylpropyl, 1-ethylpropyl, hexyl, 1, 1-dimethylpropyl, 1, 2-dimethylpropyl, 1-methylpentyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, 1, 1-dimethylbutyl, 1, 2-dimethylbutyl, 1, 3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 3,3-dimethylbutyl
- C 1 -C 6 -alkyl which may optionally be substituted by C 1 -C 4 -alkoxy, phenyl or C 3 -C 6 -cycloalkyl, stands for unsubstituted C 1 -C 6 -alkyl, as defined above, or for C 1 -C 4 -alkyl -C ⁇ -alkyl, wherein one of the hydrogen atoms is replaced by a Ci-C 4 alkoxy, phenyl or Cs-C ⁇ -cycloalkyl group.
- C 1 -C 4 -haloalkyl as used herein describes straight-chain or branched alkyl groups having 1 to 4 carbon atoms, in which the hydrogen atoms of these groups are partially or completely replaced by halogen atoms, in particular by fluorine and / or chlorine, for example chloromethyl, Dichloromethyl, trichloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl, chlorofluoromethyl, dichlorofluoromethyl, chloro difluoromethyl, 1-chloroethyl, 1-fluoroethyl, 2-fluoroethyl, 2,2-difluoroethyl, 2,2,2-trifluoroethyl, 2-chloro-2-fluoroethyl, 2-chloro-2,2-difluoroethyl, 2,2- Dichloro-2-fluoroethyl, 2,2,2-trichloroethyl, pentafluoroethyl,
- C 1 -C 6 alkoxy describes straight-chain or branched saturated alkyl groups comprising 1 to 6 carbon atoms which are bonded via an oxygen atom.
- Examples include Ci-C ⁇ -alkoxy such as methoxy, ethoxy, OCH 2 -C 2 H 5, OCH (CHs) 2, n-butoxy, OCH (CHs) -C 2 H 5, OCH 2 -CH (CHs) 2, OC (CH 3 ) 3 , n-pentoxy, 1-methylbutoxy, 2-methylbutoxy, 3-methylbutoxy, 1, 1-dimethylpropoxy, 1, 2-dimethylpropoxy, 2,2-dimethylpropoxy, 1-ethylpropoxy, n-hexoxy, 1 - Methylpentoxy, 2-methylpentoxy, 3-methylpentoxy, 4-methylpentoxy, 1, 1-dimethylbutoxy, 1, 2-dimethylbutoxy, 1, 3-dimethylbutoxy, 2,2-dimethylbutoxy, 2,3-dimethylbutoxy
- C 1 -C 4 -alkoxy-C 1 -C 6 -alkyl refers to C 1 -C 6 -alkyl in which one of the hydrogen atoms is replaced by a C 1 -C 4 -alkoxy group.
- Examples of these are CH 2 -OCH 3 , CH 2 -OC 2 H 5 , n-propoxymethyl, CH 2 -OCH (CH 2 ) 2 , n-butoxymethyl, (1-methylpropoxy) methyl, (2-methylpropoxy) methyl, CH 2 -OC (CH 3 ) 3 , 2- (methoxy) ethyl, 2- (ethoxy) ethyl, 2- (n-propoxy) ethyl, 2- (1-methylethoxy) ethyl, 2- (n-butoxy) ethyl, 2- (1-methylpropoxy) ethyl, 2- (2-methylpropoxy) ethyl, 2- (1, 1-dimethylethoxy) ethyl, 2- (methoxy) propyl, 2- (ethoxy) propyl, 2- (n- Propoxy) propyl, 2- (1-methylethoxy) propyl, 2- (n-butoxy) propyl, 2- (1-methylpropoxy) propyl,
- Ca-C ⁇ -cycloalkyl as used herein describes monocyclic saturated hydrocarbon radicals comprising from 3 to 6 carbon atoms. Examples of monocyclic radicals include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- optionally substituted phenyl as used herein means unsubstituted phenyl or phenyl which carries 1, 2, 3, 4, or 5, especially 1, 2 or 3, substituents which are inert under the conditions of the reaction are.
- inert substituents are halogen, especially fluorine, chlorine or bromine, CN, NO2, d-Ce-alkyl, Ci-C 6 alkylthio, d-Ce-alkylsulfonyl, Ci-C4-haloalkyl, Ci-C 6 - Alkoxy, Cs-Ce-cycloalkyl, and Ci-C 4 alkoxy-Ci-C 6 -alkyl.
- phenyl-C 1 -C 6 -alkyl refers to C 1 -C 6 -alkyl wherein one of the hydrogen atoms is replaced by an optionally substituted phenyl group. Examples are benzyl, 4-methylbenzyl, phenylethyl, etc.
- N-bonded 5- to 8-membered, saturated, optionally substituted heterocycle means a saturated heterocycle bonded via a ring nitrogen atom which has 5, 6, 7 or 8 ring atoms, the ring atoms also being further apart from the nitrogen atom Heteroatoms include, and is unsubstituted or 1, 2, 3, 4, or 5, in particular 1, 2 or 3 substituents which are inert under the conditions of the reaction.
- inert substituents are CN, -C 6 - alkyl, d-Ce-alkylthio, Ci-C6-Al kylsu If onyl, Ci-C 4 haloalkyl, Ci-C 6 alkoxy, C 3 -C 6 - cycloalkyl, and C 1 -C 4 -alkoxy-C 1 -C 6 -alkyl.
- the heterocycle may also contain 1 or 2 further heteroatoms selected from among N, O and S as ring atoms.
- N-linked 5- to 8-membered, saturated, optionally substituted heterocycles are pyrrolidin-1-yl, piperdin-1-yl, morpholin-4-yl, piperazin-1-yl and N-methylpiperazin-1-yl.
- a preferred embodiment of the invention relates to the preparation of pyrazole compounds of the formula I in which R 2 is a COOR 2a group in which R 2a has the abovementioned meanings and in particular C 1 -C 6 -alkyl or C 1 -C 4 -alkoxy Ci-C 6 alkyl and especially Ci-C 4 alkyl.
- the group R 2 stands for a group COOR 2a in which R 2a has the abovementioned meanings and in particular for C 1 -C 6 -alkyl or C 1 -C 4 -alkoxy-C 1 -C 6 alkyl and especially Ci-C 4 alkyl.
- R 2 is CN. Accordingly, R 2 is also in the compounds of the formulas II and VI CN.
- the inventive method is particularly suitable for the preparation of compounds of general formula I, wherein X is a group CX 1 X 2 X 3 , wherein X 1 , X 2 and X 3 have the abovementioned meanings, wherein at least one of the radicals X 1 and X 2 is different from hydrogen.
- X 1 and X 2 are fluorine.
- X 3 is preferably hydrogen, fluorine or chlorine.
- Examples of preferred radicals CX 1 X 2 X 3 are dichloromethyl, chlorofluoromethyl, difluoromethyl, chlorodifluoromethyl and trifluoromethyl. According to a specific embodiment, X stands for a group CHF2.
- R 1 in the formula I and, accordingly, in formula III is C 1 -C 4 -alkyl and in particular methyl.
- the preparation of the pyrazole compounds of the formula I is carried out using a compound of the formula II in which Y is oxygen.
- a compound of the formula II in which Y is oxygen Such compounds are also referred to below as compounds IIa.
- Compounds of the formula IIa in which R 2 is a COOR 2a group in which R 2a has the abovementioned meanings and in particular C 1 -C 6 -alkyl or C 1 -C 4 -alkoxy-C 1 -C 6 -alkyl and especially C 1 -C 4 - AIkVl stands, are referred to below as compounds I Ia.1.
- R 2 , R 2a , R 3 and X have the meanings given above.
- X in the formulas IIa and Iaa.1 stands for a group CX 1 X 2 X 3 , in which X 1 , X 2 and X 3 have the meanings given above.
- at least one of X 1 and X 2 is different from hydrogen.
- X 1 and X 2 are fluorine.
- X 3 is preferably hydrogen, fluorine or chlorine. Examples of particularly preferred group CX 1 X 2 X 3 are dichloromethyl, trifluoromethyl, chlorodifluoromethyl, fluorochloromethyl and difluoromethyl.
- X stands for a group CHF2.
- the preparation of the pyrazole compounds of the formula I is carried out using a compound of the formula II in which Y is a group [NR y2 R y3 J + Z. Such compounds are also referred to below as bonds IIb.
