以下、本発明を詳細に説明する。
重合性単量体を含有する重合性単量体組成物の粒子を水系媒体の中で形成する造粒工程と、該重合性単量体組成物の該粒子に含まれる該重合性単量体を重合させることによりトナー粒子を得る重合工程と、を有するトナー粒子の製造方法において、
該重合工程においては、第1の重合開始剤と第2の重合開始剤とを用いて重合が行われ、
該第1の重合開始剤が、
(i)過酸化物系重合開始剤であり、
(ii)下記の式(1)もしくは式(2)の構造を有し、
(iii)10時間半減期温度が70.0℃以上115.0℃以下であり、
該第2の重合開始剤が、
(i)過酸化物系重合開始剤であり、
(ii)下記の式(3)の構造を有し、
(iii)10時間半減期温度が45.0℃以上65.0℃以下であり、
該第2の重合開始剤の添加量が該重合性単量体に対して2.5mol%以上20.0mol%以下として、トナー粒子を製造することで本発明の効果が得られる。
(式中、R
1は炭素数1以上8以下のアルキル基を表し、R
2は水素原子、あるいは、炭素数1以上5以下のアルキル基を表し、R
3は水素原子、あるいは、炭素数1以上7以下のアルキル基を表す。)
(式中、R
4は炭素数1以上8以下のアルキル基を表し、R
5は水素原子、あるいは、炭素数1以上5以下のアルキル基を表し、R
6は水素原子、あるいは、炭素数1以上7以下のアルキル基を表す。)
(式中、R
7〜R
9は、それぞれ独立して、炭素数1以上5以下のアルキル基を表し、R
10は炭素数1以上6以下のアルキル基を表す。)
本発明の効果が発現する理由は必ずしも明確にはなっていないが、本発明者らは次のように考えている。
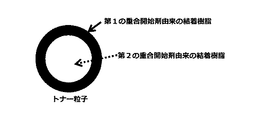
10時間半減期温度が高い重合開始剤と低い重合開始剤を併用し、重合性単量体を重合すると分子量の大きいポリマーと小さいポリマーが生成して結着樹脂となる。この時生成するポリマー1分子ずつを考えると各分子の末端には重合開始剤由来の重合開始剤残基が存在することになる。従って重合開始剤残基の性質によって各ポリマー分子の親疎水性に差が生じる。式(1)もしくは式(2)の構造を有する重合開始剤の重合開始剤残基を有するポリマー分子は、式(3)の構造を有する重合開始剤の重合開始剤残基を有するポリマー分子と比較して親水性の性質を有することになる。これは、式(3)の構造を有する重合開始剤の開始剤残基より、式(1)もしくは式(2)の構造を有する重合開始剤の重合開始剤残基の方が親水性であるためである(図1参照)。その際、式(1)もしくは式(2)の構造を有する重合開始剤は式(3)の構造を有する重合開始剤より10時間半減期温度が高いため、式(1)もしくは式(2)の構造を有する重合開始剤により生成するポリマーの分子量は、式(3)の構造を有する重合開始剤により生成するポリマーの分子量より大きくなる。また、水系媒体中で造粒、重合させトナー粒子を製造する場合、より親水性の物質がトナー粒子表面に分布することになる。従って本発明のトナー粒子製造方法の場合、分子量の大きいポリマーがトナー粒子表層側、分子量が小さいポリマーがトナー粒子中心部側に結着樹脂として分布するトナー粒子が得られる(図2参照)。その結果、残留モノマーを低減するために、重合工程もしくは蒸留工程において、より高温環境下に置かれた場合でも、トナー粒子表層側に存在する高分子量のポリマーによりトナー粒子表層の耐熱性は高いためトナー粒子同士が凝集して粗大化することが抑制される。
また、10時間半減期温度が低い重合性開始剤を用いて重合性単量体を重合すると、重合反応初期から重合開始剤が開裂してラジカルを発生して重合反応を行う。そのため、重合反応後期においては重合開始剤の大半は消費されてしまっているため、重合性単量体の減少は鈍化する。これに対して、本発明では10時間半減期温度が高い重合開始剤を併用しているため重合工程後期やその後の蒸留工程においても重合開始剤が残っているため、重合性単量体を重合することが可能で、重合工程もしくは蒸留工程において、より高温環境下にて残留モノマーがより低減できるため、残留モノマーを低減させつつ、重合工程や蒸留工程での粗大化を抑制するトナー粒子を得ることができる。更には10時間半減期温度が高い重合開始剤のみの使用では生成するポリマーが高分子量のものとなり、トナー粒子の定着性が悪くなり易いが、10時間半減期温度が低い重合開始剤を併用しているため定着性も良好なトナー粒子が得られる。
上記の効果を得るためには第1の重合開始剤が式(1)もしくは式(2)の構造を有し、10時間半減期温度が70.0℃以上115.0℃以下であり、第2の重合開始剤が式(3)の構造を有し、10時間半減期温度が45.0℃以上65.0℃以下であり、該第2の重合開始剤の添加量が該重合性単量体に対して2.5mol%以上20.0mol%以下であることが必要となる。
重合開始剤を本発明に用いられる第1の重合開始剤と第2の重合開始剤のうち、いずれか一方では本発明の効果は得られない。いずれか一方のみであると生成される結着樹脂はトナー粒子表層部側と中心部側とも同一の均質な状態となるため、重合工程や蒸留工程での粗大化の抑制と定着性が両立できないためである。
式(1)もしくは式(2)の構造を有する重合開始剤の10時間半減期温度が70.0℃未満であると式(3)の構造を有する開始剤との差が小さいため、トナー粒子表層に分布する結着樹脂の分子量が十分大きくないため耐熱性が不十分となる。また、残留モノマー低減効果も低下する。式(1)もしくは式(2)の構造を有する重合開始剤の10時間半減期温度が115.0℃超であると式(3)の構造を有する開始剤との差が大きすぎるため結着樹脂の分子量や残留モノマーの調整が困難となる。具体的には式(3)の構造を有する開始剤に最適な条件とすると、式(1)もしくは式(2)の構造を有する重合開始剤が重合反応のためにラジカルを生成する確率が低くなり重合反応自体が進行しづらくなるため残留モノマーの低減効率がかえって低下し、またトナー表層側に分布する結着樹脂の分子が十分な分子量を得られないため本発明の効果が得られない。逆の場合は式(3)の構造を有する開始剤の分子量制御が困難で、現像性と定着性の両立が困難になる。また、式(3)の構造を有する開始剤の開裂が速過ぎるため重合反応中期以降において重合が進行しづらくなり同様に残留モノマーの低減効率がかえって低下し、また残留モノマーが多いことでトナー粒子が可塑化され凝集し易くなる。
式(3)の構造を有する重合開始剤の10時間半減期温度が45.0℃未満であると、式(1)もしくは式(2)の構造を有する重合開始剤との差が大きすぎるため結着樹脂の分子量や残留モノマーの調整が困難となる。65.0℃超の場合は式(1)もしくは式(2)の構造を有する重合開始剤との差が小さいため、トナー粒子表層に分布する結着樹脂の分子量が十分大きくないため耐熱性が不十分となる。
式(1)もしくは式(2)、式(3)の構造を有する重合開始剤の構造としては、該第1の重合開始剤が式(4)もしくは式(5)の構造を有し、該第2の重合開始剤が式(6)の構造を有していると好ましい。
(式中、R
11は炭素数1以上3以下のアルキル基を表し、R
2は水素原子、あるいは、炭素数1以上5以下のアルキル基を表し、R
3は水素原子、あるいは、炭素数1以上7以下のアルキル基を表す。)
(式中、R
12は炭素数1以上3以下のアルキル基を表し、R
5は水素原子、あるいは、炭素数1以上5以下のアルキル基を表し、R
6は水素原子、あるいは、炭素数1以上7以下のアルキル基を表す。)
(式中、R
7は炭素数1以上5以下のアルキル基を表し、R
10は炭素数1以上6以下のアルキル基を表す。)
また、該第1の重合開始剤が式(7)の構造を有しているとより好ましい。
(式中、R
12は炭素数1以上3以下のアルキル基を表し、R
5は水素原子、あるいは、炭素数1以上5以下のアルキル基を表し、R
13は水素原子、あるいは炭素数1以上5以下のアルキル基を表わす。)
これは、本発明の作用効果を発現する原因である2種類の重合開始剤残基の親疎水性のバランスがよりよくなるためである。特に第1の重合開始剤が式(2)、(5)、(7)の構造を有していることが好ましい。これは、本発明の作用効果を発現する原因である2種類の重合開始剤のうち第1の重合開始剤の重合開始剤残基が酸素原子が1つ多いため親水性が強く、よりトナー粒子表面側に高分子量の結着樹脂が分布し、本発明の作用効果が強く発現するためである。
該第2の重合開始剤の添加量は該重合性単量体に対して2.5mol%以上20.0mol%以下であることが必要である。該第2の重合開始剤の添加量が2.5mol%未満であると反応の早い段階で重合開始剤がほとんど消費されてしまい第1の開始剤が残っていたとしても残留モノマーを低減しきれず、かつ残留モノマーの可塑効果によりトナー粒子の凝集もし易い。逆に20.0mol%超であると重合で生成する結着樹脂に関してオリゴマーが多く生成するため耐熱性が弱く凝集しやすい。
該第2の重合開始剤の添加量は該第1の重合開始剤より多いことが好ましい。これは、得られるトナー粒子の結着樹脂が高分子量成分と低分子量成分が適切な割合含有されるため、トナー粒子の定着性と耐熱性が双方とも良好になるためである。更に、該第1の重合開始剤の添加量が該第2の重合開始剤の添加量に対して5.0mol%以上60.0mol%以下であることが好ましい。これは残留モノマーを低減させつつ、重合工程での粗大化を抑制する効果がより大きくなるだけでなく、定着性と耐久性の両立という点でも好ましいためである。これは第1の重合開始剤由来の高分子量の結着樹脂がトナー粒子表層側に存在することで耐久性が向上するためであり、その割合が適度であるため定着性を阻害しないためである。
該重合性単量体組成物が結晶性材料を含有することが、定着性の点で優れるため好ましい。本発明に使用できる結晶性物質としては、ワックスや結晶性ポリエステル等、離型剤など公知のものを使用することができる。
本発明のトナーの場合、トナー粒子表面近傍側には結着樹脂のうち、高分子量成分が分布しているため、結晶性物質がトナー粒子表面近傍には相溶しにくいため分布しにくい。そのため、結晶性物質により定着性を良化させつつも、トナー粒子表面への露出は抑制されるため耐久性も両立できる。
本発明に使用する結晶性物質の融点Tmは、50℃以上90℃以下であることが好ましい。さらに好ましい範囲は、60℃以上85℃以下である。該結晶性物質の融点Tmが、50℃以上90℃以下であると定着性と保存性の両立の点で好ましい(融点Tmの測定方法は後述する)。
本発明のトナーは、結晶性物質として、ワックスを含有してもよい。その場合、ワックスの少なくとも1つは、融点(温度20乃至200℃の範囲におけるDSC吸熱曲線の最大吸熱ピークに対応する温度)が30℃以上120℃以下であることが好ましく、50℃以上90℃以下であることがより好ましい。また、室温で固体のワックスであることが好ましく、特に、融点が50℃以上90℃以下の固体ワックスがトナーの耐ブロッキング性、多数枚耐久性、低温定着性及び耐オフセット性の点から好ましい。
ワックスとしては、パラフィンワックス、ポリオレフィンワックス、マイクロクリスタリンワックス及びフィッシャートロプシュワックスの如きポリメチレンワックス、アミドワックス、ペトロラタム等の石油系ワックス及びその誘導体、モンタンワックス及びその誘導体、カルナバワックス及びキャンデリラワックス等の天然ワックス及びそれらの誘導体、硬化ヒマシ油及びその誘導体、植物ワックス、動物ワックス、高級脂肪酸、長鎖アルコール、エステルワックス、ケトンワックス及びこれらのグラフト化合物、ブロック化合物の如き誘導体など公知のワックスを用いることが可能である。これらは単独又は併せて用いることができる。
本発明のトナー中のワックスの含有量は、結着樹脂100質量部に対して、3質量部以上30質量部以下が好ましく、3質量部以上20質量部以下がより好ましく、4質量部以上15質量部以下が更に好ましい。ワックスの添加量が下限値以上であるとオフセット防止効果が低くならず、上限値以下の場合は耐ブロッキング効果が低下せず、耐オフセット効果にも悪影響を与え難く、トナーのドラム融着やトナーの現像スリーブ融着を起こし難い。
本発明で用いられるワックスとしては炭化水素ワックスを用いている場合はより一層、耐ブロッキング効果や耐オフセット効果に優れ、トナーのトナー層規制部材やトナー担持体への融着を起こし難い。
ワックスとしては炭化水素ワックスを用い、かつ脂肪族のジオールと脂肪族のジカルボン酸により製造された結晶性ポリエステルもトナー粒子中に含有している場合、該結晶性ポリエステルとワックスとの相互作用により、結晶化が促進されやすい。このため、よりトナー粒子表層側においてワックスや結晶性ポリエステルが結着樹脂に相溶しにくいため重合工程中にトナー粒子が凝集しにくく好ましい。
なお、上記の如き物性を求めるにあたって、ワックスをトナーから抽出することを必要とする場合には、抽出方法は特に制限されるものではなく、任意の方法を用いることができる。例えば、所定量のトナーをトルエンにてソックスレー抽出し、得られたトルエン可溶分から溶剤を除去した後、クロロホルム不溶分を得る。その後、IR法などにより同定分析をする。
また、定量に関しては、DSCなどにより定量分析を行う。本発明ではTAインスツルメンツジャパン社製DSC−2920を用いて測定を行う。
測定時の比熱変化が出る前と出た後のベースラインの中間点の線と示差熱曲線との交点をガラス転移点とする。また、得られた昇温時のDSC曲線からワックス成分の最大吸熱ピーク温度を得る。
該重合性単量体組成物が結晶性ポリエステルを含有すると定着性が優れるため更に好ましい。しかし、該重合工程において、該結晶性ポリエステルの融点以上の温度で重合反応を行う場合、結晶性ポリエステルの可塑効果により、より一層重合工程においてトナー粒子は凝集しやすくなる。それに対して本発明で用いられる第1の重合開始剤と第2の重合開始剤を用いることでトナー粒子表層側に高分子量の結着樹脂が分布することで耐熱性が向上する。そのためトナー粒子は凝集を抑制し、かつ結晶性ポリエステルの低温定着性の効果を活かせるため好ましい。その際、該第1の重合開始剤由来の高分子量の結着樹脂の方が該結晶性ポリエステルへの相溶性が低いため、該結晶性ポリエステルはトナー粒子表層側に相溶した状態では存在しにくいため、可塑化されにくく、トナー粒子表層側の耐熱性が強固に維持できるためトナー粒子は凝集しにくくより好ましい。また、結晶性ポリエステルはワックスや離型剤と比較して分子量が大きいため、より一層その効果が大きい。該結晶性ポリエステルとしては、2価以上の多価カルボン酸とジオールの反応により得ることができる。その中でも、脂肪族ジオールと脂肪族ジカルボン酸を主成分とするポリエステルが、結晶化度が高く好ましい。結晶性ポリエステルは、1種類のみを用いても、複数種を併用しても良い。更に、結晶性ポリエステルの他に非晶質のポリエステルをトナーに含有させても良い。
本発明において結晶性ポリエステルとは、示差走査熱量測定(DSC)において昇温時に吸熱ピークがあるポリエステルを指し、その測定は「ASTM D 3417−99」に準じて行う。
