CN110818760B - 一种可工业化合成地屈孕酮的生产工艺 - Google Patents
一种可工业化合成地屈孕酮的生产工艺 Download PDFInfo
- Publication number
- CN110818760B CN110818760B CN201911102923.7A CN201911102923A CN110818760B CN 110818760 B CN110818760 B CN 110818760B CN 201911102923 A CN201911102923 A CN 201911102923A CN 110818760 B CN110818760 B CN 110818760B
- Authority
- CN
- China
- Prior art keywords
- reaction
- solvent
- compound
- dydrogesterone
- tetrahydrofuran
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/0005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21
- C07J7/001—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group
- C07J7/0015—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa
- C07J7/002—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa not substituted in position 16
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Steroid Compounds (AREA)
Abstract
本发明公开了一种可工业化合成地屈孕酮的生产工艺,以易得的黄体酮为原料,通过羰基保护、溴代、脱溴、光化学开环反应、光化学闭环反应、脱保护、双键异构的步骤制得地屈孕酮,具有起始原料易得、各步骤均易实现、收率较高的优点,且操作简便、绿色环保,易于放大至工业生产。
Description
技术领域
本发明涉及药物合成技术领域,具体涉及工业化制备地屈孕酮原料药的工艺。
背景技术
地屈孕酮(Dydrogesterone)1,又名去氢孕酮,化学名为9β,10α-孕甾-4,6-二烯-3,20-二酮,CAS号:152-62-5。地屈孕酮既广泛用于保胎及预防流产,还广泛用于治疗内源性孕酮不足引起的各种疾病,如:痛经、子宫内膜异位症、继发性闭经、月经周期不规则、功能失调性子宫出血、经前期综合征、孕激素缺乏所致先兆性流产或习惯性流产、黄体不足所致不孕症等,目前有达芙通(Duphaston地屈孕酮片)和芬吗通 (Femoston雌二醇片/雌二醇地屈孕酮片复合包装)两种剂型,最初由荷兰苏威制药公司(Solvay Pharmaceuticals)研发,1961年全球上市,目前在全球60多个国家注册,150多个国家有销售。仅中国该药物近几年的年均销售额超过8亿元人民币。地屈孕酮化学式见下式 :

地屈孕酮目前公开的主要合成方法有英国专利GB929271以光甾-4,7,22-三烯-3-酮为原料经4步反应来合成(Scheme 1)。由于每步收率低、起始原料难得到等缺点,基本上不具备工业化生产的可能性。

Scheme 1
美国专利 US3198792公开了以反式孕酮为原料,以四氯苯醌为氧化剂来合成(Scheme 2).虽然路线短,但所用的原料反式孕酮在天然产物中并不存在,需要通过合成才能得到,且目前合成很困难,并无工业化产品,因此目前也不具备工业化生产的可能。

Scheme 2
比利时专利BE656770公开了以9β,10α-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛为原料,在低温下加入饱和的无水乙醇的氯化氢溶液进行脱保护,以60%左右的产率得到地屈孕酮1(Scheme 3)。其中9β,10α-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛6可以以黄体酮为原料,经羰基保护、氧化、腙化、脱腙、光化学反应后合成得到,但其合成路线及工艺均没有公开。

Scheme 3