- R 2 is a COOR 2a group in which R 2a has the abovementioned meanings and is in particular C 1 -C 6 -alkyl or C 1 -C 4 -alkoxy-C 1 -C 6 -alkyl, are also referred to below as Compounds I Ib.1 called.
- R 2 , R 2a , R ⁇ 2 , R ⁇ 3 , Z, R 3 and X have the meanings given above.
- X in the formulas IIb and IIb.1 is a group CX 1 X 2 X 3 in which X 1 , X 2 and X 3 have the meanings given above.
- at least one of X 1 and X 2 is different from hydrogen.
- X 1 and X 2 are fluorine.
- X 3 is preferably hydrogen, fluorine or chlorine. Examples of particularly preferred group CX 1 X 2 X 3 are trifluoromethyl, chlorodifluoromethyl, fluorochloromethyl and difluoromethyl.
- the group CX 1 X 2 X 3 in the formulas IIb, IIb.1 and IIb.2 stands for CHCIF or CHF 2 .
- R y2 and R y3 are in particular CrC 4 -AlkVl and especially methyl.
- Z stands for an anion or an anion equivalent which preferably consists of a Lewis acid such as MgF 2 , BF 3 , BCI 3 , AICI 3 , AIF 3 , ZnCl 2 , PF 5 , SbF 5 , BiCl 3 , GaCl 3 , SnCl 4 , or SiCl 4 , eg for fluoride, [MgF 3 ] -, [BF 4 ] -, [BCI 3 F] -, [AIF 4 ] -, [AICI 3 F] -, [ZnCl 2 F] -, [ PF6], [SbF 6 ], [BiCl 3 F], [GaCl 3 F], [SnCl 4 F] or [SiCl 4 F] -.
- a Lewis acid such as MgF 2 , BF 3 , BCI 3 , AICI 3 , AIF 3 , ZnCl 2 , PF 5 , SbF
- R 3 in the formulas II, IIa and IIa.1, IIb and IIb.1 is a group OR 3a .
- R 3a has the abovementioned meanings and is in particular C 1 -C 4 -alkyl and especially methyl or ethyl.
- R 3 in the formulas II, IIa and I Ia.1, IIb and IIb.1 is a group NR 3b R 3c .
- R 3b and R 3c have the meanings mentioned above and are in particular C 1 -C 4 -alkyl and especially for methyl or ethyl, or R 3b and R 3c form, together with the nitrogen atom to which they are bonded, an N-linked 5- to 8-membered, saturated heterocycle which, in addition to the nitrogen atom, has 1 or 2 further hetero roatome selected from N, O and S may have as ring atoms and may optionally carry 1 or 2 Ci-C4-alkyl groups.
- Examples of the latter cyclic group NR 3b R 3c are pyrrolidin-1-yl, morpholin-4-yl, piperidin-1-yl and 4-methylpiperazin-1-yl.
- hydrazone of the formula III used in the reaction is basically of minor importance. Basically, such hydrazones of formula III
- R 4 is hydrogen or C 1 -C 6 -alkyl
- R 5 is C 1 -C 6 -alkyl, C 3 -C 6 -cycloalkyl or optionally substituted phenyl, or R 4 and R 5, together with the carbon atom to which they are attached, may represent a 5- to 10-membered saturated carbocycle, which is optionally substituted one or more times, for example 1-, 2-, 3- or 4-fold, by C 1 -C 4 -alkyl groups and / or has a fused phenyl ring.
- a hydrazone of the formula III is used, in which
- R 4 is hydrogen or C 1 -C 4 -alkyl, in particular hydrogen, and R 5 is optionally substituted phenyl.
- a hydrazone of the formula III in which R 4 and R 5 are C 1 -C 4 -alkyl or together with the carbon atom to which they are attached form a 5 to 8-membered, saturated carbocycle which is optionally substituted in the manner described above.
- unsubstituted or substituted phenyl here has the abovementioned meanings and is in particular unsubstituted phenyl or phenyl which has 1, 2 or 3 substituents selected from halogen, in particular fluorine, chlorine or bromine, nitro, cyano, Ci-C 4 -alkyl, in particular methyl or ethyl, and C 1 -C 4 -alkoxy, in particular methoxy or ethoxy, for example as in 2-, 3- or 4-fluorophenyl, 2-, 3- or 4-chlorophenyl, 4-bromophenyl, 2 -, 3- or 4-methylphenyl, 2-, 3- or 4-methoxyphenyl, 4-cyanophenyl, 4-nitrophenyl.
- halogen in particular fluorine, chlorine or bromine
- nitro, cyano Ci-C 4 -alkyl
- Ci-C 4 -alkyl in particular methyl or ethyl
- the term "optionally substituted phenyl” has the abovementioned meanings and particularly preferably represents unsubstituted phenyl or phenyl which has 1 or 2 substituents selected from halogen, in particular chlorine, C 1 -C 4 -alkyl, in particular methyl or ethyl, and C 1 -C 4 -alkoxy, in particular methoxy or ethoxy, for example, as in 2-, 3- or 4-chlorophenyl, 2-, 3- or 4-methylphenyl, 2-, 3- or 4-methoxyphenyl.
- halogen in particular chlorine
- C 1 -C 4 -alkyl in particular methyl or ethyl
- C 1 -C 4 -alkoxy in particular methoxy or ethoxy
- R 5 is optionally substituted phenyl, in particular unsubstituted phenyl or phenyl which has 1 or 2 substituents, wherein the substituents are as mentioned above and are preferably selected from halogen, in particular chlorine, Ci-C 4 -AlkVl, in particular methyl or ethyl, and C 1 -C 4 -alkoxy, in particular methoxy or ethoxy.
- R 4 and R 5 together with the carbon atom to which they are bonded, represent a 5- to 10-membered, in particular 5 to 8-membered, saturated carbocycle which optionally has one or more than one, eg 1, 2, 3 or 4-fold, is substituted by C 1 -C 4 -alkyl groups.
- reaction of the compounds of the formula II with the hydrazone of the formula III in step i) of the process according to the invention is usually carried out at temperatures in the range from 0 to 180 ° C., in particular in the range from 10 to 150 ° C.
- the compounds II and III are preferably used in a ratio which corresponds to the stoichiometry of the reaction, it also being possible to deviate from the stoichiometry.
- the molar ratio of compound II to compound III is in the range of 1.5: 1 to 1: 1.5, often in the range of 1.2: 1 to 1: 1, 2, and most preferably in the range of 1.1: 1 to 1: 1, 1.
- the reaction in step i) takes place in an inert organic solvent.
- inert organic solvents are, in particular, aprotic organic solvents, such as aromatic hydrocarbons and halogenated hydrocarbons, for example benzene, toluene, xylene, cumene, chlorobenzene and tert-butylbenzene, cyclic or acyclic ethers, such as diethyl ether, diisopropyl ether, tert-butyl methyl ether (MTBE).
- the reaction is essentially anhydrous, ie the water content in the solution is below 1%, in particular below 0.1%, based on the total weight of the solvent.
- the procedure is generally such that the compound of the formula II, preferably in the form of a solution in one of the abovementioned inert organic solvents, with the hydrazone III, which is also preferably as Solution is used in one of the aforementioned inert organic solvents, together.
- the hydrazone III as a solution in an organic solvent and the compound II, preferably as a solution.
- the compound II can be initially introduced as a solution in an organic solvent and the hydrazone, preferably as a solution, added.
- the combination of the hydrazone III and the compound II can take place in the abovementioned temperature ranges.
- the procedure is such that the combination of the compounds II and III at temperatures ranging from 0 to 50 0 C, in particular 10 to 50 0 C is carried out and then the reaction mixture is heated to the desired temperature.
- the reaction time is typically in the range of 1 h to 15 h.
- the reaction mixture can also be fed without isolation of the compound VI to the reaction in step ii) of the process according to the invention.
- a procedure without isolation of the intermediate compound VI is advantageous since in this way yield losses such as occur in the separation of solids from the intermediate compound by filtration (eg losses in the mother liquor) can be reduced or avoided. In these cases, it is optionally possible to remove a partial amount of the organic solvent used in step i) and optionally replace it with another solvent.
- a mode of operation without isolation of the intermediate compound VI is particularly advantageous if, in the compound II used, the group Y stands for [NR y2 R y3 J + Z-.
