WO1994018182A1 - Morpholine derivative - Google Patents
Morpholine derivative Download PDFInfo
- Publication number
- WO1994018182A1 WO1994018182A1 PCT/JP1994/000186 JP9400186W WO9418182A1 WO 1994018182 A1 WO1994018182 A1 WO 1994018182A1 JP 9400186 W JP9400186 W JP 9400186W WO 9418182 A1 WO9418182 A1 WO 9418182A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- oxy
- compound
- acid
- group
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/30—1,4-Oxazines; Hydrogenated 1,4-oxazines not condensed with other rings
Definitions
- the present invention relates to a morpholine derivative having antidepressant activity and anxiolytic activity or a salt thereof.
- 5-HT serotonin
- Tricyclic compounds such as amitriptylin are widely used clinically as antipops.
- amine preparative Ripuchiri emissions 5 - HT re takes Write-harm or 5 - although having IIT 2 receptor antagonism, 5 one II T 2 except receptor deactivation antagonistic ⁇ to Norua Dorenari emissions reuptake inhibitory activity and anti Since it has a cholinergic effect and also exhibits a nonselective effect, it is thought to be a cause of adverse effects on the cardiovascular system (eg, increased palpitations) and adverse effects such as blood pressure and urinary retention.
- drugs that selectively inhibit 5-II— reuptake or selectively affect 5-— 9 receptivity are drugs with few side effects.
- fluoxetine for example, is a powerful drug used in clinical practice as a drug that selectively inhibits 5-II reuptake, and has an anesthetizing and insomnia effects in the course of its treatment ( [Phys ician 's Desk Reference, Medical Economics Company, Oradell, NJ (1990)].
- drugs and to e.g. Mi Ding synth Li down with selective 5 _ 11 T 2 receptor antagonism, as an antidepressant are known.
- trazodone An example of a drug having both a 5-HT reuptake inhibitory action and a 5-HT 2 receptor antagonistic action is trazodone.
- 5-HT re-uptake inhibitory activity of Trazodone is very weak, the antidepressant and anxiolytic effects 5 - HT 2 is rather report force with receptors based on antagonism is also of a [Marek GJ et al., Psychopharmacology, 109, 2-11 (1992)], and trazodone has both aforesaid effects and a ⁇ receptor affinity, so that side effects based on this are produced.
- the present invention has a structural feature in that a halogen atom is always substituted for a benzene ring in an indanyloxy group or an indenyloxy group, and is a morpholine derivative having a completely different chemical structure from the background art. is there. Further, the present invention has both selective 5_HT reuptake inhibition and selective 5-HT 2 receptor antagonism, both of which have excellent effects.
- An object of the present invention is to provide a novel morpholine derivative represented by the following general formula (I) or a salt thereof.
- Another object of the present invention is to provide a compound comprising the compound (I) of the present invention or a salt thereof as an active ingredient, which has both a selective 5-HT reuptake inhibitory action and a selective 5-HT9 receptor antagonistic action, and a compound of the present invention. () Or that And a pharmaceutically acceptable carrier.
- the present invention also encompasses the invention of the compound (I) of the present invention represented by the following general formula (II) or a salt thereof, and also has an object to provide the compound (II).
- R R3 same or different hydrogen atom or lower alkyl group
- R 2 halogen atom
- the dashed line can form a double bond. Or a salt thereof.
- R 1, R 3 same or different, hydrogen atom or lower alkyl group
- R2 a halogen atom or a salt thereof.
- the compound of the present invention is further described as follows. Unless otherwise specified in the definition of general formulas herein, the term “lower” means a straight or branched carbon chain having 1 to 6 carbon atoms.
- lower alkyl group specifically, for example, a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, an isobutyl group, a sec group 1-butyl group, tert-butyl group, pentyl group, isopentyl group, neopentyl group, tert-pentyl group.
- lower alkyl groups having 1 to 3 carbon atoms such as methyl, ethyl, propyl, and isopropyl.
- halogen atom examples include a fluorine atom, a chlorine atom, a bromine atom and a iodine atom, and a fluorine atom is preferable.
- the dotted line can form a double bond in the indane ring.
- the present compound may have stereoisomers such as optical isomers and geometric isomers based on the asymmetric carbon atom XN depending on the type of the 2-position or the substituent of the morphonyl group.
