KR920008820B1 - 진통제 1,2-벤즈이소티아졸-3-일 피페라진 유도체의 제조방법 - Google Patents
진통제 1,2-벤즈이소티아졸-3-일 피페라진 유도체의 제조방법 Download PDFInfo
- Publication number
- KR920008820B1 KR920008820B1 KR1019850006060A KR850006060A KR920008820B1 KR 920008820 B1 KR920008820 B1 KR 920008820B1 KR 1019850006060 A KR1019850006060 A KR 1019850006060A KR 850006060 A KR850006060 A KR 850006060A KR 920008820 B1 KR920008820 B1 KR 920008820B1
- Authority
- KR
- South Korea
- Prior art keywords
- benzisothiazole
- piperazinyl
- prepared
- formula
- hydrogen
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D275/00—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings
- C07D275/04—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Neurosurgery (AREA)
- Life Sciences & Earth Sciences (AREA)
- Neurology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Biomedical Technology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Anesthesiology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
내용 없음.
Description
본 발명은 일반적으로 약물특성 및 생물학적 영향을 미치는 특성이 있는 신규한 복소환식 탄소화합물 및 이들의 제조방법 및 용도에 관한 것이다. 특히, 본 발명은 하나의 치환체가 그의 벤조-환에서 치환되거나 비치환된 1,2-벤즈이소티아졸-3-일이고 다른 하나는, 그중에서도 특히 알킬, 사이클로알킬, 아랄킬 및 페녹시알킬인 1,4-이치환된 피페라진 유도체에 관한 것이다.
관련 기술은 다음 일반식(1)에 비추어 생각할 수 있다 :

상기 식에서, R 및 Z는 치환체이고 X는 헤테로환원자 또는 헤테로환원자그룹이다.
가장 밀접한 관련기술은 브리스톨 마이어스 앤드 캄파니에 양도된 두가지 등록 특허에 포함되어 있는 기술인 것 같다.
템플 및 예비치에게 1983년 10월 25일자로 허여된 미합중국 특허 제 4,411,901호에는 다음 일반식(2)의 신경이완체가 지재되어 있다 :

상기 식에서, Z는 수소 또는 할로겐이고;
R은 다음(a) 내지 (f)기중에서 선택된 하나의 기를 나타내며;

n은 3 또는 4이다.
알 수 있는 바와 같이, R-치환체중 a 내지 d는 복소환식환인 반면, 일반식(2)의 화합물에 e와 f가 붙으면 항정신병의 부티로페논 유도체가 생성된다. 합성 중간체 화합물(3)은 역시 공개되어 있고, 1984년 6월 5일자 허여된 이건에 관련된 분할 미합중국 특허 제 4,452,799호에 청구되어 있다 :

1978년 8월 1일자 허여된 웨이드(Wade) 및 키씩크(Kissick)의 미합중국 특허제 4,104,388호에 소염제로서 소개된 일련의 화합물은 다음 일반식(4)를 갖는다 :

상기 식에서, Y는 C이거나 N일 수 있고;
A는 단일결합이거나 탄소수 1 내지 4의 알킬렌쇄이며;
B는 수소, 하이드록실, 또는 최적조건으로 치환된 페닐이다.
이들 화합물은 이들이 벤즈이소티아졸의 1,1-디옥사이드환 유도체이므로 용이하게 구별된다.
관련성을 가진 다음 참조문헌들은 본 출원에 소개된 신규한 화합물과는 관련성이 적다. 1982년 10월 19일자 허여된 스트릅쥬스키(Strupczewski)등의 미합중국 특허 제4,355,037호에는 일련의 벤즈이속사졸릴 피페리딘 유도체(5)가 개시되어 있으며, 이는 진통제로서는 기재되어 있다 :

일반식(5)에서, R은 수소, 알킬, 알케닐, 사이클로알킬-알킬, 펜알킬, 하이드록시, 아미노알킬, 시아노, 시아노알킬, 알카노일 또는 카복실산 에스테르 잔기일 수 있다.
더욱 관련이 적은 것은 일반식(6)으로 표시된 일련의 벤즈이속사졸-피페리딘 화합물이다 :

이들 물질은 1983년 8월 2일자 허여된 데이비스와 클레인의 미합중국 특허제 4,396,770호에 항정신병약 및 진통제로서 기재되어 있다.
알 수 있는 바와 같이, 본 발명의 화합물을 암시하거나 명백히 제조할 수 있는 관련기술에 관한 것은 상기 참조문헌에는 전혀 없다.
다음 일반식(I)의 1-(1,2-벤즈이소티아졸-3-일)피페라진-4-일 유도체 계열 또는 이의 약제학적으로 허용되는 산부가염이 합성되었다 :
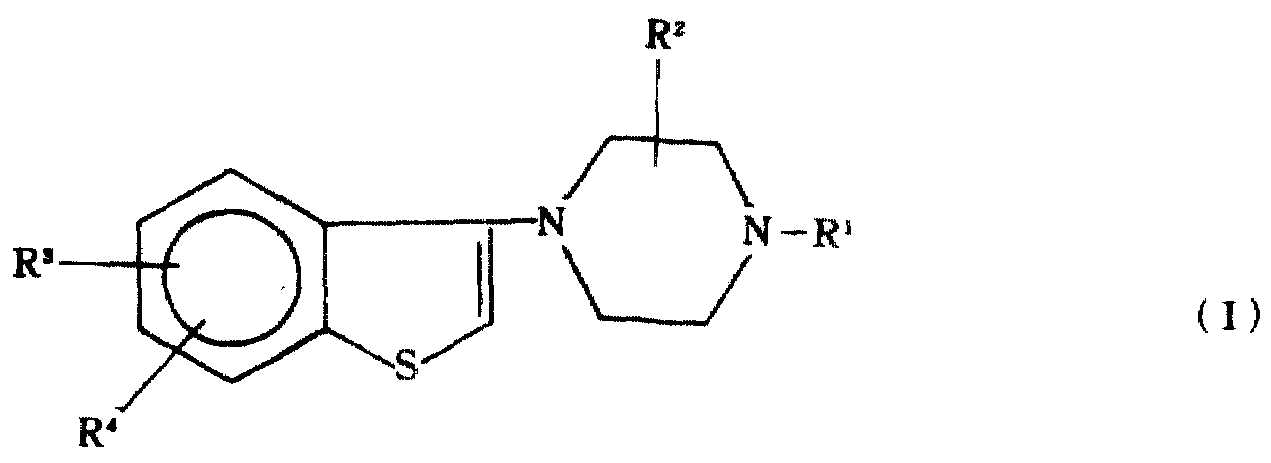
상기 식에서, R1은 수소, 저급(C1-6)알킬, 직쇄 또는 측쇄의 저급알케닐, 저급사이클로알킬, 저급사이클로알킬-저급알킬렌, 페닐-저급알킬렌, 페녹시-저급알킬렌, 페닐-저급사이클로알킬, 또는 벤조디옥산-2-일-저급알킬렌이며 ;
R2는 저급알킬 또는 수소이고;
R3와 R4수소, 저급알킬, 저급아실옥시, 저급알콕시, 저급알킬티오, 할로겐 또는 하이드록실 또는 트리플루오로메틸중에서 독립적으로 선택된다.
약리학적 시험에 의하여 이들 화합물들이 진통제 활성을 가진다는 것이 입증되었다. 그외에도, 시험관내에서 오피올로이드(opioloid) 수용체에 대해서는 친화력이 부족함을 나타내고 세로토닌성 및 아드레날린성 수용체 형태에 대해서는 고유 친화력을 나타내는 것은 이들 화합물이 신규한 비-마취성 진통제일 수 있다는 것을 암시한 것이다.
가장 넓은 관점에서 볼 때, 본 발명은 일반식(I)의 화합물임을 특징으로 하는 진통제 특성을 갖는 1,2-벤즈이소티아졸-3-일-피페라진 유도체 또는 이의 약제학적으로 허용되는 산부가염에 관한 것이다 :

상기 식에서, R1은 수소, 저급(C1-6)알킬, 직쇄 또는 측쇄의 저급알케닐, 저급사이클로알킬, 저급사이클로알킬-저급알킬렌, 페닐-저급알킬렌, 페녹시-저급알킬렌, 페닐-저급사이클로알킬, 또는 벤조디옥산-2-일-저급알킬렌이며;
R2는 저급알킬 또는 수소이고;
R3와 R4는 수소, 저급알킬, 저급아실옥시, 저급알콕시, 저급알킬티오, 할로겐, 하이드록실 또는 트리플루오로메틸중에서 독립적으로 선택된다.
R1은 R2, R3및 R4가 모두 수소일때는 수소 이외의 것이다.
본 명세서에서 사용된 할로겐은 불소, 염소, 브롬 또는 요오드를 나타내는 것으로 이해되어야 한다. 일반식(I)의 바람직한 화합물은 탄소수가 4개이상인 R1그룹을 가지며, 가장 바람직한 화합물은 R1이 사이클로알킬인 화합물이다.
본 발명은 화학기술에 숙련된 사람이면 누구나 알 수 있는 구조적 대칭의 결과로서 생길 수 있는 어떤 것이든 모든 입체이성체를 포함하는 것으로 간주된다는 것으로 이해하여야 한다. 입체 이성체가 존재할 경우 이들 각 입체이성체의 분리는 화학실무자들에게 잘 알려져 있는 여러 가지 방법을 적용시켜서 행할 수 있다.
의약용의 경우 약제학적으로 허용되는 산부가염, 즉, 음이온이 유기 양이온의 약물학적 활성에 대한 독성에 현저하게 기여하지 않는 이들 염이면 바람직할 수 있다. 산부가염은 일반식(I)의 유기염기를 유기산 또는 무기산과 바람직하게는 접촉 또는 용액에 의하여 반응시켜 얻거나 본 분야에 통상의 지식을 가진 어떤 실무자도 입수할 수 있는 문헌에 상세히 설명된 어느 표준방법에 의해서도 얻는다. 유용한 유기산의 예로는 말레인산, 초산, 주석산, 프로피온산, 푸말산, 이세티온산, 호박산, 파모인산, 사이클라민산, 피발린산등과 같은 카복실산이 있으며; 유용한 무기산은 HCl, HBr, HI와 같은 할로겐화 수소산; 황산; 인산등이다.
본 발명의 화합물은 반응도식 1에 표시된 공정을 이용하여 편리하게 제조할 수 있다.
[반응도식 1]
일반식(I) 화합물의 제조공정

