ES2758659T3 - Compuesto químico útil como intermediario para preparar un inhibidor de catecol-o-metiltransferasa - Google Patents
Compuesto químico útil como intermediario para preparar un inhibidor de catecol-o-metiltransferasa Download PDFInfo
- Publication number
- ES2758659T3 ES2758659T3 ES12806720T ES12806720T ES2758659T3 ES 2758659 T3 ES2758659 T3 ES 2758659T3 ES 12806720 T ES12806720 T ES 12806720T ES 12806720 T ES12806720 T ES 12806720T ES 2758659 T3 ES2758659 T3 ES 2758659T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- formula
- product
- hours
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- AEDVAGWYAKIOIM-UHFFFAOYSA-N COc1cc(C(O)=O)cc([N+]([O-])=O)c1O Chemical compound COc1cc(C(O)=O)cc([N+]([O-])=O)c1O AEDVAGWYAKIOIM-UHFFFAOYSA-N 0.000 description 1
- ASOADIZOVZTJSR-UHFFFAOYSA-N Cc(c(Cl)c1C)c(-c2n[o]c(-c(cc3O)cc([N+]([O-])=O)c3O)n2)c(Cl)[n+]1[O-] Chemical compound Cc(c(Cl)c1C)c(-c2n[o]c(-c(cc3O)cc([N+]([O-])=O)c3O)n2)c(Cl)[n+]1[O-] ASOADIZOVZTJSR-UHFFFAOYSA-N 0.000 description 1
- NIEYLRZZLYWRNJ-UHFFFAOYSA-N Cc1c(-c2n[o]c(-c3cc(O)cc(N)c3)n2)c(Cl)[n-]c(C)c1Cl Chemical compound Cc1c(-c2n[o]c(-c3cc(O)cc(N)c3)n2)c(Cl)[n-]c(C)c1Cl NIEYLRZZLYWRNJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Neurology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Neurosurgery (AREA)
- Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicinal Preparation (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Una composición farmacéutica en forma de dosificación unitaria para la administración oral que comprende un compuesto de la fórmula (II)**Fórmula** en forma cristalina de micropartículas micronizadas en un portador farmacéuticamente aceptable para este en donde el D10 (EDC) no es menor que 4 μm, el D50 (EDC) es de 10-45 μm y el D95 (EDC) no es mayor que 70 μm.
Description
DESCRIPCIÓN
Compuesto químico útil como intermediario para preparar un inhibidor de catecol-o-metiltransferasa
Campo de la Invención
La presente invención se refiere a una composición farmacéutica en forma de dosificación unitaria para la administración oral que comprende un compuesto de la fórmula (II)

en forma cristalina de micropartículas micronizadas en un portador farmacéuticamente aceptable para este en donde el D10 (EDC) no es menor que 4 ^m, el D50 (EDC) es de 10-45 ^m y el D95 (EDC) no es mayor que 70 ^m.
Antecedentes de la Invención
Un método preferido de tratamiento de la enfermedad de Parkinson es la administración de una combinación de levodopa y un inhibidor de la descarboxilasa de aminoácidos aromáticos selectivo periféricamente (AADCI) junto con un inhibidor de la catecol-O-metiltransferasa (COMT). Los inhibidores de COMT empleados en la actualidad son tolcapona y entacapona. Sin embargo, algunas autoridades creen que cada uno de estos inhibidores de COMT tiene problemas residuales relacionados con las propiedades farmacocinéticas o farmacodinámicas, o con la eficacia o seguridad clínica. Por tanto, no todos los pacientes obtienen el mayor beneficio de su terapia con levodopa/AADCI/inhibidor de COMT.
Nuevos inhibidores de COMT favorecidos se describieron en L. E. Kiss y otros, J. Med. Chem., 2010, 53, 3396-3411 (D1), los documentos WO 2007/013830 (D2) y WO 2007/117165 (D3) que se cree que tienen propiedades particularmente convenientes para que los pacientes puedan beneficiarse de una terapia mejorada.
D1, D2 y D3 también revelaron métodos para preparar los nuevos inhibidores de COMT. Esos procesos, aunque efectivos, se beneficiarían de un aumento en los rendimientos. Otros beneficios que serían apropiados incluyen los seleccionados de la reducción en el número de pasos del proceso, la reducción en el número de operaciones de la unidad, la reducción de los tiempos de ciclo, el aumento del rendimiento espacial, la mayor seguridad, los reactivos más fáciles de manejar y/o el aumento en la pureza del inhibidor de COMT, especialmente cuando se prevé la fabricación de grandes cantidades. Ahora se ha descubierto un proceso que procede a través de un nuevo intermediario que es adecuado para la fabricación de cantidades comercialmente útiles de un inhibidor de COMT particularmente apto con buen rendimiento. Se obtienen beneficios adicionales, como los seleccionados de un reducido número de pasos del proceso y número de operaciones de la unidad, tiempos de ciclo reducidos, mayor rendimiento espacial, mayor seguridad, con reactivos más fáciles de manipular, perfil de impurezas mejorado y/o buena pureza.
El documento WO 2008/094053 describe estos compuestos para su uso en la preparación de un medicamento para la prevención o el tratamiento de trastornos asociados con el sistema nervioso central y periférico en donde dicho medicamento se administra de acuerdo con un régimen de dosificación que tiene una periodicidad de dosificación en el intervalo de aproximadamente dos veces al día a aproximadamente una vez en días alternos.
Breve descripción de la invención
La presente invención en un aspecto proporciona una composición farmacéutica en forma de dosificación unitaria para la administración oral que comprende un compuesto de la fórmula (II)

en forma cristalina de micropartículas micronizadas en un portador farmacéuticamente aceptable para este en donde el D10 (EDC) no es menor que 4 ^m, el D50 (EDC) es de 10-45 ^m y el D95 (EDC) no es mayor que 70 ^m.
La presente descripción se refiere, además, a 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-pirid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno y sales de este, que es el compuesto de la fórmula (I):

y sales de este.
Muy acertadamente el compuesto de fórmula (I) no tiene sal. Sin embargo, se proporcionan sales del grupo hidroxilo con iones metálicos tales como los metales alcalinos o alcalinotérreos, particularmente las sales de sodio y potasio así como las de compuestos orgánicos muy básicos tales como guanidina o similares.
Particularmente adecuado, el compuesto de fórmula (I) o su sal se proporciona en una forma adecuada para usar como un intermediario químico. Esto puede ser, por ejemplo, en una forma al menos 50 % pura, en forma cristalina, en forma sólida o en un disolvente orgánico o similares. El compuesto de fórmula (I) es útil como un intermediario en la preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol, es decir el compuesto de fórmula (II):

El compuesto de fórmula (II) también puede denominarse como opicapona o 2,5-dicloro-3-(5-(3,4-dih idroxi-5-nitrofenil)-[1,2,4]-oxadiazol-3-il)-4,6-dimetilpiridina-1-óxido. Se ha encontrado que la opicapona es más potente que la tolcapona para inhibir la COMT hepática tanto a las 3 horas como a las 6 horas después de la administración oral a ratas [ED50 en mg/kg, opicapona 0,87 a las 3 horas y 1,12 a las 6 horas en comparación con tolcapona 1,28 a las 3 horas y 2,08 a las 6 horas]. Se encontró que la opicapona a una dosis de 3 mg/kg es más eficaz para inhibir la COMT hepática en ratas con una inhibición casi completa que se produce de 2 a 6 horas después de la administración oral con solo aproximadamente 90 % de la actividad enzimática recuperada después de 72 horas mientras que tolcapona proporcionó una duración más corta de la actividad con aproximadamente 84 % de recuperación después de solo 9 horas. Tanto la opicapona como la tolcapona inhiben la S-COMT humana recombinante pero la opicapona tiene una constante inhibitoria de 16 pM que es 10 veces menor que la de tolcapona. Con respecto a la propiedad conveniente de evitar la inhibición de COMT en el cerebro, se encontró que la opicapona después de la administración oral a la rata no tiene ese efecto mientras
que la tolcapona inhibió aproximadamente el 50 % de la actividad enzimática durante un periodo de 8 horas después de la administración.
En otro aspecto la presente descripción proporciona un proceso para la preparación del compuesto de la fórmula (II) como se expuso anteriormente o una sal de este que comprende la desmetilación de un compuesto de la fórmula (I) como se expuso anteriormente o una sal de este. En una modalidad, el proceso no requiere que el compuesto de la fórmula (I) se seque para la reacción de desmetilación posterior, es decir el compuesto de la fórmula (I) puede humedecerse con tolueno. Típicamente, la cantidad de tolueno en el compuesto de la fórmula (I) está en el intervalo de 1 % a 60 % p/p. Esto es ventajoso porque mejora el tiempo y la seguridad del proceso. En otra modalidad, el proceso no requiere que el compuesto de la fórmula (II) se suspenda en EtOH. Esto es ventajoso porque mejora el tiempo de ciclo del proceso.
Muy acertadamente el proceso se adapta para la preparación de un compuesto de la fórmula (II) pero pueden prepararse sales de este, por ejemplo una sal de metal alcalino o metal alcalinotérreo, preferentemente la sal de sodio o potasio, o una sal de un compuesto orgánico muy básico tal como una guanidina.
La reacción de O-desmetilación puede llevarse a cabo mediante reacción con un reactivo desmetilante. Un reactivo desmetilante adecuado es un ácido de Lewis en presencia de una base adecuada, por ejemplo, cloruro de aluminio (AlCl3) y piridina. La desmetilación se realizará generalmente a una temperatura moderadamente elevada, preferentemente entre 45 °C-70 °C, con mayor preferencia entre 55 °C-65 °C.
El compuesto de la fórmula (II) preparado mediante este proceso puede estar suficientemente puro para su uso en una composición farmacéutica para usar en el tratamiento de la enfermedad de Parkinson como se indicó anteriormente. El compuesto de fórmula (II) así preparado puede molerse con bolas o proporcionarse de cualquier otra manera en forma de micropartículas, por ejemplo micronizado a través de molinos de chorro (MC JETMILL®). Por lo tanto, en otro aspecto, la invención proporciona una composición farmacéutica de acuerdo con la reivindicación 1 para usar en el tratamiento de la enfermedad de Parkinson mediante administración oral.
Una ventaja particular del presente proceso es que el producto de la reacción de los compuestos de las fórmulas (IV) y (V) obtenidos después de precipitación con etanol puede emplearse sin la necesidad de aislar el compuesto bruto de fórmula (III) porque el procedimiento de tratamiento permite el aislamiento de un compuesto de fórmula (III) con una pureza no menor del 95 % (HPLC), preferentemente no menor del 96 % y listo para usar en la próxima etapa de la síntesis. Otra ventaja del presente proceso es la capacidad opcional para omitir el aislamiento de cualquier compuesto intermediario de la reacción de los compuestos de fórmulas (IV) y (V).
En una modalidad, el cloruro de acilo puede prepararse mediante la reacción del compuesto de fórmula (VI):

con SOCh en dioxano a 75 °C-85 °C. El uso de dioxano en esta etapa facilita el uso en reacciones posteriores, por ejemplo si se produce en un sistema disolvente que contiene dioxano (no es necesario cambiar el disolvente antes de continuar con la próxima etapa), permite un mayor rendimiento espacial, mayor salida del proceso, requiere menos recipientes de reacción, tiempos de reacción más cortos, mejor solubilidad de los reactivos (mezcla homogénea de las soluciones de reacción en lugar de una suspensión) y evita el uso de DMF (pureza aumentada).
En otra modalidad, la reacción del compuesto de fórmula (VI) con SOCl2 se realiza en DCM en presencia de una cantidad catalítica de DMF a 35-50 °C, preferentemente a temperatura de reflujo.
Cuando se prepara el compuesto de fórmula (II) en una forma para usar en una composición farmacéutica, se puede recristalizara partir de propan-2-ol y ácido fórmico y después de eso molerse en bolas o micronizarse a través de molinos de chorro en espiral para proporcionar partículas del tamaño deseado para una buena biodisponibilidad oral y/o propiedades adecuadas (por ejemplo, tamaño de partícula adecuado) para la preparación de una composición farmacéutica.
Breve descripción de las figuras
Lista de acrónimos
DMF - dimetilformamida
SOCI2 - Cloruro de tionilo
MeOH - metanol
THF -tetrahidrofurano
DMAc - Dimetil acetamida
TFAA - ácido trifluoroacético anhídrido
IPA - Isopropanol
HNO3 - Ácido nítrico
DCM - diclorometano
EtOH - Etanol
HCl - Ácido clorhídrico
UHP - Peróxido de hidrógeno de urea
AlCl3 - Tricloruro de aluminio
NMP - N-metilpirrolidona
POCl3 - Cloruro de fosforilo
(CH3)4NCl - Cloruro de tetrametilamonio
Figura 1. Proceso para preparar un compuesto de fórmula (II) mediante el uso de un compuesto de fórmula (I) como un intermediario.
1. ácido nítrico 65 %, ácido acético, 10-20 °C, recristalización; 2. SOCl2, DMF (catalítico), 50 °C;3. Hidroxilamina al 50 % en agua, cantidad catalítica de 1,10-fenantrolina hidrato, MeOH, 75-80 °C; 4. THF, DMAc, piridina, 110-120 °C; 5. TFAA, DCM, UHP, 10-20 °C; 5a. cambio de disolvente de DCM a acetonitrilo; 5b. cristalización en tolueno/ácido fórmico; 6. cloruro de aluminio, piridina, N-metilpirrolidona; 6a. Resuspensión de etanol; 6b. Recristalización en IPA/ácido fórmico.
Figura 2. Proceso para preparar un compuesto de fórmula (II) mediante el uso de un compuesto de fórmula (I) como un intermediario.
1. NH2OH al 50 % en agua, cantidad catalítica de 1,10-fenantrolina hidrato MeOH; 2. HNO3 al 65 %, ácido acético, 2a. Recristalización en ácido acético; 3. SOCh, DCM, DMF (catalítico), cambio de disolvente de DCM a THF, adición de cloruro de ácido a amidoxima en DMAc, adición de piridina, calentar a 110 °C; inactivar en HCl y DCM ac.; cristalización en DCM/EtOH; 4. DCM, UHP, TFAA, cambio de disolvente de DCM a tolueno/ácido fórmico; cristalización en tolueno/ácido fórmico; 4a. Recristalización en ácido fórmico/tolueno; 5. AlCh, NMP, piridina, el compuesto de fórmula (II) se precipita y aísla mediante adición de HCl diluido; 5a. Recristalización en IPA/ácido fórmico.
Figura 3. Proceso para preparar un compuesto de fórmula (II) mediante el uso de un compuesto de fórmula (I) como un intermediario.
1. morfolina en MeOH; 2. SOCh en acetonitrilo; 3. POCl3, (CH3)4NCl en DCM; 4. NH2OH al 50 % en agua, cantidad catalítica de 1,10-fenantrolina hidrato, MeOH; 5. HNO3 al 65 %, ácido acético, 5a. Recristalización en ácido acético; 6. SOCl2 , DCM, DMF (catalítico), cambio de disolvente de DCM a THF; 6a. adición de cloruro de ácido a amidoxima en DMAc, adición de piridina, calentar a 110 °C; precipitación de la solución de DMAc HCl ac. con aislamiento de un compuesto bruto de fórmula (III); cristalización en DCM/EtOH; 7. DCM, UHP, TFAA, cambio de disolvente de DCM a tolueno/ácido fórmico; cristalización en tolueno/ácido fórmico; 7a. Recristalización en ácido fórmico/tolueno; 8. AlCh, NMP, piridina; el compuesto de fórmula (II) se precipita y aísla mediante adición de HCl diluido, 8a. resuspensión en etanol 8b. Recristalización en IPA/ácido fórmico.
Figura 4. Proceso para preparar un compuesto de fórmula (II) mediante el uso de un compuesto de fórmula (I) como un intermediario.
1. NH2OH al 50 % en agua, cantidad catalítica de 1,10-fenantrolina hidrato MeOH; 2,65 % HNO3 , ácido acético, 2a. Recristalización en ácido acético; 3. SOCl2, dioxano, adición de cloruro de ácido a amidoxima en dioxano, adición de piridina, calentara 110 °C; Inactivaren ac. HCl y DCM ac.; cristalización en DCM/EtOH; 4. DCM, UHP, TFAA, cambio de disolvente de DCM a tolueno/ácido fórmico; cristalización en tolueno/ácido fórmico; 4a. Recristalización en ácido fórmico/tolueno; 5. AlCh, NMP, piridina; el compuesto (II) se precipita y aísla mediante adición de HCl diluido; 5a. Recristalización en IPA/ácido fórmico.
Figura 5. Proceso para preparar un compuesto de fórmula (II) mediante el uso de un compuesto de fórmula (I) como un intermediario.
1. morfolina, MeOH, 2. SOCl2, 3. POCh; 4. H2NOH, 1,10-fenantrolina; 5. HNO3, ácido acético; 6. recristalización en ácido acético; 7. SOCh, 1,4-Dioxano; 8a. 1,4-Dioxano, Piridina; 8b. EtOH; 9. DCM, UHP, TFAA; 10. tolueno, ácido fórmico; 11. AlCl3, NMP, Piridina; 12. Ácido fórmico/IPA.
Figura 6. Proceso para preparar un compuesto de fórmula (II) mediante el uso de un compuesto de fórmula (I) como un intermediario.
1. morfolina, MeOH, 22 horas, reflujo; 2a. SOCh, MeCN; 2b. 2 horas, 65 °C; 2c. 2 horas, 20 °C; 3. POCh, TMACl, 8 horas, 110 °C; 4. NH2OH/H2O, 1,10-fenantrolina monohidrato, MeOH, 6 horas, 75 °C; 5. HNO3, HOAc, 10-20 °C; 6. SOCh, DCM/DMF, 8 horas, 40 °C; 7. DMA/THF, 2 horas, 5-10 °C; 8. Urea-H2O2 , DCM/TFAA, 18 horas, 20 °C; 9. AlCh, NMP/Piridina, 2 horas, 60 °C; 10. Recristalización.
Descripción detallada de la invención
La presente invención proporciona una composición farmacéutica en forma de dosificación unitaria para la administración oral que comprende un compuesto de la fórmula (II)

en forma cristalina de micropartículas micronizadas en un portador farmacéuticamente aceptable para este en donde el D10 (EDC) no es menor que 4 ^m, el D50 (EDC) es de 10-45 ^m y el D95 (EDC) no es mayor que 70 ^m.
La presente descripción se refiere, además, al compuesto de la fórmula (I):

y sales de este.
El uso del compuesto de fórmula (I) conduce a un proceso particularmente eficaz para la preparación del compuesto de fórmula (II). Al evitar la desprotección de grupos fenólicos e hidroxilo de los procesos de la materia anterior, se pueden lograr buenos rendimientos cuando se comienza con el ácido vanílico, compuesto que está disponible fácilmente y es relativamente menos costoso.
El compuesto de fórmula (I) puede obtenerse con alta pureza, por ejemplo en forma cristalina, lo que también ayuda a lograr la preparación del compuesto de fórmula (II) en formas muy puras, por ejemplo que solo contienen muy bajas cantidades de impurezas.
Acertadamente, el compuesto de fórmula (I) se cristaliza y/o recristaliza a partir de una mezcla de disolventes orgánicos uno de los cuales es un ácido, favorablemente ácido fórmico. Un disolvente de recristalización preferido para el compuesto de fórmula (I) es una mezcla de tolueno y ácido fórmico. Otro sistema disolvente de recristalización preferido para el compuesto de fórmula (I) es ácido fórmico/isopropanol (disolvente/antidisolvente).
El compuesto de fórmula (I) o sal de este puede prepararse mediante la oxidación del compuesto de fórmula (III):

o una sal de este.
Normal y preferentemente el compuesto sin sal de fórmula (I) se prepara a partir de un compuesto de fórmula (111) pero si se necesita una sal, esta puede producirse mediante la reacción del grupo fenólico hidroxilo con una base adecuada después de la formación del compuesto de la fórmula (I).
La reacción de oxidación puede realizarse con cualquier agente oxidante adecuado pero preferentemente se emplea un peróxido. De manera adecuada el peróxido puede ser H2O2 que se emplea preferentemente como el complejo de adición H2O2-urea. La oxidación preferentemente se lleva a cabo en presencia de un ácido orgánico anhídrido tal como trifluoroacético anhídrido.
La oxidación generalmente tiene lugar en un disolvente orgánico no hidroxílico, preferentemente en disolventes halogenados tales como cloruro de metileno. La oxidación se realiza preferentemente entre 15 °C y 30 °C, con mayor preferencia de 20 °C a 25 °C.
El compuesto de fórmula (111) puede prepararse mediante la reacción de un compuesto de fórmula (IV) en donde Y es un grupo halo, tal como cloruro, u OR en el cual R puede ser hidrógeno o un C1-C6 alquilo tal como metilo o etilo:

con un compuesto de la fórmula (V):

La reacción de los compuestos de fórmula (IV) y (V) puede tener lugar en un disolvente orgánico y más generalmente en una mezcla de disolventes orgánicos al menos uno de los cuales será un disolvente básico, por ejemplo piridina. Un disolvente mixto adecuado es dimetilacetamida, tetrahidrofurano y piridina. Alternativamente, la mezcla de disolventes orgánicos es una mezcla de dioxano y piridina. La reacción de los compuestos de fórmula (IV) y (V) también puede tener lugar en presencia de una base orgánica tal como piridina o una amina terciaria.
Cuando Y es OR y R es hidrógeno en el compuesto de fórmula (IV), el compuesto tiene fórmula (IX):

El proceso de ciclación tendrá lugar a una temperatura elevada, por ejemplo a 100 °C-120 °C. Particularmente el proceso se realizará a 105 °C-115 °C.

Cuando Y es OR y R es hidrógeno en el compuesto de fórmula (IV), se necesita la adición de un reactivo de acoplamiento tal como carbodiimida, derivados del ácido fosfónico, derivados de carbonil diimidazol.
Cuando Y es OR y R es C1-C4 alquilo tal como metilo, puede requerirse la adición de un ácido de Lewis tal como tricloruro de aluminio, o un ácido de Bronstedt tal como un catalizador de ácido p-tolueno sulfónico.
Cuando Y es un cloruro, el compuesto de fórmula (VIII), puede usarse preferentemente para preparar el compuesto de fórmula (III).
Si se desea otro líquido orgánico tal como etanol puede añadirse al final de la reacción. De manera adecuada la precipitación no se afecta por la adición de este otro líquido orgánico.
Se cree que la reacción de los compuestos de fórmulas (IV) y (V) continúa a través del intermediario de cadena abierta mostrado más abajo:

Una ventaja del proceso es que no es necesario aislar este intermediario sino que se cicla al compuesto deseado de fórmula (III) en las condiciones de reacción empleadas. De manera adecuada la reacción se realiza a una temperatura de entre 100-120 °C para producir el compuesto ciclado deseado de fórmula (III).
Se ha encontrado que el uso del compuesto de fórmula (VIII) conduce a mayores rendimientos en comparación con otros análogos activados tales como los que se forman a partir de los reactivos ácidos y de acoplamiento.
En una modalidad de la presente descripción el compuesto de fórmula (V) se prepara a partir del compuesto de la fórmula (VII)

mediante una reacción con hidroxilamina en presencia de 1,10-fenantrolina monohidrato. Preferentemente la reacción se realiza en una mezcla de metanol y agua a 70-80 °C. Convenientemente, la presencia de 1,10-fenantrolina monohidrato reduce o elimina la formación de amida no deseada y favorece la formación de la amidoxima deseada de fórmula (V).
El compuesto de fórmula (IV) puede prepararse a partir del ácido carboxílico correspondiente mediante esterificación o formación de un cloruro de ácido. Acertadamente para preparar un cloruro de ácido esto puede implicar la reacción con SOCh. Dicho ácido correspondiente puede prepararse mediante nitración de ácido vanílico. Los compuestos de fórmula (IV) que son ésteres también pueden prepararse mediante nitración del éster correspondiente de ácido vanílico, por ejemplo mediante nitración del éster metílico de ácido vanílico. Las condiciones adecuadas para estas reacciones se exponen en los Ejemplos a continuación.
El compuesto de fórmula (II) en forma cristalina es particularmente apto para usar en composiciones farmacéuticas para una administración oral. En particular, tales composiciones pueden estar en la forma de dosis unitarias discretas tales como tabletas o cápsulas.
La composición farmacéutica que contiene el compuesto de fórmula (II) en forma cristalina, preparado preferentemente como se describe en la presente, también comprenderá un portador para éste. Los portadores adecuados incluyen los descritos en D1, D2 o D3, referidos anteriormente en el presente documento. La composición farmacéutica que contiene el compuesto de fórmula (II) en forma cristalina puede comprender adicionalmente levodopa (L-DOPA) y/o un inhibidor de descarboxilasa de L-aminoácidos aromáticos (AADCi) selectivo periféricamente.
El compuesto de fórmula (II) en forma cristalina o las composiciones farmacéuticas de este pueden usarse para tratar algunos trastornos del sistema nervioso central y periférico, tal como enfermedad de Parkinson, trastornos del estado de ánimo, síndrome de piernas inquietas, trastornos gastrointestinales, estados de formación de edema e hipertensión. Esto puede ser mediante la administración a un paciente que necesite levodopa, de un inhibidor de descarboxilasa de aminoácidos aromáticos selectivo periféricamente y el compuesto cristalino de fórmula (II). Dicha administración es una administración oral y emplea una dosis unitaria discreta tal como una tableta o cápsula.
El compuesto cristalino de fórmula (II) empleado en tales composiciones está microparticulado, por ejemplo como se forma mediante molienda en bolas o mediante micronización a través de molinos de chorro en espiral. La micronización adecuada puede llevarse a cabo con un equipo de molienda MCJETMILL® tipo 200. El D10 (EDC (diámetro circular equivalente)) no es menor que 4,5 o 6 pm (por ejemplo no menor que 4 pm), el D50 (EDC) es 10-45, 15-30 o 20-25 pm (por ejemplo 10-45 pm) y el D95 (EDC) no es mayor que 60 o 70 pm. Más adecuadamente el D10 (EDC) no es menor que 4 o 5 pm (por ejemplo no menor que 5 pm), el D50 (EDC) es 10-45 o 15-30 pm (por ejemplo 15-30 pm) y el D95 (EDC) no es mayor que 60 o 70 pm (por ejemplo no mayor que 60 pm).
Las siguientes preparaciones describen un proceso apto para la preparación de intermediarios útiles. Los siguientes Ejemplos ilustran procesos y productos de acuerdo con la invención. Estos ejemplos no son limitantes y pueden modificarse de acuerdo con la descripción en la presente y el conocimiento del experto.
Preparación de productos intermediarios
Preparación 1

La cianoacetamida (280 g) se hizo reaccionar con acetil acetona (352,9 g) en metanol (1015 g) y morfolina (14,9 g). La reacción se agitó en reflujo a 65 °C hasta que se completó la reacción. La suspensión de productos resultante se filtró, se lavó con metanol y se secó para proporcionar el producto deseado con aproximadamente 97 % de rendimiento.
Preparación 2

El producto de la Preparación 1 (159 g) se suspendió en acetonitrilo (749,5 g) y se enfrió a 0-5 °C. Se añadió cloruro de sulfurilo (178,9 g) y la mezcla de reacción se calentó a temperatura ambiente y se agitó hasta que se completó la reacción.
La suspensión resultante se enfrió a 0-5 °C y se filtró. El sólido se lavó con acetonitrilo, acetato de etilo y heptano. El producto se secó después al vacío a 50 °C para producir el producto deseado (82 %).
Preparación 3

El cloruro de fosforilo (973,2 g), cloruro de tetrametilamonio (67,3 g) y el compuesto de la Preparación 2 (227,1 g) se añadieron a diclorometano (500 g). La suspensión se calentó a 85 °C y se agitó durante 5 horas. El exceso de cloruro de fosforilo se eliminó mediante destilación al vacío. La mezcla de reacción se enfrió por debajo de 30 °C y se diluyó con diclorometano. La solución resultante se añadió al agua (1350 g) a temperatura ambiente y se agitó durante 30 minutos. La fase orgánica inferior se separó y la fase acuosa se extrajo con diclorometano. Las fases orgánicas se combinaron, se lavaron con agua y después se trataron con carbón. El carbón se filtró y se realizó un cambio de disolvente a heptano mediante destilación a presión atmosférica. La solución se filtró a 50 °C y después se enfrió a 30 °C. Al enfriar más a 0 °C se obtuvieron cristales. Estos se aislaron mediante filtración, se lavaron dos veces con heptano. Después de secar a 50 °C el producto deseado se obtuvo típicamente a 88-91 %.
El proceso anterior se repitió con una reducción en diclorometano durante la cristalización y adición de algo de metanol. Los cristales resultantes similares a placas se transfirieron más fácilmente para su uso posterior.
Preparación 4a

El producto de la Preparación 3 (68,6 g) y 1,10-fenantrolina monohidrato (0,9 g) se suspendieron en metanol (240 g) a temperatura ambiente. Se añadieron agua (518 g) y una solución de hidroxilamina (50 % en agua, 80,9 g), y la mezcla se calentó a 70-80 °C y se agitó durante 5-6 horas. Se añadió agua a 70-80 °C y la solución se mantuvo durante 1 hora para inducir cristalización. La cristalización terminó por enfriamiento a 15 °C durante 8 horas. El producto se filtró y lavó dos veces con agua y se secó a 50 °C a vacío. El producto fue de color blanquecino a amarillo claro y el rendimiento fue de 87,9 %.
Preparación 4b

Una suspensión de 2,5-dicloro-4,6-dimetil-nicotinonitrilo (45,0 kg) e hidroxilamina al 50 % (59,2 kg) en presencia de una cantidad catalítica de 1,10-fenantrolina monohidrato (0,680 kg) en metanol / agua (214 kg/362 kg) se calienta a 70-80 °C. La mezcla se agita a 70-80 °C. Se añade agua (353 kg) lentamente a la solución resultante mientras que la temperatura se mantiene a > 79 °C. La solución se enfría a 75 °C con agitación lo que resulta en la cristalización de (Z)-2,5-dicloro-N'-hidroxi-4,6-dimetilnicotinimidamida. La suspensión se enfría adicionalmente a 20 °C, el sólido se filtra y la torta húmeda se lava con agua (160 kg). (Z)-2,5-dicloro-N'-hidroxi-4,6-dimetilnicotinimidamida se seca a vacío a 60 °C máx. hasta que el nivel de agua residual es máx 0,15 % (KF).
Ejemplo 1a
Preparación de ácido 4-hidroxi-5-metoxi-3 nitrobenzoico

El ácido vanílico (75 g) se suspendió en ácido acético (788 g). La suspensión se enfrió de 10 °C a 15 °Cy se añadió ácido nítrico (49 g o solución al 65 %) en tres horas a una tasa que se mantuvo la temperatura entre 10 °C y 20 °C. El color naranja amarillo resultante se agitó durante otra hora de 18 °C a 23 °C. La suspensión se filtró, se lavó con ácido acético, después una mezcla de ácido acético y agua (1/2) y después agua. Se obtuvo un rendimiento de 53 % de un producto puro al 87,9 %.
El producto bruto anterior se suspendió en ácido acético y se calentó a 105 °C - 110 °C hasta obtener una solución de color marrón naranja. La solución se transfirió al recipiente de cristalización mediante un filtro de carbón (o filtración de pulido) a una temperatura por encima de 85 °C (etapa opcional). La solución se enfrió después a 80 °C - 85 °C. La mezcla se agitó durante una hora a 70 °C - 80 °C (opcionalmente a 75 °C) durante lo cual se produjo la cristalización. La suspensión producto se enfrió a 20 °C - 25 °C durante 17 horas o se agitó durante al menos 12 horas a 20 °C - 25 °C. La suspensión producto se filtró y lavó con ácido acético, después ácido acético/agua (1/2) y finalmente agua. El producto se secó al vacío a 50 °C - 55 °C. El rendimiento de 70 % corresponde a un rendimiento general de 44 % para ambas partes de esta preparación. La pureza del producto analizado fue de 99,7 %.
La etapa de cristalización anterior es opcional y la solución puede transferirse al recipiente de cristalización mediante filtración de pulido en lugar de un filtro de carbón.
La suspensión de cristalización posterior puede agitarse durante al menos 12 horas a 20 °C - 25 °C como alternativa a 17 horas.
Ejemplo 1b
Preparación de ácido 4-hidroxi-5-metoxi-3 nitrobenzoico
Un reactor se cargó con 525 kg de ácido acético glacial y 50 kg de ácido vanílico. La mezcla se calentó con agua caliente gradualmente a 50 °C en aproximadamente 75 minutos. La temperatura se ajustó a 16 °C. Después se añadió ácido nítrico, 31,4 kg, gradualmente durante un periodo de 3 horas. Cuando la administración se completó, la mezcla se agitó durante otras 3,5-4,5 horas.
La suspensión se centrifugó mientras se lavó con 25 kg de ácido acético, 50 litros de agua desionizada y 25 kg de ácido acético de nuevo. El material cristalino húmedo se suspendió en 165 kg de ácido acético y se calentó a 91 °C hasta la disolución completa. La solución se enfrió después a 19,8 °C y la mezcla se agitó durante 1 hora. Se realizó la centrifugación y el lavado con 15,2 kg de acético y 40 litros de agua desionizada. Después el material húmedo se secó en secador a vacío en bandeja entre 40-50 °C hasta un peso constante, durante 72 horas. El peso del material seco fue 28,7 kg. El rendimiento calculado fue de 45,4 %.
Ejemplo 1c
Preparación de ácido 4-hidroxi-5-metoxi-3 nitrobenzoico
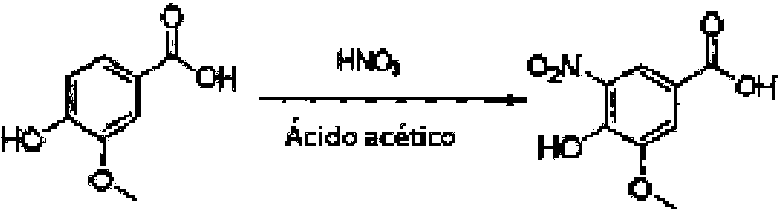
Una suspensión de ácido vanílico (68,8 kg) en ácido acético (720 kg) se enfría a 17 °C antes de añadir un exceso de ácido nítrico al 65 % (44,0 kg). Después de completar la dosificación de ácido nítrico la suspensión se agita durante 2 horas. La suspensión se filtra y la torta húmeda se lava sucesivamente con ácido acético (80,0 kg), ácido acético/agua (1:2 p/p
105 kg) y finalmente agua (80 kg — si es necesario repetir). El sólido se seca a 52 °C para NMT 12 horas antes de continuar a la próxima etapa.
Una suspensión del sólido bruto (650 kg) en ácido acético se calienta a 105 °C y se agita hasta la disolución completa del sólido bruto. Después de la filtración de pulido, la solución se enfría a 20 °C durante 3 horas lo que resulta en la cristalización y la suspensión se agita durante 2 horas a 20 °C. El sólido se filtra y la torta húmeda se lava sucesivamente con ácido acético (80 kg), ácido acético/agua (1:2 p/p - 105 kg) y finalmente agua (193 kg - si es necesario repetir). El ácido 4-hidroxi-5-metoxi-3 nitrobenzoico puro se seca a vacío a máx. 55 °C hasta alcanzar ácido acético residual a máx.
0,5 % p/p y agua a máx. 0,2 % p/p.
Ejemplo 2a
Preparación de ácido 4-hidroxi-5-metoxi-3-nitrobenzoico
El proceso del ejemplo 1a se amplió para emplear ácido vanílico (375 g) en ácido acético (3940 g) al que se añadió ácido nítrico (65 %, 245 g) a 12 °C durante 3 horas seguido por agitación durante una hora. El rendimiento general fue de 40 % de un producto puro al 99,9 %.
Ejemplo 2b
Preparación de ácido 4-hidroxi-5-metoxi-3-nitrobenzoico

El éster metílico de ácido vanílico (33 g) y nitrito de sodio (0,625 g) se cargan. Se añaden agua (158 ml) y 1,4-dioxano (158 ml) a temperatura ambiente. La mezcla de reacción se calienta a 40 °C. Se añade ácido nítrico (65 %) (15,75 g) en el transcurso de tres horas y la mezcla resultante se agita durante 4 horas después de la adición. Se toman muestras de la mezcla de reacción para la terminación.
El azeótropo de agua/ácido nítrico/dioxano se destila a vacío a 40 °C. La suspensión de productos resultante se inactiva mediante adición de solución de hidróxido de sodio (50 %, 33,2 ml) y después se agita durante 16 horas. Se tomaron muestras de la mezcla inactivada para la terminación.
Después, se añade HCl (18,5 %, 70,2 ml.) hasta que el pH esté por debajo de 1. El producto se filtra y se lava con agua (27,9 ml). El producto se seca después a vacío a 50 °C. El rendimiento general fue de 81 % de un producto puro al 97,3 %.
Ejemplo 3a
Preparación de cloruro de 4-hidroxi-5-metoxi-3-nitrobenzoilo

Una suspensión de un compuesto del ejemplo 1a (1,0 eq) en dioxano (aprox 4,5 vol) se trató con cloruro de tionilo (1,5 eq) y se calentó a 80 °C. Se formó una solución transparente a aproximadamente 75 °C. La mezcla se agitó durante 3 horas a 80 °C. El cloruro de tionilo sin reaccionar se destiló y después de la destilación el residuo se enfrió a 10 °C. Ejemplo 3B
Preparación de cloruro de 4-hidroxi-5-metoxi-3-nitrobenzoilo
Una suspensión de un compuesto del ejemplo 1a (1,0 eq) en DCM (aprox 3,4 vol) se trata con cloruro de tionilo (1,0 - 1,2 eq, por ejemplo 1,1 eq) y una cantidad catalítica (0,011 eq) de DMF y la mezcla se agita durante 16 horas a 40 °C. El DCM se destila (aprox 2,7 vol) y el residuo se diluye con Th F (aprox 1,8 vol). El exceso de cloruro de tionilo se destila con THF/DCM y el residuo después de la destilación se enfría a 10 °C.
Ejemplo 3c
Preparación de cloruro de 4-hidroxi-5-metoxi-3-nitrobenzoilo
Una suspensión del compuesto del ejemplo 1a (1,0 eq) en DCM (aprox. 4,5 vol) se trata con cloruro de tionilo (1,0 - 1,2 eq, por ejemplo 1,1 eq) y una cantidad catalítica (0,0055 eq) de DMf y la mezcla se agita durante 16 horas a reflujo. El cloruro de tionilo sin reaccionar se destila con DCM y el residuo después de la destilación se diluye con THF (aprox. 1,8 vol) y se enfría a 10 °C.
La cantidad de DCM puede ser aprox 3,4 como alternativa a aprox. 4,5 vol.
La cantidad catalítica de DMF puede ser aproximadamente 0,011 eq como alternativa a 0,0055 eq.
Ejemplo 3d
Preparación de cloruro de 4-hidroxi-5-metoxi-3-nitrobenzoilo
En un reactor se cargaron 68 kg de diclorometano, 20 kg del ácido 5-nitro-vanílico del ejemplo 1b, 76 gramos de N,N-dimetilformamida y 13,4 kg (8 L) de cloruro de tionilo, a 20,2 °C. La mezcla se calentó a 40 °C hasta que todo el material de partida se disolvió y se detuvo la evolución de HCl y SO2. Cuando se consumió todo el material de partida se destilaron 5-10 l de diclorometano a presión normal a 40 °C después la mezcla se enfrió a 20-25 °C y se continuó la destilación hasta la sequedad a vacío a 40 °C. El residuo de la evaporación se disolvió en 36 kg de THF seco. La solución de THF se usó en el ejemplo 4d.
Ejemplo 3e
Preparación de cloruro de 4-hidroxi-5-metoxi-3-nitrobenzoilo
Una suspensión del producto del ejemplo 1C (ácido 4-hidroxi-5-metoxi-3 nitrobenzoico - 160 g, 1eq) en 1,4-dioxano (720 ml, 4,5 vol) se trata con cloruro de tionilo (169,8 g, 103,7 ml, 1,5 eq) y se calienta a 80 °C. Se forma una solución transparente a aprox. 75 °C. La mezcla se agita a 80 °C (3 horas). El cloruro de tionilo sin reaccionar se destila y el residuo después de la destilación se enfría a 10 °C.
Ejemplo 4a
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno En este ejemplo el compuesto de fórmula (IV) reacciona con el compuesto de fórmula (V) para producir el compuesto de la fórmula (IIl).

El compuesto de fórmula (V) (1,24 eq) se suspendió en 1,4-dioxano (aproximadamente 4,5 vol) y la suspensión se enfrió a 10 °C. La solución de cloruro de acilo (compuesto de fórmula (IV)) del ejemplo 3a en 1,4-dioxano se añadió lentamente manteniendo la temperatura por debajo de 20 °C. Se formó una solución de color naranja transparente. Después de completar la adición, la mezcla de reacción se agitó a 20 °C durante una hora. Se añadió piridina (aproximadamente 8 eq) y la mezcla de reacción se calentó lentamente a 115 °C. La mezcla se agitó durante 6 horas a 115 °C y después se enfrió a 20 °C.
El dioxano/piridina se destiló a vacío a 70 °C. El residuo se mantuvo a 80 °C y se añadió etanol (aprox. 8 vol) para inducir cristalización. La suspensión resultante de color amarillo se enfrió a 0 °C y se agitó durante dos horas. El producto se filtró y lavó con etanol (2,5 vol) agua (3,8 vol) y etanol 2,5 vol). El producto se secó al vacío a 50 °C. Los rendimientos típicos para este proceso son de 82 a 85 %.
En una variante opcional, se empleó metanol en lugar de etanol para inducir cristalización.
Ejemplo 4b
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno En un reactor diferente, el compuesto de fórmula (V) (1,1 eq) se disuelve en DMAc (aprox. 5,8 vol) y la solución se enfría a 5 °C. La solución de cloruro de benzoilo del ejemplo 3b en THF/DCM se añade después lentamente manteniendo la temperatura por debajo de 10 °C. Después de completar la adición, la mezcla de reacción se agita a 20 ± 5 °C. La piridina (1,3 a 1,6 eq, por ejemplo 1,5 eq) se carga y la mezcla de reacción se calienta lentamente a 110 ± 5 °C eliminando los componentes de bajo punto de ebullición mediante destilación. La mezcla se agita durante otras 3 horas a 110 ± 5 °C. En otro reactor, el HCl concentrado (23,8 eq) se diluye con agua (aprox. 8,5 vol) y se enfría a 10 °C. La mezcla de reacción en piridina se dosifica lentamente a ácido clorhídrico diluido. Después de completar la adición, la suspensión resultante se agita durante otras 2 horas y el sólido se filtra. El sólido bruto se lava una vez con agua y se seca previamente en el embudo.
El sólido bruto se suspende en DCM (aprox. 28,6 vol) y la suspensión se calienta a 40 °C para lograr una solución transparente. La solución resultante se enfría a 20 °C y se extrae con agua. Después de la separación de las fases, la fase acuosa se vuelve a extraer con DCM y la fase orgánica combinada se lava una vez con agua. El DCM se destila a vacío seguido por adición de etanol. La suspensión resultante también se destila para reducirla cantidad de DCM, después se enfría a 5 °C y se agita durante otras 2 horas. Finalmente, el producto se filtra, se lava una vez con etanol frío y se seca al vacío a 45 °C.
Ejemplo 4c
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno En un segundo reactor, el compuesto de fórmula (V) (1,1 eq) se disuelve en DMAc (aprox. 7 vol) y la solución se enfría a 5 °C. La solución de cloruro de benzoilo del ejemplo 3c en THF/DCM se añade lentamente manteniendo la temperatura por debajo de 10 °C. Después de completar la adición, la mezcla de reacción se agita a 20 ± 5 °C durante 30 min. La piridina (6,9 a 7,3 eq, por ejemplo 7,14 eq) se carga y la mezcla de reacción se calienta lentamente a 110 °C eliminando componentes de bajo punto de ebullición mediante destilación. La mezcla se agita durante otras 4 horas a 110 °C y se enfría a 20 °C.
En un tercer reactor una emulsión de ácido clorhídrico diluido (preparada a partir de HCl conc. (19,6 eq) y aprox. 7,6 vol de agua destilada) y DCM (aprox. 25,5 vol) se enfría a aproximadamente 15 °C antes que la mezcla de reacción en piridina se dosifique lentamente a la emulsión. Después de completar la adición, la fase orgánica se separa y se lava con agua antes de destilar el DCM a vacío seguido por la adición de etanol. La suspensión resultante también se destila para reducir la cantidad de DCM, después se enfría a 5 °C y se agita durante otras 2 horas.
Finalmente, el producto se filtra, se lava una vez con etanol frío y se seca a vacío a 45 °C.
Ejemplo 4d
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno Se cargaron 140 kg de N,N-dimetil acetamida en el reactor. Se disolvieron 24,2 kg de amidoxima de la Preparación 4 en N,N-dimetil acetamida mientras se agitaba a 21 °C. La solución se enfrió a 5-10 °C. La solución de THF del ejemplo 3d se introdujo lentamente en la mezcla de reacción, 1,5-2 horas, mientras que la temperatura interna se mantuvo a máx.
9,5 °C por enfriamiento externo. Cuando la adición se completó, el enfriamiento externo se detuvo. La temperatura interna se dejó aumentar a 21 °C en una hora. Después de agitar durante 30 minutos, se añadió piridina 53,0 kg a la mezcla, mientras que la temperatura estaba en el intervalo de 22,4 °C - 20,6 °C. Se inició el calentamiento y la temperatura interna se elevó a 105-115 °C. La mezcla comenzó a reflujo durante 3 horas mientras que la temperatura interna alcanzó 113 °C
por destilación parcial de algo de THF. La mezcla de reacción se enfrió después y se introdujo en una mezcla de 220 kg de HCl concentrado y 170 kg de agua desionizada mientras que la temperatura interna se mantuvo entre 14-16 °C. El reactor se enjuagó con 10 kg de N,N-dimetilacetamida y 20 kg de agua desionizada. El líquido de enjuague se llevó a la mezcla. La suspensión se enfrió después adicionalmente a 5-10 °C y se agitó durante 1,5-2,0 horas. El producto se centrifugó y se lavó con 80 kg de agua desionizada. El peso húmedo bruto del producto fue de 88,6 kg.
El producto húmedo bruto, se disolvió en 460 kg (340 L) de diclorometano a 40 °C máx. Cuando se disolvió la temperatura se ajustó a 20-30 °Cy se añadieron 120 kg de agua desionizada. La fase orgánica se separó, la fase inorgánica se extrajo con 80 kg de diclorometano. La fase orgánica de 460 kg, se lavó después con 200 kg de agua desionizada y las fases se separaron. La fase inorgánica se extrajo con los 80 kg de diclorometano y las fases orgánicas se unieron. La fase orgánica así obtenida se concentró a vacío a 35 °C para 200-240 litros, después se añadió continuamente 260 kg de etanol al 96 % y la evaporación se continuó hasta un volumen final de 200-240 litros. Después la mezcla se enfrió a 5-10 °C y se agitó durante 3 horas. La centrifugación, el lavado con 20 kg de etanol dio como resultado 35,4 kg de producto húmedo. El secado a vacío durante 16 horas a 45 °C produjo 34,09 kg de producto seco. El rendimiento fue de 79,9 %.
Ejemplo 4e
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno En un segundo recipiente, se suspende (Z)-2,5-dicloro-N'-hidroxi-4,6-dimetilnicotinimidamida (201,2 g, 1,24eq) en 1,4-dioxano (720 ml, 4,5vol) y la suspensión se enfría a 10 °C. El residuo del ejemplo 3e en 1,4-dioxano se añade lentamente manteniendo la temperatura por debajo de 20 °C. Se forma una solución de color naranja transparente. Después de completar la adición, la mezcla de reacción se agita a 20 °C durante 1 hora. La piridina (483,7 ml, 8 eq) se carga después y la mezcla de reacción se calienta lentamente a 115 °C. La mezcla se agita a 115 °C durante 6 horas. La solución se enfría después a 20 °C. Se destila dioxano/piridina.
Después de la destilación, el pozo se mantiene a 80 °C y el etanol (1,28 l, 8 vol) se añade a esta temperatura para inducir cristalización. La suspensión resultante de color amarillo se enfría a 75 °C y se agita durante 1 hora a esta temperatura para permitir el crecimiento de los cristales. La suspensión producto se enfría después a 0 °C y se agita durante 2 horas a esta temperatura. El producto se filtra y se lava posteriormente con etanol (400 ml, 2,5 vol), agua (608 ml, 3,8 vol) y etanol (400 ml, 2,5 vol). El producto se seca a vacío a 50 °C hasta que la LOD es máx 1 % p/p.
Ejemplo 4f
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno Una mezcla de un compuesto de fórmula (V) (11,7 g, 50 mmol, 1,25 eq), metil 4-hidroxi-3-metoxi-5-nitrobenzoato (10 g, 40 mmol, 1 eq) y una cantidad catalítica de ácido p-toluenosulfónico (0,76 g, 4 mmol, 0,1 eq) en dimetil acetamida se calentaron a 80 °C. La reacción se siguió por HPLC. Después de 23 horas, se obtuvo un 6 % de conversión.
Ejemplo 4g
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno Una mezcla de un compuesto de fórmula (V) (11,7 g, 50 mmol, 1,25 eq), metil 4-hidroxi-3-metoxi-5-nitrobenzoato (10 g, 40 mmol, 1 eq) y una cantidad catalítica de cloruro de aluminio (0,53 g, 4 mmol, 0,1 eq) en dimetil acetamida se calentaron a 80 °C. La reacción se siguió por HPLC. Después de 20 horas, se obtuvo un 10 % de conversión.
Ejemplo 5a
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-pirid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-m etoxi-1-nitrobenceno

Una solución del producto del ejemplo 4a (24 g) se disolvió en diclorometano (388 g) a 20-40 °C. La solución amarilla se enfrió a 5 °C y se añadió peróxido de hidrógeno urea (UHP) (17,6 g) y ácido trifluoroacético anhídrido (37 g) y la agitación continuó durante 12 hr a 5 °C. La mezcla de reacción se calentó a temperatura ambiente durante una hora y la agitación continuó durante otras cinco horas. El precipitado que se formó se filtró y lavó con diclorometano. Los filtrados combinados se diluyeron adicionalmente con diclorometano, todos se lavaron y se concentraron a presión atmosférica. Se añadió tolueno y la suspensión resultante se concentró a vacío, para eliminar el diclorometano residual. Se añadió tolueno adicional y la mezcla se verificó para garantizar la presencia de menos de 0,5 % de diclorometano y menos de 0,1 % de agua. Se añadió ácido fórmico para producir ácido fórmico al 10-12 % en mezcla con tolueno. La suspensión resultante se calentó a 90 °C y se agitó hasta la disolución completa del sólido. El producto bruto se obtuvo al enfriar la solución a 5-10 °C hasta que comenzó la cristalización. La suspensión se agitó a 5-10 °C hasta que la cristalización pareció completa. El sólido se filtró, se lavó con tolueno y se secó en una corriente de nitrógeno.
El producto bruto se suspendió en solución al 10-12 % peso/peso de ácido fórmico en tolueno y se calentó a 90 °C hasta la disolución del sólido. La solución se enfrió a 5 °C y se agitó a 5 °C hasta que se produjo la cristalización. El sólido se obtuvo mediante filtración y se lavó con tolueno. Esta recristalización se repitió hasta que el producto analizado contenía menos de 0,1 % de material de partida. El producto puro se secó al vacío a 50 °C.
Ejemplo 5b
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-pirid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-m etoxi-1-nitrobenceno
Después de la disolución del producto del ejemplo 4b (24 g) en DCM (388 g) a 20-40 °C la solución amarilla se enfría a 5 °C antes de la adición del complejo peróxido de hidrógeno urea (UHP)(17,6) y trifluoroacético anhídrido (TFAA) (37 g) con control de la temperatura. Después de completar la adición de TFAA se continúa la agitación durante 12 horas a 5 °C antes de calentar la mezcla de reacción a temperatura ambiente (RT) en 1 hora y la se continúa la agitación durante otras 5 horas. El precipitado formado durante la reacción se filtra y se lava con d Cm en el filtro del embudo. Los filtrados combinados se diluyen con DCM (325 g) y después se lavan repetidamente con agua antes de concentrarlos a presión atmosférica. El DCM se reemplaza por tolueno (170 g) y la suspensión resultante se concentra de nuevo a vacío para eliminar el exceso de DCM. Los destilados se reemplazan por tolueno fresco como antes y la mezcla se analiza en cuanto al agua y DCM residuales (DCM residual después del cambio de disolvente máx. 0,5 %; agua residual después del cambio de disolvente máx. 0,1 %). El ácido fórmico (24 g) se carga, lo que resulta en ácido fórmico aprox. 10-12 % p/p en mezcla disolvente en tolueno La suspensión resultante se calienta a 90 °C y se agita hasta lograr la disolución completa del sólido. El producto bruto se cristaliza mediante enfriamiento de esta solución a 5-10 °C y posterior agitación de la suspensión resultante a 5-10 °C. El sólido se filtra y lava con tolueno y después se seca en una corriente de gas nitrógeno.
El producto bruto así obtenido se suspende en una solución aprox. 10-12 % p/p (176 g) de ácido fórmico en tolueno. La suspensión se calienta a 90 °C y se agita hasta que todo el producto se disuelve. Después de enfriar esta solución a 5 °C y agitar posteriormente a 5 °C, el producto bruto se aísla por filtración y posterior lavado del producto húmedo con tolueno.
La recristalización del producto bruto se repite (2 o más veces). El producto puro (11,8 g) se seca a 50 °C a vacío.
Ejemplo 5c
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-pirid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-m etoxi-1-nitrobenceno
Después de la disolución del producto del ejemplo 4c (24 g) en DCM (388 g) a 20-40 °C la solución amarilla se enfría a 5 °C antes de la adición con control de la temperatura del complejo peróxido de hidrógeno urea (UHP) (17,6 g) y ácido trifluoroacético anhídrido (TFAA) (37 g). Después que se completa la adición de TFAA, se continúa la agitación durante 12 horas a 5 °C antes que la mezcla de reacción se caliente a RT en 1 hora y se continúa la agitación durante otras 5 horas. El precipitado formado durante la reacción se filtra y la torta del filtro se lava con DCM. Los filtrados combinados se diluyen con DCM (325 g) y después se lavan repetidamente con agua antes de concentrarlos a presión atmosférica. El DCM se reemplaza por tolueno (170 g) y la suspensión resultante se concentra de nuevo a vacío para eliminar el exceso de DCM y agua. Los destilados se reemplazan por tolueno fresco seguido de la adición de ácido fórmico (24 g). La suspensión resultante se calienta a 80 °C y se continúa la agitación para disolver el sólido. El producto se cristaliza por enfriamiento de esta solución a 5 °C y posterior agitación de la suspensión resultante a 5 °C. El sólido se filtra, se lava con tolueno y después se seca en una corriente de gas nitrógeno.
El producto se suspende en una mezcla de ácido fórmico / tolueno (18 g/158 g) seguido por calentamiento de la mezcla de reacción a 80 °C. Después de la disolución del producto la solución se enfría a 5 °C, de manera que el producto precipita. Después de agitar adicionalmente a 5 °C la suspensión se filtra y la torta del filtro se lava con tolueno.
La recristalización del producto se repite. El producto se utiliza como material húmedo en la siguiente etapa del proceso (se obtienen 12,1 g de producto si se seca a una temperatura máxima de 60 °C).
Ejemplo 5d
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-pirid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-m etoxi-1-nitrobenceno
Se cargaron 550 kg (420 l) de diclorometano en un reactor. Se añadieron 34 kg de producto del ejemplo 4d en un periodo corto a 20 °C de temperatura interna. La solución se enfrió a 6,5 °C, después se añadieron 24,9 kg de complejo peróxido de hidrógeno urea (UHP) durante un periodo de 20-40 minutos entre 5-10 °C. La agitación continuó durante otros 20 minutos entre 6,5-7,5 °C. Se administró anhídrido trifluoroacético, 53 kg, a la mezcla de reacción, comenzando y manteniendo la temperatura a 6-7 °C durante un periodo de 2-3 horas. Cuando la administración se completó la mezcla se agitó durante otros 30 minutos. Después la temperatura interna se dejó aumentar a un máximo de 25 °C durante un periodo de 1,5 horas. La temperatura interna se mantuvo entre 20-25 °C y la mezcla se dejó reaccionar durante otras 18 20 horas. La mezcla de reacción se centrifugó y la fuga se lavó con 45 kg de diclorometano. A la solución de diclorometano separada se añadieron 460 kg (350 l) de diclorometano y 190 kg de agua desionizada. La mezcla se agitó durante 10 minutos y las fases se separaron durante 30 minutos. La fase orgánica se lavó de nuevo con 2x190 kg de agua desionizada y se separó como anteriormente. La evaporación de la solución orgánica unificada a un máximo de 35 °C a vacío se realizó hasta un volumen final de 100-120 l. Después se administró un total de 105 kg de acetonitrilo al sistema mientras que la destilación continuó para mantener el volumen a 100-120 l. Cuando se completó se añadieron 170 kg (220 L) adicionales de acetonitrilo a la mezcla a presión normal. Esta suspensión se calentó a 70-80 °C a presión normal mientras que el diclorometano se eliminó por destilación continuamente. Después la mezcla se mantuvo agitada durante una hora. La suspensión se enfrió a 20-25 °C y se agitó durante otros 30 minutos. La suspensión se centrifugó después y se lavó con 30 kg de acetonitrilo. El material húmedo, 29,7 kg, se secó a vacío durante 16 horas a 30 °C. El rendimiento del producto seco fue de 81,5 %.
Se cargaron 27,7 kg de producto, 240 kg de tolueno y 29,2 kg de ácido fórmico en el reactor y después se calentaron a 90 °C para la disolución completa durante 1 hora. Después la solución se enfrió a 7 °C y luego la suspensión se mantuvo a 7 °C durante otras 2 horas. En caso de ser necesario se aplicó una siembra con 3-5 gramos de producto puro. La suspensión se centrifugó después durante 1 hora mientras se lavaba con 28 kg de tolueno frío. El producto se suspendió en 225 kg de tolueno y se cargaron 27,2 kg de ácido fórmico. La mezcla se calentó después a 90 °C para la disolución completa durante 1 hora. Después la solución se enfrió a 20-25 °C, después la suspensión se mantuvo entre 15-25 °C durante otras 2 horas, se sembró en caso de ser necesario. Después la suspensión se centrifugó durante 60 minutos mientras se lavaba con 28 kg de tolueno frío. El proceso de recristalización puede repetirse 2-3 veces más.
El secado durante 24 horas a 38-41 °C a vacío se realizó hasta peso constante. Esto dio como resultado 16,34 kg (58,8 %) de material seco.
Ejemplo 5e
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-pirid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-m etoxi-1-nitrobenceno
Después de disolver el producto del ejemplo 4e (150 g) en DCM (2,43 kg) a reflujo, la solución amarilla se enfría a 5 °C antes de la adición con control de la temperatura de peróxido de carbamida (UHP - peróxido de hidrógeno-urea) (110 g) y ácido trifluoroacético anhídrido (TFAA) (155,1 ml en 4 porciones en 2 horas). La mezcla se agita durante 12 horas a 5 °C, después la mezcla de reacción se calienta a 25 °C durante 1,5 horas y se agita durante 5 horas. El precipitado formado durante la reacción se filtra y la torta del filtro se lava con DCM (0,36 kg). Los filtrados combinados se calientan a 30 °C y se diluyen con agua (300 g). Se añade hidróxido de sodio al 10 % hasta alcanzar pH = 4. El sistema bifásico se agita durante 10 minutos a 30 °C y después se deja que la mezcla se separe. Después la capa orgánica se lava sucesivamente con una mezcla de agua (750 g) e hidróxido de sodio al 10 % (7,5 g) (hasta pH= 4), solución de HCl al 3,2 % (300 g). El DCM se destila a presión atmosférica y después se reemplaza por tolueno (1035 g) aplicando vacío (150 mbar) y manteniendo la temperatura interna a 45 °C. Se añaden ácido fórmico (300 g) y tolueno (900 g) manteniendo la temperatura interna por encima de 40 °C. La solución resultante se destila a vacío (150 mbar, 45 °C de temperatura interna) hasta que cesa la destilación. Después de sembrar a 45 °C, la suspensión se agita durante 1 hora a 45 °C y después se enfría a 5 °C durante 2 horas. La suspensión se agita durante al menos 2 horas a 5 °C y después se filtra. La torta húmeda se lava con tolueno (195 g) y se seca en una corriente de gas nitrógeno (pureza química del producto bruto área mínima del 92 %).
Una suspensión de producto bruto en ácido fórmico (388 g, 2 en peso) se calienta a 55 °C y se agita hasta la disolución completa del producto bruto. Se añade tolueno (1242 g, 6,4 en peso) manteniendo la temperatura interna por encima de 50 °C. La reacción se agita a 150 mBar y la temperatura interna a 45 °C hasta que cesa la destilación. Se detiene el vacío y la destilación y después se añade la semilla a 45 °C. La suspensión se agita durante 1 hora a 45 °C y se enfría a 5 °C en 2 horas. La suspensión resultante se agita durante al menos 2 horas a 5 °C y después se filtra. La torta húmeda se lava con tolueno (260 g, 1,34 en peso). La torta húmeda se recoge y se carga en el reactor. Este procedimiento se repite al menos dos veces hasta que el nivel máximo de 5-[3-(2,5-dicloro-4,6-dimetil-pirid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno es 0,1 % (a/a) antes de secar a 25 °C máximo a vacío.
Ejemplo 6
El ejemplo 5a se repitió a mayor escala empleando el producto del ejemplo 3 (82 g), diclorometano (1325 g), peróxido de urea (60,1 g) y ácido trifluoroacético anhídrido (128 g).
Ejemplo 7a
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.

El producto del ejemplo 6 (15 g) se suspendió en N-metil pirrolidona (NMP) (131,5 g) y se enfrió a 5 °C. Se añadieron cloruro de aluminio (6,2 g) y piridina (12 g) a la vez que se mantenía la temperatura a 5 °C. Después de completar la adición de piridina la mezcla de reacción se calentó a 60 °C y se mantuvo durante 2 horas. Después de confirmar que quedaba menos de 0,5 material de partida, la mezcla de reacción se enfrió, y se añadió HCl acuoso (agua 233 g, HCl 123 g, 37 %). El sólido amarillo resultante se aisló mediante filtración con succión. El producto húmedo resultante se lavó con agua y propan-2-ol (67 g) y se secó al vacío.
Opcionalmente, el producto bruto se suspendió en etanol (492 g) y se calentó a reflujo. La suspensión se agitó durante 1 hora a reflujo y después se enfrió a temperatura ambiente. El sólido se obtuvo por filtración, se lavó con etanol y se secó al vacío a 50 °C. Se logró un rendimiento típico de 85 %.
Si se desea, el material cristalizado en etanol final o el producto producido inicialmente después de lavar con propan-2-ol se puede emplear en la preparación de material micronizado para usar en composiciones farmacéuticas.
Ejemplo 7b
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol. Una suspensión de aprox. 11 % p/p del producto del ejemplo 5b (20 g) en NMP (150 g) se enfría a 5 °C seguido de una adición consecutiva con control de la temperatura de cloruro de aluminio (8 g) y piridina (15,3 g). Después de completar la adición de piridina la mezcla de reacción se calienta a 60 °C seguido de otro tiempo de reacción de 2 horas. Después de la conversión completa del producto del ejemplo 5b la mezcla de reacción se enfría antes de dosificar ácido clorhídrico acuoso diluido (agua 293 g, HCl 177 g, 34 %). Mediante la adición del ácido clorhídrico, el producto bruto precipita de la matriz NMP/agua como un sólido amarillo que se aísla mediante filtración con succión. El producto húmedo resultante se lava con agua y 2-propanol en un lavado de reemplazo seguido de secado del producto bruto húmedo a vacío.
El producto bruto se suspende en etanol (282 g) seguido de calentamiento de la mezcla a reflujo. La suspensión se agita durante 1 hora en condiciones de reflujo seguido del enfriamiento a temperatura ambiente. El producto se aísla por filtración de la suspensión. El producto húmedo se lava con etanol y posteriormente se seca al vacío a aprox. 50 °C (típicamente el rendimiento corregido en peso fue del 85 %).
El producto (20 g) se suspende en ácido fórmico (725 g) antes de que la suspensión resultante se caliente a una temperatura máxima de 67 °C. La agitación continúa hasta que se logra la disolución completa del producto. La solución caliente se filtra y el filtrado se enfría a 40 - 45 °C antes de que el producto precipite primero por concentración de la solución a aprox. 40 % (v/v) de su volumen original seguido de la adición del antidisolvente 2-propanol (390 g). Después de finalizar la adición de 2-propanol la suspensión resultante se mantiene a 55-60 °C para la maduración del cristal seguido de enfriamiento a RT y filtración. La torta del filtro se lava con 2-propanol seguido de secado del material a una temperatura máxima de 58 °C hasta una pérdida por secado (LOD) máx. de 0,5 %. Típicamente, se obtuvo un rendimiento de 97-98 %.
Si se desea, el producto puede emplearse en la preparación de material micronizado para su uso en composiciones farmacéuticas.
Ejemplo 7c
Preparación de 5-[3-(2,5-didoro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
Una suspensión del producto del ejemplo 5c (20 g) o del ejemplo 6 (20 g) en NMP (153 g) se enfría a 5 °C seguido de una adición consecutiva con control de la temperatura de cloruro de aluminio (8,2 g) y piridina (15,4 g). Después de completar la adición de piridina la mezcla de reacción se calienta a 60 °C seguido de otro tiempo de reacción de 3 horas. Después de la conversión completa del producto del ejemplo 5c o del ejemplo 6 el producto bruto se precipita mediante la adición con control de la temperatura de una solución acuosa de ácido clorhídrico (agua 296 g, HCl 179 g, 34 %). La filtración del sólido seguido del lavado de la torta húmeda del filtro con agua y 2-propanol produce un material húmedo como producto bruto que se disuelve inmediatamente en ácido fórmico (536 g). Después de la filtración de pulido el filtrado se concentra a vacío seguido de la adición del antidisolvente 2-propanol (318 g). Después de envejecer la suspensión resultante a 55 60 °C la suspensión se enfría a RT y se filtra. La torta húmeda del filtro se lava con 2-propanol. El producto húmedo se seca a vacío a una temperatura máxima de 58 °C hasta una LOD máx. de 0,5 %. El rendimiento estuvo en el intervalo de 70-95 %
Si se desea, el producto puede emplearse en la preparación de material micronizado para su uso en composiciones farmacéuticas.
Ejemplo 7d
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
Se cargaron 132 kg (147 l) de N-metilpirrolidona en un reactor de 1000 l. Después se añadieron 16,3 kg de producto del ejemplo 5d. La suspensión se enfrió a 5-7 °C y se añadieron 6,5 kg de cloruro de aluminio sublimado en porciones manteniendo la temperatura interna entre 5-10 °C. La mezcla se agitó durante 10 minutos, después se añadieron 12,6 kg de piridina manteniendo la temperatura interna entre 5-10 °C. La mezcla se calentó con agua en la camisa a 20-25 °C y la mezcla se agitó durante 30 minutos. Después la mezcla se calienta a 58-62 °C y se hace reaccionar durante aproximadamente 2 horas. En un reactor separado se realizó una mezcla de 240,5 kg de agua desionizada y 146,4 kg de HCl concentrado. Esta se enfrió a 15-20 °C. La mezcla de reacción de la desmetilación se introdujo en el ácido clorhídrico diluido entre 20-25 °C. Opcionalmente, se añadieron 51,2 kg de diclorometano a la suspensión, se agitó durante 30 minutos y se centrifugó, se lavó con 60 kg de agua desionizada y 20 kg de isopropanol. El secado dio 15,9 kg de producto.
El producto se suspendió en 185,3 kg de etanol. La mezcla se agitó después a 78 °C durante una hora, después se enfrió a 20-25 °C y se agitó durante 1 hora. La suspensión se centrifugó después y la torta del filtro se lavó con 44,5 kg de etanol, 96 %. El material sólido se secó a 50 °C a vacío en un secador de bandeja de acero inoxidable. Se obtuvieron 14,35 kg (90,3 % de rendimiento) de producto seco.
Un reactor se cargó con 317,2 kg de ácido fórmico y producto seco. La mezcla se calentó a 65 °C hasta que todo el sólido se disuelva. La solución caliente se filtró después a un reactor vacío de 1000 l, se enjuagó con 20 kg de ácido fórmico, después la solución de ácido fórmico se eliminó parcialmente por destilación a vacío a aproximadamente 80-100 l. Después se introdujeron 260 kg de isopropanol a 50-60 °C y se agitó durante 30-35 minutos. La mezcla se enfrió después a 20-25 °C con agua en la camisa y se agitó un mínimo de 2 horas. La suspensión se centrifugó después y se lavó con 25 kg de isopropanol. El material húmedo se retiró de la fuga y se transfirió a un secador de bandeja a vacío y se secó hasta alcanzar un peso constante a vacío a 45-50 °C, lo que dio como resultado 13,6 kg de producto, con un rendimiento de 95,3 %.
Si se desea, el producto puede emplearse en la preparación de material micronizado para su uso en composiciones farmacéuticas.
Ejemplo 7e
Preparación de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
Una suspensión de producto del ejemplo 5e (34,1 kg) en N-metil pirrolidona (NMP) (182 kg) se calienta a 50 °C hasta su disolución y después se enfría a 5 °C seguido de una adición consecutiva con control de la temperatura de cloruro de aluminio (9,8 kg) y piridina (18,2 kg). Después de completar la adición de piridina la mezcla de reacción se calienta a 60 °C y se agita durante al menos 2 horas. La mezcla de reacción se enfría a 10-16 °C (por ejemplo 11, 13, 15 °C) antes de dosificar un ácido clorhídrico acuoso diluido (solución de 4 M, 283 l) manteniendo la temperatura por debajo de 25 °C. Durante la adición del ácido clorhídrico el producto bruto precipita de la matriz NMP/agua como un sólido amarillo. El sólido amarillo se filtra y posteriormente se lava con agua (179 kg), 2-propanol (105 kg). El sólido húmedo se seca a vacío a 55 °C.
Una suspensión del producto húmedo (25,1 kg) en ácido fórmico (813 kg) se calienta a un máx. de 67 °C. La mezcla se agita a 67 °C hasta que se logra la disolución completa del producto. La solución caliente se filtra y el filtrado se enfría a 40 -45 °C antes de que el producto precipite primero por concentración de la solución a aprox. 40 % (v/v) de su volumen original seguido por la adición del antidisolvente 2-propanol (380 kg). Después de la adición de 2-propanol, la suspensión resultante se agita a 55-60 °C para la maduración del cristal seguido de enfriamiento a RT y filtración. La torta del filtro
se lava con 2-propanol (38 kg) y después se seca a un máx. de 58 °C hasta LOD máx. de 0,5 %). El producto puede ser molido (por ejemplo mediante el uso del método del ejemplo 8).
Ejemplo 8
Micronización de 5-[3-(2,5-didoro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol con un equipo de molienda MC JETMILL® tipo 200 (micronización a través de molinos de chorro en espiral)
Equipamiento:
Molino: MC JETMILL® 200
Unidad de dosificación: K-Tron T 35
Ciclón: tipo 600
Cada prueba de micronización se realizó con al menos 2 kg de 5-(3-(2,5-d icloro-4,6-dim etil-1-oxi-p irid in-3-il)-[1.2.4] oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
Se definieron los siguientes parámetros de trabajo para la micronización:
Intervalo de velocidad de alimentación: 24,0-48,0 kg/h (200-400 g/30 seg.)
Intervalo de presión del molino: 3,0-4,0 bar
Intervalo de presión de Venturi: 3,0-4,0 bar; preferentemente la presión de Venturi es la misma que la presión del molino
Con el uso del equipo y los parámetros de trabajo anteriores, las micropartículas de 5-(3-(2,5-dicloro-4,6-dim etil-1-oxipiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol cumplen con la siguiente especificación del tamaño de partícula (tamaño de partícula determinado por microscopía óptica): D10 (EDC) no es menor que 4 o 5 pm (por ejemplo no menor que 5 pm), el D50 (EDC) es 10-45 o 15-30 pm (por ejemplo 15-30 pm) y el D95 (EDC) no es mayor que 60 o 70 pm (por ejemplo no mayor que 60 pm).
Ejemplo 9 (Figura 5)
El 2,5-dicloro-4,6-dimetil-nicotinonitrilo reacciona con hidroxilamina en presencia de cantidades catalíticas de 1,10-fenantrolina monohidrato para producir la aldoxima (Z)-2,5-dicloro-N'-hidroxi-4,6-dimetilnicotinimidamida que representa la primera pareja de acoplamiento hacia la síntesis de 5-[3-(2,5-dicloro-4,6-dim etil-p irid in-3-il)-[1.2.4] oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno. La segunda pareja de acoplamiento el ácido 5-nitro-vanílico puro se sintetiza a partir de ácido vanílico mediante nitración con ácido nítrico al 65 % seguido de recristalización del producto intermediario ácido 5-nitro-vanílico bruto a partir de ácido acético. El ensamblaje convergente del resto oxadiazol en 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno se logra mediante la primera activación del ácido 5-nitro-vanílico como su cloruro de ácido y posterior acoplamiento con la aldoxima (Z)-2,5-dicloro-N'-hidroxi-4,6-dimetilnicotinimidamida. La ciclación del producto de acoplamiento formado inicialmente se logra térmicamente para producir el resto oxadiazol mediante la eliminación de agua. La mezcla de reacción de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno, después de la reacción de cierre del anillo, se concentra y el producto se aísla de la mezcla de 1,4-dioxano/etanol en una etapa. La oxidación del anillo de piridina al correspondiente aril-N-óxido (5-[3-(2,5-dicloro-4,6-dim etil-1-oxi-p irid in-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno) se logra con ácido trifluoroperoxoacético que se forma in situ a partir de UHP (complejo peróxido de hidrógeno urea) y ácido trifluoroacético anhídrido. El 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno sin reaccionarse elimina posteriormente de 5-[3-(2,5-dicloro-4,6-dimetil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno mediante recristalización repetida en ácido fórmico/tolueno. El análogo intermediario 5-[3-(2,5-dicloro-4,6-dim etil-1-oxi-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno puro con un nivel de 5-[3-(2,5-dicloro-4,6-dimetil-piridin-3-il)-[1,2,4]oxadiazol-5-il]-2-hidroxi-3-metoxi-1-nitrobenceno por debajo de un área de 0,10 % se convierte en el análogo 5-[3-(2,5-dicloro-4,6-dimetil-1-oxipiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol bruto por escisión de éter en presencia de una cantidad estequiométrica de cloruro de aluminio y piridina. Una vez completada la reacción, el producto bruto se aísla por precipitación con un ácido clorhídrico acuoso seguido de disolución del precipitado en ácido fórmico. Después de la filtración de pulido de la solución resultante y el cambio parcial del disolvente de ácido fórmico a isopropanol, el 5-[3-(2,5-dicloro-4,6-dim etil-1-oxi-p irid in-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol se cristaliza a partir de la matriz de cristalización de ácido fórmico/IPA resultante y finalmente de manera opcional se muele al tamaño de partícula deseado.
Claims (6)
1. Una composición farmacéutica en forma de dosificación unitaria para la administración oral que comprende un compuesto de la fórmula (II)

en forma cristalina de micropartículas micronizadas en un portador farmacéuticamente aceptable para este en donde el D10 (EDC) no es menor que 4 ^m, el D50 (EDC) es de 10-45 ^m y el D95 (EDC) no es mayor que 70 ^m.
2. Una composición farmacéutica de acuerdo con cualquier reivindicación anterior en la forma de una tableta o una cápsula.
3. Una composición farmacéutica como se reivindica en cualquier reivindicación anterior en donde la forma cristalina en micropartículas micronizadas del compuesto de fórmula (II) se produce por micronización a través de molinos de chorro en espiral.
4. Una forma cristalina en micropartículas micronizadas del compuesto de fórmula (II) como se especifica en la reivindicación 1 o la reivindicación 3 para usar mediante administración oral en el tratamiento de un trastorno del sistema nervioso central y periférico, tal como enfermedad de Parkinson, trastornos del estado de ánimo, síndrome de piernas inquietas, trastornos gastrointestinales, estados de formación de edema e hipertensión.
5. El compuesto cristalino en micropartículas micronizadas para usar de acuerdo con la reivindicación 4, en donde el uso es en el tratamiento de la enfermedad de Parkinson.
6. El compuesto cristalino en micropartículas micronizadas para usar de acuerdo con la reivindicación 4 o 5, en donde el uso comprende además administrar a un paciente que padece de enfermedad de Parkinson una cantidad eficaz de levodopa y un inhibidor de la descarboxilasa de aminoácidos aromáticos selectivo periféricamente.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161570141P | 2011-12-13 | 2011-12-13 | |
| GBGB1121413.7A GB201121413D0 (en) | 2011-12-13 | 2011-12-13 | Chemical compounds and processes |
| US201261593625P | 2012-02-01 | 2012-02-01 | |
| GBGB1201758.8A GB201201758D0 (en) | 2012-02-01 | 2012-02-01 | Chemical compounds and processes |
| US201261718589P | 2012-10-25 | 2012-10-25 | |
| PCT/PT2012/000048 WO2013089573A1 (en) | 2011-12-13 | 2012-12-12 | Chemical compound useful as intermediate for preparing a catechol-o-methyltransferase inhibitor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2758659T3 true ES2758659T3 (es) | 2020-05-06 |
Family
ID=48612903
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES12806720T Active ES2758659T3 (es) | 2011-12-13 | 2012-12-12 | Compuesto químico útil como intermediario para preparar un inhibidor de catecol-o-metiltransferasa |
| ES19194756T Active ES2960805T3 (es) | 2011-12-13 | 2012-12-12 | Compuesto químico útil como intermediario para preparar un inhibidor de catecol-o-metiltransferasa |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19194756T Active ES2960805T3 (es) | 2011-12-13 | 2012-12-12 | Compuesto químico útil como intermediario para preparar un inhibidor de catecol-o-metiltransferasa |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US9126988B2 (es) |
| EP (2) | EP2791134B1 (es) |
| JP (3) | JP6456143B2 (es) |
| BR (1) | BR112014014341A2 (es) |
| CA (2) | CA3088684C (es) |
| CY (1) | CY1122580T1 (es) |
| DK (1) | DK2791134T3 (es) |
| ES (2) | ES2758659T3 (es) |
| HR (1) | HRP20192133T8 (es) |
| HU (1) | HUE047856T2 (es) |
| PL (1) | PL2791134T3 (es) |
| PT (2) | PT2791134T (es) |
| RS (1) | RS59666B1 (es) |
| SI (1) | SI2791134T1 (es) |
| WO (1) | WO2013089573A1 (es) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ565460A (en) | 2005-07-26 | 2011-06-30 | Bial Portela & Ca Sa | Nitrocatechol derivatives as COMT inhibitors |
| EP1845097A1 (en) | 2006-04-10 | 2007-10-17 | Portela & Ca., S.A. | Oxadiazole derivatives as COMT inhibitors |
| US8524746B2 (en) | 2007-01-31 | 2013-09-03 | Bial-Portela & Ca., S.A. | Dosage regimen for COMT inhibitors |
| AR070907A1 (es) | 2008-03-17 | 2010-05-12 | Bial Portela & Ca Sa | Formas cristalinas de derivado de nitrocatecol |
| ES2730678T3 (es) | 2009-04-01 | 2019-11-12 | Bial Portela & Ca Sa | Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para fabricarlas |
| US20140045900A1 (en) | 2011-02-11 | 2014-02-13 | Bial-Portela & Ca, S.A. | Administration regime for nitrocatechols |
| HUE047856T2 (hu) * | 2011-12-13 | 2020-05-28 | Bial Portela & Ca Sa | Kémiai vegyület, amely köztitermékként használható katechol-O-metil-transzferáz inhibitor elõállításához |
| WO2016083863A1 (en) | 2014-11-28 | 2016-06-02 | Bial - Portela & Ca, S.A. | Medicaments for slowing parkinson's disease |
| GB2563858A (en) * | 2017-06-27 | 2019-01-02 | Azad Pharmaceutical Ingredients Ag | New route of synthesis for opicapone |
| MX2020006283A (es) * | 2017-12-18 | 2020-12-09 | Unichem Lab Ltd | Proceso para la preparación de opicapona y sus intermediarios. |
| CA3112994A1 (en) * | 2018-10-05 | 2020-04-09 | Neurocrine Biosciences, Inc. | Methods for the administration of comt inhibitors |
| EP3860603A1 (en) | 2018-10-05 | 2021-08-11 | Neurocrine Biosciences, Inc. | Methods for the administration of comt inhibitors |
| CN109529881A (zh) * | 2018-11-12 | 2019-03-29 | 刘晓菊 | 一种治疗帕金森药物用中间体5-硝基香草酸的制备方法 |
| CN115335036A (zh) * | 2020-03-13 | 2022-11-11 | 巴尔-波特拉及康邦亚股份有限公司 | 微粉化的奥匹卡朋 |
| GB202011709D0 (en) | 2020-07-28 | 2020-09-09 | Bial Portela & Ca Sa | Solid dispersion of opicapone |
| GB202016425D0 (en) | 2020-10-16 | 2020-12-02 | Bial Portela & Ca Sa | Treatment regimens for parkinson's disease |
| KR20230118933A (ko) | 2020-12-17 | 2023-08-14 | 바이알 - 포르텔라 앤드 씨에이 에스에이 | 초기 특발성 파킨슨병에 대한 치료 요법 |
| CN112375014A (zh) * | 2020-12-17 | 2021-02-19 | 重庆柳江医药科技有限公司 | 一种奥匹卡朋工艺杂质及制备方法和用途 |
| US20240300930A1 (en) * | 2021-02-26 | 2024-09-12 | Msn Laboratories Private Limited, R&D Center | Novel process for the preparation of 2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine-1-oxide |
| CN113292489B (zh) * | 2021-06-16 | 2022-08-30 | 泓博智源(开原)药业有限公司 | 二氯代二烷基烟腈的制备方法 |
| GB202204798D0 (en) | 2022-04-01 | 2022-05-18 | Bial Portela & Ca Sa | Prodrugs of opicapone |
| GB202212082D0 (en) | 2022-08-18 | 2022-10-05 | Bial Portela & Ca Sa | Treatment regimens for parkinson's disease |
| GB202219669D0 (en) | 2022-12-23 | 2023-02-08 | Bial Portela & Ca Sa | Processes and intermediates for synthesising opicapone |
Family Cites Families (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1532178A (en) | 1921-07-25 | 1925-04-07 | Louis A Godbold | Lubricator |
| FR1260080A (fr) | 1960-03-22 | 1961-05-05 | Materiel De Forage Soc De Fab | Trépan à molettes étanche |
| US3647809A (en) | 1968-04-26 | 1972-03-07 | Chinoin Gyogyszer Es Vegyeszet | Certain pyridyl-1 2 4-oxadiazole derivatives |
| US4022901A (en) | 1975-03-05 | 1977-05-10 | E. R. Squibb & Sons, Inc. | 3-Pyridinyl-5-isothiocyanophenyl oxadiazoles |
| US4264573A (en) | 1979-05-21 | 1981-04-28 | Rowell Laboratories, Inc. | Pharmaceutical formulation for slow release via controlled surface erosion |
| US4386668A (en) | 1980-09-19 | 1983-06-07 | Hughes Tool Company | Sealed lubricated and air cooled rock bit bearing |
| US5236952A (en) | 1986-03-11 | 1993-08-17 | Hoffmann-La Roche Inc. | Catechol derivatives |
| DK175069B1 (da) | 1986-03-11 | 2004-05-24 | Hoffmann La Roche | Pyrocatecholderivater |
| YU213587A (en) | 1986-11-28 | 1989-06-30 | Orion Yhtymae Oy | Process for obtaining new pharmacologic active cateholic derivatives |
| DE3840954A1 (de) | 1988-12-05 | 1990-06-07 | Shell Int Research | Herstellung von 2-chlornicotinsaeureestern |
| EP0462639A1 (en) | 1990-06-05 | 1991-12-27 | Shell Internationale Researchmaatschappij B.V. | Preparation of 2-chloropyridine derivatives |
| DK0487774T3 (da) | 1990-11-29 | 1994-11-21 | Wei Ming Pharmaceutical Mfg Co | Hjælpestof til direkte tablettering |
| CA2126976A1 (en) | 1991-12-31 | 1993-07-08 | Hisashi Takasugi | Oxadiazole derivatives having acetylcholinesterase-inhibitory and muscarinic agonist activity |
| FR2730322B1 (fr) | 1995-02-02 | 1997-04-30 | Imago | Monture de lunettes metallique |
| DE19628617A1 (de) | 1996-07-16 | 1998-01-22 | Basf Ag | Direkttablettierhilfsmittel |
| US6206110B1 (en) | 1996-09-09 | 2001-03-27 | Smith International, Inc. | Protected lubricant reservoir with pressure control for sealed bearing earth boring drill bit |
| JP2002526482A (ja) | 1998-09-18 | 2002-08-20 | バーテックス ファーマシューティカルズ インコーポレイテッド | p38のインヒビター |
| GB2344819A (en) | 1998-12-18 | 2000-06-21 | Portela & Ca Sa | 2-Phenyl-1-(3,4-dihydroxy-5-nitrophenyl)-1-ethanones |
| FI109453B (fi) | 1999-06-30 | 2002-08-15 | Orion Yhtymae Oyj | Farmaseuttinen koostumus |
| US6660753B2 (en) | 1999-08-19 | 2003-12-09 | Nps Pharmaceuticals, Inc. | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| NZ517221A (en) | 1999-08-19 | 2004-01-30 | Nps Pharma Inc | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| FI20000635A0 (fi) | 2000-03-17 | 2000-03-17 | Orion Yhtymae Oyj | COMT-inhibiittoreiden käyttö analgeettina |
| SE0001438D0 (sv) | 2000-04-18 | 2000-04-18 | Axon Chemicals Bv | New chemical compounds and their use in therapy |
| DE10029201A1 (de) | 2000-06-19 | 2001-12-20 | Basf Ag | Verfahren zur Herstellung fester oraler Darreichungsformen mit retardierender Wirkstoffreisetzung |
| GB2363792A (en) | 2000-06-21 | 2002-01-09 | Portela & Ca Sa | Nitrocatechols |
| CN1166626C (zh) | 2000-08-30 | 2004-09-15 | 李凌松 | 三或四取代苯基化合物、其制备方法及应用 |
| US20030027253A1 (en) | 2000-11-28 | 2003-02-06 | Presnell Scott R. | Cytokine receptor zcytor19 |
| US20040097555A1 (en) | 2000-12-26 | 2004-05-20 | Shinegori Ohkawa | Concomitant drugs |
| WO2002051442A1 (fr) | 2000-12-26 | 2002-07-04 | Takeda Chemical Industries, Ltd. | Co-prescriptions |
| MXPA03007513A (es) | 2001-02-21 | 2004-07-30 | Nps Pharma Inc | Compuestos heteropoliciclicos y su uso como antagonistas del receptor metabotropico de glutamato. |
| US20030187007A1 (en) | 2001-05-30 | 2003-10-02 | Cao Sheldon Xiaodong | Inhibitors of protein kinase for the treatment of disease |
| CA2449544A1 (en) | 2001-06-08 | 2002-12-19 | Cytovia, Inc. | Substituted 3-aryl-5-aryl-[1,2,4]-oxadiazoles and analogs |
| CN1292749C (zh) | 2001-07-26 | 2007-01-03 | 默克专利股份有限公司 | 2-[5-(4-氟苯基)-3-吡啶基甲氨基甲基]苯并二氢吡喃及其生理学上可接受的盐的新用途 |
| JP4379853B2 (ja) | 2001-10-05 | 2009-12-09 | 惠民製藥股▲分▼有限公司 | 直接錠剤化用調合物および補助剤の調合方法 |
| CN100364531C (zh) | 2002-12-18 | 2008-01-30 | 西托维亚公司 | 3,5-二取代-[1,2,4]-二唑及类似物和其用途 |
| WO2005006945A2 (en) | 2003-07-03 | 2005-01-27 | The Salk Institute For Biological Studies | Methods for treating neural disorders and compounds useful therefor |
| EP1663211B1 (en) | 2003-08-06 | 2010-01-20 | Vertex Pharmaceuticals Incorporated | Aminotriazole compounds useful as inhibitors of protein kinases |
| DE10338174A1 (de) | 2003-08-20 | 2005-03-24 | Lts Lohmann Therapie-Systeme Ag | Transdermale Arzneimittelzubereitungen mit Wirkstoffkombinationen zur Behandlung der Parkinson-Krankheit |
| US7300406B2 (en) | 2003-09-30 | 2007-11-27 | Carter Vandette B | Medical examination apparatus |
| GB0325956D0 (en) | 2003-11-06 | 2003-12-10 | Addex Pharmaceuticals Sa | Novel compounds |
| ATE464303T1 (de) | 2004-04-28 | 2010-04-15 | Vertex Pharma | Als inhibitoren von rock und anderen proteinkinasen geeignete zusammensetzungen |
| GB0510143D0 (en) | 2005-05-18 | 2005-06-22 | Addex Pharmaceuticals Sa | Novel compounds A1 |
| US20060173074A1 (en) | 2004-11-10 | 2006-08-03 | Juha Ellmen | Treatment of restless legs syndrome |
| WO2006061697A1 (en) | 2004-12-06 | 2006-06-15 | Themis Laboratories Private Limited | Sulfonylurea compositions and a process for its preparation |
| US20080051441A1 (en) | 2004-12-28 | 2008-02-28 | Astrazeneca Ab | Aryl Sulphonamide Modulators |
| WO2006071184A1 (en) | 2004-12-28 | 2006-07-06 | Astrazeneca Ab | Aryl sulphonamide modulators |
| AU2006239418A1 (en) | 2005-04-26 | 2006-11-02 | Neurosearch A/S | Novel oxadiazole derivatives and their medical use |
| US20060257473A1 (en) | 2005-05-11 | 2006-11-16 | Porranee Puranajoti | Extended release tablet |
| GB0510139D0 (en) | 2005-05-18 | 2005-06-22 | Addex Pharmaceuticals Sa | Novel compounds B1 |
| JP4981794B2 (ja) | 2005-06-03 | 2012-07-25 | アボット・ラボラトリーズ | シクロブチルアミン誘導体 |
| JP2007024970A (ja) | 2005-07-12 | 2007-02-01 | Miyakawa:Kk | 液晶表示装置の開口効率を上昇させるための樹脂レンズ製造法及びその製造装置 |
| US20090000437A1 (en) | 2005-07-14 | 2009-01-01 | Provo Craft And Novelty, Inc. | Methods for Cutting |
| NZ565460A (en) | 2005-07-26 | 2011-06-30 | Bial Portela & Ca Sa | Nitrocatechol derivatives as COMT inhibitors |
| FR2889525A1 (fr) | 2005-08-04 | 2007-02-09 | Palumed Sa | Nouveaux derives de polyquinoleines et leur utilisation therapeutique. |
| US20070048384A1 (en) | 2005-08-26 | 2007-03-01 | Joerg Rosenberg | Pharmaceutical compositions |
| EP1954137A4 (en) | 2005-11-18 | 2008-12-17 | Janssen Pharmaceutica Nv | 2-KETO-OXAZOLE AS MODULATORS OF FATTY ACID AMIDHYDROLASE |
| JP4738419B2 (ja) | 2005-11-30 | 2011-08-03 | 富士フイルムRiファーマ株式会社 | アミロイドの凝集及び/又は沈着に起因する疾患の診断薬及び治療薬 |
| GB0606774D0 (en) | 2006-04-03 | 2006-05-10 | Novartis Ag | Organic compounds |
| EP1845097A1 (en) * | 2006-04-10 | 2007-10-17 | Portela & Ca., S.A. | Oxadiazole derivatives as COMT inhibitors |
| PE20080906A1 (es) | 2006-08-17 | 2008-07-05 | Kemia Inc | Derivados heteroarilo como inhibidores de citocina |
| US20080167286A1 (en) | 2006-12-12 | 2008-07-10 | Abbott Laboratories | Pharmaceutical compositions and their methods of use |
| US8486979B2 (en) | 2006-12-12 | 2013-07-16 | Abbvie Inc. | 1,2,4 oxadiazole compounds and methods of use thereof |
| US8524746B2 (en) | 2007-01-31 | 2013-09-03 | Bial-Portela & Ca., S.A. | Dosage regimen for COMT inhibitors |
| MX2010002258A (es) | 2007-08-27 | 2010-04-22 | Helicon Therapeutics Inc | Compuestos terapeuticos de isoxazol. |
| CA2715802A1 (en) | 2008-02-28 | 2009-09-03 | Bial - Portela & C.A., S.A. | Pharmaceutical composition for poorly soluble drugs |
| AR070907A1 (es) | 2008-03-17 | 2010-05-12 | Bial Portela & Ca Sa | Formas cristalinas de derivado de nitrocatecol |
| MX2011001046A (es) | 2008-07-29 | 2011-03-29 | Bial Portela & Ca Sa | Regimen de administracion para nitrocatecoles. |
| ES2915698T3 (es) | 2009-04-01 | 2022-06-24 | Bial Portela & Ca Sa | Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para preparar las mismas |
| ES2730678T3 (es) | 2009-04-01 | 2019-11-12 | Bial Portela & Ca Sa | Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para fabricarlas |
| AU2011222856B2 (en) | 2010-03-04 | 2015-10-15 | Orion Corporation | Use of levodopa, carbidopa and entacapone for treating Parkinson's disease |
| US20140045900A1 (en) | 2011-02-11 | 2014-02-13 | Bial-Portela & Ca, S.A. | Administration regime for nitrocatechols |
| HUE047856T2 (hu) * | 2011-12-13 | 2020-05-28 | Bial Portela & Ca Sa | Kémiai vegyület, amely köztitermékként használható katechol-O-metil-transzferáz inhibitor elõállításához |
-
2012
- 2012-12-12 HU HUE12806720A patent/HUE047856T2/hu unknown
- 2012-12-12 CA CA3088684A patent/CA3088684C/en active Active
- 2012-12-12 BR BR112014014341A patent/BR112014014341A2/pt not_active Application Discontinuation
- 2012-12-12 ES ES12806720T patent/ES2758659T3/es active Active
- 2012-12-12 SI SI201231703T patent/SI2791134T1/sl unknown
- 2012-12-12 WO PCT/PT2012/000048 patent/WO2013089573A1/en active Application Filing
- 2012-12-12 RS RS20191562A patent/RS59666B1/sr unknown
- 2012-12-12 EP EP12806720.4A patent/EP2791134B1/en active Active
- 2012-12-12 PT PT128067204T patent/PT2791134T/pt unknown
- 2012-12-12 EP EP19194756.3A patent/EP3604299B1/en active Active
- 2012-12-12 CA CA2858025A patent/CA2858025C/en active Active
- 2012-12-12 ES ES19194756T patent/ES2960805T3/es active Active
- 2012-12-12 PL PL12806720T patent/PL2791134T3/pl unknown
- 2012-12-12 JP JP2014547135A patent/JP6456143B2/ja active Active
- 2012-12-12 DK DK12806720T patent/DK2791134T3/da active
- 2012-12-12 PT PT191947563T patent/PT3604299T/pt unknown
- 2012-12-12 US US14/365,265 patent/US9126988B2/en active Active
-
2015
- 2015-07-31 US US14/814,603 patent/US9630955B2/en active Active
-
2017
- 2017-10-27 JP JP2017207738A patent/JP6721558B2/ja active Active
-
2019
- 2019-11-27 CY CY20191101241T patent/CY1122580T1/el unknown
- 2019-11-27 JP JP2019214566A patent/JP2020059729A/ja not_active Withdrawn
- 2019-11-27 HR HRP20192133TT patent/HRP20192133T8/hr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| JP6721558B2 (ja) | 2020-07-15 |
| PT3604299T (pt) | 2023-10-31 |
| HRP20192133T1 (hr) | 2020-02-21 |
| EP2791134A1 (en) | 2014-10-22 |
| PT2791134T (pt) | 2019-12-18 |
| JP2018052949A (ja) | 2018-04-05 |
| EP3604299A1 (en) | 2020-02-05 |
| CY1122580T1 (el) | 2021-01-27 |
| JP2020059729A (ja) | 2020-04-16 |
| BR112014014341A2 (pt) | 2017-08-22 |
| US9630955B2 (en) | 2017-04-25 |
| HUE047856T2 (hu) | 2020-05-28 |
| CA3088684A1 (en) | 2013-06-20 |
| CA2858025C (en) | 2020-09-22 |
| SI2791134T1 (sl) | 2020-01-31 |
| RS59666B1 (sr) | 2020-01-31 |
| CA3088684C (en) | 2021-10-26 |
| US9126988B2 (en) | 2015-09-08 |
| ES2960805T3 (es) | 2024-03-06 |
| CA2858025A1 (en) | 2013-06-20 |
| PL2791134T3 (pl) | 2020-03-31 |
| DK2791134T3 (da) | 2019-12-09 |
| WO2013089573A1 (en) | 2013-06-20 |
| US20140350057A1 (en) | 2014-11-27 |
| US20160009700A1 (en) | 2016-01-14 |
| EP2791134B1 (en) | 2019-09-25 |
| HRP20192133T8 (hr) | 2021-02-19 |
| JP6456143B2 (ja) | 2019-01-23 |
| JP2015500335A (ja) | 2015-01-05 |
| EP3604299B1 (en) | 2023-07-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2758659T3 (es) | Compuesto químico útil como intermediario para preparar un inhibidor de catecol-o-metiltransferasa | |
| JP4917440B2 (ja) | Gabaaアゴニストの多形性形態 | |
| PT2059513E (pt) | Processo para a preparação de ácidos 1,2,4-oxadiazolebenzóicos | |
| ES2355729T3 (es) | Nuevos polimorfos cristalinos del clopidogrel. | |
| WO2004072030A2 (en) | Process for preparing pyrrolotriazine kinase inhibitors | |
| ES2362660T3 (es) | Cristalización de formas sólidas de sales de adición de clopidogrel. | |
| AU2021277593A1 (en) | Solid forms of [(1 S)-1 -[(2S,4R,5R)-5-(5-amino-2-oxo-thiazolo[4,5-d]pyrimidin-3-yl)-4-hydroxy-te trahydrofuran-2-yl]propyl] acetate | |
| JP5523095B2 (ja) | 修飾マクロファージ遊走阻止因子阻害剤 | |
| TWI832045B (zh) | 抗病毒性1,3-二氧代茚化合物 | |
| ES2303462B1 (es) | Nuevo procedimiento para la preparacion de una forma polimorfica i de olanzapina farmaceuticamente pura. | |
| JP2009530404A5 (es) | ||
| JPH04230365A (ja) | 新規なオキサゾロピリジン化合物 | |
| EP3097097A1 (en) | Imidazoles for the treatment and prophylaxis of respiratory syncytial virus infection | |
| ES2275110T3 (es) | Procedimiento para la preparacion de zaleplon. | |
| KR101938955B1 (ko) | 시타글립틴염산염 결정형의 제조방법 | |
| WO2024069492A1 (en) | Processes for the preparation and manufacture of relugolix | |
| ES2311391B1 (es) | Forma cristalina de moxifloxacino base. | |
| JP2024504824A (ja) | 芳香族複素環化合物及びその製造方法と用途 | |
| CN102531999A (zh) | 无定形盐酸乐卡地平及其制备方法 |