BLOCKIERTE POLYURETHANPREPOLYMERE UND HITZEHÄRTENDE EPOXIDHARZZUSAMMENSETZUNGEN
Technisches Gebiet Die Erfindung betrifft das Gebiet der Schlagzähigkeitsmodifikatoren und das Gebiet der hitzehärtenden Epoxidharzzusammensetzungen.
Stand der Technik
Schlagzähigkeitsmodifikatoren werden schon seit langem dazu eingesetzt, um die Festigkeit von Klebstoffen bei schlagartiger Krafteinwirkung zu verbessern. Insbesondere Epoxidharzzusammensetzungen weisen generell zwar hohe mechanische Festigkeiten auf, sind jedoch sehr spröde, das heisst bei einer schlagartigen Krafteinwirkung, wie sie beispielsweise in einem Zusammenstoss von Fahrzeugen eintritt, bricht das gehärtete Epoxidharz und führt zu einer Zerstörung des Verbundes.
Flüssigkautschuke werden schon seit längerem zur Zähigkeitsmodifizierung eingesetzt. Beispielsweise wurden Flüssigkautschuke auf Basis von Acrylnitril/Butadien-Copolymeren, wie sie beispielsweise unter dem Namen Hycar® erhältlich sind, eingesetzt. EP-B-O 338 985 beschreibt schlagzähe Epoxidharzzusammensetzungen, welche neben Flüssigkautschuken auf Basis von Acrylnitril/Butadien- Copolymeren zusätzlich auch Flüssigkautschuke auf Basis von Polyurethan- prepolymere aufweisen, welche mit einem Phenol oder einem Lactam terminiert sind. WO-A-2005/007766 offenbart Epoxidharzzusammensetzungen, welche ein Reaktionsprodukt eines Isocyanatgruppen-terminierten Prepoly- mers und eines Blockierungsmittels, welches ausgewählt ist aus der Gruppe Bisphenol, Phenol, Benzylalkohol, Aminophenol oder Benzylamin enthalten. Derartige Epoxidharzzusammensetzungen zeigen jedoch Schwächen in der Tieftemperaturschlagzähigkeit (<0°C) auf.
WO-A-03/093387 offenbart schlagzähe Epoxidharzzusammensetzungen, welche Addukte von Dicarbonsäuren mit Glycidylethern oder von Bis(aminophenyl)sulfon-Isomeren oder aromatischen Alkoholen mit
Glycidylethern enthalten. Diese Zusammensetzungen weisen jedoch ebenfalls Defizite in der Tieftemperaturschlagzähigkeit (<0°C) auf.
WO-A-2004/055092 und WO-A-2005/007720 offenbaren Epoxidharzzusammensetzungen mit verbesserter Schlagzähigkeit, welche ein Reaktions- produkt eines Isocyanatgruppen-terminierten Polyurethanprepolymeren mit einem Monohydroxyepoxid, enthalten. Diese Epoxidharzzusammensetzungen weisen zwar gegenüber denjenigen, welche Phenol-terminierte PoIy- urethanprepolymere enthalten, eine verbesserte Tieftemperaturschlagzähigkeit auf, sind aber immer noch nicht optimal.
Darstellung der Erfindung
Aufgabe der vorliegenden Erfindung ist es daher, Schlagzähigkeitsmodifikatoren zur Verfügung zu stellen, welche gegenüber den aus dem Stand der Technik bekannten Schlagzähigkeitsmodifikatoren in Epoxidharzzusammensetzungen zu verbesserten Schlagzähigkeiten, insbesondere bei tiefen Temperaturen, führen. Überraschenderweise wurde gefunden, dass dies durch endständig blockierte Polyurethanprepolymere gemäss Anspruch 1 erreicht werden kann. Erstaunlicherweise wurde gefunden, dass mit ungleich (d.h. mit verschiedenen Blockierungsmitteln) blockierten Polyurethanprepolymeren höhere Schlagzähigkeiten als mit gleich (d.h. mit gleichem Blockierungsmittel) blockierten Polyurethanprepolymeren, wie sie aus dem Stand der Technik bekannt sind, erreicht werden können.
Die erfindungsgemässen endständig blockierten Polyurethanprepoly- meren finden als Schlagzähigkeitsmodifikatoren in Epoxidharzzusammensetzungen Anwendungen.
Somit bilden hitzehärtende Epoxidharzzusammensetzungen gemäss Anspruch 11 , welche mindestens ein erfindungsgemässes endständig blockiertes Polyurethanprepolymer enthalten, einen weiteren Aspekt der vorliegenden Erfindung. Diese Epoxidharzzusammensetzungen weisen eine besonders hohe Schlagzähigkeit sowohl bei Raumtemperatur als auch bei tiefen Temperaturen (-300C bzw. -400C) auf und eignen sich deshalb
insbesondere als Rohbauklebstoffe für Fahrzeuge und sind aufgrund ihrer Schlagzähigkeit besonders crash-tauglich.
Wege zur Ausführung der Erfindung
Die vorliegende Erfindung betrifft in einem ersten Aspekt endständig blockierte Polyurethanprepolymere der Formel (I).
Hierbei steht R1 für ein mit n+m Isocyanatgruppen terminiertes lineares oder verzweigtes Polyurethanprepolymer PU1 nach dem Entfernen aller endständigen Isocyanatgruppen. Die Reste R2 stehen unabhängig voneinander für eine Blockierungsgruppe, welche sich bei einer Temperatur über 1000C abspaltet, oder für eine Gruppe der Formel (II) und die Reste R3 stehen unabhängig voneinander für eine Blockierungsgruppe, welche sich bei einer Temperatur über 1000C abspaltet, oder für eine Gruppe der Formel (M').
Hierbei stehen R4 und R4 je für einen Rest eines eine primäre oder sekundäre Hydroxylgruppe enthaltenden aliphatischen, cycloaliphatischen, aromatischen oder araliphatischen Epoxids nach dem Entfernen der Hydroxid- und Epoxidgruppen, und p für die Werte 1 , 2 oder 3 und f für die Werte 1 ,2 oder 3.
Schliesslich stehen n und m je für Werte zwischen 1 und 7 mit der Massgabe, dass 2 < (m+n) < 8 ist. Eine weitere Massgabe ist, dass R2 von R3 verschieden ist. Somit ist das Polyurethanprepolymer „ungleich" blockiert.
Der Term „unabhängig voneinander" in der Definition von R2, respektive R3, bedeutet, dass von m Gruppen R2, respektive von n Gruppen R3, nicht alle denselben Rest darstellen müssen, sondern sie können unterschiedliche Bedeutungen aufweisen. So ist es im Extremfall möglich, dass das endständig blockierte Polyurethanprepolymer 8 voneinander verschiedene Gruppen R2, bzw. R3, aufweist.
Die möglichen Blockierungsgruppen R2 und R3 sind grundsätzlich sehr vielfältig und der Fachmann kennt eine breite Anzahl derartiger Blockierungsgruppen, zum Beispiel aus den Review Artikeln von Douglas A.
Wick in Progress in Organic Coatings 36 (1999), 148-172 und in Progress in
Organic Coatings 41 (2001 ), 1 -83.
Als Reste R2 und/oder R3 stehen insbesondere Reste, welche ausgewählt sind aus der Gruppe bestehend aus
Hierbei stehen R5, R6, R7 und R8 entweder je unabhängig voneinander für eine Alkyl- oder Cycloalkyl- oder Aryl- oder Aralkyl- oder Arylalkyl-Gruppe. Oder R5 bildet zusammen mit R6, oder R7 zusammen mit R8, einen Teil eines A- bis 7- gliedrigen Rings, welcher allenfalls substituiert ist.
Weiterhin stehen R9, R9 und R10 je unabhängig voneinander für eine Alkyl- oder Aralkyl- oder Aryl- oder Arylalkyl-Gruppe oder für eine Alkyloxy- oder Aryloxy- oder Aralkyloxy-Gruppe und R11 steht für eine Alkylgruppe.
Des Weiteren stehen R12, R13 und R14 je unabhängig voneinander für eine Alkylengruppe mit 2 bis 5 C-Atomen, welche gegebenenfalls Doppelbindungen aufweist oder substituiert ist, oder für eine Phenylengruppe oder für eine hydrierte Phenylengruppe.
R15, R16 und R17 stehen je unabhängig voneinander für H oder für eine
Alkylgruppe oder für eine Arylgruppe oder eine Aralkylgruppe und R18 steht für eine Aralkylgruppe oder für eine ein- oder mehrkernige substituierte oder unsubstituierte Aromatengruppe, welche gegebenenfalls aromatische
Hydroxylgruppen aufweist.
Die gestrichelten Linien in den Formeln dieses Dokumentes stellen jeweils die Bindung zwischen dem jeweiligen Substituenten und dem dazugehörigen Molekülrest dar.
Als R18 sind insbesondere einerseits Phenole oder Bisphenole nach Entfernung einer Hydroxylgruppe zu betrachten. Als Bespiele für derartige Phenole und Bisphenole sind insbesondere Phenol, Cardanol (3-Penta- decenylphenol (aus Cashewnuss-Schalen-Öl)), Nonylphenol, mit Styrol oder Dicyclopentadien umgesetzte Phenole, Bis-Phenol-A, Bis-Phenol-F zu nennen. Als R18 sind andererseits insbesondere Hydroxybenzylalkohol und Benzylalkohol nach Entfernung einer Hydroxylgruppe zu betrachten.
Falls R5, R6, R7, R8, R9, R9', R10, R11, R15, R16 oder R17 für eine Alkylgruppe steht, ist diese insbesondere eine lineare oder verzweigte C1-C20- Alkylgruppe.
Falls R5, R6, R7, R8, R9, R9', R10, R15, R16, R17 oder R18 für eine Aralkylgruppe steht, ist diese Gruppierung insbesondere eine über Methylen gebundene aromatische Gruppe, insbesondere eine Benzylgruppe.
Falls R5, R6, R7, R8, R9, R9' oder R10 für eine Alkylarylgruppe steht, ist diese insbesondere eine über Phenylen gebundene Cr bis C2o-Alkylgruppe, wie beispielsweise ToIyI oder XyIyI.
