WO2009084603A1 - エクオール合成に関与する酵素 - Google Patents
エクオール合成に関与する酵素 Download PDFInfo
- Publication number
- WO2009084603A1 WO2009084603A1 PCT/JP2008/073649 JP2008073649W WO2009084603A1 WO 2009084603 A1 WO2009084603 A1 WO 2009084603A1 JP 2008073649 W JP2008073649 W JP 2008073649W WO 2009084603 A1 WO2009084603 A1 WO 2009084603A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polypeptide
- dihydrodaidzein
- tetrahydrodaidzein
- amino acid
- seq
- Prior art date
Links
Images
































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0071—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14)
- C12N9/0073—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14) with NADH or NADPH as one donor, and incorporation of one atom of oxygen 1.14.13
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/93—Ligases (6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/02—Oxygen as only ring hetero atoms
- C12P17/06—Oxygen as only ring hetero atoms containing a six-membered hetero ring, e.g. fluorescein
Definitions
- the present invention relates to a polypeptide having an activity to synthesize dihydrodaidzein using daidzein as a substrate, a polypeptide having an activity to synthesize tetrahydrodaidzein using dihydrodaidzein as a substrate, and a polypeptide having an activity to synthesize equol using tetrahydrodaidzein as a substrate As well as polynucleotides encoding these polypeptides. Furthermore, this invention relates to the manufacturing method of dihydrodaidzein, tetrahydrodaidzein, and equol using the said polypeptide, and the manufacturing apparatus used therewith, and provides the technique relevant to these.
- isoflavone derivatives are considered to have various effects physiologically or pharmacologically, and are used as raw materials for foods and pharmaceuticals.
- isoflavone derivatives have estrogen-like effects as previously believed, but these derivatives are metabolized (lived) by various intestinal bacteria. Equal with a stronger estrogen-like action is produced after receiving (synthesis), and this equol is released from the bacterial body and then absorbed from the intestinal tract, so that the estrogen action is exerted systemically Has been reported.
- not all humans have the ability to produce equol in the intestine, and equol production varies from person to person. For example, there are humans who do not have equol-producing bacteria in the intestine and humans who have equol-producing bacteria in the intestine but have low equol-producing ability.
- the effective use of equol in the body is important from the standpoint of dealing with chronic senile diseases such as osteoporosis, especially in Japan and other countries that have faced an aging society. And as mentioned above, considering the reality that there are humans who have equol-producing intestinal bacteria and humans who have no equol, enter into the efficient human production of equol. You have to turn your eyes.
- An object of the present invention is to provide an enzyme involved in dihydrodaidzein synthesis, which is a raw material for equol synthesis. Specifically, an object of the present invention is to provide a polypeptide having an activity of synthesizing dihydrodaidzein using daidzein as a substrate. Furthermore, an object of the present invention is to provide a polynucleotide encoding the polypeptide, a technique relating to the synthesis of dihydrodaidzein using the polypeptide, and the like.
- Another object of the present invention is to provide an enzyme involved in tetrahydrodaidzein synthesis, which is a raw material for equol synthesis.
- an object of the present invention is to provide a polypeptide having an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate.
- an object of the present invention is to provide a polynucleotide encoding the polypeptide, a technique relating to the synthesis of tetrahydrodaidzein using the polypeptide, and the like.
- an object of the present invention is to provide an enzyme involved in equol synthesis. Specifically, an object of the present invention is to provide a polypeptide having an activity of synthesizing equol using tetrahydrodaidzein as a substrate. Furthermore, an object of the present invention is to provide a polynucleotide encoding the polypeptide, a technique relating to the synthesis of equol using the polypeptide, and the like.
- an object of the present invention is to provide a method for producing an intermediate such as dihydrodaidzein or tetrahydrodaidzein produced in the production of equol from daidzein, and a method for producing equol using the obtained intermediate. Furthermore, an object of this invention is to provide the manufacturing apparatus utilized for these manufacture.
- the present inventors synthesized tetrahydrodaidzein, an enzyme capable of synthesizing dihydrodaidzein, which is a raw material for equol synthesis, from equol-producing enteric bacteria. And the enzyme capable of synthesizing equol were successfully isolated and their structures were elucidated. Further, the inventors of the present application have further researched and succeeded in artificially producing dihydrodaidzein, tetrahydrodaidzein, and equol using the above-mentioned enzymes. The present invention has been completed by making further improvements based on such findings.
- Term A1 A polypeptide that is any of the following (Aa) to (Ac): (Aa) a polypeptide comprising the amino acid sequence set forth in SEQ ID NO: 1; (Ab) a polypeptide comprising an amino acid sequence in which one or more amino acids are substituted, deleted, inserted and / or added in the amino acid sequence of SEQ ID NO: 1 and having an activity of synthesizing dihydrodaidzein using daidzein as a substrate ; (Ac) A polypeptide comprising an amino acid sequence having 60% or more identity to the amino acid sequence of SEQ ID NO: 1 and having an activity of synthesizing dihydrodaidzein using daidzein as a substrate.
- An expression vector comprising the polynucleotide according to Item A2.
- Term A4. A recombinant cell transformed with the expression vector according to Item A3.
- Term A5. The recombinant cell according to Item A4, wherein the recombinant cell is a bacterial prokaryotic cell.
- Term A6. The recombinant cell according to Item A5, wherein the bacterial prokaryotic cell belongs to the genus Lactococcus.
- Term A7 A method for producing a polypeptide, comprising culturing the cell according to any one of Items A4 to A6 and obtaining a polypeptide having an activity of producing dihydrodaidzein using daidzein as a substrate.
- Term A9 A method for producing dihydrodaidzein, comprising a step of allowing the polypeptide of item A1 or A8 and NADPH and / or NADH to act on daidzein.
- Term A10. A method for producing dihydrodaidzein, comprising a step of causing the cell according to any one of Items A4 to A6 to act on daidzein.
- Term A11 An antibody having binding ability to the polypeptide of Item A1 or the polypeptide encoded by the polynucleotide of Item A2.
- Term A15 A15.
- Term A16. A method for detecting or measuring a polynucleotide encoding the polypeptide of Item A1 or the polynucleotide of Item A2 using the probe of Item A14.
- Term A17 The method according to Item A16, wherein the polypeptide to be detected or measured is present in bacterial prokaryotic cells.
- the method according to Item A16 comprising a step of amplifying the polynucleotide encoding the polypeptide according to Item A1, the polynucleotide according to Item A2, or a part thereof by PCR.
- Term A19 A dihydrodaidzein synthase composition comprising the polypeptide of Item A1 or the polypeptide encoded by the polynucleotide of Item A2.
- Term A20 The composition according to Item A19, further comprising NADPH and / or NADH.
- Term A21 The composition according to Item A19.
- a polypeptide according to item A1 or a polypeptide encoded by a polynucleotide according to item A2, (Aii) NADPH and / or NADH, and (Aiii) daidzein, and a dihydrodaidzein synthesis raw material composition object.
- Term A22 A dihydrodaidzein synthesis raw material composition comprising the cell according to any one of Items A4 to A6 and (Aiii) daidzein.
- Term A23 A dihydrodaidzein synthesis raw material composition comprising the cell according to any one of Items A4 to A6 and (Aiii) daidzein.
- a kit for dihydrodaidzein synthesis comprising (Ai) the polypeptide of item A1, or the polypeptide encoded by the polynucleotide of item A2, (Aii) NADPH and / or NADH, and (Aiii) daidzein.
- Term A24 (Aiv) A kit for synthesizing dihydrodaidzein, comprising the cell according to any one of items A4 to A6 and (Aiii) daidzein.
- Term A25 An immunological measurement kit for measuring the polypeptide of Item A1, or the polypeptide encoded by the polynucleotide of Item A2, comprising at least the antibody of Item A11.
- the identification kit according to Item A26 which is for identification of a polynucleotide containing the polynucleotide according to Item A1 or the polynucleotide according to Item A2.
- Term A28. The identification kit according to Item A27, which is a kit for PCR.
- Term A29. A dihydrodaidzein synthase comprising the polypeptide of Item A1.
- Term B2 A polynucleotide that is any of the following (Bd) to (Bf): (Bd) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 10; (Be) a polynucleotide encoding a polypeptide comprising the amino acid sequence of SEQ ID NO: 7 ; (Bf) encodes a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Bd) or (Be) under stringent conditions and has an activity of generating tetrahydrodaidzein using dihydrodaidzein as a substrate Polynucleotide.
- An expression vector comprising the polynucleotide according to Item B2.
- Term B4. A recombinant cell transformed with the expression vector according to Item B3.
- Term B5. The recombinant cell according to Item B4, wherein the recombinant cell is a bacterial prokaryotic cell.
- Term B6. The recombinant cell according to Item B5, wherein the bacterial prokaryotic cell belongs to the genus Lactococcus.
- Term B7 A method for producing a polypeptide, comprising culturing the cell according to any one of Items B4 to B6 to obtain a polypeptide having an activity of producing tetrahydrodaidzein using dihydrodaidzein as a substrate.
- Term B9 A method for producing tetrahydrodaidzein, comprising a step of allowing the polypeptide of Item B1 or B8 and NADPH and / or NADH to act on dihydrodaidzein.
- Term B10 A method for producing tetrahydrodaidzein, comprising a step of allowing the cell according to any one of Items B4 to B6 to act on dihydrodaidzein.
- Term B11 An antibody having binding ability to the polypeptide of Item B1 or the polypeptide encoded by the polynucleotide of Item B2.
- Term B15 A probe having a nucleotide sequence capable of hybridizing under stringent conditions to the polynucleotide encoding the polypeptide of Item B1 or the polynucleotide of Item B2.
- Term B16. A method for detecting or measuring a polynucleotide encoding the polypeptide of Item B1 or the polynucleotide of Item B2 using the probe of Item B14.
- Term B17 The method according to Item B16, wherein the polypeptide to be detected or measured is present in bacterial prokaryotic cells.
- Term B18 The method according to Item B16, wherein the polypeptide to be detected or measured is present in bacterial prokaryotic cells.
- the method according to Item B16 comprising a step of amplifying the polynucleotide encoding the polypeptide according to Item B1, the polynucleotide according to Item B2, or a part thereof by PCR.
- Term B19 A tetrahydrodaidzein synthase composition comprising the polypeptide of Item B1 or the polypeptide encoded by the polynucleotide of Item B2.
- Term B20 The composition according to Item B19, further comprising NADPH and / or NADH.
- Term B21 The composition according to Item B19.
- a tetrahydrodaidzein synthesis raw material comprising (Bi) the polypeptide according to item B1, or the polypeptide encoded by the polynucleotide of item B2, (Bii) NADPH and / or NADH, and (Biii) dihydrodaidzein Composition.
- Biv A tetrahydrodaidzein synthesis raw material composition comprising the cell according to any one of Items B4 to B6 and (Biii) dihydrodaidzein.
- Term B23 A tetrahydrodaidzein synthesis raw material comprising the cell according to any one of Items B4 to B6 and (Biii) dihydrodaidzein.
- a kit for tetrahydrodaidzein synthesis comprising (Bi) the polypeptide of item B1, or the polypeptide encoded by the polynucleotide of item B2, (Bii) NADPH and / or NADH, and (Biii) dihydrodaidzein.
- Term B24 (Biv) A tetrahydrodaidzein synthesis kit comprising the cell according to any one of Items B4 to B6 and (Biii) dihydrodaidzein.
- the identification kit according to Item B26 which is for identification of a polynucleotide containing the polynucleotide encoding the polypeptide according to Item B1 or the polynucleotide according to Item B2.
- the identification kit according to Item B27 which is a kit for PCR.
- Term B29. A tetrahydrodaidzein synthase comprising the polypeptide of Item B1.
- An expression vector comprising the polynucleotide according to Item C2.
- Term C4. A recombinant cell transformed with the expression vector according to Item C3.
- Term C5. The recombinant cell according to Item C4, wherein the recombinant cell is a bacterial prokaryotic cell.
- Term C6. The recombinant cell according to Item C5, wherein the bacterial prokaryotic cell belongs to the genus Lactococcus.
- Term C7 A method for producing a polypeptide, comprising culturing the cell according to any one of Items C4 to C6 to obtain a polypeptide having an activity of producing equol using tetrahydrodaidzein as a substrate.
- Term C9 A method for producing equol, comprising a step of allowing the polypeptide of Item C1 or C8 to act on tetrahydrodaidzein.
- Term C10 A method for producing equol, comprising a step of allowing the cell according to any one of Items C4 to C6 to act on tetrahydrodaidzein.
- Term C11 An antibody having binding ability to the polypeptide of Item C1 or the polypeptide encoded by the polynucleotide of Item C2.
- Term C15 A probe having a nucleotide sequence capable of hybridizing under stringent conditions to the polynucleotide encoding the polypeptide of Item C1, or the polynucleotide of Item C2.
- Term C16. A method for detecting or measuring a polynucleotide encoding the polypeptide of Item C1, or the polynucleotide of Item C2, using the probe of Item C14.
- Term C17 The method according to Item C16, wherein the polypeptide to be detected or measured is present in bacterial prokaryotic cells.
- Term C18 The method according to Item C16, wherein the polypeptide to be detected or measured is present in bacterial prokaryotic cells.
- the method according to Item C16 comprising a step of amplifying the polynucleotide encoding the polypeptide according to Item C1, the polynucleotide according to Item C2, or a part thereof by PCR.
- Term C19 An equol synthase composition comprising the polypeptide of Item C1 or the polypeptide encoded by the polynucleotide of Item C2.
- Term C20 An equol synthetic raw material composition comprising (Ci) the polypeptide of item C1, or the polypeptide encoded by the polynucleotide of item C2, and (Cii) tetrahydrodaidzein.
- Term C21 An equol synthetic raw material composition comprising (Ci) the polypeptide of item C1, or the polypeptide encoded by the polynucleotide of item C2, and (Cii) tetrahydrodaidzein.
- Term C25 A PCR kit for detecting a polynucleotide encoding the polypeptide of Item C1 or the polynucleotide of Item C2, comprising at least the primer of Item C15.
- Term C26 The identification kit according to Item C25, which is used for identification of a polynucleotide encoding the polypeptide according to Item C1 or a cell containing the polynucleotide according to Item C2.
- Term C27 A PCR kit for detecting a polynucleotide encoding the polypeptide of Item C1 or the polynucleotide of Item C2, comprising at least the primer of Item C15.
- the identification kit according to Item C26 which is a kit for PCR.
- Term C28 An equol synthase comprising the polypeptide of Item C1.
- Term D1 A method for producing tetrahydrodaidzein, comprising the following (first step) and (second step): (First Step) A step of producing dihydrodaidzein by causing daidzein to use an enzyme comprising a polypeptide that is any of the following (Aa) to (Ac), and NADPH and / or NADH; (Aa) a polypeptide comprising the amino acid sequence set forth in SEQ ID NO: 1; (Ab) a polypeptide comprising an amino acid sequence in which one or more amino acids are substituted, deleted, inserted and / or added in the amino acid sequence of SEQ ID NO: 1 and having an activity of synthesizing dihydrodaidzein using daidzein as a substrate ; (Ac) a polypeptide comprising an amino acid sequence having
- Term D2 A product containing tetrahydrodaidzein produced by the method according to Item D1.
- Term D3. A method for producing equol, including the following (second step) and (third step): (Second Step) A step of producing tetrahydrodaidzein by allowing dihydrodaidzein to act on an enzyme comprising a polypeptide of any one of the following (Ba) to (Bc), and NADPH and / or NADH; (Ba) a polypeptide comprising the amino acid sequence set forth in SEQ ID NO: 7; (Bb) a polypeptide comprising an amino acid sequence in which one or more amino acids are substituted, deleted, inserted and / or added in the amino acid sequence set forth in SEQ ID NO: 7 and having an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate peptide; (Bc) a polypeptide comprising an amino acid sequence having 60% or more identity
- Term D4 A product containing equol produced by the method according to Item D3.
- Term D5. A method for producing equol, comprising (first step) to (third step).
- Term D6. A product containing equol produced by the method according to Item D5.
- nucleotide (Bd) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 10; (Be) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 7; (Bf) encodes a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Bd) or (Be) under stringent conditions and has an activity of generating tetrahydrodaidzein using dihydrodaidzein as a substrate A polynucleotide; (Cd) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 16; (Ce) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 13; (Cf) a polypeptide encoding a polypeptide that hybridizes to a complementary strand of the polyn
- Term D8 A recombinant cell transformed with the expression vector according to Item D7.
- Term D9. The recombinant cell according to Item D8, wherein the recombinant cell is a bacterial prokaryotic cell.
- Term D10. The recombinant cell according to Item D9, wherein the bacterial prokaryotic cell belongs to the genus Lactococcus. Term D11.
- a method for producing dihydrodaidzein, tetrahydrodaidzein and / or equol comprising at least two steps of the following (fourth step) to (sixth step):
- (Fourth Step) A step of generating dihydrodaidzein by culturing a recombinant cell having a polynucleotide of any one of (Ad) to (Af) in a medium containing daidzein, (Ad) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 4; (Ae) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 1; (Af) a polypeptide encoding a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Ad) or (Ae) under stringent conditions and has an activity of producing dihydrodaidzein using
- (Fifth Step) A step of producing tetrahydrodaidzein by culturing a recombinant cell having a polynucleotide of any one of (Bd) to (Bf) in a medium containing dihydrodaidzein, (Bd) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 10; (Be) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 7; (Bf) encodes a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Bd) or (Be) under stringent conditions and has an activity of generating tetrahydrodaidzein using dihydrodaidzein as a substrate A polynucleotide; (Sixth Step) A step of generating equol by culturing a recombinant cell having a polynucle
- Term D12 The production method according to Item D11, wherein the recombinant cell is a bacterial prokaryotic cell.
- Term D13 The production method of Item D12, wherein the bacterial prokaryotic cell belongs to the genus Lactococcus.
- Term D14 A product containing dihydrodaidzein, tetrahydrodaidzein and / or equol, produced by the method according to any one of Items D11 to C13.
- An apparatus for producing dihydrodaidzein, tetrahydrodaidzein and / or equol comprising at least one reaction tank of the following (first reaction tank) to (third reaction tank): (First reaction tank) It has a reaction means in which an enzyme comprising a polypeptide of any one of (Aa) to (Ac) is immobilized, and dihydrodaidzein is converted from daidzein using the enzyme comprising the polypeptide.
- reaction vessel for production wherein the reaction means is arranged to be in contact with daidzein in the reaction vessel;
- (Second reaction tank) It has a reaction means in which an enzyme comprising a polypeptide of any one of (Ba) to (Bc) is immobilized, and dihydrodaidzein to tetrahydrodaidzein using the enzyme comprising the polypeptide Wherein the reaction means is arranged to contact the dihydrodaidzein in the reaction vessel;
- hird reaction tank It has a reaction means in which an enzyme comprising a polypeptide of any one of (Ca) to (Cc) is immobilized, and equol is converted from tetrahydrodaidzein using the enzyme comprising the polypeptide.
- a reaction vessel for production wherein the reaction means is arranged to be in contact with tetrahydrodaidzein in the reaction vessel;
- at least two of the reaction means may be present in one reaction tank.
- An apparatus for producing dihydrodaidzein, tetrahydrodaidzein and / or equol comprising at least one reaction tank of the following (fourth reaction tank) to (sixth reaction tank): (Fourth reaction tank) It has a reaction means to which a recombinant cell having a polynucleotide of any one of (Ad) to (Af) is fixed, and dihydrodaidzein is produced from daidzein using the reaction means
- (Fifth reaction tank) It has a reaction means to which a recombinant cell having a polynucleotide of any one of (Bd) to (Bf
- a reaction vessel for production wherein the reaction means is arranged to be in contact with dihydrodaidzein in the reaction vessel; (Sixth reaction tank) It has a reaction means to which a recombinant cell having a polynucleotide of any one of (Cd) to (Cf) is fixed, and equol is produced from tetrahydrodaidzein using the reaction means.
- at least two of the reaction means may be present together in one reaction vessel.
- dihydrodaidzein is synthesized using daidzein as a substrate
- tetrahydrodaidzein is synthesized using dihydrodaidzein as a substrate
- equol is synthesized using tetrahydrodaidzein as a substrate.
- the present invention is useful in realizing the industrial synthesis of dihydrodaidzein and / or tetrahydrodaidzein, which are intermediates that may appear in the production of equol from daidzein, and further the industrial synthesis of equol.
- dihydrodaidzein synthase (hereinafter also referred to as E1 enzyme) will be described in detail.
- E1 enzyme dihydrodaidzein synthase
- the general description regarding dihydrodaidzein synthase applies to tetrahydrodaidzein synthase and equol synthase described below, unless otherwise specified.
- polypeptides for synthesizing dihydrodaidzein using daidzein as a substrate (hereinafter referred to as "E1 polypeptide") Also provide: (Aa) a polypeptide having the amino acid sequence set forth in SEQ ID NO: 1, for example, 1 to 250, preferably 1 to 200, more preferably 1 to 150, more preferably 1 to 100, and more preferably 1 To 50, more preferably 1 to 30, more preferably 1 to 15, even more preferably 1 to 5, particularly preferably 1 to 4, more particularly preferably 1 to 3, and most preferably 1. Or two are mentioned.
- the range of “one or more” is not particularly limited as long as the polypeptide has an activity of synthesizing dihydrodaidzein using daidzein as a substrate.
- Specific examples of the polypeptide include a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 2 and a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 3. Compared to the amino acid sequence described in SEQ ID NO: 1, the amino acid sequence described in SEQ ID NO: 2 has 3 amino acid substitutions. Compared to the amino acid sequence described in SEQ ID NO: 1, the amino acid sequence described in SEQ ID NO: 3 has 10 amino acids substituted.
- amino acid sequence described in SEQ ID NO: 2 corresponds to the E1 enzyme derived from Bacteroides obatas E-23-15 strain (FERM BP-6435).
- amino acid sequence described in SEQ ID NO: 3 corresponds to the E1 enzyme derived from Streptococcus constellatus strain A6G-225 (FERM BP-6437).
- substitution of amino acids in the polypeptide (Ab) is not particularly limited, but substitution with similar amino acids is preferred from the viewpoint of not changing the phenotype of the polypeptide.
- similar amino acids can be grouped as follows: Aromatic amino acids: Phe, Trp, Tyr Aliphatic amino acids: Ala, Leu, Ile, Val Polar amino acids: Gln, Asn Basic amino acids: Lys, Arg, His Acidic amino acids: Glu, Asp Amino acids having a hydroxyl group: Ser, Thr Amino acids with small side chains: Gly, Ala, Ser, Thr, Met.
- amino acid substitutions, deletions, insertions or additions in the polypeptide (Ab) above do not significantly affect the higher order structure of the polypeptide or do not have an inhibitory effect on the active center as dihydrodaidzein synthase. It is desirable to be done in Examples of such a region include a region having low conservation between amino acid sequences described in SEQ ID NOs: 1, 2, and 3, and the vicinity thereof, or an N-terminal region or a C-terminal region.
- the “region in the vicinity” means, for example, within 5 amino acids, preferably within 4 amino acids before and after, based on the amino acid at the specific position, as long as it does not affect dihydrodaidzein synthase activity. More preferably, it means three amino acids before and after, particularly preferably two amino acids before and after, and particularly preferably one amino acid before and after.
- the 112th to 116th amino acid sequences are considered to correspond to the NADPH binding domain.
- amino acid substitutions, deletions, insertions or additions may be made in the 5 amino acid sequence.
- such mutations may be made to 3 amino acids or less, more preferably 2 amino acids or less, more preferably 1 amino acid, and most preferably the amino acid.
- the sequence is not mutated. In particular, it is preferable that no substitution, deletion, insertion or addition is made for the 112th, 115th and 116th amino acids.
- the 260th histidine in the amino acid sequence described in SEQ ID NO: 1 is also involved in the proton relay site. Therefore, as long as the function of the proton relay site is not inhibited, the histidine may be substituted with another amino acid, but is preferably not substituted.
- amino acid sequence shown in SEQ ID NO: 1 the sequence consisting of the 343th to 363rd amino acids is considered to correspond to the Fe—S cluster motif. Therefore, as long as the function of the motif is not inhibited, any amino acid may be substituted, deleted, inserted or added in the sequence, but the 343rd, 346th, 350th and 363rd cysteines Is preferably not mutated.
- an amino acid other than the three amino acids in the sequence is substituted with another amino acid, preferably 4 or less amino acids are substituted, more preferably 3 or less amino acids are substituted, and still more preferably 2 Less than one amino acid is substituted, particularly preferably one amino acid is substituted. Most preferably, no amino acid substitutions, deletions, insertions or additions are made in the sequence.
- amino acid sequence shown in SEQ ID NO: 1 the sequence consisting of the 390th to 413th amino acids is considered to correspond to the FAD binding domain. Therefore, as long as the function of the domain is not inhibited, any amino acid may be substituted, deleted, inserted or added in the sequence, but the 390th, 392rd, and 395th glycines are mutated. Preferably not.
- an amino acid is substituted, deleted, inserted or added in the sequence, it is preferably 4 amino acids or less, more preferably 3 amino acids or less, still more preferably 2 amino acids or less, particularly preferably 1 amino acid. It is preferably done for amino acids. Most preferably, no amino acid mutations are made in the sequence.
- amino acid sequence shown in SEQ ID NO: 1 the sequence consisting of the 512th to 540th amino acids is also considered to correspond to the FAD binding domain. Therefore, any amino acid may be substituted, deleted, inserted or added in the sequence as long as the function of the domain is not inhibited, but the 512th, 514th and 517th glycines are mutated. Preferably not.
- an amino acid is substituted, deleted, inserted or added in the sequence, it is preferably 4 amino acids or less, more preferably 3 amino acids or less, still more preferably 2 amino acids or less, particularly preferably 1 amino acid. It is preferably done for amino acids. Most preferably, no mutation is made in the amino acid sequence in the sequence.
- FIG. 26 shows an alignment showing amino acid sequence regions assumed to have the above functions.
- the amino acid identity may be 60% or more, for example, relative to the amino acid sequence shown in SEQ ID NO: 1, but is usually 80% or more, preferably 85% or more, more preferably Is 90% or more, more preferably 95% or more, particularly preferably 98% or more, and still more preferably 99% or more.
- polypeptide (Ac) examples include a polypeptide consisting of the amino acid sequence of SEQ ID NO: 2 and a polypeptide consisting of the amino acid sequence of SEQ ID NO: 3.
- the amino acid identity between the amino acid sequence of SEQ ID NO: 1 and the amino acid sequence of SEQ ID NO: 2 is 99.5%
- the amino acid identity between the amino acid sequence of SEQ ID NO: 1 and the amino acid sequence of SEQ ID NO: 3 is 98 .6% (Blast 2). Therefore, in a preferred embodiment of the present invention, the (Ac) polypeptide preferably has an identity of 98.6% or more, more preferably 99.5% or more to the amino acid sequence set forth in SEQ ID NO: 1.
- Amino acid sequence identity can be calculated using commercially available or analytical tools available through telecommunications lines (Internet), such as FASTA, BLAST, PSI-BLAST, SSEARCH, etc.
- polypeptides (Ab) and (Ac) above “activity for synthesizing dihydrodaidzein using daidzein as a substrate” can be confirmed as follows. That is, a polypeptide to be confirmed is added to a substrate solution having the following composition so as to be 0.001 mg / mL, incubated at 37 ° C. for 2 hours, and then the presence or absence of dihydrodaidzein is confirmed in the solution. When the presence of dihydrodaidzein is confirmed in the solution after incubation, the polypeptide is determined to have “activity for synthesizing dihydrodaidzein using daidzein as a substrate”.
- Substrate solution composition 0.1 M potassium phosphate buffer 1 mM PMSF (phenylmethylsulfonyl fluoride) 2 mM dithiothreitol 5 mM Sodium hydrosulfite 2 mM NADPH or NADH 40 ⁇ M daidzein pH 7.0
- E1 polypeptide has enzymatic activity to synthesize dihydrodaidzein using daidzein as a substrate.
- E1 polypeptide is also referred to as E1 enzyme.
- the enzyme activity of the E1 enzyme is activated by the presence of a reducing agent such as sodium hydrosulfite and metal ions such as manganese ions and iron ions.
- the E1 enzyme requires NADPH or NADH as a coenzyme, the optimum temperature is around 30 ° C., and the optimum pH is 7.0.
- the E1 enzyme can not only synthesize dihydrodaidzein using daidzein as a substrate, but can also synthesize daidzein using the reverse reaction, ie, dihydrodaidzein as a substrate.
- the E1 polypeptide can be produced by a genetic engineering technique described later, but can also be isolated and purified from a microorganism having the ability to produce E1 polypeptide.
- the E1 polypeptide can also be produced by a general chemical synthesis method according to the amino acid sequence information shown in SEQ ID NO: 1, 2, or 3.
- the chemical synthesis method includes a normal liquid phase method and a solid phase method. Phase synthesis peptide synthesis methods are included.
- the cells of a microorganism capable of producing E1 polypeptide are disrupted to obtain a crude extract of the microorganism.
- a method used in a normal microbial cell disruption process such as a treatment with a crusher such as a French press or a cell mill; an ultrasonic treatment under a hypotonic solution is used.
- the E1 polypeptide when the E1 polypeptide is acclimatized to anaerobic, it is desirable to add an appropriate reducing agent to the obtained crude extract to suppress the inactivation of the E1 polypeptide.
- the obtained crude extract may be further subjected to purification treatment such as ammonium sulfate precipitation, organic solvent precipitation using ethanol or the like, or isoelectric precipitation in order to increase the degree of purification.
- the crude extract is then subjected to treatment such as ion exchange chromatography, gel filtration chromatography, hydrophobic chromatography, various affinity chromatography, reverse phase chromatography, hydroxyapatite column chromatography, etc. A containing fraction can be obtained. Note that these chromatographic treatments may use an open column or HPLC as necessary.
- the purity of the fraction containing the E1 polypeptide thus obtained can be estimated easily and visually by electrophoresis, particularly by SDS-PAGE.
- E1 polypeptide should be confirmed by amino acid sequence analysis; mass spectrometry using mass spectrometer such as MALDI-TOF-MS, ESI Q-TOF MS or MALDI Q-TOF MS; peptide mass fingerprinting You can also.
- the microorganism having E1 polypeptide production ability can be efficiently cultured in a medium containing a predetermined amount of daidzein (though it is not particularly limited, for example, a medium containing 0.01 ⁇ g / mL or more). Is desirable in terms of producing
- the E1 polypeptide may exist as a monomer, but may exist as a dimer or higher multimer as long as it has the ability to synthesize dihydrodaidzein.
- the E1 polypeptide may be modified with polyethylene glycol or a sugar chain as necessary for the purpose of improving stability or the like.
- the E1 polypeptide can play a catalytic role to convert daidzein into dihydrodaidzein using the substrate.
- the dihydrodaidzein is converted into equol by tetrahydrodaidzein synthase (E2 enzyme) and equol synthase (E3 enzyme) described later. Equol is considered to exert various physiological actions in vivo, and in that sense, E1 polypeptide that can provide a raw material for synthesis of equol is important.
- polynucleotide encoding a polypeptide having an activity of synthesizing dihydrodaidzein using daidzein as a substrate (hereinafter, the polynucleotide may be referred to as “E1 polynucleotide”).
- polynucleotides (Ad) to (Af) are provided as E1 polynucleotides: (Ad) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 4; (Ae) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 1; (Af) a polypeptide encoding a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Ad) or (Ae) under stringent conditions and has an activity of producing dihydrodaidzein using daidzein as a substrate. nucleotide.
- amino acid sequence described in SEQ ID NO: 1 corresponds to the amino acid sequence encoded by the nucleotide sequence described in SEQ ID NO: 4.
- amino acid sequence described in SEQ ID NO: 2 corresponds to the amino acid sequence encoded by the nucleotide sequence described in SEQ ID NO: 5.
- amino acid sequence described in SEQ ID NO: 3 corresponds to the amino acid sequence encoded by the nucleotide sequence described in SEQ ID NO: 6.
- hybridizes under stringent conditions means that two polynucleotide fragments can hybridize with each other under standard hybridization conditions.
- stringent conditions means that hybridization is performed at about 45 ° C. in 6.0 ⁇ SSC and washed at 50 ° C. with 2.0 ⁇ SSC.
- Examples of the polynucleotide (Af) include the nucleotide sequence shown in SEQ ID NO: 5 and the nucleotide sequence shown in SEQ ID NO: 6.
- a polynucleotide that hybridizes under stringent conditions usually has a certain degree of homology with the nucleotide sequence of the polynucleotide used as a probe.
- the homology is, for example, 60% or more, preferably 70% or more, more preferably 80% or more, more preferably 90% or more, particularly preferably 95% or more, and still more preferably 98% or more.
- Nucleotide sequence homology can be calculated using commercially available or analytical tools available through telecommunications lines (Internet), for example, using software such as FASTA, BLAST, PSI-BLAST, SSEARCH, etc.
- the Specifically, the main initial conditions generally used for BLAST search are as follows. That is, in Advanced BLAST 2.1, blastn is used as a program, various parameters are set to default values, and a search is performed to calculate the nucleotide sequence homology value (%).
- nucleotide sequence homology between the nucleotide sequence of SEQ ID NO: 4 and the nucleotide sequence of SEQ ID NO: 5 is 99.6%
- nucleotide sequence homology between the nucleotide sequence of SEQ ID NO: 4 and the nucleotide sequence of SEQ ID NO: 6 is 97.6% (Blast 2). Therefore, in a preferred embodiment of the present invention, the polynucleotide of (Af) has a 97.6% or greater equivalent to the nucleotide sequence set forth in SEQ ID NO: 4, more preferably 99.6. % Equivalent or more is preferable.
- polypeptide (Ab) or (Ac) “activity for synthesizing dihydrodaidzein using daidzein as a substrate” is confirmed by the same method as in the case of the polypeptide (Ab) or (Ac).
- E1 polynucleotide can be produced and obtained by chemical DNA synthesis based on the sequence information of SEQ ID NO: 4, 5, or 6, but can be easily produced and obtained by general genetic engineering techniques. [Molecular Cloning 2d Ed, Cold Spring Harbor Lab. Press1989 (1989); secondary biochemistry experiment course "Gene Research Methods I, II, III", Japanese Biochemical Society (1986) etc.]
- Examples of the chemical DNA synthesis method include a solid phase synthesis method using a phosphoramidite method.
- An automatic synthesizer can be used for this synthesis method.
- a cDNA library is prepared according to a conventional method from an appropriate source from which the E1 polynucleotide is expressed, and an appropriate property specific to the E1 polynucleotide is prepared from the library. It can be carried out by selecting desired clones using various probes and antibodies [Proc. Natl. Acad. Sci., USA., 78, 6613 (1981); Science122, 778 (1983), etc.].
- the origin of cDNA is not particularly limited as long as it is an organism that expresses E1 polynucleotide.
- microorganisms having equol production ability preferably lactic acid bacteria having equol production ability, bacteria belonging to the genus Bacteroides A bacterium belonging to the genus Streptococcus, more preferably Lactococcus garvier having the ability to produce equol, Bacteroides obatas, and Streptococcus constellatus, particularly preferably a stool-derived lactococcus garvier having equol-producing ability, particularly equol-producing ability.
- Lactococcus 20-92 strain (FERM BP-10036; deposited at the Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology), Bacteroides obatas E-23-15 ( FERM BP-6435; National Institute of Advanced Industrial Science and Technology (deposited at Patent Biological Depositary Center), Streptococcus constellatus A6G-225 (FERM BP-6437; National Institute of Advanced Industrial Science and Technology, Biological Depositary Center (East Tsukuba City, Ibaraki, Japan) 1-chome No. 1-1)) is exemplified.
- the method for screening the polynucleotide of the present invention from a cDNA library is not particularly limited, and can be performed according to a usual method. Specifically, for example, for a polypeptide produced by cDNA, a method for selecting a corresponding cDNA clone by immunoscreening using the polypeptide-specific antibody, a probe that selectively binds to a target nucleotide sequence Examples thereof include plaque hybridization, colony hybridization, etc., and combinations thereof.
- Examples of the probe used here generally include DNA chemically synthesized based on information on the nucleotide sequence of the E1 polynucleotide (for example, the nucleotide sequence of SEQ ID NO: 4, 5, or 6).
- a sense primer and / or an antisense primer set based on the nucleotide sequence information of the polynucleotide of the present invention can also be used as a screening probe.
- a PCR method [Science 130, 1350 (1985)] or a modified method of DNA or RNA can be preferably used.
- the RACE method [Rapid amplification of cDNA ends; experimental medicine, 12 (6), 35 (1994)]
- the 5′-RACE method [MA Frohman, et al., Proc. Natl. Acad. Sci., USA., 8, 8998 (1988)] and the like are suitable.
- RACE method and 5'-RACE method are useful in obtaining E1 polynucleotides derived from eukaryotes.
- Primers used in adopting such a PCR method can be appropriately set based on the sequence information of the E1 polynucleotide, and can be synthesized according to a conventional method.
- isolation and purification of the amplified DNA or RNA fragment can be carried out according to a conventional method as described above, for example, by gel electrophoresis, hybridization or the like.
- the product of the polynucleotide (that is, the above-mentioned polypeptide) can be easily and stably produced in large quantities by using a normal genetic engineering technique.
- a normal genetic engineering technique Conventionally, only anaerobic strains that are difficult to handle for equol-producing bacteria have been found, but due to the successful isolation of E1 polynucleotides, industrial production of equol is possible without using conventional equol-producing bacteria. The way to is opened.
- the expression vector of the present invention is not particularly limited as long as it contains an E1 polynucleotide and can express the E1 polynucleotide, and is generally appropriately selected from the relationship with the host cell.
- the expression vector is, for example, a plasmid vector that can replicate in the host cell, and upstream of the polynucleotide so that the polynucleotide can be expressed in the vector.
- An expression plasmid to which a promoter and an SD (Shine and Dalgarno) base sequence are added can be mentioned. Specifically, mention may be made of P L promoter, the expression plasmid using T7 promoter and the lac promoter.
- Other preferable bacterial expression vectors include plasmids pKK233-2 and pKK233-3 using the tac promoter or trc promoter. However, it is not limited to these, and various known strains and vectors can also be used.
- the expression vector When eukaryotic cells are used as host cells, the expression vector usually has a promoter located upstream of the polynucleotide to be expressed, an RNA splice site, a polyadenylation site, a transcription termination sequence, etc. Which may further have an origin of replication if necessary.
- Eukaryotic vectors useful for inserting the above polynucleotides are well known.
- suitable eukaryotic vectors include pCD and pCMV.
- pMSG and pSVL using MMTV or SV40 late promoter can be mentioned as necessary.
- various known eukaryotes and vectors can also be used.
- Recombinant cell The present invention provides a recombinant cell (transformant) transformed with an expression vector containing the E1 polynucleotide.
- Either a prokaryotic cell or a eukaryotic cell may be used as the host cell used for the recombinant cell.
- bacterial prokaryotic cells such as lactic acid bacteria belonging to the genus Lactococcus can be preferably used.
- bacterial prokaryotic cells that can grow under aerobic conditions for example, E. coli , Streptomyces, Bacillus subtilis, Streptococcus, Staphylococcus, etc.).
- eukaryotic cells used as host cells include eukaryotic microorganisms such as yeast and Aspergillus; insect cells such as Drosophila S2 and Spodoptera Sf9; L cells, CHO cells, COS cells, HeLa cells, C127 cells, BALB / Examples include c3T3 cells (including mutants lacking dihydrofolate reductase and thymidine kinase), BHK21 cells, HEK293 cells, Bowes melanoma cells, and animal and plant cells such as oocytes.
- yeast and Aspergillus insect cells such as Drosophila S2 and Spodoptera Sf9
- L cells CHO cells, COS cells, HeLa cells, C127 cells, BALB / Examples include c3T3 cells (including mutants lacking dihydrofolate reductase and thymidine kinase), BHK21 cells, HEK293 cells, Bowes melanoma cells, and animal and plant
- the method for introducing the expression vector into the host cell is not particularly limited, and various general methods can be employed.
- introduction of the above expression vector into a host cell includes Davis et al. (BASIC METHODS IN MOLECULAR BIOLOGY, 1986) and Sambrook et al. (MOLECULAR CLONING: A LABORORY MANUAL, 2nd Ed.rC. Y., 1989), etc., and can be performed according to methods described in many standard laboratory manuals, including calcium phosphate transfection, DEAE-dextran-mediated transfection, and transfection. , Microinjection, cation Lipid-mediated transfection, electroporation, transduction, (scrape loading), bullet introduced (ballistic introduction), infections and the like.
- the recombinant cell can produce E1 polypeptide which is a dihydrodaidzein converting enzyme, it can be used for the production of dihydrodaidzein converting enzyme, or can be used for the production of dihydrodaidzein in the state of cells. .
- An E1 polypeptide can be produced by culturing a recombinant cell into which an E1 polynucleotide has been introduced and recovering the E1 polypeptide from the cell and / or culture.
- Culture may be performed by subculture or batch culture using a medium suitable for the host. The culture may be performed until an appropriate amount of E1 polypeptide is obtained using the amount of E1 polypeptide produced inside or outside the recombinant cell as an index.
- various media commonly used according to the employed host cells can be appropriately selected and used, and the culture can also be performed under conditions suitable for the growth of the host cells.
- the E1 polypeptide thus obtained may be subjected to various separation operations utilizing its physical properties, chemical properties, etc. ["Biochemical Data Book II", pages 1175-1259, first edition, first, as desired. Jun. 23, 1980, published by Tokyo Chemical Co., Ltd .; see Biochemistry, 25 (25), 8274 (1986); Eur. J. Biochem., 163, 313 (1987), etc.]. Specifically, a method similar to the “method for isolating and purifying E1 peptide from a microorganism having E1 polypeptide producing ability” described in the above section “A-1. Polypeptide” is exemplified.
- the present invention provides a method for producing dihydrodaidzein using E1 polypeptide. That is, in the production method, daidzein is converted to dihydrodaidzein by allowing E1 polypeptide to act on daidzein in the presence of NADPH and / or NADH. Preferably, daidzein is converted to dihydrodaidzein by allowing E1 polypeptide to act on daidzein in the presence of NADPH.
- the concentration of Mn 2+ and / or Fe 2+ in the reaction solution is not particularly limited as long as the enzyme activity of the E1 polypeptide can be activated.
- the concentration of Fe 2+ is preferably 2 mM or more, more preferably 2 mM to 100 mM, and further preferably 10 mM to 40 mM.
- the concentration of Mn 2+ is preferably 0.2 ⁇ M, more preferably 0.2 ⁇ M to 100 mM, and still more preferably 1.0 ⁇ M to 40 mM.
- the reaction employed in this production method can be performed in an appropriate buffer solution.
- the buffer solution include a phosphate buffer solution, a carbonate buffer solution, an acetate buffer solution, a Tris buffer solution, and a borate buffer solution.
- the pH condition in the reaction may be set appropriately as long as the desired enzyme activity of the E1 polypeptide is not inactivated, but preferably pH 5.0 to 10.0, more preferably 6.0 to 8.0. be able to.
