-
Diese
Erfindung bezieht sich auf die Technikgebiete Biotechnologie, Medizin,
Biologie und Biochemie. Ihre Anwendungen betreffen die Gesundheit
von Menschen, Tieren und Pflanzen. Ganz besonders erlaubt die Erfindung
die Identifizierung der Nucleinsäure-Sequenzen,
die die Entwicklung von neuen Screeningverfahren für Moleküle von therapeutischem
Interesse, von neuen Werkzeugen der Gentherapie wie auch Hinweise
auf das toxische Potenzial und das Verfolgen der Wirksamkeit von
Molekülen
und pharmakogenomischen Informationen zu liefern erlauben.
-
Die
vorliegende Erfindung beschreibt eine Reihe neuartiger Techniken
zur Identifizierung von Nucleinsäure-Sequenzen,
die auf dem Aufzeigen von qualitativen Unterschieden zwischen den
RNAs, die aus zwei verschiedenen Zusammenhängen stammen, die man zu vergleichen
wünscht
und die insbesondere von einem kranken Gewebe oder Organ und ihrem
gesunden Äquivalent
stammen. Genauer, diese Techniken sind für ein spezifisches Klonieren
der alternativen Introns und Exons bestimmt, die in einer pathologischen
Situation und einem gesunden Zustand oder in zwei physiologischen
Situationen differentiell gespleißt sind, die man zu vergleichen
wünscht.
Diese qualitativen Unterschiede innerhalb der RNAs können ebenfalls
aus Störungen
des Genoms hervorgehen, wie Insertionen oder Deletionen in den Regionen,
die in RNA transkribiert werden. Diese Reihe an Techniken wird mit
dem Akronym DATAS bezeichnet: Differential Analysis of Transcripts
with Alternative Splicing.
-
Die
Charakterisierung der genetischen Expressionsstörungen, die eine bestimmte
Krankheit beherrschen oder mit dieser verbunden sind, begründen eine
bedeutende Hoffnung, neue therapeutische Ziele und neue Diagnosemittel
zu entdecken. Jedoch liefert die Identifizierung einer genomischen
oder komplementären DNA-Sequenz,
die mittels positioneller Klonierung oder mittels quantitativer
differentieller Screening-Techniken erfolgt war, nur wenig oder
keine Information über
die Funktion und noch weniger über
die funktionellen Domänen,
die bei den mit der untersuchten Krankheit verbundenen Deregulationen
beteiligt sind. Die vorliegende Erfindung beschreibt eine Reihe
neuartiger Techniken, die darauf abzielen, die Spleißunterschiede
der RNAs zu identifizieren, die zwischen zwei verschiedenen physiopathologischen
Situationen vorliegen. Die Identifizierung dieser Unterschiede liefert
Informationen über
die qualitativen Unterschiede und nicht über die quantitativen Unterschiede,
wie es bei den bis heute beschriebenen Techniken der Fall ist. Die
Gesamtheit der in der vorliegenden Erfindung angeführten Techniken
werden folglich unter der Bezeichnung „qualitatives differentielles
Screening" oder
DATAS zusammengefasst. Zum Beispiel sind die Verfahren der Erfindung
für die Identifizierung
von neuen therapeutischen Zielen oder Produkten, für die Herstellung
von Werkzeugen für
die genetische Recherche und/oder von Diagnosemitteln, für den Aufbau
von Nucleinsäure-Banken
und in den Verfahren zur Bestimmung des toxikologischen Profils
oder der Wirkung einer Verbindung zweckdienlich.
-
Ein
erster Gegenstand der Erfindung beruht ganz besonders auf einem
Verfahren zur Identifizierung und/oder Klonierung von Nucleinsäure-Regionen,
die für
qualitative genetische Unterschiede zwischen zwei biologischen Proben
repräsentativ
sind, umfassend einen Schritt der Hybridisierung zwischen einer
Population RNA oder doppelsträngiger
cDNA, die aus einer ersten biologischen Probe stammt, und einer
Population cDNA, die aus einer zweiten biologischen Probe stammt
(1A).
-
Wie
oben angegeben, können
diese qualitativen genetischen Unterschiede in Spleißmodifikationen der
RNA oder Deletionen und/oder Insertionen in den Regionen des Genoms,
die in RNA transkribiert werden, begründet sein.
-
In
einer ersten Anwendungsform handelt es sich um eine Hybridisierung
einer RNA-Population, die von einer ersten biologischen Probe stammt,
und einer cDNA-Population
(einzelsträngig
oder doppelsträngig), die
von einer zweiten biologischen Probe stammt.
-
In
einer anderen Anwendungsform handelt es sich um eine Hybridisierung
einer doppelsträngigen
cDNA-Population, die von einer ersten biologischen Probe stammt,
und einer cDNA-Population (doppelsträngig oder bevorzugt einzelsträngig), die
von einer zweiten biologischen Probe stammt.
-
Ein
ganz besonderer Gegenstand der Erfindung beruht auf einem Verfahren
zur Identifizierung von Nucleinsäure-Regionen,
die in zwei physiologischen Situationen unterschiedlich gespleißt sind,
umfassend die Hybridisierung einer RNA-Population oder doppelsträngigen cDNA-Population,
die von einer Testsituation stammt, mit einer cDNA-Population, die
von einer Referenzsituation stammt, und die Identifizierung von
Nucleinsäuren,
die diesen differentiellen Spleißungen entsprechen.
-
Ein
anderer Gegenstand der Erfindung betrifft ein Verfahren zur Klonierung
von Nucleinsäuren,
die zwischen zwei physiologischen Situationen differentiell gespleißt sind,
umfassend die Hybridisierung einer RNA-Population oder doppelsträngigen cDNA-Population,
die von einer Testsituation stammt, mit einer cDNA-Population, die
von einer Referenzsituation stammt, und die Klonierung von Nucleinsäuren, die
diesen differentiellen Spleißungen
entsprechen.
-
In
einer besonderen Anwendungsform umfasst das Verfahren zur Identifizierung
und/oder Klonierung von Nucleinsäuren
der Erfindung zwei parallele Hybridisierungen:
- (a)
die Hybridisierung der RNA, die von einer ersten Probe (Testsituation)
stammt, mit der cDNA, die von einer zweiten Probe (Referenzsituation)
stammt;
- (b) die Hybridisierung der RNA, die von einer zweiten Probe
(Referenzsituation) stammt, mit der cDNA, die von einer zweiten
Probe (Testsituation) stammt;
- (c) die Identifizierung und/oder Klonierung von Nucleinsäuren, die
qualitativen genetischen Unterschieden entsprechen, aus den in (a)
und (b) gebildeten Hybriden.
-
Die
vorliegende Erfindung betrifft ebenfalls die Herstellung von Nucleinsäure- Banken, die so erhaltenen
Nucleinsäuren
und Banken wie auch die Verwendungen dieser Materialien in allen
Bereichen der Biologie/Biotechnologie, wie es später erläutert wird.
-
In
dieser Hinsicht bezieht sich die Erfindung ebenfalls auf ein Verfahren
zur Herstellung von Zusammensetzungen oder Banken von profilierten
Nucleinsäuren,
die für
zwischen zwei biologischen Proben vorliegende qualitative Unterschiede
repräsentativ
sind, umfassend einen Schritt der Hybridisierung einer RNA-Population, die von
einer ersten biologischen Probe stammt, mit einer cDNA-Population, die von
einer zweiten biologischen Probe stammt.
-
Die
Erfindung betrifft außerdem
ein Verfahren zur Profilierung („Profiling") einer cDNA-Zusammensetzung, umfassend
einen Schritt der Hybridisierung dieser Zusammensetzung mit einer
RNA-Population oder umgekehrt.
-
Wie
oben angegeben, betrifft die vorliegende Erfindung insbesondere
Verfahren zur Identifizierung und Klonierung von Nucleinsäuren, die
für einen
physiologischen Zustand repräsentativ
sind. Außerdem
repräsentieren
die identifizierten Nucleinsäuren
und/oder Klone die Merkmale eines physiologischen Zustands in dem
Sinne, dass diese Nucleinsäuren
im Allgemein zum großen
Teil an dem beobachteten physiologischen Zustand beteiligt sind.
Aus diesem Grund geben die qualitativen Verfahren einen direkten
Zugang zu genetischen Elementen oder zu ihrem Proteinprodukt, das
eine funktionelle Rolle bei der Entwicklung eines physiopathologischen
Zustands spielt.
-
Die
Verfahren gemäß der Erfindung
beruhen zum Teil auf einem neuartigen Schritt der Kreuzhybridisierung
zwischen RNA und cDNA von unterschiedlichen physiologischen Situationen.
Diese Kreuzybrisierung(en) erlauben vorteilhaft das Aufzeigen von
ungepaarten Regionen in den gebildeten Hybriden, nämlich Regionen,
die in den RNAs in einer bestimmten physiologischen Situation und
nicht in den RNAs in einer anderen physiologischen Situation vorhanden
sind. Diese Regionen entsprechen im Wesentlichen alternativen Spleißungen,
die für
einen physiologischen Zustand charakteristisch sind, können aber
ebenfalls genetische Störungen
wie Insertionen oder Deletionen widerspiegeln, und folglich besonders
zweckdienliche genetische Elemente für die Therapie oder Diagnose
bilden, wie es später
erklärt
wird. Die Erfindung besteht folglich insbesondere darin, die nach
Kreuzybridisierung(en) gebildeten Komplexe zu erhalten, um daraus
die Regionen zu gewinnen, die qualitativen Unterschieden entsprechen.
Diese Methodologie unterscheidet sich von quantitativen Subtraktionstechniken,
die dem Fachmann bekannt sind (Sargent and Dawid (1983), Science,
222, 135-139; Davis et al. (1984), PNAS, 81, 2194-2198; Duguid and
Dinauer (1990) Nuc. Acid Res., 18, 2789-2792; Diatchenko et al.
(1996) PNAS, 93, 6025-6030) [WO98/42781; Hubank et al., Nucleic
Acid Res. 22 (1994) Vol. 22, Nr. 25, Seiten 5640-5648], die nach
Hybridisierung(en) die gebildeten Hybride eliminieren, um nur die
nicht komplexierten Nucleinsäuren
zu erhalten. Ebenso führt
Perret et al. (Gene 208 (1998) S. 103-115), im Gegensatz zur Erfindung,
eindeutig eine Untersuchung von Expressionslevels von Genen in einer biologischen
Probe auf, und berichtet keinesfalls von der Analyse ungepaarter
Regionen in den gebildeten Hybriden zwischen zwei Proben. EP-A-0791
660 führt
die Detektion einer einzigartigen Spleißstelle auf und berichtet weder
von einem Verfahren zur Identifizierung von qualitativen Regionen
noch von Verfahren und Werkzeugen der Analyse gemäß der Erfindung.
-
Die
Erfindung betrifft folglich an erster Stelle ein Verfahren zur Identifizierung
von Nucleinsäuren
von Interesse, umfassend die Hybridisierung zwischen der RNA einer
Testprobe und der cDNA einer Referenzprobe. Diese Hybridisierung
erlaubt das Aufzeigen innerhalb der gebildeten Komplexe von qualitativen
genetischen Unterschieden zwischen den getesteten Situationen und
daher die Identifizierung und/oder Klonierung von zum Beispiel für die Testsituation
charakteristischen Spleißungen.
-
Gemäß einer
ersten Variante der Erfindung erlaubt das Verfahren folglich die
Generierung einer Population von Nucleinsäuren, die für Spleißungen des physiologischen
Testzustands im Vergleich zum Referenzzustand charakteristisch sind
(1A, 1B). Wie später angegeben, kann diese Population
für die
Klonierung und die Charakterisierung der Nucleinsäuren, für ihre Verwendung
in Diagnose, Screening, Therapie oder für die Herstellung von Antikörpern oder
Proteinfragmenten oder von ganzen Proteinen verwendet werden. Diese
Population kann ebenfalls für
die Bildung von Banken, die in verschiedenen, später angeführten Anwendungsbereichen verwendbar
sind, und für
die Herstellung von markierten Sonden (1D) dienen.
-
Gemäß einer
anderen Variante der Erfindung umfasst das Verfahren eine erste
Hybridisierung, wie sie oben beschrieben ist, und eine parallele
zweite Hybridisierung zwischen der RNA, die von der Referenzsituation
stammt, und der cDNA, die von einer Testsituation stammt. Diese
Variante ist besonders vorteilhaft, da sie die Generierung zweier
Nucleinsäure-Populationen
erlaubt, wobei eine die Merkmale der Testsituation im Vergleich
zur Referenzsituation und die andere die Merkmale der Referenzsituation
im Vergleich zur Testsituation repräsentiert (1C).
Diese beiden Populationen können
ebenfalls als Quelle von Nucleinsäuren verwendet sein, wie auch
als Marker-Bank des genetischen Abdrucks von einer bestimmten physiologischen
Situation, wie es später
genauer angegeben wird (1D).
-
Die
vorliegende Erfindung kann auf alle Arten biologischer Proben angewandt
werden. Insbesondere kann die biologische Probe jede Zelle, jedes
Organ, Gewebe, jede Entnahme, Biopsie etc. sein, das/die Nucleinsäuren enthält. Handelt
es sich um ein Organ, Gewebe oder Biopsie, werden sie gegebenenfalls
kultiviert, um einen Zugang zu den Zellen zu erlauben, aus denen
sie bestehen. Es kann sich um Proben handeln, die von Säugern (insbesondere
Menschen), von Pflanzen, Bakterien oder niederen eukaryotischen
Zellen (Hefen, Pilzzellen) stammen. Beispiele für Materialien sind insbesondere
eine Tumorbiopsie, eine Biopsie neurodegenerativer Plaques oder
von Gehirnbereichen, die neurodegenerative Schäden aufweisen, eine Hautprobe,
eine Probe von Blutzellen, die nach einer Blutentnahme erhalten
sind, eine colorektale Biopsie, Biopsien, die von Lungenspülungen stammen,
etc. Beispiele für
Zellen sind insbesondere Muskelzellen, Leberzellen, Fibroblasten,
Nervenzellen, Epidermiszellen, Hautzellen, Blutzellen wie die T-,
B-Lymphocyten, Monocyten, Mastocyten, Granulocyten, Makrophagen.
-
Wie
oben angegeben, erlaubt das qualitative differentielle Screening
gemäß der Erfindung
die Identifizierung von Nucleinsäuren,
die für
eine bestimmte physiologische Situation (Situation B) charakteristisch sind,
durch Vergleich mit einer physiologischen Referenzsituation (Situation
A) im Hinblick auf ihre Klonierung oder andere Verwendungen. Als
Veranschaulichung können
die untersuchten physiologischen Situationen A und B die Folgenden
sein:
-
RNA-Populationen
-
Für die Anwendung
der vorliegenden Erfindung ist es möglich, Gesamt-RNA oder die
Messenger-RNA zu verwenden. Diese RNAs können mit allen herkömmlichen
molekularbiologischen Verfahren präpariert sein, die dem Fachmann
gut bekannt sind. Diese Verfahren umfassen im Allgemeinen eine Lyse
der Zellen oder des Gewebes oder der Proben und die Isolierung der
RNAs mittels Extraktionsverfahren.
-
Es
kann sich insbesondere um eine Behandlung mit chaotropen Mitteln
wie beispielsweise Guanidinthiocyanat (das die Zellen zerstört und die
RNA schützt)
und anschließende
RNA-Extraktion mit Lösemitteln (zum
Beispiel Phenol, Chloroform) handeln. Derartige Verfahren sind dem
Fachmann wohl bekannt (siehe Maniatis et al., Chomczynski et al.,
Anal. Biochem. 162 (1987) 156). Diese Verfahren und können ohne
weiteres unter Verwendung von im Handel erhältlichen Kits angewandt sein
wie zum Beispiel das Kit US73750 (Amersham) oder das Kit Rneasy
(Qiagen) für
die Gesamt-RNA. Es ist nicht erforderlich, dass die eingesetzten RNAs
ganz rein sind und insbesondere stört es nicht, wenn Spuren genomischer
DNA oder anderer Zellbestandteile (Proteine, etc.) in den Präparaten
verbleiben, da sie die Stabilität
der. RNAs nicht signifikant beeinflussen und wenn die Art und Weise
der Präparation
zwischen den beiden zu vergleichenden verschiedenen Proben die gleichen
sind. Außerdem
ist es wahlweise möglich,
nicht Gesamt-RNA Präparate
sondern Messenger-RNA Präparate
zu verwenden. Diese können
entweder direkt aus einer biologischen Probe oder aus Gesamt-RNA
mit Hilfe von polyT-Sequenzen gemäß herkömmlichen Verfahren isoliert
sein. Die Gewinnung von Messenger-RNA kann in dieser Hinsicht mit
Hilfe kommerzieller Kits durchgeführt sein, wie beispielsweise
mit Kit US72700 (Amersham) oder einem oligo-(dT) Kügelchen
verwendenden Kit (Dynal). Ein vorteilhafte Art der RNA-Präparation
besteht darin, die cytosolischen RNAs, dann die cytosolischen poylA+
RNAs zu extrahieren. Kits, die die selektive Präparation von cytosolischen
RNAs erlauben, die nicht mit Prämessenger-RNAs
kontaminiert sind, die Träger
von nicht gespleißten
Exons und Introns sind, sind im Handel erhältlich. Dies sind insbesondere
die von Qiagen erhältlichen
Rneasy Kits (Bezugsbeispiel: 74103). Die RNAs können ebenfalls direkt aus Banken
oder anderen Proben gewonnen sein, die bereits hergestellt und/oder
in Sammlungen zugänglich
und unter geeigneten Bedingungen aufbewahrt sind.
-
Im
Allgemeinen umfassen die verwendeten RNA-Präparate vorteilhaft wenigstens
0,1 μg RNA,
vorzugsweise wenigstens 0,5 μg
RNA. Die Mengen können
entsprechend den verwendeten Zellen und Verfahren variieren, ohne
die Durchführung
der vorliegenden Erfindung zu verändern. Um hinreichende Mengen
an RNA zu gewinnen (vorzugsweise wenigstens 0,1 μg), ist es im Allgemeinen empfohlen,
eine biologische Probe zu verwenden, die wenigstens 105 Zellen
umfasst. In dieser Hinsicht umfasst eine klassische Biopsie allgemein zwischen
105 und 108 Zellen,
und eine Zellkultur in einer klassischen Petri-Schale (Durchmesser
6-10 cm) umfasst
größenordnungsmäßig 106 Zellen, was die leichte Gewinnung hinreichender
RNA-Mengen erlaubt.
-
Die
RNA-Präparate
können
unmittelbar verwendet werden oder für spätere Verwendungen aufbewahrt
werden, vorzugsweise in Kälte,
in Lösung
oder gefroren.
-
cDNA-Populationen
-
Die
im Rahmen der vorliegenden Erfindung verwendeten cDNAs können durch
inverse Transkription entsprechend herkömmlichen molekularbiologischen
Techniken erhalten sein. Man kann insbesondere sich auf Maniatis
et al. beziehen. Die inverse Transkription wird im Allgemeinen unter
Verwendung eines Enzyms, inverse Transkriptase („reverse Transkriptase"), und eines Primers
durchgeführt.
-
In
dieser Hinsicht sind zahlreiche inverse Transkriptasen in der Literatur
beschrieben worden und sind kommerziell erhältlich (Kit 1483188, Boehringer).
Als Beispiele kann man die am häufigsten
verwendeten inversen Transkriptasen nennen wie die des avianen Virus
AMV (Avian Myeloblastosis Virus) und des murinen Leukämie-Virus
MMLV (Moloney Murine Leukemia Virus). Man kann ebenfalls bestimmte
thermostabile DNA-Polymerasen nennen, die eine inverse Transkriptase-Aktivität besitzen,
wie diejenigen, die aus Thermus flavus und Thermus thermophilus
HB-8 isoliert sind (kommerziell erhältlich, Bezugsquelle Promega
M1941 und M2101). Gemäß einer
vorteilhaften Variante kann man zur Durchführung der vorliegenden Erfindung
die inverse Transkriptase von AMV verwenden, da dieses Enzym, das
bei 42°C
arbeitet (im Gegensatz zum MMLV-Enzym, das bei 37°C arbeitet)
bestimmte Sekundärstrukturen
der RNA destabilisiert, die die Elongation blockieren könnten, und
folglich die inverse Transkription der RNA von einer bedeutenden
Länge erlaubt,
und erlaubt, cDNA-Präparate
zu erhalten, die die RNA mit einer großen Genauigkeit und einer großen Effizienz
darstellt.
-
Gemäß einer
anderen vorteilhaften Variante der Erfindung verwendet man eine
inverse Transkriptase ohne RNase H-Aktivität. Die Verwendung dieses Enzymtyps
bietet mehrere Vorteile und insbesondere den Vorteil der Erhöhung der
Leistung der Synthese der cDNA und die Verhinderung jeglichen Abbaus
der RNA, die anschließend
in den Heteroduplices mit den neusynthetisierten cDNA beteiligt
sind, was folglich eventuell ein Verzicht auf die Phenolextraktion
von diesen erlaubt. Die inversen Transkriptasen ohne RNase N-Aktivität können aus
jeder inversen Transkriptase mittels Deletion(en) und/oder Mutagenese
hergestellt sein. Außerdem
sind derartige Enzyme ebenfalls im Handel erhältlich (zum Beispiel Life Technologies,
Referenz 18053-017).
-
Die
Anwendungsbedingungen der inversen Transkriptasen (Konzentration
und Temperatur) sind dem Fachmann gut bekannt. Insbesondere verwendet
man im Allgemeinen 10 bis 30 Enzymeinheiten pro Reaktion in Gegenwart
einer optimalen Mg2+-Konzentration von 10
mM.
-
Der
oder die für
die inverse Transkription eingesetzten Primer können unterschiedlicher Natur
sein. Es kann sich insbesondere um zufällige („random") Oligonucleotide handeln, die vorzugsweise
zwischen 4 und 10 Nucleotide, vorteilhaft; ein
Hexanucleotid, umfassen. Die Verwendung dieser Art Zufallsprimer
ist in der Literatur beschrieben worden und erlaubt die Initiation
der inversen Transkription an verschiedenen zufälligen Positionen innerhalb
der RNA-Moleküle.
Diese Technik ist besonders für
die inverse Transkription der Gesamt-RNA (nämlich die mRNA, die tRNA und
die rRNA umfassend) eingesetzt. In dem Fall wo man nur die inverse
Transkription der mRNA durchführen
möchte,
ist es von Vorteil als Primer ein oligo-dT Oligonucleotid zu verwenden, das
die Initiation der inversen Transkription von den polyA-Schwänzen aus
erlaubt, die für
Messenger-RNA spezifisch sind. Das oligo-dT Oligonucleotid kann
4 bis 20-mere, vorteilhaft ungefähr
15-mere, umfassen. Der Einsatz dieses Primer-Typs bildet eine bevorzugte
Anwendungsform der vorliegenden Erfindung. Andererseits kann es
vorteilhaft sein, für
die inverse Transkription einen markierten Primer zu verwenden.
Dies kann daher erlauben, die RNA nachträglich von der cDNA zu identifizieren
und/oder zu selektieren und/oder zu sortieren. Dies kann ebenfalls
die Isolierung der RNA/DNA-Heteroduplices erlauben, deren Bildung
einen Schlüsselschritt
der Erfindung darstellt. Die Markierung des Primers kann in einem
beliebigen System des Ligand-Empfänger-Typs bestehen, nämlich das
die Trennung der den Primer tragenden Moleküle mittels Affinität erlaubt.
Es kann sich zum Beispiel um eine Markierung mit Biotin handeln,
die mit jedem Träger (Kügelchen,
Säule,
Platte etc.) getrennt werden kann, auf dem das Streptavidin fixiert
ist. Jedes andere Markierungssystem, das diese Trennung erlaubt,
ohne die Eigenschaften des Primers zu beeinflussen, kann in äquivalenter
Weise verwendet sein.
-
Unter
den üblichen
Durchführungsbedingungen
generiert diese inverse Transkription einzelsträngige komplementäre (cDNA)
DNAs. Dies bildet eine erste vorteilhafte Form der vorliegenden
Erfindung.
-
In
einer zweiten Durchführungsvariante
wird die inverse Transkription so durchgeführt, dass doppelsträngige cDNA
hergestellt wird. Um dies zu erreichen, kann der zweite Strang,
nach Transkription des ersten cDNA-Strangs, entsprechend herkömmlichen
molekularbiologischen Techniken generiert werden, unter Verwendung
von Enzymen für
die Modifikation der DNA, wie DNA-Ligase, DNA-Polymerase I und DNA-Polymerase des
Phagen T4.
-
Die
cDNA-Präparate
können
unmittelbar verwendet werden oder für spätere Verwendungen aufbewahrt
werden, vorzugsweise in der Kälte,
in Lösung
oder gefroren.
-
Hybridisierungen
-
Wie
oben erklärt,
beruhen die Verfahren gemäß der Erfindung
zum Teil auf einem neuartigen Schritt der Kreuzhybridisierung zwischen
RNA und cDNA, die aus biologischen Proben in unterschiedlichen physiologischen
Situationen oder unterschiedlichen Ursprungs stammen. In einer bevorzugten
Anwendungsform erfolgt die Hybridisierung bevorzugt in flüssiger Phase.
Außerdem
kann die Hybridisierung in jeder geeigneten Vorrichtung durchgeführt sein,
wie beispielsweise Röhrchen
(zum Beispiel Eppendorf), Platten oder jeden anderen geeigneten
Träger,
der üblicherweise
in der Molekularbiologie Verwendung findet. Die Hybridisierung erfolgt
vorteilhafterweise in Volumina zwischen 10 und 1000 μl zum Beispiel
zwischen 10 und 500 μl.
Es ist zu verstehen, dass die verwendete Vorrichtung und die verwendeten
Volumina ohne weiteres vom Fachmann angepasst sein können. Die
für die
Hybridisierung eingesetzten Mengen von Nucleinsäuren sind ebenfalls dem Fachmann
bekannt. Im Allgemeinen sind Mikrogramm-Mengen für Nucleinsäuren hinreichend, zum Beispiel
in einer Größenordnung
von 0,1 bis 100 μg.
Ein sehr wichtiges Element bei der Durchführung der Hybridisierungen
beruht auf den jeweiligen Mengen von eingesetzten Nucleinsäuren. Daher
ist es möglich,
die Nucleinsäuren
in einem cDNA/RNA-Verhältnis,
variierend von 50 bis ungefähr
0,02, vorzugsweise von 40 bis 0,1, einzusetzen. Ganz besonders von
Vorteil ist es, ein cDNA/RNA-Verhältnis von nahe oder größer 1 zu
bevorzugen. Denn in diesen Experimenten bildet die RNA die Testverbindung
(„Tester") und die cDNA bildet
den Träger („Driver"). Aus diesem Grund
ist es wegen der Verbesserung der Spezifität des Verfahrens zu bevorzugen, dass
es unter Bedingungen durchgeführt
ist, wo der „Driver" im Verhältnis zum „Tester" im Überschuss
vorliegt. Denn unter diesen Bedingungen funktioniert das Zusammenspiel
zwischen den Nucleinsäuren
und die nicht perfekten Paarungen sind stark benachteiligt. Aus
diesem Grund sind die einzigen auftretenden Fehlpaarungen im Allgemeinen
in dem Vorhandensein von Regionen in den „Tester" RNA begründet, die nicht in der „Driver" cDNA vorkommen und
die folglich spezifisch sind. Um die Spezifität des Verfahrens zu fördern, ist
die Hybridisierung vorteilhaft bei einem cDNA/RNA-Verhältnis von
einschließlich
ungefähr
1 bis ungefähr
10 durchgeführt.
Es ist sehr wohl zu verstehen, dass das Verhältnis vom Fachmann entsprechend
den Verfahrensbedingungen (zur Verfügung stehende Mengen an Nucleinsäuren, physiologische
Situationen, verfolgte Absicht, etc.) angepasst sein kann. Die anderen
Parameter der Hybridisierung (Zeit, Temperatur, Ionenstärke) können ebenfalls
vom Fachmann angepasst sein. Im großen und ganzen erfolgt die
Hybridisierung nach Denaturierung von „Tester" und „Driver" (zum Beispiel durch Erwärmen) innerhalb
von etwa 2 bis 24 Stunden bei einer Temperatur von etwa 37°C (eventuell
Temperaturerhöhungen
unterworfen, wie später
noch beschrieben) und unter Standardbedingungen der Ionenstärke (kann
beispielsweise zwischen 0,1 bis 5 M NaCl variieren). Es ist bekannt,
dass die Ionenstärke
einer der entscheidenden Faktoren für die Stringenz einer Hybridisierung
ist, besonders im Falle einer Hybridisierung auf einem festen Träger.
-
Gemäß einer
besonderen Anwendungsform der Erfindung ist die Hybridisierung in
einer Phenol-Emulsion durchgeführt,
zum Beispiel gemäß der PERT-Technik
(„Phenol
Emulsion DNA Reassociation Technique), die von Kohne D.E. et al.
(Biochemistry, Vol. 16, Nr. 24, Seiten 5329-5341, 1977) beschrieben
ist. Vorteilhafterweise verwendet man im Rahmen der vorliegenden
Erfindung die Hybridisierung in Phenol-Emulsion, die mit Hilfe von
Thermocyclen (Temperaturerhöhungen
von etwa 37°C
auf etwa 60/65°C)
und nicht durch Rühren gemäß der von
Miller und Riblet beschriebenen Technik (NAR 23 (1995) 2339) durchgeführt ist.
Jede andere Hybridisierungstechnik in Flüssigphase, vorzugsweise in
Emulsion, kann im Rahmen der vorliegenden Erfindung eingesetzt sein.
Daher ist die Hybridisierung in einer anderen Form besonders vorteilhaft
in einer Lösung, die
80% Formamid enthält,
bei einer Temperatur von zum Beispiel 40°C durchgeführt.
-
Die
Hybridisierung kann ebenfalls mit einem auf einem Träger immobilisierten
Partner erfolgen. Vorteilhafterweise ist die cDNA immobilisiert.
Dies kann durchgeführt
sein, indem die Markierung genutzt wird, deren Ziel die cDNAs sein
können
besonders aufgrund der biotinylierten Primer. Die Biotin-Gruppen
werden mit den magnetischen Kügelchen
in Kontakt gebracht, auf denen Streptavidin-Moleküle verankert
sind. Die cDNAs können
dann mit Hilfe eines Magneten an einem Filter oder an einer Vertiefung
einer Mikrotiterplatte erhalten werden. Die RNAs werden dann unter
den erforderlichen Bedingungen hoher Ionenstärke mit den cDNAs in Kontakt
gebracht. Die ungepaarten RNAs werden durch Waschen entfernt. Die
hybridisierten RNAs wie auch die cDNAs werden durch Abschalten des
Magnetfeldes wiedergewonnen.
-
In
dem Fall wo die cDNA doppelsträngig
ist, ähneln
die angewandten Hybridisierungsbedingungen im Wesentlichen den oben
beschriebenen und können
vom Fachmann angepasst werden. Man bevorzugt in diesem Fall die
Durchführung
der Hybridisierung in Gegenwart von Formamid und setzt die Komplexe
Temperaturen aus, die zum Beispiel von 60 bis 40°C, bevorzugt von 56°C bis 44°C reichen,
um die Bildung von Komplexen des Typs R-Schleife zu fördern. Zudem
ist es erwünscht,
nach der Hybridisierung ein Mittel zur Stabilisierung der gebildeten
Triplices zuzugeben, wenn das Formamid aus dem Medium entfernt ist,
wie zum Beispiel Glyoxal (Kaback et al. (1979) Nucl. Acid Res.,
6, 2499-2517).
-
Diese
Kreuzhybridisierungen gemäß der Erfindung
generieren daher Zusammensetzungen, die cDNA/RNA-Heteroduplices
oder Heterotriplices umfassen, die die Merkmale jeder getesteten
physiologischen Situation repräsentieren.
Wie oben angegeben, können
in jeder dieser Zusammensetzungen Nucleinsäuren identifiziert und/oder
kloniert werden, die im Wesentlichen differentiellen alternativen
Spleißungen
oder anderen genetischen Störungen
entsprechen, die für
jede physiologische Situation spezifisch sind.
-
Die
Erfindung betrifft folglich vorteilhaft ein Verfahren zur Identifizierung
und/oder Klonierung von Nucleinsäure-Regionen,
die für
qualitative genetische Unterschiede zwischen zwei physiologischen
Situationen repräsentativ
sind, umfassend einen Schritt der Hybridisierung von RNA, die aus
einer biologischen Probe in einer ersten physiologischen Situation
stammt, und einzelsträngiger
cDNA, die aus einer biologischen Probe in einer zweiten physiologischen
Situation stammt, und die Identifizierung und/oder Klonierung von
ungepaarten RNA-Regionen aus den gebildeten Klonen.
-
Diese
erste Variante beruht ganz besonders auf der Heteroduplex-Bildung
zwischen RNA und einzelsträngiger
cDNA (siehe 2-4). Diese
Variante ist vorteilhaft unter Verwendung von Messenger-RNA oder
cDNA durchgeführt,
die durch inverse Transkription von Messenger-RNA gebildet ist,
nämlich
in Gegenwart eines oligo dT-Primers.
-
In
einer besonderen Anwendungsform umfasst das Verfahren zur Identifizierung
und/oder Klonierung von Nucleinsäuren
der Erfindung:
- (a) die Hybridisierung von RNA,
die von einer Testsituation stammt, mit der einzelsträngigen cDNA,
die von einer Referenzsituation stammt;
- (b) die Hybridisierung der RNA, die von einer Referenzsituation
stammt, mit der einzelsträngigen
cDNA, die von einer Testsituation stammt;
- (c) die Identifizierung und/oder Klonierung von ungepaarten
RNA-Regionen aus den in (a) und (b) gebildeten Hybriden;
-
In
einer besonderen Variante der Anwendung umfasst das Verfahren der
Erfindung die folgenden Schritte:
- (a) die Gewinnung
von RNA aus einer biologischen Probe in einer physiologischen Situation
A (rA);
- (b) die Gewinnung von RNA aus derselben biologischen Probe in
einer physiologischen Situation B (rB);
- (c) die Präparation
von cDNA aus einem Teil der in (a) gewonnenen rA RNA (cA DNA) und
aus einem Teil der in (b) gewonnenen rB RNA (cB DNA) mit Hilfe von
polyT-Primern,
- (d) die Hybridisierung in flüssiger
Phase eines Teils der rA RNA mit einem Teil der cB DNA (um rA/cB-Heteroduplices
zu generieren),
- (e) die Hybridisierung in flüssiger
Phase eines Teils der rB RNA mit einem Teil der cA DNA (um rB/cA-Heteroduplices
zu generieren),
- (f) die Identifizierung und/oder Klonierung von RNA-Regionen,
die in den in (d) und (e) erhaltenen rA/cB und rB/cA Heteroduplices
ungepaart sind.
-
In
einer anderen besonderen Variante der Anwendungsform umfasst das
Verfahren der Erfindung die Hybridisierung von RNA, die von der
Testsituation stammt, mit doppelsträngiger cDNA, die von der Referenzsituation
stammt, und die Identifizierung und/oder Klonierung von erhaltenen
doppelsträngigen
DNA-Regionen. Diese zweite Variante beruht ganz besonders auf der
Heterotriplex-Bildung zwischen der RNA und der doppelsträngigen cDNA,
die von Strukturen des Typs R-Schleife abgeleitet sind (siehe 5).
Angewandt wird diese Variante ebenfalls bevorzugt unter Verwendung
von Messenger-RNAs oder cDNAs, die hauptsächlich durch inverse Transkription
von Messenger-RNAs hergestellt sind, nämlich in Gegenwart eines polyT
Primers. Bei dieser Variante umfasst ebenfalls eine besondere Anwendungsform
zwei parallele Hybridisierungen, die zwei Populationen von Nucleinsäuren gemäß der Erfindung
generieren. In dieser Variante sind die gesuchten Regionen, die
für alternative
Spleißungen
spezifisch sind, nicht die ungepaarten RNA-Regionen, sondern doppelsträngige DNA,
die nicht von einer homologen RNA-Sequenz verdrängt werden konnten (siehe 5).
-
In
einer anderen Variante der Erfindung umfasst das Verfahren für die Isolierung
der qualitativen genetischen Unterschiede (z.B. die Spleißunterschiede),
die zwischen zwei Proben vorhanden sind, die Hybridisierung zwischen
einer Population von doppelsträngiger
cDNA, die von einer ersten biologischen Probe stammt, und einer
cDNA-Population (doppelsträngig,
vorzugsweise einzelsträngig),
die von einer zweiten biologischen Probe stammt (6).
-
Im
Unterschied zu den vorhergehend dargelegten Varianten verwendet
diese Variante weder DNAIRNA-Heteroduplices noch Heterotriplices,
sondern DNA/DNA-Homoduplices.
Diese Variante ist von Vorteil, da sie nicht nur einen Zugang zu
alternativen Exons und zu Introns erlaubt, sondern ebenfalls, und
innerhalb derselben Nucleinsäure-Bank,
zu spezifischen Verknüpfungen,
die durch Deletion eines Exons oder Introns geschaffen sind. Zudem
erlauben die Sequenzen in einer derartigen Bank den Zugang zu alternative
Exons und Introns flankierenden Sequenzen.
-
Für die Untersuchung
der beiden Proben (d.h. physiopathologische Zustände) werden die cytosolischen
polyA+ RNA entsprechend dem Fachmann bekannten und genau beschriebenen
Techniken extrahiert. Diese werden dann in cDNA mit Hilfe einer
inversen Transkriptase umgeschrieben, die keinerlei intrinsische RNase
H Aktivität
besitzt, wie bereits beschrieben. Eine dieser einzelsträngigen cDNA
wird dann in doppelsträngige
cDNA mit Hilfe von hexameren Zufallsprimern und gemäß dem Fachmann
bekannten Techniken ungewandelt. Für eine dieser untersuchten
Situationen verfügen
wir folglich über
eine einzelsträngige
cDNA (als „Driver" bezeichnet) und
für die
andere Situation über
eine doppelsträngige
cDNA (als „Tester" bezeichnet). Diese
cDNA werden durch Erhitzen denaturiert, dann so gemischt, dass der
Driver im Überschuss
bezogen auf den Tester vorliegt. Dieser Überschuss wird zwischen 1 und
50-fach, vorteilhaft 10-fach, gewählt. In einem bestimmten Versuch,
der mit beiden physiopathologischen Situationen vorgenommen wird,
ist die Situation, die den Driver bestimmt, zufällig gewählt und darf nicht die Natur
der gewonnenen Informationen beeinflussen. Daher beruht, wie im
Falle der zuvor angeführten
Versuchsansätze,
die Strategie zur Identifizierung der qualitativen Unterschiede,
die es zwischen zwei mRNA-Populationen gibt, auf der Klonierung
dieser Unterschiede, die in den gemeinsamen Messenger vorliegen:
die Strategie beruht auf der Klonierung von Sequenzen, die im Innern
des Duplex vorliegen, und nicht von einzelsträngigen Sequenzen, die den singulären oder
den überschüssigen Sequenzen
entsprechen, in einer der untersuchten Situationen. Die Mischung
der cDNA-Populationen wird gefällt,
dann in einer Formamid-haltigen Lösung (zum Beispiel 80%) wieder
aufgenommen. Die Hybridisierung wird 16 bis 48 Stunden, vorteilhaft
24 Stunden, durchgeführt.
Die Produkte dieser Hybridisierung werden gefällt, dann der Wirkung einer
Endonuclease mit einer Erkennungsstelle der doppelsträngigen DNA ausgesetzt,
die von 4 Basen bestimmt ist. Ein derartiges Restriktionsenzym wird
folglich doppelsträngige
cDNA, die bei der Hybridisierung gebildet wird, durchschnittlich
aller 256 Basen schneiden. Dieses Enzym ist vorteilhaft ausgewählt, um
klebrige Enden zu erzeugen. Beispiele für derartige Enzyme sind Restriktionsenzyme wie
Sau3Al, HpaII, TaqI und MseI. Die doppelsträngigen Fragmente, die von diesen
Enzymen geschnitten sind, sind folglich für eine Klonierungstrategie
zugänglich,
die die geschnittenen Restriktionsstellen verwendet. Diese Fragmente
bestehen aus zwei Typen: perfekt hybridisierte Fragmente, deren
beide Stränge
perfekt komplementär
sind, und Fragmente, deren Hybridisierung partiell ist, nämlich eine
einzelsträngige
Schleife umfassend, die von doppelsträngigen Bereichen flankiert
ist (6A). Diese wenigen letzteren Fragmente enthalten die
Informationen von Interesse. Um sie von der Mehrheit der perfekt
hybridisierten Fragmente zu trennen, da sie sich mehrheitlich von
der Länge
der cDNA ableiten, werden Trenntechniken auf Gelen oder anderen
geeigneten Matrices eingesetzt. Diese Techniken nutzen die verzögerte Migration
bei der Elektrophorese oder besonders Gelfiltration von DNA-Fragmenten,
die eine Schleife einzelsträngiger
DNA umfassen. Daher können
die Populationen der wenigen Fragmente, die die gewünschten
Informationen enthalten, präparativ
von der Population von mehrheitlichen Fragmenten getrennt werden,
die identischen DNA-Regionen
in beiden Populationen entsprechen. Diese Variante, die die Isolierung
der positiven und negativen Merkmale innerhalb derselben Population
erlaubt, die mit qualitativen Unterschieden verbunden sind, kann
ebenfalls mit RNA/DNA-Heteroduplices
angewandt werden. In dieser Hinsicht ist ein Beispiel einer verzögerten Migration eines
RNA/DNA-Heteroduplex, in dem ein Teil der RNA ungepaart ist, im
Vergleich zu einem homologen Heteroduplex, in dem alle Sequenzen
gepaart sind, mit dem grb2/grb33 Modell veranschaulicht, das in
den Beispielen beschrieben ist (siehe insbesondere 8,
Vertiefungen 2 und 3).
-
Identifizierung und Klonierung
-
Aus
den durch Hybridisierung generierten Nucleinsäure-Populationen können die
Regionen, die für qualitative
Unterschiede (z.B. differentielle alternative Spleißungen)
charakteristisch sind, mit jeder dem Fachmann bekannten Technik
identifiziert werden.
-
Identifizierung und Klonierung
aus RNA/DNA-Heteroduplices
-
Im
Falle von RNA/DNA-Heteroduplices (erste Variante des Verfahrens)
kommen diese Regionen daher hauptsächlich als ungepaarte RNA-Regionen
(RNA-Schleifen)
vor, wie in 3 dargestellt. Diese Regionen
können
folglich durch Abtrennung der Heteroduplices und der einzelsträngigen Nucleinsäuren (DNA, RNA)
(nichtreagierte überschüssige Nucleinsäuren), selektiven
Verdau der doppelsträngigen
RNA (an den Neteroduplices beteiligte Domänen), dann Abtrennung der resultierenden
einzelsträngigen
RNA und der einzelsträngigen
DNA identifiziert und kloniert werden.
-
In
dieser Hinsicht gemäß einem
in 3 veranschaulichten ersten Versuchsansatz werden
die ungepaarten RNA-Regionen durch Behandlung der Heteroduplices
mit Hilfe eines Enzyms identifiziert, das in der Lage ist, selektiv
die RNA-Domänen
zu verdauen, die an den RNA/DNA-Heteroduplices beteiligt sind. Enzyme mit
dieser Eigenschaft sind in der Technik bereits beschrieben und sind
im Handel erhältlich.
Dies sind die RNasen H, wie insbesondere die von E. coli in rekombinanter
Form produzierte RNase H, die im Handel erhältlich ist (Promega Ref. M4281;
Life Technologies Ref. 18021). Diese erste Behandlung erzeugt folglich
ein Gemisch, das die einzelsträngigen
ungepaarten RNA-Regionen und die einzelsträngige cDNA umfasst. Die RNAs
können
von den cDNAs mit jeder dem Fachmann bekannten Technik und insbesondere
auf Basis der Markierung für
die Präparation
der cDNA verwendeten Primer (siehe oben) getrennt werden. Diese
RNAs können
als Ausgangsmaterial für
die Identifizierung von Zielen, von genetischen Produkten von Interesse
oder für jede
andere Anwendung verwendet werden. Diese RNAs können ebenfalls in cDNAs umgeschrieben,
dann in Vektoren kloniert werden, wie es oben beschrieben ist.
-
In
dieser Hinsicht kann die Klonierung der RNAs auf verschiedene Weisen
durchgeführt
sein. Eine besteht darin, an jedem Ende der RNA Oligonucleotide
zu inserieren, die als Matrize für
eine inverse Transkription in Gegenwart entsprechender Primer dienen.
Dieses Hinzufügen
von Primern geschieht gemäß dem Fachmann
bekannten Techniken mit Hilfe eines Enzyms wie zum Beispiel die
RNA-Ligase, die
vom T4 Phagen stammt und die die Bildung von intermolekularen Phospodiester-Bindungen
zwischen dem 5'-Phosphat
eines Donator-Moleküls
und dem 3'-Hydroxyl
eines Akzeptor-Moleküls
katalysiert. Eine derartige RNA-Ligase ist im Handel erhältlich (zum
Beispiel Life Technologies – GIBCO
BRL Ref. 18003). Die cDNAs können
daher schließlich
mit herkömmlichen
Techniken (zum Beispiel PCR) unter Verwendung geeigneter Primer
amplifiziert werden, wie es in 3 veranschaulicht
ist. Diese Technik ist besonders für die Klonierung von kleiner
RNA (unter 1000 b) geeignet.
-
Ein
anderer Versuchsansatz für
die Klonierung und/oder Identifizierung der spezifischen RNA-Regionen
besteht zum Beispiel aus Durchführung
einer inversen Transkription mit dem Produkt des Verdaus mit einem
Enzym, das für
an Doppelsträngen
beteiligte RNA spezifisch ist, wie die RNase H unter Verwendung
von Zufallsprimern, die die Transkription im Innern der RNAs zufällig initiieren.
Die erhaltenen cDNAs werden folglich gemäß den herkömmlichen molekularbiologischen
Techniken amplifiziert, zum Beispiel mittels PCR, unter Verwendung
von Primern durch an den cDNA-Enden hinzugefügte Oligonucleotiden und aufgrund
der Wirkung der Phage T4 RNA-Ligase (im Handel erhältlich;
zum Beispiel bei Life Technologies – GIBCO BRL Ref. 18003). Diese
zweite Technik ist in 4 und in den Beispielen veranschaulicht.
Diese Technik ist ganz besonders für RNAs mit bedeutender Größe geeignet
und erlaubt die Gewinnung eines Teils der Sequenzinformation, der hinreichend
ist, um später
die vollständige
Ausgangssequenz wiederherzustellen.
-
Ein
anderer Versuchsansatz für
die Klonierung und/oder Identifizierung der spezifischen RNA-Regionen
beruht ebenfalls auf der Durchführung
einer inversen Transkription unter Verwendung von Zufallsprimern (4).
Jedoch sind gemäß dieser
Variante die verwendeten Primer wenigstens zum Teil halbzufällige Primer, nämlich Oligonucleotide,
umfassend:
- – eine (degenerierte) zufällige Region,
- – einen
minimalen Priming-Bereich, der ein definiertes zwingendes Maß aufweist,
und
- – einen
stabilisierenden Bereich.
-
Vorzugsweise
handelt es sich um Oligonucleotide, umfassend in 5'→3' Richtung:
- – einen
stabilisierenden Bereich, umfassend 8 bis 24 festgelegte Nucleotide,
vorzugsweise 10 bis 18 Nucleotide. Dieser stabilisierende Bereich
kann selbst einer Sequenz eines Oligonucleotids entsprechen, das für Reamplifizierung
der Fragmente verwendet ist, die von den ersten Amplifizierungen
stammen, die mit Hilfe der halbzufälligen Primer der Erfindung
durchgeführt
worden sind. Außerdem
kann der stabilisierende Bereich die Sequenz von einer oder mehreren
Stellen vorzugsweise nichtpalindromer Art umfassen, die Restriktionsenzymen
entsprechen. Dies erlaubt zum Beispiel die Klonierung der daher
amplifizierten Fragmente zu erleichtern. Ein besonderes Beispiel
für einen
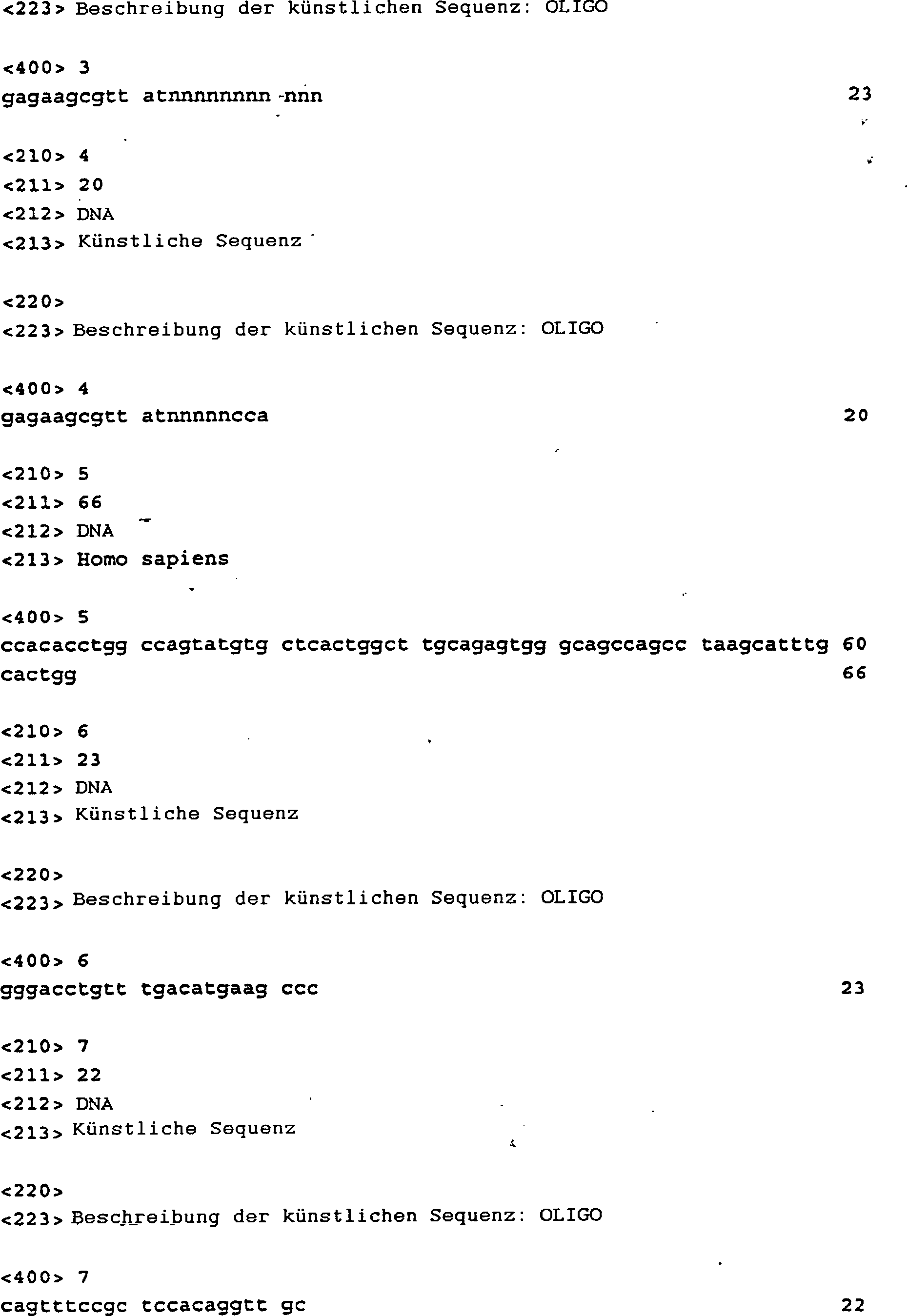
stabilisierenden Bereich ist mit der Sequenz GAG AAG CGT TAT (Reste
1 bis 12 der SEQ ID NO:1) dargestellt;
- – eine
zufällige
Region mit 3 bis 8 Nucleotiden, ganz besonders mit 5 bis 7 Nucleotiden,
und
- – einen
definierten minimalen Priming-Bereich, so dass das Oligonucleotid
im Durchschnitt wenigstens an alle ungefähr 60 bp, vorzugsweise alle
ungefähr
250 bp hybridisiert. Ganz bevorzugt umfasst der Priming-Bereich
2 bis 4 definierte Nucleotide, vorzugsweise 3 oder 4, wie zum Beispiel
AGGX, wo X für
eine der vier Basen A, C, G oder T steht. Das Vorhandensein eines
derartigen Priming-Bereichs verleiht dem Oligonucleotid die Fähigkeit,
im Schnitt alle ungefähr
256 Basenpaare zu hybridisieren.
-
Ganz
besonders bevorzugt handelt es sich um Oligonucleotide mit der Formel:
GAGAAGCGTTATNNNNNNNAGGX
(SEQ ID NO: 1) wo die festgelegten Basen so angeordnet sind, dass
das Hintergrundrauschen aufgrund von Selbstpaarungen in den PCR-Versuchen
minimiert ist, wo N anzeigt, dass die vier Basen in zufälliger Weise
an der angegebenen Position stehen können, und wo X für eine der
Basen A, C, G oder T steht. Derartige Oligonucleotide sind ebenfalls
Gegenstand der vorliegenden Erfindung.
-
In
dieser Hinsicht können,
um die Möglichkeiten
des Primings auf den zu klonierenden RNAs zu erhöhen, gleichzeitige Reaktionen
mit Oligonucleotiden durchgeführt
werden, wie
GAGAAGCGTTATNNNNNNNAGGT (Oligonucleotide A)
GAGAAGCGTTATNNNNNNNAGGA
(Oligonucleotide B)
GAGAAGCGTTATNNNNNNNAGGC (Oligonucleotide
C)
GAGAAGCGTTATNNNNNNNAGGG (Oligonucleotide G)
wobei jede
Population von Oligonucleotiden (A, B, C, D) einzeln oder zusammen
mit einer anderen verwendet sein kann.
-
Nach
dem Schritt der inversen Transkription werden die cDNAs mittels
PCR unter Verwendung der Oligonucleotide A oder B oder C oder D
amplifiziert.
-
Wie
oben angeben, kann entsprechend der erwünschten Komplexität und Spezifität der Population von
Oligonucleotiden die Anzahl an degenerierten Positionen zwischen
3 und 8, vorzugsweise zwischen 5 und 7, variieren. Unter 3 sind
die Hybridisierungen eingeschränkt
und über
8 ist die Population von Oligonucleotiden zu komplex, um eine gute
Amplifikation spezifischer Banden zu gewährleisten.
-
Überdies
kann die Länge
des festgelegten 3'-Endes
(der zwingende Priming-Bereich)
dieser Oligonucleotide ebenfalls modifiziert sein: wenn die oben
beschriebenen Primer mit 4 festen Basen im Durchschnitt die Amplifizierung
von 256 Basenpaaren erlauben, erlauben die Primer mit 3 festen Basen
die Amplifizierung von kürzeren
Fragmenten (durchschnittlich 64 Basenpaare). In einer bevorzugten
Form der Erfindung verwendet man Oligonucleotide, in denen der Priming-Bereich
4 feste Basen umfasst. In einer anderen bevorzugten Form der Erfindung
verwendet man Oligonucleotide mit einem Priming-Bereich aus drei
festgelegten Basen. Denn die Exons mit einer Größe von durchschnittlich 137
Basen sind vorteilhaft mit derartigen Oligonucleotiden amplifiziert.
In dieser Hinsicht siehe ebenfalls die Oligonucleotide zum Beispiel
mit der Sequenz SEQ ID NO: 2, 3 und 4.
-
Schließlich werden
beim Schritt der Identifizierung und/oder Klonierung der RNA die
verschiedenen PCR- und Klonierungsverfahren angewandt, um die ganze
und vollständige
Information zu erhalten.
-
Identifizierung und/oder
Klonierung aus Heterotriplices.
-
Im
Fall von Heterotriplices (andere Variante des Verfahrens) zeigen
sich Regionen der qualitativen Unterschiede (Insertionen, Deletionen,
differentielle Spleißungen)
im Wesentlichen in Form von Regionen doppelsträngiger DNA, wie in 5 dargestellt.
Diese Regionen können
folglich durch Behandlung in Gegenwart geeigneter Enzyme, wie beispielsweise
ein Enzym, das den Verdau der RNAs erlaubt, dann ein Enzym, das den
Verdau der einzelsträngigen
DNAs erlaubt, identifiziert und kloniert werden. Die so erhaltenen
Nucleinsäuren
werden folglich direkt in Form von doppelsträngiger DNA erhalten und können in
jeden geeigneten Vektor wie zum Beispiel pMos-Blue Vektor (Amersham,
RPN 5110) kloniert werden. Diese Methodologie ist von den bereits
beschriebenen Versuchsansätzen
zu unterscheiden, die RNAs oder Oligonucleotide mit vorbestimmten Sequenzen
verwenden und modifiziert sind, um eine Nuclease-Aktivität auszuüben (Landgraf
et al. (1994) Biochemistry, 33, 10607-10615).
-
Identifizierung und/oder
Klonierung aus DNA/DNA-Homoduplices (6).
-
Die
aufgrund ihrer atypischen Strukturen isolierten Fragmenten werden
anschließend
an jedem ihrer Enden Adapter oder Linker angefügt, die Restriktionsschnittstellen
an jedem ihrer Enden besitzen. Dieser Schritt kann gemäß dem Fachmann
bekannten Techniken durchgeführt
sein, zum Beispiel durch Ligation mit der DNA-Ligase des T4 Phagen.
Die so eingeführten
Restriktionsstellen sind mit den Stellen der cDNA-Fragmente kompatibel
gewählt.
Die eingeführten
Linker sind doppelsträngige
cDNA-Sequenzen, bekannte Sequenzen, die die Ableitung von Primern
erlauben, um enzymatische Amplifikationen (PCR) durchzuführen. Da der
nachfolgende Schritt aus der Amplifikation der beiden Stränge besteht,
die beiden Stränge
zu amplifizieren, die zwischen denen die zu identifizierenden qualitativen
Unterschiede vorliegen, ist es erforderlich Linker zu verwenden,
deren 5'-Enden phosphoryliert
sind. Daher ist nach thermischer Denaturierung der doppelsträngigen cDNA
mit angefügten
Linkern jedes der Enden dieser cDNA kovalent mit einer spezifischen
Priming-Sequenz verbunden. Nach PCR mit Hilfe geeigneter spezifischer
Primer werden zwei Kategorien doppelsträngiger cDNA erhalten: Fragmente,
die Sequenzen enthalten, die für
qualitative Unterschiede, die die beiden physiopathologischen Situationen
unterscheiden, spezifisch sind, und Fragmente, die den negativen
Abdruck dieser Spleißereignisse
umfassen. Die Klonierung dieser Fragmente erlaubt die Bildung einer
Bank von alternativen Spleißungen,
in der für
jedes Spleißereignis
positive und negative Abdrücke
vorhanden sind. In dieser Bank sind folglich nicht nur die alternativen
Exons und Introns sondern auch die spezifischen Verknüpfungen zugänglich,
die durch Ausschneiden dieser gespleißten Sequenzen gebildet sind.
In einer selben Bank können die
verschiedenen genetischen Informationen von zwei physiopathologischen
Situationen ohne Unterschied stammen. Überdies können, um den unterschiedlichen
Charakter der identifizierten Spleißungen zu verifizieren und
um zu bestimmen, von welcher Situation diese spezifisch stammen,
die Klone der Bank mit Sonden hybridisiert werden, die von jeder
der mRNA-Gesamtpopulationen stammen.
-
Zwei
Hauptverwendungen können
für die
cDNA-Fragmente in Betracht gezogen werden, die von identifizierten
qualitativen Unterschieden stammen:
- – Ihre Klonierung
in geeigneten Vektoren, um Banken herzustellen, die für qualitative
Unterschiede repräsentativ
sind, die zwischen zwei untersuchten physiopathologischen Situationen
vorhanden sind,
- – Ihre
Verwendung als Sonden, um eine DNA-Bank zu durchmustern, was die
Identifizierung der Spleißereignisse
differentieller Art erlaubt.
-
Die
in der Erfindung verwendeten Vektoren können insbesondere Plasmide,
Cosmide, Phagen, YAC, HAC etc. sein. Diese Nucleinsäuren können daher
als solche konserviert sein oder in Mikroorganismen eingeführt werden,
die für
den verwendeten Klonierungsvektor geeignet sind, um als Kulturen
vervielfacht und/oder aufbewahrt zu werden.
-
Die
wie oben beschriebenen Verfahren werden im Allgemeinen für jede Probe
in einem Zeitraum von weniger als zwei Monate, insbesondere weniger
als 6 Wochen, durchgeführt. Überdies
können
diese verschiedenen Verfahren automatisiert werden, um den Gesamtdauer
zu verkürzen
und die Behandlung vieler Proben zu erleichtern.
-
In
dieser Hinsicht betrifft ein anderer Gegenstand der Erfindung die
Nucleinsäuren,
die mit diesen Verfahren der Erfindung identifiziert und/oder kloniert
sind. Wie oben angegeben, können
diese Nucleinsäuren RNA
oder cDNA sein.
-
Allgemeiner,
die Erfindung betrifft eine Zusammensetzung von Nucleinsäuren, umfassend
hauptsächlich
Nucleinsäuren,
die zwei physiologische Situationen unterscheidende alternative
Spleißungen
entsprechen. Ganz besonders entsprechen diese Nucleinsäuren den
in einer biologischen Testprobe identifizierten alternativen Spleißungen und
die nicht in derselben biologischen Probe in einer Referenzsituation
vorhanden sind. Ebenfalls Gegenstand der Erfindung ist die Verwendung
von klonierten Nucleinsäuren
als therapeutisches oder diagnostisches Produkt oder als Werkzeug
für das
Screening von aktiven Molekülen,
wie unten angegeben.
-
Die
oben dargelegten verschiedenen Verfahren führen folglich alle zu einer
Klonierung von cDNA-Sequenzen, die die differentiell gespleißte genetische
Information zwischen zwei physiopathologischen Situationen repräsentieren.
Die Gesamtheit der Klone, die aus einem dieser Verfahren hervorgegangen
sind, erlaubt folglich die Bildung einer Bank, die für qualitative
Unterschiede repräsentativ
ist, die zwischen zwei untersuchten Situationen vorhanden sind.
-
Generierung qualitativer
Banken
-
In
dieser Hinsicht betrifft die Erfindung außerdem ein Verfahren zur Herstellung
einer Bank von Nucleinsäuren,
die für
einen bestimmten physiologischen Zustand einer biologischen Probe
repräsentativ
sind. Dieses Verfahren umfasst vorteilhaft die Klonierung von Nucleinsäuren, die
für qualitative
Marker der genetischen Expression (zum Beispiel alternative Spleißungen)
des besagten physiologischen Zustands repräsentativ sind und nicht in
einem Referenzzustand gezeigt sind, in Banken, die für qualitative
Unterschiede, die zwischen den beiden untersuchten Zuständen vorhanden
sind, spezifisch sind.
-
Diese
Banken sind aus cDNAs gebildet, die in Plasmid- oder Phagenvektoren
inseriert sind. Diese Banken können
auf Nitrocellulose-Filtern oder jedem anderen dem Fachmann bekannten
Träger
wie Chips oder Biochips präsentiert
sein.
-
Ein
Merkmal und gleichzeitig die Neuartigkeit des qualitativen differentiellen
Screenings ist, dass diese Technik nicht zu einer sondern vorteilhaft
zu zwei unterschiedlichen Banken führt, die die Gesamtheit der
qualitativen Unterschiede repräsentieren,
die zwischen zwei bestimmten Situationen bestehen: ein Banken-Paar (siehe 1D).
-
Daher
betrifft die Erfindung bevorzugt jede Zusammensetzung oder Bank
von Nucleinsäuren,
die durch Hybridisierung von einer Population von RNA, die von einer
biologischen Probe stammt, und einer Population von cDNA, die von
einer zweiten biologischen Probe stammt, erhalten werden kann. Bevorzugter
umfassen die Banken oder die Zusammensetzungen der Erfindung Nucleinsäuren, die
für qualitative
Unterschiede der Expression zwischen zwei biologischen Proben repräsentativ
sind, und mit einem Verfahren hergestellt sind, umfassend (i) einen
Schritt wenigstens der Hybridisierung von einer Population von RNA,
die von einer ersten biologischen Probe stammt, und einer Population
von cDNA, die von einer zweiten biologischen Probe stammt, (ii)
die Selektion der für
qualitative Unterschiede der Expression repräsentativen Nucleinsäuren und eventuell
(iii) die Klonierung besagter Nucleinsäuren.
-
Außerdem ist
es nach Bildung derartiger Banken möglich, einen Schritt der Selektion
der Klone durchzuführen,
um die Spezifität
der erhaltenen Banken zu verbessern. Daher ist es möglich, dass
bestimmte beobachtete Fehlpaarungen aufgrund der qualitativen Unterschiede
(z.B. differentielle alternative Spleißungen) nicht einzigartig sind,
sondern zum Beispiel aus einem Fehler oder Fehlern der inversen
Transkription resultieren könnten.
Obwohl diese Ereignisse im Allgemein nicht signifikant sind, werden
sie vorzugsweise vor der Klonierung der Nucleinsäuren vermindert oder eliminiert.
Hierfür
können
die Klone der Bank mit den cDNA-Populationen
der beiden untersuchten physiopathologischen Situationen hybridisiert
werden (siehe Schritt (c) oben). Die Klone, die nicht mit den beiden
Populationen unterschiedlich hybridisieren, können als nichtspezifisch betrachtet
und eventuell eliminiert werden oder mit verringerter Priorität behandelt
werden (denn das Auftreten einer neuen Isoform in der Testprobe
besagt nicht immer, dass die Isoform, die anfangs in der Referenzprobe
vorlag, aus dieser Testprobe verschwunden ist.) Die Klone, die nur
mit einer der beiden Populationen oder bevorzugt mit einer der Populationen
hybridisieren, werden als spezifisch betrachtet und können mit
erster Priorität
ausgewählt
werden, um angereicherte oder gesäuberte Banken zu bilden.
-
Eine
Säuberung
kann ebenfalls mittels Hybridisierung und Validierung von Klonen
mit Sonden durchgeführt
werden, die von einer statistisch relevanten Anzahl an pathologischen
Proben abstammen.
-
Die
vorliegende Anmeldung beschreibt ebenfalls jede Nucleinsäure-Bank,
die Nucleinsäuren
umfasst, die für
eine physiologische Situation charakteristische alternative Spleißungen spezifisch
sind. Im Allgemeinen sind diese Banken vorteilhaft aus doppelsträngiger cDNA
gebildet, die RNA-Regionen entsprechen, die für eine alternative Spleißung spezifisch
ist. Diese Banken können
aus Nucleinsäuren
gebildet sein, im Allgemeinen in einem Klonierungsvektor oder in
Zellkulturen, die besagte Nucleinsäuren enthalten.
-
Die
Auswahl der Ausgangs-RNA bestimmt zum Teil die Charakteristika der
erhaltenen Banken:
- – die RNAs der beiden Situationen
A und B sind mRNAs oder reife Gesamt- RNAs, die gemäß dem Fachmann bekannten Techniken
isoliert sind.
-
Die
Banken sind dann Banken des qualitativen differentiellen Screenings
und als beschränkt
bezeichnet, denn sie sind auf qualitative Unterschiede beschränkt, die
die reifen RNAs der beiden physiopathologischen Situationen charakterisieren.
-
Die
RNAs der einen Situation sind reife mRNAs oder Gesamt-RNAs, während die
RNAs der anderen Situation Prämessenger-RNAs
sind, die durch Spleißung
gereift ist und gemäß dem Fachmann
bekannten Techniken aus Zellkernen isoliert ist. In diesem Fall
werden die erhaltenen Banken des differentiellen Screenings als
komplex bezeichnet, da sie nicht auf Unterschiede zwischen reifen
RNAs beschränkt
sind, sondern sie umfassen das gesamte Repertoire der Spleißungen,
die in einer Situation transkribiert und in der anderen eliminiert
sind, darunter die gesamten Introns.
-
schließlich können die
RNAs von einer einzigen physiopathologischen Situation stammen und
in diesem Fall umfasst das differentielle Screening die reifen mRNAs
und die Prämessenger-RNA
von einer gleichen Probe. In diesem Fall sind die erhaltenen Banken
autologe Banken des qualitativen differentiellen Screenings. Der
Nutzen dieser Banken ist, dass sie ausschließlich das Repertoire der transkribierten
Introns in einer bestimmten Situation versammeln. Ihre Hybridisierung
mit einer Sonde, die von reifer RNAs einer anderen Situation stammt,
bestimmt schnell, ob diese Situation durch Zurückbleiben von Introns charakterisiert
ist, was ihre Identifizierung leicht erlaubt.
-
Im
Allgemeinen sind die Banken durch Ausbreiten einer mit diesen klonierten
Nucleinsäuren
transformierten Zellkultur auf einem festen Medium (besonders gelartigen
Medium) generiert. Die Transformation wird mit jeder dem Fachmann
bekannten Technik (Transfektion, Calciumphosphat, Elektroporation,
Infektion mit Bakteriophagen, etc.) durchgeführt. Die Zellkultur ist im
Allgemeinen eine Bakterienkultur wie zum Beispiel E. coli Bakterien.
Es kann sich ebenfalls um eukaryotische Zellkulturen, insbesondere
um niedere eukaryotische Zellen (zum Beispiel Hefen) handeln. Dieses
Ausbreiten kann in einer Schale oder auf jedem anderen geeigneten
Träger
unter Sterilbedingungen erfolgen. Außerdem können diese auf gelartigem Medium
ausgebrachte Kulturen zum Beispiel in gefrorener Form (zum Beispiel
in Glycerin oder einem anderen geeigneten Agens) aufbewahrt sein.
Diese Banken können
natürlich
für die
Herstellung von „Replika", nämlich von
Kopien entsprechend üblicher
Techniken, die unten genauer beschrieben werden, verwendet sein.
Außerdem
dienen diese Banken im Allgemeinen zur Herstellung amplifizierter
Banken, nämlich
jeden Klon in amplifizierter Form enthaltende Banken. Eine amplifizierte
Bank wird folgendermaßen
hergestellt: aus den ausgebreiteten Kulturen werden alle Zellklone
wiedergewonnen und werden konditioniert, um gefroren oder in der
Kälte in
jedem geeigneten Medium konserviert zu werden. Diese amplifizierte
Bank wird vorteilhaft aus E. coli Bakterienkulturen hergestellt
und bei 4°C
unter Sterilbedingungen konserviert. Diese amplifizierte Bank erlaubt
die Herstellung und unbegrenzte Reproduktion jeder späteren Bank,
die diese Klone umfasst, auf unterschiedlichen Trägern für verschiedene
Anwendungen. Eine derartige Bank erlaubt außerdem die Isolierung und Charakterisierung eines
jeden Klons von Interesse. Jeder der Klone, die die Banken der Erfindung
bilden, ist daher ein chrakteristisches Element einer physiologischen
Situation und bildet folglich ein besonders interessantes Ziel für verschiedene
Untersuchungen wie die Suche nach Markern, die Herstellung von Antikörpern, die
Diagnose, die Behandlung für
den Gentransfer, etc. Diese unterschiedlicher Anwendungen werden
noch genauer weiter unten diskutiert. Die Bank wird im Allgemeinen,
wie oben beschrieben, durch Ausbreiten der Kulturen in einem gelartigen
Medium auf einem geeigneten Träger
(zum Beispiel Petri-Schale) hergestellt. Der Vorteil bei der Verwendung
eines gelartigen Mediums ist der, dass jede Kolonie separiert und
vereinzelt werden kann. Aus dieser Kultur können identische Replika in
großen
Mengen durch einfaches „replizieren" auf jedem geeigneten Träger gemäß dem Fachmann
bekannten Techniken hergestellt werden. Daher kann die Replik mittels
Filter, Membranen (Nylon, Nitrocellulose, etc.) hergestellt werden,
was das Anheften der Kulturen erlaubt. Die Filter können schließlich bei
4°C zum
Beispiel in getrockneter Form unter beliebigen Bedingungen aufbewahrt
werden, die nicht die Nucleinsäuren
verändern.
Die Filter können
ebenfalls behandelt sein, um die Zellen, Proteine etc, auszuschließen und
um nur Bestandteile wie die Nucleinsäuren zu konservieren. Diese
Behandlungen können
besonders Proteasen, Detergentien etc umfassen. Die behandelten
Filter können
ebenfalls in jeder Vorrichtung oder unter jeder Bedingung konserviert
werden, die für
Nucleinsäuren
geeignet sind.
-
Die
Nucleinsäure-Banken
können
ebenfalls direkt aus Nucleinsäuren
durch Anlagerung auf Biochips oder jeder anderen geeigneten Vorrichtung
hergestellt sein.
-
Die
vorliegende Anmeldung beschreibt ebenfalls jede Bank, umfassend
Oligonucleotide, die für
zwei physiologische Situationen unterscheidende alternative Spleißungen charakteristisch
sind, Es handelt sich vorteilhaft um einzelsträngige Oligonucleotide, umfassend
5 bis 100-mere, vorzugsweise wenigstens 50-mere zum Beispiel ungefähr 25-mere.
-
Diese
Oligonucleotide sind für
alternative Spleißungen
spezifisch, die für
eine Situation oder einen Typ einer physiologischen Situation repräsentativ
sind. Daher können
derartige Oligonucleotide zum Beispiel Oligonucleotide sein, die
für Apoptosesituationen
charakteristische alternative Spleißungen repräsentativ sind. Es ist nämlich in
der Literatur beschrieben worden, dass bestimmte alternative Spleißungen im
Rahmen apoptotischer Situationen beobachtet wurden. Es handelt sich
zum Beispiel um Spleißungen
in den Genen Bclx, Bax, Fas und insbesondere Grb2. Aus veröffentlichten
Daten und in der Literatur und/oder Datenbanken zugänglichen
Sequenzen ist es möglich,
für gespleißte und
ungespleißte
Formen spezifische Oligonucleotide herzustellen. Diese Oligonucleotide
können
zum Beispiel gemäß folgender
Strategie hergestellt sein:
- (a) Identifizierung
eines Proteins oder eines Spleißereignisses,
das für
eine Apoptosesituation und die Sequenz der gespleißten Domäne charakteristisch
ist. Diese Identifizierung kann auf den veröffentlichten Daten beruhen
oder durch Kompilation von zugänglichen
Sequenzen in Datenbanken;
- (b) künstliche
Synthese eines oder mehrerer Oligonucleotide, die einer oder mehreren
Regionen dieser Domäne
entsprechen, die folglich durch Hybridisierung das Aufzeigen der
ungespleißten
Form in den RNA einer Testprobe erlauben;
- (c) künstliche
Synthese eines oder mehrerer Oligonucleotide, die der Verknüpfungsregion
zwischen den beiden Domänen,
die durch die gespleißte
Domäne
getrennt sind, entsprechen. Diese Oligonucleotide erlauben folglich
durch Hybridisierung das Aufzeigen der gespleißten Form in den RNA einer
Testprobe;
- (d) Reproduktion der Schritte (a) bis (c) oben mit anderen Proteinen
oder Spleißereignissen,
die für
eine Apoptosesituation charakteristisch sind;
- (e) Übertragung
auf einen ersten geeigneten Träger
des oder der Oligonucleotide, die für apoptotische Formen der oben
identifizierten Messenger spezifisch sind, und Übertragung auf einen anderen
geeigneten Träger
des oder der Oligonucleotide, die für nicht-apoptotische Formen
spezifisch sind.
-
Die
beiden so erhaltenen Träger
können
verwendet werden, um den physiologischen Zustand von Zellen oder
Testproben und besonders ihren apoptotischen Zustand durch Hybridisierung
eines Nucleinsäure-Präparats von
diesen Zellen oder Proben zu testen.
-
Andere ähnliche
Banken können
mit Oligonucleotiden, die für
verschiedene physiopathologische Zustände spezifisch sind (Neurodegeneration,
Toxizität, Proliferation,
etc.) generiert werden und daher eine Vergrößerung des Anwendungsbereichs
erlauben.
-
Banken
von alternativen Introns und Exons können ebenfalls EDV-Datenbanken
sein, die mittels systematischer Analyse von Datenbanken gebildet
werden, die die auf das Genom irgendeines Organismus, Gewebes oder
irgendeine Zellkultur bezogene Information zusammenfassen. In diesem
Fall können
die durch Bildung derartiger virtueller Banken gewonnen Daten verwendet
werden, um Oligonucleotid-Primer
zu generieren, die zum parallelen Testen beider physiopathologischer
Situationen verwendet werden.
-
Die
EDV-Datenbanken können
ebenfalls verwendet werden, um allgemeine Nucleotidsonden abzuleiten,
die für
eine Klasse von Proteinen repräsentativ
sind oder für
eine definierte Sequenz noch spezifisch sind. Diese Sonden können schließlich auf
Banken von Klonen angewandt werden, die von verschiedenen Klonierungstechniken
von alternativen Introns und Exons stammen, um ein Bild der Komplexizität dieser
molekularen Banken zu erhalten und um schnell zu bestimmen, ob diese
oder jene Klasse von Proteinen oder diese oder jene bestimmte Sequenz
zwischen zwei verschiedenen physiopathologischen Zuständen differentiell
gespleißt ist.
-
Eine
andere Bank oder Zusammensetzung von Nucleinsäuren gemäß der Erfindung ist eine Antisense-Bank,
die aus gemäß den Verfahren
der Erfindung (DATAS) identifizierten Sequenzen hergestellt ist.
Für die
Herstellung dieses Bankentyps werden diese Sequenzen so kloniert,
dass sie in RNA-Fragmenten exprimiert werden, die einer Antisense-Orientierung
bezogen auf die Messenger-RNA
entsprechen, mit denen eine DATAS durchgeführt worden ist. Man erhält daher
eine als Antisense bezeichnete Bank. Dieser Versuchsansatz verwendet
vorzugsweise die Klonierungsvariante, die eine Orientierung der
klonierten Fragmente erlaubt. Der Nutzen einer derartigen Antisense-Bank
ist, dass sie die Transfektion von Zelllinien und die Störung jeden Phänotyps,
der entweder morphologischer Art, enzymatischer Art ist oder folglich
durch Verwendung von Reportergenen oder Resistenzgenen gegen ein
Selektionsmittel zu verfolgen erlaubt. Die Analyse der Phänotypvariationen,
die aus der Einführung
eines Antisense-Expressionsvektors herrühren, erfolgen im Allgemeinen nach
Selektion stabiler Klone, nämlich
die eine koordinierte Vermehrung des Expressionsvektors und des
Genoms des Wirts erlauben. Diese Koordination wird durch die Integration
des Expressionsvektors in das zelluläre Genom oder, wenn der Expressionsvektor
episomal ist, durch Selektionsdruck ermöglicht. Dieser Selektionsdruck
entsteht durch Behandlung der transfizierten Zellkultur mit einem
toxischen Agens, das nur unschädlich
gemacht werden kann, wenn das Produkt eines Gens, das vom Expressionsvektor
getragen wird, in der Zelle exprimiert wird. Daraus resultiert eine
Synchronisierung zwischen der Replikation des Wirts und der transgenen
Zelle. Vorteilhaft verwendet man episomale Vektoren, die vom Epstein-Barr-Virus
abstammen, die die Expression in derselben Zelle von 50 bis 100
Kopien des Vektors erlauben (Deiss et al., 1996, EMBO J., 15, 3861-3870;
Kissil et al., 1995, J. Biol. Chem, 270, 27932-27936).
-
Der
Nutzen dieser Antisense-Banken verbunden mit den DATAS-Sequenzen,
die sie enthalten, ist nicht nur zu identifizieren, welches Gen
in seiner Expression inhibiert worden ist, um den selektierten Phänotyp herbeizuführen, sondern
auch um zu identifizieren, durch welche Spleißisoform dieses Gen beeinträchtigt worden
ist. Während
das Antisense-Fragment auf ein bestimmtes Exon zielt, kann das Fragment
davon abgeleitet sein, dass die Proteindomäne und folglich die Funktion,
die diese Domäne
impliziert, im Gegensatz zum beobachteten Phänotyp steht. Daher stellt die
Kopplung von DATAS mit einem Antisense-Versuchsansatz eine Abkürzung verglichen
mit dem funktionellen genomischen Ansatz dar.
-
DNA-Biochips
-
Die
Erfindung betrifft ebenfalls jeden Träger (Membran, Filter, Biochip,
Chip, etc.), der eine Bank oder eine Zusammensetzung von wie oben
definierten Nucleinsäuren
umfasst. Es kann sich ganz besonders um eine Zellbank oder Nucleinsäure-Bank
handeln. Die Erfindung betrifft ebenfalls jedes Kit oder jeden Träger, der mehrere
Banken gemäß der Erfindung
umfasst. Insbesondere kann es von Vorteil sein, eine Bank, die für Merkmale
eines physiologischen Testzustands im Vergleich zu einem physiologischen
Referenzzustand repräsentativ
ist, und als Kontrolle eine Bank, die für Merkmale des physiologischen
Referenzzustands im Vergleich zum physiologischen Testzustand repräsentativ
ist, parallel zu verwenden („Banken-Paar"). Ein vorteilhaftes Kit
gemäß der Erfindung
umfasst folglich zwei differentielle qualitative Banken von zwei
physiologischen Situationen (ein „Banken-Paar").
Gemäß einer
besonderen Anwendungsform umfassen die Kits der Erfindung mehrere
Banken-Paare, wie sie oben definiert sind, die zum Beispiel verschiedenen
physiologischen Zuständen
oder verschiedenen biologischen Proben entsprechen.
-
Die
Kits können
zum Beispiel diese verschiedenen Banken-Paare umfassen, die in Reihe
auf demselben Träger
angelagert sind.
-
Herstellung
von Sonden
-
Eine
andere Verwendung der cDNA-Zusammensetzungen gemäß der Erfindung, die für zwischen zwei
physiopathologischen Zuständen
vorhandene qualitative Unterschiede repräsentativ sind, besteht in der Ableitung
von Sonden. Denn derartige Sonden können zum Screening auf gespleißte Ereignisse
verwendet sein, die zwischen zwei physiopathologischen Zuständen verschieden
sind.
-
Diese
Sonden (siehe 1D) können durch Markierung der Nucleinsäure-Populationen oder
Banken gemäß herkömmlichen
Techniken, die dem Fachmann bekannt sind, hergestellt werden. So
kann es sich um enzymatische, radioaktive, fluoreszierende, immunologische
etc. Markierungen handeln. Vorzusweise handelt es sich um eine radioaktive
oder fluoreszierende Markierung. Diese Art von Markierung kann zum
Beispiel durch Einführen
von markierten Nucleotiden in die Nucleinsäure-Population (entweder nach
Synthese oder im Verlauf ihrer Synthese) erfolgen, was ihre Erkennung
mit herkömmlichen
Verfahren erlaubt.
-
Eine
Anwendung ist folglich das Screening einer klassischen genomischen
Bank. Eine derartige Bank kann je nach Vektor, der von einem Phagen
oder einem Cosmid stammt, DNA-Fragmente von 10 kb bis 40 kb umfassen.
Die Anzahl an Klonen, die mit den mittels DATAS generierten Sonden
hybridisieren und für
Spleißunterschiede
repräsentativ
sind, spiegelt folglich ungefähr
die Anzahl an Genen wider, die durch diese Spleißvariationen beeinträchtigt sind,
je nachdem sie in der einen oder anderen untersuchten Situation
exprimiert sind.
-
Vorzugsweise
werden die Sonden der Erfindung für das Screening einer genomischen
cDNA-Bank (im Allgemeinen human) verwendet, die für die Identifizierung
von Spleißereignissen
geeignet ist. Vorzugsweise ist eine derartige genomische Bank aus
DNA-Fragmenten mit begrenzter Größe zusammengesetzt
(im Allgemeinen in Vektoren kloniert), um statistisch nur ein differentiell
spleißbares
Element abzudecken, nämlich
ein einziges Exon oder ein einziges Intron. Die genomische cDNA-Bank
ist folglich durch Verdau von genomischer DNA mit einem Enzym mit
einer auf 4 Basen beschränkten
Erkennungsstelle hergestellt, was folglich die Möglichkeit sicherstellt, durch
Verdau Fragmente mit einer durchschnittlichen Größe von 1 kb zu erhalten. Derartige Fragmente
erfordern die Gewinnung von 107 Klonen,
um eine DNA-Bank zu bilden, die für ein Genom eines höheren eukaryotischen
Organismus repräsentativ
ist. Eine derartige Bank ist ebenfalls ein Gegenstand der vorliegenden
Anmeldung. Diese Bank wird anschließend mit Sonden hybridisiert,
die aus dem qualitativen differentiellen Screening stammen. Genauer,
von jeder betrachteten Probe und die zwei physiopathologische Situationen
A und B vergleicht, werden zwei Sonden (Sonden-Paar) erhalten. Eine
Sonde, die an charakteristischen Spleißereignissen der Situation
A angereichert ist, und eine Sonde, die an Spleißmarkern von B angereichert
ist. Die Klone der genomischen Bank, die bevorzugt mit der einen
oder der anderen Sonde hybridisieren, tragen in den entsprechenden
physiopathologischen Situationen bevorzugt gespleißte Sequenzen.
-
Die
Verfahren der Erfindung erlauben daher die systematische Identifizierung
von qualitativen Unterschieden der Genexpression. Diese Verfahren
weisen zum Beispiel bei der Identifizierung und/oder Klonierung von
Molekülen
von Interesse, in der Toxikologie, in der Pharmakologie oder auch
in der Pharmakogenomie zahlreiche Anwendungen auf.
-
Anwendungen
-
Die
Erfindung betrifft ebenfalls die Anwendung von oben beschriebenen
Verfahren, Nucleinsäuren oder
Banken zur Identifizierung von Molekülen von therapeutischem oder
diagnostischen Nutzen. Die Erfindung betrifft ganz besonders die
Anwendung von oben beschriebenen Verfahren, Nucleinsäuren oder
Banken zur Identifizierung von Proteinen oder Protein-Domänen, die
von einer Krankheit beeinträchtigt
sind.
-
Einer
der Trümpfe
dieser Techniken ist daher, innerhalb eines Messengers und infolgedessen
des entsprechenden Proteins, die funktionellen Domänen zu identifizieren,
die von einer gegebenen Krankheit beeinträchtigt sind. Dies erlaubt,
einer bestimmten Domäne
eine Bedeutung bei der Entwicklung oder Aufrechterhaltung eines
pathologischen Zustands zuzuschreiben. Der unmittelbare Vorteil,
die Wirkung einer pathologischen Deregulation auf eine genaue Domäne eines
Proteins zu beschränken,
ist diese als ein wichtiges Ziel für ein Screening von kleinen
Molekülen
für therapeutische
Zwecke vorzuschlagen. Diese Informationen schaffen ebenfalls die
Voraussetzung, Polypeptide für
eine mittels Gentechnik zu überbringende
therapeutische Aktivität
zu konstruieren. Diese Polypeptide können besonders einzelkettige
Antikörper
sein, die von neutralisierenden Antikörpern abstammen und gegen die
mit den oben beschriebenen Techniken identifizierten Domänen gerichtet
sind.
-
Spezifischer,
die Verfahren gemäß der Erfindung
erlauben die Gewinnung von Molekülen,
wobei diese können:
- – codierende
Sequenzen sein, die von alternativen Exons abstammen,
- – nicht-codierenden
Sequenzen entsprechen, die von differentiell gespleißten Introns
von einem physiopathologischen Zustandes auf einen anderen getragen
werden.
-
Aus
diesen beiden Punkten können
verschiedene Lehren gezogen werden.
-
Die
alternativen Spleißungen
von Exons, die beide physiopathologische Zustände unterscheiden, geben ein
neues Regulationsniveau der genetischen Expression wieder, das erlaubt,
eine oder mehrere Funktionen eines bestimmten Proteins zu modulieren
(genauer abschalten oder einschalten). Wobei daher die meisten strukturellen
oder funktionellen Domänen
(SH2, SH3, PTB, PDZ und die katalytischen Domänen von verschiedenen Enzymen)
von mehreren benachbarten Exons codiert sind, können sich zwei Konfigurationen
ergeben:
- i) Die Domänen sind in der pathologischen
Situation verkürzt
(Zhu, Q. et al., 1994, J. Exp. Med., vol. 180, Nr. 2, Seiten 461-470);
was darauf hinweist, dass die Signalwege, die diese Domänen umfassen,
für therapeutische
Zwecke wiederhergestellt sein müssen.
- ii) Die Domänen
sind im Verlauf einer Krankheit aufrechterhalten, während sie
in einer gesunden Situation fehlen; diese Domänen können als Ziele für ein Screening
kleiner chemischer Moleküle
betrachtet werden, die als Antagonisten für die auf dem Weg über diese
Domänen
transduzierten Signale bestimmt sind.
-
Die
differentiell gespleißten
Domänen
können
den nicht-codierenden Regionen, die in 5' oder 3' von der codierenden Sequenz liegen,
oder Introns entsprechen, die zwischen zwei codierenden Exons liegen.
In den nicht-codierenden Regionen können diese differentiellen
Spleißungen
eine Modifikation der Stabilität
oder der Translatierbarkeit des Messengers wiedergeben (Bloom, T.
J. und Beavo, J. A., 1995, Proc. Natl. Acad. Sci. USA, vol. 93,
Nr. 24, Seiten 14188-14192; Ambartsumian, N. et al., 1995, Gene,
vol. 159, Nr. 1, Seiten 125-130). Diese Phänomene müssen nun auf Basis dieser Informationen
gesucht werden und können
aufzeigen, dass die Anhäufung
oder das Verschwinden des entsprechenden Proteins dieses folglich
zu einem Ziel von Interesse macht. Wenn ein Intron in einer codierenden
Sequenz zurückbleibt,
ergibt sich hieraus am häufigsten
eine Verkürzung
des natürlichen
Proteins durch eine Einführung
eines Stop-Codons in den Leserahmen (Varesco, L., et al., 1994,
Num. Genet., vol. 93, Nr. 3, Seiten 281-286; Canton, H., et al.,
1996, Mol. Pharmacol., vol. 50, Nr. 4, Seiten 799-807; Ion, A.,
et al., 1996, Am. J: Hum, vol. 58, Nr. 6, Seiten 1185-1191). Vor Erreichen
dieses Stop-Codons erfolgt im Allgemeinen ein Ablesen einiger zusätzlicher
Codons, was dazu führt,
dass an den bereits gelesenen Teil eine spezifische Sequenz, ein
Protein-Marker der alternativen Spleißung, angefügt wird. Diese zusätzlichen
Aminosäuren
können
zur Erzeugung von Antikörpern
verwendet sein, die für
die alternative Form, die für
die pathologische Situation charakteristisch ist, spezifisch sind.
Diese Antikörper
können
dann als Diagnosemittel verwendet sein. Das verkürzte Protein zeigt seine modifizierten,
sogar gestörten
Eigenschaften. Daher können
Enzyme um ihre katalytische Domäne
oder ihre regulatorische Domäne
verkürzt
sein und werden inaktiviert oder konstitutiv aktiviert. Adaptoren
können
ihre Fähigkeit
verlieren, verschiedene Partner einer Signalkaskade zu verbinden
(Watanabe, K. et al., 1995, J. Biol. Chem., vol 270, Nr. 23, Seiten
13733-13739). Die Spleißprodukte
des Empfängers
können
zu Rezeptoren führen,
die ihre Fähigkeit
zur Ligandbindung verloren haben (Nakajima, T. et al., 1996, Life
Sci., vol 58, Nr. 9, Seiten 761-768) und können ebenfalls Rezeptorformen
erzeugen, die durch Ausfällen
ihrer extrazellulären
Domäne
löslich
werden (Cheng J., 1994, Science, vol. 263, Nr. 5154, Seiten 1759-1762).
In diesem Fall können
die diagnostischen Tests in Betracht gezogen werden, die auf der
Zirkulation der löslichen
Form eines Rezeptors für
einen bestimmten Liganden in verschiedenen physiologischen Flüssigkeiten
beruhen.
-
Die
Erfindung betrifft ganz besonders die Anwendung von oben beschriebenen
Verfahren, Nucleinsäuren
oder Banken zur Identifizierung antigener Domänen, die für an einer Krankheit beteiligte
Proteine spezifisch sind. Die Erfindung betrifft ebenfalls die Verwendung
von oben beschriebenen Nucleinsäuren,
Proteinen oder Peptiden für
die Diagnose von Krankheiten.
-
Die
Erfindung betrifft ebenfalls ein Verfahren zur Identifizierung und/oder
Produktion von Proteinen oder Protein-Domänen, die an einer Krankheit
beteiligt sind, umfassend:
- (a) die Hybridisierung
der Messenger-RNAs einer pathologischen Probe mit den cDNAs einer
gesunden Probe, oder umgekehrt oder beides parallel,
- (b) die Identifizierung, in den gebildeten Hybriden, der Regionen,
die qualitativen Unterschieden (ungepaarte (RNA) oder gepaarte (doppelsträngige DNA))
entsprechen und für
den pathologischen Zustand verglichen mit dem gesunden Zustand spezifisch
sind,
- (c) die Identifizierung und/oder Produktion des Proteins oder
der Protein-Domäne, die
einer oder mehreren in (b) identifizierten Regionen entsprechen.
Die identifizierten Regionen entsprechen im Allgemeinen differentiellen
Spleißungen,
aber es kann sich ebenfalls um andere genetische Störungen handeln,
wie zum Beispiel Insertion(en) oder Deletion(en).
-
Das
oder die Proteine oder Protein-Domänen können isoliert, sequenziert
und in therapeutischen oder diagnostischen Anwendungen insbesondere
für die
Herstellung von Antikörpern
verwendet werden.
-
In
einem spezifischeren Beispiel erlaubt das qualitative differentielle
Screening vorteilhaft das Aufzeigen von Suppressorgenen für Tumoren.
Denn zahlreiche Beispiele weisen darauf hin, dass eine der Inaktivierungsmöglichkeiten
von Suppressorgenen im Verlauf der Tumorprogression eine Inaktivierung
durch die Modulation von alternativen Spleißformen ist.
-
Daher
ist in dem kleinzelligen Lungenkarzinom das Gen des Proteins p130,
das zur RB-Familie (Retinoblastom-Protein) gehört, an der Konsensusstelle
der Spleißung
mutiert. Die Folge dieser Mutation ist die Eliminierung des Exons
2 und eine ausbleibende Synthese des Proteins aufgrund des Vorhandenseins
eines frühen
Stop-Codons. Diese Beobachtung ist die erste gewesen, die die Bedeutung
der Mitglieder der RB-Familie in der Tumorgenese unterstreicht.
Ebenso ist bei bestimmten nichtkleinzelligen Lungenkarzinomen das
Gen des Proteins p16INK4A, das ein Inhibitor der cyclin-abhängigen Kinasen
cdk4 und cdk6 ist, in einer Donorstelle der Spleißung mutiert.
Das Resultat dieser Mutation ist die Produktion eines verkürzten Proteins
mit einer kurzen Halbwertzeit, was die Akkumulation von inaktiven
phosphorylierten Formen von RB zur Folge hat. Überdies ist WT1, das Suppressorgen
des Wilms Tumors, in mehrere Messenger-RNAs transkribiert, die durch
alternative Spleißungen
generiert werden. Beim Brustkrebs sind die relativen Verhältnisse
der verschiedenen Varianten bezogen auf gesundes Gewebe verändert, was
Diagnosemittel und Spuren liefert, um die Bedeutung der verschiedenen
funktionellen Domänen
von WT1 bei der Tumorprogression zu verstehen. Dasselbe Phänomen der
Modifikation der Verhältnisse
zwischen verschiedenen Formen von Messenger-RNA und Protein-Isoformen
während
der Zelltransformation findet sich beim Neurofibrin NF1 wieder.
Außerdem
wird diese Vorstellung von der Modulation der Spleißphänomene,
die die Tumorprogression kennzeichnet, ebenfalls von dem Beispiel
von HDM2 gestützt,
von dem 5 alternative Spleißungen
in den Ovarial- und Pankreaskarzinomen detektiert sind und deren
Expressionen entsprechend die Tumorentwicklung verstärken. Andererseits
umfasst bei den Krebsarten des Schädels und Hals einer dieser
Inaktivierungsmechanismen von p53 eine Mutation in einer Konsensusstelle
der Spleißung.
-
Diese
einige angeführten
Beispiel veranschaulichen den ganzen Nutzen der Techniken der Erfindung, die
auf der systematischen Suche von Spleißvariationen basieren, die
einen bestimmten Tumor von benachbartem gesunden Gewebe unterscheiden.
Die Ergebnisse, die sich daraus ergeben; erlauben tatsächlich nicht nur
die Charakterisierung von schon bekannten Tumorsuppressorgenen sondern
ebenfalls, unter Berücksichtigung
des neuen und systematischen Aspekts der Techniken des qualitativen
differentiellen Screenings, die Identifizierung von neuen Spleißvariationen,
die für
Tumoren spezifisch sind, die wahrscheinlich neue Tumorsuppressorgene
beeinträchtigen.
-
Ebenfalls
Gegenstand der Erfindung ist ein Verfahren zur Identifizierung und/oder
Klonierung von Tumorsuppressorgenen oder genetischen Störungen (z.B.
Spleißungen)
innerhalb der Tumorsuppressorgene, wie es oben definiert ist. Dieses
Verfahren kann vorteilhaft die folgenden Schritte umfassen:
- (a) die Hybridisierung der Messenger-RNAs einer
Tumorprobe mit den cDNAs einer gesunden Probe, oder umgekehrt oder
beide parallel,
- (b) die Identifizierung, in den gebildeten Hybriden, der Regionen,
die für
die Tumorprobe im Vergleich zum gesunden Zustand spezifisch sind,
- (c) die Identifizierung und/oder Klonierung des Proteins oder
der Protein-Domäne, die
einer oder mehreren in (b) identifizierten Regionen entsprechen.
-
Die
Tumor supprimierenden Eigenschaften von identifizierten Proteinen
oder Domänen
können
dann in verschiedenen bekannten Modellen getestet werden. Diese
Proteine oder ihre native Form (besitzen beobachtete Spleißung in
gesundem Gewebe) können
dann in therapeutischen oder diagnostischen Anwendungen insbesondere
in einer antitumoralen Gentherapie verwendet werden.
-
Gegenstand
der vorliegenden Anmeldung sind folglich nicht nur die verschiedenen
Aspekte der Anwendung der Technologie sondern auch die Nutzung der
Informationen, die sich daraus ableiten lassen, für Zwecke
der Suche nach kleinen chemischen Molekülen, der Entwicklung von Screenings
auf kleine chemische Moleküle,
der Entwicklung für
Werkzeuge der Gentherapie oder Diagnose.
-
Aus
diesem Grund betrifft die Erfindung ebenfalls die Anwendung von
oben beschriebenen Verfahren, Nucleinsäuren oder Banken in der Gentoxikologie,
nämlich
um das toxische Potenzial von Testverbindungen vorwegzunehmen (vorherzusagen).
-
Die
genetischen Programme, in die während
der Behandlung von Zellen oder Geweben mit toxischen Mitteln eingegriffen
wird, sind größtenteils
mit Phänomenen
der Apoptose oder dem programmierten Zelltod korreliert. Die Bedeutung
der Phänomene
der alternativen Spleißung
bei Regulation dieser Apoptosewege ist in der Literatur gut erläutert. Doch
bis heute erlaubte keine beschriebene genomische Technologie die
systematische Suche und die erschöpfende die Isolierung der Sequenzvariationen
aufgrund alternativer Spleißungen
und die Unterscheidung zwischen zwei bestimmten physiopathologischen
Situationen. Die in der vorliegenden Erfindung entwickelten Techniken
des qualitativen differentiellen Screenings erlauben die Zusammenfassung
der gesamten Spleißunterschiede,
die zwischen zwei Situationen in cDNA-Banken bestehen. Der Vergleich
der RNA-Sequenzen
(zum Beispiel Messenger-RNA) eines mit einer toxischen Referenzverbindung
behandelten oder nichtbehandelten Gewebes (oder einer Zellkultur)
erlaubt die Etablierung von cDNA-Banken, die die qualitativen Unterschiede
der Genexpression zusammenfassen, die die untersuchte toxische Wirkung kennzeichnen.
Diese cDNA-Banken können
dann mit Sonden hybridisiert werden, die von RNA-Extrakt derselben
Gewebe oder Zellen abgeleitet sind, die mit einem chemischen Produkt
behandelt sind, dessen toxisches Potenzial man bestimmen möchte. Die
mehr oder weniger große
Fähigkeit
dieser Sonden, sich mit den genetischen Informationen zu hybridisieren,
die für
eine toxische Referenzsituation spezifisch sind, erlaubt dieser
ein toxisches Potenzial zuzuweisen. Außer der Anwendung von DATAS
zur Generierung und Verwendung von Banken von qualitativen Unterschieden,
die von toxischen Agentien induziert sind, besteht überdies
ein Teil der Erfindung ebenfalls darin, zu zeigen, dass Deregulationen
in der Spleißung
von bestimmten Messenger-RNAs von bestimmten toxischen Agentien
induziert werden können,
und zwar in niederen Dosen als IC50, die in dem Fachmann bekannten
Cytotoxizitätstests
und Apoptosetests gemessen werden. Derartige Deregulationen (oder
Fehlregulationen) können
als Marker für
das Verfolgen der Toxizität
und/oder Wirksamkeit von (chemischen oder genetischen) Molekülen verwendet
sein.
-
Die
Erfindung betrifft folglich ebenfalls jedes Verfahren zur Detektion
oder zum Verfolgen des toxischen und/oder therapeutischen Potenzials
einer Verbindung, beruhend auf der Detektion von Spleißformen und/oder
Spleißmustern,
die von dieser Verbindung in einer biologischen Probe induziert
sind. Die Erfindung betrifft außerdem
die Verwendung jeder Modifikation von Spleißformen und/oder Spleißmustern
als Marker für das
Verfolgen der Toxizität
und/oder Wirksamkeit von Molekülen.
-
Die
Bewertung oder das Verfolgen des toxischen Potenzials kann ganz
besonders gemäß zwei Versuchsansätzen erfolgen:
Gemäß dem ersten
Versuchsansatz kann das qualitative differentielle Screening durchgeführt sein
zwischen einem Gewebe oder einer Zellkultur als Referenz, einerseits
unbehandelt und andererseits mit dem Produkt behandelt, dessen Toxizität man zu
bewerten wünscht.
Die Analyse der Klone, die die qualitativen Unterschiede repräsentieren,
die spezifisch von dem Produkt induziert sind, erlauben dann möglicherweise
die Detektion von Ereignissen in diesen Klonen, die für cDNA charakteristisch
sind, die an den mit der Toxizität
verbundenen Phänomenen
wie die Apoptose beteiligt sind.
-
Das
Auftreten dieser Marker hängt
von der Dosis und der Dauer der Behandlung mit dem Produkt ab und
erlaubt eine Näherung
seines toxikologischen Profils.
-
Gegenstand
der vorliegenden Anmeldung ist folglich ebenfalls ein Verfahren
zur Identifizierung, mittels qualitativen differentiellen Screenings
gemäß den oben
angeführten
Techniken, von Toxizitätsmarkern,
die in einem biologischen Modellsystem von einer chemischen Verbindung
induziert sind, deren toxisches Potenzial man testen möchte. In
dieser Hinsicht betrifft die vorliegende Erfindung besonders ein
Verfahren zur Identifizierung und/oder Klonierung von Nucleinsäuren, die
für einen
toxischen Zustand einer bestimmten biologischen Probe spezifisch
sind, umfassend die Präparation
qualitativer differentieller Banken zwischen den cDNAs und RNAs
der Probe nach oder ohne Behandlung mit einer Testverbindung und
die Suche nach Toxizitätsmarkern,
die für
Eigenschaften der Probe nach Behandlung spezifisch sind.
-
Gemäß einem
zweiten Versuchsansatz sammeln Nomogramme für unterschiedliche Klassen
toxischer Produkte deren Toxizitätsprofil
gemäß den eingesetzten
Dosen und gemäß den Behandlungszeiträumen für ein Gewebe
oder ein zelluläres
Modell als Referenz: Zu jedem Punkt dieser Nomogramme können cDNA-Banken etabliert
sein, die für
die qualitativen genetischen Unterschiede charakteristisch sind.
Diese Banken sind qualitative differentielle Banken, d.h. sie sind
durch Gewinnung von genetischen Informationen des in den Nomogrammen
gewählten
Punkts und des entsprechenden Punkts im Gewebe- oder Zellkontrollmodell erhalten.
Wie in den Beispielen veranschaulicht, beruht das qualitative differentielle
Screening auf der Hybridisierung von mRNA-Extrakten einer Situation
mit den cDNAs, die von einer anderen Situation stammen. Wie weiter
oben angegeben, kann das qualitative differentielle Screening ebenfalls
aus Gesamt-RNA oder nukleärer
RNA, die Prämessenger
enthält,
durchgeführt
werden.
-
In
dieser Hinsicht betrifft die Erfindung ein Verfahren zur Bestimmung
oder Bewertung der Toxizität
einer Testverbindung in einer bestimmten biologischen Probe, umfassend
die Hybridisierung:
- – von differentiellen Banken
zwischen den cDNAs und den RNAs besagter biologischen Probe in gesundem Zustand
und in verschiedenen Toxizitätsstadien,
die aus Behandlung besagter Probe mit einer toxischen Referenzverbindung
resultieren, mit
- – einem
Nucleinsäure-Präparat der
mit besagter Testverbindung behandelten biologischen Probe und
- – die
Bewertung des toxischen Potenzials der Testverbindung durch Analyse
des Hybridisierungsgrades mit verschiedenen Banken.
-
Gemäß diesem
Verfahren werden für
jede Situation (Dosis der Verbindung und/oder Inkubationszeit) zwei
reziproke Hybridisierungen vorteilhaft durchgeführt, zwischen:
- – den
RNAs der Situation A (Test) und den cDNAs der Situation B (Referenz)
(rA/cB)
- – den
RNAs der Situation B (Referenz) und den cDNAs der Situation A (Test)
(rB/cA).
-
Zu
jeder toxischen Referenzsituation, zu jedem Punkt der Nomogramme
gibt es folglich zwei entsprechende Banken des qualitativen differentiellen
Screenings. Eine dieser Banken fasst die qualitativen Variationen
zusammen, nämlich
besonders alternative Spleißungen,
die für
die normale Referenzsituation spezifisch sind, während die andere Bank die für toxische
Ereignisse spezifischen Spleißungen
sammelt. Diese Banken werden auf festen Trägern wie Filter aus Nylon oder
Nitrocellulose oder vorteilhaft auf Chips repliziert. Diese anfangs
von cDNA-Fragmenten
unterschiedlicher Länge
gebildeten Banken (entsprechend den betrachteten Spleißereignissen)
können
durch Verwendung von Oligonucleotiden, die von anfangs isolierten
Sequenzen abgeleitet sind, optimiert werden.
-
Wenn
eine chemische Verbindung für
eine pharmazeutische Entwicklung vorgeschlagen wird, kann man sie
in denselben Gewebe- oder Zellmodellen einsetzen, wie diejenigen,
die im Repertoire der Toxizitätsnomogramme
enthalten sind. Molekularsonden können dann aus mRNA-Extrakten
der mit der Verbindung von Interesse behandelten biologischen Proben
hergestellt werden. Diese Sonden werden dann auf Filtern hybridisiert,
die die cDNA der Banken rA/cB und rB/cA tragen. Zum Beispiel kann
die rA/cB Bank die Sequenzen enthalten, die in der normalen Situation
vorhanden sind, und die rB/cA Bank die alternativen Spleißelemente enthalten,
die für
die toxische Situation spezifisch sind. Die Unschädlichkeit
oder die Toxizität
der chemischen Verbindung wird nun einfach in Abhängigkeit
des Hybridisierungsmusters einer Sonde bewertet, die von mRNA-Extrakten
des mit der Testverbindung behandelten Gewebe- oder Zellreferenzmodells
abgeleitet ist:
- – eine wirksame Hybridisierung
mit der rA/cB-Bank und kein Signal in der rB/cA-Bank zeigt eine fehlende Toxizität der Verbindung
im untersuchten Modell an,
- – die
Hybridisierung der Sonde mit Klonen der rB/cA-Bank weist auf eine
von der Testverbindung induzierte Toxizität.
-
Anwendungsbeispiele
für die
Bildung derartiger Banken können
von den Hepatocytenkulturmodellen, wie die HepG2-Linie, von Nierenepithelzellen,
wie die HK-2-Linie, oder von Endothelzellen wie die ECV304-Linie
bereitgestellt sein, die mit toxischen Mitteln wie Ethanol, Camptothecin
oder PMA behandelt sind.
-
Ein
Beispiel der Wahl kann ebenfalls mit der Verwendung in Kosmetologie
von Hautkulturmodellen bereitgestellt sein, die mit toxischen Agentien
oder Irritantien behandelt sind oder nicht.
-
Gegenstand
der vorliegenden Anmeldung sind folglich Banken des differentiellen
Screenings (zwischen cDNA und RNA), die aus Organen, Geweben oder
Zellkulturen als Referenz hergestellt sind, die mit chemischen Verbindungen
behandelt sind, die für
große
Klassen toxischer Agentien gemäß den in
der Literatur beschriebenen Nomogrammen repräsentativ sind. Die Erfindung
betrifft auch die Ausbreitung dieser Banken auf Filtern oder Trägern, die
dem Fachmann bekannt sind (Nitrocellulose, Nylon...). Vorteilhaft
können
diese Träger
Chips oder Biochips sein, die daher Biochips der Gentoxizität definieren.
Die Erfindung betrifft außerdem
die Nutzung, die aus der Sequenzierung der verschiedenen Klone gewonnen
werden kann, die diese Banken mit dem Zweck bilden, den Mechanismus
aufzuklären,
der durch die Wirkung der verschiedenen toxischen Agentien in Gang
gesetzt wird. wie auch die Verwendung dieser Banken, um sie mit
Sonden zu hybridisieren, die von Zellen oder Geweben stammen, die
mit einer chemischen Verbindung oder einem pharmazeutischen Produkt
behandelt sind, dessen Toxizität
man bewerten möchte.
Vorteilhaft betrifft die Erfindung wie oben definierte Nucleinsäure-Banken,
die aus Hautzellen präpariert
sind, die unter verschiedenen toxischen Bedingungen behandelt worden
sind. Die Erfindung betrifft außerdem
ein Kit, das diese verschiedenen differentiellen Banken der Haut
umfasst.
-
Die
Erfindung betrifft ebenfalls die Anwendung der wie oben beschriebenen
Verfahren, Nucleinsäuren oder
Banken für
die Bewertung (Vorhersage) oder für die Verbesserung des therapeutischen
Potenzials von Testverbindungen (Genopharmakologie).
-
Bei
dieser Anwendung ist das in der Praxis angewandte Prinzip sehr eng
mit dem zuvor beschriebenen Prinzip verwandt. Differentielle Referenz-Banken
werden zwischen den cDNAs und den RNAs einer Zellkultur oder eines
Organs in einer Kontrollsituation und ihrem Äquivalent, das ein Krankheitsmodell
imitiert, etabliert. Die therapeutische Wirksamkeit eines Produkts
kann nun bewertet werden, indem seine Fähigkeit verfolgt wird, den
qualitativen Variationen der Genexpression entgegenzuwirken, die
für dieses
pathologische Modell spezifisch sind. Dies wird durch die Modifikation
des Hybridisierungsmusters einer Sonde, die von einem pathologischen
Modell stammt, mit einer Referenz-Bank aufgezeigt: ohne Behandlung
hybridisiert die Sonde nur mir der Bank, die die für die Krankheit
spezifischen Signaturen enthält.
Nach Behandlung mit einem wirksamen Produkt hybridisiert die Sonde,
obwohl von einem pathologischen Modell abstammend, bevorzugt mit
der anderen Bank, die die Signaturen des gesunden äquivalenten
Modells trägt.
-
In
dieser Hinsicht betrifft die Erfindung ebenfalls ein Verfahren zur
Bestimmung oder Bewertung der therapeutischen Wirksamkeit einer
Testverbindung auf eine bestimmte biologische Probe, umfassend die
Hybridisierung:
- – von differentiellen Banken
zwischen den cDNAs und den RNAs besagter biologischer Probe in gesundem Zustand
und in (verschiedenen Entwicklungsstadien des) pathologischen Zustand(s),
mit
- – einem
Nucleinsäure-Präparat der
biologischen Probe, die mit besagter Testverbindung behandelt ist,
und
- – die
Bewertung des therapeutischen Potenzials der Testverbindung durch
Analyse des Hybridisierungsgrades mit den verschiedenen Banken.
-
Ein
Beispiel einer derartigen Anwendung kann ein Apoptosemodell liefern,
das bestimmte Aspekte der Neurodegeneration imitiert, denen trophische
Referenzfaktoren entgegenwirken. Die Zellen, die vom PC12-Phäochromocytom
abstammen und im Nervenkörper
in Gegenwart von NGF differenziert sind, treten in die Apoptose
bei Entfernen des Wachstumsfaktors ein. Diese Apoptose wird von
der Expression zahlreicher Marker des programmierten Zelltods begleitet,
wovon die meisten durch alternative Spleißungen reguliert sind und deren
Auftreten von der IGF1-Wirkung inhibiert wird. Zwei Banken, die
von einem qualitativen differentiellen Screening stammen, werden
aus mRNA-Extrakten einerseits von differenzierten PC12-Zellen, die
durch das Entfernen von NGF in die Apoptose eintreten, und andererseits
aus differenzierten PC12-Zellen, die vor der Apoptose durch Zugabe
von IGF1 geschützt
sind, etabliert. Mit diesen Banken können Sonden hybridisiert werden,
die aus mRNA-Extrakten von differenzierten PC12 hergestellt sind,
die in die Apoptose eingetreten sind und deren Überleben durch die Behandlung
mit einem zu testenden neuroprotektiven Produkt verbessert ist.
Die Wirksamkeit der Inversion der qualitativen Charakteristika,
die von einer Testverbindung induziert sind, kann folglich mit der
Fähigkeit
der Sonde abgeschätzt
werden, Klone spezifisch zu hybridisieren, die für eine Bank spezifisch sind,
die für
Zellen repräsentativ
ist, deren Überleben
verbessert wird. Dieser Test kann später zum Testen der Wirkung
von Derivaten von Verbindungen oder von jeder anderen neuen Familie
von neuroprotektiven Verbindungen und zur Verbesserung des pharmakologischen
Profils gemacht werden.
-
In
einer besonderen Anwendungsform erlaubt das Verfahren der Erfindung
die Wirkung einer neuroprotektiven Testverbindung durch Hybridisierung
mit einer
-
differentiellen
Bank gemäß der Erfindung
zwischen einer gesunden Nervenzelle und dieser Zelle, die ein neurodegeneratives
Modell darstellt zu bewerten.
-
In
einer anderen Form wird eine antitumorale Verbindung mit differentiellen
Banken getestet, die aus einer Probe von Tumorzellen und einer gesunden
Probe etabliert sind.
-
Wie
oben angegeben, kann das Verfahren der Erfindung außerdem zur
Verbesserung der Eigenschaften einer Verbindung verwendet sein,
indem verschiedene Derivate auf ihre Fähigkeit getestet werden, ein
Hybridisierungsmuster zu induzieren, das dem Muster der Bank, die
für die
gesunde Probe repräsentativ
ist, ähnlich
ist.
-
Die
Erfindung betrifft ebenfalls die Anwendung von oben beschriebenen
Verfahren, Nucleinsäuren oder
Banken in der Pharmakogenomie d.h. zur Bewertung (Vorhersage) der
Reaktion eines Patienten auf eine Testverbindung oder eine Testbehandlung.
-
Die
Pharmakogenomie hat das Ziel, genetische Profile von Patienten zu
erstellen, um zu bestimmen, welche Behandlung in der Lage ist, eine
bestimmte Krankheit erfolgreich zu überwinden. Die in der vorliegenden
Erfindung beschriebenen Techniken erlauben in dieser Hinsicht die
Etablierung von cDNA-Banken, die für qualitative Unterschiede
repräsentativ
sind, die zwischen einer pathologischen Situation, die auf eine
bestimmte Behandlung antwortet, und einer anderen Situation bestehen,
die nur wenig oder schlecht antwortet und in der Lage ist, Gegenstand
einer anderen Therapiestrategie zu sein. Diese etablierten Referenz-Banken
können mit
Sonden hybridisiert werden, die aus Messenger-RNAs des Patienten
hergestellt sind. Die Hybridisierungsergebnisse ermöglichen
herauszufinden, welcher Patient ein Hybridisierungsmuster hat, das
der reagierenden Situation oder der nicht reagierenden Situation
entspricht und daher so die Therapie auszuwählen.
-
In
dieser Anwendung ist einerseits das Ziel, abhängig vom Patienten die Behandlung
vorzuschlagen, die am geeignetsten ist und die am besten in der
Lage ist, erfolgreich zu sein, und andererseits für eine Behandlung
die Patienten zu rekrutieren, die am geeignetsten sind, erfolgreich
darauf zu reagieren. Wie in den anderen Anwendungen werden zwei
Banken des qualitativen differentiellen Screenings hergestellt:
eine Bank aus einem Modell oder einer pathologischen Probe, die/das
bekanntermaßen
auf eine Behandlung reagiert, die andere Bank aus einem anderen
Modell oder einer anderen pathologischen Probe, die/das wenig oder schlecht
auf die Therapie anspricht. Diese beiden Banken werden dann mit
Sonden hybridisiert, die von mRNA-Extrakten von Biopsien von verschiedenen
Patienten stammen. Je nachdem diese Sonden mit den alternativen
Spleißungen
bevorzugt hybridisieren, die für
die eine oder die andere Situation spezifisch sind, können die
Patienten in Reagierende oder Nichtreagierende auf die Referenzbehandlung
unterteilt werden, die die Ausgangsmodelle definiert hat.
-
In
dieser Hinsicht betrifft die Erfindung ebenfalls ein Verfahren zur
Bestimmung oder Bewertung der Reaktion eines Patienten auf eine
Testverbindung oder Testbehandlung, umfassend die Hybridisierung:
- – von
differentiellen Banken zwischen den cDNAs und der RNAs einer biologischen
Probe, die auf besagte Verbindung/Behandlung reagiert, und einer
biologischen Probe, die auf besagte Verbindung/Behandlung nicht
reagiert oder schlecht reagiert, mit
- – einem
Nucleinsäure-Präparat einer
biologischen Probe des Patienten, und
- – die
Bewertung des Reaktionspotenzials des Patienten durch Analyse des
Hybridisierungsgrades mit den verschiedenen Banken.
-
Ein
ausgewähltes
Beispiel für
den Beitrag des qualitativen differentiellen Screenings in der Pharmakogenomie
bildet ein qualitatives differentielles Screening zwischen zwei
Tumoren desselben histologischen Ursprungs, wobei der eine sich
bei der Behandlung mit einer antitumoralen Verbindung zurückbildet
(zum Beispiel eine Übertragung
einer cDNA, die für
das Wildtyp p53-Protein codiert, mit Hilfe der Gentherapie) und
der andere sich als behandlungsresistent zeigt. Der erste Schritt
bei der Bildung von Banken von qualitativen Unterschieden zwischen
zwei Situation ist die Bestimmung mittels Analyse der Klone, die
diese Banken bilden, welche molekularen Mechanismen bei der Regression
des ersten Modells ablaufen und welche im zweiten Modell nicht vorhanden
sind.
-
Außerdem erlaubt
die Verwendung von Filtern oder jedem anderen Träger, der die cDNA dieser Banken
präsentiert,
die Durchführung
von Hybridisierungen mit Sonden, die von mRNA von Tumorbiopsien
stammen, von denen man die Reaktion auf besagte Behandlung vorhersagen
möchte.
Diese Ergebnisse erlauben daher eine optimierte Rekrutierung von
Patienten in einem klinischen Protokoll.
-
Ein
besonderes Beispiel für
dieses Verfahren ist die Bestimmung der Reaktion des Tumors auf
eine Behandlung mit dem Tumorsuppressorgen p53. Denn es ist beschrieben
worden, das bestimmte Patienten und bestimmte Tumoren mehr oder
weniger auf diese Behandlungsart reagieren (Roth et al., Nature
Medicine, 2 (1995) 958). Es ist folglich wichtig, bestimmen zu können, welche
Tumorarten und welche Patienten auf eine Behandlung mit der Gentherapie
mit Wildtyp p53 ansprechen, um die Behandlung zu optimieren und
die Rekrutierung der Patienten in den laufenden klinischen Versuchen
zu unterstützen.
Das Verfahren der Erfindung erlaubt vorteilhaft, diese Schritte
zu erleichtern, indem Banken vorgeschlagen werden, die für Merkmale
von Zellen spezifisch sind, die auf p53 reagieren und nicht reagieren.
Beispiele für
p53 sensible oder resistente Zellmodelle sind zum Beispiel von Sabbatini
et al. (Genes Dev. 9 (1995) 2184) oder von Roemer et al. (Oncogene
12 (1996) 2069) beschrieben. Die Hybridisierung dieser Banken mit
Sonden, die von Biopsien von Patienten stammen, erlaubt eine einfache
Bewertung deren Reaktionspotenzials. Außerdem erlauben die spezifischen
Banken ebenfalls die Identifizierung der Nucleinsäuren, die
an der Reaktion auf p53 beteiligt sind.
-
Die
vorliegende Anmeldung betrifft ebenfalls die Etablierung von Banken
des differentiellen Screenings aus pathologischen Proben oder Krankheitsmodellen,
die auf wenigstens ein pharmakologisches Agens unterschiedlich reagieren.
Diese Banken können
beschränkte,
komplexe oder autologe Banken sein, wie sie oben beschrieben sind.
Die Erfindung betrifft auch das Ausbreiten dieser Banken auf Filter
oder Träger,
die dem Fachmann bekannt sind (Nitrocellulose, Nylon...). Vorteilhaft
können
diese Träger
Chips oder Biochips sein; die daher pharmakogenomische Biochips
definieren. Die Erfindung bezieht sich auch auf die Nutzung, die
aus der Sequenzierung der verschiedenen Klone erhalten werden kann,
die diese Banken mit dem Ziel bilden, die Mechanismen aufzuklären, die
zu den unterschiedlichen der Reaktionen von pathologischen Proben auf
verschiedene Behandlungen führen
wie auch die Verwendung dieser Banken, um sie mit Sonden zu hybridisieren,
die von Biopsien von pathologischen Situationen stammen, von denen
man die Reaktion auf die Referenzbehandlung vorhersagen möchte, die
die Banken definiert.
-
Die
vorliegende Erfindung beschreibt daher, dass Variationen in den
Spleißformen
und/oder Spleißmustern
Quellen für
pharmakogenomische Marker bilden, nämlich Quellen von Markern,
die das Aufzeigen der Fähigkeit
und der Art und Weise eines Patienten, auf Behandlungen zu reagieren,
erlauben. In dieser Hinsicht ist ebenfalls Gegenstand der Erfindung
die Verwendung der Intervariabilität, zwischen Individuen, der
von alternativen Spleißungen
generierten Isoformen (Analyse des Spleißosoms) als Quelle von pharmakogenomischen
Markern. Die Erfindung betrifft ebenfalls die Verwendung von Spleißmodifikationen,
die von Behandlungen induziert sind, als Quelle von pharmakogenomischen
Markern. Daher erlauben, wie oben erklärt, die DATAS Methodologien
der Erfindung die Generierung der Nucleinsäuren, die für qualitative Unterschiede
zwischen zwei biologischen Proben repräsentativ sind. Diese Nucleinsäuren oder
abgeleitete Formen (Sonden, Primer, komplementäre Säuren, etc.) können zur
Analyse des Spleißosoms
des Individuums im Hinblick auf ein Aufzeigen ihrer Fähigkeit/Art
auf Behandlungen zu reagieren oder ihre Prädisposition für eine derarige
Behandlung/Krankheit. etc. verwendet sein.
-
Diese
verschiedenen allgemeinen Beispiele veranschaulichen den Nutzen
von Banken des qualitativen differentiellen Screenings bei genotoxischen,
genopharmakologischen, pharmakogenomischen Untersuchungen wie auch
bei der Suche von diagnostischen oder therapeutischen Zielen von
Interesse. Diese Banken stammen von der Klonierung der qualitativen
Unterschiede, die zwischen zwei physiopathologischen Zuständen vorliegen.
Da eine andere Verwendung der für
diese qualitativen Unterschiede repräsentativen cDNAs die Bildung
von Sonden ist, die für
ein Screening einer genomischen DNA-Bank bestimmt sind, deren Charakteristika
oben beschrieben sind, kann ein derartiger Versuchsansatz ebenfalls
für jede
genotoxische, genopharmakologische, pharmakogenomische Untersuchung
wie auch zur Identifizierung des Gens verwendet sein. Zum Beispiel
bei den genotoxischen Untersuchungen werden die genomischen Klone,
die durch die Größe ihrer
Insertionen statistisch auf ein einziges Intron oder auf ein einziges
Exon beschränkt
sind, auf den Filtern in Abhängigkeit
ihrer Hybridisierung mit den DATAS-Sonden klassifiziert, die von
der qualitativen differentiellen Analyse zwischen einer Zellpopulation
oder einem Gewebe als Referenz und denselben mit einer toxischen Referenzverbindung
behandelten Zellen oder Gewebe stammen. Wobei diese für verschiedene
Toxizitätsklassen
repräsentativen
Klone selektiert werden, kann dann eine Hybridisierung dieser Klone
mit einer Sonde durchgeführt
werden, die von Gesamt-mRNA
derselben Zellpopulation oder desselben Gewebes durchgeführt werden,
die mit einer Verbindung behandelt worden sind, deren toxisches
Potenzial man vorhersagen möchte.
-
Weitere
Vorteile und Anwendungen der vorliegenden Erfindung sind durch Lektüre der folgenden
Beispiel zu erkennen, die als Veranschaulichung und nicht als Beschränkung betrachtet
werden sollen. Die Anwendungsbereiche der Erfindung sind in 2 dargestellt.
-
LEGENDE DER
FIGUREN
-
1. Schematische Darstellung des differentiellen
Screenings gemäß der Erfindung
(1A) unter Verwendung einer (1B)
oder zwei (1C) Hybridisierungen und Verwendung
von Nucleinsäuren (1D).
-
2.
Schematische Darstellung, die die Gewinnung von RNA/DNA-Hybriden
beschreibt und die Charakterisierung der einzelsträngigen RNA-Sequenzen,
für den
pathologischen oder den gesunden Zustand spezifische Signaturen
erlaubt.
-
3.
Schematische Darstellung, die ein anderes Mittel beschreibt, das
die Isolierung und Charakterisierung mittels Sequenzierung der einzelsträngigen RNA-Sequenzen erlaubt,
die für
eine pathologische Situation oder eine gesunde Situation spezifisch
sind.
-
4.
Schematische Darstellung, die ein anderes Mittel beschreibt, das
die Charakterisierung mittels Sequenzierung der ganzen oder eines
Teils der einzelsträngigen
RNA-Sequenzen erlaubt, die für
eine pathologische Situation oder eine gesunde Situation spezifisch
sind.
-
5.
Schematische Darstellung, die eine Isolierung der Produkte der alternativen
Spleißungen
aufgrund der Strukturen der R-Schleife erlaubt.
-
6. Schematische Darstellung des qualitativen
differentiellen Screenings durch Restriktion von Schleifen (Bildung
von doppelsträngiger
cDNA/cDNA-Homoduplices
und Gewinnung der Informationen, 6A) und
Beschreibung der gewonnenen Informationen (6B).
-
7.
Beiträge
des qualitativen differentiellen Screenings für die verschiedenen Schritte
bei der Suche und der Entwicklung von Pharmazeutika.
-
8.
Isolierung einer differentiell gespleißten Domäne im grb2/grb33-Modell. A)
Herstellung synthetischer RNAs von grb2 und grb33. B) Abfolge der
ersten Schritte von DATAS, die zur Charakterisierung eines RNA-Fragments
führen,
das der gespleißten
Domäne
entspricht: 1: RNA von grb2; 2: Hybridisierung der RNA von grb2
und der cDNA von grb33; 3: Hybridisierung der RNA von grb2 und der
cDNA von grb2; 4: Hybridisierung der RNA von grb2 und Wasser; 5: Überstand
nach Passieren von Streptavidin-Kügelchen von (2); 6: Überstand
nach Passieren von Streptavidin-Kügelchen von (3); 7: Überstand
nach Passieren von Streptavidin-Kügelchen
von (4); 8: Verdau des grb2-RNA/grb33-cDNA-Duplex mit RNase H; 9:
Verdau des grb2-RNA/grb2-cDNA-Duplex mit RNase H; 10: Verdau von
grb2-RNA mit RNase H; 11: ähnlich
wie (8) nach Passieren einer Ausschluss-Säule; 12: ähnlich wie (9) nach Passieren
einer Ausschluss-Säule;
13: ähnlich
wie (10) nach Passieren einer Ausschluss-Säule.
-
9.
Darstellung der Populationen von ungepaarten RNAs, die von dem Verdau
mit RNase H aus den RNA/einzelsträngiger cDNA-Duplices stammen,
von Ethanol-behandelten oder nichtbehandelten HepG2-Zellen stammend.
-
10.
Darstellung der Populationen doppelsträngiger cDNA, die mit einer
der DATAS-Varianten generiert sind. 1 bis 12: PCR von Populationen
von RNAs-Schleifen,
die von dem Verdau mit RNase H stammen; 13: PCR von Gesamt-cDNA.
-
11.
Anwendung der DATAS-Variante, die die doppelsträngige cDNA im grb2/grb33-Modell
einsetzt. A) Analyse der Komplexe auf Agarosegel nach Hybridisierung:
1: doppelsträngige
grb2-cDNA/grb33-RNA; 2: doppelsträngige grb2-cDNA/grb2-RNA; 3: doppelsträngige grb2-cDNA/Wasser.
B) Verdau der Proben 1, 2 und 3 von A) mit der S1-Nuclease und der „Mung Bean" Nuclease: 1 bis
3: Komplexe 1 bis 3 vor der Glyoxal-Behandlung; 4 bis 6: Komplexe
nach der Glyoxal-Behandlung;
7 bis 9: Verdau 1 bis 3 mit S1-Nuclease; 10 bis 12: Verdau 1 bis
3 mit „Mung
Bean" Nuclease.
-
12.
Anwendung der DATAS-Variante, die die einzelsträngige cDNA und die RNase H
in einem System mit HepG2-Zellen einsetzt, die 18 Stunden mit 0,1
M Ethanol behandelt oder unbehandelt sind. Die klonierten Inserts
sind auf die Membran nach Elektrophorese auf Agarosegel transferiert
und einer Hybridisierung mit Hilfe den behandelten (Tr) oder nichtbehandelten
Situationen (NT) entsprechenden Sonden unterzogen worden.
-
13. Vorgehensweise zur Bewertung des toxischen
Potenzials eines Produkts.
-
14. Vorgehensweise zum Verfolgen der Wirksamkeit
eines Produkts.
-
15. Vorgehensweise zur Untersuchung der Empfindlichkeit
einer pathologischen Situation auf eine Behandlung.
-
16. Differentielle Hybridisierungsanalyse von
Klonen, die von DATAS aus RNA-Extrakten von induzierten Zellen und
von cDNA-Extrakten von nicht induzierten Zellen stammen. A) Verwendung
von Bakterienkolonien, die an einer Membran angelagert und lysiert
sind. B) Southern Blot, aus einer Selektion von Klonen von A durchgeführt.
-
17. Nucleotid- und Peptidsequenz von ΔSHC (SEQ
ID NR: 9 und 10).
-
18. Cytotoxizitäts- und Apoptosetests mit HepG2-Zellen,
die A) mit Ethanol; B) mit Camptothecin; C) mit PMA behandelt sind.
-
19. Reaktionen von RT-PCR, die aus RNA-Extrakten
von HepG2-Zellen, die nicht behandelt (N.T.) oder mit Ethanol (Eth.),
Camptothecin (Camp.) und PMA behandelt sind und die Amplifikation
von Fragmenten erlauben, die den Domänen von MACH-a, BCL-X, FASR
und beta-Actin als Kontrolle des Grundwerts entsprechen.
-
In
den Beispielen und der Beschreibung der Erfindung wird Bezug auf
die Sequenzen der Sequenz-Liste genommen, die den folgenden freien
Text enthält:
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
<223> OLIGO
-
BEISPIELE
-
1. DIFFERENTIELLE KLONIERUNG
DER ALTERNATIVEN SPLEISSUNGEN
-
UND ANDEREN QUALITATIVEN
MODIFIKATIONEN DER RNAs UNTER VERWENDUNG VON EINZELSTRÄNGIGEN cDNAs
-
Die
Messenger-RNAs, die beiden Situationen entsprechen, einer normalen
(mN) und einer anderen pathologischen (mP), werden aus Biopsien
oder Zellkulturen isoliert. Diese Messenger-RNAs werden in komplementäre DNA (cN)
und (cP) mit Hilfe der reversen Transkriptase (RT) umgeschrieben.
mN/cP und cN/mP-Hybride werden dann in flüssiger Phase hergestellt (siehe
Schema der 2, das einen der beiden Fälle veranschaulicht,
der zur Bildung von cN/mP führt).
-
Diese
Hybride sind vorteilhaft in Phenol-Emulsion hergestellt (PERT-Technik
oder Phenol Emulsion DNA Reassociation Technique) und mit Thermocyclen
erhalten (Miller, R.D. und Riblet, R., 1995, Nucleic Acids Research,
Vol. 23, Nr. 12, Seiten 2339-2340). Typischerweise wird dieser Hybridisierungsschritt
mit 0,1 bis 1 μg
polyA+ RNA und 0,1 bis 2 μg
komplementärer
DNA in einer Emulsion, die aus einer wässrigen Phase (Puffer aus 120
mM Natriumphosphat, 2,5 M NaCl, 10 mM EDTA) und einer organischen
Phase gebildet ist, die 8% der wässrigen
Phase beinhaltet und aus bidestilliertem Phenol gebildet ist.
-
Eine
andere Technik ist ebenfalls vorteilhaft eingesetzt, um Heteroduplices
zu erhalten: am Ende der reversen Transkription wird die neu synthetisierte
cDNA von dem biotinylierten oligo-dT Primer auf einer Ausschluss-Säule getrennt.
0,1 bis 2 μg
dieser cDNA wird mit 0,1 bis 1 μg
polyA+ RNA in Gegenwart von 0,3 M Natrium acetat und zwei Volumina
Ethanol mitgefällt.
Diese mitgefällten
Nucleinsäuren
werden in 30 μl
Hybridisierungspuffer wieder aufgenommen, der 80% Formamid, 40 mM
PIPES (Piperazin-bis(2-ethansulfonsäure) pH 6,4, 0,4 M NaCl und
1 mMEDTA enthält.
-
Die
Nucleinsäuren
in Lösung
werden durch Erhitzen auf 85°C
10 min denaturiert, dann wird ihre Hybridisierung in wenigstens
16 h und bis 48 h bei 40°C
durchgeführt.
-
Der
Vorteil der Hybridisierungstechnik in Formamid ist, dass diese stärkere selektive
Bedingungen während
der Paarung der cDNA- und RNA-Stränge erlaubt.
-
Am
Ende dieser beiden Hybridisierungstechniken verfügen wir über RNA/DNA-Heteroduplex, dessen perfekte Paarung
von der Effizienz der RT abhängt,
die gesamte Länge
der cDNA zu synthetisieren. Es verbleiben ebenfalls die RNA-Regionen
in einzelsträngiger
Form (und DNA), die alternativen Spleißungen entsprechen, die die
beiden untersuchten physiopathologischen Zustände unterscheiden.
-
Das
Ziel des Verfahrens ist dann, die genetische Information zu charakterisieren,
die die Spleiß-Schleifen
enthalten.
-
Hierfür werden
die Heteroduplices durch Einfangen der cDNA (mit biotinylierten
oligo-dT Primern versehen) mit Hilfe von Kügelchen, die Streptavidin-Gruppen
tragen, gereinigt. Vorteilhafterweise sind diese Kügelchen
magnetisch, was erlaubt, sie mit Hilfe eines Magnetseparators von
RNAs zu trennen, die nicht an den Heteroduplices beteiligt sind.
Derartige Kügelchen
und Separatoren sind im Handel erhältlich.
-
In
diesem Stadium des Verfahrens sind die Heteroduplices und die an
diesen Hybridisierungen mit den RNAs nicht beteiligten cDNAs getrennt.
Dieses Material wird dann einer RNase H-Behandlung unterzogen, die spezifisch
die mit den cDNAs hybridisierten RNA-Regionen hydrolysiert. Die
aus dieser Hydrolyse resultierenden Produkte sind einerseits die
cDNAs und andererseits die RNA-Fragmente, die den Spleiß-Schleifen
oder, wegen der partiellen Wirkung der inversen Transkriptase, nicht
hybridisierten Regionen entsprechen. Die RNA-Fragmente werden von
der cDNA durch magnetische Trennung gemäß derselben weiter oben erwähnten Arbeitsweise
und durch Verdau mit der DNase getrennt, die von jeglicher Kontamination
mit einer RNase-Aktivität
befreit ist.
-
1.1. Validierung der DATAS-Technologie
bei Spleißvarianten
des Grb2-Gens
-
Eine
Darstellung der Durchführbarkeit
dieses Versuchsansatzes ist mit einem in vitro System unter Verwendung
einer RNA durchgeführt
worden, die einerseits der Grb2 codierenden Region entspricht, und
einer einzelsträngigen
cDNA entspricht, die zu einer Grb3.3 codierenden Region komplementär ist. Grb2
ist ein Gen, das 651 codierende Basenpaare besitzt. Grb33 ist eine
Isoform von Grb2, die durch alternatives Spleißen generiert ist und eine
Deletion von 121 Basenpaaren in der funktionellen SH2-Domäne von Grb2
umfasst (Fath et al., Science (1994), 264, 971-4). Die RNAs von Grb2 und Grb33 werden
gemäß dem Fachmann
bekannten Techniken aus einem Plasmid, das die von Grb2 oder Grb33
codierende Sequenz unter Kontrolle des T7-Promotors enthält, mit
Hilfe des RiboMax Kit (Promega) synthetisiert. Die Analyse der Produkte
zeigt eine homogene Synthese (8A).
Zur Visualisierung wurde ebenfalls Grb2 mittels Einbau einer markierten
Base während
der in vitro Transkription mit Hilfe des RiboProbe Kits (Promega)
radioaktiv gemacht. Die cDNAs von Grb2 und Grb3.3 sind mittels inverser
Transkription aus oben hergestellten synthetischen RNAs des Kit
Superscript II (Life Technologies) und dem für Grb2 und Grb3.3 gemeinsamen
biotinylierten Oligonucleotid-Primer, der der Komplementärsequenz
(618-639) von Grb2 entspricht, synthetisiert worden. Die RNAs und
cDNAs sind gemäß Herstellerangaben
(Promega, Life Technologies) behandelt, auf einer Ausschluss-Säule (RNase-freie
Sephadex G25 oder G50, 5 Prime, 3 Prime) gereinigt und mittels Spektrophotometrie
quantifiziert worden.
-
Angewandt
worden sind die ersten Schritte von DATAS in Suspension von 10 ng
markierter RNA von Grb2, zusammen mit:
- 1. 100
ng cDNA von biotinyliertem Grb33,
- 2. 100 ng cDNA von biotinyliertem Grb2,
- 3. Wasser
in 30 μl
Nybridisierungspuffer, der 80% Formamid, 40 mM PIPES (pH 6,4), 0,4
M NaCl, 1 mM EDTA enthält. Die
Nucleinsäuren
werden durch 10 min Erhitzen auf 85°C denaturiert, dann wird die
Hybridisierung 16 Stunden bei 40°C
durchgeführt.
Nach Einfangen mit Hilfe der Streptavidin-Kügelchen werden die Proben mit
der RNase H, wie bereits beschrieben, behandelt.
-
Die
Analyse dieser Schritte erfolgt mittels Elektrophorese auf Acrylamidgel
mit Hilfe eines Instant Imagers (Packard Instruments), der die Charakterisierung
und Quantifizierung der von der markierten Grb2-RNA stammenden Spezies
erlaubt (8B). Daher zeigen die Spuren
2, 3 und 4, dass die grb2/grb33 und grb2/grb2-Duplices sich quantitativ gebildet haben.
Die Migration des grb2/grb33-Komplexes ist verglichen mit dem der
grb2-RNA (Spur 2) verzögert,
während
die Migration des grb2/grb2-Komplexes erhöht ist (Spur 3). Die Spuren
5, 6 und 7 entsprechen den Proben, die von den Streptavidin-Kügelchen
nicht zurückgehalten
werden, was zeigt; dass 80% der grb2/grb33 und grb2/grb2-Komplexe
von den Kügelchen
zurückgehalten
werden, während
die RNA von nur grb2, die nicht biotinyliert ist, sich ausschließlich im Überstand
der Kügelchen wiederfindet.
Die Behandlung mit RNase H setzt, außer den freien Nucleotiden,
die schneller als Bromphenol-Blau (BPB) migrieren, eine Spezies
frei, die diesseits von Xylen-Cyanol-Blau (XC) (mit Pfeil in der
Figur markiert) migriert und dies spezifisch in Spur 8, die dem
grb2/grb33-Komplex
entspricht, verglichen mit den Spuren 9 und 10, die den grb2/grb2-Komplexen und der
grb2-RNA entsprechen. Die Spuren 11, 12 und 13 entsprechen den Spuren
8, 9 und 10, nachdem die Proben eine Ausschluss-Säule passiert
haben, um die freien Nucleotide zu eliminieren. Die beobachtete
Migration in den Spuren 8 und 11 entspricht derjenigen, die für ein RNA-Molekül erwartet
wird, das einer Deletion von 121 Nucleotiden entspricht, die grb2
von grb33 unterscheidet.
-
Dieses
Ergebnis zeigt gut, dass es möglich
ist, Schleifen von RNA zu erhalten, die mit der Heteroduplex-Bildung
von zwei Sequenzen generiert werden, die von zwei Spleißisoformen
abstammen.
-
1.2. Anwendung der DATAS-Technik
zur Generierung von qualitativen Banken von Hepatocyten in gesundem und
toxischem Zustand.
-
Es
wurde eine komplexere Situation untersucht. Im Rahmen der Anwendung
der DATAS-Technologie als ein Vorhersage-Instrument der Toxizität von Molekülen wurden
die humanen Hep2G-Hepatocyten 18 Stunden mit 0,1 M Ethanol behandelt.
Die RNAs sind aus behandelten oder nichtbehandelten Zellen extrahiert
worden. Die oben beschriebene DATAS-Variante (Präparation biotinylierter einzelsträngiger cDNA,
Kreuzhybridisierungen in flüssiger
Phase, Anwendung eines Magnetfeldes zur Trennung der Spezies, Behandlung
mit RNase H) ist mit nicht behandelten Zellen in einer Referenzsituation
(oder Situation A) und den behandelten Zellen in Testsituation (oder
Situation B) angewandt worden (9). Die
RNA-Extrakte sind nichtradioaktiv markiert worden und die Visualisierung
der mittels RNase H-Verdau generierten RNA-Population erfolgt durch eine
Austauschreaktion des 5'-Phosphates der
RNAs durch ein markiertes Phosphat mit Hilfe der T4-Polynucleotid-Kinase
und gamma 32P ATP. Diese Markierungen werden
dann auf einem Harnstoff/Acrylamidgel aufgetragen und mittels Exposition
mit Hilfe eines Instant Imager (Packard Instruments) analysiert.
Die von A/B und B/A-Hybridisierungen stammenden komplexen Signaturen
können
nun mit einer ersten Gruppe von Signalen, die schlecht im Gel migrieren
und den Nucleinsäure-Sequenzen
entsprechen, die eine beträchtliche Größe besitzen,
und mit einer zweiten Gruppe von Signalen sichtbar gemacht werden,
die zwischen 25 und 500 Nucleotiden migrieren. Diese Signaturen
aus der Situation A/A haben eine viel geringere Intensität, was vermuten
lässt,
dass das Ethanol eine Reprogrammierung von Spleißungen der RNAs induzieren
kann, die von dem Vorhandensein von Signalen in A/B und B/A wiedergegeben
werden.
-
1.3. Klonierung und Präparation
von Banken aus identifizierten Nucleinsäuren
-
Mehrere
experimentelle Varianten sind ferner denkbar, um diese RNase H resistenten
RNA-Fragmente zu klonieren:
A. Ein erster Versuchsansatz besteht
aus der Isolierung und Klonierung dieser Schleifen (3).
-
Gemäß diesem
Versuchsansatz wird eine Ligation von Oligonucleotiden an jedem
Ende mit Hilfe der RNA-Ligase unter dem Fachmann bekannten Bedingungen
durchgeführt.
Diese Oligonucleotide werden dann als Primer verwendet, um eine
RT PCR durchzuführen.
Die PCR-Produkte werden kloniert und mit vollständig komplementären Sonden,
die den beiden Situationen von Interesse entsprechen, durchmustert.
Nur die Klone, die bevorzugt mit nur einer der beiden Sonden hybridisieren,
enthalten die Spleiß-Schleifen,
die dann sequenziert und/oder zur Generierung der Banken verwendet
werden.
-
B.
Der zweite Versuchsansatz (4) besteht
aus einer Durchführung
der mit Hilfe wenigstens zum Teil zufälliger Primer initiierten inversen
Transkription der einzelsträngigen
RNA, die aus Heteroduplices nach Behandlung mit RNase H freigesetzt
ist. Es kann sich daher um in 3' und
5' Zufallsprimer.
um in 3' zufällige und 5' definierte Primer
oder auch um halbzufällige
Primer handeln, die nämlich
einen degenerierten Bereich und einen definierten Bereich umfassen.
-
Gemäß dieser
Strategie sind die Primer in der Lage, entweder irgendwo an der
einzelsträngigen
RNA oder an jeder mit der Wahl des halbzufälligen Primers festgelegten
Basenfolge zu hybridisieren. Eine PCR mit Primern, die oben beschriebenen
Oligonucleotiden entsprechen, erlaubt dann, von den Spleiß-Schleifen
abstammende Sequenzen zu erhalten.
-
Die 10 (Spur
1 bis 12) zeigt die Analyse auf einem Acrylamidgel der PCR-Fragmente, die aus mehreren
DATAS-Versuchen erhalten sind und für die Verwendung mit folgenden
halbzufälligen
Primern gekoppelt sind:
GAGAAGCGTTATNNNNNNNAGGT (SEQ ID NR:
1, X=T)
GAGAAGCGTTATNNNNNNNAGGA (SEQ ID NR: 1, X=A)
GAGAAGCGTTATNNNNNNNAGGC
(SEQ ID NR: 1, X=C)
GAGAAGCGTTATNNNNNNNAGGG (SEQ ID NR: 1,
X=G)
-
Der
Vergleich mit der Komplexität
der Signaturen, die unter Verwendung derselben Oligonucleotide aber
der Gesamt-cDNA als Matrize (Spur 13) erhalten sind, zeigt, dass
DATAS erlaubt hat, die den qualitativen Unterschieden entsprechenden
Informationen zu filtern („profilieren").
-
Diese
Variante ist verwendet worden, um ein Ereignis zu klonieren, das
der RNA-Domäne
von grb2 entspricht, die durch Behandlung mit RNase H aus dem Duplex
grb2-RNA/einzelsträngige
cDNA von grb33 gemäß dem bereits
beschriebenen Protokoll (Beispiel 1.1) generiert ist. Zu diesem
Zweck ist ein Oligonucleotid mit der Sequenz: GAGAAGCGTTATNNNNNNNNTCCC
(SEQ ID NR: 2) verwendet worden, das aus dem Modell GAGAAGCGTTATNNNNNNNWXYZ
(in dem N wie vorhergehend definiert ist, W, X und Y für jede feststehend
bestimmte Base stehen und Z entweder für eine bestimmte Base oder
eine 3' OH-Gruppe
steht, SEQ ID NR: 3) ausgewählt
und selektiert ist, um ein Fragment in der Deletion von grb2 zu
amplifizieren, was die Generierung eines PCR-Fragments erlaubt,
dessen Klonierung und Sequenzierung gezeigt hat, dass es tatsächlich von
der von grb2 deletierten Domäne
abstammt.
-
Die
beiden Versuchsansätze
erlauben folglich die Herstellung von Zusammensetzungen von Nucleinsäuren, die
für differentielle
Spleißungen
in den beiden getesteten Situationen repräsentativ sind, die als Sonden
oder zur Konstruktion von cDNA-Banken von qualitativen Unterschieden
eingesetzt sein können.
Die Tauglichkeit der DATAS Technologie zur Generierung der profilierten
Banken von cDNA, die für
qualitative Unterschiede repräsentativ
sind, wird ebenfalls mit nachfolgenden Beispiel 1.4. veranschaulicht.
-
1.4. Herstellung von profilierten
Banken, die für
humane Endothelzellen repräsentativ
sind
-
Dieses
Beispiel ist mit einer humanen Endothelzelllinie (ECV304) durchgeführt worden.
Die qualitative Analyse der Genexpression ist mit cytosolischen
RNA-Extrakten einerseits
von proliferierenden Zellen und andererseits von Zellen in Anoikis
(Apoptose durch Entfernen des Anhaftungsträgers) durchgeführt worden.
-
Die
ECV-Zellen sind in 199 Medium, supplementiert mit Earle Salzen (Life
Sciences), kultiviert worden. Durch 4 stündige Passage in polyHEMA behandelten
Kulturschalen werden die Zellen in den Anoikiszustand gebracht.
Bei der RNA-Präparation
sind die Zellen in einem Nonidet P-40 haltigen Puffer lysiert worden.
Die Kerne sind dann durch Zentrifugation entfernt worden. Die Lösung des
cytoplasmatischen Extrakts ist dann so eingestellt worden, um auf
spezifische Weise die RNA auf der Rneasy Siliciumdioxid-Matrize
gemäß der Qiagen
Herstelleranleitung zu fixieren. Nach Waschen werden die Gesamt-RNAs
in Wasser eluiert, das DEPC-behandelt
ist. Die Messenger-RNA wird aus der Gesamt-RNA durch Trennung auf
magnetischen Dynabeads oligo (dT)25 Kügelchen
(Dynal) präpariert.
Nachdem die Kügelchen
in Fixierungspuffer suspendiert sind, wird die Gesamt-RNA 5 min
bei Raumtemperatur inkubiert. Nach magnetischer Trennung und Waschen
werden die Kügelchen
in einem Elutionspuffer für
eine Inkubation bei 65°C
wieder aufgenommen, die die Messenger-RNAs freisetzt.
-
Die
Synthesen des ersten DNA-Stranges sind mit Messenger-RNAs unter
Verwendung der Reversen Transkriptase SuperScript II oder ThermoScript
(Life Technologies) mit Hilfe von oligo (dT) Primern durchgeführt. Nach
RNase H-Behandlung
werden die freien Nucleotide durch Passieren einer Sephadex G50
Säule (5
Prime- 3 Prime) eliminiert. Nach Extraktion in Phenol/Chloroform
und Fällung
in Ethanol werden die Proben mittels UV-Absorption quantitativ bestimmt.
-
Die
erforderlichen RNA und cDNA-Mengen (in diesem Fall jeweils 200 ng)
werden vereint und in Ethanol gefällt. Die Proben werden in einem
30 μl Volumen
Hybridisierungspuffer (40 mM Hepes (pH 7,2), 400 mM NaCl, 1 mM EDTA),
supplementiert mit deionisiertem Formamid (80% (v/v) außer anderes
ist angegeben) wieder aufgenommen. Nach 5 min Denaturierung bei
70°C werden
die Proben über
Nacht bei 40°C
inkubiert.
-
Die
Streptavidin-Kügelchen
(Dynal) werden gewaschen, dann in Fixierungspuffer (2X = 10 mM Tris-HCl
(pH 7,5), 2 M NaCl, 1 mM EDTA) rekonditioniert. Die Hybridisierungsproben
werden auf ein 200 μl Volumen
mit Wasser aufgefüllt,
dann werden 200 μl
Kügelchen
für eine
60 min Inkubation bei 30°C
zugegeben. Nach Einfangen auf einem Magneten und Waschen der Kügelchen,
werden diese in 150 μl
RNase H Puffer wieder aufgenommen, dann 20 min bei 37°C inkubiert.
Nach Einfangen auf einem Magneten sind die nichthybridisierten Regionen
in den Überstand
freigesetzt, der mit Dnase behandelt ist, dann mit Phenol/Chloroform extrahiert,
dann in Ethanol gefällt
worden. Die Ethanol-Fällungen
der geringen Mengen an Nucleinsäuren
werden mit Hilfe eines im Handel erhältlichen Enzyms, SeeDNA (Amersham
Pharmacia Biotech), durchgeführt, das
die quantitative Wiedergewinnung der Nucleinsäuren aus sehr verdünnten Lösungen (Größenordnung ng/ml)
erlaubt.
-
Die
cDNA-Synthese aus Proben von RNAs, die aus der RNase H-Behandlung
hervorgehen, wird mit zufälligen
Hexanucleotiden mit Hilfe von Superscript II Reverse Transcriptase
durchgeführt.
Die RNA wird dann mit Hilfe einer Mischung aus RNase H und RNase
T1 abgebaut. Der Primer, die nicht eingebauten Nucleotide und die
Enzyme werden von der cDNA mit Hilfe einer „GlassMAX Spin" Kartusche abgetrennt.
Die den Spleiß-Schleifen
entsprechende cDNA wird dann einer PCR-Reaktion unter Verwendung von halbzufälligen Oligonucleotiden,
die bereits weiter oben in der Erfindung beschrieben sind, unterzogen.
In diesem Fall sind die gewählten
Oligonucleotide:
GAGAAGCGTTATNNNNNCCA (SEQ ID NR: 4)
-
Die
PCR-Reaktion wird mit Hilfe der Taq Polymerase über 30 Cyclen durchgeführt:
- • Initiale
Denaturierung: 94°C
für 1 min
- • 94°C für 30 s
- • 55°C für 30 s
- • 72°C für 30 s
- • Finale
Extension 72°C
für 5 min
-
Die
PCR-Produkte sind in den pGEM-T Vektor (Promega) kloniert worden,
der ein an den 3'-Enden hängendes
T besitzt, um die Klonierung von der Behandlung mit Taq-Polymerase
stammenden Fragmenten zu erleichtern. Nach Transformation in die
kompetenten JM109 Bakterien (Promega) werden die erhaltenen Kolonien
auf Nitrocellulose-Filter übertragen
und mit Sonden hybridisiert, die von PCR-Produkten stammen, die
einerseits mit Gesamt-cDNA von prolifierierenden Zellen und andererseits
mit Zellen in Anoikis durchgeführt
wurden. Für
diese PCR werden dieselben GAGAAGCGTTATNNNNNCCA Oligonucleotide
verwendet. In einer ersten Versuchsdurchführung sind 34 Klone, die bevorzugt
mit der Sonde der Zellen in Apoptose hybridisieren, und 13 Klone,
die bevorzugt mit der Sonde von proliferierenden Zellen hybridisieren,
isoliert worden.
-
Unter
diesen 13 Klonen enthalten 3 Klone dasselbe cDNA-Fragment, das von
der SH2-Domäne
des SHC-Proteins stammt.
-
Die
Sequenz dieses Fragments ist die folgende:
CCACACCTGGCCAGTATGTGCTCACTGGCTTGCAGAGTGGGCAGCCAGCC
TAAGCATTTGCACTGG (SEQ ID NR: 5)
-
Die
Verwendung von PCR-Primern, die die SH2-Domäne von SHC flankieren (oligo5': GGGACCTGTTTGACATGAAGCCC
(SEQ ID NR: 6); oligo3':
CAGTTTCCGCTCCACAGGTTGC (SEQ ID NR: 7)), hat ermöglicht, die Deletion der SH2-Domäne von SHC
zu charakterisieren, die spezifisch in den ECV-Zellen in Anoikis
beobachtet ist. Mit diesem Primer-Paar wird ein einziges Amplifikationsprodukt,
das einem cDNA-Fragment mit 382 Basenpaaren entspricht, das die
vollständige
SH2-Domäne
enthält,
aus RNA von ECV-Zellen in exponentieller Phase erhalten. Ein zusätzliches
Fragment mit 287 Basenpaaren wird beobachtet, wenn die PCR aus RNA
von Zellen in Anoikis durchgeführt
wird. Dieses weitere Fragment stammt von einer Messenger-RNA, die
von Messenger-RNA von SHC stammt, aber eine Deletion aufweist.
-
Die
Sequenz dieser Deletion ist die folgende:
GTACGGGAGAGCACGACCACACCTGGCCAGTATGTGCTCACTGGCTTGCA
GAGTGGGCAGCCTAAGCATTTGCTACTGGTGGACCCTGAGGGTGTG (SEQ ID NR: 8).
-
Diese
Deletion entspricht den Basen 1198 bis 1293 des offenen Rahmens
des Messengers, der für
die Formen 52 kDa und 46 kDa des SHC-Proteins codiert (Pelicci,
G. et a., 1992, Cell, Seiten 93-104).
-
Die
Strukturdaten der SH2-Domänen
wie auch die Literatur weisen darauf hin, dass eine derartige Deletion
zu einem Verlust der Affinität
für die
Phosphotyrosine führt,
da sie die Aminosäuren
umfasst, die an den Wechselwirkungen mit den phosphorylierten Tyrosinen
beteiligt sind (Waksman, G. et al., 1992, Nature, vol. 358, Seiten
646-653). Wobei die SHC-Proteine Adapter sind, die verschiedene
Partner über
ihre SH2 und PTB (Phosphotyrosin-bindende Domäne)-Domänen verbinden, generiert diese
Deletion folglich eine natürliche
negative Dominanz von SHC, die wir als ΔSHC bezeichnen. Da die SH2-Domänen der
Proteine, deren Gene sequenziert sind, von zwei Exons getragen sind,
ist es wahrscheinlich, dass die mit dem DATAS-Verfahren identifizierte Deletion einem
alternativen Exon des SHC-Gens entspricht.
-
Die
Protein- und Nucleinsequenzen von ΔSHC sind in den 17 dargestellt
(SEQ ID NR: 9 und 10).
-
Da
die SH2 und SHC-Domäne
an der Transduktion zahlreicher Signale beteiligt sind, die an der
Zellproliferation und Zellviabilität beteiligt sind, erlaubt die
Untersuchung der ΔSHC-Sequenz
ihre Eigenschaften der negativen Dominanz über das SHC-Protein und ihrer
Fähigkeit,
mit verschiedenen zellulären
Signalen zu interferieren, vorherzusagen.
-
Die
Erfindung betrifft ebenfalls diese neue gespleißte Form von SHC, die der Spleißung entsprechende
Proteindomäne,
jeden Antikörper
oder jede Nucleinsonde, die ihre Detektion in einer biologischen
Probe erlaubt, und ihre beispielsweise diagnostische oder therapeutische
Verwendung.
-
Die
Erfindung betrifft insbesondere jede Variante von SHC, umfassend
wenigstens eine Deletion, die den Basen 1198 bis 1293 entspricht,
ganz besonders eine Deletion der Sequenz SEQ ID NR: 8. Die Erfindung betrifft
spezifischer die ΔSHC-Variante,
die die Sequenz SEQ ID NR: 9 besitzt, die von der SEQ ID NR: 10
codiert ist.
-
Die
Erfindung betrifft ebenfalls jede Nucleinsonde, jedes Oligonucleotid
oder jeden Antikörper,
die die Identifizierung der oberen ΔSHC-Variante und/oder jede Änderung
des SHC/ΔSHC
Verhältnisses
in einer biologischen Probe erlauben. Es kann sich besonders um
eine Sonde oder Oligonucleotid, die zu der ganzen oder einem Teil
der Sequenz SEQ ID NR: 8 komplementär ist, oder um einem Antikörper handeln,
der gegen die von dieser Sequenz codierten Proteindomäne gerichtet
ist. Derartige Sonden, Oligonucleotide oder Antikörper erlauben
das Vorliegen der nicht gespleißten
Form (z.B. SHC) in einer biologischen Probe zu detektieren.
-
Die
Materialien können
außerdem
mit Sonden, Oligonucleotiden und/oder Antikörpern parallel verwendet sein,
die für
Spleißformen
(z.B. ΔSHC)
spezifisch sind, nämlich
zum Beispiel der Verknüpfungsregion entsprechen,
die aus einer Spleißung
resultiert (befindet sich in der Nähe des Nucleotids 1198 der
Sequenz SEQ ID NR: 10).
-
Derartige
Materialien können
für die
Diagnose von Krankheiten verwendet sein, die mit einer Immundepression
(Krebs, Behandlung mit Immunsuppressoren, AIDS, etc.) verbunden
sind.
-
Die
Erfindung betrifft auch jedes Verfahren für das Screening von Molekülen, die
auf der Blockierung (i) der gespleißten Domäne in dem SHC-Protein (besonders
zur Induktion eines immuntoleranten Zustands zum Beispiel bei Autoimmunkrankheiten
oder Abstoßung
von Transplantaten oder Krebs) oder (ii) der Funktionsgewinne, die
vom ΔSHC-Protein
erworben sind, beruhen.
-
Die
Erfindung betrifft außerdem
die therapeutische Verwendung von ΔSHC und insbesondere zur Behandlung
von Krebszellen oder Krebs (ex vivo oder in vivo), in denen eine
Hyperphosphorylierung des ΔSHC-Proteins
gezeigt werden kann. In dieser Hinsicht betrifft die Erfindung auch
jeden Vektor, insbesondere viralen Vektor, der eine für ΔSHC codierende
Sequenz umfasst. Es handelt sich bevorzugt um einen Vektor, der
in der Lage ist, Krebszellen oder proliferierende Zellen zu transfizieren,
wie glatte Muskelzellen, Endothelzellen (Restenose), Fibroblasten
(Fibrosen) vorzugsweise Säugerursprungs,
insbesondere humanen Ursprungs. Als viralen Vektor kann man besonders
die Adenovirusvektoren, retroviralen Vektoren, AAV-Vektoren, Herpes-Vektoren
etc. nennen.
-
2. DIFFERENTIELLE KLONIERUNG
DER ALTERNATIVEN SPLEISSUNGEN UND ANDEREN QUALITATIVEN MODIFIKATIONEN
DER RNAs UNTER VERWENDUNG DER DOPPELSTRÄNGIGEN cDNAs (5).
-
Die
den normalen (mN) und pathologischen (mP) Situationen entsprechenden
Messenger-RNAs werden wie die komplementären doppelsträngigen DNAs
(dsN und dsP) nach klassischen molekularbiologischen Protokollen
hergestellt. Strukturen vom Typ „R-Schleife" werden nun durch
Hybridisierung von mN mit dsP und mP mit dsN in einer 70% Formamid-haltigen
Lösung
erhalten. Die zwischen der Situation N und P differentiell gespleißten Nucleinsäure-Domänen werden
in Form von doppelsträngiger
DNA bleiben. Die verdrängten
einzelsträngigen
cDNAs werden nun mit Glyoxal behandelt, um die Wiederanlagerung
des RNA-Stranges während
der Entfernung von Formamid zu vermeiden. Nach der Entfernung von
Formamid und Glyoxal, sowie anschließender Behandlung mit RNase
H finden wir Bienen-ähnliche
Strukturen vor, wobei die ungepaarten einzelsträngigen DNAs die Flügel der
Biene darstellen und die gepaarte doppelsträngige Domäne von Interesse den Bienenkörper darstellt.
Die Verwendung von Enzymen, die die einzelsträngige DNA wie die S1 Nuclease oder
die Mung Bean Nuclease spezifisch abbauen, erlaubt die Isolierung
von zurückbleibender
doppelsträngiger
DNA, die anschließend
kloniert und dann sequenziert wird. Diese zweite Technik erlaubt
die direkte Gewinnung eines Abdrucks von doppelsträngiger DNA
der Domäne
von Interesse im Vergleich zum ersten Protokoll, das einen RNA-Abdruck
dieser Domäne
liefert.
-
Dieser
Versuchsansatz ist mit einem zuvor beschriebenen grb2/grb33 Modell
durchgeführt
worden. Die doppelsträngige
DNA von grb2 ist mittels PCR-Amplifikation
aus der einzelsträngigen
cDNA von grb2 und zwei Nucleotid-Primern, die der Sequenz (1-22)
von grb2 und der komplementären
Sequenz (618-639) von grb2 entsprechen, hergestellt worden. Dieses
PCR-Fragment ist auf einem Agarosegel und auf einer Affinitätssäule (JetQuick,
Genomed) gereinigt und mittels spektrophotometrisch quantifiziert
worden. Zur selben Zeit sind zwei synthetische RNAs, die den Leserahmen
von grb2 und grb33 entsprechen, aus Plasmid-Vektoren, die die cDNAs
von grb2 und grb33 unter Kontrolle des T7-Promotors tragen, mit
Hilfe des RiboMax Kit (Promega) hergestellt worden. Die RNAs sind
nach Herstelleranleitung gereinigt und auf einer Auschluss-Säule (Sephadex
G50, 5 prime-3 prime) gereinigt worden. 600 ng doppelsträngige DNA
von grb2 (1-639) sind assoziiert mit:
- 1. 3 μg RNA von
grb33
- 2. 3 μg
RNA von grb2
- 3. Wasser
in drei verschiedenen Reaktionen in folgendem
Puffer: 100 mM PIPES (pH 7,2), 35 mM NaCl, 10 mM EDRA, 70% deionisiertes
Formamid (Sigma).
-
Die
Proben werden auf 56°C
erwärmt,
dann auf 44°C
in –0,2°C Stufen
alle 10 Minuten heruntergekühlt.
Dann werden sie bei 4°C
konserviert. Die Analyse auf Agarosegel zeigt Migrationsänderungen
in den Spuren 1 und 2 im Vergleich zur Kontrollspur 3 (11A), was die Bildung neuer Komplexe andeutet.
Die Proben werden dann mit deionisiertem Glyoxal (Sigma) (5% v/v
oder 1 M) 2 h lang bei 12°C
behandelt. Die Komplexe werden dann in 70% Ethanol gefällt, dann
in Wasser wieder aufgenommen. Sie werden schließlich mit RNase H (Life Technologies) behandelt,
dann mit einem Enzym behandelt, das spezifisch die einzelsträngige DNA
abbauen kann. Die S1-Nuclease und die "Mung bean" Nuclease weisen diese Eigenschaft auf
und sind im Handel erhältlich
(Life Technologies, Amersham). Derartige Verdaue (5 Minuten Inkubation
in Puffer vom Hersteller mit diesen Enzymen) sind auf Agarosegel
analysiert worden (11B). Signifikante Verdaue sind nur
von den Komplexen erhalten worden, die von der Reaktion 1 (grb2/grb33)
stammen (11B, Spuren 7 und 10). Diese
Verdaue scheinen mit der S1-Nuclease
(Spur 7) vollständiger
zu sein als mit der „Mung
bean" Nuclease (Spur
10). Daher ist die Bande, die einer Größe von etwas mehr als 100 Basenpaaren
entspricht (Pfeil in Spur 7 zeigt darauf) gereinigt, in den pMos-Blue
Vektor (Amersham) kloniert, dann sequenziert worden. Dieses Fragment
entspricht der Domäne
mit 120 Basenpaaren von grb2, das in grb33 deletiert ist.
-
Dieser
Versuchsansatz kann nun mit einer Messenger-RNA-Gesamtpopulation
und einer doppelsträngigen
cDNA-Gesamtpopulation gemäß dem Fachmann
bekannten Techniken durchgeführt
werden. Die RNA-Population der Referenzsituation wird an die doppelsträngige cDNA-Population
der Testsituation und umgekehrt hybridisiert. Nach Anwendung des
oben beschriebenen Protokolls werden die Verdaue auf ein Agarosegel
aufgetragen, um die Banden zu isolieren und zu reinigen, deren Größe zwischen
50 und 300 Basenpaaren entspricht. Diese Banden werden dann in einen
Vektor (pMos-Blue Vektor, Amersham) kloniert, um eine Bank von Inserts
zu schaffen, die mit Ereignissen von qualitativen Unterschieden
angereichert ist.
-
3. KONSTRUKTION VON BANKEN,
DIE VON QUALITATIVEN DIFFERENTIELLEN SCREENINGS ABSTAMMEN
-
Die
beiden oben beschrieben Beispiele führen zu Klonierungen von cDNAs,
die für
die ganze oder einen Teil der Sequenzen repräsentativ sind, die zwischen
zwei bestimmten physiopathologischen Situationen differentiell gespleißt sind.
Diese cDNAs erlauben die Bildung von Banken durch Insertion dieser
cDNAs in Plasmid- oder
Phagenvektoren. Diese Banken können
auf Nitrocellulose-Filtern oder jedem anderen Träger, der dem Fachmann bekannt
ist, wie Chips oder Biochips oder Membranen präsentiert sein. Diese Banken
können
in der Kälte
lichtgeschützt
konserviert sein. Diese Banken, wenn sie einmal auf einem Träger mit
Hilfe klassischer Techniken angelagert und fixiert sind, können mit
Verbindungen behandelt werden, um die Wirtsbakterien zu eliminieren,
die die Produktion der Plasmide oder Phagen erlauben. Diese Banken
können
ebenfalls vorteilhaft aus cDNA-Fragmenten gebildet sein, die den
klonierten cDNAs entsprechen, aber so mit PCR hergestellt sind,
dass nur die Sequenzen auf dem Filter anlagern, die von alternativen
Spleißereignissen
stammen.
-
Ein
Merkmal und gleichzeitig das Neue des qualitativen differentiellen
Screenings ist, dass diese Technik vorteilhafterweise nicht nur
zu einer sondern zu zwei differentiellen Banken („Banken-Paar") führt, die
die Gesamtheit der qualitativen Unterschiede repräsentieren,
die zwischen zwei bestimmten Situationen vorliegen. Insbesondere
repräsentiert
eine der Banken die Signatur der Merkmale der physiologischen Testsituation im
Vergleich zur physiologischen Referenzsituation, und die andere
Bank repräsentiert
die Signatur der Merkmale der physiologischen Referenzsituation
im Vergleich zur physiologischen Testsituation. Dieses Banken-Paar wird ebenfalls
als Paar von Banken oder „differentielle
Bank von Spleißungen„ bezeichnet.
-
Da
einer der Beiträge
des qualitativen differentiellen Screenings ist, die Bewertung des
toxischen Potenzials einer Verbindung zu erlauben, wie es im folgenden
Kapitel angeführt
ist, ist ein gutes Beispiel für
die Anwendung der Technologie die Gewinnung von cDNA-Klonen mit
Hilfe von DATAS, die Sequenzen entsprechen, die einerseits für naive
HepG2-Zellen und andererseits für
Ethanolbehandelte Zellen spezifisch sind. Diese Zellen zeigen Anzeichen
einer Cytotoxizität
und eines Abbaus ihrer DNA durch intranukleosomale Fragmentierung
ab 18 h in Gegenwart von 1 M Ethanol. Um frühe Marker der Ethanol-Toxizität zu erhalten,
werden die Messenger-RNAs aus naiven Zellen und 18 h mit 0,1 M konzentriertem
Ethanol behandelten Zellen präpariert.
Nach Durchführung
der DATAS-Variante, die die einzelsträngige cDNA und die RNase H
verwendet, sind entsprechend dem Fachmann bekannten Techniken die
erhaltenen klonierten cDNAs mit PCR amplifiziert, einer Elektrophorese
auf Agarosegel unterzogen und dann auf einen Nylon-Filter transferiert
worden. Jede Gesamtheit von Klonen, die einerseits für qualitativen
Unterschiede, die für
den nativen Zustand spezifisch sind, und andererseits für Sequenzen
spezifisch sind, die für
Ethanol-behandelte Zellen spezifisch sind, werden zwei identische
Repliken auf Filtern durchgeführt.
Daher werden die Abdrücke
von jeder Gesamtheit von Klonen einerseits mit einer Sonde, die
für nicht
behandelte Zellen spezifisch ist, und andererseits mit einer Sonde hybridisiert,
die für
mit 0,1 M Ethanol 18 h behandelte Zellen spezifisch ist.
-
Das
erhaltene und in 12 gezeigte differentielle Hybridisierungsmuster
erlaubt die Qualität
der durchgeführten
Subtraktion bei der Durchführung
der DATAS-Technik zu bewerten. Daher hybridisieren die Klone, die
von der Hybridisierung von der mRNA von nicht behandelten Zellen
(NT) mit cDNA von behandelten Zellen (Tr) stammen und die qualitativen
Unterschieden entsprechen müssen,
die für
die naive Situation spezifisch sind, bevorzugt mit einer Sonde,
die die Gesamtpopulation der Messenger-RNAs der nicht behandelten Zellen
repräsentiert.
Umgekehrt hybridisieren die Klone, die von Produkten, die einer
Behandlung mit RNase H widerstehen, mit der die (Tr)RNA/(NT)DNA-Heterodulices
behandelt wurden, bevorzugt mit einer Sonde, die von der Gesamtpopulation
der Messenger-RNAs der behandelten Zellen abstammt.
-
Die
beiden Gruppen von Klonen, die einerseits für die behandelte Situation
und andererseits für
die nicht behandelte Situation spezifisch sind, repräsentieren
zum Beispiel eine Bank von qualitativen Unterschieden, die für beide
verschiedene zelluläre
Zustände
charakteristisch sind.
-
4. VERWENDUNGEN UND BEITRÄGE DER QUALITATIVEN
DIFFERENTIELLEN BANKEN.
-
Die
Einsatzmöglichkeiten
der Banken von differentiellen Spleißungen sind insbesondere in
den 13 bis 15 veranschaulicht.
Daher können
die Banken verwendet sein für:
-
4.1. Die Bewertung des
toxischen Porenzials einer Verbindung (13):
-
In
diesem Beispiel wird die Referenzsituation als A bezeichnet und
die toxische Situation als B bezeichnet. Nomogramme der Toxizität werden
durch Behandlung der Situation A in Gegenwart von verschiedener
Konzentrationen einer toxischen Verbindung als Referenz innerhalb
variabler Zeiträume
erhalten. An verschiedenen Punkten der Nomogramme der Toxizität werden
qualitative differentielle Banken (Banken-Paare), in diesem Beispiel
rA/cB und rB/cA beschränkte
Banken, gebildet. Die Banken-Paare werden vorzugsweise auf einem
Träger
angebracht. Der Träger
wird dann mit Sonden hybridisiert, die von Proben stammen, die zunächst mit verschiedenen
Dosen der Testverbindung behandelten werden: Produkte X, Y und Z.
Die Hybridisierung wird durchgeführt
und macht das toxische Potenzial der Testprodukte sichtbar: in diesem
Beispiel zeigt das Produkt Z eine starke Toxizität und das Produkt Y stellt
ein mittleres Profil dar. Die Durchführbarkeit dieser Erstellung
von Nomogrammen für
die Toxizität
ist mit dem Beispiel der weiter oben beschriebenen Bildung von Banken
des qualitativen differentiellen Screenings gut veranschaulicht,
wo Ethanol und Hep2G-Zellen eingesetzt sind
-
4.2. Die Bewertung der
Wirksamkeit einer pharmazeutischen Zusammensetzung (14):
-
In
diesem Beispiel wird ein beschränktes
Banken-Paar gemäß der Erfindung
aus einem pathologischen Modell B und einem gesunden Modell A (oder
aus einem pathologischen Modell, das mit einem Wirkstoff als Referenz
behandelt ist) hergestellt. Die differentiellen Banken rA/cB und
rB/cA sind gegebenenfalls auf einem Träger angebracht. Dieses Banken-Paar
fasst die Spleißunterschiede
zwischen den beiden Situationen zusammen. Dieses Banken-Paar erlaubt
die Bewertung der Wirksamkeit einer Testverbindung, nämlich deren Fähigkeit
zu bestimmen, ein Muster „gesund" (rA/cB) aus dem
pathologischen Profil (rB/cA) zu erzeugen. In diesem Beispiel wird
das Banken-Paar mit Sonden hybridisiert, die von den Situationen
A und B mit und ohne Behandlung der Testverbindung hergestellt sind.
Das Hybridisierungsmuster, das erhalten werden kann, ist in 14 dargestellt. Die Durchführbarkeit dieser Anwendung
ist dieselbe wie die bei der weiter oben gezeigten Bildung der Banken
für qualitative
Unterschiede, die für
gesunde und toxische Situationen charakteristisch sind. Die toxische
Situation wird durch den pathologischen Zustand ersetzt und es ist
möglich,
die Fähigkeit einer
Testverbindung abzuschätzen,
eine Sonde zu produzieren, die vorzugsweise mehr oder weniger mit
der Referenzsituation oder der pathologischen Situation hybridisiert.
-
4.3. Die Vorhersage der
Antwort einer biologischen Probe auf eine Behandlung (15):
-
In
diesem Beispiel wird ein beschränktes
Banken-Paar gemäß der Erfindung
aus zwei pathologischen Modellen hergestellt, wo das eine auf eine
Behandlung mit einem bestimmten Produkt antwortet (zum Beispiel Wildtyp-p53):
Situation A; und das andere Modell hierfür resistent ist: Situation
B. Dieses Banken-Paar (rA/cB; rB/cA) ist auf einem Träger angebracht.
-
Dieses
Banken-Paar wird anschließend
zur Bestimmung der Empfindlichkeit einer biologischen Testprobe
auf dasselbe Produkt verwendet. Hierfür wird dieses Banken-Paar mit
Sonden hybridisiert, die von Biopsien von Patienten stammen, deren
Antwort auf die Referenzbehandlung man vorhersagen möchte. Das Hybridisierungsmuster
einer Biopsie eines reagierenden Patienten und einer Biopsie eines
nichtreagierenden Patienten ist in 15 dargestellt.
-
4.4. Die Identifizierung
von Liganden für
Orphan-Rezeptoren
-
Die
Aktivierung von Membran-oder Kernrezeptoren durch ihre Liganden
könnte
Fehlregulationen in der Spleißung
bestimmter RNAs spezifisch induzieren. Die Identifizierung dieser
Ereignisse mit den DATAS-Verfahren der Erfindung erlaubt, über ein
Werkzeug zu verfügen
(Marker, Banken, Kits, etc.), um die Aktivierung von Rezeptoren
zu verfolgen, die für
die Suche von natürlichen
oder synthetischen Liganden von Rezeptoren insbesondere Orphan-Rezeptoren
zweckdienlich sind. Gemäß dieser
Anwendung werden mit den Fehlregulationen verbundene Marker identifiziert
und auf Trägern
angebracht. Die Gesamt-RNA von Zellen, die den zu untersuchenden
Rezeptor (über)exprimieren
und mit verschiedenen Zusammensetzungen und/oder Testverbindungen
behandelt oder nichtbehandelt sind, wird extrahiert und als Sonde
in einer Hybridisierung mit den Trägern verwendet. Das Aufzeigen
einer Hybridisierung mit bestimmten, sogar mit der Gesamtheit der
auf dem Träger
angebrachten Marker weist darauf hin, dass der zu untersuchende
Rezeptor aktiviert worden ist und folglich dass die entsprechende
Zusammensetzung/Verbindung einen Liganden besagten Rezeptors bildet
oder umfasst.
-
4.5. Die Identifizierung
von therapeutischen Zielen von Interesse:
-
Dies
erfolgt durch die Identifizierung von Gene; deren Spleißung bei
einer Erkrankung oder in einem pathologischen Modell modifiziert
ist, und genauer durch die Identifizierung der modifizierten Exons
und Introns. Dieser Versuchsansatz muss einen Zugang zu Sequenzen
erlauben, die die funktionellen Domänen codieren, die bei Erkrankungen
oder jedem physiopathologischen Phänomen gestört sind, die zum Beispiel Phänomene der
Proliferation, Differenzierung oder Apoptose betreffen.
-
Ein
Beispiel für
den Beitrag des qualitativen differentiellen Screenings für die Identifizierung
von differentiell gespleißten
Genen wird von der DATAS-Anwendung bei einem Modell der Apoptoseinduktion
durch die Expression von Wildtyp-p53 geliefert. Dieses Zellmodell
ist durch Transfektion eines für
das Tumorsuppressorgen p53 induzierbaren Expressionssystems etabliert
worden. Um die qualitativen Unterschiede zu identifizieren, die
mit der von p53 induzierten Apoptose spezifisch verbunden sind,
ist DATAS mit den Messenger-RNA-Extrakten der induzierten und nicht
induzierten Zellen durchgeführt
worden. Für
diese Versuche sind 200 ng polyA+ RNA und 200 ng cDNA für die Bildung
der Heteroduplices verwendet worden. Einhundert Klone sind aus jeder
Kreuzhybridisierung erhalten worden. Die Hybridisierung dieser bakteriellen
Klone, dann der cDNA-Fragmente, die sie enthalten, mit Sonden, die
für Gesamt-Messenger-RNA
der Anfangssituationen repräsentativ
sind hat die Identifizierung von Sequenzen erlaubt, die bei der
starken Induktion von p53 spezifisch exprimiert sind, die zum Zelltod
führt (16).
-
Diese
Fragmente stammen von Exon- oder Intronsequenzen, die die Eigenschaft
der vorliegenden Botschaft modulieren und erlauben, die funktionellen
Domänen
vorzuschlagen, an denen sie beteiligt sind oder die sie als Interventionsziele
unterbrechen, um den Zelltod zu induzieren oder zu inhibieren.
-
Ein
derartiger Versuchsansatz führt
ebenfalls zur Bildung eines Banken-Paars, das differentiell gespleißte Ereignisse
zwischen einer Nichtapoptosesituation und einer Apoptosesituation
sammelt. Dieses Banken-Paar kann ebenfalls zum Testen der Hybridisierungsleistung
einer Sonde, die von einer anderen physiopathologischen Situation
oder einer besonderen Behandlung stammt, verwendet sein. Das Ergebnis
einer derartigen Hybridisierung wird Hinweise auf eine mögliche Beteiligung
des Genexpressionsprogramms der getesteten Situation versus Apoptosesituation
liefern.
-
Wie
es aus der vorhergehenden Beschreibung hervorgeht, betrifft die
Erfindung ebenfalls:
- – jede Nucleinsonde, jedes
Oligonucleotid, jeden Antikörper,
der gegen eine Sequenz gerichtet ist, die mit der in der vorliegenden
Anmeldung beschriebenen Technik identifiziert ist, und dadurch gekennzeichnet sind,
dass sie eine Charakterisierung einer pathologischen Situation erlauben,
- – die
Verwendung der Informationen, die aus der Anwendung der beschriebenen
Techniken gewonnen sind, für
die Suche nach organischen Molekülen
für therapeutische
Zwecke durch die Bereitstellung von Screenings, die dadurch gekennzeichnet
sind, dass sie auf die zwischen einer gesunden Situation und einer
pathologischen Situation differentiell gespleißten Domänen abzielen, oder dadurch
gut gekennzeichnet sind, dass sie auf der Hemmung der von dem Protein
erworbenen Funktionsgewinne beruhen, das aus einer differentiellen
Spleißung
resultiert.
- – die
Verwendung der Informationen, die aus den in der vorliegenden Anmeldung
beschriebenen Techniken gewonnen sind, für gentherapeutische Anwendungen,
- – die
Verwendung von cDNAs, die mit Hilfe der Gentherapie transferiert
und dadurch gekennzeichnet sind, dass sie antagonistische oder agonistische
Eigenschaften auf definierte zelluläre Signalisationswege besitzen,
- – jede
Bildung und jede Verwendung molekularer Banken von alternativen
Exons oder Introns zum Zwecke:
der Diagnose oder für kommerzielle
Reagenzien für
die Forschung
der Bildung oder Suche von Molekülen, Polypeptiden,
Nucleinsäuren
für eine
therapeutische Anwendung,
- – jede
Bildung und jede Verwendung von virtuellen Datenbanken, die alternative
Exons und Introns zusammenfassen, dadurch gekennzeichnet, dass diese
Banken erlauben, Nucleinsonden oder Oligonucleotid-Primer mit dem
Ziel zu erhalten, die alternativen Spleißungen zu charakterisieren,
die zwei verschiedene physiopathologische Zustände unterscheiden,
- – jede
pharmazeutische oder diagnostische Zusammensetzung, umfassend Polypeptide,
Sense- oder Antisense-Nucleinsäuren
oder chemische Moleküle,
die in der Lage sind, mit alternativen Spleißprodukten zu interferieren,
die mit den Techniken der Erfindung aufgezeigt und kloniert werden.
- – jede
pharmazeutische oder diagnostische Zusammensetzung, umfassend Polypeptide,
Sense- oder Antisense-Nucleinsäuren
oder chemische Moleküle,
die in der Lage sind, eine Spleißung wiederherzustellen, die
für eine
normale Situation repräsentativ
ist, im Gegensatz zu dem alternativen Ereignis, das für eine pathologische
Situation charakteristisch ist.
-
5. DEREGULATIONEN
DER SPLEISSMECHANISMEN DER RNAs MIT TOXISCHEN AGENTIEN
-
Dieses
Beispiel zeigt, dass die Unterschiede von Spleißformen und/oder Spleißmustern
als Marker für das
Verfolgen und/oder die Detektion der Toxizität und/oder der Wirksamkeit
von Verbindungen verwendet sein kann.
-
Die
Wirkungen toxischer Agentien auf die Deregulationen von Spleißungen der
RNAs sind folgendermaßen
getestet worden. HepG2-Hepatocyten sind mit verschiedenen Dosen
dreier toxischer Verbindungen (Ethanol, Camptothecin, PMA (Phorbol
12-Myristat 13-Acetat)) behandelt worden. Zwei Cytotoxizitätstest (Trypan-Blau, MTT) sind mit
verschiedenen Zeiten durchgeführt
worden: 4 h und 18 h mit Ethanol; 4 h und 18 h mit Camptothecin;
18 h und 40 mit PMA.
-
Das
Trypan-Blau ist ein Farbstoff, der von lebenden Zellen inkorporiert
wird. Ein einfacher Vergleich der „blauen" und „weißen" Zellen unter dem Mikroskop erlaubt
den Prozentsatz lebender Zellen nach Behandlung oder die Überlebensrate
zu bestimmen. Die Punkte sind dreifach bestimmt.
-
Der
MTT-Test ist ein kolorimetrischer Test, der die Fähigkeit
lebender Zellen misst, die löslichen
Tetrazoliumsalze (MTT) in einen unlöslichen Formazan-Niederschlag umzuwandeln.
Diese dunkelblauen Formazan-Kristalle können gelöst werden und ihre Konzentration
durch Absorptionsmessungen bei 550 nm bestimmt werden. Daher werden
nach Beimpfen der Platten mit 24 Vertiefungen mit je 150000 Zellen über Nacht,
dann Behandlung der Zellen mit den toxischen Verbindungen werden
50 μl MTT
(Sigma) (in 5 mg/ml in PBS) zugegeben. Die Bildungsreaktion von
Formazan-Kristallen vollzieht sich in 5 h in einem CO2-Inkubator
(37°C, 5% CO2, 95% Luftfeuchtigkeit). Nach Zugabe von
500 μl Solubilisierungslösung (0,1
N HCl in Isopropanol-Triton X-100 (10%)) werden die Kristalle unter
Rühren
gelöst
und die Absorption bei 550 bis 660 nm gemessen. Die Punkte sind
dreifach mit den geeigneten Kontrollen (Viabilität, Zelltod, Kontrolle) gemessen
worden.
-
Ein
Test der Apoptose oder des programmierten Zelltods ist ebenfalls
durch Messen der DNA-Fragmentierung unter Verwendung von anti-Histon
Antikörpern
und ELISA-Messungen durchgeführt
worden. Der angewandte Test ist der Cell Death ELISA Plus von Roche.
-
Die
Ergebnisse dreier Tests (18A,
B, C) haben die Bestimmung folgender Dosen erlaubt:
- • Ethanol:
0,1 M
- • Camptothecin:
1 μg/ml
- • PMA:
50 ng/ml
die viel niedriger waren als die gemessenen IC50
-
Die
HepG2-Zellen sind daher mit diesen drei Verbindungen mit drei Dosen
4 h mit Ethanol und Camptothecin und 18 h mit PMA behandelt worden.
Die Messenger-RNAs
sind mit Dynal-Oligo-(dT) Kügelchen
aus mit dem Rneasy Kit (Qiagen) gereinigten Gesamt-RNAs aufgereinigt
worden. cDNA-Synthesen sind aus diesen Messenger-RNAs und mit der
inversen Transkriptase Superscript (Life Technologies) unter Verwendung von
zufälligen
Hexameren als Primer durchgeführt
worden.
-
Die
ersten Stränge
haben als Matrizen für
die Amplifikationsreaktionen mittels PCR (94°C 1 min 55°C 1 min, 72°C 1 min, 30 Cyclen) mit Hilfe
folgender Oligonucleotid-Primer gedient:
-
MACH-α:
-
- 5'-TGCCCAAATCAACAAGAGC-3' (SEQ ID NR: 11)
- 5'-CCCCTGACAAGCCTGAATA-3' (SEQ ID NR: 12)
-
Diese
Primer entsprechen Regionen, die verschiedenen beschriebenen Isoformen
von MACH-α gemeinsam
sind (1, 2 und 3, die jeweils 595, 550 und 343 Basenpaare amplifizieren).
MACH-α (Caspase-8)
ist eine am programmierten Zelltod beteiligte Protease (Boldin et
al., Cell (1996), 85, 803-815).
-
BCL-X
-
- 5'-ATGTCTCAGAGCAACCGGGAGCTG-3' (SEQ ID NR: 13)
- 5'-GTGGCTCCATTCACCGCGGGGCTG-3' (SEQ ID NR: 14)
-
Diese
Primer entsprechen Regionen, die verschiedenen beschriebenen Isoformen
von bcl-x (bcl-XI, bcl-Xs, BCL-Xβ)
gemeinsam sind (Boise et al., Cell (1993) 74, 597-608; U72398 (Genbank))
und ein einziges Fragment für
diese drei Isoformen von 204 Basenpaaren amplifizieren müssen.
-
FASR
-
- 5'-TGCCAAGAAGGGAAGGAGT-3' (SEQ ID NR: 15)
- 5'-TGTCATGACTCCAGCAATAG-3' (SEQ ID NR: 16)
-
Diese
Primer entsprechen Regionen, die bestimmten Isoformen von FASR gemeinsam
sind, und ein Fragment von 478 Basenpaaren für die Wildform von FASR, 452
für die Δ8-Isoform
und 415 für
die ΔTM-Isoform
amplifizieren müssen.
-
Die
Ergebnisse mit Bezug auf 19 zeigen,
dass:
- • Camptothecin
eine Verringerung der Expression der MACH-α1 Isoform und eine Erhöhung de
MACH-α3 Isoform
induziert.
- • Camptothecin
das Auftreten einer neuen Isoform von bcl-X induziert (obere Bande
des Doublets, das in Richtung 200 Basenpaare migriert).
- • Camptothecin
eine Verringerung der Wildform des fas-Rezeptors induziert, die
durch eine Expression der kürzeren
Isoform ersetzt ist, die Fas ΔTM
entsprechen kann.
- • Ethanol
das Verschwinden von bcl-X induziert, das durch eine kürzere Isoform
ersetzt ist.
- • Ethanol
eine Vermehrung der langen, Wildform des fas-Rezeptors auf Kosten
der kürzeren
Isoform induziert.
-
Diese
Ergebnisse zeigen, dass Behandlungen mit toxischen Agentien mit
geringen Dosen Fehlregulationen von alternativen Spleißungen von
bestimmten RNAs auf spezifische Weise induzieren können. Die Identifizierung
dieser Fehlregulationen auf posttranskriptionaler Ebene, insbesondere
durch Anwendung der DATAS-Technologie, erlaubt daher ein Instrument
zur Vorhersage der Toxizität
von Molekülen
zu definieren.
-
-
-
-
-
-