このような結晶性ポリエステルを得るためのアルコール単量体としては、エチレングリコール、ジエチレングリコール、トリエチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、ジプロピレングリコール、トリメチレングリコール、テトラメチレングリコール、ペンタメチレングリコール、ヘキサメチレングリコール、オクタメチレングリコール、ノナメチレングリコール、デカメチレングリコール、ネオペンチルグリコール、1,4−ブタジエングリコールその他が挙げられる。
また、本発明においては上記の如きアルコール単量体が主成分として用いられるが、上記成分の他に、ポリオキシエチレン化ビスフェノールA、ポリオキシプロピレン化ビスフェノールA、1,4−シクロヘキサンジメタノール等の二価のアルコール、1,3,5−トリヒドロキシメチルベンゼン等の芳香族アルコール、ペンタエリスリトール、ジペンタエリスリトール、トリペンタエリスリトール、1,2,4−ブタントリオール、1,2,5−ペンタントリオール、グリセリン、2−メチルプロパントリオール、2−メチル−1,2,4−ブタントリオール、トリメチロールエタン、トリメチロールプロパン等の三価のアルコール等を用いても良い。
上記結晶性ポリエステルを得るためのカルボン酸単量体としては、シュウ酸、マロン酸、コハク酸、グルタル酸、アジピン酸、ピメリン酸、スペリン酸、グルタコン酸、アゼライン酸、セバシン酸、ノナンジカルボン酸、デカンジカルボン酸、ウンデカンジカルボン酸、ドデカンジカルボン酸、マレイン酸、フマル酸、メサコン酸、シトラコン酸、イタコン酸、イソフタル酸、テレフタル酸、n−ドデシルコハク酸、n−デドセニルコハク酸、シクロヘキサンジカルボン酸、これらの酸の無水物又は低級アルキルエステル等が挙げられる。
また、本発明においては上記の如きカルボン酸単量体を主成分として用いるが、上記の成分の他に三価以上の多価カルボン酸を用いても良い。
三価以上の多価カルボン酸成分としては、トリメリット酸、2,5,7−ナフタレントリカルボン酸、1,2,4−ナフタレントリカルボン酸、ピロメリット酸、1,2,4−ブタントリカルボン酸、1,2,5−ヘキサントリカルボン酸、1,3−ジカルボキシル−2−メチル−2−メチレンカルボキシプロパン、及びこれらの酸無水物又は低級アルキルエステル等の誘導体等が挙げられる。
特に好ましい結晶性ポリエステルとしては、1,4−シクロヘキサンジメタノールとアジピン酸とを反応して得られるポリエステル、テトラメチレングリコール及びエチレングリコールとアジピン酸とを反応させて得られるポリエステル、ヘキサメチレングリコールとセバシン酸とを反応して得られるポリエステル、エチレングリコールとコハク酸とを反応して得られるポリエステル、エチレングリコールとセバシン酸とを反応して得られるポリエステル、テトラメチレングリコールとコハク酸とを反応して得られるポリエステル、ジエチレングリコールとデカンジカルボン酸とを反応して得られるポリエステルを挙げることができる。該結晶性ポリエステルは飽和ポリエステルであると一層好ましい。該結晶性ポリエステルが不飽和部分を有する場合と比較して、該過酸化物系重合開始剤との反応で架橋反応が起こらないため、該結晶性ポリエステルの溶解性の点で有利なためである。
本発明に用いられる結晶性ポリエステルは、通常のポリエステル合成法で製造することができる。例えば、ジカルボン酸成分とジアルコ−ル成分をエステル化反応、又はエステル交換反応せしめた後、減圧下又は窒素ガスを導入して常法に従って重縮合反応させることによって得ることができる。
結晶性ポリエステルの融点としては、50.0℃以上90.0℃以下であることが好ましい。結晶性ポリエステルの融点が、この範囲であると、トナー粒子が凝集しにくく、トナー粒子の保存性、定着性が維持でき、かつ重合法によりトナー粒子を製造する場合に重合性単量体への溶解性が高くなり、好ましい。結晶性ポリエステルの融点は、示差走査熱量測定(DSC)によって測定することができる。また結晶性ポリエステルの融点は、使用するアルコール単量体やカルボン酸単量体の種類、重合度等によって調整することができる。
結晶性ポリエステルの重量平均分子量(Mw)は5,000以上35,000以下であることが好ましい。この範囲に重量平均分子量(Mw)を有する結晶性ポリエステルによれば、得られるトナー粒子において、結晶性ポリエステルの分散性が向上され、耐久安定性が向上するため好ましい。すなわち、結晶性ポリエステルの重量平均分子量(Mw)が5,000以上の場合では、結晶性ポリエステルの密度が高くなり、耐久安定性が向上する。一方、結晶性ポリエステルの重量平均分子量(Mw)が35,000以下の場合には、結晶性ポリエステルの溶融が迅速に行われ、分散状態が均一になるために、現像安定性が向上する。結晶性ポリエステルの重量平均分子量(Mw)は、使用するアルコール単量体やカルボン酸単量体の種類、重合時間や重合温度等によって調整することができる。
結晶性ポリエステルの酸価(AV)は0.0mgKOH/g以上20.0mgKOH/g以下であることが好ましく、0.0mgKOH/g以上10.0mgKOH/g以下であるとより好ましく、0.0mgKOH/g以上5.0mgKOH/g以下であると特に好ましい。酸価を下げることにより、画像形成時におけるトナーと紙との接着性は向上する。また重合法によりトナー粒子を製造する場合、結晶性ポリエステルの酸価(AV)が20.0mgKOH/g以下であると、トナー粒子同士の凝集が起こりにくくなる傾向にあり、また、トナー中における該結晶性ポリエステルの分布状態に偏りが出にくくなるため、帯電安定性及び耐久安定性が向上するため好ましい。
該重合性単量体組成物が荷電制御樹脂を含有し、重合性単量体中に含有されるスチレンの割合が60質量%以上であり、該荷電制御樹脂のスチレン−ヘキサン溶解度指数が10.0以上42.0以下であるとより好ましい。
本発明のトナー粒子製造方法の場合、分子量の大きいポリマーがトナー粒子表層側、分子量が小さいポリマーがトナー粒子中心部側に結着樹脂として分布するトナー粒子が得られる。そのため、トナー粒子表面側に分布する荷電制御樹脂は、トナー粒子表層側の分子量の大きいポリマーには相溶しにくく、荷電制御樹脂がよりトナー粒子最表面に露出して存在する量が増加するため、トナーの帯電性が良好なものとなり、好ましい。
該荷電制御樹脂のスチレン−ヘキサン溶解度指数が10.0以上42.0以下であると、該荷電制御樹脂が適度に高い極性を有するためトナー粒子最表面に分布することになる。更に重合性単量体中に含有されるスチレンの割合が60質量%以上である状態で、該荷電制御樹脂のスチレン−ヘキサン溶解度指数が10.0以上42.0以下であると、該荷電制御樹脂の極性が十分でありトナー粒子の際表面側に分布し、該荷電制御樹脂が結着樹脂側に相溶する程度が適切であるため、該荷電制御剤がトナー粒子最表面に均一に分布するのでトナーの帯電性に優れるため好ましい。
これは、スチレン−ヘキサン溶解度指数の算出においては、該荷電制御樹脂の貧溶媒として疎水性で無極性の炭化水素であるヘキサンを用いているためスチレン−ヘキサン溶解度指数が低いほど、極性が高く、親水性ということになる。また、スチレン−ヘキサン溶解度指数が低くなる程、トナー粒子表面に存在する分子量の大きいポリマーとの親和性が低くなるため該荷電制御樹脂は凝集し易くなる。また、スチレン−ヘキサン溶解度指数が高くなる程、トナー粒子表面に存在する分子量の大きいポリマーとの親和性が高くなるため荷電制御樹脂は均一に分布し易くなるが、相溶し易くなるため、荷電制御樹脂のトナー粒子最表面での存在量が減少し易くなるため適正な範囲のものを用いると効果が大きい。
従って、該荷電制御樹脂のスチレン−ヘキサン溶解度指数が10.0以上42.0以下であると、該荷電制御樹脂のスチレンモノマーへの溶解性が適切なため、該荷電制御樹脂適度に結着樹脂に相溶することで均一に分布し、且つ該荷電制御樹脂のトナー粒子表面での存在量が十分となるため、高温高湿環境下での多数枚プリントアウト時における帯電安定性の点で優れるため好ましい。
なお、該荷電制御樹脂のスチレン−ヘキサン溶解度指数は、15.0以上27.0以下であるとより好ましく、17.0以上25.0以下であると更に好ましい。
該荷電制御樹脂のモノマー組成によりスチレン−ヘキサン溶解度指数を調整する場合の具体例としては、例えば、極性の高いモノマーを用い、その含有割合を調整することで該荷電制御樹脂の極性を変えることや、その含有割合を調整することで該荷電制御樹脂のエステル濃度を変えることが挙げられる。
本発明において、モノマー中のスチレンの含有量は、60質量%以上であることが好ましい。該スチレンの含有量が60質量%以上の場合、該荷電制御樹脂のスチレン−ヘキサン溶解度指数と本発明の効果の発現との相関性が良く、トナーにおけるシェル形成が最適状態となる。上記モノマー中のスチレンの含有量は、70質量%以上であることがより好ましい。
<スチレン−ヘキサン溶解度指数の測定>
本発明で規定するスチレン−ヘキサン溶解度指数とは、荷電制御樹脂を良溶媒であるスチレンに溶解させたものに貧溶媒であるヘキサンを添加していったときに荷電制御樹脂成分が析出し始めるヘキサンの添加量で規定する。
本発明のトナーの製造方法においては、モノマーに含有されるスチレンの割合が60質量%以上とスチレンが多く含まれていることが好ましいため、スチレンを多く含むモノマー中に荷電制御樹脂が溶解している状態から重合反応が進行し、重合反応の進行とともにスチレンを含むモノマーが減少していく。荷電制御樹脂のスチレン溶液にヘキサンを添加していくことは、荷電制御樹脂のスチレン溶液におけるスチレンの割合が減少していくことを意味する。すなわち、該スチレン−ヘキサン溶解度指数の測定方法は、実際に重合反応が進行した場合に生じるスチレンを含むモノマーの減少を再現したものである。
従って、スチレン−ヘキサン溶解度指数が小さいほど、重合反応初期に荷電制御樹脂が析出してくることを表す。
また、この測定において無極性溶媒であるヘキサンを用いることで、極性が高い荷電制御樹脂ほどヘキサンの添加量が少ない段階で析出することになる。従って、スチレン−ヘキサン溶解度指数が小さいほど、水相側のトナー表層側に重合反応の進行に伴い析出した荷電制御樹脂が存在することを示す。
(サンプル作製)
4.0質量部の荷電制御樹脂をスチレン100.0質量部に溶解したスチレン溶液(液温25℃)を調製する。樹脂を溶解して12時間以上24時間未満放置したものをサンプル処理フィルター(ポアサイズ0.45μm)で濾過してスチレン不溶分を濾別し、濾液を測定用試料とする。
(測定方法)
測定装置として、(株)レスカ社製の粉体濡れ性試験機(WET−101P)を用いる。
スチレン−ヘキサン溶解度指数は、作製した測定試料をトールビーカー容器中に入れ、滴下試薬としてはn−ヘキサンとし、得られたn−ヘキサン滴下透過率曲線から決定する。なお、該トールビーカーは、直径5cmの円形で、厚さ1.75mmのガラス製のものを用い、スターラーとして、長さ25mm、最大径8mmの紡錘形でありフッ素樹脂コーティングを施されたマグネティックスターラーを用いた。
具体的な測定操作は以下の通りである。
測定用試料を280〜300rpmの速度で撹拌しながら、n−ヘキサンを0.8ml/minの滴下速度で連続的に添加し、波長780nmの光で透過率を測定し、n−ヘキサン滴下透過率曲線を作成する。
得られたn−ヘキサン滴下透過率曲線から光の透過率が50%の時点におけるn−ヘキサン濃度(体積%)を荷電制御樹脂のスチレン−ヘキサン溶解度指数とする。
上記荷電制御樹脂としては上記の範囲の物性を満たすものであれば、公知のものを使用して良く、例えば、スルホン酸基、スルホン酸塩基、4級アンモニウム塩基、カルボキシル基、水酸基、オキシカルボン酸基などを有する樹脂が用いられる。特に、該荷電制御樹脂が、スルホン酸基、スルホン酸塩基、あるいはスルホン酸エステルを有する重合体であると好ましい。特に好ましくはスルホン酸基を有する重合体である。これは、トナー粒子表面に該荷電制御樹脂が分布しやすいため帯電性の点で好ましいためである。
該荷電制御樹脂が式(8)で表わされる構造を有する重合体であると更に好ましい。これは、特に帯電性に優れるためである。
[前記式(8)中、R
1は、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、hは0以上3以下の整数を表し、hが2または3の場合、R
1はそれぞれ独立して選択できる。]
また、該荷電制御樹脂はスチレンとの共重合体であることが好ましい。これは、トナー粒子を形成する結着樹脂の構成成分である重合性単量体(モノマー)としてモノマー中のスチレンの含有量は、60質量%以上としているためである。つまり、懸濁重合によりトナー粒子を製造する場合、まず重合性単量体組成物中に該荷電制御樹脂を溶解させるため、該荷電制御樹脂がスチレンとの共重合体であると十分な溶解性を得られ好ましいためである。
本発明において、該荷電制御樹脂は、式(8)で表される構造を有する重合体であり、摩擦帯電により生じた電荷を安定に付与することができる。そのメカニズムは明確ではないが、式(8)で表される構造に存在する酸素原子やアリール基などの共役系の広がりにより、式(8)で表される構造に存在するサリチル酸部位で発生した電荷は、他のトナーを構成する材料に電荷授受され、帯電の立ち上がりを速くする効果を生み出していると考えている。また、余剰の帯電(過帯電)が生じた場合は速やかに電荷を放出し、局部的な過帯電を防止する効果も期待される。
該荷電制御樹脂中の、式(8)で示される構造のモル含有量は、該荷電制御樹脂の質量を基準として、250μmol/g以上650μmol/g以下であることが好まい。該モル含有量が、250μmol/g以上であれば、トナーに安定した電荷を付与することができる。一方、該モル含有量が650μmol/g以下であれば、式(8)で示される構造の持つ吸湿性の影響をより小さく抑えることができるため、本発明の効果をより得ることができる。
本発明において、荷電制御樹脂は、下記式(9)で表される構造を有する重合体であることが好ましい。
[前記式(9)中、R
2は、それぞれ独立して、ヒドロキシ基、カルボキシ基、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、R
3は、水素原子、ヒドロキシ基、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、jは1以上3以下の整数を表し、iは0以上3以下の整数を表す。]
上記式(9)で表される構造には、電子伝導に有利なアルキルエーテルを介して、芳香環とサリチル酸構造とが結合する構造を有している。このサリチル酸構造から伸びる大きな共役系構造が、外部の温湿度の影響を最小限に抑えつつ、摩擦帯電により生じた電荷を分子内部に保持する役割を果たし、安定して電荷を付与すると考えている。
また、該荷電制御樹脂は、下記式(10)で表されるモノマーユニットを有する重合体であってもよい。
[前記式(10)中、R
4は、それぞれ独立して、ヒドロキシ基、カルボキシ基、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、R
5は、水素原子、ヒドロキシ基、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、R
6は水素原子又はメチル基を表し、lは1以上3以下の整数を表し、kは0以上3以下の整数を表す。]
該荷電制御樹脂が、上記式(10)で表されるモノマーユニットを有する重合体である場合には、ビニル系樹脂を主成分とするトナーにおいて、本発明の効果がより好適に発揮される。ここで、「ビニル系樹脂を主成分とする」とは、トナーに含有される樹脂の50質量%以上がビニル系樹脂であることを意味する。
また、該荷電制御樹脂は、式(10)で示されるモノマーユニット、及び、ビニル系のモノマーユニットを有するビニル系共重合体であってもよい。これは、該荷電制御樹脂がビニル系共重合体であることにより、ビニル系樹脂を主成分とするトナー中では、相溶されやすくなる。相溶化により最適な分子配置を取ることが可能となり、本発明の効果がより顕著になるものと考えている。
また、該荷電制御樹脂をビニル系共重合体とすることにより、該荷電制御樹脂のガラス転移温度(Tg)を容易に制御できることから、トナーの定着性を維持しつつ、本発明の効果を発現でき、好ましい態様となる。
一方、該荷電制御樹脂は、ポリエステル構造を有する重合体とすることも可能である。この場合、多価アルコール成分と多価カルボン酸成分とを重縮合させて生成されるポリエステル構造を主鎖とし、上記式(8)で表される構造を含有させればよい。また、ポリエステル構造を有する重合体として、ポリエステル構造をビニル系モノマーで変性させたハイブリッド樹脂を用いることも可能である。
ハイブリッド樹脂を用いる場合、ハイブリッド樹脂中のビニル変性比の調整には公知の方法が使用可能である。具体的には使用するポリエステル構造成分とビニル系モノマー成分の仕込み量比を変えることで任意の変性比が調整可能である。
本発明において、該荷電制御樹脂の製造方法としては特に限定されず、公知の手法により製造することができる。
該帯電制御樹脂が、ビニル系(共)重合体の場合には、一例として、式(8)で表される構造を含有するモノマー(下記式(11))を、重合開始剤を用いて重合させればよい。
[前記式(11)中、R
7は、それぞれ独立して、ヒドロキシ基、カルボキシ基、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、R
8は、水素原子、ヒドロキシ基、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、R
9は、水素原子又はメチル基を表し、nは1以上3以下の整数を表し、mは0以上3以下の整数を表す。]
また、上記モノマー(式(11))の具体例としては、以下のものを挙げることができる。ここに示す例は、あくまで一例であり、これらに限定されるものではない。
また、該荷電制御樹脂がビニル系共重合体である場合、上記モノマー(式(11))と共に用いることができる、その他のビニル系モノマーとしては、特に制限されない。
一方、上記荷電制御樹脂が、ポリエステル構造を有する重合体である場合には、種々の公知の製造方法が利用可能である。例えば、
(A)ポリエステル構造に含まれるカルボキシ基や水酸基の反応残基を利用して、有機反応により、式(8)で表される構造を置換基として有する構造に変換する方法;
(B)式(8)で表される構造を有する多価アルコール又は多価カルボン酸を用いてポリエステルを作製する方法;
また、ハイブリッド樹脂である場合には、
(C)式(8)で表される構造を含有する不飽和基含有ポリエステルを、ビニルモノマーを用いてハイブリッド化する方法;
(D)式(8)で表される構造を有するビニル系モノマーを用いて不飽和基含有ポリエステルをハイブリッド化する方法;
などが挙げられる。
上記(A)の具体的方法としては、式(8)で表される構造を有機反応で導入する場合、ポリエステル構造に存在するカルボキシ基を下記式(12)のような構造をもつ化合物を用いてアミド化する方法を挙げることができる。
[前記式(12)中、COOHとOHは隣り合う位置に結合し、R
10は、それぞれ独立して、ヒドロキシ基、カルボキシ基、炭素数1以上18以下のアルキル基、又は、炭素数1以上18以下のアルコキシ基を表し、pは0以上3以下の整数を表す。]
上記(D)の具体的方法としては、前述の式(11)で表される構造を有するビニル系モノマーを用いて不飽和基含有ポリエステルをハイブリッド化する方法を挙げることができる。
本発明において、該荷電制御樹脂のゲルパーミエーションクロマトグラフィー(GPC)により算出した重量平均分子量(Mw)は、1000以上100000以下であることが好ましい。該荷電制御樹脂の重量平均分子量(Mw)が上記範囲である場合、本発明の効果がより好適に発揮される。
すなわち、該荷電制御樹脂の重量平均分子量(Mw)が1000以上であれば、該荷電制御樹脂中の式(8)で表される構造の、樹脂間での偏在が抑制される傾向にある。一方、100000以下であれば、該荷電制御樹脂のトナー間での偏在が抑制される傾向にある。そのことで、トナーとして安定した帯電安定性が得られ、本発明の効果をより発揮することができる。
本発明において、該荷電制御樹脂の重量平均分子量(Mw)の調整方法としては公知の方法が使用可能である。例えば、以下の通りである。
該荷電制御樹脂がビニル系(共)重合体である場合、ビニル系モノマーと重合開始剤の仕込み比、重合温度などにより調整可能である。
一方、該荷電制御樹脂がポリエステル構造を有する重合体である場合、酸成分とアルコール成分の仕込み比や重合時間により任意に調整可能である。また、ハイブリッド樹脂である場合は、ポリエステル構造の分子量調整に加えて、ビニル変性ユニットの分子量の調整でも可能となる。具体的には、ビニル変性の反応工程において重合開始剤量や重合温度などにより任意に調整可能である。本発明において、ポリエステル構造のハイブリッド化に用いることのできるビニルモノマーとしては、前述したビニル系モノマーを用いることができる。
また、帯電安定性や定着性の観点から、該荷電制御樹脂の分子量分布は狭いことが好ましい。該荷電制御樹脂のゲルパーミエーションクロマトグラフィー(GPC)により算出された重量平均分子量(Mw)と数平均分子量(Mn)との比(Mw/Mn)は、1.0以上6.0以下であることが好ましく、より好ましくは1.0以上4.0以下である。
また、該荷電制御樹脂の含有量は、スチレンを含むモノマーから形成される樹脂(すなわち、結着樹脂)100質量部に対して、0.010質量部以上20.000質量部以下であることが好ましく、0.025質量部以上10.000質量部以下であることがより好ましい。
本発明に係るトナーは、ガラス転移温度が60.0℃以上100.0℃以下で酸価2.5mgKOH/g以上25.0mgKOH/g以下の極性樹脂を0.50質量%以上30.0質量%以下有していると好ましく、該極性樹脂は非晶性ポリエステルであるとより好ましい。これは、トナー粒子の表層が該極性樹脂となり、トナー粒子表層の耐熱性が向上するためである。また、該極性樹脂が非晶性ポリエステルであるとトナーの帯電性の点で優れているためである。
特に水系媒体中でトナーを製造するため、該非晶性ポリエステルの場合、含有量は0.50質量%以上20.0質量%以下が好ましく、より好ましくは0.50質量%以上15.0質量%以下、更に好ましくは1.0質量%以上10.0質量%以下であり、1.0質量%以上8.0質量以下であると特に好ましい。0.50質量%以上では該非晶性ポリエステル層が十分な厚みを持ってトナー表面全域を覆え、特に機械的特性、帯電性の点で効果が大きい。また、ワックスの内包化が十分なため現像性、耐久性に優れる。20.0質量%以下の場合、トナーの低温定着性の点で優れ、更にワックスによる迅速な離型層の形成がなされるため、耐オフセット性の点でも好ましい。更には粒径分布がシャープになり、帯電分布もシャープになること、加えて湿度のトナーへの影響が小さく、トナーの帯電安定性に優れる。
また、該非晶性ポリエステルの酸価は2.5mgKOH/g以上25.0mgKOH/g以下であると好ましく、2.5mgKOH/g以上20.0mgKOH/g以下であると更に好ましく、3.0mgKOH/g以上15.0mgKOH/g以下であるとより一層好ましく、3.0mgKOH/g以上10.0mgKOH/g以下であると特に好ましい。酸価が2.5mgKOH/g以上では該非晶性ポリエステルはトナー表面に均一な層を形成する点で好ましく、25.0mgKOH/g以下ではトナー化した際に湿度の影響を受けにくく帯電安定性の点で好ましい。
加えて、該非晶性ポリエステルはトナーと水系媒体との中間の極性を持つことでトナー粒子製造時において分散安定成分としての作用が得られるが、酸価が2.5mgKOH/g以上25.0mgKOH/g以下では、トナー表層に安定した状態で均一に分布するため効果が大きく、異形粒子発生が抑えられ、トナーの帯電分布において均一とな。
本発明のトナーにおいては該非晶質ポリエステルの酸価は該結晶性ポリエステルの酸価より高いことが好ましい。これは、非晶性ポリエステルの酸価が結晶性ポリエステルの酸価がより高い場合は該結晶性ポリエステルの大半が非晶性ポリエステルよりトナー内部に分布することになり、該結晶性ポリエステルの融点以上での重合反応工程においてトナー表層側が過疎化されにくくトナー粒子が凝集しにくいためである。
また、該荷電制御樹脂の酸価は該非晶性ポリエステルの酸価より高い方が好ましい。これは該荷電制御樹脂がトナー粒子の最表面に位置すると帯電性に最も有効に作用するためである。
<結晶性ポリエステル、非晶性ポリエステル、荷電制御樹脂やスチレン−アクリル樹脂の酸価の測定>
酸価は試料1gに含まれる酸を中和するために必要な水酸化カリウムのmg数である。本発明における酸価は、JIS K 0070−1992に準じて測定されるが、具体的には、以下の手順に従って測定する。
0.1モル/L水酸化カリウムエチルアルコール溶液(キシダ化学社製)を用いて滴定を行う。前記水酸化カリウムエチルアルコール溶液のファクターは、電位差滴定装置(京都電子工業株式会社製 電位差滴定測定装置AT−510)を用いて求めることができる。0.100モル/L塩酸100mLを250mLトールビーカーに取り、前記水酸化カリウムエチルアルコール溶液で滴定し、中和に要した前記水酸化カリウムエチルアルコール溶液の量から求める。前記0.100モル/L塩酸は、JIS K 8001−1998に準じて作成されたものを用いる。
下記に酸価測定の際の測定条件を示す。
滴定装置:電位差滴定装置AT−510(京都電子工業株式会社製)
電極:複合ガラス電極ダブルジャンクション型(京都電子工業株式会社製)
滴定装置用制御ソフトウエア:AT−WIN
滴定解析ソフト:Tview
滴定時における滴定パラメータ並びに制御パラメータは下記のように行う。
(滴定パラメータ)
滴定モード:ブランク滴定
滴定様式:全量滴定
最大滴定量:20mL
滴定前の待ち時間:30秒
滴定方向:自動
(制御パラメーラ)
終点判断電位:30dE
終点判断電位値:50dE/dmL
終点検出判断:設定しない
制御速度モード:標準
ゲイン:1
データ採取電位:4mV
データ採取滴定量:0.1mL
(本試験)
測定サンプル0.100gを250mLのトールビーカーに精秤し、トルエン/エタノール(3:1)の混合溶液150mLを加え、1時間かけて溶解する。前記電位差滴定装置を用い、前記水酸化カリウムエチルアルコール溶液を用いて滴定する。
(空試験)
試料を用いない(すなわちトルエン/エタノール(3:1)の混合溶液のみとする)以外は、上記操作と同様の滴定を行う。
得られた結果を下記式に代入して、酸価を算出する。
A=[(C−B)×f×5.61]/S
(式中、A:酸価(mgKOH/g)、B:空試験の水酸化カリウム溶液の添加量(mL)、C:本試験の水酸化カリウム溶液の添加量(mL)、f:水酸化カリウム溶液のファクター、S:試料(g)である。)
<結晶性ポリエステル、非晶性ポリエステル、荷電制御樹脂やスチレン−アクリル樹脂の水酸基価の測定>
水酸基価は、試料1gをアセチル化するとき、水酸基と結合した酢酸を中和するのに要する水酸化カリウムのmg数である。本発明における水酸基価はJIS K 0070−1992に準じて測定されるが、具体的には、以下の手順に従って測定する。
特級無水酢酸25.0gをメスフラスコ100mLに入れ、ピリジンを加えて全量を100mLにし、十分に振りまぜてアセチル化試薬を得る。得られたアセチル化試薬は、湿気、炭酸ガスなどに触れないように、褐色びんにて保存する。
1.0モル/L水酸化カリウムエチルアルコール溶液(キシダ化学社製)を用いて滴定を行う。水酸化カリウムエチルアルコール溶液のファクターは、電位差滴定装置(京都電子株式会社製 電位差滴定測定装置AT−510)を用いて求める。具体的には、1.00mol/L塩酸100mLを250mLトールビーカーに取り、水酸化カリウムエチルアルコール溶液で滴定し、中和に要した水酸化カリウムエチルアルコール溶液の量から求める。1.00mol/L塩酸は、JIS K 8001−1998に準じて作製されたものを用いる。
以下に、水酸基価測定の際の測定条件を示す。
滴定装置:電位差滴定装置AT−510(京都電子工業株式会社製)
電極:複合ガラス電極ダブルジャンクション型(京都電子工業株式会社製)
滴定装置用制御ソフトウエア:AT−WIN
滴定解析ソフト:Tview
滴定時における滴定パラメータ並びに制御パラメータは下記のように行う。
(滴定パラメータ)
滴定モード:ブランク滴定
滴定様式:全量滴定
最大滴定量:80mL
滴定前の待ち時間:30秒
滴定方向:自動
(制御パラメータ)
終点判断電位:30dE
終点判断電位値:50dE/dmL
終点検出判断:設定しない
制御速度モード:標準
ゲイン:1
データ採取電位:4mV
データ採取滴定量:0.5mL
(本試験)
測定サンプル2.00gを200mL丸底フラスコに精秤し、これに上記アセチル化試薬5.00mLを、ホールピペットを用いて正確に加える。この際、試料がアセチル化試薬に溶解しにくいときは、特級トルエンを少量加えて溶解する。
フラスコの口に小さな漏斗をのせ、97℃のグリセリン浴中にフラスコ底部1cmを浸して加熱する。このときフラスコの首の温度が浴の熱を受けて上昇するのを防ぐため、丸い穴をあけた厚紙をフラスコの首の付根にかぶせることが好ましい。
1時間後、グリセリン浴からフラスコを取り出して放冷する。放冷後、漏斗から水1.00mLを加えて振り動かして無水酢酸を加水分解する。さらに完全に加水分解するため、再びフラスコをグリセリン浴中で10分間加熱する。放冷後、エチルアルコール5.00mLで漏斗及びフラスコの壁を洗う。
