EP1114883A1 - Verfahren zur elektrochemischen Herstellung von Alkalimetall aus Alkalimetallamalgam - Google Patents
Verfahren zur elektrochemischen Herstellung von Alkalimetall aus Alkalimetallamalgam Download PDFInfo
- Publication number
- EP1114883A1 EP1114883A1 EP99124037A EP99124037A EP1114883A1 EP 1114883 A1 EP1114883 A1 EP 1114883A1 EP 99124037 A EP99124037 A EP 99124037A EP 99124037 A EP99124037 A EP 99124037A EP 1114883 A1 EP1114883 A1 EP 1114883A1
- Authority
- EP
- European Patent Office
- Prior art keywords
- alkali metal
- amalgam
- potassium
- sodium
- anode
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25C—PROCESSES FOR THE ELECTROLYTIC PRODUCTION, RECOVERY OR REFINING OF METALS; APPARATUS THEREFOR
- C25C3/00—Electrolytic production, recovery or refining of metals by electrolysis of melts
- C25C3/02—Electrolytic production, recovery or refining of metals by electrolysis of melts of alkali or alkaline earth metals
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25C—PROCESSES FOR THE ELECTROLYTIC PRODUCTION, RECOVERY OR REFINING OF METALS; APPARATUS THEREFOR
- C25C7/00—Constructional parts, or assemblies thereof, of cells; Servicing or operating of cells
Definitions
- the present invention relates to an improved method for electrochemical Production of alkali metal from alkali metal amalgam.
- alkali metal sodium and potassium.
- the invention further relates to a method suitable for carrying out this method Electrolysis cell and the principle of a production plant.
- Sodium is an important inorganic basic product that is used, for example, for Production of sodium amide, sodium alcoholates and sodium borohydride used becomes. It is made technically after the Downs process by electrolysis of molten Table salt won. This process has a high energy consumption of> 10 kWh / kg sodium (Büchner et al., Industrial Inorganic Chemistry, 2nd edition, Verlag Chemie, p. 228 f). Furthermore, the method has the serious disadvantage that the electrolysis cells are destroyed when the salt melt solidifies become. Furthermore, the sodium metal obtained by the Downs process has the Disadvantage that it is contaminated with calcium due to the process, the residual content of subsequent cleaning steps are only reduced, but never completely excluded can be.
- Potassium is also an important inorganic basic product, which is used, for example, for the production of potassium alcoholates, potassium amides and potassium alloys is used.
- a good yield is achieved in that potassium vapor from the Reaction zone is withdrawn, creating balance on the potassium side postponed (Ullmann's Encyclopedia of Industrial Chemistry, 6th edition 1998, Electronic release).
- the disadvantage is that the process at high temperatures (870 ° C) is working.
- the resulting potassium contains about 1% sodium as an impurity and must therefore be cleaned up by a further rectification.
- the biggest The disadvantage is that the sodium used is expensive. This is also because sodium technically according to the Downs process by electrolysis of molten table salt is obtained, whereby an energy expenditure of at least 10 kWh / kg sodium is necessary is. This corresponds to about 5.3 kWh / kg of potassium (with 100% yield).
- Sodium amalgam and potassium amalgam are intermediates used in chlor-alkali electrolysis accrued in large quantities by the amalgam process and in the Usually converted to alkali metal solution with water immediately after production become.
- the alkali metal-low or alkali metal-free alkali metal amalgam is normally immediately returned to chlor-alkali electrolysis.
- the sodium concentration of this must Solution kept at values of less than 1 wt .-%, preferably 0.2 to 0.5 wt .-% become.
- the Potassium concentration of the solution at less than 1.5% by weight, preferably 0.3 to 0.6 % By weight.
- the amalgams obtained on an industrial scale contain in essential metallic impurities in the concentration range from 1 to 30 ppm such as copper, iron, potassium (or sodium in potassium amalgam), lead and Zinc.
- GB 1,155,927 describes a process according to which sodium metal can be obtained from sodium amalgam electrochemically using a solid sodium ion conductor such as beta-Al 2 O 3 with amalgam as the anode and sodium as the cathode.
- the execution of the method described in GB 1,155,927 does not lead to the results described there with regard to sodium conversion, product purity and current density. Furthermore, the system described behaves unstably over the course of a few days if the claimed temperature range is maintained.
- the task was an improved process for electrochemical production of alkali metal from an alkali metal amalgam to provide an energetic cheaper sodium production allowed than the Downs process or one Energy-efficient production of potassium allowed than that discussed at the beginning technical processes.
- the method described under GB 1,155,927 be significantly improved so that the new process in the existing network chlorine-alkali electrolysis can be integrated using the amalgam process and the disadvantages found when performing the method according to GB 1,155,927 be avoided.
- the alkali metal conversion on the anode side must meet the balance requirements of Product combination with chlor-alkali electrolysis are sufficient. That is, the Drain concentration of alkali metal in the amalgam of chlor-alkali electrolysis corresponds to the feed concentration in the alkali metal electrolysis according to the invention. Furthermore, those between chlor-alkali electrolysis and the invention Alkaline metal electrolysis circulating amounts of amalgam in one technically and economically justifiable size. Usually this is achieved when the alkali metal content of the incoming amalgams is implemented to 50%. The sodium metal must primarily in one such purity arise that further process steps for mercury separation can be eliminated and the disadvantage given in the Downs process Calcium contamination is avoided.
- the potassium metal must primarily be in one Purity arise that further process steps for mercury separation are omitted can and the sodium content is lower than in the reduction with sodium, where the Primarily produced potassium contains 1% sodium.
- the process is intended in industrial Scale can be realized and must therefore have sufficiently high current densities and space-time yields enable. Due to the statics of the production building, the Security, environmental protection and capital commitment is an instrumental concept required, which gets by with a relatively small mercury content.
- the process should be stable in continuous operation and the usual in technical Alkaline metal amalgam occurring metallic contaminants undamaged tolerate.
- alkali metal amalgam refers to a solution of one Alkali metal in mercury, which is liquid at the reaction temperature.
- the present invention thus relates to a method for producing alkali metal starting from alkali metal amalgam by electrolysis with an alkali metal amalgam as an anode, an alkali metal ion-conducting solid electrolyte and liquid alkali metal as a cathode, characterized in that the alkali metal amalgam moves as an anode becomes.
- the anode potential is kept that anodically only alkali metal is oxidized to the alkali metal ion, the ion is transported through the solid electrolyte in the electric field and eventually is reduced cathodically to alkali metal.
- the present invention also relates to a specially adapted electrolytic cell comprising a tubular solid electrolyte (1/31) which is closed on one side and which is in a Concentric stainless steel tube (33) is installed, in which the inventive method can be operated particularly preferably on an industrial scale.
- the process according to the invention is carried out in an electrolysis cell with a moving liquid alkali metal amalgam anode operated.
- This is a moving liquid anode, which during operation with regard to its Alkaline metal content is depleted, so that it is by alkali metal-rich amalgam, that in a normal amalgam cell of a chlor-alkali production or by Electrolysis of sodium or potassium salts with an Hg or amalgam cathode, such as e.g. NaOH or KOH that can be obtained can be replaced.
- the concentrated amalgam flow becomes one normal amalgam cell in a heat exchanger to the operating temperature of the inventive method heated and the hot, moving liquid anode fed. This is expediently carried out in a countercurrent heat exchanger so that the hot, depleted amalgam heats the feed.
- the replacement of depleted amalgam can be both discontinuous as well done continuously.
- the continuous operation is, however, easier to carry out in operational terms.
- the Disadvantage that the incoming concentrate is already circulated depleted alkali metal amalgam can be balanced be that the process is carried out in several stages.
- the liquid anode is expediently stirred and / or with a pump in a circuit under atmospheric pressure or slightly positive pressure emotional.
- the caused by the sales-related exchange of amalgam Movement or thermal convection is compared to that in the invention Procedure required movement is negligible and is not enough, the preferred To achieve current densities.
- the anode-side power supply is advantageously carried out via the stainless steel housing the electrolytic cell, which is stable under the reaction conditions.
- the anode side is suitably electrically insulated from the cathode side.
- the cathode is made of alkali metal, which is used at the temperatures required for stabilization of the anode process are required.
- the alkali metal is advantageously in the form of a solid reservoir in the electrolysis cell Introduced cathode compartment. At the beginning of the electrolysis, the alkali metal is then melted.
- the alkali metal can also be in liquid form at the beginning of the Electrolysis are introduced into the cathode compartment. In a technically simple way can the alkali metal formed in the process of the invention by an overflow be removed from the cathode compartment, by throttling the Alkali metal flow is ensured that the pressure on the alkali metal side higher is as the pressure on the amalgam side.
- the overpressure of the cathode compared to the anode in the process according to the invention is 0.1 to 5 bar, preferably 0.5 to 1 bar.
- the cathodic power supply is advantageously carried out via the Alkali metal filling and the drain pipes or connecting flanges.
- the anode and cathode compartments are separated from one another by a helium-tight alkali metal ion-conducting solid electrolyte.
- ceramic materials such as NASICON®, the composition of which is specified in EP-A 0 553 400, come into consideration in the production of sodium. Glasses which conduct sodium ions are also suitable, as are zeolites and feldspar. A large number of materials can also be used in the production of potassium. Both the use of ceramics and the use of glasses are possible.
- the following materials can be used: KBiO 3 (TN Nguyen et al., Chem. Mater.
- Potassium- ⁇ "aluminum oxide, potassium- ⁇ -aluminum oxide or potassium- ⁇ / ⁇ " aluminum oxide can be prepared from sodium- ⁇ "aluminum oxide, sodium- ⁇ -aluminum oxide or sodium- ⁇ / ⁇ " aluminum oxide by cation exchange become.
- the solid electrolyte expediently has the form of a thin-walled, yet pressure-resistant, tube closed on one side (EP-B 0 424 673), on the open end of which an electrically insulating ring is applied by means of a helium-tight, likewise electrically insulating glass solder connection (GB 2 207 545, EP-B 0 482 785).
- the wall thickness of the electrolyte which conducts alkali metal ions is 0.3 to 5 mm, preferably 1 to 3 mm, particularly preferably 1 to 2 mm.
- the cross-sectional shape of the tube closed on one side is circular in the preferred embodiment, in a further embodiment cross-sectional shapes with an enlarged surface are used, which can be derived, for example, from the combination of several circular surfaces, as shown in FIG. 5.
- the design of the solid electrolyte which conducts alkali metal ions with regard to its leak tightness has a decisive influence on the process according to the invention, since mercury can only get into the sodium produced via leaks in the solid electrolyte or sealing system, since in the process according to the invention the anode potentials are set such that the formation of mercury ions is excluded becomes.
- solid electrolytes are used which have leak rates of less than 1 * 10 -9 mbar * liter * sec -1 in a helium leak test, i.e. are helium-tight within the detection limit.
- the releasable sealing connections are preferably carried out so that Alkali metal and amalgam are each sealed off from the ambient atmosphere. If possible, it is avoided to dissolve between alkali metal and amalgam To have seals because the detachable seals are usually liquid-tight but not gas-tight. In the excluded case, mercury vapor could be caused by the Diffuse removable seal and undesirably contaminate the alkali metal.
- a preferred embodiment come as releasable sealing connections Flat seals are used, preferably made of graphite, for example unreinforced GRAPHIFLEX®.
- the ceramic tubes are expediently under vacuum after sintering packed in diffusion-proof aluminum / plastic composite films. For storage the originally packed ceramic tubes are filled with tightly filled argon Metal container included.
- the ceramic resistance can be significantly reduced, for example, if the cell is first operated with reversed polarity, that is to say the anode is first operated as a cathode.
- the cathode like the anode, can consist of sodium amalgam and mercury.
- the current density in the reversed state is linear over a period of 1 to 44 h, preferably 2 to 6 h, from 50 A / m 2 to 3000 A / m 2 (sodium) or from 30 A / m 2 to 1000 A / m 2 (Potassium) increased.
- the lowest ceramic resistances are obtained when starting for 1 to 24 Hours at an operating temperature of 300 ° C to 350 ° C (sodium) or 250 ° C to 350 ° C (potassium) first liquid alkali metal is used as the anode, which then replaced by amalgam. This embodiment of conditioning is particularly preferred.
- the action of Water vapor on the ceramics that conduct alkali metal ions is also essential be excluded.
- the water traces lead to amalgam heated, the water vapor removed and only then the water-free amalgam-mercury mixture fed to the liquid anode. Removal of water vapor is conveniently by stripping with inert gas or by applying Negative pressure supported.
- reaction temperature is kept in the temperature range of 250 ° C to 300 ° C described in GB 1,155,927, which forms a safety distance from the boiling point of mercury, a reduction in the initially stable current density of 1000 is observed over a period of 1 to 5 days at constant cell voltage A / m 2 to 3000 A / m 2 to values from 100 A / m 2 to 300 A / m 2 (sodium) or from 500 A / m 2 to 1000 A / m 2 to 50 A / m 2 to 70 A / m 2 (potassium).
- the increase in cell voltage leads only to an insignificant increase in current, but in the course of 2 to 5 further days to the destruction of the ceramic electrolyte which conducts alkali metal ions. In this case, the increase in the flow velocity in the moving liquid anode made of alkali metal amalgam and mercury unexpectedly leads to a further drop in the current density.
- the current density is generally 0.5 to 10 kA / m 2 , preferably 1.0 to 3 kA / m 2 (sodium) or 0.3 to 3 kA / m 2 , preferably 0.5 to 1, 5 kA / m 2 (potassium).
- the current density is specifically set at the external power source, usually a line rectifier.
- the electrolysis cell according to the invention is shown in integrated the power supply of the amalgam-supplying chlorine cell, so that a additional line rectifier can be omitted ( Figure 6).
- the alkali metal ion conductive ceramic is as tube closed on one side, which is concentric in the interior of a larger outer tube is introduced.
- the outer tube is made of a material that is very dense and resistant to hot amalgam. In particular come as Materials stainless steel and graphite in question.
- the annular gap between the outer tube and The liquid anode flows through the ceramic tube in the longitudinal direction.
- the gap width of the annular gap is expediently 1 to 10 mm, preferably 2 to 5 mm, particularly preferably 2.5 to 3 mm.
- the flow rate is 0.03 to 1.0 m / s, preferably 0.05 to 0.6 m / s, particularly preferably 0.1 to 0.3 m / s.
- a higher one Flow velocity usually allows higher current densities.
- Another design advantage of the anode in the form of an annular gap lies in the relatively small size anode volume related to the anode area. This will make it possible Demand for moderate apparatus weights and an acceptable mercury working capital to fulfill.
- the cell voltage is essentially composed of the following two individual contributions: the electrochemical potential of the redox system alkali metal to alkali metal amalgam and the ohmic voltage drop via the electrical resistance of the ceramic electrolyte.
- the cell voltage is therefore a function of the current density.
- the electrochemical potential can be measured in the de-energized state. It is set according to the alkali metal concentration in the liquid anode. At an alkali metal concentration of 0.4% by weight, a cell voltage of, for example, 0.82 V (sodium) or 1.01 V (potassium) is established in the currentless state. With a current density of 3000 A / m 2 , for example, a cell voltage of 1.9 V (sodium) is established. For potassium, for example, a cell voltage of 2.01 V results at a current density of 1000 A / m 2 .
- the cell voltage is monitored and is limited so that anode potentials in which those based on the electrochemical voltage series are excluded nobler metallic impurities are oxidized in the moving anode could.
- the value of cell voltage can be an indicator of mass transfer in the liquid moving anode to the ceramic surface and will usually do so supervised.
- the mass transfer limitation can be caused by a too low alkali metal concentration in the anode and or insufficient flow and or too high current density.
- the current direction is in time intervals of 1 up to 24 hours for 1 to 10 minutes reversed by using the anode and Cathode are short-circuited via an external resistor.
- the resistance is like that dimensioned that the current when reversing the polarity about 1.5 times the current in Operation corresponds.
- the yield of alkali metal obtained is in the The method according to the invention completely in relation to the anode side Alkali metal.
- the current yield of the alkali metal obtained is normally polarized Operating mode within the measurement accuracy 100%. By the interval Reverse polarity reduces the average current yield to values of 95% to 98%.
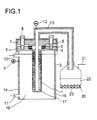
- the cell according to FIG. 1 consists of its core from a tube (1) closed on one side and made of sodium beta "aluminum oxide (32 mm Outer diameter, 210 mm length) whose wall thickness is 1.7 mm instead of is 5 mm described there.
- a tube (1) closed on one side was made of sodium beta "aluminum oxide (32 mm Outer diameter, 210 mm length) whose wall thickness is 1.7 mm instead of is 5 mm described there.
- At the open end was an alpha alumina ring (2) attached helium-tight using a glass solder connection.
- This ring was the sodium ion conductive tube made of beta "alumina with the opening upwards into a cylindrical stainless steel container (3) (with a Inside diameter of approx. 55 mm and a length of approx. 250 mm from austentic Stainless steel 1.4571) installed and sealed.
- Alpha Alumina Ring (2) was done with a flat gasket at the bottom (4) and at the top (5) over the housing (6) and the cover
- An anode power supply (9) was attached to the stainless steel container.
- For the Amalgam was supplied at the top with a pipe socket (10) for the drain at the side welded on a pipe socket (11) below.
- the same tube (13) is guided through the cover flange and for Removal of liquid sodium drilled laterally at the top.
- the apparatus was with electrical heating tapes (14) wrapped and thermally insulated (15).
- the anode is a sodium amalgam filling (16) between the housing and the Outer wall of the sodium ion-conducting solid electrolyte tube.
- the cathode (17) is one Liquid sodium filling within the solid electrolyte tube which conducts sodium ions.
- the Liquid sodium formed was heated with reaction-induced pressure over the Drain pipe into an argon (21) inert, partially filled with paraffin oil (22) Vessel (20) discharged and solidified in paraffin oil (22) in the form of small balls (23).
- the experimental setup of the apparatus according to Comparative Example 1 was supplemented by a stirrer (18) (length 38 mm, diameter 8 mm) installed on the bottom of the vessel (FIG. 2).
- the stirrer was driven by a standard magnetic stirrer.
- a special device prevented the stirrer from floating in the amalgam with its high density of 13,600 kg / m 3 .
- the stirrer was held on the bottom of the electrolytic cell by means of a bolt and a ball bearing.
- the stirrer speed was a maximum of 100 min -1 .
- the experiment was carried out as in Comparative Example 1, but with stirring of the anode. Furthermore, the polarity was first reversed when starting, so that the exterior was operated with the amalgam filling as the cathode and the interior of the ceramic with liquid sodium as the anode. Over a period of 25 minutes, the current was increased once from 5 A to 30 A in increments of 5 A each.
- the cell voltage followed the step steps of the current in the following way: 0.8V / O, OA; -0.2V / 5A; 0.1 V / 10A; O, OV / 15A; -O, 1V / 20A; -0.2V / 25A; -0.5V / 30A.
- the solidified sodium was dissolved in ethanol and analyzed by atomic absorption spectrometry for other metals (Al, Bi, Ca, Cd, Co, Cr, Cu, Fe, Li, Mg, Mn, Mo, Ni, Pb, Sb, Sn, Ti, V, Zn, Zr, Hg, K) with a detection limit of 1 ppm, with the exception of Hg 0.1 ppm. Only the following metallic impurities were found: 0.3 ppm Hg, 50 ppm K.
- the depleted amalgam was transferred from the 255 ° C hot cell to a cooled one Submitted template.
- a decrease in the sodium concentration in the amalgam of 0.40 % By weight to 0.14% by weight could be detected titrimetrically.
- the experimental setup corresponded to the apparatus from Example 1.
- the anode compartment heated to 255 ° C, was 0.4 kg with 15 kg Amalgams preheated to approx. 200 ° C. In a de-energized state there was always a cell voltage of 0.82 V at the beginning of the reaction.
- the Output voltage of a DC power supply was always limited to 2 volts and the circuit with the cell closed.
- the current setpoint was set to 25 A. Over a test period of 120 minutes was a constant current of 25 A at a cell voltage of 1.0 V to 1.1 V observed until the end of the reaction. This is for industrial use of the Process excellent. There were an average of 42.7 g of sodium per batch carried out. This corresponds to Faraday's accuracy Law. The analysis results of Example 1 were confirmed. A decrease in Sodium concentration in the amalgam from 0.40 to 0.11 wt .-% could be titrimetrically be detected.
- the core of the cell according to FIG. 3 consisted of a tube closed on one side (31) made of beta "aluminum oxide (32 mm outer diameter, 210 mm length, Wall thickness 1.7 mm). At the open end was a ring made of alpha aluminum oxide (32) attached helium-tight using a glass solder connection. By means of this ring (32) was the sodium ion conductive tube made of beta "aluminum oxide with the opening after into a concentric stainless steel tube (33) (with an inner diameter of 37 mm and a length of approx. 215 mm). The inside diameter of the steel pipe was matched to the outer diameter of the ceramic tube, so that an annular gap with a gap width of 2.5 mm.
- the one over the annular gap and the pipe length defined anode space met the demand for an apparatus Concept that got by with a relatively small mercury content.
- the ring cross section is a very effective flow through the current density Anode space in the axial direction.
- the ring was made of alpha aluminum oxide for sealing (32) with a flat gasket each below (36) and above (34) over the Housing (36) and the cover flange (37) with three or four clamping screws (38) pressed.
- An anode power supply (39) was attached to the stainless steel container.
- For the Amalgam was supplied at the bottom with a pipe socket (40) for the drain at the side a pipe socket (41) welded on top.
- the same tube (43) was passed through the cover flange and was used for the free removal of liquid sodium.
- the cell could use electrical Heating tapes (44) are wrapped and insulated or together with several pipes be installed in a heated chamber.
- the anode was the amalgam filling in the annular space between the inner tube wall and Outer wall of the sodium ion-conducting solid electrolyte tube.
- the cathode was that Liquid sodium filling within the solid electrolyte tube which conducts sodium ions.
- the Liquid sodium formed was generated with pressure generated by the reaction over the heated drain pipe (43) in an inertized container partially filled with paraffin oil discharged and solidified in paraffin oil in the form of small balls.
- the commercial tube made of sodium beta "aluminum oxide was installed immediately in the laboratory atmosphere within one hour after there was one Vacuum packaging was removed. Then both chambers of the cell were with Flooded with argon and sealed the cell. Installation in the apparatus took place 2 to 5 Days later. The apparatus was heated to 330 ° C at 20 ° C / h. After that the Cathode space within the ceramic tube closed on one side via a Feed line filled with molten sodium, the anode compartment outside of the ceramic tube was also filled with liquid sodium. Over a Over a period of 35 minutes, the current was increased from 5 A to 40 A in increments increased by 5 A each and then held at 40 A for 4 hours.
- the Cell voltage followed the step steps of the current in the following way: 0.0V / 0.0A, 0.03V / 5A; 0.05V / IOA; 0.08V / 15A; 0.10V / 20A; 0.13V / 25A; 0.16V / 30A; 0.18V / 35A; 0.22V / 40A. After 4 hours the voltage / current ratio was 0.18V / 40A broken in.
- the amalgam circuit was then filled with 39 kg of amalgam. The The contents of the amalgam circuit were heated to 330 ° C. with the pump switched off and then started the circuit. This was the one in the anode compartment Rinsed out sodium and distributed in the amalgam itself.
- This first filling was discarded and the circuit was filled with fresh amalgam heated to 330 ° C. and containing 0.4% by weight of sodium.
- An average flow rate of 0.3 m / s was set, corresponding to a circulation volume flow of 0.29 m 3 / h.
- a cell voltage of 0.82 V was set in the de-energized state.
- the output voltage of a DC power supply was limited to 2 volts and the circuit with the cell was closed.
- the current was increased linearly from 0 to 40 A over a period of 3 hours. Thereafter, 7.8 kg of amalgam were drained from the circulatory contents at intervals of 30 minutes and replaced by fresh amalgam. It was observed that the cell voltage fluctuated between 1.1 volts after filling and 1.12 volts before draining.
- a current density of 2000 A / m 2 is calculated from a current of 40 A with an anode area of 200 cm 2 , this is twice as high as is required for industrial use of the method.
- Example 1 There was a constant discharge of sodium. The sodium discharge and the depletion of the amalgam corresponded to Faraday's law. The analysis results of Example 1 were confirmed.
- the cell according to FIG. 1 consisted of its core from a tube (1) closed on one side and made of potassium beta "aluminum oxide (32 mm Outer diameter, 100 mm length) whose wall thickness was 1.2 mm.
- a ring made of alpha alumina (2) using a glass solder joint attached helium-tight.
- the potassium ion conductive tube became made of potassium beta "alumina with the opening upwards in a cylindrical Stainless steel container (3) (with an inner diameter of approx. 80 mm and a length of approx. 150 mm made of austenitic stainless steel 1.4571) and sealed.
- the ring Alpha aluminum oxide (2) was fitted with a flat gasket at the bottom (4) and above (5) over the housing flange (6) and the cover flange (7) with three clamping screws (8) pressed.
- An anode power supply (9) was attached to the stainless steel container.
- For the Amalgam was supplied at the top with a pipe socket (10) for the drain at the side welded on a pipe socket (11) below.
- the same tube (13) is passed through the cover flange and Drilled on the side at the top for the removal of liquid potassium.
- the apparatus was with electrical heating tapes (14) wrapped and thermally insulated (15).
- the anode is the amalgam filling (16) between the housing and the outer wall of the potassium ion-conducting solid electrolyte tube.
- the cathode (17) is the liquid potassium filling inside the potassium ion-conducting solid electrolyte tube.
- the liquid potassium formed was discharged at a pressure caused by the reaction through the heated drain pipe into a vessel (20) partially filled with paraffin oil (22) which had been inertized with argon (21) and solidified in the form of small balls (23) in the paraffin oil (22). Because of the density of potassium of 0.86 g / cm 3 , the potassium balls swim just below the surface of the paraffin oil.
- the experimental setup of the apparatus according to Comparative Example 2 was supplemented by a stirrer (18) (length 42 mm, diameter 5 mm) installed on the bottom of the vessel (FIG. 2).
- the stirrer was driven by a standard magnetic stirrer.
- the same device as in Example 1 prevented the stirrer from floating in the amalgam with its high density of 13,600 kg / m 3 .
- the stirrer speed was a maximum of 100 min -1 .
- the experiment was carried out as in Comparative Example 2, but with stirring of the anode. Furthermore, the polarity was first reversed when starting, so that the exterior with the amalgam filling as the cathode and the interior of the ceramic with liquid potassium as the anode was operated. Over a period of 27 minutes, the current was increased once from 1 A to 10 A in increments of 1 A each. The experiment was then carried out as described in Comparative Example 2. An average current of 10 A was set over a test time of 90 minutes (initially 12 A, at the end of the reaction 9 A). The cell voltage was limited to a maximum of 2.1 V. After disconnecting the power supply, a cell voltage of 1.08 V was measured in the de-energized state.
- the depleted amalgam was transferred from the 250 ° C cell into a cooled receiver drained.
- a decrease in the potassium concentration in the amalgam of 0.40% by weight 0.11% by weight could be detected titrimetrically.
- the experimental setup corresponded to the apparatus from Example 4.
- the anode space heated to 250 ° C, was 0.4 kg with 8 kg approx. 200 ° C preheated amalgams refilled.
- Delivered in de-energized condition Always start the reaction with a cell voltage of 1.01 V.
- the output voltage a DC power supply was always limited to 2.2 volts and the circuit with the cell closed.
- the current setpoint was set to 10 A. Over a trial period of 90 minutes was a constant current of 10 A at a cell voltage of 2.0 V to 2.1 V observed until the end of the reaction. This is for industrial use of the Process excellent.
- the core of the cell according to FIG. 3 consisted of a tube closed on one side (31) made of potassium beta "aluminum oxide (32 mm outer diameter, 100 mm long, Wall thickness 1.2 mm). At the open end is a ring made of alpha aluminum oxide (32) attached helium-tight using a glass solder connection.
- the potassium ion conductive tube made of beta "alumina with the opening facing downwards in a concentric stainless steel tube (33) (with an inner diameter of 37 mm and a length of approx. 105 mm). It is crucial that the inside diameter of the steel tube is matched to the outer diameter of the ceramic tube, so that an annular gap with a gap width of 2.5 mm is created.
- the one over the annular gap and the one Anode space defined on the one hand fulfills the requirement for a apparatus concept, which manages with a relatively small mercury content.
- the ring cross-section to be very effective in terms of current density Flow through the anode compartment in the axial direction.
- the ring is used for sealing made of alpha aluminum oxide (32), each with a flat seal at the bottom (36) and at the top (34) over the housing (36) and the cover flange (37) with three or four clamping screws (38) pressed.
- An anode power supply (39) is attached to the stainless steel container.
- For the Amalgam is supplied at the bottom with a pipe socket (40) for the drain at the side a pipe socket (41) welded on top.
- a pipe protrudes from the cover flange Stainless steel (43) as cathodic power supply in the opening of the tube made of potassium beta "aluminum oxide.
- the same tube (43) is passed through the cover flange and is used for the free removal of liquid potassium.
- the cell can be used with electrical Heating tapes (44) are wrapped and insulated or together with several pipes be installed in a heated chamber.
- the anode is the amalgam filling in the annular space between the steel tube inner wall and Outer wall of the potassium ion-conducting solid electrolyte tube.
- the cathode is the one liquid potassium filling inside the potassium ion-conducting solid electrolyte tube.
- the liquid potassium formed is heated with the reaction-generated pressure Drain pipe 43 is discharged into an inertized vessel partially filled with paraffin oil and solidifies in paraffin oil in the form of small balls.
- the cell voltage followed the step levels the current in the following way: 0.0 V / 0.0 A; 0.40 V / 4 A; 0.81 V / 8 A; 1.23 V / 12 A; 1.62 V / 16 A; 2.03 V / 20 A. After 4 hours the voltage / current ratio was up 1.99 V / 20 A run in. Then the amalgam cycle with 26 kg of amalgam filled. The content of the amalgam circuit was raised to 270 ° C. when the pump was switched off warmed up and then the circuit started. It was in the anode room Potassium is rinsed out and distributed dissolved in the amalgam.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Organic Chemistry (AREA)
- Electrolytic Production Of Metals (AREA)
- Primary Cells (AREA)
- Manufacture And Refinement Of Metals (AREA)
Abstract
Description
- Dauerversorgung (51) mit trockenem vorgeheizten Na-reichem Amalgam.
- Beheizung (52) ausgestaltet für Beheizung im Bereich von 310 °C bis 360 °C.
- Gleichstromversorgung (53).
- Definierte Strömungsgeschwindigkeit in der Anode durch einen internen mit einer Pumpe (55) getriebenen Amalgamkreislauf (54), stufenlos einstellbar zwischen 0,02 m/sec und 0,8 m/sec.
- Ableitung von flüssigem Natrium (56).
- Dauerentsorgung von Na-armem Amalgam (57).
- Abgasbehandlung (58).
- Sicherheitsüberwachung besonders hinsichtlich Hg-Emission (59).
- Dauerversorgung (51) mit trockenem vorgeheizten K-reichem Amalgam.
- Beheizung (52) ausgestaltet für Beheizung im Bereich von 265°C bis 400°C.
- Gleichstromversorgung (53)
- Definierte Strömungsgeschwindigkeit in der Anode durch einen internen mit einer Pumpe (55) getriebenen Amalgamkreislauf (54), stufenlos einstellbar zwischen 0,02 m/sec und 0,8 m/sec.
- Ableitung von flüssigem Kalium (56).
- Dauerentsorgung von K-armem Amalgam (57).
- Abgasbehandlung (58).
- Sicherheitsüberwachung besonders hinsichtlich Hg-Emission (59).
Claims (12)
- Verfahren zur Herstellung eines Alkalimetalls ausgehend von Alkalimetallamalgam durch Elektrolyse mit einer Alkalimetallamalgam enthaltenden Anode, einem Alkalimetall ionenleitenden Festelektrolyt und flüssigem Alkalimetall als Kathode, dadurch gekennzeichnet, daß das Alkalimetallamalgam als Anode bewegt wird.
- Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß das Alkalimetallamalgam als Anode durch Rühren und/oder mit einer Pumpe unter Atmosphärendruck oder leichtem Überdruck bewegt wird.
- Verfahren nach Anspruch 1 oder 2, dadurch gekennzeichnet, daß das Alkalimetall Natrium ist und es bei einer Temperatur im Bereich von 310 bis 400 °C durchgeführt wird.
- Verfahren nach Anspruch 1 oder 2, dadurch gekennzeichnet, daß das Alkalimetall Kalium ist und es bei einer Temperatur im Bereich von 260 bis 400 °C durchgeführt wird.
- Verfahren nach einem der Ansprüche 1 bis 4, dadurch gekennzeichnet, daß es bei Stromdichten oberhalb von 250 A/m2 durchgeführt wird.
- Verfahren nach einem der Ansprüche 1 bis 5, dadurch gekennzeichnet, daß das Alkalimetallamalgam aus der Chloralkali-Elektrolyse stammt.
- Verfahren nach einem der Ansprüche 1 bis 6, dadurch gekennzeichnet, daß der Festelektrolyt ausgewählt wird aus der Gruppe bestehend aus Natrium-β"-Aluminiumoxid, Natrium-β-Aluminiumoxid und Natrium-ß/ß"-Aluminiumoxid bzw. Kalium-β"-Aluminiumoxid, Kalium-β-Aluminiumoxid und Kalium-β/β"-Aluminiumoxid.
- Verfahren nach einem der Ansprüche 1 bis 7, dadurch gekennzeichnet, daß der Festelektrolyt vor der Durchführung des Verfahrens konditioniert wird.
- Integriertes Verfahren zur Herstellung von Chlor und Alkalimetall ausgehend von Alkalimetallchlorid, das die folgenden Stufen umfaßt:(i) Durchführung einer Chloralkali-Elektrolyse unter Erhalt von elementarem Chlor und Alkalimetallamalgam;(ii) Durchführung eines Verfahrens nach einem der Ansprüche 1 bis 8 unter Erhalt von Alkalimetall.
- Elektrolysezelle, umfassend einen einseitig geschlossenen rohrförmigen Festelektrolyten (1/31), der in ein konzentrisches Edelstahlrohr (33) derart eingebaut ist, daß ein Ringspalt entsteht.
- Elektrolysezelle nach Anspruch 10 dadurch gekennzeichnet, daß der Innendurchmesser des konzentrischen Edelstahlrohrs und der Außendurchmesser des rohrförmigen Festelektrolyten so aufeinander abgestimmt sind, daß ein Ringspalt mit einer Spaltweite von 1 bis 10 mm entsteht.
- Verfahren nach einem der Ansprüche 1 bis 9 unter Verwendung einer Elektrolysezelle nach Anspruch 10 oder 11 dadurch gekennzeichnet, daß der Ringspalt mit einer Geschwindigkeit von 0,03 m/s bis 1,0 m/s durchströmt wird.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19859563 | 1998-12-22 | ||
| DE19859563A DE19859563B4 (de) | 1998-12-22 | 1998-12-22 | Verbessertes Verfahren zur elektrochemischen Herstellung von Alkalimetall aus Alkalimetallamalgam |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP1114883A1 true EP1114883A1 (de) | 2001-07-11 |
| EP1114883B1 EP1114883B1 (de) | 2006-08-30 |
Family
ID=7892316
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP99124037A Expired - Lifetime EP1114883B1 (de) | 1998-12-22 | 1999-12-09 | Verfahren und Zelle zur elektrochemischen Herstellung von Alkalimetall aus Alkalimetallamalgam |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US6409908B1 (de) |
| EP (1) | EP1114883B1 (de) |
| JP (2) | JP2000219989A (de) |
| KR (2) | KR100717673B1 (de) |
| CN (1) | CN1195899C (de) |
| AT (1) | ATE338151T1 (de) |
| DE (2) | DE19859563B4 (de) |
| ES (1) | ES2272035T3 (de) |
| RU (1) | RU2250933C2 (de) |
| TW (1) | TW498113B (de) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006029792A2 (de) * | 2004-09-14 | 2006-03-23 | Basf Aktiengesellschaft | Elektrolysezelle zur herstellung von alkalimetall |
| WO2006029807A2 (de) * | 2004-09-14 | 2006-03-23 | Basf Aktiengesellschaft | Elektrolysevorrichtung zur herstellung von alkalimetall |
| EP1672098A2 (de) | 2004-12-16 | 2006-06-21 | Basf Aktiengesellschaft | Feste polykristalline Kaliumionenleiter mit einer Beta-Aluminiumoxidstruktur, seine Herstellung, und die Herstellung von Kaliummetall mit diesem Kaliumionenleiter. |
| US10399166B2 (en) | 2015-10-30 | 2019-09-03 | General Electric Company | System and method for machining workpiece of lattice structure and article machined therefrom |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6368486B1 (en) * | 2000-03-28 | 2002-04-09 | E. I. Du Pont De Nemours And Company | Low temperature alkali metal electrolysis |
| CN1429417A (zh) * | 2000-04-18 | 2003-07-09 | 电池技术电力有限公司 | 能量转换的电化学装置和方法 |
| EP2004869B1 (de) * | 2006-03-31 | 2010-12-15 | Basf Se | Verfahren zur wasserentfernung aus alkalimetallamalgam |
| DE202006018283U1 (de) | 2006-12-01 | 2008-04-03 | Coroplast Fritz Müller Gmbh & Co. Kg | Technisches Vliesklebeband |
| JP5220702B2 (ja) * | 2009-07-15 | 2013-06-26 | 日本碍子株式会社 | 電解装置 |
| CN104805469B (zh) * | 2015-05-11 | 2017-04-05 | 中国东方电气集团有限公司 | 一种电解制备金属钠装置的阴极电解槽 |
| AU2018333012A1 (en) * | 2017-09-18 | 2020-03-26 | Boston Electrometallurgical Corporation | Systems and methods for molten oxide electrolysis |
| CN108048871A (zh) * | 2017-12-28 | 2018-05-18 | 中国东方电气集团有限公司 | 用于制备高纯金属钠的电解单元 |
| CN108048872B (zh) * | 2017-12-28 | 2023-07-04 | 中国东方电气集团有限公司 | 一种制备高纯金属钠的电解提纯系统 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4097345A (en) * | 1976-10-15 | 1978-06-27 | E. I. Du Pont De Nemours And Company | Na5 GdSi4 O 12 and related rare earth sodium ion conductors and electrolytic cells therefrom |
| DE4034137C1 (de) * | 1990-10-26 | 1991-10-10 | Kernforschungszentrum Karlsruhe Gmbh, 7500 Karlsruhe, De |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB491880A (en) * | 1936-03-11 | 1938-09-12 | Ici Ltd | Improvements in or relating to the electrolytic manufacture of alkali metals |
| GB1155927A (en) * | 1967-02-20 | 1969-06-25 | Ici Ltd | Electrolytic manufacture of alkali metals. |
| JPS4837641B1 (de) * | 1968-07-18 | 1973-11-13 | ||
| JPS5523038A (en) * | 1978-08-03 | 1980-02-19 | Kanegafuchi Chem Ind Co Ltd | Concentrating method for aqueous solution of alkali metal hydroxide |
| DE3532956A1 (de) * | 1985-09-14 | 1987-03-19 | Metallgesellschaft Ag | Verfahren und vorrichtung zur herstellung von lithiummetall hoher reinheit durch schmelzflusselektrolyse |
| JPS6362163A (ja) * | 1986-09-01 | 1988-03-18 | Hitachi Ltd | ナトリウム−イオウ二次電池 |
| GB2207545A (en) * | 1987-07-28 | 1989-02-01 | Lilliwyte Sa | Glass seal for sodium-sulphur cells |
| DE3932037A1 (de) * | 1989-09-26 | 1991-04-04 | Asea Brown Boveri | Verfahren zur herstellung eines keramischen formkoerpers |
| US5196277A (en) * | 1990-10-25 | 1993-03-23 | Ngk Insulators, Ltd. | Sodium-sulfur cell and method of joining solid electrolyte tube and insulative ring |
| CN1229860A (zh) * | 1998-03-19 | 1999-09-29 | 克里安诺瓦特殊化学股份有限公司 | 电解金属盐制备汞齐的方法 |
-
1998
- 1998-12-22 DE DE19859563A patent/DE19859563B4/de not_active Expired - Fee Related
-
1999
- 1999-12-09 TW TW088121565A patent/TW498113B/zh not_active IP Right Cessation
- 1999-12-09 DE DE59913824T patent/DE59913824D1/de not_active Expired - Lifetime
- 1999-12-09 AT AT99124037T patent/ATE338151T1/de not_active IP Right Cessation
- 1999-12-09 EP EP99124037A patent/EP1114883B1/de not_active Expired - Lifetime
- 1999-12-09 ES ES99124037T patent/ES2272035T3/es not_active Expired - Lifetime
- 1999-12-13 US US09/459,726 patent/US6409908B1/en not_active Expired - Lifetime
- 1999-12-15 JP JP11376601A patent/JP2000219989A/ja active Pending
- 1999-12-21 RU RU99127308/02A patent/RU2250933C2/ru not_active IP Right Cessation
- 1999-12-21 KR KR1019990059560A patent/KR100717673B1/ko not_active IP Right Cessation
- 1999-12-22 CN CNB991264975A patent/CN1195899C/zh not_active Expired - Fee Related
-
2007
- 2007-01-15 KR KR1020070004132A patent/KR100719413B1/ko not_active IP Right Cessation
-
2010
- 2010-11-29 JP JP2010264573A patent/JP5469042B2/ja not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4097345A (en) * | 1976-10-15 | 1978-06-27 | E. I. Du Pont De Nemours And Company | Na5 GdSi4 O 12 and related rare earth sodium ion conductors and electrolytic cells therefrom |
| DE4034137C1 (de) * | 1990-10-26 | 1991-10-10 | Kernforschungszentrum Karlsruhe Gmbh, 7500 Karlsruhe, De |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006029792A2 (de) * | 2004-09-14 | 2006-03-23 | Basf Aktiengesellschaft | Elektrolysezelle zur herstellung von alkalimetall |
| WO2006029807A2 (de) * | 2004-09-14 | 2006-03-23 | Basf Aktiengesellschaft | Elektrolysevorrichtung zur herstellung von alkalimetall |
| WO2006029807A3 (de) * | 2004-09-14 | 2006-06-29 | Basf Ag | Elektrolysevorrichtung zur herstellung von alkalimetall |
| WO2006029792A3 (de) * | 2004-09-14 | 2006-08-03 | Basf Ag | Elektrolysezelle zur herstellung von alkalimetall |
| US7981260B2 (en) | 2004-09-14 | 2011-07-19 | Basf Aktiengesellschaft | Electrolysis cell for producing alkali metal |
| US8114258B2 (en) | 2004-09-14 | 2012-02-14 | Basf Aktiengesellschaft | Electrolysis device for the production of alkali metal |
| KR101253787B1 (ko) * | 2004-09-14 | 2013-04-15 | 바스프 에스이 | 알칼리 금속 제조용 전기분해 셀 |
| KR101274851B1 (ko) * | 2004-09-14 | 2013-06-13 | 바스프 에스이 | 알칼리 금속 제조용 전기분해 장치 |
| EP1672098A2 (de) | 2004-12-16 | 2006-06-21 | Basf Aktiengesellschaft | Feste polykristalline Kaliumionenleiter mit einer Beta-Aluminiumoxidstruktur, seine Herstellung, und die Herstellung von Kaliummetall mit diesem Kaliumionenleiter. |
| US8133378B2 (en) | 2004-12-16 | 2012-03-13 | Basf Aktiengesellschaft | Solid polycrystalline potassium ion conductor having a β″-Al2O3 structure, its production and the preparation of potassium metal using this potassium ion conductor |
| US8551319B2 (en) | 2004-12-16 | 2013-10-08 | Basf Aktiengesellschaft | Solid polycrystalline potassium ion conductor having a β″-Al2O3 structure, its production and the preparation of potassium metal using this potassium ion conductor |
| US10399166B2 (en) | 2015-10-30 | 2019-09-03 | General Electric Company | System and method for machining workpiece of lattice structure and article machined therefrom |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1114883B1 (de) | 2006-08-30 |
| KR100717673B1 (ko) | 2007-05-14 |
| JP2011058096A (ja) | 2011-03-24 |
| JP2000219989A (ja) | 2000-08-08 |
| KR20000052533A (ko) | 2000-08-25 |
| DE19859563B4 (de) | 2008-01-24 |
| RU2250933C2 (ru) | 2005-04-27 |
| DE19859563A1 (de) | 2000-06-29 |
| DE59913824D1 (de) | 2006-10-12 |
| CN1261631A (zh) | 2000-08-02 |
| ATE338151T1 (de) | 2006-09-15 |
| ES2272035T3 (es) | 2007-04-16 |
| KR20070013357A (ko) | 2007-01-30 |
| CN1195899C (zh) | 2005-04-06 |
| JP5469042B2 (ja) | 2014-04-09 |
| KR100719413B1 (ko) | 2007-05-18 |
| TW498113B (en) | 2002-08-11 |
| US6409908B1 (en) | 2002-06-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1041177B1 (de) | Verfahren zur elektrochemischen Herstellung von Lithium | |
| EP1114883B1 (de) | Verfahren und Zelle zur elektrochemischen Herstellung von Alkalimetall aus Alkalimetallamalgam | |
| WO2001014616A1 (de) | Verfahren zur elektrochemischen herstellung eines alkalimetalls aus wässriger lösung | |
| DE975587C (de) | Verfahren und Anordnung zur Herstellung von Titan in einer Elektrolysezelle | |
| EP1794352B1 (de) | Elektrolysezelle zur herstellung von alkalimetall | |
| DE2145504B2 (de) | Verfahren zur Gewinnung von sei tenen Erdmetallen aus ihren Oxyden durch Elektrolyse einer Salzschmelze, und elektroiytische Zelle zur Durch fuhrung dieses Verfahrens | |
| DE2818971A1 (de) | Verbesserte vorrichtung und verbessertes verfahren zur abtrennung eines metalles aus einem salz | |
| EP1059366B1 (de) | Elektrolysezelle zur Herstellung eines Alkalimetalls | |
| DE2240731A1 (de) | Verfahren zur herstellung von glyoxylsaeure | |
| EP2877614A1 (de) | Verfahren zur herstellung eines alkalimetalls | |
| DE102004044404A1 (de) | Elektrolysevorrichtung zur Herstellung von Alkalimetall | |
| EP0192718B1 (de) | Verfahren und vorrichtung zur herstellung von ozon | |
| DE2620780A1 (de) | Verfahren zur herstellung von metallischem zink durch schmelzelektrolyse aus zinkchlorid | |
| DE153619C (de) | Verfahren zur elektrolytshen darstellung van vanadin und dessen legierungen | |
| DE2725389C2 (de) | Verfahren zum Abscheiden von Metallen durch Schmelzflußelektrolyse | |
| DE3432684C2 (de) | ||
| DE2216383C2 (de) | Verfahren zur elektrochemischen Kupferabscheidung | |
| DE1783137C3 (de) | Elektrolytisches Verfahren zur Gewinnung eines Alkalimetalles aus einer Salzschmelze dieses Metalls | |
| DE3248153A1 (de) | Verfahren und vorrichtung zur verringerung des aluminiumgehalts von alkalischen loesungen | |
| DE1112722B (de) | Verfahren zur elektrolytischen Herstellung von Phosphin | |
| EP0911428A1 (de) | Verfahren zur Herstellung von Wismutverbindungen | |
| DE1558760C3 (de) | Verfahren und Vorrichtung zur Schmelzflußelektrolyse von Oxiden | |
| DE3412114A1 (de) | Verfahren zur herstellung von aluminium | |
| DE1179723B (de) | Verfahren und Vorrichtung zur schmelzelektro-lytischen Herstellung von eisenhaltigem Mangan | |
| DE1112304B (de) | Elektrolytisches Verfahren und Elektrolysezelle zum Herstellen von Titan |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU MC NL PT SE |
|
| AX | Request for extension of the european patent |
Free format text: AL;LT;LV;MK;RO;SI |
|
| 17P | Request for examination filed |
Effective date: 20010801 |
|
| AKX | Designation fees paid |
Free format text: AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU MC NL PT SE |
|
| 17Q | First examination report despatched |
Effective date: 20040708 |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| RTI1 | Title (correction) |
Free format text: PROCESS AND CELL FOR THE ELECTROCHEMICAL PRODUCTION OF ALKALI METALS FROM ALKALI METAL AMALGAMS |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU MC NL PT SE |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20060830 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20060830 |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D Free format text: NOT ENGLISH |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D Free format text: LANGUAGE OF EP DOCUMENT: GERMAN |
|
| REF | Corresponds to: |
Ref document number: 59913824 Country of ref document: DE Date of ref document: 20061012 Kind code of ref document: P |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 20061101 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20061130 Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20061130 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: MC Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20061231 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20070206 |
|
| ET | Fr: translation filed | ||
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2272035 Country of ref document: ES Kind code of ref document: T3 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FD4D |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed |
Effective date: 20070531 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: AT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20061209 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20061201 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20061209 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20060830 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20131219 Year of fee payment: 15 Ref country code: GB Payment date: 20131227 Year of fee payment: 15 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: IT Payment date: 20131219 Year of fee payment: 15 Ref country code: NL Payment date: 20131218 Year of fee payment: 15 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20140129 Year of fee payment: 15 Ref country code: DE Payment date: 20140228 Year of fee payment: 15 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20131227 Year of fee payment: 15 Ref country code: ES Payment date: 20140128 Year of fee payment: 15 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141231 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R119 Ref document number: 59913824 Country of ref document: DE |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: V1 Effective date: 20150701 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: V1 Effective date: 20150701 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20141209 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20150831 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20150701 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141231 Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141231 Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141209 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20150701 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141231 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141209 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20160129 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20141210 |