WO2014038224A1 - 1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 - Google Patents
1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 Download PDFInfo
- Publication number
- WO2014038224A1 WO2014038224A1 PCT/JP2013/057275 JP2013057275W WO2014038224A1 WO 2014038224 A1 WO2014038224 A1 WO 2014038224A1 JP 2013057275 W JP2013057275 W JP 2013057275W WO 2014038224 A1 WO2014038224 A1 WO 2014038224A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- organic solvent
- substituted
- ethyl
- acid ester
- carboxylic acid
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
Definitions
- the present invention relates to a process for producing 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid esters useful as synthetic intermediates for pharmaceuticals and agricultural chemicals.
- Patent Document 1 a method for producing 1,3-dialkylpyrazole-4-carboxylic acid ester in which 2-ethoxymethylene acylacetate and alkylhydrazine are reacted with a solvent such as ethyl acetate has been proposed.
- Patent Document 1 1,3-dialkylpyrazole-4-carboxylic acid ester (about 85%) and 1,5-dialkylpyrazole-4-carboxylic acid ester (about 15% ) Are mixed. Therefore, in order to obtain the desired 1,3-dialkylpyrazole-4-carboxylic acid ester, it was necessary to purify it by distillation or the like.
- 1-methyl-3-difluoromethylpyrazole is prepared by reacting ethyl 2-ethoxymethylene-4,4-difluoro-3-oxobutanoate with anhydrous methylhydrazine in the presence of a halogen-containing organic solvent such as hydrofluorocarbon.
- a halogen-containing organic solvent such as hydrofluorocarbon.
- the present invention has been made in view of such problems of the prior art, and the object of the present invention is to highly selectively select one of the two positional isomers. And a method for producing a 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester that can be synthesized in a high yield, is highly versatile, and can be easily applied to an industrial process. It is in.
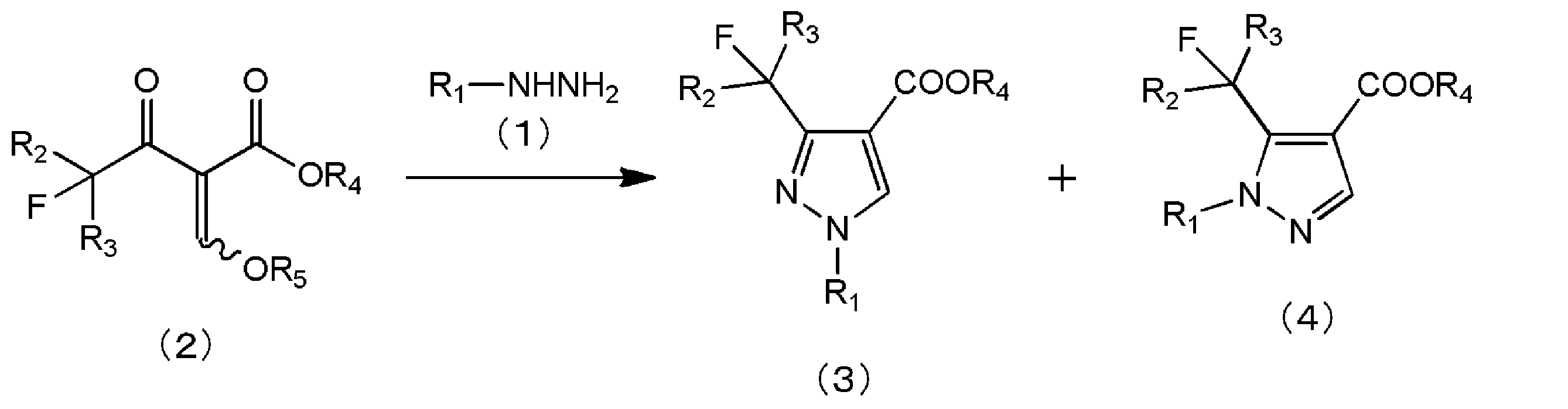
- An acylacetic acid ester derivative represented by the following general formula (2) and a second organic compound are added to the first reaction liquid containing the alkyl hydrazine represented by the following general formula (1) and the first organic solvent.
- a second reaction solution containing a solvent over 0.5 to 30 hours, stirring in the absence of a base and an acid, and reacting at a reaction temperature of ⁇ 5 to 80 ° C.
- 1 organic solvent and the second organic solvent are at least one of benzene, toluene, xylene, chlorobenzene, dichlorobenzene, ethyl acetate, butyl acetate, and dimethyl carbonate, respectively, with respect to the mass of the acyl acetate derivative
- the total mass of the first organic solvent and the second organic solvent is 1 to 60 times, and the total mass of the first organic solvent occupies the total of the first organic solvent and the second organic solvent. There is 40 to 95 mass%, the production method of 1-substituted-3-fluoroalky
- R 1 represents an optionally substituted alkyl group having 1 to 6 carbon atoms
- R 2 represents a hydrogen atom or a halogen atom
- R 3 has 1 to 12 carbon atoms which may be substituted with a hydrogen atom, a fluorine atom, or a chlorine atom or a fluorine atom.
- An alkyl group, and R 4 and R 5 each independently represents an alkyl group having 1 to 6 carbon atoms
- R 1 represents an optionally substituted alkyl group having 1 to 6 carbon atoms
- R 2 represents a hydrogen atom or a halogen atom
- R 3 represents a hydrogen atom, fluorine An atom, or an alkyl group having 1 to 12 carbon atoms which may be substituted with a chlorine atom or a fluorine atom
- R 4 represents an alkyl group having 1 to 6 carbon atoms
- one of the two positional isomers is selected with high selectivity and yield. Can be synthesized. Further, the method for producing 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester of the present invention is highly versatile and can be easily applied to industrial processes.
- FIG. 2 is a high performance liquid chromatography (HPLC) chart of white crystals obtained in Example 1.
- FIG. 2 is a high performance liquid chromatography (HPLC) chart of yellowish amber crystals obtained in Comparative Example 1.
- HPLC high performance liquid chromatography
- the present invention is a method for producing a 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester represented by the following general formula (3) (hereinafter also simply referred to as “the production method of the present invention”).
- R 1 represents an optionally substituted alkyl group having 1 to 6 carbon atoms
- R 2 represents a hydrogen atom or a halogen atom
- R 3 represents a hydrogen atom, fluorine An atom, or an alkyl group having 1 to 12 carbon atoms which may be substituted with a chlorine atom or a fluorine atom
- R 4 represents an alkyl group having 1 to 6 carbon atoms
- an acylacetic ester derivative represented by the following general formula (2) is added to the first reaction solution containing the alkyl hydrazine represented by the following general formula (1) and the first organic solvent.
- a step of adding a second reaction liquid containing a second organic solvent and reacting with stirring in the absence of a base and an acid (hereinafter also referred to as “reaction step”) is included.
- R 1 represents an optionally substituted alkyl group having 1 to 6 carbon atoms
- R 2 represents a hydrogen atom or a halogen atom
- R 3 has 1 to 12 carbon atoms which may be substituted with a hydrogen atom, a fluorine atom, or a chlorine atom or a fluorine atom.
- An alkyl group, and R 4 and R 5 each independently represents an alkyl group having 1 to 6 carbon atoms
- alkyl group having 1 to 6 carbon atoms represented by R 1 include a methyl group, an ethyl group, a propyl group, a cyclopropylmethyl group, a butyl group, and an isobutyl group. , A pentyl group, a hexyl group, and the like. These alkyl groups may be substituted with a halogen atom or the like.
- optionally substituted alkyl group having 1 to 6 carbon atoms include 2-chloroethyl group, 2-bromoethyl group, 2-hydroxyethyl group, 2,2,2-trifluoroethyl group, 3-chloro A propyl group etc. can be mentioned.
- alkyl hydrazine represented by the general formula (1) a generally available one may be used as it is, or one produced by a known method may be used.
- These alkyl hydrazines can be used in any of anhydrides, hydrates and aqueous solutions.
- halogen atom represented by R 2 include a fluorine atom, a chlorine atom, and a bromine atom.
- alkyl group having 1 to 12 carbon atoms which may be substituted with a chlorine atom or a fluorine atom represented by R 3 include a trifluoromethyl group and a difluoromethyl group.
- specific examples of the alkyl group having 1 to 6 carbon atoms represented by R 4 and R 5 are methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, pentyl, respectively. Group, hexyl group and the like.
- specific examples of the alkyl group having 1 to 6 carbon atoms represented by R 4 include methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, pentyl group, A hexyl group etc. can be mentioned.
- acyl acetate derivative represented by the general formula (2) a commercially available one may be used as it is, or one produced according to a general organic synthesis method may be used. For example, by reacting a ⁇ -ketocarboxylic acid ester obtained by Claisen condensation between a fluorine-containing carboxylic acid ester and an acetic acid ester with an orthoformate in the presence of acetic anhydride, an acyl represented by the general formula (2) Acetic acid ester derivatives can be easily produced.
- the second reaction solution is added to the first reaction solution by a method such as dropwise addition, and the alkyl hydrazine contained in the first reaction solution, The acylacetic acid ester derivative contained in the reaction solution 2 is reacted.
- the first reaction liquid contains the first organic solvent together with the alkyl hydrazine represented by the general formula (1).
- the first organic solvent for example, at least one of an aromatic hydrocarbon solvent and an ester solvent can be used.
- the aromatic hydrocarbon solvent include benzene, toluene, xylene, chlorobenzene, dichlorobenzene and the like.
- Specific examples of the ester solvent include ethyl acetate, butyl acetate, dimethyl carbonate and the like. Of these organic solvents, toluene, xylene, and ethyl acetate are preferred.
- the second reaction solution contains a second organic solvent together with the acylacetic ester derivative represented by the general formula (2).
- Specific examples of the second organic solvent include those similar to the first organic solvent, including preferred ones. Note that the types of the first organic solvent and the second organic solvent may be the same or different.
- the second reaction solution is added to the first reaction solution, and the reaction proceeds by stirring in the absence of a base and an acid.
- the hydrolysis of the resulting 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester is effectively suppressed. Can do. Therefore, the target 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester can be obtained in high yield.
- R 1 represents an optionally substituted alkyl group having 1 to 6 carbon atoms.
- R 2 represents a hydrogen atom or a halogen atom
- R 3 represents a hydrogen atom, a fluorine atom, or an alkyl group having 1 to 12 carbon atoms which may be substituted with a chlorine atom or a fluorine atom
- R 4 represents an alkyl group having 1 to 6 carbon atoms
- R 5 represents an alkyl group having 1 to 6 carbon atoms.
- the total mass of the first organic solvent and the second organic solvent is 1 to 60 times, preferably 5 to 50 times, more preferably 6 to 40 times the mass of the acyl acetate derivative. Double. That is, the reaction selectivity can be enhanced by reacting the acyl acetate derivative with alkyl hydrazine in a state of being appropriately diluted with an organic solvent.
- the amount of the first organic solvent in the total of the first organic solvent and the second organic solvent is 40 to 95% by mass, preferably 65 to 92% by mass, and more preferably 67%. ⁇ 90 mass%. That is, reaction selectivity is achieved by bringing the alkyl hydrazine contained in the first reaction liquid and the acylacetic ester derivative contained in the second reaction liquid into contact with each other in an appropriately diluted state. Can be increased.
- the target position of the two kinds of positional isomers can be obtained without reacting the alkyl hydrazine and the acyl acetate derivative in the presence of a base.
- the isomeric 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester can be produced with high selectivity.
- the second reaction solution is not added to the first reaction solution at a time, but gradually added over an appropriate time by a method such as dropping.
- the target 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester can be produced more selectively.
- the second reaction liquid containing the acyl acetate derivative is added to the first reaction liquid for 0.5 to 30 hours, preferably 1 to 25 hours.
- the time required for the addition is less than 0.5 hour, the reaction selectivity is lowered.
- the time required for the addition may exceed 30 hours, but the effect of improving the reaction selectivity tends to reach a peak.
- the amount of the acyl acetate derivative contained in the second reaction solution is usually 0.8 to 1.2 molar equivalents, preferably 0.85 to 1 with respect to the alkyl hydrazine in the first reaction solution. .15 molar equivalents.
- the reaction temperature in the reaction step is preferably ⁇ 5 to 80 ° C., more preferably 0 to 60 ° C. If the reaction temperature is less than ⁇ 5 ° C., the reaction tends to hardly proceed. On the other hand, when reaction temperature exceeds 80 degreeC, it exists in the tendency for reaction selectivity to fall. By controlling the reaction temperature within the above range, the yield and reaction selectivity can be further improved.
- the positional isomer represented by the general formula (3) (target compound) ) Is produced with high selectivity and yield.
- the target compound with high purity can be obtained by performing an extraction operation or the like after the reaction step according to a general organic synthesis technique.
- Example 1 In a 100 mL four-necked Kolben equipped with a thermometer and a stirrer, 49.55 g of toluene and 15.92 g (0.047 mol) of a 13.5% monomethylhydrazine aqueous solution were added, and stirring was started. Then, a mixed solution of 8.88 g (0.040 mol) of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate represented by the following formula (2-1) and 9.95 g of toluene was used using a metering pump. The solution was added dropwise at an internal temperature of 5 ° C. over 16 hours.
- Example 2 To a 50 mL four-necked Kolben equipped with a thermometer and a stirrer, 4.45 g of toluene and 12.0 g (0.022 mol) of an 8.8% monomethylhydrazine aqueous solution were added, and stirring was started. Thereto was added dropwise 8.90 g (0.02 mol) of a 50% toluene solution of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate at an internal temperature of 5 ° C. over 4 hours using a metering pump. After completion of dropping, the aqueous layer and the toluene layer were separated.
- the obtained toluene layer was dried under reduced pressure, and white crystals 3 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 3 .90 g (95.5% yield) was obtained.
- white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 94.1: 5.9.
- Example 3 To 50 mL four-necked Kolben equipped with a thermometer and a stirrer, 8.60 g of toluene and 12.0 g (0.022 mol) of an 8.8% monomethylhydrazine aqueous solution were added, and stirring was started. Thereto, 13.1 g (0.02 mol) of a 34% toluene solution of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate was added dropwise at an internal temperature of 5 ° C. over 4 hours using a metering pump. After completion of dropping, the aqueous layer and the toluene layer were separated.
- the obtained toluene layer was dried under reduced pressure, and white crystals 3 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 3 Obtained .95 g (yield 96.7%).
- the obtained white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 94.1: 5.9.
- Example 4 In a 50 mL four-necked Kolben equipped with a thermometer and a stirrer, 18.00 g of toluene and 8.00 g (0.022 mol) of a 13.1% monomethylhydrazine aqueous solution were added, and stirring was started. Thereto, 13.10 g (0.02 mol) of a 34% toluene solution of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate was added dropwise over 24 hours at an internal temperature of 5 ° C. using a metering pump. After completion of dropping, the aqueous layer and the toluene layer were separated.
- the obtained toluene layer was dried under reduced pressure, and white crystals 4 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 4 0.000 g (yield 98.0%) was obtained.
- the obtained white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 98.9: 1.1.
- Example 5 To a 100 mL four-necked Kolben equipped with a thermometer and a stirrer, 24.8 g of toluene and 7.96 g (0.023 mol) of a 13.5% monomethylhydrazine aqueous solution were added, and stirring was started. A mixed solution of 4.92 g (Net 4.44 g, 0.020 mol) of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate and 4.92 g of toluene was added to the solution at an internal temperature of 5 ° C. using a metering pump. Added dropwise over 5 hours. After completion of dropping, the aqueous layer and the toluene layer were separated.
- Example 6 To a 100 mL four-necked Kolben equipped with a thermometer and a stirrer, 24.8 g of toluene and 7.96 g (0.023 mol) of a 13.5% monomethylhydrazine aqueous solution were added, and stirring was started. Thereto, a mixed solution of 4.92 g of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate (Net 4.44 g, 0.020 mol) and 4.97 g of toluene was added at an internal temperature of 50 ° C. using a metering pump. It was added dropwise over time. After completion of dropping, the aqueous layer and the toluene layer were separated.
- Net 4.44 g, 0.020 mol ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate
- the obtained toluene layer was dried under reduced pressure, and a yellow oil 3 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 3 .86 g (94.5% yield) was obtained.
- a yellow oil 3 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 3 .86 g (94.5% yield) was obtained.
- HPLC high performance liquid chromatography
- Example 7 To a 100 mL four-necked Kolben equipped with a thermometer and a stirrer, 24.8 g of toluene and 7.96 g (0.023 mol) of a 13.5% monomethylhydrazine aqueous solution were added, and stirring was started. Thereto, a mixed solution of 4.92 g of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate (Net 4.44 g, 0.020 mol) and 4.97 g of toluene was added at a temperature of 5 ° C. using a metering pump. It was added dropwise over time. After completion of dropping, the aqueous layer and the toluene layer were separated.
- Net 4.44 g, 0.020 mol ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate
- the obtained toluene layer was dried under reduced pressure, and white crystals 3 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 3 .86 g (94.5% yield) was obtained.
- white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 97.6: 2.4.
- Example 8 In a 200 mL four-necked Kolben equipped with a thermometer and a stirrer, 79.92 g of toluene and 7.96 g (0.023 mol) of a 13.5% monomethylhydrazine aqueous solution were added, and stirring was started. Thereto, a mixed solution of 4.92 g of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate (Net 4.44 g, 0.020 mol) and 8.88 g of toluene was added at an internal temperature of 5 ° C. using a metering pump. It was added dropwise over time. After completion of dropping, the aqueous layer and the toluene layer were separated.
