JP2011515404A - 乾癬を治療するための方法 - Google Patents
乾癬を治療するための方法 Download PDFInfo
- Publication number
- JP2011515404A JP2011515404A JP2011500870A JP2011500870A JP2011515404A JP 2011515404 A JP2011515404 A JP 2011515404A JP 2011500870 A JP2011500870 A JP 2011500870A JP 2011500870 A JP2011500870 A JP 2011500870A JP 2011515404 A JP2011515404 A JP 2011515404A
- Authority
- JP
- Japan
- Prior art keywords
- antibody
- antigen
- binding portion
- subject
- amino acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images













































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/244—Interleukins [IL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
Abstract
Description
本願は、2008年3月18日に出願された米国仮特許出願61/069,840号、2008年9月8日に出願された米国仮特許出願61/095,275号及び2009年2月18日に出願された米国仮特許出願61/207,904号の利益を主張し、これらの各々の全内容は、参照により、本明細書に組み込まれる。
本願は、2009年3月10日に作製され、コンピュータ読み取り可能なフォーマット(CRF)でファイルされた、785kBのデータを含有するSequenceListing.txtという名前によって特定されるASCIIテキストファイルを参照によって組み込む。
a)1×10−9M又はそれ以下のIC50で、インビトロPHAアッセイにおいてフィトヘマグルチニン芽球増殖を阻害し、
b)配列番号25のアミノ酸配列を含む重鎖CDR3を有し、及び
c)配列番号26のアミノ酸配列を含む軽鎖CDR3を有する。
a)表面プラズモン共鳴によって測定された場合に、1×10−3秒−1又はそれ以下のkoff速度定数で、ヒトIL−12から解離し、
b)配列番号25のアミノ酸配列を含む重鎖CDR3を有し、及び
c)配列番号26のアミノ酸配列を含む軽鎖CDR3を有する。
配列番号26のアミノ酸配列を含む軽鎖CDR3ドメイン及び
配列番号25のアミノ酸配列を含む重鎖CDR3ドメイン
を含むヒト抗体又はその抗原結合部分である。
本発明は、乾癬の治療のために、ヒトIL−12に結合するヒト抗体又はその抗原結合部分を使用する方法及び組成物を提供する。本発明は、IL−12及びIL−23の両方を結合する抗体を使用する方法及び組成物も含む。好ましくは、本発明において使用されるヒト抗体は、組換え、中和ヒト抗hIL−12抗体である。

a)1×10−6M又はそれ以下のIC50で、インビトロPHAアッセイにおいてフィトヘマグルチニン芽球増殖を阻害し、
b)配列番号1のアミノ酸配列を含む重鎖CDR3を有し、及び
c)配列番号2のアミノ酸配列を含む軽鎖CDR3を有する、
を有する単離されたヒト抗体又はその抗原結合部分を含む方法及び組成物を提供する。
a)1×10−9M又はそれ以下のIC50で、インビトロPHAアッセイにおいてフィトヘマグルチニン芽球増殖を阻害し、
b)配列番号9のアミノ酸配列を含む重鎖CDR3を有し、及び
c)配列番号10のアミノ酸配列を含む軽鎖CDR3を有する、
を有する単離されたヒト抗体又はその抗原結合部分を含む。
a)1×10−9M又はそれ以下のIC50で、インビトロPHAアッセイにおいてフィトヘマグルチニン芽球増殖を阻害し、
b)配列番号17のアミノ酸配列を含む重鎖CDR3を有し、及び
c)配列番号18のアミノ酸配列を含む軽鎖CDR3を有する、
単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
a)インビトロでのPHAアッセイにおいて、1×10−9M又はそれ以下のIC50でフィトヘマグルチニン芽球増殖を阻害する、
b)配列番号404から配列番号469からなる群から選択されるアミノ酸配列を含む重鎖CDR3を有する、及び
c)配列番号534から配列番号579からなる群から選択されるアミノ酸配列を含む軽鎖CDR3を有する、
単離されたヒト抗体又はその抗原結合部分を特徴とする。
a)インビトロでのPHAアッセイにおいて、1×10−6M又はそれ以下のIC50でフィトヘマグルチニン芽球増殖を阻害し、
b)配列番号1のアミノ酸配列を含む重鎖CDR3、配列番号3のアミノ酸配列を含む重鎖CDR2及び配列番号5のアミノ酸配列を含む重鎖CDR1又は好ましい選択的突然変異誘発位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号1のアミノ酸配列を含む重鎖CDR3、配列番号3のアミノ酸配列を含む重鎖CDR2及び配列番号5のアミノ酸配列を含む重鎖CDR1を含む抗体より最大10倍高いkoff速度を有する。)を含み、並びに
c)配列番号2のアミノ酸配列を含む軽鎖CDR3、配列番号4のアミノ酸配列を含む軽鎖CDR2及び配列番号6のアミノ酸配列を含む軽鎖CDR1又は好ましい選択的突然変異誘発位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号2のアミノ酸配列を含む軽鎖CDR3、配列番号4のアミノ酸配列を含む軽鎖CDR2及び配列番号6のアミノ酸配列を含む軽鎖CDR1を含む抗体より最大10倍高いKoff速度を有する。)を含む、
単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
a)インビトロでのPHAアッセイにおいて、1×10−9M又はそれ以下のIC50でフィトヘマグルチニン芽球の増殖阻害し、
b)配列番号9のアミノ酸配列を含む重鎖CDR3、配列番号11のアミノ酸配列を含む重鎖CDR2及び配列番号13のアミノ酸配列を含む重鎖CDR1又は好ましい選択的突然変異誘発位置、接触位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号9のアミノ酸配列を含む重鎖CDR3、配列番号11のアミノ酸配列を含む重鎖CDR2及び配列番号13のアミノ酸配列を含む重鎖CDR1を含む抗体より最大10倍高いkoff速度を有する。)を含み、並びに
c)配列番号10のアミノ酸配列を含む軽鎖CDR3、配列番号12のアミノ酸配列を含む軽鎖CDR2及び配列番号14のアミノ酸配列を含む軽鎖CDR1又は好ましい選択的突然変異誘発位置、接触位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号10のアミノ酸配列を含む軽鎖CDR3、配列番号12のアミノ酸配列を含む軽鎖CDR2及び配列番号14のアミノ酸配列を含む軽鎖CDR1を含む抗体より最大10倍高いKoff速度を有する。)を含む、
単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
a)インビトロでのPHAアッセイにおいて、1×10−9M又はそれ以下のIC50でフィトヘマグルチニン芽球増殖を阻害し、
b)配列番号17のアミノ酸配列を含む重鎖CDR3、配列番号19のアミノ酸配列を含む重鎖CDR2及び配列番号21のアミノ酸配列を含む重鎖CDR1又は好ましい選択的突然変異誘発位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号17のアミノ酸配列を含む重鎖CDR3、配列番号19のアミノ酸配列を含む重鎖CDR2及び配列番号21のアミノ酸配列を含む重鎖CDR1を含む抗体より最大10倍高いkoff速度を有する。)を含み、並びに
c)配列番号18のアミノ酸配列を含む軽鎖CDR3、配列番号20のアミノ酸配列を含む軽鎖CDR2及び配列番号22のアミノ酸配列を含む軽鎖CDR1又は好ましい選択的突然変異誘発位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号18のアミノ酸配列を含む軽鎖CDR3、配列番号20のアミノ酸配列を含む軽鎖CDR2及び配列番号22のアミノ酸配列を含む軽鎖CDR1を含む抗体より最大10倍高いKoff速度を有する。)を含む、
単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
a)インビトロでのPHAアッセイにおいて、1×10−9M又はそれ以下のIC50でフィトヘマグルチニン芽球増殖を阻害し、
b)配列番号25のアミノ酸配列を含む重鎖CDR3、配列番号27のアミノ酸配列を含む重鎖CDR2及び配列番号29のアミノ酸配列を含む重鎖CDR1又は好ましい選択的突然変異誘発位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号25のアミノ酸配列を含む重鎖CDR3、配列番号27のアミノ酸配列を含む重鎖CDR2及び配列番号29のアミノ酸配列を含む重鎖CDR1を含む抗体より最大10倍高いkoff速度を有する。)を含み、並びに
c)配列番号26のアミノ酸配列を含む軽鎖CDR3、配列番号28のアミノ酸配列を含む軽鎖CDR2及び配列番号30のアミノ酸配列を含む軽鎖CDR1又は好ましい選択的突然変異誘発位置若しくは高頻度変異位置に1つ若しくはそれ以上のアミノ酸置換を有するこれらの変異体(前記変異体は、配列番号26のアミノ酸配列を含む軽鎖CDR3、配列番号28のアミノ酸配列を含む軽鎖CDR2及び配列番号30のアミノ酸配列を含む軽鎖CDR1を含む抗体より最大10倍高いKoff速度を有する。)を含む、
単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
本発明において使用され得る組換えヒト抗体は、ヒトリンパ球由来のmRNAから調製されたヒトVL及びVHcDNAを用いて調製された組換えコンビナトリアル抗体ライブラリー、好ましくはscFVファージディスプレイライブラリーのスクリーニングによって単離することができる。本発明の方法及び組成物において使用され得る抗体を同定するための方法は、参照により、本明細書に組み込まれる米国特許第6,914,128号に記載されている。このようなライブラリーを調製し、スクリーニングする方法は、本分野において公知である。ファージディスプレイライブラリーを作製するための市販のキット(例えば、Pharmacia Recombinant Phage Antibody System、カタログ番号27−9400−01;及びStratagene SurfZAPTMファージディスプレイキット、カタログ番号240612)に加えて、抗体ディスプレイライブラリーを作製及びスクリーニングする際に使用するのに特に適した方法及び試薬の例は、例えば、Kang他のPCT国際公開WO92/18619号;Winter他のPCT国際公開WO92/20791号;Breitling他のPCT国際公開WO93/01288号;McCafferty他のPCT国際公開WO92/01047号;Garrard他のPCT国際公開WO92/09690号;Fuchs et al.(1991)Bio/Technology 9:1370−1372;Hay et al.(1992)Hum Antibod Hybridomas3:81−85;Huse et al.(1989)Science246:1275−1281;McCafferty et al.,Nature(1990)348:552−554;Griffiths et al.(1993)EMBOJ12:725−734;Hawkins et al.(1992)JMolBiol226:889−896;Clackson et al.(1991)Nature352:624−628;Gram et al.(1992)PNAS89:3576−3580;Garrad et al.(1991)Bio/Technology9:1373−1377;Hoogenboom at al.(1999)NucAcidRes19:4133−4137;及びBarbas et al.(1991)PNAS88:7978−7982に見出すことができる。
a)ヒトIL−12に結合し、表面プラズモン共鳴によって測定された場合に0.1s−1若しくはそれ以下のKoff速度定数でヒトIL−12から解離し、又はインビトロでのフィトヘマグルチニン芽球増殖アッセイ(PHAアッセイ)において、1×10−6M又はそれ以下のIC50でフィトヘマグルチニン芽球増殖を阻害する。
b)VH3生殖系列ファミリーの一員から選択されるアミノ酸配列を含む重鎖可変領域(該重鎖可変領域は、接触又は高頻度変異位置に活性増強アミノ酸残基での変異を有する。)を有する。
c)Vλ1生殖系列ファミリーの一員から選択されるアミノ酸配列を含む軽鎖可変領域(該軽鎖可変領域は、好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に活性増強アミノ酸残基での変異を有する。)を有する。
を有する単離されたヒト抗体又はその抗原結合部分を特徴とする。
a)配列番号595から667からなる群から選択されるアミノ酸配列を含む重鎖可変領域(該重鎖可変領域は、好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に活性増強アミノ酸残基での変異を有する。)を有する。
b)配列番号669から675からなる群から選択されるアミノ酸配列を含む軽鎖可変領域(該軽鎖可変領域は、好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に活性増強アミノ酸残基での変異を有する。)を有する。
を有する単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
a)ヒトIL−12に結合し、表面プラズモン共鳴によって測定された場合に0.1s−1若しくはそれ以下のKoff速度定数でヒトIL−12から解離し、又はインビトロでのフィトヘマグルチニン芽球増殖アッセイ(PHAアッセイ)において、1×10−6M若しくはそれ以下のIC50でフィトヘマグルチニン芽球増殖を阻害する。
b)COS−3生殖系列アミノ酸配列を含む重鎖可変領域(該重鎖可変領域は、好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に活性増強アミノ酸残基での変異を有する。)を有する。
c)DPL8生殖系列アミノ酸配列を含む軽鎖可変領域(該軽鎖可変領域は、好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に活性増強アミノ酸残基での変異を有する。)を有する。
を有する単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
a)ヒトIL−12に結合し、表面プラズモン共鳴によって測定された場合に0.1s−1又はそれ以下のkoff速度定数でヒトIL−12から解離し、又はインビトロでのフィトヘマグルチニン芽球増殖アッセイ(PHAアッセイ)において、1×10−6M又はそれ以下のIC50でフィトヘマグルチニン芽球増殖を阻害する。
b)VH3生殖系列ファミリーの一員から選択されるアミノ酸配列を含む重鎖可変領域(前記重鎖可変領域は、他のVH3生殖系列ファミリーの一員から得られるCDR2と構造的に類似するCDR2及び他のVH3生殖系列ファミリーの一員から得られるCDR1と構造的に類似するCDR1を含み、並びに前記重鎖可変領域は、好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に活性増強アミノ酸残基による変異を有する。)を有する。
c)Vλ1生殖系列ファミリーの一員から選択されるアミノ酸配列を含む軽鎖可変領域(前記軽鎖可変領域は、他のVλ1生殖系列ファミリーの一員から得られるCDR2と構造的に類似するCDR2及び他のVλ1生殖系列ファミリーの一員から得られるCDR1と構造的に類似するCDR1を含み、並びに前記軽鎖可変領域は、好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に活性増強アミノ酸残基での変異を有する。)を有する。
を有する単離されたヒト抗体又はその抗原結合部分の使用を特徴とする。
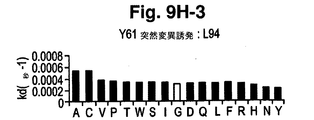
典型的には、改善された親和性を有する抗体の選択は、上記第II節及び本明細書中に組み込まれる米国特許第6,914,128号に記載されているように、ファージディスプレイ法を用いて実施することができる。これは、CDR残基の組み合わせを無作為に変異させること、及び異なる配列の抗体を含有する大規模なライブラリーを作製することによって達成することができる。しかしながら、これらの選択法が機能するために、抗体−抗原反応は、長時間にわたって、抗原に対してより高い親和性の抗体の優先的結合を可能にするために平衡状態となる傾向がなければならない。親和性のあるレベル(すなわち、抗体Y61のレベル)が達成された時点で、選択された抗IL−12抗体の親和性を改善するために、ファージディスプレイ法が使用された場合、(おそらく、抗原とファージ粒子間のさらなる非特異的相互作用のために)平衡状態を確立することができる選択条件を決定することはできない。従って、ファージディスプレイ法によって、さらに高い親和性を有する抗体を選択することはできない。従って、少なくともある種の抗体又は抗原に対しては、高度に改善された結合特異性/親和性を有する抗体を選択する能力において、ファージディスプレイ法には限界がある。従って、この限界を克服するために、抗体のファージディスプレイ親和性成熟を必要としない選択的突然変異誘発アプローチと名づけられた方法が確立され、本発明によって提供される。この選択的突然変異誘発アプローチは、ファージディスプレイ系を用いた限界を克服するために開発されたが、この方法は、ファージディスプレイ系とともに使用することもできることに注意すべきである。さらに、選択的突然変異誘発アプローチは、あらゆる抗体の活性を改善するために使用することができる。
1)好ましい選択的突然変異誘発位置、2)接触位置、3)高頻度変異位置の順序で候補位置を選択する工程、抗体の重鎖及び軽鎖可変領域内の位置の配置に基づいて前記位置に順位を付ける工程(CDR3はCDR2より好ましく、CDR2はCDR1より好ましい)、
順位の順に、候補の好ましい選択的突然変異誘発位置、高頻度変異及び/又は接触位置を、他の全ての可能なアミノ酸残基へと個別に変異させ、活性増強アミノ酸残基を決定するために、抗体の活性に対して各変異が及ぼす影響を分析する工程、
必要であれば、各活性増強アミノ酸残基の段階的な組み合わせを作製し、及び抗体の活性に対する様々な組み合わせの効果を分析する工程;活性増強アミノ酸残基を有する変異抗体を選択し、アミノ酸置換の免疫原性の可能性に関して、アミノ酸置換の位置と種類に基づいて変異抗体を順位付けする工程を含む。生殖系列データベース中に記載されている可変領域配列とほぼ同じアミノ酸配列を含む変異抗体又は他のヒト抗体と同等のアミノ酸配列を有する変異抗体に対して最も高い順位が与えられる。生殖系列配列又は別のヒト抗体の配列の何れかにおいて稀に遭遇されるアミノ酸置換を含有する変異抗体には、より低い順位が与えられる。生殖系列配列又は別のヒト抗体の配列中で遭遇されないアミノ酸置換を有する変異抗体には、最も低い順位が与えられる。上述されているように、CDR3中に配置された少なくとも1つの活性増強アミノ酸残基を含む変異抗体は、CDR2中に配置されたものより好ましく、CDR2中に配置されたものは、CDR1中に配置されたものより好ましい。重鎖可変領域のCDRは、軽鎖可変領域のCDRより好ましい。
a)親抗体又はその抗原結合部分を提供すること;
b)相補性決定領域(CDR)内の、1)好ましい選択的突然変異誘発位置、2)接触位置又は3)高頻度変異位置を順に変異のために選択することにより、選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を特定すること;
c)前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、これにより変異された抗体又はその抗原結合部分の群を作製すること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価すること;
e)少なくとも1つの他の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に対して、工程a)からd)を場合によって反復すること;
f)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された各変異を親抗体又はその抗原結合部分中で組み合わせること;並びに
g)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分に対して組み合わせ抗体又はその抗原結合部分の活性を評価すること、を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。好ましくは、選択された一又は複数の抗体は、上記親抗体の少なくとも1つの望ましい特徴又は特性を喪失せずに又は保持しながら、改善された活性を有する。望ましい特徴又は特性は、本分野で認知された技術を用いて、当業者によって測定又は観察され得る。
a)親抗体又はその抗原結合部分を提供すること;
b)相補性決定領域(CDR)内の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を変異のために選択すること;
c)これにより、変異された抗体又はその抗原結合部分の群を作製ために、前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異すること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)少なくとも1つの他の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に対して、工程a)からd)を場合によって反復すること;
f)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された2つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
g)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分と比べて、2つの活性増強アミノ酸残基を有する組み合わせ抗体又はその抗原結合部分の活性を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)親抗体又はその抗原結合部分を提供すること;
b)相補性決定領域(CDR)内の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を変異のために選択すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異すること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)少なくとも1つの他の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に対して、工程a)からd)を場合によって反復すること;
f)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された3つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
g)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分と比べて、2つの活性増強アミノ酸残基を有する組み合わせ抗体又はその抗原結合部分の活性を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)親抗体、その抗原結合部分を提供すること;
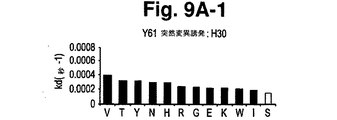
b)H30、H31、H31B、H32、H33、H52、H56、H58、L30、L31、L32、L50、L91、L92、L93、L94からなる群から選択される好ましい選択的突然変異誘発位置を選択すること;
c)変異された抗体又はその抗原結合部分の第一の群を作製するために、選択された好ましい選択的突然変異誘発位置を少なくとも2つの他のアミノ酸残基へ個別に変異すること;
d)単一の選択的突然変異誘発位置の変異が所定の目標活性又は部分的目標活性を有する抗体又はその抗原結合部分を与えるかどうかを決定するために、変異された抗体又はその抗原結合部分の前記第一の群の活性を評価すること;
e)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された各変異を、段階的な様式で、親抗体又はその抗原結合部分中に組み合わせること;
f)組み合わせ抗体又はその抗原結合部分が所定の目標活性又は部分的目標活性を有するかどうかを決定するために、組み合わせ抗体又はその抗原結合部分の活性を評価すること;
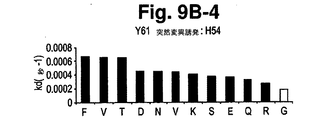
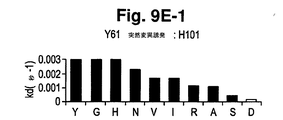
g)工程d)又はf)が所定の目標活性を有する抗体若しくはその抗原結合部分をもたらさず、又は部分活性を有するに過ぎない抗体をもたらした場合には、変異された抗体又はその抗原結合部分の第二の群を作製するために、H35、H50、H53、H54、H95、H96、H97、H98、L30A及びL96からなる群から選択されるさらなるアミノ酸残基を少なくとも2つの他のアミノ酸残基へ変異させること、
h)H35、H50、H53、H54、H95、H96、H97、H98、L30A及びL96からなる群から選択される単一のアミノ酸残基の変異が所定の目標活性又は部分活性を有する抗体又はその抗原結合部分をもたらすかどうかを決定するために、変異された抗体又はその抗原結合部分の前記第二の群の活性を評価すること;
i)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された工程g)の各変異を、段階的な様式で、親抗体又はその抗原結合部分中に組み合わせること;
j)組み合わせ抗体又はその抗原結合部分が所定の目標活性又は部分的目標活性を有するかどうかを決定するために、組み合わせ抗体又はその抗原結合部分の活性を評価すること;
k)工程h)又はj)が所定の目標活性を有する抗体若しくはその抗原結合部分をもたらさず、又は部分活性を有するに過ぎない抗体をもたらした場合には、変異された抗体又はその抗原結合部分の第三の群を作製するために、H33B、H52B及びL31Aからなる群から選択されるさらなるアミノ酸残基を少なくとも2つの他のアミノ酸残基へ変異させること;
l)H33B、H52B及びL31Aからなる群から選択される単一のアミノ酸残基の変異が所定の目標活性又は部分活性を有する抗体又はその抗原結合部分をもたらすかどうかを決定するために、変異された抗体又はその抗原結合部分の前記第三の群の活性を評価すること;
m)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された工程k)の各変異を、段階的な様式で、親抗体又はその抗原結合部分中に組み合わせること;
n)組み合わせ抗体又はその抗原結合部分が所定の目標活性を有するかどうかを決定するために、組み合わせ抗体又はその抗原結合部分の活性を評価し、これにより、所定の目標活性を有する抗体又はその抗原結合部分を与えること、を含む、所定の目標活性を達成するために、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない組換え親抗体又はその抗原結合部分を提供すること;
b)変異のために相補性決定領域(CDR)内の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を選択することにより、選択された接触又は高頻度変異位置を特定すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で前記パネルを発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価すること;
e)少なくとも1つの他の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に対して、工程b)からd)を場合によって反復すること;
f)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された各変異を親抗体又はその抗原結合部分中に組み合わせること;並びに
g)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分に対して組み合わせ抗体又はその抗原結合部分の活性を評価すること、
を含む、抗体又はその抗原結合部分の親和性を改善する方法を提供する。
a)組換え親抗体又はその抗原結合部分を提供すること;
b)変異のために相補性決定領域(CDR)内の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を選択することにより、選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を特定すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び適切な発現系中で前記パネルを発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性及び少なくとも1つの保持された特性又は特徴を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴に関して(前記特性又は特徴は、前記抗体中に保持される必要がある特性又は特徴である。)、親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない組換え親抗体又はその抗原結合部分を準備すること;
b)変異のために相補性決定領域(CDR)内の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を選択することにより、選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を特定すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で前記パネルを発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性及び少なくとも1つの保持された特性又は特徴を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴(該特性又は特徴は保持される必要がある特性又は特徴である。)に関して、親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群を評価すること、
f)少なくとも1つの他の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に対して、工程a)からe)を場合によって反復すること;
g)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性及び少なくとも1つの保持された特性又は特徴を有することが示された少なくとも2つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
h)親抗体又はその抗原結合部分と比べて改善された活性及び少なくとも1つの保持された他の特性又は特徴を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分に対して組み合わせ抗体又はその抗原結合部分の活性を評価すること、を含む、本発明は、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない組換え親抗体又はその抗原結合部分を提供すること;
b)変異のために相補性決定領域(CDR)内の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を選択することにより、選択された接触又は高頻度変異位置を特定すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で前記パネルを発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性及び少なくとも1つの保持された特性又は特徴を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴(該特性又は特徴は保持される必要がある特性又は特徴である。)に関して、親抗体又はその抗原結合部分に対して変異された抗体又はその抗原結合部分の前記群を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない組換え親抗体又はその抗原結合部分を提供すること;
b)変異のために相補性決定領域(CDR)内の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を選択することにより、選択された接触又は高頻度変異位置を特定すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された好ましい選択的突然変異誘発位置、接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で前記パネルを発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性及び少なくとも1つの保持された特徴を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴(該特性又は特徴は保持される必要がある特性又は特徴である。)に関して、親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群を評価すること;
f)少なくとも1つの他の好ましい選択的突然変異誘発位置、接触又は高頻度変異位置に対して、工程a)からe)を場合によって反復すること;
g)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性及び少なくとも1つの保持された他の特徴を有することが示された少なくとも2つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
h)親抗体又はその抗原結合部分と比べて、改善された活性及び少なくとも1つの保持された特性又は特徴を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分に対して組み合わせ抗体又はその抗原結合部分の活性を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
最終的には、ある抗体−抗原結合対中の全てのCDR残基は、何らかの手段によって、活性増強アミノ酸残基として必要とされることが同定され、及び/又は抗原への結合のために、及び/又は抗体の他の望ましい特性若しくは特徴を保持するために直接若しくは間接に必要とされることが同定される。このようなCDR残基は、「好ましい選択的突然変異誘発位置」と称される。特別な状況では、抗体及び抗原の同時結晶化並びに分子モデル化などの他の手段によっても、好ましい選択的突然変異誘発残基を同定することができることに注意すべきである。
a)親抗体又はその抗体結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;
c)変異された抗体又は変異された抗体の群又はこれらの抗原結合部分を作製するために、前記選択された位置を、例えば少なくとも2つの他のアミノ酸残基へ個別に変異すること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又は変異された抗体の群又はこれらの抗原結合部分の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴の変化に関して、親抗体又はその抗原結合部分に対して変異された抗体又は変異された抗体の群又はこれらの抗原結合部分を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)親抗体又はその抗原結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された位置を少なくとも2つの他のアミノ酸残基へ個別に変異すること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)b)の下で選択された位置でなく、並びにH30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96の位置でもない少なくとも1つの他のCDR位置に対して、工程b)からd)を反復すること;
f)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された少なくとも2つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
g)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分と比べて、2つの活性増強アミノ酸残基を有する組み合わせ抗体又はその抗原結合部分の活性を評価すること;
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない組換え親抗体又はその抗原結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;並びに
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された接触又は高頻度変異位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で前記群を発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴の変化に関して、親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない親抗体又はその抗原結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)b)の下で選択された位置でなく、並びにH30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94の位置でもない少なくとも1つの他の位置に対して、工程b)からd)を反復すること;
g)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有することが示された少なくとも2つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
h)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分と比べて、2つの活性増強アミノ酸残基を有する組み合わせ抗体又はその抗原結合部分の活性及び他の特性又は特徴を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)親抗体又はその抗原結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された位置を少なくとも2つの他のアミノ酸残基へ個別に変異すること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性及び保持された他の特性又は特徴を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴の変化に関して、親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群を評価すること;
を含む、他の特徴に影響を与えずに、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)親抗体又はその抗原結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された位置を少なくとも2つの他のアミノ酸残基へ個別に変異すること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)少なくとも1つの他の特徴又は特性の変化に関して、親抗体又はその抗原部分と比べて、変異された抗体又はその抗原結合部分の前記群を評価すること;
e)b)の下で選択された位置でなく、並びにH30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96の位置でもない少なくとも1つの他のCDR位置に対して、工程b)からe)を反復すること;
f)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有し及び少なくとも1つの他の特性又は特徴に影響を与えないことが示された少なくとも2つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
g)親抗体又はその抗原結合部分と比べて改善された活性及び少なくとも1つの保持された他の特性又は特徴を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分と比べて、2つの活性増強アミノ酸残基を有する組み合わせ抗体又はその抗原結合部分の活性及び少なくとも1つの他の特性又は特徴の保持を評価すること;
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない親抗体又はその抗原結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性を評価することにより、活性増強アミノ酸残基を同定すること;
e)親抗体又はその抗原結合部分と比べて改善された活性を有する抗体又はその抗原結合部分が得られるまで、少なくとも1つの他の特性又は特徴の変化に関して、親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群を評価すること;
を含む、抗体又はその抗原結合部分の親和性を改善する方法を提供する。
a)ファージディスプレイ系における選択によって取得されたが、前記ファージディスプレイ系中での突然変異誘発によって、その活性をさらに改善することができない親抗体又はその抗原結合部分を提供すること;
b)変異のために、H30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96以外の相補性決定領域(CDR)内のアミノ酸残基を選択すること;
c)変異された抗体又はその抗原結合部分の群を作製するために、前記選択された位置を少なくとも2つの他のアミノ酸残基へ個別に変異させ、及び非ファージディスプレイ系中で発現させること;
d)親抗体又はその抗原結合部分と比べて、変異された抗体又はその抗原結合部分の前記群の活性及び少なくとも1つの他の特性又は特徴の保持を評価することにより、活性増強アミノ酸残基を同定すること;
e)b)の下で選択された位置でなく、並びにH30、H31、H31B、H32、H33、H35、H50、H52、H52A、H53、H54、H56、H58、H95、H96、H97、H98、H101、L30、L31、L32、L34、L50、L52、L53、L55、L91、L92、L93、L94及びL96の位置でもない少なくとも1つの他のCDR位置に対して、工程b)からd)を反復すること;
f)組み合わせ抗体又はその抗原結合部分を形成するために、改善された活性を有し及び少なくとも1つの他の特性又は特徴に影響を与えないことが示された少なくとも2つの各活性増強アミノ酸残基を親抗体又はその抗原結合部分中で組み合わせること;並びに
g)親抗体又はその抗原結合部分と比べて改善された活性及び少なくとも1つの他の保持された特徴又は特性を有する抗体又はその抗原結合部分が得られるまで、親抗体又はその抗原結合部分に対して2つの活性増強アミノ酸残基を有する組み合わせ抗体又はその抗原結合部分の活性及び少なくとも1つの特性又は特徴の保持を評価すること、
を含む、抗体又はその抗原結合部分の活性を改善する方法を提供する。
本発明の抗体又は抗体部分は、宿主細胞中での、免疫グロブリン軽鎖及び重鎖遺伝子の組換え発現によって調製することが可能である。抗体を組換え的に発現させるために、軽鎖及び重鎖が宿主細胞中で発現されるように、及び好ましくは、宿主細胞がその中で培養されている培地(抗体は、この培地から回収することが可能である。)中に分泌されるように、抗体の免疫グロブリン軽鎖及び重鎖をコードするDNA断片を担持する1つ又はそれ以上の組換え発現ベクターで、宿主細胞が形質移入される。抗体重鎖及び軽鎖遺伝子を取得し、これらの遺伝子を組換え発現ベクター中に組み込み、ベクターを宿主細胞中に導入するために、「Sambrook,Fritsch and Maniatis(eds),Molecular Cloning;A Laboratory Manual, Second Edition, Cold Spring Harbor, N.Y.,(1989), Ausubel F.M. et al.(eds.)Current Protocols in Molecular Biology, Greene Publishing Associates,(1989)及びBossらによる米国特許第4,816,397号」に記載されているものなどの標準的な組換えDNA法が使用される。
a)配列番号31のアミノ酸を含む可変領域を有する抗体重鎖及び
b)配列番号32のアミノ酸配列を含む可変領域を有する抗体軽鎖
をコードする組換え発現ベクターを提供する。
本発明の抗体及び抗体部分は、対象に投与するのに適した医薬組成物中に取り込ませることが可能である。典型的には、医薬組成物は、本発明の抗体又は抗体部分と及び医薬として許容される担体とを含む。本明細書において使用される「医薬として許容される担体」には、生理的に適合性がある、全ての溶媒、分散媒、コーティング、抗菌剤及び抗真菌剤、等張剤及び吸収遅延剤などが含まれる。医薬として許容される担体の例には、水、生理的食塩水、リン酸緩衝化された生理的食塩水、デキストロース、グリセロール、エタノールなどの1つ又はそれ以上及びこれらの組み合わせが含まれる。多くの場合、組成物中に等張剤、例えば、糖、マニトール、ソルビトールなどの多価アルコール又は塩化ナトリウムを含むことが好ましい。医薬として許容される担体は、抗体又は抗体部分の保存寿命又は有効性を増大させる、湿潤剤又は乳化剤、防腐剤又は緩衝剤などの補助物質の微量をさらに含み得る。
本発明は、IL−12活性が有害である疾患に罹患している対象中のIL−12活性を阻害するための方法を提供する。一実施形態において、本発明は、IL−12抗体又はその抗原結合部分の単回用量を投与することを含む、乾癬を治療する方法を提供する。
インターロイキン−12は、関節リウマチなどの炎症性疾患において役割を果たしていると推測されている。関節リウマチ患者から得られた滑液中に誘導性IL−12p40メッセージが検出されており、関節リウマチを有する患者から得られた滑液中にIL−12が存在することが示されている(例えば、Morita et al,(1998)Arthritis and Rheumatism41:306−314参照)。関節リウマチ滑膜の下内層中に、IL−12陽性細胞が存在することが見出されている。本発明のヒト抗体及び抗体部分は、例えば、関節リウマチ、若年性関節リウマチ、ライム関節炎、リウマチ性脊椎炎、骨関節炎及び通風性関節炎を治療するために使用することができる。典型的には、抗体又は抗体部分は全身的に投与されるが、ある種の疾患に関しては、抗体又は抗体部分の局所投与が有益であり得る。本発明の抗体又は抗体部分は、自己免疫疾患の治療において有用な1つ又はそれ以上のさらなる治療剤とともに投与することもできる。
インターロイキン−12は、炎症性腸疾患であるクローン病においても役割を果たしている。IFN−γ及びIL−12の増加した発現が、クローン病を有する患者の腸粘膜中で起こる(例えば、Fais et al,(1994)J.Interferon Res.14:235−238;Parronchi et al.,(1997)Amer.J.Pathol.150:823−832;Monteleone et al,(1997)Gastroenterology112:1169−1178;Berrebi et al,(1998)Amer.J.Pathol.152:667−672参照)。抗IL−12抗体は、大腸炎のマウスモデル(例えば、TNBS誘導性大腸炎IL−2ノックアウトマウス及び最近では、IL−10ノックアウトマウス)において、疾病を抑制することが示された。従って、本発明の抗体及び抗体部分は、炎症性腸疾患の治療において使用することができる。
インターロイキン−12は、多発性硬化症の中心的媒介物質であると推測されている。誘導性IL−12p40メッセージ又はIL−12自体の発現が、多発性硬化症を有する患者の病変中に示され得る(Windhagen et al,(1995)J.Exp.Med.182:1985−1996,Drulovic et al,(1997)J.Neurol.Sci.147:145−150)。多発性硬化症を有する慢性の進行性患者は、IL−12の上昇した循環レベルを有する。多発性硬化症を有する患者から得たT細胞及び抗原提示細胞(APC)を用いた研究によって、Th1型免疫応答をもたらす進行性多発性硬化症の基礎として、自己永続的な一連の免疫相互作用が明らかとなった。T細胞からのIFN−γの増加した分泌は、APCによる増加したIL−12産生をもたらし、増加したIL−12産生は、Th1型免疫活性化及び疾病の慢性状態に至るサイクルを永続化した(Balashov et al,(1997)Proc.Natl.Acad.Sci.94:599−603)。多発性硬化症におけるIL−12の役割は、多発性硬化症のマウス及びラット実験的アレルギー性脳脊髄炎(EAE)モデルを用いて調べられてきた。マウスでの多発性硬化症の再発性寛解型EAEモデルにおいて、抗IL−12mAbでの前処理は麻痺を遅延させ、臨床スコアを低下させた。麻痺のピーク時又はその後の寛解期の間における抗IL−12mAbでの処理は、臨床スコアを低下させた。従って、本発明の抗体又はその抗原結合部分は、ヒト中の多発性硬化症を伴う症候を緩和させる役割を果たし得る。
インターロイキン−12は、インシュリン依存性糖尿病(IDDM)の重要な媒介物質として推測されてきた。IL−12の投与によって、NODマウス中にIDDMが誘導され、抗IL−12抗体はIDDMの養子免疫伝達モデルにおいて保護的であった。初期発症IDDM患者は、幾つかの残存する膵島細胞機能が維持されている所謂「ハネムーン期間」をしばしば経験する。これらの残存する膵島細胞はインシュリンを産生し、投与されたインシュリンより良好に血中グルコースレベルを制御する。これらの初期発症患者の抗IL−12抗体での処理は、膵島細胞のさらなる破壊を抑制し、これにより、インシュリンの体内源を維持し得る。
インターロイキン−12(IL−12)及び関連するサイトカインIL−23は、乾癬における中心的媒介物質として推測されている。乾癬は、TH1型サイトカイン発現プロファイルを伴う急性及び慢性の皮膚病変を伴う。(Hamid et al.(1996)J.Allergy Clin.Immunol.1:225−231;Turka et al.(1995)Mol.Med.1:690−699)。IL−12及びIL−23は何れも、乾癬におけるI型Tヘルパー細胞(Th1)免疫応答の発達に寄与する。さらに、IL−12p40及びIL−23p40メッセンジャーRNAは、乾癬の皮膚病変中で過剰発現される。従って、本発明の抗体又はその抗原結合部分は、乾癬などの慢性的皮膚疾患を緩和させる役割を果たし得る。
慢性尋常性乾癬(plaque psoriasis)(「psoriasis vulgaris」とも称される。)は、乾癬の最も一般的な形態である。慢性尋常性乾癬は、硬貨の大きさからずっと大きいサイズにわたる、皮膚の隆起した紅斑によって特徴付けられる。慢性尋常性乾癬において、斑は単一又は複数であり得、数ミリメートルから数センチメートルのサイズを変動し得る。通常、斑は、落屑性表面を有する赤であり、穏やかに引っ掻いたときに光を反射して、「銀色」効果を引き起こす。慢性尋常性乾癬から得られる(しばしば、対称性である)病変は、全身に発生するが、膝、肘、腰仙部領域、頭皮及び爪などの伸展性表面に好発する。時々、慢性尋常性乾癬は、陰茎、外陰部及び屈曲部にも発生し得るが、落屑は、通常存在しない。慢性尋常性乾癬を有する患者の診断は、上に記載されている臨床的な特徴を通常基礎とする。特に、慢性尋常性乾癬中の病変の分布、色及び典型的な銀色の落屑は、慢性尋常性乾癬の特徴である。
滴状乾癬は、特徴的な水滴形状のうろこ状斑を有する乾癬の形態を表す。感染症、特に、連鎖球菌性咽喉炎の後に、滴状乾癬の紅斑が通常発生する。滴状乾癬の診断は、皮膚の外観及び最近の咽喉痛の病歴がしばしば存在するという事実に通常基づく。
反転型乾癬は、患者が、尋常性感染に伴う落屑とは異なり、赤く炎症した皮膚の滑らかで、通常湿性の領域を有する乾癬の形態である。反転型乾癬は、間擦性乾癬(intertiginous psoriasis)又は湾曲性乾癬(flexural psoriasis)とも称される。反転型乾癬は、腋窩、鼠蹊部、乳房の下並びに性器及び臀部周囲のその他の皮膚の襞に多く発生し、発症部位の故に、摩擦及び発汗は罹患部位に刺激を与え得る。
膿疱性乾癬(掌蹠乾癬(palmar plantar psoriasis)とも称される。)は、サイズ及び位置が変動するが、手と足にしばしば発生する膿疱を引き起こす乾癬の形態である。水疱は限局し、又は身体の広範囲に広がり得る。膿疱性乾癬は、圧痛性及び有痛性であり得、発熱を引き起こし得る。
IL−12及び/又はIL−23抗体で治療することができる乾癬性疾患の他の例には、乾癬性紅皮症、尋常性、IBDを伴う乾癬及び関節リウマチを含む関節炎を伴う乾癬が含まれる。
ABT−874は、インターロイキン−12(IL−12)及びIL−23に対する完全ヒト抗体である。ABT−874は、ともに乾癬(Ps)の治療における有効性が確認された標的であるIL−12及びIL−23の両方に共通するp40サブユニットへ高い親和性で結合する。
ABT−874は、インターロイキン12(IL−12)及びIL−23に対する完全ヒト抗体である。ABT−874は、乾癬(Ps)の治療における有効性が確認された標的であるIL−12及びIL−23の両方に共通するp40サブユニットへ高い親和性で結合する。この第II相の研究の目的は、中度ないし重度の尋常性乾癬の治療におけるABT−874の皮下注射の有効性及び安全性を評価することであった。
12週の第II相無作為化対照試験及び36週の追跡期間において、ABT−874の有効性及び安全性を評価した。以下の実施例の目的は、中度ないし重度の尋常性乾癬の治療において、ABT−874の皮下注射のこの第II相試験の2番目の12週の間に治療を中断した後における応答の維持を分析することであった。
1)100mgABT−874、隔週(eow)12週間、
2)第0週目に1回200mgABT−874を投薬、
3)200mgのABT−874、毎週4週間、
4)200mgABT−874、隔週12週間、
5)200mgABT−874を毎週12週間又は
6)プラセボ。
以下の実施例の目的は、臨床的に安定な中度ないし重度の慢性尋常性乾癬を有する患者の治療において、プラセボと比較したヒトIL−12/23モノクローナル抗体(ABT−874)の広範囲の用量の有効性及び安全性を示すことであった。
A.研究デザイン:以下の研究は、米国(16ヶ所)及びカナダ(8ヶ所)内の24の施設で行われた12週の複数施設、無作為化二重盲検第II相プラセボ対照化試験であった。ABT−874(Abbott Laboratories, Abbott Park, IL)は、IL−12/23p40サブユニットタンパク質に対して高い親和性を有する遺伝子操作された相補性決定領域を有するヒトモノクローナル抗体である。6つの治療の1つを与えるために、1:1:1:1:1:1の比で患者を無作為に割り振った。ABT−874の200mg、0週目に1回投薬(200mg×1);12週間、隔週でABT−874の100mg(100mg隔週);最初の4週間毎週、ABT−874の200mg;12週間、隔週でABT−874の200mg(200mg隔週);12週間毎週、ABT−874の200mg(200mg毎週)又はプラセボ。12週後に、乾癬面積及び重度指数応答の少なくとも75%の低下(PASI75)を達成した全ての患者が、36週の盲検化された観察/再治療相を続けた。
A.患者:合計180人の患者が参加し、6つの治療群のうちの1つに無作為に振り分けた(図1)。患者の大半(プラセボ処置された患者の76.7%及びABT−874治療群の全患者の98%)は、研究の12週部分を完了した。
本実施例に記載されている第II相、複数施設無作為化二重盲検プラセボ対照化試験は、中度ないし重度の慢性尋常性乾癬の治療における統計学的に及び臨床的に有意なABT−874の有効性を示した。ABT−874200mg×1治療群を除き、全てのABT−874治療群中の患者の90%又はそれ以上が、プラセボ処理された患者の3.3%と比べて、12週までに、PASI75又はそれ以上を達成した。研究薬を1回だけ投薬された群(200mg×1)でさえ、患者の過半数(63.3%)は、12週までに、少なくともPASI75を達成した。さらに、ABT−874で処理された患者のほぼ100%が、12週までにPASI50又はそれ以上に達し、これは、臨床的に有意な改善であると考えられる(Carlin CS, Feldman SR, Krueger JG, Menter A, Krueger GG.J Am Acad Dermatol 2004; 50:859−66)。PASI90及び無又は最小限のPGAなどの他の二次的評価項目に対する結果は、一次的有効性分析と合致しており、一次的有効性分析を支持した。
12週の第II相無作為化対照試験及び36週の追跡相において、ABT−874の有効性及び安全性を評価した。以下の実施例の目的は、中度ないし重度の尋常性乾癬の治療において、ABT−874の皮下注射のこの第II相試験の2番目の12週の間に治療を中断した後における応答の維持を分析することであった。
1)100mgABT−874、隔週(eow)12週間、
2)第0週目に1回200mgABT−874投薬、
3)毎週4週間、200mgABT−874、
4)隔週で12週間、200mgABT−874
5)毎週12週間、200mgABT−874又は
6)プラセボ。
12週の初期治療相及び初期治療に対して応答した患者の36週再治療相を含む48週の第II相無作為化対照化試験において、ABT−874の有効性及び安全性を評価した。最初の12週の有効性の結果及び応答の維持の結果は、上記実施例に記載されている。以下の実施例の目的は、中度ないし重度の尋常性乾癬の治療において、ABT−874の皮下注射のこの第II相研究の初期応答を失った患者での36週再治療/追跡相の間の再治療の応答を調べることであった。以下の実施例のさらなる目的は、48週を通じて、中度ないし重度の尋常性乾癬の治療におけるABT−874の皮下注射の安全性を調べることであった。
無作為化された二重盲検プラセボ対照化用量変動研究において、ABT−874のある範囲の用量の耐容性、安全性及び薬物速度論(PK)を評価した。以下の実施例の目的は、健康なボランティアにおけるABT−874の静脈内(IV)及び皮下(SC)注射の薬物速度論を調査することであった。
・Cmax 最大血清濃度(μg/mL)
・Tmax Cmaxに到達するまでの時間(時間)
・AUC 血清濃度−時間曲線下面積(μg×時間/mL)
・t1/2 半減期(時間)
・CL クリアランス(mL/時間)(静脈内投与に関して)
・Vz 分布の容積(mL)(静脈内投与に関して)
・CL/F 見かけのCL(mL/時間)(皮下投与に関して)
・V/F 見かけのVZ(mL)(皮下投与に関して)
12週の初期治療相及び初期治療に対して応答した患者の36週再治療相を含む48週の第II相無作為化対照化試験において、ABT−874の有効性及び安全性を評価した。最初の12週の有効性の結果及び応答の維持の結果は、上記実施例1から5に記載されている。以下の実施例の目的は、中度ないし重度の尋常性乾癬の治療において、ABT−874の皮下注射のこの第II相研究の初期応答を失った患者での36週再治療/追跡相の間における再治療応答を調べることであった。以下の実施例のさらなる目的は、48週を通じて、中度ないし重度の尋常性乾癬の治療におけるABT−874の皮下注射の安全性を調べることであった。
