RU2033994C1 - Оксазинон и способ получения таксола - Google Patents
Оксазинон и способ получения таксола Download PDFInfo
- Publication number
- RU2033994C1 RU2033994C1 SU904831946A SU4831946A RU2033994C1 RU 2033994 C1 RU2033994 C1 RU 2033994C1 SU 904831946 A SU904831946 A SU 904831946A SU 4831946 A SU4831946 A SU 4831946A RU 2033994 C1 RU2033994 C1 RU 2033994C1
- Authority
- RU
- Russia
- Prior art keywords
- ethyl acetate
- mixture
- taxol
- solution
- mmol
- Prior art date
Links
- 229960001592 paclitaxel Drugs 0.000 title claims abstract description 79
- 229930012538 Paclitaxel Natural products 0.000 title claims abstract description 39
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 title claims abstract description 35
- FBXGQDUVJBKEAJ-UHFFFAOYSA-N 4h-oxazin-3-one Chemical compound O=C1CC=CON1 FBXGQDUVJBKEAJ-UHFFFAOYSA-N 0.000 title claims abstract description 30
- 238000000034 method Methods 0.000 title claims abstract description 15
- 238000003786 synthesis reaction Methods 0.000 title abstract description 13
- 230000015572 biosynthetic process Effects 0.000 title abstract description 12
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 claims abstract description 14
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 7
- 230000003213 activating effect Effects 0.000 claims abstract description 6
- 239000002253 acid Substances 0.000 claims abstract description 5
- 150000003512 tertiary amines Chemical class 0.000 claims abstract description 4
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 claims abstract 5
- -1 β-naphthyl Chemical group 0.000 claims description 67
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 36
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 12
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 10
- 125000006239 protecting group Chemical group 0.000 claims description 5
- 238000006243 chemical reaction Methods 0.000 abstract description 21
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 abstract description 10
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 abstract description 6
- 150000001875 compounds Chemical class 0.000 abstract description 5
- 239000000126 substance Substances 0.000 abstract description 5
- 239000007795 chemical reaction product Substances 0.000 abstract description 3
- 239000003153 chemical reaction reagent Substances 0.000 abstract description 3
- 230000000694 effects Effects 0.000 abstract description 2
- 239000000047 product Substances 0.000 abstract description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract 2
- 239000003814 drug Substances 0.000 abstract 1
- RUVINXPYWBROJD-UHFFFAOYSA-N para-methoxyphenyl Natural products COC1=CC=C(C=CC)C=C1 RUVINXPYWBROJD-UHFFFAOYSA-N 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 324
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 156
- 239000000203 mixture Substances 0.000 description 100
- 239000000243 solution Substances 0.000 description 80
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 62
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 41
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 37
- 229910052938 sodium sulfate Inorganic materials 0.000 description 37
- 235000011152 sodium sulphate Nutrition 0.000 description 37
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 34
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 34
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 29
- 239000000741 silica gel Substances 0.000 description 29
- 229910002027 silica gel Inorganic materials 0.000 description 29
- 229920006395 saturated elastomer Polymers 0.000 description 23
- 239000007864 aqueous solution Substances 0.000 description 21
- 235000017557 sodium bicarbonate Nutrition 0.000 description 20
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 20
- 125000000217 alkyl group Chemical group 0.000 description 17
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 17
- 238000001953 recrystallisation Methods 0.000 description 17
- 125000003118 aryl group Chemical group 0.000 description 16
- 125000003342 alkenyl group Chemical group 0.000 description 15
- OVMSOCFBDVBLFW-VHLOTGQHSA-N 5beta,20-epoxy-1,7beta,13alpha-trihydroxy-9-oxotax-11-ene-2alpha,4alpha,10beta-triyl 4,10-diacetate 2-benzoate Chemical compound O([C@@H]1[C@@]2(C[C@H](O)C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)O)C(=O)C1=CC=CC=C1 OVMSOCFBDVBLFW-VHLOTGQHSA-N 0.000 description 14
- 125000000304 alkynyl group Chemical group 0.000 description 14
- 229910000365 copper sulfate Inorganic materials 0.000 description 14
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 14
- 238000010898 silica gel chromatography Methods 0.000 description 14
- 238000003818 flash chromatography Methods 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 125000001072 heteroaryl group Chemical group 0.000 description 11
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 125000001216 2-naphthoyl group Chemical group C1=C(C=CC2=CC=CC=C12)C(=O)* 0.000 description 8
- 239000007787 solid Substances 0.000 description 8
- 229930014667 baccatin III Natural products 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- YWLXLRUDGLRYDR-ZHPRIASZSA-N 5beta,20-epoxy-1,7beta,10beta,13alpha-tetrahydroxy-9-oxotax-11-ene-2alpha,4alpha-diyl 4-acetate 2-benzoate Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](O)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 YWLXLRUDGLRYDR-ZHPRIASZSA-N 0.000 description 6
- 239000012230 colorless oil Substances 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 125000001088 1-naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 125000004423 acyloxy group Chemical group 0.000 description 4
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 description 4
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N ethenoxyethane Chemical compound CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 4
- 150000002431 hydrogen Chemical class 0.000 description 4
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 150000003952 β-lactams Chemical class 0.000 description 4
- XSYLUBKWRZCOQP-QIWLAUOQSA-N (3s,4r)-1-benzoyl-3-(1-ethoxyethoxy)-4-phenylazetidin-2-one Chemical compound N1([C@@H]([C@@H](C1=O)OC(C)OCC)C=1C=CC=CC=1)C(=O)C1=CC=CC=C1 XSYLUBKWRZCOQP-QIWLAUOQSA-N 0.000 description 3
- FBZSDKXFQUKDLD-SFYZADRCSA-N (3s,4r)-3-hydroxy-4-phenylazetidin-2-one Chemical compound N1C(=O)[C@@H](O)[C@H]1C1=CC=CC=C1 FBZSDKXFQUKDLD-SFYZADRCSA-N 0.000 description 3
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 3
- NNKCQQPVYUHCHF-UHFFFAOYSA-N 5-(1-ethoxyethoxy)-2-[(2-methylpropan-2-yl)oxy]-4-phenyl-4,5-dihydro-1,3-oxazin-6-one Chemical compound N1=C(OC(C)(C)C)OC(=O)C(OC(C)OCC)C1C1=CC=CC=C1 NNKCQQPVYUHCHF-UHFFFAOYSA-N 0.000 description 3
- FEYQACXBKHREMV-UHFFFAOYSA-N 5-(1-ethoxyethoxy)-5-methyl-2,4-diphenyl-4h-1,3-oxazin-6-one Chemical compound O1C(=O)C(OC(C)OCC)(C)C(C=2C=CC=CC=2)N=C1C1=CC=CC=C1 FEYQACXBKHREMV-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 3
- YTBSMLRVFPHDOB-ZJUUUORDSA-N [(3s,4r)-2-oxo-4-phenylazetidin-3-yl] acetate Chemical compound N1C(=O)[C@@H](OC(=O)C)[C@H]1C1=CC=CC=C1 YTBSMLRVFPHDOB-ZJUUUORDSA-N 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 150000004579 taxol derivatives Chemical class 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 0 C[C@@]([C@@](*)NC(O1)IC)C1=O Chemical compound C[C@@]([C@@](*)NC(O1)IC)C1=O 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 229940123237 Taxane Drugs 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- NSBIQPJIWUJBBX-UHFFFAOYSA-N n-methoxyaniline Chemical compound CONC1=CC=CC=C1 NSBIQPJIWUJBBX-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 2
- SKJSIVQEPKBFTJ-HUWILPJBSA-N taxusin Chemical compound C1[C@@H](C2(C)C)C[C@H](OC(C)=O)C(C)=C2[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@]2(C)CC[C@H](OC(=O)C)C(=C)[C@@H]12 SKJSIVQEPKBFTJ-HUWILPJBSA-N 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- HZDNNJABYXNPPV-UHFFFAOYSA-N (2-chloro-2-oxoethyl) acetate Chemical compound CC(=O)OCC(Cl)=O HZDNNJABYXNPPV-UHFFFAOYSA-N 0.000 description 1
- LWKHUSGRQDHINE-UHFFFAOYSA-N (2-chloro-2-oxoethyl) prop-2-enoate Chemical compound ClC(=O)COC(=O)C=C LWKHUSGRQDHINE-UHFFFAOYSA-N 0.000 description 1
- WSKFEANLIDSKSN-CXRLMVSZSA-N (4s,5r)-5-(1-ethoxyethoxy)-2,4-diphenyl-4,5-dihydro-1,3-oxazin-6-one Chemical compound N([C@H]([C@H](C(O1)=O)OC(C)OCC)C=2C=CC=CC=2)=C1C1=CC=CC=C1 WSKFEANLIDSKSN-CXRLMVSZSA-N 0.000 description 1
- MCTWTZJPVLRJOU-UHFFFAOYSA-N 1-methyl-1H-imidazole Chemical compound CN1C=CN=C1 MCTWTZJPVLRJOU-UHFFFAOYSA-N 0.000 description 1
- YWLXLRUDGLRYDR-SKXCCXORSA-N 10-dab iii Chemical compound O([C@H]1C2[C@@](C([C@H](O)C3=C(C)[C@@H](O)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 YWLXLRUDGLRYDR-SKXCCXORSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- LUYQINUDJUXWNB-UHFFFAOYSA-N 3-(1-ethoxyethoxy)-4-phenylazetidin-2-one Chemical compound N1C(=O)C(OC(C)OCC)C1C1=CC=CC=C1 LUYQINUDJUXWNB-UHFFFAOYSA-N 0.000 description 1
- WSKFEANLIDSKSN-UHFFFAOYSA-N 5-(1-ethoxyethoxy)-2,4-diphenyl-4,5-dihydro-1,3-oxazin-6-one Chemical compound O1C(=O)C(OC(C)OCC)C(C=2C=CC=CC=2)N=C1C1=CC=CC=C1 WSKFEANLIDSKSN-UHFFFAOYSA-N 0.000 description 1
- QMIZAASILSBRMQ-UHFFFAOYSA-N 5-(1-ethoxyethoxy)-2-[(2-methylpropan-2-yl)oxy]-4,4-diphenyl-5h-1,3-oxazin-6-one Chemical compound CCOC(C)OC1C(=O)OC(OC(C)(C)C)=NC1(C=1C=CC=CC=1)C1=CC=CC=C1 QMIZAASILSBRMQ-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- HZEJCGXSBOAQSQ-UHFFFAOYSA-N CCOC(C)OC1C(N=C(OC1=O)C(C)(C)C)C2=CC=CC=C2 Chemical compound CCOC(C)OC1C(N=C(OC1=O)C(C)(C)C)C2=CC=CC=C2 HZEJCGXSBOAQSQ-UHFFFAOYSA-N 0.000 description 1
- SVRLDWIWUZVVPC-UHFFFAOYSA-N CCOC(C)OC1C(N=C(OC1=O)C2=CC3=CC=CC=C3C=C2)C4=CC=CC=C4 Chemical compound CCOC(C)OC1C(N=C(OC1=O)C2=CC3=CC=CC=C3C=C2)C4=CC=CC=C4 SVRLDWIWUZVVPC-UHFFFAOYSA-N 0.000 description 1
- QTZHTLOZCGCIDX-UHFFFAOYSA-N CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC3=CC=CC=C32)C4=CC=CC=C4 Chemical compound CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC3=CC=CC=C32)C4=CC=CC=C4 QTZHTLOZCGCIDX-UHFFFAOYSA-N 0.000 description 1
- IZLAQBBCNJPQBV-UHFFFAOYSA-N CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C(C)(C)C Chemical compound CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C(C)(C)C IZLAQBBCNJPQBV-UHFFFAOYSA-N 0.000 description 1
- BLCZYUZBMNGXAH-UHFFFAOYSA-N CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C3=CC4=CC=CC=C4C=C3 Chemical compound CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C3=CC4=CC=CC=C4C=C3 BLCZYUZBMNGXAH-UHFFFAOYSA-N 0.000 description 1
- VUMOGUADSBOYLK-UHFFFAOYSA-N CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C3=CC=C(C=C3)OC Chemical compound CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C3=CC=C(C=C3)OC VUMOGUADSBOYLK-UHFFFAOYSA-N 0.000 description 1
- QYEKIVAVUXGBDH-UHFFFAOYSA-N CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C3=CC=CC4=CC=CC=C43 Chemical compound CCOC(C)OC1C(N=C(OC1=O)C2=CC=CC=C2)C3=CC=CC4=CC=CC=C43 QYEKIVAVUXGBDH-UHFFFAOYSA-N 0.000 description 1
- AMIMRNSIRUDHCM-UHFFFAOYSA-N Isopropylaldehyde Chemical compound CC(C)C=O AMIMRNSIRUDHCM-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 241001116500 Taxus Species 0.000 description 1
- 241001116498 Taxus baccata Species 0.000 description 1
- 235000009065 Taxus cuspidata Nutrition 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 230000000719 anti-leukaemic effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- HKVFISRIUUGTIB-UHFFFAOYSA-O azanium;cerium;nitrate Chemical compound [NH4+].[Ce].[O-][N+]([O-])=O HKVFISRIUUGTIB-UHFFFAOYSA-O 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 239000002026 chloroform extract Substances 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000005911 methyl carbonate group Chemical class 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000004893 oxazines Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical class OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 125000002456 taxol group Chemical group 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 235000007586 terpenes Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/14—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/64—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings
- C07C233/81—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/82—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/87—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom of a carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/04—1,3-Oxazines; Hydrogenated 1,3-oxazines
- C07D265/06—1,3-Oxazines; Hydrogenated 1,3-oxazines not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/56—Ring systems containing bridged rings
- C07C2603/58—Ring systems containing bridged rings containing three rings
- C07C2603/76—Ring systems containing bridged rings containing three rings containing at least one ring with more than six ring members
- C07C2603/84—Ring systems containing bridged rings containing three rings containing at least one ring with more than six ring members containing rings with more than eight members
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epoxy Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicinal Preparation (AREA)
Abstract
Использование: в производстве таксола - агента для хемотерапии рака с широким спектром антилейкемической и опухолеингибирующей активностей. Сущность изобретения: продукт - оксазинон ф-лы 1, где R1 - фенил, 1- или 2-нафтил, C1-C6 -алкил, третбутоксигруппа; R2 - фенил, 1- или 2-нафтил, C1-C6 -алкил или параметоксифенил; R3 - гидроксилзащищающая группа, такая как этоксиэтил, а также таксол. Реагент 1: оксазинон ф-лы 1. Реагент 2: 10-7-0-триэтилсилилбаккатин. Условия процесса: в присутствии активирующего агента, например третичного амина, и переводом полученного бета-амидосложного эфира действием кислоты в целевой продукт. Способ позволяет упростить процесс получения таксола за счет использования нового легкодоступного исходного - оксазинона со структурной ф-лой 1:
Description
Изобретение относится к новому оксазинону и способу получения таксола, включающему использование такого оксази- нона.
Таксановое семейство терпенов, членом которого является таксол, привлек значительный интерес как в биологической, так и в химической отраслях техники. Таксол является многообещающим агентом для хемотерапии рака с широким спектром антилейкемической и опухолеингибирующей активности и имеет следующую структуру
C

