CN1271070C - 用作腺苷A2α受体的配体的三唑基-咪唑并吡啶和三唑基嘌呤类的衍生物及其作为药物的应用 - Google Patents
用作腺苷A2α受体的配体的三唑基-咪唑并吡啶和三唑基嘌呤类的衍生物及其作为药物的应用 Download PDFInfo
- Publication number
- CN1271070C CN1271070C CNB028138481A CN02813848A CN1271070C CN 1271070 C CN1271070 C CN 1271070C CN B028138481 A CNB028138481 A CN B028138481A CN 02813848 A CN02813848 A CN 02813848A CN 1271070 C CN1271070 C CN 1271070C
- Authority
- CN
- China
- Prior art keywords
- compound
- application
- medicine
- treatment
- triazole
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid, pantothenic acid
- A61K31/198—Alpha-aminoacids, e.g. alanine, edetic acids [EDTA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
Abstract
本发明公开了通式(I)的化合物,其中的基团如本说明书中所定义,所述的化合物是腺苷A2a受体拮抗剂且可用作药物,特别用于治疗帕金森病。
Description
本发明涉及用作腺苷A2a受体的配体的三唑基-咪唑并吡啶和三唑基嘌呤类的衍生物、其制备方法、其作为药物的应用,特别是作为用于治疗得益于这种受体受到抑制的病理情况的药物的应用,并涉及包括这些化合物的药物组合物。
发明背景
目前用于帕金森病的疗法限于缓解症状,而尚未鉴定能够对抗与依次导致这种病理情况的复杂症状出现的基底神经节多巴胺水平不足相关的黑质多巴胺能神经元变性的建立和进展的活性剂。这种病理情况的特征在于:强直、震颤、运动徐缓、运动不能、姿式改变;代表对患有帕金森病个体的健康的严重威胁的表现。
在目前用于改善这些受治疗者生命质量的治疗策略中有目的在于重新补充缺失的神经递质的治疗手段。一个实例以通过使用左旋多巴与卡比多巴或苄丝肼(外周氨基酸脱羧酶抑制剂)相联合为代表。这种疗法是最有效的治疗之一且目前用于对抗在多巴胺能系统生理功能受到严重损害时表现出的运动功能改变。
然而,这种治疗手段在长期应用中遇到了功效下降的问题。实际上,由于左旋多巴本身具有神经毒性,所以用左旋多巴长期治疗的患者除出现其它副作用外还通常显示出所述表现的强化(emphasising)。
另一方面,虽然已经引入了应用多巴胺能受体兴奋剂,但是它们不表现出与左旋多巴相同的功效;或应用单胺氧化酶抑制剂和毒蕈碱性乙酰胆碱受体拮抗剂。后者的应用因这些产品在全身水平与中枢神经系统水平上建立了受体相互作用而出现了严重的副作用和认知损伤。
近年来,随着发现腺苷作为神经递质的作用、其受体及其功能表征,使用腺苷A2a受体拮抗剂作为治疗与帕金森病相关的运动障碍的治疗剂的假设已经获得了信赖。(P.J.Richardson,H.Kase和P.G.Jenner《药物科学趋势》(Trends Pharm.Sci)1997,18:338-345)。
近期实验证据已经使这种受体在中枢神经系统水平上的分布、功能和生理学得到了理解,从而可以得出这样的结论:阻断A2a受体可以调节胆碱能、γ-氨基丁酸能和谷氨酸能(glutamatergic)神经传递以便在基底神经节输出神经元水平上建立足以补偿黑质纹状体系统中急性或慢性多巴胺缺乏的神经化学指令。
此外,已经观察到A2a受体在功能上与多巴胺D2相关且刺激前者可以降低后者与多巴胺的结合能力。因此推定,阻断A2a腺苷受体增加D2受体对多巴胺的相互作用能力,甚至在突触间隙中低水平这种神经递质的存在下有利于结合。(Ferre S.等(1991)《美国国家科学院学报》(Proc.Nat.Acc.Sci.U.S.A.)88,7237-7241)。由于存在这些原因,所以已经提出将A2a受体选择性拮抗剂作为治疗运动障碍、特别是针对帕金森病的活性剂。
此外,已经证实这些活性剂可以提供与用左旋多巴或用多巴胺兴奋剂治疗的协同作用且可以用于与多巴胺替代疗法联用。在这种情况中,受体A2a选择性拮抗剂的应用代表了进一步的治疗优势,因为一般左旋多巴疗法所需的剂量可以在数量上降低或给药频率下降而保持治疗功效。
本发明涉及对腺苷A2a受体具有亲和力的、有拮抗剂活性的、用于制备治疗个体运动障碍的药物的化合物,所述的运动障碍与基底神经节功能改变相关而形成诸如帕金森病、阿尔茨海默病、亨廷顿舞蹈病和威尔逊病这样的疾病、因药物(传统安定药的帕金森神经功能障碍)、创伤、毒性剂(NOTP、锰,一氧化碳)带来的症状的组成部分。
本发明还可以用于治疗具有″开-关″现象的帕金森病和运动障碍占优势的帕金森病。
此外,已经证实在中枢神经系统缺血性损害后,腺苷A2a受体拮抗剂可以抑制这类现象后大量释放的兴奋性氨基酸诱发的毒性作用且脑缺血和与神经变性过程相关的机制代表了A2a受体拮抗剂可以发挥其治疗作用的其它″靶″。
腺苷A2a受体选择性拮抗剂描述在Boehringer Ingelheim的CA1.242.368中,其形式为咪唑并-三唑-嘧啶衍生物,其中它们对A1受体的活性超过对A2受体的活性;在Vernalis Res Ltd的WO 01/02409中,将噻吩并-和呋喃并嘧啶的衍生物描述为用于治疗运动障碍、例如帕金森病的化合物;Fusijawa Pharm Co Ltd的WO 00/24742中描述了对A1和A2受体具有双重拮抗作用的吡唑并吡啶衍生物Kyowa HakkoKKK的WO 00/17201和WO 98/42711中描述了1,2,4-三唑并-(1,5-c)嘧啶衍生物;Cerebrus Pharm Ltd的WO 00/13682、WO 00/13681和WO 99/26627中描述了4-喹啉甲醇衍生物;Cadus Pharm Corp的WO99/62518中描述了具有为A1、A2、A2a、A2b、A3受体拮抗剂的活性特征的N-6取代的7-去氮杂嘌呤;KyoWa Hakko KKK的WO 99/43678中描述了1,2,4-三唑并-(1,5-a)-嘧啶衍生物;Schering Plough SpA的WO 95/01356和WO 98/52568中描述了1,2,4-三唑并-(1,5-c)-嘧啶。
在处于进展测试期中的A2a受体拮抗剂中,我们可以提及EP 0 628311中所述的化合物KW-6002和KW-1783,其可表征为黄嘌呤衍生物。这些产品在8位上具有(3,4-二甲氧基苯基)烯基且通过烯键的光异构化失去活性(Annals New York Academy of Sciences-Ongini E.;Adami M;Ferri C;和Bertorelli R.;《药物科学趋势》(Trends inPharm.Sci.)1996第17卷364-72)。化合物KW6002目前正在进行临床试验(作为抗抑郁药的II期和作为抗帕金森病药的III期临床试验;Pharmaprojects Acc.No.23891)。另一种选择性A2a受体拮抗剂是处于临床前期试验中的产品SCH 63390(Pharmaproject Acc.No29842)。来自Astra Zeneca的ZM 241385(EP 0 459 702)是有效的选择性拮抗剂;由于这一原因而用于药理研究(Ji X.D.,Jacobson K.A.,《药物设计发现》(Drug Des.Discov.)1999,16:217-226;Pharmaproject Acc.No22730)。尽管该产品具有高选择性和亲和性,但是已经证实它在体内的生物利用度极低(El Yacouibi M.等《欧洲药物杂志》(Eur.J.Pharm.)401(2000)63-77)。
