CN105229026B - 人类IgG1 FC区变体及其用途 - Google Patents
人类IgG1 FC区变体及其用途 Download PDFInfo
- Publication number
- CN105229026B CN105229026B CN201380073913.XA CN201380073913A CN105229026B CN 105229026 B CN105229026 B CN 105229026B CN 201380073913 A CN201380073913 A CN 201380073913A CN 105229026 B CN105229026 B CN 105229026B
- Authority
- CN
- China
- Prior art keywords
- region
- antibody
- variant
- binding
- amino acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 241000282414 Homo sapiens Species 0.000 title claims description 292
- 230000004540 complement-dependent cytotoxicity Effects 0.000 claims abstract description 276
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 270
- 102000004196 processed proteins & peptides Human genes 0.000 claims abstract description 261
- 229920001184 polypeptide Polymers 0.000 claims abstract description 256
- 239000000427 antigen Substances 0.000 claims abstract description 192
- 108091007433 antigens Proteins 0.000 claims abstract description 192
- 102000036639 antigens Human genes 0.000 claims abstract description 192
- 230000035772 mutation Effects 0.000 claims description 444
- 230000027455 binding Effects 0.000 claims description 336
- 238000000034 method Methods 0.000 claims description 187
- 150000001413 amino acids Chemical class 0.000 claims description 123
- 108060003951 Immunoglobulin Proteins 0.000 claims description 91
- 102000018358 immunoglobulin Human genes 0.000 claims description 91
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 72
- 230000001965 increasing effect Effects 0.000 claims description 68
- 239000000203 mixture Substances 0.000 claims description 65
- 102100026120 IgG receptor FcRn large subunit p51 Human genes 0.000 claims description 60
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 claims description 55
- 239000003814 drug Substances 0.000 claims description 45
- 101710177940 IgG receptor FcRn large subunit p51 Proteins 0.000 claims description 43
- 238000003556 assay Methods 0.000 claims description 42
- 230000024203 complement activation Effects 0.000 claims description 41
- 238000002965 ELISA Methods 0.000 claims description 26
- 108010068617 neonatal Fc receptor Proteins 0.000 claims description 23
- 230000007423 decrease Effects 0.000 claims description 22
- 206010028980 Neoplasm Diseases 0.000 claims description 21
- 239000003053 toxin Substances 0.000 claims description 18
- 231100000765 toxin Toxicity 0.000 claims description 18
- 201000011510 cancer Diseases 0.000 claims description 15
- 201000010099 disease Diseases 0.000 claims description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 12
- 238000002560 therapeutic procedure Methods 0.000 claims description 8
- 239000012530 fluid Substances 0.000 claims description 7
- 238000011282 treatment Methods 0.000 claims description 7
- 238000002835 absorbance Methods 0.000 claims description 6
- 238000004519 manufacturing process Methods 0.000 claims description 6
- 241000124008 Mammalia Species 0.000 claims description 5
- 230000008859 change Effects 0.000 claims description 4
- 238000003384 imaging method Methods 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 2
- 230000003993 interaction Effects 0.000 abstract description 43
- 230000001976 improved effect Effects 0.000 abstract description 18
- 210000004027 cell Anatomy 0.000 description 343
- 235000001014 amino acid Nutrition 0.000 description 157
- 125000000539 amino acid group Chemical group 0.000 description 96
- 239000012636 effector Substances 0.000 description 93
- 238000006467 substitution reaction Methods 0.000 description 84
- 230000006870 function Effects 0.000 description 68
- 125000003275 alpha amino acid group Chemical group 0.000 description 58
- 230000000295 complement effect Effects 0.000 description 45
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 42
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 41
- 108090000623 proteins and genes Proteins 0.000 description 41
- 230000001404 mediated effect Effects 0.000 description 40
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 39
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 39
- 125000005647 linker group Chemical group 0.000 description 35
- 230000009089 cytolysis Effects 0.000 description 34
- 235000018102 proteins Nutrition 0.000 description 34
- 102000004169 proteins and genes Human genes 0.000 description 34
- 229940079593 drug Drugs 0.000 description 28
- 230000000694 effects Effects 0.000 description 28
- 210000002845 virion Anatomy 0.000 description 28
- 239000012634 fragment Substances 0.000 description 26
- 238000002474 experimental method Methods 0.000 description 25
- 230000002829 reductive effect Effects 0.000 description 25
- 210000002966 serum Anatomy 0.000 description 25
- 230000004044 response Effects 0.000 description 23
- 230000006698 induction Effects 0.000 description 20
- 229940002612 prodrug Drugs 0.000 description 20
- 239000000651 prodrug Substances 0.000 description 20
- 230000001225 therapeutic effect Effects 0.000 description 19
- 210000000170 cell membrane Anatomy 0.000 description 18
- 239000013598 vector Substances 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 241000699666 Mus <mouse, genus> Species 0.000 description 17
- 230000015572 biosynthetic process Effects 0.000 description 17
- 102000009109 Fc receptors Human genes 0.000 description 16
- 108010087819 Fc receptors Proteins 0.000 description 16
- 241000699670 Mus sp. Species 0.000 description 16
- 230000005888 antibody-dependent cellular phagocytosis Effects 0.000 description 16
- 150000007523 nucleic acids Chemical group 0.000 description 16
- 230000001580 bacterial effect Effects 0.000 description 15
- 239000002953 phosphate buffered saline Substances 0.000 description 15
- 230000009467 reduction Effects 0.000 description 15
- 238000006722 reduction reaction Methods 0.000 description 15
- 241000191967 Staphylococcus aureus Species 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 14
- 239000003795 chemical substances by application Substances 0.000 description 14
- 230000002147 killing effect Effects 0.000 description 14
- 238000002360 preparation method Methods 0.000 description 14
- -1 substituted amino acid Chemical class 0.000 description 14
- 238000012360 testing method Methods 0.000 description 14
- 108700012359 toxins Proteins 0.000 description 14
- 229940049595 antibody-drug conjugate Drugs 0.000 description 13
- 230000009827 complement-dependent cellular cytotoxicity Effects 0.000 description 13
- 238000012217 deletion Methods 0.000 description 13
- 230000037430 deletion Effects 0.000 description 13
- 230000014509 gene expression Effects 0.000 description 13
- 230000004048 modification Effects 0.000 description 13
- 238000012986 modification Methods 0.000 description 13
- 102000039446 nucleic acids Human genes 0.000 description 13
- 108020004707 nucleic acids Proteins 0.000 description 13
- 239000008194 pharmaceutical composition Substances 0.000 description 13
- 241000894006 Bacteria Species 0.000 description 12
- 206010057249 Phagocytosis Diseases 0.000 description 12
- 108010000499 Thromboplastin Proteins 0.000 description 12
- 102100030859 Tissue factor Human genes 0.000 description 12
- 239000000611 antibody drug conjugate Substances 0.000 description 12
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 12
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 12
- 238000001727 in vivo Methods 0.000 description 12
- 238000003780 insertion Methods 0.000 description 12
- 230000037431 insertion Effects 0.000 description 12
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 12
- 230000008782 phagocytosis Effects 0.000 description 12
- 108010073807 IgG Receptors Proteins 0.000 description 11
- 102000009490 IgG Receptors Human genes 0.000 description 11
- 238000003776 cleavage reaction Methods 0.000 description 11
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 11
- 108700019146 Transgenes Proteins 0.000 description 10
- 108010047295 complement receptors Proteins 0.000 description 10
- 102000006834 complement receptors Human genes 0.000 description 10
- 239000013078 crystal Substances 0.000 description 10
- 230000007246 mechanism Effects 0.000 description 10
- 102000005962 receptors Human genes 0.000 description 10
- 108020003175 receptors Proteins 0.000 description 10
- 230000007017 scission Effects 0.000 description 10
- 210000004881 tumor cell Anatomy 0.000 description 10
- 108020004414 DNA Proteins 0.000 description 9
- 241000588724 Escherichia coli Species 0.000 description 9
- 101710120037 Toxin CcdB Proteins 0.000 description 9
- 210000003719 b-lymphocyte Anatomy 0.000 description 9
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 9
- 230000004927 fusion Effects 0.000 description 9
- 210000004408 hybridoma Anatomy 0.000 description 9
- 238000000338 in vitro Methods 0.000 description 9
- 230000001939 inductive effect Effects 0.000 description 9
- 229960004641 rituximab Drugs 0.000 description 9
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 8
- 229930192392 Mitomycin Natural products 0.000 description 8
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 8
- 241000191963 Staphylococcus epidermidis Species 0.000 description 8
- 230000006037 cell lysis Effects 0.000 description 8
- 150000001875 compounds Chemical class 0.000 description 8
- 239000000562 conjugate Substances 0.000 description 8
- 230000002401 inhibitory effect Effects 0.000 description 8
- 229960004857 mitomycin Drugs 0.000 description 8
- 238000006384 oligomerization reaction Methods 0.000 description 8
- 230000037361 pathway Effects 0.000 description 8
- 239000013612 plasmid Substances 0.000 description 8
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 8
- 239000012190 activator Substances 0.000 description 7
- 230000006907 apoptotic process Effects 0.000 description 7
- 108010044540 auristatin Proteins 0.000 description 7
- 230000021615 conjugation Effects 0.000 description 7
- 238000000151 deposition Methods 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 230000013595 glycosylation Effects 0.000 description 7
- 238000006206 glycosylation reaction Methods 0.000 description 7
- 239000004337 magnesium citrate Substances 0.000 description 7
- 210000000440 neutrophil Anatomy 0.000 description 7
- 229910052700 potassium Inorganic materials 0.000 description 7
- 238000011160 research Methods 0.000 description 7
- 101000807561 Homo sapiens Tyrosine-protein kinase receptor UFO Proteins 0.000 description 6
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 6
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 6
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 6
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 6
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 6
- 241000607142 Salmonella Species 0.000 description 6
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 6
- 241000607768 Shigella Species 0.000 description 6
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 6
- 241000607734 Yersinia <bacteria> Species 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 238000000423 cell based assay Methods 0.000 description 6
- 230000003013 cytotoxicity Effects 0.000 description 6
- 231100000135 cytotoxicity Toxicity 0.000 description 6
- 230000008021 deposition Effects 0.000 description 6
- 238000000375 direct analysis in real time Methods 0.000 description 6
- 231100000673 dose–response relationship Toxicity 0.000 description 6
- 238000012063 dual-affinity re-targeting Methods 0.000 description 6
- 230000002708 enhancing effect Effects 0.000 description 6
- 230000002209 hydrophobic effect Effects 0.000 description 6
- 229940072221 immunoglobulins Drugs 0.000 description 6
- 230000003834 intracellular effect Effects 0.000 description 6
- 230000000670 limiting effect Effects 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 244000052769 pathogen Species 0.000 description 6
- 238000002823 phage display Methods 0.000 description 6
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 5
- 102100030595 HLA class II histocompatibility antigen gamma chain Human genes 0.000 description 5
- 101001082627 Homo sapiens HLA class II histocompatibility antigen gamma chain Proteins 0.000 description 5
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 5
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 5
- 241000588650 Neisseria meningitidis Species 0.000 description 5
- 102100037765 Periostin Human genes 0.000 description 5
- 101710199268 Periostin Proteins 0.000 description 5
- 239000012980 RPMI-1640 medium Substances 0.000 description 5
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 231100000599 cytotoxic agent Toxicity 0.000 description 5
- UQLDLKMNUJERMK-UHFFFAOYSA-L di(octadecanoyloxy)lead Chemical compound [Pb+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O UQLDLKMNUJERMK-UHFFFAOYSA-L 0.000 description 5
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 5
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 5
- 239000002158 endotoxin Substances 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 239000013604 expression vector Substances 0.000 description 5
- 210000000987 immune system Anatomy 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 239000013642 negative control Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 239000002773 nucleotide Substances 0.000 description 5
- 125000003729 nucleotide group Chemical group 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 238000002864 sequence alignment Methods 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- 230000009261 transgenic effect Effects 0.000 description 5
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 4
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 4
- 208000035143 Bacterial infection Diseases 0.000 description 4
- 102100031658 C-X-C chemokine receptor type 5 Human genes 0.000 description 4
- 241000222122 Candida albicans Species 0.000 description 4
- 241000606161 Chlamydia Species 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- 102000001301 EGF receptor Human genes 0.000 description 4
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 4
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 4
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 4
- 102000003886 Glycoproteins Human genes 0.000 description 4
- 108090000288 Glycoproteins Proteins 0.000 description 4
- 102000010956 Glypican Human genes 0.000 description 4
- 108050001154 Glypican Proteins 0.000 description 4
- 241000700721 Hepatitis B virus Species 0.000 description 4
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 4
- 101000922405 Homo sapiens C-X-C chemokine receptor type 5 Proteins 0.000 description 4
- 101000851181 Homo sapiens Epidermal growth factor receptor Proteins 0.000 description 4
- 101000959794 Homo sapiens Interferon alpha-2 Proteins 0.000 description 4
- 101000777628 Homo sapiens Leukocyte antigen CD37 Proteins 0.000 description 4
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 description 4
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 4
- 101000635938 Homo sapiens Transforming growth factor beta-1 proprotein Proteins 0.000 description 4
- 241000701024 Human betaherpesvirus 5 Species 0.000 description 4
- 101000926057 Human herpesvirus 2 (strain G) Envelope glycoprotein C Proteins 0.000 description 4
- 241000711920 Human orthopneumovirus Species 0.000 description 4
- 102100040018 Interferon alpha-2 Human genes 0.000 description 4
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 4
- 102100031586 Leukocyte antigen CD37 Human genes 0.000 description 4
- 239000004472 Lysine Substances 0.000 description 4
- 102000018697 Membrane Proteins Human genes 0.000 description 4
- 108010052285 Membrane Proteins Proteins 0.000 description 4
- 241001436793 Meru Species 0.000 description 4
- 108010008707 Mucin-1 Proteins 0.000 description 4
- 102100034256 Mucin-1 Human genes 0.000 description 4
- 241000699660 Mus musculus Species 0.000 description 4
- 108091028043 Nucleic acid sequence Proteins 0.000 description 4
- 241000283973 Oryctolagus cuniculus Species 0.000 description 4
- 102000035195 Peptidases Human genes 0.000 description 4
- 108091005804 Peptidases Proteins 0.000 description 4
- 239000004365 Protease Substances 0.000 description 4
- 238000011579 SCID mouse model Methods 0.000 description 4
- 238000000692 Student's t-test Methods 0.000 description 4
- 102100035721 Syndecan-1 Human genes 0.000 description 4
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 4
- 101001051488 Takifugu rubripes Neural cell adhesion molecule L1 Proteins 0.000 description 4
- 102100030742 Transforming growth factor beta-1 proprotein Human genes 0.000 description 4
- 241000700605 Viruses Species 0.000 description 4
- 229940045799 anthracyclines and related substance Drugs 0.000 description 4
- 150000001540 azides Chemical class 0.000 description 4
- 208000022362 bacterial infectious disease Diseases 0.000 description 4
- 210000004899 c-terminal region Anatomy 0.000 description 4
- 229940095731 candida albicans Drugs 0.000 description 4
- 210000002421 cell wall Anatomy 0.000 description 4
- 230000003828 downregulation Effects 0.000 description 4
- 229960004679 doxorubicin Drugs 0.000 description 4
- 229960005501 duocarmycin Drugs 0.000 description 4
- 229930184221 duocarmycin Natural products 0.000 description 4
- 108020001507 fusion proteins Proteins 0.000 description 4
- 102000037865 fusion proteins Human genes 0.000 description 4
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 4
- 210000004602 germ cell Anatomy 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 229940127121 immunoconjugate Drugs 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 229920006008 lipopolysaccharide Polymers 0.000 description 4
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 4
- 230000002101 lytic effect Effects 0.000 description 4
- 229960002450 ofatumumab Drugs 0.000 description 4
- 230000014207 opsonization Effects 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 4
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 4
- 229950007308 satumomab Drugs 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 238000007920 subcutaneous administration Methods 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 238000012353 t test Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 238000011830 transgenic mouse model Methods 0.000 description 4
- 229930188428 trichomycin Natural products 0.000 description 4
- 230000003612 virological effect Effects 0.000 description 4
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 3
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 3
- 206010069754 Acquired gene mutation Diseases 0.000 description 3
- 241000193830 Bacillus <bacterium> Species 0.000 description 3
- 241000193738 Bacillus anthracis Species 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- 102000014914 Carrier Proteins Human genes 0.000 description 3
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 3
- 108010092160 Dactinomycin Proteins 0.000 description 3
- 238000012286 ELISA Assay Methods 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 241000233866 Fungi Species 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 3
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 3
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 3
- 238000012450 HuMAb Mouse Methods 0.000 description 3
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 3
- FADYJNXDPBKVCA-UHFFFAOYSA-N L-Phenylalanyl-L-lysin Natural products NCCCCC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FADYJNXDPBKVCA-UHFFFAOYSA-N 0.000 description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 101100406343 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) mrp-3 gene Proteins 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 239000001825 Polyoxyethene (8) stearate Substances 0.000 description 3
- 229920001213 Polysorbate 20 Polymers 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000012979 RPMI medium Substances 0.000 description 3
- 108020004511 Recombinant DNA Proteins 0.000 description 3
- 108010083644 Ribonucleases Proteins 0.000 description 3
- 102000006382 Ribonucleases Human genes 0.000 description 3
- 108010039491 Ricin Proteins 0.000 description 3
- 241000283984 Rodentia Species 0.000 description 3
- 241000239226 Scorpiones Species 0.000 description 3
- 108010090763 Shiga Toxin 2 Proteins 0.000 description 3
- 241000191940 Staphylococcus Species 0.000 description 3
- 241000193998 Streptococcus pneumoniae Species 0.000 description 3
- 102000013127 Vimentin Human genes 0.000 description 3
- 108010065472 Vimentin Proteins 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 229960002964 adalimumab Drugs 0.000 description 3
- 230000001093 anti-cancer Effects 0.000 description 3
- 229940065181 bacillus anthracis Drugs 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 238000013357 binding ELISA Methods 0.000 description 3
- 108091008324 binding proteins Proteins 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 230000001143 conditioned effect Effects 0.000 description 3
- 238000005336 cracking Methods 0.000 description 3
- 235000018417 cysteine Nutrition 0.000 description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 3
- 229940127089 cytotoxic agent Drugs 0.000 description 3
- 239000002619 cytotoxin Substances 0.000 description 3
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 238000010494 dissociation reaction Methods 0.000 description 3
- 230000005593 dissociations Effects 0.000 description 3
- 210000001163 endosome Anatomy 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 108010087914 epidermal growth factor receptor VIII Proteins 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 3
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 3
- 210000003714 granulocyte Anatomy 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 229960000598 infliximab Drugs 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 230000002132 lysosomal effect Effects 0.000 description 3
- 210000002540 macrophage Anatomy 0.000 description 3
- 230000003211 malignant effect Effects 0.000 description 3
- 229950008001 matuzumab Drugs 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 210000004379 membrane Anatomy 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 210000004779 membrane envelope Anatomy 0.000 description 3
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 238000001543 one-way ANOVA Methods 0.000 description 3
- 230000001717 pathogenic effect Effects 0.000 description 3
- 239000004033 plastic Substances 0.000 description 3
- 229920003023 plastic Polymers 0.000 description 3
- 229960003171 plicamycin Drugs 0.000 description 3
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 3
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 3
- 239000011148 porous material Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 235000019419 proteases Nutrition 0.000 description 3
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical compound C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 description 3
- 238000003259 recombinant expression Methods 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- 238000002702 ribosome display Methods 0.000 description 3
- 238000002741 site-directed mutagenesis Methods 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 230000037439 somatic mutation Effects 0.000 description 3
- 125000006850 spacer group Chemical group 0.000 description 3
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 3
- 229960003989 tocilizumab Drugs 0.000 description 3
- 238000003146 transient transfection Methods 0.000 description 3
- 210000005048 vimentin Anatomy 0.000 description 3
- ALBODLTZUXKBGZ-JUUVMNCLSA-N (2s)-2-amino-3-phenylpropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CC1=CC=CC=C1 ALBODLTZUXKBGZ-JUUVMNCLSA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 2
- KUBWJGWIWGGEPZ-UHFFFAOYSA-N 1-[amino(ethoxy)phosphoryl]oxy-4-nitrobenzene Chemical compound CCOP(N)(=O)OC1=CC=C([N+]([O-])=O)C=C1 KUBWJGWIWGGEPZ-UHFFFAOYSA-N 0.000 description 2
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 2
- VPFUWHKTPYPNGT-UHFFFAOYSA-N 3-(3,4-dihydroxyphenyl)-1-(5-hydroxy-2,2-dimethylchromen-6-yl)propan-1-one Chemical compound OC1=C2C=CC(C)(C)OC2=CC=C1C(=O)CCC1=CC=C(O)C(O)=C1 VPFUWHKTPYPNGT-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 108091007504 ADAM10 Proteins 0.000 description 2
- 108091007507 ADAM12 Proteins 0.000 description 2
- 108091007505 ADAM17 Proteins 0.000 description 2
- DJQOOSBJCLSSEY-UHFFFAOYSA-N Acipimox Chemical compound CC1=CN=C(C(O)=O)C=[N+]1[O-] DJQOOSBJCLSSEY-UHFFFAOYSA-N 0.000 description 2
- 102100034608 Angiopoietin-2 Human genes 0.000 description 2
- 101100225890 Aplysia californica ENPP gene Proteins 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 102000015790 Asparaginase Human genes 0.000 description 2
- 108010024976 Asparaginase Proteins 0.000 description 2
- 241000228212 Aspergillus Species 0.000 description 2
- PYIXHKGTJKCVBJ-UHFFFAOYSA-N Astraciceran Natural products C1OC2=CC(O)=CC=C2CC1C1=CC(OCO2)=C2C=C1OC PYIXHKGTJKCVBJ-UHFFFAOYSA-N 0.000 description 2
- NDVRQFZUJRMKKP-UHFFFAOYSA-N Betavulgarin Natural products O=C1C=2C(OC)=C3OCOC3=CC=2OC=C1C1=CC=CC=C1O NDVRQFZUJRMKKP-UHFFFAOYSA-N 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- 108700012439 CA9 Proteins 0.000 description 2
- 101150013553 CD40 gene Proteins 0.000 description 2
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 2
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 2
- 101100381481 Caenorhabditis elegans baz-2 gene Proteins 0.000 description 2
- 101100004988 Caenorhabditis elegans cdh-3 gene Proteins 0.000 description 2
- 101100463133 Caenorhabditis elegans pdl-1 gene Proteins 0.000 description 2
- 241000282832 Camelidae Species 0.000 description 2
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 102100024423 Carbonic anhydrase 9 Human genes 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 241000606153 Chlamydia trachomatis Species 0.000 description 2
- VYZAMTAEIAYCRO-BJUDXGSMSA-N Chromium-51 Chemical compound [51Cr] VYZAMTAEIAYCRO-BJUDXGSMSA-N 0.000 description 2
- 102100038447 Claudin-4 Human genes 0.000 description 2
- 108090000601 Claudin-4 Proteins 0.000 description 2
- 108010034753 Complement Membrane Attack Complex Proteins 0.000 description 2
- 241001337994 Cryptococcus <scale insect> Species 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 101710112752 Cytotoxin Proteins 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 102000016607 Diphtheria Toxin Human genes 0.000 description 2
- 108010053187 Diphtheria Toxin Proteins 0.000 description 2
- 102100039673 Disintegrin and metalloproteinase domain-containing protein 10 Human genes 0.000 description 2
- 102100031112 Disintegrin and metalloproteinase domain-containing protein 12 Human genes 0.000 description 2
- 102100031111 Disintegrin and metalloproteinase domain-containing protein 17 Human genes 0.000 description 2
- 102000012804 EPCAM Human genes 0.000 description 2
- 101150084967 EPCAM gene Proteins 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 108050009340 Endothelin Proteins 0.000 description 2
- 102000002045 Endothelin Human genes 0.000 description 2
- 108091008794 FGF receptors Proteins 0.000 description 2
- 102000009123 Fibrin Human genes 0.000 description 2
- 108010073385 Fibrin Proteins 0.000 description 2
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 2
- 229920001917 Ficoll Polymers 0.000 description 2
- 102100035139 Folate receptor alpha Human genes 0.000 description 2
- 206010017533 Fungal infection Diseases 0.000 description 2
- 108010057784 Fusion Regulatory Protein-1 Proteins 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 2
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 2
- 108050007237 Glypican-3 Proteins 0.000 description 2
- 102100039939 Growth/differentiation factor 8 Human genes 0.000 description 2
- 108050006583 Growth/differentiation factor 8 Proteins 0.000 description 2
- 108010053491 HLA-DR beta-Chains Proteins 0.000 description 2
- 101710114832 Heat shock protein 90 homolog Proteins 0.000 description 2
- 241000711549 Hepacivirus C Species 0.000 description 2
- 241000228402 Histoplasma Species 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000924533 Homo sapiens Angiopoietin-2 Proteins 0.000 description 2
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 description 2
- 101001023230 Homo sapiens Folate receptor alpha Proteins 0.000 description 2
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 2
- 101000746373 Homo sapiens Granulocyte-macrophage colony-stimulating factor Proteins 0.000 description 2
- 101001055308 Homo sapiens Immunoglobulin heavy constant epsilon Proteins 0.000 description 2
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 2
- 101001078143 Homo sapiens Integrin alpha-IIb Proteins 0.000 description 2
- 101001046683 Homo sapiens Integrin alpha-L Proteins 0.000 description 2
- 101001046677 Homo sapiens Integrin alpha-V Proteins 0.000 description 2
- 101001054334 Homo sapiens Interferon beta Proteins 0.000 description 2
- 101000917826 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-a Proteins 0.000 description 2
- 101000917824 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-b Proteins 0.000 description 2
- 101000628547 Homo sapiens Metalloreductase STEAP1 Proteins 0.000 description 2
- 101000972282 Homo sapiens Mucin-5AC Proteins 0.000 description 2
- 101000972276 Homo sapiens Mucin-5B Proteins 0.000 description 2
- 101000622137 Homo sapiens P-selectin Proteins 0.000 description 2
- 101001126417 Homo sapiens Platelet-derived growth factor receptor alpha Proteins 0.000 description 2
- 101001136592 Homo sapiens Prostate stem cell antigen Proteins 0.000 description 2
- 101000711796 Homo sapiens Sclerostin Proteins 0.000 description 2
- 101000904724 Homo sapiens Transmembrane glycoprotein NMB Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 2
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 2
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 2
- 102100026212 Immunoglobulin heavy constant epsilon Human genes 0.000 description 2
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 2
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 2
- 102100022339 Integrin alpha-L Human genes 0.000 description 2
- 102100022337 Integrin alpha-V Human genes 0.000 description 2
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 2
- 102100026720 Interferon beta Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 102000008193 Interleukin-2 Receptor beta Subunit Human genes 0.000 description 2
- 108010060632 Interleukin-2 Receptor beta Subunit Proteins 0.000 description 2
- 238000012449 Kunming mouse Methods 0.000 description 2
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 2
- 102100029204 Low affinity immunoglobulin gamma Fc region receptor II-a Human genes 0.000 description 2
- 241000282567 Macaca fascicularis Species 0.000 description 2
- 102100027159 Membrane primary amine oxidase Human genes 0.000 description 2
- 101710132836 Membrane primary amine oxidase Proteins 0.000 description 2
- 102100026712 Metalloreductase STEAP1 Human genes 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- 102100022494 Mucin-5B Human genes 0.000 description 2
- 101100346932 Mus musculus Muc1 gene Proteins 0.000 description 2
- 101100369076 Mus musculus Tdgf1 gene Proteins 0.000 description 2
- 101100154912 Mus musculus Tyrp1 gene Proteins 0.000 description 2
- 208000031888 Mycoses Diseases 0.000 description 2
- 102100023472 P-selectin Human genes 0.000 description 2
- 101150062285 PGF gene Proteins 0.000 description 2
- IHPVFYLOGNNZLA-UHFFFAOYSA-N Phytoalexin Natural products COC1=CC=CC=C1C1OC(C=C2C(OCO2)=C2OC)=C2C(=O)C1 IHPVFYLOGNNZLA-UHFFFAOYSA-N 0.000 description 2
- 102100035194 Placenta growth factor Human genes 0.000 description 2
- 102100030485 Platelet-derived growth factor receptor alpha Human genes 0.000 description 2
- 241000276498 Pollachius virens Species 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 241000288906 Primates Species 0.000 description 2
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 2
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 2
- 102100036735 Prostate stem cell antigen Human genes 0.000 description 2
- 241000589516 Pseudomonas Species 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 101100372762 Rattus norvegicus Flt1 gene Proteins 0.000 description 2
- 108010084592 Saporins Proteins 0.000 description 2
- 102100034201 Sclerostin Human genes 0.000 description 2
- 102100030058 Secreted frizzled-related protein 1 Human genes 0.000 description 2
- 101000873420 Simian virus 40 SV40 early leader protein Proteins 0.000 description 2
- 241000700584 Simplexvirus Species 0.000 description 2
- 108091008874 T cell receptors Proteins 0.000 description 2
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 2
- 101150057140 TACSTD1 gene Proteins 0.000 description 2
- 108010000449 TNF-Related Apoptosis-Inducing Ligand Receptors Proteins 0.000 description 2
- 102000002259 TNF-Related Apoptosis-Inducing Ligand Receptors Human genes 0.000 description 2
- WDLRUFUQRNWCPK-UHFFFAOYSA-N Tetraxetan Chemical compound OC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 WDLRUFUQRNWCPK-UHFFFAOYSA-N 0.000 description 2
- 102100023935 Transmembrane glycoprotein NMB Human genes 0.000 description 2
- RTKIYFITIVXBLE-UHFFFAOYSA-N Trichostatin A Natural products ONC(=O)C=CC(C)=CC(C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-UHFFFAOYSA-N 0.000 description 2
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 2
- 240000001866 Vernicia fordii Species 0.000 description 2
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 2
- 229960003697 abatacept Drugs 0.000 description 2
- 229960003526 acipimox Drugs 0.000 description 2
- 229930183665 actinomycin Natural products 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000009824 affinity maturation Effects 0.000 description 2
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 2
- 229960000548 alemtuzumab Drugs 0.000 description 2
- 229950009106 altumomab Drugs 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 229960002235 anthrax antigen Drugs 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 230000009830 antibody antigen interaction Effects 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 238000003782 apoptosis assay Methods 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 229960003272 asparaginase Drugs 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 2
- OHDRQQURAXLVGJ-HLVWOLMTSA-N azane;(2e)-3-ethyl-2-[(e)-(3-ethyl-6-sulfo-1,3-benzothiazol-2-ylidene)hydrazinylidene]-1,3-benzothiazole-6-sulfonic acid Chemical compound [NH4+].[NH4+].S/1C2=CC(S([O-])(=O)=O)=CC=C2N(CC)C\1=N/N=C1/SC2=CC(S([O-])(=O)=O)=CC=C2N1CC OHDRQQURAXLVGJ-HLVWOLMTSA-N 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 229960004669 basiliximab Drugs 0.000 description 2
- 229960003094 belinostat Drugs 0.000 description 2
- NCNRHFGMJRPRSK-MDZDMXLPSA-N belinostat Chemical compound ONC(=O)\C=C\C1=CC=CC(S(=O)(=O)NC=2C=CC=CC=2)=C1 NCNRHFGMJRPRSK-MDZDMXLPSA-N 0.000 description 2
- 229940054066 benzamide antipsychotics Drugs 0.000 description 2
- 150000003936 benzamides Chemical class 0.000 description 2
- 229960000397 bevacizumab Drugs 0.000 description 2
- 229950002903 bivatuzumab Drugs 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 2
- 244000309466 calf Species 0.000 description 2
- 210000000234 capsid Anatomy 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 230000025084 cell cycle arrest Effects 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000022534 cell killing Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229960005395 cetuximab Drugs 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- 229940038705 chlamydia trachomatis Drugs 0.000 description 2
- 229960002173 citrulline Drugs 0.000 description 2
- 229950002595 clivatuzumab tetraxetan Drugs 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 230000008045 co-localization Effects 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 230000004154 complement system Effects 0.000 description 2
- 230000001268 conjugating effect Effects 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 2
- 239000002254 cytotoxic agent Substances 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 229960002806 daclizumab Drugs 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 229960002204 daratumumab Drugs 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 229960001251 denosumab Drugs 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- FOCAHLGSDWHSAH-UHFFFAOYSA-N difluoromethanethione Chemical compound FC(F)=S FOCAHLGSDWHSAH-UHFFFAOYSA-N 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229930188854 dolastatin Natural products 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 229960002224 eculizumab Drugs 0.000 description 2
- 229960001776 edrecolomab Drugs 0.000 description 2
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 210000003979 eosinophil Anatomy 0.000 description 2
- 210000002615 epidermis Anatomy 0.000 description 2
- 229950009760 epratuzumab Drugs 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 229940093443 fanolesomab Drugs 0.000 description 2
- 229950003499 fibrin Drugs 0.000 description 2
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 description 2
- 229960000390 fludarabine Drugs 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- MUJOIMFVNIBMKC-UHFFFAOYSA-N fludioxonil Chemical compound C=12OC(F)(F)OC2=CC=CC=1C1=CNC=C1C#N MUJOIMFVNIBMKC-UHFFFAOYSA-N 0.000 description 2
- 238000013467 fragmentation Methods 0.000 description 2
- 238000006062 fragmentation reaction Methods 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 229950002026 girentuximab Drugs 0.000 description 2
- 239000003862 glucocorticoid Substances 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- 229960001743 golimumab Drugs 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 2
- 229960000908 idarubicin Drugs 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 230000003053 immunization Effects 0.000 description 2
- 238000002649 immunization Methods 0.000 description 2
- 230000016784 immunoglobulin production Effects 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 238000007917 intracranial administration Methods 0.000 description 2
- 230000008863 intramolecular interaction Effects 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 229960005386 ipilimumab Drugs 0.000 description 2
- 229950010828 keliximab Drugs 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 229960004194 lidocaine Drugs 0.000 description 2
- 229950002950 lintuzumab Drugs 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 230000001868 lysosomic effect Effects 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 229960001924 melphalan Drugs 0.000 description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 2
- 229960005108 mepolizumab Drugs 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229910044991 metal oxide Inorganic materials 0.000 description 2
- 150000004706 metal oxides Chemical class 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- 229950003063 mitumomab Drugs 0.000 description 2
- 210000001616 monocyte Anatomy 0.000 description 2
- 108010093470 monomethyl auristatin E Proteins 0.000 description 2
- 229960005027 natalizumab Drugs 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- 229950010203 nimotuzumab Drugs 0.000 description 2
- 229960003347 obinutuzumab Drugs 0.000 description 2
- 229960000470 omalizumab Drugs 0.000 description 2
- 229950010626 pagibaximab Drugs 0.000 description 2
- 229960000402 palivizumab Drugs 0.000 description 2
- 229960001972 panitumumab Drugs 0.000 description 2
- 229960005184 panobinostat Drugs 0.000 description 2
- FWZRWHZDXBDTFK-ZHACJKMWSA-N panobinostat Chemical compound CC1=NC2=CC=C[CH]C2=C1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FWZRWHZDXBDTFK-ZHACJKMWSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 229960002087 pertuzumab Drugs 0.000 description 2
- 210000000680 phagosome Anatomy 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 239000000280 phytoalexin Substances 0.000 description 2
- 150000001857 phytoalexin derivatives Chemical class 0.000 description 2
- 229940012957 plasmin Drugs 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 229950003700 priliximab Drugs 0.000 description 2
- 229960004919 procaine Drugs 0.000 description 2
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 230000005522 programmed cell death Effects 0.000 description 2
- 210000001236 prokaryotic cell Anatomy 0.000 description 2
- 229960003712 propranolol Drugs 0.000 description 2
- 229950010131 puromycin Drugs 0.000 description 2
- 229960003876 ranibizumab Drugs 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 229950005854 regavirumab Drugs 0.000 description 2
- 230000001177 retroviral effect Effects 0.000 description 2
- 229950009092 rovelizumab Drugs 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 238000009738 saturating Methods 0.000 description 2
- 230000003248 secreting effect Effects 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- 210000004989 spleen cell Anatomy 0.000 description 2
- 230000003393 splenic effect Effects 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 2
- 229940037128 systemic glucocorticoids Drugs 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 229960002372 tetracaine Drugs 0.000 description 2
- GKCBAIGFKIBETG-UHFFFAOYSA-N tetracaine Chemical compound CCCCNC1=CC=C(C(=O)OCCN(C)C)C=C1 GKCBAIGFKIBETG-UHFFFAOYSA-N 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 229960005267 tositumomab Drugs 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- 229950007217 tremelimumab Drugs 0.000 description 2
- RTKIYFITIVXBLE-QEQCGCAPSA-N trichostatin A Chemical compound ONC(=O)/C=C/C(/C)=C/[C@@H](C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-QEQCGCAPSA-N 0.000 description 2
- 230000001960 triggered effect Effects 0.000 description 2
- 239000013638 trimer Substances 0.000 description 2
- 229950005082 tuvirumab Drugs 0.000 description 2
- 229950005208 vepalimomab Drugs 0.000 description 2
- 239000013603 viral vector Substances 0.000 description 2
- 229950003511 votumumab Drugs 0.000 description 2
- 210000005253 yeast cell Anatomy 0.000 description 2
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- MFZSNESUTRVBQX-XEURHVNRSA-N (2S)-2-amino-6-[4-[[3-[[(2S)-1-[[(1S,2R,3S,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl]oxy]-1-oxopropan-2-yl]-methylamino]-3-oxopropyl]disulfanyl]pentanoylamino]hexanoic acid Chemical compound CO[C@@H]1\C=C\C=C(C)\Cc2cc(OC)c(Cl)c(c2)N(C)C(=O)C[C@H](OC(=O)[C@H](C)N(C)C(=O)CCSSC(C)CCC(=O)NCCCC[C@H](N)C(O)=O)[C@]2(C)O[C@H]2[C@H](C)[C@@H]2C[C@@]1(O)NC(=O)O2 MFZSNESUTRVBQX-XEURHVNRSA-N 0.000 description 1
- FOIAQXXUVRINCI-LBAQZLPGSA-N (2S)-2-amino-6-[[4-[2-[bis(carboxymethyl)amino]-3-[2-[bis(carboxymethyl)amino]ethyl-(carboxymethyl)amino]propyl]phenyl]carbamothioylamino]hexanoic acid Chemical compound N[C@@H](CCCCNC(=S)Nc1ccc(CC(CN(CCN(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)cc1)C(O)=O FOIAQXXUVRINCI-LBAQZLPGSA-N 0.000 description 1
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 1
- BQQGAGGSEMLWRS-UHFFFAOYSA-N (4-aminophenyl)methyl carbamate Chemical compound NC(=O)OCC1=CC=C(N)C=C1 BQQGAGGSEMLWRS-UHFFFAOYSA-N 0.000 description 1
- BWDQBBCUWLSASG-MDZDMXLPSA-N (e)-n-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1h-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide Chemical compound C=1NC2=CC=CC=C2C=1CCN(CCO)CC1=CC=C(\C=C\C(=O)NO)C=C1 BWDQBBCUWLSASG-MDZDMXLPSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- AMRCSPITBQTAKS-UHFFFAOYSA-N 2-[4-(2-fluoroethylamino)phenyl]-1,3-benzothiazol-6-ol Chemical compound S1C2=CC(O)=CC=C2N=C1C1=CC=C(NCCF)C=C1 AMRCSPITBQTAKS-UHFFFAOYSA-N 0.000 description 1
- FBUTXZSKZCQABC-UHFFFAOYSA-N 2-amino-1-methyl-7h-purine-6-thione Chemical compound S=C1N(C)C(N)=NC2=C1NC=N2 FBUTXZSKZCQABC-UHFFFAOYSA-N 0.000 description 1
- NEAQRZUHTPSBBM-UHFFFAOYSA-N 2-hydroxy-3,3-dimethyl-7-nitro-4h-isoquinolin-1-one Chemical class C1=C([N+]([O-])=O)C=C2C(=O)N(O)C(C)(C)CC2=C1 NEAQRZUHTPSBBM-UHFFFAOYSA-N 0.000 description 1
- ZTOJFFHGPLIVKC-UHFFFAOYSA-N 3-ethyl-2-[(3-ethyl-6-sulfo-1,3-benzothiazol-2-ylidene)hydrazinylidene]-1,3-benzothiazole-6-sulfonic acid Chemical compound S1C2=CC(S(O)(=O)=O)=CC=C2N(CC)C1=NN=C1SC2=CC(S(O)(=O)=O)=CC=C2N1CC ZTOJFFHGPLIVKC-UHFFFAOYSA-N 0.000 description 1
- NZSWNNDHPOTJNH-VEJILBAHSA-N 4-[[4-[[4-[[(2s)-3-cyano-2-[[4-[[(e)-3-(4-hydroxyphenyl)-2-methylprop-2-enoyl]amino]benzoyl]amino]propanoyl]amino]benzoyl]amino]-2-hydroxy-3-methoxybenzoyl]amino]-2-hydroxy-3-methoxybenzoic acid Chemical compound COC1=C(O)C(C(O)=O)=CC=C1NC(=O)C(C(=C1OC)O)=CC=C1NC(=O)C(C=C1)=CC=C1NC(=O)[C@H](CC#N)NC(=O)C(C=C1)=CC=C1NC(=O)C(\C)=C\C1=CC=C(O)C=C1 NZSWNNDHPOTJNH-VEJILBAHSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 108010077593 ACE-011 Proteins 0.000 description 1
- 108010066676 Abrin Proteins 0.000 description 1
- 231100000729 Amatoxin Toxicity 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- JXQCOHHAAOVKOV-UHFFFAOYSA-N C1=CCN2C1=CN=C1C(=C2)C=CC=C1.C1=CCN2C1=CN=C1C(=C2)C=CC=C1 Chemical compound C1=CCN2C1=CN=C1C(=C2)C=CC=C1.C1=CCN2C1=CN=C1C(=C2)C=CC=C1 JXQCOHHAAOVKOV-UHFFFAOYSA-N 0.000 description 1
- 108010029697 CD40 Ligand Proteins 0.000 description 1
- 102100032937 CD40 ligand Human genes 0.000 description 1
- 102100022002 CD59 glycoprotein Human genes 0.000 description 1
- 101100476210 Caenorhabditis elegans rnt-1 gene Proteins 0.000 description 1
- 240000005589 Calophyllum inophyllum Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- 108050004290 Cecropin Proteins 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 108010049048 Cholera Toxin Proteins 0.000 description 1
- 102000009016 Cholera Toxin Human genes 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000014447 Complement C1q Human genes 0.000 description 1
- 108010078043 Complement C1q Proteins 0.000 description 1
- 108010028780 Complement C3 Proteins 0.000 description 1
- 102000016918 Complement C3 Human genes 0.000 description 1
- 102000016574 Complement C3-C5 Convertases Human genes 0.000 description 1
- 108010067641 Complement C3-C5 Convertases Proteins 0.000 description 1
- 108010069112 Complement System Proteins Proteins 0.000 description 1
- 102000000989 Complement System Proteins Human genes 0.000 description 1
- 102100025680 Complement decay-accelerating factor Human genes 0.000 description 1
- 108090000056 Complement factor B Proteins 0.000 description 1
- 102000003712 Complement factor B Human genes 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 108700032819 Croton tiglium crotin II Proteins 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 241000725619 Dengue virus Species 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- MBYXEBXZARTUSS-QLWBXOBMSA-N Emetamine Natural products O(C)c1c(OC)cc2c(c(C[C@@H]3[C@H](CC)CN4[C@H](c5c(cc(OC)c(OC)c5)CC4)C3)ncc2)c1 MBYXEBXZARTUSS-QLWBXOBMSA-N 0.000 description 1
- 102000013128 Endothelin B Receptor Human genes 0.000 description 1
- 108010090557 Endothelin B Receptor Proteins 0.000 description 1
- 108010055196 EphA2 Receptor Proteins 0.000 description 1
- 102100030340 Ephrin type-A receptor 2 Human genes 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 102100020715 Fms-related tyrosine kinase 3 ligand protein Human genes 0.000 description 1
- 101710162577 Fms-related tyrosine kinase 3 ligand protein Proteins 0.000 description 1
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 108010026389 Gramicidin Proteins 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 101000897400 Homo sapiens CD59 glycoprotein Proteins 0.000 description 1
- 101000856022 Homo sapiens Complement decay-accelerating factor Proteins 0.000 description 1
- 101001057612 Homo sapiens Dual specificity protein phosphatase 5 Proteins 0.000 description 1
- 101000994375 Homo sapiens Integrin alpha-4 Proteins 0.000 description 1
- 101000961414 Homo sapiens Membrane cofactor protein Proteins 0.000 description 1
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 1
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 1
- 102000012960 Immunoglobulin kappa-Chains Human genes 0.000 description 1
- 108010090227 Immunoglobulin kappa-Chains Proteins 0.000 description 1
- 102100032818 Integrin alpha-4 Human genes 0.000 description 1
- 102000015271 Intercellular Adhesion Molecule-1 Human genes 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 108010009254 Lysosomal-Associated Membrane Protein 1 Proteins 0.000 description 1
- 102100035133 Lysosome-associated membrane glycoprotein 1 Human genes 0.000 description 1
- 101800004761 Magainin-2 Proteins 0.000 description 1
- 108010036176 Melitten Proteins 0.000 description 1
- 102100039373 Membrane cofactor protein Human genes 0.000 description 1
- 201000009906 Meningitis Diseases 0.000 description 1
- 244000302512 Momordica charantia Species 0.000 description 1
- 235000009811 Momordica charantia Nutrition 0.000 description 1
- 101100066439 Mus musculus Fcgrt gene Proteins 0.000 description 1
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 1
- 102000002356 Nectin Human genes 0.000 description 1
- 108060005251 Nectin Proteins 0.000 description 1
- 241000588653 Neisseria Species 0.000 description 1
- 101710149892 Nucleoside diphosphate kinase B Proteins 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 241000577979 Peromyscus spicilegus Species 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 241000219506 Phytolacca Species 0.000 description 1
- 240000007643 Phytolacca americana Species 0.000 description 1
- 235000009074 Phytolacca americana Nutrition 0.000 description 1
- 101100413173 Phytolacca americana PAP2 gene Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 229940079156 Proteasome inhibitor Drugs 0.000 description 1
- 102100024952 Protein CBFA2T1 Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 101000762949 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Exotoxin A Proteins 0.000 description 1
- AUVVAXYIELKVAI-UHFFFAOYSA-N SJ000285215 Natural products N1CCC2=CC(OC)=C(OC)C=C2C1CC1CC2C3=CC(OC)=C(OC)C=C3CCN2CC1CC AUVVAXYIELKVAI-UHFFFAOYSA-N 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 108010079723 Shiga Toxin Proteins 0.000 description 1
- 108010091769 Shiga Toxin 1 Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 241000295644 Staphylococcaceae Species 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108700002718 TACI receptor-IgG Fc fragment fusion Proteins 0.000 description 1
- 229940126624 Tacatuzumab tetraxetan Drugs 0.000 description 1
- 108010055044 Tetanus Toxin Proteins 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 241000722921 Tulipa gesneriana Species 0.000 description 1
- GBOGMAARMMDZGR-UHFFFAOYSA-N UNPD149280 Natural products N1C(=O)C23OC(=O)C=CC(O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 GBOGMAARMMDZGR-UHFFFAOYSA-N 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 241000710886 West Nile virus Species 0.000 description 1
- XYVNHPYNSPGYLI-UUOKFMHZSA-N [(2r,3s,4r,5r)-5-(2-amino-6-oxo-3h-purin-9-yl)-4-hydroxy-2-(phosphonooxymethyl)oxolan-3-yl] dihydrogen phosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H]1O XYVNHPYNSPGYLI-UUOKFMHZSA-N 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- YGCFIWIQZPHFLU-UHFFFAOYSA-N acesulfame Chemical compound CC1=CC(=O)NS(=O)(=O)O1 YGCFIWIQZPHFLU-UHFFFAOYSA-N 0.000 description 1
- 229960005164 acesulfame Drugs 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000021917 activation of membrane attack complex Effects 0.000 description 1
- 229950004283 actoxumab Drugs 0.000 description 1
- 230000003044 adaptive effect Effects 0.000 description 1
- 229950009084 adecatumumab Drugs 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 229960003227 afelimomab Drugs 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 229950008459 alacizumab pegol Drugs 0.000 description 1
- 229960002459 alefacept Drugs 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229960004539 alirocumab Drugs 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 108010001818 alpha-sarcin Proteins 0.000 description 1
- 229950004549 alvircept sudotox Drugs 0.000 description 1
- 229950001537 amatuximab Drugs 0.000 description 1
- 150000003862 amino acid derivatives Chemical class 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229950005794 anrukinzumab Drugs 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 230000003095 anti-phagocytic effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 238000011230 antibody-based therapy Methods 0.000 description 1
- 210000000612 antigen-presenting cell Anatomy 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000003080 antimitotic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 229950003145 apolizumab Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229950005725 arcitumomab Drugs 0.000 description 1
- 150000001483 arginine derivatives Chemical class 0.000 description 1
- 229950002882 aselizumab Drugs 0.000 description 1
- 229950009925 atacicept Drugs 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229950001863 bapineuzumab Drugs 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 229950007843 bavituximab Drugs 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 229960005347 belatacept Drugs 0.000 description 1
- 229960003270 belimumab Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229950000321 benralizumab Drugs 0.000 description 1
- 229950010015 bertilimumab Drugs 0.000 description 1
- 229950010559 besilesomab Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229950008086 bezlotoxumab Drugs 0.000 description 1
- 229950001303 biciromab Drugs 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 238000001815 biotherapy Methods 0.000 description 1
- 229960005522 bivatuzumab mertansine Drugs 0.000 description 1
- 229960003008 blinatumomab Drugs 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000001754 blood buffy coat Anatomy 0.000 description 1
- 229950005042 blosozumab Drugs 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 229960000455 brentuximab vedotin Drugs 0.000 description 1
- 229960003735 brodalumab Drugs 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 1
- 229960001838 canakinumab Drugs 0.000 description 1
- 229950007296 cantuzumab mertansine Drugs 0.000 description 1
- 229950011547 cantuzumab ravtansine Drugs 0.000 description 1
- 229950002176 caplacizumab Drugs 0.000 description 1
- 108010023376 caplacizumab Proteins 0.000 description 1
- 229950001178 capromab Drugs 0.000 description 1
- 229940034605 capromab pendetide Drugs 0.000 description 1
- YZBQHRLRFGPBSL-RXMQYKEDSA-N carbapenem Chemical compound C1C=CN2C(=O)C[C@H]21 YZBQHRLRFGPBSL-RXMQYKEDSA-N 0.000 description 1
- JJWKPURADFRFRB-UHFFFAOYSA-N carbonyl sulfide Chemical compound O=C=S JJWKPURADFRFRB-UHFFFAOYSA-N 0.000 description 1
- 229950000771 carlumab Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 229960000419 catumaxomab Drugs 0.000 description 1
- 229950006754 cedelizumab Drugs 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229960003115 certolizumab pegol Drugs 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- NDAYQJDHGXTBJL-MWWSRJDJSA-N chembl557217 Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](C(C)C)NC(=O)[C@H](C)NC(=O)[C@H](NC(=O)CNC(=O)[C@@H](NC=O)C(C)C)CC(C)C)C(=O)NCCO)=CNC2=C1 NDAYQJDHGXTBJL-MWWSRJDJSA-N 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- VDQQXEISLMTGAB-UHFFFAOYSA-N chloramine T Chemical compound [Na+].CC1=CC=C(S(=O)(=O)[N-]Cl)C=C1 VDQQXEISLMTGAB-UHFFFAOYSA-N 0.000 description 1
- 230000010428 chromatin condensation Effects 0.000 description 1
- 239000013611 chromosomal DNA Substances 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 229950010905 citatuzumab bogatox Drugs 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 229950001565 clazakizumab Drugs 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 230000001149 cognitive effect Effects 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000001447 compensatory effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000010219 correlation analysis Methods 0.000 description 1
- 229930016834 coumestan Natural products 0.000 description 1
- 229950001954 crenezumab Drugs 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- GBOGMAARMMDZGR-TYHYBEHESA-N cytochalasin B Chemical compound C([C@H]1[C@@H]2[C@@H](C([C@@H](O)[C@@H]3/C=C/C[C@H](C)CCC[C@@H](O)/C=C/C(=O)O[C@@]23C(=O)N1)=C)C)C1=CC=CC=C1 GBOGMAARMMDZGR-TYHYBEHESA-N 0.000 description 1
- GBOGMAARMMDZGR-JREHFAHYSA-N cytochalasin B Natural products C[C@H]1CCC[C@@H](O)C=CC(=O)O[C@@]23[C@H](C=CC1)[C@H](O)C(=C)[C@@H](C)[C@@H]2[C@H](Cc4ccccc4)NC3=O GBOGMAARMMDZGR-JREHFAHYSA-N 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229950007409 dacetuzumab Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 229960002482 dalotuzumab Drugs 0.000 description 1
- ZVTDLPBHTSMEJZ-UPZRXNBOSA-N danoprevir Chemical compound O=C([C@@]12C[C@H]1\C=C/CCCCC[C@H](C(N1C[C@@H](C[C@H]1C(=O)N2)OC(=O)N1CC2=C(F)C=CC=C2C1)=O)NC(=O)OC(C)(C)C)NS(=O)(=O)C1CC1 ZVTDLPBHTSMEJZ-UPZRXNBOSA-N 0.000 description 1
- 229950002891 danoprevir Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 229950007998 demcizumab Drugs 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 229950008962 detumomab Drugs 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 229930191339 dianthin Natural products 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 206010013023 diphtheria Diseases 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 229950005168 dorlimomab aritox Drugs 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 229950009964 drozitumab Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 239000003118 drug derivative Substances 0.000 description 1
- 108010005794 dulaglutide Proteins 0.000 description 1
- 229960005175 dulaglutide Drugs 0.000 description 1
- 229950000006 ecromeximab Drugs 0.000 description 1
- 229950011109 edobacomab Drugs 0.000 description 1
- 229960000284 efalizumab Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229960004137 elotuzumab Drugs 0.000 description 1
- 229950002507 elsilimomab Drugs 0.000 description 1
- AUVVAXYIELKVAI-CKBKHPSWSA-N emetine Chemical compound N1CCC2=CC(OC)=C(OC)C=C2[C@H]1C[C@H]1C[C@H]2C3=CC(OC)=C(OC)C=C3CCN2C[C@@H]1CC AUVVAXYIELKVAI-CKBKHPSWSA-N 0.000 description 1
- 229960002694 emetine Drugs 0.000 description 1
- AUVVAXYIELKVAI-UWBTVBNJSA-N emetine Natural products N1CCC2=CC(OC)=C(OC)C=C2[C@H]1C[C@H]1C[C@H]2C3=CC(OC)=C(OC)C=C3CCN2C[C@H]1CC AUVVAXYIELKVAI-UWBTVBNJSA-N 0.000 description 1
- 229950003048 enavatuzumab Drugs 0.000 description 1
- 230000002121 endocytic effect Effects 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229950002798 enlimomab Drugs 0.000 description 1
- 229950000565 enlimomab pegol Drugs 0.000 description 1
- 229950007313 enokizumab Drugs 0.000 description 1
- 108010028531 enomycin Proteins 0.000 description 1
- 229950010640 ensituximab Drugs 0.000 description 1
- INVTYAOGFAGBOE-UHFFFAOYSA-N entinostat Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)OCC1=CC=CN=C1 INVTYAOGFAGBOE-UHFFFAOYSA-N 0.000 description 1
- 229950005837 entinostat Drugs 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 229950006414 epitumomab cituxetan Drugs 0.000 description 1
- 229950004292 erlizumab Drugs 0.000 description 1
- 229950008579 ertumaxomab Drugs 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- 229950009569 etaracizumab Drugs 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229950004912 etrolizumab Drugs 0.000 description 1
- 229950005562 exbivirumab Drugs 0.000 description 1
- 229950001488 faralimomab Drugs 0.000 description 1
- 229950000335 fasinumab Drugs 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229950001563 felvizumab Drugs 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 229950010512 fezakinumab Drugs 0.000 description 1
- 229950002846 ficlatuzumab Drugs 0.000 description 1
- 229950008085 figitumumab Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000834 fixative Substances 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 229950004923 fontolizumab Drugs 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 229950004003 fresolimumab Drugs 0.000 description 1
- 229950009370 fulranumab Drugs 0.000 description 1
- 239000003008 fumonisin Substances 0.000 description 1
- 229950001109 galiximab Drugs 0.000 description 1
- 229950004896 ganitumab Drugs 0.000 description 1
- 229950002508 gantenerumab Drugs 0.000 description 1
- 229950004792 gavilimomab Drugs 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960000578 gemtuzumab Drugs 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 229950003717 gevokizumab Drugs 0.000 description 1
- 229950000918 glembatumumab Drugs 0.000 description 1
- 229950009672 glembatumumab vedotin Drugs 0.000 description 1
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 1
- 229940126613 gomiliximab Drugs 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 241000411851 herbal medicine Species 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 230000009215 host defense mechanism Effects 0.000 description 1
- 102000052645 human CD38 Human genes 0.000 description 1
- 102000043381 human DUSP5 Human genes 0.000 description 1
- 235000020256 human milk Nutrition 0.000 description 1
- 210000004251 human milk Anatomy 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229950010245 ibalizumab Drugs 0.000 description 1
- 229950006359 icrucumab Drugs 0.000 description 1
- 229950007354 imciromab Drugs 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 102000028557 immunoglobulin binding proteins Human genes 0.000 description 1
- 108091009323 immunoglobulin binding proteins Proteins 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 230000001861 immunosuppressant effect Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 229950009230 inclacumab Drugs 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229950011428 indatuximab ravtansine Drugs 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 229950001014 intetumumab Drugs 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 229950003818 itolizumab Drugs 0.000 description 1
- 229960005435 ixekizumab Drugs 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 229950002183 lebrikizumab Drugs 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 229950001275 lemalesomab Drugs 0.000 description 1
- 229940121292 leronlimab Drugs 0.000 description 1
- 229950002884 lexatumumab Drugs 0.000 description 1
- 229950005173 libivirumab Drugs 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- GZQKNULLWNGMCW-PWQABINMSA-N lipid A (E. coli) Chemical compound O1[C@H](CO)[C@@H](OP(O)(O)=O)[C@H](OC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)[C@@H](NC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H]1OC[C@@H]1[C@@H](O)[C@H](OC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](OP(O)(O)=O)O1 GZQKNULLWNGMCW-PWQABINMSA-N 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 238000002714 localization assay Methods 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 229950003526 lorvotuzumab mertansine Drugs 0.000 description 1
- 229950004563 lucatumumab Drugs 0.000 description 1
- 230000006674 lysosomal degradation Effects 0.000 description 1
- MGIUUAHJVPPFEV-ABXDCCGRSA-N magainin ii Chemical compound C([C@H](NC(=O)[C@H](CCCCN)NC(=O)CNC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(O)=O)C1=CC=CC=C1 MGIUUAHJVPPFEV-ABXDCCGRSA-N 0.000 description 1
- 210000005075 mammary gland Anatomy 0.000 description 1
- 229950001869 mapatumumab Drugs 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000008774 maternal effect Effects 0.000 description 1
- 229950007254 mavrilimumab Drugs 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- VDXZNPDIRNWWCW-JFTDCZMZSA-N melittin Chemical compound NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(N)=O)CC1=CNC2=CC=CC=C12 VDXZNPDIRNWWCW-JFTDCZMZSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 230000008880 microtubule cytoskeleton organization Effects 0.000 description 1
- 229950002142 minretumomab Drugs 0.000 description 1
- 229950009686 mirococept Drugs 0.000 description 1
- 108010010621 modeccin Proteins 0.000 description 1
- 229950007699 mogamulizumab Drugs 0.000 description 1
- DASWEROEPLKSEI-UIJRFTGLSA-N monomethyl auristatin e Chemical group CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)C1=CC=CC=C1 DASWEROEPLKSEI-UIJRFTGLSA-N 0.000 description 1
- MFRNYXJJRJQHNW-NARUGQRUSA-N monomethyl auristatin f Chemical group CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)C([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-NARUGQRUSA-N 0.000 description 1
- 108010059074 monomethylauristatin F Proteins 0.000 description 1
- 229960001521 motavizumab Drugs 0.000 description 1
- 108091005763 multidomain proteins Proteins 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- 229950009793 naptumomab estafenatox Drugs 0.000 description 1
- 229950008353 narnatumab Drugs 0.000 description 1
- 229960000513 necitumumab Drugs 0.000 description 1
- 229950009675 nerelimomab Drugs 0.000 description 1
- 229960003301 nivolumab Drugs 0.000 description 1
- 231100000065 noncytotoxic Toxicity 0.000 description 1
- 230000002020 noncytotoxic effect Effects 0.000 description 1
- 229950009090 ocaratuzumab Drugs 0.000 description 1
- 229950005751 ocrelizumab Drugs 0.000 description 1
- 229950008516 olaratumab Drugs 0.000 description 1
- 229950010006 olokizumab Drugs 0.000 description 1
- 229950000846 onartuzumab Drugs 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 229950010444 onercept Drugs 0.000 description 1
- 229950007283 oregovomab Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 229950002610 otelixizumab Drugs 0.000 description 1
- 230000001151 other effect Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229950003709 oxelumab Drugs 0.000 description 1
- 229950004327 ozoralizumab Drugs 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229950003570 panobacumab Drugs 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 229950011485 pascolizumab Drugs 0.000 description 1
- 229950003522 pateclizumab Drugs 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 229950010966 patritumab Drugs 0.000 description 1
- 229960005570 pemtumomab Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 229940124633 peptidic drug Drugs 0.000 description 1
- 229950003203 pexelizumab Drugs 0.000 description 1
- 210000001539 phagocyte Anatomy 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 229930000184 phytotoxin Natural products 0.000 description 1
- 230000008884 pinocytosis Effects 0.000 description 1
- 229940126620 pintumomab Drugs 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 229950008092 placulumab Drugs 0.000 description 1
- 239000003123 plant toxin Substances 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 108700028325 pokeweed antiviral Proteins 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229950003486 ponezumab Drugs 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 239000003207 proteasome inhibitor Substances 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 210000004777 protein coat Anatomy 0.000 description 1
- 238000002818 protein evolution Methods 0.000 description 1
- 230000006916 protein interaction Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 229950003033 quilizumab Drugs 0.000 description 1
- 229950011613 racotumomab Drugs 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 229950011639 radretumab Drugs 0.000 description 1
- 229950002786 rafivirumab Drugs 0.000 description 1
- 229960002633 ramucirumab Drugs 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 229960004910 raxibacumab Drugs 0.000 description 1
- 108010003189 recombinant human tumor necrosis factor-binding protein-1 Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229960003254 reslizumab Drugs 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 229960001886 rilonacept Drugs 0.000 description 1
- 108010046141 rilonacept Proteins 0.000 description 1
- 229950003238 rilotumumab Drugs 0.000 description 1
- 229950001808 robatumumab Drugs 0.000 description 1
- 229950010699 roledumab Drugs 0.000 description 1
- 229950010968 romosozumab Drugs 0.000 description 1
- 229950005374 ruplizumab Drugs 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229950000106 samalizumab Drugs 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 210000003752 saphenous vein Anatomy 0.000 description 1
- 229950006348 sarilumab Drugs 0.000 description 1
- 229960004540 secukinumab Drugs 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229950004951 sevirumab Drugs 0.000 description 1
- 229950008684 sibrotuzumab Drugs 0.000 description 1
- 229950010077 sifalimumab Drugs 0.000 description 1
- 229960003323 siltuximab Drugs 0.000 description 1
- 229950003804 siplizumab Drugs 0.000 description 1
- 229950006094 sirukumab Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229950011267 solitomab Drugs 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 229950002894 sotatercept Drugs 0.000 description 1
- 101150065015 spa gene Proteins 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 229950002549 stamulumab Drugs 0.000 description 1
- 208000004048 staphylococcal pneumonia Diseases 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 229950001915 suvizumab Drugs 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 229950010265 tabalumab Drugs 0.000 description 1
- 229950001072 tadocizumab Drugs 0.000 description 1
- 229950004218 talizumab Drugs 0.000 description 1
- 229950008160 tanezumab Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229950008300 telimomab aritox Drugs 0.000 description 1
- CBPNZQVSJQDFBE-HGVVHKDOSA-N temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CCC2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-HGVVHKDOSA-N 0.000 description 1
- 229950000301 teneliximab Drugs 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229950010127 teplizumab Drugs 0.000 description 1
- 229940118376 tetanus toxin Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 229940126622 therapeutic monoclonal antibody Drugs 0.000 description 1
- 229950004742 tigatuzumab Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 229950001802 toralizumab Drugs 0.000 description 1
- 229950009165 torapsel Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 229950000835 tralokinumab Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229960001612 trastuzumab emtansine Drugs 0.000 description 1
- 108010075758 trebananib Proteins 0.000 description 1
- 229950001210 trebananib Drugs 0.000 description 1
- 239000003744 tubulin modulator Substances 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 229950004593 ublituximab Drugs 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 229950005972 urelumab Drugs 0.000 description 1
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 229950000386 vapaliximab Drugs 0.000 description 1
- 229950002148 vatelizumab Drugs 0.000 description 1
- 229960004914 vedolizumab Drugs 0.000 description 1
- 229950000815 veltuzumab Drugs 0.000 description 1
- 229950010789 vesencumab Drugs 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 229950004393 visilizumab Drugs 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 229950001212 volociximab Drugs 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229950006959 vorsetuzumab Drugs 0.000 description 1
- 229950008250 zalutumumab Drugs 0.000 description 1
- 229950009002 zanolimumab Drugs 0.000 description 1
- 229950009083 ziralimumab Drugs 0.000 description 1
Images