得られたサンプルを250mLのトールビーカーに移し、トルエンとエタノール(3:1)の混合溶液100mLを加え、1時間かけて溶解する。電位差滴定装置を用い、水酸化カリウムエチルアルコール溶液を用いて滴定する。
(空試験)
試料を用いない(すなわち、トルエンとエタノール(3:1)の混合溶液のみとする)こと以外は、上記操作と同様の滴定を行う。
得られた結果を下記式に代入して、水酸基価を算出する。
A=[{(B−C)×28.05×f}/S]+D
ここで、A:水酸基価(mgKOH/g)、B:空試験の水酸化カリウムエチルアルコール溶液の添加量(mL)、C:本試験の水酸化カリウムエチルアルコール溶液の添加量(mL)、f:水酸化カリウムエチルアルコール溶液のファクター、S:試料(g)、D:樹脂の酸価(mgKOH/g)である。
上記非晶性ポリエステルは、重量平均分子量(Mw)が6,000以上100,000以下であることが好ましく、より好ましくは6,500以上85,000以下、更に好ましくは6,500以上45,000以下である。
該非晶性ポリエステルの重量平均分子量が6,000以上であると、連続画像出力においてトナー表面の外添剤が耐久によって埋没し難く、転写性を維持できる。重量平均分子量が100,000以下であると、重合性単量体への該非晶性ポリエステルの溶解が容易で、重合性単量体組成物の粘度上昇が生じず、粒径が小さくかつ、粒度分布の揃ったトナーが得やすくなる。
該非晶性ポリエステルは、数平均分子量(Mn)が3,000以上80,000以下であることが好ましく、より好ましくは3,500以上60,000以下、更に好ましくは3,500以上12,000以下である。該非晶性ポリエステルは、ゲルパーミエーションクロマトグラム(GPC)における分子量分布のメインピーク値(Mp)が、分子量4,500以上40,000以下の領域が好ましく、より好ましくは分子量6,000以上30,000以下の領域に、さらに好ましくは分子量6,000以上20,000以下の領域である。上記範囲内であると重量平均分子量の場合と同様の傾向を示す。
該非晶性ポリエステルはMw/Mnが1.2以上3.0以下、より好ましくは1.5以上2.5以下が良い。Mw/Mnが1.2以上の場合にはトナーの多数枚耐久性及び耐オフセット性の点で好ましく、3.0以下の場合には低温定着性の面で好ましい。
<結晶性ポリエステル、非晶性ポリエステル、荷電制御樹脂やスチレン−アクリル樹脂の分子量および分子量分布>
試料の分子量及び分子量分布は、ゲルパーミエーションクロマトグラフィー(GPC)によって、ポリスチレン換算で算出される。酸基を有する樹脂の分子量を測定する場合は、カラム溶出速度が酸基の量にも依存してしまうため、予め酸基をキャッピングした試料を用意する必要がある。キャッピングにはメチルエステル化が好ましく、市販のメチルエステル化剤が使用できる。具体的には、トリメチルシリルジアゾメタンで処理する方法が挙げられる。
GPCによる分子量の測定は、以下のようにして行う。まず、室温で24時間かけて、測定サンプルをテトラヒドロフラン(THF)に溶解する。そして、得られた溶液を、ポア径が0.2μmの耐溶剤性メンブランフィルター「マイショリディスク」(東ソー社製)で濾過してサンプル溶液を得る。尚、サンプル溶液は、THFに可溶な成分の濃度が0.8質量%となるように調整する。このサンプル溶液を用いて、以下の条件で測定する。装置:HLC8120 GPC(検出器:RI)(東ソー社製)
カラム:Shodex KF−801、802、803、804、805、806、807の7連(昭和電工社製)
溶離液:テトラヒドロフラン(THF)
流速:1.0mL/min
オーブン温度:40.0℃
試料注入量:0.10mL
測定サンプルの分子量の算出にあたっては、標準ポリスチレン樹脂(例えば、商品名「TSKスタンダード ポリスチレン F−850、F−450、F−288、F−128、F−80、F−40、F−20、F−10、F−4、F−2、F−1、A−5000、A−2500、A−1000、A−500」、東ソー社製)を用いて作成した分子量校正曲線を使用する。
<非晶性ポリエステル、スチレン−アクリル樹脂、荷電制御樹脂やトナー粒子のガラス転移温度>
試料のガラス転移温度は、示差走査熱量計(DSC測定装置)を用いて測定する。示差走査熱量計は、示差走査熱量分析装置「Q1000」(TA Instruments社製)を用い、ASTM D3418−82に準じて以下のように測定する。測定サンプルは2から5mg、好ましくは3mgを精密に秤量する。それをアルミニウム製のパン中に入れ、対照用に空のアルミパンを用いる。20℃で5分間平衡を保った後、測定範囲20乃至180℃の間で、昇温速度10℃/minで測定を行う。本発明においては、ガラス転移温度は中点法で求めることができる。
上記非晶性ポリエステルの製造方法としては、例えば、カルボン酸化合物とアルコール化合物からの脱水縮合反応を利用する方法、エステル交換反応で製造される。触媒としては、エステル化反応に使う一般の酸性、アルカリ性触媒、例えば酢酸亜鉛、チタン化合物などでよい。その後、再結晶法、蒸留法などにより高純度化させてもよい。
特に好ましい製造方法は、原料の多様性、反応のしやすさからカルボン酸化合物とアルコール化合物からの脱水縮合反応である。
縮合系樹脂としてポリエステルを用いる際のポリエステルの組成について以下に説明する。
ポリエステルは、全成分中43〜57mol%がアルコール成分であり、57〜43mol%が酸成分であることが好ましい。
本発明で用いるポリエステルを製造する上で、本発明の効果を阻害しない限り、式(1)で示されるイソソルビドもしくはイソソルビド誘導体以外にも公知のアルコール成分を用いることができる。アルコール成分としては、エチレングリコール、ネオペンチルグリコール、2−エチル−1,3−ヘキサンジオール、水素化ビスフェノールA、下記式(I)
(式中、Rはエチレン又はプロピレン基を示し、x,yはそれぞれ1以上の整数を示し、かつx+yの平均値は2〜10を示す。)
で示されるビスフェノー誘導体、又は下記式(II)
2価のカルボン酸としてはフタル酸、テレフタル酸、イソフタル酸、無水フタル酸、ジフェニル−P・P’−ジカルボン酸、ナフタレン−2,7−ジカルボン酸、ナフタレン−2,6−ジカルボン酸,ジフェニルメタン−P・P’−ジカルボン酸、ベンゾフェノン−4,4’−ジカルボン酸,1,2−ジフェノキシエタン−P・P’−ジカルボン酸の如きベンゼンジカルボン酸類又はその無水物;こはく酸、アジピン酸、セバシン酸、アゼライン酸、グリタル酸、シクロヘキサンジカルボン酸、トリエチレンジカルボン酸、マロン酸の如きアルキルジカルボン酸類又はその無水物、またさらに炭素数6〜18のアルキル基又はアルケニル基で置換されたこはく酸もしくはその無水物;フマル酸、マレイン酸、シトラコン酸、イタコン酸の如き不飽和ジカルボン酸又はその無水物が挙げられる。
特に好ましいアルコール成分としては前記式(I)で示されるビスフェノール誘導体、エチレングリコールであり、酸成分としては、テレフタル酸、又はその無水物、こはく酸、n−ドデセニルコハク酸、又はその無水物、フマル酸、マレイン酸、無水マレイン酸の如きジカルボン酸が挙げられる。特にテレフタル酸が好ましい。
該ポリエステルユニットは、2価のジカルボン酸及び2価のジオールから合成することにより得ることが可能であるが、場合により、3価以上のポリカルボン酸又はポリオールを本発明に悪影響を与えない範囲で少量使用しても良い。
3価以上のポリカルボン酸としては、トリメリット酸、ピロメリット酸、シクロヘキサントリカルボン酸類、2,5,7−ナフタレントリカルボン酸、1,2,4−ナフタレントリカルボン酸、1,2,4−ブタントリカルボン酸、1,2,5−ヘキサントリカルボン酸、1,3−ジカルボキシル−2−メチレンカルボキシルプロパン、1,3−ジカルボキシル−2−メチル−メチレンカルボキシルプロパン、テトラ(メチレンカルボキシル)メタン、1,2,7,8−オクタンテトラカルボン酸及びそれらの無水物が挙げられる。
3価以上のポリオールとしては、スルビトール、1,2,3,6−ヘキサンテトール、1,4−ソルビタン、ペンタエリスリトール、ジペンタエリスリトール、トリペンタエリスリトール、ショ糖、1,2,4−メタントリオール、グリセリン、2−メチルプロパントリオール、2−メチル−1,2,4−ブタントリオール、トリメチロールエタン、トリメチロールプロパン、1,3,5−トリヒドロキシメチルベンゼンが挙げられる。
3価以上のポリカルボン酸は全酸モノマーユニットを基準として、10.00.mol%以下であると好ましい。また同様に、3価以上のポリオールは全アルコールモノマーユニットを基準として、10.00.mol%以下であると好ましい。これは、3価以上のポリカルボン酸は全酸モノマーユニットを基準として、10.00.mol%以下であると、架橋による不溶分が少ないため顔料分散性の点で好ましい。また、不溶分を生成しないように製法を工夫した場合でも、分岐型のポリエステル樹脂の割合が少なく、強度に優れるため耐久性の点で好ましい。
非晶性ポリエステルは芳香族系飽和ポリエステルであると好ましい。これは、該トナーの帯電性、耐久性、定着性に優れ、該トナー及び該ポリエステルの物性の制御が容易であるためである。特に芳香族の有するπ電子の相互作用により帯電性に優れる。また、不飽和ポリエステルを含有していると、トナーを作製する際に不飽和部が反応し、架橋することでトナーが硬くなるため、特に定着性において劣るため好ましくない。
<非晶性ポリエステル、結晶性ポリエステル、荷電制御樹脂、スチレンアクリル樹脂およびトナーの結着樹脂の構造分析>
非晶性ポリエステル、結晶性ポリエステル、荷電制御樹脂、スチレンアクリル樹脂およびトナーの結着樹脂の構造決定は、核磁気共鳴装置(1H−NMR、13C−NMR)並びにFT−IRスペクトルを用いて行うことができる。以下に用いる装置について記す。各樹脂サンプルはトナー中から分取することで採取し、分析しても良い。
(i)1H−NMR、13C−NMR
日本電子製FT−NMR JNM−EX400(使用溶媒 重クロロホルム)
(ii)FT−IRスペクトル
Thermo Fisher Scientific Inc.製 AVATAR360FT−IR
(顔料)
本発明のトナーは、着色剤として顔料を含有する。シアン系着色剤に用いられる顔料としては、銅フタロシアニン化合物及びその誘導体、アントラキノン化合物、並びに、塩基染料レーキ化合物が利用できる。具体的には、以下のものが挙げられる。C.I.ピグメントブルー15、15:1、15:2、15:3及び15:4。
マゼンタ系着色剤に用いられる顔料としては、縮合アゾ化合物、ジケトピロロピロール化合物、アントラキノン、キナクリドン化合物、塩基染料レーキ化合物、ナフトール化合物、ベンズイミダゾロン化合物、チオインジゴ化合物及びペリレン化合物が利用できる。具体的には、以下のものが挙げられる。C.I.ピグメントバイオレット19、C.I.ピグメントレッド31、32、122、150、254、264及び269。
イエロー系着色剤に用いられる顔料としては、縮合アゾ化合物、イソインドリノン化合物、アントラキノン化合物、アゾ金属錯体、メチン化合物及びアリルアミド化合物が利用できる。具体的には、以下のものが挙げられる。C.I.ピグメントイエロー74、93、120、139、151、155、180及び185。
黒色着色剤としては、カーボンブラック、磁性体、並びに、上記イエロー系、マゼンタ系及びシアン系着色剤を用い黒色に調色されたものが利用できる。
顔料がカーボンブラック、C.I.ピグメントブルー15:3、C.I.ピグメントレッド122、C.I.ピグメントレッド150、C.I.ピグメントレッド32、C.I.ピグメントレッド269、C.I.ピグメントイエロー155、C.I.ピグメントイエロー93、C.I.ピグメントイエロー74、C.I.ピグメントイエロー180及びC.I.ピグメントイエロー185であると本発明の効果が高く好ましい。特に好ましくはカーボンブラック、C.I.ピグメントブルー15:3、C.I.ピグメントレッド122である。カーボンブラックの場合は、pHが6以上で吸油量(DBP)が30(cc/100g)以上120(cc/100g)以下であると好ましい。これは、本発明で用いられる重合開始剤が反応阻害されにくいためである。
これら顔料の添加量は、結着樹脂100質量部に対して、1質量部以上20質量部以下であることが好ましい。
(その他の添加剤)
本発明のトナーにおいては、本発明の効果を阻害しない範囲で各種特性付与を目的として公知の様々な無機、有機の添加剤を用いることが可能である。用いる添加剤は、トナーに添加した時の耐久性の点から、トナー粒子の重量平均径の3/10以下の粒径であることが好ましい。この添加剤の粒径とは、走査型電子顕微鏡におけるトナー粒子の表面観察により求めたその平均粒径を意味する。
これら添加剤の含有量は、トナー100質量部に対して、0.01質量部以上5質量部以下であることが好ましく、0.02質量部以上3質量部以下であることがより好ましい。これらの添加剤は、単独で用いても複数併用してもよい。
また、これらの添加剤は疎水化処理されていてもよい。疎水化処理の方法としては、シランカップリング剤又はチタンカップリング剤など各種カップリング剤を用いることが可能であるが、シリコーンオイルで疎水化度を高くすることが好ましい。高湿下での無機微粉体の水分吸着を抑制することができ、さらには、規制部材や帯電部材などの汚染が抑制することができるため、高品位の画像が得られるためである。
トナーの重量平均粒径(D4)としては4.0μm以上12.0μm以下であると好ましく、4.0μm以上9.0μm以下であるとより好ましい。これは重量平均粒径が4.0μm未満であるとトナーの比表面積が大きいため長期使用において耐久性や耐熱性において問題が発生しやすく、重量平均粒径が12.0μmを超える場合はトナーの着色力及び画像の解像度の点で劣るため好ましくない。
<トナー粒子の重量平均粒径(D4)、個数平均粒径(D1)、トナー粒子の凝集による粗大化>