- the reaction takes place in the presence of an acid, in particular a Bronsted acid.
- Preferred acids have a pKa of not more than 4, in particular not more than 3 or not more than 2 (in dilute (eg 0.01 M) aqueous solution at 25 ° C).
- Preferred acids are hydrohalic acids such as HF, HCl and HBr, in particular in the form of their aqueous solutions, sulfuric acid, phosphoric acid, HBF 4 , and organic sulfonic acids, for example aromatic sulfonic acids of the formula Ar-SOsH in which Ar represents optionally substituted phenyl, such as benzenesulfonic acid and p-toluenesulfonic acid, and aliphatic sulfonic acids such as methanesulfonic acid, ethanesulfonic acid and trifluoromethanesulfonic acid.
- aromatic sulfonic acids of the formula Ar-SOsH in which Ar represents optionally substituted phenyl, such as benzenesulfonic acid and p-toluenesulfonic acid
- aliphatic sulfonic acids such as methanesulfonic acid, ethanesulfonic acid and trifluoromethanesulf
- Chlorobenzoic Of course, mixtures of the aforementioned acids are also suitable.
- the acid can also be used in stoichiometric or superstoichiometric amount.
- the acid is used in an amount of from 0.01 to 10 mol and in particular in an amount of from 0.02 to 5 mol per mol of compound VI, or in the case of in situ preparation of compound VI, in an amount from 0.01 to 10 moles and especially in an amount of 0.02 to 2 moles per mole of compound II.
- water can also be used in a stoichiometric or superstoichiometric amount. In general, water is added in an amount of 0.001 to 50 mol and in particular in an amount of 0.01 to 20 moles per mole of compound VI, or in the case of in situ preparation of compound VI, in an amount of 0.001 to 50 mol and in particular in an amount of 0.01 to 20 moles per mole of compound II.
- Y O
- at least stoichiometric amounts of water are required for complete conversion in the cyclization.
- the water is typically added in an amount of from 1 to 50 moles and more particularly in an amount of from 1.1 to 20 moles per mole of Compound VI or, in the case of in situ preparation of Compound VI, in an amount from 1 to 50 moles and especially in an amount of 1.1 to 20 moles per mole of Compound II.
- Suitable organic solvents for the reaction in step ii) are protic polar solvents, e.g. aliphatic alcohols having preferably 1 to 4 carbon atoms, such as methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol or tert-butanol, or carboxylic acids, such as acetic acid, aromatic hydrocarbons, such as benzene, toluene, xylene, cumene, chlorobenzene , Nitrobenzene or tert-butylbenzene, aprotic polar solvents, eg cyclic or acyclic ethers such as diethyl ether, diisopropyl ether, tert-butyl methyl ether (MTBE), tert-butyl ethyl ether, tetrahydr
- protic polar solvents e.g. aliphatic alcohols having preferably 1
- step ii) is generally carried out in such a way that the compound of the formula VI prepared in step i) of the process according to the invention or in
- Step i) obtained reaction mixture, optionally after a partial or complete replacement of the solvent used in step i), in a suitable organic solvent and to this acid and water. It is possible to introduce the water required for the reaction over the organic solvent. It is also possible to introduce the water required for the reaction via acid, e.g. in the form of an aqueous solution of the acid or in the form of a hydrate of the acid.
- the reaction in step ii) of the process according to the invention is usually carried out at temperatures in the range from 0 to 150 ° C., in particular in the range from 20 to 110 ° C.
- the reaction time is typically in the range from 0.1 h to 15 h.
- step ii) the desired 1,3-pyrazole compound I is obtained in high yield and high selectivity, ie with a very low or undetectable amount of undesired 1,5-isomer I '.
- the molar ratio of 1,3-isomer of the formula I to 1,5-isomer of the formula I ' is generally at least 20: 1, frequently at least 50: 1, in particular at least 80: 1 and especially at least 100: 1.
- the desired 1,3-pyrazole compound I can be isolated from the reaction mixture by customary methods by precipitation, crystallization or distillation or further processed in the form of the reaction mixture to give by-products.
- Y is a group [NR y1 R y2 ] Z-, typically by reacting ⁇ , ⁇ -difluoroamines of the formula XIV with an olefinic compound of the formula XV in the presence of a Lewis acid such as MgF2, BF3, BCb, AICb, AIF3, ZnCb, PF5, SbFs, BiCb, GaCb, SnCU, or SiCU according to the procedure outlined in Scheme 4.
- a Lewis acid such as MgF2, BF3, BCb, AICb, AIF3, ZnCb, PF5, SbFs, BiCb, GaCb, SnCU, or SiCU according to the procedure outlined in Scheme 4.
- hydrazone compounds of the formula III used in the process according to the invention are known or can be prepared in a manner known per se by reacting a carbonyl compound of the formula IV with a substituted hydrazine compound of the formula V.
- R 1 , R 4 and R 5 have the meanings given for formula III or VI.
- the reaction of the compounds IV and V to give the hydrazone III can be carried out in a manner known per se.
- reaction of the carbonyl compound IV with the hydrazine compound V is usually carried out at temperatures in the range from 10 to 180 ° C., in particular in the range from 20 to 150 ° C.
- the compounds IV and V are preferably used in a ratio which corresponds to the stoichiometry of the reaction, it also being possible to deviate from the stoichiometry.
- the molar ratio of compound IV to compound V is in the range of 1.5: 1 to 1: 1.5, often in the range of 1.2: 1 to 1: 1.2, and more preferably in the range of 1.1: 1 to 1: 1, 1.
- inert organic solvents are in particular aprotic organic solvents such as aromatic hydrocarbons and halogenated hydrocarbons, e.g. Benzene, toluene, xylene, cumene, chlorobenzene and tert-butylbenzene, cyclic or acyclic ethers, such as diethyl ether, diisopropyl ether, tert.
- aprotic organic solvents such as aromatic hydrocarbons and halogenated hydrocarbons, e.g. Benzene, toluene, xylene, cumene, chlorobenzene and tert-butylbenzene, cyclic or acyclic ethers, such as diethyl ether, diisopropyl ether, tert.
- Butyl methyl ether MTBE
- tert-butyl ethyl ether tetrahydrofuran (THF) or dioxane
- nitriles such as acetonitrile and propionitrile
- aliphatic halogenated hydrocarbons such as dichloromethane, dichloroethane, trichloromethane and mixtures thereof.
- the procedure is generally such that the compound of the formula IV preferably in the form of a solution in one of the aforementioned inert organic solvents with which hydrazine compound V, preferably as a solution in water, together.
- the compounding of the compounds IV and V can take place in the above-mentioned temperature ranges.
- the procedure is such that the combining of the compounds IV and V takes place at temperatures in the range from 0 to 50 ° C., in particular from 10 to 50 ° C., and then the reaction mixture is heated to the desired temperature.
- the reaction time is typically in the range of 0.5 h to 8 h.
- the hydrazone can be isolated from the reaction mixture obtained by reaction of IV with V or as the reaction mixture in the subsequent stage, i. used in step I of the process according to the invention.
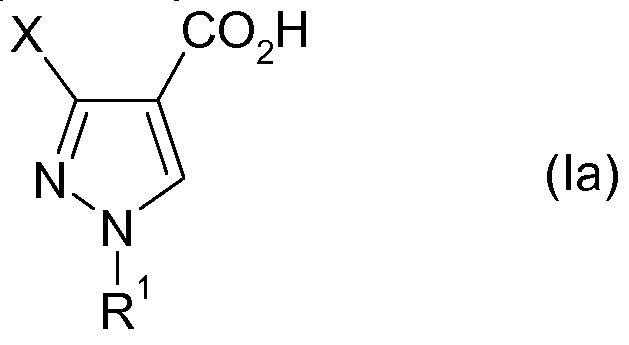
- Another object of the present invention relates to a process for the preparation of a compound of general formula Ia
- a preferred embodiment of the invention relates to a method comprising the following steps: a) the provision of a compound of the formula I by the process according to the invention as described and b) a hydrolysis of the compound I to give a 1, 3-substituted pyrazole-4-ylcarboxylic acid of the formula Ia.
- the hydrolysis can be carried out under acid catalysis or basic or otherwise.
- the compound I as such, d. H. be used after isolation.