Благодаря своей активности таксол в настоящее время проходит клинические исследования как во Франции, так и в США. Поставка таксола для этих исследований обеспечивается в настоящее время путем использования коры нескольких видов тиса. Таксол обнаружен в коре медленно растущих вечнозеленых деревьев лишь в незначительных количествах. Вследствие этого химики в последние годы пытаются найти надежный синтетический способ получения таксолов.
C
Благодаря своей активности таксол в настоящее время проходит клинические исследования как во Франции, так и в США. Поставка таксола для этих исследований обеспечивается в настоящее время путем использования коры нескольких видов тиса. Таксол обнаружен в коре медленно растущих вечнозеленых деревьев лишь в незначительных количествах. Вследствие этого химики в последние годы пытаются найти надежный синтетический способ получения таксолов.
Один из способов синтеза, который был предложен, направлен на синтез тетрациклического ядра из товарных химических веществ. Известен синтез собрата таксола таксузина [1]
Однако общий синтез таксола будет многостадийным, утомительным и дорогостоящим процессом.
Однако общий синтез таксола будет многостадийным, утомительным и дорогостоящим процессом.
Альтернативный подход к получению таксола [2] включает использование собрата таксола, 10-диацетил-баккатина III, который имеет следующую формулу:

10-Дезацетилбаккатин III является более легко доступным, чем таксол, поскольку он может быть получен из листьев Taxus baccata. Согласно способу Гриин, 10-дезацетилбаккатин III превращается в таксол с помощью присоединения С10 ацетильной группы и присоединения С13-амидосложноэфирной боковой цепи с помощью сложной этерификации С-13 спирта β-амидокарбоновой кислотой. Хотя данный подход требует относительно мало стадий, синтез β-амидокарбоновой кислоты является многостадийным процессом, который протекает с низким выходом, а реакция сочетания или конденсации является утомительной и протекает с низким выходом. Однако данная реакция является ключевой стадией, которая необходима в каждом рассматри- ваемом синтезе таксола или биологически активного производного таксола, поскольку для противоопухолевой активности требуется присутствие β-амидосложноэфирной боковой цепи в положении С13[3]
Основная трудность в синтезе таксола и других потенциальных противоопухолевых агентов заключается в недостатке или отсутствии легко доступных звеньев или молекул, которые можно было бы легко присоединить к С13 кислороду для получения β-амидосложноэфирной боковой цепи. Разработка таких веществ и способа их присоединения с достижением высокого выхода облегчило бы синтез таксола и родственных противоопухолевых агентов, имеющих модифицированный набор ядерных заместителей или модифицированной С13 боковой цепи. Данная потребность было восполнена с помощью открытия нового легкодоступного исходного продукта для боковой цепи и эффективного способа его присоединения к С13 кислороду.
10-Дезацетилбаккатин III является более легко доступным, чем таксол, поскольку он может быть получен из листьев Taxus baccata. Согласно способу Гриин, 10-дезацетилбаккатин III превращается в таксол с помощью присоединения С10 ацетильной группы и присоединения С13-амидосложноэфирной боковой цепи с помощью сложной этерификации С-13 спирта β-амидокарбоновой кислотой. Хотя данный подход требует относительно мало стадий, синтез β-амидокарбоновой кислоты является многостадийным процессом, который протекает с низким выходом, а реакция сочетания или конденсации является утомительной и протекает с низким выходом. Однако данная реакция является ключевой стадией, которая необходима в каждом рассматри- ваемом синтезе таксола или биологически активного производного таксола, поскольку для противоопухолевой активности требуется присутствие β-амидосложноэфирной боковой цепи в положении С13[3]
Основная трудность в синтезе таксола и других потенциальных противоопухолевых агентов заключается в недостатке или отсутствии легко доступных звеньев или молекул, которые можно было бы легко присоединить к С13 кислороду для получения β-амидосложноэфирной боковой цепи. Разработка таких веществ и способа их присоединения с достижением высокого выхода облегчило бы синтез таксола и родственных противоопухолевых агентов, имеющих модифицированный набор ядерных заместителей или модифицированной С13 боковой цепи. Данная потребность было восполнена с помощью открытия нового легкодоступного исходного продукта для боковой цепи и эффективного способа его присоединения к С13 кислороду.
Предлагаемое изобретение направлено на предшественник боковой цепи оксазином формулы