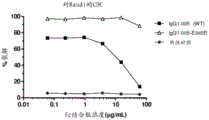













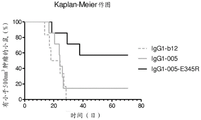

























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/283—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against Fc-receptors, e.g. CD16, CD32, CD64
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
- A61K51/1027—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody against receptors, cell-surface antigens or cell-surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2887—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against CD20
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2896—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against molecules with a "CD"-designation, not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/36—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against blood coagulation factors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/40—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against enzymes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K19/00—Hybrid peptides, i.e. peptides covalently bound to nucleic acids, or non-covalently bound protein-protein complexes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
- A61K2039/507—Comprising a combination of two or more separate antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/524—CH2 domain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/526—CH3 domain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/732—Antibody-dependent cellular cytotoxicity [ADCC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/734—Complement-dependent cytotoxicity [CDC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- General Health & Medical Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Cell Biology (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Endocrinology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Virology (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
本文中描述了包含变体Fc域的多肽和相关抗体。变体Fc域在所述多肽、一种或多种抗体与细胞表面上的其靶标、一种或多种抗原结合时提供稳定的Fc:Fc相互作用,因此提供了改进的补体依赖性细胞毒性(CDC)。
Description
发明领域
本发明涉及含有Fc域的多肽(诸如抗体),所述多肽具有提高的补体依赖性细胞毒性(CDC),并且也可以具有由Fc域中一个或多个氨基酸修饰带来的其他经修饰的效应子功能。
发明背景
由抗体Fc域介导的效应子(effector)功能允许破坏外来实体,如杀灭病原体以及清除和降解抗原。抗体依赖性细胞介导的细胞毒性(ADCC)和抗体依赖性细胞介导的吞噬(ADCP)由Fc区与具有Fc受体(FcR)的细胞的结合起始,而补体依赖性细胞毒性(CDC)由Fc区与C1q的结合起始,这起始补体激活的经典途径。
每个IgG抗体包含两个C1q结合位点,每个重链恒定(Fc)区中各一个。然而,由于单体IgG对C1q的亲和力非常弱(Kd~10-4M),溶液中的单个IgG分子不激活补体(Sledge等,1973J.Biol.Chem.248,2818-13;Hughes-Jones等, 1979Mol.Immunol.16,697-701)。抗原驱动的IgG关联可导致多价C1q分子更紧密结合(Kd~10-8M)和补体激活(Burton等,1990Mol.Immunol.22, 161-206)。相比之下,IgM以共价连接的五或六聚体天然存在,与细胞表达或固定的抗原结合后,IgM五聚体和六聚体可以有效引起CDC。抗原结合是诱导IgM构象变化以暴露C1q结合位点的要求(Feinstein等,1986,Immunology Today,169-174)。
已经提出通过Fc区CH2/CH3域的相互作用形成六聚体环结构,IgG也能完成补体激活(Burton等,1990Trends in Biochem.Sci.15,64-69)。支持此六聚体IgG结构存在的证据已经在二维(Reidler等,1986I Handbook of Experimental Immunology第4版.(Weir,D.M.编),pp17.1-17.5.Blackwell, Edinburgh;Pinteric等,1971Immunochem.8,1041-5)和三维晶体中,以及对溶液中的IgG1、IgG2a和IgG4和人Fc找到(Kuznetsov等,2000JStruct.Biol.131, 108-115)。六聚体环的形成也在针对抗HIV-1gp120的b12人类IgG1κ抗体的晶体结构(PDB中的1HZH)中观察到(Saphire等,Science 2001 Aug 10;293(5532),1155-9)。在b12六聚体环中,六个易接近的C1q结合位点位于六聚体表面,一个来自于六个抗体中的每个抗体,而其他六个结合位点面向下。
C1q类似六球头的郁金香束,包含系于六个胶原杆上的抗体结合区 (Perkins等,1985Biochem J.228,13-26;Poon等,1983J Mol Biol.168, 563-77;Reid等,1983BiochemSoc Trans 11,1-12;Weiss等,1986J.Mol.Biol. 189,573-81)。发现C1q与1HZH晶体结构的b12六聚体组装配合,以使六个球头中每一个与六个C1q结合位点中的一个相接触(Parren,FASEB Summer Research Conference,Snowmass,Co.,5-10July 2010;"CrystalStructure of an intact human IgG:implications for HIV-1neutralization andeffector Function", Erica Ollmann Saphire的论题,the Scripps ResearchInstitute,La Jolla, California.November 2000)。发现晶体结构中对称相关的b12抗体之间观察到的Fc界面中所选氨基酸的突变降低C1q结合亲合力,表明了这些氨基酸对分子间Fc:Fc相互作用的贡献。
WO 2006/104989描述了改变的抗体Fc区及其用途。
WO2005/047327描述了新生儿Fc受体(FcRn)结合多肽变体,二聚体Fc 结合蛋白和其相关方法。
WO 2010/106180描述了Fc变体,其具有与新生儿Fc受体(FcRn)增加的结合。
WO2005/070963描述了多肽Fc区变体及其用途。
WO2006/053301描述了具有改变的与FcRn的结合的Fc变体。
US 2011/0123440描述了改变的抗体Fc区及其用途。改变的Fc区有一个或多个氨基酸取代。
US 2008/0089892描述了多肽Fc区变体及包含这些Fc区变体的组合物。
US 2010/0184959描述了提供具有改变的Fc配体识别和/或效应子功能的 Fc多肽变体的方法。
US 2010/015133描述了通过调节多肽关联来生产多肽的方法。
US 2010/105873描述了生成多域蛋白治疗剂的综合方法。
US 6,737,056描述了具有改变的效应子功能的多肽变体。
之前已做出努力以识别具有增强的效应子功能或其他修饰特性的抗体 Fc变体。这样的研究集中于如在IgG同种型之间交换片段以产生嵌合IgG分子 (Natsume等,2008Cancer Res 68(10),3863-72),或者在铰链区(Dall'Acqua等, 2006J Immunol 177,1129-1138)或在CH2域的C1q结合位点处或其附近,围绕残基D270、K322、P329和P331(Idusogie等,2001J Immunol 166,2571-2575; Michaelsen等,2009Scand J Immunol 70,553-564和WO 99/51642)的氨基酸取代。例如,Moore等(2010mAbs 2(2),181-189)描述了检测S267E、H268F、 S324T、S239D、I332E、G236A和I332E的各种组合的通过CDC或ADCC的增强的效应子功能。影响与Fc受体结合(WO 2006/105062,WO 00/42072,美国专利6,737,056和美国专利7,083,784)或抗体物理特性(WO 2007/005612 Al)的其他Fc突变也已经提出。
尽管本领域中有这些和其他进步,然而,仍然有对新的和改进的基于抗体疗法的需求。
发明概述
本发明提供了相比于它们的亲本多肽/抗体,具有增强的补体依赖性细胞毒性(CDC)而且还具有其它增强的效应子功能的多肽和抗体变体。不限于理论,认为变体能在两个多肽/抗体分子Fc区之间有更稳定的结合相互作用,由此提供更亲合(avid)的表面,其能导致增强的效应子功能,如提高的或更特异的CDC应答。特定变体的特征也在于改进的ADCC应答、ADCP应答、和/或其他增强的效应子功能。如本发明所述,多肽/抗体工程的这种微妙机制可以被应用于,例如提高基于抗体的治疗剂的效力或特异性。
因此,在一个方面,本发明涉及一种提高包含免疫球蛋白的Fc域和结合区的亲本多肽的补体依赖性细胞毒性(CDC)的方法,该方法包括将一个或多个氨基酸残基中的突变引入到亲本多肽中,所述突变选自对应于人类IgG1 重链的Fc区中E430X、E345X、和S440W的组。
在另一方面,本发明涉及包含免疫球蛋白的Fc域和结合区的亲本多肽的变体,其中变体在选自对应人类IgG1重链Fc区中的E430S、E430F、E430T、 E345K、E345Q、E345R、E345Y和S440W的组的一个或多个突变,条件是变体在Fc域中不包含改变变体与新生儿Fc受体(FcRn)的结合的任何其他突变。
本发明也提供了使用一个或多个此类突变以当结合于例如抗原表达细胞、细胞膜或病毒粒子表面上的其抗原时提高由多肽或抗体介导的补体依赖性细胞毒性(CDC)。
在一个方面,本文中被称为“单突变体”的变体相比亲本多肽或抗体具有提高的CDC并也可以具有其他提高的效应子功能。
在一个方面,本文中称为“双突变体”的变体在所述片段中包含至少两个突变,并相比仅包含两个突变中一个的变体也具有改进的CDC并也可以具有其他改进的效应子功能。
在一个方面,本文中称为“混合突变体”的变体在与相同或不同多肽或抗体的在所述区段的不同氨基酸残基处包含突变的第二变体组合使用时,相比单独的变体、第二变体和亲本多肽或抗体中一种或多种,提供提高的CDC 并且任选还有也可以具有其他提高的效应子功能。
通常,突变是氨基酸取代,如下述突变,其将亲本氨基酸残基交换为具有不同大小和/或促进新分子间Fc:Fc键形成或提高现存对相互作用强度的物理化学特性的氨基酸。根据本发明的突变的示例性氨基酸残基,连同示例性氨基酸取代示于表1和2A和B中。本发明不同方面的非限制性展示在图1中提供。
本发明的这些和其他方面,具体是多肽和抗体变体的多种用途和治疗应用,在下面更详细地描述。
附图简述
图1:(A)六聚体形成中IgG分子的示意图。虚线圈显示了两个相邻IgG分子的两个接近的Fc:Fc相互作用对。框中的箭头显示了观看B、C和D中图示的方向:两个邻近的Fc分子旋转90°(在图的平面中)并以CH3域方向从Fab 臂观看。(B)观察到的寡聚增强突变对CDC的效果。示意图显示了根据本发明单突变体和双突变体方面具有增强效力的Fc:Fc相互作用对。(C)观察到的寡聚抑制突变对CDC的效果。根据本发明双突变体和混合突变体方面的示意图,其显示了相互补偿的至少两个寡聚抑制突变如何可以组合成一个分子 (双突变体方面)或者在两个分子上分开(混合突变体方面),以恢复或提高 Fc:Fc相互作用。混合突变体实现依赖于两个抗体结合的特定效应子功能,所述抗体可以识别不同的靶标。(D)C1q结合抑制突变对CDC的理论效果。 Fc:C1q相互作用的示意图,显示了由于C1q不能补偿引入抗体的缺陷,如果突变抑制C1q结合,其不能组合或混合以恢复CDC活性。
图2:使用Clustel 2.1软件的对应IgG1重链中残基P247到K447的人类 IgG1、IgG1f、IgG2、IgG3、IgG4、IgA1、IgA2、IgD、IgE和IgM Fc区段的序列比对,按Kabat中所列EU索引编号。显示的序列代表人类IgG1重链恒定区域(SEQ ID NO:1;UniProt登录号P01857)和IgG1m(f)的同种异型变体的残基130到330;IgG2重链恒定区域的残基126到326(SEQ IDNO:2;UniProt登录号P01859);IgG3重链恒定区域的残基177到377(SEQ ID NO:2;UniProt登录号P01860);和IgG4重链恒定区域的残基127到327(SEQ ID NO:4;UniProt 登录号P01861);和IgE恒定区的残基225-428(UniProt登录号P01854);和IgA1 恒定区的残基133-353(UniProt登录号P01876);和IgA2恒定区的残基120-340 (UniProt登录号P01877);和IgM抗体恒定区的残基230-452(UniProt登录号 P01871);和IgD恒定区的残基176-384(UniProt登录号P01880)。
图3A和B:IgG1(SEQ ID NO:3)、IgG4(SEQ ID NO:5)和(部分)IgG3 (SEQ ID NO:6)骨架中的抗EGFr抗体2F8的序列比对。描述了按照Kabat和按照EU索引的氨基酸编号(两者都在Kabat等,Sequences of Proteins of Immunological Interest,第5版,PublicHealth Service,National Institutes of Health,Bethesda,MD.(1991)中描述)。
图4:多聚体(如六聚体)排布中邻近分子(分别为Fc和Fc’)Fc之间 K439/S440相互作用详细视图,显示了野生型、未修饰的Fc和Fc’分子之间的相互作用。
图5:多聚体(如六聚体)排布中邻近分子(分别为Fc和Fc’)的Fc之间 K439/S440相互作用详细视图,显示了包含K439E和S440K突变的变体Fc和 Fc’分子之间的相互作用。
图6:用7D8Fc:Fc突变体的C1q结合ELISA。系列浓度的所示抗体被涂覆到微量滴定板孔上并与固定浓度的C1q温育。除了I253D,对于所有包被的变体,与C1q结合的效率可与野生型7D8相当。显示了至少3个实验的代表。
图7:7D8变体介导的对CD20阳性Raji细胞的CDC。Raji细胞与7D8变体 (K439E,S440K,K439E/S440K双突变体,K439E+S440K混合物)和系列浓度的C1q温育以通过测量细胞裂解测试CDC效力。显示了重复实验的示意图。
图8:7D8突变体(7D8-WT,K439E,S440K,K439E/S440K双突变体, K439E+S440K混合物)介导的对CD20阳性Daudi细胞的CDC。测试了系列浓度的7D8突变体的诱导CDC的效力。
图9:CD38抗体HuMAb005突变体介导的对CD38阳性细胞的CDC。(A) 005突变体系列浓度对Daudi细胞的CDC效力。(B)HuMAb 005突变体系列浓度对Raji细胞的CDC效力。(C)具有20%或50%NHS的HuMAb 005的E345R突变体对Wien133细胞的CDC效力。(D)具有20%或50%NHS的HuMAb 005和 7D8的E345R突变体对Raji细胞的CDC效力。
测试从瞬时转染中分离的未纯化抗体样品。使用模拟转染 (mock-transfected)细胞的上清作为阴性对照。
图10:与Fc结合肽的竞争性实验中,CD38抗体HuMAb 005(A)和CD20 抗体HuMAb7D8(B)的野生型和E345R突变体的CDC。对与系列浓度Fc结合 DCAWHLGELVWCT肽(SEQ IDNO:7)温育的经抗体调理的Daudi细胞的 CDC后测量细胞裂解。使用从瞬时转染中分离的未纯化抗体样品。使用模拟转染细胞的上清作为阴性对照。
图11:野生型CD38抗体HuMAb 005和突变体IgG1-005-E345R的CD38表达Daudi细胞的ADCC。显示了一个供体的PBMC的ADCC,以%裂解描绘。
图12:在pH6,如通过ELISA确定的,野生型的IgG1-7D8和突变体 IgG1-7D8-E345R与人、猕猴(cynomolgus)和小鼠FcRN的结合。
图13:在SCID小鼠中静脉注射后,野生型IgG1-7D8和-E354R,-S440K 和K322A变体的血浆浓度。
图14A、B、C和D:对CD20和CD38阳性Wien133细胞的CDC。
图15A和B:在具有Raji-luc#2D1细胞的皮下异种移植物模型中 IgG1-7D8-E345R的体内效力的评价。
图16A和B:在具有Raji-luc#2D1细胞的皮下异种移植物模型中 IgG1-005-E345R的体内效力的评价。
图17:具有E345R突变的CD38/EGFR双特异性抗体对CD38阳性、EGFR 阴性Wien133细胞的CDC。
图18A和B:具有和没有E345R突变的CD20/CD38双特异性抗体对CD20 阳性、CD38阴性的Wien133细胞或Raji细胞的CDC。
图19:具有E345R突变的EGFR抗体2F8对EGFR阳性A431细胞的CDC。
图20A和B:E345R突变体抗体介导的CDC。
图21:具有溶酶体标记LAMP1(APC)的TF抗体(FITC)的共定位分析。
图22A-D:引入E345R导致与在不同B细胞系中测试的野生型利妥昔单抗(rituximab)相比增强的CDC介导的杀伤。
图22E:引入E345R导致与野生型利妥昔单抗相比升高的最大CDC介导的杀伤,其不依赖于具有相当CD20表达水平的不同B细胞系中补体调节蛋白 CD46(A)、CD55(B)或CD59(C)的表达水平。
图23:CDC动力学。相比野生型抗体,E345R抗体导致更迅速和更大量的由CDC所致的靶细胞裂解。
图24:CDC动力学。在双特异性CD38xCD20抗体中引入E345R突变导致更迅速和更大量的CDC介导的靶细胞裂解。
图25:CDC动力学。在单价结合EGFR阴性Raji细胞的双特异性抗体 CD38xEGFR中引入E345R突变导致更迅速和更大量的 CDC介导的靶细胞裂解。
图26A-F:野生型抗体与含(A-C)E345R和Q386K或(D-F)E345R、E430G 和Q386K的突变体抗体的组合对Wien133细胞的CDC。IgG1-b12突变体不结合Wien133细胞并被用作阴性对照抗体。
图27:含E345R突变的IgG1、IgG2、IgG3和IgG4同种型抗体的CDC效力。
图28:在野生型CD38抗体005中引入Fc-Fc稳定E345R突变导致离体CDC 测定法中原代CLL细胞的杀伤增强(平均值±均值的标准偏差)。
图29:如通过ELISA测定的,在pH 6.0时野生型IgG1-005和IgG1-005突变体与人,小鼠和猕猴FcRn的FcRn结合。
图30:在Ramos和SU-DHL-4细胞系中多种利妥昔单抗突变体,野生型利妥昔单抗和不相干阴性对照抗体IgG1-B12在20%正常人血清中的CDC效力。
图31:通过Micro Vue C4d片段ELISA测定的,野生型IgG1-005、 IgG1-005-E345K、IgG1-005-E345Q、IgG1-005-E345Y、IgG1-005-E430G、 IgG1-005-E430S和IgG1-005-S440Y和热聚集的IgG(HAG)(阳性对照)在正常人血清中的C4d产生。
图32A/B:通过总人IgG ELISA(图32A)和人CD38特异性ELISA(图 32B)测定的,施用的野生型IgG1-005和抗体变体IgG1-005-E345K、 IgG1-005-E345Q、IgG1-005-E345R、IgG1-005-E345Y、IgG1-005-E430F、 IgG1-005-E430G、IgG1-005-E430S、IgG1-005-E430T和IgG1-005-S440Y在 SCID小鼠中的血浆清除率。
发明详述
如本发明中所述,令人惊奇地,不直接参与Fc:C1q结合的氨基酸中的突变还能提高抗体的CDC,并也能改进抗体的其他Fc介导的效应子功能。这支持了抗体分子如IgG1抗体能形成随后被C1q结合的寡聚体结构的假设。此外,尽管发现某些突变降低CDC诱导,但是相同或不同抗体分子中此类突变的某些组合导致CDC诱导的恢复,并显示对抗体寡聚的进一步特异性,并由此促进更特异的CDC诱导。如实施例中所示,提高CDC应答的特定突变特征也在于改进的ADCC应答,提高的亲合力,提高的内化和在鼠肿瘤模型系统中的体内效力。这些发现允许具有增强的CDC诱导能力,更有选择性的CDC诱导,和/或其他改进的效应子功能的新的基于抗体的治疗剂。
本发明的多肽变体(包含抗体变体)全包含免疫球蛋白的结合区以及全长或部分Fc域,其在对应IgG1中氨基酸残基E345至S440的区段中包含至少一个或多个突变。不限于理论,基于图1中示意并在本发明中被称为“单突变体”、“双突变体”和“混合突变体”的三个不同的原理,认为鉴定的突变导致更有效和/或更特异的CDC诱导。
本发明变体的改进的C1q和/或CDC效应主要仅是在允许抗体寡聚体形成的检测中,如在抗原没有固定但存在于流体膜上的基于细胞的检测中检测到。此外,根据图1C所示的原理验证,从更稳定的抗体寡聚体得到而不是从 C1q直接结合位点的修饰得到这些效应。
定义
术语“单突变体”应理解为相比亲本多肽或抗体具有提高的CDC并且也可以具有其他的效应子功能的本发明变体。
术语“双突变体”应理解为在所述片段中包含至少两个突变的变体,并相比仅包含所述至少两个突变中一个的变体具有改进的CDC并且也可以具有其他的增强的效应子功能。
术语“混合突变体”应理解为在与相同或不同多肽或抗体的包含所述区段的不同氨基酸残基中的突变的第二变体组合使用时,相比单独的变体、第二变体和单独亲本多肽或抗体中一种或多种,提供提高的CDC和任选地还有其他的增强的效应子功能的变体。
术语“包含免疫球蛋白Fc域和结合区的多肽”在本发明上下文中是指包含免疫球蛋白Fc域和结合区的多肽,所述结合区能与例如存在于细胞,细菌,或病毒粒子上的任何分子,如多肽结合。免疫球蛋白的Fc域定义为通常在用木瓜蛋白酶消化抗体后生成的抗体片段(这对本领域技术人员已知),其包含免疫球蛋白的两个CH2-CH3区和连接区,例如铰链区。抗体重链恒定区定义抗体同种型,例如IgG1、IgG2、IgG3、IgG4、IgA1、IgA2、IgM、IgD或IgE。 Fc域介导抗体与被称为Fc受体的细胞表面受体和补体系统蛋白的效应子功能。结合区可以是能结合细胞、细菌或病毒粒子的多肽序列,如蛋白、蛋白配体、受体、抗原结合区或配体结合区。如果结合区是例如受体,“包含免疫球蛋白Fc域和结合区的多肽”可以作为免疫球蛋白Fc域和所述结合区的融合蛋白来被制备。如果结合区是抗原结合区,“包含免疫球蛋白Fc域和结合区的多肽”可以是抗体,像嵌合抗体、人源化抗体或人抗体或仅有重链的抗体或ScFv-Fc-融合体。包含免疫球蛋白Fc域和结合区的多肽通常可以包含连接区,例如铰链区,和免疫球蛋白重链的两个CH2-CH3区,因此“包含免疫球蛋白Fc域和结合区的多肽”可以是“包含至少免疫球蛋白Fc域和结合区的多肽”。术语“免疫球蛋白Fc域”在本发明上下文中意指存在取决于抗体亚型的连接区(例如铰链),以及免疫球蛋白CH2和CH3区,例如人类IgG1、IgG2、 IgG3、IgG4、IgD、IgA1、IgGA2、IgM或IgE。多肽不限于人类来源,而是可以是任何来源,例如小鼠或猕猴来源。
本发明中使用的术语“CH2区”或“CH2域”意指免疫球蛋白的CH2区。因此例如人类IgG1抗体的CH2区对应于根据EU编号系统的氨基酸228-340。然而,CH2区也可以是本发明所述的任何其他亚型。
本发明中使用的术语“CH3区”或“CH3域”意指免疫球蛋白的CH3区。因此例如人类IgG1抗体的CH3区对应于根据EU编号系统的氨基酸341-447。然而,CH3区也可以是本发明所述的任何其他亚型。
术语“免疫球蛋白”是指一类结构上相关的糖蛋白,其由两对多肽链,即一对轻(L)低分子量链和一对重(H)链组成,所有四条潜在通过二硫键相连。免疫球蛋白的结构已经被充分表征。见例如Fundamental Immunology Ch.7 (Paul,W.,编第二版.Raven Press,N.Y.(1989))。简而言之,每条重链通常由重链可变区(本发明中缩写为VH)和重链恒定区组成。重链恒定区通常由三个域(CH1,CH2,和CH3)组成。重链之间通过所谓“铰链区”中的二硫键相连。每条轻链通常由轻链可变区(本发明中缩写为VL)和轻链恒定区组成。轻链恒定区通常由一个域CL组成。VH和VL区可以被进一步再划分为高变性区 (或在序列和/或结构限定环形式中可以高度可变的高变区),也被称为互补决定区(CDRs),其散布有更保守的区域,被称为框架区(FRs)。每条VH和VL 通常由三个CDRs和四个FRs组成,其从氨基末端到羧基末端以以下顺序排列:FR1,CDR1,FR2,CDR2,FR3,CDR3,FR4(也见Chothia和Lesk J.Mol.Biol.196,901917(1987))。除非另有说明或上下文相矛盾,本发明中恒定区序列的氨基酸根据EU-索引编号(在Kabat,E.A.等,Sequences of proteins of immunologicalinterest.第五版-US Department of Health and Human Services, NIH publicationNo.91-3242,pp 662,680,689(1991)中描述)。
本发明上下文中术语“抗体”(Ab)是指免疫球蛋白分子,免疫球蛋白分子片段,或其衍生物,其具有在典型生理条件下以相当长的时段的半衰期(如至少约30分钟,至少约45分钟,至少约1小时,至少约2小时,至少约4小时,至少约8小时,至少约12小时,约24小时或更长,约48小时或更长,约3,4, 5,6,7或更多天等),或任何其他相关功能限定时段(如足以诱导、促进、增强和/或调节与抗体结合抗原相关的生理应答的时间,和/或足以抗体募集效应子活性的时间)特异性结合抗原的能力,。本发明的抗体包含免疫球蛋白Fc 域和结合区。抗体通常含有两个CH2-CH3区和连接区,例如铰链区,例如至少Fc域。因此本发明的抗体可以包含Fc区和抗原结合区。免疫球蛋白分子的重链和轻链可变区包含与抗原相互作用的结合域。抗体的恒定或“Fc”域可介导免疫球蛋白与宿主组织或因子的结合,包含免疫系统各种细胞(如效应细胞)和补体系统的组分如C1q,补体激活经典途径的第一组分。抗体也可以是多特异性抗体,如双特异性抗体或类似分子。术语“双特异性抗体”是指具有对至少两种不同,通常是不重叠表位的特异性的抗体。这样的表位可以在相同或不同的靶标上。如果表位在不同的靶标位上,这样的靶标可以在相同细胞或不同细胞或细胞类型上。如上所述,除非另有说明或者明显与上下文相矛盾,在本发明中术语抗体包含抗体片段,其包含至少部分Fc区并保持特异性结合抗原的能力。这样的片段可以通过任何已知技术提供,如酶切、肽合成和重组表达技术。已经显示了抗体的抗原结合功能可以由全长抗体的片段来进行。涵盖在术语“Ab”或“抗体”中的结合片段实例包含,不限于,单价抗体(在WO2007059782中由Genmab描述);重链抗体,其仅由两条重链组成并在例如骆驼中天然存在(例如,Hamers-Casterman(1993)Nature 363:446);ThioMabs(Roche,WO2011069104),链交换工程域(SEED或Seed 体),其是不对称的和双特异性的抗体样分子(Merck,WO2007110205); Triomab(Fresenius,Lindhofer等(1995J Immunol 155:219));FcΔAdp (Regeneron,WO2010151792),Azymetric Scaffold(Zymeworks/Merck, WO2012/058768),mAb-FV(Xencor,WO2011/028952),双可变域免疫球蛋白(Abbott,DVD-Ig,美国专利7,612,181);双域双头抗体(Unilever;Sanofi Aventis,WO20100226923),二双抗体(ImClone/EliLilly),钮-入-孔 (knob-into-holes)抗体形式(Genentech,WO9850431);DuoBody(Genmab, WO2011/131746);静电驾驭抗体形式(Amgen,EP1870459和WO 2009089004;Chugai,US201000155133;Oncomed,WO2010129304A2);双特异性IgG1 和IgG2(Rinatneurosciences Corporation,WO11143545),CrossMAbs(Roche, WO2011117329),LUZ-Y(Genentech),Biclonic(Merus),双靶向域抗体 (GSK/Domantis),识别两个靶标的二合一抗体(Genentech,Novlmmune),交联MAbs(Karmanos Cancer Center),CovX体(CovX/Pfizer),IgG样双特异性 (ImClone/Eli Lilly,Shen,J.,等J Immunol Methods,2007.318(1-2):p.65-74),和 DIG体和PIG体(Pharmabcine),以及双亲和性再靶向分子(Macrogenics的 Fc-DART或Ig-DART,WO/2008/157379,WO/2010/080538),Zybodies(Zyngenia),普通轻链(Crucell/Merus,US7262028)或普通重链(Novlmmune 的κλ体)的方法,以及包含融合于含Fc域的抗体片段的多肽序列融合蛋白,像scFv融合体,像ZymoGenetics/BMS的BsAb,Biogen Idec的HERCULES (US007951918),EmergentBioSolutions/Trubion的SCORPIONS,Ts2Ab (Medlmmune/AZ Dimasi,N.,等J Mol Biol,2009.393(3):p.672-92),Novartis 的scFv融合体,Changzhou Adam Biotech Inc的scFv融合体(CN 102250246), Roche的TvAb(WO2012025525,WO2012025530),f-Star的mAb2(WO2008/003116)和双scFv-融合体。还应当理解,除另有指明外,术语抗体也包含多克隆抗体、单克隆抗体(如人单克隆抗体)、例如,通过Symphogen 和Merus(Oligoclonics)开发的技术生成的抗体混合物(重组多克隆),和抗体样多肽,如嵌合抗体和人源化抗体。生成的抗体可潜在地具有任何同种型。
术语“全长抗体”,当在本发明中使用时是指包含对应通常在该同种型野生型抗体中发现的那些域的所有重和轻链恒定和可变域的抗体(如亲本或变体抗体)。
本发明中所用的术语“人抗体”意图包含具有源自人种系免疫球蛋白序列的可变区和恒定区的抗体。本发明的人抗体可以包含不由人种系免疫球蛋白序列编码氨基酸残基(例如,通过体外随机或定点诱变或者通过体内体细胞突变引入的突变、插入或缺失)。然而,本发明中所用的术语“人抗体”不意欲包含其中源自另一哺乳动物种类(如小鼠)种系,的CDR序列已经被移植到人骨架序列的抗体。
本发明中所用术语“单克隆抗体”、“单克隆Ab”、“单克隆抗体组成”,“mAb”等指单分子组成的抗体分子制剂。单克隆抗体组成显示对特定表位的单结合特异性和亲和力。因此,术语“人单克隆抗体”是指显示单结合特异性且具有源自人种系免疫球蛋白序列的可变和恒定区的Ab。人mAbs可以由杂交瘤生成,所述杂交瘤包含从转基因或转染色体非人动物(如转基因小鼠)中获得并与永生细胞融合的B细胞,所述非人动物包含经过重排以产生功能性人抗体的人重链转基因全集和轻链转基因全集。
本发明中所用的“同种型”是指由重链恒定区基因编码的免疫球蛋白类 (例如IgGl、IgG2、IgG3、IgG4、IgD、IgA1、IgGA2、IgE或IgM或者其任何同种异型如IgG1m(za)和IgG1m(f))。此外,每种重链同种型可与κ或λ轻链组合。
术语“单价抗体”在本发明上下文中意为抗体分子仅能够仅以抗体结合域之一与抗原结合,例如具有单抗原-抗体相互作用,并因此不能抗原交联。
本发明中所用的术语“靶标”在本发明上下文中应理解为与包含Fc域和结合区的多肽的结合区结合的分子,当用于抗体结合的上下文中时包含针对制备抗体的任何抗原。术语“抗原”和“靶标”关于抗体时可以互换使用,并对本发明任何方面或实施方案构成相同的含义或目的。
本发明中所用的术语“结合”在抗体结合到预定抗原的上下文中通常是当使用抗原作为配体和抗体作为分析物,通过例如表面等离振子共振(SPR) 技术在BIAcore3000仪器中测定时以对应约10-6M或更小,例如10-7M或更小,如10-8M或更小,如10-9M或更小,约10- 10M或更小,或约10-11M或甚至更小的KD的亲和力结合,,,并且以对应于下述KD的亲和力结合预定的抗原,所述KD是比其结合除预定抗原或紧密相关抗原外的非特异性抗原(例如BSA,酪蛋白)的特异性低至少10倍(at least ten-fold lower),如低至少100倍,例如低至少1,000倍,如低至少10,000倍,例如低至少100,000倍。亲和力较低的量取决于抗体的KD,这样当抗体的KD非常低(即抗体高度特异)时,则对抗原亲和力相比对非特异性抗原亲和力更低的量可以是至少10,000倍。本发明中所用的术语“KD”(M)是指特定抗体-抗原相互作用的解离平衡常数。
本发明的“变体”或“抗体变体”或“亲本抗体变体”是相比“亲本抗体”包含一个或多个突变的抗体分子。不同的术语可以互换使用,并对本发明任何方面或实施方案构成相同的含义或目的。示例性的亲本抗体形式包含不限于,野生型抗体,全长抗体或含Fc的抗体片段,双特异性抗体,人抗体,或其任意组合。类似的,本发明“变体”或“包含免疫球蛋白Fc域和结合区的多肽的变体”或“包含免疫球蛋白Fc域和结合区的亲本多肽的变体”是相比“包含免疫球蛋白Fc域和结合区的亲本多肽”,包含一个或多个突变的“包含免疫球蛋白Fc域和结合区的多肽”。不同的术语可以互换使用,并对本发明任何方面或实施方案构成相同的含义或目的。示例性的突变包含亲本氨基酸序列中的氨基酸缺失、插入或氨基酸取代。氨基酸取代可将天然氨基酸替换为另一种天然存在的氨基酸,或者非天然存在的氨基酸衍生物。氨基酸取代可以是保守或非保守的。在本发明上下文中,保守取代由下面三个表的一个或多个中反映的氨基酸类之间的取代所定义:
保守取代的氨基酸残基类
| 酸性残基 | Asp(D)和Glu(E) |
| 碱性残基 | Lys(K),Arg(R),和His(H) |
| 亲水不带电荷残基 | Ser(S),Thr(T),Asn(N)和Gln(Q) |
| 脂肪族不带电荷残基 | Gly(G),Ala(A),Val(V),Leu(L)和Ile(I) |
| 非极性不带电荷残基 | Cys(C),Met(M),和Pro(P) |
| 芳香族残基 | Phe(F),Tyr(Y),和Trp(W) |
可选保守氨基酸残基取代类型
| 1 | A | S | T |
| 2 | D | E | |
| 3 | N | Q | |
| 4 | R | K | |
| 5 | I | L | M |
| 6 | F | Y | W |
氨基酸残基的可选物理和功能分类


本发明上下文中,变体中的取代表示为:
原氨基酸-位置-取代的氨基酸
使用三字母码或单字母码,包含代码Xaa和X来表示氨基酸残基。因此,符号“E345R”或“Glu345Arg”意为变体在对应亲本抗体中345位置中的氨基酸的变体氨基酸位置中包含以精氨酸取代谷氨酸的取代。
当位置本身不存在于抗体中,但变体包含氨基酸插入时,例如:
位置-取代的氨基酸;使用符号,例如“448E”。
这样的符号与同源多肽或抗体系列中的修饰特别相关联。
类似的,当取代氨基酸残基的身份不重要时:
原氨基酸-位置;或“E345”。
对于原氨基酸和/或取代的氨基酸可以包含多于1个,但非全部氨基酸的修饰,位置345处谷氨酸取代为精氨酸、赖氨酸或色氨酸:
“Glu345Arg,Lys,Trp”或“E345R,K,W”或“E345R/K/W”,或“E345至R,K或W”可以在本发明上下文中互换使用。
此外,术语“取代”包含取代为其他19个天然氨基酸的任一个,或取代为其他氨基酸,如非天然氨基酸。例如,345位置处氨基酸E的取代包含下面取代的每一个:345A,345C,345D,345G,345H,345F,345I,345K,345L, 345M,345N,345Q,345R,345S,345T,345V,345W和345Y。顺便说一下,这相当于名称345X,其中X指任意氨基酸。这些取代也可以被称为E345A,E345C等,或E345A,C等或E345A/C/等。这类似适用于本发明中涉及的每一个位置,本发明中具体地包含这样取代的任一个。
“对应于”另一个序列中的氨基酸或片段的一个序列中的氨基酸或片段是(i)使用标准序列比对程序如ALIGN,ClustalW等,通常以默认设置与另一氨基酸或片段比对,和(ii)具有与SEQ ID NO:1至少50%,至少80%,至少90%,或至少95%的序列同一性的氨基酸或片段。例如可以使用图2和3中所显示的序列比对以鉴定IgG2、IgG3或IgG4Fc序列中对应于IgG1Fc序列中特定氨基酸的任意氨基酸。
本发明涉及变体,即亲本多肽和亲本抗体,和/或变体多肽和变体抗体,其与SEQID No:1,2,3,4和5的氨基酸P247至K447具有一定程度的同一性,这样的亲本和/或变体抗体在下文中被称为“同源抗体”。
为了本发明的目的,通过Needleman-Wunsch比对(即全局比对)的程序“比对”来确定两个氨基酸序列之间的同一性程度以及两个核苷酸序列之间的同一性程度。该程序被用于多肽以及核苷酸序列的比对。默认得分矩阵 BLOSUM50被用于多肽比对,而默认同一性矩阵被用于核酸序比对,间隙 (gap)的第一个残基罚分是-12(对于多肽)和-16(对于核苷酸)。间隙的其他残基的罚分是-2(对于多肽)和-4(对于核苷酸)。
“比对”是FASTA包版本v20u6的一部分(见W.R.Pearson和D.J.Lipman (1988),"Improved Tools for Biological Sequence Analysis",PNAS 85:2444-2448,以及W.R.Pearson(1990)"Rapid and Sensitive Sequence Comparaison with FASTP andFASTA",Methods in Enzymology 183:63-98)。 FASTA蛋白比对使用Smith-Waterman算法,对间隙大小无限制(见 "Smith-Waterman algorithm",T.F.Smith和M.S.Waterman(1981)J.Mol. Biolo.147:195-197)。
本发明中所用的术语“效应细胞”是指相对免疫应答的识别(cognitive) 和激活阶段,参与免疫应答效应子阶段的免疫细胞。示例性的免疫细胞包含髓样或淋巴样来源的细胞,例如淋巴细胞(如B细胞和T细胞,包含溶细胞性T 细胞(CTLs)),杀伤细胞,天然杀伤细胞,巨噬细胞,单核细胞,嗜曙红细胞 (eosinophil),多形核细胞,如嗜中性粒细胞,粒细胞,肥大细胞,和嗜碱性粒细胞。一些效应细胞表达Fc受体(FcRs)或补体受体并执行特异免疫功能。在一些实施方案中,效应细胞如,例如天然杀伤细胞,能诱导ADCC。例如,表达FcRs的单核细胞,巨噬细胞,嗜中性粒细胞,树突状细胞和Kupffer细胞参与靶细胞的特异性杀伤和将抗原呈递到免疫系统的其他组分上,或结合到呈递抗原的细胞上。在一些实施方案中,可以通过抗体驱动经典补体激活进一步增强ADCC,导致靶细胞上的激活C3片段的沉积。C3裂解产物是表达在髓样细胞上的补体受体(CRs),如CR3的配体。效应细胞上CRs的补体片段识别可以促进增强的Fc受体介导ADCC。在一些实施方案中,抗体驱动的经典补体激活导致靶细胞上的C3片段。这些C3裂解产物可以促进直接补体依赖性细胞性细胞毒性(CDCC)。在一些实施方案中,效应细胞可以吞噬靶标抗原,靶标颗粒或靶细胞。特定FcR或补体受体在效应细胞上的表达可以受体液因子如细胞因子调节。例如,已经发现FcγRI的表达被干扰素γ(IFNγ)和/ 或G-CSF上调。此增强的表达提高了带有FcγRI的细胞对靶标的细胞毒性。效应细胞可以吞噬靶标抗原或吞噬或裂解靶细胞。在一些实施方案中,抗体驱动的经典补体激活导致靶细胞上的C3片段。这些C3裂解产物可以促进通过效应细胞的直接吞噬或者通过增强抗体介导的吞噬间接促进。
本发明中所用术语“载体”意思是指能诱导连接入载体的核酸片段转录的核酸分子。载体的一种类型是“质粒”,其为环形双链DNA环形式。载体的另一种类型是病毒载体,其中核酸片段可以被连接入病毒基因组。某些载体能在其被引入的宿主细胞中自主复制(例如有细菌复制原点的细菌载体和附加体哺乳动物载体)。其他载体(如非附加体哺乳动物载体)可以在引入宿主细胞时被整合入宿主细胞的基因组中,并由此随宿主基因组一起复制。此外,某些载体能引导可操作地与其连接的基因的表达。这样的载体本发明中被称为“重组表达载体”(或简称“表达载体”)。一般来说,用于重组DNA技术中的表达载体通常是质粒形式。在本说明书中,由于质粒是最常用的载体形式,“质粒”和“载体”可以互换使用。然而,本发明意图包含发挥等同功能的表达载体的这样其他形式,如病毒载体(如复制缺陷型逆转录病毒,腺病毒和腺伴随病毒)。
本发明中所用术语“重组宿主细胞”(或简称“宿主细胞”)意指表达载体已经被引入其中的细胞。应当理解,这样的术语意不仅指特定对象细胞,也指这样细胞的子代。由于突变或环境影响在后代中可以出现某些修饰,因此这样的子代实际上可以不与亲本细胞相同,但仍然包含在本发明中所用术语“宿主细胞”的范围内。重组宿主细胞包含,例如,转染瘤(transfectoma),如CHO细胞,HEK-293细胞,PER.C6,NS0细胞,和淋巴细胞性细胞,以及原核细胞如大肠杆菌,以及其它真核宿主如植物细胞和真菌。
本发明中所用术语“转染瘤”包含表达抗体或靶标抗原的重组宿主细胞,如CHO细胞,PER.C6,NS0细胞,HEK-293细胞,植物细胞或真菌,包含酵母细胞。
术语“制剂”是指抗体变体的制剂和不同抗体变体的混合物,它们当与细胞(例如细胞表面上表达的抗原),细胞膜,病毒粒子或其他结构关联抗原相互作用时可具有提高的形成寡聚体的能力,由此能得到提高的C1q结合,补体激活,CDC,ADCC,ADCP,其他Fc介导的效应子功能,内化,下调,凋亡,抗体-药物缀合物(ADC)摄取,亲合力或其任意组合。示例性的检测提供在实施例中,例如,C1q结合亲合力(实施例4),CDC(实施例5,6和10, 16,19,22,23,24,25和35);ADCC(实施例12),体内效力(实施例20, 21),血浆清除率(实施例37),FcRn结合(实施例34)和不依赖于靶标的流体相补体激活(实施例36)。称为“单突变体”,“双突变体”,和“混合突变体”的根据本发明方面的变体与其示例性制备过程和使用方法在下面更详细的描述。
本发明中所用的术语“亲和力(affinity)”是一个分子(例如抗体)与另一个 (例如靶标或抗原)在单个位点(如抗体的个别抗原结合位点与抗原的单价结合的结合强度。
本发明中所用的术语“亲合力(avidity)”是两个结构之间(如同时与靶标相互作用的抗体的多个抗原结合位点之间,或例如抗体与C1q之间)的多个结合位点的结合强度。当存在多于一种结合相互作用时,仅当所有结合位点解离时两个结构才会解离,并因此解离速率将会慢于单个结合位点的,并由此相比单个结合位点结合的强度(亲和力)提供了更大的有效总结合强度(亲合力)。
本发明中所用的术语“寡聚体”是指,相比至少原则上是由不限数目的单体组成的聚合物,由多于一个但有限数目的单体单元(如抗体)组成的分子。示例性的寡聚体是二聚体,三聚体,四聚体,五聚体和六聚体。希腊语前缀经常用于表示寡聚体中的单体单元数目,例如由四个单元组成的四聚体和由六个单元组成的六聚体。
本发明中所用的术语“寡聚化”意指将单体以有限程度的聚合转化的过程。本发明中,观察到Fc域的寡聚发生是在含Fc域的多肽(如抗体,优选但不限于在细胞表面上)的靶标结合后。抗体的寡聚化可以例如使用细胞表面 C1q结合检测(如实施例4和9中所述),C1q效力检测(如实施例5中所述),和实施例6,10和19中所述的补体依赖细胞毒性来评价。
本发明中所用的术语“C1q结合”意指在C1q结合与其抗原结合的抗体的上下文中的C1q结合。与其抗原结合的抗体应理解为在本发明所描述的上下文中在体内和体外两者发生。C1q结合可以例如通过使用人造表面(例如,如实施例3所描述的ELISA用板中的塑料)上固定的抗体,或者通过使用与细胞或病毒表面上预定抗原结合(如实施例4和9中所述)来评价。C1q与抗体寡聚体的结合在本文中应理解为导致高亲合力结合的多价相互作用。
本发明中所用的术语“补体激活”是指经典补体途径的激活,其是由补体组分C1q结合与其抗原结合的抗体触发的。C1q是经典补体级联的早期事件中的第一个蛋白,所述经典补体级联包含一系列裂解反应,最终导致称为 C3转化酶的酶活性的形成,该C3转化酶将补体组分C3裂解为C3b和C3a。C3b 与膜上的C5共价结合以形成C5b,其继而触发补体激活的晚期事件,其中末期补体组分C5b,C6,C7,C8和C9组装成膜攻击复合物(MAC)。补体级联导致孔的形成,由于孔的形成而引起细胞裂解,也被称为补体依赖性细胞毒性(CDC)。补体激活可以通过使用C1q效力(如实施例5中所述),CDC动力学(如实施例28,29和30中所述),CDC检测(如实施例6,10,19,25,27,33和 35中所述),或者通过Beurskens等2012年4月1日vol.188no.73532-3541中描述的C3b和C4b细胞沉积方法来评价。
本发明中所用的术语“补体依赖性细胞毒性”(“CDC”)意指由于MAC 组装所产生的膜上的孔而导致与其在细胞或病毒粒子上的抗原结合的抗体裂解的抗体介导的补体激活过程。CDC可以通过体外检测(诸如CDC检测,其中使用正常人血清作为补体源,如实施例6,10,19,25,27,33和35所述的),或者实施例5所述的C1q效力检测(其中正常人血清已经在C1q中限制) 来评价。
本发明中所用的术语“抗体依赖性细胞介导的细胞毒性”(“ADCC”) 意指通过表达能识别结合抗体恒定区的Fc受体的细胞杀伤经抗体包被的靶细胞或病毒粒子的机制。ADCC可以使用方法,如实施例12中描述的ADCC 检测来确定。
本发明中所用的术语“抗体依赖性细胞吞噬”(“ADCP”)意指通过吞噬细胞内化来消除经抗体包被的靶细胞或病毒粒子的机制。内化的抗体包被的靶细胞或病毒粒子被包含在称为吞噬体的囊泡中,其随后与一个或多个溶酶体融合,以形成吞噬溶酶体。ADCP可以通过使用van Bij等Journal of Hepatology Volume 53,Issue 4,2010年10月,677-685页中描述的那样使用巨噬细胞作为效应细胞和视觉显微术的体外细胞毒性检测来评价。或者,关于例如通过PMN的金黄色葡萄球菌吞噬,如实施例14中所述的。
本发明中所用的术语“补体依赖性细胞性细胞毒性”(“CDCC”)意指由于抗体介导的补体激活而由表达能识别与靶细胞或病毒粒子共价结合的补体3(C3)裂解产物的补体受体的细胞杀伤靶细胞或病毒粒子的机制。CDCC 可以用与对ADCC所描述的类似的方式来评价。
在这里使用的术语“血浆半衰期”表示在消除期(分布相之后)将血浆中多肽的浓度降低至其起始浓度的一半花费的时间。对于抗体,分布相通常将是1-3天,在此期间,由于血浆和组织之间的重新分配,血浆浓度降低大约50%。血浆半衰期可以通过本领域公知的方法来测量。
如本文所用的术语“血浆清除率”是在将多肽施用于活有机体后,从血液中除去所述多肽的速率的定量测量。血浆清除率可以计算为剂量/AUC (mL/天/kg),其中,按照实施例37从浓度-时间曲线确定AUC值(曲线下面积)。
本发明中所用的术语“下调”意指例如通过抗体与受体的结合,来减少细胞表面上的分子(如抗原或受体)数目的过程。
本发明中所用的术语“内化”意指抗体或含Fc的多肽被从细胞表面和/ 或从周围介质中例如通过内吞而内化入靶标表达细胞中的任何机制。抗体的内化可以使用测量内化抗体量的直接检测法(如例如实施例26中所述的溶酶体共定位检测)来评价。
本发明中所用的术语“抗体-药物缀合物”是指具有对至少一类恶性细胞的特异性的抗体或含Fc的多肽,药物,和将药物连接到例如抗体上的接头。在恶性细胞存在时接头是可裂解或不可裂解的;其中抗体-药物缀合物杀伤恶性细胞。
本发明中所用的术语“抗体-药物缀合物摄取”是指抗体-药物缀合物被连接到细胞上的靶标,后被细胞膜摄取/吞入,并由此吸入细胞的过程。如WO 2011/157741所述,抗体-药物缀合物摄取可以被评价为“抗体介导的内化和体外杀伤检测中抗TF ADC的细胞杀伤”。
本发明中所用的术语“凋亡”是指可以发生在细胞中的程序性细胞死亡 (PCD)过程。生物化学事件导致特征性细胞改变(形态学)和死亡。这些改变包含空泡化,细胞皱缩,核碎裂,染色质浓缩,和染色体DNA片段化。抗体结合到某些受体可以诱导凋亡。
Fc受体结合可以如实施例12所述间接测量。
本发明中所用的术语“FcRn”意指作为Fc受体的新生儿Fc受体。其首先在啮齿类中发现为能够将IgG从母乳中通过新生啮齿动物肠道上皮细胞输送到新生动物血流中的独特受体。进一步的研究揭示了人中的类似受体。然而,在人中,在胎盘中发现它帮助促进母体IgG向生长的胎儿中输送,并且也已经显示在检测IgG周转中发挥作用。FcRn在6.0-6.5的酸性pH,但不在中性或更高的pH结合IgG。因此,FcRn可以在微酸性pH结合来自肠腔(肠的内部)的 IgG,并确保向pH中性到碱性(pH7.0-7.5)的基底侧(身体内侧)有效单向输送。该受体在IgG成人救助中通过其在内皮细胞中内吞途径中的发生起作用。酸性内涵体中的FcRn受体结合通过胞饮作用内化的IgG,从而将其再循环到细胞表面,在血液的碱性pH时将其释放,由此防止其经历溶酶体降解。该机制也对血液中相比其他同种型更大的IgG半衰期提供了解释。实施例13和34描述了显示ELISA中在pH 6.0时IgG结合到FcRn的检测。
本发明中所用的术语“蛋白A”意指最初在细菌金黄色葡萄球菌 (Staphylococcusaureus)的细胞壁上发现的56kDa MSCRAMM表面蛋白。其由 spa基因编码,而其调节受DNA拓扑学,细胞渗透压,和称为ArlS-ArlR的双组分系统控制。由于其结合免疫球蛋白的能力,也已发现了其在生物化学研究中使用。其由折叠成三螺旋束的5个同源Ig结合域组成。每个域能结合来自许多哺乳动物种类的蛋白,最显著的是IgGs。其结合大多数免疫球蛋白的重链Fc区(与FcRn受体的保守结合位点重叠),并也与人VH3家族的Fab区域相互作用。通过血清中的这些相互作用,IgG分子通过其Fc区而不是单纯通过其Fab区域结合细菌,由此细菌破坏调理,补体激活和吞噬。
本发明中所用的术语“蛋白G”意指在组C和G链球菌细菌中表达的免疫球蛋白结合蛋白,其很类似蛋白A但有不同的特异性。其是65kDa(G148蛋白 G)和58kDa(C40蛋白G)细胞表面蛋白,其在通过其对Fc区的结合在纯化抗体中得到应用。
影响多肽CDC的方法
应当理解本发明中描述的关于亲本抗体,第一亲本抗体或第二亲本抗体的所有实施方案也可以适用于包含免疫球蛋白Fc域和结合区的其他亲本,第一亲本或第二多肽。
一方面,本发明涉及提高包含免疫球蛋白Fc域和结合域的亲本多肽的补体依赖性细胞毒性(CDC)的方法,该方法包括在一个或多个氨基酸残基中对亲本多肽引入突变,其选自对应于人类IgG1重链Fc区中E430X、E345X和 S440W的组。
在一个实施方案中,亲本多肽可以是包含免疫球蛋白的Fc域和抗原结合区的亲本抗体。
根据本发明的方法或用途向亲本多肽中引入突变得到了变体多肽(其在本文也可以称作为“变体”)。因此可以执行本发明的方法以获得本发明描述的任何变体或变体多肽。
从本发明的方法或用途获得的变体多肽相比亲本多肽具有提高的CDC。通常,多肽对效应子功能的效果可以通过EC50值确定,EC50值是获得最大裂解数值一半必需的多肽浓度。
最大裂解是使用饱和量多肽时所获得的裂解,其中饱和意思是指多肽的所有靶标都被多肽结合时的多肽量。
术语“提高CDC”“改进CDC”或“提高效应子功能”,或“改进效应自功能”在本发明上下文中指相比亲本多肽,变体多肽的EC50值有降低。EC50 值的降低可以是例如至少或约2倍,如至少或约3倍,或至少或约5倍,或至少或约10倍。可选的,“提高CDC”“改进CDC”或“提高效应子功能”或“改进效应子功能”意为在亲本多肽裂解少于所有细胞100%的条件下,裂解的细胞最大量增加(其中细胞总量被设为100%)例如所有细胞的10%到100%,如约10%,约20%,约30%,约40%,约50%,约60%,约70%,约80%,约90%,和约100%。
可通过将IgG1-005或IgG1-7D8重链可变域克隆入变体并在CDC检测中测试其效力来检测变体的提高或改进的效应子功能,如对Daudi(实施例6)和 Wien(实施例10)所描述的。使用IgG1-7D8HC可变域和Daudi细胞,提高被限定为与在研究条件下IgG1-7D8的EC50相比低超过2倍的EC50,如低约2倍,约3倍,约5倍,约10倍或超过10倍的EC50值(观察到最大裂解半数的浓度)。使用IgG1-005HC可变域和Daudi细胞,提高被限定为与在研究条件下与IgG-005的EC50相比低超过2倍的EC50,如约2倍,约3倍,约5倍,约10倍或超过10倍的EC50值(观察到最大裂解半数的浓度)。使用IgG1-7D8HC可变域和Wien133细胞,提高被限定为与在研究条件下与IgG1-7D8的EC50相比低超过2倍的EC50,如低约2倍,约3倍,约5倍,约10倍或超过10倍的EC50值(观察到最大裂解半数的浓度)。使用IgG1-005HC可变域和Wien133细胞,提高会被限定为范围为从所有细胞的10%到100%,如约10%,约20%,约30%,约40%,约50%,约60%,约70%,约80%,约90%,和约100%的最大裂解提高。CDC效力的提高也可以被限定为与在研究条件下与IgG-005的EC50相比,超过2倍的EC50,如低2倍,约3倍,约5倍,约10倍或超过10倍的EC50 值(在Wien133细胞的裂解可以检测到的条件下,观察到最大裂解半数的浓度)。
本发明的发明人惊奇地发现,在这些特定位置的突变具有对变体抗体 CDC的改进效果,所述抗体变体是根据本发明的方法向亲本抗体能够引入一个或多个突变而获得(例如实施例19中所示)。不限于理论,认为从上述位置基团取代一个或多个氨基酸会激发寡聚化。抗体以更高的亲合力结合(实施例2举例说明;IgG-7D8-E345R直接标记得到相比IgG-7D8-WT提高的与Daudi 细胞的结合),这使得抗体与细胞结合更长的时间并由此能有不同的效应子功能,例如提高的C1q结合,C1q效力CDC,ADCC,内化,ADCP,和/或体内效力。这些效果已经被实施例4(在细胞上C1q结合),实施例5(CDC检测中的C1q效力),实施例6,7,27,28和29(CDC检测),实施例12(ADCC),实施例26(内化),实施例21和22(体内效力),血浆清除率(实施例37),FcRn 结合(实施例34)和不依赖于靶标的流体相补体激活(实施例36)举例说明。
因此,选自对应于人类IgG1重链Fc区中E430X,如E430G、E430S、E430F 或E430T,E345X,如E345K、E345Q、E345R或E345Y,S440Y和S440W的那些突变的氨基酸残基的突变在本发明上下文中也可以被称为“单突变”方面或“CDC增强突变”。
因此,在一个实施方案中,在提高CDC的方法中,一个或多个氨基酸残基中的突变选自对应于人类IgG1重链Fc区中E430G、E430S、E430F、E430T、 E345K、E345Q、E345R、E345Y和S440W的组。
在优选的实施方案中,在提高CDC的方法中,一个或多个氨基酸残基中的突变选自对应于人类IgG1重链Fc区中E430G、E430S、E345K和E345Q的组。
在一个实施方案中,亲本多肽是包含免疫球蛋白Fc域和抗原结合区的亲本抗体。
在另一个方面,本发明还涉及提高包含免疫球蛋白Fc域和结合区的亲本多肽的CDC和抗体依赖性细胞介导的细胞毒性(ADCC)的方法,该方法包括在一个或多个氨基酸残基中对亲本多肽引入突变,其对应于人类IgG1重链Fc区中的E430X、E345X和S440W,其中X是任何氨基酸,例如天然存在的氨基酸。
在一个实施方案中,一个或多个氨基酸残基中的突变选自对应于人类 IgG1重链Fc区中E430G、E430S、E430F、E430T、E345K、E345Q、E345R、 E345Y和S440W的组。
在一个优选的实施方案中,一个或多个氨基酸残基中的突变选自对应于人类IgG1重链Fc区中的位置E345R、E430T和E430F的组。
在一个实施方案中,抗体的至少一个其他效应子功能,如C1q结合,补体激活,抗体依赖性细胞介导的细胞毒性(ADCC),Fc-γ受体结合,蛋白A 结合,蛋白G结合,ADCP,补体依赖性细胞性细胞毒性(CDCC),补体增强性细胞毒性,抗体介导的与受调理抗体的补体受体结合,抗体介导的细胞吞噬(ADCP),内化,凋亡,和/或与受调理抗体的补体受体结合也增强了,如 ADCC。
在一个实施方案中,亲本多肽是包含免疫球蛋白Fc域和抗原结合区的亲本抗体。
在一个实施方案中,当亲本抗体与抗原表达细胞、细胞膜或病毒粒子上的其抗原结合时,亲本抗体的CDC被提高。
在一个实施方案中,亲本抗体是单特异性,双特异性,或多特异性抗体。
在又一个方面,本发明涉及提高亲本抗体的补体依赖性细胞毒性(CDC) 的方法,所述亲本抗体为包含第一多肽和第二多肽的双特异性抗体,所述第一多肽包含免疫球蛋白的第一CH2-CH3区和第一抗原结合区,所述第二多肽包含免疫球蛋白的第二CH2-CH3区和第二抗原结合区,其中,第一和第二抗原结合区结合相同或不同抗原上的不同表位,并且其中所述方法包括将突变引入到第一和/或第二CH2-CH3区的一个或多个氨基酸残基中,所述突变选自对应于人类IgG1重链的Fc区中E430X、E345X、S440Y和S440W的组,并且其中
第一CH2-CH3区在下述位置处包含其他氨基酸突变,所述位置选自对应于人类IgG1重链Fc区中K409、T366、L368、K370、D399、F405和Y407的组;并且其中第二CH2-CH3区在下述位置包含其他氨基酸突变,所述位置选自对应于人类IgG1重链Fc区中F405、T366、L368、K370、D399、Y407和K409 的那些位置,并且其中在第一CH2-CH3区中的其他氨基酸突变不同于在第二 CH2-CH3区中的其他氨基酸突变。
在一个实施方案中,一个或多个氨基酸残基中的突变选自对应于人类 IgG1重链Fc区中E430G、E430S、E430F、E430T、E345K、E345Q、E345R、 E345Y和S440W的组。
在优选的实施方案中,一个或多个氨基酸残基中的突变选自对应于人类 IgG1重链Fc区中E430G、E430S、E345K和E345Q的组。
在一个实施方案中,该方法包括将突变引入到双特异性抗体的第一或第二多肽中的仅一个中。
在一个实施方案中,该方法包括将突变引入到双特异性抗体的第一和第二多肽两者中。
在一个优选的实施方案中,第一CH2-CH3区的其他氨基酸突变在对应于人类IgG1重链的Fc区中的K409,如K409R的位置;且其中第二CH2-CH3区的其他氨基酸突变在对应于人类IgG1重链的Fc区中的F405,如F405L的位置。
本发明发明人还已经发现在对应于人类IgG1重链Fc区中的K439或S440 的氨基酸残基上引入突变降低亲本抗体的效应子功能(实施例5、6、10)。
如实施例6中所示,作为“单突变体”的位置K439E或S440K的氨基酸取代,相比根据本发明方法的第一突变中任一个,降低了CDC。
从所述降低效应子功能的方法获得的变体抗体相比亲本抗体具有降低的效应子功能。通常,抗体对效应子功能的效果可以通过EC50值测量,EC50 值是获得最大裂解半数值必需的抗体浓度。
最大裂解是使用饱和量抗体时所获得的裂解,其中饱和意指抗体的所有抗原都被抗体结合的抗体量。
术语“降低效应子功能”本发明上下文中指相比亲本抗体,变体抗体的 EC50值有提高。EC50值的提高可以是例如至少或约2倍,如至少或约3倍,至少或约5倍,至少或约10倍。可选的,“降低效应子功能”意为在亲本抗体裂解少于所有细胞100%的条件下,裂解的细胞最大量降低例如所有细胞的 10%到100%,如约10%,约20%,约30%,约40%,约50%,约60%,约70%,约80%,约90%,和约100%。
可以通过将IgG1-005或IgG1-7D8重链可变域克隆入变体并在CDC检测中测试其效力,来检测变体的降低的效应子功能,如对Daudi细胞(实施例6) 和Wien 133细胞(实施例10)所描述的。使用IgG1-7D8HC可变域和Daudi细胞,降低会被定义为与在研究条件下与IgG1-7D8的EC50相比低超过2倍的EC50,如约2倍,约3倍,约5倍,约10倍或超过10倍的EC50值,EC50为观察到最大裂解半数的浓度。使用IgG1-005HC可变域和Daudi细胞,降低会被定义为与在研究条件下与IgG-005的EC50相比低超过2倍的EC50,如约2倍,约3倍,约5倍,约10倍或超过10倍的EC50值,EC50为观察到最大裂解半数的浓度。使用IgG1-7D8HC可变域和Wien133细胞,降低会被定义为与在研究条件下与IgG1-7D8的EC50相比低超过2倍的EC50,如约2倍,约3倍,约5倍,约10 倍或超过10倍的EC50值,EC50为观察到最大裂解半数的浓度。使用IgG1-005 HC可变域和Wien133细胞,降低会被定义为最大裂解的降低范围为所有细胞的10%到100%,如降低约10%,约20%,约30%,约40%,约50%,约60%,约70%,约80%,约90%,和约100%。CDC效力的降低也可以被定义为与在研究条件下IgG-005的EC50相比低超过2倍的EC50,如约2倍,约3倍,约5倍,约10倍或超过10倍的EC50值,EC50为在Wien133细胞的裂解可以检测到的条件下,观察到最大裂解半数的浓度。
在另一个方面,本发明涉及根据本发明和本文实施方案公开的方法,所述方法包括引入除S440Y和S440W之外的多个位置中的一个的突变,并且还引入以下中的突变:
(i)在对应于人类IgG1重链Fc区中的K439和S440的氨基酸中的每一个中,条件是S440上的突变不是S440Y或S440W,
(ii)在对应于人类IgG1重链Fc区中的K447和448的氨基酸残基中的每一个中,如在人类IgG1重链Fc区中的K447K/R/H和448E/D,优选地在人类 IgG1重链Fc区中的K447K和448E,或
(iii)在对应于人类IgG1重链Fc区中的K447,448和449的氨基酸残基中的每一个中,如在人类IgG1重链Fc区中的K447D/E、448K/R/H和449P,优选地在人类IgG1重链Fc区中的K447E、448K和449P。
对于其中如以上步骤(ii)或(iii)中所述的引入其他突变的实施方案,应注意的是,在正常情况下在细胞中的抗体产生中切去位置K447中的赖氨酸。这可以通过添加一个或多个其它氨基酸残基(如448或448/449)保护位置K447来阻止。这进一步描述于WO2013/0048411(Genmab A/S)中。
在一个实施方案中,该方法包括将突变引入到除S440Y和S440W之外的多个位置中的一个,并在对应于人类IgG1重链Fc区中的K439和/或S440的氨基酸残基中的每一个中引入突变,条件是S440中的突变不是S440Y或 S440W。
在优选的实施方案中,在对应于人类IgG1重链Fc区中的K439的位置中的突变是K439D/E,和/或在对应于人类IgG1重链Fc区中的S440的位置中的突变是S440K/R。
在一个实施方案中,亲本多肽是包含免疫球蛋白Fc域和抗原结合区的亲本抗体。
在一个实施方案中,亲本抗体是单特异性,双特异性,或多特异性抗体。双特异性抗体可以是本文所描述的实施方案中的任意一个。
此外,表1中所列的任何突变可以被引入双特异性抗体。实施例24显示了将E345R突变导入双特异性CD20xEGFR抗体增强了CDC效力。实施例23、 29和30也描述了包含根据本发明突变的一些不同的双特异性抗体。
在亲本抗体中对应于人类IgG1重链Fc区中的K439和S440的两个氨基酸残基中引入突变(条件是S440上的突变不是S440Y或S440W)在本发明中也被称为“双突变体”方面。如别处所述,S440Y或S440W突变已经被发现当引入亲本多肽时能提高CDC。
如其他地方也描述的,本发明发明人已经发现了在对应于人类IgG1重链 Fc区中的K439或S440的氨基酸残基上引入所鉴定的突变,导致效应子功能的降低(实施例5、6、10)。然而,当在对应于人类IgG1重链Fc区中的K439和S440 的两个氨基酸残基上引入抑制突变时,效应子功能的降低被恢复,由此使其与在K439和S440突变处没有突变的亲本抗体的效应子功能相似。然而,不限于任何理论,认为K439和S440突变的存在将效应子功能限于寡聚复合物,所述寡聚复合物仅对应于包含K439和S440突变两者的抗体。因此,如果治疗抗体中包含K439和S440突变,那么不限于任何理论,认为当这样的治疗抗体被施用给患者时,效应子功能诱导被限制于寡聚抗体复合物,所述寡聚抗体复合物包含含有K439/S440突变的治疗抗体但不含患者自身抗体,患者自身抗体不含K439和S440突变,由此限制了由治疗抗体与患者自身抗体相互作用引起的任何潜在副作用。
当将位置K439和/或S440的突变与第一突变组合时,获得了CDC的增强并提高了CDC的特异性。以类似的方式,可以通过引入上述实施方案(ⅱ) 和(ⅲ)中公开的突变获得CDC的增强和提高的特异性。
在另一个方面,本发明涉及提高至少第一和第二亲本多肽组合的补体依赖性细胞毒性(CDC)的方法,其中所述至少第一和第二亲本多肽中各包含免疫球蛋白的Fc域和结合区,其中所述方法包括:将一个或多个氨基酸残基中的突变引入到至少第一和/或第二亲本多肽,所述突变选自对应于人类 IgG1重链的Fc区中E430X、E345X、S440Y和S440W的组。
在一个实施方案中,该方法包括将一个或多个氨基酸残基中的突变引入到至少第一和/或第二亲本多肽,所述突变选自对应于人类IgG1重链的Fc区中E430G、E430S、E430F、E430T、E345K、E345Q、E345R、E345Y、S440Y 和S440W的组。
在一个优选的实施方案中,该方法包括将一个或多个氨基酸残基中的突变引入到至少第一和/或第二亲本多肽,所述突变选自对应于人类IgG1重链的Fc区中E430G、E430S、E345K和E345Q的组。
在一个实施方案中,该方法包括将可以相同或不同的突变引入到第一和第二亲本多肽两者中。
在其他的实施方案中,该方法包括:
(i)将突变引入到第一亲本多肽的一个或多个氨基酸残基中,所述突变选自对应于人类IgG1重链的Fc区中E430G、E430S、E430F、E430T、E345K、 E345Q、E345R、E345Y、S440Y、和S440W的组,
(ii)提供第二亲本多肽,其不包含在一个或多个氨基酸残基中的突变,所述突变选自对应于人类IgG1重链的Fc区中E430G、E430S、E430F、E430T、 E345K、E345Q、E345R、E345Y、S440Y和S440W的组。
在一个实施方案中,该方法包括将一个或多个氨基酸残基中的突变引入到第一亲本多肽,所述突变选自对应于人类IgG1重链的Fc区中E430G、 E430S、E345K或E345Q的组。
在其他的实施方案中,在一个或多个位置处的突变不是S440Y和 S440W,并且其中所述方法进一步包含如下步骤
(i)向第一亲本多肽引入对应于人类IgG1重链Fc区中的K439位置的氨基酸残基中的第二突变;和
(ii)向第二亲本多肽引入对应于人类IgG1重链Fc区中的S440位置的氨基酸残基中的第二突变,条件是突变不是S440Y或S440W;其中步骤(i)和(ii) 备选地可为
(iii)向第一亲本多肽引入对应于人类IgG1重链Fc区中的S440位置的氨基酸残基中的第二突变,条件是该突变不是S440Y或S440W;
(iv)向第二亲本多肽引入对应于人类IgG1重链Fc区中的K439位置的氨基酸残基中的第二突变。
第二亲本多肽可以是在结合到靶细胞后本身不提供足够的CDC应答的任意亲本多肽。
因此,不受理论束缚,认为所述方法提供第一变体多肽,其包含根据以上列出的一个或多个氨基酸残基中的突变,因此该变体多肽具有提高的CDC 应答,及提供第二变体多肽,所述第二变体多肽不包含此种突变,获得第二亲本多肽的CDC应答。
组合包含能提高CDC的一个所述突变的第一抗体与没有根据本发明修饰的第二抗体的方法可以如实施例31所示导致组合的提高的CDC。因此,在一个实施方案中,可以将该方法用于组合已被证明安全但不足够有效(或希望有提高的效力)的作为第二抗体的治疗抗体与包含突变的第一抗体,并由此得到有效的组合。
不包含氨基酸残基中的突变的适合的第二抗体的例子包含但不限于下列任一项,所述突变选自对应于人类IgG1重链的Fc区中E430G,E430S,E430F, E430T,E345K,E345Q,E345R,E345Y,S440Y和S440W的那些突变:(90Y) clivatuzumab tetraxetan;(90Y)tacatuzumab tetraxetan;(99mTc)fanolesomab; (99mTc)nofetumomab Merpentan;(99mTc)pintumomab;3F8;8H9; abagovomab;阿巴西普(abatacept);阿昔单抗(abciximab);Actoxumab;阿达木单抗(adalimumab);adecatumumab;阿非莫单抗(afelimomab);aflibercept; Afutuzumab;alacizumab pegol;albiglutide;ALD518;阿法赛特(alefacept);阿仑单抗(alemtuzumab);Alirocumab;阿妥莫单抗(altumomab);Altumomab pentetate;阿韦舒托(alvircept sudotox);amatuximab;AMG714/HuMax-IL15;麻安莫单抗(anatumomab mafenatox);Anrukinzumab(=IMA-638); apolizumab;阿西莫单抗(arcitumomab);aselizumab;atacicept;atinumab; Atlizumab(=托珠单抗(tocilizumab));阿西莫单抗(atorolimumab); baminercept;Bapineuzumab;巴利昔单抗(basiliximab);bavituximab;贝妥莫单抗(bectumomab);belatacept;belimumab;benralizumab;bertilimumab; besilesomab;贝伐单抗(bevacizumab);Bezlotoxumab;比西单抗(biciromab); bifarcept;比伐单抗(bivatuzumab);Bivatuzumab mertansine;blinatumomab; blosozumab;brentuximab vedotin;briakinumab;briobacept;brodalumab; canakinumab;cantuzumab mertansine;cantuzumab ravtansine;caplacizumab;卡罗单抗(capromab);卡罗单抗喷地肽(Capromab pendetide);carlumab;catumaxomab;CC49;西利珠单抗(cedelizumab);certolizumab pegol;西妥昔单抗(cetuximab);Ch.14.18;citatuzumab bogatox;cixutumumab; Clazakizumab;克立昔单抗(clenoliximab);Clivatuzumab tetraxetan; conatumumab;conbercept;CR6261;crenezumab;dacetuzumab;达利珠单抗(daclizumab);dalantercept;dalotuzumab;daratumumab;Demcizumab;地诺单抗(denosumab);地莫单抗(Detumomab);阿托度单抗(Dorlimomab aritox);drozitumab;dulaglutide;ecromeximab;依库珠单抗(eculizumab);埃巴单抗(edobacomab);依决洛单抗(edrecolomab);依法利珠单抗(efalizumab);efungumab;elotuzumab;elsilimomab;enavatuzumab;恩莫单抗(enlimomab);培戈赖莫单抗(enlimomab pegol);enokizumab;ensituximab;依匹莫单抗(epitumomab);epitumomab cituxetan;依帕珠单抗(epratuzumab); erlizumab;ertumaxomab;依那西普;etaracizumab;etrolizumab;exbivirumab; Fanolesomab;法拉莫单抗(faralimomab);farletuzumab;Fasinumab;FBTA05;非维珠单抗(felvizumab);Fezakinumab;ficlatuzumab;figitumumab; flanvolumab;fontolizumab;foralumab;foravirumab;fresolimumab;fulranumab; galiximab;ganitumab;gantenerumab;gavilimomab;吉妥珠单抗(gemtuzumab);吉妥珠单抗奥唑米(Gemtuzumab ozogamicin);gevokizumab;girentuximab; glembatumumab;Glembatumumab vedotin;戈利木单抗(golimumab); Gomiliximab;GS6624;抗-CD74抗体;如WO2011/110642公开的抗cMet抗体;如WO2011/147986或WO2011/147982公开的抗Her2抗体;如WO2004/058797 公开的抗IL-8抗体;如WO2004/045512公开的抗TAC抗体;如WO2010/066803 或WO 2011/157741公开的抗组织因子(TF)抗体;ibalizumab;替伊莫单抗 (ibritumomab tiuxetan);icrucumab;伊戈伏单抗(igovomab);英西单抗 (Imciromab);inclacumab;indatuximab ravtansine;英夫利昔单抗 (infliximab);伊诺莫单抗(inolimomab);inotuzumab ozogamcin;intetumumab;碘(I241)girentuximab;匹伊单抗(ipilimumab);iratumumab;itolizumab; ixekizumab;凯利昔单抗(keliximab);labetuzumab;lebrikizumab; lemalesomab;来那西普(lenercept);乐地单抗(lerdelimumab);lexatumumab; libivirumab;林妥珠单抗(lintuzumab);lorvotuzumab mertansine;lucatumumab; lumiliximab;mapatumumab;maslimoma;马妥珠单抗(matuzumab); mavrilimumab;美泊利单抗(mepolizumab);metelimumab;milatuzumab;明瑞莫单抗(minretumomab);mirococept;米妥莫单抗(mitumomab); mogamulizumab;莫罗木单抗(morolimumab);motavizumab;moxetumomab;pasudotox;莫罗单抗(muromonab)-CD3;他那可单抗(nacolomab tafenatox); namilumab;naptumomab estafenatox;narnatumab;那他珠单抗(natalizumab);奈巴库单抗(nebacumab);necitumumab;奈瑞莫单抗(nerelimomab);尼妥珠单抗(nimotuzumab);Nivolumab;若莫单抗(Nofetumomab);merpentan; obinutuzumab;Ocaratuzumab;ocrelizumab;奥度莫单抗(odulimomab);奥法木单抗;olaratumab;olokizumab;奥马珠单抗(omalizumab);onartuzumab;奥那西普(onercept);oportuzumab monatox;oregovomab;otelixizumab; oxelumab;ozoralizumab;pagibaximab;帕利珠单抗(palivizumab);帕尼单抗 (panitumumab);panobacumab;pascolizumab;pateclizumab;patritumab;pegsunercept;Pemtumomab;帕妥珠单抗(pertuzumab);pexelizumab; Pintumomab;Placulumab;ponezumab;普立昔单抗(priliximab);pritumumab; PRO 140;quilizumab;racotumomab;radretumab;rafivirumab;ramucirumab;兰尼单抗(ranibizumab);raxibacumab;瑞加韦单抗(regavirumab);reslizumab; RG1507/HuMax-IGFlR;RG1512/HuMax-pSelectin;rilonacept;rilotumumab;利妥昔单抗(rituximab);robatumumab;roledumab;romosozumab; rontalizumab;罗维珠单抗(rovelizumab);鲁利单抗(ruplizumab); samalizumab;sarilumab;沙妥莫单抗(satumomab);沙妥莫单抗喷地肽(Satumomab pendetide);secukinumab;司韦单抗(sevirumab);西罗珠单抗(sibrotuzumab);sifalimumab;siltuximab;siplizumab;sirukumab; solanezumab;solitomab;Sonepcizumab;sontuzumab;sotatercept;stamulumab;硫索单抗(sulesomab);suvizumab;tabalumab;Tacatuzumab tetraxetan; tadocizumab;talizumab;tanezumab;taplitumomab paptox;tefibazumab; telimomab aritox;tenatumomab;teneliximab;teplizumab;teprotumumab; TGN1412;Ticilimumab(tremelimumab);tigatuzumab;TNX-650;托珠单抗 (atlizumab);toralizumab;torapsel;托西莫单抗(tositumomab);tralokinumab;曲妥珠单抗;trastuzumab emtansine;TRBS07;trebananib;tregalizumab;tremelimumab;tucotuzumab celmoleukin;妥韦单抗(tuvirumab);ublituximab;urelumab;urtoxazumab;ustekinumab;vapaliximab;vatelizumab;vedolizumab;veltuzumab;维帕莫单抗(vepalimomab);vesencumab;visilizumab; volociximab;Vorsetuzumab mafodotin;伏妥莫单抗(votumumab);扎妥木单抗(zalutumumab);zanolimumab;ziralimumab;和阿佐莫单抗(zolimomab aritox)。
如实施例10中所示,相比任何野生型或天然存在的抗体,第一和第二变体抗体会有相互寡聚的偏好。
在一个实施方案中,在对应于人类IgG1重链的Fc区中K439的位置中的突变是K439D/E,和/或在对应于人类IgG1重链的Fc区中S440的位置中的突变是S440K/R。
因此,特异性的提高是就“CDC诱导”而言的。因此在一个实施方案中所述方法是通过至少第一和第二亲本抗体的组合来提高效应子功能的诱导特异性的方法。
通过进行通过至少第一和第二亲本多肽的组合来提高诱导效应子功能的特异性的方法,获得了第一变体和第二变体多肽的组合。
通过在亲本多肽K439或S440上引入突变,由此获得的变体多肽相比亲本多肽具有降低的效应子功能。然而,如本发明其他地方描述,K439和S440 处的突变能相互补偿以恢复包含两个突变的多肽的效应子功能。K439和S440 处突变相互补偿的能力可以被类似地用于两个多肽中。因此,当K439处的突变被引入第一亲本多肽而S440处的突变被引入第二亲本多肽时,或者反之亦然,由于组合使用第一和第二变体多肽,不再看到效应子功能的降低。术语“提高特异性”或“改进特异性”在本上下文中是指这样的事实:由包含K439 处突变的第一变体多肽和包含S440处突变的第二变体多肽的组合所诱导的效应子应答高于由包含K439处突变的第一变体多肽或包含S440处突变的第二变体多肽所诱导的效应子应答。
通过K439和S440处两个氨基酸取代的引入,使寡聚体的特异性提高。
当将位置K439和/或S440的突变与第一突变组合时,获得了CDC的增强并提高了CDC的特异性。
在一个实施方案中,至少第一和第二亲本多肽结合相同的结合位点,或者就抗体而言结合相同的表位。
在一个实施方案中,至少第一和第二亲本多肽结合相同靶标上的不同结合位点,或者就抗体而言结合相同抗原上的不同表位。
在一个实施方案中,至少第一和第二亲本多肽结合到不同靶标上的不同表位。
在一个实施方案中,第一和第二亲本多肽是具有相同或不同的VL和VH 序列的第一和第二亲本抗体。
在一个实施方案中,至少第一和第二亲本多肽的组合包含一种第一亲本多肽和一种第二亲本多肽。
在一个实施方案中,在第一和第二亲本多肽的组合与抗原表达细胞、细胞膜或病毒粒子上的其结合位点或抗原结合时,特异性提高。
因此,在另一方面,本发明也涉及使用多肽的两个或更多个氨基酸残基处突变以在与抗原表达细胞、细胞膜或病毒粒子上的其抗原结合时提高例如由多肽诱导的CDC的特异性,其中,
第一突变在对应于人类IgG1重链Fc区中的K439的氨基酸残基处;
第二突变在对应于人类IgG1重链Fc区中的S440的氨基酸残基处。
在一个实施方案中,第一和第二亲本多肽是各自包含免疫球蛋白Fc域和抗原结合区的第一和第二亲本抗体。
在一个实施方案中,第一和第二个亲本抗体是单特异性,双特异性或多特异性抗体。
在一个实施方案中,第一和/或第二亲本抗体为双特异性抗体,该双特异性抗体包含含免疫球蛋白的第一CH2-CH3区和第一抗原结合区的第一多肽,以及含第二CH2-CH3区和第二抗原结合区的第二多肽,其中第一和第二抗原结合区结合相同或不同抗原上的不同表位,和其中所述第一CH2-CH3区包含在选自对应于人类IgG1重链Fc区中K409、T366、L368、K370、D399、F405 和Y407的那些位置的位置处的进一步氨基酸突变;和其中第二CH2-CH3区包含在选自对应于人类IgG1重链Fc区中F405、T366、L368、K370、D399、 Y407和K409的那些位置的位置处的进一步氨基酸突变;而其中第一 CH2-CH3区中的进一步的氨基酸突变与第二CH2-CH3区中的所述进一步的氨基酸突变不同。
在优选的实施方案中,第一CH2-CH3区包含在对应于人类IgG1重链Fc 区中K409,如K409R的位置处的进一步氨基酸突变;并且其中第二CH2-CH3 区包含在对应于人类IgG1重链Fc区中F405,如F405L的位置处的进一步氨基酸突变。
通过进行该方法,获得了至少第一和第二变体抗体的组合。通过该方法获得的至少第一和第二变体抗体当组合时具有相比第一和第二亲本抗体的组合提高的CDC。
术语“提高的CDC”应按本发明中所描述的理解。
第一和/或第二亲本抗体可以是本发明所描述的任何亲本抗体。
具体地,可以进行提高第一和第二抗体组合的CDC的方法,以获得具有本发明所描述变体抗体的任何特征的第一和/或第二抗体。
在一个实施方案中,至少第一和第二亲本抗体结合到相同的表位。
在一个实施方案中,至少第一和第二亲本抗体结合到相同抗原上的不同表位。
在一个实施方案中,至少第一和第二亲本抗体结合到不同靶标上的不同表位。
在一个实施方案中,至少第一和第二亲本抗体具有相同或不同的VL和 VH序列。
在一个实施方案中,至少第一和第二亲本抗体的组合包含一种第一亲本抗体和一种第二亲本抗体。
在一个实施方案中,至少第一和第二亲本抗体的组合包含更多的亲本抗体,如第三,第四或第五亲本抗体。在一个实施方案中,第一和第二双特异性或多特异性亲本抗体是相同或不同的抗体。在一个实施方案中,第一和第二双特异性或多特异性抗体结合相同或不同抗原上不同的表位。因此,在一个实施方案中,所述至少第一和第二亲本抗体是结合相同抗原或不同抗原上不同表位的双特异性或多特异性抗体。
在本发明方法和/或用途的一个实施方案中,亲本抗体(无论其是亲本抗体,第一亲本抗体还是第二亲本抗体)除了已经被发现能影响效应子功能的本发明的那些突变之外还包含其他突变。这样的突变可以与影响效应子功能的本发明突变同时被引入,或者其可以被顺序引入,本发明的方法或用途不限于同时或顺序引入突变。双特异性抗体可以是任何双特异性抗体,并且本发明的方法和用途不限于任何特定的双特异性形式,因为预见可以使用不同形式。
在一个实施方案中,该方法不改变亲本多肽或亲本抗体的抗体依赖性细胞介导的细胞毒性(ADCC)。
在一个实施方案中,如通过实施例34中公开的方法测定的,该方法不改变亲本多肽或亲本抗体与新生儿Fc受体(FcRn)的结合。
在一个实施方案中,如通过实施例34中公开的方法测定的,通过在OD 405处吸光度的变化测量的,该方法不会使亲本多肽或亲本抗体与新生儿Fc 受体(FcRn)的结合提高或降低超过30%,例如超过20%,10%或5%。
在一个实施方案中,如通过实施例34中公开的方法测定的,该方法不会使亲本多肽或亲本抗体与小鼠新生儿Fc受体(FcRn)的表观亲和力提高超过 0.5倍(factor),或者不会使亲本多肽或亲本抗体与小鼠新生儿Fc受体(FcRn) 的表观亲和力降低超过2倍。
在一个实施方案中,如通过实施例37中公开的方法测定的,该方法不改变亲本多肽或亲本抗体的血浆清除率。
在一个实施方案中,如通过实施例37中公开的方法测定的,该方法不会使亲本多肽或亲本抗体的血浆清除率增加或降低超过3.0倍,如超过2.5倍, 2.0倍,1.5倍或1.2倍。
在一个实施方案中,如通过实施例36中公开的方法测定的,该方法不会改变变体的不依赖于靶标的流体相补体激活。
在一个实施方案中,该方法不改变亲本多肽或亲本抗体的血浆半衰期。
可以根据本发明的方法引入本发明所描述的任何突变或其组合。
选自示例性或优选氨基酸取代的突变可以以适当的检测方法测试,所述检测方法允许抗原结合的抗体的寡聚物形成并检测增强的C1q结合,补体激活,CDC,ADCC和/或内化,如实施例中所描述的那些。例如C1q结合亲合力可以根据类似于实施例4所描述检测方法的检测方法,使用表达抗体变体的抗原的细胞来确定。示例性示例性的CDC检测方法在实施例5、6、10、16、 19、22、23、24、25或35中提供。示例性示例性ADCC检测方法在实施例12 中提供。示例性示例性内化检测方法在实施例26中提供。最后,为区分直接参与C1q结合的氨基酸残基的突变与影响寡聚物形成的突变,根据例如实施例3的ELISA检测中的C1q结合可以与根据例如实施例4的基于细胞的检测中的C1q结合相比较,可以根据实施例37中描述的测定法比较血浆清除率,根据实施例34比较FcRn结合,根据实施例36中的测定法评估不依赖于靶标的流体相补体激活。
在一个实施方案中,在一个或多个氨基酸残基处的突变可以是氨基酸取代、氨基酸缺失或氨基酸插入。
在一个实施方案中,在一个或多个氨基酸残基处的突变可以是氨基酸缺失。
在一个实施方案中,在一个或多个氨基酸残基处的突变可以是氨基酸插入。
在一个实施方案中,在一个或多个氨基酸残基处的突变可以是氨基酸取代。
在一个实施方案中,在一个或多个氨基酸残基处的突变可以选自表1所列的任何氨基酸取代,氨基酸缺失。
因此,在一个实施方案中,E345X可以是E345R、Q、N、K、Y、A、C、 D、F、G、H、I、L、M、P、S、T、V、W或Y;具体是E345A、D、G、H、 K、N、Q、R、S、T、Y或W,或更具体是E345D、K、N、Q、R或W;或甚至更具体是E345R、Q、N、K或Y。在进一步优选的实施方案中,E345X是 E345K或E345Q。
在另一进一步的实施方式中,E430X可以是E430T、S、G、F、H、A、 C、D、I、K、L、M、N、P、Q、R、V、W或Y;具体是E430T、S、G、F 或H。在进一步优选的实施方案中,E430X是E430G或E430S。在另一个实施方案中,可选地,按通过将根据实施例3的ELISA检测中C1q结合与根据实施例4的基于细胞的检测中的C1q结合相比较来确定,突变不在直接参与C1q结合的氨基酸残基处。
在一个实施方案中,一个或多个突变是一个突变,即不多于一个突变被引入亲本抗体。
在另一个实施方案中,根据本发明的方法或用途包含引入至少两个,如两个,三个,四个,五个或更多个的表1中的氨基酸残基处的突变。
可以根据本发明的方法引入本发明所描述的任何突变组合。
在一个实施方案中,该方法包括对亲本多肽引入氨基酸残基中多于一个突变,如两个,三个,四个,或五个,具体是两个或三个突变,所述突变选自对应于人类IgG1重链Fc区中的E345X、E430X、S440Y和S440W的组。例如,可以突变至少多于一个对应于人类IgG1重链Fc区中的E345X、E430X、 S440Y和S440W的氨基酸残基,如E345X、E430X、S440Y和S440W的两个或全部,可选的与一个或多个表1所列其他氨基酸处的突变组合。至少两个突变可以是位置E345的任何氨基酸残基取代组合位置E430或S440Y或 S440W的任何氨基酸取代,或者可以是位置E430的任何氨基酸残基取代组合位置S440Y或S440W的任何氨基酸取代。在进一步的实施方案中,两个或三个突变被引入到亲本抗体的氨基酸残基,所述突变选自对应于人类IgG1重链 Fc区中的E430G、E430S、E345K和E345Q的组。
氨基酸残基中的两个突变的此种组合,所述突变选自对应于人类IgG1 重链Fc区中E345X/E430X、E345X/S440Y、E345X/S440W、E430X/S440Y和 E430X/S440W的组。
在根据本发明的方法或用途中,当抗体与其抗原结合时,提高了CDC。
不被任何理论限制,相信当抗体与其抗原结合时,提高了CDC,其中抗原在抗原表达细胞、细胞膜或病毒粒子上。在一个实施方案中,IgG1重链的 Fc区包含SEQ ID NO:1的残基130-330的序列。
亲本多肽或亲本抗体可以是本发明描述的任何亲本多肽或亲本抗体。本上下文中的亲本多肽和亲本抗体也意指第一亲本和第二亲本多肽和第一亲本和第二亲本抗体。
在一个实施方案中,亲本抗体是人类IgG1、IgG2、IgG3或IgG4、IgA1、 IgA2、IgD、IgM或IgE抗体。
在一个实施方案中,亲本抗体是人全长抗体,如人全长IgG1抗体。
在一个实施方案中,亲本抗体,第一亲本抗体和第二亲本抗体是人类 IgG1抗体,例如IgG1m(za)或IgG1m(f)同种异型体,任选地包含含SEQ ID NO: 1或5的Fc区。
在一个实施方案中,亲本抗体是人IgG2抗体,任选地包含含SEQ ID NO: 2的Fc区。
在一个实施方案中,亲本抗体是人IgG3抗体,任选地包含含SEQ ID NO: 3的Fc区。
在一个实施方案中,亲本抗体是人IgG4抗体,任选地包含含SEQ ID NO: 4的Fc区。
在一个实施方案中,亲本抗体是双特异性抗体。
在一个实施方案中,亲本抗体是本发明描述的任何抗体,例如包含至少部分Fc区的抗体片段,单价抗体(在WO2007059782中由Genmab描述);重链抗体,其仅由两条重链组成并在例如骆驼中天然存在(例如, Hamers-Casterman(1993)Nature 363:446);ThioMabs(Roche, WO2011069104),链交换改造域(SEED或seed体),其是不对称的和双特异性的抗体样分子(Merck,WO2007110205);Triomab(Fresenius,Lindhofer等 (1995J Immunol 155:219));FcΔAdp(Regeneron,WO2010151792),Azymetric Scaffold(Zymeworks/Merck,WO2012/058768),mAb-FV(Xencor, WO2011/028952),双可变域免疫球蛋白(Abbott,DVD-Ig,美国专利 7,612,181);双域双头抗体(Unilever;Sanofi Aventis,WO20100226923),二双抗体(ImClone/Eli Lilly),钮-入-孔抗体形式(Genentech,WO9850431); DuoBody(Genmab,WO2011/131746);静电驾驭抗体形式(Amgen,EP1870459 和WO 2009089004;Chugai,US201000155133;Oncomed,WO2010129304A2);双特异性IgG1和IgG2(Rinatneurosciences Corporation,WO11143545), CrossMAbs(Roche,WO2011117329),LUZ-Y(Genentech),Biclonic(Merus),双靶向域抗体(GSK/Domantis),识别两个靶标的二合一抗体(Genentech, Novlmmune),交联MAbs(Karmanos Cancer Center),CovX体(CovX/Pfizer), IgG样双特异性(ImClone/Eli Lilly,Shen,X,等J Immunol Methods,2007. 318(1-2):p.65-74),和DIG体和PIG体(Pharmabcine),以及双亲和性再靶向分子(Macrogenics的Fc-DART或Ig-DART,WO/2008/157379,WO/2010/080538), Zybodies(Zyngenia),用普通轻链(Crucell/Merus,US7262028)或普通重链 (Novlmmune的κλ体)的方法,以及融合蛋白,其包含多肽序列,所述多肽序列融合于含Fc域的抗体片段,像scFv融合体,像ZymoGenetics/BMS的 BsAb,Biogen Idec的HERCULES(US007951918),EmergentBioSolutions/Trubion的SCORPIONS,Ts2Ab(Medlmmune/AZ Dimasi,N.,等J Mol Biol,2009.393(3):p.672-92),Novartis的scFv融合,Changzhou Adam Biotech Inc的scFv融合体(CN 102250246),Roche的TvAb(WO 2012025525, WO2012025530),f-Star的mAb2(WO2008/003116)和双的scFv-融合体。还应当理解,术语抗体,除另有指明外,也包含多克隆抗体,单克隆抗体(如人单克隆抗体),抗体混合物(重组多克隆)例如,通过Symphogen和Merus (Oligoclonics)开发的技术生成的,抗体样多肽,例如嵌合抗体和人源化抗体。生成的抗体可以潜在的具有任何同种型。
在另一个实施方案中,抗原在细胞表面表达。
在另一个实施方案中,细胞是人肿瘤细胞。
在进一步的实施方案中,抗原选自下组:erbB1(EGFR),erbB2(HER2), erbB3,erbB4,MUC-1,CD4,CD19,CD20,CD38,CD138,CXCR5,c-Met, HERV-包膜蛋白,骨膜蛋白,Bigh3,SPARC,BCR,CD79,CD37,EGFrvIII, IGFr,L1-CAM,AXL,组织因子(TF),CD74,EpCAM和MRP3。
在另一个实施方案中,抗原与细胞膜相关联。
在另一个实施方案中,抗原与病毒粒子相关联,任选地其中抗原包含在病毒粒子的蛋白衣壳或脂质包膜中。
在另一个实施方案中,抗体是人抗体,任选地结合至少一个选自CD20 和CD38的抗原。
在另一个实施方案中,抗体与7D8和005中至少一个结合相同的表位,任选地包含7D8和005中至少一个的可变重和/或可变轻链区域。
在根据所公开发明的任何用途中,没有任何本发明突变的抗体可以是任何亲本抗体。因此,本发明的用途提供了这样亲本抗体的任何变体。
在一个实施方案中,效应子功能是Fc受体结合,例如包含Fc-gamma受体结合。
在一个实施方案中,效应子功能是含有Fc的多肽内在化。
在一个实施方案中,效应子功能是补体依赖性细胞毒性(CDC)和抗体依赖性细胞介导的细胞毒性(ADCC)的组合。
本发明中所用的术语“C1q结合”,当用于亲本抗体的变体或抗体的上下文中时,包含由变体或抗体与包含免疫系统各种细胞(如效应细胞)在内的宿主组织或因子结合所介导的补体激活经典途径上的第一组分的任何机制。抗体的C1q结合可以使用ELISA评价(如,例如实施例3和4中所用的C1q结合 ELISA),或者C1q效力可以通过CDC检测评价(如,例如实施例5中所用的CDC 检测)。在进一步的实施方案中,抗体的C1q结合亲合力根据实施例4中描述的检测方法来确定。
在根据所公开发明的所有方法中,没有任何本发明突变的抗体可以是任何亲本抗体。因此,本发明的方法提供了这样亲本抗体的任何变体。
通过本发明方法和/或用途获得的亲本抗体,第一亲本抗体,第二亲本抗体,或其变体可以结合本发明所描述的任何靶标。
本发明可针对的抗原或靶标的实例为:5T4;ADAM-10;ADAM-12; ADAM17;AFP;AXL;ANGPT2炭疽抗原;BSG;CAIX;CAXII;CA72-4;癌相关抗原CTAA16.88;CCL11;CCL2;CCR4;CCR5;CCR6;CD2;CD3E; CD4;CD5;CD6;CD15;CD18;CD19;CD20;CD22;CD24;CD25; CD29;CD30;CD32B;CD33;CD37;CD38;CD40;CD40LG;CD44; CD47;CD52;CD56;CD66E;CD72;CD74;CD79a;CD79b;CD80;CD86; CD98;CD137;CD147;CD138;CD168;CD200;CD248;CD254;CD257;CDH3;CEA;CEACAM5;CEACAM6;CEACAM8;Claudin4;CS-1;CSF2RA; CSPG-4;CTLA4;Cripto;DLL4;ED-B;EFNA2;EGFR;内皮缩血管肽 (Endothelin)B受体;ENPP3;EPCAM;ERBB2;ERBB3;FAPα;FcγRI; FCER2;FGFR3;纤维蛋白Ⅱβ链;FLT1;FOLH 1;FOLR1;FRP-1;GD3 神经节苷脂;GDF2;GLP1R;磷脂酰肌醇聚糖-3(Glypican-3);GPNMB; HBV(乙型肝炎病毒);HCMV(人巨细胞病毒);热休克蛋白90同源物[白色念珠菌];单纯疱疹病毒gD糖蛋白;HGF;HIV-1;HIV-1IIIB gp120 V3环; HLA-DRB(HLA-DRβ);人类呼吸道合胞病毒;糖蛋白F;ICAM-1;IFNA1;IFNA1;IFNB1双特异性;IgE Fc;IGF1R;IGHE连接区;IL12B;IL13;IL15; IL17A;IL1A;IL1B;IL2RA;IL4;IL5;IL5RA;IL6;IL6R;IL9;白细胞介素2受体β亚单位;ITGA2;ITGA2B ITGB3;ITGA4ITGB7;ITGA5; ITGAL;ITGAV_ITGB3;ITGB2;KDR;L1CAM;Lewis-y;脂质A;脂多糖LPS域;LTA;MET;MMP14;MMpl5;MST1R;MSTN;MUC1;MUC4; MUC16;MUC5AC;NCA-90粒细胞抗原;连接素(Nectin)4;NGF;NRP; NY-ESO-1;OX40L;PLAC-1;PLGF;PDGFRA;PD1;PDL1;PSCA;磷脂酰丝氨酸;PTK-7;铜绿假单胞菌血清型IATS O11;RSV(人类呼吸道合胞病毒,糖蛋白F);ROR1;RTN4;SELL;SELP;STEAP1;志贺样毒素II B 亚单位[大肠杆菌];SLAM7;SLC44A4;SOST;表皮葡萄球菌的脂磷壁酸; T细胞受体α_β;TF;TGFB1;TGFB2;TMEFF2;TNC;TNF;TNFRSF 10A;TNFRSF 10B;TNFRSF 12A;TNFSF 13;TNFSF 14;TNFSF2;TNFSF7; TRAILR2;TROP2;TYRP1;VAP-1和波形蛋白(Vimentin)。
在主要方面,本发明涉及诱导对细胞、细胞膜或病毒粒子的CDC的方法,所述细胞、细胞膜或病毒粒子表达与包含免疫球蛋白Fc域和结合区的亲本多肽结合的靶标,所述方法包括
(i)提供已经按照本文公开的实施方案中的任一项被突变的亲本多肽或者至少第一亲本多肽与第二亲本多肽的组合;和
(ii)将步骤(i)的突变亲本多肽或步骤(i)的至少第一亲本多肽与第二亲本多肽的突变组合的制剂与表达抗原的细胞、细胞膜或病毒粒子,在人补体或效应细胞的存在下接触。
在一个实施方案中,任何或所有的亲本多肽,第一亲本多肽与第二亲本多肽可以是抗体。
在另一个实施方案中,所述方法增加选自以下的进一步效应应答: ADCC,Fc-γ受体结合,蛋白A结合,蛋白G结合,ADCP,补体依赖性细胞毒性(CDCC),补体增强的细胞毒性,通过抗体介导的与受调理抗体的补体受体的结合,及其任意组合。
在进一步的实施方案中,该方法还诱导抗体依赖性细胞介导的细胞毒性 (ADCC)。
在进一步的实施方案中,该方法还诱导含Fc的多肽内化。
在一个实施方案中,细胞是人肿瘤细胞或细菌细胞。
在另一个实施方案中,IgG1亲本抗体是人类IgG1抗体。
在另一个实施方案中,第一和第二抗原分别选自erbB1(EGFR),erbB2 (HER2),erbB3,erbB4,MUC-1,CD4,CD19,CD20,CD25,CD32,CD37,CD38, CD74,CD138,CXCR5,c-Met,HERV-包膜蛋白,骨膜蛋白,Bigh3,SPARC, BCR,CD79,EGFrvIII,IGFr,L1-CAM,AXL,组织因子(TF),EpCAM和 MRP3。
在另一个实施方案中,第一和第二亲本抗体是完全人的,任选地其中所述第一和第二亲本抗体分别结合选自CD20和CD38的抗原。
在进一步的实施方案中,第一和第二亲本抗体分别选自7D8和005。
在甚至进一步的实施方案中,细胞是细菌细胞。
在另一个实施方案中,细菌细胞选自金黄色葡萄球菌(S.aureus),表皮葡萄球菌(S.Epidermidis),肺炎葡萄球菌(S.pneumonia),炭疽芽孢杆菌 (Bacillus anthracis),铜绿假单胞菌(Pseudomonas aeruginosa),衣原体属 (Chlamydia),大肠杆菌(E.coli),沙门氏菌属(Salmonella),志贺氏菌属 (Shigella),耶尔森氏菌属(Yersinia),鼠伤寒沙门氏菌(S.typhimurium),脑膜炎奈瑟氏菌(Neisseria meningitides)和结核分枝杆菌(Mycobacterium tuberculosis)。
在另一个实施方案中,第一和/或第二抗原是脂磷壁酸(LTA),任选地其中所述第一和第二亲本抗体中的至少一种的是帕昔单抗(pagibaximab)。
在另一个实施方案中,抗原在病毒粒子上表达。
在另一个实施方案中,第一和第二抗体结合相同抗原。
在另一个实施方案中,第一和第二抗体包含相同的VH序列,VL序列,或VH和VL序列两者。
对于本发明的目的来说,表达抗原或否则与抗原结合的靶细胞可以是任意原核或真核细胞。示例性抗原表达细胞包含,但不限于,哺乳动物细胞,尤其是人细胞,例如人癌细胞;和单细胞机体例如细菌,原生动物;和单细胞真菌例如酵母细胞。包含抗原或否则与抗原结合的细胞膜包含来源于抗原表达细胞的部分和/或破坏的细胞膜。与病毒粒子或病毒颗粒有关的抗原可以包含在病毒粒子的蛋白外壳和/或脂质包膜中,或在其它情况中与病毒粒子的蛋白外壳和/或脂质包膜结合。
靶细胞可以是例如人肿瘤细胞。合适的肿瘤抗原包含本发明描述的任意靶标或抗原,但不限于,erbB1(EGFR),erbB2(HER2),erbB3,erbB4,MUC-1, CD4,CD19,CD20,CD25,CD32,CD37,CD38,CD74,CD138,CXCR5,c-Met, HERV-包膜蛋白,骨膜蛋白(periostin),Bigh3,SPARC,BCR,CD79,EGFrvIII, IGFR,L1-CAM,AXL,组织因子(TF),EpCAM和MRP3。优选的抗原包含 CD20,CD38,HER2,EGFR,IGFR,CD25,CD74和CD32。示例性抗体包含WO 2004/035607公开的抗CD20抗体7D8,WO 06/099875公开的抗CD38抗体005, WO 2004/035607公开的抗CD20抗体11B8,WO 06/099875公开的抗CD38抗体 003,WO 02/100348公开的抗EGFr抗体2F8。本发明提供其他具体抗体的实例。
可选的,靶细胞可以是细菌细胞,例如,例如,金黄色葡萄球菌、表皮葡萄球菌(S.epidermidis),肺炎葡萄球菌(S.pneumonia),炭疽芽孢杆菌 (Bacillus anthracis),铜绿假单胞菌(Pseudomonas aeruginosa),衣原体属 (Chlamydia),大肠杆菌(E.coli),沙门氏菌属(Salmonella),志贺氏菌属 (Shigella),耶尔森氏菌属(Yersinia),鼠伤寒沙门氏菌(S.typhimurium),脑膜炎奈瑟菌(Neisseria meningitides)和结核分枝杆菌(Mycobacterium tuberculosis)。示例性抗原包含脂磷壁酸(LTA),和示例性抗体包含帕昔单抗。
可选的,靶标可以存在于病毒、真菌细胞或其他颗粒的表面,如例如,西尼罗河病毒,登革热病毒,丙肝病毒(HCV),人类免疫缺陷病毒(HIV),人乳头瘤病毒,埃巴病毒(Epstein-Barr virus),疱疹病毒,痘病毒,禽流感病毒, RVS,曲霉属(Aspergillus),白色念珠菌(Candida albicans),隐球菌属 (Cryptococcus),和组织胞浆菌属(Histoplasma)。
在一个实施方案中,接触步骤(ii)在体外发生。
在一个实施方案中,接触步骤(ii)在体内发生。
在另一个实施方案中,步骤(ii)包含给受试者施用变体。
在进一步的实施方案中,受试者患有癌症,细菌性感染,或病毒性感染。上述实施方案的接触步骤(ii)可以在体外或体内发生。在后一情况下,步骤(ii) 可以进一步包含给受试者(任选的患有癌症或细菌性感染的受试者)施用一种制剂或多种制剂。关于治疗应用的更多细节将在下文提供。
第一和第二抗体包含可以结合相同或不同表位的抗原结合区。这类表位可以在相同或不同的靶标上。
在一个实施方案中,第一和第二抗体结合不同靶标上的不同表位。这类靶标可以在相同细胞或细胞类型上表达,或可以在不同细胞或细胞类型上表达。在这类实施方案中,效应子功能的增强只针对表达两种靶标的细胞或细胞类型,从而降低对不是待治疗疾病病因的细胞或细胞类型的任何附带损害的风险。
不受任何理论约束,认为CDC的增强可限于同时表达两种特定靶标/抗原的靶细胞,条件是第一和第二抗体结合在相同细胞上发现的表位,从而利用靶标的组合表达来改进增强的CDC诱导的选择性。
在靶标在不同的细胞或细胞类型上表达的情况下,不受理论约束认为,按照第一和第二抗体的任意顺序的施用将通过“募集”表达第二靶标的第二细胞或细胞类型改进CDC增强和可能还有其他效应子功能。
在其中使用第一和第二抗体的组合的实施方案中,可以在人补体和/或效应细胞存在的条件下将细胞与突变的第一和第二亲本抗体同时、分开或顺序接触来进行步骤(ii)。
本发明还提供一种诱导针对与IgG1或IgG3抗体结合的抗原有关的靶细胞、细胞膜、病毒粒子或其他颗粒的CDC或其他效应应答,如ADCC的方法,包括步骤:(i)提供抗体变体,其包含在抗体Fc区的K439的突变(K439E)和S440 的突变(S440K或S440R);和(ii)在人补体和/或效应细胞存在的条件下将变体制剂与细胞接触。
本发明还提供一种诱导针对靶细胞、细胞膜或病毒粒子的CDC或其他效应应答,如ADCC的方法,所述靶细胞、细胞膜或病毒粒子表达与第一IgG1 抗体结合的第一抗原和与第二抗体结合的第二抗原,所述方法包含步骤:(i) 提供第一变体(其是包含K439E突变的第一抗体)和第二变体(其是包含S440K 或S440R突变的第二抗体);和(ii)在人补体或效应细胞存在的条件下将细胞与第一和第二变体制剂同时、分开或顺序地接触。
在单独和具体的实施方案中,第一和第二抗体结合(i)不同抗原,(ii)相同抗原上的不同表位,(iii)抗原上相同表位,和(iv)抗原上的相同表位并包含相同的VH和/或VL序列。
其他方法
在另一个主要方面,本发明涉及一种鉴定增强抗体结合C1q的效应子功能的抗体突变的方法,包括步骤:
(i)制备至少一种抗体,其包含在一个或多个氨基酸中的突变,该突变选自对应于人类IgG1重链的Fc区中的E430X、E345X、S440Y和S440W的组;
(ii)当结合抗原表达细胞的表面时,与亲本抗体比较,评估所述抗体的 C1q活性;和
(iii)挑选具有C1q亲合力增加的任意变体的突变。
在一个实施方案中,至少一种抗体包含一个或多个氨基酸取代,其选自对应于人类IgG1重链的Fc区中的E430G、E430S、E430F、E430T、E345K、 E345Q、E345R、E345Y、S440Y和S440W,如E430G、E430S、E345K和E345Q的组。
仍在另一个主要方面,本发明涉及一种鉴定增加抗体诱导CDC应答能力的亲本抗体突变的方法,包括步骤:
(i)制备至少一种亲本抗体变体,其包含在一个或多个氨基酸中的突变,该突变选自对应于人类IgG1重链的Fc区中的E430X、E345X、S440Y或 S440W的组;
(ii)在效应细胞或补体存在的条件下,当结合抗原表达细胞的表面时,与亲本抗体比较,评估所述变体诱导的CDC应答;和
(iii)挑选具有C1q应答增加的任意变体的突变。
在一个实施方案中,至少一种抗体包含一个或多个氨基酸取代,其选自对应于人类IgG1重链的Fc区中的E430G、E430S、E430F、E430T、E345K、 E345Q、E345R、E345Y、S440Y和S440W,诸如人类IgG1重链的Fc区中的 E430G,E430S,E345K,和E345Q的组。
本发明的多肽
亲本多肽
如本发明所述,本发明尤其涉及在免疫球蛋白的CH3区(例如在抗体中重链)中包含一或更多个突变的亲本多肽变体。“亲本多肽”可以是“亲本抗体”。在修饰前将用作本发明起始原料的“亲本”抗体(其可以是野生型抗体)可以例如通过Kohler等,Nature 256,495(1975)首次描述的杂交瘤方法产生,或通过重组DNA方法产生。使用描述于例如Clackson等,Nature 352,624 628(1991) 和Marks等,J.Mol.Biol.222,581 597(1991)的技术,也可以从噬菌体抗体文库分离单克隆抗体。单克隆抗体可以获自任意合适的来源。因此,例如,单克隆抗体可以获自从鼠脾B细胞制备的杂交瘤,所述鼠脾B细胞获自用感兴趣的抗原(例如以表面上表达抗原的细胞,或编码感兴趣抗原的核酸的形式) 免疫的小鼠。单克隆抗体也可以获自来源于免疫的人或非人哺乳动物(例如兔,大鼠,犬,灵长类动物,等等)的抗体表达细胞的杂交瘤。
亲本抗体可以是例如嵌合或人源化抗体。在另一个实施方案中,抗体是人抗体。可以利用转基因或转染色体小鼠(例如HuMAb小鼠,携带部分人免疫系统而不是小鼠系统)来产生人单克隆抗体。HuMAb小鼠包含编码未重排的人重链(μ和γ)和κ轻链免疫球蛋白序列的人免疫球蛋白基因迷你基因座(minilocus),以及使内源μ和κ链基因座失活的靶向突变(Lonberg,N.等, Nature 368,856859(1994))。因此,小鼠显示降低的小鼠IgM或κ表达,并且响应免疫,导入的人重链和轻链转基因经历类型转换和体细胞突变以产生高亲和力人IgG,κ单克隆抗体(Lonberg,N.等(1994),上文;综述于Lonberg,N. Handbook ofExperimental Pharmacology 113,49 101(1994),Lonberg,N.和 Huszar,D.,Intern.Rev.Immunol.Vol.13 65 93(1995)以及Harding,F.和 Lonberg,N.Ann.N.Y.Acad.Sci 764536 546(1995))。HuMAb小鼠的制备详述于Taylor,L.等,NucleicAcids Research 20,6287 6295(1992),Chen,J.等, International Immunology 5,647656(1993),Tuaillon等,J.Immunol.152,2912 2920(1994),Taylor,L.等,International Immunology 6,579591(1994),Fishwild, D.等,Nature Biotechnology14,845851(1996)。还可参见US 5,545,806,US 5,569,825,US 5,625,126,US 5,633,425,US 5,789,650,US 5,877,397,US 5,661,016,US 5,814,318,US 5,874,299,US 5,770,429,US 5,545,807,WO 98/24884,WO 94/25585,WO 93/1227,WO 92/22645,WO 92/03918和WO 01/09187。根据公知技术,可以使用来自这些转基因小鼠的脾细胞产生分泌人单克隆抗体的杂交瘤。
此外,本发明的人抗体或来自其他种的本发明抗体可以通过展示类技术来鉴定,包含但不限于,噬菌体展示,逆转录病毒展示,核糖体展示,哺乳动物展示,酵母展示和本领域已知的其他技术,并且所获分子可以进行额外的成熟,例如亲和力成熟,因为这类技术是本领域公知的。描述于实施例17 的具体策略适用于任意抗体以使用噬菌体展示来制备并获得本发明的变体。
亲本抗体不限于具有天然的例如人Fc域的抗体,但是它还可以是具有除本发明的那些突变之外的其他突变的抗体,例如例如影响糖基化或使抗体能够成为双特异性抗体的突变。术语“自然抗体”表示不包含任何遗传导入突变的任意抗体。包含天然发生的修饰(例如不同的同种异型)的抗体因此就本发明来说被理解为“天然抗体”,从而可以被理解为亲本抗体。这种抗体可以充当根据本发明的一个或更多个突变的模板,从而提供本发明的变体抗体。包含除本发明那些抗体之外的其他突变的亲本抗体的实例是描述于 WO2011/131746(Genmab)的双特异性抗体,利用还原条件以促进包含IgG4 样CH3区域的两个抗体的半分子替换,从而形成双特异性抗体而无聚集体的伴发形成。亲本抗体的其他实例包含但不限于双特异性抗体例如异二聚体双特异性∶Triomabs(Fresenius);双特异性IgG1和IgG2(Rinat neurosciences Corporation);Fc△Adp(Regeneron);钮-入-孔(Genentech);静电驾驭(Amgen, Chugai,Oncomed);SEEDbodies(Merck);Azymetric scaffold(Zymeworks);mAb-Fv(Xencor);和LUZ-Y(Genentech)。其他示例性亲本抗体形式包含,但不限于,野生型抗体,全长抗体或包含Fc的抗体片段,人抗体,或其任意组合。
亲本抗体可以结合任意靶标,本发明这种靶标或抗原的实例可以是,并且不限于,针对的是:5T4;ADAM-10;ADAM-12;ADAM 17;AFP;AXL; ANGPT2炭疽抗原;BSG;CAIX;CAXII;CA 72-4;癌相关抗原CTAA16.88; CCL11;CCL2;CCR4;CCR5;CCR6;CD2;CD3E;CD4;CD5;CD6;CD15;CD18;CD19;CD20;CD22;CD24;CD25;CD29;CD30;CD32B; CD33;CD37;CD38;CD40;CD40LG;CD44;CD47;CD52;CD56;CD66E; CD72;CD74;CD79a;CD79b;CD80;CD86;CD98;CD137;CD147; CD138;CD168;CD200;CD248;CD254;CD257;CDH3;CEA;CEACAM5; CEACAM6;CEACAM8;Claudin4;CS-1;CSF2RA;CSPG-4;CTLA4; Cripto;DLL4;ED-B;EFNA2;EGFR;内皮缩血管肽B受体;ENPP3;EPCAM; ERBB2;ERBB3;FAPα;FcγRI;FCER2;FGFR3;纤维蛋白IIβ链; FLT1;FOLH1;FOLR1;FRP-1;GD3神经节苷脂;GDF2;GLP1R;磷脂酰肌醇聚糖(Glypican)-3;GPNMB;HBV(乙型肝炎病毒);HCMV(人巨细胞病毒);热休克蛋白90同源物[白色念珠菌];单纯疱疹病毒gD糖蛋白;HGF; HIV-1;HIV-1 IIIB gpl20V3环;HLA-DRB(HLA-DRβ);人呼吸道合胞病毒,糖蛋白F;ICAM1;IFNA1;IFNA1;IFNB1双特异性;IgE Fc;IGF 1R; IGHE连接区;IL 12B;IL 13;IL 15;IL 17A;ILIA;IL 1B;IL 2RA;IL4;IL5; IL5RA;IL6;IL6R;IL9;白介素-2受体β亚单位;ITGA2;ITGA2B ITGB3; ITGA4 ITGB7;ITGA5;ITGAL;ITGAV_ITGB3;ITGB2;KDR;L1CAM; Lewis-y;脂质A,脂多糖LPS的域;LTA;MET;MMP14;MMpl5;MST1R; MSTN;MUC1;MUC4;MUC16;MUC5AC;NCA-90粒细胞抗原;连接素 4;NGF;NRP;NY-ESO-1;OX40L;PLAC-1;PLGF;PDGFRA;PD1; PDL1;PSCA;磷脂酰丝氨酸;PTK-7;铜绿假单胞菌血清型IATS O11;RSV(人呼吸道合胞病毒,糖蛋白F);ROR1;RTN4;SELL;SELP;STEAP1;志贺样毒素II B亚单位[大肠杆菌];SLAM7;SLC44A4;SOST;表皮葡萄球菌脂磷壁酸;T细胞受体α_β;TF;TGFB1;TGFB2;TMEFF2;TNC;TNF; TNFRSF 10A;TNFRSF 10B;TNFRSF 12A;TNFSF 13;TNFSF 14;TNFSF2;TNFSF7;TRAILR2;TROP2;TYRP1;VAP-1;和波形蛋白。
亲本抗体可以是任意同种型的任意人抗体,例如IgG1、IgG2、IgG3、IgG4、 IgA1、IgA2、IgE、IgM和IgD,任选的人全长抗体,例如人全长IgG1抗体。亲本抗体可以包含根据SEQID NO:1,2,3,4和5中任一项的序列。
用于本发明的单克隆抗体,例如亲本和/或变体,可以通过Kohler等, Nature256,495(1975)首先描述的杂交瘤方法产生,或可以通过重组DNA方法产生。还可以使用在例如Clackson等,Nature 352:624-628(1991)和Marks 等,J.Mol.Biol.222:581-597(1991)中描述的技术,从噬菌体抗体文库分离单克隆抗体。单克隆抗体可以获自任意合适的来源。因此,例如,单克隆抗体可以从鼠脾B细胞制备的杂交瘤获得,所述鼠脾B细胞获自用感兴趣的抗原 (例如采用以下形式:在表面表达抗原的细胞,或编码感兴趣抗原的核酸)免疫的小鼠。单克隆抗体还可以从来源于免疫的人或非人哺乳动物(例如大鼠,犬,灵长类动物,等等)的抗体表达细胞的杂交瘤获得。
在一个实施方案中,抗体是人抗体。可以利用携带部分人免疫系统而不是小鼠系统的转基因或转染色体小鼠来产生针对任意抗原的人单克隆抗体。这种转基因和转染色体小鼠分别包含在本发明中被称为 小鼠和KM 小鼠的小鼠,在本发明中统称为“转基因小鼠”。
小鼠和KM 小鼠的小鼠,在本发明中统称为“转基因小鼠”。