10%以上の体表面積を冒し、及び12以上の乾癬面積及び重度指数(PASI)スコアの乾癬を有する成人を、6つの治療群の1つに無作為に割り当てた。1)0週目に、200mg用量のABT−874を1回;2)隔週に12週間ABT−874の100mg;3)毎週4週間ABT−874の200mg;4)隔週に12週間ABT−874の200mg;5)毎週12週間ABT−874の200mg又は6)プラセボ。一次的評価項目は、12週目でのPASI75以上の応答であった。一次的評価項目に合致した患者は、36週の再治療相に進んだ。研究薬を用いた治療を中断し、12から36週の間に応答を失った(PASI50以下)患者は、最初の12週の間に割り当てられたのと同じ投薬治療計画を用いた再治療を受けた。再治療は、12週間継続した。処置に関わらず、研究の持続期間全体にわたって、又は中断まで、全ての患者をモニターした。
当業者は、定型的な実験操作のみを用いて、本明細書中に記載されている本発明の具体的な実施形態に対する多くの均等物を認識し、又は確認することができる。このような均等物は、以下の特許請求の範囲によって包含されるものとする。
Claims (69)
- IL−12及び/又はIL−23のp40サブユニットのエピトープに結合することができる抗体又はその抗原結合部分を対象に投与することを含み、前記対象が前記抗体又はその抗原結合部分の最初の投与後の第一の長期間にわたって少なくともPASI75応答を維持し、前記対象が抗体又はその抗原結合部分の投与の中断後に応答の喪失を示し、及び前記対象が前記抗体又はその抗原結合部分の再投与後の第二の長期間にわたって少なくともPASI75応答を維持し、これにより、前記対象中の乾癬を治療する、対象中の乾癬を治療する方法。
- 第一の長期間が少なくとも約12週である、請求項1に記載の方法。
- 抗体の投与が少なくとも約12週間中断される、請求項1に記載の方法。
- 第二の長期間が少なくとも約12週である、請求項1に記載の方法。
- 抗体又はその抗原結合部分が隔週に投与される、請求項1に記載の方法。
- 抗体又はその抗原結合部分が毎週投与される、請求項1に記載の方法。
- 抗体又はその抗原結合部分が単回投薬で投与される、請求項1に記載の方法。
- 抗体又はその抗原結合部分が約200mgの用量で投与される、請求項1に記載の方法。
- 抗体又はその抗原結合部分が約100mgの用量で投与される、請求項1に記載の方法。
- 乾癬が慢性の乾癬である、請求項1に記載の方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープへ結合することができる抗体又はその抗原結合部分の単回投薬を対象に投与することを含み、抗体又はその抗原結合部分の投与後に、少なくとも約3日の半減期、約4日未満又は約4日に等しいTmax及び少なくとも約40%の生物学的利用可能性からなる群から選択される少なくとも1つの薬物速度論的特徴が達成される、対象中の乾癬を治療する方法。
- 少なくとも約8日の半減期が達成される、請求項11に記載の方法。
- 約3日未満又は約3日に等しいTmaxが達成される、請求項11に記載の方法。
- 少なくとも約60%の生物学的利用可能性が達成される、請求項11に記載の方法。
- 抗体が静脈内注射を介して投与される、請求項11に記載の方法。
- 抗体が皮下注射を介して投与される、請求項11に記載の方法。
- 単回投薬が抗体又はその抗原結合部分の約0.1と約5.0mg/kgの間である、請求項11に記載の方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープに結合することができる抗体又はその抗原結合部分を対象に投与することを含み、前記対象が前記抗体又はその抗原結合部分の最初の投与後の第一の長期間にわたって少なくともPASI75応答を維持し、前記対象が抗体又はその抗原結合部分の投与の中断後に応答の喪失を示し、及び前記対象が前記抗体又はその抗原結合部分の再投与後の第二の長期間にわたって少なくともPASI50応答を維持し、これにより、前記対象中の乾癬を治療する、対象中の乾癬を治療する方法。
- 第一の長期間が少なくとも約12週である、請求項18に記載の方法。
- 抗体の投与が少なくとも約12週間中断される、請求項18に記載の方法。
- 第二の長期間が少なくとも約12週である、請求項18に記載の方法。
- 抗体又はその抗原結合部分が隔週に投与される、請求項18に記載の方法。
- 抗体又はその抗原結合部分が毎週投与される、請求項18に記載の方法。
- 抗体又はその抗原結合部分が単回投薬で投与される、請求項18に記載の方法。
- 抗体又はその抗原結合部分が約200mgの用量で投与される、請求項18に記載の方法。
- 抗体又はその抗原結合部分が約100mgの用量で投与される、請求項18に記載の方法。
- 乾癬が慢性の乾癬である、請求項18に記載の方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープに結合することができる抗体又はその抗原結合部分を対象に投与することを含み、前記対象が前記抗体又はその抗原結合部分の最初の投与後の第一の長期間にわたって少なくともPASI75応答を維持し、前記対象が抗体又はその抗原結合部分の投与の中断後に応答の喪失を示し、及び前記対象が前記抗体又はその抗原結合部分の再投与後の第二の長期間にわたって無の又は最小限のPGAスコアを維持し、これにより、前記対象中の乾癬を治療する、対象中の乾癬を治療する方法。
- 第一の長期間が少なくとも約12週である、請求項28に記載の方法。
- 抗体の投与が少なくとも約12週間中断される、請求項28に記載の方法。
- 第二の長期間が少なくとも約12週である、請求項28に記載の方法。
- 抗体又はその抗原結合部分が隔週に投与される、請求項28に記載の方法。
- 抗体又はその抗原結合部分が毎週投与される、請求項28に記載の方法。
- 抗体又はその抗原結合部分が単回投薬で投与される、請求項28に記載の方法。
- 抗体又はその抗原結合部分が約200mgの用量で投与される、請求項28に記載の方法。
- 抗体又はその抗原結合部分が約100mgの用量で投与される、請求項28に記載の方法。
- 乾癬が慢性の乾癬である、請求項28に記載の方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープへ結合することができる抗体又はその抗原結合部分の単回投薬を対象に投与することを含み、抗体又はその抗原結合部分の投与後に、約0.15と約150μg/mLの間の最大血清濃度(Cmax)及び約80と約13,000μg×時間/mLの間の血清濃度時間曲線下面積(AUC)からなる群から選択される少なくとも1つの薬物速度論的特徴が達成される、対象中の乾癬を治療する方法。
- 抗体が静脈内注射を介して投与される、請求項38に記載の方法。
- Cmaxが約1と約150μg/mLの間である、請求項39に記載の方法。
- AUCが約145と約13,000μg×時間/mLの間である、請求項39に記載の方法。
- 抗体が皮下注射を介して投与される、請求項38に記載の方法。
- Cmaxが約0.15と約20μg/mLの間である、請求項42に記載の方法。
- AUCが約80と約5,000μg×時間/mLの間である、請求項42に記載の方法。
- 単回投薬が抗体又はその抗原結合部分の約0.1と約5.0mg/kgの間である、請求項38に記載の方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープに結合することができる抗体又はその抗原結合部分の単回投薬を対象に投与することを含み、約30と約600mL/時間の間のクリアランス(CL)及び約8と約11Lの間の分布容積(Vz)からなる群から選択される少なくとも1つの薬物速度論的特徴が抗体又はその抗原結合部分の静脈内投与後に達成される、対象中の乾癬を治療する方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープに結合することができる抗体又はその抗原結合部分の単回投薬を対象に投与することを含み、約90と約250mL/時間の間の見かけのクリアランス(CL/F)及び約23と約67Lの間の見かけの分布容積(V/F)からなる群から選択される少なくとも1つの薬物速度論的特徴が抗体又はその抗原結合部分の皮下投与後に達成される、対象中の乾癬を治療する方法。
- 単回投薬が抗体又はその抗原結合部分の約0.1と約5.0mg/kgの間である、請求項46又は47に記載の方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープに結合することができる抗体又はその抗原結合部分を対象に投与することを含み、前記対象が前記抗体又はその抗原結合部分の最初の投与後にPASI75応答を示し、前記対象が抗体又はその抗原結合部分の投与の中断後に応答の喪失を示し、及び前記対象が前記抗体又はその抗原結合部分の再投与後の約25日までに、少なくともPASI75応答を示し、これにより、前記対象中の乾癬を治療する、対象中の乾癬を治療する方法。
- 対象が、抗体又はその抗原結合部分の再投与後の約50日までに、少なくともPASI75応答を示す、請求項49に記載の方法。
- 対象が、抗体又はその抗原結合部分の再投与後の約60日までに、少なくともPASI75応答を示す、請求項49に記載の方法。
- 抗体の最初の投与が少なくとも約12週間である、請求項49に記載の方法。
- 抗体の投与が少なくとも約8週間中断される、請求項49に記載の方法。
- 抗体又はその抗原結合部分が隔週に投与される、請求項49に記載の方法。
- 抗体又はその抗原結合部分が毎週投与される、請求項49に記載の方法。
- 抗体又はその抗原結合部分が単回投薬で投与される、請求項49に記載の方法。
- 抗体又はその抗原結合部分が約200mgの用量で投与される、請求項49に記載の方法。
- 抗体又はその抗原結合部分が約100mgの用量で投与される、請求項49に記載の方法。
- 乾癬が慢性の乾癬である、請求項49に記載の方法。
- IL−12及び/又はIL−23のp40サブユニットのエピトープに結合することができる抗体又はその抗原結合部分を対象に投与することを含み、前記対象が前記抗体又はその抗原結合部分の最初の投与後にPASI75応答を示し、前記対象が抗体又はその抗原結合部分の投与の中断後、約60日までに応答の喪失を示し、及び前記対象が前記抗体又はその抗原結合部分の再投与後にPASI75応答を達成し、これにより、前記対象中の乾癬を治療する、対象中の乾癬を治療する方法。
- 対象が、抗体又はその抗原結合部分の投与の中断後の約120日までに、応答の喪失を示す、請求項60に記載の方法。
- 対象が、抗体又はその抗原結合部分の投与の中断後の約180日までに、応答の喪失を示す、請求項60に記載の方法。
- 抗体の最初の投与が少なくとも約12週間である、請求項60に記載の方法。
- 抗体又はその抗原結合部分が隔週に投与される、請求項60に記載の方法。
- 抗体又はその抗原結合部分が毎週投与される、請求項60に記載の方法。
- 抗体又はその抗原結合部分が単回投薬で投与される、請求項60に記載の方法。
- 抗体又はその抗原結合部分が約200mgの用量で投与される、請求項60に記載の方法。
- 抗体又はその抗原結合部分が約100mgの用量で投与される、請求項60に記載の方法。
- 乾癬が慢性の乾癬である、請求項60に記載の方法。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6984008P | 2008-03-18 | 2008-03-18 | |
| US61/069,840 | 2008-03-18 | ||
| US9527508P | 2008-09-08 | 2008-09-08 | |
| US61/095,275 | 2008-09-08 | ||
| US20790409P | 2009-02-18 | 2009-02-18 | |
| US61/207,904 | 2009-02-18 | ||
| PCT/US2009/036765 WO2009117289A2 (en) | 2008-03-18 | 2009-03-11 | Methods for treating psoriasis |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2011515404A true JP2011515404A (ja) | 2011-05-19 |
| JP2011515404A5 JP2011515404A5 (ja) | 2012-04-26 |
Family
ID=41091468
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011500870A Ceased JP2011515404A (ja) | 2008-03-18 | 2009-03-11 | 乾癬を治療するための方法 |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US8178092B2 (ja) |
| EP (2) | EP2274333A4 (ja) |
| JP (1) | JP2011515404A (ja) |
| KR (1) | KR20100126515A (ja) |
| CN (1) | CN102037011A (ja) |
| AU (1) | AU2009225797A1 (ja) |
| BR (1) | BRPI0908715A2 (ja) |
| CA (1) | CA2717569A1 (ja) |
| HK (1) | HK1203516A1 (ja) |
| IL (1) | IL208069A0 (ja) |
| MX (1) | MX2010010265A (ja) |
| NZ (2) | NZ587765A (ja) |
| RU (2) | RU2497545C2 (ja) |
| SG (1) | SG188909A1 (ja) |
| TW (2) | TW201513883A (ja) |
| WO (1) | WO2009117289A2 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016516686A (ja) * | 2013-03-15 | 2016-06-09 | アムジェン インコーポレイテッド | 抗il23抗体を使用してクローン病を治療するための方法 |
| JP2016517408A (ja) * | 2013-03-15 | 2016-06-16 | アムジェン インコーポレイテッド | 抗il−23抗体を用いた乾癬の治療方法 |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6914128B1 (en) | 1999-03-25 | 2005-07-05 | Abbott Gmbh & Co. Kg | Human antibodies that bind human IL-12 and methods for producing |
| NZ578065A (en) * | 2007-01-16 | 2012-09-28 | Abbott Lab | Methods for treating psoriasis with an antibody which binds to an epitope |
| NZ598881A (en) * | 2007-03-29 | 2013-11-29 | Abbvie Inc | Crystalline anti-human il-12 antibodies |
| NZ587765A (en) | 2008-03-18 | 2013-02-22 | Abbott Lab | Methods for treating psoriasis |
| US20090269398A1 (en) * | 2008-04-26 | 2009-10-29 | Vilambi Nrk Reddy | Compositions for the encapsulation of natural product extracts in oil medium in hard gelatin capsules and a method of encapsulation |
| WO2010062896A1 (en) * | 2008-11-28 | 2010-06-03 | Abbott Laboratories | Stable antibody compositions and methods for stabilizing same |
| KR20140048229A (ko) * | 2009-09-14 | 2014-04-23 | 애브비 인코포레이티드 | 건선을 치료하는 방법 |
| JO3244B1 (ar) | 2009-10-26 | 2018-03-08 | Amgen Inc | بروتينات ربط مستضادات il – 23 البشرية |
| WO2011146727A1 (en) * | 2010-05-19 | 2011-11-24 | Philip Bosch | Methods of treating interstitial cystitis |
| JP2013542209A (ja) * | 2010-10-06 | 2013-11-21 | アッヴィ・インコーポレイテッド | 乾癬を治療するための方法 |
| EP3869201A1 (en) * | 2013-03-12 | 2021-08-25 | Abbott Laboratories | Reagents, systems and methods for analyzing white blood cells |
| US10858422B2 (en) | 2016-05-31 | 2020-12-08 | Abcentra, Llc | Methods for treating systemic lupus erythematosus with an anti-apolipoprotein B antibody |
| KR20210044742A (ko) * | 2018-05-29 | 2021-04-23 | 앱센트라 엘엘시 | 건선 치료용 조성물 및 방법 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005529152A (ja) * | 2002-05-17 | 2005-09-29 | プロテイン デザイン ラブス インコーポレイティド | 抗インターフェロンγ抗体を用いたクローン病又は乾癬の治療 |
| WO2006125229A2 (en) * | 2005-05-16 | 2006-11-23 | Abbott Biotechnology Ltd. | Use of tnf inhibitor for treatment of erosive polyarthritis |
| JP2007503839A (ja) * | 2003-09-04 | 2007-03-01 | アプライド リサーチ システムズ エーアールエス ホールディング ナームロゼ フェンノートシャップ | 新規UBP8rpポリペプチドおよび乾癬の治療におけるその使用 |
| JP2010515774A (ja) * | 2007-01-16 | 2010-05-13 | アボット・ラボラトリーズ | 乾せんの治療方法 |
Family Cites Families (107)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US128A (en) | 1837-02-16 | Holdback for sleds | ||
| US6914A (en) | 1849-11-27 | Propeller | ||
| US5179017A (en) | 1980-02-25 | 1993-01-12 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4399216A (en) | 1980-02-25 | 1983-08-16 | The Trustees Of Columbia University | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4634665A (en) | 1980-02-25 | 1987-01-06 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4510245A (en) | 1982-11-18 | 1985-04-09 | Chiron Corporation | Adenovirus promoter system |
| GB8308235D0 (en) | 1983-03-25 | 1983-05-05 | Celltech Ltd | Polypeptides |
| US5168062A (en) | 1985-01-30 | 1992-12-01 | University Of Iowa Research Foundation | Transfer vectors and microorganisms containing human cytomegalovirus immediate-early promoter-regulatory DNA sequence |
| US4968615A (en) | 1985-12-18 | 1990-11-06 | Ciba-Geigy Corporation | Deoxyribonucleic acid segment from a virus |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US5811523A (en) | 1988-11-10 | 1998-09-22 | Trinchieri; Giorgio | Antibodies to natural killer stimulatory factor |
| KR0184860B1 (ko) | 1988-11-11 | 1999-04-01 | 메디칼 리써어치 카운실 | 단일영역 리간드와 이를 포함하는 수용체 및 이들의 제조방법과 이용(법) |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| ES2118066T3 (es) | 1989-10-05 | 1998-09-16 | Optein Inc | Sintesis y aislamiento, exentos de celulas, de nuevos genes y polipeptidos. |
| US6683046B1 (en) | 1989-12-22 | 2004-01-27 | Hoffmann-La Roche Inc. | Purification and characterization of cytotoxic lymphocyte maturation factor and monoclonal antibodies thereto |
| US6150584A (en) | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US6713610B1 (en) | 1990-01-12 | 2004-03-30 | Raju Kucherlapati | Human antibodies derived from immunized xenomice |
| US6673986B1 (en) | 1990-01-12 | 2004-01-06 | Abgenix, Inc. | Generation of xenogeneic antibodies |
| DK0585287T3 (da) | 1990-07-10 | 2000-04-17 | Cambridge Antibody Tech | Fremgangsmåde til fremstilling af specifikke bindingsparelementer |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US6300129B1 (en) | 1990-08-29 | 2001-10-09 | Genpharm International | Transgenic non-human animals for producing heterologous antibodies |
| US5789650A (en) | 1990-08-29 | 1998-08-04 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5814318A (en) | 1990-08-29 | 1998-09-29 | Genpharm International Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US7084260B1 (en) | 1996-10-10 | 2006-08-01 | Genpharm International, Inc. | High affinity human antibodies and human antibodies against human antigens |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US6255458B1 (en) | 1990-08-29 | 2001-07-03 | Genpharm International | High affinity human antibodies and human antibodies against digoxin |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| ES2246502T3 (es) | 1990-08-29 | 2006-02-16 | Genpharm International, Inc. | Animales no humanos transgenicos capaces de producir anticuerpos heterologos. |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| ES2113940T3 (es) | 1990-12-03 | 1998-05-16 | Genentech Inc | Metodo de enriquecimiento para variantes de proteinas con propiedades de union alteradas. |
| US6284471B1 (en) | 1991-03-18 | 2001-09-04 | New York University Medical Center | Anti-TNFa antibodies and assays employing anti-TNFa antibodies |
| US5919452A (en) | 1991-03-18 | 1999-07-06 | New York University | Methods of treating TNFα-mediated disease using chimeric anti-TNF antibodies |
| US6277969B1 (en) | 1991-03-18 | 2001-08-21 | New York University | Anti-TNF antibodies and peptides of human tumor necrosis factor |
| US5698195A (en) | 1991-03-18 | 1997-12-16 | New York University Medical Center | Methods of treating rheumatoid arthritis using chimeric anti-TNF antibodies |
| US5656272A (en) | 1991-03-18 | 1997-08-12 | New York University Medical Center | Methods of treating TNF-α-mediated Crohn's disease using chimeric anti-TNF antibodies |
| DE69233750D1 (de) | 1991-04-10 | 2009-01-02 | Scripps Research Inst | Bibliotheken heterodimerer Rezeptoren mittels Phagemiden |
| WO1994004679A1 (en) | 1991-06-14 | 1994-03-03 | Genentech, Inc. | Method for making humanized antibodies |
| DE4122599C2 (de) | 1991-07-08 | 1993-11-11 | Deutsches Krebsforsch | Phagemid zum Screenen von Antikörpern |
| DK0605522T3 (da) | 1991-09-23 | 2000-01-17 | Medical Res Council | Fremgangsmåde til fremstilling af humaniserede antistoffer |
| GB9122820D0 (en) | 1991-10-28 | 1991-12-11 | Wellcome Found | Stabilised antibodies |
| DK1024191T3 (da) | 1991-12-02 | 2008-12-08 | Medical Res Council | Fremstilling af autoantistoffer fremvist på fag-overflader ud fra antistofsegmentbiblioteker |
| ATE381614T1 (de) | 1992-07-24 | 2008-01-15 | Amgen Fremont Inc | Bildung von xenogenen antikörpern |
| US5652138A (en) | 1992-09-30 | 1997-07-29 | The Scripps Research Institute | Human neutralizing monoclonal antibodies to human immunodeficiency virus |
| DK0614984T4 (da) | 1993-03-05 | 2010-12-20 | Bayer Healthcare Llc | Humane monoklonale anti-TNF-alfa-antistoffer |
| CA2161351C (en) | 1993-04-26 | 2010-12-21 | Nils Lonberg | Transgenic non-human animals capable of producing heterologous antibodies |
| US5654168A (en) | 1994-07-01 | 1997-08-05 | Basf Aktiengesellschaft | Tetracycline-inducible transcriptional activator and tetracycline-regulated transcription units |
| US5464758A (en) | 1993-06-14 | 1995-11-07 | Gossen; Manfred | Tight control of gene expression in eucaryotic cells by tetracycline-responsive promoters |
| CA2125763C (en) | 1993-07-02 | 2007-08-28 | Maurice Kent Gately | P40 homodimer of interleukin-12 |
| EP0638644A1 (en) | 1993-07-19 | 1995-02-15 | F. Hoffmann-La Roche Ag | Receptors of interleukin-12 and antibodies |
| EP0659766A1 (en) | 1993-11-23 | 1995-06-28 | Schering-Plough | Human monoclonal antibodies against human cytokines and methods of making and using such antibodies |
| ZA95960B (en) | 1994-03-14 | 1995-10-10 | Genetics Inst | Use of interleukin-12 antagonists in the treatment of autoimmune diseases |
| US5910486A (en) | 1994-09-06 | 1999-06-08 | Uab Research Foundation | Methods for modulating protein function in cells using, intracellular antibody homologues |
| US5562138A (en) * | 1994-10-13 | 1996-10-08 | The Longaberger Company | Bowing press |
| ES2304786T3 (es) | 1995-04-27 | 2008-10-16 | Amgen Fremont Inc. | Anticuerpos anti-il-8 humanos, derivados a partir de xenoratones inmunizados. |
| EP0823941A4 (en) | 1995-04-28 | 2001-09-19 | Abgenix Inc | HUMAN ANTIBODIES DERIVED FROM IMMUNIZED XENO MOUSES |
| US6685940B2 (en) | 1995-07-27 | 2004-02-03 | Genentech, Inc. | Protein formulation |
| US5853697A (en) | 1995-10-25 | 1998-12-29 | The United States Of America, As Represented By The Department Of Health & Human Services | Methods of treating established colitis using antibodies against IL-12 |
| US6297395B1 (en) | 1995-11-10 | 2001-10-02 | The Secretary Of State For Defence In Her Brittanic Majesty's Government Of The United Kingdom Of Great Britain And Northern Ireland | Calixarenes and their use for sequestration of metals |
| US6090382A (en) | 1996-02-09 | 2000-07-18 | Basf Aktiengesellschaft | Human antibodies that bind human TNFα |
| RU2270030C2 (ru) | 1996-02-09 | 2006-02-20 | Абботт Байотекнолоджи эЛтиди. | СПОСОБ ИНГИБИРОВАНИЯ АКТИВНОСТИ ЧЕЛОВЕЧЕСКОГО TNFα (ВАРИАНТЫ), ПРИМЕНЕНИЕ ВЫДЕЛЕННОГО АНТИТЕЛА ЧЕЛОВЕКА ИЛИ ЕГО АНТИГЕНСВЯЗЫВАЮЩЕГО ФРАГМЕНТА В КАЧЕСТВЕ КОМПОНЕНТА ДЛЯ ПРОИЗВОДСТВА ЛЕКАРСТВЕННОГО СРЕДСТВА (ВАРИАНТЫ) И ВЫДЕЛЕННОЕ ЧЕЛОВЕЧЕСКОЕ АНТИТЕЛО ИЛИ ЕГО АНТИГЕНСВЯЗЫВАЮЩИЙ ФРАГМЕНТ |
| AU7257696A (en) | 1996-10-11 | 1998-05-11 | Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services, The | Methods for enhancing oral tolerance and treating autoimmune disease using inhibitors of interleukin-12 |
| ATE256476T1 (de) | 1996-11-15 | 2004-01-15 | Kennedy Inst Of Rheumatology | Unterdrückung von tnfalpha und il-12 in der therapie |
| DK1500329T3 (da) | 1996-12-03 | 2012-07-09 | Amgen Fremont Inc | Humane antistoffer, der specifikt binder TNF-alfa |
| US6054487A (en) | 1997-03-18 | 2000-04-25 | Basf Aktiengesellschaft | Methods and compositions for modulating responsiveness to corticosteroids |
| PL336464A1 (en) | 1997-03-18 | 2000-06-19 | Basf Ag | Method of and compositions for modulating reactivity in respect to corticosteroids i |
| US6183744B1 (en) | 1997-03-24 | 2001-02-06 | Immunomedics, Inc. | Immunotherapy of B-cell malignancies using anti-CD22 antibodies |
| US6235883B1 (en) | 1997-05-05 | 2001-05-22 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| EP1015480A2 (en) | 1997-08-18 | 2000-07-05 | Innogenetics N.V. | Interferon-gamma-binding molecules for treating septic shock, cachexia, immune diseases and skin disorders |
| ATE274920T1 (de) | 1997-10-31 | 2004-09-15 | Wyeth Corp | Verwendung von anti-il-12 antikörpern bei transplantationsabstossungen |
| TR200002145T2 (tr) | 1998-01-23 | 2000-11-21 | F.Hoffmann-La Roche Ag | İnsan IL-12'ye karşı antikorlar |
| US20020161199A1 (en) | 1998-04-08 | 2002-10-31 | Genentech, Inc. | Compositions and methods for the diagnosis and treatment of tumor |
| EP0953639A1 (en) | 1998-04-30 | 1999-11-03 | Boehringer Ingelheim International GmbH | FAPalpha-specific antibody with improved producibility |
| DE69927520T2 (de) | 1998-12-09 | 2006-06-22 | Protein Design Labs, Inc., Fremont | Verwendung von il-12 antikörpern zur behandlung von psoriasis |
| KR101222450B1 (ko) * | 1999-03-25 | 2013-01-16 | 애보트 게엠베하 운트 콤파니 카게 | 사람 il-12에 결합하는 사람 항체 및 이의 제조방법 |
| US7883704B2 (en) | 1999-03-25 | 2011-02-08 | Abbott Gmbh & Co. Kg | Methods for inhibiting the activity of the P40 subunit of human IL-12 |
| US6914128B1 (en) | 1999-03-25 | 2005-07-05 | Abbott Gmbh & Co. Kg | Human antibodies that bind human IL-12 and methods for producing |
| BR0013231A (pt) * | 1999-08-09 | 2002-07-23 | Lexigen Pharm Corp | Complexos citocina-anticorpo múltiplos |
| US6902734B2 (en) | 2000-08-07 | 2005-06-07 | Centocor, Inc. | Anti-IL-12 antibodies and compositions thereof |
| KR20030074693A (ko) | 2000-12-28 | 2003-09-19 | 알투스 바이올로직스 인코포레이티드 | 전항체 및 이의 단편의 결정과 이의 제조 및 사용 방법 |
| WO2002097048A2 (en) | 2001-05-30 | 2002-12-05 | Centocor, Inc. | ANTI-p40 IMMUNOGLOBULIN DERIVED PROTEINS, COMPOSITIONS, METHODS AND USES |
| US20040156835A1 (en) | 2001-05-30 | 2004-08-12 | Taiji Imoto | Protein preparation |
| US20050276823A1 (en) | 2002-07-12 | 2005-12-15 | Cini John K | Methods and compositions for preventing oxidative degradation of proteins |
| US20090280065A1 (en) | 2006-04-10 | 2009-11-12 | Willian Mary K | Uses and Compositions for Treatment of Psoriasis |
| US7608260B2 (en) | 2003-01-06 | 2009-10-27 | Medimmune, Llc | Stabilized immunoglobulins |
| KR101224235B1 (ko) | 2003-04-11 | 2013-01-25 | 메디뮨 엘엘씨 | 재조합 il9 항체 및 그의 용도 |
| JP2007515939A (ja) | 2003-05-09 | 2007-06-21 | セントカー・インコーポレーテツド | IL−23p40特異的免疫グロブリン由来タンパク質、組成物、方法および用途 |
| JP4219932B2 (ja) | 2003-10-01 | 2009-02-04 | 協和発酵キリン株式会社 | 抗体の安定化方法及び安定化された溶液状抗体製剤 |
| EP1694317A4 (en) | 2003-12-19 | 2010-05-12 | Protemix Corp Ltd | COPPER ANTAGONIST COMPOUNDS |
| JP4943161B2 (ja) | 2003-12-23 | 2012-05-30 | ジェネンテック, インコーポレイテッド | 新規抗il13モノクローナル抗体での癌の処置 |
| RU2390353C2 (ru) | 2004-02-12 | 2010-05-27 | Мерк Патент Гмбх | Высококонцентрированные жидкие композиции анти-egfr антител |
| RU2292215C2 (ru) * | 2004-05-21 | 2007-01-27 | Дмитрий Васильевич Николенко | Комплекс для лечения псориаза и способ лечения |
| AR049390A1 (es) | 2004-06-09 | 2006-07-26 | Wyeth Corp | Anticuerpos contra la interleuquina-13 humana y usos de los mismos |
| JP4994232B2 (ja) | 2004-07-23 | 2012-08-08 | ジェネンテック, インコーポレイテッド | 抗体又はその断片の結晶化 |
| KR20070094927A (ko) | 2004-12-21 | 2007-09-27 | 센토코 인코포레이티드 | 항-il-12 항체, 에피토프, 조성물, 방법 및 용도 |
| JP2009501006A (ja) | 2005-06-30 | 2009-01-15 | セントカー・インコーポレーテツド | 抗il−23抗体、組成物、方法および用途 |
| AU2007212147A1 (en) | 2006-02-03 | 2007-08-16 | Medimmune, Llc | Protein formulations |