发明概述
已经发现了下列通式的化合物及其药物上可接受的盐对A2a受体具有亲和性:
其中:X是N、CH、C-R2;
R1是C1-C6直链或支链、饱和或不饱和烷基;
R2是氢、C1-C6直链或支链饱和或不饱和烷基、C6-C14芳基或C6-C14芳基(C1-C6)直链或支链饱和或不饱和烷基,其中所述的芳基任选被一个或多个相同或不同的取代基取代,这些取代基选自卤素、羟基、C1-C6直链或支链、饱和或不饱和烷氧基、氨基、一-或二-C1-C6直链或支链烷基组成的组;
R3是NH2、NHR4;
R4是C1-C6烷基或C1-C6羟基烷基、C1-C3烷氧基烷基、氨基(C1-C6)烷基,其中所述的氨基任选被一个或两个C1-C3烷基取代,所述的烷基是直链或支链、饱和或不饱和的;C6-C14芳基或C6-C14芳基(C1-C6)烷基,其中所述的芳基任选被一个或多个相同或不同的取代基修饰,这些取代基选自卤素、羟基、C1-C6直链或支链饱和或不饱和烷氧基、氨基,被C1-C6直链或支链饱和或不饱和烷基一-或二-取代。
因此,本发明的目的是上述定义的通式(I)化合物及其药物上可接受的盐。
本发明的另一个目的是上述定义的通式(I)化合物的制备方法、其作为药物特别在制备对A2a腺苷受体具有抑制活性也有选择性的药物中的应用,这类药物用于治疗对抑制腺苷A2a受体有反应的病理情况,诸如治疗运动障碍、阿尔茨海默病、亨廷顿舞蹈病、威尔逊病和帕金森病。本发明的化合物还用于制备用于治疗脑缺血和/或与神经变性过程相关的机制的药物。
本发明的其它目的是含有至少一种通式(I)化合物作为活性组分的药物组合物。
还通过附图和实施例来更具体地解释本发明的这一目的和其它目的,其中:
-附图1显示对诱发小鼠僵住症的能力的评价;
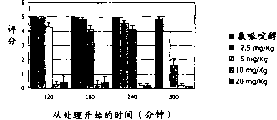
-附图2显示本发明示例化合物对CGS 21680--诱发的僵住症的作用。每一柱代表每组10只动物的僵住症评分平均值±s.e.;
-附图3显示本发明示例化合物对氟哌啶醇诱发的小鼠僵住症的作用。每一柱代表每组10只动物的僵住症评分平均值±s.e.;
-附图4显示本发明示例化合物与阈下剂量的左旋多巴+苄丝肼(分别为12.5mg/kg和6.25mg/kg腹膜内给药)对氟哌啶醇-诱发的小鼠僵住症的联合作用;
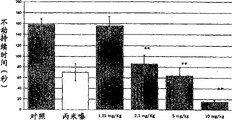
-附图5显示本发明示例化合物在小鼠强迫游泳试验中的作用。在本试验前60分钟给小鼠注射载体或测试化合物或丙米嗪。在4分钟测试期间记录不动的持续时间。表示的数据为每组10只小鼠的平均值±s.e.。相对于对照组,ANOVA和Tukey检验**=p<0.01。
发明详述
在通式(I)的化合物中,C1-C6饱和或不饱和烷基的实例是甲基、乙基、丙基、异丙基、丁基、仲丁基、叔丁基、戊基、己基、乙烯、丙烯、丁烯。链烯基和链炔基可以含有高达最大可能的不饱和度,且烷基、链烯基和链炔基可以由所有理论上可能的异构体表示。在通式(I)的化合物中,C6-C14芳基或C6-C14芳基(C1-C6)烷基其中芳基任选被取代的实例是:苯基、萘基和蒽基,在不同键位置上(例如1-或2-萘基)、苄基、苯乙基、苯基丙基、苯基丁基、苯基戊基、苯基己基,含有萘基和蒽基的芳基烷基类似物,被上述基团取代的2-、3-或4-苯基,例如2-、3-或4-羟基苯基、2-、3-或4-烷氧基苯基,其中烷基残基如上所述;2-、3-或4-卤代苯基,其中卤素是氟、氯、溴、碘;2-、3-或4-氨基苯基,其中氨基可以被如上所述的烷基单取代或二取代。本领域技术人员能够容易地表征对上述定义的通式(I)所预计的所有可能的化合物,作出对不同基团给出的定义的适宜取代。
通式(I)化合物的药物上可接受的盐是与能够使存在的碱性中心成盐且不具有任何毒性或其它不希望作用的有机或无机酸形成的所有盐。
在通式(I)的化合物中,第一优选的组包括那些X是氮且R2是2位上的丁基的化合物。
第二优选的组由那些X是氮且R2是2位上的苯乙基的化合物组成。
第三优选的组由那些X是氮且R2是2位上的戊基的化合物组成。
第四优选的组由那些X是碳且R2是6或7位上的氢的化合物组成。
特别优选下列化合物:
-6-氨基-2,9-二甲基-8-(三唑-2-基)-9(H)-嘌呤(ST 1491);
-2-丁基-9-甲基-8-(2H-1,2,3-三唑-2-基)-9H-嘌呤-6-基胺(ST 1535);
-9-甲基-2-(2-苯乙基)-8-(2H-1,2,3-三唑-2-基)-9H-嘌呤-6-基胺(ST1537);
-9-甲基-2-戊基-8-(2H-1,2,3-三唑-2-基)-9H-嘌呤-6-基胺(ST 2097)。
作为本发明的特定情况,化合物6-氨基-9-甲基-8-(三唑-2-基)-9(H)-嘌呤(ST 1490)显示出对A1腺苷受体的亲和性,由此用于制备治疗认知缺损、阿尔茨海默病、脑缺血、急性和慢性肾衰、因放射造影剂或顺铂诱发的肾衰的药物。
可以按照以下流程图中所述的合成手段制备通式(I)的化合物。
对可通过文献中已知的方法得到的化合物a)进行2位上的溴-取代,然后使溴被三唑-2-基替代。
下面的流程图1A、1Abis和2A仅通过实例显示简单命名为ST1491、ST 1536、ST 1535、ST 1537、ST 2097和ST 1680的化合物的制备过程。
反应流程1A

反应流程1°bis

(7a)R=CH3 ST 1491
(7b)R=CH(CH3)2 ST 1536
(7c)R=(CH2)3CH3ST 1535
(7d)R=CH2CH2Ph ST 1537
(7e)R=(CH2)4CH3ST 2097
反应流程2
通过文献中已知的合成步骤获得由序号(2)-(7)(流程图1A和1Abis)表示的化合物,化合物(1)可商购;化合物(8)、(9)和(10)描述在下列文献中:《杂环》(Heterocycles)1999,721-726;《药物化学杂志》(J.Med.Chem.)32,11,1989,2474-2485;《杂环化学杂志》(J.Heter.Chem.)23,3,1986,669-672;《化学协会杂志》(J.Chem.S0c.)1955,2755-2758;《药物化学杂志》(J.Med.Chem.)39,2,1996,487-493;《杂环化学杂志》(J.Heter.Chem.)27,3,1990,563-566;(11)和(12)描述在EP 0 082 369中,而本发明提供了新的制备方法。分子(13)-(14)是新的,由此特别要求保护它们作为本发明所述方法中的中间体。本领域技术人员能够根据一般知识和文献制备不同于上述流程图中例示的这些化合物的其它通式(I)的化合物。
下列实施例进一步解释本发明。
实施例1
(反应流程1A)
2-氯-6-二苄氨基-9(H)嘌呤(2)
向1g 2,6-二氯嘌呤(1)(97%,5.13mmol)溶于30ml无水乙醇所得到的溶液中加入二异丙基乙胺(1ml,5.13mmol)和蒸馏二苄胺(1.1ml,5.13mmoD。将该反应混合物保持回流20小时(1小时后形成白色沉淀)。然后在低压下除去溶剂并将残余物溶于水。
冷却并过滤后在真空中干燥固体残余物(2)。
产率:95%
Rf=0.25(环己烷/乙酸乙酯)7∶3
M.p.:250-252℃.
1H-NMR(200MHz,CDCl3):δ7.89(s,1H),7.32(s,10H),5.55(bs,2H),5.49(bs,2H).
MS(m/z):91;258-260(BP,M-苄基),349-351(<5%,M)。
2-氯-6-二苄氨基-9-甲基-嘌呤(3)
向(2)溶于热DMF所得到的溶液中加入828mg的K2CO3(6mmol)。然后将该溶液冷却并用0.46ml的CH3I(7.2mmol)处理,同时搅拌约12小时。蒸发DMF,将产物溶于水并过滤且使得到的残余物在乙醇中结晶而得到1.45g产物(3)。
产率:78%。
Rf=0.38(环己烷/乙酸乙酯)7∶3。
M.p.:144-146℃。
1H-NMR(200MHz,CDCl3):δ7.67(s,1H,H8-嘌呤);7.31(s,10H,芳族);5.5(br,2H,CH2-苄化);4.93(br,2H,CH2-苄化);3.81(s,3H,CH3)。
MS(m/z):91(BP,苄基);272-274(65%-20%,M-苄基),363-365(<5%,M)。
2-烷基-6-二苄氨基-9-甲基-9(H)-嘌呤(4)、(4c)、(4e)、(5)
(一般步骤)在充氮的烧瓶中放入700mg的2-氯-6-二苄氨基-9-甲基-9(H)-嘌呤(3)(1.93mmol)、4ml的NMP(N-甲基吡咯烷酮)、3.8mmol的烷基三丁基锡和140mg的Pd(PPh3)4。对于化合物4、4c和4e,将以上物质在120℃下搅拌8小时,而对于化合物5则搅拌2小时。将它们冷却并用水(50ml)和二氯甲烷(50ml)稀释且最终用二氯甲烷(4×50ml)提取水相。用盐水洗涤合并的有机相、用无水硫酸钠干燥并蒸发溶剂而得到深色液体。在硅胶柱上纯化产物(洗脱剂:EtOAc/环己烷1/1)而得到固体或黄色油状物形式的(4)、(4c)、(4e)和(5)。
(4):产率:63%。
M.p.:143℃
MS(m/z):278(100%,M-苄基);91(55%,苄基)。
(4c):产率:90%。
M.p.:不可测定-橡胶样物质。
MS(m/z):294(100%,M-苄基);91(65%,苄基)。
1H-NMR:200MHz,CDCl3;δ7.65(1H,s,嘌呤);7.30(10H,m,芳族);5.27(4H,br,-CH2-苄化);3.82(3H,s,N-CH3);2.88(2H,t,-CH2-CH2-CH2-CH3);1.80(2H,m,-CH2-CH2-CH2-CH3);1.37(2H,t,-CH2-CH2-CH2-CH3);0.91(3H,t,-CH2-CH2-CH2-CH3)。
(4e)产率:65%。
6-二苄氨基-9-甲基-2-戊基-9(H)-嘌呤
M.p.:不可测定-橡胶样物质。
MS:m/z=399,308,220
1H-NMR(200MHz,CDCl3)δ(ppm):7.71(s,1H),7.30(m,10H),5.30(bs,4H),3.80(s,3H),3.38(t,2H),2.38(m,2H),2.02(m,2H),1.30(m,2H),0.88(m,3H)
(5):产率:84%。
M.p.:不可测定-橡胶样物质。
MS(m/z):340(100%,M-苄基);91(70%,苄基)。
6-二苄氨基-2,9-二甲基-9(H)-嘌呤(4a)
在惰性气体环境(氮)中向冷却的三颈圆底烧瓶内放入1.07g溶于30ml无水THF中的(3)(2.94mmol)、6ml三甲基铝的2M甲苯溶液(12mmol)、27mg的PdCl2(0.15mmol)和79mg的PPh3(0.3mmol)。通过回流48小时使它们进行反应。终止该反应,将该混合物倾入烧杯、在冰浴中冷却并通过添加少量水和乙醇破坏过量的三烷基铝。通过滤纸过滤氢氧化铝沉淀并用二氯甲烷提取该混合物。在减压条件下蒸发有机相后,通过急骤层析法纯化残余物(SiO2,环己烷/EtOAc 1∶1)。得到900mg结晶形式的(4a)(2.62mmol)。
产率89%。
M.p.:117-118℃。
MS(m/z):91(80%,苄基),252(BP M-苄基),343(<5%,M)。
1H-NMR:200MHz,CDCl3 δ7.65(s,1H,H8·嘌呤);7.30(s,10H,芳族);5.30(br,4H,CH2-苄化);3.81(s,3H,N-CH3);2.62(s,3H,C-CH3)。
2-异丙基-6-二苄氨基-9-甲基-9(H)-嘌呤(4b)和2-(2-苯乙基)-6-二苄氨
基-9-甲基-9(H)-嘌呤(4d)
将100mg的(4)或(5)与5ml乙醇一起置于压热器中、加热至完全溶解且然后加入50mg的钯/石墨支持物。将该体系在4个大气压的氢气下保持搅拌过夜。通过C盐过滤催化剂并在减压条件下蒸发溶剂而得到(4b)或(4d)、为白色固体。
(4b):定量产率。
M.p.:82℃。
MS m/z:280(100%,M-苄基);91(50%,苄基)。
(4d):定量产率。
M.p.:144℃。
MS m/z:342(100%,M-苄基);91(100%,苄基)。
(反应流程1A bis)
(5a、b、c、d、e)
一般步骤
在反应烧瓶中将1.6mmol的(4a)、(4b)、(4c)、(4d)和(4e)溶于7mlMeOH、7ml THF和7ml pH=4的乙酸盐缓冲液(通过将4g乙酸钠溶于100ml水并用冰醋酸调节至pH4获得)的混合物。极缓慢地滴加0.7ml溴(13.6mmol)并在搅拌条件下将该混合物置于室温直至原料产品消失为止(约12小时)。用偏亚硫酸氢钠使过量的溴脱色并用饱和Na2CO3溶液将该反应体系碱化至pH=8。用二氯甲烷提取并在减压条件下蒸发溶剂后得到1.2g黄色油状物(5a)、(5b)、(5c)、(5d)、(5e),随后用制备型色谱柱进行纯化。
(5a):定量产率。
MS m/z:91(100%,苄基);330-332(双峰,70%,M-苄基)。
(5b):定量产率。
MS(m/z):91(100%,苄基);358-360(双峰,70%,M-苄基)。
(5c):定量产率。
MS m/z:91(100%,苄基);372-374(双峰,70%,M-苄基)。
(5d):定量产率。
MS(m/z):91(100%,苄基);420-422(双峰,45%,M-苄基)。
(5e)9-溴-6-二苄氨基-9-甲基-2-戊基-9(H)-嘌呤
M.p.:97℃
MS:m/z=479-477,388-386
1H-NMR(200MHz,CDCl3)δ(ppm):7.28(m,10H),5.16(bs,4H),3.75(s,3H),2.79(t,2H),1.79(m,2H),1.29(m,4H),0.86(m,3H)
2-烷基-6-二苄氨基-9-甲基-8-(三唑-2-基)-9(H)-嘌呤(6a、h、c、d、e)
在惰性气体环境中向反应烧瓶中加入2ml无水DMF、92mg的NaH(80%的石蜡溶液,2.5mmol)并缓慢加入0.18ml的1,2,3-三唑(2.5mmol)且在搅拌条件下保持约1小时。缓慢滴加(5a)、(5b)、(5c)、(5d)或(5e)粗品(1.7mmol)溶于5ml无水DMF所得到的溶液并在100℃下保持搅拌12小时。蒸发DMF并通过急骤层析法纯化残余物(SiO2,环己烷/EtOAc 7∶3)而得到(6a)、(6b)、(6c)、(6d)或(6e),为白色固体。
(6a):产率:20%。
M.p.:161-163℃。
MS(m/z):91(90%,苄基),319(BP,M-苄基);410(<5%,M)。
1H-NMR:200MHz,CDCl3:δ7.94(s,2H,H-三唑);7.29(s,10H芳族);5.45(br,2H,CH2-苄化);5.04(br,2H,CH2-苄化);3.96(s,3H,N-CH3);2.62(s,3H,C-CH3)。
(6b):产率20%。
M.p.:140℃。
MS(m/z):347(100%,M-苄基);91(75%,苄基)。
(6c):产率20%。
M.p.:114℃。
MS(m/z):361(100%,M-苄基);91(70%,苄基)。
1H-NMR:200MHz,CDCl3;δ7.94(2H,s,三唑);7.30(10H,m,芳族);5.49-5.21(4H,d,br,-CH2-苄化);4.12(3H,8,N-CH3);2.84(2H,t,-CH2-CH2-CH2-CH3);1.80(2H,m,-CH2-CH2-CH2-CH3);1.37(2H,m,-CH2-CH2-CH2-CH3);0.92(3H,t,-CH2-CH2-CH2-CH3)。
(6d):产率20%。
M.p.:173℃。
MS(m/z):409(65%,M-苄基),91(100%,苄基)。
(6e)6-二苄氨基-9-甲基-2-戊基-8-(三唑-2-基)-9(H)-嘌呤
M.p.:139℃
MS:m/z=466,375,348
1H-NMR(200MHz,CDCl3)δ(ppm):7.94(s,2H),7.29(m,10H),5.47(bs,2H),5.04(br,2H),3.96(s,3H),2.84(t,2H),1.82(m,2H),1.33(m,4H),0.88(m,3H)
2-烷基-6-氨基-9-甲基-8-(三唑-2-基)-9(H)-嘌呤(7a、b、c、d、e)
在氮气环境中冷却的反应烧瓶中将0.33mmol的(6a)、(6b)、(6c)、(6d)或(6e)溶于3ml无水二氯甲烷。缓慢滴加0.37ml的CF3SO3H(3.3mmol)并将该混合物保持回流6小时。然后使该混合物上载到活性铝土层析柱,首先用50ml二氯甲烷洗脱以除去强着色的芳族衍生物且然后用CH2Cl2/乙醇1∶1(40ml)洗脱,随后用乙醇(40m1)且最终用饱和的氨水的乙醇溶液(5%,40ml)洗脱。合并含有所需产物的部分并蒸发而得到黄色固体,将其通过急骤层析法纯化(SiO2,AcOEt/EtOH 95∶5),从而得到纯产物(7a、b、c、d、e),为白色固体。在乙醇中结晶而得到高纯度的极白的小结晶形式的产物。
(7c)(ST 1535)
产率:55%。
M.p.:182℃。
MS(m/z):230(100%,M-42);243(20%,M-29);257(10%,M-15);272(<10%,M)。
1H-NMR:200MHz,CDCl3;δ8.00(2H,s,三唑);5.74(2H,br,-NH2);4.07(3H,s,N-CH3);2.85(2H,t,-CH2-CH2-CH2-CH3);1.79(被水掩盖,m,-CH2-CH2-CH2-CH3);1.43(2H,m,-CH2-CH2-CH2-CH3);0.97(3H,t,-CH2-CH2-CH2-CH3)。
(7b)(ST 1536)
产率:55%。
M.p.:177℃。
1H-NMR:(200MHz,CDCl3)δ8.00(2H,s,三唑);5.70(2H,br,-NH2);4.07(3H,s,N-CH3);3.10(1H,六重峰(J=6.82Hz),CH3-CH-CH3);1.36(6H,d(J=6.82Hz),CH3-CH2-CH3)。
MS:m/z:230(100%,M-28);243(95%,M-15);216(50%,M-44);258(50%,M)。
(7d)(ST 1537)
产率:55%。
M.p.:164℃。
1H-NMR(200MHz,CDCl3):δ8.00(2H,s,三唑);7.3-7.18(5H,m,芳族);4.07(2H,s,CH2);3.17(2H,s,CH2);1.26(3H,s,CH3)。
MS m/z:91,216,243,303,320(100%,M)。
(7a)(ST 1491)
产率:43%。
M.p.:238℃。
MS(m/z):230(BP,M)。
1H-NMR(200MHz,CDCl3):δ8.00(s,2H三唑);5.63(br,2H,NH2);4.06(3H,s,N-CH3);2.64(3H,s,C-CH3)。
(7e)(ST 2097)
6-氨基-9-甲基-2-戊基-8-(三唑-2-基)-9(H)-嘌呤
M.p.:154℃
MS:m/z=286,271,257,243,230,190
1H-NMR(200MHz,CDCl3)δ(ppm):8.00(s,2H),7.26(m,10H),5.56(bs,2H),4.06(s,3H),2.83(t,2H),1.84(m,2H),1.40(m,4H),0.91(m,3H)。
实施例2
(反应流程2)
4-羟基-3-硝基吡啶(8)
将16.7ml发烟硫酸(SO3 20%的H2SO4溶液)缓慢滴加到20ml冷却至0℃的发烟硝酸中,在15分钟期限内加入7g的4-羟基吡啶。将该体系缓慢加热至开始硝化(生成红色蒸气)。然后将该反应体系冷却至所述蒸气消失为止,随后回流1小时。
将该反应混合物缓慢冷却至室温且然后倾在50g冰上。分少量加入60ml浓氨水(30%),注意温度不要超过20℃。再用氨水将pH调节至7.5且然后在4℃下保持过夜。过滤所产生的沉淀并在水中结晶而得到7.1g的(8),为透明的黄色结晶。
产率=70%。
M.p.:275-277℃。
MS(m/z):94,140。
4-氯-3-硝基吡啶(9)
在氮气环境的反应烧瓶中使51.5g的PCl5和75ml的POCl3在70℃下反应。在相同温度下谨慎加入34.6g的(8)。使温度增加至140℃并将该反应体系在氮气环境中回流4小时。向冷却的在真空中蒸发的混合物中加入100ml冰水。通过添加碳酸钠颗粒将pH调节至7.5并加入60ml二氯甲烷且将该混合物剧烈搅拌至所有残余物完全溶解。分离各相并再用二氯甲烷(5×30ml)提取含水部分。用无水硫酸钠处理合并的有机相并蒸发得到29.9g的(9)、为黄色蜡状固体。
产率=76%。
MS(m/z):85,87,100,102,112,114,158,160(M)。
4-甲氨基-3-硝基吡啶(10)
将29.9g的(9)溶于200ml热乙醇;向达到0℃的该溶液中缓慢滴加103ml的35%甲胺水溶液。将该体系保持搅拌30分钟且然后蒸发乙醇。使残余物在水中结晶而得到24g的(10),为透明的黄色结晶形式。
产率=83%。
MS(m/z):107,120,135,153(M)。
2-氯-4-甲氨基-3-氨基吡啶(11)
将10g的(10)溶于50ml的12N HCl和并使温度达到90℃。在1分钟过程中分5部分加入72.5g二SnCl2·2H2O。将该体系在90℃下保持搅拌1小时。在将该溶液冷却至室温后,加入100ml水并在减压条件下蒸发。将残余物溶于100ml水、冷却至0℃并加入浓氨水至形成白色凝胶状沉淀。将pH调节至8.5-9并离心所得的乳浊液。再次将剩余的固体残余物溶于水并离心。将该操作重复三次。将合并的固体残余物在50ml二氯甲烷中保持搅拌过夜。用二氯甲烷将离心的水相提取三次,然后合并所有的有机相,随后用无水硫酸钠干燥并在真空中蒸发而得到6.2g的粉红色结晶形式的(11)。
产率=60%。
M.p.:166-168℃。
MS(m/z):76,122,142,157(M+)。
4-氯-1-甲基-1(H)-咪唑并[45-c]吡啶(12)
将2.4g的(11)悬浮于97ml原甲酸乙酯并在搅拌下加入DMF,直到浑浊消失为止。然后向所得的澄清溶液中加入1.7ml的12N HCl。(加入该酸几分钟后该溶液变浑浊)且在氮气环境中保持搅拌12小时。然后在真空中蒸发溶剂并通过急骤层析法纯化棕色油状残余物(洗脱剂:环己烷/乙酸乙酯20∶80)而得到1.7g的(17)白色固体。
M.p.:137-38℃
产率=68%。
MS:m/z:167-169(100%-30%:M+);132(55%:M+-Cl);105(35%)。
1H-NMR(200MHz,CDCl3):δ3.91(s,3H,N-CH3);7.33(d,J=5.64Hz,1H,=N-CH=CH-),7.98(s,1H,-N=CH-N(CH3)-),8.24(d,J=5.64Hz,1H,=N-CH=CH-)。
2-溴-4-氯-1-甲基-1(H)-咪唑并[4,5-c]吡啶(13)
在氮气环境中将1.5g(9mmol)的(12)溶于25ml无水THF并将该混合物的温度调节至-78℃。缓慢加入8ml的BuLi 2.5M(20mmol)己烷溶液。该溶液呈微红色,证实在2位上形成芳香碳负离子。1小时后,在30分钟期限内谨慎滴加2ml溴(40mmol)且然后再保持搅拌2小时。使温度缓慢至0℃且然后滴加饱和偏亚硫酸氢钠溶液至完全破坏溴。用2N碳酸氢钠水溶液将该溶液的pH调节至9。用二氯甲烷提取该溶液。用盐水洗涤合并的有机相、用无水硫酸钠干燥并在真空中蒸发。得到浅棕色固体,使其在水中结晶而得到1.4g白色结晶形式的(13)。
产率=64%。
MS:m/z:245-247-249(80%-100%-25%:M+);210-212(80%-75%:M+-Cl);131(100%:M+-Cl-Br),105(50%)。
1H-NMR(200MHz,CDCl3):δ3.84(s,3H,N-CH3);7.25(d,J=6.11Hz,1H,=N-CH=CH-),8.23(d,J=6.11Hz,IH,=N-CH=CH-)。
4-氯-1-甲基-2-(三唑-2-基)-1(H)-咪唑并[45-c]吡啶(14)和4-氯-1-甲基
-2-(三唑-l-基)-1(H)-咪唑并[4,5-c]吡啶(14a)
将250mg的NaH(80%的石蜡溶液,8.6mmol)悬浮于5ml无水DMF中并加入0.5ml(8.6mmol)的1(H)-1,2,3-三唑。将该体系在室温下保持搅拌1小时,然后将温度调节至100℃。在30分钟过程中向该热溶液中滴加1.4g(5.7mmol)在15ml热的无水DMF中乳化的(13)。将该体系在100℃下保持搅拌4小时且然后使温度降至60℃并使反应保持过夜。
在该反应终止时蒸发DMF并使固体残余物在水中结晶。
通过过滤收集结晶并用二氯甲烷提取母液,合并有机相并用硫酸钠干燥、蒸发并再次在水中重结晶。得到614mg白色结晶形式的混合物(14和14a)。
总产率=46%(14+14a)。
(14):
MS(m/z):234-236(100%-30%:M+);207-209(20%-5%:M+-HCN);153-155(40%-10%)。
1H-NMR(CDCl3,200MHz):δ4.13(s,3H,N-CH3);7.35(d,J=5.62Hz,1H,=N-CH=CH-),8.05(s,2H,三唑),8.33(d,J=5.62Hz,1H,=N-CH=CH-)。
(14a):
MS(m/z):234-236(10%-3%:M+);206-208(100%-35%:M+-N2);191-193(40%-15%)。
1H-NMR(CDCl3,200MHz):δ4.23(s,3H,N-CH3);7.39(d,J=5.80Hz,1H,=N-CH=CH-),7.93(d,J=1.19Hz,1H,三唑),8.34(d,J=5.80Hz,1H,=N-CH=CH-),8.65(d,J=1.19Hz,1H,三唑)。
4-苄氨基-1-甲基-2-(三唑-2-基)-1(H)-咪唑并[4,5-c]吡啶(15)
在平底长颈反应烧瓶中将1.4g(5.5mmol)混合物(14,14a)悬浮于5ml苄胺中。将该反应体系置于微波炉中(辐射频率:2,450MHz)并以460瓦辐射至苄胺沸腾。使该体系沸腾几秒钟且然后停止辐射并使该混合物冷却。重复该操作直至通过TLC监测原料产品消失为止。冷却后得到黄色蜡状块,将其通过急骤层析法进一步纯化(梯度:环己烷/乙酸乙酯4∶6(100ml),环己烷/乙酸乙酯2∶8(100ml),乙酸乙酯)。得到390mg的(15),为黄色固体。
产率=29%。
M.p.=180-184℃。
MS(m/z):305(BP,M+);250;200,174,148。
1H-NMR(CDCl3,200MHz):δ4.02(s,3H,N-CH3);5.87(bs,2H),7.29(d,J=6.90Hz,1H),7.40-7.50(m,5H)7.88(d,J=6.90Hz,1H),8.29(s,2H,三唑)。
4-氨基-1-甲基-2-(三唑-2-基)-1(H)-咪唑并[4,5-c]吡啶并三氟甲磺酸盐
(16)(ST 1680)
将183mg的(15)(0.6mmol)溶于5ml无水二氯甲烷并缓慢滴加0.7ml三氟甲磺酸(6mmol)。将该体系保持回流1.5小时。然后使该反应混合物在氧化铝柱上层析,首先用二氯甲烷(100ml)、然后用二氯甲烷/乙醇50/50(100ml)且最终用纯乙醇洗脱。所需产物回收在乙醇部分中。蒸发溶剂后将残余物与乙醚一起研磨且然后在乙醇中结晶而得到52mg的纯ST 1680。
产率=24%。
M.p.:>290(分解)℃
MS(游离碱的):m/z:215(100%:M+);160(40%-5%);134(35%)。
1H-NMR(DMSO-d6,200MHz):δ3.99(s,3H,N-CH3);7.37(d,J=6.84Hz,IH,=NH+-CH=CH-),7.82(d,J=6.84Hz,1H,=NH+-CH=CH-),8.41(s,2H,三唑),8.62(br,1H,NH2),12.94(s,2H,=NH+CH=CH-)。
本发明的化合物是腺苷A2a受体的配体,它们特别是选择性拮抗剂且由此可用作药物,特别是治疗得益于对A2a受体的拮抗活性的病理情况。
在用本发明化合物治疗的病理情况中有运动障碍。作为本发明治疗的病理情况,我们引述了阿尔茨海默病、亨廷顿舞蹈病、威尔逊病和帕金森病。
本发明还应用于与″开-关″现象、与运动障碍占优势相关的帕金森病。
在本发明优选的实施方案中,将所述的化合物与左旋多巴或与一种或多种多巴胺兴奋剂联用。在这种情况中,本发明用于多巴胺替代疗法。
在本发明的另一个实施方案中,上述化合物用作制备治疗脑缺血和-或与神经变性过程相关的机制的药物的活性组分。
分子药理学
对腺苷A2a受体的亲和性
使用来自专门稳定表达人A2a受体亚型的HEK 293细胞(人胚胎肾细胞)的膜评价每一产物与腺苷A2a受体的相互作用能力。
在25℃下将所述膜与30nM浓度的[3H]-CGS21680一起在缓冲液中温育90′,所述的缓冲液由50mM Tris(pH7.4)、120mM NaCl、10mM MgCl2mM CaCl2、2U/ml腺苷脱氨酶组成。在有NECA(50μm)存在的情况下测定非特异性结合。
对腺苷A2h受体的亲和性
使用来自专门稳定表达人A2b受体亚型的HEK 293细胞的膜评价每一产物与腺苷A2b受体的相互作用能力。
在25℃下将这些膜与100nM浓度的[3H]-DPCPX一起在缓冲液中保温90′,所述的缓冲液由50mM Tris(pH7.4)、120mM NaCl、5mM KCl、10mM MgCl2、2mM CaCl2、2U/ml腺苷脱氨酶组成。在有NECA(50μm)存在的情况下测定非特异性结合。
对腺苷A1受体的亲和性
使用来自稳定表达人A1亚型的CHO-K1细胞的膜评价每一产物与腺苷A1受体的相互作用能力。
在25℃下将这些膜与1.66nM浓度的[3H]-DPCPX一起在缓冲液中保温90′,所述的缓冲液由50mM Tris(pH7.4)、120mM NaCl、5mM KCl、10mM MgCl2、2mM CaCl2、2U/ml腺苷脱氨酶组成。在有1μM浓度的DPCPX(8-环戊基-1,3-二丙基黄嘌呤)存在的情况下测定非特异性结合。
对腺苷A3受体的亲和性
对化合物ST 1535和ST 2097测定其对腺苷A3受体的亲和性。
为了进行本研究,按照Salvatore等在《美国国家科学院学报》(Proc.Natl.Acad.Sci.USA),1999 90:10365-10369中所述的方法使用来自稳定表达人A3亚型的HEK-293细胞的膜。实验条件需要使用0.1nM浓度的[125I]AB-MECA作为放射性配体、22℃下的保温时间为90分钟和IB-MECA(1μM)用于测定非特异性结合。
体外结果的分析和表示
在每一化合物的结合研究中,评价8种不同浓度(10-5M-10-12M)以便获得竞争曲线。通过对竞争曲线的非线性回归分析确定表示每种产物结合亲和性的IC50值。使用Cheng Prusoff等式(Ki=IC50/1+(L/Kd))计算Ki值,通过其表示所研究的每种产品对所研究受体的亲和性。
普通药理学
对小鼠自发运动活性的作用的评价
为了进行本研究使用CD1型雄性小鼠(n=8)。使用由周围有一系列监测内置动物运动的连接采集信号且随后加工的计算机系统的光电池的有机玻璃笼(40cm×40cm)组成的设备评价所研究产品和参比化合物的作用。
在腹膜内给予所述产品后进行试验。治疗后15分钟将治疗动物(与对照组交替)置于笼内以便在分为两次观察间隔(就治疗后的时间而言分别为15′-45′和45′-60′)的45分钟总期限内记录其自发运动。
为了检验所研究化合物对运动活性可能的作用,考虑下列参数:水平活动;垂直活动;总距离。
除溶于0.9%NaCl的CGS 21680(参比化合物,描述在EP 0 277917中,Ciba-Geigy)外,将所研究的产品溶于DMSO且然后用Cremofor EL和0.9%NaCl稀释(终浓度:DMSO 15%,Cremofor EL15%,NaCl 0.9%)。
所述产品诱发小鼠僵住症的能力的评价
每组使用10只CD1雄性小鼠。就本试验而言,将10cm长的钢棒放置在距支持物表面上方4.5cm高处。使动物的前腿置于该棒上。通过测定动物保持所置姿势的时间(按秒计)确定僵住症的存在。随后将该参数相对置于升高值的等级(0-5),由此可成比例表示对照组动物与用试验物质治疗的这些动物中确定的僵住症程度。
在本试验前30分钟通过腹膜内给予10ml/kg体积的参比产品和本研究中的这些产品。
除溶于0.9%NaCl的CGS 21680(参比化合物)外,将本研究中的产品和对照拮抗剂ZM 241385溶于DMSO且然后用Cremofor EL和0.9%NaCl稀释(终浓度:DMSO 15%,Cremofor EL 15%,NaCl 0.9%)。
所述产品拮抗CGS 21680诱发的僵住症的能力的评价
为了进行本研究,使用产品ST 1535。在测试僵住症评分前30分钟通过脑室内给予CGS 21680(10μg/5μl/小鼠)在动物中诱发僵住症。在用CGS 21680处理前30分钟口服给予5mg/Kg和10mg/Kg剂量的测试化合物。
在下列时间后按照用OGS 21680处理后所述的方式衍生出僵住症评分:30′、60′、120′、180′。
所述产品拮抗氟哌啶醇诱发的僵住症的能力的评价
为了进行本研究,使用产品ST 1535。在测定动物中僵住症前2小时通过腹膜内给予4mg/Kg剂量的氟哌啶醇在动物中诱发僵住症。按照前述方法测定僵住症的存在。
在对氟哌啶醇诱发的僵住症评分后,向动物口服给予等于10mg/kg和20mg/kg剂量的产品ST 1535进行治疗。治疗后60分钟对动物进行进一步的僵住症评分,该步骤在给予ST 1535后的下列时间进行:120′、240′、300′。
给予相结合的左旋多巴和A2a拮抗剂对氟哌啶醇诱发的僵住症的作用
为了进行本研究,使用产品ST 1535。
使用分成不同实验组(n=10/组)的CD1小鼠。在按照上述方法进行的僵住症试验前2小时和30′对全部动物进行氟哌啶醇处理(4mg/kg,腹膜内)。
随后根据动物的原始实验组对动物进行不同类型的治疗(参见图表)。氟哌啶醇处理后2小时和30分钟进行所有僵住症评价。
氟哌啶醇+ST 1535:ST 15352.5mg/kg,口服,测试前75′;
氟哌啶醇+苄丝肼+左旋多巴:苄丝肼3.12mg/kg腹膜内,测试前90′;
左旋多巴:12.5mg/kg,测试前60′;
氟哌啶醇+苄丝肼+左旋多巴+ST 1535:苄丝肼3.12mg/kg腹膜内,测试前90′;
ST 15351.25mg/kg或2.5mg/kg,测试前75′;
左旋多巴12.5mg/kg,测试前60′;
A2a拮抗剂和抗抑郁活性。小鼠强迫游泳试验
将小鼠个别放入含有10cm水的维持在23℃下的玻璃圆筒(高:25cm,内径10cm)中。在4分钟试验期间测定不动时间(秒)。当小鼠保持浮在水中,仅作出为使头保持在水上的必要运动时判断小鼠不动。本试验前60分钟对小鼠口服给予测试化合物ST 1535。
体内活性
表1中报导了所研究的不同化合物对腺苷A2a受体亲和性的平均值和标准偏差,表示为Ki(nM)。
能够观察到分别命名为ST 1535、ST 1537和ST 2097的产品对腺苷A2a受体均表现出升高的相互作用能力。
这些化合物的亲和值与这些化合物相对于其它具有腺嘌呤结构的产品的比较显示,用相对长的烷基链(参见ST 1535、ST 2097)或明显的位阻(参见ST 1537)在2位上取代腺嘌呤有利于对A2a受体亲和性的增加。
同一表中报导了所研究的每一化合物对腺苷受体亚型A2b和A1的亲和值和受体亲和性比值(KiA1/KiA2a),通过这一比值可以确定每一产品的选择性。
观察到化合物ST 1535、ST 1537和ST 2097对A2a受体的相互作用能力相对于对A1和A2b亚型所展示的占优势,因此,本发明的化合物对A2a受体具有选择亲和性。
此外,就化合物ST 1535和ST 2097而言,对腺苷A3受体和对属于其它神经递质的36种受体的亲和性已经得到评价。在这些结合研究中,所关注的化合物最初的测试浓度为1μM。随后,如果所述化合物替换了50%以上的特异性放射性配体,那么以8种不同产品浓度评价它以测定IC50值。
表2中报导了有关本结合研究的结果。
化合物ST 1535和ST 2097表现出对腺苷A3亚型相对低且可以忽略不计的亲和性,并且对其它受体没有相互作用能力(IC50>1000nM)。
这些结果证实本发明的化合物是选择性的,对腺苷A2a受体具有选择亲和性。
表1
| 化合物 | A2aKi(nM)±sd | A2bKi(nM)±sd | A1Ki(nM)±sd | KiA1/KiA2a |
| ST 1680ST 1491ST 1535ST 2097ST 1537ST 1490CGS 21680ZM 241385咯嗪DPCPX | 97±2370±152.29±0.580.12±0.0332.34±0.694651±130.11±0.03 | 926±55627±45153±132330±5883.8±2.1 | 1563±25210±1.4107±4026.2±6.5580±130.436.5±0.95 | 160.1547217340.009 |
表2
| 受体 | ST 1535 | ST 2097 | 参比化合物 | ||||
| 1μM | Ki(nM) | 1μM | Ki(nM) | IC50(nM) | Ki(nM) | ||
| A3(h)ADO转运蛋白α1(非选择性)α2(非选择性)β1β2BZD(中枢)D1D2D3D4.4(h)D5(h)GABAaGABAbGABA转运蛋白AMPA红藻氨酸PCPP2XP2YNMDAH1(中枢)M1M2M3M4M5胆碱转运蛋白阿片剂(非选择性)5-HT1A5-HT2A5-HT2C(h)5-HT3(h)5-HT45-HT5A(h)5-HT6(h)NE转运蛋白DA转运蛋白5-HT转运蛋白 | 24------------------------------------- | 1580 | 34------------------------------------13 | 519 | IB-MECANBTI哌唑嗪育亨宾atenololICI118551地西泮SCH23390(+)布他拉莫(+)布他拉莫氯氮平SCH23390蝇蕈醇巴氯芬3-哌啶甲酸L-谷氨酸红藻氨酸MK-801α,β-MeATdATPαSCGS 19755美吡拉敏哌仑西平(pirenzepina)美索曲明4-DAMP4-DAMP4-DAMP宓胆碱-3纳洛酮8-OH-DPAT酮色林美舒麦角MDL 722225-羟色胺5-羟色胺普罗替林GBR12909丙米嗪 | 1.20.300.86951,7702.3120.668.95.11560.61165010,100613772.014229671.322343.51.92.0121.60.662.71.99.3794211.15.04.4 | 0.84 |
就测试化合物而言,将结果表示为对于对照特异性结合的抑制百分比(平均值;n=2)。符号-表示抑制低于10%。
体内活性
为了确定所关注化合物具有的活性特征(兴奋或拮抗),检验它们对小鼠运动活性的作用。将这些结果与下列参比化合物CGS 21680(A2a受体的选择性兴奋剂,EP 0 277 917)和ZM 241385(A2a受体的选择性拮抗剂)得到的结果进行比较。注意到兴奋剂诱发运动活性抑制,而拮抗剂具有刺激作用(Nikodij evicc O.等《实验药物疗法杂志》(J.Pharm.Exp.Ther)257,286-94,1991)。
在表3中例示了所关注化合物与参比化合物诱发的对描述小鼠自发运动活性的三种参数的作用结果。报导了观察到的每一参数的平均值和标准误差。
表3
| 从治疗开始的观察间隔 | 水平活动 | 垂直活动 | 总距离 | |||
| 15’-45 | 45’-60 | 15’-45 | 45’-60’ | 15’-45’ | 45’-60’ | |
| 治疗组对照组CGS 21680(0,5mg/kg)载体(10ml/kg)ZM 241385(15mg/kg)ZM 241385(30mg/kg)ZM 241385(60mg/kg)ST 1537(2,5mg/kg)ST 1537(5mg/kg)ST 1537(10mg/kg)载体(10ml/kg) | 5992±4841608±3285890±8568706±4737035±7097790±9807494±5657203±2648242±8475184±832 | 3148±1004471±2542259±4524148±2413505±3754241±4073133±2502844±2993874±2951734±367 | 356±6227±11330±96680±64613±102570±129590±78498±77528±126123±60 | 117±4010±1119±34362±78282±37264±56210±17363±91276±5749±25 | 1394±148504±1061315±2902280±2421725±1862250±4011676±2041503±1162707±950 | 469±125121±73412±1211022±130783±1651131±158677±105532±95843±149 |
| ST 1535(2.5mg/kg)ST 1535(5mg/kg)ST 1535(10mg/kg)ST 1535(20mg/kg) | 5386±5057549±5087434±52610524±670 | 1693±2662353±1992784±3453321±363 | 282±79249±41219±64486±84 | 68±2176±1671±32148±36 |
就本发明的化合物而言,产品ST 1537特别诱发运动活性明显增加。实际上,每一检验的参数相应于对照值而言均显著增加,不依赖于所给予的产品剂量。此外,观察到化合物ST 1537比参比拮抗剂更具活性。实际上,最低剂量的ST 1537诱发与较大剂量(15mg/kg)的化合物ZM 241385相同的作用。此外,对化合物ST 1535而言,从5mg/Kg剂量开始观察到小鼠自发运动活性显著增加。因此,本发明的化合物对腺苷A2a受体具有拮抗活性。
在用所研究的产品治疗后,对动物中最终存在的僵住症的评价(图1)与这些观察结果共同证实了ST 1537和ST 1535的拮抗特征。实际上,它们中没有一种使小鼠中出现僵住症,这一结果与参比拮抗剂(ZM241385,60mg/kg)类似而与参比兴奋剂(CGS 21680,2mg/kg)证实的结果相反。
就化合物ST 1535而言,还通过对其拮抗预先通过给予选择性A2a受体兴奋剂CGS21680诱发的僵住症消失的能力的评价证实了该产品对A2a受体的拮抗活性特征(附图2)。
选择性A2a受体兴奋剂诱导动物中僵住症程度升高。口服给予产品ST 1535在所有观察时间均显著拮抗僵住症出现,特别是当给予的剂量为10mg/kg时更是如此。这一结果证实并描述了优选化合物ST1535针对腺苷A2a受体的拮抗活性特征。
还通过研究所述化合物对小鼠中氟哌啶醇僵住症的作用证实了ST 1535作为对腺苷A2a受体的选择性拮抗剂的特征。此外,通过这种评价,所述产品调节黑质纹状体系统中多巴胺能传递功能障碍的能力得到确定。在附图3中,观察到口服给药后,ST 1535减少小鼠中出现僵住症,它是急性给予氟哌啶醇后黑质纹状体系统中多巴胺能状态下降促使的行为表现。ST 1535的抗僵住症活性间接证实所关注的化合物能够补偿在用氟哌啶醇处理后黑质纹状体系统中发生的多巴胺神经传递缺乏,按照药理学特征属于腺苷A2a受体的选择性拮抗剂。
此外,就优选的化合物ST 1535而言,已经证实口服给予该产品强化了用无效剂量的左旋多巴和苄丝肼治疗的抗僵住症活性。这种评价结果报导在附图4中。用ST 1535与无效剂量的左旋多巴和苄丝肼联用
以剂量依赖性方式减轻了氟哌啶醇-僵住症。
这些结果提示可以将所关注的产品ST 1535与低剂量的左旋多巴联合给药用于治疗帕金森病。
左旋多巴常用于治疗帕金森病。然而左旋多巴的应用因作为副作用的运动障碍的出现而受到限制(Shaw K.M.等″Q.J.Med″1980 49,283)。共同给予ST 1535可以减少所给予的左旋多巴的量,从而减少所述副作用的出现。
此外,对优选的化合物ST 1535测定了抗抑郁活性。注意到A2a受体的选择性拮抗剂正被定义为新的有潜力的抗抑郁药(El Yacobui M.et al.《(英国药理学杂志》(British J.Pharmacol.)2001:134,68-77)。附图5表示ST 1535在抑郁症动物模型中的作用。该化合物以剂量依赖性方式减少了动物不动的时间,这种方式与对抗抑郁药丙米嗪所观察到的方式类似。
本发明的另一个目的是药物组合物,它包含作为活性成分的至少一种通式(I)化合物,单独或与一种或多种其它通式(I)的化合物组合,或所述通式(I)化合物与其它用于治疗本文指定的病理情况的活性组分联用,例如其它对腺苷A2a受体具有活性的产品;甚至是单独的剂型或适合于联合疗法的剂型。本发明中的活性组分是与常用于制药技术的合适的载体和/或赋形剂相混合,所述的载体和/或赋形剂例如描述在最新版《Remington氏药物科学手册》(″Remington′s PharmaceuticalSciences Handbook″)中。本发明的组合物含有治疗有效量的所述活性组分。其剂量由本领域技术人员例如临床医师和医生根据所治疗的病理情况的类型和患者条件或同时给予的其它活性组分来决定。
药物组合物的实例是允许口服或非肠道、静脉内、肌内、皮下、经皮给药的那些药物组合物。适合于该目的的药物组合物有:丸剂、硬胶囊或软胶囊、粉剂、溶液、混悬剂、糖浆剂、临时配成液体的固体形式。用于非肠道给药的组合物例如是可肌内、静脉内、皮下注射的所有剂型,为溶液、混悬剂、乳剂形式。我们还提及了脂质体制剂。还包括:口服给药用的所述活性组分的控释剂型;覆盖有合适包衣层的丸剂;微囊化粉末;含环糊精的复合物;延效型制剂,例如皮下用的长效制剂,诸如可注射的沉积物或植入物。
Claims (27)
1.具有下列通式的化合物及其药物上可接受的盐:

其中:X是N、CH;
R1是C1-C6直链或支链饱和烷基;
R2是氢、C1-C6直链或支链饱和烷基或C6-C14芳基C1-C6直链烷基;
R3是NH2、NHR4;
R4是C6-C14芳基C1-C6烷基。
2.权利要求1所述的化合物,其中X是氮且R2是2位上的正丁基。
3.权利要求1所述的化合物,其中X是氮且R2是2位上的苯乙基。
4.权利要求1所述的化合物,其中X是氮且R2是2位上的正戊基。
5.权利要求1所述的化合物,其中X是碳且R2是6或7位上的氢。
6.权利要求1所述的化合物,选自下列化合物组成的组:
-6-氨基-2,9-二甲基-8-(三唑-2-基)-9(H)-嘌呤;
-2-丁基-9-甲基-8-(2H-1,2,3-三唑-2-基)-9H-嘌呤-6-基胺;
-9-甲基-2-(2-苯乙基)-8-(2H-1,2,3-三唑-2-基)-9H-嘌呤-6基胺;
-9-甲基-2-戊基-8-(2H-1,2,3-三唑-2-基)-9H-嘌呤-6-基胺。
7.权利要求1所述的化合物,它是6-氨基-9-甲基-8-(三唑-2-基)-9(H)-嘌呤。
8.权利要求1所述化合物的制备方法,该方法包括下列反应流程的步骤:

a)在化合物a)的2位上进行溴取代;
b)用三唑-2-基替代化合物b)2位上的溴而得到化合物c),
其中R1、R2、R3和X如权利要求1中定义。
9.药物组合物,包含至少一种权利要求1-7的化合物并混有药物上可接受的赋形剂和/或载体。
10.权利要求1-6中任意一项所述的化合物在制备对腺苷A2a受体具有抑制活性的药物中的应用。
11.权利要求10的应用,其中所述的抑制活性是对腺苷A2a受体选择性的。
12.权利要求1-6中任意一项的化合物在制备用于治疗运动障碍的药物中的应用。
13.权利要求1-6中任意一项的化合物在制备用于治疗阿尔茨海默病的药物中的应用。
14.权利要求1-6中任意一项的化合物在制备用于治疗亨廷顿舞蹈病的药物中的应用。
15.权利要求1-6中任意一项的化合物在制备用于治疗威尔逊病的药物中的应用。
16.权利要求1-6中任意一项的化合物在制备用于治疗帕金森病的药物中的应用。
17.权利要求16的应用,其中所述的帕金森病具有″开-关″现象或其中所述的帕金森病是运动障碍占优势。
18.权利要求16的应用,其中将所述的化合物与左旋多巴或与一种或多种多巴胺兴奋剂联用。
19.权利要求16的应用,其中所述的药物用于多巴胺替代疗法。
20.权利要求16的应用,其中所述的疾病是因药物、创伤、毒性剂所引起。
21.权利要求1-6中任意一项的化合物在制备用于治疗脑缺血和/或与神经变性过程相关的机制的药物中的应用。
22.权利要求1-6的化合物与左旋多巴或与多巴胺兴奋剂的组合。
23.权利要求22所述的组合在制备用于多巴胺替代疗法的药物中的应用。
24.权利要求7所述的化合物在制备对腺苷A1受体具有亲和性的药物中的应用。
25.权利要求24的应用,其中所述的药物用于治疗认知缺损、阿尔茨海默病、脑缺血、急性和慢性肾衰、因放射造影剂或顺铂诱发的肾衰。
26.4-苄氨基-1-甲基-2-(三唑-2-基)-1(H)-咪唑并[4,5-c]吡啶。
27.权利要求26的化合物作为权利要求8方法中的中间体的应用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IT2001RM000465A ITRM20010465A1 (it) | 2001-07-31 | 2001-07-31 | Derivati della triazolil-imidazopiridina e delle triazolilpurine utili come ligandi del recettore a2a dell'adenosina e loro uso come medicam |
| ITRM01A000465 | 2001-07-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1525974A CN1525974A (zh) | 2004-09-01 |
| CN1271070C true CN1271070C (zh) | 2006-08-23 |
Family
ID=11455703
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB028138481A Expired - Fee Related CN1271070C (zh) | 2001-07-31 | 2002-07-25 | 用作腺苷A2α受体的配体的三唑基-咪唑并吡啶和三唑基嘌呤类的衍生物及其作为药物的应用 |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US7230102B2 (zh) |
| EP (1) | EP1412354B1 (zh) |
| JP (1) | JP4366186B2 (zh) |
| KR (1) | KR100884818B1 (zh) |
| CN (1) | CN1271070C (zh) |
| AT (1) | ATE325796T1 (zh) |
| AU (1) | AU2002326146B2 (zh) |
| BR (1) | BR0211550A (zh) |
| CA (1) | CA2451279C (zh) |
| DE (1) | DE60211343T2 (zh) |
| DK (1) | DK1412354T3 (zh) |
| ES (1) | ES2263810T3 (zh) |
| HK (1) | HK1068334A1 (zh) |
| HU (1) | HU228985B1 (zh) |
| IT (1) | ITRM20010465A1 (zh) |
| MX (1) | MXPA04000886A (zh) |
| PL (1) | PL217269B1 (zh) |
| PT (1) | PT1412354E (zh) |
| WO (1) | WO2003011864A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105622495A (zh) * | 2016-03-23 | 2016-06-01 | 叶芳 | 4-氯-3-硝基吡啶及其制备方法 |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60206756T2 (de) * | 2001-06-29 | 2006-07-13 | CV Therapeutics, Inc., Palo Alto | Purin derivate als a2b adenosin rezeptor antagonisten |
| ITRM20010465A1 (it) * | 2001-07-31 | 2003-01-31 | Sigma Tau Ind Farmaceuti | Derivati della triazolil-imidazopiridina e delle triazolilpurine utili come ligandi del recettore a2a dell'adenosina e loro uso come medicam |
| WO2003051882A1 (en) * | 2001-12-18 | 2003-06-26 | Cv Therapeutics, Inc. | A2a adenosine receptor antagonists |
| JP4472693B2 (ja) | 2004-03-26 | 2010-06-02 | 大日本住友製薬株式会社 | 9置換−8−オキソアデニン化合物 |
| WO2007109812A2 (en) * | 2006-03-23 | 2007-09-27 | Novartis Ag | Immunopotentiating compounds |
| JP2009542645A (ja) | 2006-07-05 | 2009-12-03 | アストラゼネカ・アクチエボラーグ | Tlr7のモジュレーターとして作用する8−オキソアデニン誘導体 |
| TW200831105A (en) | 2006-12-14 | 2008-08-01 | Astrazeneca Ab | Novel compounds |
| US8067413B2 (en) | 2007-03-19 | 2011-11-29 | Astrazeneca Ab | 9-substituted-8-oxo-adenine compounds as toll-like receptor (TLR7 ) modulators |
| ES2373616T3 (es) | 2007-03-19 | 2012-02-07 | Astrazeneca Ab | Compuestos de 8-oxo-adenina 9 sustituidos como moduladores del receptor de tipo toll (tlr7). |
| JPWO2008114819A1 (ja) | 2007-03-20 | 2010-07-08 | 大日本住友製薬株式会社 | 新規アデニン化合物 |
| PE20081887A1 (es) * | 2007-03-20 | 2009-01-16 | Dainippon Sumitomo Pharma Co | Nuevo compuesto de adenina |
| KR101313804B1 (ko) * | 2007-03-20 | 2013-10-01 | 쿠리스 인코퍼레이션 | Hsp90 억제제로서의 융합된 아미노 피리딘 |
| ES2393037T3 (es) * | 2007-05-08 | 2012-12-18 | Astrazeneca Ab | Imidazoquinolinas con propiedades inmunomoduladoras |
| PE20091156A1 (es) | 2007-12-17 | 2009-09-03 | Astrazeneca Ab | Sales de (3-{[[3-(6-amino-2-butoxi-8-oxo-7,8-dihidro-9h-purin-9-il)propil](3-morfolin-4-ilpropil)amino]metil}fenil)acetato de metilo |
| CA2711769A1 (en) | 2008-01-17 | 2009-07-23 | Dainippon Sumitomo Pharma Co. Ltd. | Method for preparing adenine compound |
| JPWO2009091031A1 (ja) * | 2008-01-17 | 2011-05-26 | 大日本住友製薬株式会社 | アデニン化合物の製造方法 |
| TWI473614B (zh) * | 2008-05-29 | 2015-02-21 | Kyowa Hakko Kirin Co Ltd | Anti-analgesic inhibitors |
| CN104193740A (zh) | 2008-07-03 | 2014-12-10 | 西特里斯药业公司 | 作为沉默调节蛋白调节剂的苯并咪唑类和相关的类似物 |
| BRPI0916203A2 (pt) * | 2008-07-23 | 2018-05-15 | Kyowa Hakko Kirin Co Ltd | agente terapêutico e/ou preventivo para enxaqueca, método para tratar e/ou prevenir enxaqueca, e, uso de um composto |
| EP2408775B1 (en) * | 2009-03-20 | 2015-06-17 | SIGMA-TAU Industrie Farmaceutiche Riunite S.p.A. | Oxidated derivatives of triazolylpurines useful as ligands of the adenosine a2a receptor and their use as medicaments |
| KR101830623B1 (ko) * | 2009-11-13 | 2018-02-21 | 아카데미아 시니카 | 신경퇴화 질병의 예방 및 치료를 위해 아데노신 a2a 수용기 및 아데노신 수송체를 표적화하는 이중-작용 화합물 |
| JP2013512859A (ja) * | 2009-12-03 | 2013-04-18 | 大日本住友製薬株式会社 | トール様受容体(tlr)を介して作用するイミダゾキノリン |
| US8791126B2 (en) | 2010-11-24 | 2014-07-29 | Yamasa Corporation | 2-alkynyl-N9-propargyladenine and medicinal use thereof |
| WO2012080728A1 (en) | 2010-12-16 | 2012-06-21 | Astrazeneca Ab | Imidazo [4, 5 -c] quinolin- 1 -yl derivative useful in therapy |
| JP5978226B2 (ja) | 2010-12-17 | 2016-08-24 | 大日本住友製薬株式会社 | プリン誘導体 |
| BR112015022923A2 (pt) * | 2013-03-15 | 2017-07-18 | Syngenta Participations Ag | derivados de imidazopiridina microbicidamente ativos |
| GB2555264A (en) | 2016-04-15 | 2018-04-25 | Univ Oxford Innovation Ltd | Adenosine receptor modulators for the treatment of circadian rhythm disorders |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR910700253A (ko) | 1989-01-31 | 1991-03-14 | 로버트 제이, 바란 | N_6-치환-9-메틸아데닌: 신규한 부류의 아데노신 수용체 길항질 |
| US5670501A (en) * | 1994-09-01 | 1997-09-23 | Discovery Therapeutics, Inc. | N-substituted 9-alkyladenines |
| DE69815554T2 (de) | 1998-01-05 | 2004-05-06 | Eisai Co., Ltd. | Purinderivate und antagonisten des adenosin-a2-rezeptors, welche zur vorsorge oder heilung von diabetes dienen |