- the compounds (I) and (II) or a salt thereof of the present invention is a levorotatory optical isomer.
- the compounds (I) and (H) of the present invention can form salts with acids.
- Such salts include inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, mineral acids with sulfuric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid and malonic acid.
- Acid succinic acid, fumaric acid, maleic acid, lactic acid, lingic acid, citric acid, tartaric acid, D-dibenzoyltartaric acid, carbonic acid, picric acid, methanesulfonic acid, ethanesulfonic acid, glutamic acid, etc.
- the acid addition salts with organic acids can be mentioned strongly, preferably the hydrochloride, oxalate, fumarate and D-dibenzoyl tartrate.
- the compounds of the present invention can also form hydrates, various solvates and polymorphs.
- the present compound can be produced by the method represented by the following reaction formula, and does not limit the production method of the present invention.
- R 1 is a hydrogen atom, a lower alkyl group or a ketone group.
- X is a protecting group for an amino group
- Y is a halogen atom, a mesyloxy group or a A siloxy group.
- the protecting group for the amino group is a commonly used protecting group, for example, a trityl group, a benzhydryl group, a p-methoxybenzyl group or a tert-butyl group, preferably a trityl group. is there.
- the above reaction is carried out by reacting a corresponding amount of the morpholine compound (III) and the ingole compound (IV) with stirring in an inert solvent in the presence of a base at room temperature or at elevated temperature, or After converting compound (IV) into a sodium salt or a potassium salt in advance, the compound is subjected to morpholinic compound (III) in an inert solvent at room temperature or with heating (step 1), and deprotection is carried out by a conventional method.
- the compound of the present invention (V) can be obtained by a reaction, for example, a reduction reaction such as catalytic reduction or liquor reduction or treatment with an acid (second step).
- Examples of the inert solvent used in the first step include benzene, culoform form, dimethylformamide (hereinafter referred to as DMF), dimethyl sulfoxide (hereinafter referred to as DMSO), ether, water, methanol and ethanol.
- Examples of the base in the first step include sodium hydroxide, potassium hydroxide, sodium hydride, potassium hydride, lithium hydride, potassium carbonate, and sodium carbonate. And butyllithium and potassium tert-butoxide.
- As the acid in the second step for example, acetic acid, trifluoroacetic acid, trichloroacetic acid, hydrochloric acid, sulfuric acid, hydrobromic acid-acetic acid and the like are used. This deprotection reaction is usually carried out in an inert solvent such as methanol, ethanol, or acetate or in water at room temperature or under heating (under reflux).
- the compound (X) of the present invention thus produced is prepared from hydrochloric acid, fumaric acid, Can form acids and salts with acids, succinic acid, bromic acid, D-dibenzoyl, tartaric acid, etc.
- the optically active morpholine compound In the first process or the second process, the optically active morpholine compound
- Compounds obtained by the first to fifth processes are acetonitrile monohydrate, methanol, using D-dibenzoyltartaric acid, D-ditoluoyltartaric acid, D-tartaric acid, etc. as resolving agents.
- Optical purity can be increased by recrystallization using dimethylformamide, dimethylformamide, or the like as a reconstituting solvent.
- R 4 represents a lower alkyl group in the above R 3.
- Z is a halogen atom, an arylsulfonyloxy group, a lower alkylsulfonyloxy group. Or an alkyl sulfate group.
- the above arylsulfonyloxy group includes phenylsulfonyloxy group and p-toluenesulfonyloxy group
- the lower alkyl sulfonyloxy group includes methylsulfonyloxy group and ethylsulfonyl group.
- the compound of the present invention can be obtained by a conventional N-alkylation reaction.
- N-alkylation reaction unsubstituted formolino compound (X) and its corresponding amount of alkylating agent (XI) are reacted with acetone, acetonitrile, and tetrahydrofuran (hereinafter referred to as THF).
- THF tetrahydrofuran
- the reaction is carried out at room temperature or under heating (or heating under reflux) in the presence of a base such as potassium carbonate, sodium hydride, or potassium hydride in an inert solvent such as sodium, ether or DMF.
- the following method can be used as another method for the N-alkylation reaction.
- the unsubstituted morpholino compound (X) and the corresponding amount of the lower alkyl aldehyde, sodium borohydride, sodium borohydride, sodium borohydride or sodium cyanoborohydride were reacted.
- the reaction is carried out in an inert solvent such as methanol, ethanol, THF, dioxane or the like in the presence of a rim or the like with stirring at room temperature or with heating.
- R2 means a lower alkyl group.
- the lower alkyl group for R 5 is as described above, and is preferably a methyl group or an ethyl group.
- the indene compound (XVII) of the present invention can be obtained through the following steps. First step
- the first step is a conventional reduction reaction, and the following typical reactions are exemplified.
- Ketone compound (XIII) and its corresponding reducing agent for example, borane, alan, sodium tetrahydroborate, lithium tetrahydroboride
- reducing agent for example, borane, alan, sodium tetrahydroborate, lithium tetrahydroboride
- Sodium tetrahydroammonium sodium triacetoxyborohydride, aluminum diisobutyl hydride or sodium borohydride with lower alcohols (eg, methanol) Water, ethanol, propanol, etc.), ethyl ether, THF, benzene, toluene, dichloroethane, chloroform, or water in an inert solvent to obtain the hydroxy compound (XIV) under cooling or heating to reflux.
- Method B catalytic reduction: The ketone compound (XI II) and a metal catalyst (for example, Raney nickel, nickel, or copper tetroxide) are treated with methanol or ethanol. The reaction is carried out while stirring at room temperature or in an alcohol.
- a metal catalyst for example, Raney nickel, nickel, or copper tetroxide
- Method C A ketone compound (XI II) and a corresponding amount of dithionite (eg, sodium dithionite) are reacted with DMF-water, THF-water, dioxane-monohydrate, The reaction is carried out while cooling or stirring at room temperature in a mixed solvent such as ethanol-water or ethanol water.
- dithionite eg, sodium dithionite
- This reaction converts the hydroxy compound (XIV) to an acid such as hydrochloric acid, acetic acid, trifluoroacetic acid, trichloroacetic acid, sulfuric acid, or hydrobromic acid-acetic acid in alcohol (XV) such as methanol or ethanol.
- the reaction is carried out at room temperature or with stirring under heating.
- the alkoxy compound (XVI) obtained in the second step is treated with an acid catalyst (eg, hydrochloric acid, sulfuric acid, phosphoric acid, p-toluenesulfonic acid or p-toluenesulfonic acid) in an inert solvent such as toluene, benzene, THF or 1,4-dioxane.
- an acid catalyst eg, hydrochloric acid, sulfuric acid, phosphoric acid, p-toluenesulfonic acid or p-toluenesulfonic acid
- an inert solvent such as toluene, benzene, THF or 1,4-dioxane.
- This production method provides the compound (XIX) of the present invention in which a methyl group is substituted at the 3-position of the indane ring.
- a ketone compound (XIII) and a corresponding amount of a methylating agent eg, methyllithium, Grignard reagent, methylgenogen, trimethylaluminum, methyl copper or dimethyl copper
- a methylating agent eg, methyllithium, Grignard reagent, methylgenogen, trimethylaluminum, methyl copper or dimethyl copper
- Is carried out in an anhydrous solvent such as THF, dimethyl ether, benzene, toluene, dichloromethane or 1,4-dioxane under cooling to room temperature.
- a suitable reaction is to react the ketone compound (XIII) with its corresponding amount of methyllithium or magnesium methyl bromide in THF or getyl ether under cooling to room temperature, for example, at ⁇ 78 ° C. to 0 ° C. It is performed without stirring.
- Second step (reduction reaction) Method A Hydroxymethyl compound (XVIII) obtained in the first step is subjected to a conventional catalytic reduction method, typically in the presence of a metal catalyst (for example, palladium carbon, dihydroxypalladium or platinum dioxide). Hydrogen in an inert solvent such as alcohol (eg, methanol, ethanol or propanol), THF, 1,4-dioxane, gechelether, ethyl acetate, benzene, toluene or dichloromethane. It is carried out while cooling under an atmosphere or stirring at room temperature.