トナー粒子の重量平均粒径(D4)および個数平均粒径(D1)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように前記専用ソフトの設定を行う。前記専用ソフトの「標準測定方法(SOM)を変更」画面において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。「閾値/ノイズレベルの測定ボタン」を押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、「測定後のアパーチャーチューブのフラッシュ」にチェックを入れる。前記専用ソフトの「パルスから粒径への変換設定」画面において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μm以上60μm以下までに設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250mL丸底ビーカーに前記電解水溶液200mLを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100mL平底ビーカーに前記電解水溶液30mLを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.3mL加える。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispension System Tetora150」(日科機バイオス社製)を準備する。超音波分散器の水槽内に3.3Lのイオン交換水を入れ、この水槽中にコンタミノンNを2mL添加する。(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行ない、重量平均粒径(D4)および個数平均粒径(D1)、体積基準メジアン径、個数基準メジアン径を算出する。尚、前記専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)であり、「中位径」が体積基準メジアン径(Dv50)である。また、前記専用ソフトでグラフ/個数%と設定したときの、「分析/個数統計値(算術平均)」画面の「平均径」が個数平均粒径(D1)であり、「中位径」が個数基準メジアン径(Dn50)である。
トナー粒子の凝集による粗大化はトナーの製造において反応温度70℃で5時間反応させた直後のトナー粒子の体積基準メジアン径(Dv50)であるDt70(μm)とその後の反応温度90℃で5時間反応させた後のトナー粒子の体積基準メジアン径(Dv50)であるDt90(μm)の差(Dt90−Dt70)(μm)として算出した。
トナー粒子のガラス転移温度としては53℃以上75℃以下が好ましい。トナー粒子のガラス転移温度が53℃以上75℃以下であると、保存性と定着性が両立するため好ましい。
トナー粒子の平均円形度としては平均円形度が0.975以上が好ましい。これはトナー粒子がトナー粒子間やトナー担持体、トナー層規制部材と均一に摩擦帯電する確率が高く、トナー粒子が受けるストレスも均一化されるためである。そのため、帯電性や、トナー層規制部材への融着の点で好ましい。
トナー粒子の微小粒子率は25.0%以下が好ましく、15.0%以下であると更に好ましい。これはトナー層規制部材などへのトナーの融着が抑制されるためである。
<トナー粒子の平均円形度と微小粒子率の測定方法>
トナー粒子の平均円形度は、フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)によって、校正作業時の測定及び解析条件で測定した。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中に予め不純固形物などを除去したイオン交換水約20mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で約3質量倍に希釈した希釈液を約0.2ml加える。更に測定試料を約0.02g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」(ヴェルヴォクリーア社製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2ml添加する。
測定には、対物レンズとして「LUCPLFLN」(倍率20倍、開口数0.40)を搭載した前記フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用した。前記手順に従い調製した分散液を前記フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて2000個のトナー粒子を計測する。そして、粒子解析時の2値化閾値を85%とし、解析粒子径を円相当径1.977μm以上39.54μm未満に限定し、トナー粒子の平均円形度を求めた。
解析粒子径を円相当径1.977μm以上39.54μm未満に限定し、1.977μm以上3.021μm以下の割合を微小粒子率とした。
測定にあたっては、測定開始前に標準ラテックス粒子(例えば、Duke Scientific社製の「RESEARCH AND TEST PARTICLES Latex Microsphere Suspensions 5100A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本願実施例では、シスメックス社による校正作業が行われた、シスメックス社が発行する校正証明書の発行を受けたフロー式粒子像分析装置を使用した。解析粒子径を円相当径1.977μm以上39.54μm未満に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行った。
〔重合性単量体〕
本発明に用いられる重合性単量体としては、スチレン以外にもラジカル重合が可能なビニル系重合性単量体が用いても良い。該ビニル系重合性単量体としては、単官能性重合性単量体或いは多官能性重合性単量体を使用することが出来る。単官能性重合性単量体としては、スチレン;α−メチルスチレン、β−メチルスチレン、ο−メチルスチレン、m−メチルスチレン、p−メチルスチレン、2,4−ジメチルスチレン、p−n−ブチルスチレン、p−tert−ブチルスチレン、p−n−ヘキシルスチレン、p−n−オクチルスチレン、p−n−ノニルスチレン、p−n−デシルスチレン、p−n−ドデシルスチレン、p−メトキシスチレン、p−フェニルスチレンの如きスチレン誘導体;メチルアクリレート、エチルアクリレート、n−プロピルアクリレート、iso−プロピルアクリレート、n−ブチルアクリレート、iso−ブチルアクリレート、tert−ブチルアクリレート、n−アミルアクリレート、n−ヘキシルアクリレート、2−エチルヘキシルアクリレート、n−オクチルアクリレート、n−ノニルアクリレート、シクロヘキシルアクリレート、ベンジルアクリレート、ジメチルフォスフェートエチルアクリレート、ジエチルフォスフェートエチルアクリレート、ジブチルフォスフェートエチルアクリレート、2−ベンゾイルオキシエチルアクリレートの如きアクリル系重合性単量体;メチルメタクリレート、エチルメタクリレート、n−プロピルメタクリレート、iso−プロピルメタクリレート、n−ブチルメタクリレート、iso−ブチルメタクリレート、tert−ブチルメタクリレート、n−アミルメタクリレート、n−ヘキシルメタクリレート、2−エチルヘキシルメタクリレート、n−オクチルメタクリレート、n−ノニルメタクリレート、ジエチルフォスフェートエチルメタクリレート、ジブチルフォスフェートエチルメタクリレートの如きメタクリル系重合性単量体;メチレン脂肪族モノカルボン酸エステル;酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、安息香酸ビニル、ギ酸ビニルの如きビニルエステル;ビニルメチルエーテル、ビニルエチルエーテル、ビニルイソブチルエーテルの如きビニルエーテル;ビニルメチルケトン、ビニルヘキシルケトン、ビニルイソプロピルケトンの如きビニルケトンが挙げられる。
多官能性重合性単量体としては、ジエチレングリコールジアクリレート、トリエチレングリコールジアクリレート、テトラエチレングリコールジアクリレート、ポリエチレングリコールジアクリレート、1,6−ヘキサンジオールジアクリレート、ネオペンチルグリコールジアクリレート、トリプロピレングリコールジアクリレート、ポリプロピレングリコールジアクリレート、2,2’−ビス(4−(アクリロキシ・ジエトキシ)フェニル)プロパン、トリメチロールプロパントリアクリレート、テトラメチロールメタンテトラアクリレート、エチレングリコールジメタクリレート、ジエチレングリコールジメタクリレート、トリエチレングリコールジメタクリレート、テトラエチレングリコールジメタクリレート、ポリエチレングリコールジメタクリレート、1,3−ブチレングリコールジメタクリレート、1,6−ヘキサンジオールジメタクリレート、ネオペンチルグリコールジメタクリレート、ポリプロピレングリコールジメタクリレート、2,2’−ビス(4−(メタクリロキシ・ジエトキシ)フェニル)プロパン、2,2’−ビス(4−(メタクリロキシ・ポリエトキシ)フェニル)プロパン、トリメチロールプロパントリメタクリレート、テトラメチロールメタンテトラメタクリレート、ジビニルベンゼン、ジビニルナフタリン、ジビニルエーテル等が挙げられる。
本発明においては、上記した単官能性重合性単量体を単独或いは、2種以上組み合わせて、又は、上記した単官能性重合性単量体と多官能性重合性単量体を組合せて使用する。
スチレン以外に用いる重合性単量体としてはスチレン誘導体、n−ブチルアクリレートや2−エチルヘキシルアクリレートなどのアクリル酸エステル系重合性単量体もしくはn−ブチルメタクリレートや2−エチルヘキシルメタクリレートなどのメタクリル酸エステル系重合性単量体が好ましい。これは重合性単量体を重合して得られる結着樹脂の強度や柔軟性の点で優れているためである。
(製造方法)
本発明のトナーの製造方法として懸濁重合を用いるが、以下の如き製造方法によって直接的にトナーを製造することが可能である。重合性単量体中にポリエステルなどの極性樹脂、離型剤、着色剤、架橋剤、その他の添加剤を加え、ホモジナイザー、超音波分散機等によって均一に溶解又は分散せしめた重合性単量体組成物を、分散安定剤を有する水系媒体中に通常の撹拌機またはホモミクサー、ホモジナイザーなどにより分散せしめる。その際、重合性単量体組成物の液滴が所望のトナーのサイズを有するように撹拌速度・時間を調整し、造粒して重合性単量体組成物の粒子を形成する。その後は、分散安定剤の作用により、粒子状態が維持され、且つ粒子の沈降が防止される程度の撹拌を行えば良い。重合開始剤を添加することで重合反応を進行させるが、重合温度は40℃以上、通常50〜120℃の温度に設定して重合を行う。重合温度が95℃以上の場合は重合反応を行う容器を加圧して水系媒体が蒸発するのを抑制しても良い。重合反応後半に昇温しても良く、必要に応じpHを変更しても良い。更に、定着時の臭いの原因となる未反応の重合性単量体、副生成物等を除去するために反応後半に反応温度を上げる、もしくは反応後半、又は、反応終了後に一部水系媒体を留去しても良い。反応終了後、生成したトナー粒子前駆体分散液を得る。次に該トナー粒子前駆体分散液を冷却、洗浄、ろ過により収集し、乾燥する。
造粒中の水系媒体中のpHは特に制約は受けないが、好ましくは、pH3.0〜13.0、更に好ましくは3.0〜7.0、特に好ましくは3.0〜6.0である。pHが3.0未満の場合は分散安定剤の一部に溶解がおこり、分散安定化が困難になり、造粒出来なくなることがある。またpHが13.0を超える場合はトナー中に添加されている成分が分解されてしまうことがあり、本発明の効果が弱くなることがある。造粒を酸性領域で行った場合には、分散安定剤に由来する金属のトナー中における含有量が過剰となるのを抑制することができ、本発明の規定を満たすようなトナーが得られやすくなる。
また、トナー粒子の洗浄をpH2.5以下、より好ましくはpH1.5以下の酸を用いて行うことが好ましい。トナー粒子の洗浄を酸で行うことにより、トナー粒子表面に存在する分散安定剤を低減することができる。洗浄に用いる酸としては、特に限定されるものではなく、塩酸、硫酸の如き無機酸を用いることができる。これによりトナー粒子の帯電性を所望の範囲に調整することも可能である。
本発明に用いられる分散安定剤としての難水溶性無機微粒子以外に有機系化合物、例えばポリビニルアルコール、ゼラチン、メチセルロース、メチルヒドロキシプロピルセルロース、エチルセルロース、カルボキシメチルセルロースのナトリウム塩、デンプンを併用しても構わない。これら分散安定剤は、重合性単量体100質量部に対して0.01質量部以上2.0質量部以下を使用することが好ましい。
さらに、これら分散安定剤の微細化のため0.001質量%以上0.1質量%以下の界面活性剤を併用しても良い。具体的には市販のノニオン、アニオン、カチオン型の界面活性剤が利用できる。例えばドデシル硫酸ナトリウム、テトラデシル硫酸ナトリウム、ペンタデシル硫酸ナトリウム、オクチル硫酸ナトリウム、テトラデシル硫酸ナトリウム、ペンタデシル硫酸ナトリウム、オクチル硫酸ナトリウム、オレイン酸ナトリウム、ラウリル酸ナトリウム、ステアリン酸カリウム、オレイン酸カルシウムが好ましく用いられる。
重合性単量体の重合転化率が50.0%以下において上述の2種類の過酸化物系重合開始剤を含有していると好ましく、該2種類の過酸化物系重合開始剤を該造粒工程において添加すると更に好ましい。これは、該重合性単量体の重合転化率が50.0%以下において該2種類の過酸化物系重合開始剤が含有されていると該重合性単量体組成物が十分残っているため本発明の作用効果を発現するのに十分な量の結着樹脂を生成できるためである。また、該2種類の過酸化物系重合開始剤を該造粒工程において添加すると該2種類の過酸化物系重合開始剤が該単量体組成物に均一に含有されやすく、生成されるトナー粒子の性能にムラが小さくなるため好ましい。
転化率50%以下の状態では反応温度を本発明のトナーに含有されるワックスや結晶性ポリエステルの融点未満にし、転化率50%超の状態では反応温度を本発明のトナーに含有されるワックスや結晶性ポリエステルの融点以上にしてトナー粒子を製造すると好ましい。これは、転化率が50%以下の状態では第2の重合開始剤によるポリマーの生成をメインとすることで定着性に優れた結着樹脂を中心に生成させる。この段階では10時間半減期温度が高い第1の重合開始剤の大半が残存している。転化率が50%超になったところで反応温度を本発明のトナーに含有されるワックスや結晶性ポリエステルの融点以上にすると第1の重合開始剤によるポリマーの生成がメインになり、トナーの耐熱性が向上する。これにより、トナー粒子の低温定着性と耐熱性、耐久性の両立がより効果的になるため好ましい。該ワックスや該結晶性ポリエステルは反応温度が融点以上となり結着樹脂にある程度相溶するため、トナー粒子の定着性は向上する。そのかわりトナー粒子の耐熱性が一般的には低下するが第1の重合開始剤により生成するポリマーがトナー粒子表層側に分布するため十分な耐熱性を維持できるため好ましい。また、重合反応後半において十分量の第1の重合開始剤による残留モノマーの低減が短時間で十分に行えるため残留モノマーの低減効果も大きく好ましい。
<重合性単量体の重合転化率の測定方法>
重合性単量体の重合転化率は、ガスクロマトグラフィー(GC)を用い、以下のようにして測定する。トナー粒子分散液約500mgを精秤しサンプルビンに入れる。これに精秤した約10gのアセトンを加えてフタをした後、よく混合し、発振周波数42kHz、電気的出力125Wの卓上型超音波洗浄器(商品名「B2510J−MTH」、ブランソン社製)にて超音波を30分間照射する。その後、ポア径が0.2μmの耐溶剤性メンブランフィルター「マイショリディスク」(東ソー社製)を用いて濾過を行い、濾液2μLをガスクロマトグラフィーで分析する。そして、予め使用した重合性単量体を用いて作成した検量線により、残留している重合性単量体の「残存量」を算出する。その後、下記式に従い、重合性単量体の重合転化率(質量%)を規定する。測定装置及び測定条件は、下記トナー粒子中の残留モノマーの定量方法と同じである。