世界专利WO2016154772也报道了以9β,10α-孕甾-4,6-二烯-3,20-二酮的二乙二缩6为原料,经a、b两步反应来合成地屈孕酮的方法,以5-10%稀硫酸、5-10%稀盐酸、40-60%浓度的乙酸、或对甲苯磺酸在反应温度在为0-90°C下脱保护得到中间体7,然后中间体再在碳酸钠、碳酸钾、氢氧化钠、氢氧化钾、甲醇钠、乙醇钠、乙醇钾、叔丁醇钾等碱条件下完成双键的异构化得到1(Scheme 4)。其中原料9β,10α-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛的合成也没有报道。

Scheme 4
印度专利IN 201811020593则公开了一种以双缩酮保护的黄体酮3为原料,先与二溴二甲基海因在石油醚中发生溴代反应得到中间体4, 中间体4在四丁基氟化铵为碱以甲基四氢呋喃为溶剂条件下发生脱溴反应,得到中间体9α,10β-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛5,然后以汞灯为光源,滤掉其中波长小于260纳米的光后进行光化学反应得到重要中间体9β,10α-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛。最后用HCl在无水乙醇中进行脱保护及双键异构得到地屈孕酮(Scheme 5)。

Scheme 5
世界专利WO2018109622公开了一种以黄体酮为原料,经脱氢、氧化、脱羧、成环及脱氢共五步反应来合成地屈孕酮的路线 (Scheme 6)。

Scheme 6
除此之外,一些专利则公开了其合成关键中间体如9α,10β-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛5和9β,10α-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛6的合成。如中国专利ZL201910484547.6则公布了一种以7-羟基黄体酮为原料来合成9α,10β-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛5的制备方法(Scheme 7)。

Scheme 8
世界专利WO2013078575公布了一种以黄体酮为原料经缩酮保护、氧化、腙化及脱腙共四步反应来合成5的制备方法(Scheme 8)。

Scheme 8
欧洲专利EP0558119,中国专利ZL201410085871.8和ZL201010621400.6公布以中、高压汞灯为光源,以9α,10β-孕甾-4,6-二烯-3,20-二酮的二乙二缩醛(9α,10β -去氢黄体酮二乙二缩醛)5为原料来制备9β,10α -去氢黄体酮二乙二缩酮6的方法(Scheme 9)。

Scheme 9
由于地屈孕酮全球市场很大,仅国内每年的销售额就超过8亿人民币,而目前仅有美国的雅培公司生产销售,且其技术一直保密。而现已公开的一些合成工艺均存在产率不高、污染较大、特别是不容易放大等缺陷,导致除了美国的雅培公司一直能够垄断该药品的生产,而国内因技术原因一直无法实现地区孕酮的仿制,因此研发易于放大的制备地屈孕酮的新工艺,具有重要的经济及社会价值。
发明内容
本发明目提供了一种可工业化合成地屈孕酮的生产工艺,原料易得、总产率高,容易放大至工业化生产。
为实现上述目的,本发明的技术方案为:
一种可工业化合成地屈孕酮的生产工艺,包括以下步骤:
S1. 黄体酮与乙二醇、乙酰氯和原甲酸三甲酯在有机溶剂A中于25~45℃下反应1~
2.5小时,然后淬灭反应,得化合物3 ;
;
S2. 化合物3溶解在溶剂B中,再加入溴代丁二酰亚胺或二溴海因,然后加入自由
基引发剂过氧化苯甲酰或偶氮二异丁腈,加热至65~75℃,进行溴代反应,反应完成后得化
合物4 ;
;
S3. 化合物4加入有机碱催化,在有溶剂C的条件下,氮气/惰性气体保护加热至回流,或在无溶剂的条件下,加热至115~125℃,进行脱溴反应,反应完成后,得化合物5 9α,10β-孕甾-4,6-二烯-3,20-二酮的二乙二缩酮;
S4. 将化合物5溶解于溶剂D中,在氮气/惰性气体保护下接受单一波长的LED光源照射,波长范围为265~300纳米,进行光化学开环反应,得含有开环反应中间体的溶液,然后将含有开环反应中间体的溶液在氮气/惰性气体保护下接受单一波长的LED光源照射,波长范围为305~365纳米,进行光化学闭环反应,待开环反应中间体转化后,分离得化合物6 9β,10α -去氢黄体酮二乙二缩酮;
S5. 化合物6溶解到溶剂E中,加入无机酸进行脱保护反应,反应完成后得化合物7 ;
;
S6. 化合物7溶解到溶剂F中,加入浓盐酸加热反应,或将化合物7溶解于乙酸乙酯/甲醇的饱和HCl的溶液中进行反应,反应完成后得目标产物地屈孕酮。
优选的,所述有机溶剂A为四氢呋喃、甲基叔丁基醚、异丙醚、甘醇二甲醚或甘醇二乙醚中的一种或一种以上混合溶剂;
所述溶剂B为二氧六烷、异辛烷、90-120°C沸点的石油醚、环己烷、四氢呋喃或甲基四氢呋喃中的一种或一种以上混合溶剂;
所述溶剂C为环己烷或甲基四氢呋喃中的一种或两种混合溶剂;
所述溶剂D为甲醇、乙醇、异丙醇、甘醇二甲醚、甘醇二乙醚、四氢呋喃、甲基四氢呋喃或环己烷;
所述溶剂E为四氢呋喃、乙腈、丙酮或甲醇中的一种与水的混合溶剂;
所述溶剂F为四氢呋喃、甘醇二甲醚、甲醇或乙醇中的一种或两种混合溶剂。
其中,溶剂E的更好方案为;四氢呋喃/水=3/1、乙腈/水=3/1,丙酮/水=3/1,甲醇/水=3/1或乙醇/水=3/1,上述均为体积比。
优选的,步骤S1中,所述淬灭反应是加入三乙胺、吡啶或二异丙基乙胺进行的;
淬灭反应后,还包括:在-15℃~0℃下冷却,过滤,所得固体用水洗涤,再用乙醇或丙酮洗涤,干燥得化合物3的粗产物;
优选的,化合物3的粗产物加入丙酮和吡啶,加热至回流并搅拌0.5~1.5小时后,-15~-5℃冷却,过滤,干燥得精制的化合物3。
优选的,步骤S2中,所述溴代丁二酰亚胺为1.1-1.2eq;所述二溴海因为0.55-0.6eq,所述自由基引发剂为0.01-0.05 eq;
溴代反应完成后,还包括:用溶剂B洗滤饼,滤饼再用二氯甲烷溶解,饱和碳酸氢钠溶液洗涤二氯甲烷溶液,干燥,除去干燥剂,在低于40℃下回收二氯甲烷,得化合物4。
优选的,步骤S3中,所述有机碱为三乙胺、1,8-二氮杂二环十一碳-7-烯、二异丙基乙胺或2,4,6-三甲基吡啶;
脱溴反应完成后还包括:减压回收溶剂及有机碱催化剂,然后加入乙腈和甲醇混合溶剂或丙酮和乙腈混合溶剂或丙酮和水混合溶剂,冷却至60℃以下,加二氧六烷或甲基叔丁基醚或丁醚或异丙醚,-15~-5℃冷却,析出固体,过滤,并依次用水、甲醇或乙腈或丙酮洗涤固体,干燥得化合物5。其中,更优选的,本发明提供了一种溶剂的更优体积配比,乙腈/甲醇=1/1、丙酮/乙腈=4/1或丙酮/水=4/1。
优选的,步骤S4中,反应温度为-35~35℃,整个反应过程中,控制反应温度的范围上下浮动不超过10℃。
优选的,步骤S4中,所述分离包括:将含有9β,10α -去氢黄体酮二乙二缩酮的溶液,减压蒸馏回收溶剂,再与纯化溶剂加热回流,冷却,有固体析出,分离固体和液体,取液体回收溶剂,即得9β,10α -去氢黄体酮二乙二缩酮;
所述纯化溶剂包括:乙醇和乙腈混合溶剂、甲醇和乙腈混合溶剂、甲醇和四氢呋喃混合溶剂、丙酮和四氢呋喃混合溶剂、乙腈和四氢呋喃混合溶剂、甲醇或异丙醇。其中,更优选的,本发明提供了一种溶剂的更优体积配比,乙醇/乙腈=3/1、甲醇/乙腈=4/1、甲醇/四氢呋喃=4/1、丙酮/四氢呋喃 =3/1或乙腈/四氢呋喃=4/1。
优选的,步骤S5中,脱保护反应完成后还包括:除去溶剂,用乙酸乙酯或DCM萃取,碳酸氢钠液洗涤,干燥,与丙酮和甲醇混合溶剂或丙酮和异丙醇混合溶剂加热回流后冷却结晶,分离固体即得化合物7。本发明提供了一种加热回流溶剂的更优体积配比,丙酮/甲醇=1/3或丙酮/异丙醇=1/3。
优选的,步骤S6中,反应完成后还包括:回收溶剂,二氯甲烷萃取,碳酸氢钠液洗涤,干燥,除去溶剂后,加入乙酸乙酯和异辛烷混合溶剂或乙酸乙酯和环己烷混合溶剂或乙酸乙酯和正庚烷混合溶剂重结晶,最后用异丙醇精制,得目标产物地屈孕酮。其中,重结晶溶剂的更优体积配比为,乙酸乙酯/异辛烷=1/5、乙酸乙酯/环己烷=1/5或乙酸乙酯/正庚烷=1/5。
本发明以易得的黄体酮为原料,通过羰基保护、溴代、脱溴、光化学开环反应、光化学闭环反应、脱保护、双键异构的步骤制得地屈孕酮,具有起始原料易得、各步骤均易实现、收率较高的优点,且操作简便、绿色环保,易于放大至工业生产。
本发明的关键步骤将9α,10β -去氢黄体酮二乙二缩酮利用LED光源制备出9β,10α-去氢黄体酮二乙二缩酮,在光化学开环反应中,开环反应的转化率可超过90%,进一步进行光化学闭环反应时,有20-75%左右的原料转化成目标产物,总产率高,操作简便,控制点少,容易进行放大生产,且引入的溶剂少,制备过程能耗低,是相较于现有技术汞灯制备更绿色环保、反应副产物更少的技术。
