RU2136662C1 - Производные индола и их фармацевтически приемлемые кислотно-аддитивные соли, фармацевтический препарат на их основе - Google Patents
Производные индола и их фармацевтически приемлемые кислотно-аддитивные соли, фармацевтический препарат на их основе Download PDFInfo
- Publication number
- RU2136662C1 RU2136662C1 RU94037964A RU94037964A RU2136662C1 RU 2136662 C1 RU2136662 C1 RU 2136662C1 RU 94037964 A RU94037964 A RU 94037964A RU 94037964 A RU94037964 A RU 94037964A RU 2136662 C1 RU2136662 C1 RU 2136662C1
- Authority
- RU
- Russia
- Prior art keywords
- mmol
- solution
- stirred
- ether
- residue
- Prior art date
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/32—Oxygen atoms
- C07D209/36—Oxygen atoms in position 3, e.g. adrenochrome
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Pain & Pain Management (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Hospice & Palliative Care (AREA)
- Anesthesiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Indole Compounds (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Organic Insulating Materials (AREA)
- Materials For Medical Uses (AREA)
Abstract
Изобретение относится к новым соединениям общей формулы I, где R1-R4 - водород, галоген, низший алкил или трифторметил, R5 и R6 - водород, галоген, низший алкил, трифторметил или низший алкокси и R7 - низший алкил, а также к фармацевтически приемлемым аддитивным солям кислот соединений формулы I. Эти соединения эффективны прежде всего при лечении и предупреждении расстройств центральной нервной системы: депрессии, биполярные нарушения, состояние страха, нарушения сна и расстройства в сексуальной сфере, психозы, шизофрения, мигрени и другие болезненные состояния, связанные с головными болями или болями другого типа, расстройства, связанные с личными переживаниями или навязчивыми идеями, социальная фобия или приступы паники, психические органические расстройства, нарушения психики в детском возрасте, агрессивность, нарушения памяти, связанные с возрастом, нарушения отношений функций нервной системы, вызванных травмами, кровоизлиянием в мозг, заболеваниями нервно-дегенеративного характера и т.д.; нарушений в сердечно-сосудистой системе: высокое кровяное давление, тромбоз, инсульт и т.д.; желудочно-кишечных расстройств: дисфункция моторики желудочно-кишечного тракта. 2 с.п. и 5 з.п.ф-лы, 2 табл.
Description
Настоящее изобретение относится к производным индола. В частности оно относится к производным индола общей формулы

где R1-R4 - водовод, галоген, низший алкил, или трифторметил; R5 и R6 - водород, галоген, низший алкил, трифторметил или низший алкокси и R7 - низший алкил,
а также к их фармацевтически приемлемым кислотно-аддитивным солям.
где R1-R4 - водовод, галоген, низший алкил, или трифторметил; R5 и R6 - водород, галоген, низший алкил, трифторметил или низший алкокси и R7 - низший алкил,
а также к их фармацевтически приемлемым кислотно-аддитивным солям.
Кроме вышеперечисленных обозначений для R1-R4, R5, R6 и R7 указанные радикалы могут дополнительно иметь следующие обозначения: R1-R4- циклоалкил, R5 и R6 - циклоалкил, гидрокси, R7 - водород.
Эти соединения и соли являются новыми и отличаются ценными терапевтическими свойствами.
Они эффективны прежде всего при лечении и предупреждении расстройств центральной нервной системы, таких как депрессии, биполярные нарушения, состояние страха, нарушения сна и расстройства в сексуальной сфере, психозы, шизофрения, мигрени и другие болезненные состояния, связанные с головными болями или болями другого типа, расстройства, связанные с личными переживаниями или навязчивыми идеями, социальная фобия или приступы паники, психические органические расстройства, нарушения психики в детском возрасте, агрессивность, нарушения памяти, связанные с возрастом, нарушения отношений с окружающими, болезненная страсть, ожирение, булимия и т.п.; нарушений функций нервной системы, вызванных травмами, кровоизлиянием в мозг, заболеваниями нервно-дегенеративного характера и т.д.; нарушений в сердечно-сосудистой системе: высокое кровяное давление, тромбоз, инсульт и т.д.; желудочно-кишечных расстройств: дисфункция моторики желудочно-кишечного тракта.
Объектом настоящего изобретения являются соединения формулы I и их фармацевтически приемлемые кислотно-аддитивные как таковые и в качестве фармацевтических активных веществ. Кроме того, описывается получение этих соединений и солей. Предлагаются также фармацевтические препараты, содержащие одно из соединений формулы I или его фармацевтически приемлемую кислотно-аддитивную соль. Описывается также изготовление таких препаратов и применение соединений формулы I и их фармацевтически приемлемых солей для борьбы или предупреждения болезней, прежде всего болезней и расстройств упомянутого выше типа, а также для изготовления соответствующих лекарственных средств.
Соединения общей формулы
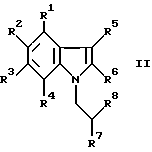
где R1-R7 имеют указанные выше значения, a R8 - переводимый в аминогруппу радикал, уходящая группа или гидроксильная группа,
представляют собой важные промежуточные продукты для получения фармацевтически ценных соединений общей формулы I.
где R1-R7 имеют указанные выше значения, a R8 - переводимый в аминогруппу радикал, уходящая группа или гидроксильная группа,
представляют собой важные промежуточные продукты для получения фармацевтически ценных соединений общей формулы I.
Употребляемое в описании заявки определение "низший" обозначает радикалы с числом атомов углерода максимум 7, предпочтительно до 4. "Алкил" обозначает линейные либо разветвленные насыщенные углеводородные радикалы, такие, как метил, этил, изопропил или трет.-бутил. "Алкокси" обозначает связанную через атом кислорода алкильную группу, такую, как метокси, этокси, пропокси, изопропокси или бутокси. "Галоген" может обозначать Cl, Br, F или J. Понятие "фармацевтически приемлемые кислотно-аддитивные соли" включает в себя соли неорганических и органических кислот, таких, как соляная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфо-кислота, п-толуолсульфокислота и т.п.
R7 может обозначать предпочтительно метил. Также предпочтительны соединения, в которых R7 - водород. Если R7 - метил, то особенно предпочтительными являются соединения, где R5 и R6 - водород.
Далее предпочтительны соединения, в которых R1 - водород или метил, R2 - водород или фтор, R3 -водород или хлор и R4 - водород.
Особенно предпочтительным является соединение, представляющее собой (S)-2-(6-xлop-5-фтopиндoл-1-ил)-1-метилэтилaмин.
Примерами других особенно предпочтительных соединений являются:
(RS) -2-(5-хлориндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(RS)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(RS)-2-(4-метилиндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-броминдол-1-ил)-1-метилэтиламин,
(RS)-2-(6-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(5,6-дифтopиндoл-1-ил)-1-метилэтилaмин,
(R)-2-(5,6-дифториндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-4-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-6-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин.
(RS) -2-(5-хлориндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(RS)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(RS)-2-(4-метилиндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-броминдол-1-ил)-1-метилэтиламин,
(RS)-2-(6-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(5,6-дифтopиндoл-1-ил)-1-метилэтилaмин,
(R)-2-(5,6-дифториндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-4-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-6-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин.
Некоторыми другими указанными в рамках данного изобретения представителями класса веществ согласно формуле 1 являются следующие:
2-(5-фториндол-1-ил)-этиламин
2-(6-хлор-5-фтор-индол-1-ил)-этиламин
2-(4-метил-3-метоксииндол-1-ил)-этиламин
2-(4-хлор-5-фтор-3-метоксииндол-1-ил)-этиламин,
2-(5-фтор-З-метоксииндол-1-ил)-этиламин,
2-(5-хлориндол-1-ил)-этиламин,
2-(4-хлор-5-фториндол-1-ил)-этиламин.
2-(5-фториндол-1-ил)-этиламин
2-(6-хлор-5-фтор-индол-1-ил)-этиламин
2-(4-метил-3-метоксииндол-1-ил)-этиламин
2-(4-хлор-5-фтор-3-метоксииндол-1-ил)-этиламин,
2-(5-фтор-З-метоксииндол-1-ил)-этиламин,
2-(5-хлориндол-1-ил)-этиламин,
2-(4-хлор-5-фториндол-1-ил)-этиламин.
Соединения общей формулы I, равно как и их фармацевтически приемлемые кислотно-аддитивные соли могут быть получены согласно изобретению следующим путем:
а) соединение общей формулы

где R1-R7 имеют указанные выше значения и R81 - переводимый в аминогруппу радикал, переводят в соответствующее аминосоединение, или
б) соединение общей формулы

где R1-R7 имеют указанные выше значения и R82 обозначает отщепляемую группу, подвергают взаимодействию с аммиаком, и
в) при необходимости полученное соединение формулы I переводят в фармацевтически приемлемую кислотно-аддитивную соль.
а) соединение общей формулы
где R1-R7 имеют указанные выше значения и R81 - переводимый в аминогруппу радикал, переводят в соответствующее аминосоединение, или
б) соединение общей формулы
где R1-R7 имеют указанные выше значения и R82 обозначает отщепляемую группу, подвергают взаимодействию с аммиаком, и
в) при необходимости полученное соединение формулы I переводят в фармацевтически приемлемую кислотно-аддитивную соль.
Соединения общей формулы II, где R81 - переводимый в аминогруппу радикал, предпочтительно азидогруппу, или нитрогруппу, могут быть получены с помощью известных методов.
Если радикал R81 представляет собой азидогруппу, то соединения формулы I получают путем восстановления. Это восстановление может осуществляться с помощью комплексных гидридов, например, литийалюминийгидридом, или посредством каталитического гидрирования с помощью металлокатализаторов, таких, как, например, платина или палладий. Если в качестве восстановителя используют литийалюминийгидрид, то в качестве растворителя наиболее пригодны простой безводный эфир или тетрагидрофуран. Целесообразно при этом процесс вести следующим образом: после добавки по каплям соединения формулы IIa, где R81 - N3, в раствор, состоящий из безводного растворителя и гидрида, кипятят с обратным холодильником, после чего гидролизуют водным раствором простого эфира или ТГФ и осадок гидроокиси алюминия и лития кипятят с ТГФ.
Каталитическое гидрирование с помощью металлокатализаторов, например, платины или палладия, целесообразно осуществлять при комнатной температуре. В качестве растворителей особенно пригодны для этой цели вода, спирты, этилацетат, диоксан, уксусная кислота или смеси этих растворителей. Гидрирование целесообразно осуществлять в атмосфере водорода, либо в автоклаве, либо в аппаратуре для встряхивания.
Соединения общей формулы I могут быть получены также из соединения формулы IIb, где R82 обозначает отщепляемую группу, например, галоген, прежде всего бром, путем взаимодействия с аммиаком.
В этом случае соединения формулы IIb целесообразно суспендировать в жидком аммиаке и перемешивать при нагреве в автоклаве. После выпаривания аммиака остаток растворяют в растворителе, предпочтительно дихлорметане, промывают и сушат.
Было найдено, что для применения этих соединений в фармацевтике особенно пригодны их кислотно-аддитивные соли. Особенно эффективны соединения формулы I в виде солей фумаровой кислоты, но и все другие кислоты, упоминаемые в описании заявки, также образуют фармацевтически приемлемые аддитивные соли.
Добавки соответствующих кислот к соединениям формулы I целесообразно производить до их окончательного выделения, соответственно после осуществления описанных вариантов получения.
Производные индола, используемые в качестве исходных веществ при получении соединений общих формул IIa и IIb, могут быть получены, например, как это указано на представленных ниже схемах, с помощью известных методов, например, путем синтеза индола по методу Фишера, где арилгидразоны альдегидов или кетонов циклизуются при действии кислот или галогенидов металлов, используемых в качестве катализаторов, с отщеплением аммиака:
Схема 1

При этом заместители R1-R6 имеют указанные выше значения. Аналогично тому, как это описано в Synthesis, 1985, стр.186, соответствующие производные индола могут быть получены согласно следующей схеме:
Схема 2


При этом R1-R4 имеют указанные выше значения, а R' - низший алкил.
Схема 1
При этом заместители R1-R6 имеют указанные выше значения. Аналогично тому, как это описано в Synthesis, 1985, стр.186, соответствующие производные индола могут быть получены согласно следующей схеме:
Схема 2
При этом R1-R4 имеют указанные выше значения, а R' - низший алкил.
Ароматические альдегиды путем одноступенчатой реакции конденсации с алкиловыми эфирами азидоуксусной кислоты, катализуемой алкоголятами, с хорошим выходом преобразуют в алкиловые эфиры α -азидокоричной кислоты.
Последующий термолиз в кипящем п-ксилоле позволяет получать с почти количественным выходом алкиловые эфиры индол-2-карбоновой кислоты. Эфирную группу можно гидролизовать с помощью известных методов, а кислотную группу отщеплять путем нагрева.
Далее, производные индола можно получать путем циклизации соответственно замещенного фенилглицина, как это представлено на приведенной ниже схеме 3.
Схема 3


R1-R4 имеют указанные выше значения, a R8 и R9 - низший алкил, причем R8 и R9 могут быть идентичными или различными. Соединения формулы V могут с помощью спиртов по известной методике преобразовываться в соединения формулы VI. Циклизацией соединения формулы VI получают индол формулы VII (см. J. Het. Chem. 16, 221 (1979)). Путем обменной реакции между индолом формулы VII и алкилирующим средством, например, диалкил сульфатом или диазометаном, получают соединения формулы VIII. Эту обменную реакцию проводят в спиртовых растворителях, предпочтительно метаноле, при комнатной температуре.
R1-R4 имеют указанные выше значения, a R8 и R9 - низший алкил, причем R8 и R9 могут быть идентичными или различными. Соединения формулы V могут с помощью спиртов по известной методике преобразовываться в соединения формулы VI. Циклизацией соединения формулы VI получают индол формулы VII (см. J. Het. Chem. 16, 221 (1979)). Путем обменной реакции между индолом формулы VII и алкилирующим средством, например, диалкил сульфатом или диазометаном, получают соединения формулы VIII. Эту обменную реакцию проводят в спиртовых растворителях, предпочтительно метаноле, при комнатной температуре.
Другая возможность получения соответствующих индолов в качестве исходных веществ для получения соединений общей формулы I представлена на схеме 4. В этом случае также исходят из соответственно замещенного фенил-глицина формулы V.
Схема 4

Значения заместителей R1-R4 соответствуют приведенным выше, R51 - алкокси и R - низший алкил.
Значения заместителей R1-R4 соответствуют приведенным выше, R51 - алкокси и R - низший алкил.
Соединение формулы V с помощью ацетата, предпочтительно ацетата натрия, в ангидриде уксусной кислоты при нагреве с обратным холодильником может переводиться в соединение формулы IX, причем затем это последнее посредством гидролиза, например, с помощью концентрированной серной кислоты, может быть переведено в соединение формулы X. Соединения формулы X известны (см. Proc. Roy. Soc., Лондон, 178В, 781 (1958)) или могут быть получены по аналогичной методике.
Алкилирование соединения формулы X может проводиться с помощью обычных алкилирующих средств, как, например, диалкилсульфаты. Метилирование до соединения формулы XI осуществляют предпочтительно с помощью диметилсульфата, аналогично тому, как это описано в представленной в Bull. Soc. Chem. France 1978, 651 публикации.
Отщепление N-ацетильной группы может осуществляться с помощью обычных методов, например, путем реакции с алкоголятом натрия в спиртовом растворителе, предпочтительно метилате натрия в метаноле.
Другая возможность получения соединений формулы XII заключается в том, что сложные индоловые эфиры общей формулы XIV гидролизуют до кислот формулы XIII. Предпочтительно применяют в этих случаях гидроокиси щелочных металлов.
Декарбоксилирование соединений формулы XIII до соединений формулы XII может осуществляться под воздействием температур в диапазоне 300 - 320oC в металлической бане.
Исходя из описанных производных индола общей формулы IV, согласно схеме 5 получают соединения формулы II.
Схема 5

Заместители R1-R6 имеют здесь указанные выше значения.
Заместители R1-R6 имеют здесь указанные выше значения.
Путем обработки соединения формулы IV соответствующим алкилирующим средством, предпочтительно 1,2-диброметаном, получают соединение формулы IIb1. В этом случае обменную реакцию целесообразно проводить в условиях межфазного катализа. Эту обменную реакцию осуществляют при перемешивании в двухфазовой системе, состоящей из воды и не смешиваемого с водой органического растворителя, в присутствии сильного основания и катализатора фазового перехода. В качестве органического растворителя может применяться предпочтительно 1,2-диброметан, если он при этом служит одновременно реагентом. В качестве сильного основания пригодны, например, гидроокись калия или гидроокись натрия. Возможно применение обычных катализаторов фазового перехода. Такими катализаторами являются, например, хлорид бензилтриметиламмония, бромид тетрабутиламмония и другие аналогичные соединения. Реакцию осуществляют предпочтительно в диапазоне температур от приблизительно 20 до 80oC.
Соединение формулы IIa1 можно получать, например, взаимодействием соединений формулы IV с эпоксидом.
Соединения формулы IV можно растворять предпочтительно в суспензии, состоящей из гидрида натрия и тетрагидрофурана, при приблизительно 0oC и затем обрабатывать алкилоксираном, и при этом образуются соединения формулы IIa1
Гидроксильная группа с помощью известных методов может быть заменена на отщепляемую группу, например, путем обменной реакции с хлорангидридом сульфокислоты, предпочтительно метансульфонилхлоридом, в результате которой получают сульфонат. Путем обработки азидом, предпочтительно азидом натрия, в полярном растворителе, например, ДМФ, соединения формул IIa2 или IIb1 могут переводиться в соответствующие азидосоединения.
Гидроксильная группа с помощью известных методов может быть заменена на отщепляемую группу, например, путем обменной реакции с хлорангидридом сульфокислоты, предпочтительно метансульфонилхлоридом, в результате которой получают сульфонат. Путем обработки азидом, предпочтительно азидом натрия, в полярном растворителе, например, ДМФ, соединения формул IIa2 или IIb1 могут переводиться в соответствующие азидосоединения.
Как уже упоминалось выше, соединения формулы I и их фармацевтически приемлемые кислотно-аддитивные соли обладают ценными фармакологическими свойствами. Они могут связываться с серотонин-рецепторами и пригодны поэтому для лечения или предупреждения болезней или функциональных нарушений упомянутого выше типа, соответственно для изготовления соответствующих лекарственных средств.
Связывание предлагаемых по изобретению соединений формулы I с серотонин-рецепторами определяли с помощью стандартных методов in vitro. Препараты испытывали в указанных ниже тестах.
Метод I
а) Для связывания с 5НТ1А-рецептором согласно тесту на связывание 3H- 8-OH-DPAT по методу S.J.Peroutka, Biol. Psychiatry 20, 971-979 (1985).
а) Для связывания с 5НТ1А-рецептором согласно тесту на связывание 3H- 8-OH-DPAT по методу S.J.Peroutka, Biol. Psychiatry 20, 971-979 (1985).
б) Для связывания с 5НТ2С рецептором согласно тесту на связывание 3H-мезулергина по методу A.Pazos и др., Europ. J. Pharmacol 106, 539-546, соответственно D.Hoyer, Receptor Research 8, 59-81 (1988).
в) Для связывания с 5НТ2A-рецептором согласно тесту на связывание 3H-кетансерина по методу J. E. Leysen, Molecular Pharmacology 21, 301-304 (1981).
В ходе экспериментов определяли значения IC5 испытуемых веществ, т.е. выявляли концентрацию в нмол., при которой вытеснялись 50% связанного с рецептором лиганда.
Определенная таким путем активность некоторых соединений согласно изобретению, равно как и активность некоторых сравнительных соединений, представлена в таблице 1.
Метод II.
а) Для выявления сродства соединения с 5НТ1A-рецептором проводили опыты по вытеснению с помощью [ЗH]-5-НТ (1 нМ) в качестве радиолиганда на рекомбинированных 5Н1A-рецепторах человека, экспремированных в клетках ЗТЗ мышей. При этом использовали мембраны, полученные из клеток 2•105, а также различные концентрации соответствующего испытуемого соединения.
б) Для связывания с 5НТ2C-рецептором согласно тесту на связывание [ЗH] -5-НТ по методу S.J.Peroutka и др., Brain Research 584, 191-196 (1992).
в) Для связывания с 5НТ2A-рецептором согласно тесту на связывание [3H] -DOB по методу T.Branchek и др., Molecular Pharmacology 38, 604-609 (1990).
Ниже указаны значения  испытуемых веществ. Значение Ki расшифровывается по следующей формуле:
испытуемых веществ. Значение Ki расшифровывается по следующей формуле:

где значения IC50 соответствуют концентрациям испытуемых соединений в нМ, при которых вытесняются 50% связанного с рецептором лиганда. [L] обозначает концентрацию лиганда, а значение KD представляет собой константу диссоциации лиганда.
где значения IC50 соответствуют концентрациям испытуемых соединений в нМ, при которых вытесняются 50% связанного с рецептором лиганда. [L] обозначает концентрацию лиганда, а значение KD представляет собой константу диссоциации лиганда.
Определенная таким путем активность некоторых соединений по изобретению представлена в таблице 2.
Эрекция полового члена (у крыс)
В ходе экспериментов было установлено, что эрекция полового члена зависит от стимулирования 5НТ2C-рецептора (ср. Berendsen & Broekkamp, Eur. Journ. Pharmacol. 135, 179-184 (1987)).
В ходе экспериментов было установлено, что эрекция полового члена зависит от стимулирования 5НТ2C-рецептора (ср. Berendsen & Broekkamp, Eur. Journ. Pharmacol. 135, 179-184 (1987)).
В течение 45 мин после введения подопытным животным испытуемого вещества определяли число эрекций полового члена. ED50 обозначает дозу, вызывающую 50% таких эрекций.
Пример - ED50 (мг/кг, подкожно)
1 - 0,49
2 - 0,23
3 - 2,7
4 - 3,3
5 - 0,27
6 - 0,3
Определение общего уровня токсичности в отношении наиболее предпочтительных соединений, например, соединения, полученного в соответствии с примером 14, показало, что предлагаемые соединения формулы I являются малотоксичными.
1 - 0,49
2 - 0,23
3 - 2,7
4 - 3,3
5 - 0,27
6 - 0,3
Определение общего уровня токсичности в отношении наиболее предпочтительных соединений, например, соединения, полученного в соответствии с примером 14, показало, что предлагаемые соединения формулы I являются малотоксичными.
Соединения формулы I и фармацевтически приемлемые кислотно-аддитивные соли соединений формулы I могут находить применение в качестве лекарственных средств, например в виде фармацевтических препаратов, обладающих способностью связываться с серотонин-рецепторами. Фармацевтические препараты могут вводиться орально, например, в виде таблеток, лаковых таблеток, драже, мягкожелатиновых и твердожелатиновых капсул, растворов, эмульсий или суспензий. Введение, однако, может проводиться также ректально, например, в виде суппозиториев, парэнтерально, например, в виде растворов для инъекций, или же через нос.
Для изготовления указанных фармацевтических препаратов в соединения формулы I и фармацевтически приемлемые кислотно-аддитивные соли соединений формулы I могут добавляться инертные в фармацевтическом отношении, неорганические или органические наполнители. В качестве таковых для таблеток, лаковых таблеток, драже и твердожелатиновых капсул используют, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.п. В качестве наполнителей для мягкожелатиновых капсул пригодны, например, растительные масла, воск, жиры, полутвердые и жидкие полиолы и т. п. Однако в зависимости от тех или иных свойств активного вещества для мягкожелатиновых капсул вообще может не потребоваться никаких наполнителей. Для изготовления растворов и сиропов в качестве наполнителей пригодны, например, вода, полиолы, глицерин, растительное масло и т.п. В качестве наполнителей для суппозиториев пригодны, например, природные или отвержденные масла, воск, жиры, полужидкие либо жидкие полиолы и т.п.
Фармацевтические препараты могут содержать, кроме того, также консерванты, вещества, способствующие растворимости, стабилизаторы, увлажняющие средства, эмульгаторы, вещества, улучшающие вкус, красители, ароматические вещества, соли для изменения осмотического давления, буферы, оболочки или антиокислители. Они могут содержать также другие терапевтически ценные вещества.
Фармацевтические препараты, обладающие способностью связываться с серотонин-рецепторами, содержащие соединение формулы I или его фармацевтически приемлемую кислотно-аддитивную соль и терапевтически инертный наполнитель, также являются объектом настоящего изобретения. Способ их получения состоит в том, что одно или несколько соединений формулы I и/или фармацевтически приемлемые кислотно-аддитивные соли и ценные в терапевтическом отношении вещества вместе с одним или несколькими терапевтически инертными наполнителями преобразуют в галеновую форму.
Согласно изобретению соединения общей формулы I, а также их фармацевтически приемлемые кислотно-аддитивные соли могут применяться для лечения или предупреждения расстройств центральной нервной системы, таких, как депрессии, биполярные нарушения, состояние страха, нарушения сна и расстройства в сексуальной сфере, психозы, шизофрения, мигрени и другие болезненные состояния, связанные с головными болями и болями другого типа, расстройства, связанные с личными переживаниями или навязчивыми идеями, социальная фобия или приступы паники, психические органические расстройства, нарушения психики в детском возрасте, агрессивность, нарушения памяти, связанные с возрастом, нарушения отношений с окружающими, болезненная страсть, ожирение, булимия и т.п.; нарушений функций нервной системы, вызванных травмами, кровоизлиянием в мозг, заболеваниями нервно-дегенеративного характера и т.д.; нарушений в сердечно-сосудистой системе, таких, как высокое кровяное давление, тромбоз, инсульт и т.д.; желудочно-кишечных расстройств, таких, как дисфункция моторики желудочно-кишечного тракта, и для изготовления соответствующих лекарственных средств. Дозировку можно варьировать в широких пределах, с учетом, естественно, в каждом отдельном случае индивидуальных особенностей. При оральном введении дозу назначают в пределах от 0,01 мг (на одну дозу) до 500 мг (суточная доза) соединения общей формулы I или соответствующего количества его фармацевтически приемлемой кислотно-аддитивной соли, причем верхняя граница, если это окажется целесообразным, также может превышаться.
Ниже настоящее изобретение поясняется более подробно на примерах, которые никоим образом не ограничивают объем изобретения
Пример 1. Раствор 9,8 г (72,6 ммоль) 5-фториндола в 360 мл 1,2-диброметана обрабатывали 180 мл 28%-ной NaOH и 0,59 г (1,82 ммоль) бромида тетрабутиламмония. Смесь перемешивали в течение 42 ч при 50oC. Затем фазы разделяли и водную фазу экстрагировали с помощью толуола. Соединенные органические фазы промывали водой и сушили над сульфатом натрия. Растворитель отгоняли и остаток суспендировали в 1,9 л жидкого аммиака и перемешивали в течение 15 ч при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в 500 мл дихлорметана и промывали 100 мл воды и 100 мл насыщенного раствора хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток хроматографировали на 350 г силикагеля с помощью этилацетата/метанола (5:1). В результате получали 8,6 г красноватого масла, которое растворяли в 970 мл простого эфира и обрабатывали активированным углем. После фильтрации раствор обрабатывали 5,6 г (48,2 ммоль) фумаровой кислоты и перемешивали в течение 2 дней. Затем кристаллы отфильтровывали и сушили. В результате получали 11,1 г (39,5%) фумарата 2-(5-фториндол-1-ил)-этиламина (1:1,8) с Т.пл. 173-175oC (распад).
Пример 1. Раствор 9,8 г (72,6 ммоль) 5-фториндола в 360 мл 1,2-диброметана обрабатывали 180 мл 28%-ной NaOH и 0,59 г (1,82 ммоль) бромида тетрабутиламмония. Смесь перемешивали в течение 42 ч при 50oC. Затем фазы разделяли и водную фазу экстрагировали с помощью толуола. Соединенные органические фазы промывали водой и сушили над сульфатом натрия. Растворитель отгоняли и остаток суспендировали в 1,9 л жидкого аммиака и перемешивали в течение 15 ч при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в 500 мл дихлорметана и промывали 100 мл воды и 100 мл насыщенного раствора хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток хроматографировали на 350 г силикагеля с помощью этилацетата/метанола (5:1). В результате получали 8,6 г красноватого масла, которое растворяли в 970 мл простого эфира и обрабатывали активированным углем. После фильтрации раствор обрабатывали 5,6 г (48,2 ммоль) фумаровой кислоты и перемешивали в течение 2 дней. Затем кристаллы отфильтровывали и сушили. В результате получали 11,1 г (39,5%) фумарата 2-(5-фториндол-1-ил)-этиламина (1:1,8) с Т.пл. 173-175oC (распад).
Пример 2. а) Суспензию 2,63 г (10,6 ммоль) N-[3-хлор-2-(гидроксикарбонил)-4-фторфенил] глицина и 2,63 г (32,06 ммоль) ацетата натрия в 25 мл ангидрида уксусной кислоты кипятили в течение 45 мин с обратным холодильником. Растворитель удаляли в вакууме и остаток обрабатывали 50 мл воды. Кристаллы отфильтровывали, промывали водой и сушили. В результате получали 2,7 г (94%) 3-ацетокси-1-ацетил-4-хлор-5-фториндола в виде желтых кристаллов с Т.пл.168-169oC.
б) В 20 мл 90%-ной серной кислоты добавляли 2,65 г (9,8 ммоль) 3-ацетокси-1-ацетил-4-хлор-5-фториндола и реакционную смесь после перемешивания в течение 3/4 ч разбавляли 100 мл ледяной воды. Осадок отфильтровывали, промывали водой и сушили. В результате получали 2 г (92,5%) 1-ацетил-4-хлор-5-фториндолин-3-она в виде светло-коричневых кристаллов с Т.пл. 203-204oC.
в) Смесь 2 г (8,78 ммоль) 1-ацетил-4-хлор-5-фториндолин-3-она, 2 г порошкообразной гидроокиси натрия и 30 мл диметилсульфата перемешивали в течение 5 ч при комнатной температуре. После добавки 250 мл насыщенного раствора бикарбоната натрия перемешивали в течение ночи и затем отфильтровывали. Остаток растворяли в 100 мл простого эфира и 100 мл этилацетата. Раствор сушили над сульфатом натрия, фильтровали и упаривали. В результате получали 2 г (95%) 1-ацетил-4-хлор-5-фтор-3-метоксииндола в виде зеленоватых кристаллов с Т.пл. 114-115oC.
г) Раствор 2 г (8,28 ммоль) 1-ацетил-4-хлор-5-фтор-3-метоксииндола и 0,48 г (8,8 ммоль) метилата натрия в 35 мл метанола перемешивали в течение 1 ч при комнатной температуре. После удаления растворителя экстрагировали с помощью воды и этилацетата, органическую фазу промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 100 г силикагеля с помощью гексана/этилацетата (3:1). В результате получали 0,88 г (53%) 4-хлор-5-фтор-3- метоксииндола.
д) Раствор 2 г (10 ммоль) 4-хлор-5-фтор-3-метоксииндола в 50 мл 1,2-диброметана обрабатывали 50 мл 28%-ной NaOH и 100 мг (0,3 ммоль) бромида тетрабутиламмония. Смесь перемешивали в течение 15 ч. Фазы разделяли и водную фазу экстрагировали с помощью толуола. Соединенные органические фазы промывали водой и сушили над сульфатом натрия. Растворитель отгоняли и остаток суспендировали в 200 мл жидкого аммиака и затем перемешивали в течение 18 ч при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в 50 мл дихлорметана и промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток растворяли в 200 мл простого эфира и обрабатывали 0,7 г (6 ммоль) фумаровой кислоты, после чего перемешивали в течение 2 дней. Кристаллы выделяли и сушили. В результате получали 2 г (55%) фумарата 2-(4-хлор-5-фтор-3-метокси-индол-1-ил)-этиламина (1: 1) в виде кристаллов бежевого цвета с Т.пл. 195-196oC.
Пример 3. а) В 20 мл 90%-ной серной кислоты добавляли 2,2 г (10 ммоль) 3-ацетокси-1-ацетил-5-фториндола и реакционную смесь после перемешивания в течение 3/4 ч разбавляли 100 мл ледяной воды. Осадок отфильтровывали, промывали водой и сушили. В результате получали 1,6 г (83%) 1-ацетил-5-фториндолин-3-она в виде кристаллов бежевого цвета с Т.пл. 143-144oC.
б) Смесь 1,6 г (8,3 ммоль) 1-ацетил-5-фториндолин-3-она, 1,9 г порошкообразной гидроокиси натрия и 28 мл диметилсульфата перемешивали в течение 2 ч при комнатной температуре. После добавки 240 мл насыщенного раствора бикарбоната натрия перемешивали в течение ночи и затем отфильтровывали. Остаток растворяли в 100 мл простого эфира и 100 мл этилацетата. Раствор сушили над сульфатом натрия, фильтровали и упаривали. В результате получали 1,47 г (86%) 1-ацетил-5-фтор-3-метоксииндола в виде зеленоватых кристаллов с Т.пл. 124-125oC.
в) Раствор 1,25 г (6,03 ммоль) 1-ацетил-5-фтор-3-метоксииндола и 0,35 г (6,5 ммоль) метилата натрия в 23 мл метанола перемешивали в течение 1 ч при комнатной температуре. После удаления растворителя экстрагировали с помощью воды и этилацетата, органическую фазу промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. Растворитель удаляли и остаток дистиллировали в трубке с шаровым расширением при температуре бани 200oC давлении 0,05 атм. В результате получали 0,76 г (76%) 5-фтор-З-метоксииндола.
г) Раствор 0,76 г (4,6 ммоль) 5-фтор-З-метоксииндола в 25 мл 1,2-диброметана обрабатывали 25 мл 28%-ной NaOH и 40 мг (0,12 ммоль) бромида тетрабутиламмония. Смесь перемешивали в течение 15 ч при 50oC. Фазы разделяли и водную фазу экстрагировали с помощью толуола. Соединенные органические фазы промывали водой и сушили над сульфатом натрия. Растворитель отгоняли и остаток хроматографировали на 60 г окиси алюминия с помощью гексана/этилацетата (5:1). В результате получали желтое масло, которое суспендировали в 80 мл жидкого аммиака и в течение 18 ч перемешивали при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в дихлорметане и промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток растворяли в 90 мл простого эфира и 3 мл метанола и обрабатывали 0,35 г (3 ммоль) фумаровой кислоты, после чего перемешивали в течение 2 дней. Кристаллы отфильтровывали и сушили. В результате получали 0,56 г (37,5%) фумарата 2-(5-фтор-3-метоксииндол-1-ил)-этиламина (1: 1) в виде кристаллов бежевого цвета с Т.пл. 167oC.
Пример 4. а) Раствор 1 г (6,6 ммоль) 5-хлориндола в 30 мл 1,2-диброметана обрабатывали 30 мл 28%-ной NaOH и 80 мг (0,24 ммоль) бромида тетрабутиламмония. Смесь перемешивали в течение 3 ч при 50oC. Затем фазы разделяли и водную фазу экстрагировали с помощью толуола. Соединенные органические фазы промывали водой и сушили над сульфатом натрия. Растворитель отгоняли и остаток хроматографировали на 150 г силикагеля с помощью гексана/этилацетата (5:1). В результате получали 1,17 г (69%) 1-(2-бромэтил)-5-хлориндола в виде белых кристаллов с Т.пл. 71oC.
