CN1293664C - 电化学元件的制造方法 - Google Patents
电化学元件的制造方法 Download PDFInfo
- Publication number
- CN1293664C CN1293664C CNB028045327A CN02804532A CN1293664C CN 1293664 C CN1293664 C CN 1293664C CN B028045327 A CNB028045327 A CN B028045327A CN 02804532 A CN02804532 A CN 02804532A CN 1293664 C CN1293664 C CN 1293664C
- Authority
- CN
- China
- Prior art keywords
- electrode active
- active material
- material layer
- layer
- solid electrolyte
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images





Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M6/00—Primary cells; Manufacture thereof
- H01M6/14—Cells with non-aqueous electrolyte
- H01M6/18—Cells with non-aqueous electrolyte with solid electrolyte
- H01M6/188—Processes of manufacture
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C14/00—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material
- C23C14/0021—Reactive sputtering or evaporation
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C14/00—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material
- C23C14/22—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material characterised by the process of coating
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C14/00—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material
- C23C14/22—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material characterised by the process of coating
- C23C14/24—Vacuum evaporation
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C14/00—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material
- C23C14/22—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material characterised by the process of coating
- C23C14/24—Vacuum evaporation
- C23C14/28—Vacuum evaporation by wave energy or particle radiation
- C23C14/30—Vacuum evaporation by wave energy or particle radiation by electron bombardment
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C14/00—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material
- C23C14/22—Coating by vacuum evaporation, by sputtering or by ion implantation of the coating forming material characterised by the process of coating
- C23C14/24—Vacuum evaporation
- C23C14/32—Vacuum evaporation by explosion; by evaporation and subsequent ionisation of the vapours, e.g. ion-plating
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0561—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of inorganic materials only
- H01M10/0562—Solid materials
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/058—Construction or manufacture
- H01M10/0585—Construction or manufacture of accumulators having only flat construction elements, i.e. flat positive electrodes, flat negative electrodes and flat separators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/04—Processes of manufacture in general
- H01M4/0402—Methods of deposition of the material
- H01M4/0421—Methods of deposition of the material involving vapour deposition
- H01M4/0423—Physical vapour deposition
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/139—Processes of manufacture
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/485—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of mixed oxides or hydroxides for inserting or intercalating light metals, e.g. LiTi2O4 or LiTi2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P70/00—Climate change mitigation technologies in the production process for final industrial or consumer products
- Y02P70/50—Manufacturing or production processes characterised by the final manufactured product
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T29/00—Metal working
- Y10T29/49—Method of mechanical manufacture
- Y10T29/49002—Electrical device making
- Y10T29/49108—Electric battery cell making
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T29/00—Metal working
- Y10T29/49—Method of mechanical manufacture
- Y10T29/49002—Electrical device making
- Y10T29/49108—Electric battery cell making
- Y10T29/49112—Electric battery cell making including laminating of indefinite length material
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T29/00—Metal working
- Y10T29/49—Method of mechanical manufacture
- Y10T29/49002—Electrical device making
- Y10T29/49108—Electric battery cell making
- Y10T29/49115—Electric battery cell making including coating or impregnating
Abstract
本发明涉及能够提供无损于材料特性的层结构的电化学元件,包含叠层的第1集电体、第1电极活性物质层、固体电解质层、第2电极活性物质层及第2集电体的电化学元件的制造方法。所述方法通过一边根据第1电极活性物质层、第2电极活性物质层或固体电解质层的组成,照射具有预定能量的电子或电磁波,一边向上述基板上供给构成第1电极活性物质层、第2电极活性物质层或固体电解质层的原子、离子或群集,以此形成第1电极活性物质层、第2电极活性物质层或固体电解质层。
Description
技术领域
本发明涉及高容量、薄型的电化学元件的制造方法。
背景技术
介绍通过导入半导体薄膜工艺技术,可利用锂聚化物电池实现薄型化的全固体电池(厚度25μm)的美国专利第5338625号。其中,具有用薄膜工艺获得薄型化的构成要素的全固体电池,通过使各构成要素连续叠层,可望获得具有已有的电池能量密度的数倍的能量密度,因而备受关注。
但是,薄型化的全固体电池中,在充放电时,必须是电极活性物质能高密度且高速地激活锂,同时固体电解质对于锂离子显示高离子传导性。
作为电极活性物质使用的材料,可举出LiCoO2、LiMn2O4、LiNiO2、V2O5、MoO2及TiS2等的结晶态物质。
另外,固体电解质包括结晶态物质及非晶态物质。结晶态的固体电解质之一的Li3PO4-Li4SiO4具有10-6~10-5Scm-1的离子传导率,耐热性优异,但其离子传导性有各向异性,所以存在难以用于电池的问题。与此相反,非晶态物质的固体电解质,基本上耐热性不高,但在离子传导性方面没有各向异性。特别是以下的硫磺系固体电解质,与氧系固体电解质相比,显示出高离子传导性。作为非晶态物质的氧系固体电解质,有例如,Li2.9PO3.3N0.36及非晶态物质Li3PO4-Li4SiO4等,它们具有10-5~10-4Scm-1的离子传导率。而作为非晶态物质的硫磺系固体电解质,有例如Li2S-SiS2、Li2S-P2S5、Li2S-B2S3及在这些化合物中添加LiI等卤化锂或Li3PO4等锂的含氧酸盐等得到的固体电解质等。它们具有10-4~10-3Scm-1的更高的离子传导率。
在制作电极活性物质的结晶膜时,为使构成原子再排列形成晶体点阵,一般采用对基板整体加热的方法。但是,如前所述,具有高离子传导率的固体电解质的耐热温度低,电极活性物质层的结晶化用的加热工序中,固体电解质层被同时加热,固体电解质层的温度超过固有的温度结晶化,其离子传导率下降。因此,在固体电解质层已经形成的基板上形成电极活性物质层时,必须不提高基板的温度,且基板上设置的固体电解质层的温度也不提高地形成电极活性物质层,从基板以外的部分供给电极活性物质层的构成原子的再排列能量。
作为与基板的加热不同的特殊的能量供给方法,以下的方法展示于日本特开平8-287901号公报。提出了这样的方法,即用蒸镀法制作含锂的结晶性的电极活性物质层时,为了提高电极活性物质层的结晶性及与基板的紧密结合性能,提出作为原子的再排列能量应在基板上照射具有100eV以上的能量的离子束,与离子束同时应在基板上照射氧,以及在基板上照射高频等离子体或紫外线等的电磁波。
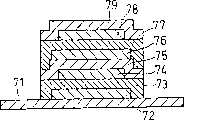
这里将全固体电池的基本构成单位展示于图3。在兼作基板的第1集电体51上,一面用金属掩膜等干式形成图案,一面依次叠层第1电极活性物质层52、固体电解质层53、第2电极活性物质层54及第2集电体55,以此形成具有如图3所示的构造的全固体电池。这里,成问题的是形成第1电极活性物质层52、固体电解质层53及第2电极活性物质层54的工序。此外,本发明对形成这些层的工序有效。
对图3的构成改进的构成示于图4及图5。图4表示具有2组并联构造的全固体电池,图5表示具有2组串联构造的全固体电池。此外,本发明对多组的全固体电解质电池、以及并联及串联组合的全固体电池等也是有效的。
现在,作为锂离子电池材料使用的主要的电极活性物质的LiCoO2、LiNiO2、LiMn2O4及LiV2O5等,需要用于结晶形成的比较高的温度,大多数固体电解质的结晶化通常都是在300~350℃附近发生的。反之,在采用锂等融点极低的电极活性物质与结晶质固体电解质的组合时,在形成固体电解质层之际,电极活性物质也熔融。在制作具有如图3~5所示构造的电池的电池工序中,需要解决各种材料的容许温度产生的矛盾。
要解决面临的问题,向来使用已有的离子照射装置。作为离子照射装置,可列举诸如加速电压为上百伏特~几十kV的离子溅射装置,以及电压更高的离子植入(plantation)装置等。但是,采用这些装置,例如按照日本特开平8-287901号公报记载的已有技术,用100eV以上的高能离子照射时,会发生以下问题。
问题1是,离子的传导率随固体电解质层的温度上升而降低。作为形成膜及层的基材的部分的表面部分的温度,因离子碰撞而急剧上升。例如,被制膜表面(基材表面)是固体电解质层时,会发生固体电解质层的表面部分的离子传导率受损,使最终得到的电池的性能下降的问题。这是由于离子具有接近原子的质量,离子撞击的原子的温度及该原子周边的温度上升,使固体电解质层发生部分结晶化。离子传导率的下降,是限制电池电流的极重要的指标,如上述的工序中的离子传导率的下降是不能忽视的,
问题2是,离子照射得到的结晶性的等级(grade)常常是有限的。结晶性的等级有限的原因主要是由于离子对电极活性物质的碰撞,在加上结晶化需要的能量的同时,结晶化了的电极活性物质层也受到离子碰撞,使晶体点阵被搞乱。特别是,电极活性物质层的晶体点阵,是锂离子插入的部分,且是锂离子扩散的通路,所以,该晶体点阵如果被搞乱,会引起电池的容量及充放电电流的降低。
此外,一般的真空制膜工艺中,氧化物膜或氮化物膜形成时,氧或氮等蒸气压高的气体的比例出现从氛围中减少的倾向。这主要是由于蒸气压差使这些气体飞散,被真空泵向外部排放所致。这种现象也会使电极活性物质层的晶体点阵搞乱,电池的容量及充放电电流下降。为了解决这样的问题,以往用氧离子或氮离子等离子进行照射。众所周知,这种方法对抑制原子缺损是有效的,但问题是,这里使用的离子限于低能离子,离子电流有上限值,因此制膜速度显然很低,只有数/分以下。
因此,本发明的目的是提供能解决上述电池制造工艺上的问题,具有无损于上述种种材料的特性的层构造的电化学元件及其制造方法。
发明内容
本发明提供一种电化学元件的制造方法,包含:
在第1集电体上形成第1电极活性物质层的第1工序;
在第1电极活性物质层上形成固体电解质层的第2工序;
在固体电解质层上形成第2电极活性物质层的第3工序;
在第2电极活性物质层上形成第2集电体的第4工序,
其中,所述第3工序是在基板上,利用使构成所述第2电极活性物质层的原子、离子或群集向所述固体电解质层的表面蒸镀的制膜工序形成所述第2电极活性物质层,
在所述制膜工序中,对构成所述第2电极活性物质层的原子、离子或群集照射具有满足以下关系式的强度E的能量,其单位为eV:
Ea≤E≤(70±α)×(ρ1d1)1/2,
其中,-7≤α≤7;Ea是第2电极活性物质层的活性化能量,单位为eV;ρ1是第2电极活性物质层的密度,单位为g/cm3;d1是第2电极活性物质层的晶格常数,单位为。
本发明还提供一种电化学元件的制造方法,包含:
在第1集电体上形成第1电极活性物质层的第1工序;
在第1电极活性物质层上形成固体电解质层的第2工序;
在固体电解质层上形成第2电极活性物质层的第3工序;
在第2电极活性物质层上形成第2集电体的第4工序,
在第2集电体上形成第3电极活性物质层的第5工序;
在第3电极活性物质层上形成固体电解质层的第6工序;
在固体电解质层上形成第4电极活性物质层的第7工序;
在第4电极活性物质层上形成第3集电体的第8工序,
其中,所述第7工序是在基板上,利用使构成所述第4电极活性物质层的原子、离子或群集向所述固体电解质层的表面蒸镀的制膜工序,形成所述第4电极活性物质层,
在所述制膜工序中,对构成所述第4电极活性物质层的原子、离子或群集照射具有满足以下关系式的强度E的能量,其单位为eV:
Ea≤E≤(70±α)×(ρ1d1)1/2,
其中,-7≤α≤7;Ea是第4电极活性物质层的活性化能量,单位为eV;ρ1是第4电极活性物质层的密度,单位为g/cm3;d1是第4电极活性物质层的晶格常数,单位为。
本发明又提供一种电化学元件的制造方法,包含:
在第1集电体上形成第1电极活性物质层的第1工序;
在第1电极活性物质层上形成固体电解质层的第2工序;
在固体电解质层上形成第2电极活性物质层的第3工序;
在第2电极活性物质层上形成第2集电体的第4工序,
其中,所述第2工序是在基板上,利用将构成所述固体电解质层的原子、离子或群集向所述第1电极活性物质层的表面蒸镀的制膜工序,形成所述固体电解质层,
在所述制膜工序中,对构成所述固体电解质层的原子、离子或群集照射具有满足以下关系式的强度E的能量,其单位为eV:
Ea≤E≤(70±α)×(ρ3d3)1/2,
其中,-7≤α≤7;Ea是固体电解质层的活性化能量,单位为eV;ρ3是第2电极活性物质层的密度,单位为g/cm3;d3是第2电极活性物质层的晶格常数,单位为。
附图说明
图1是本发明的实施例中使用的膜形成装置的概略图。
图2是本发明的实施例中使用的另一膜形成装置的概略图。
图3是本发明的实施例中制作的电池的构造的纵剖面图。
图4是本发明的实施例中制作的电池的构造的纵剖面图。
图5是本发明的实施例中制作的电池的构造的纵剖面图。
图6是用来说明电池构成要素的成图时使用的掩膜的形状及设置位置用的图。
图7是用来说明电池构成要素形成图案时使用的另一掩膜的形状及设置位置用的图。
图8是用来说明电池构成要素形成图案时使用的又一掩膜的形状及设置位置用的图。
图9是用来说明电池构成要素形成图案时使用的又一掩膜的形状及设置位置用的图。
图10表示本发明的实施例的第1电池的特性。
图11表示本发明的实施例的第1电池的特性。
图12表示本发明的实施例的第1电池的特性。
图13表示本发明的实施例的第1电池的特性。
图14表示LiCoO2的结晶化模型。
具体实施形态
本发明是包含叠层的第1集电体、第1电极活性物质层、固体电解质层、第2电极活性物质层及第2集电体的电化学元件的制造方法,其特征在于,具备
在基板上,一边根据上述第1电极活性物质层、上述第2电极活性物质层或上述固体电解质层的组成用预定的能量进行照射,一边使构成上述第1电极活性物质层、上述第2电极活性物质层或上述固体电解质层的原子、离子或群集蒸镀,从而形成上述第1电极活性物质层、上述第2电极活性物质层或上述固体电解质层的制膜工序。
上述制膜工序中形成的层的结晶温度,高于该形成的层以外的层的结晶温度或融点时特别有效。
又,上述能量的载体,最好是电子或电磁波。
又,最好是将上述制膜工序中形成的层以外的层的温度维持在根据其组成预定的范围。
此外,最好是调整上述能量的强度及上述基板的散热,使上述基板的温度不超过上述制膜工序中形成的层以外的层的结晶温度或融点。
构成在上述制膜工序中形成的层的化合物是氧化物或氮化物时,最好是蒸镀氛围采用分别包含氧或氧离子的气体氛围、或包含氮或氮离子的惰性气体氛围。
上述那样的本发明的制造方法,主要可分成以下几种情况:即形成在低温下能结晶的非晶态固体电解质层与高温下能结晶的第1电极活性物质层及/或第2电极活性物质层的情况;形成低温下能结晶的第2电极活性物质层与高温下能结晶的第1电极活性物质层的情况;以及形成低温下能结晶的第2电极活性物质层与高温下能结晶的结晶态固体电解质层的情况。此外,理想的条件也相应于这些情况而不同。以下对本发明的电化学元件的制造方法的形态进行说明。
(1)第1制造方法
本发明的第1制造方法,是将第1集电体、第1电极活性物质层、固体电解质层、第2电极活性物质层及第2集电体加以叠层的电化学元件的制造方法,其特征在于,
在基板上,利用使构成上述第1电极活性物质层或上述第2电极活性物质层的原子、离子或群集向上述固体电解质层的表面蒸镀的制膜工序,形成上述第1电极活性物质层或上述第2电极活性物质层,
在上述制膜工序中,对构成上述第1电极活性物质层或上述第2电极活性物质层的原子、离子或群集,根据上述第1电极活性物质层或第2电极活性物质层的组成以具有预定强度的能量进行照射,且将上述固体电解质层的温度维持在根据上述固体电解质层的组成预定的温度范围。
对构成上述电极活性物质层的原子、离子或群集照射的能量的载体,最好是电子或电磁波。
上述第1电极活性物质层及上述第2电极活性物质层的至少一方的结晶温度,最好高于上述固体电解质的结晶温度。
又,在形成上述第1电极活性物质层或第2电极活性物质层时,照射的能量的强度E(eV)最好是满足下述关系式(1),
Ea≤E≤(70±α)×(ρ1d1)1/2 ……(1)
式中,-7≤α≤7,Ea是电极活性物质层的活性化能量(eV),ρ1是电极活性物质层的密度(g/cm3),d1是电极活性物质层的晶格常数()。
这里,以一定的能量进行照射或碰撞的能量,不是1次照射或碰撞就使运动完全制止的能量,因此,实际上具有某种程度的宽度。这样的能量分布,大多以高斯分布的几率密度函数来表达,其宽度以中心值的e-2≈0.1的2个值代表。因此,在本发明中,取中心值为70,宽度取7(-7≤α≤7)。
这里,所谓电极活性物质层的活性化能量,是构成电极活性物质层的元素用于形成该电极活性物质层的组成所需要的最低限度的能量。结晶化,严格考虑应在结晶温度以上的状态下进行。但是,从生成速度的观点出发,通过提供暂时作为不完全的物质生成的电极活性物质再度返回到构成元素的气体状态所需要的能量,可以使结晶化顺利进行。
这里,例如,钴酸锂(LiCoO2)的情况下的结晶化开始温度最低为约400℃,所以,在这种情况下的能量可按照如下方法计算。图14表示LiCoO2的结晶化模型。图14表示将LiCoO2整体加热的情况下的固相扩散引起的再排列。
将摩尔比热C=58.8J/mol/K的钴酸锂从常温加热到400℃时需要的能量E(eV)为
E=ΔTC/eNA=0.41(eV)
这里,NA是阿伏加德罗数,e是原电荷。
但是,该能量由于结晶的生成速度低并不实用。因此,不完全生成的物质如果是例如Li2O(s)及CoO(s),最好照射
这一再分解反应中需要的9eV以上的能量。
此外,在形成上述第1电极活性物质层或上述第2电极活性物质层时,最好对照射的能量的功率及上述基板的散热功率进行调整,使上述基板的温度不超过上述固体电解质层的结晶温度。
构成上述第1电极活性物质层或第2电极活性物质层的电极活性物质是氧化物或氮化物时,最好是蒸镀氛围分别采用包含氧或氧离子的气体氛围、或包含氮或氮离子的惰性气体氛围。
(2)第2制造方法
本发明的第2制造方法,是将第1集电体、第1电极活性物质层、固体电解质层、第2电极活性物质层及第2集电体叠层的电化学元件的制造方法,其特征在于,
在基板上,利用将构成上述第1电极活性物质层的原子、离子或群集蒸镀于上述第1集电体的表面的制膜工序,形成上述第1电极活性物质层,
在上述制膜工序,对构成上述第1电极活性物质层的原子、离子或群集,照射具有根据上述第1电极活性物质层的组成预先决定的强度的能量,且将上述基板的温度维持在根据上述第2电极活性物质层的组成预先决定的温度范围。
对构成上述第1电极活性物质层的原子、离子或群集照射的能量的载体,最好是电子或电磁波。
上述第1电极活性物质层的结晶温度,最好高于第2电极活性物质层的融点。
此外,在形成上述第1电极活性物质层时,照射能量的强度E(eV)最好满足关系式(2)
Ea≤E≤(70±α)×(ρ2T2)1/2 ……(2)
式中,-7≤α≤7,Ea是第1电极活性物质层的活性化能量(eV),ρ2是第2电极活性物质层上面的层(固体电解质层或集电体层)的密度(g/cm3),T2是第2电极活性物质层上面的层(固体电解质层或集电体层)的厚度())。
又,在形成上述第1电极活性物质层时,最好是对照射的能量的功率及上述基板的散热功率进行调整,使上述基板的温度不超过上述第2电极活性物质层的融点。
在构成上述第1电极活性物质层的电极活性物质是氧化物或氮化物时,最好是蒸镀氛围分别采用包含氧或氧离子的气体氛围、或包含氮或氮离子的惰性气体氛围。
(3)第3制造方法
本发明的第3制造方法,是将第1集电体、第1电极活性物质层、固体电解质层、第2电极活性物质层及第2集电体加以叠层的电化学元件的制造方法,其特征在于,
在基板上,利用将构成上述固体电解质层的原子、离子或群集向上述第2集电体的表面蒸镀的制膜工序,形成上述固体电解质层,
在上述制膜工序中,对构成上述固体电解质层的原子、离子或群集,照射具有根据上述固体电解质层的组成预先决定的强度的能量,且将上述第2电极活性物质层的温度维持在根据上述第2电极活性物质层的组成预先决定的温度范围。
对构成上述固体电解质层的原子、离子或群集照射的能量的载体,最好是电子或电磁波。
上述固体电解质层的结晶温度最好高于第2电极活性物质层的融点。
此外,在形成上述固体电解质层时,对上述固体电解质层照射的能量的强度E(eV)最好满足关系式(3),
Ea≤E≤(70±α)×(ρ3d3)1/2 ……(3)
式中,-7≤α≤7,Ea是固体电解质层的活性化能量(eV),ρ3是第2电极活性物质层的密度(g/cm3),d3是第2电极活性物质层的晶格常数()。
又,在形成上述固体电解质层时,最好是对照射的能量的功率及上述基板的散热功率进行调整,使上述基板的温度不超过上述第2电极活性物质层的融点。
在构成上述固体电解质层的固体电解质是氧化物或氮化物时,最好是蒸镀氛围分别采用包含氧或氧离子的气体氛围、或包含氮或氮离子的惰性气体氛围。
这里,本发明的制造方法中,在对构成上述电极活性物质的原子、离子或群集进行照射的能量的载体为电子或电磁波时特别有效。与上述的以往的技术中的离子的情况不同,电子的质量与原子相比是微乎其微的,使被碰撞的原子移动的作用很小,因此能使结晶缺陷减少。而且比离子照射容易实现约大2个数量级的照射电流密度,所以能够形成结晶缺陷少的结晶层。但是,电子的穿透力比离子的穿透力强,这一点应该留意。
此外,在形成结晶态的电极活性物质层的一方,有必要使固体电解质保持非晶态,因此必须将固体电解质的温度保持在结晶温度以下。
关于使用于第1电极活性物质层、第2电极活性物质层及固体电解质层的材料的组合,本发明在电极活性物质的结晶温度高于固体电解质的结晶温度的情况下可有效适用。
能够使用于本发明的电极活性物质层中使用的材料(电极活性物质)中,作为与上述条件相应的材料,可列举出例如LiMO2(M包括Co、Ni、Mn、Fe、V)、LiMn2O4、Li1-αCoαN、V2O5、Li3N及Li(Li1/3Ti5/3)O4等。此外,作为本发明中可使用的固体电解质,可列举出例如氮化磷酸锂、Li2S-SiS2-Li3PO4、及Li2O-V2O5-SiO2等。
在本发明中,用于照射的电子的能量或光子的能量,最好能满足上述式(1)~(3)。通过使照射能量小于式(1)~(3)中规定的规定值,可将能量到达的范围限定于被照射面(基材)的最表层原子。另外,Ea是使结晶化需用的再排列发生的能量(活性化能量),通常为数eV~60eV左右的范围。利用大于该活性化能量的能量进行照射,使构成层的原子处于能够再排列的状态。
因此,用满足上式(1)~(3)的能量E进行照射,使仅存在于最表面的原子发生再排列,能量不直接传送到其最表面的下层,可大大抑制热破坏。
该方法在形成结晶态的固体电解质时同样起作用。例如,如上述第3制造方法所述,在Li等融点极低的电极活性物质层上,形成作为结晶态固体电解质层的Li3PO4-Li4SiO4结晶层,也可以用本发明的上述第3制造方法实现。
当然,如果采用本发明,即使在Li层上形成结晶态固体电解质层或集电体层,再在其上形成结晶态电极活性物质的情况下,也可不使Li层熔融,可靠地形成结晶态电极活性物质。
还有,本发明的制造方法,有使气体活性化的效果,因此能够抑制形成氧化物层或氮化物层时发生的氧或氮的缺损,如果采用含氧或氮的氛围,可有效地将氧或氮导入层中。在结晶态的电极活性物质层或固体电解质层,在形成以氛围中包含的氧或氮等作为构成要素的层时,通过使用能量照射的方法,使氛围中的氧或氮成为更具活性的离子或等离子状态,可大大改善得到的层中的化学计量比(化学量论比),还可积极地将不同的气体元素导入层中。
本发明的制造方法的特征还在于,使各层不损其特性地管理基板的温度。如上所述,保持基板温度于结晶温度以下对于抑制固体电解质的结晶化是重要的。基板的温度由加于基板的能量与基板给予外部的能量之差决定,所以根据温度的实测值等进行调整可将基板温度保持在固体电解质的结晶温度以下。例如,使具有温度调整功能的材料紧贴基板等方法是有效的方法。
以下参照附图对本发明的电化学元件制造方法进行说明。图1及图2是表示实施本发明的电化学元件制造方法可使用的设备的构造示意图。在经排气管8及主阀9连接于真空泵(未图示)的真空容器11的内部,设置着由基板架10支持的被制膜基板1、蒸镀源2及3、电子束蒸镀坩埚4、电子束蒸镀用电子枪5、基板表面温度测定用辐射温度计6、氛围气体导入口7、排气管8、对基板用电子源21、对群集及对气体用电子源22、对基板用电子源用的气体导入管23、对群集及对气体用电子源用的气体导入管24、对基板用等离子源31、对群集及对气体用等离子源32、对基板用等离子源用的气体导入管33、对群集及对气体用电子源用的气体导入管34、对基板用等离子源用的气体导入管35、对群集及对气体用电子源用的气体导入管36、对基板用光源41、以及对群集及对气体用的光源42。遮挡板(shutter)12应设置在对蒸镀源2及3、以及能量照射装置21、22、31、32、41及42,能够将基板遮盖住的位置。
图2所示的基板温度控制装置13通过控制基板的温度抑制传对基板的热损坏,并在基板表面形成结晶膜时起作用。例如,在设定高成膜速度,采用多种材料,进行多量能量照射时,基板温度控制装置13主要发挥冷却功能,在工业利用方面特别有效。
作为导入至等离子源31及32的气体,可列举出诸如Ar、He、Ne及Xe等。因而这些惰性气体的目的,主要是防止热灯丝的劣化,而不是以形成等离子体为目的。还有,等离子体气体是将构成上述层的原子即O2或N2等气体经过导入管35及36提供给真空容器11,再使电子撞击该气体而发生的。
作为光源41及42,最好采用能发出短波长端有12~62nm波长的光线的光源。由于气体要吸收该波长区域的光线,所以通常使用真空闪光源(铀阳极:15~40nm)、及激光激发等离子体光源等不利用气体放电现象的光源。
例如,日本特开平8-287901号公报记载的重氢灯,是利用重氢气体的放电光谱的灯,发射波长超过90nm(用电子伏特换算,低于13eV)的光,此外,由于需要封入气体,窗口材料是必要的。尽管如此,由于在100nm以下的短波长区域不存在透明材料,所以无论是已有的技术还是本发明,都不能有效使用。
作为本发明中能有效使用的电极活性物质(化合物),可列举诸如LiCoO2、LiMn2O4、LiNiO2、V2O5、MoO2及TiS2等、LiV2O5、Li3-αCoαN、Li3-αNiαN、Li3-αCuαN、以及Li3-αMnαN等。
在形成用MxOy组成式表示的电极活性物质层时,将金属M或氧化物MxOy作为源使用。用LiMxOy的组成式表示的电极活性物质层的情况下,由于金属中Li具有较高的蒸气压,Li与金属M间发生蒸气压差,所以将金属Li,锂氧化物或锂碳酸盐等作为Li供给源使用,将金属M或氧化物MxOy作为M供给源使用。由于蒸镀中发生氧缺损,作为从氛围气体导入口7供给的氛围气体,也可使用惰性气体加氧的混合气体。作为不活泼气体,可列举诸如He、Ar、Ne、Xe以及N2等。
作为用蒸镀法形成电极活性物质层时使用的源,可使用电极活性物质本身,或使用构成电极活性物质层的多种元素或化合物。电极活性物质层是金属氧化物时,蒸镀源可采用金属或它的氧化物。但在使用含锂的化合物的情况下,锂的蒸气压比其他金属的蒸气压高,所以蒸镀源分为Li化合物与其他金属化合物等多种,对于稳定地形成电极活性物质层是理想的。
用电子束蒸镀时,最好在蒸镀开始时的真空容器11内部,从氛围气体导入口7以1~100sccm加入氩气,并保持压强在5×10-5~1×10-3Torr左右。
以下用实施例对本发明作更加具体的说明,但是本发明不限于此。
实验例1
图3表示本实验例中制作的锂电池的构造。如图3所示,这里制作的锂电池中,具备以碳构成的第1电极活性物质层52,以Li2S-SiS2-Li3PO4构成的固体电解质层53及以LiCoO2构成的第2电极活性物质层54。用电子束蒸镀法在铜箔上依次形成这些物质的层,根据本发明的电化学元件制造方法制造锂电池。对形成第2电极活性物质层54时的基板温度、第2电极活性物质层54的X线衍射图形以及用上述基板温度制作的锂二次电池的充放电特性进行评价,根据这些评价结果确认本发明的电化学元件的制造方法是有效的。
首先,在以厚度15μm的铜箔构成的第1集电体51上,如图6所示,设置具有1.2cm边长的方形开口部的不锈钢箔制的掩膜81,将挤压成型的碳材(日本碳株式会社制的SEG-R)作为源进行电子束蒸镀,形成以厚度2μm的碳构成的第1电极活性物质层52。这里,图6表示铜箔构成的第1集电体51上设置掩膜81的情形。图6(a)为顶视图,图6(b)为侧面图。此时,用1×10-4Torr的Ar氛围,照射的电子束的加速电压及电流分别取10kV和1A/cm2。
然后,在第1电极活性物质层52上,如图7所示,设置具有1.4cm边长的方形开口部的不锈钢箔制的掩膜82,用Li2S-SiS2-Li3PO4作为靶极,实施磁控管溅射法,形成以厚度2μm的Li2S-SiS2-Li3PO4构成的固体电解质层53。这里,图7表示设置掩膜82于第1电极活性物质层52上的情形。图7(a)表示顶视图,图7(b)表示侧面图。此时,用1×10-2Torr的Ar气氛围,采用100mm直径的靶极及100W的高频输出。
此外,在固体电解质层53上,如图8所示,设置具有1cm边长的方形开口部的不锈钢箔制的掩膜83,以Li2O及Co3O4作为源使用,实施多源电子束蒸镀法,形成以厚度1μm的LiCoO2构成的第2电极活性物质层54。这里,图8表示设置掩膜83于固体电解质层53上的情形。图8(a)为顶视图,图8(b)为侧面图。此时,用1×10-4Torr的Ar50%及氧50%的混合气体氛围(由气体供给管7供给),对Li2O的电子束加速电压及电流分别取10kV和0.1A,对Co3O4的电子束的加速电压及电流分别取10kV和1A,源与基板的距离为600mm,制膜用的遮挡板(shutter)12的开放时间取60分钟左右。形成第2电极活性物质层54时的条件按表1设定。
此外,如图1所示,在基板上照射电子时,采用基板照射用电子源21,从气体导入管23导入Ar,对氛围照射电子时,采用群集及气体照射用电子源22,从气体导入管24导入Ar气。
还有,向基板照射等离子体时,采用基板照射用等离子体源31,从气体导入管33导入Ar气,对氛围照射等离子体时,采用群集及气体照射用等离子体源32,从气体导入管34导入Ar气。
此外,向基板照射光线时,采用基板照射用光源41,对氛围照射光线时,采用氛围照射用光源42。还有,电子源、等离子体源、真空闪光(spark)源及重氢灯各自与基板的距离采用100mm。此外,在第15号实验中,不使用任何照射装置(已有技术例)。
接着,在第2电极活性物质层54上,如图9所示,设置具有1cm见方开口部的不锈钢箔制的掩膜84,以铜作为源进行电子束蒸镀,形成以厚度3μm的铜箔构成的第2集电体。这里,图9表示设置掩膜84于第2电极活性物质层54上的情形。图9(a)为顶视图,图9(b)为侧面图。此时,用1×10-4Torr的Ar氛围,电子束的加速电压及电流分别取10kV和1A/cm2。这样制作锂二次电池。
[评价]
①关于第2电极活性物质层
测定形成第2电极活性物质层54时的基板最高温度(℃)及第2电极活性物质层54的X线衍射图形中的半宽度(deg)。
表1展示第2电极活性物质层54的形成条件及评价结果。还有,在表1,所谓“功率”,在电子照射时是指电子加速电压及电子电流,在等离子体照射时是指加速电压及离子电流,在光照射时是指发光波长及输出。
如表1所示,在实施第1、3、5实验时,用红外线辐射温度计6测定的基板表面温度超过350℃。此外,在实施第1~7、9、11实验中,以LiCoO2构成的第2电极活性物质层的[003]、[101]及[104]的衍射信号的半宽度小于0.2°。另一方面,在第8、10、12、13、15实验中,只能观测宽(broad)信号。
②关于锂二次电池
对如上所述制作的锂二次电池的充放电特性进行评价。使得到的电池在环境温度20℃及0.2C速率(对于以第1电极活性物质层的质量为依据的电池的理论容量(LiCoO2为150mAh/g),能够以5小时充电的电流值)的条件下充电到4.2V,接着以2C速率(对电池的理论容量,能够用0.5小时放电的电流值)放电到3.0V。第5循环的第1电极活性物质的每单位重量的放电容量在表1中表示。又,图10表示第5循环的放电曲线。
在根据X线衍射图形预测得到结晶性较好的第1电极活性物质层的实验中,第1~6实验的放电容量良好,而第7、9、11实验的放电容量减少。在这种情况下,容量的减少被认为是由于用加速电压200V的电子照射使固体电解质层53的温度上升,引起固体电解质层53结晶化,导致离子传导率减低的缘故。第8、10、12、13、14实验中,容量大幅度减低,这被认为是由于LiCoO2构成的第2电极活性物质层的结晶度减低所致。
表1
| 第二电极活性物质层54形成条件 | 评价结果 | |||||||
| 照射源 | 照射对象 | 功率 | 氛围 | 照射源引入气体(流量) | 基板温度(℃) | XRD半宽度(deg) | 容量(mAh/g) | |
| 1 | 电子 | 基板 | 40V,2A | Ar+O2 | Ar(5sccm) | 265 | 0.10 | 124 |
| 2 | 电子 | 氛围 | 40V,2A | Ar+O2 | Ar(5sccm) | 240 | 0.11 | 123 |
| 3 | 等离子体 | 基板 | 40V,2A | Ar+O2 | Ar(5sccm) | 260 | 0.12 | 119 |
| 4 | 等离子体 | 氛围 | 40V,2A | Ar+O2 | Ar(5sccm) | 243 | 0.10 | 125 |
| 5 | 光(真空闪光光源) | 基板 | 15~40nm(80W) | Ar+O2 | - | 240 | 0.12 | 118 |
| 6 | 光(真空闪光光源) | 氛围 | 15~40nm(80W) | Ar+O2 | - | 220 | 0.10 | 123 |
| 7 | 电子 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 354 | 0.09 | 68 |
| 8 | 电子 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 160 | 1.50 | 55 |
| 9 | 电子 | 氛围 | 200V,2A | Ar+O2 | Ar(5sccm) | 360 | 0.09 | 72 |
| 10 | 电子 | 氛围 | 10V,2A | Ar+O2 | Ar(5sccm) | 150 | 1.80 | 40 |
| 11 | 等离子体 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 355 | 0.08 | 70 |
| 12 | 等离子体 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 152 | 1.85 | 62 |
| 13 | 光(重氢灯) | 基板 | 250~400nm(80W) | Ar+O2 | - | 170 | 2.00 | 38 |
| 14 | 光(重氢灯) | 氛围 | 250~400nm(80W) | Ar+O2 | - | 162 | 1.85 | 42 |
| 15 | 无 | - | - | Ar+O2 | - | 89 | - | 10 |
实验例2
在本实验例,对作为形成第2电极活性物质层54时的照射源导入气体使用氧的效果进行分析。
在形成第2电极活性物质层54时,除了按表2的条件设定外,其余都与实验例1同样处理,制作了锂二次电池。
此外,在第16及17实验,分别经过气体导入管23及33以5sccm的流量导入Ar气体,从气体供给管7及35供给Ar50%及氧气50%的混合气体(10sccm),调整氛围压力到1×10-2Torr。
此外,在第18及19实验,分别经过气体导入管23及33以5sccm的流量导入Ar气体,从气体供给管7及35提供Ar50%及氧气50%的混合气体(10sccm),调整氛围压力到1×10-2Torr。
此外,与实验例1同样处理,形成以厚度3μm的Cu构成的第2集电体55,制作锂二次电池。
[评价]
①关于第2电极活性物质层的组成
通过ICP红外线发光分光分析,对本实验例中形成的第1电极活性物质层54的组成进行分析。第18及19实验中的Li∶Co∶O的原子比大约为1∶1∶2,但第16及17实验的情况下O的比例约为1.7,氧有稍微缺损的倾向。被制膜表面存在氧或氧离子作为对层的氧缺损的防止措施是有效的。
此外,在第16及17实验中没有发现明显差别,所以可以确认,在第17实验的电子照射的情况下,也与第17实验时相同,氧呈现活性状态,容易作为得到的层的构成原子取入。
②关于锂二次电池
此外,与实验例1一样对得到的锂二次电池的充放电特性进行检验。结果见表2。
第16及17实验的结果良好,但是在第18及19实验中,容量减少,这被认为是氧缺损对电池特性的影响。
表2
| 第二电极活性物质层54形成条件 | 评价结果 | ||||||||
| 照射源 | 照射方向 | 功率 | 氛围 | 照射源引入气体 | Li∶Co∶O原子比 | 容量(mAh/g) | |||
| Li | Co | O | |||||||
| 16 | 电子 | 基板 | 40V,2A | Ar | Ar+O2 | 0.96 | 1.00 | 2.01 | 125 |
| 17 | 等离子体 | 基板 | 40V,2A | Ar | Ar+O2 | 0.98 | 1.00 | 1.98 | 125 |
| 18 | 电子 | 基板 | 40V,2A | Ar | Ar | 1.10 | 1.00 | 1.70 | 106 |
| 19 | 等离子体 | 基板 | 40V,2A | Ar | Ar | 1.05 | 1.00 | 1.76 | 95 |
实验例3
在以Li2S-SiS2-Li3PO4构成的固体电解质层53上形成以LiCoO2构成的第2电极活性物质层54时,将照射的电子的加速电压变更为表3所示的值。表3表示此时的基板温度、X线衍射信号的半宽度、及最终得到的锂二次电池的充放电特性结果。还有,条件与实验例1相同。还有,照射能量的电流值在所有情况下都是2A。
从表3看出,与照射电子的加速电压的上升同时,X线衍射信号的半宽度始终较小,结晶性提高。而电池容量在低于300℃的范围随加速电压的上升而上升,但是基板温度超过300℃左右后则呈现下降倾向。这被认为是随着加速电压的上升,照射能量增大,基板温度上升引起固体电解质的结晶化,导致离子传导率减少。作为应对照射能量增大的措施,需要采取从基板散热的手段。
表3
| 电子加速电压(V) | 基板温度(℃) | XRD半宽度(deg) | 容量(mAh/g) | |
| 20 | 5 | 121 | 3.10 | 12 |
| 21 | 10 | 160 | 1.70 | 55 |
| 22 | 20 | 205 | 0.60 | 90 |
| 23 | 30 | 228 | 0.25 | 101 |
| 24 | 40 | 265 | 0.10 | 124 |
| 25 | 50 | 281 | 0.10 | 126 |
| 26 | 90 | 314 | 0.10 | 115 |
| 27 | 100 | 319 | 0.10 | 107 |
| 28 | 110 | 322 | 0.10 | 98 |
| 29 | 200 | 354 | 0.09 | 68 |
实验例4
在本实验例中,作为构成固体电解质层53的材料,采用Li2O-V2O5-SiO2,除了第2电极活性物质层54的形成条件如表4所示设定外,其他都与实验例1相同。
在以厚度15μm的铜箔构成的第1集电体51上,将挤压成型的碳材(日本碳株式会社制的SEG-R)作为源使用进行电子束蒸镀,形成以厚度2.2μm的碳构成的第1电极活性物质层52。此时,用1×10-4Torr的Ar氛围,照射电子束的加速电压及电流分别取10kV和1A。
接着,在第1电极活性物质层52上,用Li2O-V2O5-SiO2作为靶极使用,进行高频磁控管溅射,形成以厚度2μm的Li2O-V2O5-SiO2构成的固体电解质层53。此时,用1×10-2Torr的Ar氛围,采用100mm直径的靶极及100W的高频波输出功率。
接着,在固体电解质层53上,以Li2O及NiO2作为源使用,进行多源电子束蒸镀,形成以1μm的LiNiO2层构成的第2电极活性物质层54。这里,用1×10-4Torr的Ar50%及氧50%的混合气体氛围,对Li2O的电子束的加速电压及电流分别取10kV和0.08A,对NiO2的电子束加速电压及电流分别取10kV和0.9A。
将第2电极活性物质层54的形成条件按表4中的值设定,此外,与实验例1同样地制作锂二次电池。
[评价]
表4是第2电极活性物质层54的形成条件及检验结果。此外,从表4可知,在第36、38及40实验进行时,用红外线辐射温度计6测得的表面温度超过350℃。此外,在第30~36、38、40实验中,LiNiO2的[003]、[101]及[104]的衍射信号的半宽度小于0.4°。另外,在第37、39及41~43实验中,只能观测到宽平的信号。
对电池的放电容量的测定,除了放电到2.5V外,其余都与实验例1相同。第5循环的第1电极活性物质定位重量的放电容量示于表4。而图11表示第5循环的放电曲线。
在根据X线衍射图形预测可得到结晶性较好的第2电极活性物质层54的实验中,第30~35实验的容量几乎一样好,而第36、38及40实验的容量减少。容量的减少被认为是由于用加速电压200V的电子照射使固体电解质层54的温度上升,引起固体电解质层54结晶化,导致离子传导率减低的缘故。第37、39及41~43实验中,容量大幅减低,这被认为是由LiNiO2构成的第2电极活性物质层54的结晶度减低所致。
表4
| 第二电极活性物质层54形成条件 | 评价结果 | |||||||
| 照射源 | 照射对象 | 功率 | 氛围 | 照射源引入气体(流量) | 基板温度(℃) | XRD半宽度(deg) | 容量(mAh/g) | |
| 30 | 电子 | 基板 | 32V,2A | Ar+O2 | Ar(5sccm) | 205 | 0.22 | 142 |
| 31 | 电子 | 氛围 | 32V,2A | Ar+O2 | Ar(5sccm) | 198 | 0.25 | 141 |
| 32 | 等离子体 | 基板 | 32V,2A | Ar+O2 | Ar(5sccm) | 215 | 0.23 | 131 |
| 33 | 等离子体 | 氛围 | 32V,2A | Ar+O2 | Ar(5sccm) | 216 | 0.23 | 145 |
| 34 | 光(真空闪光光源) | 基板 | 15~40nm(70W) | Ar+O2 | - | 203 | 0.32 | 139 |
| 35 | 光(真空闪光光源) | 氛围 | 15~40nm(70W) | Ar+O2 | - | 200 | 0.29 | 141 |
| 36 | 电子 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 350 | 0.10 | 105 |
| 37 | 电子 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 158 | 1.69 | 92 |
| 38 | 电子 | 氛围 | 200V,2A | Ar+O2 | Ar(5sccm) | 355 | 0.09 | 106 |
| 39 | 电子 | 氛围 | 10V,2A | Ar+O2 | Ar(5sccm) | 152 | 1.82 | 90 |
| 40 | 等离子体 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 358 | 0.08 | 102 |
| 41 | 等离子体 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 148 | 2.05 | 88 |
| 42 | 光(重氢灯) | 基板 | 250~400nm(80W) | Ar+O2 | - | 166 | 1.80 | 85 |
| 43 | 光(重氢灯) | 氛围 | 250~400nm(80W) | Ar+O2 | - | 164 | 1.60 | 90 |
实验例5
在本实验例中,制作具有图3构造,包括以Li(Li1/3Ti5/3)O4构成的第1电极活性物质层52、以Li2.9PO3.3N0.36构成的固体电解质层53、以LiMn2O4构成的第2电极活性物质层54、及用Cu构成的第2集电体55的锂二次电池。将这些层用电子束蒸镀法及热蒸镀法在铜箔上依次形成,与实验例1大体相同地制作锂二次电池。
首先,最初在以厚度15μm的铜箔构成的第1集电体51上,以Li2O及TiO2作为源,用多源电子束蒸镀法,形成以厚度2.5μm的Li(Li1/3Ti5/3)O4构成的第1电极活性物质层52。此时,用1×10-4Torr的Ar+氧(混合比1∶1)氛围,对Li2O的电子束的加速电压及电流分别取10kV和0.1A,对Ti2O的电子束的加速电压及电流分别取10kV和1A这样。
接着,在上述第1电极活性物质层52上,用Li3PO4作为靶极,用磁控管溅射法形成以厚度2μm的Li2.9PO3.3N0.36构成的固体电解质层53。此时,用1×10-2Torr的N2氛围,采用100mm直径的靶极及100W的高频波输出功率。
在固体电解质层53上,以Li2O及MnO2作为源,进行多源电子束蒸镀,形成以厚度2μm的LiMn2O4构成的第2电极活性物质层54。用1×10-4Torr的Ar50%及氧50%的混合气体氛围,对Li2O的电子束的加速电压及电流分别取10kV和0.1A,对MnO2的电子束的加速电压及电流分别取10kV和1A。上述工序中图案的形成与实验例1相同。此外,与实验例1同样地制作锂二次电池。
[评价]
表5表示第2电极活性物质层54的形成条件及评价结果。从表5可知,在实施第50、52及54实验时,用红外线辐射温度计6测得的表面温度超过350℃。还有,在第44~50、52及54实验中,LiMn2O4的[111]的衍射信号的半宽度小于0.2°。另一方面,在第51、53及55~57实验,只观测到宽平的信号。
在充电到3.1V,放电到1.5V的条件以外,其他与实验例1同样地测定电池的放电容量。第5循环的第1电极活性物质单位重量的放电容量示于表5。还有,图12表示第5循环的放电曲线。
在根据X线衍射图形预测可得到结晶性较好的第2电极活性物质层54的实验中,第44~49实验的容量几乎一样好,而第50、52及54实验的容量减少。容量的减少被认为是由于用加速电压200V的电子照射使固体电解质层54的温度上升、引起固体电解质层54结晶化,导致离子传导率减低的缘故。第51、53及55~57实验中,容量大幅减低,这被认为是由LiMn2O4构成的第2电极活性物质层54的结晶度减低所致。
表5
| 第二电极活性物质层54形成条件 | 评价结果 | |||||||
| 照射源 | 照射对象 | 功率 | 氛围 | 照射源引入气体(流量) | 基板温度(℃) | XRD半宽度(deg) | 容量(mAh/g) | |
| 44 | 电子 | 基板 | 45V,2A | Ar+O2 | Ar(5sccm) | 278 | 0.10 | 96 |
| 45 | 电子 | 氛围 | 45V,2A | Ar+O2 | Ar(5sccm) | 261 | 0.14 | 90 |
| 46 | 等离子体 | 基板 | 45V,2A | Ar+O2 | Ar(5sccm) | 270 | 0.12 | 94 |
| 47 | 等离子体 | 氛围 | 45V,2A | Ar+O2 | Ar(5sccm) | 259 | 0.13 | 94 |
| 48 | 光(真空闪光光源) | 基板 | 15~40nm(80W) | Ar+O2 | - | 242 | 0.12 | 95 |
| 49 | 光(真空闪光光源) | 氛围 | 15~40nm(80W) | Ar+O2 | - | 224 | 0.16 | 88 |
| 50 | 电子 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 350 | 0.09 | 61 |
| 51 | 电子 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 163 | 1.50 | 52 |
| 52 | 电子 | 氛围 | 200V,2A | Ar+O2 | Ar(5sccm) | 361 | 0.09 | 63 |
| 53 | 电子 | 氛围 | 10V,2A | Ar+O2 | Ar(5sccm) | 147 | 1.80 | 50 |
| 54 | 等离子体 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 355 | 0.08 | 62 |
| 55 | 等离子体 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 155 | 1.85 | 48 |
| 56 | 光(重氢灯) | 基板 | 250~400nm(80W) | Ar+O2 | - | 180 | 2.00 | 44 |
| 57 | 光(重氢灯) | 氛围 | 250~400nm(80W) | Ar+O2 | - | 167 | 1.85 | 45 |
实验例6
在本实验例也进行与实验例1的第1~6实验大体相同的实验。而且,同样地制作了锂二次电池。
在以厚度15μm的铜箔构成的第1集电体51上,以Li3N及Co作为源,用电子束蒸镀,形成以厚度1.4μm的Li3-αCoαN构成的第1电极活性物质层52。此时,α最好满足0.2≤α≤0.6,其中α取0.4特别好。此时,用1×10-4Torr的N2气体氛围,对Li3N的电子束的加速电压及电流分别取10kV和0.25A,对Co的电子束的加速电压及电流分别取10kV和1A。又,一边用基板照射用电子源21进行照射一边进行制膜。对于基板照射用电子源21的条件,电子加速电压及电流分别为40V和2A,从气体导入管23来的氮流量为5sccm。
接着,在第1电极活性物质层54上,用Li2S-SiS2-Li3PO4作为靶极,进行高频磁控管溅射,形成以厚度2μm的Li2S-SiS2-Li3PO4构成的固体电解质层53。此时,用1×10-2Torr的Ar氛围,采用100mm直径的靶极及100W的高频波输出功率。
还有,在固体电解质层53上,以钒(V)作为源,进行电子束蒸镀,形成以4μm的V2O5构成的第2电极活性物质层54。在该第2电极活性物质层54形成时,使具有1cm见方开口部的不锈钢制的掩膜(厚度20μm)紧贴配置于固体电解质层53上,将c-V2O5层作成1cm见方。此时,用1×10-4Torr的Ar及氧气的混合气体(1∶1)氛围,对钒的电子束的加速电压及电流分别取2010kV和1A。
接着,在如上所述形成的具有1cm见方的第2电极活性物质层54上,以Cu作为源进行电子束蒸镀,形成厚度为3μm的Cu构成的第2集电体55。此时,使具有1cm见方开口部的不锈钢制的掩膜(厚度20μm)紧贴配置于固体电解质层53上,以第2电极活性物质层54上重叠由Cu构成的第2集电体55的状态,将第2集电体55作成1cm见方的形状。此时,用1×10-4Torr的Ar气体氛围,取电子束的加速电压及电流分别取10kV和1A。这样制作锂二次电池。
[评价]
第2电极活性物质层54的形成条件及评价,与实验例1同样进行。结果示于表6。如表6所示,在实施第59、61及63实验时,用红外线辐射温度计6测得的表面温度超过350℃。还有,在第58~64、66及68实验中,V2O5的[200]、[221]及[240]等的衍射信号的半宽度小于0.2°。另一方面,在实施第65、67、及69~71实验,只能观测到宽平的信号。
又,使制得的锂二次电池在环境温度20℃及0.2C速率(电池的理论容量以5小时的充电能够得到的情况下的电流值)的条件下充电到3.2V,以2C速率(电池的理论容量以0.5小时能够放电完的电流值)的条件放电到1.5V。第5循环的第2电极活性物质单位重量的放电容量示于表6。还有,图13表示第5循环的放电曲线。
在根据X线衍射图形预测可得到结晶性较好的第1电极活性物质的实验中,第58~63实验的放电容量几乎一样好,而第64、66及68实验的放电容量减少。
该容量的减少被认为是由于用加速电压200V的电子照射使固体电解质层54的温度上升,引起固体电解质层54结晶化,导致离子传导率减低的缘故。第65、67及69~71实验中,电流容量大幅度减低。这被认为是由c-V2O5构成的第2电极活性物质层54的结晶度低所致。
表6
| 第二电极活性物质层54形成条件 | 评价结果 | |||||||
| 照射源 | 照射对象 | 功率 | 氛围 | 照射源引入气体(流量) | 基板温度(℃) | XRD半宽度(deg) | 容量(mAh/g) | |
| 58 | 电子 | 基板 | 40V,2A | Ar+O2 | Ar(5sccm) | 263 | 0.11 | 631 |
| 59 | 电子 | 氛围 | 40V,2A | Ar+O2 | Ar(5sccm) | 245 | 0.12 | 619 |
| 60 | 等离子体 | 基板 | 40V,2A | Ar+O2 | Ar(5sccm) | 259 | 0.12 | 622 |
| 61 | 等离子体 | 氛围 | 40V,2A | Ar+O2 | Ar(5sccm) | 249 | 0.10 | 650 |
| 62 | 光(真空闪光光源) | 基板 | 15~40nm(80W) | Ar+O2 | - | 240 | 0.12 | 623 |
| 63 | 光(真空闪光光源) | 氛围 | 15~40nm(80W) | Ar+O2 | - | 225 | 0.11 | 634 |
| 64 | 电子 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 357 | 0.09 | 480 |
| 65 | 电子 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 166 | 1.60 | 430 |
| 66 | 电子 | 氛围 | 200V,2A | Ar+O2 | Ar(5sccm) | 363 | 0.09 | 485 |
| 67 | 电子 | 氛围 | 10V,2A | Ar+O2 | Ar(5sccm) | 150 | 1.90 | 413 |
| 68 | 等离子体 | 基板 | 200V,2A | Ar+O2 | Ar(5sccm) | 359 | 0.09 | 473 |
| 69 | 等离子体 | 基板 | 10V,2A | Ar+O2 | Ar(5sccm) | 158 | 1.85 | 419 |
| 70 | 光(重氢灯) | 基板 | 250~400nm(80W) | Ar+O2 | - | 173 | 2.05 | 380 |
| 71 | 光(重氢灯) | 氛围 | 250~400nm(80W) | Ar+O2 | - | 161 | 1.87 | 408 |
实验例7
在本实验例中,制作包括以c-V2O5构成的第1电极活性物质层52、以Li2S-SiS2-Li3PO4构成的固体电解质层53、以Li3-αCoαN构成的第2电极活性物质层54、以Cu构成的第2集电体55的、具有图3所示构造的锂二次电池。将这些层用电子束蒸镀法及溅射法依次形成于以铜箔构成的第1集电体51上。
首先,在以厚度15μm的铜箔构成的第1集电体51上,以钒作为源用电子束蒸镀,形成以厚度4μm的c-V2O5构成的第1电极活性物质层52。此时,用1×10-4Torr的Ar及O2的混合气体(1∶1)氛围,对钒的电子束的加速电压及电流分别取10kV和1A,用基板照射用电子源21照射电子,进行制膜。基板照射用电子源21的条件采用电子加速电压及电流分别为40V和2A,从气体导入管23来的Ar流量为5sccm。
接着,在第1电极活性物质层52上,用Li2S-SiS2-Li3PO4作为靶极进行高频磁控管溅射,形成以厚度2μm的Li2S-SiS2-Li3PO4构成的固体电解质层53。此时,用1×10-2Torr的Ar氛围,采用100mm直径的靶极及100W的高频波输出功率。
再在固体电解质层53上以Li3N及Co作为源,进行多源电子束蒸镀,形成以1.4μm厚的Li3-αCoαN构成的第2电极活性物质层54。在该形成中,使具有1cm见方开口部的不锈钢制掩膜(厚度20μm)紧贴配置于固体电解质层53上,将以Li3-αCoαN构成的第2电极活性物质层54作成1cm见方。α最好满足0.2≤α≤0.6,其中α取0.4为好。此时,用1×10-4Torr的N2气体氛围,对Li3N的电子束的加速电压及电流分别取10kV和0.25A,对Co的电子束的加速电压及电流分别取10kV和1A。
接着,在如上所述形成的具有1cm见方形状的第2电极活性物质层54上,以Cu作为源,进行电子束蒸镀,形成厚度为3μm的Cu构成的第2集电体55。此时,使具有1cm见方开口部的不锈钢制的掩膜(厚度20μm)紧贴配置于固体电解质层53上,以第2电极活性物质层54上重叠由Cu构成的第2集电体55的状态,将第2集电体55作成1cm见方的形状。此时,用1×10-4Torr的Ar气体氛围,取电子束的加速电压及电流分别为10kV和1A。这样制作锂二次电池。
[评价]
第2电极活性物质层54的形成条件及评价,与实验例1同样进行。结果示于表7。如表7所示,在实施第78、80及82实验时,用红外线辐射温度计6测得的表面温度超过350℃。又,在第72~78、80、82实验中,Li3-αCoαN的[100]、[101]及[110]等的衍射信号的半宽度小于0.2°。另外,在第79、81、及83~85实验,只能够观测到宽平的信号。
又,将得到的锂二次电池在环境温度20℃以0.2C速率(电池的理论容量能够以5小时的充电得到的情况下的电流值)的条件下充电到3.2V,以2C速率(电池的理论容量0.5小时放电完的情况下的电流值)放电到1.5V。第5循环的第2电极活性物质单位重量的放电容量示于表7。
在根据X线衍射图形预测可得到结晶性较好的第1电极活性物质的实验中,第72~77实验的放电容量几乎一样好,而第78、80及82实验的放电容量减少。该容量的减少被认为是由于加速电压200V的电子被照射,使固体电解质层54的温度上升,引起固体电解质层54的结晶化,导致离子传导率减低的缘故。第79、81及83~85的实验中,几乎不能取出电流。这被认为是由Li3-αCoαN构成的第2电极活性物质层54的结晶不充分所致。
表7
| 第二电极活性物质层54形成条件 | 评价结果 | |||||||
| 照射源 | 照射对象 | 功率 | 氛围 | 照射源引入气体(流量) | 基板温度(℃) | XRD半宽度(deg) | 容量(mAh/g) | |
| 72 | 电子 | 基板 | 40V,2A | N2+O2 | N2(5sccm) | 268 | 0.17 | 650 |
| 73 | 电子 | 氛围 | 40V,2A | N2+O2 | N2(5sccm) | 246 | 0.14 | 660 |
| 74 | 等离子体 | 基板 | 40V,2A | N2+O2 | N2(5sccm) | 257 | 0.12 | 675 |
| 75 | 等离子体 | 氛围 | 40V,2A | N2+O2 | N2(5sccm) | 248 | 0.15 | 653 |
| 76 | 光(真空闪光光源) | 基板 | 15~40nm(70W) | N2+O2 | - | 235 | 0.16 | 656 |
| 77 | 光(真空闪光光源) | 氛围 | 15~40nm(70W) | N2+O2 | - | 230 | 0.15 | 645 |
| 78 | 电子 | 基板 | 200V,2A | N2+O2 | N2(5sccm) | 354 | 0.11 | 560 |
| 79 | 电子 | 基板 | 10V,2A | N2+O2 | N2(5sccm) | 166 | 1.60 | 490 |
| 80 | 电子 | 氛围 | 200V,2A | N2+O2 | N2(5sccm) | 359 | 0.10 | 562 |
| 81 | 电子 | 氛围 | 10V,2A | N2+O2 | N2(5sccm) | 150 | 1.90 | 457 |
| 82 | 等离子体 | 基板 | 200V,2A | N2+O2 | N2(5sccm) | 356 | 1.02 | 505 |
| 83 | 等离子体 | 基板 | 10V,2A | N2+O2 | N2(5sccm) | 161 | 1.85 | 463 |
| 84 | 光(重氢灯) | 基板 | 250~400nm(80W) | N2+O2 | - | 170 | 2.05 | 442 |
| 85 | 光(重氢灯) | 氛围 | 250~400nm(80W) | N2+O2 | - | 164 | 1.87 | 449 |
实验例8
在本实验例中,制作由2单位电池叠层构成的锂二次电池。
这里制作的锂二次电池的构造示于图4。如图4所示,该锂二次电池,首先,具备:以不锈钢构成的第1集电体61、以LiCoO2构成的第1电极活性物质层62、以氮化磷酸锂构成的固体电解质层63、以金属锂构成的第2电极活性物质层64、以及以Ni构成的第2集电体65。还有,上述锂二次电池,具备:以金属锂构成的第2电极活性物质层66、以氮化磷酸锂构成的固体电解质层67、以LiCoO2构成的第1电极活性物质层68、以及以Cu构成的第1集电体69。
用金属掩膜等,又将电子束蒸镀法、溅射法以及干式图案形成方法并用,使这些层依上述顺序叠层,采用图2所示的设备形成2叠层电池。
这里,图2所示的装置与图1所示的装置大体相同,但增加了控制基板温度的机构13,该机构13在本实验例主要发挥冷却的效果,使基板背面温度保持在140℃。机构13在冷却时采用油冷却方式,加热时用电加热方式,它与外部的控制设备组合使用。
首先,利用电子束蒸镀,将1μm厚度的LiCoO2构成的第1电极活性物质层62形成于用Cu箔构成的第1集电体61上。此时,使用金属Li及Co作为源,让它们同时蒸发。在形成第1电极活性物质层62时,使具有1cm见方的正方形孔的不锈钢制掩膜(厚度20μm)紧贴配置于以Cu箔构成的第1集电体61上,将以LiCoO2构成的第1电极活性物质层62作成1cm见方的形状。此时,采用1×10-4Torr的Ar50%及氧50%的混合气体氛围,对Li的电子束的加速电压及电流分别取10kV和0.02A,对Co的电子束的加速电压及电流分别取10kV和0.2A。从源到基板的距离取200mm,制膜用的遮挡板(shutter)开放时间为5分钟左右。
与第1电极活性物质层62的形成同时,设定为表8所示的条件。此外,从气体导入管23来的Ar50%及氧50%的混合气体流量取50sccm。还有,在第96实验中,不使用能量照射装置。
接着,以Li3PO4作为靶极使用,在氮氛围中实施RF磁控管溅射,形成以厚度2μm的氮化磷酸锂构成的固体电解质层63。此时,用1×10-2Torr的Ar气体氛围,采用100mm直径的靶极及100W的高频波输出功率,成膜时间为32小时。又通过使开正方形孔的不锈钢掩膜(厚度20μm)尽可能紧贴配置于第1电极活性物质层62及第1集电体61上的方法,用固体电解质层63完全覆盖在以LiCoO2构成的第1电极活性物质层62上。
在固体电解质层63上,采用电阻加热蒸镀法形成以0.5μm厚度的Li构成的第2电极活性物质层64。此时,使开正方形孔的不锈钢掩膜(厚度20μm)尽可能紧靠着以氮化磷酸锂构成的固体电解质层63及以Cu构成的第1集电体61上配设,形成以Li构成的第2电极活性物质层64,并做到覆盖第1电极活性物质层62上且不能从固体电解质层63上露出。
接着,形成兼作引出电极的厚度0.5μm的金属Ni构成的第2集电体65。在形成厚度0.5μm的金属Li构成的第3电极活性物质层66及厚度2μm的氮化磷酸锂构成的固体电解质层67后,形成厚度2μm的LiCoO2构成的第4电极活性物质层68。
使形成第4电极活性物质层68时的条件与形成第1电极活性物质层62时的条件相同。此外,形成厚度3μm的金属Cu构成的第3集电体69。第3集电体69兼作引出电极,如图4所示,与具有同一极性的第1集电体61电气连接。但必须使其不与作为对极的第2电极活性物质层64、第2集电体65及第3电极活性物质层66接触。按各工艺条件成膜时的基板的最高到达温度示于表8。
将得到的锂二次电池在环境温度20℃以0.2C速率(以第1电极活性物质层的质量为依据的电池的理论容量可用5小时充电达到的电流值)充电到4.2V,接着2C速率(电池的理论容量可以0.5小时放电完的电流值)放电到3.0V。第5循环的第1电极活性物质每单位重量的放电容量示于表8。
表8表示形成第4电极活性物质层68时基板达到的最高温度、以及得到的锂二次电池的正极活性物质单位重量的容量。在形成第4电极活性物质层时最高到达温度低于200℃的范围的情况下,出现被认为是结晶化不足引起的XRD信号以及容量降低的现象。此外,在375℃,也出现加热引起的的容量的减低现象,而其间的温度范围能确保充分高的容量。
这里,应该注意的是,采用300V以下的照射能量,可得到充分的容量;采用超过300V的照射能量则容量减低。
在第1~85实验中,用200V的加速电压出现容量减低的现象,而在第86~94实验中,在300V的加速电压也能保证高容量。这被认为是由于,利用机构13的基板温度控制功能,可较高地保持制作中的第4电极活性物质层的表面温度,同时,可较低地保持位于基板附近的相应层的内部的温度,使固体电解质层的结晶化得到抑制。
此外,加速电压300V,接近或超过把LiCoO2的密度5.06g/cm2及LiCoO2的[003]面方向的晶格常数4代入式(1)及(2)得到的值(E1=310eV)。可以认为,如果加速电压接近或超过E1,那么,照射能量直接到达存在于最表层原子以下部分的固体电解质层67,固体电解质层67的离子传导率减低。
表8
| 第一电极活性物质层62形成条件 | 评价结果 | ||||
| 电子加速电压(V) | 照射电流(A) | 基板温度(℃) | XRD半宽度(deg) | 容量(mAh/g) | |
| 86 | 10 | 7 | 153 | - | 58 |
| 87 | 40 | 10 | 170 | 0.25 | 105 |
| 88 | 70 | 20 | 194 | 0.20 | 112 |
| 89 | 100 | 20 | 212 | 0.18 | 120 |
| 90 | 130 | 20 | 235 | 0.16 | 122 |
| 91 | 160 | 20 | 269 | 0.16 | 121 |
| 92 | 200 | 20 | 301 | 0.12 | 123 |
| 93 | 250 | 20 | 335 | 0.10 | 121 |
| 94 | 300 | 20 | 358 | 0.10 | 121 |
| 95 | 350 | 20 | 375 | 0.09 | 89 |
| 96 | - | - | 140 | - | 23 |
实验例9
在本实验例中,带Li负极而且形成结晶态固体电解质。制作具有图3所示的构造、包括以Cu构成的第1集电体51、以Li构成的第1电极活性物质层52、以c-Li3PO4-Li4SiO4构成的结晶态固体电解质层53、以c-V2O5构成的第2电极活性物质层54(c-表示结晶态)、及以Cu构成的第2集电体55的锂二次电池。
采用图2所示的设备,用金属掩膜等、电子束蒸镀法和干式图案形成方法并用,依次叠层,制作锂二次电池。
在以厚度15μm的铜箔构成的第1集电体51上,以Li作为源,进行电阻加热蒸镀,形成以厚度0.5μm的Li构成的第1电极活性物质层52。在该第1电极活性物质层52上,以Li3PO4、Li及Si作为源,进行三源电子束蒸镀,形成以Li3PO4-Li4SiO4构成的结晶态固体电解质层53。这里,作为蒸镀用电子束的条件,加速电压及电流分别取10kV和10mA(Li)、120ma(Si)及80ma(Li3PO4),此时的氛围采用Ar及氧的混合气体(混合比50∶50)。
还在形成固体电解质层53的同时,开动控制基板温度用的机构13,将基板背面的温度保持于100℃,同时设定于表9所示的条件。还有,从气体引入管23引入的Ar和氧的混合气体(混合比50∶50)的流量采用50sccm。
表9表示形成固体电解质层53时的基板表面的最高到达温度。又,以XRD观察形成的固体电解质53时,在第97~99实验出现无数极小的峰值,但在第101则没有看到。
在固体电解质层53上,将V作为源,进行电子束蒸镀,形成以厚度2μm的c-V2O5构成的第2电极活性物质层54。在形成第2电极活性物质层54时,使用基板照射用电子源21,对于此时的动作条件,电子加速电压取40V,电子电流取2A,来自气体导入管23的Ar及氧的混合气体(50∶50)的流量为5sccm。源与基板的距离为600mm,制膜用的遮挡板(shutter)的开启时间约60分钟。
在如上所述形成的具有1cm见方的第2电极活性物质层54上,以Cu作为源,进行电子束蒸镀,形成厚度为3μm的Cu构成的第2集电体55,制作具有图3所示构造的锂二次电池。在形成时,使具有1cm见方的孔的不锈钢制的掩膜(厚度20μm)紧贴配置于固体电解质层53上,以第2电极活性物质层54上重叠由Cu构成的第2集电体55的状态,将第2集电体55作成1cm见方的正方形。此时的氛围为1×10-4Torr的Ar气体氛围,取电子束的加速电压及电流分另取10kV和1A。
用上述的实验例中说明的方法为基准测定已得的锂二次电池的充放电特性,结果示于表9。按此,确认基板表面最高到达温度高于170℃时出现短路现象。放电后将短路的电池切断,观察其断面时,发现有完全没有以Li构成的第1电极活性物质层52的部分,在没有Li的部分和有Li的部份的边界附近,固体电解质层53上发生龟裂。这被认为是,在形成固体电解质层53时,基板的温度上升,因而以Li构成的第1电极活性物质层52发生暂时熔融,厚度发生偏差,伴随充放电产生的膨胀收缩导致形状变化以至形成畸变使固体电解质层53上发生龟裂,形成短路通路(bus)。
还有,短路的第100实验的电子照射装置的加速电压250V,高于把第2电极活性物质层54的密度2.5g/cm3及晶格常数4代入式(3)得到的值(E3=220V)。
因此,在第100实验的短路被认为是,能量直接到达以Li构成的第1电极活性物质层52,第1电极活性物质层52熔融,靠Li自身的表面张力凝集,导致其厚度不能保持均匀。
表9
| 固体电解质层53形成条件 | 评价结果 | ||||
| 电子加速电压(V) | 照射电流(A) | 基板温度(℃) | XRD信号 | 容量(mAh/g) | |
| 97 | 100 | 20 | 209 | 有 | 58 |
| 98 | 150 | 20 | 215 | 有 | 89 |
| 99 | 200 | 20 | 240 | 有 | 86 |
| 100 | 250 | 20 | 278 | 有 | 短路 |
| 101 | - | - | 140 | 无 | 15 |
实验例10
在本实验例,制作具有图3所示的构造,且包括以Cu构成的第1集电体51、以Li构成的第1电极活性物质层52、以c-Li3PO4-Li4SiO4构成的固体电解质层53、以LiCoO2构成的第2电极活性物质层54、及以Cu构成的第2集电体55的锂二次电池。
采用图2所示的装置,用金属掩膜等,电子束蒸镀法及干式图案成型方法并用,按照该顺序依次叠层这些层,制作锂二次电池。
首先,在以厚度15μm的铜箔构成的第1集电体51上,以Li作为源,进行电阻加热蒸镀,形成以厚度0.5μm的Li构成的第1电极活性物质层52。接着,在其上面,以Li3PO4、Li及Si作为源,进行三源电子束蒸镀,形成以结晶态的Li3PO4-Li4SiO4构成的固体电解质层53。这里,作为蒸镀用电子束的条件,加速电压及电流分别取10kV和10mA(Li)、120ma(Si)及80ma(Li3PO4),此时的氛围采用Ar及氧的混合气体(混合比50∶50)。
而且,与形成固体电解质层53同时,使控制基板温度用的机构13开动,把基板的背面温度保持在100℃,基板照射用电子源21的电子加速电压及电流分别取100V和20A,来自气体导入管23的Ar及氧的混合气体(混合比50∶50)的流量采用50sccm。
在固体电解质层53上,以Li及Co作为源,进行二源电子束蒸镀,形成以厚度2μm的LiCoO2构成的第2电极活性物质层54。用1×10-4Torr的Ar50%及氧50%的混合气体氛围(由气体供给管7供给),对Li的电子束的加速电压及电流分别取10kV和0.1A,对Co3O4的电子束的加速电压及电流分别取10kV和1A,在形成第2电极活性物质层54时,使控制基板温度用的机构13开动,把基板的背面温度保持在100℃,采用表10所示的条件。又,来自气体导入管23的Ar及氧的混合气体(混合比50∶50)的流量采用50sccm。还有,在第196实验中,不进行能量照射。源与基板的距离为200mm,制膜用的遮挡板(shutter)的开启时间约5分钟。
第2电极活性物质层54的形成中的基板表面的最高到达温度示于表10。如上所述形成的1cm见方的第2电极活性物质层54上,以Cu作为源,进行电子束蒸镀,形成厚度为3μm的Cu构成的第2集电体55层,制作具有图3构造的锂二次电池。在形成时,使具有1cm见方的孔的不锈钢制的掩膜(厚度20μm)紧贴配置于固体电解质层53上,以第2电极活性物质层54上重叠的状态,将第2集电体55作成1cm见方的形状。此时,用1×10-4Torr的Ar气体氛围,取照射电子束的加速电压及电流分别取10kV和1A。
用上述的实验例中说明的方法为基准测定已得的电池的充放电特性,结果示于表10。按此,在第102~106的实验中的锂二次电池,具有几乎同样高的容量。这被认为是,以式(3)及(4)定义的值(E2=15000eV)极高,不是用简单设备所能实现的。
表10
| 第二电极活物质层54形成条件 | 评价结果 | ||||
| 电子加速电压(V) | 照射电流(A) | 基板温度(℃) | XRD信号 | 容量(mAh/g) | |
| 102 | 200 | 20 | 263 | 有 | 123 |
| 103 | 250 | 20 | 302 | 有 | 126 |
| 104 | 300 | 20 | 322 | 有 | 124 |
| 105 | 350 | 20 | 351 | 有 | 122 |
| 106 | - | - | 100 | 无 | 5 |
此外,在上述实验例中,只将电子束蒸镀法作为主要手段使用,倘若不计能量效率(一般小于1%)等的经济问题,真空发光等的软X线领域的光源也能够在技术上有效使用。
在上述实验例中,作为电极活性物质,使用LiCoO2、LiNiO2、LiMn2O4、Li3-αCoαN、V2O5或Li(Li1/3Ti5/3)O4,如果是要求结晶性的电极活性物质,在本发明可有效使用。同样,作为低融点电极活性物质使用Li,但此外的材料,融点类近的也能够有效使用。
此外,作为照射能量源,可单独用等离子体,电子,或光(紫外线),也可同时使用其中的多个能量源。此外,蒸镀的源材料及氛围也不限于上述实验例。
此外,作为固体电解质,可使用氮化磷酸锂、Li2S-SiS2-Li3PO4、或1.7Li2O-V2O5-SiO2,但本发明不限于此。作为构成集电体的材料,只要是与Li不直接反应的良导体即可,例如,可采用Cu、Ni及不锈钢等的金属。别的金属也可用。
工业应用性
采用本发明,在形成电极活性物质层时,以等离子体、电子或光等形式,以不损坏基板的大小的能量加在形成电极活性物质层的表面,以此可对最靠近表面附近的原子提供再排列用的能量,可不使基板损坏地形成结晶化的电极活性物质层。
此时的温度的上升可通过调整冷却功率密度得到充分控制,可通过冷却功率密度与照射功率密度的平衡进行抑制。这样,可抑制工艺中基板受热的破坏,可抑制固体电解质层的离子传导率的减低,而且可抑制Li等低融点物质的熔融。此外,接触形成各种层的面的氛围中添加氧或氧离子,以此可防止已得层的氧缺损。
Claims (16)
1.一种电化学元件的制造方法,包含:
在第1集电体上形成第1电极活性物质层的第1工序;
在第1电极活性物质层上形成固体电解质层的第2工序;
在固体电解质层上形成第2电极活性物质层的第3工序;
在第2电极活性物质层上形成第2集电体的第4工序,
其中,所述第3工序是在基板上,利用使构成所述第2电极活性物质层的原子、离子或群集向所述固体电解质层的表面蒸镀的制膜工序形成所述第2电极活性物质层,
在所述制膜工序中,对构成所述第2电极活性物质层的原子、离子或群集照射具有满足以下关系式的强度E的能量,其单位为eV:
Ea≤E≤(70±α)×(ρ1d1)1/2,
其中,-7≤α≤7;Ea是第2电极活性物质层的活性化能量,单位为eV;ρ1是第2电极活性物质层的密度,单位为g/cm3;d1是第2电极活性物质层的晶格常数,单位为。
2.一种电化学元件的制造方法,包含:
在第1集电体上形成第1电极活性物质层的第1工序;
在第1电极活性物质层上形成固体电解质层的第2工序;
在固体电解质层上形成第2电极活性物质层的第3工序;
在第2电极活性物质层上形成第2集电体的第4工序,
在第2集电体上形成第3电极活性物质层的第5工序;
在第3电极活性物质层上形成固体电解质层的第6工序;
在固体电解质层上形成第4电极活性物质层的第7工序;
在第4电极活性物质层上形成第3集电体的第8工序,
其中,所述第7工序是在基板上,利用使构成所述第4电极活性物质层的原子、离子或群集向所述固体电解质层的表面蒸镀的制膜工序,形成所述第4电极活性物质层,
在所述制膜工序中,对构成所述第4电极活性物质层的原子、离子或群集照射具有满足以下关系式的强度E的能量,其单位为eV:
Ea≤E≤(70±α)×(ρ1d1)1/2,
其中,-7≤α≤7;Ea是第4电极活性物质层的活性化能量,单位为eV;ρ1是第4电极活性物质层的密度,单位为g/cm3;d1是第4电极活性物质层的晶格常数,单位为。
3.一种电化学元件的制造方法,包含:
在第1集电体上形成第1电极活性物质层的第1工序;
在第1电极活性物质层上形成固体电解质层的第2工序;
在固体电解质层上形成第2电极活性物质层的第3工序;
在第2电极活性物质层上形成第2集电体的第4工序,
其中,所述第2工序是在基板上,利用将构成所述固体电解质层的原子、离子或群集向所述第1电极活性物质层的表面蒸镀的制膜工序,形成所述固体电解质层,
在所述制膜工序中,对构成所述固体电解质层的原子、离子或群集照射具有满足以下关系式的强度E的能量,其单位为eV:
Ea≤E≤(70±α)×(ρ3d3)1/2,
其中,-7≤α≤7;Ea是固体电解质层的活性化能量,单位为eV;ρ3是第2电极活性物质层的密度,单位为g/cm3;d3是第2电极活性物质层的晶格常数,单位为。
4.如权利要求1至3中任一项所述的电化学元件的制造方法,其特征在于,
对所述基板的表面照射电子或电磁波。
5.如权利要求4所述的电化学元件的制造方法,其特征在于,
所述第1电极活性物质层及第2电极活性物质层是结晶态物质,所述固体电解质层是非晶态物质。
6.如权利要求1至3中任一项所述的电化学元件的制造方法,其特征在于,
对所述原子、离子或群集照射电子或电磁波。
7.如权利要求6所述的电化学元件的制造方法,其特征在于,
所述第1电极活性物质层及第2电极活性物质层是结晶态物质,所述固体电解质层是非晶态物质。
8.如权利要求1至3中任一项所述的电化学元件的制造方法,其特征在于,
所述制膜工序中形成的层的结晶温度高于该形成的层以外的层的结晶温度或融点。
9.如权利要求8所述的电化学元件的制造方法,其特征在于,
所述第1电极活性物质层及第2电极活性物质层是结晶态物质,所述固体电解质层是非晶态物质。
10.如权利要求1至3中任一项所述的电化学元件的制造方法,其特征在于,
将所述制膜工序中形成的层以外的层的温度维持在根据其组成预先决定的范围。
11.如权利要求10所述的电化学元件的制造方法,其特征在于,
所述第1电极活性物质层及第2电极活性物质层是结晶态物质,所述固体电解质层是非晶态物质。
12.如权利要求1至3中任一项所述的电化学元件的制造方法,其特征在于,
调整所述能量的强度及所述基板的散热,使所述基板的温度不超过所述制膜工序中形成的层以外的层的结晶温度或融点。
13.如权利要求12所述的电化学元件的制造方法,其特征在于,
所述第1电极活性物质层及第2电极活性物质层是结晶态物质,所述固体电解质层是非晶态物质。
14.如权利要求1至3中任一项所述的电化学元件的制造方法,其特征在于,
构成在所述制膜工序中形成的层的化合物是氧化物或氮化物时,蒸镀氛围分别采用包含氧或氧离子的气体氛围、或包含氮或氮离子的不活泼气体氛围。
15.如权利要求14所述的电化学元件的制造方法,其特征在于,
所述第1电极活性物质层及第2电极活性物质层是结晶态物质,所述固体电解质层是非晶态物质。
16.如权利要求1~3中的任一项所述的电化学元件的制造方法,其特征在于,
所述第1电极活性物质层及第2电极活性物质层是结晶态物质,所述固体电解质层是非晶态物质。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP265510/2001 | 2001-09-03 | ||
| JP2001265510 | 2001-09-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1498436A CN1498436A (zh) | 2004-05-19 |
| CN1293664C true CN1293664C (zh) | 2007-01-03 |
Family
ID=19091964
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB028045327A Expired - Lifetime CN1293664C (zh) | 2001-09-03 | 2002-09-02 | 电化学元件的制造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US7425223B2 (zh) |
| EP (1) | EP1359636A1 (zh) |
| JP (1) | JP4249616B2 (zh) |
| CN (1) | CN1293664C (zh) |
| WO (1) | WO2003021706A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109402562A (zh) * | 2014-01-08 | 2019-03-01 | 爱利卡技术有限公司 | 用于制造含锂薄膜层状结构的气相沉积方法 |
Families Citing this family (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8404376B2 (en) | 2002-08-09 | 2013-03-26 | Infinite Power Solutions, Inc. | Metal film encapsulation |
| US9793523B2 (en) | 2002-08-09 | 2017-10-17 | Sapurast Research Llc | Electrochemical apparatus with barrier layer protected substrate |
| US8236443B2 (en) | 2002-08-09 | 2012-08-07 | Infinite Power Solutions, Inc. | Metal film encapsulation |
| US20070264564A1 (en) | 2006-03-16 | 2007-11-15 | Infinite Power Solutions, Inc. | Thin film battery on an integrated circuit or circuit board and method thereof |
| US8394522B2 (en) | 2002-08-09 | 2013-03-12 | Infinite Power Solutions, Inc. | Robust metal film encapsulation |
| US8431264B2 (en) | 2002-08-09 | 2013-04-30 | Infinite Power Solutions, Inc. | Hybrid thin-film battery |
| US8445130B2 (en) | 2002-08-09 | 2013-05-21 | Infinite Power Solutions, Inc. | Hybrid thin-film battery |
| US8021778B2 (en) | 2002-08-09 | 2011-09-20 | Infinite Power Solutions, Inc. | Electrochemical apparatus with barrier layer protected substrate |
| JP4777593B2 (ja) * | 2002-11-29 | 2011-09-21 | 株式会社オハラ | リチウムイオン二次電池の製造方法 |
| US8728285B2 (en) | 2003-05-23 | 2014-05-20 | Demaray, Llc | Transparent conductive oxides |
| US7645373B2 (en) | 2003-06-20 | 2010-01-12 | Roche Diagnostic Operations, Inc. | System and method for coding information on a biosensor test strip |
| US8206565B2 (en) | 2003-06-20 | 2012-06-26 | Roche Diagnostics Operation, Inc. | System and method for coding information on a biosensor test strip |
| US8148164B2 (en) | 2003-06-20 | 2012-04-03 | Roche Diagnostics Operations, Inc. | System and method for determining the concentration of an analyte in a sample fluid |
| US7718439B2 (en) | 2003-06-20 | 2010-05-18 | Roche Diagnostics Operations, Inc. | System and method for coding information on a biosensor test strip |
| US7645421B2 (en) | 2003-06-20 | 2010-01-12 | Roche Diagnostics Operations, Inc. | System and method for coding information on a biosensor test strip |
| US8058077B2 (en) | 2003-06-20 | 2011-11-15 | Roche Diagnostics Operations, Inc. | Method for coding information on a biosensor test strip |
| US7569126B2 (en) | 2004-06-18 | 2009-08-04 | Roche Diagnostics Operations, Inc. | System and method for quality assurance of a biosensor test strip |
| KR101127370B1 (ko) | 2004-12-08 | 2012-03-29 | 인피니트 파워 솔루션스, 인크. | LiCoO2의 증착 |
| US7959769B2 (en) | 2004-12-08 | 2011-06-14 | Infinite Power Solutions, Inc. | Deposition of LiCoO2 |
| JP2007273349A (ja) * | 2006-03-31 | 2007-10-18 | Toyota Motor Corp | 積層型電池およびその製造方法 |
| WO2008039471A2 (en) | 2006-09-29 | 2008-04-03 | Infinite Power Solutions, Inc. | Masking of and material constraint for depositing battery layers on flexible substrates |
| US8197781B2 (en) * | 2006-11-07 | 2012-06-12 | Infinite Power Solutions, Inc. | Sputtering target of Li3PO4 and method for producing same |
| JP5239209B2 (ja) * | 2007-05-11 | 2013-07-17 | 住友電気工業株式会社 | リチウム電池用正極の製造方法 |
| US8268488B2 (en) | 2007-12-21 | 2012-09-18 | Infinite Power Solutions, Inc. | Thin film electrolyte for thin film batteries |
| EP2225406A4 (en) | 2007-12-21 | 2012-12-05 | Infinite Power Solutions Inc | PROCEDURE FOR SPUTTER TARGETS FOR ELECTROLYTE FILMS |
| CN101911367B (zh) | 2008-01-11 | 2015-02-25 | 无穷动力解决方案股份有限公司 | 用于薄膜电池及其他器件的薄膜包封 |
| JP5595377B2 (ja) | 2008-04-02 | 2014-09-24 | インフィニット パワー ソリューションズ, インコーポレイテッド | エネルギー取入れに関連したエネルギー貯蔵デバイスに対する受動的過不足電圧の制御および保護 |
| CN102119454B (zh) | 2008-08-11 | 2014-07-30 | 无穷动力解决方案股份有限公司 | 具有用于电磁能量收集的一体收集器表面的能量设备及其方法 |
| EP2332127A4 (en) | 2008-09-12 | 2011-11-09 | Infinite Power Solutions Inc | ENERGY DEVICE HAVING AN INTEGRATED CONDUCTIVE SURFACE FOR DATA COMMUNICATION VIA ELECTROMAGNETIC ENERGY AND ASSOCIATED METHOD |
| WO2010042594A1 (en) | 2008-10-08 | 2010-04-15 | Infinite Power Solutions, Inc. | Environmentally-powered wireless sensor module |
| EP2462600A2 (en) * | 2009-08-07 | 2012-06-13 | OC Oerlikon Balzers AG | All solid-state electrochemical double layer supercapacitor |
| KR101792287B1 (ko) | 2009-09-01 | 2017-10-31 | 사푸라스트 리써치 엘엘씨 | 집적된 박막 배터리를 갖는 인쇄 회로 보드 |
| JP2011142037A (ja) * | 2010-01-08 | 2011-07-21 | Sumitomo Electric Ind Ltd | 非水電解質電池の製造方法および非水電解質電池 |
| JP5692221B2 (ja) * | 2010-04-13 | 2015-04-01 | トヨタ自動車株式会社 | 固体電解質材料、リチウム電池および固体電解質材料の製造方法 |
| CN102859779B (zh) * | 2010-04-13 | 2016-10-19 | 丰田自动车株式会社 | 固体电解质材料、锂电池以及固体电解质材料的制造方法 |
