JP5634252B2 - トナー及び二成分系現像剤 - Google Patents
トナー及び二成分系現像剤 Download PDFInfo
- Publication number
- JP5634252B2 JP5634252B2 JP2010286026A JP2010286026A JP5634252B2 JP 5634252 B2 JP5634252 B2 JP 5634252B2 JP 2010286026 A JP2010286026 A JP 2010286026A JP 2010286026 A JP2010286026 A JP 2010286026A JP 5634252 B2 JP5634252 B2 JP 5634252B2
- Authority
- JP
- Japan
- Prior art keywords
- toner
- peak intensity
- wax
- mass
- absorption peak
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images

Description
特に、高画質を要求されるフルカラー複写機またはフルカラープリンタ等のフルカラー画像形成装置では、後者が好適に用いられている。
フルカラー画像形成装置は、近年POD用途やグラフィック用途に用いられている。このような分野では従来以上の高速・高画質が要求されている。高画質としては、画像濃度の安定性と画像の面内のグロス均一性が求められている。画像濃度の安定性を確保するためには、トナーの帯電性が環境(特に湿度)に影響されにくいことが必要になってくる。また、高速印刷を達成するために、トナーが低温で定着する低温定着性も同時に要求されている。そこで、低温定着性を達成しつつ、高湿環境における帯電安定性確保するために、層状複水酸化物の塩を使用したトナーの提案がなされている(特許文献1、特許文献2)。
これらのトナーは、帯電安定性は達成できているものの、画像の面内のグロス均一性は不十分であり、多数枚印刷時に画像濃度が低下する場合があった。
また、トナーの耐久性改善の目的でトナー粒子の表面から0.3μmまでの深さ領域に存在するワックス比率を規定したトナーの提案がなされている(特許文献3)。
上記提案のトナーは、トナーの耐久安定性は改善されていたが、グロスの均一性の悪化が生じる場合があり、また、画像濃度も安定しなかった。
このようにいずれの提案においても、低温定着性、耐久安定性を達成しつつ、高湿環境における画像濃度の安定性、画像のグロスの均一性を改善したトナー及び二成分系現像剤は得られていない。
すなわち、本発明は、ポリエステル樹脂、着色剤、層状複水酸化物塩及びワックスを少なくとも含有するトナー粒子を有するトナーであって、前記層状複水酸化物塩は、有機アニオン、並びに、Mg2+及びAl3+を少なくとも含有し、前記トナーは、下記式(1)の関係を満たすことを特徴とするトナー。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[式(1)中、i)P1=Pa/Pbであり、ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られた該トナーのFT−IRスペクトルにおいて、2843cm −1 以上2853cm −1 以下の範囲の吸収ピーク強度の最大値から3050cm −1 と2600cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pa”であり、1713cm −1 以上1723cm −1 以下の範囲の吸収ピーク強度の最大値から1763cm −1 と1630cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pb”であり、ii)P2=Pc/Pdであり、ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られた該トナーのFT−IRスペクトルにおいて、2843cm −1 以上2853cm −1 以下の範囲の吸収ピーク強度の最大値から3050cm −1 と2600cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pc”であり、 1713cm −1 以上1723cm −1 以下の範囲の吸収ピーク強度の最大値から1763cm −1 と1630cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pd”である。]
本発明のトナーは、ポリエステル樹脂、着色剤、層状複水酸化物塩及びワックスを少なくとも含有するトナー粒子を有するトナーであって、かつ、トナー表面からトナー中心部に向かうトナーの深さ方向におけるワックスの偏在度合いをコントロールしたトナーである。本発明のトナーは当該構成をとることで、耐久安定性と画像の面内のグロス均一性(以下単にグロス均一性ともいう)を両立することができる。
上記のように、本発明のトナーは、層状複水酸化物塩を含有する。
層状複水酸化物塩とは、一般式[M2+ 1−xM3+ x(OH)2][An− x/n・yH2O]で表される化合物である(式中、M2+は、Mg、Mn、Fe、Co、Ni、Cu、Ca、Li、及びZnなどの二価金属イオンを表し、M3+は、Al、Cr、Fe、Co、及びInなどの三価金属イオンを表し、An−は、Cl−、NO3 −、CO3 2−、及びカルボン酸などのn価のアニオン、又は後述する有機アニオンを表す。)。
本発明における層状複水酸化物塩は、上記M2+として、Mg2+、上記M3+として、Al3+を含み、上記An−として、有機アニオンを含有する。
この層状複水酸化物塩を用いることで、高湿環境下における画像濃度安定性を達成することができる。その理由は明確ではないが、筆者らは以下のように推察している。
本発明のトナーは、グロス均一性を向上させるためにワックスを含有している。しかし、含有しているワックスが過度にトナー表面に偏析すると、耐久時等にトナーに外添剤が埋め込まれ画像濃度が低下する場合がある。それに対し、層状複水酸化物塩をトナーに用いると、複水酸化物の層間にワックスの一部が取り込まれる。その結果、トナー表面への
過度のワックスの偏析が抑制される。
それと同時に、層状複水酸化物塩中に存在するMg2+及びAl3+が、トナー中のポリエステル樹脂と部分的に金属架橋を形成する。上記、ワックスの偏析抑制と、金属架橋構造形成という2つの作用により、トナーの耐久性能が向上し、多数枚印刷時の画像濃度の低下を抑制することができる。
有機アニオンとしては、安息香酸、ナフトエ酸、4−t−ブチル安息香酸、フタル酸、イソフタル酸、スルホイソフタル酸、ベンゼンスルホン酸、p−トルエンスルホン酸、p−ドデシルベンゼンスルホン酸、ベンゼンジスルホン酸、ナフタレンスルホン酸、2,2’−ジチオ安息香酸、t−ブチルサリチル酸、ジ−t−ブチルサリチル酸、3−ピロリン−2−カルボン酸、N−(4−カルボキシフェニル)ピロール、ピロール−2−カルボン酸、ピロール−3−カルボン酸、イミダゾール−1−カルボン酸、イミダゾール−2−カルボン酸、イミダゾール−4−カルボン酸、イミダゾール−4,5−ジカルボン酸、2−イミダゾリジノン−4−カルボン酸、ピラゾール−4−カルボン酸、ピラゾール−3,5−ジカルボン酸、ピリジン−2−カルボン酸、ピリジン−3−カルボン酸、ピリジン−4−カルボン酸、ピリジン−2,3−ジカルボン酸、ピリジン−2,4−ジカルボン酸、ピリジン−2,5−ジカルボン酸、ピリジン−2,6−ジカルボン酸、ピリジン−3,4−ジカルボン酸、ピリジン−3,5−ジカルボン酸、ピリミジン−2−カルボン酸、ピリミジン−2,4−ジカルボン酸、ピリダジン−3,6−ジカルボン酸、ピリダジン−3−カルボン酸、ピラジン−2−カルボン酸、ピラジン−2,3−ジカルボン酸、2−ピロリジノン−5−カルボン酸、ピペリジン−2−カルボン酸、ピペリジン−3−カルボン酸、ピペリジン−4−カルボン酸、ピペラジン−2−カルボン酸、ピペラジン−2,3−ジカルボン酸が挙げられる。
層状複水酸化物塩における無機アニオンを有機アニオンへ置換することで、高い帯電安定性を達成できる。その理由は明確ではないが、筆者らは以下のように推察している。有機アニオンが存在することで、そこが帯電サイトとなり高い帯電性を示す。それと同時に、無機アニオンの場合は吸湿性に富むために高湿環境下で帯電性が低下してしまうが、有機アニオンの場合吸湿性が低く、高湿環境下での帯電性の低下を抑制できる。
層状複水酸化物塩の使用量は、結着樹脂100質量部に対して、好ましくは0.1質量部以上10.0質量部以下であり、より好ましくは0.5質量部以上5.0質量部以下である。層状複水酸化物塩の使用量が、0.1質量部未満の場合、高湿環境下での帯電性低下の抑制が低下する傾向にあり、10.0質量部より多い場合、低温定着性を低下させる傾向にある。
しかしながら、ワックスは分子量が結着樹脂に比べ小さく軟らかいために、トナー中にワックスが多く存在するとトナー表面に外添剤が埋め込まれやすくなる。その結果、トナーとキャリア間の付着力が増加し、トナーがキャリアから像担持体へ現像されにくくなり、耐久時の画像濃度が低下してしまうことがある。
そこで、発明者らは鋭意検討し、トナー表面から約1.0μmまでの間における結着樹脂に対するワックスの存在比率をコントロールすることで、上記耐久時に現像性が低下することによる画像濃度の低下を抑制できることを見出した。
そのメカニズムは明確ではないが、発明者らは以下のように推察している。