HCo7、Hco12、Hco17和HCo20小鼠具有在其内源轻链(κ)基因中的JKD 破坏(描述于Chen等,EMBO J.12,821-830(1993)),在其内源重链基因中的 CMD破坏(描述于WO 01/14424的实施例1)和KCo5人κ轻链转基因(描述于 Fishwild等,Nature Biotechnology 14,845-851(1996))。此外,Hco7小鼠具有 HCo7人重链转基因(描述于US 5,770,429),HCo12小鼠具有HCo12人重链转基因(描述于WO 01/14424的实施例2),HCo17小鼠具有HCo17人重链转基因 (描述于WO 01/09187的实施例2)和HCo20小鼠具有HCo20人重链转基因。所获小鼠在内源小鼠重链和κ轻链基因座破坏方面纯合的背景下表达人免疫球蛋白重链和κ轻链转基因。
在KM小鼠品系中,如Chen等,EMBO J.12,811-820(1993)所描述,内源小鼠κ轻链基因已经被纯合破坏,并且如WO 01/09187的实施例1所描述,内源小鼠重链基因已经被纯合破坏。如Fishwild等,Nature Biotechnology 14, 845-851(1996)所描述,这种小鼠品系携带人κ轻链转基因KCo5。如WO 02/4347814所描述,这种小鼠品系还携带由14号染色体片段hCF(SC20)组成的人重链转染色体。如WO/2009/097006所述,通过将HCo12与KCo5[J/K](Balb) 杂交,产生HCo12-Balb/C小鼠。根据公知技术,可以使用来自这些转基因小鼠的脾细胞产生分泌人单克隆抗体的杂交瘤。
此外,可以利用本领域公知的技术从通过展示类技术(包含但不限于,噬菌体展示,逆转录病毒展示,核糖体展示和其他技术)鉴定的人抗体或来自其他种的抗体获得任意抗原结合区,并且所获分子可以经历额外的成熟,例如亲和力成熟,因为这类技术是本领域公知的(参见例如Hoogenboom等,J. Mol.Biol.227,381(1991)(噬菌体展示),Vaughan等,Nature Biotech 14,309 (1996)(噬菌体展示),Hanes和Plucthau,PNAS USA 94,4937-4942(1997)(核糖体展示),Parmley和Smith,Gene 73,305-318(1988)(噬菌体展示),ScottTIBS 17,241-245(1992),Cwirla等,PNAS USA 87,6378-6382(1990),Russel 等,Nucl.Acids Research 21,1081-1085(1993),Hogenboom等,Immunol. Reviews 130,43-68(1992),Chiswell和McCafferty TIBTECH 10,80-84(1992), 和US 5,733,743)。如果展示技术用于产生非人抗体,则这种抗体可以被人源化。
根据本发明的突变可以是,但不限于,一个或更多个氨基酸的缺失、插入或取代。氨基酸的这种取代可以利用任意天然存在或非天然氨基酸进行。
“单突变体”
应理解可以本文中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案也可以适用于包含免疫球蛋白的Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
根据本发明“单突变体”方面的抗体或多肽变体包含在表1所示的一个或多个氨基酸残基的突变,通常是氨基酸取代,表1列出了根据人类IgG1抗体的EU索引进行编号的每个氨基酸残基,连同IgG2、IgG3和IgG4亲本抗体中对应位置中的氨基酸以及“示例性”和“优选的”氨基酸取代。图2显示了对应于IgG1中残基126至326的IgG2区段,对应于残基177至377的IgG3 Fc区段,和对应于残基127至327的IgG4区段。
表1“单突变体”方面的示例突变位点和氨基酸取代

从表1可以看出,导致实施例19中的Wien133细胞的细胞裂解增加的氨基酸取代作为“优选的取代”包含在内。
在一个方面,本发明涉及一种包含免疫球蛋白的Fc域和结合区的亲本多肽的变体,其中所述变体包含选自对应于人类IgG1重链Fc区中的E430G、E430S、E430F、E430T、E345K、E345Q、E345R、E345Y和S440W的组的一个或多个突变,条件是变体在Fc域中不含有改变变体与新生儿Fc受体结合的任何其他突变。(FcRn)可以通过实施例34中公开的方法来测定。
在另一个方面,本发明涉及一种包含免疫球蛋白的Fc域和结合区的亲本多肽的变体,其中所述变体包含选自对应于人类IgG1重链Fc区中的E430G、 E430S、E430F、E430T、E345K、E345Q、E345R、E345Y和S440W的组的一个或多个突变,条件是所述变体不含有在Fc域中使变体与新生儿Fc受体 (FcRn)的结合提高或降低超过30%,例如超过20%,10%或5%的任何其他突变,如通过实施例34中公开的方法测定的OD 405nm吸光度的变化测量的。
在另一个方面,本发明涉及一种包含免疫球蛋白的Fc域和结合区的亲本多肽的变体,其中所述变体包含在选自对应于人类IgG1重链Fc区中的 E430G、E430S、E430F、E430T、E345K、E345Q、E345R、E345Y和S440W 的组的一个或多个突变,条件是变体不含有在Fc域中使亲本抗体与小鼠新生儿Fc受体(FcRn)的表观亲和力提高超过0.5倍或者不使亲本多肽或亲本抗体与小鼠FcRn的表观亲和力降低超过2倍的任何其他突变,如通过实施例34 中公开的方法确定的。
在一个实施方案中,一个或多个突变选自对应于人类IgG1重链Fc区中 E430G、E430S、E345K和E345Q的组。
在一个实施方案中,变体不含有在Fc域中改变变体的抗体依赖性细胞介导的细胞毒性(ADCC)的任何其他突变。
在一个实施方案中,变体不含有在Fc域中改变变体的血浆清除率的任何其他突变,如通过实施例37中公开的方法测定的。
在另一个实施方案中,变体不含有在Fc域中使变体的血浆清除率增加或降低超过3.0倍,如超过2.5倍,2.0倍,1.5倍或1.2倍的任何其他突变,如通过实施例37中公开的方法测定的。
在一个实施方案中,变体不含有在Fc域中改变变体的血清半衰期的任何其他突变。
在一个实施方案中,变体不含有在Fc域中改变变体的不依赖于靶标的流体相补体激活的任何其他突变,如通过实施例36中公开的方法测定的。
在一个实施方案中,变体不含有在Fc域中的任何其他突变。
在一个实施方案中,变体仅包含一个突变。
在一个实施方案中,变体多肽可以是包含免疫球蛋白的Fc域和抗原结合区的变体抗体。
在一个具体的实施方案中,氨基酸取代是E345R。
如实施例所示,包含这些氨基酸取代之一的CD38抗体HuMab-005和-003 (如WO2006/099875所述)和/或CD20抗体HuMab-7D8和-11B8(如WO 2004/035607所述)和利妥昔单抗和/或EGFR抗体HuMab-2F8(如WO 2002/100348所述)的变体分别具有比野生型HuMab 005和7D8更高的C1q结合,补体激活和/或CDC。
应理解所述变体也可以包含表1所列“示例性取代”的突变中的一个。所述变体还可以包含超过一个突变,例如表1所列任意突变中的两,三,四,五或六个。
除了指定突变外,变体可以具有针对亲本抗体描述的任意特征。具体来说,它可以是人抗体。除了突变外,变体可以进一步是任意IgG1亚型的。
当与抗原表达细胞表面、细胞膜、病毒粒子或另一颗粒上的其抗原结合时,或抗原与病毒粒子有关时,任选的其中抗原包含在病毒粒子的蛋白衣壳或脂质包膜内,与亲本抗体相比,这类抗体变体可以具有以下至少一种的增加:(i)抗体介导的CDC,(ii)抗体介导的补体激活,(iii)C1q结合,(iv)寡聚体形成,(v)寡聚体稳定性,或(i)到(v)中任意的组合。在(iv)或(v)的一个实施方案中,寡聚体是六聚体。在一个实施方案中,与亲本多肽或亲本抗体相比,变体还可以具有增加的ADCC。在进一步的实施方案中,与亲本多肽或亲本抗体相比,变体保留相同或相似的血浆清除率。在进一步的实施方案中,变体不具有与亲本多肽或亲本抗体相比时增加或降低超过3.0倍,如超过2.5倍, 2.0倍,1.5倍或1.2倍的血浆清除率,如通过实施例37中公开的方法测定的。
不受限于任何具体理论,用本发明的氨基酸残基在指定位置取代氨基酸引起的效应可以例如引起效应自身,参与直接接触另一分子的Fc域,或可以被突变以与另一Fc域直接相互作用或间接影响分子间Fc:Fc相互作用。因此,不受限于理论,取代被认为直接或间接增强寡聚形式的抗体分子间的结合强度,增强寡聚体结构(例如六聚体,五聚体,四聚体,三聚体或二聚体结构) 的稳定性。例如,氨基酸取代可以是促进或增强新分子间Fc:Fc键(例如,但不限于,范德华相互作用,氢键,电荷-电荷相互作用、或芳香族叠加相互作用)形成的氨基酸取代,或在Fc:Fc相互作用时通过水分子释放促进熵增加的氨基酸取代。此外,参考表1,可以基于参与或促进分子间Fc:Fc相互作用或分子内相互作用的大小和物化特性挑选“示例性取代”。可以基于最适合参与或激发分子间Fc:Fc相互作用或分子内相互作用的大小和物化特性挑选“示例性取代”。
在一个实施方案中,变体可以包含选自表1的其他突变。
在一个实施方案中,变体包含氨基酸残基中的两个突变的组合,其选自对应于E345X/E430X,E345X/S440Y,E345X/S440W,E430X/S440Y和 E430X/S440W的组。
在变体中包含至少两个氨基酸中的这种突变的任意实施方案中,它可以存在于变体的每一重链中,或两个中的一个可以包含在一条重链内,而另一个可以包含在另一个重链内,或反之亦然。
在一个实施方案中,至少两个氨基酸残基中的突变是缺失,插入或取代。氨基酸的这种取代可以利用任意天然存在或人工存在氨基酸进行。
根据本发明的突变可以各是但不限于一个或多个氨基酸的缺失、插入或取代。氨基酸的此类取代可以用任意天然存在或非天然存在氨基酸进行。
因此,在一个实施方案中,在至少一个氨基酸残基处的突变可以是缺失。
在另一个实施方案中,在至少一个氨基酸残基处的突变可以是插入。
在另一个实施方案中,在至少一个氨基酸残基处的突变可以是取代。
至少两个氨基酸残基中的突变的示例性具体组合是E345R/E430T, E345R/S440Y,E345R/S440W,E345R/E430G,E345Q/E430T,E345Q/S440Y, E345Q/S440W,E430T/S440Y和E430T/S440W。
除了根据本发明的实施方案中一个或更多个氨基酸的突变,IgG重链可以包含本领域已知的另外突变,例如,进一步改善效应子功能的突变。这类另外突变包含增强CDC,Fc-受体结合或FcRn结合和/或改善Fc-γ受体介导的效应子功能的已知突变。
在一个实施方案中,根据本发明的变体还包含已知的CDC增强修饰,例如,IgG同种型之间的片段交换以产生嵌合IgG分子(Natsume等,2008Cancer Res 68(10),3863-72);铰链区的一个或更多个氨基酸取代(Dall'Acqua等,2006 J Immunol 177,1129-1138),和/或围绕残基D270、K322、P329和P331,在CH2 域的C1q结合位点中或附近的一个或更多个氨基酸取代(Idusogie等,2001 J Immunol 166,2571-2575;Michaelsen等,2009Scand JImmunol 70,553-564和 WO 99/51642)。例如,在一个实施方案中,根据本发明的变体还包含氨基酸取代S267E,H268F,S324T,S239D,G236A和I332E中任一项的组合,通过CDC 或ADCC提供增强的效应子功能(Moore等,2010mAbs 2(2),181-189))。影响与Fc受体结合(描述于WO 2006/105062,WO 00/42072,美国专利US6,737,056 和美国专利US7,083,784)或抗体物理特性(描述于WO 2007/005612A1)的其他Fc突变也可用于本发明的变体。
在一个实施方案中,根据本发明的变体还包含增强Fc-γ受体结合和/或 Fc-γ受体介导的效应子功能的修饰。这种修饰包含(i)降低CH2附着的糖基化中岩藻糖的量(糖工程改造)(Umana P,等,Nat Biotechnol 1999;17:176-80; Niwa R,等,Clin Cancer Res2004;10:6248-55.)),和(ii)抗体铰链或CH2区域中氨基酸的定点诱变(蛋白工程改造)(Lazar GA,等,Proc Natl Acad Sci U S A 2006;103:4005-10)。
在一个实施方案中,根据本发明的变体在FcRn结合位点被进一步改造,例如,以延长IgG抗体的半衰期(t1/2)。这类修饰包含(i)N434A和 T307A/E380A/N434A突变(Petcova等Int Immunol.2006Dec;18(12):1759); (ii)将Pro238,Thr256,Thr307,Gln311,Asp312,Glu380,Glu382和Asn434中的一个或更多个取代为丙氨酸残基,从而改进FcRn结合(Shields RL,等J.Biol. Chem.2001;276:6591);和(iii)选自下组的氨基酸取代或氨基酸取代的组合: IgG1中的M252Y/S254T/T256E,M252W,M252Y,M252Y/T256Q, M252F/T256D,V308T/L309P/Q311S,G385D/Q386P/N389S, G385R/Q386T/P387R/N389P,H433K/N434F/Y436H,N434F/Y436H, H433R/N434Y/Y436H,M252Y/S254T/T256E-H433K/N434F/Y436H或M252Y/S254T/T256E-G385R/Q386T/P387R/N389P,从而增加对FcRn的亲和力(Dall'Acqua等,上文)。
“双突变体”
应理解可以本问中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案也可以适用于包含免疫球蛋白的Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
如上文和进一步在下文所述,本发明还涉及“双突变体”方面,其中两个突变单个各自降低效应子功能但共同将效应子功能恢复到亲本抗体水平。当一起使用时,变体的特异性增加。根据“双突变体”方面的抗体变体包含特异性氨基酸残基相互作用对K439和S440,K447和448,或K447,448和449 中的两个突变,通常是氨基酸取代。
因此,在一个方面,本发明涉及一种包含免疫球蛋白的Fc域和结合区的亲本多肽的变体,其中所述变体包含第一突变和第二突变,所述第一突变选自对应于人IgG1重链的Fc区中的E430G、E430S、E430F、E430T、E345K、 E345Q、E345R、E345Y、S440Y和S440W,如E430G、E430S、E345K或 E345Q的组,所述第二突变选自对应于以下氨基酸残基的组:
(i)对应于人类IgG1重链Fc区中的K439和S440的氨基酸残基,条件是S440的突变不是S440Y或S440W,并且如果第一突变是S440Y或S440W,那么第二突变在对应于人类IgG1重链的Fc区中的K439的氨基酸残基中,
(ii)对应于人类IgG1重链Fc区中的K447D/E或K447K/R/H和448P的突变残基;或
(iii)对应于人类IgG1重链Fc区中的K447D/E或K447K/R/H、448K/R/H和449P 的氨基酸残基。表2A和B显示“双突变体”(表A)和“混合突变体”(表2B) 方面的“示例性”和“优选的取代”。
表2A“双突变体”方面的示例突变位点和氨基酸取代