| US8945779B2 (en) * | 2010-04-13 | 2015-02-03 | Toyota Jidosha Kabushiki Kaisha | Solid electrolyte material, lithium battery, and method of producing solid electrolyte material |
| JP2013528912A (ja) | 2010-06-07 | 2013-07-11 | インフィニット パワー ソリューションズ, インコーポレイテッド | 再充電可能高密度電気化学素子 |
| JP5431257B2 (ja) * | 2010-06-30 | 2014-03-05 | ラサ工業株式会社 | 固体電解質薄膜の作製方法 |
| FR2963026B1 (fr) * | 2010-07-23 | 2013-03-15 | Univ Paul Verlaine Metz | Paroi de separation d'electrolytes pour le transfert selectif de cations a travers la paroi, procede de fabrication et procede de transfert. |
| GB2493022B (en) * | 2011-07-21 | 2014-04-23 | Ilika Technologies Ltd | Vapour deposition process for the preparation of a phosphate compound |
| US9070950B2 (en) * | 2012-03-26 | 2015-06-30 | Semiconductor Energy Laboratory Co., Ltd. | Power storage element, manufacturing method thereof, and power storage device |
| JP6170657B2 (ja) * | 2012-08-29 | 2017-07-26 | 株式会社アルバック | 薄膜リチウム二次電池製造方法、マスク、薄膜リチウム二次電池製造装置 |
| KR20140053451A (ko) * | 2012-10-25 | 2014-05-08 | 삼성에스디아이 주식회사 | 복합양극활물질, 그 제조방법 및 이를 채용한 양극과 리튬전지 |
| US9461341B2 (en) * | 2012-12-26 | 2016-10-04 | Semiconductor Energy Laboratory Co., Ltd. | Power storage device and method for charging the same |
| GB201400277D0 (en) * | 2014-01-08 | 2014-02-26 | Ilika Technologies Ltd | Vapour deposition method for preparing crystalline lithium-containing compounds |
| GB201400274D0 (en) * | 2014-01-08 | 2014-02-26 | Ilika Technologies Ltd | Vapour deposition method for preparing amorphous lithium-containing compounds |
| JP7071934B2 (ja) * | 2016-06-15 | 2022-05-19 | イリカ テクノロジーズ リミテッド | 電解質および電極保護層としてのホウケイ酸リチウムガラス |
| US10581109B2 (en) * | 2017-03-30 | 2020-03-03 | International Business Machines Corporation | Fabrication method of all solid-state thin-film battery |
| US10622680B2 (en) | 2017-04-06 | 2020-04-14 | International Business Machines Corporation | High charge rate, large capacity, solid-state battery |
| TWI634221B (zh) * | 2017-09-01 | 2018-09-01 | 行政院原子能委員會核能硏究所 | 電化學元件之製造方法 |
| WO2019164588A2 (en) * | 2018-01-05 | 2019-08-29 | University Of Maryland, College Park | Multi-layer solid-state devices and methods for forming the same |
| US11056722B2 (en) | 2018-02-08 | 2021-07-06 | International Business Machines Corporation | Tool and method of fabricating a self-aligned solid state thin film battery |
| US10679853B2 (en) | 2018-02-08 | 2020-06-09 | International Business Machines Corporation | Self-aligned, over etched hard mask fabrication method and structure |
| US10720670B2 (en) * | 2018-02-08 | 2020-07-21 | International Business Machines Corporation | Self-aligned 3D solid state thin film battery |
| GB201814039D0 (en) | 2018-08-29 | 2018-10-10 | Ilika Tech Ltd | Method |
| KR102601450B1 (ko) * | 2021-06-29 | 2023-11-13 | 충남대학교산학협력단 | 전고체 리튬이온전지의 제조방법 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5960814A (ja) * | 1982-09-29 | 1984-04-06 | 株式会社日立製作所 | 酸化リチウム系非晶質イオン導電体 |
| US5338625A (en) * | 1992-07-29 | 1994-08-16 | Martin Marietta Energy Systems, Inc. | Thin film battery and method for making same |
| US5314765A (en) * | 1993-10-14 | 1994-05-24 | Martin Marietta Energy Systems, Inc. | Protective lithium ion conducting ceramic coating for lithium metal anodes and associate method |
| JPH0845338A (ja) * | 1994-07-28 | 1996-02-16 | Mitsubishi Heavy Ind Ltd | 高分子固体電解質薄膜の製造方法 |
| JPH08236105A (ja) | 1995-02-28 | 1996-09-13 | Nissin Electric Co Ltd | リチウム二次電池正極の製造方法 |
| JPH08287901A (ja) | 1995-04-19 | 1996-11-01 | Nissin Electric Co Ltd | リチウム二次電池正極の製造方法 |
| JP3116857B2 (ja) * | 1997-04-04 | 2000-12-11 | 日本電気株式会社 | 半導体基板搭載型二次電池 |
| KR100378004B1 (ko) * | 1997-06-10 | 2003-06-09 | 삼성에스디아이 주식회사 | 유리-고분자복합전해질및그제조방법 |
| US6094292A (en) * | 1997-10-15 | 2000-07-25 | Trustees Of Tufts College | Electrochromic window with high reflectivity modulation |
| AU5095601A (en) * | 2000-03-24 | 2001-10-08 | Cymbet Corp | Thin-film battery having ultra-thin electrolyte and associated method |
-
2002
- 2002-09-02 EP EP02762971A patent/EP1359636A1/en not_active Withdrawn
- 2002-09-02 CN CNB028045327A patent/CN1293664C/zh not_active Expired - Lifetime
- 2002-09-02 US US10/432,500 patent/US7425223B2/en active Active
- 2002-09-02 JP JP2003525934A patent/JP4249616B2/ja not_active Expired - Lifetime
- 2002-09-02 WO PCT/JP2002/008897 patent/WO2003021706A1/ja not_active Application Discontinuation
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109402562A (zh) * | 2014-01-08 | 2019-03-01 | 爱利卡技术有限公司 | 用于制造含锂薄膜层状结构的气相沉积方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4249616B2 (ja) | 2009-04-02 |
| US20040058237A1 (en) | 2004-03-25 |
| JPWO2003021706A1 (ja) | 2004-12-24 |
| CN1498436A (zh) | 2004-05-19 |
| US7425223B2 (en) | 2008-09-16 |
| WO2003021706A1 (fr) | 2003-03-13 |
| EP1359636A1 (en) | 2003-11-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1293664C (zh) | 电化学元件的制造方法 | |
| CN1222064C (zh) | 锂电池和可再充电锂电池中用的电极 | |
| CN1257569C (zh) | 可再充电锂电池的电极和可再充电锂电池 | |
| CN1257568C (zh) | 可再充电锂电池的电极和可再充电锂电池 | |
| CN1266787C (zh) | 二次电池及其制造方法 | |
| CN1257567C (zh) | 锂电池和可再充电锂电池中用的电极 | |
| CN1761086A (zh) | 用于锂离子二次电池的负极、其制造方法以及包含所述负极的锂离子二次电池 | |
| CN1310357C (zh) | 锂蓄电池及其正极活性物质、正极板及它们的制造方法 | |
| CN1243384C (zh) | 生产阴极活性材料的方法和生产非水性电解质电池的方法 | |
| CN1199302C (zh) | 阴极活性材料及其制备方法,非水电解质电池及其制备方法 | |
| CN1787253A (zh) | 用于锂离子二次电池的负电极、其制造方法及含有该负电极的锂离子二次电池 | |
| CN1223030C (zh) | 正极活性材料和非水电解质电池 | |
| CN1127775C (zh) | 非水电解液二次电池 | |
| CN1300869C (zh) | 非水电解液二次电池用正极活性物质及其电池 | |
| CN1160483C (zh) | 薄膜制备方法和淀积设备 | |
| CN1870327A (zh) | 用于锂离子二次电池的负极及该负极的生产方法 | |
| CN101075670A (zh) | 负极活性材料和电池 | |
| CN1658415A (zh) | 正极活性材料和无水电解质二次电池 | |
| CN1770512A (zh) | 负极活性材料和使用该负极活性材料的电池 | |
| CN1897332A (zh) | 非水电解质二次电池 | |
| CN101047269A (zh) | 电池 | |
| CN1703370A (zh) | 制造用作阴极活性材料的锂金属化合物的方法 | |
| CN1537338A (zh) | 二次电池用负极以及使用其的二次电池、和负极的制造方法 | |
| CN1300449A (zh) | 制备正极活性材料的方法以及制备非水电解质二次电池的方法 | |
| CN101040401A (zh) | 锂离子二次电池及其固体电解质 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CX01 | Expiry of patent term |
Granted publication date: 20070103 |
|
| CX01 | Expiry of patent term |