JP7202285B2 - 活性炭の製造方法 - Google Patents
活性炭の製造方法 Download PDFInfo
- Publication number
- JP7202285B2 JP7202285B2 JP2019510164A JP2019510164A JP7202285B2 JP 7202285 B2 JP7202285 B2 JP 7202285B2 JP 2019510164 A JP2019510164 A JP 2019510164A JP 2019510164 A JP2019510164 A JP 2019510164A JP 7202285 B2 JP7202285 B2 JP 7202285B2
- Authority
- JP
- Japan
- Prior art keywords
- activated carbon
- pore volume
- less
- surface area
- specific surface
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/336—Preparation characterised by gaseous activating agents
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/306—Active carbon with molecular sieve properties
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/318—Preparation characterised by the starting materials
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/30—Active carbon
- C01B32/312—Preparation
- C01B32/318—Preparation characterised by the starting materials
- C01B32/33—Preparation characterised by the starting materials from distillation residues of coal or petroleum; from petroleum acid sludge
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/14—Pore volume
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/16—Pore diameter
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Carbon And Carbon Compounds (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
Description
さらに、水蒸気賦活においては、Mg、Mn、Fe、Y、Pt及びGdの少なくとも1種の金属成分を活性炭前駆体としてのピッチに含ませることで、得られる活性炭において30~45Åのメソ細孔モード直径を有するメソ細孔を開口する方法が知られている(特許文献2参照)。
本発明は、これらの知見に基づいて、さらに検討を重ねることにより完成された発明である。
項1.金属成分を含む活性炭前駆体を導入ガスとして炭酸ガスで賦活し、全細孔容積Aに対する直径1.0nm以下の細孔容積Bの比(細孔容積B/全細孔容積A)が0.5以上である活性炭を得る賦活工程を含み、前記金属成分を構成する金属元素が、第2族元素、第3族元素、第4族元素、第5族元素、第7族元素、及び希土類元素からなる群から選択される、活性炭の製造方法。
項2.前記金属元素が、Y、Mg、Mn、La、V、Zr、Ti及びCeからなる群から選択される、項1に記載の活性炭の製造方法。
項3.前記金属元素が、Y、Mg、Ce、Ti及びVからなる群から選択される、項1又は2に記載の活性炭の製造方法。
項4.金属成分を含む活性炭前駆体を導入ガスとして炭酸ガスで賦活し、全細孔容積Aに対する直径1.0nm以下の細孔容積Bの比(細孔容積B/全細孔容積A)が0.5以上である活性炭を得る賦活工程を含み、前記金属成分を構成する金属元素が、第6族元素及び第9族元素からなる群から選択される、活性炭の製造方法。
項5.前記金属元素が、Mo及びCoからなる群から選択される、項4に記載の活性炭の製造方法。
項6.前記活性炭の比表面積が600m2/g以上である、項1から5のいずれか1項に記載の活性炭の製造方法。
項7.前記賦活工程において、前記導入ガスの組成を変更しない、項1から6のいずれか1項に記載の活性炭の製造方法。
項8.前記導入ガスの流量が、前記活性炭前駆体1g当たり、0℃1気圧換算で1.5L/分以上である、項1から7のいずれか1項に記載の活性炭の製造方法。
項9.前記賦活工程における賦活温度が800~1000℃である、項1から8のいずれか1項に記載の活性炭の製造方法。
項10.前記活性炭前駆体中、前記金属成分の含有量が0.05~1.0質量%である、項1から9のいずれか1項に記載の活性炭の製造方法。
項11.前記活性炭前駆体が、不融化したピッチである、項1から10のいずれか1項に記載の活性炭の製造方法。
項12.前記活性炭において、全細孔容積Aに対する直径2.0nm以下の細孔容積Cの割合({細孔容積C/細孔容積A}×100)が85%以上である、項1から11のいずれか1項に記載の活性炭の製造方法。
項13.前記活性炭において、直径1.0nm以下の細孔容積Bが0.25cc/g以上である、項1から12のいずれか1項に記載の活性炭の製造方法。
[1-1.活性炭の表面構造]
以下において、細孔容積とは、QSDFT法(急冷固体密度汎関数法)によって算出される細孔容積をいう。QSDFT法とは、幾何学的・化学的に不規則なミクロポーラス・メソポーラスな炭素の細孔径解析を対象とした、約0.5nm~約40nmまでの細孔径分布の計算ができる解析手法である。QSDFT法では、細孔表面の粗さと不均一性による影響が明瞭に考慮されているため、細孔径分布解析の正確さが大幅に向上した手法である。本発明においては、Quantachrome社製「AUTOSORB-1-MP」を用いて窒素吸着等温線の測定、及びQSDFT法による細孔径分布解析をおこなう。77Kの温度において測定した窒素の脱着等温線に対し、Calculation modelとしてN2 at 77K on carbon[slit pore,QSDFT equilibrium model]を適用して細孔径分布を計算することで、特定の細孔径範囲の細孔容積を算出することができる。
また、全細孔容積A(cc/g)は、直径2nm以下のミクロ孔、好ましくは直径1nm以下の極小サイズのミクロ孔を良好に得る観点から、例えば1.50cc/g以下、好ましくは0.8cc/g以下であってよい。
また、比表面積の上限値は特に制限されないが、例えば、3000m2/g以下が挙げられ、2500m2/g以下が挙げられ、2000m2/g以下が挙げられる。
なお、本発明において比表面積とは、窒素を被吸着物質として用いたBET法(1点法)により測定される値である。
また、直径1.0nm以下の細孔容積Bの上限値は特に制限されないが、例えば0.60cc/g以下、好ましくは0.50cc/g以下が挙げられる。
また、直径1.5nm以下の細孔容積(cc/g)の上限値は特に制限されないが、例えば0.7cc/g以下が挙げられる。
また、直径2.0nm以下の細孔容積C(cc/g)の上限値は特に制限されないが、例えば0.8cc/g以下が挙げられる。
本発明の製造方法では後述のとおり金属成分を用いるため、得られる活性炭には当該金属成分が残存している。活性炭の総質量に対する、該活性炭に含有される金属成分の割合(金属元素換算)は、例えば、0.15~0.60質量%であってよく、好ましくは0.15~0.45質量%であってよく、より好ましくは0.20~0.40質量%であってよい。活性炭中の上記割合は、ICP発光分光分析装置(Varian社製型式715-ES)により測定される金属元素換算の割合である。
本発明の製造方法によって製造される活性炭の形態は特に限定されないが、例えば、粒状活性炭、粉末状活性炭、繊維状活性炭等が挙げられる。フィルター加工等して用いる場合の加工性、又は浄水器等で使用する場合吸着速度の観点から、繊維状である繊維状活性炭とすることがより好ましい。なお、本発明において、吸着速度は、例えばトリハロメタンの通水吸着試験等により評価することができる。繊維状活性炭の平均繊維径としては、好ましくは30μm以下、より好ましくは5~20μm程度が挙げられる。なお、本発明における平均繊維径は、画像処理繊維径測定装置(JIS K 1477に準拠)により測定した値である。また、粒状活性炭及び粉末状活性炭の粒径としては、レーザー回折/散乱式法で測定した積算体積百分率D50が0.01~5mmが挙げられる。
本発明の製造方法によって製造される活性炭は、気相中または液相中のいずれでも使用することができるが、特に、気相中のジクロロメタンを吸着させるために好適に用いられる。
平衡吸着量(質量%)=質量増加分/活性炭質量×100
本発明の製造方法において、活性炭の原料となる活性炭前駆体には、特定の金属成分が含まれている。
炭酸ガス賦活は、活性炭前駆体中の炭素と炭酸ガスとの反応(Cx+CO2→2CO+Cx-1)によって細孔が生成するものである。また、この炭素と炭酸ガスとの反応は、後述の金属成分による触媒的な作用により促進される。炭素と炭酸ガスとの反応及びその促進効果は、活性炭前駆体の原料種及び形態によらずに共通である。したがって、活性炭前駆体の原料種及び形態としては特に制限されるものではない。
金属成分は、炭酸ガス賦活における炭素と炭酸ガスとの反応を触媒的な作用により促進する。金属成分を構成する金属元素は、第2族元素、第3族元素、第4族元素、第5族元素、第6族元素、第7族元素及び第9族元素、並びに希土類元素からなる群から1種または複数種が選択される。
一方、炭酸ガス賦活促進効果を制御する観点からは、第2族元素、第3族元素、第4族元素、第5族元素、第7族元素、及び希土類元素のうち上述のMg、Y、Zr、V、Mn、La及びCe以外の元素を選択することもできる。
導入ガス(賦活炉に導入するガス)は、炭酸ガス(二酸化炭素)を用い、本願効果の損なわない範囲で窒素、一酸化炭素、希ガス等を含有させることもできる。
賦活工程における賦活炉内の雰囲気温度(賦活温度)は、例えば800~1000℃、好ましくは900~1000℃であってよい。
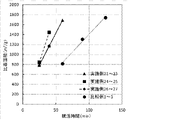
本発明の製造方法によれば、金属成分を含む活性炭前駆体を導入ガスとして炭酸ガスで賦活し、全細孔容積Aに対する直径1.0nm以下の細孔容積Bの比(細孔容積B/全細孔容積A)が0.5以上である活性炭を得る賦活工程を含み、前記金属成分を構成する金属元素が、第2族元素、第3族元素、第4族元素、第5族元素、第7族元素、及び希土類元素からなる群から選択されることから、直径1.0nm以下の極小サイズのミクロ孔の割合を高めた活性炭を効率良く製造する方法が提供される。本発明において、好ましい比表面積の発達速度としては、例えば、比表面積800m2/gに到達するまでの発達速度が25m2/g/min以上が好ましく、30m2/g/min以上がより好ましく、40m2/g/min以上が好ましく、50m2/g/min以上が特に好ましい。また、比表面積1100m2/gに到達するまでの発達速度が25m2/g/min以上が好ましく、30m2/g/min以上がより好ましく、40m2/g/min以上が好ましく、50m2/g/min以上が特に好ましい。
本発明の製造方法は、前述した賦活工程のほかに、他の工程を含むものであっても良い。他の工程としては、活性炭の製造方法で公知の工程が挙げられ、例えば、賦活工程の前に有機質材料を所定の形状に成形する成形工程(繊維状活性炭の場合は紡糸工程を含む。)や不融化工程を含むことが挙げられる。また、得られる活性炭が浄水器用途である場合は、賦活工程の後に、得られた活性炭の表面に付着している金属成分を洗浄する洗浄工程を含むことができる。
(1)不融化したピッチ繊維(活性炭前駆体)の金属含有量(質量%)
ピッチ繊維を灰化処理し、灰分を酸に溶解しICP発光分光分析装置(Varian社製型式715-ES)により測定される金属元素換算の割合を金属含有量とした。
導入ガスの組成は、JIS K 0301 5.1 オルザット式分析方法に従い測定した。
繊維状活性炭を酸に溶解しICP発光分光分析装置(Varian社製型式715-ES)により測定される金属元素換算の割合を金属含有量とした。
比表面積はBET法によって相対圧0.1の測定点から計算した。
細孔物性値は、Quantachrome社製「AUTOSORB-1-MP」を用いて77Kにおける窒素吸着等温線より測定した。全細孔容積及び下記各表に記載した各細孔径範囲における細孔容積は、測定した窒素脱着等温線に対し、Calculation modelとしてN2 at 77K on carbon[slit pore,QSDFT equilibrium model]を適用して細孔径分布を計算することで、解析した。具体的に、下記各表に記載した各細孔径における細孔容積は、窒素吸脱着等温線から得られる細孔径分布図の読み取り値である。より具体的に、細孔径1.0nm以下の細孔容積Bは、細孔径分布図の横軸Pore Widthが1.0nmにおけるCumulative Pore Volume(cc/g)の読み取り値である。同様にして、細孔径1.5nm以下の細孔容積、細孔径2.0nm以下の細孔(つまりミクロ細孔)容積Cを得た。
細孔径1.0nm以下の細孔容積比(B/A)は、細孔径1.0nm以下の細孔容積Bを、QSDFT解析により得られる全細孔容積Aで除することで計算した。ミクロ細孔容積率({C/A}×100)は、細孔径2.0nm以下の細孔容積Cを、QSDFT解析により与えられる全細孔容積Aで除し百分率で表した。メソ細孔容積率(%)は、100%からミクロ細孔容積率(%)を減ずることで計算した。
1.0nm以下の細孔発達速度は、1.0nm以下の細孔容積Bを賦活時間で除することで計算した。比表面積の発達速度は、BET比表面積を賦活時間で除することで計算した。
画像処理繊維径測定装置(JIS K 1477に準拠)により測定した。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対して金属成分としてトリスアセチルアセトナトイットリウム(CAS番号:15554-47-9)0.5質量部を添加し混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、イットリウムの含有量は0.10質量%であった。
賦活時間を44分とした以外は実施例1と同様にし、実施例2の活性炭を得た。得られた活性炭は、比表面積1237m2/g、全細孔容積Aは0.491cc/g、ミクロ細孔容積率({C/A}×100)は99%、細孔径1.0nm以下の細孔容積Bは0.383cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.779であった。
賦活時間を58分とした以外は実施例1と同様にし、実施例3の活性炭を得た。得られた活性炭は、比表面積1603m2/g、全細孔容積Aは0.654cc/g、ミクロ細孔容積率({C/A}×100)は97%、細孔径1.0nm以下の細孔容積Bは0.434cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.663であった。
金属成分(トリスアセチルアセトナトイットリウム)の添加量を1.0質量部(活性炭前駆体中の金属含有量は0.16質量%)とし、賦活時間を25分とした以外は実施例1と同様にし、実施例4の活性炭を得た。得られた活性炭は、比表面積917m2/g、全細孔容積Aは0.381cc/g、ミクロ細孔容積率({C/A}×100)は95%、細孔径1.0nm以下の細孔容積Bは0.278cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.730であった。
賦活時間を32分とした以外は実施例4と同様にし、実施例5の活性炭を得た。得られた活性炭は、比表面積1168m2/g、全細孔容積Aは0.502cc/g、ミクロ細孔容積率({C/A}×100)は92%、細孔径1.0nm以下の細孔容積Bは0.302cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.602であった。
賦活時間を40分とした以外は実施例4と同様にし、実施例6の活性炭を得た。得られた活性炭は、比表面積1338m2/g、全細孔容積Aは0.592cc/g、ミクロ細孔容積率({C/A}×100)は90%、細孔径1.0nm以下の細孔容積Bは0.352cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.595であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対してアセチルアセトンマグネシウム(II)(CAS番号:14024-56-7)2.3質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、マグネシウムの含有量は0.18質量%であった。
賦活時間を40分とした以外は実施例7と同様にし、実施例8の活性炭を得た。得られた活性炭は、比表面積1461m2/g、全細孔容積Aは0.635cc/g、ミクロ細孔容積率({C/A}×100)は87%、細孔径1.0nm以下の細孔容積Bは0.417cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.656であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対して安息香酸マンガン(CAS番号:636-13-5)1.7質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、マンガンの含有量は0.20質量%であった。
賦活時間を40分とした以外は実施例9と同様にし、実施例10の活性炭を得た。得られた活性炭は、比表面積1214m2/g、全細孔容積Aは0.484cc/g、ミクロ細孔容積率({C/A}×100)は98%、細孔径1.0nm以下の細孔容積Bは0.348cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.720であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対してアセチルアセトナトランタン(CAS番号:64424-12-0)1.3質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、ランタンの含有量は0.21質量%であった。
賦活時間を25分とした以外は実施例11と同様にし、実施例12の活性炭を得た。得られた活性炭は、比表面積758m2/g、全細孔容積Aは0.304cc/g、ミクロ細孔容積率({C/A}×100)は97%、細孔径1.0nm以下の細孔容積Bは0.256cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.841であった。
賦活時間を30分とした以外は実施例11と同様にし、実施例13の活性炭を得た。得られた活性炭は、比表面積916m2/g、全細孔容積Aは0.368cc/g、ミクロ細孔容積率({C/A}×100)は96%、細孔径1.0nm以下の細孔容積Bは0.283cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.770であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対してビス(2,4-ペンタンジオナト)バナジウム(IV)オキシド(CAS番号:3153-26-2)1.3質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、バナジウムの含有量は0.18質量%であった。
賦活時間を25分とした以外は実施例14と同様にし、実施例15の活性炭を得た。得られた活性炭は、比表面積1426m2/g、全細孔容積Aは0.569cc/g、ミクロ細孔容積率({C/A}×100)は97%、細孔径1.0nm以下の細孔容積Bは0.437cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.767であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対してアセチルアセトナトジルコニウム(CAS番号:17501-44-9)0.8質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、ジルコニウムの含有量は0.19質量%であった。
賦活時間を40分とした以外は実施例16と同様にし、実施例17の活性炭を得た。得られた活性炭は、比表面積1052m2/g、全細孔容積Aは0.445cc/g、ミクロ細孔容積率({C/A}×100)は91%、細孔径1.0nm以下の細孔容積Bは0.315cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.707であった。
有機質材料として、軟化点が280℃の粒状ピッチ100質量部に対してアセチルアセトナトセリウム(CAS番号:15653-01-7)0.8質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、セリウムの含有量は0.14質量%であった。
賦活時間を35分とした以外は実施例18と同様にし、実施例19の活性炭を得た。得られた活性炭は、比表面積1078m2/g、全細孔容積Aは0.464cc/g、ミクロ細孔容積率({C/A}×100)は89%、細孔径1.0nm以下の細孔容積Bは0.337cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.726であった。
賦活時間を45分とした以外は実施例18と同様にし、実施例20の活性炭を得た。得られた活性炭は、比表面積1249m2/g、全細孔容積Aは0.550cc/g、ミクロ細孔容積率({C/A}×100)は88%、細孔径1.0nm以下の細孔容積Bは0.352cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.641であった。
有機質材料として、軟化点が280℃の粒状ピッチ100質量部に対して(2,4-ペンタンジオナト)モリブデン(VI)ジオキシド(CAS番号:17524-05-9)0.8質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、モリブデンの含有量は0.23質量%であった。
賦活時間を40分とした以外は実施例21と同様にし、実施例22の活性炭を得た。得られた活性炭は、比表面積1171m2/g、全細孔容積Aは0.479cc/g、ミクロ細孔容積率({C/A}×100)は95%、細孔径1.0nm以下の細孔容積Bは0.365cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.761であった。
賦活時間を60分とした以外は実施例21と同様にし、実施例23の活性炭を得た。得られた活性炭は、比表面積1684m2/g、全細孔容積Aは0.714cc/g、ミクロ細孔容積率({C/A}×100)は92%、細孔径1.0nm以下の細孔容積Bは0.427cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.598であった。
有機質材料として、軟化点が280℃の粒状ピッチ100質量部に対してアセチルアセトナトコバルト(CAS番号:21679-46-9)1.5質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、コバルトの含有量は0.21質量%であった。
賦活時間を40分とした以外は実施例24と同様にし、実施例25の活性炭を得た。得られた活性炭は、比表面積1447m2/g、全細孔容積Aは0.616cc/g、ミクロ細孔容積率({C/A}×100)は89%、細孔径1.0nm以下の細孔容積Bは0.431cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.700であった。
有機質材料として、軟化点が280℃の粒状ピッチ100質量部に対してビス(2,4-ペンタンジオナト)チタン(IV)オキシド(CAS番号:14024-64-7)1.4質量部を混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、チタンの含有量は0.25質量%であった。
賦活時間を40分とした以外は実施例26と同様にし、実施例27の活性炭を得た。得られた活性炭は、比表面積1170m2/g、全細孔容積Aは0.557cc/g、ミクロ細孔容積率({C/A}×100)は80%、細孔径1.0nm以下の細孔容積Bは0.320cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.575であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量20g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。金属成分は未添加であるため、該活性炭前駆体の金属含有量は0質量%である。
賦活時間を90分とした以外は比較例1と同様にし、比較例2の活性炭を得た。得られた活性炭は、比表面積1304m2/g、全細孔容積Aは0.497cc/g、ミクロ細孔容積率({C/A}×100)は100%、細孔径1.0nm以下の細孔容積Bは0.428cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.862であった。
賦活時間を125分とした以外は比較例1と同様にし、比較例3の活性炭を得た。得られた活性炭は、比表面積1741m2/g、全細孔容積Aは0.692cc/g、ミクロ細孔容積率({C/A}×100)は100%、細孔径1.0nm以下の細孔容積Bは0.462cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.667であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対して金属成分としてカプリル酸亜鉛(CAS番号:557-09-5)1.3質量部を添加し混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、亜鉛の含有量は0.19質量%であった。
賦活時間を100分とした以外は比較例4と同様にし、比較例5の活性炭を得た。得られた活性炭は、比表面積1484m2/g、全細孔容積Aは0.577cc/g、ミクロ細孔容積率({C/A}×100)は100%、細孔径1.0nm以下の細孔容積Bは0.467cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.809であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対して金属成分としてアセチルアセトナト銅(CAS番号:13395-16-9)1.0質量部を添加し混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、銅の含有量は0.18質量%であった。
賦活時間を130分とした以外は比較例6と同様にし、比較例7の活性炭を得た。得られた活性炭は、比表面積1690m2/g、全細孔容積Aは0.681cc/g、ミクロ細孔容積率({C/A}×100)は100%、細孔径1.0nm以下の細孔容積Bは0.415cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.610であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対して金属成分としてセバシン酸銀0.7質量部を添加し混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、銀の含有量は0.27質量%であった。
賦活時間を100分とした以外は比較例8と同様にし、比較例9の活性炭を得た。得られた活性炭は、比表面積1280m2/g、全細孔容積Aは0.495cc/g、ミクロ細孔容積率({C/A}×100)は100%、細孔径1.0nm以下の細孔容積Bは0.397cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.802であった。
賦活時間を130分とした以外は比較例8と同様にし、比較例10の活性炭を得た。得られた活性炭は、比表面積1730m2/g、全細孔容積Aは0.700cc/g、ミクロ細孔容積率({C/A}×100)は100%、細孔径1.0nm以下の細孔容積Bは0.420cc/g、細孔径1.0nm以下の細孔容積比(B/A)は0.600であった。
有機質材料として、軟化点が280℃の粒状石炭ピッチ100質量部に対して金属成分としてトリスアセチルアセトナトイットリウム(CAS番号:15554-47-9)1.3質量部を添加し混合したものを、溶融押出機に供給し、溶融温度320℃で溶融混合し、吐出量16g/minで紡糸することによりピッチ繊維を得た。得られたピッチ繊維を空気中常温から354℃まで1~30℃/分の割合で54分間昇温することにより不融化処理をおこない、不融化されたピッチ繊維である活性炭前駆体を得た。該活性炭前駆体において、イットリウムの含有量は0.25質量%であった。





Claims (13)
- 活性炭前駆体に金属成分を添加する工程と、
前記金属成分が添加された活性炭前駆体を導入ガスとして炭酸ガスで賦活し、全細孔容積Aに対する直径1.0nm以下の細孔容積Bの比(細孔容積B/全細孔容積A)が0.5以上であり、直径2.0nm以下の細孔容積Cが0.35cc/g以上である活性炭を得る賦活工程を含み、
前記金属成分を構成する金属元素が、第2族元素(但し、カルシウムを除く。)、第3族元素、第4族元素、第5族元素、第7族元素、及び希土類元素からなる群から選択される、活性炭の製造方法。 - 前記金属元素が、Y、Mg、Mn、La、V、Zr、Ti及びCeからなる群から選択される、請求項1に記載の活性炭の製造方法。
- 金属成分を含む活性炭前駆体を導入ガスとして炭酸ガスで賦活し、全細孔容積Aに対する直径1.0nm以下の細孔容積Bの比(細孔容積B/全細孔容積A)が0.5以上であり、直径2.0nm以下の細孔容積Cが0.35cc/g以上である活性炭を得る賦活工程を含み、
前記金属成分を構成する金属元素が、Y、La、V、Zr、及びCeからなる群から選択される、活性炭の製造方法。 - 活性炭前駆体に金属成分を添加する工程と、
前記金属成分が添加された活性炭前駆体を導入ガスとして炭酸ガスで賦活し、全細孔容積Aに対する直径1.0nm以下の細孔容積Bの比(細孔容積B/全細孔容積A)が0.5以上である活性炭を得る賦活工程を含み、
前記金属成分を構成する金属元素が、第6族元素及び第9族元素からなる群から選択される、活性炭の製造方法。 - 前記金属元素が、Mo及びCoからなる群から選択される、請求項4に記載の活性炭の製造方法。
- 前記活性炭の比表面積が600m2/g以上である、請求項1から5のいずれか1項に記載の活性炭の製造方法。
- 前記賦活工程において、前記導入ガスの組成を変更しない、請求項1から6のいずれか1項に記載の活性炭の製造方法。
- 前記導入ガスの流量が、前記活性炭前駆体1g当たり、0℃1気圧換算で1.5L/分以上である、請求項1から7のいずれか1項に記載の活性炭の製造方法。
- 前記賦活工程における賦活温度が800~1000℃である、請求項1から8のいずれか1項に記載の活性炭の製造方法。
- 前記活性炭前駆体中、前記金属成分の含有量が0.05~1.0質量%である、請求項1から9のいずれか1項に記載の活性炭の製造方法。
- 前記活性炭前駆体が、不融化したピッチである、請求項1から10のいずれか1項に記載の活性炭の製造方法。
- 前記活性炭において、全細孔容積Aに対する直径2.0nm以下の細孔容積Cの割合({細孔容積C/細孔容積A}×100)が85%以上である、請求項1から11のいずれか1項に記載の活性炭の製造方法。
- 前記活性炭において、直径1.0nm以下の細孔容積Bが0.25cc/g以上である、請求項1から12のいずれか1項に記載の活性炭の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022206816A JP7441296B2 (ja) | 2017-03-31 | 2022-12-23 | 活性炭の製造方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017071832 | 2017-03-31 | ||
| JP2017071832 | 2017-03-31 | ||
| PCT/JP2018/013371 WO2018181778A1 (ja) | 2017-03-31 | 2018-03-29 | 活性炭の製造方法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022206816A Division JP7441296B2 (ja) | 2017-03-31 | 2022-12-23 | 活性炭の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2018181778A1 JPWO2018181778A1 (ja) | 2020-03-26 |
| JP7202285B2 true JP7202285B2 (ja) | 2023-01-11 |
Family
ID=63676198
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019510164A Active JP7202285B2 (ja) | 2017-03-31 | 2018-03-29 | 活性炭の製造方法 |
| JP2022206816A Active JP7441296B2 (ja) | 2017-03-31 | 2022-12-23 | 活性炭の製造方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022206816A Active JP7441296B2 (ja) | 2017-03-31 | 2022-12-23 | 活性炭の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| JP (2) | JP7202285B2 (ja) |
| KR (1) | KR102571710B1 (ja) |
| CN (1) | CN110461767A (ja) |
| TW (1) | TW201841831A (ja) |
| WO (1) | WO2018181778A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7300124B2 (ja) * | 2019-01-31 | 2023-06-29 | 株式会社タカギ | トリハロメタン除去用活性炭およびその製造方法 |
| US11795066B2 (en) | 2020-06-30 | 2023-10-24 | Kuraray Co., Ltd. | Carbonaceous material and method for producing same, water purification filter, and water purifier |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004182511A (ja) | 2002-12-02 | 2004-07-02 | Ad'all Co Ltd | 活性炭及びその製造方法 |
| JP2007039289A (ja) | 2005-08-04 | 2007-02-15 | Toda Kogyo Corp | 球状多孔性炭素粒子粉末及びその製造法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62149917A (ja) * | 1985-12-23 | 1987-07-03 | Gunei Kagaku Kogyo Kk | 活性炭繊維の製造方法 |
| JP3446771B2 (ja) * | 1993-12-09 | 2003-09-16 | 三菱瓦斯化学株式会社 | 高比表面積炭素材料の製造方法 |
| KR100932158B1 (ko) * | 2005-09-29 | 2009-12-16 | 쇼와 덴코 가부시키가이샤 | 활성탄 및 그것의 제조방법 |
| JP2007221108A (ja) | 2006-01-17 | 2007-08-30 | Japan Enviro Chemicals Ltd | 活性炭及びその製造方法 |
| CN101249956B (zh) * | 2007-07-09 | 2011-04-27 | 盐城市炭化工业有限公司 | 具有储能性能的炭质材料制备工艺 |
| CN102431992B (zh) * | 2011-09-22 | 2013-07-24 | 安徽工业大学 | 氧化镁模板协同氢氧化钾活化制备多孔炭材料的方法 |
| ES2655176T3 (es) | 2011-10-07 | 2018-02-19 | Teijin Pharma Limited | Adsorbente administrado por vía oral que contiene fibra de carbono activado |
| KR101874086B1 (ko) * | 2012-12-28 | 2018-08-02 | 재단법인 포항산업과학연구원 | 활성탄 제조 방법 |
| KR101588768B1 (ko) * | 2014-10-27 | 2016-01-26 | 현대자동차 주식회사 | 활성탄소 및 그 제조방법 |
| JP7155589B2 (ja) | 2017-06-22 | 2022-10-19 | 大日本印刷株式会社 | 食事用載置具 |
-
2018
- 2018-03-29 JP JP2019510164A patent/JP7202285B2/ja active Active
- 2018-03-29 KR KR1020197028686A patent/KR102571710B1/ko active IP Right Grant
- 2018-03-29 WO PCT/JP2018/013371 patent/WO2018181778A1/ja active Application Filing
- 2018-03-29 CN CN201880021396.4A patent/CN110461767A/zh active Pending
- 2018-03-30 TW TW107111406A patent/TW201841831A/zh unknown
-
2022
- 2022-12-23 JP JP2022206816A patent/JP7441296B2/ja active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004182511A (ja) | 2002-12-02 | 2004-07-02 | Ad'all Co Ltd | 活性炭及びその製造方法 |
| JP2007039289A (ja) | 2005-08-04 | 2007-02-15 | Toda Kogyo Corp | 球状多孔性炭素粒子粉末及びその製造法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2023024708A (ja) | 2023-02-16 |
| KR102571710B1 (ko) | 2023-08-28 |
| KR20190135005A (ko) | 2019-12-05 |
| CN110461767A (zh) | 2019-11-15 |
| JPWO2018181778A1 (ja) | 2020-03-26 |
| JP7441296B2 (ja) | 2024-02-29 |
| TW201841831A (zh) | 2018-12-01 |
| WO2018181778A1 (ja) | 2018-10-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7441296B2 (ja) | 活性炭の製造方法 | |
| US9156694B2 (en) | Porous carbon and method of manufacturing same | |
| JP2019108269A (ja) | 高活性表面積を有する活性炭 | |
| JP2006347864A (ja) | メソポーラスカーボンの製造方法およびメソポーラスカーボン | |
| JP6379324B1 (ja) | 活性炭及びその製造方法 | |
| WO2018116859A1 (ja) | 活性炭及びその製造方法 | |
| KR20140015985A (ko) | 다공성 탄소 및 이의 제조방법 | |
| JP2022132348A (ja) | 活性炭及びその製造方法 | |
| JP6749117B2 (ja) | 金属単体及び金属化合物の少なくとも一方を含有する活性炭の製造方法 | |
| JPWO2019244903A1 (ja) | 活性炭 | |
| JP2004182511A (ja) | 活性炭及びその製造方法 | |
| Ra et al. | Ultramicropore formation in PAN/camphor-based carbon nanofiber paper | |
| KR20180017856A (ko) | 활성탄소의 제조방법 | |
| KR101412775B1 (ko) | 다공성 탄소 및 이의 제조방법 | |
| Hunter et al. | Milling as a route to porous graphitic carbons from biomass | |
| JP7428347B2 (ja) | 活性炭 | |
| JP6719709B2 (ja) | 活性炭 | |
| JP7491506B2 (ja) | 活性炭 | |
| Tamayo et al. | Characterization of polymer-derived ceramers subjected to wet-etching and the evolution of the carbon phase during thermal conversion | |
| WO2019235044A1 (ja) | 造粒用繊維状バインダ | |
| KR20080057449A (ko) | 질소산화물 제거용 활성탄소섬유 촉매의 제조방법 및 이에따라 제조된 활성탄소섬유 촉매 | |
| JP2002138324A (ja) | 金属含有活性炭素繊維の製造方法 | |
| Tanaka et al. | Cobalt-Catalyzed Carbonization from Polyacrylonitrile for Preparing Nitrogen-Containing Ordered Mesoporous Carbon Cmk-1 Electrode with High Electric Double-Layer Capacitance | |
| JP2009191391A (ja) | 炭素繊維およびその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AA64 | Notification of invalidation of claim of internal priority (with term) |
Free format text: JAPANESE INTERMEDIATE CODE: A241764 Effective date: 20200107 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200115 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200207 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210318 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220215 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20220408 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220601 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220705 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20220824 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221025 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20221206 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20221223 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7202285 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |