ES2915698T3 - Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para preparar las mismas - Google Patents
Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para preparar las mismas Download PDFInfo
- Publication number
- ES2915698T3 ES2915698T3 ES10714386T ES10714386T ES2915698T3 ES 2915698 T3 ES2915698 T3 ES 2915698T3 ES 10714386 T ES10714386 T ES 10714386T ES 10714386 T ES10714386 T ES 10714386T ES 2915698 T3 ES2915698 T3 ES 2915698T3
- Authority
- ES
- Spain
- Prior art keywords
- api
- composition
- formulation
- binder
- weight
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4415—Pyridoxine, i.e. Vitamin B6
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Psychology (AREA)
- Inorganic Chemistry (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Una composición estable que comprende: un ingrediente farmacéutico activo (API) elegido entre 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4- oxadiazol-3-il)-4,6-dimetilpiridina y sales, ésteres, hidratos y solvatos del mismo; al menos un agente de carga; y al menos un aglutinante; en el que el ingrediente farmacéutico activo está presente en forma granular; en el que el al menos un agente de carga no es un compuesto de fosfato; en el que el al menos un aglutinante no es un compuesto de PVP; y en donde la composición tiene una densidad aparente superior a 0.5 g/mL.
Description
DESCRIPCIÓN
Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para preparar las mismas
Campo de la divulgación
La presente divulgación se refiere a composiciones y formulaciones farmacéuticas que comprenden al menos un ingrediente farmacéutico activo elegido entre los derivados de nitrocatecol y sus sales.
Antecedentes
La levodopa (L-DOPA) se ha utilizado en la práctica clínica durante varias décadas en el tratamiento sintomático de diversas afecciones, incluida la enfermedad de Parkinson. L-DOPA es capaz de atravesar la barrera hematoencefálica, donde luego se convierte en dopamina y aumenta los niveles de la misma. Sin embargo, la conversión de L-DOPA en dopamina también puede ocurrir en el tejido periférico, lo que posiblemente cause efectos adversos tras la administración de L-DOPA. Por lo tanto, se ha convertido en una práctica clínica estándar administrar conjuntamente un inhibidor de la descarboxilasa de aminoácidos periféricos (AADC), como carbidopa o benserazida, que previene la conversión a dopamina en el tejido periférico.
Esto ha llevado a un interés en el desarrollo de inhibidores de la enzima catecol-O-metiltransferasa (COMT) basado en la hipótesis de que la inhibición de la enzima puede proporcionar mejoras clínicas en pacientes que padecen la enfermedad de Parkinson en tratamiento con L-DOPA, ya que COMT cataliza la degradación de L-DOPA.
Se ha encontrado, como se establece en las Publicaciones Internacionales Nos. WO 2007/013830 y WO 2007/117165 que los compuestos de fórmula I divulgados en el presente documento, que son derivados de nitrocatecol, son inhibidores de COMT potentes y de acción prolongada. Esos compuestos son bioactivos y biodisponibles. Por tanto, los compuestos de fórmula I tienen propiedades farmacéuticas potencialmente valiosas en el tratamiento de algunos trastornos del sistema nervioso central y periférico en los que la inhibición de la O-metilación de las catecolaminas puede tener un beneficio terapéutico, como, por ejemplo, trastornos del estado de ánimo, enfermedad de Parkinson y trastornos, síndrome de piernas inquietas, trastornos gastrointestinales, estados de formación de edema e hipertensión. Además, estos compuestos también pueden tener actividad en el tratamiento de otras enfermedades y trastornos no relacionados con la inhibición de la O-metilación de las catecolaminas. El documento WO 2008/094053 A1 divulga que los compuestos de fórmula I divulgados en el presente documento se pueden administrar de acuerdo con un régimen de dosificación que tiene una periodicidad de dosificación que oscila entre aproximadamente dos veces al día y aproximadamente una vez cada dos días.
Sin embargo, también se ha encontrado que los compuestos de fórmula I son sensibles a ciertos excipientes, lo que puede provocar la descomposición de los compuestos de fórmula I y/o falta de estabilidad de las composiciones y formulaciones que contienen estos compuestos. Los compuestos de fórmula I también pueden exhibir una baja densidad aparente y/o malas características de fluidez, lo que puede aumentar la dificultad para formular y/o fabricar una formulación de dosificación estable que contenga el compuesto activo.
Resumen
Los inventores han descubierto ahora composiciones estables y formulaciones de las mismas que comprenden un ingrediente farmacéutico activo (“API”) elegido entre un derivado de nitrocatecol de fórmula I como se define aquí y sus sales, ésteres, hidratos y solvatos. El API que es un derivado de nitrocatecol es 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina. El API está presente en forma granular y la composición tiene una densidad aparente superior a 0.5 g/ml. El derivado de nitrocatecol también puede ser una mezcla del API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina y que comprende además 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
La API está presente en forma granular. En algunas realizaciones, las composiciones y/o formulaciones pueden comprender un API adicional, por ejemplo, las composiciones y/o formulaciones pueden comprender, además del API que es un derivado de nitrocatecol de fórmula I, API adicionales tales como L-DOPA, un inhibidor de la descarboxilasa de aminoácidos periféricos (AADC), como carbidopa o benserazida. Las composiciones y/o formulaciones también comprenden al menos un agente de carga y al menos un aglutinante. El agente de carga no es un derivado de fosfato. El aglutinante no es un compuesto derivado de polivinilpirrolidona (“PVP”). En diversas realizaciones cuando el API está presente en forma granular, al menos un agente de carga y al menos un aglutinante pueden ser, independientemente, intragranulares (es decir, granulados con el API y/o contenidos dentro de los mismos gránulos que el API), extragranulares (es decir, presente fuera de los gránulos de API), o parte intragranular y parte extragranular. Las composiciones exhiben una densidad aparente que es mayor que la del API solo (es decir, mayor de 0.5 g/ml), y que puede, en ciertas realizaciones, aumentar significativamente. Las composiciones también pueden exhibir mejoras en otras características tales como la compresibilidad. El uso de los métodos descritos en el presente documento también puede dar como resultado mejoras en las propiedades de los gránulos de las composiciones tales como tamaño de gránulo mejorado y uniformidad del tamaño de gránulo y/o de la masa de gránulos. Las
composiciones y/o formulaciones son estables a lo largo del tiempo y en diferentes condiciones, y pueden, en ciertas realizaciones, exhibir una estabilidad mejorada.
Descripción detallada
La presente divulgación se refiere a composiciones estables y formulaciones de las mismas que comprenden el API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina que es un derivado de nitrocatecol de fórmula I como se define aquí y sus sales, ésteres, hidratos y solvatos, al menos un agente de carga y al menos un aglutinante. El al menos un agente de carga no es un derivado de fosfato y el al menos un aglutinante no es un compuesto derivado de PVP. El API está presente en forma granular y la composición tiene una densidad aparente superior a 0.5 g/ml.
Como se usa en el presente documento, el término “gránulos”, “forma granular”, “gránulos de API” y variaciones de los mismos, se refieren a las partículas producidas por granulación húmeda o seca del API, que es un derivado de nitrocatecol 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina de fórmula I como se define aquí y sales, ésteres, hidratos y solvatos del mismo. En diversas realizaciones de la presente divulgación, el API que es un derivado de nitrocatecol de fórmula I y es 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina comprende, además 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol. Los granulados pueden comprender además al menos un agente de carga y/o al menos un aglutinante.
Como se usa en el presente documento, el término “composición” y variaciones del mismo pretende significar un compuesto que comprende el API elegido de un derivado de nitrocatecol de fórmula I como se define en el presente documento y sales, ésteres, hidratos y solvatos del mismo, al menos un agente de carga, y al menos un aglutinante. En determinadas realizaciones, la composición puede comprender dos o más derivados de nitrocatecol de fórmula I, por ejemplo, la composición puede comprender el API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina y 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol. La composición comprende gránulos del API, y al menos un agente de carga y al menos un aglutinante pueden ser independientemente intragranulares (es decir, granulados con el API y/o contenidos dentro de los mismos gránulos que el API), extragranulares (es decir, gránulos presentes fuera del API), o parte intragranular y parte extragranular. Por ejemplo, el agente de carga puede ser del 10 % al 90 % en peso, del 20 % al 80 % en peso, del 30 % al 70 % en peso, del 40 % al 60 % en peso o aproximadamente el 50 % en peso intragranular, con la porción restante siendo extragranular. El aglutinante puede ser del 10 % al 90 % en peso, del 20 % al 80 % en peso, del 30 % al 70 % en peso, del 40 % al 60 % en peso o aproximadamente el 50 % en peso intragranular, siendo la porción restante extragranular. La composición puede comprender además al menos un excipiente, que puede ser intragranular, extragranular o parcialmente intragranular y parcialmente extragranular. La composición es preferiblemente adecuada para llenar una cápsula, hacer un comprimido y/o para administrar directamente a los pacientes, por ejemplo, envasada en bolsitas.
Como se usa en el presente documento, los términos “formulación”, “formulación farmacéutica” y variaciones de los mismos pretenden incluir composiciones descritas en el presente documento que se procesan o formulan adicionalmente en una forma de dosificación. Solo a modo de ejemplo, en diversas realizaciones ejemplares, las formulaciones pueden comprender una composición descrita en el presente documento, normalmente en forma de gránulos, en una forma de dosificación adecuada para administrar a un sujeto, como una cápsula o una forma de dosificación comprimida, como un comprimido. En una realización ejemplar adicional, las formulaciones pueden comprender una composición descrita en el presente documento, normalmente en forma de gránulos, mezclada con al menos un excipiente en una forma de dosificación adecuada para administrar a un sujeto, como una cápsula o una forma de dosificación comprimida como un comprimido.
Como se usa en este documento, los derivados de nitrocatecol de fórmula I se definen como sigue:

en el que:
Ri y R2 se seleccionan independientemente de hidrógeno o un grupo que es hidrolizable en condiciones fisiológicas, alcanoílo inferior o aroílo opcionalmente sustituido;
X es un grupo metileno;
Y es un átomo de oxígeno, nitrógeno o azufre,
n se selecciona de 0, 1, 2 y 3;
m es 0 o 1;
R3 es un grupo piridina elegido de las fórmulas A, B, C, D, E y F que está conectado como se indica mediante el enlace sin marcar:

en el que:
R4, R5, Ray R7 se eligen independientemente de hidrógeno, alquilo C1-C6, tioalquilo C1-C6, alcoxi C1-C6, arilo o un grupo tioarilo C6-C12, alcanoilo C1-C6 o grupo aroilo C7-C13, amino, alquilamino C1-C6, dialquilamino C1-C6, cicloalquilamino C3-C12, heterocicloalquilamino C3-C12, alquilsulfonilo C1-C6, arilsulfonilo C6-C12, halógeno, haloalquilo
C1-C6, por ejemplo, trifluorometilo, ciano, nitro o un grupo heteroarilo; o dos o más de los residuos R4, R5, R6 y R7 tomados juntos representan anillos alifáticos o heteroalifáticos o anillos aromáticos o heteroaromáticos; y
P es una unidad central, por ejemplo, una unidad plana, como las seleccionadas entre los regioisómeros de 1,3,4-oxadiazol-2,5-diilo; 1,2,4-oxadiazol-3,5-diilo; 4-metil-4H-1,2,4-triazol-3,5-diilo; 1,3,5-triazin-2,4-diilo; 1,2,4-triazin-3,5-diilo; 2H-tetrazol-2,5-diilo; 1,2,3-tiadiazol-4,5-diilo; 1-alquil-3-(alcoxicarbonil)-1H-pirrol-2,5-diilo en el que alquilo está representado por metilo, etilo, n-propilo y n-butilo y en el que alcoxi está representado por metoxi, etoxi, n-propoxi e isopropoxi; 1-alquil-1H-pirrol-2,5-diilo en el que alquilo está representado por metilo, etilo, n-propilo y n-butilo; tiazol-2,4-diilo; 1-H-pirazol-1,5-diilo; pirimidin-2,4-diilo; oxazol-2,4-diilo; carbonilo; 1H-imidazol-1,5-diilo; isoxazol-3,5-diilo;
furan-2,4-diil; 3-alcoxicarbonilfuran-2,4-diilo en el que alcoxi está representado por metoxi, etoxi, n-propoxi e isopropoxi; benceno-1,3-diilo; y (Z)-1-cianoeteno-1,2-diilo. Los grupos adecuados que son hidrolizables en condiciones fisiológicas son bien conocidos en la técnica e incluyen grupos que forman, con el átomo de O, un enlace éter, éster, ácido carbónico o éster.
Preferiblemente, P se elige entre 1,3,4-oxadiazol-2,5-diilo y 1,2,4-oxadiazol-3,5-diilo.
El derivado de nitrocatecol de fórmula I que es el API es 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina.
El al menos un derivado de nitrocatecol de fórmula I también puede ser una mezcla del API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina y 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
En realizaciones en las que al menos un derivado de nitrocatecol es una mezcla de dos derivados de nitrocatecol, como el API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina y 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol, la proporción de los dos componentes puede ser de aproximadamente 50:50 o cualquier variación de la misma, como aproximadamente 60:40, 70:30, 80:20,
90:10, 95:5, 97:3 o 99:1, o la proporción de uno de los derivados de nitrocatecol puede estar presente en una cantidad de hasta el 5 % inclusive, hasta el 3 % inclusive o hasta el 1 % inclusive de la cantidad del otro nitrocatecol, por ejemplo, 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol puede estar presente en una cantidad de hasta el 5 % inclusive, hasta el 3 % inclusive o hasta el 1 % inclusive de la cantidad del API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina.
El API elegido del derivado de nitrocatecol 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina de fórmula I como se divulga en el presente documento, y sus sales, ésteres, hidratos y solvatos, pueden presentar una baja densidad aparente, lo que dificulta la formulación y fabricación de una forma de dosificación. Por ejemplo, el 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina API nitrocatecol de fórmula I, muestra una densidad aparente de menos de 0.1 g/ml antes de la granulación y/o formulación, y el 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol puede exhibir una densidad aparente de alrededor de 0.2 g/ml antes de la granulación y/o formulación, según se determina mediante el método descrito a continuación en el presente documento.
La formulación de API de baja densidad aparente a menudo puede dar lugar a muchos problemas. Por ejemplo, poca uniformidad de contenido, segregación de partículas, poca o ninguna fluidez, alta variabilidad de peso promedio, recubrimiento y laminación de comprimidos y alta friabilidad de los comprimidos.
En al menos una realización ejemplar, la cantidad (o dosificación) del API presente en las composiciones y/o formulaciones es preferentemente una cantidad terapéuticamente eficaz. Como se usa aquí, “cantidad terapéuticamente eficaz” significa una cantidad de un agente terapéutico suficiente para tratar, aliviar y/o prevenir cualquier afección tratable y/o prevenible mediante la administración de una composición de la divulgación, en cualquier grado. Esa cantidad puede, por ejemplo, ser una cantidad suficiente para exhibir un efecto terapéutico, preventivo o de mejora detectable. El efecto puede incluir, por ejemplo, tratamiento, alivio y/o prevención de las condiciones enumeradas en este documento. La cantidad real requerida, por ejemplo, para el tratamiento de cualquier paciente en particular, dependerá de una variedad de factores que incluyen el trastorno que se está tratando y/o previniendo; su severidad; la composición farmacéutica específica empleada; la edad, el peso corporal, la salud general, el sexo y la dieta del paciente; el modo de administración; el tiempo de administración; la vía de administración; la tasa de excreción del agente terapéutico; la duración del tratamiento; cualquier fármaco utilizado en combinación o coincidente con el agente terapéutico; y otros factores bien conocidos por los expertos en la técnica. En diversas realizaciones, por ejemplo, una formulación, es decir, una forma de dosificación de cápsula o comprimido, puede contener 1 mg o más de API, por ejemplo, 2.5 mg o más, 5 mg o más, 10 mg o más, 20 mg o más, 40 mg o más, 50 mg o más, o 100 mg o más de API. El contenido de API en la formulación puede variar de 0.02 % en peso a 90 % en peso, por ejemplo, de 0.1 % en peso a 70 % en peso, de 0.2 % en peso a 50 % en peso, o de 0.3 % en peso a 45 % en peso.
El al menos un agente de carga de la presente divulgación incluye carbonato de calcio, polvo de celulosa, celulosa microcristalina silicificada, acetato de celulosa, azúcar comprimible, azúcar glas, dextrano, dextrina, dextrosa, fructosa, caolín, lactitol, lactosa, carbonato de magnesio, óxido de magnesio, maltodextrina, maltosa, manitol, celulosa microcristalina, polidextrosa, simeticona, alginato de sodio, cloruro de sodio, sorbitol, almidones, almidón pregelatinizado, sacarosa, trehalosa y xilitol.
El al menos un agente de carga no es un derivado de fosfato. Tal como se usa en el presente documento, el término “derivado de fosfato” y variaciones del mismo se refiere a sustancias que comprenden fosfato de calcio, incluidos, entre otros: fosfato de calcio, dibásico anhidro (por ejemplo, A-TAB ™, Di-Cafos A-N ™, Emcompress ™ Anhydrous and Fujicalin ™); fosfato de calcio, dihidrato dibásico (por ejemplo, Cafos ™, Calipharm ™, Calstar ™, Di Cafos ™, Emcompress™); y fosfato de calcio tribásico (por ejemplo, Tri-Cafos ™, T R I - C a L ™ GT, TRI-TAB ™). En otra realización, El al menos un agente de carga puede elegirse entre almidones, lactosa y celulosa. En al menos una realización, pueden estar presentes al menos dos cargas, por ejemplo, una combinación de almidón, lactosa y/o celulosa.
En diversas realizaciones, por ejemplo, el al menos un agente de carga puede constituir del 0.5 % en peso al 99.5 % en peso de la composición y/o formulación, por ejemplo, del 20 % en peso al 95 % en peso, del 40 % en peso al 95 % en peso, del 40 % en peso a 85 % en peso, 40 % en peso a 70 % en peso, 60 % en peso a 95 % en peso, o 80 % en peso a 95 % en peso del peso total de la composición y/o formulación. El agente de carga puede ser intragranular, extragranular o parcialmente intragranular y parcialmente extragranular. A modo de ejemplo, una composición y/o formulación puede comprender un 85 % en peso de carga. La cantidad de al menos un agente de carga variará dependiendo, en parte, de la dosificación deseada, la densidad aparente y la estabilidad de la composición y/o formulación.
El al menos un aglutinante de la presente divulgación se puede seleccionar de acacia, ácido algínico, carbómero, carboximetilcelulosa sódica, ceratonia, aceite de semilla de algodón, dextrina, dextrosa, gelatina, goma guar, aceite vegetal hidrogenado tipo I, hidroxietilcelulosa, hidroxietilmetilcelulosa, hidroxipropil celulosa, hidroxipropilcelulosa de baja sustitución, hipromelosa, silicato de aluminio y magnesio, maltodextrina, maltosa, metilcelulosa, etilcelulosa,
celulosa microcristalina, polidextrosa, óxido de polietileno, polimetacrilatos, alginato de sodio, almidón, almidón pregelatinizado, ácido esteárico, sacarosa y zeína.
El al menos un aglutinante no es un compuesto derivado de PVP. Como se usa en el presente documento, el término “compuesto derivado de PVP” y variaciones del mismo significa sustancias que comprenden polivinilpirrolidona (PVP) y versiones sustituidas de la misma, que incluyen, pero no se limitan a: povidona (por ejemplo, plasdona y kollidon); copovidona (por ejemplo, plasdona S-630 ™ y kolidon VA-64 ™); y PVP reticulada (por ejemplo, crospovidona). En otra realización, el al menos un aglutinante puede elegirse entre almidones y, en al menos una realización, puede ser almidón 1500™.
En diversas realizaciones, el al menos un aglutinante puede constituir del 0.5 % en peso al 40 % en peso de la composición y/o formulación, por ejemplo, del 1 % en peso al 25 % en peso, del 5 % en peso al 20 % en peso, del 8 % al 15 % en peso, o 10 % en peso a 15 % en peso del peso total de la composición y/o formulación. El aglutinante puede ser intragranular, extragranular o parcialmente intragranular y parcialmente extragranular. Solo a modo de ejemplo, una composición y/o formulación puede comprender entre un 6 % en peso y un 8 % en peso de aglutinante, tal como un 7 % en peso o un 6.3 % en peso de aglutinante. La cantidad de al menos un aglutinante variará dependiendo, en parte, de la dosificación deseada, la densidad aparente y la estabilidad de la composición y/o formulación resultante.
En una realización ejemplar, la composición y/o formulación comprende de 0.2 a 50 % en peso de API, de 5 a 10 % en peso de aglutinante y de 33 a 85 % en peso de agente de carga, como las siguientes composiciones y/o formulaciones:

La invención también se refiere a formulaciones que comprenden una composición de la invención. Dichas formulaciones pueden estar en forma de dosificación como una cápsula o una forma comprimida como un comprimido.
La invención también incluye un método para hacer una composición o formulación de la invención que comprende las etapas de:
- granular el ingrediente farmacéutico activo 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina y sales de los mismos para formar gránulos;
- mezclar al menos un agente de carga con al menos un ingrediente farmacéutico activo antes, durante o después de la granulación;
- mezclar al menos un aglutinante con al menos un ingrediente farmacéutico activo antes, durante o después de la granulación; y
- preparar una formulación farmacéutica en forma de una forma de dosificación.
El agente de carga no es un derivado de fosfato. El aglutinante no es un compuesto derivado de polivinilpirrolidona (“PVP”).
El al menos un API, al menos un agente de carga y al menos un aglutinante pueden combinarse mediante mezclado (también denominado aquí como mezclado). Los expertos en la técnica pueden determinar fácilmente el aparato apropiado y el tiempo y la velocidad de mezclado basándose, por ejemplo, en la cantidad de material presente, el tipo de proceso de mezclado utilizado y otros parámetros conocidos por los expertos en la técnica. Por ejemplo, en diversas realizaciones, los componentes se pueden mezclar manualmente, utilizando un mezclador en V, un mezclador de alto cizallamiento o cualquier otro aparato y/o proceso de mezcla conocido por los expertos en la técnica. Como ejemplo adicional, los componentes se pueden mezclar durante cualquier período de tiempo apropiado, como de 1 a 20 minutos o de 2 a 10 minutos.
En diversas realizaciones ejemplares, la mezcla puede ser granulada en seco o en húmedo. Preferiblemente, los gránulos se granulan en húmedo usando al menos un líquido de granulación. A modo de ejemplo, el al menos un líquido de granulación puede elegirse entre agua, etanol, isopropanol y/o acetona. El líquido de granulación es preferiblemente agua. El aparato apropiado y el tiempo de mezclado y la velocidad para la granulación pueden ser determinados por los expertos en la técnica basándose, por ejemplo, en la cantidad de material y la cantidad de líquido de granulación, si está presente. Por ejemplo, en diversas realizaciones, los componentes se pueden granular manualmente, utilizando un mezclador de alto cizallamiento, un mezclador planetario o cualquier otro aparato y/o proceso granulador conocido por los expertos en la técnica. Como ejemplo adicional, en diversas realizaciones, los componentes pueden granularse durante cualquier período de tiempo apropiado, como de 1 a 60 minutos o de 2 a 30 minutos. La determinación del punto final de granulación está dentro de la capacidad de la persona experta, pero se puede determinar mediante la observación de la estabilización del tamaño de los gránulos y la cohesión de las partículas, lo que da como resultado una disminución del aire atrapado dentro del gránulo, o mediante el logro de un estado estacionario de determinación reológica o correlación de voltaje, torque de conductividad, consumo de energía o técnicas de IR cercano. Las velocidades de granulación pueden variar del 5 % al 100 % de la velocidad de mezclado del granulador, como del 25 % al 100 %.
Una vez que se completa el proceso de granulación en húmedo, los gránulos se pueden secar. Los gránulos se pueden secar hasta valores de pérdida por secado (LOD) por debajo del 6 %, preferentemente por debajo del 5 % e incluso más preferentemente entre el 1 y el 3 %. A continuación, se describe un método adecuado para calcular el LOD. El aparato de secado y el tiempo y la temperatura de secado apropiados pueden ser determinados por los expertos en la técnica basándose, por ejemplo, en la cantidad de material presente, el contenido de humedad del material y el líquido de granulación. Como ejemplos no limitantes, se puede usar un secador de lecho fluido o un secador de bandeja, por ejemplo, a una temperatura de 25 °C o superior, 40 °C o superior, o 70 °C o superior, para secar los gránulos. Por ejemplo, los gránulos se pueden secar a una temperatura de 66 °C.
Los gránulos se pueden tamizar. El tamizado de los gránulos separa los gránulos de un tamaño de partícula particular y puede usarse para seleccionar partículas de un tamaño ventajoso para formular una forma de dosificación o fabricar una forma de dosificación. En diversas realizaciones, los gránulos se pueden tamizar sobre un tamiz o tamiz de 0.5 mm o mayor, por ejemplo, un tamiz de 0.6 mm, 0.8 mm, 1.0 mm y 1.6 mm.
La composición puede incluir además al menos un excipiente adicional que puede mezclarse con al menos un API, al menos un agente de carga y al menos un aglutinante antes, durante o después de la granulación. Por ejemplo, en al menos una realización, el al menos un excipiente adicional puede elegirse entre excipientes tales como desintegrantes, deslizantes y lubricantes.
Los desintegrantes adecuados de la presente divulgación incluyen agar, carbonato de calcio, ácido algínico, fosfato de calcio (tribásico), carboximetilcelulosa de calcio, carboximetilcelulosa de sodio, dióxido de silicio coloidal, croscarmelosa de sodio, crospovidona, docusato de sodio, goma guar, hidroxipropilcelulosa de baja sustitución, silicato de magnesio y aluminio, metilcelulosa, celulosa microcristalina, alginato de sodio, glicolato de almidón de sodio, polacrilina potásica, celulosa microcristalina silicificada, almidón y almidón pregelatinado, y mezclas de los mismos. El desintegrante puede ser una combinación de desintegrantes y/o al menos dos desintegrantes están presentes, por ejemplo, una combinación de glicolato de almidón sódico y carboximetilalmidón sódico, como el que se vende con el nombre comercial Explotab ™.
El desintegrante puede constituir del 0.5 % al 40 % en peso de la composición y/o formulación, por ejemplo, del 1 % al 25 % en peso, del 5 % al 20 % en peso, del 10 % al 15 % en peso o del 5 % en peso al 15 % en peso. A modo de ejemplo, una composición y/o formulación puede comprender entre un 6 % en peso y un 9 % en peso de desintegrante, tal como un 6.8 % en peso de desintegrante. La cantidad de al menos un desintegrante variará dependiendo, en parte, de la dosificación deseada, la densidad aparente y la estabilidad de la composición y/o formulación resultante.
Los deslizantes adecuados de la presente divulgación incluyen silicato de calcio, celulosa, dióxido de silicio coloidal en polvo, silicato de magnesio, trisilicato de magnesio, almidón y talco, y mezclas de los mismos.
El deslizante puede constituir del 0.1 % al 15 % en peso de la composición y/o formulación, por ejemplo, del 0.5 % al 15 % en peso, del 1 % al 10 % en peso o del 2 % al 6 % en peso. La cantidad de deslizante variará dependiendo, en parte, de la dosificación deseada, la densidad aparente y la estabilidad de la composición y/o formulación resultante.
Los lubricantes de la presente divulgación incluyen estearato de calcio, monoestearato de glicerina, behenato de glicerilo, palmitostearato de glicerilo, aceite de ricino hidrogenado, aceite vegetal hidrogenado tipo I, laurilsulfato de magnesio, estearato de magnesio, triglicéridos de cadena media, poloxámero, polietilenglicol, benzoato de sodio, cloruro de sodio, lauril sulfato de sodio, estearil fumarato de sodio, ácido esteárico, talco, estearato de sacarosa y estearato de zinc, y mezclas de los mismos.
Los lubricantes pueden constituir del 0.1 % al 15 % en peso de la composición y/o formulación, por ejemplo, del 0.5 % al 15 % en peso, del 1 % al 10 % en peso, del 1 % al 2 % en peso o del 2 % en peso al 8 % en peso. La cantidad de lubricante variará dependiendo, en parte, de la dosificación deseada, la densidad aparente y la estabilidad de la composición y/o formulación resultante.
El al menos un excipiente se puede agregar antes, durante o después de mezclar el al menos un API y antes (antes de) o durante la granulación y, por lo tanto, puede ser un excipiente intragranular. Alternativamente, el al menos un excipiente se puede agregar a la composición después de la granulación, por ejemplo, mezclándolo con los gránulos y, por lo tanto, puede estar presente como un excipiente extragranular. En diversas realizaciones, se puede agregar al menos un primer excipiente antes o durante la granulación y se puede agregar al menos un segundo excipiente y/o más del al menos un primer excipiente a la composición después de la granulación. Por ejemplo, se pueden agregar disgregantes antes o durante la granulación, mientras que se pueden agregar lubricantes y deslizantes después de la granulación.
La composición que comprende al menos un API, al menos un agente de carga y al menos un aglutinante se puede usar para hacer una formulación, como, por ejemplo, para llenar cápsulas o para formar tabletas.
Las cápsulas para usar en la presente divulgación incluyen, pero no se limitan a, cápsulas de gelatina y cápsulas de hidroxipropilmetilcelulosa (hipromelosa). Los métodos adecuados para llenar dichas cápsulas con una composición de acuerdo con una realización de la divulgación son bien conocidos por los expertos en la técnica.
Los comprimidos de la presente divulgación pueden formarse mediante cualquier método conocido por los expertos en la técnica, como la compresión. En al menos una realización de la presente divulgación, los comprimidos se pueden recubrir, por ejemplo, con recubrimientos de película de base acuosa, recubrimientos de película de base solvente y/o recubrimientos de azúcar.
Las formulaciones de la invención también pueden colorearse, por ejemplo, mediante la inclusión de un colorante en la composición de la invención y/o revistiendo la composición y/o formulación.
En al menos una realización de la presente divulgación, la formulación es una cápsula que comprende al menos un API, al menos un agente de carga y al menos un aglutinante, opcionalmente en forma granular, y puede comprender además al menos un deslizante y/o al menos un desintegrante. En al menos una realización de la presente divulgación, la formulación es un comprimido que comprende al menos un API, al menos un agente de carga y al menos un aglutinante, opcionalmente en forma granular, y puede comprender además al menos un deslizante, al menos un lubricante, y/o al menos un desintegrante.
Las composiciones pueden exhibir una densidad aparente y/o propiedades de fluidez mejoradas en relación con las del API solo. Como se usa en el presente documento, los términos “densidad aparente mejorada”, “densidad aparente significativamente mejorada” y variaciones de los mismos significan que la densidad aparente de la composición es aproximadamente al menos el doble, al menos tres veces, al menos cuatro veces o al menos cinco veces la de la API sola. Está dentro de la capacidad de un experto en la técnica determinar la densidad aparente de un compuesto o composición usando métodos generalmente aceptados en la técnica. Sin embargo, los métodos adecuados incluyen, por ejemplo, la European Pharmacopeia edición 6, Prueba 2.9.15 “volumen aparente”, páginas 285-286, EDQM, 2007, y USP 31, vol. 1, prueba < 616> página 231-232, The United States Pharmacopeia Convention, 2008. A continuación, se describe un método adecuado:
Aparato:
- aparato de decantación capaz de producir en 1 minuto 250 15 compactaciones desde una altura de 3 ± 0.2 mm.
El soporte para la probeta graduada con su soporte, tiene una masa de 450 5 g
- una probeta graduada de 250 ml (intervalos de 2 ml) con una masa de 220 40 g
Método: En un cilindro seco, introducir sin compactar 100.0 g (m g) de la sustancia de ensayo. Asegure la probeta en su soporte. Lea el volumen aparente no asentado (Vo) al mililitro más cercano. Efectuar 10, 500 y 1250
compactaciones y leer los volúmenes V10, V500, V1250, correspondientes al mililitro más cercano. Si la diferencia entre V500 y V1250 es superior a 2 ml, realizar otras 1250 compactaciones.
Alternativamente, si no es posible seleccionar 100.0 g, seleccione una muestra de prueba de cualquier masa, pero con un volumen entre 50 ml y 250 ml, mida su volumen aparente, V0 como se describió anteriormente, pesar la muestra y especificar la masa en la expresión de los resultados. La densidad a granel/aparente puede entonces determinarse en g/ml usando la siguiente fórmula:
m/V0
donde m es la masa en gramos y V0 el volumen aparente no asentado.
La densidad aparente filtrada puede entonces determinarse en g/ml usando la siguiente fórmula:
M N1250
donde m es la masa en gramos y V1250 el volumen aparente después de 1250 “hubs”.
Por ejemplo, como se establece anteriormente, el API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina, un nitrocatecol de fórmula I, exhibe una densidad aparente de menos de 0.1 g/ml antes de la granulación. Composiciones de acuerdo con la presente divulgación que comprenden el API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina exhibe densidades aparentes de 0.5 g/ml o mayores. Composiciones de la presente divulgación para usar como mezclas finales para el llenado de cápsulas o la formación de comprimidos que comprenden el API 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina también puede exhibir densidades aparentes de 0.5 g/ml o mayores, y 0.6 /ml o mayores.
En ciertas realizaciones de la divulgación, las formulaciones comprimidas de la divulgación, como comprimidos, presentan una densidad aparente de 0.5 g/ml a 1.5 g/ml, como 0.6 g/ml a 1.4 g/ml, 0.7 g/ml a 1.3 g /ml, o de 0.8 g/ml a 1.2 g/ml.
La densidad aparente de una formulación comprimida se mide en términos de masa y volumen de la formulación y está dentro de las capacidades del experto en la materia.
También está dentro de la capacidad de un experto en la técnica determinar la fluidez/velocidad de flujo de un compuesto o composición usando métodos generalmente aceptados en la técnica. Sin embargo, los métodos adecuados incluyen, por ejemplo, probar la velocidad de flujo a través de un orificio descrito en USP 31, vol. 1, prueba < 1174>, The United States Pharmacopeia Convention, 2008. La fluidez se puede medir como la masa por tiempo que fluye a través de la abertura de 10 mm de diámetro de un embudo de vidrio. Una velocidad de flujo de valor superior a 10 g/segundo se considera bueno, mientras que un valor inferior a 10 g/segundo se considera deficiente.
El índice de compresibilidad y la relación de Hausner también son métodos adecuados para evaluar el compuesto o las composiciones. Por ejemplo, el índice de compresibilidad y la relación de Hausner pueden evaluarse utilizando USP 31, vol. 1, prueba < 1174>, The United States Pharmacopeia Convention, 2008, y midiendo tanto el volumen aparente (Vo) como el volumen extraído (Vf) de los gránulos. El índice de compresibilidad (CI) se puede calcular entonces usando la siguiente fórmula:
CI (%) = 100 x [(V0-Vf)/Vü]
La relación de Hausner (HR) se puede calcular utilizando la siguiente fórmula:
HR = V0/Vf
Un índice de compresibilidad se considera bueno cuando se calcula un valor inferior al 15 %. Un valor de relación de Hausner (una medida de fluidez) se considera bueno cuando se calcula un valor inferior a 1.25.
Las composiciones y/o formulaciones son estables y/o exhiben una mayor estabilidad sobre otras composiciones y/o formulaciones. Como se usa aquí, los términos “estabilidad”, “estable” y sus variaciones significan que menos del 15 % en peso de al menos un API en la composición y/o formulación se descompone durante 6 meses en condiciones de prueba de 40 °C y 75 % de humedad relativa, o más de 3 años en condiciones de prueba de 25 °C o 30 °C y 60 % de humedad relativa o más de 15 a 30 días en condiciones de prueba de 70 °C y humedad no controlada. En diversas realizaciones, por ejemplo, menos del 10 % en peso, menos del 8 % en peso, menos del 6 % en peso, menos del 5 % en peso, menos del 4 % en peso, menos del 3 % en peso, menos del 2 % en peso o menos del 1 % en peso de al menos un API puede descomponerse en estas condiciones. Está dentro de la capacidad de un experto en la técnica determinar la estabilidad de un compuesto, composición o formulación usando métodos generalmente aceptados en la técnica. Por ejemplo, la cantidad de al menos un API puede medirse mediante cualquier método adecuado, por
ejemplo, HPLC. Por ejemplo, en diversas realizaciones, el ensayo (es decir, la cantidad de API) de una composición o formulación estable puede indicar 85-115 % de API después de las condiciones de prueba, como 95-105 % de API.
La descomposición es un proceso químico compuesto por al menos una reacción, como oxidación, reducción o hidrólisis, que da como resultado un cambio químico en la sustancia en descomposición que da como resultado la generación de uno o más compuestos químicos nuevos. Estos nuevos compuestos (o impurezas) pueden dar como resultado una cantidad reducida y/o variable del API en una composición y/o formulación determinada, reduciendo su eficacia, y pueden tener efectos secundarios no deseados y/o dañinos en los pacientes. Como se usa aquí, el término “ impureza” significa cualquier compuesto nuevo que esté presente en la composición y/o formulación en una cantidad inferior al 10 % en peso del API, por ejemplo, inferior al 5 % en peso, inferior al 3 % en peso, inferior del 1 % en peso, o menos del 0.5 % en peso del API. Por lo tanto, el cambio en las impurezas totales en la composición y/o formulación en las condiciones y períodos de tiempo establecidos en este documento también puede ser indicativo de una composición o formulación estable y puede medirse mediante un método adecuado, por ejemplo, HPLC. En diversas realizaciones, por ejemplo, las impurezas totales relativas al API en una composición y/o formulación estable después de las condiciones de prueba pueden aumentar en menos del 5 % en peso, menos del 2 % en peso, menos del 1 % en peso o menos del 0.5 % en peso,
La estabilidad también se puede probar bajo la influencia de una variedad de otras condiciones de prueba, que incluyen, por ejemplo:
- 40 °C al 75 % de humedad relativa durante 6 meses;
- 25 °C o 30 °C al 60 % de humedad relativa después de 3 -5 años (condiciones a largo plazo); y
- 70 °C con humedad no controlada después de 15-30 días (condiciones de estrés).
La estabilidad también puede estar determinada por la apariencia. Como se usa en este documento, el término “estabilidad visual” y variaciones del mismo significan cambios insustanciales en el color, la integridad de una formulación comprimida (por ejemplo, que no se rompa), la forma y/o el tamaño de los gránulos, la composición y/o formulación.
Como se usa en el presente documento, el término “estabilidad mejorada”, “estabilidad mejorada” y sus variaciones, significa que la cantidad de descomposición de al menos un API en una composición y/o formulación determinada, y/o el aumento de impurezas en una determinada composición y/o formulación es menor que la de una composición y/o formulación comparativa que ha estado sujeta a las condiciones de prueba.
A menos que se indique lo contrario, todos los números utilizados en la memoria descriptiva y las reivindicaciones deben entenderse modificados en todos los casos por el término “aproximadamente”, se indique o no. También debe entenderse que los valores numéricos precisos utilizados en la especificación y las reivindicaciones forman realizaciones adicionales de la divulgación. Se han hecho esfuerzos para asegurar la precisión de los valores numéricos divulgados en los Ejemplos. Sin embargo, cualquier valor numérico medido puede contener inherentemente ciertos errores resultantes de la desviación estándar encontrada en su respectiva técnica de medición.
Tal como se usa aquí, el uso de “el”, “un” o “un” significa “al menos uno” y no debe limitarse a “solo uno” a menos que se indique explícitamente lo contrario. Así, por ejemplo, el uso de “ la formulación” o “una formulación” pretende significar al menos una formulación.
Ejemplos
Ejemplo 1
Se prepararon cuatro cápsulas de dosificación baja en una escala de lotes piloto mezclando primero el API, los almidones y la lactosa en las cantidades establecidas en la Tabla 1 a continuación (lotes A - D). El API utilizado en estos ejemplos fue 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina. A continuación, se añadió agua purificada a cada mezcla y las mezclas se granularon mediante mezclado.
A continuación, los gránulos se secaron usando un secador de lecho fluido hasta que la pérdida por secado del valor del gránulo estuvo por debajo del 6 %. Los gránulos secos se tamizaron y luego se mezclaron con los ingredientes restantes que se indican en la Tabla 1. Las cápsulas de gelatina se llenaron con la formulación utilizando una máquina de llenado de cápsulas InCAP HS.
Los gránulos y las composiciones finales se evaluaron en cuanto a densidad aparente y compactada utilizando los métodos descritos anteriormente. La fluidez/velocidad de flujo también se evaluó probando la velocidad de flujo a través de un orificio descrito en USP 31, vol. 1, prueba < 1174>, Convención de la Farmacopea de los Estados Unidos, 2008. La fluidez se midió como la masa por tiempo que fluye a través de la abertura de 10 mm de diámetro de un
embudo de vidrio. Una velocidad de flujo de valor superior a 10 g/segundo se considera bueno, mientras que un valor inferior a 10 g/segundo se considera deficiente.
El índice de compresibilidad y la relación de Hausner se evaluaron mediante USP 31, vol. 1, prueba < 1174>, The United States Pharmacopeia Convention, 2008, y midiendo tanto el volumen aparente (Vo) como el volumen extraído (Vf) de los gránulos. Luego se calculó el índice de compresibilidad (IC) utilizando la siguiente fórmula:
CI (%) = 100 x [(Vo-Vf)/Vo]
La relación de Hausner (HR) se puede calcular utilizando la siguiente fórmula:
HR = Vci/Vf
Un índice de compresibilidad se considera bueno cuando se calcula un valor inferior al 15 %. Un valor de relación de Hausner (una medida de fluidez) se considera bueno cuando se calcula un valor inferior a 1.25.
La humedad o sequedad se determinó por la pérdida por secado como se describe en USP 31, vol. 1, prueba < 731>, The United States Pharmacopeia Convention, 2008. La prueba consiste en pesar con precisión la sustancia que se va a probar (mo), (por ejemplo, utilizando una cantidad de muestra de 1 a 2 g). La muestra de ensayo se seca luego a 105 °C hasta un peso constante (mf) se consigue. La humedad se puede calcular utilizando la siguiente expresión:

Se evaluó la uniformidad de la masa y las impurezas de las cápsulas. La uniformidad de la masa se evaluó por el peso individual de 20 cápsulas; Luego se calcularon la masa promedio y la desviación estándar. La cantidad de impurezas totales se obtuvo mediante el método HPLC con un límite de cuantificación inferior al 0.05 %.
Los resultados se exponen en la Tabla 2 a continuación. Todos los lotes presentaron buenas propiedades de gránulos y cápsulas.
Ejemplo 2
Se prepararon cuatro cápsulas de dosis alta a escala de laboratorio mezclando primero el API, los almidones y la lactosa en las cantidades indicadas en la Tabla 1 a continuación (lotes EH) en un mezclador en V. El API utilizado en estos ejemplos fue 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina. Se añadió agua purificada a cada mezcla y se mezcló manualmente. A continuación, la masa húmeda así obtenida se granuló en un laboratorio de granuladores por oscilación.
A continuación, los gránulos se secaron en un secador de bandejas hasta que la pérdida por secado del gránulo estuvo por debajo del 6 %. Los gránulos secos se tamizaron. A continuación, los gránulos se mezclaron con los ingredientes restantes expuestos en la Tabla 1 en un mezclador en V. Las cápsulas de gelatina se llenaron con la formulación utilizando una máquina de llenado de cápsulas InCAP HS.
Cada Lote EH se evaluó como se indica en el Ejemplo 1 anterior y los resultados se exponen en la Tabla 3 a continuación. Todos los lotes presentaron buenas propiedades de gránulos y cápsulas.
Tabla 1: Formulaciones por lotes

Tabla 2: Resultados analíticos de los lotes A-D

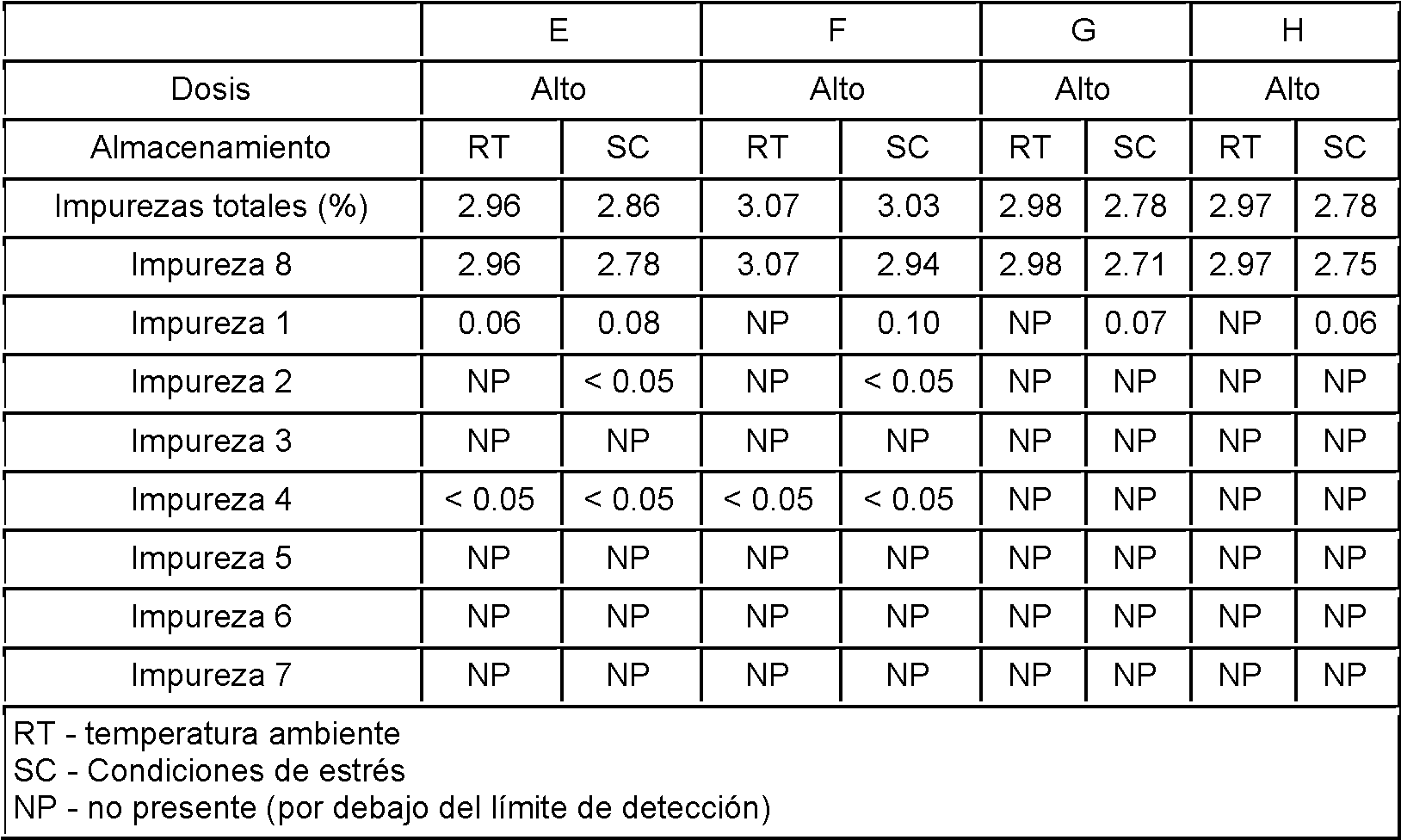
Tabla 3: Resultados analíticos de los lotes E-H

Ejemplo 3
Todos los lotes de los Ejemplos 1 y 2 se sometieron a estudios de tensión para determinar su estabilidad. Cada uno de los ocho lotes se almacenó durante 15 días a temperatura ambiente y en condiciones de estrés (70 °C sin control de humedad relativa). Se analizó el contenido de impurezas de todos los lotes para ambas condiciones de almacenamiento, cuyos resultados se muestran en las Tablas 4 y 5. Los valores de impurezas se obtuvieron mediante el método HPLC con un límite de cuantificación inferior al 0.05 %.
El API utilizado en estos lotes contenía alrededor del 3 % de impurezas antes de la formulación (compuesto por la impureza 8).
Tabla 4: Resultados de las pruebas de estabilidad en los lotes A-D


Tabla 5: Resultados de las pruebas de estabilidad en los lotes E-H
Ejemplo 4
El lote E del Ejemplo 2 se sometió a estudios de estabilidad a largo plazo para determinar su estabilidad. En un estudio, el lote se almacenó durante 6 meses a 25 °C y 60 % de humedad relativa, y en un segundo estudio, el lote se almacenó durante 6 meses a 40 °C y 75 % de humedad relativa. Después de cada prueba, se analizó el lote para determinar el contenido de impurezas y el ensayo, cuyos resultados se muestran en la Tabla 6. Los valores de ensayo y de impurezas se obtuvieron mediante el método HPLC con un límite de cuantificación inferior al 0.05 %.
Tabla 6: Datos de estabilidad para el lote E

Ejemplo comparativo
Se prepararon tres cápsulas de dosificación intermedia mezclando primero el API, los agentes de carga, el aglutinante y el desintegrante (porción más pequeña en el ejemplo comparativo y la cantidad total en los lotes I y J) en las cantidades establecidas en la Tabla 7 a continuación durante 3 minutos en un mezclador de alto cizallamiento. El API utilizado en estos ejemplos fue 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina. Se añadió agua purificada a cada mezcla durante un período de 3 minutos y las mezclas se granularon mezclando durante 3 minutos más.
A continuación, los gránulos se secaron en un secador de lecho fluido hasta que la pérdida por secado del valor del gránulo estuvo por debajo del 6 %. Los gránulos secos se tamizaron y luego se mezclaron con los ingredientes restantes indicados en la Tabla 7 en un mezclador bicónico. Las cápsulas de gelatina se llenaron con la formulación utilizando una máquina de llenado de cápsulas InCAP HS.
Tabla 7: Formulaciones para el Eemplo Comparativo 1

Se evaluaron los gránulos y las cápsulas y los resultados se muestran en la Tabla 8 a continuación. Después de dos estudios de estabilidad, uno a 25 °C y 60 % de HR y el otro a 40 °C y 75 % de HR durante 6 meses cada uno, se observó que los lotes I y J muestran una mayor estabilidad en comparación con la composición comparativa.
Tabla 8: Estabilidad de las fórmulas después de 6 meses a 40 °C 75 % de HR

Claims (18)
1. Una composición estable que comprende:
un ingrediente farmacéutico activo (API) elegido entre 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina y sales, ésteres, hidratos y solvatos del mismo;
al menos un agente de carga; y
al menos un aglutinante;
en el que el ingrediente farmacéutico activo está presente en forma granular;
en el que el al menos un agente de carga no es un compuesto de fosfato;
en el que el al menos un aglutinante no es un compuesto de PVP; y
en donde la composición tiene una densidad aparente superior a 0.5 g/mL.
2. La composición estable de la reivindicación 1, que comprende además 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
3. La composición estable de las reivindicaciones 1 o 2, en la que:
a. menos del 10 % o menos del 3 % del API se descompone:
i. más de 15 días de almacenamiento a 70 °C y humedad no controlada;
ii. más de 6 meses a 40 °C y 75 % de humedad relativa; y/o
iii. más de 3 años al 60 % de humedad relativa y 30 °C o 25 °C; y/o
b. el aumento de las impurezas totales es inferior al 5 % o inferior al 0.5 %:
i. más de 15 días de almacenamiento a 70 °C y humedad no controlada;
ii. más de 6 meses a 40 °C y 75 % de humedad relativa; y/o
iii. más de 3 años al 60 % de humedad relativa y 30 °C o 25 °C.
4. La composición estable de cualquier reivindicación anterior, en la que al menos un agente de carga se elige entre lactosa, almidón de maíz y celulosa microcristalina y en la que al menos un aglutinante se elige entre hipromelosa, hidroxipropilcelulosa, metil o etilcelulosa, almidón de maíz pregelatinizado y gelatina.
5. La composición estable de cualquier reivindicación anterior:
a. en el que los gránulos comprenden además al menos un agente de carga y/o al menos un aglutinante; o b. que comprende además al menos un excipiente adicional elegido entre desintegrantes, deslizantes y lubricantes; o c. en el que la composición presenta una densidad aparente superior a 0.6 g/ml; o
d. en el que la composición presenta una densidad aparente superior a 0.1 g/ml, o superior a 0.5 g/ml.
6. La composición estable de la reivindicación 5, en el que el lubricante se selecciona de monoestearato de glicerina, behenato de glicerilo, palmitostearato de glicerilo, aceite de ricino hidrogenado, aceite vegetal hidrogenado tipo I, triglicéridos de cadena media, estearato de calcio, lauril sulfato de magnesio, estearato de magnesio, lauril sulfato de sodio, estearilfumarato de sodio, ácido esteárico y estearato de zinc, preferiblemente estearato de calcio, laurilsulfato de magnesio, estearato de magnesio, laurilsulfato de sodio, estearilfumarato de sodio, ácido esteárico y estearato de zinc.
7. Una formulación farmacéutica que comprende la composición estable de cualquier reivindicación anterior.
8. La formulación farmacéutica de la reivindicación 7, en la que
a. el ingrediente farmacéutico activo está presente en una cantidad terapéuticamente eficaz; o
b. la composición comprende un API adicional; o
c. la formulación está en la forma de dosificación de un comprimido o cápsula; o
d. la formulación farmacéutica es estable.
9. La formulación farmacéutica de acuerdo con la reivindicación 7 o la reivindicación 8, en la que el API está presente en una cantidad de 1 mg o más, 2,5 mg o más, 5 mg o más, 10 mg o más, 20 mg o más, 40 mg o más, 50 mg o más, o 100 mg o más y en el que al menos un API está presente en forma granular.
10. La formulación farmacéutica de acuerdo con la reivindicación 9, que comprende además carbidopa o benserazida.
11. Un método para fabricar una formulación farmacéutica estable, dicho método comprende:
granular un ingrediente farmacéutico activo (API) elegido entre 1-óxido de 2,5-dicloro-3-(5-(3,4-dihidroxi-5-nitrofenil)-1,2,4-oxadiazol-3-il)-4,6-dimetilpiridina y sus sales para formar gránulos;
mezclar al menos un agente de carga con al menos un ingrediente farmacéutico activo antes, durante o después de la granulación;
mezclar al menos un aglutinante con el al menos un ingrediente farmacéutico activo antes, durante o después de la granulación;
y
preparar una formulación farmacéutica en forma de una forma de dosificación;
en el que el al menos un agente de carga no es un compuesto de fosfato;
en el que el al menos un aglutinante no es un compuesto de PVP; y
en el que la formulación tiene una densidad aparente superior a 0.5 g/mL.
12. El método de la reivindicación 11, en el que la formulación farmacéutica comprende además 5-[3-(2,5-dicloro-4,6-dimetilpiridin-3-il)-[1,2,4]oxadiazol-5-il]-3-nitrobenceno-1,2-diol.
13. El método de las reivindicaciones 11 o 12, en el que:
a. el al menos un agente de carga se elige entre lactosa, almidón de maíz y celulosa microcristalina; o
b. el al menos un aglutinante se elige entre hipromelosa, hidroxipropilcelulosa, metil o etilcelulosa, almidón de maíz pregelatinizado y gelatina.
14. El método de cualquiera de las reivindicaciones 11 a 13, en el que:
a. la granulación se lleva a cabo en un mezclador de alto cizallamiento o en un secador de lecho fluido; o b. el proceso de granulación es granulación en húmedo, que comprende además el secado de los gránulos.
15. El método de cualquiera de las reivindicaciones 11 a 14, que comprende, además:
a. tamizar los gránulos; o
b. agregar al menos un excipiente adicional antes, durante o después de la granulación.
16. El método de cualquiera de las reivindicaciones 11 a 15, en el que la forma de dosificación es un comprimido y la etapa de preparar la formulación comprende la compresión.
17. El método de cualquiera de las reivindicaciones 11 a 16, en el que la forma de dosificación es una cápsula, y la etapa de preparar la formulación comprende llenar una cápsula.
18. La composición estable o formulación farmacéutica de una cualquiera de las reivindicaciones 1 a 10, obtenible por el método de cualquiera de las reivindicaciones 11 a 17.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16579409P | 2009-04-01 | 2009-04-01 | |
| PCT/PT2010/000015 WO2010114405A2 (en) | 2009-04-01 | 2010-03-31 | Pharmaceutical formulations comprising nitrocatechol derivatives and methods of making the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2915698T3 true ES2915698T3 (es) | 2022-06-24 |
Family
ID=42229943
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES10714386T Active ES2915698T3 (es) | 2009-04-01 | 2010-03-31 | Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para preparar las mismas |
Country Status (15)
| Country | Link |
|---|---|
| US (3) | US20100256194A1 (es) |
| EP (1) | EP2413913B1 (es) |
| JP (2) | JP5864410B2 (es) |
| KR (1) | KR101824257B1 (es) |
| CN (2) | CN105816456A (es) |
| AU (1) | AU2010231962B2 (es) |
| BR (1) | BRPI1016132B8 (es) |
| CA (1) | CA2757418C (es) |
| DK (1) | DK2413913T3 (es) |
| ES (1) | ES2915698T3 (es) |
| MX (1) | MX361618B (es) |
| PL (1) | PL2413913T3 (es) |
| PT (1) | PT2413913T (es) |
| RU (1) | RU2550133C2 (es) |
| WO (1) | WO2010114405A2 (es) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUE025466T2 (en) | 2005-07-26 | 2016-02-29 | Bial-Portela & Ca S A | Nitro-pyrocatechin derivatives as comt inhibitors |
| EP1845097A1 (en) | 2006-04-10 | 2007-10-17 | Portela & Ca., S.A. | Oxadiazole derivatives as COMT inhibitors |
| DK2481410T3 (en) | 2007-01-31 | 2016-10-24 | Bial - Portela & Ca S A | Nitrocatecholderivater as COMT inhibitors administered in a specific dosage regimen |
| EP2276758B1 (en) | 2008-03-17 | 2016-01-06 | Bial-Portela & CA, S.A. | Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,4] oxadiazol-5-yl]-3-nit robenzene-1, 2-diol |
| CN102448444B (zh) * | 2009-04-01 | 2016-05-25 | 巴尔-波特拉及康邦亚股份有限公司 | 包括硝基儿茶酚衍生物的药物制剂及其制备方法 |
| US20140045900A1 (en) * | 2011-02-11 | 2014-02-13 | Bial-Portela & Ca, S.A. | Administration regime for nitrocatechols |
| DK2791134T3 (da) | 2011-12-13 | 2019-12-09 | BIAL PORTELA & Cª S A | Kemisk forbindelse, der er anvendelig som mellemprodukt til fremstilling af en catechol-o-methyltransferasehæmmer |
| NO2699580T3 (es) | 2014-01-24 | 2018-02-24 | ||
| JP2018500300A (ja) | 2014-11-28 | 2018-01-11 | ノヴィファーマ,エス.アー. | パーキンソン病を遅延させるための医薬 |
| JP2021506762A (ja) * | 2017-12-18 | 2021-02-22 | ユニケム ラボラトリーズ リミテッド | オピカポンとその中間体の製造方法 |
| JP7336450B2 (ja) * | 2018-03-01 | 2023-08-31 | アストラゼネカ・アクチエボラーグ | (2s)-n-{(1s)-1-シアノ-2-[4-(3-メチル-2-オキソ-2,3-ジヒドロ-1,3-ベンゾオキサゾール-5-イル)フェニル]エチル}-1,4-オキサゼパン-2-カルボキサミドを含む医薬組成物 |
| CA3106269A1 (en) | 2018-07-17 | 2020-01-23 | Insmed Incorporated | Certain (2s)-n-[(1s)-1-cyano-2-phenylethyl]-1,4-oxazepane-2-carboxamides for treating lupus nephritis |
| CA3112994A1 (en) | 2018-10-05 | 2020-04-09 | Neurocrine Biosciences, Inc. | Methods for the administration of comt inhibitors |
| WO2020072886A1 (en) | 2018-10-05 | 2020-04-09 | Neurocrine Biosciences, Inc. | Methods for the administration of comt inhibitors |
| KR20220154182A (ko) | 2020-03-13 | 2022-11-21 | 바이알 - 포르텔라 앤드 씨에이 에스에이 | 미분화 오피카폰 |
| GB202011709D0 (en) | 2020-07-28 | 2020-09-09 | Bial Portela & Ca Sa | Solid dispersion of opicapone |
| GB202016425D0 (en) | 2020-10-16 | 2020-12-02 | Bial Portela & Ca Sa | Treatment regimens for parkinson's disease |
| KR20230118933A (ko) | 2020-12-17 | 2023-08-14 | 바이알 - 포르텔라 앤드 씨에이 에스에이 | 초기 특발성 파킨슨병에 대한 치료 요법 |
| WO2022180649A1 (en) * | 2021-02-26 | 2022-09-01 | Msn Laboratories Private Limited, R&D Center | Novel process for the preparation of 2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine-1-oxide |
| GB202212082D0 (en) | 2022-08-18 | 2022-10-05 | Bial Portela & Ca Sa | Treatment regimens for parkinson's disease |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1532178A (en) * | 1921-07-25 | 1925-04-07 | Louis A Godbold | Lubricator |
| US3647809A (en) * | 1968-04-26 | 1972-03-07 | Chinoin Gyogyszer Es Vegyeszet | Certain pyridyl-1 2 4-oxadiazole derivatives |
| US4085563A (en) * | 1977-01-31 | 1978-04-25 | Campbell Soup Company | Cookie dispensing apparatus |
| US4264573A (en) * | 1979-05-21 | 1981-04-28 | Rowell Laboratories, Inc. | Pharmaceutical formulation for slow release via controlled surface erosion |
| US4368668A (en) * | 1981-05-15 | 1983-01-18 | Veb Kombinat Polygraph "Werner Lamberz" Leipzig | Printing plate mounting arrangement |
| US5236952A (en) * | 1986-03-11 | 1993-08-17 | Hoffmann-La Roche Inc. | Catechol derivatives |
| YU213587A (en) * | 1986-11-28 | 1989-06-30 | Orion Yhtymae Oy | Process for obtaining new pharmacologic active cateholic derivatives |
| US6361794B1 (en) * | 1996-06-12 | 2002-03-26 | Basf Corporation | Method of making ibuprofen and narcotic analgesic composition |
| US6206110B1 (en) * | 1996-09-09 | 2001-03-27 | Smith International, Inc. | Protected lubricant reservoir with pressure control for sealed bearing earth boring drill bit |
| AU762245B2 (en) * | 1998-09-18 | 2003-06-19 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 |
| GB2344819A (en) * | 1998-12-18 | 2000-06-21 | Portela & Ca Sa | 2-Phenyl-1-(3,4-dihydroxy-5-nitrophenyl)-1-ethanones |
| US6660753B2 (en) * | 1999-08-19 | 2003-12-09 | Nps Pharmaceuticals, Inc. | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| DE60140213D1 (de) * | 2000-11-28 | 2009-11-26 | Zymogenetics L L C | Zytokine rezeptor zcytor19 |
| US20040097555A1 (en) * | 2000-12-26 | 2004-05-20 | Shinegori Ohkawa | Concomitant drugs |
| JP4077320B2 (ja) * | 2001-01-29 | 2008-04-16 | 塩野義製薬株式会社 | 5−メチル−1−フェニル−2−(1h)−ピリドンを有効成分として含有する医薬製剤 |
| EP1406632A4 (en) * | 2001-06-08 | 2009-11-04 | Cytovia Inc | 3-ARYL-5-ARYL-1,2,4-OXADIAZOLES AND THEIR ANALOGUES, CASPASE ACTIVATORS AND INDUCERS OF APOPTOSIS, AND USES THEREOF |
| CA2509224A1 (en) * | 2002-12-18 | 2004-07-15 | Cytovia, Inc. | 3,5-disubstituted-[1,2,4]-oxadiazoles and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| WO2005006945A2 (en) * | 2003-07-03 | 2005-01-27 | The Salk Institute For Biological Studies | Methods for treating neural disorders and compounds useful therefor |
| EP1673071A1 (en) * | 2003-09-29 | 2006-06-28 | Novo Nordisk Femcare AG | Improved stability of progestogen formulations |
| US7300406B2 (en) * | 2003-09-30 | 2007-11-27 | Carter Vandette B | Medical examination apparatus |
| GB0325956D0 (en) * | 2003-11-06 | 2003-12-10 | Addex Pharmaceuticals Sa | Novel compounds |
| ATE464303T1 (de) * | 2004-04-28 | 2010-04-15 | Vertex Pharma | Als inhibitoren von rock und anderen proteinkinasen geeignete zusammensetzungen |
| EP1625849A1 (en) * | 2004-08-09 | 2006-02-15 | Liconsa, Liberacion Controlada de Sustancias Activas, S.A. | Pharmaceutical composition comprising drospirenone and ethynylestradiol |
| GB0510143D0 (en) * | 2005-05-18 | 2005-06-22 | Addex Pharmaceuticals Sa | Novel compounds A1 |
| US20080051441A1 (en) * | 2004-12-28 | 2008-02-28 | Astrazeneca Ab | Aryl Sulphonamide Modulators |
| US20060257473A1 (en) * | 2005-05-11 | 2006-11-16 | Porranee Puranajoti | Extended release tablet |
| JP4981794B2 (ja) * | 2005-06-03 | 2012-07-25 | アボット・ラボラトリーズ | シクロブチルアミン誘導体 |
| JP2007024970A (ja) * | 2005-07-12 | 2007-02-01 | Miyakawa:Kk | 液晶表示装置の開口効率を上昇させるための樹脂レンズ製造法及びその製造装置 |
| HUE025466T2 (en) * | 2005-07-26 | 2016-02-29 | Bial-Portela & Ca S A | Nitro-pyrocatechin derivatives as comt inhibitors |
| US20070048384A1 (en) * | 2005-08-26 | 2007-03-01 | Joerg Rosenberg | Pharmaceutical compositions |
| EP1845097A1 (en) * | 2006-04-10 | 2007-10-17 | Portela & Ca., S.A. | Oxadiazole derivatives as COMT inhibitors |
| JP2008162955A (ja) * | 2006-12-28 | 2008-07-17 | Chugai Pharmaceut Co Ltd | バリン含有高密度顆粒剤 |
| DK2481410T3 (en) * | 2007-01-31 | 2016-10-24 | Bial - Portela & Ca S A | Nitrocatecholderivater as COMT inhibitors administered in a specific dosage regimen |
| AR065802A1 (es) * | 2007-03-22 | 2009-07-01 | Schering Corp | Formulaciones de comprimidos que contienen sales de 8- [( 1- ( 3,5- bis- (trifluorometil) fenil) -etoxi ) - metil) -8- fenil -1, 7- diaza- spiro [ 4,5] decan -2- ona y comprimidos elaborados a partir de estas |
| EP2276758B1 (en) * | 2008-03-17 | 2016-01-06 | Bial-Portela & CA, S.A. | Crystal forms of 5- [3- (2, 5-dichloro-4, 6-dimethyl-1-oxy-pyridine-3-yl) [1,2,4] oxadiazol-5-yl]-3-nit robenzene-1, 2-diol |
-
2010
- 2010-03-31 PL PL10714386.9T patent/PL2413913T3/pl unknown
- 2010-03-31 AU AU2010231962A patent/AU2010231962B2/en active Active
- 2010-03-31 KR KR1020117025867A patent/KR101824257B1/ko active IP Right Grant
- 2010-03-31 EP EP10714386.9A patent/EP2413913B1/en active Active
- 2010-03-31 PT PT107143869T patent/PT2413913T/pt unknown
- 2010-03-31 BR BRPI1016132A patent/BRPI1016132B8/pt active IP Right Grant
- 2010-03-31 DK DK10714386.9T patent/DK2413913T3/da active
- 2010-03-31 ES ES10714386T patent/ES2915698T3/es active Active
- 2010-03-31 RU RU2011143619/15A patent/RU2550133C2/ru active IP Right Revival
- 2010-03-31 US US12/750,957 patent/US20100256194A1/en not_active Abandoned
- 2010-03-31 WO PCT/PT2010/000015 patent/WO2010114405A2/en active Application Filing
- 2010-03-31 MX MX2011010311A patent/MX361618B/es active IP Right Grant
- 2010-03-31 JP JP2012503351A patent/JP5864410B2/ja active Active
- 2010-03-31 CA CA2757418A patent/CA2757418C/en active Active
- 2010-03-31 CN CN201610184873.1A patent/CN105816456A/zh active Pending
- 2010-03-31 CN CN201080022455.3A patent/CN102438595B/zh active Active
-
2015
- 2015-08-27 JP JP2015168153A patent/JP6336420B2/ja active Active
-
2018
- 2018-09-05 US US16/122,643 patent/US20190008774A1/en not_active Abandoned
-
2020
- 2020-12-15 US US17/122,013 patent/US20210315824A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| CN105816456A (zh) | 2016-08-03 |
| EP2413913A2 (en) | 2012-02-08 |
| US20190008774A1 (en) | 2019-01-10 |
| BRPI1016132B8 (pt) | 2021-05-25 |
| JP6336420B2 (ja) | 2018-06-06 |
| RU2550133C2 (ru) | 2015-05-10 |
| US20100256194A1 (en) | 2010-10-07 |
| US20210315824A1 (en) | 2021-10-14 |
| BRPI1016132A2 (pt) | 2016-04-19 |
| CN102438595B (zh) | 2016-04-27 |
| CA2757418C (en) | 2021-06-15 |
| RU2011143619A (ru) | 2013-05-10 |
| CA2757418A1 (en) | 2010-10-07 |
| BRPI1016132B1 (pt) | 2020-11-24 |
| PL2413913T3 (pl) | 2022-09-26 |
| CN102438595A (zh) | 2012-05-02 |
| JP2016020369A (ja) | 2016-02-04 |
| MX361618B (es) | 2018-12-13 |
| JP2012522764A (ja) | 2012-09-27 |
| DK2413913T3 (da) | 2022-06-13 |
| WO2010114405A3 (en) | 2011-01-06 |
| AU2010231962A1 (en) | 2011-11-17 |
| KR101824257B1 (ko) | 2018-01-31 |
| KR20120008040A (ko) | 2012-01-25 |
| PT2413913T (pt) | 2022-06-09 |
| WO2010114405A2 (en) | 2010-10-07 |
| MX2011010311A (es) | 2012-03-07 |
| JP5864410B2 (ja) | 2016-02-17 |
| AU2010231962B2 (en) | 2015-05-21 |
| EP2413913B1 (en) | 2022-05-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2915698T3 (es) | Formulaciones farmacéuticas que comprenden derivados de nitrocatecol y métodos para preparar las mismas | |
| US10583130B2 (en) | Pharmaceutical formulations compromising nitrocatechol derivatives and methods of making thereof | |
| ES2957912T3 (es) | Composiciones farmacéuticas que comprenden nilotinib |