- compound I For the basic hydrolysis of compound I, typically the compound of formula I with an alkali metal hydroxide such as sodium hydroxide, potassium hydroxide or lithium thiumhydroxid, preferably with an aqueous alkali metal hydroxide solution, especially an aqueous NaOH solution or an aqueous KOH solution, until complete Treat hydrolysis of the ester, preferably with heating.
- an alkali metal hydroxide such as sodium hydroxide, potassium hydroxide or lithium thiumhydroxid
- an aqueous alkali metal hydroxide solution especially an aqueous NaOH solution or an aqueous KOH solution
- the molar ratio of compound of formula I to base is typically in the range of 1.2: 1 to 1:10, and more preferably about equimolar (ie, in the range of 1.1: 1 to 1: 1) , 5), but also a larger base excess, z. B. up to 5 moles per mole of compound I, be of advantage.
- the basic hydrolysis is carried out in a diluent or solvent.
- Suitable diluents or solvents are not only water but also organic solvents which are stable to alkali, and mixtures thereof with water.
- alkali-stable organic solvents are, in particular, the abovementioned C 1 -C 4 -alkanols and the abovementioned acyclic and cyclic ethers.
- the hydrolysis is carried out in the aqueous phase, i. H. in water or a mixture of water with one of the aforementioned organic solvents, wherein the content of organic solvent in the aqueous phase typically typically does not exceed 30 vol .-%, based on the total amount of water and organic solvent.
- the basic hydrolysis is carried out at temperatures of 20 to 100 0 C.
- the upper temperature limit is the boiling point of the solvent used in pressure-free reaction.
- a reaction temperature of 100 0 C and in particular 90 0 C will not be exceeded.
- the basic hydrolysis is carried out at a temperature below the boiling point of the alcohol component, for example at temperatures in the range of 40 to ⁇ 80 0 C, in particular in the range of 50 to 75 ° C, especially when starting from a compound of general formula I, wherein R 1 is methyl or ethyl. Higher temperatures are also possible.
- the basic hydrolysis is carried out at a temperature above the boiling point of the alcohol component of the ester.
- the hydrolysis is preferably carried out at a temperature of at least 80 0 C, z. B. in the range of 80 to 100 0 C perform, for example, when starting from a compound of general formula I, wherein R 1 is ethyl.
- the reaction time is dependent on the reaction temperature, the concentration and the stability of the respective ester bond. In general, the reaction conditions are chosen so that the reaction time is in the range of 1 to 12 hours, in particular in the range of 2 to 8 hours.
- the pyrazole compound I obtained in substep a) is, in the event that R 2 is CÜ 2 R 2a or CN, without intermediate isolation, advantageously together with the organic solvent , reacted with the aqueous alkali metal hydroxide solution.
- the alkali metal salt of the pyrazolecarboxylic acid Ia thus formed is obtained as an aqueous phase in addition to the organic phase, which can be separated off by phase separation.
- the resulting in the phase separation aqueous phase containing the alkali metal salt of the 1, 3-substituted acid Ia usually in dissolved form and. The salt can then be converted into the free acid Ia by acidifying the solution as described above.
- the acid Ia precipitates out as a solid and can be isolated by filtration and optionally dried.
- the 1,3-substituted pyrazolecarboxylic acid is obtained in high purity and in very good yield.
- the yield, based on the compound II used, is generally at least 80% and in particular at least 85%.
- the acidic hydrolysis of Compound I may be carried out in analogy to known acidic ester hydrolyses, ie in the presence of catalytic or stoichiometric amounts of an acid and water (see, for example, J. March, Advanced Organic Chemistry, 2nd Ed., 334-338, McGraw-Hill, 1977 and cited literature). Frequently, the reaction is carried out in a mixture of water and an aprotic organic solvent. agent, for example an ether as mentioned above, perform.
- acids are hydrohalic acids, sulfuric acid, organic sulfonic acids such as p-toluenesulfonic acid, methanesulfonic acid, phosphoric acid and acidic ion exchanger resins and the like.
- Suitable hydrolysis catalysts are furthermore alkali metal iodides, such as lithium iodide, trimethyliodosilane or mixtures of trimethylchlorosilane with alkali metal iodides, such as lithium, sodium or potassium iodide.
- alkali metal iodides such as lithium iodide, trimethyliodosilane or mixtures of trimethylchlorosilane with alkali metal iodides, such as lithium, sodium or potassium iodide.
- the isolation of the acid Ia is then carried out by conventional separation methods, such as precipitation by adjusting the pH or extraction.
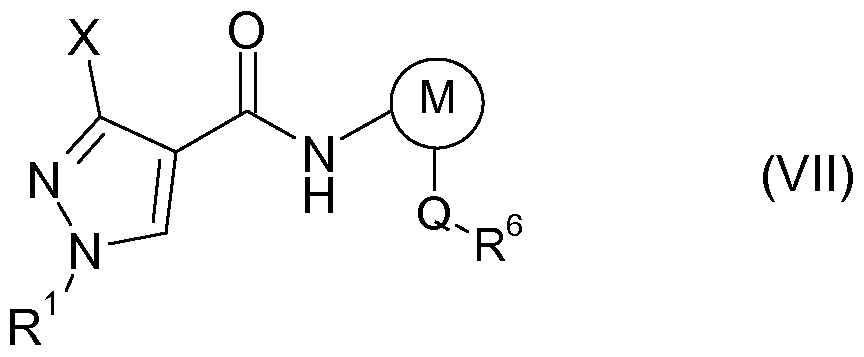
- pyrazole compounds of the formula I in particular the pyrazolecarboxylic acids of the formula Ia, are valuable intermediates in the preparation of active compounds which have a 1, 3-substituted pyrazole radical, in particular in the preparation of the fungicidal active compounds of the formula VII described below:
- R 1 and X have one of the meanings given in claim 1;
- M is thienyl or phenyl which may carry a halogen substituent
- Q is a direct bond, cyclopropylene, a fused bicyclo [2.2.1] heptane or bicyclo [2.2.1] heptene ring;
- R 6 represents hydrogen, halogen, C 1 -C 4 -alkyl, C 1 -C 4 -haloalkoxy, monosubstituted to trisubstituted phenyl, where the substituents independently of one another are selected from halogen and trifluoromethylthio, or cyclopropyl.
- the present invention also relates to a process for preparing a compound of the formula VII, comprising the following steps: a) providing a pyrazole compound of the formula I by the process according to the invention; b) conversion of the compound I into a 1,3-substituted pyrazolecarboxylic acid of
- Suitable methods for the preparation of carboxylic acids and reaction of carboxylic acids or carboxylic acid halides with aromatic amines are known in the art, for. B. from the cited prior art (see US 5,498,624, EP 545099 A1, DE 19531813 A1, EP 589301 A1, DE 19840322 A1, WO 92/12970, WO 03/066610, WO 2006/024389, WO 2007/003603, WO 2007/006806) as well as J. March, Advanced Organic Chemistry, 3rd ed. J. Wiley and Sons, New York 1985, pp. 370-386 and literature cited there as well as Organikum, 21st edition Wiley-VCH, Weinheim 2001, Pp. 481-484 and references cited therein, and can be applied to the preparation according to the invention of the compounds VII by reacting the pyrazolecarboxylic acid Ia or its acid halide with the aniline compound VIII in an analogous manner.
- the procedure is such that the pyrazolecarboxylic acid of the formula Ia first in its acid halide, z. B. converted their acid chloride and then reacting the acid halide with the amine compound of formula VIII.
- the conversion of the pyrazolecarboxylic acid into its acid chloride succeeds in analogy to standard methods of organic chemistry, for example by reaction with thionyl chloride.
- the subsequent reaction of the acid halide with the amine compound VIII is typically carried out in the presence of an auxiliary base, for example a tertiary amine.
- the pyrazolecarboxylic acid of the formula Ia can also be reacted directly with the amine compound VIII, preferably in the presence of a dehydrating agent such as 1, 1'-carbonyldiimidazole, bis (2-oxo-3-oxazolidinyl) phosphoryl chloride, N, N'-dicyclohexyl-carbodiimide or N - (3- Dimethylaminopropyl) -N'-ethylcarbodiimide in the presence of an auxiliary base, for example a tertiary amine, to convert compound VII as described, for example, in the earlier patent application PCT / EP2007 / 064390, the disclosure of which is hereby expressly incorporated by reference.