(式) 100×(1−(重合性単量体の残存量)/(使用した重合性単量体の総量))
<トナー粒子中の残留モノマーの定量方法>
トナー中の残留スチレンモノマーなどの残留モノマーの定量は、ガスクロマトグラフィー(GC)により、以下のようにして測定する。
トナー約500mgを精秤しサンプルビンに入れる。これに精秤した約10gのアセトンを加えてフタをした後、よく混合し、発振周波数42kHz、電気的出力125Wの卓上型超音波洗浄器(例えば、商品名「B2510J−MTH」、ブランソン社製)にて超音波を30分間照射する。その後、ポア径が0.2μmの耐溶剤性メンブランフィルター「マイショリディスク」(東ソー社製)を用いて濾過を行い、濾液2μlをガスクロマトグラフィーで分析する。そして、予めスチレンなど各種モノマーを用いて作成した検量線により、残留モノマーの残存量を算出する。
測定装置及び測定条件は、下記の通りである。
GC:HP社 6890GC
カラム:HP社 INNOWax(200μm×0.40μm×25m)
キャリアーガス:He(コンスタントプレッシャーモード:20psi)
オーブン:(1)50℃で10分ホールド、(2)10℃/分で200℃まで昇温、(3)200℃で5分ホールド
注入口:200℃、パルスドスプリットレスモード(20→40psi、until0.5分)
スプリット比:5.0:1.0
検出器:250℃(FID)
(製造装置)
以下、本発明における製造装置について説明する。本発明では、公知のものが使用できるが、造粒工程における撹拌手段の一例としては、パドル翼、傾斜パドル翼、三枚後退翼、アンカー翼、フルゾーン翼(神鋼パンテック社製)、マックスブレンド(住友重機社製)、スーパーミックス(佐竹化学機械工業社製)、Hi−Fミキサー(綜研化学社製)等の撹拌翼を有するものを用いることができる。他にも、高剪断力を付与できる撹拌機がより好ましい。高剪断撹拌機としては、高速回転する撹拌ロータと該撹拌ロータを囲うように設けられたスクリーンとによって形成される撹拌室を備えているものが好ましく用いられる。具体的には、ウルトラタラックス(IKA社製)、ポリトロン(キネマティカ社製)、T.K.ホモミクサー(特殊機化工業社製)、クレアミックス(エムテクニック社製)、Wモーション(エムテクニック社製)、キャビトロン(ユーロテック社製)、シャープフローミル(太平洋機工社製)等が用いられる。
上記重合性単量体組成物は、結晶性物質を含有しており、反応終了後、生成したトナー粒子前駆体分散液を得る。次に該トナー粒子前駆体分散液を任意の温度まで冷却する冷却工程が存在しており、該冷却工程における該トナー粒子前駆体分散液の冷却速度が0.3℃/sec以上であると好ましく、1.5℃/sec以上であると、より好ましい。
これは、反応終了後、生成したトナー粒子前駆体分散液を、急激に冷却することで該トナー粒子前駆体に含有される該結晶性物質のうち、結着樹脂中に相溶している成分が微細な結晶として析出する。その際、本発明のトナー粒子の場合、トナー粒子表層側には分子量の大きいポリマーが分布しているため、析出する結晶性物質は相溶しにくく、トナー粒子最表面近傍は結晶性物質の存在量は少なくなるため現像性に優れる。そのため、該結晶性物質の多くはトナー粒子最表面より内側に微細な結晶状態で析出する。この結晶性物質はトナー粒子最表面近傍での存在量は少ないが、微細な結晶で均一に分散する割合が増加するため、定着時には結晶性物質が迅速に溶融するため定着性にも優れるため好ましい。冷却速度は1.5℃/sec以上であると、より結着樹脂中に一様に結晶性物質を存在させることができるため、低温定着性がより良好となる。
水系媒体の温度を急速に冷却する手段としては、例えば冷水や氷を混合する操作や、冷風により水系媒体をバブリングする操作、熱交換器を用いて水系媒体の熱を除去する操作等を用いることが可能である。
該冷却工程における該トナー粒子前駆体分散液の冷却開始温度は、該結晶性物質の結晶化温度Tc(℃)と該トナー粒子のガラス転移温度Tg(℃)とのうちの高い方の温度以上(好ましくは、冷却開始温度は結晶性物質の結晶化温度Tc(℃)とトナー粒子のガラス転移温度Tg(℃)とのうちの高いほうの温度より5℃以上、より好ましくは10℃以上)であり、該トナー粒子前駆体分散液の冷却終了温度は該ガラス転移温度Tg(℃)以下(より好ましくは、Tg−3℃以下)であると好ましい。これは、トナー粒子を結晶性物質の結晶化温度Tc及びトナー粒子のガラス転移温度Tgより高い温度にした場合、該結晶性物質はトナー粒子中により均一に溶融する。ここで、急速に冷却した場合、その状態を維持したまま、トナー粒子中に結晶性物質が微細な結晶としてトナー粒子表面近傍以外の領域において均一に析出する。冷却終了温度を該ガラス転移温度Tg(℃)以下にすることにより、前述の状態を維持し続けることができるため、より一層、現像性と定着性の両方が優れるため好ましい。
該冷却工程における該トナー粒子前駆体分散液の冷却開始温度は、該結晶性物質の結晶化温度Tc(℃)と該トナー粒子のガラス転移温度Tg(℃)とのうちの高い方の温度以上とする場合、結晶性物質の結晶化温度Tc以上の温度で、結着樹脂及び結晶性物質を共に溶融させている。そのため、結晶性物質の融点Tmは、50℃以上90℃以下である保存性と定着性の点で好ましい。該結晶性物質の重量平均分子量Mwは600以上であると上記作用効果が大きいため好ましい。
該冷却工程を経た該トナー粒子前駆体分散液を、Tg−10℃以上Tg+10℃以下の温度で30分以上保持する保持工程を有していると更に好ましい。好ましい保持時間は90分間以上であり、さらに好ましい時間は120分間以上である。一方、該保持時間の上限値は、その効果が飽和する1440分間程度である。
急速に冷却した後、該トナー粒子のガラス転移温度の±10℃の間で30分以上保持することで、微細に析出した結晶性物質の結晶成長を促すことにより、トナー粒子内部において、結晶性物質の多数の微細な核が成長したことにより、結着樹脂中に相溶する結晶性物質の割合がより一層減少するため現像性と定着性の点で好ましい。
この工程において、トナー粒子のガラス転移温度Tg(℃)は、トナーのガラス転移温度Tg(℃)を用いても良い。
該乾燥工程が、減圧度0.67kPa以上6.7kPa以下の減圧容器内で行われると好ましい。0.67kPa以上6.7kPa以下の圧力であれば、該トナー粒子前駆体中に含まれる有機揮発物質を良好に除去できる。また、一般的には0.67kPa以下の圧力にする場合、真空容器のシールの精度を上げなくてはならないばかりか、真空ポンプの能力を大きくしなくてはならない等、装置コストが過剰となり生産性を大きく低下させることがある。また、圧力が著しく小さいことにより、該トナー粒子前駆体に含まれる離型剤等、結晶性物質が表面に析出することがある。こうした粒子をトナーとして使用すると、カートリッジの構成部材を汚染するため、結果として現像性を低下させることがある。
しかしながら、本発明のトナーの場合は、トナー表層近傍に分子量の大きいポリマーが分布しており、トナー表面近傍に相溶している結晶性物質が少ないため、該トナー粒子前駆体に含まれる離型剤等、結晶性物質が析出しにくく望ましい。なお、6.7kPaより大きい場合、該トナー粒子前駆体中の有機揮発物質が除去できず残存することがある。
該減圧乾燥工程における該トナー粒子前駆体の温度を20℃以上60℃以下にすると好ましい。この範囲であれば、有機揮発物質が揮発しやすく、該トナー粒子前駆体から除かれ易くなる。また、トナーの耐熱性や結晶性物質の染み出しが抑制されるため好ましい。
該減圧乾燥工程後の該トナー粒子前駆体の有機揮発物質の濃度が10ppm以下であると好ましい。これは、トナー粒子前駆体中に残存する有機揮発物質が少ないため、該トナー粒子前駆体に無機微粒子を外添などした際に該トナー粒子前駆体に熱が加わっても無機微粒子がトナー粒子表面に付着し易いためである。
<結晶性ポリエステルおよびワックスの融点Tm(℃)の測定>
結晶性ポリエステルおよびワックスの結晶化温度Tc(℃)及び融点Tm(℃)は、示差走査熱量分析装置「Q1000」(TA Instruments社製)を用いてASTM D3418−82に準じて測定する。
装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。具体的には、トナー粒子約10mgを精秤し、これをアルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30〜200℃の間で、昇温速度10℃/minで測定を行う。尚、測定においては、一度200℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程での温度30〜200℃の範囲におけるDSC曲線の最大の吸熱ピークとなる温度を、結晶性ポリエステルおよびワックスの融点Tm(℃)とする。
以下、具体的な製造方法、実施例、比較例をもって本発明をさらに詳細に説明するが、これは本発明を何ら限定するものではない。なお、以下の配合における部数は全て質量部である。
〔結晶性ポリエステル製造例1〕
減圧装置、水分離装置、窒素ガス導入装置、温度測定装置、撹拌装置を備えたオートクレープ中に、
・1,10−デカンジカルボン酸:210部
・1,10−デカンジカルボン酸ジメチル:50部
・1,10−デカンジオール:295部
・シュウ酸チタン酸カリウム:0.40部
上記ポリエステルモノマーを仕込み、窒素雰囲気下、常圧下で200℃で6時間反応を行い、その後更に10〜20mmHgの減圧下で220℃で1.5時間反応してポリエステル1を得た。得られたポリエステルの物性を表3に示す。
〔結晶性ポリエステル製造例2〜8〕
表2の原材料モノマー仕込み量および重縮合反応の温度条件にて、結晶性ポリエステル1と同様の操作を行い、結晶性ポリエステル2〜8を製造した。得られた樹脂の物性を表3に示す。各結晶性ポリエステルの分子量と酸価の調整に関しては適宜反応時間を調整してポリエステルの物性が達成されるようにした。
〔非晶性ポリエステル1の製造〕
減圧装置、水分離装置、窒素ガス導入装置、温度測定装置、撹拌装置を備えたオートクレープ中に、
・テレフタレート:29.9部
・ビスフェノールA−プロピレンオキサイド2モル付加物:51.7部
・エチレングリコール:4.5部
・テトラブトキシチタネート:0.125部
上記ポリエステルモノマーを仕込み、窒素雰囲気下、常圧下で200℃で5時間反応を行い、その後トリメリット酸を2.1部及びテトラブトキシチタネートを0.120部追加し、220℃で3時間反応させ、更に10〜20mmHgの減圧下で2時間反応して非晶性ポリエステル1を得た。得られた非晶性ポリエステルの物性は表5に示す。得られた非晶性ポリエステルの組成は表4に記載の仕込み量通りの組成であった。
〔非晶性ポリエステル2〜8の製造〕
表4の原材料モノマー仕込み量および重縮合反応の温度条件や反応時間にて、非晶性ポリエステル1と同様の操作を行い、非晶性ポリエステル2〜8を製造した。得られた樹脂の物性を表5に示す。各非晶性ポリエステルの分子量と酸価の調整に関しては適宜反応時間を調整して非晶性ポリエステルの物性が達成されるようにした。
〔スチレン−アクリル樹脂1の製造〕
プロピレングリコールモノメチルエーテル100部を窒素置換しながら加熱し液温120℃以上で還流させ、そこへ、下記材料を混合したものを3時間かけて滴下した。
・スチレン:91.7部
・メチルメタクリレート:2.50部
・メタクリル酸:3.30部
・2−ヒドロキシエチルアクリレート:2.50部
・ジtert−ブチルパーオキサイド(日油(株)製、商品名「パーブチルD」):1.25部
滴下終了後、溶液を3時間撹拌した後、液温170℃まで昇温しながら常圧蒸留し、液温170℃到達後は1hPaで減圧下1時間蒸留して脱溶剤し、樹脂固形物を得た。該固形物をテトラヒドロフランに溶解し、n−ヘキサンで再沈殿、析出した固体を濾別することでスチレン−アクリル樹脂1を得た。得られたスチレン−アクリル樹脂1の物性は以下の通りであった。
Mw=15000、酸価=20.0、水酸基価=10.0、Tg(中点法)=90.0℃
〔スルホン酸基を有する樹脂1の製造〕(CCR−A)
撹拌機、コンデンサー、温度計、窒素導入管の付いた2Lフラスコにトルエン100部、メタノール350部、スチレン470部、2−アクリルアミド−2−メチルプロパンスルホン酸40部、アクリル酸−2−エチルヘキシル70部、メタクリル酸ベンジル20部、ラウリルパーオキサイド10部を仕込み、撹拌、窒素導入下65℃で10時間溶液重合した。得られた内容物をフラスコから取り出し、イソプロピルアルコールで洗浄後、40℃で96時間減圧乾燥を行った。その後、ハンマーミルにて粗砕し、該粗砕物を更に40℃で48時間減圧乾燥し、スルホン酸基を有する樹脂1(CCR−A)を得た。得られた樹脂の物性は、Mw=24000、Tg=67.0℃、残留モノマー=350ppmであった。
なお、得られたスルホン酸基を有する樹脂1(CCR−A)の酸価は、20.0mgKOH/gであった。スチレン−ヘキサン溶解性指数の値は12であった。
〔スルホン酸基を有する樹脂2の製造〕(CCR−B)
撹拌機、コンデンサー、温度計、窒素導入管の付いた2Lフラスコにトルエン100部、メタノール350部、スチレン470部、2−アクリルアミド−2−メチルプロパンスルホン酸27部、アクリル酸−2−エチルヘキシル70部、メタクリル酸ベンジル20部、ラウリルパーオキサイド10部を仕込み、撹拌、窒素導入下65℃で10時間溶液重合した。得られた内容物をフラスコから取り出し、イソプロピルアルコールで洗浄後、40℃で96時間減圧乾燥を行った。その後、ハンマーミルにて粗砕し、該粗砕物を更に40℃で48時間減圧乾燥し、スルホン酸基を有する樹脂2(CCR−B)を得た。得られた樹脂の物性は、Mw=24000、Tg=66.5℃、残留モノマー=350ppmであった。
なお、得られたスルホン酸基を有する樹脂2(CCR−B)の酸価は、15.0mgKOH/gであった。スチレン−ヘキサン溶解性指数の値は15であった。
〔スルホン酸基を有する樹脂3の製造〕(CCR−C)
撹拌機、コンデンサー、温度計、窒素導入管の付いた2Lフラスコにトルエン100部、メタノール350部、スチレン470部、2−アクリルアミド−2−メチルプロパンスルホン酸52部、アクリル酸−2−エチルヘキシル70部、メタクリル酸ベンジル20部、ラウリルパーオキサイド10部を仕込み、撹拌、窒素導入下65℃で10時間溶液重合した。得られた内容物をフラスコから取り出し、イソプロピルアルコールで洗浄後、40℃で96時間減圧乾燥を行った。その後、ハンマーミルにて粗砕し、該粗砕物を更に40℃で48時間減圧乾燥し、スルホン酸基を有する樹脂3(CCR−C)を得た。得られた樹脂の物性は、Mw=24000、Tg=67.6℃、残留モノマー=350ppmであった。
なお、得られたスルホン酸基を有する樹脂3(CCR−C)の酸価は、25.0mgKOH/gであった。スチレン−ヘキサン溶解性指数の値は9であった。
〔スルホン酸基を有する樹脂4の合成〕(CCR−D)
撹拌機、コンデンサー、温度計、及び窒素導入管の付いた2Lフラスコに、トルエン100部、メタノール350部、スチレン470部、2−アクリルアミド−2−メチルプロパンスルホン酸5部、アクリル酸−2−エチルヘキシル20部、メタクリル酸ベンジル30部、及びラウリルパーオキサイド10部を仕込み、撹拌、窒素導入下65℃で10時間溶液重合した。得られた内容物をフラスコから取り出し、イソプロピルアルコールで洗浄後、40℃で96時間減圧乾燥を行った。その後、ハンマーミルにて粗砕し、該粗砕物をさらに40℃で48時間減圧乾燥し、スルホン酸基を有する樹脂4(CCR−D)を得た。得られた樹脂の物性は、Mw=24000、Tg=72.8℃、残存モノマー=350ppmであった。
なお、得られたスルホン酸基を有する樹脂4(CCR−D)の酸価は、7mgKOH/gで、スチレン−ヘキサン溶解性指数の値は40であった。
〔スルホン酸基を有する樹脂5の合成〕(CCR−E)
撹拌機、コンデンサー、温度計、及び窒素導入管の付いた2Lフラスコに、トルエン100部、メタノール350部、スチレン470部、2−アクリルアミド−2−メチルプロパンスルホン酸5部、アクリル酸−2−エチルヘキシル10部、メタクリル酸ベンジル40部、及びラウリルパーオキサイド10部を仕込み、撹拌、窒素導入下65℃で10時間溶液重合した。得られた内容物をフラスコから取り出し、イソプロピルアルコールで洗浄後、40℃で96時間減圧乾燥を行った。その後、ハンマーミルにて粗砕し、粗砕物をさらに40℃で48時間減圧乾燥し、スルホン酸基を有する樹脂5(CCR−E)を得た。得られた樹脂の物性は、Mw=28000、Tg=73.9℃、残存モノマー=350ppmであった。
なお、得られたスルホン酸基を有する樹脂5(CCR−E)の酸価は、7mgKOH/gで、スチレン−ヘキサン溶解性指数の値は45であった。
[モノマー(式(11))の合成例]
<モノマーC−1の合成例>
2,4−ジヒドロキシ安息香酸18gをメタノール150mLに溶解した。この溶解液に炭酸カリウム36.9gを加えて65℃に加熱した。4−(クロロメチル)スチレン18.7gとメタノール100mLを混合溶解させた溶解液を作製し、これをサリチル酸中間体が入った溶解液に滴下し、65℃にて3時間反応させた。得られた反応液を冷却してから、濾過し、濾液中のメタノールを減圧留去して析出物を得た。析出物をpH=2の水1.50Lに分散させ、酢酸エチルを加えて抽出した。その後、水洗してから、硫酸マグネシウムで乾燥させ、減圧下、酢酸エチルを留去することにより析出物を得た。析出物をヘキサン洗浄してから、トルエン/酢酸エチルにて再結晶し、下記式(13)で示される構造を有するモノマーC−1を20.1g得た。
<モノマーC−2の合成例>
(工程1)
2,5−ジヒドロキシ安息香酸100gと80%硫酸1441gとを50℃に加熱しながら混合し、この混合液にtert−ブチルアルコール144gを加えて50℃で30分間撹拌した。次に、混合液にtert−ブチルアルコール144gを加え50℃で30分間撹拌する操作を3回行った。反応液を室温まで冷却してから、氷水1.00kgに徐々に注ぎ、析出物を濾過した。析出物を水洗し、さらにヘキサンにより洗浄した。ここで得られた析出物をメタノール200mLに溶解させ、水3.60Lに再沈殿させた。濾過後、80℃にて乾燥させることで下記式(14)に示すサリチル酸中間体を74.9g得た。
(工程2)
上記サリチル酸中間体 25.0gをメタノール150mLに溶解した。この溶解液に炭酸カリウム36.9gを加えて65℃に加熱した。4−(クロロメチル)スチレン18.7gとメタノール100mLに混合溶解させた溶解液を作製し、これをサリチル酸中間体が入った溶解液に滴下し、65℃にて3時間反応させた。得られた反応液を冷却してから、濾過し、濾液中のメタノールを減圧留去して析出物を得た。析出物をpH=2の水1.50Lに分散させ、酢酸エチルを加えて抽出した。その後、水洗してから、硫酸マグネシウムで乾燥させ、減圧下、酢酸エチルを留去することにより析出物を得た。析出物をヘキサン洗浄してから、トルエン/酢酸エチルにて再結晶し、下記式(15)で示される構造を有するモノマーC−2を20.1g得た。
<モノマーC−3の合成例>
2,5−ジヒドロキシ安息香酸100.0gをメタノール2Lに溶解させ、炭酸カリウム88.3gを加えて67℃に加熱した。この溶解液に4−(クロロメチル)スチレン102.0gを22分間かけて滴下し、67℃にて12時間反応させた。得られた反応液を冷却し、メタノールを減圧留去し、ヘキサンで洗浄した。残渣をメタノールに溶解させ水に滴下し、再沈澱させ、析出物をろ過した。この再沈澱操作を2回繰り返し、残渣を80℃で乾燥させ、下記式(16)で示される構造を有するモノマーC−3を得た。
<モノマーC−4の合成例>
tert−ブチルアルコール144gを2−オクタノール253gに変更すること以外は、モノマーC−2の合成(工程1)と同じ方法で、サリチル酸中間体を得た。ここで得られたサリチル酸中間体32gを用いること以外は、モノマーC−2の合成(工程2)と同じ方法で、下記式(17)で示される構造を有するモノマーC−4を得た。
<モノマーC−5の合成例>
式(14)のサリチル酸中間体を2,5−ジヒドロキシ−3−メトキシ安息香酸22gに変更すること以外は、モノマーC−2の合成(工程2)と同じ方法で、下記式(18)で示される構造を有するモノマーC−5を得た。
<モノマーC−6の合成例>
式(14)のサリチル酸中間体を2,3−ジヒドロキシ安息香酸18gに変更すること以外は、モノマーC−2の合成(工程2)と同じ方法で、下記式(19)で示される構造を有する化合物C−6を得た。
<モノマーC−7の合成例>
4−(クロロメチル)スチレンを、3−(クロロメチル)メチルスチレンと4−(クロロメチル)スチレンの混合物(AGCセイケミカル社製、商品名「CMS−P」)に変更すること以外は、モノマーC−2の合成(工程2)と同じ方法で、下記式(20)で示される構造を有するモノマーの混合物を得た。この混合物をモノマーC−7とする。
[荷電制御樹脂Bの合成例]
<荷電制御樹脂B−1の合成>
式(13)に示すモノマーC−1 12.00gとスチレン88.00gをN,N−ジメチルホルムアミド(DMF)40.00mLに溶解させ、1時間撹拌した後110℃まで加熱した。この反応液に、tert−ブチルパーオキシイソプピルモノカルボネート(日本油脂株式会社製、商品名パーブチルI)3.50gをトルエン40.00mLに仕込んだ溶液を1時間撹拌して得られた溶解液を滴下した。窒素導入下、さらに110℃にて4時間反応した。その後、冷却しメタノール1.00Lに滴下し、析出物を得た。得られた析出物をTHF120mLに溶解後、メタノール1.80Lに滴下し、白色析出物を析出させ、濾過し、減圧下90℃にて乾燥させることで、モノマーC−1とスチレンとから得られた荷電制御樹脂B−1を得た。
得られた荷電制御樹脂B−1の組成分析は、前述の1H−NMRを用いて行い、モノマーC−1が重合されていることを確認した。また、荷電制御樹脂B−1の酸価は24.8mgKOH/gであり、酸価からモノマーC−1に由来する式(8)で表される構造を442μmol/g含有していることが確認された。荷電制御樹脂Bの仕込み量と組成分析を行った結果を表6に示す。また、得られた樹脂の酸価及び式(8)で表される構造のモル含有量も表6に示す。
<荷電制御樹脂B−2〜B−12の合成>
式(8)で表される構造を含むモノマーの種類及び仕込み量、ビニル系モノマーの種類及び仕込み量、重合開始剤の添加量、並びに、反応条件を表6に示すように変更すること以外は、荷電制御樹脂B−1の合成と同様の操作を行い、荷電制御樹脂B−2〜B−12を製造した。得られた樹脂の物性を表6に示す。
〔疎水性シリカ1の製造〕
シリカ(AEROSIL 200CF、日本アエロジル製)100部をヘキサメチルジシラザン10部で処理し、さらにジメチルシリコーンオイル20部で処理して疎水性シリカ1を得た。疎水性シリカ1の一次粒子の個数平均径は12nm、疎水化度は97であった。
〔疎水性酸化チタン1の製造〕
酸化チタン(P25、日本アエロジル製)100部をトルエン中でγ−メルカプトプロピルトリメトキシシラン20部で処理し、濾過、乾燥して疎水性酸化チタン1を得た。疎水性酸化チタン1の一次粒子の個数平均径は25nm、疎水化度は60であった。
(トナー製造例1)
反応容器中のイオン交換水1000部に、リン酸ナトリウム14部ならびに10%塩酸を4.5部投入し、N2パージしながら65℃で60分保温した。T.K.ホモミクサー(特殊機化工業製)を用いて、12000rpmにて撹拌しながら、イオン交換水10部に7.8部の塩化カルシウムを溶解した塩化カルシウム水溶液を一括投入し、分散安定剤を含む水系媒体を調製した。
・スチレン(分子量:104) 60部
・カーボンブラック(Orion Engineerred Carbons社製、商品名「Printex35」) 7部
・荷電制御剤(オリエント社製:ボントロンE−89) 0.25部
上記材料をアトライタ分散機(三井三池化工機株式会社)に投入し、さらに直径1.7mmのジルコニア粒子を用いて、220rpmで5時間分散させて、重合性単量体組成物Aを得た。
上記重合性単量体組成物Aに、
・スチレン(分子量:104) 20部
・n−ブチルアクリレート(分子量:128) 20部
・結晶性ポリエステル2 5部
・非晶性ポリエステル3 5部
・フィッシャートロプシュワックス
(シューマンサゾール社製、商品名「C80」:融点83.0℃) 9部
を加えた。
別容器中で上記材料を65℃に保温し、T.K.ホモミクサー(特殊機化工業製)を用いて、500rpmにて均一に溶解、分散した。これに、重合開始剤t−ヘキシルパーオキシピバレート(日油株式会社製、商品名「パーヘキシルPV」、分子量:202、10時間半減期温度:53.2℃)18.69部(重合性単量体の合計[スチレンとn−ブチルアクリレート]に対して10mol%)と重合開始剤t−ヘキシルパーオキシイソプロピルモノカルボネート(日油株式会社製、商品名「パーヘキシルI」、分子量:204、10時間半減期温度:95.0℃)5.66部(重合性単量体の合計[スチレンとn−ブチルアクリレート]に対して3mol%)とを溶解し、重合性単量体組成物Bを調製した。
造粒タンク中の上記水系媒体中に、2種類の重合開始剤が添加された上記重合性単量体組成物Bを投入し、65℃、N2パージ下において、T.K.ホモミクサーにて10000rpmで5分間撹拌し、pH5.2で造粒した。その後、重合タンクに移して、パドル撹拌翼で30回/分で撹拌しつつ70℃で6時間(転化率は90%であった)、さらに90℃に昇温し、2時間反応させた。
重合反応終了後、反応容器を冷却し、10%塩酸を加えpH=2とした状態で2時間撹拌しながら分散安定剤を溶解させた。そのエマルションを加圧濾過しさらに2000部以上のイオン交換水で洗浄した。得られたケーキを再び、1000部のイオン交換水に戻し、10%塩酸を加えpH=1以下とした状態で2時間撹拌しながら、再洗浄した。上記と同様にそのエマルションを加圧濾過し、さらに2000部以上のイオン交換水で洗浄し、充分通気をした後、乾燥して風力分級し、ブラック着色粒子(トナー粒子1)を得た。
得られたブラック着色粒子100部と、疎水性シリカ1を1.5部、及び疎水性酸化チタン1を0.3部加え、三井ヘンシェルミキサ(三井三池化工機株式会社製)で混合し、外添剤を有するトナー1を得た。得られたトナー1の物性等については表9に記載した。
(トナー製造例2〜52、55、59〜62、121〜130)
表7および8に記載される通りに各原料の種類及び含有量、重合開始剤の添加時期を変更させた以外はトナー製造例1と同様にして、外添剤を有するトナー2〜52、55、59〜62、121〜130を製造した。得られたトナー2〜52、55、59〜62、121〜130の物性等を表9に示す。
なお、これらのトナー製造例で使用の表7に記載の重合開始剤の分子量と10時間半減期温度は以下の通りである。
分子量 10時間半減期温度(℃)
パーブチルA(日油社製): 132 101.9
パーブチルE(日油社製): 246 99.0
パーブチルI(日油社製): 176 98.7
パーブチルO(日油社製): 216 72.1
ルぺロックス80(アルケマ吉富社製): 160 82.0
ルぺロックス580(アルケマ吉富社製):173 74.0
パーブチルIB(日油社製): 160 77.3
パーブチル355(日油社製): 230 97.1
パーブチルL(日油社製): 272 98.3
パーオクタO(日油社製): 272 65.3
パーヘキシルZ(日油社製): 222 99.4
パーブチルND(日油社製): 244 46.4
パーブチルPV(日油社製): 174 54.6
パーオクタND(日油社製): 301 40.7
パーヘキシルND(日油社製): 272 44.5
(トナー製造例53)
非晶性ポリエステル3をスチレン−アクリル樹脂1に変更した以外はトナー製造例1と同様にして、外添剤を有するトナー53を製造した。得られたトナー53の物性等を表9に示す。
(トナー製造例54)
非晶性ポリエステル3をポリメタクリル酸エステルマクロモノマー(東亜合成化学工業社製、商品名「AA−6」、Tg:94℃)に変更した以外はトナー製造例1と同様にして、外添剤を有するトナー54を製造した。得られたトナー54の物性等を表9に示す。
(トナー製造例56)
フィッシャートロプシュワックスをカルナバワックス(融点:83℃)に変更した以外はトナー製造例1と同様にして、外添剤を有するトナー56を製造した。得られたトナー56の物性等を表9に示す。
(トナー製造例57)
フィッシャートロプシュワックスをジペンタエリスリトールヘキサステアレート(融点:77℃)に変更した以外はトナー製造例1と同様にして、外添剤を有するトナー57を製造した。得られたトナー57の物性等を表9に示す。
(トナー製造例58)
造粒工程終了後、重合タンクに移して、パドル撹拌翼で30回/分で撹拌しつつ85℃で6時間、さらに90℃に昇温し、2時間反応させた以外はトナー製造例1と同様にして、外添剤を有するトナー58を製造した。得られたトナー58の物性等を表9に示す。
(トナー製造例63)
反応容器中のイオン交換水1000部に、リン酸ナトリウム14部ならびに10%塩酸を4.5部投入し、N2パージしながら65℃で60分保温した。T.K.ホモミクサー(特殊機化工業製)を用いて、12000rpmにて撹拌しながら、イオン交換水10部に7.8部の塩化カルシウムを溶解した塩化カルシウム水溶液を一括投入し、分散安定剤を含む水系媒体を調製した。
