ES2878087T3 - Análogos de carba-nucleósido 2-sustituidos para el tratamiento antiviral - Google Patents
Análogos de carba-nucleósido 2-sustituidos para el tratamiento antiviral Download PDFInfo
- Publication number
- ES2878087T3 ES2878087T3 ES18155061T ES18155061T ES2878087T3 ES 2878087 T3 ES2878087 T3 ES 2878087T3 ES 18155061 T ES18155061 T ES 18155061T ES 18155061 T ES18155061 T ES 18155061T ES 2878087 T3 ES2878087 T3 ES 2878087T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- pharmaceutically acceptable
- acceptable salt
- formula
- orb
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000011282 treatment Methods 0.000 title description 19
- 239000002777 nucleoside Substances 0.000 title description 7
- 230000000840 anti-viral effect Effects 0.000 title description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 168
- 150000003839 salts Chemical class 0.000 claims abstract description 63
- 238000000034 method Methods 0.000 claims abstract description 51
- 208000009620 Orthomyxoviridae Infections Diseases 0.000 claims abstract description 29
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 24
- 125000003118 aryl group Chemical group 0.000 claims abstract description 21
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 17
- 125000003710 aryl alkyl group Chemical group 0.000 claims abstract description 16
- 229910052794 bromium Inorganic materials 0.000 claims abstract description 14
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 14
- 150000002367 halogens Chemical class 0.000 claims abstract description 14
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 13
- 241000124008 Mammalia Species 0.000 claims abstract description 11
- 125000005347 halocycloalkyl group Chemical group 0.000 claims abstract description 11
- 125000005843 halogen group Chemical group 0.000 claims abstract description 11
- 229910052740 iodine Inorganic materials 0.000 claims abstract description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 10
- 125000006648 (C1-C8) haloalkyl group Chemical group 0.000 claims abstract description 8
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims abstract description 8
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 8
- 125000001424 substituent group Chemical group 0.000 claims abstract description 7
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 claims abstract description 6
- 125000000547 substituted alkyl group Chemical group 0.000 claims abstract description 6
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims abstract description 5
- 125000004648 C2-C8 alkenyl group Chemical group 0.000 claims abstract description 4
- 125000005017 substituted alkenyl group Chemical group 0.000 claims abstract description 4
- 125000004426 substituted alkynyl group Chemical group 0.000 claims abstract description 4
- 125000004649 C2-C8 alkynyl group Chemical group 0.000 claims abstract description 3
- MPVDXIMFBOLMNW-UHFFFAOYSA-N chembl1615565 Chemical compound OC1=CC=C2C=C(S(O)(=O)=O)C=C(S(O)(=O)=O)C2=C1N=NC1=CC=CC=C1 MPVDXIMFBOLMNW-UHFFFAOYSA-N 0.000 claims abstract description 3
- 239000003814 drug Substances 0.000 claims description 69
- 239000000203 mixture Substances 0.000 claims description 50
- 241000712464 Orthomyxoviridae Species 0.000 claims description 48
- 229940124597 therapeutic agent Drugs 0.000 claims description 46
- 239000003112 inhibitor Substances 0.000 claims description 26
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 21
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 claims description 13
- 229910052739 hydrogen Inorganic materials 0.000 claims description 13
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 13
- 239000003795 chemical substances by application Substances 0.000 claims description 11
- 208000015181 infectious disease Diseases 0.000 claims description 11
- 230000003612 virological effect Effects 0.000 claims description 10
- 102000005348 Neuraminidase Human genes 0.000 claims description 8
- 108010006232 Neuraminidase Proteins 0.000 claims description 8
- 239000003730 rna directed rna polymerase inhibitor Substances 0.000 claims description 8
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 claims description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 7
- 239000003172 expectorant agent Substances 0.000 claims description 7
- UBCHPRBFMUDMNC-UHFFFAOYSA-N 1-(1-adamantyl)ethanamine Chemical compound C1C(C2)CC3CC2CC1(C(N)C)C3 UBCHPRBFMUDMNC-UHFFFAOYSA-N 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 108010093857 Viral Hemagglutinins Proteins 0.000 claims description 6
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 claims description 6
- 229960003805 amantadine Drugs 0.000 claims description 6
- 230000003110 anti-inflammatory effect Effects 0.000 claims description 6
- 229940124630 bronchodilator Drugs 0.000 claims description 6
- ZCGNOVWYSGBHAU-UHFFFAOYSA-N favipiravir Chemical compound NC(=O)C1=NC(F)=CNC1=O ZCGNOVWYSGBHAU-UHFFFAOYSA-N 0.000 claims description 6
- 229950008454 favipiravir Drugs 0.000 claims description 6
- 239000001257 hydrogen Substances 0.000 claims description 6
- 229960003752 oseltamivir Drugs 0.000 claims description 6
- 229960000888 rimantadine Drugs 0.000 claims description 6
- ARAIBEBZBOPLMB-UFGQHTETSA-N zanamivir Chemical compound CC(=O)N[C@@H]1[C@@H](N=C(N)N)C=C(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO ARAIBEBZBOPLMB-UFGQHTETSA-N 0.000 claims description 6
- 229960001028 zanamivir Drugs 0.000 claims description 6
- QNRRHYPPQFELSF-CNYIRLTGSA-N Laninamivir Chemical compound OC[C@@H](O)[C@@H](OC)[C@@H]1OC(C(O)=O)=C[C@H](N=C(N)N)[C@H]1NC(C)=O QNRRHYPPQFELSF-CNYIRLTGSA-N 0.000 claims description 5
- 239000000048 adrenergic agonist Substances 0.000 claims description 5
- 239000003246 corticosteroid Substances 0.000 claims description 5
- 229950004244 laninamivir Drugs 0.000 claims description 5
- UKTIJASCFRNWCB-RMIBSVFLSA-N laninamivir octanoate hydrate Chemical compound CCCCCCCC(=O)OC[C@@H](O)[C@@H](OC)[C@@H]1OC(C(O)=O)=C[C@H](N=C(N)N)[C@H]1NC(C)=O UKTIJASCFRNWCB-RMIBSVFLSA-N 0.000 claims description 5
- 229950005327 laninamivir octanoate hydrate Drugs 0.000 claims description 5
- 229940066491 mucolytics Drugs 0.000 claims description 5
- 108700013356 oplunofusp Proteins 0.000 claims description 5
- XRQDFNLINLXZLB-CKIKVBCHSA-N peramivir Chemical compound CCC(CC)[C@H](NC(C)=O)[C@@H]1[C@H](O)[C@@H](C(O)=O)C[C@H]1NC(N)=N XRQDFNLINLXZLB-CKIKVBCHSA-N 0.000 claims description 5
- 229960001084 peramivir Drugs 0.000 claims description 5
- 229960000329 ribavirin Drugs 0.000 claims description 5
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 claims description 5
- 230000019491 signal transduction Effects 0.000 claims description 5
- 241000712431 Influenza A virus Species 0.000 claims description 4
- 241000713196 Influenza B virus Species 0.000 claims description 4
- 241000713297 Influenza C virus Species 0.000 claims description 4
- 102000014150 Interferons Human genes 0.000 claims description 4
- 108010050904 Interferons Proteins 0.000 claims description 4
- 229940126157 adrenergic receptor agonist Drugs 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 210000004969 inflammatory cell Anatomy 0.000 claims description 4
- 238000013508 migration Methods 0.000 claims description 4
- 230000005012 migration Effects 0.000 claims description 4
- 230000000770 proinflammatory effect Effects 0.000 claims description 4
- UGGITRSRFHZMKU-WEIZIPMPSA-N radavirsen Chemical compound N1([C@H]2CN(C[C@H](O2)COP(=O)(N(C)C)N2CCN(CC2)C(=O)OCCOCCOCCO)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CN(C2)P(=O)(OC[C@H]2O[C@H](CNC2)N2C(NC(=O)C(C)=C2)=O)N(C)C)N2C(NC(=O)C(C)=C2)=O)N(C)C)N2C(NC(=O)C(C)=C2)=O)N2CCNCC2)N2C(N=C(N)C=C2)=O)N(C)C)N2C(NC(=O)C(C)=C2)=O)N(C)C)N2C3=NC=NC(N)=C3N=C2)N(C)C)N2C(N=C(N)C=C2)=O)N(C)C)N2C(NC(=O)C(C)=C2)=O)N2CCNCC2)N2C(N=C(N)C=C2)=O)N(C)C)N2C3=NC=NC(N)=C3N=C2)N(C)C)N2C3=C(C(NC(N)=N3)=O)N=C2)N(C)C)N2C3=NC=NC(N)=C3N=C2)N(C)C)N2C3=NC=NC(N)=C3N=C2)N(C)C)N2C3=C(C(NC(N)=N3)=O)N=C2)N(C)C)N2C3=NC=NC(N)=C3N=C2)N(C)C)N2C(NC(=O)C(C)=C2)=O)N2CCNCC2)N2C(NC(=O)C(C)=C2)=O)N(C)C)N2C3=C(C(NC(N)=N3)=O)N=C2)N(C)C)N2C3=C(C(NC(N)=N3)=O)N=C2)N(C)C)C=CC(N)=NC1=O UGGITRSRFHZMKU-WEIZIPMPSA-N 0.000 claims description 4
- 229910052717 sulfur Inorganic materials 0.000 claims description 4
- 229910003204 NH2 Inorganic materials 0.000 claims description 3
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 claims description 3
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 claims description 3
- 230000001078 anti-cholinergic effect Effects 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 238000002663 nebulization Methods 0.000 claims description 3
- JNCMHMUGTWEVOZ-UHFFFAOYSA-N F[CH]F Chemical compound F[CH]F JNCMHMUGTWEVOZ-UHFFFAOYSA-N 0.000 claims description 2
- 108010081348 HRT1 protein Hairy Proteins 0.000 claims description 2
- 102100021881 Hairy/enhancer-of-split related with YRPW motif protein 1 Human genes 0.000 claims description 2
- VUWZPRWSIVNGKG-UHFFFAOYSA-N fluoromethane Chemical compound F[CH2] VUWZPRWSIVNGKG-UHFFFAOYSA-N 0.000 claims description 2
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 2
- 229940079322 interferon Drugs 0.000 claims description 2
- 229940126181 ion channel inhibitor Drugs 0.000 claims description 2
- 239000000076 hypertonic saline solution Substances 0.000 claims 1
- VSZGPKBBMSAYNT-RRFJBIMHSA-N oseltamivir Chemical compound CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 VSZGPKBBMSAYNT-RRFJBIMHSA-N 0.000 claims 1
- -1 [1,2,4] triazinyl Chemical group 0.000 description 58
- 239000008194 pharmaceutical composition Substances 0.000 description 57
- 239000004480 active ingredient Substances 0.000 description 43
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 40
- 238000006243 chemical reaction Methods 0.000 description 31
- 239000000243 solution Substances 0.000 description 31
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical compound C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 description 28
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 27
- 125000004432 carbon atom Chemical group C* 0.000 description 27
- 239000000651 prodrug Substances 0.000 description 23
- 229940002612 prodrug Drugs 0.000 description 23
- 229940079593 drug Drugs 0.000 description 22
- 239000000843 powder Substances 0.000 description 22
- 206010022000 influenza Diseases 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 20
- BOUIZPWBFIVJPD-UHFFFAOYSA-N 3h-1,2,4-triazin-4-amine Chemical compound NN1CN=NC=C1 BOUIZPWBFIVJPD-UHFFFAOYSA-N 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- 239000002245 particle Substances 0.000 description 17
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 16
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 16
- 239000007787 solid Substances 0.000 description 16
- 210000004027 cell Anatomy 0.000 description 15
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 14
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 14
- 239000000443 aerosol Substances 0.000 description 14
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- 230000009385 viral infection Effects 0.000 description 12
- 230000000694 effects Effects 0.000 description 11
- 239000007788 liquid Substances 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- 239000007832 Na2SO4 Substances 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- 241000700605 Viruses Species 0.000 description 9
- 150000002148 esters Chemical class 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 229910052938 sodium sulfate Inorganic materials 0.000 description 9
- 235000011152 sodium sulphate Nutrition 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- QMNWYGTWTXOQTP-UHFFFAOYSA-N 1h-triazin-6-one Chemical compound O=C1C=CN=NN1 QMNWYGTWTXOQTP-UHFFFAOYSA-N 0.000 description 8
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 8
- 239000013078 crystal Substances 0.000 description 8
- 239000003995 emulsifying agent Substances 0.000 description 8
- 235000019198 oils Nutrition 0.000 description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 8
- 241000282414 Homo sapiens Species 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- 239000000839 emulsion Substances 0.000 description 7
- 238000003818 flash chromatography Methods 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000000670 limiting effect Effects 0.000 description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 7
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 6
- 206010062106 Respiratory tract infection viral Diseases 0.000 description 6
- WUMYBFXWUJTWTB-UHFFFAOYSA-N [hydroxy(methoxy)phosphoryl] phosphono hydrogen phosphate Chemical compound COP(O)(=O)OP(O)(=O)OP(O)(O)=O WUMYBFXWUJTWTB-UHFFFAOYSA-N 0.000 description 6
- 125000003342 alkenyl group Chemical group 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 239000011575 calcium Substances 0.000 description 6
- 239000006071 cream Substances 0.000 description 6
- 239000003085 diluting agent Substances 0.000 description 6
- 239000000796 flavoring agent Substances 0.000 description 6
- 125000000524 functional group Chemical group 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- 239000006199 nebulizer Substances 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 238000004806 packaging method and process Methods 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000003765 sweetening agent Substances 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- 238000002560 therapeutic procedure Methods 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 5
- 0 CC*C1NN(C)C(CC=C[C@](*C([C@]2OCc3ccccc3)=COCc3ccccc3)[C@@]2F)C(N)N1 Chemical compound CC*C1NN(C)C(CC=C[C@](*C([C@]2OCc3ccccc3)=COCc3ccccc3)[C@@]2F)C(N)N1 0.000 description 5
- 108090000862 Ion Channels Proteins 0.000 description 5
- 102000004310 Ion Channels Human genes 0.000 description 5
- 108060004795 Methyltransferase Proteins 0.000 description 5
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical group CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 235000014113 dietary fatty acids Nutrition 0.000 description 5
- 239000002270 dispersing agent Substances 0.000 description 5
- 229940112141 dry powder inhaler Drugs 0.000 description 5
- 239000003925 fat Substances 0.000 description 5
- 235000019197 fats Nutrition 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 229930195729 fatty acid Natural products 0.000 description 5
- 150000004665 fatty acids Chemical class 0.000 description 5
- 235000011187 glycerol Nutrition 0.000 description 5
- 239000008187 granular material Substances 0.000 description 5
- 125000001188 haloalkyl group Chemical group 0.000 description 5
- 239000004615 ingredient Substances 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 5
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- NENPYTRHICXVCS-YNEHKIRRSA-N oseltamivir acid Chemical compound CCC(CC)O[C@@H]1C=C(C(O)=O)C[C@H](N)[C@H]1NC(C)=O NENPYTRHICXVCS-YNEHKIRRSA-N 0.000 description 5
- 239000003755 preservative agent Substances 0.000 description 5
- 239000000375 suspending agent Substances 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 101100296719 Caenorhabditis elegans pde-4 gene Proteins 0.000 description 4
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 4
- 239000004215 Carbon black (E152) Substances 0.000 description 4
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 4
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 206010061218 Inflammation Diseases 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 4
- 240000007472 Leucaena leucocephala Species 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 206010057190 Respiratory tract infections Diseases 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- 125000000304 alkynyl group Chemical group 0.000 description 4
- 239000007900 aqueous suspension Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 239000007859 condensation product Substances 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- 238000013461 design Methods 0.000 description 4
- 230000002255 enzymatic effect Effects 0.000 description 4
- 239000006260 foam Substances 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 235000003599 food sweetener Nutrition 0.000 description 4
- 229930195733 hydrocarbon Natural products 0.000 description 4
- 150000002430 hydrocarbons Chemical class 0.000 description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 229940125369 inhaled corticosteroids Drugs 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 229940057995 liquid paraffin Drugs 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 230000002503 metabolic effect Effects 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- 210000000214 mouth Anatomy 0.000 description 4
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 125000003835 nucleoside group Chemical group 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 239000005720 sucrose Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- QOVLJBFQXNGGDV-UHFFFAOYSA-N 1,3-dihydro-1,2,4-triazine-2,4-diamine Chemical compound NN1CN(N)C=CN1 QOVLJBFQXNGGDV-UHFFFAOYSA-N 0.000 description 3
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 3
- 241000416162 Astragalus gummifer Species 0.000 description 3
- 206010006482 Bronchospasm Diseases 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 235000019483 Peanut oil Nutrition 0.000 description 3
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 235000006708 antioxidants Nutrition 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 230000007885 bronchoconstriction Effects 0.000 description 3
- 239000000168 bronchodilator agent Substances 0.000 description 3
- 239000001506 calcium phosphate Substances 0.000 description 3
- 229910000389 calcium phosphate Inorganic materials 0.000 description 3
- 235000011010 calcium phosphates Nutrition 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 238000013270 controlled release Methods 0.000 description 3
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- UTCSSFWDNNEEBH-UHFFFAOYSA-N imidazo[1,2-a]pyridine Chemical compound C1=CC=CC2=NC=CN21 UTCSSFWDNNEEBH-UHFFFAOYSA-N 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 239000003701 inert diluent Substances 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 239000007937 lozenge Substances 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 229940071648 metered dose inhaler Drugs 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 3
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 239000000312 peanut oil Substances 0.000 description 3
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- 210000002345 respiratory system Anatomy 0.000 description 3
- 125000006413 ring segment Chemical group 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 241000712461 unidentified influenza virus Species 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- HEYCIUFBODNURY-KZNAEPCWSA-N (3r,4r,5r)-3-fluoro-4-phenylmethoxy-5-(phenylmethoxymethyl)oxolan-2-one Chemical compound O([C@H]1[C@H](C(O[C@@H]1COCC=1C=CC=CC=1)=O)F)CC1=CC=CC=C1 HEYCIUFBODNURY-KZNAEPCWSA-N 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 2
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- 125000004398 2-methyl-2-butyl group Chemical group CC(C)(CC)* 0.000 description 2
- 125000004918 2-methyl-2-pentyl group Chemical group CC(C)(CCC)* 0.000 description 2
- 125000004922 2-methyl-3-pentyl group Chemical group CC(C)C(CC)* 0.000 description 2
- 125000004917 3-methyl-2-butyl group Chemical group CC(C(C)*)C 0.000 description 2
- 125000004919 3-methyl-2-pentyl group Chemical group CC(C(C)*)CC 0.000 description 2
- 125000004921 3-methyl-3-pentyl group Chemical group CC(CC)(CC)* 0.000 description 2
- 125000004920 4-methyl-2-pentyl group Chemical group CC(CC(C)*)C 0.000 description 2
- IFGWYHGYNVGVRB-UHFFFAOYSA-N 5-(2,4-difluorophenoxy)-n-[2-(dimethylamino)ethyl]-1-(2-methylpropyl)indazole-6-carboxamide Chemical compound CN(C)CCNC(=O)C=1C=C2N(CC(C)C)N=CC2=CC=1OC1=CC=C(F)C=C1F IFGWYHGYNVGVRB-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- 206010006448 Bronchiolitis Diseases 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 2
- 239000005977 Ethylene Substances 0.000 description 2
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical class C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 239000012826 P38 inhibitor Substances 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- RUOGJYKOQBFJIG-UHFFFAOYSA-N SCH-351591 Chemical compound C12=CC=C(C(F)(F)F)N=C2C(OC)=CC=C1C(=O)NC1=C(Cl)C=[N+]([O-])C=C1Cl RUOGJYKOQBFJIG-UHFFFAOYSA-N 0.000 description 2
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 229910000831 Steel Inorganic materials 0.000 description 2
- 241000282898 Sus scrofa Species 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 230000007815 allergy Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical class C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 229940065524 anticholinergics inhalants for obstructive airway diseases Drugs 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- SLJXUJZUTPFXRJ-UHFFFAOYSA-N buzepide Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C(=O)N)CCN1CCCCCC1 SLJXUJZUTPFXRJ-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L calcium carbonate Substances [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- CTZOPIRFQQMWGR-UHFFFAOYSA-N carboxyoxymethyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OCOC(O)=O CTZOPIRFQQMWGR-UHFFFAOYSA-N 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000000812 cholinergic antagonist Substances 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000011260 co-administration Methods 0.000 description 2
- 239000012084 conversion product Substances 0.000 description 2
- 229960001334 corticosteroids Drugs 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical class C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 229940088679 drug related substance Drugs 0.000 description 2
- 229940121647 egfr inhibitor Drugs 0.000 description 2
- 210000002919 epithelial cell Anatomy 0.000 description 2
- 230000003419 expectorant effect Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 229960002848 formoterol Drugs 0.000 description 2
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000003862 glucocorticoid Substances 0.000 description 2
- 229940075507 glyceryl monostearate Drugs 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- AFQIYTIJXGTIEY-UHFFFAOYSA-N hydrogen carbonate;triethylazanium Chemical compound OC(O)=O.CCN(CC)CC AFQIYTIJXGTIEY-UHFFFAOYSA-N 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229940047124 interferons Drugs 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000004255 ion exchange chromatography Methods 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 229940043355 kinase inhibitor Drugs 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 238000003801 milling Methods 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 125000001620 monocyclic carbocycle group Chemical group 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- DPHDSIQHVGSITN-UHFFFAOYSA-N n-(3,5-dichloropyridin-4-yl)-2-[1-[(4-fluorophenyl)methyl]-5-hydroxyindol-3-yl]-2-oxoacetamide Chemical compound C1=C(C(=O)C(=O)NC=2C(=CN=CC=2Cl)Cl)C2=CC(O)=CC=C2N1CC1=CC=C(F)C=C1 DPHDSIQHVGSITN-UHFFFAOYSA-N 0.000 description 2
- OKFDRAHPFKMAJH-UHFFFAOYSA-N n-(3,5-dichloropyridin-4-yl)-4-(difluoromethoxy)-8-(methanesulfonamido)dibenzofuran-1-carboxamide Chemical compound C=12C3=CC(NS(=O)(=O)C)=CC=C3OC2=C(OC(F)F)C=CC=1C(=O)NC1=C(Cl)C=NC=C1Cl OKFDRAHPFKMAJH-UHFFFAOYSA-N 0.000 description 2
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000003883 ointment base Substances 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 2
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 230000002335 preservative effect Effects 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 150000008223 ribosides Chemical class 0.000 description 2
- MNDBXUUTURYVHR-UHFFFAOYSA-N roflumilast Chemical compound FC(F)OC1=CC=C(C(=O)NC=2C(=CN=CC=2Cl)Cl)C=C1OCC1CC1 MNDBXUUTURYVHR-UHFFFAOYSA-N 0.000 description 2
- 229960004017 salmeterol Drugs 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 230000001932 seasonal effect Effects 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910000162 sodium phosphate Inorganic materials 0.000 description 2
- 235000011008 sodium phosphates Nutrition 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000007921 spray Substances 0.000 description 2
- 238000001694 spray drying Methods 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 239000010959 steel Substances 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- HLZKNKRTKFSKGZ-UHFFFAOYSA-N tetradecan-1-ol Chemical compound CCCCCCCCCCCCCCO HLZKNKRTKFSKGZ-UHFFFAOYSA-N 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 235000010487 tragacanth Nutrition 0.000 description 2
- 239000000196 tragacanth Substances 0.000 description 2
- 229940116362 tragacanth Drugs 0.000 description 2
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- MORCTKYKWNHWLH-UHFFFAOYSA-N (8-methyl-8-propan-2-yl-8-azoniabicyclo[3.2.1]octan-3-yl) 3-[2-(diethylamino)acetyl]oxy-2-phenylpropanoate Chemical compound C1C([N+]2(C)C(C)C)CCC2CC1OC(=O)C(COC(=O)CN(CC)CC)C1=CC=CC=C1 MORCTKYKWNHWLH-UHFFFAOYSA-N 0.000 description 1
- CWVKNUVSRQRHKK-UHFFFAOYSA-N (9,9-dimethyl-3-oxa-9-azoniatricyclo[3.3.1.02,4]nonan-7-yl) 2-[2-(diethylamino)acetyl]oxy-2,2-dithiophen-2-ylacetate Chemical compound C1C([N+]2(C)C)C3OC3C2CC1OC(=O)C(OC(=O)CN(CC)CC)(C=1SC=CC=1)C1=CC=CS1 CWVKNUVSRQRHKK-UHFFFAOYSA-N 0.000 description 1
- MJLJRGGFMRCMTO-UHFFFAOYSA-N (9-ethyl-9-methyl-3-oxa-9-azoniatricyclo[3.3.1.02,4]nonan-7-yl) 3-[2-(diethylamino)acetyl]oxy-2-phenylpropanoate Chemical compound C1C([N+]2(C)CC)C3OC3C2CC1OC(=O)C(COC(=O)CN(CC)CC)C1=CC=CC=C1 MJLJRGGFMRCMTO-UHFFFAOYSA-N 0.000 description 1
- 125000004641 (C1-C12) haloalkyl group Chemical group 0.000 description 1
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 description 1
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- 125000006736 (C6-C20) aryl group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- DNIAPMSPPWPWGF-GSVOUGTGSA-N (R)-(-)-Propylene glycol Chemical compound C[C@@H](O)CO DNIAPMSPPWPWGF-GSVOUGTGSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- QMMJWQMCMRUYTG-UHFFFAOYSA-N 1,2,4,5-tetrachloro-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=C(Cl)C(Cl)=CC(Cl)=C1Cl QMMJWQMCMRUYTG-UHFFFAOYSA-N 0.000 description 1