本发明提供了能工业化生产地屈孕酮的方法,使国内自行生产地屈孕酮具备了可能性,具有非常重要经济及社会价值。
附图说明
图1是化合物5的1H-NMR谱图。
图2是化合物5的13C-NMR谱图。
图3是化合物6的1H-NMR谱图。
图4是化合物6的13C-NMR谱图。
图5是目标产物地屈孕酮的1H-NMR谱图。
图6是目标产物地屈孕酮的13C-NMR谱图。
图7是目标产物地屈孕酮的HHCOSY谱图。
图8是目标产物地屈孕酮的NOE谱图。
图9是目标产物地屈孕酮的HMQC谱图。
图10是目标产物地屈孕酮的HMBC谱图。
图11是光化学开环反应和光化学闭环反应的反应系统示意图。
具体实施方式
以下结合具体实施例对本发明作进一步说明,但本发明的保护范围不限于以下实施例。
本实施例的制备方法如Scheme 10所示;
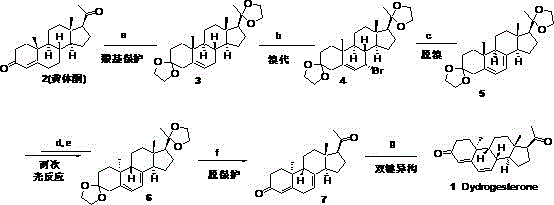
Scheme 10
实施例1
称取2kg黄体酮(化合物2)加入到20L反应罐中,再依次加入4L乙二醇、5.5L四氢呋
喃、0.1 L乙酰氯,慢慢滴加2L原甲酸三甲酯,温度控制在35±5℃,反应2小时±10分钟后,
加0.2 L吡啶淬灭反应,-10℃±5℃冷却0.5小时,过滤,并依次用6L水洗、3L丙酮洗,抽干,
40℃以下真空干燥得化合物3 的粗产物。
的粗产物。
将化合物3粗产物投入20L反应罐中,加入6L丙酮、4毫升吡啶,水浴加热至回流,搅拌1小时,-10℃±5℃冷却0.5小时,抽滤,40℃真空干燥过夜得化合物3的纯品,产率91%。
实施例 2
分别用甲基叔丁基醚、异丙醚、甘醇二甲醚或甘醇二乙醚代替四氢呋喃,其它与实施例 1相同,化合物3的产率分别为80%、83%、89%和85%。
实施例3
称取1 kg缩酮保护的黄体酮(化合物3)加入到50 L反应罐中,加入二氧六烷20 L,再加150毫升DBU,氮气(或氩气)保护下加热至55~65℃是的原料溶解完全,加入1.15当量溴代丁二酰亚胺(NBS)、偶氮二异丁腈(AIBN)5g,65~75℃下反应1 h。抽滤,固体用DCM溶解,饱和碳酸氢钠溶液洗涤,干燥,减压蒸干滤液,得类白色至淡黄色固体化合物4。
将化合物4投入反应罐中,加3.5 L 有机碱TEA,氮气保护下加热至120℃±5℃,温度反应2 h,减压回收2/3左右的TEA,然后加入3 L乙腈/甲醇=1/1,冷却至60℃以下,加500毫升二氧六烷,-10℃±5℃冷却,析晶0.5小时,过滤,并依次用1.5L水、1.5L丙酮洗,抽干,40℃干燥过夜,得淡黄色产物化合物5,产率80%。
实施例4
分别用异辛烷、90-120°C沸点的石油醚、环己烷、四氢呋喃或甲基四氢呋喃代替二氧六烷,其它与实施例3相同,化合物5的产率81%、71%、80%、83%和85%。
实施例5
分别用二异丙基乙胺或三甲基吡啶代替TEA,其它与实施例3相同,化合物5的产率分别为86%和75%。
实施例6
用二溴海因代替NBS,其它与实施例3相同,化合物5的产率79%。
实施例7
分别用3 L丙酮/乙腈=4/1或3 L丙酮/水=4/1代替实施例3中的3 L乙腈/甲醇=1/1,其它与实施例3相同,化合物5的产率非标为79%和70%。
实施例8
分别用500毫升甲基叔丁基醚、500毫升丁醚或500毫升异丙醚代替实施例 3中的500毫升二氧六烷,其它与实施例 3相同,化合物5的产率非标为78%、73%和74%。
化合物5的核磁共振结构表征数据: 1H NMR (400 MHz, CDCl3) 5.55~5.57(m,1H), 5.37~5.39(m, 1H), 3.86~4.00(m, 8H), 2.58(dd, J= 15.0, 1.5Hz, 1H), 2.27(dd, J=15.0, 2.8Hz, 1H), 2.05~ 2.17(m, 2H), 1.71~1.89(m, 9H), 1.59~1.62(m,2H), 1.42~1.48(m, 1H), 1.30(s, 3H), 1.25(dd, J= 12.8, 4.4 Hz, 1H), 0.96(s,3H), 0.72(s, 3H).13C NMR (101 MHz, CDCl3) δ 140.72, 139.19, 120.15, 116.76,112.10, 108.90, 65.15, 64.58, 64.45, 63.47, 57.88, 54.52, 46.10, 42.46,40.35, 38.98, 37.55, 37.03, 31.48, 24.64, 23.18, 22.75, 21.10, 16.30, 13.09.