表2B:“混合突变体”方面(抗体1+抗体2)的示例突变位点和氨基酸取代

在一个实施方案中,变体包含选自对应于人类IgG1重链Fc区中的 E430G、E430S、E430F、E430T、E345K、E345Q、E345R和E345Y的组的第一突变和对应于K439和S440的氨基酸残基中的第二突变,条件是S440中的突变不是S440Y和S440W。
本发明预期变体也可以只包含氨基酸残基取代之一,例如K439E或S440K,例如所述变体在K439中包含突变,任选地在S440中没有突变。
在一个实施方案中,本发明涉及变体,其中K439中的突变是氨基酸取代为选自E和D的氨基酸,例如K439E。
在另一个实施方案中,变体包含S440中的突变,任选地在K439中没有突变。
在一个实施方案中,本发明涉及变体,其中S440的突变是氨基酸取代为选自K和R的氨基酸,例如S440K。
在一个实施方案中,变体包含K439和S440两处的突变。
在另一个实施方案中,K439的突变选自K439到D、E或R,如K439D/E, S440的突变选自S440到D、E、K和R,如S440K/R。
在另一个实施方案中,K439的突变选自K439D和K439E,和S440的突变选自S440K和S440R。
在另一个实施方案中,变体包含K439E和S440K突变。
在一个实施方案中,亲本多肽是包含免疫球蛋白Fc域和抗原结合区的亲本抗体。
如实施例4-6所述,只包含K439E和S440K突变之一的抗体变体对C1q的 KD显著增加,反映降低的补体激活和/或CDC能力。出人意料的,发现包含这两个突变的HuMAb 7D8或005的抗体变体具有恢复的或增加的C1q结合或 CDC。不受任何具体理论限制,根本机制也许可以通过空间上彼此弥补的相应突变来解释,如图4和5所示。
在一个实施方案中,亲本多肽及由此其变体可以是包含免疫球蛋白Fc和抗原结合区的亲本抗体。
在另一个实施方案中,与亲本抗体或只包含K439和S440之一中的突变的抗体变体相比,如本发明所述包含位置K439和S440两者中的突变的变体具有 Fc介导的效应子功能的增加,所述效应子功能选自补体依赖性细胞毒性 (CDC),C1q结合,补体激活,抗体依赖性细胞介导的细胞毒性(ADCC),Fc 受体结合,包含Fc-γ受体结合,蛋白A结合,蛋白G结合,抗体依赖性细胞吞噬(ADCP),补体依赖性细胞性细胞毒性(CDCC),补体增强的细胞毒性,调理作用,包含Fc的多肽内化、靶标下调、ADC摄取、凋亡诱导、细胞死亡、细胞周期停滞、及其任意组合。
本发明还提供抗体中K439E和S440K突变在与亲本抗体(其可以是例如野生型抗体或只包含K439E或S440K突变之一的抗体变体)相比恢复以下一种或更多种作用中的用途:(i)抗体介导的CDC,(ii)抗体介导的补体激活,(iii) C1q结合亲合力,(iv)寡聚体形成,(v)寡聚体稳定性,或(i)到(v)中任意的组合。在(iv)或(v)的一个实施方案中,寡聚体是六聚体。
在一个实施方案中,变体选自单特异性抗体、双特异性抗体或多特异性抗体。
混合突变体
应理解本文中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案可适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
如上所述,本发明的本发明人还发现存在自身降低效应子功能但一起使用时恢复效应子功能的突变,例如人类IgG1重链Fc区中的位置K439和S440 的突变。因此,通过在一个抗体中引入K439并在另一抗体中引入S440,这一构思也可用于确保两种不同抗体的配对。因此,根据“混合突变体”方面的抗体变体包含突变,但是一个突变通常导致相同Fc分子间Fc:Fc相互作用降低或大幅降低。但是,作为“混合突变体”的本发明抗体变体能够彼此配对;与例如每种单独的变体或亲本抗体的混合相比,提供恢复或甚至增加的 CDC,C1q结合,补体激活,寡聚体形成,和/或特异性抗体变体对的寡聚体稳定性。在本发明的一个实施方案中,寡聚体是六聚体。在一个实施方案中,抗体变体对还可以或者可选的具有保留的或改善的其他效应子功能,例如 C1q结合,补体激活,抗体依赖性细胞介导的细胞毒性(ADCC),FcRn结合, Fc受体结合,包含Fc-γ受体结合,蛋白A结合,蛋白G结合,抗体依赖性细胞吞噬(ADCP),补体依赖性细胞性细胞毒性(CDCC),补体增强的细胞毒性、调理作用、包含Fc的多肽内化、靶标下调、ADC摄取、凋亡诱导、细胞死亡、细胞周期停滞、及其任意组合。本发明的这种方面提供大量应用,其中不仅 C1q结合、补体激活、CDC或其他效应子功能的强度而且还有选择性可以被调节。
“混合突变体”对中每种抗体变体的示例性突变位点显示于表2B。具体来说,本发明提供一种包含免疫球蛋白Fc域和抗原结合区的抗体变体,所述变体包含人类IgG1重链Fc区中对应于K439和S440之一的的残基中的突变。
在一个实施方案中,突变位于K439,并且是氨基酸取代为选自E或D的氨基酸,如K439E。在一个实施方案中,突变位于S440,并且是氨基酸取代为选自K或R的氨基酸,例如S440K。
在一个实施方案中,变体包含在仅对应于IgG1重链的Fc区中的K439,而不是在对应于S440位置的位置中的氨基酸突变。
在一个实施方案中,变体包含在仅对应于S440的位置中的氨基酸突变,条件是S440的突变不是S440Y或S440W,并且所述变体不包含在对应于IgG1 重链的Fc区中的K439的位置中的氨基酸突变。
因此,在一个实施方案中,本发明还涉及包含第一突变和第二突变的变体,所述第一突选自对应于人类IgG1重链的Fc区中的E430G、E430S、E430F、 E430T、E345K、E345Q、E345R、E345Y、S440Y和S440W的组,所述第二突变在对应于人类IgG1重链Fc区中的K439的氨基酸残基中。
在另一个实施方案中,本发明还涉及包含第一突变和第二突变的变体,所述第一突变选自对应于人类IgG1重链的Fc区中的E430G、E430S、E430F、 E430T、E345K、E345Q、E345R和E345Y的组,所述第二突变在对应于人类IgG1重链Fc区中的S440的氨基酸残基中,条件是第二突变不是S440Y或 S440W。
在一个实施方案中,上述两种实施方案可以组合成根据本发明的“混合突变体”对。
“混合突变体”对的每个变体可以进一步包含表1所列的氨基酸中的突变。
在本发明的一个实施方案中,“混合突变体”对包含亲本抗体的第一变体和亲本抗体的第二变体,其中所述第一变体包含免疫球蛋白的第一Fc域和抗原结合区,其中所述第一变体包含(i)除K439中突变之外的一个或多个氨基酸残基中的第一突变和在对应于人类IgG1重链Fc区的K439的位置上的第二突变,所述第一突变选自对应于人类IgG1重链Fc区中的E430X、E345X、 S440Y和S440W,如E430G、E430S、E430F、E430T、E345K、E345Q、 E345R、E345Y、S440Y和S440W的组;并且,
其中所述第二变体包含免疫球蛋白的第二Fc域和抗原结合区,其中所述第二变体包含(i)除S440中突变之外的一个或多个氨基酸残基中的第一突变,所述第一突变选自对应于人类IgG1重链Fc区的E430X和E345X,如 E430G、E430S、E430F、E430T、E345K、E345Q、E345R和E345Y的组,
和(ii)在对应于IgG1重链Fc区中的S440的位置中的第二突变,条件是 S440的突变不是S440Y或S440W。
其他示例性“混合突变体”对可以进一步包含并且不限于下列对的任意一项:包含突变K447E的第一变体和包含突变K447/P448的第二变体;包含突变K447E的第一变体和包含突变K447/K448/P449的第二变体。
在一个实施方案中,第一突变是缺失、插入或取代。氨基酸的这种取代可以利用任意天然存在的或非天然存在的氨基酸进行。
在一个实施方案中,突变是缺失。
在另一个实施方案中,突变是插入。
在另一个实施方案中,突变是氨基酸的取代。
在一个具体实施方案中,第一变体和/或第二变体包含一个或多个氨基酸残基中的突变,其选自人类IgG1重链Fc区中的E430G,E430S,E345K,和 E345Q的组。
例如,在一个实施方案中,“混合突变体”对的一个变体包含E430G, E430S,E345K或E345Q之一以及K439E突变,而另一个变体包含E430G, E430S,E345K或E345Q之一以及S440K突变,因此提供增加和更具特异性的 C1q结合亲合力,补体激活,CDC,寡聚体形成,寡聚体稳定性,和/或其他效应子相关功能例如ADCC,Fc-γ受体结合,蛋白A结合,蛋白G结合,ADCP, CDCC,补体增强的细胞毒性,抗体介导的吞噬,内化,凋亡,与受调理抗体的补体受体的结合,和/或其组合。
“混合突变体”方面还可以包含两种变体,它们各包含在人类IgG1重链 Fc区中的表2A所列的超过一种突变,例如包含突变S440K/K447E的第一变体,和包含突变K439E/K447/P448的第二变体;例如包含突变K439E/K447E 的第一变体,和包含突变S440K/K447/P448的第二变体。
本发明描述的“混合突变体”对中的变体可以衍生自相同或不同的亲本抗体。此外,“混合突变体”方面还可以用于双特异性或非对称抗体。此外,第一、第二和第三抗体可以结合相同或不同靶标上的不同表位。
此外,“混合突变体”方面通过利用具有K439E突变的针对第一抗原的第一抗体和具有S440K或S440R突变的针对第二抗原的第二抗体,提供更特异性地针对表达两种特定肿瘤抗原的肿瘤细胞的CDC或其他效应子应答。通过利用包含三种变体(任选的是双特异性抗体)的“混合突变体”方面,可以提供更特异性针对表达至少两种,例如两、三、四、五或六种特定肿瘤抗原的肿瘤细胞的CDC或其他效应子应答。
在“单突变体”,“双突变体”和“混合突变体”方面中的任一种的一个实施方案中,变体选自单特异性抗体、双特异性抗体或多特异性抗体。
在“混合突变体”方面的任意实施方案中,第一,第二和/或第三变体可以包含表1所列的任意氨基酸取代的相同或不同突变。
多特异性抗体
应理解可以本发明中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案可适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
应理解本发明描述的“单突变体”,“双突变体”和“混合突变体”方面的任意实施方案可用于如下所述的多特异性抗体方面。
因此,在一个实施方案中,变体是选自单特异性抗体、双特异性抗体或多特异性抗体的抗体。
在一个具体的实施方案中,双特异性抗体具有在WO 2011/131746中描述的形式。
在一个主要方面,本发明涉及亲本抗体的一种变体,所述亲本抗体是包含第一多肽和第二多肽的双特异性抗体,所述第一多肽包含免疫球蛋白的第一CH2-CH3区和第一抗原结合区,所述第二多肽包含免疫球蛋白的第二 CH2-CH3区和第二抗原结合区,其中第一和第二抗原结合区结合相同抗原或不同抗原上的不同表位,以及其中第一和/或第二CH2-CH3区包含一个或多个突变,所述突变选自对应于人类IgG1重链Fc区中E430G、E430S、E430F、 E430T、E345K、E345Q、E345R、E345Y、S440Y和S440W的组,并且其中
所述第一多肽包含选自对应于人类IgG1重链Fc区中的K409、T366、 L368、K370、D399、F405和Y407的那些氨基酸残基的氨基酸残基中的进一步突变;并且
所述第二多肽包含选自对应于人类IgG1重链Fc区的F405、T366、L368、 K370、D399、Y407和K409的那些氨基酸残基的氨基酸残基中的进一步突变,并且其中所述第一多肽中的所述进一步突变不同于第二多肽中的进一步突变。
在一个实施方案中,突变是缺失,插入或取代。氨基酸的这种取代可以利用任意天然存在的或非天然存在的氨基酸进行。
本发明的双特异性抗体不限于特定的形式,并且它可以是上文及本文中所描述的那些双特异性抗体中的任一个。
在本发明一个具体的实施方案中,(i)第一多肽包含对应于人类IgG1重链 Fc区K409,如K409R的氨基酸残基中的进一步突变;和
(ii)第二多肽包含对应于人类IgG1重链Fc区F405,如F405L的氨基酸残基中的进一步突变;或者其中备选地,
(iii)第一多肽包含对应于人类IgG1重链Fc区F405,如F405L的氨基酸残基的进一步突变;
和(iv)第二多肽包含对应于人类IgG1重链Fc区K409,如K409R的氨基酸残基的进一步突变。
在一个具体的实施方案中,一个或多个氨基酸残基中的突变选自对应于人类IgG1重链Fc区中E430G,E430S,E345K和E345Q的组。
可以按照实施例22的描述产生根据本发明的这种双特异性抗体。此外,可以利用实施例23使用的测定法检测所产生异二聚体蛋白对CDC杀伤的作用。
双特异性抗体可以例如包含CD20抗体的抗原结合区和CD38抗体的抗原结合区,以及表1和/或2A/B所列一个或更多个氨基酸的氨基酸取代。示例性 CD20结合区包含WO2004/035607(其因此完整引入作为参考)描述的奥法木单抗(ofatumumab)(2F2)、7D8和11B,和利妥昔单抗(WO 2005/103081)的CD20 结合区。示例性CD38结合区包含WO2006/099875(其因此完整引入作为参考) 描述的003和daratumumab(005)的那些CD38结合区。
在一个实施方案中,双特异性抗体结合相同或不同的靶标上的不同表位。
在另一个实施方案中,第一和第二多肽中的第一突变可以是相同或不同的。
在“单突变体”、“双突变体”、“混合突变体”和多特异性抗体方面的一个实施方案中,变体是人类IgG1、IgG2、IgG3、IgG4、IgA1、IgA2、IgD、 IgM或IgE抗体,任选的是人全长抗体,例如人全长IgG1抗体。
在任意“单突变体”、“双突变体”、“混合突变体”方面和多特异性抗体方面,根据实施例4描述的测定法确定抗体的C1q结合,根据实施例5、6或10 描述的测定法确定CDC,突变不位于直接参与C1q结合的氨基酸残基,任选地通过将根据实施例3ELISA测定法的C1q结合与根据实施例4的基于细胞的测定法中的C1q结合进行比较来确定,以及根据实施例12描述的测定法确定 ADCC。
此外,本发明提供如上所述的任意“单突变体”、“双突变体”、“混合突变体”和多特异性抗体方面或实施方案的变体的制品。本发明还提供包含如上所述的任意“双突变体”方面和实施方案的变体的组合物,例如,药物组合物。本发明还提供任意这种变体、制品或组合物作为药物的用途。
本发明的上述“单突变体”、“双突变体”、“混合突变体”和多特异性抗体方面尤其适用于具有IgG1重链的人抗体分子,所述IgG1重链包含对应于人类IgG1重链恒定区的下划线残基130到330(UniProt登录号P01857;SEQ ID NO:1)的相关片段P247到K447:

本发明还可以适用于具有人IgG2重链部分的抗体分子。IgG1重链的氨基酸残基P247到K447对应于IgG2重链恒定区的下划线残基126到326(登录号 P01859;SEQ ID NO:2)

本发明还可以适用于具有人IgG3重链部分的抗体分子。IgG1重链的氨基酸残基P247到K447对应于IgG3重链恒定区的残基177到377(UniProt登录号 P01860;SEQ ID NO:3),在下面用下划线表示:


本发明还可以适用于具有人IgG4重链部分的抗体分子。IgG1重链的氨基酸残基P247到K447对应于IgG4重链恒定区的下划线残基127到327(登录号 P01859,SEQ ID NO:4)

本发明还可以适用于具有人类IgG1m(f)同种异型重链部分的抗体。 IgG1m(f)同种异型的氨基酸序列(CH3序列用下划线表示)-SEQ ID NO:5

IgG1,IgG2,IgG3,IgG4和IgG1m(f)恒定区的相应片段的比对显示于图2。因此,表1或表2A和B描述的氨基酸的任意突变可以引入通过比对确定的 IgG2,IgG3,IgG4和/或IgG1m(f)的等同位置,以获得根据本发明的变体。
在一个实施方案中,本发明提供全长IgG1,IgG2,IgG3或IgG4抗体的变体,其包含根据如上所述任意方面的一个或更多个氨基酸取代。
在任意“单突变体”、“双突变体”、“混合突变体”方面和多特异性抗体中,IgG1重链的Fc区可以包含以下序列:SEQ ID NO:1的残基130到330,SEQ ID NO:2的残基126到326,SEQ ID NO:3的残基177到377,或SEQ ID NO:4 的残基127到327。
在一个实施方案中,亲本抗体包含选自SEQ ID No.:1-5的序列,例如SEQ ID No.:1,SEQ ID No.:2,SEQ ID No.:3,SEQ ID No.:4或SEQ ID No.:5。
在一个实施方案中,IgG1重链的Fc区包含SEQ ID NO:1的残基130到330 的序列。
亲本抗体可以是本发明描述的任意亲本抗体。在此上下文处亲本抗体还旨在是第一亲本和第二亲本抗体。
在一个实施方案中,亲本抗体是人类IgG1,IgG2,IgG3或IgG4,IgA1, IgA2,IgD,IgM或IgE抗体。
在一个实施方案中,亲本抗体是人全长抗体,例如人全长IgG1抗体。
在一个实施方案中,亲本抗体、第一亲本抗体和第二亲本抗体是人类 IgG1抗体,例如IgG1m(za)或IgG1m(f)同种异型,任选地其包含含有SEQ ID NO:1或5的Fc区。
在一个实施方案中,亲本抗体是人IgG2抗体,任选地其包含含有SEQ ID NO:2的Fc区。
在一个实施方案中,亲本抗体是人IgG3抗体,任选地其包含含有SEQ ID NO:3的Fc区。
在一个实施方案中,亲本抗体是人IgG4抗体,任选地其包含含有SEQ ID NO:4的Fc区。
在“单突变体”、“双突变体”、“混合突变体”和多特异性抗体方面的任意一项的具体实施方案中,变体包含与SEQ ID NOs:1,2,3,4和5的氨基酸P247到K447具有至少70%,72%,74%,76%,78%,80%,81%,82%, 83%,84%,85%,86%,87%,88%,89%,90%,91%,92%,93%,94%, 95%,96%,97%,98%或至少约99%的同一性程度的氨基酸序列,除了根据本发明引入的突变之外。
因此,除了本发明限定的任意突变之外,变体可以包含SEQ ID NO:1, SEQ ID NO:2,SEQ ID NO:3,SEQ ID NO:4或SEQ ID NO:5的序列。
根据本发明的任意上述“单突变体”、“双突变体”、“混合突变体”和多特异性方面应理解为包含下列实施方案。
在一个实施方案中,第一和/或第二亲本抗体是抗体片段,任选地选自单价抗体,重链抗体,链交换改造域(SEED),triomab,双可变域免疫球蛋白 (DVD-Ig),钮-入-孔抗体,微型抗体(mini-antibody),双亲和力再导向分子 (Fc-DART或Ig-DART);LUZ-Y抗体,Biclonic抗体,双靶向(DT)-Ig抗体,二合一(Two-in-one)抗体,交联Mab,mAb2,CovXbody,IgG样双特异性抗体, Ts2Ab,BsAb,HERCULES抗体,TvAb,ScFv/Fc融合抗体,SCORPION,与Fc域融合的scFv片段,和与Fc域融合的双重scFv片段。
在进一步的实施方案中,第一和第二亲本抗体均结合在人肿瘤细胞表面表达的抗原。
在进一步的实施方案中,针对第一和第二亲本抗体的抗原单独选自 erbB1(EGFR),erbB2(HER2),erbB3,erbB4,MUC-1,CD4,CD19,CD20, CD38,CD138,CXCR5,c-Met,HERV-包膜蛋白,骨膜蛋白,Bigh3,SPARC, BCR,CD79,CD37,EGFrvIII,L1-CAM,AXL,组织因子(TF),CD74,EpCAM 和MRP3。
在进一步的实施方案中,第一和第二亲本抗体是完全人的。
在进一步的实施方案中,针对第一和第二亲本抗体的抗原是按照任意顺序选自CD20和CD38,任选的其中第一和第二亲本抗体是按照任意顺序选自 7D8和005。
在进一步的实施方案中,第一抗体和第二抗体均结合在细菌细胞或病毒粒子表面表达的抗原。
在另一个实施方案中,细菌细胞选自金黄色葡萄球菌(S.aureus),表皮葡萄球菌(S.epidermidis),葡萄球菌肺炎(S.pneumonia),炭疽杆菌(Bacillus anthracis),绿针假单胞菌(Pseudomonas aeruginosa),沙眼衣原体(Chlamydia trachomatis),大肠杆菌(E.coli),沙门氏菌(Salmonella),志贺氏菌(Shigella),耶尔森氏菌(Yersinia),鼠伤寒沙门氏菌(S.typhimurium),脑膜炎奈瑟菌 (Neisseria meningitides)和结核分枝杆菌(Mycobacterium tuberculosis)。
在进一步的实施方案中,第一和第二亲本抗体结合相同的抗原。
在另一个实施方案中,第一和第二亲本抗体是相同的抗体。
在另一个实施方案中,亲本抗体选自7D8和005。
组合物
应理解本发明中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案可适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
本发明还涉及包含变体的组合物,并且亲本抗体可以是本发明描述的任意变体和亲本抗体。具体方面和实施方案将在下文描述。此外,可以根据本发明描述的任意方法获得这种变体
在一个方面,本发明涉及一种包含各包含免疫球蛋白的Fc域和结合区的亲本多肽的第一变体和第二变体的组合物,其中所述第一变体和/或第二变体包含一个或多个突变,所述突变选自对应于人类IgG1重链Fc区中E430X、 E345X、S440Y和S440W的组。
在一个方面,第一变体和/或第二变体包含一个或多个突变,所述突变选自对应于人类IgG1重链Fc区中E430G、E430S、E430F、E430T、E345K、E345Q、 E345R、E345Y、S440Y和S440W的组。
在一个优选的实施方案中,第一变体和/或第二变体包含一个或多个突变,所述突变选自选自对应于人类IgG1重链Fc区中E430G、E430S、E345K 和E345Q的组。
在一个实施方案中,第一变体和第二变体包含一个或多个突变,它们可以是相同或不同的。
在另一个实施方案中,第一变体包含一个或多个突变,所述突变选自对应于人类IgG1重链Fc区中E430X、E345X、S440Y和S440W,如E430G、 E430S、E430F、E430T、E345K、E345Q、E345R、E345Y、S440Y和S440W 的组,并且其中,
第二变体不包含氨基酸残基中的一个或多个突变,所述突变选自对应于人类IgG1重链Fc区中E430X、E345X、S440Y和S440W,如E430G、E430S、 E430F、E430T、E345K、E345Q、E345R、E345Y、S440Y和S440W的组。
在一个实施方案中,组合物包含至少一种分子,其包含至少一个免疫球蛋白的CH2-CH3域和根据本发明的变体,其中所述分子包含一个或多个氨基酸残基中的突变,所述突变选自对应于人类IgG1重链的Fc区中的E430X、 E345X、S440Y和S440W、如E430G、E430S、E345K和E345Q的组。
在实施方案中所描述的分子可以称为“仅Fc分子”,并且可以进一步包含例如铰链区。然而,可以不包含此种铰链区。
可以将包含仅Fc分子和根据本发明的任意变体的组合物应用于成像诊断方法,或用于调节与细胞表面结合时变体的亲合力。
仅Fc分子可以进一步包含在对应于人类IgG1重链Fc区的K439和/或S440 的氨基酸残基中的进一步突变,条件是S440的突变不是S440Y或S440W,并且如果第一突变是S440Y或S440W,那么进一步的突变在对应于人类IgG1重链的Fc区中的K439的氨基酸残基中。
在另一个实施方案中,(i)所述第一变体进一步包含在对应于人类IgG1重链Fc区中的K439的位置中的突变;和
(ii)所述第二变体进一步包含在对应于人类IgG1重链Fc区中的S440的位置中的突变,条件是突变不是S440Y或S440W;或
其中步骤(i)和(ii)备选地可为
(iii)所述第一变体进一步包含在对应于人类IgG1重链Fc区中的S440的位置中的突变,条件是突变不是S440Y或S440W,和
(iv)所述第二变体进一步包含在对应于人类IgG1重链Fc区中的K439的位置中的突变。
在一个实施方案中,在人类IgG1重链Fc区中K439位置上的突变是 K439D/E,并且在人类IgG1重链Fc区中S440位置上的突变S440K/R。
在进一步的实施方案中,本发明涉及如本文所定义的组合物,其中
(i)第一变体进一步包含前药,和
(ii)第二变体包含用于在第一变体上的前药的活化剂;或者
其中(i)和(ii)备选地可以是
(ⅲ)第二变体包含前药,和
(iv)第一变体包含用于在第二变体上的前药的活化剂。
术语“前药”根据本发明理解为一种相对非细胞毒性的药物前体,其在成为活性药理(抗癌)剂之前必须经历化学转化(例如通过代谢过程)。前药的实例和制备前药的方法是本领域公知的。一个实例是包含酶前药的抗体组合,其中通过与前药缀合的抗体结合和与用于所述前药的活化剂缀合的抗体结合相同细胞上存在的其抗原靶标提供药物递送。这使得前药和其活化剂彼此接近,由此局部释放药物,进而能够例如穿透周围的细胞,并杀死这些细胞。(Senter和Springer,2001Adv Drug Deliv Rev.2001Dec 31;53(3):247-64,Senter,1994FASEB J.1990Feb 1;4(2):188-93)。
术语“前药的活化剂”根据本发明理解为能够将前药转化为活性药物的分子。前药的活化剂的实例和制备这些的方法是本领域公知的。活化剂的实例可以是酶,其表现为用于将前药转化成活性药物的催化剂(Senter和 Springer,2001Adv Drug DelivRev.2001Dec 31;53(3):247-64,Senter,1994 FASEB J.1990Feb 1;4(2):188-93)。
在一个实施方案中,第一和/或第二亲本多肽是各自包含免疫球蛋白的 Fc域和抗原结合区的第一和第二亲本抗体。
在一个实施方案中,第一和第二抗体各是人类IgG1、IgG2、IgG3、IgG4、 IgA1、IgA2、IgD或IgE抗体,任选的各是人全长抗体,例如各是人全长IgG1 抗体。
在一个实施方案中,第一和第二抗体各选自单特异性抗体、双特异性抗体或多特异性抗体。
在进一步的实施方案中,第一和/或第二亲本抗体各是包含第一多肽和第二多肽的双特异性抗体,所述第一多肽包含免疫球蛋白的第一CH2-CH3区和第一抗原结合区,所述第二多肽包含免疫球蛋白的第二CH2-CH3区和第二抗原结合区,其中第一和第二抗原结合区结合相同抗原或不同抗原上的不同表位,其中所述第一CH2-CH3区包含在选自对应于人类IgG1重链Fc区K409、 T366、L368、K370、D399、F405和Y407的那些位置的位置中的进一步氨基酸突变;其中所述第二CH2-CH3区在选自对应于人类IgG1重链Fc区F405、 T366,L368,K370,D399,Y407和K409的那些位置的位置中的进一步氨基酸突变,并且其中在所述第一CH2-CH3区的进一步氨基酸突变与所述第二 CH2-CH3区的进一步氨基酸突变不同。
在优选的实施方案中,第一CH2-CH3区的进一步氨基酸突变在对应于人类IgG1重链Fc区K409,如K409R的位置;并且所述第二CH2-CH3区的进一步氨基酸突变在对应于人类IgG1重链Fc区F405,如F405L的位置。
在一个实施方案中,组合物的第一和第二变体结合相同或不同抗原上的不同表位。
在一个实施方案中,第一变体和第二变体中的一种或两种与药物、毒素或放射性标记缀合,例如其中第一变体和第二变体中的一种或两种通过接头与毒素缀合。
在一个实施方案中,第一变体和第二变体中的一种或两种是融合蛋白的一部分。
在一个具体的实施方案中,组合物的第一变体和第二变体仅包含一个突变。
在一个实施方案中,其中第二变体不包含本发明所述的任意所列突变,这种第二变体可包含涉及提高CDC的方法的上文列举的任意适合第二抗体实例。
在一个实施方案中,第一和第二变体中至少一个第一突变是不同的。
在一个实施方案中,第一变体和第二变体各自是人类IgG1,IgG2,IgG3, IgG4,IgA1,IgA2,IgD,IgM或IgE抗体,任选的各自是人全长抗体,如各自是人全长IgG1抗体。
在一个实施方案中,第一变体和第二变体各自选自单特异性抗体、双特异性抗体或多特异性抗体。
在进一步的实施方案中,第一和第二变体结合相同抗原或不同抗原上的不同表位。因此,在其中第一和第二抗体是双特异性抗体的实施方案中,它们可以各自结合两个不同表位。至少两种双特异性抗体可以是相同或不同的。如果双特异性抗体是不同的,则组合物包含靶向相同或不同靶标上直至 4种不同表位。
在另一个方面,本发明涉及一种包含本文所述任意变体、任意双特异性抗体或任意组合物和药学上可接受的载体的组合物。
预期根据“混合突变体”方面的任意实施方案也可以包含在任意组合物实施方案中。
在一个实施方案中,第一和第二亲本抗体的变体结合在相同细胞上表达的抗原。
在另一个实施方案中,第一亲本抗体的变体包含K439到选自E和D的氨基酸的氨基酸取代。
在另一个实施方案中,第一亲本抗体的变体的氨基酸取代是K439E。
在另一个实施方案中,第二亲本抗体的变体包含S440到选自K和R的氨基酸的氨基酸取代。
在另一个实施方案中,第二亲本抗体的变体的氨基酸取代是S440K。
在另一个方面,本发明涉及一种药物组合物,其包含上文所列任一实施方案的第一亲本多肽或亲本抗体的变体和第二多肽或亲本亲本抗体的变体。
药物组合物可以根据常规技术进行配制,例如公开于Remington:The Scienceand Practice of Pharmacy,第19版,Gennaro,Ed.,Mack Publishing Co., Easton,PA,1995的。本发明的药物组合物可以例如包含稀释剂,填充剂,盐,缓冲剂,去污剂(例如,非离子型去污剂,如Tween-20或Tween-80),稳定剂 (例如,糖或无蛋白氨基酸),防腐剂,等张剂,抗氧化剂,组织固定剂,增溶剂,和/或适于包含在药物组合物中的其他材料。可用于本发明药物组合物的合适水性和非水性载体的实例包含水,盐水,磷酸盐缓冲盐水,乙醇,右旋糖,多元醇(例如甘油,丙二醇,聚乙二醇)。
可以通过任意合适的途径和方式施用药物组合物。在一个实施方案中,肠胃外施用本发明的药物组合物。本发明使用的术语“肠胃外施用”表示除了肠和表面施用以外的施用方式,通常通过注射,并且包含表皮,静脉内,肌内,动脉内,鞘内,囊内,眶内,心内,皮内,腹腔内,腱内,经气管,皮下,表皮下,关节内,包囊下,蛛网膜下,脊柱内,颅内,胸内,硬膜外和胸骨内的注射和输注。
套组(Kit-of-parts)
应理解本发明中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案也可适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
本发明还涉及治疗中同时、分开或连续使用的套组,其包含亲本多肽和亲本抗体的变体,其中亲本多肽和亲本抗体的任意变体可以如本发明所述。具体方面和实施方案将在下文描述。此外,可以根据本发明描述的任意方法获得这种变体。
在一个方面,本发明涉及一种在治疗中同时、分开或连续使用的套组,其包含亲本多肽的第一变体和亲本多肽的第二变体,其中第一变体包含一个或多个突变,其选自对应于人类IgG1重链Fc区中E430X、E345X、S440Y和 S440W,如E430G、E430S、E430F、E430T、E345K、E345Q、E345R、E345Y、 S440Y和S440W的组,条件是变体不含有Fc域中改变变体与新生儿Fc受体 (FcRn)结合的任何其他突变,并且其中
(i)所述第一变体包含对应于人类IgG1重链Fc区的K439的位置中的突变,并且所述第二变体包含对应于人类IgG1重链Fc区中的S440的位置中的突变,条件是S440的突变不是S440Y或S440W,
(ii)所述第一变体包含对应于人类IgG1重链Fc区的K447D/E的位置中的突变;并且所述第二变体包含对应于人类IgG1重链Fc区的K447K/R/H和448P 的位置中的突变,或
(iii)所述第一变体包含对应于人类IgG1重链Fc区的K447D/E的位置中的突变;并且所述第二变体包含对应于人类IgG1重链Fc区的K447K/R/H、 448K/R/H和449P的位置中的突变。
在一个实施方案中,亲本多肽的第一变体和亲本多肽的第二变体中的一种或两种可以是包含免疫球蛋白Fc域和抗原结合区的抗体。
在一个实施方案中,对应于人类IgG1重链Fc区的K439的位置中的突变是 K439D/E,和/或对应于人类IgG1重链Fc区的S440的位置中的突变是S440K /R。
在另一个方面,本发明涉及一种治疗中的同时、分开或连续使用的套组,其包含包含免疫球蛋白的Fc域和结合区的亲本多肽的第一变体和包含免疫球蛋白的Fc域和结合区的亲本多肽的第二变体,其中
变体包含一个或多个突变,其选自对应于人类IgG1重链Fc区中E430X、 E345X、S440Y和S440W,如E430G、E430S、E430F、E430T、E345K、 E345Q、E345R、E345Y、S440Y和S440W的组,条件是变体不含有Fc域中改变变体与新生儿Fc受体(FcRn)结合的任何其他突变,并且其中,
第二变体不含有氨基酸残基中的突变,所述突变选自对应于人类IgG1 重链的Fc区中E430X、E345X、S440Y和S440W,如E430G、E430S、E430F、 E430T、E345K、E345Q、E345R、E345Y、S440Y的组。
在一个实施方案中,其中第二变体不包含本文所述的任意所列突变,这种第二变体可包含涉及效应子功能的方法的上文列举的任意适合第二抗体的实例。
在一个实施方案中,第一和第二变体中的至少一个第一突变是不同的。
在一个实施方案中,第一变体和第二变体各自是人类IgG1,IgG2,IgG3, IgG4,IgA1,IgA2,IgD,IgM或IgE抗体,任选的各自是人全长抗体,例如各自是人全长IgG1抗体。
在一个实施方案中,第一变体和第二变体各自选自单特异性抗体、双特异性抗体或多特异性抗体。
在进一步的实施方案中,第一和第二变体结合相同抗原或不同抗原上的不同表位。因此,在其中第一和第二抗体是双特异性抗体的实施方案中,它们可以各自结合两个不同表位。至少两种双特异性抗体可以是相同或不同的。如果双特异性抗体是不同的,则治疗中同时、分开或连续使用的套组包含靶向相同或不同靶标上的多至4个不同表位。
在进一步的实施方案中,第一变体和第二变体中的一种或两种与药物、毒素或放射性标记缀合,例如其中第一变体和第二变体中的一种或两种与毒素通过接头缀合。
在进一步的实施方案中,第一变体和第二变体中的一种或两种是融合蛋白的一部分。
预期根据“混合突变体”方面的任意实施方案可以包含在治疗中同时、分开或连续使用的任意套组中。
在一个实施方案中,第一和第二亲本抗体的变体结合在相同细胞上表达的抗原。
在另一个实施方案中,第一亲本抗体的变体包含K439到选自E和D的氨基酸的氨基酸取代。
在另一个实施方案中,第一亲本抗体的变体中的氨基酸取代是K439E。
在另一个实施方案中,第二亲本抗体的变体包含S440到选自K和R的氨基酸的氨基酸取代。
在另一个实施方案中,第二亲本抗体变体的变体中的氨基酸取代是 S440K。
在另一个方面,本发明涉及一种治疗中同时、分开或连续使用的药物套组,其包含上文所列任一种实施方案的第一亲本多肽或亲本抗体的变体和第二亲本多肽或亲本抗体的变体。
治疗中同时、分开或连续使用的药物套组可以通过任意合适的途径和方式进行施用。在一个实施方案中,本发明的治疗中同时、分开或连续使用的药物套组是肠胃外施用的。本发明使用的术语“肠胃外施用”表示除了肠和表面施用以外的施用方式,通常通过注射,并且包含表皮,静脉内,肌内,动脉内,鞘内,囊内,眶内,心内,皮内,腹腔内,腱内,经气管,皮下,表皮下,关节内,包囊下,蛛网膜下,脊柱内,颅内,胸内,硬膜外和胸骨内的注射和输注。
组合
此外,本发明提供一种如上所述的任意“单突变体”方面或实施方案的变体的制剂,即,包含多拷贝变体的制剂。本发明还提供包含如上所述的任意“单突变体”方面和实施方案的变体的组合物,例如,药物组合物。本发明还提供任意这种“单突变体”变体、制品或组合物作为药物的用途。
本发明还提供变体的组合,其中一种变体包含根据本发明的至少一种突变,和一种变体包含根据本发明的至少一种另一突变,以及这种变体组合的制品和药物组合物以及它们作为药物的用途。优选的是,两种变体结合通常在相同细胞、细胞膜、病毒粒子和/或其他颗粒的表面上表达的相同抗原或不同抗原。
缀合物
应理解本发明中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案也可适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
在一个方面,本发明涉及一种变体,其中所述变体与药物,毒素或放射性标记缀合,例如其中变体与毒素通过接头缀合。
在一个实施方案中,所述变体是融合蛋白的一部分。
在另一个方面,本发明的变体不在C端与另一分子(例如毒素或标记)缀合。在一个实施方案中,变体与另一分子在另一位点(通常在不干扰寡聚体形成的位点)缀合。例如,抗体变体可以在另一位点与选自毒素(包含放射性同位素)、前药或药物的化合物连接。这种化合物可以使靶细胞的杀伤更有效,例如在癌症治疗中。因此所获变体是免疫缀合物。
因此,在进一步的方面,本发明提供一种与一种或更多种治疗部分(例如细胞毒素,化疗药物,细胞因子,免疫抑制剂和/或放射性同位素)连接或缀合的抗体。这种缀合物在本发明中被称为“免疫缀合物”或“药物缀合物”。包含一种或更多种细胞毒素的免疫缀合物被称作“免疫毒素”。
细胞毒素或细胞毒剂包含对细胞有害的(例如,杀伤细胞)的任意药剂。用于形成本发明免疫缀合物的合适治疗剂包含泰素(taxol),松胞菌素 (cytochalasin)B,短杆菌肽(gramicidin)D,溴化乙啶,依米丁(emetine),丝裂霉素(mitomycin),依托泊苷(etoposide),替尼泊苷(tenoposide),长春新碱 (vincristine),长春碱(vinblastine),秋水仙碱,多柔比星,柔红霉素 (daunorubicin),二羟基炭疽菌素二酮(dihydroxyanthracin dione),美坦辛 (maytansine)或其类似物或衍生物,烯二炔(enediyene,enediyne)抗肿瘤抗生素,包含新制癌菌素(neocarzinostatin)在内,刺孢霉素(calicheamycins),埃斯波霉素(esperamicins),蒽环类抗生素(dynemicins),lidamycin,kedarcidin或其类似物或衍生物,anthracyclins,米托蒽醌(mitoxantrone),光辉霉素(mithramycin),放线菌素D(actinomycin D),1-去氢睾酮,糖皮质激素(glucocorticoids),普鲁卡因(procaine),丁卡因(tetracaine),利多卡因 (lidocaine),普萘洛尔(propranolol),和嘌呤霉素(puromycin),抗代谢物(例如甲氨蝶呤,6-巯基嘌呤,6-硫代鸟嘌呤,阿糖胞苷(cytarabine),氟达拉滨 (fludarabin,fludarabine),5-氟尿嘧啶,氨烯咪胺(decarbazine),羟基脲,天冬酰胺酶(asparaginase),吉西他滨(gemcitabine),克拉屈滨(cladribine)),烷化剂(例如氮芥(mechlorethamine),thioepa,苯丁酸氮芥(chlorambucil),美法仑 (melphalan),卡莫司汀(carmustine)(BSNU),洛莫司汀(lomustine)(CCNU),环磷酰胺(cyclophosphamide),白消安(busulfan),二溴甘露醇(dibromomannitol),链脲霉素(streptozotocin),氮烯唑胺(dacarbazine)(DTIC),甲基苄肼 (procarbazine),丝裂霉素C,顺铂和其他铂衍生物,例如卡铂(carboplatin);以及多卡米星(duocarmycin)A,多卡米星(duocarmycin)SA,CC-1065(亦称为rachelmycin),或CC-1065的类似物或衍生物,多拉司他汀(dolastatin),吡咯并[2,1-c][1,4]苯二氮卓(pyrrolo[2,1-c][1,4]benzodiazepin)(PDBs)或其类似物,抗生素(例如更生霉素(dactinomycin)(从前为放线菌素),博来霉素 (bleomycin),柔红霉素(从前为道诺霉素(daunomycin)),多柔比星 (doxorubicin),伊达比星(idarubicin),光辉霉素,丝裂霉素,米托蒽醌 (mitoxantrone),普卡霉素(plicamycin),氨茴霉素(anthramycin)(AMC)),抗有丝分裂剂(例如,微管蛋白抑制剂)例如一甲基阿里他汀E(monomethyl auristatin E),一甲基阿里他汀F,或多拉司他汀(dolastatin)10的其他类似物或衍生物;组蛋白去乙酰化酶抑制剂例如异羟肟酸曲古抑菌素A(hydroxamic acids trichostatin A),伏力诺他(vorinostat)(SAHA),贝林司他(belinostat), LAQ824,和帕比司他(panobinostat)以及苯甲酰胺(benzamides),恩替司他 (entinostat),CI994,莫西司他(mocetinostat)和脂肪酸化合物,例如苯丁酸 (phenylbutyrate)和丙戊酸(valproic acid),蛋白酶体抑制剂例如Danoprevir,硼替佐米(bortezomib),毒伞肽(amatoxins)例如α-amantin,白喉毒素和相关分子(例如白喉A链及其活性片段和杂合分子);蓖麻毒蛋白毒素(例如蓖麻毒蛋白A或去糖基化蓖麻毒蛋白A链毒素),霍乱毒素,志贺样毒素(SLT-I,SLT-II, SLT-IIV),LT毒素,C3毒素,志贺毒素,百日咳毒素,破伤风毒素,大豆 Bowman-Birk蛋白酶抑制剂,假单胞菌外毒素,alorin,皂草素(saporin),蒴莲根毒素(modeccin),gelanin,相思豆毒蛋白A链,蒴莲根毒素A链,α-sarcin,油桐(Aleurites fordii)蛋白,石竹素(dianthin)蛋白,美洲商陆(Phytolacca americana)蛋白(PAPI,PAPII,和PAP-S),苦瓜(momordica charantia)抑制剂,麻疯树毒蛋白(curcin),巴豆毒蛋白,石碱草(sapaonaria officinalis)抑制剂,白树毒素(gelonin),mitogellin,局限曲菌素(restrictocin),酚霉素(phenomycin),和伊诺霉素(enomycin)毒素。其他合适的缀合分子包含抗微生物/裂解肽例如 CLIP,爪蟾抗菌肽(Magainin)2,蜂毒素(mellitin),杀菌肽,和P18;核糖核酸酶(RNase),DNase I,葡萄球菌肠毒素-A,美洲商陆(pokeweed)抗病毒蛋白,白喉毒素,和假单胞菌属内毒素。参见,例如,Pastan等,Cell 47,641(1986) 和Goldenberg,Calif.A Cancer Journal for Clinicians44,43(1994)。可以与本发明别处描述的本发明抗体组合施用的治疗剂(如,例如,抗癌细胞因子或趋化因子)也可以是可用于缀合本发明抗体的治疗性部分的候选物。
在一个实施方案中,本发明的药物缀合物包含与阿里他汀或阿里他汀肽类似物和衍生物(US5635483;US5780588)缀合的本发明公开的抗体。阿里他汀已经显示干扰微管动力学,GTP水解以及核和细胞分裂(Woyke等(2001) Antimicrob.Agents和Chemother.45(12):3580-3584)并具有抗癌(US5663149) 和抗真菌活性(Pettit等,(1998)Antimicrob.Agents和Chemother.42:2961- 2965。阿里他汀药物部分可以利用接头,通过肽性药物部分的N(氨基)端或 C(端)粘附到抗体。
示例性阿里他汀实施方案包含N端连接的一甲基阿里他汀药物部分DE 和DF,公开于Senter等.,Proceedings of the American Association for Cancer Research.45卷,摘要编号623,2004年3月28出版并描述于US 2005/0238649。
示例性的阿里他汀实施方案是MMAE(一甲基阿里他汀E)。另一个示例性的阿里他汀实施方案是MMAF(一甲基阿里他汀F)。
在一个实施方案中,本发明的抗体包含缀合的核酸或核酸关联分子。在这种实施方案中,缀合的核酸是细胞毒性核糖核酸酶,反义核酸,抑制性RNA 分子(例如,siRNAs分子)或免疫刺激性核酸(例如,包含CpG模体的免疫刺激性DNA分子)。在另一个实施方案中,本发明的抗体与适体或核酶缀合。
在一个实施方案中,提供包含一种或更多种放射性标记的氨基酸的抗体。放射性标记的变体可用于诊断和治疗两种目的(与放射性标记分子的缀合是另一个可能的特征)。用于多肽的标记的非限制性实例包含3H,14C,15N, 35S,90Y,99Tc和125I,131I,和186Re。用于制备放射性标记的氨基酸和相关肽衍生物的方法是本领域已知的(参见,例如,Junghans等,Cancer Chemotherapy and Biotherapy 655-686(第二版.,Chafner和Longo,eds., Lippincott Raven(1996))和U.S.4,681,581,U.S.4,735,210,U.S.5,101,827,U.S.5,102,990(US RE35,500),U.S.5,648,471和U.S.5,697,902。例如,可以通过氯胺T法缀合放射性同位素。
在一个实施方案中,本发明的变体与放射性同位素或包含放射性同位素的螯合物缀合。例如,变体可以与螯合剂接头,例如DOTA、DTPA或tiuxetan 缀合,这允许抗体与放射性同位素形成复合物。变体也可以或可选包含一种或更多种放射性标记的氨基酸或其他放射性标记的分子,或与一种或更多种放射性标记的氨基酸或其他放射性标记的分子缀合。放射性标记的变体可用于诊断和治疗两种目的。在一个实施方案中,本发明的变体与α发射体缀合。放射性同位素的非限制性实例包含3H,14C,15N,35S,90Y,99Tc,125I,111In,131I, 186Re,213Bs,225Ac和227Th。
在一个实施方案中,本发明的变体可以与选自下组的细胞因子缀合:IL-2, IL-4,IL-6,IL-7,IL-10,IL-12,IL-13,IL-15,IL-18,IL-23,IL-24,IL-27,IL-28a, IL-28b,IL-29,KGF,IFNa,IFN,IFNy,GM-CSF,CD40L,Flt3配体,干细胞因子,安塞司亭(ancestim)和TNFα。
本发明的变体也可以通过共价缀合到聚合物进行化学修饰以例如增加它们的循环半衰期。示例性的聚合物,以及将它们附着到肽的方法,显示于例如US 4,766,106,US 4,179,337,US 4,495,285和US 4,609,546。其他的聚合物包含聚氧乙烯多元醇和聚乙二醇(PEG)(例如,分子量为约1,000-约40,000,例如约2,000-约20,000的PEG)。
可以使用用于将本发明的变体缀合到缀合分子的本领域已知的任何方法,例如如上所述的那些,包含以下描述的方法:Hunter等,Nature 144,945 (1962),David等,Biochemistry 13,1014(1974),Pain等,J.Immunol.Meth.40, 219(1981)和Nygren,J.Histochem.and Cytochem.30,407(1982)。可以通过将另一部分化学缀合到变体或其片段(例如,抗体H或L链)的N端侧或C端侧来产生这种变体(参见,例如,AntibodyEngineering Handbook,Osamu Kanemitsu 编辑,Chijin Shokan出版(1994))。合适情况下,通过在内部残基或糖处进行缀合也可以产生这种缀合的变体衍生物。
药剂可以直接或间接偶联到本发明的变体。第二药剂的间接偶联的一个实例是通过间隔物或接头部分偶联到双特异性抗体中的半胱氨酸或赖氨酸残基。在一个实施方案中,将变体通过间隔物或接头缀合到前药分子,所述前药分子可以在体内被活化为治疗药物。在一些实施方案中,接头在胞内条件下是可切割的,从而接头的切割在胞内环境中从抗体释放药物单元。在一些实施方案中,接头是通过可切割剂可切割的,所述可切割剂存在于胞内环境(例如,在溶酶体或内体或小窝内)。例如,间隔物或接头是通过肿瘤细胞相关酶或其他肿瘤特异性条件可切割的,借此形成活性药物。这种前药技术和接头的实例描述于Syntarga BV等的WO02083180,WO2004043493, WO2007018431,WO2007089149,WO2009017394和WO201062171。合适的抗体前药技术和多卡米星(duocarmycin)类似物还可以见于美国专利号 6,989,452(Medarex),此处引入作为参考。接头还可以或可选的是,例如肽基接头,其被胞内肽酶或蛋白酶切割,包含但不限于,溶酶体或胞内体蛋白酶。在一些实施方案中,肽基接头的长度为至少两个氨基酸或至少三个氨基酸。切割剂可以包含组织蛋白酶B和D以及纤溶酶(plasmin),已知它们全部水解二肽药物衍生物,导致靶细胞内活性药物的释放(参见例如Dubowchik和 Walker,1999,Pharm.Therapeutics 83:67-123)。在一个具体实施方案中,可被胞内蛋白酶切割的肽基接头是Val-Cit(缬氨酸-瓜氨酸)接头或Phe-Lys(苯丙氨酸-赖氨酸)接头(参见例如US6214345,其描述了具有Val-Cit接头的多柔比星的合成以及Phe-Lys接头的不同实例)。Val-Cit和Phe-Lys接头结构的实例包含但不限于下文描述的MC-vc-PAB,MC-vc-GABA,MC-Phe-Lys-PAB或 MC-Phe-Lys-GABA,其中MC是马来酰亚胺基己酰基(maleimido caproyl)的缩写,vc是Val-Cit的缩写,PAB是对氨基苄基氨基甲酸酯 (p-aminobenzylcarbamate)和GABA是γ-氨基丁酸的缩写。使用胞内蛋白水解释放治疗剂的优点是,药剂在缀合时通常是减弱的的并且缀合物的血清稳定性通常较高。
仍在另一个实施方案中,接头单元是不可切割的,并且药物通过抗体降解来释放(参见US 2005/0238649)。通常,这种接头实质上对胞外环境不敏感。如本发明使用的“实质上对胞外环境不敏感”在接头的背景下,表示当变体抗体药物缀合化合物存在于胞外环境(例如血浆)时,变体抗体药物缀合化合物的样品中不超过20%,通常不超过约15%,更通常不超过约10%,和甚至更通常不超过约5%,不超过约3%,或不超过约1%的接头被切割。通过将变体抗体药物缀合化合物与血浆温育预定时间段(例如2,4,8,16或24小时),然后定量血浆中存在的游离药物量,可以确定接头是否实质上对胞外环境不敏感。包含MMAE或MMAF和各种接头组分的示例性实施方案具有下列结构 (其中Ab表示抗体,和p表示药物载量(或每个抗体分子的细胞抑制或细胞毒性药物的平均数)是1到约8,例如p可为4-6,如3-5,或p可为1,2,3,4,5, 6,7或8)。
可切割接头与阿里他汀组合的实例包含MC-vc-PAB-MMAF(也称为 vcMMAF)和MC-vc-PAB-MMAF(也称为vcMMAE),其中MC是马来酰亚胺基己酰基的缩写,vc是基于Val-Cit(缬氨酸-瓜氨酸)接头的缩写,和PAB是对- 氨基苄基氨基甲酸酯的缩写。
其他实例包含与不可切割接头组合的阿里他汀,例如mcMMAF(mc(MC 与本上下文的mc相同)是马来酰亚胺基己酰基的缩写。
在一个实施方案中,药物接头部分是vcMMAE。vcMMAE药物接头部分和缀合方法公开于WO2004010957,US7659241,US7829531,US7851437和US 11/833,028(SeattleGenetics,Inc.),(其在此引入作为参考),和使用类似于其中公开的方法的方法将vcMMAE药物接头部分与抗体在半胱氨酸处结合。
在一个实施方案中,药物接头部分是mcMMAF。mcMMAF药物接头部分和缀合方法公开于US7498298,US 11/833,954和WO2005081711(Seattle Genetics,Inc.),(其在此引入作为参考),和使用类似于其中公开的方法的方法将mcMMAF药物接头部分与变体在半胱氨酸处结合。
在一个实施方案中,本发明的变体附着到螯合剂接头,例如tiuxetan,其允许双特异性抗体缀合到放射性同位素。
在一个实施方案中,变体的每个臂(或Fab-臂)与相同的一种或更多种治疗部分直接或间接偶联。
在一个实施方案中,只有变体的一个臂与一种或更多种治疗部分直接或间接偶联。
在一个实施方案中,变体的每个臂与不同的治疗部分直接或间接偶联。例如,在变体是双特异性抗体并通过两种不同单特异性抗体(例如本发明描述的第一和第二抗体)的受控Fab-臂交换来制备的实施方案中,可以通过使用与不同治疗部分缀合或结合的单特异性抗体来获得这种双特异性抗体。
进一步的用途
应理解本发明中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案也可以适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
在进一步的方面,本发明涉及如上所述的本发明变体,用作药物,具体来说用作治疗疾病或病症的药物,其中期望对靶细胞(例如,肿瘤,细菌或真菌细胞)或靶标生物体(例如,病毒)或细菌或病毒感染细胞的CDC介导的杀伤。这种疾病和病症的实例包含,但不限于,癌症和细菌性、病毒性或真菌性感染。
在另一个方面,本发明涉及用于治疗疾病例如癌症的本发明所述变体,双特异性抗体,组合物和套组。
在另一个方面,本发明涉及一种用于治疗人的方法,包括施用本发明描述的变体,组合物或套组。
在另一个方面,本发明涉及一种治疗人类癌症的方法,包括施用本发明描述的变体,组合物或套组。
“治疗”是指施用有效量的本发明治疗活性化合物,目的是减轻,改善,阻滞或清除(治愈)症状或疾病状态。
“有效量”或“治疗有效量”是在必要的剂量和时段对于实现期望治疗结果有效的量。抗体的治疗有效量可以根据各种因素而不同,例如个体的疾病状况,年龄,性别和体重,及所述抗体在个体中引发期望应答的能力。治疗有效量还是抗体或抗体部分的任何毒性或不利作用被治疗有益作用超过的量。
在另一个方面,本发明涉及根据本文所述的任一实施方案的变体、组合物或套组在诊断方法中使用的用途。
在另一个方面,本发明涉及一种诊断方法,其包括将根据本文所述的任一实施方案的变体、组合物或套组施用于人类或其他哺乳动物的身体的至少一部分。
在另一个方面,本发明涉及根据本文所述的任一实施方案的变体、组合物或套组用于对人类或其他哺乳动物的身体的至少一部分成像的用途。
在另一个方面,本发明涉及一种对人类或其他哺乳动物的身体的至少一部分成像的方法,其包括施用根据本文所述的任一实施方案的变体、组合物或套组。
不受理论限制,当将根据本发明的任意“单突变体”方面或实施方案引入这种治疗活性化合物时,可以降低治疗活性化合物的有效量。
用于癌症抗体的合适抗原可以是相同的,如本发明描述的。实施例15到 18描述了提供增强和/或更特异性的肿瘤细胞的补体激活或CDC的具体应用。例如,包含例如E345R突变的根据“单突变体方面”的抗肿瘤抗体可以提供肿瘤细胞的增强的CDC或ADCC、ADCP应答。此外,在这种方法的变体中,根据“单突变体”方面的突变,如例如E345R,E430或S440S/W,或表1所列的任意其他突变,可以添加到每种抗体中,从而提供特异性针对表达至少两种抗原的肿瘤细胞的增强的CDC和/或ADCC应答。
用于细菌性感染的合适抗体包含,但不限于,靶向金黄色葡萄球菌(例如嵌合单克隆IgG1帕昔单抗(BSYX-A110;Biosynexus)),靶向嵌入葡萄球菌细胞壁中的脂磷壁酸(LTA)并描述于Baker(Nat Biotechnol.2006Dec;24(12): 1491-3)和Weisman等(IntImmunopharmacol.2009May;9(5):639-44)(这两篇文献均在此完整引入作为参考)的那些抗体。实施例14描述了使用包含E345R 突变的金黄色葡萄球菌抗体变体的具体实施方案。但是,表1的其他突变,包含但不限于E430G和S440W,可以按照类似方式应用以增强抗细菌抗原的抗体的CDC介导能力。
病毒或真菌性感染的合适抗原可以是本发明描述的任一项。
在一个实施方案中,与变体结合的抗原不是人EphA2。在另一个实施方案中,变体不是来源于人EphA2 mAb 12G3H 11(描述于Dall'Acqua等,上文,其在此完整引入作为参考)。在另一个实施方案中,与变体结合的抗原不是 IL-9。在另一个实施方案中,变体不是来源于WO2007005612(在此完整引入作为参考)描述的Fa-hG1或Fa-hG4抗体,或其任意变体。在一个实施方案中,与变体结合的抗原不是HIV-1 gp120。在另一个实施方案中,变体不是来源于针对gp120的b12人类IgG1κ抗体。
在一个具体实施方案中,变体来源于双特异性亲本抗体。双特异性抗体可以属于任意同种型,如例如,IgG1,IgG2,IgG3或IgG4,并且可以是全长抗体或其包含Fc的片段。用于制备双特异性抗体的示例性方法描述于WO 2008/119353(Genmab)。
剂量
应理解本发明中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案也可适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。用于抗体的有效剂量和给药方案取决于待治疗的疾病或病征,并且可以由本领域技术人员来确定。本发明抗体的治疗有效量的示例性、非限制性范围是约0.1到100mg/kg,例如约0.1到50mg/kg,例如约0.1到 20mg/kg,例如约0.1到10mg/kg,例如约0.5,约如0.3,约1,约3,约5,或约 8mg/kg。
本发明的抗体变体还可以与一种或更多种补体因子或相关组分组合施用以增强变体的治疗功效和/或弥补补体消耗。这种补体因子和相关组分包含,但不限于,C1q,C4,C2,C3,C5,C6,C7,C8,C9,MBL和因子B。组合施用可以是同时、分开或连续的。在一个具体实施方案中,本发明提供一种试剂盒,其包含包含本发明变体的药物组合物,以及在相同或不同的药物组合物中的至少一种补体因子或相关组分,以及使用说明书。
本发明的抗体变体也可以在组合疗法中施用,即,与待治疗疾病或病征相关的其他治疗剂组合。因此,在一个实施方案中,包含抗体的药物用于与一种或更多种其他治疗剂(例如细胞毒剂、化疗剂或抗血管生成剂)组合。这种组合施用可以是同时、分开或连续的。
在进一步的实施方案中,本发明提供一种用于治疗或预防疾病(例如癌症)的方法,所述方法包括结合放疗和/或外科手术,给需要其的受试者施用治疗有效量的本发明变体或药物组合物。
制备方法
应理解本发明中涉及到亲本抗体,第一亲本抗体或第二亲本抗体描述的所有实施方案也可适用于包含免疫球蛋白Fc域和结合区的其他亲本、第一亲本或第二亲本多肽。
本发明还提供编码上述任一方面的变体的分离的核酸和载体,以及编码所述变体的载体和表达系统。用于抗体及其变体的合适核酸构建体、载体和表达系统是本领域已知的,并在实施例中描述。在变体不仅包含重链(或其包含Fc的片段)而且包含轻链的实施方案中,编码重链和轻链部分的核苷酸序列可以存在于相同或不同的核酸或载体上。
本发明还提供一种在宿主细胞中产生根据上述任一方面的抗体变体的方法,其中所述变体至少包含重链的Fc区,所述方法包括下列步骤∶
a)提供编码所述变体的所述Fc区的核苷酸构建体,
b)在宿主细胞中表达所述核苷酸构建体,
和
c)从所述宿主细胞的细胞培养物回收所述抗体变体。
在一些实施方案中,抗体是重链抗体。但是,在大部分实施方案中,抗体还将包含轻链,因此所述宿主细胞还表达在相同或不同载体上的轻链编码构建体。
适于抗体重组表达的宿主细胞是本领域公知的,包含CHO,HEK-293, PER-C6,NS/0和Sp2/0细胞。在一个实施方案中,所述宿主细胞是能够进行蛋白Asn-连接糖基化的细胞,例如真核细胞,例如哺乳动物细胞,例如人细胞。在进一步的实施方案中,所述宿主细胞是非人细胞,其被遗传改造以产生具有似人糖基化或人糖基化的糖蛋白。这种细胞的实例是遗传改造的巴斯德毕赤酵母(Pichia pastoris)(Hamilton等,Science 301(2003)1244-1246;Potgieter 等,J.Biotechnology 139(2009)318-325)和遗传改造的浮萍(Lemnaminor) (Cox等,Nature Biotechnology 12(2006)1591-1597)。
在一个实施方案中,所述宿主细胞是不能从抗体重链有效除去C端赖氨酸K447残基的宿主细胞。例如,Liu等(2008)J Pharm Sci 97:2426(在此引入作为参考)的表2列出了大量这种抗体生产系统,例如Sp2/0、NS/0或转基因乳腺(山羊),其中只获得C端赖氨酸的部分去除。在一个实施方案中,宿主细胞是具有改变的糖基化机制的宿主细胞。本领域已经描述了这种细胞并将其用做宿主细胞,在所述宿主细胞中表达本发明的变体从而产生糖基化改变的抗体。参见,例如,Shields,R.L.等(2002)J.Biol.Chem.277:26733-26740; Umana等(1999)Nat.Biotech.17:176-1,以及EP1176195;WO03/035835;和 W099/54342。用于产生改造糖型的其他方法是本领域已知的,并且包含但不限于描述于以下的那些:Davies等,2001,Biotechnol Bioeng 74:288-294; Shields等,2002,J Biol Chem 277:26733-26740;Shinkawa等,2003,J Biol Chem 278:3466-3473),US6602684,WO00/61739A1;WO01/292246A1; WO02/311140A1;WO 02/30954A1;PotelligentTM technology(Biowa,Inc.Princeton,N.J.);GlycoMAbTM糖基化工程技术(GLYCART biotechnology AG, Zurich,Switzerland);US 20030115614;Okazaki等,2004,JMB,336:1239-49。
本发明还涉及通过如上所述的本发明方法获得的或可获得的抗体。
在进一步的方面,本发明涉及一种能够产生本发明抗体变体的宿主细胞。在一个实施方案中,已经用本发明的核苷酸构建体转化或转染宿主细胞。
通过下列实施例进一步说明本发明,实施例不应被理解为进一步的限制。
实施例
实施例1:7D8突变体的设计和产生
将人单克隆抗体HuMab-7D8(描述于WO2004/035607中)用作模式抗体。其属于人抗-CD20IgG1抗体组,包含奥法木单抗(HuMax-CD20,2F2)。这些抗体靶向CD20分子上独特的近膜表位并显示出强烈的CDC。
为了测试寡聚Fc-Fc相互作用在补体激活和CDC中的功能相关性,对位于Fc:Fc界面的疏水片中的氨基酸进行突变,以潜在地破坏7D8的Fc-Fc侧向相互作用和CDC效力。在第一组突变体中(表3),在基于1HZH晶体结构而被选择以及被描述为在CH2-CH3域的疏水片中暴露的位置上引入突变以改变电荷(Burton Mol Immunol 1985 3月;22(3):161-206)。
从第一组突变体中发现,I253D和H433A对7D8的CDC丢失产生了最为强烈的影响(例如,实施例5)。1HZH晶体结构显示,I253和H433与配对抗体的相对Fc位置上的两个不同袋(pocket)相结合。基于这些数据,在晶体结构中的I253和H433位置周围合成了第二组突变以进一步研究位于Fc:Fc侧向界面的残基对于CDC的重要性。潜在地使Fc:Fc界面及随后CDC去稳定化的I253 和H433位置周围的第二组突变列于表4中。
为了排除所观察到的对于CDC的影响是由于对C1q的直接结合位点的破坏的这种可能性,基于两个显示出CDC丢失的单独突变体产生了双突变体,以测试其恢复由单突变体所造成的CDC丢失的能力。该原理图示于图1D中。双突变列于表5中,结构示意图展示在图4和图5中。
使用Quikchange定点诱变试剂盒(Stratagene,美国)来制备突变体。简言之,使用编码所期望突变的正向引物和反向引物来复制编码具有IgG1m(f)同种异型的7D8重链的全长质粒DNA模板。使用DpnI消化所得到的DNA混合物以除去来源质粒DNA,并将其用来转化大肠杆菌(E.coli)。通过DNA测序 (Agowa,德国)来验证从所得到的菌落中分离出来的突变体质粒DNA。使用 293fectin(Invitrogen,美国)基本上如制造商所述将编码抗体重链和轻链两者的质粒DNA混合物瞬时转染至Freestyle HEK293F细胞中(Invitrogen,美国)。
表3:导入7D8的CH2-CH3域的组1突变

(=)无电荷
(-)负电荷
(+)正电荷
(δ+)部分正电荷
表4,:导入7D8的CH2-CH3域的组2突变

(=)无电荷
(-)负电荷
(+)正电荷
(δ+)部分正电荷
表5:双突变导入7D8的CH2-CH3域来组合各自显示CDC 缺失的两个单突变

(=)无电荷
(-)负电荷
(+)正电荷
实施例2:7D8突变体在细胞上对CD20的结合
通过FACS分析来分析纯化的抗体样本与CD20阳性细胞的结合。在Daudi 细胞上测试第一组突变(表3),在Raji细胞上测试第二组突变(表4)。在具有 RPM1640/0.1%BSA中的系列稀释的抗体制备物(对于Daudi上的第一组,范围从0.04至10μg/ml以3倍稀释,对于Raji上的第二组,范围从0.003至10μg/ml 以3倍稀释)的聚苯乙烯96孔圆底平板(Greinerbio-one 650101)中在50μl中将 105个细胞在4℃温育30分钟。在RPM1640/0.1%BSA中洗涤两次之后,将细胞于4℃在具有二抗的100μl中温育30分钟。对于Daudi细胞上的所有实验以及 Raji细胞上使用7D8抗体的实验,都使用缀合有异硫氰酸荧光素(FITC)的兔- 抗-人IgG(F0056,Dako,Glostrup,丹麦;1/100)作为二抗。对于Raji细胞上使用纯化的7D8进行的实验,则使用缀合有R-藻红蛋白(R-PE)-的山羊F(ab’)2 抗-人κ轻链(2062-09,SouthernBiotech;1/500)作为二抗。接着,在 PBS/0.1%BSA/0.02%叠氮化物中洗涤细胞两次,重悬于100μl PBS/0.1%BSA/0.02%叠氮化物中,并在FACS Cantoll(BDBiosciences)上进行分析。使用GraphPad Prism V5.01软件(GraphPad Software,SanDiego,CA,美国)利用非线性回归(具有可变斜率的S形剂量-响应)来分析结合曲线。
7D8抗体对Daudi细胞的结合并没有受到在CH2-CH3域引入点突变的影响,对于所有测试的突变体和野生型7D8都是相同的。另外,相比起野生型 7D8,7D8抗体对Raji细胞的结合没有明显地受到在CH2-CH3域引入点突变的影响,除了E345R外。在高于0.3μg/ml的测试浓度时,在CD20-阳性Raji细胞上检测到IgG1-7D8-E345R的结合减少。在所测试的最高抗体浓度(10μg/ml),对于H433D和H433R也检测到减少的结合。IgG1-7D8-E345R、H433D和H433R的结合减少可以通过二抗表位的遮掩(shielding)所解释,这是由于 E345R和H433R的直接标记导致相似或者甚至提高的对Daudi细胞的结合。亲合力的提高可以通过与野生型IgG1-7D8相比E345R和H433R的升高的Fc-Fc 侧向结合解释。
组合K439E和S440K突变并没有影响7D8抗体对Raji细胞的结合,并且与单突变体和野生型7D8的结合是相同的。
实施例3:7D8突变体的C1q结合ELISA
在ELISA中测试7D8突变体结合C1q,其中经纯化的抗体包被在塑料表面上,引起随机的抗体多聚化。汇集的人血清用作C1q的来源。
用系列稀释于PBS中的抗体(范围0.58-10.0μg/ml以1.5-倍稀释)包被96孔Microlon ELISA板(Greiner,德国),在4℃过夜。用200μl/孔的补充有0.025%吐温20和0.1%明胶的0.5×PBS洗涤和封闭平板。平板依次用3%汇集的人血清 (Sanquin,货号#M0008)在37℃温育1小时,用100μl/孔的兔抗人C1q(DAKO,货号#A0136,1/4.000)在室温下温育1小时,以及用100μl/孔的猪抗兔IgG-HRP (DAKO,P0399,1:10000)作为检测抗体在室温下1小时,在温育之间进行洗涤。用1mg/ml的2,2’-连氮基-双(3-乙基苯并噻唑-6-磺酸)(ABTS;Roche, Mannheim,德国)显影约30分钟。通过加入100μl 2%草酸终止反应。在微板读数器(Biotek,Winooski,VT)中测量405nm处的吸光度。通过使用GraphPad Prism软件通过拟合具有可变斜率的S形剂量响应曲线来分析对数转换数据。每个平板相对于野生型IgG1-7D8标准化突变体的EC50值,并将其乘以所有野生型IgG1-7D8数据的平均值。
如图6和表6中所示,如ELISA中测量的,所测试的点突变对C1q结合具有最小的影响。对于IgG1-7D8-I253D突变体,在ELISA中测量到略微不太有效的C1q结合(更高的EC50值)。测试所有抗体的包被效力,并发现对于所有抗体是相似的。
表6:ELISA中C1q结合的EC50

1从至少3个实验计算平均和SD
2统计:对log转换的数据使用Dunnett's Multiple Comparison Test(GraphPadPrism 5.01)单路ANOVA。与野生型IgG 1-7D8比较计算显著性:(na)不适用,(ns) 不显著(*)p=0.01到0.05(**)p=0.001到0.01(***)p<0.001
实施例4:7D8突变体在细胞上与C1q的结合
将抗体包被在塑料表面上产生抗体结合和Fc-尾部呈递的人造静态系统。因此,在基于细胞的测定中也对补体结合进行测试,其中通过FACS分析来测量C1q与抗体调理的CD20-阳性B细胞的结合。在使用组1突变体的实验中,在冰上将Daudi或Raji细胞悬浮于90μl含有10%FBS的RPMI 1640培养基中(2×106个细胞/ml)。加入10μl系列浓度的C1q(Complement Technologies, Tyler,TX)(终浓度范围在0-60μg/ml和0-140μg/ml之间变化,取决于最大结合)。然后,加入10μl纯化的抗体(10μg/ml的终浓度,即饱和浓度),并立即将反应混合物转移至37℃水浴中,温育1小时。在使用组2突变体的实验中,将测试mAb批量(in bulk)加入到Daudi细胞中,然后向等分试样中加入各种浓度的C1q,如上温育混合物。用PBS/1%BSA洗涤细胞三次,与FITC标记的兔抗 C1q抗体(DakoCytomation,10 ug/ml)室温下温育30分钟。用PBS/1%BSA洗涤细胞并重悬于PBS中或者在PBS中的2%甲醛中固定。在FACSCalibur流式细胞仪(BD Biosciences)上进行流式细胞术,使用校准珠(Spherotech)将平均荧光强度转化为等同可溶性荧光分子(molecules of equivalent solublefluorescence,MESF)。使用 软件(Systat Software Inc.,Washington) 来计算C1q与经指定7D8抗体调理的CD20阳性细胞结合的解离常数(KD值)。从重复结合实验(在Daudi细胞上进行了4次,在Raji细胞中进行了3次)中计算平均KD值,并将其与经野生型7D8调理的细胞上C1q结合的KD值进行对比(表 7和表8)。
软件(Systat Software Inc.,Washington) 来计算C1q与经指定7D8抗体调理的CD20阳性细胞结合的解离常数(KD值)。从重复结合实验(在Daudi细胞上进行了4次,在Raji细胞中进行了3次)中计算平均KD值,并将其与经野生型7D8调理的细胞上C1q结合的KD值进行对比(表 7和表8)。
在Daudi和Raji细胞两者上都对组1突变体进行了测试并给出了相同的结果。与C1q ELISA结果相反,大多数测试的突变体在抗体调理的Daudi(表7A) 和Raji(表8)细胞两者上都显示出降低的C1q结合亲合力(KD增加)。与野生型 7D8相比,在经调理的Daudi或Raji细胞上的C1q结合亲合力上 IgG1-7D8-Q311A和H435A几乎没有显示出降低,I253A、I253Y和N434A显示出更为显著的降低,I253D和H433A则是非常显著的降低。在两种细胞类型上,IgG1-7D8-H435R比野生型7D8显示出稍高的C1q结合亲合力(较低KD),然而,这并不显著。
在Daudi细胞上测试组2突变体。与野生型7D8相比,IgG1-7D8-E345R、 E382R和H433R在经调理的Daudi细胞上显示出提高的结合亲合力,其通过更低的KD值反应出来(表7B)。与野生型7D8相比,所有其他的组2突变体都显示出降低的结合亲合力,G385D、Y436D、Q438D、K439E和S440K显示出急剧增加的KD值(表7B)以及H433D和Y436C显示出急剧降低的结合,从而不能测量到可靠的KD值。
双突变体IgG1-7D8-K439E/S440K在抗体调理的Daudi细胞上显示出恢复 C1q结合,而与野生型7D8相比,两个单突变体都显示出降低的C1q结合。与野生型7D8相比,K439E/S440K双突变体的结合亲合力甚至略微提高(表7C)。单突变体IgG1-7D8-K439E和IgG1-7D8-K440E的混合物能够完全恢复C1q结合,其与野生型7D8的C1q结合相当(表7C)。
IgG1-7D8突变体在ELISA中C1q结合没有变化(实施例3)与基于细胞的测定中受影响的C1q结合之间相互矛盾,显示参与抗体分子之间Fc:Fc相互作用的测试CH3位点不直接影响C1q结合,但是当结合细胞时,却是影响抗体Fc 尾部动力学定位并因此也是影响C1q结合强度的重要决定因素。
表7A:C1q与抗体调理的Daudi细胞结合的KD值(突变体组1)


*与野生型7D8比较(t-检验)
**(na)不适用
表7B:C1q与抗体调理的Daudi细胞结合的KD值(突变体组2)

*与野生型7D8比较(t-检验)
**(na)不适用
***从实验1,2,3,4,10和11计算的7D8的平均KD
****由于这些突变体的结合太弱测量不到可靠的拟合曲线和KD值
表7C:C1q与抗体调理的Daudi细胞结合的KD值(双突变体)

*与野生型7D8比较(t-检验)
**(na)不适用
***从实验1,2,3,4,10和11计算的7D8的平均KD
表8:C1q与抗体调理的Raji细胞结合的KD值(突变体组1)

*与野生型7D8比较(t-检验)
**(na)不适用
实施例5:在CD20阳性Raji细胞上进行CDC测定中7D8突变体的C1q效力
在CDC测定中使用经IgG1-7D8突变体调理的细胞测试C1q效力,以研究所观察到的C1q结合亲合力变化对于CDC活性的影响。因此,使用补充有确定浓度系列的C1q的C1q消减正常人血清来进行CDC测定。在圆底96孔板 (Nunc,Rochester,NY)中,将0.1×106个Raji细胞与10μg/ml纯化的抗体以及系列浓度的人C1q(0.005、0.025、0.1、0.3、1.0、5.0、30.0μg/ml)在总体积为 100μl的补充有0.1%BSA的RPMI1640培养基中室温下预温育15分钟。接着,加入25μl C1q消减的血清(Quidel,Sandiego,CA),在37℃的水浴中温育30 分钟或培养箱中温育45分钟。温育之后,将样品置于冰上终止反应。通过利用碘化丙啶(PI,SigmaAldrich,Zwijndrecht,荷兰)活细胞排斥测定法在FACS 上确定细胞的裂解。裂解百分比(%)如下确定:%裂解=(PI阳性细胞数量/细胞总数)×100%。
将存在30μg/ml C1q时由野生型7D8进行的裂解减去不加入C1q时的裂解设定为100%。利用GraphPad Prism软件通过在对数转换数据上拟合S形剂量响应曲线来计算CH50值(导致50%裂解的C1q浓度)。将突变体的CH50值相对于野生型7D8进行标准化(表9)。
表9中的数据显示,与C1q结合亲合力的测量一致,IgG1-7D8-Q311A、 E382R和H435A在C1q效力上没有显示出降低;I253A、I253Y、G385D、N434A 和Y436C的C1q效力显著降低;I253D、H310K、K322A、H433A、H433D、Y436D、Q438D、K439E和S440K在所有所测试的C1q浓度下几乎完全丧失诱导CDC的能力。
IgG1-7D8-H435R和H433R比起野生型7D8能略微有效地使用C1q,这导致更为有效的CDC。IgG1-7D8-E345R在C1q效力上显示出显著提高,这导致了显著高于野生型7D8的CDC裂解(表9)。
图7显示,组合K439E和S440K突变(这两者作为单一突变体导致CDC的丧失)当这两个突变都被组合在一个分子中(K439E/S440K双突变体)时或当两个单突变体被组合时(K439E+S440K混合)在C1q效力测定中恢复了CDC。
表9:对Raj细胞的CDC检测中C1q效力的CH50


(1)(n)实验数
(2)从所有进行的实验中计算平均值和SD
(3)统计:对log转换的数据使用Dunnett's Multiple Comparison Test(GraphPad Prism 5.01)的单向ANOVA(1way ANOVA)。与野生型IgG1-7D8比较计算显著性:(na)不适用(nd)未测定(ns)不显著(*)p=0.01至0.05(**) p=0.001至0.01(***)p<0.001。
(4)当裂解未达到50%时,将CH50设置为>30μg/mL。
(5)对未达到50%裂解的突变体没能测定P-值。然而,假设这些与 IgG1-7D8-WT显著不同。
实施例6:在CD20阳性细胞上进行CDC测定中7D8突变体的CDC
在圆底96孔板(Nunc,Rochester,NY)中,将0.1×106个细胞与系列浓度的抗体(0.01、0.03、0.1、0.3、1.0、3.0、10.0、30.0μg/ml)在总体积80μl中室温下在摇动器中预温育15分钟。接着,加入20μl正常人血清作为C1q来源(20%终浓度),在37℃的培养箱中温育45分钟。加入30μl补充有0.1%BSA的冰冷的 RPMI培养基终止反应。通过利用碘化丙啶在FACS上确定细胞的裂解。
对于Daudi细胞上的CDC测定,利用GraphPad Prism软件通过在对数转换数据上拟合S形剂量响应曲线来计算EC50值(导致50%裂解的抗体浓度)。将突变体的EC50值相对于野生型7D8进行标准化(表10和表11)。
表10显示,在Daudi细胞上,IgG1-7D8-I253A、Q311A、E382R、H433R 和H435A显示出的CDC与野生型7D8相比没有区别;对IgG1-7D8-I253D、 I253Y、H310K、G385D、H433A、H433D、N434A、Y436C、Y436D、Q438D、 K439E、S440K和I253D/H433A发现比野生型7D8显著更差的CDC(更高的 EC50),它们只在更高的抗体浓度下才产生CDC。C1q结合缺陷型突变体 IgG1-7D8-K322A(其作为对照包含在内)几乎完全失去诱导CDC的能力并且在所测试的浓度下没有达到EC50;IgG1-7D8-H435R在Daudi细胞上显示出比野生型7D8更加有效的CDC。重要的是,与C1q效力CDC测定一致,E345R 在Daudi细胞上显示出的CDC显著好于野生型7D8,EC50值低10倍(表10)。图8显示,组合K439E和S440K突变(这两者作为单一突变体导致CDC的丧失) 在这两个突变都被组合在一个分子(K439E/S440K双突变体)中时或当两个单突变体被组合(K439E+S440K混合)时恢复了CDC。
表11显示在Raji细胞上对IgG1-7D8突变体发现相似的数据。
表10:从对Daudi细胞的CDC测定法中计算的EC50

(1)(n)实验数
(2)从所有进行的实验中计算平均值和SD
(3)统计:对log转换的数据使用Dunnett's Multiple Comparison Test(GraphPad Prism 5.01)的单向ANOVA。与野生型IgG1-7D8比较计算显著性: (na)不适用(nd)未测定(ns)不显著(*)p=0.01至0.05(**)p=0.001至 0.01(***)p<0.001。
(4)当裂解未达到50%时,将EC50设置为>30μg/mL。
(5)对未达到EC50的突变体不能测定P-值。然而,假设这些与野生型 7D8-WT显著不同。
表11:从对Raji细胞的CDC测定法中计算的EC50

(1)(n)实验数
(2)从所有进行的实验中计算平均值和SD
(3)统计:对log转换的数据使用Dunnett's Multiple Comparison Test(GraphPad Prism 5.01)的单向ANOVA。与野生型7D8比较计算显著性:(na)不适用(nd)未测定(ns)不显著(*)p=0.01至0.05(**)p=0.001至0.01(***) p<0.001。
(4)当裂解未达到CH50时,将CH50设置为>30μg/mL。
(5)对未达到EC50的突变体不能测定P-值。然而,假设这些与野生型 7D8-WT显著不同。
实施例7:根据其诱导CDC的能力对7D8突变体的排序
对于所测试的7D8突变体,发现Daudi细胞上的C1q结合(实施例4中所述) 与Raji细胞上的C1q效力测定(实施例5中所述)之间,以及Daudi细胞上的C1q 结合与Daudi和Raji细胞上的CDC测定(实施例6中所述)之间的相关性(相关性数据表13)。因此,使用在Daudi细胞上进行的C1q结合测定的KD值根据其诱导CDC的能力对所有测试的7D8突变体进行排序,如表12所显示。
表12:根据Daudi细胞上的C1q结合的下降KD值(这充当其诱导CDC的能力的代表)对所有检测的7D8突变体排序


*无可靠的拟合曲线。由于这些突变体结合太弱不能测量斜体的KD值。
表13:Daudi细胞上的C1q结合(实施例4)和Raji细胞上的C1q效力测定 (实施例5)之间,以及Daudi细胞上的C1q结合和Daudi细胞以及Raji细胞上的CDC测定(实施例06)之间的关联。关联分析之前数据被log转化。

实施例8:CD38抗体005突变体的设计和产生
人单克隆抗体HuMab 005是全人类IgG1,κ抗体,描述于WO2006/099875 中。在本文中,将其用作验证所鉴定的Fc突变增强CDC活性的模式抗体。所测试的突变列于表14中。
使用具有IgG1m(f)同种异型的HuMab 005的重链作为诱变反应的模板,制备不同突变体的DNA构建体,并进行瞬时转染,如实施例1所述。
表14:在005(HuMax-CD38)的CH2-CH3域中导入的突变组

(=)无电荷
(-)负电荷
(+)正电荷
(δ+)部分正电荷
实施例9:HuMab-005突变体在细胞上对CD38的结合
通过FACS分析来分析未纯化的抗体样本与CD38阳性Daudi和Raji细胞的结合。在聚苯乙烯96孔圆底平板中将105个细胞温育于具有系列稀释的抗体制备物(0.01、0.03、0.1、0.3、1.0、3.0、10.0、30.0μg/ml)的100μl RPM1640/0.1% BSA中,在4℃30分钟。在RPM1640/0.1%BSA中洗涤两次之后,将细胞与缀合有FITC的兔F(ab’)2抗人IgG(货号F0056;DAKO;1:150)温育于50μl中,在 4℃30分钟。接着,在PBS/0.1%BSA/0.02%叠氮化物中洗涤细胞两次,重悬于100μl PBS/0.1%BSA/0.02%叠氮化物中,并在FACS Cantoll(BDBiosciences) 上进行分析。使用GraphPad Prism V5.01软件分析结合曲线。使用模拟转染细胞的上清液作为阴性对照。
HuMab 005对Daudi细胞的结合并没有太多地受到在CH2-CH3域中引入点突变的影响。所有测试的抗体以剂量-依赖性的方式与Daudi细胞结合。所有测试的突变体的结合与野生型HuMab-005相似,除了005-E345R,其显示出稍微降低的结合。然而,不受任何理论束缚,这种较低的结合可能是由于二抗结合的降低所造成的,与实施例2中的IgG1-7D8-E345类似。005-E345的实际结合亲合力可能与005-WT相似或者甚至比005-WT还有所提高,然而由于缺少直接标记的抗体,我们无法对此进行证实。
HuMab-005对Raji细胞的结合也没有太多地受到在CH2-CH3域中引入点突变的影响。所有测试的抗体以剂量-依赖性的方式与Raji细胞结合。 005-I253D和H433A突变体的最大结合与野生型005相似,而005-E435R、 K439E、S440K突变体以及005-K439E+005-S440K的组合却比野生型005低。然而,不受任何理论束缚,这种较低的结合可能是由于二抗结合的降低所造成的,与实施例2中的IgG1-7D8-E345类似(表位被遮蔽)。
实施例10:CD38抗体005突变体在CD38阳性细胞上的CDC测定
在圆底96孔板中,将0.1×106个Daudi或Raji细胞与系列浓度的未纯化抗体(0.01、0.03、0.1、0.3、1.0、3.0、10.0、30.0μg/ml)在总体积100μl中室温下在振荡器中预温育15分钟。接着,加入25μl正常人血清作为C1q来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入 10μl碘化丙啶并通过FACS确定细胞的裂解。
在Wien133细胞上使用不同浓度的正常人血清(NHS)来进一步分析 E435R突变增强CDC的能力,据显示该突变能增强7D8和005抗体两者在 Daudi和Raji细胞上的CDC活性。在圆底96孔板中,将0.1×106个Wien 133细胞与系列浓度的未纯化抗体(0.001、0.003、0.01、0.03、0.1、0.3、1.0、3.0、 10.0、30.0μg/ml)在总体积50μl中室温下在振荡器中预温育15分钟。接着,加入NHS作为C1q的来源使其在100μl体积中的终浓度达到20%或50%NHS。将反应混合物在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
据发现,在CH2-CH3域中导致CD20抗体7D8的丢失或增加CDC活性的鉴定突变对识别CD38的005抗体具有相同的作用。图9显示,005-I253D、H443A、 K439E和S440K在Daudi(图9A)和Raji(图9B)细胞上都显示出CDC活性完全丢失,而005-E345R突变体则在这两个细胞系上均显示出CDC活性强烈增强。 005-K439E+005-S440K(这两者作为单突变体都导致CDC丢失)的组合导致 CDC的恢复,与7D8的数据相当。令人吃惊的是,005-E435R甚至能强有力地诱导对Wien133细胞的CDC,而野生型005却不能够诱导CDC的杀伤(图 9C)。在20%和50%血清浓度下均观察到005-E345R对Wien133细胞的CDC杀伤(图9C)。在50%血清中,7D8-E345R和005-E345R在体外对Raji细胞也显示增强的CDC,这与在20%血清中的效力相似(图9D)。
由于CH2-CH3区中的E345R突变导致所测试的CD20抗体7D8和CD38抗体005两者的增强的CDC活性,因此E345R突变被认为是能够应用于诱导或增强CDC的通用抗体修饰。
实施例11:含有CDC增强型突变E345R的IgG1抗体对通过Fc结合肽 DCAWHLGELVWCT抑制CDC的敏感性小于野生型抗体
通过对IgG的Fc:Fc界面的疏水片中的氨基酸位点进行突变,发现CDC效力要么被破坏,要么被增强。进一步探究了Fc-Fc界面处的相互作用,以及因此可能如b12晶体结构中所观察到的寡聚(例如,六聚环)结构形成参与CDC 效力。因此,使用了靶向在野生型IgG Fc表面上的疏水片区域中的共有结合位点的13个残基的肽(DCAWHLGELVWCT(SEQ ID NO:7))(Delano等, Science 2000Feb 18;287(5456):1279-83)。确实,IgG Fc表面上的共有结合位点作为准备与大量不同分子相互作用的适应性区域的身份(Delano等, Science2000Feb 18;287(5456):1279-83),与IgG1b12晶体结构中参与Fc-Fc 相互作用的疏水片中的核心氨基酸的身份相一致(Saphire等,Science 2001 Aug 10;293(5532):1155-9)。在所有结合界面中存在的相互作用受到6个氨基酸的共享组(Met-252、Ile-253、Ser-254、Asn-434、His-435和Tyr-436)以及共享骨架触点(Delano等,Science 2000Feb 18;287(5456):1279-83)介导。因此,预期Fc结合肽能影响Fc-Fc相互作用,从而影响CDC效力。
在圆底96孔平板中,将0.1×106个Daudi细胞与1.0μg/ml未纯化抗体于75μl 中在摇动器中室温预温育10分钟。将25μl浓度系列(范围从0.06-60μg/ml终浓度)的Fc结合肽DCAWHLGELVWCT加入到经调理的细胞中,在摇动器中室温温育10分钟。接着,加入25μl NHS作为补体来源(20%终浓度),在37℃培养箱中温育45分钟。加入25μl冰冷RPMI培养基(补充有0.1%的BSA)以终止反应。加入15μl碘化丙啶并通过FACS分析确定细胞的裂解。
发现野生型005(图10A)或7D8(图10B)介导的CDC被Fc结合肽 DCAWHLGELVWCT以剂量响应的方式所抑制。这些竞争性数据再次表明位于IgG疏水片的Fc-Fc相互作用掺入CDC效力。相比起其相应的野生型抗体,增强CDC的IgG1-005-E345R和IgG1-7D8-E345R突变体两者对Fc结合肽的竞争都不太敏感,这表明E345R突变导致Fc-Fc相互作用稳定性提高,并因此提高CDC。
实施例12:CD38抗体HuMAb 005变体针对CD38表达细胞的抗体依赖性细胞介导的细胞毒性(ADCC)
收获Daudi细胞(5×106细胞/ml),洗涤(PBS中两次,1200rpm,5分钟)并收集在1ml补充有10%加强型小牛血清(Cosmic Calf Serum,CCS)(HyClone, Logan,UT,美国)的RPMI1640培养基中,所述RPMI1640培养基添加有 200μCi51Cr(铬-51;AmershamBiosciences Europe GmbH,Roosendaal,荷兰)。将混合物温育在37℃振荡水浴中1小时。在洗涤细胞后(PBS中两次,1200rpm, 5分钟),将细胞重悬于补充有10%CCS的RPMI1640培养基中,通过锥虫蓝拒染法计数并稀释至1×106细胞/ml的浓度。
同时,利用标准Ficoll密度离心根据制造商的手册(淋巴细胞分离培养基;Lonza,Verviers,法国)从新鲜血沉棕黄层(buffy coat)(Sanquin,Amsterdam,荷兰)中分离外周血单核细胞(PBMCs)。将细胞重悬于补充有10%CCS的 RPMI 1640培养基之后,通过锥虫蓝拒染法计数细胞,并浓缩至1×107细胞 /ml。
对于ADCC实验,在96孔微量滴定板中将50μl 51Cr-标记的Daudi细胞 (5,000个细胞)与15μg/ml CD38抗体IgG1-005或突变体IgG1-005-E345R温育于总体积为100μl的补充有10%CCS的RPMI培养基中。室温下10分钟后,加入50μl PBMCs(500,000个细胞),使得效应物与靶标的比例为100:1,。通过将 50μl 51Cr-标记的Daudi细胞(5,000个细胞)与100μl5%Triton-X100温育来确定细胞裂解的最大量。将5000个51Cr-标记的Daudi细胞在150μl培养基中温育,不加入任何的抗体或效应细胞,以确定自发裂解的量。通过将5,000个Daudi细胞与500,000个PBMCs在没有抗体的情况下温育来确定不依赖于抗体的细胞裂解的水平。随后,将细胞于5%CO2,在37℃温育4小时。为了确定细胞裂解的量,离心细胞(1200rpm,3分钟),将75μl上清液转移至微型管(micronic tube)中,在此之后使用γ计数器来计数51Cr的释放。用所测量的每分钟计数 (cpm)根据如下计算抗体介导的裂解百分比:
(cpm样本-cpm不依赖于Ab的裂解)/(cpm最大裂解-cpm自发裂解)×100%
表15显示在所进行的ADCC测定中计算得到的IgG1-005-wt和 IgG1-005-E345R的EC50值。测试了4个样本。四个所测试的样本中 IgG1-005-E345R显示出显著低于IgG1-005-wt的EC50值。
表15:从四个所进行实验计算的EC50值