이 반응도식에서, R1내지 R4는 일반식(I)에서 앞서 이것들에 관하여 부여된 의미와 동일한 의미를 갖는다. 부호A는 수소이거나 t-부틸일 수 있고, B는 수소이거나 t-부틸옥시카보닐 잔기일 수 있다. A에 대한 잔기의 선택은 A가 (V)를 (IV)으로 전화하는 중에 없어지기 때문에 공정에서는 중요하지 않다. B가 t-부틸옥시카보닐 잔기(차단그룹)일 경우, 그것은 분해되어 B가 수소인 적합한 일반식(IV) 또는 (II)의 화합물이 수득되며, 그후 추가의 반응, 예를 들면 R1X와의 반응에 의하여 생성물(I)을 수득한다.
반응도식 1에서 표시된 합성공정은 다음과 같은 1) 내지 3)의 공정이 필수적으로 포함된다 :
1) 1,2-벤즈이소티아졸-3-(2H)-온(V)을 강 염소화제, 예로서, POCl3나 PCl5로 처리하면 3-클로로-1,2-벤즈이소티아졸(IV)이 수득된다. 여러 가지 할로겐화제를 사용하여 (V)를 (IV)로 전화시키는 방법은 화학문헌[N.Davis, "Benzisothiazoles"in Advances is Heterocyclic Chemistry, Vo1. 14, Edited by A.R.Katritzky and A.J.Boulton 및 본 명세서내의 여러 가지 문헌]에 충분히 기재되어 있다.
2) 3-클로로-1,2-벤즈이소티아졸(IV)을 적합한 피페라진 중간체(III)와 반응시키면 3-(1-피페라지닐)-1,2-벤즈이소티아졸이 수득된다. 이 단계에서 사용되는 반응조건은 이런 형태의 친핵성 치환반응에 대하여 보통 사용되는 것들이다. 화합물(III)이 액체라면, 이들 반응은 가끔 순조롭게 진행된다. 일반식(IV)의 특정한 중간체 화합물의 경우에는 수율이 최적화 될 수 있고, 중간체(II)의 분리는 (III)의 N-t-부틸옥시카보닐 피페라진 중간체(여기에서 B는 t-부틸옥시카보닐이다)를 사용함으로써 더욱 쉽게 이루어질 수 있다. 이들 반응의 경우에, 차단용 N-t-부틸옥시카보닐그롭은 다음 단계에서 그것을 사용하기전에 산가수분해시켜서 (II)로부터 제거한다. 이 차단용 t-부틸옥시카보닐그룹의 제거반응은 에타놀성 HCI로 처리하면 쉽게 일어난다.
3) 3-(1-피페라지닐)-1,2-벤즈이소티아졸(II)을 R1X로 알킬화시키면 목적하는 일반식(I)의 생성물이 수득된다. 이 반응 단계에서, 중간체(II)를 불활성용매, 바람직하게는 아세토니트릴중에 용해시키고, 1당량의 보호된 염기, 바람직하게는 N,N-디이소프로필에틸아민으로 처리하고, 이어서 X가 할라이드, 토실레이트, 메실레이트 등과 같은 전형적인 이탈그룹인 R1X로 처리한다. 이 반응은 통상 수시간동안 환류하면서 가열시키면 완료된다.
단순한 알킬화 반응외에도, (II)에서 (I)로의 동일한 구조적 전화를 효과적으로 성취하는 다른 합성방법들이 이용될 수 있다. 이러한 방법의 예로는 일반식(II)(B=H)의 화합물을 카보닐 화합물로 환원적 아민화 반응시켜서 일반식(I)의 생성물을 얻는 방법이 있다. 이러한 선택적인 합성방법의 한예는 아래에 표시되어 있다 :

특정한 일반식(V)의 중간체 1,2-벤즈이소티아졸론을 용이하게 구입할 수 없는 경우 이들을 제조하기 위하여 여러 가지 합성방법을 이용할 수 있다. 이들 방법중 몇가지는 아래의 반응도식 2에 표시되어 있다 :
[반응도식 2]
일반식(V) 중간체의 제조방법

대부분의 경우에, 반응도식 2에 표시된 방법중 어느방법이든지 목적하는 중간체 화합물(V)의 제조에 이용할 수 있다. 방법이 바람직한지 여부는 출발물질의 이용 가능성과 각각의 방법에 특이한 여러 가지 중간체 화합물의 분리 용이성에 기초를 두고 있다. 방법1은 다음에 염화티오닐을 사용하여 고수율의 상응하는 산염화물(IV)로 전환되는 적합하게 치환된 디티오살리실산(VII)을 필요로 한다. 중간체(VI)를 염소와 반응시키고, 이어서 암모니아와 연속적으로 반응시키면 목적하는 중간체 생성물(V)이 수득된다.
가장 일반적이고 가장 광범위하게 적용할 수 있는 방법인 방법2는 염화티오닐로 처리하고 이어서 t-부틸아민으로 처리하면 t-부틸벤조아미드(IX)로 전화되는 적절하게 치환된 벤조산(X)을 출발물질로 한다. 이 벤즈아미드(IX)는 n-부틸리튬으로 오르토-금속화하고, 이어서 이황화메틸로 설핀화 반응시키면 화합물(VIII)이 생성된다. s-메틸 중간체(VIII)를 메타-클로로과벤존산 또는 과요오드산나트륨으로 산화시키면 상응하는 설폭시드가 생성되며, 이는 트리클로로메틸클로로포르메이트로 처리하면 목적하는 중간체 생성물(V)로 전화된다.
방법3은 방법2의 변형으로서, 벤즈아미드 중간체(IX), 예로서, 5-메톡시벤조산이 목적하는 국부 화학적인 오르토-방향의 금속화 반응되지 않을 경우에 이들(V)의 화합물을 제조하는데 이용된다. 이 경우에, 오르토-브로모 벤조산(VIII)을 t-부틸리튬으로 처리하고 금속-할로겐 교환반응시키면 오르코-음이온(XII)이 수득되면, 그다음 이를 디메틸디슬파이드와 반응시키면 중간체(XI)가 생성된다. 그 다음 이 오르토-메틸테오벤조산은 반응도식 2에 표시된 바와 같이 이 시점에서 중간체 화합물(VIII)로 전화된 다음 최종 중간체(V)로 전화된다.
방법1 내지 3의 여러가지 변형과 개량은 화학기술분야에 숙련된 사람들에게는 명백할 것이다. 적절한 중간체의 합성을 포함하는 방법1 내지 3의 예는 본 명세서에 후술할 것이다.
본 발명 화합물은 포유동물에 대하여 진통제 활성을 나타내는 유용한 약제이다. 다음의 생체내 시험(표 1)들을 본 계열의 화합물을 평가하고 분류하는데 이용된다.
[표 1]
일반식(I)의 화합물을 평가하는데 사용되는 생체내 시험
1. 조건부 회피반응(CAR)…독성이 증강된 단식 쥐에서 전기적 충격에 대한 회피반응을 감소시켜서 결정한 약물의 정신안정화 활성의 측정.
2. 노르에피네프린 치사유의 억제…노르아드레나린성 제제의 노르에피네프린의 치사율에 대한 약물억제작용은 아드레나린 차단을 가리킨다.
3. 발성한계치…전기적 충격에 의해 유발된 쥐의 발성을 억제하는 능력에 의해서 측정되는 약물의 진통제활성의 측정.
4. 페닐퀴논 몸부림…생쥐의 페닐퀴논에 의해 유발된 몸부림 증후군을 막는 능력에 의해서 측정되는 약물의 진통제활성의 측정.
또한, 다음의 시험관내의 방사능 수용체 결합 검정이 특정 결합 부위에 대한 본 계열 화합물의 고유 친화력을 측정하는데 이용된다 :
1.[3H]스피페론에 대한 선조직에서의 도파민 결합.
2.[3H]WB-4101에 대한 WB-아드레나린성 결합.
3.[3H]스피페론에 대한 수피조직에서의 세로토닌형 2 결합.
앞서 언급한 시험에 의해서 수립된 약리학적 윤곽에 따라 본 일반식(I)의 화합물은 이 계열의 화합물을 비-마취성 진통제로서 유력한 후보물이 되게하는데 충분한 잠재력이 있는 진통작용을 가진다. 상기 나열된 시험관내의 결합 연구결과는 아드레나린 및 세로토닌 기전이 이 계열에 대한 무통조정에 포함되는 것이 확실하다는 것을 나타낸 것이다.
생체내 시험자료에 관하여는, 페닐퀴논 몸부림과 발성한계치 측정이 진통제활성의 전조로서 이용된 중요한 스크린 시험이었다. 본 계열 화합물의 대부분은 발성한계치 시험에서는 10mg/kg이하이고 페닐퀴논 몸부림 시험에서는 100mg/kg미만인 성능을 가졌었다. 조건부 회피반응과 노르에피네프린 치사율 억제시험은 더 많은 항정신병 작용과 진정작용 각각의 전조가 된다. 이들 시험의 결과는 본 발명 화합물들이 이들 시험에서 부작용에 대한 부담의 감소를 나타낼 수도 있는 낮은 수준의 활성을 가짐을 나타낸다.
요약하여, 본 발명 화합물은 비-마취성 진통제로서 사용하기에 적합한 약리학적 특성을 갖는다. 그래서, 본 발명의 또다른 관점은 유효한 진통제 용량의 일반식(I)의 화합물이나 이들의 약제학적으로 허용되는 산부가염을 포유동물에 전시투여하는 것으로 구성되는 그와 같은 치료를 필요로 하는 동물에 무통을 유발시키는 방법에 관한 것이다. 일반식(I)의 화합물의 투여 및 적량 양생법은 대조 화합물 네포팜[참조예 : 힐 등의 Drugs, 19권, 249-267페이지(1980년판)]에 관한 것과 동일한 방법으로 행해질 것으로 기대된다.. 투여량 및 적량 양생법은 각각의 경우에 조절되어야만 하지만 소리의 전문적인 판단을 이용하여 수용자의 나이, 체중 및 상태, 투여경로와 질병의 성질 및 비중을 고려하여 주의 깊게 조절하여야 하지만, 일반적으로 1일 복용량은 약 30 내지 600mg을 1-3번/일 투여하게 된다. 투여량은 비경구로 투여할 경우 이 범위이하의 량으로 하고 경구투여의 경우에는 이 범위 바로 위의 량으로 투여하게 될 것으로 예상된다. 어떤 경우에는, 이보다 낮은 복용량으로도 충분한 치료효과를 얻을 수 있는 반면 다른 경우에는, 더 많은 복용량을 필요로 하게 된다.
본 명세서에서 사용된 "전신투여"란 용어는 경구, 직장 및 비경구(즉, 근육내, 정맥내 및 피하의)의 경로를 의미한다. 일반적으로, 본 발명 화합물이 바람직한 경로인 경구로 투여될 경우, 보다 적은 량을 비경구로 투여했을 때와 동일한 효과를 내도록 더 많은 량의 활성제제가 필요하다는 것을 알게된다. 우수한 임상실시에 의하여, 어떤 유해하거나 곤란한 부작용을 일으키지 않고, 유효한 진통효과를 나타내게 되는 농도수준으로 본 발명 화합물을 투여하는 것이 바람직하다. 본 발명 화합물은 비-마취성 진통제이므로, 전형적인 마취 부작용을 우회시킬뿐 아니라 이들 제제와 마취성 진통제를 동시에 진통제로서 사용하기 위한 잠재력이 존재한다.
치료적으로, 본 발명은 일반적으로 유효한 진통제 량의 일반식(I)의 화합물 또는 이의 약제학적으로 허용되는 산부가염과 약제학적으로 허용되는 담체로 구성된 약제학적 조성물로서 투여된다. 이러한 치료를 하기위한 약제학적 조성물은 95 내지 0.5%의 본 발명 화합물중 적어도 하나를 약제학적 담체와의 배합물로 대량이나 소량으로 함유하며, 그 담체는 비독성이며 불활성이고 약제학적으로 허용되는 하나 또는 그 이상의 고체, 반고체 혹은 액체의 희석제, 충전제 및 제제 보조제를 포함한다. 이들 약제학적 조성물은 투여 단위 형태, 즉, 목적하는 치료적 반응을 일으키게끔 계산된 분획용량 또는 배수용량에 상응하는 예정된 량의 약물을 함유하는 물리적으로 분리된 단위인 것이 바람직하다. 투여단위는 하나나 그 이상의 단일 복용량, 또는 단일 복용량의 2분의 1이나 3분의 1 또는 그 이하를 함유할 수도 있다. 단일 복용량은 보통 1일 투여량의 전부나 반 또는 3분의 1을 하루에 1회 내지 3회 투여하는 예정된 투여량 양생법에 따라 하나 또는 그이상의 투여단위를 한번에 투여하므로서 목적하는 치료효과를 나타내는데 충분한 량을 함유하는 것이 바람직하다. 다른 치료약제도 존재할 수 있다. 단위 복용량 당 활성성분 약 30 내지 600mg을 제공하는 약제학적 조성물이 바람직하고 통상적으로는 정제, 로젠지제, 캅셀제, 분말제, 수성 또는 오일현탁제, 시럽제, 엘릭시르제 및 수용액제로서 제조된다. 바람직한 경구용 조성물은 정제나 캅셀제의 형태이고 결합제(예:시럽, 아카시아, 젤라틴, 솔비톨, 트라가칸트 또는 폴리비닐피롤리돈)와 충전제(예:락토스, 설탕, 옥수수전분, 인산칼슘, 솔비톨이나 글리신), 윤활제(예:스테아린산마그네슘, 활석, 폴리에틸렌글리콜 또는 실리카), 봉해제(예:전분) 및 습윤제(예:라우릴황산나트륨)과 같은 통상적인 부형제를 함유할 수도 있다. 일반식(I)의 화합물과 통상적인 약제학적 비히클과의 용액이나 현탁액은 정맥내 주사용 수용액이나 근육내 주사용 오일 현탁액과 같은 비경구용 조성물용으로 사용된다.
투명성, 안정성 및 적응성을 갖는 이러한 비경구용 조성물은 글리세린, 플로필렌글리콜 및 폴리에틸렌글리콜과 같은 다가 지방족알콜 또는 이의 혼합물로 구성되는 비히클 또는 물중에 0.1 내지 10중량%의 활성 화합물을 용해시켜 수득한다. 폴리에틸렌글리콜은 물과 유기액체에 용해성이고 약 200 내지 1500의 분자량을 갖는 비-휘발성이며, 보통 액체인 폴리에틸렌글리콜의 혼합물로 구성된다.
본 발명을 구성하는 화합물들과 이들의 제조방법은 다음의 실시예들을 고찰하므로서 더욱 충분하게 이해가게 될 것이며, 이들 실시예는 단지 예시할 목적으로 주어졌으며 본 발명의 범위를 제한하는 것으로 해석해서는 안된다. 모든 온도는 특히 언급하지 않는 경우에는 섭씨온도이다.
핵자기공명(NMR) 스펙트럼 특성은 대조되는 표준물로서 테트라메틸실란(TMS)에 대한 백만분의 1(ppm)로 표시되는 화학적 변위(δ)에 관한 것이다. 프로톤(NMR) 스펙트럼 자료에서 여러가지 변위에 대하여 보고된 관련영역은 분자중에 특수 기능형의 수소원자수에 상당하는 것이다. 다양성에 관한 변위의 성질은 넓은 단일선(bs), 단일선(s), 다중선(m), 이중선(d), 이중선의 이중선(dd), 또는 4중선(q)으로서 표시되어 있다. 사용되는 약어는 DMSO-d6(제2디메틸슬폭시드) CDCl3(제 2 클로로포름)이고 다른 것들은 통상적인 것이다. 적외선(IR) 스펙트럼 설명은 기능 그룹 확인값을 갖는 흡수 파장수(cm-1)만을 포함한다. 희석제로서 브로화칼륨(KBr)을 사용하여 IR측정을 한다. 원소분석은 중량%로 표시한다.
[중간체의 합성]
A. 일반식(V)의 화합물

[실시예 1]
1,2-벤즈이소티아졸-3(2H)-온 (방법 1)
2,2-디티오살리실산, 염화티오닐(1645g, 13.826몰), 톨루엔(10리터) 및 N,N-디메틸포름아미드(40ml)의 슬러리를 약 75°에서 18시간동안 가열한다. 이 시점에서 모든 고체는 용해되고, 생성된 진한 용액을 8°로 냉각시킨다. 반응 생성물을 결정화하고 여과시켜 분리하고 약 1ℓ의 냉각 스켈리 에프(Skelly F.)를 사용하여 여과지상에서 세척한다. 건조시키면 융점 154-156도인 1619g(71%)의 2,2'-디티오-비스-벤조일클로라이드가 수득된다[문헌상 융점 155-156°; 참조 : 아이.알. 다글라스 및 비.에스. 파라, J. Org. Chem. 26권, 351-354면(1961년 판)].
염소(239g, 3137몰)로 염화 2,2,'-디티오-비스벤조일(1157g, 3.37몰) 및 염화메틸렌(8.46ℓ)의 교반된 현탁액에서 기포를 발생시킨다. 생성된 용액을 격렬하게 교반하면서 농-NH4OH(2.89ℓ)에 가한다. 그 혼합물을 첨가가 완료된 후 1시간동안 교반한다. 여과하면 습윤 고체가 수득되며, 이를 약 7ℓ의 물에 현탁시키고, 격렬하게 교반하면서 농염산을 가하여 산성화 시킨다. 고체를 여과하여 분리하고 약 3ℓ의 물로 여과지상에서 세척한다. 30°에서 진공 건조시키면 융점 155.5-157°인 902g(88.5%)의 생성물이 수득된다.
[실시예 2]
일반적 제법(방법 2)
적절한 벤조산 클로로포름(1ml의 CHCL3/1g의 산)과 두방울의 DMF중에 현탁시킨다. 단일부의 염화티오닐(4몰 당량)을 실온에서 가하고 생성된 혼합물을 교반하면서 서서히 환류하에 가온한다. 이 혼합물을 전형적으로 2 내지 4시간 범위의 환류시간으로 반응이 진행되면 투명해진다. 반응이 완료되면 실온으로 냉각시키고 진공 농축시켜 염화벤조산 화합물에 상응하는 조화합물을 약 95%의 수율로 수득한다. 중간체산염화물의 염화메틸렌 용액(50ml의 CH2Cl1/0.01몰의 산염화물)을 0°에서 염화메틸렌중의 트리에틸아민과 t-부틸아민의 용액에 첨가한다. 반응물을 실온으로 가온시키고 약 18시간동안 방치한다. 그 다음 이 혼합물을 1.5N HCl로 세척한 다음 0.5N NaOH용액으로 세척하고, 마지막으로 염수로 세척한다. 세척한 다음 반응 혼합물을 건조시키고(MGSO4로), 여과하고 진공중에서 농축하여 조아미드를 약 95%의 수율로 수득한다. 이 아미드 중간체 (IX)를 재결정화나 쿠겔로르 증류에 의해서 정제하면 71 내지 76%의 정제된 생성물이 수득될 수 있다.
벤즈아미드 중간체(IX; 0.025 내지 0.40몰)을 무수 THF(500ml)중에 용해하여 얼음/아세톤/염 욕으로 -10° 냉각하면서 질소 분위기하에 교반시킨다. 이 냉각, 교반된 반응 용액에 약 -10°의 온도를 유지하는 적정속도로 n-부틸리늄(2.5몰 당량)을 가한다. 첨가가 완료된 다음, 반응 혼합물을 15 내지 30분간 -78°(드라이아이스/아세톤욕)로 냉각한다. 각 증류된 디메틸디슬파이드(3몰 당량)을 -70°이하의 온도를 유지시킬 수 있는 속도로 적가한다. 냉각 반응액을 1시간 교반한 다음 천천히 가온시키고, 0에서 15%의 NH4Cl차 수용액(2.75몰 당량)을 사용하여 반응을 억제한다. 이 혼합물을 진공농축하고 염화메틸렌-몰 사이에 분할시킨다. 수층을 다시 염화메틸렌으로 추출하고 이들 유기부분을 합하여 건조시키고(MgSO4), 여과시키고, 진공 농축하여 조티오메틸벤즈아미드 중간체(VIII)를 수득한다. 약 90% 수율로 얻어진 조물질을 다시 정제하지 않고 사용하거나 섬광 크로마토그라피(헥산/에틸아세테이트 혹은 헥산/염화메틸렌)로 정제할 수 있다.
티오메틸벤즈아미드(VIII;0.02-0.15몰)을 메타놀(200-500ml)중에 용해하고 교반하면서 질소 분위기하에서 수성 0.05N NaIO4에 가한다. 20시간 후 반응이 완료되면 메타놀을 감압하에 제거한다. 생성된 수성상을 염화메틸렌으로 추출하고 이들 유기 추출물을 합하여 건조시키고(MgSO4), 여과하고, 진공 농축시킨다. 생성된 고체를 섬광 크로마토그라피(에틸 아세테이트)로 정제한 다음 디클로로에탄(0.002몰/8ml) 중에 용해시키고 질소 분위기하에 교반하면서 0°로 냉각시킨다. 그 용액을 약 1시간동안 실온에서 교반시키면서 트리클로로메틸글로로포르메이트(1몰 당량)을 단일 부분으로 가한다. 10%의 가성소다 용액(5몰 당량)을 2시간이하 동안 계속하여 격렬히 교반하면서 반응 혼합물에 가한다. 그 다음 이 혼합물을 염화메틸렌으로 추출하고 그추출물을 합하여 건조시키고(MgSO4), 여과 및 진공 농축하여 조생성물(V)를 얻고, 이는 재결정화시켜 정제할 수 있다.
[실시예 3]
N-t-부틸-4-메톡시-1,2-벤즈이소티아졸론
0-아니스산(0.38몰)을 60ml의 CHCl3와 3방울의 DMF중에 현탁시킨다. 단일용적의 SOCl2(4몰 당량)을 실온에서 격렬하게 교반하면서 가한 다음 그 반응 혼합물을 환류시킨다. 반응물을 1740cm-1에서의 카복실산 카보닐 신장(stretch)의 소실 및 1780cm-1에서의 아실카보닐 신장의 출현에 대하여 IR로 탐지한다. 반응이 24시간 후 완료된 것으로 판단된다. 용매와 과잉 시약을 진공증류(86mmHg)를 통하여 제거하면 오일상의 조염화 0-아니소일이 95%보다 큰 수율로 수득된다.
산염화물을 냉각시키고 50ml의 CHCl3로 희석하고, 100ml의 CHCl3중에 TEA 및 t-부틸아민(1.2몰 당량, 각각)을 용해시킨 냉각용액(얼음/물)에 적가한다. 일단 첨가가 완료되면 냉각욕을 제거하고 혼합물을 주위 온도하에 방치한다. 혼합물을 3×1.5N HCl, 2×0.5N NaOH, 및 1×염수로 세척한다. 클로로포름을 MgSO4로 건조시키고, 여과시키고 진공 농축하여 거의 93g의 오일을 얻어서 이를 쿠겔로르로 증류(110°10.3mmHg)시킨다. N-t-부틸-O-아니스아미드가 95% 수율로 황색 오일로서 수득된다.
벤즈아미드(0.05몰)을 500ml 무수 THF중에 용해시키고 N2하에 -10°(MeOH/얼음)로 냉각시킨다. 온도를 -10°와 0° 사이로 유지하기 위한 속도로 n-BuLi(2.5몰 당량)을 적가한다. 그 용액을 -10°에서 30분간 교반한 다음 -75°(CO2/아세톤)로 냉각시키고, 증류된 메틸 디슬파이드(3.0몰 당량)을 -75°와 -70°사이의 온도를 유지하는 속도로 가한다. 냉각 반응액을 서서히 가온시키고 -65°에서 NH4Cl 수용액(2.75몰 당량)을 가한다. 용액을 진공 농축하고; 잔사를 CH2Cl2및 몰 사이에 분할시키고 CH2Cl2로 3회 추출한다. 합한 유기부분을 MgSO4로 건조시키고, 여과하고, 진공 농축하면 황색고체가 수득된다. 94%의 조수율의 조물질이 TLC에 의해서 허용 가능한 것으로 판단되면 추가로 정제하지 않고 사용한다.
티오메틸벤즈아미드(0.14몰)을 200ml의 CH2Cl2중에 용해시키고 N2하에 -78°(CO2/아세톤)에서 교반한다. mCPBA(1.2몰 당량)의 700ml CH2Cl2용액을 신속히 적가한다. 1시간 후 TLC에 의해서 그 반응이 완료된 것으로 판단되면 주위 온도로 가온시킨다. 혼합물을 진공 농축한 다음 CH2Cl2와 물 사이에 분할시킨다 이 혼합물을 3×H2O와 3×10%의 수성 K2CO3로 추출한다. 유기층을 K2CO3로 건조시키고 여과하여 진공중에서 농축하여 조물질을 얻고 이를 섬광 크로마토그라피(3%, MeOHOl CH2Cl2용액)한다. 고체를 10% EtOAC의 헥산용액으로 재 결정화시켜 결정성 슬폭시드를 75%수율로 수득한다. 슬폭시드(0.08몰)를 160ml의 이염화에탄에 용해시키고 N2하에 40°로 가온시킨다. 트리클로로메틸 클로로포르메이트(1몰 당량)을 순수하게 적가한다. 반응이 격렬해지면 가온을 중단하고 남아있는 클로로포르메이트를 실온에서 첨가한다. 그 다음 혼합물을 70°로 다시 가온한다. 반응은 1/2시간 후 TLC에 의해서 완료됐음이 판단된다. 반응액을 냉각시키고 170ml의 10% NaOH(수성)중에 부어 1/2시간동안 격렬하게 교반시킨다. 이염화에탄층을 제거하고 수성상을 3×CH2Cl2로 추출한다. 합한 유기상을 MgSO4로 건조시키고, 여과하고 진공 농축시킨다. 조물질을 섬광 크로마토그라피하면(40% EtOAC/60%헥산) N-t-부틸-4-메톡시-1,2-벤즈이소티아졸론이 80%의 수율로 수득된다.
[실시예 4]
5-메톡시-N-t-부틸-1,2-벤즈이소티아졸-3(2H)-온 (방법 3)
2-브로모-5-메톡시벤조산(0.005몰)을 50ml은 무수테트라하이드로푸란중에 용해시키고 교반하면서 질소하에 -78°로 냉각시킨다. n-부틸리튬(1.1몰 당량)을 -70°미만의 온도를 유지하는 속도로 적가한다. 그 다음 황색 불용성 음이온을 -115°로 냉각(액체질소-에틸에테르욕)하고 t-부틸리튬(1.4몰 당량)을 -85°미만의 온도를 유지하기 위한 속도로 가한다. 첨가가 완료되었을 때, 불용성 이중 음이온을 -75°로 가온하여 그것이 용해되게 한다. 증류된 이황화메틸을 -70°미만의 반응 온도를 유지할 수 있는 속도로 가한다.
밝은 황색 용액이 거의 무색으로 변하다. 반응액을 서서히 가온시키고 -40°에서 15%의 수성 NH4Cl(2.75몰 당량)로 반응을 중지시킨다. 테트라하이드로푸란을 진공중에서 제거하고 수성상을 염화메틸렌으로 세 번 세척한 다음 6N HCl로 산성화시키고, 그 다음 염화메틸렌으로 세번 추출하여, 건조(MgSO4), 여과 및 진공농축하여 백색 고체를 얻는다. NMR 스펙트럼에 의하면, 백색고체는 5-메톡시-1,2-티오메틸벤조산과 m-아니스산 각각의 65:35의 혼합물로 구성되어 있다.
5-메톡시-1,2-티오메틸벤조산/m-아니스산 혼합물(S-메틸 물질로 계산하여 0.095몰)을 80ml의 클로로포름과 두방울의 DMF중에 용해시킨다. 염화티오닐(4몰 당량)을 한번에 가하고 용액을 환류하면서 가열한다. 세시간 동안 환류후 반응 혼합물을 실온으로 냉각시키고 진공 농축시켜서 조벤조산염화물 혼합물을 얻으며, 이는 실시예 2에서와 같이 t-부틸아민으로 처리하면 벤즈아미드혼합물로 전환된다. 이 시점에서 NMR은 s-메틸아니스아미드 대 아니스아미드의 비가 약 9:2임을 나타낸다.
N-t-부틸-5-메톡시-2-s-메틸벤즈아미드/N-t-부틸아니스아미드 혼합물(s-메틸 물질로서 계산하여 0.04몰)을 더 이상 정제하지 않고 사용하여, 450ml의 메타놀중에 용해시키고 420ml의 0.05M 수성(NaIO4와 질서하에 혼합한다. 5시간이나 지나 반응이 완결되면 메타놀을 감압하에 제거한다. 생성된 수성상을 염화메틸렌으로 추출하고, 이들 유기 추출물을 혼합하여, 건조(MgSO4)하고, 여과하고, 진공농축시킨다. 생성된 고체를 섬광 크로마토그라피(에틸 아세테이트)시켜 정제하면 약 76%수율의 고체 슬폭시드 중간체가 수득된다.
그 다음 N-t-부틸-5-메톡시-2-s-메틸슬폭시드 벤즈아미드를 실시예 2에서 주어진 절차를 사용하여 트리클로로포르메이트로 처리하면 융점이 92-94°인 5-메톡시-N-t-부틸-1,2-벤즈이소티아졸-3(2H)-온이 수득된다.
방법 2의 일반 합성법에 의해 제조될 수 있는 다른 치환된 벤즈이소티아졸론을 표 2에 표시한다.
[표 2]
치환된 벤즈 이소티아졸론

B. 일반식(IV)의 화합물

[실시예 19]
3-클로로-1,2-벤즈이소티아졸
1,2-벤즈이소티아졸-3(2H)-온(실시예 1, 818g, 5.414몰)과 POCl3의 혼합물을 약 2시간에 걸쳐 120°로 가열한다. 약 70°에서 HCl발생이 시작된다. 다시 1.5시간 동안 120°로 가열을 계속한다. 뜨거운 용액을 8ℓ의 H2O중에 25°에서 붓는다. 온도가 50°를 초과하지 않도록한다. 30분후, 혼합물을 25°로 냉각(얼음 첨가)하고 염화메틸렌(4ℓ)으로 추출한다. 진한 오일을 진공중에서 염화메틸렌의 증발에 의해서 수득한다. 이 오일을 스켈리비(3×1ℓ와 2×500ml)로 추출한다. 진한 추출액을 여과하기 전에 다르코 G-60(30g)과 셀라이트 A-545(20g)으로 처리한다. 여액을 진공 증발시키면 쉽게 결정화되는 743.9g(81%)의 황색오일이 수득된다. 감압하에 오일을 증류시키면, 쉽게 결정화되는 융점이 39 내지 41°이고 0.75토르에서 비점이 80 내지 85°인 707g(77%)의 무색 증류에 수득된다.
[실시예 20]
치환된 3-클로로-1,2-벤즈이소티아졸(일반적 방법)
벤즈이소티아졸론을 디클로로에탄(0.04몰/100ml)중에 용해시키고 PCl5를 가한다. TLC로 반응과정을 감지하면서 그 혼합물을 환류시킨다. 반응이 환류한지 1시간 후 완결된 것으로 판단되면, 추가로 0.1 내지 0.5몰 당량이 PCl5를 가하고 출발물질이 소모 될 때까지 환류를 계속한다. 반응 혼합물을 냉각시키고 용매와 과량의 시약을 감압하에 증류시켜 제거gks다. 잔사를 섬광 크로마토그라피(헥산/염화에틸렌)시키면 생성물이 고체로서 수득된다.
[실시예 21]
3-클로로-4-메톡시-1,2-벤즈이소티아졸
4-메톡시벤즈이소티아졸론(0.065몰)을 200ml의 디클로로에탄중에 용해시키고 PCl5(1.05몰 당량)을 순수하게 가한다. 그 반응액을 환류하면서 TLC로 탐지한다. 반응이 TCL에 의해 완료되었다고 판단되기 전에 추가로 0.25몰 당량과 2×0.10몰 당량의 PCl5를 1시간, 2-1/2시간 및 3시간 후에 가한다. 반응액을 냉각하고 용매와 과량의 시약을 실내 진공증류에 의해서 제거한다. 잔사를 섬광 크로마토그라피(30%CH2Cl2/70% 헥산)시키면 3-클로로-4-메톡시-1,2-벤즈이소티아졸이 60%의 수율로 수득된다.
실시예 20의 일반적인 방법을 통하여 제조할 수 있는 치환된 3-클로로-1,2-벤즈이소티아졸의 몇 가지 다른 예들이 표 3에 표시되어 있다.
[표 3]
치환된 3-클로로벤즈이소티아졸

C. 일반식(III)의 화합물

[실시예 31]
t-부틸옥시카보닐피페라진
벤질피페라진(0.15몰)과 분쇄된 K2CO3(0.62몰 당량을 1:1의 디옥산/몰 200ml에 혼합하여 0°로 냉각하면서 기계적으로 교반한다. 피로카보네이트(1.12몰 당량)을 가하고 반응물을 냉각 상태에서 1시간동안 교반한 다음 주위 온도에서 18시간동안 교반한다. 디옥산을 진공에서 제거하고 수성상을 추출(3×염화메틸)시키고, 건조(MgSO4), 여과 및 진공 농축하여 약 95%의 조생성물인 오일을 얻는다. 그 오일을 두 개의 동일 부분으로 나누고 각각 따뜻한 순수 에타놀(150ml)과 빙초산(2.1몰 당량) 중에 용해시키고 파르 수소화 반응병중에서 탄소 피복 팔라듐촉매 4.1g과 혼합한다. 그 혼합물을 이론량의 탄소 피복 팔라듐 촉매 4.1g과 혼합한다. 그 혼합물을 이론량의 수소가 소비해버릴때까지 약 55psi의 최초 H2압력에서 수소화 반응 시킨다. 혼합물을 셀라이에 통과시켜서 여과하고 충분한 량의 무수 에타놀로 세척한 다음 농축하여 84% 수율의 고체 생성물을 얻는다.
피페라진환이 적합하게 치환된 벤질피페라진을 사용하는 이 절차를 이용하면 R2가 수소가 아닌 목적하는 t-부틸옥시카보닐 피페라진이 생성된다.
D. 일반식(II)의 화합물

[실시예 32]
1-(1,2-벤즈이소티아졸-3-일) 피페라진
4ℓ의 흡입 플라스크를 무수 피페라진(1582g, 18.36몰)으로 충전시키고, 이어서 용융 3-클로로-1,2-벤즈이소티아졸(실시예 8, 622g, 3.672몰)로 충전시킨다. 철선이 달린 고무마개로 플라스크를 막고 짧은 길이의 압력관을 옆관에 철선으로 고정한다. 플라스크를 비우고(실내진공) 옆쪽의 압력관을 꺽쇠로 죄어 닫는다. 그후, 용융이 진행될 때 그 장치를 가끔 휘저으면서 125°로 오븐으로 가열시킨다. 이 온도로 24시간 가열한 후 오렌지색 용융물을 부서진 얼음과 물 4.8ℓ에 넣고 식힌다. 일당량의 50% NaOH(293g, 3.672몰)을 일부씩 가한다. 혼합물을 염화메틸렌으로 추출하고 이 추출물을 물로 세척한다. 진공 농축하여 734g의 조생성물을 얻고 이를 1800ml의 끓는 에틸 아세테이트로 재결정시키면 융점이 88 내지 90°인 548g(68%)이 수득된다.
[실시예 33]
치환된 3-(1-피페라지닐)-1,2-벤즈이소티아졸
이 합성은 t-BOC 보호그룹을 후속 제거하여 3-(t-부틸옥시카보닐피페라진)-벤즈이소티아졸 중간체를 거쳐 진행한다.
t-부틸옥시카보닐피페라진(실시예 15, 2.5몰 당량)을 무수 테트라하이드로푸란(0.01-.02몰/90ml)중에 용해시키고, 교반하면서 질소하에 -78°로 냉각시킨다. n-부틸리튬(2.5몰 당량)을 -70°미만의 온도를 유지하는 속도로 가한다. 3-클로로-1,2-벤즈이소티아졸의 테트라하이드로푸란(1몰 당량/60ml) 용액을 -70°미만의 온도를 유지하도록 적가한다. 첨가가 완료될 때, 용액을 0°로 가온시킨후 15%의 수성 NH4Cl(2.75몰 당량)으로 침지시킨다. 진공 농축한 다음 잔사를 염화메틸렌으로 추출하고 그추출액을 0.5N HCl로 세턱하고, 건조시키고(MgSO4), 여과 및 진공 농축하여 반고테를 얻으며, 이를 섬광 크로마토그라피(헥산/에틸 아세테이트)한다. 이들 3-(t-BOC 피페라진)벤즈이소티아졸 중간체는 일반적으로 73 내지 90% 수율로 수득된다. t-BOC 보호그룹을 제거하기 위하여, 적절한 t-BOC 피페라지닐 벤즈이소티아졸을 최소량의 따뜻한 무수에타놀중에 용해시키고 5N 에타놀성 HCl(5몰 당량)으로 산성화시킨 다음, 1/2시간동안 90°로 가열한다. 냉각시켜 용매를 진공에서 제거하고 조생성물을 무수에타놀로 재결정시키면 40-60% 범위의 수율로 수득된다.
[실시예 34]
4-메톡시-1,2-벤즈이소티아졸-3-일 피페라진 염산염
t-부틸옥시카보닐 피페라진(0.04몰)을 150ml의 무수 테트라하이드로푸란중에 용해시키고 질소하에 -78°로 냉각한다. 온도를 -65°미만으로 유지하는 속도로 n-BuLi(0.04몰)을 가한다. 10분후, 4-메톡시-3-클로로-1,2-벤즈이소티아졸(0.016몰)의 60ml 테트라하이드로푸란 용액을 -70°미만으로 유지하면서 적가한다. 그 반응은 TLC로 탐지하면 즉시 완결된 것으로 판단된다. 얼음욕을 제거하고 15% 수성 NH4Cl(0.044몰)을 가한다. 이 혼합물을 진공 농축하고 잔사를 CH2Cl2중에 용해시키고 1×H2O와 1×냉 0.5N HCl로 세척한다. 염화메틸렌을 MgSO4로 건조하고 여과 및 진공농축하여 조생성물을 얻고 이를 섬광 크로마토그라피(3:1 헥산/EtOAC)하면 목적 물질이 73%의 수율로 생성된다.
부틸옥시카보닐피페라진 벤즈이소티아졸(0.012몰)을 따뜻한 무수 에타놀중에 용해시키고 에타놀성 HCl(5몰 당량)로 산성화 시킨다. 용액을 30분간 90°에서 교반한다. 혼합물을 냉각시키고 용매를 진공하에 제거한다. 조고체를 무수 에타놀로 재결정화 시키면 4-메톡시-1,2-벤즈이소티아졸-3-일-피레라진 염산염이 62%의 수율로 수득된다.
실시예 33의 절차를 사용하여 얻을 수 있는 추가 생성물의 예는 표 4에 나타나 있다.
[표 4]
치환된 일반식(I)의 3-(1-피페라지닐)-벤즈이소티아졸(HCl 염으로서)

생성물의 합성
[실시예 42]
3-(4-에틸-1-피페라지닐)-1,2-벤즈이소티아졸
3-(1-피페라지닐)-1,2-벤즈이소티아졸(5.0g, 0.023몰)의 25ml 아세토니트릴 용액에 N,N-디이소프로필에틸아민(3.2g, 0.025)과 브로모에탄을(2.6g, 0.024몰) 20°에서 가한다. 이 혼합물을 약 3시간동안 환류한 다음 진공하에 농축시키고; 5%의 수성 K2CO3중에 용해시키고, 5% 메타놀/염화메틸렌으로 섬광 크로마토그라피한다. 유분을 진공 농축시키면 오일상의 생성물 4.6g(0.02몰, 81%)이 생성된다. 에타놀 용액을 에타놀성 HCl로 처리하여 오일상 생성물을 고체 염산염으로 전화시킨다. 에타놀로 재결정화 시키면 융점이 230 내지 232°인 백색 고체가 수득된다.
원소분석 : C13H17N3S.HCl.H2O
계산치 : C ; 51.90, H : 6.37, N : 13.97
실측치 : C ; 51.65, H ; 6.60, N ; 14.15
실시예 22의 절차 또는 이의 적절한 변형법을 사용하면 일반식(I)의 각각의 화합물을 쉽게 제조할 수 있다. 이들 화합물에 대한 추가의 예는 표 5에 나타나 있다.
[표 5]
일반식(I)의 화합물

Claims (29)
- (1) 일반식(V)의 1,2-벤즈이소티아졸-3(2H)-온율 POCl3또는 PCl로 처리하여 일반식(IV)의 3-클로로-1,2-벤즈이소티아졸 화합물을 생성시키고; (2) 상기 일반식(IV)의 화합물을 일반식(III)의 적절한 중간체 화합물과 반응시켜 일반식(II)의 3-(1-피페라지닐)-1,2-벤즈이소티아졸 중간체 화합물을 생성시키고; (3) 상기 일반식(II)의 화합물을 일반식(I)의 화합물로 전화시킴을 특징으로 하여, 일반식(I)의 화합물 또는 이의 약제학적으로 허용되는 산부가염을 제조하는 방법 :




 상기 식에서, R1은 수소, 저급(C1-6)알킬, 직쇄 또는 측쇄의 저급알케닐, 저급사이클로알킬, 저급사이클로알킬-저급알킬렌, 페닐-저급알킬렌, 페녹시-저급알킬렌, 페닐-저급사이클로알킬, 또는 벤조디옥산-2-일-저급알킬렌이며; 단 R2, R3및 R4가 모두 수소일때 R1은 수소 또는 메틸이 아니어야 하고; R2는 저급알킬 또는 수소이며; R3및 R4는 수소, 저급알킬, 저급아실옥시, 저급알콕시, 저급알킬티도, 할로겐, 하이드록실, 또는 트리플루오로메틸중에서 독립적으로 선택되고; A는 수소 또는 t-부틸이며; B는 수소 또는 t-부틸옥시카보닐잔기이다.
상기 식에서, R1은 수소, 저급(C1-6)알킬, 직쇄 또는 측쇄의 저급알케닐, 저급사이클로알킬, 저급사이클로알킬-저급알킬렌, 페닐-저급알킬렌, 페녹시-저급알킬렌, 페닐-저급사이클로알킬, 또는 벤조디옥산-2-일-저급알킬렌이며; 단 R2, R3및 R4가 모두 수소일때 R1은 수소 또는 메틸이 아니어야 하고; R2는 저급알킬 또는 수소이며; R3및 R4는 수소, 저급알킬, 저급아실옥시, 저급알콕시, 저급알킬티도, 할로겐, 하이드록실, 또는 트리플루오로메틸중에서 독립적으로 선택되고; A는 수소 또는 t-부틸이며; B는 수소 또는 t-부틸옥시카보닐잔기이다. - 제1항에 있어서, 일반식(II)의 화합물을 R1X 여기에서 R1은 제1항에서 정의한 바와 같고, X는 전형적인 이탈그룹이다 를 사용하여 알킬화 반응시킴으로서 단계 3의 전화반응을 수행하는 방법.
- 제1항에 있어서, 일반식(II)의 화합물을 일반식(XX)의 카보닐 화합물을 사용하여 환원적 아민화 반응시킴으로써 단계 3의 전화반응을 수행하는 방법:
 상기 식에서, R4는 수소, 알킬, 아르알킬 또는 아릴옥시알킬이고, R6는 알킬, 아르알킬 또는 아릴옥시알킬이며, R5-CH-R6는 R1이다.
상기 식에서, R4는 수소, 알킬, 아르알킬 또는 아릴옥시알킬이고, R6는 알킬, 아르알킬 또는 아릴옥시알킬이며, R5-CH-R6는 R1이다. - 제2항에 있어서, 다음 (a) 내지 (s)의 화합물중에서 선택된 하나의 화합물을 제조하는 방법 : (a) 3-(4-메틸-1-피페라지닐)-1,2-벤즈이소티아졸, (b) 3-(4-프로필-1-피페라지닐)-1, 2-벤즈이소티아졸, (c) 3-(4-부틸-1-피페라지닐)-1,2-벤즈이소티아졸, (d) 3-(4-사이클로펜틸-3-메틸-1-피페라지닐)-1,2-벤즈이소티아졸, (e) 3-(4-펜틸-1-피페라지닐)-1, 2-벤즈이소티아졸, (f) 3-[4-(1-메틸에틸)-1-피페라지닐]-1,2-벤즈이소티아졸, (g) 3-[4-(3-메틸부틸)-1-피페라지닐]-1,2-벤즈이소티아졸, (h) 3-[4-(2-프로페닐)-1-피페라지닐]-1,2-벤즈이소티아졸, (i) 3-[4-(사이클로프로필메틸)-[1-피페라지닐]-1, 2-벤즈이소티아졸, (i) 3-(4-사이클로펜틸-1-피페라니질)-1, 2-벤즈이소티아졸, (k) 3-[4-(페닐메틸)-1-피페라지닐]-1, 2-벤즈이소티아졸, (l) 3-[4-(2-페닐에틸)-1-피페라지닐]-1, 2-벤즈이소티아졸, (m) 3-[4-(2-페녹시에틸)-1-피페라지닐]-1, 2-벤즈이소티아졸, (n) 3-[4-(4-페녹시부틸)-1-피페라니질]-1, 2-벤즈이소티아졸, (o) 3-{4-[(1, 4-벤조디옥산-2-일)메틸]-1-피페라지닐}-1, 2-벤즈이소티아졸, (p) 3-(4-사이클로헥실-1-피페라지닐)-1, 2-벤즈이소티아졸, (q) 3-[4-(2-인다닐)-1-피페라지닐]-1, 2-벤즈이소티아졸, (r) 3-[4-(1, 2, 3, 4-테트라하이드로-2-나프틸)-1-피페라지닐]-1, 2-벤즈이소티아졸, 및 (s) 3-[4-(2-메틸사이클로헥실)-1-피페라지닐]-1, 2-벤즈이소티아졸.
- 제1항에 있어서, 기의 정의중 단서조항이 R2및 R3가 수소이고 R4가 수소 또는 할로겐일 때 R1이 수소가 아닌 경우인 방법.
- 제1항에 있어서, R1이 4개이상의 탄소원자를 함유하는 방법.
- 제1항에 있어서, R1의 저급알킬, 저급알케닐, 저급사이클로알킬 및 저급알킬렌그룹이 4 내지 6개의 탄소원자를 함유하는 방법.
- 제1항에 있어서, R1이 사이클로알킬인 방법.
- 제1항에 있어서, R1이 (C3-6)사이클로알킬인 방법.
- 제1항에 있어서, 3-(4-에틸-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-(4-메틸-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-(4-프로필-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-(4-부틸-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-(4-사이클로펜틸-3-메틸-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-(4-펜틸-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(1-메틸에틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(3-메틸부틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(2-프로페닐)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(사이클로프로필메틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-(4-사이클로펜틸-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(페닐메틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(2-페닐에틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(2-페녹시에틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(4-페녹시부틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-{4-[1,4-벤조디옥산-2-일)메틸-1-피페라지닐}-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-(4-사이클로헥실-1-피페라지닐)-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(2-인다닐)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(1,2,3,4-테트라하이드로-2-나프틸)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
- 제1항에 있어서, 3-[4-(2-메틸사이클로헥실)-1-피페라지닐]-1,2-벤즈이소티아졸을 제조하는 방법.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US06/643,614 US4590196A (en) | 1984-08-23 | 1984-08-23 | Analgesic 1,2-benzisothiazol-3-ylpiperazine derivatives |
| US643,614 | 1984-08-23 | ||
| US643.614 | 1984-08-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR860001807A KR860001807A (ko) | 1986-03-22 |
| KR920008820B1 true KR920008820B1 (ko) | 1992-10-09 |
Family
ID=24581577
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1019850006060A KR920008820B1 (ko) | 1984-08-23 | 1985-08-22 | 진통제 1,2-벤즈이소티아졸-3-일 피페라진 유도체의 제조방법 |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US4590196A (ko) |
| JP (1) | JPS61112063A (ko) |
| KR (1) | KR920008820B1 (ko) |
| AT (1) | AT396685B (ko) |
| AU (1) | AU586182B2 (ko) |
| BE (1) | BE903106A (ko) |
| CA (1) | CA1249587A (ko) |
| CH (1) | CH667268A5 (ko) |
| DE (1) | DE3530089A1 (ko) |
| DK (1) | DK381485A (ko) |
| ES (1) | ES8704485A1 (ko) |
| FI (1) | FI82039C (ko) |
| FR (1) | FR2569404B1 (ko) |
| GB (1) | GB2163432B (ko) |
| GR (1) | GR852017B (ko) |
| IE (1) | IE58608B1 (ko) |
| IT (1) | IT1187730B (ko) |
| LU (1) | LU86051A1 (ko) |
| NL (1) | NL8502310A (ko) |
| PT (1) | PT81007B (ko) |
| SE (1) | SE464304B (ko) |
| ZA (1) | ZA856387B (ko) |
Families Citing this family (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0625181B2 (ja) * | 1985-03-27 | 1994-04-06 | 住友製薬株式会社 | 新規なイミド誘導体 |
| US4999356A (en) * | 1987-08-07 | 1991-03-12 | Hoechst-Roussel Pharmaceuticals, Inc. | 1-phenyl-3-(1-piperazinyl)-1H-indazoles |
| US4880930A (en) * | 1987-11-30 | 1989-11-14 | New James S | Psychotropic acyclic amide derivatives |
| NZ230045A (en) | 1988-08-05 | 1990-11-27 | Janssen Pharmaceutica Nv | 3-piperazinylbenzazole derivatives and pharmaceutical compositions |
| US4957916A (en) * | 1988-08-05 | 1990-09-18 | Janssen Pharmaceutica N.V. | Antipsychotic 3-piperazinylbenzazole derivatives |
| US5015740A (en) * | 1988-08-05 | 1991-05-14 | Janssen Pharmaceutica N.V. | Antipsychotic 3-piperazinylbenzazole derivatives |
| HUT61760A (en) * | 1988-09-16 | 1993-03-01 | Pfizer | Process for producing bridged, bicyclic imides comprising 4-/4-(3-benzisothiazolyl)-1-piperazinyl/-butyl group and pharmaceutical compositions comprising such compounds |
| US5070090A (en) * | 1989-05-15 | 1991-12-03 | Janssen Pharmaceutica N.V. | Antipicorpaviral herterocyclic-substituted morpholinyl alkylphenol ethers |
| GB8911158D0 (en) * | 1989-05-16 | 1989-07-05 | Janssen Pharmaceutica Nv | Antiviral pyridazinamines |
| NZ233525A (en) * | 1989-05-15 | 1991-09-25 | Janssen Pharmaceutica Nv | Piperidinyl, pyrrolidinyl and piperazinyl alkylphenol ethers and pharmaceutical compositions |
| EP0402644B1 (en) * | 1989-05-19 | 1995-08-16 | Hoechst-Roussel Pharmaceuticals Incorporated | N-(aryloxyalkyl)heteroarylpiperidines and -heteroarylpiperazines,a process for their preparation and their use as medicaments |
| US5561128A (en) * | 1989-05-19 | 1996-10-01 | Hoechst-Roussel Pharmaceuticals, Inc. | N-[(4-(heteroaryl)-1-piperidinyl)alkyl]-10,11-dihydro-5H-dibenz[B,F]azepines and related compounds and their therapeutic utility |
| US5364866A (en) * | 1989-05-19 | 1994-11-15 | Hoechst-Roussel Pharmaceuticals, Inc. | Heteroarylpiperidines, pyrrolidines and piperazines and their use as antipsychotics and analetics |
| US5776963A (en) * | 1989-05-19 | 1998-07-07 | Hoechst Marion Roussel, Inc. | 3-(heteroaryl)-1- (2,3-dihydro-1h-isoindol-2-yl)alkyl!pyrrolidines and 3-(heteroaryl)-1- (2,3-dihydro-1h-indol-1-yl)alkyl!pyrrolidines and related compounds and their therapeutic untility |
| US4956368A (en) * | 1989-07-24 | 1990-09-11 | Bristol-Myers Company | Metabolites and prodrug formulations of 8-[4-[4-(1,2-benzisothiazol-3-yl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dione |
| US4954503A (en) * | 1989-09-11 | 1990-09-04 | Hoechst-Roussel Pharmaceuticals, Inc. | 3-(1-substituted-4-piperazinyl)-1H-indazoles |
| FR2671350A1 (fr) * | 1991-01-08 | 1992-07-10 | Adir | Nouveaux derives de benzisoxazole et de benzisothiazole, leur procede de preparation, et les compositions pharmaceutiques les renfermant. |
| JP2800953B2 (ja) * | 1990-07-06 | 1998-09-21 | 住友製薬株式会社 | 新規なイミド誘導体 |
| US5328920A (en) * | 1991-04-17 | 1994-07-12 | Hoechst-Roussel Pharmaceuticals Incorporated | Substituted (pyridinylamino)-indoles |
| DK0669920T3 (da) * | 1992-11-23 | 2003-05-26 | Aventis Pharma Inc | Substituerede 3-(aminoalkylamino)-1,2-benzisoxazoler og beslægtede forbindelser |
| ATE210663T1 (de) * | 1994-06-20 | 2001-12-15 | Takeda Chemical Industries Ltd | Kondensierte imidazolderivate, ihre herstellung und verwendung |
| JPH08157460A (ja) * | 1994-12-08 | 1996-06-18 | Sumitomo Seika Chem Co Ltd | イソチアゾール誘導体の製造方法 |
| JP3701044B2 (ja) * | 1995-04-24 | 2005-09-28 | 住友精化株式会社 | シアノベンゼンスルフェニルハライドおよびそれを用いる3−置換ベンゾイソチアゾールの製造方法 |
| NZ307958A (en) * | 1995-06-06 | 1999-02-25 | Hoechst Marion Roussel Inc | Benzisoxazole and indazole derivatives as long acting antipsychotic agents and particularly in the treatment of schizophrenia |
| AU698443B2 (en) * | 1995-11-07 | 1998-10-29 | Pfizer Inc. | Processes and intermediates for preparing 3-(1-piperazinyl)-1, 2-benzisothiazole |
| JP3996228B2 (ja) * | 1996-10-11 | 2007-10-24 | 住友精化株式会社 | 3−ピペラジニルベンズイソチアゾール類の製造法 |
| DE19651038A1 (de) * | 1996-12-09 | 1998-06-10 | Bayer Ag | Verfahren zur Herstellung von 3-Chlorbenzisothiazolen |
| JP2002541098A (ja) * | 1999-04-06 | 2002-12-03 | セプラコア インコーポレーテッド | ジプラシドン代謝産物を用いる神経弛緩薬性障害および関連障害を治療するための方法および組成物 |
| US7030142B1 (en) | 1999-04-06 | 2006-04-18 | Sepracor Inc. | Methods for the treatment of neuroleptic and related disorders using ziprasidone metabolites |
| US20050049295A1 (en) * | 2003-06-12 | 2005-03-03 | Dr. Reddy's Laboratories Limited | Process for the preparation of 5-(2-(4-(1,2-benzisothiazol-3-yl)-1piperazinyl) ethyl)-6-chloro-1, 3-dihydro-2h-indol-2-one hydrochloride (ziprasidone hydrochloride) and its intermediate |
| CA2598043A1 (en) * | 2005-02-25 | 2006-08-31 | Rigel Pharmaceuticals, Inc. | Benzisothiazoles useful for treating or preventing hcv infection |
| CN100387583C (zh) * | 2006-05-19 | 2008-05-14 | 浙江工业大学 | 一种3-氯-1,2-苯并异噻唑类化合物的合成方法 |
| CN102040563B (zh) * | 2009-10-16 | 2014-04-16 | 上海开拓者化学研究管理有限公司 | 一种3-芳基苯并[d]异噻唑的制备方法 |
| CN102040564B (zh) * | 2009-10-16 | 2012-11-21 | 上海开拓者化学研究管理有限公司 | 一种3-胺基苯并[d]异噻唑的制备方法 |
| US8664404B2 (en) | 2010-04-02 | 2014-03-04 | Sumitomo Seika Chemicals Co., Ltd. | Method for producing 3-halo-1,2-benzisothiazoles |
| WO2013014665A1 (en) | 2011-07-28 | 2013-01-31 | Mapi Pharma Ltd. | Intermediate compounds and process for the preparation of lurasidone and salts thereof |
| CN103204823B (zh) * | 2013-03-18 | 2015-07-15 | 寿光新泰精细化工有限公司 | 一种1,2-苯并异噻唑-3-酮的提纯方法 |
| CN105037293A (zh) * | 2015-06-04 | 2015-11-11 | 大丰跃龙化学有限公司 | 一种bit生产中固废再利用工艺 |
| AR104882A1 (es) * | 2015-06-05 | 2017-08-23 | Orion Corp | DERIVADOS DE 2-(1-HETEROARILPIPERAZIN-4-IL)METIL-1,4-BENZODIOXANO COMO ANTAGONISTAS DE a2C |
| CN106543107B (zh) * | 2016-11-04 | 2019-02-01 | 山东铂源药业有限公司 | 一种1-boc-哌嗪的合成方法 |
| CN108794424A (zh) * | 2018-07-16 | 2018-11-13 | 连云港市三联化工有限公司 | 一种bit的溶剂精制方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1670907B2 (de) * | 1967-08-16 | 1976-07-22 | Bayer Ag, 5090 Leverkusen | N-disubstituierte 3-amino-1,2benzisothiazole und verfahren in ihrer herstellung |
| US4112105A (en) * | 1977-05-04 | 1978-09-05 | Sterling Drug Inc. | Anti-inflammatory 3-(substituted-amino)-2,1-benzisothiazoles |
| US4104388A (en) * | 1977-05-23 | 1978-08-01 | E. R. Squibb & Sons, Inc. | 3-Substituted benzisothiazole, 1,1-dioxides |
| US4396770A (en) * | 1982-04-09 | 1983-08-02 | Hoechst-Roussel Pharmaceuticals Inc. | 1-[3-(6-Fluoro-1,2-benzisoxazol-3-yl)propyl]-4-hydroxy-4-phenylpiperidines |
| JPS5815965A (ja) * | 1981-07-23 | 1983-01-29 | Nippon Tokushu Noyaku Seizo Kk | ベンゾイソチアゾ−ル誘導体並びにその塩酸塩、その製法、及び農業用殺菌剤 |
| US4355037A (en) * | 1981-11-12 | 1982-10-19 | Hoechst-Roussel Pharmaceuticals | 3-(4-Piperidyl)-1,2-benzisoxales |
| US4411901A (en) * | 1981-12-23 | 1983-10-25 | Mead Johnson & Company | Benzisothiazole and benzisoxazole piperazine derivatives |
| US4452799A (en) * | 1981-12-23 | 1984-06-05 | Mead Johnson & Company | Benzisothiazole and benzisoxazole piperazine derivatives |
| US4524206A (en) * | 1983-09-12 | 1985-06-18 | Mead Johnson & Company | 1-Heteroaryl-4-(2,5-pyrrolidinedion-1-yl)alkyl)piperazine derivatives |
-
1984
- 1984-08-23 US US06/643,614 patent/US4590196A/en not_active Expired - Fee Related
-
1985
- 1985-08-16 CA CA000488887A patent/CA1249587A/en not_active Expired
- 1985-08-20 GR GR852017A patent/GR852017B/el unknown
- 1985-08-20 FI FI853193A patent/FI82039C/fi not_active IP Right Cessation
- 1985-08-22 BE BE0/215496A patent/BE903106A/fr not_active IP Right Cessation
- 1985-08-22 PT PT81007A patent/PT81007B/pt not_active IP Right Cessation
- 1985-08-22 SE SE8503922A patent/SE464304B/sv not_active IP Right Cessation
- 1985-08-22 ES ES546347A patent/ES8704485A1/es not_active Expired
- 1985-08-22 IT IT21968/85A patent/IT1187730B/it active
- 1985-08-22 JP JP60185015A patent/JPS61112063A/ja active Pending
- 1985-08-22 NL NL8502310A patent/NL8502310A/nl not_active Application Discontinuation
- 1985-08-22 DE DE19853530089 patent/DE3530089A1/de not_active Ceased
- 1985-08-22 ZA ZA856387A patent/ZA856387B/xx unknown
- 1985-08-22 AU AU46533/85A patent/AU586182B2/en not_active Ceased
- 1985-08-22 GB GB08521050A patent/GB2163432B/en not_active Expired
- 1985-08-22 LU LU86051A patent/LU86051A1/fr unknown
- 1985-08-22 IE IE206785A patent/IE58608B1/en not_active IP Right Cessation
- 1985-08-22 CH CH3606/85A patent/CH667268A5/de not_active IP Right Cessation
- 1985-08-22 DK DK381485A patent/DK381485A/da not_active Application Discontinuation
- 1985-08-22 KR KR1019850006060A patent/KR920008820B1/ko not_active IP Right Cessation
- 1985-08-22 FR FR8512635A patent/FR2569404B1/fr not_active Expired
- 1985-08-23 AT AT0246985A patent/AT396685B/de not_active IP Right Cessation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR920008820B1 (ko) | 진통제 1,2-벤즈이소티아졸-3-일 피페라진 유도체의 제조방법 | |
| CA1291135C (en) | Substituted benzamide derivatives, processes for the preparation thereof, and pharmaceutical compositions containing the same | |
| CS202515B2 (en) | Method of producing novel 4-hydroxy-2h-1,2-benzothiazine-3-carboxamide-1,1-dioxides | |
| JPH0237353B2 (ko) | ||
| US2852510A (en) | Heterocyclic compounds and process for producing same | |
| GB2147297A (en) | Antidepressant 1,2,4-triazolone compounds | |
| JPH0641095A (ja) | 置換トリアゾロンおよびトリアゾールジオンの抗うつ性3−ハロフェニルピペラジニル−プロピル誘導体 | |
| US5026717A (en) | 2-imino-6-polyfluoroalkoxybenzothiazole derivatives, and pharmaceutical compositions containing them | |
| PL157118B1 (pl) | Sposób wytwarzania zwiazków arylopiperazynylo- alkilenofenylo-p-heterocyklicznychU praw niony z patentu:Pierw szenstw o:PFIZER Inc., Nowy Jork, US17.02.1987,US.PCT/US87/00340 PL PL PL PL | |
| US4104387A (en) | 3-(Arylcycloiminoalkyl)benzisothiazole 1,1-dioxides | |
| JP2574348B2 (ja) | 新規縮合ジアゼピノン、その製造法及びそれを含有する医薬組成物 | |
| AU630085B2 (en) | 4,5,6,7-tetrahydro isothiazolo (4,5-c) pyridine derivatives | |
| HU214337B (hu) | Eljárás 1,3,4-tiadiazin-2-on-származékok és az azokat tartalmazó gyógyszer készítmények előállítására | |
| US3225037A (en) | 10-[(amino- and acylamino-1-piperidyl) lower-alkyl]-phenothiazines | |
| US3946010A (en) | 3-Phenyl-2,5-dihydro-as-triazin-6 (1H)-ones | |
| EP0364091B1 (en) | Antipsychotic 4-(4-(3-benzisothiazolyl)-1-piperazinyl)butyl bridged bicyclic imides | |
| CA1185970A (en) | Benzo-fused heterocyclic anti-ulcer agents | |
| US5424312A (en) | Aminoalkyl-substituted 2-amino-1,3,4-thiadiazoles, the preparation and use thereof | |
| US3868367A (en) | 4-Hydroxy-3-(5-isoxazolylcarbamoyl)-2H-1,2-benzothiazine 1,1-dioxides | |
| US3983106A (en) | 5-Heterocyclicalkyl-2-aryl-3-halo 1,5-benzothiazepin-4(5H)-ones | |
| HUT51609A (en) | Process for producing benzofurane derivatives and pharmaceutical preparations containing such compounds | |
| JPS63275572A (ja) | 1,5−ベンゾチアゼピン誘導体 | |
| US3419554A (en) | N-[(1-substituted piperidyl)alkylpiperidino-alkyl] derivatives of n-containing heterocyclic compounds | |
| JPH072757A (ja) | 鎮痙剤としての置換n−アミノアルキルメタンスルファニリド | |
| US3700663A (en) | Dibenzothiazepine ethers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| G160 | Decision to publish patent application | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| LAPS | Lapse due to unpaid annual fee |