ES2803513T3 - Derivados de quinazolin-4-ona sustituidos - Google Patents
Derivados de quinazolin-4-ona sustituidos Download PDFInfo
- Publication number
- ES2803513T3 ES2803513T3 ES14829331T ES14829331T ES2803513T3 ES 2803513 T3 ES2803513 T3 ES 2803513T3 ES 14829331 T ES14829331 T ES 14829331T ES 14829331 T ES14829331 T ES 14829331T ES 2803513 T3 ES2803513 T3 ES 2803513T3
- Authority
- ES
- Spain
- Prior art keywords
- fluoro
- methyl
- amino
- substituted
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 Cc1ncn*(N)c1C#N Chemical compound Cc1ncn*(N)c1C#N 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Dermatology (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Neurology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Transplantation (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Oncology (AREA)
- Otolaryngology (AREA)
- Communicable Diseases (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Un compuesto de la formula (Ib) o una sal farmaceuticamente aceptable del mismo. **(Ver fórmula)** en donde R1 se selecciona entre fenilo, que no esta sustituido o esta sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro; piridilo, que no esta sustituido o esta sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro; 1-metilpirazol-5-ilo; 2-metiltiofen-5-ilo; cicloalquilo C3-C6 que no esta sustituido o esta sustituido en la posicion 1 con metilo; tetrahidropiran-4-ilo; piperidin-1-ilo; morfolin-4-ilo; pirolidin-3-ilo, que no esta sustituido o esta sustituido en la posicion 1 con un sustituyente que se selecciona de metoxicarbonilo, metilsulfonilo, metilo o metilcarbonilo; o dimetilamina; R2 se selecciona entre heteroarilo C4-C7 que contiene un atomo de nitrogeno y 0, 1, 2 o 3 heteroatomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no esta sustituido o esta sustituido con 1-3 sustituyentes seleccionados independientemente de alquilo C1-C4, fluoroalquilo C1-C4, hidroxi-alquilo C1-C4, hidroxi-fluoroalquilo C1-C4, alcoxi C1-C4, fluoroalcoxi C1-C4, cicloalquilo C3-C6, que no esta sustituido o esta sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, heterocicloalquilo C3-C6, que no esta sustituido o esta sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, ciano, fluoro, amino, alquil C1-C4amino, o dialquil C1-C4amino; R5 y R6 se seleccionan independientemente de hidrogeno, deuterio o fluoro; R7 se selecciona de metoxi, difluorometoxi, trifluorometoxi, hidroxi , fluoro o metilsulfonilamina; y R3 se selecciona de **(Ver fórmula)** R8 se selecciona de hidrogeno, metilo, fluorometilo, difluorometilo, trideuterometilo o amino.
Description
DESCRIPCIÓN
Derivados de quinazolin-4-ona sustituidos
Campo de la invención
La presente invención se refiere a la preparación y el uso de nuevos derivados de quinazolin-4-ona sustituidos como candidatos a fármacos en forma libre o en forma de sal farmacéuticamente aceptable con propiedades valiosas similares a fármacos, tales como, p. ej., estabilidad metabólica y farmacocinética adecuada, forma para la modulación, en particular la inhibición de la actividad o función de la familia de fosfoinositida 3' OH quinasa (en adelante PI3K) .
Antecedentes de la invención
Miembros de la familia de la fosfoinositida-3 quinasa (PI3K) están implicados en el crecimiento, la diferenciación celular, la supervivencia, la remodelación del citoesqueleto y el tráfico de organelas intracelulares en muchos tipos diferentes de células (Okkenhaug y Wymann, Nature Rev. Immunol. 3:317 (2003).
Hasta la fecha, se han identificado ocho PI3K de mamíferos, divididos en tres clases principales (I, II y III ) en función de su secuencia genética, estructura, moléculas adaptadoras, expresión, modo de activación y sustrato preferido.
La familia de clase I más ampliamente comprendida (que comprende las isoformas PI3K a, p, y y 6) se subdivide adicionalmente en subclases iA e IB. Las quinasas PI3 de clase iA ( isoformas PI3Ka, PI3Kp y PI3K 6) consisten en una proteína reguladora/adaptadora de 85 kDa y tres subunidades catalíticas de 110 kDa (p110a, p110p y p110 6) que se activan en el sistema de tirosina quinasa, mientras que la clase IB consiste en una sola isoforma p110y (PI3Ky) que se activa mediante receptores acoplados a proteína G.
PI3K6 y PI3Ky son ambas lípido quinasas que pertenecen a la familia de clase I PI3K (PI3K a, p, y y 6). PI3K6 genera señales de segundo mensajero aguas abajo de los receptores enlazados a tirosina quinasa, mientras que PI3Ky es activada principalmente por receptores acoplados a proteínas G (GPCR).
PI3K6 y PI3Ky son heterodímeros compuestos de una proteína adaptadora y una subunidad catalítica p1106 o p110y, respectivamente, que convierte fosfatidilinositol-4,5-bis-fosfato (PtdInsP2) en fosfatidilinositol-3,4,5-tri-fosfato (PtdInsP3). Proteínas efectoras interactúan con PtdInsP3 y desencadenan vías de señalización específicas implicadas en la activación celular, diferenciación, migración y supervivencia de las células.
La expresión de las subunidades catalíticas p1106 y p110y es preferencial para los leucocitos. La expresión también se observa en células del músculo liso, miocitos y células endoteliales. En contraste, p110a y p110p son expresadas por todos los tipos de células (Marone et al. Biochimica et Biophysica Acta 1784:159 (2008)).
PI3K6 está asociada con el desarrollo y la función de células B (Okkenhaug et al. Science 297:1031 (2002)).
Las células B también juegan un papel crítico en la patogénesis de un cierto número de enfermedades autoinmunes y alérgicas, así como en el proceso de rechazo de trasplantes (Martin y Chan, Annu. Rev. Immunol. 24:467 (2006)).
Se ha demostrado un vínculo entre PI3Ky y procesos tales como la quimiotaxis de leucocitos y la desgranulación de mastocitos, generando con ello interés en este objetivo para el tratamiento de trastornos autoinmunes e inflamatorios (Ghigo et al., Bioessays, 2010, 32, 185-196; Reif et al., J. Immunol., 2004, 173, 2236-2240; Laffargue et al., Immunity, 2002, 16, 441-451). También existen informes que relacionan PI3Ky con el cáncer, la diabetes, la enfermedad cardiovascular y la enfermedad de Alzheimer.
La quimiotaxis está implicada en muchas enfermedades autoinmunes o inflamatorias, en la angiogénesis, invasión/metástasis, neurodegeneración o cicatrización de heridas (Gerard et al. Nat. Immunol. 2:108 (2001)). Eventos temporales distintos en la migración de leucocitos en respuesta a las quimioquinas dependen completamente de PI3K6 y PI3Ky (Liu et al. Blood 110:1191 (2007)).
PI3Ka y PI3Kp juegan un papel esencial en el mantenimiento de la homeostasis y la inhibición farmacológica de estas dianas moleculares se ha asociado con la terapia del cáncer (Maira et al. Expert Opin. Ther. Targets 12:223 (2008)). PI3Ka está implicada en la señalización de la insulina y las vías de crecimiento celular (Foukas et al. Nature 441:366 (2006)). Se espera que la inhibición selectiva de la isoforma PI3K6 y/o PI3Ky evite potenciales efectos secundarios, tales como la hiperglucemia y la desregulación metabólica o del crecimiento.
Las infecciones parasitarias todavía representan una de las causas más importantes de morbilidad y mortalidad en todo el mundo. Entre los parásitos que provocan una patología humana y animal, el filo apicomplexa comprende un grupo de parásitos portados por vectores que son responsables de una amplia diversidad de enfermedades graves, que incluyen, pero no se limitan a malaria, leishmaniasis y tripanosomiasis. La malaria por sí sola infecta al 5-10% de la humanidad y
provoca alrededor de dos millones de muertes al año. [Schofield et al, "Immunologicalprocesses in malaria pathogenesis", Nat Rev Imm 2005], [Schofiled L, "Intravascular infiltrates and organ-specific inflammation in malaria pathogenesis], [Mishra et al, "TLRs in CNS Parasitic infections", Curr Top Micro Imm 2009], [Bottieau et al, "Therapy of vector-borne protozoan infections in nonendemic settings", Expert Rev. Anti infect. Ther., 2011].
Receptores tipo Toll (TLRs) son moléculas codificadas por la línea germinal, filogenéticamente antiguas que reconocen moléculas estructurales relevantes conservados en la evolución (conocidos como patrones moleculares asociados a patógenos (PAMPs)) dentro de los patógenos microbianos. Diversos tipos diferentes de células, incluidas las células del sistema inmunitario, expresan TLRs y, con ello, pueden detectar la presencia de PAMPs. Hasta la fecha se han descrito 10 miembros de la familia TLR funcionales (TLR1-10) en seres humanos, todos los cuales reconocen moléculas específicas para PAMP. Tras el reconocimiento de estos PAMPs específicos, los TLRs inducen y organizan la respuesta inmune del huésped a infecciones con bacterias, virus, hongos y parásitos. [Hedayat et al, "Targeting of TLRs: a decade of progress in combating infectious disease", revisión, Lancet Infectious disease 2011], [Kwai et al, "TLRs and their crosstalk with other innate receptors in infection and immunity", revisión, Immunity mayo de 2011].
El sistema inmunitario del huésped infectado responde a la infección con la producción inducida por TLR de citoquinas pro-inflamatorias, principalmente del tipo T-helper 1 (Th1). Si bien las cantidades adecuadas de estas citoquinas son beneficiosas y necesarias para eliminar la infección, una sobreproducción de estos mediadores es perjudicial para el huésped y se asocia con patología mediada por el sistema inmunitario, incluida la neuropatología y el daño tisular con consecuencias graves y a menudo fatales. Un ejemplo destacado y muy relevante de dicha patología mediada por el sistema inmune es la malaria aguda y cerebral (CM), que provoca síntomas clínicos graves y a menudo es mortal. Schofield et al, "Immunological processes in malaria pathogenesis", Nat Rev Imm 2005], [Schofiled L, "Intravascular infiltrates and organ-specific inflammation in malaria pathogenesis], [Mishra et al, "TLRs in CNS Parasitic infections", Curr Top Micro Imm 2009], [Bottieau et al, "Therapy of vector-borne protozoan infections in nonendemic settings", Expert Rev. Anti infect. Ther., 2011] [Hedayat et a l," Targeting of TLRs: a decade of progress in combating infectious disease", revisión, Lancet Infectious disease 2011]. A pesar de los progresos realizados en el tratamiento y la erradicación de la malaria, la tasa de mortalidad asociada con la malaria grave, incluida la CM sigue siendo inaceptablemente alta. Estrategias dirigidas únicamente a la erradicación del parásito en el huésped podría, por lo tanto, no ser suficiente para prevenir complicaciones neurológicas y la muerte en todos los casos de CM. El desarrollo de nuevas estrategias terapéuticas complementarias innovadoras para reducir de manera eficiente la mortalidad y la morbilidad asociadas a CM que es causada, en parte, por la inmunopatología mediada por el huésped, sigue siendo, por lo tanto, una necesidad médica urgente. [Higgins et al, "Immunopathogenesis of falciparum malaria: implications for adjunctive therapy in the management of severe and cerebral malaria", Expert Rev. Anti Infect. Ther. 2011]
Recientemente se ha proporcionado evidencia adicional de que TLR9 juega un papel clave en el reconocimiento y la respuesta a los parásitos, incluidos, pero no limitados a Plasmodium, Leishmania, Trypanosoma y Toxoplasma [Gowda et al, "The Nucleosome is the TLR9-specific Immunostimulatory component of plasmodium falciparum that activates DCs", PLoS ONE, junio de 2011], [Peixoto-Rangel et al, "Candidate gene analysis of ocular toxoplasmosis in Brazil: evidence for a role for TLR9', Mem Inst Oswaldo Cruz 2009], [Pellegrini et al, "The role of TLRs and adoptive immunity in the development of protective or pathological immune response triggered by the Trypanosoma cruzi protozoan", Future Microbiol 2011] y que la interferencia con la activación de TLRs, incluido TLR9, representa una estrategia prometedora para prevenir las respuestas inflamatorias nocivas en casos de malaria graves y de malaria cerebral [Franklin et al, "Therapeutical targeting of nucleic acid-sensing TLRs prevents experimental cerebral malaria", PNAS 2011]
La malaria es una enfermedad infecciosa provocada por cuatro parásitos protozoarios: Plasmodium falciparum; Plasmodium vivax; Plasmodium ovale; y Plasmodium malaria. Estos cuatro parásitos se transmiten típicamente por la picadura de un mosquito Anopheles hembra infectado. La malaria es un problema en muchas partes del mundo y en las últimas décadas la carga de la malaria ha aumentado constantemente. Se estima que 1-3 millones de personas mueren cada año de malaria, en su mayoría niños menores de 5 años. Este aumento en la mortalidad por malaria se debe, en parte, al hecho de que Plasmodium falciparum, el parásito de la malaria más mortífero, ha adquirido resistencia contra casi todos los fármacos anti-malaria disponibles, con la excepción de los derivados de artemisinina.
La leishmaniasis es provocada por una o más de 20 variedades de protozoos parásitos que pertenecen al género Leishmania, y se transmite por la picadura de moscas de arena hembra. La leishmaniasis es endémica en aproximadamente 88 países, incluidas muchas áreas tropicales y sub-tropicales. Hay cuatro formas principales de leishmaniasis. La leishmaniasis visceral, también denominada kala-azar, es la forma más grave y es provocada por el parásito Leishmania donovani. Los pacientes que desarrollan leishmaniasis visceral pueden morir en cuestión de meses a menos que reciban tratamiento. Las dos terapias principales para la leishmaniasis visceral son los derivados de antimonio, el estibogluconato de sodio (Pentostam®) y el antimoniato de meglumina (Glucantim®). El estibogluconato de sodio se ha utilizado durante aproximadamente 70 años y la resistencia a este fármaco es un problema creciente. Además, el tratamiento es relativamente largo y doloroso, y puede provocar efectos secundarios indeseables.
La tripanosomiasis africana humana, también conocida como enfermedad del sueño, es una enfermedad parasitaria transmitida por vectores. Los parásitos afectados son los protozoos pertenecientes al género Trypanosoma. Se
transmiten a los seres humanos por picaduras de moscas tsetse (Glossina Genus) que han adquirido su infección de seres humanos o de animales que albergan los parásitos patógenos humanos.
La enfermedad de Chagas (también denominada tripanosomiasis americana) es otra enfermedad parasitaria humana que es endémica entre las poblaciones pobres del continente americano. La enfermedad es provocada por el parásito protozoario Trypanosoma cruzi, que se transmite a los seres humanos por insectos chupadores de sangre. La enfermedad humana se produce en dos etapas: la etapa aguda, que se produce poco después de la infección y la etapa crónica, que puede desarrollarse a lo largo de muchos años. Las infecciones crónicas resultan en diversos trastornos neurológicos, incluyendo demencia, daño al músculo cardíaco y, a veces, dilatación del tracto digestivo, así como pérdida de peso. Sin tratamiento, la enfermedad crónica a menudo es mortal. Los fármacos actualmente disponibles para tratar la enfermedad de Chagas son Nifurtimox y benznidazol. Sin embargo, los problemas con estas terapias actuales incluyen sus diversos efectos secundarios, la duración del tratamiento y el requisito de supervisión médica durante el tratamiento. Además, el tratamiento solo es realmente efectivo cuando se administra durante la fase aguda de la enfermedad. La resistencia a los dos fármacos de primera línea ya se ha producido. El agente antifúngico Anfotericina b se ha propuesto como un fármaco de segunda línea, pero este medicamento es costoso y relativamente tóxico.
La toxoplasmosis es endémica en muchas áreas a nivel mundial y puede infectar a una gran proporción de la población adulta. Sin embargo, su prevalencia difiere en los diferentes países. Se estima que infecta al menos al 10% de los adultos en los países templados del norte y más de la mitad de los adultos en países mediterráneos y tropicales. Toxoplasma gondii, el patógeno causante de la toxoplasmosis, es un protozoo intracelular obligatorio y ubicuo y se considera la causa más común de retinilo infecciosa en seres humanos, que depende de una diversidad de factores, incluidos el clima, la higiene y los hábitos alimenticios. El curso de la enfermedad en adultos inmunocompetentes suele ser asintomático y auto-limitante. Tan pronto como se produce la infección, el parásito forma quistes latentes en la retina y en otros órganos del cuerpo, que pueden reactivarse años después de la infección inicial, dando lugar a retinocoroiditis aguda y a la formación de nuevas lesiones retinocoroidales. [Arevalo et al, "Ocular Toxoplasmosis in the developing world', Internat. Ophthal. Clin 2010]
La neurocisticercosis es la enfermedad parasitaria más común del SNC (incidencia ~ 2,5 millones en todo el mundo) provocada por las larvas de Taenia solium. La enfermedad tiene una larga fase asintomática en seres humanos caracterizada por la ausencia de una respuesta inflamatoria detectable que rodea al parásito. La respuesta inmune general durante la fase asintomática es del fenotipo Th2. Sin embargo, la destrucción de las larvas por tratamiento terapéutico o por desgaste normal del parásito provoca una fuerte respuesta inflamatoria, que a menudo consiste en una reacción granulomatosa crónica y la manifestación de síntomas típicos de la enfermedad. La respuesta inmune en el SNC de pacientes sintomáticos consiste en un fenotipo Th1 manifiesto o una respuesta mixta Th1, Th2 y Th3, dependiendo de la ausencia o presencia de granulomas. La respuesta hiperinflamatoria que prevalece durante la fase sintomática en el SNC es la responsable de la neuropatología severa y la mortalidad asociada con la neurocisticercosis.
[Mishra et al, "TLRs in CNS Parasitic infections", Curr Top Micro Imm 2009]
El documento EP 2612 862 A2 describe inhibidores selectivos de PI3K que contienen un núcleo de quinazolinona. En particular, se informa que los compuestos del documento EP 2612862 A2 son inhibidores selectivos de PI3Kdelta (véase el documento EP 2 612 862 A2). Los compuestos ejemplificados en el documento EP 2 612 862 A2 difieren de los compuestos de la presente invención en la elección del sustituyente en la posición 5 del anillo de quinazolin-4-ona.
El documento WO 2013/032591 A1 describe compuestos de una fórmula general (I) que muestran actividad inhibidora selectiva de PI3K para PI3Kdelta y gamma (véase el documento WO 2013/032591 A1). Los compuestos ejemplificados en el documento WO 2013/032591 A1 difieren de los compuestos de la presente invención en la elección de que el enlazador utilizado en la presente invención es diferente del enlazador correspondiente en el documento WO 2013/032591 A1.
Sumario de la invención
Existe la necesidad de proporcionar nuevos inhibidores de la quinasa PI3 de clase I que sean buenos candidatos a fármacos. En particular, los compuestos de la invención deberían unirse de manera potente a las quinasas PI3 de clase I, mostrando poca afinidad por otros receptores y mostrando actividad funcional como inhibidores. Deben absorberse bien del tracto gastrointestinal, ser metabólicamente estables y poseer propiedades farmacocinéticas favorables. Cuando fijan como objetivo los receptores del sistema nervioso central, deben atravesar la barrera hematoencefálica libremente y cuando fijan como objetivo selectivamente los receptores del sistema nervioso periférico, no deben atravesar la barrera hematoencefálica. Deben ser atóxicos y demostrar pocos efectos secundarios. Además, el candidato a fármaco ideal existirá en una forma física que sea estable, no higroscópica y que se formule fácilmente.
Los compuestos de la invención muestran un determinado nivel de selectividad contra los diferentes parálogos PI3K a, p, Y y 5. En particular, muestran un determinado nivel de selectividad para la isoformas PI3K5 y PI3K y sobre la isoforma PI3Ka.
Por lo tanto, los compuestos de la presente invención son potencialmente útiles en el tratamiento de una amplia gama de trastornos, particularmente trastornos que incluyen, pero no se limitan a, trastornos autoinmunitarios, enfermedades autoinflamatorias e inflamatorias, enfermedades alérgicas, inmunopatologías asociadas a enfermedades o infecciones , enfermedades de las vías respiratorias, tales como asma. y COPD, rechazo de trasplantes, cánceres, p. ej., de origen hematopoyético o tumores sólidos.
En la presente se describen diversas realizaciones de la invención.
Dentro de determinados aspectos, se proporciona en la presente un compuesto de la fórmula (Ib) o una sal farmacéuticamente aceptable del mismo.

En otra realización, la invención proporciona una composición farmacéutica que comprende una cantidad terapéuticamente efectiva de un compuesto de acuerdo con la definición de fórmula (Ib), o una sal farmacéuticamente aceptable del mismo, o sub-fórmulas del mismo y uno o más portadores farmacéuticamente aceptables.
En otra realización, la invención proporciona una combinación, en particular una combinación farmacéutica, que comprende una cantidad terapéuticamente efectiva del compuesto de acuerdo con la definición de fórmula (Ib), o una sal farmacéuticamente aceptable del mismo, o sub-fórmulas del mismo y uno o más agentes terapéuticamente activos.
En otra realización, la invención también se refiere al tratamiento, ya sea solo o en combinación con uno o más de otros compuestos farmacológicamente activos incluyendo métodos de tratamiento de afecciones, enfermedades o trastornos en los que uno o más de las funciones de las células B, tales como la producción de anticuerpos, la presentación de antígenos, la producción de citoquinas u organogénesis linfoide son anormales o son indeseables, incluyendo la artritis reumatoide y enfermedades relacionadas (tales como espondiloartritis anquilosante, artritis psoriásica, artritis juvenil), pénfigo vulgar y enfermedades relacionadas, púrpura trombocitopénica idiopática, lupus eritematoso sistémico, esclerosis múltiple, miastenia grave, síndrome de Sjogren (tal como síndrome de Sjogren primario (PSS)) , enfermedad injerto contra huésped, anemia hemolítica autoinmune, vasculitis asociada a ANCA (tales como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg- Strauss), crioglobulinemia , púrpura trombocitopénica trombótica, lesión de isquemia por reperfusión, urticaria crónica autoinmune, alergia (dermatitis atópica, dermatitis de contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), asma, síndrome de Goodpasture, diferentes tipos de glomerulonefritis, AMR (rechazo de trasplante mediado por anticuerpos), rechazo de trasplante hiperagudo, agudo y crónico mediado por células B y cánceres de origen hematopoyético que incluyen, pero no se limitan a mieloma múltiple; leucemia mielógena aguda; leucemia mielógena crónica; leucemia linfocítica; leucemia mieloide; linfoma no Hodgkin; linfomas policitemia vera; trombocitemia esencial; mielofibrosis con metaplasia mieloide; y la enfermedad de Waldenstroem, así como en la inmunopatología asociada a enfermedades o infecciones.
Breve descripción de los dibujos
La Figura 1 es el Patrón de Difracción de rayos X en polvo de la forma cristalina del Ejemplo 7.
La Figura 2 es el termograma de Calorimetría Diferencial de Barrido (DSC) de la forma cristalina del Ejemplo 7.
Descripción detallada de la invención
La invención proporciona compuestos de quinazolin-4-ona sustituidos de fórmula (Ib) y/o sales y/o solvatos farmacéuticamente aceptables de los mismos,

R1 se selecciona de
fenilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
piridilo, que no está sustituido o está sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
1 -metilpirazol-5-ilo;
2-metiltiofen-5-ilo;
cicloalquilo C3-C6 que no está sustituido o está sustituido en la posición 1 con metilo;
tetrahidropiran-4-ilo;
piperidin-1-ilo;
morfolin-4-ilo;
pirolidin-3-ilo, que no está sustituido o está sustituido en la posición 1 con un sustituyente que se selecciona de metoxicarbonilo, metilsulfonilo, metilo o metilcarbonilo; o
dimetilamina;
R2 se selecciona entre
heteroarilo C4-C7 que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de
alquilo C1-C4 ,
fluoroalquilo C1 -C4 ,
hidroxi-alquilo C1 -C4 ,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1-C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
Heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
ciano,
fluoro,
amino,
alquil C1-C4amino, o
dialquil C-i-C4amino;
o
Alquinilo C2-C5 , que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de fluoroalquilo C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
Heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
alcoxi C1-C4 ,
fluoroalcoxi C1-C4 ,
hidroxi,
ciano,
fluoro,
amino,
alquil C1-C4amino, o
dialquil C1-C4amino;
R5 y R6 se seleccionan independientemente de hidrógeno, deuterio o fluoro;
R7 se selecciona de metoxi, difluorometoxi, trifluorometoxi, hidroxi , fluoro o metilsulfonilamina; y
R3 se selecciona de

en donde
R8 se selecciona de hidrógeno, metilo, fluorometilo, difluorometilo, trideuterometilo o amino.
A menos que se especifique lo contrario, la expresión "compuestos de la presente invención" se refiere a compuestos de fórmula (Ib) y sub-fórmulas de los mismos, sales del compuesto, así como todos los estereoisómeros (incluyendo diastereómeros y enantiómeros), rotámeros, tautómeros y compuestos isotópicamente marcados (incluyendo sustituciones de deuterio, así como restos formados inherentemente). En los casos en los que se mencionen compuestos de fórmula (Ib), se pretende que incluya también los tautómeros y N-óxidos de los compuestos de fórmula (Ib).
La invención puede apreciarse más completamente haciendo referencia a la siguiente descripción, incluyendo el siguiente glosario de términos y expresiones y ejemplos concluyentes. Como se utiliza en la presente, las expresiones "que incluye", "que contiene" y "que comprende" se utilizan en la presente en su sentido abierto y no limitativo.
Los tautómeros pueden estar presentes, por ejemplo, en la porción R3 de los compuestos de fórmula (Ib). Los residuos heterociclilo y heteroarilo que contienen nitrógeno pueden formar N-óxidos, por ejemplo en la posición R2 de los compuestos de fórmula (Ib).
En los casos en los que se utiliza la forma en plural para compuestos, sales y similares, ésta se toma para dar a entender también un solo compuesto o sal .
Los términos y expresiones generales utilizados anteriormente y en lo que sigue en la presente preferiblemente tienen dentro del contexto de esta divulgación los siguientes significados, a menos que se indique lo contrario:
Tal como se utiliza en la presente, el término "cicloalquilo C3-C6" se refiere a un anillo saturado monocíclico de 3 a 6 miembros.
En el contexto de R1, ejemplos de cicloalquilo C3-C6 incluyen ciclopropilo; ciclobutilo; ciclopentilo y ciclohexilo;
En el contexto de R1, ejemplos de cicloalquilo C3-C6 que está sustituido en la posición 1 con metilo incluyen 1-metilciclopropilo; 1 -metilciclobutilo; 1 -metilciclopentilo y 1-metilciclohexilo;
En el contexto de R2 , ejemplos de cicloalquilo C3-C6 como sustituyente en heteroarilo o alquinilo incluyen ciclopropilo y ciclobutilo.
Tal como se utiliza en la presente, el término "heterocicloalquilo C3-C6» se refiere a un anillo saturado monocíclico de 3 a 6 miembros que contiene 1,2 o 3 heteroátomos seleccionados de N, O o S.
En el contexto de R2, ejemplos de heterocicloalquilo C3-C6 como sustituyente en heteroarilo o alquinilo incluye oxetan, aziridina y morfolina.
Tal como se utiliza en la presente, el término "heteroarilo C4-C7" se refiere a un anillo monocíclico de 4 a 7 miembros con máxima saturación que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S.
En el contexto de R2 , ejemplos de heteroarilo C4-C7 incluyen pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, piridazina, pirazina, oxazol, isotiazol, tiofeno, furano, triazol, tetrazol.
Tal como se utiliza en la presente, el término "dialquil C1-C4 amino» es amino sustituido con dos grupos alquilo que se seleccionan independientemente de alquilo C1-C4.
Tal como se utiliza en la presente, el término "fluoroalquilo C1-C4" se refiere a alquilo C1-C4 que está parcial o totalmente fluorado.
Tal como se utiliza en la presente, el término "fluoroalcoxi C1-C4" se refiere a alcoxi C1-C4 que está parcial o totalmente fluorado.
Tal como se utiliza en la presente, todos los sustituyentes se escriben de manera que muestren el orden de los grupos funcionales (grupos) de los que están compuestos. Los grupos funcionales se definen anteriormente en la presente.
En la presente se describen diversas realizaciones de la invención. Se reconocerá que las características especificadas en cada realización se pueden combinar con otras características especificadas para proporcionar realizaciones adicionales de la presente invención.
En una realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En una realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R2 es heteroarilo C5-C6 que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, piridazina, pirazina, oxazol, isotiazol, tiofeno, furano, triazol o tetrazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, oxazol o isotiazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol o tiazol, que no está sustituido o está sustituido con 1 sustituyente seleccionado independientemente de
alquilo C1-C4 ,
hidroxi-alquilo C1-C4, o
alcoxi C1-C4.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1 o 2 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo u o-tolilo.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R3 se selecciona de

en donde
R8 se selecciona de metilo.
En una realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro; o
piridilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 es heteroarilo C5-C6 que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de
alquilo C1-C4 ,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4,
fluoroalcoxi C1-C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro; o
piridilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, piridazina, pirazina, oxazol, isotiazol, tiofeno, furano, triazol o tetrazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4 ,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro; o
piridilo, que no está sustituido o está sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, oxazol o isotiazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro; o
piridilo, que no está sustituido o está sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol o tiazol, que no está sustituido o está sustituido con 1 sustituyente seleccionado independientemente de
alquilo C1-C4,
hidroxi-alquilo C1-C4, o
alcoxi C1-C4 ;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En una realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1 o 2 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 es heteroarilo C5-C6 que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1 o 2 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, piridazina, pirazina, oxazol, isotiazol, tiofeno, furano, triazol o tetrazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1 o 2 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, oxazol o isotiazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1 o 2 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol o tiazol, que no está sustituido o está sustituido con 1 sustituyente seleccionado independientemente de
alquilo C1-C4,
hidroxi-alquilo C1-C4, o
alcoxi C1-C4 ;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo, que no está sustituido o está sustituido con 1 o 2 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
R2 es alqu-1-inilo C2-C5 , que está sustituido con 1 sustituyente seleccionado de hidroxi;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En una realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo u o-tolilo;
R2 es heteroarilo C5-C6 que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo u o-tolilo;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, piridazina, pirazina, oxazol, isotiazol, tiofeno, furano, triazol o tetrazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo u o-tolilo;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol, tiazol, oxazol o isotiazol, que no está sustituido o está sustituido con 1-2 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1 -C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En otra realización, la invención proporciona un compuesto de la fórmula (Ib) y/o una sal farmacéuticamente aceptable del mismo. en donde
R1 es fenilo u o-tolilo;
R2 se selecciona de pirazol, imidazol, piridina, pirimidina, isoxazol o tiazol, que no está sustituido o está sustituido con 1 sustituyente seleccionado independientemente de
alquilo C1-C4,
hidroxi-alquilo C1-C4, o
alcoxi C1-C4 ;
R5 se selecciona de hidrógeno o fluoro; y
R6 se selecciona de hidrógeno.
En una realización, la invención proporciona un compuesto de la fórmula (Ib), seleccionado de
2-amino-4-((2S,4S)-2-(6-fluoro-5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpi rimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(6-fluoro-5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
4-amino-6-((2S,4S)-2-(6-fluoro-5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)pirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(6-fluoro-5-(2-metoxitiazol-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-metoxi-2-(5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(2-metilpirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(4-oxo-3-fenil-5-(piridin-3-il)-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(5-metoxipiridin-3-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(4-oxo-3-fenil-5-(pirimidin-5-il)-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(4-metoxipiridin-3-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(1-(2-hidroxietil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(5-(2-etoxipirimidin-5-il)-6-fluoro-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-fluoro-2-(5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(6-fluoro-5-(1-metil-1H-imidazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpi rimidina-5-carbonitrilo
o
2-amino-4-((2S,4S)-4-metoxi-2-(5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
y/o una sal farmacéuticamente aceptable del mismo.
En otra realización, los compuestos individuales de acuerdo con la invención son los enumerados en la sección de Ejemplos que figura más adelante.
Dependiendo de la elección de los materiales de partida y los procedimientos, los compuestos se pueden encontrar presentes en forma de uno de los posibles isómeros o como mezclas de estos, por ejemplo, como isómeros ópticos puros, o como mezclas de isómeros tales como racematos y mezclas de diastereómeros, dependiendo del número de átomos de carbono asimétricos. Se pretende que la presente invención incluya todos los posibles isómeros de este tipo, incluidas las mezclas racémicas, mezclas diastereoméricas y formas ópticamente puras. Los isómeros (R) y (S) ópticamente activos pueden prepararse utilizando sintones quirales o reactivos quirales, o pueden resolverse utilizando técnicas convencionales. Si el compuesto contiene un doble enlace, el sustituyente puede tener la configuración E o Z. Si el compuesto contiene un cicloalquilo disustituido, el sustituyente del cicloalquilo puede tener una configuración cis o trans. También se pretende que todas las formas tautoméricas estén incluidas.
Tal como se utiliza en la presente, los términos "sal" o "sales" se refieren a una sal por adición de ácidos o una sal por adición de bases de un compuesto de la invención. El término "sales» incluye, en particular, "sales farmacéuticamente aceptables". La expresión "sales farmacéuticamente aceptables" se refiere a sales que retienen la efectividad biológica y las propiedades de los compuestos de esta invención y que típicamente no son biológicamente o de otra manera
indeseables. En muchos casos, los compuestos de la presente invención son capaces de formar sales de ácidos y/o bases en virtud de la presencia de grupos amino y/o carboxilo o grupos similares a estos.
Se pueden formar sales de adición de ácidos farmacéuticamente aceptables con ácidos inorgánicos y ácidos orgánicos.
Los ácidos inorgánicos a partir de los cuales se pueden obtener sales incluyen, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico y similares.
Los ácidos orgánicos a partir de los cuales se pueden obtener sales incluyen, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido toluenosulfónico, ácido sulfosalicílico y similares.
Se pueden formar sales de adición de bases farmacéuticamente aceptables con bases inorgánicas y orgánicas.
Las bases inorgánicas a partir de las cuales se pueden obtener sales incluyen, por ejemplo, sales de amonio y metales de las columnas I a XII de la tabla periódica. En determinadas realizaciones, las sales se obtienen a partir de sodio, potasio, amonio, calcio, magnesio, hierro, plata, zinc y cobre; algunas sales particularmente adecuadas incluyen sales de amonio, potasio, sodio, calcio y magnesio.
Las bases orgánicas a partir de las cuales se pueden obtener sales incluyen, por ejemplo, aminas primarias, secundarias y terciarias, aminas sustituidas que incluyen aminas sustituidas de origen natural, aminas cíclicas, resinas de intercambio iónico básicas y similares. Determinadas aminas orgánicas incluyen isopropilamina, benzatina, colinato, dietanolamina, dietilamina, lisina, meglumina, piperazina y trometamina.
En otro aspecto, la presente invención proporciona compuestos de fórmula (I) en forma de sal acetato, ascorbato, adipato, aspartato, benzoato, besilato, bromuro/hidrobromuro, bicarbonato/carbonato, bisulfato/sulfato, alcanforsulfonato, caprato, cloruro/hidrocloruro, clortofilonato, citrato, etandisulfonato, fumarato, gluceptato, gluconato, glucuronato, glutamato, glutarato, glicolato, hipurato, hidroyoduro/yoduro, isetionato, lactato, lactobionato, laurilsulfato, malato, maleato, malonato, mandelato, mesilato, metilsulfato, mucato, nacato, mucato, nafato, nitrato, octadecanoato, oleato, oxalato, palmitato, pamoato, fosfato/hidrógeno-fosfato/ dihidrógeno-fosfato, poligalacturonato, propionato, sebacato, estearato, succinato, sulfosalicilato, sulfato, tartrato, tosilato, trifenatato, trifluoroacetato o xinafoato.
También se pretende que cualquier fórmula proporcionada en la presente represente formas no marcadas, así como formas marcadas con isotópos de los compuestos. Compuestos marcados con isótopos tienen las estructuras representadas por las fórmulas que se proporcionan en la presente, salvo por que uno o más átomos son reemplazados por un átomo que tiene una masa atómica o un número másico seleccionado. Algunos ejemplos de isótopos que se pueden incorporar a compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro tales como 2H, 3H, 11C, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36Cl, 123I, 124I, 125I, respectivamente. La invención incluye diversos compuestos marcados isotópicamente como se definen en esta memoria, por ejemplo, aquellos en los que están presentes isótopos radiactivos, tales como 3H y 14C, o aquellos en los que están presentes isótopos no radiactivos, tales como 2H y 13C. Tales compuestos marcados isotópicamente son útiles en estudios metabólicos (con 14C), estudios de la cinética de reacción (con, por ejemplo, 2H o 3H), técnicas de detección o de obtención de imágenes, tales como la tomografía por emisión de positrones (PET) o la tomografía computarizada de emisión monofotónica (SPECT), que incluyen ensayos de distribución tisular de fármacos o sustratos, o en el tratamiento radiactivo de pacientes. En particular, un compuesto marcado o 18F puede ser particularmente deseable para estudios de PET o SPECT. Compuestos de fórmula (Ib) marcados isotópicamente generalmente pueden prepararse mediante técnicas convencionales conocidas por los expertos en la técnica o mediante procedimientos análogos a los descritos en los Ejemplos y Preparaciones que se acompañan utilizando un reactivo marcado isotópicamente apropiado en lugar del reactivo no marcado previamente empleado.
Además, la sustitución con isótopos más pesados, particularmente el deuterio (es decir, 2H o D) puede proporcionar determinadas ventajas terapéuticas que resultan de una mayor estabilidad metabólica, por ejemplo, una semivida in vivo incrementada o requisitos de dosificación reducidos o una mejora en el índice terapéutico. Se entiende que el deuterio en este contexto se considera como un sustituyente de un compuesto de la fórmula (Ib). La concentración de un isótopo más pesado,de este tipo específicamente deuterio, puede definirse por el factor de enriquecimiento isotópico. La expresión "factor de enriquecimiento isotópico", tal como se utiliza en la presente, se refiere a la relación entre la abundancia isotópica y la abundancia natural de un isótopo especificado. Si un sustituyente en un compuesto de esta invención se denomina deuterio, dicho compuesto tendrá un factor de enriquecimiento isotópico para cada átomo de deuterio designado de al menos 3500 (52,5% de incorporación de deuterio en cada átomo de deuterio designado), al menos 4000 (60% de incorporación de deuterio), al menos 4500 (67,5% de incorporación de deuterio), al menos 5000 (75% de incorporación de deuterio), al menos 5500 (82,5% de incorporación de deuterio), al menos 6000 (90% de incorporación de deuterio), al menos 6333,3 (95% de incorporación de deuterio), al menos 6466,7 (97% de incorporación de deuterio), al menos 6600 (99% de incorporación de deuterio) o al menos 6633,3 (99,5% de incorporación de deuterio).
Solvatos farmacéuticamente aceptables de acuerdo con la invención incluyen aquellos en los que el disolvente de cristalización puede estar isotópicamente sustituido, p. ej. D2O, d6-acetona, DMSO-d6 .
Compuestos de la invención, es decir, los compuestos de fórmula ( Ib ) que contienen grupos capaces de actuar como donantes y / o aceptores para enlaces de hidrógeno pueden ser capaces de formar cocristales con formadores de cocristales adecuados. Estos co-cristales pueden prepararse a partir de compuestos de fórmula (Ib) mediante procedimientos conocidos de formación de cocristales. Procedimientos de este tipo incluyen molienda, calentamiento, co-sublimación, co-fusión o contacto en solución de compuestos de fórmula (Ib) con el formador de co-cristal en condiciones de cristalización y aislando los co-cristales formados de este modo. Formadores de co-cristales adecuados incluyen los descritos en el documento WO 2004/078163. Por lo tanto, la invención proporciona, además, co-cristales que comprenden un compuesto de fórmula (Ib).
Tal como se utiliza en la presente, la expresión «portador farmacéuticamente aceptable» incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, surfactantes, antioxidantes, conservantes (por ejemplo, agentes antibacterianos, agentes antifúngicos), agentes isotónicos, agentes retardantes de la absorción, sales, conservantes, estabilizantes farmacológicos, aglutinantes, excipientes, agentes desintegrantes, lubricantes, agentes edulcorantes, agentes aromatizantes, colorantes y similares y combinaciones de estos, tal como serían conocidos por los expertos en la técnica (remítase, por ejemplo, a Remington's Pharmaceutical Sciences, 18.a ed. Mack Printing Company, 1990, págs.
1289-1329). Salvo en lo que concierne a cualquier portador convencional que sea incompatible con el principio activo, se contempla su uso en las composiciones terapéuticas o farmacéuticas.
La expresión "una cantidad terapéuticamente efectiva" de un compuesto de la presente invención se refiere a una cantidad del compuesto de la presente invención que provocará la respuesta biológica o médica de un sujeto, por ejemplo, reducción o inhibición de una actividad de una enzima o una proteína, o síntomas de mejorar, aliviar afecciones, ralentizar o demorar la progresión de la enfermedad, o prevenir una enfermedad, etc . En una realización no limitante, la expresión "una cantidad terapéuticamente eficaz" se refiere a la cantidad del compuesto de la presente invención que, cuando se administra a un sujeto, es eficaz para (1) al menos parcialmente aliviar, inhibir, prevenir y/o mejorar una afección, o un trastorno o una enfermedad (i) mediada por PI3 quinasas de clase I o (ii) asociada con la actividad de quinasa PI3 de clase I , o (iii) caracterizada por actividad (normal o anormal) de quinasas PI3 de clase I o (2) reducir o inhibir la actividad de quinasas PI3 de clase I o (3) reducir o inhibir la expresión de quinasas PI3 de clase I . En otra realización no limitante, la expresión "una cantidad terapéuticamente eficaz" se refiere a la cantidad del compuesto de la presente invención que, cuando se administra a una célula, o a un tejido, o a un material biológico no celular, o a un medio, es eficaz para reducir o inhibir al menos parcialmente la actividad de las quinasas PI3 de clase I ; o al menos reducir o inhibir parcialmente la expresión de quinasas PI3 de clase I. El significado de la expresión "una cantidad terapéuticamente eficaz" tal como se ilustra en la realización anterior para las quinasas PI3 de clase I también se aplica por el mismo medio a cualquier otra proteína/péptido/enzima relevante.
Tal como se utiliza en la presente, el término "sujeto" se refiere a un animal. Normalmente el animal es un mamífero. Un sujeto también se refiere, por ejemplo, a primates (por ejemplo, seres humanos, hombres o mujeres), vacas, ovejas, cabras, caballos, perros, gatos, conejos, ratas, ratones, peces, pájaros y similares. En ciertas realizaciones, el sujeto es un primate. En otras realizaciones más, el sujeto es un ser humano.
Tal como se utiliza en la presente, el término "inhibir", "inhibición" o la expresión "que inhibe" se refiere a la reducción o supresión de una afección, síntoma o trastorno dado, o enfermedad o una disminución significativa en la actividad basal de una actividad o proceso biológico.
Tal como se utiliza en la presente, el término "tratar", "tratando" o "tratamiento" de cualquier enfermedad o trastorno se refiere, en una realización, a mejorar la enfermedad o trastorno (es decir, ralentizar o detener o reducir el desarrollo de la enfermedad o al menos uno de los síntomas clínicos de esta). En otra realización, "tratar", "tratando" o "tratamiento" se refiere a aliviar o mejorar al menos un parámetro físico incluidos los que pueden no ser perceptibles por el paciente. En otra realización más, "tratar", "tratando" o "tratamiento" se refiere a modular la enfermedad o trastorno, ya sea de manera física (p. ej., estabilización de un síntoma perceptible), fisiológica (p. ej.,, estabilización de un parámetro físico), o ambas. En aún otra realización más, "tratar", "tratando" o "tratamiento" se refiere a prevenir o retrasar el inicio o desarrollo o la progresión de la enfermedad o trastorno.
Tal como se utiliza en la presente, un sujeto "necesita» un tratamiento si dicho sujeto se beneficiaría biológicamente, médicamente o en calidad de vida de dicho tratamiento.
Tal como se utiliza en la presente, se debe interpretar que el término "un", "una", "el", "la" y términos similares utilizados en el contexto de la presente invención (especialmente en el contexto de las reivindicaciones) cubren tanto el singular como el plural a menos que se indique lo contrario en la presente o el contexto lo contradiga claramente.
Todos los métodos descritos en la presente se pueden llevar a cabo en cualquier orden adecuado, a menos que se indique lo contrario en la presente o el contexto lo contradiga claramente. El uso de todos y cada uno de los ejemplos o el lenguaje ilustrativo (p. ej., "tal/es como") proporcionado en la presente tiene por objeto únicamente ilustrar mejor la invención y no supone ninguna limitación del alcance de la invención que por lo demás se reivindica.
Cualquier átomo asimétrico (por ejemplo, carbono o similar) del (de los) compuesto(s) de la presente invención puede estar presente en forma racémica o enriquecida en un enantiómero, por ejemplo, la configuración (R), (S) o (R,S). En determinadas realizaciones, cada átomo asimétrico tiene al menos un 50% de exceso enantiomérico, al menos un 60% de exceso enantiomérico, al menos un 70% de exceso enantiomérico, al menos un 80% de exceso enantiomérico, al menos un 90% de exceso enantiomérico, al menos un 95% de exceso enantiomérico, o al menos un 99% de exceso enantiomérico en la configuración (R) o (S). Los sustituyentes en átomos con dobles enlaces insaturados pueden estar presentes, si es posible, en forma cis (Z) o trans (E).
Por consiguiente, tal como se utiliza en la presente, un compuesto de la presente invención puede estar en forma de uno de los posibles isómeros, rotámeros, atropisómeros, tautómeros o mezclas de estos, por ejemplo, como racematos, isómeros ópticos (enantiómeros), diastereómeros, isómeros geométricos (cis o trans) sustancialmente puros o mezclas de estos. Atropisómeros diastereoméricos pueden estar presentes en determinados compuestos de fórmula (Ib), p. ej., con respecto a la rotación impedida estéricamente alrededor de enlace N-R1.
Cualesquiera mezclas de isómeros resultantes se pueden separar en base a las diferencias físico-químicas de los constituyentes, en isómeros, diastereómeros, racematos puros o sustancialmente puros, geométricos u ópticamente puros, por ejemplo, por cromatografía y/o cristalización fraccionada.
Cualquier racemato resultante de productos finales o compuestos intermedios puede resolverse en los antípodas ópticos por métodos conocidos, p. ej., por separación de sus sales diastereoméricas, obtenidas con un ácido o base ópticamente activo, y liberando el compuesto de carácter ácido o básico ópticamente activo. En particular, se puede emplear un resto de carácter básico para resolver los compuestos de la presente invención en sus antípodas ópticos, p. ej., por cristalización fraccionada de una sal formada con un ácido ópticamente activo, p. ej., ácido tartárico, ácido dibenzoil tartárico, ácido diacetil tartárico, ácido di-0,0'-p-toluoil tartárico, ácido mandélico, ácido málico o ácido alcanfor-10-sulfónico. Los productos racémicos también se pueden resolver mediante cromatografía quiral, p. ej., cromatografía líquida de alta resolución (HPLC) utilizando un adsorbente quiral.
Además, los compuestos de la presente invención, incluidas sus sales, también se pueden obtener en forma de sus hidratos o pueden incluir otros disolventes utilizados para su cristalización. Los compuestos de la presente invención pueden formar solvatos de forma inherente o por diseño con disolventes farmacéuticamente aceptables (que incluyen agua); por lo tanto, se pretende que la invención englobe tanto formas solvatadas como no solvatadas. El término "solvato" se refiere a un complejo molecular de un compuesto de la presente invención (incluidas las sales farmacéuticamente aceptables de este) con una o más moléculas de disolvente. Tales moléculas de disolvente son aquellas utilizadas comúnmente en el campo farmacéutico, que se sabe que son inocuas para el receptor, p. ej., agua, etanol y similares. El término "hidrato" se refiere al complejo en el que la molécula de disolvente es agua.
Los compuestos de la presente invención, incluidas las sales, hidratos y solvatos de estos, pueden formar polimorfos de manera inherente o por diseño.
Típicamente, los compuestos de fórmula (Ib) se pueden preparar de acuerdo con
los métodos proporcionados infra.

En una realización, la invención también describe un procedimiento para fabricar un compuesto de fórmula (I) de acuerdo con las etapas a, a', b1, b1', c1, c1', d1 y d1' (Esquema A), en donde a', b1', c1' y d1' indican etapas de funcionalización opcionales o etapas de ajuste de grupo funcionales, según se requiera.
En otra realización, la invención describe un procedimiento para fabricar un compuesto de fórmula (I) de acuerdo con las etapas a, a', b2, b2', c2, c2', d1 y d1' (Esquema A), en donde a', b2', c2' y d1' designan etapas de funcionalización o etapas de ajuste de grupo funcional opcionales, según se requiera.
En otra realización, la invención describe un procedimiento para fabricar un compuesto de fórmula (I) de acuerdo con las etapas a, a’, b2, b2’, c3, c3’, d2 y d2’ (Esquema A), en donde a’, b2’, c3’ y d2’ designan etapas de funcionalización o etapas de ajuste de grupo funcional opcionales, según se requiera.
En una realización, el compuesto de fórmula (I) se obtiene mediante la etapa de reacción de acoplamiento d1 de un

R1’ es R1, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R1mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R2’ es R2 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R2mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R5’ es R5 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R5mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R6’ es R6 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R6 mediante etapas de funcionalización o etapas de ajuste de grupo funcional; y
L’ se muestra arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a L mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
con
R3’-Hal
en donde
R3’ es R3 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R3 mediante etapas de funcionalización o etapas de ajuste de grupo funcional; y
Hal representa halógeno, tal como cloro, bromo o yodo;
en donde en una realización, la reacción de acoplamiento se lleva a cabo en presencia de una base de amina, tal como N,N-diisopropiletilamina. La reacción se lleva a cabo en presencia de un disolvente orgánico, tal como un alcohol, bajo calentamiento por microondas durante 30 minutos a 8 horas o calentamiento convencional en un baño de aceite durante 30 minutos a 6 días en un intervalo de temperaturas de 120-160°C.
Alternativamente, la reacción se lleva a cabo bajo las condiciones habituales de Buchwald-Hartwig utilizando una combinación adecuada de catalizador de Pd/ligando tal como Pd2(dba)3/2-(diciclohexilfosfino)bifenilo o Pd2(dba)3/2-diciclohexilfosfino-2’,4’,6’-triisopropil-bifenilo, Pd2(dba)3/XPhos, Pd2(dba)3/(rac)-BINAP, Pd(OAc)2/(rac)-BINAP o bis(tri-tbutilfosfina)paladio y una base adecuada, tal como NaOtBu, Cs2CO3 o K3PO4 y disolventes orgánicos, tales como tolueno, dioxano o THF. La reacción se agita a una temperatura de aproximadamente 60-140°C, por ejemplo a 100°C hasta 110°C y se realiza opcionalmente en un reactor de microondas. La reacción se lleva a cabo preferiblemente bajo un gas inerte, tal como nitrógeno o argón;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional d1 ’.
El compuesto de fórmula (E) se obtiene a través de la etapa c1 de desproteger PG del compuesto de fórmula (D),

en donde PG representa un grupo protector adecuado, tal como un grupo Boc, y los otros sustituyentes son como se definieron arriba;
en donde en una realización, en que PG es un grupo Boc, la reacción de desprotección se lleva a cabo en un disolvente orgánico, tal como THF o DCM, en presencia de un ácido orgánico, tal como ácido trifluoroacético a temperatura ambiente durante 1-18 horas; o en presencia de un ácido inorgánico, tal como HCl o H3PO4, opcionalmente en presencia de agua a temperatura ambiente durante 24 horas a 6 días;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional c1 '.
El compuesto de fórmula (D) se obtiene mediante la etapa de acoplamiento b1 del compuesto de fórmula (C),

en donde PG representa un grupo protector adecuado, tal como un grupo Boc, Hal1 representa un halógeno, tal como un bromuro o un cloruro, o un pseudohalógeno, tal como un triflato, y los otros sustituyentes son como se definieron arriba; con
R2 '-Y
en donde, cuando R2' es heteroarilo C4-C7 , como se definió arriba como R2 para un compuesto de fórmula (Ib), o un sustituyente que puede ser transferido en heteroarilo C4-C7 , a través de etapas de funcionalización o etapas de ajuste de grupo funcional.
Y representa un residuo de ácido borónico o un borolanilo cíclico o acíclico, tal como -B(OH)2 o pinacolato-boro; o un tributilestannil, tal como tributilestannilo.
Y es H (para un alquino terminal) o Y representa un alquilestannilo (alquino no terminal) en condiciones de reacción habituales de la reacción de Suzuki (Y es un residuo de ácido borónico o un éster de boronato cíclico o acíclico), la reacción de Stille (Y es alquilestannilo) o el acoplamiento de Sonogashira (Y es H de un alquino terminal), condiciones de reacción típicas se conocen en el sector y pueden aplicarse al presente procedimiento.
Condiciones típicas para la reacción de Suzuki implican, por ejemplo, la presencia de un catalizador de paladio, tal como un catalizador de Pd(0), p. ej. Pd(PPh3)4, o un catalizador de Pd (II), tal como PdCl2(PPh3)2; Pd(OAc)2 , opcionalmente en presencia de un ligando de fosfina tal como BrettPhos; o paladaciclo Brettphos, opcionalmente en presencia de uno o más coadyuvantes de reacción, tales como una base, p. ej. NaOEt, Na2CO3 sólido o en solución acuosa , opcionalmente en presencia de uno o más diluyentes, particularmente disolventes apolares, p. ej. benceno o tolueno, o disolventes polares, p. ej., acetonitrilo o N-metil-2-pirrolidona. La reacción se agita a una temperatura de aproximadamente 100-180°C, p. ej., en un horno microondas. La reacción puede llevarse a cabo bajo un gas inerte tal como nitrógeno o argón. Condiciones típicas para la reacción de Stille implican, por ejemplo, la presencia de un catalizador de paladio, tal como un catalizador de Pd(0), p. ej., bis(tri-t-butilfosfina)paladio o Pd2(dba)3, opcionalmente en presencia de uno o más auxiliares de la reacción, tales como cloruro de litio, en presencia de un disolvente, p. ej., dioxano o DMF. La reacción se agita a una temperatura de aproximadamente 80-160°C , típicamente de 80-100°C. La reacción se lleva a cabo bajo un gas inerte, tal como nitrógeno o argón.
Condiciones típicas para el acoplamiento de Sonogashira implican la presencia de un catalizador de paladio, tal como un catalizador de Pd(0), p. ej., Pd(PPh3)4 o Pd2(dba)3 y una sal de cobre(I), tal como un haluro de cobre, tal como CuI, opcionalmente en presencia de uno o más coadyuvantes de reacción, tales como una base, típicamente una base de amina, tal como dietilamina o trietilamina o carbonato de potasio o carbonato de cesio, opcionalmente en presencia de un disolvente, p. ej. DMF o un disolvente de éter. La reacción se agita a una temperatura de aproximadamente temperatura ambiente -130°C, típicamente 80°C . La reacción puede llevarse a cabo bajo un gas inerte tal como nitrógeno o argón. La etapa b1 es seguida opcionalmente por etapas de funcionalización o etapas de ajuste de grupo funcional b1'.
El compuesto de fórmula C se obtiene a través de la etapa a de hacer reaccionar un compuesto de fórmula (A),

en donde los sustituyentes son como se definieron arriba;
con un compuesto de fórmula (B),
COOH
PG‘
(B)
en donde los sustituyentes son como se definieron arriba;
seguido de reacción con R1’-NH2 ,
en donde R1’ es como se define arriba;
en donde en una realización, la etapa a se lleva a cabo en presencia de piridina y fosfito de trifenilo, a temperaturas elevadas, tales como 50-100°C , típicamente 70°C durante 0,5 a 30 h, por ejemplo, durante 2 a 18 h;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional a’.
En otra realización, el compuesto de fórmula (I) se obtiene mediante la etapa de reacción de acoplamiento d1 de un compuesto de fórmula (E),

R1 ’ es R1, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R1mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R2’ es R2 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R2mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R5 ’ es R5 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R5mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R6’ es R6 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R6 mediante etapas de funcionalización o etapas de ajuste de grupo funcional; y
L’ se muestra arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a L mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
con
R3 ’-Hal
en donde
R3’ es R3 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R3 mediante etapas de funcionalización o etapas de ajuste de grupo funcional; y
Hal representa halógeno, tal como cloro, bromo o yodo;
en donde en una realización, la reacción de acoplamiento se lleva a cabo en presencia de una base de amina, tal como N,N-diisopropiletilamina. La reacción se lleva a cabo en presencia de un disolvente orgánico, tal como un alcohol, bajo calentamiento por microondas o calentamiento convencional en un baño de aceite en un intervalo de temperaturas de 120-160°C durante 30 minutos a 6 días;
Alternativamente, la reacción se lleva a cabo bajo las condiciones habituales de Buchwald-Hartwig utilizando una combinación adecuada de catalizador de Pd/ligando tal como Pd2(dba)3/2-(diciclohexilfosfino)bifenilo o Pd2(dba)3/2-diciclohexilfosfino-2’,4’,6’-triisopropil-bifenilo, Pd2(dba)3/XPhos, Pd2(dba)3/(rac)-BINAP, Pd(OAc)2/(rac)-BINAP o bis(tri-tbutilfosfina)paladio y una base adecuada, tal como NaOtBu, Cs2CO3 o K3PO4 y disolventes orgánicos, tales como tolueno, dioxano o THF. La reacción se agita a una temperatura de aproximadamente 60-140°C, por ejemplo a 100°C hasta 110°C y se realiza opcionalmente en un reactor de microondas. La reacción se lleva a cabo preferiblemente bajo un gas inerte, tal como nitrógeno o argón;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional d1’.
El compuesto de fórmula (E) se obtiene mediante la etapa de acoplamiento c2 del compuesto de fórmula (F),

otros sustituyentes son como se definieron arriba;
con
R2'-Y
en donde, cuando R2' es heteroarilo C4-C7 , como se definió arriba como R2 para un compuesto de fórmula (Ib), o un sustituyente que puede ser transferido en heteroarilo C4-C7 , a través de etapas de funcionalización o etapas de ajuste de grupo funcional.
Y representa un residuo de ácido borónico o un borolanilo cíclico o acíclico, tal como -B(OH)2 o pinaccolato-boro; o un alquilestannilo, tal como tributilestannilo.
Y es H (para un alquino terminal) o Y representa un alquilestannilo (alquino no terminal) en condiciones de reacción habituales de la reacción de Suzuki (Y es un residuo de ácido borónico o un éster de boronato cíclico o acíclico), la reacción de Stille (Y es alquilestannilo) o el acoplamiento de Sonogashira (Y es H de un alquino terminal), condiciones de reacción típicas se conocen en el sector y pueden aplicarse al presente procedimiento. Condiciones típicas para la reacción de Suzuki implican, por ejemplo, la presencia de un catalizador de paladio, tal como un catalizador de Pd 0), p. ej., Pd(PPh3)4 , o un catalizador de Pd(II), tal como PdCh(PPh3)2 ; Pd(OAc)2 , opcionalmente en presencia de un ligando de fosfina tal como BrettPhos; o paladaciclo Brettphos, opcionalmente en presencia de uno o más coadyuvantes de reacción, tales como una base, p. ej., NaOEt, Na2CO3 sólido o en solución acuosa, opcionalmente en presencia de uno o más diluyentes, particularmente disolventes polares, p. ej., benceno, tolueno, acetonitrilo o N-metil-2-pirrolidona. La reacción se agita a una temperatura de aproximadamente 100-180°C, p. ej., en un horno microondas. La reacción puede llevarse a cabo bajo un gas inerte tal como nitrógeno o argón. Condiciones típicas para la reacción de Stille implican, por ejemplo, la presencia de un catalizador de paladio, tal como un catalizador de Pd(0), p. ej., bis(tri-t-butilfosfina)paladio o Pd2(dba)3 , opcionalmente en presencia de uno o más auxiliares de la reacción, tales como cloruro de litio, en presencia de un disolvente, p. ej., DMF. La reacción se agita a una temperatura de aproximadamente 100-160°C. La reacción se lleva a cabo bajo un gas inerte, tal como nitrógeno o argón. Condiciones típicas para el acoplamiento de Sonogashira implican la presencia de un catalizador de paladio, tal como un catalizador de Pd(0), p. ej., Pd(PPh3)4 o Pd2(dba)3 y una sal de cobre(I), tal como un haluro de cobre, tal como CuI, opcionalmente en presencia de uno o más coadyuvantes de reacción, tales como una base, típicamente una base de amina, tal como dietilamina o trietilamina o carbonato de potasio o carbonato de cesio, opcionalmente en presencia de un disolvente, p. ej. DMF o un disolvente de éter. La reacción se agita a una temperatura de aproximadamente 60-130°C . La reacción puede llevarse a cabo bajo un gas inerte, tal como nitrógeno o argón.
La etapa c2 es seguida opcionalmente por etapas de funcionalización o etapas de ajuste de grupo funcional c2'.
El compuesto de fórmula (F) se obtiene a través de la etapa b2 de desproteger PG del compuesto de fórmula (C),

en donde PG representa un grupo protector adecuado, tal como un grupo Boc, y los otros sustituyentes son como se definieron arriba;
en donde en una realización, en que PG es un grupo Boc, la reacción de desprotección se lleva a cabo en un disolvente orgánico, tal como THF o DCM, en presencia de un ácido orgánico, tal como ácido trifluoroacético, o en presencia de un ácido inorgánico, tal como HCl o H3PO4 , opcionalmente en presencia de agua. La reacción se agita a temperatura ambiente durante aproximadamente 60-140 ° C, por ejemplo, a 100°C hasta 110°C;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional b2'.
El compuesto de fórmula (C) se obtiene a través de la etapa a de hacer reaccionar un compuesto de fórmula (A),

en donde los sustituyentes son como se definieron arriba;
con un compuesto de fórmula (B),
COOH

(B)
en donde los sustituyentes son como se definieron arriba;
seguido de reacción con R1'-NH2 ,
en donde R1 ’ es como se define arriba;
en donde en una realización, la etapa a se lleva a cabo en presencia de piridina y fosfito de trifenilo, a temperaturas elevadas, tales como 50-100°C durante 0,5 a 30 h, por ejemplo, durante 2 a 18 h;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional a'.
En otra realización, el compuesto de fórmula (I) se obtiene mediante la etapa de reacción de acoplamiento d2 de un

Hal1 representa un halógeno, tal como un bromuro o un cloruro, o un pseudohalógeno, tal como un triflato;
R1 ' es R1, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R1mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R5' es R5 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R5mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R6' es R6 , según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R6 mediante etapas de funcionalización o etapas de ajuste de grupo funcional; y
L’ es L como se define arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a L mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
con
R2’-Y
en donde, cuando R2' es heteroarilo C4-C7 , como se definió arriba como R2 para un compuesto de fórmula (Ib), o un sustituyente que puede ser transferido en heteroarilo C4-C7 , a través de etapas de funcionalización o etapas de ajuste de grupo funcional.
Y representa un residuo de ácido borónico o un borolanilo cíclico o acíclico, tal como -B(OH)2 o pinaccolato-boro; o un alquilestannilo, tal como tributilestannilo.
Y es H (para un alquino terminal) o Y representa un alquilestannilo (alquino no terminal) en condiciones de reacción habituales de la reacción de Suzuki (Y es un residuo de ácido borónico o un éster de boronato cíclico o acíclico), la reacción de Stille (Y es alquilestannilo) o el acoplamiento de Sonogashira (Y es H de un alquino terminal), condiciones de reacción típicas se conocen en el sector y pueden aplicarse al presente procedimiento. Condiciones típicas para la reacción de Suzuki implican, por ejemplo, la presencia de un catalizador de paladio, tal como un catalizador de Pd 0), p. ej., Pd(PPh3)4 , o un catalizador de Pd(II), tal como PdCl2(PPh3)2 ; Pd(OAc)2 , opcionalmente en presencia de un ligando de fosfina tal como BrettPhos; o paladaciclo Brettphos, opcionalmente en presencia de uno o más coadyuvantes de reacción, tales como una base, p. ej., NaOEt, Na2CO3 sólido o en solución acuosa, opcionalmente en presencia de uno o más diluyentes, particularmente disolventes polares, p. ej., benceno, tolueno, acetonitrilo o N-metil-2-pirrolidona. La reacción se agita a una temperatura de aproximadamente 100-180°C, p. ej., en un horno microondas. La reacción puede llevarse a cabo bajo un gas inerte tal como nitrógeno o argón. Condiciones típicas para la reacción de Stille implican, por ejemplo,
la presencia de un catalizador de paladio, tal como un catalizador de Pd(0), p. ej., bis(tri-t-butilfosfina)paladio o Pd2(dba)3, opcionalmente en presencia de uno o más auxiliares de la reacción, tales como cloruro de litio, en presencia de un disolvente, p. ej., DMF. La reacción se agita a una temperatura de aproximadamente 100-160°C. La reacción se lleva a cabo bajo un gas inerte, tal como nitrógeno o argón. Condiciones típicas para el acoplamiento de Sonogashira implican la presencia de un catalizador de paladio, tal como un catalizador de Pd(0), p. ej., Pd(PPh3)4 o Pd2(dba)3 y una sal de cobre(I), tal como un haluro de cobre, tal como CuI, opcionalmente en presencia de uno o más coadyuvantes de reacción, tales como una base, típicamente una base de amina, tal como dietilamina o trietilamina o carbonato de potasio o carbonato de cesio, opcionalmente en presencia de un disolvente, p. ej. DMF o un disolvente de éter. La reacción se agita a una temperatura de aproximadamente 60-130°C . La reacción puede llevarse a cabo bajo un gas inerte, tal como nitrógeno o argón.
La etapa c2 es seguida opcionalmente por etapas de funcionalización o etapas de ajuste de grupo funcional d2'.
El compuesto de fórmula (G) se obtiene mediante la etapa de acoplamiento c3 del compuesto de fórmula (F),

en donde los sustituyentes son como se definieron arriba;
con
R3 '-Hal
en donde
R3' es R3, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R3 mediante etapas de funcionalización o etapas de ajuste de grupo funcional; y
Hal representa halógeno, tal como cloro, bromo o yodo;
en donde en una realización, la reacción de acoplamiento se lleva a cabo en presencia de una base de amina, tal como N,N-diisopropiletilamina. La reacción se lleva a cabo en presencia de un disolvente orgánico, tal como un alcohol, bajo calentamiento por microondas o calentamiento convencional en un baño de aceite en un intervalo de temperaturas de 120-160°C durante 30 minutos a 6 días;
Alternativamente, la reacción se lleva a cabo bajo las condiciones habituales de Buchwald-Hartwig utilizando una combinación adecuada de catalizador de Pd/ligando tal como Pd2(dba)3/2-(diciclohexilfosfino)bifenilo o Pd2(dba)3/2-diciclohexilfosfino-2',4',6'-triisopropil-bifenilo, Pd2(dba)3/XPhos, Pd2(dba)3/(rac)-BINAP, Pd(OAc)2/(rac)-BINAP o bis(tri-tbutilfosfina)paladio y una base adecuada, tal como NaOtBu, Cs2CO3 o K3PO4 y disolventes orgánicos, tales como tolueno, dioxano o THF. La reacción se agita a una temperatura de aproximadamente 60-140°C, por ejemplo a 100°C hasta 110°C y se realiza opcionalmente en un reactor de microondas. La reacción se lleva a cabo preferiblemente bajo un gas inerte, tal como nitrógeno o argón;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional c3'.
El compuesto de fórmula (F) se obtiene a través de la etapa b2 de desproteger PG del compuesto de fórmula (C),
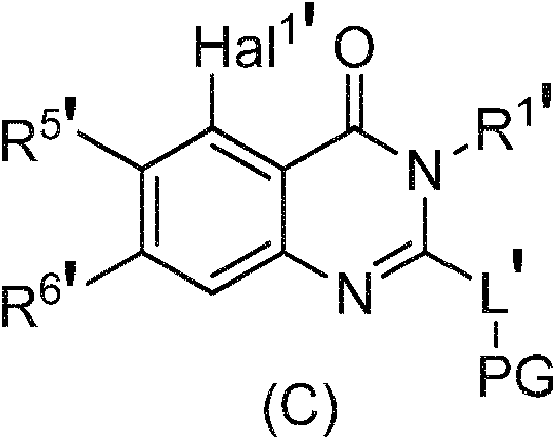
en donde PG representa un grupo protector adecuado, tal como un grupo Boc, y los otros sustituyentes son como se definieron arriba;
en donde en una realización, en que PG es un grupo Boc, la reacción de desprotección se lleva a cabo en un disolvente orgánico, tal como THF o DCM, en presencia de un ácido orgánico, tal como ácido trifluoroacético, o en presencia de un ácido inorgánico, tal como HCl o H3PO4 , opcionalmente en presencia de agua. La reacción se agita a temperatura ambiente durante aproximadamente 60-140 ° C, por ejemplo, a 100°C hasta 110°C;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional b2'.
El compuesto de fórmula (C) se obtiene a través de la etapa a de hacer reaccionar un compuesto de fórmula (A),

en donde los sustituyentes son como se definieron arriba;
con un compuesto de fórmula (B),
COOH
,L '
PG
(B)
en donde los sustituyentes son como se definieron arriba;
seguido de reacción con R1’-NH2 ,
en donde R1’ es como se define arriba;
en donde en una realización, la etapa a se lleva a cabo en presencia de piridina y fosfito de trifenilo, a temperaturas elevadas, tales como 50-100°C durante 0,5 a 30 h, por ejemplo, durante 2 a 18 h;
opcionalmente seguido por etapas de funcionalización o etapas de ajuste de grupo funcional a’.
La expresión "grupo protector", tal como se utiliza en la presente, se refiere a un grupo que protege un grupo funcional que está presente en los materiales de partida y no está destinado a participar en la reacción. En etapas adicionales del procedimiento, llevadas a cabo según se desee, los grupos funcionales de los compuestos de partida que no deberían participar en la reacción pueden estar presentes en forma desprotegida o pueden estar protegidos, por ejemplo, por uno o más grupos protectores. Los grupos protectores se separan luego total o parcialmente de acuerdo con uno de los métodos conocidos. Los grupos protectores y la forma en que se introducen y separan se describen, por ejemplo, en "Protective Groups in Organic Chemistry", Plenum Press, Londres, Nueva York 1973, y en "Methoden der organischen Chemie", Houben-Weyl, 4a edición. Vol. 15/1, Georg-Thieme-Verlag, Stuttgart 1974 y en Theodora W. Greene, "Protective Groups in Organic Synthesis", John Wiley & Sons, Nueva York 1981. Una característica de los grupos protectores es que pueden separarse fácilmente, es decir, sin la aparición de reacciones secundarias no deseadas, por ejemplo mediante solvolisis, reducción, fotólisis o, alternativamente, en condiciones fisiológicas.
Los intermedios y productos finales se pueden tratar y/o purificar de acuerdo con métodos estándar,p. ej., utilizando métodos cromatográficos, métodos de distribución, (re)cristalización y similares.
Lo siguiente se aplica en general a todos los procesos mencionados en la presente precedentemente y en lo sucesivo.
Todos las etapas de los procedimientos mencionadas anteriormente se pueden llevar a cabo en condiciones de reacción que son conocidas por los expertos en la técnica, que incluyen las mencionadas específicamente, en ausencia o, normalmente, en presencia de disolventes o diluyentes, incluidos, por ejemplo, los disolventes o diluyentes que son inertes frente a los reactivos utilizados y los disuelven, en ausencia o presencia de catalizadores, agentes de condensación o de neutralización, por ejemplo, intercambiadores de iones, tales como intercambiadores de cationes, p. ej., en la forma de H+, dependiendo de la naturaleza de la reacción y/o de los reactivos, a temperatura reducida, normal o elevada, por ejemplo, en un intervalo de temperaturas de aproximadamente -100 °C a aproximadamente 190 °C, que incluye, por ejemplo, de aproximadamente -80 °C a aproximadamente 150 °C, por ejemplo, de -80 a -60 °C, a temperatura ambiente, a una temperatura de -20 a 40 °C o a temperatura de reflujo, a presión atmosférica o en un recipiente cerrado, cuando corresponda bajo presión y/o en atmósfera inerte, por ejemplo, en atmósfera de argón o nitrógeno.
En todas las etapas de las reacciones, mezclas de isómeros que se forman pueden separarse en los isómeros individuales, por ejemplo diastereoisómeros o enantiómeros, o en cualquier mezcla deseada de isómeros, por ejemplo racematos o mezclas de diastereoisómeros, por ejemplo de manera análoga a los métodos descritos arriba en la presente.
Los disolventes a partir de los cuales se pueden seleccionar aquellos disolventes que son adecuados para cualquier reacción particular incluyen los mencionados específicamente o, por ejemplo, agua, ésteres, tales como alcanoatos C1-C8 de alquilo C1-C8, por ejemplo acetato de acetato, éteres, tales como éteres alifáticos, por ejemplo, dietiléter, o éteres cíclicos, por ejemplo, tetrahidrofurano o dioxano, hidrocarburos aromáticos líquidos, tales como benceno o tolueno, alcoholes, tales como metanol, etanol o 1- o 2-propanol, nitrilos, tales como acetonitrilo, hidrocarburos halogenados, tales como cloruro de metileno o cloroformo, amidas de ácido, tales como dimetilformamida o dimetil acetamida, bases, tales
como bases nitrogenadas heterocíclicas, por ejemplo piridina o N-metilpirrolidin-2-ona, anhídridos de ácido carboxílico, tales como anhídridos de ácido alcanoico C1-C8, por ejemplo anhídrido acético, hidrocarburos cíclicos, lineales o ramificados, tales como ciclohexano, hexano o isopentano, metilciclohexano o mezclas de esos disolventes, por ejemplo soluciones acuosas, a menos que se indique lo contrario en la descripción de los procedimientos. Dichas mezclas de disolventes también se pueden utilizar en el procesamiento, por ejemplo, mediante cromatografía o partición.
Los compuestos, incluidas sus sales, también se pueden obtener en forma de hidratos, o sus cristales pueden, por ejemplo, incluir el disolvente utilizado para la cristalización. Pueden estar presentes diferentes formas cristalinas.
En otro aspecto, la presente invención describe un compuesto intermedio de fórmula ( E )

R1’ es R1, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R1mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R2’ es R2, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R2mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R5’ es R5, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R5mediante etapas de funcionalización o etapas de ajuste de grupo funcional;
R6’ es R6, según se definió arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a R6 mediante etapas de funcionalización o etapas de ajuste de grupo funcional; y
L’ es como se muestra arriba para un compuesto de fórmula (Ib), o un sustituyente que puede transferirse a L mediante etapas de funcionalización o etapas de ajuste de grupo funcional.
En otro aspecto, la presente invención proporciona un compuesto intermedio de fórmula (D),

en donde PG representa un grupo protector adecuado, tal como un grupo Boc, y los otros sustituyentes son como se definieron arriba.
En otro aspecto, la presente invención proporciona un compuesto intermedio de fórmula (C),

en donde Hal1 representa un halógeno, tal como un bromuro o un cloruro, o un pseudohalógeno, tal como un triflato, y los otros sustituyentes son como se definieron arriba.
En otro aspecto, la presente invención proporciona un compuesto intermedio de fórmula (F),
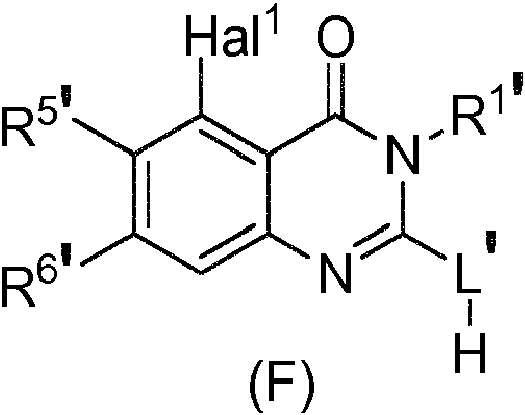
en donde los sustituyentes son como se definieron arriba.
En otro aspecto, la presente invención proporciona un compuesto intermedio de fórmula (G),

en donde los sustituyentes son como se definieron arriba.
La invención incluye además cualquier variante de los procesos de la presente, en la que se utilice un producto intermedio que se pueda obtener en cualquier etapa de estos como material de partida y se lleven a cabo los pasos restantes, o en la que los materiales de partida se formen in situ en las condiciones de reacción, o en la que los componentes de reacción se utilicen en forma de sus sales o material ópticamente puro.
Los compuestos de la invención y los intermedios también se pueden convertir los unos en los otros de acuerdo con métodos generalmente conocidos por los expertos en la técnica
En otro aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto de la presente invención, o una sal farmacéuticamente aceptable de este, y un portador farmacéuticamente aceptable. En una realización adicional, la composición comprende al menos dos portadores farmacéuticamente aceptables tales como los descritos en la presente. A efectos de la presente invención, a menos que se designen de otro modo, los solvatos e hidratos se consideran, por lo general, composiciones. Preferentemente, los portadores farmacéuticamente aceptables son estériles. La composición farmacéutica puede formularse para vías particulares de administración tales como administración oral, administración parenteral,y la administración rectal, etc . Además, las composiciones farmacéuticas de la presente invención se pueden preparar en forma sólida (incluyendo, sin limitación, cápsulas, comprimidos, píldoras, gránulos, polvos o supositorios), o en forma líquida (incluyendo, sin limitación, soluciones, suspensiones o emulsiones). Las composiciones farmacéuticas pueden ser sometidas a las operaciones farmacéuticas convencionales como esterilización y/o pueden contener diluyentes inertes, agentes lubricantes o agentes tamponantes convencionales así como adyuvantes, tales como conservantes, estabilizantes, humectantes, emulsionantes y tampones, etc.
Habitualmente, las composiciones farmacéuticas son comprimidos o cápsulas de gelatina que contienen el principio activo junto con uno o más de:
a) diluyentes, p. ej., lactosa, dextrosa, sacarosa, manitol, sorbitol, celulosa y/o glicina;
b) lubricantes, p. ej., sílice, talco, ácido esteárico, su sal de magnesio o calcio y/o polietilenglicol; para comprimidos también
c) aglutinantes, p. ej., silicato de magnesio y aluminio, pasta de almidón, gelatina, tragacanto, metilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona; si se desea
d) desintegrantes, p. ej., almidones, agar, ácido algínico o su sal de sodio, o mezclas efervescentes; y
e) absorbentes, colorantes, aromas y edulcorantes.
Los comprimidos pueden estar recubiertos con película o con recubrimiento entérico de acuerdo con métodos conocidos en la técnica.
Composiciones adecuadas para la administración oral incluyen una cantidad eficaz de un compuesto de la invención en forma de comprimidos, grageas, suspensiones acuosas u oleosas, polvos o gránulos dispersables, emulsiones, cápsulas duras o blandas, o jarabes o elixires. Composiciones destinadas para uso oral se preparan de acuerdo con cualquier método conocido en la técnica para la fabricación de composiciones farmacéuticas y composiciones de este tipo pueden contener uno o más agentes seleccionados del grupo que consiste en agentes edulcorantes, agentes saborizantes, agentes colorantes y agentes conservantes con el fin de proporcionar preparaciones farmacéuticamente elegantes y
sabrosas . Los comprimidos pueden contener el principio activo mezclado con excipientes atóxicos, farmacéuticamente aceptables, que sean adecuados para la elaboración de comprimidos. Estos excipientes son, por ejemplo, diluyentes inertes tales como carbonato de calcio, carbonato de sodio, lactosa, fosfato de calcio o fosfato de sodio; agentes granulantes y desintegrantes, por ejemplo, almidón de maíz o ácido algínico; agentes aglutinantes, por ejemplo, almidón, gelatina o goma arábiga; y agentes lubricantes, por ejemplo, estearato de magnesio, ácido esteárico o talco. Los comprimidos no están recubiertos o se recubren mediante técnicas conocidas para retrasar la desintegración y la absorción en el tubo digestivo y, por lo tanto, proporcionar una acción sostenida durante un período más prolongado. Por ejemplo, se puede utilizar un material retardante tal como monoestearato de glicerilo o diestearato de glicerilo. Formulaciones para uso oral pueden presentarse en forma de cápsulas de gelatina dura en las que el principio activo se mezcla con un diluyente sólido inerte, por ejemplo, carbonato de calcio, fosfato de calcio o caolín, o en forma de cápsulas de gelatina blanda en las que el ingrediente activo se mezcla con agua o un medio oleoso, por ejemplo, aceite de cacahuete, parafina líquida o aceite de oliva.
Determinadas composiciones inyectables son soluciones o suspensiones isotónicas acuosas, y se preparan ventajosamente supositorios a partir de emulsiones o suspensiones grasas. Dichas composiciones pueden esterilizarse y/o contener adyuvantes tales como agentes conservantes, estabilizantes, humectantes o emulsionantes, promotores de la solución, sales para regular la presión osmótica y/o tampones. Además, también pueden contener otras sustancias valiosas desde el punto de vista terapéutico. Dichas composiciones se preparan de acuerdo con métodos convencionales de mezcla, granulación o recubrimiento, respectivamente, y contienen aproximadamente un 0,1-75%, o contienen aproximadamente un 1-50%, del principio activo.
Las composiciones adecuadas para la aplicación transdérmica incluyen una cantidad eficaz de un compuesto de la invención con un portador adecuado. Los portadores adecuados para el suministro transdérmico incluyen disolventes absorbibles farmacológicamente aceptables para facilitar el paso a través de la piel del hospedador. Por ejemplo, los dispositivos transdérmicos tienen la forma de un vendaje que comprende un miembro de respaldo, un depósito que contiene el compuesto opcionalmente con portadores, opcionalmente una barrera de control de la velocidad para suministrar el compuesto a la piel del huésped a una velocidad controlada y predeterminada a lo largo de un período prolongado de tiempo y medios para asegurar el dispositivo a la piel.
Composiciones adecuadas para aplicación tópica, p. ej., a la piel y los ojos, incluyen soluciones acuosas, suspensiones, pomadas, cremas, geles o formulaciones pulverizables, p. ej., para el suministro por aerosol o similares. Sistemas de suministro tópico de este tipo serán particularmente apropiados para la aplicación dérmica, p. ej., para el tratamiento del cáncer de piel, p. ej., para el uso profiláctico en cremas de protección solar, lociones, aerosoles y similares. Por tanto, son particularmente adecuados para su uso en formulaciones tópicas, incluidas las cosméticas, muy conocidas en la técnica. Estas pueden contener solubilizantes, estabilizantes, agentes potenciadores de la tonicidad, tampones y conservantes.
Tal como se utiliza en la presente, una aplicación tópica también puede referirse a una inhalación o a una aplicación intranasal. Se pueden suministrar convenientemente en forma de un polvo seco (ya sea solo, como una mezcla, por ejemplo, una mezcla seca con lactosa, o una partícula de componentes mixtos, por ejemplo con fosfolípidos) desde un inhalador de polvo seco o una presentación en aerosol de un recipiente presurizado, bomba, pulverizador, atomizador o nebulizador, con o sin el uso de un propulsor adecuado.
Los compuestos de fórmula (Ib) en forma libre o en forma de sal farmacéuticamente aceptable exhiben propiedades farmacológicas valiosas, p. ej., propiedades de modulación de quinasa PI3 de clase I, p. ej., como se indica en ensayos in vitro e in vivo según se proporciona en las siguientes secciones y, por lo tanto, indicado para terapia o para uso como productos químicos de investigación, p. ej., como compuestos de herramientas.
Compuestos de la invención pueden ser útiles en el tratamiento de afecciones, enfermedades o trastornos que incluyen inmunopatología asociada a enfermedades o infecciones en las que una o más de las funciones de células B, tales como producción de anticuerpos, presentación de antígenos, producción de citoquinas u organogénesis linfoide son anormales o son indeseables, incluyendo artritis reumatoide y enfermedades relacionadas (tales como espondilitis anquilosante, artritis psoriásica, artritis juvenil), pénfigo vulgar y enfermedades relacionadas, púrpura idiopática trombocitopenia, lupus eritematoso sistémico, esclerosis múltiple, miastenia grave, síndrome de Sjogren (tal como síndrome de Sjogren primario (SS)), enfermedad de injerto contra huésped, anemia hemolítica autoinmune, vasculitis asociada a ANCA (tal como enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss), crioglobulinemia, púrpura trombocitopénica trombótica, lesión por isquemia-reperfusión, urticaria autoinmune crónica, alergia (dermatitis atópica, dermatitis de contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, diferentes tipos de glomerulonefritis, AMR (rechazo de trasplante mediado por anticuerpos), rechazo de trasplante hiperagudo, agudo y crónico mediado por células B y cánceres de origen hematopoyético, incluyendo pero no limitados a mieloma múltiple; leucemia mielógena aguda; leucemia mielógena crónica; leucemia linfocítica; leucemia mieloide; linfoma no Hodgkin; linfomas, policitemia vera; trombocitemia esencial; mielofibrosis con metaplasia mieloide; y enfermedad de Waldenstroem.
La invención incluye métodos para tratar afecciones, enfermedades o trastornos en los que una o más de las funciones de neutrófilos, tales como la liberación de superóxido, la exocitosis estimulada o la migración quimioatractica, son anormales o indeseables, incluyendo artritis reumatoide, trastornos pulmonares o respiratorios tales como el asma. dermatosis inflamatorias tales como la psoriasis, así como en inmunopatología asociada a enfermedades o infecciones y otras.
La invención incluye métodos para tratar afecciones, enfermedades o trastornos en los que una o más de las funciones de los basófilos y mastocitos, tales como la migración quimioatractica o la desgranulación mediada por alérgeno-IgE, son anormales o indeseables, incluyendo enfermedades alérgicas (dermatitis atópica, dermatitis de contacto, rinitis alérgica, asma alérgico, asma asociado con rinitis alérgica, urticaria alérgica crónica), así como otros trastornos tales como COPD, asma o enfisema.
La invención incluye métodos para tratar afecciones, enfermedades o trastornos en los que una o más de las funciones de las células T , tales como la producción de citoquinas o la citotoxicidad mediada por células, son anormales o indeseables, incluidas artritis reumatoide, esclerosis múltiple, rechazo agudo o crónico del tejido celular o injertos de órganos o cánceres de origen hematopoyético, así como en inmunopatología asociada a enfermedades o infecciones.
Además, la invención incluye métodos para tratar enfermedades neurodegenerativas, enfermedades cardiovasculares y agregación plaquetaria.
Además, la invención incluye métodos para tratar enfermedades de la piel tales como porfiria cutánea tardía, erupción de luz polimorfa, dermatomiositis, urticaria solar, liquen plano oral, paniculitis, esclerodermia, vasculitis urticaria.
Además, la invención incluye métodos para tratar enfermedades inflamatorias crónicas tales como sarcoidosis, granuloma anular.
En otras realizaciones, la afección o trastorno (p. ej., mediado por quinasas PI3 de clase I) se selecciona del grupo que consiste en: policitemia vera, trombocitemia esencial, mielofibrosis con metaplasia mieloide, asma, COPD, ARDs , síndrome de Loffler, neumonía eosinofílica, parasitaria (en particular, infestación de metazoos) (incluida la eosinofilia tropical), aspergilosis broncopulmonar, poliarteritis nodosa (incluido el síndrome de Churg-Strauss), granuloma eosinofílico, trastornos relacionados con eosinófilos que afectan las vías respiratorias ocasionadas por la reacción al fármaco, psoriasis, dermatitis de contacto, dermatitis atópica, alopecia areata, eritema multiforme, dermatitis herpetiforme, escleroderma, vitíligo, angiitis de hipersensibilidad, urticaria, penfigoide bulloso, lupus eritematoso, pénfigo, epidermólisis bullosa adquirida, trastornos hematológicos autoinmunes (p. ej., anemia hemolítica, anemia aplásica, anemia de glóbulos rojos pura y trombocitopenia idiopática), lupus sistémico eritematoso, policondritis, esclerodermia, granulomatosis de Wegener, dermatomiositis, hepatitis crónica activa, miastenia grave, síndrome de Steven-Johnson, enfermedad idiopática, enfermedad inflamatoria intestinal autoinmune (p. ej., colitis ulcerosa y enfermedad de Crohn), oftalmopatía endocrina, enfermedad de Grave, sarcoidosis, alveolitis, neumonitis por hipersensibilidad crónica, esclerosis múltiple, cirrosis biliar primaria, uveítis (anterior y posterior), fibrosis pulmonar intersticial, artritis psoriásica, glomerulonefritis, enfermedades cardiovasculares, aterosclerosis, hipertensión, trombosis venosa profunda, apoplejía, infarto de miocardio, angina inestable, tromboembolia, embolia pulmonar, enfermedades trombolíticas, arteriopatía aguda , oclusiones trombóticas periféricas y enfermedad de la arteria coronaria, lesiones por reperfusión, retinopatía, tales como la retinopatía diabética o la retinopatía hiperbárica inducida por oxígeno.
En otra realización, los compuestos de la presente invención son útiles en el tratamiento, la prevención o la mejora de enfermedades autoinmunes y de afecciones inflamatorias, en particular afecciones inflamatorias con una etiología que incluye un componente autoinmune, tal como artritis (por ejemplo, artritis reumatoide, artritis crónica. progrediente y artritis deformante) y enfermedades reumáticas, incluidas afecciones inflamatorias y enfermedades reumáticas que implican pérdida ósea, dolor inflamatorio, espondiloartropatías, incluyendo espondilitis anquilosante, síndrome de Reiter, artritis reactiva, artritis psoriásica y artritis enteropática, hipersensibilidad (incluyendo hipersensibilidad de las vías respiratorias e hipersensibilidad dérmica) y alergias Enfermedades autoinmunes específicas para las que se pueden emplear los compuestos de la invención incluyen trastornos autoinmunes hematológicos (incluyendo, p. ej., anemia hemolítica, anemia aplásica, anemia pura de glóbulos rojos y trombocitopenia idiopática), hemofilia A adquirida, enfermedad de la aglutinina fría, crioglobulinemia, trombótica trombocitopénica púrpura, síndrome de Sjogren, lupus eritematoso sistémico, trastornos musculares inflamatorias, policondritis, esclerodoma, vasculitis asociadas a anticuerpos anti-citoplasma de neutrófilos, neuropatía mediada por IgM, síndrome de opsoclono-mioclono, granulomatosis de Wegener, dermatomiositis, hepatitis crónica activa, miastenia grave, psoriasis, síndrome de Steven-Johnson, pénfigo vulgar, pénfigo foliáceo, esprúe idiopático, enfermedad intestinal inflamatoria autoinmune (incluyendo, p. ej., colitis ulcerosa, enfermedad de Crohn y síndrome del intestino irritable), oftalmopatía endocrina, enfermedad, de Grave, sarcoidosis, esclerosis múltiple, neuromielitis óptica, cirrosis biliar primaria, diabetes juvenil (diabetes mellitus tipo I), uveítis (anterior, intermedia y posterior, así como panuveitis), queratoconjuntivitis seca y queratoconjuntivitis vernal, fibrosis pulmonar intersticial, artritis psoriásica, espondilitis anquilosante y glomerulonefritis (con y sin síndrome nefrótico, p. ej., incluyendo nefropatía idiopática, o nefropatía de cambio mínimo), tumores, enfermedad inflamatoria de la piel y la córnea, miositis, aflojamiento de implantes óseos, trastornos metabólicos, tales como aterosclerosis, diabetes y dislipidemia.
En otra realización, los compuestos de la presente invención son útiles en el tratamiento de afecciones o trastornos seleccionados del grupo que consiste en linfoma cutáneo primario de células B, enfermedad inmunobulosa, pénfigo vulgar, pénfigo foliáceo, forma endémica de pénfigo brasileño (Fogo selvagem), pénfigo paraneoplásico, penfigoide ampolloso, penfigoide de membranas mucosas, epidermolisis ampollosa adquirida, enfermedad crónica de injerto contra huésped, dermatomiositis, lupus eritematoso sistémico, vasculitis, vasculitis de vasos pequeños, vasculitis urticarial hipocomplementémica, vasculitis de anticuerpos citoplasmáticos antineutrofilos, crioglobulinemia, síndrome de Schnitzler, macroglobulinemia de Waldenstrom, angioedema, vitiligo, lupus eritematoso sistémico, púrpura trombocitopénica idiopática, esclerosis múltiple, enfermedad de la aglutinina fría, anemia hemolítica autoinmune, vasculitis asociadas a anticuerpos anti-citoplasma de neutrófilos, enfermedad de injerto contra huésped, crioglobulinemia y tromobicitopenia trombótica.
Por lo tanto, como una realización adicional, la presente invención proporciona el uso de un compuesto de la fórmula (Ib) en terapia. En una realización adicional, la terapia se selecciona de una enfermedad que puede ser tratada por la inhibición de PI3 quinasas de clase I. En otra realización, la enfermedad se selecciona de la lista anteriormente mencionada, adecuadamente de trastornos autoinmunes, enfermedades autoinflamatorias e inflamatorias, enfermedades alérgicas, enfermedades de las vías respiratorias, tales como asma y COPD, rechazo de trasplante; producción de anticuerpos, presentación de antígenos, producción de citoquinas u organogénesis linfoide son anormales o indeseables, incluyendo la artritis reumatoide y enfermedades relacionadas (tales como espondiloartritis anquilosante, artritis psoriásica, artritis juvenil), pénfigo vulgar y enfermedades relacionadas, púrpura trombocitopénica idiopática, lupus eritematoso sistémico, esclerosis múltiple, miastenia grave, síndrome de Sjogren (tal como síndrome de Sjogren primario (pSS)), enfermedad de injerto contra huésped, anemia hemolítica autoinmune, vasculitis asociada a ANCA (tal como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss) , crioglobulinemia, púrpura trombocitopénica trombótica, la lesión por isquemia-reperfusión, urticaria crónica autoinmune, alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, diferentes tipos de glomerulonefritis, AMR (rechazo de trasplante mediado por anticuerpos), rechazo de trasplante hiperagudo, agudo y crónico mediado por células B y cánceres de origen hematopoyético que incluyen, pero no se limitan a mieloma múltiple; una leucemia; leucemia mielógena aguda; leucemia mielógena crónica; leucemia linfocítica; leucemia mieloide; linfoma no Hodgkin; linfomas; policitemia vera; trombocitemia esencial; mielofibrosis con metaplasia mieloide; y enfermedad de Waldenstroem; más adecuadamente de artritis reumatoide (RA), pénfigo vulgar (PV), púrpura trombocitopénica idiopática (ITP), púrpura trombocitopénica trombótica (TTP), anemia hemolítica autoinmune (AIHA), hemofilia adquirida tipo A (AHA), lupus eritematoso sistémico (LSE), esclerosis múltiple (MS), miastenia grave (MG), síndrome de Sjogren (SS) (tal como síndrome de Sjogren primario (pSS)), vasculitis asociada a ANCA (tales como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss), crioglobulinemia, lesión por isquemiareperfusión, urticaria autoinmune crónica (CAU), alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, rechazo de trasplantes y cánceres de origen hematopoyético, así como en la enfermedad o infección asociada a inmunopatología, por ejemplo en malaria severa y cerebral, tripanosomiasis, leishmaniasis, toxoplasmosis y neurocisticercosis.
Por lo tanto, como una realización adicional, la presente invención proporciona un compuesto de la fórmula (Ib) para uso en terapia. En una realización adicional, la terapia se selecciona de una enfermedad que puede ser tratada por la inhibición de PI3 quinasas de clase I. En otra realización, la enfermedad se selecciona de la lista anteriormente mencionada, adecuadamente de trastornos autoinmunes, enfermedades autoinflamatorias e inflamatorias, enfermedades alérgicas, enfermedades de las vías respiratorias, tales como asma y COPD, rechazo de trasplante; producción de anticuerpos, presentación de antígenos, producción de citoquinas u organogénesis linfoide son anormales o indeseables, incluyendo la artritis reumatoide y enfermedades relacionadas (tales como espondiloartritis anquilosante, artritis psoriásica, artritis juvenil), pénfigo vulgar y enfermedades relacionadas, púrpura trombocitopénica idiopática, lupus eritematoso sistémico, esclerosis múltiple, miastenia grave, síndrome de Sjogren (tal como síndrome de Sjogren primario (pSS)), enfermedad de injerto contra huésped, anemia hemolítica autoinmune, vasculitis asociada a ANCA (tal como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss) , crioglobulinemia, púrpura trombocitopénica trombótica, la lesión por isquemia-reperfusión, urticaria crónica autoinmune, alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, diferentes tipos de glomerulonefritis, AMR (rechazo de trasplante mediado por anticuerpos), rechazo de trasplante hiperagudo, agudo y crónico mediado por células B y cánceres de origen hematopoyético que incluyen, pero no se limitan a mieloma múltiple; una leucemia; leucemia mielógena aguda; leucemia mielógena crónica; leucemia linfocítica; leucemia mieloide; linfoma no Hodgkin; linfomas; policitemia vera; trombocitemia esencial; mielofibrosis con metaplasia mieloide; y enfermedad de Waldenstroem; más adecuadamente de artritis reumatoide (RA), pénfigo vulgar (PV), púrpura trombocitopénica idiopática (ITP), púrpura trombocitopénica trombótica (TTP), anemia hemolítica autoinmune (AIHA), hemofilia adquirida tipo A (AHA), lupus eritematoso sistémico (LSE), esclerosis múltiple (MS), miastenia grave (MG), síndrome de Sjogren (SS) (tal como síndrome de Sjogren primario (pSS)), vasculitis asociada a ANCA (tales como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss), crioglobulinemia, lesión por isquemia-reperfusión, urticaria autoinmune crónica (CAU), alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, rechazo de trasplantes y cánceres de origen hematopoyético, así como en la enfermedad o infección asociada a inmunopatología, por ejemplo en malaria severa y cerebral, tripanosomiasis, leishmaniasis, toxoplasmosis y neurocisticercosis.
En otra realización, la invención proporciona un método para tratar una enfermedad que se trata mediante la inhibición de quinasas PI3 de clase I que comprende la administración de una cantidad terapéuticamente aceptable de un compuesto de la fórmula (Ib). En una realización adicional, la enfermedad se selecciona de la lista anteriormente mencionada, adecuadamente de trastornos autoinmunes, enfermedades autoinflamatorias e inflamatorias, enfermedades alérgicas, enfermedades de las vías respiratorias, tales como asma y COPD, rechazo de trasplante; producción de anticuerpos, presentación de antígenos, producción de citoquinas u organogénesis linfoide son anormales o indeseables, incluyendo la artritis reumatoide y enfermedades relacionadas (tales como espondiloartritis anquilosante, artritis psoriásica, artritis juvenil), pénfigo vulgar y enfermedades relacionadas, púrpura trombocitopénica idiopática, lupus eritematoso sistémico, esclerosis múltiple, miastenia grave, síndrome de Sjogren (tal como síndrome de Sjogren primario (pSS)), enfermedad de injerto contra huésped, anemia hemolítica autoinmune, vasculitis asociada a ANCA (tal como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss), crioglobulinemia, púrpura trombocitopénica trombótica, la lesión por isquemia-reperfusión, urticaria crónica autoinmune, alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, diferentes tipos de glomerulonefritis, AMR (rechazo de trasplante mediado por anticuerpos), rechazo de trasplante hiperagudo, agudo y crónico mediado por células B y cánceres de origen hematopoyético que incluyen, pero no se limitan a mieloma múltiple; una leucemia; leucemia mielógena aguda; leucemia mielógena crónica; leucemia linfocítica; leucemia mieloide; linfoma no Hodgkin; linfomas; policitemia vera; trombocitemia esencial; mielofibrosis con metaplasia mieloide; y enfermedad de Waldenstroem; más adecuadamente de artritis reumatoide (RA), pénfigo vulgar (PV), púrpura trombocitopénica idiopática (ITP), púrpura trombocitopénica trombótica (TTP), anemia hemolítica autoinmune (AIHA), hemofilia adquirida tipo A (AHA), lupus eritematoso sistémico (LSE), esclerosis múltiple (MS), miastenia grave (MG), síndrome de Sjogren (SS) (tal como síndrome de Sjogren primario (pSS)), vasculitis asociada a ANCA (tales como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss), crioglobulinemia, lesión por isquemia-reperfusión, urticaria autoinmune crónica (CAU), alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, rechazo de trasplantes y cánceres de origen hematopoyético, así como en la enfermedad o infección asociada a inmunopatología, por ejemplo en malaria severa y cerebral, tripanosomiasis, leishmaniasis, toxoplasmosis y neurocisticercosis.
Por lo tanto, como una realización adicional, la presente invención proporciona el uso de un compuesto de la fórmula (Ib) para la fabricación de un medicamento. En una realización adicional, el medicamento es para el tratamiento de una enfermedad que puede ser tratada inhibición de quinasas PI3 de clase I. En otra realización, la enfermedad se selecciona de la lista anteriormente mencionada, adecuadamente de trastornos autoinmunes, enfermedades autoinflamatorias e inflamatorias, enfermedades alérgicas, enfermedades de las vías respiratorias, tales como asma y COPD, rechazo de trasplante; producción de anticuerpos, presentación de antígenos, producción de citoquinas u organogénesis linfoide son anormales o indeseables, incluyendo la artritis reumatoide y enfermedades relacionadas (tales como espondiloartritis anquilosante, artritis psoriásica, artritis juvenil), pénfigo vulgar y enfermedades relacionadas, púrpura trombocitopénica idiopática, lupus eritematoso sistémico, esclerosis múltiple, miastenia grave, síndrome de Sjogren (tal como síndrome de Sjogren primario (pSS)), enfermedad de injerto contra huésped, anemia hemolítica autoinmune, vasculitis asociada a ANCA (tal como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss) , crioglobulinemia, púrpura trombocitopénica trombótica, la lesión por isquemia-reperfusión, urticaria crónica autoinmune, alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgica, asma asociada con la rinitis alérgica), síndrome de Goodpasture, diferentes tipos de glomerulonefritis, AMR (rechazo de trasplante mediado por anticuerpos), rechazo de trasplante hiperagudo, agudo y crónico mediado por células B y cánceres de origen hematopoyético que incluyen, pero no se limitan a mieloma múltiple; una leucemia; leucemia mielógena aguda; leucemia mielógena crónica; leucemia linfocítica; leucemia mieloide; linfoma no Hodgkin; linfomas; policitemia vera; trombocitemia esencial; mielofibrosis con metaplasia mieloide; y enfermedad de Waldenstroem; más adecuadamente de artritis reumatoide (RA), pénfigo vulgar (PV), púrpura trombocitopénica idiopática (ITP), púrpura trombocitopénica trombótica (TTP), anemia hemolítica autoinmune (AIHA), hemofilia adquirida tipo A (AHA), lupus eritematoso sistémico (LSE), esclerosis múltiple (MS), miastenia grave (MG), síndrome de Sjogren (SS) (tal como síndrome de Sjogren primario (pSS)), vasculitis asociada a ANCA (tales como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss), crioglobulinemia, lesión por isquemia-reperfusión, urticaria autoinmune crónica (CAU), alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, rechazo de trasplantes y cánceres de origen hematopoyético, así como en la enfermedad o infección asociada a inmunopatología, por ejemplo en malaria severa y cerebral, tripanosomiasis, leishmaniasis, toxoplasmosis y neurocisticercosis.
La composición farmacéutica o combinación de la presente invención puede estar en dosis unitarias de aproximadamente 1-1000 mg de ingrediente(s) activo(s) para un sujeto de aproximadamente 50-70 kg, o aproximadamente 1-500 mg o aproximadamente 1-250 mg o aproximadamente 1-150 mg o aproximadamente 0,5-100 mg de ingredientes activos. La dosificación terapéuticamente efectiva de un compuesto, la composición farmacéutica o las combinaciones de los mismos, depende de la especie del sujeto, del peso corporal, de la edad y del estado individual, del trastorno o de la enfermedad o la gravedad del mismo que se está tratando. Un facultativo, médico o veterinario experto puede determinar fácilmente la cantidad eficaz de cada uno de los principios activos necesaria para prevenir, tratar o inhibir el avance del trastorno o la enfermedad.
Las propiedades de dosificación arriba citadas son ensayos demostrables in vitro e in vivo utilizando ventajosamente mamíferos, p. ej., ratones, ratas, perros, monos u órganos aislados, tejidos y preparaciones de los mismos. Los compuestos de la presente invención se pueden aplicar in vitro en forma de soluciones, p. ej., soluciones acuosas, e in vivo por vía enteral, parenteral, convenientemente por vía intravenosa, p. ej., en forma de una suspensión o en solución acuosa.
La dosificación in vitro puede oscilar entre concentraciones de aproximadamente 10-3 molar y 10-9 molar. Una cantidad terapéuticamente eficaz in vivo puede variar en función de la vía de administración, entre aproximadamente 0,1-500 mg/kg, o entre aproximadamente 1-100 mg/kg.
El compuesto de la presente invención se puede administrar simultáneamente con uno o más agentes terapéuticos diferentes o antes o después de estos. El compuesto de la presente invención se puede administrar por separado, mediante una vía de administración idéntica o diferente, o de forma conjunta en la misma composición farmacéutica que los otros agentes. Un agente terapéutico es, por ejemplo, un compuesto químico, péptido, anticuerpo, fragmento de anticuerpo o ácido nucleico, que es terapéuticamente activo o potencia la actividad terapéutica cuando se administra a un paciente en combinación con un compuesto de la invención.
En una realización, la invención proporciona un producto que comprende un compuesto de fórmula (Ib) y al menos otro agente terapéutico como una preparación combinada para uso simultáneo, separado o secuencial en terapia. En una realización, la terapia es el tratamiento de una enfermedad o afección mediada por la actividad de quinasas PI3 de clase I. Los productos proporcionados como una preparación combinada incluyen una composición que comprende el compuesto de fórmula (Ib) y el o los otros agentes terapéuticos juntos en la misma composición farmacéutica, o el compuesto de fórmula (Ib) y el o los otros agentes terapéuticos por separado forman, p. ej., en forma de un kit.
En una realización, la invención proporciona una composición farmacéutica que comprende un compuesto de fórmula (Ib) y otro u otros agentes terapéuticos. Opcionalmente, la composición farmacéutica puede comprender un portador farmacéuticamente aceptable, tal como se ha descrito anteriormente.
En una realización, la invención proporciona un kit que comprende dos o más composiciones farmacéuticas separadas, al menos una de las cuales contiene un compuesto de fórmula (Ib). En una realización, el kit comprende medios para mantener por separado dichas composiciones, tales como un recipiente, frasco dividido o envase de aluminio dividido. Un ejemplo de un kit de este tipo es un envase de tipo blíster, como los utilizados habitualmente para el envasado de comprimidos, cápsulas y similares.
El kit de la invención se puede utilizar para administrar diferentes formas farmacéuticas, por ejemplo, oral y parenteral, para administrar las composiciones separadas a intervalos posológicos diferentes, o para valorar las composiciones separadas entre sí. Para facilitar el cumplimiento, el kit de la invención normalmente comprende instrucciones para su administración.
En las terapias combinadas de la invención, el compuesto de la invención y el otro agente terapéutico pueden ser elaborados y/o formulados por el mismo fabricante o por fabricantes diferentes. Además, el compuesto de la invención y el otro agente terapéutico se pueden integrar en una terapia combinada: (i) antes de dispensar el producto combinado a los facultativos (p. ej., en el caso de un kit que comprende el compuesto de la invención y el otro agente terapéutico), (ii) por parte de los mismos facultativos (o bajo la supervisión del facultativo) poco antes de la administración; (iii) en los mismos pacientes, p. ej., durante la administración secuencial del compuesto de la invención y el otro agente terapéutico.
Por consiguiente, la invención proporciona el uso de un compuesto de fórmula (Ib) para tratar una enfermedad o afección mediada por la actividad de las quinasas PI3 de clase I, en donde el medicamento se prepara para la administración con otro agente terapéutico. La invención también proporciona el uso de otro agente terapéutico para tratar una enfermedad o afección mediada por la actividad de las quinasas PI3 de clase I, en donde el medicamento se administra con un compuesto de fórmula (Ib).
La invención también proporciona un compuesto de fórmula (Ib) para uso en un método de tratamiento de una enfermedad o afección mediada por la actividad de las enzimas quinasas PI3 de clase I, en donde el compuesto de fórmula (Ib) se prepara para la administración con otro agente terapéutico. La invención también proporciona otro agente terapéutico para uso en un método de tratamiento de una enfermedad o afección mediada por la actividad de las quinasas PI3 de clase I, en donde el otro agente terapéutico se prepara para la administración con un compuesto de fórmula (Ib). La invención también proporciona un compuesto de fórmula (Ib) para uso en un método de tratamiento de una enfermedad o afección mediada por la actividad de las quinasas PI3 de clase I, en donde el compuesto de fórmula (Ib) se administra con otro agente terapéutico. La invención también proporciona otro agente terapéutico para uso en un método de tratamiento de una enfermedad o afección mediada por la actividad de las quinasas PI3 de clase I, en donde el otro agente terapéutico se administra con un compuesto de fórmula (Ib).
La invención también proporciona el uso de un compuesto de fórmula (Ib) para tratar una enfermedad o afección mediada por la actividad de las quinasas PI3 de clase I, en donde el paciente ha sido tratado previamente (p. ej., dentro de las 24 horas) con otro agente terapéutico. La invención también proporciona el uso de otro agente terapéutico para tratar una enfermedad o afección mediada por la actividad de las PI3 quinasas de clase I, en donde el paciente ha sido tratado previamente (p. ej., dentro de las 24 horas) con un compuesto de fórmula (Ib).
Los compuestos de fórmula (Ib) pueden administrarse como el único ingrediente activo o junto con, p. ej., como adyuvante de otros fármacos, p. ej., agentes inmunosupresores o inmunomoduladores u otros agentes antiinflamatorios, p. ej., para el tratamiento o la prevención de rechazo por alo- o xeno-injerto agudo o crónico o trastornos inflamatorios o autoinmunes, o un agente quimioterapéutico, p. ej., un agente anti-proliferativo de células malignas. Por ejemplo, los compuestos de fórmula (Ib) pueden utilizarse en combinación con un inhibidor de calcineurina, p. ej., ciclosporina A o FK 506; un inhibidor de mTOR, p. ej., rapamicina, 40-O-(2-hidroxietil)-rapamicina, CCI779, ABT 578, AP23573, TAFA-93, biolimus-7 o biolimus-9; una ascomicina que tiene propiedades inmunosupresoras, p. ej., ABT-281, ASM981, etc .; corticosteroides; ciclofosfamida; azatiopreno; metotrexato; leflunomida; mizoribina; ácido micofenólico o sal; micofenolato de mofetilo; 15-desoxiespergualina o un homólogo inmunosupresor, análogo o derivado de la misma; un inhibidor de PKC, p. ej., tal como se describe en el documento WO 02/38561 o WO 03/82859, p. ej., el compuesto del Ejemplo 56 o 70; un inhibidor de la quinasa JAK3, p. ej. N-bencil-3,4-dihidroxi-benciliden-cianoacetamida a-ciano-(3,4-dihidroxi)-]N-bencilcinamamida (Tyrphostin AG 490), prodigiosina 25-C (PNU156804), [4-(4'-hidroxifenil) -amino-6,7-dimetoxiquinazolina] (WHI-P131), [4-(3'-bromo-4'-hidroxifenil) -amino-6,7-dimetoxiquinazolina] (WHI-P154 ), [4-(3',5'-dibromo-4'-hidroxilfenil) -amino-6,7-dimetoxiquinazolina] WHI-P97, KRX-211, 3-{(3R,4R)-4-metil-3-[metil-(7H-pirrolo[2,3-d] pirimidin-4-il)-amino]-piperidin-1-il}-3-oxo-propionitrilo, en forma libre o en forma de sal farmacéuticamente aceptable, p. ej. mono-citrato (también denominada CP-690,550), o un compuesto tal como se describe en el documento WO 04/052359 o WO 05/066156; anticuerpos monoclonales inmunosupresores, p. ej., anticuerpos monoclonales contra receptores de leucocitos, p. ej., MHC, CD2, CD3, CD4, CD7, CD8, CD25, CD28, CD40, CD45, CD52, CD58, CD80, CD86 o sus ligandos; otros compuestos inmunomoduladores, p. ej., una molécula de unión recombinante que tiene al menos una porción del dominio extracelular de CTLA4 o un mutante del mismo, p. ej., una porción al menos extracelular de CTLA4 o un mutante del mismo unido a una secuencia de proteína no CTLA4, p. ej. CTLA4Ig (p. ej., denominada ATCC 68629) o un mutante del mismo, p. ej., LEA 29Y; inhibidores de la molécula de adhesión, p. ej., antagonistas de LFA-1, antagonistas de ICAM-1 o -3, antagonistas de VCAM-4 o antagonistas de VLA-4; o antihistamínicos; o antitusivos, o un agente broncodilatador; o un bloqueador del receptor de angiotensina; o un agente anti-infeccioso.
En los casos en los que los compuestos de fórmula (Ib) se administran junto con otra terapia inmunosupresora/inmunomoduladora, antiinflamatoria, quimioterapéutica o anti-infecciosa, las dosis del compuesto inmunosupresor, inmunomodulador, antiinflamatorio, quimioterapéutico o anti-infeccioso co-administrado variarán por supuesto, en función del tipo de co-fármaco empleado, p. ej., si es un esteroide o un inhibidor de la calcineurina, en el fármaco específico empleado, en la afección a tratar, etc.
Un compuesto de la fórmula (Ib) también puede utilizarse como ventaja en combinación entre sí o en combinación con otros agentes terapéuticos, especialmente otros agentes antiproliferativos. Agentes antiproliferativos de este tipo incluyen, pero no se limitan a inhibidores de aromatasa; antiestrógenos; inhibidores de topoisomerasa I; inhibidores de topoisomerasa II; agentes activos de microtúbulos; agentes alquilantes; inhibidores de histona desacetilasa; compuestos que inducen procesos de diferenciación celular; inhibidores de la ciclooxigenasa; Inhibidores de MMP; inhibidores de mTOR; antimetabolitos antineoplásicos; compuestos de platino; compuestos que fijan como objetivo/disminuyen una actividad de proteína o lípido quinasa y otros compuestos anti-angiogénicos; compuestos que fijan como objetivo, disminuyen o inhiben la actividad de una proteína o fosfatasa lipídica; agonistas de gonadorelina; antiandrógenos; inhibidores de metionina aminopeptidasa; bifosfonatos; modificadores de respuesta biológica; anticuerpos antiproliferativos; inhibidores de heparanasa; inhibidores de las isoformas oncogénicas Ras; inhibidores de la telomerasa; inhibidores de proteasoma; agentes utilizados en el tratamiento de neoplasias hematológicas; compuestos que fijan como objetivo, disminuyen o inhiben la actividad de Flt-3; Inhibidores de Hsp90; temozolomida (TEMODAL® ); y leucovorina.
La expresión "inhibidor de aromatasa", tal como se utiliza en la presente, se refiere a un compuesto que inhibe la producción de estrógenos, es decir, la conversión de los sustratos androstenediona y testosterona en estrona y estradiol, respectivamente. La expresión incluye, pero no se limita a esteroides, especialmente atamestano, exemestano y formestano; y, en particular, no esteroides, especialmente aminoglutetimida, rogletimida, piridoglutetimida, trilostano, testolactona, cetoconazol, vorozol, fadrozol, anastrozol y letrozol. Exemestano se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada AROMASIN. Formestano se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada LENTARON. Fadrozol se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada AFEMA. Anastrozol se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada ARIMIDEX. Letrozol se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada FEMARA o FEMAR. Aminoglutetimida se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada ORIMETEN. Una combinación de la invención que comprende un agente quimioterapéutico que es un inhibidor de aromatasa es particularmente útil para el tratamiento de tumores con receptores hormonales positivos, p. ej., tumores de mama.
El término "anti-estrógeno", tal como se utiliza en la presente, se refiere a un compuesto que antagoniza el efecto de los estrógenos a nivel del receptor de estrógenos. El término incluye, pero no se limita a tamoxifeno, fulvestrant, raloxifeno e hidrocloruro de raloxifeno. Tamoxifeno se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada NOLVADEX. Raloxifeno se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada EVISTA. Fulvestrant puede formularse tal como se describe en la Patente de EE.UU. N° 4.659.516 o puede administrarse, p. ej., en la forma en que se comercializa, por ejemplo, bajo la marca registrada FASLODEX. Una combinación de la invención que comprende un agente quimioterapéutico que es un anti-estrógeno es particularmente útil para el tratamiento de tumores con receptores hormonales positivos, p. ej., tumores de mama.
El término "anti-andrógeno", tal como se utiliza en la presente, se refiere a cualquier sustancia que es capaz de inhibir los efectos biológicos de las hormonas androgénicas e incluye, pero no se limita a bicalutamida (CASODEX), que puede formularse, p. ej., como se describe en la Patente de EE.UU. N° 4.636.505.
La expresión "agonista de gonadorelina", tal como se utiliza en la presente, incluye, pero no se limita a, abarelix, goserelina y acetato de goserelina. La goserelina se describe en la Patente de EE.UU. N° 4.100.274 y se puede administrar, p. ej., en forma en que se comercializa, p. ej., bajo la marca registrada ZOLADEX. Abarelix puede formularse, p. ej., tal como se describe en la Patente de EE.UU. N° 5.843.901.
La expresión "inhibidor de topoisomerasa I", tal como se utiliza en la presente, incluye, pero no se limita a, topotecan, gimatecan, irinotecan, camptotecian y sus análogos, 9-nitrocamptotecina y el conjugado de camptotecina macromolecular PNU-166148 (compuesto A l en el documento WO 99/17804). Irinotecan se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada CAMPTOSAR. Topotecan se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada HYCAMTIN.
La expresión "inhibidor de topoisomerasa II", tal como se utiliza en la presente, incluye, pero no se limita a las antraciclinas, tales como doxorrubicina, incluida la formulación liposómica, p. ej., CAELYX; daunorrubicina; epirubicina; idarubicina; nemorubicina; las antraquinonas mitoxantrona y losoxantrona; y las podofilotoxinas etopósido y tenipósido. Etopósido se puede administrar, p. ej., en la forma en que se comercializa, por ejemplo, bajo la marca registrada ETOPOPHOS. Teniposido se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada VM 26-BRISTOL. Doxorrubicina se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada ADRIBLASTIN o ADRIAMYCIn . Epirubicina se puede administrar, p. ej., en la forma en que se comercializa, por ejemplo, bajo la marca registrada FARMORUBICIN. Idarubicina se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada ZAVEDOS. Mitoxantrona se puede administrar, p. ej., en la forma en que se comercializa, por ejemplo, bajo la marca registrada NOVANTRON.
La expresión "agente activo de microtúbulos" se refiere a estabilizadores de microtúbulos, agentes desestabilizadores de microtúbulos e inhibidores de la polimerización de microtúbulos que incluyen, pero no se limitan a, taxanos, p. ej., paclitaxel y docetaxel; alcaloides de la vinca, p. ej., vinblastina, especialmente sulfato de vinblastina; vincristina, especialmente sulfato de vincristina y vinorelbina; discodermolidas; cochicina y epotilonas y derivados de las mismas, p. ej., epotilona B o D o derivados de las mismas. Paclitaxel se puede administrar, p. ej., en la forma en que se comercializa, p. ej., TAXOL. Docetaxel se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada TAXOTERE. Vinblastina se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada VINBLASTIN R.P. Vincristina se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada FARMISTIN. Discodermolida se puede obtener, p. ej., tal como se describe en la Patente de EE.UU. N° 5,010,099. También se incluyen derivados de epotilona que se describen en los documentos WO 98/10121, Patente de EE.UU. N° 6.194.181, WO 98/25929, WO 98/08849, WO 99/43653, WO 98/22461 y WO 00/31247. Se prefieren especialmente la epotilona A y/o B.
La expresión "agente alquilante", tal como se utiliza en la presente, incluye, pero no se limita a, ciclofosfamida, ifosfamida, melfalan o nitrosourea (BCNU o Gliadel). Ciclofosfamida se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada CYCLOSTIN. Ifosfamida se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada HOLOXAN.
La expresión "inhibidores de la histona desacetilasa" o "inhibidores de HDAC" se refiere a los compuestos que inhiben la histona desacetilasa y que poseen actividad antiproliferativa. Esto incluye compuestos descritos en el documento WO 02/22577, especialmente N-hidroxi-3-[4-[[(2-hidroxietil)[2-(1H-indol-3-il)etil]-amino]metil]fenil]-2E-2-propenamida, N-hidroxi-3-[4-[[[2-(2-metil-1H-indol-3-il)-etil]-amino]metil]fenil]-2E-2-propenamida y sus sales farmacéuticamente aceptables. Además, incluye especialmente el ácido hidroxámico suberoilanilida (SAHA).
La expresión "antimetabolito antineoplásico" incluye, pero no se limita a, 5-fluorouracilo o 5-FU; capecitabina; gemcitabina; agentes de desmetilación del ADN, tales como 5-azacitidina y decitabina; metotrexato y edatrexato; y antagonistas del ácido fólico, tal como pemetrexed. Capecitabina se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada XELODA. Gemcitabina se puede administrar, p. ej., en la forma en que se comercializa, p. ej.,
bajo la marca registrada GEMZAR. También se incluye el anticuerpo monoclonal trastuzumab que puede administrarse, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada HERCEPTIN.
La expresión "compuesto de platino", tal como se utiliza en la presente, incluye, pero no se limita a, carboplatino, cis-platino, cisplatinum y oxaliplatino. Carboplatino se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada CARBOPLAT. Oxaliplatino se puede administrar, p. ej., en la forma en que se comercializa, p. ej., bajo la marca registrada ELOXATIN.
La expresión "compuestos que fijan como objetivo/disminuyen una actividad de proteína o lípido quinasa; o una actividad de proteína o lípido fosfatasa; o compuestos anti-angiogénicos adicionales", tal como se utiliza en la presente, incluye, pero no se limita a, proteína tirosina quinasa y/o serina y/o inhibidores de la treonina quinasa o inhibidores de la lípido quinasa, p. ej.,
a) compuestos que fijan como objetivo, disminuyen o inhiben la actividad de los receptores del factor de crecimiento derivado de plaquetas (PDGFR), tales como compuestos que fijan como objetivo, disminuyen o inhiben la actividad de PDGFR, especialmente compuestos que inhiben el receptor PDGF, p. ej., un derivado de N-fenil-2-pirimidina-amina, p. ej., imatinib, SU101, SU6668 y GFB-111;
b) los compuestos que fijan como objetivo, disminuyen o inhiben la actividad de los receptores del factor de crecimiento de los fibroblastos (FGFR);
c) compuestos que fijan como objetivo, disminuyen o inhiben la actividad del receptor del factor de crecimiento tipo insulina I (IGF-IR), tales como compuestos que fijan como objetivo, disminuyen o inhiben la actividad de IGF-iR, especialmente compuestos que inhiben el receptor de IGF-IR , tales como los compuestos descritos en el documento WO 02/092599;
d) compuestos que fijan como objetivo, disminuyen o inhiben la actividad de la familia de tirosina quinasa del receptor Trk;
e) compuestos que fijan como objetivo, disminuyen o inhiben la actividad de la familia del receptor tirosina quinasa Axl;
f) compuestos que fijan como objetivo, disminuyen o inhiben la actividad del receptor c-Met;
g) compuestos que fijan como objetivo, disminuyen o inhiben la actividad del receptor de Kit/SCFR tirosina quinasa;
h) compuestos que fijan como objetivo, disminuyen o inhiben la actividad del receptor C-kit (parte de la familia PDGFR), tales como compuestos que fijan como objetivo, disminuyen o inhiben la actividad de la familia tirosina quinasa del receptor c-Kit, especialmente compuestos que inhiben el receptor c-Kit, p. ej., imatinib;
i) compuestos que fijan como objetivo, disminuyen o inhiben la actividad de miembros de la familia c-Abl y sus productos de fusión génica, p. ej., BCR-Abl quinasa, tales como los compuestos que fijan como objetivo, disminuyen o inhiben la actividad de miembros de la familia c-Ab l y sus productos de fusión génica, p. ej., un derivado de N-fenil-2-pirimidina-amina, p. ej., imatinib, PD180970, AG957, NSC 680410 o PD173955 de ParkeDavis;
j) compuestos que fijan como objetivo, disminuyen o inhiben la actividad de miembros de la familia de proteína quinasa C (PKC) y Raf de serina/treonina quinasas, miembros de los miembros de la familia MEK, SRC, JAK, FAK, PDK y Ras/MAPK, o de la familia de quinasas PI(3), o de la familia de quinasas relacionadas con PI(3)-quinasa y/o miembros de la familia de quinasas dependientes de ciclina (CDK) y son especialmente aquellos derivados de estaurosporina descritos en la Patente de EE.UU. N° 5.093.330, p. ej., midostaurina; ejemplos de compuestos adicionales incluyen, p. ej., UCN-01; safingol; BAY 43-9006; Briostatina 1; Perifosina; Ilmofosina; RO 318220 y RO 320432; g O 6976; Isis 3521; LY333531/LY379196; compuestos de isoquinolina, tales como los descritos en el documento WO 00/09495; FTIs PD184352; o QAN697 (un inhibidor de P13K);
k) compuestos que fijan como objetivo, disminuyen o inhiben la actividad de los inhibidores de la proteína-tirosina quinasa, tales como los compuestos que fijan como objetivo, disminuyen o inhiben la actividad de los inhibidores de la proteína-tirosina quinasa incluyen el mesilato de imatinib (GLEEVEC) o ti rfostina. Una tirfostina es preferiblemente un compuesto de bajo peso molecular (Mr <1500), o una sal farmacéuticamente aceptable del mismo, especialmente un compuesto seleccionado de la clase de bencilidenomalonitrilo o la clase de compuestos de S-arilbencenomalonirilo o quinolina de bisustrato, más especialmente cualquier compuesto seleccionado del grupo que consiste en Tirfostina A23/RG-50810, AG 99, Tirfostina AG 213, Tirfostina AG 1748, Tirfostina AG 490, Tirofostina B44, enantiómero de Tirfostina B44 (+), Tirfostina AG 555, AG 494, Tirfostina AG 556, AG957 y adafostina éster adamantílico del ácido 4-{[(2,5-dihidroxifenil)metil]amino}-benzoico, NSC 680410, adafostina; y
l) compuestos que fijan como objetivo, disminuyen o inhiben la actividad de la familia del factor de crecimiento epidérmico de receptores de tirosina quinasas (EGFR, ErbB2, ErbB3, ErbB4 como homo o hetero-dímeros), tales como compuestos que fijan como objetivo, disminuyen o inhiben la actividad de la familia de receptores del factor de crecimiento epidérmico son especialmente compuestos, proteínas o anticuerpos que inhiben a los miembros de la familia de tirosina quinasa del receptor de e Gf , p. ej., receptor de EGF, ErbB2, ErbB3 y ErbB4 o se unen a EGF o a ligandos relacionados con EGF y son, en particular, los compuestos, proteínas o anticuerpos monoclonales descritos genérica y específicamente en el documento WO 97/02266, p. ej., el compuesto del Ejemplo 39, o en el documento EP 0564409; WO 99/03854; EP 0520722; EP 0566226; EP 0787722; EP 0 837 063; Patente de EE.UU. N° 5.747.498; WO 98/10767; WO 97/30034; WO 97/49688; WO 97/38983 y,
especialmente, WO 96/30347, p. ej., un compuesto conocido como CP 358774; documento WO 96/33980, p. ej., compuesto ZD 1839; y documento WO 95/03283, p. ej., compuesto ZM105180, p. ej., trastuzumab (HERCEPTIN), cetuximab, Iressa, Tarceva, OSI-774, CI-1033, EKB-569, GW-2016, E1.1, E2.4, E2.5, E6.2, E6.4, E2.11, E6.3 o E7.6.3; y 7H-pirrolo-[2,3-d]pirimidina que se describen en el documento WO 03/013541.
Compuestos anti-angiogénicos adicionales incluyen compuestos que tienen otro mecanismo para su actividad, p. ej., no relacionados con la inhibición de proteínas o lípidos quinasas, p. ej., talidomida (THALOMID) y TNP-470.
Compuestos que fijan como objetivo, disminuyen o inhiben la actividad de una proteína o lípido fosfatasa son, p. ej., inhibidores de fosfatasa 1, fosfatasa 2A, PTEN o CDC25, p. e., ácido ocadaico o un derivado del mismo.
Compuestos que inducen los procesos de diferenciación celular son, p. ej., ácido retinoico, a-, y- o 5-tocoferol o a-, y- o 5-tocotrienol.
La expresión inhibidor de la ciclooxigenasa, tal como se utiliza en la presente, incluye, pero no se limita a, p. ej., inhibidores de Cox-2, ácido 2-arilaminofenilacético sustituido con 5-alquilo y derivados, tales como celecoxib (CELEBr Ex ), rofecoxib (VIOXX), etoricoxib, valdecoxib o un ácido 5-alquil-2-arilaminofenilacético, p. ej., ácido 5-metil-2- (2'-cloro-6'-fluoroanilino) fenilacético o lumiracoxib.
El término "bifosfonatos", tal como se utiliza en la presente, incluye, pero no se limita a, ácido etridónico, clodrónico, tiludrónico, pamidrónico, alendrónico, ibandrónico, risedrónico y zoledrónico. "Ácido etridónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada DIDRONEL. "Ácido clodrónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada BONEFOS. "Ácido tiludrónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada SKELID . "Ácido pamidrónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada AREDIA™ "Ácido alendrónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada FOSAMAX. "Ácido ibandrónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada BONDRANAT. "Ácido risedrónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada ACTONEL. "Ácido zoledrónico" se puede administrar, p. ej., en la forma en la que se comercializa, p. ej., bajo la marca registrada ZOMETA.
La expresión "inhibidores de mTOR" se refiere a compuestos que inhiben la diana mamífero de rapamicina (mTOR) y que poseen actividad antiproliferativa, tales como sirolimus (Rapamune® ), everolimus (Certican™), CCI-779 y ABT578.
La expresión "inhibidor de heparanasa", tal como se utiliza en la presente, se refiere a compuestos que fijan como objetivo, disminuyen o inhiben la degradación de sulfato de heparina . La expresión incluye, pero no se limita a PI-88.
La expresión "modificador de respuesta biológica", tal como se utiliza en la presente, se refiere a una linfoquina o interferones, p. ej., interferón y.
La expresión "inhibidor de isoformas oncogénicas de Ras", p. ej., H-Ras, K-Ras o N-Ras, tal como se utiliza en la presente, se refiere a compuestos que fijan como objetivo, disminuyen o inhiben la actividad oncogénica de Ras, p. ej., un "inhibidor de farnesil transferasa", p. ej., L-744832, DK8G557 o R115777 (Zarnestra).
La expresión "inhibidor de telomerasa", tal como se utiliza en la presente, se refiere a compuestos que fijan como objetivo, disminuyen o inhiben la actividad de telomerasa. Compuestos que fijan como objetivo, disminuyen o inhiben la actividad de telomerasa son especialmente compuestos que inhiben el receptor de telomerasa, p. ej., telomestatina.
La expresión "inhibidor de metionina aminopeptidasa", tal como se utiliza en la presente, se refiere a compuestos que fijan como objetivo, disminuyen o inhiben la actividad de metionina aminopeptidasa. Compuestos que fijan como objetivo, disminuyen o inhiben la actividad de metionina aminopeptidasa son, p. ej., bengamida o un derivado de la misma. La expresión "inhibidor de proteasa", tal como se utiliza en la presente, se refiere a compuestos que fijan como objetivo, disminuyen o inhiben la actividad del proteosoma. Compuestos que fijan como objetivo, disminuyen o inhiben la actividad del proteosoma son, p. ej., PS-341 y MLN341.
La expresión "inhibidor de metaloproteinasa de matriz" o "inhibidor de MMP", tal como se utiliza en la presente, incluye, pero no se limita a inhibidores peptidomiméticos y no peptidomiméticos de colágeno, derivados de tetraciclina, p. ej., inhibidor peptidomimético de hidroxamato batimastat y su análogo biodisponible por vía oral marimastat (BB-2516), prinomastat (AG3340), metastat (NSC 683551) BMS - 279251, BAY 12-9566, TAA211, MMI270B o AAJ996.
La expresión "agentes usados en el tratamiento de tumores malignos hematológicos", tal como se utiliza en la presente, incluye, pero no se limita a inhibidores de tirosina quinasa de tipo FMS, p. ej., compuestos que fijan como objetivo, disminuyen o inhiben la actividad de receptores de tirosina quinasa de tipo FMS (Flt-3R); interferón, 1-b-D-arabinofuransilcitosina (ara-c) y bisulfán; e inhibidores de ALK, p. ej., compuestos que fijan como objetivo, disminuyen o inhiben la linfoma quinasa anaplásica.
Compuestos que fijan como objetivo, disminuyen o inhiben la actividad de los receptores de tirosina quinasa de tipo FMS (Flt-3R) son especialmente compuestos, proteínas o anticuerpos que inhiben a los miembros de la familia de las quinasas receptoras de Flt-3R, p. ej., PKC412, midostaurina, un derivado de estaurosporina, SU11248 y MLN518.
La expresión "inhibidores de HSP90", tal como se utiliza en la presente, incluye, pero no se limita a, compuestos fijan como objetivo, disminuyen o inhiben la actividad intrínseca de ATPasa de HSP90; degradando, fijando como objetivo, disminuyendo o inhibiendo las proteínas del cliente HSP90 a través de la vía de ubiquitina proteasoma . Compuestos que fijan como objetivo, disminuyen o inhiben la actividad intrínseca de ATPasa de HSP90 son especialmente compuestos, proteínas o anticuerpos que inhiben la actividad de ATPasa de HSP90, p. ej., 17-alilamino, 17-demetoxigeldanamicina (17AAG), un derivado de geldanamicina, otros compuestos relacionados con geldanamicina, radicicol e inhibidores de HDAC.
La expresión "anticuerpos antiproliferativos", tal como se utiliza en la presente, incluye, pero no se limita a anticuerpo trastuzumab (Herceptin™), Trastuzumab-DM1, erlotinib (Tarceva™), bevacizumab (Avastin™), rituximab (Rituxan®), PRO64553 (anti-CD40) y 2C4. Por anticuerpos se entiende, p. ej., anticuerpos monoclonales intactos, anticuerpos policlonales, anticuerpos multiespecíficos formados a partir de al menos dos anticuerpos intactos y fragmentos de anticuerpos, siempre que exhiban la actividad biológica deseada.
Para el tratamiento de la leucemia mieloide aguda (AML), los compuestos de fórmula (Ib) pueden utilizarse en combinación con terapias estándares contra la leucemia, especialmente en combinación con terapias utilizadas para el tratamiento de la AML. En particular, los compuestos de fórmula (Ib) pueden administrarse en combinación con, p. ej., inhibidores de farnesil transferasa y/u otros fármacos útiles para el tratamiento de AML, tales como Daunorubicina, Adriamicina, Ara-C, VP-16, Teniposido, Mitoxantrona, Idarubicina, Carboplatino y PKC412.
Un compuesto de la fórmula (Ib) también puede utilizarse como ventaja en combinación entre sí o en combinación con otros agentes terapéuticos, especialmente otros agentes anti-malaria. Agentes anti-malaria incluyen, pero no se limitan a proguanil, clorproguanil, trimetoprima, cloroquina, mefloquina, lumefantrina, atovacuona, pirimetamina-sulfadoxina, pirimetamina-dapsona, halofantrina, quinina, quinidina, amodiaquina, amopiroquina, sulfonamidas, artemisinina, artefleno, artemeter, primaquina, NO inhalado, L-arginina, dipropilentri-amina NONOato (donante de NO), Rosiglitzona (agonista de PPAR y), carbón vegetal activado, Eritropoyetina, Levamisol y pironaridina
Un compuesto de la fórmula (Ib) también puede utilizarse como ventaja en combinación entre sí o en combinación con otros agentes terapéuticos, tales como los utilizados para el tratamiento de la leishmaniosis, tripanosomiasis, toxoplasmosis y neurocisticercosis. Agentes de este tipo incluyen, pero no se limitan a sulfato de cloroquina, atovacuonaproguanil, arteméter-lumefantrina, sulfato de quinina, artesunato, quinina, doxiciclina, clindamicina, antimoniato de meglumina, estibogluconato de sodio, miltefosina, ketoconazol, pentamidina, amfotericina B (AmB), AmB liposomal, paromomicina, eflornitina, nifurtimox, suramin, melarsoprol, prednisolona, benznidazol, sulfadiazina, pirimetamina, clindamicina, trimetropim, sulfametoxazol, azitromicina, atovaquona, dexametasona, praziquantel, albendazol, betalactamas, fluoroquinolonas, macrolidas, aminoglicósidos, sulfadiazina y pirimetamina.
La estructura de los agentes activos identificados por los números de código, nombres genéricos o comerciales puede tomarse de la edición real del compendio estándar "The Merck Index" o de bases de datos, p. ej., Patentes Internacionales, p. ej., Publicaciones IMS World.
Los compuestos arriba mencionados, que pueden utilizarse en combinación con un compuesto de la fórmula (Ib), pueden prepararse y administrarse como se describe en la técnica, tal como en los documentos arriba citados.
Un compuesto de la fórmula (Ib) también puede utilizarse como ventaja en combinación con procesos terapéuticos conocidos, p. ej., la administración de hormonas o especialmente radiación.
Un compuesto de fórmula (Ib) puede utilizarse, en particular, como radiosensibilizador, especialmente para el tratamiento de tumores que exhiben poca sensibilidad a la radioterapia.
EJEMPLOS
Detalles experimentales:
Los siguientes ejemplos están destinados a ilustrar la invención y no deben interpretarse como limitaciones a la misma. Las temperaturas se dan en grados Celsius. Si no se menciona lo contrario, todas las evaporaciones se realizan a presión reducida, normalmente entre aproximadamente 15 mm de Hg y 100 mm de Hg (= 20-133 mbar). La estructura de los productos finales, intermedios y materiales de partida se confirma mediante métodos analíticos estándar, p. ej., microanálisis y características espectroscópicas, p. ej., MS, IR, RMN. Las abreviaturas utilizadas son las convencionales en la técnica.
Todos los materiales de partida, bloques estructurales, reactivos, ácidos, bases, agentes deshidratantes, disolventes y catalizadores utilizados para sintetizar los compuestos de la presente invención se pueden adquirir de proveedores
comerciales o se pueden producir mediante métodos de síntesis orgánica conocidos por el experto en la técnica (Houben-Weyl 4.a Ed. 1952, Methods of Organic Synthesis, Thieme, Volumen 21). Además, los compuestos de la presente invención se pueden producir mediante métodos de síntesis orgánica conocidos por un experto en la técnica tal como se muestra en los siguientes ejemplos.
Abreviaturas
ACN acetonitrilo
ac acuoso
Boc terc.-butoxicarbonilo
br. ancho
Brettphos 2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-triisopropil-1,1'-bifenilo
Paladaciclo Brettphos cloro[2-(diciclohexilfosfino)-3,6-dimetoxi-2',4',6'-triisopropiM,1'-bifenil][2-(2-aminoetil)fenil]paladio(ll)
Cul yoduro de cobre (I)
CuSO4 sulfato de cobre (II)
d día(s), doblete
DCM diclorometano
DIPEA diisopropiletilamina
DMF dimetilformamida
DMSO dimetilsulfóxido
eq. equivalente(s)
ESI ionización por electronebulización
EtaN trietilamina
EtOAc acetato de etilo
EtOH etanol
h hora(s)
HNOa ácido nítrico
HPLC cromatografía líquida de alta resolución
H3PO4 ácido fosfórico
H2SO4 ácido sulfúrico
K2CO3 carbonato de potasio
LCMS cromatografía líquida con espectrometría de masas
MeOH metanol
m multiplete
min minuto(s)
MS espectrometría de masas
mw microonda
Na2CO3 carbonato de sodio
NaHCO3 bicarbonato de sodio
Na2SO4 sulfato de sodio
nBuOH n-butanol
NMP N-metil-2-pirrolidona
NMR espectrometría por resonancia magnética nuclear
Pd(OAc)2 diacetato de paladio
Pd(PPha)4 tetrakis(trifenilfosfina)paladioO)
PdCl2(PPha)2 dicloruro de bis(trifenilfosfina)paladio(ll)
RP fase inversa
rpm revoluciones por minuto
Rt tiempo de retención
ta temperatura ambiente
sat. saturado
SFC cromatografía de fluidos supercríticos
soln. solución
t-butilo terc.-butilo
TFA ácido trifluoroacético
THF tetrahidrofurano
UPLC cromatografía líquida de ultra rendimiento
El equipo de microondas utilizado es un Biotage Initiator®
Todos los compuestos se nombran utilizando el software ChemDraw® Ultra o se han utilizado nombres comerciales.
Información general de cromatografía
Método LCMS utilizado para todos los ejemplos
Dimensiones de la columna de HPLC: 2,1 x 50 mm
Tipo de columna de HPLC: Acquity UPLC HSS T3, 1,8 pm
eluyente de HPLC: A) agua ácido fórmico al 0,05 % en vol acetato de amonio 3,75 mM B)
ACN ácido fórmico al 0,04 % en vol.
Gradiente de HPLC: 5 - 98% de B en 1,4 min, 98% de B 0,4 min, caudal = 1,0 ml / min Temperatura de la columna de HPLC: 60 °C
Difracción de rayos X en polvo
Instrumentación y Método
Instrumento Bruker D8 advantage
Geometría Reflexión
Detector Vantec
Intervalo de escaneo 2°-40° (2-Theta)
Irradiación CuKa (40 kV, 40 mA)
Medición a Temperatura ambiente
Calorimetría diferencial de barrido
Instrumentación y Método
Instrumento: Mettler Toledo DSC Star System
Intervalo de temperaturas 30-300°C
Velocidad de escaneo: 10°C/min
Flujo de nitrógeno: 45 ml/min
Preparación de ejemplos
En los casos en los que se afirma que los compuestos se prepararon de la manera descrita para un ejemplo anterior, la persona experta apreciará que los tiempos de reacción, el número de equivalentes de reactivos y las temperaturas de reacción pueden modificarse para cada reacción específica, y que, sin embargo, puede ser necesario o es deseable emplear diferentes condiciones de tratamiento o purificación. Los compuestos que muestran atropisomerismo en torno al enlace N-R1 pueden obtenerse como mezclas de atropisómeros diastereoméricos que pueden separarse por condiciones cromatográficas. Estos compuestos se ejemplifican como mezclas y, si es aplicable, además como atropisómeros diastereoméricos separados, que detallan las condiciones de separación en cada caso.
Ejemplo 1: 2-amino-4-((2S,4S)-2-(6-fluoro-5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo

a) Ácido 2-bromo-3-fluoro-6-nitrobenzoico
Una solución de ácido 2-bromo-3-fluorobenzoico (registro CAS 132715-69-6) (5 g, 22,83 mmol) en H2SO4 conc. (15,82 ml, 297 mmol) se enfrió a 0°C y se trató gota a gota con HNO3 fumante (1,36 ml, 27,4 mmol). Se formó un precipitado blanco. La suspensión resultante se agitó a 0°C durante 1,5 h. La mezcla se vertió en agua con hielo y se agitó durante 30 min. La mezcla resultante se diluyó con agua y se extrajo con DCM. Después, las fases orgánicas se secaron sobre Na2SO4 y se concentró a presión reducida. Esto proporcionó un sólido blanquecino (5,67 g, 94%) de una mezcla de ácido 2-bromo-3-fluoro-6-nitrobenzoico (regioisómero 1) y ácido 2-bromo-3-fluoro-5-nitrobenzoico (regioisómero 2 ) en una relación de aprox. 2 :1 , que se utilizó sin separación para la siguiente etapa.
HPLC 1erregioisómero (6 9 %) Rt = 0,33 min; HPLC 2° regioisómero (30%) Rt = 0,52 min.
b) Ácido 6-amino-2-bromo-3-fluorobenzoico
Una mezcla de ácido 2-bromo-3-fluoro-6-nitrobenzoico y ácido 2-bromo-3-fluoro-5-nitrobenzoico (5,60 g, 21,21 mmol, relación de aprox. 2:1) se disolvió en THF (140 ml) y sucesivamente se desgasificó y purgó con nitrógeno (5 veces). Luego, la solución amarilla se trató con paladio al 10% sobre carbono (1,13 g, 1,06 mmol). La mezcla negra se purgó, luego se volvió a llenar con gas hidrógeno y se dejó agitar bajo atmósfera de hidrógeno a ta durante 18 h. La mezcla de reacción se filtró a través de hyflo y la almohadilla se enjuagó con EtOAc (2 veces). Los filtrados recogidos se evaporaron in vacuo para proporcionar un sólido pardo claro (5,4 g, 90%) en forma de una mezcla aprox. 2:1 de ácido 6-amino-2-bromo-3-fluorobenzoico (regioisómero 1) y ácido 5-amino-2-bromo-3-fluorobenzoico (regioisómero 2). La mezcla bruta se utilizó sin purificación adicional para la siguiente etapa.
HPLC 1erregioisómero (67%) Rt = 0,50 min; ESIMS: 234, 236 [(M+H)+]
HPLC 2° regioisómero (33%) Rt = 0,51 min; ESIMS: 234, 236 [(M+H)+]
c) 2-(5-bromo-6-fluoro-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-((terc.-butildimetilsilil)oxi)pirrolidina-1-carboxilato de (2S,4S)-terc.-butilo
Una solución seca de una mezcla de ácido 6-amino-2-bromo-3-fluorobenzoico y ácido 5-amino-2-bromo-3-fluorobenzoico (5,4 g, 13,84 mmol, relación aprox. 2:1) y ácido (2S,4S)-1- (terc-butoxicarbonil)-4-((terc.-butildimetilsilil)oxi)pirrolidina-2-carboxílico (registro CAS 401564-17-8) (7,97 g, 60% de contenido, 13,84 mmol) en piridina (25 ml) se trató con fosfito de trifenilo (registro CAS 101-02-0) (9,1 ml, 34,6 mmol). La solución parda clara se agitó a 70°C durante 18 h y luego se añadió gota a gota anilina (registro CAS 62 -53-3) (1,69 ml, 18,55 mmol). Después de agitar la mezcla de reacción durante otras 3 h a 70°C, la piridina se separó a presión reducida y luego el residuo se diluyó con EtOAc y se lavó con soln. sat. ac. de NaHCO3 (2 veces) y salmuera. Las fases orgánicas se secaron sobre Na2SO4 y se concentraron a presión reducida. El producto bruto se purificó por cromatografía sobre gel de sílice (0 a 15% de EtOAc / heptano) para proporcionar el compuesto del título en forma de un aceite naranja (1,70 g, rendimiento del 17%, pureza del 85%) que se utilizó sin purificación adicional para la siguiente etapa.
HPLC Rt= 1,60 min; ESIMS: 618, 620 [(M+H)+].
d) 4-((terc.-butildimetilsilil)oxi)-2-(6-fluoro-5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidina-1-carboxilato de (2S,4S)-terc.-butilo
Una solución de 2-(5-bromo-6-fluoro-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-((terc.-butildimetilsilil)oxi)pirrolidina-1-carboxilato de (2S,4S)-terc.-butilo (160 mg, 0,22 mmol, 85% de contenido) en Dm F (2 ml) se trató con 1 -metil-4-(tributilestannil)-1 H-pirazol (registro CAS 179055-21-1) (93 pl, 0,27 mmol). Luego, la mezcla de reacción se desgasificó con argón durante 5 min y se trató con bis(tri-t-butilfosfina)paladio(0) (registro CAS 53199-31-8) (11,24 mg, 0,022 mmol). La mezcla de reacción se agitó en un recipiente sellado bajo una atmósfera de argón a 80°C durante 18 h, luego se diluyó con EtOAc, se filtró a través de un cartucho separador de fase universal Biotage y las fases orgánicas combinadas se concentraron a presión reducida. El producto bruto se purificó por cromatografía sobre gel de sílice (0 a 15% de MeOH / DCM) para proporcionar el compuesto del título en forma de una goma amarilla (127 mg, 75% de rendimiento, 80% de pureza) que se utilizó sin purificación adicional para la siguiente etapa.
HPLC Rt= 1,51 min; ESIMS: 620 [(M+H)+].
e) 6-fluoro-2-((2S,4S)-4-hidroxipirrolidin-2-il)-5-(1-metil-1H-pirazol-4-il)-3-fenilquinazolin-4(3H)-ona
A ta, una solución de 4-((terc.-butildimetilsilil)oxi)-2-(6-fluoro-5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidina-1-carboxilato de (2S,4S)-terc.-butilo (127 mg, 0,16 mmol, 80% de pureza) en THF (0,7 ml) se trató con soln. ac. de H3PO4 (0,8 ml, 85% en peso). La mezcla se agitó durante 18 h a ta. Se añadió agua y la mezcla se basificó con una soln. sat. av. de NaHCO3 a pH ~ 9. La mezcla se extrajo con EtOAc (2 veces) y DCM (2 veces), a continuación, se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró in vacuo para proporcionar una goma amarilla (80 mg, 96% de rendimiento, 80 % de pureza) que se utilizó sin purificación adicional para la siguiente etapa.
HPLC Rt= 0,49 min; ESIMS: 406 [(M+H)+].
f) 2-amino-4-((2S,4S)-2-(6-fluoro-5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo
Una solución de 6-fluoro-2-((2S, 4S)-4-hidroxipirrolidin-2-il)-5-( 1 -metil-1 H-pirazol-4-il)-3-fenilquinazolin-4 (3H)-ona (25 mg, 0,062 mmol) y 2-amino-4-cloro-6-metilpirimidina-5-carbonitrilo (registro CAS 99586-66-0) (10,40 mg, 0,062 mmol) en EtOH (1 ml) se trató con DIPEA (0,022 ml, 0,12 mmol) y se irradió a 120°C durante 45 min en un mw.
La mezcla de reacción se evaporó a presión reducida. El aceite se recogió en DCM y se lavó con soln. sat. ac. de NaHCO3 , la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó sobre RP-HPLC (columna Sunfire C18, 100 x 30 mm, 5 pm, caudal a 30 ml / min; gradiente de A c N TFA al 0,1% en H2O TFA al 0,1% / de 12% a 32% en 16 min, tiempo de ejecución total: 20 min) para proporcionar el compuesto del título en forma de una base libre
obtenida por elución a través de un cartucho Varían tipo PL-HCO3 MP (22,2 mg, 67% de rendimiento) y posterior evaporación.
HPLC Rt= 0,81 min; ESIMS: 538 [(M+H)+].
1H RMN (400 MHz, DMSO-d6): 57,75 - 7,83 (m, 1 H) 7,62 - 7,74 (m, 2 H) 7,46 - 7,60 (m, 4 H) 7,34 - 7,45 (m, 2 H) 6,33 -7,18 (a. m, 2 H) 5,09 - 5,40 (m,1 H) 4,47 - 4,76 (m, 1 H) 4,15 (d, 2 H) 3,81 (s, 3 H) 3,61 - 3,75 (m, 1 H) 2,27 (s, 3 H) 1,94 -2,09 (m, 1 H) 1,80 - 1,93 (m, 1 H).
Ejemplos 2 a 33: Los compuestos enumerados en las Tablas 1-3 se prepararon mediante un procedimiento análogo al que se utilizó en el Ejemplo 1. El acoplamiento cruzado del resto R2 se puede realizar como segunda, tercera o cuarta etapa de la secuencia (véanse los Esquemas 1 , 2 y 3, respectivamente).
Alternativa para el Ejemplo 7: 2-amino-4-((2S,4S)-2-(6-fluoro-5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroqmnazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo
a) Ácido 2-bromo-3-fluoro-6-nitrobenzoico
Una solución de ácido 2-bromo-3-fluorobenzoico (registro CAS 132715-69-6) (155 g, 0,672 mol) en H2SO4 conc. (750 ml, 14 mol) se enfrió a 0°C y se añadió gota a gota HNO3 fumante (39 ml, 0,864 mol) a lo largo de un período de 1 hora, manteniendo la temperatura entre 0-5°C. Se formó un precipitado blanco. La suspensión resultante se dejó calentar a temperatura ambiente y se agitó durante 3 h. La mezcla se vertió en agua con hielo (3 L), se agitó durante 30 min y se extrajo con 3 x 1 L de DCM. Las fases orgánicas se combinaron, se lavaron con salmuera y se secaron sobre sulfato de sodio. Después de la evaporación del disolvente, se obtuvieron 160 g de un sólido blanquecino en forma de una mezcla de ácido 2-bromo-3-fluoro-6-nitrobenzoico (regioisómero 1) y ácido 2-bromo-3-fluoro-5-nitrobenzoico (regioisómero 2) en una relación de aprox. 2:1. El producto bruto se disolvió en NaOH 2 N (2 L) y el pH se ajustó a 2 mediante la adición de HCl conc. La extracción con 4 x 2 L de acetato de etilo condujo a la separación del regioisómero 2 en la fase orgánica, mientras que el regioisómero 1 permaneció principalmente en la fase acuosa. Después de la separación del regioisómero 2, la fase acuosa se ajustó a pH 1 añadiendo más HCl conc. y se extrajo por segunda vez con 2 x 1 L de acetato de etilo. La fase orgánica se lavó con salmuera, se secó sobre sulfato de sodio y se evaporó para dar un sólido beige (92 g, 47%) que contenía 91% de regioisómero 1 y 6% de regioisómero 2.
HPLC 1er regioisómero (91%) Rt= 0,33 min; HPLC 2° regioisómero (6%) Rt= 0,52 min.
1H RMN (600 MHz, DMSO-d6): 58,34 - 8,37 (dd, 1 H) 7,73 - 7,76 (t, 1 H)
b) Ácido 6-amino-2-bromo-3-fluorobenzoico
Ácido 2-bromo-3-fluoro-6-nitrobenzoico (87 g, 0,3 mol, 91% de regioisómero 1) se disolvió en THF (870 ml) y se desgasificó y purgó sucesivamente con nitrógeno (5 veces). Luego, la solución amarilla se trató con paladio al 10% sobre carbono (26 g). La mezcla negra se purgó, luego se volvió a llenar con gas hidrógeno y se dejó agitar bajo atmósfera de hidrógeno a ta durante 16 h. La mezcla de reacción se filtró a través de hyflo y la almohadilla se enjuagó con THF (2 veces). Los filtrados recogidos se evaporaron in vacuo para proporcionar un sólido beige (76 g) que se suspendió en dietiléter (300 ml) y se agitó durante 1 h a 0°C. La suspensión se filtró y el sólido se secó in vacuo para dar 60 g (85%) de ácido 6-amino-2-bromo-3-fluorobenzoico en forma de un isómero único.
HPLC (100%) Rt= 0,47 min; ESIMS: 234, 236 [(M+H)+]
1H RMN (600 MHz, DMSO-d6): 57,14 - 7,17 (t, 1 H) 6,74 - 6,77 (dd, 1 H)
c) 2-(5-bromo-6-fluoro-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-((terc.-butildimetilsilil)oxi)pirrolidina-1-carboxilato de (2S,4S)-terc.-butilo
Una solución seca de una mezcla de ácido 6-amino-2-bromo-3-fluorobenzoico (63 g, 242 mmol) y ácido (2S,4S)-1-(terc.-butoxicarbonil)-4-((terc.butildimetilsilil)oxi)pirrolidina-2-carboxílico (registro CAS 401564-17-8) (120 g, 70% de contenido, 243 mmol) en piridina (370 ml) se trató con fosfito de trifenilo (registro CAS 101-02-0) (159 ml, 606 mmol). La solución parda clara se agitó a 80°C durante 4 h y luego se añadió gota a gota anilina (registro CAS 62 -53-3) (26,5 ml, 291 mmol). Después de agitar la mezcla de reacción durante 1 h más a 80°C, la piridina se separó a presión reducida y luego el residuo se diluyó con EtOAc, se lavó con NaOH 1 N (2 x 1 L) y salmuera. Las fases orgánicas se secaron sobre Na2SO4 y se concentraron a presión reducida. El producto bruto se purificó por cromatografía sobre gel de sílice (0 a 15% de EtOAc / heptano) para proporcionar el compuesto del título en forma de una espuma amarilla (53 g, rendimiento del 35%, pureza del 90%) que se utilizó sin purificación adicional para la siguiente etapa.
HPLC Rt= 1,57 min; ESIMS: 618, 620 [(M+H)+].
d) 4-((terc.-butildimetilsilil)oxi)-2-(6-fluoro-5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidina-1-carboxilato de (2S,4S)-terc.-butilo
A una solución de 2-(5-bromo-6-fluoro-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4- ((terc.-butildimetilsilil)oxi)pirrolidina-1-carboxilato de (2S,4S)-terc.-butilo (53 g, 86 mmol,> 90% de contenido) en NMP (600 ml) se añadió ácido (2-metoxipiridin-5-il)borónico (registro CAS 628692-15-9) (19,8 g, 129 mmol), acetato de paladio(II) (registro CAS 3375-31-3) (1,92 g, 8,6
mmol), carbonato de sodio (27,2 g, 257 mmol) y X-Phos (registro CAS 564483-18-7 ) (8,17 g, 17,1 mmol). La mezcla de reacción se calentó a 140°C y se agitó durante 1 h. Después de enfriar a temperatura ambiente, la mezcla se repartió entre acetato de etilo (1 L) y agua (1 L). La fase acuosa se extrajo con acetato de etilo (1 L) y las fases orgánicas combinadas se lavaron con salmuera (1 L), se secaron sobre sulfato de sodio y se evaporaron. El producto bruto se purificó por cromatografía sobre gel de sílice (0 a 40% acetato de etilo / hexano) para proporcionar una espuma amarilla (37 g) que se disolvió nuevamente en DCM (300 ml) y se trató con resina PL-TMT MP (0,66 mmol/g) durante 18 h. Después de filtración y evaporación in vacuo, se obtuvo el compuesto del título en forma de una espuma blanca (35 g, 63% de rendimiento).
HPLC Rt= 1,52 min; ESIMS: 648 [(M+H)+].
e) 6-fluoro-2-((2S,4S)-4-hidroxipirroNdm-2-M)-5-(2-metoxipirimidm-5-M)-3-femlqumazoNn-4(3H)-ona
A ta, una solución de 4-((terc.-butildimetilsilil)oxi)-2-(6-fluoro-5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidina-1 -carboxilato de (2S,4S)-terc.-butilo (35 g, 54 mmol) en t Hf (400 ml) se trató con soln. ac. de H3PO4 (600 ml, 85% en peso). La mezcla se agitó durante 3 h a ta. Se añadió agua y la mezcla se basificó con una soln. sat. ac. de Na2 CO3 a pH ~ 10. La mezcla se extrajo con EtOAc (2 x 5 L) y DCM (2 x 5 L), a continuación, se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró in vacuo para proporcionar un sólido amarillo pálido (22,3 g, 90% de rendimiento) que se utilizó sin purificación adicional para la siguiente etapa.
HPLC Rt= 0,60 min; ESIMS: 434 [(M+H)+].
f) 2-amino-4-((2S,4S)-2-(6-fluoro-5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo
Una solución de 6-fluoro-2-((2S,4S)-4-hidroxipirrolidin-2-il)-5-(2-metoxipirimidin-5-il)-3-fenilquinazolin-4(3H)-ona (22 g, 50,8 mmol) y 2-amino-4-cloro-6-metilpirimidina-5-carbonitrilo (registro CAS 99586-66-0) (9 g, 50,8 mmol) en EtOH (600 ml) se trató con DIPEA (17,7 ml, 102 mmol) y se irradió a 120°C durante 60 min en un mw.
La mezcla de reacción se evaporó a presión reducida. El aceite se recogió en DCM y se lavó con soln. sat. ac. de NaHCO3 , la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó por cromatografía sobre gel de sílice (10 a 100% de acetato de etilo / hexano) para proporcionar 18 g (63% de rendimiento) del compuesto del título en forma de un sólido incoloro.
HPLC Rt= 0,85 min; ESIMS: 566 [(M+H)+].
1H RMN (600 MHz, DMSO-cfe) 58,55 (s a, 2H), 7,83 - 7,78 (m, 2H), 7,73 (dd, 1H), 7,58 - 7,52 (m, 2H), 7,52 - 7,48 (t, 1H), 7,41 (d, 1H), 7,05 (s a, 1H), 6,65 (s a., 1H), 5,32 (s a., 1H), 4,61 (s a., 1H), 4,18 (m, 2H), 3,93 (s, 3H), 3,73 (s a, 1H), 2,29 (s, 3H), 2,04 (s a, 1H), 1,91 - 1,85 (m, 1H).
Cristalización del Ejemplo 7
Se pesaron 21 g del Ejemplo 7 amorfo en un matraz de fondo redondo y se agitó a 300 rpm en 400 mL de agua purificada durante 7 días. El agua se filtró y los sólidos recogidos se secaron en vacío a 60°C hasta peso constante. Se observó que el material resultante era cristalino según el análisis de difracción de rayos X en polvo, con una temperatura de inicio de fusión a 178,1°C, medida por calorimetría diferencial de barrido a 10°C/min (tamaño de muestra 2,2 mg). Se descubrió que la forma cristalina del Ejemplo 7 contenía 10,7% (p/p) de agua según se determinó mediante análisis elemental y es probable que sea un trihidrato.
Lista de los picos de 2-Theta más significativos del Patrón de Difracción de rayos X en Polvo con tolerancias ± 0,1 de la forma cristalina del Ejemplo 7:


Tabla 1: Compuestos preparados de acuerdo con el Esquema 1 (acoplamiento cruzado de arilo en la segunda etapa) Esquema 1:

R1, R2, R3, R5 son como se definieron arriba para un compuesto de fórmula (Ib), Hal1 es un halógeno, tal como un bromuro o un cloruro, R es H o R7 como se definió arriba para un compuesto de fórmula (Ib); R' es R o un sustituyente que puede transferirse a R mediante uno o más etapas de funcionalización o etapa de ajuste del grupo funcional.
a) A ta, una solución seca de derivado de ácido antranílico II (1 eq.) y de derivado de aminoácido cíclico enantioméricamente puro III (1 eq.) en piridina (0,83 M) se trató con fosfito de trifenilo (2,5 eq.) y la mezcla de reacción se agitó a 70°C durante 2 a 18 h y luego se trató con un compuesto amino IV. La mezcla de reacción se agitó a 70 a 80°C durante 2 a 18 h.
b1) Acoplamiento de Stille
Bajo una atmósfera de argón se trató una solución de compuesto intermedio de cloro o bromo V (1 eq.) en DMF (0,07 a 0,1 M) con heteroaril tributilestannano VI (1,1 a 1,5 eq.) y bis (tri-t-butilfosfina)paladio(ü) (0,1 eq.). El tubo se selló y la mezcla de reacción se agitó a 80°C a 100°C durante 3 h a 4 d.
o b2) acoplamiento de Suzuki
Una solución de compuesto intermedio de cloro o bromo V (1 eq.), ácido heteroaril borónico VI (2 eq.), Pd(OAc)2 (0,2 eq.) / BrettPhos (0,6 eq.) o paladaciclo BrettPhos (registro CAS 1148148-01-9 ) (0,2 eq.) y Na2CO3 (3 eq.) en NMP (0,094 M) se desgasificó y se rellenó de nuevo con argón (3 veces). La mezcla de reacción se irradió en un mw a 150°C a 160°C durante 1 a 3 h bajo absorción normal con recarga de catalizador y ligando si fuera necesario.
c ) A ta, una solución seca de compuesto intermedio protegido VII (1 eq.) en THF o DCM (0,1 a 0,6 M) se trató con TFA (10 eq.), soln. ac. de H3PO4 (85% en peso) o con soln. ac. de H3PO4 , seguido de TFA y la mezcla de reacción se agitó a ta durante 1 h a 5 d.
d) A ta, una solución de compuesto intermedio desprotegido (1 eq.) y un haluro de arilo VIII (1 a 2,5 eq.) en alcohol (nBuOH o EtOH, 0,08 M a 0,1 M) se trató con DIPEA (2 a 5 eq.) y se irradió en un reactor de microondas o se calentó en un baño de aceite a 120-160°C durante 0,5 h a 6 d.
La purificación de los compuestos intermedios y finales IX se realizó por cromatografía de resolución instantánea , HPLC o SFC.






Tabla 2: Compuestos preparados de acuerdo con el Esquema 2 (acoplamiento cruzado de arilo en la tercera etapa)

R1, R2, R3, R5 son como se definieron arriba para un compuesto de fórmula (Ib), Hal1 es un halógeno, tal como un bromuro o un cloruro, R es H o R7 como se definió arriba para un compuesto de fórmula (Ib); R' es R o un sustituyente que puede transferirse a R mediante uno o más etapas de funcionalización o etapa de ajuste del grupo funcional.
a) A ta, una solución seca de derivado de ácido antranílico II (1 eq.) y de derivado de aminoácido cíclico enantioméricamente puro III (1 eq.) en piridina (0,83 M) se trató con fosfito de trifenilo (2,5 eq.) y la mezcla de reacción se agitó a 70°C durante 2 a 18 h y luego se trató con un compuesto amino IV. La mezcla de reacción se agitó a 70 a 80°C durante 2 a 18 h.
b ) A ta, una solución seca de compuesto intermedio protegido (1 eq.) en THF o DCM (0,1 a 0,6 M) se trató con TFA (10 eq.), soln. ac. de H3 PO4 (85% en peso) o con soln. ac. de H3PO4, seguido de TFA y la mezcla de reacción se agitó a ta durante 1 h a 5 d.
c) Acoplamiento de Stille
Bajo una atmósfera de argón se trató una solución de compuesto intermedio de cloro o bromo X (1 eq.) en DMF (0,07 a 0,1 M) con heteroaril tributilestannano VI (1,1 a 1,5 eq.) y bis(tri-t-butilfosfina)paladio(0) (0,1 eq.). El tubo se selló y la mezcla de reacción se agitó a 80°C a 100°C durante 3 h a 4 d.
d) A ta, una solución de compuesto intermedio XI (1 eq.) y un haluro de arilo VIII (1 a 2,5 eq.) en alcohol (nBuOH o EtOH, 0,08 M a 0,1 M) se trató con DIPEA (2 a 5 eq.) y se irradió en un reactor de microondas o se calentó en un baño de aceite a 120-160°C durante 0,5 h a 6 d.
La purificación de los compuestos intermedios y finales IX se realizó por cromatografía de resolución instantánea , HPLC o SFC.

Tabla 3: Compuestos preparados de acuerdo con el Esquema 3 (acoplamiento cruzado de arilo en la cuarta etapa)

R1, R2, R3, R5 son como se definieron arriba para un compuesto de fórmula (Ib), Hal1 es un halógeno, tal como un bromuro o un cloruro, R es H o R7 como se definió arriba para un compuesto de fórmula (Ib); R' es R o un sustituyente que puede transferirse a R mediante uno o más etapas de funcionalización o etapa de ajuste del grupo funcional.
a) A ta, una solución seca de derivado de ácido antranílico II (1 eq.) y de derivado de aminoácido cíclico enantioméricamente puro III (1 eq.) en piridina (0,83 M) se trató con fosfito de trifenilo (2,5 eq.) y la mezcla de reacción se agitó a 70°C durante 2 a 18 h y luego se trató con un compuesto amino IV. La mezcla de reacción se agitó a 70 a 80°C durante 2 a 18 h.
b ) A ta, una solución seca del compuesto intermedio protegido (1 eq.) en THF o DCM (0,1 a 0,6 M) se trató con TFA (10 eq.), soln. ac. de H3PO4 (85% en peso) o con soln. ac. de H3PO4 , seguido de TFA y la mezcla de reacción se agitó a ta durante 1 h a 5 d.
c) A ta, una solución de compuesto intermedio desprotegido X (1 eq.) y un haluro de arilo VIII (1 a 2,5 eq.) en alcohol (nBuOH o EtOH, 0,08 M a 0,1 M) se trató con DIPEA (2 a 5 eq.) y se irradió en un reactor de microondas o se calentó en un baño de aceite a 120-160°C durante 0,5 h a 6 d.
d1) Acoplamiento de Stille
Bajo una atmósfera de argón se trató una solución de compuesto intermedio de cloro o bromo XII (1 eq.) en DMF (0,07 a 0,1 M) con heteroaril tributilestannano VI (1,1 a 1,5 eq.) y bis(tri-t-butilfosfina)paladio(0) (0,1 eq.). El tubo se selló y la mezcla de reacción se agitó a 80°C a 100°C durante 3 h a 4 d.
o d2) acoplamiento de Suzuki
Una solución de compuesto intermedio de cloro o bromo XII (1 eq.), un ácido heteroaril borónico o éster boronato de heteroarilo VI (1,2-2 eq.), Pd(OAc)2 (0,2 eq.) / BrettPhos (0,6 eq.), paladaciclo BrettPhos (registro CAS 1148148-01-9 ) (0,2 eq.) o PdCl2(PPh3)2 (0,07 eq.) y Na2CO3 (2,4-3 eq.) en NMp (0,094 M) o acetonitrilo (0,125 M) se desgasificó y se rellenó de nuevo con argón (3 veces). La mezcla de reacción se irradió en un mw a 150°C a 160°C durante 5 min a 3 h con recarga de catalizador y ligando si fuera necesario.
O d3) Acoplamiento de Sonogashira
Una solución de compuesto intermedio de bromo XII (1 eq.), un alquino VI (3 eq.), Pd(Ph3)4 (0,05 eq.), CuI (0,1 eq.) y Et3N (3 eq.) en DMF (0,1 M) en un vial cerrado se calentó a 100°C durante 18 h. Las mismas cantidades de Pd(Ph3)4, CuI y alquino se volvieron a cargar para completar la conversión y la mezcla de reacción se agitó durante otros 4 d a 100°C. Cualquiera de los grupos funcionales presentes en R2' se pueden funcionalizar adicionalmente en etapas adicionales como se describe en los ejemplos individuales que se muestran más adelante.
La purificación de los compuestos intermedios y finales IX se realizó por cromatografía de resolución instantánea , HPLC o SFC.




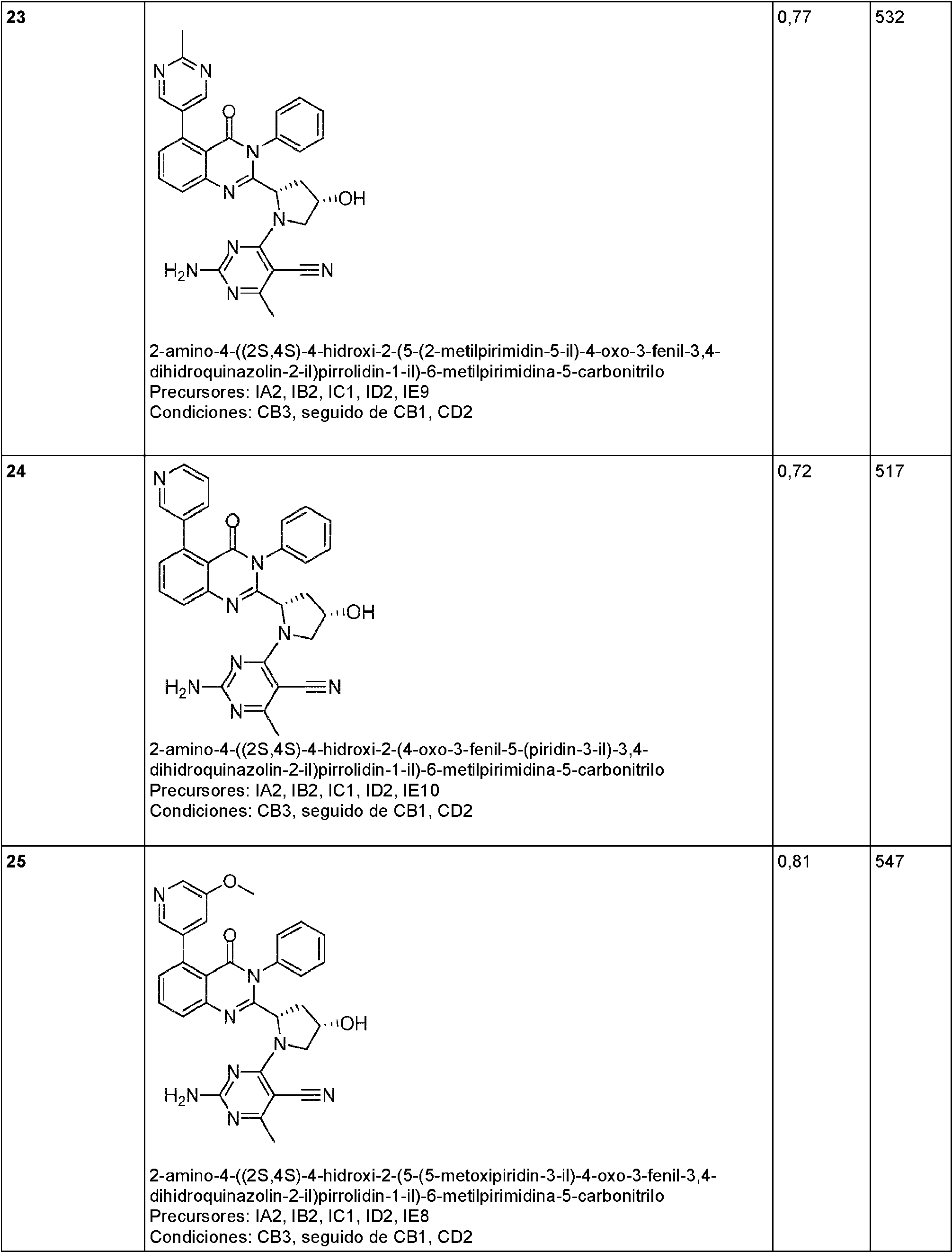




CA) Ciclación, utilizando POPh3
A ta, una solución seca de derivado de aminoácido (1 eq.) y derivado de ácido antranílico (1 eq.) en piridina (0,83 M) se trató con fosfito de trifenilo (2,5 eq.) y la mezcla de reacción se agitó a 70°C durante 2 a 18 h y luego se trató con el derivado amino o hidrazina. La mezcla de reacción se agitó a 70°C durante 2 a 18 h. La mezcla de reacción se diluyó con EtOAc, se lavó con soln. sat. ac. de NaHCO3 (2 veces) o soln. sat. ac. de CuSO4 y salmuera. La fase orgánica se secó sobre Na2SO4 y se concentró a presión reducida. El producto bruto se purificó por cromatografía sobre gel de sílice (heptano / EtOAc).
CB) Condiciones de desprotección
CB1) Utilizando TFA
A ta, una solución seca de compuesto intermedio de Boc (1 eq.) en DCM (0,1 a 0,25 M) se trató con TFA (10 eq.) y la mezcla de reacción se agitó a ta durante 1 a 18 h. La mezcla de reacción se basificó con una soln. sat. ac. de NaHCO3 a pH ~ 9. La mezcla se extrajo con DCM (2 veces), a continuación, se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró in vacuo.
CB2) Utilizando H3 PO4
b ) A ta, una solución seca de derivado de aminoácido (1 eq.) en THF (0,2 a 0,6 M) se trató con eq.), soln. ac. de H3PO4 (85% en peso, adquirido de Aldrich) (mismo volumen que el disolvente). La mezcla de reacción se agitó a ta durante 24 h a 6 d. Se añadió agua y la mezcla se basificó con una soln. sat. ac. de NaHCO3 a pH ~ 9. La mezcla se extrajo con EtOAc (2 veces) y DCM (2 veces), después se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentraron in vacuo.
CB3) Utilizando H3 PO4 y TFA
A ta, una solución seca de derivado de aminoácido (1 eq.) en THF (0,085 M) se trató con eq.), soln. ac. de H3PO4. (85% en peso, adquirido de Aldrich) (10 eq.). La mezcla de reacción se agitó a ta durante 24 h, luego se añadieron TFA (13 eq.) y DCM (0,042 M) y la mezcla de reacción se agitó a ta durante 3 d. Se realizó un tratamiento básico y, después de concentrar la capa orgánica, el residuo se diluyó con DCM (0,14 M) y se trató nuevamente con TFA (13 eq.), y se agitó a ta durante 18 h. La mezcla de reacción se basificó con una soln. sat. ac. de NaHCO3 a pH ~ 9. La mezcla se extrajo con DCM (2 veces), a continuación, se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró in vacuo. CC) Acoplamiento del resto R3
A ta, una solución de compuesto intermedio desprotegido (1 eq.) y un haluro de arilo (1 a 2,5 eq.) en alcohol (nBuOH o EtOH, 0,08 M a 0,1 M) se trató con DIPEA (2 a 5 eq.) y se calentó en un reactor de microondas o baño de aceite a 120-160°C durante 0,5 h a 6 d. La mezcla de reacción se evaporó a presión reducida. El residuo se recogió en DCM y se lavó
con soln. sat. ac. de NaHCO3 , la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó sobre cromatografía de resolución instantánea, SFC o RP-HPLC prep.
CD) Acoplamiento del resto R2
CD1) Utilizando el compuesto intermedio IE1)-IE4)
Bajo atmósfera de argón, se trató una solución de compuesto intermedio de cloro o bromo (1 eq.) en DMF (0,07 a 0,1 M) con IE (1,1 a 1,5 eq.) y bis(tri-t-butilfosfina)paladio (0) (0,1) eq.). El tubo se selló y la mezcla de reacción se agitó a 80°C a 100°C durante 3 h a 4 d. La mezcla de reacción se diluyó con EtOAc y se lavó con agua, la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó sobre cromatografía de resolución instantánea, SFC o RP-HPLC prep.
CD2) Utilizando compuestos intermedios IE5)-IE11)
Para IE5): Una solución de compuesto intermedio de cloro o bromo (1 eq.) en acetonitrilo (0,125 M) se trató con IE5) (1,2 eq.) seguido de una soln. ac. de Na2CO3 1 M. (2,4 eq.) y PdCh(PPh3)2 (0,07 eq.). La mezcla de reacción se calentó en un horno de mw a 150°C durante 5 min bajo absorción normal . La mezcla de reacción se diluyó con EtOAc y se lavó con agua, la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó sobre cromatografía de resolución instantánea, SFC o RP-HPLC prep.
Para IE6)-IE11): Una solución de compuesto intermedio de cloro o bromo (1 eq.), IE (2 eq.), Pd(OAc)2 (0,2 eq.) / BrettPhos (0,6 eq.) o paladaciclo BrettPhos (registro CAS 1148148-01-9 ) (0,2 eq.), Na2CO3 (3 eq.) en NMP (0,094 M) se desgasificó y se rellenó de nuevo con argón (3 veces). La mezcla de reacción se irradió en un mw a 150°C a 160°C durante 1 a 3 h bajo absorción normal con recarga de catalizador y ligando si fuera necesario. La mezcla de reacción se diluyó con EtOAc y se lavó con agua, la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó sobre cromatografía de resolución instantánea, SFC o RP-HPLC prep.
CD3) Utilizando el compuesto intermedio IE12)
Una solución de compuesto intermedio de bromo (1 eq.), IE12 (3 eq.), Pd(Ph3)4 (0,05 eq.), CuI (0,1 eq.), Et3N (3 eq.) en DMF (0,1 M) en un vial cerrado se calentó a 100°C durante 18 h. Las mismas cantidades de Pd(Ph3)4, CuI e IE12 se volvieron a cargar para completar la conversión y la mezcla de reacción se agitó durante otros 4 d a 100°C. La mezcla de reacción se diluyó con EtOAc y se lavó con agua, la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó sobre cromatografía de resolución instantánea, SFC o RP-HPLC prep.
CE) Desacilación para el ejemplo 28
Una solución del derivado de acetilo correspondiente (1 eq.) en MeOH seco (0,1 M) se trató con K2CO3 (3 eq.). La suspensión se agitó durante 2 h a ta. Se evaporó a presión reducida y el residuo se recogió en EtOAc y se lavó con soln. ac. de HCl 1 N, la capa orgánica se secó sobre Na2SO4 y se concentró in vacuo. El producto bruto se purificó sobre SFC.
Intermedios para ejemplos de tablas
IA) Derivados de aminoácidos
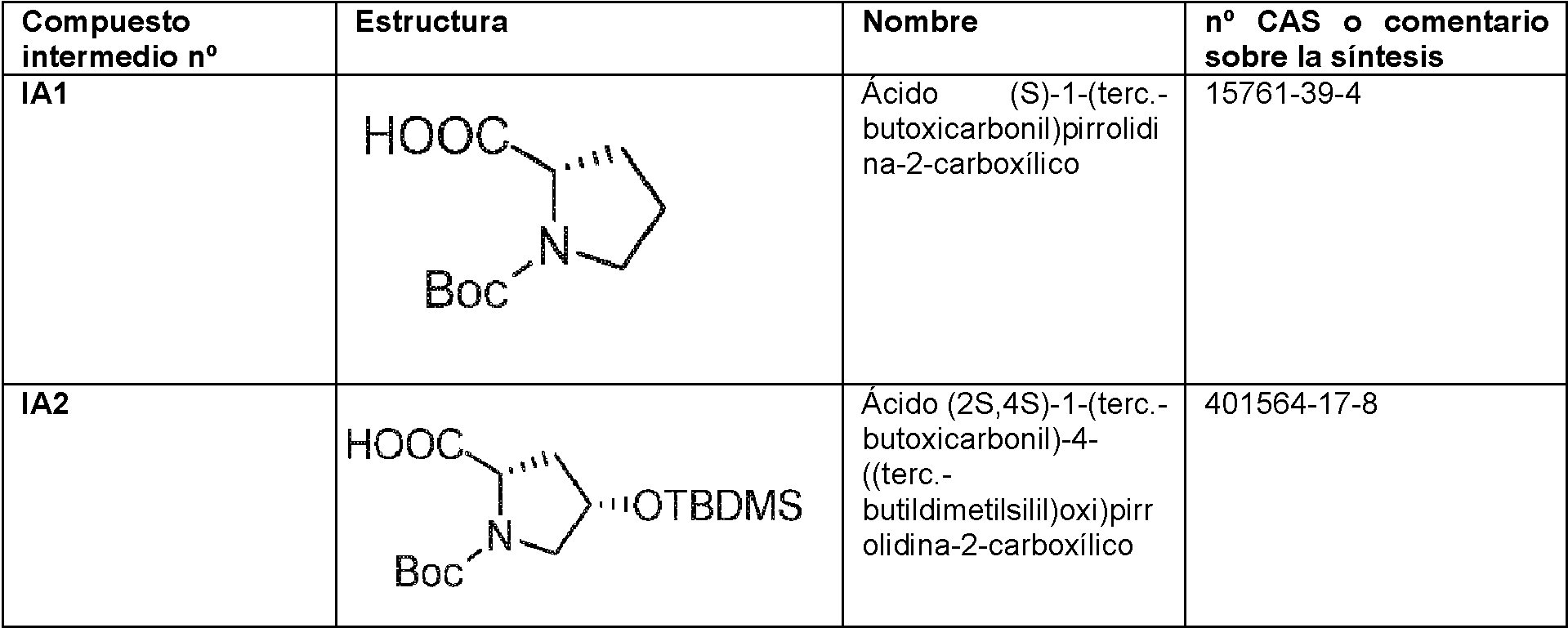

IB D riv l Á i Anr níli

I Amin Hi r zin

ID Pr r r R


IE Pr r r R2



Evaluación Biológica
La actividad de un compuesto de acuerdo con la presente invención se puede evaluar mediante los siguientes métodos in vitro e in vivo.
Ensayos biológicos
1 Determinación de la inhibición enzimática de la isoforma PI3K alfa , PI3Kbeta, PI3Kgamma y PI3K delta
1.1 Generación de construcciones génicas, expresión y purificación de proteínas
Para los ensayos enzimáticos, la preparación de construcciones de genes PI3K, la expresión de proteínas y la purificación para HTS se describen en el documento WO2012 / 004299.
1.2 Ensayos bioquímicos para PI3Kalfa, PI3Kbeta (formato Kinase-Glo)
La eficacia de los compuestos de los ejemplos 1-117 como inhibidores de la quinasa PI3 se puede demostrar de la siguiente manera:
La reacción de la quinasa se realiza en un volumen final de 50 |jl por pocillo de una placa COSTAR de zona media de 96 pocillos. Las concentraciones finales de ATP y fosfatidil inositol en el ensayo son 5 jM y 6 jg/mL, respectivamente. La reacción se inicia mediante la adición de quinasa PI3, p. ej. quinasa PI3 a.
p110S. Los componentes del ensayo se añaden por pocillo de la siguiente manera:
• 10 j l de compuesto de ensayo en DMSO al 5% por pocillo en las columnas 2-1.
• La actividad total se determina mediante la adición de 10 j l de DMSO al 5% vol/vol en los primeros 4 pocillos de la columna 1 y los últimos 4 pocillos de la columna 12.
• El fondo se determina mediante la adición de compuesto de control 10 jM a los últimos 4 pocillos de la columna 1 y los primeros 4 pocillos de la columna 12.
• Se preparan 2 mL de 'mezcla de ensayo' por placa:
1,912 mL de tampón de ensayo He PES
8,33 j l de material 3 mM de ATP, que proporciona una concentración final de 5 jM por pocillo 1 j l de [33P]ATP en la fecha de actividad, dando 0,05 j Ci por pocillo
30 j l de 1 mg/mL de material PI que proporciona una concentración final de 6 jg/m L por pocillo
5 j l de material 1 M de MgCl2 , que proporciona una concentración final de 1 mM por pocillo • Se añaden 20 j l de la mezcla de ensayo por pocillo.
• Se preparan 2 mL de 'Mezcla de enzimas' por placa (x * j l quinasa PI3 p110a en 2 mL de tampón de quinasa). La 'Mezcla de enzimas' se mantiene en hielo durante la adición a las placas de ensayo.
• Se añaden 20 j l de 'Mezcla de enzimas'/pocillo para comenzar la reacción.
• La placa se incuba a temperatura ambiente durante 90 minutos.
• La reacción termina mediante la adición de 50 |jl de perla WGA-SPA (perlas de ensayo de centelleo de proximidad recubiertas con aglutinina de germen de trigo) por pocillo.
• La placa de ensayo se sella con TopSeal-S (termosellado para microplacas de poliestireno, PerkinElmer LAS [Deutschland] GmbH, Rodgau, Alemania) y se incuba a temperatura ambiente durante al menos 60 minutos. • La placa de ensayo se centrifuga luego a 1500 rpm durante 2 minutos utilizando la centrífuga de mesa Jouan (Jouan Inc., Nantes, Francia).
• La placa de ensayo se cuenta utilizando un Packard TopCount, cada pocillo se cuenta durante 20 segundos.
* El volumen de la enzima depende de la actividad enzimática del lote en uso.
En un ensayo más preferido, la reacción de la quinasa se realiza en un volumen final de 10 j l por pocillo de una placa negra CORNING de 384 pocillos de no unión de bajo volumen (Cat. N° 3676). Las concentraciones finales de ATP y fosfatidil inositol (PI) en el ensayo son 1 jM y 10 jg/mL, respectivamente. La reacción se inicia mediante la adición de ATP.
Los componentes del ensayo se añaden por pocillo de la siguiente manera:
50 nl de compuestos de ensayo en DMSO al 90% por pocillo, en las columnas 1-20, 8 concentraciones (1/3 y 1/3,33 etapa de dilución en serie) de una vez.
• Control bajo: 50 nl de DMSO al 90% en la mitad de los pocillos de las columnas 23-24 (0,45% al final).
• Control alto: 50 nl de compuesto de referencia (p. ej., compuesto del Ejemplo 7 en el documento WO 2006/122806) en la otra mitad de las columnas 23-24 (2,5 jM al final).
• Estándar: 50 nl de compuesto de referencia como se acaba de mencionar diluido como los compuestos de ensayo en las columnas 21-22.
• Se preparan 20 mL de 'tampón' por ensayo:
200 j l de TRIS HCl 1 M pH 7,5 (10 mM al final)
60 j l de MgCl2 1 M (3 mM al final)
500 j l de NaCl 2 M (50 mM al final)
100 j l de CHAPS al 10% (0,05% al final)
200 j l de DTT 100 mM (1 mM al final)
18,94 mL de agua nanopura
• Se preparan 10 mL de 'PI' por ensayo:
200 j l de 1 mg/mL de l-alfa-fosfatidilinositol (Liver Bovine, Avanti Polar Lipids Cat. N° 840042C PM=909,12) preparado en OctilGlucósido al 3% (10 jg/m L al final)
9,8 mL de 'tampón'
• Se preparan 10 mL de 'ATP' por ensayo:
6,7 j l de material 3 mM de ATP, que proporciona una concentración final de 1 jM por pocillo 10 mL de 'tampón'
• Se preparan 2,5 mL de cada una de las construcciones PI3K por ensayo en ' PI ' con la siguiente concentración final:
PI3K alfa EMV B1075 10 nM
beta EMV BV94925 nM
• Se añaden 5 j l de 'PI/PI3K' por pocillo.
• Se añaden 5 j l de 'ATP' por pocillo para comenzar la reacción.
• Las placas se incuban luego a temperatura ambiente durante 60 minutos (alfa, beta, delta) o 120 minutos (gamma). La reacción se termina mediante la adición de 10 j l de Kinase-Glo (Promega Cat. N° 6714).
• Las placas de ensayo se leen después de 10 minutos en el lector Synergy 2 (BioTek, Vermont EE.UU.) con un tiempo de integración de 100 milisegundos y sensibilidad establecida en 191.
• Salida: El control Alto es de alrededor de 60.000 recuentos y el control Bajo es de 30.000 o menor
Este ensayo de luminiscencia proporciona una relación Z' útil entre 0,4 y 0,7.
El valor Z' es una medida universal de la robustez de un ensayo. Una Z' entre 0,5 y 1,0 se considera un excelente ensayo.
1.3 Ensayos bioquímicos para PI3Kdelta, PI3Kgamma (formato Adapta)
El kit para ensayos de cinasas universal TR-FRET Adapta™ se adquirió de Invitrogen Corporation (Carlsbad/CA, EE. UU.) (Cat. N° PV5099). El kit contiene los siguientes reactivos: anticuerpo anti-ADP-Eu de Adapta (anticuerpo anti-ADP marcado con europio en solución salina tamponada con HEPES, Cat. N° PV5097), trazador de ADP marcado con Alexa Fluor® 647 (trazador de ADP marcado con Alexa Fluor® 647 en solución salina tamponada con HEPES, Cat. N° PV5098), tampón de dilución patentado TR-FRET pH 7,5 (Cat. N° PV3574).
El sustrato PIK3CD Fosfatidilinositol se obtuvo de Invitrogen (vesículas que consisten en PI 2 mM en HEPES 50 mM pH 7,5; Cat. N° PV5371). Sustrato PIK3CG Fosfatidilinositol-4,5-bisfosfato (PIP (4,5)2 se obtuvo de Invitrogen (PIP2:PS vesículas unilamelares grandes que consisten en PIP2 1 mM: PS 19 mM en h Ep ES 50 mM pH 7,5, MgCl23 mM, EGTA 1 mM; Cat. N° PV5100).
La transferencia de energía por resonancia de fluorescencia resuelta en el tiempo (TR-FRET) es una tecnología basada en la transferencia de energía entre dos colorantes adyacentes, desde un electrón excitado en un colorante (el dador)
hasta un electrón de un colorante adyacente (el aceptor) a través de resonancia, que a continuación se libera como un fotón. Esta transferencia de energía se detecta mediante un incremento en la emisión de fluorescencia del aceptor y una reducción en la emisión de fluorescencia del dador. Los ensayos de TR-FRET para cinasas de proteínas utilizan quelatos de un lantánido de vida prolongada de terbio o europio como especie dadora, los cuales resuelven la interferencia procedente de la autofluorescencia del compuesto o la dispersión de la luz de compuestos precipitados, mediante la introducción de un retraso tras la excitación por parte de una fuente de excitación de una lámpara flash. Los resultados se expresan a menudo como una relación de las intensidades de los fluoróforos del aceptor y el dador. La naturaleza radiométrica de un valor de este tipo corrige las diferencias en los volúmenes de ensayo entre pocillos, a la vez que corrige efectos de desactivación debidos a compuestos coloreados. El ensayo Adapta™ se puede dividir en dos fases: una fase de reacción de la cinasa y una fase de detección del ADP. En la fase de reacción de la cinasa, todos los componentes de la reacción de la cinasa se añaden al pocillo y se deja incubar la reacción durante un periodo de tiempo fijo específico para cada cinasa. Después de la reacción, se añade al pocillo de ensayo una solución de detección de anticuerpo anti-ADP marcado con Eu, trazador de ADP marcado con Alexa Fluor® 647 y EDTA (para detener la reacción de la cinasa). El ADP formado por la reacción de la cinasa desplazará al trazador de ADP marcado con Alexa Fluor® 647 del anticuerpo, lo que dará como resultado una reducción en la señal de TR-FRET. En presencia de un inhibidor, la cantidad de ADP formada por la reacción de la cinasa se reduce y la interacción resultante de trazador-anticuerpo intacto mantiene una señal de TR-FRET elevada. En el ensayo Adapta™, el dador (anticuerpo anti-ADP-europio) se excita a 340 nm y transferirá su energía al aceptor (trazador de a Dp marcado con Alexa Fluor® 647). La emisión procedente de Alexa Fluor® 647 se puede monitorizar con un filtro centrado a 665 nm porque está localizada entre los picos de emisión del dador, el cual se mide a 615/620 nm.
Se dispensaron 50 nL de diluciones de compuesto en una placa blanca de poliestireno de pequeño volumen de 384 pocillos tal como se describe en la sección 2.2. Luego se incuban a TA 5 |jL de PI3Kgamma y PI3Kdelta y sustrato lipídico (PI o PIP2:PS) seguido de 5 j L de ATP (volumen de ensayo final 10 j L). El tampón de reacción estándar para el ensayo Adapta™ TR-FRET contenía Tris-HCl 10 mM, pH 7,5, MgCl2 3 mM, NaCl 50 mM, DTT 1 mM, CHAPS al 0,05%. Las reacciones se detuvieron con 5 j L de una mezcla de EDTA que contenía el anticuerpo anti-ADP marcado con Eu y Alexa Fluor® 647 - trazador ADP marcado en tampón de dilución TR-FRET (propiedad de IVG). Las placas se leyeron de 15 a 60 min más tarde en un lector Synergy2 utilizando un tiempo de integración de 0,4 segundos y un retraso de 0,05 segundos. El control para la inhibición del 100% de la reacción de la quinasa se realizó reemplazando la PI3K por el tampón de reacción estándar. El control para la inhibición de un 0% se obtuvo con el vehículo de disolvente de los compuestos (DMSO al 90% en H2O).
Los datos se analizaron utilizando el software de ajuste Excel o Graphpad Prism. Los valores de CE50 se obtuvieron ajustando una curva de dosis-respuesta sigmoidal a un gráfico de lectura de ensayo sobre la concentración de inhibidor. Todos los ajustes se llevaron a cabo con el programa XLfit4 (ID Business Solutions, Guildford, Reino Unido). La determinación de los valores de CE50 del porcentaje de inhibición de cada uno de los compuestos a 8 concentraciones (habitualmente 10, 3,0, 1,0, 0,3, 0,1, 0,030, 0,010 y 0,003 j M) n = 2 se obtuvo ajustando una curva dosis-respuesta sigmoidal a un gráfico de la lectura del ensayo sobre la concentración de inhibidor. Todos los ajustes se llevaron a cabo con el programa XLfit4 (ID Business Solutions, Guildford, Reino Unido).
En una realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma delta de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kdelta está entre 1 nM y 500 nM.
En otra realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma delta de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kdelta está entre 1 nM y 100 nM.
En otra realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma delta de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kdelta está entre 0,5 nM y 10 nM.
En una realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma gamma de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kgamma está entre 1 nM y 500 nM.
En otra realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma gamma de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kgamma está entre 1 nM y 100 nM.
En una realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma delta de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kdelta está entre 1 nM y 1000 nM.
En otra realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma delta de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kdelta está entre 1 nM y 500 nM.
En una realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma gamma de PI3K, en donde el intervalo de actividad, expresado como CI50 en el ensayo enzimático PI3Kgamma está entre 1 nM y 1000 nM.
En otra realización de la presente invención, el inhibidor de la quinasa PI3 de clase I, en donde dicho inhibidor tiene una acción inhibitoria sobre la isoforma gamma de PI3K, en donde el intervalo de actividad, expresado como CI50, en el ensayo enzimático PI3Kgamma está entre 1 nM y 500 nM.
2. Ensayos celulares
En otra realización de la presente invención, el inhibidor de la quinasa PI3 de clase I muestra una selectividad de al menos 10 veces sobre la isoforma alfa de PI3K en un ensayo celular.
En otra realización de la presente invención, el inhibidor de la quinasa PI3 de clase I muestra una selectividad de al menos 20 veces sobre la isoforma alfa de PI3K en un ensayo celular.
2.1 Fosforilación de Akt 1/2 (S473) mediada por fosfoinositida-3 quinasa (PI3K) en células Rat-1
Células Rat-1 que sobre-expresan de manera estable una forma miristoilada de la subunidad catalítica de fosfoinositida-3 quinasa (PI3K) alfa, beta o delta humana se sembraron en placas de 384 pocillos a una densidad de 7500 (PI3K alfa), 6200 (PI3K beta) o 4000 (PI3K delta) células en 30 ul de medio de crecimiento completo (medio Eagle modificado por Dulbecco (DMEM de alto contenido en glucosa) complementado con suero bovino fetal al 10% (v/v), aminoácidos no esenciales MEM al 1% (v/v), HEPES 10 mM, L-glutamina 2 mM, 10 |jg/mL de puromicina y Penicilina/Estreptomicina al 1% (v/v) y se incubaron a 37°C / 5% de CO2 / 95% de humedad durante 24 h. Los compuestos se diluyeron en placas compuestas de 384 pocillos (placas maestras compuestas), para obtener diluciones en serie de 8 puntos para 40 compuestos de ensayo en DMSO al 90%, así como 4 compuestos de referencia más 16 controles altos y 16 controles bajos (inhibidos). Las placas de predilución se prepararon dispensando mediante pipeteado 250 nl de soluciones de compuestos en placas de polipropileno de 3 8 4 pocillos (placas hijas), utilizando un dispensador de nanolitros Hummingwell. Los compuestos fueron prediluidos mediante la adición de 49,75 ul de medio de crecimiento completo. Se transfirieron 10 ul de solución de compuesto prediluido a la placa de células utilizando un pipeteador de 384 pocillos, dando como resultado una concentración final de DMSO de 0,11%. Las células se incubaron durante 1 h a 37°C / 5% de CO2 / 95% de humedad. Se retiró el sobrenadante, las células se lisaron en 20 ul de tampón de lisis para la detección por AlphaScreen® SureFire®.
Para la detección de p-AKT(Ser473), se utilizó el Kit de Ensayo SureFire® p-Akt 1/2 (Ser473) (PerkinElmer, EE.UU.). Se transfirieron 5 ul de lisado celular a Proxiplates de bajo volumen de 384 pocillos para la detección utilizando un pipeteador de 384 pocillos. La adición de reactivos AlphaScreen® SureFire® se realizó de acuerdo con el protocolo del fabricante. Primero, se añadieron 5 ul de tampón de reacción más una mezcla de tampón de activación que contenía perlas aceptoras AlphaScreen®, la placa se selló y se incubó en un agitador de placa durante 2 horas a temperatura ambiente. En segundo lugar, se añadieron 2 ul de tampón de dilución que contenía perlas donadoras AlphaScreen®, y la placa se incubó en un agitador de placa como se indicó arriba durante otras 2 horas. La placa se leyó en un lector de placas compatible con AlphaScreen®, utilizando la configuración estándar de AlphaScreen®.
Alternativamente, se detectó p-AKT (Ser473) con el Kit de Ensayo CisBio HTRF: como control positivo, 0,9 mM de 1-(3-(trifluorometil)-4-(piperazin-1-il)fenil)-8-(6-metoxipiridin-3-il)-3-metil-1H-imidazo[4,5-c]quinolin-2(3H)-ona sal de ácido maleico en DMSO al 90% (v/v) se añadió a la placa maestra del compuesto. Para los ensayos de compuestos, las células se sembraron a una densidad de 4000 (Rat-1_PI3Kdelta), 8000 (Rat-1_PI3Kalpha) o 6500 (Rat-1_PI3Kbeta) células en 30 ul de medio de crecimiento completo en placas de 384 pocillos, con una superficie de plástico fomentando la adhesión de las células y su crecimiento (placas de ensayo) y se cultivaron a 37°C / 5% de CO2 / 90% de humedad durante 24 h. Alrededor de 10 ul de las prediluciones del compuesto de la placa hija se transfirieron después a las células. Después del tratamiento con el compuesto durante 1 h, se retiró el medio y las células se lisaron mediante la adición de 20 ul de tampón de lisis complementado con tampón de bloqueo. La detección de pAKT (Ser473) se realizó con el kit de ensayo de HTRF pAKT (Ser473) de acuerdo con las instrucciones del fabricante, utilizando 16 ul de lisado de células en un volumen de detección total de 20 ul.
2.2 Determinación de la activación de células B murinas
Se ha reconocido que PI3K5 modula la función de las células B cuando las células son estimuladas a través del receptor de células B (BCR) (Okkenhaug et al. Science 297:1031 (2002). Para evaluar la propiedad inhibitoria de los compuestos
en la activación de las células B, se mide la regulación positiva de marcadores de la activación CD86 y CD69 en células B murinas derivadas del anticuerpo de bazo de ratón después de la estimulación con anti-IgM. CD69 es un marcador de activación bien conocido para las células B y T (Sancho et al. Trends Immunol. 26:136 (2005). CD86 (también conocido como B7-2) se expresa principalmente en las células presentadoras de antígenos, incluidas las células B. Las células B en reposo expresan CD86 a niveles bajos, pero lo regulan positiva después de la estimulación, p. ej., del receptor BCR o IL-4. CD86 en una célula B interactúa con CD28 en las células T. Esta interacción es necesaria para la activación óptima de las células T y para la generación de una respuesta óptima de IgG1 (Carreno et al. Annu Rev Immunol. 20:29 (2002)).
Se recogen bazos de ratones Balb/c , se aíslan los esplenocitos y se lavan dos veces con RPMI que contiene suero de bovino fetal (FBS) al 10%, HEPES 10 mM, 100 Unidades/mL de penicilina/estreptomicina. Al Rp M i complementado de esta manera se le alude posteriormente como medio. Las células se ajustan a 2,5 X 106 células/mL en medio y se añaden 200 |jl de suspensión celular (5 x 106 células) a los pocillos apropiados de placas de 96 pocillos.
Luego, las células se estimulan añadiendo 50 j l de mAb anti-IgM en medio (concentración final: 30 jg/mL). Después de incubar durante 24 horas a 37°C, las células se tiñen con los siguientes cócteles de anticuerpos: CD86-FITC anti-ratón, CD69-PerCP-Cy5.5 anti-ratón, CD19-PerCP anti-ratón para la evaluación de células B y CD3-FITC anti-ratón, CD69-PE anti-ratón para la evaluación de células T (2 j l de cada uno de los anticuerpos/pocillo). Después de una hora a temperatura ambiente (ta) en la oscuridad, las células se transfieren a placas de 96 pocillos profundos. Las células se lavan una vez con 1 mL de PBS que contiene FBS al 2% y después de la resuspensión en 200 jl, las muestras se analizan en un citómetro de flujo FACS Calibur. Los linfocitos se ligan en el gráfico de puntos FSC/SSC de acuerdo con el tamaño y la granularidad y se analizan adicionalmente para determinar la expresión de CD19, CD3 y marcadores de activación (CD86, CD69). Los datos se calculan a partir de borrones de puntos como porcentaje de células teñidas positivamente para marcadores de activación dentro de la población CD19+ o CD3+ utilizando el software BD CellQest.
Para evaluar la propiedad inhibitoria de los compuestos, los compuestos se disuelven primero y se diluyen en DMSO seguido de una dilución 1:50 en medio. Los esplenocitos de ratones Balb/c se aíslan, se resuspenden y se transfieren a placas de 96 pocillos tal se describió arriba (200 jl/pocillo). Los compuestos diluidos o el disolvente se añaden a las placas (25 j l) y se incuban a 37°C durante 1 hora. Luego, los cultivos se estimulan con 25 j l de mAb anti-IgM/pocillo (concentración final 30 jg/mL) durante 24 horas a 37°C y se tiñen con CD86-FITC anti-ratón y CD19-PerCP anti-ratón (2 j l de cada uno de los anticuerpos/pocillo). La expresión de CD86 en células B CD19 positivas se cuantifica por citometría de flujo tal como se describe arriba
2.3 Determinación de la activación de células B de rata
Se ha reconocido que PI3K5 modula la función de las células B cuando las células son estimuladas a través del receptor de células B (BCR) (Okkenhaug et al. Science 297:1031 (2002). Para evaluar la propiedad inhibitoria de los compuestos en la activación de células B, se mide la regulación positiva de marcadores de la activación CD86 en células B de rata derivadas de sangre entera después de la estimulación con anti-IgM e IL-4 recombinante. La molécula CD86 (también conocida como B7-2) se expresa principalmente en células presentadoras de antígeno, incluidas las células B. Las células B en reposo expresan CD86 a niveles bajos, pero lo regulan positiva después de la estimulación, p. ej., del receptor BCR o IL-4. CD86 en una célula B interactúa con CD28 en las células T. Esta interacción es necesaria para la activación óptima de las células T y para la generación de una respuesta óptima de IgG1 (Carreno et al. Annu Rev Immunol. 20:29 (2002)).
Recolección de sangre de rata
Se recogió sangre entera de la aorta abdominal de ratas Lewis macho adultas (LEW/HanHsd) o utilizando una jeringa de 10 ml con aguja hipodérmica previamente recubierta con heparina de sodio. La sangre se transfirió a tubos Falcon de 50 ml y la concentración de anticoagulante se ajustó a 100 U/ml.
Estimulación de células B de rata y tratamiento con inhibidor específico.
Para la evaluación de los efectos in vitro de los fármacos inmunosupresores, la sangre heparinizada se prediluyó al 50% con medio. Como medio sirvió DMEM con alto contenido de glucosa (Animed cat n° 1-26F01-I) complementado con 100 U/ml de penicilina, 100 mg/ml de estreptomicina, L-glutamina 2 mM, 50 mg/ml de dextrano 40 y suero de ternero fetal al 5% (FCS , Fetaclone I, Gibco n° 10270-106). Luego, se añadieron 190 j l de sangre prediluida con 10 j l de compuesto de ensayo diluido previamente en placas de microtitulación con fondo en U de 96 pocillos (Nunc), lo que resultó en una dilución en serie de 3 veces con un intervalo de concentraciones de 20 a 0,0003 jM . Los pocillos de control se pretrataron con DMSO para obtener una concentración final de DSM al 0,5 %. Los cultivos se establecieron por duplicado, se mezclaron bien mediante agitación en un agitador de placas (Heidolph Titramax 101; 30 s, velocidad 900), se pipetearon hacia arriba y hacia abajo y se agitaron nuevamente en el agitador de placas. Los cultivos se incubaron a 37°C, 5% de CO2 durante 1 h. Luego, se añadieron 20 j l de IgM anti-rata de cabra policlonal (Serotec, cat n° 302001) y 10 j l de rIL-4 recombinante diluida (Immunotools n° 340085) para obtener concentraciones finales de 30 jg/m l y 5 ng/ml,
respectivamente. Las placas se mezclaron por agitación en un agitador de placas como anteriormente y se incubaron durante 24 h a 37°C, 5% de CO2.
Determinación de la activación de células B por citometría de flujo.
Después de la incubación, se añadieron 15 |jl de una solución de EDTA 25 mM por pocillo y se agitó durante 15 min para separar las células adherentes. Para el análisis de marcadores de activación de superficie, las muestras se tiñeron luego con anti-ratCD45RA marcado con PE-Cy5 (BD cat n° 557015) para permitir el ligamiento de las células B en el análisis FACS. Además, las muestras se tiñeron con CD86 anti-rata marcado con PE (BD cat n° 551396). Todos los procedimientos de tinción se realizaron a ta durante 30 min en la oscuridad. Después de la incubación, las muestras se transfirieron a placas de microtitulación con fondo en V de 96 pocillos (Corning n° 396096) que contenían 2 ml/pocillo de Solución de Lisis BD (BD n° 349202). Después de la lisis de los eritrocitos, las muestras se lavaron con 2 ml de CellWASH (BD n° 349524). Los datos se obtuvieron en un citómetro de flujo LSRII o FACScalibur (BD Biosciences) utilizando el software Cellquest Plus o DIVA (versión 6.1.1), respectivamente. Los linfocitos se ligaron en los borrones de puntos FSC/SSC de acuerdo con el tamaño y la granularidad y se analizaron adicionalmente para la expresión de CD45RA y marcadores de activación. Los datos se calcularon a partir de borrones de puntos o histogramas como porcentaje de células teñidas positivamente para marcadores de activación dentro de la población CD45RA .
Evaluación estadística
El porcentaje de inhibición de la activación de células B después de la exposición al fármaco se calculó mediante la siguiente fórmula:
% Inhibición = 100 x estimulación sin fármaco - estimulación con fármaco
estimulación sin fármaco - no estimulado
Se utilizó el software ORIGIN 7 (OriginLab Corporation, Northampton, MA) para el ajuste de la curva de regresión no lineal. La concentración de fármaco que resulta en el 50% de inhibición (CI50) se obtuvo mediante el ajuste de la ecuación de Hill a los datos de inhibición.
2.4 Ensayo celular AKT U937 para PI 3-quinasa gamma
La línea celular de monocitos U937 se mantiene en un medio basal de RPMI 1640 complementado con FCS inactivado por calor al 10%, 100 U/ml de penicilina, 100 ug/ml de estreptomicina y L-glutamina 2 mM (Invitrogen). El cultivo en suspensión U937 se mantiene sembrando células a una densidad de 0,125 x 106 células por ml en medio reciente cada tres o cuatro días. Las células se incuban a 37°C, 5% de CO2. Tres o cuatro días antes del ensayo, las células se siembran a una densidad de 0,25 x 106 células por ml en un volumen total de 40 ml en un matraz de cultivo T175.
Antes de comenzar las manipulaciones celulares descritas más adelante, la placa de ensayo MSD (Meso Scale Discovery) se bloquea mediante la adición de 150 ul/pocillo de tampón de bloqueo suministrado y se incuba con agitación durante un mínimo de una hora a temperatura ambiente. Todos los pasos del ensayo se deben llevar a cabo rápidamente, con los periodos de incubación controlados con exactitud y realizando controles de temperatura cuando se indique.
Las células sembradas a razón de 0,25 x 106/ml 3 o 4 días antes del ensayo se aspiran, se transfieren a un tubo Falcon de 50 ml,
se cuentan y se centrifugan durante ocho minutos a 300 g a temperatura ambiente. Se aspira el sobrenadante y el pellet celular se suspende de nuevo y se lava una vez en HBSS (solución salina equilibrada de Hank) mediante centrifugación durante ocho minutos a 300 g a temperatura ambiente. El sedimento celular se resuspende en HBSS a una concentración de 4 x 106 por ml, y se añaden 100 j l de suspensión celular a cada uno de los pocillos de una placa de cultivo de tejidos de fondo plano de 96 pocillos. Las placas de ensayo se incuban durante 1,5 horas a 37 °C, 5% de CO2 para permitir que se reduzca la fosforilación de AKT de fondo antes del paso de estimulación con el compuesto.
Se prepara una solución patrón 5 mM del compuesto en DMSO al 100%; a partir de esta, se prepara una dilución de 1 en 125 en HBSS para obtener una concentración máxima del compuesto de 40 jM , con DMSO al 0,8%. Las titulaciones de compuesto se preparan en una placa de fondo plano de 96 pocillos reciente, por dilución en serie 10 veces de 40 jM en HBSS DMSO al 0,8%; las puntas de pipeta se reemplazan después de cada dilución. En este punto, las concentraciones del compuesto son 4 veces la concentración final requerida en la placa de ensayo. Las células se estimulan con el compuesto o HBSS con un 0,8% de DMSO mediante una transferencia directa de 50 jL/pocillo a partir de la placa de dilución del compuesto. La placa de ensayo que contiene las células tratadas con el compuesto se incuba a continuación durante 30 minutos a 37 °C. Se utiliza una disposición de la placa estándar para todos los experimentos.
Las células tratadas con el compuesto, además de los pocilios de control positivo («MIP1a máx»), se estimulan con 50 |jL por pocillo de 40 ng/mL de MIP1a (R&D Systems, número de catálogo 270-LD, patrón liofilizado reconstituido hasta 50 jg/m L con PBS con un 0,1% de BSA). Los pocillos de control negativo («HBSS mín») se estimulan con 50 jL/pocillo de HBSS en ausencia de MIP1a. Las concentraciones finales del compuesto están ahora diluidas 4 veces, lo que proporciona una concentración máxima de 10 jM ; cuando se añade, la concentración final de MIP1a es de 10 ng/mL. Las células se incuban con MIP1a durante 3 minutos a 37 °C, 5% de CO2. Después del periodo de estimulación de tres minutos, la placa de ensayo se mantiene fría con hielo en todo momento. Las placas de ensayo se centrifugan durante 2 minutos a 300 g y 4 °C, y se elimina el sobrenadante realizando una inversión cuidadosa y a continuación secando la placa sobre papel absorbente. A continuación, las células se lavan mediante la adición cuidadosa de 150 jL/pocillo de HBSS enfriado con hielo y la centrifugación a 300 g durante 5 minutos a 4 °C. El sobrenadante se aspira y la placa se seca como se ha descrito anteriormente. La placa se coloca sobre hielo y las células se tratan inmediatamente con 35 jL por pocillo de tampón de lisis enfriado con hielo, preparado de acuerdo con las instrucciones del kit (por placa de ensayo, a 5 ml de tampón de lisis Tris, añadir 100 jL de solución inhibidora de proteasas x50 y 50 jL de cada una de las soluciones inhibidoras de fosfatasas I y II x100). Las placas se incuban sobre hielo durante 20 minutos antes de realizar una centrifugación a 841 g durante 5 minutos a 4 °C.
Se aspira el tampón de bloqueo de la placa de MSD y la placa se lava cuatro veces con 300 jL/pocillo de tampón de lavado Tris. A continuación, se transfieren 25 jL del lisado celular de la placa de ensayo a la placa de MSD lavada, la cual se sella y se incuba a temperatura ambiente durante una hora con agitación. La placa se lava cuatro veces con 300 jL por pocillo de tampón de lavado Tris antes de la adición de 25 jL por pocillo de anticuerpo de detección anti-AKT total/pAKT con marca sulfo (se diluyen 60 jL de patrón de anticuerpo x50 en 1 mL de tampón de bloqueo mezclado con 2 mL de tampón de lavado) y se incuba a temperatura ambiente durante una hora con agitación. La placa se lava cuatro veces con 300 jL por pocillo de tampón de lavado Tris y se añaden 150 jL por pocillo de tampón de lectura, teniendo cuidado de evitar la introducción de burbujas. La placa se lee inmediatamente utilizando un generador de imágenes MSD SECTOR 6000.
Los resultados se exportan a Excel y el porcentaje de AKT fosforilada se calcula utilizando la ecuación: % de fosfoproteína = ((2* señal fosfo) / (señal fosfo señal total))* 100. La inhibición mediada por compuestos de la fosforilación de AKT se analiza utilizando el software Prizm V Graphpad.
2.5 Determinación de IL-6 inducida por TLR9 en esplenocitos de ratón
Preparación de suspensión de células individuales a partir de bazo de ratón
Se diseccionaron bazos de ratones C57BL/6 inmediatamente después de la eutanasia. Se recortó el exceso de grasa de los bazos antes de machacar el bazo a través de un filtro de células de 0,4 jM utilizando un émbolo de una jeringa de 5 ml. Se preparó una suspensión de células individuales y el volumen se ajustó a 15 ml en un tubo Falcon de 50 ml utilizando PBS frío. Las células se centrifugaron a 1500 rpm durante 5 minutos a 4°C grados antes de la separación del sobrenadante y la resuspensión en 5 ml de tampón de lisis de glóbulos rojos por bazo e incubación durante 5 minutos a temperatura ambiente. Se añadió PBS helado (30 ml) a las células antes de la centrifugación a 1500 rpm durante 5 minutos a 4°C. El sobrenadante se separó y las células se lavaron dos veces con 40 ml de medio de cultivo de esplenocitos murinos (MSCM). El MSCM consistía en RPMI complementado con 100 unidades/ml de Penicilina y 100 jg/m l de Estreptomicina, 1 x aminoácidos no esenciales, piruvato de sodio 1 mM, p-mercaptoetanol 0,05 mM y suero de bovino fetal (FBS) activado por calor al 10%. Las células se resuspendieron en 10-20 ml de MSCM y se contaron utilizando un contador de células Countess. Aproximadamente se obtuvieron 60x106 esplenocitos de una sola C57BL/6 de bazo de ratón.
Estimulación de esplenocitos murinos y tratamiento con inhibidor específico.
Esplenocitos se sembraron en placas a una densidad final de 2x105 células/pocillo en un volumen de 100 j l en placas de 96 pocillos de fondo plano y se incubaron en una incubadora humidificada con 37°C durante 2-4 horas. Posteriormente, los compuestos a ensayar se dispensaron utilizando una máquina de manipulación de líquidos automatizada utilizando placas madres de compuestos preparadas previamente. Las placas madres consistían en compuestos (en DMSO al 90% / 10% /ddH20) dispuestos en 8-10 puntos utilizando diluciones dobles o triples. La máquina de manipulación de líquidos dispensó 1 j l de cada una de las diluciones de la placa fuente de compuesto preparada previamente en el pocillo de destino apropiado en la placa de 96 pocillos. La concentración de partida final de los compuestos en el cultivo celular fue de 10 jM . La concentración final de DMSO en los cultivos celulares fue del 0,5%. Las células se incubaron con compuestos durante 1 hora antes de la adición del ligando TLR. Luego, se añadió una concentración de CEs010x de CpG1826 en un volumen de 20 j l (para un volumen de cultivo final de 200 jl), después de lo cual se incubaron los cultivos durante la noche en una incubadora humidificada a 37°C.
Determinación de interleucina-6 mediante ELISA
Después del cultivo durante la noche, las placas se centrifugaron a 2000 rpm durante 5 minutos a temperatura ambiente. Posteriormente, se transfirieron 150 j l de cada uno de los cultivos a placas con fondo en V de 96 pocillos y se midieron
los niveles de IL-6 utilizando el kit ELISA sándwich IL-6 de ratón disponible comercialmente. Brevemente, las placas se revistieron durante la noche con el anticuerpo de captura antes del bloqueo durante 1 hora con PBS/BSA al 0,1%. Se añadieron muestras y patrones en un volumen de 50 |jl y la placa se incubó durante 2 horas a temperatura ambiente. Después de la separación de los patrones/muestras, la placa se lavó utilizando PBS/Tween al 0,05% antes de la adición de 50 j l del anticuerpo de detección biotinilado, tras lo cual la placa se incubó durante 2 horas a temperatura ambiente con agitación. Las placas se lavaron nuevamente antes de la adición de 50 j l de estreptavidina-peroxidasa de rábano picante por pocillo durante 20 minutos. Después de lavados de placa adicionales, se añadieron 50 j l de sustrato TMB a cada uno de los pocillos y las placas se incubaron durante 20 minutos antes de la adición de 25 jl/solución de parada de pocillo. Los niveles de IL-6 se midieron utilizando un lector de placas SpectraMax 190 (450 nm) y se analizaron utilizando el software SoftMax Pro y GraphPad Prism.
2.6. Determinación de IFNalfa inducido por TLR9 en células mononucleares de sangre periférica humana (PBMC)
Preparación de PBMC a partir de sangre humana reciente
Se recogió sangre humana (aproximadamente 75 ml) en 10 tubos S-Monovette que contenían heparina (S-Monovette 7,5 mL de NH Heparina 16 UI/mL de sangre; Starstedt). Se prepararon tubos Leucosep™ (30 ml n° 227290; Greiner Bio-one) mediante la adición de 15 ml de medio de separación de linfocitos LSM1077™ por tubo (n° J15-004; PAA Laboratories) y se centrifugación durante 30 s a 1000 g. Se transfirieron unos 25 ml de sangre a tubos Leucosep™ después de la dilución con partes iguales de PBS (sin Ca2+//Mg2+; n° 14190-094). Las muestras se centrifugaron a 800 g durante 20 min a 22 °C utilizando una centrífuga Eppendorf 5810R sin freno. La capa de PBMC se retiró cuidadosamente de la interfaz plasma:medio de separación y se transfirió a un tubo limpio de 50 ml. Las células se lavaron una vez mediante la adición de PBS (hasta 45 ml) y se centrifugaron (1400 rpm, 10 min a 22 °C) con freno (ajustado a la velocidad 9) utilizando un Eppendorf 5810R. Las células sedimentadas se resuspendieron cuidadosamente en medio (RPMI 1640 GlutaMAX-I, 2-mercaptoetanol 0,05 mM, HEPES 10 mM y FCS al 5% v/v) y las muestras se agruparon. Los componentes del medio 2-mercaptoetanol (n° 31350-010; 50 mM), Hepes (n° 15630-056, 1M) y RPMI 1640 (1x) GlutaMAX-I (n° 61870-010) se obtuvieron de Gibco. FCS (n° 2-01F36-1) se obtuvo de Amimed. Las PBMC se contaron utilizando un contador de células automatizado Countess® (la muestra se diluyó previamente 1:10 en medio, antes de la adición de un volumen igual (10 j l) de azul de tripano). Las células se diluyeron a 4 x 106 células/ml y se sembraron en placas de 384 pocillos (n° 353962; Becton Dickinson AG) para dar un volumen final de 25 j l (es decir, 1 x 105 células/pocillo).
Estimulación de PBMC y tratamiento con inhibidor específico.
Los compuestos se diluyeron previamente en DMSO al 100 % v/ v (n° 41640-100 mL; Sigma-Aldrich), seguido de transferencia en medio (para lograr una concentración final de DMSO de 0,25 %). Las células se trataron con dilución apropiada de compuesto (5 j l) o control de vehículo (5 j l) y se incubaron durante 30 min a 37 °C en una incubadora humidificada en aire con 5 % (v/v) de CO2. Las células fueron estimuladas con CpG2216 (0,3 jM ; n° tlrl-hodna; Invivogen) o control de vehículo (10 jl/pocillo) y se incubaron durante 20 h. Las placas se centrifugaron brevemente (200 x g durante 2 min a 22 °C) y se retiraron las muestras de sobrenadante (30 j l) para cuantificar los niveles de IFNa.
Cuantificación de IFNa utilizando la tecnología AlphaLisa
Para la cuantificación de IFNalfa se utilizó el kit de interferón humano AlphaLISA (n° AL264F) de PerkinElmer. Se prepara una mezcla de anticuerpos que contiene perlas aceptoras anti-IFNa (5 jg/m l final) y anticuerpo biotinilado anti-IFNa (0,5 nM final) reciente y se dispensa (5 j l) en Optiplates™ de 384 pocillos (n° 6007299; PerkinElmer). Se preparó la dilución de patrones conocidos de IFNa (IFNa B humano (2b)) y junto con los sobrenadantes celulares (5 j l) se añadieron a las placas anteriores. Las placas se centrifugaron brevemente (pulso a 200 g), se cubrieron con una película de sellado adhesiva, se agitaron en vórtice y se incubaron durante 1 h a temperatura ambiente en la oscuridad. Se prepararon perlas de donadores recubiertas con estreptavidina (20 jg/m l final) y se añadieron a cada uno de los pocillos (5 j l) en un área de luz oscura (mezcla sensible a la luz). Las placas se incubaron durante 30 min a temperatura ambiente (las placas no se deben centrifugar ni cubrir). Después de la incubación, las placas se leyeron con un lector multiplacas EnVision™ equipado con la opción ALPHA utilizando los "ajustes estándar AlphaScreen" del propio instrumento (p. ej., tiempo total de medición: 550 ms, tiempo de excitación por Láser 680 nm: 180 ms, espejo: D640, filtro de emisión: M570w, longitud de onda central 570 nm, ancho de banda 100 nm, transmitancia 75%). Los datos fueron recolectados para el análisis y la cuantificación de los niveles de IFNa.
Evaluación y análisis de datos.
Los datos se analizaron utilizando Excel XL fit 4.0 (Microsoft) con el complemento XLfit (IDBS; versión 4.3.2). Las concentraciones específicas de IFNa se determinaron después de la extrapolación a curvas patrón utilizando IFNa B humano (2b). Los valores individuales de CI50 de los compuestos se determinaron por regresión no lineal después de ajustar las curvas a los datos experimentales.
3 Determinación de la producción de anticuerpos contra glóbulos rojos de oveja (SRBC).
En resumen, a ratas OFA se les inyectaron i.v. eritrocitos de oveja el d 0 y se trataron por vía oral durante cuatro días consecutivos (d 0 a d 3) con los compuestos bajo investigación. Se prepararon suspensiones de células de bazo el d 4 y los linfocitos se colocaron en placas en agar blando en presencia de células indicadoras (SRBC) y complemento. La lisis de las células indicadoras debida a la secreción de anticuerpos específicos para SRBC (predominantemente de la subclase IgM) y la presencia de complemento proporcionó halos. Se contó el número de halos por placa y se expresó como número de halos por bazo.
Inmunización Se inmunizaron grupos de cinco ratas OFA hembras el día 0 con 2 x 108/ml de SRBC (obtenido de Laboratory Animal Services LAS, Novartis Pharma AG ) en un volumen de 0,5 ml por rata mediante inyección i.v.
Tratamiento del compuesto Los animales fueron tratados con compuesto suspendido en CMC al 0,5%, Tween80 al 0,5% durante 4 días consecutivos (días 0, 1,2 y 3) a partir del día de la inmunización. El compuesto se administró por vía oral dos veces al día con intervalos de 12 horas entre dosis en un volumen de aplicación de 5 ml/kg de peso corporal.
Preparación de suspensiones de células del bazo
El día 4, los animales fueron sacrificados con CO2. Se extrajeron los bazos, se pesaron y se depositaron en tubos de plástico que contenían 10 ml de solución salina equilibrada de Hank fría (4 °C) (HBSS; Gibco, pH 7,3, que contenía 1 mg de rojo fenol /100 ml ) para cada uno de los bazos de ratas. Los bazos se homogeneizaron con un homogeneizador de vidrio, se dejaron en hielo durante 5 minutos y 1 ml de sobrenadante se transfirió a un nuevo tubo. Las células se lavaron una vez en 4 ml de HBSS, luego se descartaron los sobrenadantes y los sedimentos se volvieron a suspender en 1 ml de HBSS. Los números de linfocitos por bazo se determinaron mediante un contador automatizado de células y suspensiones de células de bazo se ajustaron a una concentración celular de 30 x 106/ml.
Ensayo formador de halos:
Se prepararon placas de Petri de agar blando con agarosa al 0,7% (SERVA) en HBSS .
Además, se preparó un ml de agarosa al 0,7% en tubos de plástico y se mantuvo a 48°C en un baño de agua. Unos 50 |jl de una suspensión 30x106/ml de células de bazo y 50 |jl de SRBC en 40 x 108/ml se añadieron, se mezclaron rápidamente (Vórtice) y se vertieron en las placas de agarosa preparadas. Las placas de Petri se inclinaron ligeramente para lograr una distribución uniforme de la mezcla de células en la capa de agarosa. Las placas se dejaron a la temperatura ambiente durante 15 minutos y luego se incubaron a 37°C durante 60 minutos. Entonces se añadieron 1,4 ml de complemento de cobaya (Harlan; 10%) y la incubación continuó durante otros 60 minutos a 37°C . Anticuerpos específicos para SRBC liberados por las células B sembradas fueron unidos al antígeno (SRBC) en su vecindad. Estos complejos de antígeno-anticuerpo activaron el complemento y condujeron a la lisis de SRBC dejando un punto brillante (halo) dentro de la capa de eritrocitos rojos. Los halos se contaron con un microscopio.
Se utilizó la siguiente fórmula para determinar la inhibición de la formación de halos: % de inhibición = C * 100/V-100 con : V = número medio de halos/bazo para el grupo de vehículo; C = número medio de halos/bazo para el grupo tratado con compuesto
Referencias
N.K. Jerne y A.A. Nordin (1963) Plaque formation in agar by single antibody-producing cells Science 140:405.
N.K. Jerne, A.A Nordin y C. Henry (1963) The agar plaque technique for recognizing antibody-producing cells. En: "Cell Bound Antibodies", B. Amos y H. Koprowski, Eds., Wistar Inst. Press, Philadelphia págs.109-125
Datos biológicos
Ensa o Enzimático


Ensa os Celulares


*Datos biológicos para el ejemplo 15 en el ensayo enzimático y celular se cree que son un artefacto.
Claims (10)
1. Un compuesto de la fórmula (Ib) o una sal farmacéuticamente aceptable del mismo.

R1 se selecciona entre
fenilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
piridilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro;
1 -metilpi razol-5-ilo;
2-metiltiofen-5-ilo;
cicloalquilo C3-C6 que no está sustituido o está sustituido en la posición 1 con metilo; tetrahidropiran-4-ilo; piperidin-1-ilo;
morfolin-4-ilo;
pirolidin-3-ilo, que no está sustituido o está sustituido en la posición 1 con un sustituyente que se selecciona de metoxicarbonilo, metilsulfonilo, metilo o metilcarbonilo; o
dimetilamina;
R2 se selecciona entre
heteroarilo C4-C7 que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de
alquilo C1-C4,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4 ,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1-C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
ciano,
fluoro,
amino,
alquil C1-C4amino, o
dialquil C1-C4amino;
R5 y R6 se seleccionan independientemente de hidrógeno, deuterio o fluoro;
R7 se selecciona de metoxi, difluorometoxi, trifluorometoxi, hidroxi , fluoro o metilsulfonilamina; y
R3 se selecciona de 
R8 se selecciona de hidrógeno, metilo, fluorometilo, difluorometilo, trideuterometilo o amino.
2. Un compuesto de acuerdo con la reivindicación 1 , en donde
R3 se selecciona entre

en donde
R8 se selecciona de metilo.
3. Un compuesto de acuerdo con la reivindicación 1 o 2 o una sal farmacéuticamente aceptable del mismo, en donde
R2 es heteroarilo C5-C6 que contiene un átomo de nitrógeno y 0, 1, 2 o 3 heteroátomos adicionales seleccionados independientemente de N, O o S , en donde heteroarilo C4-C7 no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de
alquilo C1-C4 ,
fluoroalquilo C1-C4 ,
hidroxi-alquilo C1-C4 ,
hidroxi-fluoroalquilo C1-C4 ,
alcoxi C1-C4 ,
fluoroalcoxi C1-C4 ,
cicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro,
heterocicloalquilo C3-C6 , que no está sustituido o está sustituido con 1-3 sustituyentes seleccionados independientemente de metilo, o fluoro, o
fluoro.
4. Un compuesto de acuerdo con la reivindicación 1 o 2 o una sal farmacéuticamente aceptable del mismo, en donde
R1 es fenilo, que no está sustituido o está sustituido con 1,2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro; o
piridilo, que no está sustituido o está sustituido con 1, 2 o 3 sustituyentes seleccionados independientemente de metilo, etilo, difluorometilo, metoxi, difluorometoxi, ciclopropilo, cloro o fluoro.5
5. Un compuesto de acuerdo con la reivindicación 1, seleccionado de
2-amino-4-((2S,4S)-2-(6-fluoro-5-(1-metil-1 H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(6-fluoro-5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
4-amino-6-((2S,4S)-2-(6-fluoro-5-(1-metil-1 H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)pirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(6-fluoro-5-(2-metoxitiazol-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-metoxi-2-(5-(1-metil-1 H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(1-metil-1 H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(2-metilpirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(4-oxo-3-fenil-5-(piridin-3-il)-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(5-metoxipiridin-3-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(4-oxo-3-fenil-5-(pirimidin-5-il)-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(4-metoxipiridin-3-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-hidroxi-2-(5-(1-(2-hidroxietil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(5-(2-etoxipirimidin-5-il)-6-fluoro-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-4-fluoro-2-(5-(1-metil-1H-pirazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
2-amino-4-((2S,4S)-2-(6-fluoro-5-(1-metil-1H-imidazol-4-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)-4-hidroxipirrolidin-1-il)-6-metilpi rimidina-5-carbonitrilo
o
2-amino-4-((2S,4S)-4-metoxi-2-(5-(2-metoxipirimidin-5-il)-4-oxo-3-fenil-3,4-dihidroquinazolin-2-il)pirrolidin-1-il)-6-metilpirimidina-5-carbonitrilo,
o una sal farmacéuticamente aceptable de este.
6. Un compuesto de acuerdo con la reivindicación 1, en forma cristalina.
7. Una composición farmacéutica que comprende una cantidad terapéuticamente efectiva de un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo y uno o más portadores farmacéuticamente aceptables.
8. Una combinación que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable de este y uno o más coagentes terapéuticamente activos.
9. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable de este, para su uso como un medicamento.
10. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 o una sal farmacéuticamente aceptable del mismo, para uso en el tratamiento de un trastorno o una enfermedad seleccionada de artritis reumatoide (RA, pénfigo vulgar (PV), forma endémica de pénfigo brasileño (Fogo selvagem), púrpura trombocitopénica idiopática (ITP), púrpura trombocitopénica trombótica (TTP), anemia hemolítica autoinmune (AIHA), hemofilia adquirida tipo A (AHA), lupus eritematoso sistémico (SLE), esclerosis múltiple (MS), miastenia grave (MG), síndrome de Sjogren (SS) (tal como síndrome de Sjogren primario (pSS)), vasculitis asociada a ANCA (tal como la enfermedad de Wegener, poliangeitis microscópica o síndrome de Churg-Strauss), crioglobulinemia, lesión por isquemia-reperfusión, urticaria autoinmune crónica (CAU ), alergia (dermatitis atópica, dermatitis por contacto, rinitis alérgica, asma alérgico, asma asociado con la rinitis alérgica), síndrome de Goodpasture, rechazo de trasplantes, cánceres de origen hematopoyético, malaria grave y cerebral, tripanosomiasis, leishmaniasis, toxoplasmosis y neurocisticercosis.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13177827 | 2013-07-24 | ||
| PCT/CN2014/082946 WO2015010641A1 (en) | 2013-07-24 | 2014-07-24 | Substituted quinazolin-4-one derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2803513T3 true ES2803513T3 (es) | 2021-01-27 |
Family
ID=48832822
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14829331T Active ES2803513T3 (es) | 2013-07-24 | 2014-07-24 | Derivados de quinazolin-4-ona sustituidos |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US9840498B2 (es) |
| EP (1) | EP3024827B1 (es) |
| JP (1) | JP6437544B2 (es) |
| KR (1) | KR20160033697A (es) |
| CN (1) | CN105452235B (es) |
| AR (1) | AR097024A1 (es) |
| AU (1) | AU2014295538B2 (es) |
| CA (1) | CA2917433A1 (es) |
| EA (1) | EA031430B1 (es) |
| ES (1) | ES2803513T3 (es) |
| MX (1) | MX368066B (es) |
| PL (1) | PL3024827T3 (es) |
| PT (1) | PT3024827T (es) |
| TW (1) | TW201534598A (es) |
| UY (1) | UY35675A (es) |
| WO (1) | WO2015010641A1 (es) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| NZ587051A (en) | 2008-01-04 | 2012-12-21 | Intellikine Llc | Isoquinolinone derivatives, compositions and methods of inhibiting phosphatidyl inositol-3 kinase (pi3 kinase) |
| ES2593256T3 (es) | 2010-05-21 | 2016-12-07 | Infinity Pharmaceuticals, Inc. | Compuestos químicos, composiciones y métodos para las modulaciones de cinasas |
| CN103648499B (zh) | 2011-01-10 | 2017-02-15 | 无限药品股份有限公司 | 用于制备异喹啉酮的方法及异喹啉酮的固体形式 |
| WO2013032591A1 (en) | 2011-08-29 | 2013-03-07 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CA2925944C (en) | 2013-10-04 | 2023-01-10 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| EP4066834A1 (en) | 2014-03-19 | 2022-10-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders |
| WO2015160975A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| EP3154960A1 (en) | 2014-06-13 | 2017-04-19 | Gilead Sciences, Inc. | Phosphatidylinositol 3-kinase inhibitors |
| EA201692249A1 (ru) | 2014-06-13 | 2017-05-31 | Джилид Сайэнс, Инк. | Ингибиторы фосфатидилинозитол-3-киназы |
| CN106660994A (zh) | 2014-06-13 | 2017-05-10 | 吉利德科学公司 | 磷脂酰肌醇3‑激酶抑制剂 |
| ES2763104T3 (es) | 2014-06-13 | 2020-05-27 | Gilead Sciences Inc | Derivados de quinazolinona como inhibidores de fosfatidilinositol 3-quinasa |
| ES2900812T3 (es) * | 2014-06-24 | 2022-03-18 | Gilead Sciences Inc | Inhibidores de fosfatidilinositol 3-quinasa |
| WO2016054491A1 (en) | 2014-10-03 | 2016-04-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN108349985A (zh) | 2015-09-14 | 2018-07-31 | 无限药品股份有限公司 | 异喹啉酮的固体形式、其制备方法、包含其的组合物及其使用方法 |
| CN106995438B (zh) * | 2016-01-22 | 2019-09-20 | 中国科学院上海药物研究所 | 一类取代喹唑啉-4-酮类化合物及其制备方法和医药用途 |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2017223422A1 (en) | 2016-06-24 | 2017-12-28 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| JOP20190052A1 (ar) | 2016-09-22 | 2019-03-21 | Astrazeneca Ab | 5-[2-(بيريدين-2-يلامينو )-3،1 ثيازول-5-يال]-3،2 – ثنائي هيدرو- 1h- إيزوإندول 1--مشتق واحد واستخدامها كمثبطات مزدوجة للدلتا وغاما فوسفاتيديلينوسيتول 3-كيناز |
| US10751339B2 (en) | 2018-01-20 | 2020-08-25 | Sunshine Lake Pharma Co., Ltd. | Substituted aminopyrimidine compounds and methods of use |
| US10442799B1 (en) | 2018-04-07 | 2019-10-15 | Fuqiang Ruan | Heterocyclic compounds and uses thereof |
| CN113512042A (zh) * | 2020-04-09 | 2021-10-19 | 成都赜灵生物医药科技有限公司 | 取代喹唑啉-4-酮类化合物及其制备方法和用途 |
| KR20240045472A (ko) * | 2022-09-30 | 2024-04-08 | 주식회사 베노바이오 | 퀴나졸린염 화합물을 포함하는 관절염 치료용 조성물 및 이의 제조 방법 |
Family Cites Families (94)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3301855A (en) | 1963-04-22 | 1967-01-31 | Ciba Geigy Corp | Derivatives of 4-nu-(2-nu, nu-dimethylaminolower alkyl)-amino quinazoline |
| GB1524747A (en) | 1976-05-11 | 1978-09-13 | Ici Ltd | Polypeptide |
| ATE28864T1 (de) | 1982-07-23 | 1987-08-15 | Ici Plc | Amide-derivate. |
| GB8327256D0 (en) | 1983-10-12 | 1983-11-16 | Ici Plc | Steroid derivatives |
| US5093330A (en) | 1987-06-15 | 1992-03-03 | Ciba-Geigy Corporation | Staurosporine derivatives substituted at methylamino nitrogen |
| US5010099A (en) | 1989-08-11 | 1991-04-23 | Harbor Branch Oceanographic Institution, Inc. | Discodermolide compounds, compositions containing same and method of preparation and use |
| NZ243082A (en) | 1991-06-28 | 1995-02-24 | Ici Plc | 4-anilino-quinazoline derivatives; pharmaceutical compositions, preparatory processes, and use thereof |
| GB9300059D0 (en) | 1992-01-20 | 1993-03-03 | Zeneca Ltd | Quinazoline derivatives |
| TW225528B (es) | 1992-04-03 | 1994-06-21 | Ciba Geigy Ag | |
| GB9314893D0 (en) | 1993-07-19 | 1993-09-01 | Zeneca Ltd | Quinazoline derivatives |
| EP0817775B1 (en) | 1995-03-30 | 2001-09-12 | Pfizer Inc. | Quinazoline derivatives |
| GB9508538D0 (en) | 1995-04-27 | 1995-06-14 | Zeneca Ltd | Quinazoline derivatives |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US5843901A (en) | 1995-06-07 | 1998-12-01 | Advanced Research & Technology Institute | LHRH antagonist peptides |
| MX9800215A (es) | 1995-07-06 | 1998-03-31 | Novartis Ag | Pirrolopirimidas y procesos para su preparacion. |
| US5760041A (en) | 1996-02-05 | 1998-06-02 | American Cyanamid Company | 4-aminoquinazoline EGFR Inhibitors |
| GB9603095D0 (en) | 1996-02-14 | 1996-04-10 | Zeneca Ltd | Quinazoline derivatives |
| DE69710712T3 (de) | 1996-04-12 | 2010-12-23 | Warner-Lambert Co. Llc | Umkehrbare inhibitoren von tyrosin kinasen |
| CA2258548C (en) | 1996-06-24 | 2005-07-26 | Pfizer Inc. | Phenylamino-substituted tricyclic derivatives for treatment of hyperproliferative diseases |
| WO1998008849A1 (de) | 1996-08-30 | 1998-03-05 | Novartis Aktiengesellschaft | Verfahren zur herstellung von epothilonen und zwischenprodukte innerhalb des verfahrens |
| AU4141697A (en) | 1996-09-06 | 1998-03-26 | Obducat Ab | Method for anisotropic etching of structures in conducting materials |
| AU4342997A (en) | 1996-09-13 | 1998-04-02 | Sugen, Inc. | Use of quinazoline derivatives for the manufacture of a medicament in the reatment of hyperproliferative skin disorders |
| EP0837063A1 (en) | 1996-10-17 | 1998-04-22 | Pfizer Inc. | 4-Aminoquinazoline derivatives |
| EP1367057B1 (de) | 1996-11-18 | 2008-09-17 | Gesellschaft für biotechnologische Forschung mbH (GBF) | Epothilone E und F |
| US6441186B1 (en) | 1996-12-13 | 2002-08-27 | The Scripps Research Institute | Epothilone analogs |
| CO4940418A1 (es) | 1997-07-18 | 2000-07-24 | Novartis Ag | Modificacion de cristal de un derivado de n-fenil-2- pirimidinamina, procesos para su fabricacion y su uso |
| GB9721069D0 (en) | 1997-10-03 | 1997-12-03 | Pharmacia & Upjohn Spa | Polymeric derivatives of camptothecin |
| US6194181B1 (en) | 1998-02-19 | 2001-02-27 | Novartis Ag | Fermentative preparation process for and crystal forms of cytostatics |
| EP1058679B1 (en) | 1998-02-25 | 2005-10-19 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues therof |
| EP1107964B8 (en) | 1998-08-11 | 2010-04-07 | Novartis AG | Isoquinoline derivatives with angiogenesis inhibiting activity |
| KR100851418B1 (ko) | 1998-11-20 | 2008-08-08 | 코산 바이오사이언시즈, 인코포레이티드 | 에포틸론 및 에포틸론 유도체의 생산을 위한 재조합 방법 및 물질 |
| WO2001070737A2 (en) | 2000-03-20 | 2001-09-27 | Sepracor, Inc. | Therapeutic heterocyclic compounds for the treatment of asthma and allergy and use thereof |
| WO2001081346A2 (en) | 2000-04-25 | 2001-11-01 | Icos Corporation | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| US6667300B2 (en) | 2000-04-25 | 2003-12-23 | Icos Corporation | Inhibitors of human phosphatidylinositol 3-kinase delta |
| US6608053B2 (en) | 2000-04-27 | 2003-08-19 | Yamanouchi Pharmaceutical Co., Ltd. | Fused heteroaryl derivatives |
| PE20020354A1 (es) | 2000-09-01 | 2002-06-12 | Novartis Ag | Compuestos de hidroxamato como inhibidores de histona-desacetilasa (hda) |
| WO2002026718A2 (en) | 2000-09-29 | 2002-04-04 | Millennium Pharmaceutical, Inc. | Bicyclic pyrimidin-4-one based inhibitors of factor xa |
| TWI238824B (en) | 2001-05-14 | 2005-09-01 | Novartis Ag | 4-amino-5-phenyl-7-cyclobutyl-pyrrolo[2,3-d]pyrimidine derivatives |
| GB0119249D0 (en) | 2001-08-07 | 2001-10-03 | Novartis Ag | Organic compounds |
| TW200306839A (en) | 2002-02-06 | 2003-12-01 | Novartis Ag | Quinazolinone derivatives and their use as CB agonists |
| AU2003228283A1 (en) | 2002-03-07 | 2003-09-22 | X-Ceptor Therapeutics, Inc. | Quinazolinone modulators of nuclear receptors |
| WO2004009036A2 (en) | 2002-07-23 | 2004-01-29 | Cytokinetics, Inc. | Compounds compositions and methods |
| EP1407774A1 (en) | 2002-09-10 | 2004-04-14 | LION Bioscience AG | 2-Amino-4-quinazolinones as LXR nuclear receptor binding compounds |
| AU2003277079A1 (en) | 2002-09-30 | 2004-05-04 | Cytokinetics, Inc. | Compounds, compositions, and methods |
| KR20050084224A (ko) | 2002-12-09 | 2005-08-26 | 더 보드 오브 리전츠, 더 유니버시티 오브 텍사스 시스템 | 야누스 티로신 키나제 3을 선택적으로 저해하는 방법 |
| JP4887139B2 (ja) | 2003-03-25 | 2012-02-29 | 武田薬品工業株式会社 | ジペプチジルペプチダーゼインヒビター |
| WO2005016348A1 (en) | 2003-08-14 | 2005-02-24 | Icos Corporation | Method of inhibiting immune responses stimulated by an endogenous factor |
| US20050054614A1 (en) | 2003-08-14 | 2005-03-10 | Diacovo Thomas G. | Methods of inhibiting leukocyte accumulation |
| JP2005097276A (ja) * | 2003-08-22 | 2005-04-14 | Takeda Chem Ind Ltd | 縮合ピリミジン誘導体およびその用途 |
| WO2005067901A2 (en) | 2004-01-08 | 2005-07-28 | Michigan State University | Methods for treating and preventing hypertension and hypertension-related disorders |
| EP1704145B1 (en) | 2004-01-12 | 2012-06-13 | YM BioSciences Australia Pty Ltd | Selective kinase inhibitors |
| WO2005091711A2 (en) | 2004-03-24 | 2005-10-06 | Orchid Chemicals & Pharmaceuticals Ltd | Novel condensed pyrimidones |
| US7932260B2 (en) * | 2004-05-13 | 2011-04-26 | Icos Corporation | Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta |
| WO2005112935A1 (en) | 2004-05-13 | 2005-12-01 | Vanderbilt University | Phosphoinositide 3-kinase delta selective inhibitors for inhibiting angiogenesis |
| EP1755609A1 (en) | 2004-05-25 | 2007-02-28 | Icos Corporation | Methods for treating and/or preventing aberrant proliferation of hematopoietic cells |
| CN101123968A (zh) | 2004-06-04 | 2008-02-13 | 艾科斯有限公司 | 肥大细胞病的治疗方法 |
| GB0413618D0 (en) | 2004-06-17 | 2004-07-21 | Novartis Ag | Organic compounds |
| US7939538B2 (en) | 2004-06-28 | 2011-05-10 | Amgen Inc. | Compounds, compositions and methods for prevention and treatment of inflammatory and immunoregulatory disorders and diseases |
| CA2598409A1 (en) | 2005-02-17 | 2006-08-24 | Icos Corporation | Phosphoinositide 3-kinase inhibitors for inhibiting leukocyte accumulation |
| WO2006116401A1 (en) * | 2005-04-28 | 2006-11-02 | Bristol-Myers Squibb Company | C-5 substituted quinazolinone derivatives as selective estrogen receptor beta modulators |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| GB0525068D0 (en) * | 2005-12-08 | 2006-01-18 | Novartis Ag | Organic compounds |
| NZ571182A (en) | 2006-04-04 | 2010-09-30 | Univ California | Pyrazolo[3,4-d]pyrimidines |
| AU2007323836B2 (en) | 2006-11-13 | 2013-04-18 | Icos Corporation | Thienopyrimidinones for treatment of inflammatory disorders and cancers |
| WO2008064244A2 (en) | 2006-11-20 | 2008-05-29 | The Trustees Of Columbia University In The City Of New York | Phosphoinositide modulation for the treatment of neurodegenerative diseases |
| WO2008071974A2 (en) | 2006-12-14 | 2008-06-19 | Medical Research Council | Use of pi3k m-tor and akt inhibitors to induce f0xp3 expression and generate regulatory t cells |
| WO2008076447A2 (en) | 2006-12-15 | 2008-06-26 | Ordway Research Institute | Treatments of therapy-resistant diseases comprising drug combinations |
| WO2009036768A2 (en) | 2007-09-19 | 2009-03-26 | H. Lundbeck A/S | Diagnosing potential weight gain in a subject |
| US20090131512A1 (en) | 2007-10-31 | 2009-05-21 | Dynavax Technologies Corp. | Inhibition of type I in IFN production |
| NZ587051A (en) | 2008-01-04 | 2012-12-21 | Intellikine Llc | Isoquinolinone derivatives, compositions and methods of inhibiting phosphatidyl inositol-3 kinase (pi3 kinase) |
| CA2975473C (en) | 2008-11-13 | 2021-01-19 | Gilead Calistoga Llc | Therapies for hematologic malignancies |
| WO2010059593A1 (en) | 2008-11-18 | 2010-05-27 | Intellikine, Inc. | Methods and compositions for treatment of ophthalmic conditions |
| WO2010065923A2 (en) | 2008-12-04 | 2010-06-10 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Phosphatidylinositol-3-kinase p110 delta-targeted drugs in the treatment of cns disorders |
| WO2010083163A1 (en) | 2009-01-13 | 2010-07-22 | Philadelphia Health And Education Corporation | The role of p110 delta signaling in morbidity and lung pathology induced by influenza virus infection |
| JP2012521994A (ja) | 2009-03-24 | 2012-09-20 | ギリアード カリストガ エルエルシー | 2−プリニル−3−トリル−キナゾリノン誘導体のアトロプ異性体および使用方法 |
| ES2548253T3 (es) | 2009-04-20 | 2015-10-15 | Gilead Calistoga Llc | Métodos para el tratamiento de tumores sólidos |
| CA2768843A1 (en) * | 2009-07-21 | 2011-01-27 | Gilead Calistoga Llc | Treatment of liver disorders with pi3k inhibitors |
| SG179085A1 (en) | 2009-09-09 | 2012-04-27 | Avila Therapeutics Inc | Pi3 kinase inhibitors and uses thereof |
| US20120231015A1 (en) | 2009-11-06 | 2012-09-13 | Emory University | Fragile x mental retardation protein (fmrp), compositions, and methods related thereto |
| MX2012007684A (es) | 2009-12-30 | 2012-10-05 | Avila Therapeutics Inc | Modificacion covalente ligando dirigida de proteina. |
| ES2593256T3 (es) | 2010-05-21 | 2016-12-07 | Infinity Pharmaceuticals, Inc. | Compuestos químicos, composiciones y métodos para las modulaciones de cinasas |
| UA112517C2 (uk) | 2010-07-06 | 2016-09-26 | Новартіс Аг | Тетрагідропіридопіримідинові похідні |
| WO2012028757A1 (es) | 2010-09-02 | 2012-03-08 | Universitat De Barcelona | Tiazoles fluorados útiles para el tratamiento del cáncer |
| IT1403156B1 (it) | 2010-12-01 | 2013-10-04 | Università Degli Studi Di Torino | Inibitori di fosfatidilinositol 3-chinasi, relative composizioni ed usi. |
| CN102838600A (zh) | 2011-06-24 | 2012-12-26 | 山东亨利医药科技有限责任公司 | 苯基喹唑啉类PI3Kδ抑制剂 |
| CN102838601A (zh) | 2011-06-24 | 2012-12-26 | 山东亨利医药科技有限责任公司 | 选择性磷酰肌醇3-激酶δ抑制剂 |
| EP2734520B1 (en) | 2011-07-19 | 2016-09-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2013032591A1 (en) * | 2011-08-29 | 2013-03-07 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9695156B2 (en) | 2012-07-16 | 2017-07-04 | Brown University | Compounds for the treatment and prevention of infections |
| WO2014015523A1 (en) | 2012-07-27 | 2014-01-30 | Hutchison Medipharma Limited | Novel heteroaryl and heterocycle compounds, compositions and methods |
| CA2895782C (en) | 2012-12-21 | 2017-08-22 | Gilead Calistoga Llc | Substituted pyrimidine aminoalkyl-quinazolones as phosphatidylinositol 3-kinase inhibitors |
| JP6207100B2 (ja) | 2012-12-21 | 2017-10-04 | ギリアード カリストガ エルエルシー | イソキノリノンまたはキナゾリノンホスファチジルイノシトール3−キナーゼ阻害剤 |
| WO2014128612A1 (en) * | 2013-02-20 | 2014-08-28 | Novartis Ag | Quinazolin-4-one derivatives |
-
2014
- 2014-07-22 UY UY35675A patent/UY35675A/es not_active Application Discontinuation
- 2014-07-23 AR ARP140102728A patent/AR097024A1/es unknown
- 2014-07-24 AU AU2014295538A patent/AU2014295538B2/en not_active Ceased
- 2014-07-24 PT PT148293319T patent/PT3024827T/pt unknown
- 2014-07-24 KR KR1020167001639A patent/KR20160033697A/ko not_active Application Discontinuation
- 2014-07-24 EA EA201690268A patent/EA031430B1/ru not_active IP Right Cessation
- 2014-07-24 CN CN201480041540.2A patent/CN105452235B/zh active Active
- 2014-07-24 PL PL14829331T patent/PL3024827T3/pl unknown
- 2014-07-24 EP EP14829331.9A patent/EP3024827B1/en active Active
- 2014-07-24 CA CA2917433A patent/CA2917433A1/en not_active Abandoned
- 2014-07-24 WO PCT/CN2014/082946 patent/WO2015010641A1/en active Application Filing
- 2014-07-24 US US14/907,225 patent/US9840498B2/en active Active
- 2014-07-24 TW TW103125395A patent/TW201534598A/zh unknown
- 2014-07-24 ES ES14829331T patent/ES2803513T3/es active Active
- 2014-07-24 JP JP2016528336A patent/JP6437544B2/ja active Active
- 2014-07-24 MX MX2016001011A patent/MX368066B/es active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| EP3024827A4 (en) | 2017-04-26 |
| CN105452235B (zh) | 2019-08-09 |
| JP2016527238A (ja) | 2016-09-08 |
| AU2014295538B2 (en) | 2016-07-28 |
| JP6437544B2 (ja) | 2018-12-12 |
| PT3024827T (pt) | 2020-07-15 |
| CA2917433A1 (en) | 2015-01-29 |
| TW201534598A (zh) | 2015-09-16 |
| PL3024827T3 (pl) | 2020-10-19 |
| UY35675A (es) | 2015-02-27 |
| AR097024A1 (es) | 2016-02-10 |
| EP3024827A1 (en) | 2016-06-01 |
| EA031430B1 (ru) | 2018-12-28 |
| EP3024827B1 (en) | 2020-04-15 |
| US20160168131A1 (en) | 2016-06-16 |
| MX2016001011A (es) | 2016-05-09 |
| KR20160033697A (ko) | 2016-03-28 |
| WO2015010641A1 (en) | 2015-01-29 |
| AU2014295538A1 (en) | 2016-01-21 |
| CN105452235A (zh) | 2016-03-30 |
| MX368066B (es) | 2019-09-18 |
| US9840498B2 (en) | 2017-12-12 |
| EA201690268A1 (ru) | 2016-05-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2803513T3 (es) | Derivados de quinazolin-4-ona sustituidos | |
| RU2683793C2 (ru) | Замещенные аминопиримидиновые соединения и способы их использования | |
| US9763952B2 (en) | Dihydro-benzo-oxazine and dihydro-pyrido-oxazine derivatives | |
| US20180265509A1 (en) | Tetrahydro-Pyrido-Pyrimidine Derivatives | |
| ES2691650T3 (es) | 3-(quinolin-6-il-tio)-[1,2,4]-triazolo-[4,3-a]-piridinas 6-sustituidas como inhibidores de tirosina quinasa c-Met | |
| WO2014128612A1 (en) | Quinazolin-4-one derivatives | |
| WO2016149160A1 (en) | Substituted aminopyrimidine compounds and methods of use | |
| KR102184069B1 (ko) | 디히드로-피리도-옥사진 유도체의 고체 형태 | |
| KR20200112900A (ko) | 치환된 아미노피리미딘 화합물 및 이의 사용 방법 | |
| WO2015175579A1 (en) | Alkynyl compounds and methods of use | |
| ES2643902T3 (es) | Forma sólida de un derivado de dihidro-pirido-oxazina | |
| US20150148377A1 (en) | Quinoline Derivatives | |
| NZ624872B2 (en) | Dihydro-benzo-oxazine and dihydro-pyrido-oxazine derivatives |