图11显示,相比起野生型抗体HuMab-005,突变体IgG1-005-E345R证明了ADCC能力的增强的效力,能够在较低浓度时诱导ADCC。
实施例13:与野生型7D8相比7D8突变体的FcRn结合以及药代动力学分析
新生儿Fc受体(FcRn)通过保护IgG不被降解而造成IgG的长血浆半衰期。在抗体内在化之后,FcRn在核内体中与抗体Fc区结合,而核内体中在略微酸性的环境下(pH6.0)相互作用是稳定的。当回收至质膜(在那里环境是中性的 (pH7.4))时,相互作用丢失,抗体被释放回循环中。这影响了IgG的血浆半衰期。
在ELISA中测试7D8突变体IgG1-7D8-E354R与来自于小鼠、猕猴和人的 FcRn相互作用的能力。所有温育在室温下完成。将5μg/ml(100μl/孔)重组生产的生物素化的FcRn胞外域(小鼠、人或猕猴)(FcRnECDHis-B2M-BIO)包被 96孔板,稀释于PBST加0.2%BSA中;1小时。平板用PBST洗涤3次,加入 3-倍系列稀释(于PBST/0.2%BSA,pH6.0)的野生型IgG1-7D8或 IgG1-7D8-E354R,并将平板温育1小时。用pH6.0的PBST/0.2%BSA洗涤平板。加入稀释于pH6.0的PBST/0.2%BSA中的山羊-抗-人IgG(Fab’2)-HRP(Jackson Immuno Research,货号:109-035-097),并将平板温育1小时。洗涤之后,加入ABTS作为底物,并将平板在黑暗中温育30分钟。使用EL808 ELISA酶标仪读取405处的吸光度。
本研究中的小鼠安置在中央实验动物设施(Utrecht,荷兰)的屏障单元中,并以随意提供的水和食物饲养在滤盖饲养笼中。所有的实验都由乌德勒支大学动物伦理委员会批准。
为了分析7D8突变体在体内的药代动力学,用100μg(5mg/kg)野生型 7D8、IgG1-7D8-E354R、-S440K或K322A静脉注射SCID小鼠 (C.B-17/IcrCrl-scid-BR,Charles-River);每组3只小鼠。
在施用抗体后10分钟、4小时、24小时、2天、7天、14天和21天从隐静脉血管中收集50μl血样。将血液收集进含有肝素的小瓶中,并以10,000g离心5分钟。将血浆储藏于-20℃直至确定mAb浓度。
使用夹心式ELISA来确定人IgG浓度。将以2μg/ml的浓度包被在96孔 MicrolonELISA平板(Greiner,德国)上的小鼠mAb抗-人IgG-κ克隆MH16 (#M1268,CLB Sanquin,荷兰)用作捕获抗体。将平板用补充有2%鸡血清的 PBS封闭之后,加入样本,系列稀释于ELISA缓冲液中(补充有0.05%吐温20 和2%鸡血清的PBS),室温下(RT)温育于平板摇动器中1小时。随后用山羊抗 -人IgG免疫球蛋白(#109-035-098,Jackson,West Grace,PA)温育平板,并用 2,2’-连氮-双-(3-乙基苯并噻唑啉-6-磺酸)(ABTS,Roche,Mannheim,德国) 显影。在微板酶标仪上(Biotek,Winooski,VT)测量405nm处的吸光度。
选择SCID小鼠是因为它们的血浆IgG浓度低,并因此具有相对低的IgG 清除。这提供了对检测清除的变化(由于Fcγ-部分与新生儿Fc受体(FcRn)结合的减少)的非常敏感的PK模型。
使用GraphPad PRISM版本4(Graphpad Software)来进行统计学测试。
图12显示野生型HuMab-7D8和IgG1-7D8-E345R两者都与小鼠、人和猕猴FcRn结合良好。IgG1-7D8-E345R的结合稍好于野生型7D8的结合。
图13随时间显示血浆浓度。野生型HuMab-7D8相比起IgG1-7D8-E345R、 -S440K或K322A中任何一个在血浆浓度(清除)随时间变化方面并没有差异。
实施例14:利用稳定Fc-Fc的突变E345R来提高IgG抗体针对表达Fc结合表面蛋白的细菌的杀菌活性
补体级联系统是重要的针对病原体的宿主防御机制,其可以被分为三种不同的激活途径以识别病原体:i)抗体介导的经典途径,其在C1q结合至与病原体结合的抗体后被激活,ii)凝集素和iii)替代途径,其中在没有抗体存在的情况下补体系统直接识别病原体并由病原体激发。这三条途径汇聚于C3切割和C3b沉积的步骤。微生物已经进化出多种补体逃避机制,其中一种是由蛋白A介导的(Joiner Ann.Rev.Microbiol.(1988)42:201-30;Foster Nat Rev Microbiol(2005)Dec;3(12):948-58)。蛋白A是在金黄色葡萄球菌(staphylococcus aureus)的细胞壁中被首次鉴定出来,并因其与IgG的Fc区结合而被人所熟知(Deisenhofer等.,Biochem(1981)20,2361-70;Uhlen等.,J.Biol. Chem(1984)259,1695-1702)。迄今为止,蛋白A的抗噬菌效果及其在金黄色葡萄球菌发病机制中的作用可由蛋白A和IgG之间的相互作用来解释,所述相互作用导致要由嗜中性粒细胞Fc受体识别的不正确的抗体方向(Foster Nat Rev Microbiol(2005)Dec;3(12):948-58)。
实施例11显示,由B细胞特异性IgG1抗体所介导的CDC被竞争的Fc-结合肽DCAWHLGELVWCT所抑制。该肽靶向IgG Fc上与蛋白A、蛋白G和类风湿因子的结合位点相一致的共有结合位点(Delano等,Science 2000Feb 18; 287(5456):1279-83)。基于这些数据,相信蛋白质A介导的细菌补体逃逸机制可通过竞争Fc结合来起作用,导致微生物特异性抗体的Fc-Fc相互作用不稳定,并因此抑制抗体介导的补体激活。此外,实施例11还显示含有CDC-增强的E345R突变的B细胞特异性IgG1抗体对于通过竞争性Fc-结合肽 DCAWHLGELVWCT抑制CDC的敏感性小于亲本野生型抗体。通过将这些结果外推至微生物表达的Fc结合蛋白,由E345R突变而提高的IgG1 Fc-Fc相互作用稳定性使得微生物特异性抗体更不易于借助微生物表面蛋白(如蛋白A) 的Fc结合竞争通过病原体逃逸策略来抑制补体。因此,与亲本野生型抗体相比,将E345R突变引入到针对细菌的IgG抗体将导致C3b在细菌上的沉积增加以及杀菌活性的提高。
作为补体介导的细菌杀伤的体外测量,可以如下文描述的那样测定嗜中性粒细胞的吞噬以及血浆中C3a的生成两者(其与C3b在细菌上的沉积一致)。实际上,已经描述了C3b在金黄色葡萄球菌上的沉积导致增强的细胞吞噬作用并与细胞杀伤有关(Rooijakkers等,Nature Immunology 2005:6,920-927)。
通过将指数生长的细菌培养物与100μg/ml FITC在0.1M碳酸盐缓冲液 (pH 9.6)中在37℃温育1小时来用FITC标记金黄色葡萄球菌。利用Ficoll梯度来分离人多形核细胞(PMN)。用系列浓度的具有或不具有E345R的特定抗体来调理FITC标记的细菌。通过使1×108个经过调理的FITC标记细菌与人PMN 在存在25%IgG消减血清作为补体来源的情况下在总体积200μl中在37℃在强烈振荡的条件下温育25分钟来体外进行吞噬作用。固定细胞并通过在室温与BD FACS裂解溶液温育15分钟来进行红细胞溶解。洗涤之后,通过FACS 来测量吞噬。通过前向(forward)和杂乱散射(side scatter)门控来选择嗜中性粒细胞群体,而吞噬则以嗜中性粒细胞群体中的平均荧光表示。或者,通过 ELISA作为补体激活和C3b沉积的测量来对样品中的C3a生成进行测量。
预期含有E345R突变的金黄色葡萄球菌特异性抗体比起亲本野生型抗体将诱导更多的补体激活以及嗜中性粒细胞的吞噬。可用于此类实验的抗体的例子为嵌合单克隆IgG1帕吉昔单抗(BSYX-A110;Biosynexus),其靶向嵌入 (embed)在葡萄球菌细胞壁之中的脂磷壁酸(LTA)(Baker,Nat Biotechnol.2006 Dec;24(12):1491-3;Weisman等,IntImmunopharmacol.2009May;9(5): 639-44)。
实施例15:将CDC激活限于同时被两种不同治疗性单克隆抗体的混合物结合的靶细胞的CDC-抑制性突变的用途
如实施例6所述,CD20抗体7D8突变K439E和S440K作为单克隆抗体降低了CDC效力。混合含有这些突变的7D8抗体恢复CDC。因此有效CDC受限于同时结合有两个突变体抗体的细胞。如实施例10所述,CD38抗体005突变 K439E和S440K作为单克隆抗体降低了CDC效力。混合含有这些突变的005 抗体恢复CDC。因此有效CDC受限于同时结合有两个突变体抗体的细胞。
可以有利的是将有效CDC的诱导限于同时表达两种特定抗原的靶细胞,这利用了它们的组合表达来改善CDC诱导的选择性。为了将CDC诱导限于同时结合有CD20和CD38抗体的细胞,在CDC实验中如下单独加入或以1:1混合 7D8-K439E和005-S440K的对或者7D8-S440K和005-K439E的对。在圆底96孔板中,将0.1×106个Daudi或Raji细胞与系列浓度的未纯化抗体或抗体混合物 (0.01、0.03、0.1、0.3、1.0、3.0、10.0、30.0μg/ml)在总体积100μl中在室温在摇床中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl 碘化丙啶并在FACS上确定细胞的裂解。预期7D8-K439E、005-S440K、 7D8-S440K和005-K439E将表现出有限的CDC效力。预期同时加入 7D8-K439E和005-S440K将恢复特异性在表达CD20和CD38两者的细胞上的有效CDC。同样,预期7D8-S440K和005-K439E的混合物将恢复特异性在表达CD20和CD38两者的细胞上的有效CDC。
实施例16:通过将E345R与互补的抑制性突变K439E和S440K组合在两种不同单克隆抗体的混合物中来提高增强型CDC的特异性
如实施例6所述,CD20抗体7D8突变K439E和S440K作为单克隆抗体降低了CDC效力。混合含有这些突变的7D8抗体恢复CDC。因此有效CDC限于同时结合有两个突变体抗体的细胞。如实施例10所述,CD38抗体005突变K439E 和S440K作为单克隆抗体降低了CDC效力。混合含有这些突变的005抗体恢复CDC。因此有效CDC受限于同时结合有两个突变体抗体的细胞。
可以有利的是将CDC诱导的增强限于同时表达两种特定抗原的靶细胞,这利用了它们的组合表达来改善增强的CDC诱导的选择性。还可以有利的是将CDC诱导的增强限于同时结合至少两种不同抗体的混合物的靶细胞,所述抗体在两个不同的表位上同时,或在两个交叉竞争、相似或相同的表位上结合相同的细胞表面抗原。
因此,为了将增强的CDC诱导限于同时结合有CD20和CD38抗体两者的细胞,将CDC增强突变E345R与CDC抑制性突变组合在抗体 7D8-E345R/K439E、7D8-E345R/S440K、005-E345R/S440K和 005-E345R/K439E中。在CDC实验中如下单独加入或以1:1混合这些抗体。在圆底96孔平板中,将0.1×106个Wien133细胞(也可以使用其它的细胞类型,例如Daudi或Raji细胞)与系列浓度的未纯化抗体(终浓度0.056-10,000ng/ml以3 倍稀释的7D8-E345R/K439E、7D8-E345R/S440K、005-E345R/S440K或 005-E345R/K439E)或抗体混合物(终浓度为0.01μg/ml CD20抗体与0-333 ng/ml以3倍稀释的CD38抗体混合;或3.3μg/ml的CD38抗体与0.0056-1,000 ng/ml以3倍稀释的CD20抗体混合)在总体积100μl中在室温在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
将系列浓度的005-E345R/K439E或005-E345R/S440K抗体与固定浓度为 0.01μg/ml的7D8双突变体抗体混合(如从图14A中所确定的作为单一药剂对 Wien133细胞具有最小CDC的最大浓度)以制备互补性组合 005-E345R/K439E+7D8-E345R/S440K或 005-E345R/S440K+7D8-E345R/K439E。图14C显示,在分别存在固定浓度的互补性7D8-E345R/K439E或7D8-E345R/S440K CD20抗体时,005双突变 CD38抗体剂量依赖性地诱导CDC。这些互补性组合的CDC效力(图14C)与作为单一药剂的005-E345R单突变体(增强物)抗体相当(图14B)。相反,在存在无关抗体b12时,005-E345R/K439E或005-E345R/S440K两者在测试的系列浓度中几乎没有显示出任何CDC(与作为单一药剂的005-E345R/K439E或 005-E345R/S440K相当,图14B中所示)。
将系列浓度的7D8-E345R/K439E或7D8-E345R/S440K抗体与固定浓度为3.3μg/ml的005双突变体抗体混合(如从图14B中所确定的作为单一药剂对Wien133细胞显示出少许但有限的CDC)以制备互补性组合 7D8-E345R/K439E+005-E345R/S440K或 7D8-E345R/S440K+005-E345R/K439E。图14D显示在分别存在互补性 005-E345R/K439E或005-E345R/S440K CD38抗体的情况下,7D8双突变体 CD20抗体非常有效地诱导了CDC,甚至是在测试的最低浓度时,就像每个细胞只有几个7D8双突变体抗体分子。为了消除细胞膜上Fc尾部密度的提高对于所观察到的由具有互补性K439E和S440K突变的7D8和005抗体混合物增强的CDC的贡献,还测试了具有非互补性突变的抗体组合。图14D显示,非互补性组合显示出远低于互补性组合的CDC效力,这是由于比互补性组合效率更低的Fc-Fc相互作用。
这些数据表明,治疗性抗体的(增强的)CDC诱导可限于同时结合两个互补性抗体混合物的细胞,在这种情况下具有不同的抗原特异性,从而通过需要这两种抗原共表达来提高靶细胞特异性。
如图14A和14B中可以看到,与单独的7D8-E345R相比, 7D8-E345R/K439E、005-E345R/S440K、7D8-E345R/S440K和 005-E345R/K439E展现出有限的CDC效率。可进一步见到的是, 7D8-E345R/K439E和7D8-E345R/S440K的混合物与作为单一药剂的野生型 7D8抗体相比实现具有增强的效力的CDC。同样地,也观察到 005-E345R/K439E和005-E345R/S440K的混合物与作为单一药剂的野生型 005抗体相比实现具有增强的效力的CDC(数据未显示)。
实施例17:将有效CDC激活限于专门由治疗性施用的抗体组成的抗体复合物的CDC抑制性突变的用途
如实施例6中所述,CD20抗体7D8双突变体K439E/S440K恢复了由 K439E或S440K单点突变体减少的CDC效力。如实施例10中所述,CD38抗体 005双突变体K439E/S440K恢复了由K439E或S440K单点突变体抑制的CDC 效力。正如所观察到的,单点突变在Fc:Fc界面的面向侧(facing side)上具有未突变氨基酸的情况下破坏Fc:Fc相互作用。在Fc:Fc界面的面向侧引入补偿性突变恢复了CDC效力。因此有效的CDC显然受限于专门由含有这两个突变的抗体组成的抗体复合物。
在另一个实施例中,CDC的诱导限于专门由治疗性施用的抗体组成的抗体复合物。为了将CDC诱导限于仅结合有治疗性CD20或CD38抗体的细胞,将CDC抑制性突变K439E和S440K组合到抗体7D8-K439E/S440K或 005-K439E/S440K中。这些抗体在存在或不存在非靶标特异性的IgG时如下单独加入到CDC实验中。在圆底96孔板中,将0.1×106个Daudi或Raji细胞与未纯化的抗体或抗体混合物(0.01、0.03、0.1、0.3、1.0、3.0、10.0、30.0μg/ml) 在总体积100μl中在室温在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
预计7D8-K439E/S440K将以与野生型7D8抗体相似的效力诱导CDC。预计向7D8-K439E/S440K加入非特异性IgG不会影响该抗体的CDC诱导的效力。同样地,预计005-K439E/S440K将以与野生型HuMAb 005相似的效力实现CDC。预计向005-K439E/S440K加入非特异性IgG不会影响该抗体的CDC 诱导的效力。
实施例18:将增强的CDC激活限于专门由治疗性施用的抗体组成的抗体复合物的CDC抑制性突变的用途
如实施例6中所述,CD20抗体7D8双突变体K439E/S440K恢复了由 K439E或S440K单点突变体所减少的CDC效力。如实施例10中所述,CD38 抗体HuMAb 005双突变体K439E/S440K恢复了由K439E或S440K单点突变体所抑制的CDC效力。正如所观察到的,单点突变在Fc:Fc界面面向侧上具有未突变氨基酸的情况下破坏了Fc:Fc相互作用。在Fc:Fc界面的面向侧引入补偿性突变恢复了CDC效力。因此有效的CDC显然受限于专门由含有这两个突变的抗体组成的抗体复合物。
在另一个实施例中,CDC诱导的增强限于专门由治疗性施用的抗体组成的抗体复合物。通过筛选和选择刺激用于CDC刺激的Fc:Fc相互作用的突变,可以利用对于感兴趣抗原靶标非特异性的血清抗体鉴定能够形成CDC诱导抗体复合物的突变。为了将增强的CDC诱导限于专门由CD20复合物或CD38 抗体结合的细胞,将CDC增强性突变E345R与CDC抑制性突变组合到抗体 7D8-E345R/K439E/S440K或005-E345R/K439/S440K中。这些抗体在存在或不存在非靶标特异性的IgG时如下单独加入到CDC实验中。在圆底96孔板中,将0.1×106个Daudi或Raji细胞与未纯化的抗体或抗体混合物(0.01、0.03、0.1、 0.3、1.0、3.0、10.0、30.0μg/ml)在总体积100μl中在室温在摇动器中预温育 15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上终止反应。加入10μl碘化丙啶并通过 FACS确定细胞的裂解。
预计与野生型HuMAb 7D8相比,7D8-E345R/K439E/S440K会以增强的效率诱导CDC。与野生型7D8抗体相比,向7D8-E345R/K439E/S440K加入非特异性IgG预计不会影响CDC诱导的效率。同样地,预计与野生型005抗体相比,005-E345R/K439E/S440K将实现具有增强的效率的CDC。相对于野生型 005抗体,向005-E345R/K439E/S440K加入非特异性IgG预计不会影响CDC的效率。
实施例19:使用突变体筛选方法鉴定能刺激通过CDC测定法检测的Fc:Fc相互作用介导的抗体寡聚化的突变
如实施例6和10所述,在表达各种水平的所述抗原的多个细胞系上对于识别两种不同靶标抗原(CD20和CD38)的抗体,鉴定了刺激CDC的氨基酸突变。令人惊奇的是,单点突变E345R证实足以将Wien133细胞的CDC依赖性细胞裂解赋予抗CD38抗体005,其以野生型IgG1形式不能通过CDC来裂解这些细胞。
Fc:Fc界面上或周围的其他突变能够以类似的方式刺激寡聚化和CDC。或者,突变能够间接刺激寡聚化,例如通过变构性诱导Fc:Fc相互作用。
为了确定其他氨基酸突变是否能够刺激Fc介导的抗体寡聚化,使用CDC 测定法对抗CD38IgG1-005突变体文库进行筛选,单独地及以成对的方式混合以选择例如在Fc:Fc界面间相互作用的氨基酸对。然而,可以将相同的策略用于其他的抗体,例如另一种IgG1或IgG3抗体。
产生了位于表16中所示位置处的突变的集中文库。使用Quikchange定点诱变试剂盒(Stratagene,美国)将突变引入至IgG1-005Fc区中。简而言之,对于每个所期望的突变位置,使用在所期望位置上编码简并密码子的正向和反向引物来复制具有IgG1m(f)同种异型的005重链的全长质粒DNA模板。用 DpnI消化所得到的DNA混合物以移除来源质粒DNA并用于转化大肠杆菌。汇集并培养所得到的菌落,从这些汇集物中分离质粒DNA并再转化入大肠杆菌中以获得克隆菌落。通过DNA测序(LGC genomics,Berlin,德国)来验证从所得到的菌落中分离出来的突变体质粒DNA。通过PCR从质粒DNA中扩增表达盒,并使用293fectin(Invitrogen,美国)基本上如制造商所述将同时含有 IgG1-005突变体重链和野生型轻链两者的DNA混合物瞬时转染至Freestyle HEK293F细胞(Invitrogen,美国)。收集含有抗体突变体的转染细胞的上清液。在CDC测定法中单独地以及在配对混合物中如下筛选突变体抗体上清液。
在圆底96孔板中,将0.1×106个Daudi或Wien-133细胞(也可使用其它的细胞类型,例如Raji细胞)与1.0μg/ml未纯化抗体在总体积100μl中在室温在摇床中预温育15分钟。接着,加入30μl正常人血清作为补体来源(30%终浓度) 并在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl 碘化丙啶并通过FACS确定细胞的裂解。
针对其增强寡聚化的能力(如通过CDC效力检测的),无论是作为单个突变体还是与其他突变体混合时(例如面对穿过Fc:Fc界面的突变),对描述于表 16、表17和表18中的突变进行选择。针对其不会损坏FcRn、蛋白-A或蛋白-G 结合、ADCC、ADCP或其他由Fc域介导的效应子功能的能力,可选择进一步对突变进行筛选。将此类刺激性点突变组合到一个Fc域中甚至能够更进一步地刺激寡聚化和CDC效力。
针对其抑制寡聚化的能力(如对于Daudi细胞通过CDC确定的),对引入 CD38抗体005中的CH2-CH3区中的突变进行测试。突变体抗体的裂解与野生型005(其裂解被设置为100%)相比较。抑制的截断值(cut-off)被设定为≤66%裂解。用这种方式进行测量,大部分所测试的突变都抑制CDC(见表16)。
针对其抑制寡聚化的能力(如对于Wien133细胞通过CDC确定的),对引入CD38抗体005中的CH2-CH3区中的突变进行测试(表17)。野生型CD38抗体 005不能够诱导针对Wien133细胞的CDC。将显示出≥39%细胞裂解的突变体评价为增强。完全预料不到的是,几乎所有获得的氨基酸E345和E430取代刺激通过CDC的细胞裂解。为了证实这个结果,通过定点诱变用每种可能的突变对氨基酸E345、E430和S440进行取代,并测试它们增强寡聚化的能力(如使用新鲜人血清批次通过Wien133细胞的CDC确定的),产生略微更有效的裂解(表18)。再一次,所有的E345和E430取代都诱导了Wien133细胞的有效 CDC。
下述优选的突变引起了Wien133细胞的≥39%细胞裂解:P247G、I253V、 S254L、Q311L、Q311W、E345A、E345C、E345D、E345F、E345G、E345H、 E345I、E345K、E345L、E345M、E345N、E345P、E345Q、E345R、E345S、 E345T、E345V、E345W、E345Y、D/E356G、D/E356R、T359R、E382L、 E382V、Q386K、E430A、E430C、E430D、E430F、E430G、E430H、E430I、 E430L、E430M、E430N、E430P、E430Q、E430R、E430S、E430T、E430V、 E430W、E430Y、Y436I、S440Y和S440W。



实施例20:皮下B细胞淋巴瘤异种移植物模型中IgG1-7D8-E345R的体内效力
在使用Raji-luc#2D1细胞的皮下模型中评估IgG1-7D8-E345R抗体的体内抗肿瘤效力。这些细胞显示出~300,000个CD20分子每个细胞(由QIFIKIT 分析确定,数据未显示)以及高补体防御受体表达。将细胞培养在具有10%加强型小牛血清(HyClone,Logan,UT),青霉素和链霉素,1%(v/v)丙酮酸钠和1μg/ml嘌呤霉素(P-8833,Sigma,Zwijndrecht)的RPMI中。在对数期(大约 70%汇合度)收集细胞。使用6至11周龄的雌性SCID小鼠 (C.B-17/IcrPrkdc-scid/CRL)(Charles-River)。在第0天时,将200μl PBS中的 5×106个Raji-luc#2D1细胞皮下注射到每只小鼠的右侧腹中。用测径器(caliper) 测量监控肿瘤形成。当平均肿瘤体积为100mm3时(第7天左右),将小鼠分组 (n=9)并通过向每只小鼠腹膜内(i.p.)注射单剂50μg的抗体(2.5mg/kg)来进行处理。所有的抗体样本都补充有无关抗体b12以获得总抗体浓度为0.5mg/ml。处理组显示于表18中。在处理后7天,获得血样样本以确定人IgG血清水平来检查正确的抗体施用。利用测径器(PLEXX)测量肿瘤每周至少两次直至终点肿瘤体积为1500mm3,肿瘤显示出溃疡或直至观察到严重的临床体征。
表18:处理组和剂量给药

图15A显示第22天当所有组仍然完整时的平均肿瘤生长。相比起阴性对照抗体IgG1-b12,野生型抗体IgG1-7D8轻微抑制肿瘤生长,尽管这在统计学上并不显著。相比起阴性对照抗体IgG1-b12,只有IgG1-7D8-E345R显著地抑制了肿瘤生长(单向ANOVA分析p<0.01)。
图15B显示了肿瘤大小小于700mm3的小鼠百分比的Kaplan-Meier作图。与用阴性对照抗体IgG1-b12处理的小鼠相比,用IgG1-7D8-E345R抗体处理的小鼠中的肿瘤形成被显著延迟(Mantel-Cox分析,p<0.01),但在用野生型 IgG1-7D8处理的小鼠中却没有。
这些数据显示,E345R突变增强了CD20抗体7D8的体内抗肿瘤效力。
实施例21:皮下B细胞淋巴瘤异种移植物模型中IgG1-005-E345R的体内效力
在使用Raji-luc#2D1细胞的皮下模型中评估IgG1-7D8-E345R的体内抗肿瘤效力。这些细胞显示出~150,000个CD38分子每个细胞(由QIFIKIT分析确定,数据未显示)以及高补体防御受体表达。肿瘤接种和测量的方案基本上与实施例20中所述的相同。在第0天时,将200μl PBS中的5×106个Raji-luc #2D1细胞皮下(s.c.)注射到SCID小鼠的右侧腹中。当平均肿瘤体积为100mm3时(第7天左右),将小鼠分组(n=7)并通过向每只小鼠i.p.注射单剂500μg抗体 (25mg/kg)来进行处理。处理组示于表19中。测量肿瘤直至终点肿瘤体积为1500mm3或直到肿瘤显示出溃疡或直至观察到严重的临床体征以避免重大的不适。
图16A显示第21天当所有组仍然完整时的平均肿瘤生长。野生型抗体 IgG1-005轻微抑制肿瘤生长,尽管这在统计学上并不显著。在第21天,相比起无关抗体对照,只有IgG1-005-E345R显著地抑制了肿瘤生长(单向ANOVA p<0.05)。
图16B显示了肿瘤大小小于500mm3的小鼠百分比的Kaplan-Meier作图。用IgG1-005-E345R抗体处理的小鼠中的肿瘤形成与用阴性对照抗体 IgG1-b12处理的小鼠(Mantel-Cox分析,p<0.001)或者用野生型IgG1-005处理的小鼠(p<0.05)相比被显著延迟。
这些数据显示,在CD38抗体005中引入E345R突变导致增强的体内抗肿瘤活性。
表19治疗组和剂量给药
| 组 | 抗体 | 剂量 |
| 1野生型 | IgG1-005-WT | 500μg(=25mg/kg) |
| 2CDC-增强突变体 | IgG1-005-E345R | 500μg(=25mg/kg) |
| 3无关抗体对照 | IgG1-b12 | 500μg(=25mg/kg) |
实施例22:单价靶标结合进一步增强E345R抗体的CDC效力
在b12晶体结构中观察到的IgG1六聚环的分子表面表明,对于六聚环中的每个IgG,两个C1q结合位点中的一个面朝环结构的上方而另外一个则面朝环结构的下方,并且每个抗体的一个Fab-臂也是方向朝上,而一个方向朝下,这导致每个抗体只有一个Fab-臂参与到抗原结合中,这暗示了六聚抗体环中每个抗体分子的单价结合。单价性可以对抗体带来六聚化相容性方向的抗原结合。为了测试这个设想,在CD38阳性、EGFR-阴性的Wien133细胞上(对于该细胞,这个双特异性抗体仅能通过CD38而进行单价结合)测试具有E345R 突变的双特异性CD38/EGRF抗体的CDC效力,并与同样具有E345R突变的双价结合CD38的抗体的CDC效力进行比较。将人单克隆抗体HuMax-EGFr (2F8,描述于WO2004/056847中)用作本实施例中所述EGFR抗体的基础。
根据DuoBodyTM平台,即如WO2011/147986中所述的2-MEA-诱导的Fab- 臂交换,体外产生双特异性抗体。该方法的基础是互补性CH3域(其在特定的测定条件下促进杂二聚体的形成)的使用。为了能够通过这个方法来进行双特异性抗体的生产,产生了在CH3域中携带某些突变的IgG1分子:在亲本 IgG1抗体之一上是F405L突变,而在另一个亲本IgG1抗体上是K409R突变。为了产生双特异性抗体,将这两个亲本抗体(每个抗体的终浓度为0.5mg/ml) 与25mM的2-巯基乙胺-HCl(2-MEA)在总体积100μl的TE中于37℃温育90分钟。当根据制造商规程使用离心柱(Microcon离心过滤器,30k,Millipore)移除还原剂2-MEA时,还原反应终止。
对于CDC测定,在圆底96孔板中,将0.1×106个Wien133细胞与系列浓度的抗体(0.01至10.0μg/ml)在总体积100μl中在室温在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上终止反应。加入10μl碘化丙啶并通过FACS 确定细胞的裂解。
图17显示,如预期的,不具有E345R突变的CD38抗体(野生型IgG1-005 和IgG-b12-K409R×IgG1-005-F405L)并没有诱导Wien133细胞的杀伤。如预期的,EGFR抗体IgG1-2F8-E345R/F405L(其并不结合EGFR-阴性Wien133细胞(数据未显示))也没有诱导CDC。K408R突变的引入并没有影响 IgG1-005-E345R抗体在Wien133细胞上诱导约60%杀伤的能力(描述于实施例 10中)。有趣的是,双特异性CD38/EGFR抗体IgG1-005-E345R/K409R× IgG1-2F8-E345R/F405L(其只能单价地结合CD38阳性、EGFR-阴性Wien133 细胞)显示出提高的最大CDC杀伤(从~60%至~100%杀伤)。
这些数据显示,单价靶向能够进一步增强含有CDC增强型E345R突变的抗体的最大杀死能力。此外,这些数据显示,E345R寡聚化增强突变(如通过增强CDC活性来测量)能够应用于其他抗体形式中,如DuoBody。
实施例23:寡聚化增强型E345R突变能够应用于其他抗体形式如DuoBodyTM
在DuoBody形式的双特异性抗体中测试E345R突变的效果。利用 CD20/CD38双特异性抗体在CD20-阳性、CD38-阳性Wien133和Raji细胞上实施CDC测定。
如实施例22中所述产生双特异性抗体。对于CDC测定,在圆底96孔板中,将0.1×106个Wien133或Raji细胞与系列浓度的抗体(0.01至30.0μg/ml)在总体积100μl中在室温在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
图18显示引入E345R突变增强了IgG1-005-F405L×IgG1-7D8-K409R抗体对Wien133(图18A)和Raji(图18B)细胞的CDC。这些数据显示,E345R寡聚化增强型突变能够应用于其他抗体形式以增强CDC活性。
实施例24:E345R拯救EGFR抗体2F8的CDC,其能够通过单价靶标结合进一步增强
如实施例6、10和26中所述,E345R增强或拯救了识别不同血液肿瘤靶标 (CD20和CD38)的抗体的CDC。为了将该分析延伸至实体瘤抗原,在A431表皮癌细胞上测试E345R对EGFR抗体2F8的CDC能力的影响。此外,利用双特异性EGFR×CD20抗体(IgG1-2F8-E345R/F405L×IgG1-7D8-E345R/K409R)在 EGFR-阳性、CD20阴性的A431细胞上测试单价性EGFR靶向对于E345R介导的CDC诱导的影响。
如实施例22所述产生双特异性抗体。对于CDC测定法,用100μCi 51Cr 对5×106个A431细胞/ml在37℃标记1小时。用PBS洗涤细胞三次,并以1×105个细胞/ml的浓度重悬于培养基中。在圆底96孔板中,将25,000个标记细胞与系列浓度的未纯化抗体(0-30μg/ml以3倍稀释)在总体积100μl中在室温温育 15分钟。接着,加入50μl正常人血清稀释物作为补体来源(25%终浓度),在 37℃的培养箱中温育1小时。将细胞向下旋转(300×g,3分钟),并在白色96 孔Optiplate(PerkinElmer)中向100μl Microscint加入25μl上清液以在摇床(750rpm)上温育15分钟。在闪烁计数器上以每分钟计数(cpm)来确定51Cr释放。通过在经TritonX-100处理细胞上清液中测量的51Cr水平来确定最大裂解 (100%)。通过在未经抗体温育的细胞上清液中测量的51Cr水平来确定自发裂解。根据公式:特异性裂解(specific lysis)=100×(cpm样本-cpm自发)/(cpm最大-cpm自发)来计算特异性的细胞裂解。
图19显示IgG1-2F8-E345R/F405L能够通过CDC裂解A431细胞,而野生型 2F8却不能杀死A431细胞。这些数据显示,通过引入E345R突变能够拯救 EGFR抗体2F8的CDC活性。这可能将CDC增强型E345R突变的应用扩展至靶向实体瘤抗原的抗体。
双特异性EGFR×CD20抗体IgG-2F8-E345R/F405L× IgG1-7D8-E345R/K409R,在EGFR-阳性、CD20阴性的A431细胞上显示CDC 的进一步增强。
这些数据进一步支持了该设想,即单价性促进Fc-Fc相互作用的形成及随后的CDC诱导,如对于实施例22中所述的CD38结合抗体所提出的。
实施例25:E345R增强或拯救CD38抗体003以及CD20抗体11B8和利妥昔单抗的CDC
正如实施例6、10和24所述,E345R增强或诱导了具有不同靶标特异性 (CD20、CD38和EGFR)的几个抗体的CDC活性,如在表达可变水平的所述抗原的多个细胞系上所测试的。因此,E345R突变的引入认为是对现有抗体进行增强或拯救CDC的普遍性机制。为了进一步支持这个,对更多的对Daudi 和Wien133细胞具有不同的固有CDC效力的抗体测试E345R突变对CDC的影响:CD38抗体003,描述于WO 2006/099875中以及CD20抗体利妥昔单抗(I 型)和11B8(II型),描述于WO 2005/103081中。CD20抗体可被分为两个亚群 (Beers等Seminarsin Hematology 47,(2)2010,107-114)。I型CD20抗体展示出卓越的通过将质膜中的CD20分子重新分布于脂筏上(其使得抗体Fc区聚簇并能够改进C1q结合)从而激活补体以及激发CDC的能力。II型CD20抗体并没有可感知地改变CD20的分布,并且也没有伴随的聚簇,它们在CDC方面是相对无效的。
在圆底96孔板中,将0.1×106个Daudi或Raji细胞与系列浓度的未纯化抗体(0.001、0.003、0.01、0.03、0.1、0.3、1.0、3.0、10.0μg/ml)在总体积70μl 中在室温在摇动器中预温育15分钟。接着,加入30μl正常人血清作为C1q来源(30%终浓度),并在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
图20显示,E345R突变增强了所有测试的抗体对Daudi(A)和Wien133(B) 细胞两者的CDC。有趣的是,在所使用的浓度,以野生型形式存在时没有诱导CDC的所有抗体,在引入E345R突变之后有效地诱导了CDC:CD38 mAb 003和CD20II型mAb 11B8对Daudi细胞,以及CD38mAbs 005和003以及CD20 II型mAb11B8对Wien133细胞。这些数据表明抗体寡聚化的增强,更具体地通过E345R突变的引入而增强,是增强或拯救现有抗体的CDC的普遍性机制。
实施例26:E345R增强了组织因子抗体的内在化
为了测试增强的寡聚化能否诱导提高的抗体内在化,利用溶酶体标记物 LAMP1通过共聚焦显微镜来进行野生型和E345R突变型组织因子(TF)抗体的共定位研究。
SK-OV-3细胞在玻璃盖玻片上(厚度1.5微米,Thermo Fisher Scientific,Braunschweig,德国)于标准组织培养基中在37℃培养1天。将细胞与50μg/ml 亮肽酶素(leupeptin)(Sigma)预温育1小时以封闭溶酶体活性,其后加入10 μg/ml的组织因子(TF)抗体(WO2010/066803)。在37℃将细胞再温育1、3或16 个小时。之后,用PBS洗涤细胞,并用4%甲醛(Klinipath)在室温下(RT)温育 30分钟。用封闭缓冲液(补充有0.1%的皂苷[Roche]和2%BSA[Roche]的PBS) 洗涤盖玻片,并与含有20mM NH4Cl的封闭缓冲液温育20分钟以淬灭甲醛。用封闭缓冲液再次洗涤载玻片,将其与用于鉴定溶酶体LAMP1的小鼠-抗-人 CD107a-APC(BD Pharmingen)和用于鉴定TF抗体的山羊-抗-人IgG-FITC (Jackson)的混合物在室温下温育45分钟。用封闭缓冲液再次洗涤载玻片并使用20μl封固介质(将6克甘油[Sigma]和2.4克Mowiol 4-88[Omnilabo]溶解于6 ml蒸馏水中,向其中加入12mL 0.2MTris[Sigma]pH8.5,随后在50-60℃温育10分钟;将封固介质分装并储存于-20℃)封固在显微镜载玻片上过夜。用配备63×1.32-0.6油浸物镜和LAS-AF软件的Leica SPE-II共聚焦显微镜(Leica Microsystems)对载玻片成像。
使用 软件(版本Meta Series 6.1,Molecular Devices Inc,Sunnyvale California,美国)对12-位灰度级的TIFF图像分析共定位。以栈(stack) 输入图像并扣除背景。对于所有的FITC图像和所有的APC图像都使用相同的阈值设定(手动设置)。将共定位描绘为感兴趣区域(ROI)中的FITC的像素强度,而ROI由所有的APC阳性区组成。为了对用不同TF抗体染色的不同载玻片进行比较,用APC的像素强度对图像标准化。使用小鼠抗人CD107a-APC 对溶酶体标记LAMP1(CD107a)进行染色。LAMP1的像素强度在所成像的各个TF抗体之间不应当是不同的。
软件(版本Meta Series 6.1,Molecular Devices Inc,Sunnyvale California,美国)对12-位灰度级的TIFF图像分析共定位。以栈(stack) 输入图像并扣除背景。对于所有的FITC图像和所有的APC图像都使用相同的阈值设定(手动设置)。将共定位描绘为感兴趣区域(ROI)中的FITC的像素强度,而ROI由所有的APC阳性区组成。为了对用不同TF抗体染色的不同载玻片进行比较,用APC的像素强度对图像标准化。使用小鼠抗人CD107a-APC 对溶酶体标记LAMP1(CD107a)进行染色。LAMP1的像素强度在所成像的各个TF抗体之间不应当是不同的。
FITC和APC共定位的标准化数值根据公式[(TPI FITC×共定位百分比)/100]×[1/TPI APC]以任意单位表示。
共定位百分比=与APC像素共定位的TPI FITC/TPI APC
TPI:总像素强度
图21描绘了与APC标记的溶酶体标记物重叠的野生型和E345R突变型 TF抗体的FITC像素强度的量。对于所测试的每个抗体或条件,分析来自含有~1、3或>5个细胞的一个载玻片的三张不同图像。在每个玻片中的不同图像之间观察到了差异。然而,明显的是当与野生型011和098相比时,抗体011 和098的E345R突变在1小时的温育之后导致溶酶体共定位的提高。这些结果指示突变E345R诱导了更为迅速的内在化和溶酶体共定位,并因此能够增强抗体药物缀合物。
实施例27:在具有相似CD20表达但不同水平的膜结合补体调控蛋白的不同B 细胞系中利妥昔单抗中的E345R突变增强了CDC
实施例25和28显示,野生型利妥昔单抗对于Daudi和Wien133细胞的CDC 效力通过引入E345R突变而被增强。这种增强的CDC效力是由于E345R介导的Fc-Fc相互作用稳定化造成的。然后,在靶细胞膜上伴随形成的六聚抗体环结构可以通过有助于捕获和浓缩靠近细胞膜的激活补体组分来促进膜攻击复合体的有效产生。由于这种有效的补体激活,能够部分地克服膜结合补体调控蛋白(mCRP)的抑制效果。mCRPs(如CD55、CD46和CD59)的过表达认为是利用单克隆抗肿瘤抗体的成功免疫治疗的障碍(Jurianz等,Mol Immunol 199936:929-39;Fishelson等,Mol Immunol 200340:109-23,Gorter 等,Immunol Today 199920:576-82,Zell等,Clin Exp Immunol.2007 Dec 150(3):576-84)。因此,比较利妥昔单抗-E345R与野生型利妥昔单抗对一系列具有不同水平的mCRPs CD46、CD55和CD59但是具有相当水平的CD20靶标表达的B细胞系的效力。
B细胞系Daudi、WIL2-S、WSU-NHL、MEC-2和ARH-77表达了相当量的CD20分子(~250.000特异性抗体结合能力-sABC),如通过QIFIKIT分析确定的(数据未显示)。为了比较这些细胞系之间补体调控蛋白的表达水平,实施 QIFIKIT分析以确定CD46(小鼠抗人CD46、CBL488、克隆J4.48Chemicon)、 CD55(小鼠抗人CD55、CBL511、克隆BRIC216、Chemicon)和CD59(小鼠抗人CD59、MCA1054x、克隆MEM-43、Serotec)的水平。
对于CDC测定,在圆底96孔板中,将0.1×106个细胞与饱和抗体系列浓度 (0.002-40.0μg/ml以4倍稀释)在总体积100μl中室温下在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过 FACS确定细胞的裂解。在GraphPad PRISM 5中利用非线性拟合的最佳拟合值的顶点从两个独立实验中计算最大CDC介导的杀伤。
图22A-D显示在野生型利妥昔单抗中引入E345R导致CDC效力增强,如通过对所有测试的B细胞系的提高的最大裂解和降低的EC50所观察到。
图22E显示,由利妥昔单抗-E345R突变体诱导的最大CDC-介导的杀伤总是比野生型利妥昔单抗要高,这不依赖于膜结合补体调控蛋白的表达水平。这些数据指示E345R的引入增强了单克隆抗体的治疗潜能,这是由于肿瘤细胞在逃避由含有E345R的抗体所导致的抗体介导的补体攻击方面不太有效。
实施例28:野生型和E345R抗体的CDC动力学的比较
已经显示引入稳定Fc:Fc相互作用的E345R突变增强或拯救CDC,如通过实施例6(CD20抗体7D8对Daudi和Raji)、实施例10(CD38抗体005对Daudi、 Raji和Wien133)和实施例25中(CD38抗体003和CD20抗体利妥昔单抗和11B8 对Daudi和Wien133)所述的不同细胞系上不同抗体的EC50值降低以及最大裂解提高所观察到的。接着,对CDC反应的动力学进行分析以进一步揭示野生型和E345R抗体之间的CDC效力的差别。
在圆底96孔板中,将0.1×106个Raji细胞与饱和浓度的抗体(10.0μg/ml)在总体积100μl中室温下在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育不同的时间段,0-60 分钟变化。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
图23A显示野生型CD20抗体IgG1-7D8显示80%Raji细胞的CDC介导的最大杀伤,这在测试的条件下5分钟后已经达到。然而,对于IgG-7D8-E345R,甚至更快(3分钟后)观察到Raji细胞的80%杀伤。IgG-7D8-E345R的最大裂解 (95%)也在5分钟之后达到。
图23B显示对于野生型CD20抗体利妥昔单抗也同样(该抗体针对所使用的Raji细胞诱导CDC的能力比7D8弱),E345R突变的引入导致靶细胞的更快杀伤。野生型利妥昔单抗显示出CDC介导的最大杀死为32%,这是在20分钟之后达到的。利妥昔单抗-E345R则在大约3分钟之后就达到32%杀伤并且显著地,在20分钟后也达到了利妥昔单抗的最大裂解(85%)。
图23C+D显示能够通过引入E345R突变更快地杀伤所使用的Raji细胞(该细胞对于野生型CD38抗体IgG1-003和IgG1-005的CDC介导的杀伤具有抗性)。IgG1-003-E345R和IgG1-005-E345R在5分钟后已经显示出最大CDC(分别是50%和60%)。
总而言之,E345R抗体要比它们的野生型对应物更加有效,这是由于更高效力(更低的EC50)、增加的最大裂解以及更快的CDC反应动力学的组合造成的。
实施例29:具有或不具有E345R突变的双特异性抗体的CDC动力学的比较
在实施例23中描述了可将E345R突变应用于由DuoBody平台所产生的 CD38×CD20双特异性抗体IgG1-005-F405L×IgG1-7D8-K409R,导致增强的杀伤能力,如通过在CDC测定法中对于Raji和Wien133细胞的降低的EC50所观察到的。接着,对CDC反应的动力学进行分析以进一步揭示具有或不具有 E345R突变的CD38×CD20双特异性抗体之间CDC效力的差异。
在圆底96孔板中,将0.1×106个Raji细胞与饱和浓度的抗体(10.0μg/ml)在总体积100μl中室温下在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育不同的时间段,0-60 分钟变化。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
图24显示,双特异性抗体IgG1-005-F405L×IgG1-7D8-K409R诱导了 CDC介导的最大杀伤83%,这在10分钟之后达到。E345R突变的引入导致 IgG1-005-F405L×IgG1-7D8-K409R的最大杀伤提高(98%),这在2分钟之后就已经达到。这些数据指示在双特异性抗体中引入使Fc-Fc相互作用稳定的 E345R突变导致CDC介导的靶细胞杀伤加速。
实施例30:具有或不具有E345R的单价结合抗体的CDC动力学的比较
实施例22显示,单价靶标结合进一步增强E345R抗体的CDC效力,如通过CD38×EGFR双特异性抗体对CD38阳性、EGFR阴性的Wien133细胞的最大裂解提高所观察到的。接着,对CDC反应的动力学进行分析以进一步揭示具有或不具有E345R突变的单价结合抗体之间CDC介导的杀伤能力的差异。
如实施例22所述的,根据DuoBody平台体外产生具有或不具有E345R突变的双特异性CD38×EGFR和CD20×EGFR抗体。测试CD38×EGFR双特异性抗体针对CD38阳性、EGFR阴性Raji细胞(双特异性抗体只能通过CD38单价地结合该细胞)的CDC效力。在圆底96孔板中,将0.1×106个Raji细胞与饱和浓度的抗体(10.0μg/ml)在总体积100μl中室温下在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育不同的时间段,0-60分钟变化。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
图25显示,双特异性抗体CD38×EGFR(IgG1-005-K409R× IgG1-2F8-F405L)诱导了CDC介导的最大杀伤55%,这在大约10分钟后达到。 E345R的引入导致最大杀伤提高(96%),这在5分钟之内就已经达到。
双特异性抗体CD20×EGFR(IgG1-7D8-K409R× IgG1-2F8-F405L)诱导了CDC介导的最大杀死为85%,这在大约5分钟后达到。然而,使用具有E345R引入的CD20×EGFR抗体,更快观察到85%裂解(2分钟后)。E345R CD20×EGFR抗体的最大裂解(97%)同样在5分钟后达到。
总而言之,在这些单价结合抗体中引入E345R突变导致更为有力的抗体,这是由于提高的最大裂解以及更快的CDC反应动力学所造成的。
实施例31:治疗性和E345R/Q386K抗体组合的CDC
如实施例19中所述,衍生自IgG1-005的突变体CD38抗体当野生型抗体的 E345位置被除谷氨酸(E)之外的任何其他氨基酸取代时能针对Wien 133细胞诱导有效的CDC。这意味着,寡聚化(作为CDC的先决条件)由于抗体345位置处存在谷氨酸侧链而被阻碍。由于在六聚抗体环结构中一条Fc上的E345紧邻着对面第二条Fc部分上的Q386,因此有可能通过将第二个抗体的Q386位置进行取代而移除第一个抗体中E345-介导的寡聚化阻碍。在两个抗体组合的情况下这将使得第一个抗体的E345能够与第二个抗体的突变386位置更好地相互作用。为了测试这个假设,在Wien133上进行CDC测定法,其中将野生型抗体(IgG1-003、IgG1-005或IgG1-11B8)与IgG1-005-E345R/Q386K或 IgG1-005-E345R/Q386K/E430G混合作为实例。
在圆底96孔板中,将0.1×106个Wien133细胞与系列浓度的未纯化的 IgG1-005-E345R/Q386K、IgG1-005-E345R/Q386K/E430G或对照抗体 (0.0001-20.0μg/ml以3.33倍稀释)在存在或不存在1.0或10.0μg/ml野生型 IgG1-003、IgG1-005或IgG1-11B8抗体的情况下在总体积100μl中室温下在摇动器中预温育15分钟。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
图26A/B/C显示CD38抗体IgG1-005-E345R/Q386K以剂量依赖的方式诱导Wien133细胞的CDC介导的裂解(虚线)。将IgG1-005-E345R/Q386K与1或10 μg/ml野生型CD38抗体IgG1-003(图26A)或野生型CD20抗体IgG1-11B8(图 26B)组合导致提高的最大细胞裂解。将IgG1-005-E345R/Q386K与野生型 IgG1-005组合以剂量依赖的方式抑制CDC,可能是通过竞争结合位点实现(图 26C)。
图26D/E/F显示了CD38抗体IgG1-005-E345R/Q386K/E430G的相似的结果。
这些数据指示,野生型抗体IgG1-003和IgG1-11B8当与 IgG1-005-E345R/Q386K或IgG1-005-E345R/Q386K/E430G组合时参与了抗体的寡聚化和CDC激活。在这些组合中,存在于野生型抗体中的由E345-位置造成的寡聚化阻碍能够至少部分被突变抗体中的Q386K取代消除。本申请对改善使用E345位置为野生型的抗体(例如利妥昔单抗、奥法木单抗、daratumumab或曲妥珠单抗)的治疗特别感兴趣。此外,这种寡聚化诱导抗体能够促进与针对靶细胞(像肿瘤细胞或细菌)的患者自身抗体形成细胞结合复合体。
实施例19描述了除E345之外突变后增强CDC的多个氨基酸,例如E430 和S440,其中特定突变在掺入CD38抗体IgG1-005中时诱导了针对Wien133细胞的有效CDC。除了I253和Y436突变体外,在六聚环结构中所鉴定的寡聚化增强突变与对面第二Fc部分的未突变氨基酸相接触。因此,预计所鉴定的寡聚化增强突变(单独或组合两者)也可以促进与未突变抗体的寡聚化,并且可通过与实施例19中所应用的相似的选择策略来完成这种突变体的进一步优化。
实施例32:IgG2、IgG3和IgG4抗体同种型中E345R诱导的CDC
为了测试引入促进寡聚化的突变是否能够刺激非IgG1抗体同种型的 CDC活性,通过本领域已知方法来产生具有人IgG2、IgG3或IgG4恒定区的 CD38抗体IgG1-005同种型变体,生成IgG2-005、IgG3-005和IgG4-005。此外,将增强寡聚化的E345R突变引入所有这些抗体中,生成IgG2-005-E345R、 IgG3-005-E345R和IgG4-005-E345R。从CD38抗体IgG1-003以类似的方式产生 IgG2-003和IgG2-003-E345R。在体外CDC测定中比较不同同种型的CDC效力。
在圆底96孔板中,将0.1×106个Wien133细胞与10μg/ml的未纯化抗体在总体积100μl中室温下在摇动器中预温育15分钟。以3.0μg/ml加入 IgG1-005-E345R。接着,加入25μl正常人血清作为补体来源(20%终浓度),在37℃的培养箱中温育45分钟。将平板置于冰上以终止反应。加入10μl碘化丙啶并通过FACS确定细胞的裂解。
图27显示,IgG2-005、IgG2-003、IgG3-005和IgG4-005在所测试的条件下不能有效裂解(A)Daudi或(B)Wien133细胞(所观察到~20%的裂解被认为是背景)。E345R突变的引入实现所有所测试的IgG同种型的针对Daudi细胞的有力CDC。使用针对Wien133细胞的CDC证实了这些结果,尽管相对于其他的同种型变体IgG3-005-E345R展示出有限的CDC活性。这些数据指示除了 IgG1,增强寡聚化的突变,如E345也能用于促进IgG2、IgG3和IgG4抗体的CDC活性。
实施例33:在离体CDC测定中IgG1-005和IgG1-005-E345R的针对患者来源 CD38阳性B细胞慢性淋巴细胞性白血病(CLL)细胞的CDC
来自于CLL患者样本的冷冻保存的原代细胞获自CDB-IDIBAPS-临床医院(Dr.Elias Campo,血液科,病理部,临床医院,Institutd’Investigacions BiomediquesAugust Pi I Sunyer(IDIBAPS),巴塞罗那大学,巴塞罗那,西班牙)的血液病理生物库,或国家心、肺和血液研究所(NHLBI)的临床研究(Dr. Adrian Wiestner,NHLBI,国家健康研究所的血液学分支机构(Hematology Branch of the Naional Insitutes of Health(NIH)),Bethesda)。根据临床医院的机构伦理委员会(巴塞罗那,西班牙)或NIH的机构审查委员会和赫尔辛基宣言,从所有患者获得知情同意书。所有样本都进行遗传和免疫表型表征。
将CLL样本根据如FACS所确定的它们的CD38表达分类成两组:5个样品包含于CD38高组(Daudi细胞上50%-98%的CD38表达),4个样品包含于CD38 低组(Daudi细胞上0.5%-3%的CD38表达)。
将荧光标记的CLL细胞(用5μM钙黄绿素(Calcein)AM标记)与系列浓度的抗体(0.01-10μg/ml以10倍稀释)一起温育。接着,将正常人类血清作为补体来源(10%终浓度)加入至抗体调理的细胞(100,000细胞/孔)中并在37℃温育 45分钟。回收上清液,在SynergyTM HT荧光计数器上读取荧光作为细胞裂解的测量。如下计算细胞杀伤:特异性裂解=100×(样品自发裂解)/(最大裂解- 自发裂解),其中最大裂解由用1%Triton处理的细胞样品确定,而自发裂解则由在存在不含抗体的10%NHS的情况中温育细胞的样品确定。
图28显示,相比起野生型IgG1-005,IgG1-005-E345R强烈地增强了针对具有高CD38表达的CLL原代细胞以及具有低CD38表达的CLL原代细胞两者的CDC效力。
实施例34:与野生型IgG1-005相比IgG1-005突变体的FcRn结合
新生儿Fc受体(FcRn)通过保护IgG免于降解而负责IgG的长血浆半衰期。抗体内化后,FcRn在内体中与抗体Fc区结合,其中相互作用在温和的酸性环境(pH为6.0)是稳定的。再循环至质膜后,其中环境是中性(pH 7.4)的,所述相互作用会丧失并且抗体可以释放回循环中。这影响IgG的血浆半衰期。
IgG1-005突变体E345K,E345Q,E345R,E345Y,E430F,E430G,E430S, E430T,S440Y,K439E和S440K与来自小鼠,猕猴和人的FcRn相互作用的能力在ELISA中进行了测试。在小鼠FcRn的ELISA中,突变体P247G 和I253D也被测试。对于与FcRn的结合,将I253D用作阴性对照。所有温育在室温下进行。用稀释在PBST加0.2%BSA中的5μg/mL(100μL/孔)重组产生的生物素化的FcRn胞外域(小鼠,人或猕猴)(FcRnECDHis-B2M-BIO) 包被96孔板,并温育1小时。用PBST洗涤板3次,并且加入3-倍系列稀释(在PBST/0.2%BSA,pH值6.0)的野生型IgG1-005或005的突变体,并且将平板温育1小时。用PBST/0.2%BSA,pH值6.0洗涤板。加入在PBST/0.2%BSA,pH值6.0中稀释的山羊抗-人IgG(Fab'2)-HRP(Jackson Immuno Research,目录号:109-035-097),并将板温育1小时。洗涤后,加入作为底物的ABTS溶液,并在黑暗中温育板30分钟。用EL808ELISA 酶标仪在405nm读取吸光度。使用对数转换浓度,使用非线性激动剂的剂量–响应拟合的最佳拟合值在GraphPad PRISM5中分析在小鼠FcRn的ELISA 中产生的数据,并计算表观亲和力(EC 50)(表20)。实验表明,与野生型IgG1-005相比,任何IgG1-005突变体未改变与FcRn的结合。
表20:IgG1-005和突变体与小鼠FcRn的表观亲和力(EC 50)(μg/ml)