- a dehydrating agent such as 1, 1'-carbonyldiimidazole, bis (2-oxo-3-oxazolidinyl) phosphoryl chloride, N, N'-dicyclohexyl-carbodiimide or N
- 13 C-NMR 190.1, 181, 4, 166.6, 164.7, 148.9, 146.1, 145.5, 133.9, 130.3, 128.8, 127.7, 110 , 4, 108.5, 107.0, 99.23, 60.46, 59.66, 39.43, 13.81.
- Benzaldehyde The lower aqueous phase contained as the main component the sodium salt of the desired 3-difluoromethyl-1-methyl-1H-pyrazole-4-carboxylic acid.
- the separated aqueous phase was acidified (pH ⁇ 2) with 29.7 g (0.26 mol) of concentrated hydrochloric acid to give the title compound. After filtration, 18.2 g of the moist 3-difluoromethyl-1-methyl-1H-pyrazole-4-carboxylic acid were obtained.
- the solution contained 14.9 wt .-% of the desired 3-difluoromethyl-1-methyl-1 H-pyrazole-4-carboxylic acid ethyl ester (HPLC- Analysis, quantification with external standard)
- the proportion of the isomeric ethyl 5-difluoromethyl-1-methyl-1H-pyrazole-4-carboxylate is only 0.069% by weight (corresponding to an isomer ratio of> 200: 1).
- the reaction mixture was stirred at 60 0 C for 3 hours. After cooling to 25 0 C, the phases were separated.
- the toluene upper phase contained mainly the released benzaldehyde.
- the lower aqueous phase contained as the main component the potassium salt of the desired 3-difluoromethyl-1-methyl-1H-pyrazole-4-carboxylic acid.
- the toluene phase was washed twice with 50 g of water.
- the combined water phases were at 55 0 C with 66 g (0.579 mol) of concentrated hydrochloric acid (32%) acidified (pH ⁇ 2) to give the desired title compound precipitated.
- the toluene upper phase contained mainly the released benzaldehyde.
- the lower aqueous phase contained as the major component the potassium salt of the title compound.
- the toluene phase was washed twice with 50 g of water.
- the combined aqueous phases were acidified (pH ⁇ 2) at 55 ° C. with 80 g (0.7 mol) of concentrated hydrochloric acid (32%) to precipitate the desired pyrazolecarboxylic acid.
- the solid was filtered at 3 0 C and washed with 160 g of cold water.
- the toluene upper phase contained mainly the released benzaldehyde.
- the lower aqueous phase contained as the main component the potassium salt of the desired 3-difluoromethyl-1-methyl-1H-pyrazole-4-carboxylic acid.
- the toluene phase was washed twice with 200 g of water. The combined water phases were concentrated at 55 0 C with 265 g (2.32 mol) of conc. Hydrochloric acid (32%) acidified (pH ⁇ 2), wherein the desired pyrazolecarboxylic acid precipitated.
- Example 7 Preparation of S-difluoromethyl-i-methyl-1H-pyrazolemcarboxylic acid from benzaldehyde, aqueous methylhydrazine solution and ethoxymethylene-4,4-difluoro-3-oxobutyric acid ethyl ester without isolation / purification of the intermediates (one-pot - wise).
- the solution were metered in at 60 0 C 1620 g (4.05 mol) of 10 wt .-% sodium hydroxide solution and the mixture was stirred for 3 hours at 60 0 C. After cooling to 25 ° C the phases were separated.
- the toluene upper phase contained mainly the released benzaldehyde.
- the lower aqueous phase contained as the main component the sodium salt of the desired 3-difluoromethyl-1-methyl-1H-pyrazole-4-carboxylic acid.
- the toluene phase was washed with 540 g of water. To the combined water phases was added another 1125 g of water.
- the yield based on the molar amount of 2-ethoxymethylene-4,4-difluoro-3-oxo-butyric acid ethyl ester was 84.2%.
- the undesired carboxylic acid isomer was no longer detectable.
- 13 C-NMR 190.3, 181, 8, 164.4, 148.1, 143.5, 142.9, 140.0, 139.8, 128.4, 124.0, 110.3, 108 , 2, 108, 1, 100, 8, 60, 67, 59, 89, 39, 65, 14, 15.
- Example 15 to 20 The compounds of Examples 15 to 20 were reacted in analogy to Example 1, step 1.2 to 3-difluoromethyl-1-methyl-1 H-pyrazole-4-carboxylic acid ethyl ester, which then to 3-difluoromethyl-1-methyl-1 H-pyrazole 4-carboxylic acid was saponified.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description


















Claims





Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| UAA201014232A UA106470C2 (uk) | 2008-05-05 | 2009-05-04 | Спосіб одержання 1,3,4-заміщених піразольних сполук |
| CN200980116288.6A CN102015654B (zh) | 2008-05-05 | 2009-05-04 | 制备1,3,4-取代的吡唑化合物的方法 |
| EP09742026.9A EP2288597B1 (de) | 2008-05-05 | 2009-05-04 | Verfahren zur herstellung von 1,3,4-substituierten pyrazolverbindungen |
| ES09742026.9T ES2525807T3 (es) | 2008-05-05 | 2009-05-04 | Procedimiento de preparación de compuestos de pirazol sustituidos en las posiciones 1, 3, 4 |
| US12/990,340 US8598222B2 (en) | 2008-05-05 | 2009-05-04 | Method for preparing 1,3,4-substituted pyrazol compounds |
| CA2725446A CA2725446A1 (en) | 2008-05-05 | 2009-05-04 | Method for preparing 1,3,4-substituted pyrazol compounds |
| JP2011507880A JP5587293B2 (ja) | 2008-05-05 | 2009-05-04 | 1,3,4−置換ピラゾール化合物の調製方法 |
| MX2010011433A MX2010011433A (es) | 2008-05-05 | 2009-05-04 | Procedimiento para la preparacion de compuestos pirazolicos sustituidos en posiciones 1,3,4. |
| DK09742026.9T DK2288597T3 (en) | 2008-05-05 | 2009-05-04 | PROCESS FOR THE PREPARATION OF 1,3,4-SUBSTITUTED pyrazole |
| BRPI0911767-9A BRPI0911767A2 (pt) | 2008-05-05 | 2009-05-04 | Processo, e, composto. |
| EA201001736A EA019312B1 (ru) | 2008-05-05 | 2009-05-04 | Способ получения 1,3,4-замещённых пиразольных соединений |
| AU2009243550A AU2009243550B2 (en) | 2008-05-05 | 2009-05-04 | Method for preparing 1,3,4-substituted pyrazol compounds |
| IL208541A IL208541A (en) | 2008-05-05 | 2010-10-07 | Method for making 4,3,1-pyrazole compounds |
| ZA2010/08637A ZA201008637B (en) | 2008-05-05 | 2010-12-01 | Method for preparing 1,3,4-substituted pyrazol compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08155657 | 2008-05-05 | ||
| EP08155657.3 | 2008-05-05 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009135808A2 true WO2009135808A2 (de) | 2009-11-12 |
| WO2009135808A3 WO2009135808A3 (de) | 2010-01-21 |
Family
ID=39831714
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2009/055328 Ceased WO2009135808A2 (de) | 2008-05-05 | 2009-05-04 | Verfahren zur herstellung von 1,3,4-substituierten pyrazolverbindungen |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US8598222B2 (de) |
| EP (1) | EP2288597B1 (de) |
| JP (1) | JP5587293B2 (de) |
| KR (1) | KR20110011652A (de) |
| CN (1) | CN102015654B (de) |
| AR (1) | AR071673A1 (de) |
| AU (1) | AU2009243550B2 (de) |
| BR (1) | BRPI0911767A2 (de) |
| CA (1) | CA2725446A1 (de) |
| DK (1) | DK2288597T3 (de) |
| EA (1) | EA019312B1 (de) |
| ES (1) | ES2525807T3 (de) |
| IL (1) | IL208541A (de) |
| MX (1) | MX2010011433A (de) |
| UA (1) | UA106470C2 (de) |
| WO (1) | WO2009135808A2 (de) |
| ZA (1) | ZA201008637B (de) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8288563B2 (en) | 2009-03-16 | 2012-10-16 | Basf Se | Process for the preparation of pyrazole derivatives |
| US8314233B2 (en) | 2008-05-02 | 2012-11-20 | Basf Se | Process for preparing 2-(aminomethylidene)-4,4-difluoro-3-oxobutyric esters |
| US8344157B2 (en) | 2008-07-21 | 2013-01-01 | Basf Se | Process for preparing 1,3-disubstituted pyrazolecarboxylic esters |
| EP2644593A1 (de) * | 2012-03-30 | 2013-10-02 | Solvay Sa | Dialkoxymethyloxobuttersäureester, ihre Herstellung und Verwendung |
| US8586750B2 (en) | 2008-05-02 | 2013-11-19 | Basf Se | Method for the production of halogen-substituted 2-(aminomethylidene)-3-oxobutyric acid esters |
| WO2014038224A1 (ja) * | 2012-09-05 | 2014-03-13 | タマ化学工業株式会社 | 1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 |
| CN104016920A (zh) * | 2014-06-16 | 2014-09-03 | 联化科技(上海)有限公司 | 一种含氟甲基吡唑类化合物的联产方法 |
| JP2015071615A (ja) * | 2008-02-25 | 2015-04-16 | バイエル・クロップサイエンス・アーゲーBayer Cropscience Ag | 1−アルキル−3−ハロアルキル−ピラゾール−4−カルボン酸誘導体を位置選択的に合成する方法 |
| EP3178813A1 (de) | 2015-12-09 | 2017-06-14 | Basf Se | Verfahren zur herstellung halogenierter 3-oxocarboxylate mit einer 2-alkoxymethyliden- oder 2-dialkylaminomethyliden-gruppe |
| WO2019122194A1 (en) * | 2017-12-22 | 2019-06-27 | Solvay Sa | Process for the manufacture of iminium compounds and their application in the manufacture of pyrazole derivatives |
| WO2019122164A1 (en) * | 2017-12-22 | 2019-06-27 | Solvay Sa | Process for the manufacture of pyrazole carboxylic derivatives and precursors thereof |
| EP3650443A1 (de) | 2018-11-07 | 2020-05-13 | Fujian Yongjing Technology Co., Ltd. | Kontinuierliche flusssynthese von fluorierten oder nicht fluorierten pyrazolen |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102015654B (zh) | 2008-05-05 | 2014-03-12 | 巴斯夫欧洲公司 | 制备1,3,4-取代的吡唑化合物的方法 |
| EP2671873A1 (de) * | 2012-06-06 | 2013-12-11 | Solvay Sa | Verfahren zur Zyklisierung von Hydrazinacrylsäurederivaten |
| CN102702104B (zh) * | 2012-06-08 | 2015-02-25 | 巨化集团公司 | 一种3-二氟甲基-1-甲基吡唑-4-甲酸乙酯的连续化合成方法 |
| JP5232335B1 (ja) * | 2013-01-29 | 2013-07-10 | タマ化学工業株式会社 | 1−置換−3−フルオロアルキルピラゾール−4−カルボン酸エステルの製造方法 |
| US8871947B2 (en) | 2013-02-04 | 2014-10-28 | KingChem LLC | Preparation of alkyl 3-difluoromethyl-1-methyl-1H-pyrazole-4-carboxylic acid ester |
| CN103351339A (zh) * | 2013-07-12 | 2013-10-16 | 雅本化学股份有限公司 | 3-氟代烷基-1-取代吡唑-4-羧酸及其制备方法 |
| WO2015085464A1 (en) | 2013-12-09 | 2015-06-18 | King Chem, Llc | Process for preparing alkyl 3-difluoromethyl-1-methyl-1h-pyrazole-4-carboxylate and its analogs |
| CN103880749B (zh) * | 2014-02-26 | 2019-01-01 | 南通大学 | 一种3-甲基-5-吡唑甲酸甲酯的化学合成方法 |
| US9701640B2 (en) * | 2014-03-24 | 2017-07-11 | Bayer Cropscience Aktiengesellschaft | Process for preparing 3,5-bis(haloalkyl)pyrazole derivatives from α,α-dihaloamines and ketimines |
| RU2712192C2 (ru) * | 2014-07-31 | 2020-01-24 | Басф Се | Способ получения пиразолов |
| CN104262256B (zh) * | 2014-08-01 | 2016-08-17 | 江苏大学 | 利用α,β-不饱和羰基化合物制备多取代吡唑类化合物的方法 |
| CN104496902A (zh) * | 2014-12-19 | 2015-04-08 | 浙江泰达作物科技有限公司 | 高区域选择性制备1,3,4-取代的吡唑化合物的方法 |
| AU2015401326B2 (en) * | 2015-07-03 | 2018-07-26 | China Agricultural University | Benzylhydrazone compounds and preparation method and application thereof |
| PE20181539A1 (es) * | 2016-02-02 | 2018-09-26 | Basf Se | Proceso de hidrogenacion catalitico para preparar pirazoles |
| CN107663172B (zh) * | 2016-07-27 | 2021-04-16 | 宿迁市科莱博生物化学有限公司 | 一种吡唑衍生物的制备方法 |
| JP2019524765A (ja) * | 2016-08-02 | 2019-09-05 | ソルヴェイ(ソシエテ アノニム) | ピラゾールカルボン酸および誘導体の製造に有用なヒドラジニル化合物の製造、ヒドラジニル化合物ならびにその使用 |
| CN114195678A (zh) * | 2021-12-24 | 2022-03-18 | 江苏七洲绿色科技研究院有限公司 | 一种中间体、其制备方法及用其制备3-二氟甲基-1-甲基吡唑-4-甲酸乙酯的方法 |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992012970A1 (en) | 1991-01-28 | 1992-08-06 | Monsanto Company | 3-difluoromethylpyrazolecarboxamide fungicides |
| EP0545099A2 (de) | 1991-11-22 | 1993-06-09 | BASF Aktiengesellschaft | Säureanilid-Derivate und ihre Verwendung zur Bekämpfung von Botrytis |
| EP0589301A1 (de) | 1992-09-21 | 1994-03-30 | BASF Aktiengesellschaft | Carbonsäureanilide, Verfahren zu ihrer Herstellung und sie enthaltende Mittel zur Bekämpfung von Schadpilzen |
| US5498624A (en) | 1995-05-03 | 1996-03-12 | Monsanto Company | Selected pyrazolyl derivatives |
| WO2003051820A1 (de) | 2001-12-17 | 2003-06-26 | Bayer Chemicals Ag | Verfahren zur herstellung von 2-halogenacyl-3-amino-acrylsäure- derivaten |
| WO2003066610A1 (de) | 2002-02-04 | 2003-08-14 | Bayer Cropscience Aktiengesellschaft | Difluormethyl thiazolyl carboxanilide |
| WO2005042468A1 (de) | 2003-10-23 | 2005-05-12 | Bayer Cropscience Aktiengesellschaft | Verfahren zum herstellen von 2-dihalogenacyl-3-amino-acrylsäureestern und 3-dihalogenmethyl-pyrazol-4-carbonsäureestern |
| WO2006024389A2 (de) | 2004-08-27 | 2006-03-09 | Bayer Cropscience Ag | Biphenylthiazolcarboxamide |
| WO2007003603A2 (de) | 2005-07-05 | 2007-01-11 | Basf Aktiengesellschaft | Fungizide mischungen auf der basis von 3-monosubstituierten pyrazolcarbonsäurebiphenylamiden |
| WO2007006806A2 (de) | 2005-07-14 | 2007-01-18 | Basf Aktiengesellschaft | Fungizide mischungen auf der basis von 1-methyl-pyrazol-4-yl-carbonsäureaniliden |
| WO2008022777A2 (de) | 2006-08-25 | 2008-02-28 | Bayer Cropscience Ag | Verfahren zum herstellen von 3-dihalomethyl-pyrazol-4-carbonsäurederivaten |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0822853B2 (ja) | 1987-10-23 | 1996-03-06 | 三井東圧化学株式会社 | ピラゾール−4−カルボン酸類およびその製造方法 |
| JPH0266612A (ja) | 1988-08-31 | 1990-03-06 | Toshiba Corp | 冷蔵庫の温度制御回路 |
| US5223526A (en) | 1991-12-06 | 1993-06-29 | Monsanto Company | Pyrazole carboxanilide fungicides and use |
| TW268881B (de) | 1992-07-07 | 1996-01-21 | Ciba Geigy Ag | |
| IL111793A (en) | 1993-11-30 | 1998-12-06 | Lilly Co Eli | Process for preparing 2,2-Difluoroketene silyl acetals and alpha,alpha-difluoro-beta-silyloxy-1,3-dioxolane-4-propanoic acid esters |
| JP2000212166A (ja) | 1999-01-26 | 2000-08-02 | Mitsui Chemicals Inc | 1,3―ジアルキルピラゾ―ル―4―カルボン酸エステルの製造法 |
| DE10215292A1 (de) | 2002-02-19 | 2003-08-28 | Bayer Cropscience Ag | Disubstitutierte Pyrazolylcarbocanilide |
| RU2006101337A (ru) | 2003-06-18 | 2006-06-10 | Е.И.Дюпон де Немур энд Компани (US) | Способы применения фторкетонов для пожаротушения, предотвращения возгарания и снижения или ликвидации воспламеняемости легковоспламеняющейся рабочей жидкости |
| WO2005003077A1 (de) | 2003-07-01 | 2005-01-13 | Bayer Cropscience Aktiengesellschaft | Verfahren zum herstellen von difluoracetessigsäurealkylestern |
| DE10351088A1 (de) | 2003-10-31 | 2005-06-02 | Bayer Cropscience Gmbh | Verfahren zum Herstellen von fluormethyl-substituierten Heterocyclen |
| US20050234244A1 (en) | 2004-04-20 | 2005-10-20 | Wilmin Bartolini | Synthesis of COX-2 and FAAH inhibitors |
| JP2008502636A (ja) | 2004-06-18 | 2008-01-31 | ビーエーエスエフ アクチェンゲゼルシャフト | N−(オルト−フェニル)−1−メチル−3−ジフルオロメチルピラゾール−4−カルボキシアニリドおよびそれらの殺菌剤としての使用 |
| JP4114754B2 (ja) | 2005-02-25 | 2008-07-09 | 財団法人相模中央化学研究所 | 1−置換−3−フルオロアルキルピラゾール−4−カルボン酸エステルの製造方法 |
| BRPI0616238B1 (pt) | 2005-09-16 | 2016-01-19 | Syngenta Ltd | processo para a produção de amidas |
| KR101440162B1 (ko) | 2006-06-22 | 2014-09-12 | 바스프 에스이 | 말로노니트릴 화합물 |
| WO2008053043A1 (de) | 2006-11-03 | 2008-05-08 | Basf Se | Verfahren zur herstellung von difluormethylpyrazolylcarboxylaten |
| BRPI0720410A2 (pt) | 2006-12-21 | 2013-12-31 | Basf Se | Processos para a preparação de composto, para a preparação de amidas, e, compostos. |
| CN101631767A (zh) | 2007-03-16 | 2010-01-20 | 巴斯夫欧洲公司 | 使用二氯-氟-三氟甲基苯的混合物制备2,6-二氯-4-(三氟甲基)苯基肼的方法 |
| WO2008145740A1 (de) | 2007-06-01 | 2008-12-04 | Basf Se | Verfahren zur herstellung n-substituierter (3-dihalomethyl-1-methyl-pyrazol-4-yl)carboxamide |
| WO2008152138A2 (de) | 2007-06-15 | 2008-12-18 | Basf Se | Verfahren zur herstellung difluormethylsubstituierter pyrazolverbindungen |
| EP2042482A1 (de) | 2007-09-26 | 2009-04-01 | Bayer CropScience AG | Verfahren zur Herstellung von 2-Dihalogenacyl-3-amino-acrylsäure-Derivaten |
| EP2072497A1 (de) | 2007-12-21 | 2009-06-24 | Bayer CropScience AG | Verfahren zum Herstellen von 2-Fluoracyl-3-amino-acrylsäure-Derivaten |
| EP2133341A1 (de) * | 2008-02-25 | 2009-12-16 | Bayer CropScience AG | Verfahren zur regioselektiven Synthese von 1-Alkyl-3-haloalkyl-pyrazol-4-carbonsäure-Derivaten |
| JP5602131B2 (ja) | 2008-05-02 | 2014-10-08 | ビーエーエスエフ ソシエタス・ヨーロピア | ハロゲン置換2−(アミノメチリデン)−3−オキソ酪酸エステルの製造方法 |
| AR073932A1 (es) | 2008-05-02 | 2010-12-15 | Basf Se | Compuestos de esteres de acido 2-(aminometiliden)-4,4-difluoro-3- oxobutirico y procedimiento para su preparacion |
| CN102015654B (zh) | 2008-05-05 | 2014-03-12 | 巴斯夫欧洲公司 | 制备1,3,4-取代的吡唑化合物的方法 |
| ATE555098T1 (de) | 2008-07-21 | 2012-05-15 | Basf Se | Verfahren zur herstellung 1,3-disubstituierter pyrazolcarbonsäureester |
-
2009
- 2009-05-04 CN CN200980116288.6A patent/CN102015654B/zh active Active
- 2009-05-04 JP JP2011507880A patent/JP5587293B2/ja not_active Expired - Fee Related
- 2009-05-04 WO PCT/EP2009/055328 patent/WO2009135808A2/de not_active Ceased
- 2009-05-04 BR BRPI0911767-9A patent/BRPI0911767A2/pt not_active Application Discontinuation
- 2009-05-04 MX MX2010011433A patent/MX2010011433A/es active IP Right Grant
- 2009-05-04 EA EA201001736A patent/EA019312B1/ru not_active IP Right Cessation
- 2009-05-04 CA CA2725446A patent/CA2725446A1/en not_active Abandoned
- 2009-05-04 ES ES09742026.9T patent/ES2525807T3/es active Active
- 2009-05-04 DK DK09742026.9T patent/DK2288597T3/en active
- 2009-05-04 US US12/990,340 patent/US8598222B2/en active Active
- 2009-05-04 AU AU2009243550A patent/AU2009243550B2/en not_active Ceased
- 2009-05-04 UA UAA201014232A patent/UA106470C2/uk unknown
- 2009-05-04 KR KR1020107027189A patent/KR20110011652A/ko not_active Ceased
- 2009-05-04 EP EP09742026.9A patent/EP2288597B1/de active Active
- 2009-05-05 AR ARP090101622A patent/AR071673A1/es unknown
-
2010
- 2010-10-07 IL IL208541A patent/IL208541A/en not_active IP Right Cessation
- 2010-12-01 ZA ZA2010/08637A patent/ZA201008637B/en unknown
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992012970A1 (en) | 1991-01-28 | 1992-08-06 | Monsanto Company | 3-difluoromethylpyrazolecarboxamide fungicides |
| EP0545099A2 (de) | 1991-11-22 | 1993-06-09 | BASF Aktiengesellschaft | Säureanilid-Derivate und ihre Verwendung zur Bekämpfung von Botrytis |
| EP0589301A1 (de) | 1992-09-21 | 1994-03-30 | BASF Aktiengesellschaft | Carbonsäureanilide, Verfahren zu ihrer Herstellung und sie enthaltende Mittel zur Bekämpfung von Schadpilzen |
| US5498624A (en) | 1995-05-03 | 1996-03-12 | Monsanto Company | Selected pyrazolyl derivatives |
| WO2003051820A1 (de) | 2001-12-17 | 2003-06-26 | Bayer Chemicals Ag | Verfahren zur herstellung von 2-halogenacyl-3-amino-acrylsäure- derivaten |
| WO2003066610A1 (de) | 2002-02-04 | 2003-08-14 | Bayer Cropscience Aktiengesellschaft | Difluormethyl thiazolyl carboxanilide |
| WO2005042468A1 (de) | 2003-10-23 | 2005-05-12 | Bayer Cropscience Aktiengesellschaft | Verfahren zum herstellen von 2-dihalogenacyl-3-amino-acrylsäureestern und 3-dihalogenmethyl-pyrazol-4-carbonsäureestern |
| WO2006024389A2 (de) | 2004-08-27 | 2006-03-09 | Bayer Cropscience Ag | Biphenylthiazolcarboxamide |
| WO2007003603A2 (de) | 2005-07-05 | 2007-01-11 | Basf Aktiengesellschaft | Fungizide mischungen auf der basis von 3-monosubstituierten pyrazolcarbonsäurebiphenylamiden |
| WO2007006806A2 (de) | 2005-07-14 | 2007-01-18 | Basf Aktiengesellschaft | Fungizide mischungen auf der basis von 1-methyl-pyrazol-4-yl-carbonsäureaniliden |
| WO2008022777A2 (de) | 2006-08-25 | 2008-02-28 | Bayer Cropscience Ag | Verfahren zum herstellen von 3-dihalomethyl-pyrazol-4-carbonsäurederivaten |
Non-Patent Citations (4)
| Title |
|---|
| CHEM. BER., vol. 115, 1982, pages 2766 |