外添剤の埋め込みは、トナー表層だけではなく、その下層のやわらかさも関与している。例えば、トナーの最表層のワックス比率が高かったとしても、その下層が固い樹脂の層で構成されていれば、外添剤はその機能を失うほどには埋め込まれない。この外添剤の埋め込みに関与するのがトナー表層から約1.0μmの範囲であり、この範囲に存在するワックス比率をコントロールすることで、耐久時の画像濃度の低下を抑制できる。このため、トナー表面からトナー中心部に向かうトナーの深さ方向において、トナー表面から約0.3μmの間における結着樹脂に対するワックスの存在比率を、約1.0μmの間における結着樹脂に対するワックスの存在比率より多くすることが重要である。
また、当該構成を採用することで、トナー表面に多く存在するワックスが、定着時においてトナー中からのワックスの染み出しをさらに促進する。これは、定着時に定着部材と接することでトナー表面が加熱され、トナー表面に存在するワックスが溶解する。この溶解したワックスは、定着部材とトナーの間に離型性を有するワックスの膜を形成する。この時に、トナー表層の溶解したワックスが、トナー内部からのワックスの染み出しを促進し、離型性をより高められるため、グロス均一性が向上する。
一方、トナー表面にワックスを偏析させないトナーの場合、ワックスを多量に添加しなくてはならず、その結果、トナーの耐久性が低下する場合がある。
さらに、トナー表面よりも内側のワックス比率をトナー表面のワックス比率より少なくすることで、耐久性もさらに向上する。これは、トナー表層はワックス比率が多いため、外添剤が埋め込まれやすい。それに対し、トナー表面の内側はトナー表層に比べワックス比率が少ないために、外添剤が埋め込まれにくい。このため、ワックス比率が高いトナー表層からワックス比率が低いトナー内側にいくにしたがって、外添剤の埋め込みが抑制される。
上記理由から、トナー表面から約0.3μmの間における結着樹脂に対するワックスの存在比率[A]、及び、約1.0μmの間における結着樹脂に対するワックスの存在比率[B]を調整し、当該[A]の[B]に対する比([A]/[B])を制御することで、初めて耐久性とグロスの均一性を確保できる。
ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られたFT−IRスペクトルにおいて、2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度をPc、1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度をPdとしたときに、下記式(1)の関係を満たしていることを特徴とする。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[該式(1)において、該最大吸収ピーク強度Paは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、該最大吸収ピーク強度Pbは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、該最大吸収ピーク強度Pcは、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引いた値であり、該最大吸収ピーク強度Pdは、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引いた値であり、P1=Pa/Pb、P2=Pc/Pdである。]
このため、1713cm−1以上1723cm−1以下の範囲の吸収ピークは、主に結
着樹脂由来の−CO―の伸縮振動に起因するピークである。
結着樹脂由来のピークとしては、上記以外にも芳香環のCHの面外変角振動等様々なピークが検出されるが、1500cm−1以下の範囲には、ピークが数多く存在し、結着樹脂のピークだけを分離することが困難であり、正確な数値を算出できない。このため、他のピークとの分離が容易な1713cm−1以上1723cm−1以下の範囲の吸収ピークを結着樹脂由来のピークとして用いる。
また、2843cm−1以上2853cm−1以下の範囲の吸収ピークは、主にワックス由来の−CH2−の伸縮振動(対称)に起因するピークである。
ワックスのピークとしては、上記以外にも1450cm−1以上1500cm−1以下にCH2の面内変角振動のピークが検出されるが、結着樹脂由来のピークとも重なり合ってしまい、ワックスのピークを分離することが困難である。このため、他のピークとの分離が容易な2843cm−1以上2853cm−1以下の範囲の吸収ピークをワックス由来のピークとして用いる。
本発明において、上記結着樹脂由来の最大吸収ピーク強度(Pb、Pd)及びワックス由来の最大吸収ピーク強度(Pa、Pc)が、結着樹脂及びワックスの存在量に相関することを見出した。そこで、ワックス由来の最大吸収ピーク強度を結着樹脂由来の最大吸収ピーク強度で割ることで、結着樹脂に対するワックスの存在比率を算出している。
ここで、Pa及びPcを求めるに当たり、2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値から3050cm−1と2600cm−1の吸収ピーク強度の平均値を差し引く理由は、ベースラインの影響を排除し、真のピーク強度を算出するためである。3050cm−1と2600cm−1付近には吸収ピークがないため、この2点の平均値を算出することで、ベースライン強度を算出できる。
一方、Pb及びPdを求めるに当たり、1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値から1763cm−1と1630cm−1の吸収ピーク強度の平均値を差し引く理由も、ベースラインの影響を排除し、真のピーク強度を算出するためである。3050cm−1と2600cm−1付近には吸収ピークがないため、この2点の平均値を算出することで、ベースライン強度を算出できる。
先に述べたとおり、P1の値をコントロールすることで、グロス均一性を改善することができる。具体的には、P1は0.20以上1.50以下であることが好ましく、より好ましくは、0.30以上1.20以下である。
P1は、ワックスの種類及び結着樹脂に対するワックスの添加量の調整、トナーの製造工程における熱風を用いたトナーの改質処理の実施により、上記範囲に制御することが可能である。これらの具体例は後述する。
ることができる。具体的には、P2は0.10以上0.70以下であることが好ましく、より好ましくは、0.20以上0.60以下である。
P2は、ワックスの結着樹脂に対する添加量を調整することで上記範囲に制御することが可能である。
当該ワックスの添加量は、結着樹脂100質量部に対して、3.0質量部以上20.0質量部以下であり、好ましくは3.5質量部以上16.0質量部以下である。
P1/P2が2.00より大きい場合、トナーの表面近傍に偏在するワックス比率が高すぎる。このようなトナーの場合、表面近傍に偏在するワックスが多すぎるために、外添剤がトナー表面に埋め込まれやすくなる。その結果、トナーとキャリア間の付着力が増加し、トナーがキャリアから像担持体へ現像されにくくなり、耐久時の画像濃度が低下する。一方、P1/P2が1.05未満のトナーは、トナーの内部に偏在するワックス比率が高く、耐久時の外添剤の埋め込みを抑制することが出来ないことに加えて、グロス均一性が顕著に低下する。
P1/P2は、ワックスの種類及び結着樹脂に対するワックスの添加量の調整、層状複水酸化物塩の添加、トナーの製造工程における熱風を用いたトナーの改質処理の実施により、上記範囲に制御することが可能である。
層状複水酸化物塩を添加することにより、P1/P2を制御できる理由は明確ではないが筆者らは以下のように推察している。層状複水酸化物塩は、層状構造を有している。この層間に毛細管現象によりワックスの一部が保持され、ワックスの偏在状態をコントロールすることができるものと考えている。
なかでも、炭化水素系ワックス、特に脂肪族炭化水素系ワックスが層状複水酸化物塩と併用したときに、ワックスの偏在状態のコントロールでき、好ましい。
本発明に用いられるポリエステル樹脂はアルコールモノマーとカルボン酸モノマーが縮重合したものが用いられる。アルコールモノマーとしては以下のものが挙げられる。
ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシプロピレン(3.3)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシエチレン(2.0)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシプロピレン(2.0)−ポリオキシエチレン(2.0)−2,2−ビス(4−ヒドロキシフェニル)プロパン、ポリオキシプロピレン(6)−2,2−ビス(4−ヒドロキシフェニル)プロパン等のビスフェノールAのアルキレンオキシド付加物、エチレングリコール、ジエチレングリコール、トリエチレングリコール、1,2−プロピレングリ
コール、1,3−プロピレングリコール、1,4−ブタンジオール、ネオペンチルグリコール、1,4−ブテンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,4−シクロヘキサンジメタノール、ジプロピレングリコール、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコール、ビスフェノールA、水素添加ビスフェノールA、ソルビトール、1,2,3,6−ヘキサンテトロール、1,4−ソルビタン、ペンタエリスリトール、ジペンタエリスリトール、トリペンタエリスリトール、1,2,4−ブタントリオール、1,2,5−ペンタントリオール、グリセロール、2−メチルプロパントリオール、2−メチル−1,2,4−ブタントリオール、トリメチロールエタン、トリメチロールプロパン、1,3,5−トリヒドロキシメチルベンゼン。
一方、カルボン酸モノマーとしては、以下のものが挙げられる。
フタル酸、イソフタル酸及びテレフタル酸の如き芳香族ジカルボン酸類又はその無水物;コハク酸、アジピン酸、セバシン酸及びアゼライン酸の如きアルキルジカルボン酸類又はその無水物;炭素数6〜18のアルキル基又はアルケニル基で置換されたコハク酸もしくはその無水物;フマル酸、マレイン酸及びシトラコン酸の如き不飽和ジカルボン酸類又はその無水物。
また、その他のモノマーとしては、以下のものが挙げられる。
グリセリン、ソルビット、ソルビタン、さらには例えばノボラック型フェノール樹脂のオキシアルキレンエーテル等の多価アルコール類;トリメリット酸、ピロメリット酸、ベンゾフェノンテトラカルボン酸やその無水物等の多価カルボン酸類。
それらの中でも、特に、下記一般式(1)で表されるビスフェノール誘導体を2価アルコールモノマー成分とし、2価以上のカルボン酸又はその酸無水物、又はその低級アルキルエステルとからなるカルボン酸成分(例えば、フマル酸、マレイン酸、無水マレイン酸、フタル酸、テレフタル酸、トリメリット酸、ピロメリット酸等)を酸モノマー成分として、これらのポリエステルユニット成分で縮重合した樹脂が良好な帯電特性を有するので好ましい。

そのメカニズムは明確ではないが、筆者らは以下のように推察している。
まず、トナーの耐久性を高めるためには少なくとも分子量が10000以上の高分子成分が樹脂中に存在することが好ましいことを筆者らは見出した。すなわち、層状複水酸化物塩のAl3+やMg2+と高分子量の樹脂が、ストレスに強い架橋構造を形成し、耐久性が向上する。それに対し、分子量が10000未満の場合、分子量が小さいためにストレスに強い架橋構造を形成しにくく、耐久性が低下する傾向にある。また、該層状複水酸化物塩は吸湿性が低いために、このようにして形成された架橋構造は高湿環境下でも耐久性を維持することが可能である。
それと同時に、低温定着性を確保するためには、少なくともシャープメルト性の高い分
子量が5000以下の低分子量成分が樹脂中に存在することが好ましい。
このため、トナーのTHF可溶分中に分子量10000以上の高分子量成分を20%以上含むことが好ましく、30%以上含むことがより好ましい。分子量5000以下の低分子量成分を30%以上含むことが好ましい。このような比率にすることは、トナーの低温定着性を達成しつつ、耐久性を両立させることに貢献する。
黒色着色剤としては、カーボンブラック;磁性体;イエロー着色剤とマゼンタ着色剤及びシアン着色剤とを用いて黒色に調色したものが挙げられるが、中でもカーボンブラックが好ましい。
カーボンブラックとしては、BET比表面積が20m2/g以上300m2/g以下のものが好ましい。BET比表面積が20m2/g未満の場合、カーボンブラックの粒子の影響で画像状に小さな凹凸ができ、その結果、グロスが低下する場合がある。BET比表面積が300m2/gより大きい場合、カーボンの分散が難しく、画像濃度の低下を生じる場合がある。
また、本発明において層状複水酸化物塩は該カーボンブラックと併用したときに特に帯電性が高まる。その理由は明確ではないが、筆者らは以下のように推察している。
通常、カーボンブラックを着色剤として用いた場合、カーボンが導電性であるため、電荷がリークし、帯電量が低下しやすい。特に、高湿環境下でこの現象が顕著である。しかしながら、本発明の層状複水酸化物塩は、吸湿性が低く、かつ、カーボン近傍に分散する性質があるため、カーボンの周囲に絶縁体として分散する。その結果、特に高湿環境下で電荷のリークを抑制し、トナーを安定的に帯電させることができる。
着色剤の使用量は、結着樹脂100質量部に対して、好ましくは0.1質量部以上30質量部以下であり、より好ましくは0.5質量部以上20質量部以下である。
外添剤としては、個数平均粒径が5nm以上300nm以下の外添剤を用いることが好ましいが、耐ストレス性を向上させるためには、80nm以上300nm以下の外添剤を用いることがより好ましい。個数平均粒径が80nm以上300nm以下の外添剤は、耐久時等にトナー表面に埋め込まれたとしても、粒径が大きいために流動性及び転写性の低下を低減することができる。このため、耐久時の画像濃度の低下を抑制できる。
外添剤は、トナー粒子100質量部に対して0.1質量部以上5.0質量部以下使用されることが好ましい。トナー粒子と外添剤との混合は、ヘンシェルミキサーの如き公知の混合機を用いることができる。
ここでは、粉砕法を用いたトナー製造の手順について説明する。
原料混合工程では、トナー粒子を構成する材料として、ポリエステル樹脂、着色剤、層状複水酸化物塩及びワックス、並びに必要に応じて、荷電制御剤等の他の成分を所定量秤量して配合し、混合する。混合装置の一例としては、ダブルコン・ミキサー、V型ミキサー、ドラム型ミキサー、スーパーミキサー、ヘンシェルミキサー、ナウタミキサ、メカノハイブリッド(日本コークス工業社製)が挙げられる。
次に、混合した材料を溶融混練して、ポリエステル樹脂中にワックス等を分散させる。
その溶融混練工程では、加圧ニーダー、バンバリィミキサーの如きバッチ式練り機や、連続式の練り機を用いることができる。連続生産できる優位性から、1軸又は2軸押出機が主流となっている。例えば、KTK型2軸押出機(神戸製鋼所社製)、TEM型2軸押出機(東芝機械社製)、PCM混練機(池貝鉄工製)、2軸押出機(ケイ・シー・ケイ社製)、コ・ニーダー(ブス社製)、ニーデックス(日本コークス工業社製)が挙げられる。
更に、溶融混練することによって得られる樹脂組成物は、2本ロール等で圧延され、冷却工程で水などによって冷却してもよい。
ついで、樹脂組成物の冷却物は、粉砕工程で所望の粒径にまで粉砕される。粉砕工程では、クラッシャー、ハンマーミル、フェザーミルの如き粉砕機で粗粉砕した後、更に、クリプトロンシステム(川崎重工業社製)、スーパーローター(日清エンジニアリング社製)、ターボ・ミル(ターボ工業製)やエアージェット方式による微粉砕機で微粉砕する。
その後、必要に応じて慣性分級方式のエルボジェット(日鉄鉱業社製)、遠心力分級方式のターボプレックス(ホソカワミクロン社製)、TSPセパレータ(ホソカワミクロン社製)、ファカルティ(ホソカワミクロン社製)の如き分級機や篩分機を用いて分級し、トナー粒子を得る。
本発明においては、粉砕後、または、分級後に、熱風によりトナー粒子の表面処理を行うことが、トナー表面近傍のワックス比率をコントロールし、耐久性能と画像の面内のグロス均一性を両立させるために、好ましい。
上記熱風を用いた表面処理としては、熱風でトナー粒子の表面を溶融状態にすることができ、かつ、熱風で処理されたトナー粒子を冷風で冷却できる方式を採用できる手段であればどのようなものでもかまわない。例えば、メテオレインボー MR Type(日本ニューマチック社製)などを用いることが可能である。
また、本発明においては、上記微粉砕物を得た後、熱風により表面処理を行い、続いて分級をすることにより得られたトナー粒子であることが好ましい。若しくは、予め分級したものを、熱風により表面処理を行っても良い。
ここで、上記熱風を用いた表面処理の方法の概略を、図1を用いて説明するが、これに限定されるものではない。図1は本発明で用いた表面処理装置の一例を示した断面図である。具体的には、上記微粉砕物(ここでは、トナー粒子ともいう)を得た後、当該表面処理装置に供給する。そして、トナー粒子供給口(100)から供給されたトナー粒子(114)は、高圧エア供給ノズル(115)から噴射されるインジェクションエアにより加速され、その下方にある気流噴射部材(102)へ向かう。気流噴射部材(102)からは拡散エアが噴射され、この拡散エアによりトナー粒子が外側方向へ拡散する。この時、インジェクションエアの流量と拡散エアの流量とを調節することにより、トナーの拡散状態をコントロールすることができる。また、トナー粒子の融着防止を目的として、トナー粒子供給口(100)の外周、表面処理装置外周及び移送配管(116)の外周には冷却ジャケット(106)が設けられている。尚、該冷却ジャケットには冷却水(好ましくはエチレングリコール等の不凍液)を通水することが好ましい。一方、拡散エアにより拡散したトナー粒子は、熱風供給口(101)から供給された熱風により、トナー粒子の表面が処理される。この時、熱風の吐出温度は100℃以上、300℃以下であることが好ましく、150℃以上、250℃以下であることがより好ましい。熱風の温度が100℃未満の場合にはトナー粒子の表面を溶融状態にすることができない場合がある。また、300℃を超える場合には溶融状態が進みすぎる事で、ワックスを過度にトナー表面に偏析させる場合や、トナー粒子同士の合一に起因する、トナー粒子の粗大化や融着が生じる場合がある。
熱風により表面が処理されたトナー粒子は、装置上部外周に設けた冷風供給口(103)から供給される冷風により冷却される。この時、装置内の温度分布の制御、トナー粒子の表面状態をコントロールする目的で、装置の本体側面に設けた第二の冷風供給口(104)から冷風を導入することが好ましい。第二の冷風供給口(104)の出口はスリット形状、ルーバー形状、多孔板形状、メッシュ形状等を用いる事ができ、導入方向は中心方向へ水平、装置壁面に沿う方向が、目的に応じて選択可能である。
この時、上記冷風温度は−50℃以上、10℃以下であることが好ましく、−40℃以上、8℃以下であることがより好ましい。また、上記冷風は除湿された冷風であることが好ましい。具体的には、冷風の絶対水分量が5g/m3以下であることが好ましい。更に好ましくは、3g/m3以下である。
