ES2827233T3 - Compuesto heterocíclico de cinco y seis miembros, y método de preparación, composición farmacéutica y uso del mismo - Google Patents
Compuesto heterocíclico de cinco y seis miembros, y método de preparación, composición farmacéutica y uso del mismo Download PDFInfo
- Publication number
- ES2827233T3 ES2827233T3 ES14740886T ES14740886T ES2827233T3 ES 2827233 T3 ES2827233 T3 ES 2827233T3 ES 14740886 T ES14740886 T ES 14740886T ES 14740886 T ES14740886 T ES 14740886T ES 2827233 T3 ES2827233 T3 ES 2827233T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- heterocycloalkyl
- mmol
- compound
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCS(N(C1)CC1(CN)*1*=CC(c2c3[s]ccc3nc(Nc3ccc(*C=N4)c4c3)n2)=C1)(=O)=O Chemical compound CCS(N(C1)CC1(CN)*1*=CC(c2c3[s]ccc3nc(Nc3ccc(*C=N4)c4c3)n2)=C1)(=O)=O 0.000 description 15
- PYBWJPOEILSJDM-UHFFFAOYSA-N CCS(N(C1)CC1(CN)[n](cc1)cc1-c1nc(C(C)c2ccccc2)cc2c1CC=C2)(=O)=O Chemical compound CCS(N(C1)CC1(CN)[n](cc1)cc1-c1nc(C(C)c2ccccc2)cc2c1CC=C2)(=O)=O PYBWJPOEILSJDM-UHFFFAOYSA-N 0.000 description 1
- LUKLSBOFMHVLPO-ZETCQYMHSA-N C[C@@H](C[n]1ncc(N=C)c1)[O]=C Chemical compound C[C@@H](C[n]1ncc(N=C)c1)[O]=C LUKLSBOFMHVLPO-ZETCQYMHSA-N 0.000 description 1
- OQRQOAZHSLGMEZ-DLBZAZTESA-N C[C@@H](Cc1c[n]([C@H](C)CO)nc1)c(nc1-c2c[n](C3(CC#N)COC3)nc2)cc2c1[s]cc2 Chemical compound C[C@@H](Cc1c[n]([C@H](C)CO)nc1)c(nc1-c2c[n](C3(CC#N)COC3)nc2)cc2c1[s]cc2 OQRQOAZHSLGMEZ-DLBZAZTESA-N 0.000 description 1
- PAFZNILMFXTMIY-UHFFFAOYSA-N NC1CCCCC1 Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Un compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables, **(Ver fórmula)** en donde: X es O, o S; L1 es un enlace químico, un alquilo, un alquileno, un cicloalquilo o un heterocicloalquilo; en donde el alquilo, alquileno, cicloalquilo o heterocicloalquilo pueden estar sustituidos independientemente con los sustituyentes seleccionados del grupo que consiste en un halógeno, un ciano, alquil(C1-C6)sulfonilo, cicloalquil(C3-C6)sulfonilo, un acilo, un cicloalquilo y un heterocicloalquilo; L2 es un alquilo, un acilo, un cicloalquilo, un heterocicloalquilo, un arilo o un heteroarilo; M es un halógeno, un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un arilo, un heteroarilo, un ciano, un sulfonilo o un acilo; en donde el alquilo, alquileno, alcoxi, heterocicloalquilo, arilo, heteroarilo, sulfonilo o acilo pueden estar sustituidos independientemente con los sustituyentes seleccionados del grupo que consiste en: un halógeno, un hidroxilo, un ciano, un amino, un acilamino, un nitro, un carboxilo, un sulfonilo, un acilo, un alcoxi, un cicloalquilo, un heterocicloalquilo, un arilo y un heteroarilo; R1 es un hidrógeno, un deuterio, un halógeno, un ciano, un alquilo, un cicloalquilo, un sulfonilo, un "alquil-NH-CO-" o un "alquil-NHSO2-"; en donde el alquilo puede estar sustituido con los sustituyentes seleccionados del grupo que consiste en un halógeno, un hidroxilo, un ciano, un amino, un nitro, un carboxilo, un sulfonilo, un acilo, un alcoxi, un cicloalquilo, un alquenilo y un alquinilo; R2 se selecciona independientemente entre un hidrógeno, un deuterio, un halógeno y un alquilo; n es 1, 2, 3 o 4; p es 0, 1, 2, 3, 4 o 5.
Description
DESCRIPCIÓN
Compuesto heterocíclico de cinco y seis miembros, y método de preparación, composición farmacéutica y uso del mismo
Campo de la invención
La presente invención se refiere a un compuesto heterocíclico de cinco y seis miembros, y un método de preparación, composición farmacéutica, y uso del mismo.
Técnicas anteriores
La ruta de transducción JAK-STAT (quinasa Janus - transductor de señales y activador de la transcripción) es una ruta de transducción de señales que se ha encontrado en los últimos años causada por citocinas, y está implicada en muchos procesos importantes tales como la proliferación, diferenciación, apoptosis celulares y regulación inmunitaria (Aaronson, D.S. et al. Science 2002, 296, 1653-1655; O'Shea, J.J. et al. Nat. Rev. Drug Discovery 2004, 3, 555-564). En comparación con otra ruta de señalización, este proceso de transferencia de la ruta es relativamente simple, está compuesto principalmente por tres componentes: receptor tirosina quinasa, tirosina quinasa JAK y factor de transcripción STAT.
Existen receptores de citocinas correspondientes (tales como inteferón IFN e interleucina (IL) y factores de crecimiento (tales como factor de crecimiento epidérmico EGF, factor de crecimiento derivado de plaquetas PDGF etc.) sobre la membrana celular. El rasgo común de estos receptores es que el propio receptor no tiene actividad quinasa, pero el segmento intracelular tiene un sitio de unión para la tirosina quinasa JAK. Una vez que el receptor y el ligando se combinan, los restos de tirosina de las proteínas diana son fosforilados por la activación de JAK para lograr la señal a partir de suministro extracelular a intracelular. La familia de JAK representa tirosina quinasas no receptoras (PTK), y hasta ahora se han identificado cuatro quinasas de la familia de JAK, que incluyen JAK1, JAK2, JAK3 y TYK2. Existen 7 dominios JAK (dominio de homología de JAK JH) en la estructura, en donde el dominio JH1 es una región quinasa y el dominio JH2 es una región "pseudo" quinasa, JH6 y JH7 son regiones receptoras.
En la transducción de señales y la activación transcripcional, STAT juega un papel clave. STAT es una proteína de unión a ADN, es un importante sustrato de JAK. Existen siete miembros: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, y STAT6. La estructura de la proteína STAT se puede dividir en varias regiones funcionales: dominio N-terminal, dominio de hélice, dominio de unión a ADN, región conectora, dominio SH3, dominio SH2 y región activadora transcripcional C-terminal. De ellas, el segmento más conservativo y funcionalmente importante de la secuencia es el dominio SH2, que tiene la misma secuencia central "GTFLLRFSS", que el dominio SH2 de la tirosina quinasa Src.
La transmisión de la ruta de señalización JAK-STAT es relativamente simple. El proceso de transferencia de señales es el siguiente: la unión de la citoquina y su receptor induce la dimerización de la molécula receptora, lo que hace que en el acoplamiento la quinasa JSK al receptor se aproximen entre sí y sean activados por la interacción de la fosforilación de la tirosina. Después de la activación de JAK, la fosforilación de los restos de tirosina sobre el receptor es catalizada para que se produzca, después estos restos de tirosina fosforilados y las secuencias de aminoácidos circundantes forman "sitios de anclaje", al mismo tiempo, las proteínas STAT que contienen el dominio SH2 son reclutadas hacia los "sitios de anclaje". Finalmente, la quinasa JAK cataliza las proteínas STAT combinadas con el receptor para llevar a cabo la fosforilación, las proteínas STAT activadas con forma de dímero penetran en el núcleo para combinarse con los genes diana y regular la transcripción génica (Ihie, J.J. Nature 1995, 377, 591 -594). Vale la pena mencionar que una clase de quinasa JAK puede estar implicada en el proceso de transducción de señales de muchas clases de citocinas, una clase de rutas de señalización de citocinas también puede activar muchas quinasas JAK, pero las citocinas tienen cierta selectividad sobre las moléculas STAT activadas. Por ejemplo, IL-4 activa STAT6, mientras IL-12 activa específicamente STAT4.
La ruta JAK-STAT existe ampliamente en todos los tejidos de histiocitos, juega un papel especialmente importante en la diferenciación, proliferación, anti-infección de células linfoides, y participa en una variedad de interacciones de citocinas inflamatorias y transducción de señales (Kiesseleva T. et al. J. Gene, 2002, 285, 1 -24). La activación anormal de la ruta está íntimamente relacionada con una variedad de enfermedades, la búsqueda y escrutinio de inhibidores de JAK pueden contribuir a un estudio en profundidad del mecanismo de regulación de JAK-STAT, con el fin de proporcionar nuevos fármacos y métodos para la prevención y el tratamiento de enfermedades relacionadas. La interleucina IL-6, IL-15, el interferón (IFN), el factor estimulador de las colonias de granulocitos y macrófagos (GM-CSF) y otros niveles de expresión de tejido sinovial en artritis reumatoide aumentan significativamente. Juegan un papel importante en la aparición y el desarrollo de enfermedades, y las citoquinas juegan un papel importante a través de la ruta de transducción de señales JAK-STAT. Por lo tanto, la inhibición de la ruta JAK-STAT a propósito puede mejorar el proceso patofisiológico de la artritis reumatoide (Joel M.K. et al. Arthritis Rheum. 2009, 60, 1859-1905).
La aparición, el crecimiento, la invasión y la metástasis del tumor están relacionados con la ruta de transducción de señales de JAK-STAT. La activación de los STAT en la transducción de señales normal es rápida y transitoria, y la activación persistente de los STAT está íntimamente relacionada con la transformación maligna de las células (Buettner R. et al. Clin. Cancer Res. 2002, 8(4), 945-954). STAT3 es la posición clave de EGFR, IL-6/JAK, Src y otras rutas der señalización de tirosina quinasa carcinogénica, que es activada en una variedad de células y tejidos tumorales, tales como cáncer de mama, cáncer de ovario, carcinoma de células escamosas de cabeza y cuello,
melanoma maligno, mieloma múltiple, linfoma, tumores de cerebro, cáncer de pulmón de células no pequeñas y leucemia etc. (Niu G. et al. Oncogene 2002, 21(13), 2000-2008). Los inhibidores de la ruta JAK-STAT pertenecen a los inhibidores de PTK, y esta enzima es un miembro de la familia de las proteínas oncogénicas, y juega un importante papel en la proliferación celular normal y anormal. La aparición y el crecimiento de tumores no se pueden separar de las PTK, por lo tanto, los inhibidores de la ruta JAK-STAT pueden inhibir el crecimiento tumoral al inhibir las PTK, y tienen un efecto anti-tumoral obvio (Mora L.B. et al. J. Cancer Res. 2002, 62(22), 6659-6666).
La enfermedad inflamatoria intestinal está íntimamente relacionada con la autoinmunidad. La ruta JAK-STAT está implicada en una variedad de importantes procesos patogénicos de transducción y regulación de señales de citocinas inflamatorias y anti-inflamatorias, especialmente relacionados de manera íntima con IFN-y, IL-1, IL-6, IL-10 and IL-4. Y los mediadores inflamatorios y las citocinas también pueden inducir la activación de múltiples rutas de señalización, de manera que dan como resultado directa o indirectamente la expresión de mediadores inflamatorios, lo que conduce a la lesión de la mucosa intestinal, pero todavía quedan por dilucidar muchos mecanismos de señalización complejos. En teoría, la inhibición de la activación excesiva de la ruta de transducción de señales JAK-STAT puede inhibir la expresión de muchas citocinas inflamatorias aguas arriba, con el fin de lograr la prevención y el tratamiento de la enteropatía inflamatoria (Riley, J.K. et al. J. Biol. Chem. 1999, 274, 16513-16521).
El documento relacionado con patentes WO 2010/090764 A1 describe inhibidores de Pirrolopirimidil Axl quinasa, respectivamente, derivados de tienopirimidina para su uso en el tratamiento del cáncer.
Además, los últimos estudios muestran que el rechazo en el trasplante de órganos, la psoriasis, la fibrosis de tejidos y órganos, el asma bronquial, la cardiomiopatía isquémica, la insuficiencia cardíaca congestiva, el infarto de miocardio, las enfermedades del sistema sanguíneo y las enfermedades del sistema inmunitario están íntimamente relacionados con la ruta de transducción de señales JAK-STAT, esta ruta de señalización no solamente es importante para el mantenimiento de la función fisiológica normal de las células, pero también tiene un importante efecto regulador sobre la aparición y el desarrollo de las enfermedades.
Contenido de la presente invención
El problema técnico que se va a resolver en la presente invención consiste en proporcionar un compuesto heterocíclico de cinco a seis miembros representado por la fórmula IV-1-1, que es completamente diferente de los de la técnica anterior, o una sal farmacéuticamente aceptable del mismo. El compuesto heterocíclico de cinco o seis miembros representado por la fórmula IV-1-1 en la presente invención es un inhibidor de la quinasa Janus (JAK) eficaz que se puede utilizar para prevenir o tratar enfermedades con proliferación celular tales como cáncer, infecciones, inflamación y enfermedades autoinmunitarias.
La presente invención proporciona un compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1, o una de sus sales farmacéuticamente aceptables,

en donde:
X es O o S;
L1 es un enlace químico, un alquilo (preferiblemente un alquilo C1-C4), un alquileno, un cicloalquilo (preferiblemente un cicloalquilo C3-C6 , más preferiblemente un ciclopropilo) o un heterocicloalquilo (preferiblemente un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de nitrógeno o un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de oxígeno);
En donde el alquilo, alquileno, cicloalquilo o heterocicloalquilo pueden estar sustituidos independientemente con los sustituyentes seleccionados del grupo que consiste en un halógeno (tal como F, Cl, Br o I, preferiblemente F), un ciano, un alquil(C1-C6)sulfonilo, un cicloalquil(C3-C6)sulfonilo, en donde el alquil(C1-C6)sulfonilo es preferiblemente un
o, o2
, 'x A O
alquil(Ci-C3)sulfonilo, más preferiblemente 0 ’ un acilo (preferiblemente un formilo o un alquil(Ci-C4)acilo, en donde el alquil(Ci-C4)acilo es preferiblemente un alquil(C2)acilo), un cicloalquilo (preferiblemente un cicloalquilo C3-C6 , más preferiblemente un ciclopentilo) y un heterocicloalquilo;
L2 es un alquilo (preferiblemente un alquilo C1-C4 , más preferiblemente un metilo), un acilo (un formilo o un carbonilo), un cicloalquilo (preferiblemente un cicloalquilo C3-C6 , más preferiblemente un ciclopropilo o un ciclopentilo), un heterocicloalquilo (preferiblemente un heterocicloalquilo C3-C6 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno; más preferiblemente un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de oxígeno, lo más preferiblemente
tetrahidropiranilo (tal como un arilo (preferiblemente un arilo C6-C10, más preferiblemente un fenilo (tal como
un arilo (preferiblemente un arilo C6-C10, más preferiblemente un fenilo (tal como

más preferiblemente un heteroarilo C4-C8 que contiene de 1 a 4 átomos de nitrógeno; lo más preferiblemente un
pirazolilo (tal como ), un imidazolilo, un piridilo (tal como ), un
), un
benzimidazolilo (tal zopirazolilo (tal como
zopirazolilo (tal como  a piridazinilo (tal como
a piridazinilo (tal como

M es un halógeno (preferiblemente flúor), un alquilo (preferiblemente un alquilo C1-C6 , más preferiblemente un metilo, un etilo, un propilo o un isopropilo), un alquileno, un cicloalquilo (preferiblemente un cicloalquilo C3-C6, más preferiblemente un ciclopropilo), un alcoxi (preferiblemente un alcoxi C1-C4 sustituido con un heterocicloalquilo, más preferiblemente un alcoxi C2 sustituido con un heterocicloalquilo; en donde el heterocicloalquilo de "un alcoxi C1-C4 sustituido con un heterocicloalquilo" es preferiblemente un heterocicloalquilo C2-C10 que contiene de 1 a 4 átomos de
AN
oxígeno y/o nitrógeno; más preferiblemente un morfolinilo (tal como O™ ) o un pirrolidinilo), un heterocicloalquilo (preferiblemente a C3-8 heterocicloalquilo que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno; más preferiblemente a C4-6 heterocicloalquilo que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno; lo más preferiblemente un morfolinilo ^ 0 N
V
^isr ( n
(tal como ‘"í™ ), un tetrahidropiranilo (tal como un azetidinilo (tal como N H ) i Un piperidilo (tal como , / — \ X* n=N' n ' N'
un azetidinilo (tal como N H ) i Un piperidilo (tal como , / — \ X* n=N' n ' N'
- K N— V ”i ' NH "N
\ — / ), un oxetanilo (tal como ° ) , un tetrazolilo (tal como ^ N o N ), un piperazinilo o un pirrolidinilo), un arilo (preferiblemente un arilo C6-C10, más preferiblemente un fenilo), un heteroarilo, un ciano, un sulfonilo o un acilo; en donde el alquilo, alquileno, alcoxi, heterocicloalquilo, arilo, heteroarilo, sulfonilo o acilo definidos in M pueden estar opcionalmente sustituidos con los sustituyentes del grupo que consiste en un halógeno (F, Cl, Br o
V / CF 3 I, preferiblemente F; el alquilo C1-C6 sustituido con el halógeno es preferiblemente a trifluorometilo o ), a hidroxilo el alquilo C1-C6 sustituido con el hidroxilo es preferiblemente

ciano es preferiblemente , un amino, un acilamino, un nitro, un carboxilo, un sulfonilo, un metilsulfonilo (el
alquilo C rC 6 sustituido con el metilsulfonilo es preferiblemente Un acilo, un alcoxi (preferiblemente un alcoxi C rC 6, más preferiblemente un alcoxi C1-C3 , tal como un metoxi, un etoxi, un propoxi o un isopropoxi; el alquilo C1-C6 sustituido con el alcoxi es preferiblemente un cicloalquilo te un cicloalquilo C3-C6 , más preferiblemente un ciclopropilo), un heterocicloalquilo (preferiblemente un uilo C3-C8 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno; más preferiblemente un heterocic
Un acilo, un alcoxi (preferiblemente un alcoxi C rC 6, más preferiblemente un alcoxi C1-C3 , tal como un metoxi, un etoxi, un propoxi o un isopropoxi; el alquilo C1-C6 sustituido con el alcoxi es preferiblemente un cicloalquilo te un cicloalquilo C3-C6 , más preferiblemente un ciclopropilo), un heterocicloalquilo (preferiblemente un uilo C3-C8 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno; más preferiblemente un heterocic 6 que contiene de 1 a 4
6 que contiene de 1 a 4
átomos de oxígeno y/o nitrógeno; lo más preferiblemente un morfolinilo (tal c , un tetrahidropiranilo (tal

como  un azetidinilo (tal como
un azetidinilo (tal como Un piperidilo (tal como — ), un oxetanilo (tal como ° ) , N =N N s N
Un piperidilo (tal como — ), un oxetanilo (tal como ° ) , N =N N s N
. L ,NH . /
un piperazinilo, un pirrolidinilo o un tetrazolilo (tal como o ^ N )), Un arilo y un heteroarilo;
R1 es un hidrógeno, un deuterio, un halógeno (tal como F, Cl, Br o I, preferiblemente F), un ciano, un alquilo (preferiblemente un alquilo C1-C4), un cicloalquilo, un sulfonilo, un "alquil-NH-CO-" o un "alquil-NHSO2-"; en donde el alquilo definido en R1 pueden estar opcionalmente sustituidos con los sustituyentes del grupo que consiste en un halógeno, un hidroxilo, un ciano, un amino, un nitro, un carboxilo, un sulfonilo, un acilo, un alcoxi, un cicloalquilo, un alquenilo y un alquinilo;
R2 se selecciona independientemente entre un hidrógeno, un deuterio, un halógeno y un alquilo (preferiblemente un alquilo C1-C4);
n es 1, 2, 3 o 4;
p es 0, 1, 2, 3, 4 o 5.
Cada sustituyente en el compuesto representado por la fórmula IV-1 -1 es preferiblemente como los siguientes grupos: en donde,
X es O o S
R2 es un hidrógeno, un deuterio, un halógeno (tal como F, Cl, Br o I) o un alquilo;
n es 1 o 2;
p es 0 o 1.
En la presente invención, el compuesto representado por la fórmula IV-1 -1 tiene preferiblemente la estructura representada por la fórmula V-1 -1:

R1 es un hidrógeno, un alquilo o un halógeno; en donde el alquilo es sustituido con los sustituyentes seleccionados del grupo que consiste en un halógeno, un hidroxilo, un ciano, un amino, un nitro, un carboxilo, un sulfonilo y un acilo;
R2 es un hidrógeno, un deuterio, un halógeno o un alquilo;
n es 1 o 2;
p es 0 o 1. En la presente invención, cuando R1 es un halógeno, el halógeno es F, Cl, Br o I;
y/o, cuando R2 es un halógeno, el halógeno es F, Cl, Br o I;
y/o, cuando L1 es un alquilo, el alquilo es un alquilo C1-C4 ;
o, cuando L1 es un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando L1 es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de nitrógeno o un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de oxígeno;
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 están sustituidos con los sustituyentes seleccionados del grupo que consiste en un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 están sustituidos con los sustituyentes seleccionados del grupo que consiste en un acilo, el acilo es un formilo o un alquil(C1-C4)acilo;
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 están sustituidos con los sustituyentes seleccionados del grupo que consiste en un halógeno, el halógeno es F, Cl, Br o I;
el alquilo definido en R1 es un alquilo C1-C4 ;
cuando R2 es un alquilo, el alquilo es un alquilo C1-C4.
En la presente invención, cuando L2 es un alquilo, el alquilo es un alquilo C1-C4 ;
cuando L2 es un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando L2 es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C6 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno;
cuando L2 es un arilo, el arilo es un arilo C6-C10;
cuando L2 es un heteroarilo, el heteroarilo es un heteroarilo C3-C10 que contiene de 1 a 4 átomos de nitrógeno. En la presente invención, cuando M es un halógeno, el halógeno es flúor;
cuando M es un alquilo, el alquilo es un alquilo C1-C6 ;
cuando M es un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando M es un alcoxi, el alcoxi es un alcoxi C1-C4 sustituido con un heterocicloalquilo; el heterocicloalquilo no está sustituido adicionalmente; o el heterocicloalquilo es un heterocicloalquilo C2-C10 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno;
cuando M es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C8 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno.
En la presente invención, cuando M es un arilo, el arilo es un arilo C6-C10;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un halógeno, el halógeno es F, Cl, Br o I;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un alcoxi, el alcoxi es un alcoxi C1-C6 ;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C8 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno.
En la presente invención, cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 están sustituidos con los sustituyentes seleccionados del grupo que consiste en un cicloalquilo, el cicloalquilo C3-C6 es un ciclopentilo; cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 están sustituidos con los sustituyentes
seleccionados del grupo que consiste en un alquil(Ci-C6)sulfonilo, el alquil(Ci-C6)sulfonilo es un alquil(Ci-C3)sulfonilo; cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 están sustituidos con los sustituyentes seleccionados del grupo que consiste en un alquil(C1-C4)acilo, el alquil(C1-C4)acilo es un alquil(C2)acilo; cuando L1 es un cicloalquilo C3-C6, el cicloalquilo C3-C6 es un ciclopropilo.
En la presente invención, cuando L2 es un alquilo C1-C4 , el alquilo C1-C4 es un metilo;
cuando L2 es un cicloalquilo C3-C6 , el cicloalquilo C3-C6 es un ciclopropilo o un ciclohexilo;
cuando L2 es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de oxígeno;
cuando L2 es un arilo C6-C10, el arilo C6-C10 es un fenilo;
cuando L2 es un heteroarilo C3-C10, el heteroarilo C3-C10 es un heteroarilo C4-C8 que contiene de 1 a 4 átomos de nitrógeno.
En la presente invención, cuando M es un alquilo C1-C6 , el alquilo C1-C6 es un metilo, un etilo, un propilo o un isopropilo; cuando M es un alquilo C1-C6 sustituido con un halógeno, el alquilo C1-C6 sustituido con un halógeno es un / s/ C F 3 .
trifluorometilo o >
cuando M es un alquilo C1-C6 sustituido con un hidroxilo, el alquilo C1-C6 sustituido con un hidroxilo es

cuando M es un alquilo C1-C6 sustituido con un ciano, el alquilo C1-C6 sustituido con un ciano es
cuando M es un alquilo C1-C6 sustituido con un metilsulfonilo, el alquilo C1-C6 sustituido con un metilsulfonilo es

cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un alcoxi C1-C6 , el alcoxi C1-C6 es un alcoxi C1-C3 ;
cuando M es un alcoxi C1-C4 sustituido con un heterocicloalquilo C2-C10, el alcoxi C1-C4 es un alcoxi C2 ; cuando M es un alcoxi C1-C4 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un heterocicloalquilo C3-C8 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno;
cuando M es un heterocicloalquilo C3-C8 , el heterocicloalquilo C3-C8 es un heterocicloalquilo C4-C6 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno.
En la presente invención, cuando M es un arilo C6-C10, el arilo C6-C10 es un fenilo;
cuando M es un alquilo, un alquileno, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un cicloalquilo C3-C6, el cicloalquilo C3-C6 es un ciclopropilo;
cuando M es un alcoxi C1-C4 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un heterocicloalquilo C4-C6 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno.
En la presente invención, cuando L2 es un heterocicloalquilo C3-C6 , el heterocicloalquilo C3-C6 es un tetrahidropiranilo; cuando L2 es un heteroarilo C4-C8 , el heteroarilo C4-C8 es un pirazolilo, un imidazolilo, un piridilo, un benzimidazolilo, a benzopirazolilo, un piridazinilo o un pirimidinilo.
En la presente invención, cuando M es un alcoxi C2 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un heterocicloalquilo C3-C8 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno;
cuando M es un heterocicloalquilo C4-C6 , el heterocicloalquilo C4-C6 es un morfolinilo, un tetrahidropiranilo, un azetidinilo, un piperidilo, un oxetanilo, un tetrazolilo, un piperazinilo o un pirrolidinilo;
cuando M es un alquilo C1-C6 sustituido con un alcoxi C1-C3, el alquilo C1-C6 sustituido con un alcoxi C1-C3 es
En la presente invención, cuando M es un alcoxi C2 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un heterocicloalquilo C3-C8 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno.
En la presente invención, cuando un alcoxi C2 definido en M está sustituido con un heterocicloalquilo C4-C6 , el heterocicloalquilo C4-C6 es un morfolinilo o un pirrolidinilo.
En la presente invención, cuando M es un alcoxi C2 sustituido con un heterocicloalquilo C4-C6 , el heterocicloalquilo C4-C6 es un morfolinilo, un tetrahidropiranilo, un azetidinilo, un piperidilo, un oxetanilo, un piperazinilo o un pirrolidinilo. En la presente invención, el compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1 se selecciona más preferiblemente del grupo que consiste en:





La presente invención también proporciona un método para preparar el compuesto representado por la fórmula I, que se selecciona del grupo que consiste en:
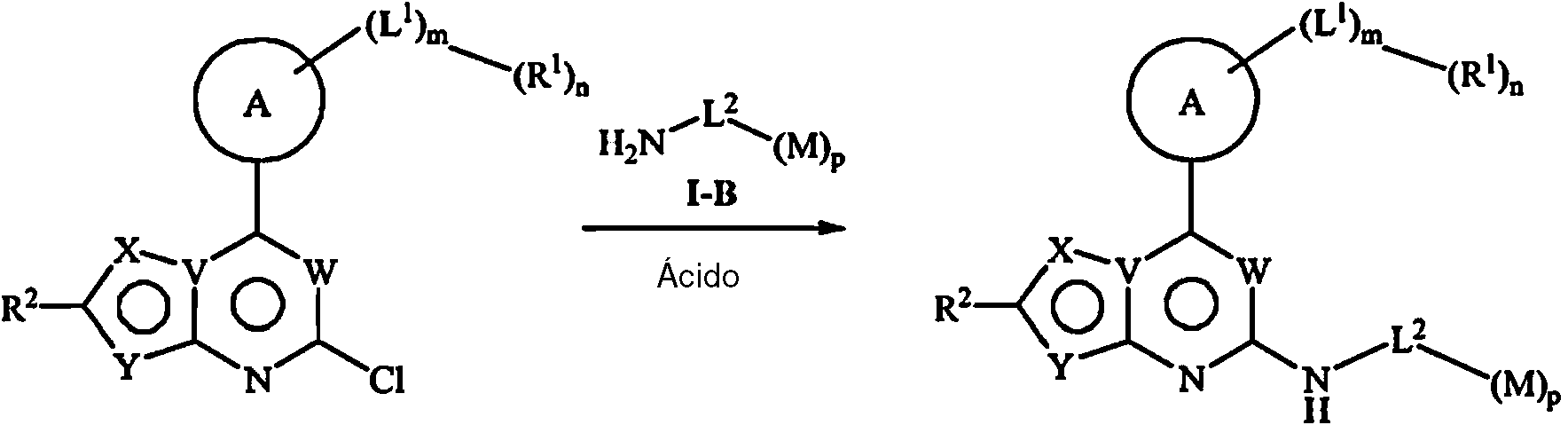
Método 1: en condiciones alcalinas, realizar una reacción de sustitución nucleofílica entre un compuesto representado por la fórmula I-A y un compuesto representado por la fórmula I-B,

Método 2: en condiciones ácidas, realizar una reacción de sustitución nucleofílica entre un compuesto representado por la fórmula I-A y un compuesto representado por la fórmula I-B; o
I-A I
Método 3: realizar una reacción de acoplamiento entre un compuesto representado por la fórmula I-A y un compuesto representado por la fórmula I-B,

En los métodos 1, 2 y 3, excepto por una explicación especial, cada letra y sustituyente tiene el significado proporcionado anteriormente.
En el método 1, el método y las condiciones para la reacción de sustitución nucleofílica son las convencionales en la técnica, el método preferido y las condiciones en la presente invención son: en un disolvente, realizar la reacción de sustitución nucleofílica entre el compuesto representado por la fórmula I-A y el compuesto representado por la fórmula I-B en condiciones alcalinas. El disolvente se selecciona preferiblemente del grupo que consiste en DMSO, 1,4-dioxano y DMF, más preferiblemente DMSO. Las condiciones alcalinas son proporcionadas preferiblemente por una base inorgánica, que se selecciona preferiblemente del grupo que consiste en KF, NaF, Cs2CO3 y K2CO3 , más preferiblemente KF. La razón de masa volumétrica del disolvente y el compuesto representado por la fórmula I-A es preferiblemente 10 mL/g-100 mL/g. La razón molar del compuesto representado por la fórmula I-A respecto al compuesto representado por la fórmula I-B es preferiblemente 0,1:1-1:1, más preferiblemente 0,3:1 -0,9:1. La razón molar de la base respecto al compuesto representado por la fórmula I-B es preferiblemente 1:1-10:1, más preferiblemente 1:1 -2:1. La reacción de sustitución nucleofílica se lleva a cabo preferiblemente a 50°C-150°C, más preferiblemente 70°C-110°C. La reacción de sustitución nucleofílica se lleva a cabo preferiblemente hasta que se detecta la finalización de la reacción, en la presente invención se prefiere 5h-24h.
En el método 2, el método y las condiciones para la reacción de sustitución nucleofílica son las convencionales en la técnica, el método preferido y las condiciones en la presente invención son: en un disolvente, realizar la reacción de sustitución nucleofílica entre el compuesto representado por la fórmula I-A y el compuesto representado por la fórmula I-B en condiciones ácidas. El disolvente es preferiblemente un disolvente orgánico, que se selecciona preferiblemente del grupo que consiste en isobutanol, n-butanol, DMSO y DMF, más preferiblemente isobutanol. Las condiciones ácidas son proporcionadas preferiblemente por un ácido orgánico y un ácido inorgánico, más preferiblemente un ácido orgánico. El ácido orgánico es preferiblemente ácido p-toluenosulfónico, mientras que el ácido inorgánico es preferiblemente HCl y/o H2SO4. La razón de masa volumétrica del disolvente y el compuesto representado por la fórmula I-A es preferiblemente 10 mL/g-100 mL/g. La razón molar del compuesto representado por la fórmula I-A respecto al compuesto representado por la fórmula I-B es preferiblemente 0,1:1 -1:1, más preferiblemente 0,3:1 -0,9:1. La razón molar del ácido respecto al compuesto representado por la fórmula I-B es preferiblemente 0,1:1 -3:1, más preferiblemente 0,6:1 -1,2:1. La reacción de sustitución nucleofílica se lleva a cabo preferiblemente a 50°C-150°C, más preferiblemente 80°C-120°C. La reacción de sustitución nucleofílica se lleva a cabo preferiblemente hasta que se detecta la finalización de la reacción, en la presente invención se prefiere 5h-24h.
En el método 3, el método y las condiciones para la reacción de acoplamiento son las convencionales en la técnica, el método preferido y las condiciones en la presente invención son: en un disolvente, en una atmósfera de gas inerte, realizar una reacción de acoplamiento entre el compuesto representado por la fórmula I-A y el compuesto representado por la fórmula I-B en presencia de una base y un catalizador de Pd, en donde el gas inerte es preferiblemente argón y/o nitrógeno; el disolvente es preferiblemente un disolvente orgánico y/o agua, el disolvente orgánico se selecciona preferiblemente del grupo que consiste en 1,4-dioxano, tolueno y dimetiléter de glicol, más preferiblemente 1,4-dioxano. La razón de masa volumétrica del disolvente y el compuesto representado por la fórmula I-A es preferiblemente 20 mL/g-100 mL/g. La base es preferiblemente una base inorgánica, y la base inorgánica se selecciona preferiblemente del grupo que consiste en K2CO3, Cs2CO3, Na2CO3 y K3 PO4. La razón molar de la base y el compuesto representado por la fórmula I-A es preferiblemente 1:1-10:1, más preferiblemente 3:1-5:1. El catalizador de Pd es el convencional en la técnica, que se selecciona preferiblemente del grupo que consiste en Pd2(dba)3, Pd(OAc)2 , Pd(Pph3)4 y Pd(dppf)Cl2. Es más preferiblemente Pd2(dba)3. La razón molar del catalizador de Pd respecto al compuesto representado por la fórmula I-A es preferiblemente 0,005:1 -0,5:1, más preferiblemente 0,01:1 -0,10:1. La razón molar del compuesto representado por la fórmula I-A respecto al compuesto representado por la fórmula I-B es preferiblemente 0,5:1 -2:1, más preferiblemente 0,9:1 -1,5:1. La reacción de acoplamiento se lleva a cabo preferiblemente a 50°C-150°C, más preferiblemente 90°C-130°C. La reacción de acoplamiento se lleva a cabo preferiblemente hasta que se detecta la finalización de la reacción, En la presente invención se prefiere 0,5h-3h.
En la presente invención, se muestra a continuación, una ruta de reacción para preparar el compuesto representado por la fórmula I:

Los expertos en la técnica pueden entender, de acuerdo con los compuestos anteriores descritos en la presente invención, que se pueden utilizar materiales bien conocidos a través de clases de métodos familiares para preparar los compuestos descritos en la presente invención, por ejemplo, a través de síntesis química o la extracción de plantas, todos estos métodos están incluidos en la presente invención. A no ser que se describa de otro modo o se proporcione un método de preparación, todos los materiales utilizados en la preparación de los compuestos o intermedios de los mismos en la presente invención son conocidos en la técnica o asequibles comercialmente.
Cada condición preferida para los métodos en la presente invención se puede combinar libremente para proporcionar mejores ejemplos.
En la presente invención, el compuesto que tiene la estructura representada por la fórmula IV-A:

en donde,
L1 es un enlace químico, un alquilo, un alquileno, un cicloalquilo o un heterocicloalquilo; en donde el alquilo, alquileno, cicloalquilo o heterocicloalquilo puede estar sustituido independientemente con los sustituyentes seleccionados del grupo que consiste en un halógeno, un ciano, un sulfonilo, un acilo, un cicloalquilo y un heterocicloalquilo;
R1 es un hidrógeno, un deuterio, un halógeno, un ciano, un alquilo, un cicloalquilo, un sulfonilo, un “alquil-NH-CO-" o un “alquil-NHSO2-"; en donde el alquilo puede estar sustituido con los sustituyentes seleccionados del grupo que consiste en un halógeno, un hidroxilo, un ciano, un amino, un nitro, un carboxilo, un sulfonilo, un acilo, un alcoxi, un cicloalquilo, un alquenilo y un alquinilo;
R2 se selecciona independientemente entre un hidrógeno, un deuterio, un halógeno y un alquilo;
m es 1; y
n es 1,2, 3 o 4.
En la presente invención, el compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-A se selecciona más preferiblemente del grupo que consiste en:

La presente invención también proporciona un método para preparar el compuesto representado por la fórmula I-A, que comprende: realizar una reacción de acoplamiento entre un compuesto representado por la fórmula I-C y un compuesto representado por la fórmula I-D;

