ES2735355T3 - Inhibidores macrocíclicos y bicíclicos de virus de hepatitis C - Google Patents
Inhibidores macrocíclicos y bicíclicos de virus de hepatitis C Download PDFInfo
- Publication number
- ES2735355T3 ES2735355T3 ES14723582T ES14723582T ES2735355T3 ES 2735355 T3 ES2735355 T3 ES 2735355T3 ES 14723582 T ES14723582 T ES 14723582T ES 14723582 T ES14723582 T ES 14723582T ES 2735355 T3 ES2735355 T3 ES 2735355T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- preparation
- concentrated
- vacuo
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *C1(CC1)S(N)(=O)=O Chemical compound *C1(CC1)S(N)(=O)=O 0.000 description 8
- OHNNZOOGWXZCPZ-CYDAGYADSA-N C(C1)C2[C@@H]3O[C@@H]3C1C2 Chemical compound C(C1)C2[C@@H]3O[C@@H]3C1C2 OHNNZOOGWXZCPZ-CYDAGYADSA-N 0.000 description 1
- KZGAXTMNECCEQV-UHFFFAOYSA-N C=CC(c1nc(ccc(OC(F)F)c2)c2nc1O)(F)F Chemical compound C=CC(c1nc(ccc(OC(F)F)c2)c2nc1O)(F)F KZGAXTMNECCEQV-UHFFFAOYSA-N 0.000 description 1
- YCGNJFWMEASNEB-UHFFFAOYSA-N C=CC(c1nc(cccc2)c2nc1Cl)(F)F Chemical compound C=CC(c1nc(cccc2)c2nc1Cl)(F)F YCGNJFWMEASNEB-UHFFFAOYSA-N 0.000 description 1
- SHGPPNKYUMVQPJ-HBIQZDMRSA-N C=CCCCC([C@H]1CC2CC1)[C@@H]2O Chemical compound C=CCCCC([C@H]1CC2CC1)[C@@H]2O SHGPPNKYUMVQPJ-HBIQZDMRSA-N 0.000 description 1
- MZMNEDXVUJLQAF-SFYZADRCSA-N CC(C)(C)OC(N(C[C@@H](C1)O)[C@@H]1C(OC)=O)=O Chemical compound CC(C)(C)OC(N(C[C@@H](C1)O)[C@@H]1C(OC)=O)=O MZMNEDXVUJLQAF-SFYZADRCSA-N 0.000 description 1
- YNKDNGKIJFMWKH-CJNGLKHVSA-N CC(C)(C)OC(N(C[C@@H](C1)Oc2c(C(C=C)(F)F)nc(cccc3)c3n2)[C@@H]1C(OC)=O)=O Chemical compound CC(C)(C)OC(N(C[C@@H](C1)Oc2c(C(C=C)(F)F)nc(cccc3)c3n2)[C@@H]1C(OC)=O)=O YNKDNGKIJFMWKH-CJNGLKHVSA-N 0.000 description 1
- CAXRCRDDEDHEIB-SLCMMARKSA-N CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc(cc(cc3)OC)c3nc2CCCCC[C@H](CC2=N[C@H]22)[C@@H]2O2)[C@@H]1C(N[C@@](C1)(C(NS(C3(C)CC3)(=O)=O)=O)C1=C(F)F)=O)=O)NC2=O Chemical compound CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc(cc(cc3)OC)c3nc2CCCCC[C@H](CC2=N[C@H]22)[C@@H]2O2)[C@@H]1C(N[C@@](C1)(C(NS(C3(C)CC3)(=O)=O)=O)C1=C(F)F)=O)=O)NC2=O CAXRCRDDEDHEIB-SLCMMARKSA-N 0.000 description 1
- HXLQQSUFBGPABO-IALVXYMDSA-N CC(C/C(/O[C@H](C1)CN[C@@H]1C(OC)=O)=N\c1c(C)cccc1)(C=C)F Chemical compound CC(C/C(/O[C@H](C1)CN[C@@H]1C(OC)=O)=N\c1c(C)cccc1)(C=C)F HXLQQSUFBGPABO-IALVXYMDSA-N 0.000 description 1
- JWLZDNDLXFFVDL-UHFFFAOYSA-N CCOC(C(C(C=C)(F)F)O)=O Chemical compound CCOC(C(C(C=C)(F)F)O)=O JWLZDNDLXFFVDL-UHFFFAOYSA-N 0.000 description 1
- HJTXXQZTAPAGEW-UHFFFAOYSA-N CCOC(C(C(C=C)F)(O)O)=O Chemical compound CCOC(C(C(C=C)F)(O)O)=O HJTXXQZTAPAGEW-UHFFFAOYSA-N 0.000 description 1
- YMIAJALIVUPLFE-UHFFFAOYSA-N COc(cc1)cc2c1NC(C(C=C)(F)F)C(O)=N2 Chemical compound COc(cc1)cc2c1NC(C(C=C)(F)F)C(O)=N2 YMIAJALIVUPLFE-UHFFFAOYSA-N 0.000 description 1
- QFMJFXFXQAFGBO-UHFFFAOYSA-N COc(cc1[N+]([O-])=O)ccc1N Chemical compound COc(cc1[N+]([O-])=O)ccc1N QFMJFXFXQAFGBO-UHFFFAOYSA-N 0.000 description 1
- VMTGPNORNAXZBK-UHFFFAOYSA-N COc(ccc1n2)cc1nc(C(C=C)(F)F)c2O Chemical compound COc(ccc1n2)cc1nc(C(C=C)(F)F)c2O VMTGPNORNAXZBK-UHFFFAOYSA-N 0.000 description 1
- QKQRSVWXCZPVOD-LCYFTJDESA-N CS/C(/Nc1cccc2ccccc12)=C\[N+]([O-])=O Chemical compound CS/C(/Nc1cccc2ccccc12)=C\[N+]([O-])=O QKQRSVWXCZPVOD-LCYFTJDESA-N 0.000 description 1
- MFHCHQCAGVHOLU-UHFFFAOYSA-N CSc(nc(c1ccccc1cc1)c1n1)c1Cl Chemical compound CSc(nc(c1ccccc1cc1)c1n1)c1Cl MFHCHQCAGVHOLU-UHFFFAOYSA-N 0.000 description 1
- FPAKXBHHKVXYQL-UHFFFAOYSA-N Cc1nc(cc(cc2)OC(F)F)c2nc1C(C=C)(F)F Chemical compound Cc1nc(cc(cc2)OC(F)F)c2nc1C(C=C)(F)F FPAKXBHHKVXYQL-UHFFFAOYSA-N 0.000 description 1
- RWZUYAHDGINOQQ-UHFFFAOYSA-N Cc1nc(cc(cc2)OC)c2nc1C(C=C)(F)F Chemical compound Cc1nc(cc(cc2)OC)c2nc1C(C=C)(F)F RWZUYAHDGINOQQ-UHFFFAOYSA-N 0.000 description 1
- NPHZYAUZXKZURW-UHFFFAOYSA-N Cc1nc2ccc(cccc3)c3c2nc1S(C)(=O)=O Chemical compound Cc1nc2ccc(cccc3)c3c2nc1S(C)(=O)=O NPHZYAUZXKZURW-UHFFFAOYSA-N 0.000 description 1
- GRBDLKBKKHPPQX-UHFFFAOYSA-N Nc(ccc(OC(F)F)c1)c1N Chemical compound Nc(ccc(OC(F)F)c1)c1N GRBDLKBKKHPPQX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/64—Cyclic peptides containing only normal peptide links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D203/00—Heterocyclic compounds containing three-membered rings with one nitrogen atom as the only ring hetero atom
- C07D203/02—Preparation by ring-closure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/22—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains four or more hetero rings
Abstract
Un compuesto seleccionado del grupo que consiste en:**Fórmula** y o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Inhibidores macrocíclicos y bicíclicos de virus de hepatitis C
CAMPO
[0001] Se describen nuevos inhibidores de moléculas pequeñas de la replicación viral, también se describen las composiciones que contienen dichos compuestos y los métodos terapéuticos que comprenden la administración de dichos compuestos.
ANTECEDENTES
[0002] El virus de la hepatitis C (VHC), un miembro de los géneros de hepacivirus dentro de la familia Flaviviridae, es la principal causa de enfermedad hepática crónica en todo el mundo (Boyer, N. et al. J Hepatol. 2000, 32, 98-112). En consecuencia, un enfoque importante de la investigación antiviral actual se dirige hacia el desarrollo de métodos mejorados para el tratamiento de infecciones crónicas por VHC en humanos (Ciesek, S., von Hahn T. y Manns, MP., Clin. Liver Dis., 2011), 15, 597-609; Soriano, V. y otros, J. Antimicrob. Chemother., 2011, 66, 1573-1686; Brody, H., Nature Outlook, 2011,474, S1-S7; Gordon, CP, et al. al., J. Med. Chem. 2005, 48, 1-20; Maradpour, D., et al., Nat. Rev. Micro. 2007, 5, 453-463).
[0003] Las curaciones virológicas de pacientes con infección crónica por VHC son difíciles de lograr debido a la prodigiosa producción diaria de virus en pacientes con infección crónica y la alta mutabilidad espontánea del VHC (Neumann, et al., Science 1998, 282, 103-7). Fukimoto, et al., Hepatology, 1996, 24, 1351-4; Domingo, et al., Gene 1985, 40, 1-8; Martell, et al., J. Virol. 1992, 66, 3225-9). El tratamiento del VHC se complica aún más por el hecho de que el VHC es genéticamente diverso y se expresa como varios genotipos diferentes y numerosos subtipos. Por ejemplo, el VHC se clasifica actualmente en seis genotipos principales (designados 1-6), muchos subtipos (denominados a, b, c, etc.) y aproximadamente 100 cepas diferentes (numeradas 1, 2, 3, etc.). VHC se distribuye en todo el mundo con los genotipos 1, 2 y 3 predominantes en los Estados Unidos, Europa, Australia y Asia Oriental (Japón, Taiwán, Tailandia y China). El genotipo 4 se encuentra principalmente en Medio Oriente, Egipto y África central, mientras que los genotipos 5 y 6 se encuentran principalmente en Sudáfrica y el sudeste asiático respectivamente (Simmonds, P. et al. J Virol. 84: 4597-4610, 2010).
[0004] La combinación de ribavirina, un análogo de nucleósido e interferón alfa (a) (IFN), se utiliza para el tratamiento de múltiples genotipos de infecciones crónicas por VHC en humanos. Sin embargo, la variable respuesta clínica observada en los pacientes y la toxicidad de este régimen han limitado su utilidad. La adición de un inhibidor de la proteasa del VHC (telaprevir o boceprevir) al régimen de ribavirina y IFN mejora las tasas de respuesta virológica post tratamiento (RVS12) de 12 semanas sustancialmente. Sin embargo, el régimen actualmente solo está ensayado para pacientes con genotipo 1 y la toxicidad y otros efectos secundarios permanecen.
[0005] El uso de antivirales de acción de dirección para tratar múltiples genotipos de infección por VHC ha demostrado ser un reto debido a la actividad variable de los antivirales contra los diferentes gentoypes. Los inhibidores de la proteasa del VHC con frecuencia tienen una actividad in vitro comprometida contra los genotipos 2 y 3 del VHC en comparación con el genotipo 1 (Ver, por ejemplo, la Tabla 1 de Summa, V. et al., Antimicrobial Agents and Chemotherapy, 2012, 56, 4161-4167; Gottwein, J. et al, Gastroenterology, 2011, 141, 1067-1079). En consecuencia, la eficacia clínica también ha demostrado ser muy variable en los genotipos del VHC. Por ejemplo, las terapias que son altamente efectivas contra los genotipos 1 y 2 del VHC pueden tener una eficacia clínica limitada o nula contra el genotipo 3, (Moreno, C. et al., Poster 895, 61a reunión de AASLD, Boston, MA, EE.UU., 29 de octubre)-2 de noviembre de 2010; Graham, F., et al, Gastroenterology, 2011, 141, 881-889; Foster, GR et al., EASL 45a Reunión Anual, 14 a 18 abril de 2010, Viena, Austria.) En En algunos casos, los agentes antivirales tienen una buena eficacia clínica contra el genotipo 1, pero son más bajos y más variables contra los genotipos 2 y 3, (Reiser, M. et al., Hepatology, 2005, 41,832-835). Para superar la eficacia reducida en el genotipo 3, pacientes, se pueden requerir dosis sustancialmente más altas de agentes antivirales para lograr reducciones sustanciales de la carga viral (Fraser, IP et al., Abstract N° 48, HEP DART 2011, Koloa, HI, diciembre de 2011).
[0006] Los agentes antivirales que son menos susceptibles a la resistencia viral también son necesarios. Por ejemplo, las mutaciones de resistencia en las posiciones 155 y 168 en la proteasa del VHC a menudo causan una disminución sustancial de la eficacia antiviral de los inhibidores de la proteasa del VHC (Mani, N. Ann Forum Collab VIH Res., 2012, 14, 1-8; Romano, KP et al. al, PNAS, 2010, 107, 20986-20991; Lenz O, Agentes antimicrobianos y quimioterapia, 2010, 54,1878-1887,)
[0007] Los documentos WO2012/040040, WO2007/131966 y WO2010/011566 describen compuestos macrocíclicos que tienen actividad anti-VHC. El documento WO2009/005676 describe compuestos lineales o ramificados que tienen actividad anti-VHC.
[0008] En vista de las limitaciones de la terapia actual contra el VHC, existe la necesidad de desarrollar terapias más eficaces contra el VHC. También sería útil proporcionar terapias que sean efectivas contra múltiples genotipos y
subtipos de VHC.
RESUMEN
[0009] Se describen nuevos compuestos que inhiben la proteasa NS3 del virus de la hepatitis C (VHC). En ciertas realizaciones, los compuestos descritos inhiben múltiples genotipos del virus de la hepatitis C. Estos compuestos son útiles para el tratamiento de la infección por VHC y los síntomas relacionados.
[0010] En una realización, se proporciona un compuesto seleccionado del grupo que consiste en:




o una sal farmacéuticamente aceptable del mismo.
[0011] En otra realización, se proporciona un compuesto seleccionado del grupo que consiste en:

o una sal farmacéuticamente aceptable del mismo.
[0012] En otra realización, se proporciona una composición farmacéutica que comprende un compuesto o una sal farmacéuticamente aceptable del mismo como se definió anteriormente, y un excipiente farmacéuticamente aceptable.
[0013] En otra realización, se proporciona un compuesto o sal farmacéuticamente aceptable del mismo como se define anteriormente, para uso en el tratamiento del VHC en un paciente que lo necesite.
DESCRIPCIÓN DETALLADA
[0014] Aunque la presente invención se puede realizar de varias formas, la siguiente descripción de varias realizaciones se realiza con el entendimiento de que la presente divulgación debe considerarse como un ejemplo de la materia reivindicada, y no pretende limitarla. Las reivindicaciones adjuntas a las realizaciones específicas ilustradas. Los encabezados utilizados a lo largo de esta divulgación se proporcionan solo por conveniencia y no deben interpretarse como limitantes de las Reivindicaciones de ninguna manera. Las realizaciones ilustradas en cualquier partida pueden combinarse con las realizaciones ilustradas en cualquier otra partida.
Abreviaturas
[0015] Las siguientes abreviaturas se usan a lo largo de la especificación, y tienen los siguientes significados:



Definiciones
[0016] A menos que se indique lo contrario, los siguientes términos y frases, como se usan en este documento, pretenden tener los siguientes significados:
"Grupo saliente" (LG) se refiere a un resto de un compuesto que es activo hacia el desplazamiento o sustitución en una reacción química. Los ejemplos en los que se produce el desplazamiento o la sustitución incluyen, entre otros, la sustitución nucleofílica bimolecular (SN2), la sustitución nucleófila unimolecular (SN1), la sustitución aromática nucleófila (SNAr) y los acoplamientos cruzados catalizados por metales de transición. Los ejemplos de grupos salientes incluyen, pero no se limitan a, un átomo de halógeno (por ejemplo, -Cl, -Br, -I) y sulfonatos (por ejemplo, mesilato (-OMs), tosilato (-OT) o triflato (-OTf)). El experto en la materia conocerá los diversos grupos salientes químicos y las estrategias para la activación y apreciará el resto adecuado que actuará como grupos salientes, en función de la reacción química particular, la funcionalidad a la que está unido el grupo y los reactivos químicos utilizados para Afecta la reacción de desplazamiento o sustitución. Como ejemplo no limitativo, en algunas situaciones, un átomo de halógeno (por ejemplo, -Cl, -Br o -I) sirve como grupo saliente en una reacción catalizada por un metal de transición (por ejemplo, un acoplamiento Suzuki catalizado por Pd entre un haluro de arilo y y ácido aril borónico) y otros reactivos, como una base.
Estereoisómeros
[0017] Las definiciones y convenciones estereoquímicas usadas en el presente documento generalmente siguen a SP Parker, DE., Diccionarios de McGraw-Hill de Chemical Terms (1984), McGraw-Hill Book Company, Nueva York; y Eliel, E. y Wilen, S., StereoChemistry of Organic Compounds (1994) John Wiley & Sons, Inc., Nueva York.
[0018] El término "quiral" se refiere a las moléculas que tienen la propiedad de no superponibilidad de la pareja de la imagen especular, mientras que el término "aquiral" se refiere a las moléculas que son superponibles en su pareja de la imagen especular.
[0019] Los "isómeros" son compuestos diferentes que tienen la misma fórmula molecular. Los isómeros incluyen estereoisómeros, enantiómeros y diastereómeros.
[0020] Los "diastereoisómeros" son estereoisómeros que tienen al menos dos átomos asimétricos, pero que no son imágenes especulares entre sí.
[0021] Los "enantiómeros" son un par de estereoisómeros que son imágenes especulares no superponibles entre sí. Una mezcla 1:1 de un par de enantiómeros es una mezcla "racémica". El término "(±)" se utiliza para designar una
mezcla racémica cuando sea apropiado.
[0022] El término "estereoisómeros" se refiere a compuestos que tienen una constitución química idéntica, pero difieren con respecto a la disposición de los átomos o grupos en el espacio. El término "atropisómeros" se refiere a estereoisómeros que tienen impedida la rotación alrededor de un enlace sencillo.
[0023] Los compuestos descritos en el presente documento pueden tener centros quirales, por ejemplo, átomos de carbono quirales. Tales compuestos incluyen, por lo tanto, mezclas racémicas de todos los estereoisómeros, incluidos enantiómeros, diastereómeros y atropisómeros. Además, los compuestos descritos en el presente documento incluyen isómeros ópticos enriquecidos o resueltos en cualquiera o todos los átomos quirales asimétricos. En otras palabras, los centros quirales que se desprenden de las representaciones se proporcionan como isómeros quirales o mezclas racémicas. Las mezclas tanto racémicas como diastereoméricas, así como los isómeros ópticos individuales aislados o sintetizados, sustancialmente libres de sus socios enantioméricos o diastereoméricos, están todos dentro del alcance de la invención. Las mezclas racémicas se pueden separar en sus isómeros individuales, sustancialmente ópticamente puros a través de técnicas bien conocidas como, por ejemplo, la separación de sales diastereoméricas formadas con adjuntos ópticamente activos, por ejemplo, ácidos o bases seguidos por la conversión de nuevo a las sustancias ópticamente activas. El isómero óptico deseado también se puede sintetizar por medio de reacciones estereoespecíficas, comenzando con el estereoisómero apropiado del material de partida deseado.
[0024] Se debe entender que para los compuestos descritos en el presente documento cuando un enlace se dibuja de una manera no estereoquímica (por ejemplo, plana), el átomo al que está unido el enlace incluye todas las posibilidades estereoquímicas. También debe entenderse que cuando un enlace se dibuja de forma estereoquímica (por ejemplo, negrita, cuña en negrita, guión discontinua o guión discontinua), el átomo al que está unido el enlace estereoquímico tiene la estereoquímica que se muestra a menos que se indique lo contrario. Por consiguiente, en una realización, un compuesto descrito en la presente memoria es superior al 50% de un solo enantiómero. En otra realización, un compuesto descrito en este documento es al menos el 80% de un solo enantiómero. En otra realización, un compuesto descrito en el presente documento es al menos el 90% de un solo enantiómero. En otra realización, un compuesto descrito en el presente documento es al menos el 98% de un solo enantiómero. En otra realización, un compuesto descrito aquí es al menos el 99% de un solo enantiómero. En otra realización, un compuesto descrito en el presente documento es mayor que 50% de un solo diastereómero. En otra realización, un compuesto descrito en este documento es al menos el 80% de un solo diastereómero. En otra realización, un compuesto descrito en el presente documento es al menos el 90% de un solo diastereómero. En otra realización, un compuesto descrito en el presente documento es al menos el 98% de un solo diastereómero. En otra realización, un compuesto descrito en el presente documento es al menos el 99% de un solo diastereómero.
Tautómeros
[0025] Los compuestos descritos en el presente documento también pueden existir como isómeros tautoméricos en ciertos casos. Aunque solo se puede representar una estructura de resonancia deslocalizada, todas estas formas se contemplan dentro del alcance de la invención. Por ejemplo, los tautómeros de enoamina pueden existir para los sistemas de purina, pirimidina, imidazol, guanidina, amidina y tetrazol y todas sus formas tautoméricas posibles están dentro del alcance de la invención.
Isótopos
[0026] Un experto en la técnica entenderá que esta invención también incluye cualquier compuesto reivindicado que pueda enriquecerse en cualquiera o en todos los átomos por encima de las relaciones isotópicas naturales con uno o más isótopos, tales como, pero sin limitarse a, deuterio (2H o D). Como ejemplo no limitativo, un grupo -CH3 puede ser reemplazado por -CD3,
[0027] Los valores específicos enumerados a continuación para radicales, sustituyentes y rangos son solo para ilustración; no excluyen otros valores definidos u otros valores dentro de rangos definidos para los radicales y sustituyentes.
Grupos protectores
[0028] En ciertas realizaciones, los grupos protectores incluyen restos de profármacos y grupos protectores químicos. Los grupos de protección pueden estar representados por la abreviatura "PG".
[0029] "Grupo de protección" ("PG") se refiere a un resto de un compuesto que enmascara o altera las propiedades de un grupo funcional o las propiedades del compuesto en su totalidad. Los grupos de protección química y las estrategias para la protección/desprotección son bien conocidos en la técnica. Ver, por ejemplo, Peter GM Wuts y Theodora W. Greene, Protective Groups in Organic Synthesis, 4a edición; John Wiley & Sons, Inc.: Nueva Jersey, 2007, Los grupos protectores se utilizan a menudo para enmascarar la reactividad de ciertos grupos funcionales, para ayudar a la eficiencia de las reacciones químicas deseadas, por ejemplo, al crear y romper enlaces químicos de forma ordenada y planificada.
[0030] La protección de los grupos funcionales de un compuesto altera otras propiedades físicas además de la reactividad del grupo funcional protegido, como la polaridad, la lipofilicidad (hidrofobicidad) y otras propiedades que pueden medirse mediante herramientas analíticas comunes. Los intermedios protegidos químicamente pueden ser biológicamente activos o inactivos.
[0031] En ciertas realizaciones, los grupos protectores se emplean opcionalmente para evitar reacciones secundarias con el grupo protegido durante los procedimientos sintéticos. Selección de los grupos apropiados para proteger, cuándo hacerlo, y la naturaleza del grupo protector químico "PG" depende de la química de la reacción que se va a proteger (por ejemplo, condiciones ácidas, básicas, oxidativas, reductoras u otras) y la dirección prevista de la síntesis. Los PG no necesitan ser, y generalmente no lo son, lo mismo si el compuesto está sustituido con múltiples PG. En general, el PG se utilizará para proteger grupos funcionales tales como grupos carboxilo, hidroxilo, tio o amino y, por lo tanto, para prevenir reacciones secundarias o para facilitar de otro modo la eficacia sintética. El orden de desprotección para obtener grupos desprotegidos libres depende de la dirección deseada de la síntesis y las condiciones de reacción que se encontrarán, y puede ocurrir en cualquier orden según lo determinado por el experto.
Sales e Hidratos
[0032] Los ejemplos de sales farmacéuticamente aceptables de los compuestos descritos en este documento incluyen sales derivadas de una base apropiada, tal como un metal alcalino (por ejemplo, sodio), un metal alcalinotérreo (por ejemplo, magnesio), amonio y NX4+ (en donde X es alquilo C1-C4). Las sales farmacéuticamente aceptables de un átomo de nitrógeno o un grupo amino incluyen, por ejemplo, sales de ácidos carboxílicos orgánicos tales como ácidos acético, benzoico, láctico, fumárico, tartárico, maleico, malónico, málico, isetiónico, lactobiónico y succínico; ácidos sulfónicos orgánicos, tales como ácidos metanosulfónico, etanosulfónico, benzenosulfónico y p-toluenosulfónico; y ácidos inorgánicos, tales como ácidos clorhídrico, bromhídrico, sulfúrico, fosfórico y sulfámico. Las sales farmacéuticamente aceptables de un compuesto de un grupo hidroxi incluyen el anión de dicho compuesto en combinación con un catión adecuado tal como Na+ y NX4+ (en donde cada X se selecciona independientemente de H o un grupo alquilo C1-C4).
[0033] Para uso terapéutico, las sales de los ingredientes activos de los compuestos descritos en el presente documento típicamente serán farmacéuticamente aceptables, es decir, serán sales derivadas de un ácido o base fisiológicamente aceptable. Sin embargo, las sales de ácidos o bases que no son farmacéuticamente aceptables también pueden encontrar uso, por ejemplo, en la preparación o purificación de un compuesto de la invención u otro compuesto descrito en el presente documento. Todas las sales, derivadas o no de un ácido o base fisiológicamente aceptable, están dentro del alcance de la presente invención.
[0034] Las sales metálicas se preparan típicamente haciendo reaccionar el hidróxido metálico con un compuesto descrito en el presente documento. Ejemplos de sales metálicas que se preparan de esta manera son sales que contienen Li+, Na+ y K+. Una sal metálica menos soluble puede precipitarse a partir de la solución de una sal más soluble mediante la adición del compuesto metálico adecuado.
[0035] Además, las sales pueden formarse a partir de la adición de ácidos de ciertos ácidos orgánicos e inorgánicos, por ejemplo, HCl, HBr, H2SO4, H3PO4 o ácidos orgánicos sulfónicos, a centros básicos, como las aminas. Finalmente, debe entenderse que las composiciones en el presente documento comprenden compuestos descritos en este documento en su forma no ionizada, así como bipolar, y combinaciones con cantidades estequiométricas de agua como en hidratos.
Realizaciones
[0036] Una realización proporciona cualquiera de los siguientes compuestos:
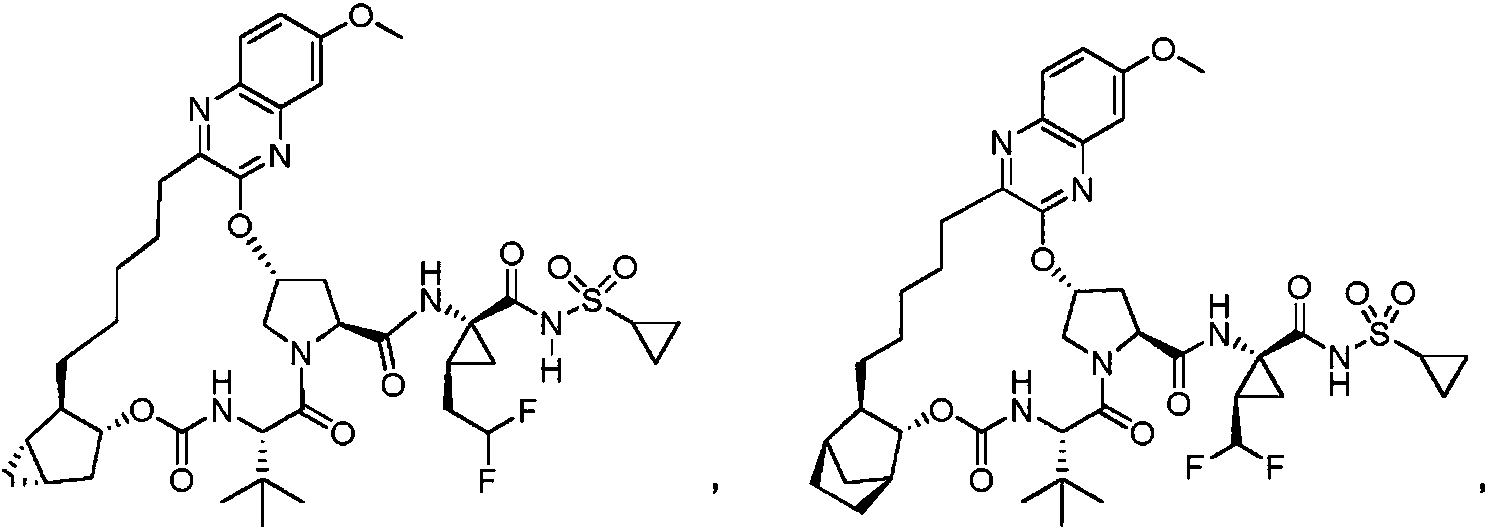



o una sal farmacéuticamente aceptable del mismo.
[0037] Una realización proporciona los siguientes compuestos:

o una sal farmacéuticamente aceptable del mismo.
Métodos de tratamiento
[0038] En este documento se describe un método para tratar una infección viral por Flaviviridae (por ejemplo, una infección viral por VHC) en un paciente que lo necesita (por ejemplo, un mamífero tal como un humano). El método incluye administrar un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, al paciente.
[0039] En este documento se describe un método para inhibir la proliferación del virus del VHC, tratar la infección por el VHC o retrasar la aparición de los síntomas del VHC en un paciente que lo necesita (por ejemplo, un mamífero como un ser humano). El método incluye administrar un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, al paciente.
[0040] Una realización proporciona un compuesto de la invención o una sal farmacéuticamente aceptable del mismo para uso en terapia médica (por ejemplo, para uso en el tratamiento de una infección viral por Flaviviridae (por ejemplo, una infección viral por VHC) o la proliferación del virus del VHC o su retraso. el inicio de los síntomas del VHC en un paciente (por ejemplo, un mamífero como un humano).
[0041] Una realización proporciona un compuesto de la invención o una sal farmacéuticamente aceptable del mismo para uso en la fabricación de un medicamento para tratar una infección viral por Flaviviridae (por ejemplo, una infección viral por VHC) o la proliferación del virus del VHC o retrasar la aparición de los síntomas del VHC en un paciente que lo necesita (por ejemplo, un mamífero como un ser humano).
[0042] Una realización proporciona un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, para uso en el tratamiento profiláctico o terapéutico de la proliferación de un virus Flaviviridae, un virus del VHC o para uso en el tratamiento terapéutico para retrasar la aparición de Los síntomas del VHC.
[0043] Una realización proporciona un compuesto de la invención o una sal farmacéuticamente aceptable del mismo, para uso en el tratamiento profiláctico o terapéutico de una infección por el virus Flaviviridae (por ejemplo, una infección por el virus del VHC).
[0044] Una realización proporciona el uso de un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, para la fabricación de un medicamento para una infección por el virus Flaviviridae (por ejemplo, una infección por el virus del VHC) en un mamífero (por ejemplo, un ser humano).
[0045] En este documento se describe un método para tratar la infección crónica por hepatitis C. El método incluye administrar a un paciente que lo necesite, un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, al paciente.
[0046] En este documento se describe un método para tratar la infección por hepatitis C en pacientes sin tratamiento previo. El método incluye la administración a un paciente sin tratamiento previo, un compuesto de la invención o una sal farmacéuticamente aceptable del mismo.
[0047] En este documento se describe un método para tratar la infección por hepatitis C en pacientes con experiencia en el tratamiento. El método incluye administrar a un paciente experimentado en el tratamiento, un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo.
[0048] En este documento se describe un método para tratar la infección por hepatitis C en un paciente inelegible con interferón o intolerante a interferón. El método incluye administrar, un compuesto de la invención, o una sal
farmacéuticamente aceptable del mismo, al paciente.
[0049] En el presente documento se describen métodos de tratamiento que incluyen la administración del compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, al paciente durante un período fijo de duración. En algunas realizaciones, el período fijo de duración es de 4 semanas, 6 semanas, 8 semanas, 10 semanas o 12 semanas. En otras realizaciones, el período fijo de duración no es más de 12 semanas.
[0050] En algunas realizaciones, el compuesto se administra durante aproximadamente 12 semanas. En realizaciones adicionales, el compuesto se administra durante aproximadamente 12 semanas o menos, durante aproximadamente 10 semanas o menos, durante aproximadamente 8 semanas o menos, durante aproximadamente 6 semanas o menos, o durante aproximadamente 4 semanas o menos.
[0051] En ciertas realizaciones, los métodos de tratamiento descritos en este documento incluyen la administración de un compuesto de la invención, o una de sus sales farmacéuticamente aceptables, para infectarse con el genotipo VHC (GT) 1,2, 3, 4, 5 o 6 (es decir, un método para tratar una infección por VHC GT 1, 2, 3, 4, 5 o 6).
[0052] En este documento se describe un método para tratar una infección por VHC en un paciente que lo necesite (por ejemplo, un mamífero tal como un ser humano), en el que el paciente está infectado con el genotipo 1 del VHC. El método incluye administrar un compuesto de la invención. o una sal farmacéuticamente aceptable del mismo, para el paciente.
[0053] En este documento se describe un método para tratar una infección por VHC en un paciente que lo necesite (por ejemplo, un mamífero tal como un ser humano), en el que el paciente está infectado con el genotipO2 del VHC. El método incluye administrar un compuesto de la invención. o una sal farmacéuticamente aceptable del mismo, para el paciente.
[0054] En este documento se describe un método para tratar una infección por VHC en un paciente que lo necesita (por ejemplo, un mamífero tal como un ser humano), en el que el paciente está infectado con el genotipo 3 del VHC. El método incluye administrar un compuesto de la invención. o una sal farmacéuticamente aceptable del mismo, para el paciente.
[0055] En este documento se describe un método para tratar una infección por VHC en un paciente que lo necesita (por ejemplo, un mamífero como un ser humano), en el que el paciente está infectado con el genotipo 4 del VHC. El método incluye administrar un compuesto de la invención. o una sal farmacéuticamente aceptable del mismo, para el paciente.
[0056] En este documento se describe un método para tratar una infección por VHC en un paciente que lo necesita (por ejemplo, un mamífero tal como un ser humano), en el que el paciente está infectado con el genotipo 5 del VHC. El método incluye administrar un compuesto de la invención. o una sal farmacéuticamente aceptable del mismo, para el paciente.
[0057] En este documento se describe un método para tratar una infección por VHC en un paciente que lo necesite (por ejemplo, un mamífero tal como un ser humano), en el que el paciente está infectado con el genotipo 6 del VHC. El método incluye administrar un compuesto de la invención. o una sal farmacéuticamente aceptable del mismo, para el paciente.
[0058] En los métodos de tratamiento descritos en este documento, el paso de administración incluye la administración de una cantidad terapéuticamente eficaz de un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo, al paciente que necesita tratamiento.
[0059] En este documento se describen métodos para inhibir la actividad del VHC. Tales métodos incluyen el paso de tratar una muestra sospechosa de contener VHC con un compuesto o composición que se describe en el presente documento.
[0060] En una realización, los compuestos descritos en el presente documento actúan como inhibidores del VHC, como intermedios para tales inhibidores o tienen otras utilidades como se describe a continuación.
[0061] En ciertas realizaciones, los compuestos que se enlazan en el hígado pueden unirse con diversos grados de reversibilidad.
[0062] En este documento se describe un método para tratar el VHC que incluye añadir un compuesto descrito en el presente documento a la muestra. El paso de adición comprende cualquier método de administración como se describe anteriormente.
[0063] Si se desea, la actividad del VHC después de la aplicación del compuesto puede observarse mediante cualquier método, incluidos los métodos directos e indirectos para detectar la actividad del VHC. Se contemplan todos los
métodos cuantitativos, cualitativos y semicuantitativos para determinar la actividad del VHC. Típicamente, uno de los métodos de selección descritos anteriormente se aplica, sin embargo, cualquier otro método, como la observación de las propiedades fisiológicas de un organismo vivo, también es aplicable.
[0064] Muchos organismos contienen VHC. Los compuestos de esta invención son útiles en el tratamiento o profilaxis de afecciones asociadas con la activación del VHC en animales o en seres humanos.
Formulaciones farmacéuticas
[0065] "Farmacéuticamente aceptable" significa adecuado para su uso en preparaciones farmacéuticas, generalmente consideradas como seguras para tal uso, oficialmente ensayado por una agencia reguladora de un gobierno nacional o estatal para tal uso, o incluido en la Farmacopea de los EE. Farmacopea reconocida para su uso en animales, y más particularmente en humanos.
[0066] "Portador farmacéuticamente aceptable" se refiere a un diluyente, adyuvante, excipiente o portador, u otro ingrediente que es farmacéuticamente aceptable y con el que se administra un compuesto de la invención.
[0067] Los compuestos de esta invención se formulan con vehículos convencionales (por ejemplo, ingrediente inactivo o material excipiente), que se seleccionarán de acuerdo con la práctica habitual. Las tabletas contendrán excipientes que incluyen deslizantes, rellenos, aglutinantes y similares. Las formulaciones acuosas se preparan en forma estéril y, cuando están destinadas a ser administradas por una administración diferente a la oral, generalmente serán isotónicas. Todas las formulaciones contendrán opcionalmente excipientes como los que se exponen en e1Handbook of Pharmaceutical Excipients (1986). Los excipientes incluyen ácido ascórbico y otros antioxidantes, agentes quelantes tales como ácido etilendiaminotetraacético, carbohidratos tales como dextrina, hidroxialquilcelulosa, hidroxialquilmetilcelulosa, ácido esteárico y similares. Una realización proporciona la formulación como una forma de dosificación sólida que incluye una forma de dosificación oral sólida. El pH de las formulaciones varía de aproximadamente 3 a aproximadamente 11, pero normalmente es de aproximadamente 7 a 10.
[0068] Aunque es posible que los ingredientes activos se administren solos, puede ser preferible presentarlos como formulaciones farmacéuticas (composiciones). Las formulaciones, tanto para uso veterinario como para uso humano, de la invención comprenden al menos un ingrediente activo, como se define anteriormente, junto con uno o más vehículos aceptables para el mismo y opcionalmente otros ingredientes terapéuticos. El (los) portador(es) deben ser "aceptables" en el sentido de ser compatibles con los otros ingredientes de la formulación y fisiológicamente inocuos para el receptor de la misma.
[0069] Las formulaciones incluyen aquellas adecuadas para las rutas de administración anteriores. Las formulaciones pueden presentarse convenientemente en forma de dosis unitaria y pueden prepararse por cualquiera de los métodos bien conocidos en la técnica de la farmacia. Las técnicas y formulaciones generalmente se encuentran en Remington's Pharmaceutical Sciences (Mack Publishing CO, Easton, PA). Tales métodos incluyen el paso de asociar el ingrediente activo con ingredientes inactivos (por ejemplo, un portador, excipiente farmacéutico, etc.) que constituye uno o más ingredientes accesorios. En general, las formulaciones se preparan asociando de manera uniforme e íntima el ingrediente activo con portadores líquidos o portadores sólidos finamente divididos o ambos, y luego, si es necesario, dando forma al producto.
[0070] En ciertas realizaciones, las formulaciones adecuadas para administración oral se presentan como unidades discretas, tales como cápsulas, sellos o tabletas, cada una de las cuales contiene una cantidad predeterminada del ingrediente activo.
[0071] En ciertas realizaciones, las formulaciones farmacéuticas incluyen uno o más compuestos de la invención junto con uno o más vehículos o excipientes farmacéuticamente aceptables y opcionalmente otros agentes terapéuticos. Las formulaciones farmacéuticas que contienen el ingrediente activo pueden estar en cualquier forma adecuada para el método de administración deseado. Cuando se usan para uso oral, por ejemplo, pueden prepararse tabletas, trociscos, pastillas, suspensiones acuosas o de aceite, polvos o gránulos dispersables, emulsiones, cápsulas duras o blandas, jarabes o elixires. Las composiciones destinadas a uso oral pueden prepararse de acuerdo con cualquier método conocido en la técnica para la fabricación de composiciones farmacéuticas y tales composiciones pueden contener uno o más agentes que incluyen agentes edulcorantes, agentes aromatizantes, agentes colorantes y agentes conservantes, con el fin de proporcionar un sabor agradable. preparación. Las tabletas que contienen el ingrediente activo en mezcla con un excipiente farmacéuticamente aceptable no tóxico que son adecuados para la fabricación de tabletas son aceptables. Estos excipientes pueden ser, por ejemplo, diluyentes inertes, tales como calcio o carbonato de sodio, lactosa, lactosa monohidrato, croscarmelosa de sodio, povidona, calcio o fosfato de sodio; agentes de granulación y desintegración, tales como almidón de maíz o ácido algínico; agentes aglutinantes, tales como celulosa, celulosa microcristalina, almidón, gelatina o goma arábiga; y agentes lubricantes, como estearato de magnesio, ácido esteárico o talco. Los comprimidos pueden no estar recubiertos o pueden recubrirse mediante técnicas conocidas, incluida la microencapsulación para retrasar la desintegración y la adsorción en el tracto gastrointestinal y, por lo tanto, proporcionar una acción sostenida durante un período más largo. Por ejemplo, se puede emplear un material de retardo de tiempo tal como monoestearato de glicerilo o diestearato de glicerilo solo o con una cera.
[0072] La cantidad de ingrediente activo que se combina con los ingredientes inactivos para producir una forma de dosificación variará dependiendo del huésped tratado y el modo particular de administración. Por ejemplo, en algunas realizaciones, una forma de dosificación para administración oral a seres humanos contiene aproximadamente 1 a 1000 mg de material activo formulado con una cantidad apropiada y conveniente de material portador (por ejemplo, ingrediente inactivo o material excipiente). En ciertas realizaciones, el material portador varía de aproximadamente 5 a aproximadamente 95% del total de composiciones (peso: peso). En algunas realizaciones, las composiciones farmacéuticas descritas en este documento contienen aproximadamente 1 a 800 mg, 1 a 600 mg, 1 a 400 mg, 1 a 200 mg, 1 a 100 mg o 1 a 50 mg del compuesto de la invención. En algunas realizaciones, las composiciones farmacéuticas descritas en el presente documento no contienen más de aproximadamente 400 mg del compuesto de la invención. En algunas realizaciones, las composiciones farmacéuticas descritas en este documento contienen aproximadamente 100 mg del compuesto de la invención.
[0073] Debe entenderse que además de los ingredientes particularmente mencionados anteriormente, las formulaciones descritas en el presente documento pueden incluir otros agentes convencionales en la técnica teniendo en cuenta el tipo de formulación en cuestión, por ejemplo, los adecuados para la administración oral pueden incluir agentes aromatizantes.
[0074] Se proporcionan además composiciones veterinarias que comprenden al menos un ingrediente activo como se define anteriormente junto con un vehículo veterinario.
[0075] Los portadores veterinarios son materiales útiles para el fin de administrar la composición y pueden ser materiales sólidos, líquidos o gaseosos que, de otro modo, son inertes o aceptables en la técnica veterinaria y son compatibles con el ingrediente activo. Estas composiciones veterinarias se pueden administrar por vía oral, parenteral o por cualquier otra vía deseada.
[0076] La dosis efectiva de ingrediente activo depende al menos de la naturaleza de la condición que se está tratando, de la toxicidad, de si el compuesto se está usando profilácticamente (dosis más bajas), del método de administración y de la formulación farmacéutica y será determinado por el clínico utilizando estudios de aumento de dosis convencionales.
Vías de administracion
[0077] Se administran uno o más compuestos de la invención (denominados en el presente documento ingredientes activos) por cualquier vía apropiada para la afección que se va a tratar. Las rutas adecuadas incluyen oral, rectal, nasal, tópica (incluyendo bucal y sublingual), vaginal y parenteral (incluyendo subcutánea, intramuscular, intravenosa, intradérmica, intratecal y epidural), y similares. Se apreciará que la ruta preferida puede variar, por ejemplo, con la condición del destinatario. Una ventaja de los compuestos de esta invención es que son biodisponibles por vía oral y pueden dosificarse por vía oral. Por consiguiente, en una realización, las composiciones farmacéuticas descritas en el presente documento son formas de dosificación oral. En ciertas realizaciones, las composiciones farmacéuticas descritas en este documento son formas de dosificación sólidas orales.
Terapia de combinación
[0078] En otra realización más, la presente solicitud describe composiciones farmacéuticas que comprenden un compuesto de la invención, o una sal farmacéuticamente aceptable de las mismas, en combinación con al menos un agente terapéutico adicional (es decir, ingrediente activo), y un portador o excipiente farmacéuticamente aceptable. En ciertas realizaciones, los agentes terapéuticos adicionales incluyen agentes antivirales adicionales.
[0079] El agente terapéutico adicional usado en combinación con los compuestos descritos en el presente documento incluye, sin limitación, cualquier agente que tenga un efecto terapéutico cuando se usa en combinación con el compuesto de la presente invención. Dichas combinaciones se seleccionan en función de la condición a tratar, las reactividades cruzadas de los ingredientes y las propiedades farmacológicas de la combinación. Por ejemplo. En ciertas realizaciones, el agente terapéutico usado en combinación con los compuestos de la invención incluye, sin limitación, uno o más de los siguientes: interferones, análogos de ribavirina, inhibidores de la proteasa NS3, inhibidores de NS5a, inhibidores de NS5b, alfa-glucosidasa 1 inhibidores, hepatoprotectores, inhibidores no nucleósidos del VHC, análogos de los nucleósidos y otros fármacos para tratar la infección por el VHC. En algunas realizaciones, los agentes terapéuticos adicionales incluyen, sin limitación, inhibidores de proteasa NS3, inhibidores de NS5a y/o inhibidores de NS5b. En algunas realizaciones, se proporciona una composición farmacéutica que incluye un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo y uno o más de un inhibidor de la proteasa NS3, un inhibidor de NS5a y/o un inhibidor de NS5b. En algunas realizaciones, se proporciona una composición farmacéutica que incluye un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo y uno o más de un inhibidor de NS5a y/o un inhibidor de NS5b. En ciertas realizaciones, se proporcionan composiciones farmacéuticas que incluyen un compuesto de la invención y uno o más agentes antivíricos adicionales, en donde el agente antiviral adicional no es un interferón, ribavirina o un análogo de ribavirina. En realizaciones adicionales, se proporcionan composiciones farmacéuticas que incluyen un compuesto de la invención, y uno o más agentes antivíricos adicionales, en donde el agente antiviral adicional no es ribavirina o un análogo de ribavirina.
[0080] En ciertas realizaciones, los compuestos descritos en el presente documento se combinan con uno o más ingredientes activos (por ejemplo, uno o más agentes antivirales adicionales) en una forma de dosificación unitaria para la administración simultánea o secuencial a un paciente. La terapia de combinación puede administrarse como un régimen simultáneo o secuencial. Cuando se administra de forma secuencial, la combinación se administra en dos o más administraciones. En ciertas realizaciones, los ingredientes activos son: (1) co-formulados y administrados o administrados simultáneamente en una composición farmacéutica combinada; (2 ) entregados por alternancia o en paralelo como composición farmacéutica separada; o (3) por algún otro régimen. Cuando se administran en terapia de alternancia, los ingredientes activos se administran o administran secuencialmente, por ejemplo, en tabletas, píldoras o cápsulas separadas, o mediante diferentes inyecciones en jeringas separadas. En general, durante la terapia de alternancia, una dosis efectiva de cada ingrediente activo se administra secuencialmente, es decir, en serie, mientras que en la terapia de combinación, las dosis efectivas de dos o más ingredientes activos se administran juntas.
[0081] Los inferferones ejemplares incluyen, sin limitación, rIFN-alfa 2b pegilado (PEG-Intron), rIFN-alfa 2a pegilado (Pegasys), rIFN-alfa 2b (Intron A), rIFN-alfa 2a (Roferon-A), interferón alfa (MOR-22, OPC-18, Alfaferona, Alfanativo, Multiferon, subalin), interferon alfacon-1 (Infergen), interferon alfa-n1 (Wellferon), interferon alfa-n3 (Al-feron), interferon-beta (Avonex, DL-8234), interferón-omega (omega DUROS, Biomed 510), albinterferón alfa-2b (Al-buferón), IFN alfa XL, BLX-883 (Locteron), DA-3021, interferón glicosilado alfa-2b (AVI- 005), PEG-Infergen, interferón lambda PEGilado (IL-29 PEGilado), o belerofon, IFN alfa-2b XL, RIFN-alfa 2a, IFN alfa consenso, infergen, rebif, IFN-beta pegilado, interferón oral, alfa, feron, reaferon, intermax alfa, r-IFN-beta e infergen actimmune.
[0082] Los ejemplos de análogos de ribavarina incluyen, sin limitación, ribavirina (Rebetol, Copegus), levovirina VX-497 y taribavirina (Viramidina).
[0083] Los inhibidores de NS5A ejemplares incluyen, sin limitación, ledipasvir (GS-5885), GS-5816, JNJ-47910382, daclatasvir (BMS-790052), ABT-267, MK-8742, EDP-239, IDX-719, PPI-668, GSK-2336805, ACH-3102, A-831, A-689, AZD-2836 (A-831), AZD-7295 (A-689) y BMS-790052,
[0084] Los inhibidores de NS5B ejemplares incluyen, sin limitación, inhibidor de polimerasa es sofosbuvir (GS-7977), tegobuvir (GS-9190), GS-9669, TMC647055, ABT-333, ABT-072, setrobuvir (ANA-598), filibuvir (PF-868554), VX-222, IDX-375, IDX-184, IDX-102, BI-207127, valopicitabina (NM-283), R1626, PSI-6130 (R1656), PSI-7851, BCX-4678, nesbuvir (VHC-796), BILB 1941, MK-0608, NM-107, R7128, VCH- 759, GSK625433, XTL-2125, VCH-916, JTK-652, MK-3281, VBY-708, A848837, GL59728, A-63890, A-48773, A-48547. BC-2329, BMS-791325 y BILB -1941.
[0085] Los inhibidores de proteasa NS3 ejemplares incluyen, sin limitación, GS-9451, GS-9256, simeprevir (TMC-435), ABT-450, boceprevir (SCH-503034), narlaprevir (s Ch -900518), vaniprevir (MK-7009)), MK-5172, danoprevir (ITMN-191), sovaprevir (ACH-1625), neceprevir (ACH-2684), Telaprevir (VX-950), VX-813, VX-500, faldaprevir (BI-201335), asunaprevir (BMS-650032), BMS-605339, VBY-376, PHX-1766, YH5531, BILN-2065, y BILN-2061.
[0086] Los inhibidores de la alfa-glucosidasa 1 ejemplares incluyen, sin limitación, celgosivir (MX-3253), Miglitol y UT-231B.
[0087] Los hepatoprotectores ejemplares incluyen, sin limitación, IDN-6556, ME 3738, MitoQ y LB-84451,
[0088] Los inhibidores no nucleósidos ejemplares del VHC incluyen, sin limitación, derivados de bencimidazol, derivados de benzo-1,2,4-tiadiazina y derivados de fenilalanina.
[0089] Los ejemplos de análogos de nucleósidos incluyen, sin limitación, ribavirina, viramidina, levovirina, un L-nucleósido o isatoribina y dicho interferón es un interferón a o un interferón pegilado.
[0090] Otros fármacos ejemplares para tratar la infección por VHC incluyen, sin limitación, imiquimod, 852A, GS-9524, ANA-773, ANA-975, AZD-8848 (DSP-3025), PF-04878691 y SM-360320, ciclofilina. inhibidores (por ejemplo, DEBIO-025, SCY-635 o NIM811) o inhibidores de IRES del VHC (por ejemplo, MCI-067); emericasan (IDN-6556), ME-3738, GS-9450 (LB-84451), silibilin o MitoQ. BAS-100, SPI-452, PF-4194477, TMC-41629, GS-9350, GS-9585 y roxitromicina.
[0091] Otros fármacos de ejemplo adicionales para tratar la infección por VHC incluyen, sin limitación, zadaxina, nitazoxanida (alinea), BIVN-401 (virostat), DEBIO-025, VGX-410C, EMZ-702, AVI 4065, bavituximab, oglufanida, PYN -17, KPE02003002, actilon (CPG-10101), KRN-7000, civacir, GI-5005, ANA-975 (isatoribina), XTL-6865, ANA 971, NOV-205, tarvacin, EHC-18 y NIM811,
[0092] Otros ejemplos de otros fármacos para tratar la infección por VHC incluyen, sin limitación, timosina alfa 1 (Zadaxin), nitazoxanida (Alinea, NTZ), BIVN-401 (virostat), PYN-17 (altirex), KPE02003002, actilon (CPG-10101), GS-9525, KRN-7000, civacir, GI-5005, XTL-6865, BIT225, PTX-111, ITX2865, TT-033i, ANA 971, NOV-205, tarvacina, EHC-18, VGX -410C, EMZ-702, AVI 4065, BMS-650032, Bavituximab, MDX-1106 (ONO-4538), Oglufanida, FK-788, VX-497 (merimepodib), DEBIO-025, ANA-975 (isatoribine), XTL -6865, o NIM811,
Procedimientos Sintéticos Generales
[0093] Los esquemas, procedimientos y ejemplos proporcionados en el presente documento describen la síntesis de los compuestos descritos en el presente documento, así como los intermedios utilizados para preparar los compuestos. Debe entenderse que los pasos individuales descritos en este documento pueden combinarse. También debe entenderse que los lotes separados de un compuesto pueden combinarse y luego llevarse a cabo en el siguiente paso sintético.
[0094] Los siguientes esquemas describen métodos que son útiles para preparar compuestos descritos en el presente documento.
Esquema 1
Donde:

S1-1 S1-2 S1-3
El esquema 1 muestra una síntesis general del compuesto intermedio de sulfonamida S1-3 que es útil para preparar los compuestos descritos en el presente documento. La ciclopropilsulfonamida S1-1 incluye el grupo protector p G. Un ejemplo no limitante de grupo protector PG es Boc. La ciclopropilsulfonamida S1-1 protegida se desprotona (p.ej., N-butil litio) y se trata con un electrófilo que contiene un grupo saliente apropiado, LG para dar la sulfonamida S1-2 sustituida. Los reactivos útiles para la desprotonación incluyen, sin limitación, n-butil litio. Los electrofilos ejemplares incluyen, sin limitación, haluros de alquilo. La desprotección con ácido (por ejemplo, 4N HCl en dioxano) proporciona el intermedio S1-3.
Esquema 2

El Esquema 2 resume los métodos para preparar los intermedios S2-3, S2-4, S2-6 y S2-7, que son útiles para preparar los compuestos descritos en el presente documento. El material de partida S2-1 incluye el grupo protector PG. Ejemplos no limitantes de grupos protectores PG son Boc y Cbz. R en el material de partida S2-1 es alquilo, que se escinde durante la hidrólisis para producir el ácido carboxílico de S2-3, S2-4, S2-6 y S2-7, Los grupos R apropiados a modo de ejemplo incluyen, sin limitación, -metilo, -etilo y -bencil. Una protección adicional de la amina en S2-1 (por ejemplo, Boc2¿>) seguida de sujeción a la escisión oxidativa (por ejemplo, OsO4) proporciona un aldehído intermedio S2-2, que luego se fluorina (por ejemplo, DAST) seguido de hidrólisis del éster (por ejemplo, LiOH) para Proporcionar difluorometilo intermedio S2-3, El intermedio S2-4 se logra directamente mediante la reducción del resto olefínico de S2-1, seguido de hidrólisis del éster (por ejemplo, H2, Rh/Al2O3, luego LiOH). Alternativamente, S2-1 experimenta una hidroboración y oxidación (por ejemplo, B^^THF, luego NaBO3) para dar alcohol S2-5. La fluoración de S2-5 seguida de hidrólisis (p.ej., DAST, seguido de LiOH) produce la especie S2-6 de monofluoroetilo. El intermedio S2-5 también se oxida a un aldehído (por ejemplo, periodinano Dess-Martin), se fluoriza (por ejemplo, DAST o Deoxofluor) y finalmente se hidroliza (por ejemplo, LiOH) para proporcionar difluoroetilo S2-7.
Esquema 3

El esquema 3 muestra una ruta general al intermedio S3-3 que es útil para preparar ciertos compuestos descritos en el presente documento. El aminoácido protegido S1-3 se prepara como se demuestra en el Esquema 2, donde E es como se define aquí. Como se muestra en el Esquema 2, los ejemplos específicos de S1-3 incluyen, sin limitación, 52- 3, S2-4, S2-6 y S2-7, Por consiguiente, los ejemplos de grupos E para S3-1 incluyen, sin limitación, etilo, 1-fluoroetilo, 1-difluoroetilo y difluoro metilo. La sulfonamida S1-3 se acopla a un aminoácido protegido S3-1 a través de un agente de acoplamiento en presencia de una base apropiada (por ejemplo, CDI con DBU) para producir el péptido 53- 2, El grupo protector de amino se elimina mediante tratamiento con un reactivo apropiado (por ejemplo, 4N HCl en dioxano cuando PG es Boc) para proporcionar el intermedio S3-3.
Esquema 4

compuestos descritos en el presente documento. La metalación del bromuro de alilo S4-1 (p.ej., N-BuLi), seguida de un tratamiento con un oxalato S4-2 (p.ej., Oxalato de dietilo) proporciona el cetoéster S4-3, La metalación alternativa de S4-1 (por ejemplo, indio), seguida de un tratamiento con glioxilato S4-4 proporciona a-hidroxi éster S4-5, La hidrólisis (por ejemplo, LiOH) de S4-5 proporciona un ácido a-hidroxi intermedio S4-6, El tratamiento de S4-5 con condiciones oxidativas (p.ej., TEMPO/lejía), seguido de hidrólisis (p.ej., LiOH), proporciona cetoácido S4-7, que puede aislarse en a-ceto (Xi = X2 = O), hidratado (X1 = X2 = -OH), o forma de hemi-acetal (X1 = -OH, X2 = - OR, donde R es -metilo, -etilo y bencilo, dependiendo de las condiciones de preparación).
Esquema 5

El esquema 5 muestra un método general para la síntesis de dicloroquinoxalina S5-3 que es útil para preparar ciertos compuestos descritos en el presente documento. El tratamiento de la diamina S5-1 con oxilato de dietilo S4-2 proporciona quinoxalina S5-2, La deshidro-halogenación (por ejemplo, POCh) de este intermedio proporciona el intermedio dicloroquinoxalino S5-3.
Esquema 6

El esquema 6 muestra una ruta general al intermedio S6-5 que es útil para preparar ciertos compuestos descritos en el presente documento. El intercambio de acetal entre S6-1 y S6-2 proporciona acetal intermedio mixto S6-3, La condensación de S6-4 con halogenación concomitante (por ejemplo, PoCh) proporciona tio éter S6-4. La oxidación de sulfuro (por ejemplo, m-CPBA) de S6-4 proporciona sulfona S6-5,
Esquema 7

El esquema 7 demuestra una ruta general al intermedio S7-3 que es útil para preparar ciertos compuestos descritos en el presente documento, donde Wn es como se define en el presente documento. En general, el Esquema 7, un acetoácido S4-7, o el hidrato de dicho compuesto, se combina con nitroanilina S7-1 en condiciones de ciclación reductora (por ejemplo, Fe, AcOH) para producir S7-2, La activación (por ejemplo, deshidrohalogenación con POCl3 o TF2O/DIPEA) del alcohol de hidroxiquinoxalina S7-2 a un grupo saliente apropiado (LG) proporciona el intermedio S7-3. Los grupos salientes ejemplares incluyen, sin limitación, -Cl, -F, -Br, -I, -SO2Me y -OTf.
Esquema 8

El esquema 8 demuestra una ruta general alternativa al intermedio S7-3 que es útil para preparar ciertos compuestos descritos aquí, donde Wn es como se define aquí. En general, el esquema 8, el cetoácido S4-7 o el cetoéster S4-3, o el hidrato de dichos compuestos, se calienta con diamina S5-1 (por ejemplo, cuando R = alquilo) o en presencia de un reactivo de acoplamiento (por ejemplo, HATU cuando R = H) y base (por ejemplo, DIPEA) para proporcionar una ruta alternativa al intermedio S7-2, La activación (por ejemplo, deshidro-halogenación con POCl3 o TF2O/DIPEA) del alcohol de hidroxiquinoxalina S7-2 a un grupo saliente apropiado (LG) proporciona el intermedio S7-3. Los ejemplos de grupos salientes de LG incluyen, sin limitación, -Cl, -F, -Br, -I, -SO2Me y -OTf.
Esquema 9
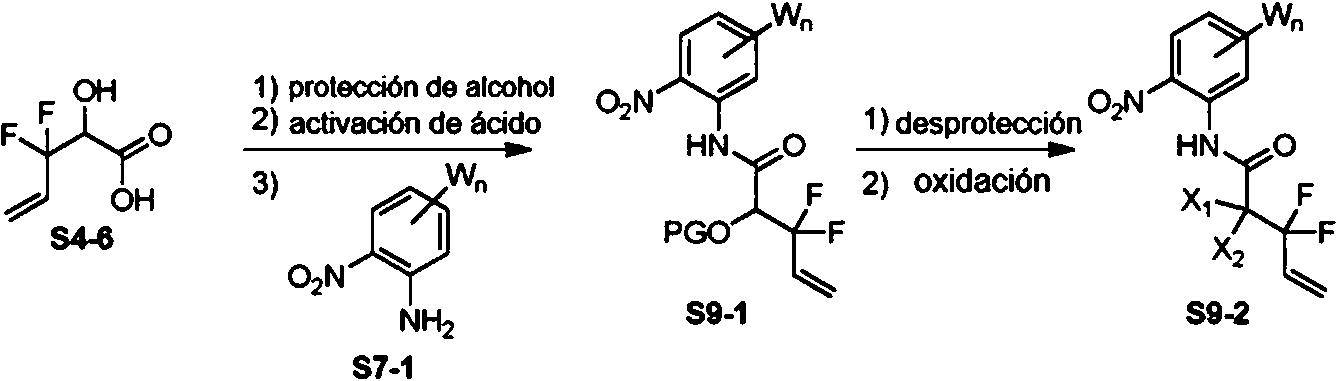

El Esquema 9 demuestra una ruta general alternativa adicional al intermedio S7-3 que es útil para preparar ciertos compuestos descritos en el presente documento, donde Wn es como se define en el presente documento. En general, el Esquema 9, el ácido a-hidroxi S4-6 está protegido como un éter (por ejemplo, TMSCI), seguido de la activación del ácido (por ejemplo, HATU), y el acoplamiento a la anilina S7-1, para llegar al intermedio S9-1, donde PG es un grupo protector apropiado. La desprotección del producto S9-1 (por ejemplo, HCl, MeOH), seguida de oxidación (por ejemplo, TEMPO, NCS) proporciona el compuesto S9-2, que se aísla como una mezcla de hidrato y hemicetal. La reducción subsiguiente (por ejemplo, Fe, AcOH) de la funcionalidad nitro aromática conduce a una ciclocondensación in situ para proporcionar hidroxiquinoxolina S7-2, La activación (por ejemplo, deshidrohalogenación con POCl3 o TF2O/DIPEA) del alcohol de hidroxiquinoxalina S7-2 a un grupo saliente apropiado (LG) proporciona el intermedio S7-3, Ejemplos de grupos de abandono de LG incluyen, sin limitación, -Cl, -F, -Br, -I, -SO2Me y -OTf.
Esquema 10

El Esquema 10 resume dos métodos diferentes para preparar como grupo ©, como se define aquí, con una relación transé,2 de M (cuando M = -O-) y grupos J unidos a átomos adyacentes del grupo © de un material de partida de epóxido común. Como se muestra en el Esquema 10, ©, M, J y LF se definen en otra parte en este documento. Un epóxido de como grupo precursor S10-1, puede abrirse al alcohol S10-2 con un nucleófilo organometálico (por ejemplo, Grignard u reactivo de organocuprate). El epóxido S10-1 también puede activarse (por ejemplo, ácido de Lewis) y abrirse con un fragmento del grupo J-LF (por ejemplo, 1 -hidroxi-y-alquenilo) para proporcionar el intermedio S10-3.
[0095] Lf es un "fragmento enlazador" (es decir, un precursor de L) en el que un enlace carbono-carbono insaturado unido (por ejemplo, alqueno o alquino) en la porción de Lf distal a © facilita, como ejemplo no limitante, una reacción catalizada por metal que da como resultado la conexión de Lf a U para formar un grupo L. Los ejemplos no limitantes de reacciones catalizadas por metal que resultan en tal conexión incluyen la metátesis de cierre del anillo catalizado por Ru o una reacción de acoplamiento cruzado catalizada por Pd (por ejemplo, acoplamientos de Negishi, Heck o Sonagashira).
Esquema 11

En el esquema general 11, se ilustran dos métodos adicionales para preparar un grupo con una relación trans -1,2 de M (cuando M = -O-) y grupos J unidos a átomos adyacentes del grupo ©. Como se muestra en el Esquema 10, © y Lf son como se definen en otra parte en este documento. Comenzando con la cetona común S11-1, se forma un enolato a través del tratamiento con una base apropiada (por ejemplo, LDA o LiHMDS) que después del tratamiento con un electrófilo apropiado (por ejemplo, bromuro de alquilo) produce una cetona funcionalizada S11-2 después del tratamiento. Esta cetona se reduce (por ejemplo, NaBH4) para proporcionar una mezcla racémica del fragmento intermedio S10-2 después de la separación de los diastereómeros cis mediante cromatografía o recristalización. Alternativamente, el enolato generado a partir de la cetona S11-1 queda atrapado (p.ej., LDA, luego TF2O) para formar el triflato de vinilo S11-3, Esto se somete a un acoplamiento cruzado catalizado por paladio (por ejemplo, acoplamiento Suzuki o Heck) para instalar el grupo Lf en el intermedio S11-4, La hidroboración de la olefina seguida de oxidación (por ejemplo, b Hs^ DMS, luego NaOH/H2O2) produce S11-5, La hidrólisis del acetal (por ejemplo, HCl acuoso), seguida de la olefinación (por ejemplo, el reactivo de Wittig o Tebbe) proporciona una mezcla racémica de olefina intermedia S10-2.
Esquema 12
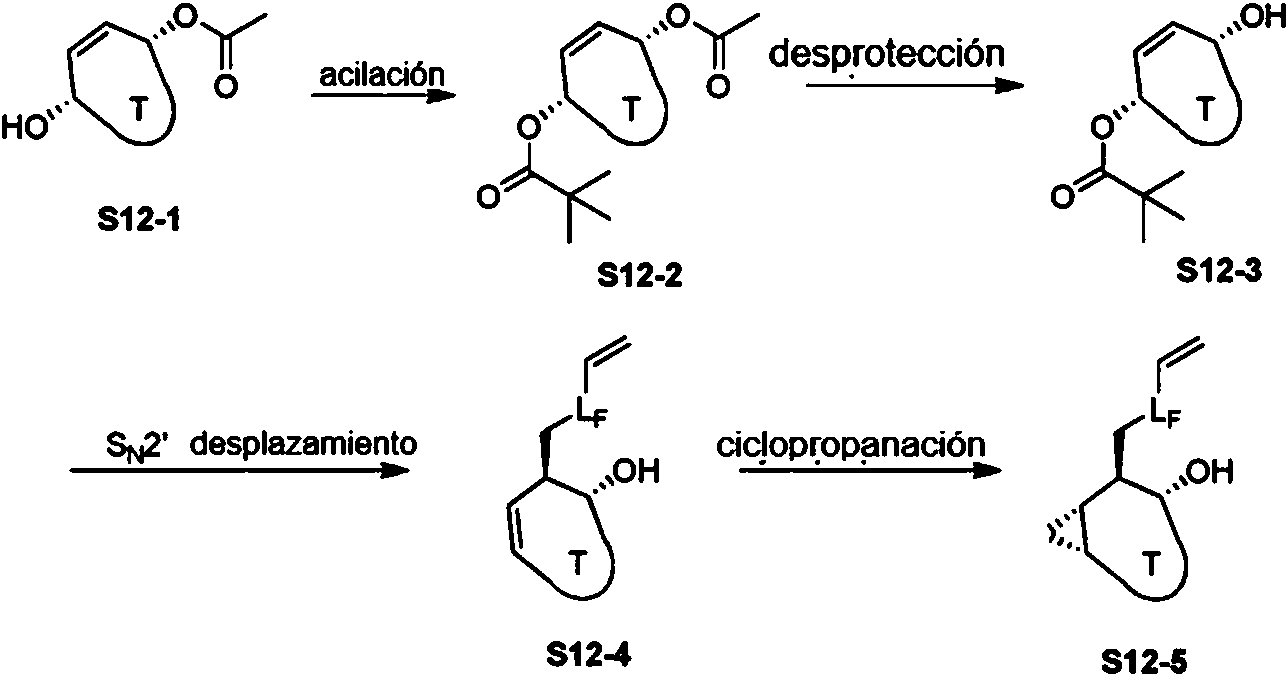
El Esquema 12 demuestra una ruta general al intermedio S12-5 que es útil para preparar ciertos compuestos descritos aquí, donde Lf es como se define aquí. El esquema general 12 representa una ruta estereoselectiva a grupos como S12-5, El alcohol alílico S12-1 puede protegerse (p.ej., Piv-Cl) para producir un diacetato mixto S12-2, El grupo acetilo se puede hidrolizar selectivamente en condiciones suaves (por ejemplo, K2CO3, MeOH) para proporcionar alcohol alílico S12-3, Este intermedio experimenta entonces el desplazamiento de SN2 (por ejemplo, reactivo de organocupación) para producir el alcohol alílico S12-4. La ciclopropanación (por ejemplo, las condiciones de Simmons-Smith) proporciona el intermedio bicíclico fusionado S12-5.
Esquema 13


[0096] El Esquema 13 demuestra una ruta general al intermedio S13-4 que es útil para preparar ciertos compuestos descritos en el presente documento, donde Lf es como se define en el presente documento. En el Esquema 13, el ciclopenta-1,3-dieno se metala (por ejemplo, Na) y luego se trata con un fragmento enlazador que contiene una funcionalidad de oxígeno protegido (PG, por ejemplo, un silil éter o un dialquil acetal) y un grupo saliente (LG, por ejemplo, un grupo saliente halógeno o pseudohalógeno) para proporcionar el intermedio S l3 -1 , La posterior oxidación y robo (por ejemplo, BHâ DMS, NaOH/H2O2) proporciona el alcohol S13-2, que se somete a una cicloadición estereoselectiva (por ejemplo, ciclopropanación de Simmons-Smith) para producir S13-3 fusionada[3,1,0]biciclo. La desprotección de la funcionalidad de oxígeno protegido de Lf (por ejemplo, ácido acuoso para un acetal o TBAF para un silil éter) es seguida por una oxidación al estado de oxidación del aldehído (por ejemplo, periodinano de Dess-Martin) si es necesario. Finalmente, la olefinación (por ejemplo, bromuro de metil trifenilfosfonio, NaHMDS) proporciona el intermedio S13-4.
Esquema 14

Donde is
r = alquilo
LG = imidazol, N-OH succimmida, etc 
El Esquema 14 demuestra una ruta general al intermedio S14-5 que es útil para preparar ciertos compuestos descritos en este documento, donde J, Lf y T son como se definen en este documento. En el Esquema 14, el alqueno S14-1 se trata con un reactivo de acoplamiento (por ejemplo, DSC) y una base (por ejemplo, piridina) para producir el intermedio S14-2 activado, en donde LG es un grupo saliente adecuado. Los ejemplos de grupos salientes LG incluyen, sin limitación, imidazol y succinimida N-OH. El intermedio S14-2 se trata posteriormente con un aminoéster S14-3 en presencia de una base (por ejemplo, K3PO4) para producir el intermedio S14-4. La hidrólisis (por ejemplo, LiOH) del éster proporciona el aminoácido S14-5.
Esquema 15

El Esquema 15 demuestra una ruta general al intermedio S15-4 que es útil para preparar ciertos compuestos descritos en este documento, donde J, Lf, T y Q son como se definen en este documento. En el Esquema 15, el alquino S15-1 se trata con un reactivo de acoplamiento (por ejemplo, DSC) y una base (por ejemplo, piridina) para producir el intermedio S15-2 activado, donde LG es un grupo saliente apropiado. Los grupos salientes ejemplares incluyen, sin limitación, imidazol y succinimida N-OH. El intermedio S15-2 se trata posteriormente con un aminoéster S14-3 en presencia de una base (por ejemplo, K3PO4) para producir el intermedio S15-3. La hidrólisis (por ejemplo, LiOH) del éster proporciona el aminoácido Sl5-4.
Esquema 16

El Esquema 16 demuestra una ruta general al intermedio S16-5 que es útil para preparar ciertos compuestos descritos en el presente documento, donde Lf, T, M y Q son como se definen en el presente documento. El alcohol S16-1 incluye un grupo protector, PG. Los ejemplos de grupos protectores adecuados incluyen, sin limitación, -TBS, -TIPS, -Bn, -PMB y -Ac. El Esquema 16 comienza con el tratamiento del alcohol protegido S16-1 con un reactivo de acoplamiento (por ejemplo, DSC) y una base (por ejemplo, piridina) para producir el intermedio activado S16-2. Este intermedio se acopla luego con un aminoéster S14-3 en presencia de una base (por ejemplo, K3PO4) para producir el intermedio S16-3. La desprotección del alcohol (por ejemplo, TBAF cuando PG es un grupo protector de silicio) seguido de un tratamiento con un bromuro de alquilo o alquenilo apropiado (por ejemplo, bromuro de alilo) produce el alqueno S16-4. La hidrólisis del éster (por ejemplo, LiOH) de S16-4 proporciona el ácido S16-5.
Esquema 17

El Esquema 17 demuestra una ruta general a los intermedios S17-4 y S17-6 que es útil para preparar ciertos compuestos descritos en el presente documento, donde U yW „ son como se definen en el presente documento. En el Esquema 17, las especies de prolina protegidas S17-2 incluyen dos grupos salientes, LG1 y LG2, que pueden ser iguales o diferentes. Los grupos salientes ejemplares LG1 y LG2 incluyen, sin limitación, cloro u OH. S17-2 experimenta una reacción de eterificación a través de condiciones de reacción tales como SNAr (por ejemplo, R2 = H, CS2CO3 tratado con S17-1 donde LG = -CI), desplazamiento SN2 de un brosilato de prolinol (S17-2 donde R2 = Bs) por S17-1 donde LG = -OH, o reacción de Mitsunobu (por ejemplo, DIAD y trifenilfosfina tratamiento de un prolinol apropiado (por ejemplo, S17-2 donde R2 = H) seguido de la adición de S17-1 donde LG = -OH para producir prolina éter Sl7-3, donde PG es un grupo protector adecuado. Eliminación del grupo protector (por ejemplo, TFA cuando PG = Boc) produce S17-4.
Alternativamente, el intermedio S17-3 experimenta un acoplamiento cruzado catalizado por metal (por ejemplo, Suzuki con viniltrifluoroborato de potasio) para proporcionar la especie S17-5 vinilada, en donde la eliminación posterior del grupo protector (por ejemplo, TFA cuando Pg = Boc) produce S17-6.
Esquema 18

on e
R ’ alquik)R: = H o Bs. PG = grupo protector; LG = grupo saliente O -OH;
El Esquema 18 demuestra una ruta general al intermedio S18-3 que es útil para preparar ciertos compuestos descritos en este documento, donde U y Wn son como se definen en este documento. En el Esquema 18, S18-1 incluye un grupo saliente, LG. Los grupos salientes ejemplares de LG incluyen, sin limitación, cloro u OH. La especie de prolina protegida S17-2 (donde PG es un grupo protector adecuado) sufre una reacción de eterificación a través de condiciones de reacción tales como SNAr (por ejemplo, R2 = H, CS2CO3 tratado con S18-1 donde LG = -CI), desplazamiento SN2 de un brosilato de prolinol (S17-2 donde R2 = Bs) por S18-1 donde LG = -OH, o reacción de Mitsunobu (por ejemplo, tratamiento con DIAD y trifenilfosfina de un prolinol apropiado (por ejemplo, S17-2 donde R2 = H) seguido de la adición de S18-1 donde LG = -OH para producir prolina éter S18-2. La eliminación del grupo protector (p.ej., TFA cuando PG = Boc) produce S18-3.
Esquema 19

El Esquema 19 demuestra una ruta general al intermedio S19-4 que es útil para preparar ciertos compuestos descritos en este documento, donde J, Lf, T, U, Wn y Q son como se definen en este documento. En el esquema 19, la prolina S17-4 (LG es un grupo saliente apropiado) y el ácido S16-5 se acoplan en presencia de un agente de acoplamiento (por ejemplo, HATU) y una base (por ejemplo, DIPEA) para proporcionar el intermedio S19-1. El acoplamiento cruzado catalizado por metal (por ejemplo, Suzuki con viniltrifluoroborato de potasio) produce la especie S l9-2 vinilada. La posterior metátesis del cierre del anillo (por ejemplo, Zhan 1B) produce el intermedio del macrociclo S19-3. La reducción del doble enlace macrocíclico (por ejemplo, H2, Pd/C o Rh/A^Oa) produce S19-4.
Esquema 20

El Esquema 20 demuestra otra ruta general al intermedio S19-4 que es útil para preparar ciertos compuestos descritos en el presente documento, donde J, Lf, T, U, Wn y Q son como se definen en el presente documento. El Esquema 20 comienza con el acoplamiento de la prolina S17-6 y el ácido S16-5 en presencia de un agente de acoplamiento (por ejemplo, HATU) y una base (por ejemplo, DIPEA) para proporcionar el intermedio S19-2. La posterior metátesis del cierre del anillo (por ejemplo, Zhan 1B) produce el intermedio del macrociclo S19-3. La reducción del doble enlace macrocíclico (por ejemplo, H2, Pd/C o Rh/AhOa) produce S19-4.
Esquema 21

El Esquema 21 demuestra una ruta general al intermedio S21-3 que es útil para preparar ciertos compuestos descritos aquí, donde J, Lf, T, U, Wn y Q son como se definen aquí. El esquema 21 comienza con el acoplamiento de prolina S18-2 y el ácido S16-5 en presencia de un agente de acoplamiento (por ejemplo, HATU) y una base (por ejemplo, DIPEA) para proporcionar el intermedio S21-1, La posterior metátesis de cierre del anillo (por ejemplo, Zhan 1B) produce el intermedio del macrociclo S21-2, La reducción del doble enlace macro-cíclico (por ejemplo, H2, Pd/C o Rh/Al2O3) produce S21-3.
Esquema 22

El esquema 22 muestra una secuencia alternativa de pasos para ensamblar el intermedio S19-4 que es útil para preparar ciertos compuestos descritos en el presente documento, donde J, LF, T, U, Wn y Q son como se definen en
el presente documento. En el esquema 22, la prolina S17-4 incluye un grupo saliente LG1, La prolina S17-4 y el aminoácido protegido S22-1 se acoplan mediante el tratamiento con un agente de acoplamiento (por ejemplo, HATU) y una base (por ejemplo, DIPEA) para proporcionar el intermedio S22-2, La desprotección de la amina (por ejemplo, HCl cuando PG = Boc) produce el intermedio S22-3 que se trata con el intermedio S14-2 en presencia de una base adecuada (por ejemplo, TEA) para producir el intermedio S22-4, El acoplamiento cruzado catalizado por metal (por ejemplo, Suzuki con viniltrifluoroborato de potasio) proporciona el intermedio de dieno S19-2 que posteriormente se somete a condiciones de metátesis de cierre del anillo (por ejemplo, Zhan 1B) para proporcionar el intermedio S19-3. La reducción del doble enlace macrocíclico (por ejemplo, H2, Pd/C o Rh/A^Oa) produce S19-4.
Esquema 23

El Esquema 23 muestra una síntesis general del intermedio S19-4 que es útil para preparar ciertos compuestos descritos en el presente documento, donde J, Lf, T, U, Wn y Q son como se definen en el presente documento. En el esquema 23, el intermedio S22-2 incluye el grupo protector PG y el grupo saliente LG1, El intermedio S22-2 experimenta un acoplamiento cruzado catalizado por metal (por ejemplo, Sonagashira) con un fragmento de enlazador alquinilo activado S15-2 para dar el intermedio S23-1. La reducción del alquino (por ejemplo, H2, Pd/C o Rh/A^Oa) proporciona el intermedio S23-2. La desprotección de la amina (p.ej., HCl cuando PG = Boc) seguida de macrociclización a través de la formación de carbamato en presencia de una base (p.ej., TEA) da S23-3.
Esquema 24


El esquema 24 muestra una síntesis general de los compuestos intermedios S24-3 y S24-4 que son útiles para preparar ciertos compuestos descritos en el presente documento, donde J, Lf, T, U, Wn y Q son como se definen en el presente documento. En el esquema 24, la desprotección de S19-3 o S19-4 para eliminar Wn (por ejemplo, H2, Pd/C cuando PG = bencil) produce un alcohol heteroarílico intermedio S24-1, La activación del alcohol resultante (por ejemplo, TF2O) a un grupo saliente adecuado (LG) como el triflato produce el intermedio S24-2. El posterior acoplamiento cruzado catalizado por metal (por ejemplo, Suzuki) proporciona el intermedio S24-4. El intermedio S24-1 también se puede alquilar alternativamente (por ejemplo, haluro de alquilo) para dar el intermedio S24-3,
Esquema 25

El Esquema 25 demuestra una ruta general a S25-1, donde J, Lf, T, U, Wn y Q son como se definen en este documento. En el Esquema 25, el intermedio de éster de prolina S19-4 se hidroliza a su correspondiente ácido carboxílico y posteriormente se acopla con el intermedio S3-3 en presencia de un agente de acoplamiento (por ejemplo, HATU) y una base (por ejemplo, DIPEA) para proporcionar compuestos del tipo general S25-1.
[0097] Las siguientes Preparaciones y Ejemplos no limitativos ilustran la preparación de diversas realizaciones descritas en el presente documento.
[0098] Los espectros de resonancia magnética nuclear (RMN) 1H fueron en todos los casos consistentes con las estructuras propuestas. Los desplazamientos químicos característicos (8) se dan en partes por millón de campo bajo desde tetrametilsilano utilizando abreviaturas convencionales para la designación de los picos principales: por ejemplo, s, singlete; d, doblete; t, triplete; q, cuarteto; m, multiplete; br, amplio Las siguientes abreviaturas se han utilizado para solventes comunes utilizados en experimentos de resonancia magnética nuclear: CD3, deuterocloroform; CD3OD, perdeuterometanol; CD3CN, perdeuteroacetonitrilo; cfe-DMSO, perdeuterodimetilsulfóxido. Los espectros de masas se obtuvieron utilizando espectrómetros de masas Thermo Scientific o Agilent Technologies equipados con ionización por electropulverización (ESI). Las masas se informan como relaciones de masa a cargar (m/z). Las mediciones de HPLC analíticas se realizaron en HPLC Agilent Technologies serie 1100 utilizando Phenomenex Kinetex C18, 2,6 um 100 A, 4,6 x 100 mm con un programa de elución de 2% de disolvente B durante 0,55 min, gradiente a 98% de disolvente B durante 8 min. mantenido al 98% del disolvente B durante 0,40 minutos antes de regresar al 2% del
disolvente B durante 0,02 minutos y al 2% del disolvente B durante 2,03 minutos a un caudal de 1,5 ml/min (Disolvente A = MiliQ filtró H2O 0,1% de TFA, Disolvente B = MeCN 0,1% de TFA). El término "cromatografía en capa fina (TLC)" se refiere a cromatografía en gel de sílice utilizando placas de gel de sílice 60 F254, El factor de retención ("Rf") de un compuesto es la distancia recorrida por un compuesto dividida por la distancia recorrida por el frente del solvente en una placa de TLC. Términos tales como "elución temprana" y "elución tardía" se refieren al orden en que un compuesto se eluye o se recupera de un método de cromatografía en fase móvil de fase estacionaria/disolvente líquido sólido (por ejemplo, cromatografía en gel de sílice en fase normal o líquido de alta presión en fase inversa cromatografía (HPLC)).
EJEMPLOS
Preparación de intermedios seleccionados
Grupo intermedio A
Preparación del Intermedio A1.
[0099]
vin ilc iclopropanocarbozilato
[0100] Pasos 1-3. Preparación del Intermedio A1: El Intermedio A1 se preparó siguiendo el procedimiento detallado en el Ejemplo 2,12 de la Publicación de Patente Internacional No. WO 2008/064066 (p. 75-76) sustituyendo (1R, 2S)-metil 1-(terc-butoxicarbonilamino)-2-vinilciclopropano-carboxilato (preparado de acuerdo con Beaulieu, PL, y col., J. Org. Chem. 2005, 70, 5869) para (1R, 2S)-etilo 1-(terc-butoxicarbonilamino)-2-vinilciclopropano-carboxilato.
Preparación del Intermedio A2.
[0101]

[0102] El Intermedio A2 se preparó de manera similar al Intermedio A1, sustituyendo 1-metilciclopropano-1-sulfonamida (preparado de acuerdo con el Ejemplo 1,2 de la Publicación de Patente Internacional No. WO 2008/064066, p. 47) por ciclopropanosulfonamida.
Preparación del Intermedio A3.
[0103]
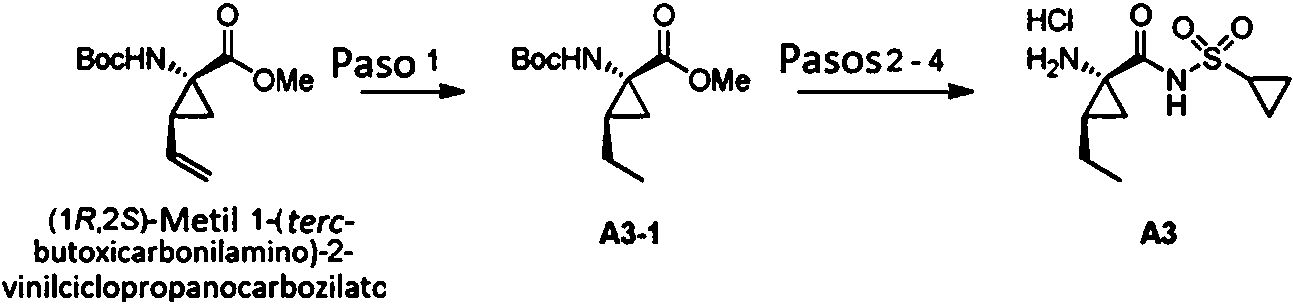
[0104] Paso 1. Preparación de A3-1: se preparó ciclopropano éster A3-1 a partir de (1R, 2S)-metil 1-(terc-butoxicarbonilamino)-2-vinilciclopropanocarboxilato (preparado de acuerdo con Beaulieu, PL, et al.., J. Org. Chem. 2005, 70, 5869) usando el procedimiento detallado en el Ejemplo 26 de la Publicación Internacional de Patente No. WO2009005677 (p. 176).
[0105] Pasos 2-4, Preparación del Intermedio A3: El Intermedio A3 se preparó de manera similar al hidrocloruro de (1R, 2S)-1-amino-N-(cidopropilsulfonil)-2-vinilcidopropanocarbox-amida del Ejemplo 2,12 de la Publicación de Patente Internacional No. WO 2008/064066 (p 75-76) sustituyendo A3-1 por (1R, 2S)-etilo-(terc-butoxicarbonilamino)-2-vinilciclopropano-carboxilato.
Preparación del Intermedio A4.
[0106]

[0107] El Intermedio A4 se preparó de manera similar al Intermedio A3, sustituyendo 1-metilciclopropano-1-sulfonamida (preparado de acuerdo con el Ejemplo 1,2 de la Publicación de Patente Internacional No. WO 2008/064066, p. 47) por ciclopropanosulfonamida.
Preparación del intermedio A5.
[0108]

[0109] Pasos 1-3. Preparación del Intermedio A5: El Intermedio A5 se preparó de manera similar al clorhidrato de (1R, 2S)-1-amino-N-(ciclopropilsulfonil)-2-vinilciclopropano-carboxamida del Ejemplo 2,12 de la Publicación de Patente Internacional No. w O 2008/064066 (p 75-76) sustituyendo A5-1 (preparado de acuerdo con el Ejemplo 104 de la Publicación de Patente Internacional No. WO 2009/005677, pág. 265) por 1-(t-butoxicarbonilamino)-2-vinilciclopropanocarboxilato de etilo (1R, 2S).
Preparación del Intermedio A6.
[0110]

[0111] El Intermedio A6 se preparó de manera similar al Intermedio A5, sustituyendo 1-metilciclopropano-1-sulfonamida (preparado de acuerdo con el Ejemplo 1,2 de la Publicación de Patente Internacional N° WO 2008/064066, pág. 47) por ciclopropanosulfonamida.
Preparación del Intermedio A7.
[0112]

[0113] El compuesto intermedio A7 se preparó de acuerdo con el Ejemplo 97,1,6 de la Publicación de Patente de EE.UU. N° 2009/274652, pág. 72-73,
Preparación del intermedio A8.
[0114]

[0115] Pasos 1-2, Preparación del Intermedio A8: El Intermedio A8 se preparó de manera similar al clorhidrato de (1R, 2S)-1-amino-N-(ciclopropilsulfonil)-2-vinilciclopropano-carboxamida del Ejemplo 2,12 de la Publicación de Patente Internacional No. WO 2008/064066 (p 75-76) sustituyendo A8-1 (preparado de acuerdo con el procedimiento detallado en el Ejemplo 97,1,4 de la Publicación de Patente de EE.UU. N° 2009/274652, pág. 72-3) por (1R, 2S)-1-(tercbutoxicarbonilamino). Ácido -2-vinilciclo-propancarboxílico y sustituyendo 1-metilciclopropano-1-sulfonamida (preparado de acuerdo con el Ejemplo 1,2 de la Publicación de Patente Internacional No. WO 2008/064066, pág. 47) por ciclopropanosulfonamida.
Preparación del intermedio A9.
[0116]

[0117] Paso 1-2. Preparación del Intermedio A9: El Intermedio A9 se preparó de manera similar al clorhidrato de (1R, 2s)-1-amino-N-(ciclopropilsulfonil)-2-vinilciclopropano-carboxamida del Ejemplo 2,12 de la Publicación de Patente Internacional No. w O 2008/064066 (p 75-76) sustituyendo A9-1 (preparado de acuerdo con el Ejemplo 1, Pasos 1L-1O de la Publicación de Patente Internacional No. WO 2009/134987, pág. 75-77) por (1r , 2S)-1-(tercbutoxicarbonilamino)-2-ácido vinilciclopropanocarboxílico.
Preparación del intermedio A10.
[0118]

[0119] Paso 1. Preparación de A10-1: Una solución de A9-1 (25 g, 100 mmol) y 1-metilciclopropano-1-sulfonamida (preparada de acuerdo con el Ejemplo 1,2 de la Publicación de Patente Internacional No. WO 2008/064066, Pág. 47; 15 g, 110 mmol) en DCM (330 ml) se trató con DMAP (24,4 g, 200 mmol) seguido de una adición lenta de EDC (38,3 g, 200 mmol) a 0°C. Una vez completada la adición, la mezcla se agitó vigorosamente a 0°C y se dejó calentar a ta durante 36 h. La reacción se diluyó con EtOAc (300 ml). La capa orgánica se lavó con ácido cítrico al 10% (3 x 30 ml) y se saturó. NaHCO3 (2X20 mL). Los lavados acuosos combinados se extrajeron una vez con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera (2 x 25 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron al vacío. Después de la trituración con hexano/EtOAc (10/1), se obtuvo acil sulfonamida A10-1 (31 g) como un sólido blanco.
[0120] Paso 2. Preparación del intermedio A10. A una suspensión de acil sulfonamida A10-1 (18,5 g, 50 mmol) en
DCM (300 ml) se añadió lentamente HCl 4 M en dioxano (150 ml, 600 mmol). Una vez completada la adición, la mezcla se agitó vigorosamente a temperatura ambiente durante 4 h. La reacción se concentró luego al vacío y el residuo se trituró con Et2O para proporcionar el Intermedio A10 (14,7 g) como un sólido blanco amorfo. 1H-RMn (400 MHz, CD3OD) 85,94 (m, 1H), 2,26 (m, 2H), 1,76 (m, 1H), 1,62 (m, 1H), 1,60 (m, 1H), 1,53 (s, 3H), 0,93 (m, 2H).
Grupo intermedio B
Preparación de la mezcla intermedia B1 y B2.
[0121]

[0122] Pasos 1 y 2: Preparación de racemato B1-1: Se añadió metal de magnesio (1,32 g, 54,3 mmol) a un matraz de 2 bocas equipado con un condensador de reflujo y el recipiente se lavó con Ar. Se añadió THF (42 ml) seguido de yodo (aprox. 5 mg). La suspensión agitada se calentó a 45°C y se añadió 5-bromopent-1-eno (1,2 g, 8,1 mmol) en una porción. Después de agitarse durante varios minutos, se añadió 5-bromopent-1-eno adicional (5,5 g, 37 mmol) a una velocidad suficiente para mantener un reflujo suave. La mezcla resultante se agitó a 50°C durante 15 minutos y luego se enfrió a temperatura ambiente y se usó inmediatamente en el siguiente paso. Una suspensión de Cul (630 mg, 3,3 mmol) en THF (24 ml) bajo Ar se enfrió a -5°C. Se añadió una parte alícuota de bromuro de pent-4-enilmagnesio (aprox. 0,95 M, 20 ml, 19 mmol) preparada en el paso 1 durante 5 minutos, y la mezcla resultante se agitó durante 15 minutos adicionales. La mezcla de reacción se enfrió luego a -20°C, y se añadió (6)-exo-2,3-epoxinORbornano (1,5 g, 14 mmol) como una solución en THF (5 ml) durante 1 minuto. Se utilizaron dos porciones adicionales de THF (2,5 ml cada una) para asegurar la transferencia completa, y la mezcla resultante se agitó durante 20 min. La reacción se retiró del baño frío y se calentó a temperatura ambiente. Después de agitarse durante 1,75 h adicionales, la reacción se detuvo con NH4Cl acuoso saturado (5 ml) y se filtró con EtOAc (100 ml) y H2O (100 ml) a través de Celite. Las fases se separaron y la fase orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar (6)-B1-1. 1H-RMN (300 MHz, CDCla) 85,90 - 5,67 (m, 1H), 5,04 - 4,86 (m, 2H), 3,12 (s, 1H), 2,20 - 1,92 (m, 5H), 1,69 -1,57 (m, 1H), 1,55 -1,12 (m, 9H), 1,03-0,84 (m, 1H).
[0123] Paso 3, Preparación de la mezcla intermedia diastereomérica B1 y B2: Se disolvió la mezcla de alcohol (6)-B1-1 (813 mg, 4,51 mmol) en DMF (4,5 ml). Se añadió piridina (370 ml, 4,5 mmol) seguido de DSC (1,5 g, 5,8 mmol). La mezcla de reacción se calentó a 45°C y se agitó durante 4 h. La mezcla de reacción se enfrió luego a 0°C y se añadió agua (4,5 ml) gota a gota durante 2 min. La mezcla de reacción se agitó durante 5 min y se retiró del baño frío. Después de 5 minutos adicionales, la mezcla de reacción se enfrió a 0°C y se agregaron L-terc-leucina (835 mg, 6,37 mmol) y K3PO4 (2,70 g, 12,7 mmol). La mezcla se agitó durante 10 minutos y se retiró del baño frío. Después de agitarse durante 24 h adicionales, la mezcla se diluyó con EtOAc (30 ml), se acidificó con HCl acuoso 1 M (15 ml) y se diluyó con HCl acuoso 0,2 M (15 ml). Las fases se separaron y la fase orgánica se lavó con HCl acuoso 0,2 M (2 x 20 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar la mezcla de intermedios diastereoisómeros B1 y B2 (1,64 g). LCMS-ESI-(m/z): [M-H]- calculado para C19H30NO4: 336,2; encontrado: 336,0.
Preparación de la mezcla intermedia B3 y B4.
[0124]

[0125] Paso 1: Preparación de B3-1: A una solución de K2CR2O7 (121 g, 0,41 mol) en H2O (1,5 L) se agregó gota a gota H2SO4 (143 g, 1,46 mol) a temperatura ambiente y la mezcla se agitó durante 1 hora. h. La mezcla se enfrió luego a 0°C y (1R, 3r, 5S)-biciclo[3,1,0]hexan-3-ol (80 g, 0,814 mol; preparada de acuerdo con la Sección A, Intermedio 1 de la patente de EE.UU. 8,178,491 B2, p 192) en MTBE (1,5 l) se añadió gota a gota. La mezcla de reacción se agitó a temperatura ambiente durante 2 h. La fase acuosa se extrajo con MTBE (3 x 500 ml), se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El producto bruto se purificó por destilación (20 mmHg, pe: 60 - 62°C) para proporcionar B3-1, 1H-RMN (400 MHz, CDCla) 82,57 - 2,63 (m, 2H), 2,14 - 2,19 (d, J = 20 Hz, 2H), 1,52 - 1,57 (m, 2H), 0,89 - 0,94 (m, 1H), -0,05 --0,02 (m, 1H).
[0126] Paso 2: Preparación de (±)-B3-2: Bajo Ar, se enfrió una mezcla de THF (4,4 ml) y HMPA (1,8 ml) a -78°C. Se añadió una solución 1 M de LiHMDS en THF (2,2 ml, 2,2 mmol). Se añadió cetona B3-1 (202 mg, 2,10 mmol) como una solución en THF (2 ml) durante 1 minuto, lavando con THF adicional (2 x 1 ml) para asegurar la transferencia completa. Después de 25 min, se añadió 5-yodopent-1-eno (preparado de acuerdo con Jin, J. y otros, J. Org. Chem.
2007, 72, 5098-5103) (880 mg, 4,5 mmol) durante 30 s mediante jeringuilla. Después de 10 minutos, la reacción se colocó en un baño frío a -45°C y se calentó a -30°C durante 1,5 h. La reacción se detuvo con NH4Cl acuoso saturado (15 ml) y se diluyó con EtOAc (30 ml) y H2O (15 ml). Las fases se separaron y la fase acuosa se extrajo con EtOAc (30 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a vacío para proporcionar un residuo bruto que se purificó mediante cromatografía en gel de sílice (0% a 15% de EtOAc en hexanos) para proporcionar (±)-B3-2, 1H-RMN (400 MHz, CDCla) 85,82 -5,67 (m, 1H), 5,03-4,87 (m, 2H), 2,61 -2,51 (m, 1H), 2,11 (d, J = 19,1 Hz, 1H), 2,08 - 1,99 (m, 3H), 1,61 - 1,40 (m, 5H), 1,36 - 1,28 (m, 1H), 0,92 - 0,81 (m, 1H), -0,03--0,11 (m, 1H).
[0127] Paso 3: Preparación de (±)-B3-3 y (±)-B3-4: Una solución de (±)-B3-2 (142 mg, 0,865 mmol) en THF (4 ml) se enfrió a -78°C. Se añadió gota a gota una solución 1 M de THF de LiBHEt3 (1,3 ml, 1,3 mmol) durante 30 s. La reacción se agitó durante 15 minutos y se retiró del baño frío. Después de calentar a temperatura ambiente (15 min), la reacción se detuvo con NH4Cl acuoso saturado (1 ml). La mezcla resultante se diluyó con Et2O (20 ml) y H2O (20 ml). Las fases se separaron y la fase acuosa se extrajo con Et2O (20 ml). Los extractos orgánicos combinados se secaron sobre MgSO4 anhidro, se filtraron y se concentraron al vacío. La purificación por cromatografía en gel de sílice (0% a 10% de EtOAc en hexanos) proporcionó 133 mg de una mezcla de diastereómeros (±)-B3-3 y (±)-B3-4, El material combinado de dos experimentos (253 mg) se purificó adicionalmente mediante cromatografía en gel de sílice (0% a 15% de EtOAc en hexanos) para proporcionar (±)-B3-3 (150 mg) y (±)-B3-4 (58 mg). 1H-RMN para (±)-B3-3 (300 MHz, CDCh) 85,91 - 5,69 (m, 1H), 5,07 - 4,88 (m, 2H), 3,97 (d, J = 6,7 Hz, 1H), 2,19 - 1,99 (m, 3H), 1,84 - 1,73 (m, 1H), 1,62 (d, J = 14,1 Hz, 1H), 1,54 - 1,40 (m, 2H), 1,32 - 1,17 (m, 3H), 1,16 - 1,06 (m, 1H), 0,60 - 0,43 (m, 2H). 1H-RMN para (±)-B3-4 (300 MHz, CDCh) 85,95 - 5,73 (m, 1H), 5,09 - 4,88 (m, 2H), 4,05 - 3,86 (m, 1H), 2,17 - 1,84 (m, 4H), 1,72 - 1,34 (m, 5H), 1,28 - 1,08 (m, 3H), 0,49 -0,36 (m, 1H), 0,21 -0,11 (m, 1H).
[0128] Paso 4: Preparación de la mezcla intermedia diastereomérica B3 y B4: se disolvió una mezcla de (±)-B3-3 (150 mg, 0,90 mmol) en DMF (1,0 ml). Se añadieron piridina (75 ml, 0,92 mmol) y DSC (302 mg, 1,18 mmol) y la reacción se agitó a 45°C durante 21,5 h. La reacción se colocó luego en un baño de agua con hielo y se añadió H2O gota a gota a través de una jeringa durante 1 minuto. La mezcla se retiró del baño frío y se dejó agitar durante 5 min. La mezcla se volvió a enfriar en un baño de agua con hielo y se añadió L-terc-leucina (154 mg, 1,17 mmol) seguido de K3PO4 (502 mg, 2,36 mmol). La mezcla de reacción se retiró del baño frío y se dejó agitar a temperatura ambiente durante 24 h. La mezcla se diluyó luego con EtOAc (40 ml) y HCl acuoso 1 M (20 ml). Las fases se separaron, y la fase acuosa se extrajo con EtOAc (30 ml). La fase orgánica combinada se lavó con HCl acuoso 0,2 M (2 x 20 ml), se secó sobre MgSO4 anhidro, se filtró y se concentró a vacío para proporcionar la mezcla de intermedios diastereoméricos B3 y B4, LCMS-ESI-(m/z): [M-H]- calculado para C18H28NO4: 322,2; encontrado: 322,0).
Preparación del Intermedio B3.
[0129]

[0130] Paso 1: Preparación de B3-5: A una solución de (1S, 4R)-cis-4-acetoxi-2-ciclopent-1-ol (Aldrich, 10 g, 70,4 mmol), trietilamina (48,8 mL, 350 mmol) y se añadió DMAP (4,29 g, 35,2 mmol) en diclorometano (352 ml) cloruro de pivaloilo (10,8 ml, 87,75 mmol) gota a gota a través de una jeringa a 0°C bajo una atmósfera de Ar. Después de 2 h, la mezcla de reacción se diluyó con una solución acuosa saturada de bicarbonato de sodio (500 ml) y se extrajo con diclorometano (23500 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 anhidro y se concentraron
al vacío para dar B3-5. 1H-RMN (300 MHz, CDCl3) 86,08 (br s, 2H), 5,54 (td, J = 8,0, 4,1 Hz, 2H), 2,88 (dt, J = 14,9, 7,5 Hz, 1H), 2,07 (s, 3H), 1,69 (dt, J = 14,7, 4,1 Hz, 1H), 1,20 (s, 9H).
[0131] Paso 2: Preparación de B3-6: A una solución de B3-5 (15,0 g, 70,4 mmol) en metanol (352 ml) se le añadió carbonato de potasio (9,73 g, 70,4 mmol) a temperatura ambiente bajo una atmósfera de argón. Después de 5 h, la mezcla de reacción se filtró y se concentró al vacío. El residuo se disolvió en acetato de etilo (500 ml) y la mezcla resultante se lavó con agua (500 ml) y salmuera (500 ml), se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo crudo se purificó por cromatografía en gel de sílice (acetato de etilo al 0-100%/hexanos) para proporcionar
B3-6, 1H-RMN (300 MHz, CDCl3) 86,11 (br d, J = 5,5 Hz, 1H), 5,97 (br d, J = 5,6 Hz, 1H), 5,48 (br s, 1H), 4,73 (br s, 1H), 2,82 (dt, J = 14,6, 7,3 Hz, 1H), 1,67 (s, 1H), 1,61 (dt, J = 14,5, 4,0 Hz, 1H), 1,20 (s, J = 3,8 Hz, 9H).
[0132] Paso 3: Preparación de B3-7: A una solución de cobre (I) cianuro (5,10 g, 57,0 mmol) en dietil éter (95 ml) se
le añadió bromuro de pent-4-enilmagnesio (Novel Chemical Solutions, 0,5 M en THF, 114 ml, 57,0 mmol) gota a gota
a través de una cánula durante un período de 30 minutos a 0°C bajo una atmósfera de argón. Después de 10 minutos, se añadió lentamente una solución de B3-6 (3,50 g, 19,0 mmol) en dietil éter (10 ml) a través de una cánula. A continuación, la mezcla de reacción se dejó calentar lentamente a ta. Después de 16 h, la mezcla resultante se inactivó
con una solución acuosa saturada de cloruro de amonio (400 ml) y la mezcla resultante se extrajo en acetato de etilo
(2 x 400 ml). Las fases orgánicas combinadas se lavaron con salmuera (400 ml), se secaron sobre Na2SO4 anhidro y
se concentraron al vacío. El residuo crudo se purificó por cromatografía en gel de sílice (acetato de etilo al 0-100%/hexanos) para proporcionar B3-7. 1H-RMN (400 MHz, CDCl3) 85,80 (ddt, J = 16,9, 10,2, 6,7 Hz, 1H), 5,69 (dd,
J = 5,8, 1,7 Hz, 1H), 5,65 (d, J = 7,2 Hz, 1H), 5,00 (dd, J = 17,1, 1,3 Hz, 1H), 4,94 (d, J = 10,2 Hz, 1H), 4,12 -4,05 (m,
1H), 2,69 (ddd, J = 17,2, 6,4, 1,5 Hz, 1H), 2,54 -2,45 (m, 1H), 2,24 (d, J = 17,2 Hz, 1H), 1,69 (br s, 1H), 1,52 -1,19 (m,
6H).
[0133] Paso 4: Preparación de (1S,2R,3R,5S)-B3-3: En una solución de B3-7 (20 mg, 0,13 mmol) y dietil cinc (1 M en hexanos, 132 pl, 0,132 mmol) en dietil éter (0,66 ml) se añadió diiodometano (21 pl, 0,26 mmol) a temperatura ambiente bajo una atmósfera de argón. Después de 2 h, la mezcla de reacción se inactivó con una solución acuosa
de 1 N HCl (0,66 ml). Después de 5 minutos, la mezcla amarilla resultante se diluyó con una solución acuosa saturada
de bicarbonato de sodio (5 ml) y la mezcla resultante se extrajo con diclorometano (3 x 5 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El residuo crudo se purificó por cromatografía en gel de sílice (acetato de etilo al 0-100%/hexanos) para proporcionar (1S,2R,3R,5S)-B3-3, 1H-RMN
(400 MHz, CDCl3) 85,83 (ddt, J = 16,9, 10,2, 6,7 Hz, 1H), 5,02 (d, J = 17,2 Hz, 1H), 4,96 (d, J = 11,3 Hz, 1H), 4,00 (d,
J = 6,7 Hz, 1H), 2,19 -2,02 (m, 3H), 1,82 (t, J = 7,2 Hz, 1H), 1,64 (d, J = 14,2 Hz, 1H), 1,55 - 1,42 (m, 2H), 1,38 -1,20
(m, 4H), 1,19 -1,08 (m, 1H), 0,62 - 0,47 (m, 2H).
[0134] Paso 5: Preparación del intermedio B3: Se recogió alcohol (1S,2R,3R,5S)-B3-3 (0,450 g, 2,7 mmol) en DMF (2,7 ml) y se trató posteriormente con DSC (0,92 g, 3,52 mmol) y piridina (0,22 ml, 2,8 mmol). La reacción se calentó luego a 50°C durante la noche. La reacción se enfrió luego a 0°C y se añadió agua (5,5 ml) gota a gota durante 1 minuto. La suspensión opaca resultante se agitó a temperatura ambiente durante 10 min antes de volver a enfriar a 0°C. La reacción se trató posteriormente con L-terc-leucina (0,462 g, 3,5 mmol) y K3PO4 (1,5 g, 7,0 mmol) y se dejó calentar a temperatura ambiente durante la noche con agitación vigorosa. La suspensión opaca resultante se diluyó con EtOAc y HCl acuoso 1M. Se añadió gota a gota HCl adicional (12 M) para ajustar el pH ~ 3, La capa acuosa se extrajo con EtOAc y los extractos orgánicos combinados se lavaron con salmuera y se secaron sobre MgSO4 anhidro. Tras la concentración al vacío, se obtuvo el Intermedio B3 que se contaminó con pequeñas cantidades de DMF y EtOAc. El material se usó en reacciones posteriores sin purificación adicional. LCMS-ESI+ (m/z): [M+H] calculado para C18H30NO4: 324,2; encontrado 324,7.
Preparación del intermedio B5:
[0135]

[0136] El Intermedio B5 se preparó de manera similar a la preparación del Intermedio B3, sustituyendo bromuro de but-3-enilmagnesio por bromuro de pent-4-enilo magnesio en el Paso 3, LCMS-ESI+ (m/z): [M+H]+ calc. para C17H28NO4: 310,2; encontrado 310,8.
Preparación de la mezcla intermedia B6 y B7:
[0137]

[0138] Paso 1: Preparación de B6-1: se diluyó una solución de KHMDS con THF 1,0 M (10 ml, 10 mmol) con THF (10 ml) en Ar y la solución resultante se enfrió a -78°C. Se añadió biciclo[3,1,1]heptan-2-ona (1,0 g, 9,1 mmol, preparado de acuerdo con Yin, y otros, J. Org. Chem. 1985, 50, 531) como una solución en THF (5 ml) sobre 2 min, lavado con THF adicional (2 x 2,5 ml) para asegurar la transferencia completa. La mezcla resultante se agitó durante 30 minutos y se añadió N-(5-cloro-2-piridil)bis(trifluorometanosulfonimida) (3,8 g, 9,7 mmol) como una solución en THF (10 ml) durante 2 minutos, lavado. con THF adicional (2 x 2,5 mL). La mezcla resultante se agitó durante 5 minutos y se retiró del baño frío. Después de agitarse 30 min adicionales, la reacción se diluyó con Et2O (70 ml) y HCl acuoso 1 M (50 ml). Las fases se separaron y la fase orgánica se lavó con NaOH acuoso 1 M (2 x 30 ml). La fase orgánica combinada se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo resultante se filtró a través de un tapón de sílice con 30% de EtOAc en hexanos para proporcionar un residuo crudo que se usó directamente en el siguiente paso.
[0139] Paso 2: Preparación de B6-1: A una solución de dietilacetal 3-butenal (1,4 ml, 8,3 mmol) bajo Ar a 0°C, se añadió una solución 0,5 M de THF de 9-borabiciclo[3,3,1]nonano (15,9 ml, 7,95 mmol) durante 3 min. La reacción se
dejó calentar a temperatura ambiente con agitación durante 20 h. Luego se añadió una solución acuosa 3 M de NaOH (2,9 ml, 8,7 mmol). Después de agitarse 20 min, la solución resultante se transfirió a un matraz que contenía el triflato de vinilo (aprox. 5,16 mmol) descrito anteriormente y PdCl2 (dppf)^CH2Cl2 (420 mg, 0,51 mmol). La mezcla resultante se agitó a 60°C durante 14 h, se dejó enfriar a temperatura ambiente y se diluyó con Et2O (50 ml) y H2O (50 ml). Las fases se separaron y la fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. La purificación por cromatografía en gel de sílice (EtOAc del 0% al 10% en hexanos después del pre-equilibrio con Et3N al 1% en EtOAc) proporcionó el intermedio B6-1, 1H-RMN (300 MHz, CDCla) 85,36 - 5,28 (m, 1H), 4,59 (t, J = 5,6 Hz, 1H), 3,73 3,58 (m, 2H), 3,54 -3,39 (m, 2H), 2,72 -2,60 (m, 1H), 2,45 -2,34 (m, 3H), 2,23-2,08 (m, 4H), 1,89 - 1,76 (m, 2H), 1,67 (dt, J = 16,1,6,9 Hz, 2H), 1,58 - 1,47 (m, 2H), 1,23 (t, J = 7,0 Hz, 6H).
[0140] Paso 3: Preparación de B6-2: una solución de olefina B6-1 (660 mg, 2,77 mmol) en THF (25 ml) a 0°C se trató con BH3^SMe2 como una solución 1 M en CH2Cl2 (2,9 ml, 2,9 mmol) durante 1 min. La solución resultante se agitó durante 2 horas a 0°C y se dejó calentar a temperatura ambiente. Después de agitarse durante 3 h adicionales, la mezcla de reacción se volvió a enfriar a 0°C y se diluyó con NaOH acuoso 2 M (7 ml) seguido de H2O2 acuoso al 30% (7 ml). La mezcla resultante se dejó calentar a temperatura ambiente con agitación durante 16 h adicionales. La mezcla se repartió entre Et2O (100 ml) y H2O (50 ml). Las fases se separaron y la fase orgánica se lavó con NaOH acuoso 0,5 M (50 ml). La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró a vacío para proporcionar un residuo bruto que se purificó mediante cromatografía en gel de sílice (15% a 40% de EtOAc en hexanos) para proporcionar el compuesto deseado. 1H-RMN (300 MHz, CDCh) 84,60 (t, J = 5,6 Hz, 1H), 3,76 - 3,60 (m, 3H), 3,58 -3,42 (m, 2H), 2,39 - 2,05 (m, 4H), 1,91 - 1,48 (m, 9H), 1,43- 1,35 (m, 1H), 1,25 (t, J = 7,0 Hz, 6H), 1,06 - 0,98 (m, 1H).
[0141] Pasos 4 y 5: Preparación de (±)-B6-3: Se disolvió acetal B6-2 (360 mg, 1,4 mmol) en THF (8 ml) y H2O (2 ml). Se añadió monohidrato de ácido para-toluensulfónico (40 mg, 0,2 mmol) y la solución resultante se agitó 16 h a temperatura ambiente. La reacción se diluyó con Et2O (50 ml) y H2O (30 ml) y las fases se separaron. La fase acuosa se extrajo con Et2O (30 ml) y la fase orgánica combinada se lavó con NaHCO3 acuoso saturado (15 ml). La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró a vacío para proporcionar un residuo bruto que se usó inmediatamente en el siguiente paso. Se suspendió bromuro de metil trifenilfosfonio (1,66 g, 4,6 mmol) en THF (40 ml) bajo Ar y se enfrió a -78°C. Se añadió gota a gota una solución 1 M de NaHMDS en THF (4,2 ml, 4,2 mmol) y la suspensión amarilla resultante se agitó durante 5 min. La mezcla fue eliminada desde el baño frío y la agitación se continuó durante 30 min adicionales. Luego, la mezcla se volvió a enfriar a -78°C y el residuo crudo del Paso anterior (aproximadamente 1,4 mmol) se añadió como una solución en THF (5 ml) durante 5 minutos, lavando con THF adicional (2 x 2,5 ml). para asegurar la transferencia completa. La mezcla resultante se agitó durante 5 min y luego se colocó en un baño de agua con hielo y se agitó durante 1 h adicional. La reacción se detuvo con NH4O acuoso saturado (20 ml) y se diluyó con Et2O (30 ml) y H2O (20 ml). Las fases se separaron y la fase orgánica se secó sobre MgSO4 anhidro, se filtró y se adsorbió en 5 g de gel de sílice. La purificación por cromatografía en gel de sílice (10% a 30% de EtOAc en hexanos) proporcionó (±)-B6-3, 1H-RMN (300 MHz, CDCh) 6,01 - 5,81 (m, 1H), 5,22 - 5,05 (m, 2H), 3,79 -3,66 (m, 1H), 2,43-2,25 (m, 2H), 2,24 -2,04 (m, 4H), 1,83-1,16 (m, 10H).
[0142] Paso 6: Preparación de la mezcla intermedia B6 y B7, El alcohol (±)-B6-3 (270 mg, 1,5 mmol) se disolvió en DMF (2,0 mL). Se añadieron piridina (125 ml, 1,5 mmol) y DSC (500 mg, 1,9 mmol) y la reacción se agitó a 45°C durante 15 h. La reacción se colocó luego en un baño de agua con hielo y se añadió H2O gota a gota a través de una jeringa durante 30 s. La mezcla se retiró del baño frío y se dejó agitar durante 10 min. La mezcla se volvió a enfriar en un baño de agua con hielo y se añadió L-terc-leucina (259 mg, 1,97 mmol) seguida de K3PO4 (835 mg, 3,93 mmol). La mezcla de reacción se retiró del baño frío y se dejó agitar a temperatura ambiente durante 5,25 h. La mezcla se diluyó luego con EtOAc (40 ml) y HCl acuoso 1 M (20 ml) y H2O (15 ml). Las fases se separaron y la fase acuosa se extrajo con EtOAc (30 ml). La fase orgánica combinada se lavó con HCl acuoso 0,2 M (2 x 25 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar la mezcla intermedia B6 y B7, LCMS-ESI+ (m/z):
[M+H] calculado para C19H32NO4: 338,2; encontrado: 337,8.
Preparación del intermedio B8.
[0143]

[0144] Pasos 1 y 2: Preparación de B8-1: A una solución de HMPA (1,8 ml) en THF (4,4 ml) bajo Ar enfriado a -78°C, se agregó una solución 1 M de LiHMDS (2,2 ml, 2,2 mmol). (1R, 5S)-biciclo[3,1,0]hexan-2-ona (200 mg, 2,08 mmol, preparado de acuerdo con Hodgson, DM et al. Synthesis, 2005, 2264) se añadió como una solución en THF (2 ml)
durante 1 minuto, lavar con THF adicional (2 x 1 ml) para asegurar la transferencia completa. Después de 50 min, se añadió 5-yodopent-1-eno (870 mg, 4,4 mmol, preparado como se describe en: Jin, J. y otros, J. Org. Chem. 2007, 72, 5098) durante 30 s. La reacción se agitó durante 1 h y se calentó a -50°C. Después de 2 h, la temperatura del baño había alcanzado los -35°C y se volvió a enfriar a -50°C. Después de 2 h adicionales, la temperatura del baño había alcanzado 0°C. La reacción se vertió entonces en NH4Cl acuoso saturado (50 ml) y se diluyó con EtOAc. Las fases se separaron y la fase orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío. La purificación por cromatografía en gel de sílice (0% a 15% de EtOAc en hexanos) proporcionó un aceite incoloro (245 mg). El residuo mencionado anteriormente (200 mg, 1,22 mmol) se disolvió en MeOH (5 ml) y se enfrió a un baño a -50°C. Se añadió NaBH4 (188 mg, 4,97 mmol) en una porción y la mezcla resultante se agitó durante 30 minutos y se retiró del baño frío. Después de 30 minutos adicionales, la reacción se inactivó con NH4Cl acuoso saturado (15 ml) y se diluyó con EtOAc (25 ml) y H2O (20 ml). Las fases se separaron y la fase orgánica se extrajo con EtOAc (30 ml). La fase orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío. La purificación por cromatografía en gel de sílice (10% a 30% de EtOAc en hexanos) proporcionó B8-1, 1H-RMN (300 MHz, CDCla) 85,88 -5,71 (m, 1H), 5,04 - 4,88 (m, 2H), 4,01 (dd, J = 7,4, 4,8 Hz, 1H), 2,08 - 1,99 (m, 2H), 1,93 (dd, J = 12,3, 6,9 Hz, 1H), 1,67 - 1,09 (m, 9H), 0,60 - 0,52 (m, 1H), 0,41 - 0,31 (m, 1H) ppm.
[0145] Paso 3: Preparación del intermedio B8: se disolvió el alcohol B8-1 (180 mg, 1,08 mmol) en DMF (1,5 ml). Se añadieron piridina (90 ml, 1,1 mmol) y DSC (349 mg, 1,36 mmol) y la reacción se agitó a 45°C durante 50 minutos. Se añadió DSC adicional (115 mg, 0,449 mmol) y la reacción se agitó durante 15 h adicionales. La reacción se colocó luego en un baño de agua con hielo y se añadió H2O gota a gota a través de una jeringa durante 30 s. Se añadió DMF adicional (2,5 ml) para facilitar la agitación. Se añadió L-terc-leucina (174 mg, 1,33 mmol) seguido de K3PO4 (550 mg, 2,6 mmol). La mezcla de reacción se retiró del baño frío y se dejó agitar a temperatura ambiente durante 2 h. Se añadieron L-terc-leucina adicional (50 mg, 0,38 mmol) y K3PO4 (162 mg, 0,76 mmol) y la mezcla de reacción se agitó durante 2 h adicionales. La mezcla luego se diluyó con EtOAc (30 ml) y H2O (30 ml) y la fase acuosa se acidificó con HCl acuoso 3 M a pH ~ 3, Las fases se separaron y la fase orgánica se lavó con HCl acuoso 0,2 M (3 x 20 ml) y se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío. El residuo bruto resultante del Intermedio B8 se usó posteriormente sin purificación adicional.
Grupo Intermedio C
Preparación del Intermedio C1.
[0146]

[0147] Paso 1: Preparación de C1-1: Se añadió paladio al 10% sobre carbón activado (10,0 g, 0,300 mol) a temperatura ambiente a una solución agitada de 4-metoxi-2-nitroanilina (46,0 g, 275 mmol) en metanol (1 l). La mezcla se agitó bajo una atmósfera de 50 PSI H2 durante 17 h, luego se filtró a través de Celite. El disolvente se eliminó al vacío para proporcionar C1-1, 1H-RMN (DMSO-cfe, 400 MHz): 86,42 (d, J = 8,4 Hz, 1H), 6,16 (d, J = 2,8 Hz, 1H), 5,97 (dd, J = 8,4, 2,8 Hz, 1H), 4,45 (s, 2H), 3,94 (s, 2H), 3,57 (s, 3H).
[0148] Paso 2: Preparación de C1-2: se trató una suspensión de C1-1 en dietil oxalato (235 g) con Et3N (54,6 g) y se calentó a 155°C durante 2 h. La mezcla se dejó enfriar a temperatura ambiente y se filtró. El sólido recogido se lavó con H2O y EtOH, luego se secó para dar C1-2, 1H-RMN (DMSO-cfe, 400 MHz): 811,81 (d, J = 18 Hz, 2H), 7,04 (d, J = 8,8 Hz, 1H), 6,72 (dd, J = 8,8, 2,8 Hz, 1H), 6,68 (d, J = 2,8 Hz, 1H), 3,72 (s, 3H).
[0149] Paso 3: Preparación del Intermedio C1: En un matraz seco bajo una atmósfera seca de N2, se colocaron 41,0 g (213 mmol) de C1-2, 200 mL de POCl3 y 41 mL de tri-n-propilamina (41 mL, 217 mmol). La mezcla de reacción exotérmica se dejó agitar a temperatura ambiente durante 1 h y luego se calentó a reflujo durante la noche. La mezcla se enfrió de nuevo a temperatura ambiente y se vertió lentamente sobre hielo/agua. La mezcla acuosa resultante se agitó a temperatura ambiente durante 20 min, luego se filtró. El precipitado recuperado se lavó con agua y se disolvió
en cloroformo. La solución de cloroformo se filtró para eliminar el material insoluble y el filtrado se lavó sucesivamente con agua, solución saturada de bicarbonato de sodio y salmuera. La concentración de la solución lavada al vacío fue seguida por recristalización del residuo en etanol. El producto se purificó mediante cromatografía en gel de sílice para producir el Intermedio C1, 1H-RMN (DMSO-de, 400 MHz): 67,99 (d, J = 9,2 Hz, 1H), 7,59 (dd, J = 9,2, 2,8 Hz, 1H), 7,50 (d, J = 2,8 Hz, 1H), 3,96 (s, 1H).
Preparación del intermedio C2.
[0150]

El compuesto intermedio C2 (2-cloro-6-metoxi-3-(metilsulfonil)quinoxalina) se preparó de acuerdo con Mahata, PK, et al. Org. Letón. 2005, 7, 2169.
Preparación del intermedio C3.
[0152]

[0153] Paso 1: Preparación de C3-1: 3-fluoroanilina (50 g, 0,45 mol) y 1,1 -bis(metiitio)-2-nitroetileno (74 g, 0,45 mol) en 500 ml de etanol se sometió a reflujo 24 h con agitación constante. La mezcla de reacción se enfrió luego con agua helada, se filtró y se lavó con éter para dar el compuesto C3-1.
[0154] Paso 2: Preparación de C3-2: A una suspensión de C3-1 (25 g, 110 mmol) en 200 ml de CH3CN, se agregó POCla (51 g, 330 mmol) gota a gota a 0°C durante un período de 15 min con agitación constante. Una vez completada la adición, la mezcla de reacción se calentó a 80°C durante 3 h. El disolvente se eliminó a vacío y el residuo resultante se neutralizó con una solución saturada de NaHCO3 enfriada con hielo. Las capas acuosas se extrajeron con CH2O 2 y los extractos orgánicos combinados se lavaron con agua, salmuera y se secaron sobre Na2SO4 anhidro. El disolvente se evaporó al vacío y el residuo se purificó por cromatografía en gel de sílice (PE/EtOAc: 30/1) para proporcionar C3-2.
[0155] Paso 3: Preparación del intermedio C3: se añadió gota a gota una solución de mCPBA (5,6 g, 32 mmol) en CH2Cl2 (100 ml) a una solución agitada de C3-2 (3,0 g, 13 mmol) en CH2O 2 (100 mL) a 0°C durante un período de 30 minutos y luego se deja calentar a temperatura ambiente con agitación durante la noche. Luego se lavó con NaOH acuoso 1 N (33100 ml), agua (100 ml) y salmuera (100 ml) y luego se secó sobre Na2SO4 anhidro. El disolvente se
evaporó al vacío y el residuo se purificó por cromatografía en gel de sílice (PE/EtOAc = 10/1) para proporcionar el Intermedio C3. 1H-RMN (400 MHz, CDCh) 83,55 (s, 3H), 7,73-7,82 (m, 2H), 8,15 (dd, J = 9,2, 5,6 Hz, 1H).
Preparación del intermedio C4.
Método A:
[0156]

[0157] Paso 1. Preparación de C4-1: A una solución de 3-bromo-3,3-difluoroprop-1-eno (25,0 g, 159 mmol) y oxalato de dietilo (21,6 mL, 159 mmol) en THF (380 mL), se añadió éter dietílico (90 mL) y n-pentano (90 mL) a -100°C gota a gota n-butillitio (2,5 M en hexano, 67 mL, 167,6 mmol) durante 30 min. La mezcla de reacción se agitó a -95°C durante 1 h y -78°C durante 2 h, y se inactivó con ac. NH4Cl (11 g en 150 ml de agua). La mezcla se extrajo con éter (tres veces). Las capas orgánicas se lavaron con HCl acuoso 1 N, salmuera y se secaron sobre Na2SO4 anhidro y se concentraron al vacío para dar el residuo crudo, que se purificó por cromatografía en gel de sílice (EtOAc en hexanos: 0% a 40%) para dar C4, -1, 1H-RMN (300 MHz, CDCla) 85,98-6,18 (m, 1H), 5,78 (dd, J = 0,9 Hz, 13 Hz, 1H), 5,60 (dd, J = 0,9 Hz, 11 Hz, 1H), 4,38 (q, J = 6,9 Hz, 2H), 1,37 (t, J = 7,2 Hz, 3H).
[0158] Paso 2. Preparación de C4-2 y C4-3: a una solución de C4-1 (14,0 g, 78,6 mmol) y dihidrocloruro de 4-metoxibenceno-1,2-diamina (15,08g, 71,4 mmol) en EtOH. (360 ml) a temperatura ambiente se añadió trietilamina (19,9 ml, 142,8 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla se concentró al vacío. La suspensión en diclorometano (30 ml) y la filtración dieron una cierta separación de regioisómeros con C4-2 como la especie precipitante. 1H-RMN (400 MHz, CDCla) 811,940 (br s, 1H), 7,850 (d, J = 9 Hz, 1H), 6,985 (dd, J = 3 Hz, 9 Hz, 1H), 6,754 (d, J = 2 Hz, 1H), 6,625-6,498 (m, 1H), 5,907 (dt, J = 17, 2 Hz, 1H), 5,601 (d, J = 11 Hz, 1H), 3,938 (s, 3H). La mezcla se suspendió, se filtró y se concentró al vacío una vez más, luego se purificó por cromatografía en gel de sílice (EtOAc en hexanos: 5% a 34%) para dar C4-3 como el primer componente eluyente.
1H-RMN (400 MHz, CDCh) 812,05 (br s, 1H), 7,850 (d, J = 9 Hz, 1H), 6,986 (dd, J = 3 Hz, 9 Hz, 1H), 6,761 (d, J = 3 Hz, 1H), 6,597-6,526 (m, 1H), 5,91 (dt, J = 17, 2 Hz, 1H), 5,601 (d, J = 11 Hz, 1H), 3,939 (s, 3H).
[0159] Paso 3. Preparación del intermedio C4: se trató una solución de C4-3 (2,07 g, 8,2 mmol en 1 ml de DMF con POCl3 (0,8 ml) y se calentó a 65°C durante 2,5 h. La reacción se diluyó con EtOAc y se detuvo vertiendo en agua con hielo. La fase orgánica se lavó posteriormente con bicarbonato de sodio acuoso saturado y salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío para dar 2,1 g de Intermedio C4. 1H-RMN (400 MHz, CDCh) 8,028 (d, J = 10 Hz, 1H), 7,46 (dd, J = 3 Hz, 9 Hz, 1H), 7,32 (d, J = 3 Hz, 1H), 6,549-6,478 (m, 1H), 5,86 (dt, J = 17, 2 Hz, 1H), 5,67 (d, J = 11 Hz, 1H), 3,981 (s, 3H).
Método B:
[0160]

[0161] Paso 1. Preparación de C4-4: Se cargó un matraz de fondo redondo de 3 bocas de 1 l con una solución de 3-bromo-3,3-difluoroprop-1-eno (25 g, 159,3 mmol) en DMF (360 ml) y agua (90 ml). La solución resultante se trató con 2-oxoacetato de etilo (33 ml, 1 M en tolueno) e In (25 g). La mezcla de reacción se agitó durante la noche a temperatura ambiente y luego se extrajo con 3 x 300 ml de éter. Las capas orgánicas se combinaron, se lavaron con 1 x 100 ml de NH4Cl acuoso saturado y 1 x 100 ml de salmuera, se secaron sobre Na2SO4 anhidro y se concentraron al vacío para proporcionar 25 g de C4-4 que se usaron posteriormente sin purificación adicional.
[0162] Paso 2. Preparación de C4-5: se agregaron THF (20 ml) y agua (4 ml) a C4-4 (2,0 g, 11,10 mmol) en un matraz de fondo redondo de 250 ml. Se añadió L iO H ^ O (0,7 g, 16,7 mmol, 1,5 equiv.) en una porción a 20-25°C. La reacción se agitó a temperatura ambiente hasta que se juzgó completa por análisis de TLC. Al finalizar, se agregaron agua (10 ml) y MTBE (10 ml). La capa acuosa (pH >9) se separa y la capa orgánica se extrae una vez con agua (4 ml). Las capas acuosas se combinaron y se añadió MTBE (10 ml). La mezcla bifásica se agitó mientras el pH se ajustó a 1 con 1N HCl. La capa acuosa se extrajo con MTBE (10 ml). Las capas de MTBE combinadas se lavaron una vez con solución de NaCl al 25% (4 ml), luego se secaron con Na2SO4 anhidro y se concentraron al vacío para proporcionar C4-5, 1H-RMN (400 MHz, CDCh) 85,97-6,07 (m, 1H), 5,76-5,82 (m, 1H), 5,60 (dd, J = 0,6, 10,9 Hz, 1H), 4,4 (dd, J = 8,0, 13,1 Hz, 1H).
[0163] Paso 3. Preparación de C4-6: el hidroxiácido C4-5 (27,7 g, 182,1 mmol) se agregó a un matraz de 1 L equipado con una sonda de temperatura y agitación general. Se añadieron DCM (280 ml), DMAP (2,0 g, 16,5 mmol) y piridina (29,4 ml, 364,1 mmol) al sustrato a 20-25°C. Se añadió TMSCI (46,0 ml, 364,1 mmol) durante 1 h a una velocidad para mantener la temperatura interna entre 18-28°C. La suspensión se agitó luego durante 1,5 h a 20°C. La reacción se enfrió a 0° y se trató con DMF (0,1 ml) y (COCl)2 (15,6 ml, 182,1 mmol) que se añadió como una velocidad para mantener la temperatura interna por debajo de 10°C. Esta suspensión se agitó durante 1 h a 0°C y 30 min a 20°C. La temperatura interna de la suspensión se redujo luego a 0°C y se añadió piridina (20,0 ml, 248,3 mmol) mientras se mantenía la temperatura interna por debajo de 10°C. Después de la adición de piridina, se formaron sólidos grandes y fue necesaria una mayor agitación. Se añadió 5-metoxi-2-nitroanilina (27,8 g, 165,5 mmol) en porciones, mientras se mantenía la temperatura interna por debajo de 10°C. Una vez completada la adición, la temperatura de reacción se elevó a 20°C. Una vez completada la reacción (controlada por UPLC), la suspensión se filtró y los sólidos se lavaron con DCM. La solución de d Cm se lavó con 1 M HCl y luego se suspendió con gel de sílice durante 15 min. La suspensión se filtró y se lavó con DCM. Se añadió HCl en MeOH (preparado a partir de MeOH y AcCl (1,3 equiv.) A 0°C) y la reacción se controló mediante UPLC hasta que se completó. La solución de DCM se neutralizó con NaHCO3 acuoso saturado. La capa acuosa se extrajo con DCM y las capas de DCM combinadas se concentraron en una espuma. La espuma se recogió en DCM y se calentó a 35°C. Se añadió heptano lentamente, se añadió semilla y la suspensión se agitó durante 30 min. Se añadió heptano lentamente durante 1 h, la suspensión se envejeció durante 1 h y luego se enfrió a 25°C durante 1 h. La suspensión se agitó durante 2 h, se filtró y se lavó con DCM/heptano (mezcla 1:3) para producir C4-6.
[0164] 1H-RMN (400 MHz, CDCla): 11,64 (s, 1H), 8,36 (d, J = 2,3 Hz, 1H), 8,20 (d, J = 9,4 Hz, 1H), 6,69 (dd, J = 9,4, 2,1 Hz, 1H), 6,19 -5,95 (m, 1H), 5,78 (br-d, J = 17,4 Hz, 1H), 5,58 (d, J = 11,1 Hz, 1H), 4,53 (t, J = 10,2 Hz, 1H), 3,90 (s, 3H) Paso 4, Preparación de C4-7: Nitroanilina C4-6 (10,96 g, 36,3 mmol) se diluyó en DCM (75 ml) y se trató con TEMPO (569 mg, 3,64 mmol) y Bu4NCl (1,0 g, 3,6 mmol) en un matraz de 3 ml de 500 ml equipado con agitación superior y una sonda de temperatura interna. Se añadió solución tampón (NaHCO30,5 M, Na2CO30,05 M, 90 ml) y la mezcla se agitó vigorosamente. Se añadió NCS (5,85 g, 43,8 mmol) en una porción. Después de 2,25 h, se añadió EtOH (2,5 ml) para ayudar a la disolución de los sólidos. La capa acuosa se retiró y se extrajo una vez con DCM. Las capas de DCM combinadas se lavaron con tiosulfato de sodio saturado, agua y luego se suspendieron con gel de sílice (15 g). La sílice se separó por filtración y se lavó con DCM. Tras la concentración al vacío, se recogió C4-7. Este material se disolvió en 45 ml de DCM caliente. Se añadió gota a gota heptano (25 ml) durante 3 minutos y la solución resultante se sembró con 30 mg de producto hidratado. La cristalización se agitó 5 min a -45°C y luego se enfrió a temperatura ambiente. Se añadió un agitador mecánico para facilitar la agitación. Se añadió heptano adicional (35 ml) a través de una bomba de jeringa a una velocidad de 20 ml/min y la suspensión resultante se agitó durante la noche. Los sólidos se filtraron al vacío a través de un embudo de vidrio fritado de porosidad media y la torta del filtro se lavó con 4 x 9 ml 2:1 de heptano: DCM. El secado proporcionó una mezcla de formas de hidrato y etilo hemicetal. 1H-RMN
(400 MHz, d e -acetona): 68,41 (d, J = 2,8 Hz, 1H), 8,30 (d, J = 9,4 Hz, 1H), 6,90 (dd, J = 9,4, 2,7 Hz, 1H), 6,34 -6,16 (m, 1H), 5,75 (br-d, J = 17,4 Hz, 1H), 5,62 (d, J = 11,1 Hz, 1H), 3,97 (s, 3H) ppm. (nota: las especies de etilcetal/hemicetal correspondientes también se observarán en cierta medida junto con el producto deseado. Estos subproductos serán evidentes a partir de duplicación de picos y multipletes en el rango de 3,4-3,9 ppm y 0,9-1,3 ppm. Los valores listados corresponden al producto principal.)
[0165] Paso 5. Preparación de C4-3, La nitroanilina C4-6 (32,9 g, 103 mmol) se suspendió en EtOH (460 ml) y HOAc (190 ml) en un matraz de 3 bocas equipado con agitación superior, sonda de temperatura y entrada de N2. Se añadió polvo de hierro (37,5 g, 672 mmol) y la mezcla heterogénea agitada vigorosamente se calentó en un bloque térmico precalentado a 55°C. Después de 20 minutos, la temperatura de reacción interna fue de aproximadamente 50°C, y se juzgó que estaba experimentando una exotermia en función de la tasa de aumento de la temperatura. Se eliminó el calentamiento y la temperatura interna continuó aumentando a 57°C durante 10 min. Después de que la temperatura comenzó a descender, la fuente de calor se volvió a aplicar. Una temperatura interna relativamente constante de 51°C se observó a partir de entonces. Después de 3H, la reacción se diluyó con EtOAc (300 ml) y se añadió Celite (50 g). La mezcla de reacción espesa resultante se filtró a través de una almohadilla corta de Celite, lavando con suficiente EtOAc para asegurar la elución de color amarillo/rojo. El filtrado se concentró al vacío y se repartió entre EtOAc (300 ml), HCl acuoso 0,2 M (250 ml) y salmuera (50 ml). Las capas se separaron y la fase orgánica se lavó con 2 x 300 ml de salmuera saturada al 15%, 1 x 300 ml de H2O 1:1: NaHCO3 acuoso saturado y 200 ml de salmuera. La fase orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró hasta 50 g de un residuo sólido dorado. Se agregaron 30 ml adicionales de EtOAc y la mezcla heterogénea se calentó en un bloque térmico a 65°C. Se añadió gota a gota hexano (500 ml) mediante un embudo de adición durante 1 h, y la suspensión resultante se enfrió a temperatura ambiente y se agitó durante 5 h adicionales. La filtración a través de un embudo de vidrio fritado de porosidad media proporcionó el producto deseado. 1H-RMN (400 MHz, CDCla): 12,43 (s, 1H), 7,83 (d, J = 9,0 Hz, 1H), 6,98 (d, J = 8,9 Hz, 1H), 6,81 (s, 1H), 6,64 -6,42 (m, 1H), 5,90 (br-d, J = 17,4 Hz, 1H), 5,59 (d, J = 11,0 Hz, 1H), 3,93 (s, 3H) ppm.
[0166] Paso 6. Preparación del Intermedio C4. Se disolvió hidroxiquinoxalina (22,4 g, 88,8 mmol) en DMF (45 ml) en un matraz de fondo redondo equipado con una sonda de temperatura y una entrada de N2. Se añadió POCl3 (12,5 ml, 134 mmol, 1,5 equiv.) A través de una jeringa a una velocidad para mantener la temperatura interna por debajo de 50°C. Se calentó entonces la solución de color rojo oscuro a través del bloque de calor pre-calentado a 75°C (temperatura interna ~ 74°C). Después de 2,5 h, la reacción se transfirió a través de una cánula a 370 ml de H2O agitada en un matraz de 3 bocas equipado con sonda de temperatura, agitación en cabeza y ventilación a la atmósfera. La velocidad de enfriamiento se controló de tal manera que la temperatura interna permaneció por debajo de 35°C. Se utilizaron tres porciones adicionales de DMF (3 ml cada una) para asegurar la transferencia completa. Una vez que la temperatura interna disminuyó a 30°C, se añadió NaOH acuoso 3 M hasta que se obtuvo un pH de ~ 6-7 (160 ml en total). La mezcla heterogénea parda se enfrió luego a una temperatura interna de 15°C y se filtró a través de una frita de grado M. La torta del filtro se lavó con H2O (2 x 30 ml) y 3:1 H2O: MeCN (3 x 20 ml). La torta del filtro se suspendió en CH2O 2 (200 ml) y la mezcla se secó con MgSO4 anhidro y se filtró a través de una capa corta de Celite. La concentración al vacío proporcionó 19 g de un aceite rojo oscuro. Este aceite se disolvió en CH2Cl2 (100 ml) y se suspendió con gel de sílice (40 g) durante 20 min. La suspensión se filtró a través de una pequeña almohadilla de gel de sílice fresco (20 g), lavando con 6 x 40 ml de CH2O 2, El filtrado se concentró para proporcionar el producto deseado. Este material se recristalizó en hexanos calientes para proporcionar 14,5 g de Intermedio C4 como agujas amarillas.
1H-RMN (400 MHz, CDCla): 8,02 (d, J = 9,2 Hz, 1H), 7,45 (dd, J = 9,3, 2,8 Hz, 1H), 7,32 (d, J = 2,7 Hz, 1H), 6,59 -6,43 (m, 1H), 5,86 (dt, J = 17,3, 2,5 Hz, 1H), 5,67 (d, J = 11,0 Hz, 1H), 3,98 (s, 3H) ppm.
Método C:
[0167]

[0168] Paso 1. Preparación de C4-8, Al hidroxiéster C4-4 (58,1 g, 323 mmol) se le añadió DCM (700 ml) en un matraz de 3 litros de 3 bocas equipado con agitación elevada y una sonda de temperatura interna. Luego se secuenciaron
TEMPO (5,4 g, 35 mmol), solución tampón (preparada disolviendo 4,2 g de NaHCO3 y 0,53 g de Na2CO3 por 100 ml de agua, 700 ml, 7v) y NaOCl (Clorox 6,15% en peso, 422 ml, 395 mmol) Añadido al matraz a 20°C. Después de 2 h, la capa orgánica se separó y la fase acuosa se extrajo con acetato de etilo (2 x 300 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentró al vacío para proporcionar C4-8. 1H-RMn (300 MHz, CDCl3) 85,98-6,18 (m, 1H), 5,78 (dd, J = 0,9 Hz, 13 Hz, 1H), 5,60 (dd, J = 0,9 Hz, 11 Hz, 1H), 4,38 (q, J = 6,9 Hz, 2H), 1,37 (t, J = 7,2 Hz, 3H).
[0169] Paso 2. Preparación de C4-9, A una solución de etilo-3,3-difluoro-2,2-dihidroxipent-4-enoato C4-8 (57,4 g, 292 mmol) en THF (725 mL) y agua (131 mL) se añadió L iO H ^ O (22 g, 529 mmol) a 20°C. Después de 2,5 h, la mezcla de reacción se concentró al vacío. El residuo sólido se suspendió en agua (300 ml) y la mezcla resultante se acidificó a pH=1 con una solución acuosa concentrada de ácido clorhídrico. La mezcla resultante se agitó hasta que se disolvieron todos los sólidos (~ 1,5 h) y luego se añadió cloruro de sodio hasta que la solución se saturó. La solución resultante se extrajo con MTBE (2 x 500 mL) y acetato de etilo (2 x 500 mL), y las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El residuo sólido anaranjado en bruto se suspendió en DCM (100 ml) y se agitó hasta que los sólidos se distribuyeron finamente antes de que los hexanos (75 ml) se agregaran lentamente a través de un embudo de adición. Los sólidos resultantes se recogieron por filtración al vacío a través de un embudo fritado medio y se lavaron con 1:1 diclorometano/hexanos (2 x 10 ml) para proporcionar el producto deseado. 1H-RMN (400 MHz, DMSO-de) 813,17 (bs, 1H), 6,18-6,01 (m, 1H), 5,64-5,52 (m, 2H).
[0170] Paso 3. Preparación de C4-2 y C4-3, Se añadió 4-metoxi-2-nitroanilina (10,0 g, 59,5 mmol), ácido cetal C4-9 (11,1 g, 74,4 mmol) y BHT (1,31 g, 5,95 mmol) a un matraz de 3 boquillas de 3 litros equipado con agitación superior, sonda de temperatura y entrada de Ar. El etanol (200 ml) y el ácido acético (100 ml) se añadieron secuencialmente a 20°C y el recipiente se purgó con Ar. Se añadió polvo de hierro (16,6 g, 297 mmol) y la mezcla heterogénea agitada vigorosamente se calentó en un bloque térmico precalentado a 65°C. Después de 10 minutos, la temperatura interna alcanzó una temperatura máxima de 70°C, y se juzgó que estaba experimentando una exotermia basada en la tasa de aumento de la temperatura. Se eliminó el calentamiento y la temperatura interna bajó a 65°C, momento en el cual se volvió a aplicar la fuente de calor. Después de 30 minutos, la mezcla de reacción contenía un precipitado sólido marrón y se dejó enfriar a temperatura ambiente. La mezcla se diluyó luego con acetato de etilo (1 l) y se añadió gel de sílice (200 ml) y se agitó. La suspensión resultante se filtró a través de un embudo sinterizado, lavando con suficiente acetato de etilo para asegurar la elución del producto deseado. El filtrado rojo claro/naranja claro resultante se concentró al vacío y el residuo bruto se repartió entre acetato de etilo (300 ml) y una solución acuosa saturada de bicarbonato de sodio (300 ml). Las fases se separaron y la fase orgánica se secó sobre Na2SO4 anhidro, se filtró, y se concentró a vacío para proporcionar un ~ proporción de 4: El residuo sólido se suspendió en acetato de etilo (150 ml) y se agitó mediante agitación magnética hasta que la mezcla era una suspensión finamente dispersada (~ 10 min). Se añadieron lentamente hexanos (1,5 l) durante un período de aproximadamente 10 minutos y los sólidos resultantes se recogieron de la suspensión mediante filtración al vacío a través de un embudo de frita medio para proporcionar el producto deseado en forma de un polvo de color tostado después del secado.
[0171] Paso 5. Preparación del intermedio C-4, Mezcla de hidroxiquinoxalina C4-2 y C4-3 (46,8 g, 97:3 mezcla regioisomérica, 186 mmol) se disolvió en DMF (93 ml) en un matraz de fondo redondo de 3 l de 1 cuello equipado con un agitador superior, sonda de temperatura, y la entrada de Ar. Se añadió lentamente POCl3 (20,2 ml, 217 mmol) a través de una jeringa a una velocidad para mantener la temperatura interna por debajo de 45°C. La solución de color rojo oscuro se calentó luego a través de un bloque de calor precalentado a 65°C. Después de 4 h, la mezcla de reacción se dejó enfriar a temperatura ambiente. La reacción se transfirió luego lentamente a través de una cánula a 1 L de agua agitada vigorosamente. La velocidad de enfriamiento se controló de tal manera que la temperatura interna permaneció por debajo de 35°C. Sólidos marrones formados al apagar la mezcla de reacción. La mezcla resultante se basificó luego a pH=8 mediante la adición lenta de una solución acuosa de KOH al 50% en peso a una velocidad tal que la temperatura interna se mantuvo por debajo de 35°C. Los sólidos se recogieron por filtración al vacío a través de un embudo fritado de curso para proporcionar un sólido. Los sólidos se recogieron en diclorometano (500 ml, 10 v) y la mezcla resultante se suspendió con gel de sílice (50 g, 10 s). La suspensión se filtró a través de una almohadilla de gel de sílice (50 g, 10 s) en un embudo fritado de curso seguido de lavado con 3 x 10 ml de diclorometano. El filtrado se concentró al vacío para proporcionar un sólido que se recristalizó en hexanos calientes (50 ml, 65°C) para proporcionar el Intermedio C-4.
Preparación del intermedio C5.
[0172]
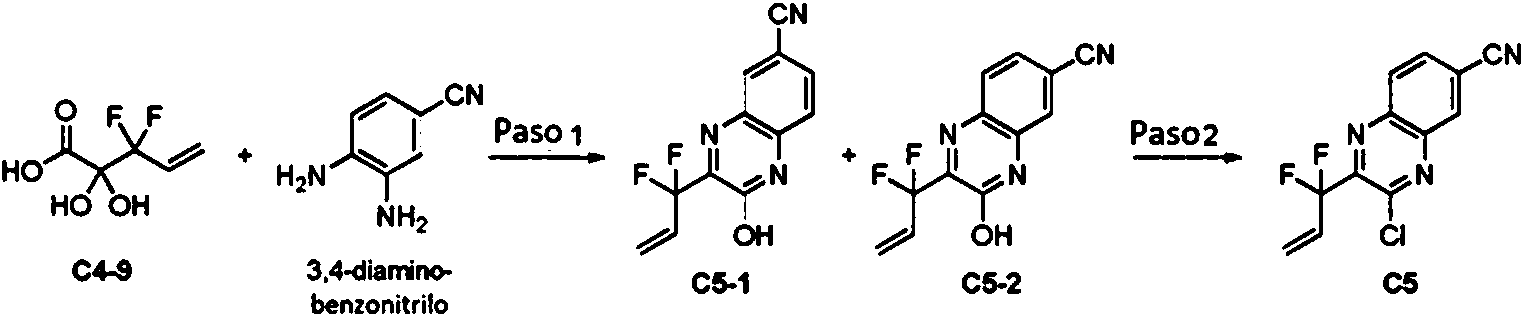
[0173] Paso 1. Preparación de C5-1 y C5-2: se trató una solución de C4-9 (0,5 g, 3,3 mmol) en EtOH (12 mL) con 3,4-diaminobenzonitrilo (0,47g, 3,5 mmol). La mezcla de reacción se calentó a 80°C durante 1 h, luego se concentró al vacío. El residuo resultante se absorbió en gel de sílice, luego se purificó por cromatografía en columna para dar C5-2 como el primer componente de elución 1H-RMN (400 MHz, CD3OD) 88,01 (d, 1H), 7,65 (dd, 2H), 6,49 (m, 1H), 5,80 (dt, 1H), 5,60 (d, 1H). C5-1 se recuperó como el segundo componente eluyente. 1H-RMN (400 MHz, CD3OD) 88,25 (d, 1H), 7,87 (dd, 1H), 7,41 (d, 1H), 6,49 (m, 1H), 5,80 (dt, 1H), 5,59 (d, 1H).
[0174] Paso 2. Preparación del intermedio C5: una solución de C5-2 (0,5 g, 2 mmol en 4,5 mL de DMF se trató con POCl3 (3 mL) y se calentó a 65°C durante 3 h. La reacción se diluyó con Se suspendió EtOAc y se vertió en agua con hielo..La fase orgánica se lavó posteriormente con NaHCO3 acuoso saturado y salmuera, se secó sobre Na2SO4 anhidro y se concentró a vacío para dar el Intermedio C5.
Preparación del intermedio C6.
[0175]

[0176] Paso 1. Preparación del Intermedio C6: A una solución de C4-1 (2,1 g, 11,79 mmol) y 4,5-difluorobenceno-1,2-diamina (1,715 g, 11,9 mmol) en EtOH (50 ml) A la temperatura ambiente se agregó trietilamina (3,0 ml, 21,5 mmol). La mezcla de reacción se sometió a reflujo durante 2 h. Tras la concentración de la mezcla de reacción al vacío, el residuo se purificó mediante cromatografía en gel de sílice para proporcionar el Intermedio C6, LCMS-ESI+ (m/z):
[M+H] calc. para C11H7F4 N2O: 259,0; encontrado: 259,0.
Preparación del intermedio C7.
[0177]

[0178] Paso 1. Preparación de C7-1: En una solución de C4-1 (1,84 g, 10,93 mmol) y 4-(difluorometoxi) benceno-1,2-diamina (1,90 g, 10,93 mmol, preparada de acuerdo con la referencia Ejemplo 30 y de la Publicación de Patente Internacional N° WO 2003/035065, página 511,) en DMF (40 ml) a temperatura ambiente se añadió DIPEA (9,5 ml, 54,65 mmol) y HATU (6,23 g, 16,4 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 24 h, se diluyó con acetato de etilo (100 ml), se lavó con agua (100 ml) y salmuera (50 ml). La mezcla se concentró al vacío. La purificación mediante cromatografía en gel de sílice (EtOAc en hexanos: 20% a 60%) proporcionó C7-1 como la fracción de elución posterior de dos con espectros de masas similares. LCMS-ESI+ (m/z): [M+H] calc. para C12H9F4N2O: 289,2; encontrado: 289,0.
[0179] Paso 2: Preparación del Intermedio C7: Se combinan hidroxiquinoxalina C7-1 (800 mg, 2,8 mmol), POCl3 (1,65 ml, 3,0 mmol) y d Mf (10 ml) a temperatura ambiente y luego se calientan a 65°C a 2,5°C. h, momento en el que se añadió POCl3 adicional (0,2 ml, 0,36 mmol). La reacción se calentó durante 3 h más a 65°C y luego se enfrió a temperatura ambiente. La reacción se detuvo mediante la adición de agua fría (30 ml) y se recogió en acetato de etilo (50 ml), se lavó con Na2CO3 acuoso saturado (100 ml) seguido de salmuera (50 ml) y se secó sobre MgSO4 anhidro. La solución resultante se concentró al vacío para dar el Intermedio C7 que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H] calc. para C12H8C F 4N2O: 307,0; encontrado: 307,0.
Preparación del intermedio C8.
[0180]

[0181] El Intermedio C8 se preparó de manera similar al Método A para preparar el Intermedio C4, sustituyendo el 1,2-diaminobenceno por dihidrocloruro de 4-metoxibenceno-1,2-diamina en el Paso 2.
Preparación del Intermedio C9.
[0182]

[0183] El compuesto intermedio C9 se preparó de acuerdo con Venkatesh, C., y col. Org. Letón. 2005, 7, 2169, Preparación del Intermedio C10.
[0184]

[0185] El Intermedio C10 se preparó de acuerdo con el método descrito en los Pasos 1 y 2 del Intermedio C11 a partir de la Publicación de Patente de EE.UU. N° 2010/0099695, pág. 31,
Preparación del Intermedio C11.
[0186]

[0187] Paso 1. Preparación de C11 -1: en un matraz de fondo redondo, 3-(benciloxi) anilina (4,025 g, 20,20 mmol) y 1,1 -bis(metiltio)-2-nitroetMeno (3,338 g, 20,20 mmol) en etanol (40 ml) se sometió a reflujo durante 24 h con agitación constante. La mezcla de reacción se enfrió luego en un baño de hielo y se diluyó con éter (150 ml). La mezcla se filtró y se lavó con éter para proporcionar C11-1 (3,32 g) en forma de un sólido amarillo que se usó directamente en el siguiente paso. Lc Ms -e S i+ (m/z): [M+H] calc. para C16H17N2O3S: 317,1; encontrado: 317,1.
[0188] Paso 2. Preparación de C11-2: A una suspensión de C11-1 (3,32 g, 10,49 mmol) en 25 ml de MeCN, se añadió gota a gota POCh (2,93 ml, 31,5 mmol) durante 15 minutos con agitación constante. La mezcla de reacción se calentó a 80°C y se agitó durante 5 h. La reacción se enfrió luego a temperatura ambiente y se neutralizó con una solución acuosa saturada de NaHCO3 enfriada con hielo, se extrajo tres veces con CH2Ch (100 ml), se lavó con agua, salmuera y se secó sobre Na2SO4 anhidro. El disolvente se eliminó a presión reducida. El material bruto se eluyó a través de un tapón de sílice con CH2Cl2, El disolvente se eliminó a presión reducida y el sólido se lavó con MeCN para proporcionar c 11-2 (1,56 g) como un sólido blanquecino. LCMS-ESI+ (m/z): [M+H] calc. para C^H-mC I^O S: 317,1; encontrado: 317,3.
[0189] Paso 3. Preparación del intermedio C11. Se añadió gota a gota una solución de mCPBA (1,87 g, 10,83 mmol) en CH2Cl2 (40 ml) a una solución agitada de C11-2 (1,56 g, 4,92 mmol) en CH2Cl2 (40 ml) a 0°C durante un período de 30 min. La mezcla de reacción se agitó adicionalmente a temperatura ambiente durante 5 h. Luego se vertió en hielo, se pudo saturar con NaHCO3 acuoso y se repartió con CH2O 2, La capa orgánica se lavó posteriormente con agua, salmuera y se secó sobre Na2SO4 anhidro. El disolvente se eliminó a presión reducida y el material bruto se purificó por cromatografía de fase normal con CH2Cl2 para proporcionar el compuesto del título Intermedio C11 en forma de un sólido amarillo pálido. LCMS-ESI+ (m/z): [M+H] calc. para C16H-mCIN203S: 349,0; encontrado: 349,0.
Preparación del Intermedio C12.
[0190]

[0191] Se preparó 2,7-dicloro-3-(prop-2-en-1-il) quinazolin-4(3H)-ona (Intermedio C12) de acuerdo con el Paso 3 del Intermedio D5 del documento WO12040040 p. 53-4.
Preparación del Intermedio C13
[0192]

[0193] Paso 1. En un matraz de fondo redondo de 1 l purgado y mantenido con una atmósfera inerte de nitrógeno se colocó naftalen-1-amina (15 g, 104,76 mmol, 1,00 equiv.), 1,1-bis(metilsulfanilo)-2-nitroeteno C13-1 (17,3 g, 104,70 mmol, 1,00 equiv.) y etanol (520 mL). La solución resultante se agitó a 80°C durante 18 h. Los sólidos se recogieron por filtración y se lavaron con 1 x 100 ml de éter para proporcionar 20 g (73%) de N-[(Z)-1-(metilsulfanil)-2-nitroetenil]naftalen-1-amina C13-2 como amarillo sólido.
(ES, m/z): 261 [M+H]+
[0194] Paso 2. En un matraz de fondo redondo de 3 bocas y 250 ml purgado y mantenido con una atmósfera inerte de nitrógeno, se colocó N-[(Z)-1-(metilsulfanil)-2-nitroetenil]naftalen-1, amina C13-2 (10 g, 38,42 mmol, 1,00 equiv.), CH3CN (92 ml), seguido de la adición de tricloruro de fosfoilo (10,8 ml, 3,00 equiv.) gota a gota con agitación a 0°C. La mezcla se agitó a 0°C durante 15 min y a 80°C durante 4 h. La mezcla resultante se concentró al vacío y el residuo se diluyó con 300 ml de DCM. La mezcla resultante se lavó con 2 x 200 ml de bicarbonato de sodio (sat.) y 1 x 100 ml de salmuera, se secó sobre sulfato de sodio anhidro y se concentró al vacío. El residuo se aplicó sobre una columna de gel de sílice eluyendo con acetato de etilo: PE (1:20) para proporcionar 5,5 g (27%) de 3-cloro-2-(metilsulfanil)benzo[f]quinoxalina C13-3 como un Sólido amarillo.
[0195] Paso 3. En un matraz de fondo redondo de 1 l purgado y mantenido con una atmósfera inerte de nitrógeno, se colocó una solución de 3-cloro-2-(metilsulfanil)benzo[f]quinoxalina C13-3 (5,0 g, 19,18 mmol, 1,00 equiv.) En diclorometano (116 ml). A la mezcla se le añadió una solución de ácido 3-clorobenceno-1-carboperoxo (16,6 g, 96,20 mmol, 5,00 equiv.) En diclorometano (263 mL) a 0°C. La mezcla se agitó a 0°C durante 30 minutos y a temperatura ambiente durante 3 horas. A continuación, la reacción se detuvo mediante la adición de 200 ml de agua. La mezcla resultante se lavó con 2x300 ml de NaHCO3 (10%), 2x200 ml de H2O y 2x200 ml de salmuera, se secó sobre sulfato de sodio anhidro y se concentró al vacío. El residuo se trituró con 30 ml de CH3CN. Los sólidos se recogieron por filtración para proporcionar 5,0 g (89%) de 3-cloro-2-metanosulfonilbenzo[f]quinoxalina C13 como un sólido amarillo. (ES, m/z): 293 [M+H]+ H-RMN: (CD3, 300MHz, ppm): h 9,03-9,00 (m, 1H), 8,26 (d, J = 2,7 Hz, 1H), 8,05-8,03 (m, 1H), 7,97 (d, J = 9,3 Hz, 1H), 7,88-7,85 (m, 2H), 3,68 (s, 3H).
Grupo intermedio D
Preparación del intermedio D1.
[0196]

[0197] El compuesto intermedio D1 se preparó de acuerdo con el método descrito en el Ejemplo 4A de la Publicación Internacional de Patente No. WO 2006/007700, pág. 87.
Preparación de ejemplos
Ejemplo 1. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil) carbamoil)-2-(difluorometil)cidopropil]-15-metoxi-4,7-dioxo-1a, 2,2a, 4,5,6,7,10,11,19,20,21,22,23,23a,23bhexadecahidro-1H,9H-8,l1-metanocidopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazacidononadecino[11,12-b]quinoxalina-9-carboxamida.
[0198]

[0199] Paso 1. Preparación de 1-1, Se combinó quinoxalina C1 (2,29 g, 10,0 mmol) con N-Boc-trans-4-hidroxi-L-prolina metil éster (2,65 g, 11,0 mmol) y CS2CO3 (3,59 g, 11,0 mmol) como una suspensión en MeCN (20 mL). La mezcla de reacción agitada se calentó a 85°C durante 16 h, luego se filtró sobre Celite y se concentró al vacío. El residuo crudo se purificó por cromatografía en gel de sílice (5% a 50% de EtOAc/Hex) para proporcionar 1-1, LCMS-ESI+ (m/z): [M-Boc+2H]+ calculado para C15H17CIN3O4: 338,09; encontrado: 337,94.
[0200] Paso 2. Preparación de 1-2. El carbamato 1-1 (401 mg, 0,916 mmol) se disolvió en DCM (10 ml) y se trató con HCl (4,0 M en dioxano, 2 ml, 8 mmol). Después de agitarse a temperatura ambiente durante 16 h, la mezcla de reacción se concentró al vacío para proporcionar hidrocloruro de amina 1 -2 que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H] calc. para C15H17ON3O4: 338,09; encontrado: 338,30.
[0201] Paso 3. Preparación de 1-3, El clorhidrato de amina 1-2 (0,916 mmol) y el compuesto intermedio B3 (413 mg, 1,28 mmol) se combinaron y se trataron con BEP (351 mg, 1,28 mmol), EtOAc (9 mL), NMP (1 mL) y DIPEA (0,80 mL, 4,6 mmol). La mezcla agitada se calentó a 50°C durante 3 h. Después de enfriarse a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc y se lavó sucesivamente con NaHCO3 acuoso saturado y salmuera. La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo bruto se purificó mediante cromatografía en gel de sílice (25% a 40% de EtOAc/Hex) para proporcionar la amida 1-3. LCMS-ESI+ (m/z): [M+H] calc. para C33H44ClN4O7: 643,29; encontrado: 644,15.
[0202] Paso 4. Preparación de 1-4. La cloroquinoxalina 1-3 (367 mg, 0,571 mmol) se trató con viniltrifluoroborato de potasio (115 mg, 0,856 mmol), Pd(dppf)C^DCM (47 mg, 0,057 mmol), EtOH (6 ml) y Et3N (0,12) mL, 0,86 mmol). La mezcla de reacción se agitó a reflujo durante 1 h, luego se enfrió a temperatura ambiente y se diluyó con EtOAc. La fase orgánica se lavó con agua y salmuera, luego se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo crudo se purificó por cromatografía en gel de sílice (25% a 40% de AcOEt/Hex) para proporcionar vinil quinoxalina 1-4. lCm S-ESI+ (m/z): [M+H] calc. para C35H47N4O7: 635,34; encontrado: 635,43.
[0203] Paso 5. Preparación de 1-5. La vinil quinoxalina 1-4 (327 mg, 0,515 mmol) se disolvió en DCE (103 ml) y se trató con Zhan Catalyst-1B (35 mg, 0,052 mmol). La mezcla se desgasificó con N2 burbujeante durante 18 min, se calentó a reflujo durante 1,75 h. La mezcla de reacción se concentró al vacío y el residuo bruto se purificó por cromatografía en gel de sílice (25% a 40% de EtOAc/Hex) para proporcionar el macrociclo 1-5, LCMS-ESI+ (m/z):
[M+H] calculado para C33H43N4O7: 607,31; encontrado: 607,46.
[0204] Paso 6. Preparación de 1-6. El macrociclo 1-5 (236 mg, 0,389 mmol) se disolvió en EtOH (20 ml) y EtOAc (5 ml). Se añadió Pd/C al 10% (56 mg) y se burbujeó H2 a través de la suspensión durante 4 min. La mezcla de reacción agitada se mantuvo bajo 1 atm de H2 durante 50 minutos antes de filtrarse sobre Celite y se concentró a vacío para proporcionar 1-6 que se llevó a cabo sin más purificación. LCMS-ESI+ (m/z): [M+H] calc. para C33H45N4O7: 609,33; encontrado: 609,34.
[0205] Paso 7. Preparación de 1-7. El compuesto 1-6 (aprox. 0,389 mmol) se trató con THF (10 ml) y LiOH (1,0 M en H2O, 10 ml, 10 mmol). La mezcla se agitó durante 15 h, luego se vertió en un embudo de separación que contenía 40 ml al 10% HCl acuoso. La capa acuosa se extrajo con DCM. Los extractos orgánicos combinados se secaron sobre MgSO4 anhidro, se filtraron y se concentraron a vacío para proporcionar ácido carboxílico 1-7 que se llevó a cabo sin purificación adicional. LCMS-ESI+ (m/z): [M+H] calc. para C32H43N4O7: 595,31; encontrado: 595,38.
[0206] Paso 8. Preparación del Ejemplo 1. El ácido carboxílico 1-7 (95 mg, 0,160 mmol) se trató con el Intermedio A9 (60 mg, 0,21 mmol), TBTU (62 mg, 0,19 mmol), DMAP (23 mg, 0,19 mmol), DCM (2 ml) y DIPEA (0,14 ml, 0,80 mmol). La mezcla de reacción se agitó durante 1 h, luego se agregaron más Intermedio A9 (35 mg, 0,12 mmol), TBTU (20 mg, 0,062 mmol) y DIPEA (0,14 mL, 0,80 mmol). Después de 34 minutos adicionales a temperatura ambiente, la mezcla de reacción se concentró al vacío. El residuo bruto se purificó por HPLC para proporcionar el Ejemplo 1 en forma de una sal TFA. Tiempo de retención de HPLC analítico: 8,89 min. LCMS-ESI+ (m/z): [M+H] calc. para C40H53F2N6O9S: 831,36; encontrado: 831,58, 1H-RMN (400 MHz, CD3OD) 8 9,36 (s, 1H), 7,79 (dd, J = 7,7, 1,9 Hz, 1H), 7,27 - 7,18 (m, 2H), 6,09 (t, J = 3,5 Hz, 1H), 5,90 (td, J = 55,9, 6,7 Hz, 1H), 5,00 (d, J = 7,5 Hz, 1H), 4,53- 4,37 (m, 2H), 4,33 (s, 1H), 4,12 (dd, J = 11,8, 3,8 Hz, 1H), 3,93 (s, 3H), 3,05 -2,89 (m, 2H), 2,84-2,70 (m, 1H), 2,49 (dd, J = 13,8, 6,2 Hz, 1H), 2,31-2,13 (m, 2H), 2,09 - 1,91 (m, 3H), 1,79 (dd, J = 28,6, 9,7 Hz, 2H), 1,68 - 1,46 (m, 5H), 1,46 -1,19 (m, 7H), 1,17 -1,09 (m, 2H), 1,09 - 1,02 (m, 11H), 0,63-0,45 (m, 2H).
Ejemplo 2. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilo)-clopropil)sulfonil]carbamoil} ciclopropil]-15-metoxi-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b -hexadecahidro-1H9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0207]

[0208] El Ejemplo 2 se preparó de manera similar al Ejemplo 1, sustituyendo al Intermedio A10 por el Intermedio A9 en el Paso 8. El Ejemplo 2 (107 mg) se aisló como una sal de TFA. Tiempo de retención de HPLC analítico: 8,85 min. LCMS-ESI+ (m/z): [M+H]+ calc. Para C41H55F2N6O9S: 845,37; encontrado: 845,67, 1H-RMN (400 MHz, CD3OD) 69,31 (s, 1H), 7,78 (d, J = 8,8 Hz, 1H), 7,20 (dt, J = 3,9, 2,6 Hz, 2H), 6,06 (t, J = 3,5 Hz, 1H), 5,87 (td, J = 55,8, 6,7 Hz, 1H), 5,00 (d, J = 7,5 Hz, 1H), 4,51 -4,38 (m, 2H), 4,34 (s, 1H), 4,11 (dd, J = 11,8, 3,8 Hz, 1H), 3,92 (s, 3H), 3,04 -2,85 (m, 1H), 2,85 - 2,67 (m, 1H), 2,49 (dd, J = 13,9, 6,4 Hz, 1H), 2,24 (ddd, J = 20,1, 12,0, 6,2 Hz, 2H), 2,08 - 1,90 (m, 3H), 1,90 - 1,66 (m, 2H), 1,66 - 1,46 (m, 10H), 1,46 - 1,20 (m, 6H)), 1,05 (s, 9H), 0,98 - 0,82 (m, 3H), 0,58 (q, J = 4,1 Hz, 1H), 0,55 -0,45 (m, 1H).
Ejemplo 3. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2S)-1-[(ciclopropilsulfonil) carbamoil)-2-(2,2-difluoroetil)ciclopropil]-15-metoxi-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23bhexadeca-hidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0209]

Ejemplo 3
[0210] El Ejemplo 3 se preparó de manera similar al Ejemplo 1, sustituyendo al Intermedio A7 por el Intermedio A9 en el Paso 8. El Ejemplo 3 (52 mg) se aisló como una sal de TFA. Tiempo de retención de HPLC analítico: 8,82 min. LCMS-ESI+ (m/z): [M+H] calc. Para C41H55F2N6O9S: 845,37; encontrado: 845,96, 1H-RMN (400 MHz, CD3OD) 69,18 (s, 1H), 7,78 (dd, J = 8,1, 1,5 Hz, 1H), 7,26 -7,16 (m, 2H), 6,07 (t, J = 3,6 Hz, 1H), 5,89 (tt, J = 56,5, 4,1 Hz, 1H), 4,99 (d, J = 7,5 Hz, 1H), 4,45 (dd, J = 11,1, 5,9 Hz, 2H), 4,32 (s, 1H), 4,11 (dd, J = 11,9, 3,8 Hz, 1H), 3,92 (s, 3H), 3,04 -2,86 (m, 2H), 2,86 -2,68 (m, 1H), 2,47 (dd, J = 13,8, 6,3 Hz, 1H), 2,34 -2,07 (m, 4H), 1,95 (dd, J = 11,0, 4,4 Hz, 1H), 1,79 (dd, J = 27,1, 9,7 Hz, 3H), 1,71 -1,47 (m, 6H), 1,47 -1,18 (m, 8H), 1,18 - 0,92 (m, 12H), 0,64 - 0,43 (m, 2H).
Ejemplo 4. Preparación de (1R, 4S, 4aR, 8S, 11S, 13R, 25aR)-8-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil) carbamoil]-2-(difluorometilo)ciclopropil]-17-metoxi-6,9-d-diox-2,3,4,4a,6,7,8,9,12,13,21,22,23,24,25,25ahexadecahidro-1H,11H-1,4:10,13-dimetanoquinoxalino[2,3-k][1,10,3,6]benzodioxadiaza-ciclononadecina-11 -carboxamida.
[0211]

[0212] Paso 1 - 3. Preparación de 4-1 y 4-2: se disolvió clorhidrato de prolina 1-2 (1,3 mmol) junto con 1:1, mezcla intermedia B1 y B2 (1,29 mmol) y DIPEA (1,0 ml, 5,7 mmol) en DMF (6,5 ml). Se agregó HATU (597 mg, 1,57 mmol) en una porción. La reacción se agitó durante 80 min a temperatura ambiente y se diluyó con NaHCO3 acuoso saturado (30 ml) y EtOAc (50 ml). Las fases se separaron y la fase orgánica se lavó con agua (30 ml) y salmuera (30 ml). La fase orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío hasta un residuo bruto que se purificó por cromatografía en gel de sílice para proporcionar un residuo (732 mg; LCMS-ESI+ (m/z): [M+H]+ calc. para Ca4H46ClN4Oy: 657,3; encontrado: 657,0). Una mezcla agitada de este residuo, PdCl2 (dppf)^CH2Cl2 (68 mg, 0,083 mmol) y viniltrifluoroborato de potasio (304 mg, 2,27 mmol) en EtOH (10 ml) se roció con Ar durante varios minutos. Se añadió trietilamina (330 ml, 2,35 mmol) y la mezcla se calentó a 75°C durante 60 min. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con EtOAc y se lavó con agua y salmuera. Los extractos orgánicos se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron al vacío para producir un residuo bruto que se purificó mediante cromatografía en gel de sílice (20% a 30% de AcOEt en hexanos) para proporcionar un aceite amarillo (676 mg; LCMS-ESI+ (m/z): [M+H]+ calculado para C36H49N4O7: 649,4; encontrado: 649,1). Este residuo (626 mg, 0,965 mmol) se disolvió en DCE (250 ml) y la solución se roció con Ar durante 15 min. Se añadió catalizador Zhan 1B (75 mg, 0,050 mmol) como una solución en DCE (10 ml) y la solución resultante se agitó a 85°C bajo Ar durante 1,5 h. La mezcla de reacción se concentró luego al vacío y el residuo resultante se purificó por cromatografía en gel de sílice (20% a 40% de EtOAc en hexanos) para proporcionar el intermedio 4-1 (LCMS-ESI+ (m/z): [M+H]+ calc. para C34H45N4O7: 621,3; encontrado: 621,1) y 4-2 (LCMS-ESI+ (m/z): [M+H]+ calculado para C34H45N4O7: 621,3; encontrado: 621,1).
[0213] Pasos 4 y 5: Preparación de 4-3: A una solución de 4-1 (205 mg, 0,330 mmol) en 1:1 EtOAc: EtOH (5 ml) se añadió Pd/C (10% en peso de Pd, 68 mg). El recipiente de reacción se purgó dos veces con H2 y se agitó a temperatura ambiente bajo 1 atm de H2 durante 3 h. La mezcla de reacción se filtró a través de una capa de Celite con EtOAc y se concentró a vacío para proporcionar un residuo bruto (188 mg, LCMS-ESI+ (m/z): [M+H]+ calc. Para C34H47N4O7: 623,3; encontrado: 623,2). Este residuo se disolvió en THF (4,3 ml) y H2O (1,7 ml). Se añadió L iO H ^ O (79 mg, 1,9 mmol) y la mezcla se agitó a temperatura ambiente durante 14,5 h. La reacción se detuvo con HCl acuoso 1 M (2 ml) y se diluyó con EtOAc (30 ml) y HCl acuoso 1 M (20 ml). Las fases se separaron y la fase acuosa se extrajo con EtOAc (30 ml). La fase orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar 4 1 que se usó directamente en el siguiente paso sin purificación adicional. LCMS-ESI-(m/z): [M-H]- calculado para C33H43N4O7: 607,3; encontrado: 607,0.
[0214] Paso 6: Preparación del Ejemplo 4: A una suspensión de ácido 4-3 (66,5 mg, 0,109 mmol) e Intermedio A9 (35 mg, 0,12 mmol) en MeCN (1,6 mL) se añadió DIPEA (80 mL, 0,46). mmol). A la solución resultante se le añadió HATU (52 mg, 0,137 mmol). La reacción se agitó a temperatura ambiente durante 60 min y se diluyó con EtOAc (15 ml), HCl acuoso 1 M (10 ml) y salmuera (5 ml). Las fases se separaron y la fase acuosa se extrajo con EtOAc (20 ml). La fase orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar un residuo bruto. La purificación por cromatografía en gel de sílice (15% a 30% de acetona en hexanos) proporcionó un residuo amorfo que se liofilizó en agua y MeCN para proporcionar el Ejemplo 4, Tiempo de retención de HPLC analítico: 8,95 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C41H55F2N6O9S: 845,4; encontrado: 845,2, 1H-RMN (300 MHz, CDCh) 810,28 (s, 1H), 7,82 (d, J = 9,1 Hz, 1H), 7,19 (dd, J = 9,1, 2,8 Hz, 1H), 7,11 (d, J = 2,3 Hz, 2H), 6,19 - 5,72 (m, 2H), 5,37 (d, J =
10,1 Hz, 1H), 4,48 (d, J = 10,1 Hz, 1H), 4,42 -4,24 (m, 3H), 4,05 (dd, J = 11,6, 3,7 Hz, 1H), 3,92 (s, 3H), 2,96 -2,81 (m, 2H), 2,73- 2,59 (m, 2H), 2,43- 2,30 (m, 1H), 2,20 - 2,04 (m, 3H), 1,96 - 1,70 (m, 3H), 1,70 - 0,96 (m, 27H).
Ejemplo 5. Preparación de (1R, 4S, 4aR, 8S, 11S, 13R, 25aR)-8-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilo -clopropil)sulfonil]carbamoil} ciclopropil]-17-metoxi-6,9-dioxo-2,3,4,4a,6,7,8,9,12,13,21,22,23,24,25,25a-hexadecano-1H,11H-1,4:10,13-dimetanoquinoxalino[2,3-k][1,10,3,6]benzodioxadiazaciclononadecina-11 -carboxamida.
[0215]

Ejemplo 5
[0216] El Ejemplo 5 (59,3 mg) se preparó de manera similar al Ejemplo 4, sustituyendo al Intermedio A10 por el Intermedio A9 en el Paso 6. Tiempo de retención de HPLC analítico: 8,86 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C42H57F2N6O9S: 859,4; encontrado: 859,3, 1H-RMN (400 MHz, CDCla) 89,85 (s, 1H), 7,83 (d, J = 9,1 Hz, 1H), 7,29 (s, 1H), 7,19 (dd, J = 9,1, 2,7 Hz, 1H), 7,12 (d, J = 2,7 Hz, 1H), 6,17 - 5,67 (m, 2H), 5,40 (d, J = 10,1 Hz, 1H), 4,53-4,26 (m, 4H), 4,04 (dd, J = 12,1,4,5 Hz, 1H), 3,93 (s, 3H), 2,95 -2,83 (m, 1H), 2,74 -2,60 (m, 2H), 2,46 -2,32 (m, 1H), 2,20 - 2,09 (m, J = 9,0 Hz, 2H), 2,00 -1,17 (m, 22H), 1,06 (s, 9H), 0,92 - 0,77 (m, 3H).
Ejemplo 6 Preparación de (1S, 4R, 4aS, 8S, 11S, 13R, 25aS)-8-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil) carbamoil]-2-(difluorometilo)ciclopropil]-17-metoxi-6,9-dioxo-2,3,4,4a,6,7,8,9,12,13,21,22,23,24,25,25a-hexadecahidro-1H,11H-1,4:10,13-dimetanoquinoxalino[2,3-k][1,10,3,6]benzodioxadiaza-cyclononadecine-11-carboxamida.
[0217]

[0218] Pasos 1-3: Preparación del Ejemplo 6: A una solución de 4-2 (160 mg, 0,26 mmol) en 1:1 EtOAc: EtOH (4 ml) se añadió Pd/C (10 % en peso de Pd, 55 mg). El recipiente de reacción se purgó dos veces con H2 y se agitó a temperatura ambiente bajo 1 atm de H2 durante 5,5 h. La mezcla de reacción se filtró a través de una capa de Celite con EtOAc y se concentró a vacío para proporcionar un residuo bruto (155 mg, LCMS-ESI+ (m/z): [M+H]+ calc. Para C34H47N4O7: 623,3; encontrado: 623,2). Este residuo se disolvió luego en THF (4,3 ml) y H2O (1,7 ml). Se añadió L iO H ^ O (64 mg, 1,5 mmol) y la mezcla se agitó a temperatura ambiente durante 14,5 h. La reacción se detuvo con HCl acuoso 1 M (2 ml) y se diluyó con EtOAc (30 ml) y HCl acuoso 1 M (20 ml). Las fases se separaron y la fase acuosa se extrajo con EtOAc (30 ml). La fase orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar un residuo (139 mg; LCMS-ESI+ (m/z): [M+H]+ calc. Para C33H45N4O7: 609,3; encontrado: 608,9) Utilizado directamente en el siguiente paso sin purificación adicional. A una suspensión del producto del paso anterior (54 mg, 0,089 mmol) y al Intermedio A9 (28,4 mg, 0,098 mmol) en MeCN (1,5 mL) se le añadió DIPEA (70 mL, 0,40 mmol). A la solución resultante se le añadió HATU (43,5 mg, 0,114 mmol). La reacción se agitó a temperatura ambiente durante 120 min y se diluyó con EtOAc (15 ml), HCl acuoso 1 M (10 ml), agua (10 ml) y salmuera (5 ml). Las fases se separaron y la fase acuosa se extrajo con EtOAc (20 ml). La fase orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar un residuo bruto. La purificación por cromatografía en gel de sílice (17% a 40% de acetona en hexanos) proporcionó un residuo amorfo que se liofilizó en agua y MeCN para proporcionar el Ejemplo
6 , Tiempo de retención de HPLC analítico: 8,85 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C41H55F2N6O9S: 845,4; encontrado: 845,2, 1H-RMN (300 MHz, CDCla) 89,80 (s, 1H), 9,05 (s, 1H), 7,88 - 7,78 (m, 1H), 7,24 - 7,17 (m, 2H), 6,95 (d, J = 9,9 Hz, 1H), 6,08 - 5,96 (m, 1H), 5,95 - 5,50 (m, 1H), 4,66 (dd, J = 8,3, 3,8 Hz, 1H), 4,41 - 4,30 (m, 2H), 4,00 (s, 1H)), 3,95 (s, 3H), 3,81 (dd, J = 10,5, 4,1 Hz, 1H), 3,12 - 2,86 (m, 2H), 2,79 (t, J = 6,1 Hz, 2H), 2,57 - 2,41 (m, 1H), 2,31 (d, J = 4,5 Hz, 1H), 2,13-1,91 (m, 3H), 1,87 - 0,96 (m, 28H), 0,93- 0,81 (m, 1H).
Ejemplo 7. Preparación de (1aR, 1bS, 5S, 8S, 10R, 22aR, 23aR)-5-terc-butil-N-[(1R, 2R)-1-[(cidopropNsulfoml)carbamoN)-2-(difluorometN)cidopropN]-14-metoxi-3,6-dioxo-1a,1b,3,4,5,6,9,10,18,19,20,21,22,22a, 23,23a-hexadecahidro-1H,8H-7,10-metanoddopopa[3',4']ddopenta[1',2':18,19][1,10,3,6]dioxadiazaddononadedno[11,12-b]quinoxalina-8-carboxamida.
[0219]

[0220] Paso 1: Preparación de 7-1: A una solución de clorhidrato de amina 1-2 (268 mg, 0,65 mmol) e intermedio B8 (0,54 mmol) se agregó iPR2NEt (0,43 ml, 2,46 mmol) seguido de HATU (271 mg, 0,713 mmol). La solución resultante se agitó durante 1,5 horas a temperatura ambiente y se diluyó con EtOAc (30 ml) y NaHCO3 acuoso saturado (20 ml). Las fases se separaron y la fase orgánica se lavó con H2O (2 x 20 ml), se secó sobre Na2SO4 anhidro, se filtró y se concentró al vacío. La purificación por cromatografía en gel de sílice (10% a 30% de acetona en hexanos) proporcionó 7-1, LCMS-ESI+ (m/z): [M+H]+ calc. para C3aH44ClN4Oy: 643,3; encontrado: 643,3.
[0221] Paso 2: Preparación de 7-2: Una mezcla agitada de 7-1 (155 mg, 0,24 mmol), PdCl2 (dppf)^CH2Cl2 (18 mg, 0,022 mmol) y vinilflorifluoroborato de potasio (84 mg, 0,63 mmol) en EtOH (3 ml) se roció con Ar durante 5 minutos. Se añadió trietilamina (90 pl, 0,64 mmol) y la mezcla se agitó a 70°C durante 65 min. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con EtOAc (30 ml) y se lavó con agua (30 ml). La fase orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar un residuo bruto que se purificó por cromatografía en gel de sílice (20% a 40% de EtOAc en hexanos) para proporcionar 7-2, LCMS-ESI+ (m/z): [M+H]+ calculado para C35H47N4O7: 635,3; encontrado: 635,2.
[0222] Paso 3: Preparación de 7-3: Vinil quinoxalina 7-2 (119 mg, 0,187 mmol) se disolvió en DCE (60 ml) y la solución se roció con Ar durante 10 min. Se añadió catalizador Zhan 1B (16 mg, 0,022 mmol) como una solución en DCE (1 ml) y la solución resultante se agitó a 85°C bajo Ar durante 45 minutos. La mezcla de reacción se concentró luego al vacío y se purificó por cromatografía en gel de sílice (20% a 40% de EtOAc en hexanos) para proporcionar 7-3, LCMS-ESI+ (m/z): [M+H]+ calculado para C33H43N4O7: 607,3; encontrado: 607,3.
[0223] Pasos 4 y 5: Preparación de 7-4: A una solución de 7-3 (74 mg, 0,12 mmol) en 1:1 EtOAc:EtOH (3 ml) se añadió Pd/C (10% en peso de Pd, 45 mg). El recipiente de reacción se purgó dos veces con H2 y se agitó a temperatura ambiente bajo 1 atm H2 durante 2 h. La mezcla de reacción se filtró a través de una capa de Celite con EtOAc y se concentró a vacío para proporcionar un residuo crudo utilizado directamente en el siguiente paso. (LCMS-ESI+ (m/z):
[M+H]+ calculado para C33H45N4O7: 609,3; encontrado: 608,9). Este residuo se disolvió en THF (1,5 ml), H2O (0,75 ml) y MeOH (0,75 ml). Se añadió L iO H ^ O (52 mg, 1,2 mmol) y la mezcla se agitó a 45°C durante 70 min. La reacción
se detuvo luego con HCl acuoso 1 M (1,1 ml) y se diluyó con CH2CI2 (20 ml) y HCl acuoso 0,3 M (20 ml). Las fases se separaron y la fase acuosa se extrajo con CH2Ch (30 ml). La fase orgánica combinada se secó sobre MgSO4 anhidro, se filtró y se concentró a vacío para dar 7-4. LCMS-ESI+ (m/z): [M+H]+ calculado para C32H43N4O7: 595,3; encontrado: 595,2.
[0224] Paso 6 : Preparación del Ejemplo 7: A una suspensión de ácido 7-4 (36,6 mg, 0,0615 mmol) y el Intermedio A9 (25 mg, 0,086 mmol) en MeCN (1,5 mL) se añadió DIPEA (56 j L, 0,32). mmol). A la solución resultante se le añadió HATU (35,5 mg, 0,093 mmol). La reacción se agitó a temperatura ambiente durante 90 min y se diluyó con EtOAc (20 ml) y HCl acuoso 0,2 M (20 ml). Las fases se separaron y la fase acuosa se extrajo con EtOAc (20 ml). La fase orgánica combinada se secó sobre Na2SO4 anhidro, se filtró y se concentró a vacío para proporcionar un residuo bruto. La purificación por cromatografía en gel de sílice (10% a 45% de acetona en hexanos) proporcionó un residuo amorfo que se liofilizó en agua y MeCN para proporcionar el Ejemplo 7. Tiempo de retención de HPLC analítico: 8,67 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C40H53F2N6O9S: 831,4; encontrado: 831,3, 1H-RMN (300 MHz, CDCh) 8 10,28 (s, 1H), 7,82 (d, J = 9,1 Hz, 1H), 7,20 (dd, J = 9,1, 2,8 Hz, 1H), 7,13 (d, J = 2,7 Hz, 1H), 7,04 (s, 1H), 6,20 - 5,72 (m, 2H), 5,45 (d, J = 9,8 Hz, 1H), 5,21 -5,10 (m, 1H), 4,41 (d, J = 9,8 Hz), 1H), 4,34 (dd, J = 10,9, 6,6 Hz, 1H), 4,20 (d, J = 12,0 Hz, 1H), 4,04 (dd, J = 11,8, 3,3 Hz, 1H), 3,94 (s, 3H), 3,05 -2,84 (m, 2H), 2,70 -2,55 (m, 2H), 2,47 -2,31 (m, 1H), 2,14 - 2,07 (m, 1H), 1,99 - 0,80 (m, 28H), 0,66 - 0,56 (m, 1H), 0,42 (dd, J = 13,0, 7,8 Hz, 1H).
Ejemplo 8. Preparación de (1aR, 1bS, 5S, 8S, 10R, 22aR, 23aR)-5-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilciclopropil)sulfonil]carbamoil} ciclopropil]-14-metoxi-3,6-dioxo-1a,1b,3,4,5,6,9,10,18,19,20,21,22,22a,23,23ahexadecahidro-1H,8H-7,10-metanociclopopa[3',4']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-8-carboxamida.
[0225]

Ejemplo 8
[0226] El Ejemplo 8 se preparó de manera similar al Ejemplo 7, sustituyendo al Intermedio A10 por el Intermedio A9 en el Paso 6 , Tiempo de retención de HPLC analítico: 8,79 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C41H55F2N6O9S: 845,4; encontrado: 845,2. 1H-RMN (300 MHz, CDCh) 89,85 (s, 1H), 7,82 (d, J = 9,0 Hz, 1H), 7,20 (dd, J = 9,1, 2,7 Hz, 1H), 7,13 (d, J = 2,7 Hz, 1H), 7,08 (s, 1H), 6,17 - 5,68 (m, 2H), 5,51 (d, J = 9,8 Hz, 1H), 5,17 (t, J = 5,9 Hz, 1H), 4,47 -4,33 (m, 2H), 4,24 (d, J = 11,9 Hz, 1H), 4,03 (dd, J = 11,9, 3,5 Hz, 1H), 3,94 (s, 3H), 3,05 - 2,88 (m, 1H), 2,72 -2,57 (m, 2H), 2,48 -2,33 (m, 1H), 2,11 -2,04 (m, 1H), 2,01 -1,84 (m, 2H), 1,82 -1,13 (m, 17H), 1,06 (s, 9H), 0,93-0,77 (m, 3H), 0,65 - 0,55 (m, 1H), 0,49 - 0,37 (m, 1H).
Ejemplo 9. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil)carbamoil)-2-(difluorometil)ciclopropil]-19,19-difluoro-15-metoxi-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0227]

[0228] Paso 1: Preparación de 9-1: Intermedio C4 (361 mg, 1,33 mmol), N-Boc-trans-4-hidroxi-L-prolina metil éster (489 mg, 2,00 mmol, productos químicos finos AIMS) y se tomó cesio carbonato (651 mg, 2,00 mmol) en CH3CN (5 ml) y se calentó a 80°C durante la noche. La reacción se enfrió a temperatura ambiente, se diluyó con acetato de etilo (15 ml) y se filtró a través de una capa de Celite. El residuo se lavó con acetato de etilo y el filtrado se concentró al vacío. El residuo se purificó mediante cromatografía en gel de sílice (0-100% de EtOAc/hex) para producir 9-1. LCMS-ESI+ (m/z): [M+H]+ calc. para C23H28F2N3O6: 480,19; encontrado: 480,30.
[0229] Paso 2: Preparación de 9-2: A una solución de 9-1 (392 mg, 0,82 mmol) en diclorometano (3 ml) se añadió lentamente trimetilsilil trifluorometanosulfonato de trimetilsililo (212 pl, 1,23 mmol) a 23°C a una temperatura de 23°C. Ar ambiente. Después de 20 minutos, la mezcla resultante se concentró al vacío para dar el intermedio 9-2 como una sal de ácido tríflico, que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C18H20F2N3O4: 380,14; encontrado: 380,22.
[0230] Paso 3: Preparación de 9-3: a una solución de 9-2 (432 mg, 0,82 mmol) en DMF (2 ml), se agregó el intermedio B5 (275 mg, 0,89 mmol), HATU (467 mg, 1,23 mmol) y iPR2NEt (0,71 ml, 4,09 mmol) a 23°C. Después de 40 horas, la mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con agua (50 ml) y salmuera (25 ml), se secaron sobre MgSO4 anhidro y se concentraron al vacío. El residuo resultante se purificó por cromatografía en gel de sílice (EtOAc al 0-100%/hex) para dar 9-3, LCMS-ESI+ (m/z): [M+H]+ calc. para C35H45F2N4O7: 671,32; encontrado: 671,51.
[0231] Paso 4: Preparación de 9-4: A una solución de 9-3 (300 mg, 0,45 mmol) en DCE desgasificado (100 ml) se agregó catalizador Zhan 1B (33 mg, 0,045 mmol) seguido de desgasificación adicional para 10 minutos. La solución resultante se sometió a reflujo bajo Ar durante 75 min. La mezcla de reacción se concentró luego al vacío y se purificó por cromatografía en gel de sílice con 0-100% de EtOAc/hex) para producir 9-4, LCMS-ESI+ (m/z): [M+H]+ calculado para C33H41F2N4O7: 643,29; encontrado: 643,44.
[0232] Pasos 5 y 6: Preparación de 9-5: A una solución de 9-4 (220 mg, 0,34 mmol) en EtOH (7 ml) se añadió Pd/C (10% en peso de Pd, 38 mg). La atmósfera de la reacción se reemplazó con H2 y la reacción se agitó a temperatura ambiente bajo 1 atm H2 durante la noche. La mezcla de reacción se filtró a través de una capa de Celite y se lavó con EtOH y se concentró a vacío para proporcionar un residuo bruto que se volvió a someter a las condiciones de reacción
durante 2 d adicionales. La mezcla de reacción se filtró a través de una capa de Celite y se lavó con EtOH y se concentró a vacío para proporcionar un residuo bruto que se usó posteriormente sin purificación adicional. (LCMS-ESI+ (m/z): [M+H]+ calculado para C33H43F2N4O7: 645,30; encontrado: 645,51). Este residuo se disolvió en Th F (3 ml), H2O (i ml) y MeOH (1 ml). Se añadió L iO H ^ O (71 mg, 1,7 mmol) y la mezcla se agitó a 23°C durante 2 h. Los disolventes se eliminaron al vacío y el residuo se recogió en acetato de etilo (10 ml) y 1 M HCl (10 ml). La capa acuosa se extrajo con acetato de etilo (10 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 anhidro y se concentraron a vacío para proporcionar un residuo de 9-5 que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C32H41F2N4O7: 631,29; encontrado: 631,46).
[0233] Paso 7: Preparación del Ejemplo 9: A una suspensión de 9-5 (56,8 mg, 0,090 mmol) y al Intermedio A9 (39,3 mg, 0,135 mmol) en MeCN (3 mL) se agregó HATU (55 mg, 0,144 mmol)) y DIPEA (78 ml, 0,45 mmol) a 23°C. Después de 20 min, la solución se purificó directamente mediante HPLC de fase inversa (columna Gemini 5u C18 110A, 50-100% ACN/H2O TFA al 0,1%) y se liofilizó para proporcionar la sal TFA del Ejemplo 9, Tiempo de retención de HPLC analítico: 8,87 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C40H51F4N6O9S: 867,33; encontrado: 867,45, 1H-RMN (400 MHz, CD3OD) 89,46 (s, 1H), 7,93 (d, J = 9,1 Hz, 1H), 7,33 (d, J = 2,8 Hz, 1H), 7,30 (d, J = 2,8 Hz, 1H), 7,28 (d, J = 2,7 Hz, 1H), 6,16 (t, J = 3,6 Hz, 1H), 5,89 (td, J = 55,7, 6,8 Hz, 1H), 4,93 (t, J = 9,8 Hz, 1H), 4,49 -4,37 (m, 2H), 4,32 (s, 1H), 4,13 (dd, J = 11,9, 3,9 Hz, 1H), 3,96 (d, J = 4,9 Hz, 3H), 3,04 - 2,91 (m, 1H), 2,64 - 2,51 (m, 1H), 2,64 -2,51 (m, 1H), 2,32 -2,15 (m, 2H), 2,11 - 1,90 (m, 4H), 1,87 - 1,62 (m, 4H), 1,61 - 1,36 (m, 4H), 1,37 - 1,23 (m, 4H), 1,14 -1,02 (m, 11H), 0,60 -0,47 (m, 2H).
Ejemplo 10. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilo-ciclopropil)sulfonil]carbamoil} ciclopropil]-19,19-difluoro-15-metoxi-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0234]

[0235] El Ejemplo 10 se preparó de manera similar al Ejemplo 9, sustituyendo al Intermedio A10 por el Intermedio A9 en el Paso 7. El Ejemplo 10 se aisló como una sal de TFA. Tiempo de retención de HPLC analítico: 8,91 min. LCMS-ESI+ (m/z): [M-H]+ calculado para C41H51F2N6O9S: 879,35; encontrado: 879,63, 1H-RMN (400 MHz, CD3OD) 89,43 (s, 1H), 7,93 (d, J = 9,1 Hz, 1H), 7,32 (d, J = 2,8 Hz, 1H), 7,30 (d, J = 2,8 Hz, 1H), 7,27 (d, J = 2,7 Hz, 1H), 6,15 (t, J = 3,6 Hz, 1H), 5,87 (td, J = 55,7, 6,9 Hz, 1H), 4,94 (d, J = 7,5 Hz, 1H), 4,51 -4,38 (m, 2H), 4,32 (s, 1H), 4,13 (dd, J = 11,9, 3,9 Hz, 1H), 3,96 (s, 3H), 2,65 -2,39 (m, 2H), 2,34 -2,14 (m, 2H), 2,12 -1,88 (m, 4H), 1,87 - 1,63 (m, 4H), 1,62 -1,43 (m, 8H), 1,62 - 1,43 (m, 2H), 1,09 - 1,03 (m, 10H), 0,96 - 0,87 (m, 2H), 0,54 (qd, J = 8,3, 4,9 Hz, 2H).
Ejemplo 11, Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilo-ciclopropil)sulfonil]carbamoil} ciclopropil]-15-fluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23bhexa-decahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0236]

[0237] Paso 1. Preparación de 11-1. El intermedio C3 (506 mg, 1,94 mmol) se combinó con el éster metílico de N-Boc-trans-4-hidroxi-L-prolina (524 mg, 2,13 mmol) y CS2CO3 (759 mg, 2,33 mmol) y luego se suspendió en MeCN (10 ml). La mezcla de reacción agitada se agitó a temperatura ambiente durante 72 h, luego se filtró sobre Celite y se concentró al vacío. El residuo crudo se purificó por cromatografía en gel de sílice para proporcionar 11-1, LCMS-ESI+ (m/z): [M-Boc+2H]+ calculado para C14H14CFN3O3: 326,07; encontrado: 326,68.
[0238] Paso 2. Preparación de 11-2. El carbamato 11-1 (409 mg, 0,960 mmol) se disolvió en DCM (10 ml) y se trató con HCl (4,0 M en dioxano, 5 ml, 20 mmol). Después de agitarse a temperatura ambiente durante 1,5 h, la mezcla de reacción se concentró a vacío para proporcionar clorhidrato de amina 11 - 2 que se llevó a cabo sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calculado para C14H14CFN3O3: 326,07; encontrado: 326,52.
[0239] Paso 3. Preparación de 11-3. Se combinaron clorhidrato de amina 11-2 (0,960 mmol) e Intermedio B3 (229 mg, 0,704 mmol) y se trataron con BEP (231 mg, 0,845 mmol), EtOAc (4,5 mL), NMP (0,5 mL) y DIPEA (0,61 mL, 3,5 mmol). La mezcla agitada se calentó a 50°C durante 2,5 h. Después de enfriarse a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc y se lavó sucesivamente con NaHCO3 acuoso saturado y salmuera. La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo bruto se purificó mediante cromatografía en gel de sílice para proporcionar la amida 11-3, LCMS-ESI+ (m/z): [M+H]+ calc. para C32H41CFN4O6: 631,27; encontrado: 631,98.
[0240] Paso 4. Preparación de 11-4. La cloroquinoxalina 11-3 (336 mg, 0,532 mmol) se trató con vinil-trifluoroborato de potasio (107 mg, 0,799 mmol), Pd(dppf)C^DCM (43 mg, 0,053 mmol), EtOH (5 ml) y Et3N (0,11 mL, 0,80 mmol). La mezcla de reacción se agitó a reflujo durante 3,5 h, luego se enfrió a temperatura ambiente y se diluyó con EtOAc. La fase orgánica se lavó con agua y salmuera, luego se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo crudo se purificó por cromatografía en gel de sílice para proporcionar vinilquinoxalina 11-4. LCMS-ESI+ (m/z): [M+H]+ calc. para C34H44FN4O6: 623,32; encontrado: 623,45.
[0241] Paso 5, Preparación de 11-5, La vinilquinoxalina 11-4 (191 mg, 0,307 mmol) se disolvió en DCE (61 ml) y se trató con Zhan Catalyst-1B (21 mg, 0,031 mmol). La mezcla se desgasificó con N2 burbujeante durante 20 min, se calentó a reflujo durante 1,5 h. La mezcla de reacción se concentró al vacío y el residuo bruto se purificó por cromatografía en gel de sílice para proporcionar el macrociclo 11-5, LCMS-ESI+ (m/z): [M+H]+ calc. para C32H40FN4O6: 595,29; encontrado: 595,46.
[0242] Pasos 6 y 7. Preparación de 11-6. El macrociclo 11-5 (155 mg, 0,261 mmol) se disolvió en EtOH (20 ml) y EtOAc (5 ml). Se añadió Pd/C (10% en peso de Pd, 47 mg) y se burbujeó H2 a través de la suspensión durante 4 min.
La mezcla de reacción agitada se mantuvo bajo 1 atm de H2 durante 52 minutos antes de filtrarse sobre Celite y concentrar al vacío. Este residuo se usó posteriormente sin purificación adicional. (LCMS-ESI+ (m/z): [M+H]+ calculado para C32H42FN4O6: 597,31; encontrado: 597,36). Este residuo (0,261 mmol teórico) se trató con THF (10 ml) y LiOH (1,0 M en H2O, 10 ml, 10 mmol). La mezcla se agitó durante 12,5 h, luego se vertió en un separador que contenía 40 ml de HCl acuoso al 10%. La capa acuosa se extrajo 3x con DCM. Los extractos orgánicos combinados se secaron sobre MgSO4 anhidro, se filtraron y se concentraron a vacío para proporcionar ácido carboxílico 11-6 que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C31H40FN4O6: 583,29; encontrado: 583,29.
[0243] Paso 8. Preparación del Ejemplo 11. El ácido carboxílico 11-6 (110 mg, 0,189 mmol) se trató con el Intermedio A10 (86 mg, 0,28 mmol), TBTU (73 mg, 0,23 mmol), DMAP (28 mg, 0,23 mmol), DCM (2 ml) y DIPEA (0,16 ml, 0,95 mmol). La mezcla de reacción se agitó durante 2,5 h, seguido de concentración al vacío. El residuo bruto se purificó por HPLC para proporcionar el Ejemplo 11 en forma de una sal TFA. Tiempo de retención de HPLC analítico: 9,05 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C40H52F3N6O8S: 833,35; encontrado: 833,17, 1H-RMN (400 MHz, CD3OD) 6 9,31 (s, 1H), 7,94 (dd, J = 9,1, 5,8 Hz, 1H), 7,47 (dd, J = 9,6, 2,7 Hz, 1H), 7,40 (td, J = 8,8, 2,8 Hz, 1H), 6,08 (t, J = 3,6 Hz, 1H), 5,87 (td, J = 55,7, 6,7 Hz, 1H), 4,99 (d, J = 7,6 Hz, 1H), 4,51 -4,38 (m, 2H), 4,33 (s, 1H), 4,12 (dd, J = 11,9, 3,8 Hz, 1H), 3,08 -2,91 (m, 1H), 2,89 -2,73 (m, 1H), 2,50 (dd, J = 13,9, 6,6 Hz, 1H), 2,25 (ddd, J = 18,5, 12,7, 5,4 Hz, 2H), 2,10 - 1,92 (m, 3H), 1,92 - 1,72 (m, 2H), 1,72 - 1,15 (m, 15H)), 1,06 (d, J = 10,1 Hz, 10H), 0,97 - 0,88 (m, 2H), 0,58 (q, J = 4,1 Hz, 1H), 0,55 - 0,45 (m, 1H).
Ejemplo 12. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil)carbamoil)-2-(difluorometil)ciclopropil]-15-fluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiaza-ciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0244]

[0245] El Ejemplo 12 se preparó de manera similar al Ejemplo 11, sustituyendo al Intermedio A9 por el Intermedio A10 en el Paso 8, Después de la purificación por HPLC preparativa, se aisló la sal de TFA del Ejemplo 12 (24 mg). Tiempo de retención de HPLC analítico: 8,95 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C39H50F3N6O8S: 819,34; encontrado: 819,34, 1H-RMN (400 MHz, CD3OD) 69,35 (s, 1H), 7,94 (dd, J = 9,1, 5,8 Hz, 1H), 7,46 (dd, J = 9,6, 2,7 Hz, 1H), 7,39 (td, J = 8,7, 2,8 Hz, 1H), 6,08 (t, J = 3,5 Hz, 1H), 5,90 (td, J = 55,9, 6,8 Hz, 1H), 4,99 (d, J = 7,5 Hz, 1H), 4,50 -4,36 (m, 2H), 4,32 (s, 1H), 4,12 (dd, J = 11,8, 3,8 Hz, 1H), 3,05 -2,91 (m, 2H), 2,88 -2,69 (m, 1H), 2,49 (dd, J = 14,1, 6,3 Hz, 1H), 2,33-2,11 (m, 2H), 2,11 - 1,91 (m, 3H), 1,84 (dd, J = 11,6, 7,1 Hz, 1H), 1,75 (d, J = 14,6 Hz, 1H), 1,70 -1,15 (m, 12H), 1,15 -1,09 (m, 2H), 1,06 (d, J = 11,2 Hz, 11H), 0,57 (q, J = 4,2 Hz, 1H), 0,54 - 0,45 (m, 1H).
Ejemplo 13. Preparación de (1aS, 2aR, 6S, 9S, 11R, 20E, 23aR, 23bS)-6-terc-butil-15-ciano-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilciclopropil)sulfonil]carbamoil} ciclopropil]-19,19-difluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,22,23,23a,23b-tetradecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazacy-clononadecino[11,12-b]quinoxalina-9-carboxamida.
[0246]

[0247] Paso 1: Preparación de 13-1: Intermedio C5 (213 mg, 0,8 mmol), N-Boc-trans-4-hidroxi-L-prolina metil éster (394 mg, 1,2 mmol) y carbonato de cesio (391 mg, 1,2 mmol) se combinaron en CH3CN (4 ml) y se calentaron a 80°C durante 3 h. La reacción se enfrió a temperatura ambiente, se diluyó con acetato de etilo (15 ml) y se filtró a través de una capa de Celite. La almohadilla de Celite se lavó con acetato de etilo y el filtrado se concentró al vacío. El residuo resultante se purificó por cromatografía en gel de sílice para producir 13-1. LCMS-ESI+ (m/z): [M+H]+ calc. para C23H25F2N4O5: 475,47; encontrado: 475,10.
[0248] Paso 2: Preparación de 13-2: A una solución de 13-1 (300 mg, 0,63 mmol) en DCM (2 ml) se añadió lentamente HCl en dioxano (4 M, 1 ml, 4 mmol) a temperatura ambiente. Después de 2,5 h, la mezcla resultante se concentró al vacío para dar prolina 13-2 en forma de una sal de HCl, que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calculado para C18H17F2N4O3: 375,35; encontrado: 375,10.
[0249] Paso 3: Preparación de 13-3: a una solución de 13-2 (320 mg, 0,8 mmol) en DMF (2 ml), se añadió el intermedio B5 (275 mg, 0,89 mmol), HATU (467 mg, 1,23 mmol)) y iPR2NEt (0,71 ml, 4,09 mmol) a temperatura ambiente. Después de 16 h, la mezcla de reacción se diluyó con agua (50 ml) y se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas combinadas se lavaron con agua (50 ml), salmuera (25 ml), se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo resultante se purificó por cromatografía en gel de sílice para dar 13-3. LCMS-ESI+ (m/z):
[M+H]+ calculado para C35H41F2N5O6: 665,73; encontrado: 665,93.
[0250] Paso 4: Preparación de 13-4: A una solución de dieno 13-3 (290 mg, 0,43 mmol) en DCE desgasificado (80 ml) se agregó catalizador Zhan 1B (32 mg, 0,043 mmol) seguido de desgasificación adicional durante 10 min. La solución resultante se sometió a reflujo bajo Ar durante 2 h. La mezcla de reacción se enfrió luego a temperatura ambiente, se concentró al vacío y se purificó por cromatografía en gel de sílice para producir el macrociclo 13-4. LCMS-ESI+ (m/z):
[M+H]+ calculado para C33H37F2N5O6: 637,67; encontrado: 637,95.
[0251] Pasos 5 y 6 : Preparación del Ejemplo 13: A una solución de 13-4 (200 mg, 0,31 mmol) en THF (1 ml), se añadió 1 N LiOH (1 ml) y la mezcla se agitó a 23°C durante 2 h. Los disolventes se eliminaron al vacío y el residuo se recogió en EtOAc (10 ml) y 1 M HCl (10 ml). La capa acuosa se extrajo con EtOAc (10 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 anhidro y se concentraron a vacío para proporcionar un residuo que se usó posteriormente sin purificación adicional. A una suspensión de este residuo (50 mg, 0,08 mmol) e Intermedio A10 (36 mg, 0,12 mmol) en MeCN (1 mL) se añadió HATU (46 mg, 0,12 mmol) y DIPEA (70 mL, 0,4 mmol) a temperatura ambiente. Después
de 1 h, la solución se purificó directamente mediante HPLC de fase inversa y se liofilizó para proporcionar la sal TFA del Ejemplo 13. Tiempo retención de HPLC analítico: 8,55 min. LCMS-EST (m/z): [M+H]+ calculado para C41H47F4N7O8S: 873,91; encontrado: 873,89, 1H-RMN (400 MHz, CD3OD) 89,38 (s, 1H), 8,30 (d, 1H), 8,18 (d, 1H), 7,90 (dd, 1H), 6,30-6,15 (m, 3H), 5,83 (m, 1H), 4,93 (d, 1H), 4,61 (d, 1H), 4,45 -4,38 (m, 2H), 4,12 (m, 1H), 2,55 -2,48 (m, 2H), 2,28 - 2,18 (m, 3H)), 2,01 - 1,90 (m, 3H), 1,79 (m, 1H), 1,59 - 1,33 (m, 10H), 1,05 - 0,91 (m, 11H), 0,88 (m, 2H). 0,62 -0,47 (m, 2H).
Ejemplo 14. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-15-ciano-N-[(1R, 2R)-2-(difluorometil)-1 -{[(1 -metilciclopropil)sulfonil]carbamoil} ciclopropil]-19,19-difluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0252]

[0253] Paso 1: Preparación del Ejemplo 14: A una solución del Ejemplo 13 (88 mg, 0,1 mmol) en EtOH (1 ml) se añadió Pd/C (10% en peso de Pd, 20 mg). El recipiente de reacción se purgó dos veces con H2 y se agitó a temperatura ambiente bajo 1 atm de H2 durante 6 h. La mezcla de reacción se filtró a través de una capa de Celite y el filtrado se concentró al vacío. El material bruto se redisolvió en dioxano (2,5 ml) y se trató con DDQ (34 mg, 0,15 mmol). Después de 1 h, la solución se purificó directamente mediante HPLC de fase inversa y se liofilizó para proporcionar la sal TFA del Ejemplo 14. Tiempo de Retención de HPLC Analítico: 8,64 min. LCMS-ESI+ (m/z): [M+H]+ calculado para C41H49F4N7O8S: 875,93; encontrado: 875,98, 1H-RMN (400 MHz, CD3OD) 88,32 (s, 1H), 8,20 (d, 1H), 7,90 (d, 1H), 6,90 (d, 1H), 6,17 (d, 1H), 5,87 (m, 1H)), 4,90 (m, 1H), 4,45 (m, 2H), 4,28 (m, 1H), 4,12 (m, 2H), 3,70-3,50 (m, 3H), 2,55 -2,48 (m, 2H), 2,28 -2,18 (m, 3H), 2,01 - 1,90 (m, 3H), 1,79 (m, 1H), 1,59 - 1,33 (m, 10H), 1,05 - 0,91 (m, 11H), 0,89 (m, 2H). 0,57 -0,51 (m, 2H).
Ejemplo 15. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil)carbamoil)-2-(difluorometil)ciclopropil]-15,16,19,19-tetrafluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0254]

[0255] Paso 1: Preparación de 15-1: el intermedio C6 (575 mg, 2,22 mmol), el intermedio D1 (1,55 g, 3,34 mmol) y el carbonato de cesio (2 g, 6,14 mmol) se combinaron en NMP (10 ml) y Se calienta a 70°C durante la noche. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con acetato de etilo (50 ml), se lavó con agua (100 ml), salmuera (50 ml) y se secó sobre MgSO4 anhidro. Después de la filtración, la solución resultante se concentró al vacío. El residuo resultante se purificó mediante cromatografía en gel de sílice para producir 15-1, LCMS-ESI+ (m/z): [M+Na]+ calculado para C22H23F4N3NaO5: 508,15; encontrado: 508,2.
[0256] Paso 2: Preparación de 15-2: A una solución de 15-1 (392 mg, 0,82 mmol) en diclorometano (3 ml) se le añadió 4n HCl en dioxano (3 ml). La mezcla resultante se dejó agitar durante la noche a temperatura ambiente y se concentró al vacío para dar 15-2 en forma de una sal de HCl, que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C17H16F4N3O3: 386,11; encontrado: 386,1,
[0257] Paso 3: Preparación de 15-3: A una solución de 15-2 (393 mg, 0,93 mmol) en DMF (3 ml) se agregó el intermedio B5 (240 mg, 0,78 mmol), HATU (324 mg, 0,853 mmol) y iPR2NEt (1,35 mL, 7,76 mmol) a 23°C. Después de 18 horas, la mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las capas orgánicas combinadas se lavaron con agua (50 ml) y salmuera (50 ml), se secaron sobre MgSO4 anhidro y se concentraron al vacío. El residuo resultante se purificó mediante cromatografía en gel de sílice para dar 15-3. LCMS-ESI+ (m/z): [M+H]+ calc. para C34H41F4N4O6: 677,30; encontrado: 677,04.
[0258] Paso 4: Preparación de 15-4: A una solución de 15-3 (450 mg, 0,665 mmol) en DCE desgasificado (133 ml) se le añadió catalizador Zhan 1B (52 mg, 0,067 mmol). La solución resultante se sometió a reflujo bajo N2 durante 3 horas. La mezcla de reacción se concentró luego al vacío y se purificó por cromatografía en gel de sílice para producir 15-4. LCMS-ESI+ (m/z): [M+H]+ calculado para C32H37F4N4O6: 649,25; encontrado: 649,2.
[0259] Pasos 5 y 6 : Preparación de 15-5: A una solución de 15-4 (270 mg, 0,42 mmol) en EtOH (8 ml) se añadió Pd/C (10% en peso de Pd, 270 mg). La atmósfera de la reacción se reemplazó con H2 y la reacción se agitó a temperatura ambiente bajo 1 atm H2 durante 3 h. La mezcla de reacción se filtró a través de una capa de Celite y se lavó con EtOH y se concentró a vacío para proporcionar un residuo bruto que se usó posteriormente sin purificación adicional. (LCMS
ESI+ (m/z): [M+H]+ calculado para C32H37F4N4O6: 651,28; encontrado: 651,3). Este residuo se disolvió en THF (2 ml) y MeOH (1 ml). Se añadió una solución acuosa de hidróxido de litio 2 N (1 ml) y la mezcla se agitó a 23°C durante 2 h. Los disolventes se eliminaron al vacío y el residuo se recogió en acetato de etilo (10 ml) y 1 M HCl (10 ml). La capa acuosa se extrajo con acetato de etilo (10 ml). Las capas orgánicas combinadas se secaron sobre MgSO4 anhidro y se concentraron a vacío para proporcionar un residuo de 15-5 que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calculado para C31H37F4N4O6: 637,26; encontrado: 637,3).
[0260] Paso 7: Preparación del Ejemplo 15: A una suspensión de 15-5 (37 mg, 0,058 mmol) y al Intermedio A9 (21,2 mg, 0,073 mmol) en DMF (3 ml) se añadió HATU (28 mg, 0,73 mmol)) y DiPEA (126 ml, 0,73 mmol) a 23°C. La mezcla de reacción T se dejó agitar durante la noche, luego se purificó por HPLC y se liofilizó para proporcionar la sal TFA del Ejemplo 15. Tiempo de Retención de HPLC analítico: 8,87 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C39H47F6N6O8S: 873,31; encontrado: 873,09, 1H-RMN (400 MHz, CDCla) 8 10,39 (s, 1H), 7,88 (dd, J = 9,4, 8,8 Hz, 1H), 7,61 (dd, J = 10,3, 7,7 Hz, 1H), 6,73 (s, 1H), 6,15 (s, 1H), 5,97 (td, J = 55,5, 6,9 Hz, 1H), 5,23 (d, J = 9,5 Hz, 1H), 4,90 (d, J = 7,4 Hz, 1H), 4,50 (d, J = 12,2 Hz, 1H), 4,37 -4,20 (m, 2H), 4,07 (dd, J = 11,8, 3,6 Hz, 1H), 2,94 (td, J = 8,3, 4,1 Hz, 1H) 2,62 - 2,25 (m, 2H), 2,62 - 2,25 (m, 2H), 2,29 - 2,08 (m, 2H), 2,07 - 1,69 (m, 6 H), 1,70 - 1,50 (m, 2H), 1,51 -1,15 (m, 6 H), 1,07 (s, 9H), 1,05 - 0,77 (m, 2H), 0,57 - 0,46 (m, 1H), 0,46 - 0,35 (m, 1H).
Ejemplo 16. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil)carbamoil)-2-(difluorometil)ciclopropil]-15-(difluorometoxi)-19,19-difluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0261]

[0262] Paso 1: Preparación de 16-1: A una solución de Intermedio C7 (425 mg, 1,39 mmol) en MeCN (5 mL) se le añadió carbonato de cesio (906 mg, 2,78 mmol) y N-Boc-trans-4 -hidroxi-L-prolina metil éster (410 mg, 1,67 mmol). La mezcla resultante se dejó agitar 40 ha temperatura ambiente y se diluyó con acetato de etilo (25 ml), se lavó con agua (15 ml) y salmuera (15 ml). La solución resultante se secó luego sobre MgSO4 anhidro y se concentró al vacío. El residuo resultante se purificó mediante cromatografía en gel de sílice para producir quinoxalina 16-1 sustituida. LCMS-ESI+ (m/z): [M+Na]+ calculado para C23H25F4N3NaO6: 538,16; encontrado: 537,93.
[0263] Paso 2: Preparación de 16-2: A una solución de quinoxalina sustituida 16-1 (590 mg, 1,14 mmol) en DCM (5 ml) se le añadió 4N HCl en dioxano (5 ml). La mezcla resultante se dejó agitar durante la noche a temperatura ambiente, luego se concentró al vacío para proporcionar 16-2, que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C18H18F4N3O4: 416,12; encontrado: 415,96.
[0264] Paso 3: Preparación de 16-3: A una solución de clorhidrato de prolina 16-2 (420 mg, 0,93 mmol) en DMF (3 ml) se agregó el intermedio B5 (240 mg, 0,78 mmol), HATU (324 mg, 0,853 mmol) y iPR2NEt (1,35 ml, 7,76 mmol) a 23°C. Después de 18 h, la mezcla de reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 25 ml). Las capas orgánicas combinadas se lavaron con agua (50 ml) y salmuera (50 ml), se secaron sobre MgSO4 anhidro y se concentraron al vacío. El residuo resultante se purificó mediante cromatografía en gel de sílice para dar la amida 16 3, LCMS-ESI+ (m/z): [M+H]+ calc. para C35H43F4N4O7: 707,3; encontrado: 707,3.
[0265] Paso 4: Preparación de 16-4: A una solución de prolina amida 16-3 (350 mg, 0,49 mmol) en DCE desgasificado (100 ml) se añadió catalizador Zhan 1B (50 mg, 0,0,65 mmol). La solución resultante se sometió a reflujo bajo N2 durante 1 h. La mezcla de reacción se concentró luego al vacío y se purificó por cromatografía en gel de sílice para producir olefina 16-4. LCMS-ESI+ (m/z): [M+H]+ calc. para C33H39F4N4O7: 679,3; encontrado: 679,2.
[0266] Paso 5: Preparación de 16-5: A una solución de olefina 16-4 (220 mg, 0,32 mmol) en EtOH (25 ml) se añadió Pd/C (10% en peso de Pd, 220 mg). La atmósfera de la reacción se reemplazó con H2 y la reacción se agitó a temperatura ambiente bajo 1 atm H2 durante 3 h. La mezcla de reacción se filtró a través de una capa de Celite que se lavó con EtOH y los orgánicos combinados concentrados al vacío. El residuo resultante se purificó por HPLC para dar 16-5. LCMS-ESI+ (m/z): [M+H]+ calc. para C33H41F4N4O7: 681,3; encontrado: 681,3.
[0267] Paso 6 : Preparación de 16-6. El éster de prolina 16-5 se disolvió en THF (2 ml) y MeOH (1 ml). Se añadió una solución acuosa 2 N de LiOH (1 ml) y la mezcla se agitó a 23°C durante 1 h. Los disolventes se eliminaron al vacío y el residuo se recogió en acetato de etilo (10 ml) y 1 M HCl (10 ml). Después de la extracción con acetato de etilo (10 ml), las capas orgánicas combinadas se secaron sobre MgSO4 anhidro y se concentraron al vacío para proporcionar un residuo de 16-6 que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C32H39F4N4O7: 667,3; encontrado: 667,3.
[0268] Paso 7: Preparación del Ejemplo 16: a una suspensión de ácido carboxílico 16-6 (67,9 mg, 0,102 mmol) y al Intermedio A9 (37 mg, 0,128 mmol) en DMF (1 ml) se le añadió HATU (49 mg, 0,128 mmoles) y DIPEA (177 ml, 1,02 mmoles) a 23°C. La mezcla de reacción se dejó agitar durante la noche, luego se purificó por HPLC y se liofilizó para proporcionar la sal de TFA del Ejemplo 16, Tiempo de Retención de HPLC analítico: 8,75 min. LCMS-ESI+ (m/z):
[M+H]+ calc. para C40H49F6N6O9S: 903,32; encontrado: 903,14, 1H-RMN (400 MHz, CDCh) 8 10,22 (s, 1H), 7,95 -7,77 (m, 2H), 7,57 (dd, J = 9,1, 2,4 Hz, 1H), 7,06 (s, 1H), 6,77 (t, J = 72,9 Hz, 1H), 6,16 (s, 1H), 5,92 (td, J = 55,7, 7,2 Hz, 1H), 5,26 (d, J = 9,2 Hz, 1H), 4,89 (d, J = 7,5 Hz, 1H), 4,53 (d, J = 11,7 Hz, 1H), 4,33 (dd, J = 11,0, 6,2 Hz, 1H), 4,26 (d, J = 9,1 Hz, 1H), 4,08 (dd, J = 11,7, 3,7 Hz, 1H), 2,98 -2,78 (m, 1H), 2,57 -2,27 (m, 2H), 2,25 -2,12 (m, 1H), 2,12 -2,04 (m, 1H), 2,03-1,94 (m, 3H), 1,94 -1,81 (m, 2H), 1,82 - 1,66 (m, 2H), 1,64 - 1,42 (m, 3H), 1,42 -1,18 (m, 6 H), 1,07 (s, 9H), 1,05 - 0,93 (m, 2H), 0,58 - 0,46 (m, 1H), 0,46 - 0,36 (m, 1H).
Ejemplo 17, Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil)carbamoil)-2-(difluorometil)ciclopropil]-19,19-difluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexa-decahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiaza-ciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0269]


[0270] Paso 1. Preparación de 17-1: una mezcla que contiene el Intermedio C8 (0,94 g, 3,92 mmol), N-Boc-trans-4-hidroxi-L-prolina metil éster (1,48 g, 4,71 mmol) y CS2CO3, (1,92 g, 5,88 mmol) en MeCN (10 mL) se agitó vigorosamente a 85°C bajo una atmósfera de Ar durante 2 h. La reacción se filtró luego a través de una capa de Celite y el filtrado se concentró al vacío. El material bruto se purificó por cromatografía en gel de sílice para proporcionar 17 1. LCMS-ESI+ (m/z): [M+H]+ calc. para C22H25F2N3O5: 449,2; encontrado: 450,7.
[0271] Paso 2. Preparación de 17-2: A una solución de quinoxalina sustituida 17-1 en DCM (10 ml) se le añadió ácido clorhídrico en dioxano (4 M, 25 ml, 98,4 mmol) y la reacción se agitó a temperatura ambiente durante 5 h. La reacción bruta se concentró al vacío para dar 17-2 que se usó en etapas posteriores sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calculado para C17H17F2N3O5: 349,1; encontrado: 350,1.
[0272] Paso 3. Preparación de 17-3: A una solución de sal de clorhidrato de prolina 17-2 (1,00 g, 2,59 mmol) en DMF (26 ml), se agregó Intermedio B5 (0,82 g, 2,85 mmol) en DMF, HATU (1,18 g, 3,11 mmol) y DIPEA (2,48 mL, 14,26 mmol) a 23°C. Después de 40 h, la reacción se diluyó con agua (50 ml) y se extrajo con acetato de etilo (3 x 20 ml). Las capas orgánicas combinadas se lavaron con agua (50 ml) y salmuera (25 ml), se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El material bruto se purificó mediante cromatografía en gel de sílice para proporcionar 17 3, LCMS-ESI+ (m/z): [M+H]+ calc. para C34H42F2N4O6: 640,31; encontrado: 641,36.
[0273] Paso 4. Preparación de 17-4: A una solución de dieno 17-3 (1,38 g, 2,25 mmol) en DCE desgasificado (430 mL) se añadió catalizador Zhan 1B (158 mg, 0,215 mmol) y se desgasificó para una 1 h adicional La mezcla de reacción resultante se sometió a reflujo bajo Ar durante 1 h, se enfrió a temperatura ambiente y se concentró al vacío. El material bruto se purificó mediante cromatografía en gel de sílice para proporcionar el macrociclo 17-4, LCMS-ESI+ (m/z):
[M+H]+ calc. para C32H38F2N4O6: 612,28; encontrado: 613,31.
[0274] Paso 5. Preparación de 17-5: A una solución de macrociclo 17-4 (1,18 g, 1,92 mmol) en EtOH (7 ml) se añadió Pd/C (10% en peso de Pd, 500 mg). El recipiente de reacción se purgó dos veces con H2 y se agitó a temperatura ambiente bajo 1 atm H2 durante la noche. La mezcla de reacción se calentó luego a 50°C y se agitó durante 24 h adicionales. La mezcla de reacción se filtró a través de una capa de Celite y se concentró al vacío. El material bruto se redisolvió en dioxano (25 ml) y se trató con DDQ (525 mg, 2,31 mmol). Después de 1 h, el disolvente se eliminó al vacío y el residuo resultante se purificó por cromatografía en gel de sílice para proporcionar el macrociclo 17-5. LCMS-ESI+ (m/z): [M+H]+ calc. para C32H40F2N4O6: 614,29; encontrado: 615,17).
[0275] Paso 6. Preparación de 17-6: se disolvió el macrociclo 17-5 (1,0 g, 1,63 mmol) en THF (25 ml). Se añadió LiOH (1,0 M, 25 ml, 25 mmol) y la mezcla de reacción se agitó durante 5 horas a temperatura ambiente. La reacción se concentró al vacío y luego se diluyó con acetato de etilo (100 ml) y 1 M HCl (60 ml). Las capas se separaron y la capa acuosa se extrajo con acetato de etilo (50 ml). La capa orgánica combinada se secó sobre Na2SO4 anhidro y se concentró a vacío para proporcionar 17-6 como un residuo que se usó sin purificación adicional. LCMS-ESI+ (m/z):
[M+H]+ calc. para C31H38F2N4O6: 600,28; encontrado: 601,16).
[0276] Paso 7. Preparación del Ejemplo 17: A una suspensión de ácido carboxílico 17-6 (350 mg, 0,583 mmol) e Intermedio A10 (259 mg, 0,850 mmol) en MeCN (7 ml) se añadió HATU (323). mg, 0,850 mmol) y DIPEA (542 ml, 3,11 mmol) a 23°C. Después de 45 minutos, la solución se concentró y se purificó por HPLC de fase inversa, luego por cromatografía en gel de sílice. Se añadió TFA y la muestra se liofilizó para proporcionar la sal de TFA del Ejemplo 17, Tiempo de retención de HPLC analítico: 8,77 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C40H51F4N6O8S: 851,93; encontrado: 851,31, 1H-RMN (400 MHz, CDCla) 69,83 (s, 1H), 8,13 (d, 1H), 7,86 (d, 1H), 7,77 (dd, 1H), 7,67 (dd, 1H), 7,17 (s, 1H), 6,22 (t, 1H), 5,91 (td, 1H), 4,89 (d, 1H), 4,52 (d, 1H), 4,38 -4,33 (m, 1H), 4,26 (d, 1H), 4,08 (dd, 1H), 2,51 - 2,34 (m, 3H), 2,10 -1,94 (m, 5H), 1,87 - 1,29 (m, 15H), 1,08 -1,01 (m, 10H), 0,87 (m, 2H), 0,53- 0,50 (m, 1H), 0,44 -0,43 (m, 1H).
Ejemplo 18. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilo-ciclopropil)sulfonil]carbamoil} ciclopropil]-19,19-difluoro-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23, 23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0277]

[0278] El Ejemplo 18 se preparó de manera similar al Ejemplo 17, sustituyendo al Intermedio A9 por el Intermedio A10 en el Paso 7. El Ejemplo 18 (234 mg) se aisló como una sal de TFA. Tiempo de retención de HPLc analítico: 8,87 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C39H49F4N6O8S: 837,33; encontrado: 837,26, 1H-RMN (400 MHz, CDCh) 6 10,21 (s, 1H), 8,13 (d, 1H), 7,86 (d, 1H), 7,77 (dd, 1H), 7,67 (dd, 1H), 7,24 (s, 1H), 6,22 (t, 1H), 5,91 (dt, 1H), 5,36 (d, 1H), 4,89 (d, 1H), 4,51 (d, 1H), 4,37 -4,33 (m, 1H), 4,27 (d, 1H), 4,08 (dd, 1H), 2,93-2,87 (m, 1H), 2,50 -2,32 (m, 3H), 2,10 - 1,91 (m, 5H), 1,80 - 1,25 (m, 13H), 1,08 -1,01 (m, 10H), 0,53-0,50 (m, 1H), 0,44-0,43 (m, 1H).
Ejemplo 19. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-tert-butil-15-cloro-N-[(1R, 2R)-1-[(ciclopropilsulfonil)carbamoilo]-2-(difluorometil)ciclopropil]-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23bhexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0279]

[0280] Paso 1. Preparación de 19-1, El intermedio C9 (1 g, 3,61 mmol), el éster metílico de N-Boc-trans-4-hidroxi-L-prolina (1,06 g, 4,33 mmol) y CS2CO3 (1,41 g, 4,33 mmol) se suspendieron en MeCN (18 ml). La mezcla de reacción se agitó a temperatura ambiente durante 18 h, luego se filtró a través de una capa de Celite. Los sólidos se enjuagaron con EtOAc. El filtrado y los lavados se combinaron y se concentraron al vacío. El residuo crudo se purificó por cromatografía en gel de sílice para proporcionar quinoxalina sustituida 19-1, LCMS-ESI+ (m/z): [M-Boc+2H]+ calculado para C-mH-mC^NsOs: 342,04; encontrado: 342,04.
[0281] Paso 2. Preparación de 19-2, Se disolvió quinoxalina sustituida 19-1 (425 mg, 0,961 mmol) en DCM (10 ml) y se trató con HCl (4,0 M en dioxano, 5 ml, 20 mmol). Después de agitarse a temperatura ambiente durante 2,5 h, la mezcla de reacción se concentró al vacío para proporcionar sal de clorhidrato de prolina 19-2, que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C14H14O 2N3O3: 342,04; encontrado: 342,11.
[0282] Paso 3. Preparación de 19-3, La sal de clorhidrato de amina 19-2 (0,961 mmol) se trató con el Intermedio B3 (348 mg, 1,08 mmol), BEP (315 mg, 1,15 mmol), EtOAc (9 ml), NMP (1 ml) y DIPEA (0,84 ml, 4,81 mmol). Después de agitarse a 50°C durante 1,5 h, la mezcla de reacción se diluyó con EtOAc. La capa orgánica se lavó con NaHCO3 acuoso saturado y salmuera, luego se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo bruto se purificó mediante cromatografía en gel de sílice para proporcionar la amida 19-3, LCMS-ESI+ (m/z): [M+H]+ calculado para C32H4iC l2N4O6: 647,24; encontrado: 647,35.
[0283] Paso 4. Preparación de 19-4, La cloroquinoxalina 19-3 (281 mg, 0,434 mmol) se trató con trifluoroborato de vinilo potásico (87 mg, 0,651 mmol), Pd(dppf)C^Aducto de Dc M (35 mg, 0,043 mmol), EtOH (4 ml) y EtsN (0,091 ml, 0,65 mmol). La mezcla de reacción agitada se calentó a reflujo durante 1 h, luego se enfrió a temperatura ambiente y se diluyó con EtOAc. La mezcla orgánica se lavó con H2O y salmuera, luego se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo crudo se purificó por cromatografía en gel de sílice para proporcionar vinil quinoxalina 19-4, LCMS-ESI+ (m/z): [M+H]+ calculado para C34H44ClN4O6: 639,29; encontrado: 639,86.
[0284] Pasos 5 y 6. Preparación de 19-5. La vinil quinoxalina 19-4 (203 mg, 0,318 mmol) se combinó con Zhan Catalyst-IB (21 mg, 0,032 mmol) en DCE (64 ml). La suspensión se desgasificó durante 20 minutos con N2 burbujeante, luego se calentó a reflujo durante 40 minutos. Después de enfriarse a temperatura ambiente, la mezcla se concentró al vacío. La mezcla cruda se purificó por cromatografía en gel de sílice para proporcionar el macrociclo 19-5 (110 mg; LCMS-ESI+ (m/z): [M+H]+ calc. para C32H40ClN4O6: 611,26; encontrado: 611,30). Este residuo (109 mg, 0,178 mmol) se disolvió en EtOAc (40 ml) y se trató con 5% de Rh/Al2O3 (40 mg). Se hizo burbujear H2 a través de la
suspensión durante 1 min, luego la mezcla de reacción se dejó agitar bajo una atmósfera de H2 durante 1 h. Después de este período, se añadió más 5% de Rh/AhOs (80 mg) a la mezcla de reacción. Se hizo burbujear H2 a través de la suspensión durante 1 min y la mezcla de reacción se dejó agitar bajo una atmósfera de H2 durante 1 h adicional. La mezcla de reacción se filtró a través de Celite, luego se concentró al vacío para producir el éster metílico 19-5 que se llevó a cabo sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para Ca2H42ClN4O6: 613,28; encontrado: 613,29.
[0285] Paso 7. Preparación de 19-6, El metil éster 19-5 (0,187 mmol) se trató con THF (10 ml) y LiOH (1,0 M en H2O, 10 ml, 10 mmol). La mezcla de reacción se agitó durante 3 h, luego se concentró al vacío para eliminar el THF. La suspensión restante se vertió en HCl al 10%. La capa acuosa se extrajo luego 3x con DCM. Los extractos orgánicos combinados se secaron sobre MgSO4 anhidro, se filtraron y se concentraron a vacío para proporcionar ácido carboxílico 19-6 que se llevó a cabo sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C31H40ClN4O6: 599,26; encontrado: 599,04.
[0286] Paso 8. Preparación del Ejemplo 19. En DMF (2 ml), se combinó ácido carboxílico 19-6 (110 mg, 0,184 mmol) con A9 (80 mg, 0,28 mmol), HAt U (84 mg, 0,22 mmol) y DIPEA (0,16 ml, 0,92 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 h, luego se detuvo mediante la adición de 1 ml de H2O. La suspensión acuosa se filtró y se purificó por HPLC para proporcionar el Ejemplo 19 en forma de una sal de TFA. Este material contenía una impureza, por lo tanto, se disolvió en EtOAc y la solución orgánica se lavó 2x con NaHCO3 acuoso saturado para liberar la base del material. La capa orgánica se secó sobre MgSO4, se filtró y se concentró al vacío. El residuo resultante se purificó mediante cromatografía en gel de sílice para proporcionar el Ejemplo 19. Tiempo de retención de HPLC analítico: 9,30 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C39H50C F 2N6O8S: 835,31; encontrado: 835,44, 1H-RMN (400 MHz, CD3OD) 87,93-7,84 (m, 1H), 7,83-7,76 (m, 1H), 7,61-7,51 (m, 1H), 6,87 (d, J = 9,3 Hz, 1H), 5,95 (m, J = 62,1, 31,3, 5,0 Hz, 2H), 4,99 (d, J = 7,5 Hz, 1H), 4,52 - 4,37 (m, 2H), 4,32 (d, J = 9,3 Hz, 1H), 4,11 (dd, J = 11,9, 3,8 Hz, 1H), 3,08 -2,90 (m, 2H), 2,81 (ddd, J = 14,0, 11,6, 4,6 Hz, 1H), 2,50 (dd, J = 13,9, 6,2 Hz, 1H), 2,37 -2,12 (m, 2H), 1,97 (ddd, J = 15,4, 10,2, 3,6 Hz, 3H), 1,85 (dd, J = 12,0, 7,7 Hz, 1H), 1,75 (d, J = 14,6 Hz, 1H), 1,71 - 1,20 (m, 13H), 1,17 - 0,97 (m, 12H), 0,97 - 0,79 (m, 1H), 0,57 (dd, J = 8 ,6 , 4,1 Hz, 1H), 0,54 - 0,45 (m, 1H)).
Ejemplo 20. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2S)-2-(2,2-difluoroetil)-1-{[(1-metilciclopropil)sulfonil]carbamoil} ciclopropil]-19,19-difluoro-15-metoxi-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinoxalina-9-carboxamida.
[0287]

[0288] El Ejemplo 20 se preparó de manera similar al Ejemplo 9, sustituyendo al Intermedio A8 por el Intermedio A9 en el Paso 7. El Ejemplo 20 (9,4 mg) se aisló como una sal de TFA. Tiempo de retención de HPLC analítico: 8,90 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C42H55F2N6O9S: 895,36; encontrado: 895,64. 1H-RMN (400 MHz, CD3OD) 89,23 (s, 1H), 7,93 (d, J = 9,0 Hz, 1H), 7,39 - 7,24 (m, 2H), 6,15 (s, 1H), 5,89 (tt, J = 57,4, 4,2 Hz, 1H), 4,94 (d, J = 7,5 Hz, 1H), 4,45 (dd, J = 11,2, 5,8 Hz, 2H), 4,32 (s, 1H), 4,13 (dd, J = 12,0, 3,7 Hz, 1H), 3,96 (s, 3H), 2,63-2,44 (m, 2H), 2,31 - 1,91 (m, 6 H), 1,84 - 1,70 (m, 2H), 1,71 - 1,25 (m, 15H), 1,06 (s, 10H), 0,96 - 0,84 (m, 2H), 0,59 - 0,48 (m, 2H).
Ejemplo 21. Preparación de (1aS, 2aR, 6S, 9S, 11R, 23aR, 23bS)-6-terc-butil-N-[(1R, 2R)-1-[(ciclopropilsulfonil)carbamoil)-2-(difluorometil)ciclopropil]-15-metoxi-4,7-dioxo-1a,2,2a,4,5,6,7,10,11,19,20,21,22,23,23a,23b-hexadecahidro-1H,9H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6]dioxadiazaciclononadecino[11,12-b]quinolina-9-carboxamida.
[0289]

[0290] Paso 1. Preparación de 21-1. El Intermedio C10 (1 g, 3,92 mmol) se combinó con el Intermedio D1 (1,51 g, 3,24 mmol) y Cs2CÜ3 (1,92 g, 5,88 mmol) como una suspensión en NMP (30 mL) y se calentó a 40°C. Después de 16 h, la reacción se enfrió a temperatura ambiente, se diluyó con EtOAc y se lavó sucesivamente con H2O, NaHCÜ3 acuoso saturado y salmuera. La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró a vacío para proporcionar 21-1, que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C2-iH26BrN2O6: 481,10; encontrado: 480,95.
[0291] Paso 2. Preparación de 21-2. La bromoquinolina 21-1 (1,48 g, 3,07 mmol) se trató con viniltrifluorobororoboro potasio (618 mg, 4,61 mmol), Pd(dppf)C^DCM (125 mg, 0,15 mmol), EtOH (30 ml) y Et3N (0,63) mL, 4,61 mmol). La mezcla de reacción se agitó a reflujo durante 1,5 h, luego se enfrió a temperatura ambiente y se diluyó con EtOAc. La fase orgánica se lavó con agua y salmuera, luego se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo crudo se purificó mediante cromatografía en gel de sílice (10 % a 40% de EtOAc/Hex) para proporcionar vinilquinolina 21-2. LCMS-ESI+ (m/z): [M+H]+ calc. para C23H29N2O6: 429,20; encontrado: 429,44.
[0292] Paso 3. Preparación de 21-3. Se disolvió vinil quinolina 21-2 (920 mg, 2,15 mmol) en DCM (5 ml) y MeOH (1 ml) y se trató con Hc I (4,0 M en dioxano, 5 ml). Después de agitarse a temperatura ambiente durante 3H, la mezcla de reacción se concentró en vacío para proporcionar clorhidrato de amina 21-3 que se llevó a cabo sin purificación adicional. LCMS-ESI+ (m/z): [M+H]+ calc. para C18H21N2O4: 329,15; encontrado: 329,2.
[0293] Paso 4. Preparación de 21-4. El clorhidrato de amina 21-3 (358 mg, 0,96 mmol) y el intermedio B3 (310 mg, 0,96 mmol) se combinaron y se trataron con BEP (289 mg, 1,06 mmol), EtOAc (9 ml), NMp (1 ml) y DIPEA (0,50 ml, 2,88 mmol). Después de agitarse a 40°C durante 1,5 h, se añadió DIPEA adicional (0,2 ml, 1,15 mmol) y la mezcla se agitó durante 30 minutos adicionales. Después de enfriarse a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc y se lavó sucesivamente con NaHCO3 acuoso saturado y salmuera. La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró a vacio. El residuo crudo se purificó por cromatografía en gel de sílice (10% a 40% de EtOAc/Hex) para proporcionar la amida 21-4. LCMS-ESI+ (m/z): [M+H]+ calculado para C36H48N3O7: 634,35; encontrado: 634,47.
[0294] Paso 6. Preparación de 21-5. La vinilquinolina 21-4 (358 mg, 0,56 mmol) se disolvió en DCE (100 ml) y se trató con Zhan Catalyst-1B (41 mg, 0,06 mmol). La mezcla se desgasificó con N2 burbujeante durante 30 minutos y se calentó a reflujo durante 45 minutos. La mezcla de reacción se concentró al vacío y el residuo bruto se purificó por cromatografía en gel de sílice (0% a 30% de EtOAc/Hex) para proporcionar el macrociclo 21-5. LCMS-ESI+ (m/z):
[M+H]+ calculado para C34H44N3O7: 606,32; encontrado: 606,16.
[0295] Pasos 6 y 7. Preparación de 21-6. El macrociclo 21-5 (235 mg, 0,39 mmol) se disolvió en EtOH (6 ml). Se añadió Pd/C (10% en peso de Pd, 200 mg) y se burbujeó H2 a través de la suspensión durante 2 min. La mezcla de reacción agitada se mantuvo bajo 1 atm de H2 durante 45 minutos antes de filtrarse sobre Celite y concentrar al vacío. Este residuo se usó posteriormente sin purificación adicional. (LCMS-ESI+ (m/z): [M+H]+ calculado para C32H42FN4O6: 597,31; encontrado: 597,36). Este residuo (0,39 mmol teórico) se trató con THF (3 ml), H2O (3 ml) y LiOH (28 mg, 1,17 mmol). La mezcla se agitó durante 1 h, luego se diluyó con EtOAc. La mezcla se acidificó a pH 3 con 1 N HCl y se extrajo 3x con EtOAc. Los extractos orgánicos combinados se lavaron con salmuera y se secaron sobre MgSO4 anhidro, se filtraron y se concentraron a vacío para proporcionar ácido carboxilico 21-6 que se usó posteriormente sin purificación adicional. LCMS-ESI+ (m/z): [M+h ]+ calc. para C33H44N3O7: 594,32; encontrado: 594,50.
[0296] Paso 8. Preparación del Ejemplo 21. El ácido carboxilico 21-6 (175 mg, 0,30 mmol) se trató con el Intermedio A9 (113 mg, 0,39 mmol), TBTU (140 mg, 0,44 mmol), DMAP (55 mg, 0,45 mmol), DCM (5 ml) y DIPEA (0,16 ml, 0,90 mmol). La mezcla de reacción se agitó durante 1,5 h, y se añadió DIPEA adicional (0,10 ml, 0,55 mmol). Después de agitarse durante 30 minutos, la mezcla se diluyó con EtOAc, se lavó con NaHCO3 acuoso saturado y salmuera. La fase orgánica se secó sobre MgSO4 anhidro, se filtró y se concentró al vacío. El residuo bruto se purificó por HPLC para proporcionar el Ejemplo 21 en forma de una sal de TFA. Tiempo de retención de HPLC analítico: 9,17 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C41H54F2N5O9S: 830,36; encontrado: 830,55, 1H-RMN (400 MHz, CD3OD) 89,36 (s, 1H), 7,84 (s, 1H), 7,62 (d, J = 8,9 Hz, 1H), 7,19 (d, J = 2,4 Hz, 1H), 7,02 (dd, J = 8,9, 2,5 Hz, 1H), 6,11 (t, J = 3,6 Hz, 1H), 5,90 (td, J = 55,8, 6,6 Hz, 1H), 5,49 (s, 1H), 5,03 (d, J = 7,6 Hz, 1H), 4,40 (dd, J = 12,0, 6,9 Hz, 3H), 4,10 (dd, J = 11,6, 3,9 Hz, 1H), 3,90 (s, 3H), 3,06 - 2,91 (m, 1H), 2,74 (m, 1H), 2,60 - 2,40 (m, 2H), 2,30 - 2,13 (m, 2H), 2,09 - 1,89 (m, 5H), 1,81 -1,18 (m, 13H), 1,16 -1,09 (m, 2H), 1,05 (d, J = 14,4 Hz, 9H), 0,62 - 0,45 (m, 2H).
Ejemplo 22. Preparación de (33R, 35S, 91S, 93R, 94R, 95S, 5S)-5-(terc-butil)-N-((1R, 2R)-2-(difluorometil)-1-(((1 -metil-ciclopropil)sulfonil)carbamoil)ciclopropil)-4,7-dioxo-2,8-dioxa-6-aza-1 (2,3)-benzo[f]quinoxalina-3 (3,1)-pirrolidina 9 (3,4)-biciclo[3,1,0]hexanaciclotetradecafano-35-carboxamida.
[0297]

Ejemplo 22
[0298] El Ejemplo 22 se preparó de manera similar al ejemplo 11, sustituyendo al intermedio C13 por el intermedio C3 en el paso 1. El Ejemplo 22 se aisló como una sal de TFA (53 mg). Tiempo de retención de HPLc analítico: 9,63 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C44H55F2N6O8S: 865,38; encontrado: 865,51.
Ejemplo 23. Preparación de (33R, 35S, 91S, 93R, 94R, 95S, 5S)-5-(terc-butil)-17-ciano-N-((1R, 2R)-2-(difluorometil)-1-(((1-metilciclopropil)sulfonil)carbamoil)ciclopropil)-4,7-dioxo-2,8-dioxa-6-aza-1(2,3)quinoxalina-3(3,1)-pirrolidin-9(3,4)-biciclo[3,1,0]hexanaciclotetradecafano-35-carboxamida.
[0299]

El Intermedio 23-1 se preparó como para el Intermedio 11-5, utilizando el intermedio C11 en lugar del Intermedio C3 en el Paso 1. LCMS-ESI+ (m/z): [M+H]+ calculado para C39H47N4O2: 683,34; encontrado: 683,39.
[0300] Paso 1. Preparación de 23-2: se hidrogenó una mezcla de Intermedio 23-1 (318 mg, 0,466 mmol), en 46 ml de etanol y 46 ml de acetato de etilo sobre 318 mg de paladio al 10% sobre carbono. Después de 4 horas, la mezcla se filtró sobre Celite y el filtrado se concentró a presión reducida. El residuo resultante fue purificado por cromatografía en gel de sílice (acetato de etilo al 5-60% en hexanos) para obtener 23-2 (245 mg). LCMS-ESI+ (m/z): [M+H]+ calc. para C32H43N4O7: 595,31; encontrado: 595,23.
[0301] Paso 2. Preparación de 23-3: se enfrió en un baño de hielo una mezcla de Intermedio 23-2 (245 mg, 0,412 mmol), en 1,6 mL de diclorometano antes de la adición de trietilamina (0,459 mL, 3,3 mmol). y luego anhídrido trifluorometanosulfónico (0,104 ml, 0,618 mmol). La mezcla se dejó agitar y llegar a temperatura ambiente. Una vez completada, la reacción se detuvo con agua y el producto se extrajo en acetato de etilo. La capa orgánica se secó sobre sulfato de sodio anhidro, se filtró y se concentró a presión reducida. El residuo resultante se purificó por cromatografía en gel de sílice (acetato de etilo al 25-75% en hexanos) para dar 23-3 (245 mg). LCMS-ESI+ (m/z):
[M+H]+ calc. para C33H42F3N4O9S: 727,26; encontrado: 727,33.
[0302] Paso 3. Preparación de 23-4: una mezcla de Intermedio 23-3 (158 mg, 0,217 mmol), tetraquis (trifenilfosfina) paladio 2,0 M en éter (25,12 mg, 0,02 mmol) y cianuro de zinc. El 98% (51,07 mg, 0,43 mmol) en 1 ml de dimetilformamida se desgasificó con argón durante 10 minutos, luego se calentó a 80°C durante 30 minutos. La mezcla se concentró luego a presión reducida. El residuo resultante se purificó por cromatografía en gel de sílice (acetato de etilo al 5-70% en hexanos) para dar 23-4 (122 mg). LCMS-ESI+ (m/z): [M+H]+ calc. para C33H42N5O6: 604,31; encontrado: 604,03.
[0303] Paso 4. Preparación de 23-5: una mezcla de Intermedio 23-4 (120 mg, 0,199 mmol) en 1,5 mL de tetrahidrofurano y 1,0 mL de agua fue tratada con hidróxido de litio monohidrato (33,36 mg, 0,8 mmol). Después de 6 horas, se añadió 1 ml de ácido clorhídrico 2 N y la mezcla se concentró a presión reducida. El residuo resultante se repartió entre agua y acetato de etilo, añadiendo gota a gota ácido clorhídrico 2 N para asegurar la acidez. La fase orgánica se secó sobre sulfato de sodio anhidro, se filtró y se concentró para dar 23-5 (101 mg). LCMS-ESI+ (m/z):
[M+H]+ calc. para C32H46N5O6: 590,30; encontrado: 590,15.
[0304] Paso 5. Preparación de 23-6: A una suspensión de ácido carboxílico 23-5 (95 mg, 0,161 mmol) e Intermedio
A10 (59 mg, 0,193 mmol) en DMF (0,8 ml) se agregó HATU (74 mg), 0,193 mmol) y DIPEA (113 ml, 0,644 mmol) a 23°C. Después de 10 min, la solución se trató con 0,5 ml de ácido fórmico y se purificó por HPLC de fase inversa para proporcionar la sal de TFA del Ejemplo 23, Tiempo de Retención de HPLC analítico: 8,86 min. LCMS-ESI+ (m/z):
[m H]+ calc. para C41H52F4N7O8S: 840,177; encontrado: 840,29.
Ejemplo 24. Preparación de (1aS, 2aR, 6S, 9S, 11R, 24aR, 24bS)-6-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilo-ciclopropil)sulfonil]carbamoil} ciclopropil]-4,7,18-trioxo-1a,2,2a,4,5,6,7,10,11,20,21,22,23,24,24a,24bhexadecahidro-1H, 9H, 18H-8,11-metanociclopropa[4',5']ciclopenta[1',2':18,19][1,10,3,6,12]dioxatriazaciclonona decino[11,12-b]quinazolina-9-carboxamida.
[0305]

[0306] El Intermedio 24-1 se preparó como para el Intermedio 17-4, utilizando el intermedio C12 en lugar del Intermedio C8 en el Paso 1. LCMS-ESI+ (m/z): [M+H]+ calc. para C32H40ClN4O7: 627,26; encontrado: 627,10.
[0307] Paso 1: Preparación de 25-1 y 24-2. La olefina macrocíclica 24-1 (0,574 g, 0,915 mmol) se disolvió en 100 ml de acetato de etilo. Después de la desgasificación con Ar, se añadió Rh/Al al 5% (0,12 g, 115 mmol) y la mezcla se hidrogenó durante 24 horas a 1 atm. La filtración a través de Celite, la concentración y la cromatografía en gel de sílice (10% - 25% de acetato de etilo en gradiente de hexanos) proporcionó el intermedio 24-2 (24 mg) y el intermedio 25-1 (385 mg). Para 25-1, LCMS-ESI+ (m/z): [M+H]+ calculado para C32H42ClN4Oy: 629,27; encontrado: 629,28. Para 24-2, LCMS-ESI+ (m/z): [M+H]+ calculado para C32H43N4O7: 595,31; encontrado: 594,91.
[0308] Paso 2. Preparación de 24-3: una mezcla de Intermedio 24-2 (24 mg) en 1 ml de metanol se trató con 0,25 ml de hidróxido de litio 1N. Después de 2 horas, la mezcla se concentró a presión reducida y se repartió entre agua y acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro, se filtró y se concentró para producir 24-3 (25 mg). LCMS-ESI+ (m/z): [M+H]+ calculado para C31H41N4O7: 581,30; encontrado: 581,03.
[0309] Paso 3. Preparación del Ejemplo 24: A una suspensión de ácido carboxílico 24-3 (25 mg, 0,044 mmol) e Intermedio A10 (16 mg, 0,053 mmol) en DMF (0,2 ml) se añadió HATU (20 mg, 0,053 mmol) y DIPEA (31 ml, 0,176 mmol) a 23°C. Después de 10 minutos, la solución se trató con 0,5 ml de ácido fórmico y se purificó por HPLC de fase inversa para proporcionar la sal de TFA del Ejemplo 24 (11,7 mg). Tiempo de retención de HPLC analítico: 8,72 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C40H53F2N6O9S: 831,95; encontrado: 831,36.
[0310] Ejemplo 25. Preparación de (1aS, 2aR, 6S, 9S, 11R, 24aR, 24bS)-6-terc-butil-15-ciano-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilciclopropil)sulfonil]carbamoil} ciclopropil]-4,7,18-trioxo-1a,2,2a,4,5,6,7,10,11,20,21,22,23,24,24a,24b-hexadecahidro-1H,9H,18H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6,12]dioxatriazaciclononadecino[11,12-b]quinazolina-9-carboxamida.
[0310]

[0311] Paso 1. Preparación de 25-2. El intermedio 25-1 (0,315 g, 0,501 mmol), Bis (Pinacolato) Diboron (0,25 g, 1 mmol) y acetato de potasio (0,15 g, 1,5 mmol) se disolvió en 5 ml de 1,4-dioxano y se desgasificó con Ar para 15 minutos. Luego se añadió tris(dibencilidenacetona) dipaladio (o) (0,02 g, 0,02 mmol) y 2-(diciclohexilfosfino)-2',4',6'-triisopropil-bifenilo (0,02 g, 0,05 mmol) y la mezcla se calentó a 90°C durante 45 minutos. La mezcla se concentró y se purificó mediante cromatografía en gel de sílice (5% - 80% de acetato de etilo en gradiente de hexanos) para proporcionar el intermedio 25-2 (0,456 g). LCMS-ESI+ (m/z): [M+H]+ calculado para C38H54BN4O9: 721,40; encontrado: 721,20.
[0312] Paso 2. Preparación de 25-3. Una solución de 25-2 (0,360 g, 0,5 mmol) en 4 ml de THF y 4 ml de hidróxido de sodio 0,5 N se trató con peróxido de hidrógeno (35%, 485,48 mg, 5 mmol) y trietilamina (0,81 ml, 5,81 mmol). Después de 5 minutos, la reacción se detuvo con 1 N HCl y se extrajo con acetato de etilo. La capa orgánica se secó sobre Na2SO4, se filtró y se concentró. Cromatografía en gel de sílice con un gradiente de acetato de etilo del 5% al 100% en hexanos dio 25-3 (224 mg). LCMS-ESI+ (m/z): [M+H]+ calc. para C32H43N4O8: 611,31; encontrado: 611,14.
[0313] Paso 3. Preparación de 25-4. Una solución helada de 25-3 (0,104 g, 0,17 mmol) y trietilamina (0,19 ml, 1,362 mmol) en 1 ml de DCM se trató con una solución de anhídrido trifluorometanosulfónico, 1M en cloruro de metileno (0,043 ml, 0,26 mmol) gota a gota. Después de agitarse durante 3 horas, la reacción se inactivó con agua y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y salmuera, se secó sobre Na2SO4, se filtró y se concentró. La cromatografía en gel de sílice usando un gradiente de acetato de etilo del 25% - 75% en hexanos dio 25-4 (126 mg). LCMS-ESI+ (m/z): [M+H]+ calc. para C33H42F3N4O10S: 743,26; encontrado: 743,06.
[0314] Paso 4. Preparación de 25-5. Se desgasificó una mezcla de triflato de macrociclo 25-4 (126 mg, 0,17 mmol), tetraquis(trifenilfosfina)paladio (19,6 mg, 0,017 mmol), cianuro de zinc, 98% (39,9 mg, 0,34 mmol) en 1,7 mL DM para 10 minutos. La reacción se calentó a 80°C durante 30 minutos. La reacción se concentró. El producto bruto se purificó por cromatografía en gel de sílice usando un gradiente de acetato de etilo al 5% -70% en hexanos para dar el intermedio 25-5 (94 mg). LCMS-ESI+ (m/z): [M+H]+ calc. para C33H42N5O7: 620,31; encontrado: 620,09.
[0315] Pasos 5 y 6. Preparación del Ejemplo 25. Se trató una solución de 25-5 (94 mg, 0,015 mmol) en 1,5 mL de THF y 1 mL de agua con hidróxido de litio monohidrato (25 mg, 0,061 mmol) y se agitó durante 1,5 horas a temperatura ambiente. La mezcla se acidificó con ácido clorhídrico 1N y se extrajo con acetato de etilo. La fase orgánica se separó,
se secó sobre sulfato de sodio anhidro, se filtró y se concentró para dar 87 mg del ácido carboxílico en bruto. Este residuo se trató con HATU (65,5 mg, 0,172 mmol) y 0,8 mL de DMF, luego se agregaron A10 (53 mg, 0,172 mmol) y DIPEA (0,1 ml, 0,58 mmol). Después de 25 minutos, se agregaron varias gotas de ácido fórmico y metanol hasta un volumen total de 1,2 ml y el producto se purificó por HPLC de fase inversa para dar la sal de TFA del Ejemplo 25 (75 mg). Tiempo de retención de HPLC analítico: 8,65 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C41H52F2N7O9S: 856,35; encontrado: 855,95.
Ejemplo 26. Preparación de (1aS, 2aR, 6S, 9S, 11R, 24aR, 24bS)-6-terc-butil-N-[(1R, 2R)-2-(difluorometil)-1-{[(1-metilo-ciclopropil)sulfonil]carbamoil} ciclopropil]-15-metoxi-4,7,18-trioxo-1a,2,2a,4,5,6,7,10,11,20,21,22,23,24, 24a,24b-hexadecahidro-1H,9H,18H-8,11-metanociclopopa[4',5']ciclopenta[1',2':18,19][1,10,3,6,12]dioxatriazaciclonona decino[11,12-b]quinazolina-9-carboxamida.
[0316]

[0317] Paso 1. Preparación de 26-1. A una solución de 25-3 (110 mg, 0,18 mmol) en 2 ml de metanol se le añadió yodometano (0,03 ml, 0,54 mmol) y carbonato de potasio (74,68 mg, 0,54 mmol). La mezcla se calentó a 80°C durante 30 minutos, luego se diluyó con acetato de etilo y se lavó con agua y salmuera. La fase orgánica se concentró para dar 26-1 en bruto, 105 mg (93,3%) que se llevó directamente a el Paso 2. LCMS-ESI+ (m/z): [M+H]+ calc. para C33H45N4O8: 625,32; encontrado: 625,18.
[0318] Pasos 2 y 3. Preparación del Ejemplo 26. A una solución de 26-1 (105 mg, 0,17 mmol) en 1 ml de THF, 1 ml de metanol y 1 ml de agua se añadió hidróxido de litio monohidrato (28 mg, 0,67 mmol). Después de 3,5 horas, la reacción se acidificó con 1 N HCl y se extrajo en acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro, se filtró y se concentró, luego se secó a vacío para dar 103 mg del ácido carboxílico en bruto. Este residuo se trató con HAt U (76 mg, 0,20 mmol) y 1,7 mL de DMF, luego se agregaron A10 (61 mg, 0,20 mmol) y DIPEA (0,117 ml, 0,668 mmol). Después de 1 hora, se agregaron varias gotas de ácido fórmico y metanol hasta un volumen total de 1,2 ml y el producto se purificó por HPLC de fase inversa para dar la sal de TFA del Ejemplo 26 (23 mg). Tiempo de retención de HPLC analítico: 8,67 min. LCMS-ESI+ (m/z): [M+H]+ calc. para C41H55N6O10S: 861,37; encontrado: 861,08.
Actividad biológica
Expresión y purificación de la proteasa NS3 de los genotipos 1a, 2a y 3 Generación de plásmidos de expresión de proteasa NS3
[0319] La secuencia de codificación del genotipo 1b (cepa con-1) del dominio de proteasa NS3 de VHC se amplificó por PCR a partir de un plásmido que codifica el replicón I389luc-ubi-neo/NS3-3'/ET (Reblikon, Mainz, Alemania). El cebador 5'-PCR se diseñó para codificar una etiqueta de hexahistidina K3 N-terminal e insertar un sitio de escisión de la proteasa del Virus del Grabado del Tabaco (rTEV) recombinante en el marco en la secuencia codificante de NS3.
El fragmento de ADN resultante se clonó en el vector de expresión de la proteína pET28 (Invitrogen, Carlsbad, CA) produciendo el p28-N6H-Tev-NS3(181) 1b.
[0320] Las secuencias codificantes para el genotipo 3 del dominio de la proteasa del VHC se amplificaron mediante RT-PCR utilizando un kit Titan OneTube RT-PCR (Roche, Indianapolis, iN) y ARN extraído de suero humano positivo para el VHC (BBI Diagnostics, MA) utilizando un kit de virus QIAmp UltraSens (Qiagen, Valencia, CA). Los cebadores de 5' PCR se diseñaron para codificar etiquetas hexa-histidina N-terminales e insertar sitios de escisión de proteasa rTEV en el marco en las secuencias codificantes de la proteasa NS3. Los fragmentos de ADN resultantes se clonaron en pET28 produciendo los vectores de expresión p28-N6H-Tev-NS3 (181) 1a y p28-N6H-Tev-NS3 (181)3, respectivamente.
Expresión de proteína de proteasa NS3
[0321] Las bacterias BL21AI (Invitrogen, Carlsbad, CA) se transformaron con vectores de expresión NS3 de genotipo 1b o 3 y se usaron para inocular un recipiente de fermentación de 20 L (Sartorius BBI System Inc., Bethlehem, PA), que contenía 18 L de medio 2YT reciente suplementado con 50 pg/ml de kanamicina. Cuando las densidades celulares alcanzaron un OD600 de 1, la temperatura de los cultivos se redujo de 37°C a 28°C y la inducción se inició inmediatamente mediante la adición de concentraciones finales de ZnSO430 pM L-arabinosa 14 mM e Isopropil 1 mM p-D-tiogalactosido (IPTG). Las células se recogieron por centrifugación cuatro horas después de la inducción y se almacenaron como gránulos congelados a -80°C antes de la purificación de la proteína NS3.
Purificación de proteasas NS3
Purificación de la proteasa NS3 del genotipo 1b
[0322] Los sedimentos celulares se descongelaron y se resuspendieron a 10 ml/g de células en un tampón de lisis que contenía tris 50 mM, pH 7,6, NaCl 300 mM, 0,1% 3-[(3-Cholamidopropil) dimetilamonio]-1-propanosulfonato (CHAPS), 5% de glicerol y 2 mM de p-mercaptoetanol. Las suspensiones celulares se sometieron a sonicación, se filtraron a través de gasa y se pasaron tres veces a través de un microfluidizador a 18.000 libras/pulgada2 Los lisados resultantes se centrifugaron a 15.500 rpm durante 45 minutos y los sobrenadantes se cargaron en una columna HisTrap HP (GE Lifesciences) previamente equilibrada con cinco volúmenes de Tampón A (50 mM tris pH 7,6, NaCl300 mM, CHAPS al 0,1%, 5% de glicerol, p-mercaptoetanol 2 mM, imidazol-HCl 50 mM). Las proteínas se eluyeron con un gradiente de 0-100% de tampón A de Ni más 500 mM de imidazol-HCl y las fracciones se recogieron y se agruparon. El grupo HisTrap se diluyó 1:10 con tampón SP-A (tris 50 mM pH 7,0, glicerol al 10%, ditiotreitol 2 mM (DTT)) y se cargó en una columna HiTrap SP-HP (GE Lifesciences) equilibrada con tampón SP-A. La proteasa NS3 se eluyó con un gradiente de tampón SP-B al 0-100% (tampón SP-A más NaCl 1 M). Los grupos concentrados de fracciones de SP que contenían NS3 se dividieron en partes alícuotas, se congelaron instantáneamente en nitrógeno líquido y se almacenaron a -80°C.
Purificación de la proteasa NS3 del genotipo 3
[0323] Los sedimentos bacterianos recogidos de la expresión de la proteasa NS3 del VHC del genotipo 3 se homogeneizaron en tampón de lisis (tris 25 mM, tampón de pH 7,5 que contiene NaCl 150 mM y fluoruro de fenilmetanosulfonilo 1 mM (PMSF)) y se pasa a través de un microfluidizador a 18.000 libras/pulgada2. Los lisados celulares homogeneizados se centrifugaron a 30.000 x g durante 30 minutos a 4°C. Los sedimentos de P1 resultantes se lavaron con tampón de lavado I (tris 25 mM, pH 7,5 que contenía CHAPS al 1%) seguido de centrifugación a 10.000 x g durante 30 minutos 4°C. Los sedimentos de P2 resultantes se lavaron con tampón de lavado II (tampón CAPS 50 mM, pH 10,8, que contenía NaCl 2 M y urea 2 M) seguido de centrifugación a 30.000 x g durante minutos a 4°C. Los sedimentos de P3 resultantes fueron resuspendido en tampón de solubilización (20 ml de tris 25 mM, pH 7,5 que contiene NaCl 150 mM y urea 8 M) e incubado a 4°C durante una hora. Las proteínas solubilizadas se pasaron a través de un filtro de 0,45 micrones. Las concentraciones de proteína se midieron y las soluciones se ajustaron a 40 mM de DTT, se incubaron durante 30 minutos a 4°C y luego se diluyeron rápidamente en tampón de replegamiento (tris 25 mM, pH 8,5, guanidina-HCl 0,8 M, L-arginina 0,4 M, 10). mM ZnSO4) mientras se agita. Las soluciones de proteínas se incubaron a 4°C durante la noche para permitir el replegamiento. Las proteasas replegadas se centrifugaron a 30.000 x g durante 10 minutos para eliminar los precipitados residuales. Las concentraciones finales de proteína se midieron y las proteasas NS3 se dividieron en alícuotas, se congelaron instantáneamente en nitrógeno líquido y se almacenaron a -80°C.
Determinación de Ki para la proteasa NS3 de los genotipos 1b y 3a.
[0324] El dominio de proteasa NS3 purificada (aminoácidos 1-181) del virus de genotipo 1b y 3a se generó como anteriormente. El sustrato depsipeptídico fluorogénico inactivado internamente Ac-DED (Edans)-EEAbuy[COO]ASK(Dabcil)-NH2 y un péptido sintético que contiene los residuos del núcleo hidrofóbico del cofactor de proteína NS4A (KKGSW iVg RIiLs g Rk K; péptido NS4A) se obtuvieron de Anaspec, Inc. (San José, CA). Otros productos químicos y productos bioquímicos fueron de grado reactivo o mejor y se compraron a proveedores estándar.
[0325] Las reacciones se realizaron a temperatura ambiente en un tampón que consistía en HEPES 50 mM, glicerol al 40%, Triton X-100 al 0,05%, DTT 10 mM y DMSO al 10%. Las soluciones finales del ensayo contenían 50 pM de proteasa de genotipo 1b de NS3 o 200 pM de proteasa de genotipo 3a, péptido de NS4A 20 pM y sustrato de 4 mM (genotipo 1b) o sustrato de 2 pM (genotipo 3a). Las concentraciones de inhibidor variaron de 100 nM a 5 pM en diluciones triples, y se incluyeron controles sin inhibidores.
[0326] Las diluciones de los compuestos se realizaron en DMSO a 20 x concentración final. Las mezclas de reacción se prepararon en placas de ensayo de 96 pocillos. Se mezcló una solución de enzima y péptido NS4A en el tampón de ensayo (25 pl de volumen con ambos reactivos a 4 x concentración final) con 45 pl de tampón de ensayo y 5 pl de inhibidor o DMSo , y se incubó previamente a temperatura ambiente durante 1 hora. La reacción se inició mediante la adición de 25 pl de solución de sustrato a 4 x concentración final. Las placas se mezclaron vigorosamente durante 5 10 segundos y se dejó que las reacciones transcurrieran durante 90 minutos. La fluorescencia se midió cada 30 s entre 90 y 120 minutos de tiempo de reacción utilizando un lector de placas multimodo Tecan Infinite M1,000 o PerkinElmer Envision con una longitud de onda de excitación de 340 nm y una longitud de onda de emisión de 490 nm.
[0327] Las tasas se calcularon a partir de las curvas de progreso en estado estable, en el marco de tiempo de 90-120 minutos después de la adición del sustrato. Para determinar la Ki, las tasas se representaron en función de la concentración del inhibidor y los datos se ajustaron a la ecuación 1 (Morrison, JF, Biochimica et Biophysica Acta 1969, 185, 269-286) para calcular Kiapp utilizando GraphPad Prism 5. La fracción activa de la enzima se determinó mediante la titulación del sitio activo con inhibidores potentes conocidos. Ki se calculó a partir de K¡app/(1 [[S]/Km]).

Evaluación de la actividad anti-VHC basada en células:
[0328] La potencia antiviral (CE50) se determinó tanto en líneas celulares de replicón subgenómico de VHC subgenómicas como en células de replicón de VHC transfectadas transitoriamente. El término media concentración máxima efectiva (CE50) se refiere a la concentración de un fármaco que induce una respuesta a medio camino entre la línea de base y el máximo después del tiempo de exposición especificado a continuación.
[0329] Se establecieron replicones subgenómicos estables del VHC para los genotipos 1a, 1b, 2a, 3a y 4a en células derivadas de Huh-7 como lo describen Lohmann y otros (Lohmann V, Korner F, Koch J, y otros. Replicación de hepatitis subgenómica ARN del virus C en una línea celular de hepatoma (Science 1999; 285: 119-3). Cada línea celular estable contiene un replicón bicistrónico del VHC que codifica un gen informador de luciferasa de Renilla humanizado (hRLuc) fusionado con un gen seleccionable de resistencia a la neomicina, seguido de un IRES de EMCV y la región de codificación NS3-NS5B del VHC. La selección de las células que expresan constitutivamente el replicón del VHC se logró en presencia del antibiótico de selección, neomicina (G418). La actividad de la luciferasa se midió como un marcador para los niveles de replicación intracelular del VHC.
[0330] El replicón estable de genotipo 1a se derivó de la cepa H77 VHC y contenía las mutaciones adaptativas P1496L y S2204I. El replicón estable del genotipo 1b se derivó de la cepa Con1 VHC y contenía las mutaciones adaptativas E1202G, T1280I y K1846T. El replicón estable del genotipo 2a se derivó de la cepa VHC JFH-1 y no requirió mutaciones adaptativas. El replicón estable del genotipo 3a se derivó de la cepa S52 VHC y contenía mutaciones adaptativas P1121L, A1198T y S2210I (equivalente a S2204 I en el genotipo 1). El replicante estable del genotipo 4a se derivó de la cepa ED43 VHC y contenía mutaciones adaptativas Q1691R y S2204l. Todas las líneas celulares de replicón se propagaron en células derivadas de Huh-7 y se mantuvieron en medio de Eagle modificado por Dulbecco (DMEM) suplementado con suero bovino fetal al 10% (Fb S) y 0,5 pg/ml de G418.
[0331] Se establecieron replicones de VHC transfectados transitoriamente para las variantes D168A resistentes al inhibidor de proteasa genotipo 1a,1b,3a y NS3/4a en el genotipo 1b o R155K en el genotipo 1a. Los replicones transfectados transitorios también son replicones subgenómicos biscistrónicos, pero no contienen el marcador seleccionable de neomicina presente en los replicones estables. Estos replicones codifican el IRES de poliovirus seguido del gen informador hRLuc, el IRES de EMCV y, finalmente, la región de codificación NS3-NS5B del VHC. Los genones 1a (H77) y 1b (Con1) de tipo silvestre se derivaron de la misma cepa y contenían las mismas mutaciones adaptativas que se enumeraron anteriormente. El replicón transitorio del genotipo 3a se derivó de la cepa S52 VHC como anteriormente, pero contenía mutaciones adaptativas ligeramente diferentes P1112L, K1615E y S2210I. Específicamente, la mutación adaptativa secundaria A1198T (A166T) en el dominio de proteasa del genotipo estable 3a replicón se reemplazó con K1615E (K583E) en la helicasa NS3, sin efecto en la eficiencia de la replicación. La eliminación de A166T ubicada en el dominio de proteasa minimiza el impacto de esta variante en los inhibidores que se dirigen al dominio de proteasa y representa un dominio de proteasa más cercano al tipo silvestre para el genotipo 3a. Los replicones resistentes que codifican mutaciones del inhibidor de la proteasa NS3/4 se introdujeron en el gen NS3 de tipo silvestre 1b o 1a mediante mutagénesis dirigida. Los ARN transcritos in vitro de todos los replicones
transitorios se transfectaron en líneas celulares derivadas de Huh-7 sin tratamiento previo mediante electroporación. La actividad de la luciferasa se midió como un marcador para los niveles de replicación intracelular del VHC.
[0332] Para realizar ensayos de CE50, las células de cada replicón de VHC se dispensaron en placas de 384 pocillos. Los compuestos se disolvieron en DMSO a una concentración de 10 mM y se diluyeron en DMSO utilizando un instrumento de pipeteo automático. Se añadieron directamente a las células compuestos triples diluidos en serie utilizando un instrumento automatizado. El DMSO se usó como control negativo (solvente, sin inhibición) y una combinación de tres inhibidores de VHC que incluyen un inhibidor de proteasa; se usaron un inhibidor de NS5A y un inhibidor de nucleósidos en concentraciones >100 x CE50 como control positivo (inhibición del 100%). Setenta y dos horas después, las células se lisaron y la actividad de luciferasa de Renilla se cuantificó según lo recomendado por el fabricante (Promega-Madison, WI). Se realizó una regresión no lineal para calcular los valores de CE50.
[0333] Los resultados se muestran en las Tablas 1 y 2:
Tabla 1: Valores de actividad biológica para líneas celulares de replicación de VHC subgenónicas estables

Tabla 2: Valores de actividad biológica para líneas celulares de replicón de VHC transfectadas transitorias

[0334] Los datos en las Tablas 1 y 2 representan un promedio a lo largo del tiempo de cada compuesto, se han realizado múltiples ensayos durante la vida del proyecto.
Composiciones farmacéuticas
[0335] Lo siguiente ilustra formas de dosificación farmacéutica representativas, (“Compuesto X”), para uso terapéutico o profiláctico en humanos.




[0336] Las formulaciones anteriores pueden obtenerse mediante procedimientos convencionales bien conocidos en la técnica farmacéutica.
[0337] La invención se ha descrito con referencia a diversas realizaciones y técnicas específicas y preferidas. El uso de los términos "un", “una”, “el” y "la" y referencias similares en el contexto de esta divulgación (especialmente en el contexto de las siguientes reivindicaciones) debe interpretarse de modo que cubra tanto el singular como el plural, a menos que se indique lo contrario aquí indicado o claramente contradicho por el contexto. Todos los métodos descritos en el presente documento pueden realizarse en cualquier orden adecuado a menos que se indique lo contrario en el presente documento o el contexto lo contradiga claramente. El uso de cualquiera y todos los ejemplos, o lenguaje ejemplar (por ejemplo, tal como, preferido, preferiblemente) proporcionado en este documento, está destinado simplemente a ilustrar más el contenido de la divulgación y no plantea una limitación en el alcance de las reivindicaciones. Ningún lenguaje en la especificación debe interpretarse como que indica que cualquier elemento no reivindicado es esencial para la práctica de la presente divulgación.
[0338] Las realizaciones alternativas de la divulgación reivindicada se describen en el presente documento, incluyendo el mejor modo conocido por los inventores para poner en práctica la invención reivindicada. De estas, las variaciones de las realizaciones descritas serán evidentes para los expertos en la técnica al leer la descripción anterior. Los inventores esperan que los expertos en la materia empleen dichas variaciones según sea apropiado (por ejemplo, alterando o combinando características o realizaciones), y los inventores pretenden que la invención se ponga en práctica de manera diferente a la descrita específicamente en este documento.
[0339] Además, cualquier combinación de los elementos descritos anteriormente en todas las posibles variaciones de los mismos está incluida en la invención a menos que se indique lo contrario en el presente documento o esté claramente contradicha por el contexto.
[0340] El uso de valores numéricos individuales se establece como aproximaciones como si los valores estuvieran precedidos por la palabra "aproximadamente” o “alrededor de". De manera similar, los valores numéricos en los diversos rangos especificados en esta solicitud, a menos que se indique expresamente lo contrario, se presentan como aproximaciones, como si los valores mínimo y máximo dentro de los rangos establecidos estuvieran precedidos por la palabra "aproximadamente” o “alrededor de". De esta manera, las variaciones por encima y por debajo de los rangos indicados pueden usarse para lograr sustancialmente los mismos resultados que los valores dentro de los rangos. Tal como se usa en el presente documento, los términos "aproximadamente" y "alrededor de" cuando se refieren a un valor numérico tendrán un significado simple y corriente para una persona con experiencia ordinaria en la técnica con la cual el tema divulgado está más estrechamente relacionado o la técnica relevante para el rango o elemento en cuestión. La cantidad de ampliación desde el límite numérico estricto depende de muchos factores. Por ejemplo, algunos de los factores que pueden considerarse incluyen la criticidad del elemento y/o el efecto que tendrá una cantidad determinada de variación en el desempeño del tema reivindicado, así como otras consideraciones conocidas por los expertos en la materia. Tal como se usa en el presente documento, el uso de diferentes cantidades de cifras significativas para diferentes valores numéricos no pretende limitar la forma en que el uso de las palabras "aproximadamente” o “alrededor de" servirá para ampliar un valor o rango numérico particular. Por lo tanto, como cuestión general, "aproximadamente” o “alrededor de" amplían el valor numérico. Además, la divulgación de los rangos está pensada como un rango continuo que incluye cada valor entre los valores mínimo y máximo más la ampliación del rango permitido por el uso del término "aproximadamente” o “alrededor de". Por lo tanto, la recitación de los rangos de valores en este documento simplemente pretende servir como un método abreviado para referirse individualmente a cada valor separado que cae dentro del rango, a menos que se indique lo contrario en este documento, y cada valor separado se incorpora a la especificación como si se hubiera recitado individualmente en este documento.
[0341] Debe entenderse que cualquier intervalo, relación e intervalo de relación que puede formarse o derivarse de cualquiera de los datos descritos en el presente documento representa realizaciones adicionales de la presente divulgación y se incluyen como parte de la divulgación como si fueran explícitamente establecidos. Esto incluye rangos que pueden formarse que incluyen o no un límite superior y/o inferior finitos. Por consiguiente, un experto en la técnica más estrechamente relacionado con un intervalo, relación o intervalo de relaciones en particular apreciará que tales valores pueden derivarse de forma no ambigua de los datos presentados en este documento.
Claims (4)
1. Un compuesto seleccionado del grupo que consiste en:




o una sal farmacéuticamente aceptable del mismo.
2. Un compuesto seleccionado del grupo que consiste en:

o una sal farmacéuticamente aceptable del mismo.
3. Una composición farmacéutica que comprende un compuesto o una sal farmacéuticamente aceptable del mismo como se define en cualquiera de las reivindicaciones 1-2, y un excipiente farmacéuticamente aceptable.
4. Un compuesto o sal farmacéuticamente aceptable del mismo como se define en cualquiera de las reivindicaciones 1-2, para uso en el tratamiento del VHC en un paciente que lo necesite.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361798961P | 2013-03-15 | 2013-03-15 | |
| PCT/US2014/029765 WO2014145095A1 (en) | 2013-03-15 | 2014-03-14 | Macrocyclic and bicyclic inhibitors of hepatitis c virus |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2735355T3 true ES2735355T3 (es) | 2019-12-18 |
Family
ID=50694007
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14723582T Active ES2735355T3 (es) | 2013-03-15 | 2014-03-14 | Inhibidores macrocíclicos y bicíclicos de virus de hepatitis C |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US9617310B2 (es) |
| EP (1) | EP2970335B1 (es) |
| JP (2) | JP6511432B2 (es) |
| KR (1) | KR102215400B1 (es) |
| CN (1) | CN105073758B (es) |
| AU (1) | AU2014233390B2 (es) |
| BR (1) | BR112015021768A2 (es) |
| CA (1) | CA2902569A1 (es) |
| EA (1) | EA029088B1 (es) |
| ES (1) | ES2735355T3 (es) |
| HK (1) | HK1217196A1 (es) |
| IL (1) | IL240662A0 (es) |
| MX (1) | MX2015013224A (es) |
| SG (1) | SG11201507223TA (es) |
| WO (1) | WO2014145095A1 (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA119315C2 (uk) | 2012-07-03 | 2019-06-10 | Гіліад Фармассет Елелсі | Інгібітори вірусу гепатиту с |
| EP3939985A1 (en) | 2014-12-26 | 2022-01-19 | Emory University | Pharmaceutical compositions comprising n4-hydroxycytidine derivatives for the treatment or prevention of influenza or coronavirus infections |
| CN105503751B (zh) * | 2015-12-22 | 2018-06-05 | 广西师范大学 | 一种喹喔啉衍生物的高效合成方法 |
| RU2020116571A (ru) | 2017-12-07 | 2021-11-22 | Эмори Юниверсити | N4-гидроксицитидин и производные и связанные с этим противовирусные применения |
| CN108329332A (zh) * | 2018-03-16 | 2018-07-27 | 安徽华昌高科药业有限公司 | 一种制备Glecaprevir的方法 |
| CN111004188B (zh) * | 2019-12-18 | 2021-04-20 | 安徽红杉生物医药科技有限公司 | Ns3/4a蛋白酶抑制剂中间体及其合成方法、应用 |
| CN111018795B (zh) * | 2019-12-25 | 2023-03-28 | 上海彩迩文生化科技有限公司 | 一种碱性条件下合成喹喔啉-3-酮的方法 |
Family Cites Families (107)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUP0303436A2 (hu) | 2000-12-12 | 2004-01-28 | Schering Corp. | Diaril-peptidek mint a hepatitis C vírus NS3-szerin proteáz inhibitorai, ezeket tartalmazó gyógyszerkészítmények és alkalmazásuk |
| BR0213562A (pt) | 2001-10-26 | 2004-08-31 | Aventis Pharma Inc | Benzimidazóis e análogos e seu uso como inibidores de cinases de proteìna |
| WO2004032827A2 (en) | 2002-05-20 | 2004-04-22 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| ATE413411T1 (de) | 2002-05-20 | 2008-11-15 | Bristol Myers Squibb Co | Substituierte cycloalkyl p1' hepatitis c virus inhibitoren |
| DE60334205D1 (en) | 2002-05-20 | 2010-10-28 | Bristol Myers Squibb Co | Heterocyclische sulfonamid-hepatitis-c-virus-hemmer |
| ES2386161T3 (es) | 2003-04-16 | 2012-08-10 | Bristol-Myers Squibb Company | Proceso para separar una mezcla de enantiómeros de éster alquílico usando una enzima |
| SI1615613T1 (sl) | 2003-04-18 | 2010-03-31 | Enanta Pharm Inc | Kinoksalinilni makrociklični inhibitorji serinske proteaze hepatitisa C |
| US7176208B2 (en) | 2003-04-18 | 2007-02-13 | Enanta Pharmaceuticals, Inc. | Quinoxalinyl macrocyclic hepatitis C serine protease inhibitors |
| ES2297424T3 (es) | 2003-05-21 | 2008-05-01 | Boehringer Ingelheim International Gmbh | Compuestos inhibidores de la hepatitis c. |
| WO2005018330A1 (en) * | 2003-08-18 | 2005-03-03 | Pharmasset, Inc. | Dosing regimen for flaviviridae therapy |
| US7135462B2 (en) | 2003-11-20 | 2006-11-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| PL1778702T3 (pl) | 2004-07-16 | 2011-12-30 | Gilead Sciences Inc | Związki przeciwwirusowe |
| UY29016A1 (es) | 2004-07-20 | 2006-02-24 | Boehringer Ingelheim Int | Analogos de dipeptidos inhibidores de la hepatitis c |
| WO2007001406A2 (en) | 2004-10-05 | 2007-01-04 | Chiron Corporation | Aryl-containing macrocyclic compounds |
| AU2006242475B2 (en) | 2005-05-02 | 2011-07-07 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US7470664B2 (en) | 2005-07-20 | 2008-12-30 | Merck & Co., Inc. | HCV NS3 protease inhibitors |
| JP5249028B2 (ja) | 2005-07-25 | 2013-07-31 | インターミューン・インコーポレーテッド | C型肝炎ウイルス複製の新規大環状阻害剤 |
| PE20070211A1 (es) | 2005-07-29 | 2007-05-12 | Medivir Ab | Compuestos macrociclicos como inhibidores del virus de hepatitis c |
| CA2615896C (en) | 2005-08-01 | 2012-11-13 | Merck & Co., Inc. | Macrocyclic peptides as hcv ns3 protease inhibitors |
| DK1999129T3 (da) | 2005-10-11 | 2011-02-07 | Intermune Inc | Forbindelser og fremgangsmåder til inhibering af replikationen af hepatitis C-virus |
| US7772183B2 (en) | 2005-10-12 | 2010-08-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| GB0609492D0 (en) * | 2006-05-15 | 2006-06-21 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| GB0612423D0 (en) | 2006-06-23 | 2006-08-02 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| KR20090024834A (ko) | 2006-07-05 | 2009-03-09 | 인터뮨, 인크. | C형 간염 바이러스 복제의 신규 억제제 |
| CA2667165A1 (en) | 2006-10-24 | 2008-05-02 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| US8309540B2 (en) | 2006-10-24 | 2012-11-13 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| EP2079479B1 (en) | 2006-10-24 | 2014-11-26 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| CN101568346B (zh) | 2006-10-27 | 2015-11-25 | 默沙东公司 | Hcv ns3蛋白酶抑制剂 |
| KR101615500B1 (ko) | 2006-10-27 | 2016-04-27 | 머크 샤프 앤드 돔 코포레이션 | Hcv ns3 프로테아제 억제제 |
| US7888464B2 (en) | 2006-11-16 | 2011-02-15 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20080287449A1 (en) | 2007-04-26 | 2008-11-20 | Deqiang Niu | Aza-tripeptide hepatitis c serine protease inhibitors |
| CL2008001274A1 (es) | 2007-05-03 | 2008-11-03 | Intermune Inc Y Array Biopharma Inc | Compuestos macrolidos derivados de 5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahidrociclopropan[e]pirrolo[1,2-a][1,4]diazaciclopentadecinilo, inhibidores de la actividad de la proteasa ns3-ns4; su procedimiento de preparacion; composicion farmaceutica; compuestos intermediarios y procedimiento de preparacion; y uso en el tratamiento de..... |
| BRPI0811447A2 (pt) | 2007-05-10 | 2014-10-29 | Intermune Inc | Compostos, composição farmacêutica e métodos de inibição da atividade da protease ns3/ns4, de tratamento da fibrose hepática e de intensificação da função hepática num indivíduo tendo infecção de vírus da hepatite c. |
| US20090005387A1 (en) | 2007-06-26 | 2009-01-01 | Deqiang Niu | Quinoxalinyl macrocyclic hepatitis c virus serine protease inhibitors |
| NZ582096A (en) * | 2007-06-29 | 2012-05-25 | Gilead Sciences Inc | Antiviral compounds that inhibit hepatitis c virus (hcv) |
| BRPI0813733A2 (pt) | 2007-06-29 | 2019-11-05 | Gilead Sciences Inc | composto com atividade inibidora de hcv, sua composição farmacêutica e seu uso. |
| US8927569B2 (en) | 2007-07-19 | 2015-01-06 | Merck Sharp & Dohme Corp. | Macrocyclic compounds as antiviral agents |
| CA2700383A1 (en) | 2007-09-24 | 2009-04-02 | Achillion Pharmaceuticals, Inc. | Urea-containing peptides as inhibitors of viral replication |
| WO2009055467A2 (en) | 2007-10-24 | 2009-04-30 | Virobay, Inc. | Compounds that inhibit protease cathepsin s and hcv replication |
| US20090111757A1 (en) | 2007-10-25 | 2009-04-30 | Taigen Biotechnology Co., Ltd. | Hcv protease inhibitors |
| CL2008003384A1 (es) | 2007-11-14 | 2009-12-11 | Enanta Pharm Inc | Compuestos derivados de quinoxalina macrocíclica, inhibidores de serina proteasa; composicion farmaceutica que los comprende; y su uso en el tratamiento de la hepatitis c. |
| US8263549B2 (en) | 2007-11-29 | 2012-09-11 | Enanta Pharmaceuticals, Inc. | C5-substituted, proline-derived, macrocyclic hepatitis C serine protease inhibitors |
| US8030307B2 (en) | 2007-11-29 | 2011-10-04 | Enanta Pharmaceuticals, Inc. | Bicyclic, C5-substituted proline derivatives as inhibitors of the hepatitis C virus NS3 protease |
| CN101932242A (zh) | 2007-12-05 | 2010-12-29 | 益安药业 | 喹喔啉基衍生物 |
| WO2009079353A1 (en) | 2007-12-14 | 2009-06-25 | Enanta Pharmaceuticals, Inc. | Triazole-containing macrocyclic hcv serine protease inhibitors |
| US8202996B2 (en) | 2007-12-21 | 2012-06-19 | Bristol-Myers Squibb Company | Crystalline forms of N-(tert-butoxycarbonyl)-3-methyl-L-valyl-(4R)-4-((7-chloro-4-methoxy-1-isoquinolinyl)oxy)-N- ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-L-prolinamide |
| US8293705B2 (en) | 2007-12-21 | 2012-10-23 | Avila Therapeutics, Inc. | HCV protease inhibitors and uses thereof |
| CN101951770B (zh) | 2007-12-21 | 2014-12-31 | 阿维拉制药公司 | Hcv蛋白酶抑制剂和其用途 |
| US8309685B2 (en) | 2007-12-21 | 2012-11-13 | Celgene Avilomics Research, Inc. | HCV protease inhibitors and uses thereof |
| US8501682B2 (en) | 2008-01-24 | 2013-08-06 | Enanta Pharmaceuticals, Inc. | Difluorinated tripeptides as HCV serine protease inhibitors |
| KR20100118991A (ko) | 2008-02-04 | 2010-11-08 | 아이데닉스 파마슈티칼스, 인코포레이티드 | 매크로시클릭 세린 프로테아제 억제제 |
| EP2268285B1 (en) | 2008-02-25 | 2018-06-27 | Merck Sharp & Dohme Corp. | Therapeutic compounds |
| WO2009114633A1 (en) | 2008-03-12 | 2009-09-17 | Virobay, Inc. | Process for the preparation of (3s)-3-amino-n-cyclopropyl-2-hydroxyalkanamide derivatives |
| US8372802B2 (en) | 2008-03-20 | 2013-02-12 | Enanta Pharmaceuticals, Inc. | Fluorinated macrocyclic compounds as hepatitis C virus inhibitors |
| US8048862B2 (en) | 2008-04-15 | 2011-11-01 | Intermune, Inc. | Macrocyclic inhibitors of hepatitis C virus replication |
| US8163921B2 (en) | 2008-04-16 | 2012-04-24 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8461107B2 (en) | 2008-04-28 | 2013-06-11 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| WO2009134987A1 (en) | 2008-04-30 | 2009-11-05 | Enanta Pharmaceuticals, Inc. | Difluoromethyl-containing macrocyclic compounds as hepatitis c virus inhibitors |
| ME02024B (me) * | 2008-07-22 | 2015-05-20 | Merck Sharp & Dohme | Makrociklična jedinjenja hinoksalina kao inhibitori hcv ns3 proteaze |
| KR101647520B1 (ko) | 2008-08-07 | 2016-08-10 | 에프. 호프만-라 로슈 아게 | 거대환식 화합물의 제조 방법 |
| EP2334680A2 (en) | 2008-08-20 | 2011-06-22 | Sequoia Pharmaceuticals, Inc. | Hcv protease inhibitors |
| UY32099A (es) | 2008-09-11 | 2010-04-30 | Enanta Pharm Inc | Inhibidores macrocíclicos de serina proteasas de hepatitis c |
| WO2010033466A1 (en) | 2008-09-16 | 2010-03-25 | Phenomix Corporation | Macrocyclic inhibitors of hepatitis c protease |
| EP2687526A1 (en) | 2008-09-16 | 2014-01-22 | Boehringer Ingelheim International Gmbh | Crystalline forms of a 2-thiazolyl- 4-quinolinyl-oxy derivative, a potent HCV inhibitor |
| US8563505B2 (en) | 2008-09-29 | 2013-10-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8044087B2 (en) | 2008-09-29 | 2011-10-25 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US20100080770A1 (en) | 2008-09-29 | 2010-04-01 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| WO2010045266A1 (en) | 2008-10-15 | 2010-04-22 | Intermune, Inc. | Therapeutic antiviral peptides |
| WO2010048468A1 (en) | 2008-10-23 | 2010-04-29 | Concert Pharmaceuticals, Inc. | Deuterated macrocyclic inhibitors of viral ns3 protease |
| JP5689810B2 (ja) | 2008-11-20 | 2015-03-25 | アキリオン ファーマシューティカルズ,インコーポレーテッド | C型肝炎ウイルス用の環状カルボキサミド化合物およびその類似体 |
| ES2445516T3 (es) | 2008-11-21 | 2014-03-03 | Boehringer Ingelheim International Gmbh | Composición farmacéutica de un potente inhibidor de HCV para su administración oral |
| US20100272674A1 (en) | 2008-12-04 | 2010-10-28 | Bristol-Myers Squibb Company | Hepatitis C Virus Inhibitors |
| NZ592705A (en) | 2008-12-10 | 2013-02-22 | Achillion Pharmaceuticals Inc | 4-amino-4-oxobutanoyl peptide cyclic analogues, inhibitors of viral replication |
| US8283310B2 (en) | 2008-12-15 | 2012-10-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CA2747636A1 (en) | 2008-12-19 | 2010-07-15 | Gilead Sciences, Inc. | Hcv ns3 protease inhibitors |
| BRPI0923393B1 (pt) | 2008-12-23 | 2018-06-19 | Ortho-Mcneil-Janssen Pharmaceuticals, Inc. | "processos para preparar um inibidor de protease de hcv macrocíclico e intermediários e uso". |
| MX2011012155A (es) | 2009-05-13 | 2012-02-28 | Enanta Pharm Inc | Compuestos macrociclicos como inhibidores del virus de hepatitis c. |
| EP2432318A4 (en) | 2009-05-22 | 2012-11-21 | Sequoia Pharmaceuticals Inc | NS3 PROTEASE INHIBITORS OF BIPACROCYCLIC HEPATITIS C VIRUS |
| US8828930B2 (en) | 2009-07-30 | 2014-09-09 | Merck Sharp & Dohme Corp. | Hepatitis C virus NS3 protease inhibitors |
| CA2770338A1 (en) | 2009-08-27 | 2011-03-03 | Merck Sharp & Dohme Corp. | Processes for preparing protease inhibitors of hepatitis c virus |
| WO2011038293A1 (en) | 2009-09-28 | 2011-03-31 | Intermune, Inc. | Cyclic peptide inhibitors of hepatitis c virus replication |
| TW201116540A (en) | 2009-10-01 | 2011-05-16 | Intermune Inc | Therapeutic antiviral peptides |
| WO2011049908A2 (en) | 2009-10-19 | 2011-04-28 | Enanta Pharmaceuticals, Inc. | Bismacrokyclic compounds as hepatitis c virus inhibitors |
| AU2011224558B2 (en) | 2010-03-10 | 2014-02-27 | Abbvie Ireland Unlimited Company | Solid compositions |
| US8530497B2 (en) | 2010-03-11 | 2013-09-10 | Boehringer Ingelheim International Gmbh | Crystalline salts of a potent HCV inhibitor |
| CA2801584A1 (en) | 2010-06-07 | 2011-12-15 | Abbvie Inc. | Macrocyclic hepatitis c serine protease inhibitors |
| WO2012019299A1 (en) | 2010-08-11 | 2012-02-16 | Boehringer Ingelheim International Gmbh | Hepatitis c inhibitor compounds |
| US20120094897A1 (en) | 2010-09-15 | 2012-04-19 | Intermune, Inc. | Novel inhibitors of hepatitis c virus replication |
| EP2618665A4 (en) | 2010-09-21 | 2014-08-20 | Merck Sharp & Dohme | HCV NS3 PROTEASE INHIBITORS |
| EA023009B1 (ru) | 2010-09-21 | 2016-04-29 | Энанта Фармасьютикалз, Инк. | Ингибитор hcv сериновой протеазы, полученный из макроциклического пролина, и фармацевтическая композиция, предназначенная для лечения вирусной инфекции |
| WO2012040242A1 (en) | 2010-09-22 | 2012-03-29 | Intermune, Inc. | Substituted proline inhibitors of hepatitis c virus replication |
| WO2012047764A1 (en) | 2010-10-04 | 2012-04-12 | Intermune, Inc. | Therapeutic antiviral peptides |
| WO2012054874A1 (en) | 2010-10-22 | 2012-04-26 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| AU2011323658A1 (en) | 2010-11-01 | 2013-05-23 | Genoscience Pharma | Novel specific HCV NS3 protease inhibitors |
| AU2011344075A1 (en) | 2010-12-14 | 2013-05-09 | Merck Sharp & Dohme Corp. | Process and intermediates for preparing macrolactams |
| BR112013016480A2 (pt) | 2010-12-30 | 2016-09-20 | Abbvie Inc | macrocíclo da fenantridina inibadores da protease da serina da hepatite c |
| JP2014502620A (ja) | 2010-12-30 | 2014-02-03 | エナンタ ファーマシューティカルズ インコーポレイテッド | 大環状c型肝炎セリンプロテアーゼ阻害剤 |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EP2726475B1 (en) | 2011-05-27 | 2017-10-25 | Bristol-Myers Squibb Company | Tripeptides incorporating deuterium as inhibitors of hepatitis c virus |
| US8691757B2 (en) | 2011-06-15 | 2014-04-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2012171332A1 (zh) | 2011-06-16 | 2012-12-20 | 爱博新药研发(上海)有限公司 | 抑制丙型肝炎病毒的大环状杂环化合物及其制备和应用 |
| WO2012176149A1 (en) | 2011-06-23 | 2012-12-27 | Panmed Ltd. | Treatment of hepatitis c virus |
| MX2014001945A (es) | 2011-08-19 | 2014-03-27 | Merck Sharp & Dohme | Procedimiento e intermedios para la preparacion de macrolactamas. |
| EP2773342A4 (en) | 2011-10-31 | 2015-08-26 | Merck Sharp & Dohme | COMPOSITIONS USEFUL FOR THE TREATMENT OF VIRAL DISEASES |
| EP2780026B1 (en) | 2011-11-15 | 2019-10-23 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
| CA2861041A1 (en) | 2012-01-12 | 2013-07-18 | Boehringer Ingelheim International Gmbh | Stabilized pharmaceutical formulations of a potent hcv inhibitor |
| UA119315C2 (uk) * | 2012-07-03 | 2019-06-10 | Гіліад Фармассет Елелсі | Інгібітори вірусу гепатиту с |
-
2014
- 2014-03-14 EP EP14723582.4A patent/EP2970335B1/en active Active
- 2014-03-14 KR KR1020157028437A patent/KR102215400B1/ko active IP Right Grant
- 2014-03-14 MX MX2015013224A patent/MX2015013224A/es unknown
- 2014-03-14 CA CA2902569A patent/CA2902569A1/en not_active Abandoned
- 2014-03-14 CN CN201480015589.0A patent/CN105073758B/zh active Active
- 2014-03-14 BR BR112015021768A patent/BR112015021768A2/pt not_active Application Discontinuation
- 2014-03-14 SG SG11201507223TA patent/SG11201507223TA/en unknown
- 2014-03-14 JP JP2016503218A patent/JP6511432B2/ja active Active
- 2014-03-14 ES ES14723582T patent/ES2735355T3/es active Active
- 2014-03-14 US US14/214,477 patent/US9617310B2/en active Active
- 2014-03-14 AU AU2014233390A patent/AU2014233390B2/en active Active
- 2014-03-14 WO PCT/US2014/029765 patent/WO2014145095A1/en active Application Filing
- 2014-03-14 EA EA201591456A patent/EA029088B1/ru not_active IP Right Cessation
-
2015
- 2015-08-18 IL IL240662A patent/IL240662A0/en unknown
-
2016
- 2016-05-05 HK HK16105156.1A patent/HK1217196A1/zh unknown
-
2017
- 2017-03-10 US US15/455,508 patent/US20170313745A1/en not_active Abandoned
-
2018
- 2018-12-13 JP JP2018233621A patent/JP2019059775A/ja not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| CN105073758B (zh) | 2017-08-11 |
| JP6511432B2 (ja) | 2019-05-15 |
| EP2970335A1 (en) | 2016-01-20 |
| CA2902569A1 (en) | 2014-09-18 |
| SG11201507223TA (en) | 2015-10-29 |
| AU2014233390B2 (en) | 2018-03-01 |
| IL240662A0 (en) | 2015-10-29 |
| WO2014145095A1 (en) | 2014-09-18 |
| EA201591456A1 (ru) | 2016-02-29 |
| JP2016516069A (ja) | 2016-06-02 |
| EP2970335B1 (en) | 2019-05-08 |
| CN105073758A (zh) | 2015-11-18 |
| HK1217196A1 (zh) | 2016-12-30 |
| EA029088B1 (ru) | 2018-02-28 |
| JP2019059775A (ja) | 2019-04-18 |
| KR20150130456A (ko) | 2015-11-23 |
| MX2015013224A (es) | 2015-12-11 |
| US9617310B2 (en) | 2017-04-11 |
| AU2014233390A1 (en) | 2015-09-03 |
| US20140274884A1 (en) | 2014-09-18 |
| BR112015021768A2 (pt) | 2016-02-02 |
| KR102215400B1 (ko) | 2021-02-10 |
| US20170313745A1 (en) | 2017-11-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2017203984B2 (en) | Inhibitors of Hepatitis C virus | |
| ES2735355T3 (es) | Inhibidores macrocíclicos y bicíclicos de virus de hepatitis C |