谱图如图1和2所示。
实施例9
本实施例的光化学反应,是最关键的技术,所采用的反应系统包括进料罐、第一光化学反应器、第二光化学反应器和接收罐。结合图11所示,反应装置中设置有流量计以在反应过程中对溶液流速进行控制,流量计的位置可以适当设置,本实施例是以设置在进料罐和第一光化学反应器之间为例。溶液流速通过利用进料罐与接收罐的高度差或相对角度大小来调整,当然也可以通过阀门来调整。进料罐、第一光化学反应器、第二光化学反应器、接收罐中均匹配有冷凝系统。第一光化学反应器和第二光化学反应器的光源均为通过光电技术产生的的LED光源。第一光化学反应器和第二光化学反应器直接相连,在第一光化学反应器进行光化学开环反应,在第二光化学反应器中进行光化学闭环反应,气体钢瓶中有氮气或是惰性气体,与进料罐连接,使反应系统充入氮气或惰性气体保护光化学开关反应和光化学闭环反应。
将100克化合物5溶解于8 L反应溶剂甲醇,氩气保护,光化学开环反应波长为275纳米,光化学闭环反应的波长为335纳米,然后打开流量计,反应温度控制在0±5°C。
反应时间约30小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约40%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约57%,其它杂质含量约3%。。
实施例10
光化学开环反应波长改为280纳米,光化学闭环反应的波长改为345纳米,反应溶剂为10L四氢呋喃,其它条件同实施例9。
反应时间约8小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约41%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约54%,其它杂质含量约5%。
实施例11
光化学开环反应的波长改为295纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L乙醇,其它条件同实施例9。
反应时间约12小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约60%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约25%,其它杂质含量约15%。
实施例12
光化学开环反应波长改为265纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L乙醇,其它条件同实施例9。
反应时间约10小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约50%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约30%,其它杂质含量约20%。
实施例13
光化学开环反应波长改为280纳米,光闭环反应的波长改为350纳米,反应溶剂为10L四氢呋喃,其它条件同实施例9。
反应时间约12小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约51%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约40%,其它杂质含量约9%。
实施例14
光化学开环反应波长改为280纳米,光化学闭环反应的波长改为360纳米,反应溶剂为10L四氢呋喃,反应温度控制在-0±5°C,其它条件同实施例9。
反应时间约15小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约65%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约30%,其它杂质含量约5%。
实施例15
光化学开环反应波长改为280纳米,光化学闭环反应的波长改为365纳米,反应溶剂为10L四氢呋喃,其它条件同实施例9。
反应时间约24小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约65%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约20%,其它杂质含量约15%。
实施例16
光化学开环反应波长改为280纳米,光闭环反应的波长改为365纳米,反应溶剂为10L甲基四氢呋喃,其它条件同实施例9。
反应时间约20小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约65%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约20%,其它杂质含量约15%。
实施例17
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L甲醇,将反应温度控制在-15±5°C,其它条件同实施例9。
反应时间约4小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约30%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约62%,其它杂质含量约8%。
实施例18
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L甲醇,将反应温度控制在30±5°C,其它条件同实施例9。
反应时间约3.2小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约50%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约43%,其它杂质含量约7%。
实施例19
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应原料为50g,溶剂用量为4L,其它条件同实施例9。
反应时间约28小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约40%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约55%,其它杂质含量约5%。
实施例20
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L乙醇,其它条件同实施例9。
反应时间约3.6小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约46%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约50%,其它杂质含量约4%。
实施例21
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L异丙醇,其它条件同实施例9。
反应时间约4.2小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约50%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约45%,其它杂质含量约5%。
实施例22
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L甘醇二甲醚,其它条件同实施例9。
反应时间约5小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约50%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约43%,其它杂质含量约7%。
实施例22
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L甘醇二甲乙醚,其它条件同实施例9。
反应时间约5小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约51%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约43%,其它杂质含量约6%。
实施例23
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L甲基四氢呋喃,其它条件同实施例9。
反应时间约5小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约50%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约44%,其它杂质含量约6%。
实施例24
光化学开环反应波长改为270纳米,光化学闭环反应的波长改为330纳米,反应溶剂为10L环己烷,其它条件同实施例9。
反应时间约5小时,HPLC检测结果显示粗产物中原料9α,10β -去氢黄体酮二乙二缩酮的含量约48%,目标产物9β,10α -去氢黄体酮二乙二缩酮的含量约45%,其它杂质含量约7%。
实施例25
将实施例9的反应液出料,减压蒸馏回收溶剂,然后加入150毫升乙醇/乙腈=3/1(体积比)的混合溶剂,加热回流10分钟,-5℃下冷却20分钟,大部分9α,10β -去氢黄体酮二乙二缩酮直接从混合物中析出,抽滤回收得38克化合物5。滤液减压回收溶剂,然后0℃下冷却20分钟,抽滤回收得61克9β,10α -去氢黄体酮二乙二缩酮的粗产品,粗品收率61%。可用异丙醇进一步重结晶纯化。
化合物6的核磁共振结构表征数据:1H NMR (400 MHz, CDCl3) 5.61(dd, J=5.2,2.4Hz, 1H), 5.43~5.44(m, 1H), 3.84~4.01(m, 8H), 2.48~2.56(m, 2H), 2.26~2.29(m, 2H),2.02~2.10(m, 2H), 1.71~1.84(m, 6H), 1.40~1.60(m, 5H),1.30(s, 3H),0.76(s, 3H), 0.70(s, 3H).13C NMR (101 MHz, CDCl3) δ 140.55, 140.31, 120.64,115.90, 111.83, 109.31, 65.51, 64.68, 64.44, 63.37, 59.46, 49.83, 45.70,41.35, 40.46, 37.30, 36.76, 36.60, 31.41, 24.54, 23.30, 22.17, 19.72, 19.22,13.63.
