EP0902846B1 - Process for preparing phthalides - Google Patents
Process for preparing phthalides Download PDFInfo
- Publication number
- EP0902846B1 EP0902846B1 EP97921810A EP97921810A EP0902846B1 EP 0902846 B1 EP0902846 B1 EP 0902846B1 EP 97921810 A EP97921810 A EP 97921810A EP 97921810 A EP97921810 A EP 97921810A EP 0902846 B1 EP0902846 B1 EP 0902846B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- phthalic acid
- alkyl
- anodic
- hydrogen
- acid derivatives
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/20—Processes
- C25B3/25—Reduction
Definitions
- the present invention relates to a new method of manufacture of phthalides by cathodic reduction of phthalic acid or phthalic acid derivatives.
- Phthalides are especially used as intermediates for manufacturing of pesticides needed.
- An electrochemical process for the production of the phthalides is known from DE-A-2 144 419.
- phthalides can be produced in satisfactory yields if you split the reduction in Electrolysis cells.
- a disadvantage of the described method is that with the Use of apparatus connected to split electrolysis cells Effort, since 2 cell circles are required in this case. In addition is working with 2 cell circles with the following further disadvantages connected:
- the cell circles must be separated by a membrane or a diaphragm; this means a loss of energy through ohmic heat.
- at least one chamber is usually charged with an aqueous (> 80% H 2 O) conductive salt solution. In the case of cathodic reductions, this is the anolyte.
- the constraint on this approach severely limits the scope for using the anode reaction. Usually only hydrogen is produced as an anode product.
- a process for producing phthalides was carried out by cathodic reduction of phthalic acid or phthalic acid derivatives, in which the carboxy groups can be replaced by units, that can be derived from carboxy groups in a condensation reaction are and one or more of the hydrogen atoms of the o-phenylene unit phthalic acid can be substituted by inert residues, found, taking the reduction in an organic Solvent containing less than 50 wt .-% water and one undivided electrolytic cell.
- ammonium salts and in particular the ammonium salt of phthalamic acid are particularly preferred.
- Suitable as electrode materials (both cathode and anode) commercially available electrodes made of graphite or carbon.
- the electrolyte is usually a 2 to 40% by weight solution of phthalic acid or a phthalic acid derivative in an organic solvent, preferably less contains more than 25, particularly preferably less than 5% by weight of water.
- Suitable organic solvents are in particular aliphatic C 1 to C 4 alcohols, in particular methanol or ethanol, or a mixture of such alcohols with a carboxamide such as dimethylformamide or t-butylformamide.
- the electrolytes generally contain alkyl sulfates, for example methyl sulfate, or quaternary ammonium salts, in particular tetra (C 1 -C 4 -alkyl) ammonium halogens or tetrafluoroborates, usually in amounts of 0.4 to 10% by weight, based on the electrolyte, as conductive salts .
- alkyl sulfates for example methyl sulfate
- quaternary ammonium salts in particular tetra (C 1 -C 4 -alkyl) ammonium halogens or tetrafluoroborates, usually in amounts of 0.4 to 10% by weight, based on the electrolyte, as conductive salts .
- anodic Depolarizer For the anodic coupling process, it is recommended to be anodic Depolarizer to use usual organic compounds whose Suitability for electrochemical oxidation to the person skilled in the art in general is known. Some of the anodic coupling processes are preferred performed in the presence of a mediator. Possible anodic coupling processes and their mediatization for example in D. Kyriakou, Modern Electroorganic Chemistry, Springer, Berlin 1994, described in chapter 4.2.
- the anodic coupling processes are particularly suitable Oxidations of C-O or C-N single or double bonds, e.g. the oxidation of carboxylic acids, arylmethanes, aldehydes, carboxamides, Alcohols and heterocycles, or the oxidative C-C linkage especially of naphthalenes or activated CH groups.
- Halogen compounds are particularly suitable as mediators all bromides or iodides.
- the type of workup of the electrolyte mixture depends in particular according to the type of anodic coupling product and can according to generally known separation methods such as distillation, precipitation or recrystallization. Can be particularly easy most of the phthalides of many in a basic, aqueous environment separate insoluble organic by-products by removing the Phthalides in ammoniacal aqueous solutions that dissolves separates aqueous phase and the phthalide by acidification the aqueous phase fails again (see also here DE-A-2 510 920).
- Phthalides are obtained by the process according to the invention technically simple way in high yield and purity. At the same time it is possible by coupling with anodic oxidation reactions to produce different types of valuable products without the current and material yield at the cathode would decrease.
- a solution of 500 g dimethyl phthalate (2.56 mol), 1600 g t.-butylformamide is placed in an electrolytic cell, consisting of ten bipolar graphite ring disks, area per side: 147 dm 2 , with an electrode spacing of 0.7 mm and 375 g of methanol were electrolyzed with 25 g of tetrabutylammonium tetrafluoroborate at a current of 2.5 A for 11.5 hours at 60 ° C.
- the solvent t-butylformamide is recovered without decomposition, the anode process is methanol oxidation with the main product Methyl formate.
Description
Die vorliegende Erfindung betrifft ein neues Verfahren zur Herstellung von Phthaliden durch kathodische Reduktion von Pthalsäure oder Phthalsäurederivaten.The present invention relates to a new method of manufacture of phthalides by cathodic reduction of phthalic acid or phthalic acid derivatives.
Phthalide werden insbesondere als Zwischenprodukte für die Herstellung von Pflanzenschutzmitteln benötigt.Phthalides are especially used as intermediates for manufacturing of pesticides needed.
Ein elektrochemisches Verfahren zur Herstellung der Phthalide ist aus der DE-A-2 144 419 bekannt. Hierbei wird Ammoniumphthalamat in wässeriger Lösung mit einem Anteil organischer Lösungsmittel bis 50% bei Temperaturen bis 65°C an Metallen mit einer Wasserstoffüberspannung größer als Cu, z.B. Blei, kathodisch reduziert. Unter diesen Bedingungen gelingt die Herstellung von Phthaliden in befriedigenden Ausbeuten, wenn man die Reduktion in geteilten Elektrolysezellen vornimmt.An electrochemical process for the production of the phthalides is known from DE-A-2 144 419. Here, ammonium phthalamate in aqueous solution with a proportion of organic solvents up to 50% at temperatures up to 65 ° C on metals with a hydrogen overvoltage larger than Cu, e.g. Lead, reduced cathodically. Under these conditions, phthalides can be produced in satisfactory yields if you split the reduction in Electrolysis cells.
Die Herstellung von besonders reinen Phthaliden ist in der DE-A-2 510 920 beschrieben. Nach dieser Lehre reduziert man ammoniakalische wässerige Lösungen von Phthalsäure oder Phthalsäureanhydrid kathodisch bei Temperaturen von bis zu 100°C an Metallen mit einer Wasserstoffüberspannung größer als Cu. Das Verfahren erfordert ebenfalls die Anwendung geteilter Elektrolysezellen. Zur Abtrennung des Phthalids aus dem Elektrolysegemisch wird es gegebenenfalls nach Abtrennung von überschüssigem Ammoniak bei einer Temperatur von 35 bis 100°C angesäuert und das ausgefallene Phthalid abgetrennt.The production of particularly pure phthalides is in the DE-A-2 510 920. After this teaching you reduce ammoniacal aqueous solutions of phthalic acid or phthalic anhydride cathodic at temperatures up to 100 ° C Metals with a hydrogen overvoltage greater than Cu. The The process also requires the use of split electrolysis cells. To separate the phthalide from the electrolysis mixture if necessary, it is removed after excess Acidified ammonia at a temperature of 35 to 100 ° C and that failed phthalide separated.
Nachteilig an den beschriebenen Verfahren ist jedoch der mit der Verwendung von geteilten Elektrolysezellen verbundene apparative Aufwand, da in diesem Fall 2 Zellkreise erforderlich sind. Zudem ist das Arbeiten mit 2 Zellkreisen mit folgenden weiteren Nachteilen verbunden:A disadvantage of the described method, however, is that with the Use of apparatus connected to split electrolysis cells Effort, since 2 cell circles are required in this case. In addition is working with 2 cell circles with the following further disadvantages connected:
Die Zellkreise müssen durch eine Membran oder ein Diaphragma getrennt werden; dies bedeutet einen Verlust an Energie durch ohmsche Wärme. Um diesen Verlust zu minimieren, wird meist wenigstens eine Kammer mit einer wäßrigen (> 80 % H2O) Leitsalzlösung beschickt. Bei kathodischen Reduktionen ist dies der Anolyt. Der Zwang zu diesem Vorgehen engt die Freiräume zur Nutzung der Anodenreaktion stark ein. Normalerweise wird als Anodenprodukt lediglich Wasserstoff erzeugt. The cell circles must be separated by a membrane or a diaphragm; this means a loss of energy through ohmic heat. In order to minimize this loss, at least one chamber is usually charged with an aqueous (> 80% H 2 O) conductive salt solution. In the case of cathodic reductions, this is the anolyte. The constraint on this approach severely limits the scope for using the anode reaction. Usually only hydrogen is produced as an anode product.
Weiterhin besteht bei den vorbekannten Verfahren die Gefahr, daß eine Anodenkorrosion sowie eine Vergiftung der Kathoden auftritt.Furthermore, there is a risk with the known methods that anode corrosion and poisoning of the cathodes occur.
Die der Erfindung zugrunde liegende technische Aufgabe bestand deshalb darin, ein technisch einfacheres Verfahren zur Herstellung von Phthaliden in hoher Reinheit und guten Ausbeuten zur Verfügung zu stellen, das die Nachteile des Standes der Technik nicht aufweist und insbesondere die Möglichkeit der Nutzung der Anodenreaktion zur Herstellung von anderen Produkten als Wasserstoff eröffnet.The technical problem underlying the invention was therefore in it, a technically simpler method of manufacture of phthalides in high purity and good yields to provide the disadvantages of the prior art Technology does not have and in particular the possibility of use the anode reaction to produce products other than Hydrogen opened.
Demgemäß wurde ein Verfahren zur Herstellung von Phthaliden durch kathodische Reduktion von Phthalsäure oder Phthalsäurederivaten, bei denen die Carboxygruppen durch Einheiten ersetzt sein können, die von Carboxygruppen in einer Kondensationsreaktion ableitbar sind und eines oder mehrere der Wasserstoffatome der o-Phenylen-Einheit der Phthalsäure durch inerte Reste substituiert sein können, gefunden, wobei man die Reduktion in einem organischen Lösungsmittel, das weniger als 50 Gew.-% Wasser enthält und einer ungeteilten Elektrolysezelle vornimmt.Accordingly, a process for producing phthalides was carried out by cathodic reduction of phthalic acid or phthalic acid derivatives, in which the carboxy groups can be replaced by units, that can be derived from carboxy groups in a condensation reaction are and one or more of the hydrogen atoms of the o-phenylene unit phthalic acid can be substituted by inert residues, found, taking the reduction in an organic Solvent containing less than 50 wt .-% water and one undivided electrolytic cell.
Als Ausgangsverbindungen für die Herstellung der Phthalide sind
insbesondere solche der allgemeinen Formel I

- R1, R2, R3 und R4:
- unabhängig voneinander Wasserstoff, C1- bis C4-Alkyl oder Halogen
- R5 und R6:
-
- a) unabhängig voneinander -COOH oder COOX, wobei X für C1- bis C4-Alkyl steht,
- b) einer der Substituenten R5 oder R6 -COONY4 und der andere Substituent CONH2, wobei Y für C1- bis C4-Alkyl oder Wasserstoff steht,
- c) R5 und R6 zusammen -CO-O-CO-.
- R 1 , R 2 , R 3 and R 4 :
- independently of one another hydrogen, C 1 - to C 4 -alkyl or halogen
- R 5 and R 6 :
-
- a) independently of one another -COOH or COOX, where X is C 1 -C 4 -alkyl,
- b) one of the substituents R 5 or R 6 -COONY 4 and the other substituent CONH 2 , where Y is C 1 -C 4 -alkyl or hydrogen,
- c) R 5 and R 6 together -CO-O-CO-.
Besonders bevorzugt sind die Derivate der Phthalsäure, bei denen R1, R2, R3 und R4 Wasserstoff bedeutet und darunter insbesondere die Pththalsäuredi(C1- bis C3-alkyl)-ester, vor allem der Phthalsäuredimethylester.Particularly preferred are the derivatives of phthalic acid in which R 1 , R 2 , R 3 and R 4 are hydrogen and in particular the phthalic acid di (C 1 - to C 3 -alkyl) esters, especially the dimethyl phthalate.
Bei den Verbindungen der Formel I, bei denen R5 und R6 die unter b) aufgeführte Bedeutung haben, sind die Ammoniumsalze und insbesondere das Ammoniumsalz der Phthalamidsäure besonders bevorzugt.In the compounds of the formula I in which R 5 and R 6 have the meaning given under b), the ammonium salts and in particular the ammonium salt of phthalamic acid are particularly preferred.
Als Elektrodenmaterialien (sowohl Kathode als auch Anode) eignen sich vor allem handelsübliche Elektroden aus Graphit oder Kohle.Suitable as electrode materials (both cathode and anode) commercially available electrodes made of graphite or carbon.
Bei dem Elektrolyten handelt es sich üblicherweise um eine 2 bis 40 gew.-%ige Lösung der Phthalsäure oder eines Phthalsäurederivates in einem organischen Lösungsmittel, das bevorzugt weniger als 25, besonders bevorzugt weniger als 5 Gew.-% Wasser enthält.The electrolyte is usually a 2 to 40% by weight solution of phthalic acid or a phthalic acid derivative in an organic solvent, preferably less contains more than 25, particularly preferably less than 5% by weight of water.
Als organische Lösungsmittel eignen sich insbesondere aliphatische C1- bis C4-Alkohole, insbesondere Methanol oder Ethanol oder eine Mischungen derartiger Alkohole mit einem Carbonsäureamid wie Dimethylformamid oder t-Butylformamid.Suitable organic solvents are in particular aliphatic C 1 to C 4 alcohols, in particular methanol or ethanol, or a mixture of such alcohols with a carboxamide such as dimethylformamide or t-butylformamide.
Als Leitsalze enthalten die Elektrolyte im allgemeinen Alkylsulfate, z.B. Methylsulfat, oder quarternäre Ammoniumsalze, insbesondere Tetra(C1- bis C4-alkyl)ammoniumhalogene oder -tetrafluoroborate, üblicherweise in Mengen von 0,4 bis 10 Gew.-% bezogen auf den Elektrolyt.The electrolytes generally contain alkyl sulfates, for example methyl sulfate, or quaternary ammonium salts, in particular tetra (C 1 -C 4 -alkyl) ammonium halogens or tetrafluoroborates, usually in amounts of 0.4 to 10% by weight, based on the electrolyte, as conductive salts .
Für den anodischen Koppelprozeß empfiehlt es sich, als anodischen Depolarisator übliche organische Verbindungen einzusetzen, deren Eignung für die elektrochemische Oxidation dem Fachmann allgemein bekannt ist. Einige der anodischen Koppelprozessen werden bevorzugt in Anwesenheit eines Mediators durchgeführt. Mögliche anodische Koppelprozesse und deren Mediatisierung werden beispielsweise in D. Kyriakou, Modern Electroorganic Chemistry, Springer, Berlin 1994, in Kapitel 4.2 beschrieben.For the anodic coupling process, it is recommended to be anodic Depolarizer to use usual organic compounds whose Suitability for electrochemical oxidation to the person skilled in the art in general is known. Some of the anodic coupling processes are preferred performed in the presence of a mediator. Possible anodic coupling processes and their mediatization for example in D. Kyriakou, Modern Electroorganic Chemistry, Springer, Berlin 1994, described in chapter 4.2.
Als anodische Koppelprozesse eignen sich insbesondere die Oxidationen von C-O- oder C-N-Einfach- oder Doppelbindungen, z.B. die Oxidation von Carbonsäuren, Arylmethanen, Aldehyden, Carbonsäureamiden, Alkoholen sowie Heterocyclen, oder die oxidative C-C-Verknüpfung insbesondere von Naphthalinen oder aktivierten CH-Gruppen.The anodic coupling processes are particularly suitable Oxidations of C-O or C-N single or double bonds, e.g. the oxidation of carboxylic acids, arylmethanes, aldehydes, carboxamides, Alcohols and heterocycles, or the oxidative C-C linkage especially of naphthalenes or activated CH groups.
Als Mediatoren eignen sich insbesondere Halogenverbindungen, vor allem Bromide oder Iodide. Halogen compounds are particularly suitable as mediators all bromides or iodides.
Was die sonstigen Verfahrensparameter wie Temperatur und Stromdichte betrifft, so sind diese unkritisch, solange sie sich im für elektrochemische Umsetzung organischer Verbindungen üblichen Rahmen bewegen. Sie sind beispielsweise in der DE-A-2 510 920 näher spezifiziert.As for the other process parameters such as temperature and current density , these are not critical as long as they are in the usual for electrochemical conversion of organic compounds Move frame. They are, for example, in DE-A-2 510 920 specified in more detail.
Die Art Aufarbeitung des Elektrolytgemisches richtet sich insbesondere nach der Art des anodischen Koppelproduktes und kann nach allgemein bekannten Trennmethoden wie Destillation, Fällung oder Umkristallisation erfolgen. Besonders einfach lassen sich die meisten Phthalide von vielen in basisch-wässerigem Milieu unlöslichen organischen Nebenprodukten abtrennen, indem man die Phthalide in ammoniakalischen wässerigen Lösungen löst, die wässerige Phase abtrennt und das Phthalid durch Ansäuern aus der wässerigen Phase wieder ausfällt (s. hierzu ebenfalls DE-A-2 510 920).The type of workup of the electrolyte mixture depends in particular according to the type of anodic coupling product and can according to generally known separation methods such as distillation, precipitation or recrystallization. Can be particularly easy most of the phthalides of many in a basic, aqueous environment separate insoluble organic by-products by removing the Phthalides in ammoniacal aqueous solutions that dissolves separates aqueous phase and the phthalide by acidification the aqueous phase fails again (see also here DE-A-2 510 920).
Nach dem erfindungsgemäßen Verfahren erhält man Phthalide auf technisch einfache Weise in hoher Ausbeute und Reinheit. Gleichzeitig ist es möglich, durch Kopplung mit anodischen Oxidationsreaktionen verschiedenartige Wertprodukte herzustellen, ohne daß die Strom- und Materialausbeute an der Kathode sinken würde.Phthalides are obtained by the process according to the invention technically simple way in high yield and purity. At the same time it is possible by coupling with anodic oxidation reactions to produce different types of valuable products without the current and material yield at the cathode would decrease.
In einer Elektrolysezelle, bestehend aus zehn bipolar geschalteten Ringscheiben aus Graphit, Fläche pro Seite: 147 dm2, mit einem Elektrodenabstand von 0,7 mm, wird eine Lösung aus 500 g Phthalsäuredimethylester (2,56 Mol), 1600 g t.-Butylformamid und 375 g Methanol mit 25 g Tetrabutylammoniumtetrafluoroborat bei einer Stromstärke von 2,5 A 11,5 h lang bei 60°C elektrolysiert.A solution of 500 g dimethyl phthalate (2.56 mol), 1600 g t.-butylformamide is placed in an electrolytic cell, consisting of ten bipolar graphite ring disks, area per side: 147 dm 2 , with an electrode spacing of 0.7 mm and 375 g of methanol were electrolyzed with 25 g of tetrabutylammonium tetrafluoroborate at a current of 2.5 A for 11.5 hours at 60 ° C.
Nach Abdestillation des Lösemittelgemisches konnten in einer Vakuumdestillation bei 10 mbar 2,18 Mol Phthalid entsprechend 85 % gewonnen werden.After distilling off the solvent mixture in a Vacuum distillation at 10 mbar corresponding to 2.18 mol of phthalide 85% can be won.
Das Lösemittel t-Butylformamid wird unzersetzt zurückgewonnen, der Anodenprozeß ist die Methanoloxidation mit dem Hauptprodukt Methylformiat. The solvent t-butylformamide is recovered without decomposition, the anode process is methanol oxidation with the main product Methyl formate.
In einer Elektrolysezelle gemäß Beispiel 1 wurden 2,56 Mol
Phthalsäuredimethylester, 750 g Methanol, 1225 g Dimethylformamid
(DMF) und 25 g Triethylmethylammoniummethosulfat bei 5 A für
6,9 h bei 50°C elektrolysiert. Es entstanden 4,1 Mol (Stromausbeute:
64 %)
N-Methoxymethyl-N-Methylformamid neben 2,1 Mol Phthalid
(Materialausbeute: 82 %).In an electrolysis cell according to Example 1, 2.56 mol of dimethyl phthalate, 750 g of methanol, 1225 g of dimethylformamide (DMF) and 25 g of triethylmethylammonium methosulfate at 5 A for 6.9 h at 50 ° C. were electrolyzed. 4.1 moles were produced (current yield: 64%)
N-methoxymethyl-N-methylformamide in addition to 2.1 moles of phthalide (material yield: 82%).
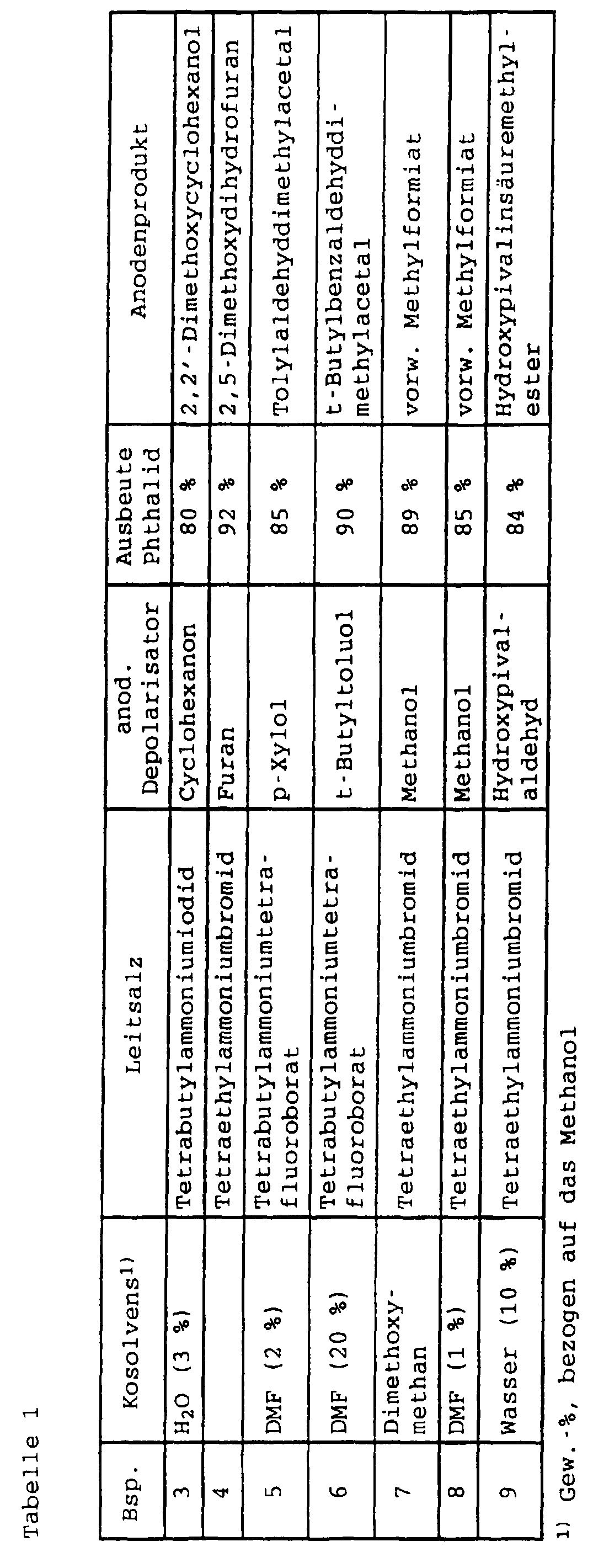
Analog zu Beispiel 2 wurde mit den jeweils in Tabelle 1 angegebenen
Ausgangsstoffen Phthalid und unterschiedliche anodische
Koppelprodukte hergestellt.
Claims (9)
- A process for preparing phthalides by cathodic reduction of phthalic acid or phthalic acid derivatives in which the carboxyl groups may be replaced by units which can be derived from carboxyl groups by a condensation reaction and one or more of the hydrogens of the o-phenylene unit of the phthalic acid may be replaced by inert radicals, the reduction being carried out in an organic solvent containing less than 50% by weight of water in an undivided electrolytic cell.
- A process as claimed in claim 1, wherein phthalic acid or phthalic acid derivatives of the general formula Iare employed where the substituents have the following meanings:

- R1, R2, R3 and R4:
- are each, independently of one another, hydrogen, C1- to C4-alkyl or halogen
- R5, R6:
-
- a) are each, independently of one another, -COOH or COOX, where X is C1- to C4-alkyl,
- b) one of the substituents R5 or R6 is -COONY4 and the other substituent is CONH2, where Y is C1- to C4-alkyl or hydrogen,
- c) R5 and R6 are together -CO-O-CO-.
- A process as claimed in claim 1 or 2, wherein the phthalic acid derivatives used are di(C1- to C3-alkyl) phthalates.
- A process as claimed in any of claims 1 to 3, wherein graphite or carbon electrodes are used.
- A process as claimed in any of claims 1 to 4, wherein the organic solvent used is an aliphatic C1- to C4-alcohol or a mixture of such an alcohol with a carboxamide.
- A process as claimed in any of claims 1 to 5, wherein the supporting electrolyte used is a quaternary ammonium salt.
- A process as claimed in any of claims 1 to 6, wherein the anodic depolarizer used for the anodic coproduction process is a conventional organic compound suitable for electrochemical oxidation.
- A process as claimed in any of claims 1 to 7, wherein the mediator used for the anodic coproduction process is a halogen compound.
- A process as claimed in claim 8, wherein the mediator used is bromide or iodide.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19618854 | 1996-05-10 | ||
| DE19618854A DE19618854A1 (en) | 1996-05-10 | 1996-05-10 | Process for the production of phthalides |
| PCT/EP1997/002185 WO1997043464A1 (en) | 1996-05-10 | 1997-04-28 | Process for preparing phthalides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0902846A1 EP0902846A1 (en) | 1999-03-24 |
| EP0902846B1 true EP0902846B1 (en) | 2000-07-26 |
Family
ID=7793943
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP97921810A Expired - Lifetime EP0902846B1 (en) | 1996-05-10 | 1997-04-28 | Process for preparing phthalides |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US6063256A (en) |
| EP (1) | EP0902846B1 (en) |
| JP (1) | JP3946260B2 (en) |
| CN (1) | CN1058302C (en) |
| CA (1) | CA2254788C (en) |
| DE (2) | DE19618854A1 (en) |
| ES (1) | ES2150770T3 (en) |
| WO (1) | WO1997043464A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1017688B1 (en) * | 1997-09-19 | 2002-04-10 | Basf Aktiengesellschaft | Method for producing phthalides |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR018507A1 (en) | 1997-09-19 | 2001-11-28 | Basf Se | RECOVERY PROCESS OF A COMPOUND DERIVED FROM THE PHTALIC ACID OF A REACTION MIXTURE IN WHICH THIS COMPOUND IS SYNTHESIZED |
| DE19808296A1 (en) * | 1998-02-27 | 1999-09-02 | Basf Ag | Process for the selective hydrolysis of acetals or ketals in the presence of phthalides |
| DE19944990A1 (en) | 1999-09-20 | 2001-03-22 | Basf Ag | Process for the electrolytic conversion of organic compounds |
| DE19944989A1 (en) | 1999-09-20 | 2001-03-22 | Basf Ag | Process for the electrolytic conversion of furan derivatives |
| CN1182127C (en) * | 2000-06-28 | 2004-12-29 | 中国医学科学院药物研究所 | Substituted 2-benzo [c] furanone compound, its preparing process and medicinal composition containing it |
| DE10057888A1 (en) | 2000-11-22 | 2002-05-23 | Basf Ag | Production of butanetetracarboxylic acid derivatives useful as intermediates for e.g. plant protectants, dyes, complexing agents, polymers by cathodic reduction of maleate or fumarate esters and producing co-product at anode |
| DE10058304A1 (en) * | 2000-11-24 | 2002-05-29 | Basf Ag | Process for the preparation of alkoxylated carbonyl compounds by an anodic oxidation process using the cathodic coupling reaction for organic synthesis |
| JP2015527483A (en) * | 2012-06-15 | 2015-09-17 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | Anodization of organic substrates in the presence of nucleophiles |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2510920A1 (en) * | 1975-03-13 | 1976-09-30 | Basf Ag | Electrochemical prepn of phthalide - from phthalic acid or anhydride, with final acidificn at controlled temp |
| DE2630927A1 (en) * | 1976-07-09 | 1978-01-19 | Basf Ag | METHOD FOR PRODUCING PHTHALIDOCARBONIC ACID- (5) |
-
1996
- 1996-05-10 DE DE19618854A patent/DE19618854A1/en not_active Withdrawn
-
1997
- 1997-04-28 JP JP54044397A patent/JP3946260B2/en not_active Expired - Fee Related
- 1997-04-28 DE DE59702087T patent/DE59702087D1/en not_active Expired - Fee Related
- 1997-04-28 WO PCT/EP1997/002185 patent/WO1997043464A1/en active IP Right Grant
- 1997-04-28 US US09/125,019 patent/US6063256A/en not_active Expired - Fee Related
- 1997-04-28 CN CN97192040A patent/CN1058302C/en not_active Expired - Fee Related
- 1997-04-28 ES ES97921810T patent/ES2150770T3/en not_active Expired - Lifetime
- 1997-04-28 CA CA002254788A patent/CA2254788C/en not_active Expired - Fee Related
- 1997-04-28 EP EP97921810A patent/EP0902846B1/en not_active Expired - Lifetime
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1017688B1 (en) * | 1997-09-19 | 2002-04-10 | Basf Aktiengesellschaft | Method for producing phthalides |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2000511592A (en) | 2000-09-05 |
| DE19618854A1 (en) | 1997-11-13 |
| CA2254788C (en) | 2005-03-01 |
| ES2150770T3 (en) | 2000-12-01 |
| CN1210564A (en) | 1999-03-10 |
| WO1997043464A1 (en) | 1997-11-20 |
| CN1058302C (en) | 2000-11-08 |
| EP0902846A1 (en) | 1999-03-24 |
| US6063256A (en) | 2000-05-16 |
| JP3946260B2 (en) | 2007-07-18 |
| DE59702087D1 (en) | 2000-08-31 |
| CA2254788A1 (en) | 1997-11-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0012215B1 (en) | 2-Hydroxybutanesulfonic acid choline and its use as conducting salt | |
| EP0902846B1 (en) | Process for preparing phthalides | |
| EP2318569A1 (en) | Method for anodic dehydrodimerisation of substituted arylalcohols | |
| EP0072914B1 (en) | Process for the production of alkyl-substituted benzaldehydes | |
| EP1619273A1 (en) | Process for the synthesis of 2 Alkyne-1-acetales | |
| DE2855508A1 (en) | METHOD FOR PRODUCING BENZALDEHYDES | |
| EP1863781A1 (en) | Method for producing alkoxylated 2,5-dihydrofuran but-2-ene derivatives or tetra-1,1,4,4-alkoxylated but-2-ene derivatives | |
| EP1339664A1 (en) | Production of butane tetracarboxylic acid derivatives by means of coupled electrosynthesis | |
| WO2008145627A1 (en) | Electrochemical oxidation at allyl groups | |
| EP0339523B1 (en) | Process for manufacturing hydroxycarboxylic-acid esters | |
| EP1769103B1 (en) | Electrochemical process for preparing cyclopropylbenzylamines | |
| EP0638665A1 (en) | Process for the preparation of benzaldehyde dialkyl acetals | |
| EP0029995A1 (en) | Process for the preparation of 4-tert.-butylbenzaldehyde | |
| EP0085158B1 (en) | Process for the preparation of cycloalkenon derivatives | |
| DE2710420C2 (en) | Process for the electrolytic production of 2,5-dialkoxy-2,5-dihydrofurans | |
| EP1913178A1 (en) | Process for preparing 1,1,4,4-tetraalkoxybut-2-ene derivatives | |
| EP1017688B1 (en) | Method for producing phthalides | |
| DE2403446C2 (en) | Process for the preparation of hydrogenated indoles | |
| EP0179377B1 (en) | Process for the preparation of 1-alkoxyisochromanes, and 1-alkoxy alkylisochromanes | |
| DE2434845C3 (en) | Electrochemical production of aromatic or aromatic-heterocyclic alkanoic acid esters | |
| DE2851732A1 (en) | Substd. benzaldehyde di:alkyl acetal cpds. prodn. - by electrochemical oxidn. of substd. toluene in alcohol contg. tetra:alkyl-ammonium aryl-sulphonate | |
| DE3045370A1 (en) | METHOD FOR PRODUCING KETALES OF TRIMETHYL-P-BENZOCHINONE | |
| DE1961364A1 (en) | Monosubst hydrazines by electrochemical - prodn | |
| DE3619656A1 (en) | NEW 2,6-DIMETHYL-P-BENZOCHINONE TETRAALKYLKETALES AND THEIR PRODUCTION | |
| DE2428878A1 (en) | Alkyl or omega-hydroxyalkyl para-hydroxymethyl-benzoate prodn - by electrochemical reduction of dialkyl or dihydroxy-alkyl terephthalate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 19980602 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): BE CH DE ES FR GB IT LI NL SE |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| 17Q | First examination report despatched |
Effective date: 19990713 |
|
| GRAG | Despatch of communication of intention to grant |
Free format text: ORIGINAL CODE: EPIDOS AGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAH | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOS IGRA |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): BE CH DE ES FR GB IT LI NL SE |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REF | Corresponds to: |
Ref document number: 59702087 Country of ref document: DE Date of ref document: 20000831 |
|
| GBT | Gb: translation of ep patent filed (gb section 77(6)(a)/1977) |
Effective date: 20000810 |
|
| ITF | It: translation for a ep patent filed |
Owner name: ING. C. GREGORJ S.P.A. |
|
| ET | Fr: translation filed | ||
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2150770 Country of ref document: ES Kind code of ref document: T3 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: IF02 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20090508 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: SE Payment date: 20090407 Year of fee payment: 13 Ref country code: NL Payment date: 20090405 Year of fee payment: 13 Ref country code: IT Payment date: 20090424 Year of fee payment: 13 Ref country code: FR Payment date: 20090417 Year of fee payment: 13 Ref country code: DE Payment date: 20090428 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20090428 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 20090416 Year of fee payment: 13 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 20090422 Year of fee payment: 13 |
|
| BERE | Be: lapsed |
Owner name: *BASF A.G. Effective date: 20100430 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: V1 Effective date: 20101101 |
|
| EUG | Se: european patent has lapsed | ||
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20100428 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20101230 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20101101 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20101103 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100428 Ref country code: BE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100428 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20110715 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20110705 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100429 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100430 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: SE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20100429 |