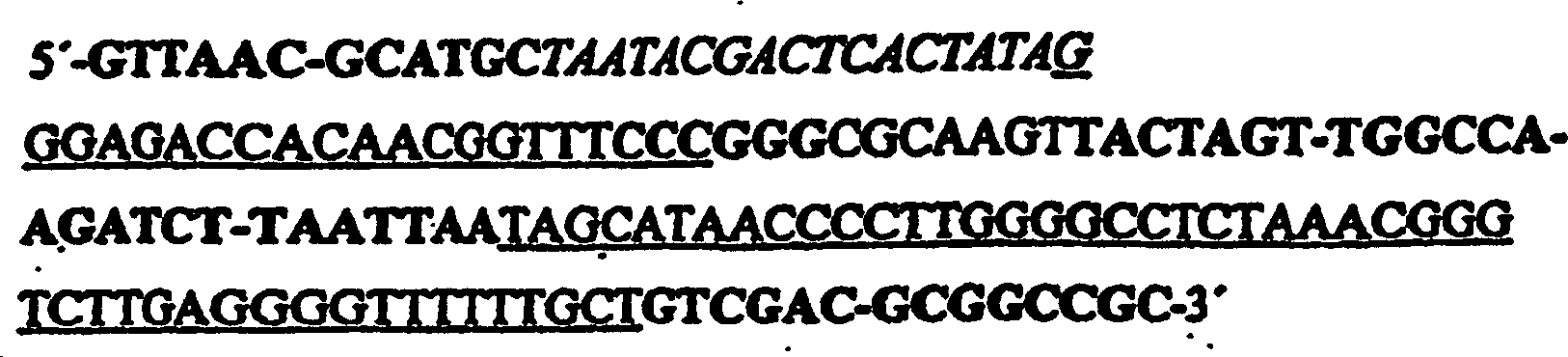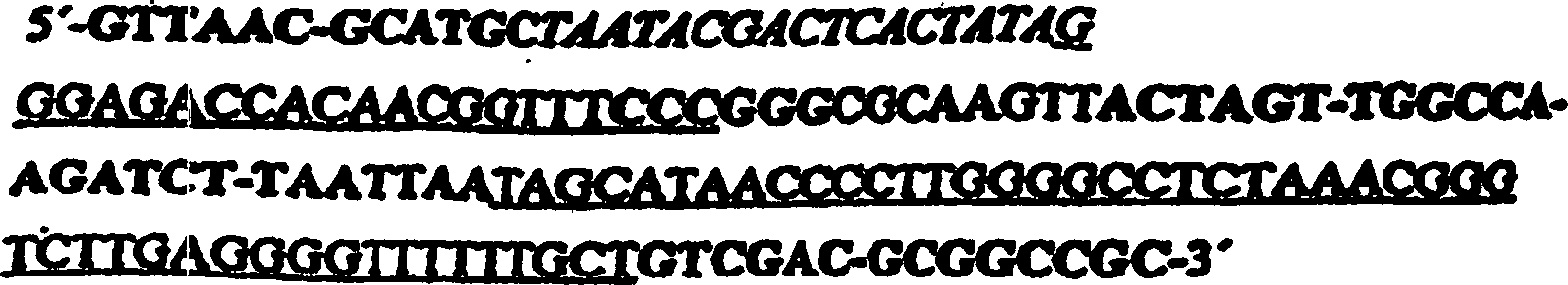-
Die
vorliegende Erfindung betrifft ein Verfahren zum Identifizieren
eines Intramers, das in der Lage ist, an ein funktionales intrazelluläres Zielmolekül zu binden
und dessen Funktion zu modifizieren, und die intrazelluläre Anwendung
von funktionalen Nukleinsäuren,
die als „Intramere" bezeichnet werden,
die die biologische Funktion einer intrazellulären Komponente beeinflussen
(z. B. ihre Funktion innerhalb des Kontextes einer lebenden Zelle
inhibieren, indem die Komponente spezifisch komplexiert wird). Es
konnte gezeigt werden, dass a) Intramere innerhalb von Zellen in
einen funktionellen Kontext plaziert werden können, unabhängig davon, ob das Zielmolekül natürlicherweise
Nukleinsäuren
bindet oder nicht; b) Intramere Wirkungen auf intrazelluläre Stellen
vermitteln können,
an denen eine Nukleinsäure
normalerweise nicht gefunden wird. Dieses Verfahren ist nützlich,
um die Aufklärung
der biologischen Rolle einer großen Vielzahl von intrazellulären Komponenten
zu ermöglichen.
Die vorliegende Erfindung betrifft weiterhin Verfahren für die Expression
von randomisierten Nukleinsäurebibliotheken
innerhalb von Zellen, um funktionale Intramere zu identifizieren,
die den Phänotyp
der Zelle ändern,
in der sie exprimiert werden. Dieses Verfahren ist, z. B., dafür nützlich,
ein intrazelluläres
Zielmolekül
als für
einen bestimmten zellulären
Phänotyp
funktional verantwortlich oder daran beteiligt zu identifizieren,
ohne dass vorher die biologische Funktion der Komponente bekannt
ist. Schließlich
betrifft die vorliegende Erfindung eine T7-RNA-Expressionskassette,
die einen T7-Promotor umfasst.
-
In
den vergangenen Jahren ist ein erheblicher Fortschritt bei der Identifizierung
des vollständigen
Satzes der genetischen Information verschiedener Organismen erzielt
worden. Das Genom von Prokaryonten, wie beispielsweise E. coli,
oder Eukaryonten, wie beispielsweise S. cerevisiae (Goffeau et al.,
Science 274 (1996), 546–567)
und C. elegans (The C. Elegans Sequencing Consortium, Science 282
(1998); 2012–2018) ist
vollständig
sequenziert worden. Man hat geschätzt, dass das vollständige humane
Genom in drei bis fünf Jahren
bekannt sein wird. Die Herausforderung, der man sich nach diesen
Erfolgen gegenübersieht,
besteht darin, ein jedes Gen mit einer Funktion zu versehen. Zum
Beispiel besteht ein wesentliches Ziel darin, jene Genprodukte als
potentielle Wirkstoffzielmoleküle
zu identifizieren, die Schlüsselrollen
in dem komplexen Netzwerk aus Proteinwechselwirkungen spielen, die
letztendlich zu Erkrankungen führen
(siehe z. B. Friedrich, Nat. Biotechnol. 14 (1996), 1234-7). Um
diese Schlüsselmoleküle unter
den 100 000 humanen Genen zu identifizieren, wird die Anzahl möglicher
Kandidatenmoleküle
definiert, indem die Proteinzusammensetzung verschiedener Entwicklungszustände oder
Erkrankungszustände
einer Zelle durch differenzielle Genexpression verglichen wird.
Der tatsächliche
Status der Proteinzusammensetzung einer Zelle kann erhalten werden,
indem die mRNA einer Zelle analysiert wird (für Übersichten der verschiedenen
Verfahren siehe Wan et al., Nat. Biotechnol. 14 (1996), 1685–1691).
Dieses Verfahren liefert jedoch nur ein indirektes und nicht sehr
genaues Maß für den gegenwärtigen Proteomstatus
der Zelle, da die mRNA schon lange abgebaut sein kann, während das
Protein noch vorhanden ist, oder eine große Menge an mRNA transkribiert
worden ist, aber aus irgendwelchen Gründen nicht translatiert werden
kann. Es ist auch nicht möglich,
posttranslationale Modifikationen der exprimierten Proteine abzudecken.
Ein alternativer Weg, um den Proteomstatus einer Zelle vermittels „proteomics" abzudecken, besteht
in der 2D-Gelelektrophorese.
Unter Verwendung dieses Verfahrens kann man versuchen, die Zusammensetzung
aller exprimierten Proteine in einer Zelle zu einem gegebenen Zeitpunkt
in der Form von getrennten Spots auf einem Gel abzudecken, wobei
jeder Spot einem einzelnen Protein entspricht. Indem verschiedene
Gele verglichen werden, können
verschiedene Proteine oder Mengen an Proteinen identifiziert werden.
Dieses Verfahren erlaubt jedoch derzeit bestenfalls, etwa 20 % der
exprimierten Proteine in einem höheren
Eukaryonten zu identifizieren. Der Nachweis von Proteinen mit einer
geringen Kopienzahl ist dabei problematisch. Zusätzlich erlauben beide Verfahren
lediglich, die Anwesenheit oder Abwesenheit von Genprodukten in
einem speziellen Entwicklungszustand oder Erkrankungszustand zu
betrachten. Sie erlauben nicht, festzustellen, ob oder ob nicht
die identifizierten Unterschiede in dem Expressionsmuster den zellulären Status
verursachen oder nur eine Konsequenz daraus sind.
-
Die
direkte Analyse der Funktion, die ein bestimmter Proteinkomplex
oder eine seiner Untereinheiten oder Domänen in einem zellulären Prozess
aufweist, kann vermittels des genetischen „Knock-out" (homologe Rekombination, Antisense-Technologien),
oder vermittels Überexpression
oder Mutation des Proteins erreicht werden. Dies führt jedoch
immer zu einer Änderung
der genetischen Information eines Organismus, was die Interpretation
der Ergebnisse schwierig macht. Z. B. erlaubt der Knock-out eines
Gens nicht, Rückschlüsse zu ziehen, welcher
Teil oder welche Domäne
eines Proteins für
seine Funktion wichtig ist. Zusätzlich
ist die Expression von anderen Genen in den meisten Fällen beeinflusst
(für eine Übersicht
siehe: Proteom Research: New Functions in Functional Genomics, Springer-Verlag
(1997), 1–30).
-
Es
besteht somit ein großer
Bedarf für
spezifische intrazelluläre
Inhibitoren oder Modulatoren, die in einem bestimmten Zeitfenster
angewandt werden können
und die die Analyse eines bestimmten unveränderten Proteins innerhalb
seines natürlichen
Expressionsstatus erlauben [Spencer et al., Science 262 (1993), 1019–1024; Huang & Schreiber, Proc.
Natl. Acad. Sci. USA 94 (1997), 13396–13401]. Die meisten der derzeit bekannten
Inhibitoren oder Modulatoren basieren auf membrangängigen kleinen
organischen Molekülen,
die oft eine beschränkte
Spezifität
aufweisen und nur für
bestimmte Proteine verfügbar
sind. Diese Beschränkung kann überwunden
werden durch intrazelluläre
Antikörper
(„Intrabodies"; siehe Richardson,
Tibtech 13 (1995), 306–10)
oder Peptidaptamere (siehe Colas et al., Nature 380 (1996) 548 –550). Das
Problem mit intrazellulären
Antikörpern
besteht jedoch darin, dass, beispielsweise, der extrazelluläre Antikörper auf
das reduktive intrazelluläre
Kompartiment angepasst werden muss. Verfahren, wie beispielsweise
Dimerisierung von schwerer und leichter Kette und die Stabilisierung
durch Disulfidbrücken
oder Glykosylierung erfolgen intrazellulär mit einer sehr geringen Effizienz
oder gar nicht und müssen
durch kostenintensive Konstruktion der Polypeptidketten ausgeglichen
werden.
-
Funktionale
Nukleinsäuren,
die innerhalb des Kontextes einer lebenden Zelle funktionieren,
können dabei
helfen, diesen Bedarf für
spezifische intrazelluläre
Inhibitoren oder Modulatoren zu befriedigen, die in einem bestimmten
Zeitfenster angewandt werden können
und die die Analyse eines bestimmten nicht veränderten Proteins innerhalb
seines natürlichen
Expressionsstatus erlauben.
-
Funktionale
Nukleinsäuren
sind eine einzelsträngige
DNA (ssDNA) oder RNA (ssRNA) oder chemisch modifizierte Nukleinsäuren (ssNAmod),
doppelsträngige
DNA (dsDNA) oder RNA (dsRNA) oder chemisch modifizierte Formen davon
(dsNAmod), die in der Lage sind, ein intrazelluläres Zielmolekül zu binden,
zu modulieren oder katalytisch zu modifizieren, wodurch seine biologische
Funktion beeinflusst wird. Eine funktionale Nukleinsäure kann
beispielsweise durch in vitro-Selektion oder „SELEX" (Systematic Evolution of Ligands by Exponential
Enrichment; siehe Tuerk und Gold, Science 249 (1990), 505–510; Ellington
und Szostak, Nature 346 (1990), 818–822) oder durch Ribozyme,
die allosterisch aktiviert wer den können (Aptazyme; siehe Robertson
und Ellington, Nat. Biotechnol. 17, (1999), 62–66) identifiziert werden.
Es ist wohl bekannt, dass funktionale Nukleinsäuren, beispielsweise Aptamere,
die durch in vitro-Selektion erzeugt wurden, an eine große Vielzahl
von Liganden binden können,
die von kleinen Molekülen
und biologischen Cofaktoren bis hin zu natürlichen und synthetischen Polymeren
reichen, einschließlich
Proteinen, Polysacchariden, Glykoproteinen, Hormonen, Rezeptoren
und Zelloberflächen.
Im Allgemeinen können
Nukleinsäuren
in Zellen eingeführt
werden durch Verfahren, die den Fachleuten üblicherweise bekannt sind,
wie beispielsweise Lipofektion, Elektroporation oder vermittels
Vektoren, die hohe Transkriptionsraten erlauben, z. B. Plasmide
mit RNA-Polymerase III-Promotoren, T7-RNA-Polymerase/Vakzinia-Virus-basierten
Systemen oder Replikation, z. B. Semliki-Forst-Virus.
-
Beispiele
für kleine,
intrazellulär
verabreichte Nukleinsäuren
sind bisher auf Antisense-Nukleinsäuren, katalytische
Antisense-Nukleinsäuren,
wie beispielsweise Hammerhead-Ribozyme
(Birikh et al., Eur. J. Biochem. 245 (1997), 1–16; Bramlage et al., Trends
Biotechnol. 16 (1998), 434–438)
und Nukleinsäureliganden beschränkt, die
vermittels eines Aptamermechanismus an bestimmte Zielmoleküle binden,
die auf natürlicherweise
Nukleinsäure
bindende Proteine beschränkt
sind, wie beispielsweise das Rev-Protein von HIV-1 (Good et al.,
Gene Ther. 4 (1997), 45–54;
Symensma et al., J. Virol. 70 (1996), 179–187), RNA-Polymerase II von Hefe
(Thomas et al., J. Biol. Chem. 272 (1997), 27980–27986), das SelB-Protein von
E. coli (Klug et al., Proc. Natl. Acad. Sci. USA 94 (1997), 6676–6681).
Diese Beispiele erfüllen
jedoch nicht die Forderung nach spezifischen intrazellulären Inhibitoren
oder Modulatoren, die routinemäßig in bestimmten
Zeitfenstern angewendet werden können
und die die Analyse einer bestimmten unveränderten zellulären Komponente
erlauben, wie beispielsweise ein Protein innerhalb seines natürlichen
Expressionsstatus. Bisher ist nur festgestellt worden, dass in vitro
selektierte Aptamere, die an ein natürlicherweise Nukleinsäure bindendes
Protein binden und üblicherweise
eng verwandt sind mit entsprechenden natürlichen Nukleinsäuresequenzen,
dies noch innerhalb der Zelle machen. Es ist noch nicht festgestellt
worden, dass, beispielsweise, ein Aptamer allgemein verwendet werden
kann, um die biologische Funktion von intrazellulären Zielmolekülen zu beeinflussen,
einschließlich,
beispielsweise, Hormone, biologischer Cofaktoren, nicht-Nukleinsäure bindender
Proteine, Biopolymere etc.
-
Die
internationale Patentanmeldung PCT/DK96/00231 beschreibt ein Verfahren
zum Identifizieren von biologisch aktiven Peptiden und Nukleinsäuren. Dieses
Dokument offenbart die grundsätzlichen
Schritte, wie beispielsweise das Herstellen eines Pools von geeigneten
Vektoren, die jeweils vollständig
oder teilweise zufällige
DNA-Sequenzen enthalten, die wirksame Transduktion der Vektoren
in eine Anzahl von identischen eukaryontischen Zellen, das Screenen
der transduzierten Zellen, um festzustellen, ob eine von ihnen ein
geändertes
bestimmtes phänotypisches
Merkmal aufweist, das Auswählen
und Klonieren der veränderten
Zellen, das Isolieren und Sequenzieren der Vektor-DNA in den phänotypisch
veränderten
Zellen und das Ableiten der RNA-Sequenz aus der DNA-Sequenz. Die
besagten Vektoren werden jedoch nicht weiter hinsichtlich der verwendeten
Expressionskassetten charakterisiert.
-
Die
der vorliegenden Erfindung somit zugrundeliegende technische Aufgabe
besteht darin, Mittel bereitzustellen zum Identifizieren und Validieren
von intrazellulären
Zielmolekülen,
einschließlich
Zielmolekülen, die
natürlicherweise
nicht an Nukleinsäuren
binden, und zum Modifizierung der Funktion des Zielmoleküls.
-
Die
Lösung
dieser technischen Aufgabe wird erreicht, indem die in den Ansprüchen gekennzeichneten Ausführungsformen
bereitgestellt werden.
-
Die
vorliegende Erfindung basiert auf der einzigartigen Einsicht, dass
Nukleinsäureliganden
gegen praktisch jegliche zelluläre
Komponente gerichtet und leicht intrazellulär verwendet werden können, wobei
- a) sie noch ihr Zielmolekül in der intrazellulären Umgebung
erkennen;
- b) sie dies selbst mit Zielmolekülen machen können, die
von Natur aus keine Nukleinsäure
binden;
- c) sie ihr Zielmolekül
in subzellulären
Kompartimenten, wie beispielsweise der zytoplasmatischen Phase der
Zellmembran, lokalisieren können,
wo Nukleinsäuren
normalerweise nicht gefunden werden;
- d) sie die biologische Funktion des Zielmoleküls modulieren
können,
was Rückschlüsse hinsichtlich
dessen biologischer Rolle erlaubt; und
- e) sie den Phänotyp
der Zelle ändern
können.
-
Dies
erlaubt die allgemeine Anwendung von intrazellulären funktionalen Nukleinsäuren zum
Validieren der Funktion von intrazellulären Zielmolekülen, insbesondere
für die
Auswahl oder Validierung von neuen Wirkstoff-Zielmolekülen. Sofern
erwünscht,
kann die funktionale Nukleinsäure
innerhalb einer Expressionskassette oder eines anderen Sequenzkontextes
plaziert werden, der nützlich
sein kann, um
- a) die Stabilität der funktionalen
Nukleinsäure
innerhalb des zellulären
Kompartiments zu erhöhen
oder Signale bereitzustellen, z. B. für die richtige Termination
einer funktionalen RNA-Sequenz, die unter der Kontrolle von RNA-Polymerasen exprimiert
wird; und
- b) zusätzliche
Sequenzinformation bereitzustellen, die für die richtige Lokalisierung
der funktionalen Nukleinsäure
erforderlich ist, beispielsweise, um die Nukleinsäure von
dem Nukleus in das Zytoplasma zu überführen.
-
Nachdem
sie einmal in die Zelle eingebracht sind, werden diese Nukleinsäuren ihre
biologischen Wirkungen entfalten, die studiert werden können. Das
Verfahren könnte
somit einen erheblichen Beitrag auf dem Gebiet des „functional
Genomics" leisten.
-
Entsprechend
betrifft in einem ersten Aspekt die vorliegende Erfindung ein Verfahren
zum Identifizieren eines Intramers, das in der Lage ist, ein funktionales
intrazelluläres
Zielmolekül
zu binden und dessen Funktion zu modifizieren, welches umfasst:
- a) Herstellen einer Kandidatenmischung aus
Nukleinsäuren;
- b) Kontaktieren der Kandidatenmischung aus Nukleinsäuren mit
dem intrazellulären
Zielmolekül
oder einem Teil davon;
- c) Auswählen
und Isolieren von Nukleinsäuren
mit einer verglichen mit der Kandidatenmischung erhöhten Affinität zu dem
Zielmolekül;
- d) reverses Transkribieren, sofern die Kandidatenmischung RNAs
umfasst und Amplifizieren der in Schritt c) erhaltenen Nukleinsäuren;
- e) optional Wiederholen der Schritte b) bis d);
- f) Isolieren und Sequenzieren der Klone (Intramere), die in
Schritt e) erhalten werden; und
- g) Testen, ob das Expressionsprodukt des Inserts des in Schritt
f) erhaltenen Klons an das intrazelluläre Zielmolekül in vivo
bindet und dessen Funktion beeinflusst, wobei eine T7-RNA-Expressionskassette
als ein Zytoplasma-Expressionssystem
verwendet wird, das einen T7-Promotor, eine stabilisierende 5'-Stamm-Schleife und
einen 3'-Terminator
Tϕ umfasst, wobei die Sequenz der T7-RNA-Expressionskassette
ohne Insert wie folgt ist: wobei
das Insert zwischen der 5'-Stamm-Schleife
und dem Terminator Tϕ vermittels der XmaI- und PacI-Restriktionsstellen
eingeführt
ist.
-
Bevorzugterweise
wird Schritt g) ausgeführt,
indem ein zytoplasmatisches Expressionssystem verwendet wird.
-
In
einer bevorzugteren Ausführungsform
umfasst das Verfahren der vorliegenden Erfindung weiterhin
- h) Kartieren der Bindungsstelle des Aptamers
auf dem Zielmolekül.
-
Die
vorliegende Erfindung betrifft weiterhin Verfahren für die Expression
randomisierter Nukleinsäurebibliotheken
innerhalb von Zellen für
die Identifizierung von funktionalen Intrameren, die den Phänotyp der
Zelle ändern,
in der sie exprimiert werden. Auf der Grund lage der Erkenntnis des
Erfinders hinsichtlich der allgemeinen Anwendbarkeit von funktionalen
Nukleinsäuren,
um intrazelluläre
Zielmoleküle
zu beeinflussen, ist es auch denkbar, Bibliotheken aus randomisierten
RNA-Sequenzen innerhalb von Zellen zu exprimieren. Diese komplexen
Bibliotheken aus randomisierten Nukleinsäuren enthalten funktionale
Moleküle,
die an zelluläre Komponenten,
wie beispielsweise Enhancer oder Repressoren von Signaltransduktionswegen,
binden und ihre biologische Funktion modulieren, was zu einem geänderten
Phänotyp
der Zelle führt.
Diese Moleküle
können
mit geeigneten Selektionsverfahren identifiziert werden. Die Modulation
umfasst nicht notwendigerweise Mechanismen, die lediglich auf einer
Bindung beruhen, sondern können
auch, beispielsweise, katalytische Verfahren umfassen, wie beispielsweise
ribozymatische Modifikation eines intrazellulären Zielmoleküls.
-
Nachdem
einmal ein bestimmter Phänotyp
beobachtet wird, ist es möglich,
das/die zellulären
Zielmolekül(e),
die für
die phänotypische
Veränderung
verantwortlich sind, zu identifizieren. Dies kann durch Variationen
verschiedener Technologien erfolgen, wie beispielsweise das „Drei-Hybrid-System" (SenGupta et al., Proc.
Natl. Acad. Sci. USA, 93 (1996), 8496–8501) oder andere in den nachfolgenden
Beispielen beschriebene Verfahren. Im Allgemeinen ist es möglich, das
zelluläre
Zielmolekül
dadurch zu identifizieren, dass die Wechselwirkung zwischen der
aktiven RNA aus der Bibliothek und dem Zielmolekül ausgenutzt wird. Dieses System ist
für die
targetierte Identifizierung von Schlüsselmolekülen nützlich, die an einem bestimmten
Phänotyp
beteiligt sind, bevorzugterweise einem Phänotyp, der mit bestimmten Erkrankungen
verbunden ist. Es ist anderen, derzeit verfügbaren Verfahren überlegen,
da es die Notwendigkeit vermeidet, eine große Anzahl von potentiellen
Kandidaten zu screenen, die indirekt identifiziert worden sind,
beispielsweise durch Proteomics, es identifiziert vielmehr die Schlüsselmoleküle in einem
direkten phänotypischen
Selektionsverfahren. Die identifizierten Schlüsselmoleküle sind potentielle Wirkstoffziehnoleküle; die
Nukleinsäwemodulatoren
sind nicht nur Zielmolekül-Validatoren,
sondern auch potentielle Wirkstoff-Leads, die selbst direkt als
Wirkstoffe, beispielsweise bei gentherapeutischen Ansätzen, verwendet
werden könnten.
-
Entsprechend
betrifft die vorliegende Erfindung in einem weiteren Aspekt ein
Verfahren für
die Identifizierung eines funktionalen intrazellulären Zielmoleküls, das
mit einem besonderen Phänotyp
verbunden ist und des entsprechenden Intramers, das in der Lage
ist, an das Zielmolekül
zu binden und dessen Funktion zu modifizieren, umfassend:
- a) Herstellen einer Kandidatenmischung aus
Nukleinsäuren;
- b) Klonieren der Kandidatenmischung aus Nukleinsäuren in
einen Vektor unter der Kontrolle eines geeigneten Promotors, der
optional einen selektierbaren Marker enthält, wobei der Vektor ein zytoplasmatisches Expressionssystem
ist, das eine T7-RNA-Expressionskassette enthält, wobei die T7-RNA-Expressionskassette
einen T7-Promotor, eine stabilisierende 5'-Stamm-Schleife und einen 3'-Terminator Tϕ umfasst, wobei die
Sequenz der T7-RNA-Expressionskassette
ohne Insert wie folgt lautet: wobei
das Insert zwischen der 5'-Stamm-Schleife
und dem Terminator Tϕ vermittels der XmaI- und PacI-Restriktionsstellen
eingefügt
ist,
- c) Einführen
des in Schritt b) erhaltenen Vektors in eine Reporter-Zelllinie,
die positive oder negative phänotypische
Selektion erlaubt;
- d) Auswählen
von Zellen mit einem geänderten
Phänotyp;
und
- e) Bestimmen der Sequenz der in den Vektor von Schritt b) eingeführten Nukleinsäure (Intramer)
und der Komponente, an die sie bindet.
-
Kurze Beschreibung der
Figuren
-
A: Aufeinanderfolgende
Kurzdarstellung der 1 bis 5 (für Beispiel
1)
-
1:
Auswahl und Charakterisierung von CD 18cyt-spezifischen RNA-Aptameren.
A*, Konstruktion des synthetischen DNA-Pools
und der primären
46-mer Aminosäuresequenz
der kompletten zytoplasmatischen Domäne von β2-Integrin (CD18cyt), auf Sepharose
immobili siert und in der Selektion verwendet. B*,
Sequenzen und vorhergesagte Sekundärstrukturen der einzelnen Aptamerklone
D20, D28 D31 und D42. In Fettdruck gezeigte Nukleotide sind Teil
der randomisierten Region. Man hat in einem Schadensselektionsexperiment
gezeigt, dass die in Grauschattierung gehaltenen Aptamerregionen
in den Klonen D28 und D20 die Minimalerfordernisse für das Beibehalten
der CD 18cyt-bindenden Fähigkeit
darstellen. Die Sequenz D42 wurde als Negativkontrolle verwendet;
diese Sequenzen zeigen keine nachweisbare Bindung an CD18cyt.
-
2:
RNA Aptamer-Expressionssystem basierend auf Doppelinfektion mit
rekombinanten Vaccinia Viren. A. Design der T7-RNA-Expressionskassette.
B. Schematische Darstellung des auf Vaccinia Virus basierenden zytoplasmatischen
RNA-Aptamer-Expressionssystems. C. Verlauf der Expression TR-kodierten
Aptamers in vT7 koinfizierten Jurkat E6-Zellen veranschaulicht durch
eine repräsentative „Dot-Blot"-Analyse für das Aptamer
TR-D31 (rechtes Feld). 7 h nach Infektion werden maximale Niveaus
der Aptamerexpression beobachtet. Die Quantifizierung (linkes Feld)
wurde wie folgt durchgeführt:
Totale zelluläre
RNA wurde isoliert, auf die Blotmembran überführt und mit einem 5'-32P-markiertem
und zu der in allen TR-Konstrukten
vorhandenen 3'-„Stemloop"-Struktur komplementären Oligonukleotid
hybridisiert. In vitro transkribiertes Aptamer TR-D20 wurde zum
Vergleich und zur Quantifizierung in gleicher Weise behandelt. D. „Dot-Blot"-Analyse und Quantifizierung
maximal exprimierter Aptamer-RNA. Jeder Punkt wurde auf einem Phosphoimager
quantifiziert. Die Quantifizierung wurde wie für 2c beschrieben
durchgeführt.
-
3:
Bestimmung der Gelmobilitäts-„Shift" von endogenem, in
Jurkat E6-Zelllysaten enthaltenem CD18, das an sein kognates Aptamer
TR-D20 bindet. A. Gel-„Shift"-Experiment 1. Spur
1: Freie TR-D20-Aptamer-RNA; Spur 2: Veränderte Bande, die in Gegenwart
von Jurkat E6-Zelllysat und 25 μM
unspezifischer Kompetitor-tRNA erhalten wurde; Spur 3: Mit der Negativkontrollsequenz
TR-D42 wurde kein „Shift" erhalten; Spur 4:
Gleich wie Spur 2 mit 40 μM
unspezifischer Kompetitor-tRNA; Spur 6: Spezifische, überveränderte Bande,
die in Gegenwart des Antikörpers
MHM23, der die extrazelluläre β2-Domäne von LFA-1
erkennt, erhalten wurde; Spur 7: Spezifische, überveränderte Bande, die in Gegenwart
des Antikörpers
MEM170, der die extrazelluläre
Domäne
der βL-Untereinheit
von LFA-1 erkennt, erhalten wurde. Das Bandenmuster dürfte Aptamer/Integrin-Komplexe
von unterschiedlicher Stöchiometrie
widerspiegeln. B. Gel-„Shift"-Experiment 2 mit zusätzlichen
Kontrollen. Alle Gel-„Shift"-Experimente wurden
in Gegenwart von 30 μM
tRNA als nicht-spezifischem Kompeti tor durchgeführt. Spur 1: Freie TR-D20-Aptamer-RNA;
Spur 2: Veränderte
Bande, die in Gegenwart von Jurkat E6-Zelllysat erhalten wurde;
Spur 3: Spezifische, überveränderte Bande,
die in Gegenwart des Antikörpers
MHM23 erhalten wurde; Spur 4: kein verändertes Aptamer wurde unter
Bedingungen identisch zu denen in Spur 3 in Abwesenheit von Jurkat
E6-Zelllysat erhalten;
Spur 5: Spezifische, überveränderte Bande,
die in Anwesenheit des Antikörpers
MEM170 erhalten wurde; Spur 6: Kein Super-„Shift" wurde in Gegenwart des Antikörpers OKT3
erhalten, der gegen den T-Zellrezeptor gerichtet ist. Die experimentellen
Bedingungen waren ansonsten die gleichen wie in Spur 2; Spur 7:
Kein verändertes
Aptamer wurde unter Bedingungen identisch zu denen in Spur 6 in
Abwesenheit von Jurkat E6 Zelllysat erhalten.
-
4:
Inhibition der PMA-stimulierten Zelladhäsion als Funktion der Aptamerexpression.
A. Der CD18cyt-Binder TR-D20 reduziert die von Phorbolester aktivierte
Jurkat-Zelladhäsion
an ICAM-1. Jurkat E6-Zellen, die mit rekombinanten Vaccinia Viren
infiziert wurden, wurden an Plastikschalen anheften gelassen, die
wie in dem Abschnitt Beispiel 1 beschrieben mit einer rekombinanten
ICAM 1-Chimäre
beschichtet waren. Die Jurkat E6-Zelladhäsion an ICAM-1 war in Anwesenheit
mehrerer Kontrollviren (vT7/vTR, Wildtyp-Vaccinia Virus, vT7, vTR-D20)
durch PMA überinduzierbar.
Jedoch reduzierte die Induktion der Expression der CD 18cyt-spezifischen
Aptamere (vT7/vTR-D20, rechtes Feld) die PMA-stimulierte Jurkat
E6 Zelladhäsion,
aber nicht die Basisadhäsion.
Diese Resultate wurden mindestens dreimal unabhängig reproduziert. Der Prozentsatz
reflektiert die Werte, die normalisiert wurden gegen stimulierte
vT7/vTR Doppelinfektionen, die auf 100% gesetzt wurden. B. Humane,
periphäre,
mononukleäre,
Blutzellen sind gute Ziele für
Proteinüberexpression durch
rekombinante Vaccinia Viren. Zytoplasmatische Immunglobulin-(cIg)-Fusionen
von Cytohesin-1-Subdomänen wurden
in permeabilisierten PBMC mit der Hilfe einer FITC-konjugierten
antihuman Ig-Antikörperpräparation
detektiert. Oberes Feld, links: Kontrollinfektion, keine rekombinanten
Moleküle
exprimiert; oberes Feld, rechts: cIg-Domänen allein; unteres Feld, links:
cIg-PH; unteres Feld, rechts: cIg-PH+c-Domäne. C. TR-D20 reduziert spezifisch
die PMA-stimulierte Adhäsion
von PBMC an ICAM-1. Aptamere oder Kontrollproteine wurden in PBMC
durch rekombiante Vaccinia Viren wie oben beschrieben exprimiert.
Die PMA-stimulierte
Adhäsion
dieser Zellen an ICAM-1 war gleich, wenn das TR-D42-Aptamer, cIg
oder cIg-PH Kontrollproteine exprimiert wurden, aber die stimulierte
Zelladhäsion
war nach der Expression der intakten PH+c-Domäne von Cytohesin-1 (cIg-PH+c)
oder des TR-D20-Aptamers
dramatisch reduziert.
-
5:
Bestimmung der Bindungsstelle der Aptamere TR-D20, TR-D28, TR-D31
und der Negativkontrollsequenz TR-D42 an CD18cyt unter Verwendung
synthetischer, biotinylierter Peptidfragmente. Alle Gel-„Shifts" wurden in Anwesenheit
von 25 μM
Streptavidin durchgeführt,
um die Trennung der verschobenen gegenüber der nicht-verschobenen
RNA zu verstärken.
Gel-„Shift"-Experimente wurden
in Anwesenheit von 4 nM radioaktiv markierter RNA und 40 μM nicht-spezifischem
tRNA-Kompetitor durchgeführt.
Spur 1: Freies Aptamer in Gegenwart von 25 μM Streptavidin und 40 μM unspezifischer
Kompetitor-tRNA; Spuren 2, 4, 6 oder 3, 5, 7: Gleich wie Spur 1
in Gegenwart von jeweils 25 μM
oder 12,5 μM
der Peptide A23 (Spur 2,3), B16 (Spur 4,5) und C17 (Spur 6, 7).
-
B: Aufeinanderfolgende
Kurzdarstellung er 6–12 (für Beispiel
2)
-
6:
Vektoren für
die phänotypische
Selektion. A. Schema wichtiger Eigenschaften des Plasmids pP1; CMV
P: Promotorelement: unmittelbarer-früher Promotor des humanen Cytomegalovirus.
Beispiel eines konstitutiven Promotorelements, das in einer großen Vielzahl
von Zellen aktiv ist. In Abhängigkeit
von den Zelllinien, der Anwendung usw. können andere häufig gebrauchte
Promotoren oder Kombinationen von Promotorelementen (z.B. SV40 früher Promotor,
Rous Sarkoma Virus unmittelbarer-früher Promotor usw.) verwendet werden;
IL-2 P: IL-2-Promotor; neo: Selektierbarer Marker: Resistenzgen
(Aminoglykosid-Phosphotransferase),
das allgemein verwendet wird, um nach Zellen, die ein transfiziertes
Plasmid enthalten, über
Detoxifikation von Aminoglykosidantibiotika zu selektieren; hsv-thymidinkinase: Gen
für die
Thymidinkinase des Herpes Simplex Virus. B. Schema wichtiger Eigenschaften
des Plasmids pP2; CMV P: vergleiche pP1; Pol III P: Promotorelement:
RNA-Polymerase-III-Promotor;
vai random vai: Zufallsbibliothek von Nukleinsäuren mit den adenoviralen VAI-RNA-Sequenzen,
die den Export der funktionalen Nukleinsäure in das Zytoplasma erlauben. OriP:
Episomale Replikation: Der Ausgangspunkt der Replikation des Epstein
Barr Virus (EBV) und das nukleäre
Antigen (EBNA-1) ermöglichen
die episomale (extrachromosomale) Hochkopie-Replikation in und die
Erhaltung in Primaten- und Hundezelllinien; EBNA-1: Gen für das nukleäre Antigen
1 des Epstein Barr Virus; hygromycin: Resistenz gegen Hygromycin
kann genutzt werden, um für
Zellen zu ko-selektieren die zwei Plasmide enthalten (z.B. ein zusätzliches
Plasmid mit der Neo-Resistenz).
-
7:
Schema der funktionalen Selektion gegen die Induktion des Interleukin-2-Promotors
in T-Zellen nach Stimulierung mit Phorbol-12-Myristat-13-Acetat
(PMA). Die Proteinkaskade, die schematisch in diesem Beispiel gezeigt
ist und in einer T-Zelle von Protein X über Y nach Z führt (gerade
Pfeile), führt
letztendlich zu der Aktivierung des Interkeukin-2-Promotors (I1-2P)
des Reporterkonstrukts IL2 P/HSV-Thymidinkinase. Transkription und
Translation der mRNA der HSV-Thymidinkinase (mRNA HSV TK) führt zu der
Expression der Herpes-Simplex-Thymidinkinase (HSV TK) (gestrichelte
Pfeile repräsentieren
Genexpression). Konstitutive Transkription der Sequenzen einer Nukleinsäurebibliothek
(vai/random/vai) unter der Kontrolle eines RNA-Polymerase-III-Promotors
(Pol III P) führt
zur Bildung einer Zufalls-RNA-Bibliothek, die aufgrund ihrer adenoviralen
VAI-Sequenz in das Zytoplasma exportiert wird. Wie in dieser Figur
angegeben beeinflusst die Expression einer nicht-funktionalen Nukleinsäure die
Induktion des I1-2P durch die Signaltransduktionskaskade X, Y, Z und
die Expression der HSV TK nicht. Die Anwesenheit der HSV TK und
von Ganciclovir wird durch die Produktion toxischer Stoffe zum Tod
der T-Zelle führen.
Kein Zelltod wird auftreten, wenn die exprimierte RNA-Bibliothek
funktionale Sequenzen enthält,
welche die Expression der HSV TK beeinflussen, beispielsweise durch Unterbrechung
der Signaltransduktionskaskade indem eines ihrer Proteine inhibiert
wird. Diese Zellen können dann
selektiert werden (vergleiche 8).
-
8:
Gleiches phänotypisches
Selektionsschema wie in 7, jedoch wird in diesem Fall
eine funktionale Nukleinsäure
von Pol III/vai/random/vai exprimiert (gestrichelte Pfeile repräsentieren
Genexpression). Die funktionale Nukleinsäure bindet in diesem Beispiel
an Protein X und blockiert die Signalübertragung in der Kaskade von
X zu Y (gerade Pfeile). Folglich wird die Induktion des I1-2-Promotors
inhibiert und es wird keine HSV TK exprimiert. Die T-Zelle überlebt
in Gegenwart von Ganciclovir und das Gen, das für die funktionale Nukleinsäure kodiert,
kann isoliert werden.
-
9:
Vektoren für
die funktionale Targetidentifizierung. A. Schema von wichtigen Eigenschaften
des Plasmids pF1; CMV P: Promotorelement „unmittelbarer-früher Promotor
des humanen Cytomegalovirus".
Beispiel eines konstitutiven Promotorelements, dass in einer großen Vielfalt
an Zelltypen aktiv ist. IL-2 P: I1-2-Promotor; OriP: Episomale Replikation.
Der Ausgangspunkt der Replikation des Epstein Barr Virus und das
nukleäre
Antigen (EBNA-1) ermöglichen
die episomale (extrachromosomale) Hochkopie-Replikation in und die
Erhaltung in Primaten- und Hundezelllinien; EBNA-1: Gen für das nukleäre Antigen
1 des Epstein Barr Virus; cDNA-Bibliothek: Die cDNA-Bibliothek ist
in diesem Beispiel von T-Zellen
abgeleitet. Die Bibliothek wird benutzt, um für die Komplementation der inhibierten
Signaltransduktionskaskade zu selektieren. Überexpression des Faktors,
der durch die funktionale Nukleinsäure inhibiert ist, umgeht den
Block in der Signalleitung, wodurch der IL-2-Promotor wieder in denjenigen Zellen
induziert wird, die das Zielgen von Interesse enthalten; fNS-hsv thymidinkinase:
Dieses Konstrukt erlaubt die Identifizierung des Produkts des Zielgens
durch die Spezifität
der Nukleinsäure-Protein-Wechselwirkung.
Das Gen der funktionalen Nukleinsäure (fNS), in diesem Beispiel
eine Aptamersequenz, wird in den 5'-UTR der Herpes-Simplex-Thymidinkinase
kloniert. Nach Bindung des Aptamerliganden (der funktionalen Nukleinsäure) wird
die Expression des Reporters HSV-TK mutmaßlich durch die Blockierung
der Translation inhibiert; neo: Selektierbarer Marker und Resistenzgen
(Aminoglykosid-Phosphotransferase), das allgemein verwendet wird,
um nach Zellen, die ein transfiziertes Plasmid enthalten, über Detoxifikation
von Aminoglykosidantibiotika zu selektieren. B. Schema wichtiger
Eigenschaften des Plasmids pF2. CMV P: vergleiche Plasmid pF1; Pol
III P: Promotorelement: RNA-Polymerase-III-Promotor; vai fNS vai: Funktionale
Nukleinsäure
(fNS) mit den adenoviralen VAI-RNA-Sequenzen, die den Export der
funktionalen Nukleinsäure
in das Zytoplasma erlauben; OriP: Vergleiche pF1; hygromycin: Resistenz
gegen Hygromycin kann genutzt werden, um für Zellen zu ko-selektieren
die zwei Plasmide enthalten (z.B. ein zusätzliches Plasmid mit der Neo-Resistenz);
mIg: Oberflächenmarker,
der die Affinitätsselektion
transformierter Zellen erlaubt. Beispielsweise werden in diesem
Beispiel die konstanten CH2- und CH3-Domänen des humanen IgG1 als eine
Transmembranversion (mIg), die auf der Zelloberfläche präsentiert
wird, exprimiert.
-
10:
Schema für
die funktionale Zielmolekülidentifizierung.
Die von Pol III/vai/fNS/vai exprimierte (gestrichelte Pfeile repräsentieren
Genexpression) und in der phänotypischen
Selektion identifizierte funktionale Nukleinsäure bindet an Protein X und
blockiert die Signalleitung von X nach Y (gerade Pfeile) in der
Kaskade nach Stimulierung mit Phorbol-12-Myristat-13-Acetat (PMA) (vergleiche
auch 8). Da Protein M (Klon einer T-Zell-cDNA-Bibliothek), das
unter der Kontrolle des Promotors CMV P (vergleiche Beschreibung
der 9) exprimiert wird, keinen Effekt auf die blockierte
Kaskade hat, ist die Induktion des IL-2-Promotors (IL-2 P) inhibiert und deshalb
kommt es nicht zur Expression der Reporterkonstrukte fNS-HSV-Thymidinkinase
und mIg (vergleiche Beschreibung der 9). Da die
T-Zellen das mIg
nicht auf ihrer Oberfläche
exprimieren, können
sie nicht beispielsweise durch Verwendung von Antikörpern gegen
mIg isoliert werden.
-
11:
Schema für
die funktionale Zielmolekülidentifizierung.
Gleich wie 10, außer dass in diesem Beispiel
das kognate Zielmolekül
Protein X für
die funktionale Nukleinsäure
(fNS) von einer cDNA-Bibliothek (gestrichelte Pfeile repräsentieren
Genexpression) exprimiert wird. Die Überexpression von Protein X
umgeht den Block des endogenen Protein X durch die fNS. Daher wird
die Signaltransduktionskaskade X, Y, Z (gerade Pfeile) nach Stimulierung
mit PMA gerettet und die mRNA wird von IL-2-Promotor-Reproterkonstrukten
transkribiert. Die Expression des Oberflächen mIg (vergleiche auch 9)
erlaubt die Affinitätsisolierung der
T-Zellen. Gleichzeitig bindet Protein X an die fNS, die in den 5'-UTR des fNS-HSV-TK-Konstrukts
inseriert ist. Das resultiert in der Blockade der Translation der
HSV TK. Die Abwesenheit des Enzyms Herpes-Simplex-Thymidinkinase
ermöglicht
den T-Zellen in der Gegenwart von Ganciclovir zu überleben
und die positiven cDNA-Klone können
isoliert werden.
-
12:
Schema der funktionalen Zielmolekülidentifizierung. Obwohl Protein
X in diesem schematischen Beispiel durch die funktionale Nukleinsäure (fNS)
blockiert wird, induziert das Protein N, das von der cDNA-Bibliothek
exprimiert wird, die I1-2-Promotor-Reporterkonstrukte durch Deregulation
der endogenen Signaltransduktionskaskade, indem es direkt auf Protein
Z wirkt (vergleiche auch 10, 11).
Diese falsch positive Komplementation führt zu der Expression des mIg,
welches die Isolierung der Zellen mit mIg-spezifischen Antikörpern erlauben würde. Da
Protein N nicht der kognate Ligand für die funktionale Nukleinsäure ist, kann
es nicht an die fNS in dem fNS-TK-mRNA-Konstrukt binden (vergleiche
auch 10, 11). Daher wird die Herpes-Simplex-Thymidinkinase
exprimiert und die Zellen werden in Gegenwart von Ganciclovir getötet. Dieser
Schritt eliminiert falsch positive cDNA-Klone, die durch Deregulierung
der Signaltransduktion wirken.
-
Die
Kandidaten-RNAs oder DNAs umfassen funktionale Nukleinsäuren, z.B.
einzelsträngige
DNA (ssDNA) oder RNA (ssRNA) oder chemisch modifizierte Formen davon
(ssNAmod), doppelsträngige
DNA (dsDNA) oder RNA (dsRNA) oder davon chemisch modifizierte Formen
davon (dsNAmod), die in der Lage sind ein intrazelluläres Zielmolekül zu binden,
zu modulieren oder katalytisch zu modifizieren und dadurch seine
biologische Funktion zu beeinflussen. Die funktionalen Nukleinsäuren können beispielsweise
durch das „Screening" großer, kombinatorischer
Nukleinsäurebibliotheken
(in vitro Selektion von Ribozymen, Aptazymen, Aptameren usw.) identifiziert
werden. Kombinatorische Nukleinsäurebibliotheken,
ob DNA oder RNA, enthalten gewöhnlich
eine randomisierte Sequenz, die von feststehenden Regionen flankiert
sind, die als Primer-Anellierungsstellen für die enzymatische Amplifikation über PCR
oder im Fall von RNA über
reverse Transkription und PCR dienen. Die Bibliotheken können beispielsweise
als ssDNA an einem automatisierten DNA-Synthesiser unter Verwendung
von 3'-Phosphoramidit-Chemie
synthetisiert werden, wobei die randomisierte Region mit 3'-Phosphoramiditlösungen synthetisiert
wird, die Mischungen der vier Basen A,C,G,T enthalten (Hermes et
al. Proc. Natl. Acad. Sci. U.S.A. 87 (1990), 696–700. Die Verhältnisse
zwischen den vier Monomeren können in
Abhängigkeit
von der Anwendung variiert werden. Die feststehenden Regionen enthalten
für die
Präparation der
RNA einen RNA-Polymerase-Promotor,
z.B. den T7-RNA-Polymerase-Promotor, der die Transkription der Templat-DNA
in RNA ermöglicht.
Falls nötig
kann die ssDNA unter Verwendung enzymatischer Protokolle, z.B. der
PCR, die einem Fachmann wohl bekannt sind, doppelsträngig gemacht
werden. Für
einen Überblick
vergleiche Abelson, Methods in Enzymology 267 (1996), Seiten 275–426, Academic
Press, San Diego. Bevorzugterweise enthält die Bibliothek 8 bis 200
randomisierte Nukleotide (Tuerk und Gold, Science 249 (1990), 505–510; Bartel
und Szostak, Science 261 (1993), 1411–1418) mit Komplexitäten von
6 × 105 bis 1 × 1016, welche in erfolgreichen Selektionsexperimenten
eingesetzt wurden. Falls gewünscht
kann die funktionale Nukleinsäure
an andere Moleküle
wie zum Beispiel chemisch reaktive Gruppen, Lipide, Peptide, Proteine
usw., die der funktionalen Nukleinsäure zusätzliche Eigenschaften hinzufügen, kovalent
angehängt
oder nicht-kovalent gebunden werden. Funktionale Nukleinsäuren umfassen
nicht Nukleinsäuren,
die durch Nukleinsäurewechselwirkungen
wirken, wie zum Beispiel Antisensemechanismen oder natürliche Ribozyme
oder modifizierte und mutierte Formen davon, die Antisenseerkennung
mit Phosphodiesterspaltungsaktivität kombinieren und beispielsweise
in Gentherapieansätzen
angewendet werden. Eine funktionale Nukleinsäure kann Teil eines Konstrukts
mit zusätzlichen
Sequenzen sein, ob natürlich
oder nicht natürlich,
die zusätzliche
Funktionen hinzufügen,
z.B. erhöhte
Stabilität,
Lokalisierungssignale oder enzymatische Aktivitäten. Darüber hinaus kann die funktionale
Nukleinsäure
selbst nach Bindung an ihr intrazelluläres Zielmolekül in einer
zweiten Funktion, ob intrinsisch oder in einem Konstrukt mit zusätzlichen
Sequenzen hinzugefügt,
moduliert werden, z.B. Bindung eines dritten Liganden oder katalytisch
aktiv zu sein.
-
Aptamere
sind spezifische ligandenbindende Nukleinsäurerezeptoren (ssDNA, RNA,
modD-NA or modRNA),
die durch „Screening" des „Formraums" von enormen kombinatorischen
Bibliotheken einzelsträngiger
Nukleinsäuren
durch in vitro Selektion isoliert werden können.
-
Die
in vitro Selektion von Aptameren kann erreicht werden durch in Kontakt
bringen einer Nukleinsäurebibliothek
mit einem Zielmolekül,
z.B. einem Protein, und dem Abtrennen der gebundenen Nukleinsäuren vom
Rest der Nukleinsäuremischung.
Die Abtrennung kann erreicht werden durch Zurückhalten der Nukleinsäure/Protein-Komplexe
auf Nitrozellulosefiltern (Tuerk und Gold, Science 249 (1990), 505–510), Immobilisierung
des Zielmoleküls
auf einem festen Träger
vor der Inkubation mit der Bibliothek und Entfernung nicht-gebundener
Spezies durch Spülungen
mit einem geeigneten Puffer (Nieuwielandt et al., Biochemistry 34
(1995), 5651–5659)
oder jeder anderen Methode, die die Abtrennung der Zielmolekül/Nukleinsäure-Komplexe
von ungebundenen Spezies erlaubt. Nach Isolierung der gebundenen
Spezies kann die DNA unter Verwendung von PCR amplifiziert werden
oder im Fall von RNA durch reverse Transkription, PCR und Transkription.
Dieser Zyklus kann mehrere Male wiederholt werden, um eine Bibliothek
mir erhöhter
Affinität
für das
Zielmolekül
hervorzubringen. Darauffolgend können
individuelle Aptamersequenzen durch Klonierung und Sequenzierung
der PCR-Produkte unter Verwendung von Standardmethoden identifiziert
werden.
-
Nukleinsäureaptamere
sind für
weit mehr als hundert verschiedene Zielmoleküle beschrieben worden, was
zeigt, dass Aptamere jetzt routinemäßig für beinahe jedes gewünschte Zielmolekül erhalten
werden können
(für Übersichtsartikel
vergleiche z.B.: Osborne und Ellington, Chem. Rev. 97 (1997), 349–370; Famulok und
Jenne, Curr. Opin. Chem. Biol. 2 (1998), 320–327.
-
Der
Ausdruck Ribozyme wird in der vorliegenden Erfindung für katalytisch
aktive Nukleinsäuren
verwendet, die auf ein intrazelluläres Zielmolekül wirken,
wodurch sie seine Funktion modifizieren. Nicht eingeschlossen sind
in der vorliegenden Erfindung natürlich vorkommende Ribozyme,
wie zum Beispiel Hammerhead-Ribozyme, Hairpin-Ribozyme oder Derivate
davon, die durch Watson-Crick-Bindung an Nukleinsäuresubstrate
und Phosphodiesterspaltung wirken und beispielsweise in Gentherapieansätzen verwendet
werden (Bramlage, Tibtech 16 (1998), 435–438). Ribozyme mit neuen katalytischen
Eigenschaften können
unter Verwendung von in vitro Selektionsexperimenten isoliert werden.
Zum Beispiel sind Ribozyme isoliert worden, die in der Lage sind
RNA-Alkylierung, Bildung von Amidbindungen oder Porphyrinmetallierung
zu katalysieren (Breaker, Chem. Rev. 97 (1997), 371–390).
-
Aptazyme
können
als Chimären
zwischen einer katalytischen RNA und einem Aptamer betrachtet werden.
Diese Chimären
können
rationell konstruiert werden in vitro von bestimmten kombinatorischen
Bibliotheken selektiert werden (vergleiche Robertson und Ellington,
Nature Biotechnol. 17 (1999), 62–66). Gewöhnlich wird die Aptamerdomäne eines
Aptazyms als regulatorisches Modul für die allosterische Ribozymaktivierung
oder- inhibition eingesetzt (Tang und Breaker, Chem. Biol. 4 (1997),
453–459;
Tang und Breaker, RNA 3 (1997), 914–925, Tang und Breaker, Nucleic
Acids Res. 26 (1998), 4214–4221;
Araki et al., Nucleic Acids Res. 26 (1998), 3379–3384). Ebenso und konsequenterweise
enthält
ein Aptazym, wenn es selektiert wird, um von einem regulatorischen
Molekül
beeinflusst zu werden, eine Bindungsdomäne für dieses Molekül und kann sich
daher auch wie ein Aptamer verhalten.
-
Intramere
sind funktionale Nukleinsäuren
(Aptamere, Aptazyme usw.), die exogen oder endogen in prokaryotische
oder eukaryotische Zellen eingebracht werden und ein intrazelluläres Zielmolekül binden,
modulieren oder enzymatisch modifizieren.
-
Modulation
eines intrazellulären
Zielmoleküls
kann zum Beispiel die Inhibition oder Stabilisierung einer Protein/Protein-Interaktion
oder Proteindomäne/Protein-Interaktion
oder Protein/Nukleinsäure-Interaktion
oder Protein/"Kleinmolekül"-Interaktionen usw.
sein. Modulierung kann auch die Aktivierung oder Inhibition von
Enzymen über
verschiedene Mechanismen umfassen wie zum Beispiel Sequestrierung
des Substrat (der Substrate), Bindung an die aktive Stelle, Induktion
von Konformationsänderungen,
allosterische Regulierung usw. Modulation eines intrazellulären Zielmoleküls kann
durch beides, nicht-kovalente Interaktionen oder kovalente Modifikationen
wie zum Beispiel Biotinylierung, erfolgen (Wecker et al., RNA 2
(1996), 982–94).
Weitere Methoden, die zum Beispiel für auf Antikörpern basierende Ansätze entwickelt
wurden, könnten
auf das funktionale Nukleinsäuresystem
adaptiert werden, wobei das Aptamer mit einem Molekül verbunden
werden könnte, das
in der Lage ist das gebundene Zielmolekül chemisch zu inaktivieren.
Beispielsweise werden die intrazellulären, funktionalen Nukleinsäuren über eine
kurze „Annealing"-Sequenz, die kovalent
an dem Farbstoff Malachitgrün
(MG) angehängt
ist, markiert. Diese kurzen Oligonukleotide könnten exogen angewendet werden. MG
absorbiert Licht bei einer sichtbaren Wellenlänge, die nicht signifikant
durch Zellen absorbiert wird. Nach der Bestrahlung mit einem Laser
wird das MG aktiviert und erzeugt kurzlebige Hydroxylradikale, die
Proteine höchstwahrscheinlich
durch Modifizierung der Aminosäureseitenketten
inaktivieren [„Chromophore
assisted laser inactivation (CALI)", vergleiche: Liao et al., Proc. Natl.
Aad. Sci. 91 (1994), 2659–63].
Der Inakti vierungsprozess tritt nur innerhalb einer kurzen Distanz
von der Farbstoffgruppe auf. Daher kann das als Ziel gesetzte Protein
oder die Untereinheit generell geschädigt werden ohne seine Nachbarn
zu beeinflussen. Dies hat zwei Vorteile: Erstens würde das
Aptamer nicht notwendigerweise an die aktive Stelle des Proteins
binden müssen, sondern
könnte
mit einem benachbarten Epitop interagieren, das besser für die Bindung
an eine Nukleinsäure geeignet
sein könnte.
Sogar obwohl das gebundene Epitop entfernt von der aktiven Stelle
ist, wird das Protein immer noch spezifisch inaktiviert. Zweitens
könnten
mehrere Bestrahlungspulse Proteine mit hoher Abundanz, die anfangs
in höheren
Kopienzahlen als das spezifische Aptamer vorhanden sind, inaktiveren,
da das Protein irreversibel inaktiviert wird und das Aptamer konstitutiv
exprimiert werden kann.
-
Intrazelluläre Zielmoleküle umfassen
kleine Moleküle,
biologische Kofaktoren, natürliche
Polymere wie zum Beispiel Proteine, Proteindomänen, Peptide und Nukleinsäuren, Glykoproteine,
Hormone, Rezeptoren, Proteinkomplexe, Protein/Nukleinsäure-Komplexe,
Toxine, Viren und andere Substanzen, die natürlicherweise in Zellen gefunden
werden.
-
Phänotyp kann
beispielsweise die Induktion oder Repression der Transkription eines
bestimmten Gens, Prozesse, die mit Signaltransduktionskaskaden verbunden
sind wie zum Beispiel Adhäsion
als Antwort auf bestimmte Reize, Zelldifferenzierung, Apoptose usw.
sein.
-
Phänotypische
Selektion ist ein Prozess, in dem Nukleinsäure-Bibliotheken durch verschiedene
Methoden, die dem Fachmann bekannt sind (Elektroporation, Llipofektion,
Expression mit viralen Vektorsystemen, Expression von RNA-Polymerase-Promotoren
enthaltenden Plasmiden usw.), in Zellen eingeführt werden. Diejenigen Zellen,
die eine funktionale Nukleinsäure
enthalten, die auf ein Zielmolekül
wirkt, das in einen bestimmten Phänotyp involviert ist, werden
auf der Basis ihres veränderten
Phänotyps
von dem Großteil
zellulären
Materials, das keinen veränderten
Phänotyp
zeigt, abgetrennt. Zellen, die in der phänotypischen Selektion verwendet
werden umfassen Prokaryoten, Hefe, Insekten- oder Pflanzenzellen
und bevorzugterweise Säugerzellen
(z.B. CHO, HeLa, COS, MDCK, 293, WI38 und Jurkat E6-Zellen). Falls
eine klare Trennung auf Basis des veränderten Phänotyps nicht möglich oder
schwierig ist, können
Reporterkonstrukte angewendet werden, die eine deutlichere oder
bessere Unterscheidung erlauben. In einem positiven phänotypischen
Selektionsexperiment, zum Beispiel der Induktion eines zellulären Promotor-
oder Enhancer/Promotor-Konstrukts, wird ein Reportergen, das die
Identifizierung des veränderten
Phänotyps
(hier die Induktion des Promo tors) erlaubt, unter der Kontrolle
eines induzierbaren Promotors in dem Reporterkonstrukt eingefügt. Reportergene,
die in dieser Art von Selektion verwendet werden, sind beispielsweise
Resistenzgene wie die Aminoglykosidphosphotransferase (Southern
und Berg, J. Mol. Appl. Genet. 1 (1982), 327–341), die Aminoglykosidantibiotika
(Neomycin, G418) inaktiviert und Zellen resistent gegen diesen Wirkstoff
macht. Folglich werden die Zellen, die das Gen für die funktionale Nukleinsäure beinhalten
in Anwesenheit des Wirkstoffs überleben, wenn
der induzierbare Promotor durch die Wirkung einer funktionalen Nukleinsäure (fNS)
angeschaltet wird, und die Gene für die fNS können identifiziert werden.
Beispielsweise könnten
Säugerpromotoren,
die von der Aktivierung durch den Transkriptionsfaktor NF B abhängig sind,
durch die Inhibition des I B Proteins, das normalerweise den Import
von NF B in den Zellkern verhindert, aktiviert werden (Rolfe et
al., J. Mol. Med. 75 (1997), 5–17;
Fuchs et al., Oncogene 17 (1998), 1483–90. Andere Beispiele von Promotoren,
die durch einen inhibitorischen Mechanismus reguliert werden, sind
bakterielle Transkriptionseinheiten, zum Beispiel das E.coli lac-Operon,
das durch den lac-Repressor reguliert wird (Gilbert und Muller-Hill,
Proc. Nat. Acad. Sci. 56 (1966), 1891–1898).
-
Negative
phänotypische
Selektion kann beispielsweise durch die Induktion eines Promotors
stattfinden. In diesem Fall könnte
das Reportergen ein Toxin sein [z.B. Herpes-Simplex-Virus-Thymidinkinase
(Fife et al., Gene Ther. 5 (1998), 614–620] oder Diphtherie-Toxin
(Massuda et al., Proc. Natl. Acad. Sci. U.S.A. 94 (1997), 14701–14706),
das den Tod von Zellen fördert,
wo keine funktionale Nukleinsäure
den zellulären
Signaltransduktionsweg inhibiert, der den Promotor induziert. Induzierbare
Gene umfassen bakterielle, virale und eukaryontische Transkriptionseinheitem,
z.B. im Arabinose-Operon (Wilcox et al., J. Biol. Chem. 249 (1974), 2946–2952),
die frühen
und späten
Gene des Vaccinia Virus (Moss. Annu. Rev. Biochem. 59 (1990), 661–688) oder
Promotoren der Immunglobulingene (Staudt et al., Ann. Rev. Immunol.
10 (1991), 373–398).
Die in phänotypischen
Selektionen verwendeten Promotoren können natürlich vorkommende Promotor/Enhancer-Elemente
oder jegliche Kombinationen oder Modifikationen davon sein.
-
Reporterkonstrukte
können
positive und negative phänotypische
Selektionen umfassen. Zum Beispiel kann eine negative phänotypische
Selektion über
die Expression eines Toxins (wie zum Beispiel dem Diphtherie-Toxin)
unter der Kontrolle eines induzierbaren zellulären Promotors, wie dem Interleukin-2-Promotor
in T-Lymphozyten, entstehen. Diese Genkonstrukte können in
Zellen z.B. über
Plasmide eingebracht werden und die Expression des Toxingens wird
von der natürlichen,
den Promotor induzierenden Maschinerie der Zelle vorangetrieben.
Nur in denjenigen Zellen, in denen eine funktionale Nukleinsäure die
Induktion des Toxin-Promotors
inhibiert, wird das Toxin nicht exprimiert werden und die Zellen überleben
und können
selektiert werden.
-
Die
Vektoren (Vektorelemente) und Protokolle für das Klonieren, das Transfizieren,
die transiente Genexpression und das Erhalten stabiler transfizierter
Zelllinien sind gut bekannt und in der Literatur beschrieben (Maniatis
et al., (1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, New York; Spector et
al., Cells: A Laboratory Manual (1998), Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, New York). Vektoren für die Expression der Nukleinsäure-Bibliotheken
können
jegliche allgemein verwendeten Konstrukte sein um fremde Gene in
Zellen einzuschleusen, zum Beispiel lineare PCR-Fragmente, Plasmide,
DNA-Viren wie M13 oder Vaccinia, RNA-Vieren wie Retroviren oder
das Semliki Forest Virus. Die Gene für die Nukleinsäure-Bibliothek
werden unter die Kontrolle geeigneter Promotoren gestellt, bevorzugt
solche, die eine hohe Transkription der Nukleinsäuren ermöglichen.
-
Beispiele,
die aber nicht darauf beschränkt
sind, umfassen Polymerase-III-abhängige Promotoren, wie zum Beispiel
der Hefe-RNAse-P-Promotor (Good und Engelke, Gene 151 (1994), 209–214), Säuger-U6-
oder -tRNAmet-Promotoren (Bertrand et al.,
RNA 3 (1997), 75–88)
retrovirale „long
terminal repeats" (Rossi,
Tibtech 13 (1995), 301–306),
T7-RNA-Polymerase-Promotoren,
wie in Beispiel 1 beschrieben, oder der subgenomische Promotor für die Semliki-Forest-Virus-Replikase
(Tubulekas et al., Gene 190 (1997), 191–95).
-
Vektoren,
die als Reporterkonstrukte benutzt werden, können Plasmide (z.B. pUC18,
pYES2, pCDM8) oder virale Genome, wie zum Beispiel Adenivirus, AAV
oder retrovirale Vektoren (Moloney Murine Leukemia Virus (MoMuLV),
Gibbon Ape Leukemia Virus (GaLV) usw.) sein. Die Verwendung von
stabil transfizierten Reporterzelllinien ist lediglich optional,
sie stellt jedoch ein zuverlässiges
System dar, um Zellen zu selektieren, welche die negative Selektion
in Gegenwart eines Toxins überleben.
-
Unter
Zielmolekülvalidierung
versteht man die Identifizierung eines bestimmten zellulären Zielmoleküls als das
für einen
bestimmten Phänotyp
verantwortliche Schlüsselmolekül. Sie umfasst
die intrazelluläre Anwendung
eines Nukleinsäuremodulators,
der das Zielmolekül beeinflusst,
um dessen Rolle in dem komplexen Netzwerk zellulärer Komponenten zu untersuchen.
Das bevorzugte Ziel ist es zu untersuchen, ob das Zielmolekül für einen
bestimmten zellulären
Status oder Krankheitszustand verantwortlich ist und daher ein gutes Zielmolekül für Medikamente
ist oder ob dies nicht der Fall ist.
-
Unter
Zielmolekülidentifizierung
versteht man die Verbindung einer zellulären Komponente mit einem bestimmten
Phänotyp,
gleichgültig
ob seine biologische Funktion bekannt ist oder nicht. Die Zielmolekülidentifizierung
kann mittels eines funktionalen Nukleinsäwemodulators erfolgen, der
durch phänotypische
Selektion identifiziert worden ist. Das Zielmolekül, dass
durch solch einen Modulator beeinflusst wird, stellt wahrscheinlich
das eigentliche Schlüsselmolekül oder ein
Schlüsselmolekül, dar das
für einen
bestimmten Phänotyp
verantwortlich ist. Das Zielmolekül kann durch seine Interaktion
mit der funktionalen Nukleinsäure
identifiziert werden. Beispielsweise wird das Hefe-Zwei-Hybrid-System,
eine Modifikation des Zwei-Hybrid-Systems, verwendet, um RNA-Protein-Interaktionen
nachzuweisen. Das Drei-Hybrid-System benutzt ein RNA-Hybrid als
Zwischenglied zwischen zwei Hybrid-Proteinen. Die Interaktion der drei
Komponenten dient zur Rekonstitution der transkriptionellen Aktivierung
von Reportergenen durch einen Mechanismus ähnlich dem Zwei-Hybrid-System (SenGupta
et al., Proc Natl. Acad: Sci. 93 (1996), 8496–8501). Die funktionale Nukleinsäure, die
in das RNA-Hybrid kloniert ist wird mit ihrem Zielprotein interagieren,
das als Teil einer Protein-Hybrid-Bibliothek von zellulären Proteinen
exprimiert wird. Daher werden die Reportergene in denjenigen Hefezellen
exprimiert werden, welche beides enthalten, die funktionale Nukleinsäure und
ihr Zielmolekül,
und die Plasmide mit den Zielgenen können isoliert werden.
-
Ein
anderer Ansatz ist es, das Zielmolekül über Komplementations-Klonierung
zu identifizieren, wie in Beispiel 2 dargestellt. Diese Technik
wurde erfolgreich angewandt, um Proteine zu isolieren, die Zelllinien,
welche mutierte endogene Gene enthalten, funktional zu komplementieren,
z.B. die Komplementations-Klonierung von NEMO, einer Komponente
des I B-Kinase-Komplexes
(Yamaoko et al., Cell 93 (1998), 1231–1240).
-
Hochspezifische
Nukleinsäwemodulatoren
zellulärer
Zielmoleküle
können
beispielsweise routinemäßig durch
Anwendung der Technologien SELEX, Aptazyme, Zwei- und Drei-Hybnd-Technologien
oder durch die hier beschriebenen phänotypischen Selektionsmethode
usw. erhalten werden. Die resultierenden Nukleinsäuremodulatoren
können
exo- oder endo gen durch Methoden wie Elektroporation, Lipofektion,
Expression mit viralen Vektorsystemen, Expression von Plasmiden,
die RNA-Polymerase-Promotoren enthalten, usw. in Zellen eingebracht
werden. Funktionale Nukleinsäuren
können
auf ihr Zielmolekül über verschiedene
Mechanismen wirken; sie können
ihr Zielmolekül
entweder in Antikörper-ähnlicher
Weise binden, wodurch sie seine biologische Funktion modulieren.
Zum Beispiel sind chemisch modifizierte, nukleaseresistente Aptamere
für die
Bindung an extrazelluläre
Wachstumsfaktoren wie den „basic
fibroblast growth factor" selektiert
worden (bFGF; vergleiche: [Jellinek et al., Proc. Natl. Acad. Sci
USA 90 (1993), 11227–11231;
Jellinek et al., Biochemistry 34 (1995), 11363–11372.], „human keratinocyte growth
factor" (hKGF; vergleiche:
[Pagratis et al., Nature Biotech. 15 (1997), 68–73]), "platelet derived growth factor" (PDGF; vergleiche:
[Green et al., Biochemistry 35 (1996), 14413–14424]) oder "vascular endothelial
growth factor" (VEGF;
vergleiche: [Green et al., Chem. Biol. 2 (1995), 683–695; Jellinek
et al., Biochemistry 33 (1994), 10450–10456; Ruckman et al., J.
Biol. Chem. 273 (1998), 20556–0567]).
Diese Aptamere blockierten die Bindung ihres extrazellulären Zielmoleküls an seinen natürlichen
Proteinrezeptor und modellierten dadurch die biologische Funktion
des Wachstumsfaktors.
-
Während es
Stand der Technik ist, die biologische Funktion eines extrazellulären Proteins
zu modulieren, ist ein zentraler Aspekt der vorliegenden Erfindung,
das enorme Potential von hochaffinen und hochspezifischen Nukleinsäurereagenzien
zu nutzen, um ein gegebenes Zielmolekül in intrazelluläre Anwendungen
zu modulieren. Dass dies in der Tat gemacht werden kann, wurde repräsentativ
in dem System gezeigt, das beispielhaft in Beispiel 1 erläutert wurde:
Es
wird ein Ansatz beschrieben, der auf der Erkennung zytoplasmatischer
Integrindomänen
durch synthetische, ligandenbindende RNA-Aptamere, die aus einer
kombinatorischen RNA-Bibliothek
isoliert wurden, beruht. Ein auf Vaccinia Virus basierendes System
ermöglichte
hohe zytoplasmatische Expression von RNA-Aptameren (Intrameren),
die gegen die intrazelluläre
Domäne
des β2-Integrins
LFA-1 gerichtet waren, ein Transmembranprotein, das die Zelladhäsion an
intazelluläre
Adhäsionsmolekül 1 (ICAM-1)
vermittelt. Die zytoplasmatische Expression von Integrin bindenden
Intrameren reduzierte die induzierbare Zelladhäsion an ICAM-1 in zwei verschiedenen
Zelltypen. Die Aptamere zielen spezifisch auf eine funktionale zytoplasmatische
Subdomäne
und definieren diese dadurch als wichtig für die Regulation der Zelladhäsion in
Leukozyten.
-
Integrine
sind vielseitige, heterodimere Transmembranproteine, die adhäsive Interaktionen
mit der extrazellulären
Matrix und zellspezifischen Gegenrezeptoren vermitteln. Sie sind
in diverse biologische Prozesse eingebunden, die Apoptose, Zellzyklusregulierung,
Zellwanderung, Blutgerinnung, Gedächtnis und Leukozytenfunktion
umfassen (Hynes, Cell 69 (1992), 11–25). Die zytoplasmatischen
Domänen
der Integrine spielen wahrscheinlich durch die Vernetzung von Signalereignissen
innerhalb der Zelle zu den extrazellulären Domänen eine wichtige Rolle in
der Regulation der Zelladhäsion,
die genauen Mechanismen sind jedoch unklar (Diamond und Springer,
Curr. Biol. 4 (1994), 506–517;
Dedhar und Hannigan, Curr. Opin. Cell Biol. 8 (1996), 657–669). Das
beta 2-Integrin LFA-1 (αL/β2, Cdl1a/CD18)
vermittelt die Adhäsion
von Leukozyten in Immun- und Entzündungsantworten durch Bindung
an zelluläre
Liganden (Lub et al., Immunol. Today 16 (1995), 479–483), die
inteazellulären
Adhäsionsmoleküle ICAM-1,
-2, -3, -4. Eine große
Datenmenge legt nahe, daß die
zytoplasmatischen Domänen
der beta 2-Kette wahrscheinlich durch ihre Interaktionen mit intrazellulären Proteinen
und/oder dem membrannahen Zytoskelett (Stewart etal., J. Cell Biol.
140 (1998), 699–707)
mit der Regulierung der durch LFA-1 vermittelten Haftfähigkeit
in Verbindung steht; einige Kandidaten für die zytoplasmatischen Liganden
sind kürzlich
identifiziert worden (Hibbs et al., J. Exp. Med. 174 (1991), 1227–1238; Pavalko
und LaRoche, J. Immunol. 151 (1993), 3795–3807; Sharma et al., J. Immunol.
154 (1995), 3461–3470
; Kolanus et al., Cell 86 (1996), 233–242). Um weitere Einsichten
in diese Mechanismen zu erlangen, wurde ein System entwickelt, das
die spezifische Inhibition der von LFA-1 vermittelten „Inside-out"-Signaltransduktion in vivo erlaubt.
Die Ziele waren die Selektion von Aptameren, die die zytoplasmatische
Domäne
von CD18 binden, die Entwicklung und Anwendung eines Systems, das
hohe Expression von Aptameren im Zytoplasma von Leukozyten erlaubt
und die Untersuchungen ihrer biologischen Effekte im Kontext lebender
Zellen.
-
Kurzum,
wie in Beispiel 1 unten genau beschrieben, waren die Autoren in
der Lage
- a) RNA-Aptamere zu isolieren, die
an ein immobilisiertes Peptid binden, welches der zytoplasmatischen Domäne des integralen
Transmembranproteins LFA-1 (CD18, beta 2-Integrin) entspricht.
- b) zu zeigen, dass die Aptamere ihr Zielmolekül spezifisch
erkennen, sogar wenn sie in einen andern Sequenzkontext in einer
RNA-Expressionskassette kloniert sind. Diese Kassette wurde von
einem T7-RNA-Polymerase-abhängigen
Expressionssystem (Fuerst und Moss, J. Mol. Biol. (1989), 333–348) abgeleitet
und enthielt eine 5'-„Stern-Loop-Struktur", von der vorher
gezeigt werden konnte, dass sie RNA-Moleküle im Zytoplasma durch Verhinderung
ihres Abbaus stabilisiert. Des weiteren enthielt das 3'-Ende das T-Terminator-„Hairpin-Loop-Motiv" als einen Terminator
für die
von der T7-RNA-Polymerase abhängigen Transkription.
Ein anderes Beispiel für
ein zytoplasmatisches System, das für RNA-Expression geeignet sein
dürfte,
basiert aus der RNA-abhängigen
RNA-Replikase viraler Vektoren, wie dem Semliki Forest Virus (SFV)
(Tubulekas et al. Gene 190 (1997), 191–195). Am bedeutsamsten war,
dass gezeigt werden konnte, dass die von der T7-RNA-Expressionskassette
entstehenden Aptamertranskripte richtig terminiert waren und ihre
Bindungsaktivität
für CD18cyt
vollständig
behielten. Nicht richtig terminierte Aptamersequenzen, die zusätzliche
Sequenzinformation aus dem viralen Genom enthielten, wurden ebenfalls
auf Bindung an CD18cyt getestet. Es wurde festgestellt, dass diese
unerwünschte
zusätzliche
Sequenzinformation in vitro wahrscheinlich wegen der Störung der
korrekten Aptamerfaltung zu einer drastisch reduzierten Bindungsaktivität dieser
Aptamervarianten an CD18cyt führte.
Dieses Ergebnis etabliert zwei sehr wichtige und überraschende
Punkte. Es zeigt, dass es möglich
ist, (i) das Aptamer im Kontext zusätzlicher Sequenzinformation,
wie zum Beispiel von stabilisierenden Sequenzmotiven oder von RNA-Lokalisierungssignalen
usw., zu exprimieren, was entscheidend für die breite Anwendung von
endogener Aptamerexpression ist und (ii) kleine kompakte Aptamere,
die in vitro selektiert wurden, auf modulare Art zu konstruieren,
um die Aptamere mit zusätzlichen
Eigenschaften zu versehen, was die angemessene und effiziente intrazelluläre Anwendbarkeit
sicher stellt, ohne die Aptamerbindung an das Zielmolekül zu beeinflussen.
- c) zu zeigen, dass die selektierten Aptamere endogenes LFA-1
spezifisch in Rohextrakten von Jurkat-E6-Zellen erkennen. Sie tun
dies auch im Kontext der RNA-Expressionskassette.
- d) ein auf Vaccinia Viren basierendes RNA-Expressionssystem
zu entwickeln, das hohe zytoplasmatische Expression von RNA-Aptameren,
die gegen die zytoplasmatische Domäne des Integrins LFA-1 gerichtet sind,
ermöglichte.
- e) den Phänotyp
von zwei verschiedenen Zelltypen, Jurkat E6 und periphären mononukleären Blutzellen (PBMC),
durch die zytoplasmatische Expression von Aptameren, die in diesen
Zellen die zytoplasmatische Domäne
von LFA-1 binden, zu verändern.
In diesem Fall reduzierte die zytoplasmatische Aptamerexpression
spezifisch die induzierbare Zelladhäsion an das interzelluläre Adhäsionsmolekül 1 (ICAM
1). Dieses Ergebnis ist unerwartet und repräsentiert aus mehreren Gründen einen
der zentralen Aspekte dieser Erfindung: i) Zum ersten Mal ist das
intrazelluläre
Zielmolekül
ein Protein oder eine Proteindomäne
ohne jede bekannte Affinität
für Nukleinsäuren. Der
neue und unerwartete Aspekt dieses Befunds ist, dass das in vitro selektierte
Aptamer das Zielmolekül,
für das
es selektiert wurde, spezifisch in einem intrazellulären Kompartiment
findet, das sehr verschieden von anderen Kompartimenten ist, weil
es eine große
Zahl von professionellen, RNA-bindenden Proteinen enthält (wie
zum Beispiel Helikasen, ribosomale Proteine, hnRNP-Proteine usw.),
die in hohen Kopienzahlen von mehreren Millionen pro Zelle vorkommen.
Diese professionellen, RNA-bindenden Proteine können mit sehr hoher Affinität (Kd 10–9 – 10–8)
an spezielle RNA-Motive binden, aber sie können auch mit RNAs in einer
mehr von der Sequenz unabhängigen
Art mit immer noch relativ hoher Affinität interagieren [z. B. die hnRNP-Proteine
A1 und C1); Kd 10–7 – 10–6 M;
vergleiche: Nagai und Mattaj (Eds.), RNA-Protein Interactions (1994),
Oxford University Press, 138–140].
Trotz der Erwartung, dass diese professionellen, RNA-bindenden Proteine
mit dem kognaten Aptamerziehnolekül um die Bindung an das Aptamer
kompetitieren, wurde hier gezeigt, dass das Aptamer sein Zielmolekül innerhalb
der Zelle finden kann und den Phänotyp
der Zelle ändern
kann. ii) Obwohl die in vitro Affinität des Aptamers für seine
kognate CD18cyt-Proteindomäne
verhältnismäßig niedrig
war (Kd 10–6 – 5 × 10–7 M),
erkennt es sein Zielmolekül
mit unerwarteter Spezifität.
Dies zeigt, dass dieses System nicht auf Zielmoleküle beschränkt ist,
die durch ein Aptamer mit Affinitäten erkannt werden, die signifikant
höher sind,
als die unspezifischen Interaktionen zwischen einem professionellen,
RNA-bindenden Protein
und einer Nukleinsäure.
Dies eröffnet
die hoch interessante Möglichkeit,
Aptamere generell anzuwenden, sogar wenn sie ihre Zielmoleküle mit relativ
niedriger Affinität
binden. iii) Es wurde zu ersten mal gezeigt, dass funktionale Nukleinsäureliganden
in zellulären
Subkompartimenten wirken können,
in denen Nukleinsäuren
normalerweise nicht vorkommen. In diesem speziellen Fall findet
das Aptamer sein Zielmolekül,
obwohl es sogar in der negativ geladenen Plasmamembran inklusive
dessen kompakten zytoskelettalen Proteinkortex verankert ist. Es
konnte im Voraus nicht erwartet werden, dass das Zielmolekül für das Aptamer
an diesen Stellen zugänglich
sein würde.
- f) die Bindungsstelle an die zytoplasmatische Domäne von LFA-1
(CD18cyt) für
das Aptamer unter Verwendung synthetischer Peptide, die überlappende
Regionen der zytoplasmatischen Domäne von LFA-1 enthalten, zu
entschlüsseln.
Dadurch wurde die Aptamerbindungsstelle auf CD18cyt als funktional
wichtig für
die stimulierte Zelladhäsion
von LFA-1 an ICAM-1 bestimmt.
-
Das
meiste unseres derzeitigen Wissens über die Faktoren und Mechanismen,
welche die Integrinaktivierung kontrollieren, stammt bis heute von
Mutationsanalysen der Integrindomänen. Diese Studie zeigt, das es
nunmehr möglich
ist, dieses Thema unter Erhaltung der genetischen Integrität der Wildtyp-CD18-Domäne zu adressieren.
Zum ersten Mal wurde gezeigt, dass die intrazelluläre Expression
von CD18cyt-spezifischen Aptameren den funktionalen Zustand endogener
LFA-1-Moleküle
beeinflussen kann. Des weitern wird der Nachweis erbracht, dass
diese „Intramere" auf eine spezifische
Subdomäne
des zytoplasmatischen β2-Rest abzielen,
wodurch die Wichtigkeit dieser Region für die korrekte Funktion des
endogenen LFA-1-Rezeptors hinsichtlich seiner Adhäsion an
ICAM-1 nahegelegt wird.
-
Der
gegenwärtige
Stand der Technik ist in der Lage, Nukleinsäureliganden hervorzubringen,
die beinahe jedes gegebene Zielmolekül mit hoher Affinität und Spezifität binden.
Die Ausweitung auf die Automatisierung der Selektionsprozeduren
dürfte
die Herstellung solcher Verbindungen sogar beschleunigen. Obwohl der
Prozess der in vitro Selektion von Nukleinsäureaptameren derzeit weitgehend
manuell bewerkstelligt wird, ist von dem ersten voll automatisierten
Selektionsprotokoll berichtet worden und es kann vermutet werden, dass
es in Zukunft möglich
sein wird, Hunderte Nukleinsäureliganden
innerhalb von wenigen Tage zu isolieren, was den Prozess viel effizienter
machen würde,
als Verbindungsbibliotheken kleiner Moleküle nach möglichen Effektoren zu durchsuchen
(vergleiche Cox. et al., Biotechnol. Prog. 14 (1998), 845–50).
-
Die
oben beschriebene Methode wendet diese spezifischen Nukleinsäuremodulatoren
an, um die Rolle einer intrazellulären Komponente in dem komplexen,
intrazellulären,
regulatorischen Netzwerk zu bestimmen oder herauszufinden, ob es
der kausale Grund für
einen gegebenen Phänotyp
ist. Des weiteren ist es ein leistungsfähiges System, das die Validierung
potentieller Medikamentenzielmoleküle ermöglicht.
-
Die
intrazelluläre
Anwendung von funktionalen Nukleinsäureliganden kann leicht erreicht
werden. In Abhängigkeit
von der Eignung und den Fragestellungen kann die funktionale Nukleinsäure entweder
endogen oder exogen verabreicht werden. In letzterem Fall werden
bevorzugt chemisch modifizierte Formen angewendet, die den Abbau
der funktionalen Nukleinsäu re
verhindern. Stabilisierende Modifizierungen umfassen beispielsweise
RNA-Moleküle,
in denen die 2'-Hydroxy-Funktion
der Pyrimidinbasen durch 2'-Amino-
oder 2'-Fluoro-Reste
ersetzt ist. Endogene Transkriptionssysteme, wie zum Beispiel das
in Beispiel 1 beschriebene, ermöglichen
die effiziente intrazelluläre
Expression funktionaler Nukleinsäuren.
In Abhängigkeit
von der Anwendung und der Wirkungsstelle werden geeignete Vektoren
und Promotoren gewählt,
welche dem Fachmann bekannt sind. Die funktionalen Nukleinsäuren können mit
zusätzlichen
Nukleinsäuredomänen konstruiert
werden um die korrekte Faltung und Lokalisierung sicherzustellen.
-
In
einem zweiten Aspekt betrifft die vorliegende Erfindung Verfahren
für die
Expression von randomisierten Nukleinsäurebibliotheken in Zellen für die Identifizierung
von funktionalen Intrameren, die den Phänotyp der Zelle ändern, in
der sie exprimiert werden. Dass dies möglich ist, ist eine direkte
Konsequenz der Wirksamkeit des oben beschriebenen Systems. Dieser
Zielmolekül-Identifizierungsansatz
ist nützlich
bei der direkten Identifizierung einer zellulären Komponente als für einen
besonderen Phänotyp
wichtig oder als daran beteiligt, wie beispielsweise ein bestimmter
Zustand, der mit einer Erkrankung verbunden ist. Auf der Grundlage des
derzeitigen Wissens kann abgeschätzt
werden, dass eine Nukleinsäurebibliothek,
die in Zellen exprimiert wird, einzelne funktionale Nukleinsäuren enthält, die
in der Lage sind, spezifisch eine zelluläre Komponente zu erkennen.
Auf der Grundlage dieser Feststellungen, die im ersten Teil dieser
Erfindung beschrieben worden sind, kann auch erwartet werden, dass
dieses spezifische Erkennen beibehalten wird oder allgemein innerhalb des
zellulären
Kompartiments möglich
ist. Zum Beispiel sollte die Bibliothek Moleküle enthalten, die in der Lage sind,
Aktivatoren oder Inhibitoren von Signaltransduktionswegen zu binden
oder zu modifizieren, wodurch der Phänotyp moduliert wird, der mit
der Kaskade verbunden ist. In einem derartigen Fall kann die Zelle,
die die funktionale Nukleinsäure
enthält,
durch phänotypische
Selektion (siehe Definition oben) isoliert werden. Sofern eine klare
Abtrennung auf der Grundlage des geänderten Phänotyps nicht möglich oder
schwierig ist, können Reporter-Konstrukte verwendet
werden, die eine genauere oder bessere Unterscheidung erlauben.
Dies kann erreicht werden ohne vorherige Kenntnis der Natur des
Zielmoleküls
oder seiner biologischen Funktion. Somit ist der erste Schritt dieses
Verfahrens die phänotypische
Selektion. Die Nukleinsäurebibliothek
wird in die Zelle beispielsweise vermittels Expressionsvektoren
eingeführt.
Eine phänotypische
Selektion kann dann bevorzugterweise vermittels Reporterkonstrukte
durch entweder positive oder negative Selektion erreicht werden.
Positive phänotypische
Selektion könnte,
beispielsweise, durch Expression eines geeigneten Zelloberflä chenmarkers
und nachfolgender Affinitätsisolierung
der Zelle mit einem Oberflächenmarkerspezifischen
Antikörper erfolgen.
Negative phänotypische
Selektion kann erfolgen vermittels der Expression eines Toxins (wie
beispielsweise des Diphtherie-Toxins oder Herpes simplex-Virus-Thymidin-Kinase)
unter der Kontrolle eines induzierbaren zellulären Promotors. Diese Genkonstrukte
können
in Zellen beispielsweise durch Plasmide eingeführt werden und die Expression
der Toxin-Gene wird durch die natürliche Promotor-induzierende
Maschinerie der Zelle angetrieben werden. Nur bei jenen Zellen,
in denen eine funktionale Nukleinsäure die Induktion des Toxin-Promotors
inhibiert wird, wird das Toxin nicht exprimiert werden und die Zellen überleben
und können
selektiert werden. Nach der Anreicherung des selektierten Phänotyps werden
die funktionalen Nukleinsäuremodulatoren
durch Sequenzieren identifiziert und einzeln hinsichtlich ihrer
Funktionalität
getestet. Nachdem die funktionale Nukleinsäure identifiziert und hergestellt
worden ist, kann sie wiederum verwendet werden, um ihren intrazellulären Wechselwirkungspartner
zu identifizieren. Dies kann, beispielsweise, durch Standardverfahren
zur Untersuchung von Protein/Nukleinsäure-Wechselwirkungen erreicht
werden, wie beispielsweise das Screenen von Proteinen, die von cDNA-Bibliotheken
gemäß dem Hefe-Drei-Hybrid-System
exprimiert werden, um RNA/Protein-Wechselwirkungen in vivo nachzuweisen
(siehe: SenGupta et al., Proc. Natl. Acad. Sci. 93 (1996), 8496–8501).
Bevorzugterweise erfolgt die Identifizierung vermittels Komplementierung in
homologen Zellen. Durch Expression von cDNA-Bibliotheken in homologen
Zellsystemen können
Klone isoliert werden, die die Signaltransduktionskaskade komplementieren,
die durch die funktionale Nukleinsäure moduliert worden ist. Zur
gleichen Zeit können
Reportersysteme in Zellen konstruiert werden, bei denen die Aptamer/Zielmolekül-Wechselwirkung
durch Systeme bestätigt
wurde, wie beispielsweise das Drei-Hybrid-System (siehe: SenGupta
et al., Proc. Natl. Acad. Sci. 93 (1996), 8496–8501) oder den in Beispiel
2 beschriebenen Ansatz (siehe unten) für die positive Selektion, die
negatives Selektionskorrekturlesen umfasst. Dies liefert ein zweifaches
Ablesergebnis für
die verlässliche
Identifizierung jener Proteine, die eine Schlüsselrolle für einen bestimmten Phänotypen
in einem Wildtyp-Kontext spielen.
-
Zusätzlich identifizieren
die phänotypischen
Selektionssysteme auch Nukleinsäureinhibitoren,
die einfach für
die Targetvalidierung verwendet werden könnten (siehe erster Teil dieser
Erfindung). Dieses Verfahren liefert somit einen erheblichen Beitrag
für das
Gebiet des functional genomics/proteomics, da es Zielmoleküle direkt
in einem unveränderten
zellulären
Status durch ihre Funktion und nicht ihre Gene oder das Vorhandensein
in einer Zelle identifiziert. Deshalb ist das Verfahren der vorliegenden
Erfindung dem Stand der Technik überlegen.
-
Die
identifizierten funktionalen Nukleinsäuren können als Wirkstoff-Leads dienen
oder können
selbst als Therapeutika verwendet werden, insbesondere bei gentherapeutischen
Ansätzen,
oder in einer pharmazeutischen Zusammensetzung, die eine funktionale
Nukleinsäure
umfasst, die durch die Verfahren der vorliegenden Erfindung identifiziert
worden und die in der Lage ist, an funktionale intrazelluläre Zielmoleküle zu binden
und deren Funktion zu modifizieren, optional in Kombination mit
einem geeigneten pharmazeutischen Träger.
-
Beispiele
für geeignete
pharmazeutische Träger
sind in der Technik gut bekannt und umfassen Phosphat-gepufferte
Salinelösungen,
Wasser, Emulsionen, wie beispielsweise Öl/Wasser-Emulsionen, verschiedene Arten von Benetzungsagenzien,
sterile Lösungen
etc. Derartige Träger
können
durch herkömmliche
Verfahren formuliert und können
einem Lebewesen in einer geeigneten Dosis verabreicht werden. Die
Verabreichung von geeigneten Zusammensetzungen kann auf verschiedenen
Wegen erfolgen, beispielsweise durch intravenöse, intraperitoneale, subkutane,
intramuskuläre,
topische oder intradermale Verabreichung. Der Verabreichungsweg
hängt natürlich von
der Art der Erkrankung und der Art der in der pharmazeutischen Zusammensetzung
enthaltenen Verbindung ab. Das Dosierungsregime wird durch den behandelnden
Arzt oder durch andere klinische Faktoren bestimmt. Wie in der Medizin
gut bekannt, hängt
die Dosierung für
einen Patienten von vielen Faktoren ab, einschließlich der
Größe des Patienten,
der Körperoberfläche, des
Alters, des Geschlechts, der speziell zu verabreichenden Verbindung,
der Zeit und Art der Verabreichung, der Art der Erkrankung, des
allgemeinen Gesundheitszustandes und anderen gleichzeitig verabreichten
pharmazeutischen Wirkstoffen.
-
Bevorzugterweise
wird die funktionale Nukleinsäure
(z. B. Intramer) direkt an die Zielstelle, beispielsweise durch
biolistische Abgabe, wie beispielsweise ein kolloidales Dispersionssystem,
oder durch Katheter an eine Stelle in einer Arterie verabreicht.
Die kolloidalen Dispersionssysteme, die zur Abgabe der pharmazeutischen
Zusammensetzung verwendet werden können, umfassen Makromolekülkomplexe,
Nanokapseln, Mikrosphären,
Kügelchen
und Lipid-basierte Systeme einschließlich Öl-in-Wasser-Emulsionen, (gemischte)
Mizellen, Liposomen und Lipoplexe. Das bevorzugte kolloidale System
ist ein Liposom.
-
Die
Zusammensetzung des Liposoms ist gewöhnlicherweise eine Kombination
aus Phospholipiden und Steroiden, insbesondere Cholesterin. Der
Fachmann ist in der Lage, derartige Li posomen auszuwählen, die
für die
Abgabe des erwünschten
Nukleinsäuremoleküls geeignet
sind. Organspezifische oder Zell-spezifische Liposomen können verwendet
werden, um eine Abgabe nur an der erwünschten Stelle zu erreichen.
Das Targetieren von Liposomen kann von den Fachleuten durchgeführt werden,
indem allgemein bekannte Verfahren angewendet werden. Dieses Targetieren
umfasst passives Targetieren (unter Verwendung der natürlichen Tendenz
der Liposomen, sich auf Zellen des RES in Organen zu verteilen,
die sinusoidale Kapillaren enthalten) oder aktives Targetieren (z.
B. durch das Koppeln der Liposomen mit einem spezifischen Liganden,
z. B. einem Antikörper,
einem Rezeptor, Zucker, Glykolipid, Protein etc., durch gut bekannte
Verfahren). Bei der vorliegenden Erfindung werden monoklonale Antikörper bevorzugterweise
verwendet, um Liposomen an spezifische Stellen vermittels spezifischer
Zell-Oberflächenliganden
zu targetieren.
-
In
einer noch bevorzugteren Ausführungsform
ist das funktionale Nukleinsäuremolekül (z. B.
Intramer) in einen rekombinanten Vektor integriert, bevorzugterweise
einen Expressionsvektor. Um die Expression nur in dem Zielorgan
zu erreichen, wird das funktionale Nukleinsäuremolekül, das DNA codiert, funktionsfähig mit einem
Gewebe-spezifischen Promotor verbunden und für die Gentherapie verwendet.
Zum Beispiel kann es mit dem CD4-Gen-Promotor (Zhao-Emonet et al., Biochim.
Biophys. Acta. 1442 (1998), 109–119),
sekretorischen Leukoprotease-Inhibitorgen-Promotor (Robertson et
al., Cancer Gene Ther. 5 (1998), 331–336) oder dem karzinoembryonischen
Antigen-Promotor (Fichera et al., Dis. Colon. Rectum. 41 (1998),
747–754)
verbunden sein. Andere Gewebe-spezifische Promotoren sind den Fachleuten
auf dem Gebiet gut bekannt.
-
Bevorzugte
rekombinante Vektoren sind virale Vektoren, beispielsweise Adenovirus,
Herpesvirus, Vakzinia oder bevorzugtererweise ein RNA-Virus, wie
beispielseweise ein Retrovirus. Noch bevorzugterer ist der retrovirale
Vektor ein Derivat eines murinen oder eines Vogel-Retrovirus. Beispiele
für derartige
retrovirale Vektoren, die bei der vorliegenden Erfindung verwendet
werden können,
sind murines Moloney-Leukämie-Virus
(MoMuLV), murines Harvey-Sarkom-Virus (HaMuSV), marines Säugetiervirus
(MuMtV) und Rous-Sarkoma-Virus
(RSF). Am bevorzugtesten wird ein nicht humaner retroviraler Primatenvektor
verwendet, wie beispielsweise der Gibbon Ape Leakemia Virus (GaLV),
der einen breiteren Wirtsbereich bereitstellt verglichen mit murinen
Vektoren. Da rekombinante Retroviren defekt sind, ist eine Unterstützung erforderlich,
um infektiöse
Partikel zu produzieren. Eine derartige Unterstützung kann, beispielsweise,
durch Verwendung von Helferzelllinien bereitgestellt wer den, die
Plasmide enthalten, die alle Strukturgene des Retrovirus unter der
Kontrolle von regulatorischen Sequenzen innerhalb des LTR enthalten.
Geeignete Helferzelllinien sind den Fachleuten auf dem Gebiet gut
bekannt. Diese Vektoren können
zusätzlich
ein Gen enthalten, das für
einen selektierbaren Marker codiert, so dass die transduzierten
Zellen identifiziert werden können.
Darüber
hinaus können die
retroviralen Vektoren so modifiziert sein, dass sie Target-spezifisch
werden. Dies kann, beispielsweise, erreicht werden, indem ein Polynukleotid
eingeführt
wird, das für
einen Zucker, ein Glyolipid oder ein Protein, bevorzugterweise einen
Antikörper,
codiert. Die Fachleute kennen zusätzliche Verfahren zum Erzeugen
von Ziel-spezifischen Vektoren. Weitere geeignete Vektoren und Verfahren
für in
vitro- oder in vivo-Gentherapie sind in der Literatur beschrieben
und sind den Fachleuten auf dem Gebiet bekannt; siehe, beispielsweise,
WO 94/29469 oder WO 97/00957.
-
Eine
pharmazeutische Zusammensetzung kann eine Verbindung enthalten,
die die gleiche oder sogar eine verglichen mit der wsprünglichen
funktionalen Nukleinsäure
(Intramer) erhöhte
Wirkung aufweist, die durch die Verfahren der vorliegenden Erfindung
identifiziert wurde. Eine Strategie zur Erhöhung der Stabilität und der
Verbesserung der Bindungsspezifitäten der funktionalen Nukleinsäuren, die
durch die Verfahren hierin identifiziert worden sind, ist als post-SELEX-Modifikation
oder Entwicklung von post-SELEX-modifizierten Aptameren (Eaton,
Cur. Opin. Chem. Biol. 1 (1997), 10–16) beschrieben worden. Nach
der Identifizierung der funktionalen Nukleinsäure wird eine zweite Nukleinsäurebibliothek
von der Elternsequenz durch kombinatorische chemische Synthese und
zufälligen
Einbau von modifizierten Nukleosiden abgeleitet. Z. B. könnten die Purin-Reste
mit Mischungen aus 2'-OH- und 2'-O-Methyl-substituierten
Nukleosidphosphoramiditen synthetisiert werden. Die Verhältnisse
können
variiert werden und werden von der erwünschten Anwendung abhängen. Modifizierte
Nukleoside, die in der chemischen Synthese enthalten sein können, sind
den Fachleuten auf dem Gebiet ebenso bekannt und schließen, beispielsweise,
Modifikationen an der 5'-Position
von Pyrimidinen, 8'-Position
von Purinen oder der 2'-Position
von allen Nukleotiden ein. Modifikationen schließen ein, sind aber nicht beschränkt auf
Vinyl-, Prenyl-, Fukose-, Biotin- oder Arginin-Gruppen, die den
Nukleinsäuren
zusätzliche chemische
Eigenschaften verleihen. In einem zweiten SELEX-Experiment wird
die modifizierte zweite Bibliothek mit dem Liganden in Kontakt gebracht
und jene Klone identifiziert, die die gleichen oder verbesserte
Bindungseigenschaften verglichen mit der Ausgangssequenz aufweisen.
Die Identifizierung der modifizierten Positionen in den gewinnenden
Oligonukleotiden kann erreicht werden, beispielsweise, durch alkalische
Hydrolyseinterferenzexperimente, da die 2'-O- Methyl-Substitutionen
die benachbarte Phosphodiesterbindung gegenüber alkalischer Hydrolyse schützen. Unter
Verwendung dieses Verfahrens sind Sekundäraptamere gegen den vaskulären endothelialen
Wachstumsfaktor (VEGF) identifiziert worden, die VEGF mit einer
17-fach erhöhten
Affinität
binden, wahrscheinlich infolge zusätzlicher hydrophober Kontakte,
die durch Dimethylsubstitutionen vermittelt werden, und eine 8-fach
erhöhte
Stabilität
gegenüber
Abbau (t1/2 = 171 h) in unverdünntem Rattenurin
(Green et al., Cur. Biol. 2 (1995), 683–695) aufweisen. Die solchermaßen erhöhte Wirksamkeit
(z. B. bessere Affinitäten
oder bessere zelluläre
Aufnahme) kann den Nukleinsäuren
durch chemische Substitution zugefügt werden und die stabilisierten
Formen könnten
für die
exogene Abgabe der funktionalen Nukleinsäuren als pharmazeutische Formulierungen
nützlich
sein, was die Notwendigkeit beseitigen würde, die Gene für die funktionalen
Nukleinsäuren
in die Zielzellen einzuführen.
-
Darüber hinaus
könnten
die funktionalen Nukleinsäuren
als funktionale Leads verwendet werden, um Bibliotheken kleiner
Moleküle
auf neue Wirkstoffe zu screenen. Wenn beispielsweise die funktionale
Nukleinsäure
als ein Inhibitor für
ein Enzym durch die Bindung der aktiven Stelle wirkt, kann diese
Wechselwirkung verwendet werden, um auf kleine Moleküle zu screenen,
die an der gleichen Stelle binden und als ein Mimetikum der funktionalen
Nukleinsäure
wirken. Ein weiteres Binden an die selbe Stelle legt nahe, dass
das Molekül in
der gleichen Art und Weise wie die funktionale Nukleinsäure wirken
kann, nämlich
durch Inhibieren der Enzymfunktion. Diese Mimetika aus kleinen Moleküle könnten, beispielsweise,
identifiziert werden durch Variation des Drei-Hybrid-Systems und
des reversen Zwei-Hybrid-Systems.
Wie in dem Zielidentifizierungsverfahren beschrieben, bilden in
dem Drei-Hybrid-System
zwei Hybrid-Proteine und eine Hybrid-RNA einen Komplex und aktivieren
Gentranskription in Hefe. In dem reverse Zwei-Hybrid-System, das
von Leanna und Hannik, Nucleic Acid Res. 24 (1996), 3341–3347, vorgestellt
worden ist, wird das Reportergen CHY2 die Aktivierung der Transkription
transkribiert, die durch die Wechselwirkung der zwei Hybrid-Proteine
vermittelt wird. Dies führt
zum Zelltod von Hefezellen in Gegenwart von Cycloheximid. In einem
Drei-Hybrid-System mit ähnlichen
Reporterkonstruktionen, die für
die Wechselwirkung der funktionalen Nukleinsäure und in diesem Beispiel
nur dem Zielenzym konstruiert sind, wird die Inhibierung der Anordnung
der drei Hybridkomponenten das Überleben
der Zelle fördern.
Entsprechend kann dieser Test verwendet werden, um Bibliotheken
aus kleinen Molekülen
auf Mimetika zu screenen, die mit der funktionalen Nukleinsäure um die
Bindung an dem Zielenzym im Wettbewerb stehen und deshalb die Bildung
des transkriptionsaktivierenden Komplexes verhindern. Diese Verbindungen
können
weiter auf ihre Fähigkeit
getestet werden, das Enzym funktional zu inhibieren und können als
Wirkstoffe oder Wirkstoff-Leads dienen.
-
Die
funktionalen Nukleinsäuren
der Erfindung oder die oben beschriebenen Verbindungen sind besonders
nützlich
für die
Herstellung eines Medikaments für
die Gentherapie. Sie könnten
neben anderen Beispielen für
die Behandlung von Erkrankungen verwendet werden, von denen vorgeschlagen
wurde, dass sie Ziele für
die Anwendung von intrazellulären
Antikörpern
wären,
beispielsweise HIV-Infektionen (Chen et al., Human Gene Ther. 5
(1994), 595–601)
oder Leukämien,
Brust- und Ovarialkarzinoma (Richardson und Marasco, Tibtech 13
(1995), 306–310).
-
Schließlich können die
funktionalen Nukleinsäuren,
die durch das Verfahren gemäß der vorliegenden Erfindung
identifiziert worden sind, in einem Kit für die Diagnose von Erkrankungen
enthalten sein, die mit einem speziellen intrazellulären Ziel,
wie oben diskutiert, verbunden sind. Ein derartiger Kit kann verwendet
werden, um zu bestimmen, ob dieses Ziel vorhanden ist, eine anormale
Konzentration etc. aufweist.
-
Beispiel 1: Cytoplasmatische
RNA-Modulatoren einer inside-out Signaltransductionskaskade
-
(a) In vitro Selektion
von CD18-bindenden RNA Aptameren
-
Eine
RNA Bibliothek mit einer Komplexität von 5 × 1014 unterschiedlichen
Molekülen
wurde synthetisiert durch in vitro T7-Transkription von dem PCR-amplifizierten
DNA Pool MF76.1: 5'-
TCTAATACGACTCACTATAGGGCGCTA AGTCCTCGCTCA-N40-ACGCGCGACTCGGATCCT-3'; Primer MF39.1: 5'-TCTAATACGACTCACTATAGGGCGCTAA
GTCCTCGCTCA-3' (kursiv:
T7-Promotor, fett: BamHI Restriktionsschnittstelle); Primer Mic20.1:
5'-GTAGGATTCGAGTCGCGCGT-3' (fett: BamHI Restriktionsschnittstelle)
wie zuvor beschrieben (Klug et al., Proc. Natl. Acad. Sci. USA 94
(1997), 6676–6681).
CNBr-aktivierte Sepharose wurde mit synthetischen Peptiden derivatisiert
(CD18cyt, MF2G) oder mit Tris-HCl (pH 8.0) alleine geblockt, gemäß dem Protokoll
des Herstellers (Pharmacia Biotech AB, Uppsala, Schweden). Für die Präselektion wurden
entweder 50–75 μl Tris-blockierte
oder MF2G-derivatisierte Sepharose mit 2–3nmol -32P-markierter
RNA in 150 μl Bindungspuffer
(Puffer B: 4,3 mM K2HPO4,
1,4 mM NaH2PO4,
150 mM NaCl, 1,0 mM MgCl2, 0,1 μM CaCl2) für 1
h bei 23° C
inkubiert. Die Aufschlämmung
wurde in eine Plastiksäule überführt und
in kleinen Fraktionen (75 μl)
mit Puffer B eluiert. Für
die eigentliche Selektion wurden die initialen vier Fraktionen gefällt und
mit 25–50 μl CD18cyt-Sepharose
(4,5 mg/ml Gel) in 100 μl
Puffer B für
1 h bei 23° C
inkubiert. Die Entfernung von nicht-bindenden RNAs wurde erreicht
durch Waschen mit 200 Sepharose-Volumen von Bindungspuffer (5–10 ml).
Gebundene RNAs wurden durch Waschen mit auf 6M Guanidinium-HCl eingestellten
Puffer B eluiert. Die eluierten Fraktionen wurden durch „Cherenkov-Zählung" quantifiziert, mit
Ethanol in Anwesenheit von 10–20 μg Glycogen
gefällt
und wie zuvor beschrieben revers transkribiert (Famulok, J. Am.
Chem. Soc. 116 (1994), 1698–1706);
die cDNA wurde PCR-amplifiziert, gevolgt von in vitro Transkription.
Für den
ersten Zyklus wurden 29 nmol RNA und 120 μl CD18cyt-Sepharose in einem
Gesamtvolumen von 475 μl
benutzt. Die RNA-Bibliothek wurde durch Bindung an ein auf einer
Sepharosematrix immobilisiertes 46-mer Peptid, welches der kompletten
zytoplasmatischen Domäne
von CD18 (CD18cyt) entspricht, 11 Zyklen der in vitro Selektion
unterworfen (1A). Nach Zyklus 11 wurden bindende
Sequenzen zur Einfügung
einer PstI-Restriktionsschnittstelle
mit den Primern Mic20.1 und SK40.4 (5'-TCTAATACGACTCACTATAGGGCTGCAGAGTCCTCGCTCA-3'; kursiv: T7-Promotor,
fett: PstI-Restriktionsschnittstelle) PCR-amplifiziert, kloniert
und sequenziert wie zuvor beschrieben (Famulok, J. Am. Chem. Soc.
116 (1994), 1698–1706).
Vom angereicherten Pool wurden 80 Aptamere kloniert und sequenziert.
Basierend auf Sequenzmotiven konnten diese Aptamere entwederin vier
Klassen oder in verwaiste Sequenzen ohne jede offensichtliche Beziehung
zueinander unterteilt werden.
-
(b) Konstruktion der zytoplasmatischen
Antamer-Expressionskasette
-
Vier
Sequenzen wurden für
die zytoplasmatischen Expressionsstudien ausgewählt. Drei von ihnen, D20, D28,
und D31 sind CD18-Binder, die vierte Sequenz, D42, ist eine nicht-bindende, negative
Kontrollsequenz der selben Länge
und mit Primerbindungsstellen identisch zu den spezifischen Aptameren
(1B). Zuerst, wurde ein T7-RNA-Expressionskasettenvektor (TR) konstruiert,
wo der T7 Promotor der Aptamer-codierenden DNA vorgeschaltet zwischen
dem 5'- und 3'-„Stemloop"-Struktur lokalisiert ist, welche als
RNA-stabilisierende Motive fungieren und für die Terminierung des T7-Transkripts
benötigt werden
(Fuerst and Moss, J. Mol. Biol. 206 (1989), 333–348) (2A). Die
Sequenz der T7-RNA
Expressionskassette, TR, hier gezeigt ohne eingefügtes Aptamer
(5'-GTTAACGCATGCTAATACGACTCACTATAG
GGAGACCACAACGGTTTCCCGGGCGCAAGTTACTAGT-TGGCCA-AGATCTTAATTAATAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTGC
TGTCGAC-GCGGCCGC-3';
fett: Restriktionsschnittstellen in der Reihenfolge in der sie auftreten
HpaI, SphI, XmaI, SpeI, BalI, BglII, PacI, SalI, NotI; Kursiv: T7-Promotor; unterstrichen:
stabilisierender 5'-„Stemloop" und 3'-Terminator T für korrekte
Terminierung der TR-Aptamere) wurden eingefügt in den Vektor pTkg (Romeo
und Seed, Cell 64 (1991), 1037–1046;
Falkner und Moss, Virol. 62 (1988), 1849–1854) über Hpal- und Notl- Restriktionsschnittstellen,
was in dem Transfer-TR-Vektor resultierte. Aptamer-kodierende Sequenzen
wurden via XmaI und PacI Restriktionsschnittstellen eingefügt (TR-Aptamer;
zur Übersicht
siehe 2A). Die resultierenden Transkripte
werden TR-Aptamere genannt. Als Negativkontrolle wurde auch ein
TR-Vektor, dem die Aptamersequenz fehlt, und einer, der die nicht-bindende negative
Kontrollsequenz exprimierte, benutzt (TR-D42).
-
(c) In vitro Bindung von
Aptameren und TR-Aptameren
-
In
vitro Bindung an das CD18cyt-Peptid, immobilisiert auf einer Sepharose
Matrix, wurde für
mehrere Aptamere bestätigt
(1B). Hierfür
wurde das zonale Elutions-, quantitative Affinitätschromatographie-Experiment
verwendet wie in Dean et al., Affmity chromatography a practical
approach (1991), pp 169–174,
IRL Press, Oxford, UK beschrieben. Die Bindungsaffinität der monoklonalen
Aptameren an das immobilisierte CD18cyt-Peptid betrug zwischen 500
und 6000 nM (Tabelle 1). Bindung von individuellen Aptameren an
underivatisierte Sepharose wurde nicht beobachtet. Wichtigerweise
wurde bestätigt,
dass korrekt terminierte Aptamere das selbe in vitro Bindungsverhalten
zum CD18cyt-Peptid zeigten, wenn sie im Kontext der flankierenden „Stemloop"-Strukturen exprimiert
wurden. Jedoch zeigten nicht terminierte TR-Aptamere mit zusätzlichen 166
Basen nach dem Terminator T abgeleitet vom Transfer-TR-Vektor eine
2–3 fache
Reduktion der Bindungsaffinität
für CD18cyt.
Vermutlich resultiert dieser Effekt aus der Beeinträchtigung
der zusätzlichen
Sequenz mit der korrekten Faltung der Aptamere. Diese Ergebnisse
zeigen, dass es möglich
ist die Aptamere im Kontext von definierten, funktionellen Sequenzen
zu platzieren, welche zusätzliche
Funktionalitäten
wie erhöhte
Stabilität
hinzufügen.
Korrekte Terminierung ist ebenso notwendig, da die funktionellen
Nukleinsäuren
eine reduzierte Fähigkeit
ihr Ziel zu binden zeigten, wenn zu umfangreiche zusätzliche
Sequenzen in dem Molekül
anwesend sind.
-
(d) Zytoplasmatische Expression
der funktionalen Nukleinsäuren
-
Zytoplasmatische
RNA-Expression beruht auf der Koinfektion der Zellen mit zwei rekombinanten
Vaccinia Viren (Fuerst und Moss, J. Mol. Biol. 206 (1989), 333–348). Ein
Virus, Vaccinia-T7 (vT7), kodiert für die Baktieriophage T7 RNA
Polymerase; das andere, vTR-Aptamer,
kodiert die Sequenz des TR-Aptamer und ist hergeleitet aus der homologe
Rekombination zwischen Vaccinia Virus und dem TR-Aptamer Vektor.
Die Vaccinia-Expressionskonstrukte
(vTR-Aptamer) wurden hergeleitet über die Rekombination zwischen
dem TR-Vektor und dem Wildtyp-Vaccinia-Virus (WR Stamm), hochtitrige
Virus-Stocklösungen wurden
wie beschrieben (Asubel et al. Cunent protocols in molecular biology
(1987), Wiley, New York) hergestellt und Doppelinfektionen mit rekombinanten
Vaccinia Viren und einem Vaccinia Virus kodierend für T7-RNA-Polymerase (vT7)
wurden durchgeführt
wie zuvor beschrieben (Kolanus et al., Cell 86 (1996), 233–242). In
Kürze wurden 2 × 106 Zellen mittels Zentrifugation gesammelt,
mit PBS gewaschen und in 300μl
RPMI-Medium resuspendiert.
100–150μl von jedem
Virus-Stock (MOI von ~10pfu/Zelle, beschallt für 10s) wurde zugegeben und
für 2 h
bei 37° C
inkubiert, gefolgt von der Zugabe von 4 ml RPMI/10% Kälberserum
und zusätzlicher
Inkubation zwischen 1 bis 7 h.
-
Der
Verlauf der RNA-Expression nach Koinfektion einer Jurkat E6-Zelllinie
mit vT7 und vTR-D31 ist in 2C gezeigt.
Maximalmengen an RNA werden 7 h nach Infektion erreicht, wie mittels „Dot-Blot"-Analyse unter Verwendung
einer DNA-Hybridisierung-Probe komplementär zum 3'-Terminus der Aptamer-Expressionskasette
quantifiziert wurde. Ungefähr
4 × 106 Jurkat E6-Zellen wurden mit Vaccinia Virus,
welcher TR-Aptamer-RNA und T7-RNA-Polymerase exprimiert, infiziert. Die
RNA wurde isoliert wie beschrieben (Asubel et al. Current protocols
in molecular biology (1997), Wiley, New York) und auf eine Hybond
N Membran (NEN Du Pont) als Punkt „geblottet" gemäß dem Protokoll
des Anbieters. Hybridisierung von virus-exprimierter RNA wurde für die Dauer
von 1 h bei 55° C
durch Inkubation von zellulärer
RNA in Prä-Hybridisierungspuffer
durchgeführt
(5x SSC, 0,1% SDS, 5x Denhardt's
Reagenz, 5% Dextransulfat, 100 μg/ml
tRNA), gefolgt von Inkubation in Gegenwart von 20 pmol radioaktiv
markierter Probe MicTR1/28.1 (5'-CAAAAAACCCCTCAAGACCCGTTTAGAG-3') in einem Gesamtvolumen
von 4.0 ml für 1
h. Die „Dot-Blots" wurden dann bei
55° C für 30 min
mit ~20 ml 2x SSC, 0,1% SDS (2x), 1x SSC, 0,1% SDS (2x) und 0,5x
SSC, 0,1% (2x) gewaschen und durch Phosphor-Bildgebung quantifiziert. Die Analyse
von allen TR-Transkripten in vivo bei diesem Peak ergab vergleichbare
Aptamer-RNA-Niveaus von ungefähr
106 Kopien pro Zelle, jedoch nur in Anwesenheit
von vt7 (2D).
-
(e) Spezifität der TR-Aptamere
gegenüber
endogenem CD18/beta 2-Integrin
-
Bevor
der biologische Effekt der TR-Aptamer-Expression untersucht wurde,
musste bestätigt
werden, dass die Aptamere ebenso in der Lage sind spezifisch an
endogenes LFA-1 zubinden. Dies wurde gezeigt, indem radioaktiv markierte,
in vitro vom TR-Aptamer Vektor transkribierte Aptamere mit zytoplasmatischem Rohlysat
von Jurkat E6-Zellen inkubiert wurden und anschließender Gelelektrophorese
unter nativen Bedingunge. 4 × 107 Jurkat E6-Zellen wurden in 400 ml Lysepuffer
(PBS, 0,5% Triton × 100,
0,5 μg/ml
Aprotenin, 0,5 μg/ml
Leupeptin, 1mM PMSF, 1mM MgCl2) lysiert
und bei 0° C
für die
Dauer von 20 min inkubiert. Die Zellkerne wurden durch 10 min Zentrifugation
bei 3000 g entfernt und der Überstand
wurde durch 10 min Zentrifugation bei 10000 g geklärt. Zu 3
ml dieses Überstandes
wurden 2 mg von gereinigtem monoklonalem Antikörper (MHM23, OKZ3) oder 2 ml
Ascites-Flüssigkeit
(MEM170) gegeben und auf 1x PBS in einem Gesamtvolumen von 7 μl eingestellt.
Nach Inkubation bei 0° C
für 2–3 h, wurden
5 μl PBS,
das 3 mM DTT, 1 mM MgCl2, 30 U Rnasin, 75–120 μM tRNA, 20%
Glycerol und 7,5 mg BSA enthielt, zugegeben und für 15 min
bei 0° C
inkubiert. Nach der Inkubation wurden 30–60 fmol 5'-32P-markierte
TR-Aptamere in 3 μl
PBS, 1mM MgCl2, für 30s auf 95° C erhitzt
und für
10 min auf 23° C
abgekühlt,
zugegeben und bei 23° C
für 30
min inkubiert. Das Lysat/Aptamer/Antikörper-Gemisch wurde auf ein
natives, 2% Glycerin enthaltendes 4.5% Polyacrylamidgel geladen (Acryamid/Bis-Acrylamid
60:1) und bei 150 V für
2,5 h in 0.,5 × TAE
Elektrophoresepuffer der Elektrophorese unterzogen.
-
Wie
in 3 in zwei stellvertretenden Experimenten gezeigt,
ergab dies eine Muster von veränderten Banden,
welche atamer-spezifisch waren. Aptamer TR-D20 zeigte ein Bandenmuster
mit signifikant reduzierter elektrophoretischer Mobilität (3A,
Spuren 2, 4) im Vergleich zu Negativkontrollen der lysatfieien RNA (Spur
1) und nicht-bindendem TR-D42 (Spuren 3, 5) in Gegenwart eines 104-fachen Überschusses
an tRNA als nicht-spezifischem Kompetitor. Um zu testen, ob die „Shifts" ebenso LFA-1 spezifisch
waren, führten
wir eine Über-„Shift"-Analyse mit den
zwei unabhängigen,
LFA-1 spezifischen Antikörpern
MHM23 und MEM170 durch, welche entsprechend gegen die extrazelluläre Domäne der β- und α-Untereinheiten von
LFA-1 gerichtet waren und dem Antikörper OKT3 als Negativkontrolle,
welcher die ε-Kette
des T-Zell-Antigenrezeptors erkennt. Die Zugabe sowohl von entweder
MHM23 oder MEM170 (Spuren 6, 7) resultierte in einer überveränderten
Bande. In 3B zeigen wieder beide LFA-1-spezifischen
Antikörper
MHM23 und MEM170 (3B, Spuren 3, 5) die überveränderten
Banden, während
mit dem nicht-verwandten Antikörper
OKT3 kein Über-„Shift" erhalten wurde (Spur
6). Die Experimente, welche in den Spuren 4 und 7 gezeigt werden,
weisen nach, dass das Aptamer TR-D20 keine direkte Affinität entweder
gegenüber
dem spezifischen Antikörper MHM23
oder zu dem unspezifischen Antikörper
OKT3 aufweist. Wenn das in Spur 2 gezeigten Gel-„Shift"-Experimente in Gegenwart von 2 mM unmarkiertem
Aptamer TR-D20 als spezifischer Kompetitor wiederholt wurde, bewegte
sich die gesamte TR-D20 RNA auf dem Niveau von ungebundener RNA
(Daten nicht gezeigt). Analoge Resultate wurden mit TR-D28 und TR-D31
erhalten, was zeigt, dass die Aptamere endogenes CD 18 in Zellrohlysaten
mit bemerkenswerter Spezifität
erkennen.
-
(f) Bestimmung der Bindungsstelle
der TR-Aptamere an der intrzellulären Domäne von CD18
-
Zur
Bestimmung der Bindungsstelle der Aptamers auf CD18cyt wurden Gel-„Shift"-Experimente mit drei synthetischen CD18cyt
Peptidfragmenten durchgeführt
(5). Gel-„Shifts" unter Einsatz der
synthetischen, biotinylierten Peptide wurden wie folgt durchgeführt: 5 μl von 3 nM
radioaktiv markierter Aptamer-RNA in PBS pH7,4 wurden für 30 s auf
95° C erhitzt
und für
10 min bei 23° C
renaturiert. Die biotinylierten Peptide wurden mit 2 mg/ml Streptavidin
in 20 mM Tris-HCl pH 7.4 in einem Gesamtvolumen von 6 μl inkubiert.
Zu dieser Lösung
wurde ein Prämix
gegeben um die Konzentrationen im finalen 20 μl Reaktionsvolumen auf 1 × PBS, 1
mM DTT, 1 mM MgCl2, 7,5 % Glycerin, 2,5
mg/ml BSA, 40 μM
tRNA, 2 U/ml RNasin einzustellen und für 20 min bei 23° C vorinkubiert.
Die Aptamerlösung
(5 μl) wurde
zugegeben, 20 min bei 23° C
inkubiert und auf einem nativen 6% Polyacrylamidgel wie oben beschrieben
einer Gelelektrophorese unterzogen.
-
Bindung
an die Carboxy-terminale Hälfte
von CD18cyt (Peptid A23) wurde bei keinem der Aptamere beobachtet,
jedoch banden TR-D20, TR-D28 und schwächer TR-D31 alle sowohl an
das N-terminale Peptid B 16 und den mittleren Teil C17. B16 und
C17 überlappen
um 8 Aminosäuren
inklusive einer Anhäufung
von drei basischen Argininresten. Das diese hoch positiv geladene
Anhäufung
von den Aptameren als Bindungsstelle auf CD18cyt ausgewählt wurde
ist nicht überraschend,
da ein Aptamer ein stark negativ geladenes Molekül darstellt. Wie erwartet zeigte
die Negativkontrolle Aptamer TR-D42 keinne Bindung an jedes der
drei Fragmente, was bestätigte,
dass die Peptidfragmenterkennung aptamer-spezifisch ist.
-
(g) Effekte der intrazellulären TR-Aptamere
auf die integrin-vermittelte zelluläre Adhäsion an das interzelluläre Adhäsionsmolekül 1 (ICAM1)
-
Um
zu überprüfen, ob
die zytoplasmatische Expression von CD18-spezifischen Aptameren
einen Effekt auf die biologische Aktivität dieses Integrins hat, wurden
Adhäsionsexperimente
mit Jurkat E6-Zellen oder mit periphären mononukleären Blutzellen
(PBMC) durchgeführt
(Coligan et al., (1994) Current protocols in immunology (John Wiley & Sons, New York).
Die Zellen wurden mit Vaccinia Viren, die CD18-spezifischen Aptamere
oder unterschiedlichen Kontrollsequenzen kodierenden, infiziert
und die Bindung dieser Zellen an einen wichtigen Liganden von LFA-1,
das ICAM-1-Molekül,
wurde in vitro festgestellt. Dieses System hat einen zusätzlichen
Kontrolllevel, da bekannt war, dass die TR-Aptamer-Expression gänzlich von
der Ko-Infektion von Zellen mit einer Virus exprimierenden T7-Polymerase
abhängig
ist (2D). Das ICAM-1-RG Fusionsprotein wurde in COS
Zellen exprimiert, von Kulturüberständen durch
Protein A-Sepharose gereinigt, eluiert, in PBS resuspendiert und,
wie beschrieben, auf Plastikschalen beschichtet (Kolanus et al.,
Cell 86 (1996), 233–242). 2 × 106 Jurkat oder PBM-Zellen wurden mit rekombinanten
Vaccinia Viren infziert und für
4–8 h
bei 37° C
inkubiert. Nach Zentrifugation wurden die Zellen in RPMI-Medium
resuspendiert und für
5 min bei 37° C
mit oder ohne den Zusatz von 40 ng/ml PMA inkubiert. Den Zellen
wurde anschließend
für 10
min bei 37°C
ermöglicht an
die ICAM-1-Rg beschichteten Schalen zu adhärieren und der gebundenene
Anteil wurde mit Hilfe eines Fadenkreuzokulars bestimmt. 4A zeigt,
dass infizierte Jurkat E6-Zellen eine bemerkenswerte Hintergrundbindung
an ICAM-1 zeigten, aber dies war nichtsdestotrotz überinduzierbar
durch Phorbol-12-Myristate-13-Acetat
(PMA), einem wohl bekannten Förderer
von LFA-1-vermittelter Leukozytenadhäsion (Lub et al., Immunol.
Today 16 (1995), 479–483).
Bemerkenswerterweise resultierte die Expression des TR-D20 Aptamers
in Jurkat Zellen in einer fast vollständigen Blockade der induzierbaren
Adhäsion,
wohingegen der TR ohne Insert und Einzelinfektionen mit Wildtyp-Vaccinia Virus (wt),
vT7 oder vTR-D20 nahezu keinen Effekt hatten.
-
Die
Reproduzierbarkeit und die Erweiterung der Methode auf andere Systeme
wurde mittels Durchführung
von ähnlichen
Studien mit humanen peripheren mononukleären Blutzellen (PBMC) getestet.
PBMC, die normalerweise schwer zu transfizieren sind, erwiesen sich
als hervorragende Ziele für
die durch rekombinate Vaccinia Viren vermittelte Genexpression.
Eine durchflusszytometrische Analyse von mit Vaccinia exprimierten
Kontrollproteinen, Derivaten von Subdomänen von Cytohesin-1, zeigt
eindeutig, dass alle, Ig-Fusionsproteine mit der Plextrin-Homlogiedomäne (PH)
von Cytohesin-1 (c-Ig-PH) und den PH+c-Domänen (cIg-PH+c) oder der isolierter
Ig-Anteil selbst (cIg), auf gleich hohen Niveaus in humanen PBMC
exprimiert wurden (4B). Diese Kontrollen, die gewählt wurden,
weil von dem zytoplasmatischen Cytohesin-1 vorher gezeigt werden
konnte, dass es ein wichtiger Regulator der adhäsiven Funktion von beta 2-Integrinen
ist (Kolanus et al., Cell 86 (1996), 233–242; Nagel et al., J. Biol.
Chem. 273 (1998), 14853–14861;
Nagel et al., Mol. Biol. Cell 9 (1998), 1981–1994),
und die Aptamer-Viren wurden in Adhäsionsuntersuchungen wie oben
beschrieben mit diesen Zellen getestet. Die PH+c-Domänen (cIg-PH+c)
von Cytohesin-1 hatten einen stark unterdrückenden Effekt auf die PMA-induzierbare
Adhäsion,
wohingegen beide, die isolierte PH-Domäne (cIg-PH), die beträchtlich
reduzierte Phospholipidbindung und Membranassoziation zeigt (Nagel
et al., Mol. Biol. Cell 9 (1998), 1981–1994), oder das Kontrollprotein
keinen Effekt hatten (4C). Diese Ergebnisse stimmen
mit veröffentlichten
Daten überein,
die in Jurkat E6-Zellen erhalten wurden (Nagel et al., J. Biol.
Chem. 273 (1998), 14853–14861).
Am wichtigsten zeigt 4C auch, dass die von dem TR-D20-Aptamer
vermittelte Reduktion der PMA-induzierbaren Adhäsion in PBMC ähnlich zu
der mit cIg-Ph+c beobachteten ist, wohingegen die Negativkontrollsequenz
TR-D42 keinen Effekt auf die beta 2-integrin-vermittelte Adhäsion an
ICAM-1 hatte. Zusammengenommen deuten diese Ergebnisse darauf hin,
dass die adhäsive
Funktion von LFA-1 durch die zytoplasmatische Expression eines Aptamers,
das in vitro für
Bindung an den zytoplasmatischen Teil des Rezeptors selektiert wurde,
spezifisch in Jurkat E6 und PBM-Zellen reduziert wurde (4A, 4C)
(Lub et al., Immunol. Today 16 (1995), 479–483).
-
Beispiel 2: Funktionale
Zielmolekül-Identifizierung
von intrazellulären
Komponenten, die in die Induktion des IL2-Promotors involviert sind.
-
Beispiel
2 veranschaulicht ein positives Selektionsschema, das eine Korrektur
durch negative Selektion einschließt um intrazelluläre Zielmoleküle zu isolieren,
die an Signaltransduktionse reignissen teilnehmen, welche zur Induktion
von Genen unter der Kontrolle des Interleukin-2-Promotors (I1-2 P) in T-Zellen führen. Nach
Stimulierung des T-Zell-Rezeptors (TCR) werden Gene durch eine Signaltransduktionskaskade
unter der Kontrolle des Interleukin-2-Promotors induziert. An der Signaltransduktionskaskade
nehmen unter anderen Proteine teil wie zum Beispiel Proteinkinase
C, Calcineurin, NF B usw. (vergleiche: Abbas et al. (Eds.), Cellular and
molecular immunology 2nd ed. (1994), p. 162, W.B. Saunders Company).
Die Blockade von Calcineurin durch immunsupprimierende Pharmazeutika
wie zum Beispiel Cyclosporin A, FK 506 kann beispielsweise die Induktion
des Promotors abschalten und folglich die Produktion zellulärer Zytokine.
Dies wird in der Klinik genutzt um die Abstoßung von Organtransplantaten
zu verhindern. Der I1-2-Promotor ist ein hervorragendes Modellsystem
mit Calcineurin als einem Beispiel für klinisch relevante, funktionale
Ziel- oder Schlüsselmoleküle, die
identifiziert werden sollten.
-
a) Phänotypische Selektion
-
(1) Konstruktion der IL-2-Promotor
Renorterzelllinie
-
Ein
Reportergen, kodierend für
ein Toxin (Herpes Simplex Virus Thymidin Kinase, HSV-TK) (Fife et al., Gene
Ther. 5 (1998), 614–620)
unter der Kontrolle des IL-2-Promotors ist in einen Vector geklont,
der einen selektierbaren Marker (Aminoglycosid-Phosphotransesterferase, Neo) (Southern
und Berg, J. Mol. Appl. Genet. 1 (1982), 327–341) enthält, der es erlaubt, diejenigen
Zellen durch Detoxifikation der Aminoglycosid Antibiotica (Neomycin,
G418) zu selektieren, welche die betreffenden Gene in ihren Chromosomen
insertiert haben (1, Plasmid pP1). In T-Zellen
eingeführt,
wird der IL-2-Promotor nach Stimulation mit Phorbolestern oder durch
den T-Zell-Rezeptor (TCR) induziert, und die Herpes Simplex Thymidin
Kinase (HSV-TK) wird exprimiert. Die Zugabe von Ganciclovir bewirkt
die Bildung von toxischen Verbindungen (z.B. Kanai et al., Cancer Res.
56 (1996), 1341–5)
und nachfolgend den Zelltod.
-
Nach
Transfektion einer Zelllinie (Jurkat E6 Zellen) und deren Kultivierung
in Medium (RPMI, 10 % FCS, Gentamycin 50 μl/ml), welches (in Abhängigkeit
von der minimalen letalen Konzentration für die verwendete Zelllinie)
0,1–1,5
mg/ml Neomycin enthält,
sowie Isolierung von einzelnen Transfektanten werden die Zellen
auf die Anwesenheit und Expression des Reportergens mittels Standardmethoden
(z.B. Hybridisierung nach Northern, „Western Blotting") untersucht.
-
(2) Konstruktion der RNA
Expressionsvektoren
-
Eine
Bibliothek einzelsträngiger
DNA (ssDNA) wird z.B. durch Standard-Festphasensynthese synthetisiert,
wobei die Region der Zufallsequenz von unveränderlichen Sequenzen, die als
Primer-Hybridisierungsstellen dienen und Restriktionsschnittstellen
für Endonukleasen
zur Klonierung in die Expressionsvektoren enthalten, flankiert wird.
Die dopplelsträngige
DNA (dsDNA) wird beispielsweise enzymatisch durch PCR oder Klenow-Protokolle
hergestellt. Dies entspricht den Standartprotokollen zur Herstellung
von Nukleinsäure-Bibliotheken
für in
vitro Selektionsexperimente (siehe z. B. Klug et al., Proc. Natl.
Acad. Sci. 94 (1997), 6676–81; Abelson,
Methods in Enzymology 267 (1996), 275–81, Academic Press, San Diego).
-
Die
Bibliothek der Zufallssequenzgene ist unter der Kontrolle eines
RNA-Polymerase-III-Promotors (Pol
III P) kloniert, welcher hohe Level an RNA-Expression erlaubt (6,
Plasmid pP2). Zusätzlich
ist die Bibliothek flankiert von konservierten Sequenzen, die in
der Lage sind, eine Stammstruktur („Stern") auszubilden, ähnlich der adenoviralen VAI-RNA
endständigen
Stammstruktur (VAI Zufallssequenz VAI) (Furtado et al., J. Virol.
63 (1989), 3423–3434).
Die resultierenden RNA-Transkripte bestehen somit aus der folgenden
Sequenz: 5'-GGGCACUCUUCCGUG-N40-AACGGGGGAGCGCUCCUUUU-3', wobei „N40" die vierzig aufeinanderfolgenden
Zufallspositionen mit gleicher Stöchometrie für alle vier Basen (A, C, G,
U) repräsentiert.
Es wurde vormals gezeigt, dass diese Konstrukte sehr effizient vom
Zellkern in das Zytoplasma von eukaryotischen Zellen exportiert
werden (Bertrand et al., Program & Abstracts
RNA '98: The third
annual meeting of the RNA Society 26. – 31.5.1998, University of
Winsconsin, Madison). In dieser Ausführungsform wird der überwiegende.
-
Teil
der RNA-Bibliothek in das Zytoplasma exportiert, wo sich die meisten
Zielmoleküle
befinden, die an der Induktion des IL-2-Promotors beteiligt sind.
-
Das
Plasmid (6, Plasmid pP2) enthält zusätzlich einen
selektierbaren Marker (Hygromycin; das Antibiotikum Hygromycin und
Resistenzmarkergene sind käuflich
und beschrieben, z.B. bei Invitrogen BV, Groningen, Niederlande)
und Elemente für
die episomale Replikation, um den Beibehalt des Plasmids sicherzustellen
(OriP, EBNA1-Protein-Gen) (Yates et al., Na ture 313 (1985), 812–815; Chittenden
et al., J. Virol. 63 (1989), 3016–25). Dies könnte notwendig
werden, wenn der Selektionsprozess über einen längeren Zeitraum andauert. Das
Bibliotheksplasmid ist in die Reporterzelllinie transfiziert und
der zeitliche Verlauf, die maximale RNA-Bibliotheksexpression und
die RNA-Lokalisierung werden mit Hilfe von Standarttechniken, wie „Northern blots" und in situ Hybridisierung
bestimmt. In einem typischen Transfektionsexperiment werden 107 bis 108 Zellen
durch Elektroporation oder Lipofektion transfiziert. RNA-Proben,
die von transfizierten Zellen präpariert wurden,
werden gemäß den in
Beispiel 1 beschriebenen Methoden ((d) cytoplasmatische Expression
von funktionalen Nukleinsäuren)
isoliert und untersucht und alsdann mit einem radioaktiv-markierten „Antisense-Oligonukleotid" (5'-AAAAGGAGCGCTCCCCCGTT-3') nachgewiesen. Die
Untersuchung ergab Expression-Level von etwa 5 × 105 RNA-Kopien
pro Zelle.
-
(3) Selektion
-
Nach
Transfektion und Expression der RNA-Bibliothek werden die Reporterzellen
(stabil transfizierte Jurkat-E6-Zellen) nun für den Phänotyp der inhibierten IL-2
Promotorinduktion selektiert. Die Zellen werden stimuliert mit Phorbol-12-myristat-13-acetat
(PMA) (50 ng/ml) (Kolanus et al., Cell 86 (1996), 233–242) und
in der Anwesenheit von Ganciclovir (Obaru et al., Hum. Gene Ther.
7 (1996), 2203–08)
inkubiert. Nur diejenigen Zellen, die eine funktionale Nukleinsäure aus
der Bibliothek, welche die Induktion oder Expression des HSV-TK-Gens
(siehe 8: hemmendes Protein X der schematisch gezeichneten
Signaltransduktionskaskade X, Y, Z) inhibiert, wird diese Selektion überleben.
Die anderen Zellen werden absterben (7). Um die Anreicherung
von Zellen zu ermöglichen,
kann die Stringenz der Selektion variiert werden, falls notwendig
unter Verwendung von verschiedenen Konzentrationen an Ganciclovir
und verschiedenen Inkubationszeiten oder wiederholten Zyklen von
Stimulation und Inkubation mit dem Wirkstoff. Die verbleibenden
Zellen können geerntet
werden und die Plasmide, die das Gen für das inhibierende RNA-Molekül tragen,
können
zur Amplifikation in E.coli mit Hilfe „Hirt Supernatant Methode" transfiziert werden.
Die Zellen werden für
1–20 min.
in 0,4 ml 0,6 % SDS, 10 mM EDTA aufgeschlossen. Die viskose Mischung
wird mit einer Pipette in ein Zentrifugenröhrchen überführt. Nach Zugabe von 0,1 ml
5 M NaCl und Mischung werden die Proben für 5 Stunden auf Eis inkubiert.
Nach 4 min. Zentrifugation wird der Überstand vorsichtig abgenommen,
mit Phenol extrahiert und mit 10 μg
linearem Polyacrylamid versetzt. Zweitens können alle funktionalen Nukleinsäuren eliminiert
werden, die nur aufgrund der Inhibition des Toxins wirken und die
Signaltransduktion nicht direkt beeinflussen.
-
(c) Identifizierung von
funktionalen zellulären
Zielmolekülen
-
(1) Konstruktion des Vektors
für die
Identifizierung des funktionalen Zielmoleküls:
-
Die
wesentlichen funktionalen Elemente des geeigneten Plasmids sind
in 9 dargestellt.
-
Plasmid pF1:
-
- – Promotor-Elemente:
CMV
P: humaner Cytomegalovirus sehr früher ("immediate early") Promotor. Beispiel eines konstitutiven
Promotors, der in einer Vielzahl von Zelltypen aktiv ist. In Abhängigkeit
von Zelltyp, Anwendung etc. können
andere häufig
verwendete Promotoren oder Kombinationen von Promotorelementen (z.B.
SV 40 früher
Promotor, Rous Sarcoma Virus sehr früher Promotor etc.) benutzt
werden (Spector et al., Celle: A Laboratory Manual (1998), Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 67.2–67.3) IL-2
P: IL-2-Promotor.
- – Episomale
Replikation:
Der Ursprung der Replikation (OriP) und das nucleare
Antigen (EBNA-1) des Epstein Barr Virus (EBV) ermöglichen
die episomale Replikation (außerhalb
des Chromosoms) in hoher Kopienzahl und Erhaltung in Primär- und Kanninchenzelllinien
(Yates et al., Nature 63 (1989), 3016–25). Dies ist ein optionales
Element, was es jedoch ermöglicht,
die Selektion, wenn notwendig, über
einen längeren
Zeitraum fortzusetzen.
- – cDNA-Bibliothek:
Die
cDNA-Bibliothek in diesem Beispiel ist von T-Zellen abgeleitet.
Die Bibliothek wird verwendet, um auf die Komplementierung der inhibierten
Signaltransduktionskaskade zu selektieren. Die Überexpression des Faktors,
der durch die funktionale Nukleinsäure inhibiert wird, sollte
die Hemmung der Signalweiterleitung bewirken und der IL-2-Promotor
sollte wieder in Zellen, die das betreffende Zielgen beinhalten,
induziert werden. Die Konstruktion und Expression der cDNA-Bibliotheken
ist dem Fachmann gut gekannt.
- – fNS-hsv
Thymidine-Kinase:
Dieses Konstrukt erlaubt die Identifizierung
des Zielgenprodukts durch die Spezifität der Nukleinsäure-Protein-Interaktion.
Das Gen der funktionalen Nukleinsäure (fNS) ist in die 5'-untranslatierte Führungssequenz („leader") der Thymidin Kinase
des Herpes Simplex Virus kloniert. Werstuck und Green konnten zeigen,
dass Konstrukte, die Aptamersequenzen in der 5'-untranslatierten Führungssequenz enthalten, sehr
nützlich
für die Kontrolle
der Genexpression sind (Science 282 (1998), 296–298). Infolge Bindung des
Aptamer-Liganden (funktionale Nukleinsäure) ist die Expression wahrscheinlich
aufgrund der Hemmung der Translation unterbunden.
- – Selektierbare
Marker:
Neo: gebräuchliches
Resistenzgen (Aminoglycosid-Phosphotransesterferase), um mittels
Detoxifikation der Aminoglycosid-Antibiotika Zellen zu selektieren,
die das transfizierte Plasmid enthalten (Southern und Berg, J. Mol.
Appl. Genet. 1 (1982), 327–341).
-
Plasmid pF2:
-
- – Promotorelemente:
siehe
pF1. Zusätzlich
RNA-Polymerase-III-Promotor-(Pol III P)-VAI/fNS/VAI-Konstrukt. Dieses
Konstrukt ermöglicht,
wie im funktionalen Selektionsschritt beschrieben, die Transkription
hoher Level und den Export von funktionalen Nukleinsäuren ins
Cytoplasma.
- – Episomale
Replikation:
siehe pF1.
- – Selektierbare
Marker:
Die Resistenz gegenüber
Hygromycin kann genutzt werden, um Zellen co-zuselektieren, die
zwei Plasmide (z.B. ein zusätzliches
Plasmid mit der Resistenz gegenüber
Neo) beinhalten.
- – mIg:
Dies
ist ein Oberflächen-Marker,
der die Affinitätsselektion
von transformierten Zellen erlaubt. In diesem Beispiel sind die
konstanten CH2- und CH3-Domänen
von humanem IgG1 als Transmembranversion (mIg) exprimiert, was zu
deren Präsentation
an der Zelloberfläche
führt.
Die Zellen können
beispielsweise mit Hilfe von immobilisierten, käuflichen Antikörpern (z.B.
Affinipure Ziegen-Anti-humanIgG, Fc γ-Fragment, Dianova, Hamburg)
isoliert werden. Fusionsproteine mit konstanten CH2- und CH3-Domänen von
humanen Ig-Molekülen sind
gut etablierte Techniken zur Expression von Proteinen (Kolanus,
et al., Cell 86 (1996), 233–242).
-
(2) Der Selektionsprozess:
-
Nach
Transformation der Plasmide pF1 und pF2 in eine T-Zelllinie (z.B
Jurkat-E6-Zellen) werden die Zellen in Gegenwart von Neomycin und
Hygromycin co-selektiert. Die Pol-III-Transkripte der funktionalen Nukleinsäuren (fNS)
werden die Signaltransduktionskaskade (schematische Proteine X,
Y, Z) durch Bindung an das endogene Protein X hemmen. Die Überexpression
der meisten Proteine (schematisch Protein M) aus der cDNA-Bibliothek
wird keinen Effekt auf die Hemmung der Signaltransduktionskaskade
haben und die IL2-Promotor-Konstrukte
werden nicht induziert (10). Die
Expression des betreffenden Zielmoleküls (Protein X, 11)
für die
funktionale Nukleinsäure
(fNS) aus der cDNA-Bibliothek
führt zu
der Komplementierung der Signalweiterleitung an den IL2-Promotor,
indem die Hemmung der funktionalen Nukleinsäure umgangen wird. Die Reportergene
für mIg
und HSV-TK werden angeschaltet. mIg wird genutzt, um diejenigen
Zellen durch Affinitätisolation
abzutrennen, bei denen der Signaltransduktionsweg wieder hergestellt
ist. Weil die Level von Protein X durch stark exprimierende Promotoren
innerhalb der T-Zellen auf gesättigten
Konzentrationen gehalten werden, bindet das Protein X auch an die
mRNA der HSV-TK
und verhindert deren Expression durch Hemmung ihrer Translation.
Die Zellen werden die Kultivierung in Gegenwart von Ganciclovir überleben.
Falls notwendig können
mehrere Runden der Geninduktion und -selektion wiederholt werden;
schließlich
können die
Plasmide mit den cDNA-Genen isoliert werden, z.B. durch die „Hirt-Supernatant-Methode".
-
Die
Eleganz dieses Systems liegt in dessen zweifacher Auslesung. Das
Zielgen wird durch zwei verschiedene Eigenschaften identifiziert.
Sein Genprodukt muss in der Lage sein, die Funktion der Signaltrasduktion
in einem homologen System zur Affinitätsselektion der Zellen mittels
mIg wieder herzustellen. Darüber
hinaus schützt
das Zielprotein die Zellen davor, ab getötet zu werden, da es spezifisch
an die funktionale Nukleinsäure
(RNA-Aptamer) bindet, welche in die 5'-UTR der HSV-TK-mRNA kloniert ist, welche
wiederum durch das Zielprotein selbst induziert wird. Dieser Ansatz
bedient sich alternativen Konformationen von RNA-Aptameren, die in Abhängigkeit
von der An- oder Abwesenheit des betreffenden Aptamer-Liganden kontrolliert
werden. Dieses ist Allgemeinwissen; detaillierte Strukturuntersuchungen
von vielen Aptamer/Ligand-Komplexen mittels multi-dimensionaler
NMR-Spektroskopie haben gezeigt, dass die RNA/Ligand-Komplexbildung
stets begleitet wird von drastischen Konformationsänderungen
und der konformellen Stabilisierung des bindenden RNA-Rezeptors, die nur
in Gegenwart des betreffenden Liganden erfolgt (Patel et al., J.
Mol. Biol. 272 (1997), 645–664;
Partel, Curr. Opin. Chem. Biol. 1 (1997), 32–46; Fan et al., J. Mol. Biol.
258 (1996), 480–500;
Dieckmann et al., RNA 2 (1996), 628–640; Yang et al., Science
272 (1996), 1343–47).
Darüber
hinaus haben Werstuck und Green (Science 282 (1998), 296–298) gezeigt,
dass die Translation eines Gens verhindert werden kann, wenn eine
Aptamersequenz in die untranslatierte Region eines Expressionsplasmids
insertiert ist und wenn der betreffende Ligand des Aptamers zugegen
ist, jedoch in der Abwesenheit des Liganden normal weiter läuft. Die
Notwendigkeit, funktional und spezifisch in zwei unabhängigen Systemen
zu sein, vermindert die Gefahr, falsch-positive cDNA-Klone zu erhalten.
Klone, die den IL2-Promotor durch Deregulation der Signalweiterleitung
aktivieren (z.B. Protein N, welches Protein Z beinflußt, 12)
sollten nicht in der Lage sein, die fNS in der HSV-TK-mRNA spezifisch
zu binden und ihre Expression zu hemmen. Daher werden diese die
Selektion aufgrund des Gancilovir-vermittelten Zelltods nicht überleben.
Letztendlich liefert das System spezifische Modulatoren die einfach
in der hier beschriebenen Validierung von Zielmolekülen eingesetzt
werden können,
um komplexe Proteinnetzwerke innerhalb von Zellen zu erhellen.
-
-
-
Tabelle
1: Kds (μM)
von CD 18cyt-bindenden Aptameren an die cytoplasmatische Domäne von CD18. TR
zeigt die richtig terminierten TR-Aptamere im Kontext der TR-Vektor-Expressionskassette
an. RT: Abschrift der nicht richtig terminierten TR-Aptamere mit
zusätzlichen
166 Nukleotiden.