JP4988552B2 - 炭化水素の蒸気改質によって水素を生成するための触媒 - Google Patents
炭化水素の蒸気改質によって水素を生成するための触媒 Download PDFInfo
- Publication number
- JP4988552B2 JP4988552B2 JP2007508378A JP2007508378A JP4988552B2 JP 4988552 B2 JP4988552 B2 JP 4988552B2 JP 2007508378 A JP2007508378 A JP 2007508378A JP 2007508378 A JP2007508378 A JP 2007508378A JP 4988552 B2 JP4988552 B2 JP 4988552B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- support
- feed stream
- atmosphere
- heating
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images









Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/02—Boron or aluminium; Oxides or hydroxides thereof
- B01J21/04—Alumina
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/02—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the alkali- or alkaline earth metals or beryllium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/46—Ruthenium, rhodium, osmium or iridium
- B01J23/468—Iridium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/58—Platinum group metals with alkali- or alkaline earth metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen; Reversible storage of hydrogen
- C01B3/02—Production of hydrogen; Production of gaseous mixtures containing hydrogen
- C01B3/32—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air
- C01B3/34—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents
- C01B3/38—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents using catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen; Reversible storage of hydrogen
- C01B3/02—Production of hydrogen; Production of gaseous mixtures containing hydrogen
- C01B3/32—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air
- C01B3/34—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents
- C01B3/38—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents using catalysts
- C01B3/40—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents using catalysts characterised by the catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/89—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with noble metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/60—Catalysts, in general, characterised by their form or physical properties characterised by their surface properties or porosity
- B01J35/61—Surface area
- B01J35/613—10-100 m2/g
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/02—Processes for making hydrogen or synthesis gas
- C01B2203/0205—Processes for making hydrogen or synthesis gas containing a reforming step
- C01B2203/0227—Processes for making hydrogen or synthesis gas containing a reforming step containing a catalytic reforming step
- C01B2203/0233—Processes for making hydrogen or synthesis gas containing a reforming step containing a catalytic reforming step the reforming step being a steam reforming step