- HNSDLXPSAYFUHK-UHFFFAOYSA-N 1,4-bis(2-ethylhexyl) sulfosuccinate Chemical compound CCCCC(CC)COC(=O)CC(S(O)(=O)=O)C(=O)OCC(CC)CCCC HNSDLXPSAYFUHK-UHFFFAOYSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- NPDFULBIQZLKSB-UHFFFAOYSA-N 1-[4-hydroxy-1-[3,3,3-tris(4-fluorophenyl)propanoyl]pyrrolidine-2-carbonyl]-n-[(1-methylpiperidin-4-yl)methyl]pyrrolidine-2-carboxamide Chemical compound C1CN(C)CCC1CNC(=O)C1N(C(=O)C2N(CC(O)C2)C(=O)CC(C=2C=CC(F)=CC=2)(C=2C=CC(F)=CC=2)C=2C=CC(F)=CC=2)CCC1 NPDFULBIQZLKSB-UHFFFAOYSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- FCXWHPXCAHMELT-UHFFFAOYSA-N 1-azabicyclo[2.2.2]octan-3-yl 1-cyclohexyl-3,4-dihydro-1h-isoquinoline-2-carboxylate Chemical compound C1N(CC2)CCC2C1OC(=O)N1CCC2=CC=CC=C2C1C1CCCCC1 FCXWHPXCAHMELT-UHFFFAOYSA-N 0.000 description 1
- PZNPLUBHRSSFHT-RRHRGVEJSA-N 1-hexadecanoyl-2-octadecanoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[C@@H](COP([O-])(=O)OCC[N+](C)(C)C)COC(=O)CCCCCCCCCCCCCCC PZNPLUBHRSSFHT-RRHRGVEJSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- LGEZTMRIZWCDLW-UHFFFAOYSA-N 14-methylpentadecyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCC(C)C LGEZTMRIZWCDLW-UHFFFAOYSA-N 0.000 description 1
- VTDVDWDFUXABTM-UHFFFAOYSA-N 2-(diethylamino)ethyl 4-cyano-4-(3-cyclopentyloxy-4-methoxyphenyl)cyclohexane-1-carboxylate Chemical compound C1CC(C(=O)OCCN(CC)CC)CCC1(C#N)C1=CC=C(OC)C(OC2CCCC2)=C1 VTDVDWDFUXABTM-UHFFFAOYSA-N 0.000 description 1
- GASJAMXNMLWWGD-UHFFFAOYSA-N 2-[2-(diethylamino)acetyl]oxy-2,2-dithiophen-2-ylacetic acid Chemical compound C=1C=CSC=1C(C(O)=O)(OC(=O)CN(CC)CC)C1=CC=CS1 GASJAMXNMLWWGD-UHFFFAOYSA-N 0.000 description 1
- SFAAOBGYWOUHLU-UHFFFAOYSA-N 2-ethylhexyl hexadecanoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(CC)CCCC SFAAOBGYWOUHLU-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- 125000006201 3-phenylpropyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- HWCBLDPYKRZMKJ-UHFFFAOYSA-N 4,4-bis(4-fluorophenyl)-1-[1-[(3-fluorophenyl)methyl]piperidin-4-yl]imidazolidin-2-one Chemical compound C1=CC(F)=CC=C1C1(C=2C=CC(F)=CC=2)NC(=O)N(C2CCN(CC=3C=C(F)C=CC=3)CC2)C1 HWCBLDPYKRZMKJ-UHFFFAOYSA-N 0.000 description 1
- JMSDQUDVBLZSMZ-UHFFFAOYSA-O 4,4-bis(4-fluorophenyl)-1-[1-methyl-1-(2-oxo-2-pyridin-2-ylethyl)pyrrolidin-1-ium-3-yl]imidazolidin-2-one Chemical compound C1CC(N2C(NC(C2)(C=2C=CC(F)=CC=2)C=2C=CC(F)=CC=2)=O)C[N+]1(C)CC(=O)C1=CC=CC=N1 JMSDQUDVBLZSMZ-UHFFFAOYSA-O 0.000 description 1
- UTUUPXBCDMQYRR-HSZRJFAPSA-N 4-[(2r)-2-(3-cyclopentyloxy-4-methoxyphenyl)-2-phenylethyl]pyridine Chemical compound COC1=CC=C([C@H](CC=2C=CN=CC=2)C=2C=CC=CC=2)C=C1OC1CCCC1 UTUUPXBCDMQYRR-HSZRJFAPSA-N 0.000 description 1
- UTUUPXBCDMQYRR-UHFFFAOYSA-N 4-[2-(3-cyclopentyloxy-4-methoxyphenyl)-2-phenylethyl]pyridine Chemical compound COC1=CC=C(C(CC=2C=CN=CC=2)C=2C=CC=CC=2)C=C1OC1CCCC1 UTUUPXBCDMQYRR-UHFFFAOYSA-N 0.000 description 1
- QSUSKMBNZQHHPA-UHFFFAOYSA-N 4-[4-(4-fluorophenyl)-1-(3-phenylpropyl)-5-pyridin-4-ylimidazol-2-yl]but-3-yn-1-ol Chemical compound C=1C=CC=CC=1CCCN1C(C#CCCO)=NC(C=2C=CC(F)=CC=2)=C1C1=CC=NC=C1 QSUSKMBNZQHHPA-UHFFFAOYSA-N 0.000 description 1
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- PSGQCCSGKGJLRL-UHFFFAOYSA-N 4-methyl-2h-chromen-2-one Chemical group C1=CC=CC2=C1OC(=O)C=C2C PSGQCCSGKGJLRL-UHFFFAOYSA-N 0.000 description 1
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 102000055025 Adenosine deaminases Human genes 0.000 description 1
- 229940124225 Adrenoreceptor agonist Drugs 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 108090000531 Amidohydrolases Proteins 0.000 description 1
- 102000004092 Amidohydrolases Human genes 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- PJFHZKIDENOSJB-UHFFFAOYSA-N Budesonide/formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1.C1CC2=CC(=O)C=CC2(C)C2C1C1CC3OC(CCC)OC3(C(=O)CO)C1(C)CC2O PJFHZKIDENOSJB-UHFFFAOYSA-N 0.000 description 1
- 239000004358 Butane-1, 3-diol Substances 0.000 description 1
- 125000003358 C2-C20 alkenyl group Chemical group 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- 101100296726 Caenorhabditis elegans pde-5 gene Proteins 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 208000002177 Cataract Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 108090000322 Cholinesterases Proteins 0.000 description 1
- 102000003914 Cholinesterases Human genes 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- LUKZNWIVRBCLON-GXOBDPJESA-N Ciclesonide Chemical compound C1([C@H]2O[C@@]3([C@H](O2)C[C@@H]2[C@@]3(C[C@H](O)[C@@H]3[C@@]4(C)C=CC(=O)C=C4CC[C@H]32)C)C(=O)COC(=O)C(C)C)CCCCC1 LUKZNWIVRBCLON-GXOBDPJESA-N 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 241000710781 Flaviviridae Species 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- 208000002705 Glucose Intolerance Diseases 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- HSRJKNPTNIJEKV-UHFFFAOYSA-N Guaifenesin Chemical group COC1=CC=CC=C1OCC(O)CO HSRJKNPTNIJEKV-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 108010055717 JNK Mitogen-Activated Protein Kinases Proteins 0.000 description 1
- 102000019145 JUN kinase activity proteins Human genes 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 229940121948 Muscarinic receptor antagonist Drugs 0.000 description 1
- VYTMBMLLTQRIFQ-RKEPMNIXSA-N N/C(/c1ncc(C([C@@H]2F)O[C@H](CO)[C@H]2O)[nH]1)=N/C(N)=N Chemical compound N/C(/c1ncc(C([C@@H]2F)O[C@H](CO)[C@H]2O)[nH]1)=N/C(N)=N VYTMBMLLTQRIFQ-RKEPMNIXSA-N 0.000 description 1
- HXJARZZKHMXFLU-GTXYJYIYSA-N NC1NCNN2C1NCC2[C@@H]([C@@H]1F)OC(COCc2ccccc2)=C1OCc1ccccc1 Chemical compound NC1NCNN2C1NCC2[C@@H]([C@@H]1F)OC(COCc2ccccc2)=C1OCc1ccccc1 HXJARZZKHMXFLU-GTXYJYIYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- YYOUILVHRQFVCN-XUTVFYLZSA-N Nc1ncn[n]2c1ncc2[C@@H]([C@@H]1F)O[C@H](CO)[C@H]1O Chemical compound Nc1ncn[n]2c1ncc2[C@@H]([C@@H]1F)O[C@H](CO)[C@H]1O YYOUILVHRQFVCN-XUTVFYLZSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 102000038030 PI3Ks Human genes 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 241000711504 Paramyxoviridae Species 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000015439 Phospholipases Human genes 0.000 description 1
- 108010064785 Phospholipases Proteins 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 206010038731 Respiratory tract irritation Diseases 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 101710141795 Ribonuclease inhibitor Proteins 0.000 description 1
- 229940122208 Ribonuclease inhibitor Drugs 0.000 description 1
- 102100037968 Ribonuclease inhibitor Human genes 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- CDMGBJANTYXAIV-UHFFFAOYSA-N SB 203580 Chemical compound C1=CC(S(=O)C)=CC=C1C1=NC(C=2C=CC(F)=CC=2)=C(C=2C=CN=CC=2)N1 CDMGBJANTYXAIV-UHFFFAOYSA-N 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229940122286 Sphingosine 1-phosphate receptor antagonist Drugs 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 101150110875 Syk gene Proteins 0.000 description 1
- 229940122202 Thromboxane receptor antagonist Drugs 0.000 description 1
- 229940111979 Thromboxane synthase inhibitor Drugs 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- VGXACJMXDYPFDB-SXMMONRFSA-N [(3r)-1-azabicyclo[2.2.2]octan-3-yl] (2s)-2-(hydroxymethyl)-4-[(r)-methylsulfinyl]-2-phenylbutanoate Chemical compound C1([C@@](CO)(C(=O)O[C@@H]2C3CCN(CC3)C2)CC[S@](=O)C)=CC=CC=C1 VGXACJMXDYPFDB-SXMMONRFSA-N 0.000 description 1
- YYAZJTUGSQOFHG-IAVNQIGZSA-N [(6s,8s,10s,11s,13s,14s,16r,17r)-6,9-difluoro-17-(fluoromethylsulfanylcarbonyl)-11-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl] propanoate;2-(hydroxymethyl)-4-[1-hydroxy-2-[6-(4-phenylbutoxy)hexylamino]eth Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)C1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(OC(=O)CC)[C@@]2(C)C[C@@H]1O YYAZJTUGSQOFHG-IAVNQIGZSA-N 0.000 description 1
- BZEFPLNVTDBXTM-UHFFFAOYSA-N [1-(3-phenoxypropyl)-1-azoniabicyclo[2.2.2]octan-3-yl] 2-[2-(diethylamino)acetyl]oxy-2,2-dithiophen-2-ylacetate Chemical compound C=1C=CSC=1C(C=1SC=CC=1)(OC(=O)CN(CC)CC)C(=O)OC(C(CC1)CC2)C[N+]21CCCOC1=CC=CC=C1 BZEFPLNVTDBXTM-UHFFFAOYSA-N 0.000 description 1
- ULCAFXOHVMHABN-UHFFFAOYSA-N [2-[3-[di(propan-2-yl)amino]-1-phenylpropyl]-4-methylphenyl] 2-(dimethylamino)acetate Chemical compound C=1C(C)=CC=C(OC(=O)CN(C)C)C=1C(CCN(C(C)C)C(C)C)C1=CC=CC=C1 ULCAFXOHVMHABN-UHFFFAOYSA-N 0.000 description 1
- WREOTYWODABZMH-DTZQCDIJSA-N [[(2r,3s,4r,5r)-3,4-dihydroxy-5-[2-oxo-4-(2-phenylethoxyamino)pyrimidin-1-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N(C=C\1)C(=O)NC/1=N\OCCC1=CC=CC=C1 WREOTYWODABZMH-DTZQCDIJSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 230000003044 adaptive effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 229940090167 advair Drugs 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000005205 alkoxycarbonyloxyalkyl group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- JBDGDEWWOUBZPM-XYPYZODXSA-N ambroxol Chemical group NC1=C(Br)C=C(Br)C=C1CN[C@@H]1CC[C@@H](O)CC1 JBDGDEWWOUBZPM-XYPYZODXSA-N 0.000 description 1
- 229960005174 ambroxol Drugs 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 150000005840 aryl radicals Chemical class 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 238000000889 atomisation Methods 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 239000004305 biphenyl Chemical class 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000004452 carbocyclyl group Chemical group 0.000 description 1
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 229940082500 cetostearyl alcohol Drugs 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- CFBUZOUXXHZCFB-OYOVHJISSA-N chembl511115 Chemical compound COC1=CC=C([C@@]2(CC[C@H](CC2)C(O)=O)C#N)C=C1OC1CCCC1 CFBUZOUXXHZCFB-OYOVHJISSA-N 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229960003728 ciclesonide Drugs 0.000 description 1
- 229950001653 cilomilast Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001485 cycloalkadienyl group Chemical group 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- HXGBXQDTNZMWGS-RUZDIDTESA-N darifenacin Chemical compound C=1C=CC=CC=1C([C@H]1CN(CCC=2C=C3CCOC3=CC=2)CC1)(C(=O)N)C1=CC=CC=C1 HXGBXQDTNZMWGS-RUZDIDTESA-N 0.000 description 1
- 229960002677 darifenacin Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- SASYSVUEVMOWPL-NXVVXOECSA-N decyl oleate Chemical compound CCCCCCCCCCOC(=O)CCCCCCC\C=C/CCCCCCCC SASYSVUEVMOWPL-NXVVXOECSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 231100000223 dermal penetration Toxicity 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960002344 dexamethasone sodium phosphate Drugs 0.000 description 1
- PLCQGRYPOISRTQ-FCJDYXGNSA-L dexamethasone sodium phosphate Chemical compound [Na+].[Na+].C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)COP([O-])([O-])=O)(O)[C@@]1(C)C[C@@H]2O PLCQGRYPOISRTQ-FCJDYXGNSA-L 0.000 description 1
- 230000001079 digestive effect Effects 0.000 description 1
- 210000002249 digestive system Anatomy 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229940042406 direct acting antivirals neuraminidase inhibitors Drugs 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000036267 drug metabolism Effects 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 229950009769 etabonate Drugs 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 229940066493 expectorants Drugs 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 1
- 229960000556 fingolimod Drugs 0.000 description 1
- AAXVEMMRQDVLJB-BULBTXNYSA-N fludrocortisone Chemical compound O=C1CC[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 AAXVEMMRQDVLJB-BULBTXNYSA-N 0.000 description 1
- 229960003469 flumetasone Drugs 0.000 description 1
- WXURHACBFYSXBI-GQKYHHCASA-N flumethasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]2(C)C[C@@H]1O WXURHACBFYSXBI-GQKYHHCASA-N 0.000 description 1
- 229940042902 flumethasone pivalate Drugs 0.000 description 1
- JWRMHDSINXPDHB-OJAGFMMFSA-N flumethasone pivalate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)COC(=O)C(C)(C)C)(O)[C@@]2(C)C[C@@H]1O JWRMHDSINXPDHB-OJAGFMMFSA-N 0.000 description 1
- 229960000676 flunisolide Drugs 0.000 description 1
- 229940043075 fluocinolone Drugs 0.000 description 1
- FEBLZLNTKCEFIT-VSXGLTOVSA-N fluocinolone acetonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O FEBLZLNTKCEFIT-VSXGLTOVSA-N 0.000 description 1
- 229960001629 fluorometholone acetate Drugs 0.000 description 1
- YRFXGQHBPBMFHW-SBTZIJSASA-N fluorometholone acetate Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@]2(F)[C@@H](O)C[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 YRFXGQHBPBMFHW-SBTZIJSASA-N 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960000289 fluticasone propionate Drugs 0.000 description 1
- WMWTYOKRWGGJOA-CENSZEJFSA-N fluticasone propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(OC(=O)CC)[C@@]2(C)C[C@@H]1O WMWTYOKRWGGJOA-CENSZEJFSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000004051 gastric juice Anatomy 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 229960002146 guaifenesin Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 244000052637 human pathogen Species 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 229920013821 hydroxy alkyl cellulose Polymers 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 208000037797 influenza A Diseases 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 230000003434 inspiratory effect Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000006662 intracellular pathway Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 229940078545 isocetyl stearate Drugs 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 238000010902 jet-milling Methods 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 239000006101 laboratory sample Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- KKMJGUDIUVOQTI-STWSTGMMSA-N methyl (2s)-2-(1-adamantylmethylamino)-3-(4h-imidazol-4-yl)propanoate Chemical compound C([C@@H](C(=O)OC)NCC12CC3CC(CC(C3)C1)C2)C1C=NC=N1 KKMJGUDIUVOQTI-STWSTGMMSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 229960002744 mometasone furoate Drugs 0.000 description 1
- WOFMFGQZHJDGCX-ZULDAHANSA-N mometasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(Cl)[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@H]1C)C(=O)CCl)C(=O)C1=CC=CO1 WOFMFGQZHJDGCX-ZULDAHANSA-N 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 150000004712 monophosphates Chemical class 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940043348 myristyl alcohol Drugs 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- GYCKQBWUSACYIF-UHFFFAOYSA-N o-hydroxybenzoic acid ethyl ester Natural products CCOC(=O)C1=CC=CC=C1O GYCKQBWUSACYIF-UHFFFAOYSA-N 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229950000175 oglemilast Drugs 0.000 description 1
- 239000012053 oil suspension Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 210000003254 palate Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- SHVLMMLZDSMPQJ-UHFFFAOYSA-N phosphonato phosphate;triethylazanium Chemical compound CC[NH+](CC)CC.CC[NH+](CC)CC.CC[NH+](CC)CC.CC[NH+](CC)CC.[O-]P([O-])(=O)OP([O-])([O-])=O SHVLMMLZDSMPQJ-UHFFFAOYSA-N 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 230000004800 psychological effect Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 238000000163 radioactive labelling Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229950000915 revatropate Drugs 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 239000002151 riboflavin Substances 0.000 description 1
- 239000003161 ribonuclease inhibitor Substances 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 229960002586 roflumilast Drugs 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000011452 sequencing regimen Methods 0.000 description 1
- 230000009919 sequestration Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 239000002911 sialidase inhibitor Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- NASFKTWZWDYFER-UHFFFAOYSA-N sodium;hydrate Chemical compound O.[Na] NASFKTWZWDYFER-UHFFFAOYSA-N 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- FBOUYBDGKBSUES-VXKWHMMOSA-N solifenacin Chemical compound C1([C@H]2C3=CC=CC=C3CCN2C(O[C@@H]2C3CCN(CC3)C2)=O)=CC=CC=C1 FBOUYBDGKBSUES-VXKWHMMOSA-N 0.000 description 1
- 229960003855 solifenacin Drugs 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000008347 soybean phospholipid Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 229940035073 symbicort Drugs 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 210000001138 tear Anatomy 0.000 description 1
- IOGXOCVLYRDXLW-UHFFFAOYSA-N tert-butyl nitrite Chemical compound CC(C)(C)ON=O IOGXOCVLYRDXLW-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 1
- OULAJFUGPPVRBK-UHFFFAOYSA-N tetratriacontyl alcohol Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCO OULAJFUGPPVRBK-UHFFFAOYSA-N 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 239000002396 thromboxane receptor blocking agent Substances 0.000 description 1
- 239000003768 thromboxane synthase inhibitor Substances 0.000 description 1
- 229940100611 topical cream Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229940100615 topical ointment Drugs 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000440 toxicity profile Toxicity 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- RLPRKIMZISRVFC-UHFFFAOYSA-N triazolo[5,1-f][1,2,4]triazine Chemical compound N1=CN=CC2=CN=NN21 RLPRKIMZISRVFC-UHFFFAOYSA-N 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- WVLBCYQITXONBZ-UHFFFAOYSA-N trimethyl phosphate Chemical compound COP(=O)(OC)OC WVLBCYQITXONBZ-UHFFFAOYSA-N 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 229950008396 ulobetasol propionate Drugs 0.000 description 1
- BDSYKGHYMJNPAB-LICBFIPMSA-N ulobetasol propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H](C)[C@@](C(=O)CCl)(OC(=O)CC)[C@@]2(C)C[C@@H]1O BDSYKGHYMJNPAB-LICBFIPMSA-N 0.000 description 1
- 238000002255 vaccination Methods 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 210000002845 virion Anatomy 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/23—Heterocyclic radicals containing two or more heterocyclic rings condensed among themselves or condensed with a common carbocyclic ring system, not provided for in groups C07H19/14 - C07H19/22
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/12—Triazine radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M15/00—Inhalators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61M—DEVICES FOR INTRODUCING MEDIA INTO, OR ONTO, THE BODY; DEVICES FOR TRANSDUCING BODY MEDIA OR FOR TAKING MEDIA FROM THE BODY; DEVICES FOR PRODUCING OR ENDING SLEEP OR STUPOR
- A61M2202/00—Special media to be introduced, removed or treated
- A61M2202/30—Vaccines
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Virology (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pulmonology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Un compuesto de Fórmula I: **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo; en donde: cada uno de R1 y R7 es independientemente H, halógeno, ORa, (C1-C8) haloalquilo, (C3-C8) halocicloalquilo, CN, N3, (C1-C8) alquilo, (C3-C8) cicloalquilo, (C1-C8) alquilo sustituido, (C3-C8) cicloalquilo sustituido, (C2-C8) alquenilo, (C2-C8) alquenilo sustituido, (C2-C8) alquinilo o (C2-C8) alquinilo sustituido, en donde el sustituyente se selecciona del grupo que consiste de -X, -Rb, -OH, =O, -ORb, -SRb, -S-, -NRb2, -N+Rb3, =NRb, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NHC(=O)Rb, -OC(=O)Rb, -NHC(=O)NRb2, -S(=O)2-, -S(=O)2OH, - S(=O)2Rb, -OS(=O)2ORb, -S(=O)2NRb2, -S(=O)Rb, -OP(=O)(ORb)2, -P(=O)(ORb)2, - P(=O)(O)2, -P(=O)(OH)2, -P(O)(ORb)(O-), -C(=O)Rb, -C(=O)X, -C(S)Rb, -C(O)ORb, -C(O)O-, -C(S)ORb, -C(O)SRb, -C(S)SRb, -C(O)NRb2, -C(S)NRb2, -C(=NRb)NRb2, donde cada X es independientemente un halógeno: F, Cl, Br o I; y cada Rb es independientemente H, alquilo, cicloalquilo, arilo, arilalquilo o un heterociclo; R2 es ORa; R3 es halógeno o N3; cada Ra es independientemente H, arilalquilo, arilo, (C1-C8) alquilo, o (C3-C8) cicloalquilo; R4 es H, =O, ORa, N(Ra)2, N3, CN, S(O)nRa, halógeno, (C1-C8) haloalquilo, o (C3-C8) halocicloalquilo; R5 es H, =O, ORa, N(Ra)2, N3, CN, S(O)nRa, halógeno, (C1-C8) haloalquilo, o (C3-C8) halocicloalquilo; cada n es 0, 1 o 2; y cada R6 es H, arilo, arilalquilo, o **(Ver fórmula)** en donde W1 y W2 son cada uno, independientemente, ORa o un grupo de la Fórmula la: **(Ver fórmula)** en donde: cada Y es independientemente un enlace u O; M2 es 0, 1 o 2; y cada Rx es H, halógeno o OH, para su uso en un método para tratar una infección Orthomyxoviridae en un mamífero.
Description
DESCRIPCIÓN
Análogos de carba-nucleósido 2-sustituidos para el tratamiento antiviral
CAMPO DE LA INVENCIÓN
[0001] La invención se refiere en general a compuestos con actividad antiviral, más particularmente nucleósidos para el uso contra infecciones de virus Orthomyxoviridae.
ANTECEDENTES DE LA INVENCIÓN
[0002] Los virus de influenza de la familia Orthomyxoviridae que pertenecen a los géneros A y B son responsables de epidemias de gripe estacionales cada año, que causan infecciones respiratorias agudas contagiosas. Los niños, los ancianos y las personas con enfermedades crónicas tienen un alto riesgo de desarrollar complicaciones graves que conducen a altas tasas de morbilidad y mortalidad (Memoli et al., Drug Discovery Today 2008, 13, 590- 595). Entre los tres géneros de influenza, los virus de tipo A son los patógenos humanos más virulentos que causan la enfermedad más grave, pueden transmitirse a otras especies y dan lugar a pandemias de influenza humana. El reciente brote de influenza humana de la cepa porcina agresiva A/H1N1 en 2009 ha enfatizado la necesidad de nuevas terapias antivirales. Si bien los programas de vacunación anual se utilizan actualmente para proteger a las poblaciones de la infección por influenza, estos programas deben anticipar las cepas de virus que prevalecerán durante los brotes estacionales para ser efectivos y no abordan el problema de las pandemias de influenza repentinas e imprevistas. Una vez más, el reciente brote de influenza humana de la agresiva cepa porcina A/H1N1 en 2009 es un ejemplo de este problema.
[0003] Actualmente se encuentran disponibles varias terapias contra la influenza y otras están en desarrollo (Hedlund et al., Virus 2010, 2, 1766-1781). Entre las terapias contra la influenza disponibles actualmente se encuentran los bloqueadores de los canales iónicos M2 amantadina y rimantadina y los inhibidores de la neuraminidasa oseltamivir y zanamivir. Sin embargo, se ha desarrollado resistencia a todos estos medicamentos. Por lo tanto, existe una necesidad continua de nuevos tratamientos contra la influenza.
[0004] Nuevos y prometedores agentes anti-influenza con nuevos mecanismos de acción están ahora en desarrollo. Entre estos nuevos agentes se encuentra el favipiravir, que se dirige a la replicación del gen viral inhibiendo la Polimerasa de influenza ARN. Sin embargo, todavía no está claro si este candidato a fármaco en investigación estará disponible para la terapia. Por lo tanto, existe una necesidad continua de desarrollar compuestos adicionales que inhiban la influenza a través de este mecanismo de acción.
[0005] Ciertos ribósidos de las nucleobases pirrolo[1,2-f][1,2,4]triazina, imidazo[1,5-f][1,2,4]triazina, imidazo[1,2-f][1,2,4]triazina y[1,2,4]triazolo[4,3-f][1,2,4]triazina se han descrito en Carbohydrate Research 2001, 331 (1), 77-82; Nucleosides & Nucleotides 1996, 15 (1-3), 793-807; Tetrahedron Letters 1994, 35 (30), 5339-42; Heterocycles 1992, 34 (3), 569-74; J. Chem. Soc. Perkin Trans. 1 1985, 3, 621-30; J. Chem. Soc. Perkin Trans. 1 1984, 2, 229-38; WO 2000056734; Organic Letters 2001, 3 (6), 839-842; J. Chem. Soc. Perkin Trans. 11999, 20, 2929-2936; y J. Med. Chem.
1986, 29 (11), 2231-5. Sin embargo, estos compuestos no se han descrito como útiles para el tratamiento de infecciones por Orthomyxoviridae.
[0006] Ribósidos de nucleobases pirrolo[1,2-f][1,2,4]triazinilo, imidazo[1,5-f][1,2,4]triazinilo, imidazo[1,2-f][1,2,4]triazinil y[1,2,4]triazolo[4,3-f][1,2,4]triazinil con actividad antiviral, anti-HCV y anti-RdRp han sido descritas por Babu, WO2008/089105 y WO2008/141079, Cho et al., WO2009/132123 y Francom et al. WO2010/002877. Butler et al., WO2009/132135, describe pirrolo[1,2-f][1,2,4]triazinil, imidazo[1,5-f][1,2,4]triazinil, imidazo[1,2-f][1,2,4]triazinilo y [1,2,4]triazolo[4,3-f][1,2,4]triazinil nucleósidos en los que la posición 1' del azúcar nucleósido está sustituida con un grupo ciano o metilo. Sin embargo, no se ha descrito la eficacia de estos compuestos para el tratamiento de infecciones por Orthomyxoviridae.
[0007] WO 02/32920 A2 (Stuyver et al.) describe una gama de nucleósidos con núcleos mono y bicíclicos útiles en el tratamiento de infecciones virales por Flaviviridae, Orthomyxoviridae y Paramyxoviridae.
RESUMEN DE LA INVENCIÓN
[0008] En este documento se proporcionan compuestos que inhiben los virus de la familia Orthomyxoviridae. La invención también comprende compuestos de Fórmula I que inhiben las polimerasas de ácidos nucleicos virales, particularmente la polimerasa ARN dependiente de ARN de Orthomyxoviridae (RdRp), en lugar de las polimerasas de ácidos nucleicos celulares. Los compuestos de Fórmula I son útiles para tratar infecciones por Orthomyxoviridae en humanos y otros animales.
[0009] La primera realización de la invención está dirigida a un compuesto de Fórmula I:
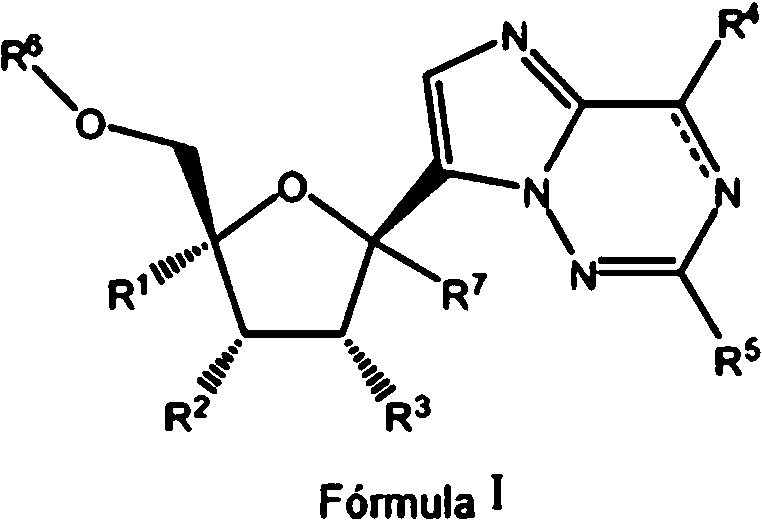
l farmacéuticamente aceptable del mismo; en donde:
cada uno de R1 y R7 es independientemente H, halógeno, ORa, (C1-C8) haloalquilo, (C3-C8) halocicloalquilo, CN, N3, (C1-C8) alquilo, (C3-C8) cicloalquilo, (C1-C8) alquilo sustituido, (C3-C8) cicloalquilo sustituido, (C2-C8) alquenilo, (C2-C8) alquenilo sustituido, (C2-C8) alquinilo o (C2-C8) alquinilo sustituido,
en donde el sustituyente se selecciona del grupo que consiste en -X, -Rb, -OH, =O, -ORb, -SRb, -S-, -NRb2, -N+Rb3, = NRb, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NHC(=O)Rb, -OC(=O)Rb, -NHC(=O)NRb2, -S(=O)2-, -S(=O)2OH, -S(=O)2Rb, -OS(=O)2ORb, -S(=O)2NRb2, -S(=O)Rb, -OP(=O)(ORb)2, -P(=O)(ORb)2, -P(=O)(O-)2, -P(=O)(OH)2, -P(O)(ORb)(O‘), -C(=O)Rb, -C(=O)X, -C(S)Rb, -C(O)ORb, -C(O)O‘, -C(S)ORb, -C(O)SRb, -C(S)SRb, -C(O)NRb2, -C(S)NRb2, -C(=NRb)NRb2, donde cada X es independientemente un halógeno: F, Cl, Br o I; y cada Rb es independientemente H, alquilo, cicloalquilo, arilo, arilalquilo o un heterociclo; R2 es ORa;
R3 es halógeno o N3;
cada Ra es independientemente H, arilo, arilalquilo, (C1-C8) alquilo, o (C3-C8) cicloalquilo;
cada uno de R4 y R5 es independientemente H, =O, ORa, N(Ra)2, N3, CN, S(O)nRa, halógeno, (C1-C8) haloalquilo, o (C3-C8) halocicloalquilo;
n es 0, 1 o 2; y
R6 es H, arilo, arilalquilo, o
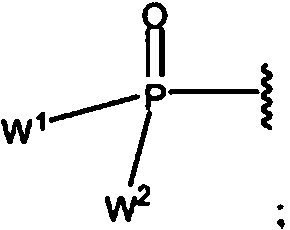
en donde W1 y W2 son cada uno, independientemente, ORa o un grupo de la Fórmula la:

Fórmula |a
en donde:
cada Y es independientemente un enlace u O;
M2 es 0, 1 o 2; y
cada Rx es H, halógeno u OH,
para uso en un método para tratar una infección por Orthomyxoviridae en un mamífero.
[0010] En una realización preferida, el compuesto de Fórmula I está representado por la Fórmula II:

o una sal farmacéuticamente aceptable del mismo. En otras realizaciones preferidas, R1 es H, R2 es OH u O-bencilo y/o R3 es F o N3 y, más preferiblemente, R3 es F. En una determinada realización de la presente invención, R4 es NH2 y R5 es H, F, Cl, Br, N3, CN, CF3, NH2, SMe o SO2Me y, en otra realización, R5 es NH2 y R4 es = O, OH, OMe, Cl, Br, I, NH2, NHMe, NHcPr o SMe. En otras realizaciones preferidas, R4 y R5 son ambos NH2 o SMe, R5 es H o R4 es = O. En otras realizaciones preferidas, R6 es H, bencilo, o

en donde W2 es OH y W 1 es un grupo de la fórmula Ia:

en donde cada Y es O; M2 es 2; y cada Rx es H. En otra realización, R7 es H u OH.
[0011] En algunas realizaciones, la infección por Orthomyxoviridae a tratar es una infección por Influenza de virus A, una infección por Influenzael de virus B, o una infección por Influenza de virus C. En otra realización, el método comprende tratar una infección por Orthomyxoviridae en un mamífero que lo necesite mediante la administración de una cantidad terapéuticamente eficaz de una composición farmacéutica que comprende una cantidad eficaz de un compuesto de Fórmula I, II o III o una sal farmacéuticamente aceptable del mismo en combinación con un diluyente o portador farmacéuticamente aceptable.
[0012] En otra realización, el método de tratamiento de una infección por el virus Orthomyxoviridae comprende además administrar una cantidad terapéuticamente eficaz de al menos un agente terapéutico adicional seleccionado del grupo que consiste de un corticosteroide, un antiinflamatorio modulador de transducción de señales, un broncodilatador agonista de p2-adrenorreceptores, un anticolinérgico, un agente mucolítico, solución salina hipertónica, un agente que inhibe la
migración de células proinflamatorias al sitio de infección y mezclas de los mismos. En determinadas realizaciones, el agente terapéutico adicional es un inhibidor de la hemaglutinina viral, un inhibidor de la neuramidasa viral, un inhibidor del canal iónico M2, un inhibidor de la polimerasa ARN dependiente de ARN de Orthomyxoviridae o una sialidasa. En otra realización, el agente terapéutico adicional se selecciona del grupo que consiste en ribavirina, oseltamivir, zanamivir, laninamivir, peramivir, amantadina, rimantadina, CS-8958, favipiravir, AVI-7100, inhibidor de la proteasa alfa-1 y DAS181.
[0013] En otra realización de la invención, el compuesto de Fórmula I, II o III y/o al menos un agente terapéutico adicional es para ser administrado por inhalación.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
[0014] La primera realización de la presente invención está dirigida a un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo; en donde:
cada uno de R1 y R7 es independientemente H, halógeno, ORa, (Ci-Ca) haloalquilo, (C3-C8) halocicloalquilo, CN, N3, (Ci-Ca) alquilo, (C3-C8) halocicloalquilo, (Ci-Ca) alquilo sustituido, (C3-C8) cicloalquilo sustituido, (C2-Ca) alquenilo, (C2-Ca) sustituido, alquenilo (C2-Ca) alquinilo o (C2-Ca) alquinilo sustituido, en donde el sustituyente se selecciona del grupo que consiste de-X, -Rb, -OH, =O, -ORb, -SRb, -S-, -NRb2, -N+Rb3, =NRb, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NHC(=O)Rb, -OC(=O)Rb, -NHC(=O)NRb2, -S(=O)2-, -S(=O)2OH, -S(=O)2Rb, -OS(=O)2ORb, S(=O)2NRb2, -S(=O)Rb, -OP(=O)(ORb)2, -P(=O)(ORb)2, -P(=O)(O')2, -P(=O)(OH)2, P(O)(ORb)(O'), - C(=O)Rb, -C(=O)X, -C(S)Rb, -C(O)ORb, -C(O)O-, -C(S)ORb, -C(O)SRb, -C(S)SRb, -C(O)NRb2, C(S)NRb 2 -C(=NRb)NRb2,
donde cada X es independientemente un halógeno: F, Cl, Br o I; y cada Rb es independientemente H, alquilo, cicloalquilo, arilo, arilalquilo o un heterociclo;
R2 es ORa;
R3 es halógeno o N3;
cada Ra es independientemente H, arilo, arilalquilo, (Ci-Ca) alquilo, o (C3-C8) cicloalquilo;
cada uno de R4 y R5 es independientemente H, =O, ORa, N(Ra)2, N3, CN, S(O)nRa, halógeno, alquilo (Ci-Ca) haloalquilo, o (C3-Ca) halocicloalquilo;
cada n es 0, 1 o 2; y
R6 es H, arilo, arilalquilo, o

en donde W1yW 2 son cada uno, independientemente, O una o un grupo de la fórmula Ia:

en donde:
cada Y es independientemente un enlace u O;
M2 es 0, 1 o 2; y
cada Rx es H, halógeno u OH,
para usar en un método para tratar una infección por Orthomyxoviridae en un mamífero. Con respecto a la Fórmula Ia, cuando Y es O, Rx no es halógeno.
[0015] A menos que se indique lo contrario, los siguientes términos y frases tal como se utiliza en la presente memoria están destinados a tener los siguientes significados.
[0016] Cuando los nombres comerciales se utilizan en el presente documento, los solicitantes pretenden incluir de manera independiente el producto nombre comercial y el (los) ingrediente(s) activo(s) farmacéutico(s) del producto de nombre comercial.
[0017] "Alquilo" es un hidrocarburo que contiene átomos normales, secundarios, o terciarios de carbono. Por ejemplo, un grupo alquilo puede tener de 1 a 20 átomos de carbono (es decir, C1-C20 alquilo), de 1 a 8 átomos de carbono (es decir, C1-C85 alquilo) o de 1 a 6 átomos de carbono (es decir, C1-C6 alquilo). Los ejemplos de grupos alquilo adecuados incluyen, pero no se limitan a metilo (Me, -CH3), etilo (Et, -CH2CH3), 1 -propilo (n-Pr, n-propilo, -CH2CH2CH3), 2-propilo (i-Pr, i-propilo, -CH(CHa)2), 1 -butilo (n-Bu, n-butilo, -CH2CH2CH2CH3), 2-metil-1-propilo (i-Bu, i-butilo, -CH2CH(CH3)2), 2-butilo (s-Bu, sbutilo, -CH(CH3)CH2CH3), 2-metil-2-propilo (t-Bu, t-butilo, -C(CH3)3), 1-pentilo (n-pentilo, -CH2CH2CH2CH2CH3), 2-pentilo (-CH(CH3)CH2CH2CH3), 3-pentilo (-CH(CH2CH3)2), 2-metil-2-butilo (-C(CH3)2CH2CH3), 3-metil-2-butilo (-CH(CH3)CH(CH3)2), 3-metil-1-butilo (-CH2CH2CH(CH3)2), 2-metil-1-butilo (-CH2CH(CH3)CH2CH3), 1-hexilo (-CH2CH2CH2CH2CH2CH3), 2-hexilo (-CH(CH3)CH2CH2CH2CH3), 3-hexilo (-CH(CH2CH3) (CH2CH2CH3)), 2-metil-2-pentilo (-C(CH3)2CH2CH2CH3), 3- metil-2-pentilo (-CH(CH3)CH(CH3)CH2CH3), 4-metil-2-pentilo (-CH(CH3)CH2CH(CH3)2), 3-metilo-3-pentilo (-C(CH3)(CH2CH3)2), 2-metil-3-pentil (-CH(CH2CH3)CH(CH3)2), 2,3-dimetil-2-butilo (-C(CH3)2CH(CH3)2), 3,3-dimetil-2-butilo (-CH(CH3)C(CH3)3 y octilo (-(CH2)7CH3).
[0018] "Cicloalquilo" es un hidrocarburo que contiene átomos de carbono cíclicos. Por ejemplo, un grupo cicloalquilo que puede tener de 3 a 20 átomos de carbono (es decir, C3-C20 cicloalquilo), de 3 a 8 átomos de carbono (es decir, C3-C8 cicloalquilo), o 3 a 6 átomos de carbono (es decir, C3-C6 cicloalquilo). Los ejemplos de grupos cicloalquilo adecuados incluyen ciclopropilo (c-propilo, cPr).
[0019] "Alquenilo" es un hidrocarburo que contiene, átomos de carbono terciarios o cíclicos normales, secundarios, con al menos un sitio de insaturación, es decir, un carbono-carbono, doble enlace sp2. Por ejemplo, un grupo alquenilo puede tener de 2 a 20 átomos de carbono (es decir, C2-C20 alquenilo), 2 a 8 átomos de carbono (es decir, C2-C8 alquenilo) o 2 a 6 átomos de carbono (es decir, C2-C6 alquenilo). Los ejemplos de grupos alquenilo adecuados incluyen, pero no se limitan a etileno o vinilo (-CH=CH2), alilo (-CH2CH=CH2), ciclopentenilo (-C5H7) y 5-hexenilo (-CH2CH2CH2CH2CH=CH2).
[0020] "Alquinilo" es un hidrocarburo que contiene, átomos de carbono terciarios o cíclicos normales, secundarios, con al menos un sitio de insaturación, es decir, un enlace carbono-carbono, sp triple enlace. Por ejemplo, un grupo alquinilo puede tener de 2 a 20 átomos de carbono (es decir, alquinilo C2-C20), 2 a 8 átomos de carbono (es decir, alquino C2-C8), o 2 a 6 átomos de carbono (es decir, C2-C6 alquinilo). Los ejemplos de grupos alquinilo adecuados incluyen, pero no se limitan a acetilénico (-C=CH), propargilo (-CH2CECH), y similares.
[0021] "Arilo" significa un radical hidrocarburo aromático derivado de la eliminación de un átomo de hidrógeno de un solo átomo de carbono de un sistema de anillo aromático progenitor. Por ejemplo, un grupo arilo puede tener de 6 a 20 átomos de carbono, de 6 a 14 átomos de carbono o de 6 a 10 átomos de carbono. Los grupos arilo típicos incluyen, pero no se
limitan a radicales derivados de benceno (por ejemplo, fenilo), benceno sustituido, naftaleno, antraceno, bifenilo y similares.
[0022] "Arilalquilo" se refiere a un radical alquilo acíclico en dondeuno de los átomos de hidrógeno unidos a un átomo de carbono, típicamente un terminal o átomo de carbono sp3, se sustituye con un radical arilo. Los grupos arilalquilo típicos incluyen, pero no se limitan a bencilo, 2-feniletan-1-ilo, naftilmetilo, 2-naftiletan-1-ilo, naftobencilo, 2-naftofeniletan-1-ilo y similares. El grupo arilalquilo puede comprender de 7 a 20 átomos de carbono, por ejemplo, el resto alquilo es de 1 a 6 átomos de carbono y el resto arilo es de 6 a 14 átomos de carbono.
[0023] "Carbociclo" o "carbociclilo" se refiere a un saturado (es decir, cicloalquilo), parcialmente insaturado (por ejemplo, cicloalquenilo, cicloalcadienilo, etc.) o un anillo aromático que tiene de 3 a 7 átomos de carbono como un monociclo, de 7 a 12 átomos de carbono como una biciclo, y hasta aproximadamente 20 átomos de carbono como un policiclo. Los carbociclos monocíclicos tienen de 3 a 7 átomos en el anillo, aún más típicamente 5 o 6 átomos en el anillo. Los carbociclos bicíclicos tienen de 7 a 12 átomos en el anillo, por ejemplo, dispuestos como un sistema biciclo [4,5], [5,5], [5,6] o [6,6], o 9 o 10 átomos del anillo dispuestos como un sistema de biciclo [5,6] o [6,6], o anillos espirocondensados. Los ejemplos no limitantes de carbociclos monocíclicos incluyen ciclopropilo, ciclobutilo, ciclopentilo, 1-ciclopent-1-enilo, 1-ciclopent-2-enilo, 1-ciclopent-3-enilo, ciclohexilo, 1-ciclohex-1-enilo, 1-ciclohex-2-enilo, 1-ciclohex-3-enilo y fenilo. Los ejemplos no limitantes de biciclocarbociclos incluyen naftilo, tetrahidronaftaleno y decalina.
[0024] "Haloalquilo" es un grupo alquilo, como se definen anteriormente, en dondeuno o más átomos de hidrógeno del grupo alquilo es reemplazado con un átomo de halógeno. La porción alquilo de un grupo haloalquilo puede tener de 1 a 20 átomos de carbono (es decir, C1-C20 haloalquilo), de 1 a 12 átomos de carbono (es decir, haloalquilo C1-C12) o de 1 a 6 átomos de carbono (es decir, C1-C6 haloalquilo). Los ejemplos de grupos haloalquilo adecuados incluyen, pero no se limitan a -CF3, -CHF2, -CFH2, -CH2CF3 y similares.
[0025] "Heterociclo" o "heterociclilo" como se usa en este documento incluye a modo de ejemplo y no de limitación esos heterociclos descritos en Paquette, Leo A.; Principles of Modern Heterocyclic Chemistry (W.A. Benjamin, Nueva York, 1968), en particular los capítulos 1, 3, 4, 6, 7 y 9; The Chemistry of Heterocyclic Compounds, A Series of Monographs" (John Wiley & Sons, Nueva York, 1950 hasta el presente), en particular los volúmenes 13, 14, 16, 19 y 28; y J. Am. Chem. Soc. (1960) 82:5566. En una realización específica de la invención, "heterociclo" incluye un "carbociclo" como se define en el presente documento, en dondeuno o más (p. ej., 1, 2, 3 o 4) átomos de carbono han sido reemplazados por un heteroátomo (p. ej., O, N, o S). Los términos "heterociclo" o "heterociclilo" incluye anillos saturados, anillos parcialmente insaturados, y anillos aromáticos (es decir, anillos heteroaromáticos).
[0026] El término "sustituido" en referencia a alquilo, cicloalquilo, alquenilo, arilo, etc., por ejemplo, "alquilo sustituido", "cicloalquilo sustituido", "alquenilo sustituido", "arilo sustituido", respectivamente, en los que uno o más átomos de hidrógeno se reemplazan cada uno independientemente con un sustituyente que no es hidrógeno. Los sustituyentes típicos incluyen, pero no se limitan a -X, -Rb, -OH, =O, -ORb, -SRb, -S-, -NRb2, -N+Rb3, =NRb, CX3, -CN, -OCN, - SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NHC(=O)Rb, -OC(=O)Rb, -NHC(=O)NRb2, -S(=O)2-, -S(=O)2OH, -S(=O)2Rb, -OS(=O)2ORb, -S(=O)2NRb2, -S(=O)Rb, -OP(=O)(ORb)2, -P(=O)(ORb)2, -P(=O)(O)2, -P(=O)(OH)2, -P(O)(ORb)(O‘), -C(=O)Rb, -C(=O)X, -C(S)Rb, -C(O)ORb, -C(O)O-, -C(S)ORb, -C(O)SRb, -C(S)SRb, -C(O)NRb2, -C(S)NRb2, -C(=NRb)NRb2, donde cada X es independientemente un halógeno: F, Cl, Br o I; y cada Rb es independientemente H, alquilo, cicloalquilo, arilo, arilalquilo o un heterociclo.
[0027] El término "profármaco" tal como se utiliza aquí, se refiere a cualquier compuesto que cuando se administra a un sistema biológico genera la sustancia fármaco, es decir, ingrediente activo, como resultado de una(s) reacción(es) química(s) espontánea(s), reacción(es) química(s) catalizada(s) por enzima, fotólisis y/o reacciones químicas metabólicas. Por tanto, un profármaco es un análogo modificado covalentemente o una forma latente de un compuesto terapéuticamente activo.
[0028] "Grupo protector" se refiere a un resto de un compuesto que enmascara o altera las propiedades de un grupo funcional o las propiedades del compuesto como un todo. La subestructura química de un grupo protector varía ampliamente. Una función de un grupo protector es servir como intermediario en la síntesis de la sustancia farmacológica parental. Los grupos protectores químicos y las estrategias de protección/desprotección son bien conocidos en la técnica. Ver: "Protective Groups in Organic Chemistry", Theodora W. Greene (John Wiley & Sons, Inc., Nueva York, 1991. Los grupos protectores se utilizan a menudo para enmascarar la reactividad de ciertos grupos funcionales, para ayudar en la eficiencia de la sustancia química deseada, por ejemplo, hacer y romper enlaces químicos de una manera ordenada y planificada. La protección de los grupos funcionales de un compuesto altera otras propiedades físicas además de la reactividad del grupo funcional protegido, como la polaridad, la lipofilicidad (hidrofobicidad) y otras propiedades que se puede medir por herramientas analíticas comunes. Los intermedios químicamente protegidos pueden ser ellos mismos biológicamente activos o inactivos.
[0029] Los compuestos protegidos también pueden exhibir propiedades alteradas, y en algunos casos, optimizadas in vitro y in vivo, tales como paso a través de las membranas celulares y la resistencia a la degradación o secuestro enzimático. En esta función, los compuestos protegidos con efectos terapéuticos previstos pueden denominarse profármacos. La función de un grupo protector es convertir el fármaco parental en un profármaco, por lo que el fármaco
parental se libera tras la conversión del profármaco in vivo. Debido a que los profármacos activos pueden absorberse más eficazmente que el fármaco original, los profármacos pueden poseer una mayor potencia in vivo que el fármaco original. Los grupos protectores se eliminan in vitro, en el caso de productos químicos intermedios, o in vivo, en el caso de profármacos. Con los intermedios químicos, no es particularmente importante que los productos resultantes después de la desprotección, por ejemplo, los alcoholes, sean fisiológicamente aceptables, aunque en general es más deseable si los productos son farmacológicamente inocuos.
[0030] El "resto de profármaco" significa un grupo funcional lábil que se separa del compuesto inhibidor activo durante el metabolismo, sistémicamente, dentro de una célula, mediante hidrólisis, escisión enzimática, o mediante algún otro proceso (Bundgaard, Hans, "Design and Application of Prodrugs" in Textbook of Drug Design and Development (1991), P. Krogsgaard-Larsen y H. Bundgaard, Eds. Harwood Academic Publishers, págs. 113-191). Las enzimas que son capaces de un mecanismo de activación enzimática con los compuestos profármacos de fosfonato de la invención incluyen, pero no se limitan a amidasas, esterasas, enzimas microbianas, fosfolipasas, colinesterasas y fosfases. Restos de profármaco pueden servir para mejorar la solubilidad, absorción y lipofilicidad para optimizar el suministro de fármacos, biodisponibilidad y eficacia. Un resto de profármaco puede incluir un metabolito activo o el propio fármaco.
[0031] Los ejemplos de restos de profármaco incluyen los ésteres hidrolíticamente sensibles o lábiles aciloximetilo -CH2OC(=O)R30 y carbonatos de aciloximetilo -CH2OC(=O)OR30 en donde R30 es C1-C6 alquilo, C1-C6 alquilo sustituido, C6 -C20 arilo o C6-C20 arilo sustituido. El éster de aciloxialquilo se utilizó como estrategia de profármaco para ácidos carboxílicos y luego se aplicó a fosfatos y fosfonatos por Farquhar et al., (1983) J. Pharm. Sci. 72:324; también las Patentes de EE.UU. Nos 4816570, 4968788, 5663159 y 5792756. En ciertos compuestos de la invención, un resto de profármaco es parte de un grupo fosfato. El éster de aciloxialquilo se puede usar para administrar ácidos fosfóricos a través de las membranas celulares y para mejorar la biodisponibilidad oral. Una variante cercana del éster de aciloxialquilo, el éster de alcoxicarboniloxialquilo (carbonato), también puede mejorar la biodisponibilidad oral como resto de profármaco en los compuestos de las combinaciones de la invención. Un éster de aciloximetilo ejemplar es pivaloiloximetoxi, (POM) -CH2OC(=O)C(CH3)3. Una fracción de profármaco de carbonato de aciloximetilo ejemplar es pivaloiloximetilcarbonato (POC)-CH2OC(=O)OC(CH3)3.
[0032] El grupo fosfato puede ser un resto de profármaco de fosfato. El resto de profármaco puede ser sensible a la hidrólisis, tal como, pero sin limitarse a los que comprenden un grupo de carbonato de pivaloiloximetilo (POC) o POM. Alternativamente, el resto de profármaco puede ser sensible a la escisión potenciada enzimáticamente, tal como un éster de lactato o un grupo éster de fosfonamidato.
[0033] Un experto en la técnica reconocerá que los sustituyentes y otros restos de los compuestos de Fórmula I deberían ser seleccionados para proporcionar un compuesto que es suficientemente estable para proporcionar un compuesto farmacéuticamente útil que se puede formular en una composición farmacéutica aceptablemente estable.
[0034] Un compuesto de sus sales farmacéuticamente aceptables de Fórmula I y puede existir como diferentes polimorfos o pseudopolimorfos. Como se usa en este documento, polimorfismo cristalino significa la capacidad de un compuesto cristalino de existir en diferentes estructuras cristalinas. El polimorfismo cristalino puede resultar de diferencias en el empaquetamiento de cristales (polimorfismo de empaquetamiento) o diferencias en el empaquetamiento entre diferentes confórmeros 8577608-1-AFAIRBAI de la misma molécula (polimorfismo conformacional). Como se usa en este documento, pseudopolimorfismo cristalino significa la capacidad de un hidrato o solvato de un compuesto para existir en diferentes estructuras cristalinas. Los pseudopolimorfos de la presente invención pueden existir debido a diferencias en el empaquetamiento de cristales (pseudopolimorfismo de empaquetamiento) o debido a diferencias en el empaquetamiento entre diferentes confórmeros de la misma molécula (pseudopolimorfismo conformacional).
[0035] Un compuesto de sus sales farmacéuticamente aceptables de Fórmula I y también puede existir como un sólido amorfo. Como se usa en este documento, un sólido amorfo es un sólido en dondeno existe un orden de largo alcance de las posiciones de los átomos en el sólido. Esta definición también se aplica cuando el tamaño del cristal es de dos nanómetros o menos. Pueden usarse aditivos, incluidos disolventes, para crear las formas amorfas de la presente invención.
[0036] En una cierta realización de la invención, el compuesto de Fórmula I está representado por la Fórmula II:

o una sal farmacéuticamente aceptable del mismo;
en donde las variables son como se definen para la Fórmula I. Preferiblemente, R1 en la Fórmula II es H, R4 es NH2 o =
[0037] En una realización preferida de los compuestos de Fórmula I, R1 es H, CH2OH, CH2F, CHF2, CH=CH2, C=CH, CN, CH2CH=CH2, N3, CH3 o CH2CH3, y, más preferiblemente, R1 es H.
[0038] En una realización adicional de la invención, R2 es OH o o-bencilo y, más preferiblemente es OH.
[0039] En una realización adicional de la invención, R3 es F o N3, y, más preferiblemente, R3 es F.
[0040] En una realización preferida adicional, R4 y R5 se seleccionan de H, NH2, =O, NHMe, NHcPr, OH, OMe, Cl, Br, I, SMe, F, N3, CN, CF3 y SO2Me, y más preferiblemente R4 es = O o NH2 y/o R5 es H o NH2.
[0041] En una realización adicional, R6 es H, bencilo, o

en donde W2 es OH y W 1 es un grupo de la fórmula la:

Fórmula |a
en donde:
Y es O;
M2 es 2; y cada Rx es H, y, más preferiblemente, R6 es H.
[0042] En una realización adicional de la invención, R7 es H o OH, y, más preferiblemente, R7 es H.
[0043] En otra realizaciones preferidas de la invención, R1 es H, R2 es OH y R3 es F. En otra realización preferida, R1 es H, R2 es OH, R3 es F, R4 y RS son NH2, H o = O, y R6 y R7 son hidrógeno.
[0044] En todavía otra realización preferida, R1 es H, R2 es O-bencilo o OH, R3 es F, R4 es SMe, NH2 o = O, R5 es SMe, SO2Me, H o NH2, R6 es bencilo o

en donde W2 es OH y W 1 es un grupo de Fórmula la:

Fórmula la
en donde:
Y es O;
M2 es 2; y
cada Rx es H, y R7 es H o OH.
[0045] En otras ciertas realizaciones de la invención, R4 es NH2 y R5 es H, F, Cl, Br, N3, CN, CF3, NH2, SMe, o SO2Me, o R5 es NH2 y R4 es = O, OH, OMe, Cl, Br, I, NH2, NHMe, NHcPr o SMe. En realizaciones preferidas del mismo, R4 y R5 son ambos NH2 o SMe, R5 es H o R4 es = O.
[0046] En otra realización de la invención, R1 es H, R2 es O-bencilo, R3 es F, R4 es SMe, NH2, OMe o OCH2CH3, R5 es H, SMe, SO2Me, NH2, N3 o F, R6 es bencilo y R7 es H u OH.
[0047] En una realización preferida de la invención, el compuesto de fórmula I es:


o una sal farmacéuticamente aceptable del mismo.
[0048] En otra realización de la invención, el compuesto de Fórmula I es:


o una sal farmacéuticamente aceptable del mismo.
[0049] En una realización aún más preferida de la invención, el compuesto de fórmula I es:



sal farmacéuticamente aceptable del mismo.
[0050] En una cierta realización de la invención, la presente invención se dirige a compuestos de Fórmula III:

en donde
R8 es NH2, OMe, OCH2CH3 o = O y
R9 es NH2, H, o F,
o una sal farmacéuticamente aceptable del mismo.
[0051] Preferiblemente, el compuesto de Fórmula III se selecciona del grupo que consiste en

o una sal farmacéuticamente aceptable del mismo. Mas preferiblemente, el compuesto de Formula III se selecciona del grupo que consiste en

una sal farmacéuticamente aceptable del mismo.
[0052] Los términos "composición farmacéutica" y "formulación farmacéutica" se usan indistintamente en el presente documento. Las composiciones farmacéuticas de la presente invención contienen compuestos de esta invención y se pueden formular usando cualquier vehículo y excipiente convencional, que se seleccionará de acuerdo con la práctica habitual. Los comprimidos contendrán excipientes, deslizantes, cargas, aglutinantes y similares. Las formulaciones farmacéuticas acuosas se preparan en forma estéril y, cuando están destinadas a ser administradas por una vía distinta a la oral, generalmente serán isotónicas. Todas las formulaciones farmacéuticas contendrán opcionalmente excipientes tales como los expuestos en el "Handbook of Pharmaceutical Excipients" (1986). Los excipientes adecuados incluyen, pero no se limitan a ácido ascórbico y otros antioxidantes, agentes quelantes, tales como EDTa , carbohidratos, tales como dextrano, hidroxialquilcelulosa, hidroxialquilmetilcelulosa, ácido esteárico y similares. El pH de las formulaciones farmacéuticas puede variar preferiblemente de aproximadamente 3 a aproximadamente 11, y más preferiblemente de aproximadamente 7 a aproximadamente 10.
[0053] Si bien es posible que los ingredientes activos sean administrados solos, puede ser preferible presentarlos como formulaciones farmacéuticas. Las formulaciones farmacéuticas, tanto para uso veterinario como para uso humano, de la invención comprenden al menos un ingrediente activo, como se definió anteriormente, junto con uno o más vehículos o excipientes y opcionalmente agentes terapéuticos adicionales.
[0054] Una cantidad terapéuticamente eficaz o dosis eficaz se utilizan indistintamente en este documento y se entiende que significa la cantidad de ingrediente activo necesaria para lograr el resultado deseado. La dosis eficaz de ingrediente activo depende, al menos, de la naturaleza de la afección que se está tratando, la toxicidad, si el compuesto se usa profilácticamente (dosis más bajas) o contra una infección viral activa, el método de administración y la formulación
farmacéutica y puede ser determinado fácilmente por el médico usando estudios convencionales de aumento de dosis. La cantidad eficaz puede ser de aproximadamente 0,0001 a aproximadamente 100 mg/kg de peso corporal por día; preferiblemente, de aproximadamente 0,01 a aproximadamente 10 mg/kg de peso corporal por día; más preferiblemente, de aproximadamente 0,01 a aproximadamente 5 mg/kg de peso corporal por día; y lo más preferiblemente, de aproximadamente 0,05 a aproximadamente 0,5 mg/kg de peso corporal por día. Por ejemplo, la dosis candidata diaria para un ser humano adulto de aproximadamente 70 kg de peso corporal puede oscilar entre aproximadamente 1 mg y aproximadamente 1000 mg, preferiblemente entre aproximadamente 5 mg y aproximadamente 500 mg, y puede tomar la forma de dosis únicas o múltiples.
[0055] En otra realización, la composición farmacéutica comprende además al menos un agente terapéutico adicional. El agente terapéutico adicional puede ser otro compuesto de Fórmula I o cualquier agente terapéutico adecuado para usar con el compuesto de Fórmula I. Por ejemplo, el agente terapéutico adicional puede seleccionarse del grupo que consiste en un corticosteroide, un modulador de la transducción de señales antiinflamatorias, un broncodilatador agonista de los receptores adrenérgicos p2, un anticolinérgico, un agente mucolítico, solución salina hipertónica, un agente que inhibe la migración de células proinflamatorias al sitio de infección y mezclas de las mismas. El agente terapéutico adicional también puede incluir otros fármacos para tratar infecciones por virus Orthomyxoviridae. En otras realizaciones, el agente terapéutico adicional puede ser inhibidores de hemaglutinina viral, inhibidores de neuramidasa viral, bloqueadores de canales de iones M2, inhibidores de polimerasa ARNs dependientes de ARN de Orthomyxoviridae, sialidasas y otros fármacos para tratar infecciones por Orthomyxoviridae. En otra realización más, el agente terapéutico adicional es un interferón, ribavirina, oseltamivir, zanamivir, laninamivir, peramivir, amantadina, rimantadina, CS-8958, favipiravir, AVI-7100, inhibidor de la proteasa alfa-1 o DAS 181.
[0056] En otras ciertas realizaciones de la invención, la infección Orthomyxoviridae a tratar por la composición farmacéutica es causada por un virus de influenza A, un virus de influenza B o un virus de influenza C.
[0057] Las composiciones farmacéuticas incluyen las adecuadas para cualquier vía de administración apropiada para la afección a tratar. Las rutas adecuadas incluyen oral, inhalación, rectal, nasal, tópica (incluyendo bucal y sublingual), vaginal y parenteral (incluyendo subcutánea, intramuscular, intravenosa, intradérmica, intratecal y epidural) y similares. Se apreciará que la ruta preferida puede variar, por ejemplo, con la condición del receptor.
[0058] Las composiciones farmacéuticas se pueden presentar convenientemente en forma de dosificación unitaria y se pueden preparar mediante cualquier método conocido en la técnica de la farmacia. Las técnicas y composiciones farmacéuticas se encuentran generalmente en Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA). Dichos métodos incluyen la etapa de asociar el ingrediente activo con el vehículo que constituye uno o más ingredientes accesorios. En general, las composiciones farmacéuticas se preparan asociando uniforme e íntimamente el ingrediente activo con vehículos líquidos o vehículos sólidos finamente divididos o ambos, y luego, si es necesario, dando forma al producto.
[0059] Las composiciones farmacéuticas de la presente invención adecuadas para administración oral pueden presentarse como unidades discretas tales como cápsulas, sellos o comprimidos conteniendo cada uno una cantidad predeterminada del ingrediente activo; como polvo o gránulos; como una solución o suspensión en un líquido acuoso o no acuoso; o como una emulsión líquida de aceite en agua o una emulsión líquida de agua en aceite. El ingrediente activo también se puede administrar en forma de bolo, electuario o pasta.
[0060] Un comprimido se fabrica mediante compresión o moldeo, opcionalmente con uno o más ingredientes accesorios. Los comprimidos pueden prepararse comprimiendo en una máquina adecuada el ingrediente activo en una forma fluida tal como un polvo o gránulos, opcionalmente mezclado con un aglutinante, lubricante, diluyente inerte, conservante, tensioactivo o agente dispersante. Los comprimidos moldeados se pueden preparar moldeando en una máquina adecuada una mezcla del ingrediente activo en polvo humedecido con un diluyente líquido inerte. Los comprimidos pueden opcionalmente recubrirse o ranurarse y opcionalmente se formulan de modo que proporcionen una liberación lenta o controlada del ingrediente activo de los mismos.
[0061] Para las infecciones del ojo u otros tejidos externos, por ejemplo boca y piel, las composiciones farmacéuticas se aplican preferiblemente como una pomada o crema tópica que contiene el ingrediente activo en una cantidad de, por ejemplo, 0,075 a 20% p/p (incluyendo activo(s) ingrediente(s) en un intervalo entre 0,1% y 20% en incrementos de 0,1% p/p tal como 0,6% p/p, 0,7% p/p, etc.), preferiblemente 0,2 a 15% p/p y lo más preferiblemente de 0,5 a 10% p/p. Cuando se formula en una pomada, los ingredientes activos se pueden emplear con una base de pomada parafínica o miscible en agua. Alternativamente, los ingredientes activos se pueden formular en una crema con una base de crema de aceite en agua.
[0062] Si se desea, la fase acuosa de la base de crema puede incluir, por ejemplo, al menos 30% p/p de un alcohol polihídrico, es decir, un alcohol que tiene dos o más grupos hidroxilo tales como propilenglicol, butano 1,3-diol, manitol, sorbitol, glicerol y polietilenglicol (incluido PEG 400) y mezclas de los mismos. Las composiciones farmacéuticas tópicas pueden incluir deseablemente un compuesto que mejore la absorción o penetración del ingrediente activo a través de la piel u otras áreas afectadas. Ejemplos de tales potenciadores de la penetración dérmica incluyen dimetilsulfóxido y análogos relacionados.
[0063] La fase oleosa de las emulsiones de esta invención puede estar constituida por ingredientes conocidos de una manera conocida. Si bien la fase puede comprender simplemente un emulsionante (también conocido como emulgente), es deseable que comprenda una mezcla de al menos un emulsionante con una grasa o un aceite o con una grasa y un aceite. Preferiblemente, se incluye un emulsionante hidrófilo junto con un emulsionante lipófilo, que actúa como estabilizador. También se prefiere incluir tanto un aceite como una grasa. Juntos, el (los) emulsionante(s) con o sin estabilizador(es) forman la denominada cera emulsionante, y la cera junto con el aceite y la grasa forman la denominada base de ungüento emulsionante que forma la fase oleosa dispersa de las composiciones farmacéuticas de crema.
[0064] Los emulgentes y estabilizadores de emulsión adecuados para su uso en la composición farmacéutica de la invención incluyen, entre otros, Tween® 60, Span® 80, alcohol cetoestearílico, alcohol bencílico, alcohol miristílico, monoestearato de glicerilo y laurilsulfato de sodio..
[0065] La elección de aceites o grasas para la composición farmacéutica adecuados se basa en lograr las propiedades cosméticas deseadas. La crema debe ser preferiblemente un producto no graso, que no manche y lavable con una consistencia adecuada para evitar fugas de tubos u otros recipientes. Ésteres de alquilo mono o dibásicos de cadena lineal o ramificada, tales como diisoadipato, estearato de isocetilo, diéster de propilenglicol de ácidos grasos de coco, miristato de isopropilo, oleato de decilo, palmitato de isopropilo, estearato de butilo, palmitato de 2-etilhexilo o una mezcla de ésteres de cadena ramificada conocidos como se puede utilizar Crodamol CAP, siendo los tres últimos ésteres preferidos. Estos pueden usarse solos o en combinación dependiendo de las propiedades requeridas. Alternativamente, se utilizan lípidos de alto punto de fusión tales como parafina blanda blanca y/o parafina líquida u otros aceites minerales.
[0066] Las composiciones farmacéuticas según la presente invención comprenden una combinación de acuerdo con la invención junto con uno o más vehículos o excipientes farmacéuticamente aceptables y agentes terapéuticos opcionalmente adicionales. Las composiciones farmacéuticas que contienen el ingrediente activo pueden estar en cualquier forma adecuada para el método de administración pretendido. Cuando se usa para uso oral, por ejemplo, se pueden preparar tabletas, grageas, suspensiones acuosas o oleosas, polvos o gránulos dispersables, emulsiones, cápsulas duras o blandas, jarabes o elixires. Las composiciones farmacéuticas destinadas a uso oral se pueden preparar de acuerdo con cualquier método conocido en la técnica para la fabricación de composiciones farmacéuticas y dichas composiciones pueden contener uno o más agentes, incluidos agentes edulcorantes, aromatizantes, colorantes y conservantes, para proporcionar una preparación agradable al paladar. Son aceptables los comprimidos que contienen el ingrediente activo mezclado con un excipiente no tóxico farmacéuticamente aceptable que sea adecuado para la fabricación de comprimidos. Estos excipientes pueden ser, por ejemplo, diluyentes inertes, tales como carbonato cálcico o sódico, lactosa, fosfato cálcico o sódico; agentes de granulación y desintegración, tales como almidón de maíz o ácido algínico; agentes aglutinantes, como almidón, gelatina o goma arábiga; y agentes lubricantes, tales como estearato de magnesio, ácido esteárico o talco. Los comprimidos pueden no estar recubiertos o pueden recubrirse mediante técnicas conocidas que incluyen microencapsulación para retrasar la desintegración y adsorción en el tracto gastrointestinal y, por lo tanto, proporcionar una acción sostenida durante un período más largo. Por ejemplo, puede emplearse un material de retardo temporal tal como monoestearato de glicerilo o diestearato de glicerilo solo o con una cera.
[0067] Las composiciones farmacéuticas para uso oral pueden también presentarse como cápsulas de gelatina dura donde el activo ingrediente se mezcla con un diluyente inerte sólido, por ejemplo fosfato de calcio o caolín, o como cápsulas de gelatina blanda en las que el ingrediente activo se mezcla con agua o un medio oleoso, como aceite de cacahuete, parafina líquida o aceite de oliva.
[0068] Las suspensiones acuosas de la invención contienen los materiales activos en mezcla con excipientes adecuados para la fabricación de suspensiones acuosas. Dichos excipientes incluyen un agente de suspensión, como carboximetilcelulosa de sodio, metilcelulosa, hidroxipropilmetilcelulosa, alginato de sodio, polivinilpirrolidona, goma de tragacanto y goma arábiga, y agentes dispersantes o humectantes como un fosfátido natural (por ejemplo, lecitina), un producto de condensación de un óxido de alquileno con un ácido graso (p. ej., estearato de polioxietileno), un producto de condensación de óxido de etileno con un alcohol alifático de cadena larga (p. ej., heptadecaetilenoxicetanol), un producto de condensación de óxido de etileno con un éster parcial derivado de un ácido graso y un hexitol anhídrido (por ejemplo, monooleato de polioxietilensorbitán). La suspensión acuosa también puede contener uno o más conservantes tales como p-hidroxibenzoato de etilo o n-propilo, uno o más agentes colorantes, uno o más agentes aromatizantes y uno o más agentes edulcorantes, tales como sacarosa o sacarina.
[0069] Las suspensiones de aceite se pueden formular suspendiendo el ingrediente activo en un aceite vegetal, tal como aceite de cacahuete, aceite de oliva, aceite de sésamo o aceite de coco, o en un aceite mineral tal como parafina líquida. Las suspensiones orales pueden contener un agente espesante, como cera de abejas, parafina dura o alcohol cetílico. Pueden añadirse agentes edulcorantes, tales como los expuestos anteriormente, y agentes aromatizantes para proporcionar una preparación oral agradable. Estas composiciones pueden conservarse mediante la adición de un antioxidante como el ácido ascórbico.
[0070] Los polvos y gránulos de la invención adecuados para la preparación de una suspensión acuosa mediante la adición de agua dispersable proporcionan el ingrediente activo en mezcla con un agente dispersante o humectante, un agente de suspensión, y uno o más conservantes. Los agentes dispersantes o humectantes y los agentes de suspensión
adecuados se ejemplifican por los descritos anteriormente. También pueden estar presentes excipientes adicionales, por ejemplo, agentes edulcorantes, aromatizantes y colorantes.
[0071] Las composiciones farmacéuticas de la invención también pueden estar en forma de emulsiones de aceite-en agua. La fase oleosa puede ser un aceite vegetal, como aceite de oliva o aceite de cacahuete, un aceite mineral, como parafina líquida, o una mezcla de estos. Los agentes emulsionantes adecuados incluyen gomas de origen natural, como goma arábiga y goma de tragacanto, fosfátidos de origen natural, como lecitina de soja, ésteres o ésteres parciales derivados de ácidos grasos y anhídridos de hexitol, como monooleato de sorbitán, y productos de condensación de estos ésteres parciales con óxido de etileno, tales como monooleato de polioxietilensorbitán. La emulsión también puede contener agentes edulcorantes y aromatizantes. Los jarabes y elixires se pueden formular con agentes edulcorantes, como glicerol, sorbitol o sacarosa. Tales composiciones farmacéuticas también pueden contener un demulcente, un conservante, un aromatizante o un colorante.
[0072] Las composiciones farmacéuticas de la invención pueden estar en la forma de una preparación inyectable estéril, tal como una suspensión acuosa u oleaginosa inyectable estéril. Esta suspensión se puede formular de acuerdo con la técnica conocida usando aquellos agentes dispersantes o humectantes y agentes de suspensión adecuados que se han mencionado anteriormente. La preparación inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o solvente parenteralmente aceptable no tóxico, tal como una solución en 1,3-butanodiol o preparada como un polvo liofilizado. Entre los vehículos y disolventes aceptables que se pueden emplear se encuentran el agua, la solución de Ringer y la solución isotónica de cloruro de sodio. Además, los aceites fijos, estériles, convencionalmente se pueden emplear como medio disolvente o de suspensión. Para este propósito, puede emplearse cualquier aceite fijo suave, incluidos mono o diglicéridos sintéticos. Además, también se pueden utilizar ácidos grasos como el ácido oleico en la preparación de inyectables.
[0073] La cantidad de ingrediente activo que se puede combinar con un vehículo, por ejemplo, para producir una dosis única forma variará dependiendo del huésped tratado y del modo particular de administración. Por ejemplo, una composición farmacéutica de liberación prolongada destinada a la administración oral a seres humanos puede contener de aproximadamente 1 a aproximadamente 1000 mg de ingrediente activo combinado con una cantidad apropiada y conveniente de vehículo que puede variar de aproximadamente 5 a aproximadamente 95% de las composiciones totales (peso:peso). La composición farmacéutica se puede preparar para proporcionar cantidades fácilmente mensurables para la administración. Por ejemplo, una solución acuosa destinada a la infusión intravenosa puede contener de aproximadamente 3 a aproximadamente 500 |jg del ingrediente activo por mililitro de solución con el fin de lograr una velocidad de infusión de alrededor de 30 ml/h.
[0074] Las composiciones farmacéuticas adecuadas para la administración tópica en el ojo también incluyen gotas oculares en las que el ingrediente activo se disuelve o suspende en un vehículo adecuado, especialmente un disolvente acuoso para el ingrediente activo. El ingrediente activo está presente preferiblemente en tales composiciones farmacéuticas en una concentración de aproximadamente 0,5 a aproximadamente 20% p/p.
[0075] Las composiciones farmacéuticas adecuadas para la administración tópica en la boca incluyen pastillas que comprenden el ingrediente activo en una base aromatizada, normalmente sacarosa y acacia o tragacanto; pastillas que comprenden el ingrediente activo en una base inerte, tal como gelatina y glicerina, o sacarosa y goma arábiga; y enjuagues bucales que comprenden el ingrediente activo en un vehículo líquido adecuado.
[0076] Las composiciones farmacéuticas para administración rectal pueden presentarse como un supositorio con una base adecuada que comprende por ejemplo manteca de cacao o un salicilato.
[0077] Las composiciones farmacéuticas adecuadas para la administración intrapulmonar o nasal tienen un tamaño de partícula por ejemplo en el intervalo de aproximadamente 0,1 a aproximadamente 500 micrómetros, tal como aproximadamente 0,5, aproximadamente 1, aproximadamente 30, aproximadamente 35 etc., que se administra por inhalación rápida a través del pasaje nasal o por inhalación por la boca para llegar a los sacos alveolares. Las composiciones farmacéuticas adecuadas incluyen soluciones acuosas u oleosas del ingrediente activo. Las composiciones farmacéuticas adecuadas para la administración de aerosol o polvo seco se pueden preparar de acuerdo con métodos convencionales y se pueden administrar con agentes terapéuticos adicionales tales como compuestos usados hasta ahora en el tratamiento o profilaxis de infecciones por Orthomyxoviridae como se describe a continuación.
[0078] En otro aspecto, la invención es un nuevo, eficaz, seguro, no irritante y la composición inhalable fisiológicamente compatible que comprende un compuesto de Fórmula I, II, o una sal farmacéuticamente aceptable del mismo, adecuado para el tratamiento de infecciones Orthomyxoviridae y bronquiolitis potencialmente asociada. Las sales farmacéuticamente aceptables preferidas son las sales de ácidos inorgánicos, incluidas las sales de clorhidrato, bromhidrato, sulfato o fosfato, ya que pueden causar menos irritación pulmonar. Preferiblemente, la composición farmacéutica inhalable se administra al espacio endobronquial en un aerosol que comprende partículas con un diámetro aerodinámico medio de masa (MMAD) entre aproximadamente 1 y aproximadamente 5 jm . Preferiblemente, el compuesto de Fórmula I se formula para la administración en aerosol usando un nebulizador, un inhalador de dosis medida presurizado (pMDI) o un inhalador de polvo seco (DPI).
[0079] Los ejemplos no limitantes de nebulizadores incluyen atomización, jet, ultrasónica, presión, vibración placa porosa, o nebulizadores equivalentes incluyendo aquellos nebulizadores que utilizan la tecnología de suministro de aerosol adaptativo (Denyer, J. Aerosol Pulmonary Medicine Drug Delivery 2010, 23 Supl 1, S1-S10). Un nebulizador de chorro utiliza presión de aire para romper una solución líquida en gotitas de aerosol. Un nebulizador ultrasónico funciona mediante un cristal piezoeléctrico que corta un líquido en pequeñas gotas de aerosol. Un sistema de nebulización presurizado fuerza la solución a presión a través de pequeños poros para generar gotas de aerosol. Un dispositivo de placa porosa vibrante utiliza una vibración rápida para cortar una corriente de líquido en tamaños de gota apropiados.
[0080] En una realización preferida, la composición farmacéutica para la nebulización se entrega al espacio endobronquial en un aerosol que comprende partículas con un MMAD predominantemente entre aproximadamente 1 pm y aproximadamente 5 pm usando un nebulizador capaz de aerosolizar la composición farmacéutica del compuesto de Fórmula I en partículas del MMAD requerido. Para ser óptimamente eficaces desde el punto de vista terapéutico y evitar efectos secundarios sistémicos y de las vías respiratorias superiores, la mayoría de las partículas aerosolizadas no deberían tener un MMAD superior a aproximadamente 5 pm. Si un aerosol contiene una gran cantidad de partículas con un MMAD mayor de 5 pm, las partículas se depositan en las vías respiratorias superiores disminuyendo la cantidad de fármaco administrada al sitio de inflamación y broncoconstricción en el tracto respiratorio inferior. Si el MMAD del aerosol es menor de aproximadamente 1 pm, entonces las partículas tienden a permanecer suspendidas en el aire inhalado y posteriormente se exhalan durante la espiración.
[0081] Cuando se formula y administra de acuerdo con el método de la invención, la composición farmacéutica de aerosol para la nebulización suministra una dosis terapéuticamente eficaz del compuesto de Fórmula I al sitio de infección Orthomyxoviridae suficiente para tratar la infección Orthomyxoviridae. La cantidad de fármaco administrado debe ajustarse para reflejar la eficacia de la administración de una dosis terapéuticamente eficaz del compuesto de Fórmula I. En una realización preferida, una combinación de la composición farmacéutica en aerosol acuoso con el poroso atomizador, de chorro, presurizado y placa vibrante o nebulizador ultrasónico permite, dependiendo del nebulizador, aproximadamente, al me nos, 20, a aproximadamente 90%, típicamente aproximadamente 70% de suministro de la dosis administrada del compuesto de Fórmula I, II o III en las vías respiratorias. En una realización preferida, se administra al menos de aproximadamente un 30 a aproximadamente un 50% del ingrediente activo. Más preferiblemente, se administra de aproximadamente un 70 a aproximadamente un 90% del ingrediente activo.
[0082] En otra realización de la presente invención, un compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo, se entrega como un polvo inhalable seco. Los compuestos de la invención se administran endobronquialmente como una composición farmacéutica de polvo seco para administrar eficazmente partículas finas de compuesto en el espacio endobronquial usando polvo seco o inhaladores de dosis medidas. Para el suministro por DPI, el compuesto de Fórmula I se procesa en partículas con, predominantemente, MMAD entre aproximadamente 1 pm y aproximadamente 5 pm mediante molienda, secado por pulverización, procesamiento de fluidos críticos o precipitación de la solución. Los dispositivos y procedimientos de molienda de medios, molienda por chorro y secado por pulverización capaces de producir tamaños de partícula con un MMAD entre aproximadamente 1 pm y aproximadamente 5 pm son bien conocidos en la técnica. En una realización, se añaden excipientes al compuesto de Fórmula I antes de procesarlo en partículas de los tamaños requeridos. En otra realización, los excipientes se mezclan con las partículas del tamaño requerido para ayudar en la dispersión de las partículas de fármaco, por ejemplo, usando lactosa como excipiente.
[0083] Determinaciones del tamaño de partícula se realizan mediante dispositivos bien conocidos en la técnica. Por ejemplo, un impactador en cascada Anderson de múltiples etapas u otro método adecuado como los citados específicamente en el Capítulo 601 de la Farmacopea de EE. UU. Como dispositivos de caracterización para aerosoles dentro de inhaladores de dosis medidas y de polvo seco.
[0084] En otra realización preferida, un compuesto de Fórmula I se suministra como un polvo seco usando un dispositivo de este tipo como un inhalador de polvo seco u dispositivos de otro tipo de dispersión de polvo seco. Los ejemplos no limitantes de dispositivos e inhaladores de polvo seco incluyen los descritos en el documento US 5,458,135; US5,740,794; US5775320; US5,785,049; US3.906.950; US4,013,075; US4,069,819; US4,995,385; US5,522,385; US4,668,218; US4,667,668; US4,805,811 y US5,388,572. Hay dos diseños principales de inhaladores de polvo seco. Un diseño es un dispositivo dosificador en donde se coloca un depósito para el fármaco dentro del dispositivo y el paciente añade una dosis del fármaco en la cámara de inhalación. El segundo diseño es un dispositivo medido en fábrica en donde cada dosis individual se ha fabricado en un recipiente separado. Ambos sistemas dependen de la composición farmacéutica del fármaco en pequeñas partículas de MMAD de 1 pm y aproximadamente 5 pm, y a menudo implican la coformulación con partículas de excipiente más grandes como, pero sin limitarse a lactosa. El polvo del fármaco se coloca en la cámara de inhalación (ya sea mediante la medición del dispositivo o mediante la rotura de una dosis medida de fábrica) y el flujo inspiratorio del paciente acelera el polvo fuera del dispositivo y hacia la cavidad oral. Las características de flujo no laminar de la trayectoria del polvo hacen que los agregados excipiente-fármaco se descompongan, y la masa de las partículas grandes del excipiente provoca su impacto en la parte posterior de la garganta, mientras que las partículas más pequeñas del fármaco se depositan profundamente en los pulmones. En realizaciones preferidas, un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, se administra como un polvo seco usando cualquier tipo de inhalador de polvo seco como se describe en este documento, en donde el MMAD del polvo seco, excluyendo cualquier excipiente, está predominantemente en el intervalo de aproximadamente 1 pm a aproximadamente 5 pm.
[0085] En otra realización preferida, un compuesto de Fórmula I se suministra como un polvo seco usando una inhalador de dosis medida. Los ejemplos no limitantes de inhaladores y dispositivos de dosis medidas incluyen los descritos en el documento US5,261.538; US5,544,647; US5,622,163; US4,955,371; US3,565,070; US3,361306 y US6,116,234. En realizaciones preferidas, un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, se administra como un polvo seco usando un inhalador de dosis medida en donde el MMAD del polvo seco, excluyendo cualquier excipiente, está predominantemente en el intervalo de aproximadamente 1 a aproximadamente 5 |jm.
[0086] Las composiciones farmacéuticas adecuadas para la administración vaginal pueden presentarse como pesarios, tampones, cremas, geles, pastas, espumas o aerosoles composiciones farmacéuticas que contienen, además del ingrediente activo los portadores como son conocidos en la técnica por ser apropiados.
[0087] Las composiciones farmacéuticas adecuadas para administración parenteral incluyen soluciones para inyección estériles acuosas y no acuosas que pueden contener antioxidantes, tampones, bacteriostáticos y solutos que hacen la composición farmacéutica isotónica con la sangre del receptor previsto; y suspensiones estériles acuosas y no acuosas que pueden incluir agentes de suspensión y agentes espesantes.
[0088] Las composiciones farmacéuticas se presentan en dosis unitarias o recipientes multidosis, por ejemplo ampollas selladas y viales, y pueden almacenarse en una condición secada por congelación (liofilizada) que requiere sólo la adición del vehículo líquido estéril, por ejemplo agua para inyección, inmediatamente antes de su uso. Las soluciones y suspensiones para inyección extemporánea se preparan a partir de polvos, gránulos y comprimidos estériles del tipo descrito anteriormente. Las composiciones farmacéuticas de dosificación unitaria preferidas son aquellas que contienen una dosis diaria o una subdosis diaria unitaria, como se ha mencionado anteriormente, o una fracción apropiada de la misma, del ingrediente activo.
[0089] Debe entenderse que además de los ingredientes particularmente mencionados anteriormente las composiciones farmacéuticas de esta invención pueden incluir otros agentes convencionales en la técnica teniendo en cuenta el tipo de composición farmacéutica en cuestión, por ejemplo aquellas adecuadas para administración oral pueden incluir agentes aromatizantes.
[0090] La invención proporciona además composiciones veterinarias que comprenden al menos un ingrediente activo como antes se define, junto con un vehículo veterinario para el mismo.
[0091] Los vehículos veterinarios son materiales útiles para el propósito de administrar la composición y pueden ser materiales sólidos, líquidos o gaseosos que son de otro modo inertes o aceptables en la técnica veterinaria y son compatibles con el ingrediente activo. Estas composiciones veterinarias se pueden administrar por vía oral, parenteral o por cualquier otra vía deseada.
[0092] Los compuestos de la invención se puede usar para proporcionar formulaciones farmacéuticas de liberación controlada que contiene como un ingrediente activo o más compuestos de la invención ("formulaciones de liberación controlada") en las que la liberación del ingrediente activo se controlan y regulan para permitir menos frecuencia de dosificación o para mejorar el perfil farmacocinético o de toxicidad de un ingrediente activo dado.
[0093] En otra realización, la presente solicitud describe composiciones farmacéuticas que comprenden un compuesto de la presente invención, o una sal farmacéuticamente aceptable del mismo, en combinación con al menos un adicional agente terapéutico, y un vehículo o excipiente farmacéuticamente aceptable.
[0094] Para el tratamiento de infecciones de virus Orthomyxoviridae, preferiblemente, el agente terapéutico adicional es activo contra infecciones por virus Orthomyxoviridae, particularmente infecciones de virus de influenza. Ejemplos no limitantes de estos agentes terapéuticos activos son inhibidores de hemaglutinina viral, inhibidores de neuramidasa viral, bloqueadores de canales de iones M2, inhibidores de polimerasa ARNs dependientes de ARN de Orthomyxoviridae y sialidasas. Los ejemplos no limitantes de inhibidores de la neuramidasa incluyen oseltamivir, zanamivir, laninamivir, peramivir y CS-8958. Los ejemplos no limitantes de inhibidores del canal M2 viral incluyen amantadina y rimantadina. Ejemplos no limitantes de inhibidores de polimerasa ARNs dependientes de ARN de Orthomyxoviridae son ribavirina y favipiravir. Un ejemplo no limitante de sialidasas es DAS 181. En otra realización, el agente terapéutico adicional se selecciona del grupo que consiste en ribavirina, oseltamivir, zanamivir, laninamivir, peramivir, amantadina, rimantadina, CS-8958, favipiravir, AVI-7100, inhibidor de la proteasa alfa-1 y DAS181.
[0095] Muchas de las infecciones de los virus Orthomyxoviridae son las infecciones respiratorias. Por lo tanto, pueden usarse agentes terapéuticos activos adicionales usados para tratar síntomas respiratorios y secuelas de infección en combinación con los compuestos de Fórmula I, II o III. Por ejemplo, otros agentes terapéuticos adicionales preferidos en combinación con los compuestos de Fórmula I, II o III para el tratamiento de infecciones respiratorias víricas incluyen, pero no se limitan a broncodilatadores y corticosteroides.
[0096] Los glucocorticoides, que se introdujeron por primera vez como una terapia de asma en 1950 (Carryer, Journal of Allergy, 21,282-287, 1950), siguen siendo el tratamiento más potente y consistentemente eficaz para esta enfermedad, aunque su mecanismo de acción no es aún entendido completamente (Morris, J. Allergy Clin. Immunol., 75 (1 Pt) 1-13,
1985). Desafortunadamente, las terapias con glucocorticoides orales se asocian con profundos efectos secundarios indeseables como obesidad del tronco, hipertensión, glaucoma, intolerancia a la glucosa, aceleración de la formación de cataratas, pérdida de minerales óseos y efectos psicológicos, todos los cuales limitan su uso como agentes terapéuticos a largo plazo (Goodman y Gilman, décima edición, 2001). Una solución a los efectos secundarios sistémicos es administrar esteroides directamente al sitio de la inflamación. Los corticosteroides inhalados (CSI) se han desarrollado para mitigar los efectos adversos graves de los esteroides orales. Ejemplos no limitativos de corticosteroides que pueden usarse en combinaciones con los compuestos de Fórmula I son dexametasona, dexametasona fosfato sódico, fluorometolona, acetato de fluorometolona, loteprednol, loteprednol etabonato, hidrocortisona, prednisolona, fludrocortisonas, triamcinometasona, triamcinometasona, metilprednisolona, fluocinolona, acetónido de fluocinolona, flunisolida, fluocortin-21-butilato, flumetasona, pivalato de flumetasona, budesonida, propionato de halobetasol, furoato de mometasona, propionato de fluticasona, ciclesonida; o sus sales farmacéuticamente aceptables.
[0097] Otros agentes anti-inflamatorios de trabajo a través de mecanismos de cascada anti-inflamatorios son también útiles como agentes terapéuticos adicionales en combinación con los compuestos de fórmula I para el tratamiento de infecciones respiratorias virales. Aplicar "moduladores de la transducción de señales antiinflamatorios" (a los que se hace referencia en este texto como AISTm ), como inhibidores de la fosfodiesterasa (p. ej., PDE-4, PDE-5 o PDE-7 específicos), inhibidores del factor de transcripción (p. ej., Bloqueo de NPkB mediante inhibición IKK), o inhibidores de la cinasa (por ejemplo, bloqueo de P38 MAP, JNK, PI3K, EGFR o Syk) es un enfoque lógico para apagar la inflamación, ya que estas pequeñas moléculas se dirigen a un número limitado de vías intracelulares comunes, aquellas vías de transducción de señales que son la intervención terapéutica antiinflamatoria (ver revisión de PJ Barnes, 2006). Estos agentes terapéuticos adicionales no limitantes incluyen: ácido 5-(2,4-difluoro-fenoxi)-1-isobutil-1H-indazol-6-carboxílico (2-dimetilamino-etil)-amida (inhibidor de cinasa P38 Map ARRY-797); 3-ciclopropilmetoxi-N-(3,5-dicloro-piridin-4-il)-4-difluorometoxibenzamida (inhibidor de PDE-4 Roflumilast); 4-[2-(3-ciclopentiloxi-4-metoxifenil)-2-fenil-etil]-piridina (inhibidor de PDE-4 CDP-840); N-(3,5-dicloro-4-piridinil)-4-(difluorometoxi)-8-[(metilsulfonil) amino]-1-dibenzofurancarboxamida (inhibidor de PDE-4 Oglemilast); N-(3,5-Dicloro-piridin-4-il)-2-[1-(4-fluorobencil)-5-hidroxi-1H-indol-3-il]-2-oxo-acetamida (PDE-4 inhibidor AWD 12-281); (3,5-dicloro-1-oxipiridin-4-il)-amida del ácido 8-metoxi-2-trifluorometil-quinolin-5-carboxílico (inhibidor de PDE-4 SCH351591); 4-[5-(4-fluorofenil)-2-(4-metanosulfinil-fenil)-1H-imidazol-4-il]-piridina (inhibidor de P38 SB-203850); 4-[4-(4-fluoro-fenil)-1-(3-fenil-propil)-5-piridin-4-il-1H-imidazol-2-il]-but-3-in-1-ol (Inhibidor de P38 RWJ-67657); éster 2-dietilaminoetílico del ácido 4-ciano-4-(3-ciclopentiloxi-4-metoxi-fenil)-ciclohexanocarboxílico (profármaco éster 2-dietil-etílico de Cilomilast, inhibidor de PDE-4); (3-cloro-4-fluorofenil)-[7-metoxi-6-(3-morfolin-4-il-propoxi)-quinazolin-4-il]-amina (Gefitinib, inhibidor de EGFR); y 4-(4-Metil-piperazin-1-ilmetil)-N-[4-metil-3-(4-piridin-3-il-pirimidin-2-ilamino)-fenil]-benzamida (Imatinib, inhibidor de EGFR).
[0098] Los agentes que inhiben la migración de células proinflamatorias al sitio de la infección también son útiles como agentes terapéuticos adicionales en combinación con los compuestos de Fórmula I para el tratamiento de infecciones respiratorias víricas. Ejemplos no limitativos de tales agentes que actúan a través de este mecanismo y han demostrado utilidad en animales, por ejemplo, reduciendo la mortalidad final causada por la influenza son EV-077 (un inhibidor dual de tromboxano sintasa/antagonista del receptor de tromboxano) y Fingolimod® (un antagonista del receptor esfingosina-1-fosfato).
[0099] Las combinaciones que comprenden broncodilatadores agonistas de los receptores adrenérgicos p2 inhalados tales como formoterol, albuterol o Salmeterol con los compuestos de Fórmula I también son combinaciones adecuadas, pero no limitantes, útiles para el tratamiento de infecciones virales respiratorias.
[0100] Las combinaciones de broncodilatadores agonistas p2-adrenérgicos inhalados tales como formoterol o Salmeterol con ICS también se utilizan para tratar tanto la broncoconstricción como la inflamación (Symbicort® y Advair®, respectivamente). Las combinaciones que comprenden estas combinaciones de agonistas de los receptores adrenérgicos p2 y ICS junto con los compuestos de Fórmula I también son combinaciones adecuadas, pero no limitantes, útiles para el tratamiento de infecciones virales respiratorias.
[0101] Para el tratamiento o la profilaxis de bronco-constricción pulmonar, los anticolinérgicos son de uso potencial y, por lo tanto, útiles como agentes terapéuticos adicionales en combinación con los compuestos de fórmula I para el tratamiento de las infecciones respiratorias virales. Estos anticolinérgicos incluyen, pero no se limitan a antagonistas del receptor muscarínico (particularmente del subtipo M3) que han demostrado eficacia terapéutica en el hombre para el control del tono colinérgico en la EPOC (Witek, 1999); ácido 1-{4-hidroxi-1-[3,3,3-tris-(4-fluorofenil)-propionil]-pirrolidin-2-carbonil} -pirrolidin-2-carboxílico (1-metil-piperidin-4-ilmetilo)-amida; 3-[3-(2-dietilamino-acetoxi)-2-fenil-propioniloxi]-8-isopropil-8-metil-8-azonia-biciclo[3,2,1]octano (ipratropio-N,N-dietilglicinato); éster 1-aza-biciclo[2,2,2]oct-3-ilo del ácido 1-ciclohexil-3,4-dihidro-1H-isoquinolin-2-carboxílico (solifenacina); ácido 2-hidroximetil-4-metanosulfinil-2-fenil-butírico éster de 1-azabiciclo[2,2,2]oct-3-ilo (Revatropate); 2-{1-[2-(2,3-dihidro-benzofuran-5-il)-etil]-pirrolidin-3-il}-2,2-difinil-acetamida (darifenacina); 4-azepan-1-il-2,2-difenil-butiramida (Buzepida); 7-[3-(2-dietilamino-acetoxi)-2-fenil-propioniloxi]-9-etil-9-metil-3-oxa-9-azonia-triciclo[3,3,1.02,4]nonano (Oxitropio-N,N-dietilglicinato); 7-[2-(2-dietilamino-acetoxi)-2,2-di-tiofen-2-il-acetoxi]-9,9-dimetil-3-oxa-9-azonia-triciclo[3,3,1.02,4]nonano (Tiotropio-N,N-dietilglicinato); éster 2-(3-diisopropilamino-1-fenilpropil)-4-metil-fenílico del ácido dimetilamino-acético (tolterodina-N,N-dimetilglicinato); 3-[4,4-Bis-(4-fluoro-fenil)-2-oxo-imidazolidin-1 -il]-1-metil-1 -(2-oxo-2-piridin-2-il-etil)-pirrolidinio; 1-[1-(3-fluoro-bencil)-piperidin-4-il]-4,4-bis-(4-fluorofenil)-imidazolidin-2-ona; 1-ciclooctil-3-(3-metoxi-1-aza-biciclo[2,2,2]oct-3-il)-1-fenil-prop-2-in-1-ol; 3-[2-(2-dietilamino-acetoxi)-2,2-di-tiofen-2-il-acetoxi]-1-(3-fenoxipropil)-1-azonia-biciclo[2,2,2]octano (Aclidmio-N,Ndietilglicinato); o 1 -metil-1 -(2-fenoxi-etil)-piperidin-4-ilo del ácido (2-dietilaminoacetoxi)-di-tiofen-2-il-acético.
[0102] Los compuestos de Fórmula I también pueden combinarse con agentes mucolíticos para tratar tanto la infección como los síntomas de las infecciones respiratorias. Un ejemplo no limitante de un agente mucolítico es el ambroxol. De manera similar, los compuestos de Fórmula I se pueden combinar con expectorantes para tratar tanto la infección como los síntomas de las infecciones respiratorias. Un ejemplo no limitante de expectorante es la guaifenesina.
[0103] La solución salina hipertónica nebulizada se usa para mejorar la eliminación inmediata y a largo plazo de las pequeñas vías respiratorias en pacientes con enfermedades pulmonares (Kuzik, J. Pediatrics 2007, 266). Los compuestos de Fórmula I también pueden combinarse con solución salina hipertónica nebulizada, particularmente cuando la infección por el virus Orthomyxoviridae se complica con bronquiolitis. La combinación de los compuestos de Fórmula I con solución salina hipertónica también puede comprender cualquiera de los agentes adicionales discutidos anteriormente. En un aspecto preferido, se usa solución salina hipertónica nebulizada al 3% aproximadamente.
[0104] También es posible combinar cualquier compuesto de la invención con uno o más de otros agentes terapéuticos activos en una forma de dosificación unitaria para la administración simultánea o secuencial a un paciente. La terapia de combinación se puede administrar como un régimen simultáneo o secuencial. Cuando se administra secuencialmente, la combinación se puede administrar en dos o más administraciones.
[0105] La coadministración de un compuesto de la invención con uno o más de otros agentes terapéuticos activos generalmente se refiere a la administración simultánea o secuencial de un compuesto de la invención y uno o más de otros agentes terapéuticos activos, de manera que cantidades terapéuticamente eficaces del compuesto de la invención y uno o más de otros agentes terapéuticos activos están presentes en el cuerpo del paciente.
[0106] La coadministración incluye la administración de dosis unitarias de los compuestos de la invención antes o después de la administración de dosis unitarias de uno o más de otros agentes terapéuticos activos, por ejemplo, la administración de los compuestos de la invención en segundos, minutos u horas. de la administración de uno o más de otros agentes terapéuticos activos. Por ejemplo, puede administrarse primero una dosis unitaria de un compuesto de la invención, seguida en segundos o minutos por la administración de una dosis unitaria de uno o más de otros agentes terapéuticos activos.
[0107] Alternativamente, una dosis unitaria de uno o más agentes terapéuticos adicionales se puede administrar primero, seguido por la administración de una dosis unitaria de un compuesto de la invención en cuestión de segundos o minutos. En algunos casos, puede ser deseable administrar una dosis unitaria de un compuesto de la invención primero, seguido, después de un período de horas (por ejemplo, 1-12 horas), por la administración de una dosis unitaria de uno o más de otros agentes terapéuticos activos. En otros casos, puede ser deseable administrar una dosis unitaria de uno o más de otros agentes terapéuticos activos primero, seguido, después de un período de horas (por ejemplo, 1-12 horas), por la administración de una dosis unitaria de un compuesto del invención.
[0108] En la presente memoria se describe un método para tratar una infección Orthomyxoviridae en un mamífero en necesidad del mismo. El método comprende la etapa de administrar una cantidad terapéuticamente eficaz de un compuesto de Fórmula I, como se definió anteriormente con respecto a la primera realización de la invención. En una determinada realización del mismo, el compuesto de Fórmula I está representado por la Fórmula II o la Fórmula III, como se definió anteriormente con respecto a la primera realización de la invención. Los términos en el método aquí descrito se definen como anteriormente con respecto a la primera realización de la invención. Las realizaciones preferidas de R1, R2, R3, R4, R5, R6 y R7 en el método aquí descrito son las mismas que para la primera realización de la invención.



o una sal farmacéuticamente aceptable del mismo, y más preferiblemente es

o una sal farmacéuticamente aceptable del mismo.
[0110] En otra realización preferida de la invención, el compuesto de fórmula I es:



o una sal farmacéuticamente aceptable del mismo.
[0111] En otra realización del método descrito, una cantidad terapéuticamente eficaz de un racemato, enantiómero, diastereómero, tautómero, polimorfo, pseudopolimorfo, forma amorfa, o hidrato de un compuesto de fórmula descrita I o una sal farmacéuticamente aceptable del mismo se administra a un mamífero que lo necesite.
[0112] En otra realización, el presente documento describe el uso de un compuesto de Fórmula I o una sal farmacéuticamente aceptable del mismo para tratar una infección viral causada por un virus Orthomyxoviridae.
[0113] En otro aspecto de este método descrito, la infección Orthomyxoviridae que se está tratando es una infección de influenza de virus A. En otro aspecto de este método descrito, la infección por Orthomyxoviridae es una infección por el virus B de la influenza. En otro aspecto de este método descrito, la infección por Orthomyxoviridae es una infección por el virus C de la influenza.
[0114] En una realización preferida, el método descrito aquí comprende el tratamiento de una infección Orthomyxoviridae en un mamífero en necesidad de la misma mediante la administración de una cantidad terapéuticamente eficaz de un compuesto de Fórmula I o una sal farmacéuticamente aceptable o éster del mismo.
[0115] En otra realización, el método descrito aquí comprende el tratamiento de una infección Orthomyxoviridae en un mamífero en necesidad del mismo mediante la administración de una cantidad terapéuticamente eficaz de una composición farmacéutica que comprende una cantidad eficaz de un compuesto de Fórmula I, o una sal farmacéuticamente aceptable del mismo, en combinación con un diluyente o portador farmacéuticamente aceptable. Puede usarse cualquier vehículo o diluyente conocido en la técnica para su uso en composiciones farmacéuticas, que también sea compatible con los otros ingredientes de la formulación y fisiológicamente inocuo para el receptor de la misma. Los diluyentes adecuados incluyen, pero no se limitan a carbonato de calcio o sodio, lactosa, fosfato de calcio o sodio.
[0116] En otra realización, el método descrito aquí comprende el tratamiento de una infección Orthomyxoviridae en un mamífero en necesidad del mismo mediante la administración de una cantidad terapéuticamente eficaz de una composición farmacéutica que comprende una cantidad eficaz de un compuesto de Fórmula I, o una sal farmacéuticamente aceptable o éster del mismo, en combinación con al menos un agente terapéutico adicional. El agente terapéutico adicional puede ser cualquier agente terapéutico adecuado para su uso con el compuesto de Fórmula I. Por ejemplo, el agente terapéutico puede seleccionarse del grupo que consiste en inhibidores de hemaglutinina viral, inhibidores de neuramidasa viral, bloqueadores de canales de iones M2, inhibidores de polimerasa ARNs dependientes de ARN de Orthomyxoviridae, sialidasas y otros fármacos para tratar infecciones por Orthomyxoviridae.
[0117] En todavía otra realización más, se describe en el presente documento procedimientos de tratamiento de infecciones Orthomyxoviridae en un paciente, que comprende: administrar al paciente una cantidad terapéuticamente eficaz de un compuesto de Fórmula I, II, o una sal farmacéuticamente aceptable del mismo.
[0118] En todavía otra realización más, se describen aquí métodos de tratamiento de infecciones Orthomyxoviridae en un paciente, que comprende: administrar al paciente una cantidad terapéuticamente eficaz de un compuesto de Fórmula I, II, o una sal farmacéuticamente aceptable del mismo, y al menos un agente terapéutico activo adicional, mediante el cual
se inhibe la polimerasa de Orthomyxoviridae.
[0119] En todavía otra realización más, se describen aquí métodos de tratamiento de infecciones Orthomyxoviridae en un paciente, que comprende: administrar al paciente una cantidad terapéuticamente eficaz de un compuesto de Fórmula I, II, o una sal farmacéuticamente aceptable del mismo, y al menos un agente terapéutico activo adicional seleccionado del grupo que consiste en interferones, análogos de ribavarina, un inhibidor de la hemaglutinina viral, un inhibidor de la neuramidasa viral, un bloqueador del canal iónico M2, un inhibidor de las polimerasas de ARNs dependientes de ARN de Orthomyxoviridae, una sialidasa y otros medicamentos para tratar infecciones Orthomyxoviridae.
[0120] En todavía otra realización más, se describe en el presente documento el uso de un compuesto de la presente invención, o una sal farmacéuticamente aceptable del mismo, para la preparación de un medicamento para tratar infecciones por Orthomyxoviridae en un paciente.
[0121] Los procesos se describen a continuación, que pueden usarse para preparar compuestos de Fórmula I de la invención.
[0122] Aquí se describen métodos para inhibir la actividad de polimerasa Orthomyxoviridae que comprende la etapa de tratar una muestra sospechosa de contener virus Orthomyxoviridae con una composición de la invención.
[0123] Las composiciones de la invención pueden actuar como inhibidores de la polimerasa de Orthomyxoviridae, como intermedios para tales inhibidores, o tener otras utilidades como se describe a continuación. Los inhibidores se unirán a ubicaciones en la superficie o en una cavidad de la polimerasa de Orthomyxoviridae que tiene una geometría única de la polimerasa de Orthomyxoviridae. Las composiciones que se unen a la polimerasa de Orthomyxoviridae pueden unirse con diversos grados de reversibilidad. Los compuestos que se unen de manera sustancialmente irreversible son candidatos ideales para su uso en este método de la invención. Una vez marcadas, las composiciones de unión sustancialmente irreversible son útiles como sondas para la detección de la polimerasa de Orthomyxoviridae. Por consiguiente, la invención se refiere a métodos para detectar polimerasa de Orthomyxoviridae en una muestra sospechosa de contener polimerasa de Orthomyxoviridae que comprenden las etapas de: tratar una muestra sospechosa de contener polimerasa de Orthomyxoviridae con una composición que comprende un compuesto de la invención unido a un marcador; y observar el efecto de la muestra sobre la actividad de la etiqueta. Los marcadores adecuados son bien conocidos en el campo del diagnóstico e incluyen radicales libres estables, fluoróforos, radioisótopos, enzimas, grupos quimioluminiscentes y cromógenos. Los compuestos de la presente invención se marcan de manera convencional usando grupos funcionales tales como hidroxilo, carboxilo, sulfhidrilo o amino.
[0124] En el contexto de la invención, las muestras sospechosas de contener polimerasa Orthomyxoviridae incluyen materiales naturales o hechos por el hombre tales como organismos vivos; cultivos de tejidos o células; muestras biológicas tales como muestras de material biológico (sangre, suero, orina, líquido cefalorraquídeo, lágrimas, esputo, saliva, muestras de tejido y similares); muestras de laboratorio; muestras de comida, agua o aire; muestras de bioproductos tales como extractos de células, particularmente células recombinantes que sintetizan una glicoproteína deseada; y similares. Normalmente, se sospechará que la muestra contiene un organismo que produce polimerasa de Orthomyxoviridae, frecuentemente un organismo patógeno como el virus Orthomyxoviridae. Las muestras pueden estar contenidas en cualquier medio, incluyendo agua y mezclas de disolventes orgánicos/agua. Las muestras incluyen organismos vivos como los seres humanos y materiales artificiales como los cultivos celulares.
[0125] La etapa de tratamiento comprende la adición de la composición de la invención a la muestra o añadir un precursor de la composición a la muestra. La etapa de adición comprende cualquier método de administración como se describe en este documento.
[0126] Si se desea, la actividad de polimerasa Orthomyxoviridae después de la aplicación de la composición se puede observar mediante cualquier método incluyendo métodos directos e indirectos de detección de la actividad de la polimerasa Orthomyxoviridae. Se contemplan todos los métodos cuantitativos, cualitativos y semicuantitativos para determinar la actividad polimerasa de Orthomyxoviridae. Normalmente se aplica uno de los métodos de cribado descritos anteriormente, sin embargo, también es aplicable cualquier otro método, como la observación de las propiedades fisiológicas de un organismo vivo.
[0127] Los organismos que contienen polimerasa Orthomyxoviridae incluyen el virus Orthomyxoviridae. Los compuestos de esta invención son útiles en el tratamiento o profilaxis de infecciones por Orthomyxoviridae en animales o en el hombre.
[0128] En todavía otra realización más, se describen aquí métodos para inhibir la Orthomyxoviridae dependiente de polimerasa ARN de ARN en una célula, que comprende: poner en contacto una célula infectada con virus Orthomyxoviridae con una cantidad eficaz de un compuesto de Fórmula I, II, o una sal farmacéuticamente aceptable del mismo, por lo que se inhibe la polimerasa de Orthomyxoviridae. En un aspecto de esta realización, la célula también se pone en contacto con al menos un agente terapéutico adicional. En determinadas realizaciones del método descrito en este documento, la polimerasa ARN dependiente de ARN de Orthomyxoviridae puede ser una polimerasa ARN dependiente de ARN del virus de la influenza A, una polimerasa ARN dependiente de ARN del virus de la influenza B, una polimerasa ARN dependiente de ARN del virus de la influenza C o mezclas del mismo.
[0129] En todavía otra realización más, se describen aquí métodos para inhibir la polimerasa Orthomyxoviridae en una célula, que comprende: poner en contacto una célula infectada con virus Orthomyxoviridae con una cantidad eficaz de un compuesto de Fórmula I, II, o una sal farmacéuticamente aceptable del mismo, y al menos un agente terapéutico activo adicional seleccionado del grupo que consiste en interferones, análogos de ribavirina, inhibidores de neuramidasa viral, inhibidores de neuramidasa viral, bloqueadores del canal iónico M2, inhibidores de polimerasa ARNs dependientes de ARN de Orthomyxoviridae, sialidasas y otros medicamentos usados para tratar infecciones por virus de Orthomyxoviridae.
[0130] En otro aspecto, en el presente documento se describen procesos y nuevos compuestos intermedios descritos en este documento que son útiles para la preparación de compuestos de fórmula I de la invención.
[0131] En otros aspectos, están descritos en este documento métodos novedosos para la síntesis, el análisis, la separación, aislamiento, purificación, caracterización, y ensayo de los compuestos de esta invención.
[0132] También se describen aquí los productos metabólicos in vivo de los compuestos descritos en el presente documento, en la medida en que tales productos son nuevos y no evidentes respecto a la técnica anterior. Dichos productos pueden resultar, por ejemplo, de la oxidación, reducción, hidrólisis, amidación, esterificación y similares del compuesto administrado, principalmente debido a procesos enzimáticos. Por consiguiente, en el presente documento se describen compuestos nuevos y no obvios producidos mediante un proceso que comprende poner en contacto un compuesto de esta invención con un mamífero durante un período de tiempo suficiente para producir un producto metabólico del mismo. Dichos productos se identifican típicamente preparando un compuesto radiomarcado (p. ej., 14C o 3H) de la invención, administrándolo por vía parenteral en una dosis detectable (p. ej., superior a aproximadamente 0,5 mg/kg) a un animal como rata, ratón, cobaya., mono, o al hombre, permitiendo suficiente tiempo para que ocurra el metabolismo (típicamente alrededor de 30 segundos a 30 horas) y aislando sus productos de conversión de la orina, sangre u otras muestras biológicas. Estos productos se aíslan fácilmente porque están marcados (otros se aíslan mediante el uso de anticuerpos capaces de unirse a los epítopos que sobreviven en el metabolito). Las estructuras de metabolitos se determinan de forma convencional, por ejemplo, mediante análisis de EM o RMN. En general, el análisis de metabolitos se realiza de la misma manera que los estudios de metabolismo de fármacos convencionales bien conocidos por los expertos en la técnica. Los productos de conversión, siempre que no se encuentren de otro modo in vivo, son útiles en ensayos de diagnóstico para la dosificación terapéutica de los compuestos de la invención incluso si no poseen actividad inhibidora de la polimerasa de Orthomyxoviridae propia.
[0133] Recetas y métodos para determinar la estabilidad de los compuestos en secreciones gastrointestinales sustitutas son conocidos. Los compuestos se definen en el presente documento como estables en el tracto gastrointestinal donde menos de aproximadamente 50 por ciento en moles de los grupos protegidos se desprotegen en jugo intestinal o gástrico sustituto tras la incubación durante 1 hora a 37°C. El simple hecho de que los compuestos sean estables en el tracto gastrointestinal no significa que no puedan hidrolizarse in vivo. Los profármacos normalmente serán estables en el sistema digestivo pero pueden hidrolizarse sustancialmente al fármaco original en el lumen digestivo, el hígado u otro órgano metabólico, o dentro de las células en general.
EJEMPLOS
[0134] Se usan ciertas abreviaturas y acrónimos para describir los detalles experimentales. Aunque la mayoría de estos los entenderá un experto en la técnica, la Tabla 1 contiene una lista de muchas de estas abreviaturas y acrónimos.
Tabla 1. Lista de abreviaturas y acrónimos.

Preparación de compuestos
Compuesto 1: (2S, 3R, 4R, 5R)-4-(bencnoxi)-5-(benciloximetil)-2-(2,4-bis(metiltío)imidazo[1,2-f][1,2,4]triazina-7-n)-3-fluorotetrahidrofuran-2-ol
[0135]

[0136] a una mezcla de 7-bromo-2,4-bis(metiltio)imidazo[1,2-f][1,2,4]triazina (2,5 g, 7,57 mmol) en THF (30 ml) a -78°C se añadió gota a gota nBuLi (1,6 M en hexano, 6,15 ml, 9,84 mmol). Después de agitar a -78°C durante 30 minutos, (3R, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorodihidrofuran-2(3H)-ona (2,43 g, 8,33 mmol) en THF (5 ml) gota a gota. Después de agitar a -78°C durante 3 horas, se dejó que la mezcla se calentara a temperatura ambiente. Después, la mezcla se agitó a temperatura ambiente durante 30 minutos y luego se inactivó con NH4Cl saturado. La reacción se extrajo con acetato de etilo. Las capas se separaron y las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 y se concentró para proporcionar un crudo, que se purificó mediante cromatografía en columna flash con acetato de etilo/hexanos para proporcionar el compuesto deseado (2S, 3R, 4R, 5RM-(benciloxi)-5-(benciloximetil)-2-(2,4-bis(metiltio)imidazo[1,2-f][1,2,4]triazina-7-il)-3-fluorotetrahidrofuran-2-ol (1) (2 g, 48%) como espuma amarilla. EM (m/z): 543,2 [M+H]+.
Compuesto 2: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-2,4-bis(metiltio)imidazo[1,2-f][1,2,4]triazina
[0137]

[0138] A una solución de (2S, 3R, 4R, 5RJ-4-(benciloxi)-5-(benciloximetil)-2-(2,4-bis(metiltio)imidazo[1,2-f][1,2,4]triazina-7-il)-3-fluorotetrahidrofuran-2-ol (1) (300 mg, 0,55 mmol) en diclorometano (3 ml) a -78°C se añadió gota a gota a BF3-OEt2 (1,20 ml, 8,81 mmol), seguido de la adición de EtaSiH (1,52 ml, 8,81 mmol). La reacción se dejó calentar a temperatura ambiente y se agitó durante 2 horas y después se inactivó con solución saturada de NaHCO3 y después se extrajo con diclorometano. Las capas orgánicas se separaron, se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron para proporcionar el crudo, que se purificó mediante cromatografía en columna flash con acetato de etilo/hexanos para proporcionar el compuesto deseado 7-((2S, 3S, 4R, 5RJ-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-2,4-bis(metiltio)imidazo[1,2-f][1,2,4]triazina (2) (218 mg, 75%). EM (m/z):527,2 [M+H]+.
Compuesto 3: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-2-(metilftio)imidazo[1,2-f][1,2,4]triazina-4-amina
[0139]

[0140] 7-((2S, 3S, 4R, 5R/)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetramdrofuran-2-il)-2,4-bis(metiltio)imidazo[1,2-f][1,2,4]triazina (2) (390 mg, 0,74 mmol) en amoniaco líquido (120 ml) se calentó a 60°C en una bomba de acero durante 18 horas. La bomba se enfrió a temperatura ambiente y la reacción se purificó mediante cromatografía en columna flash
con acetato de etilo/hexanos para proporcionar el compuesto deseado 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrahidrofuran-2-il)-2-(metiltio)imidazo[1,2-f][1,2,4]triazina-4-amina (3) (330 mg, 89%). EM (m/z):496,2 [M+H]+
Compuesto 4: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-ilo)-2-(metilsulfonil)imidazo[1,2-f][1,2,4]triazina-4-amina
[0141]

[0142] Para una solución de 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)-2-(metilt¡o)¡m¡dazo[1,2-f][1,2,4]tr¡azina-4-am¡na (3) (300 mg, 0,57 mmol) en d¡clorometano (10 ml) a 0°C se añad¡ó ác¡do 3-cloroperbenzo¡co (MCpBA, 77%) (627 mg, 3,42 mmol) en una porc¡ón. Se dejó que la reacc¡ón se calentara a temperatura amb¡ente y se ag¡tó durante 2 horas. La reacc¡ón se detuvo con una soluc¡ón de NaS2O3 al 20% en H2O (15 ml) y se dejó en ag¡tac¡ón durante 20 m¡nutos. Las capas se separaron y la soluc¡ón acuosa se extrajo con d¡clorometano. Las capas orgán¡cas comb¡nadas se lavaron con NaHCO3 saturado y salmuera, y luego se secaron sobre Na2SO4 y se concentraron para proporc¡onar una mezcla bruta que se pur¡f¡có ad¡c¡onalmente med¡ante cromatografía en columna de gel de síl¡ce con acetato de et¡lo/d¡clorometano para proporc¡onar el producto deseado 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)-2-(met¡lsulfon¡l)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na-4-am¡na (4) (276 mg, 87%) como ace¡te transparente. EM (m/z): 528,1 [M+H]+.
Compuesto 5: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)imidazo[1,2-f][1,2,4]triazina-2,4-diamina
[0143]

[0144] 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)-2-(met¡lsulfon¡l)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na-4-am¡na (4) (276 mg, 0,52 mmol) en amon¡aco líquido (100 ml) se calentó a 110°C durante 26 horas en una bomba de acero. La bomba se enfr¡ó a temperatura amb¡ente y la reacc¡ón bruta se pur¡f¡có med¡ante cromatografía en columna flash con acetato de et¡lo/d¡clorometano para proporc¡onar el compuesto deseado 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na-2,4-d¡am¡na (5) (185 mg, 78%). EM (m/z): 465,3 [M+H]+
Compuesto 6: (2S, 3S, 4R, 5R)-5-(2,4-diaminoimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil) tetrahidrofurano-3-ol
[0145]

[0146] A una soluc¡ón de 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na-2,4-d¡am¡na (5) (145 mg, 0,31 mmol) en ác¡do acét¡co (10 ml) se añad¡ó Pd/C al 10% Degussa t¡po E101 Ne /W (290 mg). La atmósfera de reacc¡ón se ¡ntercamb¡ó por H2 (g) y la reacc¡ón se agitó durante 18 horas. El catal¡zador
se eliminó por filtración y la mezcla se concentró a presión reducida. El crudo se secó para proporcionar el producto deseado (2S, 3S, 4R, 5Rj-5-(2,4-diaminoimidazo[1,2íí[1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil) tetrahidrofuran-3-ol (6) (85 mg, 96%) como un sólido blanco. EM (m/z): 285,2 [M+H]+.
1H RMN (400 MHz, CD3OD): 87,45 (s, 1H), 5,44-5,38 (m, 1H), 5,24-5,11 (d, J =, 1H), 4,38-4,33 (m, 1H), 3,98 (s, 1H), 3,91 - 3,70 (m, 2H).
19F (376 MHz, CD3OD): 8 (-199,86)-(-200,13) (m)
Compuesto 7: 2-amino-7-(2S, 3S, 4R, 5R)-3-fluoro-4-hidroxi-5-(hidroximetil)tetrahidrofurano-2-il)imidazo[1,2-f][1,2,4]triazina-4 (3H)-ona
[0147]

[0148] A una solución de (2S, 3S, 4R, 5RJ-5-(2,4-diaminoimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofuran-3-ol (6) (310 mg, 1,09 mmol) en 800 ml de agua se añadió adenosina desaminasa de bazo bovino tipo IX (CAS N° 9026-93-1,205 ml). La solución se colocó en un baño de agua a 37°C durante 16 horas. La solución se concentró y el compuesto final se cristalizó por separado de las impurezas usando agua como disolvente de cristalización. Los sólidos se recogieron y secaron para proporcionar 2-amino-7-(2S, 3S, 4R, 5R)-3-fluoro-4-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il)imidazo [1,2-f][1,2,4]triazina-4(3H)-ona (7) (246 mg, 80%) como un sólido puro de color blanquecino. EM (m/z): 286,2 [M+H]+. 1H RMN (400 MHz, DMSO-d6): 811,27 (s, 1H), 7,41 (s, 1H), 6,24 (s, 2H), 5,43-5,42 (m, 1H), 5,26-5,20 (d, J = 22,8 Hz, 1H), 5,09-4,85 (m, 2 H), 4,14-4,09 (m, 1H), 3,77 (s, 1H), 3,69 - 3,51 (m, 2H). 19F (376 MHz, DMSO-d6): 8 (-196,68)-(-196,94) (m)
Compuesto 8: ((2R, 3R, 4R, 5S)-5-(2-amino-4-oxo-3,4-dihidroimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-3-hidroxi-3-hidroxitetrahidrofuran-2-il)metilo trifosfato de tetrahidrógeno
[0149]
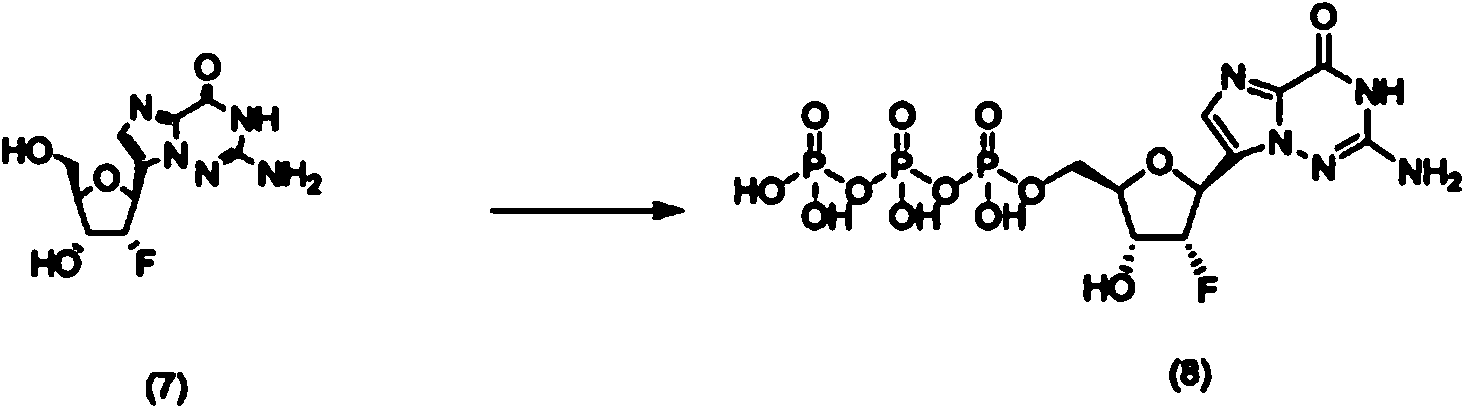
[0150] 2-am¡no-7-(2S, 3S, 4R, 5R)-3-fluoro-4-mdroxi-5-(mdroximetil)tetramdrofuran-2-il)imidazo[1,2-f][1,2,4]triazina-4(3HJ ona (7) (12 mg, 0,042 mmol) se disolvió en fosfato de trimetilo (1 ml) bajo una atmósfera inerte (N2). Se añadió oxicloruro de fósforo (58 mg, 0,378 mmol) y la mezcla se agitó a 0°C durante 2 horas y a temperatura ambiente durante 2 horas. El seguimiento mediante columna analítica de intercambio iónico determinó el tiempo en donde se formó >80% de monofosfato. La solución se enfrió a 0°C y se añadió una solución de tributilamina (0,15 ml, 0,63 mmol) y pirofosfato de trietilamonio (0,25 g, 0,55 mmol) en DMF anhidra (1 ml). La mezcla de reacción se agitó a 0°C durante 2,5 horas y luego se inactivó mediante la adición de una solución de bicarbonato de trietilamonio 1 N en H2O (6 ml). La mezcla se concentró a presión reducida y el residuo se volvió a disolver en H2O. La solución se sometió a cromatografía de intercambio iónico para producir el producto deseado ((2R, 3R, 4R, 5S)-5-(2-amino-4-oxo-3,4-dihidroimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-3-hidroxi-3-hidroxitetrahidrofuran-2-il)metilo trifosfato de tetrahidrógeno (8) (como la sal de tetratrietilamonio) (11 mg, 28% de rendimiento). EM (m/z): 526,0 [M+H]+.
1H RMN (400 MHz, D2O): 87,53 (s, 1H), 5,43-5,37 (d, J = 24,8 Hz, 1H), 5,29-5,15 (d, J = 55,2, 1H), 4,52-3,47 (m, 4H).
19F (376 MHz, D2O): 8 (-197,33)-(-197,60) (m, IF)
31P (162 MHz, D2O) 8 (-10,66)-(-10,78) (d, J = 48,4 Hz, IP), (-11,070)-(-11,193) (d, J = 49,2 Hz, 1P), (-22,990)-(-23,236) (m, IP).
Intercambio iónico HPLC: Disolvente A: Agua; Disolvente B: bicarbonato de trietilamonio 1M. 0-50% durante 12 minutos, luego 100% durante 5 minutos, luego de nuevo al 0% en 5 minutos. Columna: Dionex, DNAPac PA-100, 4x250 mm. Tr = 12,04 min
Compuesto 9: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)imidazo[1,2-
f][ 1,2,4]triazina-4-amina
[0151]

(4) (9)
[0152] A una solución de 7-((2S, 3S, 4R, 5RJ-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-2-(metilsulfonil)imidazo[1,2-f][1,2,4]triazina-4-amina (4) (63 mg, 0,12 mmol) en THF (5 ml) a -78°C se añadió gota a gota LiBHEt3 (1,0 M en t Hf , 4,78 ml, 4,78 mmol). La reacción se calentó a temperatura ambiente y se agitó a temperatura ambiente durante 31 horas. La mezcla de reacción se inactivó con agua helada y se extrajo con acetato de etilo. La solución orgánica se lavó con salmuera y se concentró para dar una mezcla bruta que se disolvió en CH3OH y se concentró al vacío (3x). El crudo se purificó mediante cromatografía en gel de sílice con acetato de etilo/diclorometano para proporcionar el producto deseado 7-((2S, 3S, 4R, 5RJ-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-ilo)imidazo[1,2-f][1,2,4]triazina-4-amina (9) (50 mg, rendimiento del 95%). EM (m/z): [M+H]+ 450,3.
Compuesto 10: (2S, 3S, 4R, 5R)-5-(4-aminoimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofurano-3-ol
[0153]

[0154] A una solución de 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)imidazo[1,2-f][1,2,4]triazina-4-amina (9) (50 mg, 0,11 mmol) en ácido acético (5 ml) se añadió Pd al 10%/C (100 mg). La atmósfera de los recipientes de reacción se cambió por hidrógeno y la reacción se agitó a temperatura ambiente durante la noche. La reacción se filtró a través de celite y se lavó con CH3OH. El filtrado se concentró para dar una mezcla bruta que se purificó mediante cromatografía en columna de gel de sílice usando CH3OH/diclorometano para proporcionar el producto deseado (2S, 3S, 4R, 5R)-5-(4-aminoimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofuran-3-ol (10) como un sólido blanco (23 mg, 77% de rendimiento). EM (m/z): 270,2 [M+H]+.
1H RMN (400 MHz, DMSO-da): 88,22 (d, J = 26 Hz, 2H), 8,07 (s, 1H), 7,67 (s, 1H), 5,50 -5,48 (d, J = 6,4,1 H), 5,42 -5,36 (m, 1H), 5,19 -5,03 (m, 1H), 4,88 -4,85 (m, 1H), 4,19-4,11 (m, 1H), 3,83 -3,81 (m, 1H), 3,72-3,67 (m, 1H), 3,54 -3,50 (m, 1H).
19F (376 MHz, CD3OD): 8 (-196,69)-(-196,95) (m)
Compuesto 11: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetilo)-3-fluorotetrahidrofuran-2-il)-2-fluoroimidazo[1,2-f][1,2,4]triazina-4-amina
[0155]
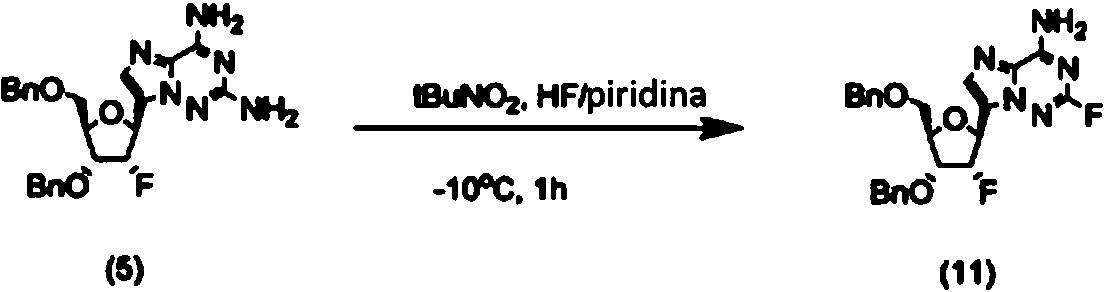
[0156] 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)imidazo[1,2-f][1,2,4]triazina-2,4-diamina (5) (140 mg, 0,30 mmol) en 4 ml de HF/piridina al 50% se agitó en un baño a -10°C y se añadieron 45 pl (0,38 mmol) de nitrito de t-butilo. La reacción se agitó a baja temperatura durante 1 hora. La reacción se inactivó mediante la adición de 50 ml de H2O y la capa acuosa se extrajo 2x 50 ml de diclorometano. Los extractos orgánicos combinados se secaron sobre Na2SO4 y se concentraron. El residuo se cromatografió en 6 g de gel de sílice para proporcionar el compuesto deseado 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-2-fluoroimidazo[1,2-f][1,2,4]triazina-4-amina (11) (50 mg, 36%). EM (m/z): 468,2 [M+H]+.
1H RMN (400 MHz, CDCI3): 7,60 (s, 1H), 7,3-7,2 (bm, 10H), 7,14 (bs, 1H), 6,37 (bs, 1H), 5,46 -5,48 (dd, J = 23,2, 2,4 Hz, 1H), 5,29 -5,15 (m, 1H), 4,72 (m, 1H), 4,56 -4,52 (m, 2H), 4,29 (m, 2H), 3,83 -3,81 (m, 1H), 3,65 -3,63 (m, 1H). 19F (376 MHz, CDCI3): 8-69,1 (s), (-197,7)-(-198,0) (m).
Compuesto 12: (2R, 3R, 4R, 5S)-5-(4-amino-2-fluoroimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofurano-3-ol
[0157]

[0158] A una solución de 7-((2S, 3S, 4R, SR) 4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)imidazo[1,2-f][1,2,4]triazina-4-amina (11) (50 mg, 0,11 mmol) en ácido acético (8 ml), se añadió Pd al 10%/C (100 mg). La atmósfera del recipiente de reacción se cambió por hidrógeno y la reacción se agitó a temperatura ambiente durante la noche. La reacción se filtró a través de celite y se lavó con ácido acético y luego con CH3OH. El filtrado se concentró para dar una mezcla bruta que se purificó mediante HPLC de fase inversa para proporcionar el producto deseado (2R, 3R, 4R, 5S)-5-(4-amino-2-fluoroimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofuran-3-ol (12) como un sólido blanco (26 mg, 84%). EM (m/z): 288,1 [M+H]+.
1H RMN (400 MHz, CD3OD): 87,66 (s, 1H), 5,47 -5,41 (dd, J = 23,6, 2,4 Hz, 1H), 5,21 -5,06 (m, 1H), 4,35 -4,27 (m, 1H), 3,93 (bm, 1H), 3,88 (m, 1H), 3,68 (m, 1H).
19F (376 MHz, CD3OD): 8 -72,15 (s), (-199,39)-(-196,69) (m)
Compuesto 13: 7-bromo-4-etoxi-2-(metil tio)imidazo[1,2-f][1,2,4]triazina
[0159]

[0160] A una mezcla de 7-bromo-2,4-bis(metiltio)imidazo[1,2-f][1,2,4]triazina (1,0 g, 3,45 mmol) en EtOH (25 ml) a temperatura ambiente, se añadió NaOEt (21% en EtOH, 1,28 ml, 3,45 mmol). Después de agitar a temperatura ambiente durante 1 hora, la reacción se detuvo con AcOH (1 ml). Los disolventes se eliminaron a presión reducida y la mezcla se repartió entre CH2Ch y A salmuera saturada. Los orgánicos se separaron, se secaron sobre Na2SO4, los sólidos se eliminaron por filtración y el disolvente se eliminó a presión reducida. El material bruto se purificó mediante cromatografía en columna flash con acetato de etilo/hexanos para proporcionar el compuesto deseado 7-bromo-4-etoxi-2-(metiltio)imidazo[1,2-f][1,2,4]triazina (13) (791 mg, 79%) como una espuma amarilla. EM (m/z): 288,9/290,8 [M+H]+.
Compuesto 14: (2S, 3R, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-2-(4-etoxi-2-(metiltio)imidazo[1,2-f][1,2,4]triazina-7-il)-3-fluorotetrahidrofuran-2-ol
[0161]

[0162] A una mezcla de 7-bromo-4-etoxi-2-(metiltio)imidazo[1,2-f][1,2,4]triazina (13) (917 mg, 3,17 mmol) en THF (15 ml) a -78°C se añadió LaCh * 2LiCl (0,6 M en THF, 5,28 ml, 3,17 mmol) seguido de la adición gota a gota de nBuLi (2,5 M en hexano, 1,27 ml, 3,17 mmol). Después de agitar a -78°C durante 30 minutos, (3R, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorodihidrofuran-2 (3 H)-ona (805 mg, 2,44 mmol) en Se añadió gota a gota THF (10 ml). Después de agitar a -78°C durante 30 min y dejar que la mezcla se caliente a temperatura ambiente, la mezcla se agitó a temperatura ambiente durante 30 minutos y luego se inactivó con AcOH. La reacción se extrajo con acetato de etilo. Las capas se separaron y las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4y se concentraron para proporcionar el crudo, que se purificó mediante cromatografía en columna flash con acetato de etilo/hexanos, para proporcionar el compuesto deseado (2S, 3R, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-2-(4-etoxi-2-(metiltio)imidazo[1,2-f][1,2,4]triazina-7-il)-3-fluorotetrahidrofuran-2-ol (14) (244 mg, 19%) en forma de espuma amarilla. e M (m/z): 541,1 [M+H]+.
Compuesto 15: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-4-etoxi-2-(metiltio)imidazo[1,2-f][1,2,4]triazina
[0163]

[0164] A una solución de (2S, 3R, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-2-(4-etox¡-2-(met¡lt¡o)¡m¡dazo[1,2-f][1,2,4]triazina-7-il)-3-fluorotetrahidrofuran-2-ol (X) (244 mg, 0,45 mmol) en CH2Ch (5 ml) a 0°C se añad¡ó gota a gota BF3-OEt2 (900 |jl, 3,5 mmol), segu¡do de la ad¡c¡ón de Et3S¡H (600 jl, 3,5 mmol). Se dejó que la reacc¡ón se calentara a temperatura amb¡ente y se ag¡tó durante 3 horas. La reacc¡ón se ¡nact¡vó con NaHCO3 saturado y se extrajo con CH2Ch. Las capas orgán¡cas se separaron, se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron para dar el crudo, que se pur¡f¡có por cromatografía en columna flash con acetato de et¡lo/hexanos, para proporc¡onar el compuesto deseado 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)-4-etox¡-2-(met¡lt¡o)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na (15) (107 mg, 46%). EM (m/z): 525,1 [M+H]+.
Compuesto 16: 7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-4-etoxi-2-(metilsulfonil)imidazo[1,2-f][1,2,4]triazina
[0165]

[0166] A una soluc¡ón de 7-((2S, 3S, 4R, SR)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofurano-2-¡l)-4-etox¡-2-(met¡lt¡o)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na (15) (107 mg, 0,204 mmol) en CH2Ch (3 ml) a temperatura amb¡ente se añad¡ó ác¡do 3-cloroperbenzo¡co (MCPBA, 77%) (100 mg, 0,443 mmol) en una porc¡ón. La reacc¡ón se ag¡tó a temperatura amb¡ente durante 4 horas. La reacc¡ón se ¡nact¡vó con una soluc¡ón de NaS2O3 al 20% en H2O (5 ml) y se dejó en ag¡tac¡ón durante 20 m¡nutos. Las capas se separaron y la soluc¡ón acuosa se extrajo con CH2Ch. Las capas orgán¡cas comb¡nadas se lavaron con NaHCO3 saturado, salmuera, se secaron sobre Na2SO4 y se concentraron para proporc¡onar 7-((2S, 3S, 4R, 5RJ-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)-4-etox¡-2-(met¡lsulfon¡l)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na en bruto (16), que se llevó adelante s¡n pur¡f¡cac¡ón. EM (m/z): 557,1 [M+H]+.
Compuesto 17: 2-azido-7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidrofuran-2-il)-4-etoxiimidazo[1,2-f][1,2,4]triazina
[0167]

[0168] A una soluc¡ón de NaN3 (66 mg, 1,01 mmol) en DMSO (5 ml) se añad¡ó 7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran-2-¡l)-4-etox¡-2-(met¡lsulfon¡l)¡m¡dazo[1,2-f][1,2,4]tr¡az¡na (16) (113 mg, 0,203 mmol) en una porc¡ón. Se dejó ag¡tar la reacc¡ón a temperatura amb¡ente durante 16 horas. La mezcla se repart¡ó entre EtOAc/H2O. Los orgán¡cos se separaron y se secaron sobre Na2SO4, y se pur¡f¡caron por cromatografía de gel de síl¡ce con EtOAc/hexanos para proporc¡onar 2-az¡do-7-((2S, 3S, 4R, 5R)-4-(benc¡lox¡)-5-(benc¡lox¡met¡l)-3-fluorotetrah¡drofuran2-il)-4-etoxiimidazo[1,2-f][1,2,4]triazina (17) (93 mg, 88%) como un sólido blanquecino. EM (m/z): 520,05 [M+H]+.
Compuesto 18: (2R, 3R, 4R, 5S)-5-(2-amino-4-etoxiimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofurano-3-ol
[0169]

[0170] Una solución de 2-azido-7-((2S, 3S, 4R, 5R)-4-(benciloxi)-5-(benciloximetil)-3-fluorotetrahidromran-2-il)-4-etoxiimidazo[1,2-f][1,2,4]triazina (17) (93 mg, 0,18 mmol) en CH3OH (5 ml) se purgó con argón y se añadió Pd/C al 10% (100 mg). El recipiente de reacción se evacuó y se vuelve a cargar con H2 tres veces. A continuación, se dejó agitar la mezcla de reacción en una atmósfera de hidrógeno durante 16 horas. Los sólidos se separaron por filtración y los orgánicos se eliminaron a presión reducida para dar material bruto que se purificó por HPLC para dar (2R, 3R, 4R, 5S)-5-(2-amino-4-etoxiimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofuran-3-ol (18) (27 mg, 48%) como un sólido blanco. EM (m/z): 314,10 [M+H]+. 1H RMN (400 MHz, DMSO-da): 87,526 (s, 1H), 8,07 (s, 1H), 6,55 (s, 2H), 5,43 5,41 (d, J = 6,46 Hz, 1H), 5,33 -5,27 (dd, J = 2,25 y 22,69 Hz, 1H), 5,11 - 4.96 (m, 1H), 4,84 (t, J = 5,58 Hz, 1H), 4,52 -4,47 (q, J = 7,04 Hz, 2H), 4,17-4.07 (m, 1H), 3,78-3,76 (m, 1H), 3,69-3,65 (m, 1H), 3,511-3,45 (m, 1H), 1,37 (t, J = 7,04 Hz, 3 H). 19F (376 MHz, DMSO-d6): 8 (-196,79)-(-197,05) (m)
Compuesto 19: (2R, 3R, 4R, 5S)-5-(2-amino-4-metoxiimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofurano-3-ol
[0171]

[0172] (2R, 3R, 4R, 5S)-5-(2-amino-4-metoxiimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofuran-3-ol (19) se preparó de una manera directamente análoga a la utilizada para la preparación de (2R, 3R, 4R, 5S)-5-(2-amino-4-etoxiimidazo[1,2-f][1,2,4]triazina-7-il)-4-fluoro-2-(hidroximetil)tetrahidrofuran-3-ol, excepto que se usó NaOMe en MeOH en lugar de NaOEt en EtOH en el primer paso de la síntesis. EM (m/z): 300,18 [M+H]+. 1H RMN (400 MHz, CD3OD): 8 7,56 (s, 1H), 5,46 (dd, J = 24, 2,4 Hz, 1H), 5,15 (ddd, J = 54,8, 4,4, 2,4 Hz, 1H), 4,34 (ddd, J = 4,4, 8, 20,4 Hz, 1H), 4,15 (s, 3 H), 3,97 (m, 1H), 3,91 (dd, J = 2,4, 12,4 Hz, 1H), 3,72 (dd, J = 4,4, 12 Hz, 1H). 19F (376 MHz, CD3OD): 8 (-198,98)-(-199,25) (m).
Ensayos anti-influenza
[0173] Ensayo de inhibición polimerasa ARN de influenza (CI50)
[0174] El virus purificado por Influenza A/PR/8/34 (H1N1) se obtuvo a partir de Advanced Biotechnologies Inc. (Columbia, MD) como suspensión en tampón PBS. Los viriones se rompieron por exposición a un volumen igual de Triton X-100 al 2% durante 30 minutos a temperatura ambiente en un tampón que contenía Tris-HCl 100 mM, pH 8, KC1 200 mM, ditiotreitol [DTT] 3 mM, glicerol al 10%, MgCh 10 mM, 2 U/ml de inhibidor de ribonucleasa RNasin, y 2 mg/mL lisolequitina tipo V (Sigma, Saint Louis, MO). El lisado de virus se almacenó a -80°C en alícuotas.
[0175] Las concentraciones se refieren a las concentraciones finales a menos que se indique lo contrario. Los inhibidores de análogos de nucleótidos se diluyeron en serie 3 veces en agua y se añadieron a la mezcla de reacción que contenía lisado de virus al 10% (v/v), Tris-HCl 100 mM (pH 8,0), KC1 100 mM, DTT 1 mM, 10% de glicerol, 0,25% Triton-101 (reducido), 5 mM MgCh, 0,4 U/ml RNasin, y 200 pM ApG dinucleótido cebador (TriLink, San Diego CA). Las reacciones se iniciaron mediante la adición de una mezcla de sustrato de ribonucleótido trifosfato (NTP) que contenía un NTP marcado con a-33P y 100 pm de los otros tres NTP naturales (PerkinElmer, Shelton, CT). El radiomarcaje utilizado para cada ensayo coincidió con la clase de análogo de nucleótidos examinado. Las concentraciones del NTP natural limitante
son 20, 10, 2 y 1 |jm para ATP, CTP, UTP y GTP, respectivamente. La relación molar de NTP no radiomarcado: radiomarcado estaba en el intervalo de 100-400:1.
[0176] Las reacciones se incubaron a 30°C durante 90 minutos y luego se mancharon sobre papel de filtro DE81. Los filtros se secaron al aire, se lavaron con Na2HPO4 0,125 M (3x), agua (1x) y EtOH (1x), y se secaron al aire antes de exponerlos al generador de imágenes de fósforo Typhoon y se cuantificó la radiactividad en un Typhoon Trio (GE Healthcare, Piscataway NJ). Los valores de CI50 se calcularon para los inhibidores ajustando los datos en GraphPad Prism con una respuesta a la dosis sigmoidal con la ecuación de pendiente variable, la fijación de los valores Ymax y Ymin al 100% y 0%. El CI50 para ((2R, 3R, 4R, 5SJ-5-('2-amino-4-oxo-3,4-dihidroimidazo[1,21][1,2,4]triazina-7-il)-4-fluoro-3-hidroxi-3-hidroxitetrahidrofurano-2-il)metilo trifosfato de tetrahidrógeno (Compuesto 8) se determinó que era 2,8 jm .
Ensayo de infección de la influenza de células epiteliales bronquiales/traqueales humanas normales (CE50)
[0177] Las células epiteliales bronquiales/traqueales humanas normales (Lonza, Basilea Suiza) se siembran en placas de 384 pocillos a una densidad de 4.000 células por pocillo en medio BEGM suplementado con factores de crecimiento (Lonza, Basilea Suiza). El medio se retira al día siguiente y las células se lavan tres veces con 100 j l de RPMI BSA al 1% (RPMI-BSA). A continuación, se añaden 30 j l de RPMI-BSA a las células. Los compuestos se diluyen en serie 3 veces en DMSO y se estampan en placas 0,4 j l de diluciones de compuesto. El virus de la influenza A HK/8/68 (Advanced Biotechnology Inc, Columbia, Md , 13,5 MOI), PC/1/73 (ATCC Manassas, VA, 0,3 MOI) y virus de la influenza B B/Lee/40 ((ATCC Manassas, VA, 10 MOI) a las células en 10 j l de medio RPMI-BSA suplementado con 8 ug/mL de tripsina (Worthington, Lakewood, NJ). Después de una incubación de cinco días, 40 j l de tampón que contiene 66 mM Mes pH 6,5, CaCl28 mM, NP-40 al 0,5% y 100 jm de sustrato neuramidasa hidrato de sal sódica del ácido (2'-(4-metilumbeliferil)-a-DN-acetilneuramínico, Sigma Aldrich, St. Luis, MO) se añade a las células. Se lee la fluorescencia del producto de la hidrólisis usando excitación a 360 nm y emisión a 450 nm después de una 1 hora de incubación a 37°C. Los valores CE50 se calculan por regresión no lineal de múltiples conjuntos de datos.
[0178] La siguiente tabla resume CEsqs determinados por este ensayo:

Claims (28)
1. Un compuesto de Fórmula I:

o una sal farmacéuticamente aceptable del mismo;
en donde:
cada uno de R1 y R7 es independientemente H, halógeno, ORa, (C1-C8) haloalquilo, (C3-C8) halocicloalquilo, CN, N3, (C1-C8) alquilo, (C3-C8) cicloalquilo, (C1-C8) alquilo sustituido, (C3-C8) cicloalquilo sustituido, (C2-C8) alquenilo, (C2-C8) alquenilo sustituido, (C2-C8) alquinilo o (C2-C8) alquinilo sustituido, en donde el sustituyente se selecciona del grupo que consiste de -X, -Rb, -OH, =O, -ORb, -SRb, -S-, -NRb2, -N+Rb3,
=NRb, -CX3, -CN, -OCN, -SCN, -N=C=O, -NCS, -NO, -NO2, =N2, -N3, -NHC(=O)Rb, -OC(=O)Rb, -NHC(=O)NRb2, -S(=O)2-, -S(=O)2OH, - S(=O)2Rb, -OS(=O)2ORb, -S(=O)2NRb2, -S(=O)Rb, -OP(=O)(ORb)2, -P(=O)(ORb)2, -P(=O)(O)2, -P(=O)(OH)2, -P(O)(ORb)(O‘), -C(=O)Rb, -C(=O)X, -C(S)Rb, -C(O)ORb, -C(O)O‘, -C(S)ORb, -C(O)SRb, -C(S)SRb, -C(O)NRb2, -C(S)NRb2, -C(=NRb)NRb2, donde cada X es independientemente un halógeno: F, Cl, Br o I; y cada Rb es independientemente H, alquilo, cicloalquilo, arilo, arilalquilo o un heterociclo;
R2 es ORa;
R3 es halógeno o N3; cada Ra es independientemente H, arilalquilo, arilo, (C1-C8) alquilo, o (C3-C8) cicloalquilo; R4 es H, =O, ORa, N(Ra)2, N3, CN, S(O)nRa, halógeno, (C1-C8) haloalquilo, o (C3-C8) halocicloalquilo; R5 es H, =O, ORa, N(Ra)2, N3, CN, S(O)nRa, halógeno, (C1-C8) haloalquilo, o (C3-C8) halocicloalquilo; cada n es 0, 1 o 2; y
cada R6 es H, arilo, arilalquilo, o

en donde W1 y W2 son cada uno, independientemente, ORa o un grupo de la Fórmula la:

en donde:
cada Y es independientemente un enlace u O;
M2 es 0, 1 o 2; y
cada Rx es H, halógeno o OH,
para su uso en un método para tratar una infección Orthomyxoviridae en un mamífero.
2. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde el compuesto de Fórmula I está representado por la Fórmula II:

3. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R1 es H, CH2OH, CH2F, CHF2, CH=CH2, CeCH, CN, CH2CH=CH2, N3, CH3 o CH2CH3.
4. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 2 o la reivindicación 3, en donde R1 es H.
5. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R2 es OH u O-bencilo.
6. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R3 es F o N3.
7. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R4 se selecciona del grupo que consiste en H, NH2, =O, NHMe, NHcPr, OH, OMe, Cl, Br, I, SMe, F, N3, CN, CF3 y SO2Me; y R5 se selecciona del grupo que consiste en H, NH2, =O, NHMe, NHcPr, OH, OMe, Cl, Br, I, SMe, F, N3, CN, CF3 y SO2Me.
8. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 7, en donde R5 es H o NH2.
9. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 7, en donde R4 es = O o NH2.
10. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R6 es H, bencilo o

en donde W2 es OH y W 1 es un grupo de Fórmula la:

Fórmula la
en donde:
cada Y es O;
M2 es 2; y
cada Rx es H.
11. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R7 es H u OH.
12. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R1 es H; R2 es OH; y R3 es F.
13. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 12, en donde R4 es NH2, H o =O; R5 es NH2 o H; y R6 yR 7 son hidrógeno.
14. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1,
en donde R1 es H; R2 es O-bencilo o OH; R3 es F; R4 es SMe, NH2 o =O; R5 es SMe, SO2Me, H o NH2; R6 es bencilo o

donde W2 es OH y W1 es un grupo de Fórmula 1 a:

Fórmula ja
en donde:
Y es O;
M2 es 2; y
cada Rx es H;
y R7 es H o OH.
15. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde el compuesto es un compuesto de Fórmula III:

en donde
R8 es NH2, OMe, OCH2CH3 o =O; y
R9 es NH2, H o F.
16. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde el compuesto es





17. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, 2 o 15, en donde el método comprende además administrar un vehículo o excipiente farmacéuticamente aceptable.
18. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, 2 o 15, en donde el método comprende además administrar una cantidad terapéuticamente eficaz de al menos un agente terapéutico adicional o composición del mismo seleccionado del grupo que consiste en un corticosteroide, un modulador de transducción de señales antiinflamatorias, un broncodilatador agonista de los receptores adrenérgicos p2, un anticolinérgico, un agente mucolítico, una solución salina hipertónica, un agente que inhibe la migración de células proinflamatorias al sitio de infección y mezclas de los mismos.
19. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 18, en donde el compuesto de Fórmula I, Fórmula II, Fórmula III y/o al menos un agente terapéutico o mezclas de los mismos debe administrarse por inhalación; o debe administrarse mediante nebulización.
20. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1 ,2 o 15, en donde la infección por Orthomyxoviridae es causada por un virus de Influenza A; o es causada por un virus de influenza B; o es causada por un virus de influenza C.
21. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R2 es OH.
22. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R3 es F.
23. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R4 es NH2 y R5 es H, F, Cl, Br, N3, CN, CF3, NH2, SMe, o SO2Me.
24. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R5 es NH2
y R4 es =O, OH, OMe, Cl, Br, I, NH2, NHMe, NHcPr o SMe.
25. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R6 es H.
26. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde R7 es H.
27. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, en donde el compuesto es

28. El compuesto o la sal farmacéuticamente aceptable del mismo para el uso de la reivindicación 1, 2 o 15, en donde el método comprende además administrar una cantidad terapéuticamente eficaz de al menos un agente terapéutico adicional o composición del mismo seleccionado del grupo que consiste en un inhibidor de la hemaglutinina viral, un inhibidor de la neuramidasa viral, un inhibidor del canal iónico M2, un inhibidor de la polimerasa ARN dependiente de ARN de Orthomyxoviridae o una sialidasa; o es un interferón, ribavirina, oseltamivir, zanamivir, laninamivir, peramivir, amantadina, rimantadina, CS-8958, favipiravir, AVI-7100, inhibidor de la proteasa alfa-1 o DAS181.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261610411P | 2012-03-13 | 2012-03-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2878087T3 true ES2878087T3 (es) | 2021-11-18 |
Family
ID=47997868
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16203290T Active ES2709071T3 (es) | 2012-03-13 | 2013-03-11 | Análogos de carba-nucleósido 2'-sustituidos para tratamiento antiviral |
| ES18155061T Active ES2878087T3 (es) | 2012-03-13 | 2013-03-11 | Análogos de carba-nucleósido 2-sustituidos para el tratamiento antiviral |
| ES13712043.2T Active ES2621217T3 (es) | 2012-03-13 | 2013-03-11 | Análogos de carba-nucleósido 2'-sustituidos para tratamiento antivírico |
| ES21168626T Active ES3005115T3 (en) | 2012-03-13 | 2013-03-11 | A nebulizer comprising a pharmaceutical composition comprising 2'-substituted carba-nucleoside analogs for antiviral treatment |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16203290T Active ES2709071T3 (es) | 2012-03-13 | 2013-03-11 | Análogos de carba-nucleósido 2'-sustituidos para tratamiento antiviral |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13712043.2T Active ES2621217T3 (es) | 2012-03-13 | 2013-03-11 | Análogos de carba-nucleósido 2'-sustituidos para tratamiento antivírico |
| ES21168626T Active ES3005115T3 (en) | 2012-03-13 | 2013-03-11 | A nebulizer comprising a pharmaceutical composition comprising 2'-substituted carba-nucleoside analogs for antiviral treatment |
Country Status (18)
| Country | Link |
|---|---|
| US (4) | US9481704B2 (es) |
| EP (4) | EP3351552B1 (es) |
| JP (3) | JP6242378B2 (es) |
| KR (1) | KR102068856B1 (es) |
| CN (2) | CN106749272B (es) |
| AU (2) | AU2013232378B2 (es) |
| CA (1) | CA2866381C (es) |
| EA (2) | EA201791916A1 (es) |
| ES (4) | ES2709071T3 (es) |
| IL (2) | IL234586A (es) |
| IN (1) | IN2014DN08505A (es) |
| MD (1) | MD4496C1 (es) |
| MX (1) | MX355267B (es) |
| NZ (1) | NZ629996A (es) |
| PL (1) | PL2834258T3 (es) |
| PT (1) | PT2834258T (es) |
| SI (1) | SI2834258T1 (es) |
| WO (1) | WO2013138236A1 (es) |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010064146A2 (en) | 2008-12-02 | 2010-06-10 | Chiralgen, Ltd. | Method for the synthesis of phosphorus atom modified nucleic acids |
| CA2767253A1 (en) | 2009-07-06 | 2011-01-13 | Ontorii, Inc. | Novel nucleic acid prodrugs and methods of use thereof |
| WO2012039448A1 (ja) | 2010-09-24 | 2012-03-29 | 株式会社キラルジェン | 不斉補助基 |
| CN103796657B (zh) | 2011-07-19 | 2017-07-11 | 波涛生命科学有限公司 | 合成官能化核酸的方法 |
| US8633198B1 (en) | 2011-09-20 | 2014-01-21 | Nant Holdings Ip, Llc | Small molecule inhibitors of influenza A RNA-dependent RNA polymerase |
| SI2794627T1 (sl) | 2011-12-22 | 2019-02-28 | Alios Biopharma, Inc. | Substituirani nukleozidi, nukleotidi in njihovi analogi |
| JP6242378B2 (ja) | 2012-03-13 | 2017-12-06 | ギリアード サイエンシーズ, インコーポレイテッド | 抗ウイルス処置のための2’−置換カルバヌクレオシド類似体 |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| ME03502B (me) * | 2012-03-21 | 2020-04-20 | Janssen Biopharma Inc | Supstituisani nukleozidi, nukleotidi i njihovi analozi |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| WO2014010250A1 (en) | 2012-07-13 | 2014-01-16 | Chiralgen, Ltd. | Asymmetric auxiliary group |
| UA119050C2 (uk) * | 2013-11-11 | 2019-04-25 | Ґілеад Саєнсиз, Інк. | ПІРОЛО[1.2-f][1.2.4]ТРИАЗИНИ, ЯКІ ВИКОРИСТОВУЮТЬСЯ ДЛЯ ЛІКУВАННЯ РЕСПІРАТОРНО-СИНЦИТІАЛЬНИХ ВІРУСНИХ ІНФЕКЦІЙ |
| WO2015088516A1 (en) * | 2013-12-11 | 2015-06-18 | Nant Holdings Ip, Llc | Small molecule inhibitors of influenza a rna-dependent rna polymerase |
| KR20230152178A (ko) | 2014-01-16 | 2023-11-02 | 웨이브 라이프 사이언시스 리미티드 | 키랄 디자인 |
| PH12016501567B1 (en) * | 2014-02-06 | 2023-12-06 | Riboscience Llc | 4`-difluoromethyl substituted nucleoside derivatives as inhibitors of influenza rna replication |
| WO2015143712A1 (en) * | 2014-03-28 | 2015-10-01 | Merck Sharp & Dohme Corp. | 4'-substituted nucleoside reverse transcriptase inhibitors |
| TWI678369B (zh) | 2014-07-28 | 2019-12-01 | 美商基利科學股份有限公司 | 用於治療呼吸道合胞病毒感染之噻吩並[3,2-d]嘧啶、呋喃並[3,2-d]嘧啶及吡咯並[3,2-d]嘧啶化合物類 |
| PE20170698A1 (es) | 2014-09-26 | 2017-06-05 | Riboscience Llc | Derivados de nucleosidos sustituidos con 4'-vinilo como inhibidores de la replicacion del arn del virus respiratorio sincitial |
| CN118286245A (zh) | 2014-12-26 | 2024-07-05 | 埃莫里大学 | N4-羟基胞苷和衍生物及与其相关的抗病毒用途 |
| RU2720811C2 (ru) | 2015-09-23 | 2020-05-13 | Мерк Шарп И Доум Лимитед | 4'-замещенные нуклеозидные ингибиторы обратной транскриптазы и их получение |
| US10202412B2 (en) | 2016-07-08 | 2019-02-12 | Atea Pharmaceuticals, Inc. | β-D-2′-deoxy-2′-substituted-4′-substituted-2-substituted-N6-substituted-6-aminopurinenucleotides for the treatment of paramyxovirus and orthomyxovirus infections |
| TWI778052B (zh) | 2017-04-24 | 2022-09-21 | 美商共結晶製藥公司 | 流感病毒複製之抑制劑 |
| RS66222B1 (sr) | 2017-12-07 | 2024-12-31 | Univ Emory | N4-hidroksicitidin i derivati i antivirusne upotrebe povezane sa njim |
| US11040975B2 (en) | 2017-12-08 | 2021-06-22 | Merck Sharp & Dohme Corp. | Carbocyclic nucleoside reverse transcriptase inhibitors |
| EP3829719B1 (en) | 2018-07-27 | 2025-04-02 | Cocrystal Pharma, Inc. | Pyrrolo[2,3-b]pyridin derivatives as inhibitors of influenza virus replication |
| TWI844564B (zh) | 2018-09-10 | 2024-06-11 | 美商共結晶製藥公司 | 流感病毒複製之抑制劑 |
| EP3887355A1 (en) | 2018-11-26 | 2021-10-06 | Cocrystal Pharma, Inc. | Inhibitors of influenza virus replication |
| TWI775313B (zh) | 2020-02-18 | 2022-08-21 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| TWI883391B (zh) | 2020-02-18 | 2025-05-11 | 美商基利科學股份有限公司 | 抗病毒化合物 |
| AU2021224588B2 (en) | 2020-02-18 | 2024-07-18 | Gilead Sciences, Inc. | Antiviral compounds |
| EP4125810A1 (en) * | 2020-03-22 | 2023-02-08 | InspirMed Corp. | Composition of antiviral agent for use in prophylactic or post-exposure treatment of infectious or respiratory diseases |
| EP4129291A4 (en) * | 2020-03-30 | 2024-04-10 | FUJIFILM Toyama Chemical Co., Ltd. | MEDICINES FOR THE TREATMENT OF CORONAVIRUS INFECTIONS |
| CN111956630A (zh) * | 2020-08-20 | 2020-11-20 | 大连理工大学 | 一种瑞德西韦供雾化器用的液体制剂、制备方法及其应用 |
| KR20230170745A (ko) | 2021-04-16 | 2023-12-19 | 길리애드 사이언시즈, 인코포레이티드 | 아미드를 사용한 카르바뉴클레오시드를 제조하는 방법 |
| US12116380B2 (en) | 2021-08-18 | 2024-10-15 | Gilead Sciences, Inc. | Phospholipid compounds and methods of making and using the same |
| US20250170155A1 (en) * | 2021-08-20 | 2025-05-29 | Shionogi & Co., Ltd. | Nucleoside derivatives and prodrugs thereof having viral growth inhibitory action |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3361306A (en) | 1966-03-31 | 1968-01-02 | Merck & Co Inc | Aerosol unit dispensing uniform amounts of a medically active ingredient |
| US3565070A (en) | 1969-02-28 | 1971-02-23 | Riker Laboratories Inc | Inhalation actuable aerosol dispenser |
| FR2224175B1 (es) * | 1973-04-04 | 1978-04-14 | Isf Spa | |
| US4069819A (en) * | 1973-04-13 | 1978-01-24 | Societa Farmaceutici S.P.A. | Inhalation device |
| IT1017153B (it) | 1974-07-15 | 1977-07-20 | Isf Spa | Apparecchio per inalazioni |
| SE438261B (sv) | 1981-07-08 | 1985-04-15 | Draco Ab | Anvendning i dosinhalator av ett perforerat membran |
| US4816570A (en) | 1982-11-30 | 1989-03-28 | The Board Of Regents Of The University Of Texas System | Biologically reversible phosphate and phosphonate protective groups |
| US4805811A (en) | 1985-03-29 | 1989-02-21 | Aktiebolaget Draco | Dosage device |
| SE448277B (sv) | 1985-04-12 | 1987-02-09 | Draco Ab | Indikeringsanordning vid en doseringsanordning for lekemedel |
| US4968788A (en) | 1986-04-04 | 1990-11-06 | Board Of Regents, The University Of Texas System | Biologically reversible phosphate and phosphonate protective gruops |
| US5388571A (en) * | 1987-07-17 | 1995-02-14 | Roberts; Josephine A. | Positive-pressure ventilator system with controlled access for nebulizer component servicing |
| IT1228459B (it) | 1989-02-23 | 1991-06-19 | Phidea S R L | Inalatore con svuotamento regolare e completo della capsula. |
| US4955371A (en) | 1989-05-08 | 1990-09-11 | Transtech Scientific, Inc. | Disposable inhalation activated, aerosol device for pulmonary medicine |
| ES2118069T3 (es) * | 1990-09-14 | 1998-09-16 | Acad Of Science Czech Republic | Profarmacos de fosfonatos. |
| CA2112674C (en) * | 1991-07-02 | 2005-10-04 | John S. Patton | Method and device for delivering aerosolized medicaments |
| US5261538A (en) | 1992-04-21 | 1993-11-16 | Glaxo Inc. | Aerosol testing method |
| US5785049A (en) | 1994-09-21 | 1998-07-28 | Inhale Therapeutic Systems | Method and apparatus for dispersion of dry powder medicaments |
| US5388572A (en) | 1993-10-26 | 1995-02-14 | Tenax Corporation (A Connecticut Corp.) | Dry powder medicament inhalator having an inhalation-activated piston to aerosolize dose and deliver same |
| US5522385A (en) | 1994-09-27 | 1996-06-04 | Aradigm Corporation | Dynamic particle size control for aerosolized drug delivery |
| US5544647A (en) | 1994-11-29 | 1996-08-13 | Iep Group, Inc. | Metered dose inhalator |
| US5622163A (en) | 1994-11-29 | 1997-04-22 | Iep Group, Inc. | Counter for fluid dispensers |
| US6116234A (en) | 1999-02-01 | 2000-09-12 | Iep Pharmaceutical Devices Inc. | Metered dose inhaler agitator |
| DE19912636A1 (de) * | 1999-03-20 | 2000-09-21 | Aventis Cropscience Gmbh | Bicyclische Heterocyclen, Verfahren zu ihrer Herstellung und ihre Verwendung als Herbizide und pharmazeutische Mittel |
| EP1411954B1 (en) | 2000-10-18 | 2010-12-15 | Pharmasset, Inc. | Modified nucleosides for treatment of viral infections and abnormal cellular proliferation |
| JP2004532184A (ja) * | 2001-01-22 | 2004-10-21 | メルク エンド カムパニー インコーポレーテッド | Rna依存性rnaウィルスポリメラーゼ阻害薬としてのヌクレオシド誘導体 |
| GB0112617D0 (en) * | 2001-05-23 | 2001-07-18 | Hoffmann La Roche | Antiviral nucleoside derivatives |
| WO2003062256A1 (en) * | 2002-01-17 | 2003-07-31 | Ribapharm Inc. | 2'-beta-modified-6-substituted adenosine analogs and their use as antiviral agents |
| JP4398631B2 (ja) * | 2002-07-12 | 2010-01-13 | 富山化学工業株式会社 | 新規なピラジン誘導体またはその塩並びにそれらを含有する抗ウイルス剤 |
| US20050196382A1 (en) | 2002-09-13 | 2005-09-08 | Replicor, Inc. | Antiviral oligonucleotides targeting viral families |
| CN101084232A (zh) * | 2004-10-19 | 2007-12-05 | 里普利科股份有限公司 | 抗病毒寡核苷酸 |
| EP2114980B1 (en) | 2007-01-12 | 2012-06-27 | BioCryst Pharmaceuticals, Inc. | Antiviral nucleoside analogs |
| EP2155758B1 (en) | 2007-05-10 | 2012-08-22 | Biocryst Pharmaceuticals, Inc. | Tetrahydrofuro[3,4-d]dioxolane compounds for use in the treatment of viral infections and cancer |
| CN102046626A (zh) * | 2008-04-23 | 2011-05-04 | 吉里德科学公司 | 用于抗病毒治疗的carba-核苷类似物 |
| WO2010036407A2 (en) * | 2008-05-15 | 2010-04-01 | Biocryst Pharmaceuticals, Inc. | Antiviral nucleoside analogs |
| EP2313102A2 (en) | 2008-07-03 | 2011-04-27 | Biota Scientific Management | Bycyclic nucleosides and nucleotides as therapeutic agents |
| US8455451B2 (en) * | 2009-09-21 | 2013-06-04 | Gilead Sciences, Inc. | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment |
| DK2480559T3 (da) * | 2009-09-21 | 2013-08-05 | Gilead Sciences Inc | Fremgangsmåder og mellemprodukter til fremstillingen af 1'-cyano-carbanukleosid-analoger |
| KR20180006499A (ko) * | 2009-09-21 | 2018-01-17 | 길리애드 사이언시즈, 인코포레이티드 | 항바이러스 치료용 2'―플루오로 치환된 카바-뉴클레오사이드 유사체 |
| MA34470B1 (fr) * | 2010-07-22 | 2013-08-01 | Gilead Sciences Inc | Procédés et composés pour traiter des infections à virus paramyxoviridae |
| TW201305185A (zh) | 2010-09-13 | 2013-02-01 | Gilead Sciences Inc | 用於抗病毒治療之2’-氟取代之碳-核苷類似物 |
| JP6242378B2 (ja) | 2012-03-13 | 2017-12-06 | ギリアード サイエンシーズ, インコーポレイテッド | 抗ウイルス処置のための2’−置換カルバヌクレオシド類似体 |
-
2013
- 2013-03-11 JP JP2015500493A patent/JP6242378B2/ja active Active
- 2013-03-11 AU AU2013232378A patent/AU2013232378B2/en active Active
- 2013-03-11 ES ES16203290T patent/ES2709071T3/es active Active
- 2013-03-11 EP EP18155061.7A patent/EP3351552B1/en active Active
- 2013-03-11 CN CN201611121597.0A patent/CN106749272B/zh active Active
- 2013-03-11 MX MX2014011009A patent/MX355267B/es active IP Right Grant
- 2013-03-11 EA EA201791916A patent/EA201791916A1/ru unknown
- 2013-03-11 US US13/793,557 patent/US9481704B2/en active Active
- 2013-03-11 EA EA201491548A patent/EA028928B9/ru unknown
- 2013-03-11 EP EP16203290.8A patent/EP3210993B1/en active Active
- 2013-03-11 CA CA2866381A patent/CA2866381C/en active Active
- 2013-03-11 EP EP21168626.6A patent/EP3932931B1/en active Active
- 2013-03-11 MD MDA20140112A patent/MD4496C1/ro active IP Right Grant
- 2013-03-11 SI SI201330567A patent/SI2834258T1/sl unknown
- 2013-03-11 EP EP13712043.2A patent/EP2834258B1/en active Active
- 2013-03-11 ES ES18155061T patent/ES2878087T3/es active Active
- 2013-03-11 CN CN201380014318.9A patent/CN104185638A/zh active Pending
- 2013-03-11 KR KR1020147028523A patent/KR102068856B1/ko active Active
- 2013-03-11 PL PL13712043T patent/PL2834258T3/pl unknown
- 2013-03-11 PT PT137120432T patent/PT2834258T/pt unknown
- 2013-03-11 NZ NZ629996A patent/NZ629996A/en unknown
- 2013-03-11 ES ES13712043.2T patent/ES2621217T3/es active Active
- 2013-03-11 WO PCT/US2013/030196 patent/WO2013138236A1/en not_active Ceased
- 2013-03-11 ES ES21168626T patent/ES3005115T3/es active Active
-
2014
- 2014-09-11 IL IL234586A patent/IL234586A/en active IP Right Grant
- 2014-10-10 IN IN8505DEN2014 patent/IN2014DN08505A/en unknown
-
2016
- 2016-09-30 US US15/282,492 patent/US20170114086A1/en not_active Abandoned
-
2017
- 2017-06-21 JP JP2017121389A patent/JP6525441B2/ja active Active
- 2017-12-18 IL IL256397A patent/IL256397A/en unknown
- 2017-12-19 AU AU2017279590A patent/AU2017279590A1/en not_active Abandoned
-
2018
- 2018-06-22 US US16/016,369 patent/US10941177B2/en active Active
- 2018-12-26 JP JP2018242679A patent/JP2019069986A/ja not_active Withdrawn
-
2021
- 2021-01-28 US US17/161,065 patent/US11787832B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2878087T3 (es) | Análogos de carba-nucleósido 2-sustituidos para el tratamiento antiviral | |
| ES2910382T3 (es) | Tieno [3,2-d]pirimidinas útiles para el tratamiento de infecciones por el virus sincitial respiratorio | |
| US20120107274A1 (en) | 2'-fluoro substituted carba-nucleoside analogs for antiviral treatment | |
| HK40066069B (en) | A nebulizer comprising a pharmaceutical composition comprising 2'-substituted carba-nucleoside analogs for antiviral treatment | |
| HK40066069A (en) | A nebulizer comprising a pharmaceutical composition comprising 2'-substituted carba-nucleoside analogs for antiviral treatment | |
| HK1256856B (en) | 2'- substituted carba-nucleoside analogs for antiviral treatment | |
| HK1242328B (en) | 2'- substituted carba-nucleoside analogs for antiviral treatment | |
| HK1242328A1 (en) | 2'- substituted carba-nucleoside analogs for antiviral treatment | |
| BR122016003311A2 (pt) | Análogos de carbanucleosídeo 2'-substituído, seu uso e composição farmacêutica que os compreende | |
| HK1207083B (en) | 2'- substituted carba-nucleoside analogs for antiviral treatment | |
| HK1238237A1 (en) | 2'- substituted carba-nucleoside analogs for antiviral treatment | |
| HK1238237B (zh) | 用於抗病毒治疗的2‘-取代的卡巴-核苷类似物 |