これらの冷風温度が−50℃未満の場合には装置内の温度が下がりすぎてしまい、本来の目的である熱による処理が十分に為されず、トナーの表面を溶融状態にすることができない場合がある。また、10℃を超える場合には、装置内における熱風ゾーンの制御が不十分になり、表面処理時にワックスを過度にトナー表面に偏析させることがある。
その後、冷却されたトナー粒子は、ブロワーで吸引され、移送配管(116)を通じて、サイクロン等で回収される。
本発明の二成分系現像剤において、トナーと磁性キャリアの混合比率は、磁性キャリア100質量部に対してトナーを2質量部以上35質量部以下とすることが好ましく、4質量部以上25質量部以下とすることがより好ましい。トナーを上記範囲とすることで、高い画像濃度を達成し、かつ、トナーの飛散を低減することができる。
<P1及びP2の算出方法>
FT−IRスペクトルは、ユニバーサルATR測定アクセサリー(Universal
ATR Sampling Accessory)を装着したフーリエ変換赤外分光分析装置(Spectrum One;PerkinElmer社製)を用い、ATR法で測定する。具体的な測定手順と、P1、P2及びP1をP2で除した[P1/P2]の算出方法は以下の通りである。
赤外光の入射角は45°に設定する。ATR結晶としては、GeのATR結晶(屈折率=4.0)、KRS5のATR結晶(屈折率=2.4)を用いる。その他の条件は以下の通りである。
Range
Start :4000cm−1
End :600cm−1(GeのATR結晶)
400cm−1(KRS5のATR結晶)
Duration
Scan number:16
Resolution:4.00cm−1
Advanced :CO2/H2O補正あり
[P1の算出方法]
(1)GeのATR結晶(屈折率=4.0)を装置に装着する。
(2)Scan typeをBackground、UnitsをEGYに設定し、バックグラウンドを測定する。
(3)Scan typeをSample、UnitsをAに設定する。
(4)トナーをATR結晶の上に、0.01g精秤する。
(5)圧力アームでサンプルを加圧する。(Force Gaugeは90)
(6)サンプルを測定する。
(7)得られたFT−IRスペクトルを、Automatic Correctionでベースライン補正をする。
(8)2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pa1)
(9)3050cm−1と2600cm−1の吸収ピーク強度の平均値を算出する。(Pa2)
(10)Pa1−Pa2=Paとする。当該Paを2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度と規定する。
(11)1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pb1)
(12)1763cm−1と1630cm−1の吸収ピーク強度の平均値を算出する(Pb2)
(13)Pb1−Pb2=Pbとする。当該Pbを1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度と規定する。
(14)Pa/Pb=P1とする。
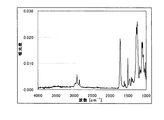
図2にATR結晶としてGeを用い測定したFT−IRスペクトルの一例を示す。
[P2の算出方法]
(1)KRS5のATR結晶(屈折率=2.4)を装置に装着する。
(2)トナーをATR結晶の上に、0.01g精秤する。
(3)圧力アームでサンプルを加圧する。(Force Gaugeは90)
(4)サンプルを測定する。
(5)得られたFT−IRスペクトルを、Automatic Correctionでベースライン補正をする。
(6)2843cm−1以上2853cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pc1)
(7)3050cm−1と2600cm−1の吸収ピーク強度の平均値を算出する。(Pc2)
(8)Pc1−Pc2=Pcとする。当該Pcを2843cm−1以上2853cm−1以下の範囲の最大吸収ピーク強度と規定する。
(9)1713cm−1以上1723cm−1以下の範囲の吸収ピーク強度の最大値を算出する。(Pd1)
(10)1763cm−1と1630cm−1の吸収ピーク強度の平均値を算出する(Pd2)
(11)Pd1−Pd2=Pdとする。当該Pdを1713cm−1以上1723cm−1以下の範囲の最大吸収ピーク強度と規定する。
(12)Pc/Pd=P2とする。
[P1/P2の算出方法]
上記のようにして求めたP1とP2を用い、P1/P2を算出する。
トナーの平均円形度は、フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)によって、校正作業時の測定及び解析条件で測定する。
フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)の測定原理は、流れている粒子を静止画像として撮像し、画像解析を行うというものである。試料チャンバーへ加えられた試料は、試料吸引シリンジによって、フラットシースフローセルに送り込まれる。フラットシースフローに送り込まれた試料は、シース液に挟まれて扁平な流れを形成する。フラットシースフローセル内を通過する試料に対しては、1/60秒間隔でストロボ光が照射されており、流れている粒子を静止画像として撮影することが可能である。また、扁平な流れであるため、焦点の合った状態で撮像される。粒子像はCCDカメ
ラで撮像され、撮像された画像は512×512画素の画像処理解像度(一画素あたり0.37×0.37μm)で画像処理され、各粒子像の輪郭抽出を行い、粒子像の投影面積Sや周囲長L等が計測される。
次に、上記面積Sと周囲長Lを用いて円相当径と円形度を求める。円相当径とは、粒子像の投影面積と同じ面積を持つ円の直径のことであり、円形度Cは、円相当径から求めた円の周囲長を粒子投影像の周囲長で割った値として定義され、次式で算出される。
円形度C=2×(π×S)1/2/L
粒子像が円形の時に円形度は1.000になり、粒子像外周の凹凸の程度が大きくなればなるほど円形度は小さい値になる。各粒子の円形度を算出後、円形度0.200乃至1.000の範囲を800分割し、得られた円形度の相加平均値を算出し、その値を平均円形度とする。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中に予め不純固形物などを除去したイオン交換水20mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.2ml加える。更に測定試料を0.02g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」(ヴェルヴォクリーア社製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に該コンタミノンNを2ml添加する。
測定には、標準対物レンズ(10倍)を搭載した該フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用する。該手順に従い調製した分散液を該フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて3000個のトナー粒子を計測する。そして、粒子解析時の2値化閾値を85%とし、解析粒子径を円相当径1.985μm以上、39.69μm未満に限定し、トナー粒子の平均円形度を求める。
測定にあたっては、測定開始前に標準ラテックス粒子(例えば、Duke Scientific社製の「RESEARCH AND TEST PARTICLES Latex Microsphere Suspensions 5200A」をイオン交換水で希釈)を用いて自動焦点調整を行う。その後、測定開始から2時間毎に焦点調整を実施することが好ましい。
なお、本願実施例では、シスメックス社による校正作業が行われた、シスメックス社が発行する校正証明書の発行を受けたフロー式粒子像分析装置を使用する。解析粒子径を円相当径1.985μm以上、39.69μm未満に限定した以外は、校正証明を受けた時の測定及び解析条件で測定を行う。
トナーの重量平均粒径(D4)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように専用ソフトの設定を行う。
専用ソフトの「標準測定方法(SOM)を変更」画面において、コントロールモードの
総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。「閾値/ノイズレベルの測定ボタン」を押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、「測定後のアパーチャーチューブのフラッシュ」にチェックを入れる。
専用ソフトの「パルスから粒径への変換設定」画面において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μmから60μmまでに設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250ml丸底ビーカーに該電解水溶液200mlを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100ml平底ビーカーに該電解水溶液30mlを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で3質量倍に希釈した希釈液を0.3ml加える。
(3)発振周波数50kHzの発振器2個を、位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispersion System Tetora150」(日科機バイオス社製)を準備する。超音波分散器の水槽内に3.3lのイオン交換水を入れ、この水槽中にコンタミノンNを2ml添加する。(4)該(2)のビーカーを該超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)該(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー10mgを少量ずつ該電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した該(1)の丸底ビーカーに、ピペットを用いてトナーを分散した該(5)の電解質水溶液を滴下し、測定濃度が5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の該専用ソフトにて解析を行い、重量平均粒径(D4)を算出する。尚、専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)である。
ピーク分子量(Mp)、数平均分子量(Mn)、重量平均分子量(Mw)、分子量分布は、ゲルパーミエーションクロマトグラフィー(GPC)により、以下のようにして測定する。
まず、室温で24時間かけて、試料(樹脂またはトナー)をテトラヒドロフラン(THF)に溶解する。そして、得られた溶液を、ポア径が0.2μmの耐溶剤性メンブランフィルター「マエショリディスク」(東ソー社製)で濾過してサンプル溶液を得る。尚、サンプル溶液は、THFに可溶な成分の濃度が0.8質量%となるように調整する。このサンプル溶液を用いて、以下の条件で測定する。
装置 :HLC8120 GPC(検出器:RI)(東ソー社製)
カラム :Shodex KF−801、802、803、804、805、
806、807の7連(昭和電工社製)
溶離液 :テトラヒドロフラン(THF)
流速 :1.0ml/min
オーブン温度 :40.0℃
試料注入量 :0.10ml
試料の分子量の算出にあたっては、標準ポリスチレン樹脂(例えば、商品名「TSKスタンダード ポリスチレン F−850、F−450、F−288、F−128、F−80、F−40、F−20、F−10、F−4、F−2、F−1、A−5000、A−2500、A−1000、A−500」、東ソ−社製)を用いて作成した分子量校正曲線を使用する。
分子量分布は、全ピーク面積に対する、分子量10000以上のピーク面積の比率、および、分子量5000以下のピーク面積の比率(百分率)を算出した。
ワックスの最大吸熱ピークのピーク温度は、示差走査熱量分析装置「Q1000」(TA Instruments社製)を用いてASTM D3418−82に準じて測定する。装置検出部の温度補正はインジウムと亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。具体的には、ワックスを10mg精秤し、これをアルミニウム製のパンの中に入れ、リファレンスとして空のアルミニウム製のパンを用い、測定温度範囲30以上200℃以下の間で、昇温速度10℃/minで測定を行う。尚、測定においては、一度200℃まで昇温させ、続いて30℃まで降温し、その後に再度昇温を行う。この2度目の昇温過程での温度30以上200℃以下の範囲におけるDSC曲線の最大の吸熱ピークを、ワックスの最大吸熱ピークとする。
また、樹脂のガラス転移温度(Tg)は、ワックスの最大吸熱ピークのピーク温度測定と同様に、樹脂を10mg精秤し測定する。温度40℃以上100℃以下の範囲において比熱変化が得られる。このときの比熱変化前と比熱変化後のベースラインの中間点の線と示差熱曲線との交点を、樹脂のガラス転移温度Tgとする。
カーボンブラックのBET比表面積の測定は、JIS Z8830(2001年)に準じて行なう。具体的な測定方法は、以下の通りである。
測定装置としては、定容法によるガス吸着法を測定方式として採用している「自動比表面積・細孔分布測定装置 TriStar3000(島津製作所社製)」を用いる。測定条件の設定および測定データの解析は、本装置に付属の専用ソフト「TriStar3000 Version4.00」を用いて行い、また装置には真空ポンプ、窒素ガス配管、ヘリウムガス配管が接続される。窒素ガスを吸着ガスとして用い、BET多点法により算出した値を本発明におけるBET比表面積とする。
尚、BET比表面積は以下のようにして算出する。
まず、カーボンブラックに窒素ガスを吸着させ、その時の試料セル内の平衡圧力P(Pa)とカーボンブラックの窒素吸着量Va(モル・g−1)を測定する。そして、試料セル内の平衡圧力P(Pa)を窒素の飽和蒸気圧Po(Pa)で除した値である相対圧Prを横軸とし、窒素吸着量Va(モル・g−1)を縦軸とした吸着等温線を得る。次いで、カーボンブラックの表面に単分子層を形成するのに必要な吸着量である単分子層吸着量Vm(モル・g−1)を、下記のBET式を適用して求める。
Pr/Va(1−Pr)=1/(Vm×C)+(C−1)×Pr/(Vm×C)
(ここで、CはBETパラメーターであり、測定サンプル種、吸着ガス種、吸着温度により変動する変数である。)
BET式は、X軸をPr、Y軸をPr/Va(1−Pr)とすると、傾きが(C−1)/(Vm×C)、切片が1/(Vm×C)の直線と解釈できる(この直線をBETプロットという)。
直線の傾き=(C−1)/(Vm×C)
直線の切片=1/(Vm×C)
Prの実測値とPr/Va(1−Pr)の実測値をグラフ上にプロットして最小二乗法により直線を引くと、その直線の傾きと切片の値が算出できる。これらの値を用いて上記
の傾きと切片の連立方程式を解くと、VmとCが算出できる。
さらに、上記で算出したVmと窒素分子の分子占有断面積(0.162nm2)から、下記の式に基づいて、カーボンブラックのBET比表面積S(m2・g−1)を算出する。
S=Vm×N×0.162×10−18
(ここで、Nはアボガドロ数(モル−1)である。)
本装置を用いた測定は、装置に付属の「TriStar3000 取扱説明書V4.0」に従うが、具体的には、以下の手順で測定する。
充分に洗浄、乾燥した専用のガラス製試料セル(ステム直径3/8インチ、容積約5ml)の風袋を精秤する。そして、ロートを使ってこの試料セルの中に0.3gのカーボンブラックを入れる。
カーボンブラックを入れた前記試料セルを真空ポンプと窒素ガス配管を接続した「前処理装置 バキュプレップ061(島津製作所社製)」にセットし、23℃にて真空脱気を約10時間継続する。尚、真空脱気の際には、カーボンブラックが真空ポンプに吸引されないよう、バルブを調整しながら徐々に脱気する。セル内の圧力は脱気とともに徐々に下がり、最終的には約0.4Pa(約3ミリトール)となる。真空脱気終了後、窒素ガスを徐々に注入して試料セル内を大気圧に戻し、試料セルを前処理装置から取り外す。そして、この試料セルの質量を精秤し、風袋との差からカーボンブラックの正確な質量を算出する。尚、この際に、試料セル内のカーボンブラックが大気中の水分等で汚染されないように、秤量中はゴム栓で試料セルに蓋をしておく。
次に、カーボンブラックが入った前記の試料セルのステム部に専用の「等温ジャケット」を取り付ける。そして、この試料セル内に専用のフィラーロッドを挿入し、前記装置の分析ポートに試料セルをセットする。尚、等温ジャケットとは、毛細管現象により液体窒素を一定レベルまで吸い上げることが可能な、内面が多孔性材料、外面が不浸透性材料で構成された筒状の部材である。
続いて、接続器具を含む試料セルのフリースペースの測定を行なう。フリースペースは、23℃においてヘリウムガスを用いて試料セルの容積を測定し、続いて液体窒素で試料セルを冷却した後の試料セルの容積を、同様にヘリウムガスを用いて測定して、これらの容積の差から換算して算出する。また、窒素の飽和蒸気圧Po(Pa)は、装置に内蔵されたPoチューブを使用して、別途に自動で測定される。
次に、試料セル内の真空脱気を行った後、真空脱気を継続しながら試料セルを液体窒素で冷却する。その後、窒素ガスを試料セル内に段階的に導入してカーボンブラックに窒素分子を吸着させる。この際、平衡圧力P(Pa)を随時計測することにより前記した吸着等温線が得られるので、この吸着等温線をBETプロットに変換する。尚、データを収集する相対圧Prのポイントは、0.05、0.10、0.15、0.20、0.25、0.30の合計6ポイントに設定する。得られた測定データに対して最小二乗法により直線を引き、その直線の傾きと切片からVmを算出する。さらに、このVmの値を用いて、前記したようにカーボンブラックのBET比表面積を算出する。
〔結着樹脂の製造例1〕
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 22.6質量部
無水トリメリット酸 1.7質量部
ポリオキシプロピレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.6質量部
チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反
応させ、その後、10mmHgに減圧し1時間反応させ、樹脂1を合成した。GPCで求めた樹脂1の分子量は、重量平均分子量(Mw)6200、数平均分子量(Mn)2500であり、ピーク分子量(Mp)2900、ガラス転移点は55℃、軟化点は93℃であった。
