ES2851177T3 - Tetrahidrobiopterina para el tratamiento de afecciones asociadas con niveles elevados de fenilalanina - Google Patents
Tetrahidrobiopterina para el tratamiento de afecciones asociadas con niveles elevados de fenilalanina Download PDFInfo
- Publication number
- ES2851177T3 ES2851177T3 ES12005352T ES12005352T ES2851177T3 ES 2851177 T3 ES2851177 T3 ES 2851177T3 ES 12005352 T ES12005352 T ES 12005352T ES 12005352 T ES12005352 T ES 12005352T ES 2851177 T3 ES2851177 T3 ES 2851177T3
- Authority
- ES
- Spain
- Prior art keywords
- acid
- tetrahydrobiopterin
- formulation
- formulations
- administration
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
- A61K9/0065—Forms with gastric retention, e.g. floating on gastric juice, adhering to gastric mucosa, expanding to prevent passage through the pylorus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0087—Galenical forms not covered by A61K9/02 - A61K9/7023
- A61K9/0095—Drinks; Beverages; Syrups; Compositions for reconstitution thereof, e.g. powders or tablets to be dispersed in a glass of water; Veterinary drenches
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/2027—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4808—Preparations in capsules, e.g. of gelatin, of chocolate characterised by the form of the capsule or the structure of the filling; Capsules containing small tablets; Capsules with outer layer for immediate drug release
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4891—Coated capsules; Multilayered drug free capsule shells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/02—Nutrients, e.g. vitamins, minerals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B63/00—Purification; Separation; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6848—Methods of protein analysis involving mass spectrometry
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N30/00—Investigating or analysing materials by separation into components using adsorption, absorption or similar phenomena or using ion-exchange, e.g. chromatography or field flow fractionation
- G01N30/02—Column chromatography
- G01N30/62—Detectors specially adapted therefor
- G01N30/72—Mass spectrometers
- G01N30/7233—Mass spectrometers interfaced to liquid or supercritical fluid chromatograph
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01J—ELECTRIC DISCHARGE TUBES OR DISCHARGE LAMPS
- H01J49/00—Particle spectrometers or separator tubes
- H01J49/004—Combinations of spectrometers, tandem spectrometers, e.g. MS/MS, MSn
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
- Y10T436/145555—Hetero-N
- Y10T436/147777—Plural nitrogen in the same ring [e.g., barbituates, creatinine, etc.]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Nutrition Science (AREA)
- Hematology (AREA)
- Physiology (AREA)
- Molecular Biology (AREA)
- Physics & Mathematics (AREA)
- Dermatology (AREA)
- Urology & Nephrology (AREA)
- Diabetes (AREA)
- Psychiatry (AREA)
- Immunology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Pain & Pain Management (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Analytical Chemistry (AREA)
- Food Science & Technology (AREA)
- Spectroscopy & Molecular Physics (AREA)
Abstract
Tetrahidrobiopterina (BH4) o una sal farmacéuticamente aceptable de la misma para uso en el tratamiento de afecciones asociadas con niveles elevados de fenilalanina, en donde la BH4 o una sal de la misma es para administrar por vía oral una vez al día, 5 a 60 minutos después de una comida y en donde la afección se selecciona a partir del grupo que consiste en fenilcetonuria leve (PKU), PKU clásica e hiperfenilalaninemia.
Description
DESCRIPCIÓN
Tetrahidrobiopterina para el tratamiento de afecciones asociadas con niveles elevados de fenilalanina Antecedentes
Campo
La presente invención se dirige en general a composiciones para tratar trastornos que responden a BH4.
Antecedentes de la tecnología relacionada
La tetrahidrobiopterina (a la que se hace referencia en esta memoria como BH4) es una amina biogénica de la familia de las pterinas de origen natural, que es un cofactor para una variedad de enzimas diferentes, que incluyen hidroxilasa de fenilalanina (PAH), hidroxilasa de tirosina, hidroxilasa de triptófano y sintasa de óxido nítrico. Las pterinas están presentes en los tejidos y fluidos fisiológicos en formas reducidas y oxidadas, sin embargo, solo la 5,6,7,8, tetrahidrobiopterina es biológicamente activa. Es una molécula quiral y se sabe que el enantiómero 6R del cofactor es el enantiómero biológicamente activo. Para una revisión detallada de la síntesis y los trastornos de BH4, véase Blau et al., 2001 (Disorders of tetrahydrobiopterin and related biogenic amines. En: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, compiladores. The Metabolic and Molecular Bases of Inherited Disease. 8a ed. New York: McGraw-Hill, 2001: 1275-1776).
Fiege, et al., Molecular Genetics and Metabolism 81:45-51 (2004) han estudiado la farmacocinética de la tetrahidrobiopterina (BH4) administrada por vía oral y han sugerido una "variabilidad bastante elevada de la BH4 administrada por vía oral, probablemente debido a una absorción diferente en el intestino y/o al efecto del primer paso". El documento US 4.550.109 describe biopterinas lipoideas y tetrahidrobiopterinas que son solubles en aceite y se pueden formular como un producto farmacéutico a base de aceite, útil para el tratamiento de la fenilcetonuria, parkinsonismo, depresión, demencia senil, enfermedad de Alzheimer y enfermedades relacionadas con una carencia de biopterina.
El documento WO 2005/049000 A2 describe métodos y composiciones para la intervención terapéutica en la hiperfenilalaninemia para tratar varios tipos de fenilcetonurias usando composiciones que comprenden BH4.
Se ha propuesto el uso de tetrahidrobiopterina para tratar una variedad de estados patológicos diferentes y existe una necesidad de métodos alternativos y mejorados para administrar este fármaco.
Compendio de la invención
La presente invención se refiere a 6R-(L-eritro)-5,6,7,8-tetrahidrobiopterina (BH4), o una sal farmacéuticamente aceptable de la misma, para uso en el tratamiento de afecciones asociadas con niveles elevados de fenilalanina, de una manera que se mejora o maximiza su biodisponibilidad oral desde una administración a la siguiente. Dicha afección se selecciona a partir del grupo que consiste en fenilcetonuria leve (PKU), PKU clásica e hiperfenilalaninemia. Dichos usos se pueden aplicar en el tratamiento de cualquier otro trastorno que responda a BH4, incluyendo enfermedades metabólicas, enfermedades cardiovasculares, anemia y trastornos neurosiquiátricos. Los usos de la invención permiten ventajosamente un mejor control de los síntomas clínicos, p. ej., una disminución de la fluctuación de los niveles plasmáticos de fenilalanina, presión arterial, niveles de neurotransmisores u otros parámetros clínicos.
Tal y como se usa en esta memoria, BH4 se refiere a 6R-(L-eritro)-5,6,7,8-tetrahidrobiopterina. El término BH4 tal y como se usa en esta memoria, también debe entenderse que significa opcionalmente una sal farmacéuticamente aceptable de 6R-(L-eritro)-5,6,7,8-tetrahidrobiopterina, a menos que el contexto indique lo contrario.
En un primer aspecto, la invención proporciona tetrahidrobiopterina (BH4) o una sal farmacéuticamente aceptable de la misma, para uso en el tratamiento de afecciones asociadas con niveles elevados de fenilalanina, en donde la BH4 o una sal de la misma es para administración oral una vez al día, 5 a 60 minutos después de una comida y en donde la afección se selecciona a partir del grupo que consiste en fenilcetonuria leve (PKU), PKU clásica e hiperfenilalaninemia.
En una realización ejemplar, se informa al paciente de que la absorción de tetrahidrobiopterina aumenta cuando se ingiere con alimentos, en comparación con cuando se ingiere sin alimentos. En algunas realizaciones, se informa al paciente de que la ingestión poco después de una comida, por ejemplo, una comida rica en grasas y calorías, da como resultado un aumento en uno, dos, tres o todos los siguientes parámetros: concentración plasmática media, Cmáx, AUC, AUC (0-t) y/o AUC (inf). En realizaciones ejemplares, se informa al paciente de que la administración de BH4 con una comida rica en grasas aumenta la Cmáx y la AUC, en comparación con la administración de BH4 sin alimentos (en ayunas). En algunas realizaciones, el aumento relativo puede ser de al menos el 20% o el 30% o más. En realizaciones alternativas o además de las realizaciones precedentes, el método de administración de tetrahidrobiopterina comprende informar al paciente de que la absorción de tetrahidrobiopterina aumenta cuando se ingiere como un comprimido intacto, en comparación con cuando se ingiere después de disolver en un líquido. En
algunas realizaciones, se informa al paciente de que la ingestión de comprimidos intactos da como resultado un aumento de cualquiera de los siguientes parámetros: concentración plasmática media, Cmáx, AUC, AUC (0-t) o AUC (inf). En realizaciones ejemplares, se informa al paciente de que la administración de BH4 como un comprimido intacto aumenta la Cmáx y la AUC, en comparación con la administración de BH4 después de disolverla en un líquido. En algunas realizaciones, el aumento relativo puede ser de al menos un 20% o más.
Cualquiera de las realizaciones anteriores se puede llevar a cabo proporcionando o administrando tetrahidrobiopterina en un envase que contiene etiquetas impresas que informan al paciente del cambio en los parámetros de absorción descritos anteriormente.
La presente invención comprende la etapa de proporcionar al paciente que lo necesite, una cantidad terapéuticamente eficaz de tetrahidrobiopterina. La cantidad terapéuticamente eficaz variará dependiendo de la afección que se va a tratar, y el médico que realiza el tratamiento puede determinarla fácilmente basándose en la mejora de los síntomas clínicos deseados.
En una realización ejemplar, tales usos implican la administración de BH4 en forma disuelta, en donde la formulación se disuelve en un líquido que incluye pero no se limita a agua, zumo de naranja y zumo de manzana. Por tanto, BH4 se puede ingerir como un producto líquido o se puede disolver previamente a partir de una forma de dosificación sólida o semisólida antes de la ingestión. En una realización adicional, BH4 también se puede disolver en la cavidad oral a partir de una forma de dosificación sólida o semisólida antes de tragar la solución disuelta.
En otra realización ejemplar, esos usos implican la administración de BH4 en una forma de dosificación sólida que incluye, pero no se limita a, comprimidos, cápsulas, caramelos, pastillas, polvos y gránulos, o en forma semisólida, que incluye, pero no se limita a, un rociado oral en gelatina, que se ingiere sin disolver en un líquido, incluyendo, pero no limitado a, agua, zumo de naranja y zumo de manzana, antes de tragar.
En otra realización, esos usos implican la administración de BH4, ya sea ingerida en forma de dosificación sólida o semisólida, o disuelta en un líquido, con alimentos, p. ej., requiere una comida con alto contenido en grasas o una comida con alto contenido en grasas y/o calorías. La invención contempla además que BH4, ya sea ingerida o disuelta, se administra en un momento específico que incluye, pero no se limita a, la mañana, el día, la noche, a la misma hora del día, con alimentos, p. ej., una comida rica en grasas o una comida rica en grasas y/o rica en calorías, una vez al día. En una realización ejemplar, BH4 se ingiere una vez al día como una forma de dosificación sólida justo después de las comidas. En una realización preferida, la forma de dosificación sólida es un comprimido o una cápsula formulada. En realizaciones más ejemplares, BH4 se ingiere 5 a 20 minutos después de una comida. Independientemente de si se ingiere como una forma de dosificación sólida, una forma de dosificación líquida o como una solución disuelta, la exposición in vivo (o biodisponibilidad) de BH4 es mayor cuando se ingiere justo después de las comidas, en comparación con controles en ayunas.
La BH4 se ingiere después de los alimentos. El período de tiempo entre el consumo de los alimentos y la ingestión de BH4, ya sea tragada o disuelta, puede ser de al menos 5 minutos. Por ejemplo, BH4 se puede administrar 60 minutos, 30 minutos, 25 minutos, 20 minutos, 15 minutos, 10 minutos o 5 minutos después de una comida.
En otra realización, para algunos pacientes, p. ej., adultos, los usos de la invención implican administrar un comprimido intacto en lugar de disolver el comprimido en un líquido, con el fin de mejorar la biodisponibilidad.
También se describe en esta memoria un método para estabilizar BH4 en el tracto intestinal de un paciente mediante la disminución del pH intestinal, p. ej., utilizando polímeros de intercambio de protones. También se contemplan productos correspondientes que comprenden BH4 y excipientes acidificantes, tales como polímeros de intercambio de protones.
En esta memoria se proporciona un método para aumentar el tiempo de residencia de BH4 en el intestino, que incluye pero no se limita a ralentizar la motilidad intestinal usando un agente que ralentiza la motilidad intestinal, tal como un ácido graso y/o un éster de ácido graso de glicerol. Esos agentes hidrófobos pueden aumentar el tiempo que BH4 permanece en el intestino y pueden aumentar la cantidad de BH4 que se absorbe. El tiempo que BH4 permanece en el intestino, cuando se formula con ese o esos agentes, puede ser al menos una y media veces, al menos dos veces, al menos tres veces, al menos cuatro veces o al menos cinco veces más largo que el de una formulación de BH4 que no tiene un agente de ese tipo. Los ácidos grasos adecuados incluyen ácido oleico, ácido esteárico, ácido araquídico, ácido palmítico, ácido araquidónico, ácido linoleico, ácido linolénico, ácido erucídico, ácido mirístico, ácido láurico, ácido miristólico y ácido palmitolítico. También se contempla para aumentar el tiempo de residencia intestinal de BH4, inducir la retención gástrica usando ácido algínico y la bioadhesión usando policarbofilo. Se contemplan productos correspondientes que comprenden BH4 y agentes que ralentizan la motilidad intestinal.
En esta memoria se proporciona un método para modificar la liberación de BH4 usando una formulación de liberación sostenida tal como HPMC, carbómero, etc. Se contemplan productos correspondientes que sean formulaciones de liberación sostenida.
También se describe en esta memoria una formulación líquida de tetrahidrobiopterina (BH4) o una sal
farmacéuticamente aceptable de la misma, que incluye una solución acuosa de BH4 o una sal farmacéuticamente aceptable de la misma, un antioxidante y un tampón del pH.
También se describe en esta memoria un método para preparar una formulación líquida de tetrahidrobiopterina (BH4) o una sal farmacéuticamente aceptable de la misma, que incluye proporcionar una solución acuosa que contiene BH4 o una sal farmacéuticamente aceptable de la misma, añadir un antioxidante y un tampón del pH a la solución que contiene BH4 o una sal farmacéuticamente aceptable de la misma, burbujeando la solución acuosa que contiene BH4 o una sal farmacéuticamente aceptable de la misma, antes o después de la adición de antioxidante y tampón del pH, con un gas inerte o dióxido de carbono, y sellando la solución burbujeada que contiene BH4 o una sal farmacéuticamente aceptable de la misma, antioxidante y tampón del pH en un recipiente.
También se describe en la presente memoria un método mejorado para medir BH4 utilizando espectrometría de masas en tándem y calcular la cantidad de biopterina reducida. Esos métodos pueden proporcionar una detección de BH4 hasta con una sensibilidad para BH4 en el intervalo de 5 - 1000 ng/mL, con una exactitud y precisión tal y como se ejemplifica mediante un % de coeficiente de variación (CV) por debajo del 15% (20% en el límite inferior de cuantificación, LLOQ). Por ejemplo, un método para medir BH4 usando HPLC (RP) acoplada con espectrometría de masas en tándem (LC/MS/MS) comprende las etapas de: (1) someter a oxidación muestras de sangre, plasma, homogeneizados de tejido u orina; (2) someter a yodometría las muestras oxidadas; (3) hacer pasar dichas muestras oxidadas a través de una columna de intercambio iónico; (4) medir la biopterina total y oxidada en dichas muestras usando HPLC y espectrometría de masas en tándem; y calcular la cantidad de biopterina reducida como la diferencia entre dichas biopterinas totales menos dicha forma oxidada. Por ejemplo, las muestras se tratan con oxidación ácida, en donde el método comprende las etapas de (1) tratar dichas muestras con KCl, HCl o TCA; (2) someter dichas muestras oxidadas con ácido a yodometría; (3) hacer pasar dichas muestras oxidadas a través de una columna de intercambio iónico; (4) medir la biopterina total que comprende 6R-BH4, R-q-DHBP (que in vivo se reduce inmediatamente a 6R-BH4, de manera que la biopterina reducida medida se basa principalmente en 6R-BH4), DHBP y BP en dichas muestras, usando HPLC y espectrometría de masas en tándem. Por ejemplo, las muestras se tratan mediante oxidación alcalina, en donde el método comprende: (1) tratar dichas muestras con KI, I o NaOH; (2) someter dichas muestras oxidadas alcalinas a una acidificación con HCl o TCA; (3) someter dichas muestras oxidadas a yodometría; (4) hacer pasar dichas muestras a través de una columna de intercambio iónico; (5) medir la biopterina oxidada que comprende DHBP y BP usando HPLC y espectrometría de masas en tándem; y (6) calcular la cantidad de biopterina reducida (6R-BH4 R-q-DHBP) como la diferencia entre las biopterinas totales menos la forma oxidada.
También se describe en esta memoria una solución en fase móvil para la separación mediante HPLC de fase inversa de dihidrobiopterina, biopterina y análogos de las mismas, incluyendo una solución acuosa que incluye metanol, acetato de sodio, ácido cítrico, EDTA y 1,4-ditioeritritol. De manera similar, se contempla un método para separar dihidrobiopterina y biopterina, o análogos de las mismas, a partir de una mezcla que contiene tanto las formas base como dihidro, que incluye realizar una HPLC de fase inversa usando una fase móvil que comprende una solución acuosa que incluye metanol, acetato de sodio, ácido cítrico, EDTA y 1,4-ditioeritritol, sobre una mezcla que contiene dihidrobiopterina y biopterina, o un análogo de dihidrobiopterina y un análogo de biopterina.
En esta memoria se describe un método para cuantificar las biopterinas en una mezcla de especies de biopterina, que incluye proporcionar una mezcla que comprende biopterina y al menos una entre dihidrobiopterina y tetrahidrobiopterina, o análogos de biopterina y al menos una entre dihidrobiopterina y tetrahidrobiopterina, separando las especies de biopterina en la mezcla mediante HPLC de fase inversa, y, en el caso de tetrahidrobiopterina y análogos de la misma, realizar una detección electroquímica oxidando la tetrahidrobiopterina y los análogos de la misma presentes, a través de un primer electrodo a formas quinonoides de dihidrobiopterina, seguido de una reducción de las formas quinonoides de nuevo a tetrahidrobiopterina y análogos de la misma presentes en un segundo electrodo, y medir la corriente generada por la reacción de reducción para determinar la concentración de las especies, y/o en el caso de dihidrobiopterina, o análogos de la misma, biopterina o análogos de la misma, midiendo esas especies mediante una detección de la fluorescencia después de una oxidación después de la columna de las especies de dihidrobiopterina a biopterina.
Para las composiciones y usos descritos en esta memoria, los componentes preferidos y los intervalos de composición de los mismos, se pueden seleccionar a partir de los diversos ejemplos proporcionados en esta memoria.
Otras características y ventajas de la invención resultarán evidentes a partir de la siguiente descripción detallada. Breve descripción de los dibujos
La Figura 1 muestra un patrón de difracción de rayos X en polvo característico de la forma B polimorfa cristalina del 6R-(L-eritro)-5,6,7,8-tetrahidrobiopterina.
La Figura 2 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma A del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 3 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma F del diclorhidrato
de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 4 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma J del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 5 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma K del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 6 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma C hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 7 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma D hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 8 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma E hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 9 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma H hidrato H del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 10 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma O hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 11 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma G solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 12 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma I solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 13 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma L solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 14 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma M solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 15 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma N solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La Figura 16 es un diagrama de flujo para la medición de biopterina.
La Figura 17 es un resumen de la validación del ensayo de biopterina.
La Figura 18 es una tabla que muestra los parámetros farmacocinéticos de las biopterinas totales en plasma después de una única administración oral de sapropterina (BH4) a ratas.
La Figura 19 muestra la concentración de biopterina en plasma y la proporción de la forma reducida después de la administración de una dosis única de sapropterina (BH4) a ratas.
La Figura 20 muestra la concentración de biopterina en plasma y la proporción de la forma reducida después de la administración de una dosis única de sapropterina (BH4) a monos.
La Figura 21 es una tabla que muestra los parámetros farmacodinámicos de las biopterinas totales en plasma después de la administración de una dosis única de sapropterina (BH4) a monos.
La Figura 22 muestra la pauta de eventos para la evaluación de la seguridad.
La Figura 23 muestra las concentraciones plasmáticas medias de BH4 después de una administración oral de 10 mg/kg de BH4 en forma de comprimidos disueltos e intactos en ayunas y comprimidos intactos bajo condiciones de alimentación a voluntarios sanos - ejes lineales.
La Figura 24 muestra las concentraciones plasmáticas medias de BH4 después de una administración oral de 10 mg/kg de BH4 en forma de comprimidos disueltos e intactos en ayunas y comprimidos intactos bajo condiciones de alimentación a voluntarios sanos - ejes semilogarítmicos.
La Figura 25 muestra una tabla que resume los parámetros farmacocinéticos de BH4 después de una administración oral de 10 mg/kg de BH4 en forma de comprimidos disueltos e intactos en ayunas y comprimidos intactos bajo condiciones de alimentación a voluntarios sanos.
La Figura 26 muestra una comparación estadística de los parámetros farmacocinéticos para BH4 después de una administración oral de 10 mg/kg de BH4 como comprimidos disueltos e intactos en ayunas y comprimidos intactos
bajo condiciones de alimentación a voluntarios sanos.
La Figura 27 muestra un estudio de la estabilidad de BH4 formulada con manitol al 5% en una solución acuosa tanto antes como después de dos semanas almacenada a -20°C.
La Figura 28 muestra un perfil de disolución de una formulación de cápsulas de BH4 tanto antes como después de un almacenamiento durante 54 días a 40°C.
La Figura 29 muestra un perfil de disolución de dos formulaciones de BH4: un comprimido bioadhesivo de BH4 y gránulos bioadhesivos de BH4.
La Figura 30 muestra un perfil de disolución de varias formulaciones de liberación sostenida de BH4.
La Figura 31 muestra un perfil de disolución de varias formulaciones de liberación sostenida de BH4.
La Figura 32 muestra un diagrama esquemático de formulaciones de dosificación flotante de BH4.
La Figura 33 muestra un perfil de disolución de varias formulaciones de dosificación flotante.
La Figura 34 muestra un diagrama esquemático de formas de dosificación de BH4 que generan gas.
La Figura 35 muestra un perfil farmacocinético de varias formulaciones de BH4.
La Figura 36 muestra un estudio de la estabilidad de formulaciones de BH4 intravenosas a pH 4 durante 35 días. La Figura 37 muestra un estudio de la estabilidad de varias formulaciones de BH4 intravenosas durante 350 horas. La Figura 38 muestra un estudio de la estabilidad de formulaciones de BH4 intravenosas con diversas concentraciones de BH4.
Descripción de las realizaciones preferidas
La invención proporciona tetrahidrobiopterina (BH4) o una sal farmacéuticamente aceptable de la misma para uso en el tratamiento de afecciones asociadas con niveles elevados de fenilalanina, en donde la BH4 o una sal de la misma se administra por vía oral una vez al día, 5 a 60 minutos después de una comida y en donde la afección se selecciona a partir del grupo que consiste en fenilcetonuria leve (PKU), PKU clásica e hiperfenilalaninemia. La invención se basa en el descubrimiento de que la tetrahidrobiopterina (BH4) administrada por vía oral tiene una baja absorción gastrointestinal, lo que es un factor importante que contribuye a la baja biodisponibilidad de BH4.
La estructura química de 6R-(L-eritro)-5,6,7,8-tetrahidrobiopterina (BH4) se muestra a continuación:

La tetrahidrobiopterina es un compuesto orgánico soluble en agua, con baja solubilidad en lípidos. Basándose en un análisis experimental in silico utilizando el programa informático BioLoom (versión 1.5 de Biobyte Corp en Claremont California), se determinó que el coeficiente de partición octanol-agua de BH4 era -1,17. Una penetración óptima de las membranas biológicas tal y como se aproxima por el coeficiente de partición octanol/agua, se produce aproximadamente a un log P de una solubilidad en lípidos 2 o 100 veces mayor. Aunque un ClogP bajo permite que ese sustrato se solubilice fácilmente en condiciones fisiológicas, la capacidad del sustrato para penetrar en las bicapas lipídicas dentro de las membranas biológicas está restringida, lo que puede limitar la disponibilidad oral. En estudios in vivo en ratas y monos descritos en esta memoria, se ha mostrado que solo el 8-11% de BH4 es absorbido en el intestino y la mayoría se excreta con las heces, en comparación con la administración intravenosa de BH4 en dosis similares. Esa variabilidad en la absorción de BH4 también se ha mostrado en un estudio descrito en esta memoria sobre el efecto de los alimentos en la biodisponibilidad de BH4 en seres humanos sanos. Aunque la administración de BH4 en agua y zumo de naranja en ayunas, daba como resultado concentraciones plasmáticas medias y valores medios de Cmáx y AUC (0-t) comparables, la administración de BH4 junto con una comida rica en grasas y calorías, daba lugar a un aumento significativo de las concentraciones plasmáticas medias y los valores medios de Cmáx y AUC (0-t) que cuando se administraba BH4 en agua.
Aunque existen muchas publicaciones que describen una biodisponibilidad incrementada en condiciones de alimentación, ese efecto de los alimentos se observa típicamente con fármacos lipófilos insolubles en agua (es decir, solubles en lípidos) y no habitualmente con una sustancia activa altamente soluble en agua, tal como BH4. La explicación habitual de los aumentos de la biodisponibilidad de compuestos lipófilos en condiciones de alimentación, es que las comidas ricas en grasas ayudan a solubilizar el fármaco ya que "similar disuelve similar" y esto lo pone disponible para la absorción. Otra posible explicación es que las comidas ricas en grasas estimulan la secreción de ácidos biliares que son biotensioactivos naturales que ayudan a solubilizar y emulsionar las grasas que ingerimos para facilitar su digestión. También se cree que esos ácidos biliares solubilizan compuestos insolubles en agua, lo que los pone disponibles para la absorción. Sin embargo, BH4 no necesita una solubilización para ser absorbida, ya que su solubilidad es superior a 1000 mg/mL y el compuesto es uno de los fármacos más solubles conocidos. Por lo tanto, la mejora de su biodisponibilidad a través de comidas ricas en grasas y energéticas no es coherente con ese mecanismo conocido.
Sin embargo, la administración como una forma de dosificación sólida o semisólida y/o con una comida rica en grasas, puede maximizar la biodisponibilidad al aumentar el tiempo de residencia de BH4 en el medio ácido del estómago y el tracto gastrointestinal superior (TGI), en donde BH4 es químicamente estable. La estabilidad de BH4 disminuye al aumentar el pH y su semivida en una solución tampón a pH 6,8, que es aproximadamente el pH del intestino delgado, es de aproximadamente 15 minutos. A pH 3,1, que se encuentra dentro del ámbito del pH normal del estómago en voluntarios normales, la estabilidad de BH4 a una concentración de 1 mg/mL es de más de 3 horas. La estabilidad química de BH4 puede aumentar aún más cuando el pH del estómago se reduce por debajo de 3,1. Por lo tanto, un tiempo de residencia prolongado en el estómago proporciona un fármaco intacto a la pared del estómago para la absorción, mientras que un vaciado rápido en el intestino degrada la BH4 y, por tanto, no está disponible para ser absorbida.
Por tanto, para maximizar la biodisponibilidad oral de BH4 en cada administración, la BH4 debe tomarse con alimentos, por ejemplo, un alimento con alto contenido en grasas o con una comida con alto contenido en grasas y/o calorías.
Tal y como se usa en esta memoria, el término "biodisponibilidad" se refiere a la fracción de una dosis administrada de un fármaco que entra en la circulación sistémica. Si el fármaco se administra por vía intravenosa, entonces su biodisponibilidad teóricamente sería del 100%. Sin embargo, si el fármaco se administra por otras vías (tal como por vía oral), su biodisponibilidad será inferior al 100% como resultado, por ejemplo, de una absorción incompleta en el tracto GI, una degradación o un metabolismo antes de la absorción y/o un efecto de primer paso hepático.
La expresión "comida rica en grasas" se refiere generalmente a una comida de al menos aproximadamente 700 kcal y al menos aproximadamente un 45% de grasa (porcentaje relativo de kcal que son grasas), o alternativamente al menos aproximadamente 900 kcal y al menos aproximadamente un 50% de grasa. La expresión "alimento rico en grasas" se refiere generalmente a un alimento que comprende al menos 20 g de grasa, o al menos 25, 30, 35, 40, 45 o 50 g de grasa, y/o al menos aproximadamente un 45% o 50% grasa. Una guía de la FDA define una "comida rica en grasas" como aproximadamente el 50% del contenido calórico total de la comida, mientras que una "comida rica en calorías" tiene aproximadamente de 800 a 1000 calorías. La FDA recomienda una comida rica en grasas y calorías como comida de prueba para determinar la biodisponibilidad del efecto de los alimentos y los estudios de bioequivalencia de la alimentación. Esa comida de prueba debe obtener aproximadamente 150, 250 y 500-600 calorías a partir de proteínas, carbohidratos y grasas, respectivamente. Un ejemplo de comida de prueba consiste en dos huevos fritos en mantequilla, dos lonchas de tocino, 114 g (cuatro onzas) de patatas salteadas y 225 g (ocho onzas) de leche entera. Una sustitución es posible si una cantidad similar de calorías de proteínas, carbohidratos y grasas tiene un volumen de comida y una viscosidad comparables (Guidance for Industry, Food-Effect Bioavailability and Fed Bioequivalence Studies, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), diciembre 2002).
En un primer aspecto, la invención proporciona tetrahidrobiopterina (BH4) o una sal farmacéuticamente aceptable de la misma para uso en el tratamiento de afecciones asociadas con niveles elevados de fenilalanina, en donde la BH4 o una sal de la misma se administra por vía oral una vez al día, 5 a 60 minutos después de una comida y en donde la afección se selecciona a partir del grupo que consiste en fenilcetonuria leve (PKU), PKU clásica e hiperfenilalaninemia.
En algunas realizaciones, se informa al paciente de que la administración de tetrahidrobiopterina con alimentos tiene un efecto sobre la farmacocinética. En una realización ejemplar, se informa al paciente de que la absorción de tetrahidrobiopterina aumenta cuando se ingiere con alimentos, en comparación con cuando se ingiere sin alimentos. En algunas realizaciones, se informa al paciente de que una ingestión poco después de una comida, por ejemplo, una comida rica en grasas y calorías, da como resultado un aumento de uno, dos, tres o todos los siguientes parámetros: concentración plasmática media, Cmáx, AUC, AUC (0-t) y/o AUC (inf). En realizaciones ejemplares, se informa al paciente de que la administración de BH4 con una comida rica en grasas aumenta la Cmáx y la AUC, en comparación con la administración de BH4 sin comida (en ayunas). En algunas realizaciones, el aumento relativo puede ser de al menos un 20% o un 30% o superior.
En realizaciones alternativas o además de las realizaciones anteriores, se informa al paciente de que la absorción de
tetrahidrobiopterina aumenta cuando se ingiere como un comprimido intacto, en comparación con cuando se ingiere después de disolverla en líquido. En algunas realizaciones, se informa al paciente de que la ingestión de comprimidos intactos da como resultado un aumento de cualquiera de los siguientes parámetros: concentración plasmática media, Cmáx, AUC, AUC (0-t) o AUC (inf). En realizaciones ejemplares, se informa al paciente de que la administración de BH4 como un comprimido intacto aumenta la Cmáx y la AUC, en comparación con la administración de BH4 después de disolverla en un líquido. En algunas realizaciones, el aumento relativo puede ser de al menos un 20% o superior.
Cualquiera de las realizaciones anteriores se puede llevar a cabo proporcionando o administrando tetrahidrobiopterina en un envase que contiene etiquetas impresas que informan al paciente del cambio en los parámetros de absorción descritos anteriormente.
La presente invención comprende la etapa de proporcionar al paciente que lo necesite una cantidad terapéuticamente eficaz de tetrahidrobiopterina. La cantidad terapéuticamente eficaz variará dependiendo de la afección que se va a tratar, y el médico que indica el tratamiento puede determinarla fácilmente basándose en una mejora de los síntomas clínicos deseados.
En una realización ejemplar, esos usos implican la administración de BH4 en forma disuelta, en donde la formulación se disuelve en un líquido que incluye pero no se limita a agua, zumo de naranja y zumo de manzana. Por tanto, BH4 se puede ingerir como un producto líquido o se puede disolver previamente a partir de una forma de dosificación sólida o semisólida antes de la ingestión. En una realización adicional, BH4 también se puede disolver en la cavidad oral a partir de una forma de dosificación sólida o semisólida antes de tragar la solución disuelta.
Estos enfoques maximizan la tasa de absorción y la biodisponibilidad al garantizar que BH4 se disuelve completamente en una solución o fluidos biológicos, antes de que se administre a sus sitios de absorción, que son principalmente el estómago y el intestino. La disolución de principios activos farmacéuticos o un fármaco en solución es un requisito previo para la absorción en la circulación sistémica (sanguínea y linfática). Cuando formas de dosificación sólidas, como comprimidos y cápsulas, se administran por vía oral, pasan a través de una serie secuencial de etapas, tales como la desintegración en gránulos, la desagregación en polvos y la disolución antes de la absorción en la circulación sistémica. Esa serie de etapas se pasa por alto mediante la administración de formas de dosificación líquidas, semisólidas y sólidas de disolución rápida. Por lo tanto, la sustancia activa está disponible antes para la absorción, y debido a que no existe una garantía de que una forma de dosificación sólida libere toda la sustancia activa que contiene antes de que transite por los sitios de absorción, las formulaciones en las que la sustancia activa está presente en forma disuelta antes de que llegue a los sitios de absorción, suelen presentar la mayor biodisponibilidad.
Esas formas de dosificación reducen la variabilidad en los niveles sanguíneos debido a que se elimina la desintegración y disolución in vivo de la forma de dosificación en humanos. La tasa de desintegración y disolución in vivo de una forma de dosificación sólida de BH4 destinada a una liberación inmediata en el estómago, depende de la variabilidad de persona a persona en el pH del fluido gástrico - que se ha alimentado y sin alimentar (en ayunas) - y la fuerza de la intensidad de la agitación del estómago, determinada por la fuerza de la motilidad gástrica y las tasas de vaciado gástrico en el intestino delgado. Dado que las formas de dosificación líquidas, semisólidas, pastillas/caramelos y sólidas de disolución rápida, no se tienen que someter a una desintegración y disolución, sus niveles en sangre son menos variables que cuando se administra BH4 como formas de dosificación sólidas de liberación inmediata (comprimidos y cápsulas).
En otra realización ejemplar, esos usos implican la administración de BH4 en una forma de dosificación sólida que incluye, entre otros, comprimidos, cápsulas, caramelos, pastillas, polvos y gránulos, o en forma semisólida, que incluyen, pero no se limitan a, rociar oralmente en gelatina que se mastica o se ingiere sin disolver en un líquido que incluye, entre otros, agua, zumo de naranja y zumo de manzana, antes de tragar.
En otra realización, esos usos implican la administración de BH4, ya sea de forma tragada como una forma de dosificación sólida o semisólida, o disuelta en un líquido, con alimentos, p. ej., un alimento con alto contenido en grasas o una comida con alto contenido en grasas y/o calorías. La invención contempla además que BH4, ya sea tragada o disuelta, es para una administración en un momento específico que incluye, pero no se limita a, mañana, día, noche, a la misma hora del día, con alimentos, p. ej., un alimento con alto contenido en grasas o una comida con alto contenido en grasas y/o calorías, una o más veces al día. En una realización ejemplar, BH4 se ingiere una vez al día como una forma de dosificación sólida justo después de las comidas. En una realización preferida, la forma de dosificación sólida es un comprimido o una cápsula formulados. En más realizaciones ejemplares, BH4 se ingiere 5 a 60 minutos o 5 a 20 minutos después de una comida. Independientemente de si se ingiere como una forma de dosificación sólida, una forma de dosificación líquida o como una solución disuelta, la exposición in vivo (o biodisponibilidad) de BH4 es mayor cuando se ingiere justo después de las comidas que en comparación con los controles en ayunas.
La BH4 se ingiere después de la comida. El período de tiempo entre la ingesta de alimentos, por ejemplo, un alimento con alto contenido en grasas o una comida con alto contenido en grasas y/o calorías y la ingestión de BH4, ya sea tragada o disuelta, puede ser de al menos 5 minutos. BH4 se puede administrar 60 minutos, 30 minutos, 25
minutos, 20 minutos, 15 minutos, 10 minutos o 5 minutos después de la ingestión de una comida.
En otra realización, para algunos pacientes, p. ej., adultos, los usos de la invención implican administrar un comprimido intacto en lugar de disolver el comprimido en un líquido, con el fin de mejorar la biodisponibilidad.
La administración de BH4 de acuerdo con los usos de la invención da como resultado concentraciones plasmáticas medias y/o tasa de absorción gastrointestinal y/o valores medios para Cmáx y/o AUC (0-t) y/o AUC (inf), que exceden los valores de cuando BH4 se administra en ayunas.
La administración de un comprimido intacto en ayunas daba como resultado un aumento promedio del 20% en la Cmáx y la AUC, en relación con los comprimidos disueltos. La administración de un comprimido disuelto en agua o zumo de naranja o un comprimido intacto, después de una comida rica en grasas/calorías, daba lugar a aumentos en la Cmáx y la AUC que variaban desde aproximadamente un 30% (comprimido intacto) a un 80% (agua). La administración de BH4 como un comprimido intacto después de una comida rica en grasas y calorías daba como resultado un aumento de aproximadamente un 30% en el grado de absorción, en comparación con la administración sin alimentos. La administración de BH4 como un comprimido intacto daba lugar a un aumento de aproximadamente un 20% en el grado de absorción en comparación con la administración de comprimidos disueltos.
"Concentración plasmática media" significa el promedio de las lecturas de concentración en una serie de muestras de plasma.
"Cmáx" significa la concentración plasmática máxima observada.
"AUC" significa el área bajo la curva de concentración plasmática-tiempo.
"AUC 0-t" significa el área bajo la curva de concentración plasmática-tiempo desde el momento 0 hasta el momento de la última concentración medible.
"AUC (inf)" significa el área calculada bajo la curva de concentración plasmática-tiempo desde el tiempo 0 hasta el infinito.
La "tasa de absorción gastrointestinal" de BH4 se estima a partir del área bajo la curva de aumento de la concentración plasmática total de biopterina (ACp)-tiempo (AAUC) después de la administración de BH4 utilizando la siguiente fórmula:
Tasa de absorción (%)=
(AAUC después de la dosis p.o. / AAUC después de la dosis i.v.) x (dosis i.v. / dosis p.o. x 100) Preferiblemente, se usa 6R-BH4 pura al menos en un 99,5%. Se puede utilizar cualquier sal, incluida la sal diclorhidrato, y cualquier forma cristalina de BH4 de acuerdo con los métodos y composiciones de la invención. Una variedad de sales y formas cristalinas se describen en el documento de publicación de patente de EE.UU. n° 2006/0040946, y/o la formulación sólida estable descrita en el documento de publicación internacional n° WO 06/55511. Las diversas formas cristalinas se pueden formar convenientemente como un comprimido, polvo u otro sólido para administración oral.
También se describe en esta memoria un método para estabilizar BH4 mediante una disminución del pH intestinal usando polímeros de intercambio de protones. BH4 se administra por vía oral a diario como una forma de dosificación sólida o líquida que comprende ingredientes inactivos que mejoran la estabilidad de BH4 más allá del estómago al reducir el pH del intestino y así evitar que BH4 se oxide rápidamente. Dado que BH4 es más estable en medios ácidos que en medios básicos, se incluyen excipientes/ingredientes inactivos acidificantes en formulaciones de dosificaciones sólidas (comprimidos, cápsulas, etc.) de BH4 para reducir el pH de los fluidos intestinales y, por lo tanto, mejorar la estabilidad química. El área o ventana más grande del tracto gastrointestinal (TGI) disponible para la absorción, optimiza la consistencia de la absorción al expandir la ventana de absorción limitada actual que se cree que está limitada al estómago y al duodeno hasta el intestino. Esas formas de dosificación incluyen, pero no se limitan a, comprimidos efervescentes, polvos y gránulos (para resuspender en un líquido antes de la administración) y materiales acidulantes. A diferencia de los ácidos de molécula pequeña, los ácidos poliméricos voluminosos permanecen más tiempo en el TGI y no son absorbidos por el TGI, pero donan sus protones a los fluidos del TGI para reducir el pH del entorno. Ejemplos de excipientes/ingredientes inactivos que comprenden la formulación, son moléculas pequeñas de ácido carboxílico tales como los ácidos maleico, fumárico y cítrico o moléculas pequeñas inorgánicas tales como ácido fosfórico, ácido acético y sus formas de sal. Otros ejemplos son ácidos farmacéuticamente aceptables, tales como clases de ácidos carboxílicos poliméricos que incluyen poli(ácidos metacrílicos), carbómeros, policarbofilo, Eudragits, formas ácidas de croscarmelosa y ácido glicólico de almidón, etc. Las formulaciones también contienen excipientes adicionales para aumentar la estabilidad, tales como antioxidantes (por ejemplo, tioles tales como cisteína, cisteína de N-acetilo, etc.; ácido ascórbico, metionina; etc.) y otros excipientes conocidos en el mercado para permitir su preparación, y mejorar los atributos de calidad y rendimiento de la formulación.
También se describe en esta memoria un método para aumentar el tiempo de residencia intestinal de BH4, que incluye, pero no se limita a, la ralentización de la motilidad intestinal utilizando un agente que es capaz de ralentizar la motilidad intestinal de BH4, tal como un ácido graso y/o un ácido graso de glicerol. Los ácidos grasos pueden incluir ácido oleico, ácido esteárico, ácido araquídico, ácido palmítico, ácido araquidónico, ácido linoleico, ácido linolénico, ácido erucídico, ácido mirístico, ácido láurico, ácido miristólico y ácido palmítico. También se contempla para aumentar el tiempo de residencia intestinal de BH4, la inducción de una retención gástrica usando ácido algínico y la bioadhesión usando policarbofilo. En una realización, las formas de dosificación de BH4 se administran como formulaciones flotantes orales que flotan y liberan BH4 de una manera definida en el líquido gástrico y se retienen más tiempo en el estómago porque son más resistentes al vaciado gástrico desde el estómago que las formulaciones que no son flotantes o que se disuelve rápidamente en el estómago. Este enfoque de diseño se basa en la retención gástrica de la forma de dosificación mediante el uso de un excipiente generador de gas dentro de la forma de dosificación, excipientes de baja densidad que hacen que la forma de dosificación flote en fluidos del TGI o una combinación de un gas y materiales de baja densidad en una forma de dosificación, para permitir la flotación de la forma de dosificación en el contenido fluido del TGI. Una retención y liberación prolongadas de la forma de dosificación en el medio estomacal, en donde BH4 es más estable en sus fluidos ácidos, mejorará tanto el tiempo de residencia de la forma de dosificación en el estómago como la estabilidad de BH4 y, por lo tanto, hará que BH4 esté disponible durante un período de absorción más prolongado en el estómago y el duodeno que las formas de dosificación convencionales de comprimidos y cápsulas. Las formulaciones de BH4 comprenderán uno o más antioxidantes, excipientes conocidos en el campo para permitir la preparación y la desintegración/disolución de la forma de dosificación sólida y excipientes adicionales que generan un gas o una mezcla de gases (por ejemplo, dióxido de carbono) al entrar en contacto la formulación con medios acuosos y/o los fluidos del TGI. Se prefieren los antioxidantes solubles en agua, por ejemplo, ácido ascórbico, metionina y tioles (cisteína, N-acetilcisteína y glutatión) o antioxidantes que se convierten en un antioxidante soluble en el TGI, por ejemplo, palmitato de ascorbilo que se convierte en ácido ascórbico en el TGI. Los excipientes añadidos a la formulación incluyen carbonatos y bicarbonatos que reaccionan directamente con BH4 para formar dióxido de carbono y ácidos poliméricos y pequeños, descritos anteriormente para reaccionar con los carbonatos y bicarbonatos para producir dióxido de carbono adicional, según sea necesario.
En otra realización, se administran formas de dosificación de BH4 que se adhieren durante un tiempo prolongado a las superficies mucosas del TGI (es decir, una formulación bioadhesiva), preferiblemente en, pero de ninguna manera limitado a, el estómago en donde debido a la acidez de los fluidos gástricos, BH4 es más estable que en el intestino. BH4 se libera de manera controlada desde la forma de dosificación bioadhesiva. La forma de dosificación sólida está diseñada para contener BH4, uno o varios antioxidantes, excipientes conocidos en el campo para permitir la preparación de formas de dosificación de calidad y controlar la desintegración/disolución de la forma de dosificación y un aditivo bioadhesivo tal como policarbofilo en su forma de ácido libre o en forma de sal. Otros ácidos poliméricos tales como poli(ácidos metacrílicos), carbómeros y derivados de celulosa, por ejemplo, HPMC, HPC, etc., se pueden combinar o sustituir por policarbofilo. Los antioxidantes son preferiblemente solubles, por ejemplo, ácido ascórbico, metionina, cisteína, N-acetilcisteína y glutatión o se pueden convertir en un antioxidante soluble tal como ácido ascórbico en el TGI, por ejemplo, palmitato de ascorbilo. En una realización, los componentes de la formulación se mezclan juntos y se preparan como una forma de dosificación sólida, por ejemplo, comprimidos o cápsulas. La forma de dosificación sólida puede tener un recubrimiento entérico para administrar BH4 más allá del estómago en el intestino o no tener un recubrimiento entérico diseñado para liberar BH4 en el estómago. En otra realización, los componentes de la forma de dosificación sólida se pueden subdividir en diferentes porciones y las diversas porciones se mezclan por separado antes de procesarlas para formar formas de dosificación multicapa. La forma de dosificación multicapa puede contener el bioadhesivo y algunos excipientes en la capa más externa de un comprimido, envuelto sobre otras capas que contienen BH4 (es decir, región activa dentro de una envoltura bioadhesiva) o como un tapón cilíndrico envolvente que introducido en una cápsula, en donde una o varias de las otras capas se ensamblan debajo o dentro de la envoltura bioadhesiva. Alternativamente, el bioadhesivo y otras capas en los tapones del comprimido o la cápsula se pueden colocar en una configuración en paralelo de dos o múltiples capas. Esos diseños permiten que el bioadhesivo interaccione con la membrana GI o la membrana mucosa GI para anclar la forma de dosificación a la membrana, lo que ralentiza su tránsito a través del tracto GI y aumenta así el tiempo de residencia. Esas formas de dosificación también pueden tener un recubrimiento entérico. Otra realización adicional de la invención es emplear ingredientes poliméricos inactivos (excipientes) con grupos funcionales que se unen a la mucosa del TGI para retrasar el tránsito de la forma de dosificación a través del TGI. Las formas de dosificación de BH4 se formulan con excipientes poliméricos tiolados (polímero-SH) tales como policarbofilo-cisteína, poli(ácido metacrílico)-cisteína, carboximetilcelulosa-cisteína, derivados de quitosano-cisteína, etc. Esos polímeros tiolados confieren propiedades bioadhesivas y antioxidantes a BH4 que mejoran considerablemente la absorción. Otros excipientes incluidos en esas formulaciones son antioxidantes y excipientes que ayudan al rendimiento y a la preparación.
En todavía otra realización, se usan formas de dosificación oral que contienen excipientes inactivos o ingredientes activos para ralentizar la motilidad gástrica. La ralentización del tránsito de la forma de dosificación de BH4 a través del tracto gastrointestinal aumentará el tiempo de residencia de la molécula y, por lo tanto, permitirá que se absorba una fracción superior de la dosis administrada. Los excipientes generalmente considerados seguros (GRAS) empleados en formulaciones orales para retrasar el vaciado gástrico y/o retrasar la motilidad intestinal, comprenden preferiblemente grasas dietéticas, tales como ácidos grasos, glicéridos de ácidos grasos y derivados de ácidos
grasos y glicéridos tales como Cremophor™ (derivados de aceite de ricino de polioxilo), etc. Los excipientes activos incluyen agentes que ralentizan la motilidad intestinal, tales como agentes antimuscarínicos o anticolinérgicos generales o selectivos (M3).
También se describe en esta memoria un método para modificar la liberación de BH4, usando una formulación de liberación sostenida tal como HPMC, carbómero, etc. Este concepto comprende la administración de formas de dosificación de BH4 en el tracto gastrointestinal, modificando o alterando la liberación de BH4 desde una liberación inmediata a una liberación lenta, prolongada, controlada o programada. La liberación lenta, prolongada y controlada se logra utilizando excipientes conocidos en la técnica y BH4 se protege dentro del sistema de administración de una degradación química por la presencia de potenciadores de la estabilidad, tales como antioxidantes. Esto puede maximizar la biodisponibilidad ya que BH4 se estabiliza dentro de la formulación y en el entorno que rodea la formulación para permitir que la molécula activa se absorba intacta en la circulación sistémica, a medida que la formulación transita a lo largo de toda la longitud del TGI. Este enfoque proporciona una ventana más grande del TGI para la absorción y lo hace al evitar la degradación de BH4 en el medio con pH más alto, de modo que BH4 está disponible para ser absorbida. Se incluirán antioxidantes en la formulación para evitar que el fármaco se degrade en los fluidos intestinales debido al pH casi neutro de los fluidos intestinales. Una liberación lenta, prolongada y controlada también entregará BH4 en las regiones de baja tensión de oxígeno del TGI. La liberación programada se logra usando excipientes conocidos en la técnica, tales como polímeros sensibles al pH que se disuelven solo cuando el pH alcanza un valor en el que el polímero es soluble.
En otra realización, la invención contempla un recubrimiento entérico de la forma de dosificación de BH4 para determinar si la inclusión de excipientes ácidos en una formulación de BH4 realmente aumenta la absorción de BH4 al reducir el pH del intestino y, por lo tanto, al estabilizar BH4 en el intestino para que esté disponible para la absorción. Por tanto, un recubrimiento entérico se utilizará para mantener los excipientes y el fármaco juntos en el sitio donde se espera que el excipiente proteja a BH4. Si se permite que la forma de dosificación de BH4 se desintegre en el estómago, es posible que los excipientes ácidos no se vacíen juntos en el estómago y no proporcionen protección.
Un recubrimiento entérico protege a los compuestos susceptibles de una degradación catalizada con ácido en el estómago de ser degradados por el ácido en el estómago. Los materiales del recubrimiento entérico evitan que el comprimido o la cápsula libere el compuesto activo en el estómago ya que los materiales de recubrimiento entérico son insolubles en ácido. Una vez que la forma de dosificación con recubrimiento entérico llega al intestino, en donde el valor del pH varía de 5 - 8, los materiales se vuelven solubles y liberan la sustancia activa en el intestino. Por el contrario, las formulaciones de liberación sostenida están diseñadas para liberar medicamentos a lo largo de una longitud/área del TGI lo más amplia posible. Puede ser necesario recubrir una formulación de liberación sostenida para que se libere justo después del estómago, solo si los medicamentos que contiene son lábiles frente al ácido.
También se describe en esta memoria la administración de BH4 en una forma de dosificación sólida estéril o líquida estéril mediante vías de administración oral distintas, incluidas, pero no limitadas a, las vías de administración tópica, intravenosa, subcutánea, intramuscular, intratecal, oftálmica e inhalatoria. BH4 se formula como una forma de dosificación sólida o líquida estéril con la concentración apropiada deseada.
Las ventajas de una forma de dosificación líquida estéril de BH4 para una administración intravenosa pueden incluir: (1 ) cinéticas más predecibles, con el potencial de niveles séricos más altos; (2) sin requerimientos de un tracto gastrointestinal funcional; (3) sin requerimientos de una participación del paciente; y (4) ausencia de un problema de incumplimiento. Las formulaciones intravenosas de BH4 pueden ser particularmente beneficiosas para controlar afecciones que requieren una administración rápida de líquidos y medicamentos en todo el cuerpo o en compartimentos corporales de difícil acceso por vía oral u otras formas de administración, que incluyen, pero no se limitan a, rabia, meningitis, trasplante/conservación de órganos, hemorragias subaracnoideas, traumatismo cerebral, accidente cerebrovascular, cirugía de derivación de arterias coronarias, vasoespasmo cerebrovascular, transfusión/conservación de sangre, hipertensión pulmonar, enfermedad de células falciformes, preeclamsia y enfermedad vascular posquimioterapia.
BH4 es muy susceptible a la oxidación en solución acuosa y en soluciones acuosas de pH fisiológico (Davis, et al., Eur. J. Biochem. 173, 345-351 (1988); Kirsch, et al., J. Biol. Chem. 278, 24481-24490 (2003)). La mayoría de las determinaciones de la estabilidad de BH4 se han llevado a cabo en soluciones con pH 7,4 de neutro a ligeramente alcalino para imitar el comportamiento probable de la estabilidad de BH4 en condiciones de pH fisiológico plasmático. Aunque el documento de Solicitud de Patente Europea n° 1757293 A describe formulaciones líquidas o en forma de jarabe, esas formulaciones consisten en mezclas de polvo o granulaciones en estado sólido que requieren una reconstitución con agua antes de la ingestión oral. El presente aspecto de la invención contempla formulaciones líquidas no limitadas a polvos o a granulaciones para constituir. La invención también contempla formulaciones líquidas mezcladas capaces de permanecer estables a temperatura ambiente durante un período de tiempo suficiente para permitir un procesamiento en instalaciones de llenado/acabado de productos estériles para llenar ampollas, frascos o viales como un producto líquido o para llenar viales para congelar-secar como productos liofilizados.
Las formulaciones líquidas y liofilizadas para reconstituir también se pueden administrar a través del canal nasal,
oftálmico y auditivo para efectos terapéuticos. La formulación de un producto liofilizado requiere una disolución previa de BH4 en un líquido, preferiblemente acuoso, y el procesamiento del producto líquido en una instalación estéril (es decir, mezcla del compuesto, filtración estéril y llenado del líquido filtrado estéril en viales antes de cargar los viales llenos en un liofilizador para la liofilización). Mantener la estabilidad de BH4 solubilizada durante un procesamiento estéril y evitar su degradación, son requisitos previos clave para fabricar un producto liofilizado que satisfaga la especificación de impurezas para el producto con llenado y acabado. Por consiguiente, la composición del producto liofilizado contiene agentes estabilizantes adecuados que minimizan o evitan la degradación de BH4 durante el proceso de llenado y acabado. Las formulaciones descritas en esta memoria estabilizarán las soluciones de BH4 durante la fabricación de llenado/acabado estéril, un proceso que requiere un mínimo de seis horas, y también proporcionarán un producto comercialmente estable.
Las formulaciones incluyen BH4, preferiblemente en una concentración en un intervalo de 0,1 mg/mL a 10 mg/mL. Debido a la alta solubilidad de BH4, también se pueden preparar formulaciones con concentraciones de hasta aproximadamente 100 mg/mL, por ejemplo. La elaboración constitutiva relativa general y los métodos descritos en esta memoria son aplicables para preparar soluciones altamente concentradas.
Las formulaciones líquidas de BH4 se formulan preferiblemente en soluciones tampón con pH de 1 a 8, preferiblemente en soluciones tampón con pH de 2 a 7. Los tampones de pH elegidos son compuestos tamponadores capaces de proporcionar una capacidad tamponadora sustancial a un pH particular deseado, a juzgar por la proximidad de la constante o constantes de ionización del tampón al pH deseado de la formulación líquida. Por tanto, se puede emplear cualquier compuesto tamponador, siempre que una o varias de las constantes de ionización del compuesto estén cerca del pH deseado de la formulación. Ejemplos de tampones que se pueden emplear en el intervalo de pH 1 - 8, comprenden varios ácidos/bases y sus respectivos conjugados ácidos/bases o formas de sal, que incluyen pero no se limitan a: ácido clorhídrico (pH 1 - 2), ácido maleico (pH 1 - 3), ácido fosfórico (pH 1 - 3), ácido cítrico (pH 3 - 6), ácido acético (pH 4,7 ± 1,0), fosfato de sodio dibásico (pH 6 - 8), trometamina (TRIS, pH 8,3 ± 1,0), y similares.
Formulaciones intravenosas
Las formulaciones intravenosas se estabilizan usando un antioxidante o una combinación de 2 o más antioxidantes. Las combinaciones de antioxidantes pueden ser sinérgicas para evitar una inestabilidad de la formulación. El burbujeo con gases inertes y/o dióxido de carbono para eliminar el oxígeno disuelto de la solución es opcional, pero se prefiere cuando se usan concentraciones bajas de antioxidantes, y más preferiblemente cuando se usan concentraciones bajas de BH4 y antioxidantes. La estabilización de BH4 en una solución acuosa está influenciada por las interacciones de la concentración de BH4 con el antioxidante y el pH. Así, por ejemplo, unas concentraciones altas de BH4 requieren menos concentraciones de antioxidantes que concentraciones bajas de BH4. Además, BH4 es más estable a pH bajo que a pH alto. Por lo tanto, las formulaciones de pH alto deseadas tienen preferiblemente concentraciones de antioxidante más altas, más preferiblemente un
Los intervalos a modo de ejemplo para formulaciones líquidas de BH4 se proporcionan en las Tablas 1 y 2. Las soluciones formuladas o mezcladas se burbujean opcionalmente con un gas inerte (por ejemplo, argón o nitrógeno) o dióxido de carbono en el tanque de mezcla y los envases primarios preferiblemente se sellan en una capa de gas inerte o dióxido de carbono para eliminar el oxígeno del espacio superior del envase. La formulación se puede aumentar a cualquier volumen, multiplicando las cantidades de los componentes por un factor de escala apropiado. Tabla 1 Ejemplos generales de intervalos de composición en una formulación con pH bajo (por ejemplo, pH 4,0)

Tabla 2 Ejemplos generales de intervalos de composición de una formulación con pH neutro (p. ej., pH 7,0)


Los antioxidantes empleados para formulaciones líquidas se seleccionan preferiblemente a partir de uno o varios compuestos basados en tiol (por ejemplo, L-cisteína), ácido ascórbico y basados en sulfito (por ejemplo, metabisulfito de sodio). Las soluciones se burbujean preferiblemente con gases inertes o dióxido de carbono para expulsar el oxígeno de las soluciones de BH4 y luego se sellan herméticamente en ampollas o viales y frascos con tapón hermético, usando chapas metálicas tipo cerveza para bebidas en una capa de gases inertes (por ejemplo, argón, nitrógeno) o gas no inerte como el dióxido de carbono, para evitar que se escapen los gases burbujeados en los espacios superiores del envase. Las formulaciones líquidas orales preferiblemente contienen de forma adicional edulcorantes y aromatizantes que mejoran el sabor de las formulaciones.
En una realización, como forma de dosificación líquida, BH4 se estabiliza con antioxidantes y/o burbujeando gases no oxidantes, preferiblemente esterilizados, tales como gases inertes (por ejemplo, nitrógeno, argón, helio, etc.) y/o un gas no inerte como dióxido de carbono, para eliminar el oxígeno molecular de la formulación. El producto se introduce preferiblemente bajo una capa de gases inertes para minimizar o evitar que el oxígeno molecular se vuelva a disolver en la formulación. El líquido se introduce en un envase (por ejemplo, viales, ampollas, etc.) y se sella herméticamente para evitar que el oxígeno entre en el envase. En otra realización, como forma de dosificación sólida estéril para administración parenteral, una solución de BH4 se liofiliza y se reconstituye en el ambulatorio antes de la administración. En otra realización adicional, la sustancia farmacéutica en forma de polvo estéril de BH4 se envasa directamente en envases estériles (por ejemplo, viales, bolsas, frascos o ampollas) en una instalación de llenado de polvo seco estéril. Por lo tanto, otro aspecto de la invención es una formulación en forma de polvo seco de tetrahidrobiopterina (BH4) o una sal farmacéuticamente aceptable de la misma para constituir en una solución acuosa, que incluye una mezcla de polvo seco de BH4 o una sal farmacéuticamente aceptable de la misma, un antioxidante y un tampón del pH.
Composiciones de formulaciones líquidas orales
Las formulaciones líquidas orales comprenden además de los componentes empleados en las formulaciones líquidas e intravenosas generales, agentes edulcorantes y aromatizantes. Los agentes edulcorantes y aromatizantes se añaden en cantidades suficientes para producir un dulzor y sabor aceptables. Las formulaciones líquidas orales contienen uno o varios agentes estabilizantes. Opcionalmente, contienen conservantes antimicrobianos. Preferiblemente se tamponan a pH bajo, por ejemplo, pH 1 - 4 y los agentes tamponadores se seleccionan para que combine con el agente aromatizante, mejorando así las propiedades organolépticas de la formulación líquida oral. Ejemplos de tampones preferidos (ácidos y bases conjugadas) son: ácido cítrico, ácido tartárico, ácido málico en combinación con sus bases conjugadas o formas de sal.
Ejemplos de edulcorantes incluyen azúcares (p. ej., sacarosa, glucosa, sorbitol, manitol, fructosa, etc.), edulcorantes intensos sin azúcar (p. ej., aspartamo, acesulfamo K, ciclamato, sacarina, sucralosa, glicirricina, alitamo, neotamo, neohesperidina DC, taumatina, monelina y similares).
Como también se describe en esta memoria, para las administraciones nasales, oftálmicas y óticas, BH4 se formula como se ha descrito para las formas de dosificación parenteral y es opcionalmente un producto estéril. Esas formas de dosificación se pueden proporcionar en una presentación como una caja tipo kit con provisiones para varios días. Cada unidad dentro del kit puede estar compuesta por un vial o ampolla y un pulverizador (para la forma de dosificación nasal) o un gotero (en el caso de las formas de dosificación oftálmica y ótica). Una vez abierto el vial o la ampolla, se enrosca el atomizador o el gotero en el vial o ampolla y se desecha el tapón anterior. El producto en forma de dosificación se usa dentro de un período de caducidad prescrito y luego se desecha y se abre un nuevo vial o ampolla para el uso. Otra realización es introducir las soluciones en envases estériles, herméticos, desechables de plástico, de un solo uso, producidos mediante un proceso de fabricación según el sistema de formado, llenado y sellado. Esos envases se abren y las soluciones se administran utilizando la vía de administración deseada presionando para extraer el líquido contenido en ellos. Esas formas de dosificación se administran una vez al día y se administran a través de las fosas nasales (producto nasal), o a través de los ojos (oftálmico) o se instilan gotitas en el canal auditivo (producto ótico). Con respecto a una medicación envasada según un envase de formado, llenado y sellado, la medicación se presiona para que se introduzca por la vía de
administración.
Tal y como se describe también en esta memoria, BH4 se administra por vía bucal y transdérmica usando lonchas, parches o películas formuladas o como productos tópicos que se colocan en el sitio de la administración. Los comprimidos sublinguales se colocan debajo de la lengua. Esas formas de dosificación se administran una vez al día y o bien se fijan a la membrana del lugar de administración (vía bucal y transdérmica) o se colocan como una forma sólida o de semisodosificación en el sitio sublingual. Para evitar una irritación del sitio de administración, se recubre con un compuesto básico, tal como carbonato o bicarbonato de sodio y se mezcla con BH4 para evitar una interacción con BH4 que la volvería inestable. Alternativamente, el compuesto básico se añade justo antes del uso para elevar el pH de BH4, que es bastante bajo. Añadir el excipiente básico en el momento de la preparación sin recubrir las partículas alcalinas para evitar una interacción con BH4, conducirá a la inestabilidad de BH4. Otra realización es recubrir un comprimido sublingual con núcleo de BH4 con una solución de recubrimiento que contiene una sustancia básica o alcalina. En el compartimento sublingual, el compuesto básico se disuelve primero e interacciona con BH4 para elevar el pH del medio.
Envasado del envase primario para formulaciones líquidas de BH4
Los recipientes de envasado primario para formulaciones líquidas de BH4 son preferiblemente impermeables al oxígeno, dióxido de carbono, nitrógeno y gases inertes. Después del llenado con formulaciones líquidas de BH4 burbujeadas en el recipiente primario, preferiblemente bajo una capa de nitrógeno, los envases se sellan de forma preferible herméticamente para mantener el gas burbujeado en el líquido y el espacio superior del envase y evitar la pérdida del gas burbujeado y la entrada de oxígeno en el envase.
Los envases primarios preferidos son ampollas selladas herméticamente así como frascos y viales sellados herméticamente con tapones metálicos como los empleados para sellar botellas de bebidas gaseosas y de cerveza. Durante el uso, las ampollas se abren cortándolas y se usan en unas pocas horas, por ejemplo, aproximadamente 12 horas. Las ampollas se pueden utilizar para productos intravenosos y estériles para inyecciones. Los líquidos inyectables estériles y los productos liofilizados también se pueden envasar en viales sellados con cierre de goma que se aseguran con una tapa de aluminio rizada. Los antioxidantes en las formulaciones protegen a los productos líquidos y liofilizados de una pérdida imperceptiblemente lenta del gas burbujeado o de una entrada de oxígeno en el vial durante la vida útil del producto.
Las formulaciones líquidas de BH4 envasadas en frascos o viales para uso oral, oftálmico u ótico, se aseguran preferiblemente de forma hermética con una tapa metálica para bebidas o un tapón de goma asegurado con un sello de aluminio rizado. Los canales de los frascos o los viales se pueden ranurar para aceptar un tapón de rosca. Cuando se quita el sello hermético, se reemplaza con un tapón de rosca con o sin gotero. La presencia de antioxidantes en la formulación puede permitir que la formulación con tapón de rosca sea estable para el uso durante al menos dos semanas, por ejemplo, después de que se ha roto el sello hermético.
I. Síntesis de tetrahidrobiopterina
Se conocen en la técnica una variedad de métodos para la síntesis de tetrahidrobiopterinas, precursores, derivados y análogos. Los documentos de patente de EE.UU. n° 5.698.408; 2.601.215; 3505329; 4.540.783; 4.550.109; 4.587.340; 4.595.752; 4.649.197; 4.665.182; 4.701.455; 4.713.454; 4.937.342; 5.037.981; 5.198.547; 5.350.851; 5.401.844; 5.698.408, la solicitud canadiense CA 2420374, las solicitudes europeas n° EP 079 574, EP 191 335 y publicaciones de patentes japonesas de Suntory JP 4-082888, JP 59-021685 y JP 9-157270, y de Sugimoto and Matsuura, Bull. Chem. Soc. Japan, 48(12):3767-3768 (1975), Sugimoto and Matsuura, Bull. Chem. Soc. Japan, 52(1 ):181-183 (1979), Matsuura et al., Chem. Lett. (Japan), 735-738 (1984), Matsuura et al., Heterocycles, Vol. 23, No. 12, 3115-3120, 1985 y Whiteley et al., Anal Biochem. 137(2):394-6 (1984), cada uno describe métodos para preparar dihidrobiopterinas, BH4 y sus derivados que se pueden emplear como composiciones para la presente invención.
Los documentos de publicación internacional n° WO2005049614, patente de EE.UU. 4.540.783, patente japonesa n° 59-021685, Schircks et al., Helv. Chim. Acta, 60: 211 (1977), Sugimoto et al., Bull. Chem. Soc. Jp, 52(1 ):181 (1979), Sugimoto et al., Bull. Chem. Soc. Jp, 48(12):3767 (1975), Visontini et al., Helv. Chim. Acta, 52:1225 (1969), y Matsuura et al., Chem. Lett., p 735 (1984), describen métodos para sintetizar BH4.
II. Formas cristalinas de la sal clorhidrato de 6R-tetrahidrobiopterina
El diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina existe en diferentes formas cristalinas, incluidas formas polimórficas y solvatos, algunas de las cuales son más estables que otras.
Formas cristalinas polimorfas de la sal diclorhidrato de (6R)-L-tetrahidrobiopterina
Forma B polimorfa
El polimorfo cristalino que se ha encontrado que es el más estable, se denomina en esta memoria "forma B" o, alternativamente, "polimorfo B". Los resultados obtenidos durante una investigación y el desarrollo de diclorhidrato
de (6R)-L-eritro-tetrahidrobiopterina revelaron que se han preparado varios sólidos cristalinos conocidos, pero en ninguno se ha reconocido el polimorfismo y su efecto sobre la estabilidad de los cristales de BH4.
El polimorfo B es un anhidrato ligeramente higroscópico con la mayor estabilidad termodinámica, por encima de aproximadamente 20°C. Además, la forma B se puede procesar y manipular fácilmente debido a su estabilidad térmica, la posibilidad de preparación en condiciones específicas, su morfología y tamaño de partícula adecuados. El punto de fusión está cerca de 260°C (AHf > 140 J/g), pero no se puede detectar un punto de fusión claro debido a la descomposición antes y durante la fusión. Esas excelentes propiedades hacen que la forma B polimorfa sea especialmente factible para aplicaciones farmacéuticas, que se preparan a temperaturas elevadas. El polimorfo B se puede obtener como un polvo fino con un tamaño de partícula que puede oscilar entre 0,2 gm y 500 gm.
La forma B muestra un patrón de difracción en polvo de rayos X, expresado en valores d (Á) de: 8,7 (vs), 6,9 (w), 5,90 (vw), 5,63 (m), 5,07 (m), 4,76 (m), 4,40 (m), 4,15 (w), 4,00 (s), 3,95 (m), 3,52 (m), 3,44 (w), 3,32 (m), 3,23 (s), 3,17 (w), 3,11 (vs), 3,06 (w), 2,99 (w), 2,96 (w), 2,94 (m), 2,87 (w), 2,84 (s), 2,82 (m), 2,69 (w), 2,59 (w), 2,44 (w). La Figura 1 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma B del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
Tal y como se usa en esta memoria, las siguientes abreviaturas entre paréntesis significan: (vs) = intensidad muy fuerte; (s) = intensidad fuerte; (m) = intensidad media; (w) = intensidad débil; y (vw) = intensidad muy débil. En la Figura 1 se muestra un patrón de difracción de polvo de rayos X característico.
Se ha encontrado que otros polimorfos de BH4 tienen una estabilidad química y física satisfactoria para un manejo seguro durante la producción y la formulación, además de proporcionar una alta estabilidad durante el almacenamiento en su forma pura o en formulaciones. Además, se ha encontrado que la forma B y otros polimorfos de BH4 se pueden preparar en cantidades muy elevadas (p. ej., escala de 100 kilos) y se almacenan durante un período prolongado de tiempo.
Todas las formas cristalinas (polimorfos, hidratos y solvatos), incluida la forma B cristalina, se pueden utilizar para la preparación del polimorfo B más estable. El polimorfo B se puede obtener mediante un equilibrio de fases de suspensiones de formas amorfas u otras formas distintas del polimorfo B, tales como el polimorfo A, en disolventes polares y no acuosos adecuados. Por tanto, las preparaciones farmacéuticas descritas en esta memoria se refieren a una preparación de la forma B polimorfa de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
Otras formas de BH4 se pueden convertir en la forma B, dispersando la otra forma de BH4 en un disolvente a temperatura ambiente, agitando la suspensión a temperatura ambiente durante un tiempo suficiente para producir la forma B polimorfa, aislando después la forma B cristalina y eliminando el disolvente de la forma B aislada. Las temperaturas ambientales, tal y como se usan en esta memoria, significan temperaturas en un intervalo de 0°C a 60°C, preferiblemente de 15°C a 40°C. La temperatura aplicada se puede cambiar durante el tratamiento y mientras que se agita al disminuir la temperatura de forma escalonada o continua. Los disolventes adecuados para la conversión de otras formas a la forma B incluyen, pero no se limitan a, metanol, etanol, isopropanol, otros alcoholes C3 y C4, ácido acético, acetonitrilo, tetrahidrofurano, éter metil-t-butílico, 1,4-dioxano, acetato de etilo, acetato de isopropilo, otros acetatos C3-C6, metiletilcetona y otras metil-alquil C3-C5-cetonas. El tiempo para completar un equilibrio de fases puede ser de hasta 30 horas y preferiblemente de hasta 20 horas o menos de 20 horas.
El polimorfo B también se puede obtener mediante cristalización a partir de mezclas de disolventes que contienen hasta aproximadamente un 5% de agua, especialmente a partir de mezclas de etanol, ácido acético y agua. Se ha encontrado que la forma B poliforma del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar mediante disolución, opcionalmente a temperaturas elevadas, preferiblemente de una forma sólida de menor energía que la forma B o de la forma del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina, en una mezcla de disolventes que comprende etanol, ácido acético y agua, adición de semillas a la solución, enfriamiento de la suspensión obtenida y aislamiento de los cristales formados. La disolución se puede llevar a cabo a temperatura ambiente o hasta 70°C, preferiblemente hasta 50°C. Se puede utilizar la mezcla final de disolventes para la disolución o el material de partida se puede disolver primero en agua y los otros disolventes se pueden añadir juntos o uno después del otro. La composición de la mezcla de disolventes puede comprender una relación en volumen de agua : ácido acético : tetrahidrofurano de1 : 3 : 2 a 1 : 9 : 4 y preferiblemente 1 : 5 : 4. La solución se agita preferiblemente. El enfriamiento puede significar temperaturas que se reducen hasta -40°C a 0°C, preferiblemente que se reducen hasta 10°C a 30°C. Las semillas adecuadas son la forma B poliforma procedente de otro lote o de cristales que tienen una morfología similar o idéntica. Después del aislamiento, la forma B cristalina se puede lavar con un no disolvente, tal como acetona o tetrahidrofurano y secar de la manera habitual.
El polimorfo B también se puede obtener mediante cristalización en soluciones acuosas mediante la adición de no disolventes tales como metanol, etanol y ácido acético. El proceso de cristalización y aislamiento se puede realizar ventajosamente a temperatura ambiente sin enfriar la solución. Por tanto, este procedimiento es muy adecuado para llevarlo a cabo a escala industrial.
En una realización de las composiciones y usos descritos en esta memoria, se prepara una composición que incluye la forma B polimorfa de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina, mediante la disolución de una forma sólida
distinta de la forma B o de la forma B de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en agua a temperatura ambiente, añadiendo un no disolvente en una cantidad suficiente para formar una suspensión, agitando opcionalmente la suspensión durante un cierto tiempo, y posteriormente aislando los cristales formados. La composición se modifica adicionalmente a una composición farmacéutica tal y como se describe a continuación. La concentración de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en la solución acuosa puede ser del 10 al 80 por ciento en peso, más preferiblemente del 20 al 60 por ciento en peso, en relación con la solución. Los no disolventes preferidos (es decir, los disolventes útiles para preparar suspensiones de BH4) son metanol, etanol y ácido acético. El no disolvente se puede añadir a la solución acuosa. Más preferiblemente, la solución acuosa se añade al no disolvente. El tiempo de agitación después de la formación de la suspensión puede ser de hasta 30 horas y preferiblemente de hasta 20 horas o menos de 20 horas. Un aislamiento mediante filtración y secado se lleva a cabo de manera conocida tal y como se ha descrito anteriormente.
La forma B poliforma es una forma cristalina muy estable, que se puede separar por filtración, secar y moler fácilmente hasta obtener los tamaños de partícula deseados para las formulaciones farmacéuticas. Estas propiedades sobresalientes hacen que la forma B polimorfa sea especialmente viable para una aplicación farmacéutica.
Forma A poliforma
Se ha encontrado que otro polimorfo cristalino de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma A" o "polimorfo A". El polimorfo A es ligeramente higroscópico y adsorbe agua hasta un contenido de aproximadamente 3 por ciento en peso, que se libera continuamente entre 50°C y 200°C, cuando se calienta a una tasa de 10°C/minuto. El polimorfo A es un anhidrato higroscópico, que es una forma metaestable con respecto a la forma B; sin embargo, es estable durante varios meses en condiciones ambientales, si se mantiene en un envase herméticamente cerrado. La forma A es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma A polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado, que normalmente oscila entre 1 gm y aproximadamente 500 gm. El polimorfo A que muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 15,5 (vs.), 12,0 (m), 6,7 (m), 6,5 (m), 6,3 (w), 6,1 (w), 5,96 (w), 5,49 (m), 4,89 (m), 3.79 (m), 3,70 (s), 3,48 (m), 3,45 (m), 3,33 (s), 3,26 (s), 3,22 (m), 3,18 (m), 3,08 (m), 3,02 (w), 2,95 (w), 2,87 (m), 2.79 (w), 2,70 (w). La Figura 2 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma A de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
El polimorfo A muestra bandas del espectro Raman características, expresadas en números de onda (cm-1) en: 2934 (w), 2880 (w), 1692 (s), 1683 (m), 1577 (w), 1462 (m), 1360 (w), 1237 (w), 1108 (w), 1005 (vw), 881 (vw), 813 (vw), 717 (m), 687 (m), 673 (m), 659 (m), 550 (w), 530 (w), 492 (m), 371 (m), 258 (w), 207 (w), 101 (s), 87 (s) cm-1. La forma A polimorfa se puede obtener mediante liofilización o eliminación de agua de soluciones de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en agua. La forma A polimorfa del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar disolviendo diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina a temperatura ambiente en agua, (1) enfriando la solución a temperaturas bajas para solidificar la solución, y eliminar el agua a presión reducida, o (2) eliminando el agua de dicha solución acuosa.
La forma A cristalina se puede aislar mediante filtración y luego secar para evaporar el agua absorbida del producto. Las condiciones y los métodos de secado son conocidos y el secado del producto aislado o la eliminación de agua de acuerdo con la variante (2) descrita en esta memoria, se puede llevar a cabo aplicando temperaturas elevadas, por ejemplo de hasta 80°C, preferiblemente en el intervalo de 30°C a 80°C, al vacío o a temperaturas elevadas y vacío. Antes del aislamiento de un precipitado obtenido en la variante (2), la suspensión se puede agitar durante un cierto tiempo para equilibrar las fases. La concentración de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en la solución acuosa puede ser del 5 al 40 por ciento en peso, en relación con la solución.
Se prefiere un enfriamiento rápido para obtener soluciones sólidas como material de partida. Se aplica una presión reducida hasta que el disolvente se elimina por completo. La liofilización es una tecnología bien conocida en la técnica. El tiempo para completar la eliminación del disolvente depende del vacío aplicado, que puede oscilar entre 0,01 y 1 mbar, el disolvente utilizado y la temperatura de congelación.
La forma A polimorfa es estable a temperatura ambiente o por debajo de la temperatura ambiente en condiciones sustancialmente exentas de agua, lo que se demuestra con pruebas de equilibrio de fases de suspensiones en tetrahidrofurano o éter terc-butil metílico agitado durante cinco días y 18 horas respectivamente, bajo atmósfera de nitrógeno a temperatura ambiente. La filtración y el secado al aire a temperatura ambiente producen una forma A polimorfa sin cambios.
Forma F polimorfa
Se ha encontrado que otro polimorfo cristalino de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma
estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma F" o "polimorfo F". El polimorfo F es ligeramente higroscópico y adsorbe agua hasta un contenido de aproximadamente el 3 por ciento en peso, que se libera continuamente entre 50°C y 200°C, cuando se calienta con una tasa de 10°C/minuto. El polimorfo F es una forma metaestable y un anhidrato higroscópico, que es más estable que la forma A a temperaturas ambientales más bajas y menos estable que la forma B a temperaturas más altas y la forma F es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma F polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente oscila entre 1 pm y aproximadamente 500 pm.
El polimorfo F muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 17,1 (vs.), 12,1 (w), 8,6 (w), 7,0 (w), 6,5 (w), 6,4 (w), 5,92 (w), 5,72 (w), 5,11 (w), 4,92 (m), 4,86 (w), 4,68 (m), 4,41 (w), 4,12 (w), 3,88 (w), 3,83 (w), 3,70 (m), 3,64 (w), 3,55 (m), 3,49 (s), 3,46 (vs), 3,39 (s), 3,33 (m), 3,31 (m), 3,27 (m), 3,21 (m), 3,19 (m), 3,09 (m), 3,02 (m) y 2,96 (m). La Figura 3 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma F de diclorhidrato de (6R)-L-eritrotetrahidrobiopterina.
El polimorfo F se puede obtener mediante equilibrio de fases de suspensiones de la forma A poliforma en disolventes polares y no acuosos adecuados, que apenas disuelven dichas formas de energía inferior, especialmente alcoholes tales como metanol, etanol, propanol e isopropanol. La forma F poliforma de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina también se puede preparar dispersando partículas de la forma A sólida de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en un disolvente no acuoso que apenas disuelve dicho diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina por debajo de la temperatura ambiente, agitando la suspensión a dichas temperaturas durante un tiempo suficiente para producir la forma F polimorfa, aislando después la forma F cristalina y retirando el disolvente de la forma F aislada. Se puede realizar la eliminación del disolvente y el secado al aire, aire seco o un gas protector seco como nitrógeno o gases nobles y a temperatura ambiente o por debajo de ella, por ejemplo hasta 0°C. La temperatura durante el equilibrio de fases es preferiblemente de 5 a 15°C y lo más preferiblemente de aproximadamente 10°C.
Forma J polimorfa
Se ha encontrado que otro polimorfo cristalino de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma J" o "polimorfo J". El polimorfo J es ligeramente higroscópico y absorbe agua cuando se manipula con humedad del aire. El polimorfo J es una forma metaestable y un anhidrato higroscópico, y se puede transformar de nuevo en la forma E descrita a continuación, de la cual se obtiene tras una exposición a condiciones de humedad relativa alta, tal como superior al 75% de humedad relativa. La forma J es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma J polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente oscila entre 1 pm y aproximadamente 500 pm.
La forma J muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 14,6 (m), 6,6 (w), 6,4 (w), 5,47 (w), 4,84 (w), 3,29 (vs) y 3,21 (vs). La Figura 4 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma J de diclorhidrato de (6R)-L-eritrotetrahidrobiopterina.
El polimorfo J se puede obtener mediante deshidratación de la forma E a temperaturas moderadas al vacío. En particular, la forma J polimorfa del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar tomando la forma E y eliminando el agua de la forma E, tratando la forma E en un secador de vacío para obtener la forma J a temperaturas moderadas, que puede ser una temperatura en el intervalo de 25 a 70°C, y más preferiblemente de 30 a 50°C.
Forma K polimorfa
Se ha encontrado que otro polimorfo cristalino del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria como "forma K" o "polimorfo K". El polimorfo K es ligeramente higroscópico y adsorbe agua hasta un contenido de aproximadamente un 2,0 por ciento en peso, que se libera continuamente entre 50°C y 100°C, cuando se calienta con una tasa de 10°C/minuto. El polimorfo K es una forma metaestable y un anhidrato higroscópico, que es menos estable que la forma B a temperaturas más altas y la forma K es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables, en particular la forma B. La forma K polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado, que normalmente oscila entre 1 pm y aproximadamente 500 pm.
La forma K muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 14,0 (s), 9,4 (w), 6,6 (w), 6,4 (w), 6,3 (w), 6,1 (w), 6,0 (w), 5,66 (w), 5,33 (w), 5,13 (vw), 4,73 (m), 4,64 (m), 4,48 (w), 4,32 (vw), 4,22 (w), 4,08 (w), 3,88 (w), 3,79 (w), 3,54 (m), 3,49 (vs), 3,39 (m), 3,33 (vs), 3,13 (s), 3,10 (m), 3,05 (m), 3,01 (m), 2,99 (m) y 2,90 (m). La Figura 5 es un gráfico del patrón de difracción de rayos X
característico, mostrado por la forma K del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
El polimorfo K se puede obtener mediante cristalización a partir de mezclas de disolventes polares que contienen pequeñas cantidades de agua y en presencia de pequeñas cantidades de ácido ascórbico. Los disolventes para la mezcla de disolventes se pueden seleccionar a partir de ácido acético y un alcohol tal como metanol, etanol, n- o isopropanol. En particular, la forma K poliforma de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar disolviendo diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en una mezcla de ácido acético y un alcohol o tetrahidrofurano que contiene pequeñas cantidades de agua y una pequeña cantidad de ácido ascórbico a temperaturas elevadas, bajando la temperatura por debajo de la temperatura ambiental para cristalizar dicho diclorhidrato, aislando el precipitado y secando el precipitado aislado a temperatura elevada, opcionalmente al vacío. Los alcoholes adecuados son, por ejemplo, metanol, etanol, propanol e isopropanol, en donde se prefiere el etanol. La proporción de ácido acético a alcohol o a tetrahidrofurano puede ser de 2:1 a 1:2 y preferiblemente aproximadamente 1:1. La disolución de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede llevar a cabo en presencia de un mayor contenido en agua y se puede añadir más mezcla de antidisolvente para obtener una precipitación completa. La cantidad de agua en la composición final puede ser del 0,5 al 5 por ciento en peso y la cantidad de ácido ascórbico puede ser del 0,01 al 0,5 por ciento en peso, ambas referidas a la mezcla de disolventes. La temperatura para la disolución puede estar en el intervalo de 30 a 100 y preferiblemente de 35 a 70°C y la temperatura de secado puede estar en el intervalo de 30 a 50°C. El precipitado se puede lavar con un alcohol, tal como etanol, después del aislamiento, por ejemplo, mediante filtración. El polimorfo K se puede convertir fácilmente en la forma B más estable mediante equilibrio de fases, p. ej., en isopropanol y, opcionalmente, sembrar con cristales de la forma B a una temperatura superior a la ambiental, tal como temperaturas de 30 a 40°C.
Formas hidrato de la sal diclorhidrato de (6R)-L-tetrahidrobiopterina
Tal y como se describe adicionalmente a continuación, se ha encontrado que el diclorhidrato de (6R)-L-eritrotetrahidrobiopterina existe como varios hidratos cristalinos, que se describirán y definirán en esta memoria como formas C, D, E, H y O. Esas formas de hidratos son útiles como una forma estable de BH4 para las preparaciones farmacéuticas descritas en esta memoria y en la preparación de composiciones que incluyen polimorfos cristalinos estables de BH4.
Forma C hidrato
Se ha encontrado que una forma cristalina de hidrato de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma C" o " hidrato C". La forma C hidrato es ligeramente higroscópica y tiene un contenido en agua de aproximadamente 5,5 por ciento en peso, lo que indica que la forma C es un monohidrato. El hidrato C tiene un punto de fusión cercano a 94°C (AHf es aproximadamente 31 J/g) y la forma C hidrato es especialmente adecuada como material intermedio y de partida para producir formas polimórficas estables. La forma C polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado, que normalmente oscila entre 1 pm y aproximadamente 500 pm.
La forma C muestra un patrón de difracción de rayos X en polvo característico con picos característicos expresados en valores d (Á) de: 18,2 (m), 15,4 (w), 13,9 (vs), 10,4 (w), 9,6 (w), 9,1 (w), 8,8 (m), 8,2 (w), 8,0 (w), 6,8 (m), 6,5 (w), 6,05 (m), 5,77 (w), 5,64 (w), 5,44 (w), 5,19 (w), 4,89 (w), 4,76 (w), 4,70 (w), 4,41 (w), 4,25 (m), 4,00 (m), 3,88 (m), 3,80 (m), 3,59 (s), 3,50 (m), 3,44 (m), 3,37 (m), 3,26 (s), 3,19 (vs), 3,17 (s), 3,11 (m), 3,06 (m), 3,02 (m), 2,97 (vs), 2,93 (m), 2,89 (m), 2,83 (m) y 2,43 (m). La Figura 6 es un gráfico del patrón de difracción de rayos X característico mostrado por la forma C hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma C hidrato se puede obtener mediante equilibrio de fases a temperatura ambiente de una forma polimorfa tal como una suspensión del polimorfo B en un no disolvente, que contiene agua en una cantidad de preferiblemente aproximadamente un 5 por ciento en peso, en relación con el disolvente. La forma C hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar suspendiendo diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en un no disolvente tal como heptano, alcoholes C1-C4, tales como metanol, etanol, 1- o 2-propanol, acetatos, tales como acetato de etilo, acetonitrilo, ácido acético o éteres tales como terahidrofurano, dioxano, éter terc-butilmetílico, o mezclas binarias o ternarias de esos no disolventes, a las que se añade suficiente agua para formar un monohidrato, y agitando la suspensión a temperatura ambiente o por debajo de ella (p. ej., de 0 a 30°C) durante un tiempo suficiente para formar un monohidrato. Suficiente agua puede significar del 1 al 10 y preferiblemente del 3 al 8 por ciento en peso de agua, con referencia a la cantidad de disolvente. Los sólidos se pueden separar por filtración y secar al aire aproximadamente a temperatura ambiente. El sólido puede absorber algo de agua y, por lo tanto, posee un contenido en agua superior al valor teórico del 5,5 por ciento en peso. La forma C hidrato es inestable con respecto a las formas D y B, y se convierte fácilmente en la forma B polimorfa a temperaturas de aproximadamente 40°C al aire y una menor humedad relativa. La forma C se puede transformar en el hidrato D más estable mediante equilibrio en suspensión a temperatura ambiente.
Forma D hidrato
Se ha encontrado que otra forma cristalina de hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una
forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma D" o "hidrato D". La forma D hidrato es ligeramente higroscópica y puede tener un contenido en agua de aproximadamente 5,0 a 7,0 por ciento en peso, lo que sugiere que la forma D es un monohidrato. El hidrato D tiene un punto de fusión cercano a 153°C (AHf es de aproximadamente 111 J/g) y tiene una estabilidad mucho mayor que la forma C e incluso es estable cuando se expone a la humedad del aire a temperatura ambiente. Por lo tanto, la forma D hidrato se puede usar para preparar formulaciones o como material intermedio y de partida para producir formas polimorfas estables. La forma D polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente oscila entre 1 pm y aproximadamente 500 pm.
La forma D muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 8,6 (s), 6,8 (w), 5,56 (m), 4,99 (m), 4,67 (s), 4,32 (m), 3,93 (vs), 3,88 (w), 3,64 (w), 3,41 (w), 3,25 (w), 3,17 (m), 3,05 (s), 2,94 (w), 2,92 (w), 2,88 (m), 2,85 (w) 2,80 (w), 2,79 (m), 2,68 (w), 2,65 (w), 2,52 (vw), 2,35 (w), 2,34 (w), 2,30 (w) y 2,29 (w). La Figura 7 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma D hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma D hidrato se puede obtener añadiendo a aproximadamente temperatura ambiente, soluciones acuosas concentradas de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina a un exceso de un no disolvente tal como hexano, heptano, diclorometano, 1- o 2-propanol, acetona, acetato de etilo, acetonitrilo, ácido acético o éteres tales como tetrahidrofurano, dioxano, éter terc-butilmetílico, o mezclas de esos no disolventes, y agitando la suspensión a temperatura ambiente. El sólido cristalino se puede separar por filtración y luego secar bajo atmósfera de nitrógeno seco a temperatura ambiente. Un no disolvente preferido es isopropanol. La adición de la solución acuosa se puede realizar gota a gota para evitar una precipitación repentina. La forma D hidrato del diclorhidrato de (6R)-L-eritrotetrahidrobiopterina se puede preparar añadiendo a aproximadamente temperatura ambiente soluciones acuosas concentradas de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina hasta un exceso de un no disolvente y agitando la suspensión a temperatura ambiente. Un exceso de no disolvente puede significar una relación entre el disolvente acuoso y el no disolvente de 1:10 a 1:1000. La forma D contiene un pequeño exceso de agua, relacionado con el monohidrato, y se cree que es agua absorbida debido a la naturaleza ligeramente higroscópica de ese hidrato cristalino. Se considera que la forma D hidrato es la más estable entre los hidratos conocidos a temperatura ambiente y una humedad relativa inferior al 70%. La forma D hidrato se puede usar para formulaciones preparadas en condiciones en las que ese hidrato es estable. La temperatura ambiente puede significar entre 20 y 30°C.
Forma E hidrato
Se ha encontrado que otra forma cristalina de hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma E" o "hidrato E". La forma E hidrato tiene un contenido en agua de aproximadamente 10 a 14 por ciento en peso, lo que sugiere que la forma E es un dihidrato. El hidrato E se forma a temperaturas inferiores a la temperatura ambiente. La forma E hidrato es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. Es especialmente adecuada producir la forma J exenta de agua, después de secar bajo atmósfera de nitrógeno u opcionalmente al vacío. La forma E no es higroscópica y es estable a humedades relativas bastante altas, es decir, a humedades relativas por encima de aproximadamente el 60% y hasta aproximadamente el 85%. La forma E polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente oscila entre 1 pm y aproximadamente 500 pm.
La forma E muestra un patrón de difracción de rayos X en polvo característico con picos característicos expresados en valores d (Á) de: 15,4 (s), 6,6 (w), 6,5 (w), 5,95 (vw), 5,61 (vw), 5,48 (w), 5,24 (w), 4,87 (w), 4,50 (vw), 4,27 (w), 3,94 (w), 3,78 (w), 3,69 (m), 3,60 (w), 3,33 (s), 3,26 (vs), 3,16 (w), 3,08 (m), 2,98 (w), 2,95 (m), 2,91 (w), 2,87 (m), 2,79 (w), 2,74 (w), 2,69 (w) y 2,62 (w). La Figura 8 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma E hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma E hidrato se puede obtener añadiendo soluciones acuosas concentradas de diclorhidrato de (6R)-L-eritrotetrahidrobiopterina a un exceso de un no disolvente enfriado a temperaturas de aproximadamente 10 a -10°C y preferiblemente de 0 a 10°C y agitando la suspensión a dichas temperaturas. El sólido cristalino se puede separar por filtración y luego secar bajo atmósfera de nitrógeno seco a temperatura ambiente. Los no disolventes son, por ejemplo, hexano, heptano, diclorometano, 1- o 2-propanol, acetona, acetato de etilo, acetonitrilo, ácido acético o éteres tales como tetrahidrofurano, dioxano, éter terc-butilmetílico, o mezclas de tales no disolventes. Un no disolvente preferido es isopropanol. La adición de la solución acuosa se puede realizar gota a gota para evitar una precipitación repentina. La forma E hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar añadiendo soluciones acuosas concentradas de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina a un exceso de un no disolvente, que se enfría a temperaturas de aproximadamente 10 a -10°C, y agitando la suspensión a temperatura ambiente. El exceso de no disolvente puede significar una relación entre el disolvente acuoso y el no disolvente de 1:10 a 1:1000. Un no disolvente preferido es tetrahidrofurano. Otro procedimiento de preparación comprende exponer la forma B polimorfa a una atmósfera de aire con una humedad relativa del 70 al 90%, preferiblemente alrededor del 80%. Se considera que la forma E hidrato es un dihidrato, por lo que puede absorber algo de agua adicional. La forma E polimorfa se puede transformar en polimorfo J al secarse al vacío a temperaturas
moderadas, lo que puede significar entre 20°C y 50°C a presiones entre 0 y 100 mbar. La forma E es especialmente adecuada para formulaciones en formas semisólidas debido a su estabilidad con humedades relativas elevadas. Forma H hidrato
Se ha encontrado que otra forma cristalina de hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma H" o " hidrato H". La forma H hidrato tiene un contenido en agua de aproximadamente 5,0 a 7,0 por ciento en peso, lo que sugiere que la forma H es un monohidrato higroscópico. La forma H hidrato se forma a temperaturas inferiores a la temperatura ambiente. La forma H hidrato es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma H polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado, que normalmente oscila entre 1 gm y aproximadamente 500 gm.
La forma H muestra un patrón de difracción en polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 8,615,8 (vs), 10,3 (w), 8,0 (w), 6,6 (w), 6,07 (w), 4,81 (w), 4,30 (w), 3,87 (m), 3,60 (m), 3,27 (m), 3,21 (m), 3,13 (w), 3,05 (w), 2,96 (m), 2,89 (m), 2,82 (w) y 2,67 (m). La Figura 9 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma H hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina. La forma H hidrato se puede obtener disolviendo a temperatura ambiente el diclorhidrato de (6R)-L-eritrotetrahidrobiopterina en una mezcla de ácido acético y agua, añadiendo luego un no disolvente para precipitar un sólido cristalino, enfriando la suspensión obtenida y agitando la suspensión enfriada durante un tiempo determinado. El sólido cristalino se separa por filtración y luego se seca al vacío a temperatura ambiente. Los no disolventes son, por ejemplo, hexano, heptano, diclorometano, 1 - o 2-propanol, acetona, acetato de etilo, acetonitrilo, ácido acético o éteres como tetrahidrofurano, dioxano, éter terc-butilmetílico, o mezclas de tales no disolventes. Un no disolvente preferido es tetrahidrofurano. La forma H hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar disolviendo a temperatura ambiente el diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en una mezcla de ácido acético y una cantidad menor que la de ácido acético de agua, añadiendo un no disolvente y enfriando la suspensión obtenida a temperaturas en el intervalo de -10 a 10°C, y preferiblemente de -5 a 5°C, y agitando la suspensión a dicha temperatura durante un cierto tiempo. Cierto tiempo puede significar de 1 a 20 horas. La relación en peso entre ácido acético y agua puede ser de 2:1 a 25:1 y preferiblemente de 5:1 a 15:1. La relación en peso entre ácido acético/agua y no disolvente puede ser de 1:2 a 1:5. La forma H hidrato parece ser un monohidrato con un ligero exceso de agua absorbida debido a la naturaleza higroscópica.
Forma O hidrato
Se ha encontrado que otra forma cristalina de hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma O" o "hidrato O". La forma O hidrato se forma a temperaturas cercanas a la temperatura ambiente. La forma O hidrato es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma O polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado, que normalmente oscila entre 1 gm y aproximadamente 500 gm.
La forma O muestra un patrón de difracción en polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 15,9 (w), 14,0 (w), 12,0 (w), 8,8 (m), 7,0 (w), 6,5 (w), 6,3 (m), 6,00 (w), 5,75 (w), 5,65 (m), 5,06 (m), 4,98 (m), 4,92 (m), 4,84 (w), 4,77 (w), 4,42 (w), 4,33 (w), 4,00 (m), 3,88 (m), 3,78 (w), 3,69 (s), 3,64 (s), 3,52 (vs), 3,49 (s), 3,46 (s), 3,42 (s), 3,32 (m), 3,27 (m), 3,23 (s), 3,18 (s), 3,15 (vs), 3,12 (m), 3,04 (vs), 2,95 (m), 2,81 (s), 2,72 (m), 2,67 (m) y 2,61 (m). La Figura 10 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma O hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma O hidrato se puede preparar mediante una exposición de la forma F polimorfa a una atmósfera de nitrógeno que contiene vapor de agua, con una humedad relativa resultante de aproximadamente 52% durante aproximadamente 24 horas. El hecho de que la forma F, que es un anhidrato ligeramente higroscópico, se pueda utilizar para preparar la forma O con un 52% de humedad relativa, sugiere que la forma O es un hidrato, que es más estable que la forma F en condiciones de temperatura y humedad ambiental.
Formas de solvato de la sal diclorhidrato de (6R)-L-tetrahidrobiopterina
Tal y como se describe adicionalmente a continuación, se ha encontrado que el diclorhidrato de (6R)-L-eritrotetrahidrobiopterina existe como una serie de formas de solvatos cristalinos, que se describirán y definirán en esta memoria como formas G, I, L, M y N. Esas formas de solvato son útiles como forma estable de BH4 para las preparaciones farmacéuticas descritas en esta memoria y en la preparación de composiciones que incluyen polimorfos cristalinos estables de BH4.
Forma G solvato
Se ha encontrado que una forma cristalina de solvato de etanol del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se
denominará en esta memoria "forma G" o "hidrato G." La forma G solvato de etanol tiene un contenido en etanol de aproximadamente 8,0 a 12,5 por ciento en peso, lo que sugiere que la forma G es un solvato de monoetanol higroscópico. La forma G solvato se forma a temperaturas por debajo de la temperatura ambiente. La forma G es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma G polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que varía típicamente de 1 pm a aproximadamente 500 pm.
La forma G muestra un patrón de difracción de rayos X en polvo característico con picos característicos expresados en valores d (Á) de: 14,5 (vs), 10,9 (w), 9,8 (w), 7,0 (w), 6,3 (w), 5,74 (w), 5,24 (vw), 5,04 (vw), 4,79 (w), 4,41 (w), 4,02 (w), 3,86 (w), 3,77 (w), 3,69 (w), 3,63 (m), 3,57 (m), 3,49 (m), 3,41 (m), 3,26 (m), 3,17 (m), 3,07 (m), 2,97 (m), 2,95 (m), 2,87 (w) y 2,61 (w). La Figura 11 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma G solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma G solvato de etanol se puede obtener mediante una cristalización del diclorhidrato de L-eritrotetrahidrobiopterina disuelto en agua y añadiendo un gran exceso de etanol, agitando la suspensión obtenida a temperatura ambiente o por debajo de ella y secando el sólido aislado al aire o bajo atmósfera de nitrógeno a aproximadamente la temperatura ambiente. En este caso, un gran exceso de etanol significa una mezcla resultante de etanol y agua con menos del 10% de agua, preferiblemente aproximadamente del 3 al 6%. La forma G etanolato de diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede preparar disolviendo aproximadamente a temperatura ambiente hasta temperaturas de 75°C, diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina en agua o en una mezcla de agua y etanol, enfriando una solución calentada a temperatura ambiente y además hasta 5 a 10°C, añadiendo opcionalmente etanol para completar la precipitación, agitando la suspensión obtenida a temperaturas de 20 a 5°C, separando por filtración el sólido blanco cristalino y secando el sólido al aire o bajo un gas protector como el nitrógeno a temperaturas cercanas a la temperatura ambiente. El procedimiento se puede llevar a cabo en una primera variante disolviendo el diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina aproximadamente a temperatura ambiente en una cantidad menor de agua y luego añadiendo un exceso de etanol y agitando después la suspensión obtenida durante un tiempo suficiente para obtener un equilibrio de fases. En una segunda variante, el diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina se puede suspender en etanol, opcionalmente añadiendo una cantidad menor de agua y calentando la suspensión y disolviendo el diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina, enfriando la solución. a temperaturas de aproximadamente 5 a 15°C, añadiendo etanol adicional a la suspensión y luego agitando la suspensión obtenida durante un tiempo suficiente para obtener un equilibrio de fases.
Forma I solvato
Se ha encontrado que una forma cristalina de solvato de ácido acético del diclorhidrato de (6R)-L-eritrotetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma I" o "hidrato I". La forma I solvato de ácido acético tiene un contenido en ácido acético de aproximadamente 12,7 por ciento en peso, lo que sugiere que la forma I es un monosolvato de ácido acético higroscópico. La forma I solvato se forma a temperaturas por debajo de la temperatura ambiente. La forma I solvato de ácido acético es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma I polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente está comprendido entre 1 pm y aproximadamente 500 pm.
La Forma I muestra un patrón de difracción en polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 14,5 (m), 14,0 (w), 11,0 (w), 7,0 (vw), 6,9 (vw), 6,2 (vw), 5,30 (w), 4,79 (w), 4,44 (w), 4,29 (w), 4,20 (vw), 4,02 (w), 3,84 (w), 3,80 (w), 3,67 (vs), 3,61 (m), 3,56 (w), 3,44 (m) 3,27 (w), 3,19 (w), 3,11 (s), 3,00 (m), 2,94 (w), 2,87 (w) y 2,80 (w). La Figura 12 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma I solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma I del solvato de ácido acético se puede obtener mediante una disolución del diclorhidrato de L-eritrotetrahidrobiopterina en una mezcla de ácido acético y agua a temperatura elevada, añadiendo más ácido acético a la solución, enfriando hasta una temperatura de aproximadamente 10°C y luego calentando la suspensión formada hasta aproximadamente 15°C, y luego agitando la suspensión obtenida durante un tiempo suficiente para obtener un equilibrio de fases, que puede durar hasta 3 días. A continuación, el sólido cristalino se separa por filtración y se seca al aire o bajo un gas protector como nitrógeno, a temperaturas cercanas a la temperatura ambiente.
Forma L solvato
Se ha encontrado que una forma cristalina mixta de solvato/hidrato de etanol del diclorhidrato de (6R)-L-eritrotetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma L" o "hidrato L". La forma L puede contener 4% pero hasta 13% de etanol y 0% a aproximadamente 6% de agua. La forma L se puede transformar en la forma G cuando se trata en etanol a temperaturas desde aproximadamente 0°C a 20°C. Además, la forma L se puede transformar en la forma B cuando se trata en un disolvente orgánico a temperatura ambiente (10°C a 60°C). La forma L polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente está comprendido entre 1 pm y aproximadamente 500 pm.
La forma L muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 14,1 (vs), 10,4 (w), 9,5 (w), 9,0 (vw), 6,9 (w), 6,5 (w), 6,1 (w), 5,75 (w), 5,61 (w), 5,08 (w), 4,71 (w), 3,86 (w), 3,78 (w), 3,46 (m), 3,36 (m), 3,06 (w), 2,90 (w) y 2,82 (w), La Figura 13 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma L solvato del diclorhidrato de (6R)-L-eritrotetrahidrobiopterina.
La forma L se puede obtener suspendiendo la forma E hidrato a temperatura ambiente en etanol y agitando la suspensión a temperaturas de 0 a 10°C, preferiblemente aproximadamente 5°C, durante un tiempo suficiente para que se produzca un equilibrio de fases, que puede ser de 10 a 20 horas. Después, el sólido cristalino se separa por filtración y se seca preferiblemente a presión reducida a 30°C o bajo atmósfera de nitrógeno. Un análisis mediante TG-FTIR sugiere que la forma L puede contener cantidades variables de etanol y agua, es decir, puede existir como un polimorfo (anhidrato), como una mezcla de solvato/hidrato de etanol, o incluso como un hidrato.
Forma M solvato
Se ha encontrado que una forma cristalina de solvato de etanol del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma M" o "hidrato M". La forma M puede contener 4% pero hasta 13% de etanol y 0% a aproximadamente 6% de agua, lo que sugiere que la forma M es un solvato de etanol ligeramente higroscópico. La forma M solvato se forma a temperatura ambiente. La forma M es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables, ya que la forma M se puede transformar en la forma G cuando se trata en etanol a temperaturas entre aproximadamente -10° a 15°C, y en la forma B cuando se trata con disolventes orgánicos, tales como etanol, alcoholes C3 y C4, o éteres cíclicos tales como THF y dioxano. La forma M poliforma se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente oscila entre 1 gm y aproximadamente 500 gm.
La forma M muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 18,9 (s), 6,4 (m), 6,06 (w), 5,66 (w), 5,28 (w), 4,50 (w), 4,23 (w) y 3,22 (vs). La Figura 14 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma M solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma M del solvato de etanol se puede obtener mediante una disolución de diclorhidrato de L-eritrotetrahidrobiopterina en etanol y una evaporación de la solución bajo atmósfera de nitrógeno a temperatura ambiente, es decir, entre 10°C y 40°C. La forma M también se puede obtener secando la forma G bajo un ligero flujo de nitrógeno seco con una tasa de aproximadamente 20 a 100 mL/min. Dependiendo del grado de secado bajo atmósfera de nitrógeno, la cantidad restante de etanol puede ser variable, es decir, de aproximadamente 3% a 13%. Forma N solvato
Se ha encontrado que otra forma cristalina de solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina es una forma estable preferida de BH4 para uso en una preparación farmacéutica descrita en esta memoria, que se denominará en esta memoria "forma N" o "hidrato N". La forma N puede contener en total hasta un 10% de isopropanol y agua, lo que sugiere que la forma N es un solvato de isopropanol ligeramente higroscópico. La forma N se puede obtener lavando la forma D con isopropanol y secando posteriormente al vacío a aproximadamente 30°C. La forma N es especialmente adecuada como material intermedio y de partida para producir formas polimorfas estables. La forma N polimorfa se puede preparar como un polvo sólido con un intervalo de tamaño de partícula medio deseado que normalmente oscila entre 1 gm y aproximadamente 500 gm.
La forma N muestra un patrón de difracción de polvo de rayos X característico con picos característicos expresados en valores d (Á) de: 19,5 (m), 9,9 (w), 6,7 (w), 5,15 (w), 4,83 (w), 3,91 (w), 3,56 (m), 3,33 (vs), 3,15 (w), 2,89 (w), 2,81 (w), 2,56 (w) y 2,36 (w). La Figura 15 es un gráfico del patrón de difracción de rayos X característico, mostrado por la forma N solvato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina.
La forma N de isopropanol se puede obtener mediante una disolución del diclorhidrato de L-eritrotetrahidrobiopterina en 4,0 mL de una mezcla de isopropanol y agua (mezclando una relación en volumen de, por ejemplo, 4:1). A esta solución se añade lentamente isopropanol (IPA, por ejemplo, aproximadamente 4,0 mL) y la suspensión resultante se enfría a 0°C y se agita durante varias horas (por ejemplo, aproximadamente de 10 a 18 horas) a esa temperatura. Se filtra la suspensión y se lava el residuo sólido con isopropanol a temperatura ambiente. El material cristalino obtenido se seca luego a temperatura ambiente (por ejemplo, aproximadamente 20 a 30°C) y presión reducida (aproximadamente 2 a 10 mbar) durante varias horas (por ejemplo, aproximadamente 5 a 20 horas). TG-FTIR muestra una pérdida de peso del 9,0% entre 25 y 200°C, que se atribuye tanto al isopropanol como al agua. Este resultado sugiere que la forma N puede existir en forma de solvato de isopropanol, o en forma de solvato/hidrato de isopropanol mixto, o como una forma no solvatada que contiene una pequeña cantidad de agua. Para la preparación de las formas polimorfas, se pueden usar técnicas de cristalización bien conocidas en la técnica, tales como agitación de una suspensión (equilibrio de fases en), precipitación, recristalización, evaporación, métodos de sorción de agua como disolventes o descomposición de solvatos. Se pueden usar soluciones diluidas, saturadas o sobresaturadas para la cristalización, con o sin siembra con agentes de nucleación adecuados. Se pueden aplicar
temperaturas de hasta 100°C para formar soluciones. Se puede aplicar un enfriamiento para iniciar la cristalización y precipitación hasta -100°C y preferiblemente hasta -30°C. Se pueden usar polimorfos metaestables o formas seudopolimórficas para preparar soluciones o suspensiones para la preparación de formas más estables y para lograr concentraciones más altas en las soluciones.
Se ha encontrado sorprendentemente que la forma D hidrato es la forma más estable entre los hidratos y las formas B y D son especialmente adecuadas para uso en formulaciones farmacéuticas. Las formas B y D presentan algunas ventajas como una fabricación focalizada, buen manejo debido al tamaño y la morfología convenientes de los cristales, muy buena estabilidad en condiciones de producción de varios tipos de formulación, estabilidad durante el almacenamiento, mayor solubilidad y biodisponibilidad elevada. Por consiguiente, una realización de las composiciones y métodos descritos en esta memoria es una composición farmacéutica que incluye la forma B polimorfa y/o la forma D hidrato del diclorhidrato de (6R)-L-eritro-tetrahidrobiopterina y un vehículo o diluyente farmacéuticamente aceptable.
III. Formulaciones farmacéuticas
Las formulaciones descritas en esta memoria se administran preferiblemente como formulaciones orales. Las formulaciones orales son preferiblemente formulaciones sólidas tales como cápsulas, comprimidos, píldoras y grageas, o formulaciones líquidas tales como suspensiones acuosas, elixires y jarabes. Las diversas formas de BH4 descritas en esta memoria se pueden usar directamente como polvo (partículas micronizadas), gránulos, suspensiones o soluciones, o se pueden combinar junto con otros ingredientes farmacéuticamente aceptables para mezclar por adición los componentes y opcionalmente dividirlos finamente y después rellenar cápsulas, compuestas, por ejemplo, a partir de gelatina dura o blanda, comprimidos de compresión, píldoras o grageas, o suspenderlos o disolverlos en vehículos para suspensiones, elixires y jarabes. Se pueden aplicar recubrimientos después de la compresión para formar píldoras.
Los ingredientes farmacéuticamente aceptables son bien conocidos para los diversos tipos de formulación y pueden ser, por ejemplo, agentes aglutinantes tales como polímeros naturales o sintéticos, excipientes, lubricantes, tensioactivos, edulcorantes y aromatizantes, materiales de recubrimiento, conservantes, colorantes, espesantes, adyuvantes, agentes antimicrobianos, antioxidantes y vehículos para los diversos tipos de formulación. La expresión "farmacéutica o farmacológicamente aceptable" se refiere a entidades moleculares y composiciones que están aprobadas por la Administración de Alimentos y Fármacos de los Estados Unidos o una agencia reguladora extranjera correspondiente para su administración a humanos. Tal y como se usa en esta memoria, "vehículo farmacéuticamente aceptable" incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardadores de la absorción y similares. El uso de tales medios y agentes para sustancias farmacéuticamente activas, es bien conocido en la técnica. Excepto en la medida en que cualquier medio o agente convencional sea incompatible con las composiciones terapéuticas, se contempla su uso en composiciones terapéuticas. También se pueden incorporar a las composiciones ingredientes activos complementarios.
La cantidad inicial de (6R)-L-eritro-tetrahidrobiopterina usada para preparar la formulación puede estar, por ejemplo, en el intervalo de aproximadamente 30% en peso a aproximadamente 40% en peso de la formulación, o en el intervalo de aproximadamente 32% en peso hasta aproximadamente 35% en peso, o aproximadamente 33% en peso. Cantidades específicas de BH4 en una formulación contemplada en esta memoria incluyen 80 mg, 100 mg, 200 mg, 300 mg, 400 mg y 500 mg.
Los agentes aglutinantes ayudan a conservar una formulación sólida. En algunos casos, se utilizan aglutinantes anhidros para preservar el estado anhidro de las formas polimorfas. En algunos casos, el agente aglutinante puede actuar como un agente de secado. Los aglutinantes ejemplares incluyen fosfato cálcico dibásico anhidro y su monohidrato. Otros ejemplos no limitantes de agentes aglutinantes útiles en una composición descrita en esta memoria, incluyen goma de tragacanto, goma arábiga, almidón, gelatina y polímeros biológicos degradables tales como homopoliésteres o copoliésteres de ácidos dicarboxílicos, alquilenglicoles, polialquilenglicoles y/o ácidos hidroxilcarboxílicos alifáticos; homopoliamidas o copoliamidas de ácidos dicarboxílicos, alquilendiaminas y/o ácidos aminocarboxílicos alifáticos; copolímeros de poliéster-poliamida correspondientes, polianhídridos, poliortoésteres, polifosfaceno y policarbonatos. Los polímeros biológicos degradables pueden ser lineales, ramificados o reticulados. Ejemplos específicos son poli(ácido glicólico), poli(ácido láctico) y poli-d,1 -láctido/glicólido. Otros ejemplos de polímeros son polímeros solubles en agua tales como polioxaalquilenos (polioxaetileno, polioxapropileno y sus polímeros mixtos, poliacrilamidas y poliacrilamidas hidroxilalquiladas, poli(ácido maleico) y ésteres o amidas de los mismos, poli(ácido acrílico) y ésteres o amidas de los mismos, poli(alcohol vinílico) y ésteres o éteres de los mismos, polivinilimidazol, polivinilpirrolidona y polímeros naturales como el quitosano.
Los agentes de desintegración ayudan a la desintegración rápida de formulaciones sólidas al absorber agua y expandirse. Ejemplos de agentes de desintegración incluyen polivinilpirrolidona (PVP, por ejemplo, comercializada bajo el nombre POVIDONE), una forma reticulada de povidona (CPVP, por ejemplo, comercializada bajo el nombre CROSPOVIDONE), una forma reticulada de carboximetilcelulosa de sodio (NaCMC, por ejemplo, comercializada bajo el nombre AC-DI-SOL), otras celulosas modificadas y almidón modificado. Los comprimidos formulados con CPVP mostraban una desintegración mucho más rápida que los comprimidos formulados con PVP.
Se pueden incluir antioxidantes y ayudar a estabilizar el producto de tetrahidrobiopterina, especialmente después de la disolución. Las soluciones acuosas de API de pH bajo son más estables que las soluciones de pH neutro o alto. Los antioxidantes se incluyen en una formulación descrita en esta memoria para evitar un deterioro por oxidación. Los antioxidantes generalmente se pueden clasificar en 3 grupos.
El primer grupo se conoce como antioxidantes verdaderos e inhiben la oxidación al reaccionar con radicales libres que bloquean la reacción en cadena. Ejemplos incluyen antioxidantes fenólicos, que incluyen hidroxianisol butilado (BHA), hidroxitolueno butilado (BHT), terc-butilhidroquinona (TBHQ), 4-hidroximetil-2,6-di-terc-butilfenol (HMBP) y 2,4,5-trihidroxibutirofenona (THBP); galatos de alquilo, que incluyen galato de propilo; ácido gálico; ácido nordihidroguaiarético; y tocoferoles, que incluyen el alfa-tocoferol.
El segundo grupo, que consiste en agentes reductores, tiene potenciales redox más bajos que el fármaco que se pretende proteger y, por lo tanto, se oxida más fácilmente. Los agentes reductores también pueden actuar reaccionando con radicales libres. Los ejemplos incluyen ácido ascórbico, ácido tioglicólico (TGA), palmitato de ascorbilo, sulfitos, incluyendo sales de potasio y sodio de ácido sulfuroso (por ejemplo, sulfito de potasio, sulfito de sodio, metabisulfito de sodio y bisulfito de sodio) y tioglicerol.
El tercer grupo consiste en sinergistas antioxidantes que normalmente tienen un efecto antioxidante moderado pero probablemente mejoran la acción de los antioxidantes en el primer o el segundo grupo, al reaccionar con iones de metales pesados que catalizan la oxidación. Ejemplos de agentes quelantes y sinergistas antioxidantes de ese tipo incluyen ácido cítrico, ácido málico, ácido edítico y sus sales, lecitina y ácido tartárico.
Los antioxidantes ácidos ejemplares incluyen ácido ascórbico, ésteres de ácidos grasos de ácido ascórbico tales como palmitato de ascorbilo y estearato de ascorbilo, y sales de ácido ascórbico tales como ascorbato de sodio, calcio o potasio. También se pueden usar antioxidantes no ácidos en las formulaciones de comprimidos estables. Ejemplos no limitantes de antioxidantes no ácidos incluyen betacaroteno y alfa tocoferol. Se pueden añadir aditivos ácidos para mejorar la estabilidad de la formulación del comprimido, incluyendo ácido cítrico o ácido málico. Los antioxidantes de molécula pequeña incluyen, pero no se limitan a, tioles, por ejemplo, cisteína, N-acetilcisteína, glutatión, etc., o polímeros tiolados (polímero-SH), por ejemplo, policarbofilo-cisteína, polimetacrílico-SH, carboximetilcelulosa-cisteína, etc., o antioxidantes de molécula pequeña como ácido ascórbico, metionina, palmitato de ascorbilo, etc. Esos antioxidantes confieren estabilidad a la forma de dosificación durante el tránsito a través del TGI, particularmente a medida que el pH del TGI aumenta al alejarse del estómago.
En una realización, se prefiere una combinación de al menos dos agentes reductores antioxidantes. En otra realización, se prefiere una combinación de al menos dos agentes antioxidantes reductores junto con un sinergista antioxidante ácido y/o un agente quelante.
Los lubricantes mejoran la estabilidad, la dureza y la uniformidad de las formulaciones sólidas. Los lubricantes ejemplares incluyen fumarato de estearilo y estearato de magnesio. Otros ejemplos no limitantes de lubricantes incluyen aceites, grasas, ceras o sales de ácidos grasos naturales o sintéticos, tales como estearato de magnesio. Opcionalmente, las formulaciones estables de la invención también pueden comprender otros excipientes tales como manitol, hidroxilpropilcelulosa, celulosa microcristalina u otros azúcares no reductores, tales como sacarosa, trehalosa, melecitosa, planteosa y rafinosa. Los azúcares reductores pueden reaccionar con BH4. Otros ejemplos no limitantes de excipientes útiles en una composición descrita en esta memoria, incluyen fosfatos tales como fosfato dicálcico.
Los tensioactivos para uso en una composición descrita en esta memoria pueden ser aniónicos, aniónicos, anfóteros o neutros. Ejemplos no limitantes de tensioactivos útiles en una composición descrita en esta memoria incluyen lecitina, fosfolípidos, sulfato de octilo, sulfato de decilo, sulfato de dodecilo, sulfato de tetradecilo, sulfato de hexadecilo y sulfato de octadecilo, oleato de Na o caprato de Na, ácidos 1-acilaminoetano-2-sulfónicos, tal como ácido 1 -octanoilaminoetano-2-sulfónico, ácido 1 -decanoilaminoetano-2-sulfónico, ácido 1 -dodecanoilaminoetano-2-sulfónico, ácido 1 -tetradecanoilaminoetano-2-sulfónico, ácido 1 -hexadecanoilaminoetano-2-sulfónico y ácido 1 -octadecanoilaminoetano-2-sulfónico, y ácido taurocólico y ácido taurodesoxicólico, ácidos biliares y sus sales tales como ácido cólico, ácido desoxicólico y glicocolatos de sodio, caprato de sodio o laurato de sodio, oleato de sodio, lauril sulfato de sodio, cetil sulfato de sodio, aceite de ricino sulfatado y dioctilsulfosuccinato de sodio, cocamidopropilbetaína y laurilbetaína, alcoholes grasos, colesteroles, mono o diestearato de glicerol mono o dioleato de glicerol y mono o dipalmitato de glicerol y estearato de polioxietileno.
Ejemplos no limitantes de agentes edulcorantes útiles en una composición descrita en esta memoria, incluyen sacarosa, fructosa, lactosa o aspartamo. Ejemplos no limitantes de agentes aromatizantes para uso en una composición descrita en esta memoria, incluyen menta, aceite de gaulteria o aromas de frutas tales como aroma de cereza o naranja. Ejemplos no limitantes de materiales de recubrimiento para uso en una composición descrita en esta memoria, incluyen gelatina, cera, goma laca, azúcar u otros polímeros biológicos degradables. Ejemplos no limitantes de conservantes para uso en una composición descrita en esta memoria, incluyen metil o propil parabenos, ácido sórbico, clorobutanol, fenol y timerosal.
La forma de BH4 también se puede formular como un comprimido efervescente o polvo, que se desintegra en un
ambiente acuoso para proporcionar una solución para beber. También se pueden preparar formulaciones de liberación lenta para lograr una liberación controlada del agente activo en contacto con los fluidos corporales en el tracto gastrointestinal, y para proporcionar un nivel sustancial constante y eficaz del agente activo en el plasma sanguíneo. La forma cristalina se puede incluir para este fin en una matriz polimérica de un polímero biodegradable, un polímero soluble en agua o una mezcla de ambos, y tensioactivos opcionalmente adecuados. La inclusión puede significar en este contexto la incorporación de micropartículas en una matriz de polímeros. Las formulaciones de liberación controlada también se obtienen mediante una encapsulación de micropartículas dispersas o microgotitas emulsionadas mediante tecnologías conocidas de recubrimiento mediante dispersión o emulsión.
La BH4 usada en una composición descrita en esta memoria se formula preferiblemente como una sal diclorhidrato, sin embargo, se contempla que otras formas de sal de BH4 posean la actividad biológica deseada y, en consecuencia, se pueden usar otras formas de sal de BH4. Específicamente, se prefieren las sales de BH4 con ácidos orgánicos o inorgánicos. Ejemplos no limitantes de formas alternativas de sales de BH4, incluyen sales de BH4 de ácido acético, ácido cítrico, ácido oxálico, ácido tartárico, ácido fumárico y ácido mandélico.
Las sales de adición de base farmacéuticamente aceptables se pueden formar con metales o aminas, tales como metales alcalinos y alcalinotérreos o aminas orgánicas. También se pueden preparar sales de compuestos farmacéuticamente aceptables con un catión farmacéuticamente aceptable. Los cationes adecuados farmacéuticamente aceptables son bien conocidos por los expertos en la técnica e incluyen cationes alcalinos, alcalinotérreos, de amonio y de amonio cuaternario. También son posibles carbonatos o hidrogenocarbonatos. Ejemplos de metales usados como cationes son sodio, potasio, magnesio, amonio, calcio o hierro y similares. Ejemplos de aminas adecuadas incluyen isopropilamina, trimetilamina, histidina, N,N'-dibenciletilendiamina, cloroprocaína, colina, dietanolamina, diciclohexilamina, etilendiamina, N-metilglucamina y procaína.
Las sales de adición de ácido farmacéuticamente aceptables incluyen sales de ácidos orgánicos o inorgánicos. Ejemplos de sales ácidas adecuadas incluyen los clorhidratos, acetatos, citratos, salicilatos, nitratos, fosfatos. Otras sales adecuadas farmacéuticamente aceptables son bien conocidas por los expertos en la técnica e incluyen, por ejemplo, ácidos acético, cítrico, oxálico, tartárico o mandélico, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico o ácido fosfórico; con ácidos orgánicos carboxílicos, sulfónicos, sulforados o fosforados o sulfámicos sustituidos en N, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido succínico, ácido maleico, ácido hidroximaleico, ácido metilmaleico, ácido fumárico, ácido málico, ácido tartárico, ácido láctico, ácido oxálico, ácido glucónico, ácido glucárico, ácido glucurónico, ácido cítrico, ácido benzoico, ácido cinámico, ácido mandélico, ácido salicílico, ácido 4-aminosalicílico, ácido 2-fenoxibenzoico, ácido 2-acetoxibenzoico, ácido embónico, ácido nicotínico o ácido isonicotínico; y con aminoácidos, como los 20 aminoácidos alfa implicados en la síntesis de proteínas en la naturaleza, por ejemplo, ácido glutámico o ácido aspártico, y también con ácido fenilacético, ácido metanosulfónico, ácido etanosulfónico, ácido 2-hidroxietanosulfónico, ácido etano 1,2-disulfónico, ácido bencenosulfónico, ácido 4-metilbencenosulfónico, ácido naftalen 2-sulfónico, ácido naftalen 1,5-disulfónico, 2 o 3 fosfoglicerato, glucosa 6-fosfato, ácido N-ciclohexilsulfámico (con formación de ciclamatos), o con otros compuestos orgánicos ácidos, tales como ácido ascórbico.
Las formulaciones orales estables ejemplares contienen uno o varios de los siguientes ingredientes adicionales que mejoran la estabilidad u otras características de la formulación: aglutinante, agente desintegrante, antioxidante ácido o lubricante o combinaciones de los mismos. Las formulaciones de comprimidos estables a modo de ejemplo incluyen un aglutinante y un agente desintegrante, opcionalmente con un antioxidante ácido, y opcionalmente incluyen además un lubricante. Las concentraciones ejemplares de aglutinante están entre aproximadamente 1% en peso y aproximadamente 5% en peso, o entre aproximadamente 1,5 y 3% en peso; una relación en peso ejemplar entre aglutinante y BH4 está en el intervalo de aproximadamente 1:10 a aproximadamente 1:20. Las concentraciones ejemplares de agente desintegrante están entre aproximadamente 1% en peso y aproximadamente 20% en peso; una relación en peso ejemplar entre agente desintegrante y BH4 está en el intervalo de aproximadamente 1:5 a aproximadamente 1:10. Las concentraciones ejemplares de antioxidante están entre aproximadamente 1% en peso y aproximadamente 3% en peso; una relación en peso ejemplar entre antioxidante y BH4 está en el intervalo de aproximadamente 1:5 a 1:30. En un ejemplo, el ácido ascórbico es el antioxidante y se usa en una relación con BH4 de menos de 1:1, p. ej., 1:2 o menos, 1:10 o menos. Las concentraciones ejemplares de lubricante en una formulación de comprimido estable de la presente invención están entre aproximadamente 0,1% en peso y aproximadamente 2% en peso; una relación en peso ejemplar entre lubricante y BH4 está en el intervalo de aproximadamente 1:25 a 1:65.
La formulación sólida estable puede incluir opcionalmente otros agentes terapéuticos adecuados para la afección que se va a tratar, p. ej., folatos, incluyendo precursores de folato, ácidos fólicos o derivados de folato; y/o arginina; y/o vitaminas, tales como vitamina C y/o vitamina B2 (riboflavina) y/o vitamina B12; y/o precursores de neurotransmisores tales como L-dopa o carbidopa; y/o 5-hidroxitriptófano.
Los folatos ejemplares, incluyendo precursores de folato, ácidos fólicos o derivados de folato, se describen en los documentos de patente de EE.UU. n° 6.011.040 y 6.544.994, e incluyen ácido fólico (pteroilmonoglutamato), ácido dihidrofólico, ácido tetrahidrofólico, ácido 5-metiltetrahidrofólico, ácido 5,10-metilentetrahidrofólico, ácido 5,10-meteniltetrahidrofólico, ácido 5,10-formiminotetrahidrofólico, ácido 5-formiltetrahidrofólico (leucovorina), ácido 10-formiltetrahidrofólico, ácido 10-metiltetrahidrofólico, uno o varios de los folilpoliglutamatos, compuestos en los que el
anillo de pirazina del resto pterina del ácido fólico o de los folilpoliglutamatos se reduce para proporcionar dihidrofolatos o tetrahidrofolatos, o derivados de todos los compuestos anteriores en los que las posiciones N-5 o N-10 son portadoras de una unidad de carbono con varios niveles de oxidación, o sales farmacéuticamente compatibles de los mismos, o una combinación de dos o más de los mismos. Ejemplos de tetrahidrofolatos incluyen ácido 5-formil-(6S)-tetrahidrofólico, ácido 5-metil-(6S)-tetrahidrofólico, ácido 5,10-metilen-(6R)-tetrahidrofólico, ácido 5,10-metenil-(6R)-tetrahidrofólico, ácido 10-formil-(6R)-tetrahidrofólico, ácido 5-formimino-(6S)-tetrahidrofólico o ácido (6S)-tetrahidrofólico, y sales farmacéuticamente aceptables de los mismos. Las sales ejemplares incluyen sales de sodio, potasio, calcio o amonio.
Las relaciones en peso relativas a modo de ejemplo entre BH4, folatos y arginina pueden ser desde aproximadamente 1:10:10 a aproximadamente 10:1: 1.
Las formulaciones estables de la invención se pueden proporcionar, p. ej., en forma de comprimidos o píldoras o cápsulas en frascos de HDPE provistos de una cápsula o bolsa desecante; o en envases de tipo blíster de lámina sobre lámina, o en envases tipo blíster que comprenden una película polimérica transparente, si es comercialmente deseable.
IV. Tratamiento de enfermedades que responden a BH4
Hiperfenilalaninemia, trastornos neurosicológicos o neurosiquiátricos
Los compuestos de la invención se pueden usar para el tratamiento de afecciones asociadas con niveles elevados de fenilalanina o niveles disminuidos de tirosina o triptófano, que se pueden deber, por ejemplo, a una actividad reducida de la hidroxilasa de fenilalanina, hidroxilasa de tirosina o hidroxilasa de triptófano. Las afecciones asociadas con niveles elevados de fenilalanina incluyen específicamente fenilcetonuria, tanto leve como clásica, e hiperfenilalaninemia tal y como se describe en esta memoria, y las poblaciones de pacientes ejemplares incluyen los subgrupos de pacientes descritos en esta memoria así como cualquier otro paciente que presente niveles de fenilalanina por encima de lo normal.
Las afecciones asociadas con niveles disminuidos de tirosina o triptófano incluyen deficiencia de neurotransmisores, trastornos neurológicos y siquiátricos tales como Parkinson, distonía, degeneración espinocerebelosa, dolor, fatiga, depresión, otros trastornos afectivos y esquizofrenia. Una producción en exceso de NO mediante nNOS se ha relacionado con accidentes cerebrovasculares, migrañas, enfermedad de Alzheimer y con tolerancia y dependencia de la morfina. Se puede administrar BH4 para cualquiera de esas afecciones. Otros trastornos neurosiquiátricos ejemplares para los que se puede administrar BH4, incluyen la enfermedad de Parkinson, enfermedad de Alzheimer, esquizofrenia, trastorno esquizofreniforme, trastorno esquizoafectivo, trastorno sicótico breve, trastorno delirante, trastorno sicótico compartido, trastorno sicótico debido a una afección médica general, trastorno sicótico inducido por sustancias, otros trastornos sicóticos, discinesia tardía, enfermedad de Machado-Joseph, degeneración espinocerebelosa, ataxia cerebelosa, distonía, síndrome de fatiga crónica, depresión aguda o crónica, síndrome de estrés crónico, fibromialgia, migraña, trastorno por déficit de atención con hiperactividad, enfermedad bipolar y autismo.
Las formulaciones estables también se pueden usar para tratar pacientes que padecen una carencia de BH4, por ejemplo, debido a un defecto en la ruta de su síntesis, que incluye, sin limitarse a, distonía sensible a la dopa (DRD), carencia de reductasa de sepiapterina (SR) o carencia de reductasa de dihidropteridina (DHPR).
Los sujetos adecuados para un tratamiento con las formulaciones estables de la invención, incluyen sujetos con una concentración plasmática elevada de Phe en ausencia del agente terapéutico, p. ej., superior a 1800 pM/L o superior a 1600 pM, superior a 1400 pM, superior a 1200 pM, superior a 1000 pM, superior a 800 pM o superior a 600 pM, superior a 420 pM, superior a 300 pM, superior a 200 pM o superior a 180 pM. Una PKU leve se clasifica generalmente como concentraciones plasmáticas de Phe de hasta 600 pM/L, una PKU moderada como concentraciones plasmáticas de Phe entre 600 pM/L y aproximadamente 1200 pM/L y una PKU clásica o grave como concentraciones plasmáticas de Phe superiores a 1200 pM/L. Preferiblemente, un tratamiento con las formulaciones estables solas o con una dieta restringida en proteínas disminuye la concentración plasmática de fenilalanina del sujeto, a menos de 600 pM, o menos de 500 pM, o 360 pM ± 15 pM o menos, o menos de 200 pM, o menos de 100 pM. Otros sujetos adecuados incluyen sujetos a los que se ha diagnosticado una actividad hidroxilasa de fenilalanina (PAH) reducida, fenilcetonuria atípica o maligna asociada con carencia de BH4, hiperfenilalaninemia asociada con trastorno hepático e hiperfenilalaninemia asociada con malaria. Una actividad PAH reducida puede ser el resultado de una mutación en la enzima PAH, por ejemplo, una mutación en el dominio catalítico de PAH o una o varias mutaciones seleccionadas a partir del grupo que consiste en F39L, L48S, I65T, R68S, A104D, S110C, D129G, E178G, V190A, P211T, R241C, R261Q, A300S, L308F, A313T, K320N, A373T, V388M, E390G, A395P, P407S y Y414C; o sujetos que sean mujeres embarazadas, mujeres en edad fértil que estén considerando un embarazo o niños entre 0 y 3 años de edad, o 0-2, 0-1,5 o 0-1; o sujetos diagnosticados como que no responden en 24 horas a una prueba de carga de BH4 de dosis única o una prueba de carga de dosis múltiple, tal como una prueba de carga de 4 dosis o 7 días. Poblaciones de pacientes ejemplares y pruebas de carga de BH4 ejemplares se describen en el documento de publicación de patente internacional n° WO 2005/049000.
Los documentos de patente de EE.UU. n24.752.573; 4.758.571; 4.774.244; 4.920.122; 5.753.656; 5.922.713; 5.874.433; 5.945.452; 6.274.581; 6.410.535; 6.441.038; 6.544.994; y los documentos de publicaciones de patentes de EE.UU. US 20020187958; US 20020106645; US 2002/0076782; US 20030032616, cada uno describe métodos para administrar composiciones de BH4 para tratamientos sin PKU. Cada una de esas patentes proporciona una descripción general de métodos de administración de composiciones de BH4 conocidos por los expertos en la técnica, que se pueden adaptar para el tratamiento tal y como se describe en esta memoria.
Aunque las necesidades individuales varían, una determinación de los intervalos óptimos de cantidades eficaces de cada componente está dentro del conocimiento de la técnica. Las dosificaciones típicas de BH4 comprenden desde aproximadamente 1 a aproximadamente 20 mg/kg de peso corporal por día, que normalmente ascenderán hasta aproximadamente 5 (1 mg/kg x 5 kg de peso corporal) a 3000 mg/día (30 mg/kg x 100 kg de peso corporal). Aunque se contempla una administración diaria continua, para HPA puede ser deseable interrumpir la terapia con BH4 cuando los síntomas de los niveles de Phe se reducen por debajo de un cierto nivel umbral. Por supuesto, la terapia puede reiniciarse en caso de que los niveles de Phe aumenten nuevamente. Las dosificaciones apropiadas se pueden determinar mediante el uso de ensayos establecidos para determinar los niveles sanguíneos de Phe junto con los datos de respuesta a dosis relevantes.
En realizaciones ejemplares, se contempla que los métodos de la presente invención proporcionarán a un paciente que lo necesite, una dosis diaria de entre aproximadamente 10 mg/kg y aproximadamente 20 mg/kg de BH4. Por supuesto, un experto en la técnica puede ajustar esa dosis aumentándola o reduciéndola, dependiendo de la eficacia que se logre mediante la administración. La dosis diaria se puede administrar en una sola dosis o alternativamente se puede administrar en múltiples dosis a intervalos convenientemente espaciados. En realizaciones ejemplares, la dosis diaria puede ser de 1 mg/kg, 2 mg/kg, 3 mg/kg, 4 mg/kg, 5 mg/kg, 6 mg/kg, 7 mg/kg, 8 mg/kg, 9 mg/kg, 10 mg/kg, 11 mg/kg, 12 mg/kg, 13 mg/kg, 14 mg/kg, 15 mg/kg, 16 mg/kg, 17 mg/kg, 18 mg/kg, 19 mg/kg, 20 mg/kg, 22 mg/kg, 24 mg/kg, 26 mg/kg, 28 mg/kg, 30 mg/kg, 32 mg/kg, 34 mg/kg, 36 mg/kg, 38 mg/kg, 40 mg/kg, 42 mg/kg, 44 mg/kg, 46 mg/kg, 48 mg/kg, 50 mg/kg o más mg/kg.
Regímenes de dosis baja
En un método terapéutico de dosis baja de la invención, se contemplan dosis bajas, por ejemplo, dosis de 0,1 a 5 mg/kg por día, incluyendo dosis de 0,1 a 2 mg/kg, o 0,1 a 3 mg/kg, o 1 mg/kg a 5 mg/kg. Se prefieren dosis de menos de 5 mg/kg por día. De acuerdo con la invención, se espera que tales dosis proporcionen mejoras con puntos finales de estudio relevantes, y se espera que los derivados de BH4 tengan propiedades biológicas mejoradas en relación con la BH4 natural a esas dosis. En particular, la invención contempla que cualquiera de las 1 ',2'-diacil-(6R,S)-5,6,7,8-tetrahidro-L-biopterinas o tetrahidrobiopterinas lipoidales descritas en esta memoria, muestren propiedades biológicas mejoradas con dosis bajas.
La invención también contempla específicamente el uso de BH4, o un precursor o derivado de la misma, para el tratamiento de enfermedades que responden a BH4 con una dosis en el intervalo de 0,1 a 5 mg/kg de peso corporal/día, a través de cualquier vía de administración que incluye pero no se limita a la administración oral, con una dosis una vez al día o en dosis múltiples (p. ej., 2, 3 o 4) divididas por día, durante una duración de al menos 1, 2, 3 o 4 semanas o más, o 1, 2, 3, 4, 5, 6 meses o más. Las dosis ejemplares incluyen menos de 5 mg/kg/día, 4,5 mg/kg/día o menos, 4 mg/kg/día o menos, 3,5 mg/kg/día o menos, 3 mg/kg/día o menos, 2,5 mg/kg/día o menos, 2 mg/kg/día o menos, 1,5 mg/kg/día o menos, 1 mg/kg/día o menos o 0,5 mg/kg/día o menos. También se contemplan dosis equivalentes por área de superficie corporal.
Para una persona de peso/área de superficie corporal promedio (por ejemplo, 70 kg), la invención también contempla una dosis diaria total de menos de 400 mg. Ejemplos de esas dosis diarias totales incluyen 360 mg/día, 350 mg/día, 300 mg/día, 280 mg/día, 210 mg/día, 180 mg/día, 175 mg/día, 150 mg/día o 140 mg/día. Por ejemplo, 350 mg/día o 175 mg/día se pueden administrar fácilmente con una formulación de dosificación oral de 175 mg, una o dos veces al día. Otras dosis diarias totales ejemplares incluyen 320 mg/día o menos, 160 mg/día o menos u 80 mg/día o menos. Esas dosis se administran fácilmente con una formulación de dosificación oral de 80 o 160 mg. Otras dosis diarias totales ejemplares incluyen 45, 90, 135, 180, 225, 270, 315 o 360 mg/día o menos, fácilmente administrables con una formulación de dosificación oral de 45 o 90 mg. Todavía otras dosis diarias totales ejemplares incluyen 60, 120, 180, 240, 300 o 360 mg/día, fácilmente administrables con una formulación de dosificación oral de 60 o 120 mg. Otras dosis diarias totales ejemplares incluyen 70, 140, 210, 280 o 350 mg/día, fácilmente administrables con una formulación de dosificación oral de 70 o 140 mg. Las dosis diarias totales ejemplares también incluyen 55, 110, 165, 220, 275 o 330 mg/día, fácilmente administrables con una formulación de dosificación oral de 55 mg. Otras dosis diarias totales ejemplares incluyen 65, 130, 195, 260 o 325 mg/día, o 75, 150, 225, 300 o 375 mg/día, p. ej., en formulaciones de dosificación de 65 mg o 75 mg.
Se entiende que la dosis adecuada de una composición según la presente invención, dependerá de la edad, la salud y el peso del receptor, el tipo de tratamiento concurrente, si lo hay, la frecuencia del tratamiento y la naturaleza del efecto deseado (es decir, la cantidad de disminución deseada de la concentración plasmática de Phe). La frecuencia de la dosificación también depende de los efectos farmacodinámicos sobre los niveles de Phe. Si el efecto dura 24 horas a partir de una dosis única. Sin embargo, la dosificación más preferida se puede adaptar a un sujeto individual, como entenderá y determinará un experto en la técnica, sin una experimentación indebida. Normalmente, esto
implica el ajuste de una dosis estándar, por ejemplo, la reducción de la dosis si el paciente tiene un peso corporal bajo.
La frecuencia de la dosificación de BH4 dependerá de los parámetros farmacocinéticos del agente y de las vías de administración. La formulación farmacéutica óptima la determinará un experto en la técnica dependiendo de la vía de administración y la dosificación deseada. Véase, por ejemplo, Remington's Pharmaceutical Sciences, 18a ed. (1990, Mack Publ. Co, Easton PA 18042) págs. 1435 1712. Tales formulaciones pueden influir en el estado físico, la estabilidad, la tasa de liberación in vivo y la tasa de aclaramiento in vivo de los agentes administrados. Dependiendo de la vía de administración, se puede calcular una dosis adecuada según el peso corporal, la superficie corporal o el tamaño de los órganos. Los expertos en la técnica habitualmente realizan un ajuste adicional de los cálculos necesarios para determinar la dosis de tratamiento adecuada, sin una experimentación indebida, especialmente de cara a la información sobre la dosificación y los ensayos descritos en esta memoria, así como los datos farmacocinéticos observados en ensayos clínicos en animales o seres humanos.
El régimen de dosificación final lo determinará el médico que realiza el tratamiento, teniendo en cuenta factores que modifican la acción de los fármacos, por ejemplo, la actividad específica del fármaco, la gravedad del daño y la capacidad de respuesta del paciente, la edad, el estado, el peso corporal, el sexo y la dieta del paciente, la gravedad de cualquier infección, el momento de la administración y otros factores clínicos. A medida que se realicen los estudios, surgirá una información adicional sobre los niveles de dosificación apropiados y la duración del tratamiento para enfermedades y afecciones específicas.
V. Terapia de combinación
Ciertos usos de la invención implican el uso combinado de las formulaciones estables de la invención y uno o varios otros agentes terapéuticos.
En una terapia de combinación de ese tipo, la administración de las formulaciones estables de la invención puede ser simultánea o puede preceder o seguir a la administración del segundo agente terapéutico, p. ej., a intervalos que van de minutos a horas, siempre que ambos agentes puedan ejercer su efecto terapéutico en períodos de tiempo solapantes. Por tanto, la invención contempla las formulaciones estables de la invención para uso con un segundo agente terapéutico. La invención también contempla el uso de un segundo agente terapéutico en la preparación de un medicamento para la administración con las formulaciones estables de tetrahidrobiopterina, precursores, derivados o análogos de la invención.
La terapia con tetrahidrobiopterina se puede combinar con una restricción de proteínas en la dieta para lograr un resultado terapéutico en pacientes con diversas formas de HPA. Por ejemplo, se podría administrar al sujeto la composición de BH4 y una composición proteica médica baja en fenilalanina, en una cantidad combinada eficaz para producir el resultado terapéutico deseado (es decir, una disminución de la concentración plasmática de Phe y/o la capacidad de tolerar mayores cantidades de ingesta de Phe/proteína sin producir un aumento concomitante de las concentraciones plasmáticas de Phe). Este procedimiento puede implicar la administración de la composición de BH4 y la composición terapéutica de proteína dietética al mismo tiempo. Esto se puede lograr mediante la administración de una única composición o una formulación de proteína farmacológica que incluya todos los requerimientos proteicos de la dieta y también incluya BH4 dentro de dicha formulación proteica. Alternativamente, la proteína dietética (complemento o comida proteica normal) se toma aproximadamente al mismo tiempo que una formulación farmacológica (comprimido, inyección o bebida) de BH4.
En algunas realizaciones, la dieta restringida en proteínas es una que se complementa con aminoácidos, tales como tirosina, valina, isoleucina y leucina. Al paciente se le puede coadministrar un complemento proteico bajo en Phe, que puede incluir L-tirosina, L-glutamina, L-carnitina con una concentración de 20 mg/100 g de complemento, L-taurina con una concentración de 40 mg/100 g de complemento y selenio. Puede comprender además las dosis diarias recomendadas de minerales, por ejemplo, calcio, fósforo y magnesio. El complemento puede comprender además la dosis diaria recomendada de uno o varios aminoácidos seleccionados a partir del grupo que consiste en L-leucina, L-prolina, L-acetato de lisina, L-valina, L-isoleucina, L-arginina, L-alanina, glicina, L-asparagina monohidrato, L-triptófano, L-serina, L-treonina, L-histidina, L-metionina, ácido L-glutámico y ácido L-aspártico. Además, el complemento se puede reforzar con la dosificación diaria recomendada de vitaminas A, D y E. Opcionalmente, el complemento comprende un contenido en grasas que proporciona al menos un 40% de la energía del complemento. Esos complementos se pueden proporcionar en forma de complemento en polvo o en forma de barra proteica. En determinadas realizaciones, la dieta restringida en proteínas comprende un complemento proteico y BH4 se proporciona en la misma composición que el complemento proteico.
En otras alternativas, el tratamiento con BH4 puede preceder o seguir a la terapia con proteínas dietéticas, en intervalos que van desde minutos a horas. En las realizaciones en las que la proteína y las composiciones de BH4 se administran por separado, generalmente se asegura que no expire un período de tiempo significativo entre el momento de cada administración, de modo que BH4 aún pueda ejercer un efecto ventajoso sobre el paciente. En esos casos, se contempla que se administre BH4 aproximadamente 2-6 horas (antes o después) de la ingesta de proteína dietética, por ejemplo, con un tiempo de retraso de solo aproximadamente 1 hora o menos. En determinadas realizaciones, se contempla que la terapia con BH4 sea una terapia continua en la que se administra
una dosis diaria de BH4 al paciente de forma indefinida. En otras situaciones, por ejemplo, en mujeres embarazadas que solo tienen las formas más leves de PKU y HPA, es posible que la terapia con BH4 solo continúe mientras que la mujer esté embarazada y/o amamantando.
Además, en adición a las terapias basadas únicamente en la administración de BH4 y una regulación de las proteínas de la dieta, los usos de la presente invención también contemplan una terapia de combinación con una tercera composición que se dirige específicamente a uno o varios de los síntomas de HPA. Por ejemplo, se sabe que un déficit en tirosina causado por HPA, da como resultado una carencia de los neurotransmisores dopamina y serotonina. Por tanto, en el contexto de la presente invención, se contempla que los usos basados en proteínas dietéticas y BH4 podrían combinarse adicionalmente con la administración de los neurotransmisores L-dopa, carbidopa y 5-hidroxitriptófano para corregir los defectos que resultan de una disminución de las cantidades de tirosina en la dieta.
Además, la terapia génica con PAH (Christensen et al., Mol. Gent. And Metabol. 76: 313-318, 2002; Christensen et al., Gene Therapy, 7:1971-1978, 2000) y fenilalanina amoniaco liasa (PAL Liu et al., Arts. Cells. Blood. Subs and Immob. Biotech. 30(4)243-257, 2002) se ha contemplado por los expertos en la técnica. Esas técnicas de terapia génica se pueden usar en combinación con las terapias combinadas de la invención basadas en BH4/estricción de proteína dietética. En terapias de combinación adicionales, se contempla que se pueda proporcionar fenilasa como una enzima inyectable para acabar con concentraciones de Phe más bajas en el paciente. Como la administración de fenilasa no generaría tirosina (a diferencia de la administración de PAH), ese tratamiento aún seguirá dando como resultado que la tirosina sea un aminoácido esencial para esos pacientes. Por lo tanto, una complementación dietética con tirosina puede ser deseable para pacientes que reciben fenilasa en combinación con la terapia con BH4.
BH4 se puede coadministrar para trastornos neurosicológicos o neurosiquiátricos, de acuerdo con los usos de la invención, con uno o varios otros agentes activos neurosiquiátricos, que incluyen antidepresivos, precursores de neurotransmisores tales como triptófano, tirosina, serotonina, agentes que activan sistemas noradrenérgicos, tales como lofepramina, desipramina, reboxetina, tirosina, agentes que actúan preferentemente sobre la serotonina, inhibidores combinados de la captación de noradrenalina y serotonina, tales como venlafaxina, duloxetina o milnaciprán, o fármacos que son inhibidores combinados de la recaptación de dopamina y noradrenalina, tales como el bupropión.
En una realización relacionada, BH4 se administra con otros agentes terapéuticos comúnmente usados para tratar la diabetes, la enfermedad vascular, la hiperlipidemia. Los agentes utilizados para tratar la diabetes incluyen, entre otros, agentes que mejoran la sensibilidad hacia la insulina, tales como ligandos gamma de PPAR (tiazolidindonas, glitazonas, troglitazonas, rosiglitazona (Avandia), pioglitazona), estimuladores de la secreción de insulina, tales como sulfonilureas (gliquidona, tolbutamida, glimeprida, clorpropamida, glipizida, gliburida, acetohexamida) y meglitinidas (meglitinida, repaglinida, nateglinida) y agentes que reducen la producción hepática de glucosa, tales como metformina. Un agente utilizado para tratar la enfermedad vascular incluye, pero no se limita a, antagonistas del receptor de endotelina utilizados habitualmente para el tratamiento de la hipertensión y otros trastornos relacionados con la disfunción endotelial, tales como bosentán, darusentán, enrasentán, tezosentán, atrasentán, ambrisentán, sitaxsentán; relajantes del músculo liso, tales como inhibidores de PDE5 (de acción indirecta) y minoxidil (de acción directa); inhibidores de la enzima convertidora de angiotensina (ECA), tales como captopril, enalapril, lisinopril, fosinopril, perindopril, quinapril, trandolapril, benazepril, ramipril; bloqueantes del receptor de angiotensina II, tales como irbesartán, losartán, valsartán, eprosartán, olmesartán, candesartán, telmisartán; betabloqueantes, tales como atenolol, metoprolol, nadolol, bisoprolol, pindolol, acebutolol, betaxolol, propranolol; diuréticos, tales como hidroclorotiazida, furosemida, torsemida, metolazona; bloqueadores de los canales de calcio, tales como amlodipina, felodipina, nisoldipina, nifedipina, verapamilo, diltiazem; bloqueadores del receptor alfa, doxazosina, terazosina, alfuzosina, tamsulosina; y agonistas alfa centrales, tales como clonidina. Los agentes utilizados para tratar la hiperlipidemia incluyen, pero no se limitan a, agentes que reducen el LDL, tales como estatinas (atorvastatina, fluvastatina, lovastatina, pravastatina, rosuvastatina cálcica, simvastatina) y ácido nicotínico, inhibidores de la proteína de transferencia de éster de colesterilo (como torcetrapib), agentes que estimulan PPAR alfa, tales como fibratos, gemfibrozil, fenofibrato, bezafibrato, ciprofibrato, agentes que se unen y evitan la readsorción de ácidos biliares y reducen los niveles de colesterol como secuestradores de ácidos biliares, colestiramina y colestipol e inhibidores de la absorción de colesterol.
BH4 también se puede administrar con un factor o una combinación de factores que potencia o normaliza la producción del vasodilatador óxido nítrico (NO) solo o en combinación con un agente terapéutico. En una realización, ese factor o factores potencian la actividad o la expresión de la biosíntesis de novo de BH4 y se seleccionan a partir del grupo que consiste en ciclohidrolasa I de trifosfato de guanosina (GTPCH1), sintasa de 6-piruvoiltetrahidropterina (PTPS) y reductasa de sepiapterina. En una realización preferida de la invención, la síntesis de BH4 se incrementa aumentando la expresión de GTPCH1 mediante el uso de cualquiera o varios análogos o agonistas de monofosfato de adenosina cíclica (AMPc), que incluyen forskolina, 8-bromo AMPc u otros agentes que actúan aumentando la señalización celular mediada por AMPc, por ejemplo, citocinas y factores de crecimiento que incluyen interleucina-1, interferón-gamma (IFN-y), factor de necrosis tumoral alfa (TNF-a), proteína c reactiva, HMG-CoA-reductasas (estatinas tales como atorvastatina), factor de crecimiento nervioso (NGF), factor de crecimiento epidérmico (EGF), hormonas que incluyen adrenomedulina y benzoato de estradiol, y otros compuestos como
NADPH y análogos de NADPH, cafeína, ciclosporina A metilxantinas que incluyen 3-isobutil-l-metil xantina, teofilina, reserpina, peróxido de hidrógeno.
Por tanto, una realización de la invención se refiere al aumento de los niveles de GTPCH1 inhibiendo la degradación de nucleótidos cíclicos 3',5' usando inhibidores de las once familias de fosfodiesterasas (PDE1-11) que incluyen PDE1, PDE3, PDE5. Los inhibidores de PDE de la presente invención incluyen viagra/sildanefil, cialis/tadalafil, vardenafil/levitra, udenafil, 8-metoximetil-IBMX, UK-90234, dexametasona, hesperetina, hesperedinas, irsogladina, vinpocetina, cilostamida, rolipram, beta-carbolina-3-carboxilato de etilo (beta-CCE), derivados de tetrahidro-betacarbolina, 3-O-metilquercetina y similares.
Otra realización de la invención se refiere al aumento de los niveles de BH4 aumentando los niveles de enzimas que sintetizan BH4 mediante terapia génica o una administración dirigida al endotelio de polinucleótidos de la maquinaria sintética de BH4. Otra realización adicional de la invención se refiere al aumento de los niveles de BH4 mediante complementación con enzimas que sintetizan BH4, GTPCH1, PTPS, SR, PCD, DHPR y DHFR. Se contempla que las enzimas que sintetizan BH4 incluyan todas las formas naturales y no naturales de las enzimas, incluyendo mutantes de las proteínas.
Otra realización de la invención se refiere al aumento de los niveles de BH4 desviando el sustrato 7,8-dihidroneopterina trifosfato hacia la enzima PTPS que sintetiza BH4, en lugar de la fosfatasa alcalina (AP), mediante una inhibición de la actividad de AP. Los agentes o compuestos que inhiben la actividad de AP incluyen análogos de fosfato, levamisol y L-Phe. Otra realización de la invención se refiere a agentes o compuestos que inhiben la fosfatasa alcalina, que incluyen ARN inhibidor pequeño (ARNip), ARN antisentido, ADN bicatenario, moléculas pequeñas, anticuerpos neutralizantes, fragmentos de anticuerpos de cadena simple, quiméricos, humanizados para inhibir la síntesis de fosfatasa alcalina.
Otra realización de la invención incluye agentes o compuestos que mejoran la actividad de catalizadores o cofactores necesarios para la síntesis de enzimas de la ruta de síntesis de novo de síntesis de BH4.
Otra realización de la invención incluye agentes o compuestos que evitan la degradación de las enzimas necesarias para la síntesis de BH4. Otra realización adicional de la invención incluye agentes o compuestos que evitan la degradación de los catalizadores necesarios para la síntesis de BH4 y sus enzimas sintéticas, incluyendo GTPCH1, PTPS y SR.
Otra realización de la invención se refiere al aumento de los niveles de BH4 incrementando la reducción de BH2 a través de la ruta de recuperación. In vivo, BH4 se oxida a BH2. BH2 existe como la forma quinoide (qBH2) y como 7,8-dihidropterina que se reduce a BH4 mediante DHPR y DHFR respectivamente. Una realización de la invención se refiere a incrementar la regeneración o recuperación de BH4 a partir de BH2, modulando la actividad y la síntesis de las enzimas PCD, DHPR y DHFR usando agentes o compuestos de la ruta NADPH, tioles, percloromercuribenzoato, peróxido de hidrógeno y similares.
Otra realización de la invención se refiere a agentes que estabilizan BH4 al disminuir la oxidación de BH4 usando agentes o compuestos tales como antioxidantes que incluyen ácido ascórbico (vitamina C), alfa tocoferol (vitamina E), tocoferoles (por ejemplo, vitamina A), selenio, betacarotenos, carotenoides, flavonas, flavonoides, folatos, flavonas, flavanonas, isoflavonas, catequinas, antocianidinas y calconas.
En una realización adicional, ese factor o factores pueden aumentar la actividad o la expresión de la sintasa de óxido nítrico y, por lo tanto, mejorar la generación de NO.
En otra realización adicional, la invención contempla factores que inhiben la proteína reguladora de la retroalimentación de GTPCH, GFRP. Una realización de la invención se refiere a agentes o compuestos que inhiben la unión de BH4 al complejo GTPCH1/GFRP, evitando de ese modo la inhibición de la retroalimentación mediante BH4. Los agentes o compuestos de esta invención incluyen inhibidores competitivos, tales como formas alternativas de BH4 con afinidades alteradas hacia el complejo, análogos estructurales, etc. Otra realización adicional de la invención incluye agentes o compuestos que mejoran la unión de L-fenilalanina a CTPCH1/GFRP, induciendo la síntesis de BH4. Otra realización de la invención incluye agentes o compuestos que aumentan los niveles de L-Phe, tales como precursores de L-fenilalanina, lo que sirve para inhibir la inhibición de la retroalimentación de GTPCH1 mediante GFRP y BH4.
Otra realización adicional de la invención se refiere a agentes o compuestos que modulan la actividad o la síntesis de GFRP. Una realización de la invención incluye agentes o compuestos que inhiben la actividad de GFRP. Otra realización de la invención incluye el uso de ARNsi, moléculas pequeñas, anticuerpos, fragmentos de anticuerpos y similares para inhibir la síntesis de GFRP.
VI. Ensayos con biopterina
La concentración de biopterina total y biopterina oxidada en plasma, sangre y otros tejidos se determina según el método de Fukishima y col. (Anal. Biochem. 102:176 (1980). La biopterina tiene cuatro formas diferentes que incluyen dos formas de biopterina reducida, R-tetrahidrobiopterina (BH4) y R-dihidrobiopterina quinonoide (q-BH2) y
dos formas de biopterina oxidada, dihidrobiopterina (BH2) y biopterina (B). De esas cuatro formas, solo las formas reducidas de biopterina tienen actividad coenzimática. La biopterina reducida se convierte en B mediante yodilación en condiciones ácidas, mientras que en condiciones alcalinas se convierte en pterina. La biopterina oxidada se convierte en B mediante yodilación en condiciones ácidas y alcalinas. Aprovechando esta propiedad, la cantidad de biopterina total se determina mediante yodilación en condiciones ácidas y la de biopterina oxidada se determina mediante yodilación en condiciones alcalinas, de modo que la cantidad de biopterina reducida se calcula a partir de la diferencia en la cantidad de las mismas. Cuando se usa como coenzima, BH4 se convierte en q-BH2. q-BH2 se convierte inmediatamente a BH4 mediante la reductasa de dihidropterina o, si no se reduce, se oxida a BH2 o DHPT. Debido a que es difícil que la biopterina exista en la forma de q-BH2 in vivo, la biopterina reducida se puede cambiar bien con BH4.
Las muestras de plasma y sangre completa recogidas se someten inmediatamente a una oxidación con una solución oxidante ácida (solución de HCl 0,6 N en agua que contiene yoduro de potasio (KI) al 0,6%, yodo al 0,3% (I2) y ácido tricloroacético (TCA) 0,6 N) y una solución oxidante alcalina (hidróxido de sodio 0,7 N (NaOH)). La determinación de B se realiza mediante HPLC y la radiactividad se mide haciendo un recuento del centelleo líquido.
Medición de BH4 usando HPLC de fase inversa (RP) acoplada con espectrometría de masas en tándem (LC/MS/MS): se mostró que el uso combinado de cromatografía líquida de alta resolución de fase inversa (RP) y espectrometría de masas en tándem (LC/MS/MS), es selectivo para BH4 en plasma humano, sensible para BH4 en el intervalo de 5 - 1000 ng/mL. El método está asociado con aproximadamente un 50% de conversión de BH4 debido a la oxidación durante la recogida y el almacenamiento. Las muestras son estables durante más de 3 meses en plasma con sal dipotásica de ácido etilendiaminotetraacético (K2EDTA). Una recuperación a partir de las etapas previas del tratamiento es de aproximadamente el 75%. Se determinó que la exactitud y la precisión del método tienen un % de coeficiente de variación (CV) por debajo del 15% (20% en el límite inferior de cuantificación, LLOQ). El uso combinado de HPLC y espectrometría de masas en tándem mostró ser una mejora con respecto a solo una HPLC para determinar el artículo de la prueba, BH4 debido a: (1) su mayor selectividad hacia el fármaco-BH4 (mientras que la HPLC mide la biopterina total), (2) intervalo cualitativo más amplio, (3) proporción de conversión establecida, (4) caracterización amplia y utilidad probada en sujetos humanos y (5) medición novedosa y útil en diferentes especies y matrices.
El método mejorado comprende las siguientes etapas. Las muestras de sangre, plasma, homogeneizados de tejidos u orina se someten a una oxidación ácida o alcalina. Con oxidación ácida, (1) las muestras se tratan con cloruro de potasio (KCl), ácido clorhídrico (HCl) o TCA durante una hora; (2) las muestras oxidadas con ácido se someten luego a yodometría; (3) las muestras pasan a través de una columna de intercambio iónico; (4) la biopterina total que comprende BH4, q-BH2 (que se reduce inmediatamente in vivo a BH4, de manera que la biopterina reducida medida se basa principalmente en BH4), BH2 y B se miden usando HPLC y espectrometría de masas en tándem. Con oxidación alcalina, (1) las muestras se tratan con KI, I2 o NaOH durante una hora; (2) las muestras oxidadas alcalinas se someten luego a una acidificación con HCl o TCA; (3) se someten a yodometría; (4) las muestras pasan a través de una columna de intercambio iónico; (5) se mide la biopterina oxidada que comprende BH2 y B; (6) se miden diferentes especies usando HPLC y espectrometría de masas en tándem; y (7) la cantidad de biopterina reducida (BH4 q-BH2) se calcula como la diferencia entre las biopterinas totales menos la forma oxidada.
Los diagramas de flujo para la medición de biopterina y el resumen de la validación del ensayo se proporcionan en las Figuras 16 y 17.
Ensayo optimizado
Un método de HPLC que utiliza detección electroquímica (ECD) y detección de fluorescencia (FL), es ventajoso ya que permite la medición de cada uno de los compuestos de biopterina discretos (BH4, BH2 y B) así como de los análogos.
BH4 es un cofactor del sistema enzimático de la sintasa de óxido nítrico (NOS), que produce óxido nítrico (NO). La producción de NO es importante para mantener la homeostasis vascular. Cuando los niveles intracelulares de BH4 están limitados, la producción de NO disminuye (debido a una disminución de la actividad de la NOS) y conduce a la generación del superóxido (O2-), un radical libre dañino. Un exceso de O2- puede provocar una disfunción endotelial y puede contribuir a la oxidación de BH4 a BH2. Una relación baja entre BH4 y BH2 puede promover una lesión endotelial, mientras que una relación alta entre BH4 y BH2 puede promover la salud endotelial. Por lo tanto, caracterizar la relación entre BH4 y BH2 puede servir como un indicador de salud endotelial.
Las concentraciones de diferentes biopterinas (BH4, BH2 y B) o de análogos se determinan utilizando inicialmente la HPLC de fase inversa para la separación, seguida de detección mediante ECD y FL.
BH4, que es una molécula no fluorescente, sensible a redox, se mide usando ECD. BH4 (y sus análogos) se miden usando ECD en donde BH4 (o un análogo) es oxidada por el electrodo 1 hasta una forma quinonoide de dihidrobiopterina (p. ej., qBH2), un producto intermedio de dihidrobiopterina de vida corta, que luego se reduce de nuevo a BH4 (o un análogo) en el electrodo 2. El detector usa entonces la corriente generada por esa reacción de reducción para determinar la concentración de BH4 o de un análogo de la misma (la qBH2 endógena es
despreciable).
BH2, B y sus análogos se pueden medir en la misma inyección mediante detección de la fluorescencia. Una oxidación posterior al ECD de BH2 o un análogo de la misma usando una celda de protección de acondicionamiento con el potencial óptimo, oxida BH2 o un análogo de la misma a B o al análogo de biopterina correspondiente. Esto es deseable porque BH2 no es activa de forma fluorescente ni se mide fácilmente y se tiene que convertir a B, que se mide fácilmente usando fluorescencia. La BH2 endógena, una vez convertida a B, y la B endógena se distinguen entre sí por dos picos fluorescentes distintos, debido a los diferentes tiempos de retención en la columna de HPLC para cada molécula.
En total, los métodos se pueden usar para medir las especies BH4, BH2 y B, y análogos de las mismas. Las biopterinas se miden preferiblemente usando una fase móvil que contiene MeOH al 2%, tal y como se describe en esta memoria. Los análogos de biopterina, tales como los derivados de biopterina de valina, pueden ser más adecuados para contenidos más altos en metanol en la fase móvil, p. ej., una fase móvil que contiene MeOH al 10%. Por tanto, un método para detectar biopterinas en una mezcla de especies de biopterina puede incluir (a) separar las especies de biopterina en la mezcla mediante HPLC de fase inversa; y en el caso de BH4 y sus análogos, (b1) realizar una detección electroquímica oxidando BH4 y sus análogos presentes en un primer electrodo a formas quinonoides de dihidrobiopterina, seguida por una reducción de las formas quinonoides de nuevo a BH4 y sus análogos, presentes en un segundo electrodo y medir la corriente generada por la reacción de reducción para determinar la concentración de las especies; y/o (b2) en el caso de BH2, análogos de la misma, biopterina o análogos de la misma, midiendo esas especies mediante una detección de la fluorescencia después de la oxidación post-columna de las especies de BH2 a biopterina. Preferiblemente, la fase móvil es una de las descritas en esta memoria.
En una realización, la fase móvil preferida incluye acetato de sodio, ácido cítrico, EDTA y 1,4-ditioeritritol (DTE) con metanol. Las concentraciones preferidas son acetato de sodio 50 mM, ácido cítrico 5 mM, EDTA 48 gM y DTE 160 pM con metanol al 2%.
VII. EJEMPLOS
Los siguientes ejemplos se incluyen para demostrar realizaciones preferidas de la invención. Los expertos en la técnica deben apreciar que las técnicas descritas en los ejemplos que siguen, representan técnicas que el inventor ha descubierto que funcionan bien en la puesta en práctica de la invención y, por tanto, se puede considerar que constituyen modos preferidos para su puesta en práctica.
Ejemplo 1
Curva de tiempo y concentración para biopterina en plasma después de una única dosis oral en la rata
Los fines de este estudio eran evaluar la farmacocinética de BH4 después de una única administración oral en ratas. Se administraron por vía oral dosis únicas de BH4 (10 y 100 mg/kg) a ratas macho Sprague Dawley (6 semanas de edad) en ayunas.
Resultados
Las concentraciones máximas de biopterina total en plasma 2 horas y 1 hora después de la dosificación eran 108 ng/mL (es decir, aproximadamente 3 veces el nivel endógeno) y 1227 ng/mL (es decir, aproximadamente 30 veces el nivel endógeno), respectivamente (Figura 18). A partir de entonces, la biopterina tenía una semivida de eliminación (t1/2) de aproximadamente 1,1 horas, volviendo al nivel endógeno 9 horas después de la dosificación para la dosis de 10 mg/kg y 24 horas después de la dosificación para la dosis de 100 mg/kg (Figura 18).
La biodisponibilidad (F) después de una administración oral de 10 y 100 mg/kg era de 6,8% y 11,8%, respectivamente, en función del área bajo la curva de la concentración plasmática-tiempo (AAUC), obtenida al restar el nivel endógeno durante una administración intravenosa de 10 mg/kg. La tasa de absorción GI era del 8,8% cuando se medía empleando marcadores radiactivos en la orina. Una estimación del valor real sería aproximadamente un 10% de la biodisponibilidad oral basada en esos datos.
La relación entre biopterina reducida y biopterinas totales en plasma (es decir, la relación de forma reducida) era relativamente estática (73% -96%) (Figura 19).
Ejemplo 2
Curva de tiempo y concentración para biopterina en plasma después de una dosis oral única a monos
Los fines de este estudio eran evaluar la farmacocinética de sapropterina después de una única administración oral en monos cynomolgus. Se administró una dosis única de sapropterina (10 mg/kg) por vía oral a monos de tipo cynomolgus hembra (3/grupo) en ayunas.
Resultados
La concentración plasmática total de biopterina (AC) alcanzó su valor máximo 3 horas después de la dosificación (344 ng/mL, aproximadamente 20x los niveles endógenos) (Figura 20). La semivida de eliminación plasmática de la biopterina era de aproximadamente 1,4 horas, volviendo al nivel endógeno 24 horas después de la administración. La relación entre biopterina reducida y biopterinas totales era casi constante durante el período de prueba. La biodisponibilidad (F) después de una administración oral de 10 mg/kg a monos hembras era de aproximadamente el 9%, medida como las relaciones AAUC oral/iv (Figura 21).
Ejemplo 3
Biodisponibilidad relativa de tetrahidrobiopterina (BH4) administrada después de una disolución de uno o varios comprimidos en agua o administrada como uno o varios comprimidos intactos, y efecto de los alimentos sobre la absorción en sujetos sanos
Objetivos
Los objetivos principales del estudio eran: (1) evaluar la biodisponibilidad relativa de la tetrahidrobiopterina (BH4, clorhidrato de sapropterina) cuando se administraba después de la disolución de uno o varios comprimidos en agua o se administraba como comprimidos intactos; (2) comparar el efecto de los alimentos sobre la biodisponibilidad de BH4 en sujetos sanos. El objetivo secundario del estudio era evaluar la seguridad y la tolerabilidad de dosis orales únicas de BH4 en sujetos sanos.
Metodología
Este estudio era un estudio cruzado, abierto, aleatorizado, de tres tratamientos, seis secuencias y tres períodos en el que 30 sujetos debían completar 3 períodos de dosificación de dosis única y se aleatorizaron en uno de los seis grupos de secuencias (Grupos 1,2, 3, 4, 5 y 6):
Grupo 1: a, b, c
Grupo 2: b, c, a
Grupo 3: c, a, b
Grupo 4: a, c, b
Grupo 5: b, a, c
Grupo 6: c, b, a
en donde todos los grupos de dosificación recibieron 10 mg/kg de BH4 por vía oral de la siguiente manera:
a: administrada después de una disolución del o de los comprimidos en agua, proporcionada en ayunas en condiciones de ayuno
b: administrada en forma de uno o varios comprimidos intactos, proporcionada en ayunas en condiciones de ayuno c: administrada como uno o varios comprimidos intactos, proporcionada 30 minutos después de comenzar a ingerir una comida rica en calorías y grasas en condiciones prandiales.
Cada sujeto recibió una dosis única de 10 mg/kg de BH4 durante cada período de tratamiento. Un período de aclaramiento de al menos siete días separaba cada administración de la dosis. Se realizó una evaluación posterior al estudio 5-7 días después del término del tercer período de tratamiento. Las muestras de sangre para el análisis farmacocinético (PK) se extrajeron en tiempos de recogida programados durante cada período de estudio: 30 minutos antes de la dosis y 0,5, 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 5,0, 6,0, 8,0, 10,0, 12,0, 18,0 y 24,0 horas después de la dosis.
Dosis y modo de administración
Los comprimidos de BH4 se administraron como dosificaciones de 10 mg/kg por cada período de tratamiento. Los comprimidos se administraron mediante a) disolución en agua administrada en ayunas, b) como comprimidos intactos administrados en ayunas, o c) como comprimidos intactos administrados en estado prandial.
Cada dosis del fármaco en estudio se preparó y se administró en forma líquida (solución) mezclada con agua. El agua suministrada era agua corriente a temperatura ambiente. Las soluciones de la dosificación se prepararon dentro de los 15 minutos del tiempo de dosis pautado. La disolución del comprimido en líquido duraba aproximadamente de 1 a 3 minutos. Los comprimidos se rompieron o trituraron en la copa dosificadora antes de la disolución para mejorar la tasa de disolución.
A la hora designada para la dosificación de la mañana, se administró BH4 por vía oral como la cantidad de comprimidos equivalente a una dosis de 10 mg/kg, disuelta en 120 mL de agua o zumo de naranja. Se observó de cerca a cada sujeto, ya que la dosis completa de 120 mL se consumía 15 minutos después de la preparación. Inmediatamente después de consumir la dosis, se lavó la copa dosificadora con 60 mL de agua y el sujeto consumió el agua del lavado. Se añadió un segundo lavado con agua de 60 mL a la taza dosificadora y luego el sujeto consumió el segundo lavado. Todo el procedimiento de dosificación se completó en un período de tiempo de 1 minuto. Una persona capacitada inspeccionó la copa dosificadora y la boca de cada sujeto inmediatamente después de completar la dosis para asegurarse de que se había consumido la dosis completa. Alternativamente, el sujeto ingirió una píldora que contenía BH4 en lugar de disolverla en agua. Para cada individuo, los períodos de dosificación tenían lugar con un mínimo de 7 días entre las dosis.
Pauta de ingesta de los alimentos
La noche del ingreso se sirvió un refrigerio. Luego se requirió que todos los sujetos ayunaran durante al menos 10 horas antes de la dosificación.
Condiciones de ayuno
Los sujetos que recibieron tratamientos administrados en ayunas, recibieron la dosis después de completar un ayuno nocturno mínimo de 10 horas.
Los sujetos continuaron ayunando durante 4 horas después de la dosis. Se permitió agua a voluntad durante el estudio, excepto desde 1 hora antes hasta 1 hora después de la dosis. Se proporcionaron comidas estandarizadas aproximadamente 4 y 10 horas después de la administración del fármaco y en momentos apropiados a partir de entonces.
Condiciones de no ayuno
A los sujetos que recibieron tratamientos administrados en condiciones que no eran de ayuno, se les administró la dosis después de consumir una comida en el desayuno rica en calorías y grasas. Los sujetos recibieron el siguiente desayuno estándar rico en grasas (aproximadamente 50% del contenido calórico total de la comida), rico en calorías (aproximadamente 1000 calorías) que comenzó 30 minutos antes de la administración pautada de la dosis y terminó (último bocado tomado) 5 minutos antes de la dosificación.
2 huevos fritos en mantequilla
2 lonchas de tocino
2 tostadas con mantequilla
114 g (4 onzas) de patatas salteadas
225 g (8 onzas) de leche entera.
Esta comida contenía aproximadamente 150 calorías de proteínas, 250 calorías de carbohidratos y 500-600 calorías de grasas. Se sustituyó una comida equivalente con una documentación del menú y el contenido calórico.
Después, los sujetos ayunaron durante 4 horas después de la dosis. Se permitió agua a voluntad durante el estudio, excepto desde 1 hora antes hasta 1 hora después de la dosis. Las comidas estándar se proporcionaron aproximadamente 4 y 10 horas después de la administración del fármaco y en momentos apropiados a partir de entonces.
Duración del tratamiento
Tres períodos de tratamiento de dosis única se separaron entre sí con un mínimo de 7 días.
Se realizó una consulta de seguimiento 5 a 7 días después de la última consulta médica del tratamiento.
Variables de seguridad: evaluación y métodos
Se evaluó la seguridad para todos los sujetos que tomaban al menos una dosis de BH4.
Mediciones de la eficacia y la seguridad evaluadas y diagrama de flujo
La seguridad se evaluó registrando la incidencia de eventos adversos, los cambios en los parámetros de un ECG de 12 derivaciones, los signos vitales y los resultados del examen físico, y los cambios en los valores iniciales de las pruebas de laboratorio. El cronograma de esas evaluaciones se muestra en la Figura 22.
Exámenes físicos y signos vitales
Cada sujeto se sometió a un examen físico de rutina por parte del investigador del estudio. El examen físico incluía una evaluación de la cabeza, ojos, oídos, nariz, garganta, cuello, corazón, tórax, pulmones, abdomen, extremidades, pulsos periféricos, estado neurológico, piel, y se evaluaron otros estados físicos notables. Este protocolo del estudio no requería exámenes genitourinarios.
Se midieron la altura (en centímetros) y el peso (en kilogramos) y se calculó el índice de masa corporal (IMC) (IMC = peso (kg)/[altura (m)]2).
La presión arterial se midió en posición sentada de acuerdo con las recomendaciones de la American Heart Association. Los sujetos estaban en reposo con los pies en el suelo durante 5 minutos en posición sentada cuando se midió la presión arterial.
La frecuencia cardíaca (pulso) se midió mientras que el sujeto estaba sentado.
Se realizó un registro de un electrocardiograma (ECG) estandarizado de 12 derivaciones en la clasificación al término del estudio. Los ECGs fueron evaluados por un investigador capacitado. Se guardaron copias del ECG y los informes de la evaluación como parte del expediente de cada sujeto.
El investigador principal revisó y evaluó el historial médico, los resultados de las pruebas del laboratorio clínico y los registros del ECG para determinar la capacidad de elección clínica de cada sujeto para participar en el estudio. Evaluaciones de laboratorio clínico
Hematología:
Se evaluó lo siguiente: hemoglobina, hematocrito, recuento de leucocitos total y diferencial, recuento de glóbulos rojos (RBC) y recuento de plaquetas.
Además, se analizó la sangre para detectar el antígeno de superficie de la hepatitis B, el anticuerpo de la hepatitis C y el virus de la inmunodeficiencia humana (VIH).
Química:
Se evaluó lo siguiente: albúmina, nitrógeno ureico en sangre (BUN), creatinina, bilirrubina total, fosfatasa alcalina (ALP), aspartato transaminasa (AST), alanina transaminasa (ALT), sodio (Na+), potasio (K+), cloruro (Cl-), deshidrogenasa láctica (LDH), ácido úrico y glucosa.
Análisis de orina:
Se evaluó lo siguiente mediante el método de la tira reactiva de orina: pH, densidad específica, proteína, glucosa, cetonas, bilirrubina, sangre, nitrito y urobilinógeno. Si los valores de proteína, sangre oculta o nitrito están fuera de rango, se realiza un examen microscópico.
También se analizaron muestras de orina en busca de drogas de abuso (anfetaminas, barbitúricos, benzodiazepinas, cannabinoides, cocaína y opiáceos).
Eventos adversos
En este estudio, un evento adverso (EA) se definió como cualquier evento médico adverso en un sujeto o un sujeto en investigación clínica al que se le administra BH4, en cualquier dosis, tenga o no una relación causal con el evento. Por lo tanto, un EA puede ser cualquier signo desfavorable e involuntario (incluido un hallazgo anormal de laboratorio), síntoma o enfermedad asociados temporalmente con el uso de BH4, estén o no relacionados con BH4. Esta definición incluía enfermedades o lesiones intercurrentes y exacerbación (aumento de la frecuencia, gravedad o especificidad) de afecciones preexistentes.
El período de notificación de EAs comenzaba con la primera administración de BH4. El período de notificación de eventos adversos graves (EAGs) comenzaba antes, desde el momento de la firma del consentimiento informado. Los EAGs se definirán más adelante en esta sección. El investigador realizó un seguimiento de todos los EAs hasta la resolución o, si se determinó que el EA era crónico, se identificó una causa. Si un EA seguía sin resolverse al final del estudio, el PI y el monitor médico realizaban una evaluación clínica para determinar si se justificaba un seguimiento continuado del EA y se documentaban los resultados. Una evaluación de la gravedad era una de las responsabilidades del investigador en la evaluación de los EAs y EAGs. El investigador era responsable de aplicar su juicio clínico para evaluar la relación causal de cada EA con BH4.
Eventos adversos graves
Un evento adverso grave (EAG) se definió como cualquier EA que tuviera al menos uno de los siguientes resultados:
Dio como resultado la muerte
Era potencialmente mortal, es decir, puso a los sujetos en riesgo inmediato de muerte a partir de que tuvo lugar el evento
Esta definición no incluía una reacción que, si hubiera ocurrido en una forma más grave, podría causar la muerte.
Hospitalización necesaria del paciente o prolongación de una hospitalización existente
El ingreso de un sujeto en el hospital como paciente internado como consecuencia de un EA, incluso si el sujeto era dado de alta el mismo día, se calificó como hospitalización. Una consulta en urgencias no constituía una hospitalización.
Daba como resultado una discapacidad o incapacidad persistente o significativa
Un evento se calificaba como que da lugar a una discapacidad o incapacidad persistente o significativa, si implicaba una interrupción sustancial de la capacidad del sujeto para llevar a cabo las funciones de una vida normal. Esa definición no pretendía incluir experiencias de importancia médica relativamente menor o temporal.
Era una anomalía congénita o un defecto congénito, es decir, un EA que había tenido lugar en la infancia o el feto de un sujeto expuesto al fármaco del estudio antes de la concepción o durante el embarazo.
Era un evento médico importante que no satisfacía ninguno de los criterios anteriores, pero que podía poner en peligro al sujeto o requerir una intervención médica o quirúrgica para prevenir uno de los resultados enumerados anteriormente.
Más de uno de los resultados anteriores se podía aplicar a cualquier evento específico.
Idoneidad de las mediciones
Las mediciones de seguridad en ese estudio eran exámenes físicos de rutina, signos vitales, incidencia y gravedad de eventos adversos y procesos clínicos y de laboratorio.
Mediciones de la concentración de fármacos
Las características farmacocinéticas (PK) en sangre (plasma) se evaluaron después de cada dosis del medicamento en estudio. Todos los sujetos permanecieron sentados en posición erguida durante 4 horas después de la dosis. Las muestras de sangre se extrajeron 30 minutos antes de la dosis y 0,5, 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 5,0, 6,0, 8,0, 10,0, 12,0, 18,0 y 24,0 horas después de la dosis. Las muestras se recogieron en tubos de 6 mL Vacutainer® con tapa morada de K2-EDTA, adecuadamente etiquetados. Las muestras de sangre se centrifugaron a aproximadamente 3000 rpm a 4°C durante 10 minutos. A partir del plasma resultante, se extrajo exactamente 1 mL de cada muestra usando una pipeta y se colocó en un tubo de partes alícuotas que contenía 0,1% de p/v de ditioeritritol. La muestra se tapó y se agitó en vórtex durante aproximadamente 10 segundos usando un VWR Mini Vortexer a velocidad 6. Después de completar estos pasos, la muestra se congeló instantáneamente en un baño de isopropilo/hielo seco y se colocó en un congelador a -70°C hasta el análisis.
Se extrajeron aproximadamente 80 mL de sangre durante cada período de tratamiento (5 mL por punto temporal) para el análisis PK.
Farmacocinética:
El análisis farmacocinético (PK) de los datos de concentración-tiempo de BH4 en plasma se realizó utilizando métodos no compartimentalizados para obtener estimaciones de los siguientes parámetros PK:
Concentración plasmática máxima (Cmáx) y tiempo hasta la concentración máxima (Tmáx), obtenido directamente a partir de los datos sin interpolación;
Az, la constante de la tasa de eliminación terminal aparente, determinada por regresión logarítmica lineal de las concentraciones plasmáticas terminales;
Área bajo la curva de concentración plasmática-tiempo desde el momento cero hasta el momento de la última concentración medible [AUC (0-t)], calculada por el método trapezoidal lineal;
La semivida de eliminación aparente (t1/2), calculada como 0,693/Az;
Área bajo la curva de concentración plasmática-tiempo desde el tiempo 0 hasta el infinito [AUC (inf)] en donde AUC (inf) = AUC (0-t) Ct/Az y Ct es la última concentración medible.
Estimación de la tasa de absorción
A los sujetos se les administró una dosis oral o intravenosa de 10 mg/kg de BH4, seguida de mediciones en serie de
la concentración plasmática de biopterina total, para determinar la tasa de absorción de BH4 a partir del tracto gastrointestinal desde el área bajo la curva del aumento de la concentración plasmática de biopterina total (ACp)-tiempo (AAUC). Se había anticipado que se requería una dosis menor de BH4 cuando se administraba por vía intravenosa, en comparación con la BH4 administrada por vía oral para lograr el mismo nivel de biodisponibilidad. Por ejemplo, se pueden requerir 10 mg/kg de BH4 administrados por vía oral para lograr el mismo nivel de biodisponibilidad que 1 mg/kg de BH4 administrado por vía intravenosa. Debido a que la forma de administración mejoraba la biodisponibilidad, es posible que solo se requieran 5 mg/kg de BH4 para alcanzar el mismo nivel de biodisponibilidad que una dosis iv de 1 mg/kg de BH4.
La tasa de absorción de BH4 desde el tracto gastrointestinal se estimó a partir del área bajo la curva del aumento de la concentración plasmática de biopterina total (ACp)-tiempo (AAUC) después de la administración de BH4, utilizando las siguientes fórmulas:
Estimación de AUC
Tasa de absorción (%)=
(AAUC después de una dosis p.o. / AAUC después de una dosis i.v.) x (dosis i.v. / dosis p.o. x 100)
Métodos estadísticos:
La comparación de los parámetros farmacocinéticos Cmáx, AUC (0-t) y AUC (inf) para BH4 se llevó a cabo utilizando un modelo de análisis de varianza (ANOVA) con secuencia, sujeto dentro de secuencia, tratamiento y período como las variables de clasificación, utilizando los logaritmos naturales de los parámetros como las variables dependientes. Las comparaciones de interés eran entre el comprimido disuelto e intacto en estado de ayunas y el comprimido intacto en los estados de alimentación y en ayunas.
Los datos procedentes de todos los sujetos que completaron al menos dos períodos de estudio, se incluyeron en los análisis estadísticos de la PK. Todos los sujetos que recibieron al menos una dosis del fármaco del estudio, se incluyeron en los análisis de seguridad.
Todos los análisis de la PK y estadísticos asociados se realizaron utilizando SAS® para Windows® versión 9.1.3 o superior.
Para proporcionar una potencia suficiente para cumplir los objetivos del estudio, se consideró adecuado un tamaño de muestra de aproximadamente 30 sujetos, cada uno con 3 períodos de tratamiento, para proporcionar estimaciones de las diferencias comparativas de interés. No se realizó ningún cálculo formal del tamaño de la muestra.
Resultados
Farmacocinética
Comprimidos intactos frente a disueltos
Las concentraciones plasmáticas medias de BH4 eran menores cuando se administraba BH4 como un comprimido disuelto en comparación con un comprimido intacto (Figuras 23 y 24). La Cmáx media era mayor para el comprimido intacto al igual que los valores medios para AUC (0-t) y AUC (inf) (Figura 25). Las relaciones medias geométricas, entre comprimido intacto y disuelto, variaron de 118% a 121% y los límites superiores de los intervalos de confianza del 90% asociados eran superiores al 125% (Figura 26), lo que indica un aumento estadísticamente significativo en la absorción cuando el comprimido intacto se administraba con una comida rica en calorías y grasas en la diferencia de la absorción entre la administración del comprimido disuelto e intacto. La mediana y el intervalo de Tmáx eran esencialmente los mismos para los comprimidos disueltos e intactos (Figura 25), lo que sugiere que el aumento observado con el comprimido intacto era en el grado pero no en la tasa de absorción.
Efecto de los alimentos ricos en calorías y grasas en la absorción de fármacos
Como era de esperar, la administración del comprimido intacto con una comida estándar rica en grasas y rica en calorías daba como resultado un aumento sustancial de las concentraciones medias en plasma de BH4 (Figura 23) y los valores medios para Cmáx, AUC (0-t) y AUC (inf) (Figura 25). Las relaciones medias geométricas (entre con alimentación y en ayunas) oscilaron entre el 126% y el 139% (Figura 26) y, en consecuencia, los límites superiores de los intervalos de confianza asociados del 90% eran superiores al 125%, lo que indica una diferencia estadísticamente significativa en el efecto de los alimentos sobre la absorción, en comparación con comprimidos intactos. La mediana y el intervalo de Tmáx eran esencialmente los mismos en condiciones de alimentación y en ayunas (Figura 25), lo que sugiere que el aumento observado con la comida era en el grado pero no en la tasa de absorción.
Seguridad:
No hubo eventos adversos graves (EAGs) en este estudio. Cinco (5) sujetos informaron de un total de 9 eventos adversos (EAs). Ocho (8) de esos 9 EAs se evaluaron como leves y 1 se evaluó como de gravedad moderada. El EA más común era dolor de cabeza; 1 sujeto experimentó dolor de cabeza moderado que se evaluó como no relacionado con el fármaco del estudio, y un sujeto experimentó dolor de cabeza leve en dos ocasiones, las cuales se evaluaron como posiblemente relacionadas. En total, se consideró que cinco eventos no estaban relacionados y cuatro estaban posiblemente relacionados con el fármaco del estudio. Las evaluaciones de salida del estudio, las evaluaciones de ECG y los exámenes físicos se completaron sin hallazgos clínicamente significativos.
Conclusiones:
La administración de BH4 como un comprimido intacto daba lugar a un aumento de aproximadamente el 20% en el grado de absorción, en comparación con un comprimido disuelto.
La administración de BH4 como un comprimido intacto con una comida rica en calorías y grasas en condiciones de alimentación, daba como resultado un aumento de aproximadamente un 30% en el grado de absorción, en comparación con las condiciones de ayuno.
No se identificaron problemas clínicamente significativos ni problemas de seguridad relacionados con los parámetros de seguridad en esa población del estudio. No hubo EAs considerados graves en este estudio. Entre los 9 EAs comunicados, todos menos uno, un caso de dolor de cabeza, eran leves y se determinó que no estaban relacionados con el fármaco del estudio. Los casos de fatiga y dolor de cabeza eran los únicos EAs que estaban posiblemente relacionados con el fármaco del estudio, pero se evaluaron como de gravedad leve.
Ejemplo 4
Enfoques de formulación para mejorar la biodisponibilidad de BH4
Se seleccionaron dos formulaciones de control (formulación intravenosa de BH4 y comprimido de BH4 para solución oral) y seis formulaciones de prueba para someterlas a ensayo en estudios con animales. Cada prototipo de formulación contenía 80 mg o 100 mg de BH4.
Formulaciones intravenosas de BH4
La Tabla 3 especifica la composición de una formulación intravenosa. BH4 se hizo pasar a través de un tamiz de acero inoxidable de malla n° 20 antes del uso, mientras que se utilizó manitol tal como se recibió. Esa formulación se introdujo en forma de polvo en un frasco y se reconstituyó con agua estéril para inyección antes de la administración. Cada frasco contenía 100 mg de BH4 y 5 g de manitol en un frasco transparente de copoliéster de tereftalato de polietileno (PETG) con un tapón blanco de rosca de polietileno de alta densidad (HDPE). Antes de la administración, la formulación se constituyó con 100 mL de agua estéril para inyección para obtener una concentración final de 1 mg/mL. La formulación IV se suministró como un polvo seco en un frasco, y cada frasco contenía el API y manitol. El polvo se disolvió en agua estéril para inyección y se filtró antes de la administración por vía IV.
Tabla 3 Composición de la formulación iv de BH4

Comprimido de BH4 para solución oral
La Tabla 4 especifica la composición de una formulación de solución oral. Se colocaron diez (10) comprimidos de BH4 (100 mg) en un frasco de PETG graduado de 125 mL con un cierre de HDPE blanco. Antes de la administración, la formulación se constituyó con 100 mL de agua estéril para inyección para obtener una concentración final de 10 mg/mL.
Tabla 4 Composición de un comprimido de BH4, 100 mg


Prototipo de formulación para ralentizar la motilidad gastrointestinal
La Tabla 5 especifica la composición de un prototipo de tiempo de vaciado gástrico retardado. BH4 se hizo pasar a través de un tamiz de acero inoxidable de malla n° 20 antes del uso. El Capmul GMO-50 se fundió en un baño de agua a 37°C. Se pesaron BH4 y ácido ascórbico y se añadieron lentamente al Capmul fundido mientras se agitaba vigorosamente. La dispersión sólida se añadió gota a gota a una cápsula de tamaño n° 2 usando una pipeta. Se colocaron tres cápsulas llenas en un frasco de polietileno de alta densidad (HDPE) de 100 cc con un cierre sellado mediante inducción térmica.
Tabla 5 Composición de formulación oral de cápsulas de BH4 con tiempo de vaciado gástrico retardado

Prototipo bioadhesivo
La Tabla 6 especifica la composición de un prototipo bioadhesivo. Todos los materiales, excepto Carbopol 71G, se hicieron pasar a través de un tamiz de acero inoxidable de malla n° 20. Todos los materiales se pesaron y se añadieron a una bolsa de plástico que tenía un cierre de cremallera, que luego se agitó durante unos minutos hasta que la mezcla pareció uniforme. El polvo se comprimió en un comprimido utilizando un instrumental B estándar, redondo, cóncavo y de cara plana de 0,6 cm (1/4“) en una prensa manual de Globe Pharma MTCM-I a 41 bar (600 psi). Tres comprimidos junto con una bolsa desecante de gel sílice se envasaron en 100 cc de HDPE con cierre sellado mediante inducción térmica.
Tabla 6 Composición de la formulación de comprimidos orales bioadhesivos de BH4

Prototipo de liberación sostenida
La Tabla 7 especifica la composición de un prototipo de liberación sostenida sometido a ensayo en el mono. Todos los materiales, excepto Methocel K100M Premium CR, se hicieron pasar a través de un tamiz de acero inoxidable de malla n° 20. Todos los materiales se pesaron y se añadieron a una bolsa de plástico con cierre de cremallera, que luego se agitó durante unos minutos hasta que la mezcla pareció uniforme. El polvo se comprimió formando un comprimido usando un instrumental B estándar, redondo, cóncavo y de cara plana de 0,6 cm (1/4“) en una prensa manual de Globe Pharma MTCM-I a 82 bar (1200 psi). Los comprimidos junto con una bolsa desecante de gel sílice se envasaron en 100 cc de HDPE con cierre sellado mediante inducción térmica.
Tabla 7 Composición de la formulación de comprimidos de liberación sostenida de BH4

Prototipo de polímero donante de protones
La Tabla 8 especifica la composición de un prototipo de polímero donante de protones sometido a ensayo en el mono. Todos los materiales, excepto Eudragit L100-55 y Kollidon CL, se tamizados previamente utilizando un tamiz de acero inoxidable de malla n° 20. Todos los materiales se pesaron y se añadieron a una bolsa de plástico con cierre de cremallera, que luego se agitó durante unos minutos hasta que la mezcla pareció uniforme. Se introdujo una cantidad de polvo previamente pesada en una cápsula de tamaño n° 2.
Se preparó una solución de recubrimiento disolviendo Eudragit L100-55 y Carbowax PEG 4600 en alcohol etílico. Se pesaron Eudragit L100-55 y Carbowax PEG 4600 y se añadieron a un frasco graduado de copoliéster de tereftalato de polietileno (PETG) de 125 mL. El alcohol etílico se añadió al frasco de PETG y se colocó en un baño de agua a 40°C sometiendo a ultrasonidos hasta que la solución era transparente.
Las cápsulas rellenas de polvo se sumergieron manualmente en la solución de recubrimiento y se dejaron secar a 40°C durante 20 minutos. Las cápsulas secas se pesaron y luego se hicieron rodar sobre Syloid FP244 para eliminar la pegajosidad residual. Se envasaron tres cápsulas en un frasco de HDPE de 100 cc con un cierre sellado mediante inducción térmica.
Tabla 8 Composición de la formulación de cápsulas donantes de protones para BH4

1 Después del recubrimiento de la cápsula y el secado en el horno a 40°C, la cápsula gana aproximadamente del 1 al 3% en peso con el recubrimiento de polímero,
ND = No determinado
Sistema de administración flotante
La Tabla 9 especifica la composición de un sistema de administración flotante. Todos los materiales, excepto el Eudragit L100-55, se hicieron pasar a través de un tamiz de acero inoxidable de malla n° 20. Este prototipo de comprimido comprendía tres capas; la capa intermedia contenía la sustancia farmacéutica, que estaba intercalada entre dos capas externas insolubles en agua. Los materiales internos y externos se pesaron y se añadieron por separado a bolsas de plástico con cierres de cremallera, que luego se agitaron hasta que las mezclas parecieron uniformes.
Se pesaron las dos capas externas (12 mg cada una) y la capa interna (14,5 mg). Una de las capas externas se añadió a la prensa, seguida de la capa interna y luego la última capa externa. Las capas se comprimieron formando un comprimido usando un instrumental B, redondo, cóncavo y de cara plana de 0,5 cm (3/16“) en una prensa manual de Globe Pharma MTCM-I a 14 bar (200 psi).
Se preparó una solución de recubrimiento disolviendo Ethocel y PEG 4600 en una mezcla de alcohol etílico y agua purificada. Los ingredientes se añadieron a un frasco de PETG, que se mezcló y se colocó en un baño de agua a 40°C sometido a ultrasonidos hasta que la solución se volvió transparente.
Los comprimidos se sumergieron manualmente en la solución de recubrimiento y se dejaron secar durante 20 minutos a 40°C. Cada comprimido se volvió a pesar después del recubrimiento. Se colocaron siete (7) comprimidos dentro de cada una de las cápsulas alargadas de tamaño n° 2. Se envasaron tres cápsulas en un frasco de HDPE de 100 cc con un cierre de sellado mediante inducción térmica.
Tabla 9 Composición de la formulación de dosificación flotante de BH4

1 Después del recubrimiento de la cápsula y el secado en el horno a 40°C, la cápsula gana aproximadamente de un 3 a un 8% en peso en el recubrimiento de polímero,
ND = No determinado
Sistema de administración flotante generador de gas
La Tabla 10 especifica la composición de un sistema de administración flotante generador de gas. Esta formulación estaba compuesta por un comprimido con núcleo que contenía el fármaco rodeado por una capa externa generadora de gas. Todos los materiales, excepto el bicarbonato de sodio y Methocel K100M CR, se tamizaron previamente usando un tamiz de acero inoxidable de malla n° 20. Los materiales del núcleo interior y de la capa externa se pesaron y se añadieron por separado a bolsas de plástico con cierres de cremallera, que se cerraron y agitaron hasta que la mezcla se volvió uniforme. El polvo mezclado para el núcleo interno (35 mg) se comprimió formando un comprimido usando un instrumental B redondo, biselado y de cara plana de 0,3 cm (1/8“) en una prensa manual de Globe Pharma MTCM-I a 55 bar (800 psi).
Se preparó una solución de recubrimiento disolviéndola usando Ethocel y PEG 4600 en alcohol etílico. Los comprimidos del núcleo interno se sumergieron manualmente en la solución de recubrimiento y se dejaron secar durante 20 minutos a 40°C. Se pesó el polvo mezclado para la capa externa (40 mg). Se añadió la mitad a la prensa, seguida del comprimido con núcleo interior y luego la segunda mitad de la capa externa. El comprimido se comprimió usando un instrumental B redondo, biselado, de cara plana de 0,5 cm (3/16“) en una prensa manual de Globe Pharma MTCM-I a 55 bar (800 psi). Se colocaron cuatro (4) comprimidos en cada cápsula de tamaño n° 2. Tabla 10 Composición de la formulación para una dosificación de BH4 flotante generadora de gas

1 Después del recubrimiento de la cápsula y el secado en el horno a 40°C, la cápsula gana peso en el recubrimiento de polímero.
ND = No determinado
Prototipo de gránulos bioadhesivos
La Tabla 11 especifica la composición de un prototipo de gránulos bioadhesivos. Todos los materiales, excepto Methocel K100M CR, se tamizaron previamente utilizando un tamiz de acero inoxidable de malla n° 20. Todos los materiales, excepto el estearil fumarato de sodio (PRUV), se pesaron y se colocaron en un recipiente granulador de tamaño n° 1 (LB Bohle Mini Granulator BMG). El polvo se mezcló a una velocidad impulsora de 300 rpm y una velocidad del triturador de 2500 rpm, durante cinco minutos, hasta que la mezcla se volvió uniforme. Manteniendo las velocidades del impulsor y del triturador, se añadió a la mezcla gota a gota 5 mL de alcohol etílico hasta que se formaron gránulos. La masa húmeda se retiró del recipiente de granulación y se tamizó a través de un tamiz de acero inoxidable de malla 18. Los gránulos se recogieron y se colocaron en un horno a 40°C para que se secaran durante una hora. Se determinó que la pérdida por secado de los gránulos era del 1,93% después de una hora de secado. Los gránulos se pesaron y se colocaron en una bolsa de plástico con cierre de cremallera. Se añadió estearil fumarato de sodio (PRUV) a los gránulos secos en la bolsa. La bolsa se cerró y se agitó hasta que el estearil fumarato de sodio (PRUV) aparecía distribuido uniformemente entre los gránulos. Se pesaron los gránulos (134 mg).
Se llenaron cápsulas alargadas de tamaño 2 con porciones de los gránulos alternando con gotas de aceite vegetal parcialmente hidrogenado (350 pl). Se envasaron tres cápsulas en un frasco de HDPE de 100 cc con un cierre de sellado mediante inducción térmica.
Tabla 11 Composición de la formulación de cápsulas de gránulos bioadhesivos de BH4

Liberación de los fármacos in vitro
Las pruebas de liberación in vitro de los fármacos a partir de comprimidos, se realizaron de acuerdo con las especificaciones del aparato II de USP 27, utilizando un medidor de disolución Distek 2100C (Distek, Inc., North Brunswick, Nueva Jersey), junto con un sistema de espectroscopia de UV visible de Agilent (Agilent Technologies, Santa Clara, CA). El medio de disolución usado para la prueba de liberación de BH4 eran 900 mL de HCl 0,1 N. Durante la prueba de disolución, el medio de cada recipiente se mantuvo a 37°C ± 0,5°C y se agitó a 50 rpm. Se tomó un volumen de muestra de 5 mL en puntos de tiempo predeterminados. Para determinar la concentración de BH4 en las muestras, se diluyeron 250 pL de cada muestra con 500 pL de HCl 0,1 N y se midió la absorción a 265 nm utilizando un espectrómetro UV (8453 Espectrofotómetro UV-Visible, Agilent Technologies, Santa Clara, CA). Los datos se recopilaron utilizando el programa informático ChemStation (Rev. A.09.01 [76], Agilent Technologies, Santa Clara, CA). Todas las pruebas de disolución se realizaron por triplicado.
Prueba de flotabilidad del comprimido
La flotabilidad de los comprimidos del prototipo flotante se determinó en primer lugar colocando los comprimidos en vasos de plástico con 25-50 mL de HCl 0,1 N. Esa prueba determinaba el tiempo necesario para que los comprimidos flotaran, asi como la duración de su flotación sin agitación. Los prototipos que flotaban durante al menos cuatro horas, se sometieron a pruebas de disolución. Durante la prueba de disolución, se determinó la flotabilidad de los comprimidos usando el método de paleta con una tasa de rotación de 50 rpm. El estado de los comprimidos se comprobó visualmente en varios momentos.
Prueba de desintegración
La prueba de desintegración se llevó a cabo de acuerdo con las especificaciones de la prueba de desintegración de USP-27, utilizando un medidor de la desintegración de la serie Distek 3100 (Distek Inc., North Brunswick, NJ). El medio de desintegración utilizado era 900 mL de HCl 0,1 N o 900 mL de fosfato potásico 0,2 M pH 5,8. Durante la prueba de desintegración, el medio en los recipientes se mantuvo a 37° ± 0,5°C. Los comprimidos y las cápsulas se inspeccionaron visualmente en busca de desintegración.
Prueba de dureza de los comprimidos
La dureza de los comprimidos se determinó usando un medidor de la dureza de comprimidos, Dr. Schleuniger Pharmatron 8M (Dr. Schleuniger® Pharmatron Inc., Manchester, NH). Los comprimidos se colocaron en la abrazadera del medidor de dureza y se midió la dureza en kilopondios (Kp).
Espesor del comprimido
El espesor de los comprimidos se midió utilizando un indicador Mitutoyo Digimatic (Mitutoyo Absolute, Dr. Schleuniger Pharmatron Inc., Manchester, NH). Los comprimidos se colocaron debajo del medidor de espesor y el valor indicado se registró en milimetros (mm).
Resultados y discusión
Se desarrollaron varios prototipos basados en tres conceptos: retención gástrica, polímero donante de protones para cambiar el pH intestinal y formas de dosificación de liberación sostenida. Las secciones siguientes describen el desarrollo de la formulación de cada prototipo.
Formulación intravenosa de BH4 - Después de la constitución con agua estéril, la solución resultante era isotónica, pH 3,2 y contenía 1 mg/mL de BH4, y era apta para una administración intravenosa después de una filtración estéril a través de un filtro de 0,22 micras. La estabilidad de la solución de 1 mg/mL almacenada a temperatura ambiente se analizó mediante HPLC cada hora, durante tres horas. Las muestras de soluciones más antiguas se almacenaron luego a -20°C y se analizaron mediante HPLC después de 2 semanas. La Figura 27 indica que la solución era estable a temperatura ambiente durante al menos 3 horas después de la constitución y que era estable durante al menos 2 semanas durante un almacenamiento a -20°C.
Comprimido de BH4 para solución oral
Cada frasco estaba envasado de forma que contuviera diez (10) comprimidos de BH4, 100 mg. Se añadieron cien (100) mL de agua purificada o agua estéril para inyección al contenido de cada frasco. Tras agitar vigorosamente el frasco, los comprimidos se desintegraron rápidamente en 5 minutos. La solución resultante contenía 10 mg/mL de BH4 para administración oral. No todos los ingredientes del comprimido eran solubles y, aunque la solución final parecía turbia o traslúcida, el ingrediente farmacéutico activo estaba completamente disuelto y las partículas finas eran ingredientes inactivos poco solubles.
Prototipo de formulación para ralentizar la motilidad gastrointestinal
Esta formulación para cápsula se compone de BH4 y ácido ascórbico disperso en un derivado de ácido graso semisólido (mono/dioleato de glicerilo, punto de fusión de 86°F (30°C)). También se seleccionó el mono/dioleato de glicerilo (MOG) porque el MOG es químicamente compatible con BH4. El perfil de disolución presentado en la Figura 28 mostraba que más del 90% del fármaco se liberaba en 2 horas y el perfil de disolución permanecía sin cambios después de almacenar las cápsulas a 40°C durante 57 días.
La dispersión del fármaco en OMG fundido, un semisólido, se introdujo manualmente en cápsulas de gelatina dura. La densidad del semisólido es superior a 1 g/mL y era posible introducir al menos una dosis de 80 mg con una carga de fármaco del 25% en una cápsula de tamaño n° 2. Se espera que una cápsula de tamaño n° 0 pueda contener al menos 200 mg de fármaco usando la misma formulación. Se observó una fuga de ácido graso desde la cápsula durante el almacenamiento a 40°C. Preferiblemente, las cápsulas o las formulaciones de cápsulas de gelatina blanda se fijaron para evitar fugas de ácido graso durante el almacenamiento.
Prototipo bioadhesivo
Muchos bioadhesivos se preararn a base de polímeros sintéticos o naturales. La mayoría de los polímeros bioadhesivos sintéticos actuales son derivados del poli(ácido acrílico) o de la celulosa. Ejemplos de polímeros a base de poli(ácido acrílico) incluyen pero no se limitan a carbopol, policarbofilo, poli(ácido acrílico) (PAAc), etc. Los celulósicos incluyen pero no se limitan a hidroxipropilcelulosa e hidroxipropilmetilcelulosa (HPMC). Se desarrollaron dos prototipos bioadhesivos para someterlos a ensayo en estudios con animales. El primer prototipo era una formulación de comprimido bioadhesivo y el segundo una cápsula que contenía gránulos bioadhesivos.
Se seleccionaron polímeros de policarbofilo y carbómero para el desarrollo del primer prototipo de comprimido bioadhesivo. Carbopol 71 G es una forma granular de carbómero y tiene buenas propiedades de flujo en polvo. Todos los lotes de comprimidos fabricados eran de buena calidad, con un contenido en fármaco aceptable (evidente por una liberación de fármaco cercana al 100% en los perfiles de disolución) y una dureza aceptable. La Tabla 12 especifica el peso, el espesor y la dureza de un comprimido representativo del prototipo bioadhesivo que contiene carbómero y policarbofilo.
Tabla 12 Peso, espesor y dureza de un comprimido representativo para el prototipo bioadhesivo que contiene carbómero y policarbofilo

Se utilizaron polímeros de carbómero y HPMC para el desarrollo de los segundos gránulos bioadhesivos. Se seleccionó HPMC porque se utiliza como un sistema hidrocoloide de baja densidad y de liberación controlada del fármaco, independientemente del pH. Los gránulos se prefirieron a los comprimidos para aumentar la posibilidad de una bioadhesión al aumentar el área de superficie de la forma de dosificación. Para facilitar la separación de la cápsula llena de gránulos en medio de disolución, los gránulos se recubrieron parcialmente con aceite hidrogenado. Sin el recubrimiento de aceite, los gránulos se hidrataban y formaban una matriz en forma de cápsula sin desintegrarse en gránulos individuales.
Los perfiles de liberación de los dos prototipos bioadhesivos (comprimido y gránulos) se muestran en la Figura 29, que muestra que el perfil de liberación del comprimido era más largo que el de los gránulos. La liberación de fármaco era aproximadamente del 90% en cuatro horas y del 95% en una hora para las formas de dosificación bioadhesivas en comprimidos y gránulos, respectivamente. Después de almacenar a 40°C con humedad ambiental, durante un mes sin protección contra la humedad (sin sellado mediante inducción térmica), el prototipo de comprimido mostraba una desaceleración en la disolución del fármaco (Figura 29). Para los prototipos que contenían carbómero, se debían tomar precauciones para proteger contra la humedad, para proteger el comprimido de una posible hidratación prematura.
Prototipo de liberación sostenida
La hidroxipropilmetilcelulosa (HPMC) se utiliza como vehículo hidrófilo para la preparación de sistemas de administración oral controlada de fármacos (Colombo, Adv. Drug Deliv. Rev., 1993, 11, 37). Se sabe que las matrices HPMC controlan la liberación de una variedad de fármacos (Chattaraj, et al. Drug Develop. Ind. Pharm., 1996, 22, 555; Pabon, et al., Drug Develop. Ind. Pharm., 1992, 18, 2163; Lee, et al., Drug Develop. Ind. Pharm., 1999, 25, 493; Basak, et al., Indian J. Pharm. Sci., 2004, 66, 827; Rajabi-Siabhoomi, et al., J. Pharm. Pharmacol., 1992, 44, 1062). En este estudio se evaluaron varios grados de viscosidad de HPMC (K4M, K15M y K100 M) para controlar la liberación de BH4. Los perfiles de disolución de los comprimidos realizados con varios grados de HPMC, se muestran en la Figura 30. Los perfiles de liberación de fármaco eran similares al 20% de HPMC, independientemente del grado de viscosidad; más del 80% del fármaco se liberaba en 2 horas. Cuando el polímero de HPMC se exponía a un medio acuoso, experimentaba una rápida hidratación y relajación de la cadena para formar una capa de gel (Naruhashi et al., Pharm Res. 2003,19: 1415-1421). Es posible que la HPMC al 20% no forme una capa de barrera de gel sustancial para ralentizar significativamente la liberación de BH4.
Los perfiles de disolución de comprimidos producidos con concentraciones variables (20% a 40%) con un grado de viscosidad elevada de HPMC (Methocel K100M CR), se presentan en la Figura 30. Se encontró que un comprimido que contenía de un 35% a 40% de Methocel K100M CR, retardaba la liberación del fármaco hasta cuatro horas, mientras que con un 20% de HPMC se liberaba el fármaco en dos horas (Figura 31). Se seleccionó un comprimido que contenía 35% de HPMC (Methocel K100M) como prototipo para la prueba en estudios con animales porque contenía la menor cantidad de HPMC requerida para ralentizar la liberación del fármaco hasta cuatro horas. Como tales, los comprimidos eran de buena calidad con un contenido en fármaco aceptable, como lo demuestra la liberación de fármaco cercana al 100% en los perfiles de disolución.
Prototipo de polímero donante de protones
Para aumentar la absorción oral de BH4, una estrategia consiste en estabilizar el fármaco disminuyendo el pH del intestino delgado proximal. Para manipular el pH luminal intestinal, se seleccionó Eudragit L100-55, un polímero liberador de protones, comúnmente utilizado para el recubrimiento entérico. Este polímero no es soluble en condiciones ácidas y se vuelve soluble y libera protones en condiciones desde débilmente ácidas (pH > 5,5) a alcalinas, debido a sus grupos carboxilo, controlando de ese modo que el pH luminal intestinal sea ácido. Naruhashi y col. (2003) encontraron que el pH en el lumen disminuía de una manera dependiente de la concentración de Eudragit L100-55 y que la absorción de cefadroxil y cefixima desde el asa ileal aumentaba en presencia del polímero ácido (Nozawa et al., J. Pharm Sci. 2003, 92 (11), 2208-2216). Nozawa, et al (2003) mostraron que Eudragit disminuía el pH en las asas intestinales y aumentaba la cinética de desaparición tanto de cefadroxil como de cefixima desde las asas.
Las formulaciones en polvo que contenían BH4 y Eudragit L100-55, como se muestran en la Tabla 8, se comprimieron formando comprimidos y se introdujeron en cápsulas. La formulación del comprimido liberaba aproximadamente un 27% de fármaco en una hora en un fluido gástrico simulado (SGF) durante la prueba de disolución. Sin embargo, durante la prueba de desintegración, el comprimido permanecía intacto en el SGF y tampón fosfato (PB) a pH 5,8 durante al menos 2 horas. Incluso en presencia de un superdesintegrante (crospovidona o croscarmelosa), el comprimido no se desintegraba. Es posible que el fármaco acidifique Eudragit, creando un entorno de micropH bajo, de modo que el polímero permanece unido e insoluble.
La formulación de fármaco-Eudragit con polvo introducido en la cápsula se desintegraba rápidamente en SGF. Para dirigir la liberación de protones en el intestino proximal, se aplicó un recubrimiento entérico a la cápsula. Después del recubrimiento de la cápsula y el secado en el horno a 40°C, la cápsula ganaba aproximadamente del 1 al 3% en peso en recubrimiento de polímero. Cuando se sometía a ensayo usando el aparato II de disolución USP (paleta), medio de disolución HCl 0,1 N mantenido a 37°C con una velocidad de rotación de 50 rpm, la cápsula recubierta liberaba aproximadamente el 25% de fármaco en una hora. Después de 1 hora de tratamiento previo con ácido (HCl 0,1 N), la cápsula recubierta se colocó en un aparato de desintegración USP con 500 mL de tampón fosfato a pH 5,8 mantenido a 37°C, la cápsula recubierta se desintegraba en aproximadamente 1 hora. El prototipo de cápsula con recubrimiento entérico se prefería al comprimido o la cápsula sin recubrimiento porque era más probable que la cápsula con recubrimiento entérico liberara el polímero liberador de protones en el sitio diana.
Sistema de entrega flotante
Se desarrollaron dos sistemas de entrega flotantes. El primer prototipo era una forma de dosificación unitaria múltiple flotante; los fines de esa forma de dosificación eran aumentar la posibilidad de que una de las unidades permaneciera en la región gástrica y, por lo tanto, prolongara el tiempo de residencia gástrica de los fármacos. Esa forma de dosificación consistía en siete comprimidos de capa triple en una cápsula; la capa intermedia contenía el fármaco, que estaba intercalado entre dos capas externas insolubles en agua (Figura 32). Las capas externas contenían ácido esteárico, un ácido graso hidrófobo e insoluble en agua, que proporcionaba la flotabilidad necesaria al comprimido flotante. Cada comprimido se revistió manualmente con una solución alcohólica de etilcelulosa y polietilenglicol PM 4600 (PEG). La etilcelulosa formaba una película insoluble en agua alrededor del comprimido y el PEG, que actuaba como un formador de poros, modulaba la tasa de liberación. Los perfiles de disolución de comprimidos recubiertos con etilcelulosa y diversas concentraciones (20% a 40%) de soluciones de PEG, se presentan en la Figura 33. Se observó que el comprimido de capa triple recubierto lograba una cinética de liberación cercana al orden cero. Como se esperaba, la tasa de disolución del fármaco aumentaba a medida que aumentaba la concentración de PEG. Los comprimidos flotaban en un medio gástrico simulado durante al menos cuatro horas durante los estudios de disolución. La Tabla 9 muestra la composición de la formulación sometida a ensayo en estudios con animales.
El segundo prototipo era una forma de dosificación generadora de gas. Se había formulado de tal manera que cuando entraba en contacto con contenidos gástricos ácidos, se liberaba dióxido de carbono y quedaba atrapado en los hidrocoloides hinchados, lo que proporcionaba flotabilidad a la forma de dosificación (Figura 33). Esa formulación flotaba en un medio gástrico simulado durante al menos cuatro horas durante los estudios de disolución. Sin embargo, para que un sistema de ese tipo funcione de manera uniforme, los comprimidos deben producirse en un entorno de humedad reducida para evitar una reacción ácida y básica prematura. Podía haber una interacción potencial entre la BH4 y el bicarbonato de sodio en el comprimido durante el almacenamiento. Por estas razones, esa forma de dosificación no se sometió a ensayo en estudios con animales.
Se desarrollaron seis prototipos de formulaciones de prueba para estudios de biodisponibilidad animal que incorporaban varios enfoques de formulación, incluyendo un polímero donante de protones para disminuir el pH intestinal, formas de dosificación de retención gástrica y formulaciones de liberación sostenida,.
Ejemplo 5
Biodisponibilidad de nuevas formulaciones de BH4
El objetivo de este estudio era mejorar la absorción de BH4 mediante el desarrollo de formas de dosificación que aumentan el tiempo de residencia del fármaco en el tracto gastrointestinal (GI).
Métodos: Se utilizaron tres monos de tipo cynomolgus, sanos que pesaban entre 3 y 4 kg en un estudio abierto, no cruzado de 8 períodos, para determinar la biodisponibilidad de siete formulaciones en comparación con una formulación de BH4 de control disuelta. Después de un ayuno nocturno, los monos recibieron, en distintas ocasiones, una dosis única de 80 mg de la misma nueva formulación por vía oral o intravenosa, con un intervalo de al menos un período de aclaramiento de 1 semana entre las diversas nuevas formulaciones estudiadas. Para la administración intravenosa, se recogieron muestras de sangre antes de la dosificación y luego 5, 15 y 30 minutos y 1,0, 2,0, 4,0, 6,0, 8,0, 12 y 24 horas después de la dosis. Para la administración oral, se tomaron muestras de sangre antes de la dosificación y luego 15 y 30 minutos y 1,0, 2,0, 3,0, 4,0, 6,0, 8,0, 12 y 24 horas después de cada dosis. Después de la separación del plasma mediante centrifugación, partes alícuotas de 200 pL de cada muestra, se transfirieron rápidamente a tubos individuales que contenían DTE al 0,1% y se congelaron a -70°C hasta que estuvieran listas para el ensayo de L-biopterina total.
Formulaciones del estudio: Las formulaciones administradas se encuentran en la Tabla 13. Tres de las formulaciones se diseñaron conceptualmente para tener retención gástrica mediante mecanismos bioadhesivos o flotantes para aumentar el tiempo de residencia GI (gránulos flotantes de múltiples partículas a base de carbómero y gránulos bioadhesivos). Otros conceptos se basaban en ralentizar la motilidad GI para aumentar el tiempo de residencia de la formulación (monooleato de glicerilo), reducir el pH del intestino delgado y, por lo tanto, mejorar la estabilidad química de BH4 para permitir una absorción del fármaco intacto (bomba de protones) o una formulación de liberación sostenida para averiguar si mejora la absorción.
Tabla 13


Ensayo en plasma para biopterina: Las concentraciones de BH4 en plasma se determinaron mediante el uso de un método de LC/MS/MS de fase inversa, específico y validado. La curva estándar era lineal en el intervalo de concentración de 50 ng/mL a 2500 ng/mL. El límite inferior de cuantificación para L-biopterina era de 50 ng/mL con precisión intradiaria mostrada por coeficientes de variación menor al 5%. La L-biopterina es estable en plasma de mono congelado, estabilizado con DTE al 0,1% a -70°C hasta que se analiza. Las concentraciones de BH4 se calcularon a partir de las concentraciones de L-biopterina determinadas.
Análisis farmacocinético y estadístico: Se determinaron los parámetros farmacocinéticos para la BH4 plasmática después de la administración de las formulaciones orales e intravenosas. Los parámetros farmacocinéticos se proporcionan en la Tabla 14.
Tabla 14

Resultados
El objetivo de este estudio era identificar formulaciones que mejoran la biodisponibilidad de BH4 en comparación con la formulación de comprimido disuelto de control. Los perfiles de concentración plasmática media de BH4-tiempo de las diversas formas de dosificación y la formulación de control después de la administración oral de BH4, se muestran en la Figura 35, y los parámetros farmacocinéticos de BH4 obtenidos a partir de los perfiles de concentración plasmática de fármaco-tiempo se proporcionan en la Tabla 14. La formulación de control (fase 2) es el comprimido disuelto.
Tal y como se muestra en la Figura 35, la formulación de monooleato de glicerilo proporcionaba la AUCúltima más
elevada y la AUC~ que eran 716 ng-h/mL y 858 ng-h/mL respectivamente. La formulación de comprimido de BH4 de control disuelto mostraba una AUCúltima y una AUC que eran 641 ng-h/mL y 805 ng-h/mL respectivamente (Tabla 14). El orden de clasificación de las formulaciones, desde la más disponible a la menos biodisponible, es: monooleato de glicerilo > comprimido disuelto > comprimido de polímero bioadhesivo > comprimido de liberación sostenida > formas de dosificación flotantes > producto en cápsula de granulaciones bioadhesivas > producto en cápsula de donante de protones.
Ejemplo 6
Preparación de una formulación intravenosa de tetrahidrobiopterina
Evaluación de la estabilidad de la formulación previa
En general, el objetivo de este estudio era evaluar la estabilidad de BH4 en soluciones tampón con pH de 1 a 7 (véase la Tabla 15) y en presencia y ausencia de antioxidantes y con o sin gas inerte en las soluciones de reacción (véase la Tabla 16).
Tabla 15 Componentes y composición de soluciones tampón que se van a utilizar en los estudios de estabilidad de la formulación previa de BH4


Tabla 16 Composición de las soluciones tampón para los estudios de estabilidad que contienen BH4 con o sin antioxidante y si están sometidas a burbujeo de gas o no 5*10

Más específicamente, se evaluó la influencia de combinar dos antioxidantes en presencia o ausencia de gas inerte a pH 4 para auxiliar a la formulación de un producto líquido, y a pH 7 para determinar la contribución de la inestabilidad a pH fisiológico a la baja biodisponibilidad del compuesto en monos y seres humanos (véanse las Tablas 17 y 18). Se espera que la estabilidad de BH4 dependa de la temperatura. Por lo tanto, la estabilidad del compuesto se evaluó a 2-8°C, 25°C, 30°C y 37°C para ayudar a determinar la vida útil predictiva a largo plazo del compuesto a diferentes temperaturas. Una determinación de la estabilidad del compuesto a una temperatura fisiológica de 37°C, proporciona datos para ayudar a estimar la vida útil de la estabilidad de una forma de dosificación oral formulada en las regiones de absorción del tracto GI.
Tabla 17 Composición de soluciones tampón para el estudio de la estabilidad de BH4 a pH 4

T abla 18 Composición de soluciones tampón para el estudio de la estabilidad de BH4 a pH 7
Los tiempos de muestreo propuestos para los estudios que se van a realizar en varias soluciones tampón, se estimaron comparando la semivida de un solo estudio a pH 3,1 con los datos obtenidos por Davis et al. (1988; Eur. J. Biochem. 173, 345-351, (1988)), en tampones Tris y fosfato a pH 6,8. El estudio de estabilidad de una solución a pH 3,1 proporcionaba un t1/2 estimado de 17769 min (12,3 días) y el trabajo de Davis et al. proporcionaba un t1/2 de 10 min en tampón fosfato a pH 6,8 y 14 min en tampón Tris a pH 6,8. Estos dos estudios sugieren una reducción de un orden de magnitud en la semivida (es decir, un aumento de la reactividad de un orden de magnitud) de BH4 para cada vez que aumenta el pH (véase la Tabla 19). Basándose en esta aproximación, inicialmente se tomaron muestras de soluciones desde pH 1,2 a pH 3 semanalmente y se hicieron correcciones en el tiempo de muestreo si era necesario después de recoger los 2 primeros puntos de datos. Los tiempos de muestreo estimados a 25°C se proporcionan en la Tabla 19.
Tabla 19 Tiempos de muestreo sugeridos con diversos pH basados en la semivida medida de BH4 y las semividas teóricas obtenidas a partir de las mismas

a t1/2 estimado se basa en cambiar por un orden de magnitud, la semivida obtenida a pH 3,0 para cada cambio de una vez en el pH. El pH <3 aumenta ascendiendo mientras que el pH >3 disminuye reduciendo en un orden de magnitud de manera escalonada para coincidir aproximadamente con los datos de pH 6,8 obtenidos por Davis et al. b Datos obtenidos por Davis et al. 1988; EUR. J. Biochem., 173, 345-351, (1988)
c El muestreo se puede modificar
d Las soluciones de la reacción se muestrean y se extinguen lo más rápido posible y requieren un cronómetro y 2 personas, una para tomar muestras/extinguir y la otra para registrar con precisión el tiempo en un cuaderno en minutos y/o segundos
Los estudios se realizaron en soluciones tampón a pH 1 - 7 y a 5°C, 25°C, 30°C y 37°C. Aunque esos estudios se realizaron en recipientes no sellados herméticamente, los antioxidantes solos (ácido ascórbico o L-cisteína) o combinados (ácido ascórbico L-cisteína) reducían la tasa de pérdida o degradación de BH4 (véanse la Figura 36 y la Figura 37). Burbujear una solución que contiene ácido ascórbico y L-cisteína mejoraba sustancialmente la estabilidad de BH4.
La tasa de degradación de BH4 depende de la concentración (véase la Figura 38). Por lo tanto, se mostró que las formulaciones de BH4 en dosis altas y altamente concentradas requieren una concentración más baja de estabilizantes para una estabilización sinérgica de las formulaciones.
Estos resultados demuestran que la formulación de formulaciones líquidas estables y de larga vida útil se puede producir de acuerdo con los métodos y composiciones descritos en esta memoria, incluyendo líquidos inyectables estériles, líquidos orales y polvos liofilizados y estériles para formulaciones de constitución.
Ejemplo 7
Formulaciones líquidas y liofilizadas de tetrahidrobiopterina para uso oral y parenteral
Ejemplo de composiciones de formulaciones
Tabla 20 Formulación específica tamponada a pH 4 que tiene ácido ascórbico como estabilizante

Tabla 21 Formulación tamponada a pH 4,0 que contiene una combinación de dos estabilizantes: ácido ascórbico y metabisulfito de sodio

Tabla 22 Formulación tamponada a pH 4,0 que contiene una combinación de tres estabilizantes: L-cisteína, ácido ascórbico y metabisulfito de sodio

Tabla 23 Formulación tamponada a pH 7,0 que contiene solo ácido ascórbico como único estabilizante


Tabla 24 Formulación tamponada a pH 7,0 que contiene ácido ascórbico y metabisulfito de sodio como estabilizantes

Tabla 25 Formulación tamponada a pH 7,0 que contiene ascórbico, metabisulfito de sodio y L-cisteína como estabilizantes

Formulaciones líquidas de dosis elevada
Tabla 26 Formulación tamponada a pH 6,0 que contiene solo ácido ascórbico como estabilizante

Tabla 27 Formulación tamponada a pH 6,0 que contiene una combinación de dos estabilizantes: ácido ascórbico y metabisulfito de sodio


Tabla 28 Formulación tamponada a pH 6,0 que contiene una combinación de tres estabilizantes: L-cisteína, ácido ascórbico y metabisulfito de sodio
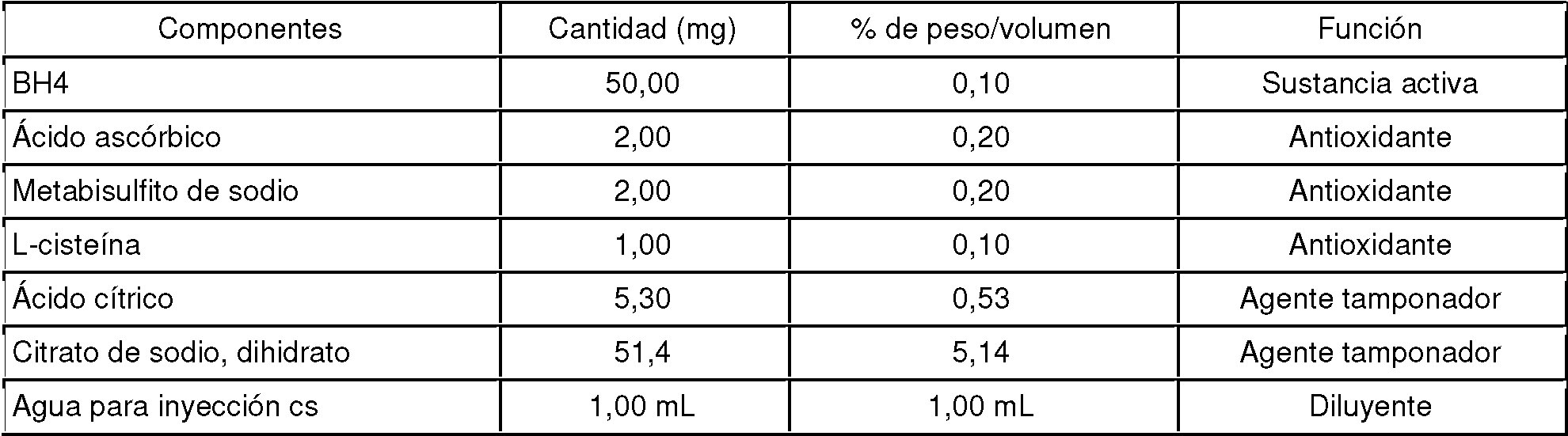
Tabla 29 Formulación oral tamponada a pH 3,0 con tampón citrato y que contiene solo ácido ascórbico como estabilizante

Tabla 30: Formulación oral tamponada a pH 3,5 con tampón tartrato y que contiene ácido ascórbico y metabisulfito de sodio como estabilizantes

Tabla 31: Formulación oral tamponada a pH 3,5 en tampón a base de ácido málico y que contiene ácido ascórbico y metabisulfito de sodio como estabilizantes

Las soluciones formuladas o mezcladas anteriores se burbujean opcionalmente con un gas inerte (por ejemplo, argón o nitrógeno) o dióxido de carbono en el tanque de mezcla, y los recipientes primarios se sellan preferiblemente con una capa de gas inerte o dióxido de carbono para eliminar el oxígeno del espacio superior del recipiente. Las formulaciones se pueden aumentar progresivamente hasta cualquier volumen multiplicando las cantidades de los componentes por un factor multiplicador apropiado.
Ejemplo 8
Determinación mediante LC/MS/MS de tetrahidrobiopterina (BH4) en plasma humano midiendo la concentración de L-biopterina después de una oxidación en condiciones básicas
La tetrahidrobiopterina (BH4) es una molécula terapéutica pequeña para el tratamiento de pacientes con fenilcetonuria (PKU). Es importante tener un método preciso y específico para medir las concentraciones de BH4 en el plasma humano. Sin embargo, es un desafío cuantificar BH4 en plasma humano debido a su baja concentración endógena y su inestabilidad. En condiciones básicas, BH4 se oxida a dihidrobiopterina (BH2) y finalmente a L-biopterina. Además, la relación de conversión de oxidación entre BH4 y L-biopterina es casi constante hasta 23 semanas. Por lo tanto, midiendo la concentración de L-biopterina tras la oxidación en condiciones básicas y aplicando un índice de conversión molar, podemos determinar de manera confiable las concentraciones de BH4 en plasma humano.
Los métodos publicados se basan en el método clásico desarrollado por Fukushima y Nixon (Anal. Biochem., 102, 176-188 (1980)) usando HPLC con detección de fluorescencia. En el método LC/MS/MS, la muestra de plasma humano se estabilizó con antioxidante, se enriqueció con una solución de patrón interno (IS) y se basificó con solución de hidróxido de sodio, después se oxidó con solución de yodo. Después de incubar en la oscuridad a temperatura ambiente, se añade ácido ascórbico para reducir el exceso de yodo. Las muestras oxidadas se extrajeron mediante precipitación de proteínas. La L-biopterina en los extractos reconstituidos se analizó utilizando HPLC de fase inversa con detección MS/MS Turbo Ion Spray®. Los iones negativos para L-biopterina se controlaron en modo MRM. Las relaciones entre el fármaco y el área de pico de IS para los patrones, se utilizaron para crear una curva de calibración lineal, utilizando análisis de regresión de mínimos cuadrados ponderados 1/x2.
La tasa de conversión de oxidación de BH4 a L-biopterina se evaluó en múltiples puntos de tiempo: 0, 1,2, 4, 8, 12 y 23 semanas, y se encontró que era compatible en todos los puntos de tiempo sometidos a ensayo con una tasa de conversión molar nominal del 47,3%, determinada a partir de los tres primeros puntos de tiempo consecutivos. La diferencia entre la tasa de conversión en otros puntos de tiempo y el valor nominal varía de -2,3 a 6,3%. El método LC/MS/MS se validó para cuantificar L-biopterina en plasma humano con K2 EDTA en el intervalo de calibración lineal de 5 a 1000 ng/mL (equivalente a 11 a 2114 ng/mL para BH4). La precisión y la exactitud del ensayo se evaluó con muestras de control de calidad (QCs) y los resultados mostraban una precisión intradiaria entre 4,7 y 14,5% de CV; una precisión intradiaria entre -7,1 a 7,4% de los valores nominales; y una precisión intradiaria y una exactitud de 7,4 a 16,4% de CV y de -8,3 a 3,7% de los valores nominales, respectivamente. La recuperación media de extracción de L-biopterina era del 65,3%. En plasma humano con K2 EDTA se encontró que la L-biopterina, era estable a temperatura ambiente durante al menos 4 horas y después de 4 ciclos de congelación y descongelación, y a -70°C durante al menos 275 días.
Ejemplo 9
Determinación de BH4/BH2/B usando HPLC con detección electroquímica y de fluorescencia
Se realizó un estudio para desarrollar un método para determinar las concentraciones de tetrahidrobiopterina (BH4), dihidrobiopterina (BH2) y biopterina (B) en plasma humano usando cromatografía líquida de alta resolución (HPLC) de fase inversa, con detección de fluorescencia (FD) y detección electroquímica (ECD). El método se basa en Cai, et al. (Cardiovascular Research 55: 838-849, 2002).
Se prepararon soluciones madre de BH4 (en HCl 20 mM), BH2 y B (en DMSO) hasta tener una concentración final de 10 mM y se almacenaron a -80°C. Se prepararon soluciones de trabajo estándar de calibración a partir de una solución madre 100, 10, 7,5, 5, 2,5 y 1 nM en plasma humano con K2 EDTA modificado con 1,4-ditioeritritol (DTE) al 0,1% (p/v). Se prepararon soluciones de trabajo de control de calidad de BH4, BH2 y B 5, 8, 25 y 50 nM en plasma humano con K2 EDTA modificado con DTE al 0,1% (p/v) y se almacenaron a -80°C.
Para el procesamiento de las muestras, el plasma se diluyó 1:10 en tampón de resuspensión. A 180 pl del plasma diluido, se añadieron 20 pl del tampón de precipitación 10X. Este procedimiento de dilución y precipitación del plasma se aplicó a todos los patrones de plasma, muestras de plasma y QCs de plasma. Después de la adición del tampón de precipitación 10X, la muestra se centrifugó a velocidad máxima a 4°C durante 5 min para eliminar restos de plasma no específicos. A continuación se transfirieron 150 mL de material sobrenadante a un vial de muestra y luego se colocaron en un muestreador automático para una inyección de 100 mL.
La fase móvil (2 L) se preparó con 13,6 g de acetato de sodio (50 mM), 2,1 g de ácido cítrico (5 mM) 36 mg de EDTA (48 mM), 49,4 mg de dTe (160 mM) y metanol al 2% en volumen en agua. El pH se ajustó a 5,22. Se preparó tampón de resuspensión (20 mL) con 20 mL de PBS pH 7,4 (50 mM), 20 uL de DTE 1 M (1 mM) y 100 mL de EDTA 100 uM. El tampón de precipitación 10X (25 mL) se preparó en el momento con 2,88 mL de ácido fosfórico (1 M), 9,39 g de ácido tricloroacético (2 M) y 20 mL de DTE 1 M (1 mM).
La tetrahidrobiopterina (BH4), la dihidrobiopterina (BH2) y la biopterina (B) se separaron empleando separación con HPLC de fase inversa. La BH4 se midió mediante detección electroquímica en la que BH4 se oxida con el electrodo 1 a dihidrobiopterina quinonoide (qBH2) y luego se reduce a BH4 en el electrodo 2. El detector utiliza después la corriente generada por esta reacción de reducción para determinar la concentración de BH4. BH2 y B se pueden medir en la misma inyección usando detección de la fluorescencia. La oxidación posterior de BH2 en la columna utilizando una celda protectora acondicionadora con el potencial óptimo, oxida BH2 a biopterina.
La separación con HPLC se llevó a cabo en una columna ACE C-18 (250 mm x 4,6 mm), 5 pM, con un caudal de 1,3 mL/min, con un tiempo de ejecución de 13 minutos. Los ajustes de la detección electroquímica eran E1: 100 mV (corriente de fondo 500 nA a 600 nA) y E2: -300 mV (corriente de fondo -50 nA a -60 nA). La oxidación posterior a la columna se fijó en 900 mV. Los ajustes de la detección de fluorescencia eran longitud de onda de excitación: 350 nM y longitud de onda de emisión: 450 nM.
La linealidad y el intervalo del método se evaluaron en función de la precisión y la exactitud de los patrones en plasma y tampón. La concentración de la curva estándar se estableció usando al menos 4-6 concentraciones distintas de cero para cada analito. La concentración de los patrones era 1, 2,5, 5, 7,5, 10 y 100 nM. Los resultados mostraban un ajuste lineal de 1 a 100 nM para BH4, BH2 y B con R2 de >0,99.
La exactitud se determinó mediante un análisis replicado de muestras de control de calidad que contenían cantidades conocidas (2, 8, 25 y 50 nM) del analito y se expresaba como un porcentaje de exactitud. La precisión también se calcula en función de los datos de los controles de calidad. La precisión intraensayo y la precisión interensayo se evaluaron en función del % de CV. En tres ciclos experimentales distintos, se prepararon y analizaron concentraciones de cada analito en plasma. Además, se “enriqueció” BH4, BH2 y B en 10 nM en muestras de plasma humano para determinar la exactitud y la recuperación. Las mediciones de BH4, BH2 y B 8, 25 y 50 nM mostraron ser exactas dentro del 112%-89% y demostraron una precisión (% de CV) de 2,5%-20%. Los experimentos de recuperación enriquecidos usando BH4, BH2 y B 10 nM en muestras clínicas de plasma humano, demostraron recuperaciones entre 70% y 130%. Los resultados demuestran que el método es exacto y preciso para muestras con concentraciones superiores a 2 nM.
Para comprobar la presencia de una interferencia endógena en seis lotes diferentes de plasma, se enriqueció con 10 nM de BH4, BH2 y B a seis lotes diferentes de plasma y se determinó la exactitud y la precisión para cada muestra de plasma. Los experimentos de selectividad muestran que los seis individuos tenían unos niveles basales de BH4 endógenos entre el límite cuantificable inferior y 2,48 nM. De manera similar, las concentraciones de BH2 y B variaban de 0,02 a 10 nM. La recuperación de los analitos enriquecidos 10 nM oscilaba entre 69% y 87%. La variabilidad (% de CV) entre las muestras de plasma individuales y los analitos cuando se enriquecían a 10 nM, oscilaba entre el 23% y el 37%. La variabilidad de los niveles endógenos de BH4, BH2 y B oscilaba entre 0 y 9,96 nM. Juntos, los resultados indican una tendencia que sugiere una interferencia de la matriz o una pérdida durante la extracción, pero no indican una fuerte selectividad entre los individuos.
Para medir el efecto de la matriz, se compararon las curvas estándar preparadas en plasma o tampón en cuanto a exactitud (recuperación), linealidad y correlación. La comparación de los patrones preparados en plasma frente a los patrones preparados en tampón, demuestra un efecto de matriz modesto y una correlación generalmente buena. Los tres analitos tenían excelentes ajustes lineales para plasma y tampón. BH4 y B no demostraron efectos de matriz significativos en todo el intervalo de concentración. Sin embargo, BH2 tenía menos recuperación con la concentración estándar más alta (100 nM). Las muestras de control de calidad preparadas en tampón y plasma demostraron una buena exactitud. En general, los efectos de la matriz parecen mínimos, con una tendencia hacia una menor recuperación en tampón, en comparación con el plasma. Debido a que BH4 y BH2 se oxidan fácilmente, el plasma recogido y los tampones de muestras deben contener antioxidantes y tener un pH bajo cuando sea
posible.
Para someter a ensayo la capacidad de diluir con exactitud una muestra de tampón y plasma enriquecida con 250 nM de BH4, BH2 y B, el plasma se diluyó usando plasma en blanco en una serie de diluciones por triplicado. Las muestras diluidas se analizaron y compararon con el valor nominal después de aplicar el factor de dilución. La dilución de concentraciones elevadas de BH4, BH2 y B se puede realizar con exactitud. Para BH4, las concentraciones observadas después de la dilución tenían una exactitud de entre 83% y 104% para concentraciones entre 83,33 nM y 3,07 nM. BH2 tenía una exactitud de 74%-80% en todo el intervalo cuantitativo (83 nM-3 nM). B tenía una exactitud de 119%-113% en todo el intervalo cuantitativo (83 nM-3 nM). Por lo tanto, una muestra que está por encima del límite cuantitativo, se puede diluir con exactitud.
Se prepararon cuatro concentraciones de analitos (2, 8, 25 y 50 nM) en plasma y se congelaron durante un mínimo de 24 horas para un ciclo y un mínimo de 12 horas para otros ciclos durante un mínimo de tres ciclos. Las muestras se descongelaron sin ayuda a temperatura ambiente entre los períodos de congelación. Se evaluó la exactitud y la variabilidad después de todos y cada uno de los ciclos de descongelación sin ayuda, para establecer el número máximo de ciclos a los que se podía someter una muestra. Las muestras que contienen BH4, BH2 y B se pueden someter a hasta 3 ciclos de congelación-descongelación sin un cambio significativo en la exactitud o la precisión de la medición. Las muestras de plasma con BH48 nM-50 nM tienen una exactitud de 121 %-91 % y un % de CV inferior al 10%. De manera similar, las mediciones de BH2 tenían una exactitud de 77%-88% en todo el intervalo cuantitativo del ensayo. Las mediciones de B tenían una exactitud de 98% al 99% en todo el intervalo cuantitativo con una precisión (% de CV) del 5% al 8%. La muestra 2 nM de BH4, BH2 y B no resultó ser exacta ni precisa después de repetidos procesos de congelación y descongelación. Por lo tanto, los patrones, los controles de calidad y las muestras del estudio se pueden congelar y descongelar hasta 3 veces.
Debido a que los analitos son sensibles a la oxidación, examinamos la estabilidad en estado congelado a largo plazo para imitar las condiciones de almacenamiento esperadas. Se prepararon cuatro niveles de concentración (2, 8, 50 y 100 nM) de BH4, BH2 y B en plasma y se almacenaron a -70°C durante 8 semanas. Las muestras de estabilidad se analizaron con material de nuevo aporte y en las semanas 3, 5, 6 y 8. BH4 y B tenían una buena estabilidad en estado congelado a largo plazo. BH2 demostró una concentración de muestra reducida después de un almacenamiento prolongado. Durante las 8 semanas de almacenamiento, las muestras de plasma con BH4 tenían una exactitud de 93% al 94% y un % de CV entre el 31% y el 0,21%, observándose la mayor variación con la concentración 2 nM. Las mediciones de BH2 tenían una exactitud de 63% al 85% en todas las concentraciones sometidas a ensayo, con una exactitud reducida con concentraciones 2 nM y 100 nM. La precisión (% de CV) oscilaba entre el 37% y el 18% para esas muestras. Las mediciones de B tenían una exactitud de 88%-101% con las concentraciones sometidas a ensayo, con una precisión (% de CV) del 23%-0,14%, con la mayor variabilidad con la concentración 2 nM. Juntos, estos datos respaldan la recomendación de almacenar las muestras durante un máximo de 8 semanas, sin una pérdida apreciable de concentración de analito. BH2 parece ser la más susceptible a la degradación (oxidación).
Para medir la estabilidad de BH4, BH2 y B en el muestreador automático, 8 nM de cada analito en disolvente de reconstitución permanecieron en el muestreador automático durante 0,25, 4 y 11 horas. Se comparó la exactitud y la precisión de las mediciones. La medición de BH4 observada era exacta dentro del 5% del valor teórico en cada punto de tiempo, con exactitud y precisión en las tres mediciones de 102% y 0,054% respectivamente. La medición de BH2 tenía una exactitud decreciente y una variabilidad creciente después de 4 horas. Después de 11 horas en el muestreador automático, se midió aproximadamente el 50% de BH2. Esto indica una baja estabilidad del muestreador automático en tampón de ejecución. La medición de B siguió siendo exacta dentro del 125% del valor teórico después de 11 horas. Por lo tanto, se recomiendan tiempos de ejecución de no más de 4 horas.
Para determinar la contaminación de la inyección, se insertó una muestra de plasma de referencia extraída después de la concentración estándar más alta de 100 nM. Esto se hizo para imitar la posibilidad de sobrestimar la concentración de analito en una muestra de baja concentración, debido a una contaminación. La contaminación de la inyección de BH4, BH2 y B es mínima y no representa más del 1% del área del pico del límite superior de cuantificación de 100 nM. La contaminación de la inyección representa aproximadamente un 5%-20% del límite inferior de cuantificación, basado en el área de pico promedio obtenida a partir del control de baja calidad (2 nM). Por lo tanto, preferiblemente las muestras deben ordenarse de menor a mayor (es decir, primero muestras antes de la dosis, seguido de muestras posteriores a la dosis) y preferiblemente se realizarán lavados adicionales para limpiar la columna periódicamente durante una ejecución para minimizar una contaminación potencial.
Se desarrolló un método cualificado que era robusto, específico, exacto y preciso. Este método es apropiado para cuantificar los niveles de BH4, BH2 y B en plasma para estudios farmacocinéticos y farmacológicos.
Claims (7)
1. Tetrahidrobiopterina (BH4) o una sal farmacéuticamente aceptable de la misma para uso en el tratamiento de afecciones asociadas con niveles elevados de fenilalanina, en donde la BH4 o una sal de la misma es para administrar por vía oral una vez al día, 5 a 60 minutos después de una comida y en donde la afección se selecciona a partir del grupo que consiste en fenilcetonuria leve (PKU), PKU clásica e hiperfenilalaninemia.
2. La BH4 o la sal para uso según la reivindicación 1, en donde la BH4 o una sal de la misma es para administrar entre 5 y 20 minutos después de una comida.
3. La BH4 o la sal para uso según la reivindicación 1 o 2, en donde la BH4 o una sal de la misma es para administrar por la mañana.
4. La BH4 o la sal para uso según una cualquiera de las reivindicaciones 1 -3, en donde la comida es una comida rica en grasas y rica en calorías que contiene al menos aproximadamente 700 kcal y al menos aproximadamente un 45% de grasa.
5. La BH4 o la sal para uso según una cualquiera de las reivindicaciones 1-4, en donde la BH4 o una sal de la misma es para administrar con una dosis diaria total de 1 mg/kg a 20 mg/kg.
6. La BH4 o la sal para uso según una cualquiera de las reivindicaciones 1-5, en donde la BH4 o una sal de la misma es para administrar disuelta en un líquido o como una forma de dosificación sólida, preferiblemente en donde la forma de dosificación sólida es un comprimido o una cápsula.
7. La BH4 o la sal para uso según una cualquiera de las reivindicaciones 1-6, en donde la BH4 o una sal de la misma es para administrar con la comida para incrementar la absorción de BH4.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US92282107P | 2007-04-11 | 2007-04-11 | |
| US1975308P | 2008-01-08 | 2008-01-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2851177T3 true ES2851177T3 (es) | 2021-09-03 |
Family
ID=39620159
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES08745614T Active ES2397746T3 (es) | 2007-04-11 | 2008-04-11 | Métodos de administración de tetrahidrobiopterina, composiciones asociadas y métodos de medición |
| ES12005352T Active ES2851177T3 (es) | 2007-04-11 | 2008-04-11 | Tetrahidrobiopterina para el tratamiento de afecciones asociadas con niveles elevados de fenilalanina |
| ES18205446T Active ES2906582T3 (es) | 2007-04-11 | 2008-04-11 | Métodos para administrar tetrahidrobiopterina, composiciones asociadas y métodos de medida |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES08745614T Active ES2397746T3 (es) | 2007-04-11 | 2008-04-11 | Métodos de administración de tetrahidrobiopterina, composiciones asociadas y métodos de medición |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18205446T Active ES2906582T3 (es) | 2007-04-11 | 2008-04-11 | Métodos para administrar tetrahidrobiopterina, composiciones asociadas y métodos de medida |
Country Status (23)
| Country | Link |
|---|---|
| US (6) | US7612073B2 (es) |
| EP (4) | EP4029519A1 (es) |
| JP (2) | JP2010523708A (es) |
| KR (5) | KR20150080026A (es) |
| CN (1) | CN101678025A (es) |
| AU (1) | AU2008240259C1 (es) |
| BR (1) | BRPI0809470A2 (es) |
| CA (1) | CA2682598C (es) |
| CY (3) | CY1113543T1 (es) |
| DE (1) | DE22154751T1 (es) |
| DK (3) | DK2545939T3 (es) |
| ES (3) | ES2397746T3 (es) |
| HK (1) | HK1139864A1 (es) |
| HR (3) | HRP20220188T3 (es) |
| HU (2) | HUE058030T2 (es) |
| IL (1) | IL201101A (es) |
| LT (2) | LT2545939T (es) |
| MX (1) | MX2009010977A (es) |
| PL (3) | PL2139485T3 (es) |
| PT (3) | PT2139485E (es) |
| RU (1) | RU2486899C2 (es) |
| SI (2) | SI2545939T1 (es) |
| WO (1) | WO2008128049A2 (es) |
Families Citing this family (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SI3138566T1 (sl) | 2003-11-17 | 2022-04-29 | Biomarin Pharmaceutical Inc. | Postopki in sestavki za zdravljenje presnovnih motenj |
| ES2397746T3 (es) | 2007-04-11 | 2013-03-11 | Biomarin Pharmaceutical Inc. | Métodos de administración de tetrahidrobiopterina, composiciones asociadas y métodos de medición |
| RU2011109210A (ru) * | 2008-08-12 | 2012-09-20 | Орфа Свисс Гмбх (Ch) | Лекарственная форма, содержащая тетрагидробиоптерин |
| US9089573B2 (en) | 2009-07-22 | 2015-07-28 | University Of Massachusetts | Methods and compositions to reduce oxidative stress |
| WO2011132435A1 (ja) | 2010-04-22 | 2011-10-27 | 学校法人日本大学 | 脳機能障害予防・改善用の薬剤及び飲食物 |
| KR102105857B1 (ko) * | 2010-12-03 | 2020-05-04 | 오렉시젠 세러퓨틱스 인크. | 날트렉손 요법에서 약물 생체이용률의 증가 |
| US9492451B2 (en) * | 2011-03-01 | 2016-11-15 | Dipharma S.A. | Stable compositions of tetrahydrobiopterin |
| US9549563B2 (en) | 2011-10-24 | 2017-01-24 | Kickass Candy Llc | Sweet tart energy tablet |
| US9216178B2 (en) * | 2011-11-02 | 2015-12-22 | Biomarin Pharmaceutical Inc. | Dry blend formulation of tetrahydrobiopterin |
| SI2806877T1 (sl) | 2012-01-23 | 2020-01-31 | Sage Therapeutics, Inc. | Nevroaktivne steroidne formulacije, ki obsegajo kompleks alopregnanolona in sulfobutil etra beta-ciklodekstrina |
| ES2646197T3 (es) | 2012-01-26 | 2017-12-12 | Vanda Pharmaceuticals Inc. | Tratamiento de trastornos del ritmo circadiano |
| US11918557B2 (en) | 2012-01-26 | 2024-03-05 | Vanda Pharmaceuticals Inc. | Treatment of circadian rhythm disorders |
| CN102650621B (zh) * | 2012-05-02 | 2014-06-25 | 安徽农业大学 | 一种从蚕体提取的墨蝶呤的鉴定方法 |
| WO2014124392A1 (en) * | 2013-02-08 | 2014-08-14 | University Of Iowa Research Foundation | Diagnostic tools to predict onset of preeclampsia |
| US11090285B2 (en) | 2013-11-12 | 2021-08-17 | Vanda Pharmaceuticals Inc | Treatment of circadian rhythm disorders |
| US10376487B2 (en) | 2013-11-12 | 2019-08-13 | Vanda Pharmaceuticals Inc. | Method of treatment |
| US20150204834A1 (en) * | 2014-01-22 | 2015-07-23 | Sarfaraz K. Niazi | Thermodynamic equivalence surrogate test (test) for bioequivalence |
| EP2926805B1 (en) | 2014-03-31 | 2016-05-18 | Vasopharm GmbH | Solid pharmaceutical compositions comprising biopterin derivatives and uses of such compositions |
| MA41124A (fr) * | 2014-12-05 | 2017-10-10 | Sun Pharmaceutical Ind Ltd | Compositions en suspension à libération prolongée à rétention gastrique |
| US10744088B2 (en) | 2015-10-06 | 2020-08-18 | G-Treebnt Co., Ltd. | Method for preparing ophthalmic preparation containing thymosin beta-4 |
| JOP20170059B1 (ar) | 2016-03-08 | 2021-08-17 | Sage Therapeutics Inc | ستيرويدات وتركيبات نشطة عصبيًا واستخداماتها |
| WO2018102314A1 (en) * | 2016-11-29 | 2018-06-07 | Censa Pharmaceuticals Inc. | Polymorphic form of sepiapterin |
| IL266955B2 (en) | 2016-11-29 | 2023-09-01 | Ptc Therapeutics Mp Inc | Polymorphs of spiafatrin and its salts |
| CN108627576A (zh) * | 2017-03-17 | 2018-10-09 | 武汉宏韧生物医药科技有限公司 | 一种人血浆中他达拉非的定量分析方法 |
| CN110944689B (zh) | 2017-06-07 | 2022-12-09 | 施菲姆德控股有限责任公司 | 血管内流体运动设备、系统和使用方法 |
| JP7277005B2 (ja) * | 2017-07-06 | 2023-05-18 | ツリーウェイ ティーダブリュー001 ビー.ブイ. | 酸化ストレス媒介性の神経変性障害の経口治療におけるエダラボンの使用 |
| US20200268657A1 (en) * | 2017-07-13 | 2020-08-27 | Michael Gulyas | Supplement tablet and packaging |
| MX2020002271A (es) | 2017-09-01 | 2020-10-05 | Ptc Therapeutics Mp Inc | Composiciones farmaceuticas que comprenden sepiapterina y sus usos de las mismas. |
| CN111556763B (zh) | 2017-11-13 | 2023-09-01 | 施菲姆德控股有限责任公司 | 血管内流体运动装置、系统 |
| EP3713577A4 (en) * | 2017-11-20 | 2021-08-18 | MediBeacon Inc. | PROCESS FOR THE PREPARATION AND ANALYSIS OF FLUORESCENT COMPOUNDS IN PLASMA |
| CN109946412B (zh) * | 2017-12-21 | 2021-10-15 | 上海产业技术研究院 | 一种体液蝶呤谱检测试剂盒及其使用 |
| EP3746149A4 (en) | 2018-02-01 | 2021-10-27 | Shifamed Holdings, LLC | INTRAVASCULAR BLOOD PUMPS AND METHODS OF USE AND METHODS OF MANUFACTURING |
| EP3766356A4 (en) * | 2018-03-16 | 2021-11-24 | Eco-Geo Bio-Technology Company Limited | MULTI-PH BUFFER FORMULATION AND COMPOSITION TO FACILITATE PROTEIN DIGESTION, AND USE OF THEM |
| CN110269099B (zh) * | 2018-03-16 | 2023-01-31 | 共生地球生物科技有限公司 | 多重pH缓冲配方与蛋白质消化助剂的组合物及其用途 |
| EP3807279A4 (en) * | 2018-05-30 | 2022-04-06 | PTC Therapeutics MP, Inc. | PHARMACEUTICALLY ACCEPTABLE SALTS OF SEPIAPTERINE |
| JP2021525729A (ja) * | 2018-05-30 | 2021-09-27 | ピーティーシー セラピューティクス エムピー,インコーポレイテッド | セピアプテリン血漿曝露を増加させるための方法 |
| EP3801534A4 (en) * | 2018-05-30 | 2022-03-16 | PTC Therapeutics MP, Inc. | COMPOSITIONS AND METHODS FOR INCREASE TETRAHYDROBIOPTERINE PLASMA EXPOSURE |
| CN108813410B (zh) * | 2018-07-06 | 2021-12-14 | 浙江工商大学 | 一种降低发酵香肠中生物胺的方法及其应用 |
| JP2020068734A (ja) * | 2018-11-01 | 2020-05-07 | 国立大学法人東京工業大学 | Sprをコードするポリヌクレオチドを含む組換えベクター、及びそれを含む組成物 |
| US20200276200A1 (en) * | 2019-02-28 | 2020-09-03 | Clark Gerald Sullivan | Treatment of cns and developmental disorders using high-dose 5-formyl-(6s)-tetrahydrofolate |
| US11964145B2 (en) | 2019-07-12 | 2024-04-23 | Shifamed Holdings, Llc | Intravascular blood pumps and methods of manufacture and use |
| WO2021011177A1 (en) * | 2019-07-15 | 2021-01-21 | R.P. Scherer Technologies, Llc | Enteric proton pump inhibitor softgel capsule |
| US11654275B2 (en) | 2019-07-22 | 2023-05-23 | Shifamed Holdings, Llc | Intravascular blood pumps with struts and methods of use and manufacture |
| TR201914416A1 (tr) | 2019-09-23 | 2021-04-21 | Sanovel Ilac Sanayi Ve Ticaret Anonim Sirketi | Sapropteri̇n di̇hi̇droklorürün efervesan formülasyonlari |
| EP4034192A4 (en) | 2019-09-25 | 2023-11-29 | Shifamed Holdings, LLC | INTRAVASCULAR BLOOD PUMP SYSTEMS AND METHODS OF USE AND CONTROL THEREOF |
| CN111505179B (zh) * | 2020-04-07 | 2021-07-13 | 厦门大学 | 海洋水体中生物蝶呤的检测方法 |
| CN112057423A (zh) * | 2020-10-21 | 2020-12-11 | 兆科药业(广州)有限公司 | 一种含有盐酸沙丙蝶呤的颗粒剂药物及其制备方法 |
| CN112697919B (zh) * | 2020-12-22 | 2023-02-28 | 北京和合医学诊断技术股份有限公司 | 度洛西汀的检测方法 |
| WO2022204517A1 (en) * | 2021-03-26 | 2022-09-29 | Emory University | Managing the acute and long-term effects of coronaviral infections and compositions related thereto |
| CN113607870A (zh) * | 2021-07-22 | 2021-11-05 | 中国食品药品检定研究院 | 一种头孢羟氨苄原料药及其制剂中聚合物杂质的检测方法 |
| CN114814036B (zh) * | 2022-05-09 | 2024-02-20 | 上海谱锐赛思生物技术有限公司 | 血浆中阿齐沙坦和氨氯地平浓度的测定方法 |
| WO2024084421A1 (en) * | 2022-10-18 | 2024-04-25 | Qrgenetics Ltd. | Treatment of vascular diseases associated with genetic variations in acta2 |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6021A (en) * | 1849-01-09 | Cast-iron cab-wheel | ||
| US6019A (en) * | 1849-01-09 | Cast-iron car-wheel | ||
| US2601215A (en) | 1948-03-17 | 1952-06-17 | American Cyanamid Co | Process of preparing dihydropterins |
| US3505329A (en) | 1968-02-06 | 1970-04-07 | Smithkline Corp | Process for the synthesis of biopterin |
| JPS5883691A (ja) | 1981-11-13 | 1983-05-19 | Kanegafuchi Chem Ind Co Ltd | 1′,2′−ジアシル−(6r,s)−5,6,7,8−テトラヒドロ−l−ビオプテリンおよびその製法 |
| JPS5925323A (ja) | 1982-03-03 | 1984-02-09 | 鐘淵化学工業株式会社 | プテリン誘導体からなる脳内神経伝達物質の関わる疾病の治療剤 |
| JPS5921685A (ja) | 1982-07-28 | 1984-02-03 | Suntory Ltd | L―エリスロ―バイオプテリンの製造法 |
| ZA836957B (en) | 1982-09-20 | 1985-04-24 | Wellcome Found | Neurologically active chemical compounds |
| GB8318833D0 (en) | 1983-07-12 | 1983-08-10 | Wellcome Found | Chemical compounds |
| US5196533A (en) | 1983-04-11 | 1993-03-23 | South Alabama Medical Science Foundation, Usa | Cyclization of 5 amino-pyrimidines to quinoid 6,6 disubstituted dihydropteridines |
| US4587340A (en) | 1983-09-19 | 1986-05-06 | Burroughs Wellcome Co. | Biopterin analogs |
| JPS60178887A (ja) | 1984-02-23 | 1985-09-12 | Kanegafuchi Chem Ind Co Ltd | 5,6,7,8−テトラヒドロ−l−ビオプテリンの製造法 |
| JPS60199889A (ja) | 1984-03-24 | 1985-10-09 | Kanegafuchi Chem Ind Co Ltd | 5,6,7,8−テトラヒドロ−l−エリスロ−ビオプテリンの硫酸塩およびその製法 |
| US4550109A (en) * | 1984-05-31 | 1985-10-29 | The Board Of Regents, The University Of Texas System | Lipoidal biopterin compounds |
| DE3437944A1 (de) | 1984-10-17 | 1986-07-31 | Biotest-Serum-Institut Gmbh, 6000 Frankfurt | Verwendung von pterinen zur steigerung der aktivitaet von lymphokinen und anderen blutfaktoren, sowie ein diagnostisches oder therapeutisches praeparat, das pterine in kombination mit lymphokinen enthaelt |
| CA1262347A (en) | 1985-01-28 | 1989-10-17 | Suntory Limited | Preparation process of (6r)-tetrahydro-l-biopterin |
| JPS61277618A (ja) | 1985-06-04 | 1986-12-08 | Suntory Ltd | 自閉症治療剤 |
| JPS621688A (ja) * | 1985-06-28 | 1987-01-07 | Nippon Kokan Kk <Nkk> | ばら積船 |
| WO1987001038A1 (en) * | 1985-08-22 | 1987-02-26 | Commonwealth Scientific And Industrial Research Or | Peptide analogues of mammalian insulin-like growth factor-1 |
| DE3853711T2 (de) | 1987-11-30 | 1996-01-11 | Vitamin Kenkyusho Kk | Zwischenverbindungen für die Synthese von 5,6,7,8-Tetrahydro-L-erythro-biopterin und seiner Derivate. |
| JP3137333B2 (ja) | 1990-07-21 | 2001-02-19 | サントリー株式会社 | テトラヒドロビオプテリンの製法およびそれに用いる酵素 |
| JP2534423B2 (ja) | 1991-12-26 | 1996-09-18 | コーネル・リサーチ・ファンデーション・インコーポレイテッド | 酸化窒素の過剰生産から生じる血管失調を阻止する阻害剤 |
| US5198547A (en) | 1992-03-16 | 1993-03-30 | South Alabama Medical Science Foundation, Usa | Process for N5-formylating tetrahydropteridines |
| DE4308739C1 (de) | 1993-03-19 | 1994-06-23 | Henning Berlin Gmbh | Pterinderivate, ihre Herstellung und ihre Verwendung |
| US5945452A (en) | 1993-06-11 | 1999-08-31 | The Board Of Trustees Of The Leland Stanford Junior University | Treatment of vascular degenerative diseases by modulation of endogenous nitric oxide production or activity |
| US5502050A (en) | 1993-11-29 | 1996-03-26 | Cornell Research Foundation, Inc. | Blocking utilization of tetrahydrobiopterin to block induction of nitric oxide synthesis |
| ATE218345T1 (de) | 1994-08-05 | 2002-06-15 | Suntory Ltd | Arzneimittel gegen spinocerebellare degeneration |
| JP2711828B2 (ja) | 1996-06-25 | 1998-02-10 | 白鳥製薬株式会社 | (6r)−テトラヒドローl−バイオプテリン塩酸塩の製造法 |
| US20020076782A1 (en) | 1996-07-05 | 2002-06-20 | John P.N. Rosazza | Purified nitric oxide synthase |
| GB9617990D0 (en) | 1996-08-29 | 1996-10-09 | Scotia Holdings Plc | Treatment of pain |
| CH693255A5 (de) | 1997-06-13 | 2003-05-15 | Eprova Ag | Verwendung von Tetrahydrofolaten in der natürlichenstereoisomeren Form zur Herstellung einer pharmazeutischenZubereitung geeignet zur Beeinflussung des Homocystein-Spiegels. |
| US5922713A (en) | 1997-06-26 | 1999-07-13 | Werner; Ernst | Inhibition of nitric oxide synthase |
| US6723560B2 (en) * | 1998-10-08 | 2004-04-20 | Mayo Foundation For Medical Education And Research | Using polyamide nucleic acid oligomers to engender a biological response |
| EP1004308B1 (en) | 1998-02-27 | 2005-05-11 | Daiichi Suntory Pharma Co., Ltd. | Preventives or remedies for drug-induced renal disturbance |
| JP4306825B2 (ja) | 1998-02-27 | 2009-08-05 | アスビオファーマ株式会社 | インスリン抵抗性が関与する血管機能異常を伴う疾患の予防または治療剤 |
| US6200758B1 (en) | 1999-02-19 | 2001-03-13 | New York State Office Of Mental Health | Phenylalanine hydroxylase gene variants, and amino acid and pterin homeostasis, in the definition, detection, treatment and prevention of psychotic, mood and personality disorders |
| WO2000056328A1 (en) | 1999-03-19 | 2000-09-28 | Enos Pharmaceuticals, Inc. | Increasing cerebral bioavailability of drugs |
| GB2355191A (en) | 1999-10-12 | 2001-04-18 | Laxdale Ltd | Combination formulations for fatigue, head injury and strokes |
| US6544994B2 (en) | 2000-06-07 | 2003-04-08 | Eprov Ag | Pharmaceutical preparation for treating or preventing cardiovascular or neurological disorders by modulating of the activity of nitric oxide synthase |
| US20040014167A1 (en) * | 2000-08-31 | 2004-01-22 | Masayuki Yabuta | Process for producing biopterin |
| RU2180233C1 (ru) * | 2001-06-26 | 2002-03-10 | Общество с ограниченной ответственностью "Протеиновый контур" | Способ получения жидких лекарственных форм рекомбинантных белков |
| US20040058962A1 (en) * | 2002-06-14 | 2004-03-25 | Amedeo Leonardi | Phenylalkylamines and pyridylalkylamines |
| DE10260263A1 (de) * | 2002-12-20 | 2004-07-15 | Biocrates Life Sciences Gmbh | Verwendung von Tetrahydrobiopterinderivaten zur Behandlung und Ernährung von Patienten mit Aminosäurestoffwechselstörungen |
| EP1667656A4 (en) * | 2003-09-01 | 2011-12-28 | Mayne Pharma Int Pty Ltd | COMPOSITIONS AND METHODS OF DISTRIBUTING BIOLOGICALLY ACTIVE AGENTS |
| SI3138566T1 (sl) | 2003-11-17 | 2022-04-29 | Biomarin Pharmaceutical Inc. | Postopki in sestavki za zdravljenje presnovnih motenj |
| CA2678165C (en) | 2003-11-17 | 2013-11-05 | Merck Eprova Ag | Crystalline forms of (6r)-l-erythro-tetrahydrobiopterin dihydrochloride |
| AU2004290692A1 (en) | 2003-11-17 | 2005-06-02 | Biomarin Pharmaceutical Inc. | Processes for preparing tetrahydrobiopterin, and analogs of tetrahydrobiopterin |
| ES2393785T3 (es) | 2004-05-11 | 2012-12-28 | Daiichi Sankyo Company, Limited | Remedios para hiperfenilalaninemia sensible a BH4 |
| CA2601716C (en) | 2004-06-25 | 2011-05-31 | Board Of Regents, The University Of Texas System | Methods and compositions for the treatment of attention deficit hyperactivity disorder and hyperphenylalanemia |
| AU2005286763A1 (en) | 2004-09-17 | 2006-03-30 | Biomarin Pharmaceutical, Inc. | Variants and chemically-modified variants of phenylalanine ammonia-lyase |
| JP2008520574A (ja) | 2004-11-17 | 2008-06-19 | バイオマリン ファーマシューティカル インコーポレイテッド | テトラヒドロビオプテリンの安定性錠剤処方物 |
| US20060194808A1 (en) * | 2005-01-14 | 2006-08-31 | Chronorx Llc, An Alaska Limited Liability Company | Clinical applications of tetrahydrobiopterin, lipoic acid and their salts and methods of preparing tetrahydrobiopterin bis-lipoate |
| WO2007072911A1 (ja) | 2005-12-22 | 2007-06-28 | Asubio Pharma Co., Ltd. | 塩酸サプロプテリンの生体内吸収性を向上させた製剤 |
| US20080075666A1 (en) | 2006-08-25 | 2008-03-27 | Dudley Samuel C Jr | Methods and compositions for treating diastolic dysfunction |
| EP2131679B1 (en) | 2007-02-22 | 2019-03-27 | Children's Hospital & Research Center at Oakland | Fatty acid formulations and methods of use thereof |
| ES2397746T3 (es) | 2007-04-11 | 2013-03-11 | Biomarin Pharmaceutical Inc. | Métodos de administración de tetrahidrobiopterina, composiciones asociadas y métodos de medición |
-
2008
- 2008-04-11 ES ES08745614T patent/ES2397746T3/es active Active
- 2008-04-11 PT PT87456141T patent/PT2139485E/pt unknown
- 2008-04-11 PT PT120053525T patent/PT2545939T/pt unknown
- 2008-04-11 CN CN200880011594A patent/CN101678025A/zh active Pending
- 2008-04-11 KR KR1020157016858A patent/KR20150080026A/ko not_active Application Discontinuation
- 2008-04-11 DK DK12005352.5T patent/DK2545939T3/da active
- 2008-04-11 DK DK18205446.0T patent/DK3461503T3/da active
- 2008-04-11 KR KR1020097023532A patent/KR20100016445A/ko active Application Filing
- 2008-04-11 HR HRP20220188TT patent/HRP20220188T3/hr unknown
- 2008-04-11 CA CA2682598A patent/CA2682598C/en active Active
- 2008-04-11 JP JP2010503234A patent/JP2010523708A/ja active Pending
- 2008-04-11 PL PL08745614T patent/PL2139485T3/pl unknown
- 2008-04-11 PT PT182054460T patent/PT3461503T/pt unknown
- 2008-04-11 DK DK08745614.1T patent/DK2139485T3/da active
- 2008-04-11 MX MX2009010977A patent/MX2009010977A/es active IP Right Grant
- 2008-04-11 BR BRPI0809470-5A patent/BRPI0809470A2/pt not_active Application Discontinuation
- 2008-04-11 HU HUE18205446A patent/HUE058030T2/hu unknown
- 2008-04-11 SI SI200832155T patent/SI2545939T1/sl unknown
- 2008-04-11 ES ES12005352T patent/ES2851177T3/es active Active
- 2008-04-11 RU RU2009141618/15A patent/RU2486899C2/ru active
- 2008-04-11 EP EP22154751.6A patent/EP4029519A1/en not_active Withdrawn
- 2008-04-11 EP EP12005352.5A patent/EP2545939B1/en not_active Revoked
- 2008-04-11 ES ES18205446T patent/ES2906582T3/es active Active
- 2008-04-11 KR KR1020177007999A patent/KR20170037676A/ko not_active Application Discontinuation
- 2008-04-11 EP EP18205446.0A patent/EP3461503B9/en active Active
- 2008-04-11 WO PCT/US2008/060041 patent/WO2008128049A2/en active Application Filing
- 2008-04-11 LT LTEP12005352.5T patent/LT2545939T/lt unknown
- 2008-04-11 PL PL18205446T patent/PL3461503T3/pl unknown
- 2008-04-11 DE DE22154751.6T patent/DE22154751T1/de active Pending
- 2008-04-11 LT LTEP18205446.0T patent/LT3461503T/lt unknown
- 2008-04-11 HU HUE12005352A patent/HUE053107T2/hu unknown
- 2008-04-11 KR KR1020167014034A patent/KR101721198B1/ko active IP Right Review Request
- 2008-04-11 KR KR1020167014024A patent/KR20160065998A/ko not_active Application Discontinuation
- 2008-04-11 SI SI200832189T patent/SI3461503T1/sl unknown
- 2008-04-11 AU AU2008240259A patent/AU2008240259C1/en active Active
- 2008-04-11 PL PL12005352T patent/PL2545939T3/pl unknown
- 2008-04-11 EP EP08745614A patent/EP2139485B1/en not_active Revoked
- 2008-12-08 US US12/329,838 patent/US7612073B2/en active Active
-
2009
- 2009-09-22 IL IL201101A patent/IL201101A/en active IP Right Revival
- 2009-10-12 US US12/577,509 patent/US7947681B2/en not_active Ceased
-
2010
- 2010-07-05 HK HK10106506.2A patent/HK1139864A1/xx not_active IP Right Cessation
-
2011
- 2011-04-29 US US13/097,223 patent/US20110281880A1/en not_active Abandoned
- 2011-11-18 US US13/299,499 patent/USRE43797E1/en active Active
-
2012
- 2012-12-27 HR HRP20121072AT patent/HRP20121072T1/hr unknown
-
2013
- 2013-01-09 CY CY20131100024T patent/CY1113543T1/el unknown
- 2013-02-06 US US13/760,156 patent/US20130237543A1/en not_active Abandoned
- 2013-05-20 JP JP2013105957A patent/JP2013163691A/ja active Pending
-
2016
- 2016-04-06 US US15/091,813 patent/US20170000793A1/en not_active Abandoned
-
2021
- 2021-02-01 CY CY20211100082T patent/CY1123764T1/el unknown
- 2021-02-11 HR HRP20210239TT patent/HRP20210239T1/hr unknown
-
2022
- 2022-03-08 CY CY20221100115T patent/CY1124976T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2851177T3 (es) | Tetrahidrobiopterina para el tratamiento de afecciones asociadas con niveles elevados de fenilalanina | |
| US20190160070A1 (en) | Dry blend formulation of tetrahydrobiopterin | |
| ES2596827T3 (es) | Tratamiento de la fenilcetonuria con BH4 | |
| CN104434894A (zh) | 使用l-鸟氨酸苯乙酸盐治疗门静脉高压和恢复肝功能 | |
| ES2736730T3 (es) | Forma de administración farmacéutica que contiene tetrahidrobiopterina | |
| WO2021061066A1 (en) | Effervescent formulations of sapropterin dihydrochloride | |
| CN116808224A (zh) | 用于治疗血管疾病的化合物组合 | |
| BR112020024346A2 (pt) | métodos para aumentar a exposição de sepiapterina no plasma |