・スチレン(分子量:104) 60部
・カーボンブラック(Orion Engineerred Carbons社製、商品名「Printex35」) 7部
・荷電制御剤(オリエント社製:ボントロンE−89) 0.25部
上記材料をアトライタ分散機(三井三池化工機株式会社)に投入し、さらに直径1.7mmのジルコニア粒子を用いて、220rpmで5時間分散させて、重合性単量体組成物Aを得た。
上記重合性単量体組成物Aに、
・スチレン(分子量:104) 20部
・n−ブチルアクリレート(分子量:128) 20部
・非晶性ポリエステル3 5部
・スルホン酸基を有する樹脂1
0.3部
・フィッシャートロプシュワックス
(シューマンサゾール社製、商品名「C80」:融点83.0℃) 9部
を加えた。
別容器中で上記材料を65℃に保温し、T.K.ホモミクサー(特殊機化工業製)を用いて、500rpmにて均一に溶解、分散した。これに、重合開始剤t−ヘキシルパーオキシピバレート(日油株式会社製、商品名「パーヘキシルPV」、分子量:202、10時間半減期温度:53.2℃)18.69部(重合性単量体の合計[スチレンとn−ブチルアクリレート]に対して10mol%)と重合開始剤t−ヘキシルパーオキシイソプロピルモノカルボネート(日油株式会社製、商品名「パーヘキシルI」、分子量:204、10時間半減期温度:95.0℃)5.66部(重合性単量体の合計[スチレンとn−ブチルアクリレート]に対して3mol%)とを溶解し、重合性単量体組成物Bを調製した。
造粒タンク中の上記水系媒体中に、2種類の重合開始剤が添加された上記重合性単量体組成物Bを投入し、65℃、N2パージ下において、T.K.ホモミクサーにて10000rpmで5分間撹拌し、pH5.2で造粒した。その後、その後、重合タンクに移して、パドル撹拌翼で30回/分で撹拌しつつ70℃で6時間(転化率は90%であった)、さらに90℃に昇温し、2時間反応させた。
重合反応終了後、反応容器を冷却し、10%塩酸を加えpH=2とした状態で2時間撹拌しながら分散安定剤を溶解させる。そのエマルションを加圧濾過しさらに2000部以上のイオン交換水で洗浄する。得られたケーキを再び、1000部のイオン交換水に戻し、10%塩酸を加えpH=1以下とした状態で2時間撹拌しながら、再洗浄する。上記と同様にそのエマルションを加圧濾過し、さらに2000部以上のイオン交換水で洗浄し、充分通気をした後、乾燥して風力分級し、ブラック着色粒子(トナー粒子63)を得た。
得られたブラック着色粒子100部と、疎水性シリカ1を1.5部、及び疎水性酸化チタン1を0.3部加え、三井ヘンシェルミキサ(三井三池化工機株式会社製)で混合し、外添剤を有するトナー63を得た。得られたトナー63の物性等については表9に記載した。
(トナー製造例64〜82)
表7および8に記載される通りに各原料の種類及び含有量、撹拌装置の種類や条件を変更させた以外はトナー製造例1と同様にして、外添剤を有するトナー64〜82を製造した。得られたトナー64〜82の物性等を表9に示す。
(トナー製造例83)
トナー製造例55における、濾別されたトナー粒子前駆体を乾燥する乾燥工程において、該トナー粒子前駆体30kgをレーディゲミキサー VT−130(中央機工社製)に入れ、真空容器内を0.67kPaし、3時間処理を行った。処理中は、ベッカーショベル(撹拌翼)を80rpmで回転させ、該トナー粒子前駆体を撹拌した。該トナー粒子前駆体の温度は、真空容器に具備されたジャケットに冷水または温水を入れることで40℃に調節した以外はトナー製造例55と同様にして、外添剤を有するトナー83を製造した。得られたトナー83の物性等を表9に示す。
(トナー製造例84〜91)
濾別された該トナー粒子前駆体を乾燥する乾燥工程において、表7および8に記載される通りに乾燥条件を変更した以外はトナー製造例83と同様にして、外添剤を有するトナー84〜91を製造した。得られたトナー84〜91の物性等を表9に示す。
(トナー製造例92)
重合開始剤t−ヘキシルパーオキシイソプロピルモノカルボネート(日油株式会社製、商品名「パーヘキシルI」、分子量:204、10時間半減期温度:95.0℃)5.66部を重合開始剤t−ブチルパーオキシ−2−エチルヘキシルモノカルボネート(日油株式会社製、商品名「パーブチルE」、分子量:246、10時間半減期温度:95.0℃)6.83部に変更した以外はトナー製造例83と同様にして外添剤を有するトナー92を製造した。得られたトナー92の物性等を表9に示す。
(トナー製造例93)
重合開始剤t−ヘキシルパーオキシイソプロピルモノカルボネート(日油株式会社製、商品名「パーヘキシルI」、分子量:204、10時間半減期温度:95.0℃)5.66部を重合開始剤t−アミルパーオキシイソブチレート(アルケマ吉富社製、商品名「ルペロックス580T75」、分子量:173、10時間半減期温度:74.0℃)4.80部に変更した以外はトナー製造例83と同様にして外添剤を有するトナー93を製造した。得られたトナー93の物性等を表9に示す。
(トナー製造例94)
重合開始剤t−ヘキシルパーオキシイソプロピルモノカルボネート(日油株式会社製、商品名「パーヘキシルI」、分子量:204、10時間半減期温度:95.0℃)5.66部を重合開始剤t−ヘキシルパーオキシアセテート(日油株式会社製、商品名「パーブチルA」、分子量:132、10時間半減期温度:101.9℃)3.66部に変更した以外はトナー製造例83と同様にして外添剤を有するトナー94を製造した。得られたトナー94の物性等を表9に示す。
(トナー製造例95)
表7および8に記載のトナー製造例55と各原料の種類及び含有量、撹拌装置の種類や条件を同様にして、90℃で2時間反応さる重合反応まで行った。
重合反応終了後、冷却工程を行った。パドル撹拌翼で30回/分で撹拌し86℃まで冷却した。この時の冷却速度Aは、0.017℃/secであった。86℃のトナー粒子前駆体分散液に5℃の水を混合し冷却速度Bを5℃/secで61℃まで冷却した。その後、61℃で120分温度を保持した後、5℃の水を混合し冷却速度Cを5℃/secで30℃まで冷却した。ここで、冷却速度Aは、本発明の冷却開始温度まで冷却した時の冷却速度である。冷却速度Bは、本発明の冷却開始温度からトナー粒子前駆体分散液の温度を保持するまでの冷却速度である。冷却速度Cはトナー粒子前駆体分散液の温度を保持した後、再度冷却する際の冷却速度である。
以降、トナー製造例55と同様にして外添剤を有するトナー95を製造した。得られたトナー95の物性等を表9に示す。
(トナー製造例96〜120)
冷却工程や温度保持条件を表8に記載の条件に変更した以外はトナー製造例95と同様にしてトナー96〜120を製造した。得られたトナー96〜120の物性等を表9に示す。
(*)1:ポリエステル未使用、スチレン−アクリル樹脂1を使用(トナー製造例53)
(*)2:ポリエステル未使用、ポリメタクリル酸エステルマクロモノマー(東亜合成化学工業社製、商品名「AA−6」、Tg:94℃)を使用(トナー製造例54)
(注)トナー製造例1〜61、63〜130において、反応温度を70℃から90℃に昇温を開始したタイミングでは重合性単量体の転化率は50%超であった。
[実施例1〜120]
トナー1〜120をそれぞれ評価機を用いて下記の各種画像評価を行った。評価結果は表18に示す。
[比較例1〜10]
トナー121〜130をそれぞれ評価機を用いて下記の各種画像評価を行った。評価結果は表18に示す。
<濡れ性変化率>
本発明においては、結晶性物質のトナー表面への染み出しの程度を測る指標として、被処理トナー粒子等の濡れ性を用いるが、下記のようにして得たメタノール滴下透過率曲線から求める。
まず、メタノール60体積%と水40体積%とからなる含水メタノール液70mlを、直径5cm、厚さ1.75mmの円筒型ガラス容器中に入れ、その測定用サンプル中の気泡等を除去するために超音波分散器で5分間分散を行う。
次いで、トナーを目開き150μmのメッシュで振るい、メッシュを通ったトナー0.1gを精秤して、上記含水メタノール液が入れられた容器の中に添加し、測定用サンプル液を調製する。そして、測定用サンプル液を粉体濡れ性試験機「WET−100P」(レスカ社製)にセットする。この測定用サンプル液を、マグネティックスターラーを用いて、6.7s−1(400rpm)の速度で撹拌する。尚、マグネティックスターラーの回転子として、フッ素樹脂コーティングされた、長さ25mm、最大胴径8mmの紡錘型回転子を用いる。
次に、この測定用サンプル液中に、上記装置を通して、メタノールを1.3ml/minの滴下速度で連続的に添加しながら波長780nmの光で透過率を測定し、図3に示したようなメタノール滴下透過率曲線を作成する。メタノール滴下透過率曲線の透過率50%でのメタノールの体積濃度を濡れ性半値とした。
トナー粒子の濡れ性半値をI、該トナー粒子を55℃雰囲気下に8時間放置した後の被処理トナー粒子の濡れ性半値をEとし、下記式に基づき濡れ性変化率 M(%)を算出する。
濡れ性変化率 M(%)=(E−I)/I×100
トナー粒子中の離型剤といった結晶性物質が被処理トナー粒子表面に析出してきたことが示唆される。
<カブリ>
カブリの測定は、画像形成装置として後述の評価機を用い、下記の環境下で印字率1%にて2枚印刷する度に1分休止する方式で耐久試験を行い、初期から耐久13000枚印字後に各環境下において6日間放置した。
その後の1枚目の画像サンプルのカブリ量を東京電色社製のREFLECT METER MODELTC−6DSを使用して測定し、下記式より算出した。耐久試験に用いた記録材としてはA4サイズの普通紙(キヤノンマーケティングジャパン社製、GF−C081A4)を用いた。
カブリ量(%)=(プリントアウト前の記録材の白色度)−(プリント後の記録材の非画像形成部(白地部)の白色度)
<定着性>
低温低湿環境下(L/L:温度10℃、湿度10%RH)で、後述の評価機を用い、マシン及びトナーを充填したカートリッジが環境になじんだ状態(該環境下に24時間放置後)から電源を入れた。ウェイトアップ直後に200μm幅の横線パターン(横幅200μm、間隔200μm)をプリントアウトし、50枚目のプリント画像を定着性の評価に用いた。定着性の評価は画像をシルボン紙で5往復100g荷重でこすり、画像のはがれを反射濃度の低下率(%)の平均で評価した。
評価に用いる転写材としてはは表面平滑度10〔sec〕以下のボンド紙を用いた。以下に評価基準を示す。
<トナー担持体およびトナー層規制部材へのトナーの融着や固着>
トナー担持体およびトナー層規制部材へのトナーの融着や固着は常温常湿環境下(N/N:温度23.5℃,湿度60%RH)、高温高湿環境下(H/H:温度32.5℃,湿度80%RH)で、後述の評価機を用いて評価した。印字率1%にて2枚印刷する度に1分休止する方式で耐久試験を行い、初期から耐久8000枚目の画像サンプルについて目視にて評価した。記録材として、A4サイズの普通紙(キヤノンマーケティングジャパン社製、GF−C081A4)を用いた。以下に評価基準を示す。
<トナーの保存安定性評価>
トナーの保存安定性評価は、10gのトナーを100mLの樹脂製カップに量り取り、50℃および55℃の恒温層の中へ3日間放置した後、200メッシュ(目開き)の篩性により評価した。測定装置として、デジタル振動計(DEGITAL VIBLATIONMETERMODEL 1332 SHOWA SOKKI CORPORATION製)を有するパウダーテスター(細川ミクロン社製)を用いた。
測定法としては、セットした200メッシュふるい(目開き75μm)上に評価用のトナーのせ、デジタル振動計の変位の値を0.50mm(peak−to−peak)になるように調整し、30秒間振動を加えた。その後、各ふるい上に残ったトナーの凝集塊の状態から保存安定性を評価した。以下に評価基準を示す。
<初期画像濃度>
初期画像濃度は、高温高湿環境下(H/H:温度32.5℃,湿度80%RH)、特別高温高湿環境下(SH/H:温度35.0℃,湿度80%RH)で、後述の評価機を用い、紙上のトナーの載り量が0.38(mg/cm2)にした全面ベタチャートを1枚印字し、各画像の画像濃度を測定した。画像サンプルの濃度については東京電色社製のREFLECT METER MODELTC−6DSを使用して濃度を測定した。記録材としてはA4サイズの普通紙(キヤノンマーケティングジャパン社製、GF−C081A4)を用いた。
<画像濃度低下>
画像濃度低下は、低温定湿環境下(SL/L:温度10.0℃,湿度10%RH)、特別高温高湿環境下(SH/H:温度35.0℃,湿度80%RH)で、後述の評価機を用い、まず紙上のトナーの載り量が0.38(mg/cm2)にした全面ベタチャートを1枚印字し、その後印字率1%にて2枚印刷する度に1分休止する方式で耐久試験を行い、初期から耐久8000枚印字後に、再度紙上のトナーの載り量が0.38(mg/cm2)にした全面ベタチャートを1枚印字し、各画像の画像濃度を測定し、初期画像と比較して耐久後の画像濃度の低下率を算出した。画像サンプルの濃度については東京電色社製のREFLECT METER MODELTC−6DSを使用して濃度を測定した。記録材としてはA4サイズの普通紙(キヤノンマーケティングジャパン社製、GF−C081A4)を用いた。
<耐オフセット性>
耐オフセット性は、高温高湿環境下(H/H:温度32.5℃,湿度80%RH)および特別高温高湿環境下(SH/H:温度35.0℃,湿度80%RH)で、後述の評価機を用いて評価した。マシン及びトナーを充填したカートリッジが環境になじんだ状態(該環境下に24時間放置後)から電源を入れ、ウェイトアップ直後に全面ベタ画像を100枚プリントアウトし、その画像サンプルについて評価を行った。
評価にはOHPフィルム(CG3700、住友スリーエム株式会社製)を用いた。以下に評価基準を示す。
<潜像担持体へのフィルミング>
潜像担持体へのフィルミングは苛酷環境下(SH/SH:温度40.0℃,湿度95%RH)で、トナーを30日放置した後、高温高湿環境下(H/H:温度32.5℃,湿度80%RH)および特別高温高湿環境下(SH/H:温度35.0℃,湿度80%RH)において後述の評価機を用い、印字率1%にて連続印字にて耐久試験を行った。初期から耐久2000枚目の画像サンプルについて目視にて評価した。記録材として、A4サイズの普通紙(キヤノンマーケティングジャパン社製、GF−C081A4)を用いた。以下に評価基準を示す。
(評価機)
市販のLBP−2710(キヤノン株式会社製)のプロセススピードを220mm/sに改造し、市販のマゼンタカートリッジからトナーを抜き取り、エアーブローにて内部を清掃した。その後、評価するトナーを260g充填し、マゼンタ用のステーションに装着した。シアン、イエロー及びブラックのカートリッジは、トナーを抜き取り、空の状態として、各ステーションに装着した。