谱图如图3和4所示。
实施例26
分别用甲醇/乙腈=4/1、甲醇/四氢呋喃=4/1、丙酮/四氢呋喃=3/1、乙腈/四氢呋喃=4/1、甲醇、异丙醇代替实施例25中的乙醇/乙腈=3/1,粗品收率分别为:60%、56%、54%、51%、56%、63%。
实施例27
将5克化合物6的粗产物溶解到30毫升四氢呋喃/水=3:1的混合溶剂中,加入10毫升醋酸,回流反应,反应完成后,减压除去溶剂,用DCM萃取,碳酸氢钠液洗涤,干燥,减压蒸馏回收溶剂,再用丙酮/甲醇=1:3回流5~10分钟后冷却结晶,抽滤即可得到中间体7,可直接用于下一步反应。
这一步脱缩酮保护反应可以用乙腈/水=3:1、丙酮/水=3:1、甲醇/水=3:1或乙醇/水=3:1的混合溶剂代替四氢呋喃/水=3:1的混合溶剂;或用三氟乙酸、2N盐酸等代替醋酸,其它实验操作相同。
实施例28
将5克化合物7的溶解于50毫升四氢呋喃,再加入10毫升浓盐酸,-5℃下搅拌,反应完成后,减压除去溶剂,粗产品溶解于DCM,碳酸氢钠液洗涤,干燥,用乙酸乙酯/正己烷=1:5重结晶,然后再用异丙醇精制,即可得到地屈孕酮,产率85%。
实施例 29
用甘醇二甲醚、甲醇代替四氢呋喃,其它条件与实施例28相同,产率分别为82%,80%。
实施例 30
用乙酸乙酯、四氢呋喃或甲醇的饱和HCl的溶液代替四氢呋喃加浓盐酸,-20±5℃下搅拌反应,其它条件与实施例28相同,产率分别为86%、88%、85%。
实施例 31
改用乙酸乙酯/异辛烷=1/5或乙酸乙酯/环己烷=1/5或乙酸乙酯/正庚烷=1/5重结晶,其它条件与实施例 28相同,产率分别为83%、85%和81%。
目标产物(化合物1)的核磁共振结构表征数据:1H NMR (400 MHz, CDCl3) δ6.13~6.19(m, 1H), 5.66(s, 1H), 2.41~2.56(m, 4H), 2.19~2.29(m, 2H), 2.12(s, 3H),1.94~1.98(m, 2H), 1.62~1.87(m, 7H), 1.31~1.55(m, 1H), 1.29(s, 3H), 0.69(s,3H).13C NMR (101 MHz, CDCl3) δ 209.03, 199.48, 163.06, 140.52, 127.18, 123.99,63.48, 49.96, 44.33, 39.77, 38.68, 37.82, 37.28, 35.68, 34.05, 31.60, 25.25,22.66, 22.39, 20.65, 12.17.
谱图如图7-10所示。
Claims (10)
1.一种可工业化合成地屈孕酮的生产工艺,其特征在于包括以下步骤:
S1. 黄体酮与乙二醇、乙酰氯和原甲酸三甲酯在有机溶剂A中于25~45℃下反应1~2.5
小时,然后淬灭反应,得化合物3 ;
;
S2. 化合物3溶解在溶剂B中,再加入溴代丁二酰亚胺或二溴海因,然后加入自由基引
发剂过氧化苯甲酰或偶氮二异丁腈,加热至65~75℃,进行溴代反应,反应完成后得化合物4 ;
;
S3. 化合物4加入有机碱催化,在有溶剂C的条件下,氮气/惰性气体保护加热至回流,或在无溶剂的条件下,加热至115~125℃,进行脱溴反应,反应完成后,得化合物5 9α,10β-孕甾-4,6-二烯-3,20-二酮的二乙二缩酮;
S4. 将化合物5溶解于溶剂D中,在氮气/惰性气体保护下接受波长范围为265~300纳米的LED光源照射,进行光化学开环反应,得含有开环反应中间体的溶液,然后将含有开环反应中间体的溶液在氮气/惰性气体保护下接受波长范围为305~365纳米的LED光源照射,进行光化学闭环反应,待开环反应中间体转化后,分离得化合物6 9β,10α -去氢黄体酮二乙二缩酮;
S5. 化合物6溶解到溶剂E中,加入无机酸进行脱保护反应,反应完成后得化合物7 ;
;
S6. 化合物7溶解到溶剂F中,加入浓盐酸加热反应,或将化合物7溶解于乙酸乙酯、THF或甲醇的饱和HCl的溶液中进行反应,反应完成后得目标产物地屈孕酮。
2.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
所述有机溶剂A为四氢呋喃、甲基叔丁基醚、异丙醚、甘醇二甲醚或甘醇二乙醚中的一种或一种以上混合溶剂;
所述溶剂B为二氧六烷、异辛烷、90-120°C沸点的石油醚、环己烷、四氢呋喃或甲基四氢呋喃中的一种或一种以上混合溶剂;
所述溶剂C为环己烷或甲基四氢呋喃;
所述溶剂D为甲醇、乙醇、异丙醇、甘醇二甲醚、甘醇二乙醚、四氢呋喃、甲基四氢呋喃或环己烷;
所述溶剂E为四氢呋喃、乙腈、丙酮或甲醇中的一种或两者的混合溶剂;
所述溶剂F为四氢呋喃、甘醇二甲醚、甲醇或乙醇中的一种或两种混合溶剂。
3.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
步骤S1中,所述淬灭反应是加入三乙胺、吡啶或二异丙基乙胺进行的;
淬灭反应后,还包括:在-15℃~0℃下冷却,过滤,所得固体用水洗涤,再用乙醇或丙酮洗涤,干燥得化合物3的粗产物。
4.根据权利要求3所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
化合物3的粗产物加入丙酮和吡啶,加热至回流并搅拌0.5~1.5小时后,-15~-5℃冷却,过滤,干燥得精制的化合物3。
5.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
步骤S2中,所述溴代丁二酰亚胺为1.1-1.2eq;所述二溴海因为0.55-0.6 eq,所述自由基引发剂为0.01-0.05 eq;
溴代反应完成后,还包括:用反应溶剂B洗滤饼,滤饼再用二氯甲烷溶解,饱和碳酸氢钠溶液洗涤二氯甲烷溶液,干燥,在低于40℃下回收二氯甲烷,得化合物4。
6.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
步骤S3中,所述有机碱为三乙胺、1,8-二氮杂二环十一碳-7-烯、二异丙基乙胺或2,4,6-三甲基吡啶;
脱溴反应完成后还包括:减压回收溶剂及有机碱催化剂,然后加入乙腈和甲醇混合溶剂或丙酮和乙腈混合溶剂或丙酮和水混合溶剂,冷却至60℃以下,加二氧六烷或甲基叔丁基醚或丁醚或异丙醚,-15~-5℃冷却,析出固体,过滤,并依次用水、甲醇或乙腈或丙酮洗涤固体,干燥得化合物5。
7.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
步骤S4中,反应温度为-35~35℃,整个反应过程中,控制反应温度的范围上下浮动不超过10℃。
8.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
步骤S4中,所述分离包括:将含有9β,10α -去氢黄体酮二乙二缩酮的溶液,减压蒸馏回收溶剂,再与纯化溶剂加热回流,冷却,有固体析出,分离固体和液体,取液体回收溶剂,即得9β,10α -去氢黄体酮二乙二缩酮;
所述纯化溶剂包括:乙醇和乙腈混合溶剂、甲醇和乙腈混合溶剂、甲醇和四氢呋喃混合溶剂、丙酮和四氢呋喃混合溶剂、乙腈和四氢呋喃混合溶剂、甲醇或异丙醇。
9.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
步骤S5中,所述无机酸为醋酸、三氟乙酸或2N盐酸;
脱保护反应完成后还包括:除去溶剂,用乙酸乙酯或DCM萃取,碳酸氢钠液洗涤,干燥,与丙酮和甲醇混合溶剂或丙酮和异丙醇混合溶剂加热回流后冷却结晶,分离固体即得化合物7。
10.根据权利要求1所述的可工业化合成地屈孕酮的生产工艺,其特征在于:
步骤S6中, 反应完成后还包括:回收溶剂,二氯甲烷萃取,碳酸氢钠液洗涤,干燥,除去溶剂后,加入乙酸乙酯和异辛烷混合溶剂或乙酸乙酯和环己烷混合溶剂或乙酸乙酯和正庚烷混合溶剂重结晶,最后用异丙醇精制,得目标产物地屈孕酮。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911102923.7A CN110818760B (zh) | 2019-11-12 | 2019-11-12 | 一种可工业化合成地屈孕酮的生产工艺 |
| PCT/CN2020/120276 WO2021093494A1 (zh) | 2019-11-12 | 2020-10-12 | 一种可工业化合成地屈孕酮的生产工艺 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201911102923.7A CN110818760B (zh) | 2019-11-12 | 2019-11-12 | 一种可工业化合成地屈孕酮的生产工艺 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN110818760A CN110818760A (zh) | 2020-02-21 |
| CN110818760B true CN110818760B (zh) | 2021-06-25 |
Family
ID=69554535
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201911102923.7A Active CN110818760B (zh) | 2019-11-12 | 2019-11-12 | 一种可工业化合成地屈孕酮的生产工艺 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN110818760B (zh) |
| WO (1) | WO2021093494A1 (zh) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110818760B (zh) * | 2019-11-12 | 2021-06-25 | 广西师范大学 | 一种可工业化合成地屈孕酮的生产工艺 |
| CN111171101B (zh) * | 2020-01-03 | 2023-04-11 | 宁波东隆智能科技有限公司 | 一种地屈孕酮中间体的制备方法 |
| CN113387991B (zh) * | 2020-03-13 | 2024-06-18 | 苏州朗科生物技术股份有限公司 | 一种合成地屈孕酮的方法及化合物 |
| CN113880904B (zh) * | 2020-07-01 | 2024-04-12 | 苏州朗科生物技术股份有限公司 | 合成地屈孕酮的新方法及化合物 |
| CN112409434B (zh) * | 2020-11-27 | 2021-10-26 | 厦门欧瑞捷生物科技有限公司 | 一种去氢孕酮的合成方法 |
| CN112812146A (zh) * | 2021-01-20 | 2021-05-18 | 江苏诺维尔医药科技有限公司 | 一种合成地屈孕酮的方法 |
| CN115215917B (zh) * | 2021-04-16 | 2024-01-26 | 扬州奥锐特药业有限公司 | 一种硫醚类化合物、其制备方法和应用 |
| CN113666981A (zh) * | 2021-08-27 | 2021-11-19 | 江西百思康瑞药业有限公司 | 一种地屈孕酮的合成方法 |
| CN114249790B (zh) * | 2021-09-03 | 2022-10-18 | 扬州奥锐特药业有限公司 | 一种甾体化合物的制备方法 |
| WO2023035906A1 (zh) * | 2021-09-08 | 2023-03-16 | 湖南醇康医药科技有限公司 | 中间体化合物及其制备方法和应用 |
| CN114057820B (zh) * | 2021-11-15 | 2023-06-27 | 湖南科瑞生物制药股份有限公司 | 一种地屈孕酮的精制方法 |
| CN114380878A (zh) * | 2021-12-15 | 2022-04-22 | 河南利华制药有限公司 | 一种氟米松的合成方法 |
| CN114437164B (zh) * | 2022-01-26 | 2024-01-30 | 西安国康瑞金制药有限公司 | 一种地屈孕酮及其制备方法 |
| CN115894593B (zh) * | 2022-11-12 | 2024-03-08 | 药康众拓(江苏)医药科技有限公司 | 一种地屈孕酮及其中间体的制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101668739A (zh) * | 2007-04-24 | 2010-03-10 | 帝斯曼知识产权资产管理有限公司 | 用于制造前维生素d的光化学方法 |
| IN201811020593A (zh) * | 2018-06-01 | 2018-06-22 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101318982A (zh) * | 2007-08-03 | 2008-12-10 | 台州市万福制药有限公司 | 3-乙酰氧基-9β,10α-孕甾-5,7-二烯-20-酮的合成方法 |
| EP2566482A1 (en) * | 2010-05-07 | 2013-03-13 | Institut National de la Santé et de la Recherche Médicale | Progesterone receptor antagonists and uses thereof |
| CN102558272B (zh) * | 2010-12-24 | 2014-04-23 | 中国科学院理化技术研究所 | 光化学异构化反应合成9-β,10-α-去氢黄体酮缩酮的方法 |
| CN110818760B (zh) * | 2019-11-12 | 2021-06-25 | 广西师范大学 | 一种可工业化合成地屈孕酮的生产工艺 |
-
2019
- 2019-11-12 CN CN201911102923.7A patent/CN110818760B/zh active Active
-
2020
- 2020-10-12 WO PCT/CN2020/120276 patent/WO2021093494A1/zh active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101668739A (zh) * | 2007-04-24 | 2010-03-10 | 帝斯曼知识产权资产管理有限公司 | 用于制造前维生素d的光化学方法 |
| IN201811020593A (zh) * | 2018-06-01 | 2018-06-22 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110818760A (zh) | 2020-02-21 |
| WO2021093494A1 (zh) | 2021-05-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN110818760B (zh) | 一种可工业化合成地屈孕酮的生产工艺 | |
| CN110790807B (zh) | 利用LED光源制备9β,10α-去氢黄体酮二乙二缩酮的方法 | |
| MX2007006701A (es) | Proceso para la preparacion de drospirenona. | |
| CN109096122B (zh) | 制备亚精胺的方法 | |
| CN107531746A (zh) | 9β,10α‑孕甾‑4,6‑二烯‑3,20‑二酮的制备方法 | |
| CN114524795B (zh) | 一种改进的Rhodomyrtone制备方法 | |
| CN113816841A (zh) | 一种环丙基甲基酮的制备方法 | |
| CN105175317B (zh) | 一种制备匹可硫酸钠的方法 | |
| CN114014903B (zh) | 麦角甾醇及其衍生物的合成方法 | |
| CN113278021B (zh) | 1,7-二氮杂螺[3.5]壬烷-7-甲酸叔丁酯及其草酸盐的制备方法 | |
| CN114105872B (zh) | 一种用于制备盐酸丙卡特罗的中间体及其制备方法 | |
| CN105384790A (zh) | 一种泼尼松龙的制备方法 | |
| CN111592491B (zh) | 左旋盐酸去甲基苯环壬酯的制备方法 | |
| CN110872225B (zh) | 一种巴洛沙韦中间体的制备方法 | |
| CN114478837A (zh) | 一种舒更葡糖钠衍生物的制备方法 | |
| CN112679512B (zh) | 曲贝替定中间体及其制备方法 | |
| CN109232222B (zh) | 一种(e)-辛-4-烯-1,8-二酸的制备方法 | |
| CN104804008B (zh) | 一种工业化生产甲磺酸特拉替尼的方法 | |
| CN114249790B (zh) | 一种甾体化合物的制备方法 | |
| CN112159358A (zh) | 一种噁拉戈利中间体的制备方法 | |
| CN105884687A (zh) | 一种5-苄基苄达明的制备方法 | |
| CN111217709A (zh) | 一种(1-氟环丙基)甲胺盐酸盐的制备方法 | |
| CN114213323B (zh) | 一种盐酸丙卡特罗的合成新工艺 | |
| CN118652255B (zh) | 一种己酮可可碱的工艺改进方法 | |
| CN109912552B (zh) | 一种布格呋喃及其中间体的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PB01 | Publication | ||
| PB01 | Publication | ||
| SE01 | Entry into force of request for substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |