WO2001098249A1 - Verfahren zur herstellung von hydroxyphenylcarbonsäureestern - Google Patents
Verfahren zur herstellung von hydroxyphenylcarbonsäureestern Download PDFInfo
- Publication number
- WO2001098249A1 WO2001098249A1 PCT/CH2001/000370 CH0100370W WO0198249A1 WO 2001098249 A1 WO2001098249 A1 WO 2001098249A1 CH 0100370 W CH0100370 W CH 0100370W WO 0198249 A1 WO0198249 A1 WO 0198249A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- formula
- radical
- butyl
- alkyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/03—Preparation of carboxylic acid esters by reacting an ester group with a hydroxy group
Definitions
- the invention relates to a process for the preparation of hydroxyphenyl carboxylic acid esters using selected catalysts.
- the invention also relates to new hydroxyphenylcarboxylic acid esters which can be prepared by the process according to the invention.
- Hydroxyphenyl carboxylic acid esters are preferably used as antioxidants.
- Numerous compounds of the formula (I) are known. They can be produced, for example, by transesterification with suitable catalysts. Such transesterification processes are e.g. in US-A-4,716,244; US-A-5,481,023; US-A-5, 563, 291.
- Compounds of the formula (I) are in part important commercial products. For example, they protect organic materials such as plastics and lubricants from thermal, oxidative and / or actinic degradation. There is still a need for new such compounds for use as antioxidants and for improved processes for their preparation.
- the present invention relates to a process for the preparation of compounds of the formula (I)
- reaction takes place at a practically neutral acid value (pH) and in the presence of at least one alkali salt of an organic carboxylic acid, or a mixture of such alkali salts, dissolved or suspended in the reaction mixture, where (i) this alkali salt consists of an alkali metalation and a Anion of an organic carboxylic acid is formed and (ii) the organic carboxylic acid is at least partially volatile under the reaction conditions used.
- pH practically neutral acid value
- the reaction conditions used in the reaction are preferably temperatures in the range from 50 ° C. to 250 ° C., preferably 80 ° C. to 220 ° C., preferably 140 ° C. to 220 ° C. and
- the salt-forming carboxylic acids preferably have a boiling point which is within the temperature and pressure ranges mentioned.
- Particularly suitable salt-forming cations of alkali metals are lithium, sodium or potassium cations.
- Prefers are sodium, potassium and / or lithium cations or sodium, potassium and / or lithium salts of organic carboxylic acids.
- carboxylic acids which are at least partially volatile in their acid form under the reaction conditions used are aliphatic saturated or unsaturated carboxylic acids with preferably 2 to 10 C atoms, preferably with 2 to 6 C atoms, such as e.g. Formic acid, acetic acid, propionic acid, n-butyric acid, isobutyric acid, n-valeric acid, trimethyl acetic acid, caproic acid, n-heptylic acid or pelargonic acid. Further examples are malonic acid, maleic acid, fumaric acid or also monomethyl malonate.
- Halogenated acids for example fluoroacetic acid, chloroacetic acid, bromoacetic acid, difluoroacetic acid, dichloroacetic acid, trifluoroacetic acid, trichloroacetic acid, alfa-chloropropionic acid or beta-choropropionic acid, are also suitable.
- Sodium acetate, potassium acetate, lithium acetate, sodium formate, potassium formate or lithium formate or a mixture of these compounds are preferred.
- the catalyst is preferably added in amounts of 0.05 to 5 mol%, based on the molar amount of the compound of the formula (I) to be reacted.
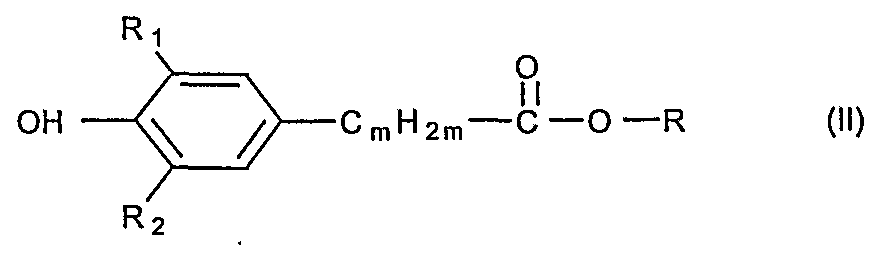
- R_ is preferably Ci-Cg-alkyl, where this alkyl radical can be linear or branched.