- Net 4.44 g, 0.020 mol ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate
- the obtained toluene layer was dried under reduced pressure, and white crystals 4 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 4 0.000 g (yield 98.0%) was obtained.
- the obtained white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 98.9: 1.1.
- Example 9 In a 300 mL four-necked Kolben equipped with a thermometer and a stirrer, 155.40 g of toluene and 7.96 g (0.023 mol) of a 13.5% monomethylhydrazine aqueous solution were added, and stirring was started. Thereto, a mixed solution of 4.92 g of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate (Net 4.44 g, 0.020 mol) and 22.20 g of toluene was added at a temperature of 5 ° C. using a metering pump. It was added dropwise over time. After completion of dropping, the aqueous layer and the toluene layer were separated.
- Net 4.44 g, 0.020 mol ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate
- the obtained toluene layer was dried under reduced pressure, and white crystals 4 consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 4 0.04 g (yield 99.0%) was obtained.
- white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 99.0: 1.0.
- Example 10 In a 50 mL four-necked Kolben equipped with a thermometer and a stirrer, 24.78 g of ethyl acetate and 7.96 g (0.023 mol) of a 13.5% monomethylhydrazine aqueous solution were added, and stirring was started. Then, a mixed solution of 4.92 g of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate (Net 4.44 g, 0.020 mol) and 4.97 g of ethyl acetate was added at an internal temperature of 5 ° C. using a metering pump. It was added dropwise over 16 hours.
- Net 4.44 g, 0.020 mol ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate
- Example 11 A 50 mL four-necked Kolben equipped with a thermometer and a stirrer was charged with 24.8 g of o-xylene and 7.96 g (0.023 mol) of a 13.5% monomethylhydrazine aqueous solution, and stirring was started. Then, a mixed solution of 4.92 g (Net 4.44 g, 0.020 mol) of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate and 4.97 g of o-xylene was added at an internal temperature of 5 ° C. using a metering pump. The solution was added dropwise over 22 hours.
- Example 12 To 50 mL four-necked Kolben equipped with a thermometer and a stirrer, 8.60 g of toluene and 3.78 g (0.022 mol) of 35% monoethylhydrazine aqueous solution were put, and stirring was started. Thereto, 13.1 g (0.02 mol) of a 34% toluene solution of ethyl 2-ethoxymethylene-4,4-difluoroacetoacetate was added dropwise at an internal temperature of 5 ° C. over 4 hours using a metering pump. After completion of dropping, the aqueous layer and the toluene layer were separated.
- the obtained toluene layer was dried under reduced pressure to obtain white crystals 4 consisting of ethyl 1-ethyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-ethyl-5-difluoromethylpyrazole-4-carboxylate 4 .15 g (95.1% yield) was obtained.
- white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 96.5: 3.5.
- Example 13 To a 50 mL four-necked Kolben equipped with a thermometer and a stirrer, 8.60 g of toluene and 12.0 g (0.022 mol) of an 8.8% monomethylhydrazine aqueous solution were added, and stirring was started. Then, 14.1 g (0.02 mol) of a 34% toluene solution of ethyl 2-ethoxymethylene-4,4,4-trifluoroacetoacetate represented by the following formula (2-2) was used using a metering pump. The solution was added dropwise at an internal temperature of 5 ° C. over 4 hours. After completion of dropping, the aqueous layer and the toluene layer were separated.
- the obtained toluene layer was evaporated to dryness under reduced pressure to obtain a white color composed of ethyl 1-methyl-3-trifluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-trifluoromethylpyrazole-4-carboxylate. 4.30 g (yield 96.8%) of crystals were obtained. When the obtained white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 96.1: 3.9.
- HPLC high performance liquid chromatography
- Example 14 To 50 mL four-necked Kolben equipped with a thermometer and a stirrer, 8.60 g of toluene and 3.78 g (0.022 mol) of 35% monoethylhydrazine aqueous solution were put, and stirring was started. Then, 14.1 g (0.02 mol) of a 34% toluene solution of ethyl 2-ethoxymethylene-4,4,4-trifluoroacetoacetate was dropped over 4 hours at an internal temperature of 5 ° C. using a metering pump. did. After completion of dropping, the aqueous layer and the toluene layer were separated.
- the obtained toluene layer was dried under reduced pressure to obtain a white color composed of ethyl 1-ethyl-3-trifluoromethylpyrazole-4-carboxylate and ethyl 1-ethyl-5-trifluoromethylpyrazole-4-carboxylate. 4.50 g (yield 95.3%) of crystals were obtained. When the obtained white crystals were analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 96.4: 3.6.
- HPLC high performance liquid chromatography
- the obtained toluene layer was dried under reduced pressure, and yellowish orange consisting of ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 4.04 g (yield 81.9%) of crystals were obtained.
- the obtained yellowish blue crystal was analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 83.7: 16.3.
- HPLC chart is shown in FIG.
- reddish brown oil containing ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and ethyl 1-methyl-5-difluoromethylpyrazole-4-carboxylate 3.6 g of oil (45% yield) was obtained.
- reddish brown oil was analyzed by high performance liquid chromatography (HPLC), the production ratio (isomer ratio) of the former and the latter was 77.8: 22.2.
- the aqueous layer and the toluene layer were separated, and the obtained toluene layer was dried under reduced pressure to obtain ethyl 1-methyl-3-difluoromethylpyrazole-4-carboxylate and 1-methyl-5-difluoromethylpyrazole.
- 2.94 g of yellowish white crystals composed of ethyl -4-carboxylate were obtained.
- the obtained yellowish white crystals were analyzed by high performance liquid chromatography (HPLC) and quantified by the absolute calibration curve method. As a result, the yield was 72.0%, and the production ratio (isomer ratio) of the former and the latter was 99. .1: 0.9.
- the aqueous layer obtained by liquid separation was analyzed by HPLC.
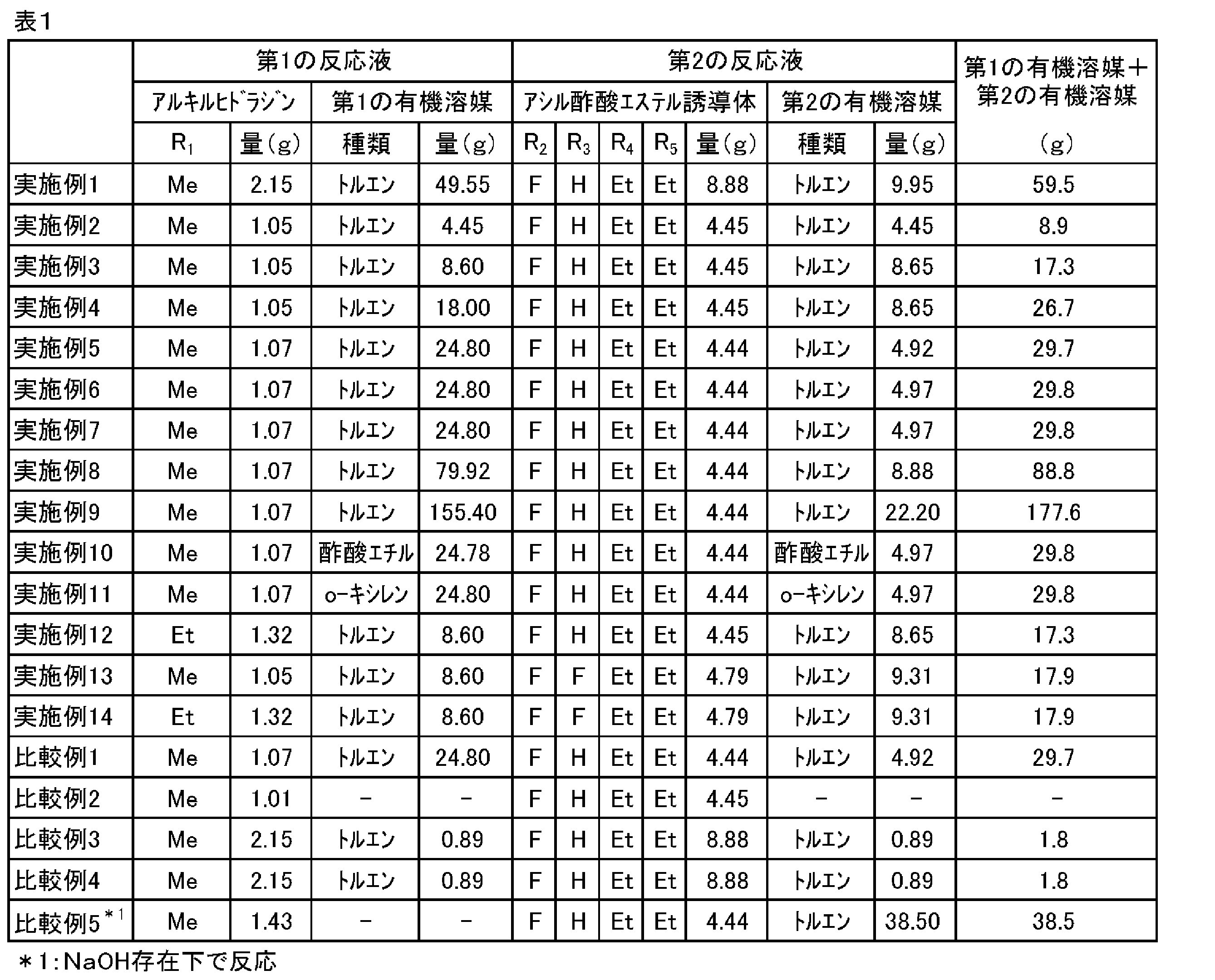
- Tables 1 and 2 summarize the reaction conditions, yields, and isomer ratios of Examples 1 to 14 and Comparative Examples 1 to 5.
- the production method of the present invention is suitable as a method for industrially producing 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid esters useful as synthetic intermediates for pharmaceuticals and agricultural chemicals.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
アルキルヒドラジン及び第1の有機溶媒を含有する第1の反応液に、アシル酢酸エステル誘導体及び第2の有機溶媒を含有する第2の反応液を0.5~30時間かけて添加し、塩基及び酸の非存在下で撹拌して-5~80℃の反応温度で反応させる工程を有し、第1の有機溶媒及び第2の有機溶媒が、それぞれ、ベンゼン、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、酢酸エチル、酢酸ブチル、及び炭酸ジメチルの少なくともいずれかであり、アシル酢酸エステル誘導体の質量に対する、第1の有機溶媒と第2の有機溶媒の合計質量が1~60倍であり、第1の有機溶媒と第2の有機溶媒の合計に占める、第1の有機溶媒の量が40~95質量%である1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法である。
Description
本発明は、医薬品及び農薬の合成中間体等として有用な1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法に関する。
2-アルコキシメチレンアシル酢酸エステルと置換ヒドラジン類を反応させると、2-アルコキシメチレンアシル酢酸エステルに複数の反応点が存在するため、位置異性体である1,3-二置換ピラゾール-4-カルボン酸エステルと、1,5-二置換ピラゾール-4-カルボン酸エステルの二種類のピラゾール誘導体が生成する。このため、目的とするピラゾール誘導体のみを得るためには、工業的に実施が困難なシリカゲルカラムクロマトグラフィーなどによる精製工程が必要となる。
関連する従来技術として、2-エトキシメチレンアシル酢酸エステル類と、アルキルヒドラジン類とを、酢酸エチル等の溶媒で反応させる1,3-ジアルキルピラゾール-4-カルボン酸エステルの製造方法が提案されている(特許文献1)。しかしながら、特許文献1に記載の製造方法によれば、1,3-ジアルキルピラゾール-4-カルボン酸エステル(約85%)と、1,5-ジアルキルピラゾール-4-カルボン酸エステル類(約15%)とが混在した混合物が得られる。このため、目的とする1,3-ジアルキルピラゾール-4-カルボン酸エステルを得るには蒸留等によって精製する必要があった。
また、2-エトキシメチレン-4,4-ジフルオロ-3-オキソブタン酸エチルと、無水メチルヒドラジンとを、ハイドロフルオロカーボン等の含ハロゲン系有機溶媒の存在下で反応させる1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルの製造方法が提案されている(特許文献2)。しかしながら、特許文献2に記載の製造方法であっても、目的とする化合物の位置異性体を相当量含有する混合物が得られるため、異性体比率についてさらなる改善の余地がある。さらに、この製造方法では特殊な含ハロゲン系溶媒を使用することが必須であるため、汎用性の面においても必ずしも十分であるとは言えなかった。
異性体比率を向上させるべく、モノメチルヒドラジンをアルデヒドやケトンと反応させてヒドラゾンとしておき、このヒドラゾンと、2-エトキシメチレン-4,4-ジフルオロ-3-オキソ酪酸エチルとを反応させてピラゾール環を形成する方法が提案されている(特許文献3)。また、水酸化ナトリウムや水酸化カリウム等の塩基の存在下、水又は水と有機溶媒の混合溶媒中で、メチルヒドラジンと2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルとを反応させる方法が提案されている(特許文献4)。
しかしながら、特許文献3に記載の方法では、ヒドラゾンを得るために事前に用いたアルデヒドやケトンが副生成物となり、目的物であるピラゾール誘導体と混在することになる。このため、ピラゾール誘導体をアルデヒドやケトンと分離して精製する工程が必要となるため、工業化の面では必ずしも満足できる方法であるとは言えなかった。また、特許文献4に記載の方法では、目的物であるカルボン酸エステルの加水分解が進行してしまい、収率が低下してしまうといった課題がある。さらには、塩基の存在下で反応を行うためにフッ素が脱離しやすく、廃液中のフッ素イオン濃度が上昇してしまい、反応装置の腐食が進行する、或いは廃液処理が煩雑になるといった課題がある。
本発明は、このような従来技術の有する問題点に鑑みてなされたものであり、その課題とするところは、二種類の位置異性体のうち、目的とする一方の位置異性体を高選択的及び高収率で合成することができるとともに、汎用性が高く、かつ、工業的プロセスに容易に適用可能な1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法を提供することにある。
本発明者らは上記課題を達成すべく鋭意検討した結果、以下の構成とすることによって上記課題を達成することが可能であることを見出し、本発明を完成するに至った。すなわち、本発明によれば、以下に示す1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法が提供される。
[1]下記一般式(1)で表されるアルキルヒドラジン及び第1の有機溶媒を含有する第1の反応液に、下記一般式(2)で表されるアシル酢酸エステル誘導体及び第2の有機溶媒を含有する第2の反応液を0.5~30時間かけて添加し、塩基及び酸の非存在下で撹拌して-5~80℃の反応温度で反応させる工程を有し、前記第1の有機溶媒及び前記第2の有機溶媒が、それぞれ、ベンゼン、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、酢酸エチル、酢酸ブチル、及び炭酸ジメチルの少なくともいずれかであり、前記アシル酢酸エステル誘導体の質量に対する、前記第1の有機溶媒と前記第2の有機溶媒の合計質量が1~60倍であり、前記第1の有機溶媒と前記第2の有機溶媒の合計に占める、前記第1の有機溶媒の量が40~95質量%である、下記一般式(3)で表される1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。


[2]前記第1の有機溶媒と前記第2の有機溶媒の合計に占める、前記第1の有機溶媒の量が65~92質量%である前記[1]に記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
[3]前記第1の有機溶媒及び前記第2の有機溶媒が、それぞれ、トルエン、キシレン、及び酢酸エチルの少なくともいずれかである前記[1]又は[2]に記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
[4]前記アシル酢酸エステル誘導体の質量に対する、前記第1の有機溶媒と前記第2の有機溶媒の合計質量が5~60倍である前記[1]~[3]のいずれかに記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
[5]前記第2の反応液に含有される前記アシル酢酸エステル誘導体の量が、前記アルキルヒドラジンに対して0.8~1.2モル当量である前記[1]~[4]のいずれかに記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
[3]前記第1の有機溶媒及び前記第2の有機溶媒が、それぞれ、トルエン、キシレン、及び酢酸エチルの少なくともいずれかである前記[1]又は[2]に記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
[4]前記アシル酢酸エステル誘導体の質量に対する、前記第1の有機溶媒と前記第2の有機溶媒の合計質量が5~60倍である前記[1]~[3]のいずれかに記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
[5]前記第2の反応液に含有される前記アシル酢酸エステル誘導体の量が、前記アルキルヒドラジンに対して0.8~1.2モル当量である前記[1]~[4]のいずれかに記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
本発明の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法によれば、二種類の位置異性体のうち、目的とする一方の位置異性体を高選択的及び高収率で合成することができる。また、本発明の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法は、汎用性が高く、工業的プロセスに容易に適用することができる。
以下、本発明の実施の形態について説明するが、本発明は以下の実施の形態に限定されるものではない。本発明は、下記一般式(3)で表される1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法(以下、単に「本発明の製造方法」とも記す)である。

本発明の製造方法は、下記一般式(1)で表されるアルキルヒドラジン及び第1の有機溶媒を含有する第1の反応液に、下記一般式(2)で表されるアシル酢酸エステル誘導体及び第2の有機溶媒を含有する第2の反応液を添加し、塩基及び酸の非存在下で撹拌して反応させる工程(以下、「反応工程」とも記す)を有する。

一般式(1)及び(3)中、R1で表される炭素数1~6のアルキル基の具体例としては、メチル基、エチル基、プロピル基、シクロプロピルメチル基、ブチル基、イソブチル基、ペンチル基、ヘキシル基等を挙げることができる。これらのアルキル基は、ハロゲン原子等で置換されていてもよい。置換されていてもよい炭素数1~6のアルキル基の具体例としては、2-クロロエチル基、2-ブロモエチル基、2-ヒドロキシエチル基、2,2,2-トリフルオロエチル基、3-クロロプロピル基等を挙げることができる。
一般式(1)で表されるアルキルヒドラジンは、一般的に入手可能なものをそのまま用いてもよいし、公知の方法により製造したものを用いてもよい。また、これらのアルキルヒドラジンは、無水物、含水物、及び水溶液のいずれであっても使用することができる。
一般式(2)及び(3)中、R2で表されるハロゲン原子の具体例としては、フッ素原子、塩素原子、臭素原子等を挙げることができる。
一般式(2)及び(3)中、R3で表される塩素原子又はフッ素原子で置換されていてもよい炭素数1~12のアルキル基の具体例としては、トリフルオロメチル基、ジフルオロメチル基、クロロジフルオロメチル基、ペンタフルオロエチル基、ペルフルオロプロピル基、ペルフルオロペンチル基、1,1,2,2,3,3,4,4,5,5-デカフルオロペンチル基、ペルフルオロヘキシル基、ペルフルオロノニル基、ペルフルオロデシル基、ペルフルオロドデシル基等を挙げることができる。
一般式(2)中、R4及びR5で表される炭素数1~6のアルキル基の具体例としては、それぞれメチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、ペンチル基、ヘキシル基等を挙げることができる。また、一般式(3)中、R4で表される炭素数1~6のアルキル基の具体例としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、ペンチル基、ヘキシル基等を挙げることができる。
一般式(2)で表されるアシル酢酸エステル誘導体は、市販されているものをそのまま用いてもよいし、通常の有機合成の手法に従って製造したものを用いてもよい。例えば、含フッ素カルボン酸エステルと酢酸エステルとをクライゼン縮合して得られるβ-ケトカルボン酸エステルを、無水酢酸の存在下、オルトギ酸エステルと作用させることによって、一般式(2)で表されるアシル酢酸エステル誘導体を容易に製造することができる。
本発明の製造方法の反応工程においては、第1の反応液に対して、例えば滴下等の方法によって第2の反応液を添加して、第1の反応液に含有されるアルキルヒドラジンと、第2の反応液に含有されるアシル酢酸エステル誘導体とを反応させる。第1の反応液には、一般式(1)で表されるアルキルヒドラジンとともに、第1の有機溶媒が含有される。第1の有機溶媒としては、例えば、芳香族炭化水素系溶媒及びエステル系溶媒の少なくともいずれかを用いることができる。芳香族炭化水素系溶媒の具体例としては、ベンゼン、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン等を挙げることができる。また、エステル系溶媒の具体例としては、酢酸エチル、酢酸ブチル、炭酸ジメチル等を挙げることができる。これらの有機溶媒のなかでも、トルエン、キシレン、及び酢酸エチルが好ましい。
第2の反応液には、一般式(2)で表されるアシル酢酸エステル誘導体とともに、第2の有機溶媒が含有される。第2の有機溶媒の具体例としては、好ましいものを含めて第1の有機溶媒と同様のものを挙げることができる。なお、第1の有機溶媒と第2の有機溶媒の種類は、同一であっても異なっていてもよい。
本発明の製造方法の反応工程においては、第1の反応液に対して第2の反応液を添加し、塩基及び酸の非存在下で撹拌して反応を進行させる。塩基及び酸の非存在下でアルキルヒドラジンとアシル酢酸エステル誘導体を反応させることで、生成する1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルが加水分解されるのを有効に抑制することができる。このため、目的物である1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルを高収率で得ることができる。さらに、第1の反応液に対して第2の反応液を添加すること、すなわち、アシル酢酸エステル誘導体に比してアルキルヒドラジンが過剰となる条件下で両者の反応を進行させることで、下記一般式(3)で表される目的化合物が生成する割合(反応選択性)を高めることができる。
また、アシル酢酸エステル誘導体の質量に対する、第1の有機溶媒と第2の有機溶媒の合計質量(有機溶媒の合計質量)を1~60倍、好ましくは5~50倍、さらに好ましくは6~40倍とする。すなわち、アシル酢酸エステル誘導体を有機溶媒で適度に希釈した状態でアルキルヒドラジンと反応させることで、反応選択性を高めることができる。
さらに、第1の有機溶媒と第2の有機溶媒の合計(有機溶媒の合計)に占める、第1の有機溶媒の量を40~95質量%、好ましくは65~92質量%、さらに好ましくは67~90質量%とする。すなわち、第1の反応液に含有されるアルキルヒドラジンと、第2の反応液に含有されるアシル酢酸エステル誘導体を、それぞれ適度に希釈した状態で相互に接触させて反応させることで、反応選択性を高めることができる。このように、使用する有機溶媒の量を適切に制御することで、塩基の存在下でアルキルヒドラジンとアシル酢酸エステル誘導体を反応させなくても、二種類の位置異性体のうち、目的とする位置異性体である1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルを高選択的に生成させることができる。
なお、本発明の製造方法では、第1の反応液に対して第2の反応液を一時に添加するのではなく、滴下等の方法により適度な時間をかけて徐々に添加する。これにより、目的とする1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルがより高選択的に生成可能となる。具体的には、アシル酢酸エステル誘導体を含有する第2の反応液を、第1の反応液に対して0.5~30時間、好ましくは1~25時間かけて添加する。添加に要する時間が0.5時間未満であると、反応選択性が低下する。一方、添加に要する時間が30時間を超えてもよいが、反応選択性の向上効果については頭打ちとなる傾向にある。なお、第2の反応液に含有されるアシル酢酸エステル誘導体の量は、第1の反応液中のアルキルヒドラジンに対して、通常0.8~1.2モル当量、好ましくは0.85~1.15モル当量である。
反応工程における反応温度は、-5~80℃とすることが好ましく、0~60℃とすることがさらに好ましい。反応温度が-5℃未満であると、反応が進行しにくくなる傾向にある。一方、反応温度が80℃を超えると、反応選択性が低下する傾向にある。反応温度を上記の範囲に制御することで、収率及び反応選択性をさらに向上させることができる。
上記の反応工程によれば、前記一般式(3)と前記一般式(4)で表される二種類の位置異性体のうち、前記一般式(3)で表される位置異性体(目的化合物)が高選択的及び高収率で生成される。このため、反応工程の後は通常の有機合成の手法に従って抽出操作等を行えば、高純度の目的化合物を得ることができる。なお、より高純度の目的化合物を得ようとする場合には、必要に応じて再結晶、洗浄、蒸留等を行ってもよい。
以下、本発明を実施例に基づいて具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、実施例、比較例中の「部」及び「%」は、特に断らない限り質量基準である。
(実施例1)
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン49.55g、及び13.5%モノメチルヒドラジン水溶液15.92g(0.047mol)を入れ、撹拌を開始した。そこに、下記式(2-1)で表される2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル8.88g(0.040mol)とトルエン9.95gの混合溶液を、定量ポンプを使用して内温5℃で16時間かけて滴下した。滴下終了後、内温5℃でさらに1時間撹拌した。水層とトルエン層を分液して得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶7.98g(収率92.8%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析し、絶対検量線法によって定量したところ、前者と後者の生成比(異性体比)は97.4:2.6(HPLC面積比)であった。なお、HPLCチャートを図1に示す。また、HPLCの条件を以下に示す。
・カラム:商品名「Inertsil ODS-3」(4.6×150mm、ジーエルサイエンス社製)
・温度:40℃
・流速:1.0mL/min
・流動相:A液;アセトニトリル、B液;0.2体積%酢酸水溶液、A:B=45:55
・検出器(波長):220nm
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン49.55g、及び13.5%モノメチルヒドラジン水溶液15.92g(0.047mol)を入れ、撹拌を開始した。そこに、下記式(2-1)で表される2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル8.88g(0.040mol)とトルエン9.95gの混合溶液を、定量ポンプを使用して内温5℃で16時間かけて滴下した。滴下終了後、内温5℃でさらに1時間撹拌した。水層とトルエン層を分液して得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶7.98g(収率92.8%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析し、絶対検量線法によって定量したところ、前者と後者の生成比(異性体比)は97.4:2.6(HPLC面積比)であった。なお、HPLCチャートを図1に示す。また、HPLCの条件を以下に示す。
・カラム:商品名「Inertsil ODS-3」(4.6×150mm、ジーエルサイエンス社製)
・温度:40℃
・流速:1.0mL/min
・流動相:A液;アセトニトリル、B液;0.2体積%酢酸水溶液、A:B=45:55
・検出器(波長):220nm
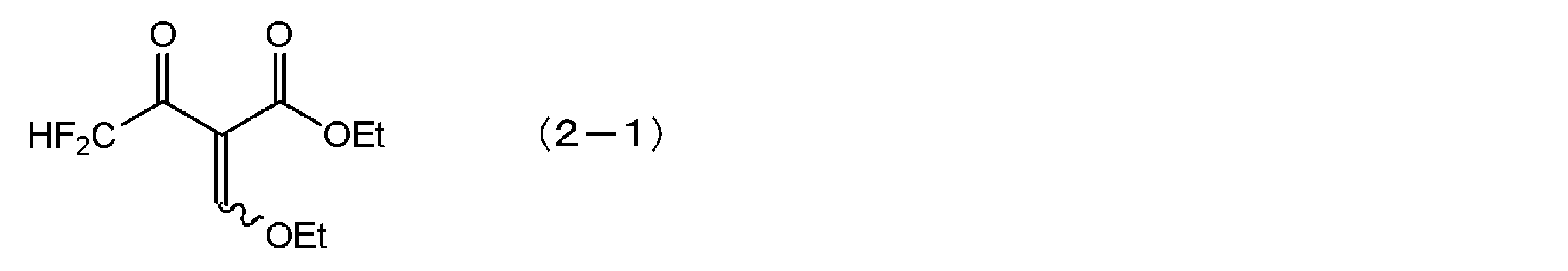
得られた白色結晶7.00gを30mL側管付ナス型フラスコに仕込み、ヘプタン10g及びアセトン1.7gを加えた後、マグネチックスターラーで撹拌しながら70℃に加温して白色結晶を溶解させた。加温停止して25℃まで徐々に空冷したところ、白色結晶が析出した。析出した白色結晶を減圧ろ過した後、減圧乾燥して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルの白色結晶5.99gを得た。得られた白色結晶の1H-NMRによる分析結果を以下に示す。
1H-NMR(CDCl3,TMS,ppm):δ1.35(t,J=7.2Hz,3H),3.96(s,3H),4.31(q,J=7.2,2H),7.11(t,J=54,1H),7.90(s,1H)
1H-NMR(CDCl3,TMS,ppm):δ1.35(t,J=7.2Hz,3H),3.96(s,3H),4.31(q,J=7.2,2H),7.11(t,J=54,1H),7.90(s,1H)
(実施例2)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン4.45g、及び8.8%モノメチルヒドラジン水溶液12.0g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの50%トルエン溶液8.90g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.90g(収率95.5%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.1:5.9であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン4.45g、及び8.8%モノメチルヒドラジン水溶液12.0g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの50%トルエン溶液8.90g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.90g(収率95.5%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.1:5.9であった。
(実施例3)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g、及び8.8%モノメチルヒドラジン水溶液12.0g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの34%トルエン溶液13.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.95g(収率96.7%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.1:5.9であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g、及び8.8%モノメチルヒドラジン水溶液12.0g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの34%トルエン溶液13.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.95g(収率96.7%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.1:5.9であった。
(実施例4)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン18.00g、及び13.1%モノメチルヒドラジン水溶液8.00g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの34%トルエン溶液13.10g(0.02mol)を、定量ポンプを使用して内温5℃で24時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.00g(収率98.0%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は98.9:1.1であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン18.00g、及び13.1%モノメチルヒドラジン水溶液8.00g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの34%トルエン溶液13.10g(0.02mol)を、定量ポンプを使用して内温5℃で24時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.00g(収率98.0%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は98.9:1.1であった。
(実施例5)
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.92gの混合溶液を、定量ポンプを使用して内温5℃で0.5時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.68g(収率90.0%)を得た。得られた黄色オイルを高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は91.5:8.5であった。
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.92gの混合溶液を、定量ポンプを使用して内温5℃で0.5時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.68g(収率90.0%)を得た。得られた黄色オイルを高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は91.5:8.5であった。
(実施例6)
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.97gの混合溶液を、定量ポンプを使用して内温50℃で22時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる黄色オイル3.86g(収率94.5%)を得た。得られた黄色オイルを高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.2:5.8であった。