Bevorzugt sind die Reste R2 und R3 derart gewählt, dass sich die Deblockie- rungstemperaturen der Blockierungsgruppen R2 und R3 deutlich voneinander unterscheiden. Insbesondere ist es von Vorteil, wenn die Differenz der De- blockierungstemperaturen von R2 und R3 mindestens 20°C, bevorzugt mindestens 30 °C, beträgt. Dies erlaubt es gezielt mehrstufige Vernetzungsprozesse zu gestalten, welche eine Vielzahl von Möglichkeiten in Klebstoffen eröffnet.
Zudem ist es vorteilhaft, wenn die Reste R2 und R3 von unterschiedlicher Klasse sind, das heisst, dass sie aus unterschiedlichen Gruppen, wie sie oben aufgezeigt sind, stammen. Beispielsweise ist es vorteilhaft, einerseits hydroxylfunktionelle Epoxide der Formel (II) oder (M') und Phenole oder andererseits Phenole und Oxazolinone als Blockierungsmittel zu verwenden. Selbstverständlich sind auch alle weiteren Kombinationen der beschriebenen Blockierungsmittel, sowie ternäre oder quaternäre Mischungen von Blockierungsmitteln, denkbar.
In einer meist bevorzugten Ausführungsform stellt R2 eine Gruppe der
Formel (M) dar.
Das Polyurethanprepolymer PU1 , auf dem R1 basiert, lässt sich aus mindestens einem Diisocyanat oder Triisocyanat sowie aus einem Polymeren QPM mit endständigen Amino-, Thiol- oder Hydroxylgruppen und / oder aus einem, gegebenenfalls substituierten, Polyphenol QPP herstellen.
In der gesamten hier vorliegenden Schrift werden mit der Vorsilbe „Poly" in „Polyisocyanat", „Polyol", „Polyphenol" und „Polymerkaptan" Moleküle bezeichnet, die formal zwei oder mehr der jeweiligen funktionellen Gruppen enthalten.
Geeignete Diisocyanate sind aliphatische, cycloaliphatische, aromatische oder araliphatische Diisocyanate, insbesondere handelsübliche Produkte wie Methylendiphenyldiisocyanat (MDI), Hexamethylendiisocyanat (HDI), Toluoldiisocyanat (TDI), Tolidindiisocyanat (TODI), Isophorondiisocyanat (IPDI), Trimethylhexamethylendiisocyanat (TMDI), 2,5- oder 2,6-Bis-(iso- cyanatomethyl)-bicyclo[2.2.1]heptan, 1 ,5-Naphthalindiisocyanat (NDI), Dicyclo- hexylmethyldiisocyanat (Hi2MDI), p-Phenylendiisocyanat (PPDI), m-Tetra- methylxylylen diisocyanat (TMXDI), etc. sowie deren Dimere. Bevorzugt sind HDI, IPDI, MDI oder TDI. Geeignete Triisocyanate sind Trimere oder Biurete von aliphatischen, cycloaliphatischen, aromatischen oder araliphatischen Diisocyanaten, insbesondere die Isocyanurate und Biurete der im vorherigen Absatz beschriebenen Diisocyanate.
Selbstverständlich können auch geeignete Mischungen von Di- oder Triisocyanaten eingesetzt werden.
Als Polymere QPM mit endständigen Amino-, Thiol- oder Hydroxylgruppen sind insbesondere geeignet Polymere QPM mit zwei oder drei endständigen Amino-, Thiol- oder Hydroxylgruppen.
Die Polymere QPM weisen vorteilhaft ein Equivalenzgewicht von 300 - 6000, insbesondere von 600 - 4000, bevorzugt von 700 - 2200 g/Equivalent NCO-reaktiver Gruppen auf.
Als Polymere QPM geeignet sind Polyole, beispielsweise die folgenden handelsüblichen Polyole oder beliebige Mischungen davon:
-Polyoxyalkylenpolyole, auch Polyetherpolyole genannt, welche das Polymerisationsprodukt von Ethylenoxid, 1 ,2-Propylenoxid, 1 ,2- oder 2,3- Butylenoxid, Tetra hydrofu ran oder Mischungen davon sind, gegebenenfalls polymerisiert mit Hilfe eines Startermoleküls mit zwei oder drei aktiven H- Atomen wie beispielsweise Wasser oder Verbindungen mit zwei oder drei OH- Gruppen. Eingesetzt werden können sowohl Polyoxyalkylenpolyole, die einen niedrigen Ungesättigtheitsgrad aufweisen (gemessen nach ASTM D-2849-69
und angegeben in Milliequivalent Ungesättigtheit pro Gramm Polyol (mEq/g)), hergestellt beispielsweise mit Hilfe von sogenannten Double Metal Cyanide Complex Katalysatoren (kurz DMC-Katalysatoren), als auch Polyoxyalkylen- polyole mit einem höheren Ungesättigtheitsgrad, hergestellt beispielsweise mit Hilfe von anionischen Katalysatoren wie NaOH, KOH oder Alkalialkoholaten. Besonders geeignet sind Polyoxypropylendiole und -triole mit einem Ungesättigtheitsgrad tiefer als 0.02 mEq/g und mit einem Molekulargewicht im Bereich von 1000 - 30O00 Dalton, Polyoxybutylendiole und -triole, Polyoxypropylendiole und -triole mit einem Molekulargewicht von 400 - 8'00O Dalton, sowie sogenannte „EO-endcapped" (ethylene oxide-endcapped) Polyoxypropylendiole oder -triole. Letztere sind spezielle Polyoxypropylenpolyoxyethylen- polyole, die beispielsweise dadurch erhalten werden, dass reine Polyoxypro- pylenpolyole nach Abschluss der Polypropoxylierung mit Ethylenoxid alkoxy- liert werden und dadurch primäre Hydroxylgruppen aufweisen. -Hydroxyterminierte Polybutadienpolyole, wie beispielsweise solche, die durch Polymerisation von 1 ,3-Butadien und Allylalkohol oder durch Oxidation von Polybutadien hergestellt werden, sowie deren Hydrierungsprodukte;
-Styrol-Acrylnitril gepfropfte Polyetherpolyole, wie sie beispielsweise von Elastogran unter dem Namen Lupranol® geliefert werden;
-Polyhydroxyterminierte Acrylnitril/Butadien-Copolymere, wie sie beispielsweise aus carboxylterminierten Acrylnitril/Butadien-Copolymeren (kommerziell erhältlich unter dem Namen Hycar® CTBN von Nanoresins AG, Deutschland) und Epoxiden oder Aminoalkoholen hergestellt werden können; -Polyesterpolyole, hergestellt beispielsweise aus zwei- bis dreiwertigen
Alkoholen wie beispielsweise 1 ,2-Ethandiol, Diethylenglykol, 1 ,2-Propandiol, Dipropylenglykol, 1 ,4-Butandiol, 1 ,5-Pentandiol, 1 ,6-Hexandiol, Neopentyl- glykol, Glycerin, 1 ,1 ,1 -Tπmethylolpropan oder Mischungen der vorgenannten Alkohole mit organischen Dicarbonsäuren oder deren Anhydriden oder Estern wie beispielsweise Bernsteinsäure, Glutarsäure, Adipinsäure, Korksäure, Sebacinsäure, Dodecandicarbonsäure, Maleinsäure, Fumarsäure, Phthalsäure, Isophthalsäure, Terephthalsäure und Hexahydrophthalsäure oder
Mischungen der vorgenannten Säuren, sowie Polyesterpolyole aus Lactonen wie beispielsweise ε-Caprolacton;
-Polycarbonatpolyole, wie sie durch Umsetzung beispielsweise der oben genannten - zum Aufbau der Polyesterpolyole eingesetzten - Alkohole mit Dialkylcarbonaten, Diarylcarbonaten oder Phosgen zugänglich sind.
Vorteilhaft sind die Polymere QPM di- oder höherfunktioneller Polyole mit OH-Equivalentsgewichten von 300 bis 6000 g/OH-Equivalent, insbesondere von 600 bis 4000 g/OH-Equivalent, vorzugsweise 700 - 2200 g/OH- Equivalent. Weiterhin vorteilhaft sind die Polyole ausgewählt aus der Gruppe bestehend aus Polyethylenglycolen, Polypropylenglycolen, Polyethylenglycol- Polypropylenglycol-Block-Co-polymeren, Polybutylenglycolen, hydroxyl- terminierten Polybutadienen, hydroxylterminierten Butadien/Acrylnitril- Copolymeren, hydroxylterminierten synthetischen Kautschuken, deren Hydrierungsprodukten und Gemischen dieser genannten Polyole. Im Weiteren können als Polymere QPM auch di- oder höherfunktionelle aminoterminierte Polyethylenether, Polypropylenether, wie sie zum Beispiel unter dem Namen Jeffamine® von Huntsman vertrieben werden, Polybutylenether, Polybutadiene, Butadien/Acrylnitril-Copolymere, wie sie zum Beispiel die unter dem Namen Hycar® ATBN von Nanoresins AG, Deutschland vertrieben werden, sowie weitere aminoterminierte synthetische Kautschuke oder Gemische der genannte Komponenten verwendet werden.
Für gewisse Anwendungen sind als Polymere QPM insbesondere Hydroxyl-Gruppen aufweisende Polybutadiene oder Polyisoprene oder deren partiell oder vollständig hydrierte Reaktionsprodukte geeignet.
Es ist weiterhin möglich, dass die Polymere Q
PM auch kettenverlängert sein können, wie es in dem Fachmann bekannter Art und Weise durch die Reaktion von Polyaminen, Polyolen und Polyisocyanaten, insbesondere von Diaminen, Diolen und Diisocyanaten, durchgeführt werden kann. Am Beispiel eines Diisocyanates und eines Diols bildet sich daraus, wie im Folgenden gezeigt, je nach gewählter Stöchiometrie eine Spezies der Formel (VI) oder (VII)
Die Reste Y1 und Y2 stellen einen divalenten organischen Rest dar und die Indizes u und v variieren je nach Stöchiometrieverhältnis von 1 bis typischerweise 5.
