WO2006022323A1 - ホスホロアミダイト化合物及びオリゴrnaの製法 - Google Patents
ホスホロアミダイト化合物及びオリゴrnaの製法 Download PDFInfo
- Publication number
- WO2006022323A1 WO2006022323A1 PCT/JP2005/015420 JP2005015420W WO2006022323A1 WO 2006022323 A1 WO2006022323 A1 WO 2006022323A1 JP 2005015420 W JP2005015420 W JP 2005015420W WO 2006022323 A1 WO2006022323 A1 WO 2006022323A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- general formula
- chemical
- following general
- Prior art date
Links
- -1 Phosphoramidite compound Chemical class 0.000 title claims abstract description 106
- 238000004519 manufacturing process Methods 0.000 title claims description 23
- 125000006239 protecting group Chemical group 0.000 claims abstract description 43
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 37
- 125000001424 substituent group Chemical group 0.000 claims abstract description 31
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 18
- 125000004434 sulfur atom Chemical group 0.000 claims abstract description 17
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 16
- 150000007523 nucleic acids Chemical group 0.000 claims abstract description 16
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 16
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 15
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 15
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 15
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 12
- 125000006575 electron-withdrawing group Chemical group 0.000 claims abstract description 11
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 9
- 125000004430 oxygen atom Chemical group O* 0.000 claims abstract description 7
- 239000001257 hydrogen Substances 0.000 claims abstract description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 5
- 125000004429 atom Chemical group 0.000 claims abstract description 5
- 150000001875 compounds Chemical class 0.000 claims description 196
- 238000006243 chemical reaction Methods 0.000 claims description 104
- 238000000034 method Methods 0.000 claims description 73
- 239000003153 chemical reaction reagent Substances 0.000 claims description 60
- 229920002477 rna polymer Polymers 0.000 claims description 55
- 239000000126 substance Substances 0.000 claims description 54
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 46
- WFDIJRYMOXRFFG-UHFFFAOYSA-N acetic acid anhydride Natural products CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims description 39
- 150000008300 phosphoramidites Chemical class 0.000 claims description 39
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 36
- 239000000203 mixture Substances 0.000 claims description 36
- 239000002253 acid Substances 0.000 claims description 27
- 239000003795 chemical substances by application Substances 0.000 claims description 24
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 21
- 230000002152 alkylating effect Effects 0.000 claims description 21
- 239000007790 solid phase Substances 0.000 claims description 19
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 18
- 229910052736 halogen Inorganic materials 0.000 claims description 17
- 150000002367 halogens Chemical class 0.000 claims description 17
- 150000003833 nucleoside derivatives Chemical class 0.000 claims description 17
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 14
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 claims description 14
- 230000002140 halogenating effect Effects 0.000 claims description 13
- 239000000178 monomer Substances 0.000 claims description 11
- 230000007935 neutral effect Effects 0.000 claims description 11
- 239000007800 oxidant agent Substances 0.000 claims description 11
- BWZVCCNYKMEVEX-UHFFFAOYSA-N 2,4,6-Trimethylpyridine Chemical compound CC1=CC(C)=NC(C)=C1 BWZVCCNYKMEVEX-UHFFFAOYSA-N 0.000 claims description 10
- 239000012190 activator Substances 0.000 claims description 9
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 claims description 8
- 125000004423 acyloxy group Chemical group 0.000 claims description 8
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 claims description 8
- 229940104302 cytosine Drugs 0.000 claims description 7
- 239000012071 phase Substances 0.000 claims description 7
- 150000003573 thiols Chemical class 0.000 claims description 7
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 claims description 6
- 229930024421 Adenine Natural products 0.000 claims description 6
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 claims description 6
- 229960000643 adenine Drugs 0.000 claims description 6
- 125000004414 alkyl thio group Chemical group 0.000 claims description 6
- 125000005360 alkyl sulfoxide group Chemical group 0.000 claims description 5
- 125000005110 aryl thio group Chemical group 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- 150000003973 alkyl amines Chemical class 0.000 claims description 4
- 239000000470 constituent Substances 0.000 claims description 4
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 4
- GFYHSKONPJXCDE-UHFFFAOYSA-N sym-collidine Natural products CC1=CN=C(C)C(C)=C1 GFYHSKONPJXCDE-UHFFFAOYSA-N 0.000 claims description 4
- 229940113082 thymine Drugs 0.000 claims description 4
- 229940035893 uracil Drugs 0.000 claims description 4
- GONFBOIJNUKKST-UHFFFAOYSA-N 5-ethylsulfanyl-2h-tetrazole Chemical compound CCSC=1N=NNN=1 GONFBOIJNUKKST-UHFFFAOYSA-N 0.000 claims description 3
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 claims description 3
- 150000001409 amidines Chemical class 0.000 claims description 3
- 230000001590 oxidative effect Effects 0.000 claims description 3
- AUHZEENZYGFFBQ-UHFFFAOYSA-N 1,3,5-Me3C6H3 Natural products CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 claims description 2
- JNJFONBBNLVENC-UHFFFAOYSA-N 1h-imidazole;trifluoromethanesulfonic acid Chemical compound C1=CNC=N1.OS(=O)(=O)C(F)(F)F JNJFONBBNLVENC-UHFFFAOYSA-N 0.000 claims description 2
- XECYTQKOWWBNNU-UHFFFAOYSA-N 2h-benzotriazole;trifluoromethanesulfonic acid Chemical compound OS(=O)(=O)C(F)(F)F.C1=CC=C2NN=NC2=C1 XECYTQKOWWBNNU-UHFFFAOYSA-N 0.000 claims description 2
- QAJJXHRQPLATMK-UHFFFAOYSA-N 4,5-dichloro-1h-imidazole Chemical compound ClC=1N=CNC=1Cl QAJJXHRQPLATMK-UHFFFAOYSA-N 0.000 claims description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 2
- 230000002378 acidificating effect Effects 0.000 claims description 2
- 230000003213 activating effect Effects 0.000 claims description 2
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical group OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 claims description 2
- 238000002360 preparation method Methods 0.000 claims description 2
- 241000670727 Amida Species 0.000 claims 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 claims 1
- 125000006413 ring segment Chemical group 0.000 claims 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 claims 1
- 238000003786 synthesis reaction Methods 0.000 abstract description 9
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 93
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 51
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 48
- 239000000243 solution Substances 0.000 description 48
- 239000002904 solvent Substances 0.000 description 43
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 42
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 34
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 32
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 30
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 24
- 238000010898 silica gel chromatography Methods 0.000 description 23
- 230000035484 reaction time Effects 0.000 description 22
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 22
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 21
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 21
- 229940029575 guanosine Drugs 0.000 description 21
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 18
- 239000002585 base Substances 0.000 description 17
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 17
- 239000002994 raw material Substances 0.000 description 17
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 17
- 229940045145 uridine Drugs 0.000 description 17
- 229920006395 saturated elastomer Polymers 0.000 description 16
- 235000017557 sodium bicarbonate Nutrition 0.000 description 16
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 16
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 16
- 125000001572 5'-adenylyl group Chemical group C=12N=C([H])N=C(N([H])[H])C=1N=C([H])N2[C@@]1([H])[C@@](O[H])([H])[C@@](O[H])([H])[C@](C(OP(=O)(O[H])[*])([H])[H])([H])O1 0.000 description 15
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 14
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 14
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 14
- 108020004414 DNA Proteins 0.000 description 13
- 102000053602 DNA Human genes 0.000 description 13
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 13
- 238000005481 NMR spectroscopy Methods 0.000 description 13
- 239000012046 mixed solvent Substances 0.000 description 13
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 12
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 12
- 125000003118 aryl group Chemical group 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 10
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 10
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 9
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 9
- 229960005305 adenosine Drugs 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 239000011734 sodium Substances 0.000 description 9
- NXXJKXHLERAGKZ-UHFFFAOYSA-N 3-chloro-2-[(3-chloro-2-cyanopropoxy)methyl]propanenitrile Chemical compound ClCC(C#N)COCC(CCl)C#N NXXJKXHLERAGKZ-UHFFFAOYSA-N 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- 239000002808 molecular sieve Substances 0.000 description 8
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 8
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 8
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 7
- 239000012300 argon atmosphere Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 6
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 150000008065 acid anhydrides Chemical class 0.000 description 6
- 125000002252 acyl group Chemical group 0.000 description 6
- 125000003710 aryl alkyl group Chemical group 0.000 description 6
- 239000013065 commercial product Substances 0.000 description 6
- 239000005289 controlled pore glass Substances 0.000 description 6
- 125000004663 dialkyl amino group Chemical group 0.000 description 6
- RJGHQTVXGKYATR-UHFFFAOYSA-L dibutyl(dichloro)stannane Chemical compound CCCC[Sn](Cl)(Cl)CCCC RJGHQTVXGKYATR-UHFFFAOYSA-L 0.000 description 6
- 239000011630 iodine Substances 0.000 description 6
- 229910052740 iodine Inorganic materials 0.000 description 6
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 6
- 229910052751 metal Inorganic materials 0.000 description 6
- 239000002184 metal Substances 0.000 description 6
- 229910052698 phosphorus Inorganic materials 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 6
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- CZKMPDNXOGQMFW-UHFFFAOYSA-N chloro(triethyl)germane Chemical compound CC[Ge](Cl)(CC)CC CZKMPDNXOGQMFW-UHFFFAOYSA-N 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical group CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 5
- HKAIIKVJIRPASO-UHFFFAOYSA-N 1-butylpyrrolidine-2,5-dione Chemical compound CCCCN1C(=O)CCC1=O HKAIIKVJIRPASO-UHFFFAOYSA-N 0.000 description 4
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 4
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 238000006482 condensation reaction Methods 0.000 description 4
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 4
- ANPWLBTUUNFQIO-UHFFFAOYSA-N n-bis(phenylmethoxy)phosphanyl-n-propan-2-ylpropan-2-amine Chemical compound C=1C=CC=CC=1COP(N(C(C)C)C(C)C)OCC1=CC=CC=C1 ANPWLBTUUNFQIO-UHFFFAOYSA-N 0.000 description 4
- 125000004437 phosphorous atom Chemical group 0.000 description 4
- ISPYQTSUDJAMAB-UHFFFAOYSA-N 2-chlorophenol Chemical compound OC1=CC=CC=C1Cl ISPYQTSUDJAMAB-UHFFFAOYSA-N 0.000 description 3
- QWTBDIBOOIAZEF-UHFFFAOYSA-N 3-[chloro-[di(propan-2-yl)amino]phosphanyl]oxypropanenitrile Chemical compound CC(C)N(C(C)C)P(Cl)OCCC#N QWTBDIBOOIAZEF-UHFFFAOYSA-N 0.000 description 3
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 3
- 239000007853 buffer solution Substances 0.000 description 3
- 238000007865 diluting Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 238000001668 nucleic acid synthesis Methods 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 3
- 235000019345 sodium thiosulphate Nutrition 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000004809 thin layer chromatography Methods 0.000 description 3
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 3
- AVBGNFCMKJOFIN-UHFFFAOYSA-N triethylammonium acetate Chemical compound CC(O)=O.CCN(CC)CC AVBGNFCMKJOFIN-UHFFFAOYSA-N 0.000 description 3
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- CCSBNBKMACZDGN-UHFFFAOYSA-N (2-phenoxyacetyl) 2-phenoxyacetate Chemical compound C=1C=CC=CC=1OCC(=O)OC(=O)COC1=CC=CC=C1 CCSBNBKMACZDGN-UHFFFAOYSA-N 0.000 description 2
- AVQQQNCBBIEMEU-UHFFFAOYSA-N 1,1,3,3-tetramethylurea Chemical compound CN(C)C(=O)N(C)C AVQQQNCBBIEMEU-UHFFFAOYSA-N 0.000 description 2
- PMBXCGGQNSVESQ-UHFFFAOYSA-N 1-Hexanethiol Chemical compound CCCCCCS PMBXCGGQNSVESQ-UHFFFAOYSA-N 0.000 description 2
- ZRKMQKLGEQPLNS-UHFFFAOYSA-N 1-Pentanethiol Chemical compound CCCCCS ZRKMQKLGEQPLNS-UHFFFAOYSA-N 0.000 description 2
- JBWYRBLDOOOJEU-UHFFFAOYSA-N 1-[chloro-(4-methoxyphenyl)-phenylmethyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC(OC)=CC=1)C1=CC=CC=C1 JBWYRBLDOOOJEU-UHFFFAOYSA-N 0.000 description 2
- DCTOHCCUXLBQMS-UHFFFAOYSA-N 1-undecene Chemical compound CCCCCCCCCC=C DCTOHCCUXLBQMS-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- CNDCQWGRLNGNNO-UHFFFAOYSA-N 2-(2-sulfanylethoxy)ethanethiol Chemical compound SCCOCCS CNDCQWGRLNGNNO-UHFFFAOYSA-N 0.000 description 2
- NHPBDVZRYPODPF-UHFFFAOYSA-N 2-[(2-cyano-3-methylsulfanylpropoxy)methyl]-3-methylsulfanylpropanenitrile Chemical compound CSCC(C#N)COCC(C#N)CSC NHPBDVZRYPODPF-UHFFFAOYSA-N 0.000 description 2
- FDIUZJXCMKZLCA-HJQYOEGKSA-N 3-[[(2r,3r,4r,5r)-2-(2,4-dioxopyrimidin-1-yl)-4-hydroxy-5-(hydroxymethyl)oxolan-3-yl]oxymethoxy]propanenitrile Chemical compound N#CCCOCO[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 FDIUZJXCMKZLCA-HJQYOEGKSA-N 0.000 description 2
- CFITVYNSTRPJBA-IDTAVKCVSA-N 3-[[(2r,3r,4r,5r)-2-(6-aminopurin-9-yl)-4-hydroxy-5-(hydroxymethyl)oxolan-3-yl]oxymethoxy]propanenitrile Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1OCOCCC#N CFITVYNSTRPJBA-IDTAVKCVSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- 125000001999 4-Methoxybenzoyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C(*)=O 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 239000008351 acetate buffer Substances 0.000 description 2
- HOPRXXXSABQWAV-UHFFFAOYSA-N anhydrous collidine Natural products CC1=CC=NC(C)=C1C HOPRXXXSABQWAV-UHFFFAOYSA-N 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- UTBIMNXEDGNJFE-UHFFFAOYSA-N collidine Natural products CC1=CC=C(C)C(C)=N1 UTBIMNXEDGNJFE-UHFFFAOYSA-N 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 229960005215 dichloroacetic acid Drugs 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000012156 elution solvent Substances 0.000 description 2
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethanethiol Chemical compound CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000012025 fluorinating agent Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- HXJZHJLLMIGFCM-UHFFFAOYSA-N hydroxy-imino-di(propan-2-yloxy)-$l^{5}-phosphane Chemical compound CC(C)OP(N)(=O)OC(C)C HXJZHJLLMIGFCM-UHFFFAOYSA-N 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 238000006386 neutralization reaction Methods 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- DPBLXKKOBLCELK-UHFFFAOYSA-N pentan-1-amine Chemical compound CCCCCN DPBLXKKOBLCELK-UHFFFAOYSA-N 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 150000003222 pyridines Chemical class 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000010532 solid phase synthesis reaction Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 229960002317 succinimide Drugs 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- CIHOLLKRGTVIJN-UHFFFAOYSA-N tert‐butyl hydroperoxide Chemical compound CC(C)(C)OO CIHOLLKRGTVIJN-UHFFFAOYSA-N 0.000 description 2
- 150000003536 tetrazoles Chemical class 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 238000000108 ultra-filtration Methods 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- KZKRRZFCAYOXQE-UHFFFAOYSA-N 1$l^{2}-azinane Chemical group C1CC[N]CC1 KZKRRZFCAYOXQE-UHFFFAOYSA-N 0.000 description 1
- LOSXTWDYAWERDB-UHFFFAOYSA-N 1-[chloro(diphenyl)methyl]-2,3-dimethoxybenzene Chemical compound COC1=CC=CC(C(Cl)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1OC LOSXTWDYAWERDB-UHFFFAOYSA-N 0.000 description 1
- BMVXCPBXGZKUPN-UHFFFAOYSA-N 1-hexanamine Chemical compound CCCCCCN BMVXCPBXGZKUPN-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- LDXJRKWFNNFDSA-UHFFFAOYSA-N 2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound C1CN(CC2=NNN=C21)CC(=O)N3CCN(CC3)C4=CN=C(N=C4)NCC5=CC(=CC=C5)OC(F)(F)F LDXJRKWFNNFDSA-UHFFFAOYSA-N 0.000 description 1
- BIVIXXXEPPIGSS-UHFFFAOYSA-N 2-[amino(propan-2-yloxy)phosphanyl]oxypropane Chemical compound CC(C)OP(N)OC(C)C BIVIXXXEPPIGSS-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- FINYOTLDIHYWPR-UHFFFAOYSA-N 2-cyanoethoxy-n,n-di(propan-2-yl)phosphonamidous acid Chemical compound CC(C)N(C(C)C)P(O)OCCC#N FINYOTLDIHYWPR-UHFFFAOYSA-N 0.000 description 1
- VKIGAWAEXPTIOL-UHFFFAOYSA-N 2-hydroxyhexanenitrile Chemical compound CCCCC(O)C#N VKIGAWAEXPTIOL-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- SOFPIAMTOZWXKT-UHFFFAOYSA-N 2h-1,2,4-triazine-3-thione Chemical compound SC1=NC=CN=N1 SOFPIAMTOZWXKT-UHFFFAOYSA-N 0.000 description 1
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 1
- IFVYLGBXAXTHSC-UHFFFAOYSA-N 3-(3-sulfanylpropoxy)propane-1-thiol Chemical compound SCCCOCCCS IFVYLGBXAXTHSC-UHFFFAOYSA-N 0.000 description 1
- BIIYXLIZNQFLAZ-UHFFFAOYSA-N 3-(methylsulfanylmethoxy)propanenitrile Chemical compound CSCOCCC#N BIIYXLIZNQFLAZ-UHFFFAOYSA-N 0.000 description 1
- RKVHNYJPIXOHRW-UHFFFAOYSA-N 3-bis[di(propan-2-yl)amino]phosphanyloxypropanenitrile Chemical compound CC(C)N(C(C)C)P(N(C(C)C)C(C)C)OCCC#N RKVHNYJPIXOHRW-UHFFFAOYSA-N 0.000 description 1
- 125000002103 4,4'-dimethoxytriphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)(C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H])C1=C([H])C([H])=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- VWAMCRGFVJJNHC-UHFFFAOYSA-N 4-(4-sulfanylbutoxy)butane-1-thiol Chemical compound SCCCCOCCCCS VWAMCRGFVJJNHC-UHFFFAOYSA-N 0.000 description 1
- SHOYSUHLFYQDTR-UHFFFAOYSA-N 4-methylbenzenethiol Chemical compound CC1=CC=C(C=C1)S.CC1=CC=C(C=C1)S SHOYSUHLFYQDTR-UHFFFAOYSA-N 0.000 description 1
- NEJMTSWXTZREOC-UHFFFAOYSA-N 4-sulfanylbutan-1-ol Chemical compound OCCCCS NEJMTSWXTZREOC-UHFFFAOYSA-N 0.000 description 1
- JXHLORIBHSZXGL-UHFFFAOYSA-N 5-(5-sulfanylpentoxy)pentane-1-thiol Chemical compound SCCCCCOCCCCCS JXHLORIBHSZXGL-UHFFFAOYSA-N 0.000 description 1
- NQIIORIRYQSRAL-UHFFFAOYSA-N 6-(6-sulfanylhexoxy)hexane-1-thiol Chemical compound SCCCCCCOCCCCCCS NQIIORIRYQSRAL-UHFFFAOYSA-N 0.000 description 1
- DEXFNLNNUZKHNO-UHFFFAOYSA-N 6-[3-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperidin-1-yl]-3-oxopropyl]-3H-1,3-benzoxazol-2-one Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C1CCN(CC1)C(CCC1=CC2=C(NC(O2)=O)C=C1)=O DEXFNLNNUZKHNO-UHFFFAOYSA-N 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- PNKUSGQVOMIXLU-UHFFFAOYSA-N Formamidine Chemical compound NC=N PNKUSGQVOMIXLU-UHFFFAOYSA-N 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 108020004518 RNA Probes Proteins 0.000 description 1
- 239000003391 RNA probe Substances 0.000 description 1
- 230000006819 RNA synthesis Effects 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- QQGWBRJQPRTJDA-UHFFFAOYSA-N [Li].CC(O)=O Chemical compound [Li].CC(O)=O QQGWBRJQPRTJDA-UHFFFAOYSA-N 0.000 description 1
- FAZPBIKOJRZHAX-UHFFFAOYSA-N [Na].[Na].C(CCC)[Sn]CCCC Chemical compound [Na].[Na].C(CCC)[Sn]CCCC FAZPBIKOJRZHAX-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 125000003435 aroyl group Chemical group 0.000 description 1
- 150000001504 aryl thiols Chemical class 0.000 description 1
- PXXJHWLDUBFPOL-UHFFFAOYSA-N benzamidine Chemical compound NC(=N)C1=CC=CC=C1 PXXJHWLDUBFPOL-UHFFFAOYSA-N 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 1
- WQAQPCDUOCURKW-UHFFFAOYSA-N butanethiol Chemical compound CCCCS WQAQPCDUOCURKW-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- IKCMAHCPTMKAAB-UHFFFAOYSA-N chloro-n,n-di(propan-2-yl)phosphonamidous acid Chemical compound CC(C)N(C(C)C)P(O)Cl IKCMAHCPTMKAAB-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 235000009508 confectionery Nutrition 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 239000012351 deprotecting agent Substances 0.000 description 1
- 238000011033 desalting Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical compound O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- JXBPSENIJJPTCI-UHFFFAOYSA-N ethyl cyanate Chemical compound CCOC#N JXBPSENIJJPTCI-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000004508 fractional distillation Methods 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 230000009368 gene silencing by RNA Effects 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- IKGLACJFEHSFNN-UHFFFAOYSA-N hydron;triethylazanium;trifluoride Chemical compound F.F.F.CCN(CC)CC IKGLACJFEHSFNN-UHFFFAOYSA-N 0.000 description 1
- POCUPXSSKQAQRY-UHFFFAOYSA-N hydroxylamine;hydrate Chemical compound O.ON POCUPXSSKQAQRY-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005524 levulinyl group Chemical group 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- CQRPUKWAZPZXTO-UHFFFAOYSA-M magnesium;2-methylpropane;chloride Chemical compound [Mg+2].[Cl-].C[C-](C)C CQRPUKWAZPZXTO-UHFFFAOYSA-M 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 229940050176 methyl chloride Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001038 naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000006384 oligomerization reaction Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- CCVKPWUMYBYHCD-UHFFFAOYSA-N oxolane;pyridine Chemical compound C1CCOC1.C1=CC=NC=C1 CCVKPWUMYBYHCD-UHFFFAOYSA-N 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical class OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- SUVIGLJNEAMWEG-UHFFFAOYSA-N propane-1-thiol Chemical compound CCCS SUVIGLJNEAMWEG-UHFFFAOYSA-N 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- GAPYKZAARZMMGP-UHFFFAOYSA-N pyridin-1-ium;acetate Chemical compound CC(O)=O.C1=CC=NC=C1 GAPYKZAARZMMGP-UHFFFAOYSA-N 0.000 description 1
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 1
- YWVYZMVYXAVAKS-UHFFFAOYSA-N pyridin-1-ium;trifluoromethanesulfonate Chemical compound C1=CC=[NH+]C=C1.[O-]S(=O)(=O)C(F)(F)F YWVYZMVYXAVAKS-UHFFFAOYSA-N 0.000 description 1
- 238000006462 rearrangement reaction Methods 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- TZGODTCAKVHMFG-UHFFFAOYSA-N sulfanylmethoxymethanethiol Chemical compound SCOCS TZGODTCAKVHMFG-UHFFFAOYSA-N 0.000 description 1
- 230000001502 supplementing effect Effects 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical compound OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/01—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms
- C07C255/11—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound oxygen atoms bound to the same saturated acyclic carbon skeleton
- C07C255/13—Carboxylic acid nitriles having cyano groups bound to acyclic carbon atoms containing cyano groups and singly-bound oxygen atoms bound to the same saturated acyclic carbon skeleton containing cyano groups and etherified hydroxy groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/067—Pyrimidine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/167—Purine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the present invention relates to a novel phosphoramidite complex compound in which a novel protecting group is introduced into the hydroxyl group at the 2'-position, and a reagent for introducing the protecting group.
- Oligoribonucleic acid is useful as an RNA probe for gene analysis, RNA pharmaceutical material (antisense RNA, ribozyme, gene expression control using RNAi), artificial enzyme, and aptamer.
- Non-patent Document 1 Solid-phase synthesis of oligo RNA was established in the late 1980s. First reported was the use of phosphoramidite compounds using a tert-butyldimethylsilyl (TBDMS) group or a triisoprovirsilyl (TIPS) group as a protective group for the hydroxyl group at the 2-position. This was a solid phase synthesis method (Non-patent Document 1).
- oligo RNA oligodeoxyribonucleic acid
- DNA deoxyribonucleic acid
- TBDMS group when a TBDMS group is used as a protecting group for the hydroxyl group at the 2′-position, for example, when the hydroxyl group at the 3′-position is phosphoramidite-coated, the TBDMS group that protected the hydroxyl group at the 2′-position is 3 A side reaction that rearranges to the 'position may occur.
- a substituent having a large steric hindrance such as a TBDMS group
- a stereo group around the phosphorus atom bonded to the 3 ′ position is crowded, so that an internucleotide bond is formed.
- the rate of the condensation reaction is reduced, or after oligomerization, when the 2′-position hydroxyl protecting group is removed, cleavage of the internucleotide bond or rearrangement reaction can occur.
- the 1- (2-cyanoethoxy) ethyl (CEE) group is It is known as a group capable of leaving at the same time as the biskeline protecting group under the conditions (neutral conditions) for removing the biskeline protecting group protecting the 5 'position (Non-patent Document 2). From this finding, Wada has found a phosphoramidite complex compound in which a CEE group that can be deprotected under neutral conditions is introduced into the 2′-position hydroxyl group as a compound for producing oligo RNA (Non-patent Document 3, Non-patent document 4).
- Non-Patent Document 1 N. A. Usman et al., Journal American Chemical Society, Vol. 109, 7845 (1987)
- Non-Patent Document 2 discloses a method for modifying the working environment in accordance with the following requirements: a method for modifying the working environment in accordance with the following requirements: a procedure for modifying the working environment in accordance with the following requirements: a procedure for modifying the working environment in accordance with the following requirements: a procedure for modifying the working environment in accordance with the following requirements: a procedure for modifying the working environment in accordance with the following requirements:
- Non-Patent Document 3 Takeshi Wada, BIO INDUSTRY, Vol. 21, No. 1, 17 (2004)
- Non-Patent Document 4 T. Umemoto et al., Tetrahedron Letters, Vol. 45, 9529 (2004) Disclosure of Invention
- An object of the present invention is to provide a useful new phosphoramidite complex, mainly for the purpose of a simple and high yield synthesis method of oligo RNA.
- Another object of the present invention is to provide a novel ether compound capable of introducing a protecting group that can be easily removed under a neutral condition into a hydroxyl group at the 2′-position.
- Examples of the present invention include phosphoramidite compounds represented by the following general formula (1) (hereinafter referred to as “the present phosphoramidite compound”).
- B represents a nucleobase optionally having a protecting group.
- R 1 represents a substituent represented by the following general formula (2).
- R 11 , R 12 and R 13 are the same or different and each represents hydrogen or alkoxy.
- R 2A and R 2B are the same or different and represent a alkyl group or a 5- to 6-membered saturated amino ring group formed by R 2A and R 2B together with the adjacent nitrogen atom.
- the powerful saturated amino ring group may have one oxygen atom or sulfur atom as a ring constituent atom in addition to the nitrogen atom.
- WG 2 is the same or different and represents an electron-withdrawing group.
- nucleobase related to X is not particularly limited as long as it is used for nucleic acid synthesis, and examples thereof include adenine, guanine, cytosine, uracil, and modified products thereof.
- a “modified product” of a nucleobase is a compound obtained by substituting a nucleobase with an arbitrary substituent.
- Examples of the substituent related to the “modified product” of B include halogen, acyl, alkyl, aryl
- Nucleobase according to B is a nucleobase having an amino group, whether it is protected or not.
- adenine, guanine, and cytosine preferably have a protected amino group.
- the “amino group protecting group” is not particularly limited as long as it is used as a nucleic acid protecting group. Specifically, for example, benzoyl, 4-methoxybenzoyl, acetyl, propionyl, butyryl. , Isobutyryl, phenylacetyl, phenoxyacetyl, 4 tert butyl phenoxyacetyl, 4 isopropylphenoxyacetyl, and (dimethylamino) methylene.
- Examples of the “saturated amino ring group” according to R 2 include pyrrolidine-1-yl, piperidine-1-yl, morpholine-1-yl, and thiomorpholine-1-yl.
- Examples of the “electron-withdrawing group” according to WG 2 include silane-containing nitro, alkylsulfonyl, and halogen. Of these, Ciano is preferred.
- halogen examples include fluorine, chlorine, bromine and iodine.
- alkanols having 1 to 6 carbon atoms
- aroyl having 7 to 13 carbon atoms.
- formyl, Asechiru, n- propionate - le, Isopuropio - Le, n Buchiriru, isobutyryl, tert- butyryl, valeryl, to Kisanoiru, Benzoiru, naphthoyl, can be mentioned levulinyl.
- substituent that may be substituted with the alkyl include, but are not limited to, nitrogen, alkyl, alkoxy, and nitro-containing nitro, and 1 to 3 of them are substituted.
- aryl alkyl In the phosphoramidite compound of the present invention, “aryl alkyl”, “alkoxyalkyl” Examples of the “alkyl” part of the “alkyl”, “monoalkylamino”, “dialkylamino” and “alkylsulfol” are the same as the above “alkyl”.
- alkoxy in the phosphoramidite complex of the present invention examples include linear or branched alkoxy having 1 to 4 carbon atoms. Specific examples include methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy and tert-butoxy. Of these, those having 1 to 3 carbon atoms are preferred, and methoxy is particularly preferred.
- alkoxy part of “alkoxyalkyl” in the phosphoramidite compound of the present invention include the same as the above “alkoxy”.
- aryl of “aryl alkyl” in the phosphoramidite compound of the present invention include aryl having 6 to 12 carbon atoms. Specific examples include phenol, 1-naphthyl, 2-naphthyl and biphenyl. Examples of the substituent that can be substituted with the aryl include halogen, alkyl, alkoxy, and nitro-containing nitro, and 1 to 3 of these are substituted.
- halogen alkyl
- alkoxy substituents of “alkyl” and “aryl” in the phosphoramidite compound of the present invention are the same as those described above.
- the phosphoramidite toy compound of the present invention can be used as a reagent for producing oligo RNA.
- the phosphoramidite toy compound of the present invention is a phosphoramidite toy compound having an ether-type protecting group that can be removed under a neutral condition at the 2′-position hydroxyl group.
- the group introduced into the hydroxyl group at the 2 'position is a linear substituent and the steric structure around the phosphorus atom bonded to the hydroxyl group at the 3' position is crowded, it has been used conventionally!
- the synthesis of oligo RNA is characterized in that the condensation reaction proceeds in a very short time and the condensation yield is good.
- oligo DNA refers to an oligo nucleic acid that can only have deoxyribonucleic acid (DNA).
- oligo RNA refers to oligonucleic acid such as ribonucleic acid (RNA) and doxyribonucleic acid (DNA), and at least one oligonucleic acid containing ribonucleic acid (RNA).
- Specific examples of the phosphoramidite toy compound of the present invention include the following compounds 1. to 5.
- N 2 Phenoxyacetyl—5, -0- (4, 4, —Dimethoxytrityl) —2, -0- (2 —Cyanethoxymethyl) guanosine 3, 1 O— (2-Chanoethyl N, N-diisopropylpropyl phosphoramidite)
- FIG. 1 shows a chromatogram obtained by reverse phase HPLC analysis.
- the vertical axis represents time (minutes) and the horizontal axis represents absorption intensity.
- the phosphoramidite toy compound of the present invention can be produced as follows.
- the reaction is performed after the raw material has been thoroughly protected according to a known method and protected with an appropriate protecting group. It is common to do.
- the protecting group can be removed after the reaction according to a known method such as catalytic reduction, alkali treatment, acid treatment, etc.
- the phosphoramidite complex of the present invention is a known compound or an intermediate that can be easily produced. From the body, it can be produced, for example, by performing the following steps a to h. This will be described in detail below.
- An ether-type protecting group that is eliminated under neutral conditions is introduced into the 2′-position hydroxyl group by acting an alkyl ⁇ reagent on the nucleoside derivative represented by the following general formula (14).
- alkylating reagent examples include ether compounds represented by the following general formula (13).
- L represents a halogen, an arylthio group, an alkyl sulfoxide group or an alkylthio group.
- WG 1 has the same meaning as described above.
- etheric compound (13) examples include the following compounds 1 to 2.
- the ether compound (13) is a novel alkyl reagent that can introduce an ether-type substituent that can be eliminated under neutral conditions into a hydroxyl group at the 2′-position under basic conditions. It is useful as a reagent for producing the invention phosphoramidite toy compound.
- the ether compound (13) can be produced by carrying out the following steps 1 to 4.
- WG 1 has the same meaning as described above.
- R 3 represents alkyl or aryl.
- the compound (24) is an ethereal compound (13) in which L is an alkylthio group.
- Examples of the “alkyl” related to R 3 include the same “alkyl” in the phosphoramidite compound of the present invention.
- examples of the alkylthiomethylating reagent include a mixed solution of dimethyl sulfoxide, acetic anhydride and acetic acid.
- the amount of “dimethyl sulfoxide” used is suitably 10 to 200-fold molar amount relative to compound (20), preferably 20 to: L00-fold molar amount.
- the amount of “acetic acid” used is suitably 10 to 150-fold molar amount relative to compound (20), and preferably 20 to: L 00-fold molar amount.
- the amount of acetic anhydride used is the compound ( For 20), 10 to 150 times the mono amount is appropriate, 20 to: LOO times the mono amount is preferred! /.
- the reaction temperature is suitably 0 ° C to 100 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 1 to 48 hours is appropriate.
- Process 2 is suitably 0 ° C to 100 ° C. The reaction time varies depending on the type of raw materials
- Compound (25) is a compound in which L in the etheric compound (13) is halogen.
- Examples of “norogen” according to X 2 include the same “norogen” in the phosphoramidite toy compound of the present invention.
- This step can be performed by a known method (for example, T. Benneche et al., Synthesis 762 (1983)).
- the solvent to be used is not particularly limited as long as it does not participate in the reaction.
- 1S Examples include halogenated hydrocarbons such as dichloromethane, chlorophenol, carbon tetrachloride, and 1,2-dichloroethane.
- Examples of the halogenating reagent include sulfuryl chloride and phosphorus oxychloride.
- the amount of the “halogen reagent” to be used is suitably 1 to 20 times by mole, preferably 1 to 10 times by mole, relative to the compound (24).
- the reaction temperature is suitably from 0 ° C to 100 ° C.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- Process 3 Process 3:
- the compound (25a) is a compound in which L in the ether compound (13) is an arylthio group.
- Examples of the “aryl” related to R 3A include the same “aryl” in the phosphoramidite compound of the present invention.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane and acetonitrile.
- the arylthiolation reagent for example, thienol and 4-methylbenzene thiol can be mentioned.
- the amount of the “arylthiol reagent” used is suitably 1 to 20 times the molar amount, preferably 1 to 5 times the molar amount relative to the compound (25).
- the reaction temperature is suitably from 0 ° C to 100 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but 1 to 48 hours is usually appropriate.
- Compound (24a) is a compound in which L in the ether compound (13) is an alkyl sulfoxide group.
- Examples of the “alkyl” related to R 3 include the same “alkyl” in the phosphoramidite compound of the present invention.
- This step can be performed by a known method.
- the solvent used is involved in the reaction If not, it is not particularly limited, but examples thereof include dichloromethane, chloroform, and methanol.
- the oxidizing agent include methacroperoxybenzoic acid, metaperiodic acid salt, and hydrogen peroxide.
- the amount of the “oxidant” to be used is suitably 1 to 10-fold mol amount, preferably 1 to 2-fold mol amount based on Compound (24).
- the reaction temperature is suitably from 0 ° C to 100 ° C.
- the reaction time varies depending on the type of raw materials used, the reaction temperature, etc. Power is usually 1 to 48 hours.
- This step can be performed by reacting an alkylating reagent and a base with compound (14) which is available as a commercial product or can be synthesized according to a method described in the literature, according to a known method.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include halogen-based hydrocarbons such as dichloromethane, chlorophenol, carbon tetrachloride, and 1,2-dichloroethane.
- the amount of the “alkylating reagent” to be used is suitably 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (14).
- an alkylating reagent can be allowed to act after passing through an intermediate produced by reacting compound (14) with a metal reagent and a base.
- the “metal reagent” that can be used include disodium dibutyltin.
- the amount of the “metal reagent” to be used is suitably 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (14).
- “Bases” include pyridine, 2,6-dimethylpyridine, 2,4,6-trimethylpyridine, N-methylimidazole, triethylamine, tributylamine, N, N-diisopropylethylamine, 1 , 8-diazabicyclo [5.
- the amount of the “base” to be used is appropriately 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (14).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- the compound (24) or (25a) is used as the “alkylating reagent”, it can be carried out as follows. This step is carried out according to a known method (for example, M. Matteucci, Tetrahedron Letters, Vol. 31, 2385 (1990)), and is commercially available or can be synthesized according to the method described in the literature (14)
- the reaction can be carried out by reacting an alkylating reagent, an acid, and a halogenating agent for the sulfur atom.
- the amount of the “alkyli reagent” used is suitably 1 to 5 times the molar amount relative to the compound (14), and preferably 1.05 to 3 times the molar amount.
- Examples of the “acid” include trifluoromethanesulfonic acid, silver trifluoromethanesulfonate, and trimethylsilyl trifluoromethanesulfonate.
- the amount of the “acid” used is suitably 0.01 to 20-fold molar amount, preferably 0.02 to 10-fold molar amount relative to compound (14).
- the solvent to be used is not particularly limited as long as it does not participate in the reaction. For example, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, benzene, toluene, xylene, tetrahydrofuran, acetonitrile, or a mixed solvent thereof. Can be mentioned.
- halogenating agent for sulfur atom examples include N-butyl succinimide (NBS) and N-odosuccinimide (NIS).
- NBS N-butyl succinimide
- NMS N-odosuccinimide
- the amount of the “halogenating agent for sulfur atom” to be used is suitably 1 to 10 times by mole, preferably 1.05 to 5 times by mole, relative to compound (14).
- the reaction temperature is suitably 78 ° C to 30 ° C.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc., but usually 5 minutes to 5 hours is appropriate.
- compound (24a) is used as the “alkylating reagent”, it can be carried out as follows.
- This step is carried out according to a known method by allowing an alkyl reagent, an acid anhydride and a base to act on the compound (14) which is available as a commercial product or can be synthesized according to a method described in the literature. be able to.
- the amount of the “alkylating reagent” to be used is suitably 1 to 5-fold mol amount, preferably 1.05 to 3-fold mol amount based on Compound (14).
- Examples of the “acid anhydride” include trifluoromethanesulfonic acid anhydride and acetic anhydride.
- the amount of the “acid anhydride” to be used is suitably 0.01 to 20-fold mol amount, preferably 0.02 to 10-fold mol amount based on Compound (14).
- Examples of the base include tetramethyl urea and collidine.
- the amount of “base” used is 0.01 to 20 times the amount of compound (14).
- a suitable amount is 0.02 to: LO molar amount is preferable.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, and mixed solvents thereof.
- the reaction temperature is suitably from ⁇ 78 ° C. to 30 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 24 hours is appropriate.
- a step of isolating and purifying the nucleoside derivative (15) produced in step a This step can be isolated and purified by using a usual separation and purification means such as thin layer chromatography, silica gel chromatography, etc. from the mixture produced in step a.
- a usual separation and purification means such as thin layer chromatography, silica gel chromatography, etc. from the mixture produced in step a.
- step b Separately from step b, by reacting the ribonucleic acid compound represented by the following general formula (16) with an alkylating reagent, the ether-type protecting group that can be eliminated under neutral conditions is located at the 2 ′ position.
- A represents a silicon substituent represented by the following general formula (18a) or (18b).
- R 6 represents alkyl.
- Examples of the “alkyl” related to R 6 include the same “alkyl” as in the phosphoramidite compound of the present invention.
- alkylating reagent examples include the same as described above.
- This step can be performed according to a known method by reacting an alkylating reagent and a base with compound (16) which is available as a commercial product or can be synthesized according to a method described in the literature.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include halogen-based hydrocarbons such as dichloromethane, chlorophenol, carbon tetrachloride, and 1,2-dichloroethane.
- the amount of the “alkylating reagent” to be used is suitably 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (14).
- an alkylating reagent can be allowed to act after passing through an intermediate produced by reacting compound (16) with a metal reagent and a base.
- a metal reagent and a base examples include dibutyltin dichloride and t-butylmagnesium chloride.
- the amount of the “metal reagent” to be used is suitably 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (16).
- the amount of the “base” to be used is suitably 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (16).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc., but usually 30 minutes to 24 hours is appropriate.
- This step is carried out according to a known method (for example, M. Matteucci, Tetrahedron Letters, Vol. 31, 2385 (1990)), and is commercially available or can be synthesized according to the method described in the literature.
- Alkylating reagents and halogenating agents for acids and sulfur atoms It is possible to carry out from the above.
- the amount of the “alkyly reagent” used is suitably 1 to 5 times the molar amount relative to the compound (16), and preferably 1.05 to 3 times the molar amount.
- the “acid” include trifluoromethanesulfonic acid, silver trifluoromethanesulfonate, and trimethylsilyl trifluoromethanesulfonate.
- the amount of the “acid” used is suitably 0.01 to 20-fold molar amount, preferably 0.02 to 10-fold molar amount relative to compound (16).
- the solvent to be used is not particularly limited as long as it does not participate in the reaction.
- Examples of the “halogenating agent for sulfur atom” used in this step include N-butyl succinimide (NBS) and N-odosuccinimide (NIS).
- the amount of the “halogenating agent for sulfur atom” to be used is suitably 1 to 10 times by mole, preferably 1.05 to 5 times by mole, relative to compound (16).
- the reaction temperature is suitably 78 ° C to 30 ° C.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc., but usually 5 minutes to 5 hours is appropriate.
- compound (24a) is used as the “alkylating reagent”, it can be carried out as follows.
- This step is carried out according to a known method by allowing an alkyl reagent, an acid anhydride and a base to act on the compound (16) which is available as a commercial product or can be synthesized according to a method described in the literature. be able to.
- the amount of the “alkylating reagent” to be used is appropriately 1 to 5-fold mol amount, preferably 1.05 to 3-fold mol amount based on Compound (16).
- Examples of the “acid anhydride” include trifluoromethanesulfonic acid anhydride and acetic anhydride.
- the amount of the “acid anhydride” to be used is suitably 0.01 to 20-fold mol amount, preferably 0.02 to 10-fold mol amount based on Compound (16).
- Examples of the base include tetramethyl urea and collidine.
- the amount of the “base” used is suitably 0.01 to 20-fold mol amount relative to compound (16), and preferably 0.02 to: L0-fold mol amount.
- the solvent used is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, chloroform, carbon tetrachloride, 1,2 dichloroethane, and a mixed solvent thereof.
- Reaction temperature is-7 8 ° C-30 ° C is suitable.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 24 hours is appropriate.
- ribonucleic acid compound (16) is reacted with dimethyl sulfoxide, acetic acid and acetic anhydride to produce a ribonucleic acid compound represented by the following general formula (19). Process.
- This step is carried out by allowing dimethyl sulfoxide, acetic acid and acetic anhydride to act on a ribonucleic acid compound (16) which is available as a commercial product or can be synthesized according to a method described in the literature according to a known method. can do.
- the amount of “dimethyl sulfoxide” used is 10 to 200 times the molar force S of the compound (16), and 20 to: LOO times the molar amount is preferable.
- the amount of “acetic acid” to be used is suitably 10 to 150-fold mol amount, preferably 20 to 100-fold mol amount based on Compound (16).
- the amount of “acetic anhydride” used is suitably 10 to 150 times the molar amount relative to compound (16), and preferably 20 to: LOO times the molar amount.
- the reaction temperature is suitably 10 ° C to 50 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- This step can be performed by reacting the ribonucleic acid compound (19) with an alcoholic compound (20), an acid, and a halogenating agent for a sulfur atom according to a known method.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction.
- dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, benzene, toluene, xylene, tetrahydrofuran, acetonitrile, or a mixed solvent thereof can be used.
- the amount of the “alcohol compound (20)” used is suitably 1 to 20-fold molar amount, preferably 1 to 10-fold molar amount, relative to Compound (19).
- Examples of the “acid” include trifluoromethanesulfonic acid, silver trifluoromethanesulfonate, and trimethylsilyl trifluoromethanesulfonate.
- Examples of the “halogenating agent for sulfur atom” include N-bromosuccinimide (NBS) and N-odosuccinimide (NIS).
- the amount of the “halogenating agent for sulfur atom” to be used is suitably 0.1 to 20-fold mol amount, preferably 0.2 to 10-fold mol amount based on Compound (19).
- the reaction temperature is suitably from 100 ° C to 20 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 5 minutes to 12 hours is appropriate. (6) Process f:
- step c or step e the following general formula (21) The process of manufacturing the ribonucleic acid compound represented by this.
- This step can be carried out by dissolving compound (17) in an organic solvent and reacting the fluorinating agent and acid as a mixed reagent having an arbitrary mixing ratio.
- fluorinating agents that can be used in this step include ammonium fluoride, tetra-n-butylammonium fluoride (TBAF), triethylamine trihydrofluoride, and hydrogen fluoride pyridine.
- Power S can be.
- the amount of the “fluorinated agent” used is suitably 0.1 to 20 times the molar amount relative to compound (17), and preferably 0.2 to 10 times the molar amount.
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc., but usually 30 minutes to 24 hours is appropriate. (7) Step g:
- Examples of “norogen” according to X 3 include the same “norogen” in the phosphoramidite toy compound of the present invention.
- This step can be carried out by reacting compound (21) with R 3 according to a known method.
- the amount of R 3 used is suitably 1 to 20-fold molar amount, preferably 1 to 10-fold molar amount relative to compound (21).
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include acetonitrile and tetrahydrofuran.
- base Includes pyridine, 2,6-dimethylpyridine, 2,4,6-trimethylpyridine, N-methylimidazole, triethylamine, tributylamine, N, N-diisopropylethylamine, 1,8- Examples include organic bases such as diazabicyclo [5. 4. 0] -7-undecene.
- the amount of the “base” to be used is suitably 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (21).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw materials used, reaction temperature, etc. Usually, 30 minutes to 24 hours is appropriate.
- step b or step f The nucleoside derivative (15) produced in step b or step f is reacted with a phosphoramidite reagent and, if necessary, an activator, so that the hydroxyl group at the 3 ′ position is phosphorylated.
- a phosphoramidite reagent and, if necessary, an activator, so that the hydroxyl group at the 3 ′ position is phosphorylated.
- Examples of the “phosphoramidite sputum reagent” include compounds represented by the following general formulas (22) and (23).
- R 2a , R 2b and WG 2 are as defined above.
- X 1 represents halogen.
- the “norogen” according to X 1 includes “norogen” in the phosphoramidite toy compound of the present invention. The same thing can be mentioned.
- This step is a reaction in which a phosphoramidite reagent is allowed to act on compound (15) to form a 3′-position hydroxyl group, and can be performed according to a known method. If necessary, an activator can be used.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include acetonitrile and tetrahydrofuran.
- the amount of the “phosphoramidite candy reagent” used is suitably 1 to 20 times the molar amount relative to the compound (15), and preferably 1 to: LO times the molar amount.
- Activators include, for example, 1H-tetrazole, 5-ethylthiotetrazole, 4,5-dichloroimidazole, 4,5-disianomidazole, benzotriazole triflate, imidazole triflate, pyridinium triflate, N, Mention may be made of N-diisopropylethylamine, 2, 4, 6-collidine / N-methylimidazole.
- the amount of the “activator” to be used is suitably 1 to 20-fold mol amount, preferably 1 to 10-fold mol amount based on Compound (15).
- the reaction temperature is suitably from 0 ° C to 120 ° C.
- the reaction time varies depending on the type of raw material used, reaction temperature, etc., but usually 30 minutes to 24 hours is appropriate.
- the produced phosphoramidite complex of the present invention can be produced by a method known per se, for example, concentration, liquid conversion, phase transfer, solvent extraction, crystallization, recrystallization, fractional distillation, chromatodara. Separation and purification can be carried out by using the first method.
- concentration, liquid conversion, phase transfer, solvent extraction, crystallization, recrystallization, fractional distillation, chromatodara Separation and purification can be carried out by using the first method.
- Examples of the present invention include a method for producing an oligo RNA represented by the following general formula (3), characterized by using the phosphoramidite complex of the present invention.
- each B independently represents adenine, guanine, cytosine, uracil, thymine, or a modified form thereof.
- Each R independently represents H or a hydroxyl group, but at least one represents a hydroxyl group.
- Z represents H or a phosphate group.
- n represents an integer in the range from 1 to: LOO. ]
- n is preferably an integer in the range of 10 to 50, and more preferably an integer in the range of 15 to 30.
- substituent related to the modified form of B include halogen, acyl, alkyl, arylalkyl, alkoxy, hydroxy, amino-containing monoalkylamino-containing dialkylamino, carboxyl, and cyanated nitro. 1-3 are substituted.
- the production method of the oligo RNA (3) using the phosphoramidite toy compound of the present invention is a force that can be performed according to a known method, for example, by performing the following operations of Step A to Step G. In particular, it can be carried out by condensing the nucleic acid monomer compound in the 3 ′ to 5 ′ direction.
- those other than the phosphoramidite compounds of the present invention are not particularly limited as long as they are generally used for the synthesis of oligo RNA or oligo DNA.
- all steps can be manufactured using a manual or a commercially available DNA automatic synthesizer. It is desirable to use an automatic synthesizer in order to simplify the operation method by using an automatic synthesizer and to ensure the accuracy of synthesis.
- nucleic acid monomer compound is not particularly limited as long as they are generally used for the synthesis of oligo DNA or oligo RNA.
- Each Bx independently represents zazanine, guanine, cytosine, uracil, thymine or a modified form thereof.
- Each R 4 independently represents H, acyloxy, or a substituent represented by the following general formula (6).
- E represents a substituent represented by isyl or the following general formula (7).
- T represents H, acyloxy, or a substituent represented by the general formula (6) or (7).
- R 41 Represents a substituent (7).
- R 2 is Asil
- R 4 represents H, acyloxy or a substituent (6).
- solid phase carrier examples include controlled pore glass (CPG), oxalylated-constant glass (eg, Alul et al., Nucleic Acids Research, Vol. 19, 15 27 (1991)). ), TentaGel support-aminoaminoglycol derivative support (see, for example, Wright et al., Tetrahedron Letters, Vol. 34, 3373 (1993)),
- linker examples include 3-aminopropyl, succiol, and 2,2′-diethanol. Examples include sulfol and long chain alkylamino (LCAA).
- the compound (26a) and the compound (26b) are compounds supported on a solid phase carrier produced according to a known method or available as a commercial product.
- a solid phase carrier produced according to a known method or available as a commercial product.
- Compounds (27) and (28) in which R 4 is substituent (6) can be produced from the phosphoramidite compounds of the present invention according to known methods.
- the “acid” that can be used in this step include trifluoroacetic acid, dichloroacetic acid, and trichloroacetic acid.
- the acid that can be used in this step can be used after diluting with a suitable solvent so as to have a concentration of 1 to 5%.
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, acetonitrile, water, and a mixed solvent thereof.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of acid used and the reaction temperature. Usually, 1 minute to 1 hour is appropriate.
- the amount of reagent used is supported on a solid support!
- the molar amount is preferably 1 to 100 times that of the compound to be obtained, more preferably 1 to
- This step can be carried out by using a nucleic acid monomer compound and an activator on a compound supported on a solid phase carrier.
- nucleic acid monomer compound examples include the compound represented by the following general formula (29), which is available as a phosphoramidite compound or a commercially available product of the present invention.
- nucleobase is not particularly limited as long as it is used for nucleic acid synthesis.
- adenine, guanine, cytosine, thymine or a modified form thereof can be mentioned.
- the “modified product” has the same meaning as B.
- substituents for the modified form of B include, for example, halogen, alkyl, arylalkyl
- Nucleobase according to B is a nucleobase having an amino group, whether it is protected or not.
- adenine, guanine, and cytosine preferably have a protected amino group.
- amino-protecting group examples include the same as those described above for B.
- Examples of the “activator” include the same as described above.
- the reaction solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include acetonitrile and tetrahydrofuran.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of activator used and the reaction temperature, but usually 1 minute to 1 hour is appropriate.
- the amount of the reagent to be used is 1 to 100 times the molar amount, more preferably 1 to 10 times the molar amount with respect to the compound supported on the solid phase carrier.
- step B A step of cabling the 5′-position hydroxyl group of the compound (5) which has not been reacted in the step B.
- R 5 is methyl, phenoxymeth
- This step is a reaction for protecting the hydroxyl group at the 5 ′ position, which has not been reacted in Step B, and can be carried out by acting a capping agent on the compound supported on the solid phase carrier. .
- capping agent examples include acetic anhydride or phenoxyacetic anhydride. Can do.
- the capping agent can be used by diluting with a suitable solvent so as to have a concentration of 0.05 to 1M.
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include pyridine, dichloromethane, acetonitrile, tetrahydrofuran, and a mixed solvent thereof.
- 4-dimethylaminopyridine or N-methylimidazole can be used as a “reaction accelerator” as necessary.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of capping agent used and the reaction temperature, but usually 1 to 30 minutes is appropriate.
- the amount of the reagent to be used is 1 to 100 times the molar amount, more preferably 1 to 10 times the molar amount with respect to the compound supported on the solid phase carrier.
- This step is a reaction for converting trivalent phosphorus to pentavalent phosphorus using an oxidizing agent, and can be performed by allowing an oxidizing agent to act on the compound supported on the solid phase carrier. .
- the “oxidant” examples include iodine and tert-butyl hydroperoxide.
- the oxidizing agent used in this step can be diluted with an appropriate solvent so as to have a concentration of 0.05 to 2M.
- the solvent is not particularly limited as long as it does not participate in the reaction.
- pyridine, tetrahydrofuran, water or a mixed solvent thereof may be mentioned.
- iodine Z water Z pyridine-tetrahydrofuran or iodine Z pyridine monoacetic acid or a peroxide (such as t-ptyl hydride mouth peroxide z methylene chloride) can be used.
- the reaction temperature is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of oxidizing agent used and the reaction temperature, but usually 1 to 30 minutes is appropriate.
- the amount of the reagent to be used is preferably 1 to: more than 10 to 50 times the molar amount of the compound supported on the solid phase carrier. (5) X @ E:
- step D the compound (11) produced in the step D is excised from the solid phase carrier force, and each nucleobase and each 2′-hydroxyl protecting group are removed.
- the cleaving step is a reaction in which oligo RNA having a desired chain length is removed from the solid phase carrier and the linker by a cleaving agent, and the cleaving agent is added to the solid carrier carrying the oligo RNA having the desired chain length. Can be implemented.
- the protecting group in the nucleobase portion can be removed.
- Examples of the “cutting agent” include concentrated aqueous ammonia and methylamine.
- the “cutting agent” that can be used in this step can be used by diluting with water, methanol, ethanol, isopropyl alcohol, acetonitrile, tetrahydrofuran, or a mixed solvent thereof, for example. Of these, ethanol is preferred.
- the reaction temperature is suitably 15 ° C to 75 ° C, preferably 15 ° C to 30 ° C, and preferably 18 ° C to 25 ° C. More preferred.
- the deprotection reaction time is suitably 1-30 hours, 1-24 hours is preferred 1-4 hours is more preferred U ,.
- the concentration of hydroxyammonium hydroxide in the solution used for deprotection is suitably 20-30% by weight, preferably 25-30% by weight, more preferably 28-30% by weight.
- the amount of the reagent to be used is suitably 1 to 100 times the molar amount, preferably 10 to 50 times the molar amount of the compound supported on the solid phase carrier! /.
- the “reagent for removing the protecting group at the 2′-position” acts, for example, by using tetraptyl ammonium fluoride, hydrogen trifluoride′-triethylamine salt. Can be performed.
- the solvent to be used is not particularly limited as long as it does not participate in the reaction, and examples thereof include tetrahydrofuran, N-methylpyrrolidone, pyridine, dimethyl sulfoxide or a mixed solvent thereof. If necessary, for example, alkylamine, amidine, thiol, thiol derivative or a mixture thereof can be added as a compound supplementing acrylonitrile which is a by-product in this step.
- alkylamine examples include linear alkylamine having 1 to 6 carbon atoms. Specifically, for example, methylamine, ethylamine, n-propylamine, n-butylamine, n-pentylamine, and n-hexylamine can be mentioned.
- amidine examples include benzamidine and formamidine.
- thiol examples include linear thiols having 1 to 6 carbon atoms. Specifically, for example, methanethiol, ethanethiol, 1-propanthiol, 1-butanethiol, 1-pentanethiol, and 1-hexanethiol can be listed.
- thiol derivative examples include alcohols or ethers having the same or different linear alkyl thiol group having 1 to 6 carbon atoms. Specifically, for example, 2 mercaptoethanol, 4 mercapto 1-butanol, 6 mercapto 1 monohexanol, mercaptomethyl ether, 2 mercaptoethyl ether, 3-mercaptopropyl ether, 4 mercaptobutyl ether, 5-mercaptopentyl ether, Mention may be made of 6-mercaptohexyl ether.
- the reaction temperature is preferably 20 ° C to 80 ° C.
- the reaction time varies depending on the type of deprotecting agent used and the reaction temperature. Usually 1 to 100 hours is appropriate.
- the amount of the reagent used is preferably 50 to 500-fold molar amount relative to the protecting group to be removed, more preferably 50 to: L00-fold mole Normal separation and purification means such as extraction, concentration, neutralization, filtration, centrifugal separation, recrystallization, silica gel column chromatography, thin layer chromatography, reverse layer o
- step E A step of removing the 5′-position hydroxyl group of the compound (12) produced in step E.
- This step is a reaction for finally removing the protecting group for the 5′-position hydroxyl group of the oligo RNA, and can be carried out by allowing an acid to act on the oligo RNA that has been cut out as a solid carrier.
- Examples of the “acid” that can be used in this step include triclonal acetic acid, dichloroacetic acid, and acetic acid.
- the acid that can be used in this step can also be diluted with an appropriate solvent.
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include dichloromethane, acetonitrile, water, a buffer solution having a pH of 2 to 5 or a mixed solvent thereof.
- Examples of the buffer solution include an acetate buffer solution.
- the reaction temperature in the above reaction is preferably 20 ° C to 50 ° C.
- the reaction time varies depending on the type of acid used and the reaction temperature, but usually 1 minute to 1 hour is appropriate.
- the amount of the reagent to be used is 1 to: more than 1 to 10 times the molar amount of the compound supported on the solid support, more preferably 1 to 10 times. [0032] (7) Process G:
- Step F A step of separating and purifying the compound (3) produced in Step F.
- the “separation and purification step” refers to usual separation and purification means from the above reaction mixture, such as extraction, concentration, neutralization, filtration, centrifugation, recrystallization, C-force C reverse phase column chromatography, C
- the desired oligo RNA is isolated and purified by using means such as column chromatography, gel filtration column chromatography, high performance liquid chromatography, dialysis, and ultrafiltration.
- Examples of the “elution solvent” include acetonitrile, methanol, ethanol, isopropyl alcohol, water alone, or a mixed solvent in an arbitrary ratio.
- examples of additives include sodium phosphate, potassium phosphate, sodium chloride salt, potassium salt salt, ammonium acetate, triethylammonium acetate, sodium acetate, acetic acid lithium, Tris-HCl, Ethylenediamine tetraacetic acid can be added at a concentration of lmM to 2M, and the pH of the solution can be adjusted in the range of 1 to 9.
- an oligo RNA (3) having a desired chain length can be produced.
- R 4 is a compound (26a) in which substituent (6) is present, y compound (26a) in which R 4 is H or acyloxy, Alternatively, a compound (26b) or the like in which R 2 is alkyloxy can be used.
- a compound (26a) in which R 4 is H or acyloxy or a compound (26b) in which R 2 is alkyloxy is used as a starting material
- at least one of the phosphoramidite compounds of the present invention is used as a nucleic acid monomer compound. It is necessary to use a compound.
- step F is performed before the operation of step E, and then the operation of step E is performed, and then the operation of step G is performed in the next step to isolate and purify oligo RNA.
- Example 2 5′-0- (4 ⁇ 4′-dimethoxyhytyl _ ⁇ ⁇ 2′-0- ⁇ 2-cyanoe]: xymethyl L) uridine 3, 1 O— (2 cyanoethyl N. N diisopropyl phosphoramidite) Step 1 5′— O— (4.4′-dimethoxytrityl) 2′— O— (2 cyanoethoxymethyl) uridine
- Step 1 3, .5'—O— (Tetraisopropyldisiloxane 1.3 gil) ⁇ 2, ⁇ 0— (
- reaction solution was filtered through celite and washed with methylene chloride.
- organic phase was washed with 1M aqueous sodium hydrogensulfate solution, washed with saturated aqueous sodium hydrogencarbonate solution, dried over anhydrous magnesium sulfate, and the solvent was distilled off. Left.
- the resulting residue was purified by thin layer chromatography to obtain 3 ′, 5′—O— (tetraisopropyldisiloxane mono-1,
- the residue was dissolved in ethyl acetate and separated with a saturated aqueous sodium hydrogen carbonate solution.
- the organic phase was washed with a saturated sodium chloride aqueous solution, dried over anhydrous magnesium sulfate, and the solvent was distilled off.
- the obtained residue was purified by silica gel chromatography to obtain the target compound (26.5 g; yield 98%).
- Step 1 N A acetyl 3,5'—O— (tetraisopropyldisiloxane 1.3 diol) 2′—O— (2-cyanethoxymethyl) cytidine
- N 4 acetyleno 3,5,1 O tetraisopropyldisiloxane 1,3 diyl
- cytidine 1.
- 975 mg (3.79 mmol) of silver trifluoromethanesulfonate was added, and Molecular Sieves 4A was added and dried. Under ice cooling, 370 mg (2.08 mmol) of N-bromosuccinimide was added, and the reaction vessel was protected from light and stirred for 10 minutes.
- N-promosuccinimide 7 Omg (0.39 mmol) was added and stirred for 25 minutes. After completion of the reaction, dilute with methylene chloride, wash with saturated aqueous sodium hydrogen carbonate, dry over anhydrous sodium sulfate, evaporate the solvent, and purify the resulting mixture by silica gel column chromatography. N 4 —acetylyl 3,5,1 O— (tetraisopropyldisiloxane 1,1,3 diyl) -1,2, -0- (2 cyanoethoxymethyl) cytidine was obtained. (936 mg; 81% yield). — NMR (CDC1): 0.90- 1.11 (m, 28H); 2. 28 (s, 3H); 2. 62— 2. 79 (
- Step 1 N 2 —acetylene 5, -0- ⁇ 4._4'—dim I I: xytil) _— 2, -0- and 2—shear,
- N 2 acetylyl 5, -0- (4, 4, -dimethoxytrityl) -2, -0 obtained in step 1 -Dissolve 400 mg (0. 563 mmol) of (2 cyanoethoxymethyl) guanosine in 2 mL of methylene chloride, and add 181 mg (l. 4 mmol) of diisopropylethylamine to give 2 cyanoethyl N, N diisopropylchlorophosphoramidite 161 mg (0.668 mmol) was added dropwise and reacted at room temperature for 1 hour.
- Og (36. Om mol) is dissolved in 170 mL of 1,2 dichloroethane, and 16.3 g (126 mmol) of diisopropyl etheramine is dissolved.
- 12. lg (39.7 mmol) of dibutyltin dichloride was added and reacted at room temperature for 1 hour. Thereafter, the mixture was stirred at 80 ° C. for 15 minutes, and 4.30 g (36. Ommol) of chloromethyl 2-cyanoethyl ether was added dropwise, followed by stirring for 30 minutes.
- Step 1 N a —Acetyl 1 3 ′. 5′— O— (Tetraisopropyldisiloxane 1 1.3 Dil) 2′— O— (2-Cyanethoxymethyl) adenosine
- Step 1 N 2 —Phenoxyacetyl- 5 '-0- (4.4'-dimethoxytrityl) 2' -0- (2-Cyanethoxymethyl) guanosine
- N 2 full enoki Xia cetyl over 5 'O (4, 4'- dimethoxytrityl) was dissolved guanosine 720mg of (Lmmol) in 1, 2 Jikuroroetan 4 mL, diisopropyl E chill ⁇ Min 452mg of (3.5 mmol) was added, followed by 365 mg ( l.2 mmol) of dibutyltin dichloride was added and reacted at room temperature for 1 hour. Thereafter, the temperature was raised to 80 ° C., and 55.4 mg (l.3 mmol) of chloromethyl 2-cyanoethyl ether 1 was added dropwise, and the mixture was stirred as it was for 60 minutes.
- N 2 phenoxycetyl-5, -0- (4,4, -dimethoxytrityl) — 2, 1, O (2 cyanoethoxymethyl) guanosine obtained in Step 1 was mixed with methyl chloride. Dissolve in 4 mL of Len, add 128.8 mg (0. 996 mmol) of diisopropylethylamine, add 2—Cyanoethyl N, N-diisopropylchlorophosphoramidite 141.5 mg (0.598 mmol), and add 1 at room temperature. Reacted for hours. After the reaction, the solvent was distilled off and the resulting mixture was purified by 3 Og silica gel column chromatography to obtain the target compound (316 mg; yield 79%).
- Example 15 N a —Acetyl mono 3 ′. 5′— O— (tetraisopropyldisiloxane mono 1.3-diyl) — 2, — O— (2-cyanethoxymethyl) adenosine
- Step 1 N a Asechiru 3 ,. 5'-O- (tetraisopropyldisiloxane 1.3 Jie
- N 440 succinimide (440 mg, 1.96 mmol) was added, trifluoromethanesulfonic acid (490 mg, 3.26 mmol) was added, and the mixture was stirred at 45 ° C for 15 min.
- nucleic acid monomer compounds 5 O- (4, 4, -dimethoxytrityl) -2, O- (2-cyanoethoxymethyl) uridine 3, mono-O- (2-cyanoethyl N, N-diisopropyl phosphoro Amidite), tetrazole as the condensation catalyst, iodine solution as the oxidizing agent, and acetic anhydride and N-methylimidazole solution as the cabbing solution.
- reaction mixture was concentrated under reduced pressure, unnecessary peaks were removed with a reverse phase column (ODS), and then purified using an elution solvent (acetonitrile—50 mM triethylamine—acetic acid buffer). After concentrating the residue under reduced pressure, the reaction was carried out for 1 hour at room temperature using a THF solution of 1M tetraptylammonium fluoride to remove the protecting group for the 2′-position hydroxyl group. After desalting the solution, the protecting group at the 5 ′ end was removed with 80% acetic acid (10 minutes at room temperature). After concentration under reduced pressure, the aqueous layer was washed with ether to obtain the desired compound with high purity without purification.
- ODS reverse phase column
- Liquid feeding unit LC— 6 A (manufactured by Shimadzu Corporation)
- Solution A 50 mM triethylamine-acetic acid buffer solution containing 5% acetonitrile
- Liquid B 50 mM triethylamine acetate buffer containing 90% acetonitrile
- nucleic acid monomer compounds 5, —O— (4,4, -dimethoxytrityl) -2, —O— (2—cyanethoxymethyl) uridine 3,1 O— (2 cyanethyl N, N diisopropyl phosphoramidite) , N 4 -Acetyl-5, -0- (4, 4, -dimethoxytrityl) -2,-O- (2 cyanoethoxymethyl) cytidine 3, -O- (2 cyanoethinole N, N diisopropyl phosphoramidite ), N 6- acetyl--5, —O— (4, 4, -dimethoxytrityl) — 2, 1 O— (2 cyanoethoxymethyl) adenosine 3, 1 O— (2 cyanoethyl N, N diisopropyl phosphoramidite) , N 2 —phenoxyacetyl-5, -0- (4, 4, —Dimethoxytrity
- the absorbance (OD) of ultraviolet light with a wavelength of 260 nm was used.
- the absorbance (OD) was similarly used as the yield of the target compound.
- Example 20 Adenylyl “3, ⁇ 5 ⁇ —Adenylyl“ 3, ⁇ 5 ⁇ —Uridylyl “3, ⁇ 5,
- the ether-type protecting group introduced into the 2′-position hydroxyl group is a linear substituent, and the three-dimensional structure around the phosphorus atom bonded to the 3′-position hydroxyl group is crowded. Therefore, compared to the conventionally used phosphoramidite complex, when synthesizing oligo RNA, the condensation reaction proceeds in a very short time and the condensation yield is good. Therefore, by using the phosphoramidite toy compound of the present invention, it has become possible to produce high-purity oligo RNA using a technique almost the same as that of oligo DNA.
Abstract
Description
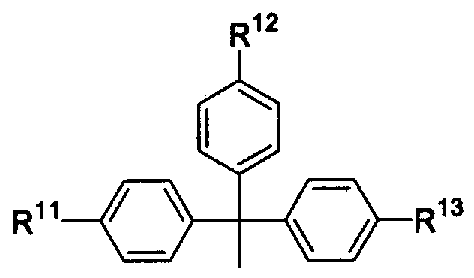


















Claims











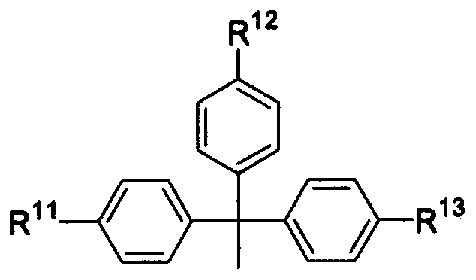








Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2005800363733A CN101048423B (zh) | 2004-08-26 | 2005-08-25 | 亚磷酰胺化合物及低聚核糖核酸的制备方法 |
| BRPI0514644-5A BRPI0514644A (pt) | 2004-08-26 | 2005-08-25 | composto fosforamidita, composto ácido ribonucléico, e método para produzir oligo-rna |
| JP2006531972A JP5157168B2 (ja) | 2004-08-26 | 2005-08-25 | ホスホロアミダイト化合物及びオリゴrnaの製法 |
| CA2577922A CA2577922C (en) | 2004-08-26 | 2005-08-25 | Phosphoramidite compound and method for producing oligo-rna |
| ES05780944T ES2386709T3 (es) | 2004-08-26 | 2005-08-25 | Compuesto de fosforamidita y método para producir un oligo-ARN |
| MX2007002245A MX2007002245A (es) | 2004-08-26 | 2005-08-25 | Compuesto de fosforamidita y metodo para la produccion de un acido oligorribonucleico. |
| AU2005275801A AU2005275801B2 (en) | 2004-08-26 | 2005-08-25 | Phosphoramidite compound and method for producing oligo-RNA |
| EP05780944A EP1795536B1 (en) | 2004-08-26 | 2005-08-25 | Phosphoramidite compound and method for producing oligo-rna |
| KR1020077006676A KR101281836B1 (ko) | 2004-08-26 | 2005-08-25 | 포스포라미다이트 화합물 및 올리고 rna의 제조 방법 |
| AT05780944T ATE556085T1 (de) | 2004-08-26 | 2005-08-25 | Phosphoramiditverbindung und verfahren zur herstellung von oligo-rna |
| US11/574,308 US8691970B2 (en) | 2004-08-26 | 2005-08-25 | Phosphoramidite compound and method for producing oligo-RNA |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004246185 | 2004-08-26 | ||
| JP2004-246185 | 2004-08-26 | ||
| JP2005110817 | 2005-04-07 | ||
| JP2005-110817 | 2005-04-07 | ||
| JP2005-193313 | 2005-07-01 | ||
| JP2005193313 | 2005-07-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006022323A1 true WO2006022323A1 (ja) | 2006-03-02 |
Family
ID=35967530
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2005/015420 WO2006022323A1 (ja) | 2004-08-26 | 2005-08-25 | ホスホロアミダイト化合物及びオリゴrnaの製法 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8691970B2 (ja) |
| EP (1) | EP1795536B1 (ja) |
| JP (1) | JP5157168B2 (ja) |
| KR (1) | KR101281836B1 (ja) |
| CN (1) | CN101048423B (ja) |
| AT (1) | ATE556085T1 (ja) |
| AU (1) | AU2005275801B2 (ja) |
| BR (1) | BRPI0514644A (ja) |
| CA (1) | CA2577922C (ja) |
| ES (1) | ES2386709T3 (ja) |
| MX (1) | MX2007002245A (ja) |
| WO (1) | WO2006022323A1 (ja) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007097447A1 (ja) * | 2006-02-27 | 2007-08-30 | Nippon Shinyaku Co., Ltd. | 核酸保護基の脱離方法 |
| WO2007099896A1 (ja) * | 2006-02-27 | 2007-09-07 | Nippon Shinyaku Co., Ltd. | 核酸保護基の脱離方法 |
| WO2008016079A1 (en) | 2006-08-02 | 2008-02-07 | Nippon Shinyaku Co., Ltd. | Method for introducing nucleic-acid-protecting group |
| WO2008090829A1 (ja) * | 2007-01-22 | 2008-07-31 | Nippon Shinyaku Co., Ltd. | リボ核酸化合物の製造方法 |
| WO2011034072A1 (ja) | 2009-09-16 | 2011-03-24 | 株式会社キラルジェン | Rna及びその誘導体合成のための新規保護基 |
| WO2011108682A1 (ja) * | 2010-03-05 | 2011-09-09 | 国立大学法人 東京大学 | リボヌクレオシドホスホロチオエートの製造方法 |
| WO2012073857A1 (ja) * | 2010-11-30 | 2012-06-07 | 株式会社キラルジェン | 2'-o-修飾rna |
| WO2013027843A1 (ja) * | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| US9394333B2 (en) | 2008-12-02 | 2016-07-19 | Wave Life Sciences Japan | Method for the synthesis of phosphorus atom modified nucleic acids |
| US9598458B2 (en) | 2012-07-13 | 2017-03-21 | Wave Life Sciences Japan, Inc. | Asymmetric auxiliary group |
| US9605019B2 (en) | 2011-07-19 | 2017-03-28 | Wave Life Sciences Ltd. | Methods for the synthesis of functionalized nucleic acids |
| US9617547B2 (en) | 2012-07-13 | 2017-04-11 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant |
| US9744183B2 (en) | 2009-07-06 | 2017-08-29 | Wave Life Sciences Ltd. | Nucleic acid prodrugs and methods of use thereof |
| US9982257B2 (en) | 2012-07-13 | 2018-05-29 | Wave Life Sciences Ltd. | Chiral control |
| US10144933B2 (en) | 2014-01-15 | 2018-12-04 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having immunity induction activity, and immunity induction activator |
| US10149905B2 (en) | 2014-01-15 | 2018-12-11 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having antitumor effect and antitumor agent |
| US10160969B2 (en) | 2014-01-16 | 2018-12-25 | Wave Life Sciences Ltd. | Chiral design |
| US10322173B2 (en) | 2014-01-15 | 2019-06-18 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having anti-allergic activity, and anti-allergic agent |
| US10428019B2 (en) | 2010-09-24 | 2019-10-01 | Wave Life Sciences Ltd. | Chiral auxiliaries |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021193954A1 (ja) | 2020-03-27 | 2021-09-30 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021210408A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021210409A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
| WO2022009959A1 (ja) | 2020-07-09 | 2022-01-13 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022064908A1 (ja) | 2020-09-24 | 2022-03-31 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2023282120A1 (ja) | 2021-07-06 | 2023-01-12 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2924186C (en) * | 2013-09-14 | 2023-03-28 | Chemgenes Corporation | Highly efficient synthesis of long rna using reverse direction approach |
| CN104211741B (zh) * | 2014-09-05 | 2017-05-31 | 河南师范大学 | 一种氘代核苷亚磷酰胺单体的合成方法 |
| JP7280248B2 (ja) | 2018-04-24 | 2023-05-23 | 住友化学株式会社 | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 |
| WO2020050411A1 (ja) | 2018-09-07 | 2020-03-12 | 住友化学株式会社 | 配糖体化合物の製造方法 |
| JP7452549B2 (ja) * | 2019-10-18 | 2024-03-19 | 富士フイルム和光純薬株式会社 | ホスホロアミダイト活性化剤 |
| KR20220086589A (ko) | 2019-10-23 | 2022-06-23 | 스미또모 가가꾸 가부시끼가이샤 | 배당체 화합물, 아미다이트 화합물, 및 이들 화합물을 사용한 폴리뉴클레오티드의 제조 방법 |
| CN115010769B (zh) * | 2022-08-04 | 2022-11-15 | 上海百力格生物技术有限公司 | 固相亚磷酰胺三酯法合成长链rna核酸的方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS49126825A (ja) | 1973-04-03 | 1974-12-04 | ||
| US4629814A (en) | 1985-08-27 | 1986-12-16 | Rohm And Haas Company | Process for preparing bis-bromoalkyl ethers |
| US4925856A (en) | 1987-07-28 | 1990-05-15 | Sri International | Aldoxime-substituted imidazolium derivatives useful in the treatment of poisoning by phosphorus-containing chemicals |
| JPH0374398A (ja) | 1989-08-17 | 1991-03-28 | Yuki Gosei Kogyo Co Ltd | ホスホアミダイト化合物及びそれを用いたオリゴリボヌクレオチドの固相合成法 |
| JPH07502037A (ja) | 1991-12-11 | 1995-03-02 | インペリアル・ケミカル・インダストリーズ・ピーエルシー | α−フルオロエーテルからハイドロフルオロカーボンの製造 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5637776A (en) | 1992-12-10 | 1997-06-10 | Imperial Chemical Industries Plc | Production of hydrofluorocarbons |
| AR037328A1 (es) * | 2001-10-23 | 2004-11-03 | Dow Agrosciences Llc | Compuesto de [7-bencil-2,6-dioxo-1,5-dioxonan-3-il]-4-metoxipiridin-2-carboxamida, composicion que lo comprende y metodo que lo utiliza |
-
2005
- 2005-08-25 EP EP05780944A patent/EP1795536B1/en active Active
- 2005-08-25 MX MX2007002245A patent/MX2007002245A/es active IP Right Grant
- 2005-08-25 US US11/574,308 patent/US8691970B2/en active Active
- 2005-08-25 WO PCT/JP2005/015420 patent/WO2006022323A1/ja active Application Filing
- 2005-08-25 AT AT05780944T patent/ATE556085T1/de active
- 2005-08-25 BR BRPI0514644-5A patent/BRPI0514644A/pt not_active IP Right Cessation
- 2005-08-25 CN CN2005800363733A patent/CN101048423B/zh active Active
- 2005-08-25 ES ES05780944T patent/ES2386709T3/es active Active
- 2005-08-25 KR KR1020077006676A patent/KR101281836B1/ko active IP Right Grant
- 2005-08-25 CA CA2577922A patent/CA2577922C/en not_active Expired - Fee Related
- 2005-08-25 AU AU2005275801A patent/AU2005275801B2/en not_active Ceased
- 2005-08-25 JP JP2006531972A patent/JP5157168B2/ja active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS49126825A (ja) | 1973-04-03 | 1974-12-04 | ||
| US3956500A (en) | 1973-04-03 | 1976-05-11 | Union Carbide Corporation | Carbamate pesticidal compositions |
| US4629814A (en) | 1985-08-27 | 1986-12-16 | Rohm And Haas Company | Process for preparing bis-bromoalkyl ethers |
| JPS62116533A (ja) | 1985-08-27 | 1987-05-28 | ロ−ム アンド ハ−ス コンパニ− | ビスブロモアルキルエ−テルの製造方法 |
| US4925856A (en) | 1987-07-28 | 1990-05-15 | Sri International | Aldoxime-substituted imidazolium derivatives useful in the treatment of poisoning by phosphorus-containing chemicals |
| JPH0374398A (ja) | 1989-08-17 | 1991-03-28 | Yuki Gosei Kogyo Co Ltd | ホスホアミダイト化合物及びそれを用いたオリゴリボヌクレオチドの固相合成法 |
| JPH07502037A (ja) | 1991-12-11 | 1995-03-02 | インペリアル・ケミカル・インダストリーズ・ピーエルシー | α−フルオロエーテルからハイドロフルオロカーボンの製造 |
| US5504263A (en) | 1991-12-11 | 1996-04-02 | Imperial Chemical Industries Plc | Production of hydrofluorocarbons |
Non-Patent Citations (13)
| Title |
|---|
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 2001:675264 |
| DATABASE CHEMICAL ABSTRACTS. [online] KITANO H ET AL: "Preparation of some fluoroethanol derivatives.", XP008050277, Database accession no. (4063g-i) * |
| HELVETICA CHIMICA ACTA, vol. 81, no. 8, pages 1545 - 1566 |
| HELVETICA CHIMICA ACTA, vol. 83, 1 January 2000 (2000-01-01), pages 1127 - 1144 |
| J CHEM SOC JAPAN IND CHEM SECT., vol. 58, 1955, pages 355 - 357 * |
| J. ORG CHEM., vol. 46, no. 3, 1981, pages 571 - 577 |
| NORMANT JF ET AL: "Reactivite des vinylcuivres. Application a la synthese d' alcools allyliques substitues stereospecifiquement.", TETRAHEDRON LETTERS., vol. 26, 1973, pages 2407 - 2408, XP002994024 * |
| OLAH GA ET AL: "Carcinogen chemistry. 4. (Haloalkyl) oxonium and (haloalkyl) carboxonium ions. Preparation, NMR structural study, and alkylating ability.", J ORG CHEM., vol. 46, no. 3, 1981, pages 571 - 577, XP002994023 * |
| ORGANIC LETTERS, vol. 7, no. 16, 1 January 2005 (2005-01-01), pages 3477 - 3480 |
| See also references of EP1795536A4 |
| TET. LETTERS, vol. 26, 1973, pages 2407 - 2408 |
| TET. LETTERS, vol. 45, no. 52, 20 December 2004 (2004-12-20), pages 9529 - 9531 |
| UMEMOTO ET AL., TETRAHEDRON LETTERS, vol. 45, no. 9529, 2004 |
Cited By (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007099896A1 (ja) * | 2006-02-27 | 2007-09-07 | Nippon Shinyaku Co., Ltd. | 核酸保護基の脱離方法 |
| WO2007097447A1 (ja) * | 2006-02-27 | 2007-08-30 | Nippon Shinyaku Co., Ltd. | 核酸保護基の脱離方法 |
| JP5187189B2 (ja) * | 2006-02-27 | 2013-04-24 | 日本新薬株式会社 | 核酸保護基の脱離方法 |
| JP5168145B2 (ja) * | 2006-08-02 | 2013-03-21 | 日本新薬株式会社 | 核酸保護基の導入方法 |
| WO2008016079A1 (en) | 2006-08-02 | 2008-02-07 | Nippon Shinyaku Co., Ltd. | Method for introducing nucleic-acid-protecting group |
| KR101405632B1 (ko) * | 2006-08-02 | 2014-06-10 | 니뽄 신야쿠 가부시키가이샤 | 핵산 보호기의 도입 방법 |
| US8158774B2 (en) | 2006-08-02 | 2012-04-17 | Nippon Shinyaku Co., Ltd. | Method for introducing a nucleic-acid protecting group |
| CN101522701B (zh) * | 2006-08-02 | 2012-05-23 | 日本新药株式会社 | 核酸保护基的导入方法 |
| WO2008090829A1 (ja) * | 2007-01-22 | 2008-07-31 | Nippon Shinyaku Co., Ltd. | リボ核酸化合物の製造方法 |
| US9695211B2 (en) | 2008-12-02 | 2017-07-04 | Wave Life Sciences Japan, Inc. | Method for the synthesis of phosphorus atom modified nucleic acids |
| US9394333B2 (en) | 2008-12-02 | 2016-07-19 | Wave Life Sciences Japan | Method for the synthesis of phosphorus atom modified nucleic acids |
| US10329318B2 (en) | 2008-12-02 | 2019-06-25 | Wave Life Sciences Ltd. | Method for the synthesis of phosphorus atom modified nucleic acids |
| US9744183B2 (en) | 2009-07-06 | 2017-08-29 | Wave Life Sciences Ltd. | Nucleic acid prodrugs and methods of use thereof |
| US10307434B2 (en) | 2009-07-06 | 2019-06-04 | Wave Life Sciences Ltd. | Nucleic acid prodrugs and methods of use thereof |
| US8470987B2 (en) | 2009-09-16 | 2013-06-25 | Chiralgen, Ltd. | Protective group for synthesis of RNA and derivative |
| JP5878758B2 (ja) * | 2009-09-16 | 2016-03-08 | 株式会社Wave Life Sciences Japan | Rna及びその誘導体合成のための新規保護基 |
| JP2016164141A (ja) * | 2009-09-16 | 2016-09-08 | 株式会社Wave Life Sciences Japan | Rna及びその誘導体合成のための新規保護基 |
| WO2011034072A1 (ja) | 2009-09-16 | 2011-03-24 | 株式会社キラルジェン | Rna及びその誘導体合成のための新規保護基 |
| JP5847700B2 (ja) * | 2010-03-05 | 2016-01-27 | 株式会社Wave Life Sciences Japan | リボヌクレオシドホスホロチオエートの製造方法 |
| JP2016074701A (ja) * | 2010-03-05 | 2016-05-12 | 株式会社Wave Life Sciences Japan | リボヌクレオシドホスホロチオエートの製造方法 |
| US8859755B2 (en) | 2010-03-05 | 2014-10-14 | Chiralgen, Ltd. | Method for preparing ribonucleoside phosphorothioate |
| WO2011108682A1 (ja) * | 2010-03-05 | 2011-09-09 | 国立大学法人 東京大学 | リボヌクレオシドホスホロチオエートの製造方法 |
| CN102918052A (zh) * | 2010-03-05 | 2013-02-06 | 国立大学法人东京大学 | 硫代磷酸核糖核苷的制造方法 |
| US10428019B2 (en) | 2010-09-24 | 2019-10-01 | Wave Life Sciences Ltd. | Chiral auxiliaries |
| WO2012073857A1 (ja) * | 2010-11-30 | 2012-06-07 | 株式会社キラルジェン | 2'-o-修飾rna |
| JP2016175933A (ja) * | 2010-11-30 | 2016-10-06 | 株式会社Wave Life Sciences Japan | 2’−o−修飾rna |
| JP6093924B2 (ja) * | 2010-11-30 | 2017-03-15 | 株式会社Wave Life Sciences Japan | 2’−o−修飾rna |
| JPWO2012073857A1 (ja) * | 2010-11-30 | 2014-05-19 | 株式会社キラルジェン | 2’−o−修飾rna |
| US10280192B2 (en) | 2011-07-19 | 2019-05-07 | Wave Life Sciences Ltd. | Methods for the synthesis of functionalized nucleic acids |
| US9605019B2 (en) | 2011-07-19 | 2017-03-28 | Wave Life Sciences Ltd. | Methods for the synthesis of functionalized nucleic acids |
| WO2013027843A1 (ja) * | 2011-08-25 | 2013-02-28 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| US9988415B2 (en) | 2011-08-25 | 2018-06-05 | Bonac Corporation | Glycoside compound, method for producing thioether, ether, method for producing ether, method for producing glycoside compound, method for producing nucleic acid |
| US9481702B2 (en) | 2011-08-25 | 2016-11-01 | Bonac Corporation | Glycoside compound, method for producing thioether, ether, method for producing ether, method for producing glycoside compound, method for producing nucleic acid |
| JP5554881B2 (ja) * | 2011-08-25 | 2014-07-23 | 株式会社ボナック | 配糖体化合物、チオエーテルの製造方法、エーテル、エーテルの製造方法、配糖体化合物の製造方法、核酸の製造方法 |
| US9982257B2 (en) | 2012-07-13 | 2018-05-29 | Wave Life Sciences Ltd. | Chiral control |
| US9617547B2 (en) | 2012-07-13 | 2017-04-11 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant |
| US10167309B2 (en) | 2012-07-13 | 2019-01-01 | Wave Life Sciences Ltd. | Asymmetric auxiliary group |
| US9598458B2 (en) | 2012-07-13 | 2017-03-21 | Wave Life Sciences Japan, Inc. | Asymmetric auxiliary group |
| US10590413B2 (en) | 2012-07-13 | 2020-03-17 | Wave Life Sciences Ltd. | Chiral control |
| US10144933B2 (en) | 2014-01-15 | 2018-12-04 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having immunity induction activity, and immunity induction activator |
| US10149905B2 (en) | 2014-01-15 | 2018-12-11 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having antitumor effect and antitumor agent |
| US10322173B2 (en) | 2014-01-15 | 2019-06-18 | Shin Nippon Biomedical Laboratories, Ltd. | Chiral nucleic acid adjuvant having anti-allergic activity, and anti-allergic agent |
| US10160969B2 (en) | 2014-01-16 | 2018-12-25 | Wave Life Sciences Ltd. | Chiral design |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| KR20220079832A (ko) | 2019-10-11 | 2022-06-14 | 스미또모 가가꾸 가부시끼가이샤 | 핵산 올리고머의 제조 방법 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021193954A1 (ja) | 2020-03-27 | 2021-09-30 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021210408A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021210409A1 (ja) | 2020-04-14 | 2021-10-21 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
| WO2022009959A1 (ja) | 2020-07-09 | 2022-01-13 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| KR20230034969A (ko) | 2020-07-09 | 2023-03-10 | 스미또모 가가꾸 가부시끼가이샤 | 핵산 올리고머의 제조 방법 |
| WO2022064908A1 (ja) | 2020-09-24 | 2022-03-31 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| KR20230074205A (ko) | 2020-09-24 | 2023-05-26 | 스미또모 가가꾸 가부시끼가이샤 | 핵산 올리고머의 제조 방법 |
| WO2023282120A1 (ja) | 2021-07-06 | 2023-01-12 | 住友化学株式会社 | 核酸オリゴマーを含む組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| ATE556085T1 (de) | 2012-05-15 |
| MX2007002245A (es) | 2007-04-20 |
| EP1795536B1 (en) | 2012-05-02 |
| ES2386709T3 (es) | 2012-08-27 |
| US20070282097A1 (en) | 2007-12-06 |
| CN101048423A (zh) | 2007-10-03 |
| KR20070054688A (ko) | 2007-05-29 |
| EP1795536A1 (en) | 2007-06-13 |
| KR101281836B1 (ko) | 2013-07-03 |
| AU2005275801A2 (en) | 2006-03-02 |
| AU2005275801A1 (en) | 2006-03-02 |
| BRPI0514644A (pt) | 2008-06-17 |
| US8691970B2 (en) | 2014-04-08 |
| CA2577922A1 (en) | 2006-03-02 |
| EP1795536A4 (en) | 2009-04-08 |
| CA2577922C (en) | 2013-06-25 |
| AU2005275801B2 (en) | 2012-05-10 |
| CN101048423B (zh) | 2011-06-08 |
| JPWO2006022323A1 (ja) | 2008-05-08 |
| JP5157168B2 (ja) | 2013-03-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006022323A1 (ja) | ホスホロアミダイト化合物及びオリゴrnaの製法 | |
| WO2007099896A1 (ja) | 核酸保護基の脱離方法 | |
| WO2007097447A1 (ja) | 核酸保護基の脱離方法 | |
| WO2007097446A1 (ja) | オリゴ核酸のキャッピング法 | |
| JP5168145B2 (ja) | 核酸保護基の導入方法 | |
| RU2415862C2 (ru) | Производное фосфорамидита и способ получения олиго-рнк | |
| WO2011090052A1 (ja) | リン酸化試薬 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006531972 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2577922 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/002245 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 811/CHENP/2007 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005780944 Country of ref document: EP Ref document number: 2005275801 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077006676 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2007110823 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2005275801 Country of ref document: AU Date of ref document: 20050825 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005275801 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580036373.3 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005780944 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11574308 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 11574308 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0514644 Country of ref document: BR |