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1064—Platinum group metal catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1082—Composition of support materials
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/36—Hydrogen production from non-carbon containing sources, e.g. by water electrolysis
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Combustion & Propulsion (AREA)
- Inorganic Chemistry (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Catalysts (AREA)
- Hydrogen, Water And Hydrids (AREA)
Description
CH4 + H2O → CO + 3H2
この反応は、触媒の存在下で起き、非常に吸熱性である。反応の進行度は低温では低い。従来の改質処理では、満足できる量の炭化水素燃料を一酸化炭素および水素に変換するには、800℃程度の温度がたいてい必要とされる。
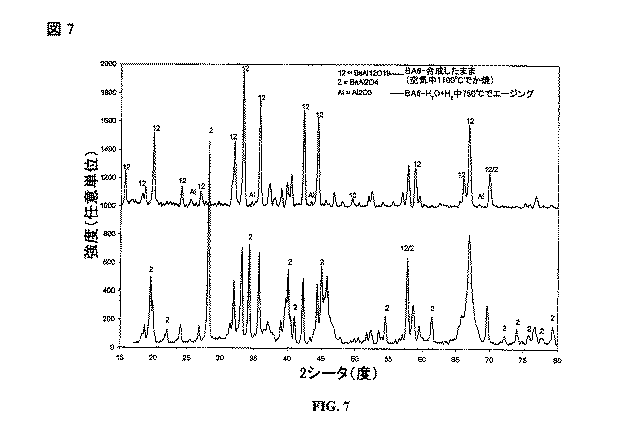
(例1)
Rh、PtまたはIrを含む未使用の触媒の活性を、エージング後の類似の触媒のそれと比較した。
様々な触媒担体上にIrを含む触媒の活性を比較した。
様々な触媒のエージング時の安定性を調べた。
バリウムアルミネート触媒担体の安定性を調べた。
様々な触媒担体材料の安定性を比較した。
本発明の触媒のサイクル運転(すなわち、反応器の運転、続いて運転停止、続いて再運転)に耐え得る能力を調べた。
Claims (36)
- アルカリ土類金属ヘキサアルミネート担体を95vol%またはそれ以上含む担持触媒であって、前記触媒担体は、6m2/gまたはそれ以上の表面積と、前記触媒担体上に堆積したIr、PdおよびPtからなる群より選択される1種以上の活性金属とを有し、前記担持触媒は炭化水素の蒸気改質による水素の製造のために使用される触媒。
- アルカリ土類金属ヘキサアルミネートを98vol%またはそれ以上含んだ請求項1記載の触媒。
- 12m2/gまたはそれ以上の表面積を有した請求項1記載の触媒。
- 18m2/gまたはそれ以上の表面積を有した請求項1記載の触媒。
- 前記アルカリ土類金属ヘキサアルミネートは、Ca、SrおよびBaからなる群より選ばれる少なくとも1つのアルカリ土類金属を含んだ請求項1記載の触媒。
- 前記アルカリ土類金属ヘキサアルミネートは、BaO・6Al2O3を含んだ請求項1記載の触媒。
- 請求項1に記載の触媒用の触媒担体を製造する方法であって、
少なくとも1つの前駆体酸化物を、0.20atmまたはそれ以下のO2分圧を有し、H2、H2Oおよび不活性ガスからなる群より選択される少なくとも1つを少なくとも50vol%含んだ雰囲気中で加熱すること
を含んだ方法。 - 前記不活性ガスは、He、Ne、Ar、Kr、XeおよびN2からなる群より選択される請求項7記載の方法。
- 前記雰囲気は、N2を少なくとも50vol%含んだ請求項7記載の方法。
- 前記加熱を、1atmの全圧で行う請求項7記載の方法。
- 前記加熱を、1atm未満の全圧で行う請求項7記載の方法。
- 前記加熱を、1atmより高い全圧で行う請求項7記載の方法。
- 前記前駆体酸化物を、0.10atmまたはそれ以下の分圧のO2を含んだ雰囲気で加熱する請求項7記載の方法。
- 前記加熱を、1100℃以下の温度で行う請求項7記載の方法。
- 前記加熱を、950℃以下の温度で行う請求項7記載の方法。
- 前記加熱を、800℃以下の温度で行う請求項7記載の方法。
- 前記少なくとも1つの前駆体酸化物は、アルカリ土類金属酸化物からなる群の要素を含んだ請求項7記載の方法。
- 前記少なくとも1つの前駆体酸化物を、0.20atmより高い分圧のO2を有した他の雰囲気で加熱することをさらに含んだ請求項7記載の方法。
- 前記他の雰囲気は、空気であり;
前記他の雰囲気の全圧は、1atmである請求項18記載の方法。 - 0.20atmまたはそれ以下の分圧のO2を有した前記雰囲気での前記加熱と0.20atmより高い分圧のO2を有する前記他の雰囲気での加熱とを、各々一回よりも多く繰り返す請求項18記載の方法。
- 請求項1に記載の触媒を使用してH 2 を製造する方法であって、
請求項1記載の触媒を用意することと、
前記触媒の上に、気体の炭化水素と気体のH2Oとを含んだ活性な原料流を通すことと、
前記気体の炭化水素と前記気体のH2Oとを前記触媒を使用して反応させ、H2を製造することと
を含んだ方法。 - 前記活性な原料流は、10質量ppmまたはそれ以上のSを含んだ請求項21記載の方法。
- 前記活性な原料流は、100質量ppmまたはそれ以上のSを含んだ請求項21記載の方法。
- Irが、前記触媒担体上にある請求項21記載の方法。
- 前記気体の炭化水素は、メタン、エタン、プロパンおよびブタンからなる群より選ばれる少なくとも1つを含んだ請求項21記載の方法。
- 前記活性な原料流は、100質量ppmまたはそれ以上のO2をさらに含んだ請求項21記載の方法。
- 前記活性な原料流は、10質量ppmまたはそれ以上のSを含んだ請求項26記載の方法。
- 前記活性な原料流は、100質量ppmまたはそれ以上のSを含んだ請求項26記載の方法。
- Irが、前記触媒担体上にある請求項26記載の方法。
- 前記気体の炭化水素は、メタン、エタン、プロパンおよびブタンからなる群より選ばれる少なくとも1つを含んだ請求項26記載の方法。
- 前記触媒の上に、空気および気体のH2Oの少なくとも1つを含んだ不活性な原料流を通すことをさらに含み、前記不活性な原料流は100質量ppm未満の気体の炭化水素を含んだ請求項21記載の方法。
- 前記不活性な原料流は、100質量ppmまたはそれ以上のO2を含んだ請求項31記載の方法。
- 前記不活性な原料流は、1vol%またはそれ以上のO2を含んだ請求項31記載の方法。
- Irが、前記触媒担体上にある請求項31記載の方法。
- 前記活性な原料流および前記不活性な原料流の各々を、前記触媒上に一回よりも多く通す請求項31記載の方法。
- 前記活性な原料流は、1質量ppmまたはそれ以上のSを含んだ請求項21記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/825,150 US7309480B2 (en) | 2004-04-16 | 2004-04-16 | Catalyst for hydrogen generation through steam reforming of hydrocarbons |
| US10/825,150 | 2004-04-16 | ||
| PCT/US2005/011037 WO2005105664A2 (en) | 2004-04-16 | 2005-04-01 | Catalyst for hydrogen generation through steam reforming of hydrocarbons |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007532305A JP2007532305A (ja) | 2007-11-15 |
| JP4988552B2 true JP4988552B2 (ja) | 2012-08-01 |
Family
ID=35096486
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007508378A Expired - Fee Related JP4988552B2 (ja) | 2004-04-16 | 2005-04-01 | 炭化水素の蒸気改質によって水素を生成するための触媒 |
Country Status (8)
| Country | Link |
|---|---|
| US (3) | US7309480B2 (ja) |
| EP (1) | EP1771376A4 (ja) |
| JP (1) | JP4988552B2 (ja) |
| KR (1) | KR101166943B1 (ja) |
| CN (1) | CN101001695B (ja) |
| AU (1) | AU2005238426B2 (ja) |
| CA (1) | CA2561427C (ja) |
| WO (1) | WO2005105664A2 (ja) |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7309480B2 (en) * | 2004-04-16 | 2007-12-18 | H2Gen Innovations, Inc. | Catalyst for hydrogen generation through steam reforming of hydrocarbons |
| JP5241488B2 (ja) * | 2005-05-06 | 2013-07-17 | オハイオ ユニバーシティ | 固体燃料スラリーから水素を生成する方法 |
| US7309479B2 (en) * | 2005-06-29 | 2007-12-18 | Samsung Engineering Co., Ltd. | Cobalt oxide catalysts |
| US8142756B1 (en) * | 2006-03-28 | 2012-03-27 | The United States Of America As Represented By The U.S. Department Of Energy | Methods of reforming hydrocarbon fuels using hexaaluminate catalysts |
| CN101679027B (zh) * | 2007-04-18 | 2013-01-02 | 乔治洛德方法研究和开发液化空气有限公司 | 氢气生产方法 |
| US20080260631A1 (en) * | 2007-04-18 | 2008-10-23 | H2Gen Innovations, Inc. | Hydrogen production process |
| US20090108238A1 (en) * | 2007-10-31 | 2009-04-30 | Sud-Chemie Inc. | Catalyst for reforming hydrocarbons |
| US8119558B2 (en) * | 2008-03-14 | 2012-02-21 | Süd-Chemie Inc. | Ultra high temperature shift catalyst with low methanation |
| US8293805B2 (en) * | 2008-05-29 | 2012-10-23 | Schlumberger Technology Corporation | Tracking feedstock production with micro scale gas-to-liquid units |
| JP5572855B2 (ja) * | 2009-03-24 | 2014-08-20 | 昭和シェル石油株式会社 | バイオマスガス化改質触媒及びこれを用いた合成ガスの製造方法 |
| US20100292076A1 (en) * | 2009-05-18 | 2010-11-18 | Sud-Chemie Inc. | Ultra high temperature shift catalyst with low methanation |
| US8735023B2 (en) * | 2009-12-14 | 2014-05-27 | GM Global Technology Operations LLC | Fuel cell with layered electrode |
| CN102451696B (zh) * | 2010-10-22 | 2014-03-05 | 中国石油化工股份有限公司 | 用于烃类蒸汽转化制取氢气或羰基合成气反应的催化剂 |
| CN102674413B (zh) * | 2011-03-16 | 2014-04-30 | 中国科学院过程工程研究所 | 一种用于co和h2甲烷化的催化剂及其制备方法 |
| CN102836718B (zh) * | 2011-06-20 | 2014-06-04 | 中国科学院过程工程研究所 | 一种介孔六铝酸盐负载镍基的甲烷化催化剂及其制备方法 |
| US20130071764A1 (en) * | 2011-09-15 | 2013-03-21 | John R. Budge | Systems and methods for steam reforming |
| JP5984620B2 (ja) * | 2011-11-29 | 2016-09-06 | 大阪瓦斯株式会社 | 炭化水素化合物類の水蒸気改質触媒およびその製造方法 |
| WO2013135656A1 (de) * | 2012-03-13 | 2013-09-19 | Bayer Intellectual Property Gmbh | Verfahren zur reduktion von kohlendioxid bei hohen temperaturen an mischmetalloxid-katalysatoren in form von hexaaluminaten |
| US20130270483A1 (en) * | 2012-04-12 | 2013-10-17 | Primus Green Energy Inc. | Catalytical gasifier configuration for biomass pyrolysis |
| CA2877956A1 (en) | 2012-06-29 | 2014-01-03 | Basf Se | High-pressure process for carbon dioxide reforming of hydrocarbons in the presence of iridium-containing active masses |
| WO2014135642A1 (de) * | 2013-03-07 | 2014-09-12 | Basf Se | Nickelhexaaluminathaltiger katalysator zur reformierung von kohlenwasserstoffen in gegenwart von kohlendioxid |
| JP6293416B2 (ja) * | 2013-03-07 | 2018-03-14 | 大阪瓦斯株式会社 | 炭化水素化合物類のオートサーマル改質方法 |
| EP2813286A1 (de) * | 2013-06-11 | 2014-12-17 | Evonik Industries AG | Reaktionsrohr und Verfahren zur Herstellung von Cyanwasserstoff |
| MX2016004436A (es) | 2013-10-11 | 2016-06-21 | Evonik Degussa Gmbh | Tubo de reaccion y metodo para producir cianuro de hidrogeno. |
| US10369549B2 (en) | 2015-03-20 | 2019-08-06 | Sabic Global Technologies B.V. | Use of nickel-manganese olivine and nickel-manganese spinel as bulk metal catalysts for carbon dioxide reforming of methane |
| KR101828791B1 (ko) * | 2016-07-27 | 2018-02-13 | 한국화학연구원 | 개질반응용 내열성 개선 모노리스 촉매 |
| CN109641203B (zh) * | 2016-08-08 | 2025-11-18 | 国立大学法人东京工业大学 | 氨合成用催化剂的制造方法以及氨的制造方法 |
| EP3301075A1 (en) | 2016-09-28 | 2018-04-04 | Evonik Degussa GmbH | Method for producing hydrogen cyanide |
| CN108262046A (zh) * | 2016-12-30 | 2018-07-10 | 中国石油天然气股份有限公司 | 掺杂六铝酸盐氧化物催化剂、其制备方法及其应用 |
| EP3553045B1 (en) * | 2018-04-09 | 2024-09-04 | Kemijski Institut | Process to produce methacrylic acid monomer from biomass-derived carboxylic acids |
| CN111389405B (zh) * | 2020-01-21 | 2023-09-15 | 天津大学 | 一种预活化甲烷水蒸气制氢催化剂的方法 |
| JP7643836B2 (ja) * | 2020-03-30 | 2025-03-11 | Eneos株式会社 | 水素供給システム、制御装置、及び水素製造方法 |
| CN114077519B (zh) * | 2020-08-21 | 2022-11-18 | 荣耀终端有限公司 | 一种系统服务恢复方法、装置和电子设备 |
| CN113509926B (zh) * | 2021-09-14 | 2021-12-03 | 清华大学 | 燃料重整催化剂前驱体、催化剂以及制备方法和应用 |
| KR102819305B1 (ko) * | 2021-11-26 | 2025-06-11 | 전남대학교산학협력단 | 헥사알루미네이트 지지체 포함 촉매 및 그 제조방법 |
| CN118002149B (zh) * | 2024-04-08 | 2024-07-26 | 中自科技股份有限公司 | 一种天然气水蒸气重整催化剂及反应器 |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4459372A (en) * | 1982-08-25 | 1984-07-10 | Uop Inc. | Surface-metallated refractory inorganic oxides, method of their preparation and catalyst supported on the oxides |
| US4572904A (en) * | 1983-09-27 | 1986-02-25 | Signal Applied Technologies Inc. | Lead-tolerant catalyst system for treating exhaust gas containing lead compounds |
| US4868148A (en) * | 1987-08-24 | 1989-09-19 | Allied-Signal Inc. | Layered automotive catalytic composite |
| US5015617A (en) * | 1988-04-14 | 1991-05-14 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Catalyst for purifying exhaust gas and method for production thereof |
| US5026536A (en) * | 1988-12-20 | 1991-06-25 | Nippon Oil Co., Ltd. | Hydrogen production from hydrocarbon |
| DE4106535A1 (de) * | 1991-03-01 | 1992-09-03 | Degussa | Monoklines zirkonoxid, verfahren zu seiner herstellung und verwendung |
| JPH07187665A (ja) * | 1993-11-10 | 1995-07-25 | Denki Kagaku Kogyo Kk | バリウムヘキサアルミネート粉末の製造方法 |
| FR2721837B1 (fr) * | 1994-07-01 | 1996-08-30 | Inst Francais Du Petrole | Catalyseur d'oxydation resistant a des temperatures elevees, son procede de preparation et procede de combustion utilisant un tel catalyseur |
| JP3377676B2 (ja) * | 1996-04-05 | 2003-02-17 | ダイハツ工業株式会社 | 排ガス浄化用触媒 |
| US5786294A (en) * | 1996-05-10 | 1998-07-28 | Northwestern University | Crystalline mesoporous zirconia catalysts having stable tetragonal pore wall structure |
| DE19653629A1 (de) * | 1996-12-20 | 1998-06-25 | Basf Ag | Monoklines Zirconiumoxid mit hoher Oberfläche |
| DE19654391A1 (de) * | 1996-12-27 | 1998-07-02 | Basf Ag | Katalysator zur selektiven Herstellung von Propylen aus Propan |
| JPH10287419A (ja) * | 1997-04-09 | 1998-10-27 | Kawai Sekkai Kogyo Kk | バリウムヘキサアルミネートの製造方法 |
| US6413489B1 (en) * | 1997-04-15 | 2002-07-02 | Massachusetts Institute Of Technology | Synthesis of nanometer-sized particles by reverse micelle mediated techniques |
| GB9806199D0 (en) * | 1998-03-24 | 1998-05-20 | Johnson Matthey Plc | Catalytic generation of hydrogen |
| DE19937107A1 (de) * | 1999-08-06 | 2001-02-08 | Basf Ag | Katalysator mit bimodaler Porenradienverteilung |
| US6607678B2 (en) * | 1999-08-17 | 2003-08-19 | Battelle Memorial Institute | Catalyst and method of steam reforming |
| US6497856B1 (en) * | 2000-08-21 | 2002-12-24 | H2Gen Innovations, Inc. | System for hydrogen generation through steam reforming of hydrocarbons and integrated chemical reactor for hydrogen production from hydrocarbons |
| US6509000B1 (en) * | 2000-08-31 | 2003-01-21 | Council Of Scientific And Industrial Research | Low temperature process for the production of hydrogen |
| US20020061277A1 (en) * | 2000-09-25 | 2002-05-23 | Engelhard Corporation | Non-pyrophoric water-gas shift reaction catalysts |
| US7166268B2 (en) * | 2000-11-08 | 2007-01-23 | Idemitsu Kosan Co., Ltd. | Catalyst for hydrocarbon reforming and method of reforming hydrocarbon with the same |
| DK1356863T3 (da) * | 2000-11-17 | 2009-06-02 | Osaka Gas Co Ltd | Fremgangsmåde til rensning af methan-holdig spildgas |
| US6562747B2 (en) * | 2000-12-19 | 2003-05-13 | Delphi Technologies, Inc. | Gas sensor electrolyte |
| US20020174603A1 (en) * | 2001-03-23 | 2002-11-28 | Shabbir Ahmed | Method for generating hydrogen for fuel cells |
| US6713040B2 (en) * | 2001-03-23 | 2004-03-30 | Argonne National Laboratory | Method for generating hydrogen for fuel cells |
| DE60223016T2 (de) * | 2001-05-23 | 2008-07-17 | Ecaps Ab | Sinterbeständiges katalysatormaterial und verfahren zu dessen herstellung |
| US7442669B2 (en) * | 2002-03-05 | 2008-10-28 | Tda Research, Inc. | Oxidation catalysts comprising metal exchanged hexaaluminate wherein the metal is Sr, Pd, La, and/or Mn |
| EP1487740A1 (en) * | 2002-03-13 | 2004-12-22 | Conocophillips Company | Controlled-pore catalyst structures and process for producing synthesis gas |
| US7214643B2 (en) * | 2002-03-22 | 2007-05-08 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Metal oxide and method for producing the same, and catalyst |
| US20040086432A1 (en) * | 2002-10-31 | 2004-05-06 | Labarge William J. | Reformer, systems utilizing the reformer, and methods for operating the systems |
| US7094730B2 (en) * | 2002-10-31 | 2006-08-22 | Delphi Technologies, Inc. | Gas treatment device, methods for making and using the same, and a vehicle exhaust system |
| US6774080B2 (en) * | 2002-11-25 | 2004-08-10 | Delphi Technologies, Inc. | Gas treatment device comprising SMSI material and methods for making and using the same |
| CN1729139A (zh) * | 2002-12-20 | 2006-02-01 | 本田技研工业株式会社 | 用于氢产生的催化剂配方 |
| US7399728B2 (en) * | 2003-12-19 | 2008-07-15 | Umicore Ag & Co Kg | Catalyst formulation, exhaust system, and gas treatment device |
| US7309480B2 (en) * | 2004-04-16 | 2007-12-18 | H2Gen Innovations, Inc. | Catalyst for hydrogen generation through steam reforming of hydrocarbons |
-
2004
- 2004-04-16 US US10/825,150 patent/US7309480B2/en not_active Expired - Lifetime
-
2005
- 2005-04-01 JP JP2007508378A patent/JP4988552B2/ja not_active Expired - Fee Related
- 2005-04-01 CN CN2005800114603A patent/CN101001695B/zh not_active Expired - Fee Related
- 2005-04-01 WO PCT/US2005/011037 patent/WO2005105664A2/en not_active Ceased
- 2005-04-01 CA CA2561427A patent/CA2561427C/en not_active Expired - Fee Related
- 2005-04-01 KR KR1020067023447A patent/KR101166943B1/ko not_active Expired - Fee Related
- 2005-04-01 AU AU2005238426A patent/AU2005238426B2/en not_active Ceased
- 2005-04-01 EP EP05732800A patent/EP1771376A4/en not_active Withdrawn
-
2006
- 2006-12-18 US US11/612,094 patent/US7645440B2/en not_active Expired - Lifetime
-
2007
- 2007-10-18 US US11/874,265 patent/US7923409B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| US20080038186A1 (en) | 2008-02-14 |
| CA2561427A1 (en) | 2005-11-10 |
| US7923409B2 (en) | 2011-04-12 |
| KR101166943B1 (ko) | 2012-07-19 |
| WO2005105664A2 (en) | 2005-11-10 |
| CA2561427C (en) | 2013-02-12 |
| US20050232857A1 (en) | 2005-10-20 |
| EP1771376A4 (en) | 2011-06-15 |
| CN101001695A (zh) | 2007-07-18 |
| US7645440B2 (en) | 2010-01-12 |
| CN101001695B (zh) | 2010-06-09 |
| AU2005238426B2 (en) | 2008-05-08 |
| WO2005105664A3 (en) | 2006-09-14 |
| JP2007532305A (ja) | 2007-11-15 |
| US7309480B2 (en) | 2007-12-18 |
| US20070116639A1 (en) | 2007-05-24 |
| KR20070044802A (ko) | 2007-04-30 |
| EP1771376A2 (en) | 2007-04-11 |
| AU2005238426A1 (en) | 2005-11-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4988552B2 (ja) | 炭化水素の蒸気改質によって水素を生成するための触媒 | |
| KR101994152B1 (ko) | 탄소 침적의 감소를 위해, 금속이온이 치환된 페로브스카이트 금속산화물 촉매 및 이의 제조 방법, 그리고 이를 이용한 메탄 개질 반응 방법 | |
| CN106362735B (zh) | 用于蒸汽重整过程的催化剂 | |
| US7378370B2 (en) | Process for the preparation of promoted calcium-aluminate supported catalysts | |
| JP5327323B2 (ja) | 炭化水素系ガス改質用触媒、その製造方法、および合成ガスの製造方法 | |
| US10010876B2 (en) | Catalyst for high temperature steam reforming | |
| US7906098B2 (en) | Method for making hydrogen using a gold containing water-gas shift catalyst | |
| US7378369B2 (en) | Nickel supported on titanium stabilized promoted calcium aluminate carrier | |
| US20100304236A1 (en) | Catalysts and methods including steam reforming | |
| KR102092736B1 (ko) | 탄소 침적을 감소시킬 수 있는, 금속이온이 치환된 페로브스카이트 금속산화물 촉매의 제조 방법 및 이를 이용한 메탄 개질 반응 방법 | |
| Tri et al. | High activity and stability of nano‐nickel catalyst based on LaNiO3 perovskite for methane bireforming | |
| JP4525909B2 (ja) | 水性ガスシフト反応用触媒及びその製造方法、並びに水性ガスの製造方法 | |
| JP5619598B2 (ja) | 銅−亜鉛−アルミニウム触媒、その製造方法、一酸化炭素変成方法、及び水素製造方法 | |
| KR20170027674A (ko) | 수증기 메탄 개질용 저온 고효율 니켈계 촉매 및 이의 이용 | |
| JP7347541B2 (ja) | 炭化水素改質触媒、炭化水素改質装置、および、炭化水素改質触媒の硫黄劣化回復方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080326 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20100317 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20100317 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20100827 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20100827 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110705 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111005 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111129 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120227 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120327 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120426 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150511 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |