EP0902846B1 - Verfahren zur herstellung von phthaliden - Google Patents
Verfahren zur herstellung von phthaliden Download PDFInfo
- Publication number
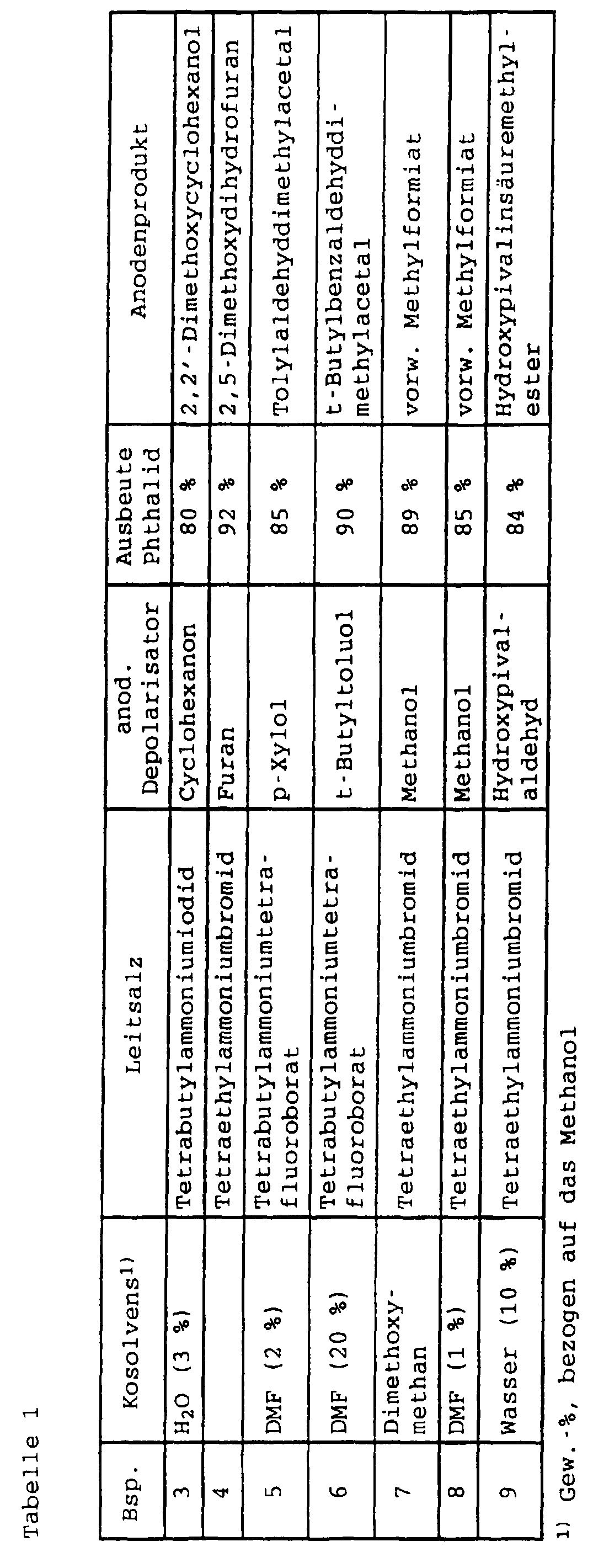
- EP0902846B1 EP0902846B1 EP97921810A EP97921810A EP0902846B1 EP 0902846 B1 EP0902846 B1 EP 0902846B1 EP 97921810 A EP97921810 A EP 97921810A EP 97921810 A EP97921810 A EP 97921810A EP 0902846 B1 EP0902846 B1 EP 0902846B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- phthalic acid
- alkyl
- anodic
- hydrogen
- acid derivatives
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 125000005506 phthalide group Chemical group 0.000 title claims description 13
- 238000004519 manufacturing process Methods 0.000 title claims description 9
- 238000000034 method Methods 0.000 claims description 20
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 claims description 18
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- 239000001257 hydrogen Substances 0.000 claims description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 7
- 150000003021 phthalic acid derivatives Chemical class 0.000 claims description 7
- 230000009467 reduction Effects 0.000 claims description 7
- 239000003960 organic solvent Substances 0.000 claims description 6
- 125000001424 substituent group Chemical group 0.000 claims description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 4
- 229910052736 halogen Inorganic materials 0.000 claims description 4
- 239000000203 mixture Substances 0.000 claims description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 150000002894 organic compounds Chemical class 0.000 claims description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 3
- 125000002030 1,2-phenylene group Chemical group [H]C1=C([H])C([*:1])=C([*:2])C([H])=C1[H] 0.000 claims description 2
- 125000001931 aliphatic group Chemical group 0.000 claims description 2
- 238000006482 condensation reaction Methods 0.000 claims description 2
- 238000006056 electrooxidation reaction Methods 0.000 claims description 2
- 229910002804 graphite Inorganic materials 0.000 claims description 2
- 239000010439 graphite Substances 0.000 claims description 2
- 150000002366 halogen compounds Chemical class 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 claims description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 claims 1
- 125000005498 phthalate group Chemical class 0.000 claims 1
- 239000003115 supporting electrolyte Substances 0.000 claims 1
- WNZQDUSMALZDQF-UHFFFAOYSA-N 2-benzofuran-1(3H)-one Chemical compound C1=CC=C2C(=O)OCC2=C1 WNZQDUSMALZDQF-UHFFFAOYSA-N 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- 238000010168 coupling process Methods 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- NIQCNGHVCWTJSM-UHFFFAOYSA-N Dimethyl phthalate Chemical compound COC(=O)C1=CC=CC=C1C(=O)OC NIQCNGHVCWTJSM-UHFFFAOYSA-N 0.000 description 6
- -1 C 4 alcohols Chemical class 0.000 description 5
- 238000005868 electrolysis reaction Methods 0.000 description 5
- 239000003792 electrolyte Substances 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- FBSAITBEAPNWJG-UHFFFAOYSA-N dimethyl phthalate Natural products CC(=O)OC1=CC=CC=C1OC(C)=O FBSAITBEAPNWJG-UHFFFAOYSA-N 0.000 description 3
- 229960001826 dimethylphthalate Drugs 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 150000003857 carboxamides Chemical class 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- AITXBHMOGHXWFR-UHFFFAOYSA-N n-(methoxymethyl)-n-methylformamide Chemical compound COCN(C)C=O AITXBHMOGHXWFR-UHFFFAOYSA-N 0.000 description 2
- SDLAKRCBYGZJRW-UHFFFAOYSA-N n-tert-butylformamide Chemical compound CC(C)(C)NC=O SDLAKRCBYGZJRW-UHFFFAOYSA-N 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- GVTLFGJNTIRUEG-ZHACJKMWSA-N (e)-n-(3-methoxyphenyl)-3-phenylprop-2-enamide Chemical compound COC1=CC=CC(NC(=O)\C=C\C=2C=CC=CC=2)=C1 GVTLFGJNTIRUEG-ZHACJKMWSA-N 0.000 description 1
- QLAJNZSPVITUCQ-UHFFFAOYSA-N 1,3,2-dioxathietane 2,2-dioxide Chemical compound O=S1(=O)OCO1 QLAJNZSPVITUCQ-UHFFFAOYSA-N 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000008051 alkyl sulfates Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- SARKQAUWTBDBIZ-UHFFFAOYSA-N azane;2-carbamoylbenzoic acid Chemical compound [NH4+].NC(=O)C1=CC=CC=C1C([O-])=O SARKQAUWTBDBIZ-UHFFFAOYSA-N 0.000 description 1
- 150000003842 bromide salts Chemical class 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000007772 electrode material Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 150000002790 naphthalenes Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 239000000575 pesticide Substances 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 238000005185 salting out Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- SEACXNRNJAXIBM-UHFFFAOYSA-N triethyl(methyl)azanium Chemical compound CC[N+](C)(CC)CC SEACXNRNJAXIBM-UHFFFAOYSA-N 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/20—Processes
- C25B3/25—Reduction
Definitions
- the present invention relates to a new method of manufacture of phthalides by cathodic reduction of phthalic acid or phthalic acid derivatives.
- Phthalides are especially used as intermediates for manufacturing of pesticides needed.
- An electrochemical process for the production of the phthalides is known from DE-A-2 144 419.
- phthalides can be produced in satisfactory yields if you split the reduction in Electrolysis cells.