- alcohol eg, methanol, ethanol or propanol
- THF 1,4-dioxane
- gechelether 1,4-dioxane
- benzene toluene or dichloromethane
- Method B Hydroxymethyl compound (XVIII) obtained in the first step and a corresponding amount of trialkylsilane (for example, trimethylsilane or triethylsilane) are reacted with acetic acid, trifluoroacetic acid, hydrochloric acid, In the presence of an acid such as hydrobromic acid, sulfuric acid or phosphoric acid, in an inert solvent such as methanol, ethanol, THF, dioxane, ether or acetonitrile under ice cooling to room temperature This is carried out with stirring under the water.
- trialkylsilane for example, trimethylsilane or triethylsilane
- the compound of the present invention extremely selectively inhibits 5-HT reuptake and also selectively has an antagonistic effect on 5-HT2 receptor, it exhibits depression, anxiety, psychosomatic disorders, and autonomic nervous system.
- the compound of the present invention has an antireserpine action, a blood viscosity improving action, an antihypoxic action, and an antioxidant action, and is also useful as a cerebral circulation / metabolism improving drug, a brain function improving drug, and as an analgesic. Is also useful. Furthermore, the compounds of the present invention can also be used for improving dementia in Alzheimer's disease with cerebral dysfunction.
- the 5-HT reuptake inhibitory activity was tested by examining how the test compound inhibited the binding of [ 3 H] -citalopram to the 5-HT reuptake site.
- 5-5-HT a precursor of 5-HT
- Male ICR mice weighing 30-40 g were used.
- the test drug was intraperitoneally administered, and 30 minutes later, 90 mg / kg of 1-5 hydroxyhydrazine phantom was administered intravenously, and observation was performed for 5 minutes after 5 minutes.
- the observation items were tremor, swinging behavior, and hindlimb abduction. Evaluation of the test drug, and have use the ED 5Q value required for the expression of each action ⁇ Tsuta.
- the compounds of the present invention 5 an in vitro test - showed by both of an HT 2 receptor antagonism and both act both excellent effect - HT re takes narrowing inhibitory activity and 5.
- the control compound has 5-HT reuptake inhibitory activity, but 5-HT 2 receptor antagonism is extremely weak (see Table 1 below). table 1
- Control compound 1 2 — [[(4—Indanyl) oxy] methyl] morpholine (compound described in JP-A-46-73333)
- Control compound 2 2 — [[(7— Indenyl) oxy] methyl] morpholine (compound described in JP-A-52-87373)
- 5-inhibition of reuptake of HT In an in vivo test, for example, each action of the compound of Example 4 The ED 5G value required for expression is 6. 3 mg / kg, swinging behavior was 7.2 mgZkg, and hindlimb abduction was 14.1 mgZkg, indicating a strong inhibitory effect on 5-HT reuptake.
- a preparation containing one or more of the compounds of the present invention or a salt thereof as an active ingredient is prepared by using a carrier (form) commonly used for production and other additives. You.
- the suspension or shaping of the production river can be either solid or liquid, such as milk, magnesium stearate, starch, talc, gelatin, agar, pectin, gum arabic, and orange. Oil, sesame oil, potato oil, ethylene glycol, and other commonly used oils.
- Administration can be in the form of tablets, pills, capsules, granules, powders, liquids, etc., or intravenous injections, such as intravenous or intramuscular injections, or suppositories, transdermals, etc. You may.
- the dosage varies depending on age, resting weight, symptom treatment effect, administration method, treatment time, etc., but is usually in the range of 1 to 100 mg, preferably 10 to 300 mg per adult per day. 1 It is orally administered once or several times. Of course, as described above, the dosage varies under various conditions, so that a smaller amount than the above dosage range may be sufficient.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Steroid Compounds (AREA)
Description












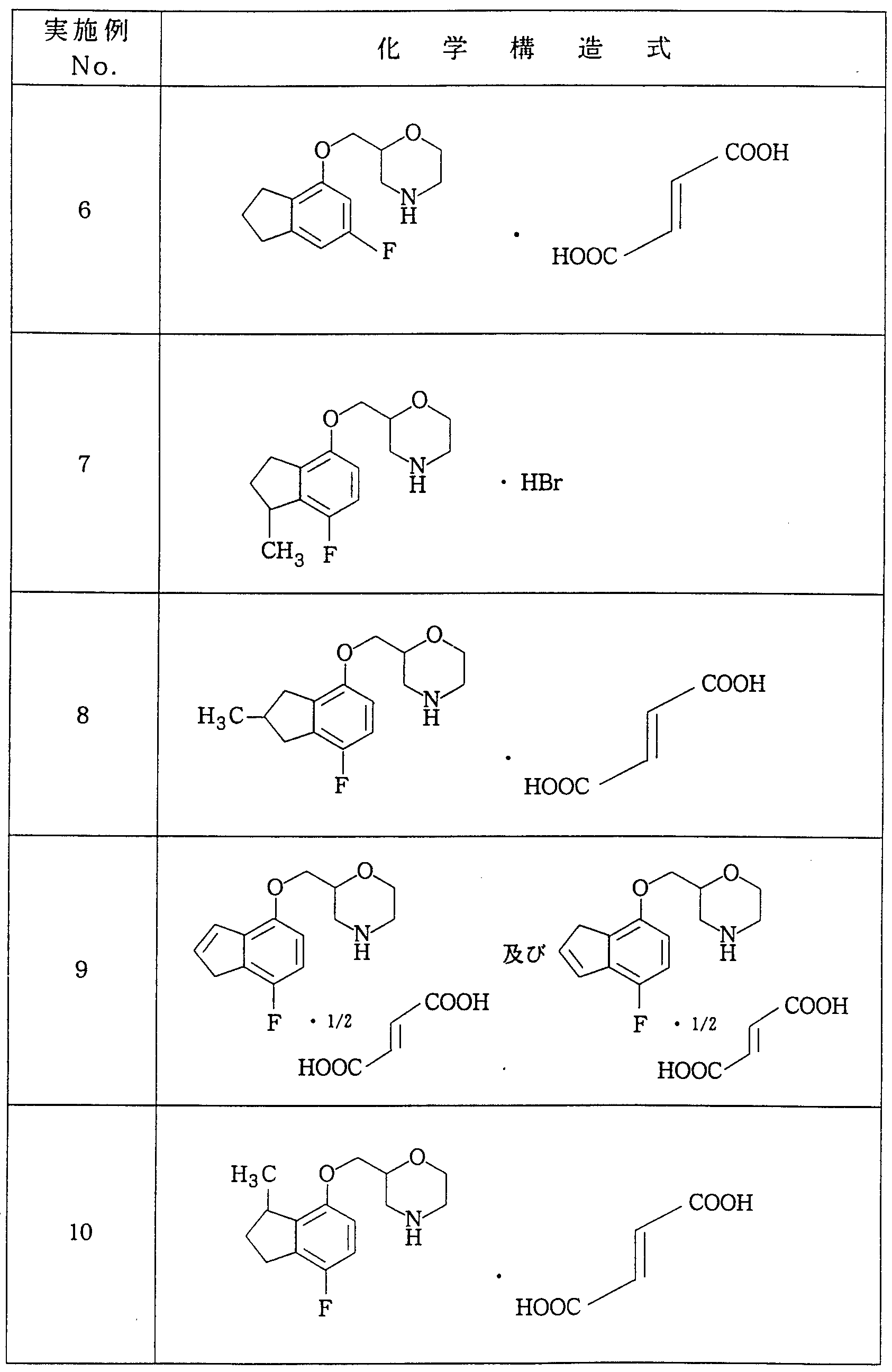

Claims




Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69422021T DE69422021T2 (de) | 1993-02-10 | 1994-02-08 | Morpholinderivate |
| US08/495,619 US5521180A (en) | 1993-02-10 | 1994-02-08 | Morpholine derivative |
| DK94905865T DK0684237T3 (da) | 1993-02-10 | 1994-02-08 | Morpholinderivater |
| UA95083737A UA41350C2 (uk) | 1993-02-10 | 1994-02-08 | Похідна морфоліну або її фармацевтично прийнятна сіль та фармацевтична композиція на її основі |
| AU59800/94A AU670675B2 (en) | 1993-02-10 | 1994-02-08 | Morpholine derivative |
| KR1019950703293A KR100302819B1 (ko) | 1993-02-10 | 1994-02-08 | 모르폴린유도체및이를포함하는약제 |
| HU9502359A HU220383B (hu) | 1993-02-10 | 1994-02-08 | Morfolinszármazékok és az ezeket tartalmazó gyógyászati készítmények |
| EP94905865A EP0684237B1 (en) | 1993-02-10 | 1994-02-08 | Morpholine derivative |
| NO953118A NO305170B1 (no) | 1993-02-10 | 1995-08-09 | Morfolinderivat |
| FI953793A FI953793A0 (fi) | 1993-02-10 | 1995-08-10 | Morfoliinijohdannainen |
| GR20000400523T GR3032816T3 (en) | 1993-02-10 | 2000-02-29 | Morpholine derivative. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP5/45691 | 1993-02-10 | ||
| JP4569193 | 1993-02-10 | ||
| JP5/240147 | 1993-09-27 | ||
| JP24014793 | 1993-09-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994018182A1 true WO1994018182A1 (en) | 1994-08-18 |
Family
ID=26385743
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1994/000186 WO1994018182A1 (en) | 1993-02-10 | 1994-02-08 | Morpholine derivative |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US5521180A (ja) |
| EP (1) | EP0684237B1 (ja) |
| KR (1) | KR100302819B1 (ja) |
| CN (1) | CN1042028C (ja) |
| AT (1) | ATE187448T1 (ja) |
| AU (1) | AU670675B2 (ja) |
| CA (1) | CA2155183A1 (ja) |
| DE (1) | DE69422021T2 (ja) |
| DK (1) | DK0684237T3 (ja) |
| ES (1) | ES2142397T3 (ja) |
| FI (1) | FI953793A0 (ja) |
| GR (1) | GR3032816T3 (ja) |
| HU (1) | HU220383B (ja) |
| NO (1) | NO305170B1 (ja) |
| PT (1) | PT684237E (ja) |
| TW (1) | TW397829B (ja) |
| UA (1) | UA41350C2 (ja) |
| WO (1) | WO1994018182A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003529563A (ja) * | 2000-02-22 | 2003-10-07 | セプラコア インコーポレーテッド | ビュープロピオン代謝産物並びにその合成および使用方法 |
| WO2006006617A1 (ja) * | 2004-07-14 | 2006-01-19 | Astellas Pharma Inc. | 中枢神経疾患発症後の機能障害の回復促進剤 |
| WO2009128057A2 (en) | 2008-04-18 | 2009-10-22 | UNIVERSITY COLLEGE DUBLIN, NATIONAL UNIVERSITY OF IRELAND, DUBLIN et al | Psycho-pharmaceuticals |
| KR101165996B1 (ko) | 2010-04-23 | 2012-07-18 | 주식회사 녹십자 | 프탈라지논 유도체, 이의 제조방법 및 이를 함유하는 약학적 조성물 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA77650C2 (en) * | 1999-12-06 | 2007-01-15 | Lundbeck & Co As H | Use of serotonin reuptake inhibitor in combination with deramcyclane |
| AU9087301A (en) * | 2000-09-11 | 2002-03-26 | Sepracor Inc | Ligands for monoamine receptors and transporters, and methods of use thereof |
| US7294637B2 (en) * | 2000-09-11 | 2007-11-13 | Sepracor, Inc. | Method of treating addiction or dependence using a ligand for a monamine receptor or transporter |
| JP2005034165A (ja) * | 2002-06-26 | 2005-02-10 | Mu-Hyun Cho | マット一体型のストーンベッド |
| TW200846003A (en) * | 2007-03-15 | 2008-12-01 | Astellas Pharma Inc | Novel prophylactic and/or therapeutic agent for diabetic neuropathy |
| TW200846002A (en) | 2007-03-15 | 2008-12-01 | Astellas Pharma Inc | Novel prophylactic and/or therapeutic agent for diabetic neuropathy |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5283773A (en) * | 1976-01-01 | 1977-07-12 | Yamanouchi Pharmaceut Co Ltd | Novel indene derivative |
| JPS52111580A (en) * | 1976-03-15 | 1977-09-19 | Yamanouchi Pharmaceut Co Ltd | Novel indene derivatives and salts thereof |
| JPS55113717A (en) * | 1979-02-24 | 1980-09-02 | Yamanouchi Pharmaceut Co Ltd | Antidepressive drug |
| JPS61145119A (ja) * | 1984-12-19 | 1986-07-02 | Yamanouchi Pharmaceut Co Ltd | 脳障害による精神症状の改善,治療剤 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1138405A (en) * | 1966-12-28 | 1969-01-01 | Ici Ltd | Morpholine derivatives |
| AU5136385A (en) * | 1984-12-20 | 1986-07-17 | Medicament P.F | 2-arylalkyloxymethyl-morpholine derivatives |
| GB2177085A (en) * | 1985-06-18 | 1987-01-14 | Ici Plc | Fungicidal compounds and compositions |
-
1994
- 1994-02-08 EP EP94905865A patent/EP0684237B1/en not_active Expired - Lifetime
- 1994-02-08 PT PT94905865T patent/PT684237E/pt unknown
- 1994-02-08 US US08/495,619 patent/US5521180A/en not_active Expired - Lifetime
- 1994-02-08 AT AT94905865T patent/ATE187448T1/de not_active IP Right Cessation
- 1994-02-08 WO PCT/JP1994/000186 patent/WO1994018182A1/ja active IP Right Grant
- 1994-02-08 DK DK94905865T patent/DK0684237T3/da active
- 1994-02-08 CN CN94191150A patent/CN1042028C/zh not_active Expired - Fee Related
- 1994-02-08 UA UA95083737A patent/UA41350C2/uk unknown
- 1994-02-08 DE DE69422021T patent/DE69422021T2/de not_active Expired - Lifetime
- 1994-02-08 HU HU9502359A patent/HU220383B/hu not_active IP Right Cessation
- 1994-02-08 CA CA002155183A patent/CA2155183A1/en not_active Abandoned
- 1994-02-08 AU AU59800/94A patent/AU670675B2/en not_active Ceased
- 1994-02-08 KR KR1019950703293A patent/KR100302819B1/ko not_active IP Right Cessation
- 1994-02-08 ES ES94905865T patent/ES2142397T3/es not_active Expired - Lifetime
- 1994-02-14 TW TW083101126A patent/TW397829B/zh not_active IP Right Cessation
-
1995
- 1995-08-09 NO NO953118A patent/NO305170B1/no not_active IP Right Cessation
- 1995-08-10 FI FI953793A patent/FI953793A0/fi unknown
-
2000
- 2000-02-29 GR GR20000400523T patent/GR3032816T3/el not_active IP Right Cessation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5283773A (en) * | 1976-01-01 | 1977-07-12 | Yamanouchi Pharmaceut Co Ltd | Novel indene derivative |
| JPS52111580A (en) * | 1976-03-15 | 1977-09-19 | Yamanouchi Pharmaceut Co Ltd | Novel indene derivatives and salts thereof |
| JPS55113717A (en) * | 1979-02-24 | 1980-09-02 | Yamanouchi Pharmaceut Co Ltd | Antidepressive drug |
| JPS61145119A (ja) * | 1984-12-19 | 1986-07-02 | Yamanouchi Pharmaceut Co Ltd | 脳障害による精神症状の改善,治療剤 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003529563A (ja) * | 2000-02-22 | 2003-10-07 | セプラコア インコーポレーテッド | ビュープロピオン代謝産物並びにその合成および使用方法 |
| WO2006006617A1 (ja) * | 2004-07-14 | 2006-01-19 | Astellas Pharma Inc. | 中枢神経疾患発症後の機能障害の回復促進剤 |
| WO2009128057A2 (en) | 2008-04-18 | 2009-10-22 | UNIVERSITY COLLEGE DUBLIN, NATIONAL UNIVERSITY OF IRELAND, DUBLIN et al | Psycho-pharmaceuticals |
| KR101165996B1 (ko) | 2010-04-23 | 2012-07-18 | 주식회사 녹십자 | 프탈라지논 유도체, 이의 제조방법 및 이를 함유하는 약학적 조성물 |
Also Published As
| Publication number | Publication date |
|---|---|
| NO953118L (no) | 1995-10-10 |
| DK0684237T3 (da) | 2000-05-08 |
| KR100302819B1 (ko) | 2001-11-22 |
| CN1117729A (zh) | 1996-02-28 |
| ATE187448T1 (de) | 1999-12-15 |
| AU5980094A (en) | 1994-08-29 |
| EP0684237A4 (en) | 1996-02-14 |
| HU220383B (hu) | 2002-01-28 |
| EP0684237B1 (en) | 1999-12-08 |
| DE69422021T2 (de) | 2000-07-13 |
| NO953118D0 (no) | 1995-08-09 |
| DE69422021D1 (en) | 2000-01-13 |
| US5521180A (en) | 1996-05-28 |
| HUT72674A (en) | 1996-05-28 |
| NO305170B1 (no) | 1999-04-12 |
| FI953793A (fi) | 1995-08-10 |
| EP0684237A1 (en) | 1995-11-29 |
| TW397829B (en) | 2000-07-11 |
| UA41350C2 (uk) | 2001-09-17 |
| PT684237E (pt) | 2000-04-28 |
| HU9502359D0 (en) | 1995-10-30 |
| AU670675B2 (en) | 1996-07-25 |
| FI953793A0 (fi) | 1995-08-10 |
| CN1042028C (zh) | 1999-02-10 |
| GR3032816T3 (en) | 2000-06-30 |
| ES2142397T3 (es) | 2000-04-16 |
| CA2155183A1 (en) | 1994-08-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0912552B1 (en) | 2,4-diaminopyrimidine derivates as dopamine d4 receptor antagonists | |
| RU2265013C2 (ru) | 3-замещенные 4-(фенил-n-алкил)-пиперидины, фармацевтическая композиция на их основе и способы лечения | |
| KR101364365B1 (ko) | 트랜스-1-((1r,3s)-6-클로로-3-페닐인단-1-일)-3,3-디메틸피페라진의 제조 방법 | |
| JP2000505795A (ja) | 置換シクロアザンから誘導された新規芳香族ピペラジン、それらの調製法、医薬組成物、および薬剤としての使用 | |
| EP0099148A1 (en) | Piperazine derivatives, processes for their preparation and pharmaceutical preparations containing them | |
| WO1994018182A1 (en) | Morpholine derivative | |
| TW593290B (en) | Alkylaminobenzothiazole and -benzoxazole derivatives | |
| KR101386387B1 (ko) | 트랜스-1-((1r,3s)-6-클로로-3-페닐인단-1-일)-3,3-디메틸피페라진의 결정성 염기 | |
| AU644081B2 (en) | Novel 8-substituted-2-aminotetralines | |
| JP2008530039A (ja) | トランス−1−((1r,3s)−6−クロロ−3−フェニルインダン−1−イル)−3,3−ジメチルピペラジンの酒石酸塩およびリンゴ酸塩 | |
| CA1335289C (en) | Piperidinyl benzisoxazole derivatives, their production and pharmaceutical use | |
| CN101014573A (zh) | 5-ht7受体拮抗剂 | |
| JPS63502348A (ja) | 縮合ベンゾアゼピン | |
| JPH0747585B2 (ja) | イソオキサゾール誘導体 | |
| JPH0225481A (ja) | 2―[(4―ピペリジニル)メチル]―1,2,3,4―テトラヒドロイソキノリン誘導体、その製造方法およびその治療への適用 | |
| JP2618609B2 (ja) | モルホリン誘導体 | |
| KR20150065190A (ko) | 피라진 유도체 | |
| JPH0940646A (ja) | ベンゼン縮合環誘導体又はその塩 | |
| Michaelides et al. | Synthesis and pharmacological evaluation of 1-(aminomethyl)-3, 4-dihydro-5-hydroxy-1H-2-benzopyrans as dopamine D1 selective ligands | |
| JP2007513197A6 (ja) | 統合失調症の治療のための複素環式置換インダン誘導体および関連化合物 | |
| JP2007513197A (ja) | 統合失調症の治療のための複素環式置換インダン誘導体および関連化合物 | |
| RU2123496C1 (ru) | Производное морфолина или его фармацевтически приемлемая соль и фармацевтическая композиция на его основе | |
| MX2007007893A (es) | Antagonistas del receptor 5-ht7. | |
| KR20070107043A (ko) | 트랜스-1-((1r,3s)-6-클로로-3-페닐인단-1-일)-3,3-디메틸피페라진의 타르트레이트 및 말레이트 염 | |
| JPH0386876A (ja) | ベンゾイソキノリン誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CN CZ FI HU JP KR KZ LK LV MG MN MW NO NZ PL PT RO RU SD SK UA US UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2155183 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08495619 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994905865 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 953793 Country of ref document: FI |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994905865 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1994905865 Country of ref document: EP |