 (1) (1) где R1 арил, гетероарил, алкил, алкенил, алкинил или ОR7, где R7 алкил, алкенил, алкинил, арил или гетероарил;
(1) (1) где R1 арил, гетероарил, алкил, алкенил, алкинил или ОR7, где R7 алкил, алкенил, алкинил, арил или гетероарил;
R2 и R5 независимо выбраны из водорода, алкила, алкенила, алкинила, арила, гетероарила и ОR8, где R8 алкил, алкенил, алкинил, арил, гетероарил или гидроксилзащищающая группа;
R3 и R6 независимо выбраны из водорода, алкила, алкенила, алкинила, арила и гетероарила.
R2 и R5 независимо выбраны из водорода, алкила, алкенила, алкинила, арила, гетероарила и ОR8, где R8 алкил, алкенил, алкинил, арил, гетероарил или гидроксилзащищающая группа;
R3 и R6 независимо выбраны из водорода, алкила, алкенила, алкинила, арила и гетероарила.
Изобретение направлено также на процесс получения таксольного промежуточного продукта, включающий контактирование спирта с оксазином формулы I в присутствии достаточного количества активирующего агента с тем, чтобы вызвать взаимо- действие оксазинов со спиртом с образованием β-амидосложного эфира, который может использоваться в качестве проме- жуточного продукта в синтезе таксола.
Оксазин (1) и его производные имеют следующую структуру

 где R1 арил, гетероарил, алкил, алкенил, алкинил или ОR7, где R7 алкил, алкенил, алкинил, арил или гетероарил;
где R1 арил, гетероарил, алкил, алкенил, алкинил или ОR7, где R7 алкил, алкенил, алкинил, арил или гетероарил;
R2 и R5 независимо выбираются из водорода, алкила, алкенила, алкинила, арила, гетероарила и OR8, где R8 алкил, алкенил, алкинил, арил, гетероарил или гидроксилзащитная группа;
R3 и R6 независимо выбираются из водорода, алкила, алкенила, алкинила, арила и гетероарила.
R2 и R5 независимо выбираются из водорода, алкила, алкенила, алкинила, арила, гетероарила и OR8, где R8 алкил, алкенил, алкинил, арил, гетероарил или гидроксилзащитная группа;
R3 и R6 независимо выбираются из водорода, алкила, алкенила, алкинила, арила и гетероарила.
Предпочтительно оксазинон (1) имеет структуру

 (1a) где R1, R3 и R8 имеют указанные значения, наиболее предпочтительно R8 этоксиэтил или 2,2,2-трихлорэтоксиметил. Таким образом, структура наиболее предпочтительного осазинона, в котором R1 и R3 фенил; R5 водород; R2-OR8, где R8 этоксиэтил, следующая
(1a) где R1, R3 и R8 имеют указанные значения, наиболее предпочтительно R8 этоксиэтил или 2,2,2-трихлорэтоксиметил. Таким образом, структура наиболее предпочтительного осазинона, в котором R1 и R3 фенил; R5 водород; R2-OR8, где R8 этоксиэтил, следующая

 (2)
(2)
Согласно правилам EUPAC оксазиноном (2) является 2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-он.
Согласно правилам EUPAC оксазиноном (2) является 2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-он.
В соответствии с настоящим изобретением представляется способ получения таксольных промежуточных продуктов, природного таксола и не встречающихся в природе таксолов, имеющих следующую структурную формулу



 где А и В водород или низший алканоилокси, алкеноилокси, алкиноалокси или арилоилокси или А и В вместе образуют оксо.
где А и В водород или низший алканоилокси, алкеноилокси, алкиноалокси или арилоилокси или А и В вместе образуют оксо.
Оксазиноны (1) превращаются в β-амидосложные эфиры в присутствии спирта и активирующего агента, предпочтительно третичного амина, такого как триэтиламин, диизопропилэтиламин, пиридин, N-метил-имидазол и 4-диметиламинопиридин(ДМАР). Например, оксазиноны (1) взаимодействуют с соединениями, имеющими таксановое тетрациклическое ядро и С13 гидроксильную группу в присутствии 4-диметиламинопиридина, давая вещества, имеющие β-амидосложноэфирную группу у С13.
Наиболее предпочтительно спиртом является 7-0-триэтилсилилбаккатин III, который может получить по способу [2] или другими способами. Дезацетилбаккатин III превращается в 7-0-триэтилсилилбаккатин III в соответствии со следующей схемой реакции

В условиях, которые тщательно оптимизированы, 10-дезацетилбаккатин III подвергается взаимодействию с 20 экв (С2Н5) SiCl при 23оС в атмосфере аргона в течение 20 ч в присутствии 50 мл пиридина/ммоль 10-дезацетилбаккатина III, давая 7-триэтилсилил-10-дезацетилбаккатин III (3 а) в виде реакционного продукта с выходом 84-86% после очистки. Продукт реакции затем ацетилируется 5 экв. СН3СОСl и 25 мл пиридина/ммоль 3 а при 0оС в атмосфере аргона в течение 48 ч, давая с выходом 86% 7-0-триэтилсилилбаккатин III (3 b).
В условиях, которые тщательно оптимизированы, 10-дезацетилбаккатин III подвергается взаимодействию с 20 экв (С2Н5) SiCl при 23оС в атмосфере аргона в течение 20 ч в присутствии 50 мл пиридина/ммоль 10-дезацетилбаккатина III, давая 7-триэтилсилил-10-дезацетилбаккатин III (3 а) в виде реакционного продукта с выходом 84-86% после очистки. Продукт реакции затем ацетилируется 5 экв. СН3СОСl и 25 мл пиридина/ммоль 3 а при 0оС в атмосфере аргона в течение 48 ч, давая с выходом 86% 7-0-триэтилсилилбаккатин III (3 b).
Как показано на следующей схеме реакции, 7-0-триэтилсилилбаккатин III (3 b) может подвергаться реакции с оксазиноном при комнатной температуре, давая таксольный промежуточный продукт, в котором С-7- и С-2-гидроксильные группы защищены триэтилсилильной и этоксиэтильной защитными группами соответственно. Эти группы затем гидролизуются в мягких условиях так, чтобы не затронуть сложноэфирную связь или заместители таксола. Синтез таксола из оксазинона (2) осуществляется следующим образом:
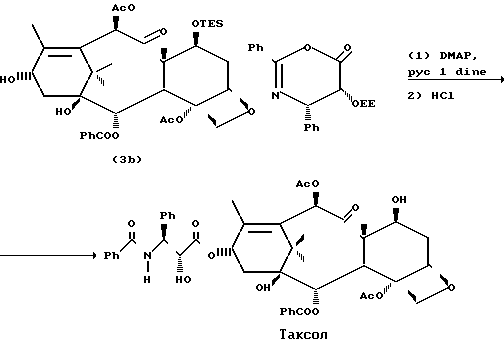
Хотя указанная схема направлена на синтез природного продукта таксола, процесс может использоваться с модификациями в оксазиноне или тетрациклическом спирте, который может быть получен из природных или неприродных источников для производства других синтетических таксолов.
Хотя указанная схема направлена на синтез природного продукта таксола, процесс может использоваться с модификациями в оксазиноне или тетрациклическом спирте, который может быть получен из природных или неприродных источников для производства других синтетических таксолов.
Оксазинон (1) может превращаться в β-амидосложный эфир в присутствии активирующего агента и спирта, иного чем 7-0-триэтилсилилбаккатин III, образуя промежуточный продукт таксола. Синтез таксола может затем протекать с использованием таксольного промежуточного продукта со соответствующей схеме реакции.
Оксазиноновыми алкильными группами (одним или с различными заместителями) предпочтительно являются низший алкил, содержащий 1-6 атомов углерода в главной цепи и до 15 атомов углерода. Они могут быть прямыми или разветвленными и включают метил, этил, пропил, изопропил, бутил, изобутил, третбутил, арил, гексил и др.
Оксазиноновыми алкенильными группами (одни или с различными заместителями) предпочтительно являются низший алкенил, содержащий 2-6 атомов углерода в главной цепи и до 15 атомов углерода. Они могут быть с прямой или разветвленной цепью и включают этенил, пропенил, изопропенил, бутенил, изобутенил, арил, гексанил и др.
Оксазиноновыми алкинильными группами (одни или с различными заместителями), предпочтительно являются низший алкинил, содержащий 2 6 углеродных атомов в главной цепи и до 15 углеродных атомов. Они могут быть с прямой или разветвленной цепью и включают этинил, пропинил, бутинил, изобутинил, арил, гексинил и др.
Примеры алканоилокси оксазинона включают ацетат, пропионат, бутират, валерат, изобутирал и др. Более предпочтительным алканоилокси является ацетат.
Указанные оксазиноновые арильные фрагменты, один или в сочетании с различными заместителями, содержат 6-15 атомов углерода и включают фенил, α-нафтил или β-нафтил и пр. Заместители включают алканокси, гидрокси, галоген, алкил, арил, алкенил, ацил, ацилокси, нитро, амино, амидо и др. Фенил является более предпочтительным арилом.
Как отмечалось выше, R2 и R5 оксазинона (1) могут быть ОR8, где R8 алкил, ацил, кеталь, этоксиэтил (ЕЕ),
2,2,2-трихлорэтоксиметил или другая гидроксилзащищающая группа, такая как ацетали и простые эфиры, т. е. метоксиметил (МОМ), бензилоксиметил, сложные эфиры, такие как ацетаты, карбонаты, такие как метилкарбонаты, и др. Известно множество разнообразных защитных групп для гидроксильной группы в органическом синтезе [4] Выбранная гидроксилзащищающая группа должна легко удаляться в условиях, которые являются достаточно мягкими, чтобы не затрагивать сложноэфирную связь или другие заместители таксольного промежуточного продукта. Однако R8предпочтительно представляет собой этоксиэтил или 2,2,2-трихлорэтоксиметил, наиболее предпочтителен этоксиэтил.
2,2,2-трихлорэтоксиметил или другая гидроксилзащищающая группа, такая как ацетали и простые эфиры, т. е. метоксиметил (МОМ), бензилоксиметил, сложные эфиры, такие как ацетаты, карбонаты, такие как метилкарбонаты, и др. Известно множество разнообразных защитных групп для гидроксильной группы в органическом синтезе [4] Выбранная гидроксилзащищающая группа должна легко удаляться в условиях, которые являются достаточно мягкими, чтобы не затрагивать сложноэфирную связь или другие заместители таксольного промежуточного продукта. Однако R8предпочтительно представляет собой этоксиэтил или 2,2,2-трихлорэтоксиметил, наиболее предпочтителен этоксиэтил.
Предпочтительные значения оксазиноновых заместитетелей R1-R3, R5,-R8 следующие: R1=OR7, R1=Ar, R1=n -MeOPh, R1 алкил, R1 алкенил, R1 алкинил, R1= H; R2=ОR8; R3=Rh, R3=Ar, R3 n -MeOPh, R3 алкил, R3= алкенил, R3 алкинил, R3= H; R5=H; R6=H; R7 алкил, R7 алкенил, R7=алкинил, R7=арил, R7-гетероарил; R8= ЕЕ, R8=алкил, R8=OCOR, R8 MOM, R8=Cl3CCH2OCH2.
Поскольку оксазинон имеет несколько асимметричных атомов углерода, соединения настоящего изобретения, имеющие асимметричные атомы углерода, могут существовать в диастереомерных, рацемической или оптической активной формах. Более конкретно данное изобретение включает энантиомеры, диастереомеры, рацемические смеси и иные смеси соединений.
Оксазиноны (1) могут быть получены из легко доступных материалов в соответствии со следующей схемой реакции

 R
R OH
OH 

Карбоновую кислоту (5) может получить в соответствии с известным [2] способом. Бета-лактамы (4) могут быть получены из легко доступных материалов, как показано на следующей схеме реакции, где R1 и R3 фенил;
R5 и R6 водород;
R2-OR8, причем R8 этоксиэтил:


 реагенты: а) триэтиламин, метиленхлорид, 25оС, 18 ч; b) 4 экв. цериевого нитрата аммония, СН3СN, 10оС, 10 мин; с) КОН, ТГФ, вода, 0оС, 30 мин; d) этилвиниловый эфир, ТГФ, толуолсульфоновая кислота (катализатор), 0оС, 1,5 ч; е) СН3Li, простой эфир, 78оС, 10 мин, бензоилхлорид, 78оС, 1 ч.
реагенты: а) триэтиламин, метиленхлорид, 25оС, 18 ч; b) 4 экв. цериевого нитрата аммония, СН3СN, 10оС, 10 мин; с) КОН, ТГФ, вода, 0оС, 30 мин; d) этилвиниловый эфир, ТГФ, толуолсульфоновая кислота (катализатор), 0оС, 1,5 ч; е) СН3Li, простой эфир, 78оС, 10 мин, бензоилхлорид, 78оС, 1 ч.
Карбоновую кислоту (5) может получить в соответствии с известным [2] способом. Бета-лактамы (4) могут быть получены из легко доступных материалов, как показано на следующей схеме реакции, где R1 и R3 фенил;
R5 и R6 водород;
R2-OR8, причем R8 этоксиэтил:
Исходные материалы являются легко доступными. Альфа-акрилоксиацетилхлорид получают из гликолевой кислоты в присутствии третичного амина, она циклоконденсируется с аминами, полученными из альдегидов и n-метоксианилина, давая 2,1-n-метоксифенил-3-ацилокси-4-арилазети-дин-2 оны.
N-Метоксифенильная группа может легко удаляться при окислении цериевым нитратом аммония, ацилокси группа может гидролизоваться в стандартных увловиях, аналогичных условиям, известным в технике для получения 3-гидрокси-4-арилазетидин-2-онов.
3-Гидроксильная группа может защищаться большим разнообразием стандартных защитных групп, таких как 1-этоксиэтильная группа. Предпочтительно, рацемический 3-гидрокси-4-арилазетидин-2-он расщепляется на чистые энантиомеры, перед защитой с помощью перекристаллизации соответствующих 2-метокси-2-(трифторметил)-фенилуксусных сложных эфиров и при получении таксола используется только правовращающий энантиомер. В любом случае 3-(1-этоксиэтокси)-4-фенил-азетидин-2-он может превращаться в β-лактам с помощью обработки основанием, предпочтительно н-бутиллитием, и ароилхлоридом при 78оС или ниже.
П р и м е р 1. Приготовление цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- он-2. Цис-1-n-метоксифенил-3-ацетокси-4-фенилазетидин-2-он. К раствору 962 мг (4,56 ммоль) имина, полученного из бензальдегида, и n-метокси анилина и 0,85 мл (6,07 ммоль) триэтиламина в 15 мл СН2Cl2 при 20оС добавлялся по каплям раствор 413 мг (3,04 ммоль α-ацетоксиацетилхлорида в 15 мл в метиленхлориде. Реакционную смесь оставляли нагреваться до 25оС в течение 18 ч. Затем ее разбавляли 100 мл метиленхлорида и раствор экстрагировали 30 мл 10%-ного водного раствора НСl. Органический слой промывали 30 мл воды и 30 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали для получения твердой массы. Твердое вещество обрабатывали 50 мл гексана и смесь фильтровали. Оставшееся твердое вещество перекристаллизовывали из смеси этилацетата (гексан, давая 645 мг (68%) цис-1-n-метоксифенил-3-ацетокси-4-фенилазетидин-2-она в виде белых кристаллов, т.пл. 163оС.
Цис-3-ацетокси-4-фенилазетидин-2-он. К раствору 20,2 г цис-1-n-метоксифенил-3-ацетокси-4-фенилазетидин-2-она в 700 мл ацетонитрила при -10оС медленно добавляли раствор цериевого нитрата аммония в 450 мл воды в течение часа. Смесь перемешивали в течение 30 мин при 10оС и разбавляли 500 мл эфира. Водный слой экстрагировали двумя (100 мл) порциями воды, двумя (100 мл) порциями насыщенного водного раствора бисульфата натрия, двумя (100 мл) порциями насыщенного водного раствора бикарбоната натрия и концентрировали, введя 18,5 г твердого вещества. Перекристаллизация твердого вещества из смеси ацетон/гексан давала 12,3 г (92%) цис-3-ацетокси-4-фенилазетидин-2-она в виде белых кристаллов, т.пл. 152-154оС.
Цис-3-гидрокси-4-фенилазетидин-2-он.
К смеси 200 мл тетрагидрофурана и 280 мл раствора 1 М водной гидроокиси калия при 0оС добавляли к раствору 4,59 г (22,4 ммоль) цис-3-ацетокси-4-фенилазетидин-2-она в 265 мл тетрагидрофурана с помощью капельной воронки в течение 40 мин. Раствор перемешивали при 0оС в течение часа и добавляли 100 мл воды и 100 мл насыщенного бикарбоната натрия. Смесь экстрагировали 4 порциями по 200 мл этилацетата; объединенные органические слои сушили над сульфатом натрия и концентрировали, давая 3,54 г (97%) рацемического цис-3-гидрокси-4-фенилазетидин-2-она в виде белых кристаллов, т.пл. 147-149оС. Это вещество разделялось на его анантиомеры путем перекристаллизации его 2-метокси-2-(трифторметил) фенилуксусного эфира из смеси гексан/ацетон с последующим гидролизом (α)25 Нg 177.
Цис-3-(1-этоксиэтокси)-4-фенилазети-дин-2-он. К раствору 3,41 г (20,9 ммоль) цис-3-гидрокси-4-фенилазетидин-2-она в 15 мл тетрагидрофурана при 0оС добавляли 5 мл этилвинилового эфира и 20 мг (0,2 ммоль) метансульфоновой кислоты. Смесь перемешивали при 0оС в течение 20 мин, разбавляли 20 мл насыщенного водного бикарбоната натрия и экстрагировали тремя (40 мл) порциями этилацетата. Объединенные этилацетатные слои сушили над сульфатом натрия и концентрировали, давая 4,87 г (99%) цис-3-(1-этоксиэтокси)-5-фенилазетидин-2-она в виде бесцветного масла.
Цис-1-бензоил-3-(1-этоксиэтокси)-4-фе- нилазетидин-2-он. К раствору 2,35 г (10 ммоль) цис-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в 40 мл ТГФ при 78оС добавляли 6,1 мл (10,07 ммоль) 1,65 М раствора н-бетиллития в гексане. Смесь перемешивали в течение 10 мин при 78оС и добавляли раствор 1,42 г (10,1 ммоль) бензоилхлорида в 10 мл ТГФ. После этого смесь перемешивали при 78оС в течение 1 ч, разбавляли 70 мл насыщенного водного бикарбоната натрия и экстрагировали тремя порциями по 50 мл этилацетата. Объединенные этилацетатные экстракты сушили над сульфатом натрия и концентрировали, давая 3,45 г масла. Хроматография масла на силикагеле с элюированием смесью этилацетата и гексана получали 3,22 г (95%) цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в виде бесцветного масла.
2R, 3S1= N-бензоил-n-(1-этоксиэтил)-3 фенилизосерин. К раствору 460 мг (1,36 ммоль) цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в 20 мл ТГФ при 0оС добавляли 13,5 мл 1 М водного раствора (13,5 ммоль) гидроокиси калия. Смесь перемешивали при 0оС в течение 10 мин и ТГФ выпаривался. Смесь распределяли между 12 мл 1 н. водного раствора НСl и 30 мл хлороформа. Водный слой экстрагировали двумя дополнительными порциями по 30 мл хлороформа. Объединенные хлороформные экстракты сушили над сульфатом натрия и концентрировали, давая 416 мг (86%) 2R, 3S1=N-бензоил-0-(1-этоксиэтил)-3-фенили- зосерина формулы (5), где R1 и R3 фенил; R2 этоксиэтил.
Цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-он-2. К раствору 416 мг (1,16 ммоль) 2R, 3S1=N-бензоил-0-(1-этоксиэтил)-3-фенилизомерина в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Затем добавляли раствор 134 мг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата и данный раствор экстрагировался 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 265 мг (65%) цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигид-ро-1,3-оксазин-6-он-2 в виде бесцветного масла, (α)25 Hg 22 (СНСl3, с.1.55).
П р и м е р 2. Получение таксола.
В небольшой реакционный сосуд добавляли 77 мг (0,218 ммоль) (-)-цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 2,40 мг (0,057 ммоль) 7-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-го водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля и элюировали этилацетатом. Мгновенная хроматография на силикагеле с элюированием смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 46 мг (77%) 21-0(1-этоксиэтил)-7-0-триэтилсилилтаксола в виде смеси приблизительно 2:1 диастереомеров и 9,3 мг (23% ) 7-0-триэтилсилилбаккатина III. Выход в расчете на потребленный 7-0-триэтилсилилбаккатин III был количественный.
Образец в 5 мг 21-(1-этоксиэтил)-7-0-триэтилсилилтаксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ного водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Затем раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонке из силикагеля с элюированием смесью этилацетата и гексана, давая 3,8 мг (приблизительно 90%) таксола, который был идентичен аутентичному образцу во всех отношениях.
П р и м е р 3. Получение N-дибензол-N-третбутоксикарбонил-таксола.


2-Третбутокси-4-фенил-5-(1-этоксиэток- си)-4,5-дигидро-1,3-оксазин-6-он. К раствору 409 мг (1,16 ммоль) N-третбутоксикарбонил-С-(1-этоксиэтил)-3-фенилизосерина (3) в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Далее добавляли раствор 134 мг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата и данный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 235 мг (70%) 2-третбутокси-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она в виде бесцветного масла.
N-дебензоил-N-третбутоксикарбонил-таксол. В небольшой реакционный сосуд добавляли 73 мг (0,218 ммоль) 2-третбутокси-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,2-оксазин-6-она, 40 мг (0,057 ммоль) 7-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ного водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля и элюировали этилацетатом. Мгновенной хроматографией на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана получали 44 мг (73%) N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтокси)-7-0-триэтилсилил- таксола в виде приблизительно 1:1 смеси диастереомеров и 9,3 мг (23%) 7-0-триэтилсилилбаккатина III.
Образец в 5 мг N-дебензоил-N-третбутоксикарбонил-21(1-этоксиэтокси)-7-0-три-этилсилилтаксол а растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ного водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонке из силикагеля с элюированием смесью этилацетата и гексана, давая 3,8 мг (приблизительно 90%) N-дебензоил-N-третбутоксикарбонилтаксола.
П р и м е р 4. Получение N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтил)-3-фе- нил-таксола

2-Третбутокси-4,5-дифенил-5-(1-этокси- этокси)-4,5-дигидро-1,3-оксазин-6-он. К раствору 497 мг (1,16 ммоль) N-третбутоксикарбонил-0-(1-этоксиэтил)-3,3-дифенилизосери- на (3) в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Затем добавляли раствор 134 кг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата и данный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 243 мг (59%) 2-третбутокси-4,4, -дифенил-5-(1-этоксиэтокси)-4,4-дигидро-1,3-оксазин-6-она в виде бесцветного масла.
2-Третбутокси-4,5-дифенил-5-(1-этокси- этокси)-4,5-дигидро-1,3-оксазин-6-он. К раствору 497 мг (1,16 ммоль) N-третбутоксикарбонил-0-(1-этоксиэтил)-3,3-дифенилизосери- на (3) в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Затем добавляли раствор 134 кг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата и данный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 243 мг (59%) 2-третбутокси-4,4, -дифенил-5-(1-этоксиэтокси)-4,4-дигидро-1,3-оксазин-6-она в виде бесцветного масла.
N-Дебензоил-N-третбутоксикарбонил-3-фенил-таксол. В небольшой реакционный сосуд добавляли 90 мг (0,218 ммоль) 2-третбутокси-4,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 40 мг (0,057 ммоль) 7-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через средство из силикагеля и элюировали этилацетатом. Мгновенной хроматографией на силикагеле с элюированием смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана получали 44 мг (66%) N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтил)-3-фенил-7-0-триэтил-силил -такв виде приблизительно 3:1 смеси диастереомеров.
Образец в 5 мг N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтил)-31-фенил- 7-0-триэтилсилил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ного водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на силикагеле с элюированием смесью этилацетата и гексана, давая 4,0 мг (приблизительно 90%) N-дебензоил-N-третбутоксикарбонил-3-фенилтак-сола.
П р и м е р 5. Получение 2,4-дифенил-5-(1-этоксиэтокси)-5-метил-4,5 дигидро-1,3-оксазин-6-она.


К раствору 480 мг (1,16 ммоль) N-бензоил -С-(1-этоксиэтил)-2-метил-3-фенилизосерина в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Добавляли раствор 134 мг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата, данный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 270 мг (76%0 2,4-дифенил-5-(1-этоксиэтокси)-5-метил-4,5-дигидро-1,3-оксазин-6-она в виде бесцветного масла.
П р и м е р 6. 3-метил-таксол.
В небольшой реакционный сосуд добавляли 77 мг (0,218 ммоль) 2,4-дифенил-5-(1-этоксиэтокси)-5-метил-4,5-дигидро-1,3-окса- зин-6-она, 40 мг (0,057 ммоль) 7-0-триэтилсилилбаккатина III 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через средство из силикагеля и элюировали этилацетататом. Мгновенная хроматография на силикагеле с элюированием смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана дает 32 мг (53%) 2-(1-этокси-этил)-3'-метил-7-0-триэтилсилил-таксола в виде приблизительно 1:1 смеси диастереомеров.
Образец 2'-(1-этоксиэтил)-3'-метил-7-0-триэтилсилил-таксола (5 мг) растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонке из силикагеля с элюированием смесью этилацетата и гексана, давая 3,9 кг (приблизительно 90%) 3'-метил-таксола.
Дополнительные примеры.
N-дебензоил-N-(1-нафтоил)-таксол. В небольшой реакционный сосуд добавляли 125 мг (0,320 ммоль) 2-(1-нафтил)-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 62 мг (90% ) N-дебензоил-N-(1-нафтоил)-2'-(1-этоксиэто- кси)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-(1-нафтоил)-2'-(1-этоксиэтокси)-7-0-триэтилсилил- таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора HCl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,9 мг (приблизительно 95%) N-дебензоил-N-(1-нафтоил)-таксола.
N-дебензоил-N-(2-нафтоил)-таксол. В небольшой реакционный сосуд добавляли 125 мг (0,320 ммоль) 2-(2-нафтил)-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 64 мг (93% ) N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэто- кси)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэтокси)-7-0-триэтилсилил- таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,5 мг (приблизительно 90%) N-дебензоил-N-(2-нафтоил)-таксола.
N-дебензоил-N-пивалоил-таксол. В небольшой реакционный сосуд добавляли 102 мг (0,320 ммоль) 2-третбутил-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 55 мг (85% ) N-дебензоил-N-пивалоил-2'-(1-этоксиэток-си)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-пивалоил-2'-(1-этоксиэтокси)-7-0-триэтилсилил-та- ксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 92%) N-дебензоил-N-пивалоил-таксола.
N-дебензоил-N-пентаноил-таксол. В небольшой реакционный сосуд добавляли 102 мг (0,320 ммоль) 2-n-бутил-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 58 мг (89%) N-дебензоил-N-пентаноил-2'-(1-этоксиэток- си)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-пентаноил-2'-(1-этоксиэтокси)-7-0-триэтилсилил-та- ксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 92%) N-дебензоил-N-пентаноил-таксола.
3-Десфенил-3'-(1-нафтил)-таксол. В небольшой реакционный сосуд добавляли 125 мг (0,320 ммоль) 2-фенил-4-(1-нафтил)-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 62 мг (90%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-(1-наф- тил)-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-(1-нафтил)-7-0-триэтилсилил-так- сола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,9 мг (приблизительно 95%) 3'-десфенил-3'-(1-нафтил)-таксола.
3'-десфенил-3'-(2-нафтил)-таксол. В небольшой реакционной сосуд добавляли 125 мг (0,320 ммоль) 2-фенил-4-(2-нафтил)-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 62 мг (90%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-(2-наф- тил)-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-(2-нафтил)-7-0-триэтилсилил-так- сола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,9 мг (приблизительно 95%) 3'-десфенил-3'-(2-нафтил)-таксола.
3'-Десфенил-3'-t-бутил-таксол. В небольшой реакционный сосуд добавляли 102 мг (0,320 ммоль) 2-фенил-4-третбутил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография из силикагеля, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 58 мг (89%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-t-бутил-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-t-бутил-7-0-триэтилсилил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 92%) 3'-десфенил-3'-t-бутил-таксола.
3'-Десфенил-3'-р-метоксифенил-таксол. В небольшой реакционный сосуд добавляют 118 мг (0,320 ммоль) 2-фенил-4-р-метокси-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 58 мг (87%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-р-метоксифенил-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-р-метоксифенил-7-0-триэтилси-лил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора HCl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 90%) 3'-десфенил-3'-р-метоксифенил-таксола.
N-Дебензоил-N-(2-нафтоил)-3'-десфе-нил-3'-(1-нафтил)таксол. В небольшой реакционный сосуд добавляли 140 мгл (0,320 ммоль) 2-(2-нафтил)-4-(1-нафтил)-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мгл (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 51 мг (70%) N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэто- кси)-3'-десфенил-3'-(1-нафтил)-7-0-триэтил- силил-таксола.
Образец в 5 мг N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэтокси)-3'-десфенил-3'-(1- нафтил)-7-0-триэтилсилил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора HCl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,3 мг (приблизительно 80%) N-дебензоил-N-(2-нафтоил)-3'-десфенил-3'-(1-нафтил)-таксола.
N-дебензоил-N-третбутоксикарбонил-10-деацетил-таксол (таксотер). В небольшой реакционный сосуд добавляли 73 мг (0,218 ммоль) 2-третбутокси-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 44 мг (0,057 ммоль) 7,10-бис-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (DMAP) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10% -ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 45 мг (68%) N-дебензоил-N-третбутоксикарбонил-2'-(1-этоксиэтокси)-3'-7,10-бис-0-триэтилс илилв виде приблизительно 1:1 смеси диастериомеров.
Образец в 5 мг N-дебензоил-N-третбутоксикарбонил-2'-(1-этоксиэтокси)-3'-7,10-бис-0-триэтилс илилтаксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,8 мг (приблизительно 90%) N-дебензоил-N-третбутоксикарбонил-10-деацетил-таксола.
Claims (3)
1. Оксазинон общей формулы