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部
ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部
チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。さらに、無水トリメリット酸4.5質量部を加え、180℃に加熱し、2時間反応させ、樹脂2を合成した。GPCで求めた樹脂2の分子量は、重量平均分子量(Mw)45000、数平均分子量(Mn)4800、ピーク分子量(Mp)12800、ガラス転移点は61℃、軟化点は122℃であった。
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部
ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部
チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。さらに、無水トリメリット酸10.2質量部を加え、180℃に加熱し、2時間反応させ、樹脂3を合成した。GPCで求めた樹脂3の分子量は、重量平均分子量(Mw)91000、数平均分子量(Mn)5800、ピーク分子量(Mp)13500、ガラス転移点は61℃、軟化点は133℃であった。
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 17.2質量部
ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
76.6質量部
チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、220℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させた。さらに、無水トリメリット酸17.2質量部を加え、180℃に加熱し、2時間反応させ、樹脂4を合成した。GPCで求めた樹脂4の分子量は、重量平均分子量(Mw)125000、数平均分子量(Mn)6400、ピーク分子量(Mp)14800、ガラス転移点は62℃、軟化点は141℃であった。
冷却管、攪拌機、及び、窒素導入管のついた反応槽中に、以下の材料を秤量した。
テレフタル酸 19.6質量部
無水トリメリット酸 5.4質量部
ポリオキシエチレン(2.2)−2,2−ビス(4−ヒドロキシフェニル)プロパン
75.3質量部
チタニウムジヒドロキシビス(トリエタノールアミネート) 0.2質量部
その後、200℃に加熱し、窒素を導入しながら生成する水を除去しながら10時間反応させ、その後、10mmHgに減圧し1時間反応させ、樹脂5を合成した。GPCで求
めた樹脂5の分子量は、重量平均分子量(Mw)6900、数平均分子量(Mn)2300であり、ピーク分子量(Mp)3600、ガラス転移点は58℃、軟化点は112℃であった。
ビーカー中に下記材料を秤量した。
Mg−Al水酸化物炭酸塩(化学量論のMg:Al比=2:1)(Syntal HSA
696 Sudchemie,Germany) 10g
イオン交換水 100g
その後、70℃で1時間、マグネチックスターラーで攪拌し、水酸化物炭酸塩懸濁液1を作成した。
次に、下記材料をビーカー中に調製し、攪拌した。
イオン交換水 100g
安息香酸 3g
その後、4Nの水酸化ナトリウム水溶液をpH8になるように滴下し、安息香酸水溶液1を作成した。
Mg−Al水酸化物炭酸塩懸濁液1に安息香酸水溶液1を添加し、70℃で6時間攪拌した。その後、メンブレンフィルターを用い懸濁液を濾過し、イオン交換水で洗浄した。さらに、洗浄された固体は真空において70℃で6時間乾燥し、層状複水酸化物塩1を得た。
安息香酸の代わりに、4−トルエンスルホン酸を用いた以外は、層状複水酸化物塩の製造例1と同様にして、層状複水酸化物塩2を得た。
同様にして、ジチオジサリチル酸を用い層状複水酸化物塩3、クエン酸を用い層状複水酸化物塩4を得た。
樹脂1 50.0質量部
樹脂2 50.0質量部
層状複水酸化物塩1 2.0質量部
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 7.0質量部
カーボンブラック(BET比表面積 72m2/g) 7.0質量部
上記材料をヘンシェルミキサー(FM−75型、三井三池化工機(株)製)でよく混合した後、温度130℃に設定した二軸混練機(PCM−30型、池貝鉄工(株)製)にて混練した。得られた混練物を冷却し、ハンマーミルにて1mm以下に粗粉砕し、粗砕物を得た。得られた粗砕物を、高圧気体を用いた衝突式気流粉砕機を用いて微粉砕した。
次に、得られた微粉砕物を図1に示す表面改質装置により表面改質を行った。表面改質時の条件として、原料供給速度が2.0kg/hr、熱風流量が4.5m3/min、熱風の吐出温度が210℃、冷風温度が3℃、冷風流量が3.0m3/min、絶対水分量が3g/m3で表面改質を行った。次に、コアンダ効果を利用した風力分級機(エルボジェットラボEJ−L3、日鉄鉱業社製)で分級しで微粉及び粗粉を同時に分級除去、トナー粒子1を得た。
得られたトナー粒子1 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が200nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を2.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー1を得た。
樹脂1 50.0質量部
樹脂3 50.0質量部
層状複水酸化物塩2 2.0質量部
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 7.0質量部
カーボンブラック(BET比表面積 287m2/g) 7.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にトナー2を得た。
樹脂1 50.0質量部
樹脂4 50.0質量部
層状複水酸化物塩3 2.0質量部
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 7.0質量部
カーボンブラック(BET比表面積 43m2/g) 7.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にトナー3を得た。
樹脂1 50.0質量部
樹脂2 50.0質量部
層状複水酸化物塩4 2.0質量部
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 5.0質量部
カーボンブラック(BET比表面積 72m2/g) 7.0質量部
混練する材料を上記材料とし、表面改質時の熱風の吐出温度を230℃、冷風温度を10℃とする以外は、トナー製造例1と同様にトナー4を得た。
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃)7.0質量部をパラフィンワックス(DSC最大吸熱ピーク65℃)10.0質量部、表面改質時の熱風の吐出温度を210℃から190℃に変更した以外は、トナー製造例1と同様にトナー5を得た。
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃)7.0質量部をフィッシャー・トロプシュワックス(DSC最大吸熱ピーク90℃)5.0質量部、表面改質時の熱風の吐出温度を210℃から250℃に変更した以外は、トナー製造例1と同様にトナー6を得た。
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃)7.0質量部を3.0質量部に、カーボンブラックのBET比表面積を72m2/gから28m2/gに変更した以外は、トナー製造例1と同様にしてトナー7を得た。
樹脂5 100.0質量部
層状複水酸化物塩1 2.0質量部
フィッシャー・トロプシュワックス(DSC最大吸熱ピーク78℃) 7.0質量部
カーボンブラック(BET比表面積を72m2/g) 7.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にしてトナー8を得た。
樹脂5 100.0質量部
層状複水酸化物塩1 2.0質量部
パラフィンワックス(DSC最大吸熱ピーク75℃) 7.0質量部
カーボンブラック(BET比表面積を18m2/g) 7.0質量部
混練する材料を上記材料とする以外は、トナー製造例1と同様にしてトナー9を得た。
トナーの製造例9のうち、表面改質を行わずに、乾式分級機(エルボジェット分級機、EJ−L−3(LABO)型、日鉄鉱業社製)を用いて分級を行ったこと以外は、トナーの製造例9と同様にしてトナー10を得た。
樹脂5 100.0質量部
層状複水酸化物塩1 1.0質量部
カルナウバワックス 3.0質量部
Pigment Red 57:1 5.0質量部
混練する材料を上記材料とし、表面改質を行わずに、気流分級装置(100ATP(ホソカワミクロン社製)で分級した以外は、トナー製造例1と同様にしてトナー11を得た。
[混練工程]
樹脂5 100.0質量部
パラフィンワックス(融点85℃) 8.0質量部
着色剤(KET.BLUE111、大日本インキ株式会社製) 9.0質量部
層状複水酸化物塩1 2.0質量部
上記材料をヘンシェルミキサー(FM−75型、三井三池化工機(株)製)でよく混合した後、温度130℃に設定した二軸混練機(PCM−30型、池貝鉄工(株)製)にて混練した。得られた混練物を冷却し、ハンマーミルにて1mm以下に粗粉砕し、粗砕物を得た。
[微粒子化工程]
粗砕物 24.0質量部
イオン交換水 50.7質量部
エアロール(エアロールCT−1p、東邦化学工業社製) 0.05質量部
ポリアクリル酸(ディスロールH14−N、日本乳化剤社製) 4.0質量部
キサンタンガム 1.3質量部
以上を泡レスミキサー(株式会社美粒製)にて回転数2000rpmで5分間攪拌して溶融混練物の分散液を作製した。音レス高圧乳化装置NANO3000(株式会社美粒製)によって、さらに微粒子化処理して、溶融混練物微粒子分散液を得た。
[脱気工程]
溶融混練物微粒子分散液をT.K.ハイビスダッパー(登録商標)3型(プライミクス株式会社製)によって脱気した。
[合一工程]
溶融混練物微粒子分散液に、3重量%の塩化ナトリウムを加えてよく攪拌した後、合一化装置へ投入した。加熱は配管周りに配したラインヒーターによって行った。T.K.パイプラインホモミクサー(プライミクス株式会社製)に外部温度制御装置を装着した装置に分散液を流入させ剪断力を付加、加熱しながら、溶融混練物微粒子を凝集、合一させて合一粒子を得た。その後、常温まで冷却した。
[乾燥工程]
冷却工程で得られたスラリーを吸引ろ過し、ろ紙上に残ったトナーを35℃/5%RH
の恒温恒湿槽内で乾燥させて、トナー粒子12を得た。
得られたトナー粒子12 100.0質量部に、無機微粒子として、個数平均粒径が40nmであり、i−ブチルトリメトキシシランで処理された酸化チタン微粉体1.0質量部、個数平均粒径が200nmであり、ヘキサメチルジシラザンで処理されたゾルゲル法シリカ微粉体を2.0質量部添加し、ヘンシェルミキサー(FM−75型、三井三池化工機(株)製)で混合し、トナー12を得た。
層状複水酸化物塩1の代わりに、Mg−Al水酸化物炭酸(Syntal HSA 696 Sudchemie,Germany)を用いたこと以外は、トナー製造例1と同様にしてトナー13を得た。
層状複水酸化物塩1の代わりに、水酸化アルミニウム(キシダ化学社製)を用いたこと以外は、トナー製造例1と同様にしてトナー14を得た。
層状複水酸化物塩1の代わりに、水酸化マグネシウム(キシダ化学社製)を用いたこと以外は、トナー製造例1と同様にしてトナー15を得た。
層状複水酸化物塩1を用いないこと以外は、トナー製造例1と同様にしてトナー16を得た。
トナー1乃至16の物性を表1に示す。
体積基準の50%粒径(D50)が32μmのマグネタイト粒子100質量部に、シリコーン樹脂(信越化学社製:KR271)1質量部、γ―アミノプロピルトリエトキシシラン0.5質量部、トルエン98.5質量部の混合液を、添加し、さらに溶液減圧ニーダーで撹拌混合しながら70℃、5時間減圧乾燥を行い、溶剤を除去した。その後、140℃で2時間焼き付け処理して、篩振とう機(300MM−2型、筒井理化学機械:75μm開口)で篩い、磁性キャリアを得た。キャリアのD50は35μmであった。
トナー1と上記磁性キャリアを用いて二成分系現像剤を作製した。二成分系現像剤は、磁性キャリア100質量部に対して、トナー9質量部の配合割合とし、V型混合機で5分間混合した。
<二成分系現像剤の評価>
得られた二成分系現像剤の低温定着性、グロス均一性、画像濃度、画像濃度変動の各種評価を、温度23℃/湿度50%RH(以下「N/N」)と、温度30℃/湿度80%RH(以下「H/H」)の2つの環境で実施した。
詳細な評価方法及び評価基準は後述する。上記各種評価用の画像形成装置としては、キヤノン製カラー複写機imageRUNNER iRC3580改造機を用いた。改造箇所は、現像ドラムに対する現像スリーブ周速を標準機に比して1.5倍としたこと、及び、プロセススピードが245mm/secとなるようしたことである。なお、上記二成分系現像剤は、画像形成装置のシアン用現像器に入れて評価を行った。評価結果を表3及び表4に示す。
紙は、カラー複写機・プリンタ用普通紙 CS−814(A4、81.4g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。FFH画像(以下、ベタ部)のトナーの紙上への載り量が0.6mg/cm2となるように直流電圧VDCを調整し、未定着のFFH画像を得た。その後、キヤノン製プロダクション向け複写機imagePRESS C1+の外部定着器(ベルト&ローラ定着器)をプロセススピードが300mm/secとなるように改造したものを定着性評価に用いた。
上記外部定着器における定着温度を100〜200℃の範囲で10℃おきに調整し、各温度で上記未定着のFFH画像を定着し定着画像を得た。得られた定着画像を4.9kPaの荷重をかけたレンズクリーニングワイパー(ダスパー 小津産業株式会社製)で5往復摺擦し、摺擦前後の画像濃度の濃度低下率が10%以下になる点を定着温度とし、下記の評価基準に従って評価した。
A:定着温度120℃以下
B:定着温度130℃
C:定着温度140℃
D:定着温度150℃以上
紙は、CLC用 厚口用紙 NS−700(A4、157g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。低温定着性評価と同様に、ベタ部のトナーの紙上への載り量が0.6mg/cm2となるように直流電圧VDCを調整し、紙の四隅及び中央部に、4cm×4cmの正方形のベタ画像を印刷した未定着画像を得た。
その後、キヤノン製プロダクション向け複写機imagePRESS C1+の外部定着器をプロセススピード100mm/secとなるように改造したものを用い、グロス均一性評価を行った。具体的には、外部定着器における定着温度を180℃に調整し、未定着画像を定着した。当該定着画像の四隅及び中央部にあるベタ画像の各グロス値を測定し、その最大値と最小値の差を以下の基準で判断した。
A:1.0未満
B:1.0以上2.0未満
C:2.0以上3.0未満
D:3.0以上
現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VD
Cを印加した。直流電圧VDCはベタ部のトナーの紙上への載り量が0.6mg/cm2となるように調整した。FFH画像を印刷し、画像濃度(反射濃度)を求めた(初期)。その後、画像比率5%で50000枚印刷し、50000枚印刷後に、再びFFH画像を印刷し、画像濃度(反射濃度)を測定した(50000枚印刷後)。紙は、CS−814(A4、81.4g/m2)(キヤノンマーケティングジャパン株式会社より販売)を用いた。画像濃度(反射濃度)は、分光濃度計500シリーズ(X−Rite社製)を用いて測定し、以下の基準で判断した。
A:1.50以上
B:1.40以上1.50未満
C:1.30以上1.40未満
D:1.30未満
現像スリーブには、周波数2.0kHz、Vpp1.3kVの交流電圧と直流電圧VDCを印加した。直流電圧VDCはベタ部のトナーの紙上への載り量が0.6mg/cm2となるように調整した。FFH画像を印刷し、画像濃度(反射濃度)を求めた。
画像濃度は、分光濃度計500シリーズ(X−Rite社製)を用いて測定した。
その後、現像器内の現像剤のトナー比率が9質量部で一定になるようにトナーを補給しながら、画像比率20%で100枚印刷し、初期と同様にFFH画像を印刷し、画像濃度(反射濃度)を測定した。
画像比率20%で100枚印刷する前後のFFH画像の画像濃度(反射濃度)の差を算出し、以下の基準で評価を行った。
A:0.10未満
B:0.10以上0.20未満
C:0.20以上0.30未満
D:0.30以上
トナー1を表2に記載のように、トナー2乃至16に変更する以外は、実施例1と同様にして二成分系現像剤を作製し、その評価を実施した。評価結果を表3及び表4に示す。
Claims (4)
- ポリエステル樹脂、着色剤、層状複水酸化物塩及びワックスを少なくとも含有するトナー粒子を有するトナーであって、
前記層状複水酸化物塩は、有機アニオン、並びに、Mg2+及びAl3+を少なくとも含有し、
前記トナーは、下記式(1)の関係を満たすことを特徴とするトナー。
1.05≦P1/P2≦2.00 ・・・ 式(1)
[式(1)中、
i)P1=Pa/Pbであり、
ATR法を用い、ATR結晶としてGe、赤外光入射角として45°の条件で測定し得られた該トナーのFT−IRスペクトルにおいて、
2843cm −1 以上2853cm −1 以下の範囲の吸収ピーク強度の最大値から3050cm −1 と2600cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pa”であり、
1713cm −1 以上1723cm −1 以下の範囲の吸収ピーク強度の最大値から1763cm −1 と1630cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pb”であり、
ii)P2=Pc/Pdであり、
ATR法を用い、ATR結晶としてKRS5、赤外光入射角として45°の条件で測定し得られた該トナーのFT−IRスペクトルにおいて、
2843cm −1 以上2853cm −1 以下の範囲の吸収ピーク強度の最大値から3050cm −1 と2600cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pc”であり、
1713cm −1 以上1723cm −1 以下の範囲の吸収ピーク強度の最大値から1763cm −1 と1630cm −1 の吸収ピーク強度の平均値を差し引いた値である、最大吸熱ピーク強度が“Pd”である。] - 前記トナー粒子は、熱風により表面処理されていることを特徴とする請求項1に記載のトナー。
- 前記着色剤は、BET比表面積が20m2/g以上300m2/g以下のカーボンブラックであることを特徴とする請求項1または2に記載のトナー。
- トナーと磁性キャリアを含む二成分系現像剤において、前記トナーは請求項1乃至3のいずれか1項に記載のトナーであることを特徴とする二成分系現像剤。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010286026A JP5634252B2 (ja) | 2010-12-22 | 2010-12-22 | トナー及び二成分系現像剤 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010286026A JP5634252B2 (ja) | 2010-12-22 | 2010-12-22 | トナー及び二成分系現像剤 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2012133193A JP2012133193A (ja) | 2012-07-12 |
| JP2012133193A5 JP2012133193A5 (ja) | 2014-02-13 |
| JP5634252B2 true JP5634252B2 (ja) | 2014-12-03 |
Family
ID=46648856
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010286026A Expired - Fee Related JP5634252B2 (ja) | 2010-12-22 | 2010-12-22 | トナー及び二成分系現像剤 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5634252B2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10101681B2 (en) | 2016-04-11 | 2018-10-16 | Canon Kabushiki Kaisha | Toner |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6107258B2 (ja) * | 2013-03-15 | 2017-04-05 | 株式会社リコー | 静電荷像現像用トナー、現像剤および画像形成装置 |
| JP6272026B2 (ja) * | 2013-12-27 | 2018-01-31 | キヤノン株式会社 | トナー |
| JP6779623B2 (ja) * | 2016-01-13 | 2020-11-04 | キヤノン株式会社 | トナー及びトナーの製造方法 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10235571A1 (de) * | 2002-08-03 | 2004-02-12 | Clariant Gmbh | Verwendung von Salzen schichtartiger Doppelhydroxide als Ladungssteuermittel |
| JP4255846B2 (ja) * | 2003-01-20 | 2009-04-15 | 株式会社リコー | トナー、現像剤、画像形成装置、プロセスカートリッジ及び画像形成方法 |
| JP4143506B2 (ja) * | 2003-09-12 | 2008-09-03 | キヤノン株式会社 | 黒トナー |
| DE102004028493A1 (de) * | 2004-06-11 | 2005-12-29 | Clariant Gmbh | Hydrophobierte Salze schichtartiger Metallhydroxide |
| JP4955965B2 (ja) * | 2004-09-07 | 2012-06-20 | 株式会社リコー | 画像定着方法、及び画像形成方法 |
| JP2006243301A (ja) * | 2005-03-02 | 2006-09-14 | Ricoh Co Ltd | 画像定着方法及び画像定着装置、並びに、画像形成方法及び画像形成装置 |
| JP5020549B2 (ja) * | 2005-06-21 | 2012-09-05 | 株式会社リコー | 画像形成装置 |
| JP4616782B2 (ja) * | 2006-03-17 | 2011-01-19 | 株式会社リコー | 静電荷像現像用トナー、画像形成装置及びプロセスカートリッジ |
| JP2008076452A (ja) * | 2006-09-19 | 2008-04-03 | Ricoh Co Ltd | トナー定着方法及びトナー定着装置並びに画像形成装置 |
| JP2008233411A (ja) * | 2007-03-19 | 2008-10-02 | Ricoh Co Ltd | 静電荷像現像用トナー、画像形成装置、トナー容器およびプロセスカートリッジ |
| US8012659B2 (en) * | 2007-12-14 | 2011-09-06 | Ricoh Company Limited | Image forming apparatus, toner, and process cartridge |
| JP5153792B2 (ja) * | 2007-12-27 | 2013-02-27 | キヤノン株式会社 | トナー及び二成分系現像剤 |
| JP5300295B2 (ja) * | 2008-03-26 | 2013-09-25 | キヤノン株式会社 | トナー及びトナーの製造方法 |
| JP5546271B2 (ja) * | 2010-02-02 | 2014-07-09 | キヤノン株式会社 | トナー及び二成分系現像剤 |
-
2010
- 2010-12-22 JP JP2010286026A patent/JP5634252B2/ja not_active Expired - Fee Related
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10101681B2 (en) | 2016-04-11 | 2018-10-16 | Canon Kabushiki Kaisha | Toner |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2012133193A (ja) | 2012-07-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101445049B1 (ko) | 토너 | |
| JP5911235B2 (ja) | トナー | |
| JP5865032B2 (ja) | トナー | |
| JP5615156B2 (ja) | トナー | |
| TWI471707B (zh) | 調色劑 | |
| JP6462999B2 (ja) | トナー、その製造方法及び画像形成方法 | |
| JP6124671B2 (ja) | トナー及びその製造方法 | |
| JP5546271B2 (ja) | トナー及び二成分系現像剤 | |
| JP2018092154A (ja) | トナー | |
| JP2017003990A (ja) | トナー及びトナーの製造方法 | |
| JP2013015830A (ja) | トナー、二成分系現像剤及び画像形成方法 | |
| JP2013092652A (ja) | トナー | |
| JP5634252B2 (ja) | トナー及び二成分系現像剤 | |
| JP2015121579A (ja) | トナー | |
| JP6727803B2 (ja) | トナーおよびトナーの製造方法 | |
| EP2680082A1 (en) | Toner for electrostatic latent image development and method for producing toner for electrostatic latent image development | |
| JP6218373B2 (ja) | トナー及び画像形成方法 | |
| JP7395276B2 (ja) | トナーの製造方法 | |
| JP2019078781A (ja) | 画像形成方法 | |
| JP5822618B2 (ja) | 非磁性トナーおよび画像形成方法 | |
| JP2020060703A (ja) | トナー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131224 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20131224 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20140908 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140916 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20141014 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 5634252 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| LAPS | Cancellation because of no payment of annual fees |