ES2945560T3 - Análogos de rapamicina unidos en C40, C28 y C-32 como inhibidores de mTOR - Google Patents
Análogos de rapamicina unidos en C40, C28 y C-32 como inhibidores de mTOR Download PDFInfo
- Publication number
- ES2945560T3 ES2945560T3 ES19723571T ES19723571T ES2945560T3 ES 2945560 T3 ES2945560 T3 ES 2945560T3 ES 19723571 T ES19723571 T ES 19723571T ES 19723571 T ES19723571 T ES 19723571T ES 2945560 T3 ES2945560 T3 ES 2945560T3
- Authority
- ES
- Spain
- Prior art keywords
- cancer
- compound
- disease
- independently
- compounds
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940124302 mTOR inhibitor Drugs 0.000 title abstract description 12
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 title abstract description 12
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical class C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 title description 39
- 150000001875 compounds Chemical class 0.000 claims abstract description 491
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 172
- 201000010099 disease Diseases 0.000 claims abstract description 140
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 claims abstract description 124
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 claims abstract description 124
- 239000000203 mixture Substances 0.000 claims abstract description 63
- 230000001404 mediated effect Effects 0.000 claims abstract description 29
- 150000003839 salts Chemical class 0.000 claims description 87
- 206010028980 Neoplasm Diseases 0.000 claims description 73
- 201000011510 cancer Diseases 0.000 claims description 46
- 208000032839 leukemia Diseases 0.000 claims description 37
- 208000035475 disorder Diseases 0.000 claims description 28
- 206010025323 Lymphomas Diseases 0.000 claims description 27
- 238000011282 treatment Methods 0.000 claims description 27
- 239000008194 pharmaceutical composition Substances 0.000 claims description 25
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 24
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 23
- 206010060862 Prostate cancer Diseases 0.000 claims description 22
- 201000001441 melanoma Diseases 0.000 claims description 19
- 201000006417 multiple sclerosis Diseases 0.000 claims description 18
- 208000030836 Hashimoto thyroiditis Diseases 0.000 claims description 17
- 210000003734 kidney Anatomy 0.000 claims description 17
- 206010038389 Renal cancer Diseases 0.000 claims description 16
- 210000000813 small intestine Anatomy 0.000 claims description 16
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 16
- 210000004185 liver Anatomy 0.000 claims description 15
- 208000024827 Alzheimer disease Diseases 0.000 claims description 14
- 201000001320 Atherosclerosis Diseases 0.000 claims description 14
- 206010018364 Glomerulonephritis Diseases 0.000 claims description 14
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 14
- 206010046851 Uveitis Diseases 0.000 claims description 14
- 210000004072 lung Anatomy 0.000 claims description 14
- 206010028417 myasthenia gravis Diseases 0.000 claims description 14
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 14
- 210000003491 skin Anatomy 0.000 claims description 13
- 210000004556 brain Anatomy 0.000 claims description 12
- 208000005017 glioblastoma Diseases 0.000 claims description 12
- 210000002216 heart Anatomy 0.000 claims description 12
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 12
- 206010006187 Breast cancer Diseases 0.000 claims description 11
- 208000026310 Breast neoplasm Diseases 0.000 claims description 11
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 11
- 206010057644 Testis cancer Diseases 0.000 claims description 11
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 11
- 206010012601 diabetes mellitus Diseases 0.000 claims description 11
- 210000000496 pancreas Anatomy 0.000 claims description 11
- 201000003120 testicular cancer Diseases 0.000 claims description 11
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 11
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 claims description 10
- 206010005003 Bladder cancer Diseases 0.000 claims description 10
- 201000003741 Gastrointestinal carcinoma Diseases 0.000 claims description 10
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 10
- 239000003937 drug carrier Substances 0.000 claims description 10
- 201000002313 intestinal cancer Diseases 0.000 claims description 10
- 206010012289 Dementia Diseases 0.000 claims description 9
- 206010014733 Endometrial cancer Diseases 0.000 claims description 9
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 9
- 208000023105 Huntington disease Diseases 0.000 claims description 9
- 206010027406 Mesothelioma Diseases 0.000 claims description 9
- 206010033128 Ovarian cancer Diseases 0.000 claims description 9
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 9
- 210000001185 bone marrow Anatomy 0.000 claims description 9
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 9
- 210000004087 cornea Anatomy 0.000 claims description 9
- 239000003085 diluting agent Substances 0.000 claims description 9
- 210000003205 muscle Anatomy 0.000 claims description 9
- 206010061424 Anal cancer Diseases 0.000 claims description 8
- 208000007860 Anus Neoplasms Diseases 0.000 claims description 8
- 206010004593 Bile duct cancer Diseases 0.000 claims description 8
- 208000019838 Blood disease Diseases 0.000 claims description 8
- 206010005949 Bone cancer Diseases 0.000 claims description 8
- 208000018084 Bone neoplasm Diseases 0.000 claims description 8
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 8
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 claims description 8
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 8
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 8
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 8
- 208000032271 Malignant tumor of penis Diseases 0.000 claims description 8
- 208000002231 Muscle Neoplasms Diseases 0.000 claims description 8
- 208000008589 Obesity Diseases 0.000 claims description 8
- 208000002471 Penile Neoplasms Diseases 0.000 claims description 8
- 206010034299 Penile cancer Diseases 0.000 claims description 8
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 8
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 8
- 208000032383 Soft tissue cancer Diseases 0.000 claims description 8
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 8
- 208000006011 Stroke Diseases 0.000 claims description 8
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 8
- 206010047741 Vulval cancer Diseases 0.000 claims description 8
- 208000004354 Vulvar Neoplasms Diseases 0.000 claims description 8
- 201000011165 anus cancer Diseases 0.000 claims description 8
- 208000026900 bile duct neoplasm Diseases 0.000 claims description 8
- 238000010322 bone marrow transplantation Methods 0.000 claims description 8
- 201000010881 cervical cancer Diseases 0.000 claims description 8
- 210000001198 duodenum Anatomy 0.000 claims description 8
- 201000002595 endometriosis of ovary Diseases 0.000 claims description 8
- 210000003414 extremity Anatomy 0.000 claims description 8
- 208000024519 eye neoplasm Diseases 0.000 claims description 8
- 206010017758 gastric cancer Diseases 0.000 claims description 8
- 208000024908 graft versus host disease Diseases 0.000 claims description 8
- 201000010536 head and neck cancer Diseases 0.000 claims description 8
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 8
- 208000014951 hematologic disease Diseases 0.000 claims description 8
- 208000018706 hematopoietic system disease Diseases 0.000 claims description 8
- 210000004153 islets of langerhan Anatomy 0.000 claims description 8
- 201000010982 kidney cancer Diseases 0.000 claims description 8
- 201000007270 liver cancer Diseases 0.000 claims description 8
- 208000014018 liver neoplasm Diseases 0.000 claims description 8
- 201000005202 lung cancer Diseases 0.000 claims description 8
- 208000020816 lung neoplasm Diseases 0.000 claims description 8
- 210000005036 nerve Anatomy 0.000 claims description 8
- 230000000926 neurological effect Effects 0.000 claims description 8
- 235000020824 obesity Nutrition 0.000 claims description 8
- 201000008106 ocular cancer Diseases 0.000 claims description 8
- 208000030747 ovarian endometriosis Diseases 0.000 claims description 8
- 230000002062 proliferating effect Effects 0.000 claims description 8
- 206010038038 rectal cancer Diseases 0.000 claims description 8
- 201000001275 rectum cancer Diseases 0.000 claims description 8
- 201000010174 renal carcinoma Diseases 0.000 claims description 8
- 201000000849 skin cancer Diseases 0.000 claims description 8
- 201000011549 stomach cancer Diseases 0.000 claims description 8
- 238000002054 transplantation Methods 0.000 claims description 8
- 201000005102 vulva cancer Diseases 0.000 claims description 8
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 7
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 claims description 6
- 208000000277 Splenic Neoplasms Diseases 0.000 claims description 6
- 230000004064 dysfunction Effects 0.000 claims description 6
- 201000007028 gastrointestinal neuroendocrine tumor Diseases 0.000 claims description 6
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 claims description 6
- 230000036961 partial effect Effects 0.000 claims description 6
- 230000002829 reductive effect Effects 0.000 claims description 6
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 5
- 208000024806 Brain atrophy Diseases 0.000 claims description 5
- 206010007572 Cardiac hypertrophy Diseases 0.000 claims description 5
- 208000006029 Cardiomegaly Diseases 0.000 claims description 5
- 208000002177 Cataract Diseases 0.000 claims description 5
- 208000028698 Cognitive impairment Diseases 0.000 claims description 5
- 206010011878 Deafness Diseases 0.000 claims description 5
- 206010052337 Diastolic dysfunction Diseases 0.000 claims description 5
- 206010014561 Emphysema Diseases 0.000 claims description 5
- 208000010228 Erectile Dysfunction Diseases 0.000 claims description 5
- 208000036119 Frailty Diseases 0.000 claims description 5
- 208000001132 Osteoporosis Diseases 0.000 claims description 5
- 206010062237 Renal impairment Diseases 0.000 claims description 5
- 206010039966 Senile dementia Diseases 0.000 claims description 5
- 206010040799 Skin atrophy Diseases 0.000 claims description 5
- 206010071436 Systolic dysfunction Diseases 0.000 claims description 5
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 5
- 206010003549 asthenia Diseases 0.000 claims description 5
- 230000000747 cardiac effect Effects 0.000 claims description 5
- 208000010877 cognitive disease Diseases 0.000 claims description 5
- 231100000895 deafness Toxicity 0.000 claims description 5
- 230000006735 deficit Effects 0.000 claims description 5
- 208000016354 hearing loss disease Diseases 0.000 claims description 5
- 201000001881 impotence Diseases 0.000 claims description 5
- 201000008482 osteoarthritis Diseases 0.000 claims description 5
- 231100000857 poor renal function Toxicity 0.000 claims description 5
- 230000002265 prevention Effects 0.000 claims description 5
- 230000000750 progressive effect Effects 0.000 claims description 5
- 208000001076 sarcopenia Diseases 0.000 claims description 5
- 210000002435 tendon Anatomy 0.000 claims description 5
- 206010028289 Muscle atrophy Diseases 0.000 claims description 4
- 206010027175 memory impairment Diseases 0.000 claims description 4
- 230000020763 muscle atrophy Effects 0.000 claims description 4
- 201000000585 muscular atrophy Diseases 0.000 claims description 4
- 230000009467 reduction Effects 0.000 claims description 4
- 208000037849 arterial hypertension Diseases 0.000 claims description 3
- 150000002919 oxepanes Chemical class 0.000 claims description 3
- 238000000034 method Methods 0.000 abstract description 64
- 230000002194 synthesizing effect Effects 0.000 abstract description 3
- 238000013519 translation Methods 0.000 abstract description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 140
- -1 heterocyclylene Chemical group 0.000 description 131
- 125000005549 heteroarylene group Chemical group 0.000 description 114
- 235000002639 sodium chloride Nutrition 0.000 description 84
- 125000000732 arylene group Chemical group 0.000 description 83
- 230000000694 effects Effects 0.000 description 64
- 125000001072 heteroaryl group Chemical group 0.000 description 62
- 125000002993 cycloalkylene group Chemical group 0.000 description 59
- 201000009030 Carcinoma Diseases 0.000 description 57
- 125000000217 alkyl group Chemical group 0.000 description 55
- 229910052736 halogen Inorganic materials 0.000 description 54
- 150000002367 halogens Chemical class 0.000 description 54
- 239000003112 inhibitor Substances 0.000 description 54
- 230000001588 bifunctional effect Effects 0.000 description 49
- 125000003118 aryl group Chemical group 0.000 description 48
- 108010035196 Mechanistic Target of Rapamycin Complex 1 Proteins 0.000 description 43
- 102000008135 Mechanistic Target of Rapamycin Complex 1 Human genes 0.000 description 43
- 125000001424 substituent group Chemical group 0.000 description 39
- 239000003795 chemical substances by application Substances 0.000 description 37
- 108090000623 proteins and genes Proteins 0.000 description 36
- 108010034057 Mechanistic Target of Rapamycin Complex 2 Proteins 0.000 description 33
- 102000009308 Mechanistic Target of Rapamycin Complex 2 Human genes 0.000 description 33
- 229910052760 oxygen Inorganic materials 0.000 description 32
- 235000018102 proteins Nutrition 0.000 description 32
- 102000004169 proteins and genes Human genes 0.000 description 32
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 30
- 229960002930 sirolimus Drugs 0.000 description 30
- 238000006467 substitution reaction Methods 0.000 description 30
- 125000005647 linker group Chemical group 0.000 description 29
- 125000000623 heterocyclic group Chemical group 0.000 description 28
- 210000004027 cell Anatomy 0.000 description 26
- 125000000753 cycloalkyl group Chemical group 0.000 description 26
- 229910052717 sulfur Inorganic materials 0.000 description 26
- 206010039491 Sarcoma Diseases 0.000 description 24
- 108010006877 Tacrolimus Binding Protein 1A Proteins 0.000 description 22
- 230000001594 aberrant effect Effects 0.000 description 21
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 description 19
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 description 19
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 18
- 102100027913 Peptidyl-prolyl cis-trans isomerase FKBP1A Human genes 0.000 description 18
- 108010027179 Tacrolimus Binding Proteins Proteins 0.000 description 18
- 102000018679 Tacrolimus Binding Proteins Human genes 0.000 description 18
- 150000001412 amines Chemical class 0.000 description 18
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 17
- 108091008611 Protein Kinase B Proteins 0.000 description 17
- 230000002401 inhibitory effect Effects 0.000 description 17
- 150000003335 secondary amines Chemical class 0.000 description 17
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 16
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 16
- 125000001188 haloalkyl group Chemical group 0.000 description 15
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 14
- 125000003545 alkoxy group Chemical group 0.000 description 13
- 230000001363 autoimmune Effects 0.000 description 13
- 230000035772 mutation Effects 0.000 description 13
- 230000037361 pathway Effects 0.000 description 13
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 12
- 208000007465 Giant cell arteritis Diseases 0.000 description 12
- 208000010125 myocardial infarction Diseases 0.000 description 12
- 229910052757 nitrogen Inorganic materials 0.000 description 12
- 206010043207 temporal arteritis Diseases 0.000 description 12
- 230000009424 thromboembolic effect Effects 0.000 description 12
- 102100022466 Eukaryotic translation initiation factor 4E-binding protein 1 Human genes 0.000 description 11
- 125000004432 carbon atom Chemical group C* 0.000 description 11
- 238000006243 chemical reaction Methods 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 10
- 241000282414 Homo sapiens Species 0.000 description 10
- 208000031888 Mycoses Diseases 0.000 description 10
- 206010052779 Transplant rejections Diseases 0.000 description 10
- 125000004122 cyclic group Chemical group 0.000 description 10
- 125000005842 heteroatom Chemical group 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 150000003254 radicals Chemical class 0.000 description 10
- 230000019491 signal transduction Effects 0.000 description 10
- 208000011580 syndromic disease Diseases 0.000 description 10
- 125000000041 C6-C10 aryl group Chemical group 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 9
- 241000721454 Pemphigus Species 0.000 description 9
- 208000007718 Stable Angina Diseases 0.000 description 9
- 208000007536 Thrombosis Diseases 0.000 description 9
- 208000002552 acute disseminated encephalomyelitis Diseases 0.000 description 9
- 150000001413 amino acids Chemical class 0.000 description 9
- 230000002526 effect on cardiovascular system Effects 0.000 description 9
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 9
- 201000000306 sarcoidosis Diseases 0.000 description 9
- 239000007787 solid Substances 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- 208000024172 Cardiovascular disease Diseases 0.000 description 8
- 108090000144 Human Proteins Proteins 0.000 description 8
- 102000003839 Human Proteins Human genes 0.000 description 8
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 8
- 208000021386 Sjogren Syndrome Diseases 0.000 description 8
- 210000001744 T-lymphocyte Anatomy 0.000 description 8
- 235000001014 amino acid Nutrition 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 208000029078 coronary artery disease Diseases 0.000 description 8
- 208000027866 inflammatory disease Diseases 0.000 description 8
- 208000023275 Autoimmune disease Diseases 0.000 description 7
- 206010017533 Fungal infection Diseases 0.000 description 7
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 7
- 208000002569 Machado-Joseph Disease Diseases 0.000 description 7
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 7
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 7
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000002246 antineoplastic agent Substances 0.000 description 7
- 239000002502 liposome Substances 0.000 description 7
- 206010025135 lupus erythematosus Diseases 0.000 description 7
- 208000030159 metabolic disease Diseases 0.000 description 7
- 230000004770 neurodegeneration Effects 0.000 description 7
- 208000015122 neurodegenerative disease Diseases 0.000 description 7
- 208000008795 neuromyelitis optica Diseases 0.000 description 7
- 108020004707 nucleic acids Proteins 0.000 description 7
- 102000039446 nucleic acids Human genes 0.000 description 7
- 150000007523 nucleic acids Chemical class 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 6
- 208000026872 Addison Disease Diseases 0.000 description 6
- 208000008190 Agammaglobulinemia Diseases 0.000 description 6
- 208000031277 Amaurotic familial idiocy Diseases 0.000 description 6
- 206010002388 Angina unstable Diseases 0.000 description 6
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 6
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 6
- 208000031212 Autoimmune polyendocrinopathy Diseases 0.000 description 6
- 208000009137 Behcet syndrome Diseases 0.000 description 6
- 208000009299 Benign Mucous Membrane Pemphigoid Diseases 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 6
- 208000030939 Chronic inflammatory demyelinating polyneuropathy Diseases 0.000 description 6
- 208000015943 Coeliac disease Diseases 0.000 description 6
- 206010009900 Colitis ulcerative Diseases 0.000 description 6
- 208000011231 Crohn disease Diseases 0.000 description 6
- 208000004986 Diffuse Cerebral Sclerosis of Schilder Diseases 0.000 description 6
- 108050000946 Eukaryotic translation initiation factor 4E-binding protein 1 Proteins 0.000 description 6
- 201000011240 Frontotemporal dementia Diseases 0.000 description 6
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 6
- 206010020983 Hypogammaglobulinaemia Diseases 0.000 description 6
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 6
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 description 6
- 208000005615 Interstitial Cystitis Diseases 0.000 description 6
- 208000003456 Juvenile Arthritis Diseases 0.000 description 6
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 6
- 208000012309 Linear IgA disease Diseases 0.000 description 6
- 208000016604 Lyme disease Diseases 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 6
- 208000003250 Mixed connective tissue disease Diseases 0.000 description 6
- 208000000733 Paroxysmal Hemoglobinuria Diseases 0.000 description 6
- 206010034277 Pemphigoid Diseases 0.000 description 6
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 6
- 208000031845 Pernicious anaemia Diseases 0.000 description 6
- 102100036050 Phosphatidylinositol N-acetylglucosaminyltransferase subunit A Human genes 0.000 description 6
- 201000004681 Psoriasis Diseases 0.000 description 6
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 6
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 6
- 206010042276 Subacute endocarditis Diseases 0.000 description 6
- 206010043561 Thrombocytopenic purpura Diseases 0.000 description 6
- 201000006704 Ulcerative Colitis Diseases 0.000 description 6
- 208000007814 Unstable Angina Diseases 0.000 description 6
- 208000018756 Variant Creutzfeldt-Jakob disease Diseases 0.000 description 6
- 206010047115 Vasculitis Diseases 0.000 description 6
- 206010047249 Venous thrombosis Diseases 0.000 description 6
- 206010047642 Vitiligo Diseases 0.000 description 6
- 206010000891 acute myocardial infarction Diseases 0.000 description 6
- 229940034982 antineoplastic agent Drugs 0.000 description 6
- 206010003119 arrhythmia Diseases 0.000 description 6
- 208000006673 asthma Diseases 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 208000005881 bovine spongiform encephalopathy Diseases 0.000 description 6
- 201000005795 chronic inflammatory demyelinating polyneuritis Diseases 0.000 description 6
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 6
- 201000001981 dermatomyositis Diseases 0.000 description 6
- 208000016097 disease of metabolism Diseases 0.000 description 6
- 206010014599 encephalitis Diseases 0.000 description 6
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 208000031225 myocardial ischemia Diseases 0.000 description 6
- 201000003631 narcolepsy Diseases 0.000 description 6
- 210000000056 organ Anatomy 0.000 description 6
- 230000002611 ovarian Effects 0.000 description 6
- 201000003045 paroxysmal nocturnal hemoglobinuria Diseases 0.000 description 6
- 230000026731 phosphorylation Effects 0.000 description 6
- 238000006366 phosphorylation reaction Methods 0.000 description 6
- 208000002574 reactive arthritis Diseases 0.000 description 6
- 208000037803 restenosis Diseases 0.000 description 6
- 208000008467 subacute bacterial endocarditis Diseases 0.000 description 6
- 238000001356 surgical procedure Methods 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 5
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 5
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 5
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 5
- 208000021866 Dressler syndrome Diseases 0.000 description 5
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 5
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 5
- 101000678280 Homo sapiens Eukaryotic translation initiation factor 4E-binding protein 1 Proteins 0.000 description 5
- 206010061218 Inflammation Diseases 0.000 description 5
- 208000034578 Multiple myelomas Diseases 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 208000002537 Neuronal Ceroid-Lipofuscinoses Diseases 0.000 description 5
- 206010035226 Plasma cell myeloma Diseases 0.000 description 5
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 5
- 229940124639 Selective inhibitor Drugs 0.000 description 5
- 208000036834 Spinocerebellar ataxia type 3 Diseases 0.000 description 5
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 5
- 208000026911 Tuberous sclerosis complex Diseases 0.000 description 5
- 208000025851 Undifferentiated connective tissue disease Diseases 0.000 description 5
- 208000017379 Undifferentiated connective tissue syndrome Diseases 0.000 description 5
- 208000009956 adenocarcinoma Diseases 0.000 description 5
- 101150045355 akt1 gene Proteins 0.000 description 5
- 125000003342 alkenyl group Chemical group 0.000 description 5
- 125000000304 alkynyl group Chemical group 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- 229960004679 doxorubicin Drugs 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 125000004404 heteroalkyl group Chemical group 0.000 description 5
- 230000004054 inflammatory process Effects 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 208000017476 juvenile neuronal ceroid lipofuscinosis Diseases 0.000 description 5
- 230000003211 malignant effect Effects 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 239000000178 monomer Substances 0.000 description 5
- 201000007607 neuronal ceroid lipofuscinosis 3 Diseases 0.000 description 5
- 239000002773 nucleotide Substances 0.000 description 5
- 125000003729 nucleotide group Chemical group 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 230000000306 recurrent effect Effects 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 4
- 201000000724 Chronic recurrent multifocal osteomyelitis Diseases 0.000 description 4
- 208000011038 Cold agglutinin disease Diseases 0.000 description 4
- 206010009944 Colon cancer Diseases 0.000 description 4
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 4
- 208000032612 Glial tumor Diseases 0.000 description 4
- 206010018338 Glioma Diseases 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 208000017604 Hodgkin disease Diseases 0.000 description 4
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 4
- 229940124647 MEK inhibitor Drugs 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 4
- 208000009525 Myocarditis Diseases 0.000 description 4
- 208000033383 Neuroendocrine tumor of pancreas Diseases 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- 206010067517 Pancreatic neuroendocrine tumour Diseases 0.000 description 4
- 208000018737 Parkinson disease Diseases 0.000 description 4
- 206010039710 Scleroderma Diseases 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 230000001154 acute effect Effects 0.000 description 4
- 230000032683 aging Effects 0.000 description 4
- 206010003246 arthritis Diseases 0.000 description 4
- 208000027625 autoimmune inner ear disease Diseases 0.000 description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 4
- 210000000481 breast Anatomy 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 210000001072 colon Anatomy 0.000 description 4
- 210000004351 coronary vessel Anatomy 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 210000003128 head Anatomy 0.000 description 4
- 230000001771 impaired effect Effects 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- VDOCQQKGPJENHJ-UHFFFAOYSA-N methyl n-[4-[4-morpholin-4-yl-1-[1-(pyridin-3-ylmethyl)piperidin-4-yl]pyrazolo[3,4-d]pyrimidin-6-yl]phenyl]carbamate Chemical compound C1=CC(NC(=O)OC)=CC=C1C1=NC(N2CCOCC2)=C(C=NN2C3CCN(CC=4C=NC=CC=4)CC3)C2=N1 VDOCQQKGPJENHJ-UHFFFAOYSA-N 0.000 description 4
- 239000004005 microsphere Substances 0.000 description 4
- 208000005264 motor neuron disease Diseases 0.000 description 4
- 210000003739 neck Anatomy 0.000 description 4
- 208000021010 pancreatic neuroendocrine tumor Diseases 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 229920000728 polyester Polymers 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 229960003387 progesterone Drugs 0.000 description 4
- 239000000186 progesterone Substances 0.000 description 4
- 208000005069 pulmonary fibrosis Diseases 0.000 description 4
- 229960001302 ridaforolimus Drugs 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 208000002320 spinal muscular atrophy Diseases 0.000 description 4
- 206010041823 squamous cell carcinoma Diseases 0.000 description 4
- 210000002784 stomach Anatomy 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 239000000829 suppository Substances 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 230000003827 upregulation Effects 0.000 description 4
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 4
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 4
- 229960004528 vincristine Drugs 0.000 description 4
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 3
- GYLDXIAOMVERTK-UHFFFAOYSA-N 5-(4-amino-1-propan-2-yl-3-pyrazolo[3,4-d]pyrimidinyl)-1,3-benzoxazol-2-amine Chemical compound C12=C(N)N=CN=C2N(C(C)C)N=C1C1=CC=C(OC(N)=N2)C2=C1 GYLDXIAOMVERTK-UHFFFAOYSA-N 0.000 description 3
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 208000002874 Acne Vulgaris Diseases 0.000 description 3
- 208000032194 Acute haemorrhagic leukoencephalitis Diseases 0.000 description 3
- 208000011403 Alexander disease Diseases 0.000 description 3
- 208000032671 Allergic granulomatous angiitis Diseases 0.000 description 3
- 206010001935 American trypanosomiasis Diseases 0.000 description 3
- 208000028185 Angioedema Diseases 0.000 description 3
- 206010003130 Arrhythmia supraventricular Diseases 0.000 description 3
- 206010003267 Arthritis reactive Diseases 0.000 description 3
- 206010003594 Ataxia telangiectasia Diseases 0.000 description 3
- 102000007371 Ataxin-3 Human genes 0.000 description 3
- 102000014461 Ataxins Human genes 0.000 description 3
- 108010078286 Ataxins Proteins 0.000 description 3
- 206010003658 Atrial Fibrillation Diseases 0.000 description 3
- 206010003662 Atrial flutter Diseases 0.000 description 3
- 206010071576 Autoimmune aplastic anaemia Diseases 0.000 description 3
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 3
- 208000023328 Basedow disease Diseases 0.000 description 3
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 3
- 208000003170 Bronchiolo-Alveolar Adenocarcinoma Diseases 0.000 description 3
- 206010068597 Bulbospinal muscular atrophy congenital Diseases 0.000 description 3
- 208000011691 Burkitt lymphomas Diseases 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- 208000022526 Canavan disease Diseases 0.000 description 3
- 208000009458 Carcinoma in Situ Diseases 0.000 description 3
- 206010007559 Cardiac failure congestive Diseases 0.000 description 3
- 208000031229 Cardiomyopathies Diseases 0.000 description 3
- 208000014882 Carotid artery disease Diseases 0.000 description 3
- 208000005024 Castleman disease Diseases 0.000 description 3
- 206010008025 Cerebellar ataxia Diseases 0.000 description 3
- 206010008088 Cerebral artery embolism Diseases 0.000 description 3
- 208000024699 Chagas disease Diseases 0.000 description 3
- 206010008609 Cholangitis sclerosing Diseases 0.000 description 3
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 description 3
- 208000006344 Churg-Strauss Syndrome Diseases 0.000 description 3
- 208000033647 Classic progressive supranuclear palsy syndrome Diseases 0.000 description 3
- 208000010200 Cockayne syndrome Diseases 0.000 description 3
- 208000010007 Cogan syndrome Diseases 0.000 description 3
- 206010009868 Cold type haemolytic anaemia Diseases 0.000 description 3
- 208000013586 Complex regional pain syndrome type 1 Diseases 0.000 description 3
- 206010070954 Congenital hypercoagulation Diseases 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical class [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- 206010011091 Coronary artery thrombosis Diseases 0.000 description 3
- 208000011990 Corticobasal Degeneration Diseases 0.000 description 3
- 206010011258 Coxsackie myocarditis Diseases 0.000 description 3
- 208000020406 Creutzfeldt Jacob disease Diseases 0.000 description 3
- 208000003407 Creutzfeldt-Jakob Syndrome Diseases 0.000 description 3
- 208000010859 Creutzfeldt-Jakob disease Diseases 0.000 description 3
- 206010051055 Deep vein thrombosis Diseases 0.000 description 3
- 201000004624 Dermatitis Diseases 0.000 description 3
- 206010012438 Dermatitis atopic Diseases 0.000 description 3
- 206010048768 Dermatosis Diseases 0.000 description 3
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 3
- 208000005189 Embolism Diseases 0.000 description 3
- 206010014513 Embolism arterial Diseases 0.000 description 3
- 206010049020 Encephalitis periaxialis diffusa Diseases 0.000 description 3
- 206010060742 Endocrine ophthalmopathy Diseases 0.000 description 3
- 208000018428 Eosinophilic granulomatosis with polyangiitis Diseases 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 208000004332 Evans syndrome Diseases 0.000 description 3
- 208000001640 Fibromyalgia Diseases 0.000 description 3
- 206010016654 Fibrosis Diseases 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- 208000003736 Gerstmann-Straussler-Scheinker Disease Diseases 0.000 description 3
- 206010072075 Gerstmann-Straussler-Scheinker syndrome Diseases 0.000 description 3
- 208000010055 Globoid Cell Leukodystrophy Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 208000024869 Goodpasture syndrome Diseases 0.000 description 3
- 208000015023 Graves' disease Diseases 0.000 description 3
- 206010019280 Heart failures Diseases 0.000 description 3
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 description 3
- 201000004331 Henoch-Schoenlein purpura Diseases 0.000 description 3
- 206010019617 Henoch-Schonlein purpura Diseases 0.000 description 3
- 206010019939 Herpes gestationis Diseases 0.000 description 3
- 101000623857 Homo sapiens Serine/threonine-protein kinase mTOR Proteins 0.000 description 3
- 208000031814 IgA Vasculitis Diseases 0.000 description 3
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 3
- 206010021263 IgA nephropathy Diseases 0.000 description 3
- 208000014919 IgG4-related retroperitoneal fibrosis Diseases 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical class C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 206010022557 Intermediate uveitis Diseases 0.000 description 3
- 208000027747 Kennedy disease Diseases 0.000 description 3
- 208000028226 Krabbe disease Diseases 0.000 description 3
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 3
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 3
- 201000010743 Lambert-Eaton myasthenic syndrome Diseases 0.000 description 3
- 208000032514 Leukocytoclastic vasculitis Diseases 0.000 description 3
- 108010000817 Leuprolide Proteins 0.000 description 3
- 201000002832 Lewy body dementia Diseases 0.000 description 3
- 206010024434 Lichen sclerosus Diseases 0.000 description 3
- 108090001030 Lipoproteins Proteins 0.000 description 3
- 102000004895 Lipoproteins Human genes 0.000 description 3
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 3
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- 208000007054 Medullary Carcinoma Diseases 0.000 description 3
- 208000027530 Meniere disease Diseases 0.000 description 3
- 208000001145 Metabolic Syndrome Diseases 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 3
- 208000024599 Mooren ulcer Diseases 0.000 description 3
- 208000012192 Mucous membrane pemphigoid Diseases 0.000 description 3
- 201000002481 Myositis Diseases 0.000 description 3
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 3
- 206010029240 Neuritis Diseases 0.000 description 3
- 206010052057 Neuroborreliosis Diseases 0.000 description 3
- 206010071579 Neuronal neuropathy Diseases 0.000 description 3
- 206010053869 POEMS syndrome Diseases 0.000 description 3
- 206010048705 Paraneoplastic cerebellar degeneration Diseases 0.000 description 3
- 208000004788 Pars Planitis Diseases 0.000 description 3
- 208000031481 Pathologic Constriction Diseases 0.000 description 3
- 208000008223 Pemphigoid Gestationis Diseases 0.000 description 3
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- 208000000609 Pick Disease of the Brain Diseases 0.000 description 3
- 208000000766 Pityriasis Lichenoides Diseases 0.000 description 3
- 206010048895 Pityriasis lichenoides et varioliformis acuta Diseases 0.000 description 3
- 206010065159 Polychondritis Diseases 0.000 description 3
- 208000007048 Polymyalgia Rheumatica Diseases 0.000 description 3
- 208000004347 Postpericardiotomy Syndrome Diseases 0.000 description 3
- 208000032319 Primary lateral sclerosis Diseases 0.000 description 3
- 208000024777 Prion disease Diseases 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 208000010378 Pulmonary Embolism Diseases 0.000 description 3
- 208000012322 Raynaud phenomenon Diseases 0.000 description 3
- 201000001947 Reflex Sympathetic Dystrophy Diseases 0.000 description 3
- 208000005587 Refsum Disease Diseases 0.000 description 3
- 208000033464 Reiter syndrome Diseases 0.000 description 3
- 206010063837 Reperfusion injury Diseases 0.000 description 3
- 208000005793 Restless legs syndrome Diseases 0.000 description 3
- 206010038979 Retroperitoneal fibrosis Diseases 0.000 description 3
- 208000021811 Sandhoff disease Diseases 0.000 description 3
- 208000021235 Schilder disease Diseases 0.000 description 3
- 206010039705 Scleritis Diseases 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 208000009415 Spinocerebellar Ataxias Diseases 0.000 description 3
- 206010072148 Stiff-Person syndrome Diseases 0.000 description 3
- 241000194017 Streptococcus Species 0.000 description 3
- 208000005716 Subacute Combined Degeneration Diseases 0.000 description 3
- 206010042600 Supraventricular arrhythmias Diseases 0.000 description 3
- 208000002286 Susac Syndrome Diseases 0.000 description 3
- 206010042742 Sympathetic ophthalmia Diseases 0.000 description 3
- 208000001871 Tachycardia Diseases 0.000 description 3
- 208000001106 Takayasu Arteritis Diseases 0.000 description 3
- 206010071574 Testicular autoimmunity Diseases 0.000 description 3
- 206010051526 Tolosa-Hunt syndrome Diseases 0.000 description 3
- 208000032109 Transient ischaemic attack Diseases 0.000 description 3
- 208000030886 Traumatic Brain injury Diseases 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 3
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 3
- 208000026928 Turner syndrome Diseases 0.000 description 3
- 206010064996 Ulcerative keratitis Diseases 0.000 description 3
- 206010046298 Upper motor neurone lesion Diseases 0.000 description 3
- 208000035868 Vascular inflammations Diseases 0.000 description 3
- 208000006269 X-Linked Bulbo-Spinal Atrophy Diseases 0.000 description 3
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 3
- 206010000496 acne Diseases 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 208000030597 adult Refsum disease Diseases 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 201000009961 allergic asthma Diseases 0.000 description 3
- 208000004631 alopecia areata Diseases 0.000 description 3
- 206010002022 amyloidosis Diseases 0.000 description 3
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 3
- 230000002280 anti-androgenic effect Effects 0.000 description 3
- 239000000051 antiandrogen Substances 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 230000006793 arrhythmia Effects 0.000 description 3
- 230000003126 arrythmogenic effect Effects 0.000 description 3
- 206010003230 arteritis Diseases 0.000 description 3
- 201000008937 atopic dermatitis Diseases 0.000 description 3
- 208000010928 autoimmune thyroid disease Diseases 0.000 description 3
- 208000029407 autoimmune urticaria Diseases 0.000 description 3
- 201000004562 autosomal dominant cerebellar ataxia Diseases 0.000 description 3
- 206010003882 axonal neuropathy Diseases 0.000 description 3
- 208000007469 bidirectional tachycardia Diseases 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 206010061592 cardiac fibrillation Diseases 0.000 description 3
- 230000002612 cardiopulmonary effect Effects 0.000 description 3
- 210000001627 cerebral artery Anatomy 0.000 description 3
- 229960004630 chlorambucil Drugs 0.000 description 3
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 3
- 208000013507 chronic prostatitis Diseases 0.000 description 3
- 208000024376 chronic urticaria Diseases 0.000 description 3
- 201000010002 cicatricial pemphigoid Diseases 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 201000004395 congenital heart block Diseases 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 239000010949 copper Chemical class 0.000 description 3
- 208000002528 coronary thrombosis Diseases 0.000 description 3
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 3
- 229960000975 daunorubicin Drugs 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 229910052805 deuterium Inorganic materials 0.000 description 3
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 3
- 210000003238 esophagus Anatomy 0.000 description 3
- 229940011871 estrogen Drugs 0.000 description 3
- 239000000262 estrogen Substances 0.000 description 3
- 239000000328 estrogen antagonist Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 230000002600 fibrillogenic effect Effects 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 3
- 229960005277 gemcitabine Drugs 0.000 description 3
- 238000001631 haemodialysis Methods 0.000 description 3
- 230000000322 hemodialysis Effects 0.000 description 3
- 208000007475 hemolytic anemia Diseases 0.000 description 3
- 229940088597 hormone Drugs 0.000 description 3
- 239000005556 hormone Substances 0.000 description 3
- 201000006362 hypersensitivity vasculitis Diseases 0.000 description 3
- 206010021198 ichthyosis Diseases 0.000 description 3
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 3
- 230000028993 immune response Effects 0.000 description 3
- 208000015446 immunoglobulin a vasculitis Diseases 0.000 description 3
- 230000004957 immunoregulator effect Effects 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 201000008319 inclusion body myositis Diseases 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 3
- 238000007917 intracranial administration Methods 0.000 description 3
- 201000010849 intracranial embolism Diseases 0.000 description 3
- 238000007918 intramuscular administration Methods 0.000 description 3
- 238000007912 intraperitoneal administration Methods 0.000 description 3
- 238000007913 intrathecal administration Methods 0.000 description 3
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 3
- 229960004768 irinotecan Drugs 0.000 description 3
- 208000028867 ischemia Diseases 0.000 description 3
- 230000000302 ischemic effect Effects 0.000 description 3
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 3
- 206010023497 kuru Diseases 0.000 description 3
- 201000010901 lateral sclerosis Diseases 0.000 description 3
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 3
- 229960004338 leuprorelin Drugs 0.000 description 3
- 201000011486 lichen planus Diseases 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 3
- 208000004731 long QT syndrome Diseases 0.000 description 3
- 201000011649 lymphoblastic lymphoma Diseases 0.000 description 3
- 208000003747 lymphoid leukemia Diseases 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 230000028161 membrane depolarization Effects 0.000 description 3
- 206010063344 microscopic polyangiitis Diseases 0.000 description 3
- 208000012268 mitochondrial disease Diseases 0.000 description 3
- 230000004898 mitochondrial function Effects 0.000 description 3
- 201000010879 mucinous adenocarcinoma Diseases 0.000 description 3
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 3
- 201000008383 nephritis Diseases 0.000 description 3
- 230000001537 neural effect Effects 0.000 description 3
- 208000002040 neurosyphilis Diseases 0.000 description 3
- 208000004235 neutropenia Diseases 0.000 description 3
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 3
- 208000015200 ocular cicatricial pemphigoid Diseases 0.000 description 3
- 201000005580 palindromic rheumatism Diseases 0.000 description 3
- 208000008510 paroxysmal tachycardia Diseases 0.000 description 3
- 208000033808 peripheral neuropathy Diseases 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 201000006292 polyarteritis nodosa Diseases 0.000 description 3
- 208000005987 polymyositis Diseases 0.000 description 3
- 230000035935 pregnancy Effects 0.000 description 3
- 201000000742 primary sclerosing cholangitis Diseases 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 208000032207 progressive 1 supranuclear palsy Diseases 0.000 description 3
- 201000002212 progressive supranuclear palsy Diseases 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 230000002035 prolonged effect Effects 0.000 description 3
- 210000002307 prostate Anatomy 0.000 description 3
- 201000007094 prostatitis Diseases 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 208000009954 pyoderma gangrenosum Diseases 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 208000009169 relapsing polychondritis Diseases 0.000 description 3
- 201000003068 rheumatic fever Diseases 0.000 description 3
- 229950009216 sapanisertib Drugs 0.000 description 3
- 201000000980 schizophrenia Diseases 0.000 description 3
- 208000010157 sclerosing cholangitis Diseases 0.000 description 3
- CYOHGALHFOKKQC-UHFFFAOYSA-N selumetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1Cl CYOHGALHFOKKQC-UHFFFAOYSA-N 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 208000017520 skin disease Diseases 0.000 description 3
- 235000015424 sodium Nutrition 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 210000000278 spinal cord Anatomy 0.000 description 3
- 208000002025 tabes dorsalis Diseases 0.000 description 3
- 230000006794 tachycardia Effects 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 201000005060 thrombophlebitis Diseases 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 3
- 229960000303 topotecan Drugs 0.000 description 3
- MFAQYJIYDMLAIM-UHFFFAOYSA-N torkinib Chemical compound C12=C(N)N=CN=C2N(C(C)C)N=C1C1=CC2=CC(O)=CC=C2N1 MFAQYJIYDMLAIM-UHFFFAOYSA-N 0.000 description 3
- 201000010875 transient cerebral ischemia Diseases 0.000 description 3
- 230000014616 translation Effects 0.000 description 3
- 208000009174 transverse myelitis Diseases 0.000 description 3
- 230000009529 traumatic brain injury Effects 0.000 description 3
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 3
- 229910052722 tritium Inorganic materials 0.000 description 3
- 210000003932 urinary bladder Anatomy 0.000 description 3
- 238000002255 vaccination Methods 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 206010047302 ventricular tachycardia Diseases 0.000 description 3
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- DWZAEMINVBZMHQ-UHFFFAOYSA-N 1-[4-[4-(dimethylamino)piperidine-1-carbonyl]phenyl]-3-[4-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)phenyl]urea Chemical compound C1CC(N(C)C)CCN1C(=O)C(C=C1)=CC=C1NC(=O)NC1=CC=C(C=2N=C(N=C(N=2)N2CCOCC2)N2CCOCC2)C=C1 DWZAEMINVBZMHQ-UHFFFAOYSA-N 0.000 description 2
- RGJOJUGRHPQXGF-INIZCTEOSA-N 1-ethyl-3-[4-[4-[(3s)-3-methylmorpholin-4-yl]-7-(oxetan-3-yl)-6,8-dihydro-5h-pyrido[3,4-d]pyrimidin-2-yl]phenyl]urea Chemical compound C1=CC(NC(=O)NCC)=CC=C1C(N=C1N2[C@H](COCC2)C)=NC2=C1CCN(C1COC1)C2 RGJOJUGRHPQXGF-INIZCTEOSA-N 0.000 description 2
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 2
- GFMMXOIFOQCCGU-UHFFFAOYSA-N 2-(2-chloro-4-iodoanilino)-N-(cyclopropylmethoxy)-3,4-difluorobenzamide Chemical compound C=1C=C(I)C=C(Cl)C=1NC1=C(F)C(F)=CC=C1C(=O)NOCC1CC1 GFMMXOIFOQCCGU-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- KOQIAZNBAWFSQM-UHFFFAOYSA-N 2-[4-(3-ethynylanilino)-7-(2-methoxyethoxy)quinazolin-6-yl]oxyethanol Chemical compound C=12C=C(OCCO)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 KOQIAZNBAWFSQM-UHFFFAOYSA-N 0.000 description 2
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 2
- JUSFANSTBFGBAF-IRXDYDNUSA-N 3-[2,4-bis[(3s)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl]-n-methylbenzamide Chemical compound CNC(=O)C1=CC=CC(C=2N=C3N=C(N=C(C3=CC=2)N2[C@H](COCC2)C)N2[C@H](COCC2)C)=C1 JUSFANSTBFGBAF-IRXDYDNUSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- QCXJEYYXVJIFCE-UHFFFAOYSA-N 4-acetamidobenzoic acid Chemical compound CC(=O)NC1=CC=C(C(O)=O)C=C1 QCXJEYYXVJIFCE-UHFFFAOYSA-N 0.000 description 2
- KVLFRAWTRWDEDF-IRXDYDNUSA-N AZD-8055 Chemical compound C1=C(CO)C(OC)=CC=C1C1=CC=C(C(=NC(=N2)N3[C@H](COCC3)C)N3[C@H](COCC3)C)C2=N1 KVLFRAWTRWDEDF-IRXDYDNUSA-N 0.000 description 2
- 101001082110 Acanthamoeba polyphaga mimivirus Eukaryotic translation initiation factor 4E homolog Proteins 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical class [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 206010002961 Aplasia Diseases 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000416162 Astragalus gummifer Species 0.000 description 2
- 206010003694 Atrophy Diseases 0.000 description 2
- 206010071577 Autoimmune hyperlipidaemia Diseases 0.000 description 2
- 206010064539 Autoimmune myocarditis Diseases 0.000 description 2
- 206010069002 Autoimmune pancreatitis Diseases 0.000 description 2
- 206010003840 Autonomic nervous system imbalance Diseases 0.000 description 2
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 2
- YUXMAKUNSXIEKN-BTJKTKAUSA-N BGT226 Chemical compound OC(=O)\C=C/C(O)=O.C1=NC(OC)=CC=C1C1=CC=C(N=CC2=C3N(C=4C=C(C(N5CCNCC5)=CC=4)C(F)(F)F)C(=O)N2C)C3=C1 YUXMAKUNSXIEKN-BTJKTKAUSA-N 0.000 description 2
- 206010004146 Basal cell carcinoma Diseases 0.000 description 2
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- UFKLYTOEMRFKAD-SHTZXODSSA-N C1C[C@@H](OC)CC[C@@H]1N1C2=NC(C=3C=NC(=CC=3)C(C)(C)O)=CN=C2NCC1=O Chemical compound C1C[C@@H](OC)CC[C@@H]1N1C2=NC(C=3C=NC(=CC=3)C(C)(C)O)=CN=C2NCC1=O UFKLYTOEMRFKAD-SHTZXODSSA-N 0.000 description 2
- 206010008583 Chloroma Diseases 0.000 description 2
- 208000006332 Choriocarcinoma Diseases 0.000 description 2
- 206010010741 Conjunctivitis Diseases 0.000 description 2
- 208000019707 Cryoglobulinemic vasculitis Diseases 0.000 description 2
- 229920000858 Cyclodextrin Polymers 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 102100029816 DEP domain-containing mTOR-interacting protein Human genes 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 101001082109 Danio rerio Eukaryotic translation initiation factor 4E-1B Proteins 0.000 description 2
- 101000678286 Danio rerio Eukaryotic translation initiation factor 4E-binding protein 3-like Proteins 0.000 description 2
- 206010012468 Dermatitis herpetiformis Diseases 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 101000800913 Dictyostelium discoideum Eukaryotic translation initiation factor 4E-1A-binding protein homolog Proteins 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- 101000800906 Drosophila melanogaster Eukaryotic translation initiation factor 4E-binding protein Proteins 0.000 description 2
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 2
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 2
- 102100023266 Dual specificity mitogen-activated protein kinase kinase 2 Human genes 0.000 description 2
- 101710146529 Dual specificity mitogen-activated protein kinase kinase 2 Proteins 0.000 description 2
- 102000001301 EGF receptor Human genes 0.000 description 2
- 108060006698 EGF receptor Proteins 0.000 description 2
- 201000009273 Endometriosis Diseases 0.000 description 2
- 206010014954 Eosinophilic fasciitis Diseases 0.000 description 2
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 2
- 206010015226 Erythema nodosum Diseases 0.000 description 2
- DSLZVSRJTYRBFB-UHFFFAOYSA-N Galactaric acid Natural products OC(=O)C(O)C(O)C(O)C(O)C(O)=O DSLZVSRJTYRBFB-UHFFFAOYSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 2
- 101000865183 Homo sapiens DEP domain-containing mTOR-interacting protein Proteins 0.000 description 2
- 101001060744 Homo sapiens Peptidyl-prolyl cis-trans isomerase FKBP1A Proteins 0.000 description 2
- 206010020772 Hypertension Diseases 0.000 description 2
- 208000021330 IgG4-related disease Diseases 0.000 description 2
- 206010053574 Immunoblastic lymphoma Diseases 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- 208000029462 Immunodeficiency disease Diseases 0.000 description 2
- 208000031781 Immunoglobulin G4 related sclerosing disease Diseases 0.000 description 2
- 208000004187 Immunoglobulin G4-Related Disease Diseases 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical class [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- RFSMUFRPPYDYRD-CALCHBBNSA-N Ku-0063794 Chemical compound C1=C(CO)C(OC)=CC=C1C1=CC=C(C(=NC(=N2)N3C[C@@H](C)O[C@@H](C)C3)N3CCOCC3)C2=N1 RFSMUFRPPYDYRD-CALCHBBNSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 2
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 2
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 2
- 208000009829 Lewy Body Disease Diseases 0.000 description 2
- 208000001089 Multiple system atrophy Diseases 0.000 description 2
- 241001467552 Mycobacterium bovis BCG Species 0.000 description 2
- 102100026784 Myelin proteolipid protein Human genes 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 2
- 206010029260 Neuroblastoma Diseases 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 206010031252 Osteomyelitis Diseases 0.000 description 2
- 108091007960 PI3Ks Proteins 0.000 description 2
- NVRXTLZYXZNATH-UHFFFAOYSA-N PP121 Chemical compound N1=C(C=2C=C3C=CNC3=NC=2)C=2C(N)=NC=NC=2N1C1CCCC1 NVRXTLZYXZNATH-UHFFFAOYSA-N 0.000 description 2
- 208000017493 Pelizaeus-Merzbacher disease Diseases 0.000 description 2
- 208000029082 Pelvic Inflammatory Disease Diseases 0.000 description 2
- 101710111747 Peptidyl-prolyl cis-trans isomerase FKBP12 Proteins 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical class [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 2
- 208000037534 Progressive hemifacial atrophy Diseases 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 229940078123 Ras inhibitor Drugs 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 2
- 201000001542 Schneiderian carcinoma Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 241000187391 Streptomyces hygroscopicus Species 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 2
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- 229920001615 Tragacanth Polymers 0.000 description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 description 2
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 2
- LNFBAYSBVQBKFR-UHFFFAOYSA-N [7-(6-aminopyridin-3-yl)-3,5-dihydro-2h-1,4-benzoxazepin-4-yl]-(3-fluoro-2-methyl-4-methylsulfonylphenyl)methanone Chemical compound CC1=C(F)C(S(C)(=O)=O)=CC=C1C(=O)N1CC2=CC(C=3C=NC(N)=CC=3)=CC=C2OCC1 LNFBAYSBVQBKFR-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 208000002517 adenoid cystic carcinoma Diseases 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 229960001686 afatinib Drugs 0.000 description 2
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 125000005218 alkyleneheteroaryl group Chemical group 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000003712 anti-aging effect Effects 0.000 description 2
- 229940046836 anti-estrogen Drugs 0.000 description 2
- 230000001833 anti-estrogenic effect Effects 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000037444 atrophy Effects 0.000 description 2
- 208000006424 autoimmune oophoritis Diseases 0.000 description 2
- 206010071578 autoimmune retinopathy Diseases 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 229960000190 bacillus calmette–guérin vaccine Drugs 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical compound BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 2
- 229960002092 busulfan Drugs 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical class C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 2
- 238000002619 cancer immunotherapy Methods 0.000 description 2
- 229950002826 canertinib Drugs 0.000 description 2
- OMZCMEYTWSXEPZ-UHFFFAOYSA-N canertinib Chemical compound C1=C(Cl)C(F)=CC=C1NC1=NC=NC2=CC(OCCCN3CCOCC3)=C(NC(=O)C=C)C=C12 OMZCMEYTWSXEPZ-UHFFFAOYSA-N 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000004640 cellular pathway Effects 0.000 description 2
- JROFGZPOBKIAEW-HAQNSBGRSA-N chembl3120215 Chemical compound N1C=2C(OC)=CC=CC=2C=C1C(=C1C(N)=NC=NN11)N=C1[C@H]1CC[C@H](C(O)=O)CC1 JROFGZPOBKIAEW-HAQNSBGRSA-N 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 230000007882 cirrhosis Effects 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 229960002271 cobimetinib Drugs 0.000 description 2
- BSMCAPRUBJMWDF-KRWDZBQOSA-N cobimetinib Chemical compound C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F BSMCAPRUBJMWDF-KRWDZBQOSA-N 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- LVXJQMNHJWSHET-AATRIKPKSA-N dacomitinib Chemical compound C=12C=C(NC(=O)\C=C\CN3CCCCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 LVXJQMNHJWSHET-AATRIKPKSA-N 0.000 description 2
- 229950006418 dactolisib Drugs 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 2
- 230000003210 demyelinating effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 2
- 208000019479 dysautonomia Diseases 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 229960001904 epirubicin Drugs 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 235000019439 ethyl acetate Nutrition 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- 229960000752 etoposide phosphate Drugs 0.000 description 2
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 2
- 208000002980 facial hemiatrophy Diseases 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- DBEPLOCGEIEOCV-WSBQPABSSA-N finasteride Chemical compound N([C@@H]1CC2)C(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)NC(C)(C)C)[C@@]2(C)CC1 DBEPLOCGEIEOCV-WSBQPABSSA-N 0.000 description 2
- 229960004039 finasteride Drugs 0.000 description 2
- 229960000390 fludarabine Drugs 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 2
- 229960002074 flutamide Drugs 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 2
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 2
- CHPZKNULDCNCBW-UHFFFAOYSA-N gallium nitrate Chemical compound [Ga+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O CHPZKNULDCNCBW-UHFFFAOYSA-N 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 208000018090 giant cell myocarditis Diseases 0.000 description 2
- 235000001727 glucose Nutrition 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 2
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 230000007813 immunodeficiency Effects 0.000 description 2
- 239000002955 immunomodulating agent Substances 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 201000004933 in situ carcinoma Diseases 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 230000002912 lymphogenic effect Effects 0.000 description 2
- 208000025036 lymphosarcoma Diseases 0.000 description 2
- 239000003120 macrolide antibiotic agent Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 230000036210 malignancy Effects 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 2
- 229960001924 melphalan Drugs 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 229960002900 methylcellulose Drugs 0.000 description 2
- 230000004065 mitochondrial dysfunction Effects 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 201000005987 myeloid sarcoma Diseases 0.000 description 2
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 2
- 229950008835 neratinib Drugs 0.000 description 2
- JWNPDZNEKVCWMY-VQHVLOKHSA-N neratinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 JWNPDZNEKVCWMY-VQHVLOKHSA-N 0.000 description 2
- 201000011519 neuroendocrine tumor Diseases 0.000 description 2
- 201000001119 neuropathy Diseases 0.000 description 2
- 230000007823 neuropathy Effects 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- PXQPEWDEAKTCGB-UHFFFAOYSA-N orotic acid Chemical compound OC(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229960001972 panitumumab Drugs 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- WVUNYSQLFKLYNI-AATRIKPKSA-N pelitinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 WVUNYSQLFKLYNI-AATRIKPKSA-N 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 208000030761 polycystic kidney disease Diseases 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 239000011591 potassium Chemical class 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 2
- 229960004618 prednisone Drugs 0.000 description 2
- 208000018290 primary dysautonomia Diseases 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 201000008752 progressive muscular atrophy Diseases 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- GHBFNMLVSPCDGN-UHFFFAOYSA-N rac-1-monooctanoylglycerol Chemical compound CCCCCCCC(=O)OCC(O)CO GHBFNMLVSPCDGN-UHFFFAOYSA-N 0.000 description 2
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical compound C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 201000006845 reticulosarcoma Diseases 0.000 description 2
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- 208000000649 small cell carcinoma Diseases 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- 229960003787 sorafenib Drugs 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 210000000952 spleen Anatomy 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 230000036262 stenosis Effects 0.000 description 2
- 208000037804 stenosis Diseases 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 2
- 229950007866 tanespimycin Drugs 0.000 description 2
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 2
- 229960000235 temsirolimus Drugs 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N thiocyanic acid Chemical compound SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 2
- 229960001196 thiotepa Drugs 0.000 description 2
- 201000002510 thyroid cancer Diseases 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- AKCRNFFTGXBONI-UHFFFAOYSA-N torin 1 Chemical compound C1CN(C(=O)CC)CCN1C1=CC=C(N2C(C=CC3=C2C2=CC(=CC=C2N=C3)C=2C=C3C=CC=CC3=NC=2)=O)C=C1C(F)(F)F AKCRNFFTGXBONI-UHFFFAOYSA-N 0.000 description 2
- GUXXEUUYCAYESJ-UHFFFAOYSA-N torin 2 Chemical compound C1=NC(N)=CC=C1C1=CC=C(N=CC2=C3N(C=4C=C(C=CC=4)C(F)(F)F)C(=O)C=C2)C3=C1 GUXXEUUYCAYESJ-UHFFFAOYSA-N 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- 229960004066 trametinib Drugs 0.000 description 2
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 239000002691 unilamellar liposome Substances 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 206010046766 uterine cancer Diseases 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 2
- 229960003048 vinblastine Drugs 0.000 description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 2
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 2
- 229960004355 vindesine Drugs 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- QFJCIRLUMZQUOT-KADBNGAOSA-N (1R,9S,12S,15R,16E,18R,19R,21R,23S,24Z,26E,28E,30S,32S,35R)-1,18-dihydroxy-12-[(2R)-1-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]propan-2-yl]-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20-pentone Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)\C(C)=C\C=C\C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-KADBNGAOSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- SVNJBEMPMKWDCO-KCHLEUMXSA-N (2s)-2-[[(2s)-3-carboxy-2-[[2-[[(2s)-5-(diaminomethylideneamino)-2-[[4-oxo-4-[[4-(4-oxo-8-phenylchromen-2-yl)morpholin-4-ium-4-yl]methoxy]butanoyl]amino]pentanoyl]amino]acetyl]amino]propanoyl]amino]-3-hydroxypropanoate Chemical compound C=1C(=O)C2=CC=CC(C=3C=CC=CC=3)=C2OC=1[N+]1(COC(=O)CCC(=O)N[C@@H](CCCNC(=N)N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C([O-])=O)CCOCC1 SVNJBEMPMKWDCO-KCHLEUMXSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 description 1
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- UQVNRKBFAXNOGA-OHLDGCSVSA-N (Z)-tomaymycin Chemical compound CO[C@H]1NC2=CC(O)=C(OC)C=C2C(=O)N2C\C(=C/C)C[C@@H]12 UQVNRKBFAXNOGA-OHLDGCSVSA-N 0.000 description 1
- DQXKOHDUMJLXKH-PHEQNACWSA-N (e)-n-[2-[2-[[(e)-oct-2-enoyl]amino]ethyldisulfanyl]ethyl]oct-2-enamide Chemical compound CCCCC\C=C\C(=O)NCCSSCCNC(=O)\C=C\CCCCC DQXKOHDUMJLXKH-PHEQNACWSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- SJJCQDRGABAVBB-UHFFFAOYSA-N 1-hydroxy-2-naphthoic acid Chemical compound C1=CC=CC2=C(O)C(C(=O)O)=CC=C21 SJJCQDRGABAVBB-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- RZRNAYUHWVFMIP-KTKRTIGZSA-N 1-oleoylglycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-KTKRTIGZSA-N 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- FRPZMMHWLSIFAZ-UHFFFAOYSA-N 10-undecenoic acid Chemical compound OC(=O)CCCCCCCCC=C FRPZMMHWLSIFAZ-UHFFFAOYSA-N 0.000 description 1
- 101710175516 14 kDa zinc-binding protein Proteins 0.000 description 1
- BFPYWIDHMRZLRN-UHFFFAOYSA-N 17alpha-ethynyl estradiol Natural products OC1=CC=C2C3CCC(C)(C(CC4)(O)C#C)C4C3CCC2=C1 BFPYWIDHMRZLRN-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- RWEVIPRMPFNTLO-UHFFFAOYSA-N 2-(2-fluoro-4-iodoanilino)-N-(2-hydroxyethoxy)-1,5-dimethyl-6-oxo-3-pyridinecarboxamide Chemical compound CN1C(=O)C(C)=CC(C(=O)NOCCO)=C1NC1=CC=C(I)C=C1F RWEVIPRMPFNTLO-UHFFFAOYSA-N 0.000 description 1
- VHVPQPYKVGDNFY-DFMJLFEVSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2r,4s)-2-(2,4-dichlorophenyl)-2-(1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@@H]3O[C@](CN4N=CN=C4)(OC3)C=3C(=CC(Cl)=CC=3)Cl)=CC=2)C=C1 VHVPQPYKVGDNFY-DFMJLFEVSA-N 0.000 description 1
- OIQOAYVCKAHSEJ-UHFFFAOYSA-N 2-[2,3-bis(2-hydroxyethoxy)propoxy]ethanol;hexadecanoic acid;octadecanoic acid Chemical compound OCCOCC(OCCO)COCCO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O OIQOAYVCKAHSEJ-UHFFFAOYSA-N 0.000 description 1
- MHXVDXXARZCVRK-WCWDXBQESA-N 2-[2-[4-[(e)-3,3,3-trifluoro-1,2-diphenylprop-1-enyl]phenoxy]ethylamino]ethanol Chemical compound C1=CC(OCCNCCO)=CC=C1C(\C=1C=CC=CC=1)=C(C(F)(F)F)/C1=CC=CC=C1 MHXVDXXARZCVRK-WCWDXBQESA-N 0.000 description 1
- PXJJOGITBQXZEQ-JTHROIFXSA-M 2-[4-[(z)-1,2-diphenylbut-1-enyl]phenoxy]ethyl-trimethylazanium;iodide Chemical compound [I-].C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCC[N+](C)(C)C)=CC=1)/C1=CC=CC=C1 PXJJOGITBQXZEQ-JTHROIFXSA-M 0.000 description 1
- OTLLEIBWKHEHGU-UHFFFAOYSA-N 2-[5-[[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy]-3,4-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-4-phosphonooxyhexanedioic acid Chemical compound C1=NC=2C(N)=NC=NC=2N1C(C(C1O)O)OC1COC1C(CO)OC(OC(C(O)C(OP(O)(O)=O)C(O)C(O)=O)C(O)=O)C(O)C1O OTLLEIBWKHEHGU-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- XDLYKKIQACFMJG-UHFFFAOYSA-N 2-amino-8-[4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxypyridin-3-yl)-4-methylpyrido[2,3-d]pyrimidin-7-one Chemical compound C1=NC(OC)=CC=C1C(C1=O)=CC2=C(C)N=C(N)N=C2N1C1CCC(OCCO)CC1 XDLYKKIQACFMJG-UHFFFAOYSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- CTRPRMNBTVRDFH-UHFFFAOYSA-N 2-n-methyl-1,3,5-triazine-2,4,6-triamine Chemical class CNC1=NC(N)=NC(N)=N1 CTRPRMNBTVRDFH-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-N 3-Hydroxy-2-naphthoate Chemical compound C1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-N 0.000 description 1
- RCLQNICOARASSR-SECBINFHSA-N 3-[(2r)-2,3-dihydroxypropyl]-6-fluoro-5-(2-fluoro-4-iodoanilino)-8-methylpyrido[2,3-d]pyrimidine-4,7-dione Chemical compound FC=1C(=O)N(C)C=2N=CN(C[C@@H](O)CO)C(=O)C=2C=1NC1=CC=C(I)C=C1F RCLQNICOARASSR-SECBINFHSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- QOXOZONBQWIKDA-UHFFFAOYSA-N 3-hydroxypropyl Chemical group [CH2]CCO QOXOZONBQWIKDA-UHFFFAOYSA-N 0.000 description 1
- HQAIUXZORKJOJY-UHFFFAOYSA-N 3-iodo-2h-pyrazolo[3,4-d]pyrimidin-4-amine Chemical compound NC1=NC=NC2=NNC(I)=C12 HQAIUXZORKJOJY-UHFFFAOYSA-N 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- SXIFAEWFOJETOA-UHFFFAOYSA-N 4-hydroxy-butyl Chemical group [CH2]CCCO SXIFAEWFOJETOA-UHFFFAOYSA-N 0.000 description 1
- UWXSAYUXVSFDBQ-CYBMUJFWSA-N 4-n-[3-chloro-4-(1,3-thiazol-2-ylmethoxy)phenyl]-6-n-[(4r)-4-methyl-4,5-dihydro-1,3-oxazol-2-yl]quinazoline-4,6-diamine Chemical compound C[C@@H]1COC(NC=2C=C3C(NC=4C=C(Cl)C(OCC=5SC=CN=5)=CC=4)=NC=NC3=CC=2)=N1 UWXSAYUXVSFDBQ-CYBMUJFWSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical group [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- VERUFXOALATMPS-UHFFFAOYSA-N 5,5-diamino-2-(2-phenylethenyl)cyclohex-3-ene-1,1-disulfonic acid Chemical compound C1=CC(N)(N)CC(S(O)(=O)=O)(S(O)(=O)=O)C1C=CC1=CC=CC=C1 VERUFXOALATMPS-UHFFFAOYSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- SDEAXTCZPQIFQM-UHFFFAOYSA-N 6-n-(4,4-dimethyl-5h-1,3-oxazol-2-yl)-4-n-[3-methyl-4-([1,2,4]triazolo[1,5-a]pyridin-7-yloxy)phenyl]quinazoline-4,6-diamine Chemical compound C=1C=C(OC2=CC3=NC=NN3C=C2)C(C)=CC=1NC(C1=C2)=NC=NC1=CC=C2NC1=NC(C)(C)CO1 SDEAXTCZPQIFQM-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- PLIVFNIUGLLCEK-UHFFFAOYSA-N 7-[4-(3-ethynylanilino)-7-methoxyquinazolin-6-yl]oxy-n-hydroxyheptanamide Chemical compound C=12C=C(OCCCCCCC(=O)NO)C(OC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 PLIVFNIUGLLCEK-UHFFFAOYSA-N 0.000 description 1
- GOJJWDOZNKBUSR-UHFFFAOYSA-N 7-sulfamoyloxyheptyl sulfamate Chemical compound NS(=O)(=O)OCCCCCCCOS(N)(=O)=O GOJJWDOZNKBUSR-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- OONFNUWBHFSNBT-HXUWFJFHSA-N AEE788 Chemical compound C1CN(CC)CCN1CC1=CC=C(C=2NC3=NC=NC(N[C@H](C)C=4C=CC=CC=4)=C3C=2)C=C1 OONFNUWBHFSNBT-HXUWFJFHSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 208000016557 Acute basophilic leukemia Diseases 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 206010000871 Acute monocytic leukaemia Diseases 0.000 description 1
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 1
- 241000321096 Adenoides Species 0.000 description 1
- 208000006468 Adrenal Cortex Neoplasms Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000037540 Alveolar soft tissue sarcoma Diseases 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 208000022106 Autoimmune polyendocrinopathy type 2 Diseases 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 102000008096 B7-H1 Antigen Human genes 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- GUBGYTABKSRVRQ-DCSYEGIMSA-N Beta-Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-DCSYEGIMSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 208000013165 Bowen disease Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 206010058354 Bronchioloalveolar carcinoma Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- OHQZVHZKJPUGPI-UHFFFAOYSA-N C(C1)NCC[S+]1O[S+]1CCNCC1 Chemical compound C(C1)NCC[S+]1O[S+]1CCNCC1 OHQZVHZKJPUGPI-UHFFFAOYSA-N 0.000 description 1
- HXNLKCWYIZCDCQ-UHFFFAOYSA-N C(O)(O)=S.C(O)(O)=O Chemical compound C(O)(O)=S.C(O)(O)=O HXNLKCWYIZCDCQ-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 1
- 229940124292 CD20 monoclonal antibody Drugs 0.000 description 1
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 1
- LLVZBTWPGQVVLW-SNAWJCMRSA-N CP-724714 Chemical compound C12=CC(/C=C/CNC(=O)COC)=CC=C2N=CN=C1NC(C=C1C)=CC=C1OC1=CC=C(C)N=C1 LLVZBTWPGQVVLW-SNAWJCMRSA-N 0.000 description 1
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 1
- 206010006895 Cachexia Diseases 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N Camphoric acid Natural products CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 1
- 241000222122 Candida albicans Species 0.000 description 1
- 239000005461 Canertinib Substances 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 201000005947 Carney Complex Diseases 0.000 description 1
- 108010031425 Casein Kinases Proteins 0.000 description 1
- 102000005403 Casein Kinases Human genes 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 108010004103 Chylomicrons Proteins 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000012609 Cowden disease Diseases 0.000 description 1
- 201000007336 Cryptococcosis Diseases 0.000 description 1
- 241000221204 Cryptococcus neoformans Species 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 208000011518 Danon disease Diseases 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- 206010067889 Dementia with Lewy bodies Diseases 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 208000006402 Ductal Carcinoma Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 201000009051 Embryonal Carcinoma Diseases 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010057649 Endometrial sarcoma Diseases 0.000 description 1
- 102100030013 Endoribonuclease Human genes 0.000 description 1
- 101710199605 Endoribonuclease Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 206010014958 Eosinophilic leukaemia Diseases 0.000 description 1
- 206010064212 Eosinophilic oesophagitis Diseases 0.000 description 1
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 208000032027 Essential Thrombocythemia Diseases 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- BFPYWIDHMRZLRN-SLHNCBLASA-N Ethinyl estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=C1 BFPYWIDHMRZLRN-SLHNCBLASA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 208000001382 Experimental Melanoma Diseases 0.000 description 1
- 208000009331 Experimental Sarcoma Diseases 0.000 description 1
- 201000006107 Familial adenomatous polyposis Diseases 0.000 description 1
- 201000006850 Familial medullary thyroid carcinoma Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 description 1
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 229910052688 Gadolinium Inorganic materials 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 1
- 208000008069 Geographic Atrophy Diseases 0.000 description 1
- 208000008999 Giant Cell Carcinoma Diseases 0.000 description 1
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 1
- 208000001500 Glycogen Storage Disease Type IIb Diseases 0.000 description 1
- 208000035148 Glycogen storage disease due to LAMP-2 deficiency Diseases 0.000 description 1
- 206010053185 Glycogen storage disease type II Diseases 0.000 description 1
- 229920002683 Glycosaminoglycan Polymers 0.000 description 1
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 208000002927 Hamartoma Diseases 0.000 description 1
- 206010019263 Heart block congenital Diseases 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 208000017662 Hodgkin disease lymphocyte depletion type stage unspecified Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000878213 Homo sapiens Inactive peptidyl-prolyl cis-trans isomerase FKBP6 Proteins 0.000 description 1
- 101000690268 Homo sapiens Proline-rich AKT1 substrate 1 Proteins 0.000 description 1
- 101100087590 Homo sapiens RICTOR gene Proteins 0.000 description 1
- 101000633815 Homo sapiens TELO2-interacting protein 1 homolog Proteins 0.000 description 1
- 101000597183 Homo sapiens Telomere length regulation protein TEL2 homolog Proteins 0.000 description 1
- 101001057127 Homo sapiens Transcription factor ETV7 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- DOMWKUIIPQCAJU-LJHIYBGHSA-N Hydroxyprogesterone caproate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)CCCCC)[C@@]1(C)CC2 DOMWKUIIPQCAJU-LJHIYBGHSA-N 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 206010048643 Hypereosinophilic syndrome Diseases 0.000 description 1
- 208000031309 Hypertrophic Familial Cardiomyopathy Diseases 0.000 description 1
- 210000005131 Hürthle cell Anatomy 0.000 description 1
- 102100036984 Inactive peptidyl-prolyl cis-trans isomerase FKBP6 Human genes 0.000 description 1
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 1
- 102000014429 Insulin-like growth factor Human genes 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 206010023256 Juvenile melanoma benign Diseases 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- CZQHHVNHHHRRDU-UHFFFAOYSA-N LY294002 Chemical compound C1=CC=C2C(=O)C=C(N3CCOCC3)OC2=C1C1=CC=CC=C1 CZQHHVNHHHRRDU-UHFFFAOYSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 102000004058 Leukemia inhibitory factor Human genes 0.000 description 1
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 1
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical class [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 206010049459 Lymphangioleiomyomatosis Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102000004318 Matrilysin Human genes 0.000 description 1
- 108090000855 Matrilysin Proteins 0.000 description 1
- 102000000422 Matrix Metalloproteinase 3 Human genes 0.000 description 1
- 208000009018 Medullary thyroid cancer Diseases 0.000 description 1
- 208000037196 Medullary thyroid carcinoma Diseases 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102100040243 Microtubule-associated protein tau Human genes 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 208000035489 Monocytic Acute Leukemia Diseases 0.000 description 1
- 208000026072 Motor neurone disease Diseases 0.000 description 1
- 206010057269 Mucoepidermoid carcinoma Diseases 0.000 description 1
- 241000306281 Mucor ambiguus Species 0.000 description 1
- 208000008770 Multiple Hamartoma Syndrome Diseases 0.000 description 1
- 206010073148 Multiple endocrine neoplasia type 2A Diseases 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 201000009623 Myopathy Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- MVZGYPSXNDCANY-UHFFFAOYSA-N N-[4-[3-chloro-4-[(3-fluorophenyl)methoxy]anilino]-6-quinazolinyl]-2-propenamide Chemical compound FC1=CC=CC(COC=2C(=CC(NC=3C4=CC(NC(=O)C=C)=CC=C4N=CN=3)=CC=2)Cl)=C1 MVZGYPSXNDCANY-UHFFFAOYSA-N 0.000 description 1
- LKJPYSCBVHEWIU-UHFFFAOYSA-N N-[4-cyano-3-(trifluoromethyl)phenyl]-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methylpropanamide Chemical compound C=1C=C(C#N)C(C(F)(F)F)=CC=1NC(=O)C(O)(C)CS(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 208000002454 Nasopharyngeal Carcinoma Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 208000009905 Neurofibromatoses Diseases 0.000 description 1
- 208000003019 Neurofibromatosis 1 Diseases 0.000 description 1
- 208000024834 Neurofibromatosis type 1 Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 206010029488 Nodular melanoma Diseases 0.000 description 1
- HBPQPBSTHOHSFP-UHFFFAOYSA-N OC(=O)C([Pt])=O Chemical compound OC(=O)C([Pt])=O HBPQPBSTHOHSFP-UHFFFAOYSA-N 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 206010030216 Oesophagitis Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- TUVCWJQQGGETHL-UHFFFAOYSA-N PI-103 Chemical compound OC1=CC=CC(C=2N=C3C4=CC=CN=C4OC3=C(N3CCOCC3)N=2)=C1 TUVCWJQQGGETHL-UHFFFAOYSA-N 0.000 description 1
- 239000012828 PI3K inhibitor Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 206010033701 Papillary thyroid cancer Diseases 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 101710111682 Peptidyl-prolyl cis-trans isomerase FKBP1A Proteins 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- 206010034764 Peutz-Jeghers syndrome Diseases 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 102000010752 Plasminogen Inactivators Human genes 0.000 description 1
- 108010077971 Plasminogen Inactivators Proteins 0.000 description 1
- 229920000604 Polyethylene Glycol 200 Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 102100024091 Proline-rich AKT1 substrate 1 Human genes 0.000 description 1
- 102100034733 Proline-rich protein 5 Human genes 0.000 description 1
- 101710124303 Proline-rich protein 5 Proteins 0.000 description 1
- 101710132895 Proline-rich protein 5-like Proteins 0.000 description 1
- 102100034734 Proline-rich protein 5-like Human genes 0.000 description 1
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102100032420 Protein S100-A9 Human genes 0.000 description 1
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 1
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 1
- 229940123924 Protein kinase C inhibitor Drugs 0.000 description 1
- 208000003670 Pure Red-Cell Aplasia Diseases 0.000 description 1
- ODHCTXKNWHHXJC-GSVOUGTGSA-N Pyroglutamic acid Natural products OC(=O)[C@H]1CCC(=O)N1 ODHCTXKNWHHXJC-GSVOUGTGSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 108091000106 RNA cap binding Proteins 0.000 description 1
- 102000028391 RNA cap binding Human genes 0.000 description 1
- 108700019586 Rapamycin-Insensitive Companion of mTOR Proteins 0.000 description 1
- 102000046941 Rapamycin-Insensitive Companion of mTOR Human genes 0.000 description 1
- 102000003901 Ras GTPase-activating proteins Human genes 0.000 description 1
- 108090000231 Ras GTPase-activating proteins Proteins 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 101710088998 Response regulator inhibitor for tor operon Proteins 0.000 description 1
- 208000007014 Retinitis pigmentosa Diseases 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 102000003861 Ribosomal protein S6 Human genes 0.000 description 1
- 108090000221 Ribosomal protein S6 Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- ZQUSFAUAYSEREK-WKILWMFISA-N SB-239063 Chemical compound COC1=NC=CC(C=2N(C=NC=2C=2C=CC(F)=CC=2)[C@@H]2CC[C@@H](O)CC2)=N1 ZQUSFAUAYSEREK-WKILWMFISA-N 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 208000008765 Sciatica Diseases 0.000 description 1
- 101710113029 Serine/threonine-protein kinase Proteins 0.000 description 1
- 102100023085 Serine/threonine-protein kinase mTOR Human genes 0.000 description 1
- 208000003252 Signet Ring Cell Carcinoma Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- BCKXLBQYZLBQEK-KVVVOXFISA-M Sodium oleate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC([O-])=O BCKXLBQYZLBQEK-KVVVOXFISA-M 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 235000019486 Sunflower oil Nutrition 0.000 description 1
- 206010042553 Superficial spreading melanoma stage unspecified Diseases 0.000 description 1
- 108010021188 Superoxide Dismutase-1 Proteins 0.000 description 1
- 102100038836 Superoxide dismutase [Cu-Zn] Human genes 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 1
- 102100029253 TELO2-interacting protein 1 homolog Human genes 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 102100035154 Telomere length regulation protein TEL2 homolog Human genes 0.000 description 1
- PDMMFKSKQVNJMI-BLQWBTBKSA-N Testosterone propionate Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](OC(=O)CC)[C@@]1(C)CC2 PDMMFKSKQVNJMI-BLQWBTBKSA-N 0.000 description 1
- 208000033781 Thyroid carcinoma Diseases 0.000 description 1
- 102000011923 Thyrotropin Human genes 0.000 description 1
- 108010061174 Thyrotropin Proteins 0.000 description 1
- 208000026062 Tissue disease Diseases 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- DFBIRQPKNDILPW-CIVMWXNOSA-N Triptolide Chemical compound O=C1OCC([C@@H]2C3)=C1CC[C@]2(C)[C@]12O[C@H]1[C@@H]1O[C@]1(C(C)C)[C@@H](O)[C@]21[C@H]3O1 DFBIRQPKNDILPW-CIVMWXNOSA-N 0.000 description 1
- 108700036309 Type I Plasminogen Deficiency Proteins 0.000 description 1
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 1
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 1
- 208000000208 Wet Macular Degeneration Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 201000008803 Wolff-Parkinson-white syndrome Diseases 0.000 description 1
- 208000012018 Yolk sac tumor Diseases 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical class [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- LUJZZYWHBDHDQX-QFIPXVFZSA-N [(3s)-morpholin-3-yl]methyl n-[4-[[1-[(3-fluorophenyl)methyl]indazol-5-yl]amino]-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yl]carbamate Chemical compound C=1N2N=CN=C(NC=3C=C4C=NN(CC=5C=C(F)C=CC=5)C4=CC=3)C2=C(C)C=1NC(=O)OC[C@@H]1COCCN1 LUJZZYWHBDHDQX-QFIPXVFZSA-N 0.000 description 1
- JOSCNYCOYXTLTN-GFCCVEGCSA-N [3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxyphenyl]-[(3R)-3-(hydroxymethyl)pyrrolidin-1-yl]methanone Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C=CC=1)C(=O)N1C[C@@H](CC1)CO JOSCNYCOYXTLTN-GFCCVEGCSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 208000006336 acinar cell carcinoma Diseases 0.000 description 1
- 206010000583 acral lentiginous melanoma Diseases 0.000 description 1
- 229930183665 actinomycin Natural products 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 208000036676 acute undifferentiated leukemia Diseases 0.000 description 1
- 210000002534 adenoid Anatomy 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 229960000250 adipic acid Drugs 0.000 description 1
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 1
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 1
- 230000001780 adrenocortical effect Effects 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 229940023476 agar Drugs 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 208000006431 amelanotic melanoma Diseases 0.000 description 1
- 230000002707 ameloblastic effect Effects 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229950003153 amsonate Drugs 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- ACPOUJIDANTYHO-UHFFFAOYSA-N anthra[1,9-cd]pyrazol-6(2H)-one Chemical compound C1=CC(C(=O)C=2C3=CC=CC=2)=C2C3=NNC2=C1 ACPOUJIDANTYHO-UHFFFAOYSA-N 0.000 description 1
- RGHILYZRVFRRNK-UHFFFAOYSA-N anthracene-1,2-dione Chemical compound C1=CC=C2C=C(C(C(=O)C=C3)=O)C3=CC2=C1 RGHILYZRVFRRNK-UHFFFAOYSA-N 0.000 description 1
- 239000003817 anthracycline antibiotic agent Substances 0.000 description 1
- 230000001399 anti-metabolic effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 230000002555 anti-neurodegenerative effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000025171 antigen binding proteins Human genes 0.000 description 1
- 108091000831 antigen binding proteins Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045686 antimetabolites antineoplastic purine analogs Drugs 0.000 description 1
- 229940045688 antineoplastic antimetabolites pyrimidine analogues Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 108010044540 auristatin Proteins 0.000 description 1
- 201000009780 autoimmune polyendocrine syndrome type 2 Diseases 0.000 description 1
- 230000003376 axonal effect Effects 0.000 description 1
- OISFUZRUIGGTSD-LJTMIZJLSA-N azane;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound N.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO OISFUZRUIGGTSD-LJTMIZJLSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 208000016894 basaloid carcinoma Diseases 0.000 description 1
- 201000000450 basaloid squamous cell carcinoma Diseases 0.000 description 1
- 208000003373 basosquamous carcinoma Diseases 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- 229940092782 bentonite Drugs 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- PUJDIJCNWFYVJX-UHFFFAOYSA-N benzyl carbamate Chemical compound NC(=O)OCC1=CC=CC=C1 PUJDIJCNWFYVJX-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- ACWZRVQXLIRSDF-UHFFFAOYSA-N binimetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1F ACWZRVQXLIRSDF-UHFFFAOYSA-N 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000008436 biogenesis Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000000319 biphenyl-4-yl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 210000003969 blast cell Anatomy 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 229960001467 bortezomib Drugs 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 201000009480 botryoid rhabdomyosarcoma Diseases 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- KVUAALJSMIVURS-ZEDZUCNESA-L calcium folinate Chemical compound [Ca+2].C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC([O-])=O)C([O-])=O)C=C1 KVUAALJSMIVURS-ZEDZUCNESA-L 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical compound CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 229940127093 camptothecin Drugs 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 229940095731 candida albicans Drugs 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 229940056434 caprelsa Drugs 0.000 description 1
- KHAVLLBUVKBTBG-UHFFFAOYSA-N caproleic acid Natural products OC(=O)CCCCCCCC=C KHAVLLBUVKBTBG-UHFFFAOYSA-N 0.000 description 1
- LDVVMCZRFWMZSG-UHFFFAOYSA-N captan Chemical compound C1C=CCC2C(=O)N(SC(Cl)(Cl)Cl)C(=O)C21 LDVVMCZRFWMZSG-UHFFFAOYSA-N 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 208000002458 carcinoid tumor Diseases 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 229940097647 casodex Drugs 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 1
- 229960005110 cerivastatin Drugs 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 208000021668 chronic eosinophilic leukemia Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 208000029664 classic familial adenomatous polyposis Diseases 0.000 description 1
- 229960004287 clofazimine Drugs 0.000 description 1
- WDQPAMHFFCXSNU-BGABXYSRSA-N clofazimine Chemical compound C12=CC=CC=C2N=C2C=C(NC=3C=CC(Cl)=CC=3)C(=N/C(C)C)/C=C2N1C1=CC=C(Cl)C=C1 WDQPAMHFFCXSNU-BGABXYSRSA-N 0.000 description 1
- GKIRPKYJQBWNGO-OCEACIFDSA-N clomifene Chemical class C1=CC(OCCN(CC)CC)=CC=C1C(\C=1C=CC=CC=1)=C(\Cl)C1=CC=CC=C1 GKIRPKYJQBWNGO-OCEACIFDSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 201000011050 comedo carcinoma Diseases 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 210000000795 conjunctiva Anatomy 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000013267 controlled drug release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 201000011063 cribriform carcinoma Diseases 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- 238000006352 cycloaddition reaction Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 210000004292 cytoskeleton Anatomy 0.000 description 1
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 1
- 229960002465 dabrafenib Drugs 0.000 description 1
- 229950002205 dacomitinib Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 125000004431 deuterium atom Chemical group 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- 125000002720 diazolyl group Chemical group 0.000 description 1
- ACYGYJFTZSAZKR-UHFFFAOYSA-J dicalcium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Ca+2].[Ca+2].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O ACYGYJFTZSAZKR-UHFFFAOYSA-J 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229930188854 dolastatin Natural products 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical class COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 201000002491 encephalomyelitis Diseases 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 208000001991 endodermal sinus tumor Diseases 0.000 description 1
- 238000004146 energy storage Methods 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 229960004671 enzalutamide Drugs 0.000 description 1
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 201000000708 eosinophilic esophagitis Diseases 0.000 description 1
- YJGVMLPVUAXIQN-UHFFFAOYSA-N epipodophyllotoxin Natural products COC1=C(OC)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YJGVMLPVUAXIQN-UHFFFAOYSA-N 0.000 description 1
- 208000037828 epithelial carcinoma Diseases 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 1
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 1
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 208000006881 esophagitis Diseases 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229950000206 estolate Drugs 0.000 description 1
- WCDWBPCFGJXFJZ-UHFFFAOYSA-N etanidazole Chemical compound OCCNC(=O)CN1C=CN=C1[N+]([O-])=O WCDWBPCFGJXFJZ-UHFFFAOYSA-N 0.000 description 1
- 229950006566 etanidazole Drugs 0.000 description 1
- 229960002568 ethinylestradiol Drugs 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 239000002095 exotoxin Substances 0.000 description 1
- 231100000776 exotoxin Toxicity 0.000 description 1
- 238000004880 explosion Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 229950011548 fadrozole Drugs 0.000 description 1
- 201000006692 familial hypertrophic cardiomyopathy Diseases 0.000 description 1
- 229950008696 farnesil Drugs 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 230000003328 fibroblastic effect Effects 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 229940013317 fish oils Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- YLRFCQOZQXIBAB-RBZZARIASA-N fluoxymesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1CC[C@](C)(O)[C@@]1(C)C[C@@H]2O YLRFCQOZQXIBAB-RBZZARIASA-N 0.000 description 1
- 229960001751 fluoxymesterone Drugs 0.000 description 1
- 150000002224 folic acids Chemical class 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 1
- 229940044658 gallium nitrate Drugs 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000002406 gelatinase inhibitor Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical compound N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229960005219 gentisic acid Drugs 0.000 description 1
- 230000000762 glandular Effects 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- RZRNAYUHWVFMIP-HXUWFJFHSA-N glycerol monolinoleate Natural products CCCCCCCCC=CCCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-HXUWFJFHSA-N 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 208000017750 granulocytic sarcoma Diseases 0.000 description 1
- 210000002503 granulosa cell Anatomy 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000005802 health problem Effects 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 230000002008 hemorrhagic effect Effects 0.000 description 1
- 125000006588 heterocycloalkylene group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- HYFHYPWGAURHIV-UHFFFAOYSA-N homoharringtonine Natural products C1=C2CCN3CCCC43C=C(OC)C(OC(=O)C(O)(CCCC(C)(C)O)CC(=O)OC)C4C2=CC2=C1OCO2 HYFHYPWGAURHIV-UHFFFAOYSA-N 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 230000004727 humoral immunity Effects 0.000 description 1
- 210000004276 hyalin Anatomy 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical group [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical class OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 229950000801 hydroxyprogesterone caproate Drugs 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000004857 imidazopyridinyl group Chemical group N1C(=NC2=C1C=CC=N2)* 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 229940045207 immuno-oncology agent Drugs 0.000 description 1
- 239000002584 immunological anticancer agent Substances 0.000 description 1
- 230000006054 immunological memory Effects 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 229960001438 immunostimulant agent Drugs 0.000 description 1
- 239000003022 immunostimulating agent Substances 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229960004130 itraconazole Drugs 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 201000008632 juvenile polyposis syndrome Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- 208000003849 large cell carcinoma Diseases 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- YACHGFWEQXFSBS-RJXCBBHPSA-N leptomycin Chemical class OC(=O)/C=C(C)/C[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)/C=C(\C)/C=C/C[C@@H](C)\C=C(/CC)\C=C\[C@@H]1OC(=O)C=C[C@@H]1C YACHGFWEQXFSBS-RJXCBBHPSA-N 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- 230000000610 leukopenic effect Effects 0.000 description 1
- 229960001614 levamisole Drugs 0.000 description 1
- 206010071570 ligneous conjunctivitis Diseases 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000000014 lung giant cell carcinoma Diseases 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 201000010953 lymphoepithelioma-like carcinoma Diseases 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 229940041033 macrolides Drugs 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000014380 magnesium carbonate Nutrition 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940091250 magnesium supplement Drugs 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 206010061526 malignant mesenchymoma Diseases 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical class [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 208000000516 mast-cell leukemia Diseases 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 239000003771 matrix metalloproteinase inhibitor Substances 0.000 description 1
- 229940121386 matrix metalloproteinase inhibitor Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- 229960002985 medroxyprogesterone acetate Drugs 0.000 description 1
- 210000003593 megakaryocyte Anatomy 0.000 description 1
- 229960001786 megestrol Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 210000002752 melanocyte Anatomy 0.000 description 1
- 230000000684 melanotic effect Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000015654 memory Effects 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 229960005558 mertansine Drugs 0.000 description 1
- ANZJBCHSOXCCRQ-FKUXLPTCSA-N mertansine Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)[C@@H](C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCS)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 ANZJBCHSOXCCRQ-FKUXLPTCSA-N 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229940102396 methyl bromide Drugs 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical compound CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- VKHAHZOOUSRJNA-GCNJZUOMSA-N mifepristone Chemical compound C1([C@@H]2C3=C4CCC(=O)C=C4CC[C@H]3[C@@H]3CC[C@@]([C@]3(C2)C)(O)C#CC)=CC=C(N(C)C)C=C1 VKHAHZOOUSRJNA-GCNJZUOMSA-N 0.000 description 1
- 229960003248 mifepristone Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000006682 monohaloalkyl group Chemical group 0.000 description 1
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical class CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 208000001611 myxosarcoma Diseases 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- ZYQXEVJIFYIBHZ-UHFFFAOYSA-N n-[2-[4-[3-chloro-4-[3-(trifluoromethyl)phenoxy]anilino]pyrrolo[3,2-d]pyrimidin-5-yl]ethyl]-3-hydroxy-3-methylbutanamide Chemical compound C=12N(CCNC(=O)CC(C)(O)C)C=CC2=NC=NC=1NC(C=C1Cl)=CC=C1OC1=CC=CC(C(F)(F)F)=C1 ZYQXEVJIFYIBHZ-UHFFFAOYSA-N 0.000 description 1
- RDSACQWTXKSHJT-NSHDSACASA-N n-[3,4-difluoro-2-(2-fluoro-4-iodoanilino)-6-methoxyphenyl]-1-[(2s)-2,3-dihydroxypropyl]cyclopropane-1-sulfonamide Chemical compound C1CC1(C[C@H](O)CO)S(=O)(=O)NC=1C(OC)=CC(F)=C(F)C=1NC1=CC=C(I)C=C1F RDSACQWTXKSHJT-NSHDSACASA-N 0.000 description 1
- 125000004370 n-butenyl group Chemical group [H]\C([H])=C(/[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 208000014761 nasopharyngeal type undifferentiated carcinoma Diseases 0.000 description 1
- 201000011216 nasopharynx carcinoma Diseases 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 201000004931 neurofibromatosis Diseases 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229940125745 nitric oxide modulator Drugs 0.000 description 1
- 229960005419 nitrogen Drugs 0.000 description 1
- 201000000032 nodular malignant melanoma Diseases 0.000 description 1
- 208000029809 non-keratinizing sinonasal squamous cell carcinoma Diseases 0.000 description 1
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 1
- 125000005593 norbornanyl group Chemical group 0.000 description 1
- 125000003518 norbornenyl group Chemical group C12(C=CC(CC1)C2)* 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 235000003715 nutritional status Nutrition 0.000 description 1
- 229960002446 octanoic acid Drugs 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 229960002969 oleic acid Drugs 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- HYFHYPWGAURHIV-JFIAXGOJSA-N omacetaxine mepesuccinate Chemical compound C1=C2CCN3CCC[C@]43C=C(OC)[C@@H](OC(=O)[C@@](O)(CCCC(C)(C)O)CC(=O)OC)[C@H]4C2=CC2=C1OCO2 HYFHYPWGAURHIV-JFIAXGOJSA-N 0.000 description 1
- 229960002230 omacetaxine mepesuccinate Drugs 0.000 description 1
- 235000020660 omega-3 fatty acid Nutrition 0.000 description 1
- 229940012843 omega-3 fatty acid Drugs 0.000 description 1
- 239000006014 omega-3 oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 229960005010 orotic acid Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 201000008968 osteosarcoma Diseases 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- UHHKSVZZTYJVEG-UHFFFAOYSA-N oxepane Chemical compound C1CCCOCC1 UHHKSVZZTYJVEG-UHFFFAOYSA-N 0.000 description 1
- 125000003585 oxepinyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- 125000003544 oxime group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 229940098695 palmitic acid Drugs 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 208000021255 pancreatic insulinoma Diseases 0.000 description 1
- 229950003440 panomifene Drugs 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 201000010198 papillary carcinoma Diseases 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 229950006299 pelitinib Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 210000001539 phagocyte Anatomy 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- 239000002797 plasminogen activator inhibitor Substances 0.000 description 1
- 150000003058 platinum compounds Chemical class 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229960001237 podophyllotoxin Drugs 0.000 description 1
- YJGVMLPVUAXIQN-XVVDYKMHSA-N podophyllotoxin Chemical compound COC1=C(OC)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@H](O)[C@@H]3[C@@H]2C(OC3)=O)=C1 YJGVMLPVUAXIQN-XVVDYKMHSA-N 0.000 description 1
- YVCVYCSAAZQOJI-UHFFFAOYSA-N podophyllotoxin Natural products COC1=C(O)C(OC)=CC(C2C3=CC=4OCOC=4C=C3C(O)C3C2C(OC3)=O)=C1 YVCVYCSAAZQOJI-UHFFFAOYSA-N 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920001610 polycaprolactone Polymers 0.000 description 1
- 229920002721 polycyanoacrylate Polymers 0.000 description 1
- 125000006684 polyhaloalkyl group Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 239000000583 progesterone congener Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 239000003881 protein kinase C inhibitor Substances 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 239000003806 protein tyrosine phosphatase inhibitor Substances 0.000 description 1
- 208000002815 pulmonary hypertension Diseases 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 239000000784 purine nucleoside phosphorylase inhibitor Substances 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- YUOCYTRGANSSRY-UHFFFAOYSA-N pyrrolo[2,3-i][1,2]benzodiazepine Chemical class C1=CN=NC2=C3C=CN=C3C=CC2=C1 YUOCYTRGANSSRY-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 238000011363 radioimmunotherapy Methods 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- MOODSJOROWROTO-UHFFFAOYSA-N salicylsulfuric acid Chemical compound OC(=O)C1=CC=CC=C1OS(O)(=O)=O MOODSJOROWROTO-UHFFFAOYSA-N 0.000 description 1
- 201000007416 salivary gland adenoid cystic carcinoma Diseases 0.000 description 1
- DFJSJLGUIXFDJP-UHFFFAOYSA-N sapitinib Chemical compound C1CN(CC(=O)NC)CCC1OC(C(=CC1=NC=N2)OC)=CC1=C2NC1=CC=CC(Cl)=C1F DFJSJLGUIXFDJP-UHFFFAOYSA-N 0.000 description 1
- 208000014212 sarcomatoid carcinoma Diseases 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 208000004259 scirrhous adenocarcinoma Diseases 0.000 description 1
- 201000008157 scrotal carcinoma Diseases 0.000 description 1
- 229940116353 sebacic acid Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229950010746 selumetinib Drugs 0.000 description 1
- 239000008299 semisolid dosage form Substances 0.000 description 1
- 229960003440 semustine Drugs 0.000 description 1
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 description 1
- 229960002073 sertraline Drugs 0.000 description 1
- 201000008123 signet ring cell adenocarcinoma Diseases 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 206010040882 skin lesion Diseases 0.000 description 1
- 231100000444 skin lesion Toxicity 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 208000011584 spitz nevus Diseases 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229960004274 stearic acid Drugs 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000024642 stem cell division Effects 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 208000028210 stromal sarcoma Diseases 0.000 description 1
- 108091007196 stromelysin Proteins 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229940071103 sulfosalicylate Drugs 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000002600 sunflower oil Substances 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 208000030457 superficial spreading melanoma Diseases 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 206010042863 synovial sarcoma Diseases 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960001367 tartaric acid Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 239000003277 telomerase inhibitor Substances 0.000 description 1
- 102000055501 telomere Human genes 0.000 description 1
- 108091035539 telomere Proteins 0.000 description 1
- 210000003411 telomere Anatomy 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- UYGBIHFTZHBDEQ-UHFFFAOYSA-N tert-butyl n-[[4-(bromomethyl)phenyl]methyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC1=CC=C(CBr)C=C1 UYGBIHFTZHBDEQ-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 230000002381 testicular Effects 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229960001712 testosterone propionate Drugs 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000002769 thiazolinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 208000013077 thyroid gland carcinoma Diseases 0.000 description 1
- 208000013818 thyroid gland medullary carcinoma Diseases 0.000 description 1
- 208000030045 thyroid gland papillary carcinoma Diseases 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 239000003558 transferase inhibitor Substances 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 102000028894 translation initiation factor binding proteins Human genes 0.000 description 1
- 108091009374 translation initiation factor binding proteins Proteins 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 150000004654 triazenes Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- YKUJZZHGTWVWHA-UHFFFAOYSA-N triptolide Natural products COC12CC3OC3(C(C)C)C(O)C14OC4CC5C6=C(CCC25C)C(=O)OC6 YKUJZZHGTWVWHA-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 208000009999 tuberous sclerosis Diseases 0.000 description 1
- 229940094060 tykerb Drugs 0.000 description 1
- 208000035408 type 1 diabetes mellitus 1 Diseases 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- GFNNBHLJANVSQV-UHFFFAOYSA-N tyrphostin AG 1478 Chemical compound C=12C=C(OC)C(OC)=CC2=NC=NC=1NC1=CC=CC(Cl)=C1 GFNNBHLJANVSQV-UHFFFAOYSA-N 0.000 description 1
- 229960002703 undecylenic acid Drugs 0.000 description 1
- 208000022810 undifferentiated (embryonal) sarcoma Diseases 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 229960005356 urokinase Drugs 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- 229960003862 vemurafenib Drugs 0.000 description 1
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 1
- 208000008662 verrucous carcinoma Diseases 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 208000006542 von Hippel-Lindau disease Diseases 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- QDLHCMPXEPAAMD-QAIWCSMKSA-N wortmannin Chemical compound C1([C@]2(C)C3=C(C4=O)OC=C3C(=O)O[C@@H]2COC)=C4[C@@H]2CCC(=O)[C@@]2(C)C[C@H]1OC(C)=O QDLHCMPXEPAAMD-QAIWCSMKSA-N 0.000 description 1
- QDLHCMPXEPAAMD-UHFFFAOYSA-N wortmannin Natural products COCC1OC(=O)C2=COC(C3=O)=C2C1(C)C1=C3C2CCC(=O)C2(C)CC1OC(C)=O QDLHCMPXEPAAMD-UHFFFAOYSA-N 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Chemical class 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
- 150000003952 β-lactams Chemical class 0.000 description 1
- 229930195724 β-lactose Natural products 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/545—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicinal Preparation (AREA)
Abstract
La presente divulgación se refiere a los inhibidores de mTOR. Específicamente, las realizaciones están dirigidas a compuestos y composiciones que inhiben mTOR, métodos para tratar enfermedades mediadas por mTOR y métodos para sintetizar estos compuestos. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Análogos de rapamicina unidos en C40, C28 y C-32 como inhibidores de mTOR
REFERENCIA CRUZADA A SOLICITUDES RELACIONADAS
La presente solicitud reivindica el beneficio de la solicitud provisional de EE. UU. N.º 62/665.435, presentada el 1 de mayo de 2018, y la solicitud provisional de EE. UU. N.º 62/752.874, presentada el 30 de octubre de 2018, y la solicitud provisional de EE. UU. N.º 62/836.036, presentada el 18 de abril de 2019.
REFERENCIA A UN LISTADO DE SECUENCIAS
El Listado de secuencias asociado a la presente solicitud se proporciona en formato de texto en lugar de una copia en papel. El nombre del archivo de texto que contiene el Listado de secuencias es REME_008_01WO_SeqList_ST25.txt. El archivo de texto tiene aproximadamente 40 kilobytes, se creó el 26 de abril de 2019 y se presenta electrónicamente por EFS-Web.
CAMPO DE LA DIVULGACIÓN
La presente divulgación se refiere a inhibidores de mTOR. Específicamente, las realizaciones se refieren a compuestos y composiciones que inhiben mTOR, a métodos de tratamiento de enfermedades mediadas por mTOR y a métodos de síntesis de estos compuestos.
ANTECEDENTES DE LA DIVULGACIÓN
La diana de mamífero de rapamicina (mTOR) es una serina-treonina cinasa relacionada con las cinasas lipídicas de la familia de fosfoinositida 3-cinasa (PI3K). mTOR existe en dos complejos, mTORC1 y mTORC2, que están regulados diferencialmente, tienen distintas especificidades por sustrato y son diferencialmente sensibles a la rapamicina. mTORC1 integra señales de receptores de los factores de crecimiento con estado nutricional celular y controla el nivel de traducción de ARNm dependiente de caperuza, modulando la actividad de componentes traduccionales clave, tales como la proteína de unión a caperuza y el oncogén eIF4E.
La señalización de mTOR se ha descifrado cada vez con más detalle. La farmacología diferente de los inhibidores de mTOR ha sido particularmente informativa. Se entiende ahora que el primer inhibidor de mTOR informado, la rapamicina, es un inhibidor incompleto de mTORC1. La rapamicina es un inhibidor selectivo de mTORC1 mediante la unión al dominio de unión a rapamicina (FRB) FK506 de cinasa de mTOR con la ayuda de la proteína 12 de unión a FK506 (FKBP12). El dominio de mTOR de FRB está accesible en el complejo de mTORC1, pero menos que en el complejo de mTORC2. Es interesante señalar que se sabe que la potencia de las actividades inhibidoras contra mTORC1 aguas abajo por el tratamiento de rapamicina es diversa entre los sustratos de mTORC1. Por ejemplo, la rapamicina inhibe fuertemente la fosforilación del sustrato de mTORC1 S6K e, indirectamente, la fosforilación de la proteína ribosómica aguas abajo S6 que controla la biogénesis ribosómica. Por otra parte, la rapamicina muestra solo actividad inhibidora parcial contra la fosforilación de 4E-BP1, un regulador importante de eIF4E que controla el inicio de la traducción dependiente de CAP. Como resultado, son de interés inhibidores más completos de la señalización de mTORC1.
Se informó de una segunda clase de inhibidores del "sitio de ATP" de la cinasa mTOR. Esta clase de inhibidores de mTOR se denominará TORi (inhibidor de TOR del sitio de ATP). Las moléculas compiten con ATP, el sustrato para la reacción de cinasa, en el sitio activo de la cinasa mTOR (y, por lo tanto, también son inhibidores del sitio activo de mTOR). Como resultado, estas moléculas inhiben la fosforilación aguas abajo de una variedad más amplia de sustancias.
Aunque la inhibición de mTOR puede tener el efecto de bloquear la fosforilación de 4E-BP1, estos agentes también pueden inhibir mTORC2, que conduce a un bloqueo de la activación de Akt debido a la inhibición de la fosforilación de Akt S473.
En el presente documento se desvelan, entre otras cosas, inhibidores de mTOR. En algunas realizaciones, los compuestos desvelados en el presente documento son inhibidores más selectivos de mTORC1 frente a mTORC2. En algunas realizaciones, los compuestos desvelados en el presente documento son inhibidores más selectivos de mTORC2 frente a mTORC1. En algunas realizaciones, los compuestos desvelados en el presente documento no presentan diferencia de selectividad entre mTORC1 y mTORC2.
El documento de patente US 2016/279108 A1 desvela inhibidores de mTOR dirigidos. El documento de patente WO 2009/131631 A1 desvela análogos de rapamicina como agentes antineoplásicos. El documento de patente WO 2007/068462 A2 desvela un agente para el tratamiento de enfermedad inflamatoria del intestino. El documento de patente US 5480989 A desvela carbamatos de rapamicina. Vanessa S. Rodrik-Outmezguine et al. desvelan, en NATURE, (2016), vol. 534, no. 7606, páginas 272-276, "Overcoming mTOR resistance mutations with a newgeneration mTOR inhibitor". El documento de patente WO 94/25072 A1 desvela conjugados y anticuerpos de rapamicina. El documento de patente WO 94/11380 A1 desvela ésteres de carbonato de rapamicina como inmunosupresores. El documento de patente WO 95/14023 A1 desvela análogos semisintéticos de rapamicina (macrólidos) que son inmunomoduladores. El documento de patente US 6 342 507 B1 desvela compuestos de rapamicina deuterados, método y usos de los mismos. El documento de patente WO 2009/122176 A2 desvela ésteres de carbonato de rapamicina.
SUMARIO DE LA DIVULGACIÓN
La presente invención se refiere a compuestos capaces de inhibir la actividad de mTOR, y preparados farmacéuticos que comprenden dichos compuestos. Los compuestos de la presente invención son los de los Ejemplos 1-152 que siguen, y sales, tautómeros o estereoisómeros farmacéuticamente aceptables de los mismos. La presente invención también se refiere a dichos compuestos para su uso en un método de tratamiento, prevención o reducción del riesgo de una enfermedad o trastorno mediado por mTOR. En el presente documento también se desvela un proceso para la preparación de compuestos de la presente invención.
También se desvela en el presente documento (pero no es parte de la invención) un compuesto de la fórmula Ic:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O, -OR3, -N3 o -O-C(=Z1)-R32a;
R28 es -H, alquilo (C1-C6) o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde cuando R28 y R40 son H, entonces R32 no es =O;
cada Z1 es independientemente O o S;
R28a, R32a y R40a son independientemente -A1-L1-A2-B; -A3-A2-B; -L2-A1-L1-A2-L3-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno;
A1 y A2 están independientemente ausentes o se seleccionan de


en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno;
cada L1 se selecciona independientemente de


L2 y L3 están independientemente ausentes o se seleccionan de

cada B se selecciona independientemente de


cada B1 se selecciona independientemente de
NR3-(C(R3)2)n-,
NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,
NR3-(C(R3)2)n-heteroarileno-,
NR3-(C(R3)2)n-heteroarileno-(C(R3)2)n-,
arileno (C6-C10)-,
NR3-(C(R3)2)n-NR3C(O)-,
NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,
heteroarileno-heterociclileno-arileno (C6-C10)-,


NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, y

NR3-(C(R3)2)n-S(O)2-arileno-(C(R3)2)n-, en donde el enlace

en el lado izquierdo de B1, como está dibujado, está unido a A2, L3 o L1; y en donde el heteroarileno, heterociclileno y arileno están cada uno independientemente opcionalmente sustituido con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo están cada uno independientemente opcionalmente sustituido con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada Y es independientemente -C(R3)2 o un enlace;
cada n es independientemente un número entero de uno a 12;
cada o es independientemente un número entero de cero a 30;
cada p es independientemente un número entero de cero a 12;
cada q es independientemente un número entero de cero a 30; y
cada r es independientemente un número entero de uno a 6.
También se desvela en el presente documento (pero no es parte de la invención) un compuesto de la fórmula Ia:

o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O, -OR3, -N3 o -O-C(=Z1)-R32a;
R28 es -H, alquilo (C1-C6) o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde cuando R28 y R40 son H, entonces R32 no es =O;
cada Z1 es independientemente O o S;
R28a, R32a y R40a son independientemente -A1-L1-A2-B; -A3-A2-B; -L2-A1-L1-A2-L3-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno;
A1 y A2 están independientemente ausentes o se seleccionan de

en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno;
cada L1 se selecciona independientemente de

L2 y L3 están independientemente ausentes o se seleccionan de

cada B se selecciona independientemente de


cada B1 se selecciona independientemente de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

arileno (C6-C10)-,

NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,

y

NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, en donde el enlace

en el lado izquierdo de B1, como está dibujado, está unido a A2, L3 o L1; y en donde el heteroarileno, heterociclileno y arileno están cada uno independientemente opcionalmente sustituido con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo están cada uno independientemente opcionalmente sustituido con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) está cada uno independientemente opcionalmente sustituido con -N(R3)2 o -OR3;
cada Y es independientemente -C(R3)2 o un enlace;
cada n es independientemente un número entero de uno a 12;
cada o es independientemente un número entero de cero a 30;
cada p es independientemente un número entero de cero a 12;
cada q es independientemente un número entero de cero a 30; y
cada r es independientemente un número entero de uno a 6.
También se desvela en el presente documento (pero no es parte de la invención) un compuesto de la fórmula I:

o sal o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O o -OR3;
R28 es -H o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde al menos uno de R28 y R40 no es H;
Z1 es independientemente O o S;
R28a y R40a son independientemente -A1-L1-A2-B; -A3-A2-B; ―-L2-A1-L1-A2-L3-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno;
A1 y A2 están independientemente ausentes o se seleccionan de


en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno;
cada L1 se selecciona independientemente de

L2 y L3 están independientemente ausentes o se seleccionan de


cada B se selecciona independientemente de

cada B1 se selecciona independientemente de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

arileno (C6-C10)-,

NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,

y

NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, en donde el enlace

en el lado izquierdo de B1, como está dibujado, está unido a A2, L3 o L1; y en donde el heteroarileno, heterociclileno y arileno están cada uno independientemente opcionalmente sustituido con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo están cada uno independientemente opcionalmente sustituido con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo es de alquilo (C1-C6) opcionalmente sustituido con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada Y es independientemente -C(R3)2 o un enlace;
cada n es independientemente un número entero de uno a 12;
cada o es independientemente un número entero de cero a 30;
cada p es independientemente un número entero de cero a 12;
cada q es independientemente un número entero de cero a 30; y
cada r es independientemente un número entero de uno a 6.
También se desvela en el presente documento (pero no es parte de la invención) un compuesto de la fórmula II:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O o -OR3;
R28 es -H o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde al menos uno de R28 y R40 no es H;
Z1 es independientemente O o S;
R28a y R40a son independientemente -A1-L1-A2-B; -A3-A2-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno; A1 y A2 están independientemente ausentes o se seleccionan de


en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)-; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno;
cada L1 se selecciona independientemente de

cada B se selecciona independientemente de
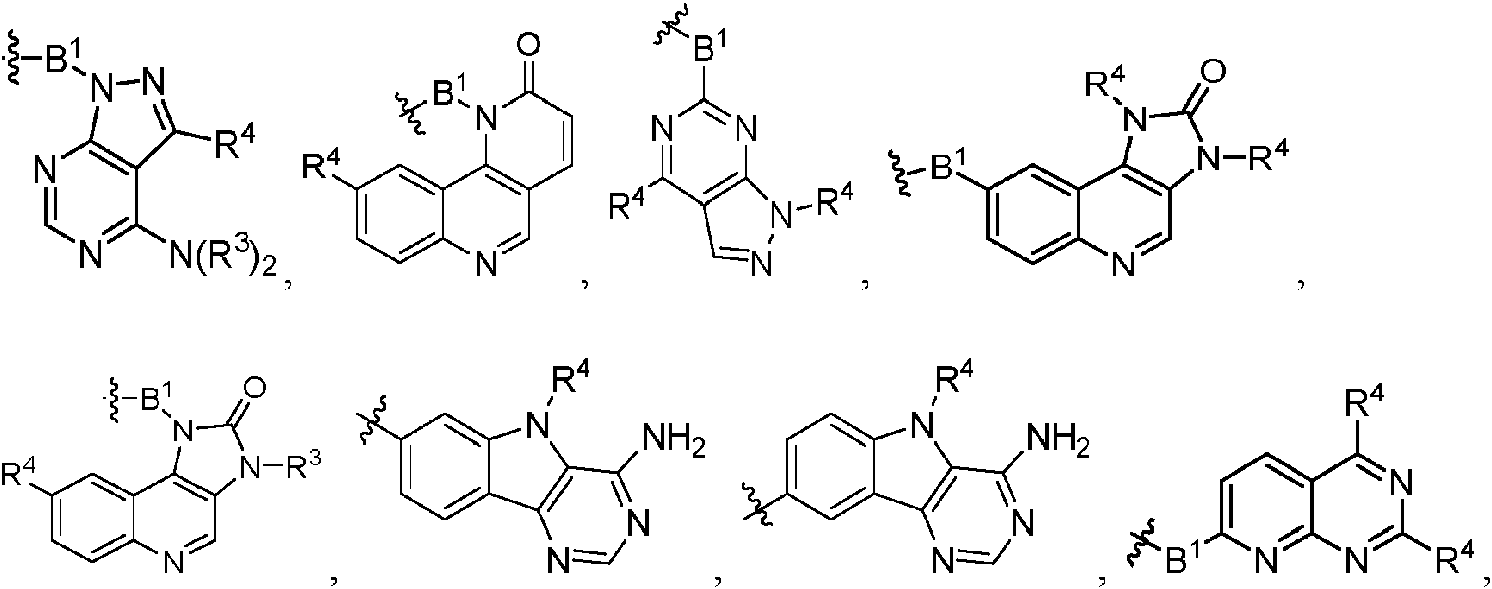

cada B1 se selecciona independientemente de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

arileno (C6-C10)-,

NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,

y

NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, en donde el enlace
en el lado izquierdo de B1, como está dibujado, está unido a A2 o L1; y en donde el heteroarileno, heterociclileno y arileno están cada uno independientemente opcionalmente sustituido con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo están cada uno independientemente opcionalmente sustituido con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada Y es independientemente C(R3)2 o un enlace;
cada n es independientemente un número entero de uno a 12;
cada o es independientemente un número entero de cero a 30;
cada p es independientemente un número entero de cero a 12;
cada q es independientemente un número entero de cero a 30; y
cada r es independientemente un número entero de uno a 6.
Un compuesto de la fórmula I o II se puede representar por la estructura de la fórmula I-28:

o una sal o tautómero farmacéuticamente aceptable del mismo.
Un compuesto de la fórmula Ia, Ic, I o II se puede representar por la estructura de la fórmula I-28b:

o una sal o un tautómero farmacéuticamente aceptable del mismo.
En algunas realizaciones, un compuesto de la fórmula I o II se representa por la estructura de la fórmula I-40:

o una sal o tautómero farmacéuticamente aceptable del mismo.
Un compuesto de la fórmula Ia, Ic, I o II se puede representar por la estructura de la fórmula I-40b:

o una sal o un tautómero farmacéuticamente aceptable del mismo.
Un compuesto de la fórmula Ia, Ic, I o II se puede representar por la estructura de la fórmula I-32b:

o una sal o un tautómero farmacéuticamente aceptable del mismo.
La presente invención también proporciona un compuesto de la presente invención para su uso en un método de tratamiento, prevención o reducción del riesgo de una enfermedad o trastorno mediado por mTOR. El método puede comprender administrar al sujeto que padece o susceptible de desarrollar una enfermedad o trastorno mediado por mTOR una cantidad terapéuticamente eficaz de uno o más de dichos compuestos.
Otro aspecto de la presente invención se refiere a una composición farmacéutica que comprende un compuesto de la presente invención o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo, y al menos uno de un vehículo, excipiente o diluyente farmacéuticamente aceptable. La composición puede comprender un tensioactivo. La composición farmacéutica puede ser eficaz para tratar, prevenir o reducir el riesgo de una enfermedad o trastorno mediado por mTOR en un sujeto en necesidad del mismo.
DESCRIPCIÓN DETALLADA DE LA DIVULGACIÓN
La presente divulgación se refiere a inhibidores de mTOR. Específicamente, las realizaciones se refieren a compuestos y composiciones que inhiben mTOR, a métodos de tratamiento de enfermedades mediadas por mTOR y a métodos de síntesis de estos compuestos.
Los detalles de la divulgación se exponen en la descripción adjunta que sigue. Aunque se pueden usar métodos y materiales similares o equivalentes a los descritos en el presente documento en la práctica o prueba de la presente divulgación, ahora se describen métodos y materiales ilustrativos. Otras características, objetos y ventajas de la divulgación serán evidentes a partir de la descripción y de las reivindicaciones. En la memoria descriptiva y las reivindicaciones adjuntas, las formas en singular también pueden incluir el plural, a menos que el contexto dicte claramente de otro modo. A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que comúnmente es entendido por un experto habitual en la técnica a la que pertenece la presente divulgación.
Términos
Los artículos "un" y "una" se usan en la presente divulgación y se refieren a uno o más de uno (es decir, a al menos uno) del objeto gramatical del artículo, a menos que se indique lo contrario. A modo de ejemplo, "un elemento" puede significar un elemento o más de un elemento, a menos que se indique lo contrario.
El término "o" significa "y/o", a menos que se indique lo contrario. El término "y/o" significa o "y" u "o", o ambos, a menos que se indique lo contrario.
El término "opcionalmente sustituido", a menos que se especifique de otro modo, significa que un grupo puede estar sin sustituir o sustituido con uno o más (por ejemplo, 0, 1, 2, 3, 4 o 5 o más, o cualquier intervalo derivable en su interior) de los sustituyentes listados para ese grupo, en el que dichos sustituyentes pueden ser iguales o diferentes. En una realización, un grupo opcionalmente sustituido tiene 1 sustituyente. En otra realización, un grupo opcionalmente sustituido tiene 2 sustituyentes. En otra realización, un grupo opcionalmente sustituido tiene 3 sustituyentes. En otra realización, un grupo opcionalmente sustituido tiene 4 sustituyentes. En otra realización, un grupo opcionalmente sustituido tiene 5 sustituyentes.
El término "alquilo", por sí mismo o como parte de otro sustituyente, significa, a menos que se establezca de otro modo, una cadena de carbono no cíclica lineal (es decir, no ramificada) o ramificada completamente saturada (o carbono), o combinación de los mismos, que puede incluir radicales di- y multivalentes, que tienen el número de átomos de carbono designado (es decir, C1-C10 significa uno a diez carbonos). Los ejemplos de radicales de hidrocarburo saturado
incluyen, pero no se limitan a, grupos tales como metilo, etilo, n-propilo, isopropilo, n-butilo, t-butilo, isobutilo, secbutilo, (ciclohexil)metilo, homólogos e isómeros de, por ejemplo, n-pentilo, n-hexilo, n-heptilo, n-octilo y similares. El término "alquileno", por sí mismo o como parte de otro sustituyente, significa, a menos que se establezca de otro modo, un radical divalente derivado de un alquilo. Normalmente, un grupo alquilo (o alquileno) tendrá desde 1 hasta 24 átomos de carbono, tal como aquellos grupos que tienen 10 o menos átomos de carbono.
El término "alquenilo" significa un grupo de hidrocarburo alifático que contiene un doble enlace carbono-carbono y que pueden ser lineal o ramificado que tiene aproximadamente 2 a aproximadamente 6 átomos de carbono en la cadena. Ciertos grupos alquenilo tienen 2 a aproximadamente 4 átomos de carbono en la cadena. Ramificado puede significar que uno o más grupos alquilo inferior, tales como metilo, etilo o propilo, están unidos a una cadena de alquenilo lineal. Los grupos alquenilo a modo de ejemplo incluyen etenilo, propenilo, n-butenilo e i-butenilo. Los ejemplos adicionales incluyen vinilo, 2-propenilo, crotilo, 2-isopentenilo, 2-(butadienilo), 2,4-pentadienilo y 3-(1,4-pentadienilo). Un grupo alquenilo C2-C6 es un grupo alquenilo que contiene entre 2 y 6 átomos de carbono.
El término "alquenileno", por sí mismo o como parte de otro sustituyente, significa, a menos que se establezca de otro modo, un radical divalente derivado de un alqueno.
El término "alquinilo" significa un grupo de hidrocarburo alifático que contiene un triple enlace carbono-carbono y que pueden ser lineal o ramificado que tiene aproximadamente 2 a aproximadamente 6 átomos de carbono en la cadena. Ciertos grupos alquinilo tienen 2 a aproximadamente 4 átomos de carbono en la cadena. Ramificado puede significar que uno o más grupos alquilo inferior, tales como metilo, etilo o propilo, están unidos a una cadena de alquinilo lineal. Los grupos alquinilo a modo de ejemplo incluyen etinilo, propinilo, n-butinilo, 2-butinilo, 3-metilbutinilo y n-pentinilo. Los ejemplos adicionales incluyen 1-propinilo, 3-propinilo y 3-butinilo. Un grupo alquinilo C2-C6 es un grupo alquinilo que contiene entre 2 y 6 átomos de carbono.
El término "alquinileno", por sí mismo o como parte de otro sustituyente, significa, a menos que se establezca de otro modo, un radical divalente derivado de un alquino.
El término "cicloalquilo" significa un anillo de carbono monocíclico o policíclico, saturado o parcialmente insaturado, que contiene 3-18 átomos de carbono. Los ejemplos de grupos cicloalquilo incluyen, sin limitación, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptanilo, ciclooctanilo, norbornanilo, norbornenilo, biciclo[2.2.2]octanilo o biciclo[2.2.2]octenilo. Un cicloalquilo C3-C8 es un grupo cicloalquilo que contiene entre 3 y 8 átomos de carbono. Un grupo cicloalquilo puede estar condensado (por ejemplo, decalina) o puenteado (por ejemplo, norbornano).
Un "cicloalquileno", solo o como parte de otro sustituyente, significa un radical divalente derivado de un cicloalquilo. Los términos "heterociclilo" o "heterocicloalquilo" o "heterociclo" se refiere a un anillo monocíclico o policíclico de 3 a 24 miembros que contiene carbono y al menos un heteroátomo seleccionado de oxígeno, fósforo, nitrógeno y azufre y en donde no hay átomos π deslocalizados (aromaticidad) compartidos entre el anillo carbono o heteroátomo(s). Los anillos de heterociclilo incluyen, pero no se limitan a, oxetanilo, azetadinilo, tetrahidrofuranilo, pirrolidinilo, oxazolinilo, oxazolidinilo, tiazolinilo, tiazolidinilo, piranilo, tiopiranilo, tetrahidropiranilo, dioxalinilo, piperidinilo, morfolinilo, tiomorfolinilo, S-óxido de tiomorfolinilo, S-dióxido de tiomorfolinilo, piperazinilo, azepinilo, oxepinilo, diazepinilo, tropanilo y homotropanilo. Un anillo de heterociclilo o heterocicloalquilo también puede estar condensado o puenteado, por ejemplo, puede ser un anillo bicíclico.
Un "heterociclileno" o "heterocicloalquileno", solo o como parte de otro sustituyente, significa un radical divalente derivado de un "heterociclilo" o "heterocicloalquilo" o "heterociclo".
El término "arilo" significa, a menos que se establezca de otro modo, un sustituyente de hidrocarburo aromático poliinsaturado, que puede ser un único anillo o múltiples anillos (preferentemente desde 1 hasta 3 anillos) que están condensados juntos (es decir, un arilo de anillo condensado) o unido covalentemente. Un arilo de anillo condensado se puede referir a múltiples anillos condensados juntos, en donde al menos uno de los anillos condensados es un anillo de arilo.
Un "arileno", solo o como parte de otro sustituyente, significa un radical divalente derivado de un arilo.
El término "heteroarilo" se refiere a un grupo arilo (o anillos) que contiene al menos un heteroátomo, tal como N, O o S, en donde el (los) átomos de nitrógeno y azufre están opcionalmente oxidados, y el (los) átomo(s) de nitrógeno están opcionalmente cuaternizados. Por lo tanto, el término "heteroarilo" incluye grupos heteroarilo de anillo condensados (es decir, múltiples anillos condensados juntos en donde al menos uno de los anillos condensados es un anillo heteroaromático). Un anillo heteroarileno 5,6-condensado se refiere a dos anillos condensados juntos, en donde un anillo tiene 5 miembros y el otro anillo tiene 6 miembros, y en donde al menos un anillo es un anillo de heteroarilo. Asimismo, un anillo heteroarileno 6,6-condensado se refiere a dos anillos condensados juntos, en donde un anillo tiene 6 miembros y el otro anillo tiene 6 miembros, y en donde al menos un anillo es un anillo heteroarilo. Y un anillo heteroarileno 6,5-condensado se refiere a dos anillos condensados juntos, en donde un anillo tiene 6 miembros y el
otro anillo tiene 5 miembros, y en donde al menos un anillo es un anillo heteroarilo. Un grupo heteroarilo se puede unir al resto de la molécula mediante un carbono o heteroátomo. Los ejemplos no limitantes de grupos arilo y heteroarilo incluyen fenilo, 1-naftilo, 2-naftilo, 4-bifenilo, 1-pirrolilo, 2-pirrolilo, 3-pirrolilo, 3-pirazolilo, 2-imidazolilo, 4-imidazolilo, pirazinilo, 2-oxazolilo, 4-oxazolilo, 2-fenil-4-oxazolilo, 5-oxazolilo, 3-isoxazolilo, 4-isoxazolilo, 5-isoxazolilo, 2-tiazolilo, 4-tiazolilo, 5-tiazolilo, 2-furilo, 3-furilo, 2-tienilo, 3-tienilo, 2-piridilo, 3-piridilo, 4-piridilo, 2-pirimidilo, 4-pirimidilo, 5-benzotiazolilo, purinilo, 2-bencimidazolilo, 5-indolilo, 1-isoquinolilo, 5-isoquinolilo, 2-quinoxalinilo, 5-quinoxalinilo, 3-quinolilo y 6-quinolilo. Los sustituyentes para cada uno de los sistemas de anillos de arilo y heteroarilo anteriormente mencionados se seleccionan del grupo de sustituyentes aceptables descritos en el presente documento.
El término "heteroarilo" también puede incluir sistemas de múltiples anillos condensados que tienen al menos dicho anillo aromático, cuyos sistemas de múltiples anillos condensados se describen además más adelante. El término también puede incluir sistemas de múltiples anillos condensados (por ejemplo, sistemas de anillos que comprenden 2, 3 o 4 anillos) en donde un grupo heteroarilo, como se ha definido anteriormente, se puede condensar con uno o más anillos seleccionados de heteroarilos (para formar, por ejemplo, un tal naftiridinilo, como 1,8-naftiridinilo), heterociclos (para formar, por ejemplo, un 1,2,3,4-tetrahidronaftiridinilo, tal como 1,2,3,4-tetrahidro-1,8-naftiridinilo), carbociclos (para formar, por ejemplo, 5,6,7,8-tetrahidroquinolilo) y arilos (para formar, por ejemplo, indazolilo) para formar el sistema de múltiples de anillos condensados. Los anillos del sistema de múltiples de anillos condensados se pueden conectar entre sí por enlaces condensados, espiro y puenteados cuando lo permitan los requisitos de valencia. Se debe entender que los anillos individuales de los sistemas de múltiples anillos condensados se pueden conectar en cualquier orden los unos con respecto a los otros. También se debe entender que el punto de unión de un sistemas de múltiples anillos condensados (como se ha definido anteriormente para un heteroarilo) puede estar en cualquier posición de los sistemas de múltiples anillos condensados que incluye una porción de heteroarilo, heterociclo, arilo o carbociclo del sistema de múltiples anillos condensados y en cualquier átomo adecuado del sistema de múltiples anillos condensados que incluye un átomo de carbono y heteroátomo (por ejemplo, un nitrógeno).
Un "heteroarileno", solo o como parte de otro sustituyente, significa un radical divalente derivado de un heteroarilo. Ejemplos no limitantes de grupos arilo y heteroarilo incluyen piridinilo, pirimidinilo, tiofenilo, tienilo, furanilo, indolilo, benzoxadiazolilo, benzodioxolilo, benzodioxanilo, tianaftanilo, pirrolopiridinilo, indazolilo, quinolinilo, quinoxalinilo, piridopirazinilo, quinazolinonilo, benzoisoxazolilo, imidazopiridinilo, benzofuranilo, benzotienilo, benzotiofenilo, fenilo, naftilo, bifenilo, pirrolilo, pirazolilo, imidazolilo, pirazinilo, oxazolilo, isoxazolilo, tiazolilo, furiltienilo, piridilo, pirimidilo, benzotiazolilo, purinilo, bencimidazolilo, isoquinolilo, tiadiazolilo, oxadiazolilo, pirrolilo, diazolilo, triazolilo, tetrazolilo, benzotiadiazolilo, isotiazolilo, pirazolopirimidinilo, pirrolopirimidinilo, benzotriazolilo, benzoxazolilo o quinolilo. Los ejemplos anteriores pueden estar sustituidos o sin sustituir y radicales divalentes de cada ejemplo de heteroarilo anterior son ejemplos no limitantes de heteroarileno. Un resto heteroarilo puede incluir un heteroátomo de anillo (por ejemplo, O, N o S). Un resto heteroarilo puede incluir dos heteroátomos de anillo opcionalmente diferentes (por ejemplo, O, N o S). Un resto heteroarilo puede incluir tres heteroátomos de anillo opcionalmente diferentes (por ejemplo, O, N o S). Un resto heteroarilo puede incluir cuatro heteroátomos de anillo opcionalmente diferentes (por ejemplo, O, N o S). Un resto heteroarilo puede incluir cinco heteroátomos de anillo opcionalmente diferentes (por ejemplo, O, N o S). Un resto arilo puede tener un único anillo. Un resto arilo puede tener dos anillos opcionalmente diferentes. Un resto arilo puede tener tres anillos opcionalmente diferentes. Un resto arilo puede tener cuatro anillos opcionalmente diferentes. Un resto heteroarilo puede tener un anillo. Un resto heteroarilo puede tener dos anillos opcionalmente diferentes. Un resto heteroarilo puede tener tres anillos opcionalmente diferentes. Un resto heteroarilo puede tener cuatro anillos opcionalmente diferentes. Un resto heteroarilo puede tener cinco anillos opcionalmente diferentes. Los términos "halo" o "halógeno," por sí mismos o como parte de otro sustituyente, significan, a menos que se establezca de otro modo, un átomo de flúor, cloro, bromo o yodo. Además, términos tales como "haloalquilo" pueden incluir monohaloalquilo y polihaloalquilo. Por ejemplo, el término "haloalquilo (C1-C4)" puede incluir, pero no se limita a, fluorometilo, difluorometilo, trifluorometilo, 2,2,2-trifluoroetilo, 4-clorobutilo, 3-bromopropilo, 1-fluoro-2-bromoetilo y similares.
El término "hidroxilo", como se usa en el presente documento, significa -OH.
El término "hidroxialquilo", como se usa en el presente documento, significa un resto alquilo como se define en el presente documento, sustituido con uno o más, tal como uno, dos o tres, grupos hidroxi. En ciertos casos, el mismo átomo de carbono no lleva más de un grupo hidroxi. Los ejemplos representativos incluyen, pero no se limitan a, hidroximetilo, 2-hidroxietilo, 2-hidroxipropilo, 3-hidroxipropilo, 1-(hidroximetil)-2-metilpropilo, 2-hidroxibutilo, 3-hidroxibutilo, 4-hidroxibutilo, 2,3-dihidroxipropilo, 2-hidroxi-1-hidroximetiletilo, 2,3-dihidroxibutilo, 3,4-dihidroxibutilo y 2-(hidroximetil)-3-hidroxipropilo.
El término "oxo", como se usa en el presente documento, significa un oxígeno que está doblemente unido a un átomo de carbono.
Un grupo sustituyente, como se usa en el presente documento, puede ser un grupo seleccionado de los siguientes restos:
1. (A) oxo, halógeno, -CF3, -CN, -OH, -OCH3, -NH2, -COOH, -CONH2, -NO2, -SH, -SO3H, -SO4H, -SO2NH2, -NHNH2, -ONH2, -NHC=(O)NHNH2, -NHC=(O)NH2, -NHSO2H, -NHC=(O)H, -NHC(O)-OH, -NHOH, -OCF3, -OCHF2, -OCH2F, alquilo sin sustituir, cicloalquilo sin sustituir, heterocicloalquilo sin sustituir, arilo sin sustituir, heteroarilo sin sustituir, y
2. (B) alquilo, cicloalquilo, heterociclilo, arilo, heteroarilo, sustituido con al menos un sustituyente seleccionado de:
(i) oxo, halógeno, -CF3, -CN, -OH, -OCH3, -NH2, -COOH, -CONH2, -NO2, -SH, -SO3H, -SO4H, -SO2NH2, -NHNH2, -ONH2, -NHC=(O)NHNH2, -NHC=(O)NH2, -NHSO2H, -NHC=(O)H, -NHC(O)-OH, -NHOH, -OCF3, -OCHF2, -OCH2F, alquilo sin sustituir, sin sustituir heteroalquilo, cicloalquilo sin sustituir, heterocicloalquilo sin sustituir, arilo sin sustituir, heteroarilo sin sustituir, y
(ii) alquilo, heteroalquilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, sustituido con al menos un sustituyente seleccionado de:
(a) oxo, halógeno, -CF3, -CN, -OH, -OCH3, -NH2, -COOH, -CONH2, -NO2, -SH, -SO3H, -SO4H, -SO2NH2, -NHNH2, -ONH2, -NHC=(O)NHNH2, -NHC=(O)NH2, -NHSO2H, -NHC=(O)H, -NHC(O)-OH, -NHOH, -OCF3, -OCHF2, -OCH2F, alquilo sin sustituir, sin sustituir heteroalquilo, cicloalquilo sin sustituir, heterocicloalquilo sin sustituir, arilo sin sustituir, heteroarilo sin sustituir, y
(b) alquilo, heteroalquilo, cicloalquilo, heterocicloalquilo, arilo, heteroarilo, sustituido con al menos un sustituyente seleccionado de: oxo, halógeno, -CF3, -CN, -OH, -OCH3, -NH2, -COOH, -CONH2, -NO2, -SH, -SO3H, -SO4H, -SO2NH2, -NHNH2, -ONH2, -NHC=(O)NHNH2, -NHC=(O)NH2, -NHSO2H, -NHC=(O)H, -NHC(O)-OH, -NHOH, -OCF3, -OCHF2, -OCH2F, alquilo sin sustituir, sin sustituir heteroalquilo, cicloalquilo sin sustituir, heterocicloalquilo sin sustituir, arilo sin sustituir, heteroarilo sin sustituir.
Una "cantidad eficaz", cuando se usa a propósito de un compuesto, es una cantidad eficaz para tratar o prevenir una enfermedad en un sujeto como se describe en el presente documento.
El término "vehículo", como se usa en la presente divulgación, engloba vehículos, excipientes y diluyentes y puede significar un material, composición o vehículo, tal como una carga, diluyente, excipiente, disolvente o material de encapsulación líquido o sólido, implicado en llevar o transportar un agente farmacéutico de un órgano, o porción del cuerpo, a otro órgano, o porción del cuerpo de un sujeto.
El término "tratar", con respecto a un sujeto, se refiere a mejorar al menos un síntoma del trastorno del sujeto. Tratar puede incluir curar, mejorar o al menos aliviar parcialmente el trastorno.
El término "previenen" o "prevenir", con respecto a un sujeto, se refiere a evitar que una enfermedad o trastorno aqueje a un sujeto. Prevenir puede incluir tratamiento profiláctico. Por ejemplo, prevenir puede incluir administrar al sujeto un compuesto desvelado en el presente documento antes de que un sujeto esté aquejado de una enfermedad y la administración evitará que el sujeto esté aquejado de la enfermedad.
El término "trastorno" se usa en la presente divulgación y significa, y se usa indistintamente con, los términos enfermedad, afección o padecimiento, a menos que se indique lo contrario.
El término "administrar", "que administra" o "administración", como se usa en la presente divulgación, se refiere a o administrar directamente un compuesto desvelado o sal o tautómero farmacéuticamente aceptable del compuesto desvelado o una composición a un sujeto, o administrar un derivado o análogo de profármaco del compuesto o sal o tautómero farmacéuticamente aceptable del compuesto o composición al sujeto, que puede formar una cantidad equivalente de compuesto activo dentro del cuerpo del sujeto.
Un "paciente" o "sujeto" es un mamífero, por ejemplo, un humano, ratón, rata, cobaya, perro, gato, caballo, vaca, cerdo o primate no humano, tal como un mono, chimpancé, babuino o macaco de la India.
Compuestos
La presente invención proporciona un compuesto seleccionado del grupo que consiste en:


























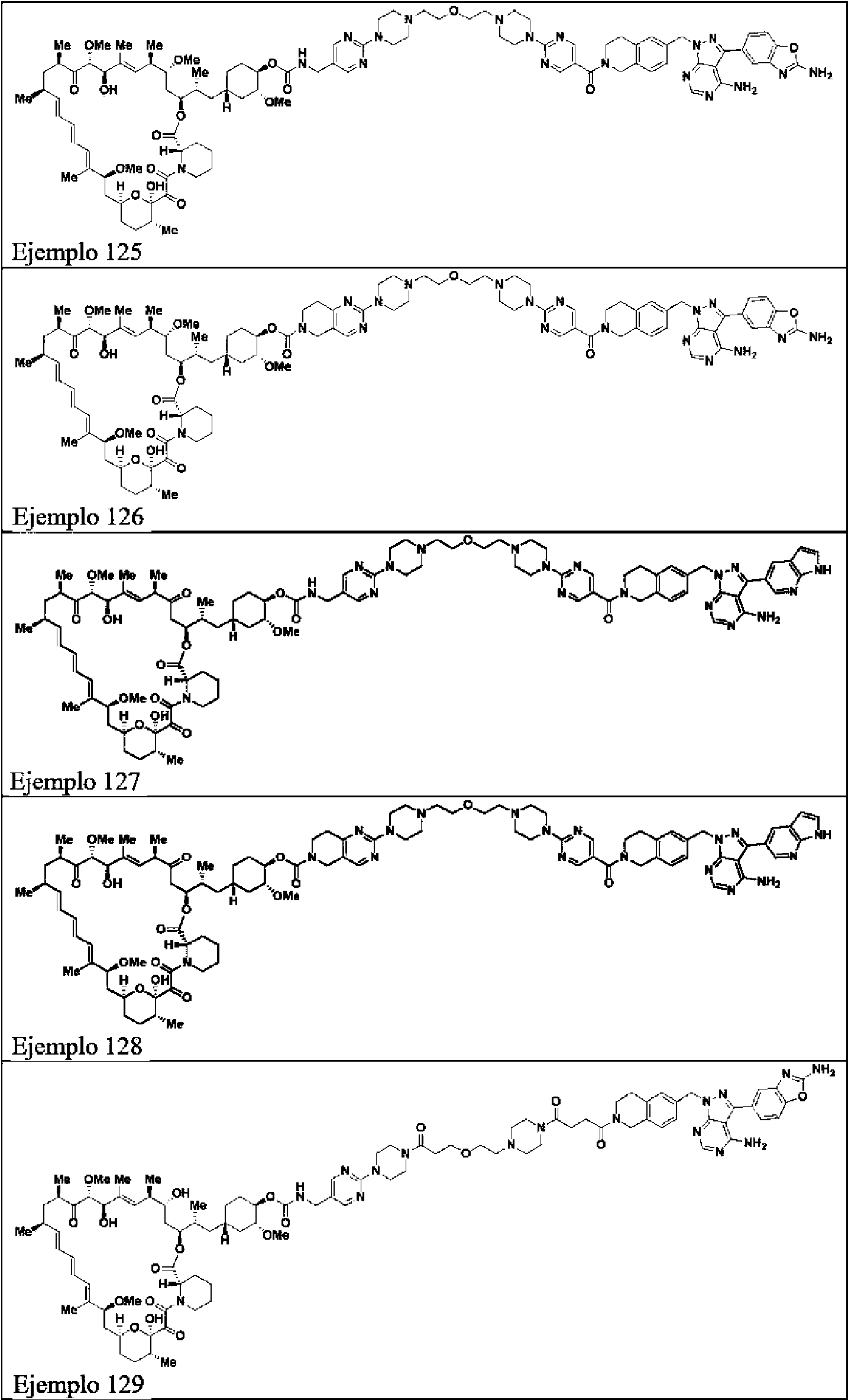





o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto tiene la fórmula

o un estereoisómero del mismo.
En algunas realizaciones, el compuesto tiene la fórmula

o un tautómero del mismo.
En algunas realizaciones, el compuesto tiene la fórmula

o un isómero de oxepano del mismo.
En algunas realizaciones, el compuesto tiene la fórmula

o un estereoisómero del mismo.
En algunas realizaciones, el compuesto tiene la fórmula

o un tautómero del mismo
En algunas realizaciones, el compuesto tiene la fórmula

En algunas realizaciones, el compuesto tiene la fórmula
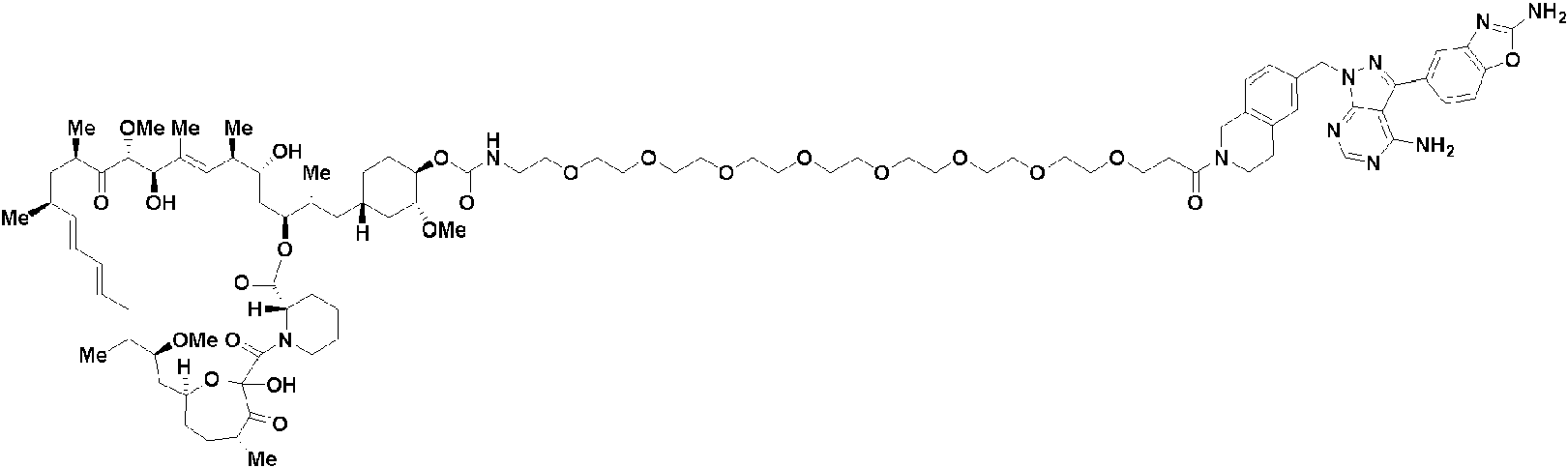
También se describe un compuesto que tiene la estructura de la fórmula Ic,

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32, R28 y R40 se describen como antes. También se describe un compuesto que tiene la estructura de la fórmula Ia,

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32, R28 y R40 se describen como antes. También se describe un compuesto que tiene la estructura de la fórmula I,

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32, R28 y R40 se describen como antes. También se describe un compuesto que tiene la estructura de la fórmula Ib:
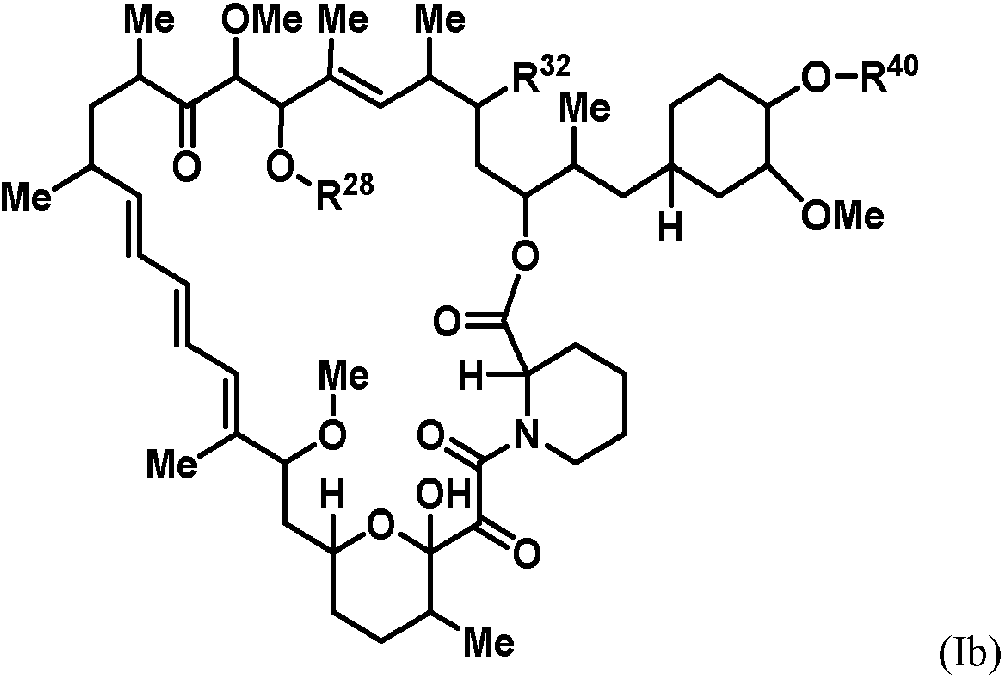
o una sal y o tautómero farmacéuticamente aceptable del mismo, en donde R32, R28 y R40 se describen como antes para la fórmula I.
También se describe un compuesto que tiene la estructura de la fórmula II,

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32, R28 y R40 se describen como antes. También se describe un compuesto que tiene la estructura de la fórmula IIb:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32, R28 y R40 se describen como antes para la fórmula II.
También se describe un compuesto que tiene la siguiente fórmula:

o una sal o tautómero farmacéuticamente aceptable del mismo.
R32 puede ser =O. R32 puede ser -OR3. R32 puede ser H. R32 puede ser -N3.
Como se ha descrito anteriormente, cada R3 es independientemente H o alquilo (C1-C6). R3 puede ser H. R3 puede ser alquilo (C1-C6). R3 puede ser metilo.
R28 puede ser H. R28 puede ser alquilo (C1-C6). R40 puede ser H.
También se describe un compuesto representado por la estructura de la fórmula I-40b:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32 y R40 se describen como antes para la fórmula Ia, Ic, I, o II.
También se describe un compuesto representado por la estructura de la fórmula I-40:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32 y R40 se describen como antes.
R40 puede ser -C(=Z1)-R40a. Z1 puede ser O. Z1 puede ser S.
R40a puede ser -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de NO2 y halógeno.
R40a puede ser -A1-L1-A2-B. R40a puede ser -A1-A2-B. R40a puede ser -L2-A1-L1-A2-L3-B.
R40a puede ser -A1-L1-A2-B, en donde A1 y A2 están ausentes. R40a puede ser -A1-L1-A2-B, en donde A2 está ausente. R40a puede ser -A1-L1-A2-B, en donde A1 está ausente. R40a puede ser - A1-L1-A2-B. R40a puede ser -A1A2-B. R40a puede ser -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes. R40a puede ser -L2-A1-L1-A2-L3-B, en donde L2 está ausente. R40a puede ser -L2-A1-L1-A2-L3-B, en donde L3 está ausente.
También se describe un compuesto representado por la estructura de la fórmula I-28b:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32 y R28 se describen como antes para la fórmula Ia, Ic, I, o II.
También se describe un compuesto representado por la estructura de la fórmula I-28:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32 y R28 se describen como antes.
R28 puede ser -C(=Z1)-R28a. Z1 puede ser O. Z1 puede ser S.
R28a puede ser -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de NO2 y halógeno.
R28a puede ser -A1-L1-A2-B. R28a puede ser -A1A2-B. R28a puede ser -L2-A1-L1-A2-L3-B.
R28a puede ser -A1-L1-A2-B, en donde A1 y A2 están ausentes. R28a puede ser -A1-L1-A2-B, en donde A2 está ausente. R28a puede ser -A1-L1-A2-B, en donde A1 está ausente. R28a puede ser - A1-L1-A2-B. R28a es -A1-A2-B. R28a puede ser -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes. R28a puede ser -L2-A1-L1-A2-L3-B, en donde L2 está ausente. R28a puede ser -L2-A1-L1-A2-L3-B, en donde L3 está ausente.
También se describe un compuesto representado por la estructura de la fórmula I-32b:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde R32 se describe como antes para la fórmula Ia, Ic, I o II.
R32 puede ser -O-C(=Z1)-R32a. Z1 es O. Z1 puede ser S.
R32a puede ser -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de NO2 y halógeno.
R32a puede ser -A1-L1-A2-B. R32a puede ser -A1-A2-B. R32a puede ser -L2-A1-L1-A2-L3-B.
R32a puede ser -A1-L1-A2-B, en donde A1 y A2 están ausentes. R32a puede ser -A1-L1-A2-B, en donde A2 está ausente. R32a puede ser -A1-L1-A2-B, en donde A1 está ausente. R32a puede ser - A1-L1-A2-B. R32a puede ser -A1-A2-B. R32a es -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes. R32a puede ser -L2-A1-L1-A2-L3-B, en donde L2 está ausente. R32a puede ser -L2-A1-L1-A2-L3-B, en donde L3 está ausente.
Como se ha descrito anteriormente, cada L1 se selecciona independientemente de

Como se ha descrito anteriormente para la fórmula Ia, cada L1 se selecciona independientemente de

Como se ha descrito anteriormente para la fórmula Ic, cada L1 se selecciona independientemente de

y

L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

. L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

L1 puede ser

Como se ha descrito anteriormente, L2 y L3 están independientemente ausentes o se seleccionan de

Como se ha descrito anteriormente para la fórmula Ia y Ic, L2 y L3 están independientemente ausentes o se seleccionan de
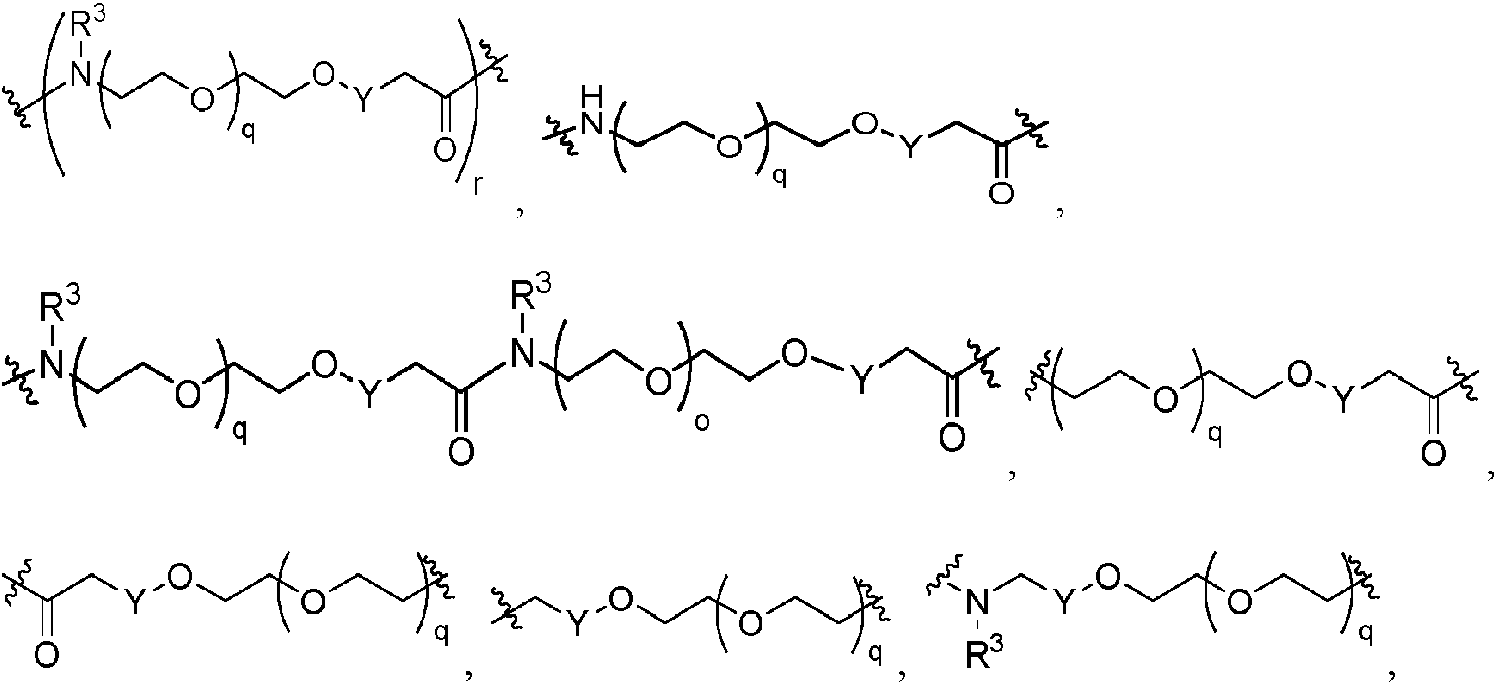
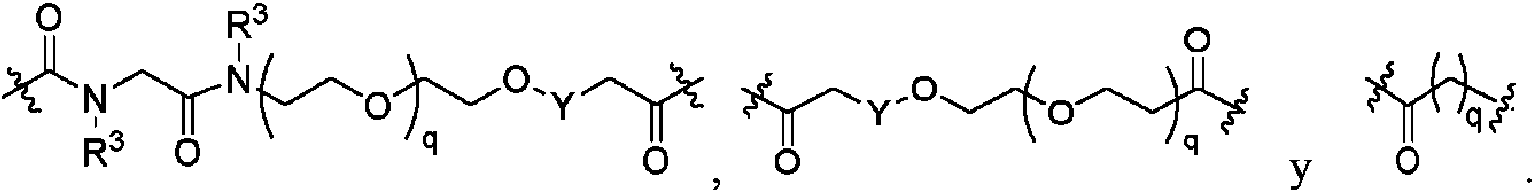
L2 puede estar ausente. L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L3 puede estar ausente. L3 puede ser

L3 puede ser

L3 puede ser

L3 puede ser

L3 puede ser

L3 puede ser

L3 puede ser

L3 puede ser

L2 puede ser

L2 puede ser

L2 puede ser

L3 puede ser

L3 puede ser

L3 puede ser

Como se ha descrito anteriormente, A1 y A2 están independientemente ausentes o se seleccionan de

cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno.
Como se ha descrito anteriormente para la fórmula Ia, A1 y A2 están independientemente ausentes o se seleccionan de


cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno.
Como se ha descrito anteriormente para la fórmula Ic, A1 y A2 están independientemente ausentes o se seleccionan de


cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno;
Para la fórmula I, el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3. Para la fórmula II, el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)-; y el enlace en el lado derecho del resto A2, como está dibujado, está unido a B. Para la fórmula Ia y Ic, el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3.
A1 puede estar ausente. A1 puede ser

A1 puede ser

A1 puede ser

A1 puede ser

en donde cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno.
A1 puede ser

en donde cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno.
A1 puede ser
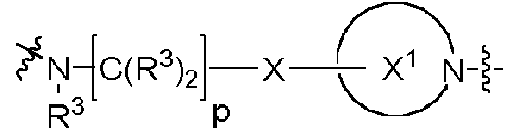
en donde cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada X1 es un anillo de heteroarileno o heterociclileno.
A1 puede ser

en donde cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada W1 es un anillo de heteroarileno o heterociclileno.
A1 puede ser

en donde cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada W1 es un anillo de heteroarileno o heterociclileno.
A1 puede ser

en donde cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno.
A1 puede ser

en donde cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno.
A1 puede ser


A1 puede ser

A1 puede ser

A1 puede ser

A1 puede ser

A2 puede estar ausente. A2 puede ser

. A2 puede ser

A2 puede ser

A2 puede ser

A2 puede ser

en donde cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno.
A2 puede ser

en donde cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno.
A2 puede ser

en donde cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada X1 es independientemente un anillo de heteroarileno o heterociclileno.
A2 puede ser

en donde cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada W1 es independientemente un anillo de heteroarileno o heterociclileno.
A2 puede ser

en donde cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada W1 es independientemente un anillo de heteroarileno o heterociclileno.
A2 puede ser

en donde cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno.
A2 puede ser

en donde cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno; y cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno.
A2 puede ser

A2 puede ser

A2 puede ser
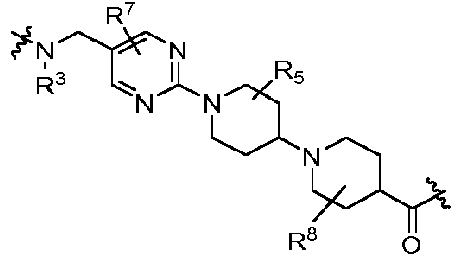
A2 puede ser

A2 puede ser

Como se ha descrito anteriormente, cada B se selecciona independientemente de

B puede ser

B puede ser

B puede ser


B puede ser

Como se ha descrito anteriormente, cada B1 se selecciona independientemente de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

arileno (C6-C10)-,

NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,


y

NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, en donde el enlace
en el lado izquierdo de B1, como está dibujado, está unido a A2 o L1; y en donde el heteroarileno, heterociclileno y arileno están cada uno independientemente opcionalmente sustituido con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo.
Como se ha descrito anteriormente para la fórmula Ic, cada B1 se selecciona independientemente de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,
N R (C(R3)2)n-heteroarileno-(C(R3)2)n-,
R (C(R3)2)n-heteroarileno-(C(R3)2)n-,

arileno (C6-C10)-,
N R3-(C(R3)2)n-NR3C(O)-,
R3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,


NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, y

NR3-(C(R3)2)n-S(O)2-arileno-(C(R3)2)n -; en donde el enlace

en el lado izquierdo de B1, como está dibujado, está unido a A2, L3 o L1; y en donde el heteroarileno, heterociclileno y arileno están cada uno independientemente opcionalmente sustituido con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo.
B1 puede ser

NR3-(C(R3)2)n-.
B1 puede ser

en donde arileno se sustituye opcionalmente con haloalquilo.
B1 puede ser

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

arileno (C6-C10)-,

NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2).-heteroarileno-heterociclileno-arileno (C6-C10)-, o

heteroarileno-heterociclileno-arileno (C6-C10)-. En ciertas realizaciones, B1 es

B1 puede ser

B1 puede ser
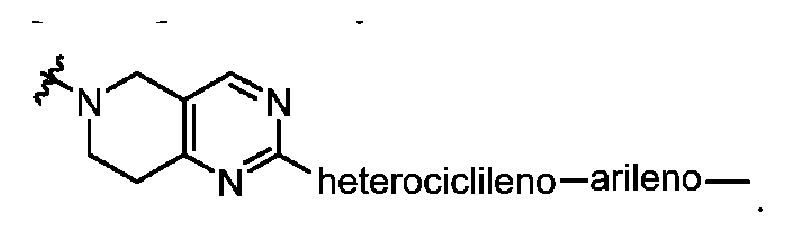
B1 puede ser

NR3-(C(R3)2)n-S(O)2-arileno-C(O)-.
B1 puede ser
NR3-(C(R3)2)n-heteroarileno-(C(R3)2)n-. B1 puede ser

