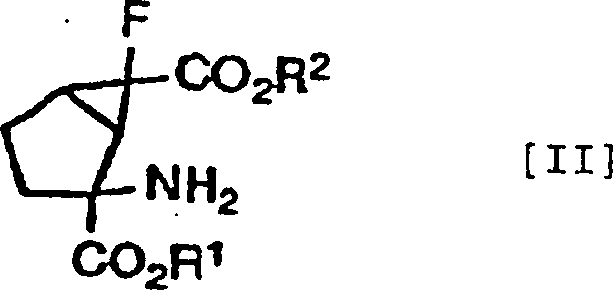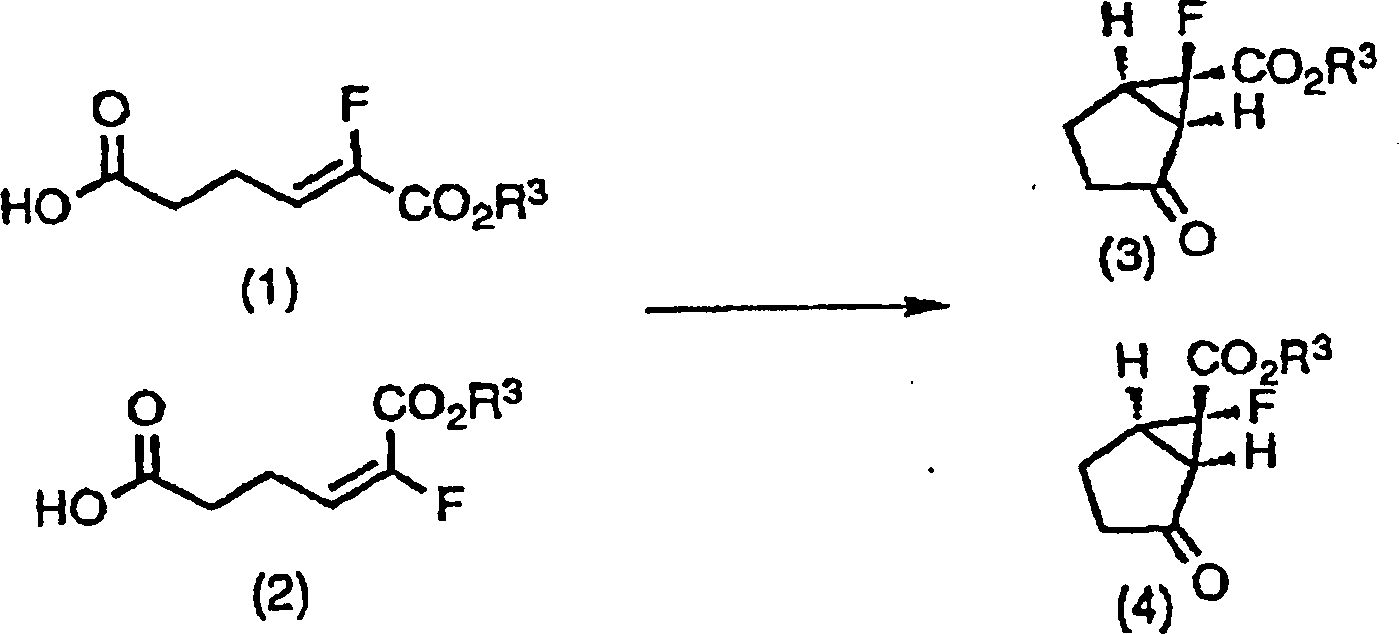-
Technologisches Gebiet
-
Diese
Erfindung betrifft 6-Fluorbicyclo[3.1.0]hexanderivate, die als Arzneimittel
verwendbar sind. Insbesondere betrifft sie neue 2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäurederivate,
die zur Behandlung und Vorbeugung von psychiatrischen Störungen verwendbar
sind, wie z.B. Schizophrenie, Angst und damit verbundene Krankheiten,
Depression, bipolare Störung
und Epilepsie; und neurologische Krankheiten wie Drogenabhängigkeit,
kognitive Störungen,
Alzheimer-Krankheit, Chorea Huntington, Parkinson-Krankheit, Dyskinesie,
verbunden mit Muskelversteifung, zerebrale Ischämie, zerebrales Versagen, Myelopathie
und Kopftrauma.
-
Stand der Technik
-
In
letzten Jahren ist mit dem wiederholten Klonieren von Glutamatrezeptorgenen
klar geworden, dass es überraschend
viele Untertypen an Glutamatrezeptoren gibt. Gegenwärtig werden
Glutamatrezeptoren grob in zwei Typen unterteilt: der "ionotrope Typ", in dem der Rezeptor
eine Ionenkanalstruktur hat, und der "metabotrope Typ", in dem der Rezeptor an G-Proteine
gekoppelt ist. Ionotrope Rezeptoren werden pharmakologisch in drei
Typen unterteilt: N-Methyl-D-asparaginsäure (NMDA), α-Amino-3-hydroxy-5-methylisoxazol-4-propionat
(AMPA) und Kynat (Science, 258, 597–603, 1992). Metabotrope Rezeptoren
werden in acht Typen, Typ 1 bis Typ 8, unterteilt (J. Neurosci.,
13, 1372–1378,
1993; Neuropharmacol., 34, 1–26,
1995).
-
Die
metabotropen Glutamatrezeptoren werden pharmakologisch in drei Gruppen
eingeteilt. Unter diesen bindet Gruppe 2 (mGluR2/mGluR3) mit Adenylcyclase
und inhibiert die Akkumulierung der Forskolinstimulation von cyclischem
Adenosinmonophosphat (cAMP) (Trends Pharmacol. Sci., 14, 13 (1993)),
was darauf hinweist, dass Verbindungen, die auf metabotrope Glutamatrezeptoren
der Gruppe 2 wirken, für
die Behandlung oder Vorbeugung von akuten oder chronischen psychiatrischen
und neurologischen Krankheiten nützlich sein
sollten. Als Substanzen, die auf metabotrope Glutamatrezeptoren
der Gruppe 2 wirken, wurde (+)-(1S,2S,5R,6S)-2-Aminobicyclo[3.1.0]hexan-2,6-dicarbonsäure in der
ungeprüften
Japanischen Patentveröffentlichung,
1. Veröffentlichungsnummer
Hei 8-188561 [1996] offenbart. Und (1S*,2S*,5R*,6R*)-2-Amino-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure, (1S*,2S*,4S*,5R*,6R*)-2-Amino-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure und
(1S*,2R*,4S*,5S*,6S*)-2-Amino-4-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure wurden
in EP-A-878,463 offenbart.
-
Fluoratome
neigen dazu, stark elektronenanziehend zu sein und hohe Fettlöslichkeit
zu verleihen, so dass Verbindungen, in denen Fluoratome eingebaut
sind, ihre physikalischen Eigenschaften stark ändern. Daher kann der Einbau
von Fluoratomen die Absorptionsfähigkeit,
Stoffwechselstabilität
und pharmakologischen Wirkungen einer Verbindung stark beeinflussen.
Aber es ist keineswegs einfach, Fluoratome einzubauen. Tatsächlich diskutiert
die ungeprüfte
Japanische Patentveröffentlichung,
1. Veröffentlichungsnummer
Hei 8-188561 [1996] nicht einmal den Einbau von Fluoratomen in (+)-(1S,2S,5R,6S)-2-Aminobicyclo[3.1.0]hexan-2,6-dicarbonsäure. Ferner
wurde (1S*,2R*,4S*,5S*,6S*)-2-Amino-4-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
in EP-A-O 878 463 offenbart wurde, durch lediglich eine Substituierung
einer Hydroxylgruppe in (1S*,2S*,4S*,5R*,6R*)-2-Amino-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure mit
einem Fluoratom unter Verwendung normaler Fluorierungsmittel hergestellt.
-
Offenbarung der Erfindung
-
Im
Hinblick auf den oben genannten gegenwärtigen Stand der Technik ist
der Zweck dieser Erfindung, Arzneimittel bereitzustellen, die für die Behandlung
und Vorbeugung psychiatrischer Störungen wirksam sind, wie z.B.
Schizophrenie, Angst und damit verbundene Krankheiten, Depression,
bipolare Störung
und Epilepsie; und neurologische Krankheiten wie Drogenabhängigkeit,
kognitive Störungen,
Alzheimer-Krankheit, Chorea Huntington, Parkinson-Krankheit, Dyskinesie,
verbunden mit Muskelversteifung, zerebrale Ischämie, zerebrales Versagen, Myelopathie
und Kopftrauma; insbesondere orale Arzneimittel, die auf metabotrope
Glutamatrezeptoren der Gruppe 2 wirken.
-
Die
Erfinder der vorliegenden Erfindung, die eine sorgfältige Studie
an 2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäurederivaten,
worin ein Fluoratom in der 6-Position von (+)-(1S,2S,5R,6S)-2-Aminobicyclo[3.1.0]hexan-2,6-dicarbonsäure, (1S*,2S*,5R*,6R*)-2-Amino-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure und
(1S*,2S*,4S*,5R*,6R*)-2-Amino-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure eingebaut
ist, durchgeführt
haben, haben neue 2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäurederivate
entdeckt, die, wenn sie oral eingenommen werden, metabotrope Glutamatrezeptoren
der Gruppe 2 beeinflussen, wodurch diese Erfindung vollendet wurde.
-
Das
heißt,
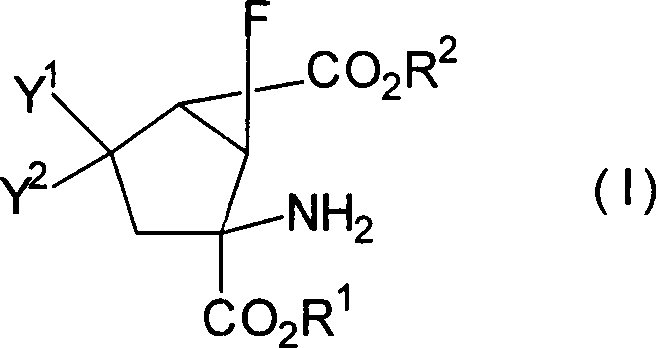
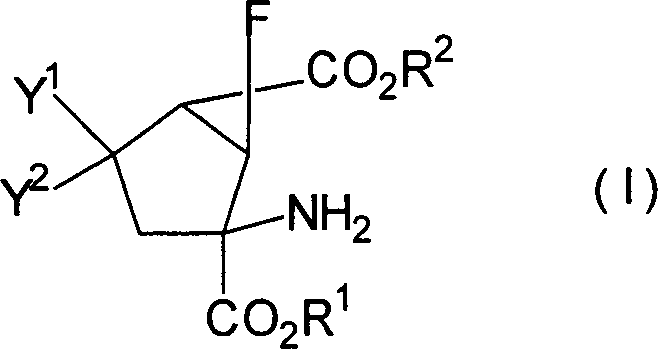
die vorliegende Erfindung betrifft 6-Fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäurederivate, dargestellt
durch die Formel [I]
[worin R
1 und
R
2 gleich oder verschieden sind und jeweils
ein Wasserstoffatom, eine C
1-10-Alkylgruppe,
eine C
3-8-Cycloalkylgruppe oder eine C
3-8-Cycloalkyl-C
1-5-alkyl-Gruppe
darstellen; Y
1 und Y
2 gleich
oder verschieden sind und jeweils ein Wasserstoffatom, eine C
1-10-Alkylthiogruppe, eine C
3-8-Cycloalkylthiogruppe,
eine C
3-8-Cycloalkyl-C
1-5-alkylthio-Gruppe,
eine C
1-5-Alkoxygruppe, eine C
3-8-Cycloalkoxygruppe
oder eine C
3-8-Cycloalkyl-C
1-5-alkoxy-Gruppe
darstellen; oder eines ein Wasserstoffatom darstellt und das andere
eine Hydroxylgruppe, eine C
1-5-Alkoxygruppe, eine
C
3-8-Cycloalkoxygruppe oder eine C
3-8-Cycloalkyl-C
1-5-alkoxy-Gruppe darstellt;
oder Y
1 und Y
2 zusammen
ein Sauerstoffatom oder -X(CH
2)
nX-Gruppe
darstellen (X bedeutet ein Sauerstoffatom oder ein Schwefelatom:
n ist 2 oder 3)], ein pharmazeutisch annehmbares Salz davon oder
Hydrate davon.
-
In
der vorliegenden Erfindung bedeutet die C1-10-Alkylgruppe
eine geradkettige oder verzweigte Alkylgruppe, deren Beispiele beinhalten
eine Methylgruppe, eine Ethylgruppe, eine Propylgruppe, eine Isopropylgruppe,
eine Butylgruppe, eine Isobutylgruppe, eine t-Butylgruppe, eine
Pentylgruppe, eine Isopentylgruppe, eine 1-Ethylpropylgruppe, eine
Hexylgruppe, eine Isohexylgruppe, eine 1-Ethylbutylgruppe, eine
Heptylgruppe, eine Isoheptylgruppe, eine Octylgruppe, eine Nonylgruppe
und eine Decylgruppe. Die C3-8-Cycloalkylgruppe
bedeutet z.B. eine Cyclopropylgruppe, eine Cyclobutylgruppe, eine
Cyclopentylgruppe, eine Cyclohexylgruppe, usw.. Die C3-8-Cycloalkyl-C1-5-alkyl-Gruppe bedeutet z.B. eine Cyclopropylmethylgruppe,
eine Cyclobutylmethylgruppe, eine Cyclopentylmethylgruppe, eine
Cyclohexylmethylgruppe, usw.. Die C1-10-Alkylthiogruppe bedeutet
eine geradkettige oder verzweigte Alkylthiogruppe, deren Beispiele
beinhalten eine Methylthiogruppe, eine Ethylthiogruppe, eine Propylthiogruppe,
eine Isopropylgruppe, eine Butylthiogruppe, eine Isobutylthiogruppe,
eine t-Butylthiogruppe, eine Pentylthiogruppe, eine Isopentylthiogruppe,
eine 1-Ethylpropylthiogruppe, eine Hexylthiogruppe, eine Isohexylthiogruppe,
eine 1-Ethylbutylthiogruppe, eine Heptylthiogruppe, eine Isoheptylthiogruppe,
eine Octylthiogruppe, eine Nonylthiogruppe und eine Decylthiogruppe.
Die C3-8-Cycloalkylthiogruppe bedeutet z.B.
eine Cyclopropylthiogruppe, eine Cyclobutylthiogruppe, eine Cyclopentylthiogruppe,
eine Cyclohexylthiogruppe, usw.. Die C3-8-Cycloalkyl-C1-5-alkylthio-Gruppe bedeutet z.B. eine Cyclopropylmethylthiogruppe,
eine Cyclobutylmethylthiogruppe, eine Cyclopentylmethylthiogruppe,
eine Cyclohexylmethylthiogruppe, usw.. Die C1-5-Alkoxygruppe
bedeutet eine geradkettige oder verzweigte Alkoxygruppe, deren Beispiele
beinhalten eine Methoxygruppe, eine Ethoxygruppe, eine Propoxygruppe,
eine Isopropoxygruppe, eine Butoxygruppe, eine Isobutoxygruppe,
eine t-Butoxygruppe, eine Pentoxygruppe, eine Isopentoxygruppe und
eine 1-Ethylpropoxygruppe. Die C3-8-Cycloalkoxygruppe
bedeutet z.B. eine Cyclopropoxygruppe, eine Cyclobutoxygruppe, eine
Cyclopentoxygruppe, usw.. Die C3-8-Cycloalkyl-C1-5-alkoxygruppe bedeutet z.B. eine Cyclopropylmethoxygruppe,
eine Cyclobutylmethoxygruppe, eine Cyclopropylethoxygruppe, usw..
-
Als
pharmazeutisch annehmbares Salz in dieser Erfindung kann z.B. genannt
werden ein Salz mit einer anorganischen Säure wie Schwefelsäure, Salzsäure und
Phosphorsäure;
ein Salz mit einer organischen Säure
wie Essigsäure,
Oxalsäure,
Milchsäure,
Weinsäure,
Fumarsäure,
Maleinsäure,
Methansulfonsäure
und Benzolsulfonsäure;
ein Salz mit einem Amin wie Trimethylamin und Methylamin; oder ein
Salz mit einem Metallion wie Natriumion, Kaliumion und Calciumion.
Die Verbindungen der vorliegenden Erfindung können als unterschiedliche Solvate
existieren, aber im Hinblick auf die Anwendbarkeit als Arzneimittel
sind Hydrate bevorzugt.
-
In
Verbindungen der Formel [I] sind, wenn sowohl Y1 als
auch Y2 Wasserstoffatome oder zusammen ein
Sauerstoffatom oder -X(CH2)nX-
(X ist ein Sauerstoffatom oder ein Schwefelatom: n ist 2 oder 3),
darstellen, oder wenn sowohl Y1 als auch
Y2 eine C1-10-Alkylthiogruppe,
eine C3-8-Cycloalkylthiogruppe, eine C3-8-Cycloalkyl-C1-5-alkylthiogruppe,
eine C1-5-Alkoxygruppe, eine C3-8-Cycloalkoxygruppe
oder eine C3-8-Cycloalkyl-C1-5-alkoxygruppe darstellen,
asymmetrische Kohlenstoffatome in der 1, 2, 5 und 6 Position anwesend.
Daher können
die Verbindungen der vorliegenden Erfindung in den obigen Fällen als
optisch aktive Substanzen, Enantiomere davon oder racemischer Stoff
davon vorliegen.
-
Wenn
ferner Y1 und Y2 unterschiedlich
voneinander ein Wasserstoffatom, eine C1-10-Alkylthiogruppe, eine
C3-8-Cycloalkylthiogruppe,
eine C3-8-Cycloalkyl-C1-5-alkylthiogruppe,
eine C1-5-Alkoxygruppe, eine C3-8-Cycloalkoxygruppe
oder eine C3-8-Cycloalkyl-C1-5-alkoxygruppe darstellen,
oder wenn eines von Y1 und Y2 ein Wasserstoffatom
darstellt und das andere eine Hydroxylgruppe, eine C1-5-Alkoxygruppe,
eine C3-8-Cycloalkoxygruppe oder eine C3-8-Cycloalkyl-C1-5-alkoxygruppe
darstellt, sind asymmetrische Kohlenstoffatome in den Positionen
1, 2, 4, 5 und 6 vorhanden. Daher können die Verbindungen der vorliegenden
Erfindung in den obigen Fällen
optisch aktive Substanzen, Enantiomere davon, racemischer Stoff
oder eine Mischung aus Diastereomeren, basierend auf Y1 und
Y2 in der 4-Position, sein.
-
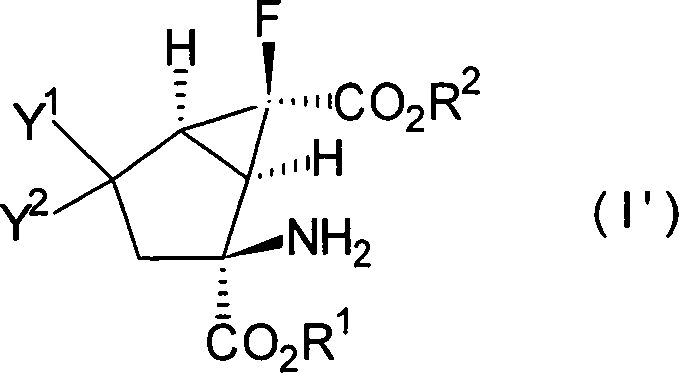
Es
ist bevorzugt, dass die Verbindungen, dargestellt in der Formel
[I], die folgende relative stereochemische Konfiguration aufweisen,
die durch die Formel [I']
dargestellt ist:
-
Eine
(+)- oder (–)-(1R*,2S*,6S*)-2-Amino-6-fluor-4-substituiertebicyclo[3.1.0]hexan-2,6-dicarbonsäure kann
als spezielles Beispiel für
besonders bevorzugte Verbindungen der Formel [I'] genannt werden.
-
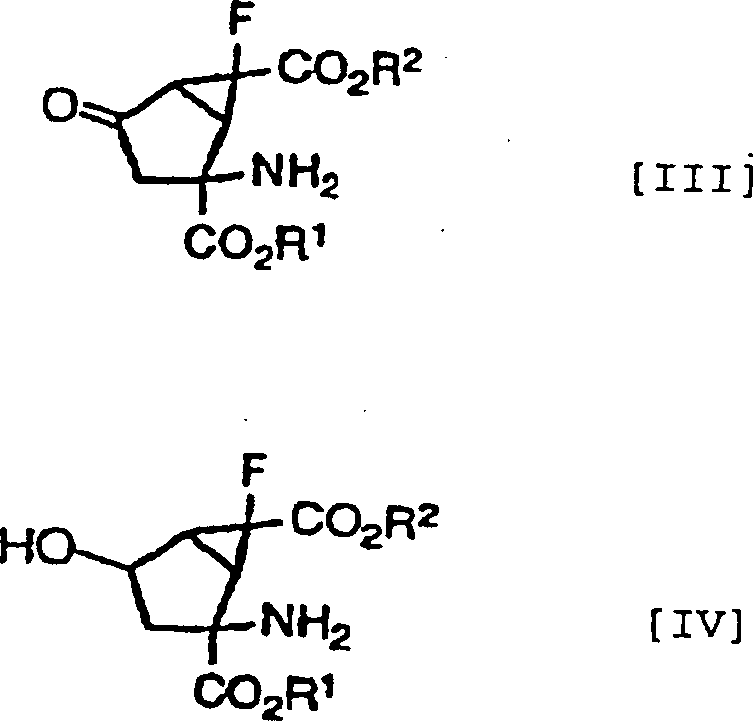
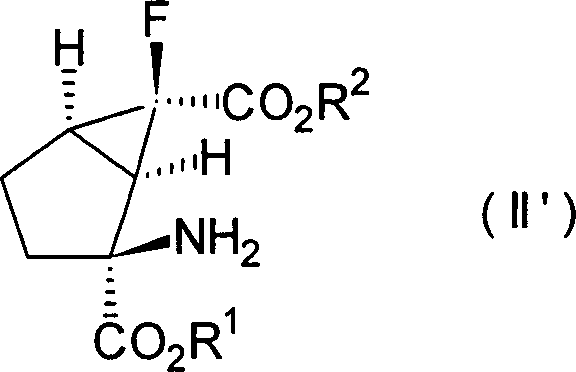
Die
anderen bevorzugten Kombinationen von Y1 und
Y2 in den Verbindungen, dargestellt durch
die Formel [I], beinhalten die Fälle,
in denen beide Wasserstoffatome darstellen, in denen sie zusammen
ein Sauerstoffatom darstellen und worin eines von ihnen ein Wasserstoffatom
darstellt und das andere eine Hydroxylgruppe darstellt, wobei diese
durch die folgenden Formeln [II], [III] bzw. [IV] gezeigt werden
können.
-
-
-
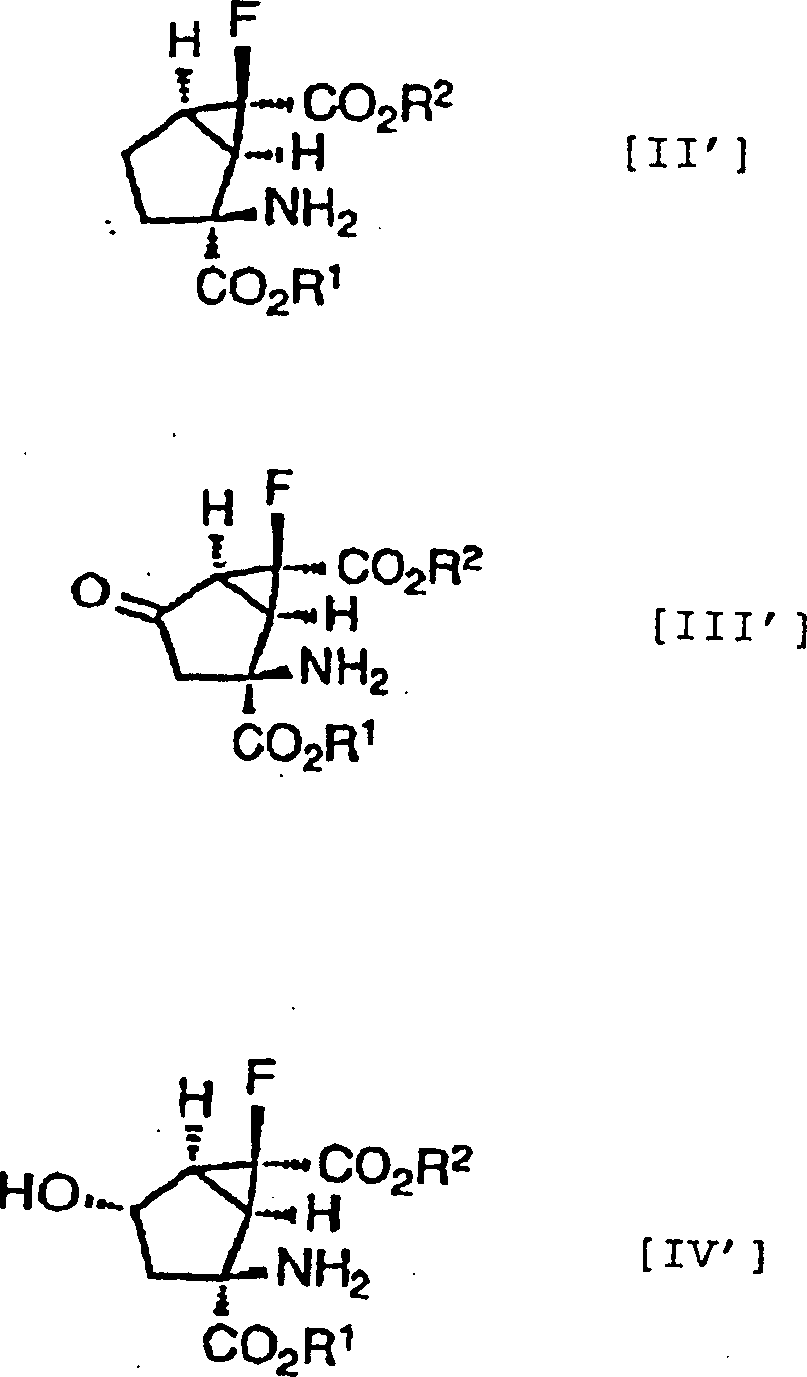
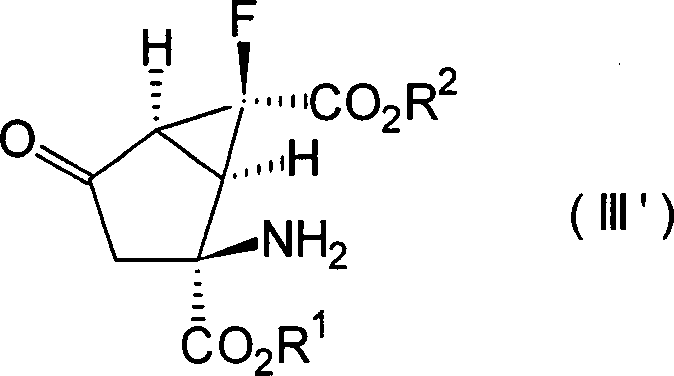
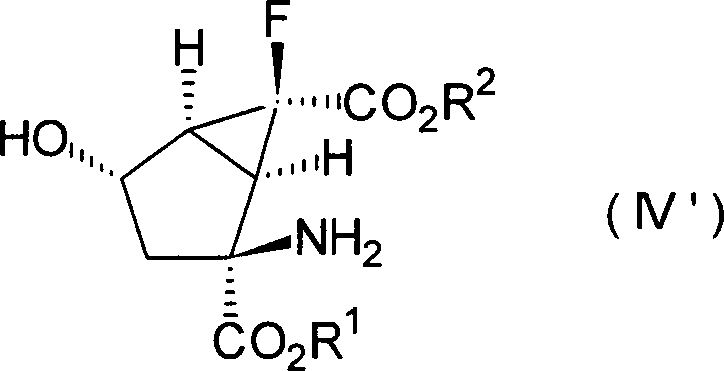
Ferner
ist es noch bevorzugter, dass die Verbindungen der obigen Formeln
[II], [III] und [IV] die folgenden relativen stereochemischen Konfigurationen
aufweisen, die durch die Formeln [II'], [III'] bzw. [IV'] dargestellt sind.
-
-
(–)-(1R*,2S*,5R*,6R*)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure, (+)-(1R*,2S*,5S*,6S*)-2-Amino-6-fluor-4-oxobicyclo [3.1.0]hexan-2,6-dicarbonsäure und
(+)- oder (–)-(1R*,2S*,4S*,5S*,6S*)-2-Amino-6-fluor-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
alle optisch aktive Substanzen sind, können als besonders bevorzugte
Verbindungen unter den Verbindungen, die durch die Formeln [II'], [III'] bzw. [IV'] dargestellt sind,
genannt werden.
-
Wenn
in den Formeln [I], [II], [III] und [IV] (einschließlich der
Fälle von
[I'], [II'], [III'] und [IV']) ein oder beide
von R1 und R2 etwas
anderes als ein Wasserstoffatom darstellen, hat dies zur Folge,
dass die Esterformen keine Wirkung auf die metabotropen Glutamatrezeptoren
der Gruppe 2 haben werden. Aber diese Esterformen werden in vivo
hydrolysiert und in eine Carbonsäure
umgewandelt, die eine Wirkung auf die metabotropen Glutamatrezeptoren
der Gruppe 2 hat. Auf diese Weise sind die Esterformen der Verbindungen,
die von der vorliegenden Erfindung umfasst werden, sehr nützlich,
weil sie als Pro-Arzneimittel wirken.
-
Die
Verbindungen der Formel [I] können
gemäß den folgenden
Reaktionen hergestellt werden. In den folgenden Reaktionsformeln
sind R1, R2, Y1 und Y2 dieselben
wie oben, R3 und R4 stellen
abgesehen von einem Wasserstoffatom R2 und
R1 dar. X' stellt ein Chloratom, ein Bromatom
oder ein Iodatom dar. Y3 und Y4 stellen zusammen
-X(CH2)nX- dar (X stellt ein Sauerstoffatom
oder ein Schwefelatom dar: n ist 2 oder 3), oder sie stellen identisch
oder unterschiedlich voneinander eine C1-10-Alkylthiogruppe,
eine C3-8-Cycloalkylthiogruppe, eine C3-8-Cycloalkyl-C1-5-alkylthiogruppe,
eine C1-5-Alkoxygruppe, eine C3-8-Cycloalkoxygruppe
oder eine C3-8-Cycloalkyl-C1-5-alkoxygruppe
dar. Ar stellt eine Arylgruppe dar, wie eine Phenylgruppe, eine
4-Chlorphenylgruppe und eine 4-Methoxyphenylgruppe. Z1 stellt
eine bekannte Schutzgruppe für
eine Hydroxylgruppe dar. Z2 stellt eine
bekannte Schutzgruppe für
eine Hydroxylgruppe dar oder ist ein Wasserstoffatom. Z3 stellt eine
bekannte Schutzgruppe für
eine Aminogruppe dar. Bekannte Schutzgruppen für Hydroxyl- und Aminogruppen
sind im Detail beschrieben in "Protective
Groups in Organic Synthesis",
von Theodora W. Greene und Peter G. M. Wuts, dessen Inhalte hiermit
durch das Zitat eingeschlossen sind.
-
-
Wie
in dem obigen Reaktionsschema gezeigt, kann das racemische Keton
(3) oder (4) oder eine Mischung von Diastereomeren aus beiden erhalten
werden durch zuerst Umwandeln eines Carbonsäurerests der Z-Form (1) oder
E-Form (2) des obigen Fluoracrylsäurederivats oder einer Mischung
aus beiden in eine aktive Form und dann Umsetzen mit Diazomethan,
gefolgt von der Umsetzung in einem inerten Lösungsmittel in Gegenwart eines
Metallkatalysators.
-
Die
hier erwähnte
aktive Form bedeutet ein Säurehalogenid
oder ein gemischtes Anhydrid. Das Säurehalogenid kann erhalten
werden durch Umsetzen bekannter Halogenierungsmittel für eine Hydroxylgruppe
in einer Carbonsäure,
wie z.B. Thionylchlorid, Oxalylchlorid und Carbontetrachloridtriphenylphosphin,
mit der Z-Form (1) oder der E-Form (2) des Fluoracrylsäurederivats
oder mit einer Mischung aus beiden. Das gemischte Anhydrid kann
erhalten werden durch Umsetzen von Halocarbonaten, wie Isobutylchlorcarbonat
und Ethylchlorcarbonat, oder organischen Anhydriden, wie Essigsäureanhydrid
und Trifluoressigsäureanhydrid,
mit der Z-Form (1) oder der E-Form (2) des Fluoracrylsäurederivats
oder einer Mischung aus beiden, mit oder ohne Anwesenheit von organischen
Basen, wie Triethylamin, N-Methylmorpholin, Diisopropylethylamin
und Pyridin, oder anorganische Basen, wie Kaliumcarbonat, Natriumhydrogencarbonat
und Natriumhydrid.
-
Der
Metallkatalysator kann z.B. Kupferagenzien sein, wie Kupfer(I)-iodid, Kupfer(II)-sulfat,
Kupfer(II)-acetat, Kupfer(II)-bis(acetylacetonat) und Kupfer(II)-bis(N-t-butylsalicylaldiimidat);
Rhodiumagenzien, wie Rhodium(II)-acetat und Rhodium(II)-trifluoracetat;
und Palladiumagenzien, wie Palladium(II)-acetat und Bis(benzonitril)dichlorpalladium(II).
-
Beispiele
für das
inerte Lösungsmittel
können
beinhalten Ether, wie Tetrahydrofuran, Dioxan und Diethylether;
Kohlenwasserstoffe, wie Toluol und Benzol; Lösungsmittel vom Halogentyp,
wie Methylenchlorid, Chloroform und 1,2-Dichlorethan; N,N-Dimethylformamid;
und Acetonitril.
-
Das
racemische Keton (3) oder das racemische Keton (4) kann direkt optisch
getrennt werden durch die Anwendung des HPLC-Verfahrens, bei dem
chirale Träger,
wie Cellulosecarbamatderivate und Amylosecarbamatderivate, verwendet
werden. Sie können
auch optisch getrennt werden, indem sie in Salze mit optisch aktiven
Aminen umgewandelt werden, wie (+)- oder (–)-1-Phenylethylamin, (+)-
oder (–)-2-Amino-1-butanol,
(+)- oder (–)-Alaninol,
Brucin, Cinchonidin, Cinchonin, Chinin, Chinidin und Dehydroabiethylamin,
nachdem ein Esterrest des racemischen Ketons (3) oder des racemischen
Ketons (4) in eine Carbonsäure
unter normalen Hydrolysebedingungen umgewandelt wurde. Ferner können sie
getrennt werden, nachdem sie in Amidoformen durch die Verwendung
eines primären
oder sekundären
optisch aktiven Amins, wie (+)- oder (–)-1-Phenylethylamin, (+)-
oder (–)-2-Amino-1-butanol
und (+)- oder (–)-Alaninol,
und normalen Amidierungsmitteln wie Dicyclohexylcarbodiimid (DCC)
umgewandelt wurden.
-
Wie
in dem obigen Reaktionsschema gezeigt, kann das Keton (3), das als
eine optisch aktive Substanz, ein Enantiomer oder racemischer Stoff
vorliegt, in das Enon (5) umgewandelt werden, das als eine optische
aktive Substanz, ein Enantiomer oder racemischer Stoff vorliegt,
durch seine Umsetzung mit z.B. Silylierungsmitteln in Gegenwart
von Basen, wodurch der Silylenolether gebildet wird, gefolgt von
seiner Umsetzung mit z.B. Palladium(II)-acetat. Das Enon (5) kann
in den Keto-Alkohol (7), der eine optisch aktive Substanz, ein Enantiomer
oder racemischer Stoff ist, umgewandelt werden, indem es zuerst
in die Epoxyform (6) mittels der Umsetzung mit Peroxiden, wie t-Butylhydroperoxid
und m-Chlorperoxybenzoesäure,
geändert
wird und dann mit z.B. Diphenyldiselenid in Gegenwart von Thiolen
reduziert wird (J. Org. Chem. 59, 5179–5183 (1994)).
-
Hier
können
Amine, wie Triethylamin und Diisopropylethylamin, Amidbasen, wie
Lithiumdiisopropylamid und Kaliumbis(trimethylsilyl)amid, und anorganische
Basen, wie Natriumhydrid, als Basen verwendet werden. Silanverbindungen,
wie Trimethylsilylchlorid, Trimethylsilyliodid und t-Butyldimethylsilylchlorid,
können
als Silylierungsmittel verwendet werden. Als Reaktionslösungsmittel
können
inerte Lösungsmittel,
wie Benzol, Toluol, Tetrahydrofuran und Acetonitril, genannt werden.
-
Der
Keto-Alkohol (7), der eine optisch aktive Substanz, ein Enantiomer
oder ein racemischer Stoff ist, kann in die Verbindung (9) umgewandelt
werden, indem er direkt oder wenn notwendig nach dem Schützen einer
Hydroxylgruppe des Keto-Alkohols (7) mit einer bekannten Schutzgruppe
für eine
Hydroxylgruppe in die Ketonform (8), die eine optisch aktive Substanz,
ein Enantiomer oder ein racemischer Stoff ist, geändert wird, gefolgt
von seiner Umsetzung mit z.B. Alkohol oder Thiol in Gegenwart von
Lewis-Säuren,
wie Bortrifluorid-Diethylether-Komplex. Der Keto-Alkohol (7) und
sein Hydroxylgruppe-geschützter
Typ sind zusammen durch die Formel (8) dargestellt. Dann ist es
möglich,
ihn in das Ketal oder Thioketal (9), das eine optisch aktive Substanz,
ein Enantiomer oder ein racemischer Stoff ist, zu ändern, wenn
Z2 ein Wasserstoffatom ist, und durch Entschützen, wenn
Z2 die bekannte Schutzgruppe für die Hydroxylgruppe
ist. Das Ketal oder Thioketal (9) mit Z2 als
Wasserstoffatom kann in die Verbindung (10), die eine optisch aktive
Substanz, ein Enantiomer oder ein racemischer Stoff ist, durch Oxidation
der Hydroxylgruppe umgewandelt werden.
-
Die
Verfahren, die in "Protective
Groups in Organic Synthesis",
von Theodora W. G. Reene und Peter G. M. Wuts beschrieben sind,
können
zum Schützen
und Entschützen
der Hydroxylgruppe und Ketalierung und Thioketalierung der Carbonylgruppe
verwendet werden. Oxidation bedeutet, dass mit z.B. Oxdationsmittel vom
Chromtyp, verkörpert
durch Jones- und Collins-Oxidationen; Oxidationsmittel vom Mangantyp,
wie Kaliumpermanganat und Mangandioxid; Oxidationsmittel vom Dimethylsulfoxidtyp
unter Verwendung von Oxalylchlorid, Essigsäureanhydrid, Phosphorpentoxid,
Schwefeltrioxid-Pyridin,
Dicyclohexylcarbodiimid (DCC) usw. als ein Aktivierungsmittel; Oxidationsmittel
vom Certyp, wie Cerdiammoniumnitrat und Cersulfat; Oxidationsmittel
vom Rutheniumtyp, wie Tetrapropylammoniumperrutheniumat und Rutheniumoxid;
und ein Dess-Martin-Mittel (siehe "Oxidations in Organic Chemistry", American Chemical
Society, Washington, D.C., 1990, Milos Hudlicky) oxidiert wird oder
dass mit Sauerstoff in Gegenwart eines Katalysators, wie Palladium
und Platin, oxidiert wird. Sie kann z.B. in inerten Lösungsmitteln,
wie Ethern, wie Tetrahydrofuran und Diethylether, Kohlenwasserstoffen,
wie Toluol und Benzol, Lösungsmitteln
vom Halogentyp, wie Dichlormethan und Chloroform, Lösungsmitteln
vom Ketontyp, wie Aceton und Ethylmethylketon, Acetonitril, N,N-Dimethylformamid,
Essigsäure,
Pyridin, Wasser und Mischungen davon, durchgeführt werden.
-
Die
racemischen Stoffe (5), (6), (7), (8), (9) oder (10) können direkt
optisch getrennt werden durch die Anwendung des HPLC-Verfahrens,
bei dem chirale Träger,
wie Cellulosecarbamatderivate und Amylosecarbamatderivate, verwendet
werden. Sie können
auch optisch getrennt werden, indem sie mit optisch aktiven Aminen,
wie (+)- oder (–)-1-Phenylethylamin,
(+)- oder (–)-2-Amino-1-butanol,
(+)- oder (–)-Alaninol,
Brucin, Cinchonidin, Cinchonin, Chinin, Chinidin und Dehydroabiethylamin,
in Salze umgewandelt werden, nachdem ein Esterrest des racemischen
Stoffes (5), (6), (7), (8), (9) oder (10) in eine Carbonsäure durch
Hydrolyse unter normalen basischen oder sauren Esterhydrolysebedingungen überführt worden
war. Ferner können
sie getrennt werden, nachdem sie in Amidoformen durch die Verwendung
von primären
oder sekundären
optisch aktiven Aminen, wie (+)- oder (–)-1-Phenylethylamin, (+)-
oder (–)-2-Amino-1-butanol
und (+)- oder (–)-Alaninol, und
normalen Amidierungsmitteln, wie Dicyclohexylcarbodiimid (DCC),
umgewandelt wurden.
-
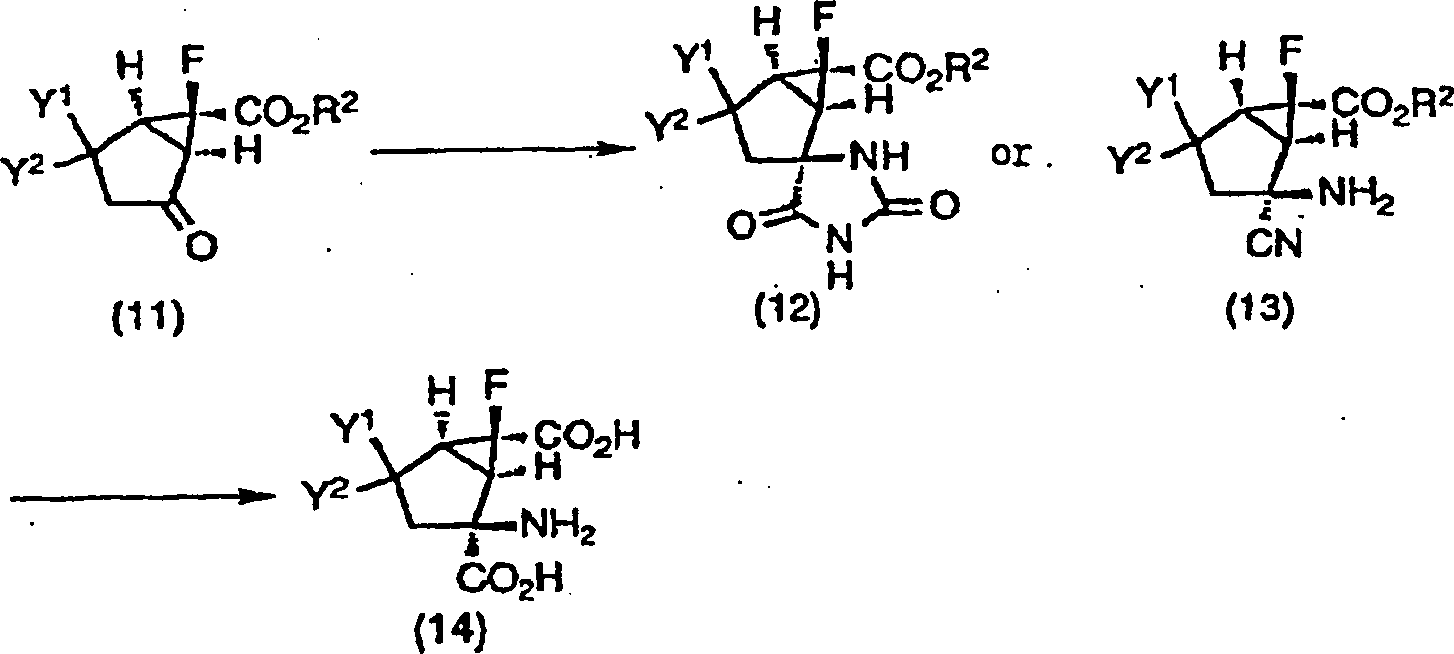
-
Die
Ketone (11) einschließlich
der Verbindungen (3), (7) und (10) sind nützlich als Intermediate, um
die Verbindungen gemäß der vorliegenden
Erfindung zu synthetisieren. Die Ketone (11), die optisch aktive
Substanzen, Enantiomere oder racemische Stoffe sind, können in
Hydantoinderivate (12) oder Aminocyanidderivate (13) durch die Strecker-Aminosäuresynthese
(Ann., 75, 27 (1850); 91, 349 (1850)), die Bucherer-Bergs-Reaktion (J. Prakt.
Chem., 140, 69 (1934)) oder Variationen davon umgesetzt werden.
Die Hydantoinderivate (12) und die Aminocyanidderivate (13) können in
die Verbindungen der vorliegenden Erfindung, die optisch aktive
Substanzen, Enantiomere oder racemische Stoffe, 4-substituierte-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäuren (14),
durch basische Hydrolyse mit z.B. Natriumhydroxid, Bariumhydroxid, usw.
umgesetzt werden. Wenn z.B. Y1 und Y2 in den Hydantoinderivaten (12) oder den
Cyanidderivaten (13) zusammen -S(CH2)nS- darstellen oder identisch oder unterschiedlich
voneinander eine C1-10-Alkylthiogruppe, eine
C3-8-Cycloalkylthiogruppe oder eine C3-8-Cycloalkyl-C1-5-alkylthiogruppe
darstellen, kann eine der Verbindungen (14) gemäß der vorliegenden Erfindung,
2-Amino-6-fluor-4,4-dialkylthiobicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
eine optisch aktive Substanz, ein Enantiomer oder ein racemischer
Stoff ist, hergestellt werden, indem für die Verbindungen (12) oder
(13) basische Hydrolyse mit Natriumhydroxid, Bariumhydroxid usw. durchgeführt wird.
Andererseits können
aus den Hydantoinderivaten (12) und den Aminocyanidderivaten (13) durch
Hydrolyse unter sauren Bedingungen unter Verwendung von z.B. Schwefelsäure oder
dergleichen eine der Verbindungen (14) gemäß der vorliegenden Erfindung,
2-Amino-6-fluor-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
eine optisch aktive Substanz, ein Enantiomer oder ein racemischer
Stoff ist, hergestellt werden. Ferner kann auch 2-Amino-6-fluor-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
eine optisch aktive Substanz, ein Enantiomer oder ein racemischer
Stoff ist, erhalten werden durch Entfernen einer Dialkylthiogruppe
aus 2-Amino-6-fluor-4,4-dialkylthiobicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
eine optisch aktive Substanz, ein Enantiomer oder ein racemischer
Stoff ist (siehe "Protective
Groups in organic Synthesis",
Theodora W. Greene und Peter G. M. Wuts). Ferner kann 2-Amino-6-fluor-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
eine optisch aktive Substanz, ein Enantiomer oder ein racemischer
Stoff ist, auch erhalten werden durch z.B. Oxidation einer Hydroxylgruppe
von 2-Amino-6-fluor-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure, die eine
optisch aktive Substanz, ein Enantiomer oder ein racemischer Stoff
ist (siehe "Oxidation
in Organic Chemistry",
American Chemical Society, Washington D.C., 1990, Milos Hudlicky).
Währendessen
ist es bevorzugt, die Carboxyl- und Aminogruppen der Verbindungen
(14), wenn notwendig, zu schützen
(siehe "Protecting Groups
in Organic Synthesis",
Theodora W. Greene, John Wiley & Sons
Inc.).
-
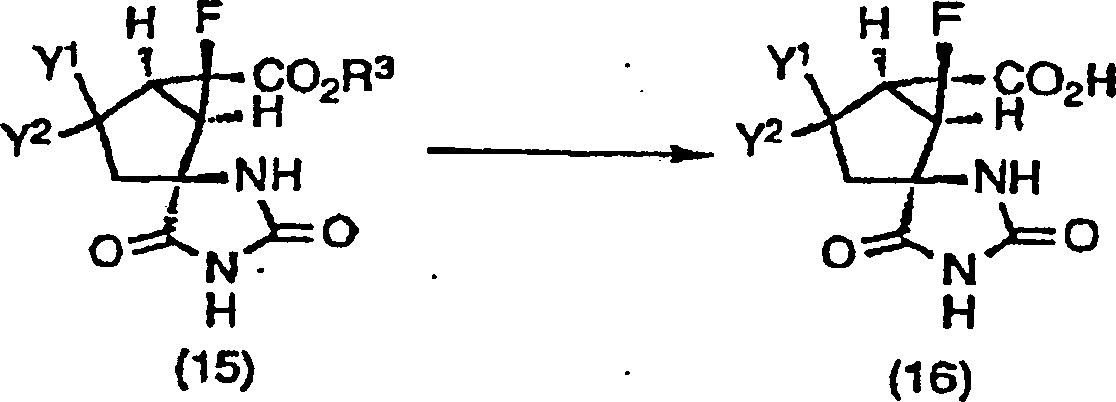
-
Der
racemische Stoff der Formel (15) kann direkt optisch getrennt werden
durch Anwendung des HPLC-Verfahrens, bei dem chirale Träger, wie
Cellulosecarbamatderivate und Amylosecarbamatderivate, verwendet
werden. Der racemische Stoff (15) kann auch optisch getrennt werden,
indem er mit optisch aktiven Aminen, wie (+)- oder (–)-1-Phenylethylamin,
(+)- oder (–)-2-Amino-1-butanol,
(+)- oder (–)-Alaninol,
Brucin, Cinchonidin, Cinchonin, Chinin, Chinidin und Dehydroabiethylamin,
in Salze umgewandelt wird, nachdem ein Esterrest des racemischen
Stoffes (15) durch Hydrolyse unter normal basischen oder sauren
Esterhydrolysebedingungen hydrolysiert wurde, um eine Carbonsäure (16)
zu bilden. Ferner kann er getrennt werden, nachdem er durch Verwendung
von primären
und sekundären
optisch aktiven Aminen, wie (+)- oder (–)-1-Phenylethylamin, (+)-
oder (–)-2-Amino-1-butanol
und (+)- oder (–)-Alaninol,
und normalen Amidationsmitteln, wie Dicyclohexylcarbodiimid (DCC),
in eine Amidoform umgewandelt wurde.
-
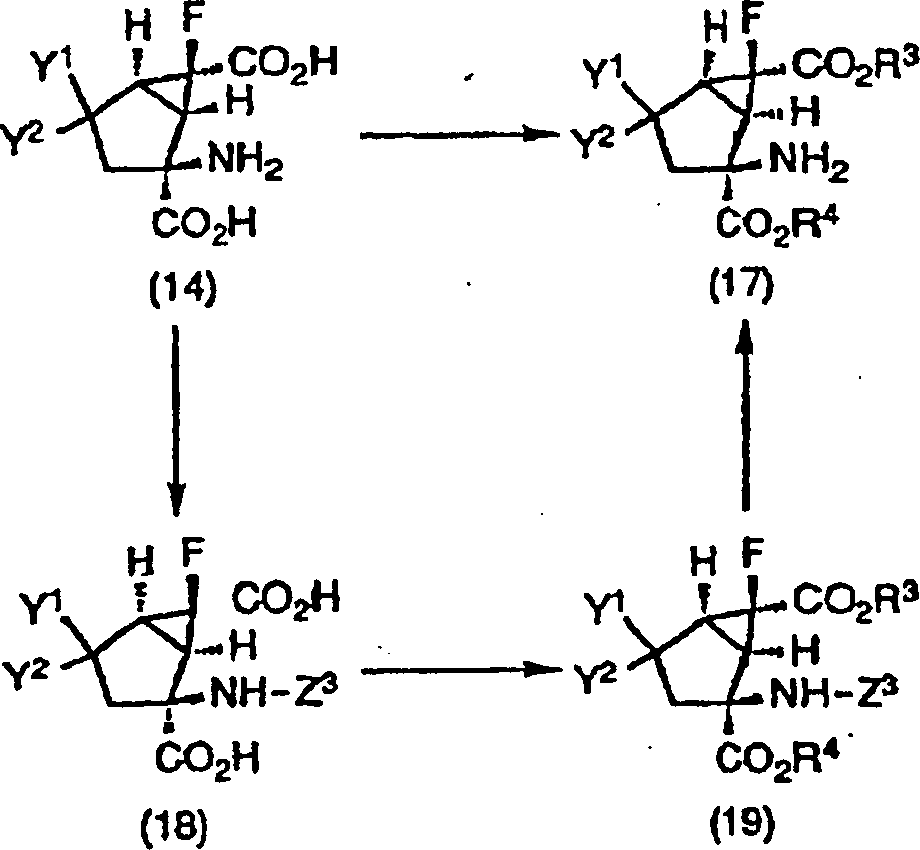
-
Wie
in dem obigen Reaktionsschema gezeigt, kann die 4-substituierte-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure (14),
die eine Verbindung gemäß der vorliegenden
Erfindung ist und als eine optisch aktive Substanz, ein Enantiomer
oder racemischer Stoff vorliegt, in die Verbindung gemäß der vorliegenden
Erfindung, dargestellt durch die Formel (17), der Esterform der
4-substituierten-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure, die
als eine optisch aktive Substanz, ein Enantiomer oder ein racemischer Stoff
vorliegt, durch Veresterung mit bekannten Verfahren unter Verwendung
von Alkoholen, dargestellt durch R3-OH oder
R4-OH, umgewandelt werden; oder durch Umwandlung
in die Verbindung der Formel (19) durch Veresterung mit bekannten
Verfahren unter Verwendung von Alkylhalogeniden, dargestellt durch
R3-X' oder R4-X' oder
Alkoholen, dargestellt durch R3-OH oder
R4-OH, nachdem sie in die Verbindung (18)
durch Schützen
einer Aminogruppe mit einer Schutzgruppe, dargestellt durch Z3, umgewandelt worden war, gefolgt von Entschützen der
Schutzgruppe Z3 für die Aminogruppe.
-
Schützen, Verestern
und Entschützen
der Aminogruppe im obigen Fall kann gemäß normalen Verfahren durchgeführt werden
("Protective Groups
in Organic Synthesis",
von Theodora W. Greene und Peter G. M. Wuts).
-
Wenn
die Verbindung (17) ein racemischer Stoff ist, kann er durch eine
allgemeine optische Trennung unter Verwendung eines sauren chiralen
Trennmittels optisch getrennt werden. Wenn die Verbindung (18) ein racemischer
Stoff ist, kann er durch eine allgemeine optische Trennung unter
Verwendung eines basischen chiralen Trennmittels optisch getrennt
werden.
-
Hier
kann man als das saure chirale Trennmittel optisch aktive organische
Säuren,
wie (+)- oder (–)-Di-p-Toluoylweinsäure, (+)-
oder (–)-Dibenzoylweinsäure, (+)-
oder (–)-Weinsäure, (+)-
oder (–)-Mandelsäure, (+)-
oder (–)-Camphersäure und
(+)- oder (–)-Camphersulfonsäure, verwenden.
Als das basische chirale Trennmittel kann man z.B. optisch aktive
Amine, wie (+)- oder (–)-1-Phenylethylamin,
(+)- oder (–)-2-Amino-1-butanol,
(+)- oder (–)-Alaninol,
Brucin, Cinchonidin, Cinchonin, Chinin, Chinidin, Dehydroabiethylamin, usw.
verwenden.
-
-
Wie
in dem obigen Reaktionsschema gezeigt, können die Z-Form (1) und E-Form
(2) des Fluoracrylsäurederivats
oder eine Mischung aus den Z- und E-Formen, dargestellt durch die
Formel (23), erhalten werden, indem die Verbindung (22) durch die
Umsetzung von γ-Butyrolactol
(20) mit dem Phosphonoessigsäurederivat
(21) hergestellt wird, gefolgt von Oxidation einer Hydroxylgruppe
zu einer Carbonsäure,
direkt oder nach Schützen
der Hydroxylgruppe dargestellt.
-
Das
Schützen
der Hydroxylgruppe kann gemäß normalen
Schutzverfahren für
eine Hydroxylgruppe durchgeführt
werden ("Protective
Groups in Organic Synthesis",
Theodora W. G. Greene und Peter G. M. Wuts). Als spezielle Ausführungsformen
für die
Oxidation können
z.B. genannt werden die Direktoxidation zu einer Carbonsäure unter
Verwendung von Oxidationsmitteln vom Chromtyp, wie Jones-Reagens,
Pyridindichromat (PDC) oder Oxidationsmittel vom Mangantyp, wie
Kaliumpermanganat; oder durch stufenweise Oxidation zu einer Carbonsäure durch
z.B. Natriumchlorit usw. nach der Umwandlung in ein Aldehyd durch
Oxidation mit z.B. Dimethylsulfoxid, wie Swern-Oxidation ("Oxidations in Organic
Chemistry", American
Chemical Society, Washington D.C., 1990, Milos Hudlicky).
-
Die
Verbindungen (22), worin Z2 eine t-Butyldimethylsilylgruppe,
eine t-Butyldiphenylsilylgruppe oder dergleichen ist, können in
zwei Isomere der Z- und E-Form durch Anwendung von Silikagel-Säulenchromatographie
usw. aufgetrennt werden.
-
-
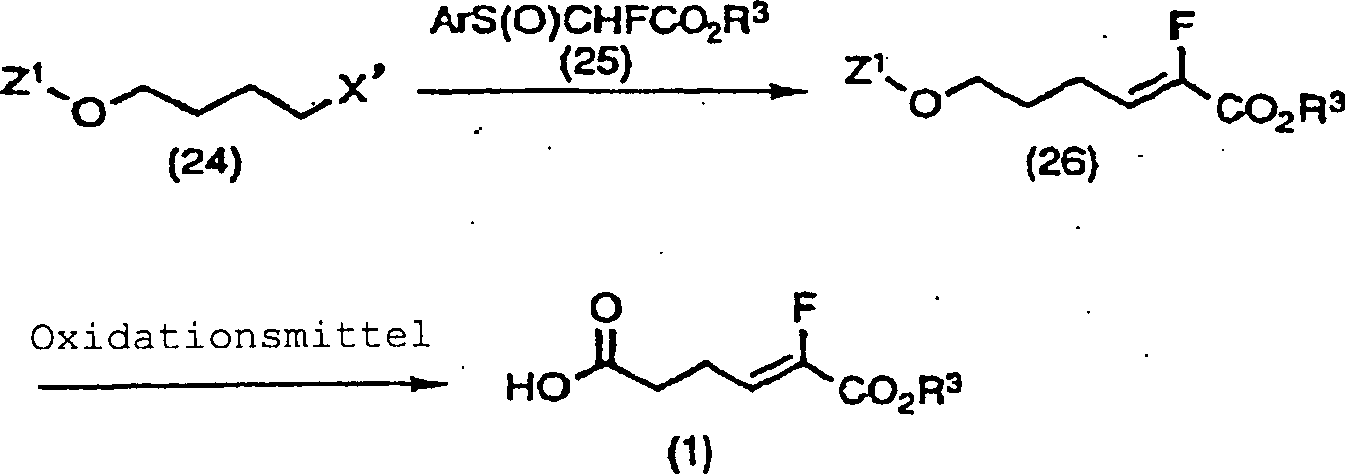
Wie
in dem obigen Reaktionsschema gezeigt, kann ferner die Z-Form (1)
des Fluoracrylsäurederivats erhalten
werden durch Oxidation mit oder ohne Entschützen der Schutzgruppe Z1 einer Hydroxylgruppe nach Herstellung der
Verbindung (26) durch Umsetzung des Halogenids der Formel (24) und
des Sulfoxidderivats (25).
-
Das
Entschützen
der Schutzgruppe Z1 kann gemäß normalen
Verfahren durchgeführt
werden (siehe "Protective
Groups in Organic Synthesis",
Theodora W. Greene und Peter G. M. Wuts). Als spezielle Ausführungsformen
für die
Oxidation können
z.B. genannt werden Direktoxidation zu Carbonsäure unter Verwendung von Oxidationsmitteln
vom Chromtyp, wie Jones-Reagens, Pyridindichromat (PDC) oder Oxidationsmitteln vom
Mangantyp, wie Kaliumpermanganat; oder stufenweise Oxidation zu
einer Carbonsäure
durch z.B. Natriumchlorit usw. nach dem Umwandeln in ein Aldehyd
durch Oxidation, wie z.B. Dimethylsulfoxid, wie Swern-Oxidation.
-
Aus
den Verbindungen gemäß der vorliegenden
Erfindung können
pharmazeutische Präparate
hergestellt werden, indem sie mit ein oder mehreren pharmazeutisch
annehmbaren Trägern,
Arzneistoffträgern
oder Verdünnungsmitteln
kombiniert werden. Beispiele für
diese Träger,
Arzneistoffträger
und Verdünnungsmittel beinhalten
Wasser, Lactose, Dextrose, Fructose, Saccharose, Sorbit, Mannit,
Polyethylenglykol, Propylenglykol, Stärke, Gummi, Gelatine, Arginat,
Calciumsilikat, Calciumphosphat, Cellulose, Wasser, Sirup, Methylcellulose,
Polyvinylpyrrolidon, Alkylparahydroxybenzoat, Talk, Magnesiumstearat,
Stearinsäure,
Glycerin und Öle,
wie Sesamöl,
Olivenöl
und Sojabohnenöl.
-
Die
Verbindungen gemäß der vorliegenden
Erfindung können,
nachdem sie mit diesen Trägern,
Arzneistoffträgern
oder Verdünnungsmitteln
und wenn notwendig mit Additiven, wie gewöhnlich verwendete Füllstoffe,
Bindemittel, Abbaumittel, pH-Regulatoren und Lösungsvermittler, vermischt
wurden, mittels gewöhnlicher
Formulierungstechnologie als pharmazeutische Präparate zur oralen oder parenteralen
Verabreichung, insbesondere als Präparate, die auf metabotrope
Glutamatrezeptoren der Gruppe 2 wirken (metabotrope Glutamatrezeptoragonisten
der Gruppe 2) und die psychiatrische oder neurologische Krankheiten
verhindern oder behandeln, in solchen Formen, wie Tabletten, Pillen,
Kapseln, Körner,
Pulver, Flüssigkeiten,
Emulsionen, Suspensionen, Salben, Injektionen und Hauptpflaster,
hergestellt werden. Die Verbindungen gemäß dieser Erfindung können einem
erwachsenen Patienten in einer Menge von 0,01 bis 500 mg als einzelne
Dosis oder aufgeteilt in mehrere Dosen pro Tag oral oder parenteral
verabreicht werden. Diese Dosis kann unter Berücksichtigung des zu behandelnden
Krankheitstyps und des Alters, Gewicht und der Symptome des Patienten
geeignet erhöht
oder verringert werden.
-
Beste Weise zur Durchführung der
Erfindung
-
Im
Folgenden beschreiben wir diese Erfindung speziell durch die Darstellung
von Arbeitsbeispielen und experimentellen Beispielen. Diese Erfindung
ist jedoch dadurch nicht auf diese Beispiele limitiert.
-
Beispiel 1
-
Synthese von (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
-
(1)
Unter einem Stickstofffluss mit Eiskühlung wurden 78,0 ml einer
1,00 M Tetrahydrofuranlösung
aus Natrium-bis(trimethylsilyl)amid tropfenweise über 40 min
zu einer Lösung
aus 18,9 g Ethyldiethylphosphonofluoracetat in 75 ml Tetrahydrofuran
zugetropft, und dann wurde die Mischung ferner für 45 min gerührt. Eine
vorpräparierte
Lösung
aus γ-Butyrolactol
(bei –78°C, unter
einem Stickstofffluss, wurden 70,3 ml einer 1,01 M Toluollösung aus
Aluminiumdiisobutylhydrid über
1,5 h tropfenweise zu 6,1 g γ-Butyrolacton
in 75 ml Tetrahydrofuran zugegeben, und dann wurde die Mischung
ferner bei dieser Temperatur für
weitere 1,5 h gerührt)
wurde tropfenweise über
30 min zu dieser Reaktionslösung
zugegeben. Das Eisbad wurde nach der Zugabe weggenommen. Die Reaktionslösung wurde
mit 120 ml 6 N Salzsäure
gequencht, nachdem sie für
2 h bei Raumtemperatur und dann für 3 h bei 30°C gerührt worden
war. Die Reaktionslösung
wurde 2 Mal mit Ethylacetat extrahiert. Die erhaltenen organischen
Schichten wurden vereinigt und über
wasserfreiem Natriumsulfat getrocknet, nachdem sie mit einer gesättigten
wässrigen
Lösung
aus Natriumchlorid gewaschen worden waren. Nach dem Abfiltrieren
des Trocknungsmittels wurde die Konzentration des Filtrats unter
reduziertem Druck durchgeführt.
Der Rückstand
wurde durch Säulenchromatographie
gereinigt (Silikagel: Wako Gel C200 (hergestellt von Wako Pure Chemical
Industries Ltd.), Eluent: Hexan-Ethylacetat = 4:1 bis 2:1), wodurch
sich 7,9 g einer Mischung der Z- und E-Formen mit dem Verhältnis von
etwa 1:3 von Ethyl-2-fluor-6-hydroxy-2-hexenoat ergaben.
-
Die
Protonen-NMR-Daten der erhaltenen Verbindungen sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,34
(3H*1/4, t, J = 7,1 Hz), 1,36 (3H*3/4, t, J = 7,1 Hz), 1,73 (2H,
quint., J = 6,6 Hz), 2,01 (1H, br. s), 2,30–2,41 (2H*1/4, m), 2,56–2,68 (2H*3/4,
m), 3,63–3,73
(2H, m), 4,30 (2H*1/4, q, J = 7,1 Hz), 4,32 (2H*3/4, q, J = 7,1
Hz), 5,94 (1H*3/4, dt, J = 21,3, 8,7 Hz), 6,16 (1H*1/4, dt, J =
33,2, 8,1 Hz)
-
(2)
7,8 g der Mischung aus den Z- und E-Formen mit dem Verhältnis von
etwa 1:3 von Ethyl-2-fluor-6-hydroxy-2-hexenoat und 14,6 g t-Butyldiphenylchlorsilan
wurden in 40 ml N,N-Dimethylformamid gelöst, und dann wurden 4,5 g Imidazol
unter Eiskühlung
zugegeben. Die Reaktionslösung
wurde auf Raumtemperatur erwärmt
und dann mit Ethylacetat verdünnt.
Nach dem aufeinanderfolgenden Waschen der organischen Schicht mit
Wasser, einer gesättigten
wässrigen
Lösung
von Ammoniumchlorid, einer gesättigten
wässrigen Lösung von
Natriumhydrogencarbonat und einer gesättigten wässrigen Lösung von Natriumchlorid wurde
sie mit wasserfreiem Natriumsulfat getrocknet. Nach dem Abfiltrieren
des Trocknungsmittels wurde Konzentration des Filtrats unter reduziertem
Druck durchgeführt.
Der Rückstand
wurde in das jeweilige geometrische Isomer aufgetrennt und durch
Säulenchromatographie
gereinigt (Silikagel: MSG D-40-60A (hergestellt von Dokai Chemicals
Ltd.), Eluent: Hexan-Ethylacetat = 50:1), wodurch sich 2,4 g Ethyl-2-fluor-6-t-butyldiphenylsilyloxy-2(Z)-hexenoat
und 7,1 g Ethyl-2-fluor-6-t-butyldiphenylsilyloxy-2(E)-hexenoat
ergaben.
-
Die
Protonen-NMR- und massenspektroskopischen Daten von Ethyl-2-fluor-6-t-butyldiphenylsilyloxy-2(Z)-hexenoat
sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm):
1,05 (9H, s), 1,33 (3H, t, J = 7,1 Hz), 1,61–1,76 (2H, m), 2,31–2,43 (2H,
m), 3,68 (2H, t, J = 6,2 Hz), 4,27 (2H, q, J = 7,1 Hz), 6,14 (1H,
dt, J = 33,4, 7,8 Hz), 7,33–7,48
(6H, m), 7,62–7,70
(4H, m), MS (Cl) (Pos) m/e: 415 (M++1),
357 (M+–57),
337 (M+–77,
100%)
-
Die
Protonen-NMR- und massenspektroskopischen Daten von Ethyl-2-fluor-6-t-butyldiphenylsilyloxy-2(E)-hexanoat
sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm):
1,05 (9H, s), 1,32 (3H, t, J = 7,1 Hz), 1,61–1,77 (2H, m), 2,56–2,69 (2H,
m), 3,69 (2H, t, J = 6,3 Hz), 4,28 (2H, q, J = 7,1 Hz), 5,92 (1H,
dt, J = 21,8, 8,1 Hz), 7,33–7,48
(6H, m), 7,62–7,70
(4H, m) MS (Cl) (Pos) m/e: 415 (M++1), 357
(M+–57),
337 (M+–77,
100%)
-
(3)
2,3 g Ethyl-2-fluor-6-t-butyldiphenylsilyloxy-2(Z)-hexenoat wurden
in 12 ml Aceton gelöst,
und dann wurden 9 ml 8 N Jones-Reagens unter Eiskühlung zugegeben.
Nach dem Rühren
der Reaktionslösung
bei Raumtemperatur für
2,5 h wurde das überschüssige Reagens
durch Zugabe von 2-Propanol zu der Reaktionslösung unter Eiskühlung gequencht.
Die Reaktionsmischung wurde mit Ethylacetat verdünnt und dann mit Wasser gewaschen.
Die wässrige
Schicht wurde mit Ethylacetat extrahiert und die organische Schicht
wurde insgesamt 2 Mal mit Wasser und mit einer gesättigten
wässrigen
Lösung
von Natriumchlorid gewaschen, und dann wurde sie über wasserfreiem
Natriumsulfat getrocknet. Nach dem Abfiltrieren des Trocknungsmittels
wurde die Konzentration des Filtrats unter reduziertem Druck durchgeführt. Der
Rückstand
wurde durch Säulenchromatographie
gereinigt (Silikagel: Wako Gel C200 (hergestellt von Wako Chemical
Industries Ltd.), Eluent: Hexan-Ethylacetat = 3:1), wodurch sich
970 mg Ethyl-2-fluor-5-carboxy-2(Z)-pentenoat ergaben.
-
Die
Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,34
(3H, t, J = 7,1 Hz), 2,46–2,60
(4H, m), 4,29 (2H, q, J = 7,1 Hz), 6,03–6,27 (1H, m)
MS (Cl)
(Pos) m/e: 191 (M++1, 100%).
-
Ebenso
wurde Ethyl-2-fluor-5-carboxy-2(E)-pentenoat erhalten. Die Protonen-NMR-
und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,36
(3H, t, J = 7,1 Hz), 2,54 (2H, t, J = 7, 3 Hz), 2, 78–2,90 (2H,
m), 4,32 (2H, q, J = 7,1 Hz), 5,98 (1H, dt, J = 20,5, 8,2 Hz)
MS
(Cl) (Pos) m/e: 191 (M++1), 173 (M+–17,
100%)
-
(4)
920 mg Ethyl-2-fluor-5-carboxy-2(Z)-pentenoat und 1,3 ml Oxalylchlorid
wurden in Hexan unter Rückfluss
für 3 h
erwärmt.
Die Reaktionslösung
wurde unter reduziertem Druck konzentriert und unter Verwendung
einer Vakuumpumpe getrocknet. Eine Etherlösung mit einem Überschuss
an Diazomethan wurde tropfenweise zu dem erhaltenen Rückstand
unter Eiskühlung
zugegeben, dies wurde bei Raumtemperatur für 1 h gerührt. Nach Filtration der Reaktionslösung wurde
das Filtrat unter reduziertem Druck konzentriert. Der erhaltene
Rückstand
wurde in 10 ml Benzol gelöst, und
dies wurde tropfenweise über
30 min zu 120 ml Benzollösung
von 40 mg Kupfer(II)-bis(N-t-butylsalicylaldiimidat) unter Wärmerefluxieren
zugegeben. Die Reaktionslösung
wurde auf Raumtemperatur abgekühlt
und unter reduziertem Druck konzentriert. Der Rückstand wurde durch Säulenchromatographie
gereinigt (Silikagel: Wako Gel C200 (hergestellt von Wako Chemical
Industries Ltd.), Eluent: Hexan-Aceton = 9:1), wodurch sich 263
mg (1RS,5RS,6RS)-Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
ergaben.
-
Die
Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,33
(3H, t, J = 7,1 Hz), 2,05–2,55
(4H, m), 2,59 (1H, d, J = 6,6 Hz), 2,70–2,77 (1H, m); 4,30 (2H, q,
J = 7,1 Hz)
MS (IonSpray) (Pos) m/e: 187 (M++1),
204 (M++18), 209 (M++23,
100%)
-
Genauso
wurde (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat erhalten.
Die Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,36
(3H, t, J = 7,1 Hz), 2,00–2,80
(6H, m), 4,32 (2H, q, J = 7,1 Hz)
MS (IonSpray) (Pos) m/e:
187 (M++1, 100%)
-
Beispiel 2
-
Synthese von (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
-
(1)
3,7 g 60%iges Natriumhydrid (ölig)
wurden in 85 ml N,N-Dimethylformamid
suspendiert und dann wurden 19,6 g Ethylphenylsulfinylfluoracetat
in 35 ml N,N-Dimethylformamid tropfenweise über 30 min unter Eiskühlung zugegeben.
Nach der Zugabe wurde das Rühren
für 30
min unter Eiskühlung
fortgesetzt, und dann wurde weiteres Rühren für 30 min bei Raumtemperatur
fortgesetzt. Unter Eiskühlung
wurden 20,2 g 1-Brom-4-tetrahydropyranyloxybutan
auf einmal dazugegeben, und dann wurde das Rühren bei Raumtemperatur für 4 h und
bei 95 bis 110°C
für 1 h
fortgesetzt. Nach dem Abkühlen
der Reaktionslösung
auf Raumtemperatur wurde sie in Eiswasser gefüllt und mit 10%igem Hexan-Ethylacetat
extrahiert. Die erhaltenen organischen Schichten wurden über wasserfreiem
Natriumsulfat getrocknet, nachdem sie mit Wasser und einer gesättigten
wässrigen
Natriumchlorid-Lösung gewaschen
worden waren. Nach dem Abfiltrieren des Trocknungsmittels wurde
Konzentration des Filtrats unter reduzierten Druck durchgeführt. Der
Rückstand
wurde durch Säulenchromatographie
gereinigt (Silikagel: Wako Gel C200 (hergestellt von Wako Pure Chemical
Industries Ltd.), Eluent: Hexan- Ethylacetat
= 15:1) und (Silikagel: MSG D-40-60A (hergestellt von Dokai Chemicals
Ltd.), Eluent: Hexan-Aceton = 20:1), wodurch sich 7,4 g Ethyl-2-fluor-6-tetrahydropyranyloxy-2(Z)-hexenoat
ergaben.
-
Die
Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,33
(3H, t, J = 7,1 Hz), 1,46–1,90
(8H, m), 2,30–2,41
(2H, m), 3,33–3,57
(2H, m), 3,72–3,90
(2H, m), 4,28 (2H, q, J = 7,1 Hz), 4,57–4,60 (1H, m), 6,17 (1H, dt,
J = 33,3, 7,8 Hz)
MS (CI) (Pos) m/e: 261 (M++1),
85 (M+–175,
100%)
-
(2)
4,7 g Ethyl-2-fluor-5-carboxy-2(Z)-pentenoat wurden gemäß Schritt
(3) aus Beispiel 1 erhalten.
-
Die
Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,34
(3H, t, J = 7,1 Hz), 2,46–2,60
(4H, m), 4,29 (2H, q, J = 7, 1 Hz), 6,03–6,27 (1H, m)
MS (CI)
(Pos) m/e: 191 (M++1, 100%)
-
(3)
2,8 g (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat wurden
gemäß Schritt (4)
aus Beispiel 1 erhalten.
-
Die
Protonen-NMR- und massenspektrometrischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,33
(3H, t, J = 7,1 Hz), 2,05–2,55
(4H, m), 2,59 (1H, d, J = 6,6 Hz), 2,70–2,77 (1H, m), 4,30 (2H, q,
J = 7,1 Hz)
MS (IonSpray) (Pos) m/e: 187 (M++1),
204 (M++18), 209 (M++23,
100%)
-
Beispiel 3
-
Synthese von (1R*,5R*,6R*)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
-
919
mg (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat, das gemäß Schritt
(4) aus Beispiel 1 erhalten wurde, wurde unter Verwendung von HPLC
mit CHIRALPAK AD (hergestellt von Daicel Chemicals Industries Ltd.,
2,0·25
cm, Eluent: n-Hexan/2-Propanol = 3:1, Flussrate: 5,0 ml/min, Temp.:
Raumtemperatur, Nachweis: UV 210 nm) getrennt, wodurch sich 423
mg (+)-(1R*,5R*,6R*)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
und 405 mg (–)-(1R*,5R*,6R*)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat ergaben.
(+)-(1R*,5R*,6R*)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
1H-NMR (CDCl3) δ (ppm): 1,33
(3H, t, J = 7,1 Hz), 2,05–2,55
(4H, m), 2,59 (1H, d, J = 6,6 Hz), 2,70–2,77 (1H, m), 4,30 (2H, q,
J = 7,1 Hz)
MS (IonSpray) (Pos) m/e: 187 (M++1),
204 (M++18), 209 (M++23,
100%
TR = 5,65 min (CHIRALPAK AD 0,46·25 cm,
Eluent: n-Hexan/2-Propanol = 3:1, Flussrate: 1,0 ml/min, Temperatur:
Raumtemperatur, Nachweis: UV 210 nm) [α]D 27 = +27,98 (c = 0,13, CHCl3)
(–)-(1R*,5R*,6R*)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
1H-NMR (CDCl3) δ (ppm): 1,33
(3H, t, J = 7,1 Hz), 2,05–2,55
(4H, m), 2,59 (1H, d, J = 6,6 Hz), 2,70–2,77 (1H, m), 4,30 (2H, q,
J = 7,1 Hz)
MS (IonSpray) (Pos) m/e: 187 (M++1),
204 (M++18), 209 (M++23,
100%
TR = 9,13 min (CHIRALPAK AD 0,46·25 cm,
Eluent: n-Hexan/2-Propanol = 3:1, Flussrate: 1,0 ml/min, Temperatur:
Raumtemperatur, Nachweis: UV 210 nm)
[α]D 27 = –30,33
(c = 0,16, CHCl3)
-
Beispiel 4
-
Synthese von (1RS,2SR,5RS,6RS)-2-Spiro-5'-hydantoin-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure
-
256
mg (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat wurden
in 2,5 ml Ethanol gelöst.
1,4 ml wässrige
1 N Natriumhydroxidlösung
wurden tropfenweise unter Eiskühlung
dazugegebe, und dann wurde das Rühren
bei dieser Temperatur für
10 min fortgesetzt. Nach dem Ansäuern
der Reaktionslösung
mit 1 N Salzsäure
(pH war fast gleich 1) wurde mit Ethylacetat verdünnt und
mit einer gesättigten
wässrigen
Natriumchloridlösung
gewaschen. Die wässrige
Schicht wurde 2 Mal mit Ethylacetat extrahiert und die erhaltenen
organischen Schichten wurden vereinigt, um sie über wasserfreiem Natriumsulfat
zu trocknen. Nach dem Abfiltrieren des Trocknungsmittels wurde Konzentration
des Filtrats unter reduziertem Druck durchgeführt. Der erhaltene Rückstand
wurde in 2 ml einer Mischung aus Wasser und Ethanol (1:1) gelöst, und
dann wurde er bei 55°C
für 8,5
h gerührt,
nachdem 796 mg Ammoniumcarbonat und 277 mg Kaliumcyanid dazugegeben
worden waren. Die Reaktionsmischung wurde eisgekühlt und dann wurde sie durch
Zugabe von konzentrierter Salzsäure
neutralisiert. Sie wurde durch Ionenaustauschchromatographie gereinigt
(AG50W-X8 Kationenaustauschharz (Bio-Rad), Eluent: Wasser), wodurch
sich 320 mg (1RS,2SR,5RS,6RS)-2-Spiro-5'-hydantoin-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure ergaben.
Die Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (DMSO-d6) δ (ppm): 1,49–1,70 (1H,
m), 1,93–2,40
(5H, m), 8,08 (1H, s), 10,71 (1H, s)
MS (CI) (Pos) m/e: 229
(M++1, 100%)
-
Genauso
wurden die folgenden Verbindungen erhalten. Die Daten der physikalischen
Eigenschaften für
jede sind unten zusammen gezeigt.
(1RS,2SR,5RS,6SR)-2-Spiro-5'-hydantoin-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure
1H-NMR (DMSO-d6) δ (ppm): 1,80–2,38 (6H,
m), 7,34 (1H, s), 10,74 (1H, s)
MS (CI) (Pos) m/e: 229 (M++1, 100%)
(+)-(1R*,2S*,6R*)-2-Spiro-5'-hydantoin-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure
1H-NMR (DMSO-d6) δ (ppm): 1,49–1,70 (1H,
m), 1,93–2,40
(5H, m), 8,08 (1H, s), 10,71 (1H, s)
MS (CI) (Pos) m/e: 229
(M++1, 100%)
[α]D 25,5 = + 77,87 (c = 0,43, 1 N NaOH)
(–)-(1R*,2S*,5R*,6R*)-2-Spiro-5'-hydantoin-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure
1H-NMR (DMSO-d6) δ (ppm): 1,49–1,70 (1H,
m), 1,93–2,40
(5H, m), 8,08 (1H, s), 10,71 (1H, s)
MS (CI) (Pos) m/e: 229
(M++1, 100%)
[α]D 25,5 = –77,30
(c = 0,41, 1 N NaOH)
-
Beispiel 5
-
Synthese von (1RS,2SR,5RS,6RS)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
-
200
mg (1RS,2SR,5RS,6RS)-2-Spiro-5'-hydantoin-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure wurden in
3,0 ml einer 60%igen Schwefelsäure
bei 140°C
für 6 Tage
gerührt.
Nach Eiskühlung
und Neutralisierung der Reaktionslösung mit wässriger 5 N Natriumhydroxidlösung wurde
sie durch Ionenaustauschchromatographie gereinigt (AG50W-X8 Kationenaustauschharz
(Bio-Rad), Eluent: Wasser-50% THF/Wasser-10% Pyridin/Wasser), wodurch
sich 61 mg (1RS,2SR,5RS,6RS)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure ergaben.
Die Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (TFA-d) δ (ppm): 2,15–2,28 (1H, m), 2,57 (1H, dd,
J = 13,5, 8,6 Hz), 2,67–2,94
(4H, m)
MS (IonSpray) (Nega) m/e: 202 (M+–1, 100%)
-
Genauso
wurden die folgenden Verbindungen erhalten. Die Daten der physikalischen
Eigenschaften für
jede sind zusammen gezeigt. (1RS,2SR,5RS,6SR)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
1H-NMR (TFA-d) δ (ppm): 2,36–2,54 (2H, m), 2,58–2,87 (4H,
m)
MS (CI) (IonSpray) (Nega) m/e: 202 (M+–1, 100%)
(–)-(1R*,2S*,5R*,6R*)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
1H-NMR (TFA-d) δ (ppm): 2,15–2,28 (1H, m), 2,57 (1H, dd,
J = 13,5, 8,6 Hz), 2,67–2,99
(4H, m)
MS (IonSpray) (Nega) m/e: 202 (M+–1, 100%)
[α]D 26 = –58,81 (c
= 0,14, H2O)
(+)-(1R*,2S*,5R*,6R*)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
1H-NMR (TFA-d) δ (ppm): 2,15–2,28 (1H, m), 2,57 (1H, dd,
J = 13,5, 8,6 Hz), 2,67–2,94
(4H, m)
MS (IonSpray) (Nega) m/e: 202 (M+–1, 100%)
[α]D 26 = +57,49 (c =
0,16, H2O)
-
Beispiel 6
-
Synthese von (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyco[3.1.0]hex-3-en-6-carboxylat
-
Unter
einer Stickstoffatmosphäre
wurden 19,5 g (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat,
gelöst
in 230 ml Tetrahydrofuran, bei –78°C tropfenweise
zu 230 ml einer Tetrahydrofuranlösung
von Lithium-bis(trimethylsilyl)amid, die aus 78 ml n-Butyllithium
(1,61 M Hexanlösung)
und 20,3 g 1,1,1,3,3,3-Hexamethyldisilazan
hergestellt wurde, zugegeben. Nach Rühren bei dieser Temperatur
für 1 h wurden
19,8 ml Chlortrimethylsilan zugegeben, und dies wurde bei Raumtemperatur
für 1,5
h gerührt.
Nach dem Durchführen
von Konzentration der Reaktionslösung
unter reduziertem Druck wurde wasserfreies Hexan zu dem Rückstand
zugegeben. Das resultierende anorganische Salz wurde abfiltriert
und das Filtrat wurde weiter unter reduziertem Druck konzentriert.
Nachdem der Rückstand
in 240 ml Acetonitril gelöst
worden war, wurden 25,9 g Palladiumacetat zugegeben, und dann wurde
dies bei Raumtemperatur für
1 Tag gerührt.
Die Reaktionslösung
wurde mit 240 ml Diethylether verdünnt, das Palladium wurde unter
Verwendung von Celite abfiltriert und das Filtrat wurde unter reduziertem
Druck konzentriert. Der Rückstand
wurde durch Säulenchromatographie
(Silikagel: Wako Gel C200 (hergestellt von Wako Pure Chemical Industries
Ltd.), Eluent: Hexan-Ethylacetat = 9:1 bis 5:1) gereinigt, wodurch
sich 17,1 g (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyclo[3.1.0]hex-3-en-6-carboxylat
ergaben.
-
Die
Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,39
(3H, t, J = 7,3 Hz), 2,78 (1H, dt, J = 0,6, 5,8 Hz), 3,22 (1H, dd,
J = 2,9, 5,8 Hz), 4,31 (2H, q, J = 7,3 Hz), 6,07 (1H, dd, J = 0,6,
5,6 Hz), 7,42 (1H, ddd, J = 0,6, 2,9, 5,6 Hz)
MS (CI) (Pos)
m/e: 185 (M++1, 100%)
-
Beispiel 7
-
Synthese von (1RS,3RS,4RS,5SR,6RS)Ethyl-3,4-epoxy-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
-
19,6
g (1RS,5RS,6RS)Ethyl-6-fluor-2-oxobicyco[3.1.0]hex-3-en-6-carboxylat wurden
in 100 ml Toluol gelöst:
30,6 ml einer wässrigen
70%igen t-Butylhydroxyperoxidlösung
und 11,5 ml einer 10%igen Benzyltrimethylammoniumhydroxid/Methanol-Lösung wurden
dazugegeben, und dies wurde bei Raumtemperatur für 4 h gerührt. Nachdem die Reaktionslösung in
Wasser gefüllt
worden war, wurde sie 2 Mal mit Ethylacetat extrahiert. Die erhaltenen
organischen Schichten wurden vereinigt, um sie mit einer gesättigten
wässrigen
Natriumchloridlösung
zu waschen und wurden über
wasserfreiem Natriumsulfat getrocknet. Nach dem Abfiltrieren des Trocknungsmittels
wurde Konzentration des Filtrats unter reduziertem Druck durchgeführt. Der
Rückstand
wurde durch Säulenchromatographie
(Silikagel: Wako Gel C200 (hergestellt von Wako Pure Chemical Industries Ltd.),
Eluent: Hexan-Ethylacetat = 8:1 bis 6:1) gereinigt, wodurch sich
13,4 g (1RS,3RS,4RS,5SR,6RS)Ethyl-3,4-epoxy-6-fluor-2-oxobicyco[3.1.0]hexan-6-carboxylat ergaben.
-
Die
Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (CDCl3) δ (ppm): 1,34
(3H, t, J = 7,3 Hz), 2,50 (1H, ddt, J = 0,8, 2,4 6,0 Hz), 3,19 (1H,
dt, J = 0,8, 6,0 Hz), 3,53 (1H, dt, J = 0,8, 2,4 Hz), 4,02 (1H,
tt, J = 0,8, 2,4 Hz), 4,32 (2H, q, J = 7,3 Hz)
MS (EI) (Pos)
m/e: 99 (M+–101, 100%), 200 (M+)
-
Beispiel 8
-
Synthese von (1RS,4SR,5SR,6RS)Ethyl-6-fluor-4-hydroxy-2-oxobicyclo[3.1.0]hexan-6-carboxylat
-
Unter
einer Stickstoffatmosphäre
wurden 23,2 g N-Acetyl-L-cystein, 54,3 g Natriumtetraboratdecahydrat
und 0,7 g Diphenyldiselenid in 450 ml einer Wasser-Ethanol(1:1)-Mischlösung, die
entgast worden war, suspendiert. 9,5 g (1RS,3RS,4RS,5SR,6RS)Ethyl-3,4-epoxy-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat, gelöst in 225
ml Tetrahydrofuran, wurden dazugegeben, und dies wurde bei Raumtemperatur
für 1 Tag
bei 38°C
für 12
h und bei 85°C
für 5 h
gerührt.
Nachdem die Reaktionslösung
auf Raumtemperatur abgekühlt
worden war, wurde sie in Wasser gefüllt und 3 Mal mit Diethylether
extrahiert. Die erhaltenen organischen Schichten wurden vereinigt
und über
wasserfreiem Natriumsulfat getrocknet. Nach Abfiltration des Trocknungsmittels wurde
Konzentration des Filtrats unter reduziertem Druck durchgeführt. Der
Rückstand
wurde durch Säulenchromatographie
(Silikagel: Wako Gel (hergestellt von Wako Pure Chemical Industries
Ltd.), Eluent: Hexan-Ethylacetat = 3:1 bis 1:1) gereinigt, wodurch
sich 3,9 g (1RS,4SR,5SR,6RS)Ethyl-6-fluor-4-hydroxy-2-oxobicyclo[3.1.0]hexan-6-carboxylat ergaben.
-
Die
Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1-NMR (CDCl3) δ (ppm): 1,34
(3H, t, J = 7,1 Hz), 2,05 (1H, d, J = 5,1 Hz), 2,30 (1H, dd, J =
3,5, 19,2 Hz), 2,63 (1H, dt, J = 5,9, 19,2 Hz), 2,72 (1H, d, J =
5,9 Hz), 2,85 (1H, dd, J = 2,1, 5,9 Hz), 4,31 (2H, q, J = 7,1 Hz),
4,76 (1H, t, J = 5,1 Hz)
MS (EI) (Pos) m/e: 129 (M+–73, 100%),
202 (M+)
-
Beispiel 9
-
Synthese von (1RS,4SR,5SR,6RS)Ethyl-6-fluor-4-t-butyldimethylsilyloxy-2-oxobicyclo[3.1.0]hexan-6-carboxylat
-
2,8
g (1RS,4SR,5SR,6RS)Ethyl-6-fluor-4-hydroxy-2-oxobicyco[3.1.0]hexan-6-carboxylat
und 2,5 g t-Butyldimethylchlorsilan wurden in 14 ml N,N-Dimethylformamid
gelöst.
1,0 g Imidazol wurde ferner unter Eiskühlung dazugegeben, und dies
wurde bei Raumtemperatur für
1 Tag gerührt.
Die Reaktionslösung
wurde in Wasser gefüllt
und mit n-Hexan-Ethylacetat (1:9) extrahiert. Die erhaltene organische
Schicht wurde nacheinander mit Wasser und einer gesättigten
wässrigen
Natriumchloridlösung
gewaschen und dann über
wasserfreiem Natriumsulfat getrocknet. Nach Abfiltration des Trocknungsmittels
wurde Konzentration des Filtrats unter einem reduziertem Druck durchgeführt. Der
Rückstand
wurde durch Säulenchromatographie
(Silikagel: Wako Gel (hergestellt von Wako Pure Chemical Industries
Ltd.), Eluent: Hexan-Ethylacetat = 15:1) gereinigt, wodurch sich
3,8 g (1RS,4SR,5SR,6RS)Ethyl-6-fluor-4-t-butyldimethylsilyloxy-2-oxobicyclo[3.1.0]hexan-6-carboxylat
ergaben. Die Protonen-NMR- und massenspektroskopischen Daten sind
unten gezeigt.
1-NMR (CDCl3) δ (ppm): 0,11
(3H, s), 0,13 (3H, s), 0,90 (9H, s), 1,33 (3H, t, J = 7,1 Hz), 2,21
(1H, dd, J = 9,0, 19,1 Hz), 2,57 (1H, dt, J = 5,6, 19,1 Hz), 2,60–2,72 (4H,
m), 4,31 (2H, q, J = 7,1 Hz), 4,66 (1H, d, J = 5,6 Hz)
MS (CI)
(Pos) m/e: 259 (M+–57, 100%), 317 (M++1)
-
Beispiel 10
-
Synthese von (1RS,4RS,5RS,6SR)Ethyl-2,2-ethylendithio-6-fluor-4-hydroxybicyclo[3.1.0]hexan-6-carboxylat
-
3,7
g (1RS,4SR,5SR,6RS)Ethyl-6-fluor-4-t-butyldimethylsilyloxy-2-oxobicyclo[3.1.0]hexan-6-carboxylat
und 1,2 ml 1,2-Ethandithiol wurden in 37 ml Chloroform gelöst. Trifluorboran-Diethylether-Komplex
wurde tropfenweise zugegeben, und dies wurde bei Raumtemperatur
für 1 Tag
gerührt.
Die Reaktionslösung
wurde nacheinander mit einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
und einer gesättigten
wässrigen
Natriumchloridlösung
gewaschen und dann über
wasserfreiem Natriumsulfat getrocknet. Nach Abfiltration des Trocknungsmittels
wurde Konzentration des Filtrats unter reduziertem Druck durchgeführt. Der
Rückstand wurde
durch Säulenchromatographie
(Silikagel: Wako Gel (hergestellt von Wako Pure Chemical Industries Ltd.),
Eluent: Hexan-Ethylacetat = 2:1) gereinigt, wodurch sich 3,2 g (1RS,4RS,5RS,6SR)Ethyl-2,2-ethylendithio-6-fluor-4-hydroxybicyclo[3.1.0]hexan-6-carboxylat
ergaben. Die Protonen-NMR- und massenspektroskopischen Daten sind
unten gezeigt.
1-NMR (CDCl3) δ (ppm): 1,32
(3H, t, J = 7,1 Hz), 2,07 (1H, d, J = 7,1 Hz), 2,38–2,69 (4H,
m), 3,33–3,45
(4H, m), 4,27 (2H, q, J = 7,1 Hz), 4,50 (1H, dd, J = 5,5, 7,1 Hz)
MS
(EI) (Pos) m/e: 131 (M+–147, 100%), 278 (M+)
-
Beispiel 11
-
Synthese von (1RS,5RS,6SR)Ethyl-4,4-ethylendithio-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
-
3,1
g (1RS,4RS,5RS,6SR)Ethyl-2,2-ethylendithio-6-fluor-4-hydroxybicyclo[3.1.0]hexan-6-carboxylat und
9,0 g Dicyclohexylcarbodiimid wurden in 116 ml Dimethylsulfoxid
gelöst.
Nacheinander wurden 1,2 ml Pyridin und 0,6 ml Trifluoressigsäure tropfenweise
zugegeben, und dies wurde bei Raumtemperatur für 1 Tag gerührt. Nach Abfiltration des
resultierenden Harnstoffs wurde er mit Ethylacetat gewaschen. Das
Filtrat wurde mit Ethylacetat verdünnt, 3 Mal mit Wasser und mit
einer gesättigten
wässrigen
Natriumchloridlösung
gewaschen und dann über
wasserfreiem Natriumsulfat getrocknet. Nach Abfiltration des Trocknungsmittels
wurde Konzentration des Filtrats unter reduziertem Druck durchgeführt. Der
Rückstand
wurde durch Säulenchromatographie
(Silikagel: Wako Gel (hergestellt vgon Wako Pure Chemical Industries
Ltd.), Eluent: Hexan-Ethylacetat
= 5:1) gereinigt, wodurch sich 2,6 g (1RS,5RS,6SR)Ethyl-4,4- ethylendithio-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
ergaben. Die Protonen-NMR- und massenspektroskopischen Daten sind
unten gezeigt.
1-NMR (CDCl3) δ (ppm): 1,35
(3H, t, J = 7,1 Hz), 2,79 (1H, d, J = 6,3 Hz), 2,86–3,08 (2H,
m), 3,18 (1H, dd, J = 1,9, 6,3 Hz), 3,38–3,53 (4H, m), 4,31 (2H, q,
J = 7,1 Hz)
MS (EI) (Pos) m/e: 131 (M+–145, 100%),
276 (M+)
-
Beispiel 12
-
Synthese von (1R*,2S*,5R*,65*)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluor-N-((R)-1-phenylethyl)bicyclo[3.1.0]hexan-6-carboxyamid
-
(1)
1,3 g (1RS,5RS,6SR)Ethyl-4,4-ethylendithio-6-fluor-2-oxobicyclo[3.1.0]hexan-6-carboxylat
wurden in 5,0 ml Ethanol gelöst.
Unter Eiskühlung
wurden 5,0 ml wässrige
1 N Natriumhydroxidlösung
tropfenweise zugegeben, und dies wurde bei Raumtemperatur für 15 min
gerührt.
Nach Erwärmen
der Reaktionslösung
auf Raumtemperatur wurden 1,1 g Ammoniumcarbonat und 350 mg Kaliumcyanid
zugegeben, und dann wurde dies bei 37°C für 3 Tage gerührt. Nach
Eiskühlung
und Einstellen des pHs der Reaktionsmischung auf 1 durch Zugabe
von konzentrierter Salzsäure
wurden 5 ml Ethanol zugegeben und Rühren bei dieser Temperatur
für 1 h
fortgesetzt. Nach Abfiltrieren der resultierenden Kristalle und
Waschen mit einer Mischlösung
aus Ethanol/Wasser (2:1) wurde dies bei 80°C getrocknet, wodurch sich 1,1
g (1RS,2SR,5RS,6SR)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure ergaben.
Die Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (DMSO-d6) δ (ppm): 2,37–2,50 (2H,
m), 2,68 (1H, dd, J = 1,9, 6,9 Hz), 2,76 (1H, dd, J = 4,2, 15,4
Hz), 3,28–3,50
(4H, m), 8,10 (1H, s), 10,78 (1H, s).
MS (ES) (Nega) m/e: 317
(M+–1,
100%)
-
(2)
5,7 g (1RS,2SR,5RS,6SR)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure und
2,6 g (R)-(+)-1-Phenylethylamin wurden in 240 ml Dimethylformamid
gelöst.
3,4 g 1-Hydroxybenzotriazol-Monohydrat und 4,1 g 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-Hydrochlorid wurden
unter Eiskühlung
dazugegeben, und Rühren
wurde bei Raumtemperatur für
1 Tag fortgesetzt. Nach Zugeben von 1 N Salzsäure zu der Reaktionslösung und
4-maligem Extrahieren mit Ethylacetat wurde sie über wasserfreiem Natriumsulfat
getrocknet. Nach Abfiltrieren des Trocknungsmittels wurde Konzentration
unter reduziertem Druck durchgeführt.
Der Rückstand
wurde durch Chromatographie behandelt (Silikagel: MSG D-40-60A (hergestellt
von Dokai Chemicals Ltd.), Eluent: Chloroform-Methanol = 50:1),
wodurch sich 3,5 g eines niedrigpolaren Diastereomers (1R*,2S*,5R*,6S*)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluor-N-((R)-1-phenylethyl)bicyclo[3.1.0]hexan-6-carboxyamid
ergaben (Rf-Wert 0,74 (TLC: Silikagel 60 F254 (hergestellt
von Merck), Eluent: Chloroform-Methanol = 9:1)), und 3,5 g eines
polaren Diastereomers (1R*,2S*,5R*,6S*)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluor-N-((R)-1-phenylethyl)bicyclo[3.1.0]hexan-6-carboxyamid
(Rf-Wert 0,69 (TLC: Silikagel 60 F254 (hergestellt von Merck), Eluent:
Chloroform-Methanol = 9:1)). Der Schmelzpunkt und der spezifische
optische Rotationswert jeder Verbindung sind unten gezeigt.
-
Das
niedrigpolare Diastereomer
Schmelzpunkt 288–289°C
[α]D 26 = +62,55 (c = 0,21, MeOH)
-
Das
polare Diastereomer
Schmelzpunkt 315–316°C
[α]D 26 = +52,58 (c = 0,24, MeOH)
-
Beispiel 13
-
Synthese von (1RS,2SR,5SR,6SR)-2-Amino-6-fluor-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure
-
500
mg (1RS,2SR,5RS,6SR)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure wurden
in 12 ml 60%ige Schwefelsäure
(W/V%) bei 145°C
für 4 Tage
gerührt.
Nach Eiskühlung
und Neutralisierung der Reaktionslösung mit wässriger 5 N Natriumhydroxidlösung wurde
sie durch Ionenaustauschchromatographie gereinigt (AG50W-X8 Kationenaustauschharz
(Bio-Rad), H+-Typ, Eluent: Wasser-50% THF/Wasser-Wasser-10%
Pyridin/Wasser). Die erhaltenen Kristalle wurden mit einer Mischlösung aus
Tetrahydrofuran-Wasser gewaschen, wodurch sich 41 mg (1RS,2SR,5SR,6SR)-2-Amino-6-fluor-4-oxobicylo[3.1.0]hexan-2,6-dicarbonsäure ergaben.
Die Protonen-NMR- und massenspektroskopischen Daten sind unten gezeigt.
1H-NMR (TFA-d) δ (ppm): 3,16 (1H, dd, J = 4,6,
19,5 Hz), 3,45 (1H, dd, J = 4,6, 19,5 Hz), 3,46 (1H, d, J = 6,6 Hz),
3,67 (1H, d, J = 6,6 Hz)
MS (ES) (Nega) m/e): 216 (M+–1)
-
Genauso
wurden die folgenden Verbindungen aus dem niedrigpolaren Diastereomer
und dem polaren Diastereomer von (1R*,2S*,5R*,6S*)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluor-N-((R)-1-phenylethyl)bicyclo[3.1.0]hexan-6-carboxyamid
erhalten. Die Daten der physikalischen Eigenschaften jeder Verbindung
sind unten gezeigt.
(–)-(1R*,2S*,5S*,6S*)-2-Amino-6-fluor-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure
Schmelzpunkt
175°C (zersetzt)
1H-NMR (TFA-d) δ (ppm): 3,16 (1H, dd, J = 4,6,
19,5 Hz), 3,45 (1H, dd, J = 4,6, 19,5 Hz), 3,46 (1H, d, J = 6,6 Hz),
3,67 (1H, d, J = 6,6 Hz)
MS (ES) (Nega) m/e: 216 (M+–1)
[α]D 26 = –97,01 (c
= 0,16, H2O)
(+)-(1R*,2S*,5S*,6S*)-2-Amino-6-fluor-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure
Schmelzpunkt
175°C (zersetzt)
1H-NMR (TFA-d) δ (ppm): 3,16 (1H, dd, J = 4,6,
19,5 Hz), 3,45 (1H, dd, J = 4,6, 19,5 Hz), 3,96 (1H, d, J = 6,6 Hz),
3,67 (1H, d, J = 6,6 Hz)
MS (ES) (Nega) m/e: 216 (M+–1)
[α]D 26 = +99,84 (c =
0,13, H2O)
-
Beispiel 14
-
Synthese von (1RS,2SR,5RS,6SR)-2-Amino-4,4-ethylendithio-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
-
120
mg (1RS,2SR,5RS,6SR)-2-Spiro-5'-hydantoin-4,4-ethylendithio-6-fluorbicyclo[3.1.0]hexan-6-carbonsäure in 1,4
ml wässrige
2 N Natriumhydroxidlösung
wurden unter Reflux für
1,5 Tage behandelt. Nachdem sie abgekühlt wurde, wurde sie durch
Ionenaustauschchromatographie gereinigt (AG50W-X8 Kationenaustauschharz
(Bio-Rad), H+-Typ, Eluent: Wasser-50% THF/Wasser-Wasser-10%
Pyridin/Wasser), wodurch sich 75 mg (1RS,2SR,5RS,6SR)-2-Amino-4,4-ethylendithio-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure ergaben. Die
Daten der physikalischen Eigenschaften sind unten gezeigt.
Schmelzpunkt
230°C (zersetzt)
1H-NMR (TFA-d) δ (ppm): 3,07 (1H, dd, J = 5,5,
16,1 Hz), 3,16 (1H, d, J = 5,5 Hz), 3,25 (1H, dd, J = 2,7, 7,1 Hz),
3,38–3,51
(5H, m)
MS (ES) (Nega) m/e: 292 (M+–1, 100%)
-
Beispiel 15
-
Synthese of (1RS,2SR,4SR,5SR,6SR)Ethyl-2-spiro-5'-hydantoin-6-fluor-4-hydroxybicyclo[3.1.0]hexan-6-carboxylat
-
1,3
g (1RS,4SR,5SR,6RS)Ethyl-6-fluor-4-hydroxy-2-oxobicyclo[3.1.0]hexan-6-carboxylat
wurden in 3,7 ml Ethanol gelöst.
Unter Eiskühlung
wurden 3,7 ml wässrige
1 N Natriumhydroxidlösung
tropfenweise zugegeben, und Rühren
wurde bei dieser Temperatur für
15 min fortgesetzt. Nach Erhöhen
der Reaktionslösungstemperatur
auf Raumtemperatur wurden 860 mg Ammoniumcarbonat und 260 mg Kaliumcyanid
zugegeben, und dies wurde bei 37°C
für 3 Tage
gerührt.
Nach Abkühlen
der Reaktionsmischung wurde der pH auf 1 durch Zugeben von konzentrierter
Salzsäure
eingestellt. Diese Lösung
wurde durch Ionenaustauschchromatographie behandelt (AG50W-X8 Kationenaustauschharz
(Bio-Rad), H+-Typ, Eluent: Wasser), wodurch
sich 450 mg rohe (1RS,2SR,4SR,5SR,6SR)-2-Spiro-5'-hydantoin-6-fluor-4-hydroxybicyclo[3.1.0]hexan-6-carbonsäure ergaben.
450 mg dieser (1RS,2SR,4SR,5SR,6SR)-2-Spiro-5'-hydantoin-6-fluor-hydroxybicyclo[3.1.0]-hexan-6-carbonsäure, 90
mg Ethanol und 20 mg 4-Dimethylaminopyridin wurden in 3,9 ml Dimethylformamid
gelöst.
Ferner wurden 380 mg 1-(3-dimethylaminopropyl)-3-ethylcarbodiimid-Hydrochlorid unter
Eiskühlung
zugegeben, und dies wurde für
1 Tag gerührt.
Die Reaktionslösung
wurde in 1 N Salzsäure
gefüllt,
mit Chloroform 6 Mal extrahiert und über wasserfreiem Natriumsulfat
getrocknet, nachdem die erhaltenen organischen Schichten vereinigt
worden waren. Nach Abfiltrieren des Trocknungsmittels wurde das
Filtrat unter reduziertem Druck konzentriert. Der Rückstand
wurde durch Säulenchromatographie
(Silikagel: MSGD75-60A (hergestellt von Dokai Chemicals Ltd.), Eluent:
Ethylacetat = 50:1) gereinigt, wodurch sich 198 mg (1RS,2SR,4SR,5SR,6SR)Ethyl-2-spiro-5'-hydantoin-6-fluor-4-hydroxybicyclo[3.1.0]hexan-6-carboxylat
ergaben. Die Protonen-NMR- und massenspektroskopischen Daten sind
unten gezeigt.
1H-NMR (DMSO-d6) δ (ppm):
1,21 (3H, t, J = 7,2 Hz), 1,90–2,08
(2H, m), 2,26 (1H, dd, J = 1,8, 7,2 Hz), 2,45 (1H, dd, J = 1,8,
7, Hz), 4,17 (2H, q, J = 7,2 Hz), 4,33 (dd, J = 5,6, 8,8 Hz), 4,75
(1H, d, J = 8,8 Hz), 8,13 (1H, s), 11,00 (1H, s)
MS (ES) (Nega)
m/e: 271 (M+–1, 100%)
-
Beispiel 16
-
Synthese von (1RS,2SR,4SR,5SR,6SR)-2-Amino-6-fluor-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure
-
140
mg (1RS,2SR,4SR,5SR,6SR)Ethyl-2-spiro-5'-hydantoin-6-fluor-4-hydroxybicyclo[3.1.0]hexan-6-carboxylat
wurden in 4 ml 60%ige Schwefelsäure
(W/V%) bei 145°C
für 2,5
Tage gerührt.
Nach Eiskühlung
und Neutralisieren der Reaktionslösung mit wässriger 5 N Natriumhydroxidlösung wurde
sie durch Ionenaustauschchromatographie (AG50W-X8 Kationenaustauschharz
Bio-Rad), H+-Typ, Eluent: Wasser-50% THF/Wasser-Wasser-10%
Pyridin/Wasser) gereinigt. Die erhaltenen Kristalle wurden mit einer
gemischten Lösung
aus Aceton-Tetrahydrofuran gewaschen, wodurch sich 17 mg (1RS,2SR,4SR,5SR,6SR)-2-Amino-6-fluor-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure ergaben.
Die Daten der physikalischen Eigenschaften sind unten gezeigt.
Schmelzpunkt
220°C (zersetzt)
1H-NMR (Pyridin-d6/D2O = 1/1) δ (ppm):
2,56–2,75
(3H, m), 2,92 (1H, dd, J = 1,2, 6,9 Hz), 4,56 (1H, d, J = 5,9 Hz)
MS
(ES) (Nega) m/e: 218 (M+–1, 100%)
-
Experimentelle Beispiele
(Wirkung von Testverbindungen auf cAMP-Akkumulation)
-
CHO-Zellen,
die metabotrope Glutamatrezeptoren mGluR2 stabil exprimieren, wurden
in eine 96-Wellplatte (1,26·10
4 Zellen/Vertiefung (0,32 cm
2/150 μl) in einem
Dulbecco-modifizierten Eagle-Medium [1% Prolin, 50 Einheiten/ml
Penicillin, 50 μg/ml
Streptomycin, 2 mM L-Glutamin (bei der Verwendung zugegeben)], das 10%
dialysiertes fötales
Rinderserum enthielt, ausgesät
und für
2 Tage bei 37°C
unter einer Atmosphäre
von 5% CO
2 kultiviert. Nachdem das Medium
durch ein L-Glutamin-freies Medium ersetzt worden war, wurden es für 4 h kultiviert
und die Überstandsflüssigkeit
abgesaugt. Nach der Zugabe von 150 μl PBS(+)-IBMX (10 mM PBS(–), 1 mM
MgCl
2, 1 mM CaCl
2,
1 mM IBMX) wurde die Inkubation bei 37°C in Gegenwart von 5% CO
2 für 20
Minuten durchgeführt.
Noch einmal wurde die Überstandsflüssigkeit
abgesaugt, 60 μl
10
–5 M
Forskolin und PBS(+)-IBMX, die in den Proben in Tabelle 1 zwischen
10
–10 und
10
–4 M
enthalten waren, wurden zugegeben, Inkubation wurde für 15 min
bei 37°C
in Gegenwart von 5% CO
2 durchgeführt, und
eine Untersuchung im Hinblick auf die Inhibitionswirkung der Agonisten
auf die Forskolin-Stimulierungs-cAMP-Akkumulationsmenge wurde durchgeführt [zur
Kontrolle wurden die Bedingungen auf Forskolin, ohne die Zugabe
der Verbindungen eingestellt (Tanabe et al., Neuron 8, 169–179 (1992))].
Die Reaktionen wurden durch die Zugabe von 100 μl eisgekühltem Ethanol unterbrochen,
die gesamte Menge der Überstandsflüssigkeit
wurde in einer separaten Platte gesammelt, dann wurde sie bei Normaltemperatur
mit einem Evaporator getrocknet und bei –20°C gehalten. In den getrockneten
Proben wurde die Menge an cAMP unter Verwendung eines cAMP EIA-Kits
(von der Amasham Company) gemessen. Der Kontrollwert wurde von jeder
cAMP-Menge abgezogen. Der Konzentrationswert der Testverbindungen
bei der cAMP-Akkumulation, der um 50% inhibiert wurde, wenn Stimulation durch
10
–5 M
Forskolin bewirkt wurde, wurde als ED
50 bestimmt.
Die Ergebnisse sind in Tabelle 1 dargestellt. Tabelle
1
- Komp. 1:
(1RS,2SR,5RS,6RS)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
- Komp. 2:
(–)-(1R*,2S*,5R*,6R*)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
- Komp. 3:
(1RS,2SR,5SR,6SR)-2-Amino-6-fluorbicyclo[3.1.0]hexan-2,6-dicarbonsäure
- Komp. 4:
(+)-(1R*,2S*,5S*,6S*)-2-Amino-6-fluor-4-oxobicyclo[3.1.0]hexan-2,6-dicarbonsäure
- Komp. 5:
(1RS,2SR,45R,5SR,6SR)-2-Amino-6-fluor-4-hydroxybicyclo[3.1.0]hexan-2,6-dicarbonsäure
- LY354740:
(+)-(1S,2S,5R,6S)-2-Aminobicyclo[3.1.0]hexan-2,6-dicarbonsäure
- DCG IV:
(2S,1'R,2'R,3'R)-2-(2',3'-Dicarboxycyclopropyl)glycin
- (1S,3R)-ACPD
(1S,3R)-1-Aminocyclopentan-1,3-dicarbonsäure
- L-CCG-I:
(2S,1'S,2'S)-2-(Carboxycyclopropyl)glycin
-
Industrielle Anwendbarkeit
-
Die
6-Fluorbicyclo[3.1.0]hexan-Derivate gemäß der vorliegenden Erfindung
sind als Arzneistoffe nützlich.
Insbesondere sind sie als Agonisten nützlich, die auf metabotrope
Glutamatrezeptoren wirken. Daher kann diese Erfindung zur Behandlung
und Vorbeugung psychiatrischer Störungen, wie z.B. Schizophrenie, Angst,
damit verbundene Krankheiten, Depression, bipolare Störung und
Epilepsie sowie neurologische Krankheiten, wie z.B. Drogenabhängigkeit,
kognitive Störung,
Alzheimer-Krankheit, Chorea Huntington, Parkinson-Krankheit, Dyskinesie,
verbunden mit Muskelversteifung, zerebrale Ischämie, zerbrales Versagen, Myelopathie
und Kopftrauma verwendet werden.