б) Суспензию 0,55 г (2,1 ммоль) 1-(2-бромэтил)-5-хлориндола в 60 мл жидкого аммиака перемешивали в течение 18 ч при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в дихлорметане и промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток растворяли в 60 мл простого эфира и 3 мл метанола и обрабатывали 0,25 г (2,1 ммоль фумаровой кислоты, после чего перемешивали в течение 2 дней. Затем кристаллы отфильтровывали и сушили. В результате получали 0,51 г (88%) фумарата 2-(5-хлориндол-1-ил)-этиламина (1: 0,7) в виде белых кристаллов с Т.пл. 167oC.
Пример 5. а) В раствор 145 мл (1,02 моль) 2-метилацетоуксусного эфира в 1 л этанола при 2-4oC в течение 90 мин по каплям добавляли 400 мл 50%-ного раствора едкого кали. Затем добавляли 2 л ледяной воды и быстро обрабатывали раствором соли диазония, который приготавливали следующим образом: при охлаждении ледяной баней 200 мл 25%-ного раствора соляной кислоты по каплям добавляли в раствор из 145,6 г (1 моль) 3-хлор-4-фторанилина в 1 л этанола. Затем при 4oC в течение 90 мин добавляли 137 мл (1,02 моль) изопентилнитрита. Оранжевую эмульсию, полученную в результате добавки соли диазония, выливали в 4 л воды и экстрагировали один раз с помощью 4 л и дважды соответственно порциями по 2 л толуола. Соединенные органические фазы сушили над сульфатом натрия, фильтровали и концентрировали до объема 1,5 л. После кипячения в течение 30 мин в водоотделителе добавляли раствор 203 г (1,07 моль) моногидрата п-толуолсульфокислоты в 1,5 л толуола и смесь продолжали нагревать в водоотделителе еще в течение 1 ч. После охлаждения экстрагировали с помощью 1,5 л 1н. соляной кислоты, 1,5 л 1н. гидроокиси натрия и 0,4 л насыщенного раствора хлорида натрия. Водные фазы промывали 1,5 л толуола и соединенные органические фазы сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на 3 кг силикагеля с помощью толуола. В результате получали 10,3 г (4,2%) сырого этилового эфира 6-хлор-5-фториндол-2-карбоновой кислоты. Пробу перекристаллизовывали из толуола и эта проба имела Т. пл. 190-191oC. Вторая фракция содержала 7 г (2,9%) этилового эфира 4-хлор-5-фториндол-2-карбоновой кислоты в виде коричневых кристаллов с Т.пл. 179-182oC.
б) Суспензию из 6,3 г (26 ммоль) этилового эфира 4-хлор-5-фториндол-2-карбоновой кислоты в 260 мл этанола обрабатывали 130 мл 2н. гидроокиси натрия, после чего перемешивали в течение 3 ч. Затем этанол выпаривали и pH реакционного раствора с помощью 25%-ной соляной кислоты доводили до pH 1. Осадок промывали водой и сушили. В результате получали 5,3 г (96%) сырой 4-хлор-5-фториндол-2-карбоновой кислоты, которую без дальнейшей очистки использовали при проведении следующей ступени.
в) Металлическую баню нагревали до 300-320oC. Затем в атмосфере аргона добавляли 4,8 г (22,47 ммоль) 4-хлор-5-фториндол-2-карбоновой кислоты и через 3 мин металлическую баню удаляли. Реакционную смесь дистиллировали в трубке с шаровым расширением при 0,15•10-3атм и 75oC температуры бани. В результате получали 3,15 г (83%) 4-хлор-5-фториндола в виде белых кристаллов с Т.пл. 41-43oC.
г) Раствор 130 мг (0,75 ммоль) 4-хлор-5-фториндола в 3,7 мл 1,2-диброметана обрабатывали 3,7 мл 28%-ной NaOH и 7,7 мг (0,003 ммоль бромида тетрабутиламмония. Затем смесь перемешивали в течение 5 ч при 50oC. Фазы разделяли и водную фазу экстрагировали с помощью толуола. Соединенные органические фазы промывали водой и сушили над сульфатом натрия. Растворитель отгоняли и остаток хроматографировали на 15 г силикагеля с помощью гексана/этилацетата (5: 1). В результате получали желтое масло, которое суспендировали в 30 мл жидкого аммиака и перемешивали в течение 16 ч при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в дихлорметане и промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток хроматографировали на 15 г силикагеля с помощью этилацетата/метанола (5:1). В результате получали 110 мг желтого масла, которое растворяли в 16 мл простого эфира, обрабатывали 60 мг (0,5 ммоль) фумаровой кислоты и перемешивали в течение 2 дней. Затем кристаллы отфильтровывали и сушили. В результате получали 150 мг (59%) фумарата 2-(4-хлор-5-фториндол-1-ил)-этиламина (1:1) с Т.пл. 200-201oC (распад).
Пример 6. а) Суспензию 21,8 г (90,2 ммоль) этилового эфира 6-хлор-5-фториндол-2-карбоновой кислоты в 450 мл этанола обрабатывали 180 мл 2н. гидроокиси натрия и перемешивали в течение 21 ч. Раствор упаривали и остаток растворяли в 450 мл воды, после чего обрабатывали 60 мл 25%-ной соляной кислоты. Осадок промывали водой и сушили. В результате получали 18,8 г (97,7%) 6-хлор-5-фториндол-2-карбоновой кислоты. Пробу перекристаллизовывали из толуола, и эта проба имела Т.пл. 274-276oC.
б) Металлическую баню нагревали до 300-320oC. Затем в атмосфере аргона добавляли 6,4 г (30 ммоль) 6-хлор-5-фториндол-2-карбоновой кислоты и через 3 мин металлическую баню удаляли. После охлаждения реакционную смесь хроматографировали на 25 г силикагеля с помощью гексан/этилацетата (4:1). В результате получали 4,17 г (85%) 6-хлор-5-фториндола в виде кристаллов бежевого цвета. Пробу тщательно перемешивали с гексаном, и эта проба имела Т.пл. 96-98oC.
в) Раствор 0,95 г (5,6 ммоль) 6-хлор-5-фториндола в 30 мл 1,2-диброметана обрабатывали 30 мл 28%-ной NaOH и 60 мг (0,18 ммоль) бромида тетрабутиламмония. Затем смесь перемешивали в течение 8 ч, фазы разделяли и водную фазу экстрагировали с помощью толуола. Соединенные органические фазы промывали водой и сушили над сульфатом натрия. Растворитель отгоняли и остаток хроматографировали на 100 г силикагеля с помощью гексана/этилацетата (6:1). В результате получали светло-коричневое масло, которое суспендировали в 120 мл жидкого аммиака и перемешивали в течение 16 ч при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в дихлорметане, после чего промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток растворяли в 120 мл простого эфира и 6 мл метанола, обрабатывали 0,5 г (4,3 ммоль) фумаровой кислоты и перемешивали в течение ночи. Затем кристаллы отфильтровывали и сушили. В результате получали 1 г (61%) фумарата 2-(6-хлор-5-фториндол-1-ил)-этиламина (1:0,7) с Т.пл. 192- 193oC (распад).
Пример 7. а) Раствор 1,76 г (7,54 ммоль) этилового эфира 4-метил-З-метоксииндол-2-карбоновой кислоты в 90 мл этанола обрабатывали 45 мл 2н. гидроокиси натрия и перемешивали в течение 17 ч при комнатной температуре. Затем спирт выпаривали и остаток обрабатывали 60 мл 2н. соляной кислоты. Выпавшие кристаллы отфильтровывали, промывали водой и сушили. В результате получали 1,2 г (78%) 4-метил-3-метоксииндол-2-карбоновой кислоты в виде кристаллов коричневого цвета с Т.пл. 136oC.
б) При 150oC 1,2 г (5,85 ммоль) 4-метил-3-метоксииндол-2-карбоновой кислоты нагревали до тех пор, пока не прекращалось улетучивание газа. В результате получали 0,94 г (колич.) сырого 4-метил-З-метоксииндола, который без дальнейшей очистки использовали при проведении следующей ступени.
в) Раствор 0,94 г (5,8 ммоль) 4-метил-З-метоксииндола в 30 мл 1,2-диброметана обрабатывали 30 мл 28%-ной NaOH и 400 мг (1,2 ммоль) бромида тетрабутиламмония. Смесь перемешивали в течение 24 ч при 40oC. Затем реакционную смесь разбавляли 150 мл толуола и промывали 50 мл воды и 25 мл насыщенного раствора хлорида натрия. Водные фазы экстрагировали с помощью 75 мл толуола. Соединенные органические фазы сушили с помощью сульфата натрия. Затем растворитель отгоняли и остаток хроматографировали на 70 г силикагеля с помощью гексана/этилацетата (6: 1). В результате получали красное масло, которое суспендировали в 20 мл жидкого аммиака и перемешивали в течение 16 ч при 80oC в автоклаве. После выпаривания аммиака остаток растворяли в дихлорметане и промывали водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом натрия и растворитель отгоняли. Остаток растворяли в 60 мл простого эфира и обрабатывали 0,3 г (2,5 ммоль) фумаровой кислоты, после чего перемешивали в течение 18 ч. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,5 г (28%) фумарата 2-(4-метил-3-метоксииндол-1-ил)-этиламина (1:0,9) с Т.пл.163-164oC.
Пример 8. а) Суспензию 0,4 г (14,3 ммоль, 80%) дисперсии гидрида натрия в 60 мл тетрагидрофурана обрабатывали при 0oC 1,74 г (11,5 ммоль) 5-хлориндола и перемешивали в течение 1 ч при этой температуре. После добавки 1,6 мл (23 ммоль) (RS)-метилоксирана реакционную смесь в течение 48 ч перемешивали при комнатной температуре и затем обрабатывали 11 мл воды. После этого разбавляли 300 мл простого эфира, промывали 140 мл воды и 70 мл насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 60 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 2,1 г (87%) (RS)-1-(5-xлopиндoл-1-ил)-пpoпaн-2-oлa в виде желтого масла.
МС: m/e (% основного пика): 209, 211 (М+, 28), 164 (100).
б) Раствор 2 г (9,6 ммоль) (RS)-1-(5-хлориндол-1-ил)-пропан-2-ола в 50 мл дихлорметана обрабатывали 5,4 мл (38,7 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 1,5 мл (19,3 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли 480 мл простого эфир, промывали 120 мл 1М раствора карбоната натрия и 60 мл насыщенного раствора хлорида натрия и водную фазу экстрагировали с помощью 240 мл простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 50 мл диметилформамида и после добавки 1,26 г (19,4 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли 500 мл простого эфира и дважды экстрагировали соответственно порциями по 240 мл воды и один раз 60 мл насыщенного раствора поваренной соли. Водную фазу экстрагировали с помощью 240 мл простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 46 г силикагеля с помощью толуола. В результате получали 2 г (90%) (RS)-1- (2-азидопропил)-5-хлориндола в виде бесцветного масла.
MC: m/e (% основного пика): 234, 236 (М+, 20), 164 (100).
в) Суспензию 0,2 г окиси платины в 40 мл этанола в течение получаса перемешивали в атмосфере водорода, после чего обрабатывали раствором 1,9 г (8,1 ммоль) (RS)-1-(2-азидопропил)-5-хлориндола в 40 мл этанола. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Затем раствор упаривали и остаток растворяли в 160 мл простого эфира, после чего обрабатывали 0,94 г (8,1 ммоль фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 1,85 г (70%) фумарата (RS)-2-(5-хлориндол-1-ил)-1-метил-этиламина (1:1,9) в виде белых кристаллов с Т. пл.183-185oC (распад).
Пример 9. а) Суспензию 0,55 г (18,5 ммоль, 80%) дисперсии гидрида натрия в 75 мл тетрагидрофурана обрабатывали при 0o 2 г (14,8 ммоль) 5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 2,1 мл (30 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 24 ч при комнатной температуре и затем обрабатывали 5 мл воды. Затем разбавляли простым эфиром, трижды промывали соответственно порциями по 75 мл воды и 70 мл насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 60 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 1,8 г (62%) (RS)-1-(5-фториндол-1-ил)-пропан-2-ола в виде желтого масла.
MC: m/e (% основного пика): 193 (М+, 22), 148 (100).
б) Раствор 1,75 г (9 ммоль) (RS)-1-(5-фториндол-1-ил)-пропан-2-ола в 45 мл дихлорметана обрабатывали 3,8 мл (27 ммоль) триэтиламина и охлаждали до 0o. В этот раствор добавляли по каплям 1,4 мл (18 ммолей) метансульфонилхлорида, после чего реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 45 мл диметилформамида и после добавки 1,18 г (18,1 ммоль) азида натрия перемешивали в течение 6 ч при 60oC. Затем разбавляли простым эфиром и экстрагировали с помощью воды и 60 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира, соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 20 г силикагеля с помощью толуола. В результате получали 1,8 г (91%) (RS)-1-(2-азидопропил)-5-фториндола в виде бесцветного масла.
MC: m/e (% основного пика): 218 (М+, 19), 148 (100).
в) Раствор 1,7 г (7,7 ммоль) (RS)-1-(2-азидопропил)-5-фториндола в 80 мл этанола гидрировали в присутствии 170 мг Pd-C (5%). Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 240 мл простого эфира, после чего обрабатывали 0,93 г (8 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 2,3 г (95,8%) фумарата (RS)-2-(5-фтopиндoл-1-ил)-1-метилэтилaминa (1: 1) в виде белых кристаллов с Т.пл. 169-170oC (распад).
Пример 10. а) Суспензию 0,75 г (24 ммоль 80%) дисперсии гидрида натрия в 100 мл тетрагидрофурана обрабатывали при 0o 3,38 г (20 ммоль) 6-хлор-5- фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 2,8 мл (40 ммолей) (RS)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали 11 мл воды. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 75 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 2,87 г (63%) (RS)- 1-(6-хлор-5-фториндол-1-ил)-пропан-2-ола в виде кристаллов бежевого цвета с Т.пл. 185-186oC.
б) Раствор 2,85 г (12,5 ммоль) (RS)-1-(6-хлор-5-фториндол-1-ил)- пропан-2-ола в 60 мл дихлорметана обрабатывали 5,2 мл (37,6 ммоль) триэтиламина и охлаждали до 0o. В этот раствор добавляли по каплям 1,9 мл (25,1 ммоль) метансульфонилхлорида, после чего реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 60 мл диметилформамида и после добавки 1,55 г (23,8 ммоль) азида натрия перемешивали в течение 3 ч при 60oC. Затем разбавляли простым эфиром и дважды экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 60 г силикагеля с помощью толуола. В результате получали 2,87 г (92%) (RS)-1-(2-азидо-пропил)-6-хлор-5-фториндола в виде желтоватого масла.
MC: m/e (% основного пика): 252, 254 (М+, 20), 182 (100).
в) Суспензию 0,26 г окиси платины в 50 мл этанола в течение получаса перемешивали в атмосфере водорода, после чего обрабатывали раствором из 2,8 г (11,7 ммоль) (RS)-1-(2-азидопропил)-6-хлор-5-фториндола в 50 мл этанола. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 380 мл простого эфира, после чего обрабатывали 1,28 г (11 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 3,6 г (95%) фумарата (RS)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламина (1: 1) в виде белых кристаллов с Т.пл. 174 -175oC (распад).
Пример 11. а) Суспензию 0,26 г (8,75 ммоль, 80%) дисперсии гидрида натрия в 35 мл тетрагидрофурана обрабатывали при 0oC 0,95 г (7 ммоль) 5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 1 мл (14 ммоль) (S)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали 7 мл воды. Затем разбавляли 180 мл простого эфира, дважды промывали соответственно порциями по 90 мл воды и 50 мл насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 28 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 1,15 г (84%) (S)-1-(5-фториндол-1-ил)-пропан-2-ола в виде светло-коричневого масла [α] = +48,4 (с 0,25, CHCl3).
б) Раствор 1,1 г (5,7 ммоль) (S)-1-(5-фториндол-1-ил)-пропан-2-ола в 30 мл дихлорметана обрабатывали 3,17 мл (38,7 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,88 мл (11,4 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли 280 мл простого эфира, после чего дважды промывали соответственно порциями по 70 мл 1М раствора карбоната натрия и 35 мл насыщенного раствора хлорида натрия и водную фазу экстрагировали с помощью 140 мл простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 30 мл диметилформамида и после добавки 0,74 г (11,4 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли 280 мл простого эфира и дважды экстрагировали соответственно порциями по 140 мл воды и один раз 70 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью 140 мл простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 25 г силикагеля с помощью толуола. В результате получали 1,14 г (95%) (R)-1-(2-азидопропил)-5-фториндола в виде масла желтоватого цвета.
[α] = -124 (с 0,25, CHCl3).
в) Суспензию 0,1 г окиси платины в 25 мл этанола в течение получаса перемешивали в атмосфере водорода, после чего обрабатывали раствором 0,92 г (4,8 ммоль (R)-1-(2-азидопропил)-5-фториндола в 25 мл этанола. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 150 мл простого эфира, после чего обрабатывали 0,58 г (5 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 1,14 г (88%) фумарата (R)-2-(5-фториндол-1-ил)-1-метилэтиламина (1: 1) в виде белых кристаллов с Т.пл. 159-161oC (распад).
[α] = -38 (c 0,25, CH3H).
Пример 12. а) Суспензию 0,26 г (8,75 ммоль, 80%) дисперсии гидрида натрия в 35 мл тетрагидрофурана обрабатывали при 0o 0,95 г (7 ммоль) 5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 1 мл (14 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали 7 мл воды. Затем разбавляли 180 мл простого эфира, дважды промывали соответственно порциями по 90 мл воды и 50 мл насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 28 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 1,09 г (80%) (R)-1-(5-фториндол-1-ил)-пропан-2-ола в виде светло-коричневого масла.
[α] = -48,8 (с 0,25 CHCl3).
Раствор 1,04 г (5,3 ммоль) (R)-1-(5-фториндол-1-ил)-пропан-2-ола в 30 мл дихлорметана обрабатывали 3,17 мл (38,7 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,88 мл (11,4 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли 280 мл простого эфира, после чего дважды промывали соответственно порциями по 70 мл 1М раствора карбоната натрия и 35 мл насыщенного раствора хлорида натрия и водную фазу экстрагировали с помощью 140 мл простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 30 мл диметилформамида и после добавки 0,74 г (11,4 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли 280 мл простого эфира и дважды экстрагировали соответственно порциями по 140 мл воды и один раз 70 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью 140 мл простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 25 г силикагеля с помощью толуола. В результате получали 1,14 г (97,4%) (S)-1-(2-азидопропил)-5-фториндола в виде масла желтоватого цвета.
[α] = -119,2 (с 0,25, CHCl3).
в) Суспензию 0,1 г окиси платины в 25 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 1,12 г (5,13 ммоль) (S)-1-(2-азидопропил)-5-фториндола в 25 мл этанола. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Затем отфильтровывали катализатор и промывали этанолом. Раствор упаривали и остаток растворяли в 150 мл простого эфира, после чего обрабатывали 0,58 г (5 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 1,42 г (90%) фумарата (S)-2-(6-хлор-5-фториндол- 1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 159-161oC (распад).
[α] = -34 )c 0,25, CH3OH).
Пример 13.
а) Суспензию 0,11 г (3,7 ммоль 80%) дисперсии гидрида натрия в 15 мл тетрагидрофурана обрабатывали при 0oC 0,5 г (3 ммоль) 6-хлор-5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,42 мл (6 ммоль) (S)-метилоксирана реакционную смесь перемешивали в течение 85 ч при комнатной температуре и затем обрабатывали водой. Затем разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 25 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 0,54 г (79%) (S)-1-(6-хлор-5-фториндол-1-ил)-пропан-2-ола в виде кристаллов бежевого цвета с Т.пл. 99-101oC.
б) Раствор 0,53 г (2,3 ммоль) (S)-1-(6-хлор-5-фториндол-1-ил)-пропан-2-ола в 30 мл дихлорметана обрабатывали 1 мл (7 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,36 мл (4,6 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 10 мл диметилформамида и после добавки 0,3 г (4,6 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 20 г силикагеля с помощью толуола/н-гексана (9: 1). В результате получали 0,54 г (92%) (R)-1-(2-азидопропил)-6-хлор-5-фториндола в виде масла желтоватого цвета.
[α] = -131,6 (с 0,25, CHCl3).
в) Суспензию 0,05 г окиси платины в 10 мл этанола в течение получаса перемешивали в атмосфере водорода, после чего обрабатывали раствором 0,51 г (2 ммоль (R)-1-(2-азидопропил)-6-хлор-5-фториндола в 10 мл этанола. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 60 мл простого эфира и обрабатывали 0,23 г (1,98 ммоль) фумаровой кислоты, после чего перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,55 г (69%) фумарата (R)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламина (1:1,5) в виде белых кристаллов с Т.пл. 153-154oC.
[α] = -28,8 (с 0,25, CH3OH).
Пример 14. а) Суспензию 0,11 г (3,7 ммоль, 80%) дисперсии гидрида натрия в 15 мл тетрагидрофурана обрабатывали при 0oC 0,5 г (3 ммоль) 6-хлор-5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,42 мл (6 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 85 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 25 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,51 r (74,6%) (R)-1-(6-хлор-5-фториндол-1-ил)-пропан-2-ола в виде белых кристаллов с Т.пл. 104-105oC.
б) Раствор 0,28 г (1,2 ммоль) (R)-1-(6-хлор-5-фториндол-1-ил)-пропан-2-ола в 6 мл дихлорметана обрабатывали 0,5 мл (3,8 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,2 мл (2,5 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 6 мл диметилформамида и после добавки 0,16 г (2,4 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 10 г силикагеля с помощью толуола. В результате получали 0,29 г (93,5%) (S)-1-(2-азидопропил)-6-хлор-5-фториндола в виде желтоватого масла.
[α] = +151,2 (с 0,25, CHCl3).
в) Суспензию 0,02 г окиси платины в 5 мл этанола в течение получаса перемешивали в атмосфере водорода, после чего обрабатывали раствором 0,26 г (1,05 ммоль) (S)-1-(2-азидопропил)-6-хлор-5-фториндола в 5 мл этанола. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 30 мл простого эфира и обрабатывали 0,12 г (1,03 ммоль) фумаровой кислоты, после чего перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,25 г (58,8%) фумарата (S)-2-(6-хлор-5-фториндол-1-ил)-1-метил-этиламина (1:1,6) в виде белых кристаллов с Т.пл. 151-152oC (распад).
[α] = +31,6 (с 0,25, CH3OH).
Пример 15. а) Суспензию 0,26 г (8,7 ммоль, 80%) дисперсии гидрида натрия в 35 мл тетрагидрофурана обрабатывали при 0oC 0,95 г (7,25 ммоль) 4-метилиндола и перемешивали в течение 1 ч при этой температуре. После добавки 1 мл (15 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 25 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 0,86 г (62,7%) (RS)-1-(4-метилиндол -1-ил)пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика): 189 (М+, 26), 144 (100).
б) Раствор 0,74 г (4 ммоль) (RS)-1-(4-метилиндол-1-ил)-пропан-2-ола в 20 мл дихлорметана обрабатывали 1,6 мл (11 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,6 мл (7,7 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 20 мл диметилформамида и после добавки 0,5 г (7,8 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. 3атем разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 30 г силикагеля с помощью толуола. В результате получали 0,68 г (81,2%) (S)-1-(2-азидопропил)-4-метилиндола в виде масла оранжевого цвета.
МС: m/e (% основного пика): 214 (М+ 20), 144 (100).
в) Раствор 0,67 г (3 ммоль) (RS)-1-(2-азидопропил)-4-метилиндола в 30 мл этанола гидрировали в присутствии 70 мг Pd-C (5%). Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 90 мл простого эфира и затем обрабатывали 0,35 г (3 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,88 г (92,3%) фумарата (RS)-2-(4-метилиндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 163-164oC (распад).
Пример 16. а) Суспензию 0,56 г (18,75 ммоль, 80%) дисперсии гидрида натрия в 75 мл тетрагидрофурана обрабатывали при 0oC 2,95 г (15 ммоль) 5-броминдола в течение 1 ч при этой температуре. После добавки 2,1 мл (30 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 60 ч при комнатной температуре и далее обрабатывали 15 мл воды. Затем разбавляли 750 мл простого эфира, дважды промывали порциями по 250 мл воды и 125 мл насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 120 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 3,4 г (89%) (RS)-1-(5-броминдол-1-ил)-пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика: 253, 255 (М+, 41), 208, 210 (100), 129 (70).
б) Раствор 3,36 г (13,2 ммоль) (RS)-1-(5-броминдол-1-ил)-пропан-2-ола в 60 мл дихлорметана обрабатывали 7,37 мл (53 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 2 мл (26,4 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли 660 мл простого эфира, после чего дважды промывали соответственно порциями по 135 мл 1М раствора карбоната натрия и 80 мл насыщенного раствора хлорида натрия и водную фазу экстрагировали с помощью 300 мл простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 60 мл диметилформамида и после добавки 1,72 г (11,4 ммоль) азида натрия перемешивали в течение 16 ч при 60oC. Затем разбавляли 660 мл простого эфира и дважды экстрагировали соответственно порциями по 330 мл воды и один раз 80 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью 330 мл простого эфира и соединенные органические фазы сушили над сульфатом натрия. Затем растворитель удаляли и остаток хроматографировали на 90 г силикагеля с помощью толуола. В результате получали 3,22 г (87,2%) (RS)-1-(2-азидопропил)-5-броминдола в виде светло-желтого масла.
МС: m/e (% основного пика): 278, 280 (М+ 16), 208, 210 (87), 129 (100).
в) Суспензию 0,1 г окиси платины в 20 мл этанола перемешивали в течение получаса в атмосфере водорода, после чего обрабатывали раствором 1,12 г (4 ммоль) (RS)-1-(2-азидопропил)-5-броминдола в 20 мл этанола и 2 мл 33%-ного раствора метиламина в этаноле. Реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 120 мл простого эфира и 2,5 мл метанола, после чего обрабатывали 0,46 г (3,96 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 1,38 г (93%) фумарата (RS)-2-(5-броминдол-1-ил)-1-метилэтиламина (1: 1) в виде белых кристаллов с Т.пл. 192-193oC (распад).
Пример 17. а) Суспензию 0,28 г (9,35 ммоль, 80%) дисперсии гидрида натрия в 40 мл тетрагидрофурана обрабатывали при 0oC 0,98 г (7,5 ммоль) 6-метилиндола и перемешивали в течение 1 ч при этой температуре. После добавки 1 мл (15 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 60 ч при комнатной температуре и обрабатывали затем 7 мл воды. Далее разбавляли 370 мл простого эфира, дважды промывали соответственно порциями по 120 мл воды и 60 мл насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 35 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 0,5 г (35%) (RS)-1-(6-метилиндол-1-ил)-пропан-2-ола в виде светло-коричневых кристаллов с Т.пл. 65-69oC.
б) Раствор 0,49 г (13,2 ммоль) (RS)-1-(6-метилиндол-1-ил)-пропан-2-ола в 10 мл дихлорметана обрабатывали 1 мл (53 ммоль) триэтиламина и охлаждали до 0oC. B этот раствор добавляли по каплям 0,5 мл (26,4 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего дважды промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 20 мл диметилформамида и после добавки 0,5 г (11,4 ммоль) азида натрия перемешивали в течение 16 ч при 60oC. Затем разбавляли 660 мл простого эфира и дважды экстрагировали с помощью воды и один раз с помощью 80 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 30 г силикагеля с помощью толуола. В результате получали 0,5 г (90%) (RS)-1-(2-азидопропил)-5-метилиндола в виде светло-желтого масла. \\ \ 2 в) Раствор 0,5 г (3 ммоль) (RS)-1-(2-азидопропил)-5-метилиндола в 20 мл этанола гидрировали в присутствии 70 мг Pd-C (5%). Затем отфильтровывали катализатор и промывали этанолом. Раствор упаривали и остаток растворяли в 100 мл простого эфира и затем обрабатывали 0,25 г (2,15 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,43 г (60,5%) фумарата (RS)-2-(6-метилиндол-1-ил)-1-метилэтиламина в виде белых кристаллов с Т. пл. 152- 153oC (распад).
Пример 18. а) Суспензию 0,26 г (8,75 ммоль, 80%) дисперсии гидрида натрия в 35 мл тетрагидрофурана обрабатывали при 0oC 0,97 г (7,2 ммоль) 6-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 1 мл (14 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали 7 мл воды. Далее разбавляли 180 мл простого эфира, дважды промывали соответственно порциями по 90 мл воды и 50 мл насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 28 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 1,1.г (79,3%) (RS)-1-(6-фториндол-1-ил)-пропан-2-ола в виде бесцветного масла.
б) Раствор 1,1 г (5,7 ммоль) (RS)-1-(6-фториндол-1-ил)-пропан-2-ола в 30 мл дихлорметана обрабатывали 3,17 мл (38,7 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,88 мл (11,4 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли 280 мл простого эфира, после чего дважды промывали порциями по 70 мл 1М раствора карбоната натрия и 35 мл насыщенного раствора хлорида натрия и водную фазу экстрагировали с помощью 140 мл простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 20 мл диметилформамида и после добавки 0,5 г (11,4 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. 3атем разбавляли 280 мл простого эфира и дважды экстрагировали соответственно порциями по 140 мл воды и один раз 70 мл насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью 140 мл простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 25 г силикагеля с помощью толуола. В результате получали 1 г (80,5%) (RS)-1-(2-азидопропил)-6-фториндола в виде желтоватого масла.
в) Раствор 1 г (3 ммоль) (RS)-1-(2-азидопропил)-6-фториндола в 30 мл этанола гидрировали в присутствии 100 мг Pd-C (5%). Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 100 мл простого эфира, после чего обрабатывали 0,5 г (4,3 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 1,1 г (78%) фумарата (RS)-2-(6-фториндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 158-159oC (распад).
Пример 19. В раствор 14,5 мл (102 ммоль) 2-метилацетоуксусного эфира в 100 мл этанола добавляли по каплям при 2-4oC 40 мл 50%-ного едкого кали. Затем добавляли 200 мл ледяной воды и обрабатывали раствором соли диазония, который приготавливали следующим образом: при охлаждении ледяной баней 20 мл 25%-ного раствора соляной кислоты по каплям добавляли в раствор 10 мл (100 ммоль) 3,4-дифторанилина в 100 мл этанола. Затем добавляли при 4oC 13,7 мл (102 ммоль) изопентилнитрита. Эмульсию, полученную при добавке раствора соли диазония, выливали в воду и экстрагировали с помощью толуола. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали до объема 160 мл. После кипячения в течение 1 ч в водоотделителе добавляли раствор 19 г (100 ммоль) моногидрата п-толуолсульфокислоты в 160 мл толуола и смесь продолжали нагревать еще в течение 1 ч. После охлаждения экстрагировали с помощью 1н. соляной кислоты, 1н. раствора гидроокиси натрия и насыщенного раствора хлорида натрия. Водные фазы промывали толуолом и соединенные органические фазы сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на 170 г силикагеля с помощью гексана/этилацетата (2:1). В результате получали 8,9 г (39%) сырого этилового эфира 4,5-дифториндол-2-карбоновой кислоты. Затем пробу перекристаллизовывали из гексана/этилацетата, и эта проба имела Т.пл. 149-150oC.
б) Раствор 2,06 г (9,15 ммоль) этилового эфира 5,6-дифториндол-2-карбоновой кислоты в 90 мл этанола обрабатывали 45 мл 2н. раствором гидроокиси натрия и перемешивали в течение 17 ч при комнатной температуре. Затем спирт выпаривали и остаток обрабатывали 50 мл 2н. соляной кислоты. Выпавшие кристаллы отфильтровывали, промывали водой и сушили. В результате получали 1,78 г (98,8%) 5,6-дифториндол-2-карбоновой кислоты в виде коричневых кристаллов с Т.пл. 279-280oC.
в) Металлическую ванну нагревали до 330oC после чего в атмосфере аргона вводили 1,74 г (8,83 ммоль) 5,6-дифториндол-2-карбоновой кислоты. Остаток черного цвета хроматографировали на 60 г силикагеля с помощью гексана/этилацетата (4:1). В результате получали 1,11 г (82%) 5,6-дифториндола в виде светло-коричневых кристаллов. Затем сублимировали пробу, которая имела Т.пл. 89-91oC.
г) Суспензию 0,12 г (4,1 ммоль, 80%) дисперсии гидрида натрия в 15 мл тетрагидрофурана обрабатывали при 0oC 0,5 г (3,2 ммоль) 5,6-дифториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,46 мл (6,5 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 60 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, дважды промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 35 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,46 г (66,5%) (R)-1-(5,6-дифториндол-1-ил)-пропан-2-ола в виде кристаллов желтоватого цвета с Т.пл. 114-116oC.
[α] = -44,4 (с 0,25, CHCl3).
д) Раствор 0,43 г (2 ммоль) (R)-1-(5,6-дифториндол-1-ил)-пропан-2-ола в 10 мл дихлорметана обрабатывали 0,86 мл (6,2 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,32 мл (4,14 ммоль) метансульфонилхлорид и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 10 мл диметилформамида и после добавки 0,26 г (4,1 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и дважды экстрагировали с помощью воды и один раз с помощью насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 15 г силикагеля с помощью толуола. В результате получали 0,4 г (83%) (S)-1-(2-азидопропил)-5,6-дифториндола в виде желтоватого масла.
[α] =+97,2 (с 0,25 CHCl3
е) Раствор 0,37 г (1,5 ммоль) (S)-1-(2-азидопропил)-5,6-дифториндола в 15 мл этанола гидрировали в присутствии 40 мг Pd-C (5%). Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 50 мл простого эфира, после чего обрабатывали 0,17 г (1,46 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,43 г (84%) фумарата (S)-2-(5,6-дифториндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 159-160oC (распад).
е) Раствор 0,37 г (1,5 ммоль) (S)-1-(2-азидопропил)-5,6-дифториндола в 15 мл этанола гидрировали в присутствии 40 мг Pd-C (5%). Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 50 мл простого эфира, после чего обрабатывали 0,17 г (1,46 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,43 г (84%) фумарата (S)-2-(5,6-дифториндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 159-160oC (распад).
[α] = +35,2 (с 0,25, CH3OH).
Пример 20. а) Суспензию 0,12 г (4,1 ммола(, 80%) дисперсии гидрида натрия в 15 мл тетрагидрофурана обрабатывали при 0oC 0,5 г (3,2 ммоль) 5,6-дифториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,46 мл (6,5 ммолей) (S)-метилоксирана реакционную смесь перемешивали в течение 60 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, дважды промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 35 г силикагеля с помощью толуола/этилацетата (19:1). В результате получали 0,5 г (74%) (S)-1-(5,6- дифториндол-1-ил)-пропан-2-ола в виде желтоватых кристаллов с Т.пл. 116- 118oC
[α] = +44,8 (с 0,25, CHCl3).
[α]
б) Раствор 0,48 г (2,3 ммоль) (S)-1-(5,6-дифториндол-1-ил)-пропан-2-ола в 10 мл дихлорметана обрабатывали 0,97 мл (6,9 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,36 мл (4,6 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 10 мл диметилформамида и после добавки 0,29 г (4,6 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и дважды экстрагировали с помощью воды и один раз с помощью насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 15 г силикагеля с помощью толуола. В результате получали 0,48 г (89,4%) (R)-1-(2-азидопропил)-5,6-дифториндола в виде желтоватого масла.
[α] = -98 (c 0,25 CHCl3.