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.97gの混合溶液を、定量ポンプを使用して内温50℃で22時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる黄色オイル3.86g(収率94.5%)を得た。得られた黄色オイルを高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.2:5.8であった。
(実施例7)
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.97gの混合溶液を、定量ポンプを使用して内温5℃で1時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.86g(収率94.5%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は97.6:2.4であった。
温度計及び撹拌機を備えた100mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.97gの混合溶液を、定量ポンプを使用して内温5℃で1時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶3.86g(収率94.5%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は97.6:2.4であった。
(実施例8)
温度計及び撹拌機を備えた200mL四つ口コルベンに、トルエン79.92g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン8.88gの混合溶液を、定量ポンプを使用して内温5℃で18時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.00g(収率98.0%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は98.9:1.1であった。
温度計及び撹拌機を備えた200mL四つ口コルベンに、トルエン79.92g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン8.88gの混合溶液を、定量ポンプを使用して内温5℃で18時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.00g(収率98.0%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は98.9:1.1であった。
(実施例9)
温度計及び撹拌機を備えた300mL四つ口コルベンに、トルエン155.40g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン22.20gの混合溶液を、定量ポンプを使用して内温5℃で23時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.04g(収率99.0%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は99.0:1.0であった。
温度計及び撹拌機を備えた300mL四つ口コルベンに、トルエン155.40g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン22.20gの混合溶液を、定量ポンプを使用して内温5℃で23時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.04g(収率99.0%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は99.0:1.0であった。
(実施例10)
温度計及び撹拌機を備えた50mL四つ口コルベンに、酢酸エチル24.78g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及び酢酸エチル4.97gの混合溶液を、定量ポンプを使用して内温5℃で16時間かけて滴下した。滴下終了後、水層と酢酸エチル層を分液した。得られた酢酸エチル層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる淡黄色結晶3.91g(収率95.8%)を得た。得られた淡黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.7:5.3であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、酢酸エチル24.78g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及び酢酸エチル4.97gの混合溶液を、定量ポンプを使用して内温5℃で16時間かけて滴下した。滴下終了後、水層と酢酸エチル層を分液した。得られた酢酸エチル層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる淡黄色結晶3.91g(収率95.8%)を得た。得られた淡黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.7:5.3であった。
(実施例11)
温度計及び撹拌機を備えた50mL四つ口コルベンに、o-キシレン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びo-キシレン4.97gの混合溶液を、定量ポンプを使用して内温5℃で22時間かけて滴下した。滴下終了後、水層とo-キシレン層を分液した。得られたo-キシレン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる燈黄色結晶3.59g(収率87.9%)を得た。得られた燈黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.5:5.5であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、o-キシレン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びo-キシレン4.97gの混合溶液を、定量ポンプを使用して内温5℃で22時間かけて滴下した。滴下終了後、水層とo-キシレン層を分液した。得られたo-キシレン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる燈黄色結晶3.59g(収率87.9%)を得た。得られた燈黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は94.5:5.5であった。
(実施例12)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g、及び35%モノエチルヒドラジン水溶液3.78g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの34%トルエン溶液13.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-エチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-エチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.15g(収率95.1%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は96.5:3.5であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g、及び35%モノエチルヒドラジン水溶液3.78g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチルの34%トルエン溶液13.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-エチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-エチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.15g(収率95.1%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は96.5:3.5であった。
(実施例13)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g及び8.8%モノメチルヒドラジン水溶液12.0g(0.022mol)を入れ、撹拌を開始した。そこに、下記式(2-2)で表される2-エトキシメチレン-4,4,4-トリフルオロアセト酢酸エチルの34%トルエン溶液14.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-トリフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-トリフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.30g(収率96.8%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は96.1:3.9であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g及び8.8%モノメチルヒドラジン水溶液12.0g(0.022mol)を入れ、撹拌を開始した。そこに、下記式(2-2)で表される2-エトキシメチレン-4,4,4-トリフルオロアセト酢酸エチルの34%トルエン溶液14.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-トリフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-トリフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.30g(収率96.8%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は96.1:3.9であった。

(実施例14)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g、及び35%モノエチルヒドラジン水溶液3.78g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4,4-トリフルオロアセト酢酸エチルの34%トルエン溶液14.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-エチル-3-トリフルオロメチルピラゾール-4-カルボン酸エチルと、1-エチル-5-トリフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.50g(収率95.3%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は96.4:3.6であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン8.60g、及び35%モノエチルヒドラジン水溶液3.78g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4,4-トリフルオロアセト酢酸エチルの34%トルエン溶液14.1g(0.02mol)を、定量ポンプを使用して内温5℃で4時間かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-エチル-3-トリフルオロメチルピラゾール-4-カルボン酸エチルと、1-エチル-5-トリフルオロメチルピラゾール-4-カルボン酸エチルとからなる白色結晶4.50g(収率95.3%)を得た。得られた白色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は96.4:3.6であった。
(比較例1)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.97gの混合溶液を、内温5℃で5分かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる黄燈色結晶4.04g(収率81.9%)を得た。得られた黄燈色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は83.7:16.3であった。なお、HPLCチャートを図2に示す。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン24.8g、及び13.5%モノメチルヒドラジン水溶液7.96g(0.023mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.92g(Net4.44g、0.020mol)及びトルエン4.97gの混合溶液を、内温5℃で5分かけて滴下した。滴下終了後、水層とトルエン層を分液した。得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる黄燈色結晶4.04g(収率81.9%)を得た。得られた黄燈色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は83.7:16.3であった。なお、HPLCチャートを図2に示す。