Diese Spezies der Formel (VI) oder (VII) können dann wiederum weiterreagiert werden. So kann beispielsweise aus der Spezies der Formel (VI) und einem Diol mit einem divalenten organischen Rest Y3 ein kettenverlängertes Polyurethanprepolymer PU1 der folgenden Formel gebildet werden:
Aus der Spezies der Formel (VII) und einem Diisocyanat mit einem divalenten organischen Rest Y4 kann ein kettenverlängertes Polyurethanprepolymer PU1 der folgenden Formel gebildet werden:
Die Indizes x und y variieren je nach Stöchiometrieverhältnis von 1 bis typischerweise 5, und sind insbesondere 1 oder 2.
Weiterhin kann auch die Spezies der Formel (VI) mit der Spezies der Formel (VII) umgesetzt werden, so dass ein NCO Gruppen aufweisendes kettenverlängertes Polyurethanprepolymer PU1 entsteht.
Für die Kettenverlängerung werden insbesondere Diole und/oder Diamine und Diisocyanate bevorzugt. Selbstverständlich ist dem Fachmann klar, dass auch höherfunktionelle Polyole, wie beispielsweise Trimethylolpro- pan oder Pentaerythrit, oder höherfunktionelle Polyisocyanate, wie Isocyanura- te von Diisocyanaten, für die Kettenverlängerung verwendet werden können.
Bei den Polyurethanprepolymeren PU1 generell und bei den kettenverlängerten Polyurethanprepolymeren im Speziellen ist vorteilhaft darauf zu achten, dass die Prepolymere nicht zu hohe Viskositäten aufweisen, insbesondere wenn höher funktionelle Verbindungen für die Kettenver- längerung eingesetzt werden, denn dies kann deren Umsetzung zu den Polymeren der Formel (I) beziehungsweise die Applikation der Zusammensetzung erschweren.
Als Polymere QPM bevorzugt sind Polyole mit Molekulargewichten zwischen 600 und 6000 Dalton ausgewählt aus der Gruppe bestehend aus
Polyethylenglykolen, Polypropylenglykolen, Polyethylenglykol-Polypropylengly- kol-Blockpolymeren, Polybutylenglykolen, hydroxylterminierte Polybutadiene, hydroxylterminierte Butadien-Acrylnitril-Copolymere sowie deren Gemische.
Als Polymere QPM sind insbesondere bevorzugt α,ω-Dihydroxypoly- alkylenglykole mit C2-C6-Alkylengruppen oder mit gemischten C2-C6- Alkylen- gruppen, die mit Amino-, Thiol- oder, bevorzugt, Hydroxylgruppen terminiert sind. Besonders bevorzugt sind Polypropylenglykole oder Polybutylenglykole. Weiterhin besonders bevorzugt sind Hydroxylgruppen-terminierte Polyoxy- butylene.
Als Polyphenol QPP sind insbesondere geeignet Bis-, Tris- und Tetraphenole. Hierunter werden nicht nur reine Phenole, sondern gegebenenfalls auch substituierte Phenole verstanden. Die Art der Substitution kann sehr vielfältig sein. Insbesondere wird hierunter eine Substitution direkt am aromatischen Kern, an den die phenolische OH-Gruppe gebunden ist, verstanden. Unter Phenolen werden weiterhin nicht nur einkernige Aromaten, sondern auch mehrkernige oder kondensierte Aromaten oder Heteroaromaten verstanden, welche die phenolische OH-Gruppe direkt am Aromaten beziehungsweise Heteroaromaten aufweisen.
Durch die Art und Stellung eines solchen Substituenten wird unter anderem die für die Bildung des Polyurethanprepolymeren PU1 nötige Reaktion mit Isocyanaten beeinflusst.
Besonders eignen sich die Bis- und Trisphenole. Als Bisphenole oder Trisphenole sind beispielsweise geeignet 1 ,4-Dihydroxybenzol, 1 ,3-Dihydroxy- benzol, 1 ,2-Dihydroxybenzol, 1 ,3-Dihydroxytoluol, 3,5-Dihydroxybenzoate, 2,2- Bis(4-hydroxyphenyl)propan (=Bisphenol-A), Bis(4-hydroxyphenyl)methan (=Bisphenol-F), Bis(4-hydroxyphenyl)sulfon (=Bisphenol-S), Naphtoresorcin, Dihydroxynaphthalin, Dihydroxyanthrachinon, Dihydroxy-biphenyl, 3,3-bis(p- hydroxyphenyl)phthalide, 5,5-Bis(4-hydroxyphenyl)hexahydro-4,7-methano- indan, Phenolpthalein, Fluorescein, 4,4'-[bis-(hydroxyphenyl)-1 ,3-Phenylene- bis-(1 -Methyl-ethyliden)] (=Bisphenol-M), 4,4'-[bis-(hydroxyphenyl)-1 ,A- Phenylenebis-(1 -Methyl-ethyliden)] (=Bisphenol-P), 2,2'-Diallyl-bisphenol-A, Diphenole und Dikresole hergestellt durch Umsetzung von Phenolen oder Kresolen mit Di-isopropylidenbenzol, Phloroglucin, Gallsäureester, Phenoloder Kresolnovolacke mit -OH-Funktionalität von 2.0 bis 3.5 sowie alle Isomeren der vorgenannten Verbindungen. Bevorzugte Diphenole und Dikresole hergestellt durch Umsetzung von
Phenolen oder Kresolen mit Di-isopropylidenbenzol weisen eine chemische Strukturformel auf, wie sie entsprechend für Kresol als Bespiel nachfolgend gezeigt ist:
Besonders bevorzugt sind schwerflüchtige Bisphenole. Als meist bevorzugt gelten Bisphenol-M, Bisphenol-S und 2,2'-Diallyl-Bisphenol-A. Bevorzugt weist das QPP 2 oder 3 phenolische Gruppen auf.
In einer ersten Ausführungsform wird das Polyurethanprepolymer PU1 aus mindestens einem Diisocyanat oder Triisocyanat sowie aus einem Polymeren QPM mit endständigen Amino-, Thiol- oder Hydroxylgruppen hergestellt. Die Herstellung des Polyurethanprepolymers PU1 erfolgt in einer
dem Polyurethan-Fachmann bekannten Art und Weise, insbesondere, indem das Diisocyanat oder Triisocyanat in einem stöchiometrischen Überschuss in Bezug auf die Amino-, Thiol- oder Hydroxylgruppen des Polymeren QPM eingesetzt wird. In einer zweiten Ausführungsform wird das Polyurethanprepolymer
PU1 aus mindestens einem Diisocyanat oder Triisocyanat sowie aus einem, gegebenenfalls substituierten, Polyphenol QPP hergestellt. Die Herstellung des Polyurethanprepolymers PU1 erfolgt in einer dem Polyurethan-Fachmann bekannter Art und Weise, insbesondere indem das Diisocyanat oder Triisocyanat in einem stöchiometrischen Überschuss in Bezug auf die phenolischen Gruppen des Polyphenols QPP eingesetzt wird.
In einer dritten Ausführungsform wird das Polyurethanprepolymer PU1 aus mindestens einem Diisocyanat oder Triisocyanat sowie aus einem Polymeren QPM mit endständigen Amino-, Thiol- oder Hydroxylgruppen sowie aus einem, gegebenenfalls substituierten, Polyphenol QPP hergestellt. Zur Herstellung des Polyurethanprepolymers PU1 aus mindestens einem Diisocyanat oder Triisocyanat sowie aus einem Polymeren QPM mit endständigen Amino-, Thiol- oder Hydroxylgruppen und / oder aus einem, gegebenenfalls substituierten, Polyphenol QPP stehen unterschiedliche Möglichkeiten zur Verfügung.
In einem ersten Verfahren, „Eintopfverfahren" genannt, wird eine Mischung von mindestens einem Polyphenol QPP und mindestens einem Polymeren QPM mit mindestens einem Diisocyanat oder Triisocyanat in einem Isocyanatüberschuss umgesetzt.
In einem zweiten Verfahren, „2-Schrittverfahren I" genannt, wird mindestens ein Polyphenol QPP mit mindestens einem Diisocyanat oder Triisocyanat in einem Isocyanatüberschuss und anschliessend mit mindestens einem Polymeren QPM in Unterschuss umgesetzt. Im dritten Verfahren schliesslich, „2-Schrittverfahren II" genannt, wird mindestens ein Polymer QPM mit mindestens einem Diisocyanat oder Triisocyanat in einem Isocyanatüberschuss und anschliessend mit mindestens einem Polyphenol QPP in Unterschuss umgesetzt.
Die drei Verfahren führen zu Isocyanat-terminierten Polyurethanpre- polymeren PU1 , die sich bei gleicher Zusammensetzung in der Sequenz ihrer Bausteine unterscheiden können. Es sind alle drei Verfahren geeignet, jedoch ist das „2-Schrittverfahren II" bevorzugt. Werden die beschriebenen Isocyanat-endständigen Polyurethanpre- polymeren PU1 aus difunktionellen Komponenten aufgebaut, zeigte sich, dass das Equivalenz-Verhältnis Polymer QPM/Polyphenol QPP bevorzugt grösser als 1.50 und das Equivalenz-Verhältnis Polyisocyanat/(Polyphenol QPP + Polymer QPM) bevorzugt grösser als 1.20 ist. Wird die durchschnittliche Funktionalität der verwendeten
Komponenten grösser als 2, so erfolgt eine raschere Molekulargewichtserhöhung als im rein difunktionellen Fall. Für den Fachmann ist klar, dass die Grenzen der möglichen Equivalenz-Verhältnisse stark davon abhängen, ob entweder das gewählte Polymer QPM, das Polyphenol QPP, das Polyisocyanat oder mehrere der genannten Komponenten eine Funktionalität >2 besitzen. Je nach dem können unterschiedliche Equivalenz-Verhältnisse eingestellt werden, deren Grenzen durch die Viskosität der resultierenden Polymere bestimmt wird und die experimentell von Fall zu Fall bestimmt werden müssen.
Das Polyurethanprepolymer PU1 weist bevorzugt elastischen Charak- ter auf und zeigt eine Glasumwandlungstemperatur Tg von kleiner als 0°C.