- an appropriate amount of a protease inhibitor such as PMSF or EDTA may be added to the reaction as necessary.
- an appropriate amount of a reducing agent such as DTT, 2ME, DET, or sodium hydrosulfite may be added.
- each component is added in a solvent so as to satisfy the following concentration range at the start of the reaction to prepare a raw material mixture, which is 20 to 45 ° C., preferably 25 to 40 ° C. It is carried out by incubating at a temperature condition of 0 ° C., more preferably 30-38 ° C., for 0.5-10 hours, preferably 1-6 hours, more preferably 2-4 hours: 0.0001-1.0% by weight of the polypeptide, preferably 0.001-0.1% by weight, more preferably 0.001-0.01% by weight; Daidzein is 0.0001-10.0 wt%, preferably 0.001-1.0 wt%, more preferably 0.001-0.1 wt%; and NADPH and / or NADH is 0.01 to 5% by weight, preferably 0.05 to 1% by weight, more preferably 0.1 to 0.5% by weight.
- the present invention also provides a dihydrodaidzein synthesis raw material composition containing (Ai) E1 polypeptide, (Aii) NADPH and / or NADH, and (Aiii) daidzein as a mixed raw material for synthesizing dihydrodaidzein. . Furthermore, the present invention contains (Ai) E1 polypeptide, (Aii) NADPH and / or NADH, (Aiii) daidzein, and (Aiv) Mn 2+ and / or Fe 2+ as a mixed raw material for synthesizing dihydrodaidzein. A dihydrodaidzein synthesis raw material composition is provided.
- the synthetic raw material composition corresponds to the raw material mixture at the start of the reaction in the production of the above-mentioned dihydrodaidzein, the compounding concentration of E1 polypeptide, the compounding concentration of NADPH and / or NADH in the synthetic raw material composition, daidzein
- the blending concentration and other components that can be blended in the synthetic raw material composition are also the same as in the case of the reaction system (raw material mixed solution at the start of the reaction) employed in the production method.
- the present invention provides a kit for synthesizing dihydrodaidzein containing (Ai) E1 polypeptide, (Aii) NADPH and / or NADH, and (Aiii) daidzein as a kit for synthesizing dihydrodaidzein.
- the present invention provides a kit for synthesizing dihydrodaidzein as a dihydro containing (Ai) E1 polypeptide, (Aii) NADPH and / or NADH, (Aiii) daidzein, and (Aiv) Mn 2+ and / or Fe 2+.
- a kit for synthesizing daidzein is provided.
- the synthesis kit only needs to be provided with each component divided as necessary so that synthesis of dihydrodaidzein from daidzein can be easily performed under the conditions described above.
- the synthesis kit may contain a buffer to be used, if necessary.
- the synthesis kit may include necessary instruments and operation manuals in order to easily synthesize dihydrodaidzein.
- Dihydrodaidzein synthase composition The present invention further provides a dihydrodaidzein synthase composition comprising an E1 polypeptide.
- the enzyme composition is suitably used as a dihydrodaidzein synthase in a method for producing dihydrodaidzein using an E1 polypeptide.
- the enzyme composition may be a crude E1 polypeptide, or a mixture of a crude or purified E1 polypeptide in a suitable carrier.
- the proportion of the E1 polypeptide is not particularly limited as long as it can be used as a dihydrodaidzein synthase in the dihydrodaidzein production method.
- the E1 polypeptide is 0.001 to 20.0% by weight, preferably 0.005 to 5.0% by weight, more preferably 0.01 to 1.0% by weight, based on the total amount of the enzyme composition.
- the enzyme composition may contain NADPH and / or NADH which acts as a coenzyme for E1 polypeptide.
- NADPH and / or NADH acts as a coenzyme for E1 polypeptide.
- the blending ratio of NADPH and / or NADH is not particularly limited, but is 0.0005 to 25.0 wt%, preferably 0.005 to 5.0 wt% per the enzyme composition. %, More preferably 0.01 to 2.5% by weight.
- the enzyme composition comprises Mn 2+ and / or Fe 2+ .
- the compounding ratio of the metal ions is not particularly limited as long as the dihydrodaidzein synthase activity of the E1 polypeptide can be activated.
- the blending ratio of Fe 2+ is preferably 2 mM or more, more preferably 2 mM to 100 mM, still more preferably 10 mM to 40 mM per composition.
- the mixing ratio of Mn 2+ is preferably 0.2 ⁇ M or more, more preferably 0.2 ⁇ M to 100 mM, and still more preferably 1.0 ⁇ M to 40 mM per composition.
- the enzyme composition may contain an antioxidant such as sulfite, ascorbic acid, ⁇ -tocopherol, cysteine, etc., in order to improve the stability of the polypeptide.
- an antioxidant such as sulfite, ascorbic acid, ⁇ -tocopherol, cysteine, etc.
- a preservative such as paraoxybenzoates, chlorobutanol, benzyl alcohol, 2-phenylethyl alcohol, dehydroacetic acid, sorbic acid or the like is added as necessary. May be.
- A-8. Method for Producing Dihydrodaidzein Using the Recombinant Cell The present invention provides a method for producing dihydrodaidzein using a recombinant cell into which an E1 polynucleotide has been introduced. That is, in the production method, daidzein is converted to dihydrodaidzein by allowing the recombinant cell to act on daidzein.
- the reaction employed in this production method is carried out in an environment where the above recombinant cells can survive and daidzein can be converted to dihydrodaidzein.
- the medium used is appropriately selected from various commonly used media depending on the type of cell employed as the host cell for the recombinant cell.
- an appropriate amount of a protease inhibitor such as PMSF or EDTA may be added to the medium as necessary.
- an appropriate amount of a reducing agent such as DTT, 2ME, DET, or sodium hydrosulfite may be added.
- NADPH and / or NADH is not essential, but NADPH and / or NADH may be added to the medium as necessary.
- Mn 2+ and / or Fe 2+ may be added to the medium.
- the above recombinant cells are inoculated into a medium containing 0.001 to 1% by weight, preferably 0.01 to 1% by weight, more preferably 0.01 to 0.5% by weight of daidzein, and the growth temperature conditions are satisfied. It is carried out by incubating under 6 to 30 hours, preferably 7 to 24 hours, more preferably 7 to 18 hours.
- the present invention also provides a dihydrodaidzein synthesis raw material composition containing (Aiv) the above recombinant cells and (Aiii) daidzein as a mixed raw material for synthesizing dihydrodaidzein.
- a dihydrodaidzein synthesis raw material composition containing (Aiv) the above recombinant cells and (Aiii) daidzein as a mixed raw material for synthesizing dihydrodaidzein.
- daidzein in the synthetic raw material composition can be converted to dihydrodaidzein.
- the synthetic raw material composition corresponds to the raw material mixture at the start of the reaction.
- the recombinant raw material and the concentration of daidzein in the synthetic raw material composition, and the synthetic raw material composition Other possible components and the like are the same as the conditions and the like adopted in the above production method.
- the present invention provides a kit for synthesizing dihydrodaidzein containing (Aiv) the above recombinant cell and (Aiii) daidzein as a kit for synthesizing dihydrodaidzein.
- the present invention also provides a kit for synthesizing dihydrodaidzein comprising (Aiv) the above recombinant cell, (Aiii) daidzein, and (Aiv) Mn 2+ and / or Fe 2+ as a kit for synthesizing dihydrodaidzein. .
- the recombinant cells and daidzein may be separated as necessary so that dihydrodaidzein can be easily synthesized from daidzein under the conditions described above.
- the synthesis kit may contain a buffer or a medium as necessary.
- the synthesis kit may include necessary instruments and operation manuals in order to easily synthesize dihydrodaidzein.
- the recombinant cells contained in the synthesis kit may be those stored by a known method.
- Techniques for storing recombinant cells are known, and examples include a method of storing recombinant cells in dimethylformamide or the like, treating the ampoule in a vacuum state with a freeze dryer, and storing at 4 to 25 ° C. it can.
- a liquid nitrogen method in which cells are suspended in a 10% glycerol-preserved medium, stored in a dedicated ampoule, and stored in a liquid nitrogen tank ( ⁇ 150 to ⁇ 196 ° C.) can be mentioned.
- the present invention further provides an antibody (IgG antibody) having binding property to E1 polypeptide.
- Monoclonal antibodies are prepared according to conventional methods. Specifically, Harlow and Lane (Harlow, H. and Lane, D.), “Antibodies: Laboratory Manual”, Cold Spring Harbor Lab (Cold Spring Harbor Lab), New York, pages 139-240 (1988). It can be made according to the described method.
- Polyclonal antibodies are also prepared according to conventional methods. Specifically, it can be prepared according to the method described in Cell Engineering Experiment Protocol (University of Tokyo Institute of Medical Science, 1992, pp. 155-173).
- Both polyclonal IgG antibodies and monoclonal IgG antibodies can be purified by the usual ammonium sulfate precipitation method, protein A chromatography, or the like.
- the present invention provides an immunological method for detecting or measuring the E1 polypeptide using the antibody.
- the immunological method is performed by bringing the antibody into contact with a test sample. That is, by bringing the antibody into contact with the test sample, when the E1 polypeptide is present in the test sample, the antibody and the E1 polypeptide are specifically bound. Subsequently, the said antibody couple
- the test sample is a sample to be detected or measured for E1 polypeptide. Since E1 polypeptide is expected to be found in bacterial prokaryotic cells, the immunological method is suitable for the purpose of detecting or measuring E1 polypeptide present in bacterial prokaryotic cells. In addition, when detecting or measuring E1 polypeptide present in cells, a test sample obtained by subjecting the cells to disruption or purifying proteins after the disruption is used. Use it.
- a technique for detecting or measuring a target polypeptide by an immunological method using an antibody is known, and those skilled in the art can appropriately set various conditions in the immunological method. For example, a radioimmunoassay method, an ELISA method or the like may be adopted and appropriate conditions may be set as appropriate.
- the present invention provides an immunological detection kit containing the antibody as a kit for detecting or measuring E1 polypeptide.
- the detection kit may contain an E1 polypeptide as a standard if necessary.
- the detection kit may be provided with other reagents and the like as required so that the E1 polypeptide can be easily detected under the conditions described above.
- the detection kit may contain necessary instruments and operation manuals in order to easily detect the E1 polypeptide.
- the present invention also provides a method for detecting or measuring an E1 polynucleotide. Specifically, the method is performed by contacting a probe that binds to an E1 polynucleotide with a test sample. That is, by contacting the probe with the test sample, when the E1 polynucleotide is present in the test sample, the probe and the E1 polynucleotide are hybridized. Subsequently, the presence or absence of the formation of this double strand is detected, and this is quantified as necessary, whereby the E1 polynucleotide in the test sample can be detected or measured.
- the test sample is a sample to be detected or measured for E1 polynucleotide. Since E1 polynucleotides are expected to be found in bacterial prokaryotic cells, the method is suitable for the purpose of detecting or measuring E1 polynucleotides present in bacterial prokaryotic cells.
- a test sample obtained by subjecting the cell to disruption or purifying nucleic acid after the disruption is used. Use it.
- the probe used in the method a probe having a nucleotide sequence that can hybridize to the polynucleotide under stringent conditions is used.
- the stringent conditions include normal conditions used as a probe or primer. Specifically, the above-mentioned A-2. The conditions shown in the section are illustrated.
- the probe may be chemically synthesized based on information on the nucleotide sequence of the E1 polynucleotide (for example, the nucleotide sequence set forth in SEQ ID NO: 4, 5, or 6). It may consist of nucleotides or fragments thereof.
- the probe is usually a labeled probe, but may be unlabeled.
- the length is preferably about 10 to 40 nucleotides, particularly about 20 to 30 nucleotides.
- Examples of the method for specifically detecting the polynucleotide using the probe include plaque hybridization, colony hybridization, Southern blotting, Northern blotting, and PCR. From this point, a method of amplifying the E1 polynucleotide or a part thereof by a PCR method using the probe as a primer is suitably employed.
- the RT-PCR method for example, the RT-PCR method is exemplified, but various modified methods used in this field can be applied. It is also possible to quantify the presence and amount of E1 polynucleotide using PCR. Such methods include competitive quantification methods such as MSSA (Kinoshita, M., et al., CCA, 228, 83-90 (1994)), or mobility associated with changes in higher-order structure of single-stranded DNA.
- MSSA Kininoshita, M., et al., CCA, 228, 83-90 (1994)
- mobility associated with changes in higher-order structure of single-stranded DNA for example, the PCR-SSCP method, which is known as a mutation detection method using the change in the above, can be exemplified.
- the present invention provides an E1 polynucleotide detection kit containing the probe as a kit for detecting or measuring E1 polynucleotide.
- the detection kit may be provided with reagents and the like used in addition to the probe as required so that the E1 polynucleotide can be easily detected under the conditions described above.
- the detection kit can also be used as a kit for identifying cells containing the E1 polynucleotide.
- the detection kit is preferably a kit for performing detection by PCR from the viewpoint of enabling highly accurate detection.
- B Tetradaidzein synthase B-1.
- Polypeptides The present invention relates to the following polypeptides (Ba) to (Bc) (hereinafter referred to as “E2 polypeptides”) as polypeptides that synthesize tetrahydrodaidzein using dihydrodaidzein as a substrate.
- the range of “one or more” is not particularly limited as long as the polypeptide has an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate, but for example 1 to 50, Preferably 1 to 30, more preferably 1 to 15, more preferably 1 to 5, still more preferably 1 to 4, particularly preferably 1 to 3, and still more preferably 1 or 2 are mentioned. It is done.
- polypeptide (Bb) examples include a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 8 and a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 9.
- the amino acid sequence described in SEQ ID NO: 8 has two amino acid sequences substituted, and an amino acid sequence consisting of 24 amino acids is added to the N-terminus.
- the amino acid sequence described in SEQ ID NO: 9 and 20 amino acids are substituted, and one amino acid is deleted.
- amino acid sequence described in SEQ ID NO: 8 corresponds to the E2 enzyme derived from Bacteroides obatas E-23-15 strain (FERM BP-6435).
- amino acid sequence described in SEQ ID NO: 9 corresponds to the E2 enzyme derived from Streptococcus constellatus A6G-225 strain (FERM BP-6437).
- the amino acid substitution, deletion, insertion or addition in the above polypeptide (Bb) can be performed according to the substitution, deletion, insertion or addition in the E1 polypeptide described in the section A-1.
- the amino acid substitution, deletion, insertion or addition in the polypeptide (Bb) is performed in a region that does not significantly affect the higher order structure of the polypeptide or a region that does not have an inhibitory effect on the active center as a tetrahydrodaidzein synthase. It is desirable. Examples of such a region include a region having low conservability between the amino acid sequences described in SEQ ID NOs: 7, 8, and 9 and the vicinity thereof, or an N-terminal region or a C-terminal region.
- the seventh valine, the eighth proline, the 26th valine, the 36th leucine, the 46th arginine, the 94th Aspartic acid 101st glutamic acid, 126th glycine, 137th isoleucine, 156th glutamine, 157th lysine, 159th aspartic acid, 160th, 171st alanine, 1st 185th cysteine, 221st serine, 233rd alanine, 241st valine, 258th serine, 266th isoleucine, 286th valine, and the vicinity of these amino acids An area can be mentioned.
- the “near region” means within a range that does not affect tetrahydrodaidzein synthase activity, based on the amino acid at the specific position, and within 5 amino acids, preferably within 4 amino acids. More preferably, the amino acid is within 3 amino acids before and after, more preferably within 2 amino acids before and after, and particularly preferably 1 amino acid before and after.
- the sequence consisting of the 38th to 45th amino acids is considered to correspond to the NADPH binding domain. Therefore, as long as the function of the domain is not inhibited, any amino acid may be substituted, deleted, inserted or added in the sequence.
- the 38th threonine, 39th glycine, The 43rd glycine and the 45th glycine are preferably not mutated.
- an amino acid is substituted, deleted, inserted or added in the sequence, it is preferably 4 amino acids or less, more preferably 3 amino acids or less, still more preferably 2 amino acids or less, particularly preferably 1 amino acid. It is preferably done for amino acids.
- amino acid sequence described in SEQ ID NO: 7 the sequence consisting of the 115th to 118th amino acids is considered to correspond to a motif highly conserved in the SDR family.
- amino acid substitutions, deletions, insertions or additions are made in the sequence, it is preferably 3 amino acids or less, more preferably 2 amino acids or less, and even more preferably 1 amino acid. Most preferably, no amino acid mutations are made in the sequence.
- the sequence consisting of the 212th to 217th amino acids is considered to be involved in the binding to the cofactor. Therefore, any amino acid may be substituted, deleted, inserted or added in the sequence as long as the function is not inhibited.
- the 212th proline, the 213th glycine, and the 217th position are used.
- the threonine is not mutated.
- amino acids are substituted, deleted, inserted or added in the sequence, it is preferably 3 amino acids or less, more preferably 2 amino acids or less, and even more preferably 1 amino acid. Particularly preferably, no amino acid mutation is made in the sequence.
- FIG. 27 shows an alignment showing amino acid sequence regions assumed to have the above functions.
- the amino acid identity may be, for example, 60% or more with respect to the amino acid sequence shown in SEQ ID NO: 7, but usually 80% or more, preferably 85% or more, more preferably Is 90% or more, more preferably 95% or more, particularly preferably 98% or more, and still more preferably 99% or more.
- polypeptide (Bc) examples include a polypeptide consisting of the amino acid sequence shown in SEQ ID NO: 8 and a polypeptide consisting of the amino acid sequence shown in SEQ ID NO: 9.
- the identity between the amino acid sequence shown in SEQ ID NO: 7 and the amino acid sequence shown in SEQ ID NO: 8 is 99.3%, and the amino acid sequence shown in SEQ ID NO: 7 is identical to the amino acid sequence shown in SEQ ID NO: 9.
- the sex is 93.0% (Blast 2).
- the (Bc) polypeptide preferably has an identity of 93.0% or more, more preferably 99.3% or more, with respect to the amino acid sequence set forth in SEQ ID NO: 7.
- polypeptides (Bb) and (Bc) above “activity for synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate” can be confirmed as follows. That is, a polypeptide to be confirmed is added to a substrate solution having the following composition so as to be 0.001 mg / mL, incubated at 37 ° C. for 2 hours, and then the presence or absence of tetrahydrodaidzein is confirmed in the solution. When the presence of tetrahydrodaidzein is confirmed in the solution after incubation, the polypeptide is determined to have “activity for synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate”.
- E2 polypeptide has enzymatic activity to synthesize tetrahydrodaidzein using dihydrodaidzein as a substrate.
- E2 polypeptide is also referred to as E2 enzyme.
- the E2 enzyme requires NADPH or NADH as a coenzyme.
- the optimum temperature for the E2 enzyme is around 37 ° C. and the optimum pH is 4.5.
- the E2 enzyme can not only synthesize tetrahydrodaidzein using dihydrodaidzein as a substrate, but also reverse reaction, that is, synthesize dihydrodaidzein using tetrahydrodaidzein as a substrate.
- the E2 polypeptide can be obtained by genetic engineering techniques using the nucleotide sequence information set forth in SEQ ID NO: 10, 11, or 12, as with the E1 peptide. It can also be obtained by chemical synthesis based on the amino acid sequence information set forth in SEQ ID NO: 7, 8, or 9. It can also be obtained by isolation and purification from a microorganism having E2 polypeptide-producing ability. These methods can be performed according to the description in the section A-1.
- the microorganism having the ability to produce E2 polypeptide may be cultured in a medium containing a desired amount of dihydrodaidzein and further daidzein. Even in this case, the E2 polypeptide can be produced from a microorganism having the ability to produce the E2 polypeptide.
- the E2 polypeptide may exist as a monomer, but may exist as a dimer or higher multimer as long as it has the ability to synthesize tetrahydrodaidzein.
- the E2 polypeptide may be modified with polyethylene glycol or a sugar chain as necessary for the purpose of improving stability or the like.
- the E2 polypeptide can play a catalytic role in converting dihydrodaidzein into tetrahydrodaidzein using dihydrodaidzein as a substrate.
- the tetrahydrodaidzein is further converted into equol by equol synthase described later. Equol is considered to exert various physiological actions in vivo, and in that sense, it can be said that an E2 polypeptide capable of providing a raw material for synthesis of equol is important.
- the present invention provides a tetrahydrodaidzein synthase containing the polypeptides (Ba) to (Bc).
- polynucleotide encoding a polypeptide having an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate (hereinafter, the polynucleotide may be referred to as "E2 polynucleotide").
- polynucleotides (Bd) to (Bf) are provided as E2 polynucleotides: (Bd) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 10; (Be) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 7; (Bf) encodes a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Bd) or (Be) under stringent conditions and has an activity of generating tetrahydrodaidzein using dihydrodaidzein as a substrate Polynucleotide.
- amino sequence described in SEQ ID NO: 7 corresponds to the amino acid sequence encoded by the nucleotide sequence described in SEQ ID NO: 10.
- amino acid sequence of SEQ ID NO: 8 corresponds to the amino acid sequence encoded by the nucleotide sequence set forth in SEQ ID NO: 11.
- amino acid sequence set forth in SEQ ID NO: 9 corresponds to the amino acid sequence encoded by the nucleotide sequence set forth in SEQ ID NO: 12.
- hybridizes under stringent conditions with respect to the polynucleotide (Bf) above is synonymous with “hybridizes under stringent conditions” described in A-2.
- Specific examples of the polynucleotide (Bf) include the nucleotide sequence set forth in SEQ ID NO: 11 and the nucleotide sequence set forth in SEQ ID NO: 12.
- the nucleotide sequence homology between the nucleotide sequence set forth in SEQ ID NO: 10 and the nucleotide sequence set forth in SEQ ID NO: 11 is 99.7%.
- the nucleotide sequence set forth in SEQ ID NO: 10 and the nucleotide sequence set forth in SEQ ID NO: 12 Is 91.0% (Blast 2).
- the polynucleotide of (Bf) has a 91.0% or greater equivalent to the nucleotide sequence set forth in SEQ ID NO: 10, more preferably 99.7. % Equivalent.
- polypeptide (Bf) “activity for synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate” is confirmed by the same method as in the case of the polypeptide (Bb) or (Bc).
- the E2 polynucleotide can be produced and obtained by a chemical synthesis method or a genetic engineering method based on the sequence information described in SEQ ID NO: 10, 11, or 12. As a specific method, the method described in section A-2. Regarding the E1 polynucleotide can be used. Such a method can be modified and changed as necessary.
- the origin of cDNA of E2 polynucleotide is not particularly limited as long as it is a microorganism that expresses E2 polynucleotide. Specifically, it belongs to the genus Bacteroides, a microorganism having equol-producing ability, preferably a lactic acid bacterium having equol-producing ability.
- Bacteria and bacteria belonging to the genus Streptococcus more preferably Lactococcus garbie, Bacteroides obatas having equol-producing ability, and Streptococcus constellatus, particularly preferably stool-derived Lactococcus garbie, particularly equol-producing ability Lactococcus 20-92 strain (FERM BP-10036; deposited at the Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology), Bacteroides obatas E-23-15 strain FERM BP-6435; National Institute of Advanced Industrial Science and Technology, Deposited at Patent Biological Depositary Center), Streptococcus constitutus A6G-225 (FERM BP-6437; National Institute of Advanced Industrial Science and Technology, Patent Biological Depository Center ( 1) 1-1, East, Tsukuba City, Ibaraki Prefecture, Japan)).
- a product of the polynucleotide ie, the above polypeptide
- a product of the polynucleotide ie, the above polypeptide
- the expression vector of the present invention is not particularly limited as long as it contains an E2 polynucleotide and can express the E2 polynucleotide. In general, similar to an expression vector containing an E1 polynucleotide, It is appropriately selected from the relationship. As specific host cells, those described in the section A-3. Can be used.
- B-4 Recombinant cell
- the present invention provides a recombinant cell (transformant) transformed with an expression vector containing the E2 polynucleotide.
- Examples of the host cell used for the recombinant cell include A-4. Those described in the section can be used without particular limitation. The method for introducing an expression vector into a host cell is described in A-4. This can be done as described in the section.
- the recombinant cell can produce an E2 polypeptide that is a tetrahydrodaidzein converting enzyme, it can be used for the production of tetrahydrodaidzein converting enzyme, or it can be used for the production of tetrahydrodaidzein in the state of cells. it can.
- E2 polypeptides can be produced by culturing recombinant cells into which an E2 polynucleotide has been introduced and recovering the E2 polypeptide from the culture.
- the culture is performed according to A-5. This can be done according to the description in the section.
- the present invention provides a method for producing tetrahydrodaidzein using E2 polypeptide. That is, in the production method, dihydrodaidzein is converted to tetrahydrodaidzein by allowing E2 polypeptide to act on dihydrodaidzein in the presence of NADPH and / or NADH.
- reaction employed in this production method is the above A-6. It can be carried out in a suitable buffer as described in the section.
- the reaction employed in this production method is such that each component is added so as to satisfy the following concentration range in a buffer solution (raw material mixture at the start of the reaction) at the start of the reaction, and the above E2 polypeptide, dihydrodaidzein, etc. It is carried out by incubating the raw material and a product such as tetrahydrodaidzein for 0.5 to 10 hours, preferably 1 to 6 hours, more preferably 2 to 4 hours, under temperature conditions that do not cause alteration or inactivation.
- the reaction temperature is not limited as long as such temperature conditions are satisfied. For example, when the temperature is set to 0 ° C. or lower, a buffer solution that does not freeze at these reaction temperatures is used.
- the reaction temperature is preferably 0 to 45 ° C, more preferably 0 to 37 ° C.
- tetrahydrodaidzein has a cis type and a trans type, and it is also possible to control the generation of the cis type and the trans type by changing the conditions such as the reaction temperature and time.
- a tetrahydrodaidzein mixed with a cis type and a trans type can be produced at a reaction temperature of 0 ° C., and a trans type can be produced with priority at 37 ° C.
- the concentration range of each component in the above production method is as follows.
- E2 polypeptide preferably 0.001 to 0.1% by weight, more preferably 0.001 to 0.01% by weight
- Dihydrodaidzein is 0.0001 to 10.0 wt%, preferably 0.001 to 1.0 wt%, more preferably 0.001 to 0.1 wt%
- NADPH and / or NADH is 0.01 to 5% by weight, preferably 0.05 to 1% by weight, more preferably 0.1 to 0.5% by weight.
- the present invention also provides a tetrahydrodaidzein synthesis raw material composition containing (Bi) E2 polypeptide, (Bii) NADPH and / or NADH, and (Biii) dihydrodaidzein as a mixed raw material for synthesizing tetrahydrodaidzein. To do. By incubating the synthetic raw material composition under the conditions described above, dihydrodaidzein in the synthetic raw material composition can be converted to tetrahydrodaidzein.
- the synthetic raw material composition corresponds to the raw material mixture at the start of the reaction in the production of tetrahydrodaidzein.
- the synthetic raw material composition contains E2 polypeptide, NADPH and / or NADH,
- the blending concentration of daidzein and other components that can be blended in the synthetic raw material composition are the same as in the case of the reaction system (raw material mixed solution at the start of the reaction) employed in the production method.
- the present invention provides a kit for synthesizing tetrahydrodaidzein containing (Bi) E2 polypeptide, (Bii) NADPH and / or NADH, and (Biii) dihydrodaidzein as a kit for synthesizing tetrahydrodaidzein.
- the synthesis kit it is sufficient that each component is divided and provided as necessary so that the synthesis of tetrahydrodaidzein from dihydrodaidzein can be easily performed under the conditions described above.
- the synthesis kit may contain a buffer to be used, if necessary.
- the synthesis kit may include necessary instruments and operation manuals in order to easily synthesize tetrahydrodaidzein.
- Tetrahydrodaidzein synthase composition The present invention further provides a tetrahydrodaidzein synthase composition comprising an E2 polypeptide.
- the enzyme composition is suitably used as a tetrahydrodaidzein synthase in a method for producing tetrahydrodaidzein using an E2 polypeptide.
- the enzyme composition may be a crude E2 polypeptide, or a mixture of a crude or purified E2 polypeptide in a suitable carrier.
- the carrier does not adversely affect the activity of the E2 polypeptide, and is mixed in an appropriate amount.
- the blending ratio of E2 polypeptide is not particularly limited as long as it can be used as a tetrahydrodaidzein synthase in the tetrahydrodaidzein production method.
- the E2 polypeptide is exemplified by 0.001 to 20.0% by weight, preferably 0.005 to 5.0% by weight, more preferably 0.01 to 1.0% by weight, based on the total amount of the enzyme composition.
- the enzyme composition may contain NADPH and / or NADH which acts as a coenzyme for E2 polypeptide.
- NADPH and / or NADH acts as a coenzyme for E2 polypeptide.
- the blending ratio of NADPH and / or NADH is not particularly limited, but is 0.005 to 50.0 wt%, preferably 0.05 to 10.0 wt% per the enzyme composition. %, More preferably 0.1 to 5.0% by weight.
- the enzyme composition includes the above A-7.
- the enzyme composition includes the above A-7.
- Various antioxidants and preservatives described in the section can also be added.
- the present invention provides a method for producing tetrahydrodaidzein using a recombinant cell into which an E2 polynucleotide has been introduced. That is, in this production method, dihydrodaidzein is converted to tetrahydrodaidzein by allowing the recombinant cells to act on dihydrodaidzein.
- the reaction employed in this production method is carried out in an environment where the above recombinant cells can survive and dihydrodaidzein can be converted to tetrahydrodaidzein.
- the medium used is appropriately selected from various commonly used media depending on the type of cell employed as the host cell for the recombinant cell.
- an appropriate amount of a protease inhibitor such as PMSF or EDTA may be added to the medium as necessary.
- an appropriate amount of a reducing agent such as DTT, 2ME, DET, or sodium hydrosulfite may be added.
- addition of NADPH and / or NADH is not essential, but NADPH and / or NADH may be added to the medium as necessary.
- the present production method comprises inoculating the above recombinant cells in a medium containing 0.001 to 1% by weight of dihydrodaidzein, preferably 0.01 to 0.5% by weight, more preferably 0.01 to 0.1% by weight, It is carried out by incubating for 7 to 30 hours, preferably 15 to 24 hours, more preferably 17 to 20 hours under conditions where the temperature can grow.
- the growth temperature conditions are not particularly limited, and the growth can be performed at the same temperature as in “B-6. Production of tetrahydrodaidzein using E2 polypeptide”.
- the present invention also provides a tetrahydrodaidzein synthesis raw material composition containing (Biv) the above recombinant cells and (Biii) dihydrodaidzein as a mixed raw material for synthesizing tetrahydrodaidzein.
- a tetrahydrodaidzein synthesis raw material composition containing (Biv) the above recombinant cells and (Biii) dihydrodaidzein as a mixed raw material for synthesizing tetrahydrodaidzein.
- the synthetic raw material composition corresponds to the raw material mixture at the start of the reaction, the concentration of the recombinant cells and dihydrodaidzein in the synthetic raw material composition, and the synthetic raw material composition
- the other components that can be blended in are the same as the conditions employed in the production method.
- the present invention provides a kit for synthesizing tetrahydrodaidzein containing (Biv) the above recombinant cell and (Biii) dihydrodaidzein as a kit for synthesizing tetrahydrodaidzein.
- the above recombinant cells and dihydrodaidzein may be separated and provided as necessary so that tetrahydrodaidzein can be easily synthesized from dihydrodaidzein under the conditions described above.
- the synthesis kit may contain a buffer or a medium as necessary. Further, the synthesis kit may include necessary instruments and operation manuals in order to easily synthesize tetrahydrodaidzein.
- the recombinant cells contained in the synthesis kit may be those stored by a known method.
- Techniques for storing recombinant cells are known, and examples include a method of storing recombinant cells in dimethylformamide or the like and treating the ampoule in a vacuum state with a lyophilizer, followed by storage at 4 to 25 ° C. Can do.
- a liquid nitrogen method in which cells are suspended in a 10% glycerol-preserved medium, stored in a dedicated ampoule, and stored in a liquid nitrogen tank ( ⁇ 150 to ⁇ 196 ° C.) can be mentioned.
- the present invention further provides an antibody (IgG antibody) having binding property to E2 polypeptide.
- Monoclonal antibodies and polyclonal antibodies are prepared according to conventional methods. Specifically, it can be prepared according to the method described in the section A-9.
- the present invention provides an immunological method for detecting or measuring the E2 polypeptide using the antibody.
- the immunological method includes A-10. This can be done according to the method described in the section.
- the present invention provides an immunological detection kit containing the antibody as a kit for detecting or measuring E2 polypeptide.
- the detection kit may contain an E2 polypeptide as a standard if necessary. Further, the detection kit may be provided with other reagents and the like as necessary so that the E2 polypeptide can be easily detected under the conditions described above. In addition, the detection kit may include necessary instruments and operation manuals in order to easily detect the E2 polypeptide.
- the present invention also provides a method for detecting or measuring an E2 polynucleotide. Specifically, this method is carried out by bringing a probe that binds to the E2 polynucleotide into contact with the test sample, and can be performed according to the method described in the section A-11.
- the present invention provides an E2 polynucleotide detection kit containing the probe as a kit for detecting or measuring an E2 polynucleotide.
- the detection kit may be provided with reagents and the like used in addition to the probe as required so that E2 polynucleotide can be easily detected under the conditions described above.
- the detection kit can also be used as a kit for identifying cells containing an E2 polynucleotide.
- the detection kit is preferably a kit for performing detection by PCR from the viewpoint of enabling highly accurate detection.
- Equol synthase C-1 Polypeptides
- the present invention relates to the following polypeptides (Ca) to (Cc) (hereinafter referred to as “E3 polypeptides”) as polypeptides that synthesize equol using tetrahydrodaidzein as a substrate.
- the range of “one or more” is not particularly limited as long as the polypeptide has an activity of synthesizing equol using tetrahydrodaidzein as a substrate.
- 1 to 200 preferably Is 1 to 150, more preferably 1 to 100, more preferably 1 to 50, more preferably 1 to 45, more preferably 1 to 40, more preferably 1 to 30, more preferably 1 To 15, more preferably 1 to 5, even more preferably 1 to 4, particularly preferably 1 to 3, and still more preferably 1 or 2.
- Such (Cb) polypeptide include a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 14 and a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 15. Compared to the amino acid sequence described in SEQ ID NO: 13, the amino acid sequence described in SEQ ID NO: 14 has two amino acids substituted. Compared with the amino acid sequence described in SEQ ID NO: 13, the amino acid sequence described in SEQ ID NO: 15 has 42 amino acids substituted, and one amino acid (glutamic acid) is added to the C-terminus.
- the amino acid sequence described in SEQ ID NO: 14 corresponds to the E3 enzyme derived from Bacteroides obatas strain E-23-15 (FERM BP-6435).
- the amino acid sequence described in SEQ ID NO: 15 corresponds to the E3 enzyme derived from Streptococcus constellatus strain A6G-225 (FERM BP-6437).
- amino acid substitution, deletion, insertion or addition in the polypeptide (Cb) above can be made in accordance with the substitution, deletion, insertion or addition in the E1 peptide described in the section A-1. .
- Amino acid substitution, deletion, insertion or addition in the above polypeptide (Cb) is performed in a region which does not significantly affect the higher order structure of the polypeptide or a region which does not affect the active center as an equol synthase. It is desirable.
- a region for example, a region having low conservation between the amino acid sequences described in SEQ ID NOs: 13, 14, and 15 and the vicinity thereof, and an N-terminal region or a C-terminal region can be mentioned.
- the “near region” means within 5 amino acids, preferably 4 amino acids before and after, preferably 4 amino acids before and after the amino acid at the specific position as a starting point within a range not inhibiting equol synthase activity.
- Amino acid more preferably two amino acids before and after, and particularly preferably one amino acid before and after.
- the region corresponding to the 25th to 35th preferably, in the amino acid sequence shown in SEQ ID NO: 13, the region corresponding to the 25th to 35th, the region corresponding to the 170th to 177th, the 201st -208th region, 242nd-248th region, 276th-289th region, 355th-385th region, 396th-409th region And a region corresponding to the 431st to 443rd regions.
- amino acid sequence described in SEQ ID NO: 13 the sequence consisting of the 14th amino acid to the 19th amino acid is considered to correspond to the FAD binding domain. Therefore, any amino acid may be substituted, deleted, inserted or added in the sequence as long as the function of the domain is not inhibited, but preferably the 14th glycine, the 16th glycine and the 19th Of glycine is not mutated.
- amino acid is substituted, deleted, inserted or added in the sequence, it is preferably 3 amino acids or less, more preferably 2 amino acids or less, still more preferably 1 amino acid, most preferably Is not mutated.
- FIG. 28 shows the alignment of the amino acid sequences described in SEQ ID NOs: 13, 14, and 15.
- the amino acid identity may be, for example, 60% or more with respect to the amino acid sequence shown in SEQ ID NO: 13, but is usually 80% or more, preferably 85% or more, more preferably Is 90% or more, more preferably 95% or more, particularly preferably 98% or more, and still more preferably 99% or more.
- polypeptide (Cc) examples include a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 14 and a polypeptide consisting of the amino acid sequence set forth in SEQ ID NO: 15.
- the identity between the amino acid sequence shown in SEQ ID NO: 13 and the amino acid sequence shown in SEQ ID NO: 14 is 99.6%, and the amino acid sequence shown in SEQ ID NO: 13 is identical to the amino acid sequence shown in SEQ ID NO: 15.
- the sex is 90.9% (Blast 2). Therefore, in a preferred embodiment of the present invention, the (Cc) polypeptide preferably has an identity of 90.9% or more, more preferably 99.6% or more, with respect to the amino acid sequence set forth in SEQ ID NO: 14.
- polypeptides (Cb) and (Cc) above “activity for synthesizing equol using tetrahydrodaidzein as a substrate” can be confirmed as follows. That is, a polypeptide to be confirmed is added to a substrate solution having the following composition so as to be 0.001 mg / mL, incubated at 37 ° C. for 2 hours, and then the presence or absence of equol is confirmed in the solution. When the presence of equol is confirmed in the solution after incubation, the polypeptide is determined to have “activity for synthesizing equol using tetrahydrodaidzein as a substrate”.
- composition of substrate solution 0.1 M potassium phosphate buffer (pH 7.0) 1 mM PMSF (phenylmethylsulfonyl fluoride) 2 mM dithiothreitol 5 mM sodium hydrosulfite 40 ⁇ M tetrahydrodaidzein
- E3 polypeptide has enzymatic activity to synthesize equol using tetrahydrodaidzein as a substrate.
- E3 polypeptide is also referred to as E3 enzyme.
- the optimum temperature for the E3 enzyme is about 23-37 ° C. and the optimum pH is 4.5.
- the E3 enzyme can not only synthesize equol using tetrahydrodaidzein as a substrate, but also reverse reaction, that is, synthesize tetrahydrodaidzein using equol as a raw material.
- the E3 polypeptide can be obtained by genetic engineering techniques based on the nucleotide sequence information set forth in SEQ ID NOs: 16, 17, or 18 as with the E1 peptide and E2 peptide. It can also be obtained by chemical synthesis based on the amino acid sequence information described in SEQ ID NOs: 13, 14, or 15. Further, it can be obtained by isolation and purification from a microorganism having the ability to produce E3 polypeptide. These methods can be performed according to the description in the section A-1.
- the microorganism having the ability to produce E3 polypeptide may be cultured in a medium containing a desired amount of tetrahydrodaidzein. Even in this case, the E3 polypeptide can be produced from a microorganism having the ability to produce the E3 polypeptide.
- the E3 polypeptide may exist as a monomer, but may exist as a dimer or higher multimer as long as it has the ability to synthesize equol.
- the E3 polypeptide may be modified with polyethylene glycol or a sugar chain as necessary for the purpose of improving stability or the like.
- the E3 polypeptide can play a catalytic role to convert to equol using tetrahydrodaidzein as a substrate. Equol is considered to exert various physiological actions in vivo, and in that sense, E3 polypeptide that can provide equol is important.
- the present invention provides an equol synthase containing the polypeptides (Ca) to (Cc).
- polynucleotide encoding a polypeptide having an activity of synthesizing equol using tetrahydrodaidzein as a substrate (hereinafter, the polynucleotide may be referred to as “E3 polynucleotide”).
- polynucleotides (Cd) to (Cf) are provided as E3 polynucleotides: (Cd) a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 16; (Ce) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 13; (Cf) a polypeptide encoding a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Cd) or (Ce) under stringent conditions and has an activity of producing equol using tetrahydrodaidzein as a substrate. nucleotide.
- amino sequence described in SEQ ID NO: 13 corresponds to the amino acid sequence encoded by the nucleotide sequence described in SEQ ID NO: 16.
- amino acid sequence described in SEQ ID NO: 14 corresponds to the amino acid sequence encoded by the nucleotide sequence described in SEQ ID NO: 17.
- amino acid sequence described in SEQ ID NO: 15 corresponds to the amino acid sequence encoded by the nucleotide sequence described in SEQ ID NO: 18.
- hybridizes under stringent conditions relating to the above (Cf) polynucleotide has the same meaning as “hybridizes under stringent conditions” described in Section A-2.
- Specific examples of the polynucleotide (Cf) include the nucleotide sequence set forth in SEQ ID NO: 17 and the nucleotide sequence set forth in SEQ ID NO: 18.
- the nucleotide sequence homology between the nucleotide sequence set forth in SEQ ID NO: 16 and the nucleotide sequence set forth in SEQ ID NO: 17 is 99.8%.
- the nucleotide sequence set forth in SEQ ID NO: 16 and the nucleotide sequence set forth in SEQ ID NO: 18 Is 85.2%.
- the polynucleotide of (Cf) has a correspondingness of 85.2% or more, more preferably 99.8, to the nucleotide sequence set forth in SEQ ID NO: 16. % Equivalent.
- the “activity for synthesizing equol using tetrahydrodaidzein as a substrate” is confirmed by the same method as in the case of the polypeptide (Cb) or (Cc).
- the E3 polynucleotide can be produced and obtained by a chemical DNA synthesis method or a genetic engineering method based on the sequence information of SEQ ID NO: 16, 17, or 18. As a specific method, the method described in the section A-2. Can be used.
- the origin of cDNA of E3 polynucleotide is not particularly limited as long as it is a microorganism that expresses E3 polynucleotide. Specifically, it belongs to the genus Bacteroides, a microorganism having equol-producing ability, preferably a lactic acid bacterium having equol-producing ability.
- Bacteria and bacteria belonging to the genus Streptococcus more preferably Lactococcus garbie, Bacteroides obatas having equol-producing ability, and Streptococcus constitutus, particularly preferably stool-derived Lactococcus garbie, particularly equol-producing ability Lactococcus 20-92 strain (FERM BP-10036; deposited at the Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology), Bacteroides obatas E-23-15 strain FERM BP-6435; National Institute of Advanced Industrial Science and Technology, Deposited at Patent Biological Depositary Center), Streptococcus constitutus A6G-225 (FERM BP-6437; National Institute of Advanced Industrial Science and Technology, Patent Biological Depository Center ( 1) 1-1, East, Tsukuba City, Ibaraki Prefecture, Japan)).
- a product of the polynucleotide ie, the above polypeptide
- a product of the polynucleotide ie, the above polypeptide
- the expression vector of the present invention is not particularly limited as long as it contains an E3 polynucleotide and can express the E3 polynucleotide, and in general, similar to an expression vector containing an E1 polynucleotide, It is appropriately selected from the relationship.
- Specific examples of the host cell include A-3. The ones described in the section can be used.
- Recombinant cell The present invention provides a recombinant cell (transformant) transformed with an expression vector containing the E3 polynucleotide.
- the host cell used for the recombinant cell those described in the section A-4. Can be used without particular limitation.
- the method for introducing an expression vector into a host cell can be performed as described in the section A-4.
- the recombinant cell can produce E3 polypeptide, which is an equol converting enzyme, it can be used for the production of equol converting enzyme, and can also be used for the production of equol in the state of cells.
- E3 polypeptide can be produced by culturing a recombinant cell into which an E3 polynucleotide has been introduced and recovering the E3 polypeptide from the culture. The culture is performed according to A-5. This can be done as described in the section.
- the present invention provides a method for producing equol using E3 polypeptide. That is, in the production method, tetrahydrodaidzein is converted to equol by allowing E3 polypeptide to act on tetrahydrodaidzein.
- the reaction employed in this production method can be performed in an appropriate buffer solution. Specifically, A-6.
- the buffers described in the section can be used.
- the reaction employed in this production method is such that each component is added in a buffer solution (raw material mixture at the start of the reaction) at the start of the reaction so as to satisfy the following concentration range, and the above E3 polypeptide, tetrahydrodaidzein, etc.
- the reaction is carried out by incubating at 0.5 to 10 hours, preferably 1 to 6 hours, more preferably 2 to 4 hours, under temperature conditions where the raw materials and products such as equol are not altered or inactivated.
- the reaction temperature is not limited as long as such temperature conditions are satisfied. For example, when the temperature is set to 0 ° C. or lower, a buffer solution that does not freeze at these reaction temperatures is used.
- the reaction temperature is preferably 0 to 45 ° C, more preferably 0 to 37 ° C.
- Tetrahydrodaidzein is 0.0001 to 10.0% by weight, preferably 0.001 to 1.0% by weight, more preferably 0.001 to 0.1% by weight.
- the present invention also provides an equol synthesis raw material composition containing (Ci) E3 polypeptide and (Cii) tetrahydrodaidzein as a mixed raw material for synthesizing equol.
- tetrahydrodaidzein in the synthetic raw material composition can be converted to equol.
- the synthetic raw material composition corresponds to the raw material mixture at the start of the reaction in the production of equol.
- the synthetic raw material composition contains E3 polypeptide, tetrahydrodaidzein, and the synthetic raw material composition.
- Other components that can be incorporated into the product are the same as in the case of the reaction system (the raw material mixed solution at the start of the reaction) employed in the above production method.
- the present invention provides an equol synthesis kit containing (Ci) E3 polypeptide and (Cii) tetrahydrodaidzein as a kit for synthesizing equol.
- the synthesis kit only needs to be provided with each component divided as necessary so that equol can be easily synthesized from tetrahydrodaidzein under the conditions described above.
- the synthesis kit may contain a buffer to be used, if necessary.
- the synthesis kit may include necessary instruments and operation manuals in order to easily synthesize equol.
- Equol synthase composition The present invention further provides an equol synthase composition comprising an E3 polypeptide.
- the enzyme composition is suitably used as an equol synthase in a method for producing equol using an E3 polypeptide.
- the enzyme composition may be a crude E3 polypeptide, or a mixture of a crude or purified E3 polypeptide in a suitable carrier.
- the carrier does not adversely affect the activity of the E3 polypeptide, and is mixed in an appropriate amount.
- the mixing ratio of the E3 polypeptide is not particularly limited as long as it can be used as an equol synthase in the equol production method.
- the E3 polypeptide is 0.001 to 20.0% by weight, preferably 0.005 to 5.0% by weight, more preferably 0.01 to 1.0% by weight, based on the total amount of the enzyme composition.
- the enzyme composition includes the above A-7.
- the antioxidants and preservatives described in the section can also be added.
- the present invention provides a method for producing equol using a recombinant cell into which an E3 polynucleotide has been introduced. That is, in this production method, tetrahydrodaidzein is converted to equol by allowing the recombinant cells to act on tetrahydrodaidzein.
- the reaction employed in this production method is carried out in an environment where the above recombinant cells can survive and tetrahydrodaidzein can be converted to equol.
- the medium used is appropriately selected from various commonly used media depending on the type of cell employed as the host cell for the recombinant cell.
- an appropriate amount of a protease inhibitor such as PMSF or EDTA may be added to the medium as necessary.
- an appropriate amount of a reducing agent such as DTT, 2ME, DET, or sodium hydrosulfite may be added.
- addition of NADPH and / or NADH is not essential, but NADPH and / or NADH may be added to the medium as necessary.
- the production method comprises inoculating the recombinant cell with a medium containing 0.001 to 1% by weight, preferably 0.01 to 0.5% by weight, more preferably 0.01 to 0.1% by weight of tetrahydrodaidzein, It is carried out by incubating for 7 to 30 hours, preferably 15 to 24 hours, more preferably 17 to 20 hours under conditions where the temperature can grow.
- the growth temperature condition is not particularly limited, and the C-6. It can be carried out at a temperature similar to that described in the section.
- the present invention provides an equol synthesis raw material composition containing (Ciii) the above recombinant cells and (Cii) tetrahydrodaidzein as a mixed raw material for synthesizing equol.
- equol synthesis raw material composition containing (Ciii) the above recombinant cells and (Cii) tetrahydrodaidzein as a mixed raw material for synthesizing equol.
- the synthetic raw material composition corresponds to the raw material mixture at the start of the reaction in the production of the equol, and the concentration of the recombinant cells and tetrahydrodaidzein in the synthetic raw material composition, and the synthetic raw material composition
- Other components and the like that can be blended are the same as the conditions and the like employed in the above production method.
- the present invention provides an equol synthesis kit containing (Ciii) the above recombinant cell and (Cii) tetrahydrodaidzein as a kit for synthesizing equol.
- the kit for synthesis the recombinant cells and tetrahydrodaidzein may be provided separately as necessary so that equol can be easily synthesized from tetrahydrodaidzein under the conditions described above.
- the synthesis kit may contain a buffer or a medium as necessary. Further, the synthesis kit may include necessary instruments and operation manuals in order to easily synthesize equol.
- the recombinant cells contained in the synthesis kit may be those stored by a known method.
- Techniques for storing recombinant cells are known, and examples include a method of storing recombinant cells in dimethylformamide or the like and treating the ampoule in a vacuum state with a lyophilizer, followed by storage at 4 to 25 ° C. Can do.
- a liquid nitrogen method in which cells are suspended in a 10% glycerol-preserved medium, stored in a dedicated ampoule, and stored in a liquid nitrogen tank ( ⁇ 150 to ⁇ 196 ° C.) can be mentioned.
- the present invention further provides an antibody (IgG antibody) having binding property to E3 polypeptide.
- Monoclonal antibodies and polyclonal antibodies are prepared according to conventional methods. Specifically, the A-9. It can be created according to the method described in the section.
- the present invention provides an immunological method for detecting or measuring an E3 polypeptide using the antibody.
- the immunological method includes the A-10. This can be done according to the method described in the section.
- the present invention provides an immunological detection kit containing the above antibody as a kit for detecting or measuring an E3 polypeptide.
- the detection kit may contain an E3 polypeptide as a standard if necessary. Further, the detection kit may be provided with other reagents and the like as necessary so that the E3 polypeptide can be easily detected under the conditions described above. In addition, the detection kit may include necessary instruments and operation manuals in order to easily detect the E3 polypeptide.
- the present invention provides a method for detecting or measuring an E3 polynucleotide. Specifically, the method is carried out by contacting a probe that binds to an E3 polynucleotide with a test sample, and the A-11. This can be done according to the method described in the section.
- the present invention provides a kit for detecting an E3 polynucleotide containing the probe as a kit for detecting or measuring an E3 polynucleotide.
- the detection kit may be provided with reagents and the like used in addition to the probe as required so that E3 polynucleotide can be easily detected under the conditions described above.
- the detection kit can also be used as a kit for identifying cells containing an E3 polynucleotide.
- the detection kit is preferably a kit for performing detection by PCR from the viewpoint of enabling highly accurate detection.
- the manufacturing method of tetrahydrodaidzein including the first step and the second step
- the present invention includes the following first step of producing dihydrodaidzein from daidzein and second step of producing tetrahydrodaidzein from dihydrodaidzein (Hereinafter, this production method may be referred to as “first production method”).
- Aa a polypeptide comprising the amino acid sequence set forth in SEQ ID NO: 1;
- Ab a polypeptide comprising an amino acid sequence in which one or more amino acids are substituted, deleted, inserted and / or added in the amino acid sequence of SEQ ID NO: 1 and having an activity of synthesizing dihydrodaidzein using daidzein as a substrate ;
- An enzyme comprising a polypeptide that is one of the following (Ba) to (Bc) (hereinafter, the polypeptide may be referred to as “E2 polypeptide”), and NADPH: And / or producing tetrahydrodaidzein by acting NADH:
- E2 polypeptide a polypeptide comprising the amino acid sequence set forth in SEQ ID NO: 7
- Bb a polypeptide comprising an amino acid sequence in which one or more amino acids are substituted, deleted, inserted and / or added in the amino acid sequence set forth in SEQ ID NO: 7 and having an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate peptide
- a polypeptide comprising an amino acid sequence having 60% or more identity to the amino acid sequence set forth in SEQ ID NO: 7, and having an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate.
- tetrahydrodaidzein can be produced from dihydrodaidzein, and equol can be produced from tetrahydrodaidzein.
- daidzein is converted to dihydrodaidzein by allowing an enzyme consisting of E1 polypeptide to act on daidzein in the presence of NADPH and / or NADH.
- the reaction employed in the first step is the production of the enzyme consisting of the E1 polypeptide, daidzein and NADPH and / or NADH and / or the raw material such as daidzein and dihydrodaidzein in a solution containing NADH. This is carried out by incubating at a temperature and for a time that does not alter or inactivate the product. Specifically, the above A-6. It can be carried out according to the conditions described in the section.
- the reaction employed is the reaction system at the start of the reaction (
- a raw material mixture is prepared so that each component in the raw material mixture at the start of the reaction satisfies the following concentration range, and this is performed at a temperature of 15 to 45 ° C, preferably 25 to 40 ° C, more preferably 30 to 38 ° C. , 0.5 to 10 hours, preferably 1 to 6 hours, more preferably 2 to 4 hours.
- enzyme comprising E1 polypeptide, preferably 0.001-0.1% by weight, more preferably 0.001-0.01% by weight; Daidzein is 0.0001 to 10.0 wt%, preferably 0.001 to 1.0 wt%, more preferably 0.001 to 0.1 wt%; and NADPH / or NADH is 0.01 to 5 wt% , Preferably 0.05 to 1% by weight, more preferably 0.1 to 0.5% by weight.
- the origin of daidzein used as a substrate in the first step is not limited.
- commercially available daidzein may be used, or daidzein that is appropriately generated or synthesized may be used.
- dihydrodaidzein synthesis containing (Ai) an enzyme consisting of E1 polypeptide, (Aii) NADPH and / or NADH, and (Aiii) daidzein
- a raw material composition may be used.
- daidzein in the synthetic raw material composition can be converted to dihydrodaidzein.
- the synthetic raw material composition comprises the above A-6. And A-7. More detailed in the section.
- dihydrodaidzein is converted to tetrahydrodaidzein by allowing an enzyme consisting of E2 polypeptide to act on dihydrodaidzein in the presence of NADPH and / or NADH.
- the reaction employed in the second step includes, for example, an enzyme comprising the E2 polypeptide, dihydrodaidzein and NADPH and / or NADH, a solution containing the E2 polypeptide, a raw material such as dihydrodaidzein, and tetrahydro. It is carried out by incubating at a temperature and for a time at which a product such as daidzein is not altered or inactivated. Specifically, the above B-6. It can be carried out according to the conditions described in the section.
- Tetrahydrodaidzein has a cis type and a trans type. It is also possible to control the generation of the cis type and the trans type by changing the conditions such as the reaction temperature and time. For example, a tetrahydrodaidzein mixed with a cis type and a trans type can be produced at a reaction temperature of 0 ° C., and a trans type can be produced with priority at 37 ° C.
- the reaction adopted from the viewpoint of efficiently performing both the first and second steps is the “first step”. It is carried out by incubating both under the conditions when the first step and the second step are performed under the same conditions.
- the concentration of the enzyme consisting of E2 polypeptide, dihydrodaidzein, and NADPH and / or NADH can be set as follows: 0.0001-1.0% by weight of the enzyme comprising E2 polypeptide, preferably 0.001-0.1% by weight, more preferably 0.001-0.01% by weight; Dihydrodaidzein is 0.0001 to 10.0% by weight, preferably 0.001 to 1.0% by weight, more preferably 0.001 to 0.1% by weight; and NADPH and / or NADH is 0.01 to 5%. % By weight, preferably 0.05 to 1% by weight, more preferably 0.1 to 0.5% by weight.
- the concentration of NADPH is determined based on the conditions described in the “first step” when the first step and the second step are performed under the same conditions.
- the concentration of NADH used in combination with NADPH is set to 0.01 to 5% by weight, preferably 0.05 to 1% by weight, and more preferably 0.1 to 0.5% by weight.
- the origin of dihydrodaidzein used as a substrate is not limited.
- dihydrodaidzein produced from daidzein in the first step may be used as a substrate in the second step.
- the solution containing the dihydrodaidzein produced in the first step may be used as it is, or may be roughly purified or purified.
- dihydrodaidzein may be used, or appropriately synthesized dihydrodaidzein may be used.
- (Bi) an enzyme composed of E2 polypeptide, (Bii) NADPH and / or NADH, and (Biii) dihydrodaidzein synthetic raw material composition containing dihydrodaidzein You may use things. By incubating the synthetic raw material composition under the conditions described above, dihydrodaidzein in the synthetic raw material composition can be converted to tetrahydrodaidzein. For the explanation of the constituent raw material composition, see B-6. And A-7. More detailed in the section.
- dihydrodaidzein produced from daidzein in the first step is preferably used as a substrate in the second step, and commercially available dihydrodaidzein, the synthetic raw material composition, the enzyme composition, etc. may be used in combination. Good.
- E1 polypeptide and E2 polypeptide In the first production method of the present invention, the A-1. And an enzyme comprising the E1 polypeptide according to B-1. An enzyme comprising the E2 polypeptide described in 1) is used.
- the product containing tetrahydrodaidzein produced by the first production method is a product containing tetrahydrodaidzein produced by a tetrahydrodaidzein production method (first production method) comprising a first step and a second step. Offer things.
- dihydrodaidzein can be produced from daidzein and tetrahydrodaidzein can be produced from dihydrodaidzein.
- the product containing tetrahydrodaidzein produced by the first production method of the present invention may contain not only tetrahydrodaidzein but also dihydrodaidzein and daidzein.
- the product of the present invention may be a solution obtained by the first production method, or may be a product obtained by roughly purifying tetrahydrodaidzein or the like from the solution. Also good.
- the product of the present invention can be blended in foods and drinks, cosmetics, pharmaceuticals, etc., and can also be used as a substrate.
- the present invention relates to a method for producing equol, which comprises the following second step of producing tetrahydrodaidzein from dihydrodaidzein and third step of producing equol from tetrahydrodaidzein.
- a method (hereinafter, this production method may be referred to as a “second production method”) is provided.
- An enzyme comprising a polypeptide that is one of the following (Ba) to (Bc) (hereinafter, the polypeptide may be referred to as “E2 polypeptide”), and NADPH: And / or producing tetrahydrodaidzein by acting NADH:
- E2 polypeptide a polypeptide comprising the amino acid sequence set forth in SEQ ID NO: 7
- Bb a polypeptide comprising an amino acid sequence in which one or more amino acids are substituted, deleted, inserted and / or added in the amino acid sequence set forth in SEQ ID NO: 7 and having an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate peptide
- a polypeptide comprising an amino acid sequence having 60% or more identity to the amino acid sequence set forth in SEQ ID NO: 7, and having an activity of synthesizing tetrahydrodaidzein using dihydrodaidzein as a substrate.
- An enzyme comprising a polypeptide which is one of the following (Ca) to (Cc) (hereinafter, the polypeptide may be referred to as “E3 polypeptide”) is allowed to act on tetrahydrodaidzein.
- a process for producing equol (Ca) a polypeptide comprising the amino acid sequence set forth in SEQ ID NO: 13; (Cb) a polypeptide comprising an amino acid sequence in which one or more amino acids are substituted, deleted, inserted and / or added in the amino acid sequence set forth in SEQ ID NO: 13, and having an activity of synthesizing equol using tetrahydrodaidzein as a substrate ; (Cc) A polypeptide comprising an amino acid sequence having 60% or more identity to the amino acid sequence set forth in SEQ ID NO: 13, and having an activity of synthesizing equol using tetrahydrodaidzein as a substrate.
- tetrahydrodaidzein can be produced from dihydrodaidzein, and equol can be produced from tetrahydrodaidzein.
- 2nd process The 2nd process contained in the 2nd manufacturing method of this invention is demonstrated similarly to the 2nd process contained in the above-mentioned 1st manufacturing method.
- adopted is a reaction system at the time of a reaction start from a viewpoint which implements both the 2nd and 3rd processes efficiently.
- a raw material mixture is prepared so that each component satisfies the following concentration range in (the raw material mixture at the start of the reaction), and this is a temperature condition of 15 to 45 ° C, preferably 25 to 40 ° C, more preferably 30 to 38 ° C.
- 0.0001-1.0% by weight of the enzyme comprising E2 polypeptide preferably 0.001-0.1% by weight, more preferably 0.001-0.01% by weight; Dihydrodaidzein is 0.0001 to 10.0% by weight, preferably 0.001 to 1.0% by weight, more preferably 0.001 to 0.1% by weight; and NADPH and / or NADH is 0.01 to 5%.
- % By weight, preferably 0.05 to 1% by weight, more preferably 0.1 to 0.5% by weight.
- tetrahydrodaidzein is converted to equol by allowing an enzyme comprising an E3 polypeptide to act on tetrahydrodaidzein.
- the reaction employed in the third step is that the E3 polypeptide enzyme, tetrahydrodaidzein-containing solution, the E3 polypeptide enzyme, tetrahydrodaidzein raw material, and equol product, etc. It is carried out by incubating at non-activated temperature conditions and time. Specifically, the above A-6. The reaction can be carried out according to the conditions described in the section.
- the reaction employed is described in the “second step” from the viewpoint of efficiently performing both the second and third steps.
- the second step and the third step are performed by incubating both under the same conditions.
- the concentration of the enzyme consisting of E3 polypeptide, tetrahydrodaidzein, and NADPH and / or NADH can be set as follows: 0.0001-1.0% by weight of an enzyme comprising an E3 polypeptide, preferably 0.001-0.1% by weight, more preferably 0.001-0.01% by weight; Tetrahydrodaidzein is 0.0001 to 10.0% by weight, preferably 0.001 to 1.0% by weight, more preferably 0.001 to 0.1% by weight; and NADPH and / or NADH is 0.01 to 5%.
- % By weight, preferably 0.05 to 1% by weight, more preferably 0.1 to 0.5% by weight.
- the origin of tetrahydrodaidzein used as a substrate is not limited.
- tetrahydrodaidzein produced from dihydrodaidzein in the second step may be used as a substrate in the third step.
- the solution containing tetrahydrodaidzein produced in the second step may be used as it is, may be roughly purified, or may be purified.
- tetrahydrodaidzein may be used, or tetrahydrodaidzein synthesized appropriately may be used.
- an equol synthetic raw material composition containing (Ci) an enzyme composed of E3 polypeptide and (Cii) tetrahydrodaidzein may be used as a mixed raw material for synthesizing equol in the third step.
- an equol synthetic raw material composition containing (Ci) an enzyme composed of E3 polypeptide and (Cii) tetrahydrodaidzein may be used as a mixed raw material for synthesizing equol in the third step.
- tetrahydrodaidzein produced from dihydrodaidzein in the second step is preferably used as a substrate in the third step, and commercially available tetrahydrodaidzein, the synthetic raw material composition, the enzyme composition, etc. are used in combination. Also good.
- E2 polypeptide and E3 polypeptide In the second production method of the present invention, the B-1.
- An enzyme comprising the E2 polypeptide described in the section and the C-1. Enzymes consisting of the E3 polypeptides described in the section are used.
- the present invention provides a product containing equol produced by the production method of equol (second production method) comprising the second step and the third step. To do.
- tetrahydrodaidzein can be produced from dihydrodaidzein, and equol can be produced from tetrahydrodaidzein.
- the product containing equol produced by the second production method of the present invention may contain not only equol but also tetrahydrodaidzein and further dihydrodaidzein.
- the product of the present invention may be a solution as obtained by the second production method, or may be a product obtained by roughly purifying equol from the solution, or a product obtained by further purification. Good.
- the product of the present invention can be blended in foods and drinks, cosmetics, pharmaceuticals, etc., and can also be used as a substrate.
- the present invention includes a method for producing equol , comprising the first step, the second step and the third step (hereinafter referred to as “third production method”). May be written).
- dihydrodaidzein can be produced from daidzein
- tetrahydrodaidzein can be produced from dihydrodaidzein
- equol can be produced from tetrahydrodaidzein.
- daidzein is converted to dihydrodaidzein by allowing an enzyme comprising E1 polypeptide to act on daidzein in the presence of NADPH and / or NADH.
- an enzyme comprising E1 polypeptide to act on daidzein in the presence of NADPH and / or NADH.
- the 1st process contained in the 3rd manufacturing method of the present invention is explained like the above-mentioned 1st process.
- the reaction adopted is the incubation under the conditions and concentrations based on the case where the first step and the second step, etc., are carried out under the same conditions as described in the “first step”. It is carried out by doing.
- Second Step In the second step contained in the third production method of the present invention, an enzyme comprising an E2 polypeptide is allowed to act on dihydrodaidzein in the presence of NADPH and / or NADH, whereby dihydrodaidzein is converted into tetrahydrodaidzein. Convert to The second step is described in the same manner as the second step described above.
- the reaction is carried out by incubating under conditions and concentrations based on the case where the first step, the second step, etc. described in the “second step” are performed under the same conditions.
- first step and the second step are performed in the presence of an enzyme comprising an E1 polypeptide and an enzyme comprising an E2 polypeptide
- first to third steps are carried out from an enzyme comprising an E1 polypeptide and an E2 polypeptide.
- reaction is performed in the coexistence of the enzyme consisting of the enzyme consisting of E1 polypeptide and the enzyme consisting of the E2 polypeptide as described in the “second step”, etc. Performed by incubating at each concentration.
- adopted is the 2nd process and the 2nd process as described in the said "2nd process.” It is carried out by incubating at the respective conditions / concentrations based on the case where the three steps are performed under the same conditions.
- tetrahydrodaidzein is converted to equol by allowing an enzyme comprising an E3 polypeptide to act on tetrahydrodaidzein.
- the third step is described in the same manner as the third step described above.
- the reaction adopted from the viewpoint of efficiently performing each step Is carried out by incubating the first step and the second step described in the “first step” under the same conditions.
- the concentration of the enzyme consisting of E3 polypeptide, tetrahydrodaidzein, and NADPH and / or NADH can be set as follows: 0.0001-1.0% by weight of an enzyme comprising an E3 polypeptide, preferably 0.001-0.1% by weight, more preferably 0.001-0.01% by weight; Tetrahydrodaidzein is 0.0001 to 10.0% by weight, preferably 0.001 to 1.0% by weight, more preferably 0.001 to 0.1% by weight; and NADPH and / or NADH is 0.01 to 5%.
- % By weight, preferably 0.05 to 1% by weight, more preferably 0.1 to 0.5% by weight.
- the first step and the third step are performed in the presence of an enzyme comprising an E1 polypeptide and an enzyme comprising an E3 polypeptide, or the first to third steps are carried out from an enzyme comprising an E1 polypeptide and an E2 polypeptide.
- NADPH is essential from the viewpoint of efficiently carrying out a dihydrodaidzein production reaction using an enzyme comprising an E1 polypeptide, as described above, in the presence of an enzyme comprising an E3 polypeptide and an enzyme comprising an E3 polypeptide.
- the concentration of NADPH is determined based on the conditions described in the “first step” when the first step and the second step are performed under the same conditions.
- the concentration of NADH used in combination with NADPH is the same as that described in the above “second step” when the enzyme is composed of an enzyme consisting of E1 polypeptide and an enzyme consisting of E2 polypeptide. Is determined based on the conditions when the second step, the third step, etc. are performed under the same conditions.
- E1 to E3 polypeptide In the third production method of the present invention, the enzyme comprising the E1 polypeptide, the enzyme comprising the E2 polypeptide, and the enzyme comprising the E3 polypeptide are used.
- the E1-E3 polypeptides are described as above.
- the present invention provides an equol-containing product produced by the equol production method (third production method) comprising the first to third steps. To do.
- dihydrodaidzein can be produced from daidzein
- tetrahydrodaidzein can be produced from dihydrodaidzein
- equol can be produced from tetrahydrodaidzein.
- the product containing equol produced by the third production method of the present invention may contain not only equol but also tetrahydrodaidzein, dihydrodaidzein, and daidzein.
- the product of the present invention may be a solution obtained by the third production method as it is, a product obtained by roughly purifying equol from the solution, or a product obtained by further purification. Good.
- the product of the present invention can be blended in foods and drinks, cosmetics, pharmaceuticals, etc., and can also be used as a substrate.
- a method for producing dihydrodaidzein, tetrahydrodaidzein and / or equol comprising at least two steps of the fourth step to the sixth step
- the present invention provides the following fourth step of producing dihydrodaidzein from daidzein, tetrahydrodaidzein from dihydrodaidzein
- a method for producing dihydrodaidzein, tetrahydrodaidzein and / or equol, comprising at least two of a fifth step for producing equol and a sixth step for producing equol from tetrahydrodaidzein (hereinafter referred to as “fourth production”). Method ”).
- (Fifth step) causing a recombinant cell having a polynucleotide (Bd) to (Bf) (hereinafter, the polynucleotide may be referred to as “E2 polynucleotide”) to act on dihydrodaidzein
- E2 polynucleotide a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 10
- (Be) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 7
- (Bf) encodes a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Bd) or (Be) under stringent conditions and has an activity of generating tetrahydrodaidzein using dihydrodaidzein as a substrate Polynucleotide.
- Step 6 Reacting a recombinant cell having a polynucleotide which is any one of (Cd) to (Cf) (hereinafter, the polynucleotide may be referred to as “E3 polynucleotide”) on tetrahydrodaidzein
- E3 polynucleotide a polynucleotide comprising the nucleotide sequence set forth in SEQ ID NO: 16
- (Ce) a polynucleotide encoding a polypeptide consisting of the amino sequence set forth in SEQ ID NO: 13
- (Cf) a polypeptide encoding a polypeptide that hybridizes to a complementary strand of the polynucleotide of (Cd) or (Ce) under stringent conditions and has an activity of producing equol using tetrahydrodaidzein as a substrate. nucleotide.
- dihydrodaidzein, tetrahydrodaidzein and / or equol can be produced according to various combinations of the fourth to sixth steps.
- the fourth manufacturing method of the present invention includes at least two steps among the fourth step, the fifth step and the sixth step.
- dihydrodaidzein is converted from dihydrodaidzein to tetrahydro Daidzein can be produced.
- the fourth production method of the present invention when the fourth production method of the present invention includes two steps of the fifth step and the sixth step and does not include the fourth step, the fourth production method of the present invention converts tetrahydrodaidzein from dihydrodaidzein to tetrahydrodaidzein. Equol can be produced from daidzein.
- dihydrodaidzein is produced from daidzein
- tetrahydrodaidzein is produced from dihydrodaidzein
- equol is produced from tetrahydrodaidzein.
- the recombinant cells used in the fourth production method of the present invention are selected from the group consisting of (Ad) to (Af), (Bd) to (Bf) and (Cd) to (Cf) in one cell. May be included, or two or more polynucleotides selected from the group may be included.
- the fourth step and the fifth step can be carried out with one type of recombinant cell.
- one cell is composed of one polynucleotide selected from (Ad) to (Af), one polynucleotide selected from (Bd) to (Bf), and (Cd) to (Cf).
- the fourth to sixth steps can be carried out with one kind of recombinant cell by using this recombinant cell.
- one kind of polynucleotide selected from (Ad) to (Af) and one kind of polynucleotide selected from (Bd) to (Bf) are used.
- the fourth step can be carried out.
- the sixth step can be performed.
- a total of 2 of one polynucleotide selected from (Ad) to (Af) and one polynucleotide selected from (Cd) to (Cf) is 2 in total.
- Step 4 the A-2. Recombinant cells having the E1 polynucleotide described in the section are converted to daidzein by acting on daidzein.
- the reaction employed in the fourth step can be performed under the same conditions as in the first step.
- the recombinant cells are inoculated into a medium containing 0.001 to 1% by weight, preferably 0.01 to 1% by weight, more preferably 0.01 to 0.5% by weight of daidzein, It is carried out by incubating at a temperature that allows growth for 6 to 30 hours, preferably 7 to 24 hours, more preferably 7 to 18 hours.
- the growth temperature condition is not particularly limited, and the growth can be performed at the same temperature as in the “first step”.
- Recombinant cells used in the fourth step have an E1 polynucleotide and are not limited as long as they can express the E1 polynucleotide. Specifically, A-4.
- the recombinant cells described in the section can be used.
- Enzymes consisting of polypeptides can be produced. Specifically, A-5.
- the E1 polypeptide can be produced by culturing the recombinant cell according to the conditions described in the section.
- Dihydrodaidzein is converted to tetrahydrodaidzein by allowing recombinant cells having the E2 polynucleotides described in the section to act on dihydrodaidzein.
- the reaction employed in the fifth step can be performed under the same conditions as in the second step. Specifically, the recombinant cells are added to a medium containing 0.001 to 1% by weight of dihydrodaidzein, preferably 0.01 to 0.5% by weight, more preferably 0.01 to 0.1% by weight. It is carried out by inoculating and incubating for 7 to 30 hours, preferably 15 to 24 hours, more preferably 17 to 20 hours under conditions where the temperature can be grown.
- the growth temperature condition is not particularly limited, and the growth can be performed at the same temperature as in the “second step”.
- Recombinant cells used in the fifth step are B-2.
- the E2 polynucleotide described in the section is not limited as long as the E2 polynucleotide can be expressed. Specifically, B-4.
- the recombinant cells described in the section can be used.
- culture of recombinant cells into which E2 polynucleotide has been introduced culture medium used for the culture, and separation and purification of E2 polypeptide are the same as those described above for "enzyme comprising E1 polypeptide using recombinant cells having E1 polynucleotide. It can be carried out in the same manner as “manufacturing”.
- the reaction employed in the sixth step can be performed under the same conditions as in the third step.
- the recombinant cells are inoculated in a medium containing 0.001 to 1% by weight of tetrahydrodaidzein, preferably 0.01 to 0.5% by weight, more preferably 0.01 to 0.1% by weight, It is carried out by incubating for 7 to 30 hours, preferably 15 to 24 hours, and more preferably 17 to 20 hours under conditions where growth is possible.
- the growth temperature condition is not particularly limited, and the growth can be performed at the same temperature as in the “third step”.
- the origin of tetrahydrodaidzein used as a substrate in the sixth step is not limited.
- tetrahydrodaidzein produced from dihydrodaidzein in the fifth step may be used as a substrate in the sixth step.
- the tetrahydrodaidzein may be a solution containing tetrahydrodaidzein produced in the fifth step, or may be a crude product or a purified product.
- tetrahydrodaidzein may be used, or tetrahydrodaidzein synthesized appropriately may be used.
- a recombinant cell having an E3 polynucleotide and an equol synthetic raw material composition containing tetrahydrodaidzein may be used as a mixed raw material for synthesizing equol in the sixth step.
- tetrahydrodaidzein in the synthetic raw material composition can be converted to equol. Concentrations of the recombinant cells and tetrahydrodaidzein in the synthetic raw material composition, other components that can be blended in the synthetic raw material composition, and reaction conditions are described in the same manner as described above.
- tetrahydrodaidzein produced from dihydrodaidzein in the fifth step is used as a substrate in the fifth step, and commercially available tetrahydrodaidzein, the synthetic raw material composition, or the like may be used in combination.
- Recombinant cells used in the sixth step are the C-2.
- the E3 polynucleotide described in the section is not limited as long as the E3 polynucleotide can be expressed. Specifically, the C-4. Recombinant cells as described in the section can be used.
- the culture of recombinant cells into which E3 polynucleotide has been introduced the culture medium used for the culture, and the separation and purification of E3 polypeptide are the same as those described above for "enzyme comprising E1 polypeptide using recombinant cells having E1 polynucleotide. It can be carried out in the same manner as “manufacturing”.
- E1-E3 polynucleotide In the fourth production method of the present invention, at least one of E1-E3 polynucleotides is used. Each of these polynucleotides is A-2. , B-2. And C-2. As described in the section.
- a product containing dihydrodaidzein, tetrahydrodaidzein and / or equol produced by the fourth production method The present invention relates to a product containing dihydrodaidzein, tetrahydrodaidzein and / or equol produced by the fourth production method. I will provide a.
- dihydrodaidzein, tetrahydrodaidzein and / or equol can be produced by various combinations of the fourth to sixth steps.
- the product produced by the fourth production method of the present invention contains dihydrodaidzein, tetrahydrodaidzein and / or equol.
- the product of the present invention may be a solution obtained by the fourth production method, or may be a product obtained by roughly purifying dihydrodaidzein, tetrahydrodaidzein and / or equol from the solution. It may be what was done.
- the product of the present invention can be blended in foods and drinks, cosmetics, pharmaceuticals, etc., and can also be used as a substrate.
- An apparatus for producing dihydrodaidzein, tetrahydrodaidzein and / or equol comprising at least one reaction tank of the first reaction tank to the third reaction tank.
- the present invention provides at least one reaction of the following first reaction tank to third reaction tank.
- An apparatus for producing dihydrodaidzein, tetrahydrodaidzein and / or equol (hereinafter sometimes referred to as “first production apparatus”) provided with a tank is provided.
- reaction means 1 A reaction vessel for producing dihydrodaidzein from daidzein using the polypeptide, wherein the reaction means is arranged to contact daidzein in the reaction vessel; (Second reaction tank) Reaction means (hereinafter referred to as “reaction means 2”) to which an enzyme comprising a polypeptide (ie, E2 polypeptide) which is any one of (Ba) to (Bc) is immobilized.
- reaction vessel for producing tetrahydrodaidzein from dihydrodaidzein using the polypeptide, wherein the reaction means is arranged to contact the dihydrodaidzein in the reaction vessel.
- reaction means 3 an enzyme comprising a polypeptide (ie, E3 polypeptide) which is any one of (Ca) to (Cc) is immobilized (hereinafter referred to as “reaction means 3”)
- reaction means 3 A reaction vessel for producing equol from tetrahydrodaidzein using the polypeptide, wherein the reaction means is arranged to contact the tetrahydrodaidzein in the reaction vessel .
- dihydrodaidzein, tetrahydrodaidzein and / or equol can be produced according to various combinations of the first reaction tank to the third reaction tank.
- the first production apparatus of the present invention includes at least one reaction tank among the first reaction tank, the second reaction tank, and the third reaction tank.
- the first production apparatus of the present invention includes the first reaction tank and does not include the second reaction tank and the third reaction tank
- the daidzein to dihydro Daidzein can be produced.
- the first production apparatus of the present invention includes the second reaction tank and does not include the first reaction tank and the third reaction tank
- the dihydrodaidzein Tetrahydrodaidzein can be produced.
- the first production apparatus of the present invention includes the third reaction tank and does not include the first reaction tank and the second reaction tank
- the tetrahydrodaidzein Equol can be manufactured.
- the first production apparatus of the present invention includes two reaction tanks, ie, a first reaction tank and a second reaction tank, and does not include a third reaction tank
- the first production apparatus of the present invention is used.
- dihydrodaidzein can be produced from daidzein
- tetrahydrodaidzein can be produced from dihydrodaidzein.
- the first manufacturing apparatus of the present invention when the first manufacturing apparatus of the present invention includes two reaction tanks, ie, the second reaction tank and the third reaction tank, and does not include the first reaction tank, the first manufacturing apparatus of the present invention is used. By doing so, tetrahydrodaidzein can be produced from dihydrodaidzein, and equol can be produced from tetrahydrodaidzein.
- the first production apparatus of the present invention includes three reaction tanks of the first reaction tank to the third reaction tank, dihydrodaidzein is converted from daidzein by using the first production apparatus of the present invention.
- Tetrahydrodaidzein can be produced from dihydrodaidzein, and equol can be produced from tetrahydrodaidzein.
- At least two means of the reaction means 1 to 3 may be present in one reaction tank.
- each reaction in the first reaction tank and the second reaction tank can be carried out in one reaction tank.
- each reaction in the first reaction tank to the third reaction tank can be carried out in one reaction tank.
- the arrangement means in the reaction tank is not limited.
- reaction layers are supplied. Connected through means.
- the first production apparatus of the present invention includes a first reaction tank and a second reaction tank, does not include a third reaction tank, and the first reaction tank and the second reaction tank are independent from each other.
- the reaction means 1 is arranged in the tank and the reaction means 2 is arranged in the second reaction tank
- the first reaction tank and the second reaction tank contain dihydrodaidzein produced in the first reaction tank. It connects via the supply means for supplying the product to be supplied to a 2nd reaction tank.
- the first production apparatus of the present invention includes the reaction means 1 and 2 in one reaction tank and the reaction means 3 in another reaction tank, the reaction in which the reaction means 1 and 2 are arranged.
- the reaction tank in which the tank and the reaction means 3 are arranged is connected via a supply means for supplying the product produced in the reaction reaction tank in which the reaction means 1 and 2 are arranged to the reaction tank in which the reaction means 3 is arranged. Connected.
- the product may be in the state of the obtained solution, dihydrodaidzein or the like may be roughly purified, or may be further purified.
- the shape, size, material, etc. of the first reaction tank to the third reaction tank used in the first production apparatus of the present invention can have the reaction means, and dihydrodaidzein, tetrahydrodaidzein and / or equol. As long as the production of is suitably carried out, there is no limitation.
- Reaction means In the reaction means 1 to 3 used in the first production apparatus of the present invention, an enzyme composed of the above-mentioned polypeptide of each reaction means is fixed, which is used for the production of dihydrodaidzein, tetrahydrodaidzein and / or equol. It is not limited as long as it can be suitably applied.
- the enzyme comprising each polypeptide immobilized on each reaction means may be in a crudely purified state or may be purified. Immobilization of the polypeptide enzyme in the reaction means is performed according to a conventionally known technique.
- the carrier when an enzyme comprising a polypeptide is immobilized on a carrier, the carrier is not limited as long as it does not interfere with the desired activity of the enzyme comprising each polypeptide.
- the carrier has a functional group such as an amino group, a carboxyl group or a hydroxyl group that can be covalently bound to the enzyme comprising the polypeptide, or can be connected to the enzyme comprising the polypeptide via a linker. Etc. are exemplified.
- the shape of the carrier is not limited. Such a carrier, a functional group, a linker and the like are appropriately selected according to a conventionally known technique for immobilizing an enzyme comprising a polypeptide to the carrier. Also, immobilization of an enzyme comprising a polypeptide on a carrier is performed according to a conventionally known technique.
- the supply means used in the first production apparatus of the present invention can connect different reaction vessels that can be used in the first production apparatus of the present invention via the supply means, and the first production of the present invention.
- the supply means is appropriately selected according to a conventionally known technique.
- E1-E3 polypeptide The E1-E3 polypeptide used in the first production apparatus of the present invention is described in the same manner as described above.
- the enzyme consisting of the E1 polypeptide immobilized on the reaction means 1 is allowed to act on daidzein in the presence of NADPH and / or NADH, whereby Convert to daidzein.
- the enzyme composed of the E1 polypeptide may be fixed to the reaction means together with NADPH and / or NADH that acts as a coenzyme for the enzyme composed of the E1 polypeptide.
- the reaction in the first reaction tank is carried out based on the aforementioned “first step”.
- the enzyme comprising the E2 polypeptide immobilized on the reaction means 2 is allowed to act on dihydrodaidzein in the presence of NADPH and / or NADH, thereby producing dihydrodaidzein. Is converted to tetrahydrodaidzein.
- the enzyme composed of the E2 polypeptide may be fixed to the reaction means together with NADPH and / or NADH that acts as a coenzyme for the enzyme composed of the E2 polypeptide.
- the reaction in the second reaction tank is carried out based on the aforementioned “second step”.
- tetrahydrodaidzein is converted to equol by allowing the enzyme comprising the E3 polypeptide immobilized on the reaction means 3 to act on tetrahydrodaidzein.
- the reaction in the third reaction tank is carried out based on the aforementioned “third step”.
- An apparatus for producing dihydrodaidzein, tetrahydrodaidzein and / or equol comprising at least one reaction tank from the fourth reaction tank to the sixth reaction tank.
- An apparatus for producing dihydrodaidzein, tetrahydrodaidzein and / or equol (hereinafter also referred to as “second production apparatus”) provided with a tank is provided.
- reaction means in which a recombinant cell having a polynucleotide (ie, E1 polynucleotide) of any one of (Ad) to (Af) is immobilized (hereinafter referred to as reaction means 4)
- reaction means A reaction vessel for producing dihydrodaidzein from daidzein using the reaction means, wherein the reaction means is arranged to be in contact with daidzein in the reaction vessel.
- reaction means 5 Reaction means in which a recombinant cell having a polynucleotide (ie, E2 polynucleotide) (Bd) to (Bf) is immobilized (hereinafter referred to as reaction means 5)
- reaction means 5 A reaction vessel for producing tetrahydrodaidzein from dihydrodaidzein using the reaction means, wherein the reaction means is arranged in contact with the dihydrodaidzein in the reaction vessel.
- reaction means 6 a recombinant cell having a polynucleotide (ie, E3 polynucleotide) (Cd) to (Cf) is fixed (hereinafter referred to as reaction means 6)
- reaction means 6 A reaction vessel for producing equol from tetrahydrodaidzein using the reaction means, wherein the reaction means is arranged in contact with the tetrahydrodaidzein in the reaction vessel. Yes.
- dihydrodaidzein, tetrahydrodaidzein and / or equol can be produced according to various combinations of the fourth reaction tank to the sixth reaction tank.
- the second production apparatus of the present invention includes at least one reaction tank among the fourth reaction tank, the fifth reaction tank, and the sixth reaction tank.
- the second production apparatus of the present invention includes the fourth reaction tank and does not include the fifth reaction tank and the sixth reaction tank
- the daidzein to dihydro Daidzein can be produced.
- the second production apparatus of the present invention includes the fifth reaction tank and does not include the fourth reaction tank and the sixth reaction tank, by using the second production apparatus of the present invention, from the dihydrodaidzein Tetrahydrodaidzein can be produced.
- the second production apparatus of the present invention includes the sixth reaction tank and does not include the fourth reaction tank and the fifth reaction tank, by using the second production apparatus of the present invention, the tetrahydrodaidzein Equol can be manufactured.
- the 2nd manufacturing apparatus of this invention when the 2nd manufacturing apparatus of this invention is equipped with two reaction tanks of the 4th reaction tank and the 5th reaction tank, and is not equipped with the 6th reaction tank, the 2nd manufacturing apparatus of this invention is used.
- dihydrodaidzein can be produced from daidzein
- tetrahydrodaidzein can be produced from dihydrodaidzein.
- the 2nd manufacturing apparatus of this invention when the 2nd manufacturing apparatus of this invention is equipped with two reaction tanks of the 5th reaction tank and the 6th reaction tank, and is not equipped with the 4th reaction tank, the 2nd manufacturing apparatus of this invention is used. By doing so, tetrahydrodaidzein can be produced from dihydrodaidzein, and equol can be produced from tetrahydrodaidzein.
- dihydrodaidzein is converted from daidzein by using the second production apparatus of the present invention.
- Tetrahydrodaidzein can be produced from dihydrodaidzein, and equol can be produced from tetrahydrodaidzein.
- At least two means of the reaction means 4 to 6 may be present in one reaction tank.
- each reaction in the fourth reaction tank and the fifth reaction tank can be carried out in one reaction tank.
- each reaction in the fourth reaction tank to the sixth reaction tank can be carried out in one reaction tank.
- the arrangement means in the reaction tank is not limited.
- the reaction when the production apparatus has at least two means of reaction means 4 to 6 and has a plurality of different reaction vessels, the reaction is the different reaction. It is connected to the tank via a supply means.
- the second production apparatus of the present invention includes a fourth reaction tank and a fifth reaction tank, does not include a sixth reaction tank, and the fourth reaction tank and the fifth reaction tank are independent from each other, and the fourth reaction is performed.
- the reaction means 4 is arranged in the tank and the reaction means 5 is arranged in the fifth reaction tank
- the fourth reaction tank and the fifth reaction tank contain dihydrodaidzein produced in the fourth reaction tank. It connects via the supply means for supplying the product to be supplied to a 5th reaction tank.
- the second production apparatus of the present invention includes the reaction means 4 and 5 in one reaction tank and the reaction means 6 in another reaction tank, the reaction in which the reaction means 4 and 5 are arranged.
- the reaction tank in which the tank and the reaction means 6 are arranged is provided via a supply means for supplying the product produced in the reaction reaction tank in which the reaction means 4 and 5 are arranged to the reaction tank in which the reaction means 6 is arranged. Connected.
- the product may be in the state of the obtained solution, dihydrodaidzein or the like may be roughly purified, or may be further purified.
- Reaction tank The shape, size, material, and the like of the fourth to sixth reaction tanks used in the second production apparatus of the present invention can have the reaction means, and dihydrodaidzein, tetrahydrodaidzein and / or equol. As long as the production of is suitably carried out, there is no limitation.
- reaction means 4 to 6 used in the second production apparatus of the present invention have the above-mentioned recombinant cells fixed in each reaction means, and are suitable for production of dihydrodaidzein, tetrahydrodaidzein and / or equol. There are no restrictions as far as applicable.
- Immobilization of the recombinant cells in the reaction means is not limited as long as it does not interfere with the desired effect of each recombinant cell, and is performed according to a conventionally known technique.
- the recombinant cell may be in a state where the cell can be cultured or cultured in the reaction means.
- the culture conditions for the recombinant cells applied here are set based on the descriptions of the above-mentioned “fourth step”, “fifth step”, “sixth step” and the like.
- the supply means used in the second production apparatus of the present invention can connect different reaction vessels that can be used in the second production apparatus of the present invention via the supply means, and the second production of the present invention.
- the supply means is appropriately selected according to a conventionally known technique.
- Recombinant cells used in the second production apparatus of the present invention are the above-mentioned “recombinant cells used in the fourth step", “recombinant cells used in the fifth step” and “6th. It is explained in the same manner as the description of “recombinant cells used in the process” and the like.
- E1-E3 polynucleotide The E1-E3 polynucleotide used in the second production apparatus of the present invention is described in the same manner as described above.
- daidzein is converted to dihydrodaidzein using recombinant cells having E1 polynucleotide immobilized on the reaction means 4.
- the reaction in the fourth reaction tank is performed based on the description of the above-mentioned “fourth step” and the like.
- An apparatus for producing dihydrodaidzein, tetrahydrodaidzein and / or equol comprising at least one reaction tank of the first reaction tank to the third reaction tank, and at least one reaction tank of the fourth reaction tank to the sixth reaction tank.
- a device for producing dihydrodaidzein, tetrahydrodaidzein and / or equol, comprising at least one reaction tank of the first to third reaction tanks and at least one reaction tank of the fourth to sixth reaction tanks (Hereinafter also referred to as “third manufacturing apparatus”).
- each reaction tank and each reaction means in the third manufacturing apparatus will be described in the same manner as in the first and second manufacturing apparatuses described above.
- the reaction vessel, reaction means, polynucleotide, recombinant cell, and reaction in each reaction vessel used in the third production apparatus are also described in the same manner as in the first and second production apparatuses described above.
- Example A1 Lactococcus 20-92 (FERM BP-10036) is inoculated into daidzein-containing growth medium (daidzein added to modified GAM bouillon medium (Nissui Pharmaceutical Co., Ltd.) in an amount of 10 ⁇ g / mL)
- daidzein-containing growth medium daidzein added to modified GAM bouillon medium (Nissui Pharmaceutical Co., Ltd.) in an amount of 10 ⁇ g / mL
- the cells were appropriately cultured at 37 ° C. for 7 to 18 hours under anaerobic conditions (using BBL Gas Pack systems). After culturing, the cells were collected by centrifugation and stored frozen and used in the following examples.
- Example A1 Confirmation of presence of dihydrodaidzein biosynthetic activity in centrifuge supernatant of cell disruption and confirmation of NADPH dependence Refrigerated frozen cells (67 mL, 2 tubes) after thawing 8,000 rpm, 4 Centrifugation was carried out at 10 ° C. for 10 minutes, and the sediment was subjected to the following test.
- the sediment is suspended in 2 mL of 0.1 M potassium phosphate solution containing 1 mM PMSF (Wako Pure Chemical Industries, Ltd.), 2 mM DTT (Wako Pure Chemical Industries, Ltd.) and 5 mM Sodium hydrosulfite (Wako Pure Chemical Industries, Ltd.). Made cloudy.
- the suspension was transferred to two 2 ml tubes with screw caps (manufactured by Assist Co., Ltd.) containing 0.1 mm zirconia / silica beads (BioSpec Products, Inc.) in advance, and FastPrep (registered trademark) FP100A (Thermo ELECTRON CORPORATION ) Was crushed (6500 rpm, 10 seconds, ice-cooled 8 times) to obtain a microbial cell disruption solution. The obtained cell disruption solution is centrifuged at about 10,000 rpm at 4 ° C.
- centrifugal supernatant is 0.1 M potassium phosphate solution containing 1 mM PMSF, 2 mM DTT, and 5 mM Sodium hydrosulfite.
- 0.1 M potassium phosphate solution containing 1 mM PMSF, 2 mM DTT, and 5 mM Sodium hydrosulfite.
- An enzyme reaction solution having the following composition was prepared and incubated at 37 ° C. for 2 hours. After incubation, 3 mL of ethyl acetate was added to the resulting enzyme reaction product for extraction treatment, and then dried to be subjected to HPLC analysis.
- a standard solution for HPLC analysis a mixed solution (2 ⁇ g / mL each) of daidzein (Funakoshi Co., Ltd.), equol (Funakoshi Co., Ltd.), dihydrodaidzein (Trend Research Chemical Co., Ltd.) was used.
- Enzyme reaction solution composition was used as a standard solution for HPLC analysis.
- Example A2 Purification of dihydrodaidzein synthase
- 8 mM DTT 0.1-dithiothreitol, Merck
- 0.1 mM potassium phosphate buffer pH 7 containing 5 mM Sodium hydrosulfite (Wako Pure Chemical Industries, Ltd.)
- protease inhibitors Complete protease inhibitor cocktail EDTA-free, Roche Diagnostics
- the suspension was transferred to 3 tubes with 2 ml screw caps (manufactured by Assist Co., Ltd.) containing 0.1 mm zirconia / silica beads (BioSpec Products, Inc.), and FastPrep (registered trademark) FP100A (Thermo ELECTRON CORPORATION)
- the cells were crushed (6500 rpm, 20 seconds 4 times), and the crushed solution was centrifuged to obtain a supernatant.
- 8 Lactococcus 20-92 strains cultured in 200 ml of the same liquid medium per culture bottle for 18 hours were similarly disrupted and centrifuged to obtain 8 supernatants.
- Buffer A 0.1 mM potassium phosphate buffer pH 7
- PMSF phenylmethylsulfonyl fluoride
- DTT 2 mM DTT
- 5 mM Sodium hydrosulfite was used.
- Buffer A 0.1 mM potassium phosphate buffer pH 7
- PMSF phenylmethylsulfonyl fluoride
- DTT 2 mM DTT
- 5 mM Sodium hydrosulfite was used.
- Buffer A containing an equal volume of 2M ammonium sulfate, pack Butyl® Sepharose 4F Fast® Flow (about 0.3% gel per gel, GE Healthcare), and add 1M M ammonium sulfate.
- the sample was applied to a microbiospin column (11 tubes, Bio-Rad® Laboratories) equilibrated with Buffer A.
- the obtained eluate was subjected to HPLC using a Mono Q column (GE Healthcare) equilibrated with eluent C having a pH of 7.5.
- the eluent C was fed to the column at a flow rate of 0.1 ml / min, and after washing, a program for linearly changing to 0.65 M NaCl in 32.5 minutes was executed by changing the mixing ratio of eluent C and eluent D.
- the HPLC conditions are shown below. Absorption at 280 nm was measured as protein absorption.
- FIG. 2 shows the results of Mono Q HPLC performed in this way and the enzyme activity.
- FIG. 3 shows the results of SDS-PAGE of fraction Nos. 27 to 31 under reducing conditions. As a result, a 70 kDa band was observed in the fraction having dihydrodaidzein synthesis activity under reducing conditions.
- Example A3 Determination of N-terminal amino acid sequence 30 ⁇ l of 0.1% trifluoroacetic acid (TFA) was added to 70 ⁇ l of the dihydrodaidzein synthetic activity fraction No. 29 obtained by Mono Q HPLC to make 100 ⁇ l.
- TFA trifluoroacetic acid
- the PVDF membrane of the ProSorb cartridge (Applied Biosystems Japan) was moistened with 10 ⁇ l of methanol, and then the previously mixed solution was added. After absorbing water using a ProSorb filter (Applied Biosystems Japan), the membrane was dried, and the membrane was cut using a membrane punch (Applied Biosystems Japan). The membrane was washed 5 times with 20% aqueous methanol and dried. The membrane was subjected to N-terminal amino acid sequence analysis using a protein sequencer (Applied Biosystems, Procise 494cLC), and the following 22-residue continuous amino acid sequence was obtained. Met Lys Asn Lys Phe Tyr Pro Lys Thr Phe Glu Arg Gly Tyr Ile Gly Asn Leu Glu Val Glu Asn (SEQ ID NO: 19)
- Example A4 Determination of Internal Amino Acid Sequence An enzyme protein showing dihydrodaidzein synthesis activity was fragmented with a digestive enzyme to obtain a peptide. N-terminal amino acid sequence analysis of the peptide was performed to obtain internal amino acid sequence information.
- Buffer A potassium phosphate buffer pH 7
- SFF phenylmethylsulfonyl fluoride
- DTT phenylmethylsulfonyl fluoride
- 5 mM Sodium hydrosulfite 0.1 M potassium phosphate buffer pH 7
- Buffer A containing 1M ammonium sulfate
- Buffer A containing 0.5M ammonium sulfate
- 0.75 ml of Buffer A was added twice to elute a fraction having dihydrodaidzein synthesis activity.
- One microbiospin column packed with 2′5′ADP Sepharose 4B was equilibrated with Buffer A, and 1.5 ml of the previously eluted solution was added. After washing 5 times with 0.75 ml of Buffer A, elution was performed with 0.75 ml of Buffer A containing 20 mM NADPH and then 0.45 ml. The eluate was mixed and desalted and concentrated to 5 ⁇ l in a microconcentration tube (NANOSEP 10K OMEGA, Pall Life Sciences).
- the collected liquid was mixed using a double concentration SDS-PAGE sample buffer containing 2-mercaptoethanol and the mixture was heated at 90 ° C. for 7 minutes, and then subjected to SDS-PAGE according to the Laemmuli method.
- Supersep® HG 10-20% (Wako Pure Chemical Industries, Ltd.) was used as the gel plate for electrophoresis. After staining with Colloidal Blue (Invitrogen) and decolorizing with milliQ water, a band migrated to about 70 kDa was excised.
- a gel piece having the same molecular weight position and size was cut out from the portion where the protein was not migrated, and the same treatment was performed thereafter.
- the cells were treated in the same manner, and N-terminal amino acid sequence analysis was performed using the band at the same position transferred to the PVDF membrane after SDS-PAGE, and the sequence was the same as that obtained with Mono Q Q HPLC fraction No. 29 Confirmed to do.
- the centrifuged supernatant was transferred to a separate tube, 60% acetonitrile-0.1% TFA aqueous solution was added to the gel, heated at 30 ° C for 20 minutes, and then vortexed for 10 minutes three times to extract the fragmented peptide.
- the collected supernatant was filtered using Ultrafree-MC (0.22 ⁇ m, Amicon), and then concentrated by centrifugation.
- Example A5 Amplification of dihydrodaidzein synthase gene from N-terminal and partial amino acid sequence of purified polypeptide
- a degenerative-primer was designed and prepared based on the N-terminal and partial amino acid sequences obtained in Examples A3 and A4 above. An attempt was made to amplify the gene encoding dihydrodaidzein synthase by performing Degenerative-PCR using the genomic DNA of Lactococcus 20-92 as a template.
- the mixture was centrifuged at 5000 rpm, 4 ° C. for 10 minutes, and the supernatant was passed through a QIAGEN-Genomic-tip-500 / G (Qiagen) column equilibrated with a QBT solution to adsorb the genomic DNA to the column. After washing the column twice with 30 mL of QC solution, genomic DNA was eluted from the column with 15 mL of QF, and 10.5 mL of isopropanol was added for salting out.
- QIAGEN-Genomic-tip-500 / G Qiagen
- genomic DNA precipitated in a filament shape was taken into a 1.5 mL microtube, washed with 75% ethanol, air-dried, and then dissolved in 250 ⁇ L of TE solution (0.4 ⁇ g / ⁇ L). The concentration of the genomic DNA solution thus obtained was measured, adjusted to 40 ng / ⁇ L with a TE solution, and used as a PCR template.
- the 5'-side sequence is the Codon Usage Database (Codon Usage Database http: //www.kazusa.or Based on .jp / codon /), the most frequently occurring codon was adopted for each amino acid.
- the 3 'side was devised so as not to cause mismatch with the dihydrodaidzein synthase gene by using a mixed base.
- E1-N-terminal-31 TGAAGAATAANTTNTAYCCNAARACNTTYGA (SEQ ID NO: 23) (However, in SEQ ID NO: 23, “N” at positions 11 and 14 represents inosine, and “N” at positions 20 and 26 represents adenine, guanine, cytosine or thymine.)
- E1-N-terminal-37 TGAAGAATAANTTNTAYCCNAARACNTTYGARRGNGG (SEQ ID NO: 24) (However, in SEQ ID NO: 24, “N” at positions 11, 14, 20, and 26 represents inosine, and “N” at position 35 represents adenine, guanine, cytosine, or thymine.)
- E1-N-terminal-F32 ATGAAGAATAAGTTTTAYCCNAA
- Degenerative-primers were designed and prepared based on the internal sequence Peptide 2: ASRMVMDAVHEGYIAG determined in Example A4, and used for Degenerative-PCR.
- E1-internal-RP1 CCTGCAATATAACCTTCATGTACNGCRTCCATNACCAT (SEQ ID NO: 26) (However, in SEQ ID NO: 26, “N” at positions 24 and 33 represents adenine, guanine, cytosine or thymine.)
- oligo DNAs used as primers in this example were prepared by Sigma Aldrich Japan.
- the agarose electrophoresis in this example was performed by staining with ethidium bromide, and ⁇ / StyI (Nippon Gene Co., Ltd.) and 100 bp ladder (Toyobo Co., Ltd.) were used as molecular weight markers.
- FIG. 5 shows the result of electrophoresis of the Degenerative-PCR product.
- the annealing temperature was 54 ° C that was most amplified.
- the amplified DNA fragment of about 1.9 kb (annealing temperature 54 ° C.) was excised from the agarose gel and purified using Gel-Extraction kit (Qiagen). The purified DNA fragment was inserted into pT7-Blue Cloning Vector (Novagen), and the nucleotide sequence was determined.
- the obtained DNA base sequence was analyzed using a DNA sequence-assembly software SEQUENCHER (Gene Codes Inc, USA). As a result, base sequences corresponding to the amino acid sequences of Peptide 1 and 3 determined in Example A4 were present.
- Example A6 Determination of the total base sequence of the dihydrodaidzein synthase gene The sequences of the 5 ′ end and 3 ′ end of the 1.9 kb dihydrodaidzein synthase gene obtained in Example A5 are determined, and the total base sequence is determined. Therefore, rapid amplification of cDNA end sequences (5′-, 3′-RACE (rapid amplification of cDNA ends)) was performed using the genomic DNA library of the Lactococcus 20-92 strain as a template.
- genomic DNA library The genomic DNA of the Lactococcus 20-92 strain purified in Example A5 was used as a restriction enzyme (BamHI, EcoRI, HindIII, KpnI, PstI, SacI, SalI, Sau3AI, XhoI) (all Takara Bio) The product was fragmented by digestion at 37 ° C. for 16 hours, and treated with phenol / chloroform and purified by ethanol precipitation.
- TaKaRa Ligation kit var.2.1 (Takara Bio Inc.) into the pUC19 cloning vector, which was previously digested with the corresponding restriction enzyme and dephosphorylated by Shrimp Alkaline Phosphatase (Takara Bio Inc.) was used for ligation to prepare a genomic DNA library.
- oligo DNAs used as primers in this example were prepared by Sigma Aldrich Japan.
- the agarose electrophoresis in this example was performed by staining with ethidium bromide (Nippon Gene Co., Ltd.), and ⁇ / StyI (Nippon Gene Co., Ltd.) and 100 bp ladder (Toyobo Co., Ltd.) were used as molecular weight markers.
- the amplified DNA fragment was excised from an agarose gel and purified using Gel-Extraction kit (Qiagen).
- the base sequence of the purified DNA fragment was determined by direct sequencing using the primers used for amplification.
- the DNA fragment of about 1.2 kb (SacI), about 1.0 kb (Sau3AI) and about 0.6 kb (SacI) of 3′-RACE was the sequence of the dihydrodaicein synthase gene in 5′-RACE. .
- E1-conf-NP TGCCGGTGCAATGGCTGACATCATGTTCAACCTG (SEQ ID NO: 35)
- E1-conf-CP TCCTCCATCGTTCCTCCAATCAGTAAGACACGCG (SEQ ID NO: 36)
- the obtained DNA fragment was purified using Gel-Extraction kit (Qiagen), and the sequence was confirmed and final determination was performed by direct sequencing.
- Example A7 Induction of expression of dihydrodaidzein synthase gene by addition of daidzein into the growth medium
- daidzein as a substrate was added to the culture medium for growth of Lactococcus 20-92 strain
- the cultured cells have dihydrodaidzein synthesis activity. From this, it is speculated that the addition of daidzein to the growth culture medium induces the transcription of the dihydrodaidzein synthase gene, which is further translated into protein.
- the medium with and without daidzein added Whether or not the expression of dihydrodaidzein synthase gene is induced by the addition of daidzein to the growth culture by preparing cDNA derived from Lactococcus 20-92 cells cultured in Was examined.
- Lactococcus 20-92 strain sealed at 4 ° C was anaerobically cultured at 37 ° C for 8 hours in modified GAM bouillon medium with or without daidzein (Funakoshi Co., Ltd.) 10mg / L.
- a 25 mL tube was transferred to a 50 mL tube, centrifuged at 3500 rpm, 4 ° C. for 10 minutes, the medium was removed by decantation, and the cells were collected.
- the collected cells were quickly frozen in liquid nitrogen, and then total RNA was extracted and purified using 1 mL of TRIzol solution (Invitrogen) according to the manual.
- the purified total RNA was treated with DNase I (Invitrogen) to remove contaminating genomic DNA and used for first-strand cDNA synthesis.
- the final reaction solutions prepared above are the following four types. 1. Reverse transcript of whole cell-derived RNA cultured in daidzein-supplemented medium (DZN (+) RT (+)) 2. Reverse transcript of total RNA derived from cells cultured in daidzein-free medium (DZN (-) RT (+)) 3. Non-reverse transcript (DZN (+) RT (-)) of total RNA derived from cells cultured in daidzein-added medium 4). Total RNA irreversible transcript (DZN (-) RT (-)) derived from bacterial cells cultured in daidzein-free medium
- RT-PCR amplification conditions and the primer sequences used for each gene amplification are shown below.
- Ex-Taq DNA polymerase (Takara Bio Inc.) is used and the amplification program: 95 ° C. for 2 min, (95 ° C. for 30 sec, 56 ° C. for 20 sec, 72 ° C. for 30 sec) ⁇ 30 cycles, 72 ° C. for 2 min.
- As the template 1 ⁇ L of the final reaction solution in (2) of this example was used.
- E1-FP CTACATCGGTAACCTAGAGGTCG
- E1-RP CCGTGCTGCTTGATGGTCTTTGC
- 16S-ribosomal RNA sequence 326 bp
- Gar-16S-Ribo-FP TGCGTAGATATATGGAGGAAC
- Gar-16S-Ribo-RP CTTATCTCTAAGGATAGCACG
- the primers used for the amplification of the 16S-ribosomal RNA sequence are base sequence databases provided by the National Center for Biotechnology Information (National Center for Biotechnology Information: http://www.ncbi.nlm.nih.gov/). GenBank) was constructed based on the sequence of Lactococcus garvieae strain FLG12 16S ribosomal RNA gene and partial sequence (Accesion No. AF352163-66).
- the agarose electrophoresis in this example was performed by staining with ethidium bromide (Nippon Gene Co., Ltd.), and ⁇ / StyI (Nippon Gene Co., Ltd.) and 100 bp ladder (Toyobo Co., Ltd.) were used as molecular weight markers.
- the gene was expressed efficiently only when a reverse transcript derived from cells cultured in daidzein-added medium was used as a template. Observed. The 16S-ribosomal RNA sequence amplified as a control was observed to have the same expression regardless of whether daidzein was added to the medium. In addition, in the irreversible transcript, neither the dihydrodaidzein synthase gene nor the 16S-ribosomal RNA sequence was amplified, so that the genomic DNA of the total RNA treated with DNase I used for the synthesis of first-strand DNA was used. It was confirmed that there was no contamination. From the above, it was shown that the expression of dihydrodaidzein synthase gene mRNA was induced by the addition of daidzein to the medium.
- Example A8 Expression of recombinant E1 polypeptide using E. coli and confirmation of dihydrodaidzein synthesis activity E1 polypeptide was expressed using pET system, a recombinant protein expression system using E. coli, and its dihydrodaidzein synthesis activity It was confirmed.
- E1 polypeptide expression vector The DNA of the E1 nucleotide open reading frame region was amplified by PCR for the purpose of preparing an E1 polypeptide expression vector (pET21-E1-His).
- exp.E1 pet F Nde AGCTCATATGAAGAACAAGTTCTATCCGAA (SEQ ID NO: 41)
- exp. E1 pet His AATCGAATTCCTACAGGTTGCAGCCAGCGATGT (SEQ ID NO: 42)
- the above-mentioned amplification primers: exp.E1 pet F Nde and exp.E1 pet His contain the restriction enzyme NdeI cleavage site and EcoRI cleavage site sequences for insertion into pET21a (Novagen), respectively.
- the recovered DNA fragment was cleaved with restriction enzymes NdeI and EcoRI, and then subjected to agarose gel electrophoresis.
- the target band was excised and purified and collected by Qiagen Gel Extraction kit (Qiagen).
- the obtained DNA fragment was ligated overnight at 16 ° C using pET21a digested with NdeI and EcoRI and DNA Ligation Kit ver.2.1 (Takara Bio Inc.), and then E. coli JM109 strain (Takara Bio) using the ligation reaction solution. Co.) was transformed.
- the transformants were grown overnight at 37 ° C. on LB medium agar (GIBCO) plates containing ampicillin (50 ⁇ g / mL) to obtain colonies.
- the obtained single colony was cultured overnight in 3 mL of LB medium (GIBCO) containing ampicillin (50 ⁇ g / mL), and then plasmid DNA was extracted using an automatic plasmid extractor PI-100 (KURABO).
- the base sequence of the DNA inserted into the plasmid was sequenced by the dye terminator method to confirm that the E1 nucleotide was correctly inserted as intended, and pET21-E1-His was obtained.
- the DNA sequence in this example was performed using a DNA sequencer ABI3700 (Applied Biosystems).
- E. coli BL21 (DE3) strain using plasmid pET21-E1-His and pET21a (negative control) expressing recombinant E1 polypeptide (Novagen) was transformed.
- the transformant was grown overnight at 37 ° C. on an LB medium agar plate containing ampicillin (50 ⁇ g / mL) to obtain a single colony.
- E. coli BL21 (DE3) transformants was cultured overnight at 37 ° C. in 3 mL of liquid LB medium containing 50 ⁇ g / mL of ampicillin. Add 0.5 mL of the culture broth to 50 mL of liquid LB medium containing ampicillin at the same concentration, pre-incubate for 3 hours (until OD630nm is about 0.4), and use IPTG (isopropyl- ⁇ -thiogalactopyra to a final concentration of 1 mM. Noside) (Wako Pure Chemical Industries, Ltd.) was added, and further cultured at 37 ° C. for 4 hours.
- IPTG isopropyl- ⁇ -thiogalactopyra to a final concentration of 1 mM. Noside
- the cells were collected by centrifugation (6000 rpm 4 ° C 15 minutes) with Avanti HP25 (beckman coulter). Subsequent operations were performed on ice. After removing the centrifuge (medium), the suspension is suspended in 1 mL of 0.1 M potassium phosphate buffer pH 7.0 (KPB-PDH) containing 1 mM PMSF, 2 mM DTT, 5 mM Sodium hydrosulfite, and 0.7 mL zirconia Place in silica gel (BioSpec Products, Inc.) and KPB-PDH 400 ⁇ L in a 2mL assist tube and use FastPrep (R) (Thermo ELECTRON CORPORATION) at 6500rpm for 20 seconds-ice-cool for 2 minutes By repeating the procedure, the cells were crushed to obtain a cell lysate.
- KPB-PDH potassium phosphate buffer pH 7.0
- Figure 7 shows the results of SDS-PAGE.
- a recombinant E1 polypeptide having a molecular weight of about 70 kDa was confirmed in the cell disruption solution derived from the pET21-E1-His transformant.
- An enzyme reaction solution having the following composition was prepared and incubated at 37 ° C. for 2 hours. Enzyme reaction solution composition After the incubation, 3 mL of ethyl acetate was added to the resulting enzyme reaction solution to carry out an extraction treatment. After drying, the solution was dissolved in a moving bed (eluent). The contents of daidzein and dihydrodaidzein in the enzyme reaction solution were measured by HPLC analysis of the lysate.
- Example B Reference Example B1 Synthesis of cis-tetrahydrodaidzein and trans-tetrahydrodaidzein Cis-tetrahydrodaidzein and trans-tetrahydrodaidzein were produced according to the following flow. Hereinafter, the following abbreviations are used for the notation of compounds.
- Reference example B2 Lactococcus 20-92 strain (FERM BP-10036) was inoculated into daidzein-containing growth liquid medium and cultured at 37 ° C. for 7 to 24 hours under anaerobic conditions. After culturing, the cells were collected by centrifugation and stored frozen and used in the following examples.
- Example B1 Confirmation of Tetrahydrodaidzein Production The following test was conducted to confirm that tetrahydrodaidzein is an intermediate metabolite of equol biosynthesis.
- composition of enzyme reaction solution 0.1M potassium phosphate buffer / 1mM PMSF / 2mM DTT / 5mM sodium hydrosulfite (pH7.0) 2 mM NADPH 2 mM NADH 10 ⁇ g / mL dihydrodaidzein or tetrahydrodaidzein
- FIG. 9 shows the results of HPLC analysis of the enzyme reaction product obtained by using dihydrodaidzein as a substrate and using the disrupted bacteria as an enzyme source. Moreover, the HPLC analysis of the tetrahydrodaidzein synthesized in Reference Example 1 is also shown in FIG. From this result, the presence of an intermediate having a retention time corresponding to the retention time of trans-tetrahydrodaidzein is recognized in the enzyme reaction product obtained by using dihydrodaidzein as a substrate and using a microbial disruption as an enzyme source. It was clarified that trans-tetrahydrodaidzein was produced.
- Example B2 Confirmation of presence of tetrahydrodaidzein biosynthetic activity in centrifuge supernatant of disrupted cells and confirmation of NADH or NADPH dependency After thawing frozen cells stored at 5000 ⁇ G, 4 ° C., 15 minutes Centrifugation was performed and the sediment was subjected to the following test.
- the sediment (wet weight 2.7 g) is suspended in 10 ml of 0.1 M monopotassium phosphate solution containing 1 mM PMSF and 5 mM Sodium hydrosulfite, pre-warmed at 37 ° C for 5 minutes, and then Lysozyme is added per 1 g of wet weight.
- the mixture was added to 100 mg / wet weight and reacted at 37 ° C. for 1.5 hours.
- the obtained cell disruption solution was centrifuged at about 10,000 ⁇ G for 15 minutes to obtain a centrifugal supernatant, which was used as an enzyme source.
- An enzyme reaction solution having the following composition was prepared and incubated at 37 ° C. for 2 hours. After incubation, 5 mL of ethyl acetate was added to the resulting enzyme reaction product for extraction treatment, and then dried and subjected to HPLC analysis.
- Example B3 Purification of tetrahydrodaidzein synthase
- Ten cells of Lactococcus 20-92 strain cultured for 20 hours in 67 ml of daidzein-containing growth medium per culture bottle were centrifuged and 1 mM PMSF (phenylmethylsulfonyl fluoride) and 4 Suspend in 0.02 M potassium phosphate buffer pH 7 (hereinafter referred to as “Buffer A”) containing mM DTT (Dithiothreitol), and disrupt the cells with a French press (SLM INSTRUMENTS INC) (1800 psi 6 times), the crushed liquid was centrifuged to obtain a supernatant.
- PMSF phenylmethylsulfonyl fluoride
- Fractions Nos. 1 to 5 having tetrahydrodaidzein biosynthetic activity were ultrafiltered and concentrated using Amicon® Ultra® Centrifugal® UFC801024® (MW Cut: 10,000) to obtain about 2.1 ⁇ m of concentrated solution.
- the concentrated solution was divided into 3 parts and mixed with Buffer A containing an equal amount of 3M ammonium sulfate, and then subjected to HPLC using TSKgel Phenyl-5PW (Tosoh Corporation). The HPLC conditions are shown below. Absorption at 280 nm was measured as protein absorption.
- FIG. 13 shows the results of SDS-PAGE of each fraction under reducing conditions.
- 28 kDa and 32 kDa bands were observed in the fraction No. 7, which is the main fraction showing tetrahydrodaidzein synthesis activity, under reducing conditions.
- Example B4 Analysis of amino acid sequence of tetrahydrodaidzein synthase MS analysis was performed using the fraction (fraction No. 7) having tetrahydrodaidzein synthesizing activity obtained in Example 3 above as a sample. Specifically, the sample was separated by SDS-PAGE, and the band was cut out. The excised band was reductively alkylated in gel and digested in gel with trypsin. Subsequently, the trypsin digested peptide was collected and purified, and analyzed by LC-MS. The amino acid sequence of the peptide was calculated and estimated by performing de novo sequencing on the data acquired by LC-MS analysis using PEAKS TM (Infocom Corp.), which is MS analysis support software. The detailed method is as follows.
- the solution was discarded, and the gel pieces were sequentially washed with a 50% acetonitrile solution, a 100% acetonitrile solution, a 100 mM ammonium bicarbonate solution, and a 100% acetonitrile solution, and dried and solidified with a SpeedVac Concentrator.
- a small amount of trypsin solution (12.5 ⁇ g / ml in 50 mM Ammonium Bicarbonate) was added to the dried gel pieces and allowed to permeate on ice for 45 minutes. After infiltration, excess trypsin solution was removed, and 50 mM ammonium bicarbonate solution was added to the extent that the gel pieces were immersed, and the mixture was reacted at 37 ° C. for 16 hours.
- TFA was added to the digested peptide solution to adjust the pH to around 3, and the autosampler HTS-PAL was set.
- the sample set in HTS-PAL was loaded onto the sample concentration column in the LC-MS injector valve, and on-column washing was performed.
- Samples of the concentrated column were separated with an analytical column by nanoHPLC-Chorus220, ionized with FortisTip set on the analytical column, and analyzed with QSTAR Pulsar i.
- LC-MS analysis conditions are as follows.
- the amino acid sequence of the digested peptide was estimated by de novo sequencing the data obtained by LC-MS for each of the bands LG1 and LG2 (see FIG. 14) using PEAKS TM software.
- Example B5 Analysis of the genomic DNA sequence surrounding the dihydrodaidzein synthesis (E1) enzyme gene (1)
- genomic DNA of Lactococcus 20-92 strain (FERM BP-10036) purified in Example A5 is represented by the following restriction enzymes (BamHI, EcoRI, HindIII, KpnI, PstI, SacI, SalI, Sau3AI, XhoI) (both Takara Bio Inc.) were digested at 37 ° C. for 16 hours, fragmented, treated with phenol / chloroform, and purified by ethanol precipitation.
- the purified genomic DNA fragment was self-ligated using TaKaRa Ligation kit var.2.1 (Takara Bio Inc.). Each ligation solution was diluted 10-fold with sterile water to prepare a genomic DNA library for inverse-PCR.
- First-PCR is 1 ⁇ PCR Buffer (Mg 2+ free), primers 0.5nM each, dNTP 0.5mM each, MgCl 2 2.5mM, 20 ⁇ L reaction solution containing TaKaRa LA Taq 0.2U diluted with genomic DNA library 1 ⁇ L (40 ng) of the solution was used as a template, and the following amplification program was performed at 98 ° C. for 1 min, (95 ° C. 10 sec, 62 ° C. 10 sec, 68 ° C. 10 min) ⁇ 35 cycles, 68 ° C. for 15 min.
- Upstream amplification primer sequence RACE-N-P3-1 ATGGAGATAGTGCCGCTGGCAAGGCAACGGCAC (SEQ ID NO: 43)
- RACE-N-P3-2 TCAACGAAGACTCGATTTGAGCGAGAGGCGAGG
- E1-Bub-N-P1 ACGGTGGAACCGGCATCGTGTTCATGGACAAC
- E1-Bub-N-P2 GCGTGACCCAGTTCCACCATGTCGGACTGTC (SEQ ID NO: 46)
- RACE-C-P3-2 AGGATCACCCATGAGCGCATCGCTATCATGGAC (SEQ ID NO: 48)
- E1-Bub-C-P1 CATCGCTCTTGCAG
- oligo DNAs used as amplification primers in this example were synthesized by Sigma-Aldrich Japan.
- agarose electrophoresis in this example, staining with ethidium bromide (Nippon Gene Co., Ltd.) was performed, and ⁇ / StyI (Nippon Gene Co., Ltd.) and 100 bp ladder (Toyobo Co., Ltd.) were used as molecular weight markers.
- the amplified DNA fragment was excised from the agarose gel and purified using a QIAGEN Gel-Extraction kit (Qiagen). The base sequence of the purified DNA fragment was determined by direct sequencing using the primers used for amplification and the walking method.
- Example B6 Verification of digested peptide sequence and genomic sequence by LC-MS analysis Peripheral genomic DNA sequence data of the dihydrodaidzein synthesis (E1) enzyme gene determined in Example B5 above for the deduced amino acid sequence obtained in Example B4 above Matched. As a result, several sequences obtained mainly from LG2 matched the polypeptide sequence deduced from the ORF-US2 nucleotide sequence. These results suggested that the ORF-US2 polypeptide may be a tetrahydrodaidzein synthase. The digested peptide sequence consistent with the ORF-US2 polypeptide is shown below. In addition, FIGS. 16-1, 16-2, and 16-3 show data acquired by LC-MS.
- m / z is the mass-to-charge ratio
- z is the number of charges
- Mass is the mass of the peptide
- Peptide is the deduced amino acid sequence.
- Score represents the probability of sequence calculation calculated on the PEAKS software as a score value (100% is the highest value). Since isoleucine (I) and leucine (L) have the same molecular weight and are indistinguishable, both are indicated as X in the digested peptide sequence that matches the ORF-US2 polypeptide.
- Example B7 Synthesis of ORF-US2 polypeptide using cell-free protein synthesis system and confirmation of its tetrahydrodaidzein synthesis activity
- ORF-US2 polypeptide was cell-free protein synthesis system (PURESYSTEM Classic II mini (Post Genome Institute) ) And the tetrahydrodaidzein synthesis activity was confirmed.
- E2-invitroTS-FP1 ACTTTAAGAAGGAGATATACCAATGGCACAGGAAGTCAAAGTCC
- E2-invitroTS-RP CTAGACCTCGATCTCGCCCTGCATGCCG
- Universal-Primer GAAATTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCA
- E2-invitroTS-FP1 and E2-invitroTS-RP were synthesized at Sigma-Aldrich Japan based on the ORF-US2 nucleotide sequence determined in Example B5.
- the Universal-Primer used was the one attached to PURESYSTEM Classic II mini (Post Genome Research Institute).
- ORF-US2 nucleotides were amplified with the above-described primers: E2-invitroTS-FP1 and E2-invitroTS-RP in Example (1-1) using the genomic DNA of Lactococcus 20-92 as a template.
- the second-stage PCR was carried out with Universal Primer and E2-invitroTS-RP.
- 300 ⁇ L of PCR products 50 ⁇ L ⁇ 6) were purified by PCR-Purification kit (Qiagen) and used as template DNA (template DNA for ORF-US2 polypeptide synthesis) for ORF-US2 polypeptide synthesis.
- the first and second stage PCR was performed under the following conditions.
- First stage 10 pmol of each amplification primer, 2.5 pmol each of dNTP, 40 ng of genomic DNA derived from Lactococcus 20-92 strain, buffer solution for Easy-A (Stratagene), Easy-A (registered trademark), PCR-Cloning, Enzyme, 2U
- a 50 ⁇ L reaction solution containing (Stratagene) was used, and the following amplification program was performed at 95 ° C. for 2 minutes (95 ° C. for 45 seconds, 58 ° C. for 20 seconds, 72 ° C. for 1 minute) ⁇ 30 cycles, 72 ° C. for 3 minutes.
- Second stage 10 pmol each of amplification primers, 2.5 pmol each of dNTP, 0.5 ⁇ L of PCR reaction solution of the first stage, Easy-A buffer solution, Easy-A (registered trademark) High-Fidelity PCR Cloning Enzyme 2 U reaction
- the following amplification program was performed at 95 ° C. for 2 min (95 ° C. for 45 sec, 45 ° C. for 20 sec, 72 ° C. for 1 min) ⁇ 5 cycles, (95 ° C. for 45 sec, 60 ° C. for 20 sec, 72 ° C. for 1 min) ⁇ 25 cycles for 72 ° C. for 3 min.
- DHFR dihydrofolate reductase
- PURESYSTEM Classic II mini 0.5 ⁇ g of template DNA for dihydrofolate reductase (DHFR) synthesis included with PURESYSTEM Classic II mini was used (2 ⁇ L of 0.2 ⁇ g / ⁇ L).
- no template DNA was added and only sterile water was used.
- activity measurement 40 ⁇ L of the above reaction solution was added to the enzyme reaction buffer having the following composition, incubated at 37 ° C. for 6 hours. After the reaction was completed, the enzyme reaction solution was extracted with 3 mL of ethyl acetate, dried and dissolved in the electrophoresis buffer. Then, tetrahydrodaidzein in the enzyme reaction solution was measured by HPLC analysis.
- Buffer composition for enzyme reaction 0.1M potassium phosphate buffer / 1mM PMSF / 2mM DTT / 5mM sodium hydrosulfite (pH7.0) 2 mM NADPH 2 mM NADH 10 ⁇ g / mL dihydrodaidzein
- DHFR hydrofolate reductase
- ORF-US2 polypeptide has a tetrahydrodaidzein biosynthesis activity from dihydrodaidzein. That is, it can be said that the ORF-US2 polypeptide corresponds to the E2 polypeptide.
- Example B8 Expression of recombinant ORF-US2 polypeptide using E. coli and confirmation of its tetrahydrodaidzein synthesis activity
- ORF-US2 polypeptide was expressed using pET system, which is a recombinant protein expression system using E. coli. Tetrahydrodaidzein synthesis activity was confirmed.
- ORF-US2 polypeptide expression vector For the purpose of preparing an ORF-US2 polypeptide expression vector (pET21-US2), DNA of the open reading frame region of the ORF-US2 polypeptide was amplified by PCR. The following amplification primers were prepared based on the ORF-US2 polypeptide sequence determined in Example B5. exp.US2 pet F Nde: TATACATATGGCACAGGAAGTCAAAGTC (SEQ ID NO: 60) exp. US2 pet : AATCGAATTCCTAGACCTCGATCTCGCCCTGC (SEQ ID NO: 61)
- exp.US2pet FNde and exp.US2pet contain a restriction enzyme NdeI cleavage site and EcoRI cleavage site sequence for insertion into pET21a (Novagen), respectively.
- the recovered DNA fragment was cleaved with restriction enzymes NdeI and EcoRI, and then subjected to agarose gel electrophoresis.
- the target band was excised and purified and collected by Qiagen Gel Extraction kit (Qiagen).
- the obtained DNA fragment was ligated overnight at 16 ° C using pET21a digested with NdeI and EcoRI and DNA Ligation Kit ver.2.1 (Takara Bio), and then Escherichia coli JM109 strain (Takara Bio) was used using the ligation reaction solution. Transformed.
- the transformants were grown overnight at 37 ° C. on LB medium agar (GIBCO) plates containing ampicillin (50 ⁇ g / mL) to obtain colonies.
- the obtained single colony was cultured overnight in 3 mL of LB medium (GIBCO) containing ampicillin (50 ⁇ g / mL), and then plasmid DNA was extracted using an automatic plasmid extractor PI-100 (KURABO).
- the nucleotide sequence of the DNA inserted into the plasmid was sequenced by the dye terminator method, and it was confirmed that the ORF-US2 polynucleotide was inserted correctly as intended.
- the DNA sequence in this example was performed using a DNA sequencer ABI3700 (Applied Biosystems).
- E. coli BL21 (DE3) transformants was cultured overnight at 37 ° C. in 3 mL of liquid LB medium containing 50 ⁇ g / mL of ampicillin. Add 0.5 mL of the culture broth to 50 mL of liquid LB medium containing ampicillin at the same concentration, pre-incubate for 3 hours (until OD630nm is about 0.4), and use IPTG (isopropyl- ⁇ -thiogalactopyra to a final concentration of 1 mM. Noside) (Wako Pure Chemical Industries, Ltd.) was added, and further cultured at 37 ° C. for 4 hours.
- IPTG isopropyl- ⁇ -thiogalactopyra to a final concentration of 1 mM. Noside
- the cells were collected by centrifugation (6000 rpm 4 ° C 15 minutes) with Avanti HP25 (beckman coulter). Subsequent operations were performed on ice. After removing the supernatant (medium), the suspension was suspended in 1 mL of 0.1 M potassium phosphate buffer pH 7.0 (KPB-PDH) containing 1 mM PMSF, 2 mM DTT, and 5 mM Sodium hydrosulfite.
- KPB-PDH potassium phosphate buffer pH 7.0
- the conversion activity from dihydrodaidzein to tetrahydrodaidzein was measured by the following method.
- An enzyme reaction solution having the following composition was prepared and incubated at 0 ° C. for 2 hours.
- Composition of enzyme reaction solution Cell disruption solution (enzyme source) 100 ⁇ l NADH (100 mM) 20 ⁇ l NADPH (100 mM) 20 ⁇ l Dihydrodaidzein (2 mg / ml) 5 ⁇ l KPB-PDH 855 ⁇ l 1000 ⁇ l total
- Example C Example C1 Confirmation of equol biosynthesis activity of bacterial cells from tetrahydrodaidzein Lactococcus 20-92 strain (FERM BP-10036) was added to a tetrahydrodaidzein-containing growth medium (cis- or trans-tetrahydrodaidzein (in-house organic). Synthesis: See Reference Example B1) in modified GAM bouillon medium (Nissui Pharmaceutical Co., Ltd.) added to an amount of 10 ⁇ g / mL) and anaerobic conditions (using BBL Gas Pack systems) at 37 ° C. Incubate for hours.
- modified GAM bouillon medium Nasui Pharmaceutical Co., Ltd.
- Standard solutions for HPLC analysis include mixed solutions of daidzein (Funakoshi Co., Ltd.), equol (Funakoshi Co., Ltd.), dihydrodaidzein (Trend Research Chemical Co., Ltd.), cis-tetrahydrodaidzein, and trans-tetrahydrodaidzein (both chemically synthesized in-house). 2 ⁇ g / mL) was used.
- daidzein is abbreviated as DZN, dihydrodaidzein as DD, cis-tetrahydrodaidzein as c-THD, trans-tetrahydrodaidzein as t-THD, and equol as EQL.
- Example C2 Search for and identification of equol synthase using a recombinant protein expression system using E. coli Dihydrohydrolysis determined in Example B5 using pET system (Novagen) which is a recombinant protein expression system using E. coli Polypeptides corresponding to the three ORF polynucleotides (ORF-US3, US1, DS1) identified on the genomic DNA sequence surrounding the daidzein synthesis (E1) enzyme gene are expressed in Escherichia coli, and the tetrahydrodaidzein is converted into equol. Equol synthesis (E3) by examining its catalytic activity Enzyme search was performed.
- ORF-US3, US1, DS1 ORF polynucleotides
- ORF polypeptide expression vector For the purpose of preparing an expression vector for each ORF polypeptide (ORF-US3, US1, DS1), the polynucleotide of each open reading frame region was amplified by PCR, and pET21a Inserted into vector (Novagen).
- Amplification primer The following amplification primer was prepared based on the genomic sequence around dihydrodaidzein synthase (E1) determined in Example B5.
- ORF-US3 polypeptide exp.US3 F TATACATATGGCAGAATTCGATGTTGAG (SEQ ID NO: 62) exp.US3 R: CCGCAAGCTTCTACATAGTGGAGATCGCGTGG (SEQ ID NO: 63)
- ORF-DS1 polypeptide exp.DS1 F ATATACATATGCAGGATATGGACTTCATGG (SEQ ID NO: 66) exp.DS1 R: GCTCGAATTCTCATAGTGACATCAGCGCTCCC (SEQ ID NO: 67)
- exp.US3 F, exp.US1 F, and exp.DS1 F contain a restriction enzyme NdeI cleavage site sequence for insertion into pET21a (Novagen)
- exp.US3 R contains a HindIII cleavage site sequence
- exp .US1 R and exp.DS1 R contain the EcoRI cleavage site sequence.
- coli JM109 using the ligation reaction solution.
- a strain (Takara Bio Inc.) was transformed by a conventional method. The transformant thus obtained was grown overnight on an LB medium agar (GIBCO) plate containing ampicillin (50 ⁇ g / mL) at 37 ° C. to form colonies. The obtained single colony was cultured overnight in 3 mL of LB medium (GIBCO) containing ampicillin (50 ⁇ g / mL), and then plasmid DNA was extracted using an automatic plasmid extractor PI-100 (KURABO).
- the nucleotide sequence of the DNA inserted into the plasmid was sequenced by the dye terminator method to confirm that each polynucleotide was inserted correctly as intended, and pET-US3, pET-US1, and pET-DS1 were obtained.
- the DNA sequence in this example was performed using a DNA sequencer ABI3700 (Applied Biosystems).
- Example C3 Measurement of Equol Synthesis Activity of Recombinant ORF Polypeptide
- the conversion activity from tetrahydrodaidzein to equol was measured using the disrupted solution of each transformant obtained in Example C2 as an enzyme source.
- the conversion activity from tetrahydrodaidzein to equol was measured by the following method.
- the standard solution for HPLC analysis is a mixed solution of daidzein (Funakoshi Co., Ltd.), equol (Funakoshi Co., Ltd.), dihydrodaidzein (Trend Research Chemical Co., Ltd.), cis-tetrahydrodaidzein, and trans-tetrahydrodaidzein (both chemically synthesized in-house). 2 ⁇ g / mL each) was used.
- daidzein is abbreviated as DZN, dihydrodaidzein as DD, cis-tetrahydrodaidzein as c-THD, trans-tetrahydrodaidzein as t-THD, and equol as EQL.
- ORF-US3 polypeptide has equol bioactivity from tetrahydrodaidzein. That is, it can be said that the ORF-US3 polypeptide corresponds to the E3 polypeptide.
- Example D Example D1 Expression and purification of recombinant His-tagged equol production-related enzyme using E. coli Using pET system (Novagen), a recombinant protein expression system using E. coli, His-tagged equol production-related enzyme [ Dihydrodaidzein synthase (E1), tetrahydrodaidzein synthase (E2), equol synthase (E3)] were expressed, and affinity purification was performed using His-tagged protein purification (Ni) column.
- E1 Dihydrodaidzein synthase
- E2 tetrahydrodaidzein synthase
- E3 equol synthase
- Amplification primer The following amplification primer was prepared based on the genomic sequence around dihydrodaidzein synthase (E1) determined in Example B5. His-tagged E1 enzyme exp.E1 pet F Nde : AGCTCATATGAAGAACAAGTTCTATCCGAA (SEQ ID NO: 41) exp.E1 pet His : AATCGAATTCGTACAGGTTGCAGCCAGCGATGT (SEQ ID NO: 42) His-tagged E2 enzyme exp.US2 pet F Nde: TATACATATGGCACAGGAAGTCAAAGTC (SEQ ID NO: 60) exp.E2 pet His : AATCGAATTCGAGACCTCGATCTCGCCCTGC (SEQ ID NO: 68) His-tagged E3 enzyme exp.US3 F: TATACATATGGCAGAATTCGATGTTGAG (SEQ ID NO: 62) exp.E3 R His : CCGCAAGCTTGTACATAGTGGAGATCGCGTGG (SEQ ID NO: 69) The above amplification primer
- PCR was performed using GeneAmpPCR System 9700 (Applied Biosystems). As a result of agarose gel electrophoresis of a part of the PCR reaction solution, a band having an expected size was detected. The entire PCR product was recovered with the QIAGEN PCR Purification kit (Qiagen).
- the transformant thus obtained was grown overnight on an LB medium agar (GIBCO) plate containing ampicillin (50 ⁇ g / mL) at 37 ° C. to form colonies.
- the obtained single colony was cultured overnight in 3 mL of LB medium (GIBCO) containing ampicillin (50 ⁇ g / mL), and then plasmid DNA was extracted using an automatic plasmid extractor PI-100 (KURABO).
- E. coli (2) Expression and affinity purification of each recombinant His-tagged enzyme polypeptide in E. coli (2-1) Preparation of E. coli BL21 transformant Escherichia coli BL21 (DE3) strain (Novagen) was transformed by a conventional method using plasmids pET-E1-His, pET-E2-His, and pET-E3-His expressing His-tagged enzyme polypeptide. The transformant was grown overnight at 37 ° C. on an LB medium agar plate containing ampicillin (50 ⁇ g / mL) to obtain a single colony.
- ampicillin 50 ⁇ g / mL
- the obtained bacterial cells were added with Bugbuster protein Extraction solution (Novagen) at 15 mL per 1 g of wet cell weight, gently suspended with a pipette, and then Lysozyme (SIGMA) at 2000 units / mL, Benzonase (Novagen) at 25 units ( 1 ⁇ L) / mL. Thereafter, the mixture was slowly stirred at room temperature for 30 minutes using a rotator (RT-50: Taitec) to obtain fungus lysate A. Furthermore, the fungus lysate A was centrifuged (8000 rpm, 4 ° C., 15 minutes) with Avanti HP25 (beckman coulter) to obtain fungus lysate B as its supernatant.
- Bugbuster protein Extraction solution Novagen
- SIGMA Lysozyme
- Benzonase Novagen
- fungus lysate A was centrifuged (8000 rpm, 4 ° C., 15 minutes)
- His-tagged protein purification column uses His GraviTrap (GE Healthcare Bioscience), and the His-tagged E1 and E2 enzyme polypeptides are partially modified according to the procedure described in the instruction manual. Affinity purification was performed. That is, equilibrate His GraviTrap with 10 mL of ice-cold binding buffer, pour the whole amount of fungus lysate B prepared in (2-2-2), and naturally drop the target His-tagged E1 or E2 enzyme. Adsorbed to His GraviTrap.
- His GraviTrap was washed twice with 10 mL of ice-cold washing buffer, and then the desired His-tagged E1 and E2 enzymes were eluted from His GraviTrap with 3 mL of elution buffer that had been ice-cooled.
- DTT dithiothreitol
- Binding buffer 20 mM Tris-HCl, 20 mM Imidazole (Wako Pure Chemical Industries, Ltd.), 0.5 M NaCl (Wako Pure Chemical Industries, Ltd.), 1 mM DTT [dithiothreitol] (Wako Pure Chemical Industries, Ltd.), 1 mM PMSF [phenylmethylsulfonyl fluoride] (Wako Pure Chemical Industries, Ltd.)
- Washing buffer 20 mM Tris-HCl, 60 mM Imidazole, 0.5 M NaCl, 1 mM DTT [dithiothreitol], 1 mM PMSF [phenylmethylsulfonyl fluoride]
- Elution buffer 20 mM Tris-HCl 500 mM Imidazole, 0.5 M NaCl, 1 mM DTT [dithiothreitol], 1 mM PMSF [phenylmethylsulfonyl fluoride]
- Elution buffer 20
- IPTG Isopropyl- ⁇ -thiogalactopyranoside
- HP25 Avanti HP25
- PMSF phenylmethylsulfonyl fluoride
- DTT mM DTT
- Sodiumhydrosulfite Wako Pure Chemical Industries, Ltd.
- 0.1 M potassium phosphate buffer pH 7.0 hereinafter referred to as buffer A
- Example D2 Tetrahydrodaidzein synthesis from daidzein using recombinant His-tagged E1 and E2 enzymes
- the recombinant His-tagged E1 enzyme and E2 enzyme obtained in (2-2-3) of Example D1 above were used.
- An enzyme reaction solution having the following composition was prepared as an enzyme source, and tetrahydrodaidzein was synthesized from daidzein by reacting at 37 ° C. for 2 hours. At the same time, a reaction using each of the recombinant His-tagged E1 and E2 enzymes alone as an enzyme source was also carried out.
- the standard solution for HPLC analysis was a mixed solution of daidzein (Funakoshi), equol (Funakoshi), dihydrodaidzein (Trend Research Chemical), cis-tetrahydrodaidzein (Reference Example B1), and trans-tetrahydrodaidzein (Reference Example B1) (each 2 ⁇ g / mL) was used.
- the result of HPLC analysis is shown in FIG. When a mixture of recombinant His-tagged E1 enzyme and E2 enzyme was used as the enzyme source, cis- and trans-tetrahydrodaidzein were confirmed in the product. However, when the recombinant His-tagged E1 enzyme or the recombinant His-tagged E2 enzyme alone was used as the enzyme source, cis- and transtetrahydrodaidzein could not be confirmed in the product.
- Example D3 Synthesis of equol from dihydrodaidzein using recombinant His-tagged E2 and E3 enzymes Recombinant His- obtained in (2-2-3) and (2-3-3) of Example D1 above Using the tagged E2 and E3 enzymes as enzyme sources, an enzyme reaction solution having the following composition was prepared and reacted at 37 ° C. for 2 hours to synthesize equol from dihydrodaidzein. At the same time, a reaction using each of the recombinant His-tagged E2 enzyme and E3 enzyme alone as an enzyme source was also carried out.
- Enzyme reaction solution composition After the incubation, 3 mL of ethyl acetate (Wako Pure Chemical Industries, Ltd.) was added to the resulting enzyme reaction solution to perform extraction treatment, and after drying, it was dissolved in a moving bed (eluent). The equol in the enzyme reaction solution was measured by HPLC analysis of the lysate. The result of HPLC analysis is shown in FIG. When a recombinant His-tagged E2 or E3 enzyme mixture was used as the enzyme source, equol was confirmed in the product. However, when the recombinant His-tagged E2 enzyme or the recombinant His-tagged E3 enzyme was used alone as the enzyme source, equol could not be confirmed in the product.
- Example D4 Synthesis of equol from daidzein using recombinant His-tagged E1 enzyme, E2 enzyme and E3 enzyme Group obtained in (2-2-3) and (2-3-3) of Example D1 above Using the His-tagged E1 enzyme, E2 enzyme and E3 enzyme as enzyme sources, an enzyme reaction solution having the following composition was prepared and reacted at 37 ° C. for 2 hours to synthesize equol from daidzein. At the same time, a reaction using each of the recombinant His-tagged E1 enzyme, E2 enzyme, and E3 enzyme alone as an enzyme source was also carried out. Enzyme reaction solution composition
- Example E Effect of Metal Ions on Dihydrodaizen Synthesis Activity of His-Tagged Recombinant E1 Enzyme From daidzein using Escherichia coli-expressed His-tagged recombinant E1 enzyme (E1-His) purified by Ni-Sepharose as an enzyme source The effect of metal ions on the conversion activity to dihydrodaidzein was investigated.
- composition of enzyme reaction solution 20 ⁇ l of recombinant His-tagged E1 enzyme NADH (100 mM) 10 ⁇ l NADPH (100 mM) 10 ⁇ l Daidzein (1 mg / ml) 10 ⁇ l Metal ion solution 100 ⁇ l 0.2M KPB-DH 850 ⁇ l 1000 ⁇ l total
- Example A1 The result of the HPLC analysis in Example A1 is shown.
- the three chromatographs shown above are HPLC analysis results of the enzyme reaction product obtained when Control (no coenzyme added), NADH added as a coenzyme, and NADPH added as a coenzyme.
- the bar graph shown below in FIG. 1 is a diagram showing a peak area corresponding to dihydrodaidzein when Control (no coenzyme added), NADH is added as a coenzyme, and NADPH is added as a coenzyme.
- the results of Mono QQ HPLC performed in Example A2 are shown in FIG. 2, and the activity of dihydrodaidzein synthase in each fraction is shown in FIG.
- the enzyme activity shows the peak area of dihydrodaidzein produced using daidzein as a substrate.
- the results of SDS-PAGE carried out in Example A2 are shown on the left in FIG. 3, and the activity of dihydrodaidzein synthase in each fraction is shown on the right in FIG.
- the enzyme activity shows the peak area of dihydrodaidzein produced using daidzein as a substrate.
- a commercially available gel plate (SuperSep TM 10-20% (Wako Pure Chemical Industries, Ltd.)) was used for SDS-PAGE.
- the result of performing the peptide mapping in Example A4 and the amino acid sequence of the peptide corresponding to each peak are shown. In FIG.
- M1 ⁇ / Styl
- M2 100bp Ladder
- the result of the agarose electrophoresis of RT-PCR product performed in Example A7 is shown.
- the upper figure is dihydrodaidzein synthase and the lower figure is ribosomal RNA.
- SDS-PAGE performed in Example A8 is shown.
- the result of the HPLC analysis performed in Example A8 is shown.
- the upper figure shows the control
- the middle figure shows pET-E1-His
- the lower figure shows pET21a.
- the result of the HPLC analysis in Example 1 is shown.
- the upper chart is the HPLC analysis result of cis-tetrahydrodaidzein (REF-000312)
- the inner chart is the enzyme reaction obtained by using dihydrodaidzein as a substrate and using crushed bacteria as an enzyme source.
- HPLC analysis results of the product the chart below shows the HPLC analysis results of trans-tetrahydrodaidzein (REF-000313), respectively.
- the result of the HPLC analysis in Example B1 is shown.
- FIG. 9 the upper chart is the HPLC analysis result of cis-tetrahydrodaidzein (REF-000312)
- the inner chart is the enzyme reaction obtained by using dihydrodaidzein as a substrate and using crushed bacteria as an enzyme source.
- HPLC analysis results of the product the chart below shows the HPLC analysis results of
- the upper two charts show the results of HPLC analysis of the enzyme reaction product obtained by using cis-tetrahydrodaidzein (REF-000312) as a substrate and using the disrupted bacteria as an enzyme source;
- Each of the charts shows the results of HPLC analysis of the enzyme reaction product obtained using trans-tetrahydrodaidzein (REF-000313) as a substrate and using a bacterial disruption as an enzyme source.
- the result of the HPLC analysis in Example B2 is shown.
- the three charts shown above are the results of HPLC analysis of the enzyme reaction product obtained when Control (no coenzyme added), NADH added as a coenzyme, and NADPH added as a coenzyme. is there.
- FIG. 11 the three charts shown above are the results of HPLC analysis of the enzyme reaction product obtained when Control (no coenzyme added), NADH added as a coenzyme, and NADPH added as a coenzyme.
- the bar graph shown below shows the peak area corresponding to tetrahydrodaidzein when Control (no coenzyme added), NADH is added as a coenzyme, and NADPH is added as a coenzyme. is there.
- the result of the gel filtration HPLC analysis in Example B3 is shown.
- the upper graph shows the results of gel filtration HPLC analysis of the standard protein;
- the middle graph shows the peak area corresponding to tetrahydrodaidzein that produced the enzyme activity of each fraction;
- the lower graph shows the protein of each fraction
- the intensity of the corresponding absorption (280 nm) is shown.
- the result of having performed SDS-PAGE in Example B3 is shown.
- the result of having performed SDS-PAGE in Example B4 is shown.
- the schematic diagram of the genome structure around the dihydrodaidzein synthesis (E1) enzyme gene determined in Example B5 is shown.
- An example of the data acquired by LC-MS analysis in Example B6 and the amino acid sequence of the digested peptide estimated from it are shown.
- the amino acid residue represented by L in FIG. 16-1 is the same as the amino acid residue represented by X in the specification.
- the upper figure is for the peptide: TPGVAASVADEXK.
- An example of the data acquired by LC-MS analysis in Example B6 and the amino acid sequence of the digested peptide estimated from it are shown.
- 16-2 is the same as the amino acid residue represented by X in the specification.
- the upper figure is for the peptide: KXXXTGTTK.
- the figure below is for VTQEXXCAHGAFVCGSGR.
- the data acquired by LC-MS analysis in Example B6 and an example (WXSPEESVGQR) of the amino acid sequence of the digested peptide estimated from it are shown.
- the amino acid residue represented by L in FIG. 16-3 is the same as the amino acid residue represented by X in the specification.
- the result of the HPLC analysis in Example B7 is shown. In FIG.
- the upper left two charts show the results of HPLC analysis (ORF-) of the enzyme reaction product obtained by using dihydrodaidzein as a substrate and using the reaction solution expressing the ORF-US2 polypeptide as the enzyme source. US2); The two charts on the right show the HPLC analysis results (NC) of the enzyme reaction products obtained using the reaction solution without protein synthesis as the enzyme source. Further, in FIG. 17, the bar graph shown below is an enzyme reaction using NC (no protein synthesis), DHFR (dihydrofolate reductase expression reaction solution), and ORF-US2 (ORF-US2 polypeptide expression reaction solution) as enzyme sources.
- NC no protein synthesis
- DHFR dihydrofolate reductase expression reaction solution
- ORF-US2 ORF-US2 polypeptide expression reaction solution
- the lower part of the upper two charts shows the HPLC analysis of the enzyme reaction product obtained by using dihydrodaidzein as a substrate and using a bacterial crush from the negative control pET21a transformant as an enzyme source.
- the result (pET21a) is shown.
- the bar graph shown below shows the enzyme reaction in the case where the enzyme reaction was performed using the reaction solution expressing the ORF-US2 polypeptide or the bacterial crush solution derived from the pET21a transformant as the enzyme source.
- FIG. 2 shows peak areas corresponding to dihydrodaidzein (DD) and cis- (c-THD) and trans-tetrahydrodaidzein (t-THD). The result of the HPLC analysis in Example C1 is shown.
- Example C2 The result of SDS-PAGE performed in Example C2 is shown.
- the result of the HPLC analysis performed in Example C3 is shown.
- the upper figure is for Standard, the middle figure is for pET-US3, and the lower figure is for pET21a.
- the result of the HPLC analysis performed in Example D2 is shown.
- the top diagram is for Standard, the second diagram from the top is E1 and E2, the third diagram from the top is E1, and the bottom diagram is for E2.
- the result of the HPLC analysis performed in Example D3 is shown.
- the result of the HPLC analysis performed in Example D4 is shown.
- the top diagram is for Standard, the second diagram from the top is E1, E2, and E3, the third diagram from the top is E1, the fourth diagram from the top is E2, and the bottom diagram is for E3.
- the influence of metal ions on the E1 enzyme is shown.
- the alignment of the amino acid sequence of sequence number 1, 2 and 3 is shown.
- the alignment of the amino acid sequences set forth in SEQ ID NOs: 7, 8, and 9 is shown.
- the alignment of the amino acid sequences set forth in SEQ ID NOs: 13, 14 and 15 is shown.
- SEQ ID NO: 23 is the base sequence of primer E1-N-terminal-31.
- SEQ ID NO: 24 is the base sequence of primer E1-N-terminal-37.
- SEQ ID NO: 25 is the base sequence of primer E1-N-terminal-F32.
- SEQ ID NO: 26 shows the base sequence of primer E1-internal-RP1.
- SEQ ID NO: 27 is the base sequence of primer pUC19-FP-1.
- SEQ ID NO: 28 is the base sequence of primer pUC19-RP-1.
- SEQ ID NO: 29 is the base sequence of primer pUC19-FP-2.
- SEQ ID NO: 30 is the base sequence of primer pUC19-RP-2.
- SEQ ID NO: 31 is the base sequence of primer E1-RACE-N-P1.
- SEQ ID NO: 32 shows the base sequence of primer E1-RACE-RP2-1.
- SEQ ID NO: 33 is the base sequence of primer E1-RACE-N-P2.
- SEQ ID NO: 34 is the base sequence of primer E1-RACE-RP2-2.
- SEQ ID NO: 35 is the base sequence of primer E1-conf-NP.
- SEQ ID NO: 36 is the base sequence of primer E1-conf-CP.
- SEQ ID NO: 37 is the base sequence of primer E1-FP.
- SEQ ID NO: 38 shows the base sequence of primer E1-RP.
- SEQ ID NO: 39 is the base sequence of primer Gar-16S-Ribo-FP.
- SEQ ID NO: 40 is the base sequence of primer Gar-16S-Ribo-RP.
- SEQ ID NO: 41 is the base sequence of primer exp.E1 pet F Nde.
- SEQ ID NO: 42 is the base sequence of primer exp. E1 pet His.
- SEQ ID NO: 43 is the base sequence of primer RACE-N-P3-1.
- SEQ ID NO: 44 is the base sequence of primer RACE-N-P3-2.
- SEQ ID NO: 45 shows the base sequence of primer E1-Bub-N-P1.
- SEQ ID NO: 46 is the base sequence of primer E1-Bub-N-P2.
- SEQ ID NO: 47 is the base sequence of primer RACE-C-P3-1.
- SEQ ID NO: 48 is the base sequence of primer RACE-C-P3-2.
- SEQ ID NO: 49 is the base sequence of primer E1-Bub-C-P1.
- SEQ ID NO: 50 is the base sequence of primer E1-Bub-C-P2.
- SEQ ID NO: 57 is the base sequence of primer E2-invitroTS-FP.
- SEQ ID NO: 58 is the base sequence of primer E2-invitroTS-RP.
- SEQ ID NO: 59 is the base sequence of primer Universal-Primer.
- SEQ ID NO: 60 is the base sequence of primer exp.US2 pet F Nde.
- SEQ ID NO: 61 is the base sequence of primer exp. US2 pet.
- SEQ ID NO: 62 is the base sequence of primer exp.US3 F.
- SEQ ID NO: 63 is the base sequence of primer exp.US3 R.
- SEQ ID NO: 64 is the base sequence of primer exp.US1 F.
- SEQ ID NO: 65 is the base sequence of primer exp.US1 R.
- SEQ ID NO: 66 is the base sequence of primer exp.DS1 F.
- SEQ ID NO: 67 is the base sequence of primer
Abstract
Description
項A1. 以下の(Aa)~(Ac)のいずれかであるポリペプチド:
(Aa)配列番号1に記載のアミノ酸配列からなるポリペプチド;
(Ab)配列番号1に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(Ac)配列番号1に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド。
項A2. 以下の(Ad)~(Af)のいずれかであるポリヌクレオチド:
(Ad)配列番号4に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ae)配列番号1に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Af)前記(Ad)又は(Ae)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つダイゼインを基質としジヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
項A3. 項A2に記載のポリヌクレオチドを有する発現ベクター。
項A4. 項A3に記載の発現ベクターによって形質転換された組換え細胞。
項A5. 組換え細胞が細菌性原核細胞である、項A4に記載の組換え細胞。
項A6. 細菌性原核細胞がラクトコッカス属に属するものである、項A5に記載の組換え細胞。
項A7. 項A4乃至A6のいずれかに記載の細胞を培養し、ダイゼインを基質としジヒドロダイゼインを生成する活性を有するポリペプチドを得る工程を含む、ポリペプチドの製造方法。
項A8. 項A7に記載の製造方法により得られるポリペプチド。
項A9. ダイゼインに対して、項A1又はA8に記載のポリペプチド、並びにNADPH及び/又はNADHを作用させる工程を含む、ジヒドロダイゼインの製造方法。
項A10. ダイゼインに、項A4乃至A6のいずれかに記載の細胞を作用させる工程を含む、ジヒドロダイゼインの製造方法。
項A11. 項A1に記載のポリペプチド、又は項A2に記載のポリヌクレオチドがコードするポリペプチドに結合性を有する抗体。
項A12. 項A11に記載の抗体を被験試料に接触させる工程を含む、項A1に記載のポリペプチド又は項A2に記載のポリヌクレオチドがコードするポリペプチドを検出又は測定する免疫学的方法。
項A13. 検出又は測定対象となるポリペプチドが、細菌性原核細胞内に存在するものである、項A12に記載の方法。
項A14. 項A1に記載のポリペプチドをコードするポリヌクレオチド、又は項A2に記載のポリヌクレオチドに対してストリンジェントな条件下でハイブリダイズできるヌクレオチド配列を有する、プローブ。
項A15. 項A1に記載のポリペプチドをコードするポリヌクレオチド、又は項A2に記載のポリヌクレオチドに対してストリンジェントな条件下でハイブリダイズできるヌクレオチド配列を有する、プライマー。
項A16. 項A14に記載のプローブを用いて、項A1に記載のポリペプチドをコードするポリヌクレオチド、又は項A2に記載のポリヌクレオチドを検出又は測定する方法。
項A17. 検出又は測定対象となるポリペプチドが、細菌性原核細胞内に存在するものである、項A16に記載の方法。
項A18. 項A1に記載のポリペプチドをコードするポリヌクレオチド、項A2に記載のポリヌクレオチド又はこれらの一部をPCRにより増幅させる工程を含む、項A16に記載の方法。
項A19. 項A1記載のポリペプチド、又は項A2のポリヌクレオチドがコードするポリペプチドを含有することを特徴とする、ジヒドロダイゼイン合成酵素組成物。
項A20. 更にNADPH及び/又はNADHを含む、項A19に記載の組成物。
項A21. (Ai)項A1記載のポリペプチド、又は項A2のポリヌクレオチドがコードするポリペプチド、(Aii)NADPH及び/又はNADH、及び(Aiii)ダイゼインを含有することを特徴とする、ジヒドロダイゼイン合成原料組成物。
項A22. (Aiv)項A4乃至A6のいずれかに記載の細胞、及び(Aiii)ダイゼインを含有することを特徴とする、ジヒドロダイゼイン合成原料組成物。
項A23. (Ai)項A1記載のポリペプチド、又は項A2のポリヌクレオチドがコードするポリペプチド、(Aii)NADPH及び/又はNADH、及び(Aiii)ダイゼインを含む、ジヒドロダイゼイン合成用キット。
項A24. (Aiv)項A4乃至6のいずれかに記載の細胞、及び(Aiii)ダイゼインを含む、ジヒドロダイゼイン合成用キット。
項A25. 少なくとも項A11に記載の抗体を含む、項A1記載のポリペプチド、又は項A2のポリヌクレオチドがコードするポリペプチドを測定するための免疫学的測定用キット。
項A26. 少なくとも項A15に記載のプライマーを含む、項A1に記載のポリペプチドをコードするポリヌクレオチド又は項A2に記載のポリヌクレオチドの検出用PCR用キット。
項A27. 項A1に記載のポリペプチドをコードするポリヌクレオチド又は項A2に記載のポリヌクレオチドを含有する細胞の同定用である、項A26に記載の同定用キット。
項A28. PCR用のキットである、項A27に記載の同定用キット。
項A29. 項A1に記載のポリペプチドからなる、ジヒドロダイゼイン合成酵素。
項B1. 以下の(Ba)~(Bc)のいずれかであるポリペプチド:
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド。
項B2. 以下の(Bd)~(Bf)のいずれかであるポリヌクレオチド:
(Bd)配列番号10に記載のヌクレオチド配列からなるポリヌクレオチド;
(Be)配列番号7に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド
;
(Bf)前記(Bd)又は(Be)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
項B3. 項B2に記載のポリヌクレオチドを有する発現ベクター。
項B4. 項B3に記載の発現ベクターによって形質転換された組換え細胞。
項B5. 組換え細胞が細菌性原核細胞である、項B4に記載の組換え細胞。
項B6. 細菌性原核細胞がラクトコッカス属に属するものである、項B5に記載の組換え細胞。
項B7. 項B4乃至B6のいずれかに記載の細胞を培養し、ジヒドロダイゼインを基質としテトラヒドロダイゼインを生成する活性を有するポリペプチドを得る工程を含む、ポリペプチドの製造方法。
項B8. 項B7に記載の製造方法により得られるポリペプチド。
項B9. ジヒドロダイゼインに対して、項B1又はB8に記載のポリペプチド、並びにNADPH及び/又はNADHを作用させる工程を含む、テトラヒドロダイゼインの製造方法。
項B10. ジヒドロダイゼインに、項B4乃至B6のいずれかに記載の細胞を作用させる工程を含む、テトラヒドロダイゼインの製造方法。
項B11. 項B1に記載のポリペプチド、又は項B2に記載のポリヌクレオチドがコードするポリペプチドに結合性を有する抗体。
項B12. 項B11に記載の抗体を被験試料に接触させる工程を含む、項B1に記載のポリペプチド又は項B2に記載のポリヌクレオチドがコードするポリペプチドを検出又は測定する免疫学的方法。
項B13. 検出又は測定対象となるポリペプチドが、細菌性原核細胞内に存在するものである、項B12に記載の方法。
項B14. 項B1に記載のポリペプチドをコードするポリヌクレオチド、又は項B2に記載のポリヌクレオチドに対してストリンジェントな条件下でハイブリダイズできるヌクレオチド配列を有する、プローブ。
項B15. 項B1に記載のポリペプチドをコードするポリヌクレオチド、又はB項2に記載のポリヌクレオチドに対してストリンジェントな条件下でハイブリダイズできるヌクレオチド配列を有する、プライマー。
項B16. 項B14に記載のプローブを用いて、項B1に記載のポリペプチドをコードするポリヌクレオチド、又は項B2に記載のポリヌクレオチドを検出又は測定する方法。
項B17. 検出又は測定対象となるポリペプチドが、細菌性原核細胞内に存在するものである、項B16に記載の方法。
項B18. 項B1に記載のポリペプチドをコードするポリヌクレオチド、項B2に記載のポリヌクレオチド又はこれらの一部をPCRにより増幅させる工程を含む、項B16に記載の方法。
項B19. 項B1記載のポリペプチド、又は項B2のポリヌクレオチドがコードするポリペプチドを含有することを特徴とする、テトラヒドロダイゼイン合成酵素組成物。
項B20. 更にNADPH及び/又はNADHを含む、項B19に記載の組成物。
項B21. (Bi)項B1記載のポリペプチド、又は項B2のポリヌクレオチドがコードするポリペプチド、(Bii)NADPH及び/又はNADH、並びに(Biii)ジヒドロダイゼインを含有することを特徴とする、テトラヒドロダイゼイン合成原料組成物。
項B22. (Biv)項B4乃至6のいずれかに記載の細胞、及び(Biii)ジヒドロダイゼインを含有することを特徴とする、テトラヒドロダイゼイン合成原料組成物。
項B23. (Bi)項B1記載のポリペプチド、又は項B2のポリヌクレオチドがコードするポリペプチド、(Bii)NADPH及び/又はNADH、並びに(Biii)ジヒドロダイゼインを含む、テトラヒドロダイゼイン合成用キット。
項B24. (Biv)項B4乃至6のいずれかに記載の細胞、及び(Biii)ジヒドロダイゼインを含む、テトラヒドロダイゼイン合成用キット。
項B25. 少なくとも項B11に記載の抗体を含む、項B1記載のポリペプチド、又は項B2のポリヌクレオチドがコードするポリペプチドを測定するための免疫学的測定用キット。
項B26. 少なくとも項B15に記載のプライマーを含む、項B1に記載のポリペプチドをコードするポリヌクレオチド又は項B2に記載のポリヌクレオチドをコードするポリヌクレオチドの検出用PCR用キット。
項B27. 項B1に記載のポリペプチドをコードするポリヌクレオチド又は項B2に記載のポリヌクレオチドを含有する細胞の同定用である、項B26に記載の同定用キット。
項B28. PCR用のキットである、項B27に記載の同定用キット。
項B29. 項B1に記載のポリペプチドからなる、テトラヒドロダイゼイン合成酵素。
項C1. 以下の(Ca)~(Cc)のいずれかであるポリペプチド:
(Ca)配列番号13に記載のアミノ酸配列からなるポリペプチド;
(Cb)配列番号13に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド;
(Cc)配列番号13に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド。
項C2. 以下の(Cd)~(Cf)のいずれかであるポリヌクレオチド:
(Cd)配列番号16に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ce)配列番号13に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Cf)前記(Cd)又は(Ce)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つテトラヒドロダイゼインを基質としエクオールを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
項C3. 項C2に記載のポリヌクレオチドを有する発現ベクター。
項C4. 項C3に記載の発現ベクターによって形質転換された組換え細胞。
項C5. 組換え細胞が細菌性原核細胞である、項C4に記載の組換え細胞。
項C6. 細菌性原核細胞がラクトコッカス属に属するものである、項C5に記載の組換え細胞。
項C7. 項C4乃至C6のいずれかに記載の細胞を培養し、テトラヒドロダイゼインを基質としエクオールを生成する活性を有するポリペプチドを得る工程を含む、ポリペプチドの製造方法。
項C8. 項C7に記載の製造方法により得られるポリペプチド。
項C9. テトラヒドロダイゼインに対して、項C1又はC8に記載のポリペプチドを作用させる工程を含む、エクオールの製造方法。
項C10. テトラヒドロダイゼインに、項C4乃至C6のいずれかに記載の細胞を作用させる工程を含む、エクオールの製造方法。
項C11. 項C1に記載のポリペプチド、又は項C2に記載のポリヌクレオチドがコードするポリペプチドに結合性を有する抗体。
項C12. 項C11に記載の抗体を被験試料に接触させる工程を含む、項C1に記載のポリペプチド又は項C2に記載のポリヌクレオチドがコードするポリペプチドを検出又は測定する免疫学的方法。
項C13. 検出又は測定対象となるポリペプチドが、細菌性原核細胞内に存在するものである、項C12に記載の方法。
項C14. 項C1に記載のポリペプチドをコードするポリヌクレオチド、又は項C2に記載のポリヌクレオチドに対してストリンジェントな条件下でハイブリダイズできるヌクレオチド配列を有する、プローブ。
項C15. 項C1に記載のポリペプチドをコードするポリヌクレオチド、又は項C2に記載のポリヌクレオチドに対してストリンジェントな条件下でハイブリダイズできるヌクレオチド配列を有する、プライマー。
項C16. 項C14に記載のプローブを用いて、項C1に記載のポリペプチドをコードするポリヌクレオチド、又は項C2に記載のポリヌクレオチドを検出又は測定する方法。
項C17. 検出又は測定対象となるポリペプチドが、細菌性原核細胞内に存在するものである、項C16に記載の方法。
項C18. 項C1に記載のポリペプチドをコードするポリヌクレオチド、項C2に記載のポリヌクレオチド又はこれらの一部をPCRにより増幅させる工程を含む、項C16に記載の方法。
項C19. 項C1記載のポリペプチド、又は項C2のポリヌクレオチドがコードするポリペプチドを含有することを特徴とする、エクオール合成酵素組成物。
項C20. (Ci)項C1記載のポリペプチド、又は項C2のポリヌクレオチドがコードするポリペプチド、並びに(Cii)テトラヒドロダイゼインを含有することを特徴とする、エクオール合成原料組成物。
項C21. (Ciii)項C4乃至C6のいずれかに記載の細胞、及び(Cii)テトラヒドロダイゼインを含有することを特徴とする、エクオール合成原料組成物。
項C22. (Ci)項C1記載のポリペプチド、又は項C2のポリヌクレオチドがコードするポリペプチド、並びに(Cii)テトラヒドロダイゼインを含む、エクオール合成用キット。
項C23. (Ciii)項C4乃至C6のいずれかに記載の細胞、及び(Cii)テトラヒドロダイゼインを含む、エクオール合成用キット。
項C24. 少なくとも項C11に記載の抗体を含む、項C1記載のポリペプチド、又は項C2のポリヌクレオチドがコードするポリペプチドを測定するための免疫学的測定用キット。
項C25. 少なくとも項C15に記載のプライマーを含む、項C1に記載のポリペプチドをコードするポリヌクレオチド又は項C2に記載のポリヌクレオチドの検出用PCR用キット。
項C26. 項C1に記載のポリペプチドをコードするポリヌクレオチド又は項C2に記載のポリヌクレオチドを含有する細胞の同定用である、項C25に記載の同定用キット。
項C27. PCR用のキットである、項C26に記載の同定用キット。
項C28. 項C1に記載のポリペプチドからなる、エクオール合成酵素。
項D1.以下の(第1工程)及び(第2工程)を含む、テトラヒドロダイゼインの製造方法:
(第1工程)ダイゼインに、以下の(Aa)~(Ac)のいずれかであるポリペプチドからなる酵素、並びにNADPH及び/又はNADHを用させることにより、ジヒドロダイゼインを生成する工程;
(Aa)配列番号1に記載のアミノ酸配列からなるポリペプチド;
(Ab)配列番号1に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(Ac)配列番号1に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(第2工程)ジヒドロダイゼインに、以下の(Ba)~(Bc)のいずれかであるポリペプチドからなる酵素、並びにNADPH及び/又はNADHを作用させることにより、テトラヒドロダイゼインを生成する工程;
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド。
項D2.項D1に記載の方法により製造された、テトラヒドロダイゼインを含有する生成物。
項D3.以下の(第2工程)及び(第3工程)を含む、エクオールの製造方法:
(第2工程)ジヒドロダイゼインに、以下の(Ba)~(Bc)のいずれかであるポリペプチドからなる酵素、並びにNADPH及び/又はNADHを作用させることにより、テトラヒドロダイゼインを生成する工程;
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(第3工程)テトラヒドロダイゼインに、以下の(Ca)~(Cc)のいずれかであるポリペプチドからなる酵素、並びにNADPH及び/又はNADHを作用させることにより、エクオールを製造する工程、
(Ca)配列番号13に記載のアミノ酸配列からなるポリペプチド;
(Cb)配列番号13に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド;
(Cc)配列番号13に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド。
項D4.項D3に記載の方法により製造された、エクオールを含有する生成物。
項D5.(第1工程)~(第3工程)を含む、エクオールの製造方法。
項D6.項D5に記載の方法により製造された、エクオールを含有する生成物。
項D7.以下の(Ad)~(Af)、(Bd)~(Bf)及び(Cd)~(Cf)からなる群より選択される少なくとも1種のポリヌクレオチドを有する発現ベクター:
(Ad)配列番号4に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ae)配列番号1に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Af)前記(Ad)又は(Ae)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つダイゼインを基質としジヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド;
(Bd)配列番号10に記載のヌクレオチド配列からなるポリヌクレオチド;
(Be)配列番号7に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Bf)前記(Bd)又は(Be)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド;
(Cd)配列番号16に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ce)配列番号13に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Cf)前記(Cd)又は(Ce)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つテトラヒドロダイゼインを基質としエクオールを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
項D8.項D7に記載の発現ベクターによって形質転換された組換え細胞。
項D9.組換え細胞が細菌性原核細胞である、項D8に記載の組換え細胞。
項D10.細菌性原核細胞がラクトコッカス属に属するものである、項D9に記載の組換え細胞。
項D11.以下の(第4工程)~(第6工程)の少なくとも2つの工程を含む、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造方法:
(第4工程)(Ad)~(Af)のいずれかであるポリヌクレオチドを有する組換え細胞をダイゼインを含有する培地で培養することにより、ジヒドロダイゼインを生成させる工程、
(Ad)配列番号4に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ae)配列番号1に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Af)前記(Ad)又は(Ae)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つダイゼインを基質としジヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド;
(第5工程)(Bd)~(Bf)のいずれかであるポリヌクレオチドを有する組換え細胞をジヒドロダイゼインを含有する培地で培養することにより、テトラヒドロダイゼインを生成させる工程、
(Bd)配列番号10に記載のヌクレオチド配列からなるポリヌクレオチド;
(Be)配列番号7に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Bf)前記(Bd)又は(Be)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド;
(第6工程)(Cd)~(Cc)のいずれかであるポリヌクレオチドを有する組換え細胞をテトラヒドロダイゼインを含有する培地で培養することにより、エクオールを生成させる工程、
(Cd)配列番号16に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ce)配列番号13に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Cf)前記(Cd)又は(Ce)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つテトラヒドロダイゼインを基質としエクオールを生成する活性を有するポリペプチドをコードするポリヌクレオチド;
ここで、(Ad)~(Af)、(Bd)~(Bf)及び(Cd)~(Cf)からなる群より選択されるポリヌクレオチドの少なくとも2つが一つの組換え細胞内にあってもよい。
項D12.組換え細胞が細菌性原核細胞である、項D11に記載の製造方法。
項D13.細菌性原核細胞がラクトコッカス属に属するものである、項D12に記載の製造方法。
項D14.項D11~C13のいずれかに記載の方法により製造された、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールを含有する生成物。
項D15.以下の(第1反応槽)~(第3反応槽)の少なくとも1つの反応槽を備える、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造装置:
(第1反応槽)(Aa)~(Ac)のいずれかであるポリペプチドからなる酵素が固定されている反応手段を有しており、該ポリペプチドからなる酵素を用いてダイゼインからジヒドロダイゼインを製造するための反応槽、ここで該反応手段は反応槽内のダイゼインと接触できるように配置されている;
(第2反応槽)(Ba)~(Bc)のいずれかであるポリペプチドからなる酵素が固定されている反応手段を有しており、該ポリペプチドからなる酵素を用いてジヒドロダイゼインからテトラヒドロダイゼインを製造するための反応槽、ここで該反応手段は反応槽内のジヒドロダイゼインと接触できるように配置されている;
(第3反応槽)(Ca)~(Cc)のいずれかであるポリペプチドからなる酵素が固定されている反応手段を有しており、該ポリペプチドからなる酵素を用いてテトラヒドロダイゼインからエクオールを製造するための反応槽、ここで該反応手段は反応槽内のテトラヒドロダイゼインと接触できるように配置されている;
ここで、前記反応手段の少なくとも2つは一つの反応槽に共に存在していてもよい。
項D16.以下の(第4反応槽)~(第6反応槽)の少なくとも1つの反応槽を備える、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造装置:
(第4反応槽)(Ad)~(Af)のいずれかであるポリヌクレオチドを有する組換え細胞が固定されている反応手段を有しており、該反応手段を用いてダイゼインからジヒドロダイゼインを製造するための反応槽、ここで該反応手段は反応槽内のダイゼインと接触できるように配置されている;
(第5反応槽)(Bd)~(Bf)のいずれかであるポリヌクレオチドを有する組換え細胞が固定されている反応手段を有しており、該反応手段を用いてジヒドロダイゼインからテトラヒドロダイゼインを製造するための反応槽、ここで該反応手段は反応槽内のジヒドロダイゼインと接触できるように配置されている;
(第6反応槽)(Cd)~(Cf)のいずれかであるポリヌクレオチドを有する組換え細胞が固定されている反応手段を有しており、該反応手段を用いてテトラヒドロダイゼインからエクオールを製造するための反応槽、ここで該反応手段は反応槽内のテトラヒドロダイゼインと接触できるように配置されている;
ここで、前記反応手段の少なくとも2つが一つの反応槽に共に存在していてもよい。
A:ジヒドロダイゼイン合成酵素
本項では、ジヒドロダイゼイン合成酵素(以下、E1酵素と表記する場合もある)について詳しく説明する。ジヒドロダイゼイン合成酵素に関する一般的な説明は、別段の記載がされる場合を除き、後述するテトラヒドロダイゼイン合成酵素及びエクオール合成酵素についても適用される。
本発明は、ダイゼインを基質として利用し、ジヒドロダイゼインを合成するポリペプチドとして、以下の(Aa)~(Ac)のポリペプチド(以下、該ポリペプチドを「E1ポリペプチド」と表記することもある)を提供する:
(Aa)配列番号1に記載のアミノ酸配列からなるポリペプチド;例えば1~250個、好ましくは1~200個、より好ましくは1~150個、より好ましくは1~100個、より好ましくは、1~50個、より好ましくは1~30個、更に好ましくは1~15個、更により好ましくは1~5個、特に好ましくは1~4個、より特に好ましくは1~3個、最も好ましくは1又は2個が挙げられる。
(Ab)配列番号1に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(Ac)配列番号1に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド。
芳香族アミノ酸:Phe、Trp、Tyr
脂肪族アミノ酸:Ala、Leu、Ile、Val
極性アミノ酸:Gln、Asn
塩基性アミノ酸:Lys、Arg、His
酸性アミノ酸:Glu、Asp
水酸基を有するアミノ酸:Ser、Thr
側鎖の小さいアミノ酸:Gly、Ala、Ser、Thr、Met。
0.1 M リン酸カリウム緩衝液
1 mM PMSF(フェニルメチルスルフォニルフルオライド)
2 mM dithiothreitol
5 mM Sodium hydrosulfite
2 mM NADPH又はNADH
40 μM ダイゼイン
pH 7.0
E1ポリペプチドは、ダイゼインを基質としてジヒドロダイゼインを合成する酵素活性を有する。よって、E1ポリペプチドは、E1酵素とも称される。E1酵素は、Sodium hydrosulfite等の還元剤やマンガンイオンや鉄イオン等の金属イオンの存在により、その酵素活性が賦活化される。E1酵素は、補酵素として、NADPH又はNADHを必要とし、至適温度は30℃付近であり、至適pHは7.0である。E1酵素は、ダイゼインを基質としてジヒドロダイゼインを合成するだけでなく、その逆反応、即ち、ジヒドロダイゼインを基質としてダイゼインを合成することも可能である。
本発明は、更に、ダイゼインを基質としてジヒドロダイゼインを合成する活性を有するポリペプチドをコードするポリヌクレオチド(以下、該ポリヌクレオチドを「E1ポリヌクレオチド」と表記することもある)を提供する。具体的には、E1ポリヌクレオチドとして、以下の(Ad)~(Af)のポリヌクレオチドを提供する:
(Ad)配列番号4に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ae)配列番号1に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Af)前記(Ad)又は(Ae)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つダイゼインを基質としジヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
本発明の発現ベクターは、E1ポリヌクレオチドを含んでおり、且つ該E1ポリヌクレオチドを発現できるものであれば特に制限されず、一般に宿主細胞との関係から適宜選択される。
本発明は、上記E1ポリヌクレオチドを含む発現ベクターによって形質転換された組み換え細胞(形質転換体)を提供する。
E1ポリヌクレオチドが導入された組み換え細胞を培養し、細胞及び又は培養物からE1ポリペプチドを回収することにより、E1ポリペプチドを製造することができる。
本発明は、E1ポリペプチドを用いたジヒドロダイゼインの製造方法を提供する。即ち、該製造方法では、E1ポリペプチドを、NADPH及び/又はNADHの存在下で、ダイゼインに作用させることにより、ダイゼインをジヒドロダイゼインに変換する。好ましくは、E1ポリペプチドを、NADPHの存在下で、ダイゼインに作用させることにより、ダイゼインをジヒドロダイゼインに変換する。
上記ポリペプチドが0.0001~1.0重量%、好ましくは0.001~0.1重量%、更に好ましくは0.001~0.01重量%;
ダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%;及び
NADPH及び/又はNADHが0.01~5重量%、好ましくは0.05~1重量%、更に好ましくは0.1~0.5重量%。
本発明は、更に、E1ポリペプチドを含むジヒドロダイゼイン合成酵素組成物を提供する。該酵素組成物は、E1ポリペプチドを用いたジヒドロダイゼインの製造方法において、ジヒドロダイゼイン合成酵素として好適に使用される。
本発明は、E1ポリヌクレオチドが導入された組み換え細胞を用いたジヒドロダイゼインの製造方法を提供する。即ち、該製造方法では、上記組み換え細胞を、ダイゼインに作用させることにより、ダイゼインをジヒドロダイゼインに変換する。
本発明は、更に、E1ポリペプチドに結合性を有する抗体(IgG抗体)を提供する。
更に、本発明は、上記抗体を用いてE1ポリペプチドを検出又は測定する免疫学的方法を提供する。具体的には、該免疫学的方法は、上記抗体を被験試料に接触させることにより実施される。即ち、上記抗体を被験試料に接触させることにより、被験試料中にE1ポリペプチドが存在する場合には、上記抗体とE1ポリペプチドが特異的に結合する。次いで、E1ポリペプチドに結合した上記抗体を検出し、必要に応じてこれを定量することにより、被験試料中の上記ポリペプチドを検出又は測定することができる。
また、本発明は、E1ポリヌクレオチドを検出又は測定する方法を提供する。該方法は、具体的には、E1ポリヌクレオチドに結合するプローブを被験試料と接触させることにより実施される。即ち、該プローブを被験試料に接触させることにより、被験試料中にE1ポリヌクレオチドが存在する場合には、該プローブとE1ポリヌクレオチドがハイブリダイズする。次いで、この二本鎖の形成の有無を検出し、必要に応じてこれを定量することにより、被験試料中のE1ポリヌクレオチドを検出又は測定することができる。
B-1.ポリペプチド
本発明は、ジヒドロダイゼインを基質として利用しテトラヒドロダイゼインを合成するポリペプチドとして、以下の(Ba)~(Bc)のポリペプチド(以下、該ポリペプチドを「E2ポリペプチド」と表記することもある)を提供する:
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド。
0.1 M リン酸カリウム緩衝液(pH7.0)
1 mM PMSF(フェニルメチルスルフォニルフルオライド)
2 mM dithiothreitol
5 mM Sodium hydrosulfite
2 mM NADPH
2 mM NADH
40 μM ジヒドロダイゼイン
E2ポリペプチドは、ジヒドロダイゼインを基質としてテトラヒドロダイゼインを合成する酵素活性を有する。よって、E2ポリペプチドは、E2酵素とも称される。E2酵素は、補酵素として、NADPH又はNADHを必要とする。E2酵素の至適温度は37℃付近であり、至適pHは4.5である。E2酵素は、ジヒドロダイゼインを基質としてテトラヒドロダイゼインを合成するだけでなく、その逆反応、即ち、テトラヒドロダイゼインを基質としてジヒドロダイゼインを合成することも可能である。
本発明は、更に、ジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチドをコードするポリヌクレオチド(以下、該ポリヌクレオチドを「E2ポリヌクレオチド」と表記することもある)を提供する。具体的には、E2ポリヌクレオチドとして、以下の(Bd)~(Bf)のポリヌクレオチドを提供する:
(Bd)配列番号10に記載のヌクレオチド配列からなるポリヌクレオチド;
(Be)配列番号7に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Bf)前記(Bd)又は(Be)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
本発明の発現ベクターは、E2ポリヌクレオチドを含んでおり、且つ該E2ポリヌクレオチドを発現できるものであれば特に制限されず、E1ポリヌクレオチドを含む発現ベクターと同様に、一般に宿主細胞との関係から適宜選択される。具体的な宿主細胞としては、前記A-3.セクションに記載のものを使用することができる。
本発明は、上記E2ポリヌクレオチドを含む発現ベクターによって形質転換された組換え細胞(形質転換体)を提供する。組換え細胞に使用される宿主細胞としては、前記A-4.セクションに記載のものを特に制限なく使用することができる。また、発現ベクターを宿主細胞に導入する方法については、上記A-4.セクションの記載に従って行うことが出来る。
E2ポリヌクレオチドが導入された組換え細胞を培養し、培養物からE2ポリペプチドを回収することにより、E2ポリペプチドを製造することができる。培養は、前記A-5.セクションの記載に準じて行うことができる。
本発明は、E2ポリペプチドを用いたテトラヒドロダイゼインの製造方法を提供する。即ち、該製造方法では、E2ポリペプチドを、NADPH及び/又はNADHの存在下で、ジヒドロダイゼインに作用させることにより、ジヒドロダイゼインをテトラヒドロダイゼインに変換する。
ジヒドロダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%;及び
NADPH及び/又はNADHが0.01~5重量%、好ましくは0.05~1重量%、更に好ましくは0.1~0.5重量%。
本発明は、更に、E2ポリペプチドを含むテトラヒドロダイゼイン合成酵素組成物を提供する。該酵素組成物は、E2ポリペプチドを用いたテトラヒドロダイゼインの製造方法において、テトラヒドロダイゼイン合成酵素として好適に使用される。
本発明は、E2ポリヌクレオチドが導入された組換え細胞を用いたテトラヒドロダイゼインの製造方法を提供する。即ち、該製造方法では、上記組み換え細胞を、ジヒドロダイゼインに作用させることにより、ジヒドロダイゼインをテトラヒドロダイゼインに変換する。
本発明は、更に、E2ポリペプチドに結合性を有する抗体(IgG抗体)を提供する。
更に、本発明は、上記抗体を用いてE2ポリペプチドを検出又は測定する免疫学的方法を提供する。具体的には、該免疫学的方法は、前記A-10.セクションに記載の方法に準じて行うことが出来る。
また、本発明は、E2ポリヌクレオチドを検出又は測定する方法を提供する。該方法は、具体的には、E2ポリヌクレオチドに結合するプローブを被験試料と接触させることにより実施され、前記A-11.セクションに記載の方法に準じて行うことが出来る。
C-1.ポリペプチド
本発明は、テトラヒドロダイゼインを基質として利用しエクオールを合成するポリペプチドとして、以下の(Ca)~(Cc)のポリペプチド(以下、該ポリペプチドを「E3ポリペプチド」と表記することもある)を提供する:
(Ca)配列番号13に記載のアミノ酸配列からなるポリペプチド;
(Cb)配列番号13に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド;
(Cc)配列番号13に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド。
0.1 M リン酸カリウム緩衝液(pH7.0)
1 mM PMSF(フェニルメチルスルフォニルフルオライド)
2 mM dithiothreitol
5 mMSodium hydrosulfite
40 μM テトラヒドロダイゼイン
E3ポリペプチドは、テトラヒドロダイゼインを基質としてエクオールを合成する酵素活性を有する。よって、E3ポリペプチドは、E3酵素とも称される。E3酵素の至適温度は約23~37℃であり、至適pHは4.5である。E3酵素は、テトラヒドロダイゼインを基質としてエクオールを合成するだけでなく、その逆反応、即ち、エクオールを原料としてテトラヒドロダイゼインを合成することも可能である。
本発明は、更に、テトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチドをコードするポリヌクレオチド(以下、該ポリヌクレオチドを「E3ポリヌクレオチド」と表記することもある)を提供する。具体的には、E3ポリヌクレオチドとして、以下の(Cd)~(Cf)のポリヌクレオチドを提供する:
(Cd)配列番号16に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ce)配列番号13に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Cf)前記(Cd)又は(Ce)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つテトラヒドロダイゼインを基質としエクオールを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
本発明の発現ベクターは、E3ポリヌクレオチドを含んでおり、且つ該E3ポリヌクレオチドを発現できるものであれば特に制限されず、E1ポリヌクレオチドを含む発現ベクターと同様に、一般に宿主細胞との関係から適宜選択される。具体的な宿主細胞としては、前記A-3.セクションに記載のものを使用することが出来る。
本発明は、上記E3ポリヌクレオチドを含む発現ベクターによって形質転換された組換え細胞(形質転換体)を提供する。組み換え細胞細胞に使用される宿主細胞としては、前記A-4.セクションに記載されるものを特に制限なくしようすることができる。また、発現ベクターを宿主細胞に導入する方法については、上記A-4.セクションの記載に従って行うことが出来る。
E3ポリヌクレオチドが導入された組換え細胞を培養し、培養物からE3ポリペプチドを回収することにより、E3ポリペプチドを製造することができる。培養は、前記A-5.セクションの記載に従って行うことができる。
本発明は、E3ポリペプチドを用いたエクオールの製造方法を提供する。即ち、該製造方法では、E3ポリペプチドを、テトラヒドロダイゼインに作用させることにより、テトラヒドロダイゼインをエクオールに変換する。
テトラヒドロダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%。
本発明は、更に、E3ポリペプチドを含むエクオール合成酵素組成物を提供する。該酵素組成物は、E3ポリペプチドを用いたエクオールの製造方法において、エクオール合成酵素として好適に使用される。
本発明は、E3ポリヌクレオチドが導入された組換え細胞を用いたエクオールの製造方法を提供する。即ち、該製造方法では、上記組み換え細胞を、テトラヒドロダイゼインに作用させることにより、テトラヒドロダイゼインをエクオールに変換する。
本発明は、更に、E3ポリペプチドに結合性を有する抗体(IgG抗体)を提供する。
更に、本発明は、上記抗体を用いてE3ポリペプチドを検出又は測定する免疫学的方法を提供する。具体的には、該免疫学的方法は、前記A-10.セクションに記載の方法に準じて行うことが出来る。
本発明は、E3ポリヌクレオチドを検出又は測定する方法を提供する。該方法は、具体的には、E3ポリヌクレオチドに結合するプローブを被験試料と接触させることにより実施され、前記A-11.セクションに記載の方法に準じて行うことができる。
D-I-1.第1工程及び第2工程を含む、テトラヒドロダイゼインの製造方法
本発明は、以下の、ダイゼインからジヒドロダイゼインを生成する第1工程、及びジヒドロダイゼインからテトラヒドロダイゼインを生成する第2工程を含む、テトラヒドロダイゼインの製造方法(以下、該製造方法を「第1製造方法」と表記することもある)を提供する。
(Aa)配列番号1に記載のアミノ酸配列からなるポリペプチド;
(Ab)配列番号1に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(Ac)配列番号1に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド。
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド。
第1工程では、E1ポリペプチドからなる酵素を、NADPH及び/又はNADHの存在下で、ダイゼインに作用させることにより、ダイゼインをジヒドロダイゼインに変換する。 第1工程に採用される反応は、前記E1ポリペプチドからなる酵素、ダイゼイン及びNADPH及び/又はNADHを含有する溶液中で、前記E1ポリペプチドからなる酵素、ダイゼイン等の原料及びジヒドロダイゼイン等の生成物が変質・不活性化されない温度条件及び時間インキュベートすることにより実施される。具体的には、上記A-6.セクションに記載の条件に従って実施することができる。
E1ポリペプチドからなる酵素が0.0001~1.0重量%、好ましくは0.001~0.1重量%、更に好ましくは0.001~0.01重量%;
ダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%;及び
NADPH/またはNADHが0.01~5重量%、好ましくは0.05~1重量%、更に好ましくは0.1~0.5重量%。
第2工程では、E2ポリペプチドからなる酵素を、NADPH及び/又はNADHの存在下で、ジヒドロダイゼインに作用させることにより、ジヒドロダイゼインをテトラヒドロダイゼインに変換する。
E2ポリペプチドからなる酵素が0.0001~1.0重量%、好ましくは0.001~0.1重量%、更に好ましくは0.001~0.01重量%;
ジヒドロダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%;及び
NADPH及び/またはNADHが0.01~5重量%、好ましくは0.05~1重量%、更に好ましくは0.1~0.5重量%。
本発明の第1製造方法では、前記A-1.に記載のE1ポリペプチドからなる酵素及び前記B-1.に記載のE2ポリペプチドからなる酵素が使用される。
本発明は、第1工程及び第2工程を含むテトラヒドロダイゼインの製造方法(第1製造方法)により製造された、テトラヒドロダイゼインを含有する生成物を提供する。
本発明は、以下の、ジヒドロダイゼインからテトラヒドロダイゼインを生成する第2工程、及びテトラヒドロダイゼインからエクオールを生成する第3工程を含む、エクオールの製造方法(以下、該製造方法を「第2製造方法」と表記することもある)を提供する。
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド。
(Ca)配列番号13に記載のアミノ酸配列からなるポリペプチド;
(Cb)配列番号13に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド;
(Cc)配列番号13に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド。
第2工程
本発明の第2製造方法に含有される第2工程は、前述の第1製造方法に含有される第2工程と同様に説明される。
E2ポリペプチドからなる酵素が0.0001~1.0重量%、好ましくは0.001~0.1重量%、更に好ましくは0.001~0.01重量%;
ジヒドロダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%;及び
NADPH及び/またはNADHが0.01~5重量%、好ましくは0.05~1重量%、更に好ましくは0.1~0.5重量%。
第3工程では、E3ポリペプチドからなる酵素を、テトラヒドロダイゼインに作用させることにより、テトラヒドロダイゼインをエクオールに変換する。
E3ポリペプチドからなる酵素が0.0001~1.0重量%、好ましくは0.001~0.1重量%、更に好ましくは0.001~0.01重量%;
テトラヒドロダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%;及び
NADPH及び/又はNADHが0.01~5重量%、好ましくは0.05~1重量%、更に好ましくは0.1~0.5重量%。
本発明の第2製造方法では、前記B-1.セクションに記載のE2ポリペプチドからなる酵素及び前記C-1.セクションに記載のE3ポリペプチドからなる酵素が使用される。
本発明は、第2工程及び第3工程を含むエクオールの製造方法(第2製造方法)により製造された、エクオールを含有する生成物を提供する。
本発明は、前記第1工程、第2工程及び第3工程を含む、エクオールの製造方法(以下、該製造方法を「第3製造方法」と表記することもある)を提供する。
本発明の第3製造方法に含有される第1工程では、E1ポリペプチドからなる酵素を、NADPH及び/又はNADHの存在下で、ダイゼインに作用させることにより、ダイゼインをジヒドロダイゼインに変換する。本発明の第3製造方法に含有される第1工程は、前述の第1工程と同様に説明される。
本発明の第3製造方法に含有される第2工程では、E2ポリペプチドからなる酵素を、NADPH及び/又はNADHの存在下で、ジヒドロダイゼインに作用させることにより、ジヒドロダイゼインをテトラヒドロダイゼインに変換する。第2工程は、前述の第2工程と同様に説明される。
本発明の第3製造方法に含有される第3工程では、E3ポリペプチドからなる酵素を、テトラヒドロダイゼインに作用させることにより、テトラヒドロダイゼインをエクオールに変換する。第3工程は、前述の第3工程と同様に説明される。
E3ポリペプチドからなる酵素が0.0001~1.0重量%、好ましくは0.001~0.1重量%、更に好ましくは0.001~0.01重量%;
テトラヒドロダイゼインが0.0001~10.0重量%、好ましくは0.001~1.0重量%、更に好ましくは0.001~0.1重量%;及び
NADPH及び/又はNADHが0.01~5重量%、好ましくは0.05~1重量%、更に好ましくは0.1~0.5重量%。
本発明の第3製造方法では、前記E1ポリペプチドからなる酵素、E2ポリペプチドからなる酵素及びE3ポリペプチドからなる酵素が使用される。E1~E3ポリペプチドは、前述と同様に説明される。
本発明は、第1工程~第3工程を含むエクオールの製造方法(第3製造方法)により製造された、エクオールを含有する生成物を提供する。
本発明は、以下の、ダイゼインからジヒドロダイゼインを生成する第4工程、ジヒドロダイゼインからテトラヒドロダイゼインを生成する第5工程、及びテトラヒドロダイゼインからエクオールを生成する第6工程のうち少なくとも2つの工程を含む、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造方法(以下、該製造方法を「第4製造方法」と表記することもある)を提供する。
(Ad)配列番号4に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ae)配列番号1に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Af)前記(Ad)又は(Ae)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つダイゼインを基質としジヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
(Bd)配列番号10に記載のヌクレオチド配列からなるポリヌクレオチド;
(Be)配列番号7に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Bf)前記(Bd)又は(Be)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
(Cd)配列番号16に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ce)配列番号13に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Cf)前記(Cd)又は(Ce)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つテトラヒドロダイゼインを基質としエクオールを生成する活性を有するポリペプチドをコードするポリヌクレオチド。
第4工程では、前記A-2.セクションに記載のE1ポリヌクレオチドを有する組換え細胞を、ダイゼインに作用させることにより、ダイゼインをジヒドロダイゼインに変換する。
第4工程で使用される組換え細胞はE1ポリヌクレオチドを有し、E1ポリヌクレオチドを発現できる限り制限されない。具体的には、前記A-4.セクションに記載の組み換え細胞を使用することが出来る。
E1ポリヌクレオチドが導入された組換え細胞を培養し、細胞及び/又は培養物からE1ポリペプチドを回収することにより、E1ポリペプチドからなる酵素を製造することができる。具体的には、前記A-5.セクションに記載の条件に従って当該組換え細胞を培養することによってE1ポリペプチドを製造することが出来る。
第5工程では、前記B-2.セクションに記載のE2ポリヌクレオチドを有する組換え細胞を、ジヒドロダイゼインに作用させることにより、ジヒドロダイゼインをテトラヒドロダイゼインに変換する。
第5工程で使用される組換え細胞は前記B-2.セクションに記載のE2ポリヌクレオチドを有し、E2ポリヌクレオチドを発現できる限り制限されない。具体的には、前記B-4.セクションに記載の組み換え細胞を使用することが出来る。
E2ポリヌクレオチドが導入された組換え細胞を培養し、細胞及び/又は培養物からE2ポリペプチドを回収することにより、E2ポリペプチドからなる酵素を製造することができる。
第6工程では、前記C-2.セクションに記載のE3ポリヌクレオチドを有する組換え細胞を、テトラヒドロダイゼインに作用させることにより、テトラヒドロダイゼインをエクオールに変換する。
第6工程で使用される組換え細胞は前記C-2.セクションに記載のE3ポリヌクレオチドを有し、E3ポリヌクレオチドを発現できる限り制限されない。具体的には、前記C-4.セクションに記載の組換え細胞を使用することが出来る。
E3ポリヌクレオチドが導入された組換え細胞を培養し、細胞及び/又は培養物からE3ポリペプチドを回収することにより、E3ポリペプチドからなる酵素を製造することができる。
本発明の第4製造方法では、E1~E3ポリヌクレオチドの少なくとも1種が使用される。これらのポリヌクレオチドは、各々前記A-2.、B-2.及びC-2.セクションに説明されるものである。
本発明は、第4製造方法により製造された、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールを含有する生成物を提供する。
本発明は、以下の第1反応槽~第3反応槽の少なくとも1つの反応槽を備える、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造装置(以下、「第1製造装置」と表記することもある)を提供する。
(第1反応槽)(Aa)~(Ac)のいずれかであるポリペプチド(すなわち、E1ポリペプチド)からなる酵素が固定されている反応手段(以下、これを「反応手段1」と表記することもある)を有しており、該ポリペプチドを用いてダイゼインからジヒドロダイゼインを製造するための反応槽、ここで該反応手段は該反応槽内のダイゼインと接触できるように配置されている;
(第2反応槽)(Ba)~(Bc)のいずれかであるポリペプチド(すなわち、E2ポリペプチド)からなる酵素が固定されている反応手段(以下、これを「反応手段2」と表記することもある)を有しており、該ポリペプチドを用いてジヒドロダイゼインからテトラヒドロダイゼインを製造するための反応槽、ここで該反応手段は該反応槽内のジヒドロダイゼインと接触できるように配置されている;
(第3反応槽)(Ca)~(Cc)のいずれかであるポリペプチド(すなわち、E3ポリペプチド)からなる酵素が固定されている反応手段(以下、これを「反応手段3」と表記することもある)を有しており、該ポリペプチドを用いてテトラヒドロダイゼインからエクオールを製造するための反応槽、ここで該反応手段は該反応槽内のテトラヒドロダイゼインと接触できるように配置されている。
本発明の第1製造装置において使用される第1反応槽~第3反応槽の形状、大きさ、素材等は、前記反応手段を有することができ、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造が好適に実施される限り制限されない。
本発明の第1製造装置において使用される反応手段1~3は、各々の反応手段が有する前記ポリペプチドからなる酵素が固定されており、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造に好適に適用できる限り制限されない。
本発明の第1製造装置において使用される供給手段は、本発明の第1製造装置において使用され得る異なる反応槽を該供給手段を介して接続させることができ、本発明の第1製造装置においてジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールを製造できる限り制限されない。該供給手段は、従来公知の技術に従い適宜選択される。
本発明の第1製造装置において使用されるE1~E3ポリペプチドは、前述と同様に説明される。
第1反応槽では、反応手段1に固定されたE1ポリペプチドからなる酵素を、NADPH及び/又はNADHの存在下で、ダイゼインに作用させることにより、ダイゼインをジヒドロダイゼインに変換する。E1ポリペプチドからなる酵素は、E1ポリペプチドからなる酵素の補酵素として作用するNADPH及び/又はNADHとともに、反応手段に固定されていても良い。第1反応槽における反応は、前述の「第1工程」に基づき実施される。
第2反応槽では、反応手段2に固定されたE2ポリペプチドからなる酵素を、NADPH及び/又はNADHの存在下で、ジヒドロダイゼインに作用させることにより、ジヒドロダイゼインをテトラヒドロダイゼインに変換する。E2ポリペプチドからなる酵素は、E2ポリペプチドからなる酵素の補酵素として作用するNADPH及び/又はNADHとともに、反応手段に固定されていても良い。第2反応槽における反応は、前述の「第2工程」に基づき実施される。
第3反応槽では、反応手段3に固定されたE3ポリペプチドからなる酵素を、テトラヒドロダイゼインに作用させることにより、テトラヒドロダイゼインをエクオールに変換する。第3反応槽における反応は、前述の「第3工程」に基づき実施される。
本発明は、以下の第4反応槽~第6反応槽の少なくとも1つの反応槽を備える、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造装置(以下、「第2製造装置」と表記することもある)を提供する。
(第4反応槽)(Ad)~(Af)のいずれかであるポリヌクレオチド(すなわち、E1ポリヌクレオチド)を有する組換え細胞が固定されている反応手段(「以下、これを反応手段4と表記することもある」)を有しており、該反応手段を用いてダイゼインからジヒドロダイゼインを製造するための反応槽、ここで該反応手段は反応槽内のダイゼインと接触できるように配置されている;
(第5反応槽)(Bd)~(Bf)のいずれかであるポリヌクレオチド(すなわち、E2ポリヌクレオチド)を有する組換え細胞が固定されている反応手段(「以下、これを反応手段5と表記することもある」)を有しており、該反応手段を用いてジヒドロダイゼインからテトラヒドロダイゼインを製造するための反応槽、ここで該反応手段は反応槽内のジヒドロダイゼインと接触できるように配置されている;
(第6反応槽)(Cd)~(Cf)のいずれかであるポリヌクレオチド(すなわち、E3ポリヌクレオチド)を有する組換え細胞が固定されている反応手段(「以下、これを反応手段6と表記することもある」)を有しており、該反応手段を用いてテトラヒドロダイゼインからエクオールを製造するための反応槽、ここで該反応手段は反応槽内のテトラヒドロダイゼインと接触できるように配置されている。
本発明の第2製造装置において使用される第4反応槽~第6反応槽の形状、大きさ、素材等は、前記反応手段を有することができ、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造が好適に実施される限り制限されない。
本発明の第2製造装置において使用される反応手段4~6は、各々の反応手段が有する前記組換え細胞が固定されており、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造に好適に適用できる限り制限されない。
本発明の第2製造装置において使用される供給手段は、本発明の第2製造装置において使用され得る異なる反応槽を該供給手段を介して接続させることができ、本発明の第2製造装置においてジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールを製造できる限り制限されない。該供給手段は、従来公知の技術に従い適宜選択される。
本発明の第2製造装置において使用される組換え細胞は、前述の「第4工程で使用される組換え細胞」、「第5工程で使用される組換え細胞」及び「第6工程で使用される組換え細胞」等の記載と同様に説明される。
本発明の第2製造装置において使用されるE1~E3ポリヌクレオチドは、前述と同様に説明される。
第4反応槽では、反応手段4に固定された、E1ポリヌクレオチドを有する組換え細胞を用いて、ダイゼインをジヒドロダイゼインに変換する。第4反応槽における反応は、前述の「第4工程」等の記載に基づき実施される。
第5反応槽では、反応手段5に固定された、E2ポリヌクレオチドを有する組換え細胞を用いて、ジヒドロダイゼインをテトラヒドロダイゼインに変換する。第5反応槽における反応は、前述の「第5工程」等の記載に基づき実施される。
第6反応槽では、反応手段6に固定された、E3ポリヌクレオチドを有する組換え細胞を用いて、テトラヒドロダイゼインをエクオールに変換する。第6反応槽における反応は、前述の「第6工程」等の記載に基づき実施される。
本発明は、前述の第1反応槽~第3反応槽の少なくとも1つの反応槽、及び第4反応槽~第6反応槽の少なくとも1つの反応槽を備える、ジヒドロダイゼイン、テトラヒドロダイゼイン及び/又はエクオールの製造装置(以下、「第3製造装置」と表記することもある)を提供する。
参考例A1
ラクトコッカス20-92株(FERM BP-10036号)を、ダイゼイン含有増殖用液体培地(ダイゼインを10μg/mLとなる量で変法GAMブイヨン培地(日水製薬株式会社)に添加したもの)に接種し、嫌気的条件下(BBL Gas Pack systemsを使用)、37℃で7から18時間適宜培養した。培養後、遠心分離により集菌して冷凍保存し、以下の実施例に使用した。
保存してある凍結菌体(67mL分、2本)を融解後8,000rpm、4℃、10分間遠心分離し、沈渣を以下の試験に供した。沈渣を、1 mM PMSF(和光純薬工業株式会社)及び2mM DTT(和光純薬工業株式会社)、5 mM Sodium hydrosulfite(和光純薬工業株式会社)を含有する0.1 M リン酸カリウム溶液2mLに懸濁させた。懸濁液をあらかじめ0.1 mm zirconia/silica beads(BioSpec Products, Inc.)を入れておいた2ml容スクリューキャップ付チューブ(株式会社アシスト製)2本に移し、FastPrep(登録商標) FP100A(Thermo ELECTRON CORPORATION)にて菌体を破砕し(6500rpm 10秒 氷冷を8回)、菌体破砕液を得た。得られた菌体破砕液を約10,000rpm、4℃にて10分間遠心して遠心上清を得、遠心上清を1 mM PMSF及び2mM DTT、5 mM Sodium hydrosulfiteを含有する0.1 M リン酸カリウム溶液で4.5mLに希釈し、これを酵素源とした。
酵素反応液組成

培養ボトル当たり67 mlの変法GAMブイヨン培地(日水製薬株式会社)にて18時間培養したラクトコッカス20-92株菌体9本を遠心後、2 mM DTT(1, 4-Dithiothreitol、Merck)、5 mM Sodium hydrosulfite(和光純薬工業株式会社)及びプロテアーゼインヒビター(コンプリートプロテアーゼインヒビターカクテルEDTA-free、Roche Diagnostics)を含有する0.1 M リン酸カリウム緩衝液pH 7に縣濁させた。懸濁液を0.1 mm zirconia/silica beads(BioSpec Products, Inc.)を入れた2ml容スクリューキャップ付チューブ(株式会社アシスト製)3本に移し、FastPrep(登録商標) FP100A(Thermo ELECTRON CORPORATION)にて菌体を破砕し(6500rpm、20秒を4回)、破砕液を遠心して上清を得た。また、培養ボトル当たり200mlの同液体培地にて18時間培養したラクトコッカス20-92株菌体8本を同様に菌体破砕後遠心し8本分の上清を得た。
カラム: TSKgel Ether-5PW
流速: サンプルアプライ時0.05~0.1 ml/分、溶出時 0.1 ml/分
溶離液A: 0.1 M リン酸カリウム緩衝液pH 7/2mM DTT/2.5 mM Sodium hydrosulfite/1% 2-プロパノール
溶離液B: 1M 硫安を含む溶離液A
カラム: Mono Q PC 1.6/5
溶離液C: 0.1 M リン酸カリウム緩衝液pH 7.5/3 mM DTT/2.5 mM Sodium hydrosulfite/1% 2-プロパノール
溶離液D: 1M NaClを含む溶離液C
酵素活性は0.4~0.46 M NaCl画分(フラクション No. 28~30)に認められた。
70μlのMono Q HPLCで得たジヒドロダイゼイン合成活性のあるフラクション、No. 29に、30μlの0.1 %トリフルオロ酢酸(TFA)を加えて100μlとした。
Met Lys Asn Lys Phe Tyr Pro Lys Thr Phe Glu Arg Gly Tyr Ile Gly Asn Leu Glu Val Glu Asn (配列番号19)
ジヒドロダイゼイン合成活性を示す酵素タンパク質を消化酵素で断片化しペプチドとした。ペプチドのN末端アミノ酸配列分析を行って、内部アミノ酸配列情報を得た。
培養ボトル当たり200 mlの変法GAMブイヨン培地(日水製薬株式会社)にて18時間培養したラクトコッカス20‐92株菌体3本を遠心後、2 mM DTT(1, 4-Dithiothreitol、Merck)、5 mM Sodium hydrosulfite(和光純薬工業株式会社)及びプロテアーゼインヒビター(コンプリートプロテアーゼインヒビターカクテルEDTA-free、Roche Diagnostics)を含有する0.1 M リン酸カリウム緩衝液pH 7に縣濁させた。懸濁液を0.1 mm zirconia/silica beads(BioSpec Products, Inc.)を入れた2ml容スクリューキャップ付チューブ(アシスト)3本に移し、FastPrep(登録商標) FP100A(Thermo ELECTRON CORPORATION)にて菌体を破砕し(6500rpm、20秒を4回)、破砕液を遠心して上清を得た。
切り出したゲル片は細断し、50 %アセトニトリル水溶液を用いて脱色した後アセトニトリルで脱水し、遠心濃縮機(SpeedVac A160、Savant)にて乾燥させた。55 mM のDTTを含む100 mM炭酸水素アンモニウム水溶液を加え、56 ℃で1時間還元した。DTT溶液を除き、100 mMのヨードアセトアミドを含む100 mM炭酸水素アンモニウム水溶液を加え、遮光下、室温で30 分間ゆるく振とうしてカルボキサミドメチル化処理を行った。反応試薬を除去後、50 %アセトニトリル水溶液、アセトニトリル、100 mM炭酸水素アンモニウム水溶液、及びアセトニトリルの順に洗浄し、ゲルを遠心濃縮機にて乾燥した。2 μgのAchromobacter protease I(和光純薬工業株式会社)を含む0.02 % Tween 20入り20 mMトリス塩酸緩衝液 pH 9を加え、37℃で7 時間消化した。遠心上清を別チューブに移し、ゲルに60 % アセトニトリル- 0.1 % TFA水溶液を加えて30 ℃で20 分間加温後、10 分間ボルテックスする操作を3回行って断片化したペプチドを抽出した。集めた上清液はUltrafree-MC(0.22μm、Amicon)を用いてろ過した後、遠心濃縮した。
逆相HPLCを行い、酵素消化後のペプチドを分離した。
カラム: μRPC C2/C18 SC2.1/10(GEヘルスケアバイオサイエンス)
流速: 0.1 ml/分
溶離液E: 0.05 % TFA
溶離液F: 90 % アセトニトリル/0.04 % TFA
溶出プログラム: 0 分 5 %F
3 分 5 %F
43 分 65 %F
48 分 100 %F
68 分 100 %F
フラクション: 30μl
検出: 215 nm
なお、例えば上記「0 分 5 %B」は、溶出0時間時には溶離液Eが95%、溶離液Fが5%含有された溶離液を使用したことを示す。
コントロールのクロマトグラムとの比較により、ジヒドロダイゼイン合成活性を示す酵素タンパク質由来のペプチドピークを選び、プロテインシーケンサ(アプライドバイオシステムズ、Procise 492HT)にてN末端アミノ酸配列分析を行った。逆相HPLCで分離開始後、20.6分から溶出し始めたピークのアミノ酸配列(以下、Peptide1とする)は次の通りであった。
PheAspGluProValTyrProGlnAlaGlu(配列番号20)
22.1分から溶出し始めたピークのアミノ酸配列(以下、Peptide2とする)は次の通りであった。
AlaSerArgMetValMetAspAlaValHisGluGlyTyrIleAlaGly(配列番号21)
26.6分から溶出し始めたピークは、非特異的に切断されたペプチドであったが、次に示すように当該酵素タンパク質のN末端から13残基目のグリシンをN末とするペプチドであった(以下のアミノ酸配列を、Peptide3とする)。
GlyTyrIleGlyAsnLeuGluValGluAsnArgAlaIleArgMetProMet(配列番号22)
図4に、実施例A4においてペプチドマッピングを行った結果及び各ピークに対応するペプチドのアミノ酸配列を示す。
ピーク1(20.6 分)
ピーク2(22.1分)
ピーク3(26.6分)
上記実施例A3及びA4で得られたN末端及び部分アミノ酸配列を基にDegenerative-プライマーを設計、作製し、ラクトコッカス20-92株のゲノムDNAを鋳型にDegenerative-PCRを行うことによりジヒドロダイゼイン合成酵素をコードする遺伝子の増幅を試みた。
変法GAMブイヨン培地(日水製薬株式会社) 40mLで嫌気培養したラクトコッカス20-92株を5000rpm、4℃、10分間で遠心し、培地をデカンテーションで除去し、菌体を集めた。集めた菌体は直ちにQIAGEN Genomic DNA Buffer Set(キアゲン)のB1溶液11mL (200μg/mL RNaseを含有)に懸濁し、更に300μLのLysozyme溶液(100mg/mL)、500μLのQIAGEN Proteinase K solution(キアゲン)を加えて、37℃、16時間インキュベーションした。次いで、B2溶液4mL加え、数回転倒混和した後、50℃、3時間インキュベーションした。
Degenerative-プライマーの設計では縮退数を小さくするため、5’側の配列はラクトコッカス ガルビエのコドンユーセイジ情報(Codon Usage Database http://www.kazusa.or.jp/codon/)に基づき、各アミノ酸において最も出現頻度が高いコドンを採用した。3’側は混合塩基を利用することにより、ジヒドロダイゼイン合成酵遺伝子とのミスマッチが起こらないように工夫した。
E1-N-terminal-31:TGAAGAATAANTTNTAYCCNAARACNTTYGA(配列番号23)
(ただし、配列番号23において、位置11及び14の「N」はイノシンを示し、位置20及び26の「N」はアデニン、グアニン、シトシンまたはチミンを示す。)
E1-N-terminal-37:TGAAGAATAANTTNTAYCCNAARACNTTYGARRGNGG(配列番号24)
(ただし、配列番号24において、位置11、14、20及び26の「N」はイノシンを示し、位置35の「N」はアデニン、グアニン、シトシンまたはチミンを示す。)
E1-N-terminal-F32:ATGAAGAATAAGTTTTAYCCNAARACNTTYGA(配列番号25)
(ただし、配列番号25において、位置21及び27の「N」はアデニン、グアニン、シトシンまたはチミンを示す。)
E1-internal-RP1:CCTGCAATATAACCTTCATGTACNGCRTCCATNACCAT(配列番号26)
(ただし、配列番号26において、位置24及び33の「N」はアデニン、グアニン、シトシンまたはチミンを示す。)
上記のDegenerative-プライマーを用いてDegenerative-PCRによるジヒドロダイゼイン合成酵素遺伝子の増幅を試みた。
Degenerative-PCRに用いたDegenerative-プライマーの組み合わせを以下に示す。
[1]E1-N-terminal-31とE1-internal-RP1
[2]E1-N-terminal-37とE1-internal-RP1
[3]E1-N-terminal-F32とE1-internal-RP1
Degenerative-PCRによるジヒドロダイゼイン合成酵素遺伝子の増幅はEx-Taq DNA polymerase(タカラバイオ株式会社)を使用し、増幅プログラム:
95℃ 2min、(95℃ 45sec、38℃-54℃ 30sec、72℃ 2min)×50cycles、72℃ 3minで行った。Degenerative-プライマーのゲノムDNAへのミスマッチを考慮し、アニーリングの温度は38℃から4℃刻みに54℃までの5段階で行った。PCR反応終了後、PCR産物に1/10量の10×Dyeを加え、6μLを0.8%のアガロースゲルで電気泳動した。本実施例におけるアガロース電気泳動ではエチジウムブロミドによる染色で行い、分子量マーカーにはλ/StyI(株式会社ニッポンジーン)、100bp ladder(東洋紡績株式会社)を使用した。
増幅した約1.9kbのDNA断片(アニーリング温度54℃)をアガロースゲルから切り出し、Gel-Extraction kit(キアゲン)を用いて精製した。精製したDNA断片はpT7-Blue Cloning Vector(Novagen)に挿入し、塩基配列を決定した。
実施例A5で得られた1.9kbのジヒドロダイゼイン合成酵素遺伝子の5’端及び3’端の配列を決定し、全塩基配列の決定をおこなうため、ラクトコカッス20-92株のゲノムDNAライブラリーを鋳型にcDNA末端配列の急速増幅(5’-、3’-RACE(rapid amplification of cDNA ends))を行った。
実施例A5で精製したラクトコカッス20-92株のゲノムDNAを制限酵素(BamHI,EcoRI,HindIII,KpnI,PstI,SacI,SalI,Sau3AI,XhoI)(いずれもタカラバイオ株式会社)により37℃、16時間消化することで断片化し、フェノール・クロロホルム処理後、エタノール沈殿にて精製した。精製したゲノムDNA断片はあらかじめ相当する制限酵素で切断処理し、Shrimp Alkaline Phosphatase(タカラバイオ株式会社)処理により脱リン酸化しておいたpUC19クローニング ベクターへTaKaRa Ligation kit var.2.1(タカラバイオ株式会社)を用いてライゲーションし、ゲノムDNAライブラリーを作製した。
ゲノムDNAライブラリー(ライゲーション反応液)を滅菌水で20倍希釈し、1μLを鋳型に使用して、cDNA末端配列の急速増幅(5’-RACE、3’-RACE)によるジヒドロダイゼイン合成酵素遺伝子の5’端及び3’端の配列の増幅を試みた。
5’-RACE、3’-RACEにそれぞれ用いたプライマーの組み合わせを以下に示す。
First-PCR:E1-RACE-N-P1とpUC19-FP-1、E1-RACE-N-P1とpUC19-RP-1
Nested-PCR:E1-RACE-N-P2とpUC19-FP-2、E1-RACE-N-P2とpUC19-RP-2
First-PCR:E1-RACE-RP2-1とpUC19-FP-1、E1-RACE-RP2-1とpUC19-RP-1
Nested-PCR:E1-RACE-RP2-2とpUC19-FP-2、E1-RACE-RP2-2とpUC19-RP-2
5’-RACE、3’-RACEにそれぞれ用いたプライマーの配列を以下に示す。
ベクター側プライマー:
pUC19-FP-1: ACACAGGAAACAGCTATGACCATGATTACG(配列番号27)
pUC19-RP-1: AGCTGGCGAAAGGGGGATGTGCTGCAAGGC(配列番号28)
pUC19-FP-2: ATGATTACGCCAAGCTTGCATGCCTGCAGG(配列番号29)
pUC19-RP-2: CCAGTCACGACGTTGTAAAACGACGGCCAG(配列番号30)
ジヒドロダイゼイン合成酵素遺伝子側プライマー
E1-RACE-N-P1: ATGCGGATCGCTCGGTTCTCGACCTCTAGGTTAC(配列番号31)
E1-RACE-RP2-1: ATCGAGGAGAAGTGCGAGGACGTCAGGGTCATC(配列番号32)
E1-RACE-N-P2: TTCTCGACCTCTAGGTTACCGATGTAGCCGC(配列番号33)
E1-RACE-RP2-2: ACGTCAGGGTCATCGGCATCGGCGACTGCAAG(配列番号34)
5’-RACE、3’-RACEにはEx-Taq DNA polymerase(タカラバイオ株式会社)を使用し、First-PCR及びNested-PCRを同増幅プログラム:
95℃ 2min、(95℃ 45sec、60℃ 30sec、72℃ 1min)×30cycles、72℃ 3minで行った。First-PCRでは鋳型に調製方法を上記記載したゲノムDNA希釈液1μL(40ng)を使用し、Nested-PCRには0.5μLのFirst-PCR産物を使用した。
実施例A5で示したDegenerative-PCR及び本実施例内(2)に示したcDNA末端配列の急速増幅により得られたDNA塩基配列をDNAシークエンス-アセンブルソフトウェアSEQUENCHER(Gene Codes Inc、USA)を使用してアセンブル解析を行った。その結果、3548bpのジヒドロダイゼイン合成酵素遺伝子周辺のゲノム構造を決定し、ジヒドロダイゼイン合成酵素遺伝子は1935ヌクレオチド、644アミノ酸からなるポリペプチドであることが判明した。
本実施例内(3)で得られた配列にはPCRでの増幅の際、DNAポリメラーゼの塩基取り込みエラーによるクローン間で塩基の異なる箇所が多数見られた。そのため、First strand cDNAを鋳型にして、High-FidelityDNA-PolymeraseであるEasy-A(R) High-Fidelity PCR Cloning Enzyme (Stratagene)を使用したPCRにより、ジヒドロダイゼイン合成酵素遺伝子のコーディング領域を含んだ領域(2368bp)を増幅させた。
使用した増幅プライマーを以下に示す。
E1-conf-NP:TGCCGGTGCAATGGCTGACATCATGTTCAACCTG(配列番号35)
E1-conf-CP:TCCTCCATCGTTCCTCCAATCAGTAAGACACGCG(配列番号36)
得られたDNA断片はGel-Extraction kit(キアゲン)を用いて精製し、ダイレクトシークエンスにより、配列の確認、最終決定を行った。
実施例A1で示したようにラクトコッカス20-92株の増殖用培養液に基質であるダイゼインを添加した場合、培養された菌体はジヒドロダイゼイン合成活性を有する。このことから増殖培養液中へのダイゼインの添加により、ジヒドロダイゼイン合成酵素遺伝子の転写が誘導され、更にタンパク質へと翻訳されることが推測されるため、ダイゼインを添加した培地及び添加していない培地でそれぞれ培養したラクトコッカス20-92株菌体由来のcDNAを作製し、RT-PCRを行うことで増殖培養液中へのダイゼインの添加によりジヒドロダイゼイン合成酵素遺伝子の発現が誘導されるか否かについて検討した。
ラクトコッカス20-92株からの全RNAの抽出、精製は以下の方法で行った。
DNaseI処理を施した全RNA 2μgからSuperScript(登録商標)First-Strand Synthesis System for RT-PCR(Invitrogen)を用いてfirst-strand cDNA(逆転写物)を合成した。DNA合成のための伸長プライマーには付属のランダムヘキサマーミックスを用いてマニュアルに従いfirst-strand cDNAの合成を行った。また、first-strand cDNAの合成に用いたDNase I処理済みの全RNAへのゲノムDNAの混入が無いことを確認するため、逆転写を行わない反応も併せて行った。RT-PCRの鋳型には最終反応液を使用した。
1.ダイゼイン添加培地で培養した菌体由来全RNAの逆転写物(DZN(+)RT(+))
2.ダイゼイン無添加培地で培養した菌体由来全RNAの逆転写物(DZN(‐)RT(+))
3.ダイゼイン添加培地で培養した菌体由来全RNAの無逆転写物(DZN(+)RT(‐))
4.ダイゼイン無添加培地で培養した菌体由来全RNA無逆転写物(DZN(‐)RT(‐))
本実施例内(2)で示した4種類の最終反応物を鋳型にしてRT-PCRを行うことにより、ジヒドロダイゼイン合成酵素遺伝子を増幅し、発現量を比較した。コントロール遺伝子として16S-リボゾームRNA配列を採用し、併せてRT-PCRを行った。
RT-PCRにはEx-Taq DNA polymerase(タカラバイオ株式会社)を使用し、増幅プログラム:
95℃ 2min、(95℃ 30sec、56℃ 20sec、72℃ 30sec)×30cycles、72℃ 2minで行った。鋳型には本実施例内(2)の最終反応液1μLを使用した。
ジヒドロダイゼイン合成酵素:239bp
E1-FP:CTACATCGGTAACCTAGAGGTCG(配列番号37)
E1-RP:CCGTGCTGCTTGATGGTCTTTGC(配列番号38)
16S-リボゾームRNA配列:326bp
Gar-16S-Ribo-FP:TGCGTAGATATATGGAGGAAC(配列番号39)
Gar-16S-Ribo-RP:CTTATCTCTAAGGATAGCACG(配列番号40)
大腸菌を用いた組換えタンパク質発現システムであるpETシステムを用いてE1ポリペプチドを発現させ、そのジヒドロダイゼイン合成活性を確認した。
E1ポリペプチド発現ベクター(pET21-E1-His)を作製する目的でPCRにより、E1ヌクレオチドのオープン・リーディング・フレーム領域のDNAを増幅した。
exp.E1 pet F Nde:AGCTCATATGAAGAACAAGTTCTATCCGAA (配列番号:41)
exp. E1 pet His:AATCGAATTCCTACAGGTTGCAGCCAGCGATGT (配列番号:42)
上記増幅プライマー:exp.E1 pet F Nde、exp.E1 pet HisにはpET21a(Novagen)へ挿入するため、それぞれ制限酵素NdeI切断部位、EcoRI切断部位配列を含ませてある。
組換えE1ポリペプチドを発現するプラスミドpET21-E1-His とpET21a(ネガティブコントロール)を用いて、大腸菌BL21(DE3)株(Novagen)を形質転換した。形質転換体はアンピシリン(50μg/mL)を含むLB培地アガープレート上で37℃、終夜生育させ、シングルコロニーを得た。
本実施例内(2)で得た菌体破砕液を酵素源として、ダイゼインからジヒドロダイゼインへの変換活性を測定し、発現させた組換えE1ポリペプチドにその活性を確認した。
酵素反応液組成

参考例B1 シス-テトラヒドロダイゼイン及びトランス-テトラヒドロダイゼインの合成
シス-テトラヒドロダイゼイン及びトランス-テトラヒドロダイゼインを、以下のフローに従って製造した。なお、以下、化合物の表記に関して、下記の略記を使用する。
化合物1(ダイゼイン): 4’,7-ジヒドロキシイソフラボン
化合物2: 4’,7-ジアセトキシイソフラボン
化合物3: 4’,7-ジアセトキシイソフラバン-4-オン
化合物4: シス-4’,7-ジアセトキシイソフラバン-4-オール
化合物5: トランス-4’,7-ジアセトキシイソフラバン-4-オール
化合物6: シス-テトラヒドロダイゼイン
化合物7: トランス-テトラヒドロダイゼイン

ダイゼイン(化合物1) 500 mg(1.97 mmol)のピリジン(5 mL)溶液に無水酢酸0.76 mL(8.0 mmol)を加え、60℃で2時間撹拌した。反応液に少量のメタノールを加えた後に、3N塩酸中に注いだ。水で希釈した後に析出した固体を濾取し、これを水洗した。得られた固体を室温にて風乾して白色粉末状の化合物2 609 mg(1.80 mmol、91%収率)を得た。
化合物2:1H NMR(250 MHz, CDCl3)δ(ppm): 2.33 (3H, s), 2.37 (3H, s), 7.13-7.22 (3H, m), 7.32 (1H, d, J = 2.3 Hz), 7.54-7.63 (2H, m), 8.01 (1H, s), 8.33 (1H, d, J = 8.8 Hz).
化合物2 400 mg(1.18 mmol)及び10%パラジウム-炭素(含水品、約50 wt%)150 mgのメタノール(6 mL)-酢酸エチル(6 mL)懸濁液を水素雰囲気下、室温で2時間撹拌した。反応液をセライト濾過し、残渣を酢酸エチルで洗浄した。濾液を濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル:ジクロロメタン/酢酸エチル= 100/0 - 19/1)で精製して、白色粉末状の化合物3 312 mg(0.917 mmol、78%収率)を得た。
化合物3:1H NMR (250 MHz, CDCl3) δ(ppm): 2.29 (3H, s), 2.32 (3H, s), 3.93-4.04 (1H, m), 4.60-4.75 (2H, m), 6.76-6.84 (2H, m), 7.04-7.13 (2H, m), 7.27-7.35 (2H, m), 7.93-8.01 (1H, m).
化合物3 100 mg(0.294 mmol)のメタノール(1 mL)-ジクロロメタン(1 mL)溶液に0℃で水素化ホウ素ナトリウム11 mg(0.29 mmol)を加え、0℃で30分撹拌した。反応液に1N塩酸2 mL、水50 mLを順に加え、酢酸エチルで抽出した。有機層を飽和重曹水、飽和食塩水の順に洗浄し、無水硫酸ナトリウムで乾燥した。溶媒を留去して得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル:ジクロロメタン/酢酸エチル= 19/1-3/1)で精製した。得られたジアスレレオマー混合物を中圧カラムクロマトグラフィー(山善製ウルトラパックSI-40B:n-へキサン/酢酸エチル = 3/2)で精製して、無色針状の化合物4 44 mg(0.13 mmol、44%収率)及び無色針状の化合物5 26 mg(75 mmol、26%収率)を得た。化合物4:1H NMR (250 MHz, CDCl3) δ(ppm): 1.80 (1H, d, J = 4.0 Hz), 2.30 (3H, s), 2.31 (3H, s), 3.32 (1H, td, J = 3.5, 11.5 Hz), 4.32 (1H, ddd, J = 1.3, 3.5, 10.5 Hz), 4.59 (1H, dd, J= 10.5, 11.5 Hz), 4.76-4.83 (1H, m), 6.64-6.73 (2H, m), 7.06-7.14 (2H, m), 7.27-7.35 (3H, m).
化合物5:1H NMR (250 MHz, CDCl3) δ(ppm): 2.02 (1H, d, J = 5.5 Hz), 2.29 (3H, s), 2.30 (3H, s), 3.11-3.24 (1H, m), 4.26 (1H, dd, J = 9.0, 11.3 Hz), 4.37 (1H, dd, J = 3.8, 11.3 Hz), 4.92 (1H, dd, J = 5.5, 7.8 Hz), 6.62 (1H, d, J = 2.3 Hz), 6.72 (1H, dd, J= 2.3, 8.5 Hz), 7.04-7.12 (2H, m), 7.21-7.30 (2H, m), 7.47 (1H, d, J = 8.5 Hz).
化合物4 116 mg(0.339 mmol)のメタノール(4 mL)溶液にナトリウムメトキシド55 mg(1.0 mmol)を加え、室温で2時間撹拌した。反応液にイオン交換樹脂(ダウエックス50×8W、アンモニウムフォーム)を加えて中和した後に、樹脂を濾去した。濾液を濃縮して得られた固体をメタノールで洗浄して、白色粉末状の化合物6 62 mg(0.24 mmol、71%収率)を得た。
化合物6:1H NMR (250 MHz, DMSO-d6) δ(ppm): 3.03 (1H, td, J = 3.3, 12.0 Hz), 4.00-4.15 (1H, m), 4.31-4.51 (2H, m, including 4.39, dd, J = 10.3, 12.0 Hz), 4.96 (1H, d, J = 5.8 Hz), 6.18 (1H, d, J = 2.3 Hz), 6.32 (1H, dd, J = 2.3, 8.3 Hz), 6.69 (2H, d, J = 8.5 Hz), 7.00 (1H, d, J = 8.3 Hz), 7.09 (2H, d, J= 8.5 Hz), 9.19 (1H, br s), 9.31 (1H, br s).
化合物5 84 mg(0.25 mmol)のメタノール(2 mL)懸濁液に1N水酸化ナトリウム0.73 mL(0.73 mmol)を加え、室温で2.5時間撹拌した。反応液に飽和塩化アンモニウム水を加え、酢酸エチルで抽出した。有機層を飽和重曹水、飽和食塩水の順に洗浄し、無水硫酸ナトリウムで乾燥した。溶媒を留去して白色粉末状の化合物7 64 mg(0.25 mmol、定量的収率)を得た。
化合物7:1H NMR (250 MHz, DMSO-d6) δ(ppm): 2.82-2.96 (1H, m), 4.04-4.22 (2H, m), 4.60 (1H, t, J = 7.0 Hz), 5.18 (1H, d, J = 7.0 Hz), 6.14 (1H, d, J = 2.3 Hz), 6.34 (1H, dd, J= 2.3, 8.5 Hz), 6.67 (2H, d, J = 8.5 Hz), 7.04 (2H, d, J = 8.5 Hz), 7.16 (1H, d, J = 8.5 Hz), 9.21 (1H, s), 9.28 (1H, s).
ラクトコッカス20-92株(FERM BP-10036号)を、ダイゼイン含有増殖用液体培地に接種し、嫌気的条件下、37℃で7から24時間培養した。培養後、遠心分離により集菌して冷凍保存し、以下の実施例に使用した。
以下の試験を行い、テトラヒドロダイゼインがエクオール生合成の中間代謝物であることを確認した。
-80℃で保存したラクトコッカス20‐92株菌体をすばやく融解、タッピングで懸濁し、8000rpm×5min,4℃で遠心分離VC‐960(タイテック株式会社(製))した。上清を取り除いた後に、0.1Mリン酸カリウム緩衝液/1mM PMSF(PhenylMethaneSulfonyl Fluoride)/2mM DTT(dithiothreitol)/5mM Sodium hydrosulfite(pH7.0)を加え、菌懸濁液を得た。次いで、予め、ジルコニア‐シリカビーズ[0.1mm 1lb(和研薬株式会社(製))を約0.8mL容入れておいた2 mlチューブに菌懸濁液を入れ、更に0.1Mリン酸カリウム緩衝液/1mM PMSF/2mM DTT/5mM Sodium hydrosulfite(pH7.0)を加え、チューブをほぼ一杯にした。次いで、チューブの蓋をして、氷冷しながらFastPrepTM‐FP100A(Thermo ELECTRON CORPORATION(製))を用いて6500rpm×20秒-氷冷の操作を合計4回繰り返して、破砕処理を行った。斯くして得られた菌破砕物を酵素源として酵素反応を行った。
上記(1)の菌破砕物を0.1ml含む下記組成の酵素反応液1mLを調製し、37℃で2時間インキュベートした。インキュベート後、得られた酵素反応生成物に3mLの酢酸エチルを添加して抽出処理を行った後、乾固して、HPLC分析に供した。
0.1Mリン酸カリウム緩衝液/1mM PMSF/2mM DTT/5mM Sodium hydrosulfite(pH7.0)
2 mM NADPH
2 mM NADH
10μg/mL ジヒドロダイゼイン又はテトラヒドロダイゼイン
基質にジヒドロダイゼインを用いて、菌破砕物を酵素源にして得られた酵素反応生成物のHPLC分析の結果を図9に示す。また、上記参考例1で合成したテトラヒドロダイゼインのHPLC分析を図9に併せて示す。この結果から、基質にジヒドロダイゼインを用いて、菌破砕物を酵素源にして得られた酵素反応生成物には、トランス-テトラヒドロダイゼインの保持時間に一致する保持時間を有する中間体の存在が認められ、トランス-テトラヒドロダイゼインが生成していることが明らかとなった。
基質にシス-テトラヒドロダイゼイン又はトランス-テトラヒドロダイゼインを用いて、菌破砕物を酵素源にして得られた酵素反応生成物のHPLC分析の結果を図10に示す。図10から明らかなように、いずれの化合物からもエクオールが生成され、テトラヒドロダイゼインはシス型及びトランス型を問わず、エクオール生合成過程において基質として利用されることが判明した。
保存してある凍結菌体を融解後5000×G、4℃、15分間遠心分離し、沈渣を以下の試験に供した。沈渣(湿重量2.7 g)を、1 mM PMSF及び5 mM Sodium hydrosulfiteを含有する0.1 M リン酸一カリウム溶液10 mlに縣濁させ、37℃で5分間予温した後、Lysozymeを湿重量1g当たり100 mg/ wet weightとなるよう加え、37℃にて1.5時間反応させた。次いで、得られた反応液に、等量の0.1Mリン酸二カリウム溶液を加えた後、更にジルコニア‐シリカビーズ3 ml量を加えVortex Mixerにて激しく攪拌し、更に超音波処理機 (Branson Sonifier Cell Disruptor 200)にて菌体破砕した(5min処理2min休憩のサイクルを3回)。得られた菌体破砕液を約10,000 ×Gにて15 分間遠心して遠心上清を得、これを酵素源とした。

培養ボトル当たり67 mlのダイゼイン含有増殖用液体培地にて20時間培養したラクトコッカス20‐92株菌体10本分を遠心後1 mM PMSF(phenylmethylsulfonyl fluoride)及び4 mM DTT(Dithiothreitol)を含有する0.02 M リン酸カリウム緩衝液pH 7 (以下、「Buffer A」と表記する)に縣濁させ、フレンチプレス(SLM INSTRUMENTS INC)にて菌体を破砕し(1800 psiにて6回)、破砕液を遠心して上清液を得た。また、培養ボトル当たり200mlの液体培地にて18時間培養したラクトコッカス20‐92株菌体5本分を同様にフレンチプレスに供して菌体破砕後遠心し上清を得た。前者及び後者の遠心上清を、それぞれ38 ml及び47mlを混合し、その混合液の82 mlを、予めBuffer Aにて平衡化しておいたRed-Sepharose(約7 ml)に供した。次いで、150 mlのBuffer AにてRed-Sepharoseを洗浄した後、10 mMのNADPHを含むBuffer Aにて溶出した(20 ml/フラクション)。各フラクションを酵素源として用いて、実施例B2に示す酵素反応条件(補酵素としてNADPHを使用)と同条件でテトラヒドロダイゼイン生合成活性について測定し、活性画分としてフラクションNo.1~5を得た。
流速: 1 ml / min
フラクション: 2 ml/2 min/フラクション
溶離液 A: 0.02 M リン酸カリウム緩衝液pH 7/1mM DTT / 2.5 mM Sodiumhydrosulfite/0.5% isoPrOH
溶離液 B: 1M 硫安を含む溶離液A
溶出プログラム: 時間(min) (溶離液B)/(溶離液A+溶離液B)
0 1
5 1
25 0
45 0
流速: 0.6 ml/min
フラクション: 1.2 ml/2 min/フラクション
溶離液: 0.05 Mリン酸カリウム緩衝液pH 7/1mM DTT/2.5 mM Sodium hydrosulfite/1% isoPrOH/0.3M NaCl
上記実施例3で得られたテトラヒドロダイゼイン合成活性を有するフラクション(フラクションNo.7)をサンプルとしてMS分析を行った。具体的には、サンプルをSDS-PAGEにて分離し、バンドを切り出した。切り出したバンドをゲル内還元アルキル化し、トリプシンでゲル内消化した。次いで、トリプシン消化ペプチドを回収・精製し、LC-MSにて分析した。LC-MS分析により取得されたデータについてMS分析支援ソフトウェアであるPEAKSTM(インフォコム株式会社)を用いたde novo シーケンシングを行うことにより、ペプチドのアミノ酸配列を計算・推定した。詳細な方法等については、以下の通りである。
本試験では、SuperSep HG 10/20% (和光純薬工業株式会社)、Flamingo gel staining kit (Bio-Rad)、TCEP(Tris[2-carboxyethyl]phosphine)(Pierce)、分子量マーカー(株式会社アプロサイエンス)、DTT(Calbiochem)、ヨードアセトアミド(和光純薬工業株式会社)、アセトニトリル(関東化学株式会社)、トリプシン(Promega)、TFA(Pierce)、Ammonium Bicarbonate(Sigma)、アンモニア水(メルク株式会社)、ギ酸(関東化学株式会社)、Empore Cation-SR Disk(住友スリーエム株式会社)、MonoCap濃縮カラム(GLサイエンス株式会社)、MonoCap for Nano Flow 0.1X150mm(GLサイエンス株式会社)、FortisTip(エーエムアール株式会社)、SpeedVac Concentrator(SAVANT)、HTS-PALオートサンプラー(CTC-Analytics)、Chorus220(CTC-Analytics)、QSTAR Pulsar i(アプライドバイオシステムズ)、PEAKSTMソフトウェア(インフォコム株式会社)を使用した。
上記実施例B3で得られた、テトラヒドロダイゼイン合成活性を有するフラクション(フラクションNo.7)20μl に100mM TCEPを1μl加え、70℃にて10分還元処理を行った。サンプルをSuperSep HGに全量アプライし、定法に従いSDS-PAGEを行った。泳動後にFlamingo Gel staining kit(Bio-Rad)にて染色した(図14参照)。染色後に確認されたバンドLG1及びLG2をそれぞれ切り出し1mm角程度に裁断した。切り出したゲルを100mM Ammonium Bicarbonate溶液にて洗浄後、アセトニトリルで脱水し、SpeedVac Concentratorにて乾燥固化した。
乾燥したゲル片にDTT溶液(1.54mg/ml in 100mM Ammonium Bicarbonate)を加え55℃にて45分間インキュベートし、還元処理を実施した。DTT溶液を捨て、ヨードアセトアミド溶液(10.1mg/ml in 100mM Ammonium Bicarbonate)を加えて室温遮光条件下で30分間インキュベーションした。溶液を捨て、ゲル片を50%アセトニトリル溶液、100%アセトニトリル溶液、100mM Ammonium Bicarbonate溶液、100%アセトニトリル溶液で順次洗浄し、SpeedVac Concentratorにて乾燥固化した。乾燥したゲル片にトリプシン溶液(12.5μg/ml in 50mM Ammonium Bicarbonate)を少量加え、氷上にて45分間浸透させた。浸透後に余分なトリプシン溶液を取り除き、ゲル片が浸る程度の50mM Ammonium Bicarbonate溶液を加え37℃にて16時間反応させた。
トリプシン消化ペプチドを回収し、Empore Cation-SR Diskをピペットチップに詰めた簡易カラムで前処理精製した。トリプシン消化ペプチドの回収は、0.1%TFA/90%アセトニトリル溶液にて洗浄回収した。簡易カラムの操作は、平衡化0.1%TFA/2%アセトニトリル溶液、サンプル吸着、カラム洗浄0.1%TFA/90%アセトニトリル溶液、サンプル溶出5%アンモニア/30%アセトニトリル溶液の順に行い、溶出させた消化ペプチドをSpeedVac Concentratorにて乾燥濃縮した。消化ペプチド溶液にTFAを加えpH3付近になるよう調整し、オートサンプラーHTS-PALにセットした。HTS-PALにセットしたサンプルを、LC-MS用インジェクターバルブにあるサンプル濃縮カラムにロードし、オンカラム洗浄を行った。濃縮カラムのサンプルを、nanoHPLC-Chorus220により分析カラムにて分離し、分析カラムにセットしたFortisTipにおいてイオン化し、QSTAR Pulsar iにて分析した。LC-MS分析条件は、次の通りである。
(1)Inverse-PCR用ゲノムDNAライブラリーの作製
実施例A5で精製したラクトコッカス20-92株(FERM BP-10036号)のゲノムDNAを以下に示す制限酵素(BamHI,EcoRI,HindIII,KpnI,PstI,SacI,SalI,Sau3AI,XhoI)(いずれもタカラバイオ株式会社)により37℃、16時間消化することで断片化し、フェノール・クロロホルム処理後、エタノール沈殿にて精製した。精製したゲノムDNA断片はTaKaRa Ligation kit var.2.1(タカラバイオ株式会社)を用いてセルフ-ライゲーションした。各ライゲーション溶液を滅菌水で10倍希釈することにより、Inverse-PCR用ゲノムDNAライブラリーを作製した。
上記(1)に記載したInverse-PCR用ゲノムDNAライブラリー1μL(40ng相当)を鋳型に使用して、Inverse-PCRにより、E1ポリヌクレオチド周辺上流及び下流域のゲノムDNAの増幅を試みた。上流側領域増加のInverse-PCRにはPstI、XhoIで処理したものを、下流側領域増加のInverse-PCRにはHindIII,PstI,SacI,XhoIで処理したものをそれぞれ鋳型DNAとして使用した。Inverse-PCRにはTaKaRa LA Taq(タカラバイオ株式会社)を使用した。First-PCRは1×PCR Buffer(Mg2+ free)、プライマー各0.5nM、dNTP 各0.5mM、MgCl2 2.5mM、TaKaRa LA Taq 0.2U を含む20μLの反応液を用いて、ゲノムDNAライブラリー希釈液1μL(40ng)を鋳型に使用し、以下の増幅プログラム: 98℃ 1min、(95℃ 10sec、62℃ 10sec、68℃ 10min)×35cycles、68℃ 15minで行った。更に、続いてNested-PCRをFirst-PCR産物0.5μLを鋳型に1×PCR Buffer(Mg2+ free)、プライマー各0.5nM、dNTP各0.5mM、MgCl2 2.5mM、TaKaRa LA Taq 0.3U を含む30μLの反応液を用いて、以下の増幅プログラム:98℃ 1min、(95℃ 10sec、62℃ 10sec、68℃ 10min)×30cycles、68℃ 15minで行った。
Inverse-PCRに用いたプライマーの組み合わせを以下に示す。
First-PCR:RACE-N-P3-1とE1-Bub-N-P1
Nested-PCR:RACE-N-P3-2とE1-Bub-N-P2
First-PCR:RACE-C-P3-1とE1-Bub-C-P1
Nested-PCR:RACE-C-P3-2とE1-Bub-C-P2
上流側及び下流側のInverse-PCRそれぞれに用いたプライマーの配列を以下に示す。
上流側の増幅プライマー配列
RACE-N-P3-1: ATGGAGATAGTGCCGCTGGCAAGGCAACGGCAC (配列番号:43)
RACE-N-P3-2: TCAACGAAGACTCGATTTGAGCGAGAGGCGAGG (配列番号:44)
E1-Bub-N-P1: ACGGTGGAACCGGCATCGTGTTCATGGACAAC (配列番号:45)
E1-Bub-N-P2: GCGTGACCCAGTTCCACCATGTCGGACTGTC (配列番号:46)
下流側の増幅プライマー配列
RACE-C-P3-1: GACATCCCGTTCGAGCGCAGGATCACCCATGAG (配列番号:47)
RACE-C-P3-2: AGGATCACCCATGAGCGCATCGCTATCATGGAC (配列番号:48)
E1-Bub-C-P1: CATCGCTCTTGCAGTCGTTGTCCAGGAAGTCC (配列番号:49)
E1-Bub-C-P2: TTGTCCAGGAAGTCCATCGCGTACACGACGGAG (配列番号:50)
本実施例(2)で実施したNested-PCR産物に1/10量の10×dayを加え、5μLを0.8%のアガロースゲルで電気泳動し、上流側領域で0.5kb(PstI),3.5kb(XhoI)のDNA断片の増幅を、下流側領域で1kb(HindIII)、1kb(SacI)、2.5kb(XhoI)のDNA断片の増幅を確認した。本実施例におけるアガロース電気泳動ではエチジウムブロミド(株式会社ニッポンジーン)による染色を行い、分子量マーカーにはλ/StyI(株式会社ニッポンジーン)、100bp ladder(東洋紡株式会社)を使用した。増幅したDNA断片をアガロースゲルから切り出し、QIAGEN Gel-Extraction kit(キアゲン)を用いて精製した。精製したDNA断片は増幅に使用したプライマーを使用したダイレクトシークエンス、及びウォーキング法により、その塩基配列を決定した。
本実施例(3)で得られたDNA配列をシーケンス-アセンブルソフトウェアSEQUENCHER(Gene Codes Inc、USA)を使用してアセンブル解析を行い、さらにORFの予測を行った。その結果、E1酵素遺伝子を含めた周辺ゲノム領域6685bpの配列を決定した。解析結果から得られたジヒドロダイゼイン合成酵素遺伝子周辺のゲノム構造の略図を図15に示す。ORF予測の結果、E1酵素遺伝子の上流に3つ(うち一つはN末端未同定)、下流に1つのORFを見出した。見出したORFはジヒドロダイゼイン合成酵素から上流に向かってUpstream(US)1、US2、US3、下流に向かってDownstream(DS)1とした。
上記実施例B4において得られた推定アミノ酸配列を上記実施例B5で決定したジヒドロダイゼイン合成(E1)酵素遺伝子の周辺ゲノムDNA配列データと照合した。その結果、主にLG2より得られたいくつかの配列がORF-US2ヌクレオチド配列から推定されるポリペプチドの配列に一致した。以上のことからORF-US2ポリペプチドがテトラヒドロダイゼイン合成酵素である可能性が示唆された。以下にORF-US2ポリペプチドと一致した消化ペプチド配列を示す。また、図16-1、16-2、16-3にLC-MSにて取得したデータを示す。

ORF-US2ポリペプチドを無細胞系タンパク質合成システム(PURESYSTEM Classic II mini(ポストゲノム研究所))を用いて合成し、そのテトラヒドロダイゼイン合成活性を確認した。
2段階PCRにより、無細胞系タンパク質合成に用いたORF-US2ポリヌクレオチド鋳型DNAを作製した。
ORF-US2用鋳型DNA作製のためのPCRに使用したプライマーを以下に示す。E2-invitroTS-FP1:ACTTTAAGAAGGAGATATACCAATGGCACAGGAAGTCAAAGTCC (配列番号:57)
E2-invitroTS-RP:CTAGACCTCGATCTCGCCCTGCATGCCG (配列番号:58)
Universal-Primer:GAAATTAATACGACTCACTATAGGGAGACCACAACGGTTTCCCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACCA (配列番号:59)
E2-invitroTS-FP1及びE2-invitroTS-RPは実施例B5で決定したORF-US2ヌクレオチド配列を基にシグマ アルドリッチ ジャパンにて合成した。また、Universal-PrimerはPURESYSTEM Classic II mini(ポストゲノム研究所)に付属されていたものを使用した。
ORF-US2ポリペプチドの合成に用いた鋳型DNAはマニュアルに従い、2段階PCRで作製した。本実施例でのPCRにはDNA-ポリメラーゼにEasy-A(登録商標) High-Fidelity PCR Cloning Enzyme (ストラタジーン)、PCR装置はGeneAmp PCR System 9700 (アプライド・バイオシステムズ)を用いた。
-80℃保存しておいたPURESYSTEM Classic II miniのA液25μLとB液10μLを氷上で融解後、混合し、そこへ2段階PCRで作製したORF-US2ポリペプチド合成用鋳型DNA(開始コドンの5‘側上流にT7‐プロモーター配列及びリボゾーム結合配列を有する)を0.6μg(60ng/μLを10μL)加え、さらに滅菌水を加えて計50μLにして37℃、90分間インキュベーションすることで目的のポリペプチドを合成した。このシステムを用いたタンパク質合成のポジティブコントロールにはPURESYSTEM Classic II miniに付属のジヒドロ葉酸レダクターゼ(DHFR)合成用鋳型DNAを0.5μg(0.2μg/μLを2μL)用いた。ネガティブコントロールには鋳型となるDNAは添加せず、滅菌水のみを使用した。活性測定は前記反応液40μLを以下の組成の酵素反応用バッファーに加え、37℃、6時間インキュベーションし、反応終了後、酵素反応液を3mLの酢酸エチルで抽出、乾固し、泳動バッファーで溶解してHPLC分析により、酵素反応液中のテトラヒドロダイゼインを測定した。
0.1Mリン酸カリウム緩衝液/1mM PMSF/2mM DTT/5mM Sodium hydrosulfite(pH7.0)
2 mM NADPH
2 mM NADH
10μg/mL ジヒドロダイゼイン
なお、前記ヒドロ葉酸レダクターゼ(DHFR)合成用鋳型DNAを用いた場合には、当該DNAを基にしてタンパク質が良好に発現されたが、鋳型となるDNAを添加しない場合には、タンパク質は発現されなかった。このことから、当該実験は適切に行われているものと判断される。
大腸菌を用いた組換えタンパク質発現システムであるpETシステムを用いてORF-US2ポリペプチドを発現させ、そのテトラヒドロダイゼイン合成活性を確認した。
ORF-US2ポリペプチド発現ベクター(pET21-US2)を作製する目的でPCRにより、ORF-US2ポリペプチドのオープン・リーディング・フレーム領域のDNAを増幅した。
実施例B5で決定したORF-US2ポリペプチド配列を基に下記の増幅プライマーを作製した。
exp.US2 pet F Nde:TATACATATGGCACAGGAAGTCAAAGTC (配列番号:60)
exp. US2 pet:AATCGAATTCCTAGACCTCGATCTCGCCCTGC (配列番号:61)
組換えORF-US2ポリペプチドを発現するプラスミドpET21-US2 とpET21a(ネガティブコントロール)を用いて、大腸菌BL21(DE3)株(Novagen)を形質転換した。形質転換体はアンピシリン(50μg/mL)を含むLB培地アガープレート上で37℃、終夜生育させ、シングルコロニーを得た。
本実施例内(2)で得た菌体破砕液を酵素源として、ジヒドロダイゼインからテトラヒドロダイゼインへの変換活性を測定し、発現させた組換えORF-US2ポリペプチドにその活性を確認した。
酵素反応液組成
菌体破砕液(酵素源) 100 μl
NADH(100 mM) 20 μl
NADPH(100 mM) 20 μl
ジヒドロダイゼイン (2 mg / ml) 5 μl
KPB-PDH 855 μl
計 1000 μl
実施例C1 菌体のテトラヒドロダイゼインからのエクオール生合成活性の確認
ラクトコッカス20-92株(FERM BP-10036号)を、テトラヒドロダイゼイン含有増殖用液体培地(シス-もしくはトランス-テトラヒドロダイゼイン(自社で有機合成:参考例B1参照)を10μg/mLとなる量に加えた変法GAMブイヨン培地(日水製薬株式会社))に接種し、嫌気的条件下(BBL Gas Pack systemsを使用)37℃で18時間培養した。培養終了後、直ちに培養液1mLを蓋付ガラス製遠沈管に分取し、3mLの酢酸エチルを添加して抽出処理を行った後、乾固し、HPLC分析を行った。HPLC分析の標準溶液にはダイゼイン(フナコシ株式会社)、エクオール(フナコシ株式会社)、ジヒドロダイゼイン(トレンドリサーチケミカル社)シス-テトラヒドロダイゼイン、トランス-テトラヒドロダイゼイン(共に自社で化学合成)の混合溶液(各2μg/mL)を用いた。
大腸菌を用いた組換えタンパク質発現システムであるpETシステム(Novagen)を用いて、実施例B5において決定したジヒドロダイゼイン合成(E1)酵素遺伝子周辺ゲノムDNA配列上に同定した3つのORFポリヌクレオチド(ORF-US3,US1,DS1)に対応するポリペプチドを大腸菌内でそれぞれ発現させ、そのテトラヒドロダイゼインからのエクオールへの変換を触媒する活性を調べることにより、エクオール合成(E3)
酵素の探索を行った。
各ORFポリペプチド(ORF-US3,US1,DS1)の発現ベクターを作製する目的でPCRにより、それぞれのオープン・リーディング・フレーム領域のポリヌクレオチドを増幅し、pET21aベクター(Novagen)に挿入した。
実施例B5で決定したジヒドロダイゼイン合成酵素(E1)周辺ゲノム配列を基に下記の増幅プライマーを作製した。
ORF-US3ポリペプチド
exp.US3 F:TATACATATGGCAGAATTCGATGTTGAG (配列番号:62)
exp.US3 R:CCGCAAGCTTCTACATAGTGGAGATCGCGTGG (配列番号:63)
ORF-US1ポリペプチド
exp.US1 F:TATACATATGTTCAAGGGTCCACAGGGC (配列番号:64)
exp.US1 R:GCTCGAATTCTTAGTGCTGCTGTGCCTTTTCAG (配列番号:65)
ORF-DS1ポリペプチド
exp.DS1 F:ATATACATATGCAGGATATGGACTTCATGG (配列番号:66)
exp.DS1 R:GCTCGAATTCTCATAGTGACATCAGCGCTCCC (配列番号:67)
上記した増幅プライマーを各5pmol、dNTP 5nmol each、実施例A5において精製したラクトコッカス20-92株のゲノムDNA 40ng 、KOD-plus DNA polymerase用10×緩衝液(東洋紡績株式会社)2.5μL、KOD-Plus DNA polymerase 0.3ユニット(東洋紡績株式会社)を含む25μLの反応液を用い、増幅プログラム:95℃ 3分、(94℃ 30秒、60℃ 30秒、68℃ 2分)×30cycles、68℃ 7分でGeneAmpPCR System 9700(アプライド・バイオシステムズ)を用いてPCRを行った。PCR反応液の一部をアガロースゲル電気泳動した結果、それぞれに予想される大きさのバンドが検出できた。PCR産物全量をQIAGEN PCR Purification kit(キアゲン)にて回収した。
(1-2)で回収したORF-US3ポリヌクレオチド断片を制限酵素NdeI及びHindIIIで、ORF-US1及びORF-DS1ポリヌクレオチド断片をNdeI及びEcoRIで切断後、アガロースゲル電気泳動を行い、目的とするバンドの部分を切り出し、Qiagen Gel Extraction kit (キアゲン)により精製、回収した。得られたポリヌクレオチド断片はDNA Ligation Kit ver.2.1(タカラバイオ株式会社)を用いて、NdeI及びHindIIIもしくはEcoRIで消化したpET21aと16 ℃、終夜でライゲーションした後、ライゲーション反応液を用いて大腸菌JM109株(タカラバイオ株式会社)を定法で形質転換した。斯くして得た形質転換体を、アンピシリン(50μg/mL)を含むLB培地アガー(GIBCO)プレート上で37℃、終夜生育させ、コロニーを形成させた。得られたシングルコロニーを、アンピシリン(50μg/mL)を含むLB培地(GIBCO)3mLで終夜培養した後、プラスミド自動抽出機PI-100(KURABO)を用いてプラスミドDNAを抽出した。
(2-1)大腸菌BL21形質転換体の作製
組換えORFポリペプチドを発現するプラスミドpET-US3、pET-US1、pET-DS1とpET21a(ネガティブコントロール)を用いて、大腸菌BL21(DE3)株(Novagen)を定法で形質転換した。形質転換体はアンピシリン(50μg/mL)を含むLB培地アガープレート上で37℃、終夜生育させ、シングルコロニーを得た。
上記の大腸菌BL21(DE3)形質転換体それぞれをアンピシリン(50μg/mL)を含む3mLの液体LB培地で終夜37℃において培養を行った。その培養液0.5mLを同濃度のアンピシリンを含む液体LB培地50mLに加え、3時間(OD630nmが約0.4-0.7になるまで)前培養し、終濃度が1mMになるようにIPTG(イソプロピル-β-チオガラクトピラノシド)(和光純薬工業株式会社)を加え、さらに37℃で4時間培養し、大腸菌内での組換えポリペプチドの発現誘導行った。
上記(2-2)での発現誘導終了後、菌体をAvanti HP25(beckman coulter)で遠心分離(6000rpm 4℃ 15分)により集菌した。以降の操作は氷上で行った。遠心上静(培地)を除いた後、1mM PMSF、2mM DTT、5mM Sodium hydrosulfiteを含む0.1Mリン酸カリウム緩衝液pH7.0(以後KPB-PDHと略す)1mLに懸濁し、あらかじ0.7mL容のジルコニア-シリカビーズ(BioSpec Products, Inc.)及びKPB-PDH 400μLを入れておいた2mL容アシストチューブへ入れ、FastPrep(R)(Thermo ELECTRON CORPORATION)を用いて6500rpm、20秒間処理-3分間氷冷を2回繰り返すことで、菌体を破砕し、菌体破砕液を得た。
大腸菌内での各組換えORFポリペプチドの発現確認はSDS-PAGEで行った。(2-3)に記した菌体破砕液20μLに5×サンプル緩衝液(125mM Tris-HCl(pH6.5)/ 25% グリセロール/5% SDS/5% 2-メルカプトエタノール/BPB 0.5%)を5μL加え、98℃ 5分間加熱変性後、氷冷し、うち4μLをSDS-PAGEで泳動した。SDS‐PAGEには市販のゲル板(SuperSepTM5-20% (和光純薬工業株式会社))を使用し、染色はクイックCBB(和光純薬工業株式会社)で行った。分子量マーカーにはPrestained XL-Ladder Broad range(株式会社アプロサイエンス)を使用した。SDS-PAGEの結果を図21に示す。pET-US3、pET-DS1形質転換体由来の菌体破砕液それぞれにおいて分子量約52kDa、50kDaの組換えORF-US3及びORF-DS1ポリペプチドの発現が確認された。pET-US1形質転換体由来の菌体破砕液では組換えORF-US1ポリペプチドの発現は確認できなかった。
実施例C2で得た各形質転換体菌破砕液を酵素源として、テトラヒドロダイゼインからエクオールへの変換活性を測定した。本実施例でのテトラヒドロダイゼインからエクオールへの変換活性の測定は以下の方法で行った。
酵素反応液組成
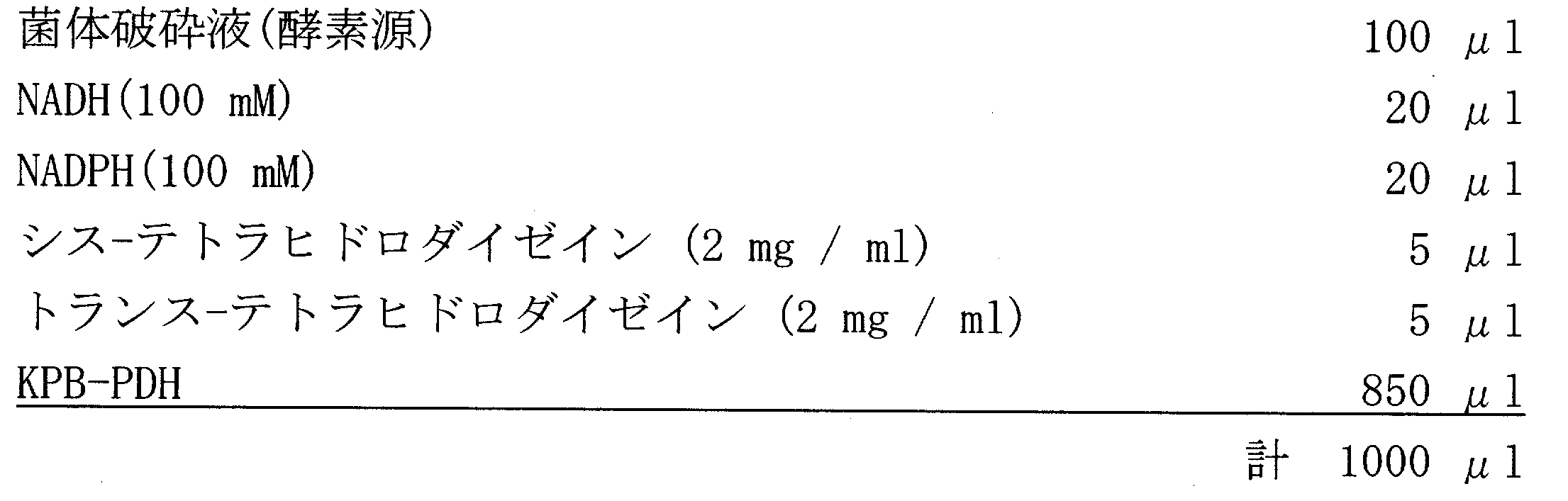
実施例D1 大腸菌を用いた組換えHis-タグ付きエクオール産生関連酵素の発現及び精製
大腸菌を用いた組換えタンパク質発現システムであるpET システム(Novagen)を用いて、His-タグ付きエクオール産生関連酵素[ジヒドロダイゼイン合成酵素(E1),テトラヒドロダイゼイン合成酵素(E2),エクオール合成酵素(E3)]を発現させ、His-タグ付きタンパク質精製用(Ni)カラムを用いてアフィニティー精製を行った。
各酵素(E1,E2,E3)の発現ベクターを作製する目的でPCRにより、それぞれのオープン・リーディング・フレーム領域のポリヌクレオチドを増幅し、pET21aベクター(Novagen)に挿入した。
実施例B5で決定したジヒドロダイゼイン合成酵素(E1)周辺ゲノム配列を基に下記の増幅プライマーを作製した。
His-タグ付きE1酵素
exp.E1 pet F Nde:AGCTCATATGAAGAACAAGTTCTATCCGAA (配列番号:41)
exp.E1 pet His:AATCGAATTCGTACAGGTTGCAGCCAGCGATGT (配列番号:42)
His-タグ付きE2酵素
exp.US2 pet F Nde:TATACATATGGCACAGGAAGTCAAAGTC (配列番号:60)
exp.E2 pet His:AATCGAATTCGAGACCTCGATCTCGCCCTGC (配列番号:68)
His-タグ付きE3酵素
exp.US3 F:TATACATATGGCAGAATTCGATGTTGAG (配列番号:62)
exp.E3 R His:CCGCAAGCTTGTACATAGTGGAGATCGCGTGG (配列番号:69)
上記増幅プライマー:exp.E1 pet F Nde、exp.US2 pet F、exp.US3 FにはpET21a(Novagen)へ挿入するため、制限酵素NdeI切断部位配列を、exp.E1 pet His、exp.E2 pet HisにはEcoRI切断部位配列をexp.E3 R HisにはHindIII切断部位配列を含ませてある。
上記した増幅プライマーを各5pmolとdNTPを各5nmolと実施例A5において精製したラクトコッカス20-92株(FERM BP-10036号)のゲノムDNA 40ng 、KOD-plus DNA polymerase用10×緩衝液(東洋紡績株式会社)2.5μL、KOD-Plus DNA polymerase 0.3ユニット(東洋紡績株式会社)を含む25μLの反応液を用い、増幅プログラム:95℃ 3分、(94℃ 30秒、60℃ 30秒、68℃ 2分)×30cycles、68℃ 7分でGeneAmpPCR System 9700(アプライド・バイオシステムズ)を用いてPCRを行った。PCR反応液の一部をアガロースゲル電気泳動した結果、それぞれに予想される大きさのバンドが検出できた。PCR産物全量をQIAGEN PCR Purification kit(キアゲン)にて回収した。
(1-2)で回収した各His-タグ付き酵素ポリヌクレオチド断片を制限酵素NdeI及びEcoRIで切断後、アガロースゲル電気泳動を行い、目的とするバンドの部分を切り出し、Qiagen Gel Extraction kit (キアゲン) により精製、回収した。得られたポリヌクレオチド断片はDNA Ligation Kit ver.2.1(タカラバイオ株式会社)を用いて、NdeI及びEcoRIで消化したpET21aと16 ℃、終夜でライゲーションした後、ライゲーション反応液を用いて大腸菌JM109株(タカラバイオ株式会社)を定法で形質転換した。斯くして得た形質転換体を、アンピシリン(50μg/mL)を含むLB培地アガー(GIBCO)プレート上で37℃、終夜生育させ、コロニーを形成させた。得られたシングルコロニーを、アンピシリン(50μg/mL)を含むLB培地(GIBCO)3mLで終夜培養した後、プラスミド自動抽出機PI-100(KURABO)を用いてプラスミドDNAを抽出した。
(2-1)大腸菌BL21形質転換体の作製
His-タグ付き酵素ポリペプチドを発現するプラスミドpET-E1-His、pET-E2-His、pET-E3-Hisを用いて、大腸菌BL21(DE3)株(Novagen)を定法で形質転換した。形質転換体はアンピシリン(50μg/mL)を含むLB培地アガープレート上で37℃、終夜生育させ、シングルコロニーを得た。
(2-2-1)大腸菌培養及び組換えHis-タグ付きE1及びE2酵素ポリペプチドの発現誘導
上記pET-E1-His又はpET-E2-Hisによる大腸菌BL21(DE3)形質転換体それぞれをアンピシリン(50μg/mL)を含む10mLの液体LB培地で終夜37℃において培養を行った。その培養液7.5mL を同濃度のアンピシリンを含む液体LB培地150mL 加え、37℃で2時間(OD600nmが約0.5になるまで)前培養し、終濃度が0.5mM になるようにIPTG(イソプロピル-β-チオガラクトピラノシド)(和光純薬工業株式会社)を加え、さらに弱い振とう条件下で30℃、4時間培養し、大腸菌内での組換えHis-タグ付きE1及びE2酵素ポリペプチドの発現誘導行った。
上記(2-2)での発現誘導終了後、菌体をAvanti HP25(beckman coulter)で遠心分離(6000rpm 4℃ 10分)により集菌し、組換えHis-タグ付きE1及びE2酵素ポリペプチド発現大腸菌をそれぞれ湿重量にして0.66g及び0.73gを得た。得た菌体はBugbuster protein Extraction solution(Novagen)が菌体湿重量1gあたり15mLになるように加え、ピペットで穏やかに懸濁し、更にLysozyme(SIGMA) を2000units/mL、Benzonase(Novagen)を 25units(1μL)/mLになるように加えた。その後、ローテーター(RT-50:タイテック)を用いて30分間室温でゆっくり攪拌し、菌ライゼートAを得た。更に菌ライゼートAをAvanti HP25(beckman coulter)で遠心分離(8000rpm 4℃ 15分)により、その上清である菌ライゼートBを得た。
His-タグ付きタンパク質精製用カラムにはHis GraviTrap(GEヘルスケアバイオサイエンス)を用い、取り扱い説明書に記載されている手順に従い、一部変更した方法でHis-タグ付きE1及びE2酵素ポリペプチドのアフィニティー精製を行った。即ち、His GraviTrapを氷冷した10mLの結合バッファーで平衡化した後、(2-2-2)で調製した菌ライゼートBを全量注ぎ込み、自然落下にて目的のHis-タグ付きE1或いはE2酵素をHis GraviTrapに吸着させた。その後、His GraviTrapを氷冷した10mLの洗浄バッファーで2回洗浄した後、氷冷しておいた3mLの溶出バッファーで目的のHis-タグ付きE1及びE2酵素をHis GraviTrapより溶出した。溶出液にDTT[dithiothreitol](和光純薬工業株式会社)を終濃度が3mMになるように加えた後、 300μLずつマイクロチューブに小分けした。それらの一部を用いて、それぞれの溶出液のE1酵素活性及びE2酵素活性を確認し、溶出液は酵素実験に使用するまで4℃で保存した。
結合バッファー:20mM Tris-HCl、20mM Imidazole(和光純薬工業株式会社)、0.5M NaCl(和光純薬工業株式会社)、1mM DTT[dithiothreitol](和光純薬工業株式会社)、1 mM PMSF[phenylmethylsulfonyl fluoride](和光純薬工業株式会社)
洗浄バッファー:20mM Tris-HCl、 60mM Imidazole、0.5M NaCl、1mM DTT[dithiothreitol]、1 mM PMSF[phenylmethylsulfonyl fluoride]
溶出バッファー:20mM Tris-HCl 500mM Imidazole、0.5M NaCl、1mM DTT[dithiothreitol]、1 mM PMSF[phenylmethylsulfonyl fluoride]
(2-3-1)大腸菌培養及び組換えHis-タグ付きE3酵素ポリペプチドの発現誘導
上記(2-1)に記載のpET-E3-Hisによる大腸菌BL21(DE3)形質転換体をアンピシリン(50μg/mL)を含む50mLの液体LB培地で終夜37℃において培養を行った。その培養液20mL を同濃度のアンピシリンを含む液体LB培地1Lに加え、37℃で3時間(OD660nmが約0.4になるまで)前培養した。終濃度が0.5mM になるようにIPTG(イソプロピル-β-チオガラクトピラノシド)(和光純薬工業株式会社)を加え、37℃で4時間振とう培養し、大腸菌内での組換えHis-タグ付きE3酵素ポリペプチドの発現誘導行った。発現誘導を行った大腸菌をAvanti HP25(beckman coulter)で遠心分離(6000rpm 4℃ 10分)をして集菌した。菌体を1 mM PMSF(phenylmethylsulfonyl fluoride)2 mM DTT(和光純薬工業株式会社)5mM Sodiumhydrosulfite(和光純薬工業株式会社)を含有する0.1 M リン酸カリウム緩衝液pH 7.0(以後バッファーAとする)を25 ml加えて懸濁し、遠心分離(6000rpm 4℃ 10分)後そのままの状態で-80℃に保存した。
上記(2-3-1)で保存した菌体を融解後、遠心(10,000xg 15 min)し、新しいバッファーAを25 ml加えて懸濁させた。その懸濁液をFastPrep(R)(Thermo ELECTRON CORPORATION)を用いて6500rpm、20秒間処理-3分間氷冷を3回繰り返すことで、菌体を破砕した。得られた菌体破砕液を遠心 (10000 X g, 15 min)し、その上清を分離し、菌ライゼートを得た。
上記(2-3-3)で得た菌ライゼート4 mLを以下に示す精製条件によりHis-タグ融合タンパク質精製用カラムであるHisTrap HP (GEヘルスケアバイオサイエンス)に供して、組換えHisタグ付きE3酵素の精製を行った。蛋白質の測定は280 nmの吸収によった。またフラクションコレクターによる分画は素通り画分の280nmにおける吸収がベースラインに低下した後開始した。
フラクション: 2 ml/2 min/フラクション
溶離液 A: 0.1 M リン酸カリウム緩衝液pH 7/0.5 M NaCl /1 % isoPrOH
溶離液 B: 0.5 M Imidazoleを含む溶離液A
溶出プログラム: 時間(min) (溶離液B)/(溶離液A+溶離液B)
0 0.05
5 0.05
25 0.5
30 1
35 1
37 0.05
上記実施例D1の(2-2-3)にて得た組換えHis-タグ付きE1酵素及びE2酵素を酵素源として、下記組成の酵素反応液を調製し、37℃で2時間反応させることでダイゼインからテトラヒドロダイゼインの合成を行った。また、同時に酵素源として組換えHis-タグ付きE1、E2酵素それぞれを単独で用いた反応も行った。
酵素反応液組成

HPLC分析の標準溶液はダイゼイン(フナコシ)、エクオール(フナコシ)、ジヒドロダイゼイン(トレンドリサーチケミカル社)シス-テトラヒドロダイゼイン(参考例B1)、トランス-テトラヒドロダイゼイン(参考例B1)の混合溶液(各2μg/mL)を用いた。
HPLC分析の結果を図23に示す。酵素源に組換えHis-タグ付きE1酵素及びE2酵素の混合を使用した場合、その生成物にシス-及びトランス-テトラヒドロダイゼインを確認した。しかしながら、酵素源に組換えHis-タグ付きE1酵素もしくは組換えHis-タグ付きE2酵素単独で使用した場合には、その生成物にシス-及びトランステトラヒドロダイゼインは確認できなかった。
上記実施例D1の (2-2-3)及び(2-3-3)にて得た組換えHis-タグ付きE2及びE3酵素を酵素源として、下記組成の酵素反応液を調製し、37℃で2時間反応させることでジヒドロダイゼインからエクオールの合成を行った。また、同時に酵素源として組換えHis-タグ付きE2酵素及びE3酵素それぞれを単独で用いた反応も行った。
酵素反応液組成

HPLC分析の結果を図24に示す。酵素源に組換えHis-タグ付きE2、E3酵素混合を使用した場合、その生成物にエクオールを確認した。しかしながら、酵素源に組換えHis-タグ付きE2酵素もしくは組換えHis-タグ付きE3酵素を単独で使用した場合には、その生成物にエク オールは確認できなかった。
上記実施例D1の(2-2-3)及び(2-3-3)にて得た組換えHis-タグ付きE1酵素、E2酵素及びE3酵素を酵素源として、下記組成の酵素反応液を調製し、37℃で2時間反応させることでダイゼインからエクオールの合成を行った。また、同時に酵素源として組換えHis-タグ付きE1酵素、E2酵素、及びE3酵素をそれぞれ単独で用いた反応も行った。
酵素反応液組成

HPLC分析の結果を図25に示す。酵素源にそれぞれの組換えHis-タグ付き酵素単独で使用した場合、その生成物にエクオールは確認できなかった。しかしながら、酵素源に組換えHis-タグ付きE1酵素、E2酵素及びE3酵素混合を使用した場合には、その生成物にエクオールが確認された。
Ni-Sepharoseにて精製した大腸菌発現His-タグ付き組換えE1酵素(E1-His)を酵素源として、ダイゼインからジヒドロダイゼインへの変換活性に対する金属イオンの影響を検討した。 各種金属 (MnCl2・2H2O、FeSO4・7H2O、CaCl2・2 H2O、Zn(CH3COO)2・2H2O、CoSO4・7H2O、MgSO4・7 H2O、NiSO4・6 H2O ) を蒸留水にて100mMとなるよう溶解し、実験に供した。活性の測定は下記組成の酵素反応液を調製し、37℃で1時間インキュベートして行った。
酵素反応液組成
組換えHis-タグ付きE1酵素 20 μl
NADH(100 mM) 10 μl
NADPH(100 mM) 10 μl
ダイゼイン (1 mg / ml) 10 μl
金属イオン溶液 100 μl
0.2M KPB-DH 850 μl
計 1000 μl
配列番号24はプライマーE1-N-terminal-37の塩基配列を示す。
配列番号25はプライマーE1-N-terminal-F32の塩基配列を示す。
配列番号26はプライマーE1-internal-RP1の塩基配列を示す。
配列番号27はプライマーpUC19-FP-1の塩基配列を示す。
配列番号28はプライマーpUC19-RP-1の塩基配列を示す。
配列番号29はプライマーpUC19-FP-2の塩基配列を示す。
配列番号30はプライマーpUC19-RP-2の塩基配列を示す。
配列番号31はプライマーE1-RACE-N-P1の塩基配列を示す。
配列番号32はプライマーE1-RACE-RP2-1の塩基配列を示す。
配列番号33はプライマーE1-RACE-N-P2の塩基配列を示す。
配列番号34はプライマーE1-RACE-RP2-2の塩基配列を示す。
配列番号35はプライマーE1-conf-NPの塩基配列を示す。
配列番号36はプライマーE1-conf-CPの塩基配列を示す。
配列番号37はプライマーE1-FPの塩基配列を示す。
配列番号38はプライマーE1-RPの塩基配列を示す。
配列番号39はプライマーGar-16S-Ribo-FPの塩基配列を示す。
配列番号40はプライマーGar-16S-Ribo-RPの塩基配列を示す。
配列番号41はプライマーexp.E1 pet F Ndeの塩基配列を示す。
配列番号42はプライマーexp. E1 pet Hisの塩基配列を示す。
配列番号43はプライマーRACE-N-P3-1の塩基配列を示す。
配列番号44はプライマーRACE-N-P3-2の塩基配列を示す。
配列番号45はプライマーE1-Bub-N-P1の塩基配列を示す。
配列番号46はプライマーE1-Bub-N-P2の塩基配列を示す。
配列番号47はプライマーRACE-C-P3-1の塩基配列を示す。
配列番号48はプライマーRACE-C-P3-2の塩基配列を示す。
配列番号49はプライマーE1-Bub-C-P1の塩基配列を示す。
配列番号50はプライマーE1-Bub-C-P2の塩基配列を示す。
配列番号57はプライマーE2-invitroTS-FPの塩基配列を示す。
配列番号58はプライマーE2-invitroTS-RPの塩基配列を示す。
配列番号59はプライマーUniversal-Primerの塩基配列を示す。
配列番号60はプライマーexp.US2 pet F Ndeの塩基配列を示す。
配列番号61はプライマーexp. US2 petの塩基配列を示す。
配列番号62はプライマーexp.US3 Fの塩基配列を示す。
配列番号63はプライマーexp.US3 Rの塩基配列を示す。
配列番号64はプライマーexp.US1 Fの塩基配列を示す。
配列番号65はプライマーexp.US1 Rの塩基配列を示す。
配列番号66はプライマーexp.DS1 Fの塩基配列を示す。
配列番号67はプライマーexp.DS1 Rの塩基配列を示す。
Claims (12)
- 以下の(Ca)~(Cc)のいずれかであるポリペプチド:
(Ca)配列番号13に記載のアミノ酸配列からなるポリペプチド;
(Cb)配列番号13に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド;
(Cc)配列番号13に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド。 - 以下の(Cd)~(Cf)のいずれかであるポリヌクレオチド:
(Cd)配列番号16に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ce)配列番号13に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Cf)前記(Cd)又は(Ce)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つテトラヒドロダイゼインを基質としエクオールを生成する活性を有するポリペプチドをコードするポリヌクレオチド。 - テトラヒドロダイゼインに対して、請求項1に記載のポリペプチドを作用させる工程を含む、エクオールの製造方法。
- 以下の(Ba)~(Bc)のいずれかであるポリペプチド:
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド。 - 以下の(Bd)~(Bf)のいずれかであるポリヌクレオチド:
(Bd)配列番号10に記載のヌクレオチド配列からなるポリヌクレオチド;
(Be)配列番号7に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Bf)前記(Bd)又は(Be)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。 - ジヒドロダイゼインに対して、請求項4に記載のポリペプチド、並びにNADPH及び/又はNADHを作用させる工程を含む、テトラヒドロダイゼインの製造方法。
- 以下の(Aa)~(Ac)のいずれかであるポリペプチド:
(Aa)配列番号1に記載のアミノ酸配列からなるポリペプチド;
(Ab)配列番号1に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(Ac)配列番号1に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド。 - 以下の(Ad)~(Af)のいずれかであるポリヌクレオチド:
(Ad)配列番号4に記載のヌクレオチド配列からなるポリヌクレオチド;
(Ae)配列番号1に記載のアミノ配列からなるポリペプチドをコードするポリヌクレオチド;
(Af)前記(Ad)又は(Ae)のポリヌクレオチドの相補鎖に対して、ストリンジェントな条件下でハイブリダイズし、且つダイゼインを基質としジヒドロダイゼインを生成する活性を有するポリペプチドをコードするポリヌクレオチド。 - ダイゼインに対して、請求項7に記載のポリペプチド、並びにNADPH及び/又はNADHを作用させる工程を含む、ジヒドロダイゼインの製造方法。
- 以下の(第1工程)及び(第2工程)を含む、テトラヒドロダイゼインの製造方法:
(第1工程)ダイゼインに、以下の(Aa)~(Ac)のいずれかであるポリペプチドからなる酵素、及びNADPHを作用させることにより、ジヒドロダイゼインを生成する工程;
(Aa)配列番号1に記載のアミノ酸配列からなるポリペプチド;
(Ab)配列番号1に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(Ac)配列番号1に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つダイゼインを基質としジヒドロダイゼインを合成する活性を有するポリペプチド;
(第2工程)ジヒドロダイゼインに、以下の(Ba)~(Bc)のいずれかであるポリペプチドからなる酵素、並びにNADPH及び/又はNADHを作用させることにより、テトラヒドロダイゼインを生成する工程;
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド。 - 以下の(第2工程)及び(第3工程)を含む、エクオールの製造方法:
(第2工程)ジヒドロダイゼインに、以下の(Ba)~(Bc)のいずれかであるポリペプチドからなる酵素、並びにNADPH及び/又はNADHを作用させることにより、テトラヒドロダイゼインを生成する工程;
(Ba)配列番号7に記載のアミノ酸配列からなるポリペプチド;
(Bb)配列番号7に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(Bc)配列番号7に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つジヒドロダイゼインを基質としテトラヒドロダイゼインを合成する活性を有するポリペプチド;
(第3工程)テトラヒドロダイゼインに、以下の(Ca)~(Cc)のいずれかであるポリペプチドからなる酵素、並びにNADPH及び/又はNADHを作用させることにより、エクオールを製造する工程、
(Ca)配列番号13に記載のアミノ酸配列からなるポリペプチド;
(Cb)配列番号13に記載のアミノ酸配列において、1若しくは複数のアミノ酸が置換、欠失、挿入及び/又は付加したアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド;
(Cc)配列番号13に記載のアミノ酸配列に対して60%以上の同一性を有するアミノ酸配列からなり、且つテトラヒドロダイゼインを基質としエクオールを合成する活性を有するポリペプチド。 - (第1工程)~(第3工程)を含む、エクオールの製造方法。
Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137027968A KR20130133893A (ko) | 2007-12-27 | 2008-12-25 | 에쿠올 합성에 관여하는 효소 |
| KR1020127030667A KR101543501B1 (ko) | 2007-12-27 | 2008-12-25 | 에쿠올 합성에 관여하는 효소 |
| CA2710812A CA2710812C (en) | 2007-12-27 | 2008-12-25 | Enzyme associated with equol synthesis |
| US12/810,742 US8399232B2 (en) | 2007-12-27 | 2008-12-25 | Enzyme associated with equol synthesis |
| EP08867315.7A EP2228441B1 (en) | 2007-12-27 | 2008-12-25 | Enzyme involved in synthesis of equol |
| SI200831018T SI2228441T1 (sl) | 2007-12-27 | 2008-12-25 | Encim, vključen v sintezo ekvola |
| KR1020127030666A KR101543500B1 (ko) | 2007-12-27 | 2008-12-25 | 에쿠올 합성에 관여하는 효소 |
| CN200880127246.8A CN101952417B (zh) | 2007-12-27 | 2008-12-25 | 与雌马酚合成有关的酶 |
| BRPI0821816-1A BRPI0821816B1 (pt) | 2007-12-27 | 2008-12-25 | Processos para produzir equol, tetraidrodaidzeína e dihidrodaidzeína |
| JP2009548072A JP5230654B2 (ja) | 2007-12-27 | 2008-12-25 | エクオール合成に関与する酵素 |
| ES08867315T ES2423937T3 (es) | 2007-12-27 | 2008-12-25 | Enzima involucrado en la síntesis de ecuol |
| DK08867315.7T DK2228441T3 (da) | 2007-12-27 | 2008-12-25 | Enzym, der er involveret i syntese af equol. |
| AU2008344416A AU2008344416B2 (en) | 2007-12-27 | 2008-12-25 | Enzyme associated with equol synthesis |
| KR1020137027981A KR20130133894A (ko) | 2007-12-27 | 2008-12-25 | 에쿠올 합성에 관여하는 효소 |
| HK10111230.5A HK1145189A1 (en) | 2007-12-27 | 2010-12-02 | Enzyme involved in synthesis of equol |
| US13/668,924 US8669088B2 (en) | 2007-12-27 | 2012-11-05 | Enzyme associated with equol synthesis |
| US14/155,557 US8993296B2 (en) | 2007-12-27 | 2014-01-15 | Enzyme associated with equol synthesis |
| US14/621,871 US20150240275A1 (en) | 2007-12-27 | 2015-02-13 | Enzyme associated with equol synthesis |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007-336227 | 2007-12-27 | ||
| JP2007336227 | 2007-12-27 | ||
| JP2008-054874 | 2008-03-05 | ||
| JP2008054874 | 2008-03-05 | ||
| JP2008-080570 | 2008-03-26 | ||
| JP2008080570 | 2008-03-26 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/810,742 A-371-Of-International US8399232B2 (en) | 2007-12-27 | 2008-12-25 | Enzyme associated with equol synthesis |
| US13/668,924 Division US8669088B2 (en) | 2007-12-27 | 2012-11-05 | Enzyme associated with equol synthesis |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009084603A1 true WO2009084603A1 (ja) | 2009-07-09 |
Family
ID=40824317
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2008/073649 WO2009084603A1 (ja) | 2007-12-27 | 2008-12-25 | エクオール合成に関与する酵素 |
Country Status (16)
| Country | Link |
|---|---|
| US (4) | US8399232B2 (ja) |
| EP (3) | EP2404995B1 (ja) |
| JP (2) | JP5230654B2 (ja) |
| KR (5) | KR101543500B1 (ja) |
| CN (5) | CN104830813A (ja) |
| AR (1) | AR070058A1 (ja) |
| CA (1) | CA2710812C (ja) |
| DK (3) | DK2228441T3 (ja) |
| ES (3) | ES2424960T3 (ja) |
| HK (3) | HK1145189A1 (ja) |
| MY (1) | MY156222A (ja) |
| PT (3) | PT2404994E (ja) |
| SG (2) | SG2013066634A (ja) |
| SI (3) | SI2404995T1 (ja) |
| TW (1) | TWI468516B (ja) |
| WO (1) | WO2009084603A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015053061A1 (ja) * | 2013-10-08 | 2015-04-16 | 東洋紡株式会社 | メチシリン耐性遺伝子の検出方法および黄色ブドウ球菌の検出方法 |
| JP2015073457A (ja) * | 2013-10-08 | 2015-04-20 | 東洋紡株式会社 | 黄色ブドウ球菌の検出方法 |
| JP2016127860A (ja) * | 2009-11-30 | 2016-07-14 | ファルマツェル、ゲーエムベーハー | 新規7β−ヒドロキシステロイドデヒドロゲナーゼおよびその使用 |
| JP2018186823A (ja) * | 2018-07-13 | 2018-11-29 | 株式会社ダイセル | イソフラバノン類の製造方法 |
| JP2020508671A (ja) * | 2017-02-28 | 2020-03-26 | ソウル大学校産学協力団Seoul National University R&Db Foundation | エクオール誘導体を生産する組換え大腸菌およびこれを利用したエクオール誘導体の合成方法 |
| WO2020090789A1 (ja) | 2018-10-29 | 2020-05-07 | 一博 舘田 | エクオールを有効成分として含む腸内細菌に対する抗菌剤 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104830813A (zh) * | 2007-12-27 | 2015-08-12 | 大塚制药株式会社 | 与雌马酚合成有关的酶 |
| AU2014226126B2 (en) | 2013-03-08 | 2019-02-14 | Genzyme Corporation | Continuous purification of therapeutic proteins |
| TWI709569B (zh) | 2014-01-17 | 2020-11-11 | 美商健臻公司 | 無菌層析樹脂及其用於製造方法的用途 |
| TWI709570B (zh) | 2014-01-17 | 2020-11-11 | 美商健臻公司 | 無菌層析法及製法 |
| CN108350508B (zh) * | 2015-10-29 | 2021-09-17 | 株式会社益力多本社 | 雌马酚产生能力的测定方法 |
| CN105861363B (zh) * | 2016-04-15 | 2019-11-12 | 河北农业大学 | 爱格氏菌、s-雌马酚产生工程菌及其构建方法和应用 |
| CN107641611B (zh) * | 2017-06-29 | 2020-09-04 | 浙江省农业科学院 | 一种具有s-雌马酚抗性的大肠杆菌突变株及应用 |
| KR102079003B1 (ko) * | 2018-08-10 | 2020-02-19 | 서울대학교산학협력단 | 활성이 증대된 테트라하이드로다이드제인 환원효소 및 에쿠올 유도체 생산에의 응용 |
| KR20210049155A (ko) | 2018-08-31 | 2021-05-04 | 젠자임 코포레이션 | 멸균 크로마토그래피 수지 및 제조 공정에서 이의 용도 |
| RU2727412C1 (ru) * | 2019-07-04 | 2020-07-21 | Юрий Феодосович Ясенчук | Способ получения антикоррозионного покрытия на изделиях из монолитного никелида титана |
| CN112442490B (zh) * | 2020-11-26 | 2022-03-01 | 湖南科技学院 | 一种转化酶及其在产s-雌马酚中的应用 |
| WO2023056413A1 (en) * | 2021-09-30 | 2023-04-06 | Board Of Regents, The University Of Texas System | Engineered vectors and organisms containing the same for isoflavone conversion in the gut |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000066576A1 (en) * | 1999-04-30 | 2000-11-09 | G.J. Consultants Pty Ltd | Isoflavone metabolites |
| JP2006296434A (ja) | 2003-06-30 | 2006-11-02 | Otsuka Pharmaceut Co Ltd | エクオール産生乳酸菌含有組成物 |
| WO2007099764A1 (ja) * | 2006-02-28 | 2007-09-07 | Kaneka Corporation | 新規カルボニル還元酵素、その遺伝子、およびそれらを利用した光学活性アルコールの製造方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2298679A1 (en) * | 1997-08-08 | 1999-02-18 | Otsuka Pharmaceutical Co., Ltd. | Isoflavone-containing compositions |
| KR20020056894A (ko) * | 2000-08-01 | 2002-07-10 | 후루타 타케시 | 신규 카르보닐 환원효소, 그의 유전자 및 그의 이용 방법 |
| US7416866B2 (en) * | 2002-02-01 | 2008-08-26 | Schering Ag | Process for the overexpression of dehydrogenases |
| US7314974B2 (en) * | 2002-02-21 | 2008-01-01 | Monsanto Technology, Llc | Expression of microbial proteins in plants for production of plants with improved properties |
| PL1649760T3 (pl) * | 2003-06-30 | 2013-10-31 | Otsuka Pharma Co Ltd | Kompozycja zawierająca bakterię kwasu mlekowego wytwarzającą ekwol |
| JP4746548B2 (ja) * | 2004-08-06 | 2011-08-10 | 株式会社カネカ | 新規カルボニル還元酵素、その遺伝子、およびその利用法 |
| EP1944021B1 (en) * | 2005-11-02 | 2014-05-21 | Kabushiki Kaisha Yakult Honsha | Equol level regulator |
| TR201807884T4 (tr) * | 2005-12-06 | 2018-06-21 | Otsuka Pharma Co Ltd | Soya fasulyesi embriyonik ekseninin ekuol-içeren fermentasyon ürünü ve bunun üretim yöntemi. |
| CN104830813A (zh) * | 2007-12-27 | 2015-08-12 | 大塚制药株式会社 | 与雌马酚合成有关的酶 |
| JP5414368B2 (ja) * | 2009-05-29 | 2014-02-12 | 大塚製薬株式会社 | ジヒドロダイゼインをラセミ化する酵素 |
-
2008
- 2008-12-25 CN CN201510191176.4A patent/CN104830813A/zh active Pending
- 2008-12-25 KR KR1020127030666A patent/KR101543500B1/ko not_active IP Right Cessation
- 2008-12-25 KR KR1020127030667A patent/KR101543501B1/ko not_active IP Right Cessation
- 2008-12-25 SG SG2013066634A patent/SG2013066634A/en unknown
- 2008-12-25 ES ES11170947T patent/ES2424960T3/es active Active
- 2008-12-25 KR KR1020137027968A patent/KR20130133893A/ko not_active Application Discontinuation
- 2008-12-25 EP EP11170947.3A patent/EP2404995B1/en not_active Not-in-force
- 2008-12-25 EP EP11170943.2A patent/EP2404994B1/en not_active Not-in-force
- 2008-12-25 SI SI200831033T patent/SI2404995T1/sl unknown
- 2008-12-25 ES ES08867315T patent/ES2423937T3/es active Active
- 2008-12-25 DK DK08867315.7T patent/DK2228441T3/da active
- 2008-12-25 US US12/810,742 patent/US8399232B2/en active Active
- 2008-12-25 ES ES11170943T patent/ES2424470T3/es active Active
- 2008-12-25 DK DK11170947.3T patent/DK2404995T3/da active
- 2008-12-25 EP EP08867315.7A patent/EP2228441B1/en active Active
- 2008-12-25 CA CA2710812A patent/CA2710812C/en active Active
- 2008-12-25 TW TW97150642A patent/TWI468516B/zh active
- 2008-12-25 PT PT111709432T patent/PT2404994E/pt unknown
- 2008-12-25 SG SG2013066642A patent/SG2013066642A/en unknown
- 2008-12-25 CN CN200880127246.8A patent/CN101952417B/zh active Active
- 2008-12-25 CN CN2012101265354A patent/CN102676463A/zh active Pending
- 2008-12-25 SI SI200831028T patent/SI2404994T1/sl unknown
- 2008-12-25 SI SI200831018T patent/SI2228441T1/sl unknown
- 2008-12-25 MY MYPI2010002883A patent/MY156222A/en unknown
- 2008-12-25 CN CN2012101265049A patent/CN102703395A/zh active Pending
- 2008-12-25 PT PT111709473T patent/PT2404995E/pt unknown
- 2008-12-25 CN CN201210126502XA patent/CN102676462A/zh active Pending
- 2008-12-25 KR KR1020107016700A patent/KR101265400B1/ko active IP Right Grant
- 2008-12-25 KR KR1020137027981A patent/KR20130133894A/ko not_active Application Discontinuation
- 2008-12-25 JP JP2009548072A patent/JP5230654B2/ja active Active
- 2008-12-25 DK DK11170943.2T patent/DK2404994T3/da active
- 2008-12-25 WO PCT/JP2008/073649 patent/WO2009084603A1/ja active Application Filing
- 2008-12-25 PT PT88673157T patent/PT2228441E/pt unknown
- 2008-12-29 AR ARP080105754A patent/AR070058A1/es not_active Application Discontinuation
-
2010
- 2010-12-02 HK HK10111230.5A patent/HK1145189A1/xx unknown
- 2010-12-02 HK HK12106834.3A patent/HK1166104A1/xx not_active IP Right Cessation
- 2010-12-02 HK HK12106833.4A patent/HK1166103A1/xx not_active IP Right Cessation
-
2012
- 2012-11-05 US US13/668,924 patent/US8669088B2/en active Active
- 2012-11-08 JP JP2012246695A patent/JP5689104B2/ja not_active Expired - Fee Related
-
2014
- 2014-01-15 US US14/155,557 patent/US8993296B2/en active Active
-
2015
- 2015-02-13 US US14/621,871 patent/US20150240275A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000066576A1 (en) * | 1999-04-30 | 2000-11-09 | G.J. Consultants Pty Ltd | Isoflavone metabolites |
| JP2006296434A (ja) | 2003-06-30 | 2006-11-02 | Otsuka Pharmaceut Co Ltd | エクオール産生乳酸菌含有組成物 |
| WO2007099764A1 (ja) * | 2006-02-28 | 2007-09-07 | Kaneka Corporation | 新規カルボニル還元酵素、その遺伝子、およびそれらを利用した光学活性アルコールの製造方法 |
Non-Patent Citations (11)
| Title |
|---|
| "IUPAC-IUB Communication on Biological Nomenclature", EUR. J. BIOCHEM., vol. 138, 1984, pages 9 |
| BIOCHEMISTRY, vol. 25, no. 25, 1986, pages 8274 |
| EUR. J. BIOCHEM., vol. 163, 1987, pages 313 |
| KINOSHITA, M. ET AL., CCA, vol. 228, 1994, pages 83 - 90 |
| M.A. FROHMAN ET AL., PROC. NATL. ACAD. SCI., USA., vol. 8, 1988, pages 8998 |
| ORITA, M. ET AL., GENOMICS, vol. 5, 1989, pages 874 - 879 |
| PROC. NATL. ACAD. SCI., USA., vol. 78, 1981, pages 6613 |
| RAPID AMPLIFICATION OF CDNA ENDS, EXPERIMENTAL MEDICINE, vol. 12, no. 6, 1994, pages 35 |
| SCIENCE, vol. 130, 1985, pages 1350 |
| SCIENCE, vol. 222, 1983, pages 778 |
| See also references of EP2228441A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016127860A (ja) * | 2009-11-30 | 2016-07-14 | ファルマツェル、ゲーエムベーハー | 新規7β−ヒドロキシステロイドデヒドロゲナーゼおよびその使用 |
| WO2015053061A1 (ja) * | 2013-10-08 | 2015-04-16 | 東洋紡株式会社 | メチシリン耐性遺伝子の検出方法および黄色ブドウ球菌の検出方法 |
| JP2015073457A (ja) * | 2013-10-08 | 2015-04-20 | 東洋紡株式会社 | 黄色ブドウ球菌の検出方法 |
| JP2020508671A (ja) * | 2017-02-28 | 2020-03-26 | ソウル大学校産学協力団Seoul National University R&Db Foundation | エクオール誘導体を生産する組換え大腸菌およびこれを利用したエクオール誘導体の合成方法 |
| JP7002557B2 (ja) | 2017-02-28 | 2022-02-10 | ソウル大学校産学協力団 | エクオール誘導体を生産する組換え大腸菌およびこれを利用したエクオール誘導体の合成方法 |
| JP2018186823A (ja) * | 2018-07-13 | 2018-11-29 | 株式会社ダイセル | イソフラバノン類の製造方法 |
| WO2020090789A1 (ja) | 2018-10-29 | 2020-05-07 | 一博 舘田 | エクオールを有効成分として含む腸内細菌に対する抗菌剤 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5689104B2 (ja) | エクオール合成に関与する酵素 | |
| JP4474518B2 (ja) | 2−ヒドロキシイソフラバノンデヒドラターゼをコードするポリヌクレオチドおよびその応用 | |
| CN110777155A (zh) | 最小霉素生物合成基因簇、重组菌及其应用 | |
| JP5414368B2 (ja) | ジヒドロダイゼインをラセミ化する酵素 | |
| JP4517100B2 (ja) | 2−ヒドロキシイソフラバノンシンターゼをコードするポリヌクレオチド | |
| BRPI0821816B1 (pt) | Processos para produzir equol, tetraidrodaidzeína e dihidrodaidzeína |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880127246.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08867315 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2009548072 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008344416 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12010501397 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4530/DELNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2710812 Country of ref document: CA Ref document number: MX/A/2010/007122 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008867315 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2008344416 Country of ref document: AU Date of ref document: 20081225 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PI 2010002883 Country of ref document: MY |
|
| ENP | Entry into the national phase |
Ref document number: 20107016700 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12810742 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0821816 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100629 |