(比較例2)
温度計及び撹拌機を備えた50mL四つ口コルベンに、35%モノメチルヒドラジン水溶液2.90g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.45gを、内温5℃で5分かけて滴下した。滴下終了後、水層を分液して除去し、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとを含む赤褐色オイル3.6g(収率45%)を得た。得られた赤褐色オイルを高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は77.8:22.2であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、35%モノメチルヒドラジン水溶液2.90g(0.022mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.45gを、内温5℃で5分かけて滴下した。滴下終了後、水層を分液して除去し、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとを含む赤褐色オイル3.6g(収率45%)を得た。得られた赤褐色オイルを高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は77.8:22.2であった。
(比較例3)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン0.89g、及び13.5%モノメチルヒドラジン水溶液15.92g(0.046mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル9.84g(Net8.88g、0.040mol)及びトルエン0.89gの混合溶液を、定量ポンプを使用して内温5℃で1時間かけて滴下した。なお、滴下終了時には反応液中にクリーム色の結晶が析出していた。滴下終了後に反応液を室温に戻したところ、結晶は溶解してエマルジョンとなった。トルエン10gを加えて抽出し、得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる燈黄色結晶7.73g(収率65.7%)を得た。得られた燈黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は89.0:11.0であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン0.89g、及び13.5%モノメチルヒドラジン水溶液15.92g(0.046mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル9.84g(Net8.88g、0.040mol)及びトルエン0.89gの混合溶液を、定量ポンプを使用して内温5℃で1時間かけて滴下した。なお、滴下終了時には反応液中にクリーム色の結晶が析出していた。滴下終了後に反応液を室温に戻したところ、結晶は溶解してエマルジョンとなった。トルエン10gを加えて抽出し、得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる燈黄色結晶7.73g(収率65.7%)を得た。得られた燈黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は89.0:11.0であった。
(比較例4)
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン0.89g、及び13.5%モノメチルヒドラジン水溶液15.92g(0.046mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル9.84g(Net8.88g、0.040mol)及びトルエン0.89gの混合溶液を、定量ポンプを使用して内温5℃で24時間かけて滴下した。なお、滴下終了時には反応液中にクリーム色の結晶が析出していた。滴下終了後に反応液を室温に戻したところ、結晶は溶解してエマルジョンとなった。トルエン10gを加えて抽出し、得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる燈黄色結晶6.98g(収率70.4%)を得た。得られた燈黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は85.0:15.0であった。
温度計及び撹拌機を備えた50mL四つ口コルベンに、トルエン0.89g、及び13.5%モノメチルヒドラジン水溶液15.92g(0.046mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル9.84g(Net8.88g、0.040mol)及びトルエン0.89gの混合溶液を、定量ポンプを使用して内温5℃で24時間かけて滴下した。なお、滴下終了時には反応液中にクリーム色の結晶が析出していた。滴下終了後に反応液を室温に戻したところ、結晶は溶解してエマルジョンとなった。トルエン10gを加えて抽出し、得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる燈黄色結晶6.98g(収率70.4%)を得た。得られた燈黄色結晶を高速液体クロマトグラフィー(HPLC)により分析したところ、前者と後者の生成比(異性体比)は85.0:15.0であった。
(比較例5)
温度計及び撹拌機を備えた100mL四つ口コルベンに、水8.67g、48%水酸化ナトリウム水溶液1.68g、及び35%モノメチルヒドラジン水溶液4.08g(0.031mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.44g(0.020mol)及びトルエン38.5gの混合溶液を、内温50℃で5分かけて滴下した。滴下終了後、内温50℃で10分間撹拌した。次いで、水層とトルエン層を分液し、得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる黄白色結晶2.94gを得た。得られた黄白色結晶を高速液体クロマトグラフィー(HPLC)により分析し、絶対検量線法により定量したところ、収率は72.0%であり、前者と後者の生成比(異性体比)は99.1:0.9であった。なお、分液して得られた水層をHPLCにより分析したところ、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルの加水分解生成物である1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸が収率23.7%で含まれていた。
温度計及び撹拌機を備えた100mL四つ口コルベンに、水8.67g、48%水酸化ナトリウム水溶液1.68g、及び35%モノメチルヒドラジン水溶液4.08g(0.031mol)を入れ、撹拌を開始した。そこに、2-エトキシメチレン-4,4-ジフルオロアセト酢酸エチル4.44g(0.020mol)及びトルエン38.5gの混合溶液を、内温50℃で5分かけて滴下した。滴下終了後、内温50℃で10分間撹拌した。次いで、水層とトルエン層を分液し、得られたトルエン層を減圧乾固して、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルと、1-メチル-5-ジフルオロメチルピラゾール-4-カルボン酸エチルとからなる黄白色結晶2.94gを得た。得られた黄白色結晶を高速液体クロマトグラフィー(HPLC)により分析し、絶対検量線法により定量したところ、収率は72.0%であり、前者と後者の生成比(異性体比)は99.1:0.9であった。なお、分液して得られた水層をHPLCにより分析したところ、1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸エチルの加水分解生成物である1-メチル-3-ジフルオロメチルピラゾール-4-カルボン酸が収率23.7%で含まれていた。
上記実施例1~14及び比較例1~5の反応条件等、収率、及び異性体比をまとめたものを表1及び2に示す。

本発明の製造方法は、医薬品及び農薬の合成中間体等として有用な1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルを工業的に製造する方法として好適である。
Claims (5)
- 下記一般式(1)で表されるアルキルヒドラジン及び第1の有機溶媒を含有する第1の反応液に、下記一般式(2)で表されるアシル酢酸エステル誘導体及び第2の有機溶媒を含有する第2の反応液を0.5~30時間かけて添加し、塩基及び酸の非存在下で撹拌して-5~80℃の反応温度で反応させる工程を有し、
前記第1の有機溶媒及び前記第2の有機溶媒が、それぞれ、ベンゼン、トルエン、キシレン、クロロベンゼン、ジクロロベンゼン、酢酸エチル、酢酸ブチル、及び炭酸ジメチルの少なくともいずれかであり、
前記アシル酢酸エステル誘導体の質量に対する、前記第1の有機溶媒と前記第2の有機溶媒の合計質量が1~60倍であり、
前記第1の有機溶媒と前記第2の有機溶媒の合計に占める、前記第1の有機溶媒の量が40~95質量%である、下記一般式(3)で表される1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
(前記一般式(1)中、R1は、置換されていてもよい炭素数1~6のアルキル基を示す)
(前記一般式(2)中、R2は、水素原子又はハロゲン原子を示し、R3は、水素原子、フッ素原子、又は塩素原子若しくはフッ素原子で置換されていてもよい炭素数1~12のアルキル基を示し、R4及びR5は、それぞれ独立に炭素数1~6のアルキル基を示す)
(前記一般式(3)中、R1は、置換されていてもよい炭素数1~6のアルキル基を示し、R2は、水素原子又はハロゲン原子を示し、R3は、水素原子、フッ素原子、又は塩素原子若しくはフッ素原子で置換されていてもよい炭素数1~12のアルキル基を示し、R4は、炭素数1~6のアルキル基を示す)
- 前記第1の有機溶媒と前記第2の有機溶媒の合計に占める、前記第1の有機溶媒の量が65~92質量%である請求項1に記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
- 前記第1の有機溶媒及び前記第2の有機溶媒が、それぞれ、トルエン、キシレン、及び酢酸エチルの少なくともいずれかである請求項1又は2に記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
- 前記アシル酢酸エステル誘導体の質量に対する、前記第1の有機溶媒と前記第2の有機溶媒の合計質量が5~60倍である請求項1~3のいずれか一項に記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
- 前記第2の反応液に含有される前記アシル酢酸エステル誘導体の量が、前記アルキルヒドラジンに対して0.8~1.2モル当量である請求項1~4のいずれか一項に記載の1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/426,294 US20150218105A1 (en) | 2012-09-05 | 2013-03-14 | Method for producing 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester |
| EP13836036.7A EP2894150B1 (en) | 2012-09-05 | 2013-03-14 | Method for producing 1-substituted-3-fluoroalkylpyrazole-4-carboxylic acid ester |
| CN201380003229.4A CN103842345B (zh) | 2012-09-05 | 2013-03-14 | 1-取代-3-氟烷基吡唑-4-羧酸酯的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-194959 | 2012-09-05 | ||
| JP2012194959A JP5140776B1 (ja) | 2012-09-05 | 2012-09-05 | 1−置換−3−フルオロアルキルピラゾール−4−カルボン酸エステルの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014038224A1 true WO2014038224A1 (ja) | 2014-03-13 |
Family
ID=47789852
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/057275 WO2014038224A1 (ja) | 2012-09-05 | 2013-03-14 | 1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20150218105A1 (ja) |
| EP (1) | EP2894150B1 (ja) |
| JP (1) | JP5140776B1 (ja) |
| CN (1) | CN103842345B (ja) |
| TW (1) | TW201410652A (ja) |
| WO (1) | WO2014038224A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3650443A1 (en) | 2018-11-07 | 2020-05-13 | Fujian Yongjing Technology Co., Ltd. | Continuous flow synthesis of fluorinated or non-fluorinated pyrazoles |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105418503A (zh) * | 2015-12-12 | 2016-03-23 | 常州大学 | 一种3-二氟甲基-1-甲基-1h-吡唑-4-羧酸乙酯的合成方法 |
| CN111458432B (zh) * | 2020-04-15 | 2023-08-01 | 山东京博农化科技股份有限公司 | 一种高效液相色谱法检测三氟乙酰乙酸乙酯的方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000212166A (ja) | 1999-01-26 | 2000-08-02 | Mitsui Chemicals Inc | 1,3―ジアルキルピラゾ―ル―4―カルボン酸エステルの製造法 |
| WO2006090778A1 (ja) * | 2005-02-25 | 2006-08-31 | Sagami Chemical Research Center | 1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 |
| WO2009135808A2 (de) * | 2008-05-05 | 2009-11-12 | Basf Se | Verfahren zur herstellung von 1,3,4-substituierten pyrazolverbindungen |
| WO2010009990A1 (en) * | 2008-07-21 | 2010-01-28 | Basf Se | Process for preparing 1,3-disubstituted pyrazolecarboxylic esters |
| WO2010130532A1 (en) * | 2009-05-15 | 2010-11-18 | Syngenta Limited | Process for purification of 1-methylpyrazole-4-carboxylic acid esters |
| WO2011012620A2 (en) * | 2009-07-31 | 2011-02-03 | Syngenta Participations Ag | Processes for the alkylation of pyrazoles |
| WO2012025469A1 (en) | 2010-08-24 | 2012-03-01 | Solvay Sa | Improved process for the preparation of esters of 1-h-pyrazole-4-carboxylic acids |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008059370A2 (en) * | 2006-11-17 | 2008-05-22 | Pfizer Japan Inc. | Substituted bicyclocarboxyamide compounds |
-
2012
- 2012-09-05 JP JP2012194959A patent/JP5140776B1/ja active Active
-
2013
- 2013-03-14 US US14/426,294 patent/US20150218105A1/en not_active Abandoned
- 2013-03-14 EP EP13836036.7A patent/EP2894150B1/en active Active
- 2013-03-14 WO PCT/JP2013/057275 patent/WO2014038224A1/ja active Application Filing
- 2013-03-14 CN CN201380003229.4A patent/CN103842345B/zh active Active
- 2013-05-13 TW TW102116857A patent/TW201410652A/zh unknown
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000212166A (ja) | 1999-01-26 | 2000-08-02 | Mitsui Chemicals Inc | 1,3―ジアルキルピラゾ―ル―4―カルボン酸エステルの製造法 |
| WO2006090778A1 (ja) * | 2005-02-25 | 2006-08-31 | Sagami Chemical Research Center | 1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 |
| JP4114754B2 (ja) | 2005-02-25 | 2008-07-09 | 財団法人相模中央化学研究所 | 1−置換−3−フルオロアルキルピラゾール−4−カルボン酸エステルの製造方法 |
| WO2009135808A2 (de) * | 2008-05-05 | 2009-11-12 | Basf Se | Verfahren zur herstellung von 1,3,4-substituierten pyrazolverbindungen |
| JP2011519889A (ja) | 2008-05-05 | 2011-07-14 | ビーエーエスエフ ソシエタス・ヨーロピア | 1,3,4−置換ピラゾール化合物の調製方法 |
| WO2010009990A1 (en) * | 2008-07-21 | 2010-01-28 | Basf Se | Process for preparing 1,3-disubstituted pyrazolecarboxylic esters |
| WO2010130532A1 (en) * | 2009-05-15 | 2010-11-18 | Syngenta Limited | Process for purification of 1-methylpyrazole-4-carboxylic acid esters |
| WO2011012620A2 (en) * | 2009-07-31 | 2011-02-03 | Syngenta Participations Ag | Processes for the alkylation of pyrazoles |
| WO2012025469A1 (en) | 2010-08-24 | 2012-03-01 | Solvay Sa | Improved process for the preparation of esters of 1-h-pyrazole-4-carboxylic acids |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2894150A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3650443A1 (en) | 2018-11-07 | 2020-05-13 | Fujian Yongjing Technology Co., Ltd. | Continuous flow synthesis of fluorinated or non-fluorinated pyrazoles |
| JP2021508669A (ja) * | 2018-11-07 | 2021-03-11 | 福建永晶科技股▲ふん▼有限公司Fujian Yongjing Technology Co., Ltd | ピラゾール又はピリミジノンの新しい製造方法 |
| US11299463B2 (en) | 2018-11-07 | 2022-04-12 | Fujian Yongjing Technology Co., Ltd. | Process for the manufacture of pyrazoles or pyrimidones |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2014051439A (ja) | 2014-03-20 |
| EP2894150B1 (en) | 2017-05-17 |
| CN103842345A (zh) | 2014-06-04 |
| CN103842345B (zh) | 2016-01-20 |
| TW201410652A (zh) | 2014-03-16 |
| US20150218105A1 (en) | 2015-08-06 |
| EP2894150A4 (en) | 2016-01-20 |
| EP2894150A1 (en) | 2015-07-15 |
| JP5140776B1 (ja) | 2013-02-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4114754B2 (ja) | 1−置換−3−フルオロアルキルピラゾール−4−カルボン酸エステルの製造方法 | |
| WO2014038224A1 (ja) | 1-置換-3-フルオロアルキルピラゾール-4-カルボン酸エステルの製造方法 | |
| WO2012176717A1 (ja) | ピラゾール化合物の製造方法 | |
| CN110891940A (zh) | 吡唑-4-甲酰胺衍生物的制备方法 | |
| JP5232335B1 (ja) | 1−置換−3−フルオロアルキルピラゾール−4−カルボン酸エステルの製造方法 | |
| JP5830957B2 (ja) | ピラゾール化合物の製造方法 | |
| JP6787331B2 (ja) | 酸ハライド溶液の製造方法、混合溶液、及びモノエステル化合物の製造方法 | |
| JP2012533581A (ja) | 新規なアルコキシエノンおよびエナミノケトンおよびその調製方法 | |
| JP5186115B2 (ja) | 2‐置換ベンジル‐3,3‐ジフルオロアクリル酸エステル誘導体及びそれらの製造方法 | |
| JP2003335735A (ja) | パーフルオロイソプロピルアニリン類の製造方法 | |
| JP2022184396A (ja) | 1-アルキル-5-ヒドロキシピラゾールの製造方法 | |
| JP2013006781A (ja) | ピラゾール化合物の製造方法 | |
| WO2016152831A1 (ja) | ピラゾール誘導体の製造方法 | |
| JP6994498B2 (ja) | 3-アミノ-1-(2,6-二置換フェニル)ピラゾール類を調製する方法 | |
| JP2012193124A (ja) | 2,3−ジクロロピリジンの製造方法 | |
| JP5605104B2 (ja) | ピラゾール化合物の製造方法 | |
| JP5271280B2 (ja) | 光学活性2,2’−ビフェノール誘導体及びその製造方法 | |
| TW201536746A (zh) | 製備3,5-雙(氟烷基)吡唑衍生物之方法 | |
| JP6809485B2 (ja) | 酸ハライド溶液の製造方法、及びモノエステル化合物の製造方法 | |
| US8242313B2 (en) | Alkoxy enones and enamino ketones and a process for preparation thereof | |
| WO2016190280A1 (ja) | O-[1-(2-ヒドロキシプロピル)]オキシム化合物の製造方法 | |
| WO2024100791A1 (ja) | 1-アルキル-5-ヒドロキシピラゾールの製造方法 | |
| JP2013028547A (ja) | ブロモアニリン誘導体の製造方法 | |
| JP2001335564A (ja) | 1−アルキル−5−置換ピラゾール−3−カルボン酸エステルの製造法 | |
| JP2002012586A (ja) | 1−アルキル−5−置換ピラゾール−3−カルボン酸の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13836036 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013836036 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013836036 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14426294 Country of ref document: US |