| GB9915437D0 (en) * | 1999-07-01 | 1999-09-01 | Cerebrus Ltd | Chemical compounds III |
| DE60206756T2 (de) * | 2001-06-29 | 2006-07-13 | CV Therapeutics, Inc., Palo Alto | Purin derivate als a2b adenosin rezeptor antagonisten |
| ITRM20010465A1 (it) | 2001-07-31 | 2003-01-31 | Sigma Tau Ind Farmaceuti | Derivati della triazolil-imidazopiridina e delle triazolilpurine utili come ligandi del recettore a2a dell'adenosina e loro uso come medicam |
-
2001
- 2001-07-31 IT IT2001RM000465A patent/ITRM20010465A1/it unknown
-
2002
- 2002-07-25 MX MXPA04000886A patent/MXPA04000886A/es active IP Right Grant
- 2002-07-25 KR KR1020047000173A patent/KR100884818B1/ko not_active IP Right Cessation
- 2002-07-25 AT AT02760555T patent/ATE325796T1/de active
- 2002-07-25 DK DK02760555T patent/DK1412354T3/da active
- 2002-07-25 WO PCT/IT2002/000489 patent/WO2003011864A1/en active IP Right Grant
- 2002-07-25 EP EP02760555A patent/EP1412354B1/en not_active Expired - Lifetime
- 2002-07-25 JP JP2003517056A patent/JP4366186B2/ja not_active Expired - Fee Related
- 2002-07-25 PT PT02760555T patent/PT1412354E/pt unknown
- 2002-07-25 HU HU0401987A patent/HU228985B1/hu not_active IP Right Cessation
- 2002-07-25 AU AU2002326146A patent/AU2002326146B2/en not_active Ceased
- 2002-07-25 US US10/484,491 patent/US7230102B2/en not_active Expired - Fee Related
- 2002-07-25 PL PL368409A patent/PL217269B1/pl unknown
- 2002-07-25 BR BR0211550-6A patent/BR0211550A/pt not_active IP Right Cessation
- 2002-07-25 CA CA2451279A patent/CA2451279C/en not_active Expired - Fee Related
- 2002-07-25 DE DE60211343T patent/DE60211343T2/de not_active Expired - Lifetime
- 2002-07-25 ES ES02760555T patent/ES2263810T3/es not_active Expired - Lifetime
- 2002-07-25 CN CNB028138481A patent/CN1271070C/zh not_active Expired - Fee Related
-
2005
- 2005-01-13 HK HK05100306A patent/HK1068334A1/xx unknown
-
2007
- 2007-05-04 US US11/797,566 patent/US7528252B2/en not_active Expired - Fee Related
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105622495A (zh) * | 2016-03-23 | 2016-06-01 | 叶芳 | 4-氯-3-硝基吡啶及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US7230102B2 (en) | 2007-06-12 |
| US7528252B2 (en) | 2009-05-05 |
| HUP0401987A2 (hu) | 2005-01-28 |
| HU228985B1 (en) | 2013-07-29 |
| PL217269B1 (pl) | 2014-06-30 |
| DE60211343D1 (de) | 2006-06-14 |
| CN1525974A (zh) | 2004-09-01 |
| JP2005500355A (ja) | 2005-01-06 |
| CA2451279A1 (en) | 2003-02-13 |
| MXPA04000886A (es) | 2004-06-03 |
| ITRM20010465A1 (it) | 2003-01-31 |
| PT1412354E (pt) | 2006-09-29 |
| DE60211343T2 (de) | 2007-05-10 |
| CA2451279C (en) | 2011-05-10 |
| US20040204428A1 (en) | 2004-10-14 |
| AU2002326146B2 (en) | 2008-05-01 |
| ITRM20010465A0 (it) | 2001-07-31 |
| US20070249638A1 (en) | 2007-10-25 |
| ES2263810T3 (es) | 2006-12-16 |
| KR20040023641A (ko) | 2004-03-18 |
| BR0211550A (pt) | 2004-07-13 |
| JP4366186B2 (ja) | 2009-11-18 |
| EP1412354A1 (en) | 2004-04-28 |
| KR100884818B1 (ko) | 2009-02-20 |
| ATE325796T1 (de) | 2006-06-15 |
| HUP0401987A3 (en) | 2012-12-28 |
| HK1068334A1 (en) | 2005-04-29 |
| DK1412354T3 (da) | 2006-09-18 |
| WO2003011864A1 (en) | 2003-02-13 |
| EP1412354B1 (en) | 2006-05-10 |
| PL368409A1 (en) | 2005-03-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1271070C (zh) | 用作腺苷A2α受体的配体的三唑基-咪唑并吡啶和三唑基嘌呤类的衍生物及其作为药物的应用 | |
| CN1173975C (zh) | 咪唑并吡啶衍生物 | |
| CN1186324C (zh) | 稠合杂芳基衍生物 | |
| CN1165537C (zh) | 作为5-ht2c兴奋剂的吡咯并吲哚、吡啶并吲哚和氮杂�并吲哚 | |
| CN1045778C (zh) | 具有p*de-ⅳ抑制活性的新型化合物 | |
| CN1166678C (zh) | 嘌呤衍生物 | |
| CN1255403C (zh) | 用作磷酸二酯酶抑制剂的β-咔啉衍生物 | |
| CN1289499C (zh) | 吡嗪并(氮杂)吲哚衍生物 | |
| CN101039944A (zh) | 具有cdk1抗增殖活性的1,5-二氮杂萘唑烷酮类 | |
| CN1278819A (zh) | 作为促肾上腺皮质素释放激素拮抗剂、可用于治疗cns障碍和紧张相关性疾病的杂环基取代的环稠合吡啶和嘧啶 | |
| CN1161341C (zh) | cGMP磷酸二酯酶的稠合吡啶并哒嗪抑制剂 | |
| CN1281458A (zh) | 具有pde iv抑制活性的嘌呤化合物及其合成方法 | |
| CN1922185A (zh) | 作为激酶抑制剂的吡唑并三嗪类化合物 | |
| CN1694886A (zh) | 作为周期素依赖性激酶抑制剂的咪唑并吡嗪 | |
| CN101056879A (zh) | 作为a3和a1腺苷受体激动剂的嘌呤衍生物 | |
| CN1582277A (zh) | 用作糖原合酶激酶3β抑制剂的酰胺衍生物 | |
| CN100338040C (zh) | 黄嘌呤氧化酶抑制化合物 | |
| CN1729192A (zh) | ( 1-苄基-4-甲基哌啶-3-基)-甲基胺的旋光拆分及其用于制备作为蛋白激酶抑制剂的吡咯并[2 , 3-d ]嘧啶衍生物的应用 | |
| CN1726216A (zh) | C-6修饰的吲唑基吡咯并三嗪 | |
| CN1701074A (zh) | 用作依赖细胞周期蛋白激酶抑制剂的吡唑并嘧啶 | |
| CN1809565A (zh) | 用于治疗癌症的吡咯并二氢异喹啉 | |
| CN1278822A (zh) | 用作磷酸二酯酶抑制剂的2-苯基取代的咪唑并三嗪酮 | |
| CN1582285A (zh) | 用作糖原合酶激酶3β抑制剂(GSK3抑制剂)的杂芳胺化合物 | |
| CN101076532A (zh) | 作为甘氨酸转运体I(GlyT-1)抑制剂用于治疗阿尔茨海默氏病的二环和三环取代的苯甲酮化合物 | |
| CN1328560A (zh) | [1,2,4]三唑并[1,5-c]嘧啶衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1068334 Country of ref document: HK |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20060823 Termination date: 20140725 |
|
| EXPY | Termination of patent right or utility model |