где R1 фенил, α- -или β -нафтил, C1 - C6-алкил или T-бутоксигруппа;
R2 фенил, a- -или β -нафтил, C1 -C6-алкил или p-метоксифенил;
R3 гидроксилзащищающая группа, такая как этоксиэтил.
где R1 фенил, α- -или β -нафтил, C1 - C6-алкил или T-бутоксигруппа;
R2 фенил, a- -или β -нафтил, C1 -C6-алкил или p-метоксифенил;
R3 гидроксилзащищающая группа, такая как этоксиэтил.
2. Способ получения таксола общей формулы

где R1 фенил, α- -или β -нафтил, C1 - C6-алкил или t-бутоксигруппа;
R2 фенил, a- -или β -нафтил, C1 C6-алкил или p-метоксифенил,
отличающийся тем, что оксазинон общей формулы

где R1, R2 и R3 имеют указанные значения, подвергают взаимодействию со спиртом формулы

в присутствии активирующего агента и полученный β -амидосложный эфир действием кислоты переводят в целевой продукт.
где R1 фенил, α- -или β -нафтил, C1 - C6-алкил или t-бутоксигруппа;
R2 фенил, a- -или β -нафтил, C1 C6-алкил или p-метоксифенил,
отличающийся тем, что оксазинон общей формулы
где R1, R2 и R3 имеют указанные значения, подвергают взаимодействию со спиртом формулы
в присутствии активирующего агента и полученный β -амидосложный эфир действием кислоты переводят в целевой продукт.
3. Способ по п.2, отличающийся тем, что в качестве активирующего агента используют третичный амин.
Приоритет по признакам:
14.11.89 при R1 и R2 фенил, a или b -нафтил, C1 C6-алкил, R3-гидроксизащищающая группа, такая как этоксиэтил, R2 p-метоксифенил;
30.10.90 при R1 фенил, a или b -нафтил, C1 - C6-алкил или t-бутоксигруппа, R2 фенил, a- или β -нафтил, C1 C6-алкил или p-метоксифенил и R3 - гидроксизащищающая группа, такая как этоксиэтил.
14.11.89 при R1 и R2 фенил, a или b -нафтил, C1 C6-алкил, R3-гидроксизащищающая группа, такая как этоксиэтил, R2 p-метоксифенил;
30.10.90 при R1 фенил, a или b -нафтил, C1 - C6-алкил или t-бутоксигруппа, R2 фенил, a- или β -нафтил, C1 C6-алкил или p-метоксифенил и R3 - гидроксизащищающая группа, такая как этоксиэтил.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US07/436,235 US5015744A (en) | 1989-11-14 | 1989-11-14 | Method for preparation of taxol using an oxazinone |
| US436235 | 1989-11-14 | ||
| US603041 | 1990-10-30 | ||
| US07/603,041 US5136060A (en) | 1989-11-14 | 1990-10-30 | Method for preparation of taxol using an oxazinone |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2033994C1 true RU2033994C1 (ru) | 1995-04-30 |
Family
ID=27030868
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| SU904831946A RU2033994C1 (ru) | 1989-11-14 | 1990-11-14 | Оксазинон и способ получения таксола |
Country Status (28)
| Country | Link |
|---|---|
| US (3) | US5136060A (ru) |
| EP (1) | EP0428376B1 (ru) |
| JP (1) | JP2507830B2 (ru) |
| KR (1) | KR0161499B1 (ru) |
| CN (1) | CN1043528C (ru) |
| AT (1) | ATE132860T1 (ru) |
| AU (1) | AU633375B2 (ru) |
| BG (1) | BG60478B1 (ru) |
| CA (2) | CA2029787C (ru) |
| CZ (2) | CZ283581B6 (ru) |
| DE (1) | DE69024757T2 (ru) |
| DK (1) | DK0428376T3 (ru) |
| EG (1) | EG19641A (ru) |
| ES (1) | ES2083438T3 (ru) |
| FI (1) | FI104487B (ru) |
| GR (1) | GR3019192T3 (ru) |
| HU (1) | HU209299B (ru) |
| IE (2) | IE74154B1 (ru) |
| IL (3) | IL109246A (ru) |
| MY (1) | MY106071A (ru) |
| NO (1) | NO177902C (ru) |
| NZ (1) | NZ235993A (ru) |
| OA (1) | OA09327A (ru) |
| PL (1) | PL167898B1 (ru) |
| PT (1) | PT95870B (ru) |
| RO (1) | RO110491B1 (ru) |
| RU (1) | RU2033994C1 (ru) |
| YU (1) | YU48232B (ru) |
Families Citing this family (101)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5175315A (en) * | 1989-05-31 | 1992-12-29 | Florida State University | Method for preparation of taxol using β-lactam |
| MY110249A (en) * | 1989-05-31 | 1998-03-31 | Univ Florida State | Method for preparation of taxol using beta lactam |
| US5136060A (en) * | 1989-11-14 | 1992-08-04 | Florida State University | Method for preparation of taxol using an oxazinone |
| US5380916A (en) * | 1990-11-02 | 1995-01-10 | University Of Florida | Method for the isolation and purification of taxane derivatives |
| US5475120A (en) * | 1990-11-02 | 1995-12-12 | University Of Florida | Method for the isolation and purification of taxol and its natural analogues |
| CA2071160A1 (en) * | 1991-07-31 | 1993-02-01 | Vittorio Farina | Asymmetric synthesis of taxol side chain |
| US5998656A (en) * | 1991-09-23 | 1999-12-07 | Florida State University | C10 tricyclic taxanes |
| US5283253A (en) * | 1991-09-23 | 1994-02-01 | Florida State University | Furyl or thienyl carbonyl substituted taxanes and pharmaceutical compositions containing them |
| US6011056A (en) * | 1991-09-23 | 2000-01-04 | Florida State University | C9 taxane derivatives and pharmaceutical compositions containing them |
| US5489601A (en) * | 1991-09-23 | 1996-02-06 | Florida State University | Taxanes having a pyridyl substituted side-chain and pharmaceutical compositions containing them |
| US5721268A (en) * | 1991-09-23 | 1998-02-24 | Florida State University | C7 taxane derivatives and pharmaceutical compositions containing them |
| US5274124A (en) * | 1991-09-23 | 1993-12-28 | Florida State University | Metal alkoxides |
| US5714513A (en) * | 1991-09-23 | 1998-02-03 | Florida State University | C10 taxane derivatives and pharmaceutical compositions |
| US6005138A (en) * | 1991-09-23 | 1999-12-21 | Florida State University | Tricyclic taxanes having a butenyl substituted side-chain and pharmaceutical compositions containing them |
| US6028205A (en) * | 1991-09-23 | 2000-02-22 | Florida State University | C2 tricyclic taxanes |
| US5284864A (en) | 1991-09-23 | 1994-02-08 | Florida State University | Butenyl substituted taxanes and pharmaceutical compositions containing them |
| US6335362B1 (en) * | 1991-09-23 | 2002-01-01 | Florida State University | Taxanes having an alkyl substituted side-chain and pharmaceutical compositions containing them |
| US5710287A (en) * | 1991-09-23 | 1998-01-20 | Florida State University | Taxanes having an amino substituted side-chain and pharmaceutical compositions containing them |
| US5728850A (en) * | 1991-09-23 | 1998-03-17 | Florida State University | Taxanes having a butenyl substituted side-chain and pharmaceutical compositions containing them |
| CA2418125C (en) * | 1991-09-23 | 2007-12-04 | Florida State University | Beta-lactams useful in the preparation of substituted isoserine esters |
| US6495704B1 (en) | 1991-09-23 | 2002-12-17 | Florida State University | 9-desoxotaxanes and process for the preparation of 9-desoxotaxanes |
| US5728725A (en) * | 1991-09-23 | 1998-03-17 | Florida State University | C2 taxane derivaties and pharmaceutical compositions containing them |
| US6521660B2 (en) | 1991-09-23 | 2003-02-18 | Florida State University | 3′-alkyl substituted taxanes and pharmaceutical compositions containing them |
| US5739362A (en) * | 1991-09-23 | 1998-04-14 | Florida State University | Taxanes having an alkoxy, alkenoxy or aryloxy substituted side-chain and pharmaceutical compositions containing them |
| US6018073A (en) * | 1991-09-23 | 2000-01-25 | Florida State University | Tricyclic taxanes having an alkoxy, alkenoxy or aryloxy substituted side-chain and pharmaceutical compositions containing them |
| US7074945B2 (en) * | 1991-09-23 | 2006-07-11 | Florida State University | Metal alkoxide taxane derivatives |
| US5654447A (en) * | 1991-09-23 | 1997-08-05 | Florida State University | Process for the preparation of 10-desacetoxybaccatin III |
| US5430160A (en) * | 1991-09-23 | 1995-07-04 | Florida State University | Preparation of substituted isoserine esters using β-lactams and metal or ammonium alkoxides |
| US6794523B2 (en) | 1991-09-23 | 2004-09-21 | Florida State University | Taxanes having t-butoxycarbonyl substituted side-chains and pharmaceutical compositions containing them |
| SG46582A1 (en) * | 1991-09-23 | 1998-02-20 | Univ Florida State | 10-Desacetoxytaxol derivatives |
| DE69333675T2 (de) * | 1992-01-15 | 2006-02-02 | E.R. Squibb & Sons, Inc. | Enzymatische Verfahren zur Trennung von Enantiomerengemischen von Verbindungen, die als Zwischenprodukte zur Herstellung von Taxanen nützlich sind |
| US5272171A (en) * | 1992-02-13 | 1993-12-21 | Bristol-Myers Squibb Company | Phosphonooxy and carbonate derivatives of taxol |
| US5254703A (en) * | 1992-04-06 | 1993-10-19 | Florida State University | Semi-synthesis of taxane derivatives using metal alkoxides and oxazinones |
| US5294637A (en) * | 1992-07-01 | 1994-03-15 | Bristol-Myers Squibb Company | Fluoro taxols |
| US5254580A (en) * | 1993-01-19 | 1993-10-19 | Bristol-Myers Squibb Company | 7,8-cyclopropataxanes |
| US5614549A (en) * | 1992-08-21 | 1997-03-25 | Enzon, Inc. | High molecular weight polymer-based prodrugs |
| FR2696463B1 (fr) | 1992-10-05 | 1994-11-25 | Rhone Poulenc Rorer Sa | Procédé d'obtention de la désacétyl-10 baccatine III. |
| FR2696462B1 (fr) * | 1992-10-05 | 1994-11-25 | Rhone Poulenc Rorer Sa | Procédé d'obtention de la désacétyl-10 baccatine III. |
| FR2696458B1 (fr) * | 1992-10-05 | 1994-11-10 | Rhone Poulenc Rorer Sa | Procédé de préparation de dérivés du taxane. |
| FR2697752B1 (fr) * | 1992-11-10 | 1995-04-14 | Rhone Poulenc Rorer Sa | Compositions antitumorales contenant des dérivés du taxane. |
| CA2149150C (en) * | 1992-11-27 | 2000-08-01 | David R. Carver | Injectable taxol composition with improved stability |
| KR100371062B1 (ko) * | 1992-11-27 | 2003-04-21 | 에프.에이치.포울딩 앤드 컴퍼니 리미티드 | 안정성이향상된주사가능한택솔조성물및이를제형화하는방법 |
| US5380751A (en) * | 1992-12-04 | 1995-01-10 | Bristol-Myers Squibb Company | 6,7-modified paclitaxels |
| FR2698871B1 (fr) | 1992-12-09 | 1995-02-24 | Rhone Poulenc Rorer Sa | Nouveau taxoïdes, leur préparation et les compositions pharmaceutiques qui les contiennent. |
| IL107950A (en) | 1992-12-15 | 2001-04-30 | Upjohn Co | b 7, b 8 - Matano - Taxols, their preparation and pharmaceutical preparations against malignant tumors containing them |
| US5973160A (en) * | 1992-12-23 | 1999-10-26 | Poss; Michael A. | Methods for the preparation of novel sidechain-bearing taxanes |
| US5646176A (en) * | 1992-12-24 | 1997-07-08 | Bristol-Myers Squibb Company | Phosphonooxymethyl ethers of taxane derivatives |
| US6339164B1 (en) | 1993-01-29 | 2002-01-15 | Florida State University | C2 substituted phenyl taxane derivatives and pharmaceutical compositions containing them |
| US6593482B2 (en) | 1993-02-01 | 2003-07-15 | Aventis Pharma S.A. | Methods for preparing new taxoids and pharmaceutical compositions containing them |
| US6187916B1 (en) * | 1993-02-01 | 2001-02-13 | Research Foundation Of State University Of New York | Process for the preparation of taxane derivatives and β-lactam intermediates therefor |
| EP1260507A1 (en) * | 1993-02-05 | 2002-11-27 | Bryn Mawr College | Synthesis of taxol, analogs and intermediates with variable A-nng side chains |
| US5684175A (en) * | 1993-02-05 | 1997-11-04 | Napro Biotherapeutics, Inc. | C-2' hydroxyl-benzyl protected, N-carbamate protected (2R, 3S)- 3-phenylisoserine and production process therefor |
| WO1994020485A1 (en) * | 1993-03-05 | 1994-09-15 | Florida State University | Process for the preparation of 9-desoxotaxanes |
| US6710191B2 (en) * | 1993-03-05 | 2004-03-23 | Florida State University | 9β-hydroxytetracyclic taxanes |
| TW467896B (en) | 1993-03-19 | 2001-12-11 | Bristol Myers Squibb Co | Novel β-lactams, methods for the preparation of taxanes and sidechain-bearing taxanes |
| ES2242807T3 (es) * | 1993-03-22 | 2005-11-16 | Florida State University | Taxanos que tienen furilo o tienilo sustituido en la cadena lateral sustituida. |
| US5965739A (en) * | 1993-06-11 | 1999-10-12 | Pharmacia & Upjohn Company | Δ6,7 -taxols antineoplastic use and pharmaceutical compositions containing them |
| TW397866B (en) | 1993-07-14 | 2000-07-11 | Bristol Myers Squibb Co | Enzymatic processes for the resolution of enantiomeric mixtures of compounds useful as intermediates in the preparation of taxanes |
| US6005120A (en) | 1993-07-20 | 1999-12-21 | Florida State University | Tricyclic and tetracyclic taxanes |
| US5442065A (en) * | 1993-09-09 | 1995-08-15 | Board Of Regents, The University Of Texas System | Synthesis of tetrahydroquinoline enediyne core analogs of dynemicin |
| US5880131A (en) * | 1993-10-20 | 1999-03-09 | Enzon, Inc. | High molecular weight polymer-based prodrugs |
| WO1995011020A1 (en) * | 1993-10-20 | 1995-04-27 | Enzon, Inc. | 2'- and/or 7- substituted taxoids |
| US5965566A (en) * | 1993-10-20 | 1999-10-12 | Enzon, Inc. | High molecular weight polymer-based prodrugs |
| US6441026B1 (en) | 1993-11-08 | 2002-08-27 | Aventis Pharma S.A. | Antitumor compositions containing taxane derivatives |
| IL127599A (en) * | 1994-01-28 | 2004-06-01 | Upjohn Co | Process for preparing isotaxol analogs |
| US5508447A (en) * | 1994-05-24 | 1996-04-16 | Board Of Regents, The University Of Texas System | Short synthetic route to taxol and taxol derivatives |
| CA2170661A1 (en) | 1995-03-22 | 1996-09-23 | John K. Thottathil | Novel methods for the preparation of taxanes using oaxzolidine intermediates |
| US5560872A (en) * | 1995-05-18 | 1996-10-01 | Lever Brothers Company | Compositions comprising oxazolidine and tetrahydrooxazine amide surfactants |
| US5675025A (en) * | 1995-06-07 | 1997-10-07 | Napro Biotherapeutics, Inc. | Paclitaxel synthesis from precursor compounds and methods of producing the same |
| US5807888A (en) * | 1995-12-13 | 1998-09-15 | Xechem International, Inc. | Preparation of brominated paclitaxel analogues and their use as effective antitumor agents |
| US5854278A (en) * | 1995-12-13 | 1998-12-29 | Xechem International, Inc. | Preparation of chlorinated paclitaxel analogues and use thereof as antitumor agents |
| US5840748A (en) * | 1995-10-02 | 1998-11-24 | Xechem International, Inc. | Dihalocephalomannine and methods of use therefor |
| US5654448A (en) * | 1995-10-02 | 1997-08-05 | Xechem International, Inc. | Isolation and purification of paclitaxel from organic matter containing paclitaxel, cephalomannine and other related taxanes |
| US6177456B1 (en) | 1995-10-02 | 2001-01-23 | Xechem International, Inc. | Monohalocephalomannines having anticancer and antileukemic activity and method of preparation therefor |
| US6107497A (en) * | 1996-02-29 | 2000-08-22 | Napro Biotherapeutics, Inc. | Intermediate for use in docetaxel synthesis and production method therefor |
| US5688977A (en) * | 1996-02-29 | 1997-11-18 | Napro Biotherapeutics, Inc. | Method for docetaxel synthesis |
| FR2745814B1 (fr) * | 1996-03-06 | 1998-04-03 | Rhone Poulenc Rorer Sa | Nouveaux taxoides, leur preparation et les compositions pharmaceutiques qui les contiennent |
| IL126856A0 (en) * | 1996-05-06 | 1999-09-22 | Univ Florida State | 1-Deoxy baccatin iii 1-deoxy taxol and 1-deoxy taxol analogs and method for the preparation thereof |
| US5750737A (en) * | 1996-09-25 | 1998-05-12 | Sisti; Nicholas J. | Method for paclitaxel synthesis |
| DE19726146A1 (de) * | 1997-06-19 | 1998-12-24 | Basf Ag | Neue ß-Amino und ß-Azidopcarbonsäurederivate, ihre Herstellung und Verwendung als Endothelinrezeptorantagonisten |
| US7288665B1 (en) * | 1997-08-18 | 2007-10-30 | Florida State University | Process for selective derivatization of taxanes |
| ATE234800T1 (de) | 1997-08-21 | 2003-04-15 | Univ Florida State | Verfahren zur synthese von taxanen |
| US6150537A (en) * | 1997-12-12 | 2000-11-21 | Emory University | Methods for the esterification of alcohols and compounds useful therefor as potential anticancer agents |
| US6156789A (en) * | 1998-03-17 | 2000-12-05 | Rhone-Poulenc Rorer S.A. | Method for treating abnormal cell proliferation in the brain |
| US6025516A (en) * | 1998-10-14 | 2000-02-15 | Chiragene, Inc. | Resolution of 2-hydroxy-3-amino-3-phenylpropionamide and its conversion to C-13 sidechain of taxanes |
| CA2385528C (en) | 1999-10-01 | 2013-12-10 | Immunogen, Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| US6452024B1 (en) | 2000-02-22 | 2002-09-17 | Chaichem Pharmaceuticals International | Process for extraction and purification of paclitaxel from natural sources |
| CZ2003837A3 (cs) | 2000-09-22 | 2004-12-15 | Bristol-Myers Squibb Company | Způsob pro snížení toxicity při kombinovaných chemoterapiích |
| JP2004536026A (ja) | 2000-11-28 | 2004-12-02 | トランスフォーム ファーマシューティカルズ,インコーポレーティッド. | パクリタキセル、その誘導体、および薬剤として許容される塩を含む医薬品製剤 |
| US6452025B1 (en) | 2001-04-25 | 2002-09-17 | Napro Biotherapeutics, Inc. | Three-step conversion of protected taxane ester to paclitaxel |
| US6479679B1 (en) | 2001-04-25 | 2002-11-12 | Napro Biotherapeutics, Inc. | Two-step conversion of protected taxane ester to paclitaxel |
| CN1596250A (zh) * | 2001-11-30 | 2005-03-16 | 布里斯托尔-迈尔斯斯奎布公司 | 紫杉醇溶剂化物 |
| AU2003273671A1 (en) * | 2002-10-09 | 2004-05-04 | Phytogen Life Sciences, Inc. | Novel taxanes and methods related to use and preparation thereof |
| US6759539B1 (en) | 2003-02-27 | 2004-07-06 | Chaichem Pharmaceuticals International | Process for isolation and purification of paclitaxel from natural sources |
| US7202370B2 (en) * | 2003-10-27 | 2007-04-10 | Conor Medsystems, Inc. | Semi-synthesis of taxane intermediates from 9-dihydro-13-acetylbaccatin III |
| WO2006116532A2 (en) * | 2005-04-28 | 2006-11-02 | University Of Massachusetts Lowell | Synthesis of oligo/poly(catechins) and methods of use |
| EP2029563A4 (en) * | 2006-06-12 | 2009-12-02 | Canada Inc 6570763 | SEMI-SYNTHETIC PATHWAY FOR THE PREPARATION OF PACLITAXEL, DOCETAXEL AND 10-DEACETYLBACCATINE III FROM 9-DIHYDRO-13-ACETYLBACCATINE III |
| US7847111B2 (en) | 2006-06-19 | 2010-12-07 | Canada Inc. | Semi-synthetic route for the preparation of paclitaxel, docetaxel, and 10-deacetylbaccatin III from 9-dihydro-13-acetylbaccatin III |
| CA2723654A1 (en) * | 2008-05-07 | 2009-11-12 | Ivax Research, Llc | Processes for preparation of taxanes and intermediates thereof |
| KR101014438B1 (ko) * | 2008-09-26 | 2011-02-14 | 동아제약주식회사 | 탁산 유도체의 제조방법 |
| RU2013103763A (ru) | 2010-07-02 | 2014-08-10 | Ангиохем Инк. | Короткие и содержащие d-аминокислоты полипептиды для терапевтических конъюгатов и их применения |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4521599A (en) * | 1983-07-11 | 1985-06-04 | The Upjohn Company | Process for the preparation of 1,3-oxazine-4-ones |
| US4549014A (en) * | 1984-09-06 | 1985-10-22 | Pennwalt Corporation | Adamantyl containing 1,4-oxazinone derivatives |
| US4631340A (en) * | 1984-09-06 | 1986-12-23 | Pennwalt Corporation | 1,4-oxazinone derivatives |
| US4552585A (en) * | 1984-10-04 | 1985-11-12 | Fmc Corporation | Herbicidal 2-(aminophenyl)methyl derivatives of 3-isoxazolidinones or 3-oxazinones |
| FR2601676B1 (fr) * | 1986-07-17 | 1988-09-23 | Rhone Poulenc Sante | Procede de preparation du taxol et du desacetyl-10 taxol |
| FR2601675B1 (fr) * | 1986-07-17 | 1988-09-23 | Rhone Poulenc Sante | Derives du taxol, leur preparation et les compositions pharmaceutiques qui les contiennent |
| US4876399A (en) * | 1987-11-02 | 1989-10-24 | Research Corporation Technologies, Inc. | Taxols, their preparation and intermediates thereof |
| GB8803114D0 (en) * | 1988-02-11 | 1988-03-09 | Bp Chem Int Ltd | Bleach activators in detergent compositions |
| FR2629819B1 (fr) * | 1988-04-06 | 1990-11-16 | Rhone Poulenc Sante | Procede de preparation de derives de la baccatine iii et de la desacetyl-10 baccatine iii |
| FR2629818B1 (fr) * | 1988-04-06 | 1990-11-16 | Centre Nat Rech Scient | Procede de preparation du taxol |
| US4960790A (en) * | 1989-03-09 | 1990-10-02 | University Of Kansas | Derivatives of taxol, pharmaceutical compositions thereof and methods for the preparation thereof |
| US5136060A (en) * | 1989-11-14 | 1992-08-04 | Florida State University | Method for preparation of taxol using an oxazinone |
-
1990
- 1990-10-30 US US07/603,041 patent/US5136060A/en not_active Expired - Lifetime
- 1990-11-07 NZ NZ235993A patent/NZ235993A/en unknown
- 1990-11-08 AU AU65904/90A patent/AU633375B2/en not_active Ceased
- 1990-11-11 IL IL10924690A patent/IL109246A/en not_active IP Right Cessation
- 1990-11-11 IL IL9630490A patent/IL96304A/en not_active IP Right Cessation
- 1990-11-12 MY MYPI90001993A patent/MY106071A/en unknown
- 1990-11-12 IE IE406190A patent/IE74154B1/en not_active IP Right Cessation
- 1990-11-12 IE IE960720A patent/IE76279B1/en not_active IP Right Cessation
- 1990-11-13 AT AT90312366T patent/ATE132860T1/de not_active IP Right Cessation
- 1990-11-13 CA CA002029787A patent/CA2029787C/en not_active Expired - Fee Related
- 1990-11-13 DK DK90312366.9T patent/DK0428376T3/da active
- 1990-11-13 FI FI905609A patent/FI104487B/fi not_active IP Right Cessation
- 1990-11-13 ES ES90312366T patent/ES2083438T3/es not_active Expired - Lifetime
- 1990-11-13 CA CA002347588A patent/CA2347588C/en not_active Expired - Fee Related
- 1990-11-13 NO NO904923A patent/NO177902C/no unknown
- 1990-11-13 PT PT95870A patent/PT95870B/pt not_active IP Right Cessation
- 1990-11-13 EP EP90312366A patent/EP0428376B1/en not_active Expired - Lifetime
- 1990-11-13 DE DE69024757T patent/DE69024757T2/de not_active Expired - Fee Related
- 1990-11-14 JP JP2308463A patent/JP2507830B2/ja not_active Expired - Fee Related
- 1990-11-14 CN CN90109112A patent/CN1043528C/zh not_active Expired - Fee Related
- 1990-11-14 YU YU216090A patent/YU48232B/sh unknown
- 1990-11-14 PL PL90287757A patent/PL167898B1/pl not_active IP Right Cessation
- 1990-11-14 OA OA59891A patent/OA09327A/xx unknown
- 1990-11-14 KR KR1019900018425A patent/KR0161499B1/ko not_active IP Right Cessation
- 1990-11-14 CZ CS905634A patent/CZ283581B6/cs not_active IP Right Cessation
- 1990-11-14 HU HU907131A patent/HU209299B/hu not_active IP Right Cessation
- 1990-11-14 RO RO146329A patent/RO110491B1/ro unknown
- 1990-11-14 EG EG68490A patent/EG19641A/xx active
- 1990-11-14 CZ CZ953136A patent/CZ283539B6/cs not_active IP Right Cessation
- 1990-11-14 BG BG93228A patent/BG60478B1/bg unknown
- 1990-11-14 RU SU904831946A patent/RU2033994C1/ru not_active IP Right Cessation
-
1992
- 1992-06-15 US US07/898,438 patent/US5384399A/en not_active Expired - Lifetime
-
1994
- 1994-04-07 IL IL10924694A patent/IL109246A0/xx unknown
- 1994-11-07 US US08/335,495 patent/US5532363A/en not_active Expired - Lifetime
-
1996
- 1996-03-05 GR GR960400595T patent/GR3019192T3/el unknown
Non-Patent Citations (4)
| Title |
|---|
| 1. R.A.Hrlton, et al, JACS, 1989, 110, С.6558. * |
| 2. T.W.Greene et al, JACS 1988, 110, с.5917. * |
| 3. Wani M.C. et al, JACS, 1971, 93, с.2325. * |
| 4. T.W. Creene et al "Protective Groups in organic synlhesis", 1981. * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2033994C1 (ru) | Оксазинон и способ получения таксола | |
| US5015744A (en) | Method for preparation of taxol using an oxazinone | |
| US5574156A (en) | β-lactams used in preparing taxol | |
| EP0400971B1 (en) | Method for preparation of taxol | |
| FI109796B (fi) | Metallialkoksidit | |
| EP0568203B1 (en) | Semi-synthesis of taxane derivatives using metal alkoxides and oxazinones | |
| JP2004043439A (ja) | β−ラクタム | |
| PL168497B1 (pl) | Sposób wytwarzania taksolu |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20081115 |