| EP2064345B1 (en) | 2006-09-11 | 2013-03-13 | Celera Corporation | Genetic polymorphisms associated with psoriasis, methods of detection and uses thereof |
| SG175622A1 (en) | 2006-10-13 | 2011-11-28 | Centocor Ortho Biotech Inc | Enhancement of hybridoma fusion efficiencies through cell synchronization |
| NZ598881A (en) | 2007-03-29 | 2013-11-29 | Abbvie Inc | Crystalline anti-human il-12 antibodies |
| NZ587765A (en) | 2008-03-18 | 2013-02-22 | Abbott Lab | Methods for treating psoriasis |
| WO2010062896A1 (en) | 2008-11-28 | 2010-06-03 | Abbott Laboratories | Stable antibody compositions and methods for stabilizing same |
| KR20140048229A (ko) | 2009-09-14 | 2014-04-23 | 애브비 인코포레이티드 | 건선을 치료하는 방법 |
| JP2013542209A (ja) | 2010-10-06 | 2013-11-21 | アッヴィ・インコーポレイテッド | 乾癬を治療するための方法 |
| AU2012204237A1 (en) | 2011-01-07 | 2013-07-04 | Abbvie Inc. | Anti-IL-12/IL-23 antibodies and uses thereof |
| WO2012097288A1 (en) | 2011-01-14 | 2012-07-19 | Abbott Laboratories | Human antibodies that bind the p40 subunit of human il-12/il-23 and uses therefor |
-
2009
- 2009-03-11 NZ NZ587765A patent/NZ587765A/xx not_active IP Right Cessation
- 2009-03-11 CN CN2009801179497A patent/CN102037011A/zh active Pending
- 2009-03-11 MX MX2010010265A patent/MX2010010265A/es active IP Right Grant
- 2009-03-11 US US12/402,342 patent/US8178092B2/en not_active Expired - Fee Related
- 2009-03-11 TW TW103129438A patent/TW201513883A/zh unknown
- 2009-03-11 KR KR1020107023101A patent/KR20100126515A/ko not_active Application Discontinuation
- 2009-03-11 JP JP2011500870A patent/JP2011515404A/ja not_active Ceased
- 2009-03-11 EP EP09723430A patent/EP2274333A4/en not_active Withdrawn
- 2009-03-11 NZ NZ602511A patent/NZ602511A/en not_active IP Right Cessation
- 2009-03-11 RU RU2010142309/10A patent/RU2497545C2/ru not_active IP Right Cessation
- 2009-03-11 EP EP14176768.1A patent/EP2810954A3/en not_active Withdrawn
- 2009-03-11 SG SG2013019617A patent/SG188909A1/en unknown
- 2009-03-11 AU AU2009225797A patent/AU2009225797A1/en not_active Abandoned
- 2009-03-11 CA CA2717569A patent/CA2717569A1/en not_active Abandoned
- 2009-03-11 WO PCT/US2009/036765 patent/WO2009117289A2/en active Application Filing
- 2009-03-11 BR BRPI0908715A patent/BRPI0908715A2/pt not_active IP Right Cessation
- 2009-03-11 TW TW098107971A patent/TWI461210B/zh not_active IP Right Cessation
-
2010
- 2010-09-07 IL IL208069A patent/IL208069A0/en unknown
-
2011
- 2011-06-29 HK HK15103080.8A patent/HK1203516A1/xx unknown
-
2012
- 2012-03-08 US US13/415,337 patent/US8945545B2/en not_active Expired - Fee Related
-
2013
- 2013-07-16 RU RU2013132928/10A patent/RU2013132928A/ru not_active Application Discontinuation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005529152A (ja) * | 2002-05-17 | 2005-09-29 | プロテイン デザイン ラブス インコーポレイティド | 抗インターフェロンγ抗体を用いたクローン病又は乾癬の治療 |
| JP2007503839A (ja) * | 2003-09-04 | 2007-03-01 | アプライド リサーチ システムズ エーアールエス ホールディング ナームロゼ フェンノートシャップ | 新規UBP8rpポリペプチドおよび乾癬の治療におけるその使用 |
| WO2006125229A2 (en) * | 2005-05-16 | 2006-11-23 | Abbott Biotechnology Ltd. | Use of tnf inhibitor for treatment of erosive polyarthritis |
| JP2010515774A (ja) * | 2007-01-16 | 2010-05-13 | アボット・ラボラトリーズ | 乾せんの治療方法 |
Non-Patent Citations (5)
| Title |
|---|
| ARCH DERMATOL., VOL.144 NO.2 P.200-7. (2008 FEB), JPN6013035937, ISSN: 0002639733 * |
| J INVEST DERMATOL., VOL.123 NO.6 P.1037-44. (2004), JPN6013035944, ISSN: 0003042392 * |
| MEDICAL NEWS TODAY, "ABBOTT'S ABT 874 SHOWS POSITIVE RESULTS FOR MAINTENANCE OF RESPONSE IN PHASE II, JPN6013035942, ISSN: 0002639735 * |
| N ENGL J MED., VOL.356 NO.6 P.580-92. (2007), JPN6013035939, ISSN: 0002639734 * |
| NAT BIOTECHNOL., VOL.25 NO.11 P.1290-7. (2007), JPN6013035947, ISSN: 0003042393 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016516686A (ja) * | 2013-03-15 | 2016-06-09 | アムジェン インコーポレイテッド | 抗il23抗体を使用してクローン病を治療するための方法 |
| JP2016517408A (ja) * | 2013-03-15 | 2016-06-16 | アムジェン インコーポレイテッド | 抗il−23抗体を用いた乾癬の治療方法 |
| JP2020063237A (ja) * | 2013-03-15 | 2020-04-23 | アムジェン インコーポレイテッド | 抗il−23抗体を用いた乾癬の治療方法 |
| JP2020073470A (ja) * | 2013-03-15 | 2020-05-14 | アムジェン インコーポレイテッド | 抗il23抗体を使用してクローン病を治療するための方法 |
| JP2021102644A (ja) * | 2013-03-15 | 2021-07-15 | アムジェン インコーポレイテッド | 抗il−23抗体を用いた乾癬の治療方法 |
| JP2021107397A (ja) * | 2013-03-15 | 2021-07-29 | アムジェン インコーポレイテッド | 抗il23抗体を使用してクローン病を治療するための方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2497545C2 (ru) | 2013-11-10 |
| US8178092B2 (en) | 2012-05-15 |
| TW200948379A (en) | 2009-12-01 |
| US8945545B2 (en) | 2015-02-03 |
| EP2274333A4 (en) | 2011-06-15 |
| WO2009117289A3 (en) | 2010-04-22 |
| CN102037011A (zh) | 2011-04-27 |
| US20120219562A1 (en) | 2012-08-30 |
| CA2717569A1 (en) | 2009-09-24 |
| AU2009225797A1 (en) | 2009-09-24 |
| NZ602511A (en) | 2014-03-28 |
| WO2009117289A2 (en) | 2009-09-24 |
| MX2010010265A (es) | 2010-09-30 |
| EP2810954A3 (en) | 2015-03-18 |
| RU2013132928A (ru) | 2015-01-27 |
| TW201513883A (zh) | 2015-04-16 |
| SG188909A1 (en) | 2013-04-30 |
| TWI461210B (zh) | 2014-11-21 |
| BRPI0908715A2 (pt) | 2016-05-03 |
| HK1203516A1 (en) | 2015-10-30 |
| EP2274333A2 (en) | 2011-01-19 |
| RU2010142309A (ru) | 2012-04-27 |
| KR20100126515A (ko) | 2010-12-01 |
| EP2810954A2 (en) | 2014-12-10 |
| US20100028363A1 (en) | 2010-02-04 |
| IL208069A0 (en) | 2010-12-30 |
| NZ587765A (en) | 2013-02-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4841038B2 (ja) | ヒトil−12に結合するヒト抗体およびその産生法 | |
| US8178092B2 (en) | Methods of treating psoriasis by administration of antibodies to the p40 subunit of IL-12 and/or IL-23 | |
| US9051368B2 (en) | Methods for treating psoriasis by administering an antibody which binds an epitope of the p40 subunit of IL-12 and/or IL-23 | |
| JP2010513529A (ja) | ヒトil−12を結合するヒト抗体及び製造方法 | |
| US8557239B2 (en) | Methods for treating psoriasis using antibodies that bind to the P40 subunit of IL-12 and/or IL-23 | |
| JP2013542209A (ja) | 乾癬を治療するための方法 | |
| AU2013202849A1 (en) | Methods for treating psoriasis | |
| AU2013202866A1 (en) | Methods for treating psoriasis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110404 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120309 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120309 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20130701 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130822 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20131001 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131227 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140826 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20141120 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20141128 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150331 |
|
| A045 | Written measure of dismissal of application [lapsed due to lack of payment] |
Free format text: JAPANESE INTERMEDIATE CODE: A045 Effective date: 20150728 |