- a disadvantage of the described method is that with the Use of apparatus connected to split electrolysis cells Effort, since 2 cell circles are required in this case. In addition is working with 2 cell circles with the following further disadvantages connected:
- the cell circles must be separated by a membrane or a diaphragm; this means a loss of energy through ohmic heat.
- at least one chamber is usually charged with an aqueous (> 80% H 2 O) conductive salt solution. In the case of cathodic reductions, this is the anolyte.
- the constraint on this approach severely limits the scope for using the anode reaction. Usually only hydrogen is produced as an anode product.
- a process for producing phthalides was carried out by cathodic reduction of phthalic acid or phthalic acid derivatives, in which the carboxy groups can be replaced by units, that can be derived from carboxy groups in a condensation reaction are and one or more of the hydrogen atoms of the o-phenylene unit phthalic acid can be substituted by inert residues, found, taking the reduction in an organic Solvent containing less than 50 wt .-% water and one undivided electrolytic cell.
- ammonium salts and in particular the ammonium salt of phthalamic acid are particularly preferred.
- Suitable as electrode materials (both cathode and anode) commercially available electrodes made of graphite or carbon.
- the electrolyte is usually a 2 to 40% by weight solution of phthalic acid or a phthalic acid derivative in an organic solvent, preferably less contains more than 25, particularly preferably less than 5% by weight of water.
- Suitable organic solvents are in particular aliphatic C 1 to C 4 alcohols, in particular methanol or ethanol, or a mixture of such alcohols with a carboxamide such as dimethylformamide or t-butylformamide.
- the electrolytes generally contain alkyl sulfates, for example methyl sulfate, or quaternary ammonium salts, in particular tetra (C 1 -C 4 -alkyl) ammonium halogens or tetrafluoroborates, usually in amounts of 0.4 to 10% by weight, based on the electrolyte, as conductive salts .
- alkyl sulfates for example methyl sulfate
- quaternary ammonium salts in particular tetra (C 1 -C 4 -alkyl) ammonium halogens or tetrafluoroborates, usually in amounts of 0.4 to 10% by weight, based on the electrolyte, as conductive salts .
- anodic Depolarizer For the anodic coupling process, it is recommended to be anodic Depolarizer to use usual organic compounds whose Suitability for electrochemical oxidation to the person skilled in the art in general is known. Some of the anodic coupling processes are preferred performed in the presence of a mediator. Possible anodic coupling processes and their mediatization for example in D. Kyriakou, Modern Electroorganic Chemistry, Springer, Berlin 1994, described in chapter 4.2.
- the anodic coupling processes are particularly suitable Oxidations of C-O or C-N single or double bonds, e.g. the oxidation of carboxylic acids, arylmethanes, aldehydes, carboxamides, Alcohols and heterocycles, or the oxidative C-C linkage especially of naphthalenes or activated CH groups.
- Halogen compounds are particularly suitable as mediators all bromides or iodides.
- the type of workup of the electrolyte mixture depends in particular according to the type of anodic coupling product and can according to generally known separation methods such as distillation, precipitation or recrystallization. Can be particularly easy most of the phthalides of many in a basic, aqueous environment separate insoluble organic by-products by removing the Phthalides in ammoniacal aqueous solutions that dissolves separates aqueous phase and the phthalide by acidification the aqueous phase fails again (see also here DE-A-2 510 920).
- Phthalides are obtained by the process according to the invention technically simple way in high yield and purity. At the same time it is possible by coupling with anodic oxidation reactions to produce different types of valuable products without the current and material yield at the cathode would decrease.
- a solution of 500 g dimethyl phthalate (2.56 mol), 1600 g t.-butylformamide is placed in an electrolytic cell, consisting of ten bipolar graphite ring disks, area per side: 147 dm 2 , with an electrode spacing of 0.7 mm and 375 g of methanol were electrolyzed with 25 g of tetrabutylammonium tetrafluoroborate at a current of 2.5 A for 11.5 hours at 60 ° C.
- the solvent t-butylformamide is recovered without decomposition, the anode process is methanol oxidation with the main product Methyl formate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Electrolytic Production Of Non-Metals, Compounds, Apparatuses Therefor (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Furan Compounds (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
Description

- R1, R2, R3 und R4:
- unabhängig voneinander Wasserstoff, C1- bis C4-Alkyl oder Halogen
- R5 und R6:
-
- a) unabhängig voneinander -COOH oder COOX, wobei X für C1- bis C4-Alkyl steht,
- b) einer der Substituenten R5 oder R6 -COONY4 und der andere Substituent CONH2, wobei Y für C1- bis C4-Alkyl oder Wasserstoff steht,
- c) R5 und R6 zusammen -CO-O-CO-.
N-Methoxymethyl-N-Methylformamid neben 2,1 Mol Phthalid (Materialausbeute: 82 %).
Claims (9)
- Verfahren zur Herstellung von Phthaliden durch kathodische Reduktion von Phthalsäure oder Phthalsäurederivaten, bei denen die Carboxygruppen durch Einheiten ersetzt sein können, die von Carboxygruppen in einer Kondensationsreaktion ableitbar sind und eines oder mehrere der Wasserstoffatome der o-Phenylen-Einheit der Phthalsäure durch inerte Reste substituiert sein können, wobei man die Reduktion in einem organischen Lösungsmittel, das weniger als 50 Gew.-% Wasser enthält und einer ungeteilten Elektrolysezelle vornimmt.
- Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß man Phthalsäure oder Phthalsäurederivate der allgemeinen Formel Ieinsetzt, in der die Substituenten die folgende Bedeutung haben:
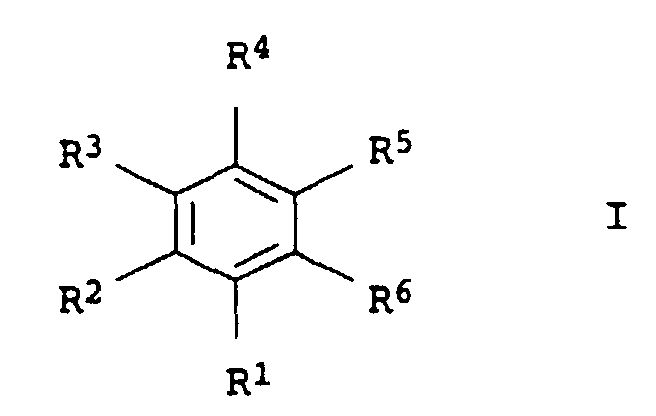
- R1, R2, R3 und R4:
- unabhängig voneinander Wasserstoff, C1- bis C4-Alkyl oder Halogen
- R5, R6:
-
- a) unabhängig voneinander -COOH oder COOX, wobei X für C1- bis C4-Alkyl steht,
- b) einer der Substituenten R5 oder R6 -COONY4 und der andere Substituent CONH2, wobei Y für C1- bis C4-Alkyl oder Wasserstoff steht,
- c) R5 und R6 zusammen -CO-O-CO-.
- Verfahren nach Anspruch 1 oder 2, wobei man als Phthalsäurederivate Phthalsäuredi(C1- bis C3-alkyl)-ester einsetzt.
- Verfahren nach den Ansprüchen 1 bis 3, wobei man Graphit- oder Kohleelektroden einsetzt.
- Verfahren nach den Ansprüchen 1 bis 4, wobei man als organisches Lösungsmittel einen aliphatischen C1- bis C4-Alkohol oder eine Mischung eines derartigen Alkohols mit einem Carbonsäureamid einsetzt.
- Verfahren nach den Ansprüchen 1 bis 5, wobei man als Leitsalz ein quarternäres Ammoniumsalz einsetzt.
- Verfahren nach den Ansprüchen 1 bis 6, wobei man für den anodischen Koppelprozeß als anodischen Depolarisator eine übliche organische Verbindung einsetzt, die sich für die elektrochemische Oxidation eignet.
- Verfahren nach den Ansprüchen 1 bis 7, wobei man als Mediator für den anodischen Koppelprozeß eine Halogenverbindung einsetzt.
- Verfahren nach Anspruch 8, wobei man als Mediator Bromid oder Iodid einsetzt.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19618854A DE19618854A1 (de) | 1996-05-10 | 1996-05-10 | Verfahren zur Herstellung von Phthaliden |
| DE19618854 | 1996-05-10 | ||
| PCT/EP1997/002185 WO1997043464A1 (de) | 1996-05-10 | 1997-04-28 | Verfahren zur herstellung von phthaliden |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0902846A1 EP0902846A1 (de) | 1999-03-24 |
| EP0902846B1 true EP0902846B1 (de) | 2000-07-26 |
Family
ID=7793943
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP97921810A Expired - Lifetime EP0902846B1 (de) | 1996-05-10 | 1997-04-28 | Verfahren zur herstellung von phthaliden |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US6063256A (de) |
| EP (1) | EP0902846B1 (de) |
| JP (1) | JP3946260B2 (de) |
| CN (1) | CN1058302C (de) |
| CA (1) | CA2254788C (de) |
| DE (2) | DE19618854A1 (de) |
| ES (1) | ES2150770T3 (de) |
| WO (1) | WO1997043464A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1017688B1 (de) * | 1997-09-19 | 2002-04-10 | Basf Aktiengesellschaft | Verfahren zur herstellung von phthaliden |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR018507A1 (es) | 1997-09-19 | 2001-11-28 | Basf Se | Proceso de recuperacion de un compuesto derivado del acido ftalico de una mezcla de reaccion en la que se sintetiza este compuesto |
| DE19808296A1 (de) * | 1998-02-27 | 1999-09-02 | Basf Ag | Verfahren zur selektiven Hydrolyse von Acetalen bzw. Ketalen in Gegenwart von Phthaliden |
| DE19944990A1 (de) | 1999-09-20 | 2001-03-22 | Basf Ag | Verfahren zur elektrolytischen Umwandlung von organischen Verbindungen |
| DE19944989A1 (de) | 1999-09-20 | 2001-03-22 | Basf Ag | Verfahren zur elektrolytischen Umwandlung von Furanderivaten |
| CN1182127C (zh) * | 2000-06-28 | 2004-12-29 | 中国医学科学院药物研究所 | 新的取代的2-苯并[c]呋喃酮化合物,其制备方法以及包含它们的药物组合物 |
| DE10057888A1 (de) | 2000-11-22 | 2002-05-23 | Basf Ag | Herstellung von Butantetracarbonsäurederivaten mittels gekoppelter Elektrosynthese |
| DE10058304A1 (de) * | 2000-11-24 | 2002-05-29 | Basf Ag | Verfahren zur Herstellung von alkoxylierten Carbonylverbindungen durch ein anodisches Oxidationsverfahren unter Nutzung der kathodischen Koppelreaktion zur organischen Synthese |
| WO2013186094A2 (en) * | 2012-06-15 | 2013-12-19 | Basf Se | Anodic oxidation of organic substrates in the presence of nucleophiles |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2510920A1 (de) * | 1975-03-13 | 1976-09-30 | Basf Ag | Verfahren zur elektrochemischen herstellung von phthalid |
| DE2630927A1 (de) * | 1976-07-09 | 1978-01-19 | Basf Ag | Verfahren zur herstellung von phthalidcarbonsaeure-(5) |
-
1996
- 1996-05-10 DE DE19618854A patent/DE19618854A1/de not_active Withdrawn
-
1997
- 1997-04-28 JP JP54044397A patent/JP3946260B2/ja not_active Expired - Fee Related
- 1997-04-28 US US09/125,019 patent/US6063256A/en not_active Expired - Fee Related
- 1997-04-28 ES ES97921810T patent/ES2150770T3/es not_active Expired - Lifetime
- 1997-04-28 DE DE59702087T patent/DE59702087D1/de not_active Expired - Fee Related
- 1997-04-28 EP EP97921810A patent/EP0902846B1/de not_active Expired - Lifetime
- 1997-04-28 CA CA002254788A patent/CA2254788C/en not_active Expired - Fee Related
- 1997-04-28 WO PCT/EP1997/002185 patent/WO1997043464A1/de not_active Ceased
- 1997-04-28 CN CN97192040A patent/CN1058302C/zh not_active Expired - Fee Related
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1017688B1 (de) * | 1997-09-19 | 2002-04-10 | Basf Aktiengesellschaft | Verfahren zur herstellung von phthaliden |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2254788C (en) | 2005-03-01 |
| DE19618854A1 (de) | 1997-11-13 |
| DE59702087D1 (de) | 2000-08-31 |
| JP2000511592A (ja) | 2000-09-05 |
| CN1058302C (zh) | 2000-11-08 |
| CN1210564A (zh) | 1999-03-10 |
| JP3946260B2 (ja) | 2007-07-18 |
| ES2150770T3 (es) | 2000-12-01 |
| EP0902846A1 (de) | 1999-03-24 |
| CA2254788A1 (en) | 1997-11-20 |
| WO1997043464A1 (de) | 1997-11-20 |
| US6063256A (en) | 2000-05-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0012215B1 (de) | 2-Hydroxybutansulfonsaures Cholin und dessen Verwendung als Leitsalz | |
| EP0902846B1 (de) | Verfahren zur herstellung von phthaliden | |
| EP2318569A1 (de) | Verfahren zur anodischen dehydrodimerisierung von substituierten arylalkoholen | |
| EP0072914B1 (de) | Verfahren zur Herstellung von alkylsubstituierten Benzaldehyden | |
| EP0339523B1 (de) | Verfahren zur Herstellung von Hydroxicarbonsäureestern | |
| DE10057888A1 (de) | Herstellung von Butantetracarbonsäurederivaten mittels gekoppelter Elektrosynthese | |
| DE2855508A1 (de) | Verfahren zur herstellung von benzaldehyden | |
| WO2008145627A1 (de) | Elektrochemische oxidation an allylgruppen | |
| WO2006100289A1 (de) | Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4-alkoxylierten but-2-enderivaten | |
| EP0638665A1 (de) | Verfahren zur Herstellung von Bezaldehyddialkylacetalen | |
| EP0029995A1 (de) | Verfahren zur Herstellung von 4-tert. Butylbenzaldehyd | |
| EP1619273A1 (de) | Verfahren zur Herstellung von 2-Alkin-1-acetalen | |
| EP0085158B1 (de) | Verfahren zur Herstellung von Cycloalkenonderivaten | |
| DE2710420C2 (de) | Verfahren zur elektrolytischen Herstellung von 2,5-Dialkoxy-2,5-dihydrofuranen | |
| EP1769103B1 (de) | Elektrochemisches verfahren zur herstellung cyclopropylbenzylaminen | |
| EP1017688B1 (de) | Verfahren zur herstellung von phthaliden | |
| DE2403446C2 (de) | Verfahren zur Herstellung hydrierter Indole | |
| EP0179377B1 (de) | Verfahren zur Herstellung von 1-Alkoxyisochromanen und neue 1-Alkoxy-alkylisochromane | |
| DE68906589T2 (de) | Elektrochemische Synthese von 2-Aryl-Hydrochinonen. | |
| DE2434845C3 (de) | Elektrochemische Herstellung aromatischer oder aromatisch-heterocyclischer Alkansäureester | |
| WO2007014932A1 (de) | Verfahren zur herstellung von 1,1,4,4-tetraalkoxy-but-2-enderivaten | |
| DE2851732A1 (de) | Verfahren zur herstellung von substituierten benzaldehyd-dialkylacetalen | |
| DE1961364A1 (de) | Verfahren zur Herstellung von substituierten Hydrazinen | |
| DE3619656A1 (de) | Neue 2,6-dimethyl-p-benzochinontetraalkylketale sowie deren herstellung | |
| DE2428878A1 (de) | Verfahren zur herstellung von p-hydroxymethyl-benzoesaeureestern |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 19980602 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): BE CH DE ES FR GB IT LI NL SE |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| 17Q | First examination report despatched |
Effective date: 19990713 |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): BE CH DE ES FR GB IT LI NL SE |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REF | Corresponds to: |
Ref document number: 59702087 Country of ref document: DE Date of ref document: 20000831 |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 20000810 |
|
| ITF | It: translation for a ep patent filed | ||
| ET | Fr: translation filed | ||
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2150770 Country of ref document: ES Kind code of ref document: T3 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: IF02 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20090508 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 20090407 Year of fee payment: 13 Ref country code: NL Payment date: 20090405 Year of fee payment: 13 Ref country code: IT Payment date: 20090424 Year of fee payment: 13 Ref country code: FR Payment date: 20090417 Year of fee payment: 13 Ref country code: DE Payment date: 20090428 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20090428 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20090416 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20090422 Year of fee payment: 13 |
|
| BERE | Be: lapsed |
Owner name: *BASF A.G. Effective date: 20100430 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: V1 Effective date: 20101101 |
|
| EUG | Se: european patent has lapsed | ||
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20100428 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20101230 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20101101 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20101103 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100428 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100428 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20110715 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20110705 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100429 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100429 |