En el método para preparar el compuesto representado por la fórmula I-A, el método y las condiciones para la reacción de acoplamiento son las convencionales en la técnica, el método preferido y las condiciones en la presente invención son: en un disolvente, en una atmósfera de gas inerte, realizar una reacción de acoplamiento entre el compuesto representado por la fórmula I-C y el compuesto representado por la fórmula I-D en presencia de una base y un catalizador de Pd. En donde el gas inerte es preferiblemente argón y/o nitrógeno; el disolvente es preferiblemente un disolvente orgánico y/o agua, el disolvente orgánico se selecciona preferiblemente del grupo que consiste en 1,4-dioxano, tolueno y dimetiléter de glicol, más preferiblemente 1,4-dioxano. La razón de masa volumétrica del disolvente y el compuesto representado por la fórmula I-C es preferiblemente 20 mL/g-100 mL/g. La base es preferiblemente una base inorgánica, y la base inorgánica se selecciona preferiblemente del grupo que consiste en K2CO3 , Cs2CO3, Na2CO3 y K3 PO4. La razón molar de la base respecto al compuesto representado por la fórmula I-C es preferiblemente 1:1-10:1, más preferiblemente 3:1-5:1. El catalizador de Pd es el convencional en la técnica, que se selecciona preferiblemente del grupo que consiste en Pd2(dba)3, Pd(OAc)2 , Pd(Pph3)4 y Pd(dppf)Cl2 , más preferiblemente Pd(dppf)Cl2. La razón molar del catalizador de Pd respecto al compuesto representado por la fórmula I-C es preferiblemente 0,005:1 -0,5:1, más preferiblemente 0,01: 1 -0,10:1. La razón molar del compuesto representado por la fórmula I-C respecto al compuesto representado por la fórmula I-D es preferiblemente 0,5:1 -2:1, más preferiblemente 0,9:1 -1,5:1. La reacción de acoplamiento se lleva a cabo preferiblemente a 20°C - 120°C, más preferiblemente 70°C-110°C. La reacción de acoplamiento se lleva a cabo preferiblemente hasta que se detecta la finalización de la reacción, en la presente invención se prefiere 4h-18h.
La presente invención también proporciona el uso del compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1 o una de sus sales farmacéuticamente aceptables para la preparación de un medicamento como inhibidor de la quinasa Janus (JAK).
La presente invención también proporciona el compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1 o una de sus sales farmacéuticamente aceptables para su uso en el tratamiento y/o la prevención de enfermedades de proliferación celular. En donde las enfermedades de proliferación celular son enfermedades convencionales causadas por la proliferación celular en la técnica, las enfermedades preferidas en la presente invención son cáncer, infección, inflamación y enfermedades autoinmunitarias.
La presente invención también proporciona una composición farmacéutica, que contiene una dosificación terapéuticamente eficaz del compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1, o una de sus sales farmacéuticamente aceptables, y una variedad de portadores y/o atenuantes farmacéuticamente aceptables.
La presente invención proporciona el uso de la composición farmacéutica para la preparación de un medicamento como inhibidor de la quinasa Janus (JAK).
La presente invención proporciona el uso de la composición farmacéutica para la preparación de un medicamento para el tratamiento y/o la prevención de enfermedades de proliferación celular. En donde las enfermedades de proliferación celular son enfermedades convencionales causadas por la proliferación celular en la técnica, las enfermedades preferidas en la presente invención son cáncer, infección, inflamación y enfermedades autoinmunitarias.
La composición farmacéutica en la presente invención puede estar en una forma adecuada para administración oral, también puede estar en forma de una solución acuosa inyectable estéril. La administración oral o la solución acuosa inyectable se pueden preparar según métodos conocidos para la preparación de una composición farmacéutica en la técnica.
A no ser que se indique de otro modo, los siguientes términos, cuando se utilizan en las descripciones y las reivindicaciones de la presente invención tienen los siguientes significados:
Como se emplea en la presente memoria, el término "alquilo" se refiere a un hidrocarbilo alifático de cadena lineal o
ramificada saturado que contiene de 1 a 20 átomos de carbono, preferiblemente de 1 a 10 átomos de carbono, más preferiblemente de 1 a 8 átomos de carbono, tal como un metilo, un etilo, un n-propilo, un isopropilo, un n-butilo, una terc- butilo, un isobutilo, un pentilo, un hexilo, un heptilo, un octilo, un nonilo, un decilo, un 4,4-dimetilpentilo, un 2,2,4-trimetilpentilo, un undecilo, un dodecilo, y diversos isómeros de los mismos etc.; así como el alquilo que contiene de 1 a 4 sustituyentes seleccionados del grupo que consiste en: un deuterio, un halógeno (se prefiere F, Br, Cl o I), un alquilo, un alcoxi, un arilo, un ariloxi, un arilo o un diarilo sustituido con un arilo, un arilalquilo, un arilalcoxi, un alquenilo, un alquinilo, un cicloalquilo, un cicloalquenilo, un cicloalquilalquilo, un cicloalquilalcoxi, un amino, un hidroxilo, un hidroxialquilo, un acilo, un grupo aldehído, un heteroarilo, un heteroariloxi, un heterocicloalquilo, un heterocicloalcoxi, un arilheteroarilo, un arilalcoxicarbonilo, un heteroariloalquilo, un heteroariloalcoxi, un ariloxialquilo, un ariloxiarilo, un alquilamino, un acilamino, un arilcarbonilamino, un nitro, un ciano, un tiol, un haloalquilo, un trihaloalquilo y/o un alquiltio.
Como se emplea en la presente memoria, el término "alquileno" (utilizado solo o como parte de otros grupos) se refiere a un hidrocarbilo alifático de cadena lineal o ramificada subsaturado que contiene de 1 a 20 átomos de carbono, preferiblemente de 1 a 10 átomos de carbono, más preferiblemente de 1 a 8 átomos de carbono, tal como a metileno, un etileno, un n-propileno, un isopropileno, un n-butileno, un terc-butileno, un isobutileno, un pentileno, un hexileno, un heptileno, un octileno, un nonileno, un decileno, un 4,4-dimetilpentileno, un 2,2,4-trimetilpentileno, un undecileno, un dodecileno, y diversos isómeros de los mismos etc.; así como el alquileno que contiene de 1 a 4 sustituyentes seleccionados del grupo que consiste en: un deuterio, un halógeno (se prefiere F, Br, Cl o I), un alquilo, un alcoxi, un arilo, un ariloxi, un arilo o un diarilo sustituido con un arilo, un arilalquilo, un arilalcoxi, un alquenilo, un alquinilo, un cicloalquilo, un cicloalquenilo, un cicloalquilalquilo, un cicloalquilalcoxi, un amino, un hidroxilo, un hidroxialquilo, un acilo, un grupo aldehído, un heteroarilo, un heteroariloxi, un heterocicloalquilo, un heterocicloalcoxi, un arilheteroarilo, un arilalcoxicarbonilo, un heteroariloalquilo, un heteroariloalcoxi, un ariloxialquilo, un ariloxiarilo, un alquilamino, un acilamino, un arilcarbonilamino, un nitro, un ciano, un tiol, un haloalquilo, un trihaloalquilo y/o un alquiltio; los sustituyentes seleccionados del grupo mencionado anteriormente también pueden formar un anillo junto con el grupo alquileno, formando de ese modo un anillo fusionado o un anillo espiro.
El término "aliciclo" o "cicloalquilo” se refiere a un grupo que tiene un anillo sencillo o anillos múltiples sólo con átomos de carbono, en donde cada anillo puede contener uno o más de un enlace doble sin un sistema de electrones n conjugado. Preferiblemente, un cicloalquilo que contiene de 1 a 3 anillos con un total de 3 a 20 átomos de carbono, más preferiblemente de 3 a 10 átomos de carbono, por ejemplo: un ciclopropilo, un ciclobutilo, un ciclopentilo, un ciclohexilo, un cicloheptilo, un ciclooctilo, un ciclodecilo y un ciclododecilo, un ciclohexenilo; el cicloalquilo puede estar opcionalmente sustituido con 1 a 4 sustituyentes seleccionados del grupo que consiste en: un deuterio, un halógeno, un alquilo, un alcoxi, un hidroxilo, un arilo, un ariloxi, un arilalquilo, un cicloalquilo, un alquilamino, un acilamino, un oxo, un acilo, un arilcarbonilamino, un amino, un nitro, un ciano, un tiol y/o un alquiltio y/o cualquier alquilo definido en la presente invención.
El término "alcoxi" se refiere a un grupo alquilo cíclico o no cíclico que contiene el número indicado de átomos de carbono y que tiene una conexión a través de un puente de oxígeno. Así, "alcoxi" incluye la definición del término "alquilo” y del término "cicloalquilo” mencionados anteriormente.
El término "alquenilo" se refiere a un hidrocarbilo no aromático de cadena lineal, ramificada o cíclico que un número indicado de átomos de carbono y al menos un enlace doble carbono-carbono. Preferiblemente hay un enlace doble carbono-carbono, y puede tener hasta cuatro enlaces dobles carbono-carbono no aromáticos. Así, "alquenilo C2-C12" se refiere a un grupo alquenilo que tiene de 2 a 12 átomos de carbono. "Alquenilo C2-C6" se refiere a un grupo alquenilo que tiene de 2 to 6 átomos de carbono, que incluye un vinilo, un propenilo, un butenilo, un 2-metil-butenilo y un ciclohexenilo. El enlace doble se puede localizar en un segmento de cadena lineal, cadena ramificada o porción cíclica del grupo alquenilo y, cuando se especifique, el grupo alquenilo puede estar sustituido, el alquenilo puede estar opcionalmente sustituido con los sustituyentes seleccionados del grupo que consiste en: un alquilo, un halógeno, un alcoxi, un hidroxilo, un arilo, un ariloxi, un arilalquilo, un cicloalquilo, un alquilamino, un acilamino, un acilo, un arilcarbonilamino, un amino, un nitro, un ciano, un tiol y/o un alquiltio y/o cualquier alquilo definido en la presente invención.
El término "alquinilo" se refiere a un grupo hidrocarbilo de cadena lineal, cadena ramificada o cíclico que tiene el número indicado de átomos de carbono y al menos un enlace triple carbono-carbono. Puede tener hasta tres enlaces triples carbono-carbono. Así, "alquinilo C2-C12" se refiere a un grupo alquinilo que tiene de 2 a 12 átomos de carbono. "Alquinilo C2-C6" se refiere a un grupo alquinilo que tiene de 2 a 6 átomos de carbono, que incluye un etinilo, un propinilo, un butinilo y un 3-metil-1 -butinilo y similares.
Como se emplea en la presente memoria, el término "arilo" se refiere a cualquier anillo carbocíclico monocíclico o bicíclico estable que contiene hasta 7 átomos en cada anillo, en donde al menos un anillo es un anillo aromático. Los ejemplos del grupo arilo anteriormente mencionados incluyen un fenilo, un naftilo, un tetrahidronaftilo, un 2,3-indanilo, un bifenilo, un fenantrilo, un antrilo o un acenaftilo. Se puede entender que, si un sustituyente arilo es un anillo bicíclico que tiene un anillo no aromático, la conexión es a través del anillo aromático. También incluye el arilo opcionalmente sustituido con 1 a 4 sustituyentes seleccionados del grupo que consiste en: un deuterio, un halógeno (se prefiere F, Br, Cl o I), un alquilo, un alcoxi, un arilo, un ariloxi, un arilo o un diarilo sustituido con un arilo, un arilalquilo, un arilalcoxi, un alquenilo, un alquinilo, un cicloalquilo, un cicloalquenilo, un cicloalquilalquilo, un cicloalquilalcoxi, un amino opcionalmente sustituido, un hidroxilo, un hidroxialquilo, un acilo, un grupo aldehído, un heteroarilo, un heteroariloxi, un heterocicloalquilo, un heterocicloalquiloxi, un arilheteroarilo, un arilalcoxicarbonilo, un heteroariloalquilo, un
heteroariloalcoxi, un ariloxialquilo, un ariloxiarilo, un alquilamino, un acilamino, un arilcarbonilamino, un nitro, un ciano, un tiol, un haloalquilo, un trihaloalquilo, y/o un alquiltio.
El término "anillo aromático" se refiere a cualquier anillo carbocíclico monocíclico o bicíclico estable que contiene hasta 7 átomos en cada anillo, en donde al menos un anillo es un anillo aromático. Los ejemplos del grupo anular aromático anteriormente mencionado incluyen un fenilo, un naftilo, un tetrahidronaftilo, un 2,3-indanilo, un bifenilo, un fenantrilo, un antrilo o un acenaftilo. Se puede entender que, si un sustituyente arilo es un anillo bicíclico que tiene un anillo no aromático, la conexión es a través del anillo aromático. También incluye el anillo aromático opcionalmente sustituido con 1 a 4 sustituyentes seleccionados del grupo que consiste en: un deuterio, un halógeno (se prefiere F, Br, Cl o I), un alquilo, un alcoxi, un arilo, un ariloxi, un arilo o un diarilo sustituido con un arilo, un arilalquilo, un arilalcoxi, un alquenilo, un alquinilo, un cicloalquilo, un cicloalquenilo, un cicloalquilalquilo, un cicloalquilalcoxi, un amino, un hidroxilo, un hidroxialquilo, un acilo, un grupo aldehído, un heteroarilo, un heteroariloxi, un heterocicloalquilo, un heterocicloalquiloxi, un arilheteroarilo, un arilalcoxicarbonilo, un heteroariloalquilo, un heteroariloalcoxi, un ariloxialquilo, un ariloxiarilo, un alquilamino, un acilamino, un arilcarbonilamino, un nitro, un ciano, un tiol, un haloalquilo, un trihaloalquilo, y/o un alquiltio.
Como se emplea en la presente memoria, el término "arilheterociclo" o "heteroarilciclo" se refiere a cualquier anillo monocíclico o bicíclico estable que contiene hasta 7 átomos en cada anillo, en donde al menos un anillo es un anillo aromático que contiene de 1 a 4 heteroátomos seleccionados del grupo que consiste en O, N, y S. El término grupos "arilheterociclo" o "heteroarilciclo" dentro del alcance de esta definición incluye, pero no se limita a, un acridinilo, un carbazolilo, un cinolinilo, un carbolinilo, un quinoxalinilo, un imidazolilo, un pirazolilo, un pirrolilo, un indolilo, un indolinilo, un benzotriazolilo, un benzimidazolilo, un furilo, un tienilo, un isotiazolilo, un benzotienilo, un dihidrobenzotienilo, un benzofuranilo, un isobenzofuranilo, un benzoxazolilo, un benzofuroxanilo, un benzopirazolilo, un quinolinilo, un isoindolilo, un isoquinolinilo, un oxazolilo, un oxadiazolilo, un isoxazolilo, un indolilo, un pirazinilo, un piridinopiridinilo, un piridinotetrazolilo, un piridazinilo, un piridinilo, un antimintilo, un pirimidinilo, un pirrolilo, un tetrazolilo, un tiadiazolilo, un tiazolilo, un tiofenilo, un triazolilo, un quinazolinilo, un tetrahidroquinolinilo, un dihidrobenzimidazolilo, un dihidrobenzofuranilo, un dihidrobenzoxazolilo, un dihidroquinolinilo. En cuanto al heterociclo definido a continuación, se debe entender que el término "heteroarilciclo" incluye derivados N-oxido de cualquier grupo heteroaromático que contiene nitrógeno. Se debe entender que si un sustituyente heteroarilo es un anillo bicíclico que tiene un anillo no aromático o un anillo sin heteroátomo la conexión es a través del anillo aromático o del heteroátomo que contiene en el anillo. El término grupos "heteroarilciclo" o "arilheterociclo" puede estar opcionalmente sustituido con 1 a 4 sustituyentes seleccionados del grupo que consiste en un deuterio, un halógeno, un alquilo, un alcoxi, un hidroxilo, un arilo, un ariloxi, un arilalquilo, un cicloalquilo, un alquilamino, un acilamino, un acilo, un arilcarbonilamino, un amino, un nitro, un ciano, un tiol y/o un alquiltio y/o cualquier alquilo definido en la presente invención.
El término "halógeno" se refiere a un flúor, un cloro, un bromo, un yodo, o una astatina.
El término "hidroxilo" se refiere
El término "amino" se refiere
El término "ciano" se refiere
El término "carboxilo" se refiere
El término "sulfonilo" se refiere
El término "acilo" se refiere a un carbonilo o un formilo, el término "carbonilo" se refiere a que hay sustituyentes en ambos lados de un acilo, y el término "formilo" se refiere a que hay a sustituyente sólo en un lado.
El término "acilamino" se refiere a una carbonilamida o una formilamida, el término "carbonilamida" se refiere a que hay sustituyentes en ambos lados de un acilamino, y el término "formilamida" se refiere a que hay un sustituyente sólo en un lado.
El término "haloalquilo” se refiere a un alquilo sustituido con un halógeno en una posición opcional. Por tanto, el "haloalquilo” incluye la definición del término "halógeno" y del término "alquilo”.
El término "haloalcoxi" se refiere a un alcoxi sustituido con un halógeno en una posición opcional. Por tanto, el "haloalcoxi" incluye la definición del término "halógeno" y del término "alcoxi".
El término "ariloxi" se refiere a un grupo arilo que contiene el número indicado de átomos de carbono y que tiene una conexión a través de un puente de oxígeno. Por tanto, el "ariloxi" incluye la definición del término "arilo".
Como se emplea en la presente memoria, el término "arilhetero" o "heteroarilo" se refiere a cualquier anillo monocíclico o bicíclico estable que contiene hasta 7 átomos en cada anillo, en donde al menos un anillo es un anillo aromático que contiene de 1 a 4 heteroátomos seleccionados del grupo que consiste en O, N, y S. El término grupos "heteroarilo" o "arilhetero" dentro del alcance de esta definición incluye, pero no se limita a, un acridinilo, un carbazolilo, un cinolinilo, un carbolinilo, un quinoxalinilo, un imidazolilo, un pirazolilo, un pirrolilo, un indolilo, un indolinilo, un benzotriazolilo, un benzimidazolilo, un furilo, un tienilo, un isotiazolilo, un benzotienilo, un dihidrobenzotienilo, un benzofuranilo, un isobenzofuranilo, un benzoxazolilo, un benzofuroxanilo, un benzopirazolilo, un quinolinilo, un isoindolilo, un isoquinolinilo, un oxazolilo, un oxadiazolilo, un isoxazolilo, un indolilo, un pirazinilo, un piridinopiridinilo, un piridinotetrazolilo, un piridazinilo, un piridinilo, un antimintilo, un pirimidinilo, un pirrolilo, un tetrazolilo, un tiadiazolilo, un tiazolilo, un tiofenilo, un triazolilo, un quinazolinilo, un quinoxalinilo, un tetrahidroquinolinilo, un dihidrobenzimidazolilo, un dihidrobenzofuranilo, un dihidrobenzoxazolilo, un dihidroquinolinilo, un metilendioxibenzoilo. En cuanto al heterociclo definido a continuación, se debe entender que el término "heteroarilo" incluye derivados N-oxido de cualquier grupo heteroaromático que contiene nitrógeno. Se debe entender que si un sustituyente heteroarilo es un anillo bicíclico que tiene un anillo no aromático o un anillo son heteroátomo, la conexión es a través del anillo aromático. El término grupos "heteroarilo" o "arilhetero" puede estar opcionalmente sustituido con 1 a 4 sustituyentes seleccionados del grupo que consiste en un deuterio, un halógeno, un alquilo, un alcoxi, un hidroxilo, un arilo, un ariloxi, un arilalquilo, un cicloalquilo, un alquilamino, un acilamino, un acilo, un arilcarbonilamino, un amino, un nitro, un ciano, un tiol y/o un alquiltio y/o cualquier alquilo definido en la presente invención.
El término "heteroaliciclo" o "heterocicloalquilo” utilizado en la presente memoria solo o como parte de otros grupos se refiere a un grupo monocíclico o policíclico de 4 a 12 miembros que contiene de 1 a 4 heteroátomos (tal como nitrógeno, oxígeno y/o azufre) que puede tener uno o más de un enlace doble en cada anillo, pero sin un sistema de electrones n conjugado en un anillo. El término grupos "heterocicloalquilo” puede incluir de 1 a 4 sustituyentes, tal como un alquilo, un halógeno, un oxo y/o cualquier alquilo expuesto anteriormente. Además, cualquier anillo de heterocicloalquilo se puede fusionar a un anillo de cicloalquilo, arilo, heteroarilo o heterocicloalquilo, y formar un anillo fusionado. El término grupos "heterocicloalquilo” dentro del alcance de esta definición incluye, pero no se limita a, un oxazolinilo, un oxetanilo, un piranilo, un tetrahidropiranilo, un azetidinilo, un 1,4-dioxanilo, un hexahidroazepanilo, un piperazinilo, un piperidinilo, un pirrolidinilo, un morfolinilo, u tiomorfolinilo, un dihidrofuranilo, un dihidroimidazolilo, un dihidroindolilo, un dihidroisoxazolilo, un dihidroisotiazolilo, un dihidroxadiazolilo, un dihidroxazolilo, un dihidropirazinilo, un dihidropirazolilo, un dihidropiridilo, un dihidropirimidinilo, un dihidropirrolilo, un dihidrotetrazolilo, un dihidrotiadiazolilo, un dihidrotiazolilo, un dihidrotienilo, un dihidrotriazolilo, un dihidroazetidinilo, un tetrahidrofuranilo y un tetrahidrotienilo and N-oxidos de los mismos. Un sustituyente heterocicloalquilo se puede conectar a otros grupos a través de un átomo de carbono o de un heteroátomo.
En la presente invención, todos los grupos alquilo, cicloalquilo, heterocicloalquilo, arilo y heteroarilo que tienen los números de carbonos indicados como "Cx1-Cy1" (x1 e y1 son números enteros), por ejemplo "alquilo C1-C12”, indican que esos grupos no están sustituidos adicionalmente con sustituyentes, de manera que "alquilo C1-C12” significa un alquilo que tiene de 1 a 12 átomos de carbono sin sustituyentes adicionales.
Basándose en la no vulneración del buen sentido del tema en cuestión, todas las condiciones preferidas anteriores se pueden combinar de cualquier manera para proporcionar las realizaciones preferidas de la presente invención.
Todos los materiales y reactivos utilizados en la presente invención son asequibles comercialmente.
El efecto positivo de la presente invención es que: el compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1 o una de sus sales farmacéuticamente aceptables en la presente invención es una clase de inhibidor de quinasa Janus (JAK) eficaz que se puede utilizar para el tratamiento o la prevención de enfermedades de proliferación celular tales como cáncer, infecciones, inflamación y enfermedades autoinmunitarias etc.
Descripción detallada de las realizaciones preferidas
Más abajo, junto con las realizaciones específicas, la presente invención se elabora adicionalmente. Las condiciones específicas de los experimentos para las siguientes realizaciones, cuando no se indique, están usualmente de acuerdo con los métodos y las condiciones convencionales, o el manual del producto.
Las estructuras de los compuestos se identifican mediante Resonancia Magnética Nuclear (RMN) o Espectro de Masas (MS). El espectro de RMN se obtuvo mediante Bruker Avance-500, utilizando dimetilsulfóxido deuterado, cloroformo deuterado y metanol deuterado etc. como disolvente, tetrametilsilano (TMS) como patrón interno. El espectro LC-MS se obtuvo mediante cromatografía líquida acoplada a espectrometría de masas (LC-MS) Agilent Technologies 6110, y se utilizó la fuente de iones ESI. La reacción de microondas se realizó en el sintetizador de microondas completamente automático Explorer producido por CEM, la frecuencia del magnetrón fue de 2450 MHz, la potencia continua de salida de microondas fue de 300W. Se utilizó Gilson 281 para la HPLC preparativa, y la columna fue Shimadazu Shim-Pack, PRC-ODS, 20x250 mm, 15 pm.
Los compuestos T-16, T-17, T-18, T-42, T-55, T-56, 16-b, 30, 42, 23, 56, 55 no entran dentro de la invención reivindicada.
Ejemplo 1
3-(4-(2-(Benc¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)butanon¡tr¡lo T-01
Ruta s¡ntét¡ca:

A una solución de 3-Bromobutanonitrilo (2,0 g, 10,3 mmoles) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1 H-pirazol (2,3 g, 15,5 mmoles) en acetonitrilo (100 mL) se le añadió carbonato de cesio (13,3 g, 41,2 mmoles). La mezcla se calentó a 90°C y se agitó durante 3 horas. Después de enfriar a temperatura ambiente, la reacción se sofocó con agua (100 mL). Se utilizó acetato de etilo (100 mLx3) para extraer la mezcla, las capas orgánicas se combinaron y lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente. Después de secar sobre sulfato de sodio anhidro, la capa orgánica se filtró, el producto filtrado se concentró a vacío para proporcionar un aceite incoloro 1-a (2,3 g), el producto bruto se utilizó directamente para la siguiente etapa. LC-MS (ESI): m/z = 262 [M+H]+.
Preparac¡ón del Compuesto 1
Bajo nitrógeno, a una solución de 2,4-diclorotieno[3,2-d]pirimidina (265 mg, 1,29 mmoles), el compuesto 1-a (261 mg, 1,0 mmoles) y Pd(dppf)Cl2 (80 mg, 0,1 mmoles) en 1,4-dioxano (10 mL) se le añadió carbonato de sodio (318 mg, 3,0 mmoles), la mezcla se calentó a 65°C y se agitó durante 18 horas. La mezcla se concentró a vacío, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1) para proporcionar el compuesto 1 (250 mg, rendimiento: 82%). LC-MS (ESI):m/z = 304 [M+H]+.
Preparac¡ón del Compuesto T-01
Una mezcla del compuesto 1 (30 mg, 0,1 mmoles), bencilamina (32 mg, 0,3 mmoles), fluoruro de potasio (17 mg, 0,3 mmoles), 1,4-dioxano (5 mL) y DMSO (1 mL) se calentó a 110°C y se agitó durante 20 horas. La mezcla se concentró a vacío, el residuo se diluyó con agua (50 mL), a continuación, se extrajo con acetato de etilo (50 mL). La capa orgánica se lavó con agua (20 mLx2) y salmuera saturada (20 mL) sucesivamente, a continuación, se secó sobre sulfato de sodio anhidro. Después de la filtración, el producto filtrado se concentró a vacío y el residuo se purificó mediante cromatografía en columna de sílice (diclorometano : metanol=100:1) para proporcionar el compuesto T-01 (20 mg, rendimiento: 53 %). LC-MS (ESI):m/z=375 [M+H]+. RMN H1 (400MHz, CDCla) 5: 8,29 (s, 1H), 8,24 (s, 1H), 7,80 (d, J=5,6Hz, 1H), 7,41-7,42 (m, 2H), 7,31-7,35 (m, 2H), 7,24-7,28 (m, 2H), 5,63 (br, 1H), 4,75 (d, J=6,0Hz, 2H), 4,73 (m, 1H), 2,93-3,06 (m, 2H), 1,76 (d, J=6,8Hz, 3H) ppm
Ejemplo 2
3-(4-(2-(Fen¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)butanon¡tr¡lo T-02
Ruta s¡ntét¡ca:

El compuesto 1 (30 mg, 0,1 mmoles), anilina (55 mg, 0,6 mmoles) y monohidrato de ácido p-tolueno sulfonico (76 mg, 0,4 mmoles) se disolvieron en isobutanol (8 mL), la mezcla se calentó a 110°C y se agitó durante 16 horas. La mezcla se concentró a vacío, y el residuo se diluyó con etanol (30 mL), a continuación, se lavó con dicarbonato de sodio acuoso saturado (30 mL), agua (30 mL) y salmuera saturada (30 mL) sucesivamente. Después de secar sobre sulfato de sodio anhidro, la mezcla se filtró, y el producto filtrado se concentró a vacío, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto T-02 (20 mg, rendimiento: 55 %). LC-MS (ESI):m/z=361 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,35 (s, 1H), 8,32 (s, 1H), 7,88 (d, J=5,6Hz, 1H), 7,75 (d, J=8,0Hz, 1H), 7,33-7,39 (m, 4H), 7,03-7,06 (m, 1H), 4,75-4,80 (m, 1H), 2,96-3,09 (m, 2H), 1,76 (d, J=6,8Hz, 3H) ppm
Ejemplo 3
3-C¡clopent¡l-3-(4-(2-(fen¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)propanon¡tr¡lo T-03
Ruta s¡ntét¡ca:

Preparac¡on del Compuesto 3-b
Bajo nitrógeno, una suspensión de bromuro de (cianometil)trifenilfosfonio (12 g, 31,49 mmoles) en THF anhidro (100 mL) se enfrió a 0°C, se añadió gota a gota una solución de n-BuLi 2,5 M en n-hexano (13 mL, 34,64 mmoles). La mezcla se agitó a 0°C durante otros 30 minutos, a continuación, se añadió ciclopentano-carbaldehído (3,1 g, 31,49 mmoles), y la mezcla se calentó a temperatura ambiente y se agitó durante 1 hora adicional. La reacción se sofocó con una solución acuosa saturada de cloruro de amonio (50 mL), se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro. La mezcla se filtró, el producto filtrado se concentró a vacío, el residuo se purificó
mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=10:1) para proporcionar un aceite incoloro 3-b (1,0 g, rendimiento: 26,2%). LC-m S (ESI):m/z=122 [M+H]+.
Preparación del Compuesto 3-a
A una solución del compuesto 3-b (1 g, 8,26 mmoles) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1 H-pirazol (2,4 g, 12,39 mmoles) en acetonitrilo (10 mL) se le añadió 1,8-diazabiciclo[5,4,0]undec-7-eno (2,5 g, 16,52 mmoles). La mezcla se agitó a 60°C durante 18 horas. La mezcla se concentró a vacío. Al residuo se le añadió agua (50 mL), a continuación, la mezcla se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro. La mezcla se filtró, el producto filtrado se concentró a vacío, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar un aceite de color amarillo 3-a (715 mg, rendimiento: 27,5 %). LC-MS (ESI):m/z=316 [M+H]+.
Preparación del Compuesto 3
Bajo nitrógeno, a una suspensión del compuesto 3-a (715 mg, 2,27 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (465 mg, 2,27 mmoles) y carbonato de sodio (72 mg, 6,80 mmoles) en 1,4-dioxano (4 mL) y agua (4 mL) se le añadió Pd(dppf)Cl2 (233 mg, 0,28 mmoles). La mezcla se agitó a 80°C durante 16 horas. La mezcla se concentró, y al residuo se le añadió agua (20 mL). A continuación, la mezcla se extrajo con cloruro de metileno (20 mLx3), las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, y a continuación, se secaron sobre sulfato de sodio anhidro, se filtraron. El producto filtrado se concentró a vacío, y el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1) para proporcionar un sólido de color amarillo claro 3 (330 mg, rendimiento: 40,7 %).LC-Ms (ESI):m/z=358 [M+H]+.
Preparación del Compuesto T-03
A una solución del compuesto 3 (50 mg, 0,14 mmoles) y anilina (39 mg, 0,42 mmoles) en isobutanol (0,5 mL) se le añadió monohidrato de ácido p-toluenosulfónico (54 mg, 0,28 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. La mezcla se enfrió a continuación, a temperatura ambiente, y se agitó durante 2 horas adicionales. La mezcla se filtró y el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar el compuesto T-03 (23 mg, rendimiento: 39,7%). LC-MS (ESI):m/z=415 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,37 (d, J=6Hz, 2H), 7,90 (d, J=6Hz, 1H), 7,76 (d, J=8Hz, 2H), 7,37 (m, 3H), 7,05 (t, J=7Hz, 1H), 4,28 (m, 1H), 3,17 (m, 1H), 2,97 (m, 1H), 2,61 (m, 1H), 1,99 (m, 1H), 1,71 (m, 7H) ppm
Ejemplo 4
3-Ciclopentil-3-(4-(2-(pirimidin-5-ilamino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)propanonitrilo T-04
Ruta sintética:

Preparación del Compuesto T-04
A una solución del compuesto 3 (50 mg, 0,14 mmoles) y 5-aminopirimidina (40 mg, 0,42 mmoles) en isobutanol (0,5 mL) se le añadió monohidrato de ácido p-toluenosulfónico (53 mg, 0,28 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. La mezcla se concentró a continuación, a vacío y el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar el compuesto T-04 (7 mg, rendimiento: 12%) en forma de un sólido de color amarillo. LC-MS
(ESI):m/z=417 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 9,37 (s, 2H), 8,76 (s, 1H), 8,65 (s, 1H), 8,41 (s, 1H), 8,21 (d, J=6Hz, 1H), 7,43 (d, J=6Hz, 1H), 4,53 (m, 1H), 3,12-3,28 (m, 2H), 2,56 (m, 1H), 1,97 (m, 1H), 1,41-1,72 (m, 7H) ppm
Ejemplo 5
2-(1-(Et¡lsulfon¡l)-3-(4-(2-(fen¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡tr¡lo T-05
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 5-c
Bajo nitrógeno, una suspensión de bromuro de (cianometil)trifenilfosfonio (13,4 g, 35,09 mmoles) en THF anhidro (100 mL) se enfrió a 0°C, se añadió gota a gota una solución de 2,5 M n-BuLi in n-hexano (15,5 mL, 38,59 mmoles). La mezcla se agitó a 0°C durante otros 30 minutos, a continuación, se añadió 3-oxoazetidin-1-carboxilato de ferc-butilo (6,0 g, 35,09 mmoles), y la mezcla se calentó a temperatura ambiente y se agitó durante 1 hora adicional. La reacción se sofocó con una solución acuosa saturada de cloruro de amonio (50 mL), se extrajo con acetato de etilo (150 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (100 mLx3) y salmuera saturada (100 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro. La mezcla resultante se filtró, el producto filtrado se concentró a vacío, y el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar un sólido de color blanco 5-c (2,5 g, rendimiento: 37%). LC-MS (ESI):m/z=217 [M+Na]+.
Preparación del Compuesto 5-b
A una solución del compuesto 5-c (6,0 g, 30,93 mmoles) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1 H-pirazol (9,2 g, 47,42 mmoles) en acetonitrilo (60 mL) se le añadió 1,8-diazabiciclo[5,4,0]undec-7-eno (10,0 g, 65,79 mmoles). La mezcla se agitó a 60°C durante 18 horas. La mezcla se concentró a vacío. Al residuo se le añadió una solución acuosa de cloruro de hidrógeno 1 N (100 mL), a continuación, la mezcla se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro. La mezcla resultante se filtró, el producto filtrado se concentró a vacío, y el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar un sólido de color blanco 5-b (7,1 g, rendimiento: 59,2%). Lc -MS (ESI):m/z=389 [M+H]+.
Preparación del Compuesto 5-a
Bajo nitrógeno, a una suspensión del compuesto 5-b (4,0 g, 10,3 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (2,52 g, 12,4 mmoles) y carbonato de sodio (3,3 g, 31,2 mmoles) en 1,4-dioxano (25 mL) y agua (25 mL) se le añadió Pd(dppf)Cl2 (1,1 g, 1,5 mmoles). La mezcla se agitó a 80°C durante 16 horas. La mezcla se concentró a vacío. Al residuo se le añadió agua (200 mL), a continuación, la mezcla se extrajo con cloruro de metileno (200 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (100 mLx3) y salmuera saturada (100 mL) sucesivamente, y a continuación, se secaron sobre sulfato de sodio anhidro, se filtraron. El producto filtrado se concentró a vacío, y el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1) para proporcionar un sólido de color amarillo claro 5-a (3,2 g, rendimiento: 63%).LC-MS (ESI):m/z=431 [M+H]+.
Preparación del Compuesto 5
A una solución del compuesto 5-a (310 mg, 0,72 mmoles) en diclorometano (2 mL) se le añadió una solución de cloruro de hidrógeno en 1,4-dioxano (4 N, 1 mL), la mezcla se agitó a temperatura ambiente durante 16 horas. La mezcla resultante se concentró y al residuo se le añadió diclorometano (10 mL) y trietilamina (2 mL). La mezcla se enfrió a continuación, a 0°C, se añadió gota a gota cloruro de etanosulfonilo (154 mg, 1,37 mmoles), y una vez completada la adición gota a gota, la mezcla se agitó a 0°C durante 30 minutos adicionales. A la mezcla resultante se le añadió agua (5 mL), y la mezcla se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro. La mezcla se filtró, el producto filtrado se concentró a vacío, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1) para proporcionar el compuesto 5 (108 mg, rendimiento: 34%). LC-MS (ESI):m/z=423 [M+H]+.
Preparación del Compuesto T-05
A una solución del compuesto 5 (50 mg, 0,12 mmoles) y anilina (33 mg, 0,36 mmoles) en isobutanol (1 mL) se le añadió monohidrato de ácido p-toluenosulfónico (45 mg, 0,24 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. La mezcla se concentró a continuación, a presión reducida y el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-05 (21 mg, rendimiento: 37 %). lC-MS (ESI):m/z=480 [M+H]+.
RMN H1 (400MHz , CDCla) 5: 8,47 (s, 1H), 8,39 (s, 1H), 7,92 (d, J=5Hz, 1H), 7,75 (d, J=8Hz, 2H), 7,38 (m, 3H), 7,06 (t, J=7Hz, 1H), 4,64 (d, J=9Hz, 2H), 4,26 (d, J= 9Hz, 2H), 3,39 (s, 2H), 3,10 (q, J= 7Hz, 2H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 6
2-(1-(Etilsulfonil)-3-(4-(2-((tetrahidro-2H-piran-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-06
Ruta sintética:
El compuesto 5 (150 mg, 0,36 mmoles), 4-aminotetrahidropiran (90 mg, 0,89 mmoles) y fluoruro de potasio anhidro (31 mg, 0,54 mmoles) se suspendieron en DMSO (2 mL), la mezcla se agitó a 80°C durante 16 horas. La mezcla se enfrió a continuación, a temperatura ambiente y se diluyó con diclorometano (50 mL), después de lavar con agua (20 mLx2), la capa orgánica se secó sobre sulfato de sodio anhidro. La mezcla se filtró, el producto filtrado se concentró a vacío, y el residuo se purificó mediante TLC preparativa (éter de petróleo : acetato de etilo=2:1) para proporcionar el compuesto T-06 (18 mg, rendimiento: 11 %). LC-MS (ESI):m/z=488 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,41 (s, 1H), 8,34 (s, 1H), 7,83 (d, J=5Hz, 1H), 7,25 (d, J=5Hz, 1H), 5,10 (br, 1H), 4,63 (d, J= 9Hz, 2H), 4,24 (d, J= 9Hz, 2H), 4,19 (m, 1H), 4,05 (m, 2H), 3,55 (m, 2H), 3,41 (s, 2H), 3,11 (q, J= 7Hz, 2H), 2,10 (m, 2H), 1,61 (m, 2H), 1,42 (t, J= 7Hz, 3H) ppm
Ejemplo 7
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((4-morfol¡nofen¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡trilo T-07
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 7-b
Se disolvieron 1 -fluoro-4-nitrobenceno (2,9 g, 20,56 mmoles) y morfolina (3,6 g, 41,13 mmoles) en DMSO (15 mL), se añadió carbonato de potasio y la mezcla se agitó a 80°C durante 18 horas. Después de enfriar a temperatura ambiente, la mezcla se vertió en agua (100 mL), precipitó un sólido de color amarillo. Después de la filtración, la torta del filtro se lavó con agua. La torta del filtro se secó a continuación, a vacío durante 24 horas para proporcionar el compuesto 7-b (3,5 g, rendimiento: 82%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparac¡ón del Compuesto 7-a
El compuesto 7-b (1,5 g, 7,21 mmoles) y cloruro de amonio (1,0 g, 18,03 mmoles) se disolvieron en etanol-agua al 50% (20 mL), a continuación, se añadió polvo de Zn (1,2 g, 18,03 mmoles). La mezcla se sometió a reflujo durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se filtró. La torta del filtro se lavó con etanol (10 mL), el producto filtrado combinado se concentró a presión reducida. El residuo se diluyó con agua (50 mL) y se extrajo con acetato de etilo (50 mLx3), la capa orgánica se secó sobre sulfato de sodio anhidro. Después de la filtración, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color pardo claro 7-a (1,1 g, rendimiento: 86%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=179 [M+H]+.
Preparac¡ón del Compuesto T-07
El compuesto 5 (150 mg, 0,36 mmoles) y el compuesto 7-a (190 mg, 1,07 mmoles) se disolvieron en isobutanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (135 mg, 0,71 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se agitó durante 30 minutos adicionales. Se produjo un precipitado sólido, después de la filtración, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar un sólido de color amarillo T-07 (21 mg, rendimiento: 26%). LC-MS (ESI): m/z=565 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,44 (s, 1H), 8,35 (s, 1H), 7,89 (d, J=5Hz, 1H), 7,61 (d, J=9Hz, 2H), 7,32 (d, J=5Hz, 1H), 6,96 (d, J=9Hz, 2H), 4,64 (d, J=9Hz, 2H), 4,24 (d, J= 9Hz, 2H), 3,89 (m, 4H), 3,41 (s, 2H), 3,08-3,16 (m, 6H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 8
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((4-(2-morfol¡noetox¡)fen¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)acetonitr¡lo T-08
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 8-c
Se disolvieron p-nitrofenol (6,0 g, 43,16 mmoles) y 1,2-dibromoetano (16,0 g, 86,02 mmoles) en acetona (60 mL), a continuación, se añadió carbonato de potasio (9,0 g, 65,22 mmoles). La mezcla se sometió a reflujo durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla resultante se filtró y la torta del filtro se lavó con acetona (30 mL). El producto filtrado se concentró a presión reducida, y el residuo se diluyó con agua (50 mL), a continuación, se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (50 mLx3) y salmuera saturada (50 mL). Después de secar sobre sulfato de sodio anhidro, la mezcla se filtró, y el producto filtrado se concentró a vacío, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar un sólido de color amarillo claro 8-c (2,1 g, rendimiento: 19%).
Preparac¡ón del Compuesto 8-b
El compuesto 8-c (1,0 g, 4,08 mmoles) y morfolina (702 mg, 8,16 mmoles) se disolvieron en acetonitrilo (5 mL), se añadió carbonato de potasio (1,2 g, 8,16 mmoles). La mezcla se sometió a reflujo durante 3 horas, y a continuación, se enfrió a temperatura ambiente, después de la filtración, la torta del filtro se lavó con acetato de etilo (30 mL). El producto filtrado se concentró a presión reducida, el residuo se diluyó con agua (50 mL) y se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron y lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente. Después de secar sobre sulfato de sodio anhidro, la mezcla se filtró, y el producto filtrado se concentró a vacío para proporcionar un sólido de color amarillo claro 8-b (900 mg), que se utilizó directamente para la siguiente etapa sin purificación adicional.
Preparac¡ón del Compuesto 8-a
Bajo hidrógeno (1 atm), a una solución del compuesto 8-b (900 mg, 3,57 mmoles) en etanol (50 mL) se le añadió Pd-C al 10% (0,5 g). La mezcla se agitó a 25°C durante 18 horas, y a continuación, se filtró. El producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo claro 8-a (760 mg, rendimiento: 96%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto T-08
El compuesto 8-a (105 mg, 0,47 mmoles) y el compuesto 5 (100 mg, 0,24 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (68 mg, 0,36 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, después de enfriar a temperatura ambiente, la mezcla se agitó durante 30 minutos adicionales. La mezcla resultante se filtró, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar un sólido de color amarillo T-08 (51 mg, rendimiento: 35 %). LC-MS (ESI): m/z=609 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,43 (s, 1H), 8,35 (s, 1H), 7,86 (d, J=5Hz, 1H), 7,61 (d, J=9Hz, 2H), 7,31 (d, J=5Hz, 1H), 7,19 (s, 1H), 6,93 (d, J=9Hz, 2H), 4,63 (d, J=9Hz, 2H), 4,16 (t, J=5Hz, 2H), 3,78 (t, J=4Hz, 4H), 3,39 (s, 2H), 3,09 (q, J=6Hz, 2H), 2,89 (t, J=4Hz, 2H), 2,69 (s, 4H), 1,43 (t, J= 6Hz, 3H) ppm
Ejemplo 9
2-(1-(Etilsulfonil)-3-(4-(2-((4-(2-(pirrolidin-1-il)etoxi)fenil)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-09
Ruta sintética:

Preparación del Compuesto 9-b
El compuesto 8-c (1,0 g, 4,08 mmoles) y pirrolidina (580 mg, 8,16 mmoles) se disolvieron en acetonitrilo (50 mL), se añadió carbonato de potasio (1,2 g, 8,16 mmoles). La mezcla se sometió a reflujo durante 3 horas, y a continuación, se enfrió a temperatura ambiente. Después de la filtración, la torta del filtro se lavó con acetato de etilo (30 mL). El producto filtrado se concentró a presión reducida, y el residuo se diluyó con agua (50 mL) y se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron y lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente. Después de secar sobre sulfato de sodio anhidro, la mezcla se filtró, y el producto filtrado se concentró a vacío para proporcionar un aceite de color amarillo 9-b (1,0 g), que se utilizó directamente para la siguiente etapa sin purificación adicional.
Preparación del Compuesto 9-a
Bajo hidrógeno (1 atm), a una solución del compuesto 9-b (1,0 g, 4,24 mmoles) en etanol (50 mL) se le añadió Pd-C al 10% (0,5 g). La mezcla se agitó a 25°C durante 18 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo claro 9-a (780 mg, rendimiento: 90%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto T-09
Se disolvieron el compuesto 9-a (73 mg, 0,35 mmoles) y el compuesto 5 (50 mg, 0,12 mmoles) en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (45 mg, 0,24 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se agitó durante 30 minutos adicionales. La
mezcla resultante se filtró, y el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar un sólido de color amarillo T-09 (51 mg, rendimiento: 36%). LC-MS (ESI): m/z=593 [M+H]+.
RMN H1 (400MHz, MeOD) 5: 8,60 (m, 1H), 8,20 (m, 2H), 7,55 (m, 1H), 7,22 (m, 1H), 7,02 (m, 2H), 4,61 (d, J=9Hz, 2H), 4,34 (m, 2H), 4,26 (d, J=9Hz, 2H), 3,77 (m, 2H), 3,68 (m, 2H), 3,60 (s, 2H), 3,17-3,24 (m, 4H), 2,04-2,20 (m, 4H), 1,38 (t, J= 6Hz, 3H) ppm
Ejemplo 10
2- (3-(4-(2-((4-(1H-1,2,4-Tr¡azol-1-¡l)fen¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(et¡lsulfon¡l)azet¡d¡n-3- ¡l)acetonitr¡lo T-10
Ruta s¡ntét¡ca:

A 0°C, a una solución de 1,2,4-triazol (2,7 g, 39,01 mmoles) en DMF (50 mL) se le añadió hidruro de sodio (1,6 g, 39,01 mmoles). La mezcla se agitó durante 30 minutos y se añadió 1-flouro-4-nitrobenceno (5,0 g, 35,46 mmoles), y la mezcla resultante se agitó durante 2 horas adicionales. Se añadió agua (150 mL) lentamente a la mezcla, se produjo un precipitado sólido. Después de la filtración, la torta del filtro se lavó con agua (50 mLx3), y el sólido se secó a vacío durante 8 horas para proporcionar un sólido de color amarillo 10-b (6,2 g, rendimiento: 91%), que se utilizó para la siguiente etapa sin purificación adicional. LC-MS (ESI): m/z=191 [M+H]+.
Preparac¡ón del Compuesto 10-a
El compuesto 10-b (3,0 g, 15,78 mmoles) y cloruro de amonio (2,1 g, 39,62 mmoles) se disolvieron en etanol-agua al 50% (60 mL). A continuación, se añadió polvo de Zn (2,6 g, 40 mmoles). La mezcla se sometió a reflujo durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se filtró, y la torta del filtro se lavó con etanol (10 mL). El producto filtrado combinado se concentró a presión reducida, y el residuo se diluyó con agua (100 mL) y se extrajo con acetato de etilo (100 mLx3). La capa orgánica se secó sobre sulfato de sodio anhidro. Después de la filtración, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color amarillo 10-a (1,1 g, rendimiento: 81 %), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=161 [M+H]+.
Preparac¡ón del Compuesto T-10
El compuesto 10-a (114 mg, 0,71 mmoles) y el compuesto 5 (100 mg, 0,24 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (180 mg, 0,95 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se agitó durante 30 minutos adicionales, y se produjo un precipitado sólido. Después de la filtración, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-10 (25 mg, rendimiento: 19%). LC-MS (ESI): m/z=547 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,58 (d, J=4Hz, 2H), 8,41 (s, 1H), 8,13 (s, 1H), 8,09 (d, J=6Hz, 1H), 7,92 (d, J=9Hz, 2H),
7,71 (d, J=9Hz, 2H), 7,49 (d, J=6Hz, 1H), 4,66 (d, J=9Hz, 2H), 4,25 (d, J=9Hz, 2H), 3,45 (s, 2H), 3,10 (q, J=6Hz, 2H), 1,43 (t, J= 6Hz, 3H) ppm
Ejemplo 11
2-(3-(4-(2-((4-(1H-P¡razol-1-¡l)fen¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(et¡lsulfon¡l)azet¡d¡n-3-¡l)acetonitr¡lo T-11
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 11-b
A 0°C, a una solución de pirazol (2,7 g, 39,01 mmoles) en DMF (50 mL) se le añadió hidruro de sodio (1,6 g, 39,01 mmoles), la mezcla se agitó durante 30 minutos. A continuación, se añadió 1-flouro-4-nitrobenceno (5,0 g, 35,46 mmoles), y la mezcla resultante se agitó durante 2 horas adicionales. Se añadió agua (250 mL) lentamente a la mezcla, y se produjo un precipitado sólido. Después de la filtración, la torta del filtro se lavó con agua (50 mLx3), y el sólido se secó a vacío durante 8 horas para proporcionar un sólido de color amarillo 11-b (6 g, rendimiento: 90%), que se utilizó para la siguiente etapa sin purificación adicional.
Preparac¡ón del Compuesto 11-a
El compuesto 11-b (1,0 g, 5,29 mmoles) y cloruro de amonio (0,7 g, 13,23 mmoles) se disolvieron en etanol-agua al 50% (20 mL). A continuación, se añadió polvo de Zn (0,9 g, 13,23 mmoles). La mezcla se sometió a reflujo durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se filtró, y la torta del filtro se lavó con etanol (10 mL). El producto filtrado combinado se concentró a presión reducida, y el residuo se diluyó con agua (50 mL) y se extrajo con acetato de etilo (50 mLx3). La capa orgánica se secó sobre sulfato de sodio anhidro. Después de la filtración, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color amarillo 11-a (670 mg, rendimiento: 80%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=160 [M+H]+.
Preparac¡ón del Compuesto T-11
El compuesto 11-a (79 mg, 0,50 mmoles) y el compuesto 5 (70 mg, 0,17 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (95 mg, 0,50 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se agitó durante 30 minutos adicionales, y se produjo un precipitado sólido. Después de la filtración, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-11 (31 mg, rendimiento: 34%). LC-MS (ESI): m/z=546 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,49 (s, 1H), 8,40 (s, 1H), 7,94 (d, J=6Hz, 1H), 7,91 (d, J=2Hz, 1H), 7,84 (d, J=9Hz, 2H), 7,68-7,73 (m, 3H), 7,40 (d, J=6Hz, 2H), 6,47 (t, J=2Hz, 1H), 4,65 (d, J=9Hz, 2H), 4,26 (d, J=9Hz, 2H), 3,43 (s, 2H), 3,10 (q, J=6Hz, 2H), 1,43 (t, J= 6Hz, 3H) ppm
Ejemplo 12
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((4-(met¡lsulfon¡l)fen¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-12
Ruta s¡ntét¡ca:

Se disolvieron 4-metilsulfonilanilina (98 mg, 0,57 mmoles) y el compuesto 5 (80 mg, 0,19 mmoles) en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (72 mg, 0,38 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se agitó durante 30 minutos adicionales, y se produjo un precipitado sólido. Después de la filtración, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 65%-95%-10%) para proporcionar un sólido de color blanco T-12 (30 mg, rendimiento: 28%). LC-Ms (ESI): m/z=558 [M+H]+.
RMN H1 (400MHz, CDCls) 5: 8,51 (s, 1H), 8,40 (s, 1H), 7,92-8,00 (m, 6H), 7,44 (d, J=6Hz, 1H), 4,66 (d, J=9Hz, 2H), 4,26 (d, J=9Hz, 2H), 3,44 (s, 2H), 3,13 (q, J=6Hz, 2H), 3,07 (m, 3H), 1,44 (t, J= 6Hz, 3H) ppm
Ejemplo 13
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((3-morfol¡nofen¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡trilo T-13
Ruta s¡ntét¡ca:

Se disolvieron 3-(4-morfolinil)anilina (102 mg, 0,57 mmoles) y el compuesto 5 (80 mg, 0,19 mmoles) en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (180 mg, 0,95 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se agitó durante 30 minutos adicionales, y se produjo un precipitado sólido. Después de la filtración, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color blanco T-13 (15 mg, rendimiento: 14%). LC-Ms (ESI): m/z=565 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 10,2 (br, 1H), 8,56 (s, 1H), 8,36 (s, 1H), 8,04 (d, J=6Hz, 1H), 7,61 (d, J=6Hz, 2H), 7,28-7,36 (m, 3H), 6,75 (m, 1H), 4,63 (d, J=9Hz, 2H), 4,25 (d, J= 9Hz, 2H), 3,90 (m, 4H), 3,41 (s, 2H), 3,24 (m, 2H), 3,08 (q, J=6Hz, 2H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 14
2-(1-(Et¡lsulfon¡l)-3-(4-(2-(p¡r¡m¡d¡n-5-¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-14 Ruta s¡ntét¡ca:

Bajo nitrógeno, a una suspensión del compuesto 5 (130 mg, 0,31 mmoles), 5-amino pirimidina (89 mg, 0,94 mmoles) y carbonato de cesio (102 mg, 0,32 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (47 mg, 0,05 mmoles) y BINAP (34 mg, 0,05 mmoles). La mezcla se calentó a 120°C mediante microondas y se agitó durante 60 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (20 mL), y a continuación, se filtró, el producto filtrado se concentró a presión reducida. El residuo se diluyó con metanol (5 mL), y se produjo un precipitado sólido. Después de la filtración, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar un sólido de color amarillo claro T-14 (16 mg, rendimiento: 10%). LC-MS (ESI): m/z=482 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 9,29 (s, 2H), 8,94 (s, 1H), 8,55 (s, 1H), 8,40 (br, 1H), 8,39 (s, 1H), 8,02 (d, J=6Hz, 1H), 7,42 (d, J=6Hz, 1H), 4,67 (d, J=9Hz, 2H), 4,27 (d, J= 9Hz, 2H), 3,44 (s, 2H), 3,11 (q, J= 7Hz, 2H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 15
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-met¡l-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡trilo T-15
Ruta s¡ntét¡ca:
Preparac¡ón del Compuesto 15-b
A 0°C, a una solución de 4-nitropirazol (3,3 g, 29,2 mmoles) en THF anhidro (30 mL) se le añadió hidruro de sodio (1,3 g, 32,1 mmoles). Después de agitar la mezcla durante 1 hora, se añadió yodometano (2 mL) lentamente, y la mezcla resultante se agitó durante 2 horas adicionales. A continuación, la mezcla se vertió en agua helada (100 mL), se extrajo con acetato de etilo (50 mLx3), y la capa orgánica se secó sobre sulfato de sodio anhidro. La mezcla se filtró, el
producto filtrado se concentró a presión reducida. El residuo se añadió a un disolvente componente (20 mL) de éter de petróleo y acetato de etilo (20:1), se agitó, y se produjo un precipitado sólido. Después de la filtración, el sólido se secó a vacío durante 8 horas para proporcionar un sólido de color blanco 15-b (2,6 g, rendimiento: 70%), que se utilizó directamente para la siguiente etapa sin purificación adicional. LC-MS (ESI): m/z=128 [M+H]+.
Preparación del Compuesto 15-a
Bajo hidrógeno (1 atm), a una solución del compuesto 15-b (1,0 g, 7,87 mmoles) en etanol (15 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 18 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar un aceite de color rojo 15-a (700 mg, rendimiento: 92 %).
Preparación del Compuesto T-15
Bajo nitrógeno, a una suspensión del compuesto 5 (200 mg, 0,47 mmoles), el compuesto 15-a (138 mg, 1,42 mmoles) y carbonato de cesio (309 mg, 0,95 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (55 mg, 0,06 mmoles) y BINAP (40 mg, 0,06 mmoles). La mezcla se calentó a 120°C mediante microondas y se agitó durante 60 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (20 mL), y a continuación, se filtró, el producto filtrado se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo claro T-15 (23 mg, rendimiento: 14%). LC-MS (ESI): m/z= 484[M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,44 (s, 1H), 8,34 (s, 1H), 8,55 (s, 1H), 7,97 (s, 1H), 7,86 (d, J=6Hz, 1H), 7,53 (s, 1H), 7,31 (d, J=6Hz, 1H), 7,19 (s, 1H), 4,63 (d, J=9Hz, 2H), 4,24 (d, J= 9Hz, 2H), 3,93 (s, 3H), 3,39 (s, 2H), 3,09 (q, J= 7Hz, 2H), 1,41 (t, J= 7Hz, 3H) ppm
Ejemplo 16
N-(Cianometil)-4-(2-(fenilamino)tieno[3,2-d]pirimidin-4-il)benzamida T-16
Ruta sintética:

Preparación del Compuesto 16-b
Bajo nitrógeno, a una suspensión de 2,4-diclorotieno[3,2-d]pirimidina (1,0 g, 4,9 mmoles), ácido 4-boronobenzoico (542 mg, 3,3 mmoles) y carbonato de sodio (2,1 g, 19,6 mmoles) en 1,4-dioxano (10 mL) y agua (10 mL) se le añadió Pd(dppf)Cl2 (400 mg, 0,5 mmoles). La mezcla se agitó a 80°C durante 16 horas, a continuación, se enfrió a temperatura ambiente. Se añadió lentamente ácido clorhídrico (1,0 N) para ajustar a pH=3. La mezcla se extrajo a continuación, con acetato de etilo (20 mLx3), la capa orgánica se lavó con dicarbonato de sodio acuoso saturado (50 mL) y salmuera saturada (50 mL). Después de secar sobre sulfato de sodio anhidro, la mezcla se filtró, y el producto filtrado se concentró a presión reducida para proporcionar un sólido de color amarillo 16-b (160 mg, rendimiento: 20%), que se utilizó directamente para la siguiente etapa sin purificación adicional. LC-MS (ESI): m/z=291 [M+H]+.
Preparación del Compuesto 16-a
A una suspensión del compuesto 16-b in diclorometano se le añadieron cloruro de oxalilo (4 mL) y DMF (0,1 mL) respectivamente, se agitó a temperatura ambiente durante 3 horas. La mezcla se concentró a continuación, a presión reducida, el residuo se diluyó con diclorometano (50 mL) y se enfrió a 0°C. Se añadieron lentamente a la mezcla sucesivamente hidrocloruro de aminoacetonitrilo (75 mg, 0,81 mmoles) y trietilamina (0,6 mL). Después de templar lentamente a temperatura ambiente, la mezcla se agitó durante 2 horas adicionales, se añadieron diclorometano (50 mL) y agua (20 mL). La capa orgánica se lavó con hidrocloruro acuoso (1 N, 10 mL) y agua (20 mL), y a continuación, se secó sobre sulfato de sodio anhidro. La mezcla se filtró, el producto filtrado se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar 16-a (125 mg, rendimiento: 68 %). LC-MS (ESI): m/z=329 [M+H]+.
Preparación del Compuesto T-16
Se disolvieron anilina (108 mg, 1,16 mmoles) y el compuesto 16-a (125 mg, 0,38 mmoles) en n-butanol (0,5 mL), se añadió monohidrato de ácido p-toluenosulfónico (133 mg, 0,7 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. La mezcla se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-16 (33 mg, rendimiento: 23%). LC-Ms (ESI): m/z=386 [M+H]+.
RMN H1 (400MHz, MeOD) 5: 8,31-8,34 (m, 2H), 8,19 (d, J=5Hz, 1H), 8,08 (dd, J=2Hz J=6Hz, 2H), 7,82 (dd, J=2Hz J=8Hz, 3H), 7,40 (d, J=6Hz, 1H), 7,33 (t, J=8Hz, 2H), 7,01 (t, J=8Hz, 1H), 4,39 (s, 2H) ppm
Ejemplo 17
N-(Terc-butil)-4-(2-((4-morfolinofenil)amino)tieno[3,2-d]pirimidin-4-il)bencenosulfonamida T-17
Ruta sintética:

Preparación del Compuesto 17-c
A 0°C, cloruro de 4-bromobencenosulfonilo (3,0 g, 11,8 mml) se le añadió a una solución de f-butilamina (2,0 g, 29,6mmoles) en diclorometano (30 mL) y se agitó durante 30 minutos. La mezcla de reacción se lavó con hidrocloruro acuoso (1 N, 20 mL) y agua (30 mLx3) sucesivamente. La capa orgánica se secó sobre sulfato de sodio anhidro. Después de la filtración, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color blanco 17-c (3,6 g, rendimiento:100%), que se utilizó directamente para la siguiente etapa sin purificación adicional.
Preparación del Compuesto 17-b
Bajo nitrógeno, a una suspensión del compuesto 17-c (500 mg, 1,72 mmoles), bis(pinacolato)diboro (524 mg, 2,06 mmoles) y acetato de potasio (505 mg, 5,15 mmoles) en 1,4-dioxano (5 mL) se le añadió Pd(dppf)Cl2 (130 mg, 0,17 mmoles). La mezcla se agitó a 80°C durante 16 horas. Después de la concentración de la mezcla a presión reducida, el residuo se diluyó con agua (50 mL), a continuación, se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron y lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente. Después de secar sobre
sulfato de sodio anhidro, la mezcla se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar un sólido de color blanco 17-b (350 mg, rendimiento: 60%). LC-MS (ESI): m/z=340 [M+H]+.
Preparación del Compuesto 17-a
Bajo nitrógeno, a una suspensión del compuesto 17-b (300 mg, 0,89 mmoles), el compuesto 5 (181 mg, 0,89 mmoles) y carbonato de sodio (281 mg, 2,67 mmoles) en etanol (2 mL) y agua (2 mL) se le añadió Pd(dppf)Cl2 (74 mg, 0,1 mmoles). La mezcla se agitó a 80°C durante 16 horas. Después de la concentración de la mezcla a presión reducida, el residuo se diluyó con agua (50 mL), y a continuación, se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron y lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente. Después de secar sobre sulfato de sodio anhidro, la mezcla se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar un sólido de color amarillo claro 17-a (105 mg, rendimiento: 32%). LC-MS (ESI): m/z=382 [M+H]+.
Preparación del Compuesto T-17
El compuesto 17-a (100 mg, 0,26 mmoles) y 4-(4-morfolinil)anilina (140 mg, 0,79 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (180 mg, 0,95 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se agitó durante otros 30 minutos y se produjo un precipitado sólido. Después de la filtración, el sólido se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-17 (15 mg, rendimiento: 25%). LC-MS (ESI): m/z=524 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,26 (d, J=9Hz, 2H), 8,08 (d, J=9Hz, 1H), 7,89 (d, J=5Hz, 1H), 7,94 (d, J=5Hz, 1H), 7,70 (br, 2H), 7,3,8 (d, J=5Hz, 1H), 7,04 (br, 2H), 4,82 (s, 1H), 3,90 (m, 4H), 3,20 (br, 4H), 1,27 (s, 9H) ppm
Ejemplo 18
2-(1-(Etilsulfonil)-3-(4-(2-((4-(metilsulfonil)fenil)amino)tieno[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-18
Ruta sintética:

Preparación del Compuesto 18-a
Bajo nitrógeno, a una suspensión de 2,4-diclorotieno[2,3-d]pirimidina (500 mg, 2,45 mmoles), el compuesto 5-a (633
mg, 1,63 mmoles) y carbonato de sodio (779 mg, 7,35 mmoles) en 1,4-dioxano (5 mL) se le añadió Pd(dppf)Cl2 (250 mg, 0,3 mmoles). La mezcla se agitó a 80°C durante 4 horas, a continuación, se enfrió a temperatura ambiente. Se añadió diclorometano (50 mL), la mezcla resultante se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1 a diclorometano:metanol=10:1) para proporcionar un sólido de color gris 18-a (360 mg, rendimiento: 52%). LC-MS (ESI): m/z=431 [M+H]+.
Preparación del Compuesto 18
El compuesto 18-a (360 mg, 0,84 mmoles) se disolvió en diclorometano (2 mL), se añadió una solución de hidrocloruro 4N en 1,4-dioxano (4 mL). La mezcla se agitó a temperatura ambiente durante 16 horas, y a continuación, se concentró a presión reducida. El residuo se diluyó con diclorometano (10 mL) y trietilamina (2 mL). La mezcla se enfrió a 0°C, se añadió gota a gota cloruro de etilsulfonilo (141 mg, 1,26 mmoles). Una vez completada la adición gota a gota, la mezcla se agitó a 0°C durante 30 minutos adicionales. Se añadió agua (5 mL), y la mezcla se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1) para proporcionar el compuesto 18 (90 mg, rendimiento: 26%). LC-MS (ESI): m/z=423 [M+H]+.
Preparación del Compuesto T-18
El compuesto 18 (80 mg, 0,19 mmoles) y 4-metilsulfonilanilina (98 mg, 0,57 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (72 mg, 0,38 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas. La mezcla se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar un sólido de color amarillo claro T-18 (13 mg, rendimiento: 13%). LC-MS (ESI): m/z=558 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 9,70 (br, 1H), 8,75 (s, 1H), 8,29 (s, 1H), 8,99 (d, J=9Hz, 2H), 7,93 (d, J=9Hz, 2H), 7,51 (d, J=6Hz, 1H), 7,43 (d, J=6Hz, 1H), 4,66 (d, J=9Hz, 2H), 4,28 (d, J= 9Hz, 2H), 3,44 (s, 2H), 3,10 (q, J= 7Hz, 2H), 3,06 (s, 3H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 19
4-((4-(1-(3-(Cianometil)-1-(etilsulfonil)azetidin-3-il)-1 H-pirazol-4-il)tieno[3,2-d]pirimidin-2-il)amino)benzonitrilo T-19
Ruta sintética:

Preparación del Compuesto T-19
El compuesto 5 (50 mg, 0,12 mmoles) y 4-aminobenzonitrilo (42 mg, 0,36 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (45 mg, 0,24 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 65%-95%-10%) para proporcionar un sólido de color amarillo claro T-19 (13 mg, rendimiento: 13%). LC-MS (ESI): m/z=505 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,49 (s, 1H), 8,39 (s, 1H), 7,98 (d, J=6Hz, 1H), 7,90 (d, J=9Hz, 2H), 7,65 (d, J=9Hz, 2H), 7,43 (d, J=6Hz, 1H), 4,66 (d, J=9Hz, 2H), 4,26 (d, J= 9Hz, 2H), 3,44 (s, 2H), 3,11 (q, J= 7Hz, 2H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 20
2-(3-(4-(2-(Fenilamino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)oxetan-3-il)acetonitrilo T-20
Ruta sintética:

Preparación del Compuesto 20-b
A temperatura ambiente, se disolvieron bromuro de (cianometil)trifenilfosfonio (10,0 g, 33,2 mmoles) y 3-oxetanona (1,2 g, 16,7 mmoles) en diclorometano (100 mL), la mezcla se agitó durante 16 horas, a continuación, se concentró a presión reducida. El residuo se añadió a un disolvente componente (50 mL) de éter de petróleo y acetato de etilo (10:1), y se produjo la precipitación de un sólido de color blanco. Después de la filtración, el producto filtrado se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 20-b (1,0 g, rendimiento: 63%).
RMN H1 (400MHz, CDCls) 5: 5,39 (m, 2H), 5,30 (m, 2H), 5,25 (m, 1H) ppm
Preparación del Compuesto 20-a
A una solución del compuesto 20-b (500 mg, 5,26 mmoles) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (1,3 g, 6,7 mmoles) en acetonitrilo (10 mL) se le añadió 1,8-diazabiciclo[5,4,0]undec-7-eno (1,6 g, 10,53 mmoles). La mezcla se agitó a 60°C durante 18 horas, a continuación, se concentró a presión reducida. Se añadió una solución acuosa de hidrocloruro 1 N al residuo para ajustar a pH=3-4, a continuación, la mezcla se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, se filtró, y el producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo 20-a (900 mg, rendimiento: 59%), que se utilizó directamente para la siguiente etapa sin purificación adicional. LC-MS (ESI): m/z=290 [M+H]+.
Preparación del Compuesto 20
Bajo nitrógeno, el compuesto 20-a (300 mg, 1,04 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (317 mg, 1,56 mmoles) y carbonato de sodio (331 mg, 3,12 mmoles) se suspendieron en 1,4-dioxano (0,5 mL) y agua (0,5 mL), se añadió Pd(dppf)Cl2 (82 mg, 0,1 mmoles). La mezcla se agitó a 80°C durante 4 horas, a continuación, se concentró a presión reducida. Al residuo se le añadió agua (20 mL), la mezcla se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1) para proporcionar un sólido de color amarillo claro 20 (102 mg, rendimiento: 30%). LC-MS (ESI): m/z=332 [M+H]+.
Preparación del Compuesto T-20
El compuesto 20 (50 mg, 0,15 mmoles) y anilina (42 mg, 0,45 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (58 mg, 0,3 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo claro T-20 (23 mg, rendimiento: 40%). LC-MS (ESI): m/z=389 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,45 (s, 1H), 8,40 (s, 1H), 7,89 (d, J=5Hz, 1H), 7,75 (d, J=8Hz, 2H), 7,37 (m, 3H), 7,20
(s, 1H), 7,05 (t, J=7Hz, 1H), 5,20 (d, J=8Hz, 2H), 4,89 (d, J= 8Hz, 2H), 3,46 (s, 2H) ppm
Ejemplo 21
2-(1-(Et¡lsulfon¡l)-3-(4-(2-(p¡r¡d¡n-4-¡lam¡no)tieno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1 H-pirazol-1-il)azetidin-3-il)acetonitrilo T-21 Ruta sintética:

Preparación del Compuesto 21-a
A una solución de 4-nitropiridina 1-oxide (600 mg, 6,46 mmoles) y formiato de amonio (622 mg, 10,92 mmoles) en etanol (10 mL) se le añadió lentamente Pd-C al 10% (0,6 g). La mezcla se agitó a temperatura ambiente durante 16 horas. Después de la filtración, el producto filtrado se concentró a presión reducida. Al residuo se le añadió agua (20 mL), la mezcla se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color amarillo claro 21-a (380 mg, rendimiento: 94%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparac¡ón del Compuesto T-21
Bajo nitrógeno, a una suspensión del compuesto 5 (100 mg, 0,24 mmoles), el compuesto 21-a (45 mg, 0,48 mmoles) y carbonato de cesio (155 mg, 0,48 mmoles) en 1,4-dioxano (2 mL) se le añadieron Pd2(dba)3 (22 mg, 0,03 mmoles) y BINAP (20 mg, 0,03 mmoles). La mezcla se calentó a 120°C mediante microondas y se agitó durante 60 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (20 mL), y a continuación, se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar un sólido de color amarillo T-21 (8 mg, rendimiento: 7%). LC-MS (ESI): m/z=481 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,72 (s, 1H), 8,37 (m, 4H), 8,30 (br, 1H), 8,23 (d, J=6Hz, 1H), 7,42 (d, J=6Hz, 1H), 4,57 (d, J=9Hz, 2H), 4,22 (d, J= 9Hz, 2H), 3,55 (s, 2H), 3,10 (q, J= 7Hz, 2H), 1,31 (t, J= 7Hz, 3H) ppm
Ejemplo 22
5-((4-(1-(3-(C¡anomet¡l)-1-(et¡lsulfon¡l)azet¡din-3-¡l)-1H-p¡razol-4-¡l)t¡eno[3,2-d]p¡r¡m¡d¡n-2-¡l)am¡no)p¡col¡non¡tr¡lo T-22
Ruta sintética:

Bajo nitrógeno, a una suspensión del compuesto 5 (100 mg, 0,24 mmoles), 2-ciano-5-aminopiridina (56 mg, 0,48 mmoles) y carbonato de cesio (155 mg, 0,48 mmoles) en 1,4-dioxano (2 mL) se le añadieron Pd2(dba)3 (22 mg, 0,03 mmoles) y BINAP (20 mg, 0,03 mmoles). La mezcla se calentó a 120°C mediante microondas y se agitó durante 60 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (20 mL), y a continuación, se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar un sólido de color amarillo T-22 (33 mg, rendimiento: 28 %). LC-MS (ESI): m/z=506 [M+H]+.
RMN H1 (400MHz, CDCh) 5: 10,49 (s, 1H), 9,10 (d, J=2Hz, 1H), 8,89 (s, 1H), 8,66 (dd, J=9Hz J=2Hz, 1H), 7,97 (s, 1H), 8,49 (d, J=6Hz, 1H), 8,45 (s, 1H), 7,96 (d, J=9Hz, 1H), 7,54 (d, J=5Hz, 1H), 4,58 (d, J=9Hz, 2H), 4,29 (d, J= 9Hz, 2H), 3,73 (s, 2H), 3,25 (q, J= 7Hz, 2H), 1,26 (t, J= 7Hz, 3H) ppm
Ejemplo 23
2-(1-(Et¡lsulfon¡l)-3-(4-(6-met¡l-2-(fen¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡din-3-¡l)aceton¡tr¡lo T-23 Ruta s¡ntét¡ca:

Preparación del Compuesto 23-a
Bajo nitrógeno, a una suspensión del compuesto 5-a (194 mg, 0,5 mmoles), 2,4-dicloro-6-metiltieno[3,2-d]pirimidina (149 mg, 0,5 mmoles) y carbonato de sodio acuoso (2,0 N, 0,75 mL) en 1,4-dioxano (7,5 mL) se le añadió Pd(dppf)Cl2 (18 mg, 0,025 mmoles). La mezcla se agitó a 80°C durante 4 horas, a continuación, se concentró, y el residuo se diluyó con acetato de etilo (50 mL), se filtró a través de celite. A continuación, la capa orgánica se lavó con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, se filtró, el producto filtrado se concentró a presión reducida, y el residuo se purificó mediante TLC preparativa (diclorometano:metanol=30:1) para proporcionar el compuesto 23-a (165 mg, rendimiento: 74%). LC-MS (ESI): m/z=445 [M+H]+.
Preparación del Compuesto 23
A una solución del compuesto 23-a (165 mg, 0,37 mmoles) en 1,4-dioxano (4 mL) se le añadió una solución de hidrocloruro en 1,4-dioxano (4 N, 0,93 mL). La mezcla se agitó a temperatura ambiente durante 16 horas, a continuación, se concentró. Al residuo se le añadieron diclorometano (10 mL) y trietilamina (0,16 mL, 1,12 mmoles). La mezcla se enfrió a continuación, a 0°C, se añadió gota a gota cloruro de etilsulfonilo (62 mg, 0,56 mmoles). Una vez completada la adición gota a gota, la mezcla se agitó a 0°C durante 30 minutos adicionales. Se añadió agua (5 mL), y la mezcla se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, se filtraron, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto 23 (60 mg, rendimiento: 37%). LC-MS (ESI): m/z=437 [M+H]+.
Preparación del Compuesto T-23
El compuesto 23 (60 mg, 0,14 mmoles) y anilina (39 mg, 0,42 mmoles) se disolvieron en isobutanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (53 mg, 0,28 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida, y el residuo se purificó mediante TLC preparativa (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto T-23 (20 mg, rendimiento: 29%). LC-MS (ESI): m/z=494 [M+H]+.
RMN H1 (400MHz, DMSO-ds) 5: 9,56(s, 1H), 8,78 (s, 1H), 8,35 (s, 1H), 7,88 (d, J=8,4Hz, 2H), 7,31 (t, J=7,8Hz, 2H), 7,18 (s, 1H), 6,94 (t, J=7,4Hz, 1H), 4,57 (d, J=9,2Hz, 2H), 4,27 (d, J= 9,2Hz, 2H), 3,72 (s, 2H), 3,22-3,28 (m, 2H), 2,67 (s, 3H), 1,25 (t, J= 7,4Hz, 3H) ppm
Ejemplo 24
2-(1-(Etilsulfonil)-3-(4-(2-((4-fluorofenil)amino)tieno[3,2-d]pirimidin-4-il)-1 H-pirazol-1-il)azetidin-3-il)acetonitrilo T-24
Ruta sintética:

El compuesto 5 (208 mg, 0,49 mmoles) y 4-fluoroanilina (164 mg, 1,48 mmoles) se disolvieron en isobutanol (12 mL), se añadió monohidrato de ácido p-toluenosulfónico (188 mg, 0,98 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida, y el residuo se disolvió en diclorometano (100 mL), se lavó con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, se filtró, el producto filtrado se concentró a presión reducida, y se formó un sólido. El sólido se lavó con diclorometano (5 mL) y acetato de etilo (25 mL) para proporcionar el compuesto T-24 (30 mg, rendimiento: 12%). LC-MS (ESI): m/z=498 [M+H]+.
RMN H1 (400MHz, DMSO-ds) 5: 9,66 (s, 1H), 8,83 (s, 1H), 8,39-8,40 (m, 2H), 7,89-7,92 (m, 2H), 7,42 (d, J=5,5Hz, 1H), 7,16 (t, J=9,0Hz, 2H), 4,58 (d, J=9,0Hz, 2H), 4,28 (d, J=9,5Hz, 2H), 3,73 (s, 2H), 3,23-3,27 (m, 2H), 1,26 (t, J= 7,5Hz, 3H) ppm
Ejemplo 25
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-met¡l-1H-p¡razol-3-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡trilo T-25
Ruta s¡ntét¡ca:

Bajo nitrógeno, a una suspensión del compuesto 5 (150 mg, 0,35 mmoles), 3-amino-1-metilpirazol (104 mg, 1,48 mmoles) y carbonato de cesio (228 mg, 0,7 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (32 mg, 0,035 mmoles) y BINAP (22 mg, 0,035 mmoles). La mezcla se calentó a 120°C mediante microondas y se agitó durante 60 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (100 mL), se lavó con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, y el producto filtrado se concentró a presión reducida. Se formó un sólido y el sólido se lavó con THF (20 mL) y metanol (10 mL) para proporcionar el compuesto T-25 (80 mg, rendimiento: 46%). LC-MS (ESI): m/z=484 [M+H]+.
RMN H1 (400MHz, DMSO) 5: 9,58(s, 1H), 8,80 (s, 1H), 8,36-8,37 (m, 2H), 7,57(d, J=1,5Hz, 1H), 7,38 (d, J=5,0Hz, 1H), 6,77 (d, J=2,0Hz, 1H), 4,57 (d, J=9,0Hz, 2H), 4,27 (d, J=9,5Hz, 2H), 3,76 (s, 3H), 3,72 (s, 2H), 3,23-3,27 (m, 2H), 1,25 (t, J= 7,5Hz, 3H) ppm
Ejemplo 26
2-(1-(Et¡lsulfon¡l)-3-(4-(2-(p¡r¡d¡n-3-¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-26
Ruta s¡ntét¡ca:

Bajo nitrógeno, a una suspensión del compuesto 5 (100 mg, 0,23 mmoles), 3-aminopiridina (22 mg, 0,23 mmoles) y carbonato de cesio (154 mg, 0,47 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (22 mg, 0,023 mmoles) y BINAP (15 mg, 0,023 mmoles). La mezcla se calentó a 125°C mediante microondas y se agitó durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (100 mL), se lavó con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil:
acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar el compuesto T-26 (18 mg, rendimiento: 16%). LC-MS (ESI): m/z=481 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,91 (d, J=2,4Hz, 1H), 8,45(s, 1H), 8,33 (s, 1H), 8,27-8,31 (m, 2H), 7,90 (d, J=7,0Hz, 1H), 7,51 (s, 1H), 7,29-7,34 (m, 2H), 4,66 (d, J=9,2Hz, 2H), 4,25 (d, J=9,6Hz, 2H), 3,42 (s, 2H), 3,08-3,14 (m, 2H), 1,42 (t, J= 7,2Hz, 3H) ppm
Ejemplo 27
2-(3-(4-(2-(C¡clohex¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(et¡lsulfon¡l)azet¡din-3-¡l)aceton¡tr¡lo T-27
Ruta s¡ntét¡ca:

Bajo nitrógeno, a una suspensión del compuesto 5 (200 mg, 0,47 mmoles), ciclohexilamina (141 mg, 1,42 mmoles) y carbonato de cesio (463 mg, 1,42 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (43 mg, 0,047 mmoles) y BINAP (30 mg, 0,047 mmoles). La mezcla se calentó a 125°C mediante microondas y se agitó durante 60 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo (100 mL), se lavó con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar el compuesto T-27 (25 mg, rendimiento: 11 %). LC-MS (ESI): m/z=486 [M+H]+.
RMN H1 (500MHz, CDCla) 5: 8,38 (s, 1H), 8,34 (s, 1H), 7,79 (d, J=5,5Hz, 1H), 7,24 (d, J=5,0Hz, 1H), 5,05 (d, J=8,5Hz, 1H), 4,62 (d, J=9,5Hz, 2H), 4,25 (d, J=9,5Hz, 2H), 3,95-3,97 (m, 1H), 3,40 (s, 2H), 3,06-3,11 (m, 2H), 2,09-2,12 (m, 2H), 1,76-1,80 (m, 3H), 1,65-1,67 (m, 1H), 1,45-1,48 (m, 1H), 1,42 (t, J=7,0Hz, 3H), 1,25-1,31 (m, 3H) ppm
Ejemplo 28
2-(3-(4-(2-(C¡cloprop¡lam¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(et¡lsulfon¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-28
Ruta s¡ntét¡ca:

Bajo nitrógeno, a una suspensión del compuesto 5 (85 mg, 0,20 mmoles), ciclopropilamina (33 mg, 0,6 mmoles) y carbonato de cesio (196 mg, 0,6 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (18 mg, 0,02 mmoles) y BINAP (12 mg, 0,02 mmoles). La mezcla se calentó a 125°C mediante microondas y se agitó durante 40 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo (100 mL), se lavó con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (diclorometano : acetato de etilo=1:1) para proporcionar el compuesto T-28 (8 mg, rendimiento: 9%). LC-MS (ESI): m/z=444 [M+H]+.
RMN H1 (500MHz, CDCla) 5: 8,33 (s, 1H), 8,28 (s, 1H), 7,76 (d, J=5,5Hz, 1H), 7,26 (d, J=5,5Hz, 1H), 5,33 (s, 1H), 4,55 (d, J=9,0Hz, 2H), 4,18 (d, J=9,5Hz, 2H), 3,33 (s, 2H), 3,00-3,04 (m, 2H), 2,80-2,84 (m, 1H), 1,34 (t, J=7,5Hz, 3H), 0,78 0,82 (m, 2H), 0,52-0,55 (m, 2H) ppm
Ejemplo 29
N-(4-(1-(3-(Cianometil)-1-(etilsulfonil)azetidin-3-il)-1H-pirazol-4-il)tieno[3,2-d]pirimidin-2-il)ciclopropanocarboxamida T-29
Ruta sintética:

Bajo nitrógeno, a una suspensión del compuesto 5 (50 mg, 0,12 mmoles), ciclopropanocarboxamida (30 mg, 0,36 mmoles) y carbonato de cesio (55 mg, 0,16 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (11 mg, 0,012 mmoles) y Xantphos (7 mg, 0,012 mmoles). La mezcla se calentó a 125°C mediante microondas y se agitó durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (100 mL), se lavó con agua (20 mLx3) y salmuera saturada (20 mL), se secó sobre sulfato de sodio anhidro, a continuación, se filtró, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar el compuesto T-29 (22 mg, rendimiento: 39%). LC-MS (ESI): m/z=472 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 10,88 (s, 1H), 8,87 (s, 1H), 8,53 (d, J=5,2Hz, 1H), 8,41 (s, 1H), 7,54 (d, J=5,2Hz, 1H), 4,57 (d, J=8,8Hz, 2H), 4,27 (d, J=9,2Hz, 2H), 3,72 (s, 2H), 3,22-3,28 (m, 2H), 2,17-2,23 (m, 1H), 1,24 (t, J=7,2Hz, 3H),
0,81-0,85 (m, 4H) ppm
Ejemplo 30
2-(1-(Et¡lsulfon¡l)-3-(4-(2-(fen¡lam¡no)furo[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-30 Ruta s¡ntét¡ca:

30 T-30
Preparac¡ón del Compuesto 30-h
Se añadieron ácido 3-furoico (2,8 g, 25 mmoles), azidofosfato de difenilo (6 ml, 27,5 mmoles) y trietilamina (5 mL, 35 mmoles) a ferc-butanol (50 mL). La mezcla se calentó a 90°C y se agitó durante 12 horas. Después de enfriar a temperatura ambiente, se añadió dicarbonato de sodio acuoso (2 N, 100 mL). La mezcla se filtró, la torta del filtro se disolvió en acetato de etilo (100 mL), y se lavó con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=10:1) para
proporcionar un sólido de color blanco 30-h (3,2 g, rendimiento: 70%). LC-MS (ESI): m/z=184 [M+H]+.
Preparación del Compuesto 30-g
A -40°C, a una solución del compuesto 30-h (1,7 g, 9,3 mmoles) en THF anhidro (50 mL) se le añadió N,N-tetrametiletilendiamina (1,8 mL, 12,1 mmoles), después de agitar durante 20 minutos, se le añadió gota a gota una solución de n-BuLi en n-hexano (2,5 N, 8,4 mL, 21 mmoles) y la temperatura de reacción se mantuvo a -40°C. Una vez completada la adición gota a gota, la mezcla se agitó durante 30 minutos adicionales. La mezcla se calentó lentamente a 0°C, y se agitó durante otra hora, a continuación, se volvió a enfriar a -40°C, se agitó durante 10 minutos, se añadió rápidamente a la mezcla carbonato de dimetilo (2,4 mL, 28 mmoles). La mezcla de reacción se calentó lentamente a temperatura ambiente, se agitó durante otra hora. Se añadieron una solución acuosa de hidrocloruro (2 N, 11 mL) y agua (100 mL) para sofocar la reacción, a continuación, la mezcla se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo= 100:1) para proporcionar un sólido de color blanco 30-g (0,56 g, rendimiento: 25%). LC-MS (ESI): m/z=142 [M+H]+.
Preparación del Compuesto 30-f
A una solución del compuesto 30-g (0,56 g, 2,3 mmoles) en diclorometano (4 mL) se le añadió ácido trifluoroacético (2,5 mL), la mezcla se agitó a temperatura ambiente durante 2 horas. Después de concentrar la mezcla a presión reducida, el residuo se trató con una solución acuosa de dicarbonato de sodio (2 N, 6 mL), a continuación, se extrajo con acetato de etilo (5 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (20 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un líquido de color amarillo 30-f (0,35 g, rendimiento: 100%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto 30-e
A -78°C, a una solución del compuesto 30-f (0,35 g, 2,5 mmoles) en diclorometano (5 mL) se le añadió gota a gota isocianato de clorosulfonilo (0,49 g, 3,5 mmoles). Una vez completada la adición gota a gota, la mezcla se calentó a temperatura ambiente, y se agitó durante 40 minutos. La mezcla se concentró a presión reducida, el residuo se trató con HCl (6 N, 3 mL, 18 mmoles), a continuación, calentó a 100°C, y se agitó durante 30 minutos. La mezcla se enfrió a temperatura ambiente, a continuación, se concentró a presión reducida, el residuo se trató con una solución acuosa de dicarbonato de sodio (2 N, 6 mL), se extrajo con acetato de etilo (5 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (20 mL) sucesivamente. Después de secar sobre sulfato de sodio anhidro, la mezcla se filtró, y el producto filtrado se concentró a presión reducida para proporcionar un sólido de color amarillo 30-e (176mg, rendimiento: 38%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=185 [M+H]+.
Preparación del Compuesto 30-d
A una solución del compuesto 30-e (0,176 g, 0,96 mmoles) en metanol (4 mL) se le añadió una solución acuosa de hidróxido de sodio (2 N, 2 mmoles), la mezcla se sometió a reflujo durante 2 horas. Después de enfriar a temperatura ambiente, se añadió una solución acuosa de hidrocloruro (6 N, 0,5 mL) para ajustar a pH=3. La mezcla se concentró a presión reducida, al residuo se le añadió metanol (5 mL), y precipitó un sólido de color gris. Después de la filtración, la torta del filtro se secó a vacío para proporcionar un sólido de color gris 30-d (110 mg, rendimiento: 75%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=153 [M+H]+.
Preparación del Compuesto 30-c
A -40°C, a una solución del compuesto 30-d (0,86 g, 5,6 mmoles) en oxicloruro de fósforo (8 mL) se le añadió N,N-diisopropiletilamina (2 mL), después de agitar durante 10 minutos, la mezcla se sometió a reflujo durante 24 horas. Después de enfriar a temperatura ambiente, la mezcla se vertió en agua helada para sofocar la reacción, y a continuación, se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, a continuación, se secaron sobre sulfato de sodio anhidro, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo= 100:1) para proporcionar un sólido de color blanco 30-c (430 mg, rendimiento: 41%). LC-MS (ESI): m/z=189 [M+H]+.
Preparación del Compuesto 30-b
Bajo nitrógeno, a una suspensión del compuesto 5-a (640 mg, 1,65 mmoles), el compuesto 30-c (280 mg, 1,49 mmoles) y carbonato de potasio (720 mg, 5,2 mmoles) en 1,4-dioxano (2 mL) y agua (6 mL) se le añadió Pd(PPh3)4 (57 mg, 0,05 mmoles), la mezcla se calentó a 80°C y se agitó durante 16 horas. La mezcla se concentró a continuación, a presión reducida, el residuo se diluyó con agua (20 mL), se extrajo con diclorometano (20 mLx3). La capa orgánica se combinó, se lavó con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, a continuación, se secó sobre sulfato de sodio anhidro, se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante
cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar un sólido de color amarillo claro 30-b (620 mg, rendimiento: 70%). LC-MS (ESI): m/z=415 [M+H]+.
Preparación del Compuesto 30-a
A una solución del compuesto 30-b (500 mg, 1,2 mmoles) en diclorometano (4 mL) se le añadió ácido trifluoroacético (1 mL), la mezcla se agitó a temperatura ambiente durante 2 horas. Después de concentrar la mezcla a presión reducida, el residuo se trató con una solución acuosa de dicarbonato de sodio (2 N, 6 mL), se extrajo con acetato de etilo (5 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, a continuación, se secaron sobre sulfato de sodio anhidro, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un líquido de color amarillo claro 30-a (370 mg, rendimiento: 97%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=315 [M+H]+.
Preparación del Compuesto 30
A 0°C, a una solución del compuesto 30-a (314 mg, 1,0 mmoles) y trietilamina (1 mL) en diclorometano (4 mL) se le añadió gota a gota cloruro de etilsulfonilo (154 mg, 1,2 mmoles). Después de agitar durante 30 minutos, la mezcla se concentró a presión reducida, y el residuo se trató con dicarbonato de sodio acuoso (2 N, 6 mL), se extrajo con acetato de etilo (5 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (20 mL) sucesivamente, a continuación, se secaron sobre sulfato de sodio anhidro, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un líquido de color amarillo 30 (280 mg, rendimiento: 70%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=407 [M+H]+.
Preparación del Compuesto T-30
Bajo nitrógeno, a una suspensión del compuesto 30 (70 mg, 0,17 mmoles), anilina (25 mg, 0,26 mmoles) y carbonato de potasio (138 mg, 1,0 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (15 mg, 0,017 mmoles) y Ruphos (10 mg, 0,07 mmoles), la mezcla se calentó a 130°C mediante microondas y se agitó durante 3 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo (10 mL), a continuación, se filtró, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar el compuesto T-30 (16 mg, rendimiento: 20%). LC-MS (ESI): m/z=464 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 9,48 (s, 1H), 8,86 (s, 1H), 8,42 (d, J=1,6Hz, 1H), 8,40 (s, 1H), 7,88 (d, J=6,4Hz, 2H), 7,30 (t, J=6,4Hz, 1H), 7,04 (d, J=1,6Hz, 1H), 6,93 (t, J=6,4Hz, 1H), 4,56 (d, J=7,2Hz, 2H), 4,27 (d, J= 7,2Hz, 2H), 3,72 (s, 2H), 3,25 (q, J= 6Hz, 2H), 1,25 (t, J= 6Hz, 3H) ppm
Ejemplo 31
2-(3-(4-(2-((1H-Pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(etilsulfonil)azetidin-3-il)acetonitrilo T-31
Ruta sintética:

Preparación del Compuesto 31-a
Bajo hidrógeno (1 atm), a una solución de 4-nitropirazol (1,13 g, 10 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 31-a (860 mg, rendimiento: 100%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto T-31
El compuesto 31-a (130 mg, 1,5 mmoles) y el compuesto 5 (222 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (344 mg, 2,0 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), a continuación, se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (0,04% ácido trifluoroacético), acetonitrilo; gradiente: 32%-62%) para proporcionar el compuesto T-31 (143 mg, rendimiento: 61 %). LC-MS (ESI): m/z=470 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 9,43 (s, 1H), 8,81 (s, 1H), 8,37 (s, 1H), 8,34 (d, J=4,0Hz, 1H), 7,87 (s, 2H), 7,39 (d, J=4,0Hz, 1H), 4,57 (d, J=8Hz, 2H), 4,27 (d, J = 8Hz, 2H), 3,91 (s, 4H), 3,24 (q, J= 6Hz, 2H), 1,25 (t, J= 6Hz, 3H) ppm
Ejemplo 32
2-(1-(Etilsulfonil)-3-(4-(2-((1-(2-metoxiacetil)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-i l)azetidin-3-il)acetonitrilo T-32
Ruta sintética:

A 0°C, a una solución del compuesto T-31 (47 mg, 0,1 mmoles) y trietilamina (100 mg, 1,0 mol) en diclorometano (10 mL) se le añadió lentamente cloruro de metoxiacetilo (11 mg, 0,2 mmoles), y se agitó durante 1 hora. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (5 mL), se extrajo con diclorometano (5 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto T-32 (32 mg, rendimiento: 59%). LC-MS (ESI): m/z=542 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 9,94 (s, 1H), 8,85 (s, 1H), 8,66 (s, 1H), 8,43 (d, J=5,2Hz, 1H), 8,39 (s, 1H), 8,06 (s, 1H), 7,51 (d, J=5,2Hz, 1H), 4,87 (s, 2H), 4,57 (d, J=9,2Hz, 2H), 4,28 (d, J=9,2Hz, 2H), 3,73 (s, 2H), 3,43 (s, 3H), 3,25 (q, J= 7,2Hz, 2H), 1,25 (t, J= 7,2Hz, 3H) ppm
Ejemplo 33
Ácido 2-(4-((4-(1-(3-(cianometil)-1-(etilsulfonil)azetidin-3-il)-1H-pirazol-4-il)tieno[3,2-d]pirimidin-2-il)amino)-1H-pirazol-1-il)acetic T-33
Ruta sintética:

Preparación del Compuesto 33-b
Se añadieron bromoacetato de etilo (1,67 g, 10 mmoles) y carbonato de potasio (2,76 g, 20 mmoles) a una solución de 4-nitropirazol (1,13 g, 10 mmoles) en DMF (15 mL), la mezcla se calentó a 90°C y se agitó durante 12 horas. Después de enfriar la mezcla a temperatura ambiente, se añadió agua (60 mL), a continuación, la mezcla se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mL) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 33-b (1,68 g, rendimiento: 84%). LC-MS (ESI): m/z=200 [M+H]+.
Preparación del Compuesto 33-a
Bajo hidrógeno (1 atm), a una solución del compuesto 33-b (1,0 g, 5 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 33-a (760 mg, rendimiento: 90%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=170 [M+H]+.
Preparación del Compuesto 33
El compuesto 33-a (170 mg, 1,0 mmoles) y el compuesto 5 (211 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (344 mg, 2,0 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 33 (116 mg, rendimiento: 21%). LC-MS (ESI): m/z=584 [M+H]+.
Preparación del Compuesto T-33
Se añadió una solución acuosa de LiOH (2 N, 0,5 mL) a una solución del compuesto 33 (83 mg, 0,15 mmoles) en MeOH (2 mL) y THF (6 mL), la mezcla se agitó a 25°C durante 1 hora. Se añadió una solución acuosa de HCl (2 N, 0,5 mL), la mezcla se concentró a presión reducida para eliminar los disolventes orgánicos. El residuo se trató con agua (2 mL), se extrajo con acetato de etilo (2 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto T-33 (36 mg, rendimiento: 47%). LC-MS (ESI): m/z=528 [M+H]+.
RMN H1 (400MHz, DMSO-ds) 5: 8,65 (s, 1H), 8,31 (s, 1H), 8,09 (s, 1H), 8,07 (d, J=4,4Hz, 1H), 7,56 (s, 1H), 7,23 (d, J=4,4Hz, 1H), 4,90 (s, 2H), 4,56 (d, J=7,2Hz, 2H), 4,22 (d, J=7,2Hz, 2H), 3,51 (s, 2H), 3,07 (q, J= 5,6Hz, 2H), 1,27 (t, J= 5,6Hz, 3H) ppm
Ejemplo 34
3-(4-((4-(1-(3-(Cianometil)-1-(etilsulfonil)azetidin-3-il)-1H-pirazol-4-il)tieno[3,2-d]pirimidin-2-il)amino)-1H-pirazol-1-il)propanoato de butilo T-34
Ruta sintética:

Se disolvieron 4-nitropirazol (1,13 g, 10 mmoles) y acrilato de etilo (1,72 g, 20 mmoles) en acetonitrilo (30 mL), se añadió DBU (2 mL). La mezcla se agitó a 90°C durante 12 horas. A continuación, la mezcla se concentró a presión reducida, el residuo se trató con agua (50 mL), se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 34-b (1,37 g, rendimiento: 69%). LC-MS (ESI): m/z=214 [M+H]+.
Preparación del Compuesto 34-a
Bajo hidrógeno (1 atm), a una solución del compuesto 34-b (1,0 g, 5 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 34-a (780 mg, rendimiento: 92%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=184 [M+H]+.
Preparación del Compuesto T-34
El compuesto 34-a (184 mg, 1,0 mmoles) y el compuesto 5 (211 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (258 mg, 1,5 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo =3:1) para proporcionar el compuesto T-34 (138 mg, rendimiento: 24,8%). LC-MS (ESI): m/z=598 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,45 (s, 1H), 8,23 (s, 1H), 8,01 (s, 1H), 7,80 (d, J=5,6Hz, 1H), 7,46 (s, 1H), 7,23 (d, J=5,6Hz, 1H), 7,05(s, 1H), 4,62 (d, J=9,6Hz, 2H), 4,38 (t, J=6,4Hz, 2H), 4,19 (d, J=9,6Hz, 2H), 4,00 (t, J=6,4Hz, 2H), 3,35 (s, 2H), 3,02 (q, J= 7,2Hz, 2H), 2,85 (t, J=6,4Hz, 2H), 1,49 (q, J=7,2Hz, 2H), 1,34 (t, J=7,2Hz, 3H), 1,24 (m, 4H), 0,79 (t, J= 7,2Hz, 3H) ppm
Ejemplo 35
Ácido 3-(4-((4-(1-(3-(Cianometil)-1-(etilsulfonil)azetidin-3-il)-1H-pirazol-4-il)tieno[3,2-d]pirimidin-2-il)amino)-1H-pirazol-1-il)propanoico T-35
Ruta sintética:

Se añadió una solución acuosa de LiOH (2 N, 0,5 mL) a una solución del compuesto T-34 (138 mg, 0,25 mmoles) en MeOH (2 mL) y THF (6 mL), la mezcla se agitó a 25°C durante 1 hora. Se añadió una solución acuosa de HCl (2 N, 0,5 mL), la mezcla se concentró a presión reducida para eliminar los disolventes orgánicos. El residuo se trató con agua (2 mL), se extrajo con acetato de etilo (2 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (0,04% ácido trifluoroacético), acetonitrilo; gradiente: 32%-62%) para proporcionar el compuesto T-35 (98 mg, rendimiento: 71 %). LC-MS (ESI): m/z=542 [M+H]+.
RMN H1 (400MHz, DMSO-ds) 5: 9,44(s, 1H), 8,81 (s, 1H), 8,41 (s, 1H), 8,34 (d, J=5,2Hz, 1H), 8,09 (s, 1H), 7,57 (s, 1H), 7,37 (d, J=5,2Hz, 1H), 4,59 (d, J=8,8Hz, 2H), 4,32 (t, J=6,4Hz, 2H), 4,27 (d, J=8,8Hz, 2H), 3,73 (s, 2H), 3,25 (q, J=7,6Hz, 2H), 2,79 (t, J=6,4Hz, 2H), 1,25 (t, J=7,6Hz, 3H) ppm
Ejemplo 36
3-(4-((4-(1-(3-(C¡anomet¡l)-1-(et¡lsulfon¡l)azet¡d¡n-3-¡l)-1H-p¡razol-4-¡l)t¡eno[3,2-d]p¡r¡m¡d¡n-2-¡l)am¡no)-)-1H-pirazol-1-il)-N,N-dimetilpropanamida T-36
Ruta sintética:

Se añadieron TBTU (200 mg, 0,62 mmoles) y N,N-diisopropiletilamina (65 mg, 0,5 mmoles) a una solución del compuesto T-35 (70 mg, 0,13 mmoles) y hidrocloruro de dimetilamina (12 mg, 0,26 mmoles) en diclorometano (5 mL). Después de agitar a 25°C durante 1 hora, la mezcla se trató con una solución acuosa de hidrocloruro (1 N, 4 mL), a continuación, se extrajo con diclorometano (3 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil : agua (0,04% ácido trifluoroacético), acetonitrilo; gradiente: 30%-60%) para proporcionar el compuesto T-36 (31 mg, rendimiento: 42%). LC-MS (ESI): m/z=569 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,86 (s, 1H), 8,47 (s, 1H), 8,26 (d, J=5,2Hz, 1H), 8,18 (s, 1H), 7,65 (s, 1H), 7,37 (d, J=5,2Hz, 1H), 4,71 (d, J=8,8Hz, 2H), 4,49 (t, J=6,0Hz, 2H), 4,32 (d, J=8,8Hz, 2H), 3,65 (s, 2H), 3,32 (q, J= 7,2Hz, 2H), 2,97 (m, 8H), 1,38 (t, J=7,2Hz, 3H) ppm
Ejemplo 37
4-(4-((4-(1-(3-(C¡anomet¡l)-1-(et¡lsulfon¡l)azet¡d¡n-3-¡l)-1H-pirazol-4-¡l)t¡eno[3,2-d]p¡r¡m¡d¡n-2-¡l)am¡no)-1H-
pirazol-1-il)-N,N-dimetilbutanamida T-37
Ruta sintética:

Se disolvieron 4-nitropirazol (1,13 g, 10 mmoles) y 4-bromobutirato de metilo (1,81 g, 10 mmoles) en DMF (15 mL), se añadió carbonato de potasio (2,76 g, 2 mmoles). La mezcla se agitó a 90°C durante 12 horas, a continuación, se trató con agua (60 mL), se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se
filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 37-f (1,42 g, rendimiento: 67%). LC-MS (ESI): m/z=214 [M+H]+.
Preparación del Compuesto 37-e
Se añadió una solución acuosa de LiOH (2 N, 1,0 mL) a una solución del compuesto 37-f (426 mg, 2,0 mmoles) en MeOH (2 mL) y THF (6 mL), la mezcla se agitó a 25°C durante 1 hora. Se añadió una solución acuosa de HCl (2 N, 1,0 mL), la mezcla se concentró a presión reducida para eliminar los disolventes orgánicos. El residuo se trató con agua (2 mL), se extrajo con acetato de etilo (2 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 37-e (368 mg, rendimiento: 92%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=200 [M+H]+.
Preparación del Compuesto 37-d
Se añadieron TBTU (800 mg, 2,4 mmoles) y N,N-diisopropiletilamina (650 mg, 5 mmoles) a una solución del compuesto 37-e (368 mg, 1,85 mmoles) e hidrocloruro de dimetilamina (110 mg, 2,4 mmoles) en diclorometano (20 mL). Después de agitar a 25°C durante 1 hora, la mezcla se trató con una solución acuosa de hidrocloruro (1 N, 4 mL), se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 37-d (320 mg, rendimiento: 76%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=227 [M+H]+.
Preparación del Compuesto 37-c
Bajo hidrógeno (1 atm), a una solución del compuesto 37-d (320 mg, 1,4 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 37-c (280 mg, rendimiento: 74%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=197 [M+H]+.
Preparación del Compuesto 37-b
El compuesto 37-c (200 mg, 1,0 mmoles) y el compuesto 5 (221 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (258 mg, 1,5 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 37-b (118 mg, rendimiento: 41%). LC-MS (ESI): m/z=612 [M+H]+.
Preparación del Compuesto 37-a
Se añadió una solución acuosa de LiOH (2 N, 0,5 mL) a una solución del compuesto 37-b (114 mg, 0,2 mmoles) en MeOH (2 mL) y THF (6 mL), la mezcla se agitó a 25°C durante 1 hora. Se añadió una solución acuosa de HCl (2 N, 0,5 mL), la mezcla se concentró a presión reducida para eliminar los disolventes orgánicos. El residuo se trató con agua (2 mL), se extrajo con acetato de etilo (2 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 37-a (68 mg, rendimiento: 61 %), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=556 [M+H]+.
Preparación del Compuesto T-37
Se añadieron TBTU (200 mg, 0,62 mmoles) y N,N-diisopropiletilamina (65 mg, 0,5 mmoles) a una solución del compuesto 37-a (68 mg, 0,12 mmoles) e hidrocloruro de dimetilamina (11 mg, 0,24 mmoles) en diclorometano (5 mL). Después de agitar a 25°C durante 1 hora, la mezcla se trató con una solución acuosa de hidrocloruro (1 N, 4 mL), se extrajo con diclorometano (3 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (0,04% ácido trifluoroacético), acetonitrilo; gradiente: 30%-60%) para proporcionar el compuesto T-37 (21 mg, rendimiento: 30%). LC-MS (ESI): m/z=583 [M+H]+.
RMN H1 (400MHz, CDaOD) 5: 8,79 (s, 1H), 8,44 (s, 1H), 8,23 (d, J=5,2Hz, 1H), 8,12 (s, 1H), 7,66 (s, 1H), 7,36 (d, J=5,2Hz, 1H), 4,67 (d, J=8,8Hz, 2H), 4,32 (d, J=8,8Hz, 2H), 4,25 (t, J=6,8Hz, 2H), 3,64(s, 2H), 3,20 (q, J= 7,2Hz, 2H), 2,97 (d, J= 35,6Hz, 6H), 2,42 (t, J= 6,4Hz, 2H), 2,19 (m, 2H), 1,38 (t, J=7,2Hz, 3H) ppm
Ejemplo 38
2-(1-(Etilsulfonil)-3-(4-(2-((1-(tetrahidro-2H-piran-4-il)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-38
Ruta sintética:
Preparación del Compuesto 38-b
A 25°C, se añadió lentamente DIAD (3,06 g, 15 mmoles) a una solución de 4-nitropirazol (1,13 g, 10 mmoles), tetrahidro-2H-piran-4-ol (1,12 g, 11 mmoles) y PPh3 (3,93 g, 15 mmoles) en THF anhidro (50 mL). Después de agitar durante 3 horas, la mezcla se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 38-b (990 mg, rendimiento: 49%). LC-MS (ESI): m/z=198 [M+H]+.
Preparación del Compuesto 38-a
Bajo hidrógeno (1 atm), a una solución del compuesto 38-b (990 mg, 49 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 38-a (620 mg, rendimiento: 75 %), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto T-38
El compuesto 38-a (170 mg, 1,0 mmoles) y el compuesto 5 (221 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (258 mg, 1,5 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (0,04% ácido trifluoroacético), acetonitrilo; gradiente: 30%-60%) para proporcionar el compuesto T-38 (38 mg, rendimiento: 13,6%). LC-MS (ESI): m/z=554 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,76 (s, 1H), 8,44 (s, 1H), 8,22 (d, J=5,2Hz, 1H), 8,14 (s, 1H), 7,69 (s, 1H), 7,35 (d, J=5,2Hz, 1H), 4,66 (d, J=9,6Hz, 2H), 4,51 (m, 1H), 4,31 (d, J=9,6Hz, 2H), 4,10 (d, J=3,6Hz, 2H), 3,64 (s, 2H), 3,62 (d, J=3,6Hz, 2H), 3,20 (q, J= 7,6Hz, 2H), 2,11 (m, 3H), 1,38 (t, J=7,6Hz, 3H) ppm
Ejemplo 39
2-(1-(Etilsulfonil)-3-(4-(2-((1-(oxetan-3-il)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-39
Ruta sintética:

Preparación del Compuesto 39-c
A 0°C, se añadió gota a gota una solución acuosa de NaOH (2 N, 15 mL) a una solución de oxetan-3-ol (1,48 g, 20 mmoles) y cloruro de p-toluenosulfonilo (4,18 g, 22 mmoles) en agua (50 mL). Después de agitar durante 3 horas, la mezcla se trató con agua (100 mL), se extrajo con diclorometano (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=10:1) para proporcionar el compuesto 39-c (3,9 g, rendimiento: 85%). LC-MS (ESI): m/z=229 [M+H]+.
Preparación del Compuesto 39-b
El compuesto 39-c (3,9 g, 17 mmoles) se añadió a una solución de 4-nitropirazol (1,13 g, 10 mmoles) y carbonato de cesio (6,5 g, 20 mmoles) en DMF (15 mL), y a continuación, se calentó a 120°C y se agitó durante 12 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (60 mL), se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 39-b (1,3 g, rendimiento: 77%). LC-MS (ESI): m/z=170 [M+H]+.
Preparación del Compuesto 39-a
Bajo hidrógeno (1 atm), a una solución del compuesto 39-b (1,3 g, 7,7 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 39-a (820 mg, rendimiento: 76%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=140 [M+H]+.
Preparación del Compuesto T-39
Bajo nitrógeno, a una suspensión del compuesto 39-a (24 mg, 0,17 mmoles), el compuesto 5 (71 mg, 0,17 mmoles) y carbonato de potasio (138 mg, 1,0 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd2(dba)3 (15 mg, 0,017 mmoles) y Ruphos (10 mg, 0,07 mmoles), la mezcla se calentó a 130°C mediante microondas y se agitó durante 3 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo (10 mL), a continuación, se filtró, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar el compuesto T-39 (13 mg, rendimiento: 14,5 %). LC-MS (ESI): m/z=526 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,74 (s, 1H), 8,43 (s, 1H), 8,31 (s, 1H), 8,13 (d, J=4,4Hz, 1H), 7,77 (s, 1H), 7,34 (d, J=4,4Hz, 2H), 5,60 (m, 1H), 5,10 (m, 4H), 4,77 (d, J=7,2Hz, 2H), 4,33(d, J=7,2Hz, 2H), 3,64 (s, 2H), 3,21 (q, J=6,0Hz, 2H), 2,51 (s, 3H), 1,40 (t, J=6,0Hz, 3H) ppm
Ejemplo 40
2-(3-(4-(2-((1-Etil-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(etilsulfonil)azetidin-3-
¡l)acetonitr¡lo T-40
Ruta sintética:

Se añadieron Bromoetano (1,1 g, 10 mmoles) y carbonato de potasio (2,76 g, 20 mmoles) a una solución de 4-nitropirazol (1,13 g, 10 mmoles) en DMF (15 mL) respectivamente, la mezcla se calentó a 90°C y se agitó durante 12 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (60 mL), se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 40-b (1,2 g, rendimiento: 85%). LC-MS (ESI): m/z=142 [M+H]+.
Preparación del Compuesto 40-a
Bajo hidrógeno (1 atm), a una solución del compuesto 40-b (1,0 g, 7,1 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 40-a (760 mg, rendimiento: 96%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=112 [M+H]+.
Preparación del Compuesto T-40
El compuesto 40-a (111 mg, 1,0 mmoles) y el compuesto 5 (221 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (258 mg, 1,5 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (0,04% ácido trifluoroacético), acetonitrilo; gradiente: 40%-70%) para proporcionar el compuesto T-40 (23 mg, rendimiento: 9,2%). LC-MS (ESl): m/z=498 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 9,44 (s, 1H), 8,82 (s, 1H), 8,39 (s, 1H), 8,34 (d, J=5,2, 1H), 8,06 (s, 1H), 7,58 (s, 1H), 7,38 (d, J=5,2Hz, 1H), 4,58 (d, J=9,2Hz, 2H), 4,27 (d, J=9,2Hz, 2H), 4,14 (q, J= 7,2Hz, 2H), 3,24 (q, J=7,2Hz, 2H), 1,39 (t, J= 7,2Hz, 3H), 1,25 (t, J=7,2Hz, 3H) ppm
Ejemplo 41
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-¡soprop¡l-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)acetonitrilo T-41
Preparación del Compuesto T-41
Ruta sintética:

Se añadieron 2-yodopropano (2,3 g, 13,27 mmoles) y carbonato de potasio (1,81 g, 13,27 mmoles) a una solución de 4-nitropirazol (1,0 g, 8,85 mmoles) en DMF (10 mL) sucesivamente, la mezcla se calentó a 60°C y se agitó durante 3 horas. La mezcla se vertió en agua helada (100 mL), se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo 41-b (1,1 g, rendimiento: 81%), que se utilizó para la siguiente etapa sin purificación.
Preparación del Compuesto 41 -a
Bajo hidrógeno (1 atm), a una solución del compuesto 41-b (1,1 g, 8,8 mmoles) en etanol (20 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 41-a (830 mg, rendimiento: 94%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=126 [M+H]+.
Preparación del Compuesto T-41
El compuesto 41-a (71 mg, 0,57 mmoles) y el compuesto 5 (80 mg, 0,19 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (71 mg, 0,38 mmoles). La mezcla se calentó a 115°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo/metanol (1:1), agua (ácido trifluoroacético al 0,05%); gradiente: 75%-95%-10%) para proporcionar el compuesto T-41 (48 mg, rendimiento: 49%). LC-MS (ESI): m/z=512 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,65 (s, 1H), 8,30 (d, J=3Hz, 1H), 8,15 (d, J=5Hz, 1H), 8,00 (d, J=3Hz, 1H), 7,67 (d, J=3Hz, 1H), 7,36 (d, J=6Hz, 1H), 4,63 (t, J=6Hz, 2H), 4,53(m, 1H), 4,28 (d, J=9Hz, 1H), 3,60 (s, 3H), 3,18 (q, J= 7Hz, 2H), 1,39 (t, J= 7Hz, 3H) ppm
Ejemplo 42
2-(3-(4-(2-((1-Metil-1H-pirazol-4-il)amino)tieno[2,3-d]pirimidin-4-il)-1H-pirazol-1-il)oxetan-3-il)acetonitrilo T-42
Ruta sintética:

Preparación del Compuesto 42
Bajo nitrógeno, 2,4-diclorotieno[2,3-d]pirimidina (317 mg, 1,56 mmoles), el compuesto 20-a (300 mg, 1,04 mmoles) y carbonato de sodio (331 mg, 3,12 mmoles) se suspendieron en 1,4-dioxano (5 mL), se añadió Pd (dppf)Cl2 (82 mg, 0,1 mmoles). La mezcla se calentó a 80°C y se agitó durante 18 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (50 mL), a continuación, se filtró, y el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1 a diclorometano:metanol=10:1) para proporcionar un sólido de color gris 42 (120 mg, rendimiento: 35%). LC-MS (ESI): m/z=332 [M+H]+.
Preparación del Compuesto T-42
El compuesto 42 (50 mg, 0,15 mmoles) y 1-metil-4-aminopirazol (44 mg, 0,45 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (58 mg, 0,31 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar el compuesto T-42 (14 mg, rendimiento: 24%). LC-MS (ESI): m/z=393 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,59 (br, 1H), 8,25 (s, 1H), 7,92 (s, 1H), 7,66 (s, 1H), 7,42 (d, J=6Hz, 1H), 7,24 (d, J=6Hz, 1H), 5,21 (d, J=9Hz, 2H), 4,87 (d, J= 9Hz, 2H), 3,93 (s, 3H), 3,45 (s, 2H) ppm
Ejemplo 43
2-(3-(4-(2-((1-Metil-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)oxetan-3-il)acetonitrilo T-43
Ruta sintética:

El compuesto 20 (108 mg, 0,33 mmoles) y 1 -metil-4-aminopirazol (95 mg, 0,98 mmoles) se disolvieron en n-butanol (3 mL), se añadió monohidrato de ácido p-toluenosulfónico (124 mg, 0,66 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato
de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante TLC preparativa (fase móvil: acetato de etilo) para proporcionar el compuesto T-43 (75 mg, rendimiento: 59%). LC-MS (ESI): m/z=393 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,42 (s, 1H), 8,36 (s, 1H), 7,96 (s, 1H), 7,87 (d, J=6Hz, 1H), 7,54 (s, 1H), 7,33 (d, J=6Hz, 1H), 7,14 (s, 1H), 5,18 (d, J=9Hz, 2H), 4,87 (d, J= 9Hz, 2H), 3,92 (s, 3H), 3,43 (s, 2H) ppm
Ejemplo 44
2-(4-(4-(2-((1-Met¡l-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)tetrah¡dro-2H-p¡ran-4-¡l)acetonitr¡lo T-44
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 44-b
A temperatura ambiente, se disolvieron cianometilen trifenilfosforano (8,0 g, 20 mmoles) y 4-oxotetrahidropirano (2,0 g, 20 mmoles) en diclorometano (20 mL), la mezcla se agitó durante 16 horas. La mezcla se concentró a presión reducida, y el residuo se añadió a un disolvente componente (50 mL) de éter de petróleo y acetato de etilo (10:1). Se produjo la precipitación de un sólido de color blanco, y la mezcla se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 44-b (2,0 g, rendimiento: 81%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparac¡ón del Compuesto 44-a
El compuesto 44-b (500 mg, 4,06 mmoles) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (1,2 g, 6,09 mmoles) se disolvieron en acetonitrilo (10 mL), se añadió 1,8-diazabiciclo(5,4,0)undec-7-eno (1,23 g, 8,12 mmoles). La mezcla se agitó a 60°C durante 18 horas. La mezcla se concentró a continuación, a presión reducida, y el residuo se trató con una solución acuosa 1 N de hidrocloruro para ajustar a pH=3-4, a continuación, se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo 44-a (700 mg, rendimiento: 54%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=318 [M+H]+.
Preparac¡ón del Compuesto 44
Bajo nitrógeno, el compuesto 44-a (500 mg, 3,65 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (745 mg, 3,65 mmoles) y carbonato de sodio (1,2 g, 10,95 mmoles) se suspendieron en 1,4-dioxano (0,5 mL) y agua (0,5 mL), se añadió Pd(dppf)Cl2 (330 mg, 0,4 mmoles). La mezcla se calentó a 80°C y se agitó durante 4 horas. La mezcla se concentró a presión reducida, el residuo se trató con agua (20 mL), se extrajo con diclorometano (20 mLx3). Las capas orgánicas
se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color amarillo claro 44 (210 mg, rendimiento: 38%). LC-MS (ESI): m/z=360 [M+H]+.
Preparación del Compuesto T-44
El compuesto 44 (120 mg, 0,34 mmoles) y 1-metil-4-aminopirazol (97 mg, 10,03 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (127 mg, 0,67 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (fase móvil: acetato de etilo) para proporcionar un sólido de color amarillo claro T-44 (121 mg, rendimiento: 86%). LC-MS (ESI): m/z=421 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,43 (s, 1H), 8,38 (s, 1H), 7,98 (s, 1H), 7,87 (d, J=6Hz, 1H), 7,54 (s, 1H), 7,33 (d, J=6Hz, 1H), 7,03 (s, 1H), 3,93 (s, 3H), 3,89 (m, 2H), 3,61 (m, 2H), 2,95 (s, 2H), 2,67 (m, 2H), 2,24 (m, 2H) ppm Ejemplo 45
2-(3-(4-(2-((4-(2H-Tetrazol-5-il)fenil)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(etilsulfonil)azetidin-3-il)acetonitrilo T-45
Ruta sintética:

El compuesto 5 (422 mg, 1,0 mmoles) y 4-(2H-tetrazol-5-il)anilina (242 mg, 1,5 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (285 mg, 1,5 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo claro T-45 (52 mg, rendimiento: 10%). LC-MS (ESI): m/z=548 [M+H]+.
RMN H1 (400MHz, DMSO-ds) 5: 10,08 (s, 1H), 8,88 (s, 1H), 8,45 (m, 2H), 8,13 (d, J=9Hz, 1H), 7,99 (d, J=9Hz, 1H), 7,50 (d, J=9Hz, 1H), 4,59 (d, J=9Hz, 2H), 4,29 (d, J= 9Hz, 2H), 3,74 (s, 2H), 3,24 (q, J= 7Hz, 2H), 1,25 (t, J= 7Hz, 3H) ppm Ejemplo 46
2-(3-(4-(2-((1-Metil-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidm-4-il)-1H-pirazol-1-il)-1-propionilazetidin-3-il)acetonitrilo T-46
Ruta sintética:

Preparación del Compuesto 46
A una solución del compuesto 5-a (250 mg, 0,58 mmoles) en diclorometano (2 mL) se le añadió una solución de hidrocloruro en 1,4-dioxano (4 N, 4 mL), la mezcla se agitó a temperatura ambiente durante 16 horas, a continuación, se concentró a presión reducida, y el residuo se trató con diclorometano (5 mL) y trietilamina (0,4 mL). La mezcla se enfrió a continuación, a 0°C, se añadió gota a gota cloruro de propionilo (100 mg, 1,09 mmoles), y la mezcla se agitó a 0°C durante 30 minutos adicionales una vez completada la adición gota a gota. Se añadió agua (15 mL), y la mezcla se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1) para proporcionar el compuesto 46 (140 mg, rendimiento: 63%). LC-MS (ESI): m/z=387 [M+H]+.
Preparación del Compuesto T-46
El compuesto 46 (140 mg, 0,36 mmoles) y 1-metil-4-aminopirazol (105 mg, 1,09 mmoles) se disolvieron en n-butanol (3 mL), se añadió monohidrato de ácido p-toluenosulfónico (137 mg, 0,73 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida, y el residuo se purificó mediante TLC preparativa (diclorometano : metanol=10:1) para proporcionar un sólido de color amarillo T-46 (65 mg, rendimiento: 40%). LC-MS (ESI): m/z=448 [M+H]+.
RMN H1 (400MHz, DMSO-ds) 5: 9,46 (s, 1H), 8,79 (s, 1H), 8,37 (s, 1H), 8,34 (d, J=5Hz, 1H), 8,05 (s, 1H), 7,56 (s, 1H), 7,37 (d, J=5Hz, 1H), 4,78 (d, J=9Hz, 1H), 4,78 (m, 2H), 4,24 (d, J=9Hz, 1H), 3,84 (s, 3H), 3,72 (s, 2H), 2,15 (q, J= 7Hz, 2H), 0,99 (t, J= 7Hz, 3H) ppm
Ejemplo 47
2-(1-(Etilsulfonil)-3-(4-(2-(piridazin-4-ilamino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-47
Ruta sintética:

El compuesto 5 (50 mg, 0,12 mmoles) y 4-aminopiridazina (34 mg, 0,36 mmoles) se disolvieron en n-butanol (3 mL), se añadió monohidrato de ácido p-toluenosulfónico (45 mg, 0,24 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-47 (6 mg, rendimiento: 11%). LC-MS (ESI): m/z=482 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,79 (d, J=9Hz, 1H), 8,54 (s, 1H), 8,45 (s, 1H), 8,08 (d, J=6Hz, 1H), 7,92 (d, J=3Hz, 1H), 7,66 (d, J=6Hz, 1H), 6,32 (m, 1H), 4,66 (d, J=9Hz, 2H), 4,26 (d, J= 9Hz, 2H), 3,44 (s, 2H), 3,12 (q, J= 7Hz, 2H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 48
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-met¡l-1H-¡m¡dazol-4-¡l)am¡no)tieno[3,2-d]pr¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡trilo T-48
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 48-a
Bajo hidrógeno (1 atm), a una solución del compuesto 1-metil-4-nitro-1H-imidazol (500 mg, 3,94 mmoles) en etanol (20 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 48-a (310 mg, rendimiento: 82%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparac¡ón del Compuesto T-48
El compuesto 5 (70 mg, 0,17 mmoles) y el compuesto 48-a (48 mg, 0,5 mmoles) se disolvieron en n-butanol (3 mL), se añadió monohidrato de ácido p-toluenosulfónico (60 mg, 0,33 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-48 (5 mg, rendimiento: 6%). LC-MS (ESI): m/z=484 [M+H]+.
RMN H1 (500MHz, CD3OD) 5: 8,80 (s, 1H), 8,64 (s, 1H), 8,47 (s, 1H), 8,28 (d, J=5Hz, 1H), 7,49 (d, J=5Hz, 1H), 7,45 (s, 1H), 4,67 (d, J=9Hz, 2H), 4,32 (d, J= 9Hz, 2H), 3,99 (s, 2H), 3,65 (s, 2H), 3,20 (q, J= 7Hz, 2H), 1,39 (t, J= 7Hz, 3H) ppm Ejemplo 49
2-(1-(2-H¡drox¡acet¡l)-3-(4-(2-((1-met¡l-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d] p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-49
Ruta s¡ntét¡ca:
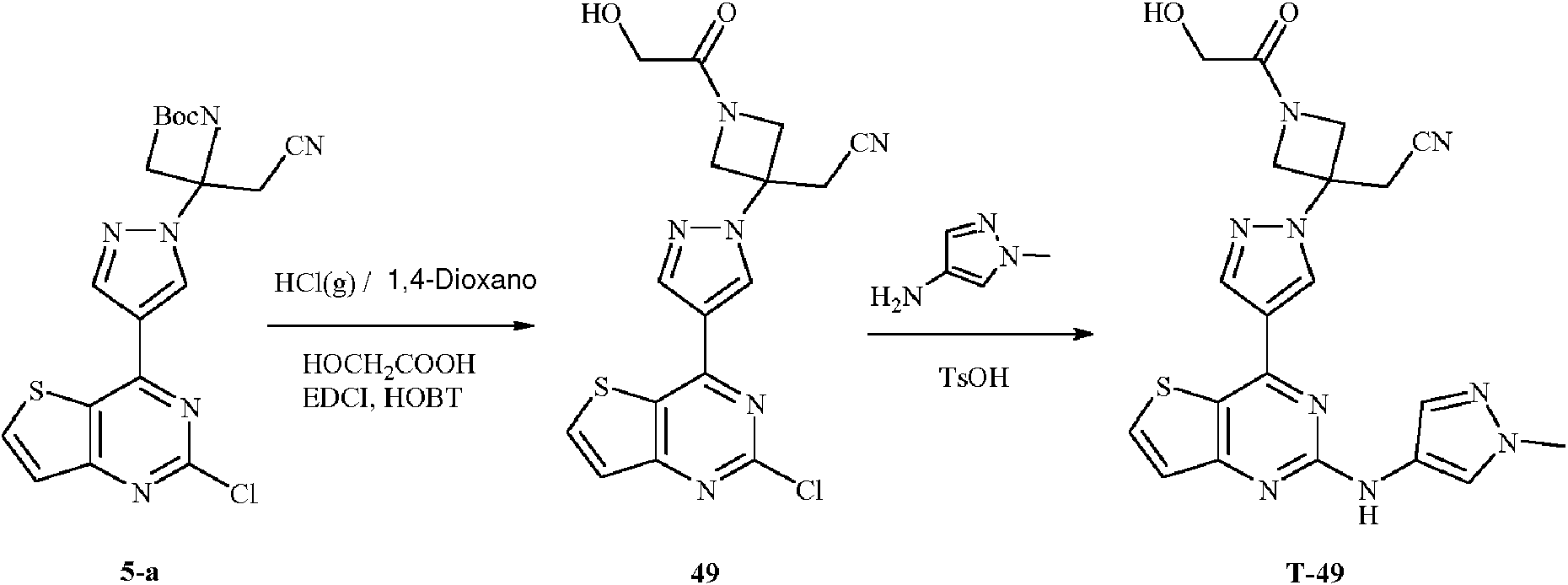
Preparación del Compuesto 49
A una solución del compuesto 5-a (70 mg, 0,16 mmoles) en diclorometano (1 mL) se le añadió una solución de hidrocloruro en 1,4-dioxano (4 N, 1 mL), la mezcla se agitó a temperatura ambiente durante 16 horas, a continuación, se concentró a presión reducida, y el residuo se trató con diclorometano (5 mL) y trietilamina (0,1 mL). La mezcla se agitó durante 15 minutos y a continuación, se trató con ácido glicólico (16 mg, 0,21 mmoles), EDCI (40 mg, 0,21 mmoles) y HOBT (3 mg, 0,02 mmoles) sucesivamente. La mezcla se agitó a temperatura ambiente durante 16 horas adicionales, a continuación, se añadió agua (15 mL), y la mezcla se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con una solución acuosa de HCl (1 N, 50 mL) y una solución acuosa saturada de NaHCO3 (50 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (diclorometano : metanol=10:1) para proporcionar el compuesto 49 (20 mg, rendimiento: 44%). LC-MS (ESI): m/z=389 [M+H]+.
Preparación del Compuesto T-49
El compuesto 49 (20 mg, 0,05 mmoles) y 1-metil-4-aminopirazol (15 mg, 0,16 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (20 mg, 0,1 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se concentró a presión reducida, el residuo se purificó mediante TLC preparativa (diclorometano : metanol=10:1) para proporcionar un sólido de color amarillo T-49 (5 mg, rendimiento: 22%). LC-MS (ESI): m/z=450 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,77 (s, 1H), 8,43 (s, 1H), 8,25 (d, J=5Hz, 1H), 8,02 (s, 1H), 7,65 (s, 1H), 7,35 (d, J=5Hz, 1H), 4,99 (d, J=10Hz, 1H), 4,76 (d, J=10Hz, 1H), 4,69 (d, J=11 Hz, 1H), 4,46 (d, J=11 Hz, 1H), 4,21 (s, 2H), 3,93 (s, 3H), 3,62 (s, 2H) ppm
Ejemplo 50
2-(4-(4-(2-((1-(2-Morfolinoetil)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)tetrahidro-2H-piran-4-il)acetonitrilo T-50
Ruta sintética:
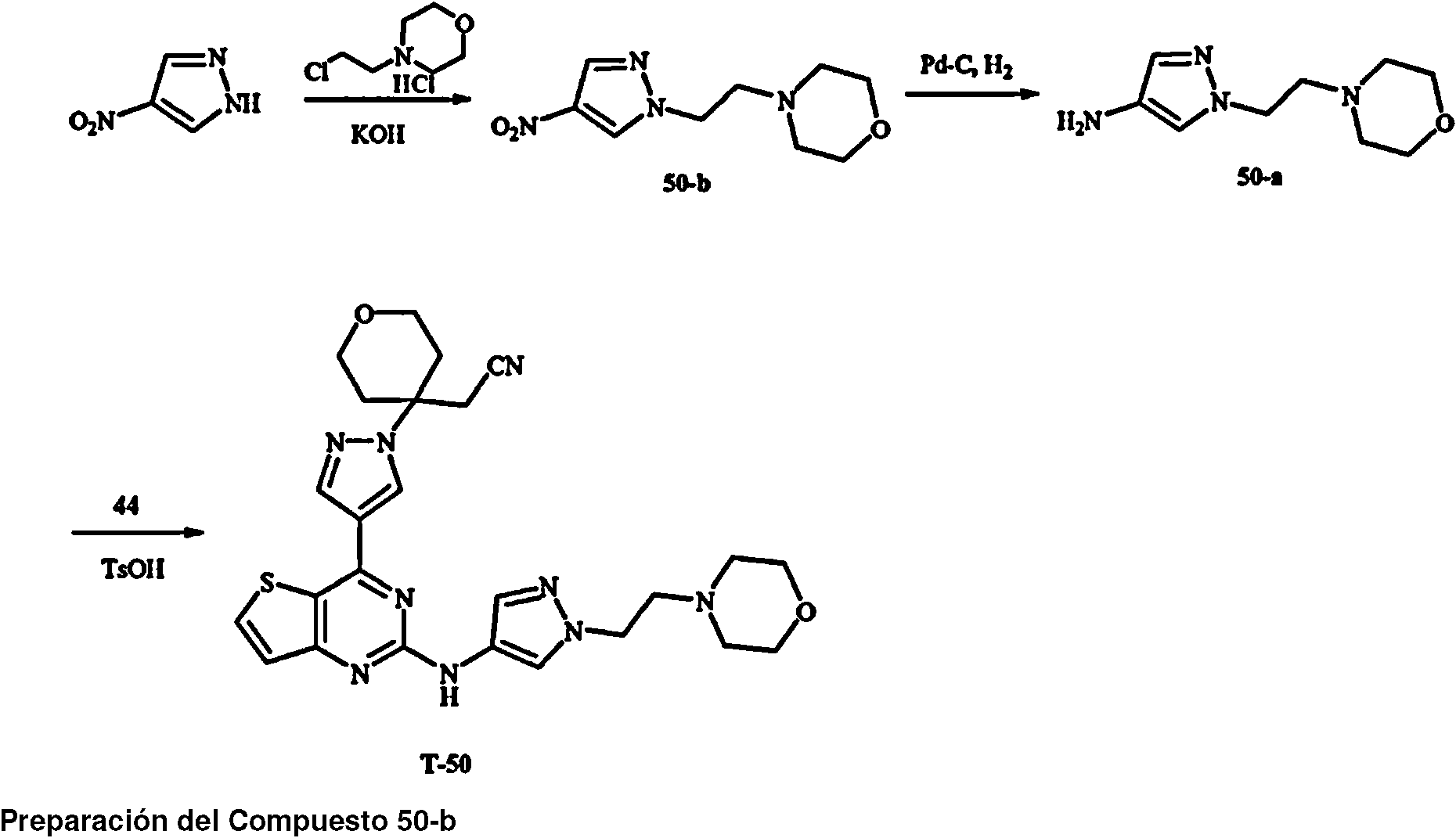
Se añadieron hidrocloruro de N-(2-cloroetil)morfolina (2,1 g, 11,06 mmoles) e hidróxido de potasio (1,24 g, 22,12 mmoles) a una solución de 4-nitropirazol (1,0 g, 8,85 mmoles) en etanol (20 mL) sucesivamente, la mezcla se calentó a 80°C y se agitó durante 3 horas. La mezcla se concentró a continuación, a presión reducida, el residuo se trató con agua (50 mL), se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto 50-b (600 mg, rendimiento: 30%). LC-MS (ESI): m/z=227 [M+H]+.
Preparación del Compuesto 50-a
Bajo hidrógeno (1 atm), a una solución del compuesto 50-b (600 mg, 2,66 mmoles) en etanol (20 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 50-a (450 mg, rendimiento: 87%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=197 [M+H]+.
Preparación del Compuesto T-50
El compuesto 50-a (80 mg, 0,41 mmoles) y el compuesto 44 (60 mg, 0,17 mmoles) se disolvieron en n-butanol (6 mL), se añadió monohidrato de ácido p-toluenosulfónico (210 mg, 1,11 mmoles). La mezcla se calentó a 130°C y se agitó durante 3 horas, a continuación, se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 60%-90%-10%) para proporcionar un sólido de color amarillo T-50 (10 mg, rendimiento: 12%). LC-MS (ESl): m/z=520 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,71 (s, 1H), 8,44 (s, 1H), 8,25 (s, 1H), 8,21 (d, J=6Hz, 1H), 7,75 (s, 1H), 7,36 (d, J=6Hz, 1H), 4,65 (t, J=6Hz, 2H), 3,91 (m, 6H), 3,57 (m, 2H), 3,32 (br, 4H), 3,18 (s, 2H), 2,70 (m, 2H), 2,25 (m, 2H) ppm
Ejemplo 51
2-(1-(Etilsulfonil)-3-(4-(2-((1-(2-hidroxietil)-1H-pirazo1-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-51
Ruta sintética:

Se añadieron 2-bromoetanol (1,9 g, 15,57 mmoles) y carbonato de potasio (2,9 g, 21,12 mmoles) a una solución de 4-nitropirazol (1,6 g, 14,16 mmoles) en acetonitrilo (20 mL) sucesivamente, la mezcla se calentó a 60°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 51-b (1,1 g, rendimiento: 49,5%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto 51-a
Bajo hidrógeno (1 atm), a una solución del compuesto 51-b (1,1 g, 7 mmoles) en etanol (20 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 51-a (740 mg, rendimiento: 83%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=128 [M+H]+.
Preparación del Compuesto T-51
El compuesto 51-a (72 mg, 0,57 mmoles) y el compuesto 5 (80 mg, 0,19 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (72 mg, 0,38 mmoles). La mezcla se calentó a 115°C y se agitó durante 18 horas, a continuación, se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar un sólido de color amarillo T-51 (45 mg, rendimiento: 46%). LC-MS (eS i): m/z=514 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 11,80 (s, 1H), 8,52 (s, 1H), 8,15 (s, 1H), 8,01 (d, J=6Hz, 1H), 7,77 (s, 1H), 7,68 (s, 1H), 7,28 (s, 1H), 7,18 (d, J=6Hz, 1H), 4,69 (d, J=9Hz, 2H), 4,24 (d, J=9Hz, 4H), 4,03 (s, 2H), 3,49 (s, 2H), 3,12 (q, J=7Hz, 2H), 1,43 (t, J=7Hz, 3H) ppm
Ejemplo 52
2-(3-(4-(2-((1-Acetil-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(etilsulfonil)azetidin-3-il)acetonitrilo T-52
Ruta sintética:

A 0°C, a una solución del compuesto T-31 (35 mg, 0,075 mmoles) y trietilamina (0,1 mL) en diclorometano (5 mL) se le añadió lentamente cloruro de acetilo (7 mg, 0,089 mmoles), y se agitó durante 30 minutos. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (5 mL), se extrajo con diclorometano (5 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 70%-95%-10%) para proporcionar el compuesto T-52 (25 mg, rendimiento: 66%). LC-MS (ESI): m/z=512 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,78 (s, 1H), 8,56 (s, 1H), 8,41 (s, 1H), 8,01 (d, J=5Hz, 1H), 7,92 (s, 1H), 7,46 (d, J=5Hz, 1H), 4,65 (d, J=9Hz, 2H), 4,28 (d, J=9Hz, 4H), 3,42 (s, 2H), 3,11 (q, J= 7Hz, 2H), 2,72 (s, 3H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 53
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-(2-morfol¡noet¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡tr¡lo T-53
Ruta s¡ntét¡ca:

El compuesto 50-a (97 mg, 0,5 mmoles) y el compuesto 5 (70 mg, 0,17 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (126 mg, 0,67 mmoles). La mezcla se calentó a 115°C y se agitó durante 18 horas, a continuación, se concentró a presión reducida, el residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar un sólido de color amarillo T-53 (41 mg, rendimiento: 43%). LC-MS (ESl): m/z=583 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 12,09 (s, 1H), 8,94 (s, 1H), 8,39 (s, 1H), 8,21 (s, 1H), 8,14 (d, J=5Hz, 1H), 7,72 (s, 1H), 7,47 (d, J=5Hz, 1H), 4,74 (m, 4H), 4,26 (d, J=9Hz, 4H), 3,93 (m, 4H), 3,71 (t, J=5Hz, 2H), 3,51 (s, 2H), 3,11 (m, 4H), 3,09 (q, J= 7Hz, 2H), 1,43 (t, J= 7Hz, 3H) ppm
Ejemplo 54
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-(1-met¡lazet¡d¡n-3-¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-54
Ruta sintética:

Se añadió lentamente borohidruro de sodio (1,01 g, 26,7 mmoles) a una solución de 1 -Boc-3-azetidinona (2,28 g, 13,3 mmoles) en etanol (30 mL), se agitó durante 2 horas, la mezcla se concentró a continuación, a presión reducida. El residuo se trató con agua (50 mL), se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 54-d (2,28 g, rendimiento: 99%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=175 [M+H]+.
Preparación del Compuesto 54-c
A 0°C, se añadió lentamente cloruro de p-toluenosulfonilo (2,12 g, 11,1 mmoles) a una solución del compuesto 54-d (1,28 g, 7,4 mmoles) y DABCO (1,66 g, 14,8 mmoles) en diclorometano (30 mL). Después de aumentar la temperatura a temperatura ambiente, la mezcla se agitó durante 40 minutos adicionales. La mezcla se diluyó con diclorometano (30 mL), se lavó con una solución acuosa de HCl (1 N, 50 mL), una solución acuosa saturada de NaHCO3 (50 mL) y agua (50 mL) sucesivamente. La capa orgánica se secó sobre sulfato de sodio anhidro, a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 54-c (1,96 g, rendimiento: 81%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=350 [M+Na]+.
Preparación del Compuesto 54-b
El compuesto 54-c (4,9 g, 15 mmoles) y carbonato de cesio (6,5 g, 2 mmoles) se añadieron a una solución de 4-nitropirazol (1,13 g, 10 mmoles) en DMF (15 mL) sucesivamente, y a continuación, la mezcla se calentó a 120°C y se agitó durante 12 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (60 mL), se extrajo con acetato de etilo (30 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 54-b (1,1 g, rendimiento: 41%). LC-MS (ESI): m/z=291 [M+Na]+.
Preparación del Compuesto 54-a
Una solución del compuesto 54-b (1,1 g, 4,1 mmoles) en THF anhidro (10 mL) se añadió lentamente a una suspensión de hidruro de litio y aluminio (360 mg, 10 mmoles) en THF anhidro (10 mL). La mezcla se sometió a reflujo durante 3 horas, a continuación, se enfrió a temperatura ambiente, después de añadió lentamente gota a gota acetato de etilo (2 mL). La mezcla se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 54-a (430 mg, rendimiento: 69%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=153 [M+H]+.
Preparación del Compuesto T-54
El compuesto 54-a (152 mg, 1 mmol) y el compuesto 5 (221 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (258 mg, 1,5 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCÜ3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar un sólido de color amarillo T-54 (28 mg, rendimiento: 10,4%). LC-MS (ESI): m/z=554 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,74 (s, 1H), 8,43 (s, 1H), 8,25 (s, 1H), 8,13 (d, J=4,0Hz, 1H), 7,74 (s, 1H), 7,35 (d, J=4,0Hz, 1H), 5,05 (m, 1H), 4,67 (d, J=7,2Hz, 2H), 4,33 (d, J=7,2Hz, 2H), 3,90 (t, J=5,6Hz, 2H), 3,66 (t, J=5,6Hz, 2H), 3,64 (s, 2H), 3,21 (q, J=6,0Hz, 2H), 2,51 (s, 3H), 1,38 (t, J=6,0Hz, 3H) ppm
Ejemplo 55
2-(1-(Etilsulfonil)-3-(4-(5-(fenilamino)pirazolo[1,5-a]pirimidin-7-il)-1H-pirazol-1-il)azetidin-3-il)acetonitrilo T-55
Ruta sintética:

Preparación del Compuesto 55-a
Bajo nitrógeno, el compuesto 5-a (400 mg, 1,04 mmoles), 5,7-dicloropirazolo[1,5-a]pirimidina (200 mg, 1,04 mmoles) y carbonato de sodio (331 mg, 3,12 mmoles) se suspendieron en 1,4-dioxano (2 mL) y agua (2 mL), se añadió Pd(dppf)Cl2 (100 mg, 0,1 mmoles). La mezcla se calentó a 80°C y se agitó durante 16 horas. La mezcla se concentró a presión reducida, el residuo se trató con diclorometano (20 mL), se lavó con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente. La mezcla se secó sobre sulfato de sodio anhidro, a continuación, se filtró, el producto filtrado se concentró a presión reducida, el residuo se purificó mediante TLC preparativa (diclorometano : acetato de etilo=30:1) para proporcionar el compuesto 55-a (230 mg, rendimiento: 54%). LC-MS (ESl): m/z=414 [M+H]+.
Preparación del Compuesto 55
A una solución del compuesto 55-a (230 mg, 0,56 mmoles) en THF (2 mL) se le añadió una solución de hidrocloruro en 1,4-dioxano (4 N, 1,5 mL), la mezcla se agitó a temperatura ambiente durante 16 horas, a continuación, se concentró a presión reducida y el residuo se trató con diclorometano (5 mL) y trietilamina (0,3 mL, 2,2 mmoles). La mezcla se enfrió a continuación, a 0°C, se añadió gota a gota cloruro de etilsulfonilo (0,15 mL, 0,84 mmoles), y la mezcla se agitó a 0°C durante 30 minutos adicionales una vez completada la adición gota a gota. Se añadió agua (5 mL), y la mezcla se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto 55(102 mg, rendimiento: 45%). LC-MS (ESI): m/z=406 [M+H]+.
Preparación del Compuesto T-55
Bajo nitrógeno, a una suspensión del compuesto 55 (70 mg, 0,17 mmoles), anilina (25 mg, 0,26 mmoles) y carbonato de cesio (102 mg, 0,32 mmoles) en 1,4-dioxano (4 mL) se le añadieron Pd(OAc)2 (15 mg, 0,07 mmoles) y BlNAP (40 mg, 0,07 mmoles), la mezcla se calentó a 125°C mediante microondas y se agitó durante 40 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo (10 mL), se lavó con agua (5 mLx3) y salmuera saturada (5 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar el compuesto T-55 (10 mg, rendimiento: 13 %). LC-MS (ESI): m/z=463 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 9,12 (s, 1H), 8,15 (s, 1H), 8,01 (d, J=2Hz, 1H), 7,46 (m, 4H), 7,26 (m, 1H), 6,61 (s, 1H), 6,39 (d, J=2Hz, 1H), 4,63 (d, J=9Hz, 2H), 4,24 (d, J= 9Hz, 2H), 3,42 (s, 2H), 3,09 (q, J= 7Hz, 2H), 1,42 (t, J= 7Hz, 3H) ppm
Ejemplo 56
2-Fluoro-4-(2-(fenilamino)tieno[3,2-d]pirimidin-4-il)benzonitrilo T-56
Ruta sintética:

Preparación del Compuesto 56-a
Bajo nitrógeno, se suspendieron 4-bromo-2-fluorobenzonitrilo (4,0 g, 20 mmoles), bis(pinacolato)diboro (3,8 g, 30 mmoles) y acetato de potasio (6,1 g, 60 mmoles) en DMSO (50 mL), se añadió Pd(dppf)Cl2 (1,5g, 0,2 mmoles). La mezcla se agitó a 80°C durante 4 horas. La mezcla se diluyó con agua (100 mL), se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=50:1 a 10:1) para proporcionar el compuesto 56-a (3,6 g, rendimiento: 73 %).
RMN H1 (400MHz, CD3OD) 5: 7,62 (m, 3H), 1,35 (s, 12H) ppm
Preparación del Compuesto 56
Bajo nitrógeno, el compuesto 56-a (1,0 g, 4 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (1,0 g, 4,9 mmoles) y carbonato de potasio (1,38 g, 10 mmoles) se suspendieron en 1,4-dioxano (20 mL) y agua (4 mL), se añadió Pd(PPh3)4 (490 mg, 0,6 mmoles). La mezcla se calentó a 100°C y se agitó durante 2 horas. La mezcla se concentró a presión reducida, el residuo se trató con agua (20 mL), se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar un sólido de color amarillo claro 56 (1,0 g, rendimiento: 87,5 %). LC-MS (ESI): m/z=290 [M+H]+.
Preparación del Compuesto T-56
El compuesto 56 (290 mg, 1,0 mmoles) y anilina (140 mg, 1,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (260 mg, 1,5 mmoles). La mezcla se calentó a 120°C y se agitó durante 6 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1 a 1:1) para proporcionar el compuesto T-56 (120 mg, rendimiento: 35 %). LC-MS (ESI): m/z=347 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 9,89 (s, 1H), 8,48 (d, J=5,2, 1H), 8,29 (m, 3H), 7,87 (d, J=7,6, 1H), 7,51 (d, J=5,2Hz, 1H), 7,33 (t, J=7,2Hz, 2H), 6,98 (t, J=7,2Hz, 1H) ppm
Ejemplo 57
2-(3-(4-(2-((1H-Indazol-5-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(etilsulfonil)azetidin-3-il)acetonitrilo T-57
Ruta sintética:

Se disolvieron 5-aminoindazol (47 mg, 0,36 mmoles) y el compuesto 5 (50 mg, 0,12 mmoles) en n-butanol (0,5 mL), se añadió monohidrato de ácido p-toluenosulfónico (43 mg, 0,24 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (5 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (5 mLx3) y salmuera saturada (5 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: metanol, agua (ácido trifluoroacético al 0,05%); gradiente: 70%-95%-10%) para proporcionar un sólido de color amarillo T-57 (20 mg, rendimiento: 33%). Lc -MS (ESI): m/z=520 [M+H]+.
RMN H1 (400MHz, CDCh) 5: 11,1 (br, 1H), 8,53 (s, 1H), 8,36 (s, 1H), 8,15 (d, J=1,2Hz, 1H), 8,09 (d, J=9Hz, 2H), 7,52 (d, J=1,2Hz, 1H), 7,48 (d, J=9Hz, 1H), 4,62 (d, J=9Hz, 2H), 4,23 (d, J=9Hz, 2H), 3,41 (s, 2H), 3,09 (q, J= 7Hz, 2H), 1,42 (t, J= 7Hz, 3H) ppm
Ejemplo 58
2-(3-(4-(2-((1H-Benzo[d]imidazol-5-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(etilsulfonil)azetidin-3-
¡l)acetonitr¡lo T-58
Ruta sintética:

Se disolvieron 5-aminobenzimidazol (47 mg, 0,36 mmoles) y el compuesto 5 (50 mg, 0,12 mmoles) en n-butanol (0,5 mL), se añadió monohidrato de ácido p-toluenosulfónico (43 mg, 0,24 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCÜ3 (10 mL), se extrajo con diclorometano (5 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (5 mLx3) y salmuera saturada (5 mL), se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: metanol, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar un sólido de color amarillo T-58 (20 mg, rendimiento: 33%). LC-MS (ESI): m/z=520 [M+H]+.
RMN H1 (500MHz, DMSO-d6) 5: 10,11 (s, 1H), 9,45 (s, 1H), 8,88 (s, 1H), 8,76 (d, J=1,5Hz, 1H), 8,52 (s, 2H), 8,47 (d, J=1,5Hz, 1H), 7,85 (dd, J=10Hz, J=1,5Hz, 1H), 7,44 (d, J=5,5Hz, 1H), 4,59 (d, J=9Hz, 2H), 4,29 (d, J=9Hz, 2H), 3,75 (s, 2H), 3,25 (q, J= 7Hz, 2H), 1,26 (t, J= 7Hz, 3H) ppm
Ejemplo 59
2-(3-(4-(2-((4-(1H-Tetrazol-1-¡l)fen¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(et¡lsulfon¡l)azet¡d¡n-3-il)acetonitrilo T-59
Ruta sintética:

Se disolvieron 4-(1 H-tetrazol-1 -il)anilina (77 mg, 0,47 mmoles) y el compuesto 5 (100 mg, 0,26 mmoles) en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (68 mg, 0,36 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 50%-80%-10%) para proporcionar un sólido de color amarillo T-59 (44 mg, rendimiento: 34%). Lc -MS (ESI): m/z=548 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 10,07 (s, 1H), 10,00 (s, 1H), 8,88 (s, 1H), 8,45 (d, =9Hz, 1H), 8,16 (d, J=9Hz, 2H),
7,84 (d, J=1,2Hz, 1H), 7,49 (d, J=10Hz, J=1,6Hz, 1H), 4,58 (d, J=9Hz, 2H), 4,28 (d, J=9Hz, 2H), 3,74 (s, 2H), 3,25 (q, J= 7Hz, 2H), 1,26 (t, J= 7Hz, 3H) ppm
Ejemplo 60
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-(3-morfol¡noprop¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡tr¡lo T-60
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 60-b
A 0°C, se añadió NaH al 60% en aceite mineral (530 mg, 13,27 mmoles) a una solución de 4-nitropirazol (1,0 g, 8,85 mmoles) en THF (10 mL), se agitó durante 30 minutos, se añadió N-(3-cloropropil)morfolina (1,73 g, 10,62 mmoles). La mezcla se calentó a temperatura ambiente y se agitó durante 16 horas adicionales. La mezcla se diluyó con agua (25 mL), se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar un aceite de color amarillo 60-b (1,2 g, rendimiento: 56,6%). LC-MS (ESI): m/z=241 [M+H]+.
Preparac¡ón del Compuesto 60-a
Bajo hidrógeno (1 atm), a una solución del compuesto 60-b (300 mg, 1,25 mmoles) en etanol (5 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color pardo rojizo 60-a (180 mg, rendimiento: 69%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=211 [M+H]+.
Preparac¡ón del Compuesto T-60
El compuesto 60-a (100 mg, 0,48 mmoles) y el compuesto 5 (100 mg, 0,24 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (68 mg, 0,36 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCÜ3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 25%-55%-10%) para proporcionar un sólido de color amarillo T-60 (60 mg, rendimiento: 43%). Lc -MS (ESI): m/z=597 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,78 (s, 1H), 8,44 (s, 1H), 8,25 (s, 1H), 8,21 (d, J=5,2Hz, 1H), 8,18 (s, 1H), 7,72 (s, 1H), 7,35 (d, J=5,6Hz, 1H), 4,66 (d, J=9Hz, 2H), 4,33 (m, 4H), 4,06 (m, 2H), 3,77 (m, 2H), 3,65 (s, 2H), 3,50 (m, 2H), 3,20 (m, 6H), 2,36 (m, 2H), 1,38 (d, J=7Hz, 2H) ppm
Ejemplo 61
2-(3-(4-(2-((1-(3-(2H-Tetrazol-5-¡l)prop¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-il)-1H-p¡razol-1-¡l)-1-(etilsulfonil)azetidin-3-il)acetonitrilo T-61
Ruta sintética:
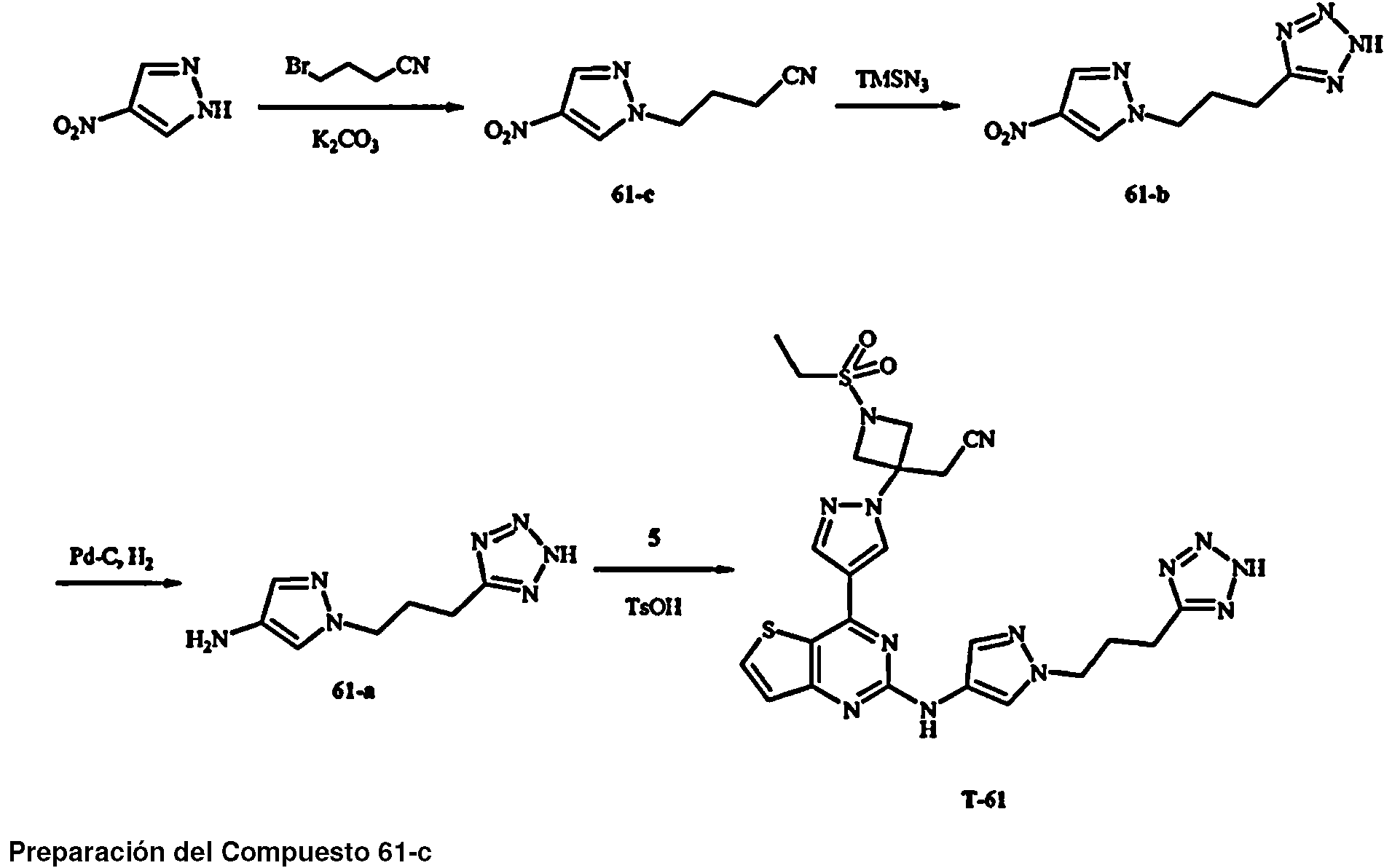
Se añadió 4-Bromobutironitrilo (4,27 g, 26,55 mmoles) a una solución de 4-nitropirazol (2,0 g, 17,69 mmoles) y carbonato de potasio (5,0 g, 35,39 mmoles) en acetonitrilo (20 mL), la mezcla se sometió a reflujo durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (60 mL), se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar el compuesto 61-c (2,5 g, rendimiento: 79%).
Preparación del Compuesto 61-b
Se añadió una solución de fluoruro de fluoruro de tetrabutilamonio 1,0 M in THF (12 mL, 12 mmoles) a una solución del compuesto 61-c (2,0 g, 11,1 mmoles) y azidotrimetilsilano (2,0 g, 17,39 mmoles) en tolueno (20 mL), la mezcla se sometió a reflujo durante 16 horas. Se añadió una porción adicional de azidotrimetilsilano (2,0 g, 17,39 mmoles), la mezcla se sometió a reflujo durante 16 horas adicionales. A continuación, la mezcla se concentró, el residuo se purificó mediante cromatografía en columna de sílice (diclorometano : metanol=10:1) para proporcionar el compuesto 61-b (2,1 g, rendimiento: 85 %). LC-MS (ESI): m/z=224 [M+H]+.
Preparación del Compuesto 61-a
Bajo hidrógeno (1 atm), a una solución del compuesto 61-b (1,0 g, 4,48 mmoles) en etanol (20 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color pardo rojizo 61-a (750 mg, rendimiento: 86%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=194 [M+H]+.
Preparación del Compuesto T-61
El compuesto 61-a (137 mg, 0,71 mmoles) y el compuesto 5 (100 mg, 0,24 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (91 mg, 0,47 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCÜ3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 25%-55%-10%) para proporcionar un sólido de color amarillo T-61 (19 mg,
rendimiento: 13,6%). LC-MS (ESI): m/z=580 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,49 (s, 1H), 8,37 (s, 1H), 8,03 (s, 1H), 7,91 (d, J=5,6Hz, 1H), 7,69 (s, 1H), 7,35 (d, J=5,6Hz, 1H), 6,97 (s, 1H), 4,66 (d, J=9Hz, 2H), 4,25 (d, J=9Hz, 4H), 4,17 (t, J=6Hz, 4H), 3,44 (s, 2H), 3,11 (q, J=7Hz, 2H), 2,91 (m, 2H), 2,28 (m, 2H), 1,42 (t, J=7Hz, 3H) ppm
Ejemplo 62
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((6-morfol¡nop¡r¡d¡n-3-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡trilo T-62
Ruta s¡ntét¡ca:
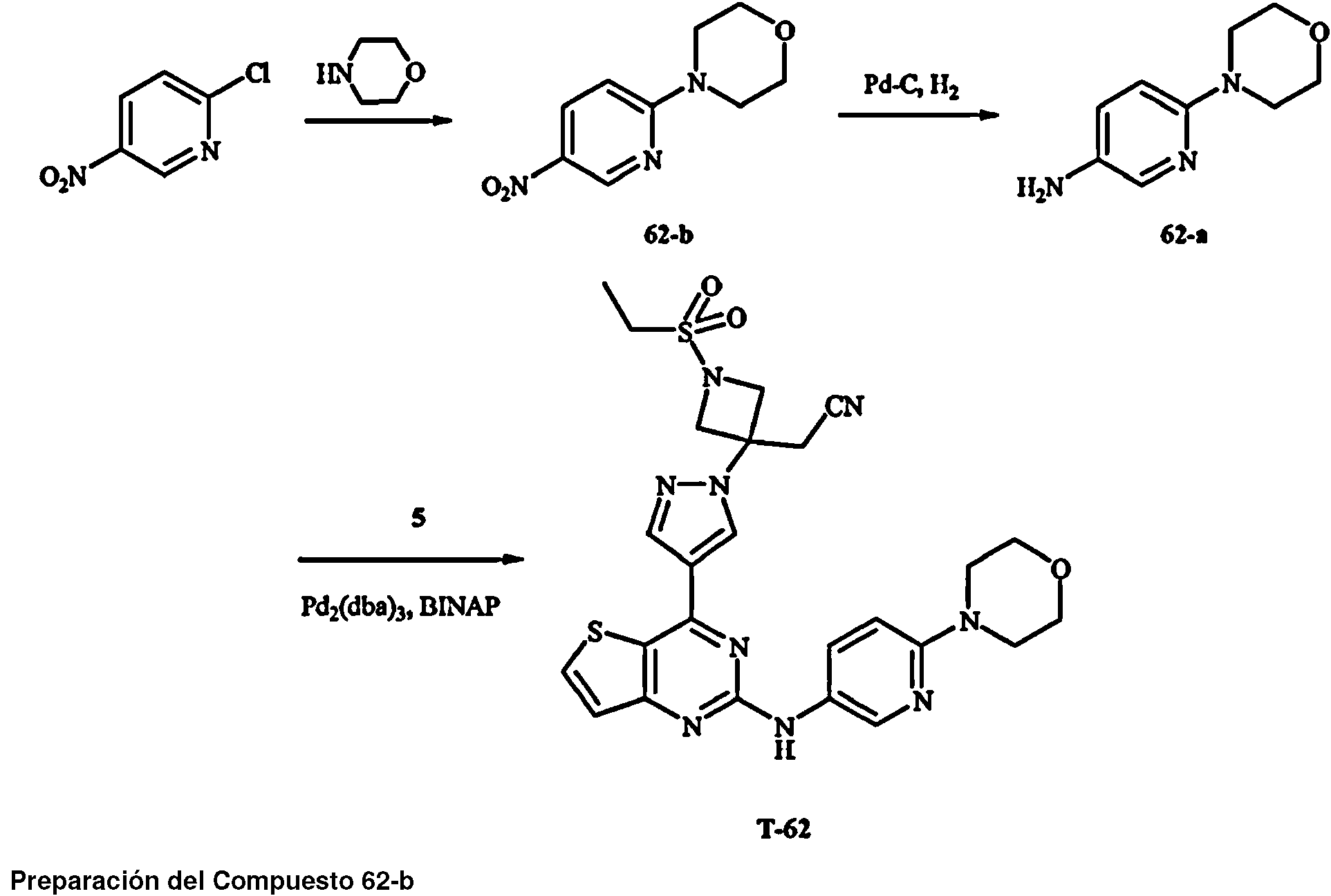
Se añadió morfolina (2,0 g, 31,7 mmoles) a una solución de 2-cloro-5-nitropiridina (2,0 g, 12,7 mmoles) en acetonitrilo (20 mL), la mezcla se agitó a temperatura ambiente durante 16 horas. Después de añadir agua (50 mL), se produjo un precipitado sólido. Después de la filtración, el sólido se secó a vacío durante 6 horas para proporcionar el compuesto 62-b (2 g, rendimiento: 75,6%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparac¡ón del Compuesto 62-a
Bajo hidrógeno (1 atm), a una solución del compuesto 62-b (2,0 g, 9,62 mmoles) en etanol (20 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color pardo 62-a (1,5 g, rendimiento: 87%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=180 [M+H]+.
Preparac¡ón del Compuesto T-62
Bajo nitrógeno, se añadieron Pd2(dba)3 (28 mg, 0,03 mmoles) y BINAP (20 mg, 0,03 mmoles) a una suspensión del compuesto 5 (100 mg, 0,24 mmoles), el compuesto 62-a (85 mg, 0,47 mmoles) y carbonato de cesio (155 mg, 0,48 mmoles) en 1,4-dioxano (2 mL), la mezcla se calentó a 110°C mediante microondas y se agitó durante 30 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con diclorometano (15 mL), se lavó con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 35%-65%-10%) para proporcionar el compuesto T-62 (10 mg, rendimiento: 8 %). LC-MS (ESI): m/z=566 [M+H]+.
RMN H1 (500MHz, CDCla) 5: 12,45 (br, 1H), 9,22 (br, 2H), 8,43 (s, 1H), 8,23 (d, J=5,2Hz, 1H), 8,04 (dd, J=9,6Hz,
J=2,8Hz, 1H), 7,55 (d, J=9,6Hz, 1H), 7,03 (d, J=9,6Hz, 1H), 4,77 (d, J=9Hz, 2H), 4,30 (d, J= 9Hz, 2H), 3,93 (m, 4H), 3,73 (m, 4H), 3,59 (s, 2H), 3,15 (q, J= 7Hz, 2H), 1,42 (t, J= 7Hz, 3H) ppm
Ejemplo 63
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((2-morfol¡nop¡r¡m¡d¡n-5-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)acetonitr¡lo T-63
Ruta s¡ntét¡ca:
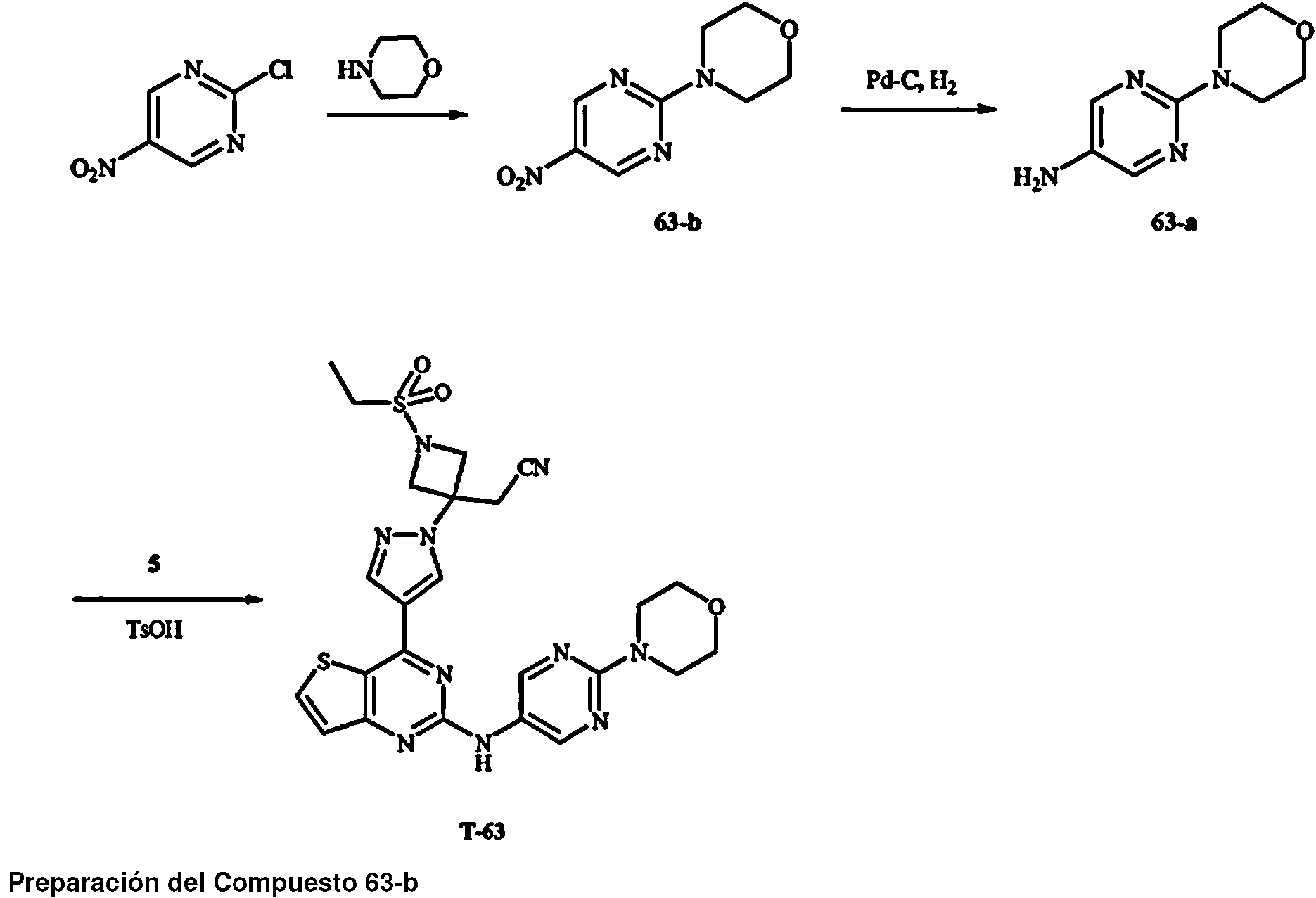
Se añadió morfolina (492 mg, 6,07 mmoles) a una solución de 2-doro-5-nitropirimidina (440 mg, 2,76 mmoles) en acetonitrilo (5 mL), la mezcla se agitó a temperatura ambiente durante 3 horas, a continuación, se concentró a presión reducida. El residuo se diluyó con acetato de etilo (50 mL) y una solución acuosa 1N de HCl (20 mL). La capa orgánica se lavó con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secó sobre sulfato de sodio anhidro, a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 63-b (350 mg, rendimiento: 61%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=211 [M+H]+.
Preparac¡ón del Compuesto 63-a
Bajo hidrógeno (1 atm), a una solución del compuesto 63-b (350 mg, 1,67 mmoles) en etanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color pardo 63-a (230 mg, rendimiento: 77%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=181 [M+H]+.
Preparac¡ón del Compuesto T-63
El compuesto 63-a (52 mg, 0,28 mmoles) y el compuesto 5 (80 mg, 0,19 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (44 mg, 0,23 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida, a continuación, se trató con una solución acuosa saturada de NaHCÜ3 (5 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 40%-70%-10%) para proporcionar un sólido de color pardo T-63 (12 mg, rendimiento: 12%). LC-MS (ESI): m/z=567 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,70 (d, J=2,4Hz, 1H), 8,48 (s, 1H), 8,37 (s, 1H), 7,97 (d, J=5,6Hz, 1H), 7,36 (d, J=5,2Hz, 1H), 4,63 (d, J=9,2Hz, 2H), 4,25 (d, J= 9,2Hz, 2H), 3,81 (s, 8H), 3,43 (s, 2H), 3,15 (q, J= 7Hz, 2H), 1,42 (t, J= 7Hz, 3H) ppm
Ejemplo 64
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-(1-met¡lp¡per¡d¡n-4-¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-i l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-64
Ruta s¡ntét¡ca:
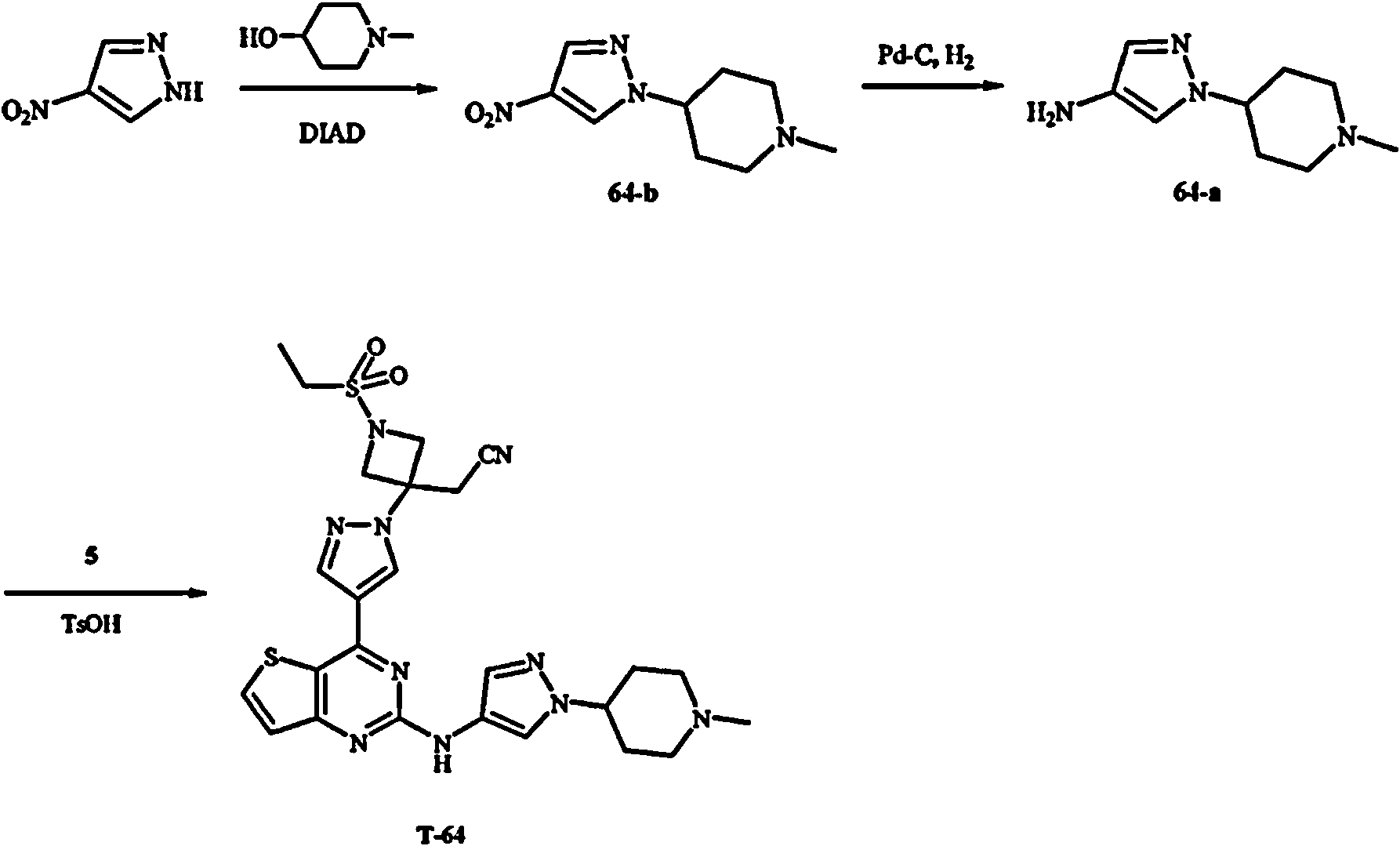
Preparac¡ón del Compuesto 64-b
A 0°C, se añadió lentamente DIAD (5,4 g, 26,54 mmoles) a una solución de 4-nitropirazol (2,0 g, 17,69 mmoles), PPh3 (6,95 g, 26,54 mmoles) y N-metil-4-hidroxipiperidina (2,4 g, 21,23 mmoles) en THF anhidro (50 mL). La mezcla se calentó a temperatura ambiente y se agitó durante 16 horas. Después de concentrar la mezcla a presión reducida, el residuo se diluyó con acetato de etilo (50 mL) y una solución acuosa 3 N de HCl (50 mL) sucesivamente. La capa acuosa se trató con una solución acuosa saturada de K2CO3 para ajustar a pH=9, a continuación, se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo 64-b (2,1 g, rendimiento: 57%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparac¡ón del Compuesto 64-a
Bajo hidrógeno (1 atm), a una solución del compuesto 64-b (500 mg, 2,38 mmoles) en etanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color pardo rojizo 64-a (420 mg, rendimiento: 98%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=181 [M+H]+.
Preparac¡ón del Compuesto T-64
El compuesto 64-a (128 mg, 0,71 mmoles) y el compuesto 5 (100 mg, 0,24 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (135 mg, 0,71 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de NaHCO3 (5 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 25%-55%-10%) para proporcionar un sólido de color pardo T-64 (62 mg, rendimiento: 47%). LC-MS (ESI): m/z=567 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,43 (s, 1H), 8,35 (s, 1H), 8,03 (s, 1H), 7,87 (d, J=5,6Hz, 1H), 7,58 (s, 1H), 7,31 (d, J=5,2Hz, 1H), 7,18 (s, 1H), 4,63 (d, J=9,2Hz, 2H), 4,25 (d, J= 9,2Hz, 2H), 4,18 (m, 1H), 3,40 (s, 2H), 3,09 (q, J= 7Hz, 2H), 3,01 (m, 2H), 2,35 (s, 3H), 2,15 (m, 6H), 1,41 (t, J= 7Hz, 3H) ppm
Ejemplo 65
2-(3-(4-(2-((1-(Azet¡d¡n-3-¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-il)-1H-p¡razol-1-¡l)-1 (etilsulfonil)azetidin-3-il)acetonitrilo T-65
Ruta sintética:
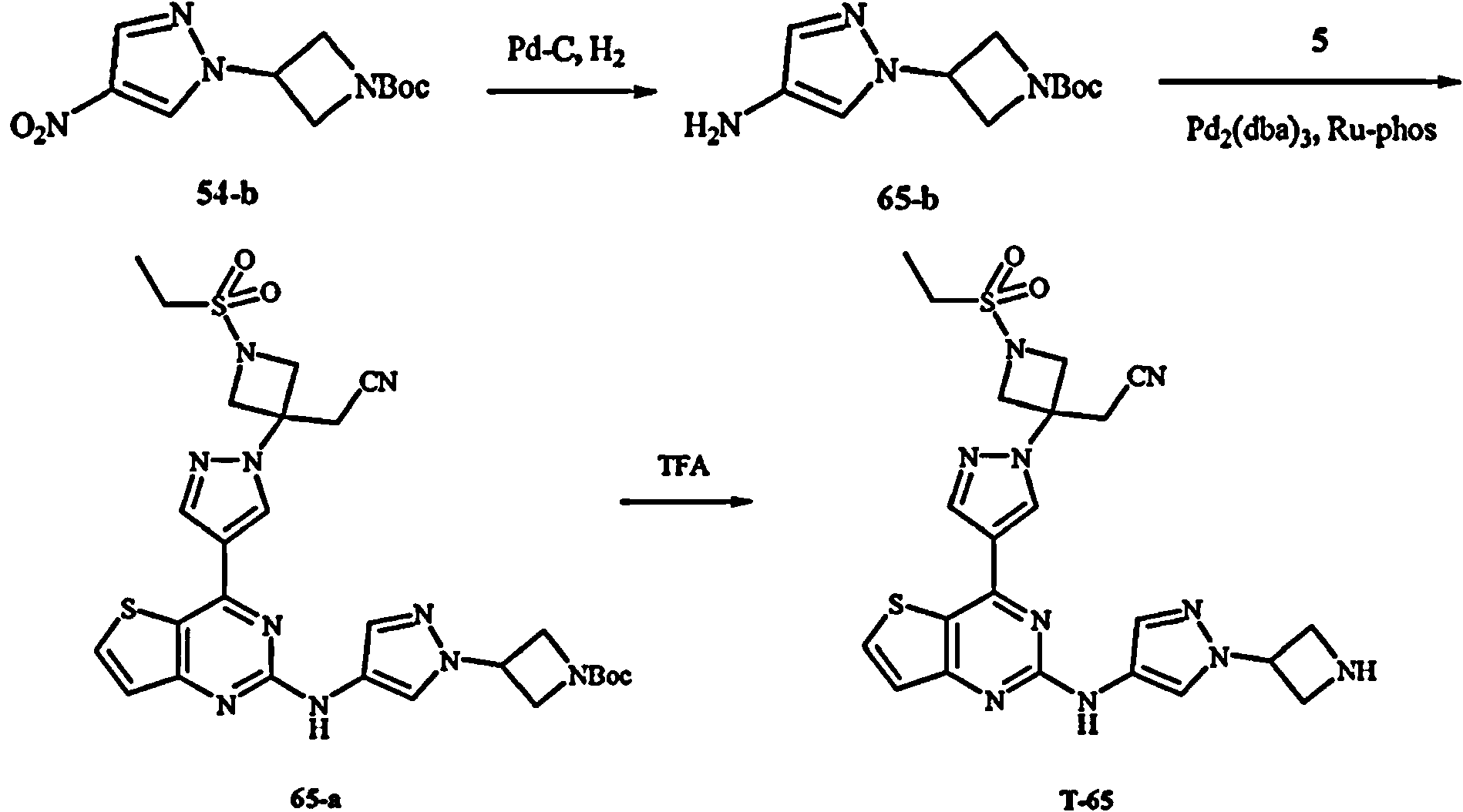
Preparación del Compuesto 65-b
Bajo hidrógeno (1 atm), a una solución del compuesto 54-b (1,33 g, 5 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 65-b (940 mg, rendimiento: 79%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=261 [M+Na]+.
Preparación del Compuesto 65-a
Bajo nitrógeno, a una suspensión del compuesto 5 (422 mg, 1,0 mmoles), el compuesto 65-b (357 mg, 1,5 mmoles) y carbonato de potasio (276 mg, 2,0 mmoles) en 1,4-dioxano (10 mL) se le añadieron Pd2(dba)3 (15 mg, 0,017 mmoles) y Ruphos (10 mg, 0,07 mmoles), la mezcla se calentó a 120°C mediante microondas y se agitó durante 40 minutos. Después de enfriar a temperatura ambiente, la mezcla se diluyó con acetato de etilo (10 mL), a continuación, se filtró, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1 a 1:1) para proporcionar el compuesto 65-a (374 mg, rendimiento: 60%). LC-MS (ESI): m/z=625 [M+H]+.
Preparac¡ón del Compuesto T-65
Se añadió ácido trifluoroacético (2 mL) a una solución del compuesto 65-a (370 mg, 0,6 mmoles) en diclorometano (6 mL) y se agitó durante 3 horas. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de NaHCÜ3 A continuación (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1 a 1:3) para proporcionar el compuesto T-65 (160 mg, rendimiento: 50%). LC-MS (ESI): m/z=525 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 9,56 (s, 1H), 8,84 (s, 1H), 8,41 (s, 1H), 8,37 (d, J=5,6, 1H), 8,22 (s, 1H), 7,74 (s, 1H), 7,81 (d, J=5,6Hz, 1H), 5,35 (m, 1H), 4,59 (d, J=9,6Hz, 2H), 4,27 (d, J=9,6Hz, 2H), 4,18 (m, 4H), 3,73 (s, 2H), 3,27 (q, J= 7,2Hz, 2H), 1,25 (t, J= 7,2Hz, 3H) ppm
Ejemplo 66
2-(3-(4-(2-((1-(1-(C¡cloprop¡lmet¡l)azet¡d¡n-3-¡l)-1H-p¡razol-4-¡l)amino)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(etilsulfon¡l)azet¡d¡n-3-¡l)acetonitr¡lo T-66
Ruta s¡ntét¡ca:

A 0°C, se añadió NaBH (AcO)3 (212 mg, 1,0 mmoles) a una solución del compuesto T-65 (120 mg, 0,23 mmoles) y ciclopropanocarbaldehído (70 mg, 1,0 mmoles) en un disolvente componente de metanol (6 mL) y diclorometano (6 mL), se agitó durante 2 horas, a continuación, se calentó a temperatura ambiente, y se agitó durante 16 horas adicionales. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1 a 1:3) para proporcionar el compuesto T-66(60 mg, rendimiento: 43 %). LC-MS (ESI): m/z=579 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 9,49 (s, 1H), 8,83 (s, 1H), 8,40 (s, 1H), 8,36 (d, J=5,2Hz, 1H), 8,20 (s, 1H), 7,65 (s, 1H), 7,38 (d, J=5,2Hz, 1H), 4,98 (m, 1H), 4,59 (d, J=9,2Hz, 2H), 4,27 (d, J=9,2Hz, 2H), 3,72 (m, 4H), 3,37 (t, J= 7,2Hz, 2H), 3,25 (q, J= 7,2Hz, 2H), 2,36 (d, J=7,2Hz, 2H), 1,25 (t, J= 7,2Hz, 3H), 0,79 (m, 1H), 0,41 (m, 2H), 0,11 (m, 2H) ppm
Ejemplo 67
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((1-(3-(4-met¡lp¡peraz¡n-1-¡l)prop¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-67
Ruta s¡ntét¡ca:

Se añadió lentamente 1,3-dibromopropano (4,0 g, 20 mmoles) a una solución de 1-boc piperazina (1,86 g, 10 mmoles) y trietilamina (4,0 g, 40 mmoles) en diclorometano (200 mL). La mezcla se agitó durante 16 horas, se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de NaHCO3 (100 mL), se extrajo con diclorometano (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1 a 1:3) para proporcionar el compuesto 67-e (1,25 g, rendimiento: 41 %).
RMN H1 (400MHz, CDCla) 5: 3,60 (t, J=6,6Hz, 2H), 3,43(t, J=5,0Hz, 4H), 2,49(t, J=7,0Hz, 2H), 2,38 (t, J=5,0Hz, 4H), 1,94 (m, 2H), 1,46 (s, 9H) ppm
Preparación del Compuesto 67-d
El compuesto 67-e (1,23 g, 4,1 mmoles) se añadió a una solución de 4-nitropirazol (650 mg, 5,0 mmoles) y carbonato de cesio (3,25 g, 10,0 mmoles) en DMF (15 mL), la mezcla se calentó a 90°C y se agitó durante 6 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (60 mL), se extrajo con acetato de etilo (20 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto 67-d (910 mg, rendimiento: 66%). LC-MS (ESI): m/z=340 [M+H]+.
Preparación del Compuesto 67-c
Se añadió ácido trifluoroacético (2 mL) a una solución del compuesto 67-d (560 mg, 1,66 mmoles) en diclorometano (6 mL) y se agitó durante 3 horas. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 67-c (360 mg, rendimiento: 90%), que se utilizó directamente para la siguiente etapa sin purificación. LC-Ms (ESI): m/z=240 [M+H]+.
Preparación del Compuesto 67-b
Se añadió lentamente p-toluenosulfonato de metilo (190 mg, 1,0 mmoles) a una solución del compuesto 67-c (239 mg, 1,0 mmoles) y carbonato de potasio (1,38 g, 10,0 mmoles) en THF. Después de agitar durante 2 horas, la mezcla se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (15 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 67-b (255 mg, rendimiento: 100%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=254 [M+H]+.
Preparación del Compuesto 67-a
Bajo hidrógeno (1 atm), a una solución del compuesto 67-b (255 mg, 1,0 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,1 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 67-a (180 mg, rendimiento: 80%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=224 [M+Na]+.
Preparación del Compuesto T-67
El compuesto 67-a (180 mg, 0,8 mmoles) y el compuesto 5 (221 mg, 0,5 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (258 mg, 1,5 mmoles). La mezcla se calentó a 120°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se trató con una solución acuosa saturada de NaHCÜ3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron. El residuo se purificó mediante HPLC preparativa (fase móvil: acetonitrilo, agua (ácido trifluoroacético al 0,05%); gradiente: 20%-50%-10%) para proporcionar el compuesto T-67 (15 mg, rendimiento: 4,9%). LC-MS (ESI): m/z=610 [M+H]+.
RMN H1 (400MHz, DMSO-d6) 5: 8,75 (s, 1H), 8,38 (s, 1H), 8,25 (d, J=5,2Hz, 1H), 8,09 (s, 1H), 7,89 (s, 1H), 7,32 (d, J=5,2Hz, 1H), 4,67 (d, J=9,2Hz, 2H), 4,33 (s, 2H), 4,30 (d, J=9,2Hz, 2H), 3,65 (s, 2H), 3,53 (s, 4H), 3,37 (s, 2H), 3,20 (q, J= 7,6Hz, 2H), 3,06 (m, 2H), 2,95 (s, 3H), 2,29 (m, 2H), 1,39 (t, J=7,6Hz, 3H) ppm
Ejemplo 68
2-(3-(4-(6-Cloro-2-((1-metil-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(etilsulfonil)azetidin-3-il)acetonitrilo T-68
Ruta sintética:
Preparación del Compuesto 68-d
A 0°C, se añadió lentamente 1,3-dihidrotiofeno[3,2-d]pirimidino-2,4-diona (1,68 g, 10 mmoles) a un disolvente componente de HNO3 conc. (15 mL) y H2SO4 conc. (15 mL), se agitó durante 1 hora, se añadió agua helada (30 mL), y la mezcla se agitó durante 3 horas adicionales. La mezcla se filtró, la torta del filtro se lavó con agua helada (5 mLx3), se secó a vacío durante 8 horas para proporcionar un sólido de color amarillo 68-d (1,36 g, rendimiento: 63,8%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=214 [M+H]+.
Preparación del Compuesto 68-c
Una mezcla del compuesto 68-d (1,36 g, 6,4 mmoles) y dicloruro fenilfosfónico (15 mL) se calentó a 180°C, se agitó durante 4 horas, la mezcla se enfrió a 90°C. Se añadió lentamente agua (200 mL). La mezcla se enfrió lentamente a 25°C, y se agitó durante 16 horas adicionales. La mezcla se filtró, la torta del filtro se lavó con agua (15 mLx3), se secó a vacío durante 8 horas para proporcionar un sólido de color blanco 68-c (1,1 g, rendimiento: 72%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=239 [M+H]+.
Preparación del Compuesto 68-b
Bajo nitrógeno, el compuesto 5-b (390 mg,1,0 mmoles), el compuesto 68-c (240 mg, 1,0 mmoles) y carbonato de potasio (280 mg, 2,0 mmoles) se suspendieron en 1,4-dioxano (8 mL) y agua (2 mL), se añadió Pd(PPh3)4 (48 mg, 0,04 mmoles), la mezcla se agitó a 80°C durante 4 horas. La mezcla se concentró a presión reducida, el residuo se diluyó con agua (10 mL), se extrajo con acetato de etilo (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (10 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 68-b (230 mg, rendimiento: 49,6%). LC-MS (ESI): m/z=465 [M+H]+.
Preparación del Compuesto 68-a
Se añadió ácido trifluoroacético (3 mL) a una solución del compuesto 68-b (230 mg, 0,5 mmoles) en diclorometano (6 mL) y se agitó durante 3 horas a temperatura ambiente. La mezcla se concentró a presión reducida para proporcionar el compuesto 68-a (160 mg, rendimiento: 88%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=365 [M+H]+.
Preparación del Compuesto 68
A temperatura ambiente, a una solución del compuesto 68-a (160 mg, 0,44 mmoles) y trietilamina (90 mg, 0,88 mmoles) en diclorometano (6 mL) se le añadió lentamente gota a gota cloruro de etilsulfonilo (65 mg, 0,5 mmoles). Después de agitar durante 3 horas, la mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (10 mLx3) y salmuera saturada (20 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 68 (150 mg, rendimiento: 74,5%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=459 [M+H]+.
Preparación del Compuesto T-68
El compuesto 68 (150 mg, 0,33 mmoles) y 1 -metil-4-aminopirazol (97 mg, 1,0 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (190 mg, 1,0 mmoles). La mezcla se calentó a 108°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida para eliminar el disolvente, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1 a 1:1) para proporcionar el compuesto T-68 (36 mg, rendimiento: 21 %). LC-MS (ESI): m/z=518 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,57 (s, 1H), 8,22 (s, 1H), 7,95 (s, 1H), 7,49 (s, 1H), 7,19 (s, 1H), 4,50 (d, J=9,2Hz, 2H), 4,20 (d, J=9,2Hz, 2H), 3,80 (s, 3H), 3,51 (s, 2H), 3,08 (q, J= 7,6Hz, 2H), 1,27 (t, J= 7,6Hz, 3H) ppm
Ejemplo 69
2-(1-(Etilsulfonil)-3-(4-(2-((1-(2,2,2-trifluoroetil)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-i l)azetidin-3-il)acetonitrilo T-69
Ruta sintética:

Se añadió 4-nitropirazol (1,13 g, 10 mmoles) a una solución de metanosulfonato de 2,2,2-trifluoroetilo (3,48 g, 15 mmoles) y carbonato de potasio (2,76 g, 20 mmoles) en DMF (10 mL), la mezcla se calentó a 40°C y se agitó durante 6 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (100 mL), se extrajo con acetato de etilo (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se lavó con éter de petróleo (20 mL) para proporcionar un sólido de color blanco 69-b (1,6 g, rendimiento: 82%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=196 [M+h ]+.
Preparación del Compuesto 69-a
Bajo hidrógeno (1 atm), a una solución del compuesto 69-b (1,5 g, 7,69 mmoles) en etanol (5 mL) se le añadió Pd-C al 10% (0,15 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color pardo rojizo 69-a (1,2 g, rendimiento: 95 %), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=166 [M+H]+.
Preparación del Compuesto T-69
El compuesto 69-a (176 mg, 1,07 mmoles) y el compuesto 5 (150 mg, 0,36 mmoles) se disolvieron en n-butanol (0,5 mL), se añadió monohidrato de ácido p-toluenosulfónico (170 mg, 0,89 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida para eliminar el disolvente, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (50 mL), se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (solución cromatográfica: acetato de etilo) para proporcionar un sólido de color amarillo T-69 (73 mg, rendimiento: 38 %). LC-MS (ESI): m/z=552 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,47 (s, 1H), 8,35 (s, 1H), 8,18 (s, 1H), 7,91 (d, J = 5,2Hz, 1H), 7,63 (s, 1H), 7,34 (d, J = 5,2Hz, 1H), 7,22 (s, 1H), 4,74 (dd, J = 16,8Hz, J = 8,4Hz, 2H), 4,64 (d, J=9Hz, 2H), 4,24 (d, J=9Hz, 2H), 3,41 (s, 2H), 3,10 (q, J= 7Hz, 2H), 1,42 (t, J= 7Hz, 3H) ppm
Ejemplo 70
2-(1-(Etilsulfonil)-3-(4-(2-((1-(2-metoxietil)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-i l)azetidin-3-il)acetonitrilo T-70
Ruta sintética:

A 0°C, se añadió lentamente DEAD (4,1 g, 19,91 mmoles) a una solución de 4-nitropirazol (1,5 g, 13,27 mmoles), 2-metoxietanol (1,5 g, 19,91 mmoles) y PPh3 (5,2 g, 19,91 mmoles) en THF anhidro (25 mL). La mezcla se calentó a temperatura ambiente y se agitó durante 3 horas, a continuación, se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar un sólido de color blanco 70-b (1,72 g, rendimiento: 76,1 %). LC-m S (ESI): m/z=198 [M+H]+.
Preparación del Compuesto 70-a
Bajo hidrógeno (1 atm), a una solución del compuesto 70-b (1,7 g, 10 mmoles) en etanol (20 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color pardo rojizo 70-a (1,3 g, rendimiento: 93%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=142 [M+H]+.
Preparación del Compuesto T-70
El compuesto 70-a (151 mg, 1,07 mmoles) y el compuesto 5 (150 mg, 0,36 mmoles) se disolvieron en n-butanol (3 mL), se añadió monohidrato de ácido p-toluenosulfónico (170 mg, 0,89 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida para eliminar el disolvente, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (50 mL), se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (solución cromatográfica: acetato de etilo) para proporcionar un sólido de color amarillo T-70 (55 mg, rendimiento: 30%). LC-MS (ESI): m/z=528 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,41 (s, 1H), 8,32 (s, 1H), 8,03 (s, 1H), 7,84 (d, J=5,2Hz, 1H), 7,58 (s, 1H), 7,45 (s, 1H), 7,27 (d, J=5,2Hz, 1H), 4,62 (d, J=9Hz, 2H), 4,30 (t, J=5,2Hz, 2H), 4,24 (d, J=9Hz, 2H), 3,79 (t, J=5,2Hz, 2H), 3,39 (s, 2H), 3,37 (s, 3H), 3,09 (q, J=7Hz, 2H), 1,41 (t, J=7Hz, 3H) ppm
Ejemplo 71
3-(4-((4-(1-(3-(Cianometil)-1-(etilsulfonil)azetidin-3-il)-1H-pirazol-4-il)tieno[3,2-d]pirimidin-2-il)amino)-1H-pirazol-1-il)propanonitrilo T-71
Ruta sintética:

A temperatura ambiente, se añadió DBU (0,1 mL) a una solución del compuesto T-31 (235 mg, 0,5 mmoles) y acrilonitrilo (50 mg, 1 mmol) en acetonitrilo (8 mL). Después de agitar durante 3 horas, la mezcla se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=5:1) para proporcionar un sólido de color amarillo claro T-71 (38 mg, rendimiento: 14,6%). LC-MS (ESI): m/z=523 [M+H]+.
RMN H1 (400MHz, DMSO-ds) 5: 9,54 (s, 1H), 8,83 (s, 1H), 8,38 (s, 1H), 8,36 (s, 1H), 8,20 (s, 1H), 7,65 (s, 1H), 4,58 (d, J=9,2Hz, 2H), 4,40 (t, J=6,0Hz, 2H), 4,27 (d, J=9,2Hz, 2H), 3,73 (s, 2H), 3,25 (q, J=7,6Hz, 2H), 3,07 (t, J=6,4Hz, 2H), 1,25 (t, J=7,6Hz, 3H) ppm
Ejemplo 72
2-(1-(Et¡lsulfon¡l)-3-(4-(2-((5-met¡l-1H-p¡razol-3-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)azet¡d¡n-3-il)aceton¡trilo T-72
Ruta s ¡ ntét ¡ ca:

Preparac¡on del Compuesto T-72
Se disolvieron 5-metil-3-aminopirazol (69 mg, 0,72 mmoles) y el compuesto 5 (100 mg, 0,24 mmoles) en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (91 mg, 0,48 mmoles). La mezcla se calentó a 110°C y se agitó durante 48 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de NaHCO3 (50 mL), se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (10 mmol/mL bicarbonato de amonio), acetonitrilo; gradiente: 22%-55%) para proporcionar un sólido de color amarillo T-72 (10 mg, rendimiento: 9%). LC-Ms (ESI): m/z=484 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,71-8,45 (2xs, 1H), 8,37-8,33 (2xs, 1H), 8,04-7,90 (2xd, J=5,6Hz, 1H), 7,90-7,57(2xd, J=5,6Hz, 1H), 7,34 (d, J=5,6Hz, 1H), 6,21-5,93 (2xbr, 1H), 5,44 (s, 1H), 4,64 (dd, J=3,6H, J=9,2Hz, 2H), 4,26 (dd, J=9Hz, J=3,6Hz, 2H), 3,41 (s, 2H), 3,10 (m, 2H), 2,31 (s, 3H), 1,42 (m, 3H) ppm
Ejemplo 73
2-(1-(4-(2-((1-(3-(Met¡lsulfon¡l)prop¡l)-1H-p¡razol-4-il)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)c¡clobut¡l)aceton¡tr¡lo T-73
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 73-f
A 0°C, una solución de cloruro de p-nitrobencenosulfonilo (2,43 g, 11 mmoles) en tolueno (20 mL) se le añadió gota a gota a una solución de 3-(metiltio)propan-1-ol (1,06 g, 10 mmoles) y DMAP (1,46 g, 12 mmoles) en tolueno (20 mL). La mezcla se calentó a 25°C y se agitó durante 16 horas. La mezcla se trató con agua (100 mL), se extrajo con acetato de etilo (100 mLx2). Las capas orgánicas se combinaron, se lavaron con salmuera saturada (100 mL), se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar un sólido de color blanco 73-f (2 g, rendimiento: 69 %). LC-MS (ESI): m/z=292 [M+H]+.
Preparación del Compuesto 73-e
Se añadió gota a gota a 0°C, una solución de Oxone (9,3 g, 15,12 mmoles) en agua (50 mL) a una solución del compuesto 73-f (2 g, 6,87 mmoles) en metanol (50 mL). La mezcla se calentó a temperatura ambiente y se agitó durante 2 horas. La mezcla se concentró a presión reducida para eliminar el disolvente, el residuo se trató con agua (50 mL), se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se lavaron con salmuera saturada (100 mL), se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 73-e (1,7 g, rendimiento: 77%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=346 [M+Na]+.
Preparación del Compuesto 73-d
Se añadió 4-nitropirazol (250 mg, 2,21 mmoles) a una solución del compuesto 73-e (790 mg, 2,45 mmoles) y carbonato de potasio anhidro (765 mg, 5,54 mmoles) en acetonitrilo (10 mL), la mezcla se sometió a reflujo durante 18 horas. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida, el residuo se trató con agua (20 mL), se extrajo con acetato de etilo (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un sólido de color amarillo 73-d (485 mg, rendimiento: 94%). LC-MS (ESI): m/z=234 [M+H]+.
Preparación del Compuesto 73-c
Bajo hidrógeno (1 atm), a una solución del compuesto 73-d (210 mg, 0,91 mmoles) en etanol (5 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 16 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color pardo rojizo 73-c (171 mg, rendimiento: 93 %), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=204 [M+H]+.
Preparación del Compuesto 73-b
A temperatura ambiente, se añadió ciclobutanona (2,5 g, 35,7 mmoles) a una solución de cianometilen trifenilfosforano (16,2 g, 53,57 mmoles) en diclorometano (30 mL), la mezcla se agitó durante 3 horas. La mezcla se concentró a presión reducida para eliminar el disolvente, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=10:1) para proporcionar un aceite de color amarillo 73-b (3 g, rendimiento: 90,3%).
Preparación del Compuesto 73-a
El compuesto 73-b (1,5 g, 15,79 mmoles) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (4,6 g, 23,68 mmoles) se disolvieron en acetonitrilo anhidro (50 mL), se añadió 1,8-diazabiciclo[5,4,0]undec-7-eno (6,1 g, 40 mmoles). La mezcla se agitó a 80°C durante 18 horas, a continuación, se concentró a presión reducida. El residuo se trató con agua (50 mL), se extrajo con acetato de etilo (50 mLx2). La capa acuosa se ajustó a pH=3 con una solución acuosa de HCl (3 N), a continuación, se extrajo con acetato de etilo (50 mLx2). Las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un aceite incoloro 73-a (2,6 g, rendimiento: 58%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=288 [M+H]+.
Preparación del Compuesto 73
Bajo nitrógeno, el compuesto 73-a (2,6 g, 9,06 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (1,85 g, 9,06 mmoles) y carbonato de sodio (2,9 g, 27,18 mmoles) se suspendieron en 1,4-dioxano (15 mL) y agua (15 mL), se añadió Pd(dppf)Cl2 (733 mg, 0,9 mmoles). La mezcla se calentó a 80°C y se agitó durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida, el residuo se trató con agua (20 mL), se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (diclorometano : metanol=100:1) para proporcionar un sólido de color amarillo claro 73 (1,6 g, rendimiento: 53,6%). LC-MS (ESI): m/z=330 [M+H]+.
Preparación del Compuesto T-73
El compuesto 73-c (46 mg, 0,227 mmoles) y el compuesto 73 (50 mg, 0,152 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (43 mg, 0,227 mmoles). La mezcla se calentó a 110°C y se agitó durante 48 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (50 mL), se extrajo con diclorometano (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida, el residuo se purificó mediante TLC preparativa (solución cromatográfica: acetato de etilo) para proporcionar un sólido de color amarillo T-73 (27 mg, rendimiento: 36%). LC-MS (ESI): m/z=497 [M+H]+.
RMN H1 (400MHz, CDCI3) 5: 8,39 (s, 1H), 8,34 (s, 1H), 8,09 (s, 1H), 7,88 (d, J=5,2Hz, 1H), 7,56 (s, 1H), 7,32 (d, J=5,2Hz, 1H), 7,14 (s, 1H), 4,32 (t, J=6,4Hz, 2H), 3,16 (s, 2H), 3,05 (t, J=6,4Hz, 2H), 2,91 (s, 3H), 2,86 (m, 2H), 2,58 (m, 2H), 2,47 (m, 2H), 2,12 (m, 2H) ppm
Ejemplo 74
2-(3-(4-(2-((1-Met¡l-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(met¡lsulfon¡l)azet¡d¡n-3-il)aceton¡trilo T-74
Ruta s¡ntét¡ca:

Preparac¡ón del Compuesto 74
A temperatura ambiente, se añadió ácido trifluoroacético (3 mL) a una solución del compuesto 5-a (250 mg, 0,58 mmoles) en diclorometano (3 mL) y se agitó durante 3 horas, a continuación, se concentró a presión reducida y el residuo se trató con diclorometano (10 mL) y trietilamina (2 mL). La mezcla se enfrió a continuación, a 0°C, se añadió gota a gota cloruro de metilsulfonilo (100 mg, 0,87 mmoles), y la mezcla resultante se agitó durante 30 minutos adicionales, a continuación, se trató con agua (5 mL), se extrajo con diclorometano (5 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 74 (135 mg, rendimiento: 57%). LC-MS (ESI): m/z=409 [M+H]+.
Preparac¡ón del Compuesto T-74
El compuesto 74 (100 mg, 0,25 mmoles) y 1 -metil-4-aminopirazol (71 mg, 0,74 mmoles) se disolvieron en n-butanol (2 mL), se añadió monohidrato de ácido p-toluenosulfónico (117 mg, 0,62 mmoles). La mezcla se calentó a 110°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente, el residuo se trató con una solución acuosa saturada de bicarbonato de sodio (50 mL), se extrajo con diclorometano (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (solución cromatográfica: acetato de etilo) para proporcionar un sólido de color amarillo T-74 (25 mg, rendimiento: 22%). LC-MS (ESI): m/z=470 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,45 (s, 1H), 8,36 (s, 1H), 7,96 (s, 1H), 7,88 (d, J=5,2Hz, 1H), 7,55 (s, 1H), 7,32 (d, J=5,2Hz, 1H), 7,12 (s, 1H), 4,60 (d, J=9,2Hz, 2H), 4,26 (d, J=9,6Hz, 2H), 3,93 (s, 3H), 3,42 (s, 2H), 3,01 (s, 3H) ppm Ejemplo 75
2-(1-(4-(2-((1-Met¡l-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)c¡cloprop¡l)aceton¡tr¡lo T-75 Ruta s¡ntét¡ca:

Preparación del Compuesto 75-e
Se añadió ácido benzoico (1,59 g, 1,3 mmoles) a una solución de (l-etoxi-ciclopropoxi)trimetilsilano (17,4 g, 100 mmoles) y (trifenil-fosforaniliden)acetato de etilo (45 g, 130 mmoles) en tolueno (250 mL). La mezcla se agitó a 90°C durante 18 horas, a continuación, se enfrió a temperatura ambiente, la mezcla se filtró a través de gel de sílice (300 g), se lavó con éter de petróleo (1000 mL), el producto filtrado se concentró hasta 100 mL a presión reducida. La solución se utilizó directamente para la siguiente etapa.
Preparación del Compuesto 75-d
La solución de 75-e (100 mL) que se obtuvo a partir de la etapa previa se añadió a una solución de 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (1,9 g, 10 mmoles) y 1,8-diazabiciclo[5,4,0]undec-7-eno (1 g, 6,6 mmoles) en acetonitrilo anhidro (200 mL). La mezcla se agitó a 80°C durante 3 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida, el residuo se trató con agua (50 mL), se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto 75-d (1,6 g, rendimiento: 50%). LC-MS (ESI): m/z=321 [M+H]+.
Preparación del Compuesto 75-c
Bajo nitrógeno, se añadió el compuesto 75-d (1,6 g, 5 mmoles) a una suspensión de 2,4-diclorotieno[3,2-d]pirimidina (1,02 g, 5 mmoles), carbonato de potasio (1,38 g, 10 mmoles) y Pd(PPh3)4 (0,56 g, 0,5 mmoles) en 1,4-dioxano (15 mL). La mezcla se calentó a 80°C y se agitó durante 4 horas, a continuación, se concentró a presión reducida, el residuo se trató con agua (20 mL), se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (60 mLx3) y salmuera saturada (60 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=2:1 a 1:1) para proporcionar un sólido de color amarillo claro 75-c (0,91 g, rendimiento: 50%). LC-MS (ESI): m/z=363 [M+H]+.
Preparación del Compuesto 75-b
Se añadió LiOH (90 mg. 3,75 mmoles) a una solución del compuesto 75-c (910 mg, 2,5 mmoles) en un disolvente componente de MeOH (1 mL), THF (4 mL) y agua (1 mL), se agitó durante 1 hora. La mezcla se trató con agua (20 mL) y una solución acuosa de HCl (2 N, 2 mL), se extrajo con acetato de etilo (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 75-b (0,79 g, rendimiento: 94,6%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=335 [M+H]+.
Preparación del Compuesto 75-a
Se añadió cloruro de tionilo (2 mL) a una solución del compuesto 75-b (800 mg, 2,3 mmoles) en tolueno (5 mL), la mezcla se calentó a 100°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida. El residuo se trató con hidróxido de amonio (3 mL), se agitó durante 1 hora, a continuación, se añadió una solución acuosa saturada de NaHCO3 (10 mL). La mezcla se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 75-a (0,68 g, rendimiento: 88,8%) de color gris. LC-MS (ESI): m/z=334 [M+H]+.
Preparación del Compuesto 75
A 0°C, se añadió lentamente anhídrido trifluoroacético (430 mg, 2,1 mmoles) a una solución del compuesto 75-a (680 mg, 2,1 mmoles) y piridina (320 mg, 4,2 mmoles) en diclorometano (10 mL). La mezcla se calentó a temperatura ambiente, se agitó durante 1 hora adicional. Se añadió una solución acuosa saturada de NaHCO3 (10 mL), la mezcla se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 75 (510 mg, rendimiento: 77%). LC-MS (ESI): m/z=316 [M+H]+.
Preparación del Compuesto T-75
Se disolvieron 1-metil-4-aminopirazol (37 mg, 0,38 mmoles) y el compuesto 75 (100 mg, 0,32 mmoles) en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (72 mg, 0,38 mmoles). La mezcla se calentó a 108°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (10 mmol/mL bicarbonato de amonio), acetonitrilo; gradiente: 26%-78%) para proporcionar el compuesto T-75 (32 mg, rendimiento: 26,6%). LC-MS (ESI): m/z=377 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,60 (s, 1H), 8,35 (s, 1H), 8,11 (d, J=5,6Hz, 1H), 8,07 (s, 1H), 7,63 (s, 1H), 7,33 (d, J=5,6Hz, 1H), 3,92 (s, 3H), 3,18 (s, 2H), 1,55 (t, J=5,6Hz, 2H), 1,36 (t, J=5,6Hz, 2H) ppm
Ejemplo 76
2-(1-(4-(2-((1-Ciclopropil-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1 H-pirazol-1-il)ciclopropil)acetonitrilo T-76
Ruta sintética:

Preparación del Compuesto 76-b
Bajo nitrógeno, se añadió 4-nitropirazol (1,0 g, 8,45 mmoles) a una solución de ácido ciclopropilborónico (1,45 g, 16,9 mmoles), CuSO4 anhidro (1,55 g, 8,45 mmoles), 4,4'-di-ferc-butil-2,2'-dipiridilo (2,26 g, 8,45 mmoles) y Na2CO3 (1,87 g, 16,9 mmoles) en 1,2-dicloroetano (30 mL). La mezcla se agitó a 70°C durante 16 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=8:1) para proporcionar el compuesto 76-b (740 mg, rendimiento: 55%). LC-MS (ESI): m/z=154 [M+H]+.
Preparación del Compuesto 76-a
Bajo hidrógeno (1 atm), a una solución del compuesto 76-b (100 mg, 0,65 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (30 mg). La mezcla se agitó a 25°C durante 1 hora, y a continuación, se filtró, el producto filtrado se trató con monohidrato de ácido p-toluenosulfónico (124 mg, 0,65 mmoles). La mezcla se concentró a presión reducida para proporcionar un p-tosilato del compuesto 76-a, que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=124 [M+H]+.
Preparación del Compuesto T-76
El p-tosilato del compuesto 76-a (94 mg, 0,3 mmoles) obtenido a partir de la etapa previa se añadió a una solución del compuesto 75 (100 mg, 0,32 mmoles) en n-butanol (3 mL), la mezcla se calentó a 108°C y se agitó durante 3 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de bicarbonato de sodio (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto T-76 (23 mg, rendimiento: 19%). LC-MS (ESI): m/z=403 [M+H]+.
RMN H1 (400MHz, CDaOD) 5: 8,68 (s, 1H), 8,41 (s, 1H), 8,28 (d, J=5,6Hz, 1H), 8,10 (s, 1H), 7,68 (s, 1H), 7,38 (d, J=5,6Hz, 1H), 3,72 (m, 3H), 3,20 (s, 2H), 1,56 (t, J=5,6Hz, 2H), 1,37 (t, J=5,6Hz, 2H), 1,19 (m, 2H), 1,11 (m, 2H) ppm
Ejemplo 77
2-(3-Fluoro-1-(4-(2-((1-metil-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)ciclobutil)acetonitrilo T-77
Ruta sintética:


Preparación del Compuesto 77-d
Se añadió 3-(benciloxi)ciclobutanona (1,0 g, 5,68 mmoles) a una solución de (cianometilen)trifenilfosforano (2,6 g, 8,52 mmoles) en tolueno, la mezcla se sometió a reflujo durante 16 horas. Después de enfriar a temperatura ambiente, la mezcla se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=10:1) para proporcionar un aceite incoloro 77-d (1,1 g, rendimiento: 97%). LC-MS (ESI): m/z=200 [M+H]+.
RMN H1 (400MHz, CDCb) 5: 7,25-7,38 (m, 5H), 5,22 (m, 1H), 4,47 (s, 2H), 4,17 (m, 1H), 3,05 (m, 1H), 2,96 (m, 1H), 2,91 (m, 2H) ppm
Preparación del Compuesto 77-c
El compuesto 77-d (1,0 g, 5 mmoles) y 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1 H-pirazol (1,16 g, 6 mmoles) se disolvieron en acetonitrilo anhidro (10 mL), se añadió 1,8-diazabiciclo[5,4,0]undec-7-eno (5 g, 33 mmoles). La mezcla se agitó a 55°C durante 18 horas. La mezcla se concentró a presión reducida, el residuo se trató con agua (50 mL), se extrajo con acetato de etilo (50 mLx2). La capa acuosa se trató con una solución acuosa de HCl (1 N) para ajustar a pH=5, se extrajo con acetato de etilo (50 mLx2). Las capas orgánicas se combinaron, se lavaron con agua (50 mLx3) y salmuera saturada (50 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 77-c (1,15 g, rendimiento: 58 %) en forma de un aceite de color amarillo claro, que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=394 [M+H]+.
Preparación del Compuesto 77
Bajo nitrógeno, el compuesto 77-c (1,1 g, 2,8 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (0,571 g, 8,51 mmoles) y carbonato de sodio (0,902 g, 8,51 mmoles) se suspendieron en un disolvente componente de 1,4-dioxano (10 mL) y agua (10 mL), se añadió Pd(dppf)Cl2 (228 mg, 0,28 mmoles). La mezcla se agitó a 80°C durante 16 horas, a continuación, se concentró a presión reducida. El residuo se trató con agua (20 mL), se extrajo con diclorometano (20 mLx3). Las capas orgánicas se combinaron, se lavaron con agua (20 mLx3) y salmuera saturada (20 mL) sucesivamente, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (diclorometano : metanol=100:1) para proporcionar un sólido de color amarillo claro 77 (1,05 g, rendimiento: 94%). LC-MS (ESI): m/z=436 [M+H]+.
Preparación del Compuesto 77-b
Se disolvieron 1 -metil-4-aminopirazol (0,667 g, 6,87 mmoles) y el compuesto 77 (1,0 g, 2,29 mmoles) en n-butanol (20 mL), se añadió monohidrato de ácido p-toluenosulfónico (1,31 g, 6,87 mmoles). La mezcla se calentó a 100°C y se agitó durante 18 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCÜ3 (100 mL), se extrajo con diclorometano (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (solución cromatográfica: diclorometano : metanol=100:1) para proporcionar un sólido de color amarillo claro 77-b (0,515 g, rendimiento: 45%). LC-MS (ESI): m/z=497 [M+H]+.
Preparación del Compuesto 77-a
A -78°C, se añadió lentamente gota a gota una solución de tribromuro de boro en diclorometano (4 N, 2 mL) a una solución del compuesto 77-b (510 mg, 1,03 mmoles) en diclorometano (20 mL), a continuación, se calentó a temperatura ambiente, y se agitó durante 30 minutos. La mezcla se añadió lentamente a una solución acuosa saturada de NaHCÜ3 (50 mL), se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (diclorometano : metanol=100:1) para proporcionar un sólido de color amarillo claro 77-a (98 mg, rendimiento: 24%). LC-MS (ESI): m/z=407 [M+H]+.
Preparación del Compuesto T-77
A 0°C, se añadió lentamente DAST (360 mg, 1,62 mmoles) a una solución del compuesto 77-a (98 mg, 0,24 mmoles) en diclorometano (10 mL), la mezcla se calentó a temperatura ambiente y se agitó durante 30 minutos adicionales. La mezcla se diluyó con agua (10 mL), se ajustó a pH = 10 con una solución acuosa saturada de NaHCO3, a continuación, se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (ácido trifluoroacético al 0,05%), acetonitrilo; gradiente: 15%-50%) para proporcionar el compuesto T-77 (7 mg, rendimiento: 12%). LC-MS (ESI): m/z=409 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,74 (d, J=0,4Hz, 1H), 8,44 (d, J=6,0Hz, 1H), 8,31 (d, J=5,6Hz, 1H), 8,04 (s, 1H), 7,68 (s, 1H), 7,38 (d, J=5,2Hz, 1H), 5,13-5,34 (m, 1H), 3,91 (s, 3H), 3,44 (s, 2H), 3,37 (m, 2H), 2,88 (m, 2H) ppm
Ejemplo 78
2-(3-(4-(2-((1-(2-Hidroxi-2-metilpropil)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(metilsulfonil)azetidin-3-il)acetonitrilo T-78
Ruta sintética:

Se añadió 4-nitropirazol (1,13 g, 10 mmoles) a una solución de 2,2-dimetiloxirano (20 mL) y carbonato de cesio (2 g, 6 mmoles) en acetonitrilo (65 mL), la mezcla se calentó a 80°C y se agitó durante 12 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (100 mL), se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto 78-b (0,99 g, rendimiento: 54%). LC-MS (ESI): m/z=186 [M+H]+.
Preparación del Compuesto 78-a
Bajo hidrógeno (1 atm), a una solución del compuesto 78-b (99 mg, 5,4 mmoles) en metanol (10 mL) se le añadió Pd-C al 10% (0,3 g). La mezcla se agitó a 25°C durante 12 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 78-a (0,8 g, rendimiento: 95%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=156 [M+H]+.
Preparación del Compuesto T-78
El compuesto 78-a (116 mg, 0,75 mmoles) y el compuesto 74 (210 mg, 0,5 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (140 mg, 0,75 mmoles). La mezcla se calentó a 100°C y se agitó durante 6 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida para eliminar el disolvente, el residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto T-78 (61 mg, rendimiento: 23%). LC-MS (ESI): m/z=528 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,73 (s, 1H), 8,43 (s, 1H), 8,20 (s, 1H), 8,11 (d, J=5,6Hz, 2H), 7,64 (s, 1H), 7,30 (d, J=5,6Hz, 1H), 4,67 (d, J=9,6Hz, 2H), 4,34 (d, J=9,6Hz, 2H), 4,12 (s, 2H), 3,65 (s, 2H), 3,09 (s, 3H), 1,23 (s, 6H) ppm
Ejemplo 79
2-(3-(4-(2-((1-(1-Hidroxi-2-metilpropan-2-il)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-
(met¡lsulfon¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-79
Ruta sintética:
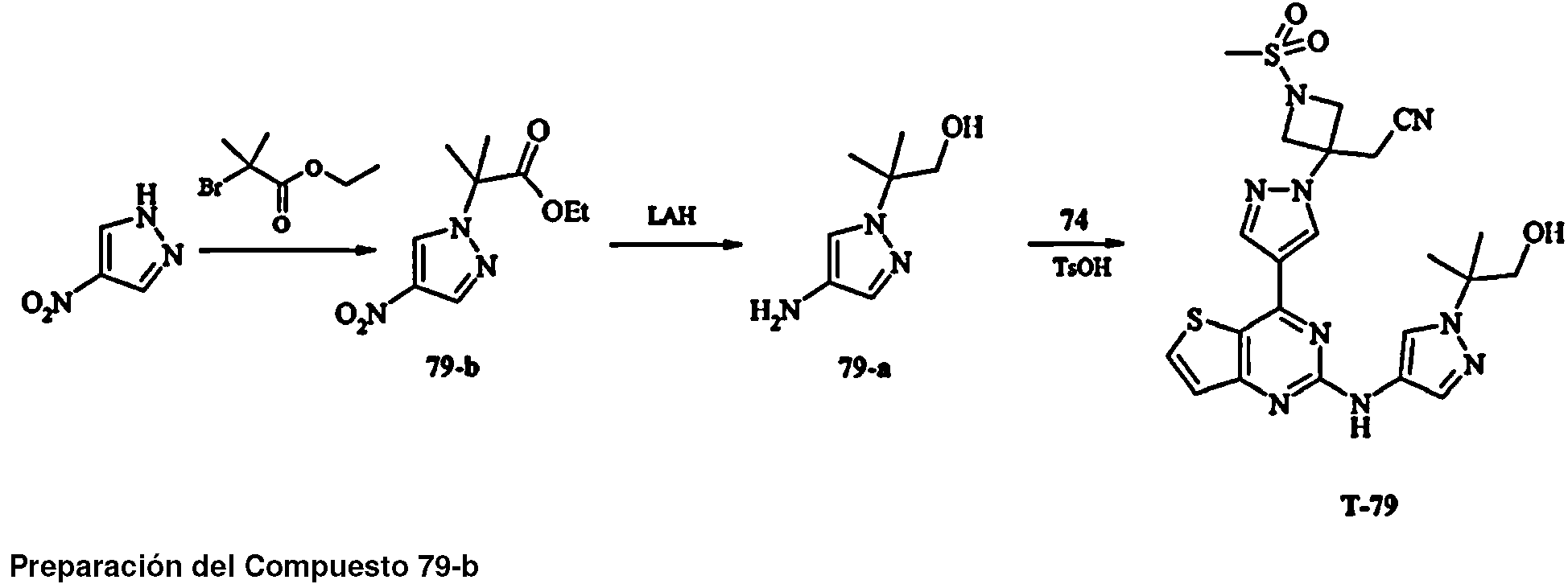
Se añadió 4-nitropirazol (1,13 g, 10 mmoles) a una solución de etilo 2-bromoisobutirato (3,88 g, 20 mmoles) y carbonato de cesio (6,5 g, 20 mmoles) en DMF (50 mL), la mezcla se calentó a 80°C y se agitó durante 4 horas. Después de enfriar a temperatura ambiente, la mezcla se trató con agua (100 mL), se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1) para proporcionar el compuesto 79-b (1,7 g, rendimiento: 75%). LC-MS (ESI): m/z=228 [M+H]+.
Preparación del Compuesto 79-a
A temperatura ambiente, una solución del compuesto 79-b (1,15 g, 5 mmoles) en THF anhidro (30 mL) se añadió lentamente gota a gota a una solución de LiAlH4 en THF (1 N, 15 mL, 15 mmoles). La mezcla se agitó durante 2 horas, a continuación, se trató en porciones con NaSÜ4^10H2O (3 g). La mezcla se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 79-a (0,6 g, rendimiento: 77%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESl): m/z=156 [M+H]+.
Preparac¡ón del Compuesto T-79
El compuesto 79-a (116 mg, 0,75 mmoles) y el compuesto 74 (210 mg, 0,5 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (140 mg, 0,75 mmoles). La mezcla se calentó a 110°C y se agitó durante 6 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente, el residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto T-79 (76 mg, rendimiento: 28%). LC-MS (ESl): m/z=528 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,75 (s, 1H), 8,45 (s, 1H), 8,21 (s, 1H), 8,13 (d, J=5,6Hz, 2H), 7,71 (s, 1H), 7,32 (d, J=5,6Hz, 1H), 4,67 (d, J=9,6Hz, 2H), 4,31 (d, J=9,6Hz, 2H), 3,79 (s, 2H), 3,65 (s, 2H), 3,09 (s, 3H), 1,62 (s, 6H) ppm
Ejemplo 80
(R)-2-(3-(4-(2-((1-(2-H¡drox¡prop¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)oxetan-3-¡l)aceton¡tr¡lo T-80
Ruta s¡ntét¡ca:

Se añadió 4-nitropirazol (2,5 g, 22,12 mmoles) a una mezcla de (R)-epoxipropano (30 mL) y carbonato de cesio (2 g, 6 mmoles), la mezcla se agitó a temperatura ambiente durante 18 horas. Se añadió agua (50 mL), la mezcla se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo claro 80-b (2,5 g, rendimiento: 66%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=172 [M+H]+.
Preparación del Compuesto 80-a
Bajo hidrógeno (1 atm), a una solución del compuesto 80-b (1,0 g, 5,85 mmoles) en etanol (25 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 18 horas, y a continuación, se filtró, el producto filtrado se concentró para proporcionar un aceite de color pardo rojizo 80-a (0,78 g, rendimiento: 95%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto T-80
El compuesto 80-a (179 mg, 1,27 mmoles) y el compuesto 20 (140 mg, 0,43 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (242 mg, 1,27 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente, el residuo se trató con una solución acuosa saturada de NaHCÜ3 (100 mL), se extrajo con diclorometano (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (ácido trifluoroacético al 0,05%), acetonitrilo; gradiente: 20%-55%) para proporcionar un sólido de color amarillo claro T-80 (25 mg, rendimiento: 14%). LC-MS (ESI): m/z=437 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,46 (s, 1H), 8,36 (s, 1H), 8,06 (s, 1H), 7,88 (d, J=5,6Hz, 1H), 7,56 (s, 1H), 7,32 (d, J=5,6Hz, 1H), 7,04 (s, 1H), 5,19 (d, J=7,6Hz, 2H), 4,87 (d, J=7,6Hz, 2H), 4,24 (br, 1H), 4,17-4,21 (m, 1H), 3,97-4,03 (m, 1H), 3,56 (s, 1H), 3,45 (s, 2H), 1,25 (d, J=6,4Hz, 3H) ppm
Ejemplo 81
(S)-2-(3-(4-(2-((1-(2-Hidroxipropil)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)oxetan-3-il)acetonitrilo T-81
Ruta sintética:

Preparación del Compuesto 81
Bajo nitrógeno, se añadió Pd(PPh3)4 (0,56 g, 0,5 mmoles) a una suspensión de 1-(tetrahidro-2H-piran-2-il)-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (2,8 g, 10 mmoles), 2,4-diclorotieno[3,2-d]pirimidina (2,05 g, 10 mmoles) y carbonato de potasio (2,76 g, 20 mmoles) en 1,4-dioxano (30 mL). La mezcla se agitó a 80°C durante 4 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida, el residuo se trató con agua (100 mL), se extrajo con acetato de etilo (100 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=3:1 a 2:1) para proporcionar un sólido de color amarillo claro 81 (2,8 g, rendimiento: 87,5%). LC-MS (ESI): m/z=321 [M+H]+.
Preparación del Compuesto 81-c
Se añadió 4-nitropirazol (1,0 g, 8,85 mmoles) a una mezcla de (S)-epoxipropano (3 mL) y carbonato de cesio (1,73 g, 5,31 mmoles), la mezcla se agitó a temperatura ambiente durante 18 horas. Se añadió agua (50 mL), la mezcla se extrajo con acetato de etilo (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color amarillo claro 81-c(1 g, rendimiento: 66%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=172 [M+H]+.
Preparación del Compuesto 81-b
Bajo hidrógeno (1 atm), a una solución del compuesto 81-c (1,0 g, 5,85 mmoles) en etanol (25 mL) se le añadió Pd-C al 10% (0,2 g). La mezcla se agitó a 25°C durante 18 horas, y a continuación, se filtró, el producto filtrado se concentró a presión reducida para proporcionar un aceite de color pardo rojizo 81-b (0,7 g, rendimiento: 85%), que se utilizó directamente para la siguiente etapa sin purificación.
Preparación del Compuesto 81-a
El compuesto 81-b (397 mg, 2,82 mmoles) y el compuesto 81 (300 mg, 0,94 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (555 mg, 2,82 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se enfrió a temperatura ambiente. La mezcla se concentró a presión reducida, el residuo se trató con una solución acuosa saturada de NaHCÜ3 (50 mL), se extrajo con diclorometano (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (diclorometano : metanol=100:1) para proporcionar un sólido de color amarillo 81-a (67 mg, rendimiento: 21%). LC-MS (ESI): m/z=341 [M+H]+.
Preparación del Compuesto T-81
El compuesto 81-a (67 mg, 0,29 mmoles) y el compuesto 20-b (31 mg, 0,32 mmoles) se disolvieron en acetonitrilo anhidro (1 mL), se añadió DBU (0,1 g, 0,6 mmoles), la mezcla se agitó a temperatura ambiente durante 1 hora. Después de concentrar la mezcla a presión reducida, el residuo se trató con agua (50 mL), se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (solución cromatográfica: acetato de etilo) para proporcionar un sólido de color amarillo claro T-81 (21 mg, rendimiento: 26%). LC-MS (ESI): m/z=437 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,44 (s, 1H), 8,36 (s, 1H), 8,06 (s, 1H), 7,87 (d, J=5,6Hz, 1H), 7,54 (d, J=0,4Hz, 1H), 7,31 (d, J=5,6Hz, 1H), 7,15 (s, 1H), 5,19 (d, J=7,6Hz, 2H), 4,87 (d, J=7,6Hz, 2H), 4,16-4,21 (m, 2H), 3,97-4,02 (m, 2H), 3,55 (br, 1H), 3,45 (s, 3H), 1,26 (d, J=6,4Hz, 3H) ppm
Ejemplo 82
(R)-2-(3-(4-(2-((1-(1-Hidroxipropan-2-il)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)oxetan-3-il)acetonitrilo T-82
Ruta sintética:

A 25°C, se añadió lentamente DIAD (10,8 g, 53,1 mmoles) a una solución de 4-nitropirazol (5 g, 44,2 mmoles), L-lactato de metilo (5,1 g, 48,6 mmoles) y PPh3 (13,9 g, 53,1 mmoles) en THF anhidro (100 mL). Después de agitar durante 3 horas, la mezcla se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=10:1) para proporcionar el compuesto 82-c (6,6 g, rendimiento: 74%). LC-MS (ESI): m/z=200 [M+H]+.
Preparación del Compuesto 82-b
A temperatura ambiente, compuesto 82-c (2,0 g, 10 mmoles) se añadió lentamente gota a gota a una solución de LÍAIH4 in THF (1 N, 26,3 mL, 26,3 mmoles). La mezcla se agitó durante 2 horas, a continuación, se trató en porciones con Na2SO4^10H2O (3 g). La mezcla se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 82-b (1,1 g, rendimiento: 78%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=142 [M+H]+.
Preparación del Compuesto 82-a
El compuesto 82-b (140 mg, 1 mmol) y el compuesto 81 (320 mg, 1 mmol) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (190 mg, 1 mmol). La mezcla se calentó a 110°C y se agitó durante 6 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar un sólido de color amarillo 82-a (38 mg, rendimiento: 10,6%). LC-MS (ESI): m/z=342 [M+H]+.
Preparación del Compuesto T-82
El compuesto 82-a (38 mg, 0,106 mmoles) y el compuesto 20-b (11 mg, 0,106 mmoles) se disolvieron en acetonitrilo anhidro (3 mL), se añadió DBU (100 mg, 0,6 mmoles). La mezcla se agitó a temperatura ambiente durante 1 hora, a continuación, se concentró a presión reducida. El residuo se trató con agua (50 mL), se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (10mmol/mL bicarbonato de amonio), acetonitrilo; gradiente: 15%-45%) para proporcionar un sólido de color amarillo claro T-82 (13 mg, rendimiento: 28%). LC-MS (ESI): m/z=437 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,72 (s, 1H), 8,43 (s, 1H), 8,16 (s, 1H), 8,13 (d, J=5,6Hz, 1H), 7,69 (s, 1H), 7,32 (d, J=5,6Hz, 1H), 5,24 (d, J=8,4Hz, 2H), 4,95 (d, J=8,4Hz, 2H), 4,43 (m, 1H), 3,86 (m, 2H), 3,64 (s, 2H), 1,53 (d, J=6,8Hz, 3H) ppm
Ejemplo 83
(S)-2-(3-(4-(2-((1-(1-Hidroxipropan-2-il)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)oxetan-3-il)acetonitrilo T-83
Ruta sintética:

A 25°C, se añadió lentamente DIAD (10,8 g, 53,1 mmoles) a una solución de 4-nitropirazol (5 g, 44,2 mmoles), D
lactato de metilo (5,1 g, 48,6 mmoles) y PPh3 (13,9 g, 53,1 mmoles) en THF anhidro (100 mL). Después de agitar durante 3 horas, la mezcla se concentró a presión reducida, el residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=10:1) para proporcionar el compuesto 83-c (6,2 g, rendimiento: 70%). LC-MS (ESI): m/z=200 [M+H]+.
Preparación del Compuesto 83-b
A temperatura ambiente, el compuesto 83-c (2,0 g, 10 mmoles) se añadió lentamente a una solución de LiAlH4 in THF (1 N, 26,3 mL, 26,3 mmoles). La mezcla se agitó durante 2 horas, a continuación, se trató en porciones con Na2SO4^10H2O (3 g). La mezcla se filtró, el producto filtrado se concentró a presión reducida para proporcionar el compuesto 83-b (1,0 g, rendimiento: 71%), que se utilizó directamente para la siguiente etapa sin purificación. LC-MS (ESI): m/z=142 [M+H]+.
Preparación del Compuesto 83-a
El compuesto 83-b (140 mg, 1 mmol) y el compuesto 81 (320 mg, 1 mmol) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (190 mg, 1 mmoles). La mezcla se calentó a 110°C y se agitó durante 6 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar un sólido de color amarillo 83-a (76 mg, rendimiento: 22,3%). LC-MS (ESI): m/z=342 [M+H]+.
Preparación del Compuesto T-83
El compuesto 83-a (76 mg, 0,223 mmoles) y el compuesto 20-b (21 mg, 0,223 mmoles) se disolvieron en acetonitrilo anhidro (5 mL), se añadió DBU (100 mg, 0,6 mmoles). La mezcla se agitó a temperatura ambiente durante 1 hora, a continuación, se concentró a presión reducida. El residuo se trató con agua (50 mL), se extrajo con diclorometano (50 mLx2). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante TLC preparativa (solución cromatográfica: acetato de etilo) para proporcionar un sólido de color amarillo T-83 (56 mg, rendimiento: 57,6%). LC-MS (ESI): m/z=437 [M+H]+.
RMN H1 (400MHz, CDaOD) 5: 8,72 (s, 1H), 8,43 (s, 1H), 8,16 (s, 1H), 8,13 (d, J=5,6Hz, 1H), 7,69 (s, 1H), 7,33 (d, J=5,6Hz, 1H), 5,24 (d, J=7,2Hz, 2H), 4,95 (d, J=7,2Hz, 2H), 4,44 (m, 1H), 3,33 (m, 2H), 1,55 (d, J=6,8Hz, 3H) ppm
Ejemplo 84
(R)-2-(3-(4-(2-((1-(1-Hidroxipropan-2-il)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazol-1-il)-1-(metilsulfonil)azetidin-3-il)acetonitrilo T-84
Ruta sintética:

Preparación del Compuesto T-84
El compuesto 74 (210 mg, 0,5 mmoles) y el compuesto 82-b (70 mg, 0,5 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (100 mg, 0,5 mmoles). La mezcla se calentó a 110°C y se agitó durante 6 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron,
el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (éter de petróleo : acetato de etilo=1:1) para proporcionar el compuesto T-84 (100 mg, rendimiento: 38%). LC-MS (ESI): m/z=514 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,83 (s, 1H), 8,50 (s, 1H), 8,27 (d, J=5,6Hz, 2H), 8,15 (s, 1H), 7,72 (s, 1H), 7,37 (d, J=5,6Hz, 2H), 4,68 (d, J=9,6Hz, 2H), 4,46 (m, 1H), 4,37 (d, J=9,6Hz, 2H), 3,87 (m, 1H), 3,66 (s, 2H), 3,09 (s, 3H), 1,54 (d, J=6,8Hz, 3H) ppm
Ejemplo 85
(S)-2-(3-(4-(2-((1-(1-H¡drox¡propan-2-¡l)-1H-p¡razol-4-¡l)am¡no)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(metilsulfonil)azetidin-3-il)acetonitrilo T-85
Ruta sintética:

Preparación del Compuesto T-85
El compuesto 74 (210 mg, 0,5 mmoles) y el compuesto 83-b (70 mg, 0,5 mmoles) se disolvieron en n-butanol (5 mL), se añadió monohidrato de ácido p-toluenosulfónico (100 mg, 0,5 mmoles). La mezcla se calentó a 110°C y se agitó durante 6 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCO3 (10 mL), se extrajo con diclorometano (10 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante HPLC preparativa (fase móvil: agua (ácido trifluoroacético al 0,05%), acetonitrilo; gradiente: 15%-65%) para proporcionar el compuesto T-85 (80 mg, rendimiento: 32%). LC-MS (ESI): m/z=514 [M+H]+.
RMN H1 (400MHz, CD3OD) 5: 8,82 (s, 1H), 8,49 (s, 1H), 8,26 (d, J=5,6Hz, 2H), 8,15 (s, 1H), 7,71 (s, 1H), 7,37 (d, J=5,6Hz, 2H), 4,68 (d, J=9,6Hz, 2H), 4,47 (m, 1H), 4,36 (d, J=9,6Hz, 2H), 3,88 (m, 1H), 3,66 (s, 2H), 3,09 (s, 3H), 1,54 (d, J=6,8Hz, 3H) ppm
Ejemplo 86
(R)-2-(3-(4-(2-((1-(2-H¡drox¡prop¡l)-1H-p¡razol-4-¡l)amino)t¡eno[3,2-d]p¡r¡m¡d¡n-4-¡l)-1H-p¡razol-1-¡l)-1-(met¡lsulfon¡l)azet¡d¡n-3-¡l)aceton¡tr¡lo T-86
Ruta s¡ntét¡ca:

Preparación del Compuesto T-86
El compuesto 80-a (311 mg, 2,21 mmoles) y el compuesto 74 (300 mg, 0,74 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (349 mg, 1,84 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCO3 (50 mL), se extrajo con diclorometano (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (acetato de etilo) para proporcionar un sólido de color amarillo T-86 (115 mg, rendimiento: 31%). LC-MS (ESI): m/z=514 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,47 (d, J=0,4Hz, 1H), 8,36 (s, 1H), 8,04 (s, 1H), 7,88 (d, J=5,6Hz, 1H), 7,57 (d, J=0,4Hz, 1H), 7,31 (d, J=5,6Hz, 1H), 6,96 (s, 1H), 4,61 (d, J=7,6Hz, 2H), 4,26 (d, J=7,6Hz, 2H), 4,03-4,18 (m, 3H), 3,43 (s, 2H), 3,02 (s, 3H), 1,26 (d, J=6,4Hz, 3H) ppm
Ejemplo 87
(S)-2-(3-(4-(2-((1-(2-Hidroxipropil)-1H-pirazol-4-il)amino)tieno[3,2-d]pirimidin-4-il)-1H-pirazoL-1-il)-1-(metilsulfonil)azetidin-3-il)acetonitrilo T-87
Ruta sintética:

Preparación del Compuesto T-87
El compuesto 81-b (311 mg, 2,21 mmoles) y el compuesto 74 (300 mg, 0,74 mmoles) se disolvieron en n-butanol (10 mL), se añadió monohidrato de ácido p-toluenosulfónico (349 mg, 1,84 mmoles). La mezcla se calentó a 110°C y se agitó durante 16 horas, a continuación, se enfrió a temperatura ambiente, se concentró a presión reducida para eliminar el disolvente. El residuo se trató con una solución acuosa saturada de NaHCO3 (50 mL), se extrajo con diclorometano (50 mLx3). Las capas orgánicas se combinaron, se secaron sobre sulfato de sodio anhidro, a continuación, se filtraron, el producto filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna de sílice (acetato de etilo) para proporcionar un aceite de color amarillo T-87 (65 mg, rendimiento: 17%). LC-MS (ESI): m/z=514 [M+H]+.
RMN H1 (400MHz, CDCla) 5: 8,47 (d, J=0,4Hz, 1H), 8,36 (s, 1H), 8,04 (s, 1H), 7,88 (d, J=5,6Hz, 1H), 7,57 (d, J=0,4Hz,
1H), 7,31 (d, J=5,6Hz, 1H), 6,96 (s, 1H), 4,61 (d, J=7,6Hz, 2H), 4,26 (d, J=7,6Hz, 2H), 4,03-4,18 (m, 3H), 3,43 (s, 2H), 3,02 (s, 3H), 1,26 (d, J=6,4Hz, 3H) ppm
Experimento sobre el efecto
Efecto biológico: ensayo de CI50 de la actividad inhibidora enzimática de JAK 1,2,3 intracitoplásmicas
Etapas experimentales:
1. Preparación de tampón
Solución tampón de JAK1: HEPES 25 mM, pH 7,5, Brij-35 al 0,01%, Triton 0,01 M. Solución tampón de JAK2,3: HEPES 50 mM, pH 7,5, Brij-35 al 0,0015%.
2. El compuesto se formuló en DMSO al 100% a un gradiente de concentración, se depositó en una placa de 384 pocillos con una concentración final de DMSO de 2%.
3. Las enzimas JAK2,3 se diluyeron para que tuvieran una concentración óptima con el siguiente tampón: HEPES 50 mM, pH 7,5, Brij-35 al 0,0015%, DTT 2 mM. La enzima JAK1 se diluyó para que tuviera una concentración óptima con el siguiente tampón: HEPES 25 mM, pH 7,5, Brij-35 al 0,01%, DTT 2 mM, Triton 0,01M. A continuación, se transfirieron a una placa de 384 pocillos y se incubaron con el compuesto durante un cierto tiempo.
4. El sustrato de JAK2,3 se diluyó para que tuviera una concentración óptima con el siguiente tampón: HEPES 50 mM, pH 7,5, Brij-35 al 0,0015%, MgCl210 mM, Km adenosina trifosfato. El sustrato de JAK1 se diluyó para que tuviera una concentración óptima con el siguiente tampón: HEPES 25 mM, pH 7,5, Brij-35 al 0,01%, MgCl210 mM, Triton 0,01 M. Se añadió Km adenosina trifosfato a placas de 384 pocillos para iniciar las reacciones, y las reacciones se llevaron a cabo a 28°C durante 1 hora.
5. Realizar la lectura de las tasas de conversión por medio de Caliper Reader, y calcular la tasa de inhibición según el promedio de dos pruebas.
Resultados
La actividad biológica para los compuestos de la presente invención se sometió a prueba de acuerdo con el método descrito anteriormente, los resultados se enumeran más abajo (tabla 1):
Tabla 1: Resultados para el ensayo de actividad inhibidora enzimática de JAK 1,2,3 de los compuestos en la presente invención


En la tabla 1, "n.d." indica: no detectado, "A" indica: CI50 á 50nM, "B" indica: 50 nM < CI50 á 500nM, "C" indica: 500 nM <CI50 á 1000nM, "D" indica: 1000 nM < CI50.
Claims (22)
1. Un compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables,

en donde:
X es O, o S;
L1 es un enlace químico, un alquilo, un alquileno, un cicloalquilo o un heterocicloalquilo; en donde el alquilo, alquileno, cicloalquilo o heterocicloalquilo pueden estar sustituidos independientemente con los sustituyentes seleccionados del grupo que consiste en un halógeno, un ciano, alquil(C1-C6)sulfonilo, cicloalquil(C3-C6)sulfonilo, un acilo, un cicloalquilo y un heterocicloalquilo;
L2 es un alquilo, un acilo, un cicloalquilo, un heterocicloalquilo, un arilo o un heteroarilo;
M es un halógeno, un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un arilo, un heteroarilo, un ciano, un sulfonilo o un acilo; en donde el alquilo, alquileno, alcoxi, heterocicloalquilo, arilo, heteroarilo, sulfonilo o acilo pueden estar sustituidos independientemente con los sustituyentes seleccionados del grupo que consiste en: un halógeno, un hidroxilo, un ciano, un amino, un acilamino, un nitro, un carboxilo, un sulfonilo, un acilo, un alcoxi, un cicloalquilo, un heterocicloalquilo, un arilo y un heteroarilo;
R1 es un hidrógeno, un deuterio, un halógeno, un ciano, un alquilo, un cicloalquilo, un sulfonilo, un “alquil-NH-CO-" o un “alquil-NHSO2-"; en donde el alquilo puede estar sustituido con los sustituyentes seleccionados del grupo que consiste en un halógeno, un hidroxilo, un ciano, un amino, un nitro, un carboxilo, un sulfonilo, un acilo, un alcoxi, un cicloalquilo, un alquenilo y un alquinilo;
R2 se selecciona independientemente entre un hidrógeno, un deuterio, un halógeno y un alquilo;
n es 1,2, 3 o 4;
p es 0, 1,2, 3, 4 o 5.
2. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 1, en donde:
X es O, o S
R1 es un hidrógeno, un alquilo o un halógeno; en donde el alquilo es sustituido con los sustituyentes seleccionados del grupo que consiste en un halógeno, un hidroxilo, un ciano, un amino, un nitro, un carboxilo, un sulfonilo y un acilo; R2 es un hidrógeno, un deuterio, un halógeno o un alquilo;
n es 1 o 2;
p es 0 o 1.
3. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 1, en donde:
cuando R1 es un halógeno, el halógeno es F, Cl, Br o I;
y/o, cuando R2 es un halógeno, el halógeno es F, Cl, Br o I;
y/o, cuando L1 es un alquilo, el alquilo es un alquilo C1-C4 ;
o, cuando L1 es un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando L1 es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de nitrógeno o un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de oxígeno;
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 es sustituido con los sustituyentes seleccionados del grupo que consiste en un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 es sustituido con los sustituyentes seleccionados del grupo que consiste en un acilo, el acilo es un formilo o un alquil(C1-C4)acilo;
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 es sustituido con los sustituyentes seleccionados del grupo que consiste en un halógeno, el halógeno es F, Cl, Br o I;
el alquilo definido en R1 es un alquilo C1-C4 ;
cuando R2 es un alquilo, el alquilo es un alquilo C1-C4.
4. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 1, en donde:
cuando L2 es un alquilo, el alquilo es un alquilo C1-C4 ;
cuando L2 es un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando L2 es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C6 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno;
cuando L2 es un arilo, el arilo es un arilo C6-C10;
cuando L2 es un heteroarilo, el heteroarilo es un heteroarilo C3-C10 que contiene de 1 a 4 átomos de nitrógeno.
5. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 1, en donde:
cuando M es un halógeno, el halógeno es flúor;
cuando M es un alquilo, el alquilo es un alquilo C1-C6 ;
cuando M es un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando M es un alcoxi, el alcoxi es un alcoxi C1-C4 sustituido con un heterocicloalquilo; el heterocicloalquilo no está sustituido adicionalmente; o el heterocicloalquilo es un heterocicloalquilo C2-C10 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno;
cuando M es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C8 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno.
6. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 1, en donde:
cuando M es un arilo, el arilo es un arilo C6-C10;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un halógeno, el halógeno es F, Cl, Br o I;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un alcoxi, el alcoxi es un alcoxi C1-C6 ;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un cicloalquilo, el cicloalquilo es un cicloalquilo C3-C6 ;
cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C8 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno.
7. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1, o una de sus sales
farmacéuticamente aceptables como se define en la reivindicación 3, en donde:
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 es sustituido con los sustituyentes seleccionados del grupo que consiste en un cicloalquilo, el cicloalquilo C3-C6 es un ciclopentilo;
cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 es sustituido con los sustituyentes seleccionados del grupo que consiste en un alquil(C1-C6)sulfonilo, el alquil(C1-C6)sulfonilo es un alquil(C1-C3)sulfonilo; cuando el alquilo, alquileno, cicloalquilo o heterocicloalquilo definidos en L1 es sustituido con los sustituyentes seleccionados del grupo que consiste en un alquil(C1-C4)acilo, el alquil(C1-C4)acilo es un alquil(C2)acilo;
cuando L1 es un cicloalquilo C3-C6, el cicloalquilo C3-C6 es un ciclopropilo.
8. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 4, en donde:
cuando L2 es un alquilo C1-C4 , el alquilo C1-C4 es un metilo;
cuando L2 es un cicloalquilo C3-C6 , el cicloalquilo C3-C6 es un ciclopropilo o un ciclohexilo;
cuando L2 es un heterocicloalquilo, el heterocicloalquilo es un heterocicloalquilo C3-C6 que contiene de 1 a 3 átomos de oxígeno;
cuando L2 es un arilo C6-C10, el arilo C6-C10 es un fenilo;
cuando L2 es un heteroarilo C3-C10, el heteroarilo C3-C10 es un heteroarilo C4-C8 que contiene de 1 a 4 átomos de nitrógeno.
9. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 5, en donde:
cuando M es un alquilo C1-C6 , el alquilo C1-C6 es un metilo, un etilo, un propilo o un isopropilo;
cuando M es un alquilo C1-C6 sustituido con un halógeno, el alquilo C1-C6 sustituido con un halógeno es un > Í / CF3.
trifluorometilo o
cuando M es un alquilo C1-C6 sustituido con un hidroxilo, el alquilo C1-C6 sustituido con un hidroxilo es


cuando M es un alquilo C1-C6 sustituido con un ciano, el alquilo C1-C6 sustituido con un ciano es cuando M es un alquilo C1-C6 sustituido con un metilsulfonilo, el alquilo C1-C6 sustituido con un metilsulfonilo es

cuando M es un alquilo, un alquileno, un cicloalquilo, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un alcoxi C1-C6 , el alcoxi C1-C6 es un alcoxi C1-C3 ;
cuando M es un alcoxi C1-C4 sustituido con un heterocicloalquilo C2-C10, el alcoxi C1-C4 es un alcoxi C2 ; cuando M es un alcoxi C1-C4 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un heterocicloalquilo C3-C8 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno;
cuando M es un heterocicloalquilo C3-C8, el heterocicloalquilo C3-C8 es un heterocicloalquilo C4-C6 que contiene de 1 a 4 átomos de oxígeno y/o nitrógeno.
10. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 6, en donde:
cuando M es un arilo C6-C10, el arilo C6-C10 es un fenilo;
cuando M es un alquilo, un alquileno, un alcoxi, un heterocicloalquilo, un heteroarilo, un sulfonilo o un acilo sustituido con un cicloalquilo C3-C6, el cicloalquilo C3-C6 es un ciclopropilo;
cuando M es un alcoxi C1-C4 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un
heterocicloalquilo C4-C6 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno.
11. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 8, en donde:
cuando L2 es un heterocicloalquilo C3-C6 , el heterocicloalquilo C3-C6 es un tetrahidropiranilo;
cuando L2 es un heteroarilo C4-C8 , el heteroarilo C4-C8 es un pirazolilo, un imidazolilo, un piridilo, un benzimidazolilo, a benzopirazolilo, un piridazinilo o un pirimidinilo.
12. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 9, en donde:
cuando M es un alcoxi C2 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un heterocicloalquilo C3-C8 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno;
cuando M es un heterocicloalquilo C4-C6 , el heterocicloalquilo C4-C6 es un morfolinilo, un tetrahidropiranilo, un azetidinilo, un piperidilo, un oxetanilo, un tetrazolilo, un piperazinilo o un pirrolidinilo;
cuando M es un alquilo C1-C6 sustituido con un alcoxi C1-C3 , el alquilo C1-C6 sustituido con un alcoxi C1-C3 es
13. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 12, en donde:
cuando M es un alcoxi C2 sustituido con un heterocicloalquilo C2-C10, el heterocicloalquilo C2-C10 es un heterocicloalquilo C3-C8 que contiene de 1 a 3 átomos de oxígeno y/o nitrógeno.
14. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 13, en donde:
cuando un alcoxi C2 definido en M está sustituido con un heterocicloalquilo C4-C6 , el heterocicloalquilo C4-C6 es un morfolinilo o un pirrolidinilo.
15. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 13, en donde:
cuando M es un alcoxi C2 sustituido con un heterocicloalquilo C4-C6 , el heterocicloalquilo C4-C6 es un morfolinilo, un tetrahidropiranilo, un azetidinilo, un piperidilo, un oxetanilo, un piperazinilo o un pirrolidinilo.
16. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en la reivindicación 1, en donde el compuesto representado por la fórmula IV-1 -1 tiene la estructura representada por la fórmula V-1 -1,

en donde, cada una de las letras y los sustituyentes tiene el significado proporcionado en una cualquiera de las reivindicaciones 1 a 15.
17. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en una cualquiera de las reivindicaciones 1 -15, en donde el compuesto representado por la fórmula IV-1 -1 se selecciona del grupo que consiste en





18. Un compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-A,

en donde:
L1 es un enlace químico, un alquilo, un alquileno, un cicloalquilo o un heterocicloalquilo; en donde el alquilo, alquileno,
cicloalquilo o heterocicloalquilo pueden estar sustituidos independientemente con los sustituyentes seleccionados del
grupo que consiste en un halógeno, un ciano, alquil(C1-C6)sulfonilo, cicloalquil(C3-C6)sulfonilo, un acilo, un cicloalquilo
y un heterocicloalquilo;
R1 es un hidrógeno, un deuterio, un halógeno, un ciano, un alquilo, un cicloalquilo, un sulfonilo, un “alquil-NH-CO-" o
un “alquil-NHSO2-"; en donde el alquilo puede estar sustituido con los sustituyentes seleccionados del grupo que
consiste en un halógeno, un hidroxilo, un ciano, un amino, un nitro, un carboxilo, un sulfonilo, un acilo, un alcoxi, un
cicloalquilo, un alquenilo y un alquinilo;
R2 se selecciona independientemente entre un hidrógeno, un deuterio, un halógeno y un alquilo;
m es 1; y
n es 1,2, 3 o 4.
19. El compuesto heterocíclico de cinco y seis miembros representado por la fórmula I-A como se define en la reivindicación 18, en donde el compuesto se selecciona del grupo que consiste en:

20. Un compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en una cualquiera de las reivindicaciones 1-17 para su uso en el tratamiento y/o la prevención de enfermedades de proliferación celular.
21. Un compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1-1, o una de sus sales farmacéuticamente aceptables como se define en una cualquiera de las reivindicaciones 1-17 para su uso en el tratamiento del cáncer, infecciones, inflamación y enfermedades autoinmunitarias.
22. Una composición farmacéutica, que comprende una cantidad terapéuticamente eficaz del compuesto heterocíclico de cinco y seis miembros representado por la fórmula IV-1 -1, o una de sus sales farmacéuticamente aceptables como se define en una cualquiera de las reivindicaciones 1-17, y al menos uno seleccionado del grupo que consiste en portadores y diluyentes farmacéuticamente aceptables.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310019856 | 2013-01-18 | ||
| PCT/CN2014/070778 WO2014111037A1 (zh) | 2013-01-18 | 2014-01-17 | 五元并六元杂环化合物、其制备方法、药物组合物和应用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2827233T3 true ES2827233T3 (es) | 2021-05-20 |
Family
ID=51184664
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14740886T Active ES2827233T3 (es) | 2013-01-18 | 2014-01-17 | Compuesto heterocíclico de cinco y seis miembros, y método de preparación, composición farmacéutica y uso del mismo |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9745320B2 (es) |
| EP (1) | EP2947084B8 (es) |
| JP (1) | JP6321039B2 (es) |
| CN (1) | CN103936757B (es) |
| DK (1) | DK2947084T3 (es) |
| ES (1) | ES2827233T3 (es) |
| HK (1) | HK1215567A1 (es) |
| WO (1) | WO2014111037A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014111037A1 (zh) | 2013-01-18 | 2014-07-24 | 上海昀怡健康管理咨询有限公司 | 五元并六元杂环化合物、其制备方法、药物组合物和应用 |
| CN104177363B (zh) * | 2013-05-24 | 2018-06-05 | 江苏先声药业有限公司 | 双环杂环胺类Hedgehog信号通路抑制剂 |
| JP2019536820A (ja) * | 2016-12-01 | 2019-12-19 | アプトセ バイオサイエンシーズ インコーポレイテッド | Brad4およびjak2の二重阻害剤としての縮合環式ピリミジン化合物、ならびにその使用に関する方法 |
| KR102398659B1 (ko) | 2017-03-17 | 2022-05-16 | 주식회사 대웅제약 | 카이네이즈 저해제로서의 피롤로트리아진 유도체 |
| CN110446713B (zh) * | 2018-06-06 | 2022-05-24 | 杭州澳津生物医药技术有限公司 | 一种吡唑嘧啶衍生物及其用途和药物组合物 |
| EP3942045A1 (en) | 2019-03-21 | 2022-01-26 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| CN111943959A (zh) * | 2019-05-17 | 2020-11-17 | 上海再极医药科技有限公司 | 一种jak抑制剂的合成方法 |
| CN110305140B (zh) * | 2019-07-30 | 2020-08-04 | 上海勋和医药科技有限公司 | 二氢吡咯并嘧啶类选择性jak2抑制剂 |
| CN110862380A (zh) * | 2019-10-24 | 2020-03-06 | 嘉兴特科罗生物科技有限公司 | 一种小分子化合物 |
| EP4054579A1 (en) | 2019-11-08 | 2022-09-14 | Institut National de la Santé et de la Recherche Médicale (INSERM) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2022020887A1 (en) * | 2020-07-27 | 2022-02-03 | Esfam Biotech Pty Ltd | Treatment of cd151 related disorders |
| WO2022020890A1 (en) * | 2020-07-27 | 2022-02-03 | Esfam Biotech Pty Ltd | Treatment of veterinary conditions associated with cd151 |
| WO2022129376A1 (en) | 2020-12-18 | 2022-06-23 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | 6-6 or 5-6 fused bicyclic compounds comprising a pyri(mi)dine ring useful in the|treatment of infectious diseases |
| CN114181076A (zh) * | 2021-12-23 | 2022-03-15 | 维思普新材料(苏州)有限公司 | 一种2,6-二卤代苯甲酸甲酯的制备方法 |
| WO2023193761A1 (zh) * | 2022-04-08 | 2023-10-12 | 深圳再极医药科技有限公司 | 一种含jak抑制剂的药物组合物及其制备方法和用途 |
| WO2024041397A1 (en) * | 2022-08-22 | 2024-02-29 | Hangzhou Highlightll Pharmaceutical Co., Ltd | Jak1/jak2/tyk2 inhibitors for topical treatment of dermatological diseases |
| CN117700428A (zh) * | 2022-09-08 | 2024-03-15 | 广州再极医药科技有限公司 | 一种五元并六元杂环化合物的晶型及其制备方法和应用 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4178449A (en) * | 1978-04-17 | 1979-12-11 | American Cyanamid Company | Pyrazolo[1,5-a]pyrimidines and imidazo-[1,5-a]pyrimidines |
| JPS60146891A (ja) * | 1984-01-05 | 1985-08-02 | Mitsubishi Chem Ind Ltd | 〔2,3−d〕チエノピリミジン誘導体およびその塩 |
| GB0100620D0 (en) * | 2001-01-10 | 2001-02-21 | Vernalis Res Ltd | Chemical cokpounds V |
| JP2004137270A (ja) * | 2002-09-26 | 2004-05-13 | Nippon Nohyaku Co Ltd | 新規除草剤、その使用方法、新規置換チエノピリミジン誘導体及びその中間体並びにそれらの製造方法 |
| BRPI0708813A2 (pt) * | 2006-03-17 | 2011-05-24 | Wyeth Corp | composto; composição; método de tratamento, inibição do crescimento ou erradicaçõ de neoplasias em um mamìfero necessitado; método de tratamento dse cáncer em um mamìfero necessitado; e mistura |
| WO2008059368A2 (en) * | 2006-11-17 | 2008-05-22 | Pfizer Products Inc. | Fused 2-amino pyrimidine compounds and their use for the treatment of cancer |
| KR20100015353A (ko) | 2007-04-02 | 2010-02-12 | 팔라우 파르마 에스에이 | Jak3 억제제로서의 피롤로피리미딘 유도체 |
| DE102007049451A1 (de) * | 2007-10-16 | 2009-04-23 | Merck Patent Gmbh | 5-Cyano-thienopyridine |
| US8461163B2 (en) * | 2008-03-31 | 2013-06-11 | Takeda Pharmaceutical Company Limited | Substituted N-(pyrazolo[1,5-a]pyrimidin-5-yl)amides as inhibitors of apoptosis signal-regulating kinase 1 |
| TW201041892A (en) * | 2009-02-09 | 2010-12-01 | Supergen Inc | Pyrrolopyrimidinyl Axl kinase inhibitors |
| TW201132644A (en) * | 2009-12-23 | 2011-10-01 | Biocryst Pharm Inc | Heterocyclic compounds as Janus kinase inhibitors |
| WO2014111037A1 (zh) | 2013-01-18 | 2014-07-24 | 上海昀怡健康管理咨询有限公司 | 五元并六元杂环化合物、其制备方法、药物组合物和应用 |
-
2014
- 2014-01-17 WO PCT/CN2014/070778 patent/WO2014111037A1/zh active Application Filing
- 2014-01-17 JP JP2015552995A patent/JP6321039B2/ja active Active
- 2014-01-17 DK DK14740886.8T patent/DK2947084T3/da active
- 2014-01-17 ES ES14740886T patent/ES2827233T3/es active Active
- 2014-01-17 EP EP14740886.8A patent/EP2947084B8/en active Active
- 2014-01-17 US US14/761,772 patent/US9745320B2/en active Active
- 2014-01-20 CN CN201410022938.3A patent/CN103936757B/zh active Active
-
2016
- 2016-03-24 HK HK16103485.8A patent/HK1215567A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| US9745320B2 (en) | 2017-08-29 |
| JP6321039B2 (ja) | 2018-05-09 |
| US20150336982A1 (en) | 2015-11-26 |
| EP2947084A4 (en) | 2016-10-05 |
| JP2016505025A (ja) | 2016-02-18 |
| HK1215567A1 (zh) | 2016-09-02 |
| EP2947084B8 (en) | 2021-03-10 |
| EP2947084A1 (en) | 2015-11-25 |
| DK2947084T3 (da) | 2020-11-02 |
| CN103936757A (zh) | 2014-07-23 |
| WO2014111037A1 (zh) | 2014-07-24 |
| EP2947084B1 (en) | 2020-09-23 |
| CN103936757B (zh) | 2019-09-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2827233T3 (es) | Compuesto heterocíclico de cinco y seis miembros, y método de preparación, composición farmacéutica y uso del mismo | |
| KR102075886B1 (ko) | 신규한 피라졸로[3,4-d]피리미딘 화합물 또는 그 염 | |
| JP5302186B2 (ja) | Pi3k阻害剤としてのピリミジン誘導体 | |
| ES2377358T3 (es) | Compuestos farmacéuticos | |
| KR101623286B1 (ko) | 3,5-이치환 벤젠알키닐 화합물 및 그의 염 | |
| ES2547447T3 (es) | Compuestos farmacéuticos | |
| KR101974254B1 (ko) | Fgfr 저해제의 간헐 투여용 항종양제 | |
| ES2347187T3 (es) | Derivado de tieno(3,2-d)pirimidina, util como un inhibidor de pi3k. | |
| ES2869048T3 (es) | Compuestos de piridinona-piridinilo sustituidos con metil/fluoro-piridinil-metoxi y compuestos de piridinona-piridinilo sustituidos con fluoro-pirimidinil-metoxi | |
| KR101828187B1 (ko) | 신규 축합 피리미딘 화합물 또는 그 염 | |
| ES2738573T3 (es) | Sal de compuesto heterocíclico que contiene nitrógeno o cristal del mismo, composición farmacéutica e inhibidor de FLT3 | |
| CN114269735A (zh) | 二氢或四氢喹唑啉类化合物及其中间体、制备方法和应用 | |
| CA2667487A1 (en) | Imidazo[1,2-b]pyridazine and pyrazolo[1,5-a]pyrimidine derivatives and their use as protein kinase inhibitors | |
| EP3546460A1 (en) | Pyrimido[5,4-b]indolizine or pyrimido[5,4-b]pyrrolizine compound, preparation method and use thereof | |
| BRPI0711045A2 (pt) | compostos de tetraidropiridotienopirimidina e métodos de uso destes | |
| CN105315285A (zh) | 2,4-二取代7H-吡咯并[2,3-d]嘧啶衍生物、其制法与医药上的用途 | |
| EP1740578A1 (en) | 4- 2- (cycloalkylamino) pyrimidin-4-yl ] - (phenyl) - imidazolin-2- one derivatives as p38 map- kinase inhibitors for the treatment of inflammatory diseases | |
| KR20150111362A (ko) | 중수소화된 페닐 아미노 피리미딘 화합물 및 이 화합물을 함유하는 약물 조성물 | |
| CN109020957B (zh) | 作为mnk抑制剂的杂环化合物 | |
| WO2014154723A1 (en) | Novel pyrrole derivatives for the treatment of cancer | |
| ES2905985T3 (es) | Compuesto de oxazino-quinazolina y de tipo oxazino-quinazolina, método de preparación y usos de los mismos | |
| ES2686828T3 (es) | Derivados de poliéteres macrocíclicos de N-aril-2-amino-4-aril-pirimidinas como inhibidores de la FLT3 y JAK | |
| WO2011078226A1 (ja) | 三環系化合物 | |
| CN111763217B (zh) | 一类噻吩并氮杂环类化合物、制备方法和用途 | |
| CN115960117B (zh) | 含硫并环类衍生物抑制剂、其制备方法和应用 |