в) Раствор 0,45 г (1,9 ммоль) (R)-1-(2-азидопропил)-5,6-дифториндола в 20 мл этанола гидрировали в присутствии 40 мг Pd-C (5%). Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 50 мл простого эфира и далее обрабатывали 0,21 г (1,8 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,51 г (82%) фумарата (R)-2-(5,6-дифториндол-1-ил)-1-метилэтиламина (1: 1) в виде белых кристаллов с Т.пл. 161-162oC (распад).
[α] = -34,4 (с 0,25, CHCl3).
Пример 21. а) В раствор 20,3 мл (143 ммоль) 2-метилацетоуксусного эфира в 140 мл этанола добавляли по каплям при 2-4oC 56 мл 50%-ного едкого кали. Далее добавляли 280 мл ледяной воды и быстро обрабатывали раствором соли диазония, который приготавливали следующим образом: При охлаждении ледяной баней 28 мл 25%-ного раствора соляной кислоты по каплям добавляли в раствор 25 г (140 ммоль) 4-фтор-3-трифторметиланилина в 140 мл этанола. Затем при 4oC добавляли 19,2 мл (143 ммоль) изопентилнитрита. Эмульсию, полученную путем добавки раствора соли диазония, выливали в воду и экстрагировали с помощью толуола. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали до получения объема 250 мл. После кипячения в течение 30 мин в водоотделителе добавляли раствор 26,6 г (140 ммоль) моногидрата п-толуолсульфокислоты в 250 мл толуола и смесь продолжали нагревать еще в течение 1 ч. После охлаждения экстрагировали с помощью 1н. соляной кислоты, 1н. гидроокиси натрия и насыщенного раствора хлорида натрия. Водные фазы промывали толуолом и соединенные органические фазы сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на 200 г силикагеля с помощью толуола. В результате получали 3,2 г (6,3%) сырого этилового эфира 5-фтор-6-трифторметилиндол-2-карбоновой кислоты. Затем пробу тщательно перемешивали с гексаном, и эта проба имела Т.пл. 159-162oC Вторая фракция содержала 1,4 г (3,6%) этилового эфира 5-фтор-4-трифторметилиндол-2-карбоновой кислоты в виде кристаллов оранжевого цвета с Т.пл. 121-124oC.
б) Раствор 2,24 г (8,14 ммоль) этилового эфира 5-фтор-4-трифторметилиндол-2-карбоновой кислоты в 80 мл этанола обрабатывали 40 мл 2н. гидроокиси натрия и перемешивали в течение 1 ч при комнатной температуре. Затем спирт выпаривали и pH раствора доводили с помощью 2н. соляной кислоты до pH 1. Выпавшие кристаллы выделяли, промывали водой и сушили. В результате получали 1,7 г (85%) 5-фтор-4-трифторметилиндол-2-карбоновой кислоты в виде кристаллов бежевого цвета с Т.пл. 204-210oC.
в) Суспензию 0,8 г (3,2 моль) 5-фтор-4-трифторметилиндол-2-карбоновой кислоты в 16 мл дифенилового эфира перемешивали в течение 4 ч при 260oC и после охлаждения до 0oC разбавляли 16 мл тетрагидрофурана. Затем добавляли 122 мг (4 ммоль, 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Далее добавляли 0,46 мл (6,5 ммоль) (S)-метилоксирана и реакционную смесь перемешивали в течение 60 ч при комнатной температуре. Смесь экстрагировали с помощью диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 40 г силикагеля сначала с помощью гексана/толуола в соотношении 1:1, а затем с помощью толуола/этилацетата в соотношении 15: 1. В результате получали 0,3 г (35,5%) (R)-1-(5-фтор-4-трифторметил-индол-1-ил)-пропан-2-ола в виде светло-коричневых кристаллов.
[α] = +34,4 (с 0,25, CHCl3).
г) Раствор 0,27 г (0,57 ммоль) (R)-1-(5-фтор-4-трифторметил-индол-1-ил)-пропан-2-ола в 5 мл дихлорметана обрабатывали 0,44 мл (3,2 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,16 мл (2,1 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего дважды промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 5 мл диметилформамида и после добавки 0,13 г (1,1 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и дважды экстрагировали с помощью воды и один раз с помощью насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 10 г силикагеля с помощью толуола/гексана (1:1). В результате получали 0,26 г (87,8%) (S)-1-(2-азидопропил)-5-фтор-4-трифторметилиндола в виде бесцветного масла.
MC: m/e (% основного пика): 286 (М+, 12), 216 (100).
д) Суспензию 26 мг окиси платины в 4,5 мл этанола в течение получаса перемешивали в атмосфере водорода, после чего обрабатывали раствором 0,26 г (0,9 ммоль) (S)-1-(2-азидопропил)-5-фтор-4-трифторметилиндола в 4,5 мл этанола. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 27 мл простого эфира и 0,3 мл метанола, после чего обрабатывали 105 мг (0,9 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,32 г (93,6%) фумарата (S)-2-(5-фтор-4-тритформетилиндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 180-181oC (распад).
[α] = + 36,8 (с 0,25, CH3OH).
Пример 22. а) Раствор 1,85 г (6,7 ммоль) этилового эфира 5-фтор-6-трифторметил-индол-2-карбоновой кислоты в 60 мл этанола обрабатывали 30 мл 2н. гидроокиси натрия и перемешивали в течение 1 ч при комнатной температуре. Затем спирт выпаривали и pH раствора с помощью 2н. соляной кислоты доводили до pH 1. Выпавшие кристаллы выделяли, промывали водой и сушили. В результате получали 1,5 г (90%) 5-фтор-6-трифторметилиндол-2-карбоновой кислоты в виде коричневых кристаллов с Т.пл. 178-180oC.
б) Суспензию 0,75 г (3 ммоль) 5-фтор-6-трифторметилиндол-2-карбоновой кислоты в 16 мл дифенилового эфира перемешивали в течение 4 ч при 260oC и после охлаждения до 0oC разбавляли 16 мл тетрагидрофурана. Затем добавляли 113 мг (3,8, ммоль 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Далее добавляли 0,43 мл (6,14 ммоль) (R)-метилоксирана и реакционную смесь перемешивали в течение 60 ч при комнатной температуре. Смесь экстрагировали с помощью диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 40 г силикагеля сначала с помощью гексана/толуола в соотношении 1:1, а затем с помощью толуола/этилацетата в соотношении 15: 1. В результате получали 0,15 г (18,9%) (R)-1-(5-фтор-6-трифторметилиндол-1-ил)-пропан-2-ола в виде светло-коричневых кристаллов.
в) Раствор 0,15 г (0,57 ммоль) (R)-1-(5-фтор-6-трифторметилиндол-1-ил)-пропан-2-ола в 3 мл дихлорметана обрабатывали 0,24 мл (1,73 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,09 мл (1,1 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 3 мл диметилформамида и после добавки 0,07 г (1,1 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Далее разбавляли простым эфиром и дважды экстрагировали с помощью воды и один раз с помощью насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 8 г силикагеля с помощью толуола/гексана (1:1). В результате получали 0,13 г (79,2%) (S)-1-(2-азидопропил)-5-фтор-6-трифторметилиндола в виде бесцветного масла.
MC: m/e (% основного пика): 286 (М+ 12), 216 (100).
г) Суспензию 13 мг окиси платины в 2,5 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 0,13 г (0,4 ммоль) (S)-1-(2-азидопропил)-5-фтор-6-трифторметилиндола в 2,5 мл этанола. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 14 мл простого эфира и 0,1 мл метанола, после чего обрабатывали 53 мг (0,45 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,16 г (93,5%) фумарата (S)-2-(5-фтор-6-трифторметилиндол-1-ил)-1- метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 170-171oC (распад).
[α] = +24 (с 0,25, CH3OH).
Пример 23. а) Суспензию 0,11 г (3,7 ммоля, 80%) дисперсии гидрида натрия в 15 мл тетрагидрофурана обрабатывали при 0oC 0,5 г (3 ммоль) 4-хлор-5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,4 мл (6 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 30 г силикагеля с помощью толуола/этилацетата (33: 1). В результате получали 0,5 г (74,5%) (R)-1-(4-хлор-5-фториндол-1-ил)-пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика): 227, 229 (М+, 32), 182, 184 (100).
б) Раствор 0,49 г (2,1 ммоль) (R)-1-(4-хлор-5-фториндол-1-ил) -пропан-2-ола в 11 мл дихлорметана обрабатывали 0,9 мл (6,5 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,33 мл (4,3 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 8 мл диметилформамида и после добавки 0,22 г (3,4 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и дважды экстрагировали с помощью воды и один раз с помощью насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 12 г силикагеля с помощью толуола. В результате получали 0,4 г (73,5%) (S)-1-(2-азидопропил)-4-хлор-5-фториндола в виде бесцветного масла.
МС: m/e (% основного пика): 252, 254 (М+, 34), 182, 184 (100).
в) Суспензию 40 мг окиси платины в 8 мл этанола в течение получаса перемешивали в атмосфере водорода и затем обрабатывали раствором 0,4 г (1,5 ммоль) (S)-1-(2-азидопропил)-4-хлор-5-фториндола в 8 мл этанола. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 46 мл простого эфира и 0,3 мл метанола, после чего обрабатывали 180 мг (1,55 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,48 г (83,8%) фумарата (S)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламина (1: 1) в виде белых кристаллов с Т.пл. 183-184oC (распад).
[α] = +32,4 (с 0,25, CH3OH).
Пример 24. а) Суспензию 0,11 г (3,7 ммоль, 80%) дисперсии гидрида натрия в 15 мл тетрагидрофурана обрабатывали при 0oC 0,5 г (3 ммоль) 4-хлор-5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,4 мл (6 ммоль) (S)-метилоксирана реакционную смесь в течение 48 ч перемешивали при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 30 г силикагеля с помощью толуола/этилацетата (33:1). В результате получали 0,53 г (78,9%) (S)-1-(4-хлор-5-фториндол-1-ил)-пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика): 227, 229 (М+, 32), 182, 184 (100).
б) Раствор 0,52 г (2,3 ммоль) (S)-1-(4-хлор-5-фториндол-1-ил)-пропан-2-ола в 11 мл дихлорметана обрабатывали 0,97 мл (6,9 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,36 мл (4,6 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 8 мл диметилформамида и после добавки 0,22 г (3,4 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Далее разбавляли простым эфиром и дважды экстрагировали с помощью воды и один раз с помощью насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия и сушили. Растворитель удаляли и остаток хроматографировали на 12 г силикагеля с помощью толуола. В результате получали 0,4 г (70%) (R)-1-(2-азидопропил)-4-хлор-5-фториндола в виде бесцветного масла.
МС: m/e (% основного пика): 252, 254 (М+, 34), 182, 184 (100).
в) Суспензию 40 мг окиси платины в 8 мл этанола в течение получаса перемешивали в атмосфере водорода и затем обрабатывали раствором 0,4 г (1,5 ммоль) (S)-1-(2-азидопропил)-4-хлор-5-фториндола в 8 мл этанола. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 46 мл простого эфира и 0,3 мл метанола, после чего обрабатывали 180 мг (1,55 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 0,48 г (83,8%) фумарата (R)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламина (1: 1) в виде белых кристаллов с Т.пл. 186-187oC (распад).
[α] = -32,4 (c 0,25, CH3OH).
Пример 25. а) Суспензию 0,26 г (8,7 ммоль, 80%) дисперсии гидрида натрия в 35 мл тетрагидрофурана обрабатывали при 0oC 0,95 г (7,25 ммоль) 5-метилиндола и перемешивали в течение 1 ч при этой температуре. После добавки 1 мл (14 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 30 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,86 г (62,7%) (RS)-1-(5-метилиндол-1-ил)-пропан-2-ола в виде масла желтого цвета.
б) Раствор 0,86 г (4,5 ммоль) (RS)-1-(5-метилиндол-1-ил)-пропан-2-ола в 20 мл дихлорметана обрабатывали 1,6 мл (11 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,6 мл (7,7 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 20 мл диметилформамида и после добавки 0,6 г (9,2 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого простым эфиром и соединение органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 30 г силикагеля с помощью толуола. В результате получали 0,68 г (71,6%) (RS)-1-(2-азидопропил)-5-метилиндола в виде желтого масла.
МС: m/e (% основного пика): 214 (М+, 20), 144 (100).
в) Раствор 0,6 г (2,6 ммоль) (RS)-1-(2-азидопропил)-5-метилиндола в 20 мл этанола гидрировали в присутствии 50 мг Pd-C (5%). Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 90 мл простого эфира, после чего обрабатывали 0,35 г (3 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,83 г (87%) фумарата (RS)-2-(5-метилиндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 165-167oC (распад).
Пример 26. а) При охлаждении ледяной баней 1,35 г (58,7 ммоль) натрия растворяли в 30 мл метанола, после чего в течение 20 мин добавляли раствор 4,65 г (29,3 моль) 3-хлор-4-фторбензальдегида и 6,75 г (58,6 ммоль) метилового эфира азидоуксусной кислоты в 10 мл метанола. Реакционную смесь перемешивали в течение 3 ч при комнатной температуре и затем нейтрализовали с помощью 2н. HCl. Далее экстрагировали с помощью этилацетата, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 250 г силикагеля с помощью н-гексана/толуола (2:1). В результате получали 2,5 г (33%) метилового эфира 3-хлор-4-фтор -α- азидокоричной кислоты в виде желтых кристаллов с Т.пл. 72-74oC.
б) Смесь 2,38 г (9,3 ммоль) метилового эфира 3-хлор-4-фтор -α- азидокоричной кислоты и 180 мл ксилола нагревали в течение 45 мин с обратным холодильником. Затем растворитель удаляли. В результате получали 2,04 г (колич. ) смеси (приблизительно в соотношении 1:1) метилового эфира 5-хлор-6-фториндол-2-карбоновой кислоты и метилового эфира 7-хлор-6-фториндол-2-карбоновой кислоты в виде светло-желтых кристаллов, которые без дальнейшей очистки использовали при проведении последующей ступени.
в) Суспензию 2,04 г (9 ммоль) смеси (приблизительно в соотношении 1:1) метилового эфира 5-хлор-6-фториндол-2-карбоновой кислоты и метилового эфира 7-хлор-6-фториндол-2-карбоновой кислоты в 90 мл этанола и 45 мл 2н. гидроокиси натрия перемешивали в течение 1 ч при комнатной температуре. Затем спирт выпаривали и остаток обрабатывали 55 мл 2н. соляной кислоты. Выпавшие кристаллы выделяли, промывали водой и сушили. В результате получали 1,92 г (колич. ) смеси 5-хлор-6-фториндол-2-карбоновой кислоты и 7-хлор-б-фториндол-2-карбоновой кислоты (примерно в соотношении 1:1) в виде желтых кристаллов.
г) Суспензию 1,92 г (9 ммоль) смеси 5-хлор-6-фториндол-2-карбоновой кислоты и 7-хлор-6-фториндол-2-карбоновой кислоты (приблизительно в соотношении 1: 1) в 45 мл дифенилового эфира перемешивали в течение 4 ч при 260oC. Затем реакционную смесь хроматографировали на 100 г силикагеля с помощью гексана и гексана/толуола (3:1). В результате получали 0,6 г (39%) 5-хлор-6-фториндола в виде светло-коричневых кристаллов с Т.пл. 88-90oC 0,22 г (14%) 7-хлор-6-фториндола в виде темно-коричневого масла.
д) Суспензию 128 мг (4,25 ммоль, 80%) дисперсии гидрида натрия в 8 мл тетрагидрофурана обрабатывали при 0oC 0,57 г (3,4 ммоль) 5-хлор-6-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,48 мл (6,8 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 25 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,59 г (77%) (R)-1-(5-хлор-6-фториндол-1-ил)-пропан-2-ола в виде кристаллов бежевого цвета с Т.пл. 108-110oC.
е) Раствор 0,57 г (2,5 ммоль) (R)-1-(5-хлор-6-фториндол-1-ил)-пропан-2-ола в 12 мл дихлорметана обрабатывали 0,4 мл (5 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор по каплям добавляли 0,2 мл (2,3 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали после этого с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 12 мл диметилформамида и после добавки 0,32 г (5 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 15 г силикагеля с помощью толуола/н-гексана (3:1). В результате получали 0,58 г (92%) (S)-1-(2-азидопропил)-5-хлор-6-фториндола в виде бесцветного масла.
МС: m/e (% основного пика): 252, 254 (М+, 10), 182, 184 (100).
ж) Суспензию 57 мг окиси платины в 10 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 0,57 г (2,25 ммоль) (S)-1-(2-азидопропил)-5-хлор-6-фториндола в 10 мл этанола. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 70 мл простого эфира и 2 мл метанола, после чего обрабатывали 262 мг (2,25 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,63 г (82%) фумарата (S)-2-(5-хлор-6-фториндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 159-161oC (распад).
Пример 27. а) Суспензию 45 мг (1,5 ммоль, 80%) дисперсии гидрида натрия в 10 мл тетрагидрофурана обрабатывали при 0oC 0,2 г (1,2 ммоль) 7-хлор-6-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,17 мл (2,4 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 4 дней при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 15 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,21 г (77,8%) (RS)-1-(7-хлор-6-фториндол-1-ил)-пропан-2-ола в виде белых кристаллов с Т.пл. 85-87oC.
б) Раствор 0,2 г (0,9 ммоль) (RS)-1-(7-хлор-6-фториндол-1-ил)-пропан-2-ола в 4,5 мл дихлорметана обрабатывали 0,4 мл (2,7 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,14 мл (1,81 ммоля) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали далее с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 6 мл диметилформамида и после добавки 0,15 г (2,3 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 10 г силикагеля с помощью толуола/н-гексана (1:1). В результате получали 0,17 г (80%) (RS)-1-(2-азидопропил)-7-хлор-6-фториндола в виде бесцветного масла.
в) Суспензию 17 мг окиси платины в 4 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 0,16 г (0,65 ммоль) (RS)-1-(2-азидопропил)-7-хлор-6-фториндола в 4 мл этанола. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 20 мл простого эфира и 0,2 мл метанола, после чего обрабатывали 74 мг (0,63 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,19 г (86,5%) фумарата (RS)-2-(7-хлор-6-фториндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 187-188oC (распад).
Пример 28. а) Суспензию 75 мг (2,5 ммоль, 80%) дисперсии гидрида натрия в 15 мл тетрагидрофурана обрабатывали при 0oC 0,33 г (2 ммоля) 5-xлop-2-метилиндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,28 мл (4 ммоль) (RS)-метилоксирана реакционную смесь перемешивали в течение 48 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 13 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,27 г (61,7%) (RS)-1-(5-хлор-2-метилиндол-1-ил)-пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика): 223, 225 (М+, 26), 178, 180 (100).
б) Раствор 0,26 г (1,1 ммоль) (RS)-1-(5-хлор-2-метил-индол-1-ил)-пропан-2-ола в 6 мл дихлорметана обрабатывали 0,5 мл (3,5 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор по каплям добавляли 0,2 мл (2,3 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали далее с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 6 мл диметилформамида и после добавки 0,15 г (2,3 ммоль) азида натрия перемешивали в течение 7 ч при 60oC. Затем разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 10 г силикагеля с помощью толуола/н-гексана (1:1). В результате получали 0,17 г (58,6%) (RS)-1-(2-азидопропил)-5-хлор-2-метилиндола в виде бесцветного масла.
МС: m/e (% основного пика): 248, 250 (М+, 16), 178, 180 (100).
в) Суспензию 16 мг окиси платины в 4 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 0,16 г (0,65 ммоль) (RS)-1-(2-азидопропил)-5-хлор-2-метилиндола в 4 мл этанола. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли 20 мл простого эфира и 0,2 мл метанола, после чего обрабатывали 74 мг (0,63 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,19 г (86,5%) фумарата (RS)-2-(5-хлор-2-метилиндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 193-194oC (распад).
Пример 29. а) Суспензию 263 мг (8,75 ммоль, 80%) дисперсии гидрида натрия в 35 мл тетрагидрофурана обрабатывали при 0oC 0,94 г (7 ммоль) 5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 1,22 мл (14 ммоль) (RS)-окиси бутилена реакционную смесь перемешивали в течение 96 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 60 г силикагеля с помощью толуола. В результате получали 1,2 г (83%) (RS)-(5-фториндол-1-ил)-бутан-2-ола в виде желтого масла.
МС: m/e (% основного пика): 207 (М+, 22), 148 (100).
б) Раствор 1,15 г (5,55 ммоль) (RS)-(5-фториндол-1-ил)-бутан-2-ола в 28 мл дихлорметана обрабатывали 3 мл (22 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,86 мл (11,1 ммоль) метансульфонилхлорида, реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали далее с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 28 мл диметилформамида и после добавки 0,72 г (11,1 ммоль) азида натрия перемешивали в течение 15 ч при 60oC. Затем разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 34 г силикагеля с помощью толуола. В результате получали 1,21 г (94%) (RS)-1-(3-азидобутил)-5-фториндола в виде масла желтоватого цвета.
МС: m/e (% основного пика): 232 (М+, 14), 148 (100).
в) Раствор 1,18 г (5,08 ммоль) (RS)-1-(3-азидобутил)-5-фториндола в 25 мл этанола гидрировали в присутствии 120 мг Pd-C (5%). Затем катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 150 мл простого эфира и 3 мл метанола, после чего обрабатывали 0,47 г (4,05 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 1,26 г (77%) фумарата (RS)-1-этил-2-(5-фториндол-1-ил)-этиламина (1: 1) в виде белых кристаллов с Т.пл. 169-171oC (распад).
Пример 30. а) В раствор 4,4 г (25,5 ммоль) 2-пропилацетоуксусного эфира в 26 мл этанола в течение 10 мин добавляли по каплям при 2-4oC 10 мл 50%-ного едкого кали. Затем добавляли 50 мл ледяной воды и быстро обрабатывали раствором соли диазония, который приготавливали следующим образом. При охлаждении ледяной баней 5 мл 25%-ного раствора соляной кислоты добавляли по каплям в раствор из 2,4 мл (25 ммоль) 4-фторанилина в 25 мл этанола. Далее при 4oC в течение 10 мин добавляли 3,4 мл (25,5 ммоль) изопентилнитрита. Оранжевую эмульсию, полученную в результате добавки раствора соли диазония, выливали в 100 мл воды и экстрагировали один раз с помощью 100 мл и дважды порциями по 50 мл толуола. Соединенные органические фазы сушили над сульфатом натрия, фильтровали и концентрировали до получения объема 50 мл. После кипячения в течение 30 мин в водоотделителе добавляли раствор 7,13 г (37,5 ммоль) моногидрата п-толуолсульфокислоты в 50 мл толуола и продолжали нагревать смесь в водоотделителе еще в течение 1 ч. После охлаждения экстрагировали с помощью 50 мл 1н. соляной кислоты, 50 мл 1н. гидроокиси натрия и 25 мл насыщенного раствора хлорида натрия. Водные фазы промывали 50 мл толуола, соединенные органические фазы сушили над сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на 105 г силикагеля с помощью толуола. В результате получали 4,27 г (72,6%) сырого этилового эфира 3-этил-5-фториндол-2-карбоновой кислоты. Пробу перекристаллизовывали из гексана, и эта проба имела Т.пл. 111-113oC.
б) Суспензию 1,4 г (6 ммоль) этилового эфира 3-этил-5-фториндол-2-карбоновой кислоты в 30 мл этанола обрабатывали 12 мл 2н. гидроокиси натрия и перемешивали в течение 16 ч. Этанол выпаривали и pH реакционного раствора доводили с помощью 25%-ной соляной кислоты до pH 1. Осадок промывали водой и сушили. В результате получали 1,22 г (98%) сырой 3-этил-5-фториндол-2-карбоновой кислоты, которую без дальнейшей очистки использовали при проведении последующей ступени.
в) Суспензию 1,21 г (5,8 ммоль) 3-этил-5-фториндол-2-карбоновой кислоты в 16 мл дифенилового эфира перемешивали в течение 2 ч при 260oC и после охлаждения до 0oC разбавляли 29 мл тетрагидрофурана. Далее добавляли 218 мг (7,28 ммоль, 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Затем добавляли 0,61 мл (8,74 ммоль) (R)-метилоксирана и реакционную смесь перемешивали в течение 90 ч при комнатной температуре. После этого смесь экстрагировали с помощью простого диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 120 г силикагеля с помощью сначала гексана/толуола (1:2), а затем толуола/этилацетата (9:1). В результате получали 0,8 г (62%) (R)-1-(3-этил-5-фториндол-1-ил)-пропан-2-ола в виде светло-коричневых кристаллов с Т.пл. 65-67oC.
г) Раствор 0,77 г (3,48 ммоль) (R)-1-(3-этил-5-фториндол-1-ил)-пропан-2-ола в 17 мл дихлорметана обрабатывали 1,9 мл (13,9 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,54 мл (6,96 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 2 ч. Далее разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали затем с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 17 мл диметилформамида и после добавки 0,45 г (6,96 ммоль) азида натрия перемешивали в течение ночи при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 15 г силикагеля с помощью толуола/гексана (2:1). В результате получали 0,78 г (91%) (S)-1-(2-азидопропил)-3-этил-5-фториндола в виде желтого масла.
МС: m/e (% основного пика): 246 (М+, 20), 176 (100).
д) Суспензию 76 мг окиси платины в 15 мл этанола в течение получаса перемешивали в атмосфере водорода и затем обрабатывали раствором 0,76 г (3,09 ммоль) (S)-1-(2-азидопропил)-3-этил-5-фториндола в 15 мл этанола. Реакционную смесь перемешивали в течение 20 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 90 мл простого эфира и 1,9 мл метанола, после чего обрабатывали 358 мг (3 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,91 г (88%) фумарата (S)-2-(2-этил-5-фториндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 164-166oC (распад).
Пример 31. а) Суспензию 0,16 г (6 ммоль, 80%) дисперсии гидрида натрия в 22 мл тетрагидрофурана обрабатывали при 0oC 0,38 г (2,14 ммоль) 4-изопропил-5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,6 мл (8,6 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 120 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 25 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,41 г (81%) (R)-1-(4-изопропил-5-фториндол-1-ил)-пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика): 235 (М+, 32), 190 (100).
б) Раствор из 0,41 г (1,74 ммоль) (R)-1-(4-изопропил-5-фториндол-1-ил)-пропан-2-ола в 10 мл дихлорметана обрабатывали 0,74 мл (5,3 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,27 мл (3,4 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 2 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали далее с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 10 мл диметилформамида и после добавки 0,22 г (3,4 ммоль) азида натрия перемешивали в течение ночи при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали затем с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и в результате получали 0,4 г (90%) (S)-1-(2-азидопропил)-4-изопропил-5-фториндола в виде желтого масла.
МС: m/e (% основного пика): 260 (М+, 18), 190 (100).
в) Раствор 0,4 г (1,5 ммоль) (S)-1-(2-азидопропил)-4-изопропил-5-фториндола в 15 мл этанола гидрировали в присутствии 40 мг Pd-C (5%). Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 45 мл простого эфира и 2 мл метанола, после чего обрабатывали 0,17 г (1,5 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,43 г (80%) фумарата (S)-2-(4-изопропил-5-фториндол-1-ил)-1-метилэтиламина (1: 1) в виде белых кристаллов с Т.пл. 166-167oC (распад).
Пример 32. а) Суспензию 0,05 г (1,6 ммоль, 80%) дисперсии гидрида натрия в 10 мл тетрагидрофурана обрабатывали при 0oC 0,23 г (2,14 ммоль) 6-изопропил-5-фториндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,18 мл (2,6 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 70 ч при комнатной температуре и затем обрабатывали водой. Далее разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 15 г силикагеля с помощью толуола/этилацетата (19: 1). В результате получали 0,08 г (27%) (R)-1-(6-изопропил-5-фториндол-1-ил)-пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика): 235 (М+, 46), 190 (100).
б) Раствор 0,08 г (0,34 ммоль) (R))-1-(6-изопропил-5-фториндол-1-ил)-пропан-2-ола в 1,7 мл дихлорметана обрабатывали 0,14 мл (1 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,05 мл (0,68 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 2 ч. Далее разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали затем с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 1,7 мл диметилформамида и после добавки 0,04 г (0,6 ммоль) азида натрия перемешивали в течение ночи при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали затем с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 10 г силикагеля с помощью толуола/гексана (1:1). В результате получали 0,06 г (75%) (S)-1-(2-азидопропил)-6-изопропил-5-фториндола в виде бесцветного масла.
МС: m/e (% основного пика): 260 (М+, 22), 190 (100).
в) Раствор 0,05 г (0,2 ммоль) (S)-1-(2-азидопропил)-6-изопропил-5-фториндола в 2 мл этанола гидрировали в присутствии 5 мг Pd-C (5%). Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 6 мл простого эфира, после чего обрабатывали 0,02 г (0,17 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,07 г (97%) фумарата (S)-2-(6-изопропил-5-фториндол-1-ил)-1-метилэтиламина (1:1,2) в виде белых кристаллов с Т.пл. 168-169oC (распад).
Пример 33. а) Суспензию 1,16 г (4,8 ммоль) 6-хлор-5-фтор-3-этил-индол-2-карбоновой кислоты в 16 мл дифенилового эфира перемешивали в течение 4 ч при 260oC и после охлаждения до 0oC разбавляли 24 мл тетрагидрофурана. Далее добавляли 180 мг (6 ммоль, 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Затем добавляли 0,5 мл (7,2 ммоль) (R)-метилоксирана и реакционную смесь перемешивали в течение 112 ч при комнатной температуре. Смесь экстрагировали с помощью простого диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 120 г силикагеля с помощью сначала гексана/толуола (1:2), а затем толуола/этилацетата (9:1). В результате получали 0,97 г (79%) (R)-1-(6-хлор-5-фтор-3-этилиндол-1-ил)-пропан-2-ола в виде светло-коричневых кристаллов с Т.пл. 112-114oC.
б) Раствор 0,93 г (3,66 ммоль) (R)-1-(6-хлор-5-фтор-3-этилиндол-1-ил)-пропан-2-ола в 18 мл дихлорметана обрабатывали 2 мл (14,6 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,57 мл (7,3 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 2 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали далее с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 18 мл диметилформамида и после добавки 0,47 г (7,3 ммоль) азида натрия перемешивали в течение ночи при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали затем с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 20 г силикагеля с помощью толуола/гексана (2:1). В результате получали 1 г (97%) (S)-1-(2-азидопропил)-6-хлор-5-фтор-3-этилиндола в виде желтого масла.
МС: m/e (% основного пика): 280 (М+, 20), 210 (100).
в) Суспензию 100 мг окиси платины в 17 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 0,97 г (3,5 ммоль) (S)-1-(2-азидопропил)-6-хлор-5-фтор-3-этилиндола в 17 мл этанола. Реакционную смесь перемешивали в течение 15 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 100 мл простого эфира и 2 мл метанола, после чего обрабатывали 400 мг (3,5 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 1,18 г (92%) фумарата (S)-2-(6-хлор-5-фтор-3-этилиндол-1-ил)-1-метил-этиламина (1:1) в виде белых кристаллов с Т.пл. 172-173oC (распад).
Пример 34. а) Суспензию 1,2 г (4,9 ммоль) 4-хлор-5-фтор-3-этилиндол-2-карбоновой кислоты в 16 мл дифенилового эфира перемешивали в течение 4 ч при 260oC и после охлаждения до 0oC разбавляли 24 мл тетрагидрофурана. Далее добавляли 186 мг (6,2 ммоль, 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Затем добавляли 0,5 мл (7,2 ммоль) (R)-метилоксирана и реакционную смесь перемешивали в течение 114 ч при комнатной температуре. Смесь экстрагировали с помощью простого диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 120 г силикагеля с помощью сначала гексана/толуола (1:2), а затем толуола/этилацетата (9:1). В результате получали 1,02 г (80%) (R)-1-(4-xлор-5-фтор-3-этилиндол-1-ил)-пропан-2-ола в виде светло-коричневых кристаллов с Т.пл. 87-88oC.
б) Раствор 0,98 г (3,85 ммоль) (R)-1-(4-хлор-5-фтор-3-этилиндол-1-ил)-пропан-2-ола в 19 мл дихлорметана обрабатывали 2 мл (14,6 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,6 мл (7,7 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 2 ч. Далее разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали затем с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 19 мл диметилформамида и после добавки 0,5 г (7,7 ммоль) азида натрия перемешивали в течение ночи при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого простым эфиром и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 20 г силикагеля с помощью толуола/гексана (2:1). В результате получали 1,04 г (96%) (S)-1-(2-азидопропил)-4-хлор-5-фтор-3-этилиндола в виде желтого масла.
МС: m/e (% основного пика): 280 (М+, 26), 210 (100).
в) Суспензию 100 мг окиси платины в 18 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 1,02 г (3,6 ммоль) (S)-1-(2-азидопропил)-4-хлор-5-фтор-3-этилиндола в 18 мл этанола. Реакционную смесь перемешивали в течение 4 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 100 мл простого эфира и 2 мл метанола, после чего обрабатывали 416 мг (3,5 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы отфильтровывали и сушили. В результате получали 1,23 г (91%) фумарата (S)-2-(4-xлop-5-фтор-3-этилиндол-1-от)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 178-181oC (распад).
Пример 35. а) Суспензию 1,16 г (4,9 ммоль) 5-фтор-3-метилиндол-2-карбоновой кислоты в 16 мл дифенилового эфира перемешивали в течение 5 ч при 260oC и после охлаждения до 0oC разбавляли 30 мл тетрагидрофурана. Далее добавляли 225 мг (7,5 ммоль, 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Затем добавляли 0,63 мл (7,2 ммоль) (R)-метилоксирана и реакционную смесь перемешивали в течение 90 ч при комнатной температуре. Смесь экстрагировали с помощью простого диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 120 г силикагеля с помощью сначала гексана/толуола (1:1), а затем толуола/этилацетата (19:1). В результате получали 0,87 г (70%) (R)-1-(5-фтop-3-мeтилиндoл-1-ил)-пpoпaн-2-ола в виде светло-коричневого масла.
МС: m/e (% основного пика): 207 (М+, 22), 207 (100).
б) Раствор 0,84 г (4,05 ммоль) (R)-1-(5-фтор-3-метилиндол-1-ил)-пропан-2-ола в 20 мл дихлорметана обрабатывали 2 мл (14,6 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,63 мл (8,1 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 2 ч. Далее разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали после этого с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 20 мл диметилформамида и после добавки 0,52 г (8,1 ммоль) азида натрия перемешивали в течение ночи при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 90 г силикагеля с помощью толуола/гексана (1:1). В результате получали 0,87 г (90%) (S)-1-(2-азидопропил)-5-фтор-3-метилиндола в виде бесцветного масла.
МС: m/e (% основного пика): 280 (М+, 26), 210 (100).
в) Раствор 0,85 г (3,66 ммоль) (S)-1-(2-азидопропил)-5-фтор-3-метилиндола в 37 мл этанола гидрировали в присутствии 85 мг Pd-C (5%). Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 100 мл простого эфира и 5 мл метанола, после чего обрабатывали 0,42 г (3,6 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 1,02 г (86%) фумарата (S)-2-(5-фтор-3-метилиндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т. пл. 167-168oC (распад).
Пример 36. а) Суспензию 0,07 г (2,3 ммоль, 80%) дисперсии гидрида натрия в 10 мл тетрагидрофурана обрабатывали при 0oC 0,33 г (1,82 ммоль) 6-хлор-5-фтор-3-метилиндола и перемешивали в течение 1 ч при этой температуре. После добавки 0,19 мл (2,7 ммоль) (R)-метилоксирана реакционную смесь перемешивали в течение 17 ч при комнатной температуре, затем смешивали с водой, разбавляли простым эфиром, промывали водой и насыщенным раствором хлорида натрия и органическую фазу сушили над сульфатом натрия. После удаления растворителя остаток хроматографировали на 20 г силикагеля с помощью толуола/этилацетата (9: 1). В результате получали 0,22 г (50%) (R)-1-(6-хлор-5-фтор-3-метилиндол-1-ил)-пропан-2-ола в виде желтого масла.
МС: m/e (% основного пика); 241 (М+, 28), 196 (100).
б) Раствор 0,2 г (0,85 ммоль) (R)-1-(6-хлор-5-фтор-3-метилиндол-1-ил)-пропан-2-ола в 4 мл дихлорметана обрабатывали 0,47 мл (3,4 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,13 мл (1,69 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 5 ч. Далее разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали затем с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 4 мл диметилформамида и после добавки 0,1 г (1,66 ммоль) азида натрия перемешивали в течение ночи при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 4,5 г силикагеля с помощью толуола/гексана (2:1). В результате получали 0,2 г (93%) (S)-1-(2-азидопропил)-6-хлор-5-фтор-3-метилиндола в виде бесцветного масла.
МС: m/e (% основного пика): 266 (М+, 20), 296 (100).
в) Суспензию 20 мг окиси платины в 3,6 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 0,19 г (0,7 ммоль) (S)-1-(2-азидопропил)-6-хлор-5-фтор-3-метилиндола в 3,6 мл этанола. Реакционную смесь перемешивали в течение 17 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 20 мл простого эфира и 0,4 мл метанола, после чего обрабатывали 76 мг (0,65 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,18 г (74%) фумарата (S)-2-(6-хлор-5-фтор-3-метилиндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 174-176oC (распад).
Пример 37. а) Суспензию 1,98 г (8,87 ммоль) 5-фтор-3-метокси-4-метилиндол-2-карбоновой кислоты в 16 мл дифенилового эфира перемешивали в течение 0,5 ч при 260oC после охлаждения до 0oC разбавляли 44 мл тетрагидрофурана. Затем добавляли 0,33 г (11 ммоль, 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Далее добавляли 1 мл (13,3 ммоль) (R)-метилоксирана и реакционную смесь перемешивали в течение 90 ч при комнатной температуре. Смесь экстрагировали с помощью диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 120 г силикагеля с помощью толуола и толуола/этилацетата (9: 1). В результате получали 1,69 г (80%) (R)-1-(5-фтор-3-метокси-4-метилиндол-1-ил)-пропан-2-ола в виде масла светло-коричневого цвета.
МС: m/e (% основного пика): 237 (М+, 56), 192 (100).
б) Раствор 1,64 г (6,91 ммоль) (R)-1-(5-фтор-3-метокси-4-метилиндол-1-ил)-пропан-2-ола в 35 мл дихлорметана обрабатывали 3,85 мл (27,6 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 1,07 мл (13,8 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 2 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали после этого с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 30 мл диметилформамида и после добавки 0,82 г (12,6 ммоль) азида натрия перемешивали в течение 3 ч при 60oC. Далее разбавляли простым эфиром и насыщенным раствором хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 50 г силикагеля с помощью толуола. В результате получали 1,82 г (77%) (S)-1-(2-азидопропил)-5-фтор-3-метокси-4-метилиндола в виде желтого масла.
МС: m/e (% основного пика): 262 (М+, 18), 192 (100).
в) Суспензию 127 мг окиси платины в 24 мл этанола перемешивали в течение получаса в атмосфере водорода и затем обрабатывали раствором 1,27 г (4,84 ммоль) (S)-1-(2-азидопропил)-5-фтор-3-метокси-4-метилиндола в 3,6 мл этанола. Реакционную смесь перемешивали в течение 15 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 145 мл простого эфира и 3 мл метанола, после чего обрабатывали 0,56 г (4,84 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 1,5 г (88%) фумарата (S)-2-(5-фтор-3-метокси-4-метилиндол-1-ил)-1-метилэтиламина (1:1) в виде белых кристаллов с Т.пл. 173-176oC (распад).
Пример 38. а) При обработке аргоном 1,4 г (6,8 ммоль) 3-метокси-4-метилиндол-2-карбоновой кислоты нагревали в течение 5 мин до 160oC и после охлаждения до 0oC разбавляли 34 мл тетрагидрофурана. Затем добавляли 0,25 г (8,5 ммоль, 80%) дисперсии гидрида натрия и перемешивали в течение 1 ч. Далее добавляли 0,72 мл (10,2 ммоль) (R)-метилоксирана и реакционную смесь перемешивали в течение 60 ч при комнатной температуре. Смесь экстрагировали с помощью диэтилового эфира, воды и насыщенного раствора хлорида натрия и органическую фазу сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 60 г силикагеля с помощью толуола/этилацетата (9:1). В результате получали 0,8 г (54%) (R)-1-(3-метокси-4-метилиндол-1-ил)-пропан-2-ола в виде светло-коричневого масла.
б) Раствор 0,8 г (3,644 ммоль) (R)-1-(3-метокси-4-метилиндол-1-ил)-пропан-2-ола в 18 мл дихлорметана обрабатывали 2 мл (14,5 ммоль) триэтиламина и охлаждали до 0oC. В этот раствор добавляли по каплям 0,5 мл (7,28 ммоль) метансульфонилхлорида и реакционную смесь перемешивали в течение 1 ч. Затем разбавляли простым эфиром, после чего промывали 1М раствором карбоната натрия и насыщенным раствором хлорида натрия и водную фазу экстрагировали после этого с помощью простого эфира. Соединенные органические фазы сушили над сульфатом натрия и растворитель удаляли. Остаток растворяли в 18 мл диметилформамида и после добавки 0,43 г (6,6 ммоль) азида натрия перемешивали в течение 3 ч при 60oC. Далее разбавляли простым эфиром и экстрагировали с помощью воды и насыщенного раствора хлорида натрия. Водную фазу экстрагировали после этого с помощью простого эфира и соединенные органические фазы сушили над сульфатом натрия. Растворитель удаляли и остаток хроматографировали на 22 г силикагеля с помощью толуола/гексана (1:1). В результате получали 0,46 г (57%) (S)-1-(2-азидопропил)-3-метокси-4-метилиндола в виде желтого масла.
МС: m/e (% основного пика): 244 (М+, 16), 174 (100).
в) Суспензию 45 мг окиси платины в 9 мл этанола в течение получаса перемешивали в атмосфере водорода и затем обрабатывали раствором 0,45 г (1,84 ммоль) (S)-1-(2-азидопропил)-3-метокси-4-метилиндола в 9 мл этанола. Реакционную смесь перемешивали в течение 3 ч при комнатной температуре. Катализатор отфильтровывали и промывали этанолом. Раствор упаривали и остаток растворяли в 55 мл простого эфира и 1 мл метанола, после чего обрабатывали 214 мг (1,8 ммоль) фумаровой кислоты и перемешивали в течение ночи. Выпавшие кристаллы выделяли и сушили. В результате получали 0,53 г (86%) фумарата (S)-2-(3-метокси-4-метилиндол-1-ил)-1-метил-этиламина (1:1) в виде светло-желтых кристаллов с Т.пл. 161-162oC (распад).
Пример А. По обычной методике изготавливают таблетки следующего состава, мг/таблетка:
Активное вещество - 100
Лактоза порошкообразная - 95
Кукурузный крахмал белый - 35
Поливинилпирролидон - 8
Na-Карбоксиметилированный крахмал - 10
Стеарат магния - 2
Вес таблетки - 250
Пример Б. По обычной методике изготавливают таблетки следующего состава, мг/таблетка:
Активное вещество - 200
Лактоза порошкообразная - 100
Кукурузный крахмал белый - 64
Поливинилпирролидон - 12
Na-Карбоксиметилированный крахмал - 20
Стеарат магния - 4
Вес таблетки - 400
Пример В. Изготавливают капсулы следующего состава, мг/капсула:
Активное вещество - 50
Лактоза кристаллическая - 60
Микрокристаллическая целлюлоза - 34
Тальк - 5
Стеарат магния - 1
Вес содержимого капсулы - 150
Активное вещество с соответствующим размером частиц, кристаллическую лактозу и микрокристаллическую целлюлозу тщательно перемешивают до получения гомогенной смеси, пропускают через сито, после чего добавляют при перемешивании тальк и стеарат магния. Из полученной смеси изготавливают твердожелатиновые капсулы соответствующего размера.
Активное вещество - 100
Лактоза порошкообразная - 95
Кукурузный крахмал белый - 35
Поливинилпирролидон - 8
Na-Карбоксиметилированный крахмал - 10
Стеарат магния - 2
Вес таблетки - 250
Пример Б. По обычной методике изготавливают таблетки следующего состава, мг/таблетка:
Активное вещество - 200
Лактоза порошкообразная - 100
Кукурузный крахмал белый - 64
Поливинилпирролидон - 12
Na-Карбоксиметилированный крахмал - 20
Стеарат магния - 4
Вес таблетки - 400
Пример В. Изготавливают капсулы следующего состава, мг/капсула:
Активное вещество - 50
Лактоза кристаллическая - 60
Микрокристаллическая целлюлоза - 34
Тальк - 5
Стеарат магния - 1
Вес содержимого капсулы - 150
Активное вещество с соответствующим размером частиц, кристаллическую лактозу и микрокристаллическую целлюлозу тщательно перемешивают до получения гомогенной смеси, пропускают через сито, после чего добавляют при перемешивании тальк и стеарат магния. Из полученной смеси изготавливают твердожелатиновые капсулы соответствующего размера.
Claims (7)
1. Производные индола общей формулы I

где R1 - R4 - водород, галоген, низший алкил или трифторметил;
R5 - R6 - водород, галоген, низший алкил, трифторметил или низший алкокси;
R7 - низший алкил,
а также их фармацевтически приемлемые кислотно-аддитивные соли.
где R1 - R4 - водород, галоген, низший алкил или трифторметил;
R5 - R6 - водород, галоген, низший алкил, трифторметил или низший алкокси;
R7 - низший алкил,
а также их фармацевтически приемлемые кислотно-аддитивные соли.
2. Соединения по п.1, где R7 - метил.
3. Соединения по п.2, где R5 и R6 - водород.
4. Соединения по пп.1 - 3, где R1 - водород или метил, R2 - водород или фтор, R3 - водород или хлор и R4 - водород.
5. Соединение по п.1, представляющее собой (S)-2-(6-хлор-5-фтор-индол-1-ил)-1-метилэтиламин.
6. Соединение по п.1, выбранное из группы, включающей:
(RS)-2-(5-хлориндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(RS)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин, (RS)-2-(4-метилиндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-броминдол-1-ил)-1-метилэтиламин,
(RS)-2-(6-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(5,6-дифториндол-1-ил)-1-метилэтиламин,
(R)-2-(5,6-дифториндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-4-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-6-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин.
(RS)-2-(5-хлориндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(RS)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(5-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(6-хлор-5-фториндол-1-ил)-1-метилэтиламин, (RS)-2-(4-метилиндол-1-ил)-1-метилэтиламин,
(RS)-2-(5-броминдол-1-ил)-1-метилэтиламин,
(RS)-2-(6-фториндол-1-ил)-1-метилэтиламин,
(S)-2-(5,6-дифториндол-1-ил)-1-метилэтиламин,
(R)-2-(5,6-дифториндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-4-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(5-фтор-6-трифторметилиндол-1-ил)-1-метилэтиламин,
(S)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин,
(R)-2-(4-хлор-5-фториндол-1-ил)-1-метилэтиламин.
7. Фармацевтический препарат, обладающий способностью связываться с серотонинрецепторами, содержащий активное вещество и терапевтически инертный наполнитель, отличающийся тем, что в качестве активного вещества он содержит производные индола общей формы I по п.1 - 6.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CH3201/93 | 1993-10-22 | ||
| CH320193 | 1993-10-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU94037964A RU94037964A (ru) | 1996-09-10 |
| RU2136662C1 true RU2136662C1 (ru) | 1999-09-10 |
Family
ID=4250849
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU94037964A RU2136662C1 (ru) | 1993-10-22 | 1994-10-20 | Производные индола и их фармацевтически приемлемые кислотно-аддитивные соли, фармацевтический препарат на их основе |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US5494928A (ru) |
| EP (1) | EP0655440A3 (ru) |
| JP (1) | JP2638752B2 (ru) |
| KR (1) | KR950011406A (ru) |
| CN (1) | CN1061341C (ru) |
| AU (1) | AU685841B2 (ru) |
| BR (1) | BR9404203A (ru) |
| CA (1) | CA2132883A1 (ru) |
| CO (1) | CO4290356A1 (ru) |
| CZ (1) | CZ260494A3 (ru) |
| FI (1) | FI944969A (ru) |
| HU (1) | HU218669B (ru) |
| IL (1) | IL111314A (ru) |
| NO (1) | NO301369B1 (ru) |
| NZ (1) | NZ264713A (ru) |
| PL (1) | PL179561B1 (ru) |
| RU (1) | RU2136662C1 (ru) |
| TW (1) | TW270114B (ru) |
| ZA (1) | ZA948094B (ru) |
Families Citing this family (76)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9523999D0 (en) | 1995-11-23 | 1996-01-24 | Lilly Co Eli | Indolyl neuropeptide y receptor antagonists |
| PL320010A1 (en) * | 1995-09-01 | 1997-09-01 | Lilly Co Eli | Indolyllic antagonists of neuropeptide y receptors |
| AU3255697A (en) * | 1996-06-12 | 1998-01-07 | F. Hoffmann-La Roche Ag | A process for the manufacture of 1-(amino-alkyl)-indoles |
| AU727654B2 (en) * | 1997-06-13 | 2000-12-21 | Yamanouchi Pharmaceutical Co., Ltd. | Tricyclic pyrazole derivative |
| IL129322A (en) * | 1997-08-07 | 2004-02-19 | Fujimoto Brothers Co Ltd | History of ethylamine and pharmaceutical preparations containing them |
| US6960579B1 (en) * | 1998-05-19 | 2005-11-01 | Alcon Manufacturing, Ltd. | Serotonergic 5HT7 receptor compounds for treating ocular and CNS disorders |
| RS50104B (sr) * | 1998-08-21 | 2009-01-22 | Novartis Ag., | Nova oralna formulacija za 5-th4 agoniste ili antagoniste |
| GB9819019D0 (en) * | 1998-09-01 | 1998-10-28 | Cerebrus Ltd | Chemical compounds II |
| GB9819032D0 (en) * | 1998-09-01 | 1998-10-28 | Cerebrus Ltd | Chemical compounds IV |
| GB9819033D0 (en) * | 1998-09-01 | 1998-10-28 | Cerebrus Ltd | Chemical compounds VI |
| US6326379B1 (en) | 1998-09-16 | 2001-12-04 | Bristol-Myers Squibb Co. | Fused pyridine inhibitors of cGMP phosphodiesterase |
| US6664286B1 (en) | 1998-09-18 | 2003-12-16 | Alcon Manufacturing, Ltd. | Serotonergic 5ht2 agonists for treating glaucoma |
| FR2788772B1 (fr) * | 1999-01-26 | 2001-03-02 | Adir | Nouveaux composes cyano-indoles inhibiteurs de recapture de serotonine, leur procede de preparation et les compositions pharmaceutiques qui les contiennent |
| FR2792313A1 (fr) * | 1999-04-13 | 2000-10-20 | Synthelabo | Derives de 2-aminoethyl-indole, leur preparation et leur application en therapeutique |
| US6465467B1 (en) | 1999-05-21 | 2002-10-15 | Biovitrum Ab | Certain aryl-aliphatic and heteroaryl-aliphatic piperazinyl pyrazines and their use in the treatment of serotonin-related diseases |
| GB9918962D0 (en) | 1999-08-11 | 1999-10-13 | Cerebrus Ltd | Chemical compounds xxii |
| GB9918965D0 (en) * | 1999-08-11 | 1999-10-13 | Cerebrus Ltd | Chemical compounds xxi |
| JP5117658B2 (ja) * | 1999-11-05 | 2013-01-16 | エミスフェアー・テクノロジーズ・インク | フェノキシカルボン酸化合物及び活性剤を送達するための組成物 |
| WO2001040183A1 (en) * | 1999-12-03 | 2001-06-07 | Alcon Universal Ltd. | 1-aminoalkyl-1h-indoles for treating glaucoma |
| JP3585796B2 (ja) | 1999-12-17 | 2004-11-04 | 新光電気工業株式会社 | 多層配線基板の製造方法、及び半導体装置 |
| EP1132389A1 (en) | 2000-03-06 | 2001-09-12 | Vernalis Research Limited | New aza-indolyl derivatives for the treatment of obesity |
| US7012090B1 (en) | 2000-03-17 | 2006-03-14 | Alcon, Inc. | Pyranoindoles for treating glaucoma |
| EP1263434A1 (en) * | 2000-03-17 | 2002-12-11 | Alcon, Inc | Compounds with 5-ht 2 and 5-ht 1a agonist activity for treating glaucoma |
| US6806285B1 (en) | 2000-03-17 | 2004-10-19 | Alcon, Inc. | 5-Hydroxyl indole derivatives for treating glaucoma |
| US7005443B1 (en) * | 2000-03-17 | 2006-02-28 | Alcon, Inc. | 5-Hydroxy indazole derivatives for treating glaucoma |
| US6956036B1 (en) | 2000-03-17 | 2005-10-18 | Alcon, Inc. | 6-hydroxy-indazole derivatives for treating glaucoma |
| AU2001219185A1 (en) | 2000-03-17 | 2001-10-03 | Alcon, Inc. | 2-acylaminobenzimidazole derivatives for treating glaucoma |
| US6903081B2 (en) * | 2000-03-31 | 2005-06-07 | Purdue Research Foundation | Phosphoramidates and methods therefor |
| TWI290136B (en) | 2000-04-05 | 2007-11-21 | Daiichi Seiyaku Co | Ethylenediamine derivatives |
| AU5256301A (en) * | 2000-04-28 | 2001-11-12 | Yamanouchi Pharmaceutical Co., Ltd. | Froindazole derivative |
| JP4047723B2 (ja) * | 2000-10-16 | 2008-02-13 | エフ.ホフマン−ラ ロシュ アーゲー | インドリン誘導体、および5−ht2受容体リガンドとしてのその使用 |
| DK1337518T3 (da) | 2000-11-20 | 2009-10-19 | Biovitrum Ab Publ | Piperazinylpyrazinforbindelser som antagonister for serotonin-5-HT2-receptoren |
| SE0004245D0 (sv) | 2000-11-20 | 2000-11-20 | Pharmacia Ab | Novel compounds and their use |
| WO2004098807A1 (en) * | 2000-11-21 | 2004-11-18 | Barsplice Products, Inc. | Method of making steel couplers for joining concrete reinforcing bars |
| GB0030710D0 (en) * | 2000-12-15 | 2001-01-31 | Hoffmann La Roche | Piperazine derivatives |
| JP2005506280A (ja) | 2000-12-20 | 2005-03-03 | ブリストル−マイヤーズ・スクイブ・ファーマ・カンパニー | アリールおよびアミノアリール置換セロトニン受容体アゴニストおよびアンタゴニストリガンド |
| JP4094950B2 (ja) | 2000-12-27 | 2008-06-04 | エフ.ホフマン−ラ ロシュ アーゲー | インドール誘導体および5−ht2bおよび5−ht2c受容体リガンドとしてのそれらの使用 |
| JP4544820B2 (ja) | 2001-03-09 | 2010-09-15 | オーソ−マクニール・フアーマシユーチカル・インコーポレーテツド | 複素環化合物 |
| WO2002098860A1 (en) * | 2001-06-01 | 2002-12-12 | Alcon, Inc. | Novel fused indazoles and indoles and their use for the treatment of glaucoma |
| CA2447479A1 (en) * | 2001-06-01 | 2002-12-12 | Alcon, Inc. | Novel arylaminopropane analogues and their use for the treatment of glaucoma |
| DE60209486T2 (de) * | 2001-06-01 | 2006-08-03 | Alcon Inc. | Pyranoindazole und ihre verwendung in der glaukombehandlung |
| US7365205B2 (en) | 2001-06-20 | 2008-04-29 | Daiichi Sankyo Company, Limited | Diamine derivatives |
| EP1405852B9 (en) | 2001-06-20 | 2013-03-27 | Daiichi Sankyo Company, Limited | Diamine derivatives |
| US6884816B2 (en) * | 2001-08-31 | 2005-04-26 | Alcon, Inc. | Hydroxy substituted fused naphthyl-azoles and fused indeno-azoles and their use for the treatment of glaucoma |
| TW593302B (en) * | 2001-12-20 | 2004-06-21 | Alcon Inc | Novel benzodifuranimidazoline and benzofuranimidazoline derivatives and their use for the treatment of glaucoma |
| AU2003243089B2 (en) | 2002-06-19 | 2010-01-07 | Biovitrum Ab (Publ) | Novel compounds, their use and preparation |
| AU2003262931B2 (en) * | 2002-08-30 | 2008-06-26 | Alcon, Inc. | Substituted 5-chroman-5-yl-ethylamine compounds and their use for the treatment of glaucoma |
| AU2003265886A1 (en) | 2002-09-06 | 2004-03-29 | Janssen Pharmaceutica N.V. | (1H-Benzoimidazol-2-yl)-(piperazinyl)-methanone derivatives and related compounds as histamine H4-receptor antagonists for the treatment of inflammatory and allergic disorders |
| US20040127395A1 (en) * | 2002-09-06 | 2004-07-01 | Desai Pragnya J. | Use of histamine H4 receptor modulators for the treatment of allergy and asthma |
| CA2506204A1 (en) * | 2002-12-13 | 2004-07-01 | Alcon, Inc. | Novel benzopyran analogs and their use for the treatment of glaucoma |
| CL2004000826A1 (es) | 2003-04-25 | 2005-03-04 | Pfizer | Uso de un agonista para el receptor 5-ht2c para preparar un medicamento util en el tratamiento de la incontinencia urinaria provocada por estres, con la condicion de que el agonista no sea 1-[6-cloro-5-(trifluorometil)-2-piridinil]piperazina (org-129 |
| GB0314967D0 (en) | 2003-06-26 | 2003-07-30 | Hoffmann La Roche | Piperazine derivatives |
| WO2005053688A1 (en) * | 2003-11-26 | 2005-06-16 | Alcon, Inc. | Substituted furo[2,3-g] indazoles for the treatment of glaucoma |
| WO2005058911A2 (en) * | 2003-12-15 | 2005-06-30 | Alcon, Inc. | Substituted [1,4]oxazino[2,3-g]indazoles for the treatment of glaucoma |
| US7338972B1 (en) | 2003-12-15 | 2008-03-04 | Alcon, Inc. | Substituted 1-alkylamino-1H-indazoles for the treatment of glaucoma |
| US7129257B1 (en) | 2003-12-15 | 2006-10-31 | Alcon, Inc. | Pyrazolo[3,4- e]benzoxazoles for the treatment of glaucoma |
| EP2400300A1 (en) | 2004-08-25 | 2011-12-28 | Takeda Pharmaceutical Company Limited | Method of screening preventives/remedies for stress urinary incontinence |
| US20060073172A1 (en) * | 2004-10-01 | 2006-04-06 | Schneider L W | Stabilized ophthalmic solution for the treatment of glaucoma and lowering intraocular pressure |
| WO2006062839A1 (en) * | 2004-12-08 | 2006-06-15 | Alcon, Inc. | Use of dioxindoindazoles and dioxoloindazoles for treating glaucoma |
| WO2006103511A1 (en) | 2005-03-31 | 2006-10-05 | Pfizer Products Inc. | Cyclopentapyridine and tetrahydroquinoline derivatives |
| TW200744567A (en) * | 2005-09-23 | 2007-12-16 | Alcon Inc | Phenylethylamine analogs and their use for treating glaucoma |
| WO2007132841A1 (ja) | 2006-05-16 | 2007-11-22 | Takeda Pharmaceutical Company Limited | 縮合複素環化合物およびその用途 |
| WO2007149728A2 (en) * | 2006-06-20 | 2007-12-27 | Alcon Research, Ltd. | Aryl and heteroaryl tetrahydrobenzazepine derivatives and their use for treating glaucoma |
| EP2044029B1 (en) * | 2006-07-14 | 2011-01-26 | Pfizer Products Inc. | Tartrate salt of (7s)-7-[(5-fluoro-2-methyl-benzyl)oxy]-2-[(2r)-2-methylpiperazin-1-yl]-6,7-dihydro-5h-cyclopenta[b]pyridine |
| WO2009063992A1 (ja) | 2007-11-15 | 2009-05-22 | Takeda Pharmaceutical Company Limited | 縮合ピリジン誘導体およびその用途 |
| DE102008009609A1 (de) | 2008-02-18 | 2009-08-20 | Freie Universität Berlin | Substituierte 4-(Indol-3-yl)chinazoline und deren Verwendung und Herstellung |
| KR101062376B1 (ko) | 2008-04-10 | 2011-09-06 | 한국화학연구원 | 신규 인돌 카르복실산 비스피리딜 카르복사마이드 유도체,이의 제조방법 및 이를 유효성분으로 함유하는 조성물 |
| JPWO2011071136A1 (ja) | 2009-12-11 | 2013-04-22 | アステラス製薬株式会社 | 線維筋痛症治療剤 |
| US20130267500A1 (en) | 2010-09-01 | 2013-10-10 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists in the treatment of disorders ameliorated by reduction of norepinephrine level |
| CN102702066A (zh) * | 2011-03-28 | 2012-10-03 | 苏州百灵威超精细材料有限公司 | 一种用于合成抗癌和减肥药物中间体6-氯-5-氟吲哚的制备新方法 |
| EP2968284B1 (en) | 2013-03-14 | 2021-04-21 | Osteoqc Inc. | Alkyl-amine harmine derivatives for promoting bone growth |
| JPWO2019131902A1 (ja) | 2017-12-27 | 2020-12-10 | 武田薬品工業株式会社 | 腹圧性尿失禁および便失禁の治療薬 |
| WO2020037001A1 (en) | 2018-08-14 | 2020-02-20 | Osteoqc Inc. | FLUORO β-CARBOLINE COMPOUNDS |
| JP2022504011A (ja) | 2018-08-14 | 2022-01-13 | オステオーク インコーポレイティド | ピロロ-ジピリジン化合物 |
| BR112021016620A2 (pt) | 2019-02-27 | 2021-11-03 | Univ California | Azepino-indóis e outros heterociclos para o tratamento de distúrbios cerebrais |
| SG11202109020QA (en) | 2019-02-27 | 2021-09-29 | Univ California | N-substituted indoles and other heterocycles for treating brain disorders |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE791899A (fr) * | 1971-11-26 | 1973-05-24 | Ciba Geigy | Procede de preparation d'indoles n-substitues |
| US3904617A (en) * | 1972-10-12 | 1975-09-09 | Ayerst Mckenna & Harrison | Process for preparing new heterocyclic derivatives |
| IE37234B1 (en) * | 1972-02-14 | 1977-06-08 | Ayerst Mckenna & Harrison | Fused-ring indole derivatives |
| DE2430682A1 (de) * | 1974-06-26 | 1976-01-15 | Bayer Ag | Indolyl-mono-azofarbstoffe |
| FR2355006A1 (fr) * | 1976-06-17 | 1978-01-13 | Labaz | Nouveaux derives d'indole et procedes pour les preparer |
| DE2824447A1 (de) * | 1978-06-03 | 1979-12-13 | Cassella Ag | Pyrazino-carbazole, verfahren zu ihrer herstellung und sie enthaltendes pharmazeutisches mittel |
| DE3022648A1 (de) * | 1979-06-29 | 1981-01-15 | Sandoz Ag | Neue organische verbindungen, ihre herstellung und pharmazeutischen praeparate, welche diese verbindungen enthalten |
| FR2564465B1 (fr) * | 1984-05-16 | 1986-10-03 | Cortial | Nouveaux diamino-1,3 propanols-2, leurs methodes de preparation et leurs applications therapeutiques |
| PH23498A (en) * | 1984-08-06 | 1989-08-16 | Sterling Drug Inc | 3-carbonyl-1-aminoalkyl-1h-indoles,composition and use thereof |
| US4660060A (en) * | 1985-06-17 | 1987-04-21 | The Hilton-Davis Chemical Co. | Imaging systems containing 3-(indol-3-yl)-3-(4-substituted aminophenyl)phthalides |
| SK278989B6 (sk) * | 1988-02-10 | 1998-05-06 | F. Hoffmann-La Roche Ag | Substituované pyroly, ich použitie na výrobu lieči |
| FR2679561A1 (fr) * | 1991-07-26 | 1993-01-29 | Schneider Marc | Nouveaux derives de l'(indolyl-1) alkylamine substituee par une chaine presentant des groupements carbonyles conjugues a un noyau aromatique. |
| US5252732A (en) * | 1991-09-09 | 1993-10-12 | Merck & Co., Inc. | D-heteroaryl, O-alkylheteroaryl, O-alkenylheteroaryl and O-alkynylheteroarylmacrolides having immunosuppressive activity |
| EP0572863A1 (de) * | 1992-06-05 | 1993-12-08 | F. Hoffmann-La Roche Ag | ZNS Pyrazinoindole |
| US5310901A (en) * | 1993-03-05 | 1994-05-10 | Merck & Co., Inc. | O-heteroaryl, O-alkylheteroaryl, O-alkenylheteroaryl and O-alkynlheteroarylrapamycin derivatives |
-
1994
- 1994-09-07 TW TW083108265A patent/TW270114B/zh active
- 1994-09-21 CA CA002132883A patent/CA2132883A1/en not_active Abandoned
- 1994-10-03 US US08/317,259 patent/US5494928A/en not_active Expired - Fee Related
- 1994-10-07 EP EP94115800A patent/EP0655440A3/de not_active Withdrawn
- 1994-10-14 AU AU75837/94A patent/AU685841B2/en not_active Ceased
- 1994-10-14 ZA ZA948094A patent/ZA948094B/xx unknown
- 1994-10-17 HU HU9402983A patent/HU218669B/hu not_active IP Right Cessation
- 1994-10-17 IL IL11131494A patent/IL111314A/xx not_active IP Right Cessation
- 1994-10-17 NZ NZ264713A patent/NZ264713A/en unknown
- 1994-10-19 CO CO94047587A patent/CO4290356A1/es unknown
- 1994-10-20 CN CN94117149A patent/CN1061341C/zh not_active Expired - Fee Related
- 1994-10-20 RU RU94037964A patent/RU2136662C1/ru active
- 1994-10-21 PL PL94305543A patent/PL179561B1/pl unknown
- 1994-10-21 BR BR9404203A patent/BR9404203A/pt not_active Application Discontinuation
- 1994-10-21 CZ CZ942604A patent/CZ260494A3/cs unknown
- 1994-10-21 NO NO943999A patent/NO301369B1/no not_active IP Right Cessation
- 1994-10-21 FI FI944969A patent/FI944969A/fi unknown
- 1994-10-22 KR KR1019940027045A patent/KR950011406A/ko not_active Application Discontinuation
- 1994-10-24 JP JP6257775A patent/JP2638752B2/ja not_active Expired - Lifetime
Non-Patent Citations (1)
| Title |
|---|
| Машковский М.Д. Лекарственные средства. - М.: Медицина, 1986, ч.1, с.117 - 138. * |
Also Published As
| Publication number | Publication date |
|---|---|
| CO4290356A1 (es) | 1996-04-17 |
| RU94037964A (ru) | 1996-09-10 |
| JP2638752B2 (ja) | 1997-08-06 |
| FI944969A0 (fi) | 1994-10-21 |
| HU218669B (hu) | 2000-10-28 |
| IL111314A (en) | 1999-08-17 |
| CN1061341C (zh) | 2001-01-31 |
| ZA948094B (en) | 1995-04-24 |
| US5494928A (en) | 1996-02-27 |
| HUT70848A (en) | 1995-11-28 |
| IL111314A0 (en) | 1994-12-29 |
| EP0655440A3 (de) | 1996-11-06 |
| BR9404203A (pt) | 1995-07-04 |
| CZ260494A3 (en) | 1995-10-18 |
| CN1105988A (zh) | 1995-08-02 |
| NO301369B1 (no) | 1997-10-20 |
| CA2132883A1 (en) | 1995-04-23 |
| PL305543A1 (en) | 1995-05-02 |
| HU9402983D0 (en) | 1995-02-28 |
| JPH07149723A (ja) | 1995-06-13 |
| EP0655440A2 (de) | 1995-05-31 |
| AU685841B2 (en) | 1998-01-29 |
| KR950011406A (ko) | 1995-05-15 |
| PL179561B1 (pl) | 2000-09-29 |
| NZ264713A (en) | 1996-05-28 |
| FI944969A (fi) | 1995-04-23 |
| NO943999D0 (no) | 1994-10-21 |
| TW270114B (ru) | 1996-02-11 |
| AU7583794A (en) | 1995-05-11 |
| NO943999L (no) | 1995-04-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2136662C1 (ru) | Производные индола и их фармацевтически приемлемые кислотно-аддитивные соли, фармацевтический препарат на их основе | |
| RU2152388C1 (ru) | Трициклические производные пиразола | |
| JP3263081B2 (ja) | バソプレシンおよび/またはオシトシン作動薬および/または拮抗薬としての窒素基によって3位において置換されている1,3−ジヒドロインドール−2−オン誘導体 | |
| RU2128649C1 (ru) | Трициклические производные пиррола, фармацевтический препарат | |
| EP0593513B1 (en) | Tryptamine analogues, their synthesis and their use as 5-ht1-like receptors or 5-ht2 receptor agonistes | |
| JP2978566B2 (ja) | ノイロキニンレセプター拮抗薬としての5−アザビシクロ[3.1.0]ヘキシルアルキル−2−ピペリドンおよび−グルタルイミド | |
| GB2081717A (en) | Hereocyclic compounds | |
| JP3327473B2 (ja) | インドール誘導体 | |
| AU2004261649A1 (en) | 3-amino choman and 2-amino tetralin derivatives | |
| JPH07149762A (ja) | ピペリジン類およびピペラジン類 | |
| MX2008003202A (es) | Derivados de carbazol. | |
| JPH08109169A (ja) | 非ペプチドタキキニン受容体拮抗物質 | |
| RU2151148C1 (ru) | 3-индолилпиперидины, способ их получения, фармацевтическая композиция и способ ее получения | |
| JPH08508251A (ja) | ドーパミン作働性活性を有するインドールテトラリン類 | |
| JPH08253474A (ja) | インドールピペリジン誘導体 | |
| JPH05230059A (ja) | ベンゾジオキサン誘導体 | |
| NZ197998A (en) | 5-amino(methyl)-1h-indol-3-ylalkaneamines | |
| EP0220066A2 (en) | Carboxamide derivatives | |
| EP0290145B1 (en) | Aliphatic carboxamides | |
| US6734301B2 (en) | 2,3,4,5-Tetrahydro-1H-[1,4]diazepino[1,7-a]indole compounds | |
| CA2720275A1 (en) | Indolinone compound | |
| US4898863A (en) | Heterocyclic carboxamides | |
| RU2712036C2 (ru) | Производные индолинона, обладающих свойствами лигандов мелатонинового рецептора и их применение | |
| US4292323A (en) | Phenyl-1,2,3,4-tetrahydrocarbazoles and use thereof | |
| GB2126230A (en) | Substituted 1-pyridyloxy-3- indolylakylamino-2-propanols, preparation, and use thereof |