图29显示野生型IgG1-005和所有测试的IgG1-005突变体在pH6.0很好地结合小鼠,人和猕猴FcRn。在pH7.4未检测到与FcRn的显著结合(数据未显示)。
实施例35:在利妥昔单抗中的不同突变在B细胞系Ramos和SU-DHL-4中增强CDC
如实施例19中所述,抗-CD38抗体IgG1-005的寡聚和CDC活性可通过Fc:Fc界面上或周围的特定残基的单个突变来刺激。寡聚也可以通过远离 Fc:Fc界面的残基处的另一种类型的突变来间接刺激,这变构增强Fc:Fc的相互作用。这也对IgG1抗CD20抗体利妥昔单抗在两个B细胞系(Ramos和 SU-DHL-4)上测试。测试了下列突变:E345K,E345Q,E345R,E345Y,E430G, E430S,E430T和S440Y(基本上如实施例19中所述)。
对于CDC测定,在圆底96孔板中使0.1x106个细胞(Ramos或SU-DHL-4) (0.0001-10.0μg/mL以3倍稀释)与饱和抗体浓度系列在100μL的总体积中在摇动器上在室温下预温育15分钟。接着,加入25μL正常人血清作为补体来源(20%的终浓度),并在37℃的培养箱中温育45分钟。通过将板放在冰上终止反应。加入10μL的碘化丙啶溶液,并且通过FACS测定细胞裂解。使用对数转换浓度,使用非线性激动剂的剂量–响应拟合的最佳拟合值在GraphPadPRISM5中分析数据。图30显示,所有测试的利妥昔单抗突变体能够在这两个B细胞系中增加CDC效力。
实施例36:不依赖于靶标的流体相补体激活:IgG1-005突变体与野生型 IgG1-005相比较
当抗体在例如血流或器官组织中激活补体时,不依赖于靶标的补体激活可能构成安全问题。这可能会导致不想要的补体激活产物或不想要的补体沉积。为了测试不依赖于靶标的流体相补体激活,将100μg/ml的IgG1-005 突变体E345K,E345Q,E345R,E345Y,E430F,E430G,E430S,E430T, S440Y,野生型IgG1-005或热聚集的IgG(HAG,阳性对照)在90%正常人血清中在37℃温育1小时。然后将样品转移到ELISA试剂盒,来测量C4d 的产生(Micro Vue C4d-fragment,Quidel,San Diego,CA,USA)。C4d是C4的活化片段,C4是经典补体途径激活的标记。
图31显示,野生型IgG1-005,IgG1-005-E345K,IgG1-005-E345Q,IgG1-005-E345Y,IgG1-005-E430G,IgG1-005-E430S和IgG1-005-S440Y展示最小的C4激活,而IgG1-005-E345R,IgG1-005-E430F和IgG1-005-E430T 展示相比于野生型IgG1-005增加的C4d的产生(C4激活)。
实施例37:IgG1-005突变体与野生型IgG1-005相比的血浆清除率
在这项研究中的小鼠被安置在Central Laboratory Animal Facility(Utrecht,The Netherlands)的屏障单位(barrier unit)中,并饲养在滤盖饲养笼中,其中水和食物无限制地提供。所有实验均得到了Utrecht University动物伦理委员会的批准。使用500μg抗体,每组使用3只小鼠,静脉内注射SCID 小鼠(C.B-17/Icr-Prkdc<Scid>/IcrIcoCrl,Charles-River)。
在抗体施用后10分钟,4小时,1天,2天,7天,14天和21天从隐静脉收集50μL的血液样品。将血液收集到含有肝素的小瓶中并在10,000g下离心5分钟。在测定抗体浓度之前,将血浆储存在-20℃。
使用总hIgG和CD38特异性夹心ELISA测定特异性的人IgG浓度。
对于总hIgG ELISA,将以2μg/mL的浓度包被到96孔Microlon ELISA 板(Greiner,Germany)上的小鼠mAb抗人IgG-kappa克隆MH16(#M1268, CLB Sanquin,The Netherlands)用作捕获抗体。用补充了0.2%牛血清白蛋白的 PBS封闭板后,加入在ELISA缓冲液(补充了0.05%Tween 20和0.2%牛血清白蛋白的PBS)中连续稀释的样品,在室温(RT)在板摇动器上温育1 小时。随后用山羊抗人IgG免疫球蛋白(#109-035-098,Jackson,West Grace, PA)温育板,用2,2'-连氮基-双(3-乙基苯并噻唑-6-磺酸)(ABTS;Roche, Mannheim,Germany)显色。在微板酶标仪(Biotek,Winooski,VT)中测定 405nm处的吸光度。
对于特异性CD38ELISA,将His标记的CD38胞外域以2μg/mL的浓度包被到96孔Microlon ELISA板(Greiner,Germany)中。用ELISA缓冲液封闭板后,加入用ELISA缓冲液系列稀释的样品,并在室温(RT)在板摇动器上培养1小时。随后用30ng/ml小鼠抗人类IgG1-HRP,(Sanquin M1328, clone MH161-1)温育板,用2,2'-连氮基-双(3-乙基苯并噻唑-6-磺酸)(ABTS; Roche,Mannheim,Germany)显色。在微板酶标仪(Biotek,Winooski,VT)中测定405nm处的吸光度。
图32A显示了野生型参照抗体IgG1-005和抗体变体IgG1-005-E345K, IgG1-005-E345Q,IgG1-005-E345R,IgG1-005-E345Y,IgG1-005-E430F, IgG1-005-E430G,IgG1-005-E430S,IgG1-005-E430T,IgG1-005-S440Y的IgG 清除率。突变体IgG1-005-E430S,IgG1-005-E430G和IgG1-005-S440Y, IgG1-005-E430T,IgG1-005-E345K,IgG1-005-E345Q和IgG1-005-E345Y显示与野生型IgG1-005相似的清除率。突变体IgG1-005-E430F和 IgG1-005-E345R显示更快的清除率。将血浆清除率以剂量/AUC(mL/day/kg) 计算。AUC值(曲线下的面积)由浓度-时间曲线来确定。
图32B显示了当腹膜内施用8.0mg无关的IgG1-B12对照抗体后一天静脉内注射时,野生型参照抗体IgG1-005以及抗体变体IgG1-005-E345K, IgG1-005-E345R,IgG1-005-E430G,IgG1-005-E430S,和IgG1-005-S440Y 的IgG清除率,如通过CD38特异性ELISA测定的。在缺乏无关b12对照的情况下,将野生型参照抗体IgG1纳入作为对照。突变体IgG1-005-E430S, IgG1-005-E430G,IgG1-005-S440Y和IgG1-005-E345K显示与野生型类似的清除率。突变IgG1-005-E345R展示更快的清除率。
等同方案
本领域的技术人员将认识到,或仅仅使用常规的实验能够确认在此描述的本发明的具体实施方案的许多等同方案。这样的等同方案意图由所附权利要求所涵盖。在从属权利要求中公开的实施方案的任一种和所有组合也意图在本发明的范围之内。
































Claims (38)
1.一种提高亲本抗体的补体依赖性细胞毒性(CDC)的方法,所述亲本抗体是包含第一多肽和第二多肽的双特异性抗体,所述第一多肽包含免疫球蛋白的第一CH2-CH3区和第一抗原结合区,所述第二多肽包含免疫球蛋白的第二CH2-CH3区和第二抗原结合区,其中所述第一和第二抗原结合区结合相同或不同抗原上的不同表位,并且其中所述方法包括将E430G突变引入到人类IgG1重链Fc区中的所述第一和第二CH2-CH3区中,并且其中
所述第一CH2-CH3区包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的K409R和F405L的那些氨基酸突变;并且其中所述第二CH2-CH3区包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的F405L和K409R的那些氨基酸突变,并且其中在所述第一CH2-CH3区中的进一步氨基酸突变不同于在所述第二CH2-CH3区中的进一步氨基酸突变。
2.根据权利要求1的方法,其中所述第一CH2-CH3区的进一步氨基酸突变是人类IgG1重链Fc区中的K409R。
3.根据权利要求1的方法,其中所述第二CH2-CH3区的进一步氨基酸突变是人类IgG1重链Fc区中的F405L。
4.一种提高第一和第二双特异性抗体的组合的补体依赖性细胞毒性(CDC)的方法,其中所述第一和第二双特异性抗体各包含第一多肽和第二多肽,所述第一多肽包含免疫球蛋白的第一CH2-CH3区和第一抗原结合区,所述第二多肽包含第二CH2-CH3区和第二抗原结合区,其中所述第一和第二抗原结合区结合相同或不同抗原上的不同表位,并且每个双特异性抗体包含免疫球蛋白的Fc域,其中所述方法包括:
将E430G突变引入所述第一和第二多肽的人类IgG1重链Fc区中,
并且其中所述第一CH2-CH3区包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的K409R和F405L的那些氨基酸突变;并且其中所述第二CH2-CH3区包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的F405L和K409R的那些氨基酸突变,并且其中在所述第一CH2-CH3区中的进一步氨基酸突变不同于在所述第二CH2-CH3区中的进一步氨基酸突变。
5.根据权利要求4的方法,其中在人类IgG1重链Fc区的所述第一CH2-CH3区中的所述进一步氨基酸突变是K409R。
6.根据权利要求4的方法,其中在人类IgG1重链Fc区的所述第二CH2-CH3区中的所述进一步氨基酸突变是F405L。
7.根据权利要求1-6中任一项的方法,其中所述方法不改变所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的抗体依赖性细胞介导的细胞毒性(ADCC)。
8.根据权利要求1-7中任一项的方法,其中根据通过ELISA的测定,所述方法不改变所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体与新生儿Fc受体(FcRn)的结合。
9.根据权利要求1-8中任一项的方法,其中
a)根据ELISA测定的OD405 nm吸光度的变化测量,所述方法不会使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体与新生儿Fc受体(FcRn)的结合提高或降低超过30%;和/或
b)根据ELISA的测定,所述方法不使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体与小鼠新生儿Fc受体(FcRn)的表观亲和力提高超过0.5倍,或者不会使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体与小鼠新生儿Fc受体(FcRn)的表观亲和力降低超过2倍;和/或
c)根据ELISA的测定,所述方法不改变所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的血浆清除率;和/或
d)根据ELISA的测定,所述方法不使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的血浆清除率增加或降低超过3.0倍;和/或
e)根据ELISA的测定,所述方法不改变变体的不依赖于靶标的流体相补体激活;和/或
f)所述方法不改变所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的血浆半衰期。
10.根据权利要求9的方法,其中所述方法不会使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体与新生儿Fc受体(FcRn)的结合提高或降低超过20%。
11.根据权利要求9的方法,其中所述方法不会使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体与新生儿Fc受体(FcRn)的结合提高或降低超过10%。
12.根据权利要求9的方法,其中所述方法不会使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体与新生儿Fc受体(FcRn)的结合提高或降低超过5%。
13.根据权利要求9的方法,所述方法不使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的血浆清除率增加或降低超过2.5倍。
14.根据权利要求9的方法,所述方法不使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的血浆清除率增加或降低超过2.0倍。
15.根据权利要求9的方法,所述方法不使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的血浆清除率增加或降低超过1.5倍。
16.根据权利要求9的方法,所述方法不使所述第一多肽和所述第二多肽或根据权利要求1-3方法的亲本抗体的血浆清除率增加或降低超过1.2倍。
17.一种亲本抗体的变体,其是包含第一多肽和第二多肽的双特异性抗体,所述第一多肽包含免疫球蛋白的第一CH2-CH3区和第一抗原结合区,所述第二多肽包含免疫球蛋白的第二CH2-CH3区和第二抗原结合区,其中所述第一和第二抗原结合区结合相同抗原或不同抗原上的不同表位,并且其中所述第一和第二CH2-CH3区包含E430G突变,并且其中
所述第一多肽包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的K409R和F405L的那些氨基酸突变;并且
所述第二多肽包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的F405L和K409R的那些氨基酸突变,并且其中所述第一多肽中的进一步突变不同于所述第二多肽中的进一步突变。
18.根据权利要求17的变体,其中所述第一多肽包含人类IgG1重链Fc区中的K409R。
19.根据权利要求17的变体,其中所述第二多肽包含人类IgG1重链Fc区中的F405L。
20.根据权利要求17的变体,其中所述第一多肽包含人类IgG1重链Fc区中的F405L。
21.根据权利要求17的变体,其中所述第二多肽包含人类IgG1重链Fc区中的K409R。
22.根据权利要求17至21中任一项的变体,其中所述变体与毒素或放射性标记缀合。
23.根据权利要求22的变体,其中所述变体通过接头与毒素缀合。
24.根据权利要求17至23中任一项的变体,其中所述变体是人类IgG1抗体,任选地人类全长抗体。
25.根据权利要求24的变体,其中所述人类全长抗体是人类全长IgG1抗体。
26.组合物,其包含第一和第二双特异性抗体,其中所述第一和第二双特异性抗体包含第一多肽和第二多肽,所述第一多肽包含免疫球蛋白的第一CH2-CH3区和第一抗原结合区,所述第二多肽包含第二CH2-CH3区和第二抗原结合区,其中所述第一和第二抗原结合区结合相同抗原或不同抗原上的不同表位,且其中所述第一和第二CH2-CH3区包含突变,其为对应于E430G的突变,并且其中所述第一CH2-CH3区包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的K409R和F405L的那些氨基酸突变;并且其中所述第二CH2-CH3区包含进一步氨基酸突变,其选自对应于人类IgG1重链Fc区中的F405L和K409R的那些氨基酸突变,并且其中在所述第一CH2-CH3区中的进一步氨基酸突变与所述第二CH2-CH3区中的进一步氨基酸突变不同。
27.根据权利要求26的组合物,其中所述第一CH2-CH3区的进一步氨基酸突变是人类IgG1重链Fc区中的K409R。
28.根据权利要求26的组合物,其中所述第二CH2-CH3区的进一步氨基酸突变是人类IgG1重链Fc区中的F405L。
29.根据权利要求26至28中任一项的组合物,其中所述第一多肽和所述第二多肽中的一种或两者与毒素或放射性标记缀合。
30.根据权利要求29的组合物,其中所述第一多肽和所述第二多肽中的一种或两者通过接头与毒素缀合。
31.一种组合物,其包含根据权利要求17至25中任一项的变体或根据权利要求26至30中任一项的组合物,和药学上可接受的载体。
32.一种套组,其包含用于治疗时同时、分开或连续使用的根据权利要求17至25中任一项中定义的所述第一多肽和所述第二多肽。
33.根据权利要求17至32中任一项的变体、组合物或套组,用于治疗疾病。
34.根据权利要求33的变体、组合物或套组,其中所述疾病是癌症。
35.根据权利要求17至32中任一项的变体、组合物或套组在制备用于治疗人类中疾病的药物中的用途。
36.根据权利要求17至32中任一项的变体、组合物或套组在制备用于治疗人类中癌症的药物中的用途。
37.根据权利要求17至32中任一项的变体、组合物或套组,用于对人类或其他哺乳动物的身体的至少一部分成像。
38.根据权利要求17至32中任一项的变体、组合物或套组在制备用于对人类或其他哺乳动物的身体的至少一部分成像的药物中的用途。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202110783172.0A CN113735967A (zh) | 2013-01-10 | 2013-01-10 | 人类IgG1 FC区变体及其用途 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2013/050429 WO2014108198A1 (en) | 2013-01-10 | 2013-01-10 | Human igg1 fc region variants and uses thereof |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110783172.0A Division CN113735967A (zh) | 2013-01-10 | 2013-01-10 | 人类IgG1 FC区变体及其用途 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN105229026A CN105229026A (zh) | 2016-01-06 |
| CN105229026B true CN105229026B (zh) | 2021-07-30 |
Family
ID=47557156
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110783172.0A Pending CN113735967A (zh) | 2013-01-10 | 2013-01-10 | 人类IgG1 FC区变体及其用途 |
| CN201380073913.XA Active CN105229026B (zh) | 2013-01-10 | 2013-01-10 | 人类IgG1 FC区变体及其用途 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN202110783172.0A Pending CN113735967A (zh) | 2013-01-10 | 2013-01-10 | 人类IgG1 FC区变体及其用途 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20150353636A1 (zh) |
| EP (2) | EP2943506B1 (zh) |
| JP (1) | JP6377635B2 (zh) |
| KR (6) | KR20160007478A (zh) |
| CN (2) | CN113735967A (zh) |
| AU (4) | AU2013372331A1 (zh) |
| BR (1) | BR112015016561B1 (zh) |
| CA (1) | CA2896955C (zh) |
| EA (1) | EA201500741A1 (zh) |
| HK (1) | HK1217719A1 (zh) |
| IL (3) | IL283173B1 (zh) |
| MX (1) | MX2015008740A (zh) |
| SG (1) | SG11201505286TA (zh) |
| WO (1) | WO2014108198A1 (zh) |
Families Citing this family (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT2560993T (pt) * | 2010-04-20 | 2024-09-16 | Genmab As | Proteínas contendo anticorpo heterodimérico fc e métodos para a produção das mesmas |
| UA117901C2 (uk) * | 2011-07-06 | 2018-10-25 | Ґенмаб Б.В. | Спосіб посилення ефекторної функції вихідного поліпептиду, його варіанти та їх застосування |
| EP3674320A3 (en) | 2011-10-27 | 2020-08-12 | Genmab A/S | Production of heterodimeric proteins |
| SG11201408646VA (en) | 2012-07-06 | 2015-01-29 | Genmab Bv | Dimeric protein with triple mutations |
| AU2016246695A1 (en) | 2015-04-07 | 2017-10-26 | Genentech, Inc. | Antigen binding complex having agonistic activity and methods of use |
| CN108368510B (zh) | 2015-09-30 | 2023-09-01 | 詹森生物科技公司 | 特异性结合人cd40的激动性抗体和使用方法 |
| SG11201803520PA (en) | 2015-11-03 | 2018-05-30 | Janssen Biotech Inc | Antibodies specifically binding pd-1 and their uses |
| WO2017093448A1 (en) * | 2015-12-01 | 2017-06-08 | Genmab B.V. | Anti-dr5 antibodies and methods of use thereof |
| WO2017165464A1 (en) | 2016-03-21 | 2017-09-28 | Elstar Therapeutics, Inc. | Multispecific and multifunctional molecules and uses thereof |
| WO2017198731A1 (en) * | 2016-05-18 | 2017-11-23 | Genmab B.V. | Antibodies and methods of use thereof in treatment of infectious disease |
| EP3497126A4 (en) | 2016-08-12 | 2020-04-08 | Janssen Biotech, Inc. | ANTIBODIES OF FC MODIFIED ANTI-TNFR SUPERFAMILY HAVING IMPROVED AGONIST ACTIVITY AND METHODS OF USE THEREOF |
| CN109863170B (zh) | 2016-08-12 | 2024-08-16 | 詹森生物科技公司 | 具有增强的激动作用和效应子功能的工程化抗体及其他含Fc结构域分子 |
| BR112019008702A2 (pt) | 2016-11-01 | 2019-07-16 | Genmab B.V. | polipeptídeo, método de diminuição de uma função fc efetora de um polipeptídeo, composição, método de tratamento, kit de partes, e, uso de um polipeptídeo ou composição. |
| EP3576793A4 (en) * | 2017-02-06 | 2021-03-31 | Dana Farber Cancer Institute, Inc. | COMPOSITIONS AND PROCEDURES FOR ENHANCING ANTIBODY-MEDIATED RECEPTOR SIGNALING |
| AU2018219887B2 (en) | 2017-02-08 | 2024-08-15 | Dragonfly Therapeutics, Inc. | Multi-specific binding proteins for activation of natural killer cells and therapeutic uses thereof to treat cancer |
| SG11201906961UA (en) | 2017-02-10 | 2019-08-27 | Genmab Bv | Polypeptide variants and uses thereof |
| WO2018151820A1 (en) | 2017-02-16 | 2018-08-23 | Elstar Therapeutics, Inc. | Multifunctional molecules comprising a trimeric ligand and uses thereof |
| EP4273258A3 (en) * | 2017-02-20 | 2024-01-17 | Dragonfly Therapeutics, Inc. | Proteins binding her2, nkg2d and cd16 |
| NZ757603A (en) * | 2017-03-31 | 2024-05-31 | Genmab Holding B V | Bispecific anti-cd37 antibodies, monoclonal anti-cd37 antibodies and methods of use thereof |
| JP2020522254A (ja) | 2017-05-31 | 2020-07-30 | エルスター セラピューティクス, インコーポレイテッド | 骨髄増殖性白血病(mpl)タンパク質に結合する多特異性分子およびその使用 |
| CN111328335A (zh) * | 2017-06-07 | 2020-06-23 | 根马布私人有限公司 | 基于突变igg六聚体的治疗性抗体 |
| WO2019035938A1 (en) | 2017-08-16 | 2019-02-21 | Elstar Therapeutics, Inc. | MULTISPECIFIC MOLECULES BINDING TO BCMA AND USES THEREOF |
| RS61883B1 (sr) | 2017-09-19 | 2021-06-30 | Tillotts Pharma Ag | Varijante antitela |
| EP3456737B1 (en) | 2017-09-19 | 2024-02-14 | Tillotts Pharma Ag | Antibody variants |
| WO2019145455A1 (en) | 2018-01-24 | 2019-08-01 | Genmab B.V. | Polypeptide variants and uses thereof |
| AU2019218136A1 (en) | 2018-02-08 | 2020-08-13 | Dragonfly Therapeutics, Inc. | Antibody variable domains targeting the NKG2D receptor |
| WO2019178362A1 (en) | 2018-03-14 | 2019-09-19 | Elstar Therapeutics, Inc. | Multifunctional molecules that bind to calreticulin and uses thereof |
| WO2019178364A2 (en) | 2018-03-14 | 2019-09-19 | Elstar Therapeutics, Inc. | Multifunctional molecules and uses thereof |
| MX2020011552A (es) | 2018-05-03 | 2020-11-24 | Genmab Bv | Combinaciones de variantes de anticuerpos y usos de las mismas. |
| WO2019235426A1 (ja) * | 2018-06-04 | 2019-12-12 | 中外製薬株式会社 | 細胞質内での半減期が変化した抗原結合分子 |
| EP3810649A1 (en) | 2018-06-22 | 2021-04-28 | Genmab A/S | Method for producing a controlled mixture of two or more different antibodies |
| WO2019243636A1 (en) | 2018-06-22 | 2019-12-26 | Genmab Holding B.V. | Anti-cd37 antibodies and anti-cd20 antibodies, compositions and methods of use thereof |
| AU2019297451A1 (en) | 2018-07-03 | 2021-01-28 | Marengo Therapeutics, Inc. | Anti-TCR antibody molecules and uses thereof |
| WO2020014526A2 (en) * | 2018-07-11 | 2020-01-16 | Anthony Manning | Compositions and methods related to engineered fc-antigen binding domain constructs targeted to cd38 |
| BR112021000415A2 (pt) * | 2018-07-11 | 2021-04-06 | Momenta Pharmaceuticals, Inc. | Composições e métodos relacionados a construtos do domínio de ligação fc-antígeno manipulados |
| US20240254252A1 (en) * | 2018-07-13 | 2024-08-01 | Genmab A/S | Trogocytosis-mediated therapy using cd38 antibodies |
| TW202019518A (zh) | 2018-07-13 | 2020-06-01 | 丹麥商珍美寶股份有限公司 | Cd38抗體之變體及其用途 |
| US20240252635A1 (en) * | 2018-10-04 | 2024-08-01 | Genmab Holding B.V. | Pharmaceutical compositions comprising bispecific anti-cd37 antibodies |
| JP2022521937A (ja) | 2019-02-21 | 2022-04-13 | マレンゴ・セラピューティクス,インコーポレーテッド | NKp30に結合する抗体分子およびその使用 |
| JP2022523197A (ja) | 2019-02-21 | 2022-04-21 | マレンゴ・セラピューティクス,インコーポレーテッド | T細胞関連のがん細胞に結合する多機能性分子およびその使用 |
| AU2020224154A1 (en) | 2019-02-21 | 2021-09-16 | Marengo Therapeutics, Inc. | Multifunctional molecules that bind to calreticulin and uses thereof |
| AU2020224680A1 (en) | 2019-02-21 | 2021-09-16 | Marengo Therapeutics, Inc. | Multifunctional molecules that bind to T cells and uses thereof to treat autoimmune disorders |
| WO2020172596A1 (en) | 2019-02-21 | 2020-08-27 | Elstar Therapeutics, Inc. | Anti-tcr antibody molecules and thereof |
| JP2022531894A (ja) | 2019-05-09 | 2022-07-12 | ゲンマブ ビー.ブイ. | がんの処置において使用するための抗dr5抗体の組み合わせの投与レジメン |
| GB201910900D0 (en) | 2019-07-31 | 2019-09-11 | Scancell Ltd | Modified fc-regions to enhance functional affinity of antibodies and antigen binding fragments thereof |
| EP4055046A1 (en) | 2019-11-06 | 2022-09-14 | Genmab B.V. | Antibody variant combinations and uses thereof |
| EP3831849A1 (en) | 2019-12-02 | 2021-06-09 | LamKap Bio beta AG | Bispecific antibodies against ceacam5 and cd47 |
| WO2021138407A2 (en) | 2020-01-03 | 2021-07-08 | Marengo Therapeutics, Inc. | Multifunctional molecules that bind to cd33 and uses thereof |
| WO2021144457A1 (en) | 2020-01-16 | 2021-07-22 | Genmab A/S | Formulations of cd38 antibodies and uses thereof |
| US20230140384A1 (en) | 2020-03-09 | 2023-05-04 | Bristol-Myers Squibb Company | Antibodies to cd40 with enhanced agonist activity |
| US20230159662A1 (en) * | 2020-04-01 | 2023-05-25 | Kyowa Kirin Co., Ltd. | Antibody composition |
| JP2023523011A (ja) | 2020-04-24 | 2023-06-01 | マレンゴ・セラピューティクス,インコーポレーテッド | T細胞関連のがん細胞に結合する多機能性分子およびその使用 |
| JP2023534726A (ja) | 2020-07-23 | 2023-08-10 | ジェンマブ ビー.ブイ. | 多発性骨髄腫の治療に使用するための抗dr5抗体と免疫調節イミド薬との併用 |
| CN116249718A (zh) | 2020-08-26 | 2023-06-09 | 马伦戈治疗公司 | 结合至钙网蛋白的多功能性分子及其用途 |
| CN116761818A (zh) | 2020-08-26 | 2023-09-15 | 马伦戈治疗公司 | 检测trbc1或trbc2的方法 |
| CN116917316A (zh) | 2020-08-26 | 2023-10-20 | 马伦戈治疗公司 | 与NKp30结合的抗体分子及其用途 |
| US20230365714A1 (en) | 2020-10-02 | 2023-11-16 | Genmab A/S | Antibodies capable of binding to ror2 and bispecific antibodies binding to ror2 and cd3 |
| CA3214757A1 (en) | 2021-04-08 | 2022-10-13 | Andreas Loew | Multifuntional molecules binding to tcr and uses thereof |
| KR20240051280A (ko) | 2021-09-06 | 2024-04-19 | 젠맵 에이/에스 | Cd27과 결합할 수 있는 항체, 그의 변이체 및 그의 용도 |
| WO2023218051A1 (en) | 2022-05-12 | 2023-11-16 | Genmab A/S | Binding agents capable of binding to cd27 in combination therapy |
| WO2023218046A1 (en) | 2022-05-12 | 2023-11-16 | Genmab A/S | Binding agents capable of binding to cd27 in combination therapy |
| WO2024094660A1 (en) | 2022-10-31 | 2024-05-10 | Genmab A/S | Cd38 antibodies and uses thereof |
| WO2024119049A1 (en) * | 2022-12-02 | 2024-06-06 | Lytica Therapeutics, Inc. | Compositions and methods for protein internalization |
Family Cites Families (105)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| JPS5896026A (ja) | 1981-10-30 | 1983-06-07 | Nippon Chemiphar Co Ltd | 新規ウロキナ−ゼ誘導体およびその製造法ならびにそれを含有する血栓溶解剤 |
| EP0098110B1 (en) | 1982-06-24 | 1989-10-18 | NIHON CHEMICAL RESEARCH KABUSHIKI KAISHA also known as JAPAN CHEMICAL RESEARCH CO., LTD | Long-acting composition |
| US4681581A (en) | 1983-12-05 | 1987-07-21 | Coates Fredrica V | Adjustable size diaper and folding method therefor |
| US4766106A (en) | 1985-06-26 | 1988-08-23 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| US5101827A (en) | 1985-07-05 | 1992-04-07 | Immunomedics, Inc. | Lymphographic and organ imaging method and kit |
| US4735210A (en) | 1985-07-05 | 1988-04-05 | Immunomedics, Inc. | Lymphographic and organ imaging method and kit |
| US5776093A (en) | 1985-07-05 | 1998-07-07 | Immunomedics, Inc. | Method for imaging and treating organs and tissues |
| US5648471A (en) | 1987-12-03 | 1997-07-15 | Centocor, Inc. | One vial method for labeling antibodies with Technetium-99m |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| ATE172879T1 (de) | 1989-08-09 | 1998-11-15 | Rhomed Inc | Direkte radioetikettierung von antikörpern und sonstigen proteinen mittels technetium oder rhenium |
| US6300129B1 (en) | 1990-08-29 | 2001-10-09 | Genpharm International | Transgenic non-human animals for producing heterologous antibodies |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5877397A (en) | 1990-08-29 | 1999-03-02 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5814318A (en) | 1990-08-29 | 1998-09-29 | Genpharm International Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5789650A (en) | 1990-08-29 | 1998-08-04 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| ATE158021T1 (de) | 1990-08-29 | 1997-09-15 | Genpharm Int | Produktion und nützung nicht-menschliche transgentiere zur produktion heterologe antikörper |
| US5874299A (en) | 1990-08-29 | 1999-02-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| AU2235992A (en) | 1991-06-14 | 1993-01-12 | Genpharm International, Inc. | Transgenic immunodeficient non-human animals |
| WO1993001227A1 (en) | 1991-07-08 | 1993-01-21 | University Of Massachusetts At Amherst | Thermotropic liquid crystal segmented block copolymer |
| US5733743A (en) | 1992-03-24 | 1998-03-31 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| US5635483A (en) | 1992-12-03 | 1997-06-03 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Tumor inhibiting tetrapeptide bearing modified phenethyl amides |
| US5780588A (en) | 1993-01-26 | 1998-07-14 | Arizona Board Of Regents | Elucidation and synthesis of selected pentapeptides |
| JPH08509612A (ja) | 1993-04-26 | 1996-10-15 | ジェンファーム インターナショナル インコーポレイテッド | 異種抗体を産生することができるトランスジェニック非ヒト動物 |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| US5663149A (en) | 1994-12-13 | 1997-09-02 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Human cancer inhibitory pentapeptide heterocyclic and halophenyl amides |
| IL132560A0 (en) | 1997-05-02 | 2001-03-19 | Genentech Inc | A method for making multispecific antibodies having heteromultimeric and common components |
| US7951917B1 (en) * | 1997-05-02 | 2011-05-31 | Genentech, Inc. | Method for making multispecific antibodies having heteromultimeric and common components |
| DE69937291T2 (de) | 1998-04-02 | 2008-07-10 | Genentech, Inc., South San Francisco | Antikörpervarianten und fragmente davon |
| DK2180007T4 (da) | 1998-04-20 | 2017-11-27 | Roche Glycart Ag | Glycosyleringsteknik for antistoffer til forbedring af antistofafhængig cellecytotoxicitet |
| KR20060067983A (ko) | 1999-01-15 | 2006-06-20 | 제넨테크, 인크. | 효과기 기능이 변화된 폴리펩티드 변이체 |
| US6737056B1 (en) | 1999-01-15 | 2004-05-18 | Genentech, Inc. | Polypeptide variants with altered effector function |
| EP2275541B1 (en) | 1999-04-09 | 2016-03-23 | Kyowa Hakko Kirin Co., Ltd. | Method for controlling the activity of immunologically functional molecule |
| DE60037896D1 (de) | 1999-07-29 | 2008-03-13 | Medarex Inc | Menschliche antikörper gegen her2/neu |
| EP3214175A1 (en) | 1999-08-24 | 2017-09-06 | E. R. Squibb & Sons, L.L.C. | Human ctla-4 antibodies and their uses |
| US7504256B1 (en) | 1999-10-19 | 2009-03-17 | Kyowa Hakko Kogyo Co., Ltd. | Process for producing polypeptide |
| EP1333032A4 (en) | 2000-10-06 | 2005-03-16 | Kyowa Hakko Kogyo Kk | METHOD FOR PURIFYING ANTIBODIES |
| EA013224B1 (ru) | 2000-10-06 | 2010-04-30 | Киова Хакко Кирин Ко., Лтд. | Клетки, продуцирующие композиции антител |
| US6946292B2 (en) | 2000-10-06 | 2005-09-20 | Kyowa Hakko Kogyo Co., Ltd. | Cells producing antibody compositions with increased antibody dependent cytotoxic activity |
| PT1354034E (pt) | 2000-11-30 | 2008-02-28 | Medarex Inc | Roedores transgénicos transcromossómicos para produção de anticorpos humanos |
| ATE489395T1 (de) | 2000-12-12 | 2010-12-15 | Medimmune Llc | Moleküle mit längeren halbwertszeiten, zusammensetzungen und deren verwendung |
| EP1243276A1 (en) | 2001-03-23 | 2002-09-25 | Franciscus Marinus Hendrikus De Groot | Elongated and multiple spacers containing activatible prodrugs |
| AU2002303929B9 (en) | 2001-05-31 | 2007-01-25 | E. R. Squibb & Sons, L.L.C. | Cytotoxins, prodrugs, linkers and stabilizers useful therefor |
| US7595378B2 (en) | 2001-06-13 | 2009-09-29 | Genmab A/S | Human monoclonal antibodies to epidermal growth factor receptor (EGFR) |
| CN1966525A (zh) | 2001-06-13 | 2007-05-23 | 根马布股份公司 | 表皮生长因子受体(egfr)的人单克隆抗体 |
| AU2002337935B2 (en) | 2001-10-25 | 2008-05-01 | Genentech, Inc. | Glycoprotein compositions |
| CA2965865C (en) | 2002-07-18 | 2021-10-19 | Merus N.V. | Recombinant production of mixtures of antibodies |
| US7659241B2 (en) | 2002-07-31 | 2010-02-09 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| MXPA05004022A (es) | 2002-10-17 | 2005-10-05 | Genmab As | Anticuerpos monoclonales humanos contra cd20. |
| JP2006507322A (ja) | 2002-11-14 | 2006-03-02 | シンタルガ・ビーブイ | 多重自己脱離放出スペーサーとして構築されたプロドラッグ |
| WO2004045512A2 (en) | 2002-11-15 | 2004-06-03 | Genmab A/S | Human monoclonal antibodies against cd25 |
| AU2003299641C1 (en) | 2002-12-16 | 2016-06-02 | Cormorant Pharmaceuticals Ab | Human monoclonal antibodies against interleukin 8 (IL-8) |
| BR122018071808B8 (pt) | 2003-11-06 | 2020-06-30 | Seattle Genetics Inc | conjugado |
| CA2545603A1 (en) | 2003-11-12 | 2005-05-26 | Biogen Idec Ma Inc. | Neonatal fc receptor (fcrn)-binding polypeptide variants, dimeric fc binding proteins and methods related thereto |
| CN1918178B (zh) * | 2004-01-12 | 2012-08-22 | 应用分子进化公司 | Fc区变体 |
| WO2005103081A2 (en) | 2004-04-20 | 2005-11-03 | Genmab A/S | Human monoclonal antibodies against cd20 |
| EP1776384B1 (en) * | 2004-08-04 | 2013-06-05 | Mentrik Biotech, LLC | Variant fc regions |
| US8802820B2 (en) * | 2004-11-12 | 2014-08-12 | Xencor, Inc. | Fc variants with altered binding to FcRn |
| EP2845865A1 (en) | 2004-11-12 | 2015-03-11 | Xencor Inc. | Fc variants with altered binding to FcRn |
| US7741568B2 (en) | 2005-01-13 | 2010-06-22 | The Wiremold Company | Downward facing receptacle assembly for cable raceway |
| CN107033243B (zh) * | 2005-03-23 | 2020-12-15 | 根马布股份公司 | 用于治疗多发性骨髓瘤的cd38抗体 |
| US20110123440A1 (en) | 2005-03-29 | 2011-05-26 | Genevieve Hansen | Altered Antibody FC Regions and Uses Thereof |
| EP3050963B1 (en) | 2005-03-31 | 2019-09-18 | Chugai Seiyaku Kabushiki Kaisha | Process for production of polypeptide by regulation of assembly |
| CA2614181A1 (en) * | 2005-07-01 | 2007-01-11 | Medimmune, Inc. | An integrated approach for generating multidomain protein therapeutics |
| AU2006277117B2 (en) | 2005-08-05 | 2013-01-10 | Syntarga B.V. | Triazole-containing releasable linkers and conjugates comprising the same |
| US7612181B2 (en) | 2005-08-19 | 2009-11-03 | Abbott Laboratories | Dual variable domain immunoglobulin and uses thereof |
| WO2007059782A1 (en) | 2005-11-28 | 2007-05-31 | Genmab A/S | Recombinant monovalent antibodies and methods for production thereof |
| AU2007210377B2 (en) | 2006-02-02 | 2012-08-09 | Georg-August-Universitat Gottingen Stiftung Offentlichen Rechts (Ohne Bereich Humanmedizin) | Water-soluble CC-1065 analogs and their conjugates |
| JP5374359B2 (ja) | 2006-03-17 | 2013-12-25 | バイオジェン・アイデック・エムエイ・インコーポレイテッド | 安定化されたポリペプチド化合物 |
| JP5474531B2 (ja) | 2006-03-24 | 2014-04-16 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | 操作されたヘテロ二量体タンパク質ドメイン |
| AT503902B1 (de) | 2006-07-05 | 2008-06-15 | F Star Biotech Forsch & Entw | Verfahren zur manipulation von immunglobulinen |
| EP2087111A2 (en) * | 2007-03-19 | 2009-08-12 | Medimmune Limited | Polypeptide variants |
| EP2626372B1 (en) | 2007-03-29 | 2018-03-21 | Genmab A/S | Bispecific antibodies and methods for production thereof |
| CN101821288A (zh) | 2007-06-21 | 2010-09-01 | 宏观基因有限公司 | 共价双抗体及其用途 |
| CA2695297C (en) | 2007-08-01 | 2017-03-21 | Syntarga B.V. | Substituted cc-1065 analogs and their conjugates |
| EP3255144A1 (en) | 2007-08-10 | 2017-12-13 | E. R. Squibb & Sons, L.L.C. | Recombineering construct for preparing transgenic mice capable of producing human immunoglobulin |
| HUE028536T2 (en) | 2008-01-07 | 2016-12-28 | Amgen Inc | Method for producing antibody to FC heterodimer molecules using electrostatic control effects |
| DE102008039957A1 (de) | 2008-08-27 | 2010-03-04 | Continental Automotive Gmbh | Verfahren zur Ermittlung eines Unfallschwerekriteriums mittels eines Beschleunigungssignals und eines Körperschallsignals |
| JP5913980B2 (ja) * | 2008-10-14 | 2016-05-11 | ジェネンテック, インコーポレイテッド | 免疫グロブリン変異体及びその用途 |
| BRPI0921687A8 (pt) | 2008-11-03 | 2022-11-08 | Syntarga Bv | Composto , conjugado , uso de um composto , composição farmacêutica, processo para preparar uma composição famacêutica , método para tratar um mamífero em necessidade do mesmo ,e, método para tratar ou prevenir um tumor em um mamífero. |
| UA109633C2 (uk) | 2008-12-09 | 2015-09-25 | Антитіло людини проти тканинного фактора | |
| KR20110104032A (ko) | 2008-12-19 | 2011-09-21 | 마크로제닉스, 인크. | 공유결합형 디아바디 및 이의 용도 |
| EP2233500A1 (en) * | 2009-03-20 | 2010-09-29 | LFB Biotechnologies | Optimized Fc variants |
| EP2424567B1 (en) | 2009-04-27 | 2018-11-21 | OncoMed Pharmaceuticals, Inc. | Method for making heteromultimeric molecules |
| EP3916011A1 (en) | 2009-06-26 | 2021-12-01 | Regeneron Pharmaceuticals, Inc. | Readily isolated bispecific antibodies with native immunoglobulin format |
| US9493578B2 (en) | 2009-09-02 | 2016-11-15 | Xencor, Inc. | Compositions and methods for simultaneous bivalent and monovalent co-engagement of antigens |
| RU2580038C2 (ru) | 2009-12-04 | 2016-04-10 | Дженентек, Инк. | Мультиспецифические антитела, аналоги антител, композиции и способы |
| SG10201501767VA (en) | 2010-03-10 | 2015-05-28 | Genmab As | Monoclonal antibodies against c-met |
| AR080794A1 (es) | 2010-03-26 | 2012-05-09 | Hoffmann La Roche | Anticuerpos bivalentes biespecificos anti- vegf/ anti-ang-2 |
| PT2560993T (pt) | 2010-04-20 | 2024-09-16 | Genmab As | Proteínas contendo anticorpo heterodimérico fc e métodos para a produção das mesmas |
| JP6022444B2 (ja) | 2010-05-14 | 2016-11-09 | ライナット ニューロサイエンス コーポレイション | ヘテロ二量体タンパク質ならびにそれを生産および精製するための方法 |
| CN107253992B (zh) | 2010-05-27 | 2022-03-11 | 根马布股份公司 | 针对her2的单克隆抗体 |
| JP6082344B2 (ja) | 2010-05-27 | 2017-02-15 | ゲンマブ エー/エス | Her2エピトープに対するモノクローナル抗体 |
| US9168314B2 (en) | 2010-06-15 | 2015-10-27 | Genmab A/S | Human antibody drug conjugates against tissue factor |
| MX340558B (es) | 2010-08-24 | 2016-07-14 | F Hoffmann-La Roche Ag * | Anticuerpos biespecificos que comprenden fragmento fv estabilizado con disulfuro. |
| BR112013002167A2 (pt) | 2010-08-24 | 2016-05-31 | Roche Glycart Ag | anticorpo biespecífico, composição farmacêutica, uso, método de tratamento de um paciente que sofre de câncer e de um paciente que sofre de inflamação |
| RS59589B1 (sr) | 2010-11-05 | 2019-12-31 | Zymeworks Inc | Dizajniranje stabilnog heterodimernog antitela sa mutacijama u fc domenu |
| JP5972915B2 (ja) * | 2011-03-16 | 2016-08-17 | アムジエン・インコーポレーテツド | Fc変異体 |
| CN102250246A (zh) | 2011-06-10 | 2011-11-23 | 常州亚当生物技术有限公司 | 抗VEGF/PDGFRβ双特异性抗体及其应用 |
| UA117901C2 (uk) * | 2011-07-06 | 2018-10-25 | Ґенмаб Б.В. | Спосіб посилення ефекторної функції вихідного поліпептиду, його варіанти та їх застосування |
| EP2729496B8 (en) | 2011-07-06 | 2017-10-18 | Genmab A/S | Modulation of complement-dependent cytotoxicity through modifications of the c-terminus of antibody heavy chains |
-
2013
- 2013-01-10 WO PCT/EP2013/050429 patent/WO2014108198A1/en active Application Filing
- 2013-01-10 KR KR1020157021402A patent/KR20160007478A/ko not_active Application Discontinuation
- 2013-01-10 KR KR1020247026237A patent/KR20240123420A/ko active Search and Examination
- 2013-01-10 KR KR1020227039306A patent/KR20220156667A/ko active Application Filing
- 2013-01-10 US US14/760,135 patent/US20150353636A1/en not_active Abandoned
- 2013-01-10 AU AU2013372331A patent/AU2013372331A1/en not_active Abandoned
- 2013-01-10 IL IL283173A patent/IL283173B1/en unknown
- 2013-01-10 EP EP13700218.4A patent/EP2943506B1/en active Active
- 2013-01-10 EA EA201500741A patent/EA201500741A1/ru unknown
- 2013-01-10 SG SG11201505286TA patent/SG11201505286TA/en unknown
- 2013-01-10 EP EP24163081.3A patent/EP4434543A2/en active Pending
- 2013-01-10 CA CA2896955A patent/CA2896955C/en active Active
- 2013-01-10 JP JP2015551994A patent/JP6377635B2/ja active Active
- 2013-01-10 CN CN202110783172.0A patent/CN113735967A/zh active Pending
- 2013-01-10 BR BR112015016561-3A patent/BR112015016561B1/pt active IP Right Grant
- 2013-01-10 CN CN201380073913.XA patent/CN105229026B/zh active Active
- 2013-01-10 MX MX2015008740A patent/MX2015008740A/es unknown
- 2013-01-10 IL IL313424A patent/IL313424A/en unknown
- 2013-01-10 KR KR1020207033594A patent/KR20200134340A/ko not_active Application Discontinuation
- 2013-01-10 KR KR1020207005732A patent/KR20200024345A/ko not_active Application Discontinuation
- 2013-01-10 KR KR1020217023772A patent/KR20210096697A/ko not_active Application Discontinuation
-
2015
- 2015-06-25 IL IL239633A patent/IL239633B/en unknown
-
2016
- 2016-05-19 HK HK16105710.0A patent/HK1217719A1/zh unknown
-
2018
- 2018-11-07 AU AU2018260863A patent/AU2018260863A1/en not_active Abandoned
-
2020
- 2020-07-13 AU AU2020205207A patent/AU2020205207A1/en not_active Abandoned
-
2023
- 2023-08-29 AU AU2023222851A patent/AU2023222851A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| IL239633A0 (en) | 2015-08-31 |
| EP2943506B1 (en) | 2024-03-13 |
| US20150353636A1 (en) | 2015-12-10 |
| MX2015008740A (es) | 2015-10-26 |
| KR20220156667A (ko) | 2022-11-25 |
| IL313424A (en) | 2024-08-01 |
| AU2023222851A1 (en) | 2023-11-16 |
| BR112015016561A2 (zh) | 2017-08-15 |
| BR112015016561B1 (pt) | 2023-01-10 |
| IL283173A (en) | 2021-06-30 |
| CN105229026A (zh) | 2016-01-06 |
| CN113735967A (zh) | 2021-12-03 |
| JP6377635B2 (ja) | 2018-08-22 |
| KR20200024345A (ko) | 2020-03-06 |
| KR20200134340A (ko) | 2020-12-01 |
| AU2018260863A1 (en) | 2018-11-22 |
| EP4434543A2 (en) | 2024-09-25 |
| IL239633B (en) | 2021-12-01 |
| IL283173B1 (en) | 2024-08-01 |
| AU2020205207A1 (en) | 2020-07-30 |
| SG11201505286TA (en) | 2015-08-28 |
| CA2896955C (en) | 2023-10-31 |
| EA201500741A1 (ru) | 2016-01-29 |
| AU2013372331A1 (en) | 2015-07-23 |
| WO2014108198A1 (en) | 2014-07-17 |
| KR20240123420A (ko) | 2024-08-13 |
| EP2943506A1 (en) | 2015-11-18 |
| KR20160007478A (ko) | 2016-01-20 |
| HK1217719A1 (zh) | 2017-01-20 |
| CA2896955A1 (en) | 2014-07-17 |
| JP2016509476A (ja) | 2016-03-31 |
| KR20210096697A (ko) | 2021-08-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105229026B (zh) | 人类IgG1 FC区变体及其用途 | |
| US12049512B2 (en) | Antibody variants and uses thereof | |
| JP7509716B2 (ja) | 抗体変異体およびその使用 | |
| JP2018138022A (ja) | ヒトIgG1 Fc領域変異体およびその使用 | |
| BR122022020571B1 (pt) | Método para aumentar a citotoxicidade dependente de complemento |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| GR01 | Patent grant | ||
| GR01 | Patent grant |