B1 puede ser

B1 puede ser

NR3-(C(R3)2)n-S(O)2-arileno-(C(R3)2)n -.
En B1, el heteroarilo, heterociclilo y arileno se pueden sustituir opcionalmente con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo.
R3 puede ser H. R3 puede ser alquilo (C1-C6).
R4 puede ser H. R4 puede ser alquilo (C1-C6). R4 puede ser halógeno. R4 puede ser heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, o arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo se sustituyen opcionalmente con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN o -C(O)NR3-heteroarilo. R4 puede ser -C(O)NR3-heterociclilo. R4 puede ser heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2 o -OR3.
Como se ha descrito anteriormente, cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R5 es H. En ciertas realizaciones, R5 es alquilo (C1-C6), en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R5 es -C(O)OR3. En ciertas realizaciones, R5 es -N(R3)2.
Como se ha descrito anteriormente, cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R6 es H. En ciertas realizaciones, R6 es alquilo (C1-C6), en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R6 es -C(O)OR3. En ciertas realizaciones, R6 es -N(R3)2.
Como se ha descrito anteriormente, cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R7 es H. En ciertas realizaciones, R7 es alquilo (C1-C6), en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R7 es -C(O)OR3. En ciertas realizaciones, R7 es -N(R3)2.
Como se ha descrito anteriormente, cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R8 es H. En ciertas realizaciones, R8 es alquilo (C1-C6), en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3. En ciertas realizaciones, R8 es -C(O)OR3. En ciertas realizaciones, R8 es -N(R3)2.
Como se ha descrito anteriormente, cada Y es independientemente C(R3)2 o un enlace. En ciertas realizaciones, Y es C(R3)2. En ciertas realizaciones, Y es CH2. En ciertas realizaciones, Y es un enlace.
n puede ser 1, 2, 3, 4, 5, 6, 7 u 8, o cualquier intervalo derivable en su interior. Por ejemplo, n puede ser 1, 2, 3 o 4. Alternativamente, n puede ser 5, 6, 7 u 8. Alternativamente, n puede ser 9, 10, 11 o 12.
o puede ser un número entero de cero a 10, o cualquier intervalo derivable en su interior. Por ejemplo, o puede ser 0, 1, 2, 3, 4 o 5. Alternativamente, o puede ser 6, 7, 8, 9 o 10. Alternativamente, o puede ser uno a 7. Alternativamente, o puede ser uno a 8. Alternativamente, o puede ser uno a 9. Alternativamente, o es 3 a 8.
o puede ser un número entero de cero a 30, o cualquier intervalo derivable en su interior. Por ejemplo, o puede ser un número entero de cero a 30, 29, 28, 27 o 26. Alternativamente, o puede ser un número entero de cero a 25, 24, 23, 22 o 21. Alternativamente, o puede ser un número entero de cero a 20, 19, 18, 17 o 16. Alternativamente, o puede ser un número entero de cero a 15, 14, 13, 12 o 11.
p puede ser 0, 1, 2, 3, 4, 5 o 6, o cualquier intervalo derivable en su interior. Por ejemplo, p puede ser 7, 8, 9, 10, 11 o 12. Alternativamente, p puede ser 0, 1, 2 o 3. Alternativamente, p puede ser 4, 5 o 6.
q puede ser un número entero de cero a 10, o cualquier intervalo derivable en su interior. Por ejemplo, q puede ser 0, 1, 2, 3, 4 o 5. Alternativamente, q puede ser 6, 7, 8, 9 o 10. Alternativamente, q puede ser uno a 7. Alternativamente, q puede ser uno a 8. Alternativamente, q puede ser uno a 9. Alternativamente, q puede ser 3 a 8.
q puede ser un número entero de cero a 30, o cualquier intervalo derivable en su interior. Por ejemplo, q puede ser un número entero de cero a 30, 29, 28, 27 o 26. Alternativamente, q puede ser un número entero de cero a 25, 24, 23, 22 o 21. Alternativamente, q puede ser un número entero de cero a 20, 19, 18, 17 o 16. Alternativamente, q puede ser un número entero de cero a 15, 14, 13, 12 u 11.
r puede ser un número entero de uno a 6. Por ejemplo, r puede ser uno. Alternativamente, r puede ser 2. Alternativamente, r puede ser 3. Alternativamente, r puede ser 4. Alternativamente, r puede ser 5. Alternativamente, r puede ser 6.
Como se ha descrito anteriormente, cuando R28 y R40 son H, entonces R32 no es =O. En ciertas realizaciones, el compuesto no es rapamicina, como se muestra a continuación:

En la fórmula Ia o Ic, R32 puede ser -O-C(=Z1)-R32a. R32 puede ser -O-C(=Z1)-R32a; en donde R32a es -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B. En Fórmula Ia o Ic, R28 puede ser -C(=Z1)-R28a. R28 puede ser -C(=Z1)-R28a; en donde R28a es -A1-L1- A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B. En Fórmula Ia o Ic, R40 puede ser -C(=Z1)-R40a. R40 puede ser -C(=Z1)-R40a. en donde R40a es -A1-L1-A 2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B.
También se describe en el presente documento un compuesto de la fórmula Ia o Ic, o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, o tres de las siguientes características:
a) R32 es -O-C=(Z1-R32a;
b) R28 es -C(=Z1)-R28a;
c) R40 es -C(=Z1)-R40a.
También se describe en el presente documento un compuesto de la fórmula Ia o Ic, o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, o tres de las siguientes características:
a) R32 es -O-C(=Z1)-R32a; en donde R32a es -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2 -L3-B;
b) R28 es -C(=Z1)-R28a; en donde R28a es -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B;
c) R40 es -C(=Z1)-R40a. en donde R40a es -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B.
También se describe en el presente documento un compuesto de la fórmula Ia o Ic, o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, o tres de las siguientes características:
a) R40 es -C(=Z1)-R40a;
b) R40a es -A1-L1-A2-B; -A1-A2-B; -L2-A1-L1-A2-L3-B;
c) R32 es -OR3, tal como -OH.
También se describe en el presente documento un compuesto de la fórmula Ia o Ic, o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, tres o cuatro de las siguientes características:
a) uno de R28a, R32a y R40a es -A1-L1-A2-B;
b) A1 está ausente;
c) A2 está ausente;
d) L1 es

e) B es

f) B1 es

NR3-(C(R3)2)n- o

g) R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2 o -OR3.
También se describe en el presente documento un compuesto de fórmula:

o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, tres o cuatro de las siguientes características:
a) Z1 es O;
b) A1 está ausente;
c) L1 es

d) B es

e) B1 es

NR3-(C(R3)2)n- o

f) R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2 o -OR3; y
g) R32 es =O.
En lo anterior, R40a puede ser -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B.
También se describe en el presente documento un compuesto de fórmula:

o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, tres o cuatro de las siguientes características:
a) Z1 es O;
b) A1 está ausente;
c) L1 es

d) B es

e) B1 es

NR3-(C(R3)2)n- o

f) R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2 o -OR3; y
g) R32 es -OH.
En lo anterior, R40a puede ser -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B.
También se describe en el presente documento un compuesto de fórmula:

o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, tres o cuatro de las siguientes características:
a) Z1 es O;
b) A1 es

c) L1 es

d) B es

e) B1 es

NR3-(C(R3)2)n- o

f) R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2 o -OR3; y
g) R32 es =O.
En lo anterior, R40a puede ser -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B.
También se describe en el presente documento un compuesto de fórmula:

o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, tres o cuatro de las siguientes características:
a) Z1 es O;
b) A1 está ausente;
c) L1 es

d) B es

e) B1 es

NR3-(C(R3)2)n- ,

o

NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, en donde el arileno se sustituye opcionalmente con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
f) R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2 o -OR3; y
g) R32 es -OH.
En lo anterior, R40a puede ser -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B.
También se describe en el presente documento un compuesto de fórmula:

o una sal o tautómero farmacéuticamente aceptable del mismo, que tiene una, dos, tres o cuatro de las siguientes características:
a) Z1 es O;
b) A1 es

c) A2 es

d) L1 es

e) B es

f) B1 es

NR3-(C(R3)2)n-,

o

NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, en donde el arileno se sustituye opcionalmente con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
g) R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2 o -OR3; y
h) R32 es -OH.
En lo anterior, R40a puede ser -A1-L1-A2-B; -A1-A2-B; o -L2-A1-L1-A2-L3-B.
En la fórmula Ia o Ic, R40a puede ser cualquier resto orgánico, que puede tener un peso molecular (por ejemplo, la suma de las masas atómicas de los átomos del sustituyente) inferior a 15 g/mol, 50 g/mol, 100 g/mol, 150 g/mol, 200 g/mol, 250 g/mol, 300 g/mol, 350 g/mol, 400 g/ moles, 450 g/mol o 500 g/mol.
Los compuestos de la divulgación pueden incluir sales farmacéuticamente aceptables de los compuestos desvelados en el presente documento. "Sales farmacéuticamente aceptables" representativas pueden incluir, por ejemplo, sales solubles en agua e insolubles en agua, tales como las sales de acetato, amsonato (4,4-diaminoestilbeno-2,2-disulfonato), bencenosulfonato, benzonato, bicarbonato, bisulfato, bitartrato, borato, bromuro, butirato, calcio, edetato de calcio, camsilato, carbonato, cloruro, citrato, clavulariato, diclorhidrato, edetato, edisilato, estolato, esilato, fumarato, gluceptato, gluconato, glutamato, glicolilarsanilato, hexafluorofosfato, hexilresorcinato, hidrabamina, bromhidrato, clorhidrato, hidroxinaftoato, yoduro, setionato, lactato, lactobionato, laurato, magnesio, malato, maleato, mandelato, mesilato, metilbromuro, metilnitrato, metil sulfato, mucato, napsilato, nitrato, sal de amonio de N-metilglucamina, 3-hidroxi-2-naftoato, oleato, oxalato, palmitato, pamoato, 1,1-meteno-bis-2-hidroxi-3-naftoato, einbonato, pantotenato, fosfato/difosfato, picrato, poligalacturonato, propionato, p-toluenosulfonato, salicilato, estearato, subacetato, succinato, sulfato, sulfosalicilato, suramato, tannato, tartrato, teoclato, tosilato, trietyoduro y valerato.
"Sal farmacéuticamente aceptable" también puede incluir tanto sales de adición de ácido como de base. "Sal de adición de ácido farmacéuticamente aceptable" puede referirse a las sales que retienen la eficacia biológica y propiedades de las bases libres, que no son biológicamente o de otro modo no deseables, y que se pueden formar con ácidos inorgánicos tales como, pero no se limitan a, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares, y ácidos orgánicos tales como, pero no se limitan a, ácido acético, ácido 2,2-dicloroacético, ácido adípico, ácido algínico, ácido ascórbico, ácido aspártico, ácido bencenosulfónico, ácido benzoico, ácido 4-acetamidobenzoico, ácido canfórico, ácido canfor-10-sulfónico, ácido cáprico, ácido caproico, ácido caprílico, ácido carbónico, ácido cinámico, ácido cítrico, ácido ciclámico, ácido dodecilsulfúrico, ácido etano-1,2-disulfónico, ácido etanosulfónico, ácido 2-hidroxietanosulfónico, ácido fórmico, ácido fumárico, ácido galactárico, ácido gentísico, ácido glucoheptónico, ácido glucónico, ácido glucurónico, ácido glutámico, ácido glutárico, ácido 2-oxo-glutárico, ácido glicerofosfórico, ácido glicólico, ácido hipúrico, ácido isobutírico, ácido láctico, ácido lactobiónico, ácido láurico, ácido maleico, ácido málico, ácido malónico, ácido mandélico, ácido metanosulfónico, ácido múcico, ácido naftaleno-1,5-disulfónico, ácido naftaleno-2-sulfónico, ácido 1-hidroxi-2-naftoico, ácido nicotínico, ácido oleico, ácido orótico, ácido oxálico, ácido palmítico, ácido pamoico, ácido propiónico, ácido piroglutámico, ácido pirúvico, ácido salicílico, ácido 4-aminosalicílico, ácido sebácico, ácido esteárico, ácido succínico, ácido tartárico, ácido tiociánico, ácido ptoluenosulfónico, ácido trifluoroacético, ácido undecilénico y similares.
"Sal de adición de base farmacéuticamente aceptable" puede referirse a las sales que retienen la eficacia biológica y propiedades de los ácidos libres, que no son biológicamente o de otro modo no deseables. Estas sales se pueden preparar a partir de la adición de una base inorgánica o una base orgánica al ácido libre. Sales derivadas de bases inorgánicas pueden incluir, pero no se limitan a, las sales de sodio, potasio, litio, amonio, calcio, magnesio, hierro, cinc, cobre, manganeso, aluminio y similares. Por ejemplo, las sales inorgánicas pueden incluir, pero no se limitan a, sales de amonio, sodio, potasio, calcio y magnesio. Las sales derivadas de bases orgánicas pueden incluir, pero no se limitan a, sales de aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas que existen de forma natural, aminas cíclicas y resinas de intercambio iónico básicas, tales como amoniaco, isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, dietanolamina, etanolamina, decanol, 2-dimetilaminoetanol, 2-dietilaminoetanol, diciclohexilamina, lisina, arginina, histidina, cafeína, procaína, hidrabamina, colina, betaína, benetamina, benzatina, etilendiamina, glucosamina, metilglucamina, teobromo, trietanolamina, trometamina, purinas, piperazina, piperidina, N-etilpiperidina, resinas de poliamina y similares.
A menos que se establezca de otro modo, las estructuras representadas en el presente documento también pueden incluir compuestos que solo se diferencian en la presencia de uno o más átomos isotópicamente enriquecidos. Por ejemplo, los compuestos que tienen la presente estructura, excepto por la sustitución de un átomo de hidrógeno por deuterio o tritio o la sustitución de un átomo de carbono por 13C o 14C, o la sustitución de un átomo de nitrógeno por 15N, o la sustitución de un átomo de oxígeno con 17O o 18O, están dentro del alcance de la divulgación. Dichos compuestos isotópicamente marcados son útiles como herramientas de investigación o diagnóstico.
En algunas realizaciones, uno o más átomos de deuterio se pueden introducir en el resto de PEG de cualquier compuesto de la presente invención. Los mecanismos de dichas modificaciones se conocen en la técnica a partir de materiales de partida comercialmente disponibles, tales como elementos estructurales de hidroxilamina
isotópicamente enriquecida. En algunas realizaciones, un tritio o un deuterio se puede introducir en la posición C32 de compuestos de la presente invención usando, por ejemplo, un agente reductor isotópicamente puro comercialmente disponible y métodos conocidos por los expertos en la técnica. En algunas realizaciones, se puede introducir 14C en el resto de carbamato C40 de los compuestos de la presente invención usando materiales comercialmente disponibles y métodos conocidos por los expertos en la técnica. En algunas realizaciones, un isótopo, tal como deuterio o tritio, se puede introducir en el sustituyente R40a de un compuesto de la fórmula Ia, Ic, I o II, usando materiales de partida comercialmente disponibles y métodos conocidos por los expertos en la técnica.
Métodos de síntesis de los compuestos desvelados
Los compuestos de la presente divulgación se pueden preparar mediante una variedad de métodos, que incluyen química habitual. Rutas de síntesis adecuadas se representan en los esquemas dados a continuación.
Los compuestos de cualquiera de las fórmulas descritas en el presente documento se pueden preparar por métodos conocidos en la técnica de la síntesis orgánica, como se expone en parte por los siguientes esquemas de síntesis y ejemplos. En los esquemas descritos a continuación, es bien entendido que grupos protectores de grupos sensibles o reactivos se emplean donde sea necesario según los principios generales o la química. Los grupos protectores se manipulan según métodos convencionales de síntesis orgánica (T. W. Greene y P. G. M. Wuts, "Protective Groups in Organic Synthesis", tercera edición, Wiley, Nueva York 1999). Estos grupos se retiran en una etapa conveniente de la síntesis del compuesto usando métodos que son fácilmente evidentes para los expertos en la técnica. Los procesos de selección, así como las condiciones de reacción y el orden de su ejecución, debe estar de acuerdo con la preparación de los compuestos de la presente invención, o una sal o tautómero farmacéuticamente aceptable de los mismos.
Los compuestos de la presente invención se pueden preparar por métodos que evitan el uso de reacciones de cicloadición catalizadas por metal que requieren el uso de compuestos que contienen azida. Los compuestos que contienen azida presentan posibles riesgos de seguridad asociados a su preparación y almacenamiento (por ejemplo, explosión debido a descomposición de alta energía). Por tanto, los esquemas de reacción en el presente documento pueden evitan el uso de metales de cobre o rutenio en la penúltima o última etapas de síntesis, que puede ser ventajoso. El evitar el uso de metales de cobre o rutenio en la penúltima o última etapas de síntesis reduce las posibilidades de contaminación de los compuestos finales con impurezas metálicas no deseables.
Como la rapamicina puede ser un material de partida caro, son ventajosos buenos rendimientos de las reacciones. Los esquemas de reacción en el presente documento proporcionan mejores rendimientos que otros esquemas de reacción. En los esquemas de reacción en el presente documento, no hay necesidad de alquilar en el C40-hidroxilo de la rapamicina, que es ventajoso para proporcionar hasta 5 veces mejor rendimiento global en la preparación de compuestos bivalentes a partir de rapamicina en comparación con otros esquemas de reacción.
Hay una mejora sintética adicional asociada a mejores rendimientos. Evitar la necesidad de alquilar en el C40-hidroxilo da hasta 5 veces mejor rendimiento global en la preparación de compuestos bivalentes partir de rapamicina.
Los expertos en la técnica reconocerán si hay un estereocentro en alguno de los compuestos de la presente divulgación. Por consiguiente, la presente divulgación puede incluir tanto posibles estereoisómeros (a menos que se especifique en la síntesis) como puede incluir no solo compuestos racémicos, sino los enantiómeros individuales y/o diaestereómeros también. Cuando un compuesto se desea como un enantiómero individual o diaestereómero, se puede obtener por síntesis estereoespecífica o por resolución del producto final o cualquier producto intermedio conveniente. La redisolución del producto final, un producto intermedio o un material de partida se puede efectuar por cualquier método adecuado conocido en la técnica. Véase, por ejemplo, "Stereochemistry of Organic Compounds" por E. L. Eliel, S. H. Wilen, y L. N. Mander (Wiley-lnterscience, 1994).
Preparación de compuestos
Los compuestos descritos en el presente documento se pueden preparar a partir de materiales de partida comercialmente disponibles o sintetizar usando procesos orgánicos, inorgánicos y/o enzimáticos conocidos.
Los compuestos de la presente divulgación se pueden preparar de varias formas bien conocidas por los expertos en la técnica de la síntesis orgánica. A modo de ejemplo, los compuestos de la divulgación se pueden sintetizar usando los métodos descritos a continuación, junto con métodos de síntesis conocidos en la técnica de la química orgánica sintética, o variaciones en ellos, como se aprecia por los expertos en la técnica. Estos métodos pueden incluir, pero no se limitan a, los métodos descritos a continuación.
El término "tautómeros" puede referirse a un conjunto de compuestos que tiene el mismo número y tipo de átomos, pero se diferencian en la conectividad del enlace y están en equilibrio entre sí. Un "tautómero" es un único miembro de este conjunto de compuestos. Normalmente, se dibuja un único tautómero, pero se puede entender que esta estructura individual puede representar todos los posibles tautómeros que podrían existir. Los ejemplos pueden incluir
tautomería enol-cetona. Cuando una cetona se dibuja, se puede entender que tanto las formas de enol como de cetona son parte de la divulgación.
Además de los tautómeros que pueden existir en todos los grupos amida, carbonilo y oxima dentro de los compuestos de la presente invención, los compuestos en esta familia se interconvierten fácilmente por una especie de anillo abierto entre dos formas isoméricas principales, conocidos como los isómeros de pirano y oxepano (mostrados a continuación). Esta interconversión se puede promover por iones magnesio, condiciones ligeramente ácidas, o sales de alquilamina, como se describe en las siguientes referencias: i) Hughes, P. F.; Musser, J.; Conklin, M.; Russo, R.1992. Tetrahedron Lett. 33(33): 4739-32. ii) Zhu, T. 2007. Patente de EE. UU. 7.241.771; Wyeth. iii) Hughes, P.F. 1994. Patente de EE. UU.5.344.833; American Home Products Corp. El esquema que sigue muestra una interconversión entre los isómeros de pirano y oxepano en compuestos de la fórmula I, Ia, Ib, Ic, II o IIb.

Como esta interconversión ocurre en condición suave, y la posición en equilibrio termodinámico puede variar entre diferentes miembros de los compuestos de la fórmula I, Ia, Ib, Ic, II o IIb, se contemplan ambos isómeros para los compuestos de la fórmula I, Ia, Ib, Ic, II o IIb. Por brevedad, se muestra la forma de isómero de pirano de todos los productos intermedios y compuestos de la fórmula I, Ia, Ib, Ic, II o IIb.
Enfoques de ensamblaje general para rapálogos bifuncionales
Con referencia a los esquemas que siguen, la rapamicina es la fórmula RAP,

donde R16 es -OCH3; R26 es =O; R28 es -OH; R32 es =O; y R40 es -OH. Un "rapálogo" se refiere a un análogo o derivado de rapamicina. Por ejemplo, con referencia a los esquemas que siguen, un rapálogo puede ser rapamicina que está sustituida en cualquier posición, tal como R16, R26, R28, R32 o R40. Un inhibidor de sitio activo (inhibidor AS) es un inhibidor de sitio activo de mTOR. En ciertas realizaciones, el inhibidor AS se representa por B, en la fórmula I, Ia, Ib, Ic, II o IIb.
Rapálogos bifuncionales de la serie 1
Una estructura general de los rapálogos bifuncionales de la serie 1 se muestra en el Esquema 1 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7 y r = 1 a
6. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 2
Una estructura general de los rapálogos bifuncionales de la serie 2 se muestra en el Esquema 2 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina preconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 3
Una estructura general de los rapálogos bifuncionales de la serie 3 se muestra en el Esquema 3 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina posconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 4
Una estructura general de los rapálogos bifuncionales de la serie 4 se muestra en el Esquema 4 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. Las aminas pre- y posconectoras pueden cada una incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 5
Una estructura general de los rapálogos bifuncionales de la serie 5 se muestra en el Esquema 5 que sigue. Para estos tipos de rapálogos bifuncionales, la amina preconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 6
Una estructura general de los rapálogos bifuncionales de la serie 6 se muestra en el Esquema 6 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. Las aminas conectoras pueden incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina posconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 7
Una estructura general de los rapálogos bifuncionales de la serie 7 se muestra en el Esquema 7 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. Las aminas pre- y posconectoras pueden cada una incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 8
Una estructura general de los rapálogos bifuncionales de la serie 8 se muestra en el Esquema 8 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina posconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I, Ia, Ib, Ic, II o IIb), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Composiciones farmacéuticas
Otro aspecto proporciona una composición farmacéutica que incluye un compuesto de la presente invención, o sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo, y al menos uno de un vehículo, diluyente o excipiente farmacéuticamente aceptable.
En realizaciones de las composiciones farmacéuticas, un compuesto de la presente invención, o una sal o tautómero farmacéuticamente aceptable del mismo, se puede incluir en una cantidad terapéuticamente eficaz.
En algunas realizaciones, la composición comprende una mezcla de

o un estereoisómero o tautómero del mismo y

o un estereoisómero o tautómero del mismo. En algunas de dichas realizaciones, la composición comprende un excipiente farmacéuticamente aceptable.
En algunas realizaciones, la composición comprende una mezcla de

En algunas de dichas realizaciones, la composición comprende un excipiente farmacéuticamente aceptable.
La administración de los compuestos desvelados o composiciones se puede llevar a cabo por cualquier modo de administración para agentes terapéuticos. Estos modos pueden incluir administración sistémica o local, tal como modos de administración oral, nasal, parenteral, transdérmica, subcutánea, vaginal, yugal, rectal, tópica, intratecal o intracraneal.
En ciertas realizaciones, la administración puede incluir administración por vía oral, administración como un supositorio, contacto tópico, administración intravenosa, parenteral, intraperitoneal, intramuscular, intralesional, intratecal, intracraneal, intranasal o subcutánea, o la implantación de un dispositivo de liberación lenta, por ejemplo, una minibomba osmótica, a un sujeto. La administración puede ser por cualquier vía, que incluye parenteral y transmucosa (por ejemplo, yugal, sublingual, palatal, gingival, nasal, vaginal, rectal o transdérmica). La administración parenteral incluye, por ejemplo, intravenosa, intramuscular, intrarterial, intradérmica, subcutánea, intraperitoneal, intraventricular e intracraneal. Otros modos de administración incluyen, pero no se limitan a, el uso de formulaciones liposomales, infusión intravenosa, parches transdérmicos, etc. Las composiciones de la presente divulgación se pueden administrar por vía transdérmica, por una vía tópica, formuladas como bastoncillos aplicadores, disoluciones, suspensiones, emulsiones, geles, cremas, pomadas, pastas, gelatinas, pinturas, polvos y aerosoles. Los preparados orales incluyen comprimidos, píldoras, polvo, comprimidos recubiertos de azúcar, cápsulas, líquidos, pastillas para chupar, sellos, geles, jarabes, suspensiones, suspensiones, etc., adecuados para ingestión por el paciente. Los preparados en forma sólida incluyen polvos, comprimidos, píldoras, cápsulas, sellos, supositorios y gránulos dispersables. Los preparados en forma líquida incluyen disoluciones, suspensiones y emulsiones, por ejemplo, agua o disoluciones de agua/propilenglicol. Las composiciones de la presente divulgación pueden incluir además componentes para proporcionar liberación sostenida y/o comodidad. Dichos componentes incluyen polímeros mucomiméticos aniónicos de alto peso molecular, polisacáridos gelificantes y sustratos para vehículos de fármaco finamente divididos. Estos componentes se tratan en mayor detalle en las patentes de EE. UU. N.º 4.911.920; 5.403.841; 5.212.162; y 4.861.760. Las composiciones de la presente divulgación también se pueden administrar como microesferas para la liberación lenta en el cuerpo. Por ejemplo, las microesferas se pueden administrar por inyección intradérmica de microesferas que contienen fármaco, que se liberan lentamente por vía subcutánea (véase Rao, J. Biomater Set Polym. Ed.7:623-645, 1995); como formulaciones en gel biodegradables e inyectables (véase, por ejemplo, Gao Pharm. Res.12:857-863, 1995); o como microesferas para administración por vía oral (véase, por ejemplo, Eyles, J. Pharm. Pharmacol.
49:669-674, 1997). En otra realización, las formulaciones de las composiciones de la presente divulgación se pueden administrar usando liposomas que se fusionan con la membrana celular o son endocitados, es decir, empleando ligandos de receptor unidos al liposoma, que se unen a receptores de la proteína de la membrana superficial de la célula, dando como resultado la endocitosis. Usando liposomas, particularmente donde la superficie del liposoma lleva ligandos receptores específicos de células diana, o se dirigen de otro modo preferentemente hacia un órgano específico, la administración de las composiciones de la presente invención se pueden centrar en las células diana in vivo (véanse, por ejemplo, Al-Muhammed, J. Microencapsul.13:293-306, 1996; Chonn, Curr. Opin. Biotechnol.6:698-708, 1995; Ostro, Am. J. Hosp. Pharm.46: 1576-1587, 1989). Las composiciones de la presente divulgación también se pueden administrar como nanopartículas.
Dependiendo del modo de administración previsto, los compuestos desvelados o composiciones farmacéuticas pueden ser una forma farmacéutica sólida, semisólida o líquida, tal como, por ejemplo, inyectables, comprimidos, supositorios, píldoras, cápsulas de liberación con el tiempo, elixires, tinturas, emulsiones, jarabes, polvos, líquidos, suspensiones o similares, algunas veces en dosis unitarias y de acuerdo con prácticas farmacéuticas convencionales. Asimismo, también se pueden administrar en forma intravenosa (tanto en embolada como en infusión), intraperitoneal, intratecal, subcutánea o intramuscular, y todas usando formas bien conocidas por los expertos en las técnicas farmacéuticas.
Composiciones farmacéuticas ilustrativas son comprimidos y cápsulas de gelatina que comprenden un compuesto de la divulgación y un vehículo farmacéuticamente aceptable, tal como a) un diluyente, por ejemplo, agua purificada, aceites de triglicéridos, tales como aceite vegetal hidrogenado o parcialmente hidrogenado, o mezclas de los mismos, aceite de maíz, aceite de oliva, aceite de girasol, aceite de alazor, aceites de pescado, tales como EPA o DHA, o sus ésteres o triglicéridos o mezclas de los mismos, ácidos grasos omega-3 o derivados de los mismos, lactosa, dextrosa, sacarosa, manitol, sorbitol, celulosa, sodio, sacarina, glucosa y/o glicina; b) un lubricante, por ejemplo, sílice, talco, ácido esteárico, su sal de magnesio o calcio, oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato sódico, cloruro sódico y/o polietilenglicol; para comprimidos también; c) un aglutinante, por ejemplo, silicato de magnesio y aluminio, pasta de almidón, gelatina, tragacanto, metilcelulosa, carboximetilcelulosa de sodio, carbonato de magnesio, azúcares naturales tales como glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como goma arábiga, tragacanto o alginato de sodio, ceras y/o polivinilpirrolidona, si se desea; d) un disgregante, por ejemplo, almidones, agar, metil celulosa, bentonita, goma xantana, ácido algínico o su sal de sodio, o mezclas efervescentes; e) absorbente, colorante, aromatizante y edulcorante; f) un emulsionante o dispersante, tal como Tween 80, Labrasol, HPMC, DOSS, caproil 909, Labrafac, Labrafil, Peceol, transcutol, Capmul MCM, Capmul PG-12, Captex 355, Gelucire, TGPS de vitamina E u otro emulsionante aceptable; y/o g) un agente que potencia la absorción del compuesto, tal como ciclodextrina, hidroxipropil-ciclodextrina, PEG400, PEG200.
Se pueden preparar composiciones líquidas, particularmente inyectables, por ejemplo, por disolución, dispersión, etc. Por ejemplo, el compuesto desvelado se disuelve en o se mezcla con un disolvente farmacéuticamente aceptable, tal como, por ejemplo, agua, solución salina, dextrosa acuosa, glicerol, etanol y similares, para así formar una disolución
o suspensión isotónica inyectable. Se pueden usar proteínas tales como albúmina, partículas de quilomicrones o proteínas séricas para solubilizar los compuestos desvelados.
Los compuestos desvelados también se pueden formular como un supositorio que se puede preparar a partir de emulsiones o suspensiones grasas; usando polialquilenglicoles, tales como propilenglicol, como vehículo.
Los compuestos desvelados también se pueden administrar en forma de sistemas de administración de liposomas, tales como vesículas unilaminares pequeñas, vesículas unilaminares grandes y vesículas multilaminares. Los liposomas se pueden formar a partir de una variedad de fosfolípidos, que contienen colesterol, estearilamina o fosfatidilcolinas. En algunas realizaciones, una película de componentes lipídicos se hidrata con una disolución acuosa de fármaco dando una forma de capa lipídica que encapsula el fármaco, como se describe, por ejemplo, en la patente de EE. UU. N.º 5.262.564.
Los compuestos desvelados también se pueden administrar usando anticuerpos monoclonales como vehículos individuales a los que se acoplan los compuestos desvelados. Los compuestos desvelados también se pueden acoplarse con polímeros solubles como vehículos de fármaco direccionable. Dichos polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamida-fenol, polihidroxietil-aspanamida-fenol, o poli(óxido de etileno)-polilisina sustituido con restos de palmitoílo. Además, los compuestos desvelados se pueden acoplar a una clase de polímeros biodegradables útil en logra la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, poliépsilon-caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polidihidropiranos, policianoacrilatos y copolímeros de bloque reticulados o anfipáticos de hidrogeles. En una realización, los compuestos desvelados no se unen covalentemente a un polímero, por ejemplo, un polímero de ácido policarboxílico, o un poliacrilato.
Se usa, en general, administración inyectable parenteral para inyecciones e infusiones subcutáneas, intramusculares o intravenosas. Los inyectables se pueden preparar en formas convencionales, ya sea como disoluciones líquidas o suspensiones o formas sólidas adecuadas para disolver en líquido antes de inyección.
Otro aspecto de la divulgación se refiere a una composición farmacéutica que comprende un compuesto, o una sal o tautómero farmacéuticamente aceptable del mismo, de la presente divulgación y un vehículo farmacéuticamente aceptable. El vehículo farmacéuticamente aceptable puede incluir adicionalmente un excipiente, diluyente o tensioactivo.
Las composiciones se pueden preparar según métodos convencionales de mezcla, granulación o recubrimiento, respectivamente, y las presentes composiciones farmacéuticas pueden contener desde aproximadamente el 0,1 % hasta aproximadamente el 99 %, desde aproximadamente el 5 % hasta aproximadamente el 90 %, o desde aproximadamente el 1 % hasta aproximadamente el 20 % del compuesto desvelado en peso o volumen.
Los compuestos descritos en el presente documento se pueden usar en combinación entre sí, con otros agentes activos que se sabe que son útiles en el tratamiento de cáncer, enfermedad autoinmunitaria, enfermedad inflamatoria, enfermedad metabólica, enfermedad neurodegenerativa, infección fúngica o rechazo de trasplante, o con agentes complementarios que pueden no ser eficaces solos, pero pueden contribuir a la eficacia del agente activo. Los compuestos descritos en el presente documento se pueden usar en combinación con otros agentes activos que se sabe que son agentes de longevidad o antienvejecimiento.
En realizaciones de las composiciones farmacéuticas, la composición farmacéutica puede incluir un segundo agente (por ejemplo, agente terapéutico). En realizaciones de las composiciones farmacéuticas, la composición farmacéutica puede incluir un segundo agente (por ejemplo, agente terapéutico) en una cantidad terapéuticamente eficaz. En ciertas realizaciones, el segundo agente es un agente antineoplásico. En ciertas realizaciones, el segundo agente es un agente inmunoterapéutico. En ciertas realizaciones, el segundo agente es un agente inmuno-oncológico. En ciertas realizaciones, el segundo agente es un agente anti-enfermedad autoinmunitaria. En realizaciones, el segundo agente es un agente anti-enfermedad inflamatoria. En ciertas realizaciones, el segundo agente es un agente anti-enfermedad neurodegenerativa. En ciertas realizaciones, el segundo agente es un agente anti-enfermedad metabólica. En ciertas realizaciones, el segundo agente es un agente anti-enfermedad cardiovascular. En ciertas realizaciones, el segundo agente es un agente antienvejecimiento. En ciertas realizaciones, el segundo agente es un agente de longevidad. En ciertas realizaciones, el segundo agente es un agente de tratamiento o prevención del rechazo de trasplante. En ciertas realizaciones, el segundo agente es un agente de tratamiento o prevención de infección fúngica. En ciertas realizaciones, el segundo agente es un represor del sistema inmunitario. En ciertas realizaciones, el segundo agente es un modulador de mTOR. En ciertas realizaciones, el segundo agente es un inhibidor de mTOR. En ciertas realizaciones, el segundo agente es un inhibidor de sitio activo de mTOR. En ciertas realizaciones, el segundo agente es una rapamicina. En ciertas realizaciones, el segundo agente es un análogo de rapamicina. En ciertas realizaciones, el segundo agente es un inhibidor de la vía de mTOR. En ciertas realizaciones, el segundo agente es un inhibidor de CDK4/6; anti-PD1/PD-L1, inhibidor de PI3K; o inhibidor de Ras.
"Agente antineoplásico" o "fármaco antineoplásico" se usa según su significado llano y corriente y se refiere a una composición (por ejemplo, compuesto, fármaco, antagonista, inhibidor, modulador) que tiene propiedades
antineoplásicas o la capacidad de inhibir el crecimiento o la proliferación de células. En algunas realizaciones, un agente antineoplásico es un quimioterapéutico. En algunas realizaciones, un agente antineoplásico es un agente autorizado por la FDA o agencia reguladora similar de un país distinto de los EE. UU. para tratar el cáncer. Los ejemplos de agentes antineoplásicos incluyen, pero no se limitan a, rapamicina, análogo de rapamicina, bevacizumab, PP242, ΓN 128, MLN0128, antiandrógenos (por ejemplo, Casodex, flutamida, MDV3100 o ARN-509), inhibidores de MEK (por ejemplo, MEK1, MEK2, o MEK1 y MEK2) (por ejemplo, XL518, CI-1040, PD035901, selumetinib/ AZD6244, GSK1120212/ trametinib, GDC-0973, ARRY-162, ARRY-300, AZD8330, PD0325901, U0126, PD98059, TAK-733, PD318088, AS703026, BAY 869766), agentes alquilantes (por ejemplo, ciclofosfamida, ifosfamida, clorambucilo, busulfán, melfalán, mecloretamina, uramustina, tiotepa, nitrosoureas, mostazas nitrogenadas (por ejemplo, mecloroetamina, ciclofosfamida, clorambucilo, melfalán), etilenimina y metilmelaminas (por ejemplo, hexametilmelamina, tiotepa), sulfonatos de alquilo (por ejemplo, busulfán), nitrosoureas (por ejemplo, cardemustina, lomustina, semustina, estreptozocina), triazenos (decarbazina)), antimetabolitos (por ejemplo, 5-azatioprina, leucovorina, capecitabina, fludarabina, gemcitabina, pemetrexed, raltitrexed, análogo de ácido fólico (por ejemplo, metotrexato), análogos de pirimidina (por ejemplo, fluorouracilo, floxuridina, citarabina), análogos de purina (por ejemplo, mercaptopurina, tioguanina, pentostatina), etc.), alcaloides de planta (por ejemplo, vincristina, vinblastina, vinorelbina, vindesina, podofilotoxina, paclitaxel, docetaxel, etc.), inhibidores de la topoisomerasa (por ejemplo, irinotecán, topotecán, amsacrina, etopósido (VP 16), fosfato de etopósido, tenipósido, etc.), antibióticos antitumorales (por ejemplo, doxorubicina, adriamicina, daunorubicina, epirubicina, actinomicina, bleomicina, mitomicina, mitoxantrona, plicamicina, etc.), compuestos basados en platino (por ejemplo, cisplatino, oxaloplatino, carboplatino), antracenodiona (por ejemplo, mitoxantrona), urea sustituida (por ejemplo, hidroxiurea), derivado de metilhidracina (por ejemplo, procarbazina), supresor corticosuprarrenal (por ejemplo, mitotano, aminoglutetimida), epipodofilotoxinas (por ejemplo, etopósido), antibióticos (por ejemplo, daunorubicina, doxorubicina, bleomicina), enzimas (por ejemplo, L-asparaginasa), inhibidores de la señalización de proteínas cinasas activadas por mitógeno (por ejemplo, U0126, PD98059, PD184352, PD0325901, ARRY-142886, SB239063, SP600125, BAY 43-9006, wortmanina o LY294002), inhibidores de mTOR, anticuerpos (por ejemplo, rituxan), 5-aza-2'-desoxicitidina, doxorubicina, vincristina, etopósido, gemcitabina, imatinib (Gleevec.RTM.), geldanamicina, 17-N-alilamino-17-demetoxigeldanamicina (17-AAG), bortezomib, trastuzumab, anastrozol; inhibidores de la angiogénesis; antiandrógeno, antiestrógeno; oligonucleótidos antisentido; moduladores de genes de la apoptosis; reguladores de la apoptosis; arginina desaminasa; antagonistas de BCR/ABL; derivados de beta-lactama; inhibidor de bFGF; bicalutamida; derivados de camptotecina; inhibidores de caseína cinasas (ICOS); análogos de clomifeno; citarabina dacliximab; dexametasona; agonistas de estrógenos; antagonistas de estrógenos; etanidazol; fosfato de etopósido; exemestano; fadrozol; finasterida; fludarabina; clorhidrato de fluorodaunorubicina; gadolinio texapirina; nitrato de galio; inhibidores de la gelatinasa; gemcitabina; inhibidores de glutatión; hepsulfam; péptidos inmunoestimulantes; inhibidor de receptores del factor de crecimiento similar a la insulina-1; agonistas de interferón; interferones; interleucinas; letrozol; factor inhibidor de leucemia; alfainterferón de leucocitos; leuprolida+estrógeno+progesterona; leuprorelina; inhibidores de matrilisina; inhibidores de metaloproteinasa de la matriz; inhibidor de MIF; mifepristona; ARN bicatenario de emparejamiento incorrecto; anticuerpo monoclonal; extracto de pared celular micobacteriana; moduladores de la óxido nítrico; oxaliplatino; panomifeno; pentrozol; inhibidores de la fosfatasa; inhibidor del activador de plasminógeno; complejo de platino; compuestos de platino; prednisona; inhibidores del proteasoma; modulador inmunitario basado en proteína A; inhibidor de la proteína cinasa C; inhibidores de la proteína tirosina fosfatasa; inhibidores de la purina nucleósido fosforilasa; inhibidores de transferasa de la proteína ras farnesil; inhibidores de ras; inhibidor de ras-GAP; ribozimas; inhibidores de la transducción de señales; moduladores de la transducción de señales; proteína de unión al antígeno monocatenario; inhibidor de células madre; inhibidores de la división de células madre; inhibidores de estromelisina; glucosaminoglicanos sintéticos; metyoduro de tamoxifeno; inhibidores de la telomerasa; hormona estimulante tiroidea; inhibidores de la traducción; inhibidores de tirosina cinasas; antagonistas de receptores de urocinasa; esteroides (por ejemplo, dexametasona), finasterida, inhibidores de la aromatasa, agonistas de la hormona liberadora de gonadotropina (GnRH), tales como goserelina o leuprolida, adrenocorticosteroides (por ejemplo, prednisona), progestinas (por ejemplo, caproato de hidroxiprogesterona, acetato de megestrol, acetato de medroxiprogesterona), estrógenos (por ejemplo, dietilestilbestrol, etinilestradiol), antiestrógeno (por ejemplo, tamoxifeno), andrógenos (por ejemplo, propionato de testosterona, fluoximesterona), antiandrógeno (por ejemplo, flutamida), inmunoestimulantes (por ejemplo, Bacillus Calmette-Guerin (BCG), levamisol, interleucina-2, alfa-interferón, etc.), anticuerpos monoclonales (por ejemplo, anticuerpos monoclonales anti-CD20, anti-HER2, anti-CD52, anti-HLA-DR y anti-VEGF), inmunotoxinas (por ejemplo, conjugado anticuerpo monoclonal anti-CD33-caliqueamicina, conjugado de anticuerpo monoclonal anti-CD22-exotoxina de pseudomonas, etc.), radioinmunoterapia (por ejemplo, anticuerpo monoclonal anti-CD20 conjugado con 111ln, 90Y o 131I, etc.), triptolida, homoharringtonina, dactinomicina, doxorubicina, epirubicina, topotecán, itraconazol, vindesina, cerivastatina, vincristina, desoxiadenosina, sertralina, pitavastatina, irinotecán, clofazimina, 5-noniloxitriptamina, vemurafenib, dabrafenib, erlotinib, gefitinib, inhibidores de EGFR, terapia dirigida al receptor del factor de crecimiento epidérmico (EGFR) o terapéutico (por ejemplo, gefitinib (Iressa™), erlotinib (Tarceva™), cetuximab (Erbitux™), lapatinib (Tykerb™), panitumumab (Vectibix™), vandetanib (Caprelsa™), afatinib/BIBW2992, CI-1033/canertinib, neratinib/HKI-272, CP-724714, TAK-285, AST-1306, ARRY334543, ARRY-380, AG-1478, dacomitinib/PF299804, OSI-420/desmetil erlotinib, AZD8931, AEE788, pelitinib/EKB-569, CUDC-101, WZ8040, WZ4002, WZ3146, AG-490, XL647, PD153035, BMS-599626), sorafenib, imatinib, sunitinib, dasatinib, pirrolobenzodiacepinas (por ejemplo, tomaimicina), carboplatino, análogos de CC-1065 y CC-1065 que incluyen amino-CBI, mostazas nitrogenadas (tales como clorambucilo y melfalán), dolastatina y análogos de dolastatina (incluyendo auristatinas: por ejemplo, monometil auristatina E), antibióticos de antraciclina (tales como doxorubicina, daunorubicina, etc.), duocarmicinas y análogos de duocarmicina, enediínas (tales como neocarcinostatina y
caliqueamicinas), derivados de leptomicina, maitansinoides y análogos de maitansinoides (por ejemplo, mertansina), metotrexato, mitomicina C, taxoides, alcaloides de la vinca (tales como vinblastina y vincristina), epotilonas (por ejemplo, epotilona B), camptotecina y sus análogos clínicos topotecán e irinotecán, GNK128, PP242, PP121, MLN0128, AZD8055, AZD2014, VP-BEZ235, BGT226, SFl 126, Torin 1, Torin 2, WYE 687, sal de WYE 687 (por ejemplo, clorhidrato), PF04691502, PI-103, CC-223, OSI-027, XL388, KU-0063794, GDC-0349, PKI-587, rapamicina, deforolimus (AP23573, MK-8669, ridaforolimus), temsirolimus (CCI-779), ABT478, everolimus (RAD001) o similares.
mTOR y métodos de tratamiento
El término "mTOR" se refiere a la proteína "diana mecanística de rapamicina (serina/treonina cinasa)" o "diana de mamífero de rapamicina". El término "mTOR" se puede referir a la secuencia de nucleótidos o secuencia de proteínas de mTOR humano (por ejemplo, Entrez 2475, Uniprot P42345, RefSeq NM_004958 o RefSeq NP_004949) (SEQ ID NO: 1). El término "mTOR" puede incluir tanto la forma natural de las secuencias de nucleótidos o proteínas, así como cualquier mutante de las mismas. En algunas realizaciones, "mTOR" es mTOR natural. En algunas realizaciones, "mTOR" es una o más formas mutantes. El término "mTOR" XYZ puede referirse a una secuencia de nucleótidos o proteína de una mTOR mutante en donde el aminoácido numerado Y de mTOR que normalmente tiene un aminoácido X en la natural, tiene en su lugar un aminoácido Z en el mutante. En realizaciones, un mTOR es mTOR humana. En realizaciones, mTOR tiene la secuencia de nucleótidos correspondiente al número de referencia GL206725550 (SEQ ID NO:2). En realizaciones, mTOR tiene la secuencia de nucleótidos correspondiente a RefSeq NM_004958.3 (SEQ ID NO:2). En realizaciones, mTOR tiene la secuencia de proteínas correspondiente al número de referencia GL4826730 (SEQ ID NO: 1). En realizaciones, mTOR tiene la secuencia de proteínas correspondiente a RefSeq NP_004949.1 (SEQ ID NO: 1). En realizaciones, mTOR tiene la siguiente secuencia de aminoácidos:


En realizaciones, mTOR es un mTOR mutante. En realizaciones, mTOR mutante está asociado con una enfermedad que no está asociada a mTOR natural. En realizaciones, mTOR puede incluir al menos una mutación de aminoácido (por ejemplo, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 o 30 mutaciones, o cualquier intervalo derivable en su interior) en comparación con la secuencia anterior.
El término "mTORC1" se refiere al complejo de proteína que incluye mTOR y Raptor (proteína reguladora asociada a mTOR). mTORC1 también puede incluir MLST8 (proteína 8 letal de mamífero con SEC 13), PRAS40 y/o DEPTOR. mTORC1 puede funcionar de sensor de nutrientes/energía/rédox y regulador de la síntesis de proteínas. El término "vía de mTORC1" o "vía de transducción de señales de mTORC1" se puede referir a una vía celular que incluye mTORC1. Una vía de mTORC1 incluye los componentes de la vía aguas arriba y aguas abajo de mTORC1. Una vía de mTORC1 es una vía de señalización que se modula por modulación de la actividad de mTORC1. En realizaciones, una vía de mTORC1 es una vía de señalización que se modula por modulación de la actividad de mTORC1, pero no por modulación de la actividad de mTORC2. En realizaciones, una vía de mTORC1 es una vía de señalización que se modula a un mayor grado por la modulación de la actividad de mTORC1 que por la modulación de la actividad de mTORC2.
El término "mTORC2" se refiere al complejo de proteína que incluye mTOR y RICTOR (acompañante insensible a la rapamicina de mTOR). mTORC2 también puede incluir GβL, mSIN1 (proteína 1 que interactúa con la proteína cinasa activada por estrés de mamífero), Protor 1/2, DEPTOR, TTI1 y/o TEL2. mTORC2 puede regular el metabolismo celular y el citoesqueleto. El término "vía de mTORC2" o "vía de transducción de señales de mTORC2" puede referirse a una vía celular que incluye mTORC2. Una vía de mTORC2 incluye los componentes de la vía aguas arriba y aguas abajo de mTORC2. Una vía de mTORC2 es una vía de señalización que se modula por modulación de la actividad de mTORC2. En realizaciones, una vía de mTORC2 es una vía de señalización que se modula por modulación de la actividad de mTORC2, pero no por modulación de la actividad de mTORC1. En realizaciones, una vía de mTORC2 es una vía de señalización que se modula a un mayor grado por modulación de la actividad de mTORC2 que por modulación de la actividad de mTORC1.
El término "rapamicina" o "sirolimus" se refiere a un macrólido producido por las bacterias Streptomyces hygroscopicus. La rapamicina puede prevenir la activación de linfocitos T y linfocitos B. La rapamicina tiene el nombre de la IUPAC (3S,6R,7E,9R,10A,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-hexadecahidro-9,27-dihidroxi-3-[(1R)-2-[(1S,3R,4R)-4-hidroxi-3-metoxiciclohexil]-1-metiletil]-10,21-dimetoxi-6,8,12,14,20,26-hexametil-23,27-epoxi-3H-pirido[2,1-c][1,4]-oxaazaciclohentriacontino-1,5,11,28,29(4H,6H,31H)-pentona. La rapamicina tiene el número CAS 53123-88-9. La rapamicina se puede producir sintéticamente (por ejemplo, por síntesis química) o mediante el uso de un método de producción que no incluye el uso de Streptomyces hygroscopicus.
"Análogo" se usa según su significado llano y corriente dentro de la química y biología y se refiere a un compuesto químico que es estructuralmente similar a otro compuesto (es decir, un denominado compuesto de "referencia"), pero se diferencia en composición, por ejemplo, en la sustitución de un átomo por un átomo de un elemento diferente, o en presencia de un grupo funcional particular o la sustitución de un grupo funcional por otro grupo funcional, o la estereoquímica absoluta de uno o más centros quirales del compuesto de referencia, que incluye isómeros del mismo.
El término "análogo de rapamicina" o "rapálogo" se refiere a un análogo o derivado (por ejemplo, un profármaco) de rapamicina.
Los términos "inhibidor de sitio activo de mTOR" y "mimético de ATP" se refieren a un compuesto que inhibe la actividad de mTOR (por ejemplo, actividad de cinasa) y se une al sitio activo de mTOR (por ejemplo, el sitio de unión a ATP, que se solapa con el sitio de unión a ATP, bloqueando el acceso por ATP al sitio de unión a ATP de mTOR). Los ejemplos de inhibidores de sitio activo de mTOR incluyen, pero no se limitan a, ΓNK128, PP242, PP121, MLN0128, AZD8055, AZD2014, NVP-BEZ235, BGT226, SF1126, Torin 1, Torin 2, WYE 687, sal de WYE 687 (por ejemplo, clorhidrato), PF04691502, PI-103, CC-223, OSI-027, XL388, KU-0063794, GDC-0349 y PKI-587. En realizaciones, un inhibidor de sitio activo de mTOR es un TORi. En algunas realizaciones, "inhibidor de sitio activo" puede referirse a "inhibidor de sitio activo de mTOR".
El término "FKBP" se refiere a la proteína peptidil-prolil cis-trans isomerasa. Para ejemplos no limitantes de FKBP, véase Cell Mol Life Sci.2013 Sep;70(18):3243-75. En realizaciones, "FKBP" puede referirse a "FKBP-12" o "FKBP 12" o "FKBP 1 A". En realizaciones, "FKBP" puede referirse a la proteína humana. En el término "FKBP" se incluyen las formas naturales y mutantes de la proteína. En realizaciones, "FKBP" puede referirse a la proteína humana natural. En realizaciones, "FKBP" puede referirse al ácido nucleico humano natural. En realizaciones, FKBP es una FKBP mutante. En realizaciones, la FKBP mutante está asociada con una enfermedad que no está asociada a FKBP natural. En realizaciones, la FKBP incluye al menos una mutación de aminoácido (por ejemplo, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 o 30 mutaciones, o cualquier intervalo derivable en su interior) en comparación con FKBP natural.
El término "FKBP-12" o "FKBP 12" o "FKBP1A" puede referirse a la proteína "peptidil-prolil cis-trans isomerasa FKBP 1 A". En realizaciones, "FKBP-12" o "FKBP 12" o "FKBP 1 A" puede referirse a la proteína humana. En el término "FKBP-12" o "FKBP 12" o "FKBP 1 A" se incluyen las formas naturales y mutantes de la proteína. En realizaciones, "FKBP-12" o "FKBP 12" o "FKBP 1 A" puede referirse a la proteína asociada a Entrez Gene 2280, OMIM 186945, UniProt P62942 y/o RefSeq (proteína) NP_000792 (SEQ ID NO:3). En realizaciones, los números de referencia inmediatamente anteriores pueden referirse a la proteína, y ácidos nucleicos asociados, conocida a partir de la fecha de presentación de la presente solicitud. En realizaciones, "FKBP-12" o "FKBP 12" o "FKBP 1 A" puede referirse a la proteína humana natural. En realizaciones, "FKBP-12" o "FKBP 12" o "FKBP1A" puede referirse al ácido nucleico humano natural. En realizaciones, la FKBP-12 es una FKBP-12 mutante. En realizaciones, la FKBP-12 mutante está asociada con una enfermedad que no está asociada con FKBP-12 natural. En realizaciones, la FKBP-12 puede incluir al menos una mutación de aminoácido (por ejemplo, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 o 30 mutaciones, o cualquier intervalo derivable en su interior) en comparación con FKBP-12 natural. En realizaciones, la FKBP-12 tiene la secuencia de proteínas correspondiente al número de referencia GE206725550. En realizaciones, la FKBP-12 tiene la secuencia de proteínas correspondiente a RefSeq NP_000792.1 (SEQ ID NO:3).
El término "4E-BP1" o "4EBP1" o "EIF4EBP1" se refiere a la proteína "proteína 1 de unión al factor de inicio de la traducción eucariota 4E". En realizaciones, "4E-BP1" o "4EBP1" o "EIF4EBP 1" puede referirse a la proteína humana. En el término "4E-BP 1" o "4EBP 1" o "EIF4EBP1" se incluyen las formas naturales y mutantes de la proteína. En realizaciones, "4E-BP1" o "4EBP1" o "EIF4EBP1" puede referirse a la proteína asociada a Entrez Gene 1978, OMIM 602223, UniProt Q13541 y/o RefSeq (proteína) NP_004086 (SEQ ID NO:4). En realizaciones, los números de referencia inmediatamente anteriores pueden referirse a la proteína, y ácidos nucleicos asociados, conocida a partir de la fecha de presentación de la presente solicitud. En realizaciones, "4E-BP 1" o "4EBP1" o "EIF4EBP1" puede referirse a la proteína humana natural. En realizaciones, "4E-BP1" o "4EBP1" o "EIF4EBP1" puede referirse al ácido nucleico humano natural. En realizaciones, la 4EBP1 es una 4EBP1 mutante. En realizaciones, la 4EBP1 mutante está asociada con una enfermedad que no está asociada con 4EBP1 natural. En realizaciones, la 4EBP1 puede incluir al menos una mutación de aminoácido (por ejemplo, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 o 30 mutaciones, o cualquier intervalo derivable en su interior) en comparación con 4EBP1 natural. En realizaciones, 4EBP1 tiene la secuencia de proteínas correspondiente al número de referencia GL4758258. En realizaciones, 4EBP1 tiene la secuencia de proteínas correspondiente a RefSeq NP_004086.1 (SEQ ID NO: 4).
El término "Akt" se refiere a la proteína cinasa específica de serina/treonina implicada en procesos celulares, tales como el metabolismo de la glucosa, la apoptosis, la proliferación y otras funciones, también conocida como "proteína cinasa B" (PKB) o "Akt1". En realizaciones, "Akt" o "AM" o "PKB" puede referirse a la proteína humana. En el término "Akt" o "Akt1" o "PKB" se incluyen las formas naturales y mutantes de la proteína. En realizaciones, "Akt" o "Akt1" o "PKB" puede referirse a la proteína asociada a Entrez Gene 207, OMIM 164730, UniProt P31749 y/o RefSeq (proteína) NP_005154 (SEQ ID NO:5). En realizaciones, los números de referencia inmediatamente anteriores pueden referirse a la proteína, y ácidos nucleicos asociados, conocida a partir de la fecha de presentación de la presente solicitud. En realizaciones, "Akt" o "Akt1" o "PKB" puede referirse a la proteína humana natural. En realizaciones, "Akt" o "Akt1" o "PKB" puede referirse al ácido nucleico humano natural. En realizaciones, Akt es una Akt mutante. En realizaciones, la Akt mutante está asociada con una enfermedad que no está asociada con una Akt natural. En realizaciones, la Akt puede incluir al menos una mutación de aminoácido (por ejemplo, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 o 30 mutaciones, o cualquier intervalo derivable en su interior) en
comparación con Akt natural. En realizaciones, la Akt tiene la secuencia de proteínas correspondiente al número de referencia GI: 62241011. En realizaciones, la Akt tiene la secuencia de proteínas correspondiente a RefSeq NP_005154.2 (SEQ ID NO:5).
La presente invención proporciona un compuesto de uno cualquiera de los Ejemplos 1-152 o una sal, tautómero o estereoisómero farmacéuticamente aceptable de los mismos para su uso en un método de tratamiento, prevención o reducción del riesgo de una enfermedad o trastorno mediado por mTOR. El método puede comprender administrar al sujeto que padece o susceptible de desarrollar una enfermedad o trastorno mediado por mTOR una cantidad terapéuticamente eficaz de uno o más de dichos compuestos.
En algunas realizaciones, la enfermedad es cáncer o una enfermedad mediada por inmunidad. En algunas realizaciones, el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos. En algunas realizaciones, el trastorno es cirrosis hepática. En algunas realizaciones, la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis. En ciertas realizaciones, la enfermedad es complejo de esclerosis tuberosa (TSC). En ciertas realizaciones, la enfermedad es tumor neuroendocrino pancreático (PNET), linfoma de células del manto (MCL), cáncer colorrectal o cáncer de intestino (CRC), cáncer uterino, cáncer de ovario, cáncer de vejiga, cáncer de las vías genitourinarias o carcinoma de células renales (RCC).
La presente invención también proporciona un compuesto de uno cualquiera de los Ejemplos 1-152 o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo para su uso en un método de tratamiento de cáncer. El método puede comprender administrar al sujeto una cantidad terapéuticamente eficaz de uno o más de dichos compuestos. En algunas realizaciones, el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos. En algunas realizaciones, el trastorno es cirrosis hepática. En ciertas realizaciones, la enfermedad es complejo de esclerosis tuberosa (TSC). En ciertas realizaciones, la enfermedad es tumor neuroendocrino pancreático (PNET), linfoma de células del manto (MCL), cáncer colorrectal o cáncer de intestino (CRC), cáncer uterino, cáncer de ovario, cáncer de vejiga, cáncer de las vías genitourinarias o carcinoma de células renales (RCC).
En ciertas realizaciones, el cáncer incluye cánceres y carcinomas humanos, sarcomas, adenocarcinomas, linfomas, leucemias, etc., que incluyen cánceres sólidos y linfoides, cáncer de riñón, mama, pulmón, vejiga, colon, ovario, próstata, páncreas, estómago, cerebro, cabeza y cuello, piel, uterino, testicular, glioma, esófago y de hígado, que incluye hepatocarcinoma, linfoma, que incluye linfoma linfoblástico agudo B, linfomas no Hodgkin (por ejemplo, linfomas de Burkitt, de células pequeñas y de células grandes), linfoma de Hodgkin, leucemia (incluyendo LMA, LLA y LMC), o mieloma múltiple. En ciertas realizaciones, la enfermedad es mieloma múltiple. En ciertas realizaciones, la enfermedad es cáncer de mama. En ciertas realizaciones, la enfermedad es cáncer de mama triple negativo.
En ciertas realizaciones, el cáncer incluye cáncer, neoplasia o tumores malignos encontrados en mamíferos (por ejemplo, seres humanos), que incluyen leucemia, carcinomas y sarcomas. Los cánceres a modo de ejemplo que se pueden tratar con un compuesto o método proporcionado en el presente documento incluyen cáncer de próstata, tiroides, sistema endocrino, cerebro, mama, cuello uterino, colon, cabeza y cuello, hígado, riñón, pulmón, pulmón no microcítico, melanoma, mesotelioma, ovario, sarcoma, estómago, útero, meduloblastoma, cáncer colorrectal, cáncer pancreático. Los ejemplos adicionales pueden incluir enfermedad de Hodgkin, linfoma no Hodgkin, mieloma múltiple, neuroblastoma, glioma, glioblastoma multiforme, cáncer de ovario, rabdomiosarcoma, trombocitosis primaria, macroglobulinemia primaria, tumores cerebrales primarios, insulinoma pancreático maligno, carcinoide maligno, cáncer de vejiga urinario, lesiones premalignas de piel, cáncer testicular, linfomas, cáncer de tiroides, neuroblastoma, cáncer de esófago, cáncer de las vías genitourinarias, hipercalcemia maligna, cáncer endometrial, cáncer corticosuprarrenal, neoplasias del páncreas endocrino o exocrino, cáncer de tiroides medular, carcinoma tiroideo medular, melanoma, cáncer colorrectal, cáncer de tiroides papilar, carcinoma hepatocelular o cáncer de próstata.
En ciertas realizaciones, la enfermedad es leucemia. El término "leucemia" se refiere ampliamente a enfermedades malignas progresivas de los órganos hematopoyéticos y, en general, se caracteriza por una proliferación y desarrollo distorsionados de leucocitos y sus precursores en la sangre y la médula ósea. La leucemia se clasifica, en general, clínicamente basándose en (1) la duración y el carácter de la enfermedad - aguda o crónica; (2) el tipo de célula implicado; mieloide (mielógena), linfoide (linfógena) o monocítica; y (3) el aumento o no aumento en el número de células aberrantes en la sangre - leucémica o aleucémica (subleucémica). Las leucemias a modo de ejemplo que se pueden tratar con un compuesto o método proporcionado en el presente documento incluyen, por ejemplo, leucemia no linfocítica aguda, leucemia linfocítica crónica, leucemia granulocítica aguda, leucemia granulocítica crónica, leucemia promielocítica aguda, leucemia de linfocitos T del adulto, leucemia aleucémica, una leucemia leucocitémica, leucemia basófila, leucemia de blastocitos, leucemia bovina, leucemia mielocítica crónica, leucemia cutánea, leucemia embrionaria, leucemia eosinofílica, leucemia de Gross, leucemia de células pilosas, leucemia hemoblástica, leucemia hemocitoblástica, leucemia histiocítica, leucemia de células madre, leucemia monocítica aguda, leucemia leucopénica, leucemia linfática, leucemia linfoblástica, leucemia linfocítica, leucemia linfógena, leucemia linfoide, leucemia de células de linfosarcoma, leucemia de mastocitos, leucemia de megacariocitos, leucemia micromieloblástica, leucemia monocítica, leucemia de mieloblastos, leucemia mielocítica, leucemia granulocítica mieloide, leucemia mielomonocítica, leucemia de Naegeli, leucemia de células plasmáticas, mieloma múltiple, leucemia plasmacítica, leucemia promielocítica, leucemia de células de Rieder, leucemia de Schilling, leucemia de células madre, leucemia subleucémica o leucemia de células no diferenciadas.
En ciertas realizaciones, la enfermedad es sarcoma. El término "sarcoma" se refiere, en general, a un tumor que está constituido por una sustancia como el tejido conjuntivo embrionario y, en general, está compuesto por células estrechamente concentradas incorporadas en una sustancia fibrilar o homogénea. Los sarcomas que se pueden tratar con un compuesto o método proporcionado en el presente documento incluyen un condrosarcoma, fibrosarcoma, linfosarcoma, melanosarcoma, mixosarcoma, osteosarcoma, sarcoma de Abernethy, sarcoma adiposo, liposarcoma, sarcoma alveolar de partes blandas, sarcoma ameloblástico, sarcoma botrioide, sarcoma cloroma, coriocarcinoma, sarcoma embrionario, sarcoma de tumor de Wilms, sarcoma endometrial, sarcoma de estroma, sarcoma de Ewing, sarcoma fascial, sarcoma fibroblastoico, sarcoma de células gigantes, sarcoma granulocítico, sarcoma de Hodgkin, sarcoma hemorrágico pigmentado idiopático múltiple, sarcoma inmunoblástico de linfocitos B, linfoma, sarcoma inmunoblástico de linfocitos T, sarcoma de Jensen, sarcoma de Kaposi, sarcoma de célula de Kupffer, angiosarcoma, leucosarcoma, sarcoma de mesenquimoma maligno, sarcoma parostal, sarcoma reticulocítico, sarcoma de Rous, sarcoma seroquístico, sarcoma sinovial o sarcoma telangiectásico.
En ciertas realizaciones, la enfermedad es melanoma. El término "melanoma" se refiere a un tumor que surge del sistema de melanocitos de la piel y otros órganos. Los melanomas que se pueden tratar con un compuesto o método proporcionado en el presente documento incluyen, por ejemplo, melanoma acral lentiginoso, melanoma amelanótico, melanoma juvenil benigno, melanoma de Cloudman, melanoma S91, melanoma de Harding-Passey, melanoma juvenil, melanoma lentigo maligno, melanoma maligno, melanoma nodular, melanoma subungueal o melanoma de extensión superficial.
En ciertas realizaciones, la enfermedad es carcinoma. El término "carcinoma" se refiere a un nuevo crecimiento maligno constituido de células epiteliales que tienden a infiltrar los tejidos circundantes y dar lugar a metástasis. Los carcinomas a modo de ejemplo que se pueden tratar con un compuesto o método proporcionado en el presente documento incluyen, por ejemplo, carcinoma tiroideo medular, carcinoma tiroideo medular familiar, carcinoma acinar, carcinoma acinoso, carcinoma adenoquístico, carcinoma quístico adenoide, carcinoma adenomatoso, carcinoma de la corteza suprarrenal, carcinoma alveolar, carcinoma de células alveolares, carcinoma de células basales, carcinoma basocelular, carcinoma basaloide, carcinoma de células basoescamosas, carcinoma broncoalveolar, carcinoma bronquiolar, carcinoma broncogénico, carcinoma cerebriforme, carcinoma colangiocelular, carcinoma coriónico, carcinoma coloide, comedocarcinoma, carcinoma del cuerpo, carcinoma cribriforme, carcinoma en coraza, carcinoma cutáneo, carcinoma cilíndrico, carcinoma de células cilíndricas, carcinoma canalicular, carcinoma durum, carcinoma embrionario, carcinoma encefaloide, carcinoma epidermoide, carcinoma epitelial adenoide, carcinoma exofítico, carcinoma ex ulcere, carcinoma fibroso, carcinoma gelatiniforme, carcinoma gelatinoso, carcinoma de células gigantes, carcinoma gigantocelular, carcinoma glandular, carcinoma de células de la granulosa, carcinoma de la matriz del pelo, carcinoma hematoide, carcinoma hepatocelular, carcinoma de células de Hurthle, carcinoma hialino, carcinoma hipernefroide, carcinoma embrionario infantil, carcinoma in situ, carcinoma intraepidérmico, carcinoma intraepitelial, carcinoma de Krompecher, carcinoma de células de Kulchitzky, carcinoma de células grandes, carcinoma lenticular, carcinoma lenticular, carcinoma lipomatoso, carcinoma linfoepitelial, carcinoma medular, carcinoma medular, carcinoma melanótico, carcinoma medular, carcinoma mucinoso, carcinoma mucíparo, carcinoma mucocelular, carcinoma mucoepidermoide, carcinoma mucoso, carcinoma mucoso, carcinoma mixomatodes, carcinoma nasofaríngeo, carcinoma de células pequeñas, carcinoma osificante, carcinoma osteoide, carcinoma papilar, carcinoma periportal, carcinoma preinvasivo, carcinoma espinocelular, carcinoma pultáceo, carcinoma de células renales de riñón, carcinoma de células de reserva, carcinoma sarcomatodes, carcinoma schneideriano, carcinoma escirroso, carcinoma del escroto, carcinoma de células en anillo de sello, carcinoma simple, carcinoma de células pequeñas, carcinoma solanoide, carcinoma de células esferoides, carcinoma de células del huso, carcinoma esponjoso, carcinoma escamoso, carcinoma de células escamosas, carcinoma en collar, carcinoma telangiectásico, carcinoma telangiectodes, carcinoma de células transitorias, carcinoma tuberoso, carcinoma tuberoso, carcinoma verrucoso o carcinoma velloso.
La presente invención también proporciona un compuesto de una cualquiera de los Ejemplos 1-152 o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo para su uso en un método de tratamiento de una enfermedad mediada por la inmunidad. El método puede comprender administrar al sujeto una cantidad terapéuticamente eficaz de uno o más de dichos compuestos. En algunas realizaciones, la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis.
En ciertas realizaciones, la enfermedad es enfermedad autoinmunitaria. Como se usa en el presente documento, el término "enfermedad autoinmunitaria" se refiere a una enfermedad o afección en la que el sistema inmunitario de un sujeto tiene una respuesta inmunitaria aberrante contra una sustancia que no causa normalmente una respuesta inmunitaria en un sujeto sano. Los ejemplos de enfermedades autoinmunitarias que se pueden tratar con un compuesto, composición farmacéutica o método descrito en el presente documento incluyen encefalomielitis aguda diseminada (ADEM), leucoencefalitis aguda hemorrágica necrotizante, enfermedad de Addison, agammaglobulinemia, alopecia areata, amiloidosis, espondilitis anquilosante, nefritis anti-GBM/anti-TBM, síndrome antifosfolípidos (APS), angioedema autoinmune, anemia aplásica autoinmune, disautonomía autoinmune, hepatitis autoinmune, hiperlipidemia autoinmune, inmunodeficiencia autoinmune, enfermedad autoinmune del oído interno (AIED), miocarditis autoinmune, ooforitis autoinmune, pancreatitis autoinmune, retinopatía autoinmune, púrpura trombocitopénica autoinmune (ATP), enfermedad tiroidea autoinmune, urticaria autoinmune, neuropatías axonales o neuronales, enfermedad de Baló, enfermedad de Behcet, pénfigo ampolloso, cardiomiopatía, enfermedad de Castleman, celiaquía, enfermedad de Chagas, síndrome de fatiga crónica, polineuropatía desmielinizante inflamatoria crónica (CIDP), osteomielitis crónica multifocal recurrente (CRMO), síndrome de Churg-Strauss, penfigoide cicatricial / penfigoide mucoso benigno, enfermedad de Crohn, síndrome de Cogans, enfermedad de aglutininas frías, bloqueo auriculoventricular congénito, miocarditis de Coxsackie, enfermedad de CREST, crioglobulinemia esencial mixta, neuropatías desmielinizantes, dermatitis herpetiforme, dermatomiositis, enfermedad de Devic (neuromielitis óptica), lupus discoide, síndrome de Dressler, endometriosis, esofagitis eosinofílica, fascitis eosinofílica, eritema nodoso, encefalomielitis alérgica experimental, síndrome de Evans, fibromialgia, alveolitis fibrosante, arteritis de células gigantes (arteritis temporal), miocarditis de células gigantes, glomerulonefritis, síndrome de Goodpasture, granulomatosis con poliangitis (GPA) (antiguamente llamada granulomatosis de Wegener), enfermedad de Graves, síndrome de Guillain-Barre, encefalitis de Hashimoto, tiroiditis de Hashimoto, anemia hemolítica, púrpura de Henoch-Schonlein, herpes gestacional, hipogammaglobulinemia, púrpura trombocitopénica idiopática (ITP), nefropatía de IgA, enfermedad esclerosante relacionada con IgG4, lipoproteínas inmunorreguladoras, miositis por cuerpos de inclusión, cistitis intersticial, artritis juvenil, diabetes juvenil (diabetes de tipo 1), miositis juvenil, síndrome de Kawasaki, síndrome de Lambert-Eaton, vasculitis leucocitoclástica, liquen plano, liquen escleroso, conjuntivitis lignea, enfermedad por IgA lineal (LAD), lupus (SLE), enfermedad de Lyme, crónica, enfermedad de Meniere, poliangitis microscópica, enfermedad mixta del tejido conjuntivo (MCTD), úlcera de Mooren, enfermedad de Mucha-Habermann, esclerosis múltiple, miastenia grave, miositis, narcolepsia, neuromielitis óptica (Devic), neutropenia, penfigoide cicatricial ocular, neuritis óptica, reumatismo palindrómico, PANDAS (trastornos pediátricos neuropsiquiátricos autoinmunes asociados a Streptococcus), degeneración cerebelosa paraneoplásica, hemoglobinuria paroxística nocturna (PNH), síndrome de Parry Romberg, síndrome de Parsonnage-Turner, pars planitis (uveítis periférica), pénfigo, neuropatía periférica, encefalomielitis perivenosa, anemia perniciosa, síndrome de POEMS, poliarteritis nodosa, síndromes poliglandulares autoinmunes de tipo I, II y III, polimialgia reumática, polimiositis, síndrome de postinfarto de miocardio, síndrome postpericardiotomía, dermatitis por progesterona, cirrosis biliar primaria, colangitis esclerosante primaria, psoriasis, artritis psoriásica, fibrosis pulmonar idiopática, pioderma gangrenoso, aplasia pura de la serie roja, fenómeno de Raynaud, artritis reactiva, distrofia simpática refleja, síndrome de Reiter, policondritis recidivante, síndrome de piernas inquietas, fibrosis retroperitoneal, fiebre reumática, artritis reumatoide, sarcoidosis, síndrome de Schmidt, escleritis, esclerodermia, síndrome de Sjogren, autoinmunidad del esperma y testicular, síndrome del hombre rígido, endocarditis subaguda bacteriana (SBE), síndrome de Susac, oftalmia simpática, arteritis de Takayasu, arteritis temporal/arteritis de células gigantes, púrpura trombocitopénica (TTP), síndrome de Tolosa-Hunt, mielitis transversa, diabetes de tipo 1, colitis ulcerosa, enfermedad indiferenciada de tejido conjuntivo (UCTD), uveítis, vasculitis, dermatosis vesiculobulosa, vitíligo o granulomatosis de Wegener (es decir, granulomatosis con poliangitis (GPA)).
La presente invención también proporciona un compuesto de uno cualquiera de los Ejemplos 1-152 o una sal, tautómero, o estereoisómero farmacéuticamente aceptable del mismo para su uso en un método de tratamiento de una afección senil. El método puede comprender administrar al sujeto una cantidad terapéuticamente eficaz de uno o más de dichos compuestos. En ciertas realizaciones, la afección senil se selecciona de sarcopenia, atrofia de la piel, atrofia muscular progresiva, atrofia cerebral, aterosclerosis, arteriosclerosis, enfisema pulmonar, osteoporosis, osteoartritis, hipertensión arterial, disfunción eréctil, demencia, enfermedad de Huntington, enfermedad de Alzheimer, cataratas, degeneración macular senil, cáncer de próstata, accidente cerebrovascular, reducida esperanza de vida, deterioro de la función renal y sordera parcial senil, incapacidad de movilidad relacionada con el envejecimiento (por ejemplo, fragilidad), deterioro cognitivo, demencia senil, deterioro de la memoria, rigidez de los tendones, disfunción cardíaca, tal como hipertrofia cardíaca y disfunción sistólica y diastólica, inmunosenescencia, cáncer, obesidad y diabetes.
En ciertas realizaciones, las composiciones o compuestos desvelados se pueden usar con respecto a la inmunosenescencia. La inmunosenescencia puede referirse a una disminución en la función inmunitaria que da como resultado una alteración de la respuesta inmunitaria, por ejemplo, al cáncer, vacunación, patógenos infecciosos, entre otros. Implica tanto a la capacidad del hospedador para responder a infecciones como al desarrollo de la memoria inmunitaria a largo plazo, especialmente por vacunación. La deficiencia inmunitaria es ubicua y se encuentra en tanto especies de larga vida como de corta en función de su edad con respecto a la esperanza de vida en vez del tiempo cronológico. Se considera un factor contribuyente importante al aumento de la frecuencia de morbilidad y mortalidad entre los ancianos. La inmunosenescencia no es un fenómeno de deterioro aleatorio, quizás parece que repite inversamente un patón evolutivo y la mayoría de los parámetros afectados por la inmunosenescencia parecen estar bajo el control genético. La inmunosenescencia también puede ser algunas veces concebida como resultado del desafío continuo de la exposición inevitable a una variedad de antígenos, tales como virus y bacterias. La inmunosenescencia es una afección multifactorial que conduce a muchos problemas de salud patológicamente significativos, por ejemplo, en la población de edad avanzada. Los cambios biológicos dependientes de la edad, tales como reducción de células madre hematopoyéticas, un aumento en linfocitos PD1+, una disminución en el número total de fagocitos y células NK y una disminución en la inmunidad humoral, contribuyen a la aparición de inmunosenescencia. En un aspecto, la inmunosenescencia se pueden medir en un individuo midiendo la longitud de los telómeros en células inmunitarias (véase, por ejemplo, la patente de EE. UU. N.º 5.741.677). La inmunosenescencia también se puede determinar documentando en un individuo un número inferior al normal de linfocitos T CD4 y/o CD8 intactos, repertorio de linfocitos T, el número de linfocitos T que expresan PD1, por ejemplo, un número inferior a normal de linfocitos T PD-1 negativos, o respuesta a la vacunación en un sujeto superior o igual a 65 años de edad. En ciertas realizaciones, la modulación selectiva de mTOR de ciertas poblaciones de linfocitos T puede mejorar la eficacia de una vacuna en la población en envejecimiento y potencia la eficacia de la inmunoterapia del cáncer. La presente divulgación proporciona un método de tratamiento de la inmunosenescencia que comprende administrar al sujeto una cantidad terapéuticamente eficaz de una o más composiciones o compuestos desvelados.
En ciertas realizaciones, una enfermedad que se puede tratar con un compuesto, composición farmacéutica o método descrito en el presente documento es rechazo de trasplante de órgano o tejido (por ejemplo, trasplantes de corazón, pulmón, corazón-pulmón combinado, hígado, riñón, páncreas, piel o córnea; enfermedad injerto contra huésped), reestenosis, síndromes de hamartomas (por ejemplo, esclerosis tuberosa o enfermedad de Cowden), linfangioleiomiomatosis, retinitis pigmentosa, encefalomielitis, diabetes mellitus dependiente de insulina, lupus, dermatomiositis, artritis, enfermedades reumáticas, leucemia linfoblástica aguda resistente a esteroides, fibrosis, esclerodermia, fibrosis pulmonar, fibrosis renal, fibrosis quística, hipertensión pulmonar, esclerosis múltiple, síndrome de VHL, complejo de Carney, poliposis adenomatosa familiar, síndrome de poliposis juvenil, síndrome de Birt-Hogg-Duke, cardiomiopatía hipertrófica familiar, síndrome de Wolf-Parkinson-White, enfermedad de Parkinson, enfermedad de Huntington, enfermedad de Alzheimer, demencias provocadas por mutaciones tau, ataxia espinocerebelosa de tipo 3, enfermedad de las neuronas motoras causada por mutaciones SOD1, lipofucinosis neuronal ceroidea / enfermedad de Batten (neurodegeneración pediátrica), degeneración macular húmeda, degeneración macular seca, atrofia muscular progresiva (atrofia, caquexia), miopatías (por ejemplo, enfermedad de Danon), infección bacteriana, infección vírica, M. tuberculosis, estreptococo del grupo A, VHS tipo I, infección por HIV, neurofibromatosis (por ejemplo, neurofibromatosis tipo 1) o síndrome de Peutz-Jeghers.
En ciertas realizaciones, la enfermedad es una enfermedad neurodegenerativa. Como se usa en el presente documento, el término "enfermedad neurodegenerativa" se refiere a una enfermedad o afección en la que se altera la función del sistema nervioso de un sujeto. Los ejemplos de enfermedades neurodegenerativas que se pueden tratar con un compuesto, composición farmacéutica o método descrito en el presente documento incluyen enfermedad de Alexander, enfermedad de Alper, enfermedad de Alzheimer, esclerosis lateral amiotrófica, ataxia telangiectasia, enfermedad de Batten (también conocida como enfermedad de Spielmeyer-Vogt-Sjogren-Batten), encefalopatía espongiforme bovina (BSE), enfermedad de Canavan, síndrome de Cockayne, degeneración corticobasal, enfermedad de Creutzfeldt-Jakob, demencia frontotemporal, síndrome de Gerstmann-Straussler-Scheinker, enfermedad de Huntington, demencia asociada al VIH, enfermedad de Kennedy, enfermedad de Krabbe, kuru, demencia con cuerpos de Lewy, enfermedad de Machado-Joseph (ataxia espinocerebelosa de tipo 3), esclerosis múltiple, atrofia multisistémica, narcolepsia, neuroborreliosis, enfermedad de Parkinson, Enfermedad de Pelizaeus-Merzbacher, enfermedad de Pick, esclerosis lateral primaria, enfermedades priónicas, enfermedad de Refsum, enfermedad de Sandhoff, enfermedad de Schilder, degeneración combinada subaguda de médula espinal secundaria a anemia perniciosa, esquizofrenia, ataxia espinocerebelosa (múltiples tipos con características variables), atrofia muscular espinal, enfermedad de Steele-Richardson-Olszewski o tabes dorsal.
En ciertas realizaciones, la enfermedad es una enfermedad metabólica. Como se usa en el presente documento, el término "enfermedad metabólica" se refiere a una enfermedad o afección en la que se altera el metabolismo o sistema metabólico de un sujeto (por ejemplo, función de almacenamiento o utilización de energía). Los ejemplos de enfermedades metabólicas que se pueden tratar con un compuesto, composición farmacéutica o método descrito en el presente documento incluyen diabetes (por ejemplo, tipo I o tipo II), obesidad, síndrome metabólico, o una enfermedad mitocondrial (por ejemplo, disfunción de mitocondrias o función mitocondrial aberrante).
En ciertas realizaciones, la enfermedad es una enfermedad fúngica. Como se usa en el presente documento, el término "enfermedad fúngica" se refiere a una enfermedad o afección asociada a una infección por hongo del sujeto. Los ejemplos de enfermedades fúngicas que se pueden tratar con un compuesto, composición farmacéutica o método descrito en el presente documento incluyen infección con Mucor circinelloides, cigomicetos, Cryptococcus neoformans, Candida albicans, levadura y Saccharomyces cerevisiae, entre otros.
En ciertas realizaciones, la enfermedad es una enfermedad inflamatoria. Como se usa en el presente documento, el término "enfermedad inflamatoria" se refiere a una enfermedad o afección caracterizada por inflamación aberrante (por ejemplo, un aumento del nivel de inflamación en comparación con un control, tal como una persona sana que no padece una enfermedad). Los ejemplos de enfermedades inflamatorias incluyen lesión cerebral traumática, artritis, artritis reumatoide, artritis psoriásica, artritis idiopática juvenil, esclerosis múltiple, lupus eritematoso sistémico (SLE), miastenia grave, diabetes juvenil, diabetes mellitus de tipo 1, síndrome de Guillain-Barre, encefalitis de Hashimoto, tiroiditis de Hashimoto, espondilitis anquilosante, psoriasis, síndrome de Sjogren, vasculitis, glomerulonefritis, tiroiditis autoinmune, enfermedad de Behcet, enfermedad de Crohn, colitis ulcerosa, pénfigo ampolloso, sarcoidosis, ictiosis, oftalmopatía de Graves, enfermedad inflamatoria del intestino, enfermedad de Addison, vitíligo, asma, asma alérgica, acné vulgar, celiaquía, prostatitis crónica, enfermedad inflamatoria del intestino, enfermedad inflamatoria pélvica, lesión por reperfusión, sarcoidosis, rechazo de trasplante, cistitis intersticial, aterosclerosis y dermatitis atópica.
En ciertas realizaciones, la enfermedad es una enfermedad cardiovascular. Como se usa en el presente documento, el término "enfermedad cardiovascular" se refiere a una enfermedad o afección en la que se altera la función del sistema cardiovascular de un sujeto. Los ejemplos de enfermedades cardiovasculares que se pueden tratar con un compuesto, composición farmacéutica o método descrito en el presente documento incluyen insuficiencia cardíaca congestiva; síndromes arritmogénicos (por ejemplo, taquicardia paroxística, retardada después de despolarizaciones, taquicardia ventricular, taquicardia súbita, arritmias inducidas por el ejercicio, síndromes de QT largo, o taquicardia bidireccional); trastornos tromboembólicos (por ejemplo, trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cardiovasculares venosos o trastornos tromboembólicos en las cámaras del corazón); aterosclerosis; reestenosis; enfermedad arterial periférica; cirugía de injerto de revascularización coronaria; enfermedad de las arterias carótidas; arteritis; miocarditis; inflamación cardiovascular; inflamación vascular; enfermedad cardíaca coronaria (CHD); angina inestable (UA); angina inestable refractaria; angina estable (SA); angina estable crónica; síndrome coronario agudo (ACS); infarto de miocardio (primer o recurrente); infarto agudo de miocardio (AMI); infarto de miocardio; infarto de miocardio sin onda Q; infarto de miocardio sin STE; enfermedad de las arterias coronarias; enfermedad cardíaca isquémica; isquemia cardíaca; isquemia; muerte súbita isquémica; ataque isquémico transitorio; accidente cerebrovascular; enfermedad arterial oclusiva periférica; trombosis venosa; trombosis venosa profunda; tromboflebitis; embolia arterial; trombosis de las arterias coronarias; trombosis de las arterias cerebrales, embolia cerebral; embolia renal; embolia pulmonar; trombosis (por ejemplo, asociada a válvulas prostéticas u otros implantes, catéteres permanentes, prótesis endovasculares, derivación cardiopulmonar, hemodiálisis); trombosis (por ejemplo, asociada a aterosclerosis, cirugía, inmovilización prolongada, fibrilación arterial, trombofilia congénita, cáncer, diabetes, hormonas o embarazo); o arritmias cardíacas (por ejemplo, arritmias supraventriculares, arritmias auriculares, aleteo auricular o fibrilación auricular).
También se describe en el presente documento un método de tratamiento de una enfermedad asociada a un nivel aberrante de actividad de mTOR en un sujeto en necesidad de dicho tratamiento. La enfermedad puede ser causada por una regulación por incremento de mTOR. El método puede incluir administrar al sujeto una o más composiciones o compuestos descritos en el presente documento. El método puede incluir administrar al sujeto una cantidad terapéuticamente eficaz de una o más composiciones o compuestos descritos en el presente documento (por ejemplo, un modulador de mTOR (por ejemplo, inhibidor) como se ha descrito anteriormente).
También se describe una o más composiciones o compuestos como se describe en el presente documento para su uso como un medicamento. En realizaciones, el medicamento es útil para tratar una enfermedad causada por una regulación por incremento de mTOR. El uso puede incluir administrar al sujeto una o más composiciones o compuestos descritos en el presente documento. El uso puede incluir administrar al sujeto una cantidad terapéuticamente eficaz de una o más composiciones o compuestos descritos en el presente documento (por ejemplo, un modulador de mTOR (por ejemplo, inhibidor) como se ha descrito anteriormente).
También se describe una o más composiciones o compuestos como se describe en el presente documento para su uso en el tratamiento de una enfermedad causada por niveles aberrantes de actividad de mTORC1 en un sujeto en necesidad de dicho tratamiento. La enfermedad puede ser causada por una regulación por incremento de mTORC1. El uso puede incluir administrar al sujeto una o más composiciones o compuestos descritos en el presente documento. El uso puede incluir administrar al sujeto una cantidad terapéuticamente eficaz de una o más composiciones o compuestos descritos en el presente documento (por ejemplo, un modulador de mTORC1 (por ejemplo, inhibidor) como se ha descrito anteriormente).
La regulación por incremento de mTOR puede dar como resultado un aumento de la cantidad de actividad de mTOR en comparación con niveles normales de actividad de mTOR en un sujeto particular o una población de sujetos sanos. El aumento de la cantidad de actividad de mTOR puede dar como resultado, por ejemplo, excesivas cantidades de proliferación celular, provocando así el estado de enfermedad.
El sujeto de tratamiento para la enfermedad es normalmente un mamífero. El mamífero tratado con el compuesto (por ejemplo, compuesto descrito en el presente documento, modulador de mTOR (por ejemplo, inhibidor)) puede ser un humano, humano no primate y/o mamífero no humano (por ejemplo, roedor, canino).
El medicamento puede ser útil para tratar un enfermedad asociada a la actividad de mTORC1 en un sujeto en necesidad de dicho tratamiento. El uso puede incluir administrar una o más composiciones o compuestos como se describe en el presente documento al sujeto.
En otro aspecto, se proporciona una o más composiciones o compuestos para su uso en el tratamiento de una enfermedad asociada a la actividad de mTOR en un sujeto en necesidad de dicho tratamiento. En realizaciones, el uso puede incluir administrar una o más composiciones o compuestos como se describe en el presente documento, que incluyen realizaciones (por ejemplo, un aspecto, realización, ejemplo, tabla, figura o reivindicación) al sujeto.
En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es cáncer. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es una enfermedad autoinmunitaria. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es una enfermedad inflamatoria. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es una enfermedad neurodegenerativa. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es una enfermedad metabólica. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es rechazo de trasplante. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es infección fúngica. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es una enfermedad cardiovascular.
En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es envejecimiento. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es la muerte de una enfermedad senil. En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es una afección senil. En ciertas realizaciones, la afección senil se selecciona del grupo que consiste en sarcopenia, atrofia de la piel, atrofia muscular progresiva, atrofia cerebral, aterosclerosis, arteriosclerosis, enfisema pulmonar, osteoporosis, osteoartritis, hipertensión arterial, disfunción eréctil, demencia, enfermedad de Huntington, enfermedad de Alzheimer, cataratas, degeneración macular senil, cáncer de próstata, accidente cerebrovascular, reducida esperanza de vida, deterioro de la función renal y sordera parcial senil, incapacidad de movilidad relacionada con el envejecimiento (por ejemplo, fragilidad), deterioro cognitivo, demencia senil, deterioro de la memoria, rigidez de los tendones, disfunción cardíaca, tal como hipertrofia cardíaca y disfunción sistólica y diastólica, inmunosenescencia, cáncer, obesidad y diabetes. En ciertas realizaciones, la modulación selectiva de mTOR de ciertas poblaciones de linfocitos T puede mejorar la eficacia de una vacuna en la población en envejecimiento y potenciar la eficacia de la inmunoterapia del cáncer. La presente divulgación proporciona un método de tratamiento de la inmunosenescencia que comprende administrar al sujeto una cantidad terapéuticamente eficaz de uno o más compuestos desvelados.
En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es el cáncer (por ejemplo, carcinomas, sarcomas, adenocarcinomas, linfomas, leucemias, cánceres sólidos, cánceres linfoides; cáncer de riñón, mama, pulmón, vejiga, colon, gastrointestinal, ovario, próstata, páncreas, estómago, cerebro, cabeza y cuello, piel, uterino, esófago, hígado; cáncer testicular, glioma, hepatocarcinoma, linfoma, que incluye linfoma linfoblástico agudo B, linfomas no Hodgkin (por ejemplo, linfomas de Burkitt, de células pequeñas y de células grandes), linfoma de Hodgkin, leucemia (incluyendo LMA, LLA y LMC), mieloma múltiple y cáncer de mama (por ejemplo, triple negative cáncer de mama).
En realizaciones, la enfermedad asociada a la actividad de mTOR o enfermedad asociada a niveles aberrantes de actividad de mTOR es encefalomielitis aguda diseminada (ADEM), leucoencefalitis aguda hemorrágica necrotizante, enfermedad de Addison, agammaglobulinemia, alopecia areata, amiloidosis, espondilitis anquilosante, nefritis anti-GBM/anti-TBM, síndrome antifosfolípidos (APS), angioedema autoinmune, anemia aplásica autoinmune, disautonomía autoinmune, hepatitis autoinmune, hiperlipidemia autoinmune, inmunodeficiencia autoinmune, enfermedad autoinmune del oído interno (AIED), miocarditis autoinmune, ooforitis autoinmune, pancreatitis autoinmune, retinopatía autoinmune, púrpura trombocitopénica autoinmune (ATP), enfermedad tiroidea autoinmune, urticaria autoinmune, neuropatías axonales o neuronales, enfermedad de Baló, enfermedad de Behcet, pénfigo ampolloso, cardiomiopatía, enfermedad de Castleman, celiaquía, enfermedad de Chagas, síndrome de fatiga crónica, polineuropatía desmielinizante inflamatoria crónica (CIDP), osteomielitis crónica multifocal recurrente (CRMO), síndrome de Churg-Strauss, penfigoide cicatricial / penfigoide mucoso benigno, enfermedad de Crohn, síndrome de Cogans, enfermedad de aglutininas frías, bloqueo auriculoventricular congénito, miocarditis de Coxsackie, enfermedad de CREST, crioglobulinemia esencial mixta, neuropatías desmielinizantes, dermatitis herpetiforme, dermatomiositis, enfermedad de Devic (neuromielitis óptica), lupus discoide, síndrome de Dressler, endometriosis, esofagitis
eosinofílica, fascitis eosinofílica, eritema nodoso, encefalomielitis alérgica experimental, síndrome de Evans, fibromialgia, alveolitis fibrosante, arteritis de células gigantes (arteritis temporal), miocarditis de células gigantes, glomerulonefritis, síndrome de Goodpasture, granulomatosis con poliangitis (GPA) (antiguamente llamada granulomatosis de Wegener), enfermedad de Graves, síndrome de Guillain-Barre, encefalitis de Hashimoto, tiroiditis de Hashimoto, anemia hemolítica, púrpura de Henoch-Schonlein, herpes gestacional, hipogammaglobulinemia, púrpura trombocitopénica idiopática (ITP), nefropatía de IgA, enfermedad esclerosante relacionada con IgG4, lipoproteínas inmunorreguladoras, miositis por cuerpos de inclusión, cistitis intersticial, artritis juvenil, diabetes juvenil (diabetes de tipo 1), miositis juvenil, síndrome de Kawasaki, síndrome de Lambert-Eaton, vasculitis leucocitoclástica, liquen plano, liquen escleroso, conjuntivitis lignea, enfermedad por IgA lineal (LAD), lupus (SLE), enfermedad de Lyme, crónica, enfermedad de Meniere, poliangitis microscópica, enfermedad mixta del tejido conjuntivo (MCTD), úlcera de Mooren, enfermedad de Mucha-Habermann, esclerosis múltiple, miastenia grave, miositis, narcolepsia, neuromielitis óptica (Devic), neutropenia, penfigoide cicatricial ocular, neuritis óptica, reumatismo palindrómico, PANDAS (trastornos pediátricos neuropsiquiátricos autoinmunes asociados a Streptococcus), degeneración cerebelosa paraneoplásica, hemoglobinuria paroxística nocturna (PNH), síndrome de Parry Romberg, síndrome de Parsonnage-Turner, pars planitis (uveítis periférica), pénfigo, neuropatía periférica, encefalomielitis perivenosa, anemia perniciosa, síndrome de POEMS, poliarteritis nodosa, síndromes poliglandulares autoinmunes de tipo I, II y III, polimialgia reumática, polimiositis, síndrome de postinfarto de miocardio, síndrome postpericardiotomía, dermatitis por progesterona, cirrosis biliar primaria, colangitis esclerosante primaria, psoriasis, artritis psoriásica, fibrosis pulmonar idiopática, pioderma gangrenoso, aplasia pura de la serie roja, fenómeno de Raynaud, artritis reactiva, distrofia simpática refleja, síndrome de Reiter, policondritis recidivante, síndrome de piernas inquietas, fibrosis retroperitoneal, fiebre reumática, artritis reumatoide, sarcoidosis, síndrome de Schmidt, escleritis, esclerodermia, síndrome de Sjogren, autoinmunidad del esperma y testicular, síndrome del hombre rígido, endocarditis subaguda bacteriana (SBE), síndrome de Susac, oftalmia simpática, arteritis de Takayasu, arteritis temporal/arteritis de células gigantes, púrpura trombocitopénica (TTP), síndrome de Tolosa-Hunt, mielitis transversa, diabetes de tipo 1, colitis ulcerosa, enfermedad indiferenciada de tejido conjuntivo (UCTD), uveítis, vasculitis, dermatosis vesiculobulosa, vitíligo, granulomatosis de Wegener (es decir, granulomatosis con poliangitis (GPA)), lesión cerebral traumática, artritis, artritis reumatoide, artritis psoriásica, artritis idiopática juvenil, esclerosis múltiple, lupus eritematoso sistémico (SLE), miastenia grave, diabetes juvenil, diabetes mellitus de tipo 1, síndrome de Guillain-Barre, encefalitis de Hashimoto, tiroiditis de Hashimoto, espondilitis anquilosante, psoriasis, síndrome de Sjogren, vasculitis, glomerulonefritis, tiroiditis autoinmune, enfermedad de Behcet, enfermedad de Crohn, colitis ulcerosa, pénfigo ampolloso, sarcoidosis, ictiosis, oftalmopatía de Graves, enfermedad inflamatoria del intestino, enfermedad de Addison, vitíligo, asma, asma alérgica, acné vulgar, celiaquía, prostatitis crónica, enfermedad inflamatoria del intestino, enfermedad inflamatoria pélvica, lesión por reperfusión, sarcoidosis, rechazo de trasplante, cistitis intersticial, aterosclerosis, dermatitis atópica, enfermedad de Alexander, enfermedad de Alper, enfermedad de Alzheimer, esclerosis lateral amiotrófica, ataxia telangiectasia, enfermedad de Batten (también conocida como enfermedad de Spielmeyer-Vogt-Sjogren-Batten), encefalopatía espongiforme bovina (BSE), enfermedad de Canavan, síndrome de Cockayne, degeneración corticobasal, enfermedad de Creutzfeldt-Jakob, demencia frontotemporal, síndrome de Gerstmann-Straussler-Scheinker, enfermedad de Huntington, demencia asociada al VIH, enfermedad de Kennedy, enfermedad de Krabbe, kuru, demencia con cuerpos de Lewy, enfermedad de Machado-Joseph (ataxia espinocerebelosa de tipo 3), esclerosis múltiple, atrofia multisistémica, narcolepsia, neuroborreliosis, enfermedad de Parkinson, Enfermedad de Pelizaeus-Merzbacher, enfermedad de Pick, esclerosis lateral primaria, enfermedades priónicas, enfermedad de Refsum, enfermedad de Sandhoff, enfermedad de Schilder, degeneración combinada subaguda de médula espinal secundaria a anemia perniciosa, esquizofrenia, ataxia espinocerebelosa (múltiples tipos con características variables), atrofia muscular espinal, enfermedad de Steele-Richardson-Olszewski, tabes dorsal, diabetes (por ejemplo, tipo I o tipo II), obesidad, síndrome metabólico, una enfermedad mitocondrial (por ejemplo, disfunción de mitocondrias o función mitocondrial aberrante), infección fúngica, rechazo de trasplante, o una enfermedad cardiovascular (por ejemplo, insuficiencia cardíaca congestiva; síndromes arritmogénicos (por ejemplo, taquicardia paroxística, retardada después de despolarizaciones, taquicardia ventricular, taquicardia súbita, arritmias inducidas por el ejercicio, síndromes de QT largo, o taquicardia bidireccional); trastornos tromboembólicos (por ejemplo, trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cardiovasculares venosos o trastornos tromboembólicos en las cámaras del corazón); aterosclerosis; reestenosis; enfermedad arterial periférica; cirugía de injerto de revascularización coronaria; enfermedad de las arterias carótidas; arteritis; miocarditis; inflamación cardiovascular; inflamación vascular; enfermedad cardíaca coronaria (CHD); angina inestable (UA); angina inestable refractaria; angina estable (SA); angina estable crónica; síndrome coronario agudo (ACS); infarto de miocardio (primer o recurrente); infarto agudo de miocardio (AMI); infarto de miocardio; infarto de miocardio sin onda Q; infarto de miocardio sin STE; enfermedad de las arterias coronarias; enfermedad cardíaca isquémica; isquemia cardíaca; isquemia; muerte súbita isquémica; ataque isquémico transitorio; accidente cerebrovascular; enfermedad arterial oclusiva periférica; trombosis venosa; trombosis venosa profunda; tromboflebitis; embolia arterial; trombosis de las arterias coronarias; trombosis de las arterias cerebrales, embolia cerebral; embolia renal; embolia pulmonar; trombosis (por ejemplo, asociada a válvulas prostéticas u otros implantes, catéteres permanentes, prótesis endovasculares, derivación cardiopulmonar, hemodiálisis); trombosis (por ejemplo, asociada a aterosclerosis, cirugía, inmovilización prolongada, fibrilación arterial, trombofilia congénita, cáncer, diabetes, hormonas o embarazo); o arritmias cardíacas (por ejemplo, arritmias supraventriculares, arritmias auriculares, aleteo auricular o fibrilación auricular).
También se describe un método de tratamiento de una enfermedad que incluye administrar una cantidad eficaz de una o más composiciones o compuestos como se describe en el presente documento. También se describe una o
más composiciones o compuestos como se describe en el presente documento para su uso como un medicamento (por ejemplo, para el tratamiento de una enfermedad). En un aspecto se proporciona una o más composiciones o compuestos como se describe en el presente documento para su uso en el tratamiento de una enfermedad (por ejemplo, que incluye administrar una cantidad eficaz de una o más composiciones o compuestos como se describe en el presente documento). En realizaciones, la enfermedad es el cáncer. En realizaciones, la enfermedad es una enfermedad autoinmunitaria. En realizaciones, la enfermedad es una enfermedad inflamatoria. En realizaciones, la enfermedad es una enfermedad neurodegenerativa. En realizaciones, la enfermedad es una enfermedad metabólica. En realizaciones, la enfermedad es infección fúngica. En realizaciones, la enfermedad es rechazo de trasplante. En realizaciones, la enfermedad es una enfermedad cardiovascular.
En realizaciones, la enfermedad es cáncer (por ejemplo, carcinomas, sarcomas, adenocarcinomas, linfomas, leucemias, cánceres sólidos, cánceres linfoides; cáncer de riñón, mama, pulmón, vejiga, colon, ovario, próstata, páncreas, estómago, cerebro, cabeza y cuello, piel, uterino, esófago, hígado; cáncer testicular, glioma, hepatocarcinoma, linfoma, que incluye linfoma linfoblástico agudo B, linfomas no Hodgkin (linfomas de Burkitt, de células pequeñas y de células grandes), linfoma de Hodgkin, leucemia (incluyendo LMA, LLA y LMC), mieloma múltiple y cáncer de mama (por ejemplo, cáncer de mama triple negativo).
En realizaciones, la enfermedad es encefalomielitis aguda diseminada (ADEM), leucoencefalitis aguda hemorrágica necrotizante, enfermedad de Addison, agammaglobulinemia, alopecia areata, amiloidosis, espondilitis anquilosante, nefritis anti-GBM/anti-TBM, síndrome antifosfolípidos (APS), angioedema autoinmune, anemia aplásica autoinmune, disautonomía autoinmune, hepatitis autoinmune, hiperlipidemia autoinmune, inmunodeficiencia autoinmune, enfermedad autoinmune del oído interno (AIED), miocarditis autoinmune, ooforitis autoinmune, pancreatitis autoinmune, retinopatía autoinmune, púrpura trombocitopénica autoinmune (ATP), enfermedad tiroidea autoinmune, urticaria autoinmune, neuropatías axonales o neuronales, enfermedad de Baló, enfermedad de Behcet, pénfigo ampolloso, cardiomiopatía, enfermedad de Castleman, celiaquía, enfermedad de Chagas, síndrome de fatiga crónica, polineuropatía desmielinizante inflamatoria crónica (CIDP), osteomielitis crónica multifocal recurrente (CRMO), síndrome de Churg-Strauss, penfigoide cicatricial / penfigoide mucoso benigno, enfermedad de Crohn, síndrome de Cogans, enfermedad de aglutininas frías, bloqueo auriculoventricular congénito, miocarditis de Coxsackie, enfermedad de CREST, crioglobulinemia esencial mixta, neuropatías desmielinizantes, dermatitis herpetiforme, dermatomiositis, enfermedad de Devic (neuromielitis óptica), lupus discoide, síndrome de Dressler, endometriosis, esofagitis eosinofílica, fascitis eosinofílica, eritema nodoso, encefalomielitis alérgica experimental, síndrome de Evans, fibromialgia, alveolitis fibrosante, arteritis de células gigantes (arteritis temporal), miocarditis de células gigantes, glomerulonefritis, síndrome de Goodpasture, granulomatosis con poliangitis (GPA) (antiguamente llamada granulomatosis de Wegener), enfermedad de Graves, síndrome de Guillain-Barre, encefalitis de Hashimoto, tiroiditis de Hashimoto, anemia hemolítica, púrpura de Henoch-Schonlein, herpes gestacional, hipogammaglobulinemia, púrpura trombocitopénica idiopática (ITP), nefropatía de IgA, enfermedad esclerosante relacionada con IgG4, lipoproteínas inmunorreguladoras, miositis por cuerpos de inclusión, cistitis intersticial, artritis juvenil, diabetes juvenil (diabetes de tipo 1), miositis juvenil, síndrome de Kawasaki, síndrome de Lambert-Eaton, vasculitis leucocitoclástica, liquen plano, liquen escleroso, conjuntivitis lignea, enfermedad por IgA lineal (LAD), lupus (SLE), enfermedad de Lyme, enfermedad de Meniere, poliangitis microscópica, enfermedad mixta del tejido conjuntivo (MCTD), úlcera de Mooren, enfermedad de Mucha-Habermann, esclerosis múltiple, miastenia grave, miositis, narcolepsia, neuromielitis óptica (Devic), neutropenia, penfigoide cicatricial ocular, neuritis óptica, reumatismo palindrómico, PANDAS (trastornos pediátricos neuropsiquiátricos autoinmunes asociados a Streptococcus), degeneración cerebelosa paraneoplásica, hemoglobinuria paroxística nocturna (PNH), síndrome de Parry Romberg, síndrome de Parsonnage-Turner, pars planitis (uveítis periférica), pénfigo, neuropatía periférica, encefalomielitis perivenosa, anemia perniciosa, síndrome de POEMS, poliarteritis nodosa, síndromes poliglandulares autoinmunes de tipo I, II y III, polimialgia reumática, polimiositis, síndrome de postinfarto de miocardio, síndrome postpericardiotomía, dermatitis por progesterona, cirrosis biliar primaria, colangitis esclerosante primaria, psoriasis, artritis psoriásica, fibrosis pulmonar idiopática, pioderma gangrenoso, aplasia pura de la serie roja, fenómeno de Raynaud, artritis reactiva, distrofia simpática refleja, síndrome de Reiter, policondritis recidivante, síndrome de piernas inquietas, fibrosis retroperitoneal, fiebre reumática, artritis reumatoide, sarcoidosis, síndrome de Schmidt, escleritis, esclerodermia, síndrome de Sjogren, autoinmunidad del esperma y testicular, síndrome del hombre rígido, endocarditis subaguda bacteriana (SBE), síndrome de Susac, oftalmia simpática, arteritis de Takayasu, arteritis temporal/arteritis de células gigantes, púrpura trombocitopénica (TTP), síndrome de Tolosa-Hunt, mielitis transversa, diabetes de tipo 1, colitis ulcerosa, enfermedad indiferenciada de tejido conjuntivo (UCTD), uveítis, vasculitis, dermatosis vesiculobulosa, vitíligo, granulomatosis de Wegener (es decir, granulomatosis con poliangitis (GPA)), lesión cerebral traumática, artritis, artritis reumatoide, artritis psoriásica, artritis idiopática juvenil, esclerosis múltiple, lupus eritematoso sistémico (SLE), miastenia grave, diabetes juvenil, diabetes mellitus de tipo 1, síndrome de Guillain-Barre, encefalitis de Hashimoto, tiroiditis de Hashimoto, espondilitis anquilosante, psoriasis, vasculitis, glomerulonefritis, tiroiditis autoinmune, enfermedad de Behcet, enfermedad de Crohn, colitis ulcerosa, pénfigo ampolloso, sarcoidosis, ictiosis, oftalmopatía de Graves, enfermedad inflamatoria del intestino, enfermedad de Addison, vitíligo, asma, asma alérgica, acné vulgar, celiaquía, prostatitis crónica, enfermedad inflamatoria del intestino, enfermedad inflamatoria pélvica, lesión por reperfusión, sarcoidosis, rechazo de trasplante, cistitis intersticial, aterosclerosis, dermatitis atópica, enfermedad de Alexander, enfermedad de Alper, enfermedad de Alzheimer, esclerosis lateral amiotrófica, ataxia telangiectasia, enfermedad de Batten (también conocida como enfermedad de Spielmeyer-Vogt-Sjogren-Batten), encefalopatía espongiforme bovina (BSE), enfermedad de Canavan, síndrome de Cockayne, degeneración corticobasal, enfermedad de Creutzfeldt-Jakob, demencia frontotemporal,
síndrome de Gerstmann-Straussler-Scheinker, enfermedad de Huntington, demencia asociada al VIH, enfermedad de Kennedy, enfermedad de Krabbe, kuru, demencia con cuerpos de Lewy, enfermedad de Machado-Joseph (ataxia espinocerebelosa de tipo 3), esclerosis múltiple, atrofia multisistémica, narcolepsia, neuroborreliosis, enfermedad de Parkinson, Enfermedad de Pelizaeus-Merzbacher, enfermedad de Pick, esclerosis lateral primaria, enfermedades priónicas, enfermedad de Refsum, enfermedad de Sandhoff, enfermedad de Schilder, degeneración combinada subaguda de médula espinal secundaria a anemia perniciosa, esquizofrenia, ataxia espinocerebelosa (múltiples tipos con características variables), atrofia muscular espinal, enfermedad de Steele-Richardson-Olszewski, tabes dorsal, diabetes (por ejemplo, tipo I o tipo II), obesidad, síndrome metabólico, una enfermedad mitocondrial (por ejemplo, disfunción de mitocondrias o función mitocondrial aberrante), infección fúngica, rechazo de trasplante, o una enfermedad cardiovascular (por ejemplo, insuficiencia cardíaca congestiva; síndromes arritmogénicos (por ejemplo, taquicardia paroxística, retardada después de despolarizaciones, taquicardia ventricular, taquicardia súbita, arritmias inducidas por el ejercicio, síndromes de QT largo, o taquicardia bidireccional); trastornos tromboembólicos (por ejemplo, trastornos tromboembólicos cardiovasculares arteriales, trastornos tromboembólicos cardiovasculares venosos o trastornos tromboembólicos en las cámaras del corazón); aterosclerosis; reestenosis; enfermedad arterial periférica; cirugía de injerto de revascularización coronaria; enfermedad de las arterias carótidas; arteritis; miocarditis; inflamación cardiovascular; inflamación vascular; enfermedad cardíaca coronaria (CHD); angina inestable (UA); angina inestable refractaria; angina estable (SA); angina estable crónica; síndrome coronario agudo (ACS); infarto de miocardio (primer o recurrente); infarto agudo de miocardio (AMI); infarto de miocardio; infarto de miocardio sin onda Q; infarto de miocardio sin STE; enfermedad de las arterias coronarias; enfermedad cardíaca isquémica; isquemia cardíaca; isquemia; muerte súbita isquémica; ataque isquémico transitorio; accidente cerebrovascular; enfermedad arterial oclusiva periférica; trombosis venosa; trombosis venosa profunda; tromboflebitis; embolia arterial; trombosis de las arterias coronarias; trombosis de las arterias cerebrales, embolia cerebral; embolia renal; embolia pulmonar; trombosis (por ejemplo, asociada a válvulas prostéticas u otros implantes, catéteres permanentes, prótesis endovasculares, derivación cardiopulmonar, hemodiálisis); trombosis (por ejemplo, asociada a aterosclerosis, cirugía, inmovilización prolongada, fibrilación arterial, trombofilia congénita, cáncer, diabetes, hormonas o embarazo); o arritmias cardíacas (por ejemplo, arritmias supraventriculares, arritmias auriculares, aleteo auricular o fibrilación auricular). En realizaciones, la enfermedad es una enfermedad poliquística. En realizaciones, la enfermedad es enfermedad renal poliquística. En realizaciones, la enfermedad es estenosis. En realizaciones, la enfermedad es reestenosis. En realizaciones, la enfermedad es proliferación de la neoíntima. En realizaciones, la enfermedad es hiperplasia de la neoíntima.
En otro aspecto, se proporciona una o más composiciones o compuestos desvelados en el presente documento para su uso en el tratamiento de envejecimiento en un sujeto en necesidad de dicho tratamiento. En realizaciones, el uso puede incluir administrar una o más composiciones o compuestos como se describe en el presente documento, que incluyen realizaciones (por ejemplo, un aspecto, realización, ejemplo, tabla, figura o reivindicación) al sujeto.
En otro aspecto, se proporciona una o más composiciones o compuestos para su uso en prolongar la esperanza de vida o inducir la longevidad en un sujeto en necesidad de dicho tratamiento. En realizaciones, el uso puede incluir administrar una o más composiciones o compuestos como se describe en el presente documento, que incluyen realizaciones (por ejemplo, un aspecto, realización, ejemplo, tabla, figura o reivindicación) al sujeto.
En un aspecto, se proporciona una o más composiciones o compuestos como se describe en el presente documento para su uso en el tratamiento de una enfermedad poliquística en un sujeto en necesidad de dicho tratamiento. La enfermedad poliquística puede ser enfermedad renal poliquística. El uso puede incluir administrar al sujeto una o más composiciones o compuestos descritos en el presente documento. El uso puede incluir administrar al sujeto una cantidad terapéuticamente eficaz de una o más composiciones o compuestos descritos en el presente documento (por ejemplo, un modulador de mTOR (por ejemplo, inhibidor) como se ha descrito anteriormente).
En un aspecto, se proporciona una o más composiciones o compuestos como se describen en el presente documento para su uso en el tratamiento de estenosis en un sujeto en necesidad de dicho tratamiento. La estenosis puede ser reestenosis. El uso puede incluir administrar al sujeto una o más composiciones o compuestos descritos en el presente documento. En realizaciones, la una o más composiciones o compuestos se administran en una prótesis endovascular que eluye fármaco. El uso puede incluir administrar al sujeto una cantidad terapéuticamente eficaz de una o más composiciones o compuestos descritos en el presente documento (por ejemplo, un modulador de mTOR (por ejemplo, inhibidor) como se ha descrito anteriormente).
En realizaciones, la enfermedad es una enfermedad descrita en el presente documento y el compuesto es un compuesto descrito en el presente documento y la composición es una composición descrita en el presente documento.
Métodos de modulación de mTOR
En algunas realizaciones, los compuestos desvelados en el presente documento son inhibidores más selectivos de mTORC1 frente a mTORC2. En algunas realizaciones, los compuestos desvelados en el presente documento son inhibidores más selectivos de mTORC2 frente a mTORC1. En algunas realizaciones, los compuestos desvelados en el presente documento no presentan diferencia de selectividad entre mTORC1 y mTORC2.
En el presente documento también se desvela un método de modulación de la actividad de mTORC1 en un sujeto en necesidad del mismo, que incluye administrar al sujeto una cantidad eficaz de un compuesto como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo. El método puede incluir inhibir la actividad de mTORC1. El método puede incluir inhibir la actividad de mTORC1 y no inhibir la actividad de mTORC2.
El método puede incluir inhibir la actividad de mTORC1 más que inhibir la actividad de mTORC2. El método puede incluir inhibir la actividad de mTORC1 al menos 1,1 veces más que inhibir la actividad de mTORC2 (por ejemplo, al menos 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000, 10000, 20000, 30000, 40000, 50000, 60000, 70000, 80000, 90000, 100000, 100000, 200000, 300000, 400000, 500000, 600000, 700000, 800000, 900000 o 1000000 veces).
También se describe un método de modulación de la actividad de mTORC2 en un sujeto en necesidad del mismo, que incluye administrar al sujeto una cantidad eficaz de un compuesto como se describe en el presente documento, o una sal farmacéuticamente aceptable del mismo. El método puede incluir inhibir la actividad de mTORC2. El método puede incluir inhibir la actividad de mTORC2 y no inhibir la actividad de mTORC1.
El método puede incluir inhibir la actividad de mTORC2 más que inhibir la actividad de mTORC1. El método puede incluir inhibir la actividad de mTORC2 al menos 1,1 veces más que inhibir la actividad de mTORC1 (por ejemplo, al menos 1,1, 1,2, 1,3, 1,4, 1,5, 1,6, 1,7, 1,8, 1,9, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000, 10000, 20000, 30000, 40000, 50000, 60000, 70000, 80000, 90000, 100000, 100000, 200000, 300000, 400000, 500000, 600000, 700000, 800000, 900000 o 1000000 veces).
En algunas realizaciones, mTOR es en una célula. En algunas realizaciones, la célula es una célula de mamífero, tal como una célula humana. La célula se pueden aislar in vitro, formar parte de un tejido in vitro, o puede formar parte de un organismo.
Divulgaciones adicionales
En el presente documento también se desvelan Compuestos I-1 a I-59, Realizaciones I-60 a I-71, Compuestos II-1 a II-71 y Realizaciones II-72 a II-83, del siguiente modo:
Compuesto I-1. Un compuesto de la fórmula I:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O o -OR3;
R28 es -H o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde al menos uno de R28 y R40 no es H;
Z1 es O o S;
R28a y R40a son independientemente -A1-L1-A2-B; -A1-A2-B; --L2-A1-L1-A2-L3-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno;
A1 y A2 están independientemente ausentes o se seleccionan de


en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno; L1 se selecciona de

L2 y L3 están independientemente ausentes o se seleccionan de


B1 se selecciona de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

arileno (C6-C10)-,

NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,

y

NR3-(C(R3)2)n-S(O)2-arileno-C(O) -, en donde el enlace

en el lado izquierdo de B1, como está dibujado, está unido a A2, L3 o L1; y en donde el heteroarileno, heterociclileno y arileno se sustituyen opcionalmente con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo se sustituyen opcionalmente con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo; o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada Y es independientemente C(R3)2 o un enlace;
cada n es independientemente un número de uno a 12;
cada o es independientemente un número de cero a 30;
cada p es independientemente un número de cero a 12;
cada q es independientemente un número de cero a 30; y
cada r es independientemente un número de uno a 6.
Compuesto I-2. Un compuesto de la fórmula II:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O o -OR3;
R28 es -H o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde al menos uno de R28 y R40 no es H;
Z1 es O o S;
R28a y R40a son independientemente -A1-L1-A2-B; -A1-A2-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno;
A1 y A2 están independientemente ausentes o se seleccionan de

en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)-; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno; L1 se selecciona de


B1 se selecciona de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

arileno (C6-C10)-,

NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,

NR3-(C(R3)2)n-S(O)2-arileno-C(O) -, en donde el enlace
en el lado izquierdo de B1, como está dibujado, está unido a A2 o L1; y en donde el heteroarileno, heterociclileno y arileno se sustituyen opcionalmente con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo se sustituyen opcionalmente con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo; o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo se sustituye opcionalmente con -N(R3)2 o -OR3;
cada Y es independientemente C(R3)2 o un enlace;
cada n es independientemente un número de uno a 12;
cada o es independientemente un número de cero a 30;
cada p es independientemente un número de cero a 12;
cada q es independientemente un número de cero a 30; y
cada r es independientemente un número de uno a 6.
Compuesto I-3. Compuesto I-1 o I-2, en donde R32 es =O.
Compuesto I-4. Compuesto I-1 o I-2, en donde R32 es -OR3.
Compuesto I-5. Uno cualquiera de los Compuestos I-1 a I-4, en donde los compuestos se representan por la estructura de la fórmula I-40:

o una sal o tautómero farmacéuticamente aceptable del mismo.
Compuesto I-6. Compuesto I-5, en donde Z1 es O.
Compuesto I-7. Compuesto I-5, en donde Z1 es S.
Compuesto I-8. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -A1-L1-A2-B, en donde A1 y A2 están ausentes.
Compuesto I-9. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -A1-L1-A2-B, en donde A2 está ausente.
Compuesto I-10. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -A1-L1-A2-B, en donde A1 está ausente.
Compuesto I-11. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -A1-L1-A2-B.
Compuesto I-12. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -A1-A2-B.
Compuesto I-13. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes.
Compuesto I-14. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -L2-A1-L1-A2-L3-B, en donde L2 está ausente.
Compuesto I-15. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es --L2-A1-L1-A2-L3-B, en donde L3 está ausente.
Compuesto I-16. Uno cualquiera de los Compuestos I-5 a I-7, en donde R40a es -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno.
Compuesto I-17. Uno cualquiera de los Compuestos I-1 a I-4, en donde los compuestos se representan por la estructura de la fórmula I-28:

o una sal o tautómero farmacéuticamente aceptable del mismo.
Compuesto I-18. Compuesto I-17, en donde Z1 es O.
Compuesto I-19. Compuesto I-17, en donde Z1 es S.
Compuesto I-20. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -A1-L1-A2-B, en donde A1 y A2 están ausentes.
Compuesto I-21. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -A1-L1-A2-B, en donde A2 está ausente.
Compuesto I-22. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -A1-L1-A2-B, en donde A1 está ausente.
Compuesto I-23. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -A1-L1-A2-B.
Compuesto I-24. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -A1-A2-B.
Compuesto I-25. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes.
Compuesto 1-26. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -L2-A1-L1-A2-L3-B, en donde L2 está ausente.
Compuesto I-27. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es --L2-A1-L1-A2-L3-B, en donde L3 está ausente.
Compuesto I-28. Uno cualquiera de los Compuestos I-17 a I-19, en donde R28a es -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno.
Compuesto I-29. Uno cualquiera de los Compuestos I-1 a I-11, I-13 a I-15, I-17 a I-23, y I-25 a I-27, en donde L1 es

Compuesto I-30. Uno cualquiera de los Compuestos I-1 a I-11, I-13 a I-15, I-17 a I-23, y I-25 a I-27, en donde L1 es

Compuesto I-31. Uno cualquiera de los Compuestos I-1 a I-11, I-13 a I-15, I-17 a I-23, y I-25 a I-27, en donde L1 es

Compuesto I-32. Uno cualquiera de los Compuestos I-1 a I-11, I-13 a I-15, I-17 a I-23, y I-25 a I-27, en donde L1 es

Compuesto I-34. Uno cualquiera de los Compuestos I-1 a I-7, I-15, I-17 a I-19, y I-27, en donde L2 es

Compuesto I-35. Uno cualquiera de los Compuestos I-1 a I-7, I-13 a I-14, I-17 a I-19, y I-25 a I-26, en donde L3 es

Compuesto I-36. Uno cualquiera de los Compuestos I-1 a I-8, I-10, I-13, I-17 a I-19, I-20, I-22, I-25 y I-29 a 1-35, en donde A1 está ausente.
Compuesto I-37. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es

Compuesto I-38. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es

Compuesto I-39. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es

Compuesto I-40. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es

Compuesto I-41. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es

Compuesto I-42. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es

Compuesto I-43. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es.

Compuesto I-44. Uno cualquiera de los Compuestos I-1 a I-7, I-9, I-11 a I-12, I-14 a I-15, 1-17 a I-19, I-21, I-23 a I-24, I-26 a I-27, y I-29 a 1-35, en donde A1 es

Compuesto I-45. Uno cualquiera de los Compuestos I-1 a I-9, I-17 a I-21, y I-29 a I-44, en donde A2 está ausente.
Compuesto I-46. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es

Compuesto I-47. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es

Compuesto I-48. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es

Compuesto I-49. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es

Compuesto I-50. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es.

Compuesto I-51. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es

Compuesto I-52. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es.

Compuesto I-53. Uno cualquiera de los Compuestos I-1 a I-7, I-10 a I-15, I-17 a I-19, I-22 a I-27 y I-29 a I-44,en donde A2 es

Compuesto I-54. Uno cualquiera de los Compuestos I-1 a I-53, en donde B es

Compuesto I-55. Uno cualquiera de los Compuestos I-1 a I-53, en donde B es

Compuesto I-56. Uno cualquiera de los Compuestos I-1 a I-53, en donde B1 es

NR3-(C(R3)2)n-.
Compuesto I-57. Uno cualquiera de los Compuestos I-1 a I-53, en donde B1 es

Compuesto I-58. Uno cualquiera de los Compuestos I-1 a I-57, en donde R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN o -C(O)NR3-heteroarilo.
Compuesto I-59. Uno cualquiera de los Compuestos I-1 a I-58, o una sal o tautómero farmacéuticamente aceptable del mismo, en donde el compuesto tiene la siguiente fórmula:

Realización I-60. Un compuesto seleccionado del grupo que consiste en:































o una sal farmacéuticamente aceptable o isómero del mismo.
Realización I-61. Una composición farmacéutica que comprende un compuesto de la Realización I-60, o una sal farmacéuticamente aceptable del mismo, y al menos uno de un vehículo, diluyente o excipiente farmacéuticamente aceptable.
Realización I-62. Un compuesto de la Realización I-60, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento, prevención o reducción del riesgo de una enfermedad o afección mediada por mTOR.
Realización I-63. El compuesto para el uso de una cualquiera de la Realización I-62, en donde la enfermedad es cáncer o una enfermedad mediada por inmunidad.
Realización I-64. El compuesto para el uso de la Realización I-63, en donde el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos.
Realización I-65. El compuesto para el uso de la Realización I-63, en donde la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis.
Realización I-66. Un compuesto de la Realización I-60, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de cáncer.
Realización I-67. El compuesto para el uso de la Realización I-66, en donde el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos.
Realización I-68. Un compuesto de la Realización I-60, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una enfermedad mediada por inmunidad.
Realización I-69. El compuesto para el uso de la Realización I-68, en donde la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis.
Realización I-70. Un compuesto de la Realización I-60, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una afección senil.
Realización I-71. El compuesto para el uso de la Realización I-70, en donde la afección senil se selecciona de sarcopenia, atrofia de la piel, atrofia muscular progresiva, atrofia cerebral, aterosclerosis, arteriosclerosis, enfisema pulmonar, osteoporosis, osteoartritis, hipertensión arterial, disfunción eréctil, demencia, enfermedad de Huntington, enfermedad de Alzheimer, cataratas, degeneración macular senil, cáncer de próstata, accidente cerebrovascular, reducida esperanza de vida, deterioro de la función renal y sordera parcial senil, incapacidad de movilidad relacionada con el envejecimiento (por ejemplo, fragilidad), deterioro cognitivo, demencia senil, deterioro de la memoria, rigidez de los tendones, disfunción cardíaca, tal como hipertrofia cardíaca y disfunción sistólica y diastólica, inmunosenescencia, cáncer, obesidad y diabetes.
Compuesto II-1. Un compuesto de la fórmula Ic:

o una sal o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O, -OR3, -N3 o -O-C(=Z1)-R32a;
R28 es -H, alquilo (C1-C6) o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde cuando R28 y R40 son H, entonces R32 no es =O;
cada Z1 es independientemente O o S;
R28a, R32a y R40a son independientemente -A1-L1-A2-B; -A1-A2-B; -L2-A1-L1-A2-L3-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno;
A1 y A2 están independientemente ausentes o se seleccionan de

en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno;
cada L1 se selecciona independientemente de

L2 y L3 están independientemente ausentes o se seleccionan de

cada B se selecciona independientemente de


cada B1 se selecciona independientemente de

NR3-(C(R3)2)n-,

NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,

NR3-(C(R3)2)n-heteroarileno-,

NR3-(C(R3)2)n-heteroarileno-(C(R3)2)n-,

arileno (C6-C10)-, -NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,

heteroarileno-heterociclileno-arileno (C6-C10)-,


NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, y

NR3-(C(R3)2)n-S(O)2-arileno-(C(R3)2)n -, en donde el enlace

en el lado izquierdo de B1, como está dibujado, está unido a A2, L3 o L1; y en donde el heteroarileno, heterociclileno y arileno están cada uno independientemente opcionalmente sustituido con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo se sustituyen opcionalmente con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada Y es independientemente C(R3)2 o un enlace;
cada n es independientemente un número entero de uno a 12;
cada o es independientemente un número entero de cero a 30;
cada p es independientemente un número entero de cero a 12;
cada q es independientemente un número entero de cero a 30; y
cada r es independientemente un número entero de uno a 6.
Compuesto II-1A. Un compuesto de la fórmula Ia:

o tautómero farmacéuticamente aceptable del mismo, en donde:
R32 es -H, =O, -OR3, -N3 o -O-C(=Z1)-R32a;
R28 es -H, alquilo (C1-C6) o -C(=Z1)-R28a;
R40 es -H o -C(=Z1)-R40a;
en donde cuando R28 y R40 son H, entonces R32 no es =O;
cada Z1 es independientemente O o S;
R28a, R32a y R40a son independientemente -A1-L1-A2-B; -A1-A2-B; -L2-A1-L1-A2-L3-B; -O-alquilo (C1-C6); o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno;
A1 y A2 están independientemente ausentes o se seleccionan de

en donde el enlace en el lado izquierdo de A1, como está dibujado, está unido a -C(=Z1)- o L2; y en donde el enlace en el lado derecho del resto A2, como está dibujado, está unido a B o L3;
cada Q es independientemente 1 a 3 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada X1 es independientemente un anillo de heteroarileno o heterociclileno;
cada W está independientemente ausente o es 1 a 2 anillos seleccionados de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada W1 es independientemente un anillo de heteroarileno o heterociclileno;
cada G está independientemente ausente o es un anillo seleccionado de arileno, cicloalquileno, heteroarileno y heterociclileno;
cada G1 y G2 son independientemente un anillo de heteroarileno o heterociclileno;
cada L1 se selecciona independientemente de

L2 y L3 están independientemente ausentes o se seleccionan de
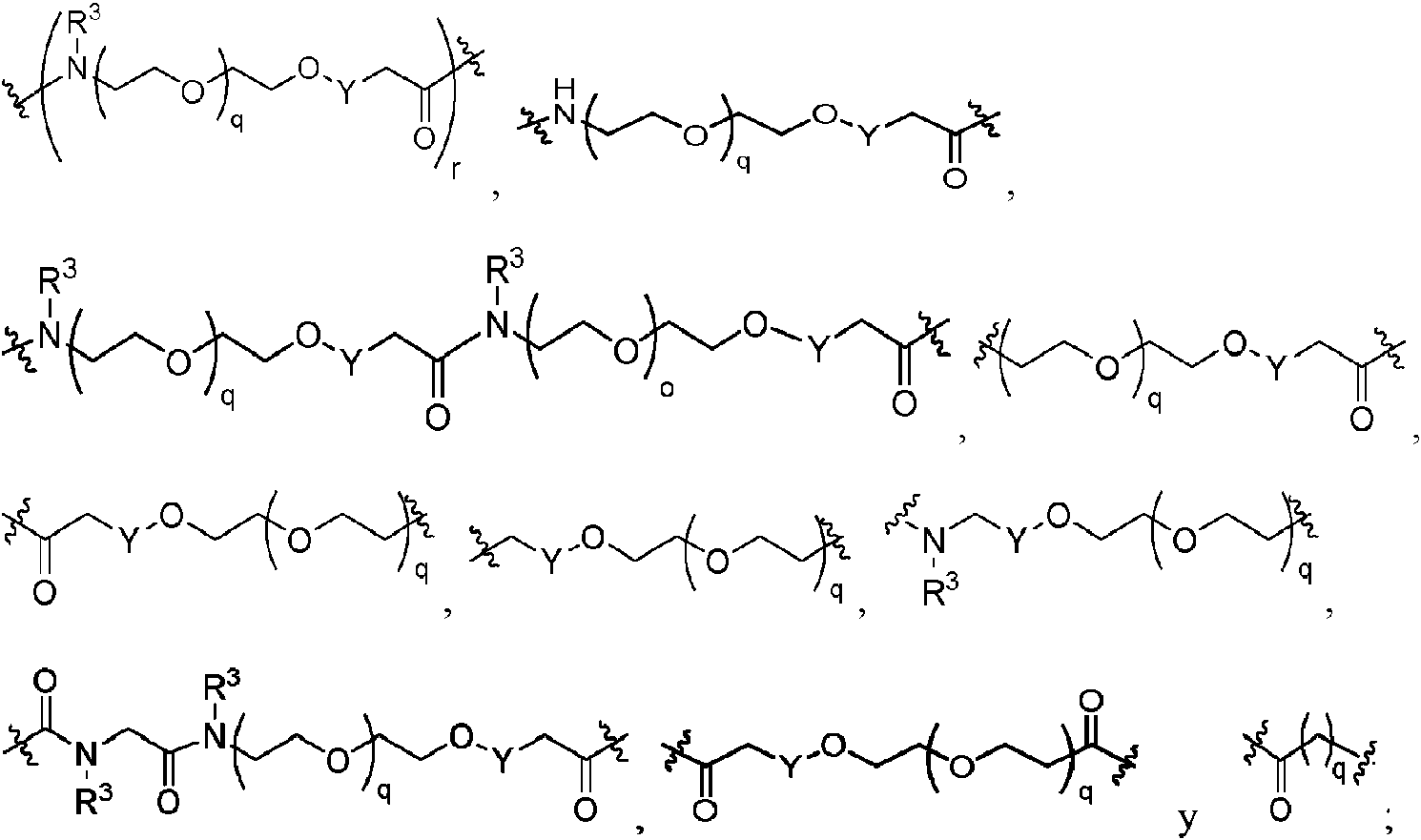
cada B se selecciona independientemente de


B1 se selecciona de
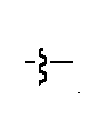
NR3-(C(R3)2)n-,
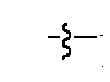
NR3-(C(R3)2)n-arileno (C6-C10)-(C(R3)2)n-,
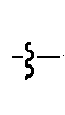
-NR3-(C(R3)2)n-heteroarileno-,
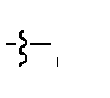
-arileno (C6-C10)-,
NR3-(C(R3)2)n-NR3C(O)-,

NR3-(C(R3)2)n-heteroarileno-heterociclileno-arileno (C6-C10)-,
heteroarileno-heterociclileno-arileno (C6-C10)-,
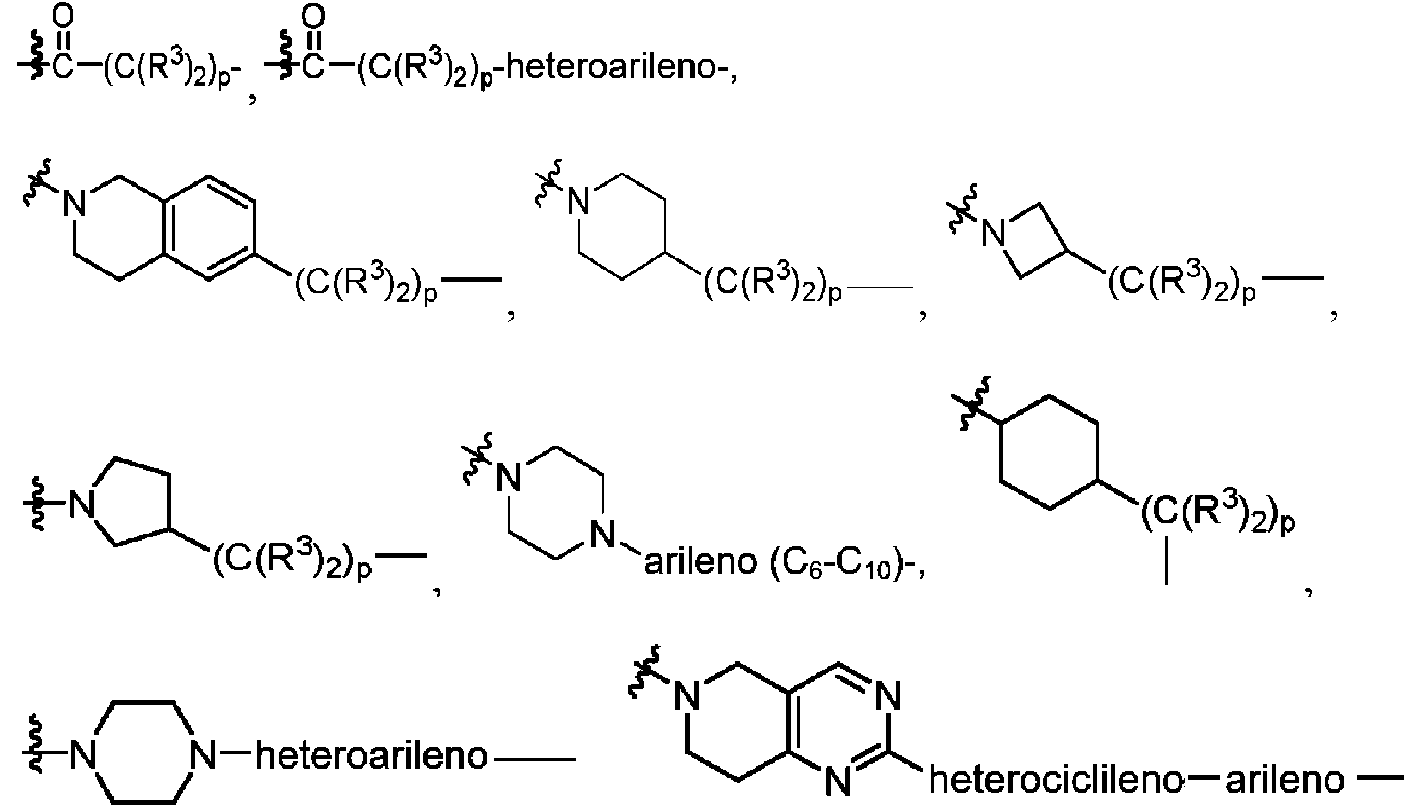
y
NR3-(C(R3)2)n-S(O)2-arileno-C(O)-, en donde el enlace
en el lado izquierdo de B1, como está dibujado, está unido a A2, L3 o L1; y en donde el heteroarileno, heterociclileno y arileno se sustituyen opcionalmente con alquilo, hidroxialquilo, haloalquilo, alcoxi, halógeno o hidroxilo;
cada R3 es independientemente H o alquilo (C1-C6);
cada R4 es independientemente H, alquilo (C1-C6), halógeno, heteroarilo de 5-12 miembros, heterociclilo de 5-12 miembros, arilo (C6-C10), en donde el heteroarilo, heterociclilo y arilo se sustituyen opcionalmente con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN, -C(O)NR3-heteroarilo o -C(O)NR3-heterociclilo;
cada R5 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R6 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R7 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada R8 es independientemente H, alquilo (C1-C6), -C(O)OR3 o -N(R3)2, en donde el alquilo de alquilo (C1-C6) se sustituye opcionalmente con -N(R3)2 o -OR3;
cada Y es independientemente C(R3)2 o un enlace;
cada n es independientemente un número entero de uno a 12;
cada o es independientemente un número entero de cero a 30;
cada p es independientemente un número entero de cero a 12;
cada q es independientemente un número entero de cero a 30; y
cada r es independientemente un número entero de uno a 6.
Compuesto II-2. Compuesto II-1, en donde R32 es =O.
Compuesto II-3. Compuesto II-1, en donde R32 es -OR3.
Compuesto II-4. Uno cualquiera de los Compuestos II-1 a II-3, o una sal o tautómero farmacéuticamente aceptable del mismo, en donde el compuesto se representa por la estructura de la fórmula (I-40b):

en donde R40 es -C(=Z1)-R40a.
Compuesto II-5. Compuesto II-4, en donde Z1 es O.
Compuesto II-6. Compuesto II-4, en donde Z1 es S.
Compuesto II-7. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -A1-L1-A2-B, en donde A1 y A2 están ausentes.
Compuesto II-8. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -A1-L1-A2-B, en donde A2 está ausente. Compuesto II-9. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -A1-L1-A2-B, en donde A1 está ausente. Compuesto II-10. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -A1-L1-A2-B.
Compuesto II-11. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -A1-A2-B.
Compuesto II-12. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes.
Compuesto II-13. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -L2-A1-L1-A2-L3-B, en donde L2 está ausente.
Compuesto II-14. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -L2-A1-L1-A2-L3-B, en donde L3 está ausente.
Compuesto II-15. Uno cualquiera de los Compuestos II-4 a II-6, en donde R40a es -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno.
Compuesto II-16. Uno cualquiera de los Compuestos II-1 a II-3, o una sal o tautómero farmacéuticamente aceptable del mismo, en donde los compuestos se representan por la estructura de la fórmula (I-28b):

en donde R28 es -C(=Z1)-R28a.
Compuesto II-17. Compuesto II-16, en donde Z1 es O.
Compuesto II-18. Compuesto II-16, en donde Z1 es S.
Compuesto II-19. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -A1-L1-A2-B, en donde A1 y A2 están ausentes.
Compuesto II-20. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -A1-L1-A2-B, en donde A2 está ausente.
Compuesto II-21. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -A1-L1-A2-B, en donde A1 está ausente.
Compuesto II-22. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -A1-L1-A2-B.
Compuesto II-23. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -A1-A2-B.
Compuesto II-24. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes.
Compuesto II-25. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -L2-A1-L1-A2-L3-B, en donde L2 está ausente.
Compuesto II-26. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -L2-A1-L1-A2-L3-B, en donde L3 está ausente.
Compuesto II-27. Uno cualquiera de los Compuestos II-16 a II-18, en donde R28a es -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno.
Compuesto II-28. Compuesto II-1, o una sal o tautómero farmacéuticamente aceptable del mismo, en donde el compuesto se representa por la estructura de la fórmula (I-32b):

en donde R32 es -O-C(=Z1)-R32a.
Compuesto II-29. Compuesto II-28, en donde Z1 es O.
Compuesto II-30. Compuesto II-28, en donde Z1 es S.
Compuesto II-31. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -A1-L1-A2-B, en donde A1 y A2 están ausentes.
Compuesto II-32. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -A1-L1-A2-B, en donde A2 está ausente.
Compuesto II-33. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -A1-L1-A2-B, en donde A1 está ausente.
Compuesto II-34. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -A1-L1-A2-B.
Compuesto II-35. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -A1-A2-B.
Compuesto II-36. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -L2-A1-L1-A2-L3-B, en donde L2 y A1 están ausentes.
Compuesto II-37. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -L2-A1-L1-A2-L3-B, en donde L2 está ausente.
Compuesto II-38. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -L2-A1-L1-A2-L3-B, en donde L3 está ausente.
Compuesto II-39. Uno cualquiera de los Compuestos II-28 a II-30, en donde R32a es -O-alquilo (C1-C6) o -O-arilo (C6-C10); en donde el arilo de -O-arilo (C6-C10) está sin sustituir o sustituido con 1-5 sustituyentes seleccionados de -NO2 y halógeno.
Compuesto II-40. Uno cualquiera de los Compuestos II-1 a II-10, II-12 a II-22, II-24 a II-35, y II-36 a II-39, en donde L1 es

Compuesto II-41. Uno cualquiera de los Compuestos II-1 a II-10, II-12 a II-22, II-24 a II-35, y II-36 a II-39, en donde L1 es

Compuesto II-42. Uno cualquiera de los Compuestos II-1 a II-10, II-12 a II-22, 24-35, y 36-39, en donde L1 es

Compuesto II-43. Uno cualquiera de los Compuestos II-1 a II-10, II-12 a II-22, II-24 a II-35, y II-36 a II-39, en donde L1 es

Compuesto II-44. Uno cualquiera de los Compuestos II- 1 a II-10, II-12 a II-22, II-24 a II-35, y II-36 a II-39, en donde L1 es

Compuesto II-45. Uno cualquiera de los Compuestos II-1 a II-10, II-12 II-22, 11-24 a II-35, y II-36 a II-39, en donde L1 es

Compuesto II-46. Uno cualquiera de los Compuestos II-1 a II-6, II-12 a II-18, II-24 a II-30, y II-36 a II-45, en donde L2 es

Compuesto II-47. Uno cualquiera de los Compuestos II-1 a II-6, II-12 a II-18, II-24 a II-30, y II-36 a II-45, en donde L3 es

Compuesto II-48. Uno cualquiera de los Compuestos II-1 a II-7, II-9, II-12, II-16 a II-19, II-21, II-24, II-28 a II-31, II-33, II-36, y II-39 a II-45, en donde A1 está ausente.
Compuesto II-49. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 a II-35, y II-37 a II-45, en donde A1 es

Compuesto II-50. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 t o II-35, y II-37 a II-45, en donde A1 es

Compuesto II-51. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 a II-35, y II-37 a II-45, en donde A1 es

Compuesto II-52. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 a II-35, y II-37 a II-45, en donde A1 es

Compuesto II-53. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 a II-35, y II-37 a II-45, en donde A1 es

Compuesto II-54. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 a II-35, y II-37 a II-45, en donde A1 es

Compuesto II-55. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 a II-35, y II-37 a II-45, en donde A1 es.

Compuesto II-56. Uno cualquiera de los Compuestos II-1 a II-6, II-8, II-10 a II-11, II-13 a II-18, II-20, II-22 a II-23, II-25 a II-30, II-32, II-34 a II-35, y II-37 a II-45, en donde A1 es

Compuesto II-57. Uno cualquiera de los Compuestos II-1 a II-8, II-15 a II-20, II-27 a II-32, y II-39 a II-45, en donde A2 está ausente.
Compuesto II-58. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a II-45, en donde A2 es

Compuesto II-59. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a II-45, en donde A2 es

Compuesto II-60. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a II-45, en donde A2 es

Compuesto II-61. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a II-45, en donde A2 es

Compuesto II-62. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a Ii-45, en donde A2 es.

Compuesto II-63. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a II-45, en donde A2 es

Compuesto II-64. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a II-45, en donde A2 es.

Compuesto II-65. Uno cualquiera de los Compuestos II-1 a II-6, II-9 a II-18, II-21 a II-30, y II-33 a II-45, en donde A2 es

Compuesto II-66. Uno cualquiera de los Compuestos II-1 a II-65, en donde B es

Compuesto II-67. Uno cualquiera de los Compuestos II-1 a II-65, en donde B es

Compuesto II-68. Uno cualquiera de los Compuestos II-1 a II-65, en donde B1 es

NR3-(C(R3)2)n-.
Compuesto II-69. Uno cualquiera de los Compuestos II-1 a II-65, en donde B1 es

Compuesto II-70. Uno cualquiera de los Compuestos II-1 a II-69, en donde R4 es heteroarilo de 5-12 miembros, opcionalmente sustituido con -N(R3)2, -OR3, halógeno, alquilo (C1-C6), -alquileno (C1-C6)-heteroarilo, -alquileno (C1-C6)-CN o -C(O)NR3-heteroarilo.
Compuesto II-71. Uno cualquiera de los Compuestos II-1 a II-70, o una sal o tautómero farmacéuticamente aceptable del mismo, en donde el compuesto tiene la siguiente fórmula:

Realización II-72. Un compuesto seleccionado del grupo que consiste en:


































o una sal o tautómero farmacéuticamente aceptable del mismo.
Realización II-73. Una composición farmacéutica que comprende un compuesto de la Realización II-72, o una sal farmacéuticamente aceptable del mismo, y al menos uno de un vehículo, diluyente o excipiente farmacéuticamente aceptable.
Realización II-74. Un compuesto de la Realización II-72, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento, prevención o reducción del riesgo de una enfermedad o afección mediada por mTOR.
Realización II-75. El compuesto para el uso de la Realización II-74, en donde la enfermedad es cáncer o una enfermedad mediada por inmunidad.
Realización II-76. El compuesto para el uso de la Realización II-75, en donde el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos.
Realización II-77. El compuesto para el uso de la Realización II-75, en donde la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis.
Realización II-78. Un compuesto de la Realización II-72, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una enfermedad mediada por inmunidad .
Realización II-79. El compuesto para el uso de la Realización II-78, en donde el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos.
Realización II-80. Un compuesto de la Realización II-72, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una enfermedad mediada por inmunidad.
Realización II-81. El compuesto para el uso de la Realización II-80, en donde la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis.
Realización II-82. Un compuesto de la Realización II-72, o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una afección senil.
Realización II-83. El compuesto para el uso de la Realización II-82, en donde la afección senil se selecciona de sarcopenia, atrofia de la piel, atrofia muscular progresiva, atrofia cerebral, aterosclerosis, arteriosclerosis, enfisema pulmonar, osteoporosis, osteoartritis, hipertensión arterial, disfunción eréctil, demencia, enfermedad de Huntington, enfermedad de Alzheimer, cataratas, degeneración macular senil, cáncer de próstata, accidente cerebrovascular, reducida esperanza de vida, deterioro de la función renal y sordera parcial senil, incapacidad de movilidad relacionada con el envejecimiento (por ejemplo, fragilidad), deterioro cognitivo, demencia senil, deterioro de la memoria, rigidez de los tendones, disfunción cardíaca, tal como hipertrofia cardíaca y disfunción sistólica y diastólica, inmunosenescencia, cáncer, obesidad y diabetes.
Ejemplos
La divulgación se ilustra además por los siguientes ejemplos y ejemplos de síntesis, que no se deben interpretar como limitantes del alcance o espíritu de la presente divulgación a los procedimientos específicos descritos en el presente documento. Se debe entender que los ejemplos se proporcionan para ilustrar ciertas realizaciones y que no pretenden limitar el alcance de la divulgación. Se debe entender además que se puede recurrir a diversas otras realizaciones, modificaciones y equivalentes de las mismas que puedan ser sugeridas ellas mismas a los expertos en la técnica sin apartarse del espíritu de la presente divulgación y/o alcance de las reivindicaciones adjuntas.
Las definiciones usadas en los siguientes ejemplos y en cualquier parte en el presente documento son:
CH2Cl2, DCM
Cloruro de metileno, diclorometano
CH3CN, MeCN
Acetonitrilo
DIPEA
Diisopropiletilamina o base de Hunig
DMA
Dimetilacetamida
DME
Dimetoxietano
DMF
N,N-Dimetilformamida
EDCI
1-Etil-3-(3-dimetilaminopropil)carbodiimida
EtOAc
Acetato de etilo
h
hora
H2O
Agua
HCl
Ácido clorhídrico
HOBt
Hidroxibenzotriazol
HPLC
Cromatografía de líquidos de alta resolución
LCMS
Cromatografía de líquidos-espectrometría de masas
MeOH
Metanol
MTBE
Metil terc-butil éter
Na2SO4
Sulfato de sodio
PEG
Polietilenglicol
TBDMS
terc-butildimetilsililo
TFA
Ácido trifluoroacético
THF
Tetrahidrofurano
TMS
Tetrametilsilano
Rapálogos bifuncionales de la serie 1
Una estructura general de los rapálogos bifuncionales de la serie 1 se muestra en el Esquema 1 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7 y r = 1 a 6. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 2
Una estructura general de los rapálogos bifuncionales de la serie 2 se muestra en el Esquema 2 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina preconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 3
Una estructura general de los rapálogos bifuncionales de la serie 3 se muestra en el Esquema 3 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina posconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 4
Una estructura general de los rapálogos bifuncionales de la serie 4 se muestra en el Esquema 4 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. Las aminas pre- y posconectoras pueden cada una incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 5
Una estructura general de los rapálogos bifuncionales de la serie 5 se muestra en el Esquema 5 que sigue. Para estos tipos de rapálogos bifuncionales, la amina preconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 6
Una estructura general de los rapálogos bifuncionales de la serie 6 se muestra en el Esquema 6 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. Las aminas conectoras pueden incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina posconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 7
Una estructura general de los rapálogos bifuncionales de la serie 7 se muestra en el Esquema 7 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. Las aminas pre- y posconectoras pueden cada una incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Rapálogos bifuncionales de la serie 8
Una estructura general de los rapálogos bifuncionales de la serie 8 se muestra en el Esquema 8 que sigue. Para estos tipos de rapálogos bifuncionales, el conector puede incluir variaciones donde q = 0 a 30, tal como q = 1 a 7. La amina conectora puede incluir sustituciones, tales como R = H y grupos alquilo C1-C6. La amina posconectora puede incluir sustituciones, tales como R2 = H, grupos alquilo C1-C6 y cicloalquilo que incluye anillos de 4 a 8 miembros. El resto carbamato, donde Z1 = O o S, se puede unir al rapálogo en R40 o R28 (fórmula I y II), que incluye variaciones que se
encuentran en la Tabla 1 en la sección de Ejemplos. Un inhibidor de sitio activo de mTOR se puede unir al conector por una amina primaria o secundaria, y puede incluir variaciones que se encuentran en la Tabla 2 en la sección de Ejemplos.

Tabla 1. Monómeros de rapálogos que contienen carbonato y tiocarbonato.




Tabla 2. Monómeros de inhibidor de sitio activo.







Tabla 3. Monómeros de inhibidor de sitio activo


Tabla 4. Pre- y posconectores que contienen amina.







Preparación de monómero de inhibidor de sitio activo
Monómero A. Sal de ácido trifluoroacético de 5-(4-amino-1-(4-(aminometil)bencil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 4-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)bencilcarbamato de terc-butilo A una disolución de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (3,8 g, 14,56 mmoles, 1,0 equiv) en DMF (20 ml) se añadió NaH (582,27 mg, 14,56 mmoles, 60 % en peso, 1,0 equiv) a 0 °C y la disolución de reacción se agitó a esta temperatura durante 30 min, entonces se añadió 4-(bromometil)bencilcarbamato de terc-butilo (4,59 g, 15,29 mmoles, 1,05 equiv) a la reacción a 0 °C y la disolución de reacción se agitó a temperatura ambiente durante 2 h. La disolución se vertió en H2O (80 ml) y se filtró el sólido que precipitó. La torta sólida se lavó con H2O (2 × 10 ml) y luego se secó a presión reducida dando 4-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)bencilcarbamato de terc-butilo (5 g, 53 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M Na] calculado para C18H21IN6O2: 503,07; observado 503,2.
Etapa 2: Síntesis de 4-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)bencilcarbamato de terc-butilo
A una suspensión bifásica de 4-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)bencilcarbamato de terc-butilo (5 g, 7,68 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (2,40 g, 9,22 mmoles, 1,2 equiv) y Pd(PPh3)4 (887,66 mg, 768,16 µmoles, 0,1 equiv) en DME (100 ml) y H2O (50 ml) se añadió Na2CO3 (1,91 g, 23,04 mmoles, 3,0 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. La mezcla de reacción se enfrió hasta temperatura ambiente y se filtró, el filtrado se extrajo con EtOAc (3 × 50 ml). Las fases orgánicas se combinaron y se lavaron con salmuera (10 ml), se secaron sobre Na2SO4, se filtraron y el filtrado se concentró a presión reducida dando un residuo. El residuo se purificó por cromatografía en gel de sílice (0→20 % de MeOH/EtOAc) dando 4-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)bencilcarbamato de terc-butilo (4,5 g, 82 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C25H26N8O3: 487,22; observado 487,2.
Etapa 3: Síntesis de 5-(4-amino-1-(4-(aminometil)bencil)-1H-pirazolo[3,4-d] pirimidin-3-il)benzo[d]oxazol-2-amina
A una disolución de 4-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)bencilcarbamato de terc-butilo (4,5 g, 6,29 mmoles, 1,0 equiv) en DCM (50 ml) se añadió TFA (30,80 g, 270,12 mmoles, 20 ml, 42,95 equiv) a 0 °C. La disolución de reacción se agitó a temperatura ambiente durante 2 h. La disolución de reacción se concentró a presión reducida dando un residuo, que se disolvió en 10 ml de MeCN, luego se vertió en MTBE (100 ml). Entonces se filtró el sólido que precipitó y la torta sólida se secó a presión reducida dando 5-[4-amino-1-[[4-(aminometil)fenil]metil]pirazolo[3,4-d]pirimidin-3-il]-1,3-benzoxazol-2-amina (2,22 g, 71 % de rendimiento, TFA) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C20H18N8O: 387,16; observado 387,1.
Monómero B. Sal de ácido trifluoroacético de 2-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-6-ol.

Etapa 1: Síntesis de N-(4-{4-amino-3-[6-(benciloxi)-1H-indol-2-il]-1H-pirazolo[3,4-d]pirimidin-1-il}butil)carbamato de terc-butilo
A una mezcla de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (300 mg, 694 µmoles, 1,0 equiv) y ácido (6-(benciloxi)-1-(terc-butoxicarbonil)-1H-indol-2-il)borónico (763 mg, 2,08 mmoles, 3,0 equiv) en DMF (2,6 ml), EtOH (525 µl) y H2O (350 µl) se añadieron Pd(OAc)2 (15,5 mg, 69 µmoles, 0,1 equiv), trifenilfosfina (36,1 mg, 138 µmoles, 0,2 equiv) y carbonato sódico (440 mg, 4,16 mmoles, 6,0 equiv). La reacción se calentó a 80 °C durante 20 h, se enfrió hasta temperatura ambiente, y se enfrió rápidamente con H2O (10 ml) y EtOAc (10 ml). La mezcla se transfirió a un embudo de decantación y la fase acuosa se extrajo con EtOAc (3 × 20 ml). La fase orgánica combinada se lavó con NaCl ac. sat. (1 × 20 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El material en bruto se purificó por cromatografía en gel de sílice (20→85 % de EtOAc/heptano) proporcionando el producto (201 mg, 46 % de rendimiento) como un sólido naranja. LCMS (ESI) m/z: [M H] calculado para C29H33N7O3: 528,27; observado 528,2.
Etapa 2: Síntesis de (4-(4-amino-3-(6-hidroxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo
A una disolución de N-(4-{4-amino-3-[6-(benciloxi)-1H-indol-2-il]-1H-pirazolo[3,4-d]pirimidin-1-il}butil)carbamato de terc-butilo (1,0 equiv) en EtOH se añade Pd/C (10 % en moles). La reacción se purga con H2 y se deja que la reacción se agite bajo una atmósfera de H2 hasta que se consuma el material de partida, como se ha determinado por LCMS. La reacción se diluye entonces con EtOAc, se filtra sobre Celite y se concentra a presión reducida. El residuo resultante se purifica por cromatografía en gel de sílice proporcionando el producto deseado.
Etapa 3: Síntesis de 2-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-6-ol
A una disolución de (4-(4-amino-3-(6-hidroxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (1,0 equiv) en DCM anhidro se añade TFA (50 equiv.) gota a gota a 0 °C. La reacción se agita a 0 °C y se calienta hasta temperatura ambiente. Una vez la reacción está completa, como se ha determinado por LCMS, la reacción se concentra a presión reducida. El residuo se tritura con MeCN, luego se añade gota a gota en MTBE durante 10 min. El sobrenadante se retira y el precipitado se recoge por filtración bajo N2 dando 2-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-6-ol.
Monómero C. Sal de ácido trifluoroacético de 5-(4-amino-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 6-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una suspensión de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (5 g, 19,16 mmoles, 1,0 equiv) en DMF (50,0 ml) se añadió NaH (766,22 mg, 19,16 mmoles, 60 % en peso, 1,0 equiv) a 4 °C. La mezcla se agitó a 4 °C durante 30 min. A la mezcla de reacción se añadió 6-(bromometil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (6,87 g, 21,07 mmoles, 1,1 equiv) en DMF (30 ml) a 4 °C. La mezcla se agitó a temperatura ambiente durante 2 h. La mezcla se enfrió entonces hasta 4 °C y se añadió H2O (400 ml) y la mezcla se agitó durante 30 min. El precipitado resultante se recogió por filtración dando 6-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (9,7 g, 76 % de rendimiento) en bruto como un sólido amarillo claro. El producto en bruto se usó para la siguiente etapa directamente.
Etapa 2: Síntesis de 6-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una suspensión bifásica de 6-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (9,7 g, 14,63 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (4,57 g, 17,55 mmoles, 1,2 equiv) y Na2CO3 (7,75 g, 73,14 mmoles, 5,0 equiv) en DME (120,0 ml) y H2O (60 ml) se añadió Pd(PPh3)4 (1,69 g, 1,46 mmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. La mezcla de reacción se enfrió entonces hasta temperatura ambiente y se repartió entre EtOAc (100 ml) y H2O (100 ml). La fase acuosa se separó y se extrajo con EtOAc (2 × 60 ml). Las fases orgánicas se combinaron, se lavaron con salmuera (80 ml) y se secaron sobre Na2SO4 anhidro, se filtraron y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1→100 % de EtOAc/éter de petróleo, luego 20-50 % de MeOH/EtOAc) proporcionando 6-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (4,5 g, 58 % de rendimiento) como un sólido amarillo claro.
Etapa 3: Síntesis de 5-(4-amino-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]piramidin-3-il)benzo[d]oxazol-2-amina
A TFA puro (32,5 ml, 438,97 mmoles, 50,0 equiv) se añadió 6-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (4,5 g, 8,78 mmoles, 1,0 equiv) a temperatura ambiente. La mezcla se agitó durante 30 min y luego se concentró a presión reducida. El residuo aceitoso se trituró con MeCN (8 ml), luego se añadió gota a gota en MTBE (350 ml) durante 10 min. El sobrenadante se retiró y entonces el precipitado se recogió por filtración bajo N2 dando 5-(4-amino-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (5,72 g, rendimiento superior al 100 %, TFA) como un sólido rosa claro. LCMS (ESI) m/z: [M H] calculado para C22H20N8O: 413,18; observado 413,2.
Monómero D. Sal de ácido trifluoroacético de 2-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-7-ol.

Etapa 1: Síntesis de 2-(4-amino-1-(4-((terc-butoxicarbonil)amino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)-7-metoxi-1H-indol-1-carboxilato de terc-butilo
A una mezcla de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (1,0 equiv) y ácido (1-(terc-butoxicarbonil)-7-metoxi-1H-indol-2-il)borónico (3,0 equiv) en DME y H2O se añade Pd(PPh3)4 (0,1 equiv) y carbonato sódico (6,0 equiv). La reacción se calienta a 80 °C hasta que se completa, como se ha determinado por LCMS y análisis por CCF. La reacción se inactiva entonces con H2O y EtOAc. La mezcla se transfiere a un embudo de decantación y la fase acuosa se extrae con EtOAc. La fase orgánica se lava con NaCl ac. sat., se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El producto deseado se aísla después de la cromatografía sobre gel de sílice.
Etapa 2: Síntesis de 2-(4-amino-1-(4-((terc-butoxicarbonil)amino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)-7-hidroxi-1H-indol-1-carboxilato de terc-butilo
A una disolución de 2-(4-amino-1-(4-((terc-butoxicarbonil)amino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)-7-metoxi-1H-indol-1-carboxilato de terc-butilo (1,0 equiv) en DCM a -10 °C se añade BBr3 (2,0 equiv). La reacción se deja con agitación hasta que se consume el material de partida, como se ha determinado por LCMS. La reacción se inactiva por adición lenta de NaHCO3 ac. sat., se transfiere a un embudo de decantación y la mezcla se extrae con DCM. La
fase orgánica se lava con NaCl ac. sat., se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El producto deseado se aísla después de la cromatografía sobre gel de sílice.
Etapa 3: Síntesis de 2-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-7-ol
A una disolución de 2-(4-amino-1-(4-((terc-butoxicarbonil)amino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)-7-hidroxi-1H-indol-1-carboxilato de terc-butilo (1,0 equiv) en DCM a 0 °C se añade TFA gota a gota. La reacción se agita a 0 °C y se calienta hasta temperatura ambiente. Una vez la reacción está completa, como se ha determinado por LCMS, la reacción se concentra a presión reducida. El residuo se tritura con MeCN, luego se añade gota a gota en MTBE durante 10 min. El sobrenadante se retira y el precipitado se recoge por filtración bajo N2 dando 2-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-7-ol.
Monómero E. Sal de ácido trifluoroacético de 5-(4-amino-1-(piperidin-4-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 4-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)piperidin-1-carboxilato de terc-butilo
A una disolución de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (3 g, 11,49 mmoles, 1,0 equiv) en DMA (30 ml) se añadió 4-(bromometil)piperidin-1-carboxilato de terc-butilo (3,36 g, 12,07 mmoles, 1,05 equiv) y K2CO3 (4,77 g, 34,48 mmoles, 3,0 equiv), entonces la reacción se agitó a 80 °C durante 3 h. La mezcla de reacción se filtró para retirar K2CO3 y el filtrado se vertió en H2O (200 ml). Entonces se filtró un sólido precipitado dando 4-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)piperidin-1-carboxilato de terc-butilo (3 g, 57 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C16H23IN6O2: 459,10; observado 459,1.
Etapa 2: Síntesis de 4-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)piperidin-1-carboxilato de terc-butilo
A una suspensión bifásica de 4-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)piperidin-1-carboxilato de tercbutilo (3 g, 6,55 mmoles, 1,0 equiv) y 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (2,04 g, 7,86 mmoles, 1,2 equiv) y Na2CO3 (3,47 g, 32,73 mmoles, 5,0 equiv) en DME (60 ml) y H2O (30 ml) se añadió Pd(PPh3)4 (756,43 mg, 654,60 µmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h y los dos lotes se combinaron juntos. La mezcla de reacción se enfrió y se repartió entre EtOAc (500 ml) y H2O (500 ml). La fase acuosa se separó y se extrajo con EtOAc (3 × 300 ml). Todos las fases orgánicas se combinaron, se lavaron con salmuera (20 ml), se secaron sobre Na2SO4 anhidro, se filtraron y el filtrado se concentró a presión reducida dando 4-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)piperidin-1-carboxilato de terc-butilo (4,5 g, 74 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C23H28N8O3: 465,24; observado 465,2.
Etapa 3: Síntesis de 5-(4-amino-1-(piperidin-4-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina Se agitó una disolución de 4-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)piperidin-1-carboxilato de terc-butilo (2,5 g, 5,38 mmoles, 1,0 equiv) en TFA (25 ml) a temperatura ambiente durante 30 min. La disolución de reacción se concentró a presión reducida para retirar el TFA. El residuo se añadió a MTBE (400 ml) y un sólido precipitado, que entonces se filtró dando 5-(4-amino-1-(piperidin-4-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (2,7 g, rendimiento superior al 100 %, TFA) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C18H20N8O: 365,18; observado 365,1.
Monómero F. Sal de ácido trifluoroacético de 2-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-5-ol.

Etapa 1: Síntesis de (4-(4-amino-3-(5-((terc-butildimetilsilil)oxi)-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo
A una disolución de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (1,0 g, 2,31 mmoles, 1,0 equiv) en dioxano (10,5 ml) y H2O (3,5 ml) se añadió ácido (1-(terc-butoxicarbonil)-5-((tercbutildimetilsilil)oxi)-1H-indol-2-il)borónico (1,54 g, 2,78 mmoles, 1,2 equiv), K3PO4 (1,47 g, 6,94 mmoles, 3,0 equiv), Pd2(dba)3 (211,84 mg, 231,34 µmoles, 0,1 equiv) y SPhos (189,95 mg, 462,69 µmoles, 0,2 equiv) a temperatura ambiente bajo N2. El tubo cerrado se calentó a 150 °C durante 20 min en un microondas. Esto se repitió para 9 lotes adicionales. Los 10 lotes se combinaron y la mezcla de reacción se enfrió y se repartió entre EtOAc (60 ml) y H2O (80 ml). La fase acuosa se separó y se extrajo con EtOAc (2 × 50 ml). Las fases orgánicas se combinaron, se lavaron con salmuera (60 ml) y se secaron sobre Na2SO4 anhidro. La suspensión se filtró y el filtrado se concentró a presión reducida. El material en bruto se purificó por cromatografía en gel de sílice (1→75% de EtOAc/éter de petróleo). Las fracciones deseadas se combinaron y se evaporaron a presión reducida dando (4-(4-amino-3-(5-((terc-butildimetilsilil)oxi)-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (10 g, 60 % de rendimiento) como un sólido amarillo claro.
Etapa 2: Síntesis de (4-(4-amino-3-(5-hidroxi-1H-indol-2-il)-1H-pirazolo[3,4-d] pirimidin-1-il)butil)carbamato de terc-butilo
A una mezcla de (4-(4-amino-3-(5-((terc-butildimetilsilil)oxi)-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (10 g, 18,12 mmoles, 1,0 equiv) en THF (100 ml) se añadió TBAF•3H2O (1 M, 54,37 ml, 3,0 equiv) en una porción a temperatura ambiente bajo N2. La mezcla se agitó durante 1 h y entonces se añadió H2O (100 ml) a la mezcla de reacción. Las fases se separaron y la fase acuosa se extrajo con EtOAc (2 × 80 ml). La fase orgánica combinada se lavó con salmuera (100 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1→67 % de EtOAc/ éter de petróleo) proporcionando (4-(4-amino-3-(5-hidroxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (7 g, 87 % de rendimiento) como un sólido rosa claro.
Etapa 3: Síntesis de 2-[4-amino-1-(4-aminobutil)pirazolo[3,4-d]pirimidin-3-il]-1H-indol-5-ol
A TFA (50,0 ml, 675,26 mmoles, 38,9 equiv) se añadió (4-(4-amino-3-(5-hidroxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (7,6 g, 17,37 mmoles, 1,0 equiv) a temperatura ambiente. La mezcla se agitó durante 40 min y entonces se concentró a presión reducida. El residuo aceitoso se trituró con MeCN (20 ml), luego se añadió gota a gota en MTBE (300 ml) durante 10 min. El sobrenadante se retiró y entonces el precipitado se recogió por filtración bajo N2 dando 2-[4-amino-1-(4-aminobutil)pirazolo[3,4-d]pirimidin-3-il]-1H-indol-5-ol (7,79 g, 91 % de rendimiento, TFA) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C17H19N7O: 338,17; observado 338,2. Monómero G. Sal de ácido trifluoroacético de 5-(4-amino-1-(azetidin-3-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 3-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)azetidin-1-carboxilato de terc-butilo
A una disolución de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (4 g, 15,32 mmoles, 1,0 equiv), 3-(hidroximetil)azetidin-1-carboxilato de terc-butilo (3,01 g, 16,09 mmoles, 1,05 equiv) y PPh3 (6,03 g, 22,99 mmoles, 1,5 equiv) en THF (80 ml) enfriada a 0 °C se añadió DIAD (4,47 ml, 22,99 mmoles, 1,5 equiv), gota a gota. Después de completarse la adición, la reacción se agitó a temperatura ambiente durante 14 h. La reacción se vertió en H2O (200 ml) y entonces se extrajo con EtOAc (3 × 50 ml). Las fases orgánicas se combinaron y se lavaron con salmuera (2 × 50 ml). La fase orgánica se secó sobre Na2SO4, se filtró, el filtrado se concentró a presión reducida dando un
residuo. El residuo se purificó por cromatografía en gel de sílice (0→100 % de EtOAc/ éter de petróleo) dando 3-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)azetidin-1-carboxilato de terc-butilo (4,2 g, 64 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C14H19IN6O2: 431,07; observado 431,0.
Etapa 2: Síntesis de 3-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)azetidin-1-carboxilato de terc-butilo
A una suspensión bifásica de 3-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)azetidin-1-carboxilato de terc-butilo (4 g, 9,30 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2 -dioxaborolan-2-il)benzo[d]oxazol-2-amina (2,90 g, 11,16 mmoles, 1,2 equiv) y Na2CO3 (4,93 g, 46,49 mmoles, 5,0 equiv) en DME (100 ml) y H2O (50 ml) se añadió Pd(PPh3)4 (1,07 g, 929,71 µmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente y se filtró y el filtrado se extrajo por EtOAc (3 × 50 ml). Las fases orgánicas se combinaron y se lavaron con salmuera (10 ml), se secaron sobre Na2SO4, se filtraron y el filtrado se concentró a presión reducida dando un residuo. El residuo se purificó por cromatografía en gel de sílice (0→20 % de MeOH/EtOAc) dando 3-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)azetidin-1-carboxilato de terc-butilo (3,5 g, 80 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C21H24N8O3: 437,20; observado 437,2.
Etapa 3: Síntesis de 5-(4-amino-1-(azetidin-3-ilmetil)-1H-pirazolo[3,4-d]pirimidin- 3-il)benzo[d]oxazol-2-amina A una disolución de 3-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)azetidin-1-carboxilato de terc-butilo (3,29 g, 6,87 mmoles, 1,0 equiv) en DCM (20 ml) se añadió TFA (7,50 ml, 101,30 mmoles, 14,7 equiv) a 0 °C. La reacción se calentó hasta temperatura ambiente y se agitó durante 2 h. La disolución de reacción se concentró a presión reducida dando un residuo. El residuo se disolvió en MeCN (6 ml) y luego se vertió en MTBE (80 ml). Precipitó un sólido, que se filtró, y la torta sólida se secó a presión reducida dando 5-[4-amino-1-(azetidin-3-ilmetil)pirazolo[3,4-d]pirimidin-3-il]-1,3-benzoxazol-2-amina (4,34 g, rendimiento superior al 100 %, TFA) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C16H16N8O: 337,15; observado 337,1.
Monómero H. Sal de ácido trifluoroacético de 5-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]-oxazol-2-amina.

Este monómero se sintetizó siguiendo los procedimientos brevemente expuestos en Nature 2015, 534, 272-276. Monómero I. Sal de ácido trifluoroacético de 5-(4-amino-1-(pirrolidin-3-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 3-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirrolidin-1-carboxilato de terc-butilo
Una suspensión de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (4,5 g, 17,24 mmoles, 1,0 equiv), 3-(bromometil)pirrolidin-1-carboxilato de terc-butilo (4,78 g, 18,10 mmoles, 1,05 equiv) y K2CO3 (7,15 g, 51,72 mmoles, 3,0 equiv) en DMA (40 ml) se calentó hasta 85 °C. La reacción se agitó a 85 °C durante 3 h, momento en el que la disolución se enfrió hasta temperatura ambiente. Entonces, se añadió H2O (80 ml) a la reacción, y precipitó un sólido. La mezcla se filtró, y la torta sólida se lavó con H2O (2 × 40 ml), y luego se secó a presión reducida dando 3-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirrolidin-1-carboxilato de terc-butilo (6 g, 78 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C15H21IN6O2: 445,08; observado 445,1.
Etapa 2: Síntesis de 3-[[4-amino-3-(2-amino-1,3-benzoxazol-5-il)pirazolo[3,4-d]pirimidin-1-il]metil]pirrolidin-1-carboxilato de terc-butilo
A una suspensión bifásica de 3-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirrolidin-1-carboxilato de terc-butilo (4 g, 9,00 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)benzo[d]oxazol-2-amina (2,81 g, 10,80 mmoles, 1,2 equiv) y Na2CO3 (4,77 g, 45,02 mmoles, 5,0 equiv) en DME (120 ml) y H2O (60 ml) se añadió Pd(PPh3)4 (1,04 g, 900,35 µmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. La mezcla de reacción se enfrió hasta temperatura ambiente y se filtró y el filtrado se extrajo con EtOAc (3 × 50 ml). Las fases orgánicas se combinaron y se lavaron con salmuera (50 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida dando un residuo. El residuo se purificó por cromatografía en gel de sílice (0→20 % de MeOH/EtOAc) dando 3-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirrolidin-1-carboxilato de terc-butilo (3 g, 64 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C22H26N8O3: 451,21, observado 451,2.
Etapa 3: Síntesis de 5-(4-amino-1-(pirrolidin-3-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina A una disolución de 3-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirrolidin-1-carboxilato de terc-butilo (3 g, 6,66 mmoles, 1,0 equiv) en DCM (40 ml) se añadió TFA (20 ml) a 0 °C, gota a gota. La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 2 h. La disolución de reacción se concentró entonces a presión reducida dando un residuo. El residuo se disolvió en MeCN (4 ml), luego se vertió en MTBE (100 ml), y precipitó un sólido. El sólido se filtró y la torta se secó a presión reducida dando 5-(4-amino-1-(pirrolidin-3-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (4,00 g, rendimiento superior al 100 %, TFA) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C17H18N8O: 351,17; observado 351,2.
Monómero J. Sal de ácido trifluoroacético 1-(4-aminobutil)-3-(7-metoxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-4-amina.

Etapa 1: Síntesis de 2-(4-amino-1-(4-((terc-butoxicarbonil)amino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)-7-metoxi-1H-indol-1-carboxilato de terc-butilo
A una mezcla de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (1,0 equiv) y ácido (1-(terc-butoxicarbonil)-7-metoxi-1H-indol-2-il)borónico (3,0 equiv) en DME y H2O se añade Pd(PPh3)4 (0,1 equiv) y carbonato sódico (6,0 equiv). La reacción se calienta a 80 °C hasta que se completa, como se ha determinado por LCMS y análisis por CCF. La reacción se inactiva entonces con H2O y EtOAc. La mezcla se transfiere a un embudo de decantación y la fase acuosa se extrae con EtOAc. La fase orgánica se lava con NaCl ac. sat., se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El producto deseado se aísla después de la cromatografía sobre gel de sílice.
Etapa 2: Síntesis de 1-(4-aminobutil)-3-(7-metoxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-4-amina
A una disolución de 2-(4-amino-1-(4-((terc-butoxicarbonil)amino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)-7-hidroxi-1H-indol-1-carboxilato de terc-butilo (1,0 equiv) en DCM a 0 °C se añade TFA gota a gota. La reacción se agita a 0 °C y se calienta hasta temperatura ambiente. Una vez la reacción está completa, como se ha determinado por LCMS, la reacción se concentra a presión reducida. El residuo se tritura con MeCN, luego se añade gota a gota en MTBE durante 10 min. El sobrenadante se retira y el precipitado se recoge por filtración bajo N2 dando 1-(4-aminobutil)-3-(7-metoxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-4-amina.
Monómero K. Síntesis de sal de ácido trifluoroacético 1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-4-amina.

Etapa 1: Síntesis de (4-(4-amino-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo
A una mezcla de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (300 mg, 694 µmoles, 1,0 equiv) en MeOH (14 ml) a 0 °C se añadió polvo de cinc (226 mg, 3,46 mmoles, 5,0 equiv). Se añadió NH4Cl ac. sat. (14 ml) a la mezcla de reacción y la reacción se calentó hasta temperatura ambiente y se agitó durante 18 h. La reacción se inactivó por EtOAc (40 ml) y H2O (10 ml) y la mezcla se transfirió a un embudo de decantación. La fase acuosa se extrajo con EtOAc (3 × 20 ml) y las fases orgánicas combinadas se lavaron con NaHCO3 ac. sat. (15 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida proporcionando el producto (210 mg, 99 % de rendimiento) como un sólido amarillo claro que se usó sin más purificación. LCMS (ESI) m/z: [M H] calculado para C14H22N6O2: 307,19; observado 307,1.
Etapa 2: Síntesis de 1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-4-amina
A una disolución de (4-(4-amino-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (210 mg, 691 µmoles) en DCM (3,5 ml) a 0 °C se añadió TFA (3,5 ml), gota a gota. Después de 3 h, la reacción se calentó hasta temperatura ambiente y se concentró a presión reducida proporcionando la sal de trifluoroacetato del producto (220 mg, 99 % de rendimiento) como un aceite marrón, que se usó sin más purificación. LCMS (ESI) m/z: [M H] calculado para C9H14N6: 207,13; observado 207,1.
Monómero L.1-[4-(piperazin-1-il)-3-(trifluorometil)fenil]-9-(quinolin-3-il)-1H,2H-benzo[h] 1,6-naftiridin-2-ona

La preparación de este monómero se ha informado previamente en la bibliografía. Véanse las siguientes referencias: i) Liu, Qingsong; Chang, Jae Won; Wang, Jinhua; Kang, Seong A.; Thoreen, Carson C.; Markhard, Andrew; Hur, Wooyoung; Zhang, Jianming; Sim, Taebo; Sabatini, David M.; et al From Journal of Medicinal Chemistry (2010), 53(19), 7146-7155. ii) Gray, Nathanael; Chang, Jae Won; Zhang, Jianming; Thoreen, Carson C.; Kang, Seong Woo Anthony; Sabatini, David M.; Liu, Qingsong de la solicitud internacional PCT (2010), WO 2010044885A2.
Monómero M. Sal de ácido trifluoroacético de 5-(1-(4-aminobutil)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 3-yodo-1-tritil-1H-pirazolo[3,4-d]pirimidin-4-amina
Se trató una suspensión de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (10,5 g, 40,23 mmoles, 1,0 equiv) en DMF (170,0 ml) con Cs2CO3 (19,7 g, 60,34 mmoles, 1,5 equiv) y [cloro(difenil)metil]benceno (13,5 g, 48,27 mmoles, 1,2 equiv) a temperatura ambiente. La mezcla de reacción se agitó a 70 °C durante 4 h bajo una atmósfera de nitrógeno. La mezcla de reacción se añadió a H2O (1200 ml). El precipitado se filtró y se lavó con H2O. El residuo se purificó por cromatografía en gel de sílice (0→60 % de EtOAc/ éter de petróleo) proporcionando 3-yodo-1-tritil-1H-pirazolo[3,4-d]pirimidin-4-amina (15,40 g, 73,5 % de rendimiento) como un sólido blanco.
Etapa 2: Síntesis de 3-yodo-N,N-dimetil-1-tritil-1H-pirazolo[3,4-d]pirirnidin-4-amina
A una suspensión de NaH (2,98 g, 74,50 mmoles, 60 % en peso, 2,5 equiv) en DMF (150 ml) se añadió la disolución de 3-yodo-1-tritil-1H-pirazolo[3,4-d]pirimidin-4-amina (15,0 g, 29,80 mmoles, 1,0 equiv) en DMF (50 ml) a 0 °C. La mezcla se agitó a 0 °C durante 10 min. A la mezcla de reacción se añadió entonces yodometano (16,92 g,
119,20 mmoles, 7,42 ml, 4,0 equiv) a 0 °C. La mezcla se agitó a temperatura ambiente durante 2 h, momento en que se añadió H2O (1400 ml) a 0 °C. La mezcla se agitó durante 10 min adicionales a 0 °C. El precipitado resultante se recogió por filtración dando el producto en bruto, que se purificó por cromatografía en gel de sílice (1 %→25 % de EtOAc/éter de petróleo) dos veces proporcionando 3-yodo-N,N-dimetil-1-tritil-1H-pirazolo[3,4-d]pirimidin-4-amina (9,0 g, 89 % de rendimiento) como un sólido blanco.
Etapa 3: Síntesis de 3-yodo-N,N-dimetil-1H-pirazolo[3,4-d]pirimidin-4-amina
A una disolución enfriada de TFA (19,1 ml, 258,1 mmoles, 15,0 equiv) en DCM (100,0 ml) se añadió 3-yodo-N,N-dimetil-1-tritil-1H-pirazolo[3,4-d]pirimidin-4-amina (9,10 g, 17,12 mmoles, 1,0 equiv) a 4 °C. La mezcla se agitó a temperatura ambiente durante 1 h. El residuo se vertió en H2O (100 ml) y la fase acuosa se extrajo con DCM (2 × 50 ml). A la fase acuosa se añadió entonces una disolución acuosa saturada de NaHCO3 hasta que la disolución estuvo a pH 8. El precipitado resultante se recogió por filtración dando 3-yodo-N,N-dimetil-1H-pirazolo[3,4-d]pirimidin-4-amina (3,40 g, 68,7 % de rendimiento) como un sólido blanco.
Etapa 4: Síntesis de (4-(4-(dimetilamino)-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo A una suspensión de 3-yodo-N,N-dimetil-1H-pirazolo[3,4-d]pirimidin-4-amina (1,7 g, 5,88 mmoles, 1,0 equiv) en DMF (20 ml) se añadió NaH (247 mg, 6,17 mmoles, 60 % en peso, 1,05 equiv) a 4 °C. La mezcla se agitó a 4 °C durante 30 min. A la mezcla de reacción se añadió entonces N-(4-bromobutil)carbamato de terc-butilo (2,22 g, 8,82 mmoles, 1,81 ml, 1,5 equiv) en DMF (10 ml) a 4 °C. La mezcla se agitó a temperatura ambiente durante 2 h. A la mezcla se añadió entonces H2O (100 ml) a 4 °C. La mezcla se agitó durante 30 min adicionales a 4 °C y el precipitado resultante se recogió por filtración dando el producto en bruto. El residuo se purificó por cromatografía en gel de sílice (0→75 % de EtOAc/éter de petróleo) proporcionando (4-(4-(dimetilamino)-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (2,0 g, 56 % de rendimiento) como un sólido blanco.
Etapa 5: Síntesis de (4-(3-(2-aminobenzo[d]oxazol-5-il)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo
A una suspensión bifásica de (4-(4-(dimetilamino)-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (4,0 g, 8,69 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (3,4 g, 13,03 mmoles, 1,5 equiv) y Na2CO3 (4,6 g, 43,45 mmoles, 5,0 equiv) en DME (80,0 ml) y H2O (40,0 ml) se añadió Pd(PPh3)4 (1,0 g, 868,98 µmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. Entonces se enfrió la mezcla de reacción y se repartió entre EtOAc (300 ml) y H2O (600 ml). La fase acuosa se separó y se extrajo con EtOAc (2 × 100 ml). Las fases orgánicas se combinaron, se lavaron con salmuera (2 × 60 ml) y se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El material en bruto se purificó por cromatografía en columna de gel de sílice (50 % de EtOAc/hexanos seguido de 20 % de MeOH/EtOAc). Las fracciones deseadas se combinaron y se concentraron a presión reducida dando (4-(3-(2-aminobenzo[d]oxazol-5-il)-4-(dimetilamino)-1H-pirazolo[3,4-d]piramidin-1-il)butil)carbamato de terc-butilo (3,2 g, 78,9 % de rendimiento) como un sólido marrón claro.
Etapa 6: Síntesis de 5-(1-(4-aminobutil)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina
A TFA (20,82 ml, 281,27 mmoles, 36,5 equiv) se añadió (4-(3-(2-aminobenzo[d]oxazol-5-il)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (3,6 g, 7,72 mmoles, 1,0 equiv) a temperatura ambiente. La mezcla se agitó durante 30 min, momento en el que la mezcla se concentró a presión reducida. El residuo aceitoso se trituró con MeCN (8 ml) y MTBE (60 ml) durante 10 min. El sobrenadante se retiró y entonces el precipitado se recogió por filtración bajo N2 dando 5-(1-(4-aminobutil)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (4,0 g, en bruto, TFA) como un sólido marrón claro.
A NaOH 1 M (107,2 ml, 14,7 equiv) se añadió 5-(1-(4-aminobutil)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (3,5 g, en bruto, TFA) a temperatura ambiente. La mezcla se agitó durante 10 min y entonces la fase acuosa se extrajo con DCM (3 × 50 ml). La fase orgánica combinada se lavó con salmuera (50 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. Se añadió TFA (539,37 µl, 7,28 mmoles, 1,0 equiv) y se concentró a presión reducida. Entonces se añadió MeCN (10 ml), seguido de MTBE (150 ml). El precipitado resultante se recogió por filtración dando 5-(1-(4-aminobutil)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (1,3 g, 36,6 % de rendimiento, TFA) como un producto marrón claro. LCMS (ESI) m/z: [M H] calculado para C18H22N8O: 367,19; observado 367,1.
Monómero N. Sal de ácido trifluoroacético de 6-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo-[d]isoxazol-3-amina.

Etapa 1: Síntesis de (6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]isoxazol-3-il)carbamato de terc-butilo A una disolución de (6-bromobenzo[d]isoxazol-3-il)carbamato de terc-butilo (1,0 equiv) en dioxano se añade Pd(PPh3)4 (0,1 equiv), carbonato sódico (6,0 equiv) y bis(pinacolato)diboro (3,0 equiv). La mezcla de reacción se agita y se calienta hasta que se completa, como se ha determinado por LCMS y análisis por CCF. La reacción se enfría hasta temperatura ambiente, se enfría rápidamente con NaHCO3 ac. sat. y la mezcla se transfiere a un embudo de decantación. La fase acuosa se extrae con EtOAc y la fase orgánica se lava con NaCl ac. sat., se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El producto deseado se aísla después de la purificación por cromatografía en gel de sílice.
Etapa 2: Síntesis de (4-(4-amino-3-(3-((terc-butoxicarbonil)amino)benzo[d]isoxazol-6-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo
A una mezcla de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (1,0 equiv) y (6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]isoxazol-3-il)carbamato de terc-butilo (3,0 equiv) en DME y H2O se añade Pd(PPh3)4 (0,1 equiv) y carbonato sódico (6,0 equiv). La reacción se calienta a 80 °C hasta que se completa, como se ha determinado por LCMS y análisis por CCF. La reacción se inactiva entonces con H2O y EtOAc. La mezcla se transfiere a un embudo de decantación y la fase acuosa se extrae con EtOAc. La fase orgánica se lava con NaCl ac. sat., se seca sobre Na2SO4, se filtra y se concentra a presión reducida. El producto deseado se aísla después de la cromatografía sobre gel de sílice.
Etapa 3: Síntesis de 6-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo-[d]isoxazol-3-amina A una disolución de (4-(4-amino-3-(3-((terc-butoxicarbonil)amino)benzo[d]isoxazol-6-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (1,0 equiv) en DCM a 0 °C se añade TFA, gota a gota. La reacción se agita a 0 °C y se calienta hasta temperatura ambiente. Una vez la reacción está completa, como se ha determinado por LCMS, la reacción se concentra a presión reducida. El residuo se tritura con MeCN, luego se añade gota a gota en MTBE durante 10 min. El sobrenadante se retira y el precipitado se recoge por filtración bajo N2 dando 6-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo-[d]isoxazol-3-amina.
Monómero O. Sal de ácido trifluoroacético de 4-(5-(4-morfolino-1-(1-(piridin-3-ilmetil)piperidin-4-il)-1H-pirazolo[3,4-d]pirimidin-6-il)-1H-indol-1-il)butan-1-amina.

La síntesis de este monómero avanza por alquilación de WAY-600 (CAS# 1062159-35-6) con (4-bromobutil)carbamato de terc-butilo en condiciones básicas, seguido de desprotección de Boc usando TFA para producir la sal de TFA. Referencia para la preparación de WAY-600: Discovery of Potent and Selective Inhibitors of the Mammalian Target of Rapamycin (mTOR) Kinase: Nowak, P.; Cole, D.C.; Brooijmans, N.; Bursavich, M.G.; Curran, K.J.; Ellingboe, J.W.; Gibbons, J.J.; Hollander, I.; Hu, Y.; Kaplan, J.; Malwitz, D.J.; Toral-Barza, L.; Verheijen, J.C.; Zask, A.; Zhang, W.-G.; Yu, K.2009; Journal of Medicinal Chemistry, Volumen 52, edición 22, 7081-89.
Monómero P. Sal de ácido trifluoroacético de 2-(4-(8-(6-(aminometil)quinolin-3-il)-3-metil-2-oxo-2,3-dihidro-1H-imidazo[4,5-c]quinolin-1-il)fenil)-2-metilpropanonitrilo.

La síntesis de este monómero avanza primero por síntesis del componente de acoplamiento de la reacción de Suzuki (3-(4,4,5,5-tetrametil-1,3,2-dioxaborolane)quinolin-6-il)-N-boc-metanamina a partir de 3-bromoquinolina-6-carboxilato de metilo. La reducción del éster metílico con hidruro de litio y aluminio seguido de reacción de Mitsunobu con ftalimida y escisión de hidracina proporciona la amina bencílica. La protección de la amina bencílica con dicarbonato de di-terc-butilo, seguido de una reacción de borilación de Miyaura proporciona (3-(4,4,5,5-tetrametil-1,3,2-dioxaborolano)quinolin-6-il)-N-boc-metanamina.
Una reacción SNAr de 2-(4-aminofenil)-2-metilpropanonitrilo con 6-bromo-4-cloro-3-nitroquinolina proporciona la amino-nitro-piridina sustituida. La reducción del grupo nitro con Ni Raney bajo una atmósfera de hidrógeno, seguido de ciclación con cloroformiato de triclorometilo proporciona la urea sustituida con arilo. La sustitución de N-H libre de la urea con yoduro de metilo mediada por bromuro de tetrabutilamonio e hidróxido sódico seguido de acoplamiento de Suzuki de (3-(4,4,5,5-tetrametil-1,3,2-dioxaborolane)quinolin-6-il)-N-boc-metanamina y luego Boc-desprotección usando TFA produce la sal de TFA.
Referencia para la preparación de 2-[4-(8-bromo-3-metil-2-oxo-2,3-dihidro-imidazo[4,5-c]quinolin-1-il)-fenil]-2-metilpropionitrilo: Vannucchi, A.M.; Bogani, C.; Bartalucci, N.2016. JAK PI3K/mTOR combination therapy. Documento de patente US9358229. Novartis Pharma AG, Incyte Corporation.
Monómero Q. 8-(6-metoxipiridin-3-il)-3-metil-1-[4-(piperazin-1-il)-3-(trifluorometil)fenil]-1H,2H,3H-imidazo[4,5-c]quinolin-2-ona

Este monómero es un producto químico disponible comercialmente como BGT226(CAS# 1245537-68-1). En el momento en que se hizo la presente solicitud, estaba disponible para su compra de varios vendedores como la amina libre.
Monómero R. Sal de ácido trifluoroacético de 3-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-N-(4,5-dihidrotiazol-2-il)benzamida.

Etapa 1: Síntesis de (4-(4-amino-3-(3-((4,5-dihidrotiazol-2-il)carbamoil)fenil)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo
A una disolución de ácido (3-((4,5-dihidrotiazol-2-il)carbamoil)fenil)borónico (500 mg, 1,15 mmoles, 1,0 equiv) y (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (575 mg, 2,30 mmoles, 2,0 equiv) en dioxano (19,1 ml), EtOH (3,8 ml), y H2O (2,3 ml) se añadió Pd(PPh3)4 (265 mg, 230 µmoles, 0,2 equiv) y carbonato sódico (730 mg, 6,89 mmoles, 6,0 equiv). La mezcla de reacción se sonicó hasta que se formó una disolución amarilla clara, que posteriormente se calentó a 80 °C durante 14 h. La reacción se diluyó entonces con NaCl ac. sat. (30 ml) y la mezcla se transfirió a un embudo de decantación. La fase acuosa se extrajo con DCM (3 × 25 ml). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El producto deseado se aisló como un sólido amarillo (324 mg, 53 % de rendimiento) después de la cromatografía en gel de sílice (0→15 % de MeOH/DCM). LCMS (ESI) m/z: [M H] calculado para C24H30N8O3S: 511,22; observado 511,2.
Etapa 2: Síntesis de 3-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)-N-(4,5-dihidrotiazol-2-il)benzamida
A una disolución de (4-(4-amino-3-(3-((4,5-dihidrotiazol-2-il)carbamoil)fenil)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (324 mg, 614 µmol) en DCM (4,1 ml) a 0 °C se añadió TFA (1,5 ml), gota a gota. Después de 1 h, la reacción se calentó hasta temperatura ambiente y se concentró a presión reducida proporcionando la sal de trifluoroacetato del producto como un sólido amarillo (320 mg, 99 % de rendimiento). Se usó sin más purificación. LCMS (ESI) m/z: [M H] calculado para C19H22N8OS: 411,16; observado 411,1
Monómero S. 2-(5-(4-morfolino-1-(1-(piridin-3-ilmetil)piperidin-4-il)-1H-pirazolo[3,4-d]pirimidin-6-il)-1H-indol-3-il)etan-1-amina.

La síntesis de este monómero avanza por condensación de 2,4,6-tricloropirimidin-5-carbaldehído con clorhidrato de 3-((4-hidracinailpiperidin-1-il)metil)piridina. La reacción del producto con morfolina seguido de una reacción de Suzuki con éster borónico da la amina protegida con Boc. La desprotección final con TFA da el monómero. Esta vía de síntesis sigue estrechamente la preparación informada de estructuras altamente relacionadas en las siguientes referencias: i) Nowak, Pawel; Cole, Derek C.; Brooijmans, Natasja; Curran, Kevin J.; Ellingboe, John W.; Gibbons, James J.; Hollander, Irwin; Hu, Yong Bo; Kaplan, Joshua; Malwitz, David J.; et al From Journal of Medicinal Chemistry (2009), 52(22), 7081-7089. ii) Zask, Arie; Nowak, Pawel Wojciech; Verheijen, Jeroen; Curran, Kevin J.; Kaplan, Joshua;
Malwitz, David; Bursavich, Matthew Gregory; Cole, Derek Cecil; Ayral-Kaloustian, Semiramis; Yu, Ker; et al. de la solicitud internacional PCT (2008), documento de patente WO 2008115974 A220080925.
Monómero T. Sal de ácido trifluoroacético de 1-(4-aminobutil)-3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina.

A una mezcla de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)carbamato de terc-butilo (496 mg, 1,14 mmoles, 1,0 equiv) en DCM (5,7 ml) a 0 °C se añadió TFA (1,5 ml) gota a gota. La reacción se dejó con agitación a 0 °C durante 1 h, momento en el que la reacción se concentró a presión reducida proporcionando un sólido amarillo (505 mg, 99 % de rendimiento) que se sacó on sin más purificación. LCMS (ESI) m/z: [M H] calculado para C9H13IN6: 333,02; observado 332,9.
Monómero U. Sal de ácido trifluoroacético de 5-(4-amino-1-(4-(metilamino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de (4-hidroxibutil)(metil)carbamato de terc-butilo
A una disolución de 4-(metilamino)butan-1-ol (0,5 g, 4,85 mmoles, 104,2 ml, 1,0 equiv) en DCM (10 ml) a temperatura ambiente se añadió BoczO (1,06 g, 4,85 mmoles, 1,11 ml, 1,0 equiv). La mezcla se agitó durante 3 h a temperatura ambiente y entonces la mezcla se concentró a presión reducida a 30 °C. El residuo se purificó por cromatografía en gel de sílice (100/1 a 3/1 de éter de petróleo/EtOAc) proporcionando (4-hidroxibutil)(metil)carbamato de terc-butilo (0,9 g, 91,4 % de rendimiento) como un aceite incoloro.
Etapa 2: Síntesis de (4-bromobutil)(metil)carbamato de terc-butilo
A una disolución de (4-hidroxibutil)(metil)carbamato de terc-butilo (0,9 g, 4,43 mmoles, 1,0 equiv) en THF (20 ml) a temperatura ambiente se añadió PPh3 (2,21 g, 8,41 mmoles, 1,9 equiv) y CBr4 (2,79 g, 8,41 mmoles, 1,9 equiv). La mezcla se agitó durante 1 h y entonces la mezcla de reacción se filtró y se concentró. El residuo se purificó por cromatografía en gel de sílice (1/0 a 4/1 de éter de petróleo/EtOAc) proporcionando (4-bromobutil)(metil)carbamato de terc-butilo (1,1 g, 93,3 % de rendimiento) como un aceite incoloro.
Etapa 3: Síntesis de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)(metil)carbamato de terc-butilo A una suspensión de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (0,9 g, 3,45 mmoles, 1,0 equiv) en DMF (10 ml) a 4 °C se añadió NaH (137,92 mg, 3,45 mmoles, 60 % en peso, 1,0 equiv). La mezcla se agitó a 4 °C durante 30 min y entonces se añadió una disolución de (4-bromobutil)(metil)carbamato de terc-butilo (1,01 g, 3,79 mmoles, 25,92 ml, 1,1 equiv) en DMF (3 ml). La mezcla se agitó a temperatura ambiente durante 3 h, momento en el que se añadió H2O (100 ml). La fase acuosa se extrajo con EtOAc (3 × 30 ml) y las fases orgánicas combinadas se lavaron con salmuera (20 ml), se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 0/1 de éter de petróleo/EtOAc) proporcionando (4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)butil)(metil)carbamato de terc-butilo (1,2 g, 78 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C15H23IN6O2: 447,10; observado 447,1.
Etapa 4: Síntesis de (4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)(metil)carbamato de terc-butilo
A una suspensión bifásica de (4-(4-amino-3-yodo-1H-pirazolo[3,4-d] pirimidin-1-il)butil)(metil)carbamato de terc-butilo (1,2 g, 2,69 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (1,19 g, 3,23 mmoles, 1,2 equiv) y Na2CO3 (1,42 g, 13,44 mmoles, 5,0 equiv) en DME (20 ml) y H2O (10 ml) a temperatura ambiente se añadió Pd(PPh3)4 (310,71 mg, 268,89 µmoles, 0,1 equiv) bajo N2. La mezcla se agitó a 110 °C durante 3
h y entonces la mezcla de reacción se enfrió y se repartió entre EtOAc (20 ml) y H2O (15 ml). La fase acuosa se separó y se extrajo con EtOAc (3 × 20 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 × 20 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El producto en bruto se purificó por cromatografía en gel de sílice (1/0 a 4/1 EtOAc/MeOH) dando (4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)(metil)carbamato de terc-butilo (0,78 g, 62,5 % de rendimiento) como un sólido naranja.
Etapa 5: Síntesis de 5-(4-amino-1-(4-(metilamino)butil)-1H-pirazolo[3,4-d] pirimidin-3-il) benzo[d]oxazol-2-amina
Una disolución de (4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)(metil)carbamato de terc-butilo (0,78 g, 1,72 mmoles, 1,0 equiv) en TFA (5 ml) a temperatura ambiente se agitó durante 30 min. La disolución se concentró a presión reducida y el residuo aceitoso se trituró con MeCN (1 ml) y entonces se añadió a MTBE (100 ml). El sobrenadante se retiró y entonces el precipitado se recogió por filtración bajo N2 dando bistrifluorosulfonato de 5-(4-amino-1-(4-(metilamino)butil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (0,959 g, 93 % de rendimiento) como un sólido naranja. LCMS (ESI) m/z: [M H] calculado para C17H20N8O: 353,18; observado 353,1.
Monómero V. 1-(4-(4-(5-(aminometil)pirimidin-2-il)piperazin-1-il)-3-(trifluorometil)fenil)-8-(6-metoxipiridin-3-il)-3-metil-1,3-dihidro-2H-imidazo [4,5-c] quinolin-2-ona.

Etapa 1: Síntesis de N-terc-butoxicarbonil-N-[(2-cloropirimidin-5-il)metil] carbamato de terc-butilo
A una disolución de N-terc-butoxicarbonilcarbamato de terc-butilo (7,33 g, 33,74 mmoles, 1,0 equiv) en DMF (80 ml) se añadió NaH (1,62 g, 40,49 mmoles, 60 % en peso, 1,2 equiv) a 0 °C. La mezcla se agitó a 0 °C durante 30 min y entonces se añadió 5-(bromometil)-2-cloropirimidina (7 g, 33,74 mmoles, 1 equiv). La mezcla de reacción se agitó a temperatura ambiente durante 1,5 h y entonces la mezcla se vertió en NH44Cl sat. (300 ml) y se agitó durante 5 min. La fase acuosa se extrajo con EtOAc (3 × 80 ml) y las fases orgánicas combinadas se lavaron con salmuera (50 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía en gel de sílice (20:1 a 1:1 de éter de petróleo/EtOAc) proporcionando de N-terc-butoxicarbonil-N-[(2-cloro pirimidin-5-il)metil]carbamato de terc-butilo (7,0 g, 60,3 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C15H22ClN3O4: 344,14; observado 344,2.
Etapa 2: Síntesis de N-terc-butoxicarbonil-N-[[2-[4-[4-[8-(6-metoxi-3-piridil)-3-metil-2-oxo-imidazo[4,5-c]quinolin-1-il]-2-(trifluorometil)fenil]piperazin-1-il]pirimidin-5-il]metil]carbamato de terc-butilo
A una disolución de 8-(6-metoxi-3-piridil)-3-metil-1-[4-piperazin-1-il-3-(trifluorometil)fenil]imidazo[4,5-c]quinolin-2-ona (0,4 g, 748,32 µmoles, 1,0 equiv) en MeCN (7 ml) se añadió N-terc-butoxicarbonil-N-[(2-cloropirimidin-5-il)metil]carbamato de terc-butilo (514,55 mg, 1,50 mmoles, 2,0 equiv) y K2CO3 (413,69 mg, 2,99 mmoles, 4 equiv) a temperatura ambiente. La mezcla de reacción se agitó a 80 °C durante 14 h y entonces la mezcla se enfrió hasta temperatura ambiente, se filtró y se concentró a presión reducida. El residuo se purificó lavando con MTBE (5 ml) dando N-terc-butoxicarbonil-N-[[2-[4-[4-[8-(6-metoxi-3-piridil)-3-metil-2-oxo-imidazo[4,5-c]quinolin-1-il]-2-(trifluorometil)fenil]piperazin-1-il]pirimidin-5-il]metil]carbamato de terc-butilo (0,57 g, 90,5 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C43H46F3N9O6: 842,36; observado 842,7
Etapa 3: Síntesis de 1-[4-[4-[5-(aminometil)pirimidin-2-il]piperazin-1-il]-3-(trifluorometil)fenil]-8-(6-metoxi-3-piridil)-3-metil-imidazo[4,5-c]quinolin-2-ona
Una disolución de N-terc-butoxicarbonil-N-[[2-[4-[4-[8-(6-metoxi-3-piridil)-3-metil-2-oxo-imidazo[4,5-c]quinolin-1-il]-2-(trifluorometil)fenil]piperazin-1-il]pirimidin-5-il]metil]carbamato de terc-butilo (0,95 g, 1,13 mmoles, 1 equiv) en TFA (10 ml) se agitó a temperatura ambiente durante 1 h, momento en el que el disolvente se concentró. El residuo se disolvió en MeCN (10 ml) y entonces la disolución se añadió a MTBE (150 ml), gota a gota. El precipitado se recogió dando trifluorometanosulfonato de 1-[4-[4-[5-(aminometil)pirimidin-2-il]piperazin-1-il]-3-(trifluorometil)fenil]-8-(6-metoxi-3-piridil)-3-metil-imidazo[4,5-c]quinolin-2-ona (0,778 g, 84,8 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C33H30F3N9O2: 642,26; observado 642,4
Monómero W.1-(4-aminobutil)-3-(1H-pirrolo[2,3-b]piridin-5-il)pirazolo[3,4-d]pirimidin-4-amina.

Etapa 1: Síntesis de N-[4-[4-amino-3-(1H-indol-5-il)pirazolo[3,4-d]pirimidin-1-il]butil]carbamato de terc-butilo A una suspensión bifásica de N-[4-(4-amino-3-yodo-pirazolo[3,4-d]pirimidin-1-il)butil]carbamato de terc-butilo (8 g, 18,51 mmoles, 1 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirrolo[2,3-b]piridina (5,42 g, 22,21 mmoles, 1,2 equiv) y Na2CO3 (9,81 g, 92,54 mmoles, 5 equiv) en diglima (160 ml) y H2O (80 ml) se añadió Pd(PPh3)4 (2,14 g, 1,85 mmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. La mezcla de reacción se enfrió hasta temperatura ambiente, se filtró y el filtrado se repartió entre EtOAc (500 ml) y H2O (500 ml). La fase acuosa se separó y se extrajo con EtOAc (3 × 300 ml). Las fases orgánicas se combinaron, se lavaron con salmuera (20 ml) y se secaron sobre Na2SO4 anhidro, entonces se filtraron y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 0/1 de éter de petróleo/EtOAc entonces 4/1 EtOAc/MeOH) dando N-[4-[4-amino-3-(1H-indol-5-il)pirazolo[3,4-d]pirimidin-1-il]butil]carbamato de terc-butilo (6,6 g, 84,6 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C22H27N7O2: 422,22; observado 423,3.
Etapa 2: Síntesis de 1-(4-aminobutil)-3-(1H-pirrolo[2,3-b]piridin-5-il)pirazolo[3,4-d]pirimidin-4-amina
A N-[4-[4-amino-3-(1H-indol-5-il)pirazolo[3,4-d]pirimidin-1-il]butil]carbamato de terc-butilo (6,6 g, 15,66 mmoles, 1 equiv) se añadió TFA (66 ml), que entonces se agitó a temperatura ambiente durante 30 min. La disolución de reacción se concentró a presión reducida para retirar el TFA y entonces se añadió MTBE (400 ml) al residuo. La suspensión se agitó durante 15 min, momento en el que se filtró el sólido amarillo, y la torta sólida se secó a presión reducida dando 1-(4-aminobutil)-3-(1H-pirrolo[2,3-b]piridin-5-il)pirazolo[3,4-d]pirimidin-4-amina (10,2 g, 97,1 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C16H18N8: 323,17; observado 323,1.
Monómero X. 2,2,2-Trifluoroacetato de 2-(4-amino-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-5-ol.

Etapa 1: Síntesis de 6-((4-amino-3-(5-((terc-butildimetilsilil)oxi)-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una disolución de 6-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (1 g, 1,97 mmoles, 1,0 equiv) en dioxano (10,5 ml) y H2O (3,5 ml) se añadió ácido (1-(terc-butoxicarbonil)-5-((tertbutildimetilsilil)oxi)-1H-indol-2-il)borónico (1,16 g, 2,96 mmoles, 1,5 equiv), K3PO4 (1,26 g, 5,92 mmoles, 3,0 equiv), Pd2(dba)3 (180,85 mg, 197,50 µmoles, 0,1 equiv) y SPhos (162,16 mg, 394,99 µmoles, 0,2 equiv) a temperatura ambiente bajo N2. El tubo cerrado se calentó a 150 °C durante 20 min bajo microondas. Entonces se enfrió la mezcla de reacción y 6 lotes separados se combinaron juntos. La mezcla de reacción se repartió entre EtOAc (100 ml) y H2O (100 ml). La fase acuosa se separó y se extrajo con EtOAc (3 × 80 ml). Las fases orgánicas se combinaron, se lavaron con salmuera (100 ml) y se secaron sobre Na2SO4 anhidro. La disolución se filtró y el filtrado
se concentró a presión reducida. El material en bruto se purificó por cromatografía en columna de gel de sílice (100/1 a 1/4 éter de petróleo/EtOAc) dando 6-((4-amino-3-(5-((terc-butildimetilsilil)oxi)-1H-indol-2-il)-1H-pirazolo [3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (6,17 g, 82,9 % de rendimiento) como un sólido amarillo claro.
Etapa 2: Síntesis de 6-((4-amino-3-(5-hidroxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una mezcla de 6-((4-amino-3-(5-((terc-butildimetilsilil)oxi)-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (6,17 g, 9,86 mmoles, 1,0 equiv) en THF (100 ml) se añadió fluoruro de tetrabutilamonio trihidratado (1 M, 10,84 ml, 1,1 equiv) en una porción a 0 °C bajo N2. La mezcla se agitó a 0 °C durante 1 h y entonces se añadió a H2O (100 ml). La fase acuosa se extrajo con EtOAc (3 × 80 ml) y la fase orgánica combinada se lavó con salmuera (2 × 80 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/1 a 0/1 de éter de petróleo/EtOAc) proporcionado 6-((4-amino-3-(5-hidroxi-1H-indol-2-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (4 g, 79,3 % de rendimiento) como un sólido rosa claro. LCMS (ESI) m/z: [M H] calculado para C28H29N7O3: 512,24; observado 512,3.
Etapa 3: Síntesis de 2,2,2-trifluoroacetato de 2-(4-amino-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-5-ol
A una disolución de 6-((4-amino-3-(5-hidroxi-1H-indol-2-il)-1H-pirazolo [3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (4,5 g, 8,80 mmoles, 1,0 equiv) en MeOH (50 ml) se añadió HCl en MeOH (4 M, 50 ml, 22,7 equiv) a temperatura ambiente. La mezcla se agitó a temperatura ambiente durante la noche y entonces se concentró a presión reducida. Al producto en bruto se añadió EtOAc (100 ml) y el precipitado resultante se recogió por filtración bajo N2 dando 2,2,2-trifluoroacetato de 2-(4-amino-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)-1H-indol-5-ol (4,1 g, 85,0 % de rendimiento, 3HCl) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C23H21N7O: 412,19; observado 412,1.
Monómero Y. 2,2,2-Trifluoroacetato de 3-(1H-pirrolo[2,3-b]piridin-5-il)-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-4-amina.

Etapa 1: Síntesis de 6-(bromometil)-3,4-dihidroisoquinolin-2(1H)- carboxilato de terc-butilo
Una disolución de NBS (34,07 g, 191,39 mmoles, 4 equiv) en THF (200 ml) se añadió en porciones a una disolución de 6-(hidroximetil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (12,6 g, 47,85 mmoles, 1,0 equiv) y trifenilfosfina (37,65 g, 143,55 mmoles, 3,0 equiv) en THF (200 ml) a 0 °C. Después de completarse la adición, la mezcla se agitó durante 1 h a temperatura ambiente. Se añadió EtOAc (150 ml) y la mezcla se lavó con H2O (200 ml) y salmuera (150 ml), se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (100/1 a 10/1 de éter de petróleo/EtOAc) proporcionando 6-(bromometil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (8,56 g, 54,8 % de rendimiento) como un sólido amarillo claro. Etapa 2: Síntesis de 6-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una suspensión de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (9,5 g, 36,40 mmoles, 1,0 equiv) en DMF (110 ml) se añadió NaH (1,46 g, 36,40 mmoles, 60 % en peso, 1,0 equiv) a 0 °C. La mezcla se agitó a 0 °C durante 30 min at que puna una disolución de terc-butil 6-(bromometil)-3,4-dihidroisoquinolin-2(1H)-carboxilato (12,47 g, 38,22 mmoles, 1,05 equiv) en DMF (40 ml) se añadió a 0 °C. La mezcla se agitó a temperatura ambiente durante 1 h y entonces H2O (1000 ml) se añadió a 0 °C. La mezcla se agitó a 0 °C durante 30 min y entonces el precipitado resultante se recogió por filtración dando 6-((4-amino-3-yodo-1H-pirazolo[3,4-d] pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (17,8 g, 76,3 % de rendimiento) como un sólido amarillo claro, que se usó la siguiente etapa directamente. LCMS (ESI) m/z: [M H] calculado para C20H23IN6O2: 507,10; observado 507,1.
Etapa 3: Síntesis de 6-((4-amino-3-(1H-pirrolo[2,3-b]piridin-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una suspensión bifásica de 6-((4-amino-3-yodo-1H-pirazolo [3,4-d] pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (6,5 g, 10,14 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirrolo [2,3-b] piridina (2,97 g, 12,16 mmoles, 1,2 equiv) y Na2CO3 (5,37 g, 50,68 mmoles, 5,0 equiv) en diglima (100 ml) y H2O (50 ml) se añadió Pd(PPh3)4 (1,17 g, 1,01 mmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. Entonces se enfrió la mezcla de reacción y se repartió entre EtOAc (100 ml) y H2O (100 ml). La fase acuosa se separó y se extrajo con EtOAc (2 × 100 ml). La fase orgánica combinada se lavó con salmuera (100 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (0/1 a 1/4 MeOH/EtOAc) proporcionando 6-((4-amino-3-(1H-pirrolo[2,3-b]piridin-5-il)-1H-pirazolo[3,4-d]piramid in-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (3,77 g, 72,1 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C27H28N8O2: 497,24; observado 497,3.
Etapa 4: Síntesis de 2,2,2-trifluoroacetato de 3-(1H-pirrolo[2,3-b]piridin-5-il)-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-4-amina
Se añadió 6-((4-amino-3-(1H-pirrolo[2,3-b]piridin-5-il)-1H-pirazolo[3,4-d] pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (3,77 g, 7,59 mmoles, 1,0 equiv) a TFA (85,36 ml, 1,15 moles, 151,8 equiv) a temperatura ambiente. La mezcla de reacción se agitó durante 1 h. Entonces se concentró a presión reducida y el residuo aceitoso se trituró con MeCN (3 ml), entonces se añadió gota a gota en MTBE (200 ml) durante 5 min. El sobrenadante se retiró y entonces el precipitado se recogió por filtración bajo N2 dando el producto, que se disolvió en MeCN (20 ml), y finalmente se concentró a presión reducida dando 2,2,2-trifluoroacetato de 3-(1H-pirrolo[2,3-b]piridin-5-il)-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-4-amina (4,84 g, 85,0 % de rendimiento, 3TFA) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C22H20N8: 397,19; observado 397,2. Monómero Z. 2,2,2-Trifluoroacetato de (4-((2-aminoetil)sulfonil)-3-fluoro-2-metilfenil)(7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)metanona.

Etapa 1: Síntesis de 3,4-difluoro-2-metilbenzoato de metilo
A una disolución de ácido 3,4-difluoro-2-metilbenzoico (2 g, 11,62 mmoles, 1,0 equiv) en DMF (20 ml) se añadió K2CO3 (4,82 g, 34,86 mmoles, 3,0 equiv) y yodometano (3,26 ml, 52,29 mmoles, 4,5 equiv) a temperatura ambiente. La mezcla se agitó a temperatura ambiente durante 3 h. La disolución de 3,4-difluoro-2-metilbenzoato de metilo en DMF (20 ml) se usó directamente en la siguiente etapa.
Etapa 2: Síntesis de 4-((2-((terc-butoxicarbonil)amino)etil)tio)-3-fluoro-2-metilbenzoato de metilo
A una disolución de 3,4-difluoro-2-metilbenzoato de metilo (2,16 g, 11,28 mmoles, 1,0 equiv) en DMF (20 ml) se añadió (2-mercaptoetil)carbamato de terc-butilo (2,0 g, 11,28 mmoles, 1 equiv) y K2CO3 (3,12 g, 22,56 mmoles, 2,0 equiv) a temperatura ambiente. La reacción se agitó a 110 °C durante 12 h, momento en el que la mezcla se añadió a H2O (50 ml). La disolución acuosa se extrajo entonces con EtOAc (3 × 30 ml) y la fase orgánica se combinó y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 3/1 de éter de petróleo/EtOAc) proporcionando 4-((2-((terc-butoxicarbonil)amino)etil)tio)-3-fluoro-2-metilbenzoato de metilo (3,0 g, 76 % de rendimiento) como un sólido amarillo claro.
Etapa 3: Síntesis de 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-3-fluoro-2-metilbenzoato de metilo
A una disolución de 4-((2-((terc-butoxicarbonil)amino)etil)tio)-3-fluoro-2-metilbenzoato de metilo (3,3 g, 9,61 mmoles, 1,0 equiv), NaOH (2 M, 4,80 ml, 1,0 equiv) y NaHCO3 (2,42 g, 28,83 mmoles, 3,0 equiv) en acetona (30 ml) se añadió peroximonosulfato de potasio (12,35 g, 20,08 mmoles, 2,1 equiv). La mezcla se agitó durante 12 h a temperatura ambiente y entonces la mezcla se acidificó hasta pH 5 mediante la adición de HCl 1 N. La fase acuosa se extrajo con EtOAc (3 × 30 ml) y la fase orgánica combinada se lavó con salmuera (20 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 3/1 de éter de petróleo/EtOAc) proporcionando 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-3-fluoro-2-metilbenzoato de metilo (2,1 g, 58,2 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M-56 H] calculado para C16H22FNO6S: 320,12; observado 320,1
Etapa 4: Síntesis de ácido 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-3-fluoro- 2-metilbenzoico
A una disolución de 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-3-fluoro-2-metilbenzoato de metilo (2,1 g, 5,59 mmoles, 1,0 equiv) en THF (20 ml), MeOH (10 ml) y H2O (10 ml) se añadió LiOH•H2O (704,16 mg, 16,78 mmoles, 3,0 equiv) a temperatura ambiente. La mezcla de reacción se agitó a 40 °C durante 4 h. La mezcla se concentró entonces a presión reducida para retirar el THF y el MeOH. La fase acuosa se neutralizó con HCl 0,5N y entonces se extrajo con EtOAc (5 × 20 ml). La fase orgánica combinada se lavó con salmuera (2 × 20 ml), se secó con Na2SO4
anhidro, se filtró y se concentró a presión reducida dando ácido 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-3-fluoro-2-metilbenzoico (2,01 g, 97,1 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M-100 H] calculado para C15H20FNO6S: 262,11; observado 262,1.
Etapa 5: Síntesis de ácido (4-(terc-butoxicarbonil)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepin-7-il)borónico A una disolución de 7-bromo-2,3-dihidrobenzo[f][1,4]oxazepino-4(5H)-carboxilato de terc-butilo (4 g, 12,19 mmoles, 1,0 equiv) en THF (80 ml) a -60 °C se añadió B(OiPr)3 (4,58 g, 24,38 mmoles, 5,60 ml, 2,0 equiv), seguido de la adición gota a gota de n-BuLi (2,5 M, 12,19 ml, 2,5 equiv) en n-hexano. La reacción se agitó a -65 °C durante 1 h. La mezcla de reacción se inactivó con HCl 1 N (12,25 ml) y se dejó que se calentara hasta temperatura ambiente. La mezcla de reacción se extrajo con EtOAc (3 × 30 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida dando ácido (4-(terc-butoxicarbonil)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepin-7-il)borónico (3,5 g, en bruto) como un aceite amarillo claro, que se usó en la siguiente etapa directamente. LCMS (ESI) m/z: [M-100 H] calculado para C14H20BNO5: 194,15; observado 194,2.
Etapa 6: Síntesis de 7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepino-4(5H)-carboxilato de terc-butilo A una disolución de ácido (4-(terc-butoxicarbonil)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepin-7-il)borónico (4,2 g, 14,33 mmoles, 1,0 equiv) en H2O (20 ml) y dioxano (60 ml) se añadió 5-bromopiridin-2-amina (2,48 g, 14,33 mmoles, 1,0 equiv), Pd(dppf)Cl2•DCM (1,17 g, 1,43 mmoles, 0,1 equiv) y Et3N (4,35 g, 42,99 mmoles, 5,98 ml, 3,0 equiv) a temperatura ambiente. La mezcla se agitó a 85 °C durante 12 h. La mezcla se enfrió entonces hasta temperatura ambiente y el residuo se vertió en H2O (15 ml). La fase acuosa se extrajo con EtOAc (3 × 40 ml) y la fase orgánica combinada se lavó con salmuera (2 × 40 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 1/8 éter de petróleo/EtOAc) proporcionando 7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepino-4(5H)-carboxilato de terc-butilo (3,3 g, 65,0 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C19H23N3O3: 342,18; observado 342,2.
Etapa 7: Síntesis de 5-(2,3,4,5-tetrahidrobenzo[f][1,4]oxazepin-7-il)piridin-2-amina
A una disolución de 7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepino-4(5H)-carboxilato de terc-butilo (3,3 g, 9,67 mmoles, 1,0 equiv) en THF (40 ml) se añadió HCl en EtOAc (4 M, 100 ml, 41,38 equiv) a temperatura ambiente. La mezcla se agitó durante 3 h. La mezcla de reacción se filtró y la torta de filtración se lavó con EtOAc (3 × 15 ml) y entonces se secó a presión reducida dando 5-(2,3,4,5-tetrahidrobenzo[f][1,4]oxazepin-7-il)piridin-2-amina (3 g, 95,1 % de rendimiento, 2HCl) como un sólido amarillo claro.
Etapa 8: Síntesis de (2-((4-(7-(6-aminopiridin-3-il)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepino-4-carbonil)-2-fluoro-3-metilfenil)sulfonil)etil)carbamato de terc-butilo
A una disolución de ácido 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-3-fluoro-2-metilbenzoico (690,08 mg, 1,91 mmoles, 1,0 equiv) en DMF (10 ml) se añadió HATU (1,09 g, 2,86 mmoles, 1,5 equiv) y DIPEA (1,66 ml, 9,55 mmoles, 5 equiv). La reacción se agitó a temperatura ambiente durante 30 min y entonces se añadió 5-(2,3,4,5-tetrahidrobenzo[f][1,4]oxazepin-7-il)piridin-2-amina (0,6 g, 1,91 mmoles, 1,0 equiv, 2HCl). La mezcla se agitó durante 2 h, momento en el que se añadió H2O (40 ml). La mezcla se agitó durante 5 min y el precipitado resultante se recogió por filtración dando el producto en bruto. El residuo se purificó por cromatografía en gel de sílice (1/0 a 10/1 EtOAc/MeOH) proporcionando (2-((4-(7-(6-aminopiridin-3-il)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepino-4-carbonil)-2-fluoro-3-metilfenil)sulfonil)etil)carbamato de terc-butilo (0,538 g, 47,4 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C29H33FN4O6S: 585,22; observado 585,3.
Etapa 9: Síntesis de 2,2,2-trifluoroacetato de (4-((2-aminoetil)sulfonil)-3-fluoro-2-metilfenil)(7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)metanona
Una disolución (2-((4-(7-(6-aminopiridin-3-il)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepino-4-carbonil)-2-fluoro-3-metilfenil)sulfonil)etil)carbamato de terc-butilo (0,538 g, 920,20 µmoles, 1,0 equiv) en TFA (10,35 ml, 139,74 mmoles, 151,85 equiv) se agitó a temperatura ambiente durante 2 h. Entonces se concentró la disolución a presión reducida. El residuo aceitoso se trituró con MeCN (1 ml) y entonces se añadió gota a gota en MTBE (30 ml) durante 10 min. El sobrenadante se retiró y entonces el precipitado se recogió por filtración bajo N2 dando 2,2,2-trifluoroacetato de (4-((2-aminoetil)sulfonil)-3-fluoro-2-metilfenil)(7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)metanona (0,50 g, 87,4 % de rendimiento) como un sólido marrón claro. LCMS (ESI) m/z: [M H] calculado para C24H25FN4O4S: 485,17; observado 485,1.
Monómero AA. Sal de ácido trifluoroacético de 5-(4-amino-1-(6-(piperazin-1-il)pirimidin-4-il)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 1-(6-cloropirimidin-4-il)-3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina
A una suspensión de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (5 g, 19,16 mmoles, 1,0 equiv) en DMF (60 ml) se añadió NaH (804,53 mg, 20,11 mmoles, 60 % en peso, 1,05 equiv) a 0 °C. La mezcla se agitó a 0 °C durante 30 min. A la mezcla de reacción entonces se añadió 4,6-dicloropirimidina (3,42 g, 22,99 mmoles, 1,2 equiv) a 0 °C. La mezcla se agitó a temperatura ambiente durante 2,5 h, momento en el que la mezcla de reacción se añadió a H2O (600 ml). La suspensión se filtró entonces dando el producto (7,1 g, 99,2 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C9H5ClIN7: 373,94; observado 373,9.
Etapa 2: Síntesis de 4-(6-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)pirimidin-4-il)piperazin-1-carboxilato de terc-butilo
A una disolución de 1-(6-cloropirimidin-4-il)-3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (5 g, 13,39 mmoles, 1,0 equiv) y piperazin-1-carboxilato de terc-butilo (2,99 g, 16,06 mmoles, 1,2 equiv) en DMF (50 ml) se añadió K2CO3 (3,70 g, 26,77 mmoles, 2,0 equiv). La mezcla de reacción se agitó a 100 °C durante 4 h, momento en el que se añadió a H2O (500 ml). La suspensión se filtró entonces dando el producto (6,2 g, 88,5 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C18H22IN9O2: 524,09; observado 524,2.
Etapa 3: Síntesis de 4-(6-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)pirimidin-4-il)piperazin-1-carboxilato de terc-butilo
A una suspensión bifásica de 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (3,08 g, 11,85 mmoles, 1,0 equiv), 4-(6-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (6,2 g, 11,85 mmoles, 1,0 equiv) y Na2CO3 (6,28 g, 59,24 mmoles, 5,0 equiv) en H2O (100 ml) y DME (200 ml) se añadió Pd(PPh3)4 (1,37 g, 1,18 mmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 24 h y entonces la mezcla se filtró dando una torta sólida. El sólido se añadió a dioxano (20 ml) y se agitó a 110 °C durante 60 min, entonces se filtró dando el producto (3,5 g, 55,8 % de rendimiento) como un sólido marrón. LCMS (ESI) m/z: [M H] calculado para C25H27N11O3: 530,24; observado 530,3.
Etapa 4: Síntesis de sal de ácido trifluoroacético de 5-(4-amino-1-(6-(piperazin-1-il)pirimidin-4-il)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina
Una disolución de 4-(6-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)pirimidin-4-il)piperazin-1-carboxilato de terc-butilo (3,5 g, 6,61 mmoles, 1,0 equiv) en TFA (35 ml) se agitó a temperatura ambiente durante 1 h. La disolución de reacción se concentró a presión reducida y el material resultante en bruto se disolvió en MeCN (20 ml) y se añadió gota a gota a MTBE (500 ml). El sólido resultante se filtró entonces dando el producto (5,5 g, 91,9 % de rendimiento) como un sólido marrón. LCMS (ESI) m/z: [M H] calculado para C20H19N11O: 430,19; observado 430,1.
Monómero AB. Sal de ácido trifluoroacético de 8-(6-metoxipiridin-3-il)-3-metil-1-(4-(4-(5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)-3-(trifluorometil)fenil)-1H-imidazo[4,5-c]quinolin-2(3H)-ona.

Etapa 1: Síntesis de 2-(4-(4-(8-(6-metoxipiridin-3-il)-3-metil-2-oxo-2,3-dihidro-1H-imidazo[4,5-c]quinolin-1-il)-2-(trifluorometil)fenil)piperazin-1-il)-7, 8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una mezcla de 8-(6-metoxipiridin-3-il)-3-metil-1-(4-(piperazin-1-il)-3-(trifluorometil)fenil)-1H-imidazo[4,5-c]quinolin-2(3H)-ona (0,3 g, 561,24 µmoles, 1,0 equiv) y 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (151,38 mg, 561,24 µmoles, 1,0 equiv) en DMF (5 ml) se añadió K2CO3 (193,92 mg, 1,40 mmoles, 2,5 equiv). La mezcla se agitó a 100 °C durante 14 h, momento en el que se añadió H2O (20 ml). La fase acuosa se extrajo con EtOAc (3 × 40 ml) y las fases orgánicas combinadas se concentraron a presión reducida. El material en bruto se purificó por cromatografía en columna (30/1 a 15/1 DCM/MeOH) dando el producto (0,30 g, 69,6 % de rendimiento) como un sólido de color amarillo claro. LCMS (ESI) m/z: [M H] calculado para C40H40F3N9O4: 768,33; observado 768,5.
Etapa 2: Síntesis de 8-(6-metoxipiridin-3-il)-3-metil-1-(4-(4-(5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)-3-(trifluorometil)fenil)-1H-imidazo[4,5-c]quinolin-2(3H)-ona
Una disolución de 2-(4-(4-(8-(6-metoxipiridin-3-il)-3-metil-2-oxo-2,3-dihidro-1H-imidazo[4,5-c]quinolin-1-il)-2-(trifluorometil)fenil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (0,8 g, 1,04 mmoles, 1,0 equiv) en TFA (8 ml) se agitó a temperatura ambiente durante 2 h. El disolvente se concentró y el residuo se disolvió en MeCN (5 ml), entonces la disolución se añadió gota a gota a MTBE (150 ml). El precipitado se filtró y el sólido se secó a presión reducida dando el producto (600 mg, 70,6 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C35H32F3N9O2: 668,27; observado 668,3.
Monómero AC. Sal de ácido trifluoroacético 5-(4-amino-1-(piperidin-4-ilmetil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 4-((metilsulfonil)oxi)piperidin-1-carboxilato de terc-butilo
A una disolución de 4-hidroxipiperidin-1-carboxilato de terc-butilo (4 g, 19,87 mmoles, 1,0 equiv) y Et3N (3,87 ml, 27,82 mmoles, 1,4 equiv) en DCM (40 ml) se añadió MsCl (2,15 ml, 27,82 mmoles, 1,4 equiv) a 0 °C. Entonces la mezcla de reacción se agitó a temperatura ambiente durante 1 h. Se añadió H2O (50 ml) y la fase acuosa se extrajo con DCM (3 × 50 ml). La fase orgánica combinada se lavó con salmuera, se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida dando el producto (5,62 g, rendimiento en bruto del 101 %) como un sólido amarillo que se usó directamente en la siguiente etapa.
Etapa 2: Síntesis de 4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)piperidin-1-carboxilato de terc-butilo A una suspensión de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (5 g, 19,16 mmoles, 1,0 equiv) y 4-((metilsulfonil)oxi)piperidin-1-carboxilato de terc-butilo (5,62 g, 20,11 mmoles, 1,05 equiv) en DMF (100 ml) se añadió K2CO3 (5,29 g, 38,31 mmoles, 2,0 equiv). La mezcla se agitó a 80 °C durante 12 h. La mezcla de reacción se añadió entonces a H2O (400 ml) a 0 °C. El precipitado resultante se filtró dando el producto (5,0 g, 58,8 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C15H21IN6O2: 445,09; observado 445,1.
Etapa 3: Síntesis de 4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)piperidin-1-carboxilato de terc-butilo
A una suspensión de 4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)piperidin-1-carboxilato de terc-butilo (5 g, 11,25 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (3,51 g, 13,51 mmoles, 1,2 equiv) y Na2CO3 (5,96 g, 56,27 mmoles, 5,0 equiv) en H2O (50 ml) y DME (100 ml) se añadió Pd(PPh3)4 (1,30 g, 1,13 mmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. La mezcla de reacción se enfrió entonces hasta temperatura ambiente y se filtró. El filtrado se repartió entre EtOAc (100 ml) y H2O (100 ml) y entonces la fase acuosa se separó y se extrajo con EtOAc (3 × 100 ml). La fase orgánica combinada se lavó con salmuera (20 ml) y se secó sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se trituró con EtOAc (30 ml) y se filtró dando el producto (3,6 g, 71 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C22H26N8O3: 451,22; observado 451,3.
Etapa 4: Síntesis de sal de ácido trifluoroacético de 5-(4-amino-1-(piperidin-4-il)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina
Una disolución de 4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)piperidin-1-carboxilato de terc-butilo (1,4 g, 3,11 mmoles, 1,0 equiv) en TFA (10 ml) se agitó a temperatura ambiente durante 30 min. La disolución de reacción se concentró a presión reducida y el sólido en bruto se disolvió en MeCN (20 ml). La disolución se añadió gota a gota a MTBE (100 ml) y el sólido resultante se filtró dando el producto (1,6 g, 85,8 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C17H18N8O3: 351,17; observado 351,1.
Monómero AD. Sal de ácido trifluoroacético de 1-(piperidin-4-il)-3-(1H-pirrolo[2,3-b]piridin-5-il)-1H-pirazolo[3,4-d]pirimidin-4-amina.

Etapa 1: Síntesis de 4-(4-amino-3-(1H-pirrolo[2,3-b]piridin-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)piperidin-1-carboxilato de terc-butilo
A una suspensión de 5-(4,4,5-trimetil-1,3,2-dioxaborolan-2-il)-1H-pirrolo[2,3-b]piridina (857,12 mg, 3,51 mmoles, 1,2 equiv), 4-(4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)piperidin-1-carboxilato de terc-butilo (1,3 g, 2,93 mmoles, 1,0 equiv) y Na2CO3 (1,55 g, 14,63 mmoles, 5,0 equiv) en DME (20 ml) y H2O (10 ml) se añadió Pd(PPh3)4 (338,13 mg, 292,62 µmoles, 0,1 equiv) a temperatura ambiente bajo N2. La mezcla se agitó a 110 °C durante 3 h. La mezcla de reacción se enfrió entonces hasta temperatura ambiente y se filtró. El filtrado se repartió entre EtOAc (50 ml) y H2O (50 ml) y la fase acuosa se separó y se extrajo con EtOAc (3 × 50 ml). La fase orgánica combinada se lavó con salmuera, se secó sobre anhidro Na2SO4, se filtró y se concentró a presión reducida. El residuo se trituró con EtOAc (10 ml), se filtró, la torta sólida se secó a presión reducida dando el producto (1,0 g, 78,7 % de rendimiento) como un sólido amarillo.
Etapa 2: Síntesis de sal de ácido trifluoroacético de 1-(piperidin-4-il)-3-(1H-pirrolo[2,3-b]piridin-5-il)-1H-pirazolo[3,4-d]pirimidin-4-amina
Una disolución de 4-(4-amino-3-(1H-pirrolo[2,3-b]piridin-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)piperidin-1-carboxilato de terc-butilo (1,5 g, 3,45 mmoles, 1,0 equiv) en TFA (10 ml) se agitó a temperatura ambiente durante 30 min. La disolución de reacción se concentró a presión reducida y el residuo en bruto se disolvió en MeCN (20 ml). La disolución se añadió gota a gota a MTBE (100 ml) y el sólido resultante se filtró dando el producto (1,19 g, 74,2 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C17H18N8: 335,18; observado 335,1.
Monómero AE. (4-((2-aminoetil)sulfonil)-2-metilfenil)(7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)metanona.

Etapa 1: Síntesis de 4-fluoro-2-metilbenzoato de metilo
A una disolución de ácido 4-fluoro-2-metilbenzoico (86 g, 557,94 mmoles, 1,0 equiv) en DMF (900 ml) se añadió K2CO3 (231,33 g, 1,67 moles, 3,0 equiv) y yodometano (79,19 g, 557,94 mmoles, 34,73 ml, 1,0 equiv). La mezcla se agitó a temperatura ambiente durante 1 h. La disolución de 4-fluoro-2-metilbenzoato de metilo en DMF (900 ml) se usó directamente en la siguiente etapa.
Etapa 2: Síntesis de 4-((2-((terc-butoxicarbonil)amino)etil)tio)-2-metilbenzoato de metilo
A una disolución de 4-fluoro-2-metilbenzoato de metilo (93,8 g, 557,94 mmoles, 1,0 equiv) en DMF (900 ml) se añadió (2-mercaptoetil)carbamato de terc-butilo (98,91 g, 557,97 mmoles, 1,0 equiv) y K2CO3 (154,23 g, 1,12 moles, 2,0 equiv). La reacción se agitó a 110 °C durante 12 h, momento en el que la mezcla se enfrió hasta temperatura ambiente y se añadió a H2O (1000 ml). La fase acuosa se extrajo entonces con EtOAc (3 × 600 ml) y las fases orgánicas combinadas se lavaron con salmuera, se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (0→25 % de EtOAc/éter de petróleo) dio el producto deseado como un aceite incoloro (144 g, 79 % de rendimiento).
Etapa 3: Síntesis de 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-2-metilbenzoato de metilo
A dos lotes separados que contenían una disolución de 4-((2-((terc-butoxicarbonil)amino)etil)tio)-2-metilbenzoato de metilo (72 g, 221,25 mmoles, 1,0 equiv), NaOH (2 M, 110,6 ml, 1,0 equiv) y NaHCO3 (55,76 g, 663,75 mmoles, 3,0 equiv) en acetona (750 ml) se añadió peroximonosulfato de potasio (284,28 g, 462,41 mmoles, 2,1 equiv). La mezcla se agitó durante 12 h a temperatura ambiente, momento en el que los dos lotes se combinaron y entonces la mezcla se acidificó hasta pH 5 mediante la adición de HCl 1 N. La fase acuosa se extrajo con EtOAc (3 × 1500 ml) y las fases orgánicas combinadas se lavaron con salmuera (2 × 500 ml), se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (0→25 % de EtOAc/éter de petróleo) dio el producto deseado como un sólido blanco (120 g, 76 % de rendimiento).
Etapa 4: Síntesis de ácido 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-2-metilbenzoico
A una disolución de 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-2-metilbenzoato de metilo (35 g, 97,92 mmoles, 1,0 equiv) en THF (200 ml), MeOH (100 ml) y H2O (100 ml) se añadió LiOH•H2O (12,33 g, 293,77 mmoles, 3,0 equiv) a temperatura ambiente. La mezcla de reacción se agitó a 40 °C durante 1 h. La mezcla se concentró entonces a presión reducida para retirar el THF y el MeOH. La fase acuosa se neutralizó con HCl 0,5 N y el precipitado resultante se aisló por filtración. La torta sólida se lavó con H2O (3 × 20 ml) proporcionando el producto deseado como un sólido blanco (25 g, 74 % de rendimiento).
Etapa 5: Síntesis de (2-((4-(7-(6-aminopiridin-3-il)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepino-4-carbonil)-3-metilfenil)sulfonil)etil)carbamato de terc-butilo
A una disolución de ácido 4-((2-((terc-butoxicarbonil)amino)etil)sulfonil)-2-metilbenzoico (9,7 g, 28,25 mmoles, 1,0 equiv) y 5-(2,3,4,5-tetrahidrobenzo[f][1,4]oxazepin-7-il)piridin-2-amina (8,88 g, 28,25 mmoles, 1,0 equiv, 2HCl) en DMF (120 ml) se añadió HATU (16,11 g, 42,37 mmoles, 1,5 equiv) y DIPEA (18,25 g, 141,24 mmoles, 24,60 ml, 5,0 equiv). La reacción se agitó a temperatura ambiente durante 1 h, momento en el que la mezcla de reacción se vertió en H2O (1000 ml). La mezcla se agitó durante 5 min y el precipitado resultante se recogió por filtración dando el producto en bruto. El producto en bruto se trituró con EtOAc (100 ml), se filtró y la torta sólida se secó a presión reducida proporcionando el producto deseado como un sólido blanco (14 g, 87 % de rendimiento).
Etapa 6: Síntesis de (4-((2-aminoetil)sulfonil)-2-metilfenil)(7-(6-aminopiridin-3-il)-2,3-dihidrobenzo[f][1,4]oxazepin-4(5H)-il)metanona
Una disolución (2-((4-(7-(6-aminopiridin-3-il)-2,3,4,5-tetrahidrobenzo[f][1,4]oxazepino-4-carbonil)-3-metilfenil)sulfonil)etil)carbamato de terc-butilo (19 g, 33,53 mmoles, 1,0 equiv) en TFA (100 ml) se agitó a temperatura ambiente durante 30 min. La disolución se concentró entonces a presión reducida. El residuo se trituró con MeCN (30 ml) y entonces se añadió gota a gota en MTBE (600 ml) y se agitó durante 20 min. La suspensión se filtró y el sólido resultante se disolvió en MeCN (30 ml) y se concentró a presión reducida proporcionando el producto deseado como un sólido amarillo claro (24 g, sal de TFA). LCMS (ESI) m/z: [M H] calculado para C24H26N4O4S:467,18; observado 467,1.
Monómero AF. 5-(4-amino-1-((5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de 3-((dimetilamino)metileno)-4-oxopiperidin-1-carboxilato de (Z)-terc-butilo
Una disolución de 4-oxopiperidin-1-carboxilato de terc-butilo (15 g, 75,28 mmoles, 1,0 equiv) y 1,1-dimetoxi-N,N-dimetilmetanamina (11,00 ml, 82,81 mmoles, 1,1 equiv) en DMF (105 ml) se agitó a 95 °C durante 12 h. La mezcla de reacción se concentró entonces a presión reducida y el residuo resultante se disolvió en EtOAc (30 ml) y se lavó con salmuera (3 × 30 ml). La fase acuosa se extrajo con EtOAc (50 ml), y las fases orgánicas combinadas se secaron y se concentraron a presión reducida proporcionando el producto deseado como un sólido amarillo (10,1 g, 53 % de rendimiento).
Etapa 2: Síntesis de 2-(hidroximetil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de NaOEt (1,98 g, 29,10 mmoles, 1,0 equiv) en EtOH (70 ml) se añadió 3-((dimetilamino)metileno)-4-oxopiperidin-1-carboxilato de (Z)-terc-butilo (7,4 g, 29,10 mmoles, 1,0 equiv) y clorhidrato de 2-hidroxiacetimidamida (3,54 g, 32,01 mmoles, 1,1 equiv). La mezcla de reacción se calentó hasta 90 °C durante 12 h, momento en el que la mezcla se enfrió hasta temperatura ambiente y se concentró a presión reducida. El residuo se repartió con EtOAc (40 ml) y se lavó con NaHCO3 sat. (40 ml). La fase acuosa se extrajo con EtOAc (3 × 20 ml) y las fases orgánicas combinadas se lavaron con salmuera (2 × 50 ml), se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (25 % de EtOAc/éter de petróleo) dio el producto deseado como un sólido amarillo (7,24 g, 94 % de rendimiento).
Etapa 3: Síntesis de 2-(bromometil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de 2-(hidroximetil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (6,24 g, 23,52 mmoles, 1,0 equiv) y PPh3 (12,34 g, 47,04 mmoles, 2,0 equiv) en DCM (140 ml) se añadió CBr4 (14,82 g, 44,69 mmoles, 1,9 equiv). La mezcla se agitó a temperatura ambiente durante 3 h, momento en el que mezcla se concentró a presión reducida. El residuo se repartió entre EtOAc (20 ml) y H2O (20 ml), la fase acuosa se extrajo con EtOAc (3 × 20 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 × 50 ml), se secaron y se concentraron
a presión reducida. La purificación por cromatografía en gel de sílice (14 % de EtOAc/éter de petróleo) dio el producto deseado como un sólido amarillo (3,6 g, 47 % de rendimiento).
Etapa 4: Síntesis de 2-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (1,59 g, 6,09 mmoles, 1,0 equiv) en DMF (15 ml) se añadió NaH (243,73 mg, 6,09 mmoles, 60 % en peso, 1,0 equiv) a 0 °C. La suspensión se agitó durante 30 min y entonces se añadió 2-(bromometil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (2,2 g, 6,70 mmoles, 1,1 equiv). La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 3 h. La mezcla se vertió en H2O a 0 °C y el precipitado se recogió por filtración proporcionando el producto deseado como un sólido marrón (2,5 g, 66 % de rendimiento).
Etapa 5: Síntesis de 2-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de 2-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (4,55 g, 8,95 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (2,79 g, 10,74 mmoles, 1,2 equiv) y Na2CO3 (4,74 g, 44,76 mmoles, 5,0 equiv) en dioxano (70 ml) y H2O (35 ml) se añadió Pd(PPh3)4 (1,03 g, 895,11 µmoles, 0,1 equiv). La mezcla de reacción se calentó hasta 110 °C durante 3 h, momento en el que la mezcla se enfrió hasta temperatura ambiente y se vertió en H2O a 0 °C. El precipitado se filtró, y la torta sólida se secó a presión reducida. El producto en bruto se lavó con EtOAc (50 ml) proporcionando el producto deseado como un sólido amarillo claro (3,14 g, 68 % de rendimiento).
Etapa 6: Síntesis de 5-(4-amino-1-((5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina
Una disolución de 2-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (3,14 g, 6,10 mmoles, 1,0 equiv) en TFA (20 ml) se agitó a temperatura ambiente durante 30 min. La mezcla se concentró a presión reducida y el residuo resultante se añadió disuelto en MeCN (7 ml) y se añadió a MTBE (700 ml). El precipitado se recogió por filtración proporcionando el producto deseado como un sólido marrón (4,25 g, 92 % de rendimiento, 3 TFA). LCMS (ESI) m/z: [M H] calculado para C20H18N10O: 415,18; observado 415,1.
Monómero AG. 5-(4-amino-1-((2-((2-aminoetil)sulfonil)-1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina.

Etapa 1: Síntesis de sal de tetrabutilamonio de N-Boc taurina
A una disolución de ácido 2-aminoetanosulfónico (10,00 ml, 79,91 mmoles, 1,0 equiv) en THF (60 ml) y NaOH acuoso (2 M, 40 ml, 1,0 equiv) se añadió Boc2O (18,31 g, 83,90 mmoles, 1,05 equiv). La mezcla se agitó a temperatura ambiente durante 15 h, momento en el que la mezcla se extrajo con EtOAc (10 ml). La fase acuosa se diluyó con H2O (450 ml), se trató con LiOH•H2O (3,35 g, 79,83 mmoles, 1,0 equiv) y nBu4NHSO4 (27,13 g 79,90 mmoles, 1,0 equiv) y se agitó durante 30 min. Esta mezcla se extrajo con DCM (3 × 80 ml), y las fases orgánicas combinadas se secaron y se concentraron a presión reducida proporcionando el producto deseado como un aceite incoloro (34,26 g, 91 % de rendimiento).
Etapa 2: Síntesis de (2-(clorosulfonil)etil)carbamato de terc-butilo
A una disolución de sal de tetrabutilamonio de N-Boc taurina (4,7 g, 10,05 mmoles, 1,0 equiv) en DCM (42 ml) se añadió DMF (77,32 µl, 1,00 mmol, 0,1 equiv) seguido de una disolución de trifosgeno (0,5 M, 8,04 ml, 0,4 equiv) en DCM a 0 °C. La mezcla se calentó hasta temperatura ambiente y se agitó durante 30 min. La disolución de (2-(clorosulfonil)etil)carbamato de terc-butilo (2,45 g, en bruto) en DCM se usó directamente en la siguiente etapa. Etapa 3: Síntesis de (2-((6-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-il)sulfonil)etil)carbamato de terc-butilo
A una disolución de 5-(4-amino-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (6,04 g, 9,44 mmoles, 1,0 equiv, 2TFA) en DMF (40 ml) se añadió Et3N (7,88 ml, 56,63 mmoles, 6,0 equiv). Se añadió una disolución de (2-(clorosulfonil)etil)carbamato de terc-butilo en DCM (42 ml) a 0 °C. La mezcla se calentó hasta temperatura ambiente y se agitó a 16 h. La mezcla de reacción se concentró a presión reducida para retirar el DCM y la disolución resultante se purificó por cromatografía de fase inversa (15→45 % de MeCN/H2O) proporcionando el producto deseado como un sólido blanco (5,8 g, 83 % de rendimiento, TFA). LCMS (ESI) m/z: [M H] calculado para C29H33N9O5S: 620,24; observado 620,3.
Etapa 4: Síntesis de 5-(4-amino-1-((2-((2-aminoetil)sulfonil)-1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina
Una disolución de (2-((6-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-il)sulfonil)etil)carbamato de terc-butilo (5,8 g, 9,36 mmoles, 1,0 equiv) en TFA (48 ml) se agitó a temperatura ambiente durante 0,5 h, momento en el que la mezcla de reacción se concentró a presión reducida. El producto en bruto se disolvió en MeCN (30 ml) y se añadió gota a gota en MTBE (200 ml). La mezcla se agitó durante 5 min y se filtró, la torta de filtración se secó a presión reducida proporcionando el producto deseado como un sólido amarillo (3,6 g, 62 % de rendimiento, 2,2TFA). LCMS (ESI) m/z: [M H] calculado para C24H25N9O3S: 520,19; observado 520,1.
Monómero AH. ((5-((4-Amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirimidin-2-il)metil)carbamato de terc-butilo

Etapa 1: Síntesis de metanosulfonato de (2-(((terc-butoxicarbonil)amino)metil)pirimidin-5-il)metilo
A una disolución de ((5-(hidroximetil)pirimidin-2-il)metil)carbamato de terc-butilo (4,2 g, 17,55 mmoles, 1,0 equiv) en DCM (42 ml) a 0 °C se añadió Et3N (7,33 ml, 52,66 mmoles, 3,0 equiv) seguido de MsCl (2,41 g, 21,06 mmoles, 1,63 ml, 1,2 equiv). La mezcla se agitó a 0 °C durante 10 min, y entonces se añadió H2O (15 ml). La mezcla de reacción se extrajo con DCM (5 × 10 ml) y las fases orgánicas combinadas se lavaron con salmuera (5 ml), se secaron, se filtraron y se concentraron a presión reducida proporcionando el producto deseado (5,5 g, 98,7 % de rendimiento) como un sólido incoloro.
Etapa 2: Síntesis de ((5-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirimidin-2-il)metil)carbamato de terc-butilo
A una disolución de metanosulfonato de (2-(((terc-butoxicarbonil)amino)metil)pirimidin-5-il)metilo (5,47 g, 17,24 mmoles, 1,2 equiv) y 3-yodo-1H-pirazolo[3,4-d]pirimidin-4-amina (3,75 g, 14,37 mmoles, 1,0 equiv) en DMF
(55 ml) a temperatura ambiente se añadió K2CO3 (5,96 g, 43,10 mmoles, 3 equiv). La mezcla se agitó a 80 °C durante 5 h, momento en el que se vertieron H2O (100 ml) y salmuera (20 ml) en la mezcla de reacción. La disolución se extrajo con EtOAc (10 × 30 ml) y las fases orgánicas combinadas se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (0→30 % de EtOAc/MeOH) dio el producto deseado (2 g, 28,9 % de rendimiento) como un sólido amarillo.
Etapa 3: Síntesis de ((5-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirimidin-2-il)metil)carbamato de terc-butilo
A una disolución de ((5-((4-amino-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirimidin-2-il)metil)carbamato de terc-butilo (2 g, 4,15 mmoles, 1,0 equiv), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1,3-benzoxazol-2-amina (1,13 g, 4,35 mmoles, 1,05 equiv) y Na2CO3 (688,39 mg, 8,29 mmoles, 2,0 equiv) en dioxano (20 ml) y H2O (10 ml) se añadió Pd(PPh3)4 (479,21 mg, 414,70 µmoles, 0,1 equiv). La mezcla se agitó a 110 °C durante 1 h, momento en el que la mezcla se enfrió hasta temperatura ambiente, se filtró y la torta sólida se lavó con MeOH (3 × 10 ml). El filtrado se concentró a presión reducida para retirar el MeOH y entonces se añadió gota a gota en H2O (50 ml). La suspensión resultante se filtró, y la torta de filtración se lavó con H2O (3 × 10 ml). La torta sólida se agitó en MeOH (20 ml) durante 30 min. La suspensión resultante se filtró, y la torta de filtración se lavó con MeOH (3 × 8 ml). La torta de filtración se secó a presión reducida proporcionando el producto deseado (1,03 g, 48,9 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C23H24N10O3: 489,21; observado 489,2.
Etapa 4: Síntesis de 5-(4-amino-1-{[2-(aminometil)pirimidin-5-il]metil}-1H-pirazolo[3,4-d]pirimidin-3-il)-1,3-benzoxazol-2-amina
A ((5-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)pirimidin-2-il)metil)carbamato de terc-butilo (100 mg, 0,205 mmoles, 1,0 equiv) se añadió HCl conc. (850 µl, 10,2 mmoles, 50 equiv). La reacción se agitó durante 1 h y entonces se vertió en acetona (3 ml). El precipitado resultante se filtró, se lavó con acetona y se secó a presión reducida proporcionando el producto deseado (80 mg, 92 % de rendimiento) como un sólido marrón. LCMS (ESI) m/z: [M H] calculado para C18H16N10O: 389,16; observado 389,0.
Monómero Al. Sal de ácido trifluoroacético de 5-(4-(dimetilamino)-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina

Etapa 1: Síntesis de 6-((4-(dimetilamino)-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una disolución de 3-yodo-N,N-dimetil-1H-pirazolo[3,4-d]pirimidin-4-amina (3,6 g, 12,45 mmoles, 1,0 equiv) en DMF (36 ml) a 0 °C se añadió NaH (523,00 mg, 13,08 mmoles, 60 % en peso, 1,05 equiv). La mezcla se agitó a 0 °C durante 30 min. A la mezcla de reacción se añadió entonces una disolución de 6-(bromometil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (4,47 g, 13,70 mmoles, 1,1 equiv) en DMF (18 ml) a 0 °C. La mezcla se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se añadió entonces a H2O fría (200 ml) y se agitó durante 30 min. El precipitado resultante se recogió por filtración proporcionando el producto deseado (6 g, 71,9 % de rendimiento) como un sólido blanco.
Etapa 2: Síntesis de 6-((3-(2-aminobenzo[d]oxazol-5-il)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo
A una disolución de 6-((4-(dimetilamino)-3-yodo-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (2 g, 2,96 mmoles, 1,0 equiv) y 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[d]oxazol-2-amina (922,81 mg, 3,55 mmoles, 1,2 equiv) en dioxano (24 ml) y H2O (12 ml) se añadió Na2CO3 (1,57 g, 14,78 mmoles, 5,0 equiv) y Pd(PPh3)4 (341,66 mg, 295,66 µmoles, 0,1 equiv). La mezcla se agitó a 110 °C durante 12 h. La mezcla de reacción se vertió entonces en H2O fría (200 ml) y se agitó durante 30 min. El precipitado resultante se recogió por filtración. La purificación por cromatografía en gel de sílice (5→100 % éter de petróleo/EtOAc) dio el producto deseado (1,2 g, 72,3 % de rendimiento) como un sólido amarillo.
Etapa 3: Síntesis de 5-(4-(dimetilamino)-1-((1,2,3,4-tetrahidroisoquinolin-6-il)metil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina
Una disolución de 6-((3-(2-aminobenzo[d]oxazol-5-il)-4-(dimetilamino)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-carboxilato de terc-butilo (1,7 g, 3,14 mmoles, 1,0 equiv) en TFA (10 ml) se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se concentró entonces a presión reducida. El residuo se añadió a MeCN (10 ml) y la disolución se añadió gota a gota en MTBE (200 ml). El sólido resultante se disolvió en MeCN (30 ml) y la disolución se concentró a presión reducida proporcionando el producto deseado (1,67 g, 92,9 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C24H24N8O: 441,22; observado 441,2.
Monómero AJ. Ácido 4-amino-5-(2-aminobenzo[d]oxazol-5-il)-5H-pirimido[5,4-b]indol-7-carboxílico

Este monómero se puede preparar a partir de 7-metil-5H-pirimido[5,4-b]indol-4-ol por oxidación bencílica al ácido carboxílico, conversión en el éster etílico, seguido de O-etilación con tetrafluoroboroato de trietiloxonio. La arilación mediada por paladio, seguida de hidrólisis de éster y amoniólisis final, proporciona el monómero.
Monómero AK. Ácido 4-amino-5-(2-aminobenzo[d]oxazo-5-il)-5H-pirimido[5,4-b]indol-8-carboxílico.

Este monómero se puede preparar siguiendo una vía similar a aquella para preparar el monómero previo, pero usando el material de partida isomérico de 8-metil-5H-pirimido[5,4-b]indol-4-ol. La oxidación bencílica al ácido carboxílico, conversión en el éster etílico, seguida de O-etilación con tetrafluoroboroato de trietiloxonio y arilación mediada por paladio, seguida de hidrólisis de éster y amoniólisis final, proporciona el monómero.
Monómero AL. Ácido 3-(2,4-bis((S)-3-metilmorfolino)-4a,8a-dihidropirido[2,3-d]pirimidin-7-il)benzoico.

Etapa 1: Síntesis de (3S)-4-[7-cloro-2-[(3S)-3-metilmorfolin-4-il]pirido[2,3-d] pirimidin-4-il] 3-metil-morfolina A una disolución de 2,4,7-tricloropirido[2,3-d]pirimidina (4,0 g, 17,06 mmoles, 1,0 equiv) en DMA (10 ml) se añadió (3S)-3-metilmorfolina (4,31 g, 42,65 mmoles, 2,5 equiv) y DIPEA (5,51 g, 42,65 mmoles, 7,43 ml, 2,5 equiv). La disolución de reacción se calentó hasta 70 °C durante 48 h. La suspensión de reacción se enfrió hasta temperatura ambiente, se vertió en H2O fría (50 ml) para precipitar un sólido. El sólido se filtró y la torta de filtración se aclaró con H2O, y se secó a presión reducida dando el producto en bruto, que se purificó por cromatografía en columna sobre gel de sílice (0→100 % éter de petróleo/EtOAc) dando (3S)-4-[7-cloro-2-[(3S)-3-metilmorfolin-4-il]pirido[2,3-d] pirimidin-4-il] 3-metil-morfolina (3,5 g, 56,4 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C17H22ClN5O2: 364,15; observado 364,2
Etapa 2: Síntesis de ácido 3-[2,4-bis[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-7-il]benzoico
A una disolución de (3S)-4-[7-cloro-2-[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-4-il]-3-metil-morfolina (2 g, 5,50 mmoles, 1,0 equiv) y ácido 3-boronobenzoico (1,09 g, 6,60 mmoles, 1,2 equiv) en 1,4-dioxano (40 ml) se añadió una disolución de K2CO3 (911,65 mg, 6,60 mmoles, 1,2 equiv) en H2O (4 ml), seguido por Pd(PPh3)4 (317,60 mg, 274,85 µmoles, 0,05 equiv). La disolución se desgasificó durante 10 min y se rellenó con N2, entonces la mezcla de reacción se calentó hasta 100 °C bajo N2 durante 5 h. La reacción se enfrió hasta temperatura ambiente y se filtró. El filtrado se acidificó por HCl (2 N) a pH 3, y la fase acuosa se lavó con EtOAc (3 × 20 ml). La fase acuosa se concentró a presión reducida dando un residuo, que se purificó por cromatografía en columna sobre gel de sílice (50 %→100 % de éter de petróleo/EtOAc) dando clorhidrato de ácido 3-[2,4-bis[(3S)-3-metilmorfolin-4-il]pirido[2,3-d]pirimidin-7-il]benzoico (2,5 g, 89,9 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C24H27N5O4: 450,21; observado 450,2.
Referencia para la preparación de este monómero: Menear, K.; Smith, G.C.M.; Malagu, K.; Duggan, H.M.E.; Martin, N.M.B.; Leroux, F.G.M. 2012. Pyrido-, pyrazo- and pyrimido-pyrimidine derivatives as mTOR inhibitors. US8101602. Kudos Pharmaceuticals, Ltd.
Monómero AM. Ácido (1r,4r)-4-[4-amino-5-(7-metoxi-1H-indol-2-il)imidazo[4,3-f][1,2,4]triazin-7-il]ciclohexano-1-carboxílico

Este monómero, también conocido como OSI-027 (CAS N.º = 936890-98-1), es un compuesto comercialmente disponible. En el momento en que se preparó la presente solicitud, estuvo disponible para compra de varios vendedores.
Monómero AN. Ácido 2-(4-(4-(8-(6-metoxipiridin-3-il)-3-metil-2-oxo-2,3-dihidro-1H-imidazo[4,5-c]quinolin-1-il)-2-(trifluorometil)fenil)piperazin-1-il)pirimidin-5-carboxílico

La preparación de este monómero avanza haciendo reaccionar BGT226 con 2-cloropirimidin-5-carboxilato de metilo, seguido de hidrólisis de éster, dando el monómero del título.
Monómero AO. Ácido 4-amino-5-{1H-pirrolo[2,3-b]piridin-5-il}-5H-pirimido[5,4-b]indol-8-carboxílico

Este monómero se puede preparar a partir de 7-metil-5H-pirimido[5,4-b]indol-4-ol por oxidación bencílica al ácido carboxílico, conversión en el éster etílico, seguido de O-etilación con tetrafluoroboroato de trietiloxonio. La arilación mediada por paladio, seguida de hidrólisis de éster y amoniólisis final, proporciona el monómero.
Preparación de pre- y posconectores
Elemento estructural A. N-[(terc-butoxi)carbonil]-N-{[2-(piperazin-1-il)pirimidin-5-il]metil}carbamato de terc-butilo.

Etapa 1: Síntesis de 5-(bromometil)-2-cloropirimidina
A una disolución de 2-cloro-5-metilpirimidina (92 g, 715,62 mmoles, 1,0 equiv) en CCl4 (1000 ml) se añadió NBS (178,31 g, 1,00 moles, 1,4 equiv) y peróxido de benzoílo (3,47 g, 14,31 mmoles, 0,02 equiv). La mezcla se agitó a 76 °C durante 18 h. La mezcla de reacción se enfrió entonces hasta temperatura ambiente y se concentró a presión reducida. La mezcla de reacción se filtró y la torta sólida se lavó con DCM (150 ml). La disolución resultante se concentró a presión reducida dando el producto en bruto. El residuo se purificó por cromatografía en gel de sílice (1/0 a 0/1 de éter de petróleo/EtOAc) dando el producto (70,8 g, rendimiento en bruto del 47,7 %) como un aceite amarillo, que se usó directamente para la siguiente etapa. LCMS (ESI) m/z: [M H] calculado para C5H4BrClN2: 206,93; observado 206,9.
Etapa 2: Síntesis de N-terc-butoxicarbonil-N-((2-piperazin-1-ilpirimidin-5-il)metil)carbamato de terc-butilo A una disolución de N-terc-butoxicarbonilcarbamato de terc-butilo (36,89 g, 169,79 mmoles, 0,74 equiv) en DMF (750 ml) se añadió NaH (6,88 g, 172,09 mmoles, 60 % en peso, 0,75 equiv) a 0 °C. La mezcla se agitó a 0 °C durante 30 min. Entonces, se añadió 5-(bromometil)-2-cloropirimidina (47,6 g, 229,45 mmoles, 1,0 equiv) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 15,5 h. La mezcla se vertió entonces en H2O (1600 ml) y la fase acuosa se extrajo con EtOAc (3 × 300 ml). La fase orgánica combinada se lavó con salmuera (2 × 200 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 0/1 de éter de petróleo/EtOAc) dando el producto (70 g, en bruto) como un sólido amarillo, que se usó en la siguiente etapa directamente.
Etapa 3: Síntesis de N-terc-butoxicarbonil-N-[(2-piperazin-1-ilpirimidin-5-il)metil]carbamato de terc-butilo A una disolución de 1-bencilpiperazina (30,44 g, 122,16 mmoles, 1,0 equiv, 2HCl) en MeCN (550 ml) se añadió N-terc-butoxicarbonil-N-((2-cloropirimidin-5-il)metil)carbamato de terc-butilo (42 g, 122,16 mmoles, 1,0 equiv) y K2CO3 (84,42 g, 610,81 mmoles, 5,0 equiv). La mezcla se agitó a 80 °C durante 61 h. La mezcla de reacción se diluyó entonces con EtOAc (150 ml) y la mezcla se filtró. La disolución resultante se concentró a presión reducida dando el producto en bruto. El residuo se purificó por cromatografía en gel de sílice (1/0 a 0/1 de éter de petróleo/EtOAc) dando el producto (45 g, 74 % de rendimiento) como un sólido blanco.
Etapa 4: Síntesis de N-terc-butoxicarbonil-N-[(2-piperazin-1-ilpirimidin-5-il)metil]carbamato de terc-butilo A una disolución de N-[[2-(4-bencilpiperazin-1-il)pirimidin-5-il]metil]-N-terc-butoxicarbonil-carbamato de terc-butilo (24 g, 49,63 mmoles, 1,0 equiv) en MeOH (600 ml) se añadió Pd/C (24 g, 47,56 mmoles, 10 % en peso, 1,0 equiv) bajo argón. La mezcla se desgasificó a presión reducida y se purgó con H2 tres veces. La mezcla se agitó bajo H2 (50 psi) a 50 °C durante 19 h. La mezcla de reacción se enfrió hasta temperatura ambiente, se filtró y la torta de filtración se lavó con MeOH (500 ml). La disolución resultante se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 0/1 de EtOAc/MeOH) dando el producto (25,5 g, 68 % de rendimiento) como un sólido blanco.
Elemento estructural B. Ácido 2-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de 2-cloropirimidin-5-carboxilato de etilo (2,37 g, 12,71 mmoles, 1,0 equiv) y N-terc-butoxicarbonil-N-((2-piperazin-1-ilpirimidin-5-il)metil)carbamato de terc-butilo (5 g, 12,71 mmoles, 1,0 equiv) en MeCN (80 ml) se añadió K2CO3 (5,27 g, 38,12 mmoles, 3,0 equiv). La mezcla se agitó a 80 °C durante 16 h. La mezcla de reacción se vertió entonces en H2O (200 ml) y la suspensión se filtró. El filtrado se lavó con H2O (80 ml) y se secó a presión reducida dando el producto (6,1 g, 87 % de rendimiento) como un sólido blanco.
Etapa 2: Síntesis de ácido 2-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxilato de etilo (5 g, 9,20 mmoles, 1,0 equiv) en H2O (50 ml), EtOH (15 ml) y THF (50 ml) se añadió LiOH•H2O (1,54 g, 36,79 mmoles, 4,0 equiv). La mezcla de reacción se agitó a 55 °C durante 16 h. La mezcla se concentró entonces para retirar el THF y el EtOH y entonces la mezcla se diluyó con H2O (55 ml) y se acidificó (pH=3) con HCl acuoso (1 N). La mezcla se filtró y la torta de filtración se lavó con H2O (36 ml). La torta de filtración se secó a presión reducida dando el producto (2,7 g, 69,3 %) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C19H25N7O4: 416,21; observado 416,1.
Elemento estructural C.2-(Piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo.

Etapa 1: Síntesis de 2-(4-bencilpiperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo A una disolución de 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (15 g, 55,61 mmoles, 1,0 equiv) en MeCN (150 ml) se añadió 1-bencilpiperazina (11,76 g, 66,73 mmoles, 1,2 equiv) y K2CO3 (46,12 g, 333,67 mmoles, 6,0 equiv). La mezcla se agitó a 80 °C durante 27 h. La mezcla de reacción se diluyó con EtOAc (200 ml), se filtró y se concentró a presión reducida. El producto en bruto se purificó por cromatografía en gel de sílice (1/0 a 0/1 de éter de petróleo/EtOAc) dando el producto (20,2 g, 80 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C23H31N5O2: 410,26; observado 410,1.
Etapa 2: Síntesis de 2-(piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de 2-(4-bencilpiperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (8 g, 19,53 mmoles, 1,0 equiv) en MeOH (200 ml) se añadió Pd/C (8 g, 19,53 mmoles, 10 % en peso, 1,0 equiv) bajo argón. La mezcla se desgasificó y se purgó con H2 tres veces. La mezcla se agitó bajo H2 (50 psi) a 50 °C durante 19 h. La mezcla de reacción se enfrió hasta temperatura ambiente, se filtró a través de una almohadilla de Celite y la torta de filtración se lavó con MeOH (150 ml). La disolución resultante se concentró a presión reducida. El producto en bruto se lavó con éter de petróleo (60 ml) dando el producto (9,25 g, 72 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C16H25N5O2: 320,21; observado 320,2.
Elemento estructural D. Ácido 2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de 2-cloropirimidin-5-carboxilato de etilo (4,09 g, 21,92 mmoles, 1,0 equiv) en dioxano (80 ml) se añadió 2-(piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (7 g, 21,92 mmoles, 1,0 equiv) y Et3N (9,15 ml, 65,75 mmoles, 3,0 equiv). La mezcla se agitó a 90 °C durante 64 h. La disolución se vertió en H2O (200 ml) y entonces la mezcla se filtró y la torta de filtración se lavó con H2O (100 ml), seguido de éter de petróleo (60 ml). La torta de filtración se secó a presión reducida dando el producto (10,1 g, 92 % de rendimiento) como un sólido marrón. LCMS (ESI) m/z: [M H] calculado para C23H31N7O4: 470,25; observado 470,4.
Etapa 2: Síntesis de ácido 2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (6,0 g, 12,78 mmoles, 1,0 equiv) en THF (40 ml), EtOH (20 ml) y H2O (40 ml) se añadió LiOH•H2O (1,07 g, 25,56 mmoles, 2,0 equiv). La mezcla de reacción se agitó a 35 °C durante 15 h. La mezcla se concentró entonces a presión reducida para retirar el THF y el EtOH. La mezcla se diluyó entonces con H2O (500 ml) y se ajustó a pH 3 con HCl acuoso (1 N). La mezcla se filtró y la torta de filtración se lavó con H2O (80 ml), seguido de éter de petróleo (80 ml). La torta de filtración se secó a presión reducida dando el producto (3,8 g, 65 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C21H27N7O4: 442,22; observado 442,3.
Elemento estructural E. Metil((2-(piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo.

Etapa 1: Síntesis de (2-cloropirimidin-5-il)metanamina
A una disolución de N-terc-butoxicarbonil-N-((2-cloropirimidin-5-il)metil)carbamato de terc-butilo (28 g, 81,44 mmoles, 1,0 equiv) en EtOAc (30 ml) se añadió HCl en EtOAc (260 ml). La mezcla de reacción se agitó a temperatura ambiente durante 5 h. La mezcla de reacción se filtró y la torta de filtración se lavó con EtOAc (100 ml). La torta sólida se secó a presión reducida dando el producto (14,3 g, 96,6 % de rendimiento, HCl) como un sólido blanco.
Etapa 2: Síntesis de ((2-cloropirimidin-5-il)metil)carbamato de terc-butilo
A una disolución de (2-cloropirimidin-5-il)metanamina (13 g, 72,21 mmoles, 1,0 equiv, HCl) en DCM (130 ml) se añadió DIPEA (20,41 ml, 144,42 mmoles, 1,8 equiv) y Boc2O (16,59 ml, 72,21 mmoles, 1,0 equiv), entonces la mezcla se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se añadió a H2O (100 ml) y entonces la fase acuosa se separó y se extrajo con DCM (2 × 100 ml). Entonces se lavó fase orgánica combinada con NH4Cl sat. (2 × 200 ml) y salmuera (2 × 200 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 1/1 de éter de petróleo/EtOAc) dando el producto (12 g, 68,2 % de rendimiento) como un sólido blanco.
Etapa 3: Síntesis de ((2-cloropirimidin-5-il)metil)(metil)carbamato de terc-butilo
A una disolución de ((2-cloropirimidin-5-il)metil)carbamato de terc-butilo (11 g, 45,14 mmoles, 1,0 equiv) y MeI (14,05 ml, 225,70 mmoles, 5,0 equiv) en THF (150 ml) se añadió NaH (1,99 g, 49,65 mmoles, 60 % en peso, 1,1 equiv) a 0 °C. La mezcla se agitó a 0 °C durante 3 h y entonces la reacción se inactivó con H2O (100 ml). La fase acuosa se extrajo con EtOAc (3 × 150 ml) y la fase orgánica combinada se lavó con salmuera (50 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 3/1 de éter de petróleo/EtOAc) dando el producto (9 g, 77,4 % de rendimiento) como un sólido blanco.
Etapa 4: Síntesis de ((2-(4-bencilpiperazin-1-il)pirimidin-5-il)metil)(metil)carbamato de terc-butilo
A una disolución de ((2-cloropirimidin-5-il)metil)(metil)carbamato de terc-butilo (9 g, 34,92 mmoles, 1,0 equiv) en MeCN (90 ml) se añadió 1-bencilpiperazina (8,70 g, 34,92 mmoles, 1,0 equiv, 2HCl) y K2CO3 (24,13 g, 174,61 mmoles, 5,0 equiv). La mezcla de reacción se agitó a 80 °C durante 20 h. Entonces se filtró la mezcla y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 1/1 de éter de petróleo/EtOAc) dando el producto (12 g, 86,4 % de rendimiento) como un aceite amarillo.
Etapa 5: Síntesis de metil((2-(piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo
A una disolución de ((2-(4-bencilpiperazin-1-il)pirimidin-5-il)metil)(metil)carbamato de terc-butilo (12 g, 30,19 mmoles, 1,0 equiv) en MeOH (120 ml) se añadió Pd/C (2 g, 10 % en peso). La suspensión se desgasificó y se purgó con H2 y entonces la mezcla se agitó bajo H2 (15 psi) a temperatura ambiente durante 3 h. La mezcla de reacción se filtró a través de Celite y el filtrado se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 1/1 de éter de petróleo/EtOAc) dando material semipuro (9 g) como un aceite amarillo. Se añadió éter de petróleo al residuo y la disolución se agitó a -60 °C hasta que apareció el sólido. La suspensión se filtró y el filtrado se concentró a presión reducida dando el producto (4,07 g, 55,6 % de rendimiento) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C15H25N5O2: 308,21; observado 308,1.
Elemento estructural F. Ácido 2-(4-(5-(((terc-butoxicarbonil)(metil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(5-(((terc-butoxicarbonil)(metil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una mezcla de metil((2-(piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo (4,3 g, 13,99 mmoles, 1,0 equiv) y 2-cloropirimidin-5-carboxilato de etilo (2,87 g, 15,39 mmoles, 1,1 equiv) en MeCN (20 ml) se añadió K2CO3 (3,87 g, 27,98 mmoles, 2,0 equiv). La mezcla se agitó a 80 °C durante 12 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente y se filtró. El filtrado se concentró a presión reducida y el producto en bruto se purificó por cromatografía en gel de sílice (1/0 a 1/1 de éter de petróleo/EtOAc) dando el producto (4,7 g, 71,3 % de rendimiento) como un sólido blanco.
Etapa 2: Síntesis de ácido 2-(4-(5-(((terc-butoxicarbonil)(metil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(5-(((terc-butoxicarbonil)(metil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxilato de etilo (6 g, 13,11 mmoles, 1,0 equiv) en THF (100 ml), EtOH (30 ml) y H2O (30 ml) se añadió LiOH•H2O (1,10 g, 26,23 mmoles, 2,0 equiv). La mezcla se agitó a temperatura ambiente durante 16 h. La mezcla se concentró entonces a presión reducida para retirar el THF y el EtOH y entonces se neutralizó mediante la adición de HCl 1 N. El precipitado resultante se recogió por filtración dando el producto (5,11 g, 90,1 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C20H27N7O4: 430,22; observado 430,2.
Elemento estructural G. N-terc-butoxicarbonil-N-((2-(2-((terc-butil(difenil)silil)oximetil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo.

Etapa 1: Síntesis de N-((2-(4-bencil-2-(hidroximetil)piperazin-1-il)pirimidin-5-il)metil)-N-terc-butoxicarbonilcarbamato de terc-butilo
A una disolución de N-terc-butoxicarbonil-N-((2-cloropirimidin-5-il)metil)carbamato de terc-butilo (18,33 g, 53,32 mmoles, 1,1 equiv) y (4-bencilpiperazin-2-il)metanol (10 g, 48,48 mmoles, 1,0 equiv) en DMF (100 ml) se añadió K2CO3 (13,40 g, 96,95 mmoles, 2,0 equiv). La mezcla se agitó a 100 °C durante 12 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente y se añadió H2O (100 ml). La fase acuosa se extrajo con EtOAc (2 × 150 ml) y la fase orgánica combinada se lavó con salmuera (20 ml), se secó con Na2SO4, se filtró y el filtrado se concentró a presión reducida dando el producto (7,3 g, 29,3 % de rendimiento) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C27H39N5O5: 514,31; observado 514,5
Etapa 2: Síntesis de N-((2-(4-bencil-2-((terc-butil(difenil)silil)oximetil)piperazin-1-il)pirimidin-5-il)metil)-N-terc-butoxicarbonil-carbamato de terc-butilo
A una disolución de N-((2-(4-bencil-2-(hidroximetil)piperazin-1-il)pirimidin-5-il)metil)-N-terc-butoxicarbonil-carbamato de terc-butilo (2,3 g, 4,48 mmoles, 1,0 equiv) en DCM (30 ml) se añadió imidazol (609,69 mg, 8,96 mmoles, 2,0 equiv) y TBDPSCl (1,73 ml, 6,72 mmoles, 1,5 equiv). La mezcla de reacción se agitó a temperatura ambiente durante 2 h. La mezcla se lavó entonces con H2O (100 ml) y la fase acuosa se extrajo con EtOAc (2 × 60 ml). La fase orgánica combinada se lavó con salmuera (20 ml), se secó con Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (20/1 a 3/1 de éter de petróleo/EtOAc) dando el producto (4 g, 59,4 % de rendimiento) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C43H57N5O5Si: 752,42; observado 752,4.
Etapa 3: Síntesis de N-terc-butoxicarbonil-N-((2-(2-((terc-butil(difenil)silil)oximetil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo
A una disolución de N-((2-(4-bencil-2-((terc-butil(difenil)silil)oximetil)piperazin-1-il)pirimidin-5-il)metil)-N-terc-butoxicarbonil-carbamato de terc-butilo (3,3 g, 4,39 mmoles, 1,0 equiv) en EtOH (10 ml) se añadió Pd(OH)2/C (1 g, 10 % en peso). La mezcla se calentó hasta 50 °C bajo H2 (30 psi) durante 30 h. Entonces se enfrió la mezcla hasta temperatura ambiente, se filtró a través de Celite y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (20/1 a 3/1 de EtOAc/EtOH) dando el producto (1,44 g, 45,6 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C36H51N5O5Si: 662,38; observado 662,3.
Elemento estructural H. Ácido 2-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)-3-(hidroximetil)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de N-terc-butoxicarbonil-N-((2-(2-(hidroximetil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo
A una disolución de N-((2-(4-bencil-2-(hidroximetil)piperazin-1-il)pirimidin-5-il)metil)-N-terc-butoxicarbonil-carbamato de terc-butilo (3 g, 5,84 mmoles, 1,0 equiv) en EtOH (40 ml) se añadió Pd/C (2 g, 10 % en peso). La suspensión se desgasificó y se purgó con H2, luego se agitó bajo H2 (50 psi) a 30 °C durante 16 h. La mezcla de reacción se enfrió hasta temperatura ambiente y se filtró a través de Celite y entonces se concentró a presión reducida dando el producto (1,6 g, en bruto) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C20H33N5O5: 424,26; observado 424,3.
Etapa 2: Síntesis de 2-(4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)-3-(hidroximetil)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de N-terc-butoxicarbonil-N-((2-(2-(hidroximetil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo (1,4 g, 3,31 mmoles, 1,0 equiv) en MeCN (20 ml) se añadió K2CO3 (2,28 g, 16,53 mmoles, 5,0 equiv) y 2-cloropirimidin-5-carboxilato de etilo (616,84 mg, 3,31 mmoles, 1,0 equiv). La disolución se agitó a 80 °C durante 4 h. La mezcla se enfrió hasta temperatura ambiente y se vertió en H2O (30 ml). La fase acuosa se extrajo con EtOAc (2 × 30 ml) y la fase orgánica combinada se lavó con salmuera (20 ml), se secó con Na2SO4, se filtró y se concentró a presión reducida. La mezcla se purificó por cromatografía en gel de sílice (20/1 a 3/1 de éter de petróleo/EtOAc) dando el producto (1,6 g, 66,7 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C27H39N7O7: 574,30; observado 574,4.
Etapa 3: Síntesis de ácido 2-(4-(5-(((terc-butoxicarbonil)amino)metil)pilimidin-2-il)-3-(hidroximetil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)-3-(hidroximetil)piperazin-1-il)pirimidin-5-carboxilato de etilo (1,4 g, 2,44 mmoles, 1,0 equiv) en THF (6 ml) y EtOH (6 ml) a 0 °C se añadió una disolución de LiOH•H2O (512,07 mg, 12,20 mmoles, 5,0 equiv) en H2O (3 ml). La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 2 h. Entonces se concentró la mezcla a presión reducida para retirar el THF y el EtOH. La fase acuosa se ajustó a pH 3 con HCl 0,1 M y la suspensión resultante se filtró. La torta sólida se secó a presión reducida dando el producto (613,14 mg, 55,6 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C20H27N7O5: 446,22; observado 446,2.
Elemento estructural I. N-[(terc-butoxi)carbonil]-N-({2-[(3R)-3-(hidroximetil)piperazin-1-il]pirimidin-5-il}metil)carbamato de terc-butilo.

Etapa 1: Síntesis de (R)-terc-butil-N-terc-butoxicarbonil-((2-(3-(((terc-butildifenilsilil)-oxi)metil)piperazin-1-il)pirimidin-5-il)metil)carbamato
A una disolución de terc-butil-N-terc-butoxicarbonil-((2-cloropirimidin-5-il)metil)carbamato (24,24 g, 70,51 mmoles, 1,0 equiv) en MeCN (300 ml) se añadió (R)-2-(((terc-butildifenilsilil)oxi)metil)piperazina (25 g, 70,51 mmoles, 1,0 equiv)
y K2CO3 (29,24 g, 211,53 mmoles, 3,0 equiv). La mezcla se agitó a 80 °C durante 16 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente, se diluyó con EtOAc (200 ml), se filtró y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→100 % de EtOAc/éter de petróleo) dio el producto deseado (46,5 g, 94 % de rendimiento) como un sólido blanco.
Etapa 2: Síntesis de N-[(terc-butoxi)carbonil]-N-({2-[(3R)-3-(hidroximetil)piperazin-1-il]pirimidin-5-il}metil)carbamato de terc-butilo
A una disolución de (R)-terc-butil-N-terc-butoxicarbonil-((2-(3-(((terc-butildifenilsilil)oxi)metil)piperazin-1-il)pirimidin-5-il)metil)carbamato (12 g, 18,13 mmoles, 1,0 equiv) en THF (120 ml) se añadió TBAF (1 M, 23,93 ml, 1,3 equiv). La mezcla se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se vertió entonces en H2O (300 ml) y la fase acuosa se extrajo con EtOAc (3 × 80 ml). Las fases orgánicas combinadas se combinaron, se lavaron con salmuera (80 ml), se secaron, se filtraron y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→20 % de MeOH/DCM) dio el producto deseado (5 g, 64 % de rendimiento) como un sólido amarillo.
Elemento estructural J. Ácido 2-{4-[5-({[(terc-butoxi)carbonil]amino)metil)pirimidin-2-il]piperazin-1-il}pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)-2-(((terc-butildifenilsilil)oxi)metil)piperazin-1-il)pirimidin-5-carboxilato de (R)-etilo
A una disolución de (R)-terc-butil-N-terc-butoxicarbonil-N-((2-(3-(((terc-butildifenilsilil)oxi)metil)piperazin-1-il)pirimidin-5-il)metil)carbamato (31,5 g, 45,21 mmoles, 1,0 equiv) en MeCN (350 ml) se añadió 2-cloropirimidin-5-carboxilato de etilo (8,44 g, 45,21 mmoles, 1,0 equiv) y K2CO3 (18,75 g, 135,63 mmoles, 3,0 equiv). La mezcla se agitó a 80 °C durante 16 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente, se diluyó con EtOAc (150 ml) y se filtró para retirar las sales inorgánicas. El filtrado se concentró entonces a presión reducida. La purificación por cromatografía en gel de sílice (0→100 % de EtOAc/éter de petróleo) dio el producto deseado (33,5 g, 89 % de rendimiento).
Etapa 2: Síntesis de 2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)-2-(hidroximetil)piperazin-1-il)pirimidin-5-carboxilato de (R)-etilo
A una disolución de 2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)-2-(((terc-butildifenilsilil)oxi)metil)piperazin-1-il)pirimidin-5-carboxilato de (R)-etilo (36,5 g, 44,95 mmoles, 1,0 equiv) en THF (300 ml) se añadió TBAF (1 M, 59,33 ml, 1,32 equiv). La mezcla se agitó a temperatura ambiente durante 6 h, momento en el que la mezcla de reacción se vertió en H2O (500 ml). La fase acuosa se separó y se extrajo con EtOAc (3 × 150 ml) y las fases orgánicas combinadas se lavaron con salmuera (150 ml), se secaron, se filtraron y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→100 % de EtOAc/éter de petróleo) dio el producto deseado (17 g, 64 % de rendimiento) como un aceite amarillo.
Etapa 3: Síntesis de ácido (R)-2-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)-2-(hidroximetil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)-2-(hidroximetil)piperazin-1-il)pirimidin-5-carboxilato de (R)-etilo (17 g, 29,64 mmoles, 1,0 equiv) en H2O (160 ml), EtOH (80 ml) y THF (160 ml) se añadió LiOH•H2O (4,97 g, 118,54 mmoles, 4,0 equiv). La mezcla de reacción se agitó a 55 °C durante 16 h. A la mezcla se añadió entonces LiOH•H2O (1,01 g, 24,00 mmoles, 0,81 equiv) y la mezcla de reacción se agitó a 55 °C durante 9 h adicionales. La mezcla se enfrió hasta temperatura ambiente, se diluyó con H2O (150 ml) y se concentró a presión reducida para retirar el THF y el EtOH. La mezcla se acidificó (pH = 5) con HCl 1 N, se filtró y la torta de filtración se lavó con H2O (2 × 30 ml). La torta de filtración se secó a presión reducida proporcionando el producto deseado (9,2 g, 67 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C20H27N7O5: 446,22; observado 446,1.
Elemento estructural K. N-[(terc-butoxi)carbonil]-N-(12-[(3S)-3-(hidroximetil)piperazin-1-il]pirimidin-5-il}metil)carbamato de terc-butilo.

Este elemento estructural se prepara por un proceso similar a aquél para el Elemento estructural I utilizando [(2S)-piperazin-2-il]metanol.
Elemento estructural L. Ácido 2-[(2S)-4-[5-({[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il]-2-(hidroximetil)piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural K por un proceso similar a aquél para el Elemento estructural J.
Elemento estructural M.2-[(3R)-3-(Hidroximetil)piperazin-1-il]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-6-carboxilato de terc-butilo.

Etapa 1: Síntesis de 2-(3-(((terc-butildifenilsilil)oxi)-metil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo
A una disolución de (R)-2-(((terc-butildifenilsilil)oxi)metil)piperazina (25 g, 70,51 mmoles, 1,0 equiv) en MeCN (250 ml) se añadió K2CO3 (29,24 g, 211,53 mmoles, 3,0 equiv) y 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (17,12 g, 63,46 mmoles, 0,9 equiv). La mezcla se agitó a 80 °C durante 17 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente, se filtró y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→100 % de EtOAc/éter de petróleo) dio el producto deseado (31 g, 73,5 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C33H45N5O3Si: 588,34; observado 588,2.
Etapa 2: Síntesis de 2-(3-(hidroximetil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo
A una mezcla de 2-(3-(((terc-butildifenilsilil)oxi)metil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo (12 g, 20,41 mmoles, 1,0 equiv) en THF (120 ml) se añadió TBAF (1,0 M, 24,50 ml, 1,2 equiv). La mezcla se agitó a temperatura ambiente durante 5 h. La mezcla se vertió en H2O (100 ml), y la fase acuosa se extrajo con EtOAc (2 × 100 ml). Las fases orgánicas combinadas se lavaron con salmuera (100 ml), se secaron, se filtraron y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→10 % de MeOH/DCM) dio el producto deseado (6 g, 84,1 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C17H27N5O3: 350,22; observado 350,2.
Elemento estructural N. Ácido 2-[(2R)-4-{6-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}-2-(hidroximetil)piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural M por un proceso similar a aquél para el Elemento estructural J.
Elemento estructural O.2-[(3S)-3-(Hidroximetil)piperazin-1-il]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-6-carboxilato de terc-butilo.

Este elemento estructural se prepara por un proceso similar a aquél para el Elemento estructural I utilizando 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo y [(2S)-piperazin-2-il]metanol.
Elemento estructural P. Ácido 2-[(2S)-4-16-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}-2-(hidroximetil)piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural O por un proceso similar a aquél para el Elemento estructural J.
Elemento estructural Q. N-[(terc-Butoxi)carbonil]-N-({2-[(3S)-3-[(dimetilamino)metil]piperazin-1-il]pirimidin-5-il}metil)carbamato de terc-butilo.

Etapa 1: Síntesis de 2-(dimetilcarbamoil)piperazin-1,4-dicarboxilato de (R)-dibencilo
A una disolución de CDI (12,21 g, 75,30 mmoles, 1,2 equiv) en DCM (300 ml) a 0 °C se añadió ácido (R)-1,4-bis((benciloxi)carbonil)piperazin-2-carboxílico (25 g, 62,75 mmoles, 1,0 equiv). La mezcla se agitó a 0 °C durante 0,5 h, momento en el que se añadió dimetilamina (8,51 ml, 92,87 mmoles, 1,5 equiv, HCl). La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 12 h. La mezcla de reacción se añadió entonces a H2O (200 ml), y la fase acuosa se separó y se extrajo con DCM (2 × 200 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 × 50 ml), se secaron, se filtraron y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (50→100 % de EtOAc/éter de petróleo) dio el producto deseado (23,5 g, 88,0 % de rendimiento) como un aceite amarillo.
Etapa 2: Síntesis de 2-((dimetilamino)metil)piperazin-1,4-dicarboxilato de (S)-dibencilo
A una disolución de 2-(dimetilcarbamoil)piperazin-1,4-dicarboxilato de (R)-dibencilo (28 g, 65,81 mmoles, 1,0 equiv) en THF (300 ml) a 0 °C se añadió BH3•Me2S (10 M, 13,16 ml, 2,0 equiv). La mezcla de reacción se agitó entonces a 80 °C durante 3 h. La mezcla de reacción se enfrió hasta temperatura ambiente y entonces se añadió MeOH (50 ml). Después de agitar durante 1 h adicional, la mezcla se concentró a presión reducida. La purificación por cromatografía en gel de sílice (50→100 % de EtOAc/éter de petróleo) dio el producto deseado (18 g, 66,5 % de rendimiento) como un aceite amarillo.
Etapa 3: Síntesis de (R)-N,N-dimetil-1-(piperazin-2-il)metanamina
A una disolución de 2-((dimetilamino)metil)piperazin-1,4-dicarboxilato de (S)-dibencilo (18 g, 43,74 mmoles, 1,0 equiv) en EtOAc (200 ml) se añadió Pd/C (1,5 g, 10 % en peso). La suspensión se desgasificó a presión reducida y se purgó con H2 tres veces. La suspensión se agitó bajo H2 (30 psi) a 30 °C durante 5 h. Entonces se filtró la mezcla de reacción a través de Celite y el filtrado se concentró a presión reducida proporcionando el producto deseado (6 g, 95,8 % de rendimiento) como un sólido amarillo.
Etapa 4: Síntesis de N-terc-butoxicarbonil-N-((2-((3S)-3-((dimetilamino)metil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo
A una disolución de (R)-N,N-dimetil-1-(piperazin-2-il)metanamina (2,8 g, 19,55 mmoles, 1,0 equiv) en MeCN (40 ml) se añadió N-terc-butoxicarbonil-N-((2-cloropirimidin-5-il)metil)carbamato de terc-butilo (6,72 g, 19,55 mmoles, 1,0 equiv) y K2CO3 (5,40 g, 39,10 mmoles, 2,0 equiv). La mezcla se agitó a 80 °C durante 24 h. Entonces se enfrió la mezcla hasta temperatura ambiente, se filtró y la torta de filtración se lavó con EtOAc (3 × 10 ml). El filtrado se concentró entonces a presión reducida. La purificación por cromatografía en gel de sílice (0→100 % de MeOH/EtOAc) dio el producto deseado (5,3 g, 57,8 % de rendimiento) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C22H38N6O4: 451,31; observado 451,2.
Elemento estructural R. Ácido 2-[(2S)-4-[5-({[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il]-2-[(dimetilamino)metil]piperazin-1-il]pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(5-(((bi-terc-butoxicarbonil)amino)metil) pirimidin-2-il)-2-((dimetilamino)metil)piperazin-1-il)pirimidin-5-carboxilato de (S)-etilo
A una disolución de ((2-(3-((dimetilamino)metil)piperazin-1-il)pirimidin-5-il)metil)carbamato de (S)-terc-butil-N-terc-butoxicarbonilo (3,26 g, 7,24 mmoles, 1,0 equiv) en DMF (30 ml) se añadió Et3N (3,02 ml, 21,71 mmoles, 3,0 equiv) y 2-cloropirimidin-5-carboxilato de etilo (1,47 g, 7,86 mmoles, 1,1 equiv). La mezcla se agitó a 50 °C durante 3 h y entonces se concentró a presión reducida proporcionando el producto deseado (4,35 g, en bruto) como una disolución en DMF (30 ml), que se usó directamente en la siguiente etapa. LCMS (ESI) m/z: [M H] calculado para C29H44N8O6: 601,35; observado 601,5.
Etapa 2: Síntesis de ácido (S)-2-(4-(5-(((terc-butoxicarbonil)amino)metil)-pirimidin-2-il)-2-((dimetilamino)metil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(5-(((bi-terc-butoxicarbonil)amino)metil)-pirimidin-2-il)-2-((dimetilamino)metil)piperazin-1-il)pirimidin-5-carboxilato de (S)-etilo (4,35 g, 7,24 mmoles, 1,0 equiv) en DMF (30 ml) se añadió DMF (50 ml), EtOH (30 ml) y H2O (30 ml). A la disolución se añadió entonces LiOH•H2O (3 g, 71,50 mmoles, 9,9 equiv) a 50 °C. La reacción se agitó a 50 °C durante 36 h. La mezcla se enfrió entonces hasta temperatura ambiente, se neutralizó con HCl 0,5 N y se concentró a presión reducida. La purificación por cromatografía de fase inversa (2→30 % de MeCN/H2O) dio el producto deseado (1,15 g, 34 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C22H32N8O4: 473,26; observado 473,3.
Elemento estructural S. N-[(terc-Butoxi)carbonil]-N-({2-[(3R)-3-[(dimetilamino)metil]piperazin-1-il]pirimidin-5-il}metil)carbamato de terc-butilo.

Este elemento estructural se prepara por un proceso similar a aquél para el Elemento estructural I utilizando dimetil({[(2S)-piperazin-2-il]metil})amina.
Elemento estructural T. Ácido 2-[(2R)-4-[5-({[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il]-2-[(dimetilamino)metil]piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural S por un proceso similar a aquél para el Elemento estructural J.
Elemento estructural U. 2-[(3S)-3-[(Dimetilamino)metil]piperazin-1-il]-SH,6H,7H,8H-pirido[4,3-d]pirimidin-6-carboxilato de terc-butilo.

A una disolución de 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (4,80 g, 17,80 mmoles, 1,4 equiv) en MeCN (45 ml) se añadió K2CO3 (10,42 g, 75,40 mmoles, 3,0 equiv) y (R)-N,N-dimetil-1-(piperazin-2-il)metanamina (3,6 g, 25,13 mmoles, 1,0 equiv). La mezcla se agitó a 80 °C durante 8 h. La mezcla se enfrió entonces hasta temperatura ambiente, se filtró y la torta de filtración se lavó con EtOAc (50 ml). A la fase orgánica se añadió H2O (50 ml) y la fase acuosa se extrajo con EtOAc (3 × 50 ml). Las fases orgánicas combinadas se lavaron con salmuera (5 ml), se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (8→67 % de EtOAc/éter de petróleo) dio el producto deseado (6,5 g, 63,5 % de rendimiento) como un aceite amarillo.
Elemento estructural V. Ácido 2-[(2S)-4-{6-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}-2-[(dimetilamino)metil]piperazin-1-il]pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(3-((dimetilamino)metil)-4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (S)-terc-butilo
A una disolución de 2-(3-((dimetilamino)metil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (S)-terc-butilo (3 g, 7,97 mmoles, 1,0 equiv) en DMF (70 ml) a 0 °C se añadió NaH (382,44 mg, 9,56 mmoles, 60 % en peso, 1,2 equiv). La suspensión se agitó a 0 °C durante 0,5 h, entonces se añadió 2-cloropirimidin-5-carboxilato de etilo (1,49 g, 7,97 mmoles, 1 equiv) en DMF (50 ml), gota a gota. La mezcla se calentó hasta temperatura ambiente y se agitó durante 5 h. La mezcla se enfrió entonces hasta 0 °C y se vertió en H2O (360 ml). La suspensión se filtró, y la torta de filtración se lavó con H2O (30 ml) y se secó a presión reducida. La purificación por cromatografía en gel de
sílice (6 %→33 % de EtOAc/éter de petróleo) dio el producto deseado (1,8 g, 39,6 % de rendimiento) como un aceite marrón.
Etapa 2: Síntesis de ácido (S)-2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)-2-((dimetilamino)metil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(3-((dimetilamino)metil)-4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (S)-terc-butilo (1,1 g, 2,09 mmoles, 1,0 equiv) en THF (5 ml), EtOH (2,5 ml) y H2O (2,5 ml) se añadió LiOH•H2O (175,30 mg, 4,18 mmoles, 2,0 equiv). La mezcla se agitó a temperatura ambiente durante 2 h, momento en el que el pH se ajustó a 7 mediante la adición de HCl 1 N a 0 °C. La mezcla se concentró a presión reducida para retirar el THF y el MeOH. La suspensión resultante se filtró, y la torta de filtración se lavó con H2O (5 ml) y se secó a presión reducida proporcionando el producto deseado (680 mg, 65,3 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C24H34N8O4: 499,28; observado 499,2.
Elemento estructural W. 2-[(3R)-3-[(Dimetilamino)metil]piperazin-1-il]-SH,6H,7H,8H-pirido[4,3-d]pirimidin-6-carboxilato de terc-butilo.

Este elemento estructural se prepara por un proceso similar a aquél para el Elemento estructural I utilizando 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo y dimetil({[(2S)-piperazin-2-il]metil})amina.
Elemento estructural X. Ácido 2-[(2R)-4-{6-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}-2-[(dimetilamino)metil]piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural W por un proceso similar a aquél para el Elemento estructural J.
Elemento estructural Y. (2R)-4-16-[(terc-Butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}piperazin-2-carboxilato de terc-butilo.

Etapa 1: Síntesis de ácido (R)-1,4-bis((benciloxi)carbonil)piperazin-2-carboxílico
A una disolución de ácido (R)-piperazin-2-carboxílico (70 g, 344,71 mmoles, 1,0 equiv, 2HCl) en dioxano (1120 ml) y H2O (700 ml) se añadió 50 % de NaOH ac. hasta que la disolución tuvo pH=11. Se añadió cloroformiato de bencilo (156,82 ml, 1,10 moles, 3,2 equiv) y la mezcla de reacción se agitó a temperatura ambiente durante 12 h. A la disolución se añadió entonces H2O (1200 ml) y la fase acuosa se lavó con MTBE (3 × 800 ml). La fase acuosa se ajustó a pH=2 con HCl concentrado (12 N) y se extrajo con EtOAc (2 × 1000 ml). Los extractos orgánicos combinados se secaron, se filtraron y el filtrado se concentró a presión reducida proporcionando el producto deseado (137 g, 99,8 %
de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C21H22N2O6: 399,16; observado 399,2.
Etapa 2: Síntesis de piperazin-1,2,4-tricarboxilato de (R)-1,4-dibencil-2-terc-butilo
A una disolución de ácido (R)-1,4-bis((benciloxi)carbonil)piperazin-2-carboxílico (50 g, 125,50 mmoles, 1,0 equiv) en tolueno (500 ml) a 80 °C se añadió 1,1-di-terc-butoxi-N,N-dimetilmetanamina (57,17 ml, 238,45 mmoles, 1,9 equiv). La disolución se agitó a 80 °C durante 2 h, momento en el que la mezcla de reacción se enfrió hasta temperatura ambiente y se repartió entre EtOAc (300 ml) y H2O (500 ml). La fase acuosa se extrajo con EtOAc (2 × 500 ml) y las fases orgánicas combinadas se secaron, se filtraron y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→25 % de EtOAc/éter de petróleo) dio el producto deseado (35 g, 61,2 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M Na] calculado para C25H30N2O6: 477,20; observado 477,1.
Etapa 3: Síntesis de piperazin-2-carboxilato de (R)-terc-butilo
A una disolución de piperazin-1,2,4-tricarboxilato de (R)-1,4-dibencil-2-terc-butilo (35 g, 77,01 mmoles, 1,0 equiv) en EtOAc (350 ml) se añadió Pd/C (10 g, 10 % en peso). La suspensión se desgasificó a presión reducida y se purgó con H2 tres veces. La mezcla se agitó bajo H2 (30 psi) a 30 °C durante 4 h. La mezcla de reacción se filtró entonces a través de Celite, el residuo se lavó con MeOH (5 × 200 ml) y el filtrado se concentró a presión reducida proporcionando el producto deseado (14 g, 79,6 % de rendimiento) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C9H19N2O2: 187,15; observado 187,1.
Etapa 4: Síntesis de 2-(3-(terc-butoxicarbonil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo
A una disolución de (2R)-piperazin-2-carboxilato de terc-butilo (12 g, 64,43 mmoles, 1,0 equiv) en MeCN (200 ml) se añadió K2CO3 (17,81 g, 128,86 mmoles, 2,0 equiv) y 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (17,38 g, 64,43 mmoles, 1,0 equiv). La mezcla de reacción se calentó hasta 80 °C y se agitó durante 12 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente y se filtró, el residuo se lavó con EtOAc (3 × 150 ml) y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→100 % de EtOAc/éter de petróleo) dio el producto deseado (19 g, 69,2 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C21H33N5O4: 420,26; observado 420,2.
Elemento estructural Z. Ácido 4-amino-2-[(2R)-2-[(terc-butoxi)carbonil]-4-{6-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}piperazin-1-il]pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(4-amino-5-(etoxicarbonil)pirimidin-2-il)-3-(terc-butoxicarbonil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo
A una disolución con agitación de 2-(3-(terc-butoxicarbonil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo (12 g, 28,60 mmoles, 1,0 equiv) en MeCN (150 ml) se añadió K2CO3 (7,91 g, 57,20 mmoles, 2,0 equiv) y 4-amino-2-cloropirimidin-5-carboxilato de etilo (6,92 g, 34,32 mmoles, 1,2 equiv). La mezcla de reacción se agitó a 80 °C durante 12 h, momento en el que la mezcla de reacción se filtró y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→17 % de EtOAc/éter de petróleo) dio el producto deseado (16 g, 91,6 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C28H40N8O6: 585,32; observado 585,1.
Etapa 2: Síntesis de ácido (R)-4-amino-2-(2-(terc-butoxicarbonil)-4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico
A dos lotes separados realizados en paralelo que contenía cada uno una disolución de 2-(4-(4-amino-5-(etoxicarbonil)pirimidin-2-il)-3-(terc-butoxicarbonil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo (7 g, 11,97 mmoles, 1,0 equiv) en THF (70 ml), EtOH (35 ml) y H2O (35 ml) se añadió LiOH•H2O (2,01 g, 47,89 mmoles, 4,0 equiv). Las mezclas se agitaron a 60 °C durante 3 h, momento en el que las dos mezclas de reacción se combinaron, y se ajustaron a pH=7 con HCl 1 N. La mezcla se concentró a presión reducida para retirar el THF y el EtOH, se filtró y el residuo se secó a presión reducida. El residuo se agitó en MTBE (100 ml) durante 10 min, se filtró y el residuo se secó a presión reducida proporcionando el producto deseado (8,02 g, 55,1 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C26H36N8O6:557,29; observado 557,3.
Elemento estructural AA. (2S)-4-{6-[(terc-Butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}piperazin-2-carboxilato de terc-butilo.

Este elemento estructural se prepara por un proceso similar a aquél para el Elemento estructural I utilizando 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato y terc-butilo (2S)-piperazin-2-carboxilato de terc-butilo.
Elemento estructural AB. Ácido 4-amino-2-[(2S)-2-[(terc-butoxi)carbonil]-4-16-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural AA por un proceso similar a aquél para el Elemento estructural J utilizando 4-amino-2-cloropirimidin-5-carboxilato de etilo.
Elemento estructural AC. Ácido 4-amino-2-(4-{6-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(4-amino-5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de 2-(piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (8,3 g, 25,99 mmoles, 1,0 equiv) y 4-amino-2-cloropirimidin-5-carboxilato de etilo (5,24 g, 25,99 mmoles, 1,0 equiv) en MeCN (100 ml) se añadió a K2CO3 (7,18 g, 51,97 mmoles, 2,0 equiv). La reacción se agitó a 80 °C durante 12 h. La reacción se enfrió entonces hasta temperatura ambiente, se añadió DCM (100 ml) y la mezcla de reacción se agitó durante 30 min. La suspensión se filtró, y la torta de filtración se lavó con DCM (6 × 100 ml). El filtrado se concentró a presión reducida y el residuo se trituró con EtOAc (30 ml), se filtró y entonces la torta de filtración se secó a presión reducida proporcionando el producto deseado (8,7 g, 65,9 % de rendimiento) como un sólido amarillo claro.
Etapa 2: Síntesis de ácido 4-amino-2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(4-amino-5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (8,7 g, 17,95 mmoles, 1,0 equiv) en THF (120 ml), EtOH (60 ml) y H2O (60 ml) se añadió LiOH•H2O (1,51 g, 35,91 mmoles, 2,0 equiv). La mezcla se agitó a 55 °C durante 12 h. La mezcla de reacción se concentró entonces a presión reducida para retirar el EtOH y el THF, y la mezcla de reacción se ajustó a pH=6 mediante la adición de HCl 1 N. El precipitado se filtró, y la torta de filtración se lavó con H2O (3 × 50 ml) y entonces se secó a presión reducida proporcionando el producto deseado (7,3 g, 89,1 % de rendimiento) como un sólido amarillo claro. LCMS (ESI) m/z: [M H] calculado para C21H28N8O4: 457,23; observado 457,2.
Elemento estructural AD. Ácido 4-amino-2-{4-[5-({[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il] piperazin-1-il} pirimidin-5-carboxílico.

Etapa 1: Síntesis de 4-amino-2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de terc-butil-N-terc-butoxicarbonil-N-((2-(piperazin-1-il)pirimidin-5-il)metil)carbamato (8,3 g, 21,09 mmoles, 1,0 equiv) en MeCN (100 ml) se añadió 4-amino-2-cloropirimidin-5-carboxilato de etilo (4,04 g, 20,04 mmoles, 0,95 equiv) y K2CO3 (8,75 g, 63,28 mmoles, 3,0 equiv). La mezcla se agitó a 80 °C durante 3 h. La reacción se enfrió entonces hasta temperatura ambiente, se añadió DCM (150 ml) y la mezcla de reacción se agitó durante 30 min. La suspensión se filtró, la torta de filtración se lavó con DCM (3 × 100 ml) y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→100 % de EtOAc/éter de petróleo) dio el producto deseado (8,35 g, 67 % de rendimiento) como un sólido blanco.
Etapa 2: Síntesis de ácido 4-amino-2-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 4-amino-2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxilato de etilo (8,3 g, 14,86 mmoles, 1,0 equiv) en H2O (70 ml), EtOH (36 ml) y THF (80 ml) se añadió LiOH•H2O (2,49 g, 59,43 mmoles, 4,0 equiv). La mezcla de reacción se agitó a 55 °C durante 16 h. La mezcla se concentró entonces a presión reducida para retirar el THF y el EtOH. La mezcla se diluyó con H2O (55 ml) y se ajustó a pH=6 mediante la adición de HCl 1 N. La mezcla se filtró, y la torta de filtración se lavó con H2O (2 × 20 ml). La torta sólida se secó a presión reducida proporcionando el producto deseado (5,5 g, 84 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C19H26N8O4: 431,22; observado 431,4.
Elemento estructural AE. Ácido 4-amino-2-[(2R)-4-16-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-ill-2-(hidroximetil)piperazin-1-il]pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(4-amino-5-(etoxicarbonil)pirimidin-2-il)-3-(((terc-butildifenilsilil)oxi)metil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo
A una disolución de 2-(3-(((terc-butildifenilsilil)oxi)metil) piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo (17,2 g, 29,26 mmoles, 1,0 equiv) en MeCN (200 ml) se añadió K2CO3 (12,13 g, 87,78 mmoles, 3,0 equiv) y 4-amino-2-cloropirimidin-5-carboxilato de etilo (6,37 g, 31,60 mmoles, 1,08 equiv). La mezcla se agitó a 80 °C durante 18 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente, se filtró y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→33 % de EtOAc/éter de petróleo) dio el producto deseado (20,3 g, 90,6 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C40H52N8O5Si: 753,39; observado 753,4.
Etapa 2: Síntesis de ácido (R)-4-amino-2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)-2-(hidroximetil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(4-amino-5-(etoxicarbonil)pirimidin-2-il)-3-(((terc-butildifenilsilil)oxi)metil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo (20,3 g, 26,96 mmoles, 1,0 equiv) en THF (200 ml) se añadió TBAF (1,0 M, 50,75 ml, 1,9 equiv). La mezcla de reacción se agitó a temperatura ambiente durante 5 h. La mezcla se vertió entonces en H2O (200 ml) y la fase acuosa se extrajo con EtOAc (2 × 150 ml). Las fases orgánicas combinadas se lavaron con salmuera (2 × 100 ml), se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (0 %→20 % de EtOAc/éter de petróleo) dio el producto deseado (12 g,
85,7 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C24H34N8O5: 515,28; observado 515,4.
Etapa 3: Síntesis de ácido (R)-4-amino-2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)-2-(hidroximetil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de ácido (R)-4-amino-2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)-2-(hidroximetil)piperazin-1-il)pirimidin-5-carboxílico (12 g, 23,32 mmoles, 1,0 equiv) en THF (100 ml), EtOH (30 ml) y H2O (30 ml) se añadió LiOH•H2O (5,87 g, 139,92 mmoles, 6,0 equiv). La mezcla se agitó a 50 °C durante 22 h. La mezcla se concentró entonces a presión reducida para retirar el THF y el EtOH. La fase acuosa se neutralizó con HCl 1 N y el precipitado resultante se filtró. La torta de filtración se lavó con H2O (50 ml) y se secó a presión reducida. El filtrado se extrajo con DCM (8 × 60 ml) y las fases orgánicas combinadas se lavaron con salmuera (2 × 50 ml), se secaron, se filtraron y se concentraron a presión reducida. El residuo resultante se combinó con la torta de filtración inicial y el sólido se disolvió en DCM (150 ml) y se concentró a presión reducida proporcionando el producto deseado (9,76 g, 85,2 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C22H30N8O5: 487,24; observado 487,2.
Elemento estructural AF. Ácido 4-amino-2-[(2S)-4-{6-[(terc-butoxi)carbonil]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}-2-(hidroximetil)piperazin-1-il]pirimidin-5-carboxílico

Este elemento estructural se prepara a partir del Elemento estructural O por un proceso similar a aquél para el Elemento estructural J utilizando 4-amino-2-cloropirimidin-5-carboxilato de etilo.
Elemento estructural AG. Ácido 2-((2-(4-(5-((di-(terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-2-oxoetil)(metil)amino)acético.

A una disolución de N-terc-butoxicarbonil-N-((2-piperazin-1-ilpirimidin-5-il)metil)carbamato de terc-butilo (4,88 g, 12,39 mmoles, 1,0 equiv) en EtOAc (40 ml) se añadió 4-metilmorfolino-2,6-diona (1,6 g, 12,39 mmoles, 1,0 equiv). La reacción se agitó a temperatura ambiente durante 2 h, entonces mezcla de reacción se concentró a presión reducida dando el producto en bruto. El residuo se trituró con EtOAc (15 ml) y se filtró dando el producto (5,65 g, 87,2 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C24H39N6O7: 523,28; observado 523,3.
Elemento estructural AH. N-terc-Butoxicarbonil-N-((2-(4-(3-(2-piperazin-1-iletoxi)propanoil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo.

Etapa 1: Síntesis de 4-(2-(3-(terc-butoxi)-3-oxopropoxi)etil)piperazin-1-carboxilato de bencilo A una disolución de 3-(2-bromoetoxi)propanoato de terc-butilo (35 g, 138,27 mmoles, 1,0 equiv) y piperazin-1-carboxilato de bencilo (31,14 ml, 138,27 mmoles, 1,0 equiv, HCl) en MeCN (420 ml) se añadió K2CO3 (57,33 g, 414,80 mmoles, 3,0 equiv). La reacción se agitó a 80 °C durante 20 h. La mezcla de reacción se enfrió hasta temperatura ambiente y la suspensión se filtró. La torta de filtración se lavó con EtOAc (3 × 50 ml) y los filtrados combinados se concentraron a presión reducida dando el producto en bruto. El residuo se purificó por cromatografía en gel de sílice (5/1 a 0/1 de éter de petróleo/EtOAc) dando el producto (46 g, 84,8 % de rendimiento) como un aceite amarillo.
Etapa 2: Síntesis de ácido 3-(2-(4-((benciloxi)carbonil)piperazin-1-il)etoxi)propanoico
Una disolución de 4-(2-(3-(terc-butoxi)-3-oxopropoxi)etil)piperazin-1-carboxilato de bencilo (21 g, 53,50 mmoles, 1,0 equiv) en TFA (160 ml) se agitó a temperatura ambiente durante 2 h y entonces se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 4/1 de EtOAc/MeOH) dando el producto (20,4 g, 84,7 % de rendimiento) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C17H24N2O5: 337,18; observado 337,1.
Etapa 3: Síntesis de 4-(2-(3-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-carboxilato de bencilo
A una disolución de ácido 3-(2-(4-((benciloxi)carbonil)piperazin-1-il)etoxi)propanoico (20,2 g, 44,85 mmoles, 1,0 equiv, TFA) en DCM (500 ml) se añadió HATU (25,58 g, 67,27 mmoles, 1,5 equiv) y DIPEA (17,39 g, 134,55 mmoles, 23,44 ml, 3,0 equiv). La reacción se agitó a temperatura ambiente durante 30 min, y entonces se añadió N-terc-butoxicarbonil-N-((2-piperazin-1-ilpirimidin-5-il)metil)carbamato de terc-butilo (14,12 g, 35,88 mmoles, 0,8 equiv). La mezcla de reacción se agitó durante 2 h y entonces se enfrió rápidamente con NH4Cl sat. (500 ml). La fase acuosa se extrajo con DCM (3 × 300 ml) y la fase orgánica combinada se lavó con salmuera (30 ml), se secó con anhidro Na2SO4, se filtró y se concentró a presión reducida dando el producto en bruto. El residuo se purificó por cromatografía en gel de sílice (0/1 de éter de petróleo/EtOAc a 10/1 DCM/MeOH) dando el producto (29 g, 90,8 % de rendimiento) como un aceite amarillo. LCMS (ESI) m/z: [M H] calculado para C36H53N7O8: 712,41; observado 712,4.
Etapa 4: Síntesis de N-terc-butoxicarbonil-N-((2-(4-(3-(2-piperazin-1-iletoxi)propanoil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo
A una disolución de 4-(2-(3-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-carboxilato (5 g, 7,02 mmoles, 1,0 equiv) en EtOAc (150 ml) se añadió Pd/C (2 g, 10 % en peso). La suspensión se desgasificó y se purgó con H2 y entonces se agitó bajo H2 (30 psi) a 30 °C durante 3 h. La suspensión se enfrió entonces hasta temperatura ambiente y se filtró a través de Celite. La torta de filtración se lavó con MeOH (15 × 100 ml) y los filtrados combinados se concentraron a presión reducida dando el producto (12 g, 89,9 % de rendimiento) como un aceite amarillo claro. LCMS (ESI) m/z: [M H] calculado para C28H47N7O6: 578,37; observado 578,5.
Elemento estructural AI.2-(Piperazin-1-il)pirimidin-5-carboxilato de etilo.

Etapa 1: Síntesis de 2-(4-(terc-butoxicarbonil)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de piperazin-1-carboxilato de terc-butilo (11,94 g, 53,59 mmoles, 1,0 equiv, HCl) y 2-cloropirimidin-5-carboxilato de etilo (10 g, 53,59 mmoles, 1,0 equiv) en MeCN (100 ml) se añadió K2CO3 (7,41 g, 53,59 mmoles, 1,0 equiv). La mezcla se agitó a 80 °C durante 17 h y luego se vertió en H2O (200 ml). La mezcla se filtró y la torta de filtración se lavó con H2O (80 ml) y se secó a presión reducida dando el producto (15,76 g, 82 % de rendimiento) como un sólido blanco.
Etapa 2: Síntesis de 2-(piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de 2-(4-(terc-butoxicarbonil)piperazin-1-il)pirimidin-5-carboxilato de etilo (15,7 g, 46,67 mmoles, 1,0 equiv) en EtOAc (150 ml) se añadió HCl/EtOAc (150 ml) a 0 °C. La mezcla resultante se agitó a temperatura ambiente durante 9 h. La mezcla de reacción se filtró y la torta de filtración se lavó con EtOAc (100 ml). El sólido se
secó a presión reducida dando el producto (12,55 g, 96 % de rendimiento, HCl) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C11H16N4O2: 237,14; observado 237,3.
Elemento estructural AJ. Ácido 2-(4-(2-(3-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(2-(3-(terc-butoxi)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo A una disolución de 2-piperazin-1-ilpirimidin-5-carboxilato de etilo (17,92 g, 75,85 mmoles, 1,2 equiv) y 3-(2-bromoetoxi)propanoato de terc-butilo (16 g, 63,21 mmoles, 1,0 equiv) en MeCN (200 ml) se añadió K2CO3 (17,47 g, 126,42 mmoles, 2,0 equiv). La reacción se agitó a 80 °C durante 12 h y entonces la mezcla de reacción se filtró, y el filtrado se concentró a presión reducida. El producto en bruto se suspendió en éter de petróleo (200 ml) y se agitó durante 20 min a 0 °C y entonces se filtró. El sólido se secó a presión reducida dando el producto (19,4 g, 75,1 % de rendimiento) como un sólido amarillo.
Etapa 2: Síntesis de ácido 3-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)etoxi)propanoico
Una disolución de 2-(4-(2-(3-(terc-butoxi)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (19,4 g, 47,49 mmoles, 1,0 equiv) en TFA (200 ml) se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se concentró entonces a presión reducida y el residuo se purificó por cromatografía en gel de sílice (50/1 a 1/1 de EtOAc/MeOH) dando el producto (18 g, 81,3 % de rendimiento) como un aceite amarillo.
Etapa 3: Síntesis de 2-(4-(2-(3-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de ácido 3-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)etoxi)propanoico (13 g, 27,87 mmoles, 1,0 equiv) en DCM (200 ml) se añadió HATU (15,90 g, 41,81 mmoles, 1,5 equiv) y DIPEA (19,42 ml, 111,49 mmoles, 4,0 equiv). La reacción se agitó entonces a temperatura ambiente durante 30 min y entonces se añadió N-terc-butoxicarbonil-N-[(2-piperazin-1-ilpirimidin-5-il)metil]carbamato de terc-butilo (10,97 g, 27,87 mmoles, 1,0 equiv). La mezcla se agitó a durante 2 h y luego se vertió en una disolución sat. de NH4Cl (200 ml). La fase acuosa se extrajo con DCM (2 × 200 ml) y la fase orgánica combinada se lavó con salmuera (2 × 20 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (100/1 a 9/1 de EtOAc/MeOH) dando el producto (17 g, 79,0 % de rendimiento) como un aceite amarillo.
Etapa 4: Síntesis de ácido 2-(4-(2-(3-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(2-(3-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (11 g, 15,11 mmoles, 1,0 equiv) en THF (40 ml), EtOH (10 ml) y H2O (20 ml) se añadió LiOH●H2O (1,27 g, 30,23 mmoles, 2,0 equiv). La mezcla se agitó entonces a 35 °C durante 1,5 h. La mezcla de reacción se extrajo con EtOAc (30 ml) y la fase acuosa se ajustó a pH = 7 mediante la adición de HCl (1 N). La mezcla se concentró entonces a presión reducida. El producto en bruto se purificó por cromatografía de fase inversa (20/1 a 3/1 de H2O/MeCN) dando el producto (6,1 g, 67,3 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C33H49N9O8: 700,38; observado 700,4.
Elemento estructural AK. Ácido 2-(4-(2-(3-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico.

A una disolución de 2-(4-(2-(3-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (5,4 g, 7,42 mmoles, 1,0 equiv) en THF (40 ml), EtOH (10 ml) y H2O (10 ml) se añadió LiOH●H2O (933,92 mg, 22,26 mmoles, 3,0 equiv). La mezcla se agitó entonces a 30 °C durante 12 h. La mezcla de reacción se extrajo entonces con EtOAc (2 × 50 ml) y la fase acuosa se ajustó a pH = 7 mediante la adición de HCl (1 N). La disolución se concentró entonces a presión reducida. El producto en bruto se purificó por cromatografía de fase inversa (20/1 a 3/1 de H2O/MeCN) dando el producto (1,01 g, 22,5 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C28H41N9O6: 600,33; observado 600,2.
Elemento estructural AL. Ácido 4-{4-[2-(3-{4-[5-({[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il]piperazin-1-il}-3-oxopropoxi)etil]piperazin-1-il}-4-oxobutanoico.

A una disolución de N-terc-butoxicarbonil-N-((2-(4-(3-(2-piperazin-1-iletoxi)propanoil)piperazin-1-il)pirimidin-5-il)metil)carbamato de terc-butilo (1,0 equiv) en DCM se añade anhídrido succínico (1,2 equiv) y Et3N (2,0 equiv). La reacción se agita a temperatura ambiente hasta que se consume el material de partida, como se ha determinado por análisis de LCMS. La mezcla de reacción se concentra entonces a presión reducida dando el producto en bruto. El residuo se purifica por cromatografía en gel de sílice proporcionando el producto.
Elemento estructural AM. Ácido 2-(4-(4-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-4-oxobutil)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(4-(terc-butoxi)-4-oxobutil)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de clorhidrato de 2-(piperazin-1-il)pirimidin-5-carboxilato de etilo (10 g, 36,67 mmoles, 1,0 equiv, HCl) y 4-bromobutanoato de terc-butilo (8,18 g, 36,67 mmoles, 1,0 equiv) en DMF (100 ml) se añadió Et3N (15,31 ml, 110,00 mmoles, 3,0 equiv). La mezcla se agitó a 130 °C durante 14 h. La mezcla se vertió entonces en H2O (400 ml) y la disolución se extrajo con EtOAc (3 × 150 ml). La fase orgánica combinada se lavó con salmuera (200 ml), se secó sobre Na2SO4 y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (5/1 a 1/1 de éter de petróleo/EtOAc) dando el producto (9,5 g, 68,5 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z:
[M H] calculado para C19H30N4O4: 379,24; observado 379,2, 380,2.
Etapa 2: Síntesis de clorhidrato de ácido 4-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)butanoico
A una disolución de 2-(4-(4-(terc-butoxi)-4-oxobutil)piperazin-1-il)pirimidin-5-carboxilato de etilo (9,5 g, 25,10 mmoles, 1,0 equiv) en EtOAc (100 ml) se añadió HCl/EtOAc (500 ml). La mezcla se agitó a temperatura ambiente durante 10 h
y entonces la disolución se concentró a presión reducida dando el producto (9,6 g, 96,8 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C15H22N4O4: 323,17; observado 323,2.
Etapa 3: Síntesis de ácido etil-2-(4-(4-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-4-oxobutil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de clorhidrato de ácido 4-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)butanoico (5 g, 15,51 mmoles, 1,0 equiv) y N-terc-butoxicarbonil-N-((2-piperazin-1-ilpirimidin-5-il)metil)carbamato de terc-butilo (6,10 g, 15,51 mmoles, 1,0 equiv) en DMF (150 ml) se añadió DIPEA (8,11 ml, 46,53 mmoles, 3,0 equiv) y HATU (7,08 g, 18,61 mmoles, 1,2 equiv). La mezcla se agitó a temperatura ambiente durante 3 h y entonces la disolución se vertió en H2O (600 ml). La fase acuosa se extrajo con EtOAc (3 × 200 ml) y entonces la fase orgánica combinada se lavó con salmuera (100 ml), se secó con Na2SO4 y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (50/1 a 15/1 de DCM/MeOH) dando el producto (6,3 g, 58,2 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C34H51N9O7: 698,40; observado 698,6.
Etapa 4: Síntesis de ácido 2-(4-(4-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-4-oxobutil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(4-(4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-4-oxobutil)piperazin-1-il)pirimidin-5-carboxilato de etilo (4,5 g, 6,45 mmoles, 1,0 equiv) en EtOH (7 ml) y THF (28 ml) se añadió una disolución de LiOH•H2O (541,17 mg, 12,90 mmoles, 2,0 equiv) en H2O (7 ml). La mezcla se agitó a 30 °C durante 8 h, entonces se añadió LiOH•H2O adicional (541 mg, 12,90 mmoles, 2,0 equiv). Después de agitar durante 8 h adicionales a 30 °C, la disolución se concentró a presión reducida. Se añadió H2O (20 ml) y disolución se ajustó a pH 3 con HCl 1 N. La suspensión se filtró y el sólido se secó a presión reducida dando el producto (3,2 g, 79,1 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C27H39N9O5: 570,32; observado 570,3.
Elemento estructural AN. Ácido 2-(4-(2-(2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 4-(2-hidroxietil)piperazin-1-carboxilato de bencilo
A una disolución de clorhidrato de piperazin-1-carboxilato de bencilo (41,09 g, 160,04 mmoles, 1,0 equiv, HCl) en MeCN (200 ml) se añadió K2CO3 (66,36 g, 480,13 mmoles, 3,0 equiv) y 2-bromoetanol (20 g, 160,04 mmoles, 1,0 equiv). La mezcla de reacción se agitó a 80 °C durante 16 h, momento en el que se enfrió hasta temperatura ambiente y se filtró. La torta de filtración se lavó con EtOAc (100 ml) y el filtrado se lavó entonces con H2O (100 ml). La fase acuosa se extrajo con EtOAc (3 × 50 ml) y las fases orgánicas combinadas se lavaron con salmuera (50 ml), se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (5→25 % de
MeOH/EtOAc) dio el producto deseado como un sólido amarillo (20 g, 47 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C14H20N2O3: 265,16; observado 264,9.
Etapa 2: Síntesis de 4-(2-hidroxietil)piperazin-1-carboxilato de terc-butilo
A una disolución de piperazin-1-carboxilato de terc-butilo (198,72 g, 1,07 moles, 1,0 equiv) en MeCN (1500 ml) se añadió 2-bromoetanol (240 g, 1,92 moles, 1,8 equiv) y K2CO3 (221,19 g, 1,60 moles, 1,5 equiv). La mezcla de reacción se agitó a 80 °C durante 16 h, momento en el que la mezcla se enfrió hasta temperatura ambiente, se filtró y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→714 % de MeOH/EtOAc) dio el producto deseado como un sólido blanco (146 g, 59 % de rendimiento).
Etapa 3: Síntesis de 4-(2-bromoetil)piperazin-1-carboxilato de terc-butilo
A una disolución de 4-(2-hidroxietil)piperazin-1-carboxilato de terc-butilo (45 g, 195,39 mmoles, 1,0 equiv) en THF (600 ml) se añadió trifenilfosfina (97,38 g, 371,25 mmoles, 1,9 equiv) y CBr4 (116,64 g, 351,71 mmoles, 1,8 equiv). La mezcla se agitó a temperatura ambiente durante 3 h. Se combinaron dos lotes separados, y la mezcla de reacción se filtró, y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (1→25 % de EtOAc/éter de petróleo) dio el producto deseado como un sólido amarillo claro (31 g, 27 % de rendimiento).
Etapa 4: Síntesis de 4-(2-(2-(4-(terc-butoxicarbonil)piperazin-1-il)etoxi)etil)piperazin-1-carboxilato de bencilo A una disolución de 4-(2-hidroxietil)piperazin-1-carboxilato de bencilo (18 g, 68,10 mmoles, 1,0 equiv) en tolueno (200 ml) se añadió NaNH2 (26,57 g, 680,99 mmoles, 10,0 equiv). Se añadió 4-(2-bromoetil)piperazin-1-carboxilato de terc-butilo (25 g, 85,27 mmoles, 1,25 equiv) y la mezcla se calentó hasta 90 °C durante 18 h. La mezcla se enfrió hasta temperatura ambiente y se vertió en H2O (700 ml) a 0 °C. La fase acuosa se extrajo con EtOAc (3 × 240 ml) y las fases orgánicas combinadas se lavaron sucesivamente con H2O (350 ml) y salmuera sat. (2 × 200 ml), se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (0→12 % de MeOH/EtOAc) dio el producto deseado como un aceite amarillo claro (20 g, 62 % de rendimiento).
Etapa 5: Síntesis de 4-(2-(2-(piperazin-1-il)etoxi)etil)piperazin-1-carboxilato de terc-butilo
A una disolución de 4-(2-(2-(4-(terc-butoxicarbonil)piperazin-1-il)etoxi)etil)piperazin-1-carboxilato de bencilo (20 g, 41,96 mmoles, 1,0 equiv) en EtOAc (180 ml) se añadió Pd/C (8 g, 10 % en peso). La suspensión se desgasificó a presión reducida y se purgó con H2 tres veces. La mezcla se agitó bajo H2 (30 psi) a 35 °C durante 12 h. La mezcla de reacción se filtró entonces, y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→100 % de MeOH/EtOAc) dio el producto deseado como un aceite incoloro (10,8 g, 75 % de rendimiento).
Etapa 6: Síntesis de 2-(4-(2-(2-(4-(terc-butoxicarbonil)piperazin-1-il)etoxi)-etil)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de 4-(2-(2-(piperazin-1-il)etoxi)etil)piperazin-1-carboxilato de terc-butilo (10,8 g, 31,54 mmoles, 1,0 equiv) en MeCN (100 ml) se añadió K2CO3 (13,08 g, 94,61 mmoles, 3,0 equiv) y 2-cloropirimidin-5-carboxilato de etilo (5,88 g, 31,54 mmoles, 1,0 equiv). La mezcla se agitó a 80 °C durante 12 h, momento en el que la reacción se enfrió hasta temperatura ambiente, se filtró y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→9 % de MeOH/DCM) dio el producto deseado como un sólido blanco (13,6 g, 85 % de rendimiento).
Etapa 7: Síntesis de ácido 2-(4-(2-(2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido-[4,3-d]pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(2-(2-(4-(terc-butoxicarbonil)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (13,6 g, 27,61 mmoles, 1,0 equiv) en MeOH (50 ml) se añadió una disolución de HCl en MeOH (4 M, 150 ml, 21,7 equiv). La reacción se agitó a temperatura ambiente durante 4 h, momento en el que la mezcla se concentró a presión reducida proporcionando el producto deseado en bruto como un sólido blanco (13,8 g, 4HCl) que se llevó directamente a la siguiente etapa. LCMS (ESI) m/z: [M H] calculado para C19H32N6O3: 393,26; observado 393,3.
Etapa 8: Síntesis de 2-(4-(2-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)-piperazin-1-il)etoxi)etil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución con agitación de ácido 2-(4-(2-(2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxílico (10,2 g, 18,95 mmoles, 1,0 equiv, 4HCl) y DIPEA (16,50 ml, 94,74 mmoles, 5,0 equiv) en DMF (100 ml) se añadió 2-cloro-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (5,11 g, 18,95 mmoles, 1,0 equiv). La mezcla de reacción se agitó a 90 °C durante 12 h. Entonces se enfrió la mezcla de reacción hasta temperatura ambiente y se añadió a EtOAc (200 ml) y H2O (400 ml). La fase acuosa se extrajo con EtOAc (2 × 100 ml) y las fases orgánicas combinadas se lavaron con NH4Cl acuoso (4 × 100 ml), salmuera (2 × 100 ml), se secaron, se filtraron y se concentraron a presión reducida. La purificación por
cromatografía en gel de sílice (0→9 % de MeOH/DCM) dio el producto deseado como un sólido blanco (5,4 g, 45 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C31H47N9O5: 626,38; observado 626,3.
Etapa 9: Síntesis de ácido 2-(4-(2-(2-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(2-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (5,4 g, 8,63 mmoles, 1,0 equiv) en THF (50 ml), EtOH (20 ml) y H2O (20 ml) se añadió LiOH•H2O (1,09 g, 25,89 mmoles, 3,0 equiv). La mezcla de reacción se agitó a 35 °C durante 12 h, momento en el que la mezcla se concentró a presión reducida para retirar el THF y el EtOH. La fase acuosa se neutralizó a pH = 7 con HCl 0,5 N y se concentró a presión reducida. La purificación por cromatografía de fase inversa dio el producto deseado como un sólido blanco (4,72 g, 92 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C29H43N9O5: 598,35; observado 598,3.
Elemento estructural AO. Ácido 1'-[(terc-butoxi)carbonil]-[1,4'-bipiperidin]-4-carboxílico.

En el momento de la presente solicitud este elemento estructural estaba comercialmente disponible (CAS N.º 201810-59-5).
Elemento estructural AP. Sal de clorhidrato de 2-((2-(piperazin-1-il)pirimidin-5-il)metil)isoindolin-1,3-diona.

Etapa 1: Síntesis de 2-cloro-5-(dibromometil)pirimidina
A una disolución de 2-cloro-5-metilpirimidina (100 g, 777,85 mmoles, 1,0 equiv) en CCl4 (1200 ml) se añadió NBS (304,58 g, 1,71 moles, 2,2 equiv) y AIBN (51,09 g, 311,14 mmoles, 0,4 equiv). La mezcla se agitó a 80 °C durante 16 h. La disolución de reacción se enfrió entonces hasta temperatura ambiente, se filtró y el filtrado se vertió en H2O (1500 ml). La disolución se diluyó con DCM (3 × 250 ml) y la fase orgánica se lavó con salmuera (300 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida dando el producto en bruto como un aceite marrón, que se usó directamente en la siguiente etapa.
Etapa 2: Síntesis de 5-(bromometil)-2-cloropirimidina
A una disolución de 2-cloro-5-(dibromometil)pirimidina (229 g, 799,72 mmoles, 1,0 equiv) en THF (600 ml) se añadió DIPEA (111,44 ml, 639,77 mmoles, 0,8 equiv) y 1-etoxifosfonoiloxietano (82,57 ml, 639,77 mmoles, 0,8 equiv). La mezcla se agitó a temperatura ambiente durante 19 h. La mezcla se vertió entonces en H2O (1200 ml) y la fase acuosa se extrajo con EtOAc (3 × 300 ml). La fase orgánica combinada se lavó con salmuera (300 ml), se secó con Na2SO4 anhidro, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (1/0 a 0/1 de éter de petróleo/EtOAc) dando el producto como un aceite marrón, que se usó directamente para la siguiente etapa.
Etapa 3: Síntesis de 2-((2-cloropirimidin-5-il)metil)isoindolin-1,3-diona
A una mezcla de isoindolin-1,3-diona (15 g, 101,95 mmoles, 1,0 equiv) en DMF (126 ml) se añadió NaH (4,89 g, 122,34 mmoles, 60 % en peso, 1,2 equiv) a 0 °C. La mezcla se agitó a 0 °C durante 30 min, entonces se añadió gota a gota una disolución de 5-(bromometil)-2-cloro-pirimidina (30,21 g, 101,95 mmoles, 1,0 equiv) en DMF (24 ml) a la mezcla anterior a temperatura ambiente. La mezcla se agitó a temperatura ambiente durante 2 h y entonces se enfrió hasta 0 °C y se enfrió rápidamente con NH4Cl sat. (600 ml). La suspensión se filtró y el sólido se secó a presión
reducida dando el producto en bruto (27,4 g, 98,2 % de rendimiento) como un sólido gris, que se usó directamente en la siguiente etapa. LCMS (ESI) m/z: [M H] calculado para C13H8ClN3O2: 274,04; observado 274,0.
Etapa 4: Síntesis de 4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-carboxilato de terc-butilo A una disolución de 2-((2-cloropirimidin-5-il)metil)isoindolin-1,3-diona (27 g, 98,66 mmoles, 1,0 equiv) y piperazin-1-carboxilato de terc-butilo (20,21 g, 108,52 mmoles, 1,1 equiv) en DMF (270 ml) se añadió K2CO3 (34,09 g, 246,64 mmoles, 2,5 equiv). La mezcla se agitó a 80 °C durante 3 h y entonces la reacción se enfrió hasta temperatura ambiente y se vertió en H2O (1200 ml). La suspensión se filtró y el sólido se secó a presión reducida dando el producto en bruto (35,58 g, 85,2 % de rendimiento) como un sólido blanco, que se usó directamente en la siguiente etapa. Etapa 5: Síntesis de 2-((2-(piperazin-1-il)pirimidin-5-il)metil)isoindolin-1,3-diona
Una disolución de 4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-carboxilato de terc-butilo (15 g, 35,42 mmoles, 1 equiv) en HCl/EtOAc (150 ml) se agitó a temperatura ambiente durante 2 h. La mezcla se filtró y entonces la torta de filtración se lavó con EtOAc (20 ml) y se secó a presión reducida dando el producto (42,53 g, 92,5 % de rendimiento) como un sólido blanco.
Elemento estructural AQ. Ácido 2-[(2-{4-[2-(3-{4-[5-({bis[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il]piperazin-1-il}-3-oxopropoxi)etil]piperazin-1-il}-2-oxoetil)(metil)amino]acético.

A una disolución de N-[(terc-butoxi)carbonil]-N-{[2-(4-{3-[2-(piperazin-1-il)etoxi]propanoil}piperazin-1-il)pirimidin-5-il]metil}carbamato de terc-butilo (300 mg, 519 µmoles, 1,0 equiv) en piridina (8 ml) a 0 °C se añadió 4-metilmorfolina-2,6-diona (80,3 mg, 622 µmoles, 1,2 equiv). La mezcla de reacción se agitó a 0 °C durante 1 h y entonces se calentó hasta temperatura ambiente y se agitó durante 12 h adicionales. El disolvente se concentró a presión reducida y el sólido se repartió entre DCM y H2O. La fase orgánica se separó, se secó sobre MgSO4 y el disolvente se concentró a presión reducida dando el producto (23,0 mg, 6,28 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C33H54N8O9: 707,41; observado 707,4.
Elemento estructural AR. Ácido 2-(4-(2-(3-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(3-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)etoxi)propanoil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo
A una disolución de ácido 3-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)etoxi)propanoico (6 g, 12,86 mmoles, 1,0 equiv, TFA) en DMF (55 ml) se añadió HATU (6,36 g, 16,72 mmoles, 1,3 equiv) y DIPEA (11,20 ml, 64,32 mmoles, 5,0 equiv). Después de 0,5 h, se añadió 2-(piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (4,11 g, 12,86 mmoles, 1,0 equiv). La mezcla se agitó durante 3 h, momento en el que se filtró y la torta sólida se secó a presión reducida proporcionando el producto deseado como un sólido blanco (7,5 g, 89 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C32H47N9O6: 654,37; observado 654,4.
Etapa 2: Síntesis de ácido 2-(4-(2-(3-(4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(3-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)etoxi)propanoil)piperazin-1-il)-7, 8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de terc-butilo (7,2 g, 11,01 mmoles, 1,0 equiv) en THF (72 ml), EtOH (36 ml) y H2O (36 ml) se añadió LiOH•H2O (1,85 g, 44,05 mmoles, 4,0 equiv). La mezcla de reacción se agitó a temperatura ambiente durante 2,5 h, momento en el que la mezcla se filtró y el filtrado se concentró a presión reducida para retirar el THF y el EtOH. La fase acuosa se neutralizó a pH = 7 con HCl 1 N, y entonces se concentró a presión reducida. La purificación por cromatografía de fase inversa (30 % de MeCN/H2O) dio el producto deseado como un sólido blanco (3,85 g, 54 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C30H43N9O6: 626,34; observado 626,3.
Elemento estructural AS. Ácido 2-(4-(2-(2-(3-(4-(5-((di-terc-butoxicarbonilamino)metil)pirimidin-2-il)piperazin-1-il)-3-oxo-propoxi)etoxi)etil)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de ácido 3-(2-(2-(4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1il)etoxi)etoxi)propanoico Una disolución de 2-(4-(2-(2-(3-(terc-butoxi)-3-oxopropoxi)etoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (4 g, 8,84 mmoles, 1,0 equiv) en TFA (12,29 ml, 166,00 mmoles, 18,8 equiv) se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→720 % de MeOH/EtOAc) dio el producto deseado como un aceite marrón (4,35 g, 95 % de rendimiento, sal de TFA).
Etapa 2: Síntesis de 2-(4-(2-(2-(3-(4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de ácido 3-(2-(2-(4-(5-etoxicarbonilpirimidin-2-il)piperazin-1-il)etoxi)etoxi)propanoico (3,8 g, 7,44 mmoles, 1,0 equiv, TFA) en DCM (30 ml) se añadió HATU (4,25 g, 11,17 mmoles, 1,5 equiv) y DIPEA (6,48 ml, 37,22 mmoles, 5,0 equiv). La reacción se agitó a temperatura ambiente durante 30 min, y entonces se añadió N-terc-butoxicarbonil-N-((2-piperazin-lilpirimidin-5-il)metil)carbamato de terc-butilo (2,93 g, 7,44 mmoles, 1,0 equiv). La mezcla se agitó a temperatura ambiente durante 3,5 h, momento en el que la mezcla de reacción se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→20 % de MeOH/EtOAc) dio el producto deseado como un aceite marrón (4,14 g, 70 % de rendimiento).
Etapa 3: Síntesis de ácido 2-(4-(2-(2-(3-(4-(5-((di-terc-butoxicarbonilamino)metil)pirimidin-2-il)piperazin-1-il)-3-oxo-propoxi)etoxi)etil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(2-(2-(3-(4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (1,4 g, 1,81 mmoles, 1,0 equiv) en THF (28 ml), EtOH (14 ml) y H2O (14 ml) se añadió LiOH•H2O (304,44 mg, 7,25 mmoles, 4,0 equiv). La mezcla se agitó a 40 °C durante 30 min, momento en el que la mezcla de reacción se concentró a presión reducida. La purificación por cromatografía de fase inversa (10→40 % de MeCN/H2O) dio el producto deseado como un sólido amarillo (500 mg, 43 % de rendimiento).
Elemento estructural AT. Ácido 2-{4-[2-(2-{4-[5-({[(terc-butoxi)carbonil]amino)metil)pirimidin-2-il]piperazin-1-il}etoxi)etil]piperazin-1-il}pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(2-(2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo
A una disolución de clorhidrato de 2-(4-(2-(2-(piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (7,3 g, 13,56 mmoles, 1,0 equiv, 4HCl) en DMF (75 ml) se añadió DIPEA (14,17 ml, 81,36 mmoles, 6,0 equiv) y terc-butil-N-terc-butoxicarbonil-N-[(2-cloropirimidin-5-il)metil]carbamato (5,59 g, 16,27 mmoles, 1,2 equiv). La mezcla se agitó a 80 °C durante 12 h. La mezcla se enfrió entonces hasta temperatura ambiente y se vertió en H2O (300 ml). La fase acuosa se extrajo con EtOAc (3 × 80 ml). Las fases orgánicas combinadas se lavaron con NH4Cl sat. (4 × 80 ml) y salmuera (150 ml), se secaron, se filtraron y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→17 % de MeOH/EtOAc) dio el producto deseado (7,7 g, 81,1 % de rendimiento) como un aceite amarillo claro. LCMS (ESI) m/z: [M Na] calculado para C34H53N9O7: 722,40; observado 722,4. Etapa 2: Síntesis de ácido 2-(4-(2-(2-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(4-(2-(2-(4-(5-(((di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)etoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de etilo (7,7 g, 11,00 mmoles, 1,0 equiv) en THF (80 ml), EtOH (20 ml) y H2O (40 ml) se añadió LiOH•H2O (2,31 g, 55,01 mmoles, 5,0 equiv). La mezcla se agitó a 50 °C durante 26 h. La mezcla se concentró entonces a presión reducida para retirar el THF y el EtOH. La fase acuosa se neutralizó con HCl 0,5 N y se concentró a presión reducida. La purificación por cromatografía de fase inversa dio el producto deseado (4,67 g, 74,3 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M - H] calculado para C27H41N9O5: 570,31; observado 570,3.
Elemento estructural AU. 4-(5-(((terc-Butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-2-carboxilato de (R)-terc-butilo.

Etapa 1: Síntesis de ácido (R)-1,4-bis((benciloxi)carbonil)piperazin-2-carboxílico
A dos lotes separados que contenían una disolución de ácido (2R)-piperazin-2-carboxílico (70 g, 344,71 mmoles, 1 equiv, 2HCl) en H2O (700 ml) y dioxano (1120 ml) se añadió 50 % de NaOH ac. hasta que pH = 11. Se añadió cloroformiato de bencilo (156,82 ml, 1,10 moles, 3,2 equiv) y la reacción se agitó a temperatura ambiente durante 12 h. Las dos mezclas de reacción se combinaron y se añadió H2O (1200 ml). La fase acuosa se extrajo con MTBE (3 × 1000 ml), se ajustó a pH = 2 con HCl conc. y luego se extrajo con EtOAc (2 × 1000 ml). Las fases orgánicas combinadas se secaron, se filtraron y se concentraron a presión reducida proporcionando el producto deseado (280 g, 86 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C21H22N2O6: 399,16; observado 399,0.
Etapa 2: Síntesis de piperazin-1,2,4-tricarboxilato de (R)-1,4-dibencil-2-terc-butilo
A una disolución de ácido (R)-1,4-bis((benciloxi)carbonil)piperazin-2-carboxílico (70 g, 175,70 mmoles, 1,0 equiv) en tolueno (700 ml) a 80 °C se añadió 1,1-di-terc-butoxi-N,N-dimetil-metanamina (80,04 ml, 333,83 mmoles, 1,9 equiv). La reacción se agitó a 80 °C durante 2 h, momento en el que se enfrió hasta temperatura ambiente y se repartió entre
EtOAc (300 ml) y H2O (500 ml). La fase acuosa se extrajo con EtOAc (2 × 500 ml) y las fases orgánicas combinadas se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (0→25 de EtOAc/éter de petróleo) dio el producto deseado como un sólido blanco (50 g, 57 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C25H30N2O6: 477,20; observado 476,9.
Etapa 3: Síntesis de piperazin-2-carboxilato de (R)-terc-butilo
A una disolución de piperazin-1,2,4-tricarboxilato de (R)-1,4-dibencil-2-terc-butilo (50 g, 110,01 mmoles, 1 equiv) en EtOAc (20 ml) se añadió Pd/C (15 g, 10 % en peso). La suspensión se desgasificó a presión reducida y se purgó con H2 tres veces. La suspensión se agitó bajo H2 (30 psi) a 30 °C durante 4 h. La mezcla de reacción se filtró entonces, el residuo se lavó con MeOH (5 × 200 ml) y el filtrado se concentró a presión reducida proporcionando el producto deseado como un aceite amarillo (17 g, 81 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C9H18N2O2: 187,15; observado 187,1.
Etapa 4: Síntesis de 4-(5-(((terc-butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-2-carboxilato de (R)-terc-butilo
A una suspensión de piperazin-2-carboxilato de (R)-terc-butilo (8 g, 23,27 mmoles, 1,0 equiv) y ((2-cloropirimidin-5-il)metil)carbamato de terc-butil-N-terc-butoxicarbonilo (5,20 g, 27,92 mmoles, 1,2 equiv) en MeCN (100 ml) se añadió K2CO3 (6,43 g, 46,54 mmoles, 2,0 equiv). La mezcla de reacción se calentó hasta 80 °C durante 12 h, momento en el que se enfrió hasta temperatura ambiente, se filtró y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→100 % de EtOAc/éter de petróleo) dio el producto deseado como un sólido amarillo (9,2 g, 73 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C24H39N5O6:494,30; observado 494,1.
Elemento estructural AV. 4-(5-(((terc-Butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-2-carboxilato de (S)-terc-butilo.

Este elemento estructural se prepara por un proceso similar a aquél para el Elemento estructural AU utilizando ácido (2S)-piperazin-2-carboxílico.
Elemento estructural AW. Ácido (R)-2-(4-(2-(3-(2-(terc-butoxicarbonil)-4-(5-(((terc-butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(4-(2-(3-(2-(terc-butoxicarbonil)-4-(5-(((terc-butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de (R)-etilo
A una disolución de 4-(5-(((terc-butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-2-carboxilato de (R)-terc-butilo (5,3 g, 11,36 mmoles, 1,0 equiv, TFA) en DCM (80 ml) se añadió HATU (6,48 g, 17,05 mmoles,
1,5 equiv) y DIPEA (7,92 ml, 45,45 mmoles, 4,0 equiv). La reacción se agitó a temperatura ambiente durante 30 min y entonces se añadió (2R)-4-(5-((bis(terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-2-carboxilato de terc-butilo (5,61 g, 11,36 mmoles, 1,0 equiv). La mezcla se agitó durante 1 h, momento en el que se añadió NH4Cl sat. (80 ml). La fase orgánica se lavó con NH4Cl sat. (5 × 80 ml), se secó, se filtró y se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→9 % de MeOH/EtOAc) dio el producto deseado como un sólido amarillo (8,4 g, 85 % de rendimiento).
Etapa 2: Síntesis de ácido (R)-2-(4-(2-(3-(2-(terc-butoxicarbonil)-4-(5-(((terc-butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico
A dos lotes separados que contenían una disolución una disolución de 2-(4-(2-(3-(2-(terc-butoxicarbonil)-4-(5-(((terc-butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxilato de (R)-etilo (3,4 g, 4,11 mmoles, 1,0 equiv) en THF (16 ml), EtOH (8 ml) y H2O (8 ml) se añadió LiOH•H2O (344,61 mg, 8,21 mmoles, 2,0 equiv). La mezcla se agitó a temperatura ambiente durante 2 h. Las dos mezclas de reacción se combinaron entonces y se ajustaron a pH = 7 con HCl 1 N. La disolución se concentró a presión reducida para retirar el THF y el EtOH. La disolución se filtró entonces, y el sólido resultante se purificó por cromatografía de fase inversa proporcionando el producto deseado como un sólido blanco (4 g, 59 % de rendimiento). LCMS (ESI) m/z:
[M H] calculado para C38H57N9O10: 800,43; observado 800,3.
Elemento estructural AX. Ácido (S)-2-(4-(2-(3-(2-(terc-butoxicarbonil)-4-(5-(((terc-butoxicarbonil-N-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)piperazin-1-il)pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural AV por un proceso similar a aquél para el Elemento estructural AW.
Elemento estructural AY. Ácido 1'-(2-(3-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)-[1,4'-bipiperidina]-4-carboxílico.

Etapa 1: Síntesis de 4-etil[1,4'-bipiperidin]-1',4-dicarboxilato de 1'-terc-butilo
A una disolución de piperidin-4-carboxilato de etilo (30 g, 150,57 mmoles, 1,0 equiv) y 4-oxopiperidin-1-carboxilato de terc-butilo (23,67 g, 150,57 mmoles, 1,0 equiv) en DCM (300 ml) se añadió HOAc (6,00 ml, 104,95 mmoles, 0,7 equiv). La mezcla se agitó a temperatura ambiente durante 30 min, entonces se añadió NaBH(OAc)3 (63,82 g, 301,13 mmoles, 2,0 equiv). La mezcla se agitó durante 16 h, momento en el que se añadió H2O (50 ml). La fase acuosa se extrajo con DCM (3 × 15 ml) y las fases orgánicas combinadas se lavaron con salmuera (10 ml), se secaron, se filtraron y se
concentraron a presión reducida. La purificación por cromatografía en gel de sílice (8→100 de MeOH/EtOAc) dio el producto deseado como un aceite amarillo (30 g, 59 % de rendimiento).
Etapa 2: Síntesis de [1,4'-bipiperidin]-4-carboxilato de etilo
A una disolución de HCl en EtOAc (200 ml) se añadió 4-etil[1,4'-bipiperidin]-1',4-dicarboxilato de 1'-terc-butilo (20 g, 58,74 mmoles, 1,0 equiv). La mezcla se agitó a temperatura ambiente durante 3 h. La mezcla se concentró entonces a presión reducida proporcionando el producto en bruto deseado como un sólido blanco (15 g, sal de HCl).
Etapa 3: Síntesis de 1'-(2-(3-(terc-butoxi)-3-oxopropoxi)etil)-[1,4'-bipiperidin]-4-carboxilato de etilo
A una disolución de 3-(2-bromoetoxi)propanoato de terc-butilo (6,46 g, 25,54 mmoles, 1,0 equiv) en DMF (240 ml) se añadió K2CO3 (10,59 g, 76,61 mmoles, 3,0 equiv) y [1,4'-bipiperidina]-4-carboxilato de etilo (8 g, 25,54 mmoles, 1,0 equiv, 2HCl). La mezcla se agitó a 120 °C durante 12 h, momento en el que la reacción se enfrió hasta temperatura ambiente, se filtró la torta de filtración, se lavó con H2O (20 ml), y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→11 % de MeOH/EtOAc) dio el producto deseado como un aceite amarillo (6,6 g, 63 % de rendimiento).
Etapa 4: Síntesis de ácido 3-(2-(4-(etoxicarbonil)-[1,4'-bipiperidin]-1'-il)etoxi)propanoico
A la disolución de HCl en EtOAc (70 ml) se añadió 1'-(2-(3-(terc-butoxi)-3-oxopropoxi)etil)-[1,4'-bipiperidin]-4-carboxilato de etilo (6,6 g, 16,00 mmoles, 1,0 equiv). La mezcla se agitó a temperatura ambiente durante 3 h, momento en el que la reacción se concentró a presión reducida proporcionando el producto deseado como un sólido blanco (6,5 g, 95 % de rendimiento, 2HCl).
Etapa 5: Síntesis de 1'-(2-(3-(4-(5-(((N,N-di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)-[1,4'-bipiperidin]-4-carboxilato de etilo
A una disolución de terc-butil-terc-butoxicarbonil((2-(piperazin-1-il)pirimidin-5-il)metil)carbamato (2,49 g, 6,33 mmoles, 1,5 equiv) en DMF (40 ml) se añadió DIPEA (9,74 ml, 55,89 mmoles, 6,0 equiv) y HATU (5,31 g, 13,97 mmoles, 1,5 equiv). La mezcla se agitó a temperatura ambiente durante 30 min, y entonces se añadió ácido 3-(2-(4-(etoxicarbonil)-[1,4'-bipiperidin]-1'-il)etoxi)propanoico (4 g, 9,32 mmoles, 1,0 equiv, 2HCl). La mezcla se agitó a durante 1,5 h, momento en el que se añadieron H2O (5 ml) y EtOAc (20 ml). La fase acuosa se extrajo con EtOAc (3 × 10 ml) y las fases orgánicas combinadas se lavaron con salmuera (5 ml), se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía de fase inversa dio el producto deseado como un aceite marrón (1,6 g, 23 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C37H61N7O8: 732,47; observado 732,6.
Etapa 6: Síntesis de ácido 1'-(2-(3-(4-(5-(((terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)-[1,4'-bipiperidina]-4-carboxílico
A una disolución de 1'-(2-(3-(4-(5-(((N,N-di-terc-butoxicarbonil)amino)metil)pirimidin-2-il)piperazin-1-il)-3-oxopropoxi)etil)-[1,4'-bipiperidin]-4-carboxilato de etilo (1,4 g, 1,91 mmoles, 1,0 equiv) en THF (7,5 ml), EtOH (3,8 ml) y H2O (3,8 ml) se añadió LiOH•H2O (321,07 mg, 7,65 mmoles, 4,0 equiv). La mezcla se agitó a temperatura ambiente durante 2 h, momento en el que la mezcla se concentró a presión reducida. La purificación por cromatografía de fase inversa (5→38 % de MeCN/H2O) dio el producto deseado como un sólido amarillo (325 mg, 22 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C30H49N7O6: 604,38; observado 604,3.
Elemento estructural AZ. Ácido 1-(4-{2-[2-(2-{[(benciloxi)carbonil]amino}etoxi)etoxi]etil}piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oico.

Etapa 1: Síntesis de (2-(2-(2-bromoetoxi)etoxi)etil)carbamato de bencilo
A una disolución de (2-(2-(2-hidroxietoxi)etoxi)etil)carbamato de bencilo (10 g, 35,30 mmoles, 1,0 equiv) en DCM (300 ml) a 0 °C se añadió PPh3 (13,79 g, 52,59 mmoles, 1,49 equiv) y CBr4 (17,44 g, 52,59 mmoles, 1,49 equiv). Entonces la mezcla se calentó hasta temperatura ambiente y se agitó durante 12 h. Entonces se filtró la mezcla de reacción, y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (1 %→25 % de EtOAc/éter de petróleo) dio el producto deseado (10,8 g, 88,4 % de rendimiento) como un aceite amarillo.
Etapa 2: Síntesis de 4-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)piperazin-1-carboxilato de terc-butilo A una disolución de (2-(2-(2-bromoetoxi)etoxi)etil)carbamato de bencilo (10,8 g, 31,19 mmoles, 1,0 equiv) y piperazin-1-carboxilato de terc-butilo (5,81 g, 31,19 mmoles, 1,0 equiv) en MeCN (100 ml) se añadió K2CO3 (4,31 g, 31,19 mmoles, 1,0 equiv). La mezcla se agitó a 80 °C durante 1 h. Entonces se filtró la mezcla de reacción, y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→50 % de MeOH/EtOAc) dio el producto deseado (13,1 g, 93,0 % de rendimiento) como un aceite amarillo.
Etapa 3: Síntesis de (2-(2-(2-(piperazin-1-il)etoxi)etoxi)etil)carbamato de bencilo
Una disolución de 4-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)piperazin-1-carboxilato de terc-butilo (5,64 g, 12,49 mmoles, 1,0 equiv) en HCl/EtOAc (50 ml, 4 M) se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se concentró entonces a presión reducida proporcionando el producto deseado (5,23 g, en bruto, HCl sal) como un aceite amarillo.
Etapa 4: Síntesis de 1-(4-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo
A una disolución de (2-(2-(2-(piperazin-1-il)etoxi)etoxi)etil)carbamato de bencilo (13,3 g, 31,34 mmoles, 1,0 equiv, 2HCl) y 1-bromo-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo en MeCN (150 ml) se añadió K2CO3 (21,66 g, 156,71 mmoles, 5,0 equiv). La mezcla se agitó a 80 °C durante 12 h. Entonces se filtró la mezcla de reacción, y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (1 %→17 % de MeOH/DCM) dio el producto deseado (5,4 g, 26,3 % de rendimiento) como un aceite amarillo.
Etapa 5: Síntesis de ácido 1-(4-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oico
Una disolución de 1-(4-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo (2,4 g, 3,66 mmoles, 1,0 equiv) en TFA (20 ml) se agitó a temperatura ambiente durante 30 min. La mezcla de reacción se concentró entonces a presión reducida proporcionando el producto deseado (3,03 g, sal de TFA) como un aceite amarillo.
Elemento estructural BA. Ácido (R)-2-(2-(terc-butoxicarbonil)-4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico.

Etapa 1: Síntesis de 2-(3-(terc-butoxicarbonil)-4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo
A dos lotes separados realizados en paralelo que contenía cada uno una disolución de 2-(3-(terc-butoxicarbonil)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo (6 g, 14,30 mmoles, 1,0 equiv) y K2CO3 (3,95 g, 28,60 mmoles, 2,0 equiv) en MeCN (80 ml) se añadió 2-cloropirimidin-5-carboxilato de etilo (3,20 g, 17,16 mmoles, 1,2 equiv). Las mezclas de reacción se agitaron a 80 °C durante 12 h. Las dos mezclas de reacción se combinaron y se filtraron, el residuo se lavó con EtOAc (3 × 50 ml) y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0 %→17 % de MeOH/EtOAc) dio el producto deseado (15 g, 91,5 % de rendimiento) como un sólido amarillo. LCMS (ESI) m/z: [M H] calculado para C28H39N7O6: 570,31; observado 570,1.
Etapa 2: Síntesis de ácido (R)-2-(2-(terc-butoxicarbonil)-4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico
A una disolución de 2-(3-(terc-butoxicarbonil)-4-(5-(etoxicarbonil)pirimidin-2-il)piperazin-1-il)-7,8-dihidropirido[4,3-d]pirimidin-6(5H)-carboxilato de (R)-terc-butilo (15 g, 26,33 mmoles, 1,0 equiv) en THF (80 ml), EtOH (40 ml) y H2O (40 ml) se añadió LiOH●H2O (2,21 g, 52,66 mmoles, 2,0 equiv). La mezcla se agitó a temperatura ambiente durante 6 h. La mezcla de reacción se ajustó entonces a pH=6 con HCl 1 N. La suspensión resultante se filtró, y la torta sólida se secó a presión reducida proporcionando el producto deseado (10,87 g, 75,9 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C26H35N7O6: 542,27; observado 542,1.
Elemento estructural BB. Ácido (S)-2-(2-(terc-butoxicarbonil)-4-(6-(terc-butoxicarbonil)-5,6,7,8-tetrahidropirido[4,3-d]pirimidin-2-il)piperazin-1-il)pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural AA por un proceso similar a aquél para el Elemento estructural BA.
Elemento estructural BC. Ácido 2-[(2R)-2-[(terc-butoxi)carbonil]-4-[5-({[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il]piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural AU por un proceso similar a aquél para el Elemento estructural BA.
Elemento estructural BD. Ácido 2-[(2S)-2-[(terc-butoxi)carbonil]-4-[5-({[(terc-butoxi)carbonil]amino}metil)pirimidin-2-il]piperazin-1-il]pirimidin-5-carboxílico.

Este elemento estructural se prepara a partir del Elemento estructural AV por un proceso similar a aquél para el Elemento estructural BA.
Elemento estructural BE. 15-(6-((4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-il)-1-((1S,4S)-5-(2-(2-(2-aminoetoxi)etoxi)etil)-2,5-diazabiciclo[2.2.1]heptan-2-il)-3,6,9,12-tetraoxapentadecan-15-ona.

Etapa 1: Síntesis de 5-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)-2,5-diazabiciclo[2.2.1]heptano-2-carboxilato de (1S,4S)-terc-butilo
A una disolución de 2,5-diazabiciclo[2.2.1]heptano-2-carboxilato de (1S,4S)-terc-butilo (2,85 g, 14,37 mmoles, 1,0 equiv) en MeCN (50 ml) se añadió K2CO3 (3,97 g, 28,75 mmoles, 2,0 equiv) y (2-(2-(2-bromoetoxi)etoxi)etil)carbamato de bencilo (4,98 g, 14,37 mmoles, 1,0 equiv). La mezcla se agitó a 80 °C durante 24 h. Entonces se filtró la mezcla de reacción, y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→710 % de MeOH/EtOAc) dio el producto deseado (6,2 g, 93,0 % de rendimiento) como un aceite incoloro. LCMS (ESI) m/z: [M H] calculado para C24H37N3O6: 464,27; observado 464,2.
Etapa 2: Síntesis de (2-(2-(2-((1S,4S)-2,5-diazabiciclo[2.2.1]heptan-2-il)etoxi)etoxi)etil)carbamato de bencilo A una disolución de 5-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)-2,5-diazabiciclo[2.2.1]heptano-2-carboxilato de (1S,4S)-terc-butilo (6,2 g, 13,37 mmoles, 1,0 equiv) en DCM (60 ml) se añadió TFA (20,7 ml, 279,12 mmoles, 20,9 equiv). La reacción se agitó durante 2 h, momento en el que la mezcla se concentró a presión reducida a 45 °C proporcionando el producto en bruto deseado (10,5 g, 4TFA) como un aceite marrón claro, que se usó la siguiente etapa directamente. LCMS (ESI) m/z: [M H] calculado para C19H29N3O4: 364,22; observado 364,2.
Etapa 3: Síntesis de 1-((1S,4S)-5-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)-2,5-diazabiciclo[2.2.1]heptan-2-il)-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo
A una disolución de (2-(2-(2-((1S,4S)-2,5-diazabiciclo[2.2.1]heptan-2-il)etoxi)etoxi)etil)carbamato de bencilo (5 g, 6,10 mmoles, 1,0 equiv, 4TFA) en MeCN (80 ml) se añadió K2CO3 (5,06 g, 36,61 mmoles, 6,0 equiv) y 1-bromo-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo (2,35 g, 6,10 mmoles, 1,0 equiv). La mezcla de reacción se agitó a 80 °C durante 12 h. Entonces se filtró la mezcla de reacción, y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→15 % de MeOH/EtOAc) dio el producto deseado (5,2 g, 92,8 % de rendimiento) como un aceite amarillo claro. LCMS (ESI) m/z: [M H] calculado para C34H57N3O10: 668,4; observado 668,4.
Etapa 4: Síntesis de ácido 1-((1S,4S)-5-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)-2,5-diazabiciclo[2.2.1]heptan-2-il)-3,6,9,12-tetraoxapentadecan-15-oico
Una disolución de 1-((1S,4S)-5-(3-oxo-1-fenil-2,7,10-trioxa-4-azadodecan-12-il)-2,5-diazabiciclo[2.2.1]heptan-2-il)-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo (5,2 g, 5,66 mmoles, 1,0 equiv) en TFA (47,3 ml, 638,27 mmoles, 112,75 equiv) se agitó a temperatura ambiente durante 30 min. Entonces se concentró la mezcla a presión reducida a 45 °C. La purificación por cromatografía de fase inversa (2→35 % de MeCN/H2O (0,05 % de NH4OH)) dio el producto deseado (1,88 g, 54,3 % de rendimiento) como un aceite marrón claro. LCMS (ESI) m/z: [M H] calculado para C30H49N3O10: 612,34; observado 612,3.
Elemento estructural BF. 21-(6-((4-Amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d] pirimidin-1-il)metil)-3,4-dihidroisoquinolin-2(1H)-il)-1-(piperazin-1-il)-3,6,9,12,15,18-hexaoxahenicosan-21-ona.

Etapa 1: Síntesis de 4-(23,23-dimetil-21-oxo-3,6,9,12,15,18,22-heptaoxatetracosil)piperazin-1-carboxilato de bencilo
A una disolución de 1-bromo-3,6,9,12,15,18-hexaoxahenicosan-21-oato de terc-butilo (5 g, 10,56 mmoles, 1,0 equiv) y piperazin-1-carboxilato de bencilo (2,62 ml, 11,62 mmoles, 1,1 equiv, HCl) en MeCN (50 ml) se añadió K2CO3 (4,38 g, 31,69 mmoles, 3,0 equiv). La mezcla de reacción se agitó a 80 °C durante 10 h. Entonces se filtró la mezcla, la torta sólida se lavó con EtOAc (3 × 3 ml) y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→10 % de MeOH/EtOAc) dio el producto deseado (4 g, 61,8 % de rendimiento) como un líquido rojo.
Etapa 2: Síntesis de ácido 1-(4-((benciloxi)carbonil)piperazin-1-il)-3,6,9,12,15,18-hexaoxahenicosan-1-oico A una disolución de 4-(23,23-dimetil-21-oxo-3,6,9,12,15,18,22-heptaoxatetracosil)piperazin-1-carboxilato de bencilo (1,8 g, 2,94 mmoles, 1,0 equiv) en DCM(10 ml) se añadió TFA (10 ml). La disolución se agitó durante 0,5 h. La disolución se concentró entonces a presión reducida. Al residuo se añadió DCM (30 ml) y entonces la disolución se concentró a presión reducida proporcionando el producto deseado (1,6 g, 2,87 mmoles, TFA) como un líquido rojo.
Preparación de monómeros de rapamicina.
Monómero 1.40(R)-O-(4-nitrofenil)carbonato rapamicina.

A una disolución de rapamicina (30,10 g, 32,92 mmoles, 1,0 equiv) en DCM (148,9 ml) se añadió piridina (29,6 ml, 367 mmoles, 11,1 equiv). La disolución se enfrió hasta -78 °C y entonces se añadió cloroformiato de p-nitrofenilo (12,48 g, 61,92 mmoles, 1,9 equiv). La reacción se agitó a -78 °C durante 2 h. A la mezcla de reacción se añadió entonces DCM y la disolución se vertió entonces en H2O. La fase acuosa se extrajo con DCM y las fases orgánicas combinadas se secaron sobre MgSO4, y se concentraron a presión reducida. El material en bruto se purificó por cromatografía en gel de sílice (0→50 % de EtOAc/hexanos) proporcionando el producto (23,1 g, 59,2 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M Na] calculado para C58H82N2O17: 1101,55; observado 1101,6.
Monómero 2.32(R)-hidroxi 40(R)-O-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 32(R)-hidroxi rapamicina
Una disolución de 32(R)-hidroxi-28,40-bistrietilsilil rapamicina (3,64 g, 3,18 mmoles, 1 equiv) en THF (41,8 ml) se trató con piridina (20,8 ml, 258 mmoles, 81 equiv) y la mezcla de reacción se enfrió hasta 0 °C. La disolución se trató gota a gota con 70 % de HF-piridina (4,60 ml, 159 mmoles, 50 equiv) y la mezcla de reacción se agitó a 0 °C durante 20 min, seguido de calentamiento hasta temperatura ambiente. Después de 5 h, la mezcla de reacción se enfrió de nuevo a 0 °C y se añadió cuidadosamente una disolución sat. helada de NaHCO3 (400 ml). La mezcla se extrajo con EtOAc (2 × 100 ml) y las fases orgánicas se lavaron con porciones de 75 ml de H2O, disolución sat. de NaHCO3 y salmuera. La disolución orgánica se secó sobre Na2SO4, se filtró y se concentró dando un aceite amarillo claro que produjo una espuma rígida a presión reducida. El material en bruto se purificó por cromatografía en gel de sílice (20→40 % de acetona/hexanos) dando el producto deseado como un sólido amorfo blanco (1,66 g, 57 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C51H81NO13: 938,56; observado 938,7; m/z: [M - H] calculado para C51H81NO13: 914,56; observado 914,7.
Etapa 2: Síntesis de 32(R)-hidroxi 40(R)-O-(4-nitrofenil)carbonato rapamicina
A una suspensión de tamices moleculares en polvo de 4Å (6,0 g) en DCM (130 ml) se añadió 32(R)-hidroxi rapamicina (6,00 g, 6,55 mmoles, 1,0 equiv). Después de agitar a temperatura ambiente durante 45 min, se añadió piridina (5,99 ml, 74,0 mmoles, 11,3 equiv). La suspensión se enfrió hasta -15 °C y entonces se añadió 4-nitrofenilcloroformiato (1,78 g, 8,84 mmoles, 1,4 equiv). La mezcla de reacción se agitó a -10 °C durante 2 h y entonces se filtró y la almohadilla de filtro se lavó con DCM (140 ml). El filtrado se lavó con NaHCO3 sat. (130 ml), H2O (130 ml) y salmuera (130 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El material en bruto se purificó por cromatografía en gel de sílice (20→50 % de EtOAc/hexanos) dando el producto (4,44 g, 63 % de rendimiento) como una espuma rígida blanquecina. LCMS (ESI) m/z: [M Na] calculado para C58H84N2O17: 1103,57; observado 1103,5.
Monómero 3.32(R)-metoxi 40(R)-O-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 32(R)-metoxi-28,40-bistrietilsilil rapamicina
A una disolución con agitación de 32(R)-hidroxi-28,40-bistrietilsilil rapamicina (3,83 g, 3,34 mmoles, 1,0 equiv) en cloroformo (95,8 ml) se añadió Proton Sponge® (7,17 g, 33,5 mmoles, 10,0 equiv) junto con tamices moleculares de 4 Å recién secados (4 g). La disolución se agitó durante 1 h antes de la adición de tetrafluoroborato de trimetiloxonio (4,95 g, 33,5 mmoles, 10,0 equiv, se secó calentando a presión reducida a 50 °C durante 1 h antes de uso) a temperatura ambiente. La mezcla de reacción se agitó durante 18 h, y entonces la mezcla de reacción se diluyó con DCM y se filtró a través de Celite. El filtrado se lavó sucesivamente con HCl acuoso 1 M (2×), disolución acuosa sat. de NaHCO3, luego se secó y se concentró a presión reducida. La purificación por cromatografía en gel de sílice (10→20 % de EtOAc/hexanos) dio el producto deseado como un aceite amarillo que estuvo contaminado con 3 % en peso de Proton Sponge®. El residuo se recogió en MTBE y se lavó con HCl acuoso 1 M, disolución acuosa sat. de NaHCO3, se secó y luego se concentró a presión reducida proporcionando una espuma amarilla (3,15 g, 81,2 % de rendimiento). LCMS (ESI) m/z: [M - TES H2O] calculado para C64H111NO13Si2: 1061,68; observado 1061,9.
Etapa 2: Síntesis de 32(R)-metoxi rapamicina
A una disolución con agitación de 32(R)-metoxi-28,40-bistrietilsilil rapamicina (1,11 g, 0,958 mmoles, 1,0 equiv) en THF (12,6 ml) y piridina (6,30 ml) en un vial de plástico se añadió 70 % de HF-piridina (2,22 ml, 76,6 mmoles, 80,0 equiv) gota a gota a 0 °C. La mezcla de reacción se agitó a 0 °C durante 20 min antes de calentarse hasta temperatura ambiente durante 3 h, cuando la HPLC mostró el consumo completo de material de partida. La mezcla de reacción se enfrió hasta 0 °C y se vertió lentamente en helado disolución acuosa sat. de NaHCO3 (50 ml). La fase acuosa se extrajo con EtOAc (3×) y los extractos orgánicos combinados se lavaron con disolución acuosa sat. de NaHCO3, salmuera, se secaron y se concentraron a presión reducida. El residuo amarillo se disolvió en MeOH (5 ml) y se añadió gota a gota a H2O (50 ml) produciendo un precipitado blanco. Después de agitar durante 15 min, la suspensión se filtró sobre un embudo de porosidad media y la torta se lavó con H2O (2×). Entonces se disolvieron los sólidos en MeCN (50 ml) y se liofilizaron durante la noche proporcionando el producto como un sólido blanco (780 mg, 87 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C52H83NO13: 952,58; observado 952,4.
Etapa 3: Síntesis de 32(R)-metoxi 40(R)-O-(4-nitrofenil)carbonato rapamicina
A una disolución de 32(R)-metoxi rapamicina (4,50 g, 4,84 mmoles, 1,0 equiv) en DCM (180 ml) se añadió tamices moleculares en polvo de 4Å (6,0 g). La mezcla se agitó a temperatura ambiente durante 1 h y entonces se añadió piridina (3,91 ml, 48,4 mmoles, 10 equiv). La mezcla se enfrió hasta -10 °C y se añadió 4-nitrofenilcloroformiato (0,990 g, 4,91 mmoles, 1,0 equiv) en una porción. La reacción se dejó calentar lentamente hasta temperatura ambiente y después de 3 h la mezcla de reacción se enfrió hasta 0 °C y se añadió 4-nitrofenilcloroformiato (250 mg, 1,24 mmoles, 0,3 equiv). La mezcla se calentó hasta temperatura ambiente y después de 1 h la mezcla de reacción se filtró a través de una almohadilla de Celite y la almohadilla se lavó con DCM (140 ml). El filtrado se lavó con H2O (120 ml) y NaHCO3 sat. (2 × 120 ml). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El material en bruto se purificó por cromatografía ultrarrápida (20→50 % de EtOAc/hex) dando una espuma blanca rígida. El material se recogió en MeCN, tiempo durante el cual se formó un sólido blanco. El sólido se filtró, se lavó con MeCN adicional y se dejó secar al aire proporcionando el producto (4,51 g, 85 % de rendimiento). LCMS (ESI) m/z [M Na] calculado para C59H86N2O17: 1117,58; observado 1117,6.
Monómeros 4.32(R)-etoxi 40(R)-O-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 32(R)-etoxi-28,40-bistrietilsilil rapamicina
Una disolución de 32(R)-hidroxi-28,40-bistrietilsilil rapamicina (773 mg, 0,675 mmoles, 1,0 equiv) en cloroformo (19 ml) se trató con N,N,N',N'-tetrametil-1,8-naftalenodiamina (1,85 g, 8,63 mmoles, 12,8 equiv) junto con tamices moleculares de 4Å recién secado. La mezcla se agitó durante 1 h a temperatura ambiente y se trató con tetrafluoroborato de trietiloxonio (1,51 g, 7,95 mmoles, 11,8 equiv) en una porción a temperatura ambiente. La mezcla de reacción se agitó durante 3 h, momento en el que la mezcla de reacción se diluyó con DCM y se filtró a través de Celite, lavando la almohadilla de filtro con DCM adicional. Los filtrados combinados se lavaron dos veces con HCl 1 M, una vez con disolución saturada de NaHCO3 y se secaron sobre Na2SO4. La disolución se filtró y se concentró a un residuo. El residuo en bruto se trató con MTBE y se filtró para retirar el material insoluble polar. El filtrado se concentró y se purificó por cromatografía en gel de sílice (5→25 % de EtOAc/hex) proporcionando el producto como una espuma (516 mg, 65 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C65H113NO13Si21194,77; observado 1194,6.
Etapa 2: Síntesis de 32(R)-etoxi rapamicina
A una disolución de 32(R)-etoxietoxi-28,40-bistrietilsilil rapamicina (131 mg, 0,112 mmoles, 1,0 equiv) en THF (1,3 ml) a 0 °C se añadió piridina (271 µl, 3,35 mmoles, 3,4 equiv), seguido de 70 % de HF-piridina (51 µl, 1,8 mmoles, 1,8 equiv). Se tapó el matraz de reacción y se guardó en el frigorífico durante 3 días, momento en el que la mezcla de reacción se vertió en NaHCO3 saturado frío (20 ml). La fase acuosa se extrajo con EtOAc (3 × 20 ml) y las fases orgánicas combinadas se lavaron con HCl 1 M (2 × 20 ml), disolución saturada de NaHCO3 (20 ml) y salmuera. La disolución se secó sobre Na2SO4, se filtró y se concentró. El residuo se recogió en MeOH (1,5 ml) y se añadió gota a
gota a H2O (20 ml). Los sólidos se filtraron y se lavaron con H2O adicional proporcionando el producto (53 mg, 51 % de rendimiento) como un polvo blanco. LCMS (ESI) m/z: [M Na] calculado para C53H85NO13: 966,59; observado 966,5.
Etapa 3: Síntesis de 32(R)-etoxi 40(R)-O-(4-nitrofenil)carbonato rapamicina
A una disolución 0,03 M de 32(R)-etoxi rapamicina (1,0 equiv) en DCM se añaden tamices moleculares en polvo de 4Å. La mezcla se agita a temperatura ambiente durante 1 h y entonces se añade piridina (10 equiv). La mezcla se enfría hasta -10 °C y se añade 4-nitrofenilcloroformiato (1,0 equiv) en una porción. La reacción se calienta hasta temperatura ambiente y se agitó hasta que se consume la 32(R)-etoxi rapamicina, como se determina por análisis de LCMS. La mezcla se filtra a través de una almohadilla de Celite y la almohadilla se lava con DCM. El filtrado se lava con H2O y NaHCO3 sat. Entonces se seca la fase orgánica sobre Na2SO4, se filtra y se concentra a presión reducida. El material en bruto se purifica por cromatografía ultrarrápida (20→50 % de EtOAc/hex) proporcionando el producto.
Monómero 5.40(R)-O-(4-nitrofenil)tiocarbonato rapamicina.

A una disolución de rapamicina (4,00 g, 4,38 mmoles, 1,0 equiv) en DCM (20 ml) a -78 °C se añadió piridina (4,0 ml, 49 mmoles, 11,2 equiv), seguido por una disolución de O-(4-nitrofenil)clorotiocarbonato (1,19 g, 5,47 mmoles, 1,3 equiv) en DCM (8,0 ml). La mezcla de reacción se calentó hasta -20 °C y se agitó durante 48 h. Entonces se añadió hexano (40 ml) y la suspensión resultante se purificó por cromatografía en gel de sílice (15/25/60 de EtOAc/DCM/hexano, luego 20/25/55 EtOAc/DCM/hexano) proporcionando el producto (3,09 g, 64,4 % de rendimiento) como un sólido blanquecino. LCMS (ESI) m/z: [M Na] calculado para C58H82N2O16S: 1117,53; observado 1117,5.
Monómero 6.28-O-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 40-O-terc-butildimetilsilil rapamicina
A una disolución de rapamicina (1,00 g, 1,09 mmoles, 1,0 equiv) en DMF (4 ml) a temperatura ambiente se añadió imidazol (0,22 g, 3,2 mmoles, 2,9 equiv), seguido de cloruro de terc-butildimetilsililo (0,176 g, 1,17 mmoles, 1,07 equiv). La mezcla de reacción se agitó durante 18 h. La mezcla de reacción se diluyó entonces con DCM (100 ml) y se lavó con 20 % de LiCl ac. (3 × 20 ml). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía en gel de sílice (20→40 % de EtOAc/hexanos) dando el producto (950 mg, 75 % de rendimiento) como un vidrio amarillo pálido. LCMS (ESI) m/z: [M H2O] calculado para C57H93NO13Si: 1045,65; observado 1045,9.
Etapa 2: Síntesis de 28-O-(4-nitrofenoxicarbonil)-40-O-(terc-butildimetilsilil) rapamicina
A una disolución de 40-O-terc-butildimetilsilil rapamicina (0,845 g, 0,822 mmoles, 1,0 equiv) en DCM (10 ml) a temperatura ambiente se añadió piridina (0,9 ml, 10 mmoles, 12,1 equiv), seguido de cloroformiato de 4-nitrofenilo (0,373 g, 1,85 mmoles, 2,25 equiv). La mezcla de reacción se agitó durante 2 h. La mezcla de reacción se diluyó entonces con DCM (150 ml) y la disolución se lavó sucesivamente con NaHCO3 sat. (20 ml), 10 % de ácido cítrico (2 × 20 ml) y salmuera (20 ml). La fase orgánica se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía de fase inversa (30-7100 % de MeCN/H2O) dando el producto (930 mg, 95 % de rendimiento) como una espuma amarilla pálida. LCMS (ESI) m/z: [M H] calculado para C64H96N2O17Si: 1193,66; observado 1193,7.
Etapa 3: Síntesis de 28-O-(4-nitrofenoxicarbonil) rapamicina
A una disolución de 28-O-(4-nitrofenoxicarbonil)-40-O-(terc-butildimetilsilil)rapamicina (0,930 g, 0,779 mmoles, 1,0 equiv) en THF (10,7 ml) se añadió piridina (3,78 ml, 46,8 mmoles, 60,1 equiv), seguido de la adición gota a gota de 70 % de HF-piridina (0,91 ml, 31,2 mmoles, 40,0 equiv). La mezcla de reacción se agitó a temperatura ambiente durante 48 h. Entonces se vertió lentamente la mezcla en NaHCO3 acuosa sat. helado (20 ml). La fase acuosa se extrajo con EtOAc (3 × 20 ml) y la fase orgánica combinada se lavó con NaHCO3 sat. (10 ml) y salmuera (10 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía de fase inversa (30→100 % de MeCN/H2O) dando el producto (200 mg, 24 % de rendimiento) como un polvo amarillo pálida. LCMS (ESI) m/z: [M Na] calculado para C58H82N2O17: 1101,55; observado 1101,3.
Monómero 7.32(R)-hidroxi 40(R)-O-(4-nitrofenil)tiocarbonato rapamicina.

A una disolución de 32(R)-hidroxi rapamicina (2,80 g, 3,06 mmoles, 1,0 equiv) en DCM (28 ml) se añadió piridina (27,6 ml, 34 mmoles, 11 equiv) y tamices moleculares de 4Å secados (2,8 g). La suspensión se agitó a temperatura ambiente durante 1 h, momento en el que la mezcla se enfrió hasta -25 °C y se añadió una disolución de O-(4-nitrofenil)clorotioformiato (0,798 g, 3,67 mmoles, 1,2 equiv) en DCM (6 ml). La reacción se calentó hasta temperatura ambiente y después de 21 h se filtró a través de Celite. El filtrado se repartió entre DCM y H2O y la fase acuosa se extrajo con DCM. Las fases orgánicas combinadas se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexanos) dio el producto deseado como un sólido blanco (2,15 g, 64 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C58H84N2O16S: 1119,54; observado 1120,0.
Monómero 8.32(R)-metoxi 40(R)-O-(4-nitrofenil)tiocarbonato rapamicina.

A una disolución de 32(R)-metoxi rapamicina (6,69 g, 7,19 mmoles, 1,0 equiv) en DCM (67 ml) se añadió piridina (6,6 ml, 81 mmoles, 11 equiv) y tamices moleculares de 4Å secados (6,7 g). La suspensión se agitó a temperatura ambiente durante 1 h, momento en el que la mezcla se enfrió hasta -25 °C y se añadió una disolución de O-(4-nitrofenil)clorotioformiato (1,88 g, 8,63 mmoles, 1,20 equiv) en DCM (13 ml). La reacción se calentó hasta temperatura ambiente y después de 21 h se filtró a través de Celite. El filtrado se repartió entre DCM y H2O y la fase acuosa se extrajo con DCM. Las fases orgánicas combinadas se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexanos) dio el producto deseado como un sólido blanco (5,1 g, 64 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C59H86N2O16S: 1133,56; observado 1134,1.
Monómero 9.32-desoxi 40(R)-O-(4-nitrofenil)carbonato rapamicina.

A una disolución de 32-desoxi rapamicina (0,623 g, 0,692 mmoles, 1,0 equiv) en DCM (24,7 ml) se añadió tamices moleculares de 4 Å (600 mg). La suspensión se agitó durante 1 h y entonces se añadió piridina (557 µL, 6,92 mmoles, 10 equiv). La mezcla de reacción se enfrió hasta 0 °C y entonces se añadió O-(4-nitrofenil)cloroformiato (175 mg, 1,03 mmoles, 1,7 equiv). La reacción se calentó hasta temperatura ambiente y se agitó durante 2 h, momento en el que la mezcla de reacción se concentró a presión reducida. La purificación por cromatografía en gel de sílice (0→10 % de MeOH/DCM) dio el producto deseado como un sólido blanco (0,61 g, 82 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C58H84N2O16: 1087,57; observado 1087,6.
Monómero 10.32-desoxi 40(R)-O-(4-nitrofenil)tiocarbonato rapamicina.

A una disolución 0,2 M de 32-desoxi rapamicina (1,0 equiv) en DCM a -78 °C se añade piridina (11,2 equiv), seguido de una disolución 0,7 M de O-(4-nitrofenil)clorotiocarbonato (1,3 equiv) en DCM. La mezcla de reacción se calienta a -20 °C y se agita hasta que se consume del material de partida, como se ha determinado por análisis de LCMS. Entonces se añade hexano y la suspensión resultante se purifica por cromatografía en gel de sílice proporcionando el producto.
Monómero 11.28(R)-metoxi 32(R)-hidroxi 40(R)-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 28(R)-metoxi 40(R)-O-terc-butildimetilsilil rapamicina
A una disolución de 40(R)-O-terc-butildimetilsilil rapamicina (4,00 g, 4,89 mmoles, 1,0 equiv) en cloroformo (67 ml) se añadió Proton Sponge (11,2 ml, 52,3 mmoles, 13 equiv) y tamices moleculares secados de 4 Å (5,8 g). La suspensión se agitó a temperatura ambiente durante 1 h, momento en el que se añadió tetrafluoroborato de trimetiloxonio (7,21 g, 48,8 mmoles, 12,5 equiv). Después de 4 h, la suspensión se filtró a través de Celite. El filtrado se lavó sucesivamente o con acuosa HCl 2 N, H2O, NaHCO3 acuoso sat., luego se secó y se concentró a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexano) dio el producto deseado como un sólido blanco (2,1 g, 52 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C58H95NO13Si: 1064,65; observado 1065,26.
Etapa 2: Síntesis de 28(R)-metoxi 32(R)-hidroxi 40(R)-O-terc-butildimetilsilil rapamicina
A una disolución de 28(R)-metoxi 40(R)-O-terc-butildimetilsilil rapamicina (2,13 g, 2,04 mmoles, 1,0 equiv) en THF (31 ml) a -20 °C se añadió una disolución de hidruro de tri-terc-butoxialuminio y litio en THF (1 M, 4,09 ml, 4,09 mmoles, 2,0 equiv), gota a gota. La mezcla de reacción se calentó hasta temperatura ambiente y después de 3 h se añadió a una disolución de H2O (4 ml), EtOAc (31 ml) y ácido cítrico acuoso 2 M (4 ml) a 0 °C. Después de 5 min, la mezcla se repartió, y la fase acuosa se extrajo con EtOAc. Las fases orgánicas combinadas se vertieron en una disolución acuosa sat. de NaHCO3 (60 ml) a 0 °C. Las fases se repartieron, y la fase orgánica se secó y se concentró a presión reducida proporcionando un sólido blanco en bruto (2,32 g). El sólido en bruto se disolvió en DCM (12 ml) y entonces se añadieron piridina (241 µl, 2,98 mmoles, 1,5 equiv), tamices moleculares secados de 4Å (2,1 g) y acetato cúprico (0,27 g, 1,49 mmoles, 0,7 equiv). La suspensión se agitó a temperatura ambiente durante 1 h. La suspensión se burbujeó con O2 y entonces se mantuvo bajo una atmósfera de O2 durante 30 min. Después de 2 h, la mezcla se filtró a través de Celite y el filtrado se concentró a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexano) dio el producto deseado como un sólido blanco (307 mg, 14 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C58H97NO13Si: 1066,66; observado 1067,0.
Etapa 3: Síntesis de 28(R)-metoxi 32(R)-hidroxi rapamicina
A una disolución de 28(R)-metoxi 32(R)-hidroxi 40(R)-O-terc-butildimetilsilil rapamicina (0,307 g, 0,294 mmoles, 1,0 equiv) en THF (4 ml) en un vial de polipropileno a 0 °C se añadió piridina (1,42 ml, 17,6 mmoles, 60,0 equiv), seguido de 70 % de HF-piridina (0,34 ml, 11,7 mmoles, 40 equiv). La disolución se calentó hasta temperatura ambiente y se agitó durante 21 h, momento en el que la disolución se vertió en NaHCO3 acuoso sat. a 0 °C. La fase acuosa se extrajo con EtOAc (2×) y las fases orgánicas combinadas se lavaron con NaHCO3 acuoso sat. y salmuera, luego se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexano) dio
el producto deseado como un sólido blanco (146 mg, 53 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C52H83NO13: 952,58; observado 952,8.
Etapa 4: Síntesis de 28(R)-metoxi 32(R)-hidroxi 40(R)-(4-nitrofenil)carbonato rapamicina
A una disolución de 28(R)-metoxi 32(R)-hidroxi rapamicina (0,66 g, 0,71 mmoles, 1,0 equiv) en DCM (3 ml) se añadió piridina (0,64 ml, 7,9 mmoles, 11 equiv) y tamices moleculares secados de 4 Å (0,66 g). La suspensión se agitó a temperatura ambiente durante 1 h, momento en el que la mezcla se enfrió hasta -35 °C y se añadió O-(4-nitrofenil)cloroformiato (0,17 g, 0,85 mmoles, 1,2 equiv). Después de 3 h, se añadió DCM (5 ml) y la suspensión se filtró a través de Celite. El filtrado se lavó con H2O, se secó y se concentró a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexanos) dio el producto deseado como un sólido blanco (0,44 g, 57 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C59H86N2O17: 1117,58; observado 1118,0.
Monómero 12.28(R)-metoxi 32(R)-metoxi 40(R)-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 28(R)-metoxi 32(R)-metoxi 40(R)-O-terc-butildimetilsilil rapamicina
A una disolución de 28(R)-metoxi 32(R)-hidroxi 40(R)-O-terc-butildimetilsilil rapamicina (1,15 g, 1,10 mmoles, 1,0 equiv) en cloroformo (19 ml) se añadió Proton Sponge (3,22 ml, 15,0 mmoles, 14 equiv) y tamices moleculares secados de 4Å (2,3 g). La suspensión se agitó a temperatura ambiente durante 1 h, momento en el que se añadió tetrafluoroborato de trimetiloxonio (2,07 g, 14,0 mmoles, 12,7 equiv). Después de 4 h, la suspensión se filtró a través de Celite y el filtrado se lavó con HCl 1 N, H2O y NaHCO3 acuoso sat., se secó y se concentró a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexano) dio el producto deseado como un sólido blanco. LCMS (ESI) m/z:
[M Na] calculado para C59H99NO13Si: 1080,68; observado 1081,2.
Etapa 2: Síntesis de 28(R)-metoxi 32(R)-metoxi rapamicina
A una disolución de 28(R)-metoxi 32(R)-metoxi 40(R)-O-terc-butildimetilsilil rapamicina en THF (4 ml) en un vial de polipropileno a 0 °C se añadió piridina (1,13 ml, 14,2 mmoles, 12,9 equiv), seguido de 70 % de HF-piridina (0,27 ml, 9,42 mmoles, 8,6 equiv). La disolución se calentó hasta temperatura ambiente y se agitó durante 41 h, momento en el que la disolución se vertió en NaHCO3 acuoso sat. a 0 °C. La fase acuosa se extrajo con EtOAc (2×) y las fases orgánicas combinadas se lavaron con NaHCO3 acuoso sat. y salmuera, luego se secaron y se concentraron a presión reducida. La purificación por cromatografía en gel de sílice (EtOAc/hexano) dio el producto deseado como un sólido blanco (516 mg, 49 % de rendimiento en 2 etapas). LCMS (ESI) m/z: [M Na] calculado para C53H85NO13: 966,59; observado 967,0.
Etapa 3: Síntesis de 28(R)-metoxi 32(R)-metoxi 40(R)-(4-nitrofenil)carbonato rapamicina
A una disolución de 28(R)-metoxi 32(R)-metoxi rapamicina (0,30 g, 0,32 mmoles, 1,0 equiv) en DCM (1,4 ml) se añadió piridina (0,29 ml, 3,5 mmoles, 11 equiv) y tamices moleculares secados de 4 Å (0,30 g). La suspensión se agitó a temperatura ambiente durante 1 h, momento en el que se enfrió hasta -35 °C y se añadió O-(4-nitrofenil)cloroformiato (0,08 g, 0,38 mmoles, 1,2 equiv). Después de 3 h, se añadió DCM (2 ml) y la suspensión se filtró a través de Celite. El filtrado se lavó con H2O, se secó y se concentró a presión reducida. La purificación por cromatografía en gel de sílice
(EtOAc/hexanos) dio el producto deseado como un sólido blanquecino (0,20 g, 57 % de rendimiento). LCMS (ESI) m/z:
[M Na] calculado para C60H88N2O17: 1131,60; observado 1132,1.
Monómero 13.32(R)-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 28,40-O-bis(trietilsilil) 32(R)-(4-nitrofenil)carbonato rapamicina
A una disolución de 28,40-O-bis(trietilsilil) 32(R)-hidroxi rapamicina (0,602 g, 0,526 mmoles, 1,0 equiv) en DCM (16 ml) a -20 °C se añadió piridina (0,82 ml, 10 mmoles, 19 equiv), seguido de O-(4-nitrofenil)cloroformiato (0,36 g, 1,8 mmoles, 3,4 equiv). La mezcla de reacción se calentó hasta temperatura ambiente y se agitó durante 1 h, momento en el que la disolución se diluyó con DCM (50 ml) y se vertió en H2O (30 ml). La fase acuosa se extrajo con DCM (50 ml) y las fases orgánicas combinadas se lavaron con salmuera (20 ml), se secaron y se concentraron a presión reducida proporcionando una espuma amarilla tenue que se usó directamente en la siguiente etapa.
Etapa 2: Síntesis de 32(R)-(4-nitrofenil)carbonato rapamicina
A una disolución de 28,40-O-bis(trietilsilil) 32(R)-(4-nitrofenil)carbonato rapamicina en THF (10 ml) en un vial de polipropileno a 0 °C se añadió piridina (1,70 ml, 21,0 mmoles, 40,0 equiv), seguido de 70 % de HF-piridina (0,46 ml, 15,8 mmoles, 30,0 equiv). La disolución se calentó hasta temperatura ambiente y se agitó durante la noche, momento en el que la disolución se vertió en NaHCO3 acuoso sat. a 0 °C. La fase acuosa se extrajo con EtOAc (3×) y las fases orgánicas combinadas se lavaron con NaHCO3 acuoso sat. y salmuera, luego se secaron y se concentraron a presión reducida. La purificación por cromatografía de fase inversa (20→100 % de MeCN/H2O) dio el producto deseado como un polvo blanquecino (420 mg, 74 % de rendimiento en 2 etapas). LCMS (ESI) m/z: [M Na] calculado para C58H84N2O17: 1103,57; observado 1104,0.
Monómero 14.32(S)-azido 40-(4-nitrofenil)carbonato rapamicina.

Etapa 1: Síntesis de 28,40-O-bis(trietilsilil) 32(R)-O-metanosulfonil rapamicina
A una disolución de 28,40-O-bis(trietilsilil) 32(R)-hidroxi rapamicina (2,50 g, 2,18 mmoles, 1,0 equiv) en DCM (25 ml) a 0 °C se añadió Et3N (0,912 ml, 6,54 mmoles, 3,0 equiv), seguido de cloruro de metanosulfonilo (0,338 ml, 4,36 mmoles, 2,0 equiv). La disolución se agitó a 0 °C durante 3 h, momento en el que se añadió EtOAc y la disolución se lavó con NaHCO3 acuoso sat. Las fases orgánicas combinadas se lavaron con salmuera, se secaron y se concentraron a presión reducida dando un aceite amarillo que se usó directamente en la siguiente etapa.
Etapa 2: Síntesis de 28-O-trietilsilil 32(S)-azido rapamicina
A una disolución de 28,40-O-bis(trietilsilil) 32(R)-O-metanosulonil rapamicina en THF (40 ml) se añadió DIPEA (0,761 ml, 4,37 mmoles, 2,0 equiv) y azida de tetrabutilamonio (3,72 g, 13,1 mmoles, 6,0 equiv). La disolución de reacción se calentó a reflujo durante 5,5 h y luego se enfrió hasta temperatura ambiente. La disolución se diluyó con EtOAc y se lavó con NaHCO3 acuoso sat. Las fases orgánicas combinadas se lavaron con salmuera, se secaron y se concentraron a presión reducida. La purificación por cromatografía de fase inversa (30→100 % de MeCN/H2O) dio el producto deseado como un vidrio claro (746 mg, 33 % de rendimiento en 2 etapas). LCMS (ESI) m/z: [M Na] calculado para C57H94N4O12Si: 1077,65; observado 1077,8.
Etapa 3: Síntesis de 28-O-trietilsilil 32(S)-azido 40(R)-(4-nitrofenil)carbonato rapamicina
A una disolución de 28-O-trietilsilil 32(S)-azido rapamicina (0,505 g, 0,478 mmoles, 1,0 equiv) en DCM (15 ml) se añadió piridina (0,75 ml, 9,3 mmoles, 19 equiv) y tamices moleculares de 4 Å. La suspensión se enfrió hasta -20 °C y se añadió O-(4-nitrofenil)cloroformiato (0,32 g, 1,6 mmoles, 3,4 equiv). La suspensión se agitó a -20 °C durante 2 h, momento en el que se diluyó con DCM (50 ml), se filtró y se vertió en H2O (20 ml). La fase acuosa se extrajo con DCM (50 ml) y las fases orgánicas combinadas se lavaron con salmuera (20 ml), se secaron y se concentraron a presión reducida dando una espuma amarilla pálida que se usó directamente en la siguiente etapa.
Etapa 4: Síntesis de 32(S)-azido 40-O-(4-nitrofenil)carbonato rapamicina
A una disolución de 28-O-trietilsilil 32(S)-azido 40(R)-(4-nitrofenil)carbonato rapamicina en THF (10 ml) en un vial de polipropileno a 0 °C se añadió piridina (1,55 ml, 19,1 mmoles, 40,0 equiv), seguido de 70 % de HF-piridina (0,42 ml, 14,4 mmoles; 30,0 equiv). La disolución se calentó hasta temperatura ambiente y se agitó durante la noche, momento en el que la disolución se vertió en NaHCO3 acuoso sat. a 0 °C. La fase acuosa se extrajo con EtOAc (3×) y las fases
orgánicas combinadas se lavaron con NaHCO3 acuoso sat. y salmuera, luego se secaron y se concentraron a presión reducida. La purificación por cromatografía de fase inversa (30→7100 % de MeCN/H2O) dio el producto deseado como un polvo blanquecino (410 mg, 77 % de rendimiento en 2 etapas). LCMS (ESI) m/z: [M Na] calculado para C58H83N5O16: 1128,57; observado 1129,0.
Monómero 15.28-metoxi-40-O-(4-nitrofenoxi)carbonil rapamicina.

Etapa 1: Síntesis de 28-metoxi rapamicina
A una disolución de 28-metoxi-40-O-(terc-butildimetil)silil rapamicina (0,500 g, 0,480 mmoles, 1,0 equiv) en MeOH (1,6 ml) a -20 °C se añadió H2SO4 (1,28 µl, 0,024 mmoles, 0,05 equiv). La mezcla de reacción se agitó a -20 °C durante 48 h. La mezcla de reacción se vertió entonces en NaHCO3 acuoso sat. (4 ml)/H2O (4 ml). La fase acuosa se extrajo con MTBE (2 × 6 ml), y las fases orgánicas combinadas se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía de fase inversa (30-7100 % de MeCN/H2O) dio el producto deseado como un polvo amarillo (270 mg, 61 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C52H81NO13: 950,5; observado 950,7.
Etapa 2: Síntesis de 28-metoxi-40-O-(4-nitrofenoxi)carbonil rapamicina
A una disolución de 28-metoxi rapamicina (0,210 g, 0,226 mmoles, 1,0 equiv) en DCM (7,1 ml) a -20 °C se añadió piridina (0,35 ml, 4,4 mmoles, 19 equiv) y luego cloroformiato de p-nitrofenilo (0,15 g, 0,76 mmoles, 3,4 equiv). La mezcla de reacción se agitó a -20 °C durante 30 min y luego se calentó hasta temperatura ambiente. Después de agitar durante la noche, se añadió cloroformiato de p-nitrofenilo (0,15 g, 0,76 mmoles, 3,4 equiv) y la reacción se agitó durante 2 h adicionales. La mezcla de reacción se diluyó con DCM (20 ml) y se vertió en H2O (10 ml). La fase acuosa se extrajo con DCM (20 ml), y las fases orgánicas combinadas se lavaron con salmuera (9 ml), se secaron, se filtraron y se concentraron a presión reducida. La purificación por cromatografía de fase inversa (50→100 % de MeCN/H2O) dio el producto deseado como un polvo amarillo pálido (200 mg, 81 % de rendimiento). LCMS (ESI) m/z: [M Na] calculado para C59H84N2O17: 1115,6; observado 1115,8.
Monómero 16.32(R),40-O,O-bis[(4-nitrofenoxi)carbonil] rapamicina.