- R- preferably denotes an alkyl radical with 1-4 C atoms, preferably methyl, ethyl, propyl or butyl.
- R x is preferably a branched radical, preferably methyl or tert. Butyl.
- R 2 is preferably C - C 8 alkyl, and this alkyl radical can be linear or branched.
- R 2 is preferably an alkyl radical having 1-4 C atoms, preferably methyl, ethyl, propyl or butyl.
- R 2 is a branched radical, preferably methyl or tert. Butyl, preferably tert. Butyl.
- R x and R 2 can both be methyl or tert. -Butyl, or R x methyl and R 2 tert. -Butyl mean.
- R 3 as an n-valent alkyl radical with 4-30 C atoms, which is optionally interrupted by oxygen, can be linear or branched or can be present as a mixture.
- Such mixtures can consist of compounds in which R 3 is a mixture of predominantly linear or branched alkyl radicals, for example having 14, 16, 18 and 20 carbon atoms, it being possible for these alkyl radicals to be branched in some cases.
- R 3 denotes a radical R 4 - [NR 5 -C m H 2m -] p
- this radical preferably denotes R 4 - [NH-C m H 2ra -] p or R 4 - [N (C m H 2m - ) 2 ] p and preferably R 4 - [NH-C m H 2ra -] p
- R 4 is preferably an alkyl radical with 4-30 C atoms, which is optionally interrupted by -NR 5 -, preferably -NH-, and can be linear or branched, for example with 14, 16, 18 and 20 C atoms.
- R 3 is preferably a monovalent radical, preferably n-butyl, isobutyl, tert. -Butyl, pentyl, isopentyl, hexyl, heptyl, octyl, 2-ethylhexyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, eicosyl and the corresponding homologous residues with increasing carbon atoms up to 30 C atoms or a mixture of these C 4 -C 30 residues.
- R 3 preferably also denotes a monovalent radical such as cycloalkyl having 5-12 C atoms, preferably cyclopentyl and cyclohexyl, in particular cyclohexyl.
- R 3 preferably denotes C 2 -C a -alkylene or C 4 -C 12 -alkylene interrupted by oxygen.
- the n-valent radicals R 3 which are interrupted by oxygen can be derived from etherified polyhydroxy compounds, preferably from polyglycerol or from polypentaerythite.
- diamines are examples of such diamines.
- Alkylenediamines such as 1,2-ethylenediamine or 1,2-propylenediamine or dialkylenediamines such as di (ethylenediamine) or di (propylenediamine).
- Di (ethylenediamine) may optionally have a [-NH-C m H 2m -] group on the central nitrogen atom.
- R 3 is a branched alkyl-alkylene group derived from a trihydric alcohol by omitting the OH groups. In this sense, R 3 is preferably methane-C 1 -C 3 -alkyltrimethylene. Examples of such trihydric alcohols are 1,1,1-tris-hydroxymethylethane (trimethylolethane), 1,1,1-tris-hydroxymethylpropane (trimethylolpropane).
- R 3 is a branched alkyl alkylene group derived from a tetravalent alcohol or polyol by omitting the OH groups.
- R 3 is preferably methane tetramethylene, for example tetrakis hydroxymethyl methane (pentaerythritol).
- the alkylene radical - (C m H 2m ) - the compound of formula (I) resp. of the formula (II) m is preferably two, or the alkylene radical is -CH 2 CH 2 -.
- R x is tert
- R 2 is methyl or tert.
- Butyl, and R 3 n-octadecyl, a mixture of higher alkyl radicals with 8-30 C atoms (C 8 -C 30 alkyl radicals) or one of 1,6-hexanediol, triethylene glycol, pentaerythritol, 1, 1, 1-tris-hydroxymethylethane, 1, 1, 1-tris-hydroxymethylpropane, or
- R 4 is a radical derived from 1,2-ethylenediamine or 1,2-propylenediamine or such as di (ethylenediamine) or di (propylenediamine).
- the invention also relates to the use of an alkali salt of an organic carboxylic acid, or a mixture of such alkali salts, as described above, as a catalyst in the preparation of compounds of the formula (I), starting from compounds of the formulas (II) and (III), as described above, the use of sodium acetate, potassium acetate, lithium acetate, sodium formate, potassium formate or lithium formate or a mixture of these compounds being preferred.
- the catalyst is added in amounts of 0.05 to 5 mol%, preferably 0.05 to 3 mol%, particularly preferably 0.1 to 1 mol%, based on the molar amount of the compound of the formula (I) to be reacted.
- the compounds of the formulas (II) and (III) together with the catalyst under inert conditions preferably at a temperature of 90-120 ° C. and with stirring, heated until there is a melt.
- the pressure is then reduced and the temperature is increased, that is to say the reaction is preferably carried out at a pressure in the range from 0.1-200 mbar and at temperatures in the range from 140-220 ° C.
- the pressure is preferably 0.1-50 mbar, particularly preferably 0.1-20 mbar.
- the reaction temperature is preferably 160-220 ° C, particularly preferably 165-185 ° C.
- the reaction time depends on the pressure and the temperature and is generally between 1 and 12 hours, preferably between 1 and 10 hours, in particular between 2 and 6 hours.
- the ratio of the compound of the formula (II) to the compound of the formula (III) [calculated in molar equivalents] is preferably in the range from 0.8: 1 to 1.5: 1, preferably in the range from 1: 1 to 1.2: 1, particularly preferably between 1.05: 1 and 1.15: 1.
- the product of formula (I) can be brought to crystallization or solidification directly by cooling, with or without seeding, whereupon it can be processed directly into the commercial form without an additional purification step, such as recrystallization.
- the melt can of course also be taken up in a suitable solvent, cooled and, with or without seeding, crystallized.
- solvents for example aliphatic hydrocarbons such as heptane or cyclohexane or mixtures thereof; aromatic hydrocarbons, such as toluene and / or xylene; Alcohols " such as methanol, ethanol, propanol and / or isopropanol and the corresponding alcohol / water mixtures (50-100% alcohol).
- the compounds of the formulas (II) and (III) used in the process according to the invention are known per se.
- R 4 is one of 1,2-ethylenediamine or 1,2-propylenediamine or of di (ethylenediamine) or di (propylenediamine ) derived radical and R lf R 2 , and m have the meanings given are new.
- the internal temperature (IT) is increased to 170-180 ° C and further evacuated to ⁇ 1 mbar.
- the reaction melt now contains ⁇ 0.2% of the starting materials and has one Content> 98% of ß- (3, 5-di-t-butyl-4-hydroxypheny-propionic acid octadecyl ester. The reaction melt is cooled and left to crystallize. Yield
- the reaction melt now contains ⁇ 0.2% of the starting materials and has a content> 98% of ß- (3, 5-di-t-butyl-4-hydroxyphenyl) propionic acid octadecyl ester.
- the reaction melt is cooled and left to crystallize. Yield 96.5%; Mp 51 ° C.
- the reaction melt now contains ⁇ 0.2% of the starting materials and has a content > 97% of triethylene glycyl-bis- [ß- (3-t-butyl-5-methyl-4-hydroxyphenyl) propionic acid] ester.
- the reaction melt is cooled and left to crystallize. Yield 94.5%; 74-77 ° C.
- the reaction melt now contains ⁇ 0.2% of ß- (3, 5-di-t-butyl-4-hydroxyphenyl) propionic acid methyl ester and has a content> 97% of 1, 1, 1-tris- [ß- (3, 5-di-t-butyl-4-hydroxyphenyl) propionyloxymethyl] propane.
- the colorless reaction melt is filtered, cooled and solidified / crystallized. Yield 95%; Mp 66-79 ° C (amorphous form)
- Reaction melt now contains ⁇ 0.2% methyl ß- (3, 5-di-t-butyl-4-hydroxyphenyl) propionate and has a content
- the reaction melt now contains ⁇ 0.2% of ß- (3,5-di-t-butyl-4-hydroxyphenyl) propionic acid methyl ester and has a content> 97% of 1, 1, 1-tris- [ß- (3, 5-di-t-butyl-4-hydroxyphenyl) propionyloxymethyl] ethane.
- the colorless reaction melt is filtered, cooled and solidified / crystallized.
- the reaction melt now contains ⁇ 0.2% of ⁇ - (3,5-di-t-butyl -4-hydroxyphenyl) propionic acid methyl ester and has a content> 96% of tetrakis [ß- (3, 5-di-t-butyl-4-hydroxyphenyl) propionyloxymethyl] methane.
- the colorless reaction melt is filtered, cooled and the Solidification / crystallization brought yield 96%, mp: 55-85 ° C (amorphous form).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description


Claims

Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/311,724 US6878843B2 (en) | 2000-06-23 | 2001-06-14 | Method for preparing hydroxyphenyl carboxylic acid esters |
| AU2001263698A AU2001263698A1 (en) | 2000-06-23 | 2001-06-14 | Method for preparing hydroxyphenyl carboxylic acid esters |
| BRPI0111876-5A BR0111876B1 (pt) | 2000-06-23 | 2001-06-14 | processo para preparação de ésteres de ácido hidroxifenilcarboxìlico. |
| MXPA02012369A MXPA02012369A (es) | 2000-06-23 | 2001-06-14 | Procedimiento para la obtencion de esteres de acido hidroxifenilcarboxilico. |
| KR1020027016517A KR100813185B1 (ko) | 2000-06-23 | 2001-06-14 | 히드록시페닐 카르복시산 에스테르의 제조방법 |
| JP2002504205A JP4925547B2 (ja) | 2000-06-23 | 2001-06-14 | ヒドロキシフェニルカルボン酸エステルの生成方法 |
| CA2412062A CA2412062C (en) | 2000-06-23 | 2001-06-14 | Method for preparing hydroxyphenyl carboxylic acid esters |
| EP01937911A EP1292560B1 (de) | 2000-06-23 | 2001-06-14 | Verfahren zur herstellung von hydroxyphenylcarbonsäureestern |
| DE50109215T DE50109215D1 (de) | 2000-06-23 | 2001-06-14 | Verfahren zur herstellung von hydroxyphenylcarbonsäureestern |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CH12512000 | 2000-06-23 | ||
| CH1251/00 | 2000-06-23 | ||
| CH0312/01 | 2001-02-22 | ||
| CH3122001 | 2001-02-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001098249A1 true WO2001098249A1 (de) | 2001-12-27 |
Family
ID=25735909
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CH2001/000370 WO2001098249A1 (de) | 2000-06-23 | 2001-06-14 | Verfahren zur herstellung von hydroxyphenylcarbonsäureestern |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US6878843B2 (de) |
| EP (1) | EP1292560B1 (de) |
| JP (1) | JP4925547B2 (de) |
| KR (1) | KR100813185B1 (de) |
| CN (1) | CN1250513C (de) |
| AT (1) | ATE320412T1 (de) |
| AU (1) | AU2001263698A1 (de) |
| BR (1) | BR0111876B1 (de) |
| CA (1) | CA2412062C (de) |
| CZ (1) | CZ301602B6 (de) |
| DE (1) | DE50109215D1 (de) |
| ES (1) | ES2259325T3 (de) |
| MX (1) | MXPA02012369A (de) |
| RU (1) | RU2272022C2 (de) |
| WO (1) | WO2001098249A1 (de) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005042463A1 (en) * | 2003-10-27 | 2005-05-12 | Great Lakes Chemical (Europe) Gmbh | Preparation of tetrakis [3-(e,5-di-tert-butyl-4-hydroxy phenyl) propionyl oxymethyl] methane |
| WO2008055832A2 (en) * | 2006-11-09 | 2008-05-15 | Ciba Holding Inc. | Process using liquid phenolic antioxidants |
| WO2020152120A1 (en) | 2019-01-23 | 2020-07-30 | Basf Se | Oxime ester photoinitiators having a special aroyl chromophore |
| WO2021175855A1 (en) | 2020-03-04 | 2021-09-10 | Basf Se | Oxime ester photoinitiators |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL207511B1 (pl) * | 2002-04-26 | 2010-12-31 | Ciba Sc Holding Ag | Związek fotoinicjatora, sposób otrzymywania związku fotoinicjatora, kompozycja zdolna do fotopolimeryzacji, sposób fotopolimeryzowania, oraz zastosowanie kompozycji zdolnej do fotopolimeryzacji |
| JP6312259B2 (ja) | 2012-07-31 | 2018-04-18 | 株式会社Adeka | 潜在性添加剤及び該添加剤を含有する組成物 |
| WO2015119721A1 (en) | 2014-02-07 | 2015-08-13 | C. R. Bard, Inc. | Eptfe valves |
| PL3067343T3 (pl) | 2015-03-10 | 2018-12-31 | Evonik Degussa Gmbh | Przeciwutleniacze do wytwarzania systemów PUR o niskiej emisji |
| CN105294441B (zh) * | 2015-12-10 | 2017-09-15 | 中国科学院新疆理化技术研究所 | 抗氧化剂八[β‑(3,5‑二叔丁基‑4‑羟基苯基)丙酸]三季戊四醇酯的合成方法 |
| CN106674002A (zh) * | 2016-12-28 | 2017-05-17 | 天津利安隆新材料股份有限公司 | 受阻酚类抗氧剂的制备方法 |
| CN114835991A (zh) * | 2022-06-10 | 2022-08-02 | 安徽顺彤包装材料有限公司 | 一种水性转印膜及其制备工艺 |
| EP4318166A1 (de) | 2022-08-05 | 2024-02-07 | Volocopter GmbH | Verfahren zum steuern eines übergangsflugzeuges und übergangsflugzeug |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4716244A (en) * | 1985-05-02 | 1987-12-29 | Ciba-Geigy Corporation | Process for the preparation of sterically hindered hydroxyphenylcarboxylic acid esters |
| US5563291A (en) * | 1994-03-11 | 1996-10-08 | Ciba-Geigy Corporation | Process for the preparation of hydroxyphenylcarboxylates |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3644482A (en) * | 1961-10-30 | 1972-02-22 | Geigy Ag J R | (4-hydroxy-5-alkylphenyl) alkanoic acid esters of polyols |
| CH454171A (de) | 1964-06-02 | 1968-04-15 | Geigy Ag J R | Verfahren zur Herstellung organischer Ester |
| CH471769A (de) * | 1965-08-02 | 1969-04-30 | Geigy Ag J R | Verfahren zur Herstellung von Hydroxyphenylfettsäureestern |
| US3435065A (en) * | 1965-10-22 | 1969-03-25 | Geigy Chem Corp | N,n,n',n' - (ethylenediamine) - tetrakis (ethylene - 3 - (3',5' - di-tert-butyl-4' - hydroxyphenyl)propionate) and compositions stabilized therewith |
| US4536593A (en) * | 1982-07-13 | 1985-08-20 | Ciba-Geigy Corporation | Process for the preparation of sterically hindered hydroxphenylcarboxylic acid esters |
| US4594444A (en) | 1983-12-22 | 1986-06-10 | Ciba-Geigy Corporation | Process for the preparation of sterically hindered hydroxyphenylcarboxylic acid esters |
| US5206414A (en) * | 1990-01-11 | 1993-04-27 | Ciba-Geigy Corporation | Process for the preparation of hydroxyphenylpropionic acid esters |
| US5136082A (en) | 1990-08-03 | 1992-08-04 | Himont Incorporated | Process for preparing organic esters and amides and catalyst system therefor |
| TW212790B (de) * | 1991-10-15 | 1993-09-11 | Ciba Geigy Ag | |
| EP0608089B1 (de) * | 1993-01-21 | 1997-10-15 | Sankyo Company Limited | Phenylalkansäureester und ihre Anwendung als Antioxydantien |
| EP0808818B1 (de) * | 1996-05-23 | 2000-09-20 | Ciba SC Holding AG | Verfahren zum Herstellen von substituierten Hydroxy-Hydrocinnamatestern |
| EP1209144A1 (de) | 1996-12-20 | 2002-05-29 | Ciba SC Holding AG | An feste Trägermaterialien fixierte Umesterungskatalysatoren |
-
2001
- 2001-06-14 DE DE50109215T patent/DE50109215D1/de not_active Expired - Lifetime
- 2001-06-14 CA CA2412062A patent/CA2412062C/en not_active Expired - Fee Related
- 2001-06-14 JP JP2002504205A patent/JP4925547B2/ja not_active Expired - Fee Related
- 2001-06-14 RU RU2003100511/04A patent/RU2272022C2/ru not_active IP Right Cessation
- 2001-06-14 BR BRPI0111876-5A patent/BR0111876B1/pt not_active IP Right Cessation
- 2001-06-14 CN CNB018115365A patent/CN1250513C/zh not_active Expired - Fee Related
- 2001-06-14 WO PCT/CH2001/000370 patent/WO2001098249A1/de active IP Right Grant
- 2001-06-14 AT AT01937911T patent/ATE320412T1/de not_active IP Right Cessation
- 2001-06-14 ES ES01937911T patent/ES2259325T3/es not_active Expired - Lifetime
- 2001-06-14 MX MXPA02012369A patent/MXPA02012369A/es active IP Right Grant
- 2001-06-14 AU AU2001263698A patent/AU2001263698A1/en not_active Abandoned
- 2001-06-14 CZ CZ20030108A patent/CZ301602B6/cs not_active IP Right Cessation
- 2001-06-14 EP EP01937911A patent/EP1292560B1/de not_active Expired - Lifetime
- 2001-06-14 KR KR1020027016517A patent/KR100813185B1/ko active IP Right Grant
- 2001-06-14 US US10/311,724 patent/US6878843B2/en not_active Expired - Lifetime
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4716244A (en) * | 1985-05-02 | 1987-12-29 | Ciba-Geigy Corporation | Process for the preparation of sterically hindered hydroxyphenylcarboxylic acid esters |
| US5563291A (en) * | 1994-03-11 | 1996-10-08 | Ciba-Geigy Corporation | Process for the preparation of hydroxyphenylcarboxylates |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005042463A1 (en) * | 2003-10-27 | 2005-05-12 | Great Lakes Chemical (Europe) Gmbh | Preparation of tetrakis [3-(e,5-di-tert-butyl-4-hydroxy phenyl) propionyl oxymethyl] methane |
| WO2008055832A2 (en) * | 2006-11-09 | 2008-05-15 | Ciba Holding Inc. | Process using liquid phenolic antioxidants |
| WO2008055832A3 (en) * | 2006-11-09 | 2008-06-26 | Ciba Holding Inc | Process using liquid phenolic antioxidants |
| WO2020152120A1 (en) | 2019-01-23 | 2020-07-30 | Basf Se | Oxime ester photoinitiators having a special aroyl chromophore |
| WO2021175855A1 (en) | 2020-03-04 | 2021-09-10 | Basf Se | Oxime ester photoinitiators |
Also Published As
| Publication number | Publication date |
|---|---|
| BR0111876B1 (pt) | 2012-09-18 |
| ES2259325T3 (es) | 2006-10-01 |
| AU2001263698A1 (en) | 2002-01-02 |
| ATE320412T1 (de) | 2006-04-15 |
| RU2272022C2 (ru) | 2006-03-20 |
| CZ2003108A3 (cs) | 2003-05-14 |
| CA2412062C (en) | 2011-09-20 |
| BR0111876A (pt) | 2003-06-24 |
| EP1292560B1 (de) | 2006-03-15 |
| EP1292560A1 (de) | 2003-03-19 |
| CA2412062A1 (en) | 2001-12-27 |
| CN1250513C (zh) | 2006-04-12 |
| US20030166962A1 (en) | 2003-09-04 |
| US6878843B2 (en) | 2005-04-12 |
| JP2004501128A (ja) | 2004-01-15 |
| MXPA02012369A (es) | 2003-04-25 |
| CN1437572A (zh) | 2003-08-20 |
| JP4925547B2 (ja) | 2012-04-25 |
| KR20030007861A (ko) | 2003-01-23 |
| CZ301602B6 (cs) | 2010-04-28 |
| DE50109215D1 (de) | 2006-05-11 |
| KR100813185B1 (ko) | 2008-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2486000B1 (de) | Verfahren zur herstellung von polyolestern | |
| DE69132892T2 (de) | Verfahren zur Herstellung von 4-Hydroxystyrol | |
| EP1292560B1 (de) | Verfahren zur herstellung von hydroxyphenylcarbonsäureestern | |
| WO2009003744A1 (de) | Verfahren zur herstellung von (meth)acrylaten | |
| WO2010003709A2 (de) | Verfahren zur herstellung von (meth)acrylsäureestern | |
| DE19940622C1 (de) | Verfahren zur Herstellung von Di(meth)acrylsäureestern | |
| EP0200685A2 (de) | Verfahren zur Herstellung von sterisch gehinderten Hydroxyphenylcarbonsäureestern | |
| EP3044200B1 (de) | Verfahren zur nachbehandlung von polyolestern | |
| DE2745982C2 (de) | Verfahren zur Herstellung von Phosphonsäuren und Phosphinsäuren | |
| DE2928944A1 (de) | Verbessertes verfahren zur herstellung von hoeheren ungesaettigten ketonen | |
| DE69804607T2 (de) | Verfahren zur herstellung und reinigung von komplexestern | |
| DE3325372A1 (de) | Verfahren zur herstellung von reinen adipinsaeuremonoestern | |
| DE2361402B2 (de) | Verfahren zur Herstellung von PoIyolestern von Carbonsäuren | |
| EP0245792B1 (de) | Verfahren zur Herstellung von Carbonsäureestern | |
| DE3013257C2 (de) | Verfahren zur Gewinnung von Essigsäure und/oder ihren Estern | |
| DE2410782C2 (de) | Phenylendiessigsäuredialkylester und Verfahren zu deren Herstellung | |
| EP0623579B1 (de) | Verfahren zur Herstellung von Ethercarbonsäuren | |
| EP0068282B2 (de) | Verfahren zur Herstellung von Bromhydrinen aus Polyolen | |
| EP0360087A2 (de) | Neue Estergruppen gebunden enthaltende Dihydroxyverbindungen, Verfahren zu ihrer Herstellung und ihre Verwendung | |
| DE112006002975B4 (de) | Verfahren zur Herstellung von Menthylbenzoat | |
| DE69405193T2 (de) | Verfahren zur Herstellung von Alkyl-3-phthalidylidenacetat | |
| DE1595628A1 (de) | Verbindungen mit zwei oder mehr Acetessigestergruppen | |
| DE2203813A1 (de) | Verfahren zur Herstellung von Carbonsäureestern | |
| DE875804C (de) | Verfahren zur Herstellung von Umwandlungsprodukten des Pentaerythrit-dichlorhydrin-monoschwefligsaeureesters | |
| DE1938460C (de) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ CZ DE DE DK DK DM DZ EC EE EE ES FI FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2001937911 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027016517 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2412062 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/012369 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2002 504205 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018115365 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2003-108 Country of ref document: CZ |
|
| ENP | Entry into the national phase |
Country of ref document: RU Kind code of ref document: A Format of ref document f/p: F Ref document number: 2003100511 Country of ref document: RU Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027016517 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001937911 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10311724 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2003-108 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001937911 Country of ref document: EP |