-
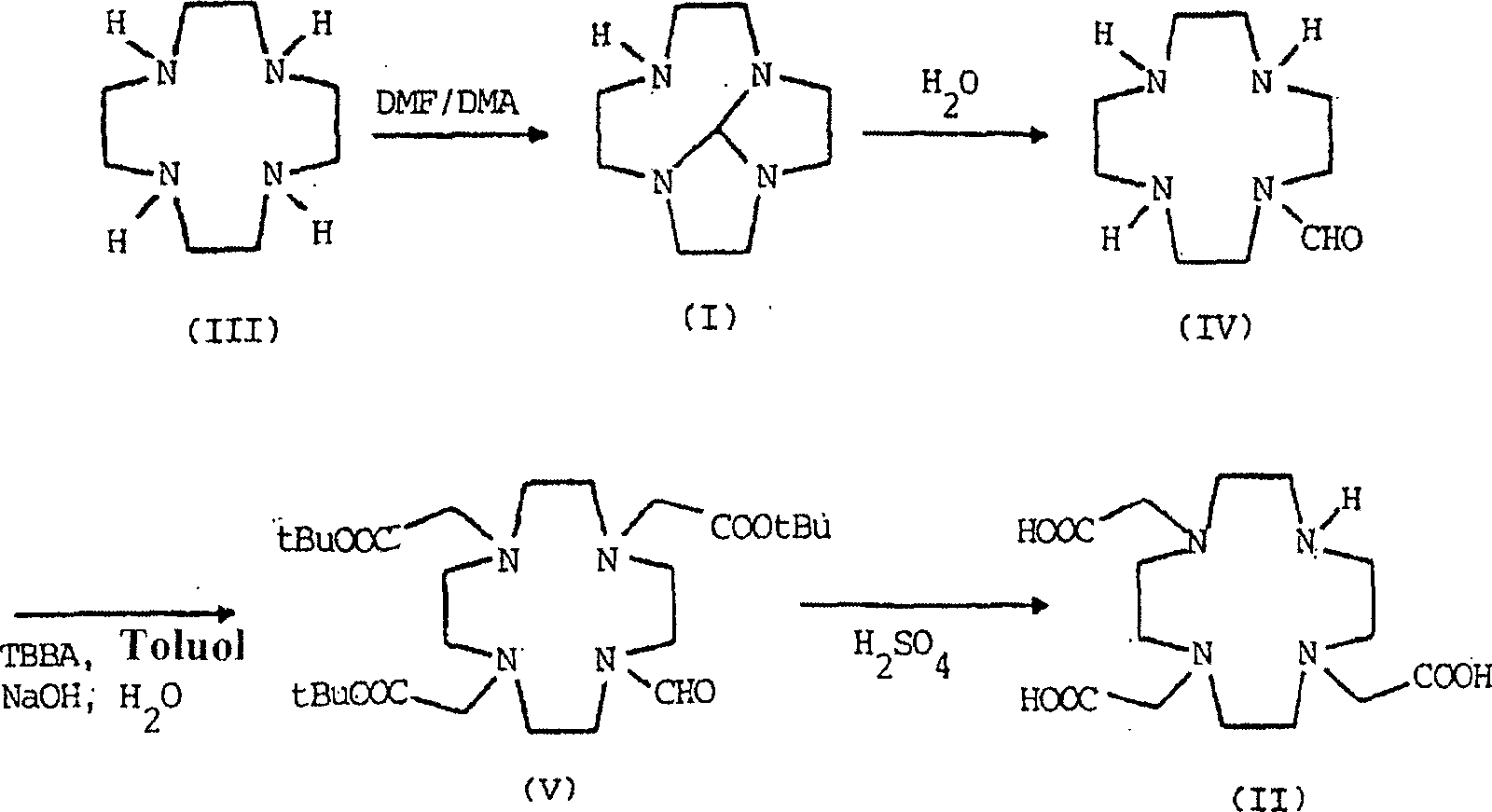
5H,9bH-2a,4a,7,9a-Octahydrotetraazacycloocta[cd]pentalen
(CAS RN 67705-42-4) der Formel (I), das unten beschrieben ist, ist
ein Intermediat für
die Herstellung von 1,4,7,10-Tetraazacyclododecan-Derivaten, worin
drei Stickstoffatome mit der gleichen funktionellen Gruppe, z.B.
einer Carboxymethylgruppe, substituiert sind, während das vierte Stickstoffatom
mit einer Gruppe, die sich von der vorherigen unterscheidet, substituiert
ist.
-
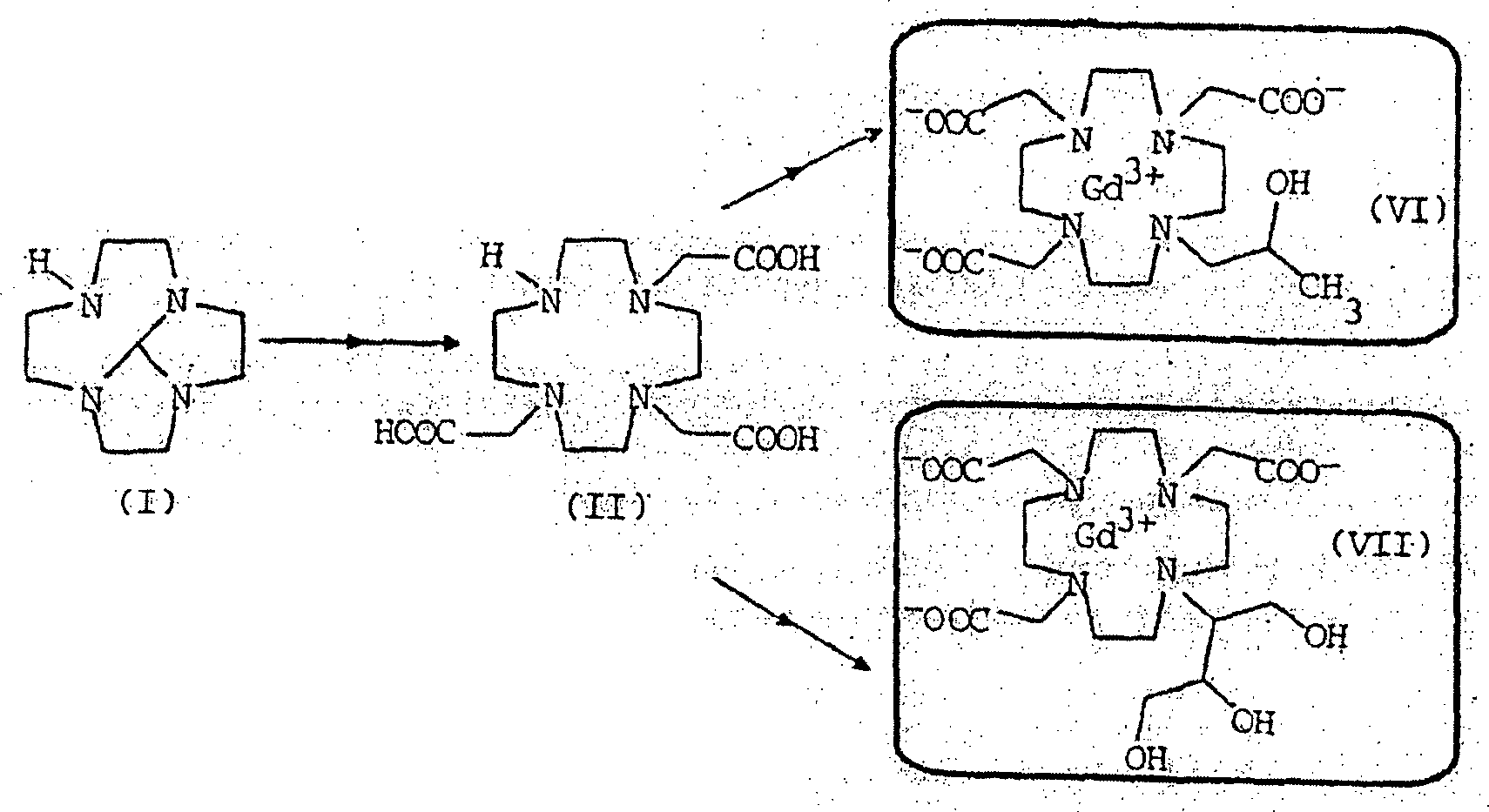
Von
besonderer Bedeutung ist z.B. die Synthese von 1,4,7,10-Tetraazacyclododecan-1,4,7-triessigsäure (allgemein
bekannt als DO3A) der Formel (II), die in verschiedenen Arbeiten
beschrieben wurde, zuerst in
EP
0 292 689 und in
EP
0 232 751 und anschließend
in dem Paper (Dischino et al., Inorg. Chem., 1991, 30, 1265), worin
die Synthese gemäß Schema
1 detailliert beschrieben ist. Schema
1
-
Der
Schritt von dem im Handel verfügbaren
Ausgangsprodukt 1,4,7,10-Tetraazacyclododecan (III) zu der Verbindung
der Formel (I) wird nach dem herkömmlichen Verfahren durchgeführt, das
in
US 4,085,106 beschrieben
ist, gefolgt von der Bildung von 1-Formyl-1,4,7,10-tetraazacyclododecan der Formel
(IV) in Wasser-Alkohol-Medium.
-
Dieses
Intermediat wird anschließend
mit tert-Butylbromacetat (TBBA) in Dimethylformamid tricarboxymethyliert,
dann mit einem Toluol-Natriumhydroxid-Zweiphasengemisch behandelt, um die
Verbindung der Formel (V), 10-Formyl-1,4,7,10-tetraazacyclododecan-1,4,7-triessigsäure-tris(1,1-dimethylethyl)ester
zu ergeben, der dann in saurer Lösung
zur Verbindung der Formel (II) hydrolysiert wird.
-
Makrocyclische
Derivate dieses Typs sind Intermediate für die Herstellung von Gadoliniumkomplexen, die
als Kontrastmittel für
magnetische Resonanz (MRI) verwendet werden können, z.B. Gadoteridol der
Formel (VI), ein Gadoliniumkomplex von 10-(2-Hydroxypropyl)-1,4,7,10-tetraazacyclododecan-1,4,7-triessigsäure, oder
Gadobutrol der Formel (VII), Gadoliniumkomplex von [10-[2,3-Dihydroxy-1-(hydroxymethyl)propyl]-1,4,7,10-tetraazacyclododecan-1,4,7-triessigsäure.
-
Die
Herstellung von Verbindung (I) mit hohem Reinheitsniveau unter einfach
reproduzierbaren Bedingungen ist eine essentielle Anforderung für die Herstellung
dieser wichtigen diagnostischen Mittel im industriellen Maßstab.
-
Verbindung
(I) und ihre Herstellung wurden zuerst in
US 4,130,715 oder in
US 4,085,106 beschrieben, ein derartiges
Verfahren wird auch in den anderen Referenzen verwendet, in denen
dieser Intermediattyp notwendig ist.
-
Die
beschriebenen Vorgehensweisen basieren auf der Verwendung von Dialkylformamid-dialkylacetalen:
beispielsweise offenbart J. Atkins (zitierte Patente) die Synthese
von Verbindung (I) in einem aromatischen Lösungsmittel (Benzol) durch
Umsetzung von 1,4,7,10-Tetraazacyclododecan und N,N-Dimethylformamid-dimethylacetal
(üblicherweise
in äquimolaren
Mengen) ohne Katalysator.
-
Aliphatische
und cycloaliphatische Kohlenwasserstoffe, chlorierte Kohlenwasserstoffe,
Dialkylether und Alkylnitrile können
als alternative Lösungsmittel
zu den aromatischen verwendet werden.
-
Atkins
selbst (J. Am. Chem. Soc. 1980, 102, 6364-6365) nennt auch die Möglichkeit
eines Arbeitens in Abwesenheit von Lösungsmitteln.
-
Obgleich
diese Bedingungen die Verbindung (I) in guten Ausbeuten liefern,
ist ihre Anwendung im industriellen Maßstab aufgrund der extremen
Reaktivität
von Dialkylformamid-dialkylacetal für solche Nucleophile wie Wasser
und die Verbindung (I) selbst schwierig.
-
Um
eine übermäßige Bildung
von Nebenprodukten zu vermeiden, die eine Abnahme in der Ausbeute und
eine Verschlechterung der Qualität
von Verbindung (I) involviert, ist es notwendig: a) unter wasserfreien Bedingungen
zu arbeiten und b) Dialkylformamid-dialkylacetal in Mengen zuzusetzen,
die zu 1,4,7,10-Tetraazacyclododecan äquimolar sind oder irgendwie
eine vollständige
Umwandlung des Letztgenannten bewirken.
-
Das
Vorliegen von Wasser in der Reaktion kann einerseits die Zerstörung von
Dialkylformamid-dialkylacetal und andererseits die Hydrolyse von
Verbindung (I) zu 1-Formyl-1,4,7,10-tetraazacyclododecan
involvieren, welches wiederum mit Dialkylformamid-dialkylacetal unter
Produktion weiterer Nebenprodukte reagieren kann.
-
Kommerzielles
1,4,7,10-Tetraazacyclododecan enthält üblicherweise Wasser in minimalen
prozentualen Gehalten, was allerdings ausreichend ist, um einen
nicht vernachlässigbaren
Teil des reaktiven Bestandteils oder der Verbindung (I) selbst zu
hydrolysieren: Für
die Reaktionsumgebung ist es daher notwendig, dass sie vor der Zugabe
von Dimethylformamid-dialkylacetal
trocken ist. Wenn das verwendete Lösungsmittel ein aromatisches
Lösungsmittel
ist, ist die Reaktionslösung
besonders schwierig zu trocknen: beispielsweise involviert die Destillation
des Wasser-Toluol-Azeotrops einen hohen Verbrauch des organischen
Lösungsmittels, und
sie erfordert lange Reaktionszeiten, was die Produktivität beeinträchtigt.
-
Andererseits
ist der Zusatz von Dialkylformamid-dialkylacetal kritisch, da ein Überschuss
desselben immerhin die schnelle Bildung von Nebenprodukten verursachen
kann, wohingegen ein Mangel an diesem bedeutet, dass noch restliches
1,4,7,10-Tetraazacyclododecan vorliegt und das Fortschreiten der
Synthese für die
Herstellung der oben genannten makrocyclischen Derivate schädigt. Die
Kontrolle der Reaktionsstöchiometrie
ist ziemlich kritisch und schwierig, wenn man auch berücksichtigt,
dass Dimethylformamid-dimethylacetal-Assay die Tendenz hat, mit
der Zeit abzunehmen. Im industriellen Maßstab können daher erfolgreiche Resultate
nur erhalten werden, wenn das Fortschreiten der Reaktion durch eine
Reihe von Verfahrenskontrollen untersucht wird: beispielsweise verschwindet
1,4,7,10-Tetraazacyclodecan tatsächlich
im Allgemeinen nur nach allmählichen
Zugaben von Dialkylformamid-dialkylacetal,
was durch gaschromatographische Kontrollen bestimmt wird.
-
Eine
weitere Komplikation, die aus der Verwendung eines Dialkylformamid-dialkylacetals
im industriellen Maßstab
resultiert, ist die, dass die Anlage mit einem geeigneten Gaswäscher ausgestattet
sein muss, wenn die oben genannte reaktive Komponente im Handel
verfügbar
ist, z.B. N,N-Dimethylformamid-dimethylacetal oder N,N-Dimethylformamid-diethylacetal. Die
mit diesen reaktiven Komponenten durchgeführte Reaktion bewirkt in der
Tat die Bildung von gasförmigem
Dimethylamin, das geeigneterweise zu entfernen ist, z.B. mit einem
Schwefelsäureabsorber.
-
Darüber hinaus
wird die Reaktion üblicherweise
in Gegenwart von eher hohen Mengen eines aromatischen Lösungsmittels
durchgeführt;
dadurch werden Produktivität
und Kosten bezüglich
Verkauf, Gewinnung und Entsorgung des verwendeten Lösungsmittels
beeinträchtigt:
Tatsache ist, dass die von Atkins beschriebene Massenreaktion eine
geringe Anwendbarkeit in industriellen Maßstab hat, da das erste Reagenz
hoch reaktiv und hoch toxisch ist und das zweite ein Feststoff ist,
was Probleme bezüglich
Arbeitsweise und thermischer Kontrolle verursacht.
-
Schließlich machen
die hohen Kosten für
Dimethylformamid-dimethylacetal das Verfahren weniger attraktiv.
-
Die
Hauptalternative zu Dialkylformamid-dialkylacetalen besteht in der
Verwendung von Trialkylorthoformiaten, die gemäß der Literatur (Weisman, Tetrahedron
Letters, 21, 3635-3638, 1980) eine niedrigere Reaktivität als die
oben genannten Dialkylformamid-dialkylacetale
haben, so dass die Reaktion trotz des Zusatzes eines sauren Katalysators
nicht beendet werden kann.
-
Die
niedrigen Ausbeuten, die von Weisman im Fall von Reaktionen, die
in aromatischen Lösungsmitteln
durchgeführt
wurden, beschrieben wurden, stützen
die Anwendbarkeit des Verfahrens im industriellen Maßstab nicht.
-
Andererseits
werden die Beispiele von Säure-katalysierten
Massenreaktionen zwischen Polyaminen und Triethylorthoformiat (Stetter,
Chem. Ber. 106, 2523-2529, 1973) auch durch Ausbeuten gekennzeichnet, die
für industrielle
Anwendungen in der Synthese der Verbindung (I) zu niedrig sind,
was extrem unökonomisch wäre.
-
Es
wurde nun überraschenderweise
gefunden, und dies ist der Gegenstand der vorliegenden Erfindung,
dass unter geeigneten Bedingungen 1,4,7,10-Tetraazacyclododecan
in hohen Ausbeuten in die Verbindung (I) umgewandelt werden kann,
indem genau Triethylorthoformiat verwendet wird, und zwar in Abwesenheit
eines Lösungsmittels
und in Gegenwart eines Säurekatalysators
bei hoher Temperatur.
-
Bedingungen,
die Sauerstoff und Licht aus der Reaktionsumgebung ausschließen, sind
weiter bevorzugt, wobei Sauerstoff z.B. ausgeschlossen wird, indem
die üblichen
Techniken mit Stickstoffdecke verwendet werden.
-
Triethylorthoformiat
kann in Mengen zugesetzt werden, die von 105% bis 200% des stöchiometrischen Werts
reichen.
-
Die
Reaktionstemperatur kann von 110 bis 150°C reichen, und die Reaktionszeit
kann von 5 bis 24 h reichen.
-
Der
Katalysator ist eine Carbonsäure,
die wenigstens 3 Kohlenstoffatome hat, C3-C18, vorzugsweise ausgewählt aus der Gruppe, bestehend
aus Propionsäure,
Buttersäure
und Pivalinsäure,
und sie wird in Mengen zugesetzt, die zwischen 4 und 42 g/kg Substrat
liegen.
-
Triethylorthoformiat
ist ein billigeres Produkt als N,N-Dimethylformamid-dimethylacetal,
produziert keine gefährlichen,
nicht-kondensierbaren gasförmigen
Nebenprodukte, sondern nur Ethanol, welches vorteilhaft für die Herstellung
von Triethylorthoformiat oder zu anderen Synthesezwecken gewonnen
werden kann.
-
Darüber hinaus
ist Triethylorthoformiat weniger reaktiv als N,N-Dimethylformamid-dimethylacetal, was es
möglich
macht, die Zusätze
der reaktiven Komponenten und die Reaktion selbst sogar in einem
großen Maßstab unter
völlig
sicheren Bedingungen durchzuführen;
das Fortschreiten der Reaktion lässt
sich auf der Basis solcher Arbeitsparameter wie Zeit und Temperatur
besser überwachen,
ohne dass das Fortschreiten durch Gaschromatographie untersucht
wird, und es macht den Zusatz des reaktiven Bestandteils dadurch
weniger kritisch, dass dieser von Anfang an zugesetzt werden kann,
ohne dass die Bildung von unerwünschten Nebenprodukten
verursacht wird: All das macht das Verfahren für die Herstellung von Verbindung
(I) unter leicht reproduzierbaren Bedingungen geeignet.
-
Wie
im Fall von N,N-Dimethylformamid-dimethylacetal, muss Wasser, das
im kommerziellen 1,4,7,10-Tetraazacyclododecan enthalten ist, entfernt
werden: Die Wasserentfer nung kann einfach durchgeführt werden,
entweder indem 1,4,7,10-Tetraazacyclododecan im Stickstoffstrom
geschmolzen wird oder indem ein geeignetes Lösungsmittel zugesetzt wird
und anschließend
eine Destillation des Lösungsmittels
zu einem Rest von trockenem geschmolzenem 1,4,7,10-Tetraazacyclododecan
bei einer Temperatur von über 110°C durchgeführt wird.
-
Ethylorthoformiat
und der Säurekatalysator
können
direkt ohne Probleme der thermischen Kontrolle oder der Sicherheit
zu diesem Rückstand
gegeben werden, da nämlich
Orthoformiat wenig reaktionsfähig
ist und die Reaktion nicht exotherm ist.
-
Das
Trocknungslösungsmittel
kann aus geradkettigen oder verzweigten (C4-C6)-Alkoholen
ausgewählt werden,
vorzugsweise aus der Gruppe, bestehend aus 1-Butanol, 2-Butanol,
Amylalkohol, Isoamylalkohol.
-
Die
Reaktion involviert eine Ethanolentwicklung: Eine erste Menge des
entwickelten Ethanols verbleibt in dem Reaktionsgemisch, bis es
eine solche Konzentration erreicht, dass der Dampfdruck des Reaktionsgemisches
den atmosphärischen
Wert erreicht: von diesem Punkt an destilliert das entwickelte Ethanol
zusammen mit einer kleinen Menge an Orthoformiat aus dem Reaktionsgemisch.
Um Verluste an Orthoformiat zu vermeiden, kann der entwickelte Dampf
leicht mit einer kleinen Rektifizierkolonne rektifiziert werden:
das Destillat aus dem Kopf der Kolonne ist im Wesentliches reines
Ethanol, wohingegen die Flüssigkeit
vom Boden, die mit Orthoformiat angereichert ist, zum Reaktor zurückgeführt wird.
-
Bei
vorher eingestellten Arbeitsbedingungen ist die Messung des Gewichts
oder des Volumens des entwickelten Ethanols ein zweckdienlicher,
genauer Index für
das Fortschreiten der Reaktion.
-
Wenn
die Reaktion beendet ist, kann die Verbindung (I) in Abhängigkeit
von den Synthesezwecken als solche eingesetzt werden oder sie kann
durch fraktionierte Destillation gereinigt werden. In beiden Fällen ist die
Ausbeute an Verbindung (I) äußerst hoch
(typischerweise 95 bis 98% für
die rohe Verbindung (I) und höher als
90% für
die gereinigte Verbindung (I)).
-
Ein
weiterer Gegenstand der vorliegenden Erfindung ist das Verfahren
zur Herstellung der Verbindung (II), 1,4,7,10-Tetraazacyclododecan-1,4,7-triessigsäure, das
die folgenden Schritte umfasst:
- a) Herstellung
von 5H,9bH-2a,4a,7,9a-Octahydrotetraazacycloocta[cd]pentalen, ausgehend
von 1,4,7,10-Tetraazacyclododecan nach dem Verfahren der Erfindung,
wobei die Verbindung, ohne dass sie isoliert wird, carboxymethyliert
und anschließend
hydrolysiert wird, und zwar nach bekannten Verfahren, um das gewünschte Produkt
zu erhalten.
-
Die
folgenden Beispiele erläutern
die besten experimentellen Bedingungen zur Durchführung des
Verfahrens der Erfindung.
-
Das
folgende gaschromatographische Verfahren wurde verwendet, um das
Fortschreiten der Reaktionen zu kontrollieren:
| Gerätschaft: | Gaschromatographieeinheit
Hewlett-Packard, Serie 5890 II Plus, ausgestattet mit einem Autosampler,
Serie 7673 und der Einheit HP-3365 |
| Säule: | 25
m Quarzkapillare, innerer Durchmesser 0,32 mm, stationäre Phase
CP Sil 19CB, Filmdicke 0,2 μm (Chrompack
Art. 7742) |
| Ofentemperaturprogramm: | zuerst
isoterm bei 120°C
für 5 min;
Temperaturanstieg 15°C/min;
abschließend
isotherm bei 260°C
für 2 min |
| Injiziertes
Volumen: | 1 μL |
| Detektor:
FID; Experimenteller Abschnitt | Temperatur
275°C |
-
Beispiel 1
-
Herstellung von Verbindung (I) durch Reaktion
zwischen 1,4,7,10-Tetraazacyclododecan und Triethylorthoformiat
in Gegenwart von Propionsäure
-
Ein
Gasreaktor, der mit einer Randompackungssäule, einem Destillationskopf
und einem Kühler
ausgestattet ist, mit einer Aluminiumfolie vor Licht geschützt ist,
wird mit 71,4 g (0,414 mol) 1,4,7,10-Tetraazacyclododecan und 71,4
g n-Butanol beschickt. Das Gemisch wird bis zur vollständigen Auflösung auf
80°C erhitzt und
die Lösung
wird durch Abdestillieren des n-Butanol-Wasser-Azeotrops (14,4 g)
bei reduziertem Druck getrocknet, dann wird das restliche n-Butanol
abdestilliert, bis die Bodentemperatur 120°C erreicht und der restliche
Druck 20 mbar erreicht. Nach Wiederherstellung des atmosphärischen
Drucks mit Stickstoff werden 73,5 g (0,498 mol) Triethylorthoformiat
und 0,6 g Propionsäure
zugegeben. Das Gemisch wird für
7 h bei 135°C erhitzt,
während
das entwickelte Ethanol kondensiert und getrennt gewonnen wurde.
Das überschüssige Triethylorthoformiat
wird bei reduziertem Druck abdestilliert, um 76,0 g der gewünschten
Verbindung zu erhalten (GC-Assay: 95% Fläche).
-
Die
Destillation bei reduziertem Druck (7 mbar) bei 128°C ergibt
68,8 g (0,377 mol) an gereinigtem 5H,9bH,2a,4a,7,9a-Octahydrotetraazacycloocta[cd]pentalen
(GC-Assay: 99% Fläche).
Gesamtausbeute:
91%
-
Die 1H-NMR-, 13C-NMR-,
IR- und MS-Spektren stimmen mit der angegebenen Struktur überein.
-
Beispiel 2
-
Herstellung der Verbindung (I) durch Reaktion
zwischen 1,4,7,10-Tetraazacyclododecan und Triethylorthoformiat
in Gegenwart von Pivalinsäure
-
Ein
Glasreaktor, ausgestattet mit einer Randompackungssäule, einem
Destillationskopf und einem Kühler,
mit Stickstoff unter einem Manometerdruck von 1 mbar bedeckt und
mit einer Aluminiumfolie vor Licht geschützt, wird mit 102,6 g (0,593
mol) 1,4,7,10-Tetraazacyclododecan, das 0,5 Gew.-% Wasser enthält, beladen,
dann wird die Verbindung bei 140°C
unter einem milden Stickstoffstrom geschmolzen. Weiße Kristalle, die
aus der sublimierten Substratform bestehen, bilden sich in der Säule. Nach
Abkühlen
auf 130°C
werden 123 g (0,829 mol) Triethylorthoformiat und danach 1 g Pivalinsäure zugesetzt.
Nach 5-stündigem Erwärmen bei
140°C, bis
eine Ethanolmenge von 90% der stöchiometrischen
Menge gewonnen ist, wird der Triethylorthoformiatüberschuss
unter Vakuum abdestilliert, wodurch 108 g der gewünschten
Verbindung als viskoses gelbes Öl
erhalten werden (GC-Assay:
96% Fläche).
Ausbeute:
96%
-
Die 1H-NMR-, 13C-NMR-,
IR- und MS-Spektren stimmen mit der angegebenen Struktur überein.
-
Beispiel 3
-
Wiederholung der Herstellung von Beispiel
2 in Gegenwart von atmosphärischem
Sauerstoff und Licht
-
Das
Verfahren des Beispiels 2 wird in einem Reaktor wiederholt, der
mit einer Decke aus trockener Luft versehen ist, ohne dass eine
Abschirmung mit einer Aluminiumfolie erfolgt. Es wird dieselbe Menge
an Produkt erhalten, allerdings ist es dunkel und hat einen deutlich
niederen GC-Assay (89%).
-
Beispiel 4
-
Herstellung von Verbindung (I) durch Reaktion
zwischen 1,4,7,10-Tetraazacyclododecan und Triethylorthoformiat
in Gegenwart von Propionsäure
-
Ein
Glasreaktor, ausgestattet mit einer Randompackungssäule, einem
Destillationskopf und einem Kühler
und mit einer Aluminiumfolie vor Licht geschützt, wird mit 110 g (0,634
mol) 1,4,7,10-Tetraazacyclododecan, das 0,7 Gew.-% Wasser enthält, beschickt,
die Verbindung wird bei 144°C
unter einem milden Stickstoffstrom geschmolzen. Nach Abkühlung auf
115°C werden
113 g (0,761 mol) Triethylorthoformiat und 1,65 g Propionsäure zugesetzt.
Das Gemisch wird für
20 h bei 115°C
umgesetzt, während
Ethanol abdestilliert wird. Schließlich wird der Triethylorthoformiatüberschuss
unter Vakuum abdestilliert, wodurch 115 g des gewünschten
Produkts erhalten werden (GC-Assay: 95% Fläche).
Ausbeute: 94%
-
Die 1H-NMR-, 13C-NMR-,
IR- und MS-Spektren sind mit der angegebenen Struktur konsistent.
-
Beispiel 5
-
Herstellung von Verbindung (I) und sofortige
Umwandlung in 1,4,7,10-Tetraazacyclododecan-1,4,7-triessigsäure (II)
-
(A) Herstellung einer wässrigen
Lösung
der Verbindung (II) als Trinatriumsalz
-
110
g (0,634 mol) 1,4,7,10-Tetraazacyclododecan, das 0,7 Gew.-% Wasser
enthält,
werden in 110 g Amylalkohol gelöst.
Das Wasser-Amylalkohol-Azeotrop und der Amylalkoholüberschuss
werden nacheinander unter Vakuum abdestilliert, dann werden 113
g (0,761 mol) Triethylorthoformiat und 1,2 g Propionsäure in Stickstoffatmosphäre zugegeben.
Das Gemisch wird für
8 h bei 135°C
erhitzt, während
der gebildete Ethanol abdestilliert wird, dann wird das Reaktionsgemisch
auf 35°C
abgekühlt,
um die rohe Verbindung (I) als flüssiges Öl zu erhalten, das zu einer
Lösung
gegeben wird, die durch Lösen
von 274 g (1,972 mol) Bromessigsäure und
263 g 30 Gew.-% NaOH in 370 g Wasser hergestellt wird, gegeben wird.
Während
der Zugabe der rohen Verbindung (I) wird der pH durch Zusatz von
NaOH bei 10 gehalten; am Ende der Zugabe wird der pH erneut durch
Zugabe von 30 Gew.-%igem NaOH, auf 11,3 eingestellt, und das Gemisch
wird für
24 h bei 30°C
umgesetzt.
-
360
g 30 Gew.-%iges NaOH werden dann zugegeben und die Lösung wird
für 9 h
auf 75°C
erwärmt. Eine
wässrige
Lösung,
die 204 g (0,589 mol) 1,4,7,10-Tetraazacyclododecan-1,4,7-triessigsäure (Gehalt
bestimmt durch HPLC) als Trinatriumsalz enthält, wird erhalten.
Ausbeute:
93%
-
B) Gewinnung von Verbindung (II) als Sulfat
-
Die
Lösung
von Schritt A) wird mit 192 g 40%iger H2SO4 angesäuert
und mit Aceton versetzt, um 70,2 g der gewünschten Verbindung (0,158 mol)
zu präzipitieren.
Ausbeute:
81%
-
Die 1H-NMR-, 13C-NMR-,
IR- und MS-Spektren sind mit der angegebenen Struktur konsistent.
-
C) Freie Säure aus dem in Schritt B) erhaltenen
Salz
-
Das
in Schritt B) erhaltene Salz wird auf ein PVP-Harz aufgebracht (entsprechend
dem Verfahren, beschrieben in Dischino et al., Inorg. Chem., 1991,
30, 1265).
-
Es
werden 49,25 g der Verbindung (II) (0,142 mol) erhalten.
Ausbeute:
90%
-
Die 1H-NMR-, 13C-NMR-,
IR- und MS-Spektren sind mit der angegebenen Struktur konsistent.
-
Beispiel 6
-
Herstellung
von Verbindung (I) und sofortige Umwandlung in 1,4,7,10-Tetraazacyclododecan-1,4,7-triessigsäure (II),
einsetzbar für
die Synthese von Gadoteridol
-
A) Herstellung von Verbindung (I)
-
23,8
kg (0,138 kmol) 1,4,7,10-Tetraazacyclododecan, das 0,7 Gew.-% Wasser
enthält,
werden in 23,8 kg Amylalkohol gelöst. Das Wasser-Amylalkohol-Azeotrop
und der Amylalkoholüberschuss
werden bei reduziertem Druck nacheinander abdestilliert, dann werden
24,5 kg (0,166 kmol) Triethylorthoformiat und 355 g Propionsäure in Stickstoffatmosphäre zugegeben.
Das Gemisch wird für
11 h auf 125°C
erwärmt,
während
das gebildete Ethanol abdestilliert wird, dann wird das Reaktionsgemisch
auf 35°C
gekühlt,
um Verbindung (I) als flüssiges Öl zu erhalten.
-
B) Herstellung von 10-Formyl-1,4,7,10-tetraazacyclododecan-1,4,7-triessigsäure-Natriumsalz
-
Verbindung
(I), erhalten in Schritt A), wird zu einer Lösung gegeben, die durch Lösen von
81,5 kg (0,469 kmol) Bromessigsäure
und etwa 62,6 kg 30 Gew.-%iges NaOH in 100 kg Wasser zu pH 5 gelöst wurde. Während der
Zugabe der rohen Verbindung (I) wird der pH durch Zugabe von NaOH
bei 11 gehalten; am Ende der Zugabe wird der pH wiederum durch Zusatz
von 30 Gew.-%igem NaOH auf pH 11,1 eingestellt; das Gemisch wird
für 24
h bei 35°C
umgesetzt.
-
C) Herstellung von 1,4,7,10-Tetraazacyclododecan-1,4,7-triessigsäure-Natriumsalz
-
Das
Gemisch von Schritt B) wird dann mit 77,3 kg 30 Gew.-%igem NaOH
versetzt und für
9 h bei 70°C erwärmt. Die
resultierende wässrige
Lösung
enthält
0,131 kmol der gewünschten
Verbindung (Gehalt bestimmt durch HPLC) als Trinatriumsalz.
-
D) Synthese von Gadoteridol
-
Der
pH wird mit konz. HCl auf 12,3 eingestellt, 15,2 kg (0,262 kmol)
Propylenoxid werden zugesetzt und das Gemisch wird für 4 h bei
40°C umgesetzt.
Danach wird die Lösung
auf 50°C
erwärmt
und mit 120 kg einer wässrigen
Lösung,
die 0,135 kmol Gadoliniumtrichlorid enthält, versetzt. Nach 1 h wird
das Gemisch auf 17°C
abgekühlt
und mit konz. HCl auf pH 1,7 angesäuert; der pH wird für 2 h bei
diesem Wert kontrolliert. Danach wird die Lösung auf 50°C erwärmt, der pH wird mit Natriumhydroxid
auf 7 eingestellt, und die Lösung
wird für
1 h bei diesen Bedingungen gehalten.
-
E) Vorreinigung der rohen Gadoteridol-Lösung
-
Die
rohe Gadoteridol-Lösung
aus dem vorangehenden Schritt wird gekühlt und durch ein "In-Line"-Filter und eine
Säule,
die mit 150 L R&H
Amberlite XAD 1600-Harz gepackt ist, zu einer Nanofiltrationseinheit transferiert,
die mit Desal DK4040F-Elementen versehen ist. Wenn der Reaktor leer
ist, werden der Reaktor, der "In-Line"-Filter und die Säule 3-mal
mit 300 L entionisiertem Wasser gewaschen. Die resultierende Waschlösung wird
mit der Produktlösung
in der Nanofiltrationseinheit kombiniert, wo das Produkt bei 32
bar und 25°C konzentriert
und teilweise entsalzt wird.
-
250
L Lösung
von rohem Gadoteridol mit einer Leitfähigkeit von 2,9 mS/cm werden
schließlich
erhalten.
-
F) Endentsalzen
-
Die
Gadoteridol-Lösung
wird dann mit 200 L/h zu einer Reihe von 4 Ionenaustauscherbetten
geleitet, wobei das erste (C1) aus 120 L stark basischem Anionenaustauscher
Relite 3ASfb in Hydrogencarbonatform besteht, das zweite (C2) aus
100 L eines schwach sauren Kationenaustauschers Relite CC in der
H+-Form besteht, das dritte (C3) aus 20
L Relite 3ASfb in der OH--Form besteht und
das vierte (C4) aus 20 L Relite CC-Harz in der H+-Form
besteht. Alle Säulen
werden in die Atmosphäre
entlüftet
und die Flüssigkeit
aus der zweiten Säule
wird durch einen Gasabtrennungstank, der an eine Vakuumpumpe angeschlossen
ist, geführt, um
das entwickelte CO2 aus der Lösung zu
entfernen. Der Auslass aus der vierten Säule ist mit einem Dichtewandler
ausgestattet, um das Produkt im Eluat zu detektieren. Die ersten
180 L Eluat werden verworfen; das Eluat wird dann in einer produktreichen
Fraktion gesammelt. Wenn die gesamte Lösung von rohem Gadoteridol
auf die Ionenaustauschereinheit aufgebracht worden ist, wird das
Produkt mit 600 L entionisiertem Wasser eluiert, das Eluat wird
dann mit der produktreichen Fraktion kombiniert, die farblos ist
und im Wesentlichen frei von ionischen Verunreinigungen ist (Leitfähigkeit
2,2 μS/cm).
-
Die
Ausbeute der Endentsalzung, bestimmt durch HPLC, ist 98%.
-
G) Isolierung des Produkts (Gadoteridol)
-
Die
produktreiche Fraktion wird dann thermisch zu einem viskosen Rückstand
konzentriert, der mit 350 kg Isopropanol mit 79°C versetzt wird.
-
Die
resultierende Suspension wird für
1 h unter Rückfluss
erhitzt, dann abgekühlt,
zentrifugiert und bei reduziertem Druck getrocknet, wodurch 68,2
kg Gadoteridol, das 10 Hydratwasser enthält, (0,111 kmol) erhalten wird,
HPLC-Assay: 98,5% (s.a.)
Gesamtausbeute: 80,7%
-
Die 1H-NMR-, IR- und MS-Spektren sind mit der
angegebenen Struktur konsistent.