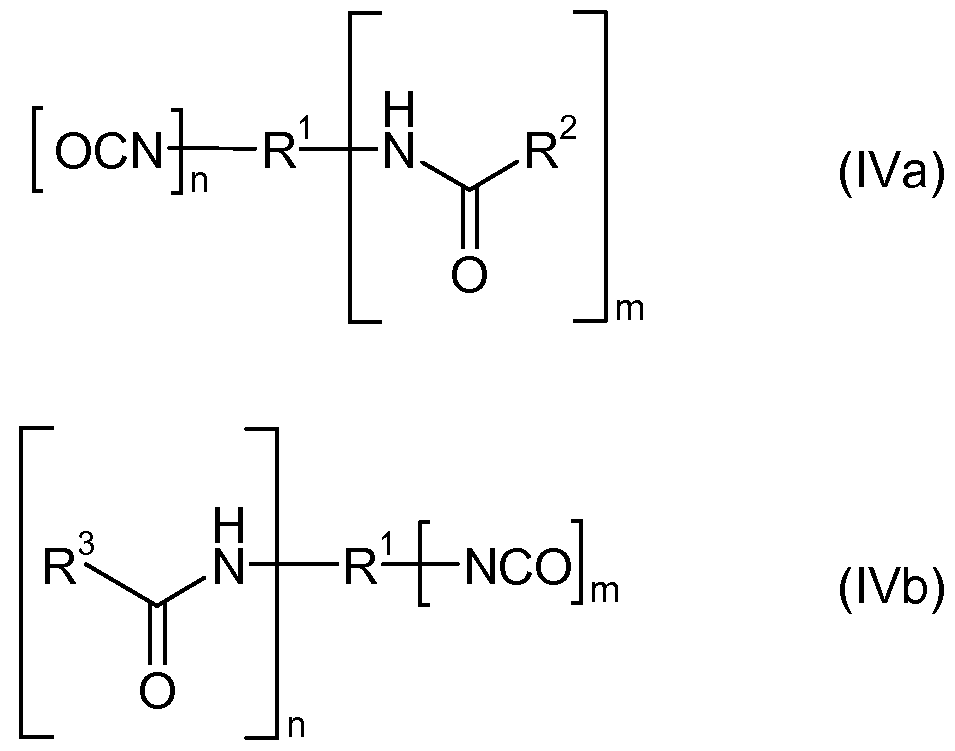
Das endständig blockierte Polyurethanprepolymer der Formel (I) kann aus einem Isocyanatgruppen aufweisenden Polyurethanprepolymer PU1 , welches die Formel (III) aufweist, und den NCO-reaktiven Verbindungen R
2-H und R
3-H hergestellt werden.
Hierbei kann die Umsetzung des Isocyanatgruppen aufweisenden Polyurethanprepolymers PU1 entweder mit einer Mischung von R
2-H und R
3-H erfolgen oder es kann eine sequentielle Umsetzung über ein Zwischenprodukt der Formel (IVa) oder (IVb) erfolgen.
In einem zweiten Schritt wird dann dieses NCO-Gruppen-haltige Zwischenprodukt der Formel (IVa) mit R3H, beziehungsweise dieses NCO- Gruppen-haltige Zwischenprodukt der Formel (IVb) mit R2H, zum endständig blockierten Polyurethanprepolymer der Formel (I) umgesetzt. Diese sequentiel- Ie Umsetzung hat den Vorteil, dass die Reaktion besser gesteuert werden kann, so dass die Bildung von symmetrischen Adduktionsprodukten („gleich" blockiert) reduziert wird. Dies ist insbesondere dann vorteilhaft, wenn die NCO- Reaktivitäten der Verbindungen R2-H und R3-H sehr unterschiedlich sind.
In einer bevorzugten Ausführungsform, in der R2 für die Gruppe der Formel (II) steht, erfolgt die entsprechende Reaktion des Polyurethanpre- polymers PU1 , welches die Formel (III) aufweist, mit einer Monohydroxyl- Epoxidverbindung der Formel (V) und einem Blockierungsmittel R3-H.
Bei einer sequentiellen Reaktion, die auch hier bevorzugt wird, entsteht ein Zwischenprodukt der Formel (IVb) oder der Formel (IVc)
Es ist bevorzugt, dass die Herstellung des endständig blockierten Polyurethan- prepolymers der Formel (I) über das Zwischenprodukt der Formel (IVc) erfolgt.
Diese sequentielle Reaktion kann einerseits dazu verwendet werden, um das blockierte Polyurethanprepolymer der Formel (I) direkt zu bilden, wel- ches dann in der Zubereitung der Epoxidharzzusammensetzung eingesetzt wird.
Andererseits kann die sequentielle Reaktion auch in einer spezifischen Ausführungsform, wie im Folgenden beschrieben, verwendet werden. Das im ersten Schritt der sequentiellen Reaktion erhaltene Zwischenprodukt, d.h. das partiell blockierte Prepolymer der Formel (IVa) oder (IVb), insbesondere der Formel (IVc), kann so mit den weiteren Bestandteilen einer hitzehärtenden Epoxidharzzusammensetzung, wie später in diesem Dokument im Detail beschrieben wird, weiter vermengt werden und zu einem Vorfabrikat gefertigt werden, welches transportier- und bei Raumtemperatur unter Ausschluss von Feuchtigkeit lagerfähig ist. Zu einem späteren Zeitpunkt kann vor der Applikation die entsprechende Verbindung R
2H oder R
3H in das Vorfabrikat eingemischt werden, zum Beispiel mit Hilfe einer Extrusion, und so in situ innerhalb der Zusammensetzung zur Bildung des blockierten Polyurethanpre- polymers der Formel (I) führen. Ein konkretes Beispiel hierfür ist die Herstel- lung eines Halbfabrikates, welches mindestens ein Epoxidharz A und mindestens ein partiell blockiertes Prepolymer der Formel (IVc) sowie mindestens einen Härter B für Epoxidharze, welcher durch erhöhte Temperatur aktiviert wird, enthält. Details zu diesen Inhaltsstoffen und weiteren mögliche Bestandteilen sind weiter unten in diesem Dokument beschrieben. Dieses Halbfabrikat ist lagerfähig und kann zu einem späteren Zeitpunkt zu einem Epoxid-Flüssig- harz oder ein derartiges Harz enthaltenden Zusammensetzung zudosiert und zugemischt werden, beispielsweise auch mit Hilfe einer Extrusion, bei einer Temperatur unter der Aktivierungstemperatur des Härters B. Das Epoxid- Flüssigharz enthält, wie im Folgenden erwähnt, eine Monohydroxyl- Epoxidverbindung der Formel (IX), welche der Verbindung R
3H, beziehungsweise einer Monohydroxyl-Epoxidverbindung der Formel (V), entspricht
Somit wird in situ aus dem partiell blockierten Prepolymer der Formel (IVc) und der Monohydroxyl-Epoxidverbindung der Formel (V) das endständig blockierte Polyurethanprepolymer der Formel (I) hergestellt. Das hier beschriebene konkrete Beispiel einer hitzehärtenden Epoxidharzzusammen- Setzung kann anschliessend als Klebstoff oder als sogenannter Reinforcer für das Verstärken von Blech- oder Rohrkonstruktionen im Fahrzeugbau verwendet werden, indem die Aushärtung bei einer Temperatur über der Aktivierungstemperatur des Härters B erfolgt.
Die Monohydroxyl-Epoxidverbindung der Formel (V), bzw. der Formel
(V), weist 1 , 2 oder 3 Epoxidgruppen auf. Die Hydroxylgruppe dieser Monohydroxyl-Epoxidverbindung (V), bzw. der Formel (V), kann eine primäre oder eine sekundäre Hydroxylgruppe darstellen.
Solche Monohydroxyl-Epoxidverbindungen lassen sich beispielsweise durch Umsetzung von Polyolen mit Epichlorhydrin erzeugen. Je nach Reaktionsführung entstehen bei der Umsetzung von mehrfunktionellen Alkoholen mit Epichlorhydrin als Nebenprodukte auch die entsprechenden Mono- hydroxyl-Epoxidverbindungen in unterschiedlichen Konzentrationen. Diese lassen sich durch übliche Trennoperationen isolieren. In der Regel genügt es aber, das bei der Glycidylisierungsreaktion von Polyolen erhaltene Produktgemisch aus vollständig und partiell zum Glycidylether reagiertem Polyol einzusetzen. Beispiele solcher hydroxylhaltigen Epoxide sind Butandiolmono- glycidylether (enthalten in Butandioldiglycidylether), Hexandiolmonoglycidyl- ether (enthalten in Hexandioldiglycidylether), Cyclohexandimethanolglycidyl- ether,Trimethylolpropandiglycidylether (als Gemisch enthalten in Trimethylol- propantriglycidylether), Glycerindiglycidylether (als Gemisch enthalten in Glycerintriglycidylether), Pentaerythrittriglycidylether (als Gemisch enthalten in Pentaerythrittetraglycidylether). Vorzugsweise wird Trimethylolpropan- diglycidylether, welcher zu einem relativ hohen Anteil in üblich hergestellten Trimethylolpropantriglycidylether vorkommt, verwendet.
Es können aber auch andere ähnliche hydroxylhaltige Epoxide, insbesondere Glycidol, 3-Glycidyloxybenzylalkohol oder Hydroxymethyl-cyclo- hexenoxid eingesetzt werden. Weiterhin bevorzugt ist der ß-Hydroxyether der Formel (IX), der in handelsüblichen flüssigen Epoxidharzen, hergestellt aus Bisphenol-A (R = CH3) und Epichlorhydrin, zu etwa 15 % enthalten ist, sowie die entsprechenden ß-Hydroxyether der Formel (IX), die bei der Reaktion von Bisphenol-F (R = H) oder des Gemisches von Bisphenol-A und Bisphenol-F mit Epichlorhydrin gebildet werden.
Weiterhin bevorzugt sind auch Destillationsrückstände, welche bei der
Herstellung von hochreinen, destillierten Epoxid-Flüssigharzen anfallen. Solche Destillationsrückstände weisen eine bis zu drei Mal höhere Konzentration an hydroxylhaltigen Epoxiden auf als handelübliche undestillierte Epoxid- Flüssigharze. Im Weiteren können auch unterschiedlichste Epoxide mit einer ß-Hydroxyether-Gruppe, hergestellt durch die Reaktion von (Poly-)Epoxiden mit einem Unterschuss von einwertigen Nukleophilen wie Carbonsäuren, Phenolen, Thiolen oder sec- Aminen, eingesetzt werden.
Die freie primäre oder sekundäre OH-Funktionalität der Monohydroxyl-
Epoxidverbindung der Formel (V) lässt eine effiziente Umsetzung mit terminalen Isocyanatgruppen von Prepolymeren zu, ohne dafür unverhältnis- mässige Überschüsse der Epoxidkomponente einsetzen zu müssen.
Zur Umsetzung der Polyurethanprepolymeren PU1 der Formel (III) können gesamthaft stöchiometrische Mengen an R3H, insbesondere der Monohydroxyl-Epoxidverbindung der Formel (V), und R2H eingesetzt werden. Falls eine sequentielle Umsetzung unter Bildung des Zwischenproduktes der Formel (IVa), (IVb) oder (IVc) durchgeführt wird, kann es von Vorteil sein, einen stöchiometrischen Überschuss der im zweiten Schritt verwendeten
Verbindung R3H, bzw. R2H, einzusetzen, um zu gewährleisten, dass alle NCO Gruppen abreagiert werden.
Das endständig blockierte Polyurethanprepolymer der Formel (I) weist vorteilhaft einen elastischen Charakter auf und ist weiterhin vorteilhaft in Epoxid-Flüssigharzen löslich oder dispergierbar.
Es wurde gefunden, dass das endständig blockierte Polyurethanprepolymer der Formel (I) in hervorragender Art und Weise als Schlagzähigkeitsmodifikator, insbesondere in Epoxidharzen, verwendet werden kann.
In einem weiteren Aspekt betrifft die vorliegende Erfindung hitzehärtbare Epoxidharzzusammensetzungen, welche - mindestens ein Epoxidharz A mit durchschnittlich mehr als einer
Epoxidgruppe pro Molekül;
- mindestens ein vorgängig beschriebenes endständig blockiertes Polyurethanprepolymer der Formel (I), sowie
- mindestens einen Härter B für Epoxidharze, welcher durch erhöhte Temperatur aktiviert wird, enthalten.
Das Epoxidharz A mit durchschnittlich mehr als einer Epoxidgruppe pro Molekül ist vorzugsweise ein Epoxid-Flüssigharz oder ein Epoxid-Festharz. Der Begriff „Epoxid-Festharz" ist dem Epoxid-Fachmann bestens bekannt und wird im Gegensatz zu „Epoxid-Flüssigharzen" verwendet. Die Glastemperatur von Festharzen liegt über Raumtemperatur, d.h. sie lassen sich bei Raumtemperatur zu schüttfähigen Pulvern zerkleinern.
Bevorzugte Epoxid-Festharze weisen die Formel (X) auf
(X)
Hierbei stehen die Substituenten R' und R" unabhängig voneinander entweder für H oder CH
3. Weiterhin steht der Index s für einen Wert von > 1.5, insbesondere von 2 bis 12.
Derartige Epoxid-Festharze sind kommerziell erhältlich beispielsweise von Dow oder Huntsman oder Hexion.
Verbindungen der Formel (X) mit einem Index s zwischen 1 und 1.5 werden vom Fachmann als Semisolid-Epoxidharze bezeichnet. Für die hier vorliegende Erfindung werden sie ebenfalls als Festharze betrachtet. Bevorzugt sind jedoch Epoxidharze im engeren Sinn, d.h. wo der Index s einen Wert von > 1.5 aufweist.
Bevorzugte Epoxid-Flüssigharze weisen die Formel (Xl) auf
Hierbei stehen die Substituenten R'" und R"" unabhängig voneinander entweder für H oder CH3. Weiterhin steht der Index r für einen Wert von 0 bis 1. Bevorzugt steht r für einen Wert von kleiner als 0.2.
Es handelt sich somit vorzugsweise um Diglycidylether von Bisphenol- A (DGEBA), von Bisphenol-F sowie von Bisphenol-A/F (Die Bezeichnung ,A/F' verweist hierbei auf eine Mischung von Aceton mit Formaldehyd, welche als Edukt bei dessen Herstellung verwendet wird). Solche Flüssigharze sind beispielsweise als Araldite® GY 250, Araldite® PY 304, Araldite® GY 282 (Huntsman) oder D.E.R.™ 331 oder D.E.R.™ 330 (Dow) oder Epikote 828 (Hexion) erhältlich.
Bevorzugt stellt das Epoxidharz A ein Epoxid-Flüssigharz der Formel (Xl) dar. In einer noch mehr bevorzugten Ausführungsform enthält die hitzehärtende Epoxidharzzusammensetzung sowohl mindestens ein Epoxid-
Flüssigharz der Formel (Xl) als auch mindestens ein Epoxid-Festharz der Formel (X).
Der Anteil von Epoxidharzes A beträgt vorzugsweise 10 - 85 Gew.-%, insbesondere 15 - 70 Gew.-%, bevorzugt 15 - 60 Gew.-%, am Gewicht der Zusammensetzung.
Der Anteil des endständig blockierten Polyurethanprepolymers der Formel (I) beträgt vorzugsweise 1 - 45 Gew.-%, insbesondere 3 - 30 Gew.-%, am Gewicht der Zusammensetzung.
Die erfindungsgemässe Zusammensetzung enthält weiterhin mindestens einen Härter B für Epoxidharze, welcher durch erhöhte Temperatur akti- viert wird. Es handelt sich hierbei vorzugsweise um einen Härter, welcher ausgewählt ist aus der Gruppe bestehend aus Dicyandiamid, Guanamine, Guanidine, Aminoguanidine und deren Derivate. Weiterhin möglich sind beschleunigend wirksame Härter, wie substituierte Harnstoffe, wie beispielsweise 3-Chlor-4-Methylphenylharnstoff (Chlortoluron), oder Phenyl-Dimethyl- harnstoffe, insbesondere p-Chlorphenyl-N,N-dimethylharnstoff (Monuron), 3- Phenyl-1 ,1 -dimethylharnstoff (Fenuron) oder 3,4-Dichlorphenyl-N,N-dimethyl- harnstoff (Diuron). Weiterhin können Verbindungen der Klasse der Imidazole und Amin-Komplexe eingesetzt werden.
Bevorzugt handelt es sich beim Härter B um einen Härter, welcher ausgewählt ist aus der Gruppe bestehend aus Dicyandiamid, Guanamine, Guanidine, Aminoguanidine und deren Derivate; substituierte Harnstoffe, insbesondere 3-Chlor-4-Methylphenylharnstoff (Chlortoluron), oder Phenyl-Di- methylharnstoffe, insbesondere p-Chlorphenyl-N,N-dimethylharnstoff (Monuron), 3-Phenyl-1 ,1 -dimethylharnstoff (Fenuron), 3,4-Dichlorphenyl-N,N-di- methylharnstoff(Diuron), sowie Imidazole und Amin-Komplexe. Besonders bevorzugt als Härter B ist Dicyandiamid.
Vorteilhaft beträgt der Gesamtanteil des Härters B 1 - 10 Gewichts-%, vorzugsweise 2 - 8 Gewichts-%, bezogen auf das Gewicht der gesamten Zusammensetzung.
Die hitzehärtende Epoxidharzzusammensetzung kann weiterhin ein
Thixotropiermittel C auf Basis eines Harnstoffderivates enthalten. Das Harnstoffderivat ist insbesondere ein Umsetzungsprodukt eines aromatischen monomeren Diisocyanates mit einer aliphatischen Aminverbindung. Es ist auch durchaus möglich, mehrere unterschiedliche monomere Diisocyanate mit einer oder mehreren aliphatischen Aminverbindungen oder ein monomeres Diisocyanat mit mehreren aliphatischen Aminverbindungen umzusetzen. Als besonders vorteilhaft hat sich das Umsetzungsprodukt von 4,4'-Diphenyl- methylen-diisocyanat (MDI) mit Butylamin erwiesen.
Das Harnstoffderivat ist vorzugsweise in einem Trägermaterial vorhanden. Das Trägermaterial kann ein Weichmacher, insbesondere ein Phthalat oder ein Adipat sein, vorzugsweise ein Diisodecylphthalat (DIDP) oder Dioctyladipat (DOA). Das Trägermittel kann auch ein nicht-diffundierendes Trägermittel sein. Dies ist bevorzugt, um möglichst eine geringe Migration nach Aushärtung von nicht regierten Bestandteilen zu gewährleisten. Bevorzugt sind als nicht-diffundierende Trägermittel blockierte Polyurethanprepolymere.
Die Herstellung von solchen bevorzugten Harnstoffderivaten und Trägermaterialien sind im Detail in der Patentanmeldung EP 1 152 019 A1 beschrieben. Das Trägermaterial ist vorteilhaft ein blockiertes Polyurethanpre- polymer PU2, insbesondere erhalten durch Umsetzung eines trifunktionellen Polyetherpolyols mit IPDI und anschliessender Blockierung der endständigen Isocyanatgruppen mit ε-Caprolactam.
Vorteilhaft beträgt der Gesamtanteil des Thixotropiermittels C 0 - 40 Gewichts-%, vorzugsweise 5 - 25 % Gewichts-%, bezogen auf das Gewicht der gesamten Zusammensetzung. Das Verhältnis vom Gewicht des Harnstoffderivates zum Gewicht des allenfalls vorhandenen Trägermittels beträgt vorzugsweise 2 / 98 bis 50 / 50, insbesondere 5 / 95 - 25 /75.
Die hitzehärtende Epoxidharzzusammensetzung enthält vorzugsweise weiterhin einen Flüssigkautschuk D, welcher vorzugsweise ein carboxyl- oder epoxidterminiertes Polymer ist.
In einer ersten Ausführungsform ist dieser Flüssigkautschuk D ein carboxyl- oder epoxidterminiertes Acrylnitril/Butadien-Copolymer oder ein
Derivat davon. Derartige Flüssigkautschuke sind beispielsweise unter dem unter dem Namen Hycar® CTBN und CTBNX und ETBN von Nanoresins AG,
Deutschland kommerziell erhältlich. Als Derivate sind insbesondere
Epoxidgruppen aufweisende Elastomer-modifizierte Prepolymere, wie sie unter der Produktelinie Polydis®, vorzugsweise aus der Produktelinie Polydis® 36.., von der Firma Struktol® (Schill+Seilacher Gruppe, Deutschland) oder unter der
Produktelinie Albipox (Nanoresins, Deutschland) kommerziell vertrieben werden, geeignet.
In einer zweiten Ausführungsform ist dieser Flüssigkautschuk D ein epoxidterminiertes Polyurethanprepolymer der Formel (XII) beziehungsweise in einer anderen Darstellungsart gemäss Formel (XM').
Derartige mit einem einzigen Blockierungsmittel blockierte, d.h. gleich blockierte, Polyurethanprepolymere sind zum Grossteil bereits aus WO-A- 2005/007720, dort als Polymer B der Formel (I) bezeichnet, bekannt. Ihre
Herstellung erfolgt, wie auch in WO-A-2005/007720 im Detail beschrieben, aus dem Isocyanatgruppen aufweisenden Polyurethanprepolymer PU1 , welches
die Formel (III) aufweist, und einem Überschuss der Monohydroxyl- Epoxidverbindung der Formel (V).
In einer dritten Ausführungsform ist dieser Flüssigkautschuk D ein
Polyacrylatflüssigkautschuk, der mit flüssigen Epoxidharzen vollständig mischbar ist und sich erst beim Aushärten der Epoxidharzmatrix zu Mikrotröpf- chen entmischt. Derartige Polyacrylatflüssigkautschuke sind beispielsweise unter der Bezeichnung 20208-XPA von Rohm und Haas erhältlich.
Es ist dem Fachmann klar, dass natürlich auch Mischungen von Flüssigkautschuken verwendet werden können, insbesondere Mischungen von carboxyl- oder epoxidterminiertes Acrylnitril/Butadien-Copolymeren oder Derivaten davon mit epoxidterminierten Polyurethanprepolymeren der Formel (XII).
Der Flüssigkautschuk D wird vorteilhaft in einer Menge von 1 - 35 Gew.-%, insbesondere 1 - 25 Gew.-%, bezogen auf das Gewicht der Zusammensetzung, eingesetzt.
Die hitzehärtende Epoxidharzzusammensetzung enthält vorzugsweise weiterhin einen festen Zähigkeitsverbesserer E. Unter einem „Zähigkeitsverbesserer" wird hierbei und im Folgenden ein Zusatz zu einer Epoxidharzmatrix verstanden, der bereits bei geringen Zuschlägen von 0.1 - 15 Gew.-%, insbesondere von 0.5 - 8 Gew.-%, eine deutliche Zunahme der Zähigkeit bewirkt und somit in der Lage ist, höherer Biege-, Zug-, Schlag- oder Stossbeanspruchung aufzunehmen, bevor die Matrix einreisst oder bricht.
Der feste Zähigkeitsverbesserer E ist in einer ersten Ausführungsform ein organisches lonen-getauschtes Schichtmineral E1.
Das lonen-getauschte Schichtmineral E1 kann entweder ein Kationengetauschtes Schichtmineral E1c oder ein Anionen-getauschtes Schichtmineral E1a sein.
Das Kationen-getauschte Schichtmineral E1c wird hierbei erhalten aus einem Schichtmineral ET, bei welchem zumindest ein Teil der Kationen durch organische Kationen ausgetauscht worden sind. Beispiele für derartige
Kationen-getauschte Schichtmineralien E1c sind insbesondere diejenigen, welche in US 5,707,439 oder in US 6,197,849 erwähnt sind. Ebenso ist dort
das Verfahren zur Herstellung dieser Kationen-getauschten Schichtminerale E1c beschrieben. Bevorzugt als Schichtmineral EV ist ein Schichtsilikat. Insbesondere bevorzugt handelt es sich beim Schichtmineral EV um ein Phyllosilikat, wie sie in US 6,197,849 Spalte 2, Zeile 38 bis Spalte 3, Zeile 5 beschrieben sind, insbesondere um einen Bentonit. Als besonders geeignet haben sich Schichtmineral EV wie Kaolinit oder ein Montmorillionit oder ein Hectorit oder ein Illit gezeigt.
Zumindest ein Teil der Kationen des Schichtminerals EV wird durch organische Kationen ersetzt. Beispiele für derartige Kationen sind n-Octyl- ammonium, Trimethyldodecylammonium, Dimethyldodecylammonium oder Bis(hydroxyethyl)octadecylammonium oder ähnliche Derivate von Aminen, die aus natürlichen Fetten und Ölen gewonnen werden können; oder Guanidinium Kationen oder Amidiniumkationen; oder Kationen der N-substituierten Derivate von Pyrrolidin, Piperidin, Piperazin, Morpholin, Thiomorpholin; oder Kationen von 1 ,4-Diazobicyclo[2.2.2]octan (DABCO) und 1 -Azobicyclo[2.2.2]octan; oder Kationen von N-substituierten Derivaten von Pyridin, Pyrrol, Imidazol, Oxazol, Pyrimidin, Chinolin, Isochinoilin, Pyrazin, Indol, Bezimidazol, Benzoxaziol, Thiazol Phenazin und 2,2'-Bipyhdin. Weiterhin sind geeignet cyclische Amidiniumkationen, insbesondere solche, wie sie in US 6,197,849 in Spalte 3 Zeile 6 bis Spalte 4 Zeile 67 offenbart werden. Cyclische Ammoniumverbindungen zeichnen sich gegenüber linearen Ammoniumverbindungen durch eine erhöhte Thermostabilität aus, da der thermische Hoffmann - Abbau bei ihnen nicht auftreten kann.
Bevorzugte Kationen-getauschte Schichtminerale E1c sind dem Fachmann unter dem Term Organoclay oder Nanoclay bekannt und sind kommerziell zum Beispiel unter den Gruppennamen Tixogel® oder Nanofil® (Südchemie), Cloisite® (Southern Clay Products) oder Nanomer® (Nanocor Inc.) erhältlich.
Das Anionen-getauschte Schichtmineral E1a wird hierbei erhalten aus einem Schichtmineral EV, bei welchem zumindest ein Teil der Anionen durch organische Anionen ausgetauscht worden sind. Ein Beispiel für ein derartig Anionen-getauschtes Schichtmineral E1a ist ein Hydrotalcit EV, bei dem
zumindest ein Teil der Carbonat-Anionen der Zwischenschichten durch organische Anionen ausgetauscht wurden. Ein weiteres Beispiel sind funktionalisierte Alumoxane wie z. B. in US Patent 6 322890 beschrieben.
Es ist durchaus auch möglich, dass die Zusammensetzung gleichzeitig ein Kationen-getauschtes Schichtmineral E1c und ein Anionen-getauschtes Schichtmineral E1a enthält.
Der feste Zähigkeitsverbesserer ist in einer zweiten Ausführungsform ein Blockcopolymer E2. Das Blockcopolymer E2 wird erhalten aus einer anionischen oder kontrollierten radikalischen Polymerisation von Methacryl- säureester mit mindestens einem weiteren eine olefinisch Doppelbindung aufweisenden Monomeren. Als eine olefinische Doppelbindung aufweisende Monomere sind insbesondere solche bevorzugt, bei denen die Doppelbindung unmittelbar mit einem Heteroatom oder mit mindestens einer weiteren Doppelbindung konjugiert ist. Insbesondere sind Monomere geeignet, welche ausgewählt sind aus der Gruppe umfassend Styrol, Butadien, Acrylnitril und Vinylacetat. Bevorzugt sind Acrylat-Styrol-Acrylsäure (ASA) Copolymere, erhältlich z.B. unter dem Namen GELOY 1020 von GE Plastics.
Besonders bevorzugte Blockcopolymere E2 sind Blockcopolymere aus Methacrylsäuremethylester, Styrol und Butadien. Derartige Blockcopolymere sind beispielsweise als Triblockcopolymere unter der Gruppenbezeichnung SBM bei Arkema erhältlich.
Der feste Zähigkeitsverbesserer E ist in einer dritten Ausführungsform ein Core-Shell Polymer E3. Core-Shell-Polymere bestehen aus einem elastischen Kernpolymer und einem starren Schalen-Polymer. Insbesondere geeignete Core-Shell-Polymere bestehen aus einem Kern (Core) aus elastischem Acrylat- oder Butadien-Polymer, den eine starre Schale (Shell) eines starren thermoplastischen Polymers umhüllt. Diese Core-Shell Struktur bildet sich entweder spontan durch Entmischen eines Blockcopolymeren oder ist durch die Polymerisationsführung als Latex oder Suspensionspolymerisation mit nachfolgender Pfropfung vorgegeben. Bevorzugte Core- Shell-Polymere sind sogenannte MBS Polymere, welche kommerziell unter
dem Handelsnamen Clearstrength™ von Atofina, Paraloid™ von Rohm and Haas oder F-351 ™ von Zeon erhältlich sind.
Besonders bevorzugt sind Core-Shell Polymerpartikel, die bereits als getrockneter Polymerlatex vorliegen. Beispiele hierfür sind GENIOPERL M23A von Wacker mit Polysiloxankern und Acrylatschale, strahlungsvernetzte
Kautschukpartikel der NEP Reihe, hergestellt von Eliokem oder Nanoprene von Lanxess oder Paraloid EXL von Rohm und Haas.
Weitere vergleichbare Beispiele für Core-Shell-Polymere werden unter dem Namen Albidur ™ von Nanoresins AG, Deutschland, angeboten. Der feste Zähigkeitsverbesserer E ist in einer vierten Ausführungsform ein festes Umsetzungsprodukt E4 eines carboxylierten festen Nitrilkautschuks mit überschüssigem Epoxidharz.
Als fester Zähigkeitsverbesserer E sind Core-Shell Polymere bevorzugt.
Die hitzehärtende Epoxidharzzusammensetzung kann das feste Core- Shell Polymer E3 enthalten, insbesondere in einer Menge von 0.1 - 15 Gew.- %, vorzugsweise 1 - 8 Gew.-%, bezogen auf das Gewicht der Zusammensetzung.
In einer weiteren bevorzugten Ausführungsform enthält die Zusammensetzung zusätzlich mindestens einen Füllstoff F. Bevorzugt handelt es sich hierbei um Glimmer, Talk, Kaolin, Wollastonit, Feldspat, Syenith, Chlorit, Bentonit, Montmorillonit, Calciumcarbonat (gefällt oder gemahlen), Dolomit, Quarz, Kieselsäuren (pyrogen oder gefällt), Cristobalit, Calciumoxid, Aluminiumhydroxid, Magnesiumoxid, Keramikhohlkugeln, Glashohlkugeln, organische Hohlkugeln, Glaskugeln, Farbpigmente. Als Füllstoff F sind sowohl die organisch beschichteten als auch die unbeschichteten kommerziell erhältlichen und dem Fachmann bekannten Formen gemeint. Vorteilhaft beträgt der Gesamtanteil des gesamten Füllstoffs F 3 - 50
Gewichts-%, vorzugsweise 5 - 35 Gewichts-%, insbesondere 5 - 25 Gewichts- %, bezogen auf das Gewicht der gesamten Zusammensetzung.
In einer weiteren bevorzugten Ausführungsform enthält die Zusammensetzung ein physikalisches oder chemisches Treibmittel, wie es beispielsweise unter dem Handelsnamen Expancel™ der Firma Akzo Nobel oder Celogen™ der Firma Chemtura erhältlich ist. Der Anteil des Treibmittels beträgt vorteilhaft 0.1 - 3 Gew.-%, bezogen auf das Gewicht der Zusammensetzung.
In einer weiteren bevorzugten Ausführungsform enthält die Zusammensetzung zusätzlich mindestens einen Epoxidgruppen-tragenden Reaktivverdünner G. Bei diesen Reaktivverdünnern G handelt es sich insbesondere um:
- Glycidylether von monofunktionellen gesättigten oder ungesättigten, verzweigten oder unverzweigten, zyklischen oder offenkettigen C4 - C30 Alkoholen, z.B. Butanolglycidylether, Hexanolglycidylether, 2-Ethyl- hexanolglycidylether, Allylglycidylether, Tetrahydrofurfuryl- und
Furfurylglycidylether, Trimethoxysilylglycidylether etc.
- Glycidylether von difunktionellen gesättigten oder ungesättigten, verzweigten oder unverzweigten, zyklischen oder offenkettigen C2 - C30 Alkolen, z.B Ethylenglykol-, Butandiol-, Hexandiol-, Oktandiolgylcidyl- ether, Cyclohexandimethanoldigylcidylether, Neopentylglycoldiglycidyl- ether etc.
- Glycidylether von tri- oder polyfunktionellen, gesättigten oder ungesättigten, verzweigten oder unverzweigten, zyklischen oder offenkettigen Akoholen wie epoxidiertes Rhizinusöl, epoxidiertes Trimethylolpropan, epoxidiertes Pentaerythrol oder Polyglycidylether von aliphatischen
Polyolen wie Sorbitol, Glycerin, Trimethylolpropan etc.
- Glycidylether von Phenol- und Anilinverbindungen wie Phenylglycidyl- ether, Kresylglycidylether, p-tert.-Butylphenylglycidylether, Nonyl- phenolglycidylether, 3-n-Pentadecenyl-glycidylether (aus Cashewnuss- Schalen-Öl), N,N-Diglycidylanilin etc.
- Epoxidierte Amine wie N, N-Diglycidylcyclohexylamin etc.
- Epoxidierte Mono- oder Dicarbonsäuren wie Neodecansäure-glycidyl- ester, Methacrylsäureglycidylester, Benzoesäureglycidylester, Phthal-
säure-, Tetra- und Hexahydrophthalsäurediglycidylester, Diglycidyl- ester von dimeren Fettsäuren etc.
- Epoxidierte di- oder trifunktionelle, nieder- bis hochmolekulare PoIy- etherpolyole wie Polyethylenglycol-diglycidylether, Polypropylenegly- col-diglycidylether etc.
Besonders bevorzugt sind Hexandioldiglycidylether, Kresylglycidyl- ether, p-te/t-Butylphenylglycidylether, Polypropylenglycoldiglycidylether und Polyethylenglycoldiglycidylether. Vorteilhaft beträgt der Gesamtanteil des epoxidgruppentragenden
Reaktivverdünners G 0.5 - 20 Gewichts-%, vorzugsweise 1 - 8 Gewichts-%, bezogen auf das Gewicht der gesamten Zusammensetzung.
Die Zusammensetzung kann weitere Bestandteile, insbesondere Katalysatoren, Hitze- und/oder Lichtstabilisatoren, Thixotropiermittel, Weichmacher, Lösungsmittel, mineralische oder organische Füllstoffe, Treibmittel, Farbstoffe und Pigmente, umfassen.
Es hat sich gezeigt, dass sich die beschriebenen hitzehärtenden Epoxidharzzusammensetzungen besonders als einkomponentige Klebstoffe eignen. Ein derartiger einkomponentiger Klebstoff weist breite Einsatzmöglichkeiten auf. Insbesondere sind hiermit hitzehärtende einkomponentige Klebstoffe realisierbar, die sich durch eine hohe Schlagzähigkeit, sowohl bei höheren Temperaturen und vor allem bei tiefen Temperaturen, insbesondere zwischen 00C bis -400C auszeichnen. Solche Klebstoffe werden für das Verkleben von hitzestabilen Materialien benötigt. Unter hitzestabilen Materialien werden Materialien verstanden, welche bei einer Aushärtetemperatur von 100 - 220 0C, vorzugsweise 120 - 2000C zumindest während der Aushärtezeit formstabil sind. Insbesondere handelt es sich hierbei um Metalle und Kunststoffe wie ABS, Polyamid, Polyphenylenether, Verbundmaterialien wie SMC, ungesättigte Polyester GFK, Epoxid- oder Acrylatverbundwerkstoffe. Bevorzugt ist die Anwendung, bei der zumindest ein Material ein Metall ist. Als besonders bevorzugte Verwendung gilt das Verkleben von gleichen oder
verschiedenen Metallen, insbesondere im Rohbau in der Automobilindustrie. Die bevorzugten Metalle sind vor allem Stahl, insbesondere elektrolytisch verzinkter, feuerverzinkter, beölter Stahl, Bonazink-beschichteter Stahl, und nachträglich phosphatierter Stahl, sowie Aluminium insbesondere in den im Autobau typischerweise vorkommenden Varianten.
Mit einem Klebstoff basierend auf einer erfindungsgemässen hitzehärtenden Zusammensetzung ist es möglich, die gewünschte Kombination von hoher Crashfestigkeit sowohl hoher als auch tiefer Einsatztemperatur zu er- reichen.
Ein solcher Klebstoff wird zuerst mit den zu verklebenden Materialien bei einer Temperatur von zwischen 10° C und 800C, insbesondere zwischen 100C und 60°C, kontaktiert und später ausgehärtet bei einer Temperatur von typischerweise 100 - 220 0C, vorzugsweise 120 - 2000C. Aus einem derartigen Verfahren zum Verkleben von hitzestabilen
Materialien resultiert ein verklebter Artikel. Ein derartiger Artikel ist vorzugsweise ein Fahrzeug oder ein Anbauteil eines Fahrzeugs.
Selbstverständlich können mit einer erfindungsgemässen Zusammen- Setzung neben hitzehärtenden Klebstoffen auch Dichtmassen oder Beschich- tungen realisiert werden. Ferner eignen sich die erfindungsgemässen
Zusammensetzungen nicht nur für den Automobilbau sondern auch für andere
Anwendungsgebiete. Besonders zu erwähnen sind verwandte Anwendungen im Transportmittel bau wie Schiffe, Lastwagen, Busse oder Schienenfahrzeuge oder im Bau von Gebrauchsgütern wie beispielsweise Waschmaschinen.
Die mittels einer erfindungsgemässen Zusammensetzung verklebten Materialien kommen bei Temperaturen zwischen typischerweise 120°C und -40°C, vorzugsweise zwischen 1000C und -40°C, insbesondere zwischen 80°C und -400C zum Einsatz. Es lassen sich Zusammensetzungen formulieren, welche typischerweise Bruchenergien, gemessen nach ISO 11343, von mehr als 10.0 J bei 23°C und mehr als 9.0 J bei -30°C und/oder von mehr als 8.0 J bei -400C aufweisen. Es lassen sich zuweilen Zusammensetzungen formulieren, welche
Bruchenergien von mehr als 13.0 J bei 23°C und von mehr als 10.0 J bei - 300C und/oder von mehr als 9.0 J bei -400C aufweisen. Besonders vorteilhafte Zusammensetzungen weisen gar Bruchenergien von mehr als 14.0 J bei 23°C und von mehr als 11.0 J bei -300C und/oder von mehr als 10.0 J bei -40°C auf. Eine besonders bevorzugte Anwendung der erfindungsgemässen hitzehärtenden Epoxidharzzusammensetzung ist die Anwendung als hitzehärtender Rohbauklebstoff im Fahrzeugbau.
Beispiele Im Folgenden sollen einige Beispiele aufgezeigt werden, welche die
Erfindung weiter veranschaulichen, den Umfang der Erfindung aber in keiner Weise beschränken sollen. Die in den Beispielen verwendeten Rohstoffe sind in Tabelle 1 aufgelistet.
Tabellei . Eingesetzte Rohstoffe.
Beispielhafte Herstellung eines monohvdroxylhaltiαen Epoxides „M1"
Trimethylolpropanglycidylether wurde gemäss dem Verfahren in Patent US 5,668,227, Beispiel 1 , aus Trimethylolpropan und Epichlorhydrin mit Tetramethylammoniumchlorid und Natronlauge hergestellt. Man erhält ein gelbliches Produkt mit einer Epoxidzahl von 7.5 eq/kg und einem Hydroxylgruppengehalt von 1.8 eq/kg. Aus dem HPLC-MS Spektrum kann geschlossen werden, dass im Wesentlichen ein Gemisch von Trimethylolpropandiglycidyl- ether und Trimethylolpropantriglycidylether vorliegt. Dieses Produkt wurde in Tabelle 2 als M1 eingesetzt.
Monohydroxylhaltiges Epoxid „M2"
1 ,3-bis(4-(2-(4-(oxiran-2-ylmethoxy)phenyl)propan-2-yl)phenoxy)- propan-2-ol) („DGEBA-Dimer"):
Entsprechend Verbindung der Formel (IX), in welcher mit R für Methyl steht. 1 ,3-bis(4-(2-(4-(oxiran-2-ylmethoxy)phenyl)propan-2-yl)phenoxy)propan-2-ol) wurde aus technischem Bisphenol-A-diglycidylether (DGEBA) (Araldite® GY 250, Hersteller Huntsman) gewonnen, in welchem es ca. zu 15 Gew.-% vorhanden ist. Es kann durch destillatives Entfernen von DGEBA aufkonzentriert werden. In einem Dünnfilmverdampfer (Hersteller: Fa Ilmag) wird bei 180
0C Heizmanteltemperatur im Ölpumpenvakuum technischer Bisphenol-A-diglycidylether (EEW = 195 g/Epoxidequivalent, bestimmt durch Titration) mit einer Membranpumpe mit 200 ml/h dosiert. Dabei destilliert reines DGEBA ab, das bei Raumtemperatur kristallisiert. Der verbleibende Sumpf weist ein EEW = 207.1 g/Epoxidequivalent auf. Mit THF als Lösungsmittel zeigt das GPC Diagramm ein Flächenverhältnis der Peaks von „DGEBA-Dimer" und DGEBA von 40:60. Dieses Produkt wurde als „M2" in Tabelle 2 eingesetzt.
Beispiele der Herstellung von Isocvanat-terminierten Polvurethanprepolvmeren
PU1-1
200.00 g Desmophen 3060 BS (OH-Zahl 57.0 mg/g KOH) wurden 30 Minuten unter Vakuum bei 1100C getrocknet. Nachdem die Temperatur auf 900C redu- ziert worden war, wurde 47.55 g IPDI und 25 mg Dibutylzinndilaurat zugegeben. Die Reaktion wurde unter Vakuum bei 90°C bis zur Konstanz des NCO- Gehaltes bei 3.64% nach 2.5 h geführt (theoretischer NCO-Gehalt: 3.73%).
PU1-2 200.00 g PoIy-THF 2000 (OH-Zahl 57.0 mg/g KOH) wurden 30 Minuten unter Vakuum bei 1100C getrocknet. Nachdem die Temperatur auf 90°C reduziert worden war, wurde 48.0 g IPDI und 25 mg Dibutylzinndilaurat zugegeben. Die Reaktion wurde unter Vakuum bei 90°C bis zur Konstanz des NCO-Gehaltes bei 3.65 % nach 2.5 h geführt (theoretischer NCO-Gehalt: 3.76 %).
Beispiel eines endständiq blockierten Polyurethanprepolymers der Formel (I) Beispielhaft wird hier im Detail die Herstellung des Polymers P2 beschrieben: Zu den 247.6 g des oben hergestellten Isocyanatterminierten Polyurethanprepolymers PU1-1 wurden nun 65.5 g des oben beschriebenen monohydroxylhaltigen Epoxides M1 zugegeben, womit molmässig die Hälfte der endständigen Isocyanate des Polymers abreagiert wurden. Es wurde bei 900C unter Vakuum gerührt, bis der NCO-Gehalt nach weiteren 2 Stunden auf ca. 1.4% gesunken war. Anschliessend wurden 38.6g Cardanol zugegeben (Hydroxylgehalt: ca. 3.33 eq/kg).
Es wurde ein klares Produkt mit einem Epoxid-Gehalt („End-Gehalt") von 1.39 eq/kg und einem NCO-Gehalt von < 0.05% erhalten.
Die anderen, in Tabelle 2 beschriebenen, blockierten Polyurethanpre- polymere wurden analog hergestellt. Bei den blockierten Polyurethanpolymeren P1, P2, P3, P-R1 und P-R2 erfolgte eine Variation der eingesetzten Menge an monohydroxylhaltigem Epoxid (M1) und Cardanol {NC). Bei den blockierten Polyurethanpolymeren P4, P5 und P6 wurde, verglichen mit P2,
das monohydroxylhaltige Epoxid M2, beziehungsweise Mischungen von M1 und M2, eingesetzt. Bei den blockierten Polyurethanpolymeren P7, P8, P-R3, P-R4 und P-R5 wurde PU1-2 als Polyurethanprepolymer eingesetzt und es wurden andere Blockierungsmittel verwendet. In PT, P-R3 und P-R4 wurde die Menge M1 und 2,2'-Diallyl-bisphenol-A [DABPA) variiert und in P8 und P-R5 wurde die Menge an M1 und 2-Hydroxybenzylalkohol (HBA) variiert.
Die Polymere P-R1, P-R2, P-R3, P-R4 und P-R5 weisen lediglich ein Blockierungsmittel auf und sind somit „gleich blockiert".
Tabelle 2: Herstellung blockierter Polyurethanprepolymere.
1 Anzahl ιmol-% der mit diesem Blockierungsmittel blockierten NCO Gruppen des PoIy- urethanprepolymers PU1-1 resp. PU1-2.
Thixotropiermittel C
Als Beispiel für ein Thixotropiermittel C auf Basis eines Harnstoffderivates in einem nicht-diffundierenden Trägermaterial wurde ein Thixotropiermittel C gemäss Patentanmeldung EP 1 152 019 A1 in einem blockierten Polyurethan- Prepolymer mit oben erwähnten Rohstoffen hergestellt:
Trägermaterial: Blockiertes Polvurethanprepolymer „BlockPU" 600.0 g eines Polyetherpolyols (Desmophen 3060BS; 3000 Dalton; OH-Zahl 57 mg/g KOH) wurden unter Vakuum und Rühren bei 900C mit 140.0 g IPDI und 0.10 g Dibutylzinndilaurat zum Isocyanat-terminierten Prepolymer umgesetzt. Die Reaktion wurde bis zur Konstanz des NCO-Gehaltes bei 3.41 % nach 2.5 h geführt (theoretischer NCO-Gehalt: 3.60%). Anschliessend wurden die freien Isocyanatgruppen bei 900C unter Vakuum mit 69.2g ε-Caprolactam (2% Über- schuss) blockiert, wobei ein NCO-Gehalt von < 0.1 % nach 3h erreicht wurde.
Harnstoffderivat („HSD") in blockiertem Polvurethan-Prepolvmer:
Unter Stickstoff und leichtem Wärmen wurden 68.7 g MDI-Flocken in 181.3 g des oben beschriebenen blockierten Prepolymeres „ß/oc/cPLT eingeschmolzen. Danach wurden während zwei Stunden unter Stickstoff und schnellem Rühren 40.1 g N-Butylamin gelöst in 219.9 g des oben beschriebenen blockierten Prepolymers „BlockPU" zugetropft. Nach Beendigung der Zugabe der Aminlösung wurde die weisse Paste für weitere 30 Minuten weitergerührt. So wurde nach dem Abkühlen eine weisse, weiche Paste erhalten, welche einen freien Isocyanatgehalt von < 0.1 % aufwies (Anteil Harnstoffderivat ca. 21 %).
Herstellung der Zusammensetzungen
Es wurden gemäss Tabelle 3 die Referenzzusammensetzungen Ref.1 - Ref. 5 sowie die erfindungsgemässen Zusammensetzungen 1 bis 8 hergestellt.
Prüfmethoden: Zugscherfestigkeit (ZSF) (DIN EN 1465)
Die Probekörper wurden aus den beschriebenen Beispiel-Zusammensetzungen und mit elektrolytisch verzinktem DC04 Stahl (eloZn) mit dem Mass 100 x 25 x 1.5 mm bzw. 100 x 25 x 0.8 mm, hergestellt, dabei betrug die Klebfläche 25 x 10mm bei einer Schichtdicke von 0.3mm. Gehärtet wurde 30 Min. bei 180°C. Die Zuggeschwindigkeit betrug 10mm/min.
Zugfestigkeit (ZF) (DIN EN ISO 527)
Eine Klebstoffprobe wurde zwischen zwei Teflonpapieren auf eine Schichtdicke von 2mm verpresst. Anschliessend wurde der Klebstoff während 30 Minuten bei 1800C gehärtet. Die Teflonpapiere wurden entfernt und die Probekörper nach DIN-Norm wurden im heissen Zustand ausgestanzt. Die Prüfkörper wurden nach 1 Tag Lagerung unter Normklima mit einer Zuggeschwindigkeit von 2 mm/min gemessen.
Die Zugfestigkeit wurde gemäss DIN EN ISO 527 bestimmt.
Schlagschälarbeit (ISO 11343)
Die Probekörper wurden aus den beschriebenen Beispiel- Zusammensetzungen und mit elektrolytisch verzinktem DC04 Stahl (eloZn) mit dem Mass 90 x 20 x 0.8mm hergestellt, dabei betrug die Klebfläche 20 x 30mm bei einer Schichtdicke von 0.3mm. Gehärtet wurde 30 Min. bei 1800C. Die Messung der Schlagschälarbeit erfolgte jeweils bei Raumtemperatur, bei minus 20°C und bei minus 400C, respektive minus 30°C. Die Schlaggeschwindigkeit betrug 2 m/s. Als Bruchenergie (BE) in Joule wird die Fläche unter der Messkurve (von 25% bis 90%, gemäss ISO 11343) angegeben.
Die Ergebnisse dieser Prüfungen sind in Tabelle 3 zusammengestellt.
Tabelle 3. Zusammensetzungen von hitzehärtenden Zusammensetzungen und Resultate. 1BE =Bruchenergie 2n.m. = nicht gemessen.