| J. MARCH: "Advanced Organic Chemistry, 2nd Ed.,", 1977, MCGRAW-HILL, pages: 334 - 338 |
| JACS, vol. 73, pages 3684 |
| JOURNAL OF MEDICINAL CHEMISTRY, vol. 43, no. 21, 2000 |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015071615A (ja) * | 2008-02-25 | 2015-04-16 | バイエル・クロップサイエンス・アーゲーBayer Cropscience Ag | 1−アルキル−3−ハロアルキル−ピラゾール−4−カルボン酸誘導体を位置選択的に合成する方法 |
| US8314233B2 (en) | 2008-05-02 | 2012-11-20 | Basf Se | Process for preparing 2-(aminomethylidene)-4,4-difluoro-3-oxobutyric esters |
| US8586750B2 (en) | 2008-05-02 | 2013-11-19 | Basf Se | Method for the production of halogen-substituted 2-(aminomethylidene)-3-oxobutyric acid esters |
| US8592578B2 (en) | 2008-05-02 | 2013-11-26 | Basf Se | Process for preparing 2-(aminomethylidene)-4,4-difluoro-3-oxobutyric esters |
| US8344157B2 (en) | 2008-07-21 | 2013-01-01 | Basf Se | Process for preparing 1,3-disubstituted pyrazolecarboxylic esters |
| US8288563B2 (en) | 2009-03-16 | 2012-10-16 | Basf Se | Process for the preparation of pyrazole derivatives |
| EP2644593A1 (de) * | 2012-03-30 | 2013-10-02 | Solvay Sa | Dialkoxymethyloxobuttersäureester, ihre Herstellung und Verwendung |
| WO2014038224A1 (ja) * | 2012-09-05 | 2014-03-13 | タマ化学工業株式会社 | 1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 |
| CN104016920A (zh) * | 2014-06-16 | 2014-09-03 | 联化科技(上海)有限公司 | 一种含氟甲基吡唑类化合物的联产方法 |
| EP3178813A1 (de) | 2015-12-09 | 2017-06-14 | Basf Se | Verfahren zur herstellung halogenierter 3-oxocarboxylate mit einer 2-alkoxymethyliden- oder 2-dialkylaminomethyliden-gruppe |
| WO2019122194A1 (en) * | 2017-12-22 | 2019-06-27 | Solvay Sa | Process for the manufacture of iminium compounds and their application in the manufacture of pyrazole derivatives |
| WO2019122164A1 (en) * | 2017-12-22 | 2019-06-27 | Solvay Sa | Process for the manufacture of pyrazole carboxylic derivatives and precursors thereof |
| EP3650443A1 (de) | 2018-11-07 | 2020-05-13 | Fujian Yongjing Technology Co., Ltd. | Kontinuierliche flusssynthese von fluorierten oder nicht fluorierten pyrazolen |
| US11299463B2 (en) | 2018-11-07 | 2022-04-12 | Fujian Yongjing Technology Co., Ltd. | Process for the manufacture of pyrazoles or pyrimidones |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009135808A3 (de) | 2010-01-21 |
| IL208541A (en) | 2014-05-28 |
| CN102015654B (zh) | 2014-03-12 |
| US8598222B2 (en) | 2013-12-03 |
| EP2288597A2 (de) | 2011-03-02 |
| UA106470C2 (uk) | 2014-09-10 |
| ES2525807T3 (es) | 2014-12-30 |
| EA019312B1 (ru) | 2014-02-28 |
| CN102015654A (zh) | 2011-04-13 |
| ZA201008637B (en) | 2014-08-27 |
| EP2288597B1 (de) | 2014-10-29 |
| EA201001736A1 (ru) | 2011-06-30 |
| BRPI0911767A2 (pt) | 2015-07-28 |
| DK2288597T3 (en) | 2015-01-26 |
| JP5587293B2 (ja) | 2014-09-10 |
| US20110172436A1 (en) | 2011-07-14 |
| AR071673A1 (es) | 2010-07-07 |
| JP2011519889A (ja) | 2011-07-14 |
| MX2010011433A (es) | 2010-11-12 |
| KR20110011652A (ko) | 2011-02-08 |
| AU2009243550A1 (en) | 2009-11-12 |
| AU2009243550B2 (en) | 2014-06-26 |
| CA2725446A1 (en) | 2009-11-12 |
| IL208541A0 (en) | 2010-12-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2288597B1 (de) | Verfahren zur herstellung von 1,3,4-substituierten pyrazolverbindungen | |
| EP2086944B1 (de) | Verfahren zur herstellung von difluormethylpyrazolylcarboxylaten | |
| EP2247577B2 (de) | Verfahren zur regioselektiven synthese von 1-alkyl-3-haloalkyl-pyrazol-4-carbonsäur e-derivaten | |
| EP2285779B1 (de) | Verfahren zur Herstellung von Arylcarboxamiden | |
| EP2164831B1 (de) | Verfahren zur herstellung n-substituierter (3-dihalomethyl-1-methyl-pyrazol-4-yl)carboxamide | |
| EP2257532B1 (de) | Verfahren zur regioselektiven synthese von l-alkvl-3-haloalkyl-pvrazol-4-carbonsäure-derivaten | |
| EP0595130B1 (de) | N-Phenylacetaminonitrile und ihre Verwendung als Zwischenprodukte zur Synthese von Insektiziden und Herbiziden 3-Aryl-pyrrolidin-2,4-dionen | |
| EP2300417B1 (de) | Verfahren zur herstellung von 2-(aminomethyliden)-4,4-difluor-3-oxobuttersäureestern | |
| EP2297111B1 (de) | Verfahren zur herstellung halogensubstituierter 2-(aminomethyliden)-3-oxobuttersäureester | |
| EP2623496A1 (de) | Verfahren zur Herstellung von 3,5-bis (fluoralkyl)-pyrazol-4-carbonsäure-Derivaten und 3,5-bis(fluoralkyl)-pyrazolen | |
| EP1644314B1 (de) | Verfahren zum herstellen von difluoracetessigs urealkylestern | |
| EP1858858A2 (de) | Verfahren zum herstellen von alkylaniliden | |
| DE2708189C2 (de) | Verfahren zur Herstellung von Phenylglyoxylsäureestern | |
| EP1517906B1 (de) | Verfahren zur herstellung von 1,2,4-triazolylmethyl-oxiranen | |
| WO2014187774A1 (de) | Verfahren zur herstellung von 3,5-bis(fluoralkyl)-pyrazol-derivaten | |
| DE10331496A1 (de) | Verfahren zum Herstellen von Difluoracetessigsäurealkylestern | |
| DE60029245T2 (de) | Verfahren zur Herstellung von Aryltriazolinonen | |
| EP0365913B1 (de) | Substituierte 2-Aminothiazole | |
| DE3612939A1 (de) | Verfahren zur herstellung von 1-aryl-5-amino-pyrazolen | |
| DE19507915A1 (de) | Verfahren zur Herstellung von Pyrazolen | |
| DE10005572A1 (de) | Verfahren zur Herstellung von 2-(1,2,4-Triazolyl-1-yl)-ethanolen | |
| WO1996012707A1 (de) | 2-chlor-n-[1-(2,6-dichlor-4-difluormethyl-phenyl)-1h-pyrazol-5-yl]-propanamid und ein verfahren zu seiner herstellung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980116288.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09742026 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2725446 Country of ref document: CA Ref document number: 3769/KOLNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/011433 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009243550 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011507880 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2009243550 Country of ref document: AU Date of ref document: 20090504 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009742026 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201001736 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20107027189 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12990340 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0911767 Country of ref document: BR Kind code of ref document: A2 Effective date: 20101027 |