A una disolución de 32(R)-O-[(4-nitrofenoxi)carbonil] rapamicina (675 mg, 0,624 mmoles, 1,0 equiv) en DCM (13 ml) se añadió tamices moleculares en polvo de 4Å (675 mg). La suspensión se agitó durante 1 h, momento en el que se añadió piridina (0,56 ml, 6,90 mmoles, 11,1 equiv). La mezcla se enfrió hasta -15 °C y entonces se añadió cloroformiato de p-nitrofenilo (132 mg, 0,655 mmoles, 1,05 equiv) en una porción. La mezcla se calentó hasta 0 °C, se agitó durante 4 h y luego se calentó hasta temperatura ambiente. La mezcla de reacción se filtró y se lavó con DCM (25 ml). El filtrado se lavó con NaHCO3 acuoso sat. (15 ml), H2O (15 ml) y salmuera (10 ml), se secó, se filtró y se concentró a presión reducida. La purificación por cromatografía en gel de sílice (25→45 % de EtOAc/hexanos) dio el producto deseado como un sólido amarillo tenue (566 mg, 73 % de rendimiento). LC-MS (ESI) m/z: [M Na] calculado para C65H87N3O21: 1268,57; observado 1269,3.
Monómero 17.28(R),32(R),40(R)-O,O,O-tris[(4-nitrofenoxi)carbonil] rapamicina.

A una disolución de 32(R)-hidroxi rapamicina (1,00 g, 1,09 mmoles, 1,0 equiv) en DCM (22 ml) se añadió tamices moleculares en polvo de 4Å (1,0 g). La suspensión se agitó durante 45 min, momento en el que se añadió piridina (0,97 ml, 12,0 mmoles, 11,0 equiv). La mezcla se enfrió hasta -15 °C y entonces se añadió cloroformiato de p-nitrofenilo (550 mg, 2,73 mmoles, 2,5 equiv) en una porción. La mezcla se calentó hasta temperatura ambiente durante 4 h y se agitó durante la noche. La mezcla se enfrió hasta 0 °C y se añadió cloroformiato de p-nitrofenilo adicional (220 mg, 1,09 mmoles, 1,0 equiv) en una porción. La mezcla de reacción se agitó durante 1 h, se calentó hasta temperatura ambiente y luego se agitó durante 2 h. La mezcla se enfrió nuevamente a 0 °C y se añadió cloroformiato de p-nitrofenilo adicional (660 mg, 3,27 mmoles, 3,0 equiv). La mezcla de reacción se agitó durante 15 min y luego a temperatura ambiente durante 1 h. La mezcla de reacción se filtró y se lavó con DCM (25 ml). El filtrado se lavó con NaHCO3 acuoso sat. (20 ml), H2O (20 ml) y salmuera (15 ml), se secó, se filtró y se concentró a presión reducida. La purificación por cromatografía en gel de sílice (5→15 % de EtOAc/DCM) dio el producto deseado como un sólido amarillo tenue (550 mg, 36 % de rendimiento). LC-MS (ESI) m/z: [M Na] calculado para C72H90N4O25: 1433,58; observado 1434,3.
Procedimientos generales y ejemplos específicos.
Procedimiento general 1: Acoplamiento de un ácido carboxílico y una amina, seguido de desprotección de N-Boc.

Etapa 1:
A una disolución 0,1 M de ácido carboxílico (1,0 equiv) en DMA se añadió una amina (1,2 equiv), DIPEA (4,0 equiv) y PyBOP (1,3 equiv). La reacción se dejó con agitación hasta que se consumió el ácido carboxílico, como se indica por LCMS. La mezcla de reacción se purificó entonces por cromatografía en gel de sílice proporcionando el producto. Etapa 2:
A una disolución 0,07 M de la amina protegida con N-Boc (1,0 equiv) en dioxano se añadió HCl (4 M en dioxano) (50 equiv). La reacción se dejó con agitación hasta que se consumió la amina protegida con N-Boc, como se indica por LCMS. Entonces se concentró la reacción a un aceite, que luego se disolvió en H2O y se liofilizó proporcionando el producto.
Producto intermedio A1-7. 1-Amino-27-(6-{[4-amino-3-(2-amino-1,3-benzoxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il]metil}-1,2,3,4-tetrahidroisoquinolin-2-il)-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-ona

Etapa 1: Síntesis de N-[27-(6-{[4-amino-3-(2-amino-1,3-benzoxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il]metil}-1,2,3,4-tetrahidroisoquinolin-2-il)-27-oxo-3,6,9,12,15,18,21,24-octaoxaheptacosan-1-il]carbamatocarbamato de terc-butilo
A una disolución de ácido 1-{[(terc-butoxi)carbonil]amino}-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-oico (102 mg, 189 µmoles, 1,0 equiv) y 6-{[4-amino-3-(2-amino-1,3-benzoxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il]metil}-1,2,3,4-tetrahidroisoquinolin-2-io (120 mg, 227 µmoles, 1,2 equiv) en DMA (1,88 ml) se añadió DIPEA (131 µL, 756 µmoles, 4,0 equiv), seguido de PyBOP (127 mg, 245 µmoles, 1,3 equiv). La reacción se agitó a temperatura ambiente. Después de 2 h, la mezcla de reacción se concentró a presión reducida y el residuo en bruto se purificó por cromatografía en gel de sílice (0→20 % de MeOH/DCM) dando el producto (161,5 mg, 91 % de rendimiento) como un aceite amarillo pálido. LCMS (ESI) m/z: [M H] calculado para C46H65N9O12: 936,49; observado 936,3.
Etapa 2: Síntesis de 1-amino-27-(6-{[4-amino-3-(2-amino-1,3-benzoxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il]metil}-1,2,3,4-tetrahidroisoquinolin-2-il)-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-ona
A una disolución de N-[27-(6-{ [4-amino-3-(2-amino-1,3-benzoxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il]metil}-1,2,3,4-tetrahidroisoquinolin-2-il)-27-oxo-3,6,9,12,15,18,21,24-octaoxaheptacosan-1-il]carbamato de terc-butilo (0,9 g, 0,9614 mmoles, 1,0 equiv) en dioxano (3,20 ml) se añadió HCl (4 M en dioxano, 2,40 ml, 9,61 mmoles, 10,0 equiv). La reacción se agitó durante 2 h y entonces se concentró a presión reducida a un aceite. El aceite se destiló azeotrópicamente con DCM (3 × 15 ml) proporcionando el producto (881 mg, 105 % de rendimiento, HCl) como un sólido de color tostado, que se usó directamente en la siguiente etapa. LCMS (ESI) m/z: [M H] calculado para C41H57N9O10: 836,43; observado 836,3.
Siguiendo el Procedimiento general 1, pero usando el inhibidor de sitio activo que contiene amina apropiado en la Tabla 2 y ácido carboxílico de PEG, se prepararon los Productos intermedios A1 en la Tabla 5:
Tabla 5. Aminas adicionales preparadas








Siguiendo el Procedimiento general 1, pero usando el Producto intermedio A1 apropiado en la Tabla 5 y ácido carboxílico de PEG, se prepararon los Productos intermedios A2 en la Tabla 6:
Tabla 6. Aminas adicionales preparadas

Procedimiento general 2: Acoplamiento de un monómero de rapamicina que contiene carbonato de 4-nitrofenilo y un producto intermedio que contiene inhibidor de sitio activo que tiene una amina primaria o secundaria.

A una disolución 0,02 M de monómero de rapamicina que contiene carbonato de 4-nitrofenilo (1,0 equiv) y un producto intermedio que contiene inhibidor de sitio activo (2,0 equiv) en DMA se añadió DIPEA (4,0 equiv). La disolución resultante se agitó a temperatura ambiente bajo nitrógeno. Tras completarse como se ha determinado por análisis de LCMS, la mezcla de reacción en bruto se purificó por HPLC preparativa proporcionando el producto.
Ejemplo 2: Síntesis del compuesto de rapamicina bivalente de la serie 1

A una disolución de 40(R)-O-(carbonato de 4-nitrofenilo) rapamicina (25 mg, 23,16 µmoles, 1,0 equiv) y Producto intermedio A1-7 (42,0 mg, 46,32 µmoles, 2,0 equiv) en DMA (1,15 ml) se añadió DIPEA (16,0 µl, 92,64 µmoles, 4,0 equiv). La reacción se agitó durante 18 h, momento en el que la mezcla de reacción se purificó por cromatografía de fase inversa (10→40→95 % de MeCN 0,1 % de ácido fórmico/H2O 0,1 % de ácido fórmico) dando el producto (9,92 mg, 24 % de rendimiento) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C93H134N10O24: 1775,97; observado 1775,7.
Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y los Productos intermedios A1 y A2 de las Tablas 5 y 6, se sintetizaron los análogos bivalentes de la serie 1 en la Tabla 7:
Tabla 7. Compuestos bivalentes de la serie 1:












Procedimiento general 3: Acoplamiento de un éster de PEG que contiene haluro y un preconector que contiene amina, seguido de desprotección del éster.

Etapa 1:
A una disolución 0,1 M de preconector que contiene amina (1,0 equiv) en MeCN se añadió K2CO3 (2,0 equiv), seguido de éster de PEG que contiene haluro (1,0 equiv). La reacción se agitó a 80 °C hasta que se consumió el preconector que contiene amina, como se indica por análisis de LCMS. La reacción se purificó entonces por cromatografía en gel de sílice proporcionando el producto.
Etapa 2:
una disolución 0,1 M de éster terc-butílico de PEG (1,0 equiv) en EtOAc se añadió una disolución de HCl en EtOAc. La suspensión resultante se agitó a temperatura ambiente hasta que se consumió el éster de PEG, como se indica por análisis de LCMS. La reacción se concentró entonces a presión reducida proporcionando el producto.
Producto intermedio B1-1. Ácido1-(4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oico

Etapa 1: Síntesis de 1-(4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo
A una mezcla de 2-((2-(piperazin-1-il)pirimidin-5-il)metil)isoindolin-1,3-diona (7,97 g, 24,66 mmoles, 1,0 equiv) en MeCN (200 ml) se añadió K2CO3 (6,82 g, 49,31 mmoles, 2,0 equiv), seguido de 1-bromo-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo (9,5 g, 24,66 mmoles, 1,0 equiv). La mezcla de reacción se calentó hasta 85 °C y se agitó durante 15 h. La mezcla se enfrió entonces hasta temperatura ambiente y se filtró. El filtrado se concentró a presión
reducida y el residuo se purificó por cromatografía en gel de sílice (0→20 % de EtOAc/MeOH) dando el producto (11,5 g, 74,3 % de rendimiento) como un líquido amarillo claro.
Etapa 2: Síntesis de ácido 1-(4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oico
A una disolución de 1-(4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oato de terc-butilo (3,5 g, 5,58 mmoles, 1,0 equiv) en EtOAc (50 ml) se añadió una disolución de HCl en EtOAc (500 ml). La mezcla se agitó a temperatura ambiente durante 3 h. La mezcla se concentró entonces a presión reducida dando el producto (5,3 g, 78,2 % de rendimiento, HCl) como un sólido blanco. LCMS (ESI) m/z: [M H] calculado para C28H37N5O8: 572,27; observado 572,4.
Siguiendo el Procedimiento general 3, pero usando el PEG que contiene haluro apropiado y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios B1 en la Tabla 8:
Tabla 8. Amina protegidas adicionales preparadas

Procedimiento general 4: Acoplamiento de un ácido carboxílico de PEG y un inhibidor de sitio activo que contiene amina, seguido de desprotección de amina.

Etapa 1:
A una disolución 0,15 M de ácido carboxílico de PEG (1,0 equiv) en DMF se añadió HATU (1,3 equiv) y DIPEA (5,0 equiv). Después de agitar durante 30 min, se añadió el inhibidor de sitio activo que contiene amina (1,2 equiv). La reacción se agitó a temperatura ambiente hasta que se consumió el ácido carboxílico de PEG, como se indica por LCMS. La reacción se purificó entonces por cromatografía de fase inversa proporcionando el producto.
Etapa 2:
A una disolución 0,1 M de amina protegida con ftalimida (1,0 equiv) en MeOH a 0 °C se añadió NH2NH2•H2O (4,0 equiv). La mezcla resultante se agitó a 60 °C hasta que se consumió la amina protegida con ftalimida, como se indica por análisis de LCMS. La reacción se purificó entonces por cromatografía de fase inversa proporcionando el producto.
Producto intermedio B2-1. N-(4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)-1-(4-(5-(aminometil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-amida

Etapa 1: Síntesis de N-(4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)-1-(4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-amida
A una mezcla de ácido 1-(4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-oico (3 g, 4,93 mmoles, 1,0 equiv, HCl) en DMF (30 ml) se añadió HATU (12,11 µl, 6,41 mmoles, 1,3 equiv) y DIPEA (4,30 ml, 24,67 mmoles, 5,0 equiv). Después de 30 min, se añadió 5-(4-amino-1-(4-aminobutil)-1H-pirazolo[3,4-d]pirimidin-3-il)benzo[d]oxazol-2-amina (4,03 g, 5,92 mmoles, 1,2 equiv, 3TFA). La mezcla se agitó a temperatura ambiente durante 3 h. Entonces se purificó la mezcla de reacción por prep-HPLC (MeCN/H2O) dando el producto (5,4 g, 81,2 % de rendimiento, 4TFA) como un sólido rojo claro. LCMS (ESI) m/z: [M 2H]/2 calculado para C44H53N13O8: 446,71; observado 447,0.
Etapa 2: Síntesis de N-(4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)-1 -(4-(5 -(aminometil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15 -amida
A una mezcla de N-(4-(4-amino-3-(2-aminobenzo[d]oxazol-5-il)-1H-pirazolo[3,4-d]pirimidin-1-il)butil)-1-(4-(5-((1,3-dioxoisoindolin-2-il)metil)pirimidin-2-il)piperazin-1-il)-3,6,9,12-tetraoxapentadecan-15-amida (4 g, 2,97 mmoles, 1,0 equiv, 4TFA) en MeOH (25 ml) a 0 °C se añadió NH2NH2•H2O (588,63 µl, 11,87 mmoles, 4,0 equiv). La mezcla se agitó a 60 °C durante 2 h. Entonces, la mezcla se enfrió hasta temperatura ambiente y se filtró y la torta de filtración se lavó con MeOH (5 ml). El filtrado se concentró a presión reducida y el residuo se purificó por prep-HPLC (MeCN/H2O) dando el producto (700 mg, 24,5 % de rendimiento, TFA) como un sólido blanco. LCMS (ESI) m/z: [M 2H]/2 calculado para C36H51N13O6: 381,71; observado 381,8.
Siguiendo el Procedimiento general 4, pero usando el Producto intermedio B1 apropiado en la Tabla 8 y los inhibidores de sitio activo que contienen amina en la Tabla 2, se prepararon los productos intermedios B2 en la Tabla 9:
Tabla 9. Aminas adicionales preparadas


Procedimiento general 5: Acoplamiento de un ácido carboxílico de PEG que contiene haluro y un inhibidor de sitio activo que contiene amina.

A una disolución 0,1 M de inhibidor de sitio activo que contiene amina (1,0 equiv) y ácido carboxílico que contiene PEG (1,2 equiv) en DMA se añadió DIPEA (4,0 equiv), seguido de PyBOP (1,3 equiv). La reacción se agitó hasta que se consumió el inhibidor de sitio activo que contiene amina, como se indica por LCMS. La reacción se purificó entonces por HPLC de fase inversa proporcionando el producto.
Producto intermedio B3-1. 18-{6-[(4-amino-3-{1H-pirrolo[2,3-b]piridin-5-il}-1H-pirazolo[3,4-d]pirimidin-1-il)metil]-1,2,3,4-tetrahidroisoquinolin-2-il}-1-bromo-3,6,9,12,15-pentaoxaoctadecan-18-ona

A una disolución de ácido 1-bromo-3,6,9,12,15-pentaoxaoctadecan-18-oico (105 mg, 282 µmoles, 1,2 equiv) y 3-{1H-pirrolo[2,3-b]piridin-5-il}-1-[(1,2,3,4-tetrahidroisoquinolin-6-il)metil]-1H-pirazolo[3,4-d]pirimidin-4-amina (120 mg, 235 µmoles, 1,0 equiv) en DMA (2,34 ml) se añadió DIPEA (163 µl, 940 µmoles, 4,0 equiv), seguido de PyBOP (158 mg, 305 µmoles, 1,3 equiv). La disolución resultante se agitó a temperatura ambiente durante 3 h, luego se purificó por HPLC de fase inversa (10→98 % de MeCN 0,1 % de ácido fórmico/H2O 0,1 % de ácido fórmico) proporcionando el producto (82,7 mg, 47 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C35H43BrN8O6: 751,26; observado 751,2.
Siguiendo el Procedimiento general 5, pero usando el ácido carboxílico de PEG que contiene haluro apropiado e inhibidores de sitio activo que contiene amina en la Tabla 2, se prepararon los Productos intermedios B3 en la Tabla 10:
Tabla 10. Haluros de PEG adicionales preparados


Procedimiento general 6: Desplazamiento de un haluro de PEG con un posconector que contiene amina y desprotección de la amina.

Etapa 1:
A una disolución 0,1 M de haluro que contiene PEG (1,0 equiv) en MeCN se añadió K2CO3 (3,0 equiv), seguido de posconector que contiene amina (1,2 equiv). La suspensión resultante se calentó hasta 80 °C y se agitó hasta que se consumió el haluro de PEG, como se indica por análisis de LCMS. La reacción se enfrió hasta temperatura ambiente y luego se purificó por cromatografía en gel de sílice proporcionando el producto.
Etapa 2:
A una disolución 0,07 M de amina protegida con N-Boc (1,0 equiv) en dioxano se añadió HCl (4 M en dioxano, 10,0 equiv). La reacción se agitó hasta que se consumió la amina protegida con N-Boc, como se indica por análisis de LCMS. La reacción se concentró entonces a presión reducida proporcionando el producto.
Producto intermedio B2-4. 18-{6-[(4-amino-3-{1H-pirrolo[2,3-b]piridin-5-il}-1H-pirazolo[3,4-d]pirimidin-1-il)metil]-1,2,3,4-tetrahidroisoquinolin-2-il}-1-(4-{5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}piperazin-1-il)-3,6,9,12,15-pentaoxaoctadecan-18-ona

Etapa 1: Síntesis de 2-[4-(18-{6-[(4-amino-3-{1H-pirrolo[2,3-b]piridin-5-il}-1H-pirazolo[3,4-d]pirimidin-1-il)metil]-1,2,3,4-tetrahidroisoquinolin-2-il}-18-oxo-3,6,9,12,15-pentaoxaoctadecan-1-il)piperazin-1-il]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-6-carboxilato de terc-butilo
A una suspensión de 18-{6-[(4-amino-3-{1H-pirrolo[2,3-b]piridin-5-il}-1H-pirazolo[3,4-d]pirimidin-1-il)metil]-1,2,3,4-tetrahidroisoquinolin-2-il}-1-bromo-3,6,9,12,15-pentaoxaoctadecan-18-ona (82,7 mg, 110 µmoles, 1,0 equiv) en MeCN
(1,09 ml) se añadió K2CO3 (45,6 mg, 330 µmoles, 3,0 equiv), seguido de 2-(piperazin-1-il)-5H,6H,7H,8H-pirido[4,3-d]pirimidin-6-carboxilato de terc-butilo (42,1 mg, 132 µmoles, 1,2 equiv). La suspensión resultante se calentó hasta 80 °C durante 8 h, luego se purificó por cromatografía en gel de sílice (0→20 % de MeOH/DCM) proporcionando el producto (75,1 mg, 70 % de rendimiento). LCMS (ESI) m/z: [M H] calculado para C51H67N13O8: 990,53; observado 990,5.
Etapa 2: Síntesis de 18-{6-[(4-amino-3-{1H-pirrolo[2,3-b]piridin-5-il}-1H-pirazolo[3,4-d]pirimidin-1-il)metil]-1,2,3,4-tetrahidroisoquinolin-2-il}-1-(4-{5H,6H,7H,8H-pirido[4,3-d]pirimidin-2-il}piperazin-1-il)-3,6,9,12,15-pentaoxaoctadecan-18-ona
A una disolución de 2-[4-(18-{6-[(4-amino-3-{1H-pirrolo[2,3-b]piridin-5-il}-1H-pirazolo[3,4-d]pirimidin-1-il)metil]-1,2,3,4-tetrahidroisoquinolin-2-il}-18-oxo-3,6,9,12,15-pentaoxaoctadecan-1-il)piperazin-1-il]-5H,6H,7H,8H-pirido[4,3-d]pirimidin-6-carboxilato de terc-butilo (75,1 mg, 75,8 µmoles, 1,0 equiv) en dioxano (1 ml) se añadió HCl (4 M en dioxano, 472 µl, 1,89 mmoles, 10,0 equiv). La disolución se agitó a temperatura ambiente durante 45 min, entonces se concentró a presión reducida proporcionando el producto. LCMS (ESI) m/z: [M Na] calculado para C46H59N13O6: 912,46; observado 912,5.
Siguiendo el Procedimiento general 6, pero usando el ácido carboxílico de PEG apropiado y los inhibidores de sitio activo que contienen amina en la Tabla 2, se prepararon los Productos intermedios B2 en la Tabla 11:
Tabla 11. Aminas adicionales preparadas

Siguiendo el Procedimiento general 1, pero usando el éster terc-butílico de PEG de ácido carboxílico apropiado e inhibidores de sitio activo que contienen amina en la Tabla 2, se prepararon los Productos intermedios B4 en la Tabla 12:
Tabla 12. Aminas adicionales preparadas

Siguiendo el Procedimiento general 1, pero usando los Productos intermedios B4 apropiados en la Tabla 12 y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios B2 en la Tabla 13: Tabla 13. Aminas adicionales preparadas

Siguiendo el Procedimiento general 1, pero usando los Productos intermedios A1 apropiados y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios B2 en la Tabla 14:
Tabla 14. Aminas adicionales preparadas







Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y los Productos intermedios B2 de las Tablas 9, 11, y 13 y 14, se sintetizaron los análogos bivalentes de la serie 2 en la Tabla 15:
Tabla 15. Compuestos bivalentes de la serie 2:






Siguiendo el Procedimiento general 1, pero usando los inhibidores de sitio activo que contienen amina apropiados en la Tabla 2 y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios C1 en la Tabla 16:
Tabla 16. Aminas adicionales preparadas



Siguiendo el Procedimiento general 1, pero usando los ácidos carboxílicos de PEG y Productos intermedios C1 en la Tabla 16, se prepararon los Productos intermedios C2 en la Tabla 17:
Tabla 17. Aminas adicionales preparadas


Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y Productos intermedios C2 de la Tabla 17, se sintetizaron los análogos bivalentes de la serie 3 en la Tabla 18:
Tabla 18. Compuestos bivalentes de la serie 3


Siguiendo el Procedimiento general 1, pero usando los Productos intermedios C2 apropiados en la Tabla 17 y preconectores que contienen amina en la Tabla 4, se prepararon los productos intermedios D1 en la Tabla 19: Tabla 19. Aminas adicionales preparadas

Siguiendo el Procedimiento general 1, pero usando los inhibidores de sitio activo que contienen amina apropiados en la Tabla 2 y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios D1 en la Tabla 20:
Tabla 20. Aminas adicionales preparadas、





Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y Productos intermedios D1 de las Tablas 19 y 20, se sintetizaron los análogos bivalentes de la serie 4 en la Tabla 21:
Tabla 21. Compuestos bivalentes de la serie 4







Siguiendo el Procedimiento general 1, pero usando los Productos intermedios C1 apropiados en la Tabla 16 y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios E1 en la Tabla 22: Tabla 22. Aminas adicionales preparadas


Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y Productos intermedios E1 de la Tabla 22, se sintetizaron los análogos bivalentes de la serie 5 en la Tabla 23:
Tabla 23. Compuestos bivalentes de la serie 5

Tabla 24. Aminas adicionales preparadas
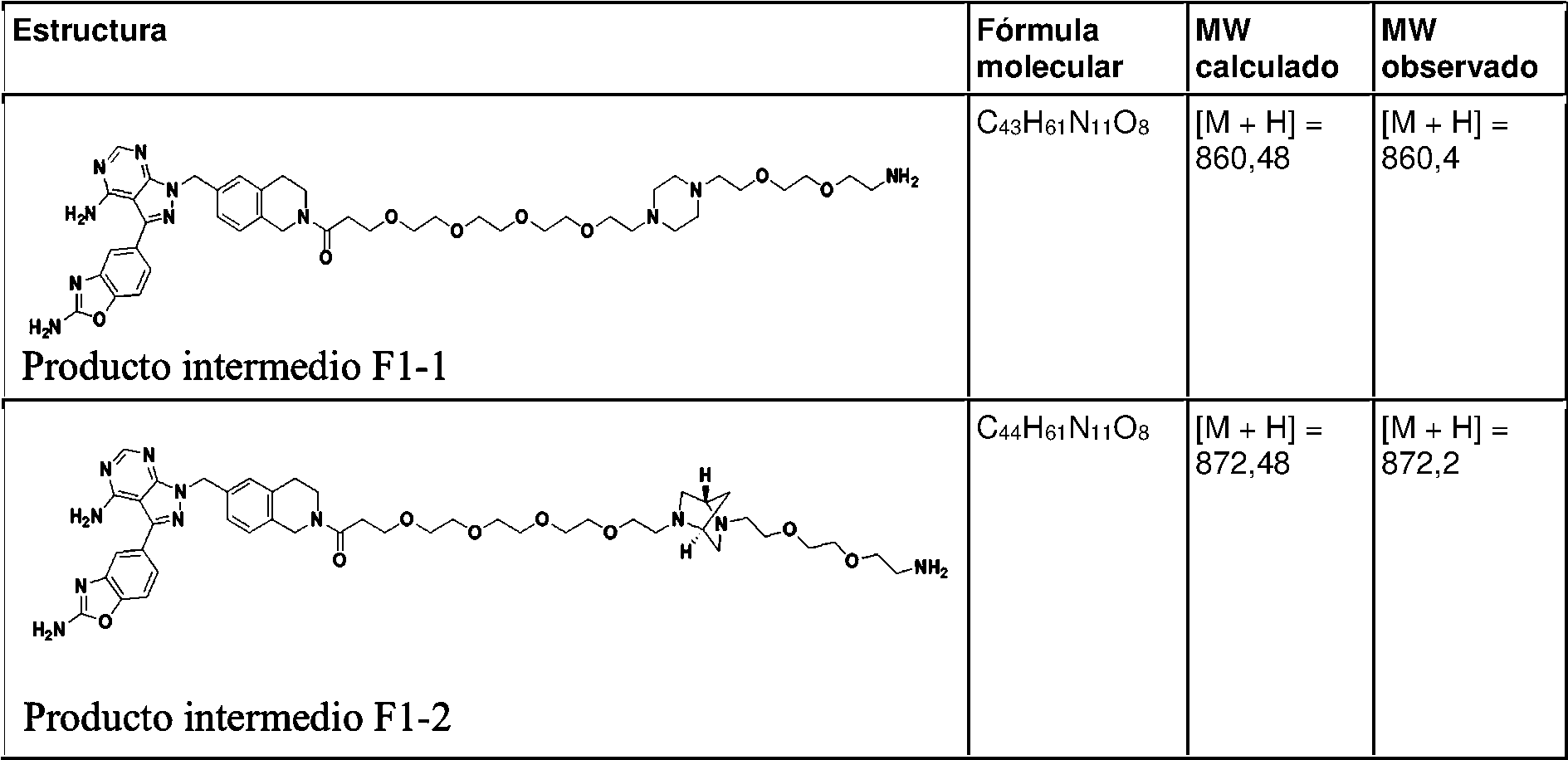
Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y Productos intermedios F1 de la Tabla 24, se sintetizaron los análogos bivalentes de la serie 6 en la Tabla 25:
Tabla 25. Compuestos bivalentes de la serie 6

Siguiendo el Procedimiento general 1, pero usando los Productos intermedios A1 apropiados en la Tabla 5 y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios G1 en la Tabla 26:
Tabla 26. Aminas adicionales preparadas

Siguiendo el Procedimiento general 6, pero usando los Productos intermedios B3 apropiados en la Tabla 10 y preconectores que contienen amina en la Tabla 4, se prepararon los Productos intermedios G1 en la Tabla 27: Tabla 27. Aminas adicionales preparadas

Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y Productos intermedios G1 de las Tablas 26 y 27, se sintetizaron los análogos bivalentes de la serie 7 en la Tabla 28:
Tabla 28. Compuestos bivalentes de la serie 7

Siguiendo el Procedimiento general 1, pero usando los Productos intermedios D1 apropiados en las Tablas 19 y 20 y ácidos carboxílicos de PEG, se prepararon los Productos intermedios H1 en la Tabla 29:
Tabla 29. Aminas adicionales preparadas
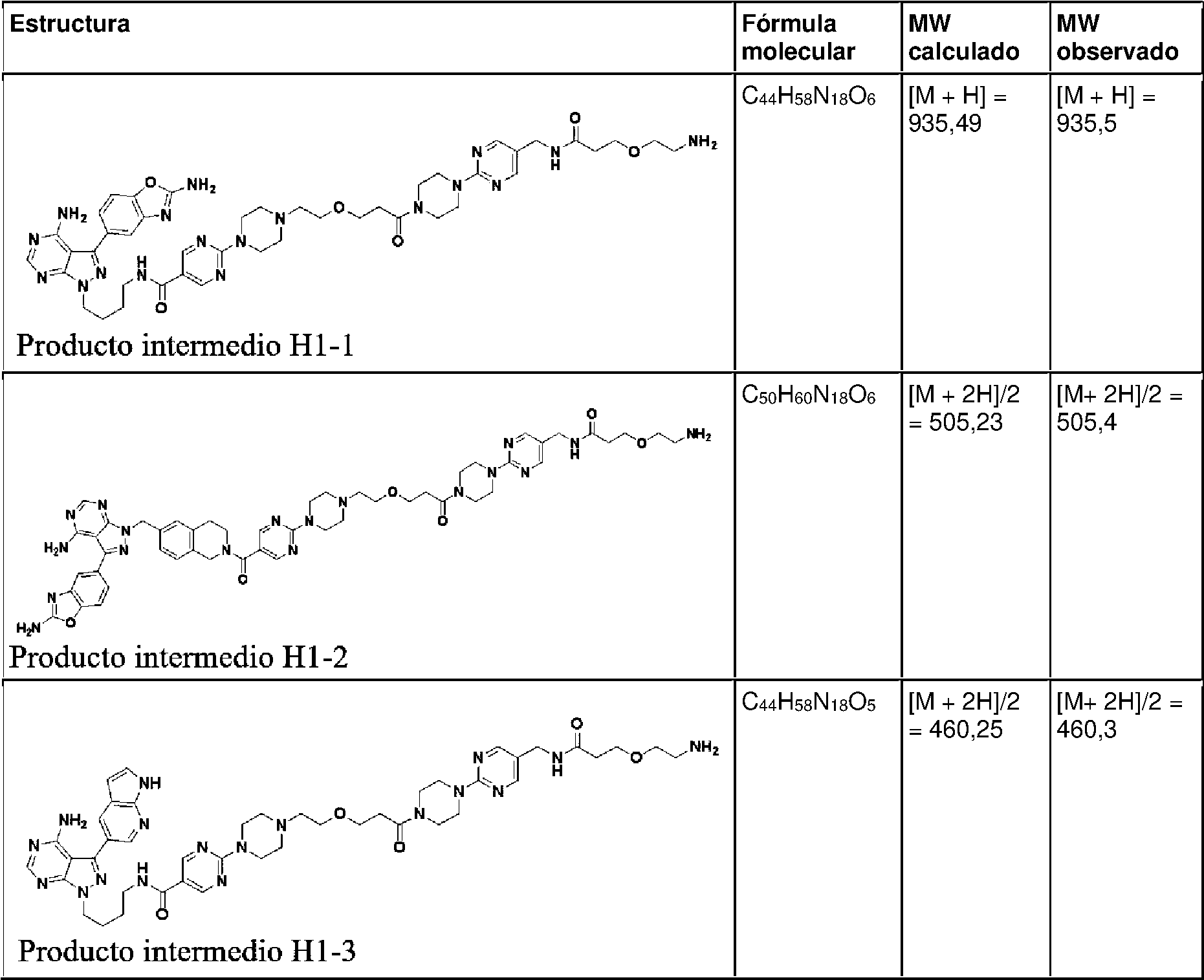
Siguiendo el Procedimiento general 2, pero usando el monómero de rapamicina que contiene carbonato de 4-nitrofenilo apropiado en la Tabla 1 y Productos intermedios H1 de la Tabla 29, se sintetizaron los análogos bivalentes de la serie 8 en la Tabla 30:
Tabla 30. Compuestos bivalentes de la serie 8:


Ejemplos biológicos
Ensayos AlphaLISA basados en células para determinar CI50 para la inhibición de P-Akt (S473), P-4E-BP1 (T37/46) y P-P70S6K (T389) en células MDA-MB-468
Ensayo celular de cinasa mTOR
Para medir la actividad funcional de mTORC1 y mTORC2 en células, se monitorizó la fosforilación de 4EBP1 (Thr37/46) y P70S6K (Thr389), y AKT1/2/3 (Ser473) usando AlphaLisa SureFire Ultra Kits (Perkin Elmer). Se cultivaron células MDA-MB-468 (ATCC® HTB-132) en placas de cultivo de tejido de 96 pocillos y se trataron con compuestos en la divulgación a concentraciones variables desde 0,017 - 1.000 nM durante dos a cuatro horas a 37 °C. Las incubaciones se terminaron por retirada de tampón de ensayo y adición de tampón de lisis proporcionados por el kit del ensayo. Se procesaron muestras según las instrucciones del fabricante. Se midió por duplicado la señal de Alpha de las fosfoproteínas respectivas usando un lector de microplacas (Envision, Perkin-Elmer o Spectramax M5, Molecular Devices). Se analizaron curvas de respuesta a concentración de inhibidor usando un ajuste de curva de regresión de CI50 normalizado con normalización basada en controles.
Como un ejemplo, se informan a continuación los valores de CI50 medidos para compuestos seleccionados:

Como un ejemplo, se informan a continuación los valores de pCI50 medidos para compuestos seleccionados:






Claims (23)
1. Un compuesto seleccionado del grupo que consiste en:



































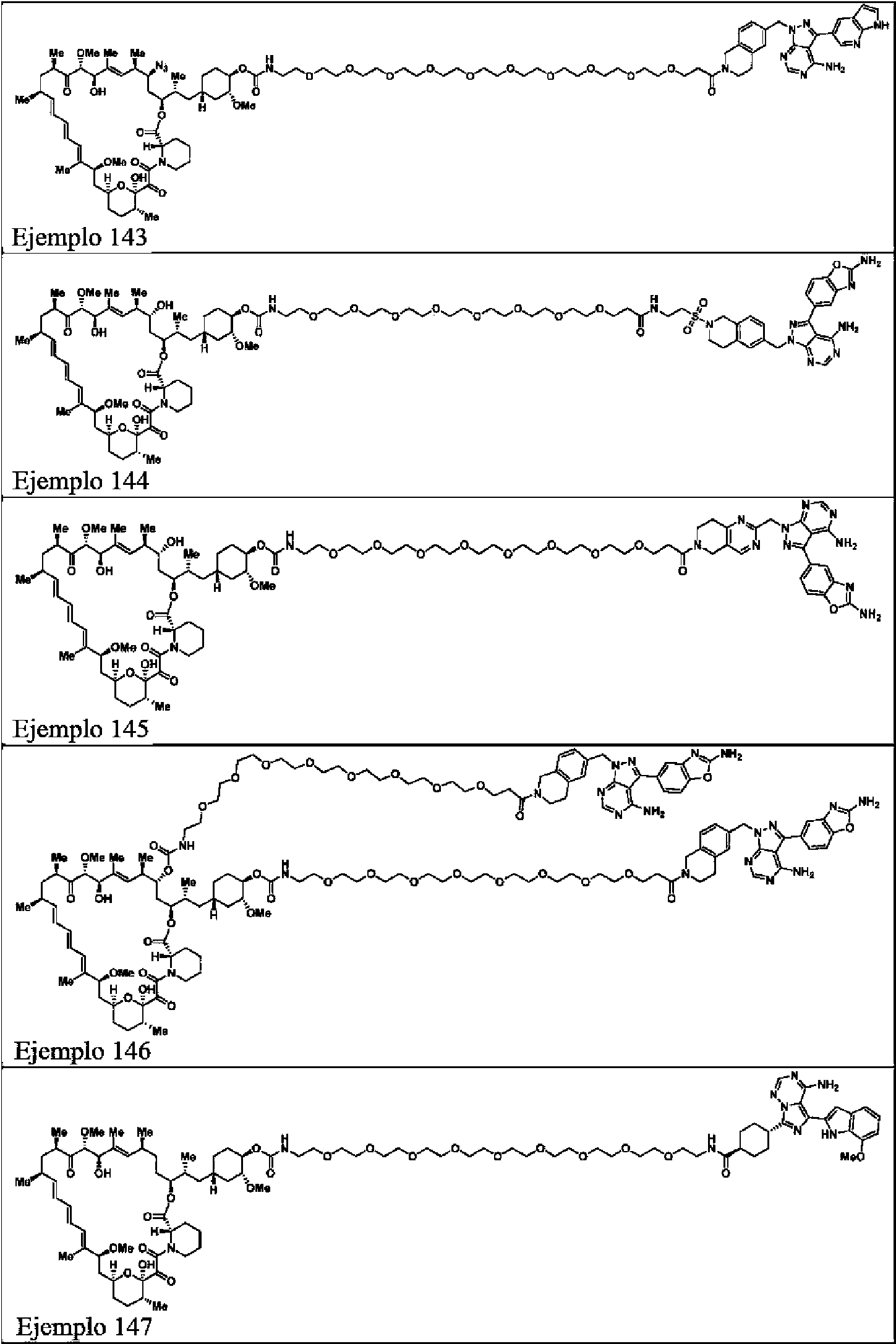


o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo.
2. El compuesto de la reivindicación 1, que tiene la fórmula

o un estereoisómero del mismo.
3. El compuesto de la reivindicación 1, que tiene la fórmula

o un tautómero del mismo.
4. El compuesto de la reivindicación 1, que tiene la fórmula

o un isómero de oxepano del mismo.
5. El compuesto de la reivindicación 1, que tiene la fórmula

o un estereoisómero del mismo.
6. El compuesto de la reivindicación 1, que tiene la fórmula

o un tautómero del mismo.
7. El compuesto de la reivindicación 1, que tiene la fórmula

8. El compuesto de la reivindicación 1, que tiene la fórmula

9. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1-8, o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo, y al menos uno de un vehículo, diluyente o excipiente farmacéuticamente aceptable.
10. La composición farmacéutica de la reivindicación 9, que comprende una mezcla de

o un estereoisómero o tautómero del mismo y

o un estereoisómero o tautómero del mismo.
11. La composición farmacéutica de la reivindicación 10, que comprende un excipiente farmacéuticamente aceptable.
12. La composición farmacéutica de la reivindicación 9, que comprende una mezcla de

13. La composición de la reivindicación 12, que comprende un excipiente farmacéuticamente aceptable.
14. Un compuesto de una cualquiera de las reivindicaciones 1-8, o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo, para su uso en el tratamiento, prevención o reducción del riesgo de una enfermedad o trastorno mediado por mTOR.
15. El compuesto para el uso de la reivindicación 14, en donde la enfermedad es cáncer o una enfermedad mediada por inmunidad.
16. El compuesto para el uso de la reivindicación 15, en donde el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos.
17. El compuesto para el uso de la reivindicación 15, en donde la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto
contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis.
18. Un compuesto de una cualquiera de las reivindicaciones 1-8, o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo, para su uso en el tratamiento del cáncer.
19. El compuesto para el uso de la reivindicación 18, en donde el cáncer se selecciona de tumores cerebrales y neurovasculares, cánceres de cabeza y cuello, cáncer de mama, cáncer de pulmón, mesotelioma, cáncer linfoide, cáncer de estómago, cáncer de riñón, carcinoma renal, cáncer de hígado, cáncer de ovario, endometriosis de ovario, cáncer testicular, cáncer gastrointestinal, cáncer de próstata, glioblastoma, cáncer de piel, melanoma, cánceres neurológicos, cánceres del bazo, cánceres pancreáticos, trastornos proliferativos de la sangre, linfoma, leucemia, cáncer endometrial, cáncer de cuello uterino, cáncer de vulva, cáncer de próstata, cáncer de pene, cánceres de huesos, cánceres musculares, cánceres de tejido blando, cáncer intestinal o de recto, cáncer anal, cáncer de vejiga, cáncer de las vías biliares, cáncer ocular, tumores del estroma gastrointestinal y tumores neuroendocrinos.
20. Un compuesto de una cualquiera de las reivindicaciones 1-8, o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una enfermedad mediada por inmunidad.
21. El compuesto para el uso de la reivindicación 20, en donde la enfermedad mediada por la inmunidad se selecciona de resistencia por trasplante de corazón, riñón, hígado, médula ósea, piel, córnea, pulmón, páncreas, intestino delgado, extremidad, músculo, nervios, duodeno, intestino delgado o células de los islotes pancreáticos; enfermedad injerto contra huésped causada por trasplante de médula ósea; artritis reumatoide, lupus eritematoso sistémico, tiroiditis de Hashimoto, esclerosis múltiple, miastenia grave, diabetes de tipo I, uveítis, encefalomielitis alérgica y glomerulonefritis.
22. Un compuesto de una cualquiera de las reivindicaciones 1-8, o una sal, tautómero o estereoisómero farmacéuticamente aceptable del mismo, para su uso en el tratamiento de una afección senil.
23. El compuesto para el uso de la reivindicación 22, en donde la afección senil se selecciona de sarcopenia, atrofia de la piel, atrofia muscular progresiva, atrofia cerebral, aterosclerosis, arteriosclerosis, enfisema pulmonar, osteoporosis, osteoartritis, hipertensión arterial, disfunción eréctil, demencia, enfermedad de Huntington, enfermedad de Alzheimer, cataratas, degeneración macular senil, cáncer de próstata, accidente cerebrovascular, reducida esperanza de vida, deterioro de la función renal y sordera parcial senil, incapacidad de movilidad relacionada con el envejecimiento (por ejemplo, fragilidad), deterioro cognitivo, demencia senil, deterioro de la memoria, rigidez de los tendones, disfunción cardíaca, tal como hipertrofia cardíaca y disfunción sistólica y diastólica, inmunosenescencia, cáncer, obesidad y diabetes.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862665435P | 2018-05-01 | 2018-05-01 | |
| US201862752874P | 2018-10-30 | 2018-10-30 | |
| US201962836036P | 2019-04-18 | 2019-04-18 | |
| PCT/US2019/029737 WO2019212990A1 (en) | 2018-05-01 | 2019-04-29 | C40-, c28-, and c-32-linked rapamycin analogs as mtor inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2945560T3 true ES2945560T3 (es) | 2023-07-04 |
Family
ID=66484166
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19723571T Active ES2945560T3 (es) | 2018-05-01 | 2019-04-29 | Análogos de rapamicina unidos en C40, C28 y C-32 como inhibidores de mTOR |
Country Status (29)
| Country | Link |
|---|---|
| US (4) | US20190336609A1 (es) |
| EP (2) | EP3788049B1 (es) |
| JP (2) | JP7358387B2 (es) |
| KR (1) | KR20210018244A (es) |
| CN (1) | CN112771054B (es) |
| AU (2) | AU2019262978B2 (es) |
| BR (1) | BR112020022201A2 (es) |
| CA (1) | CA3098692A1 (es) |
| CL (1) | CL2020002827A1 (es) |
| CO (1) | CO2020013865A2 (es) |
| CR (2) | CR20200578A (es) |
| DK (1) | DK3788049T3 (es) |
| ES (1) | ES2945560T3 (es) |
| FI (1) | FI3788049T3 (es) |
| HR (1) | HRP20230488T1 (es) |
| HU (1) | HUE062352T2 (es) |
| IL (2) | IL300091A (es) |
| LT (1) | LT3788049T (es) |
| MX (3) | MX2020011565A (es) |
| PE (1) | PE20212112A1 (es) |
| PH (1) | PH12020551795A1 (es) |
| PL (1) | PL3788049T3 (es) |
| PT (1) | PT3788049T (es) |
| RS (1) | RS64272B1 (es) |
| SA (1) | SA520420458B1 (es) |
| SG (1) | SG11202010559UA (es) |
| SI (1) | SI3788049T1 (es) |
| TW (1) | TW202014208A (es) |
| WO (1) | WO2019212990A1 (es) |
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4302834A3 (en) | 2016-07-12 | 2024-07-17 | Revolution Medicines, Inc. | 2,5-disubstituted 3-methyl pyrazines and 2,5,6-trisubstituted 3-methyl pyrazines as allosteric shp2 inhibitors |
| TWI820013B (zh) | 2017-01-23 | 2023-11-01 | 美商銳新醫藥公司 | 作為別構shp2抑制劑之雙環化合物 |
| MX2019008696A (es) | 2017-01-23 | 2019-09-13 | Revolution Medicines Inc | Compuestos de piridina como inhibidores de shp2 alostericos. |
| MX2020003579A (es) | 2017-10-12 | 2020-07-22 | Revolution Medicines Inc | Compuestos de piridina, pirazina, y triazina como inhibidores de shp2 alostericos. |
| MX2020006273A (es) | 2017-12-15 | 2020-09-14 | Revolution Medicines Inc | Compuestos policiclicos como inhibidores alostericos de shp2. |
| TW202014208A (zh) | 2018-05-01 | 2020-04-16 | 美商銳新醫藥公司 | 作為mTOR抑制劑之C40-、C28-及C-32連接之雷帕黴素類似物 |
| EP3788050B1 (en) | 2018-05-01 | 2024-08-28 | Revolution Medicines, Inc. | C26-linked rapamycin analogs as mtor inhibitors |
| US20230096028A1 (en) | 2019-03-01 | 2023-03-30 | Revolution Medicines, Inc. | Bicyclic heterocyclyl compounds and uses thereof |
| CN113767100A (zh) | 2019-03-01 | 2021-12-07 | 锐新医药公司 | 双环杂芳基化合物及其用途 |
| CR20220241A (es) | 2019-11-04 | 2022-08-03 | Revolution Medicines Inc | Inhibidores de ras |
| TW202132315A (zh) | 2019-11-04 | 2021-09-01 | 美商銳新醫藥公司 | Ras 抑制劑 |
| WO2021091956A1 (en) | 2019-11-04 | 2021-05-14 | Revolution Medicines, Inc. | Ras inhibitors |
| KR20220100903A (ko) | 2019-11-08 | 2022-07-18 | 레볼루션 메디슨즈, 인크. | 이환식 헤테로아릴 화합물 및 이의 용도 |
| EP4065231A1 (en) | 2019-11-27 | 2022-10-05 | Revolution Medicines, Inc. | Covalent ras inhibitors and uses thereof |
| WO2021257736A1 (en) | 2020-06-18 | 2021-12-23 | Revolution Medicines, Inc. | Methods for delaying, preventing, and treating acquired resistance to ras inhibitors |
| WO2022020114A2 (en) | 2020-07-10 | 2022-01-27 | Ting Therapeutics Llc | Methods for the prevention and treatment of hearing loss |
| AU2021344830A1 (en) | 2020-09-03 | 2023-04-06 | Revolution Medicines, Inc. | Use of SOS1 inhibitors to treat malignancies with SHP2 mutations |
| PE20231207A1 (es) | 2020-09-15 | 2023-08-17 | Revolution Medicines Inc | Derivados indolicos como inhibidores de ras en el tratamiento del cancer |
| AU2021409816A1 (en) | 2020-12-22 | 2023-07-06 | Qilu Regor Therapeutics Inc. | Sos1 inhibitors and uses thereof |
| CN117412954A (zh) * | 2021-04-09 | 2024-01-16 | 锐新医药公司 | 雷帕霉素类似物化合物的合成 |
| CR20230570A (es) | 2021-05-05 | 2024-01-22 | Revolution Medicines Inc | Inhibidores de ras |
| AR125787A1 (es) | 2021-05-05 | 2023-08-16 | Revolution Medicines Inc | Inhibidores de ras |
| JP2024516450A (ja) | 2021-05-05 | 2024-04-15 | レボリューション メディシンズ インコーポレイテッド | 共有結合性ras阻害剤及びその使用 |
| WO2022240947A1 (en) * | 2021-05-12 | 2022-11-17 | Revolution Medicines, Inc. | Use of sos1 inhibitors with mtor inhibitors to treat cancers |
| CN114044775B (zh) * | 2021-08-30 | 2023-04-07 | 杭州医学院 | 一种靶向泛素化诱导bcr-abl蛋白降解的化合物及其应用 |
| AR127308A1 (es) | 2021-10-08 | 2024-01-10 | Revolution Medicines Inc | Inhibidores ras |
| WO2023114954A1 (en) | 2021-12-17 | 2023-06-22 | Genzyme Corporation | Pyrazolopyrazine compounds as shp2 inhibitors |
| EP4227307A1 (en) | 2022-02-11 | 2023-08-16 | Genzyme Corporation | Pyrazolopyrazine compounds as shp2 inhibitors |
| WO2023172940A1 (en) | 2022-03-08 | 2023-09-14 | Revolution Medicines, Inc. | Methods for treating immune refractory lung cancer |
| WO2023240263A1 (en) | 2022-06-10 | 2023-12-14 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| GB202212000D0 (en) * | 2022-08-17 | 2022-09-28 | Mironid Ltd | Compounds and their use as PDE4 activators |
| WO2024206858A1 (en) | 2023-03-30 | 2024-10-03 | Revolution Medicines, Inc. | Compositions for inducing ras gtp hydrolysis and uses thereof |
Family Cites Families (165)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA737247B (en) | 1972-09-29 | 1975-04-30 | Ayerst Mckenna & Harrison | Rapamycin and process of preparation |
| GB1459571A (en) | 1974-09-12 | 1976-12-22 | Pfizer Ltd | Thiophene-2-sulphonamide derivatives and their use as therapeutic agents sheet orienting apparatus |
| US4316885A (en) | 1980-08-25 | 1982-02-23 | Ayerst, Mckenna And Harrison, Inc. | Acyl derivatives of rapamycin |
| US4513135A (en) | 1982-03-05 | 1985-04-23 | Eli Lilly And Company | Diaryl-pyrazine derivatives affecting GABA binding |
| US4911920A (en) | 1986-07-30 | 1990-03-27 | Alcon Laboratories, Inc. | Sustained release, comfort formulation for glaucoma therapy |
| FR2588189B1 (fr) | 1985-10-03 | 1988-12-02 | Merck Sharp & Dohme | Composition pharmaceutique de type a transition de phase liquide-gel |
| US4650803A (en) | 1985-12-06 | 1987-03-17 | University Of Kansas | Prodrugs of rapamycin |
| JPH0249775A (ja) | 1988-05-19 | 1990-02-20 | Nippon Soda Co Ltd | 6員環又は7員環を有する複素環化合物及びその製造方法 |
| US5120726A (en) | 1991-03-08 | 1992-06-09 | American Home Products Corporation | Rapamycin hydrazones |
| JPH04230389A (ja) | 1990-07-16 | 1992-08-19 | American Home Prod Corp | ラパマイシン誘導体 |
| US5023264A (en) | 1990-07-16 | 1991-06-11 | American Home Products Corporation | Rapamycin oximes |
| US5023263A (en) | 1990-08-09 | 1991-06-11 | American Home Products Corporation | 42-oxorapamycin |
| JPH04112877A (ja) | 1990-09-04 | 1992-04-14 | Nippon Soda Co Ltd | 新規シアノピラジン誘導体及びその製造方法 |
| PT98990A (pt) | 1990-09-19 | 1992-08-31 | American Home Prod | Processo para a preparacao de esteres de acidos carboxilicos de rapamicina |
| US5130307A (en) | 1990-09-28 | 1992-07-14 | American Home Products Corporation | Aminoesters of rapamycin |
| US5221670A (en) | 1990-09-19 | 1993-06-22 | American Home Products Corporation | Rapamycin esters |
| US5233036A (en) | 1990-10-16 | 1993-08-03 | American Home Products Corporation | Rapamycin alkoxyesters |
| DE69212850T2 (de) | 1991-01-15 | 1997-03-06 | Alcon Lab Inc | Verwendung von Karrageenan in topischen ophthalmologischen Zusammensetzungen |
| GB9103430D0 (en) | 1991-02-19 | 1991-04-03 | Smithkline Beecham Plc | Novel compound |
| US5212162A (en) | 1991-03-27 | 1993-05-18 | Alcon Laboratories, Inc. | Use of combinations gelling polysaccharides and finely divided drug carrier substrates in topical ophthalmic compositions |
| US5120842A (en) | 1991-04-01 | 1992-06-09 | American Home Products Corporation | Silyl ethers of rapamycin |
| US5100883A (en) | 1991-04-08 | 1992-03-31 | American Home Products Corporation | Fluorinated esters of rapamycin |
| US5118678A (en) | 1991-04-17 | 1992-06-02 | American Home Products Corporation | Carbamates of rapamycin |
| US5102876A (en) | 1991-05-07 | 1992-04-07 | American Home Products Corporation | Reduction products of rapamycin |
| US5118677A (en) | 1991-05-20 | 1992-06-02 | American Home Products Corporation | Amide esters of rapamycin |
| US5162333A (en) | 1991-09-11 | 1992-11-10 | American Home Products Corporation | Aminodiesters of rapamycin |
| US5151413A (en) | 1991-11-06 | 1992-09-29 | American Home Products Corporation | Rapamycin acetals as immunosuppressant and antifungal agents |
| GB9125660D0 (en) | 1991-12-03 | 1992-01-29 | Smithkline Beecham Plc | Novel compound |
| US5221740A (en) | 1992-01-16 | 1993-06-22 | American Home Products Corporation | Oxepane isomers of rapamycin useful as immunosuppressive agents |
| US5177203A (en) | 1992-03-05 | 1993-01-05 | American Home Products Corporation | Rapamycin 42-sulfonates and 42-(N-carboalkoxy) sulfamates useful as immunosuppressive agents |
| ZA935111B (en) | 1992-07-17 | 1994-02-04 | Smithkline Beecham Corp | Rapamycin derivatives |
| ZA935112B (en) | 1992-07-17 | 1994-02-08 | Smithkline Beecham Corp | Rapamycin derivatives |
| US5256790A (en) | 1992-08-13 | 1993-10-26 | American Home Products Corporation | 27-hydroxyrapamycin and derivatives thereof |
| MX9304868A (es) | 1992-08-13 | 1994-05-31 | American Home Prod | 27-hidroxirapamicina, derivados de la misma y composicion farmaceutica que la contiene. |
| GB9221220D0 (en) | 1992-10-09 | 1992-11-25 | Sandoz Ag | Organic componds |
| US5411967A (en) | 1992-10-13 | 1995-05-02 | American Home Products Corporation | Carbamates of rapamycin |
| US5480989A (en) * | 1992-10-13 | 1996-01-02 | American Home Products Corporation | Carbamates of rapamycin |
| US5480988A (en) | 1992-10-13 | 1996-01-02 | American Home Products Corporation | Carbamates of rapamycin |
| DK0593227T3 (da) | 1992-10-13 | 2006-05-01 | Wyeth Corp | Carbamater af rapamycin |
| US5302584A (en) * | 1992-10-13 | 1994-04-12 | American Home Products Corporation | Carbamates of rapamycin |
| US5489680A (en) | 1992-10-13 | 1996-02-06 | American Home Products Corporation | Carbamates of rapamycin |
| US5434260A (en) | 1992-10-13 | 1995-07-18 | American Home Products Corporation | Carbamates of rapamycin |
| US5262423A (en) | 1992-10-29 | 1993-11-16 | American Home Products Corporation | Rapamycin arylcarbonyl and alkoxycarbonyl carbamates as immunosuppressive and antifungal agents |
| US5262564A (en) | 1992-10-30 | 1993-11-16 | Octamer, Inc. | Sulfinic acid adducts of organo nitroso compounds useful as retroviral inactivating agents anti-retroviral agents and anti-tumor agents |
| US5258389A (en) * | 1992-11-09 | 1993-11-02 | Merck & Co., Inc. | O-aryl, O-alkyl, O-alkenyl and O-alkynylrapamycin derivatives |
| US5260300A (en) * | 1992-11-19 | 1993-11-09 | American Home Products Corporation | Rapamycin carbonate esters as immuno-suppressant agents |
| US5252579A (en) | 1993-02-16 | 1993-10-12 | American Home Products Corporation | Macrocyclic immunomodulators |
| AU6711994A (en) * | 1993-04-23 | 1994-11-21 | American Home Products Corporation | Rapamycin conjugates and antibodies |
| US5504091A (en) | 1993-04-23 | 1996-04-02 | American Home Products Corporation | Biotin esters of rapamycin |
| US5387680A (en) * | 1993-08-10 | 1995-02-07 | American Home Products Corporation | C-22 ring stabilized rapamycin derivatives |
| US5378836A (en) | 1993-10-08 | 1995-01-03 | American Home Products Corporation | Rapamycin oximes and hydrazones |
| US5391730A (en) | 1993-10-08 | 1995-02-21 | American Home Products Corporation | Phosphorylcarbamates of rapamycin and oxime derivatives thereof |
| US5373014A (en) | 1993-10-08 | 1994-12-13 | American Home Products Corporation | Rapamycin oximes |
| EP0729471A1 (en) * | 1993-11-19 | 1996-09-04 | Abbott Laboratories | Semisynthetic analogs of rapamycin (macrolides) being immunomodulators |
| US5527907A (en) | 1993-11-19 | 1996-06-18 | Abbott Laboratories | Macrolide immunomodulators |
| US5385910A (en) | 1993-11-22 | 1995-01-31 | American Home Products Corporation | Gem-distributed esters of rapamycin |
| US5385908A (en) | 1993-11-22 | 1995-01-31 | American Home Products Corporation | Hindered esters of rapamycin |
| US5385909A (en) | 1993-11-22 | 1995-01-31 | American Home Products Corporation | Heterocyclic esters of rapamycin |
| ATE191218T1 (de) | 1993-12-17 | 2000-04-15 | Novartis Ag | Rapamycin-derivate als immunosuppressoren |
| US5389639A (en) | 1993-12-29 | 1995-02-14 | American Home Products Company | Amino alkanoic esters of rapamycin |
| US5362718A (en) | 1994-04-18 | 1994-11-08 | American Home Products Corporation | Rapamycin hydroxyesters |
| US5463048A (en) | 1994-06-14 | 1995-10-31 | American Home Products Corporation | Rapamycin amidino carbamates |
| US5491231A (en) | 1994-11-28 | 1996-02-13 | American Home Products Corporation | Hindered N-oxide esters of rapamycin |
| US5563145A (en) | 1994-12-07 | 1996-10-08 | American Home Products Corporation | Rapamycin 42-oximes and hydroxylamines |
| US6323201B1 (en) | 1994-12-29 | 2001-11-27 | The Regents Of The University Of California | Compounds for inhibition of ceramide-mediated signal transduction |
| US5741677A (en) | 1995-06-07 | 1998-04-21 | Geron Corporation | Methods for measuring telomere length |
| ATE228135T1 (de) | 1995-06-09 | 2002-12-15 | Novartis Erfind Verwalt Gmbh | Rapamycinderivate |
| US5780462A (en) | 1995-12-27 | 1998-07-14 | American Home Products Corporation | Water soluble rapamycin esters |
| AU727708B2 (en) | 1996-06-20 | 2000-12-21 | Board Of Regents, The University Of Texas System | Compounds and methods for providing pharmacologically active preparations and uses thereof |
| EP1007042A4 (en) | 1997-06-13 | 2001-07-04 | Sugen Inc | NEW HETEROCYCLIC COMPOUNDS FOR MODULATING THE PROTEIN-TYROSIN-ENZYME-RELATED CELLULAR SIGNAL TRANSDUCTION |
| US6342507B1 (en) * | 1997-09-05 | 2002-01-29 | Isotechnika, Inc. | Deuterated rapamycin compounds, method and uses thereof |
| JP2002508971A (ja) | 1998-01-15 | 2002-03-26 | アリアド・ジーン・セラピューティクス・インコーポレーテッド | 多量体キメラ蛋白質を使用する生物学的イベントの調節 |
| IL147803A0 (en) | 1999-08-24 | 2002-08-14 | Ariad Gene Therapeutics Inc | 28-epirapalogs |
| US6277983B1 (en) | 2000-09-27 | 2001-08-21 | American Home Products Corporation | Regioselective synthesis of rapamycin derivatives |
| CA2994779C (en) | 2001-02-19 | 2020-08-25 | Novartis Ag | Use of 40-o-(2-hydroxyethyl)-rapamycin for inhibiting growth of a solid tumour of the brain other than lymphatic cancer |
| EP1539736A1 (en) | 2002-09-12 | 2005-06-15 | Pharmacia & Upjohn Company LLC | Substituted 1,4-pyrazine derivatives |
| JP2004161716A (ja) | 2002-11-15 | 2004-06-10 | Takeda Chem Ind Ltd | Jnk阻害剤 |
| US7160867B2 (en) * | 2003-05-16 | 2007-01-09 | Isotechnika, Inc. | Rapamycin carbohydrate derivatives |
| GB0314057D0 (en) | 2003-06-18 | 2003-07-23 | Astrazeneca Ab | Therapeutic agents |
| US7429596B2 (en) | 2003-06-20 | 2008-09-30 | The Regents Of The University Of California | 1H-pyrrolo [2,3-D] pyrimidine derivatives and methods of use thereof |
| CA2528173A1 (en) | 2003-07-16 | 2005-02-03 | Wyeth | Cci-779 isomer c |
| RU2006118325A (ru) | 2003-10-27 | 2007-12-10 | Астеллас Фарма Инк. (Jp) | Производные пиразина и их фармацевтическое применение |
| US7220755B2 (en) | 2003-11-12 | 2007-05-22 | Biosensors International Group, Ltd. | 42-O-alkoxyalkyl rapamycin derivatives and compositions comprising same |
| EP1571151B1 (en) | 2004-03-01 | 2008-12-24 | Terumo Kabushiki Kaisha | Process for production of O-alkylated rapamycin derivatives |
| ES2389907T3 (es) | 2004-04-30 | 2012-11-02 | Takeda Pharmaceutical Company Limited | Compuesto de amida heterocíclica y uso del mismo como un inhibidor de MMP-13 |
| CN100516195C (zh) | 2004-07-19 | 2009-07-22 | 汕头市双骏生物科技有限公司 | 一种新的菌株及应用该菌株生产紫杉醇的方法 |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| WO2006069038A1 (en) | 2004-12-20 | 2006-06-29 | Ariad Gene Therapeutics, Inc. | Therapeutic materials and methods |
| JP2008533007A (ja) | 2005-03-07 | 2008-08-21 | ワイス | 42−o−(2−ヒドロキシ)エチル−ラパマイシンのオキセパン異性体 |
| WO2006113704A2 (en) | 2005-04-18 | 2006-10-26 | Neurogen Corporation | Subtituted heteroaryl cb1 antagonists |
| US7189582B2 (en) | 2005-04-27 | 2007-03-13 | Dade Behring Inc. | Compositions and methods for detection of sirolimus |
| US7906692B2 (en) | 2005-05-20 | 2011-03-15 | Solvay (Societe Anonyme) | Method for making a chlorohydrin by chlorinating a polyhydroxylated aliphatic hydrocarbon |
| KR20080059399A (ko) | 2005-10-27 | 2008-06-27 | 코닌클리케 필립스 일렉트로닉스 엔.브이. | 심장 재동기 요법을 위한 더 좋은 도플러 조직 이미지 처리파형을 생성하기 위한 조직 가속도의 사용 |
| ATE507227T1 (de) | 2005-11-17 | 2011-05-15 | Osi Pharm Inc | Kondensierte bicyclische mtor-inhibitoren |
| EP1954269A2 (en) | 2005-11-21 | 2008-08-13 | Novartis AG | Neuroendocrine tumor treatment using mtor inhibitors |
| GB0525546D0 (en) * | 2005-12-15 | 2006-01-25 | Novartis Ag | Organic compounds |
| PT2004654E (pt) | 2006-04-04 | 2013-08-27 | Univ California | Derivados de pirazolopirimidina para utilização como antagonistas da quinase |
| US20090274739A1 (en) | 2006-04-13 | 2009-11-05 | The Trustees Of Columbia University In The City Of New York | Methods and compositions for treating neointimal hyperplasia |
| WO2007121453A2 (en) | 2006-04-17 | 2007-10-25 | The Regents Of The University Of California | 2-hydroxy-1-oxo 1,2 dihydro isoquinoline chelating agents |
| WO2008023161A1 (en) | 2006-08-23 | 2008-02-28 | Kudos Pharmaceuticals Limited | 2-methylmorpholine pyrido-, pyrazo- and pyrimido-pyrimidine derivatives as mtor inhibitors |
| EP2077267A4 (en) | 2006-10-18 | 2010-04-07 | Takeda Pharmaceutical | FUSED HETEROCYCLIC COMPOUND |
| EP1916006A1 (en) | 2006-10-19 | 2008-04-30 | Albert Schömig | Implant coated with a wax or a resin |
| EP2090580B1 (en) | 2006-11-27 | 2014-06-04 | Terumo Kabushiki Kaisha | Process for producing o-alkylated rapamycin derivative, and o-alkylated rapamycin derivative |
| US20080234262A1 (en) | 2007-03-21 | 2008-09-25 | Wyeth | Pyrazolopyrimidine analogs and their use as mtor kinase and pi3 kinase inhibitors |
| CA2678415A1 (en) | 2007-04-05 | 2008-10-16 | Wyeth | Wortmannin-rapamycin conjugate and uses thereof |
| JPWO2008156174A1 (ja) | 2007-06-21 | 2010-08-26 | 大正製薬株式会社 | ピラジンアミド化合物 |
| US20090074831A1 (en) | 2007-09-18 | 2009-03-19 | Robert Falotico | LOCAL VASCULAR DELIVERY OF mTOR INHIBITORS IN COMBINATION WITH PEROXISOME PROLIFERATORS-ACTIVATED RECEPTOR STIMULATORS |
| WO2009046436A1 (en) | 2007-10-04 | 2009-04-09 | Memorial Sloan-Kettering Cancer Center | Methods for inhibiting senescence of epithelial cells |
| MY159955A (en) | 2008-01-04 | 2017-02-15 | Intellikine Inc | Certain chemical entities, compositions and methods |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US7842475B2 (en) | 2008-01-08 | 2010-11-30 | Siemens Healthcare Diagnostics Inc. | Stabilization of solid support assay reagents |
| WO2009114874A2 (en) | 2008-03-14 | 2009-09-17 | Intellikine, Inc. | Benzothiazole kinase inhibitors and methods of use |
| US20090253733A1 (en) * | 2008-04-02 | 2009-10-08 | Biointeractions, Ltd. | Rapamycin carbonate esters |
| US20110098241A1 (en) * | 2008-04-14 | 2011-04-28 | Poniard Pharmaceuticals, Inc. | Rapamycin analogs as anti-cancer agents |
| TW200948361A (en) | 2008-05-26 | 2009-12-01 | Chunghwa Chemical Synthesis & Biotech Co Ltd | Method for synthesizing Biolimus A9 and the like and method for improving stability of the same |
| US20110224223A1 (en) | 2008-07-08 | 2011-09-15 | The Regents Of The University Of California, A California Corporation | MTOR Modulators and Uses Thereof |
| KR20110039326A (ko) | 2008-07-08 | 2011-04-15 | 인텔리카인, 인크. | 키나제 억제제 및 사용 방법 |
| US20100055145A1 (en) | 2008-08-29 | 2010-03-04 | Biosensors International Group | Stent coatings for reducing late stent thrombosis |
| CA2738429C (en) | 2008-09-26 | 2016-10-25 | Intellikine, Inc. | Heterocyclic kinase inhibitors |
| DK2358720T3 (en) | 2008-10-16 | 2016-06-06 | Univ California | Heteroarylkinaseinhibitorer fused-ring |
| EP2350053B1 (en) | 2008-10-17 | 2013-12-11 | Whitehead Institute For Biomedical Research | Modulators of MTOR Complexes |
| US8110578B2 (en) | 2008-10-27 | 2012-02-07 | Signal Pharmaceuticals, Llc | Pyrazino[2,3-b]pyrazine mTOR kinase inhibitors for oncology indications and diseases associated with the mTOR/PI3K/Akt pathway |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| WO2010059593A1 (en) | 2008-11-18 | 2010-05-27 | Intellikine, Inc. | Methods and compositions for treatment of ophthalmic conditions |
| CA2760791C (en) | 2009-05-07 | 2017-06-20 | Intellikine, Inc. | Heterocyclic compounds and uses thereof |
| KR20120087808A (ko) | 2009-05-26 | 2012-08-07 | 엑셀리시스, 인코포레이티드 | PI3K/m TOR의 억제제로서의 벤즈옥사제핀 및 이의 사용 및 제조 방법 |
| MX2012002066A (es) | 2009-08-17 | 2012-03-29 | Intellikine Inc | Compuestos heterociclicos y usos de los mismos. |
| EP2467142B1 (en) | 2009-08-17 | 2016-09-21 | Memorial Sloan-Kettering Cancer Center | 2-(Pyrimidin-5-yl)-thiopyrimidine derivatives as Hsp70 and Hsc70 modulators for the treatment of proliferative disorders |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US8604032B2 (en) | 2010-05-21 | 2013-12-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9321782B2 (en) | 2010-08-04 | 2016-04-26 | Meril Life Sciences Pvt. Ltd. | Process for preparation of novel 42-O-(heteroalkoxyalkyl) rapamicin compounds with anti-proliferative properties |
| JP5847386B2 (ja) | 2010-09-15 | 2016-01-20 | 関東化學株式会社 | アミン化合物の製造方法 |
| GB201106829D0 (en) | 2011-04-21 | 2011-06-01 | Proximagen Ltd | Heterocyclic compounds |
| ES2753529T3 (es) | 2010-11-19 | 2020-04-13 | Biocon Ltd | Procesos para la preparación de everolimús y productos intermedios del mismo |
| WO2012103959A1 (en) | 2011-02-04 | 2012-08-09 | Synthon Bv | Process for making everolimus |
| EP2678018A4 (en) | 2011-02-23 | 2015-09-30 | Intellikine Llc | COMBINATION OF CHINESE HEMMER AND USES THEREOF |
| WO2012151562A1 (en) | 2011-05-04 | 2012-11-08 | Intellikine, Llc | Combination pharmaceutical compositions and uses thereof |
| CA2835197A1 (en) | 2011-05-06 | 2012-11-15 | Intellikine Llc | Treatment of polycystic disease |
| WO2013023119A1 (en) | 2011-08-10 | 2013-02-14 | Novartis Pharma Ag | JAK P13K/mTOR COMBINATION THERAPY |
| CA2846496C (en) | 2011-09-02 | 2020-07-14 | The Regents Of The University Of California | Substituted pyrazolo[3,4-d]pyrimidines and uses thereof |
| WO2013105063A1 (en) | 2012-01-13 | 2013-07-18 | Novartis Ag | Fused piperidines as ip receptor agonists for the treatment of pulmonary arterial hypertension (pah) and related disorders |
| SG11201404432QA (en) | 2012-11-30 | 2014-10-30 | Hangzhou Zylox Pharma Co Ltd | Rafamycin analogs and methods for making same |
| US20150374687A1 (en) | 2013-02-07 | 2015-12-31 | Merck Patent Gmbh | Substituted quinoxaline derivatives and their use as positive allosteric modulators of mglur4 |
| WO2015066371A1 (en) | 2013-10-31 | 2015-05-07 | Forum Pharmaceuticals, Inc. | SPIRO-OXADIAZOLINE COMPOUNDS AS AGONISTS OF α-7-NICOTINIC ACETYLCHOLINE RECEPTORS |
| KR20220127364A (ko) | 2013-12-19 | 2022-09-19 | 씨젠 인크. | 표적화된-약물 컨쥬게이트와 함께 사용되는 메틸렌 카바메이트 링커 |
| JO3517B1 (ar) | 2014-01-17 | 2020-07-05 | Novartis Ag | ان-ازاسبيرو الكان حلقي كبديل مركبات اريل-ان مغايرة وتركيبات لتثبيط نشاط shp2 |
| CN105899491B (zh) | 2014-01-17 | 2019-04-02 | 诺华股份有限公司 | 用于抑制shp2活性的1-哒嗪-/三嗪-3-基-哌(-嗪)/啶/吡咯烷衍生物及其组合物 |
| CN105899493B (zh) | 2014-01-17 | 2019-03-29 | 诺华股份有限公司 | 用于抑制shp2活性的1-(三嗪-3-基/哒嗪-3-基)-哌(-嗪)啶衍生物及其组合物 |
| CN105461738B (zh) | 2014-06-03 | 2019-03-08 | 中国人民解放军军事医学科学院毒物药物研究所 | 一种雷帕霉素衍生物、其制备方法、其药物组合物及用途 |
| KR101668590B1 (ko) | 2014-06-19 | 2016-10-25 | 전남대학교병원 | 재협착 억제 및 재내피화 촉진을 위한 신규한 펩타이드 화합물 및 이의 제조방법 |
| MX2017001981A (es) | 2014-09-11 | 2017-12-12 | Univ California | Inhibidores de diana de rapamicina en celulas de mamifero de complejo 1 (mtorc1). |
| EP3234607B1 (en) | 2014-12-17 | 2021-04-28 | Siemens Healthcare Diagnostics Inc. | Sandwich assay design for small molecules |
| US20160279108A1 (en) * | 2015-02-24 | 2016-09-29 | University Of Kansas | Targeted mtor inhibitors |
| CN106188093B (zh) * | 2015-05-08 | 2018-06-12 | 上海医药工业研究院 | 一种雷帕霉素结构类似物及其制备方法 |
| EP3310774B1 (en) | 2015-06-19 | 2020-04-29 | Novartis AG | Compounds and compositions for inhibiting the activity of shp2 |
| US10683308B2 (en) * | 2015-09-11 | 2020-06-16 | Navitor Pharmaceuticals, Inc. | Rapamycin analogs and uses thereof |
| SG11201805869XA (en) | 2016-01-11 | 2018-08-30 | Merck Patent Gmbh | Quinolin-2-one derivatives |
| US10851071B2 (en) | 2016-07-21 | 2020-12-01 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | M-dihydroxybenzene derivative crystal and salt, and manufacturing method thereof |
| CN110770243A (zh) | 2017-05-02 | 2020-02-07 | 锐新医药公司 | 作为mtor抑制剂的雷帕霉素类似物 |
| UY37900A (es) * | 2017-09-26 | 2019-04-30 | Novartis Ag | Nuevos derivados de rapamicina |
| EP3788050B1 (en) | 2018-05-01 | 2024-08-28 | Revolution Medicines, Inc. | C26-linked rapamycin analogs as mtor inhibitors |
| TW202014208A (zh) | 2018-05-01 | 2020-04-16 | 美商銳新醫藥公司 | 作為mTOR抑制劑之C40-、C28-及C-32連接之雷帕黴素類似物 |
| KR20210125025A (ko) | 2019-02-07 | 2021-10-15 | 베이진 엘티디 | Tlr7 작용제로서의 이미다조[2,1-f][1,2,4]트리아진-4-아민 유도체 |
| WO2021257736A1 (en) | 2020-06-18 | 2021-12-23 | Revolution Medicines, Inc. | Methods for delaying, preventing, and treating acquired resistance to ras inhibitors |
| CN117412954A (zh) | 2021-04-09 | 2024-01-16 | 锐新医药公司 | 雷帕霉素类似物化合物的合成 |
-
2019
- 2019-04-29 TW TW108114977A patent/TW202014208A/zh unknown
- 2019-04-29 SI SI201930546T patent/SI3788049T1/sl unknown
- 2019-04-29 HR HRP20230488TT patent/HRP20230488T1/hr unknown
- 2019-04-29 PT PT197235716T patent/PT3788049T/pt unknown
- 2019-04-29 PL PL19723571.6T patent/PL3788049T3/pl unknown
- 2019-04-29 IL IL300091A patent/IL300091A/en unknown
- 2019-04-29 MX MX2020011565A patent/MX2020011565A/es unknown
- 2019-04-29 PE PE2020001763A patent/PE20212112A1/es unknown
- 2019-04-29 EP EP19723571.6A patent/EP3788049B1/en active Active
- 2019-04-29 CN CN201980044255.9A patent/CN112771054B/zh active Active
- 2019-04-29 AU AU2019262978A patent/AU2019262978B2/en active Active
- 2019-04-29 WO PCT/US2019/029737 patent/WO2019212990A1/en active Application Filing
- 2019-04-29 FI FIEP19723571.6T patent/FI3788049T3/fi active
- 2019-04-29 LT LTEPPCT/US2019/029737T patent/LT3788049T/lt unknown
- 2019-04-29 SG SG11202010559UA patent/SG11202010559UA/en unknown
- 2019-04-29 ES ES19723571T patent/ES2945560T3/es active Active
- 2019-04-29 DK DK19723571.6T patent/DK3788049T3/da active
- 2019-04-29 BR BR112020022201-1A patent/BR112020022201A2/pt unknown
- 2019-04-29 EP EP23164819.7A patent/EP4234031A3/en active Pending
- 2019-04-29 JP JP2020561074A patent/JP7358387B2/ja active Active
- 2019-04-29 US US16/398,011 patent/US20190336609A1/en not_active Abandoned
- 2019-04-29 HU HUE19723571A patent/HUE062352T2/hu unknown
- 2019-04-29 CR CR20200578A patent/CR20200578A/es unknown
- 2019-04-29 CA CA3098692A patent/CA3098692A1/en active Pending
- 2019-04-29 CR CR20230103A patent/CR20230103A/es unknown
- 2019-04-29 KR KR1020207034117A patent/KR20210018244A/ko unknown
- 2019-04-29 RS RS20230386A patent/RS64272B1/sr unknown
-
2020
- 2020-03-26 US US16/831,579 patent/US10980889B1/en active Active
- 2020-10-27 IL IL278334A patent/IL278334B2/en unknown
- 2020-10-28 PH PH12020551795A patent/PH12020551795A1/en unknown
- 2020-10-28 US US17/083,172 patent/US11364300B2/en active Active
- 2020-10-30 CL CL2020002827A patent/CL2020002827A1/es unknown
- 2020-10-30 MX MX2023000081A patent/MX2023000081A/es unknown
- 2020-10-30 MX MX2023011282A patent/MX2023011282A/es unknown
- 2020-11-01 SA SA520420458A patent/SA520420458B1/ar unknown
- 2020-11-06 CO CONC2020/0013865A patent/CO2020013865A2/es unknown
-
2022
- 2022-04-29 US US17/733,755 patent/US12048749B2/en active Active
-
2023
- 2023-09-27 JP JP2023164097A patent/JP2023182654A/ja active Pending
- 2023-10-05 AU AU2023241345A patent/AU2023241345A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2945560T3 (es) | Análogos de rapamicina unidos en C40, C28 y C-32 como inhibidores de mTOR | |
| JP7381492B2 (ja) | Mtor阻害剤としてのc26-連結ラパマイシン類似体 | |
| JP7348071B2 (ja) | mTOR阻害剤としてのラパマイシン類似体 | |
| US11739093B2 (en) | Substituted pyrazolopyrazines, imidazopyrazines and [1,2,4]triazolopyrazines as allosteric SHP2 inhibitors | |
| RU2826559C2 (ru) | C26-СВЯЗАННЫЕ АНАЛОГИ РАПАМИЦИНА В КАЧЕСТВЕ ИНГИБИТОРОВ mTOR | |
| RU2805211C2 (ru) | С40-, с28- и с-32-связанные аналоги рапамицина в качестве ингибиторов mtor | |
| KR20240148007A (ko) | E3 유비퀴틴 리가제 리간드 (ubiquitin ligase ligand) 화합물, 이 리간드 화합물을 기초로 하여 개발된 단백질 분해제 및 이들의 응용 |