BRPI0918268B1 - Derivados de picolinamida, seu uso, e composição farmacêutica - Google Patents
Derivados de picolinamida, seu uso, e composição farmacêutica Download PDFInfo
- Publication number
- BRPI0918268B1 BRPI0918268B1 BRPI0918268-3A BRPI0918268A BRPI0918268B1 BR PI0918268 B1 BRPI0918268 B1 BR PI0918268B1 BR PI0918268 A BRPI0918268 A BR PI0918268A BR PI0918268 B1 BRPI0918268 B1 BR PI0918268B1
- Authority
- BR
- Brazil
- Prior art keywords
- equiv
- tert
- added
- solution
- methyl
- Prior art date
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4418—Non condensed pyridines; Hydrogenated derivatives thereof having a carbocyclic group directly attached to the heterocyclic ring, e.g. cyproheptadine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
compostos derivados de picolinamida, sua composição farmacêuica e seu uso. a presente invenção refere-se a novos compostos, composições e métodos de inibição da atividade de integração do pró-vírus de maloney quinase (pim quínase) associada a tumorigênese em um individuo humano ou animal. em certas modalidades, os compostos e composições são eficazes para inibir a atividade de pelo menos uma pim quínase. os novos compostos e composições podem ser usados sejam sozinhos ou em combinação com pelo menos um agente adicional para o tratamento de um distúrbio mediado por serina/treonina quínase ou receptor tirosina quínase, tal como um câncer.
Description
[0001] Este pedido reivindica o benefício sob 35 U.S.C. §119(e) para o pedido provisório No. de série US 61/093,666, depositado em 2 de setembro de 2008, e para o pedido provisório No. de série US 61/225,660, depositado em 15 de julho de 2009, que são incorporados aqui no presente em sua totalidade por referência.
[0002] A presente invenção se refere aos novos compostos e seus tautômeros e estereoisômeros, e sais, ésteres, metabólitos ou profármacos dos mesmos farmaceuticamente aceitáveis, composições dos novos compostos junto com veículos farmaceuticamente aceitáveis, e usos dos novos compostos, sozinhos ou em combinação com pelo menos um agente terapêutico adicional, na profilaxia ou no tratamento de câncer.
[0003] Infecção com o retrovírus Maloney e integração de genoma no genoma da célula hospedeira resultam em desenvolvimento de linfomas em camundongos. A integração de Provírus de Maloney Quinase (PIM-Quinase) foi identificada como uma dos protooncogenes frequentes, capazes de ser transcripcionalmente ativados por este evento de integração de retrovírus (Cuypers HT et al., "Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region," Cell 37(1):141-50 (1984); Selten G, et al., "Proviral activation of the putative oncogene Pim-1 in MuLV induced T-cell lymphomas" EMBO J 4(7):1793-8 (1985)), dessa maneira estabelecendo uma correlação entre superexpressão desta quinase e seu potencial oncogênico. Análise da homologia da sequência demonstrou que existem 3 Pim-Quinases altamente homólogas (Pim1, 2 & 3), sendo Pim1 a proto-oncogene originalmente identificada pela integração de retrovírus. Além do mais, camundongos transgênicos superexpressando Pim1 ou Pim2 mostram aumento da incidência de linfomas de célula T- (Breuer M et al., "Very high frequency of lymphoma induction by a chemical carcinogen in pim-1 transgenic mice" Nature 340(6228):61-3 (1989)), enquanto que a superexpressão em conjunto com c-myc é associada com a incidência de linfomas de célula B- (Verbeek S et al., "Mice bearing the E mu-myc and E mu-pim-1 transgenes develop pre-B-cell leukemia prenatally" Mol Cell Biol 11(2):1176-9 (1991)). Dessa maneira, esses modelos de animais estabelecem uma forte correlação entre superexpressão de Pim e oncogêneses em malignidades hematopoiéticas. Além disso, para esses modelos de animais, a superexpressão de Pim tem sido reportada em muitas outras malignidades de ser humano. A superexpressão de Pim1, 2 & 3 é frequentemente observada em muitas malignidades hematopoiéticas (Amson R et al., "The human protooncogene product p33pim is expressed during fetal hematopoiesis and in diverse leukemias," PNAS USA 86(22):8857-61 (1989); Cohen AM et al., "Increased expression of the hPim-2 gene in human chronic lymphocytic leukemia and non-Hodgkin lymphoma," Leuk Lymph 45(5):951-5 (2004), Huttmann A et al., "Gene expression signatures separate B-cell chronic lymphocytic leukaemia prognostic subgroups defined by ZAP-70 and CD38 expression status," Leukemia 20:1774-1782 (2006)) e em câncer de próstata (Dhanasekaran SM, et al., "Delineation of prognostic biomarkers in prostate câncer," Nature 412(6849):822-6 (2001); Cibull TL, et al., "Overexpression of Pim-1 during progression of prostatic adenocarcinoma," J Clin Pathol 59(3):285-8 (2006)), enquanto que a superexpressão de Pim3 é frequentemente observada em carcinoma hepatocelular (Fujii C, et al., "Aberrant expression of serine/threonine quinase Pim-3 in hepatocellular carcinoma desenvolvimento and its role in the proliferação of human hepatoma cell lines," Int J Câncer 114:209-218 (2005)) e câncer pancreático (Li YY et al., "Pim-3, a proto-oncogene with serine/threonine quinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates bad to block bad- mediated apoptosis in human pancreatic câncer cell lines," Câncer Res 66(13):6741-7 (2006)).
[0004] Pim1, 2 & 3 são Serina/Treonina quinases que normalmente funcionam na sobrevivência e proliferação de células hematopoiéticas em resposta aos fatores de crescimento e citocinas. Citocinas que sinalizam através da via de Jak/Stat leva à ativação de transcrição de genes Pim e síntese de proteínas. Nenhuma modificação pós-traducional adicional é requerida para a atividade de Quinase Pim. Dessa maneira, a sinalização a jusante é principalmente controlada no nível de renovação transcricional/traducional e de proteína. Substratos para Pim quinases incluem reguladores de apoptose tais como o membro da família Bcl-2, BAD (Aho T et al., "Pim-1 quinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site,: FEBS Letters 571: 43-49 (2004)), reguladores de ciclos de células tais como p21WFA1/CIP1 (Wang Z, et al., "Phosphorylation of the cell cycle inhibitor p21Cip1/WAF1 by Pim-1 quinase," Biochem Biophys Acta 1593:45- 55 (2002)), CDC25A (1999), C-TAK (Bachmann M et al., "The Oncogenic Serine/Threonine Quinase Pim-1 Phosphorylates and Inhibits the Activity of Cdc25C-associated Quinase 1 (C-TAK1). A novel role for Pim-1 at the G2/M cell cycle checkpoint," J Biol Chem 179:4831948328 (2004)) e NuMA (Bhattacharya N, et al., "Pim-1 associates with protein complexes necessary for mitosis," Chromosoma 111(2):80-95 (2002)) e o regulador de síntese de proteína 4EBP1 (Hammerman PS et al., "Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival," Blood 105(11):4477-83 (2005)). Os efeitos de Pim(s) nestes reguladores são consistentes com um papel na proteção de apoptose e promoção de proliferação de crescimento de células. Dessa maneira, acha-se que a superexpressão de Pim(s) em câncer desempenha um papel promovendo a sobrevivência e proliferação de células de câncer e, dessa maneira, suas inibições devem ser uma maneira eficaz de tratar cânceres em que elas estão superexpressadas. De fato, diversos relatórios indicam que desmontar a expressão de Pim(s) com siRNA resulta em inibição de proliferação e morte das células (Dai JM, et al., "Antisense oligodeoxynucleotides targeting the serine/threonine quinase Pim-2 inhibited proliferation of DU-145 cells," Acta Pharmacol Sin 26(3):364-8 (2005); Fujii et al. 2005; Li et al. 2006). Além do mais, acredita-se que a ativação mutacional de diversos oncogenes bem conhecidos em malignidades hematopoiéticas exerce seus efeitos, pelo menos em parte, através de Pim(s). Por exemplo, a regulagem de expressão de Pim usada como alvo compromete a sobrevivência de células hematopoiéticas transformadas por Flt3 e BCR/ABL (Adam et al. 2006). Dessa maneira, inibidores para Pim1, 2 &3 serão úteis no tratamento dessas malignidades. Em adição a um papel em potencial no tratamento de câncer e doenças mieloproliferativas, tal inibidor poderá ser útil para controlar a expansão de células imunes em outras condições patológicas tais como doenças autoimunes, reações alérgicas e em síndromes de rejeição de transplante de órgãos. Esta noção é apoiada pelas constatações de que essa diferenciação de células Th1 Helper T- por IL-12 e IFN-α resulta em indução de expressão de ambos, Pim1&2 (Aho T et al., "Expression of human Pim family genes is selectively up-regulated by cytokines promoting T helper type 1, but not T helper type 2, cell differentiation," Immunology 116: 82-88 (2005)). Além do mais, a expressão de Pim(s) é inibida em ambos os tipos de célula através de TGF-β imunossupressor (Aho et al. 2005). Este resultado sugere que Pim quinases estão envolvidas no processo inicial de diferenciação de células Helper T-, que coordenam a resposta imunológica em doenças autoimunes, reação alérgica e rejeição a transplante de tecido.
[0005] Existe uma necessidade contínua por compostos que inibem a proliferação de capilares, inibem o crescimento de tumores, trata o câncer, modula a interrupção do ciclo da célula, e/ou inibe moléculas tais como Pim1, Pim2 e Pim3, formulações e medicamentos farmacêuticos que contêm tais compostos. Existe também uma necessidade de métodos de administrar tais compostos, formulações farmacêuticas, e medicamentos para pacientes ou indivíduos com necessidade dos mesmos.
[0006] Novos compostos, e seus estereoisômeros, tautômeros e sais farmaceuticamente aceitáveis, são providos da Fórmula I em que, X1, X2, X3 e X4 são independentemente selecionados de CR2 e N; desde que pelo menos um, porém não mais do que dois de X1, X2, X3 e X4 são N; Y é selecionado de um grupo consistindo em cicloalquila, cicloalquila parcialmente insaturada, e heterocicloalquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; Z2 e Z3 são independentemente selecionados de CR12 e N; desde que não mais do que um de Z2 e Z3 pode ser N; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, ciano, e nitro; R2 e R12 independentemente em cada ocorrência são selecionados do grupo consistindo em hidrogênio, halo, hidroxila, nitro, ciano, SO3H e alquila, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila substituídos ou insubstituídos, e cicloalquila parcialmente saturada; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila, cicloalquila, heterociclila, arila e heteroarila substituídos ou insubstituídos; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído e alquilamino; e R5 representa um grupo selecionado de arila substituída ou insubstituída, C3-C7 cicloalquila, heteroarila, cicloalquila e alquila parcialmente insaturada, em que cada dito grupo R5 substituído pode ser substituído com até quatro substituintes selecionados de halo, ciano, amino, C1-4 alquila, C3-6 cicloalquila, alcóxi, nitro, carbóxi, carbonila, carboalcóxi, aminocarbóxi, aminocarbonila substituída, aminossulfonila, aminossulfonila substituída e alcoxialquila.
em que, X1, X2, X3 e X4 são independentemente selecionados de CR2 e N; desde que pelo menos um, porém não mais do que dois de X1, X2, X3 e X4 são N; Y é selecionado de um grupo consistindo em cicloalquila, cicloalquila parcialmente insaturada, e heterocicloalquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; Z2 e Z3 são independentemente selecionados de CR12 e N; desde que não mais do que um de Z2 e Z3 pode ser N; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, ciano, e nitro; R2 e R12 independentemente em cada ocorrência são selecionados do grupo consistindo em hidrogênio, halo, hidroxila, nitro, ciano, SO3H e alquila, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila substituídos ou insubstituídos, e cicloalquila parcialmente saturada; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila, cicloalquila, heterociclila, arila e heteroarila substituídos ou insubstituídos; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído e alquilamino; e R5 representa um grupo selecionado de arila substituída ou insubstituída, C3-C7 cicloalquila, heteroarila, cicloalquila e alquila parcialmente insaturada, em que cada dito grupo R5 substituído pode ser substituído com até quatro substituintes selecionados de halo, ciano, amino, C1-4 alquila, C3-6 cicloalquila, alcóxi, nitro, carbóxi, carbonila, carboalcóxi, aminocarbóxi, aminocarbonila substituída, aminossulfonila, aminossulfonila substituída e alcoxialquila.
[0007] Em algumas modalidades, novos compostos de Fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que X2 é N e X1, X3 e X4 são CR2.
[0008] Em algumas modalidades, novos compostos de Fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos em que R2 é selecionado de hidrogênio, metila, etila, halo, ciano.
[0009] Em algumas modalidades, compostos de Fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos em que Z2 e Z3 são CR12.
[00010] Em algumas modalidades, compostos de Fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos em que R12 é selecionado de hidrogênio, halo, metila, etile e ciano.
[00011] Em outras modalidades, novos compostos, e seus estereoisômeros, tautômeros e sais farmaceuticamente aceitáveis, são providos a partir da Fórmula II em que, Y é selecionado de um grupo consistindo em cicloexila, cicloexila parcialmente insaturada, e heterociclo-C5-alquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, C3-4 cicloalquila, ciano, e nitro; R12 independentemente em cada ocorrência é selecionado do grupo consistindo em hidrogênio, halo, hidroxila, amino, nitro, ciano, SO3H e alquila, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila substituídos ou insubstituídos, e cicloalquila parcialmente saturada; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila, cicloalquila, heterociclila, arila e heteroarila substituídos ou insubstituídos; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído, e alquilamino; e R5 representa um grupo selecionado de hidrogênio e alquila substituída ou insubstituída, C6-cicloalquila, arila e heteroarila, em que cada um do dito grupo R5 substituído pode ser substituído com até quatro substituintes selecionados de halo, ciano, amino, C1-4 alquila, C3-6 cicloalquila, alcóxi, nitro, carbóxi, carbonila, carboalcóxi, aminocarbóxi, aminocarbonila substituída, aminossulfonila, aminossulfonila substituída e alcoxialquila.
em que, Y é selecionado de um grupo consistindo em cicloexila, cicloexila parcialmente insaturada, e heterociclo-C5-alquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, C3-4 cicloalquila, ciano, e nitro; R12 independentemente em cada ocorrência é selecionado do grupo consistindo em hidrogênio, halo, hidroxila, amino, nitro, ciano, SO3H e alquila, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila substituídos ou insubstituídos, e cicloalquila parcialmente saturada; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila, cicloalquila, heterociclila, arila e heteroarila substituídos ou insubstituídos; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído, e alquilamino; e R5 representa um grupo selecionado de hidrogênio e alquila substituída ou insubstituída, C6-cicloalquila, arila e heteroarila, em que cada um do dito grupo R5 substituído pode ser substituído com até quatro substituintes selecionados de halo, ciano, amino, C1-4 alquila, C3-6 cicloalquila, alcóxi, nitro, carbóxi, carbonila, carboalcóxi, aminocarbóxi, aminocarbonila substituída, aminossulfonila, aminossulfonila substituída e alcoxialquila.
[00012] Em algumas modalidades, compostos das Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado de um grupo consistindo em cicloalqila substituída ou insubstituída, cicloalquenila, piperidinila e piperazinila, em que cada membro do dito grupo é substituído com até quatro substituintes. Em algumas modalidades, Y é substituído com até quatro substituintes selecionados de ciano, nitro, halo, hidroxila, amino, alcóxi, amino substituído, C1-4 alquila, C1-4 halo alquila e C3-4 cicloalquila. Em ainda outras modalidades, Y é substituído com até quatro substituintes selecionados de metila, propila, i-propila, etila, hidroxila, amino, halo, mono-halo C1-3 alquila, tri-halo C1-3 alquila e di-halo C1-3 alquila.
[00013] Em algumas modalidades, novos compostos de Fórmula II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado de um grupo consistindo em ciclo-hexila, ciclo-hexinila, e piperidinila substituídas ou insubstituídas, em que cada membro do dito grupo é substituído com até quatro substituintes.
[00014] Em aglumas modalidades, novos compostos de Fórmula II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é substituído com até quatro substituintes independentemente selecionados de hidrogênio, ciano, nitro, halo, hidroxila, amino, alcóxi, amino substituído, C1-4 alquila, C1-4 halo alquila e C3-4 cicloalquila. Em algumas modalidades, os substituintes são independentemente selecionados de metila, propila, i-propila, etila, hidroxila, amino, halo, monohalo C1-3 alquila, tri-halo C1-3 alquila e di-halo C1-3 alquila.
[00015] Em algumas modalidades, compostos de Fórmula II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que R12 é selecionado de hidrogênio, halo, metila, etila e ciano.
[00016] Em algumas modalidades, compostos das Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado do grupo consistindo em ciclo-hexila substituída ou insubstituída, ciclo- hexenila, piperidinila, piperazinila, em que o dito grupo Y pode ser substituído com até três substituintes selecionados de metila, etila, hidroxila, amino, e metóxi; R1 é selecionado do grupo consistindo em hidrogênio, e amino; e R12 independentemente em cada ocorrência representa hidrogênio, halo, ou metila.
[00017] Em algumas modalidades, compostos de Fórmula II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado de um grupo consistindo em ciclo-hexila substituída, ciclo-hexenila, piperidinila e piperazinila; R1 é selecionado do grupo consistindo em hidrogênio, - NH2, halo, C1-4 alquila, C3-4 cicloalquila, e -CN; R12 independentemente em cada ocorrência é selecionado do grupo consistindo em hidrogênio, halo, C1-4 alquila, e amino; e R5 é selecionado do grupo consistindo em fenila substituída ou insubstituída, ciclo-hexila, ciclopentila, tiazol, piridila, pirimidila e pirazinila, em que o grupo R5 pode ser substituído com até três substituintes selecionados de halo, hidrogênio, metila, aminocarbonila substituída e alcóxi.
[00018] Em uma modalidade representativa, compostos das Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, selecionados do grupo consistindo em N-(4-((1R,3R,4R,5S)-3-amino-4-hidróxi-5- metilciclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida, N-(4- ((1R,3S,5S)-3-amino-5-metilciclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)- 5-flúorpicolinamida. N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida, 3- amino-N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5-metilpiperidin-1-il)piridin- 3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida e N-(4-((1R,3S)-3- aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida, e 3- amino-N-(4-((1R,3S)-3-aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)- 5-flúorpicolinamida.
[00019] Em outros aspectos, a presente invenção provê métodos para tratar Integração Provirus de Maloney Quinase (PIM Quinase) relacionada a distúrbios em um sujeito animal ou ser humano com necessidade de tal tratamento, compreendendo administrar para o dito sujeito uma quantidade de um composto de Fórmula I ou II eficaz para inibir a atividade de PIM no sujeito.
[00020] Em outros aspectos, a presente invenção provê métodos para tratar distúrbios relacionados com PIM em um sujeito ser humano ou animal com necessidade de tal tratamento, compreendendo administrar para o dito sujeito uma quantidade de um composto de Fórmula I ou II eficaz para reduzir ou prevenir o crescimento de tumor no sujeito.
[00021] Em ainda outros aspectos, a presente invenção provê métodos para tratar distúrbios relacionados a PIM em um sujeito animal ou ser humano com necessidade de tal tratamento, compreendendo administrar para o dito sujeito uma quantidade de um composto de Fórmula I ou II eficaz para reduzir ou evitar crescimento de tumor no sujeito, em combinação com pelo menos um agente adicional para o tratamento de câncer.
[00022] Em ainda outros aspectos, a presente invenção provê composições terapêuticas compreendendo pelo menos um composto de Fórmula I ou II em combinação com um ou mais agentes adicionais para o tratamento de câncer, como são comumente empregados em terapia de câncer.
[00023] Os compostos da invenção são úteis no tratamento de cânceres, incluindo malignidades hematopoiéticas, carcinomas (por exemplo, dos pulmões, fígado, pâncreas, ovários, tiroide, bexiga ou cólon), melanoma, distúrbios mieloides (por exemplo, leucemia mieloide, mieloma múltiplo e eritroleucemia), adenomas (por exemplo, adenoma do cólon viloso), sarcomas (por exemplo, osteossarcoma), doenças autoimunes, reações alérgicas e em síndromes de rejeição de transplante de órgãos.
[00024] A invenção ainda provê composições, métodos de uso, e métodos de preparação como descrito na descrição detalhada da invenção.
[00025] Os aspectos anteriores e muitas das vantagens de assistente desta invenção se tornarão mais prontamente evidentes, assim como os mesmos se tornarão melhor compreendidos pela referência à descrição detalhada a seguir, quando tomada em conjunto com os desenhos que acompanham, em que:
[00026] A figura 1 é um gráfico mostrando a eficácia do composto do Exemplo 99 a partir de uma avaliação no modelo de xenoenxerto KMS11-luc como descrito no Exemplo 144.
[00027] A figura 2 é um gráfico mostrando a eficácia do composto do Exemplo 70 a partir de uma avaliação no modelo de xenoenxerto KMS11-luc, como descrito no Exemplo 144.
[00028] A figura 3 é um gráfico mostrando a eficácia do composto do Exemplo 96 a partir de uma avaliação no modelo de xenoenxerto KMS11-luc, como descrito no Exemplo 144.
[00029] De acordo com um aspecto da presente invenção, novos compostos, e seus estereoisômeros, tautômeros e sais farmaceuticamente aceitáveis, são providos da Fórmula I: em que, X1, X2, X3 e X4 são independentemente selecionados de CR2 e N; desde que pelo menos um, mas não mais do que dois de X1, X2, X3 e X4 são N; Y é selecionado de um grupo consistindo em cicloalquila, cicloalquila parcialmente insaturada, e heterocicloalquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; Z2 e Z3 são independentemente selecionados de CR12 e N; desde que não mais do que um de Z2 e Z3 podem ser N; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, ciano, e nitro; R2 e R12 independentemente em cada ocorrência são selecionados do grupo consistindo em hidrogênio, halo, hidroxila, nitro, ciano, SO3H e alquila, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila substituídos ou insubstituídos, e cicloalquila parcialmente saturada; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila, cicloalquila, heterociclila, arila e heteroarila substituídos ou insubstituídos; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído, e alquilamino; e R5 representa um grupo selecionado de arila substituída ou insubstituída, C3-C7 cicloalquila, heteroarila, cicloalquila parcialmente insaturada e alquila, em que cada dito grupo R5 substituído pode ser substituído com até quatro substituintes selecionados de halo, ciano, amino, C1-4 alquila, C3-6 cicloalquila, alcóxi, nitro, carbóxi, carbonila, carboalcóxi, aminocarbóxi, aminocarbonila substituída, aminossulfonila, aminossulfonila substituída e alcoxialquila.
em que, X1, X2, X3 e X4 são independentemente selecionados de CR2 e N; desde que pelo menos um, mas não mais do que dois de X1, X2, X3 e X4 são N; Y é selecionado de um grupo consistindo em cicloalquila, cicloalquila parcialmente insaturada, e heterocicloalquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; Z2 e Z3 são independentemente selecionados de CR12 e N; desde que não mais do que um de Z2 e Z3 podem ser N; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, ciano, e nitro; R2 e R12 independentemente em cada ocorrência são selecionados do grupo consistindo em hidrogênio, halo, hidroxila, nitro, ciano, SO3H e alquila, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila substituídos ou insubstituídos, e cicloalquila parcialmente saturada; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila, cicloalquila, heterociclila, arila e heteroarila substituídos ou insubstituídos; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído, e alquilamino; e R5 representa um grupo selecionado de arila substituída ou insubstituída, C3-C7 cicloalquila, heteroarila, cicloalquila parcialmente insaturada e alquila, em que cada dito grupo R5 substituído pode ser substituído com até quatro substituintes selecionados de halo, ciano, amino, C1-4 alquila, C3-6 cicloalquila, alcóxi, nitro, carbóxi, carbonila, carboalcóxi, aminocarbóxi, aminocarbonila substituída, aminossulfonila, aminossulfonila substituída e alcoxialquila.
[00030] Em algumas modalidades, novos compostos de Fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos em que X2 é N e X1, X3 e X4 são CR2.
[00031] Em algumas modalidades, novos compostos de Fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que R2 é selecionado de hidrogênio, metila, etila, halo, ciano.
[00032] Em algumas modalidades, compostos de fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Z2 e Z3 são CR12.
[00033] Em algumas modalidades, compostos de fórmula I, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que R12 é selecionado de hidrogênio, halo, metila, etila e ciano.
[00034] Em outras modalidades, novos compostos, e seus estereoisômeros, tautômeros e sais farmaceuticamente aceitáveis, são providos da Fórmula II em que, Y é selecionado de um grupo consistindo em ciclohexila, ciclohexila parcialmente insaturada, e heterociclo-C5-alquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, C3-4 cicloalquila, ciano, e nitro; R12 independentemente em cada ocorrência é selecionado do grupo consistindo em hidrogênio, halo, hidroxila, amino, nitro, ciano, SO3H e alquila substituída ou insubstituído, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila, e cicloalquila parcialmente saturado; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila substituída ou insubstituída, cicloalquila, heterociclila, arila e heteroarila; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído, e alquilamino; e R5 representa um grupo selecionado de hidrogênio e alquila substituída ou insubstituída, C6-cicloalquila, arila e heteroarila.
em que, Y é selecionado de um grupo consistindo em ciclohexila, ciclohexila parcialmente insaturada, e heterociclo-C5-alquila, em que cada membro do dito grupo pode ser substituído com até quatro substituintes; R1 é selecionado do grupo consistindo em hidrogênio, -NHR3, halo, hidroxila, alquila, C3-4 cicloalquila, ciano, e nitro; R12 independentemente em cada ocorrência é selecionado do grupo consistindo em hidrogênio, halo, hidroxila, amino, nitro, ciano, SO3H e alquila substituída ou insubstituído, alquenila, alquinila, alcóxi, amino, cicloalquila, hetero cicloalquila, e cicloalquila parcialmente saturado; R3 é selecionado do grupo consistindo em hidrogênio, -CO-R4 e alquila substituída ou insubstituída, cicloalquila, heterociclila, arila e heteroarila; R4 é selecionado do grupo consistindo em alquila, alquila substituída, alcóxi, alcóxi substituído, amino, amino substituído, e alquilamino; e R5 representa um grupo selecionado de hidrogênio e alquila substituída ou insubstituída, C6-cicloalquila, arila e heteroarila.
[00035] Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou um sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado de um grupo consistindo em cicloalquila substituída ou insubstituída, cicloalquenila, piperidinila e piperazinila, em que cada membro do dito grupo é substituído com até quatro substituintes. Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado de um grupo consistindo em ciclohexila substituída ou insubstituída, ciclohexinila, e piperidinila, em que cada membro do dito grupo é substituído com até quatro substituintes. Em algumas modalidades, Y é substituído com até quatro substituintes selecionados de hidrogênio, ciano, nitro, halo, hidroxila, amino, alcóxi, amino substituído, C1-4 alquila, C1-4 halo alquila e C3-4 cicloalquila. Em ainda outras modalidades, Y é substituído com até quatro substituintes selecionados de metila, propila, i-propila, etila, hidroxila, amino, halo, mono-halo C1-3 alquila, tri-halo C1-3 alquila e dihalo C1-3 alquila.
[00036] Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que R1 é hidrogênio, amino ou flúor. Em uma modalidade são providos compostos de Fórmula II selecionados da Tabela I ou Tabela II.
[00037] Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que R5 é selecionado de arila substituída ou insubstituída, C5-C6 cicloalquila, heteroarila, C5C6 cicloalquila parcialmente insaturada e C1-C4 alquila, em que cada dito grupo pode ser substituído com até quatro substituintes selecionados de halo, ciano, amino, C1-4 alquila, C3-5 cicloalquila, alcóxi, nitro, carbóxi, carbonila, carboalcóxi, aminocarbóxi, aminocarbonila substituída, aminossulfonila, aminossulfonila substituída e alcoxialquila. Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que R5 é fenila substituída ou insubstituída, em que o grupo fenila pode ser substituído com até quatro substituintes selecionados de hidrogênio, ciano, nitro, halo, hidroxila, amino, alcóxi, amino substituído, C1-4 alquila, C1-4 halo alquila e C3-4 cicloalquila. Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos em que R5 é 2,6-di-flúorfenila.
[00038] Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que R12 é selecionado de hidrogênio, halo, metila, etila e ciano. Em algumas modalidades, compostos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado do grupo consistindo em ciclohexila substituído ou insubstituído, ciclo-hexenila, piperidinila, piperazinila, em que o dito grupo Y pode ser substituído com até três substituintes selecionados de metila, etila, hidroxila, amino, e metóxi; R1 é selecionado do grupo consistindo em hidrogênio, e amino; e R12 independentemente em cada ocorrência representa hidrogênio, halo, ou metila.
[00039] Em algumas modalidades, compostos de Fórmula II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são providos, em que Y é selecionado de um grupo consistindo em ciclo-hexila substituída, ciclo-hexenila, piperidinila, e piperazinila; R1 é selecionado do grupo consistindo em hidrogênio, - NH2, halo, C1-4 alquila, C3-4 cicloalquila, e -CN; R12 independentemente em cada ocorrência é selecionado do grupo consistindo em hidrogênio, halo, C1-4 alquila, e amino; e R5 é selecionado do grupo consistindo em fenila substituído ou insubstituído, ciclo-hexila, ciclopentila, tiazol, piridila, pirimidila e pirazinila, em que o grupo R5 pode ser substituído com até três substituintes selecionados de halo, hidrogênio, metila, aminocarbonila substituída e alcóxi.
[00040] Uma modalidade preferida da presente invenção é um composto de Fórmula (II), em que Y é ciclo-hexila, substituído com um a três substituintes, os ditos substituintes preferivelmente selecionados de hidroxila, amino, C1-4 alquila ou C1-4 halo alquila, e mais preferivelmente, selecionados de metila, hidroxila, amino, e CF3, e mais preferivelmente de metila, amino, e hidróxi; R1 é hidrogênio, NH2, ou halo (preferivelmente, R1 é hidrogênio, amino ou flúor, mais preferivelmente, R1 é hidrogênio); R12 são cada um independentemente hidrogênio ou halo (preferivelmente, cada R12 é hidrogênio, cloro ou flúor); R5 é ciclo-hexila, fenila, ou piridila, em que a dita ciclo-hexila, a dita fenila e a dita piridila são cada uma independentemente substituída com até três substituintes selecionados de halo, hidroxila, C1-4 alquila, e C1-4 alcóxi (preferivelmente, R5 é piridila ou fenila cada um independentemente substituído com até três substitutents selecionados de halo, hidroxila, C1-4 alquila ou C1-4 alcóxi, mais preferivelmente, R5 é fenila substituída com até três substituintes selecionados de halo, hidroxila, C1-4 alcóxi e C1-4 alquila, mais preferivelmente, fenila substituída com até três substituintes selecionados de flúor, hidroxila, metila, etila, metóxi, ou propóxi, mais preferivelmente, R5 é 2,6-di-flúorfenila.
[00041] Ainda outra modalidade preferida da presente invenção provê um composto de Fórmula II, em que Y é piperidinila substituída com metila, hidroxila, e amino; R1 é hidrogênio, NH2, ou flúor; R12 independentemente em cada ocorrência é selecionado do grupo consistindo em hidrogênio e halo; e R5 é piridila, flúor piridila, ciclo- hexila, ou fenila, em que a dita fenila é substituída com até três substituintes selecionados de flúor, hidroxila e metila, preferivelmente R5 sendo diflúor fenila. Em uma modalidade preferida adicional preferivelmente Y é 3-amino-4-hidróxi-5-metilpiperidin-1-ila; R1 é hidrogênio; e R5 é 2,6-diflúor fenila.
[00042] Em uma modalidade representativa, compostos preferidos de Fórmulas I ou II, ou um estereoisômero, tautômero, ou sal dos mesmos farmaceuticamente aceitáveis são selecionados do grupo consistindo em N-(4-((3S,5S)-3-amino-5-metilciclo-hexil)piridin-3-il)-6- (2,6-di-flúorfenil)-5-flúorpicolinamida; 3-amino-N-(4-((1R,3R,4S,5S)-3- amino-4-hidróxi-5-metilciclo-hexil)piridin-3-il)-6-(2,6-di- flúorfenil)picolinamida; N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N- (4-((3R,4R,5S)-3-amino-4-hidróxi-5-metilpiperidin-1-il)piridin-3-il)-6- (2,6-di-flúorfenil)-5-flúorpicolinamida; 3-amino-N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N- (4-((3R,4R,5S)-3-amino-4-hidróxi-5-metilpiperidin-1-il)piridin-3-il)-6- (2,6-di-flúor-3-metilfenil)-5-flúorpicolinamida; 3-amino-N-(4-((1R,3S)-3- aminociclo-hexil) piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N- (4-((3S)-3-aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5- flúorpicolinamida; N-(4-((1R,3R,4R,5S)-3-amino-4-hidróxi-5-metilciclo- hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N-(4- ((1R,3S,5S)-3-amino-5-metilciclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)- 5-flúorpicolinamida); N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N- (4-((1R,3S)-3-aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5- flúorpicolinamida; 3-amino-N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; e 3-amino-N-(4-((1R,3S)-3-aminociclo-hexil)piridin-3-il)-6-(2,6-di- flúorfenil)-5-flúorpicolinamida.
[00043] Em outros aspectos, a presente invenção provê métodos para tratar Integração Provirus de Maloney Quinase (PIM Quinase) relacionada a distúrbios em um sujeito humano ou animal com necessidade de tal tratamento, compreendendo administrar para o dito sujeito uma quantidade de um composto de Fórmula I ou II eficaz para inibir a atividade de PIM no sujeito. Uma modalidade preferida da presente invenção provê um método para tratar uma condição através da modulação de Integração Provirus de Maloney Quinase (PIM Quinase) compreendendo administrar para um paciente com necessidade de tal tratamento an quantidade eficaz de um composto de Fórmula I.
[00044] Em outros aspectos, a presente invenção provê métodos para tratar distúrbios relacionados a PIM em um sujeito animal ou ser humano com necessidade de tal tratamento compreendendo administrar para o dito sujeito uma quantidade de um composto de Fórmula I ou II eficaz para reduzir ou prevenir o crescimento de tumor no sujeito. Em ainda outros aspectos, a presente invenção provê métodos para tartar distúrbios relacionados a PIM em um sujeito animal ou ser humano com necessidade de tal tratamento compreendendo administrar para o dito sujeito uma quantidade de um composto de Fórmula I ou II eficaz para reduzir ou prevenir crescimento do tumor no sujeito, em combinação com pelo menos um agente adicional para o tratamento de câncer.
[00045] Em ainda outro aspecto, a presente invenção provê composições terapêuticas compreendendo pelo menos um composto de Fórmula I ou II em combinação com um ou mais agentes adicionais para o tratamento de câncer, como são comumente empregados na terapia de câncer. A presente invenção dessa maneira provê uma composição farmacêutica compreendendo a composto of Fórmula I or Fórmula II. Uma modalidade preferida deste aspecto provê uma composição farmacêutica compreendendo um composto selecionado de N-(4-((3S,5S)-3-amino-5-metilciclo-hexil)piridin-3-il)-6-(2,6-di- flúorfenil)-5-flúorpicolinamida; 3-amino-N-(4-((1R,3R,4S,5S)-3-amino-4- hidróxi-5-metilciclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)picolinamida; N- (4-((3R,4R,5S)-3-amino-4-hidróxi-5-metilpiperidin-1-il)piridin-3-il)-6- (2,6-di-flúorfenil)-5-flúorpicolinamida; 3-amino-N-(4-((1R,3S)-3- aminociclo-hexil) piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N- (4-((3S)-3-aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5- flúorpicolinamida; N-(4-((1R,3R,4R,5S)-3-amino-4-hidróxi-5-metilciclo- hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N-(4- ((1R,3S,5S)-3-amino-5-metilciclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)- 5-flúorpicolinamida); N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; N- (4-((1R,3S)-3-aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5- flúorpicolinamida; 3-amino-N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; 3- amino-N-(4-((1R,3S)-3-aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)- 5-flúorpicolinamida; N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5- metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; 3- amino-N-(4-((3R,4R,5S)-3-amino-4-hidróxi-5-metilpiperidin-1-il)piridin- 3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida; e N-(4-((3R,4R,5S)-3- amino-4-hidróxi-5-metilpiperidin-1-il)piridin-3-il)-6-(2,6-di-flúor-3- metilfenil)-5-flúorpicolinamida. Outra modalidade preferida provê uma composição farmacêutica ainda compreendendo um agente adicional para o tratamento de câncer, em que preferivelmente o agente adicional é selecionado de irinotecano, topotecano, gemcitabina, 5- flúoruracila, leucovorina carboplatina, cisplatina, taxanos, tezacitabina, ciclofosfamida, alcaloides vinca, imatinib (Gleevec), antraciclinas, rituximab, e trastuzumab.
[00046] Os compostos da invenção são úteis no tratamento de cânceres, incluindo malignidades hematopoiéticas, carcinomas (por exemplo, dos pulmões, fígado, pâncreas, ovários, tiroide, bexiga e cólon), melanoma, distúrbios mieloides (por exemplo, leucemia mieloide, mieloma múltiplo e eritroleucemia), adenomas (por exemplo, adenoma de cólon viloso), sarcomas (por exemplo, osteossarcoma), doenças autoimunes, reações alérgicas e em síndromes de rejeição de transplante de órgão.
[00047] Em ainda outro aspecto da presente invenção é provido um uso de um composto de Fórmula I ou Fórmula II para preparar um medicamento para tratar uma condição por modulação da atividade de Integração Provirus de Maloney Quinase (PIM Quinase). Em uma modalidade preferida deste aspecto da invenção, a condição é um câncer selecionado de carcinoma dos pulmões, pâncreas, tiroide, ovário, bexiga, mama, próstata, ou cólon, melanoma, leucemia mieloide, mieloma múltiplo e eritro leucemia, adenoma de cólon viloso, e osteossarcoma.
[00048] Em outro aspecto, a presente invenção se refere aos métodos de inibir a atividade de pelo menos uma quinase selecionada do grupo consistindo em Pim1, Pim2 e Pim3, em um sujeito, ou tratar uma condição biológica mediada por pelo menos um de Pim1, Pim2 e Pim3, em um sujeito animal ou ser humano com necessidade de tal tratamento, compreendendo administrar para o sujeito pelo menos um composto de Fórmula I ou II em uma quantidade eficaz para inibir a quinase no sujeito. Os compostos terapêuticos são úteis para tratar pacientes com uma necessidade de tais inibidores (por exemplo, aqueles que sofrem de câncer mediado por sinalização anormal de receptor de serina/treonina quinases).
[00049] "Inibidor de PIM" é usado aqui no presente para se referir a um composto que exibe um IC50 com respeito à atividade de PIM Quinase de não mais do que cerca de 100 μM e mais tipicamente não mais do que cerca de 50 μM, como medido nos ensaios de esgotamento de PIM descritos aqui abaixo.
[00050] A frase "alquila" se refere a grupos alquila que não contêm heteroátomos. Dessa maneira, a frase inclui grupos alquila de cadeia linear tais como metila, etila, propila, butila, pentila, hexila, heptila, octila, nonila, decila, undecila, dodecila e similares. A frase também inclui isômeros de cadeia ramificada de grupos alquila de cadeia linear, incluindo mas não limitado a, os a seguir que são providos a título de exemplo: -CH(CH3)2, -CH(CH3)(CH2CH3), -CH(CH2CH3)2, -C(CH3)3, -C(CH2CH3)3, -CH2CH(CH3)2, -CH2CH(CH3)(CH2CH3), -CH2CH(CH2CH3)2, -CH2C(CH3)3, -CH2C(CH2CH3)3, -CH(CH3)CH(CH3)(CH2CH3), -CH2CH2CH(CH3)2, -CH2CH2CH(CH3)(CH2CH3), -CH2CH2CH(CH2CH3)2, -CH2CH2C(CH3)3, -CH2CH2C(CH2CH3)3, -CH(CH3)CH2CH(CH3)2, -CH(CH3)CH(CH3)CH(CH3)2, -CH(CH2CH3)CH(CH3)CH(CH3)(CH2CH3), e outros. Dessa maneira a frase grupos alquila inclui grupos alquila primários, grupos alquila secundários e grupos alquila terciários. Grupos alquila preferidos incluem grupos alquila de cadeia linear ou ramificada tendo 1 a 12 átomos de carbono. Uma definição preferida de "alquila" se refere a grupos alquila de cadeia C1-4 linear tais como metila, etila, n-propila, e n-butila. A definição de alquila preferida também inclui grupos alquila ramificados C3-5, incluindo CH(CH3)2, CH2CH(CH3)2, CH(CH3)CH2CH3, C(CH3)3, CH(CH3)CH2CH2CH3, CH(CH3)CH(CH3)2, CH2CH(CH3)CH2CH3, CH2CH2CH(CH3)2, e CH(CH2CH3)2, etc.
[00051] O termo "alquenila" se refere aos grupos alquila como definido acima, em que existe pelo menos um ponto de insaturação, isto é, em que dois átomos de carbono adjacentes são acoplados por ligação dupla. O termo "alquinila" se refere aos grupos alquila em que dois átomos de carbono adjacentes são acoplados por uma ligação tripla. O termo 'alcóxi" se refere a -OR, em que R é alquila.
[00052] Como usado aqui no presente, o termo "halogênio" ou "halo" se refere a grupos cloro, bromo, flúor e iodo. "Haloalquila" se refere a um radical alquila substituído com um ou mais átomos de halogênio. O termo "haloalquila" dessa maneira inclui mono-halo alquila, di-halo alquila, tri-halo alquila e similares. Grupos mono-halo alquila representativos incluem -CH2F, -CH2Cl, -CH2CH2F, -CH2CH2Cl, -CH(F)CH3, -CH(Cl)CH3; grupos alquila di-halo representativos incluem CHCl2, -CHF2, -CCl2CH3, -CH(Cl)CH2Cl, -CH2CHCl2, -CH2CHF2; grupos alquila tri-halo representativos incluem -CCl3, -CF3, -CCl2CH2Cl, -CF2CH2F, -CH(Cl)CHCl2, -CH(F)CHF2; e grupos alquila per-halo representativos incluem -CCl3, -CF3, -CCl2CCl3, -CF2CF3.
[00053] "Amino" se refere, aqui no presente, ao grupo -NH2. O termo "alquilamino" se refere, aqui no presente, ao grupo -NRR' em que R e R' são cada um, independentemente, selecionado de hidrogênio ou de uma alquila inferior. O termo "arilamino" se refere, aqui no presente, ao grupo -NRR' em que R é arila e R' é hidrogênio, uma alquila inferior, ou uma arila. O termo "aralquilamino" se refere, aqui no presente, ao grupo -NRR' em que R é uma aralquila inferior e R' é hidrogênio, uma alquila inferior, uma arila ou um aralquila inferior. O termo ciano se refere ao grupo -CN. O termo nitro se refere ao grupo -NO2.
[00054] O termo "alcoxialquila" se refere ao grupo -alk1-O-alk2 em que alk1 é alquila ou alquenila, e alk2 é alquila ou alquenila. O termo "alcoxialquila inferior" se refere a uma alcoxialquila em que alk1 é alquila inferior ou alquenila inferior, e alk2 é alquila inferior ou alquenila inferior. O termo "ariloxialquila" se refere ao grupo -alquil-O-arila. O termo "aralcoxialquila" se refere ao grupo -alquilenil-O-aralquila, em que aralquila é uma aralquila inferior.
[00055] O termo "aminocarbonila" se refere, aqui no presente, ao grupo -C(O)-NH2 . "Aminocarbonila substituída " se refere, aqui no presente, ao grupo -C(O)-NRR' em que R é alquila inferior e R' é hidrogênio ou uma alquila inferior. Em algumas modalidades, R e R', junto com o átomo de N a eles acoplado, podem ser tomados juntos para formar um grupo "heterocicloalquilcarbonila". O termo "arilaminocarbonila" se refere, aqui no presente, ao grupo -C(O)-NRR' em que R é uma arila e R' é hidrogênio, alquila inferuior ou arila. "Aralquilaminocarbonila" se refere, aqui no presente, ao grupo -C(O)- NRR' em que R é aralquila inferior e R' é hidrogênio, alquila inferior, arila, ou aralquila inferior.
[00056] "Aminossulfonila" se refere, aqui no presente, ao grupo - S(O)2-NH2. "Aminossulfonila substituída " se refere, aqui no presente, ao grupo -S(O)2-NRR' em que R é alquila inferior e R' é hidrogênio ou uma alquila inferior. O termo "aralquilaminossulfonliarila" se refere, aqui no presente, ao grupo -aril-S(O)2-NH-aralquila, em que a aralquila é aralquila inferior.
[00057] "Carbonila" se refere ao grupo divalente -C(O)-. "Carbóxi" se refere a -C(=O)-OH. "Alcoxicarbonila" se refere a éster -C(=O)-OR em que R é alquila. "Alcoxicarbonila inferior" se refere a éster -C(=O)- OR em que R é alquila. "Cicloalquiloxicarbonila" se refere a -C(=O)-OR em que R é cicloalquila.
[00058] "Cicloalquila" se refere a um substituinte de alquila carbocíclica, mono- ou policíclica. Grupos carbocicloalquila são grupos cicloalquila em que todos os átomos de anel são carbono. Substituintes de cicloalquila típicos têm de 3 a 8 cadeias principais (isto é, anel) de átomos em que cada cadeia principal é carbono ou um heteroátomo. O termo "heterocicloalquila" se refere aqui no presente a substituintes de cicloalquila que têm de 1 a 5, e mais tipicamente de 1 a 4 heteroátomo na estrutura de anel. Heteroátomos apropriados empregados em compostos da presente invenção são nitrogênio, oxigênio, e enxofre. Porções representativas de heterocicloalquila incluem, por exemplo, morfolino, piperazinila, piperidinila e similares. Grupos carbocicloalquila são grupos cicloalquila em que todos os átomos de anel são carbono. Quando usado em conexão com substituintes de cicloalquila, o termo "policíclico" se refere aqui no presente em estruturas cíclicas de alquila fundidas e não fundidas. O termo "cicloalquila parcialmente insaturada", "cicloalquila parcialmente saturada", e "cicloalquenila" todos se referem a um grupo cicloalquila em que existe pelo menos um ponto de insaturação, isto é, em que os átomos de anel adjacentes são conectados por uma ligação dupla ou uma ligação tripla. Exemplos ilustrativos incluem ciclo-hexinila, ciclohexinila, ciclopropenila, ciclobutinila e similares.
[00059] Os termos "heterociclo substituído", "grupo heterocíclico" ou "heterociclo", como usados aqui no presente, se referem a qualquer anel de 3- ou 4- elementos contendo um heteroátomo selecionado de nitrogênio, oxigênio, e enxofre ou um anel de 5- ou 6- elementos contendo de um a três heteroátomos selecionados do grupo consistindo em nitrogênio, oxigênio, ou enxofre; em que o anel de 5- elementos tem 0 a 2 ligações duplas e o anel de 6- elementos tem 0 a 3 ligações duplas; em que o átomo de nitrogênio e enxofre pode estar opcionalmente oxidado; em que os heteroátomos de nitrogênio e enxofre podem estar opcionalmente quarternizados; e incluindo qualquer grupo bicíclico em que qualquer um dos anéis heterocíclicos acima é fundido a um anel de benzeno ou outro anel heterocíclico de 5- ou 6- elementos independentemente definidos acima. O termo ou "heterocicloalquila" como usado aqui no presente se refere a um anel de 5- ou 6- elementos contendo de um a três heteroátomos selecionados do grupo consistindo em nitrogênio, oxigênio, ou enxofre, em que o anel tem ligações duplas. Por exemplo, o termo heterociclo- C5-alquila se refere a um anel de 6- elementos contendo 5 átomos de carbono e um heteroátomo, tal como N. O termo "heterociclo" dessa maneira inclui anéis em que nitrogênio é o heteroátomo como também anéis parcialmente e totalmente saturados. Heterociclos preferidos incluem, por exemplo: diazapinila, pirrila, pirrolinila, pirrolidinila, pirazolila, pirazolinila, pirazolidinila, imidazoil, imidazolinila, imidazolidinila, piridila, piperidinila, pirazinila, piperazinila, N-metila piperazinila, azetidinila, N-metilazetidinila, pirimidinila, piridazinila, oxazolila, oxazolidinila, isoxazolila, isoazolidinila, morfolinila, tiazolila, tiazolidinila, isotiazolila, isotiazolidinila, indolila, quinolinila, isoquinolinila, benzimidazolila, benzotiazolila, benzoxazolila, furila, tienila, triazolila e benzotienila.
[00060] Porções heterocíclicas podem ser insubstituídas ou monossubstituídas ou dissubstituídas ou trissubstituídas com vários substituintes independentemente selecionados de hidróxi, halo, oxo (C=O), alquilimino (RN=, em que R é uma alquila inferior ou grupo alcóxi inferior), amino, alquilamino, dialquilamino, acilaminoalquila, alcóxi, tioalcóxi, polialcóxi, alquila inferior, cicloalquila ou haloalquila.
[00061] Os grupos heterocíclicos podem ser acoplados a várias posições como sera evidente para aqueles tendo qualificação nas técnicas de química orgânica e médica em conjunto com a descrição aqui no presente.
[00062] Heterocíclicos representativos incluem, por exemplo, imidazolila, piridila, piperazinila, piperidinila, azetidinila, tiazolila, furanila, triazolila benzimidazolila, benzotiazolila, benzoxazolila, quinolinila, isoquinolinila, quinazolinila, quinoxalinila, ftalazinila, indolila, naftpiridinila, indazolila, e quinolizinila.
[00063] "Arila" se refere aos grupos aromáticos monocíclicos e policíclicos opcionalmente substituídos tendo de 3 a 14 átomos de carbono ou hetero de estrutura principal, e inclui ambos, grupos arila carbocíclicos e grupos arila heterocíclicos. Grupos arila carbocíclicos são grupos arila em que todos os átomos de anel, no anel aromático, são carbono. O termo "heteroarilas" se refere, aqui no presente, a grupos arila tendo de 1 a 4 heteroátomos como átomos de anel em um anel aromático com o restante dos átomos de anel sendo átomos de carbono. Quando usado em conexão com substituintes de arila, o termo "arila policíclica" se refere, aqui no presente, a estruturas cíclicas fundidas e não fundidas em que pelo menos uma estrutura cíclica é aromática, tal como, por exemplo, benzodioxozol (que tem uma estrutura heterocíclica fundida a um grupo fenila, isto é, naftila e similares. Porções de arila exemplares empregadas como substituintes em compostos da presente invenção incluem fenila, piridila, pirimidinila, tiazolila, indolila, imidazolila, oxadiazolila, tetrazolila, pirazinila, triazolila, tiofenila, furanila, quinolinila, purinila, naftila, benzotiazolila, benzopiridila, e benzimidazolila e similares.
[00064] "Opcionalmente substituído" ou "substituído" se refere à substituição de um ou mais átomos de hidrogênio com um radical monovalente ou divalente. Grupos de substituição apropriados incluem, por exemplo, hidróxi, nitro, amino, imino, ciano, halo, tio, sulfonila, tioamido, amidino, imidino, oxo, oxamidino, metoxamidino, imidino, guanidino, sulfonamido, carboxila, formila, alquila inferior, haloalquila inferior, alquilamino inferior, haloalquilamino inferior, alcóxi inferior, haloalcóxi inferior, alcoxialquila inferior, alquilcarbonila, aminocarbonila, arilcarbonila, aralquilcarbonila, heteroarilcarbonila, heteroaralquilcarbonila, alquiltio, aminoalquila, cianoalquila, arila e similares.
[00065] O grupo de substituição pode ele próprio ser substituído. O grupo substituído no grupo de substituição pode ser carboxila, halo; nitro, amino, ciano, hidróxi, alquila inferior, alcóxi inferior, aminocarbonila, -SR, tioamido, -SO3H, -SO2R ou cicloalquila, em que R é tipicamente hidrogênio, hidroxila ou alquila inferior.
[00066] Quando o substituinte substituído inclui um grupo de cadeia linear, a substituição pode ocorrer dentro da cadeia (por exemplo, 2- hidroxipropila, 2-aminobutila, e similares) ou na terminação da cadeia (por exemplo, 2-hidroxietila, 3-cianopropila, e similares). Substituintes substituídos podem ser de cadeia linear, ramificada ou disposições cíclicas do carbono ou heteroátomos covalentemente ligados. Entende-se que as definições acima definitions não são destinadas a incluir padrões de substituição não permitidos (por exemplo, metila substituída com cinco grupos flúor ou um átomo de halogênio substituído com outro átomo se halogênio). Tais padrões de substituição não permitidos são bem conhecidos do técnico versado.
[00067] Também será evidente para aqueles versados na técnica que os compostos da invenção, ou seus estereoisômeros, como também os sais, ésteres, metabólitos e profármacos farmaceuticamente aceitáveis de qualquer um deles, podem estar sujeitos à tautomerização e podem dessa maneira existir em várias formas tautoméricas em que um próton de um átomo de uma molécula desvia para outro átomo e as ligações químicas entre os átomos das moléculas são consequentemente rearrumados. Ver, por exemplo, March, Advanced Organic Chemistry: Reactions, Mechanisms and Structures, Fourth Edition, John Wiley & Sons, páginas 69-74 (1992). Como usado aqui no presente, o termo "tautômero" se refere aos compostos produzidos pelo desvio do próton, e deve ser entendido que todas as formas tautoméricas, como elas podem existir até agora, são incluídas dentro da invenção.
[00068] Os compostos da invenção, ou seus tautômeros, como também os sais, ésteres, metabólitos e profármacos farmaceuticamente aceitáveis de qualquer um deles, podem compreender átomos de carbono assimetricamente substituídos. Tais átomos de carbono assimetricamente substituídos podem resultar nos compostos da invenção existentes em enantiômeros, diastereômeros, e outras formas estereoisoméricas que podem ser definidas, em termos de estereoquímica absoluta, tal como nas formas (R)- ou (S)-. Como um resultado, todos os isômeros possíveis, estereoisômeros individuais em suas formas opticamente puras, misturas das mesmas, misturas racêmicas (ou "racematos"), misturas de diastereômeros, como também diastereômeros únicos dos compostos da invenção são incluídos na presente invenção. Os termos da configuração "S" e "R", como usados aqui no presente, são como definido pela IUPAC 1974 RECOMMENDATIONS FOR SECTION E, FUNDAMENTAL STEREOCHEMISTRY, Pure Appl. Chem. 45:13-30 (1976). Os termos α e β são empregados para posições de anéis de compostos cíclicos. O lado α- do plano de referência é aquele lado no qual o substituinte preferido fica na posição numerada inferior. Aqueles substituintes que ficam no lado oposto do plano de referência são designados pelo descriptor β . Deve ser observado que esta utilização difere daquela para estereoparentes cíclicos, em que "α" significa "abaixo do plano" e denota configuração absoluta. Os termos de configuração α e β, como usados aqui no presente, são como definidos pelo CHEMICAL ABSTRACTS INDEX GUIDEAPPENDIX IV (1987) paragráfo 203.
[00069] Como usado aqui no presente, o termo "sais farmaceuticamente aceitáveis" se refere os sais de metais alcalinoterrosos ou ácidos não tóxicos dos compostos de Fórmula I ou II. Estes sais podem ser preparados in situ durante o isolamento e a purificação final dos compostos de Fórmula I ou II, ou por reagir, separadamente, a base ou as funções de ácido com ácido ou base orgânico ou inorgânico respectivamente, apropriados. Sais representativos incluem mas não são limitados aos seguintes: acetato, adipato, alginato, citrato, aspartato, benzoato, benzenossulfonato, bissulfato, butirato, canforato, canforsulfonato, digluconato, ciclopentanopropionato, dodecilsulfato, etanossulfonato, glucoeptanoato, glicerofosfato, hemissulfato, heptanoato, hexanoato, fumarato, cloridrato, bromidrato, iodoidrato, 2-hidroxietanossulfonato, lactato, maleato, metanossulfonato, nicotinato, 2-naftalenossulfonato, oxalato, pamoato, pectinato, persulfato, 3-fenilproionato, picrato, pivalato, propionato, succinato, sulfato, tartarato, tiocianato, p-toluenossulfonato e undecanoato. Também, os grupos contendo nitrogênio básico podem ser quaternizados comtais agentes como haletos de alquila inferior, tais como metila, etila, propila, e cloridrato, bromidrato e iodoidrato de butila; sulfatos de dialquila como dimetila, dietila, dibutila, e sulfatos de diamila, haletos de cadeia longa tais como cloretos, brometos e iodetos de decila, laurila, miristila e estearila, haletos de aralquila como brometos de benzila e fenetila e outros. Água ou produtos dispersáveis ou solúveis em óleo são, dessa maneira, obtidos.
[00070] Exemplos de ácidos que podem ser empregados para formar sais de adição de ácido farmaceuticamente aceitáveis incluem tais ácidos inorgânicos como ácido clorídrico, ácido enxofreico e ácido fosfórico, e tais ácidos orgânicos como ácido oxálico, ácido maleico, ácido metanossulfônico, ácido succínico e ácido cítrico. Sais de adição básica podem ser preparados in situ durante o isolamento e a purificação finais dos compostos de fórmula (I), ou separadamente pela reação de porção de ácido carboxílico com uma base apropriada tal como hidróxido, carbonato ou bicarbonato de um cátion de metal farmaceuticamente aceitável ou com amônia, ou uma amina primária, secundária ou terciária orgânica. Sais farmaceuticamente aceitáveis incluem, mas não são limitados a, cátions baseados em metais de álcali ou alcalinos terrosos, tais como os sais de sódio, lítio, cálcio, magnésio, alumínio e similares, como também um amônio não tóxico, amônio quaternário e cátions de amina, incluindo, mas não limitado a amônio, tetrametilamônio, tetraetilamônio, metilamina, dimetilamina, trimetilamina, trietilamina, etilamina e similares. Outras aminas orgânicas representativas úteis para a formação de sais de adição de base incluem dietilamina, etilenodiamina, etanolamina, dietanolamina, piperazina e similares.
[00071] Como usado aqui no presente, o termo "éster farmaceuticamente aceitável" se refere aos ésteres, que hidrolisam in vivo e inclui aqueles que prontamente se decompõem no corpo humano para deixar o composto original ou um sal do mesmo. Grupos éster apropriados incluem, por exemplo, aqueles derivados de ácidos carboxílicos alifáticos farmaceuticamente aceitáveis, particularmente ácidos alcanóicos, alquenóicos, cicloalcanóicos e alcanedióicos, em que cada porção de alquila ou alquenila vantajosamente têm não mais do que 6 átomos de carbono. Exemplos de ésteres particulares incluem formiatos, acetatos, propionatos, butiratos, acrilatos e etilsuccinatos.
[00072] O termo "profármacos farmaceuticamente aceitáveis" como usado aqui no presente se refere àqueles profármacos dos compostos da presente invenção que são, dentro do escopo de julgamento médico perfeito, apropriados para uso em contacto com os tecidos de seres humanos e de animais inferiores sem toxicidade indevida, irritação, resposta alérgica e similares, compatível com uma proporção de risco/benefício razoável, e eficaz para seu uso pretendido, como também as formas zwitteriônicas, quando possível, dos compostos da invenção. O termo "profármaco" se refere aos compostos que são rapidamente transformados in vivo para render o composto de origem da fórmula acima, por exemplo, por hidrólise no sangue. Uma discussão completa é provida em T. Higuchi e V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series, e em Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, ambos os quais são incorporados aqui no presente por referência.
[00073] Qualquer fórmula dada aqui no presente é também destinada a representar formas não marcadas como também formas marcadas isotopicamente dos compostos. Compostos marcados isotopicamente têm estruturas ilustradas pelas fórmulas dadas aqui no presente, exceto que aquele um ou mais átomos são substituídos por um átomo tendo uma massa atômica selecionada ou número de massa. Exemplos de isótopos que podem ser incorporados nos compostos da invenção incluem isótopos de hidrogênio, carbono, nitrogênio, oxigênio, fósforo, flúor e cloro, tais como 2H, 3H, 11C, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36Cl, 125I respectivamente. A invenção inclui vários compostos isotopicamente marcados como definido aqui no presente, por exemplo, aqueles dentro dos quais isótopos radioativos, tais como 3H, 13C, e 14C, estão presentes. Tais compostos isotopicamente marcados são úteis em estudos metabólicos (com 14C), estudos de reação cinética (com, por exemplo 2H ou 3H), técnicas de detecção ou de imagem, tais como tomografia de emissão de pósitron (PET) ou tomografia computadorizada de emissão de fóton único (SPECT) incluindo ensaios de distribuição de fármaco ou tecido de substrato, ou em tratamento radioativo de pacientes. Em particular, um 18F ou composto marcado pode ser particularmente desejável para estudos de PET ou SPECT. Compostos isotopicamente marcados desta invenção e profármacos dos mesmos podem geralmente ser preparados conduzindo os procedimentos descritos nos esquemas ou nos exemplos e preparações descritas abaixo, substituindo um reagente isotopicamente marcado prontamente disponível por um reagente marcado não isotopicamente.
[00074] Além disso, substituição com isótopos mais pesados, particularmente deutério (isto é, 2H ou D) pode produzir certas vantagens terapêuticas resultantes de estabilidade metabólica maior, por exemplo média de vida in vivo aumentada, ou requisitos de dosagem reduzidos, ou um melhoramento no índice terapêutico. Entende-se que deutério, neste contexto, é visto como um substituinte de um composto da fórmula (I). A concentração de tal isótopo mais pesado, especificamente deutério, pode ser definida prlo fator de enriqucimento isotópico. O termo "fator de enriquecimento isotópico", como usado aqui no presente, significa a relação entre a abundância isotópica e a abundância natural do isótopo especificado. Se um substituinte em um composto desta invenção é denotado deutério, tal composto tem um fator de enriquecimento isotópico para cada átomo de deutério designado de pelo menos 3500 (52,5% de incorporação de deutério em cada átomo de deutério designado), pelo menos 4000 (60% de incorporação de deutério), pelo menos 4500 (67,5% de incorporação de deutério), pelo menos 5000 (75% de incorporação de deutério), pelo menos 5500 (82,5% de incorporação de deutério), pelo menos 6000 (90% de incorporação de deutério), pelo menos 6333.3 (95% de incorporação de deutério), pelo menos 6466.7 (97% de incorporação de deutério), pelo menos 6600 (99% de incorporação de deutério), ou pelo menos 6633.3 (99,5% de incorporação de deutério).
[00075] Compostos isotopicamente marcados de fórmula (I) podem geralmente ser preparados pelas técnicas convencionais conhecidas daqueles versados na técnica, ou por processos análogos àqueles descritos nos Exemplos e Preparações que acompanham usando um reagente marcado isotopicamente apropriado no lugar do reagente não marcado empregado anteriormente.
[00076] Será evidente para aqueles versados na técnica que os compostos da invenção, ou seus tautômeros, profármacos e estereoisômeros, como também sais, ésteres e profármacos de qualquer um deles farmaceuticamente aceitáveis, podem ser processados in vivo através de metabolismo em um corpo ou célula de ser humano ou animal para produzir metabólitos. O termo "metabólito", como usado aqui no presente, se refere à fórmula de qualquer derivado produzido em um sujeito depois da administração de um composto de origem. Os derivados podem ser produzidos a partir do composto de origem por várias transformações bioquímicas no sujeito tais como, por exemplo, oxidação, redução, hidrólise ou conjugação e incluem, por exemplo, óxidos e derivados demetilados. Os metabólitos de um composto da invenção podem ser identificados usando ténicas rotineiras conhecidas na técnica. Ver, por exemplo, Bertolini, G. et al., J. Med. Chem. 40:2011-2016 (1997); Shan, D. et al., J. Pharm. Sci. 86(7):765-767; Bagshawe K., Drug Dev. Res. 34:220-230 (1995); Bodor, N., Advances in Drug Res. 13:224-331 (1984); Bundgaard, H., Design of Prodrugs (Elsevier Press 1985); e Larsen, I. K., Design and Application of Prodrugs, Drug Design and Desenvolvimento (Krogsgaard-Larsen et al., eds., Harwood Academic Publishers, 1991). Deverá ser entendido que compostos químicos individuais que são metabólitos dos compostos de fórmula I, fórmula II, ou seus tautômeros, profármacos e estereoisômeros, como também os sais, ésteres e profármacos de qualquer um deles farmaceuticamente aceitáveis, são incluídos dentro da invenção.
[00077] O termo "câncer" se refere às doenças de câncer que podem ser beneficamente tratadas pela inibição de Pim quinase, incluindo, por exemplo, cânceres sólidos, tais como carcinomas (por exemplo, dos pulmões, pâncreas, tiroide, ovário, bexiga, mama, próstata ou cólon), melanomas, distúrbios mieloides (por exemplo, leucemia mieloide, mieloma múltiplo e eritroleucemia), adenomas (por exemplo, adenoma do cólon viloso) e sarcomas (por exemplo, osteossarcoma).
[00078] Os compostos da invenção podem ser obtidos através de procedimentos conhecidos daqueles versados na técnica. Por exemplo, como mostrado no Esquema 1, ciclohexanodionas podem ser convertidos através de monotriflatos para os ésteres de ciclo- hexenonaboronato correspondentes que podem se submeter a formação de ligação de carbono mediada por paládio com 4-cloro, 3- nitro piridina para render ciclo-hexenonas I substituídas por nitropiridina. A redução da funcionalidade de enona pode render um ciclohexenol II que, mediante proteção de álcool, redução de nitro e alceno, amida acoplada e desproteção podem render ciclo-hexanol amidas III. Ciclohexenol II pode também se submeter a reação de Mitsunobu com ftalimida para render um aminociclohexeno IV protegido. Em seguida à redução de nitro e alceno, ftalimida protegida por aminociclo-hexila piridila anilina Va pode ser submetida à acoplamento e desproteção de amida, para render amidas Vi de aminociclo-hexano. A anilina Vb de aminociclo-hexano piridila protegida por Boc pode também ser preparada de ciclo-hexenol II da maneira a seguir: proteção de álcool, alceno e redução de nitro, proteção de piridil amina Cbz, desproteção de silil éter, oxidação de Dess-Martin para a ciclohexanona, aminação redutora com benzilamina, desproteção de Cbz e Bn e proteção de Boc de amina alilfática primária. Nos produtos de amida III e VI, se R2 é halo ou triflato, as amidas III e VI podem ainda ser modificadas pelas modificações padrão para introduzir arilas, alquilas e heteroarilas substituídas em R2. Por exemplo, se R2 é Br, pela reação com ácidos borônicos ou reagentes organometálicos, ou conversão para o éster de boronato correspondente e reação com haletos de arila/heteroarila ou triflatos, uma variedade de modificações de R2 é possível. Esquema 1.
[00079] Alternativamente, como mostrado no Esquema 2,ciclohexenol II pode ser desidratado rendendo um ciclo-hexadieno que, mediante epoxidação (através da formação de bromo-hidrina e eliminação de HBr ou de mCPBA diretamente) e abertura de epóxido de azida rende álcool VI de ciclohexenil azida. Álcool VI de ciclo- hexenil azida pode ser convertido para anilina VIIa de hidroxi amino trans protegida pela redução de azida, proteção de álcool e redução de alceno e nitro. Alternativamente, o álcool VI ciclohexenil azida pode ser convertido para cis amino hidroxi anilina VIIb protegido através da redução de azida e proteção de Boc, mesilação de álcool e ciclização intramolecular para o cis carbamato cíclico, seguido pela proteção de Boc e redução de alceno e nitro. As ciclo-hexilpiridil anilines VIIa e VIIb resultantes podem ser convertidas para as piridina amidas correspondentes pela acoplagem de amida, acetato ou clivagem de carbamato cíclico e desproteção de Boc. Se R2 é halo ou triflato, as amidas VIIIa e VIIIb podem ser ainda modificadas pelas modificações padrão para introduzir arilas, alquilas e heteroarilas substituídas em R2, depois da formação da ligação de amida e antes da desproteção total. Por exemplo, se R2 é Br, através de reação com ácidos borônicos ou reagentes organometálicos, ou conversão para o éster de boronato correspondente e reação com haletos ou triflatos de arila/heteroarila, uma variedade de modificações de R2 é possível. Esquema 2.
[00080] Alternativamente, como mostrado no Esquema 3, 5-alquila, 4-hidróxi, 3-aminopiperidinas trissubstituídos podem ser preparados e modificados para render 5-alquila, 4-hidróxi, 3-aminopiperidinila piridina amidas IX trissubstituídos como a seguir. Reação de aldeído de Garner com (R)-4-benzil-3-propioniloxazolidin-2-ona seguida por proteção de TBS do álcool resultante produz o composto X. Redução de oxazolidinona seguida pela introdução do grupo azida rende o intermediário XI. Desproteção sob condições acídicas revela o álcool amino correspondente, que mediante proteção com o grupo Boc seguida pela mesilação do álcool primário rende o intermediário XII. Redução de azida produz a formação da piperidina que é subsequentemente reagida com 4-cloro-3-nitropiridina, reduzida para amina, acoplada com o ácido carboxílico correspondente e desprotegida para prover 5-metil,4-hidróxi-3-aminopiperidinila piridina amidas IX trissubstituído. Se R1 é halo ou triflato, a amida IX pode ser ainda modificada pelas modificações padrão para introduzir arilas, alquilas e heteroarilas substituídas em R1 depois da formação da ligação de amida e antes da desproteção total. Por exemplo, se R1 é Br, através da reação com ácidos borônicos ou reagentes organometálicos, ou conversão para o éster de boronato correspondente e reação com haletos de arila/heteroarila ou triflatos, uma variedade de modificações de R1 é possível. Esquema 3.
[00081] Alternativamente, como mostrado no Esquema 4, 5-metila, 4-hidróxi, 3-aminopiperidinas trissubstituídas podem também ser preparadas e modificadas para render 5-metila, 4-hidróxi, 3- aminopiperidinila amidas XIII trissubstituídas como a seguir. Reação de ésteres de boronato de crotila com SerOBn aldeído seguida pela formação de carbamato cíclico, clivagem oxidativa de alceno e redução rende o composto XIV de hidroxila. A desproteção de benzila seguida pela bistosilação e reação com p-metoxibenzilamina e desproteção de amina rende piperidina XV. Reação de piperidina XV substituída com arenos ou heteroarenos substituídos por halo nitro seguidos por proteção de carbamato, redução de nitro, acoplagem de amida, abertura e desproteção de carbamato cíclico rende 5-metila, 4- hidróxi, 3-aminopiperidinil amidas XIII trissubstituídos. Se R3 é halo ou triflato, a amida XV pode ser ainda modificada pelaas modificações padrão para introduzir arilas, alquilas e heteroarilas substituídas em R3. Por exemplo, se R3 é Br, através da reação com ácidos borônicos ou reagentes organometálicos, ou conversão para o éster de boronato correspondente e reação com haletos ou triflatos de arila/heteroarila, uma variedade de modificações de R3 é possível. Esquema 4. NMP. 100*C, 2 horas 4. H2, 20%N(OHVC MeOH, 2 horas 3a. CSJCOJ, MeO] 1 3 b- 4M [ ICV dioxano
NMP. 100*C, 2 horas 4. H2, 20%N(OHVC MeOH, 2 horas 3a. CSJCOJ, MeO] 1 3 b- 4M [ ICV dioxano
[00082] Os compostos da invenção são úteis in vitro e/ou in vivo por inibir o crescimento de células de câncer. Os compostos podem ser usados sozinhos ou em composições junto com um veículo ou excipiente farmaceuticamente aceitável. Veículos ou excipientes farmaceuticamente aceitáveis apropriados incluem, por exemplo, agentes de processamento e modificadores e intensificadores de distribuição de fármacos, tais como, por exemplo, fosfato de cálcio, estearato de magnésio, talco, monossacarídeos, dissacarídeos, amido, gelatina, celulose, metil celulose, carboximetil celulose de sódio, dextrose, hidroxipropil-β-ciclodextrina, polivinilpirrolidinona, ceras de ponto de fusão baixo, resinas de troca de íons, e similares, como também combinações de quaisquer dos dois ou mais dos mesmos. Outros excipientes farmaceuticamente aceitáveis apropriados são descritos em "Remington's Pharmaceutical Sciences," Mack Pub. Co., New Jersey (1991), incorporados aqui no presente por referência.
[00083] Quantidades eficazes dos compostos da invenção geralmente incluem qualquer quantidade suficiente para detectavelmente inibir a atividade de Pim por qualquer uma dos ensaios descritos aqui no presente, através de outros ensaios de atividade de Pim quinase conhecidos daqueles que têm conhecimento comum na técnica, ou detectando uma inibição ou alívio de sintomas de câncer. A quantidade do ingrediente ativo que pode ser combinado com os materiais de veículo para produzir uma forma de dosagem única, vão variar dependendo do hospedeiro tratado e o modo particular de administração. Ficará entendido, entretanto, que o nível de dose específico para qualquer paciente particular dependerá de uma variedade de fatores, incluindo a atividade do composto específico empregado, a idade, peso corporal, saúde em geral, sexo, dieta, hora de administração, via de administração, taxa de excreção, combinação de fármacos, e a gravidade da doença particular sendo submetida a terapia. A quantidade terapeuticamente eficaz para uma dada situação pode ser prontamente determinada por experimentação de rotina e dentro da habilidade e julgamento do clínico comum.
[00084] Para os propósitos da presente invenção, uma dose terapeuticamente eficaz geralmente será uma dose diária total administrada para um hospedeiro, em doses únicas ou divididas, pode ser em quantidades, por exemplo, de 0,001 a 1000 mg/kg de peso corporal diariamente e mais preferido de 1,0 a 30 mg/kg de peso corporal diariamente. Composições de dosagem unitária podem conter tais quantidades de submúltiplos das mesmas para perfazer a dose diária.
[00085] Os compostos da presente invenção podem ser administrados oralmente, parenteralmente, sublingualmente, por aerossolização ou inalação spray, retalmente, ou topicamente em formulações de unidade de dosagem contendo carreadores, adjuvantes e veículos convencionais não tóxicos farmaceuticamente aceitáveis como desejado. A administração tópica pode também envolver o uso de administração transdérmica tal como emplastro transdérmico ou dispositivos de ionoforese. O termo parenteral, como usado aqui no presente, inclui injeções subcutâneas, intravenosas, intramuscular, injeção intrasternal ou técnicas deinfusão.
[00086] Preparações injetáveis, por exemplo, suspensões aquosas ou oleaginosas injetáveis estéreis podem ser formuladas de acordo com o conhecido na técnica usando agentes dispersantes ou umidificantes apropriados e agentes de suspensão. A preparação injetável estéril pode também ser uma solução ou suspensão injetável estéril em um diluente ou solvente aceitável parenteralmente não tóxico, por exemplo, como uma solução em 1,3-propanediol. Entre os veículos e solventes aceitáveis que podem ser empregados estão água, solução de Ringer e solução de cloreto de sódio isotônico. Além disso, óleos estéreis, fixos convencionalmente empregados são como um solvente ou meio de suspensão. Para este propósito qualquer óleo fixo suave pode ser empregado incluindo mono- ou di-glicerídeos sintéticos. Adicionalmente, ácidos graxos, tais como ácido oléico, encontram uso na preparação de injetáveis.
[00087] Supositórios para administração retal do fármaco podem ser preparados misturando o fármaco com um excipiente não irritante apropriado tal como manteiga de cacau e polietileno glicóis, que são sólidos em temperaturas comuns, porém líquidos a temperatura retal e dessa maneria serão fundidos no reto e liberam o fármaco.
[00088] Formas de dosagem sólida para administração oral podem incluir cápsulas, comprimidos, pilulas, pós e grânulos. Em tais formas de dosagem sólida, o composto ativo pode ser admisturado com pelo menos um diluente inerte tal como sacarose, lactose ou amido. Tais formas de dosagem podem também compreender, como é prática normal, substâncias adicionais em vez de diluentes inertes, por exemplo, agentes lubrificadores tais como estearato de magnésio. No caso de cápsulas, comprimidos e pilulas, as formas de dosagem podem também compreender agentes de tampão. Comprimidos e pílulas podem adicionalmente ser preparados com revestimentos entéricos. Formas de dosagem líquida para administração oral podem incluir emulsões, soluções, suspensões, xaropes e elixires farmaceuticamente aceitáveis contendo diluentes inertes comumente usados na técnica, tal como água. Tais composições podem também compreender adjuvantes, tais como agentes de umidificação, agentes emulsificantes e de suspensão, ciclodextrinas, e adoçantes, flavorizantes e agentes de perfumar.
[00089] Os compostos da presente invenção podem também ser administrados na forma de lipossomas. Como é conhecido na técnica, lipossomas são geralmente derivados de fosfolipídeos ou outras substâncias lipídicas. Lipossomas são formados por cristais líquidos hidratados mono- ou multi-lamelares que são dispersos em um meio aquoso. Qualquer lipídeo não tóxico, fisiologicamente aceitável e metabolizável capaz de formar lipossomas pode ser usado. As composições presentes em forma de lipossoma podem conter, em adição a um composto da presente invenção, estabilizadores, conservantes, excipientes, e similares. Os lipídeos preferidos são os fosfolipídeos e fosfatidil colina (lecitinas), ambos naturais e sintéticos. Métodos para formar lipossomas são conhecidos na técnica. Ver, por exemplo, Prescott, Ed., Métodos in Cell Biology, Volume XIV, Academic Press, New York, N.W., p. 33 et seq. (1976).
[00090] Enquanto os compostos da invenção podem ser administrados como o único agente farmacêutico ativo, eles podem também ser usados em combinação com um ou mais outros agentes usados no tratamento de câncer. Os compostos da presente invenção são também úteis em combinação com agentes terapêuticos conhecidos e agentes anticâncer, e combinações dos compostos presentemente descritos com outros agentes anticâncer ou quimioterápicos estão dentro do escopo da invenção. Exemplos de tais agentes podem ser encontrados em Câncer Principles and Practice of Oncology, V. T. Devita e S. Hellan (editores), 6a edição (15 de fevereiro de 2001), Lippincott Williams & Wilkins Publishers. Uma pessoa de conhecimento comum na técnica sera capaz de discernir quais combinações de agentes serão úteis com base nas características particulares dos fármacos e o câncer envolvido. Tais agentes anticâncer incluem, mas não são limitados ao seguinte: moduladores de receptor de estrogênio, moduladores de receptor de androgênio, moduladores de receptor retinoide, agentes citotóxicos / citostáticos, agentes antiproliferativos, inibidores de transferase de proteína prenil, inibidores de redutase de HMG-CoA e outros inibidores de angiogênese, inibidores de proliferação de células e sinalização de sobrevivência, apoptose induzindo agentes e agentes que interferem com pontos de verificação de ciclo de células. Os compostos da invenção são também úteis quando coadministrados com radioterapia.
[00091] Dessa maneira, em uma modalidade da invenção, os compostos da invenção são também usados em combinação com agentes anticâncer conhecidos incluindo, por exemplo, moduladores de receptor de estrogênio, moduladores de receptor de androgênio, moduladores de receptor retinoide, agentes citotóxicos, agentes antiproliferativo, inibidores de transferase de prenil-proteína, inibidores de redutase de HMG-CoA, inibidores de protease de HIV, inibidores de transcriptase reversa e outros inibidores de angiogênese.
[00092] Em certas modalidades da invenção presentemente preferidas, agentes representativos úteis em combinação com os compostos da invenção para o tratamento de câncer incluem, por exemplo, irinotecano, topotecano, gemcitabina, 5-flúoruracila, leucovorin carboplatina, cisplatina, taxanos, tezacitabina, ciclofosfamida, alcaloides vinca, imatinib (Gleevec), antraciclinas, rituximab, trastuzumab, como também outros agentes quimioterápicos de câncer.
[00093] Os compostos acima para serem empregados em combinação com os compostos da invenção serão úteis em quantidades terapêuticas como indicado no Physicians' Desk Reference (PDR) 47a Edição (1993), que é incorporado aqui no presente por referência, ou tais quantidades terapeuticamente úteis como será conhecido da pessoa de habiidade comum na técnica.
[00094] Os compostos da invenção e outros agentes anticâncer podem ser administrados na dosagem clínica máxima recomendada ou em doses mais baixas. Os níveis de dosagem dos compostos ativos nas composições da invenção podem ser variados de maneira obter uma resposta terapêutica desejada, dependendo da via de administração, da gravidade da doença e da resposta do paciente. A combinação pode ser administrada como composições separadas ou côo uma forma de dosagem única contendo ambos os agentes. Quando administrados como uma combinação, os agentes terapêuticos podem ser formulados como composições separadas, que são dadas ao mesmo tempo ou em tempos diferentes, ou os agentes terapêuticos podem ser dados como uma composição única.
[00095] Em uma modalidade, a invenção provê um método de inibir Pim1, Pim2 ou Pim3 em um ser humano ou sujeito animal. O método inclui administrar uma quantidade eficaz de um composto, ou um sal do mesmo farmaceuticamente aceitável, de qualquer uma das modalidades dos compostos de Fórmula I ou II para um sujeito com necessidade do mesmo.
[00096] A presente invenção sera compreendida mais prontamente pela referência aos exemplos a seguir, que são providoa a título de ilustração e não são destinados a ser limitantes da presente invenção. EXEMPLOS
[00097] Com referência aos exemplos que estão a seguir, os compostos das modalidades preferidas foram sintetizados usando os métodos descritos aqui no presente, ou outros métodos, que são conhecidos na técnica.
[00098] Os compostos e/ou intermediários foram caracterizados por cromatografia íquida de alto desempenho (HPLC) usando um sistema cromatográfico Waters Millenium com um 2695 Separation Module (Milford, MA). As colunas analíticas foram Fenomenex Luna C18 -5 μ de fase reversa, 4,6 x 50 mm, de Alltech (Deerfield, IL). Uma purificação de gradiente foi usada (2,5 mL/min de fluxo), tipicamente iniciando com 5% de acetonitrila/95% de água e progredindo para 100% de acetonitrila durante um período de 10 minutos. Todos os solventes contendo 0,1% de ácido triflúoracético (TFA). Compostos foram detectados por absorção de luz ultravioleta (UV) em 220 ou 254 nm. Solventes de HPLC foram os de Burdick e Jackson (Muskegan, MI), ou Fisher Scientific (Pittsburgh, PA).
[00099] Em alguns casos, a pureza foi avaliada por cromatografia de camada fina (TLC), usando placas de sílica-gel sustentadas por vidro ou plástico, tais como, por exemplo, folhas flexíveis Baker-Flex Sílica Gel 1B2-F. Os resultados de TLC foram prontamente detectados visualmente sob luz ultravioleta, ou empregando vapor de iodo bemconhecido e outras diversas técnicas de coloração.
[000100] Análise espectrométrica de massa foi realizada em um dos três instrumentos de LCMS: Um Waters System (Alliance HT HPLC e um espectrometro de massa Micromass ZQ; Coluna: Eclipse XDB-C18, 2.1 x 50 mm; gradiente: 5-95% (ou 35-95%, ou 65-95% ou 95-95%) acetonitrila em água com 0,05% de TFA durante um período de 4 min; taxa de fluxo 0,8 mL/min; variação do peso molecular 200-1500; cone Voltage 20 V; temperatura da coluna 40°C), outro Waters System (sistema ACQUITY UPLC e um sistema ZQ 2000; Coluna: ACQUITY UPLC HSS-C18, 1,8um, 2,1 x 50mm; gradiente: 5-95% (ou 35-95%, ou 65-95% ou 95-95%) acetonitrila em água com 0,05% de TFA durante um período de 1,3 min; taxa de fluxo 1,2 mL/min; variação de peso molecular 150-850; cone Voltage 20 V; temperatura da coluna 50°C) ou um Hewlett Packard System (Série 1100 HPLC; Coluna: Eclipse XDB-C18, 2.1 x 50 mm; gradiente: 5-95% de acetonitrila em água com 0,05% de TFA durante um período de 4 min; taxa de fluxo 0,8 mL/min; variação do peso molecular 150-850; cone Voltage 50 V; temperatura da coluna 30°C). Todas as massas foram reportadas como aquelas de íons de origem protonados.
[000101] Análise de ressonância magnética nuclear (NMR) foi realizada de alguns compostos com um Varian 400 MHz NMR (Palo Alto, CA). A referência espectral foi TMS ou o desvio químico conhecido do solvente.
[000102] Separações preparadoras são realizadas usando sistema de cromatografia instantânea 40 e KP-Sil, 60A (Biotage, Charlottesville, VA), ou por cromatografia de coluna instantânea usando sílica-gel (230-400 de malha) material de embalagem, ou por HPLC usando um Waters 2767 Sample Manager, coluna de fase reversa C-18, 30X50 mm, fluxo de 75 mL/min. Solventes típicos empregados para o sistema Biotage Instantâneo 40 e cromatografia de coluna instantânea são diclorometano, metanol, etil acetato, hexano, acetona, amônia aquosa (ou hidróxido de amonio), e trietil amina. Solventes típicos empregados para a HPLC de fase reversa são concentrações variando de acetonitrila e água com 0,1% de ácido triflúoracético.
[000103] Deverá ficar entendido que os compostos orgânicos, de acordo com as modalidades preferidas, podem exibir o fenômeno de tautomerismo. Como as estruturas químicas dentro deste relatório descritivo podem representar somente uma das possíveis formas tautoméricas, deverá ficar entendido que as modalidades preferidas abrangem qualquer forma tautomérica da estrutura lançada.
[000104] Entende-se que a invenção não é limitada às modalidades mencionadas aqi no presente para ilustração, mas abrange totas tais formas das mesmas como vêm dentro do escopo da descrição acima.
[000105] Os exemplos abaixo como também por todo o pedido, as abreviaturas a seguir têm os significados a seguir. Se não definidos, os termos têm seus significados geralmente aceitos.
 Síntese de triflúormetanossulfonato de 3-oxociclo-hex-1-enila
Síntese de triflúormetanossulfonato de 3-oxociclo-hex-1-enila 
[000106] Para uma solução de ciclo-hexano-1,3-diona (equiv. 1) em DCM (0,4 M) foi adicionado Na2CO3 (equiv. 1,0) e refrigerado até 0 oC. Adicionado Tf2O (equiv. 1,0) em DCM (5 M) em gotas durante 1 hora a temperatura ambiente sob uma atmosfera de nitrogênio. Mediante a adição, a reação foi agitada por 2 horas (solução vermelha escura). A solução foi filtrada e o filtrado foi adicionado a NaHCO3 saturado (cuidadosamente), depois extraídos os orgânicos, secos com salmoura, depois Na2SO4, e concentrada. O bruto foi usado para a etapa seguinte sem purificação adicional. Triflúormetanossulfonato de 3-oxociclo-hex-1-enila foi obtido em 67% de rendimento. O triflato se decompõe mediante armazenamento e deverá ser usado imediatamente para a próxima reação. LC/MS=244,9/286,0 (M+H and M+CH3CN); Rt = 0,88 min. Síntese de 3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)ciclo-hex-2- enona
[000107] Para uma solução de triflúormetanossulfonato de 3- oxociclo-hex-1-enila (equiv. 1,0) em dioxano desgaseificado (0,3 M) foi adicionado bi(pinacolato)diboro (equiv. 2,0), KOAc (equiv. 3,0), e Pd(dppf)Cl2-DCM (equiv. 0,05). A reação foi aquecida até 80 oC por 2 horas, depois filtrada. A solução de dioxano foi usada para a próxima etapa sem purificação adicional. LC/MS = 140,9 (M+H de ácido borônico). Síntese de 3-(3-nitropiridin-4-il)ciclo-hex-2-enona
[000108] Para uma solução de 3-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)ciclo-hex-2-enona (equiv. 1,0) em dioxano desgaseificado e 2M Na2CO3 foi adicionado 4-cloro-3-nitropiridina (1,2 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.). A reação foi aquecida em um banho de óleo a 110 oC por 2 horas. Resfriada à temperatura ambiente, depois diluída com EtOAc, adicionado H2O - solução escura, um grande número de emulsões. Filtrada para ficar livre dos sólidos, depois extraída a fase orgânica, seca com Na2SO4, e concentrada. O produto bruto foi purificado através de cromatografia em sílica-gel eluindo com acetato de etila e hexanos (1:1) para render 3-(3- nitropiridin-4-il)ciclo-hex-2-enona (55%, 2 etapas). LC/MS = 219 (M+H), LC = 2.294 min. Síntese de 3-(3-nitropiridin-4-il)ciclo-hex-2-enol
[000109] Para uma solução de 3-(3-nitropiridin-4-il)ciclo-hex-2-enona (equiv. 1,0) foi adicionado EtOH (0,2 M) e CeCIs-THaO (1,3 equiv.). A reação foi resfriada a 0 oC, depois NaBH4 (1,3 equiv.) foi adicionado em porções. Agitada por 2 h a 0 oC, depois extinta por adição de água, concentrada para remover o EtOH, adicionado EtOAc, extraídos os produtos orgânicos, seca com salmoura, depois Na2SO4, e concentrada para render 3-(3-nitropiridin-4-il)ciclo-hex-2-enol (99%). LC/MS = 221,1 (M+H), LC = 2,235 min. Síntese de 2-(3-(3-nitropiridin-4-il)ciclo-hex-2-enil)isoindolina-1,3-diona
[000110] Para uma solução de 3-(3-nitropiridin-4-il)ciclo-hex-2-enol (equiv. 1,0), trifenilfosfina (equiv. 1,5) e ftalimida (equiv. 1,5) em THF (0.3 M) a 0 oC foi adicionado (E)-di-terc-butil diazeno-1,2-dicarboxilato (equiv. 1,5) em gotas. A reação foi agitada a 0 oC por 2 horas. Concentrada até a secura sob vácuo, depois purificado o produto bruto através de cromatografia em coluna de sílica-gel eluindo com EtOAc e hexanos (1:1 com metanol 5%) para render a 2-(3-(3-nitropiridin-4- il)ciclo-hex-2-enil)isoindolina-1,3-diona (rendimento de 63%). LC/MS = 350,3 (M+H), LC = 3,936 min. Síntese de 2-(3-(3-aminopiridin-4-il)ciclo-hex-2-enil)isoindolina-1,3- diona
[000111] Para uma solução de 2-(3-(3-nitropiridin-4-il)ciclo-hex-2- enil)isoindolina-1,3-diona (equiv. 1,0) em AcOH (0,38 M) foi adicionado Fe (equiv. 6,0) e a reação foi agitada à temperatura ambiente por 2 h. Filtrada, depois lavada com metanol e o filtrado concentrado. Para o produto bruto foram adicionados acetato de etila e NaHCO3 saturado, os produtos orgânicos foram secos com Na2SO4, e concentrados para dar 2-(3-(3-aminopiridin-4-il)ciclo-hex-2-enil)isoindolina-1,3-diona como uma goma espessa amarela em rendimento de 96%. LC/MS = 320,0 (M+H), LC = 2,410 min. Síntese de 2-(3-(3-aminopiridin-4-il)ciclo-hexil)isoindolina-1,3-diona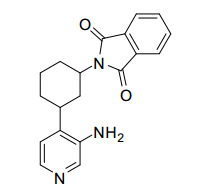
[000112] Para uma solução de 2-(3-(3-nitropiridin-4-il)ciclo-hex-2- enil)isoindolina-1,3-diona (equiv. 1,0) em ácido acético (0,1 M) foi adicionado Pd/C 10% (equiv. 0,2) e a reação foi agitada sob um balão de H2. Após 3 dias, a reação foi filtrada através de Celite, lavada com acetato de etila e metanol, o filtrado foi diluído com acetato de etila e lavado duas vezes com 2M Na2CO3 saturado. A fase orgânica foi seca com sulfato de magnésio, filtrada e concentrada. O material bruto foi triturado com hexanos e acetato de etila para render 2-(3-(3- aminopiridin-4-il)ciclo-hexil)isoindolina-1,3-diona em 73% de rendimento. LC/MS = 322,2 (M+H), temperatura ambiente = 0,64 min. Síntese de triflúormetanossulfonato de 5,5-dimetil-3-oxociclo-hex-1-enila
[000113] Em um balão de fundo redondo de 3 gargalos, 5,5- dimetilciclohexano-1,3-diona (eq. 1,1) foi dissolvido em DCM (0,2 M). Carbonato de sódio (eq. 1,1) foi adicionado e a mistura foi resfriada com agitação magnética em um banho de gelo/água salgada a ~ -5 °C sob N2. Anidrido tríflico (equiv. 1,05) diluído em DCM foi adicionado em gotas através de um funil de adição durante 90 minutos. Após a conclusão da adição, a reação foi agitada a ~ 0 °C por 1h. A partir de LCMS e 1H NMR, ainda havia material de partida restante. Carbonato de sódio adicional (eq. 0,51) e anidrido tríflico (eq. 0,50) foram adicionados. Após 2 horas, a mistura foi filtrada através de um funil de frita de vidro grosso (a torta foi lavada com DCM), transferida para um balão Erlenmeier, extinta através de cuidadosa adição de bicarbonato de sódio aquoso saturado com vigorosa agitação até que o pH =7, transferida para um funil separador e as camadas separadas. A camada orgânica foi lavada com salmoura, seca sobre MgSO4, filtrada e concentrada para dar triflúormetanossulfonato de 5,5-dimetil-3- oxociclo-hex-1-enila, o qual foi usado para a próxima etapa sem purificação posterior. LC/MS (m/z): MH+=273,1, Rt=1,03. Síntese de 5,5-dimetil-3-(4,4,5,5-tetrametil- 1,3,2-dioxaborolan-2-il)ciclo-hex-2-enona
[000114] Todos os reagentes triflúormetanossulfonato de 5,5-dimetil- 3-oxociclo-hex-1-enila (eq. 1,0), acetato de potássio (eq. 3,0), e bis(pinacolato)diboro (eq. 2,0) foram adicionados a 1,4-dioxano (0.2 M) em um balão de fundo redondo e desgaseificado por borbulhamento de N2 através da mistura por 10 min. Aduto de PdCl2(dppf) - DCM (eq. 0,03) foi adicionado e a reação aquecida a 80 °C equipado com um condensador de refluxo em um banho de óleo sob N2 durante a noite. A mistura foi resfriada à temperatura ambiente, filtrada através de um funil de frita de vidro grosso, a torta lavada com 1,4-dioxano para dar o 5,5-dimetil-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il) ciclo-hex-2- enona em 1,4-dioxano o qual foi usado para a próxima etapa sem purificação posterior. LC/MS (m/z): MH+(ácido borônico) =169,1, Rt=0,50. Síntese de 5,5-dimetil-3-(3-nitropiridin-4-il)ciclo-hex-2-enona
[000115] O éster boronato de 5,5-dimetil-3-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)ciclo-hex-2-enona (eq. 1,0) foi dissolvido em 1,4- dioxano em um balão de fundo redondo e desgaseificado por borbulhamento de N2 através da solução por 30 minutos. 4-cloro-3- nitro-piridina (eq. 1,3) e 2M carbonato de sódio (aq.) (eq. 2,0) foram adicionados e N2 foi borbulhado por 10 minutos e depois PdCl2(dppf) - DCM (eq. 0,05) foi adicionado. A mistura de reação foi agitada a 110 °C por 2 h. A mistura foi adicionada a EtOAc e água. A mistura resultante foi filtrada através de celite, a torta foi lavada com EtOAc. A camada orgânica foi separada e a parte aquosa foi extraída com EtOAc. As camadas orgânicas combinadas foram lavadas com salmoura, secas sobre MgSO4, filtradas e concentradas. O resíduo foi purificado através de cromatografia em sílica-gel (eluída com EtOAc:Hexanos = 1:10 a 2:1) para dar 5,5-dimetil-3-(3-nitropiridin-4- il)ciclo-hex-2-enona (46.7% para três etapas). LC/MS (m/z): MH+=247,2, Rt=0,79. Síntese de 5,5-dimetil-3-(3-nitropiridin-4-il)ciclo-hex-2-enol
[000116] Para uma solução de 5,5-dimetil-3-(3-nitropiridin-4-il)ciclo- hex-2-enona (eq. 1,0), e CeCl3-7H2O (eq. 1,2) em MeOH (0,2 M) foi adicionado NaBH4 (eq. 1,0) a 0 °C. A solução foi agitada por 1 hora, e depois extinta pela adição de 5 mL de água. Os voláteis foram removidos em vácuo e o resíduo foi particionado entre EtOAc e H2O. A camada orgânica foi separada e lavada com salmoura. A parte aquosa combinada foi extraída de volta com EtOAc e o produto orgânico foi lavado com salmoura. Os produtos orgânicos combinados foram secos sobre MgSO4, filtrados e concentrados. O resíduo foi purificado através de coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar 5,5-dimetil-3-(3-nitropiridin-4-il)ciclo-hex-2-enol (74%). LC/MS (m/z): MH+=249,2, Rt=0,76. Síntese de 2-(5,5-dimetil-3-(3-nitropiridin-4-il)- ciclo-hex-2-enil) isoindolina-1,3-diona
[000117] Para uma solução homogênea de 5,5-dimetil-3-(3- nitropiridin-4-il) ciclo-hex-2-enol (eq. 1,1), trifenil fosfina (eq. 1,5), e ftalimida (eq. 1,5) em THF (0.2 M) resfriada até 0 °C, azodicarboxilato de ditercbutila (eq. 1,5) em THF foi adicionado para a solução. A mistura foi agitada a 0 °C por 2 horas. A reação foi concentrada a vácuo. O resíduo foi purificado através de coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar 2-(5,5-dimetil-3-(3-nitropiridin-4- il)ciclo-hex-2-enil)isoindolina-1,3-diona (99%). LC/MS (m/z): MH+=378,2, Rt=1,10. Síntese de 2-(3-(3-aminopiridin-4-il)-5,5-dimetil-ciclo-hex-2-enil) isoindolina-1,3-diona
[000118] Uma solução de 2-(5,5-dimetil-3-(3-nitropiridin-4-il)ciclo-hex- 2-enil) isoindolina-1,3-diona (eq. 1) em ácido acético (0.1 M) foi purgada com nitrogênio por 10 min. Depois Pd/C 10% (eq. 0,10) foi adicionado. A mistura de reação foi agitada à temperatura ambiente durante a noite sob uma atmosfera de hidrogênio. Os sólidos foram removidos por filtração sobre celite, depois lavados com EtOAc e MeOH. O filtrado foi concentrado, diluído com EtOAc e lavado 2x com 2M Na2CO3 aquoso saturado. A camada orgânica foi seca com MgSO4, filtrada, e concentrada. O resíduo foi purificado por coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar 2-(3-(3-aminopiridin-4- il)-5,5-dimetilciclo-hex-2-enil)isoindolina-1,3-diona (89%). LC/MS (m/z): MH+=348,3, Rt=0,79. Síntese de 2-(5-(3-aminopiridin-4-il)-3,3-dimetilciclo-hexil) isoindolina- 1,3-diona
[000119] Uma solução de 2-(3-(3-aminopiridin-4-il)-5,5-dimetilciclo- hex-2-enil) isoindolina-1,3-diona (eq. 1,0) em ácido acético (0.1 M) foi purgada com nitrogênio por 10 min. Depois Pd/C 10% (eq. 0,1) foi adicionado. A mistura de reação foi agitada a 45 °C, atmosfera de hidrogênio de 2,068 MPa (300 psi) em uma bomba de aço durante a noite e a 65 °C, 2,068 MPa (300 psi) por 5 horas. Os sólidos foram removidos por filtração sobre celite, depois lavados com EtOAc e MeOH. O filtrado foi concentrado, diluído com EtOAc e lavado 2x com 2M Na2CO3 aquoso saturado. A camada orgânica foi seca com MgSO4, filtrada, e concentrada. O resíduo foi purificado por coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar 2-(5-(3-aminopiridin-4- il)-3,3-dimetilciclo-hexil)isoindolina-1,3-diona (53%). LC/MS (m/z): MH+=350,3, Rt=0,78. Uma 2-((1R,5R)-5-(3-aminopiridin-4-il)-3,3- dimetilciclo-hexil)isoindolina-1,3-diona enantiomericamente pura e 2- ((1S,5S)-5-(3-aminopiridin-4-il)-3,3-dimetilciclo-hexil)isoindolina-1,3- diona foram resolvidos por HPLC quiral (para análise temperatura ambiente = 7,526 min e 13,105 min respectivamente; hexanos:etanol= 80:20 (v:v), Chiralcel OJ-H 100 x 4,6 mm a 1 mL/min. Para a preparação separativa, hexanos:etanol = 80:20 (v:v), Chiralcel OJ-H (250 x 20 mm at 20 mL/min ). 1H NMR (CDCl3): δ 8,04 (s, 1H), 8,00 (d, 1H), 7,82 (m, 2H), 7,71 (m, 2H), 7,06 (d, 1H), 4,54 (m, 1H), 3,71 (m, 2H), 2,89 (m, 1H), 2,23-2,44 ( m, 2H), 1,90 (m, 1H), 1,20-1,60 (m, 3H), 1,18 (s, 3H), 1,07 (s, 3H). Síntese de 4-(ciclo-hexa-1,3-dienil)-3-nitropiridina
[000120] Para uma solução de 3-(3-nitropiridin-4-il)ciclo-hex-2-enol (equiv. 1,0) foi adicionado dioxano (0.18 M) e p-TSA (equiv. 1,1). A solução foi aquecida a 100 oC por 4 h. Resfriada à temperatura ambiente, "manipulada" com NaHCO3 saturado e acetato de etila, a fase orgânica foi seca com Na2SO4 e concentrada. O produto bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com DCM a 100% para dar 4-(ciclo-hexa-1,3-dienil)-3-nitropiridina como um óleo amarelo (27% rendimento). LCMS (m/z): 203,1 (MH+), LC temperatura ambiente = 3,53 min, 1H-NMR (CDCl3): 9,02 (s, 1H), 8,70 (d, J=5,3, 1H), 7,30 (d, J=5,3, 1H), 6,15-6,17 (m, 1H), 6,02-6,11 (m, 2H), 2,35-2,38 (m, 4H). Síntese de (+/-)-2-azido-4-(3-nitropiridin-4-il)ciclo-hex-3-enol
[000121] Para uma solução de 4-(ciclo-hexa-1,3-dienil)-3-nitropiridina (equiv. 1,0) em DCM (0.1 M) foi adicionado NaHCO3 (equiv. 1,2) para dar uma solução amarela. Resfriada para 0 °C, depois adicionado m- CPBA (equiv. 1,0) Para uma solução de uma vez como um sólido. A reação foi agitada a 0 °C por 3,5 h. Monitorado por ambos TLC e LC/MS. O produto ioniza como M+H = 237 (diol); Rt=0,41 min sobre UPLC. A reação foi extinta com NaHCO3 saturado, depois extraída com DCM (3 vezes). A fase orgânica foi ainda seca com salmoura, depois Na2SO4, filtrada e concentrada para dar o produto epóxido bruto como um óleo amarelo, o qual foi usado sem purificação posterior.
[000122] Para uma solução do material bruto acima em EtOH e água (3:1) (solução amarela turva) foi adicionado NaN3 (equiv. 2,0) e NH4Cl (equiv. 2,0) para dar uma solução laranja clara. A reação foi agitada por 16 h, depois concentrada. EtOAc e água foram adicionados, a fase orgânica foi ainda seca com MgSO4 e concentrada para dar um óleo marrom. O óleo foi carregado em sílica-gel e purificado através de cromatografia em coluna (ISCO, 0-50% EtOAc) para dar (+/-)-2-azido- 4-(3-nitropiridin-4-il)ciclo-hex-3-enol como um óleo amarelo (44% para 2 etapas). LCMS (m/z) = 262 (MH+), LC temperatura ambiente = 2,35 min. Síntese de (+/-)-4-(3-azido-4-(terc-butildimetilsililóxi)ciclo-hex-1-enil)-3- nitropiridina
[000123] Para uma solução de (+/-)-2-azido-4-(3-nitropiridin-4-il)ciclo- hex-3-enol (equiv. 1,0) em DCM (0.15 M) foi adicionado TBSCl (equiv. 2,0), imidazol (equiv. 2,0) e DMAP (equiv. 0,1) à temperatura ambiente. Após 18 h, água foi adicionada, os produtos orgânicos foram secos com salmoura, depois Na2SO4, e concentrados. O material bruto foi carregado para sílica-gel e purificado através de cromatografia em coluna (ISCO) eluindo com acetato de etila e hexanos (20%). (+/-)-4-(3-azido-4-(terc-butildimetilsililóxi)ciclo-hex-1- enil)-3-nitropiridina obtido como um óleo amarelo em 60% de rendimento. LCMS (m/z): 376,3 (MH+), LC temperatura ambiente =5,848 min. Síntese de 6-(terc-butildimetilsililóxi)-3-(3-nitropiridin-4-il)ciclo-hex-2- enilcarbamato de (+/-)-terc-butila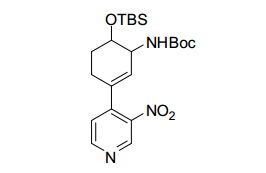
[000124] Em um balão de fundo redondo foi adicionado (+/-)-4-(3- azido-4-(terc-butildimetilsililóxi)ciclo-hex-1-enil)-3-nitropiridina (equiv. 1,0) e piridina (0,1 M) para dar uma solução amarela. Hidróxido de amônio (10:1 piridina: hidróxido de amônio) foi adicionado seguido por PMe3 (equiv. 3,0). A reação tornou-se marrom escura após 10 min. Agitada à temperatura ambiente por 1,5 h. Extinta pela adição de EtOH, e concentrada. Repetida 2 vezes mais. Para o produto bruto foram adicionados NaHCO3 saturado e dioxano (1:1, 0,1M). Boc2O (equiv. 1,0) foi adicionado. Agitada por uma hora à temperatura ambiente. Lavada com H2O e EtOAc, a fase orgânica foi seca com MgSO4, filtrada e concentrada. O resíduo foi purificado através de cromatografia em coluna de sílica-gel (ISCO, 5:1 Hex/EtOAc). Coletadas as frações puras e concentradas para dar 6-(terc- butildimetilsililóxi)-3-(3-nitropiridin-4-il)ciclo-hex-2-enilcarbamato de (+/- )-terc-butila como uma espuma. LCMS (m/z): 450,3 (MH+), LC temperatura ambiente = 5,83 min. Síntese de 3-(3-aminopiridin-4-il)-6-(terc-butildimetilsililóxi) ciclo-hex-2- enilcarbamato de (+/-)-terc-butila
[000125] Para uma solução de 6-(terc-butildimetilsililóxi)-3-(3- nitropiridin-4-il)ciclo-hex-2-enilcarbamato de (+/-)-terc-butila (equiv. 1,0) em AcOH (0,18 M) foi adicionado Fe (6,0 equiv.) e a reação foi agitada por 20 h. Manipulada pela diluição da reação com metanol, filtrada, e o filtrado concentrado. Para o produto bruto foram adicionados acetato de etila e NaHCO3 saturado, os produtos orgânicos foram secos com sulfato de sódio e concentrados para dar 3-(3-aminopiridin-4-il)-6-(terc- butildimetilsililóxi)ciclo-hex-2-enilcarbamato (+/-)-terc-butila como um óleo amarelo em 94% de rendimento. LCMS (m/z): 420,3 (MH+), LC temperatura ambiente = 3,88 min. Síntese de 5-(3-aminopiridin-4-il)-2-(terc-butildimetilsililóxi) ciclo- hexilcarbamato (+/-)-terc-butila
[000126] Para uma solução de 3-(3-aminopiridin-4-il)-6-(terc- butildimetilsililóxi)ciclo-hex-2-enilcarbamato de (+/-)-terc-butila (equiv. 1,0) em MeOH (0,1 M) foi adicionado Pd/C (20% em peso) e a reação foi agitada sob um balão de hidrogênio por 18 h. LC/MS da reação indicou mistura de diaestereômeros, a reação foi filtrada, lavada com EtOAc e o filtrado concentrado. O material bruto foi purificado através de prep-HPLC (em DMSO), e as frações puras foram combinadas, neutralizadas com NaHCO3 sólido, extraídas com acetato de etila, lavadas com salmoura, secas sob Na2SO4, e concentradas para dar o produto A (8% de rendimento) e produto B (51% de rendimento).
[000127] Produto A: LCMS (m/z): 422,4 (MH+), LC temperatura ambiente = 3,75 min.
[000128] Produto B: LCMS (m/z): 422,4 (MH+), LC temperatura ambiente =3,94 min. Síntese de triflúormetanossulfonato de 1,4-dioxaespiro[4.5]dec-7-en-8- ila
[000129] 1,4-Dioxaespiro[4.5]decan-8-ona (equiv. 1,0) foi dissolvida em éter (0,1 M) e agitada a -15 °C depois 1M NaHMDS (equiv. 1,05) foi adicionado e agitada por 70 min depois Tf2O (equiv. 1,05) adicionado e a reação deixada para aquecer lentamente até a temperatura ambiente. A mistura foi agitada por 28 h, lavada com NaHCO3 aquoso saturado e depois água. As camadas aquosas combinadas e extraídas com éter. As camadas orgânicas combinadas, secas sobre MgSO4, filtradas, e concentradas. O resíduo foi purificado por coluna (éter etila : hexanos = 1 : 4) para dar triflúormetanossulfonato de 1,4-dioxaspiro[4,5]dec-7-en-8-ila (65%). LC/MS (m/z): MH+=289,0, Rt=0,97. HPLC Rt=3,77. Síntese de 4,4,5,5-tetrametil-2-(1,4-dioxaespiro[4,5]dec-7-en-8-il)- 1,3,2-dioxaborolano
[000130] Uma solução de triflúormetanossulfonato de 1,4- dioxaespiro[4,5]dec-7-en-8-ila (equiv. 1,0) em dioxano (0.5 M) foi purgada com nitrogênio por 30 min. Depois 4,4,4',4',5,5,5',5'-octametil- 2,2'-bi(1,3,2-dioxaborolano) (equiv. 1,0), KOAc (equiv. 3,0), Pd(dppf)Cl2-DCM (0,2 equiv.) foram adicionados e a solução foi agitada em uma bomba lacrada a 80 °C. A reação foi filtrada sobre uma almofada de celite, depois ao filtrado foi adicionado acetato de etila, e lavada com salmoura, seca sobre MgSO4, filtrada, e concentrada. O resíduo foi purificada por coluna (acetato de etila : hexanos = 1 : 1) para dar 4,4,5,5-tetrametil-2-(1,4-dioxaespiro[4.5]dec- 7-en-8-il)-1,3,2-dioxaborolano (95%). LC/MS (m/z): MH+=267,1, Rt=0,95. Síntese de 3-nitro-4-(1,4-dioxaespiro[4.5]dec-7-en-8-il)piridina
[000131] Uma solução de DME (0.2 M) e 2M carbonato de sódio aquoso (equiv. 1,7) foi purgada com nitrogênio por 20 min. Depois 4- cloro-3-nitropiridina (equiv. 1,6), 4,4,5,5-tetrametil-2-(1,4- dioxaespiro[4.5]dec-7-en-8-il)-1,3,2-dioxaborolano (equiv. 1,0), Pd(dppf)Cl2-DCM (equiv. 0,05) foram adicionados e agitados em uma bomba lacrada a 110 °C. A reação foi agitada naquela temperatura por 3,5 horas. A reação foi diluída com acetato de etila, lavada com água, seca sobre MgSO4, filtrada, e concentrada. O resíduo foi purificado por coluna (acetato de etila : hexanos = 1 : 1 com metanol 10%) para dar 3-nitro-4-(1,4-dioxaespiro[4.5]dec-7-en-8-il)piridina (83%). LC/MS (m/z): MH+=263,2, Rt=0,71. Síntese de 4-(3-nitropiridin-4-il)ciclo-hex-3-enona
[000132] A mistura de 3-nitro-4-(1,4-dioxaespiro[4.5]dec-7-en-8- il)piridina (equiv. 1,0) em TFA a 20% em CH2CI2 (0,2 M) foi agitada à temperatura ambiente durante a noite. Os solventes foram removidos sob pressão reduzida. O resíduo foi dissolvido com acetato de etila (200 mL), e lavado com NaHCO3 saturado (30mL), e NaCl saturado (30mL). O produto orgânico foi sêco com MgSO4, filtrada e concentrada para dar 4-(3-nitropiridin-4-il)ciclo-hex-3-enona (85%). O produto bruto foi usado para a próxima etapa sem purificação posterior. LC/MS (m/z): MH+=218,9, Rt=0,60 Síntese de 4-(3-nitropiridin-4-il)ciclo-hex-3-enol
[000133] Para uma solução de 4-(3-nitropiridin-4-il)ciclo-hex-3-enona (eq. 1,0) em metanol (0.2 M) foi adicionado boro-hidreto de sódio (equiv. 1,8) at 0 °C. A mistura de reação foi agitada a 0 °C por 2 h. Metanol foi removido sob pressão reduzida. O resíduo foi dissolvido com acetato de etila (200 mL), e lavado com NaCl saturado (30mL). O produto orgânico foi seco com MgSO4, filtrado e concentrado para dar 4-(3-nitropiridin-4-il)ciclo-hex-3-enol (85%). O produto bruto foi usado na próxima etapa sem purificação posterior. LC/MS (m/z): MH+=221,0, Rt=0,55 Síntese de metanossulfonato de 4-(3-nitropiridin-4-il)ciclo-hex-3-enila
[000134] Para uma solução de 4-(3-nitropiridin-4-il)ciclo-hex-3-enol (equiv. 1,0) e DIPEA (2,5 equiv.) em CH2Cl2 (0,15 M) foi adicionado cloreto de metanossulfonila (equiv. 1,8) a 0 °C. A mistura de reação foi agitada a 0 °C por 1 h. A mistura de reação foi diluída com acetato de etila (200 mL), e lavada com NaCl saturado (30 mL). O produto orgânico foi seco com MgSO4, filtrado e concentrado para dar metanossulfonato de 4-(3-nitropiridin-4-il)ciclo-hex-3-enila (93%). O resíduo foi usado na etapa seguinte sem purificação posterior. LC/MS (m/z): MH+=299,0, Rt=0,70 Síntese de 4-(ciclo-hexa-1,3-dienil)-3-nitropiridina
[000135] Para uma solução de metanossulfonato 4-(3-nitropiridin-4- il)ciclo-hex-3-enila (equiv. 1,0) em tetrahidrofurano (0,1 M) foi adicionado DBU (equiv. 1,8) à temperatura ambiente. A mistura de reação foi agitada à temperatura ambiente durante a noite. A mistura de reação foi diluída com acetato de etila (200 mL), e lavada com NaCl saturado (30mL). O produto orgânico foi seco com MgSO4, filtrado e concentrado. O resíduo foi purificado por coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar 4-(ciclo-hexa-1,3-dienil)-3- nitropiridina. LC/MS (m/z): MH+=203,2, Rt=0,85. Síntese de 6-hidróxi-3-(3-nitropiridin-4-il)ciclo-hex-2-enilcarbamato de (+/-)-terc-butila
[000136] Para uma solução de (+/-)-2-azido-4-(3-nitropiridin-4-il)ciclo- hex-3-enol (equiv. 1,0) em Piridina e NH4OH (8:1, 0,23 M) foi adicionada trimetilfosfina (equiv. 3,0) à temperatura ambiente. A mistura foi agitada à temperatura ambiente por 3 horas. Os solventes foram removidos. Ao resíduo foi adicionado etanol. Depois o etanol foi removido a vácuo para garantir a remoção total da amônia. O resíduo foi dissolvido em 1,4-dioxano e bicarbonato de sódio aquoso saturado, e depois Boc2O (eq. 1,0) em THF foram adicionados para uma mistura. A mistura resultante foi agitada à temperatura ambiente por 2 horas. A mistura de reação foi diluída com acetato de etila, e lavada com NaCl saturado. O produto orgânico foi seco com MgSO4, filtrado e concentrado. O resíduo foi purificado por coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar 6-hidróxi-3-(3-nitropiridin-4-il)ciclo- hex-2-enilcarbamato de (+/-)-terc-butila (82%). LC/MS (m/z): MH+=336,0, Rt=0,71. Síntese de metanossulfonato de (+/-)-2-(terc-butoxicarbonilamino)- 4-(3-nitropiridin-4-il)ciclo-hex-3-enila
[000137] Para uma solução de 6-hidróxi-3-(3-nitropiridin-4-il)ciclo- hex-2-enilcarbamato de (+/-)-terc-butila (equiv. 1,0) e trietil amina (equiv. 1,5) em CH2CI2 (0,2 M) foi adicionado cloreto de metanossulfonila (equiv. 1,2) a 0 °C. A mistura foi agitada por 2 horas nessa temperatura. A mistura de reação foi diluída com acetato de etila, e lavada com NaCl saturado. O produto orgânico foi seco com MgSO4, filtrado e concentrado para dar metanossulfonato de (+/-)-2- (terc-butoxicarbonilamino)-4-(3-nitropiridin-4-il)ciclo-hex-3-enila (85%), o qual foi usado na etapa seguinte sem purificação posterior. LC/MS (m/z): MH+=414,0, Rt=0,82 Síntese de(+/-)-5-(3-nitropiridin-4-il)-3,3a,7,7a- tetrahidrobenzo[d]oxazol-2(6H)-ona
[000138] A mistura de metanossulfonato de (+/-)-2-(terc- butoxicarbonilamino)-4-(3-nitropiridin-4-il)ciclo-hex-3-enila (equiv. 1,0) em piridina (0.21 M) foi agitada a 110 °C oor 10 min em micro-ondas. A piridina foi removida sob pressão reduzida. O resíduo foi dissolvido em acetato de etila, e lavado com NaCl saturado. O produto orgânico foi seco com MgSO4, filtrado e concentrado para dar (+/-)-5-(3- nitropiridin-4-il)-3,3a,7,7a-tetra-hidrobenzo[d]oxazol-2(6H)-ona (85%), o qual foi usado na etapa seguinte sem purificação posterior. LC/MS (m/z): MH+=262,1, Rt=0,49 Síntese de 5-(3-nitropiridin-4-il)-2-oxo-3a,6,7,7a-tetra- hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila
[000139] Para uma solução de (+/-)-5-(3-nitropiridin-4-il)-3,3a,7,7a- tetrahidrobenzo[d]oxazol-2(6H)-ona (equiv. 1,0), TEA (equiv. 1,8), e uma quantidade catalítica de DMAP em CH2CI2 (0,19 M) foi adicionado dicarbonato de di-terc-butila (equiv 1,2) à temperatura ambiente. A mistura de reação foi agitada por 1 hora. A mistura de reação foi diluída com acetato de etila (100 mL), e lavada com NaCl saturado (30mL). O produto orgânico foi seco com MgSO4, filtrado e concentrado. O resíduo foi purificado por coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar 5-(3-nitropiridin-4-il)-2-oxo- 3a,6,7,7a-tetra-hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc- butila (98%). LC/MS (m/z): MH+=306,0, Rt=0,75 Síntese de 5-(3-aminopiridin-4-il)-2-oxo-hexa-hidrobenzo[d]Oxazol- 3(2H)-carboxilato de (+/-)-terc-butila
[000140] Para uma solução de 5-(3-nitropiridin-4-il)-2-oxo-3a,6,7,7a- tetra-hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila (equiv. 1,0) em metanol e acetato de etila (1:1, 0,1 M) foi adicionado Pd/C 10% (equiv. 0,1). A mistura resultante foi agitada sob atmosfera de H2 por 6 horas. O sólido foi removido por filtração. O filtrado foi concentrado sob pressão reduzida para dar 5-(3-aminopiridin-4-il)-2- oxo-hexa-hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila (87%), o qual foi usado na etapa seguinte sem purificação posterior. LC/MS (m/z): MH+=334,1, Rt=0,51. Síntese de 5-metil-3-oxociclo-hex-1-eniltriflúormetanossulfonato
[000141] Para uma solução de 5-metilciclo-hexano-1,3-diona (equiv. 1,0) em DCM (0.5M) foi adicionado Na2CO3 (equiv. 1,1) e resfriada a 0 oC. Adicionado Tf2O (equiv. 1,0) em DCM (5.0 M) em gotas durante 1 h a 0oC sob uma atmosfera de nitrogênio. Após a adição, a reação foi agitada por 1 h à temperatura ambiente (solução vermelho escura). A solução foi filtrada e o filtrado foi extinto por adição cuidadosa de NaHCO3 saturado com vigorosa agitação até pH=7. A solução foi transferida para um funil separador e as camadas foram separadas. A camada orgânica foi lavada com salmoura, seca com Na2SO4, filtrada, concentrada sob vácuo e seca sob alto vácuo por 15 min para render triflúormetanossulfonato de 5-metil-3-oxociclo-hex-1-enila como um óleo amarelo claro em 78% rendimento. O triflato se decompõe durante o armazenamento e deve ser usado imediatamente para a reação seguinte. LC/MS=259,1/300,1 (M+H e M+CH3CN); temperatura ambiente = 0,86 min, LC = 3,84 min. 1H-NMR (400 MHz, CDCl3) δ ppm: 6,05 (s, 1H), 2.70 (dd, J=17,2, 4,3, 1H), 2,53 (dd, J=16,6, 3,7, 1H), 2,48-2,31 (m, 2H), 2,16 (dd, J=16,4, 11,7, 1H), 1,16 (d, J=5,9, 3H). Síntese de 5-metil-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)ciclo- hex-2-enona
[000142] Para uma solução de triflúormetanossulfonato de 5-metil-3- oxociclo-hex-1-enila (equiv. 1,0) em dioxano desgaseificado (0.7 M) foram adicionados bis(pinacolato)diboro (equiv. 2,0), KOAc (equiv. 3,0), e Pd(dppf)Cl2-DCM (equiv. 0,03). A reação foi aquecida a 80 o C por 10 h (aquecimento inicial em resultados de larga escala na formação exotérmica de uma espuma laranja no topo da solução, o banho de aquecimento deve ser removido até que a espuma se retraia, reaquecimento a 80 ° C neste ponto parece estar bem), em seguida, resfriada à temperatura ambiente e filtrada através de um funil de vidro de frita grosso. A torta foi lavada com mais dioxano e a solução filtrada foi utilizada para a próxima etapa sem purificação adicional. LC/MS = 155,1 (M+H de ácido borônico); temperatura ambiente = 0,41 min, LC = 1,37 min. Síntese de 5-metil-3-(3-nitropiridin-4-il)ciclo-hex-2-enona
[000143] Para uma solução de 5-metil-3-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)ciclo-hex-2-enona (equiv. 1,0) em dioxano desgaseificado (0,5 M) e 2M Na2CO3 (2 equiv.) foram adicionados 4- cloro-3-nitropiridina (equiv. 1,3) e Pd(dppf)Cl2-DCM (equiv. 0,05). A reação foi colocada sob um condensador de refluxo e aquecida em um banho de óleo a 110o C por 1 h. Resfriada à temperatura ambiente, filtrada através de uma almofada de Celite, lavada a almofada com acetato de etila e concentrado o filtrado sob vácuo. O resíduo foi ainda bombeado a 80 oC em um evaporador rotativo por uma hora para remover os subprodutos de boronato (M+H = 101) através de sublimação. O resíduo foi particionado entre salmoura e acetato de etila, e as camadas foram separadas, a fase aquosa foi depois extraída com acetato de etila (4x), os orgânicos foram combinados, secos sobre sulfato de sódio, filtrados, e concentrados. O produto bruto foi purificado através de cromatografia em sílica-gel carregado em DCM e eluindo com 2-50% de acetato de etila e hexanos. As frações puras foram concentradas em vácuo para render um óleo laranja. O óleo foi colocado sob alto vácuo (~500 mtorr) com cristais de sementes durante a noite para render um sólido laranja. O sólido foi ainda purificado através de trituração em hexanos para render 5-metil- 3-(3-nitropiridin-4-il)ciclo-hex-2-enona (48% 2 etapas). LC/MS = 233,2 (M+H); temperatura ambiente = 0,69 min, LC = 2,70 min. 1H-NMR (400 MHz, CdCl3) δ ppm: 9,31 (s, 1H), 8,88 (d, J=5,1, 1H), 7,30 (d, J=5,1, 1H), 6,00 (d, J=2,4, 1H), 2,62 (dd, J=16,4, 3,5, 1H), 2,53-2,34 (m, 3H), 2,23 (dd, J=16,1, 11,7, 1H), 1,16 (d, J=6,3, 3H). Síntese de cis-(+/-)-5-metil-3-(3-nitropiridin-4-il)ciclo-hex-2-enol
[000144] Para uma solução de 5-metil-3-(3-nitropiridin-4-il)ciclo-hex- 2-enona (equiv. 1,0) em EtOH (0,3 M) foi adicionado CeCl3-7H2O (1,2 equiv.). A reação foi resfriada a 0o C, depois NaBH4 (1,2 equiv.) foi adicionado em porções. Agitada por 1 h a 0o C, depois extinta por adição de água, concentrada para remover o EtOH, adicionado EtOAc, extraídos os orgânicos, lavada com salmoura, depois seca com Na2SO4, filtrada e concentrada para render cis-(+/-)-5-metil-3-(3- nitropiridin-4-il)ciclo-hex-2-enol (94%). LC/MS = 235,2 (M+H), LC = 2,62 min. Síntese de 4-(3-(terc-butildimetilsililóxi)-5-metilciclo-hex-1-enil)-3- nitropiridina NO2
[000145] Para uma solução de 5-metil-3-(3-nitropiridin-4-il)ciclo-hex- 2-enol (equiv. 1,0) em DMF (0,5 M) foram adicionados imidazol (equiv. 4,0) e TBDMSCl (equiv. 2,5). Após agitação por 18 horas a solução foi parcionada entre EtOAc e H2O e separada. Após lavagem posterior com H2O (3x) e NaCl(sat.), secagem sobre MgSO4, filtragem e remoção de solventes, 4-(3-(terc-butildimetilsililóxi)-5-metilciclo-hex-1-enil)-3- nitropiridina foi obtida (85%). LC/MS = 349,2 (M+H), LC = 5,99 min. Síntese de 4-(3-(terc-butildimetilsililóxi)-5-metilciclo-hex-1-enil)piridin-3- amina
[000146] Uma solução heterogênea de 4-(3-(terc-butildimetilsililóxi)- 5-metilciclo-hex-1-enil)-3-nitropiridina (eq. 1,0) e ferro (eq. 6,0) em ácido acético, a uma concentração de 0.4 M, foi agitada vigorosamente por 2 horas. A mistura foi depois passada através de uma almofada de celite, eluindo com MeOH. Após a remoção dos voláteis a vácuo, o resíduo foi dissolvido em EtOAc, lavado com Na2CO3 (sat.), NaCl(sat.), foi seco sobre MgSO4, foi filtrado e os voláteis foram removidos a vácuo rendendo 4-(3-(terc-butildimetilsililóxi)-5- metilciclo-hex-1-enil)piridin-3-amina (78%). LCMS (m/z): 319,3 (MH+); LC temperatura ambiente = 3,77 min. Síntese de 4-(3-(terc-butildimetilsililóxi)-5-metilciclo-hexil)piridin-3- amina
[000147] Para uma solução de 4-(3-(terc-butildimetilsililóxi)-5- metilciclo-hex-1-enil)-3-nitropiridina (equiv. 1,0) em metanol, a uma concentração de 0,1 M, foi adicionado paládio sobre carbono 10% (eq. 1,0). A solução heterogênea resultante foi posta sob uma atmosfera de hidrogênio e foi agitada por 15 horas. Neste momento a mistura foi filtrada através de uma almofada de celite eluindo com metanol. Os voláteis foram removidos a vácuo rendendo 4-(3-(terc- butildimetilsililóxi)-5-metilciclo-hexil)piridin-3-amina (90%). LCMS (m/z): 321,3 (MH+); LC temperatura ambiente = 3,85 min. Síntese de 4-3-(terc-butildimetilsililóxi)-5-metilciclo-hexil) piridin-3- ilcarbamato de cis (+/-) benzila
[000148] Para uma solução de cis-(+/-)-4-(3-(terc-butildimetilsililóxi)- 5-metilciclo-hexil)piridin-3-amina em diclorometano a uma concentração de 0,5 M foi adicionado carbonato de benzil 2,5- dioxopirrolidin-1-ila (equiv. 1,1) e DMAP (equiv. 0,05). Após agitação por 16 horas à temperatura ambiente, carbonato de benzil 2,5- dioxopirrolidin-1-ila adicional (equiv. 0,55) e DMAP (equiv. 0,03) foram adicionados. Após agitação por 24 horas adicionais à temperatura ambiente, carbonato de benzil 2,5-dioxopirrolidin-1-ila adicional (equiv. 0,1) e DMAP (equiv. 0,03) foram adicionados. Após agitação por mais 18 horas a solução foi particionada entre EtOAc e Na2CO3(sat.) e separada. Após lavagem adicional com Na2CO3(sat.) (2x) e NaCl(sat.), secagem sobre MgSO4, filtragem e remoção de solventes, 4-3-(terc- butildimetilsililóxi)-5-metilciclo-hexil)piridin-3-ilcarbamato de cis (+/-) benzila foi obtido. O material bruto foi usado desse jeito. LC/MS = 455,3 (M+H), LC = 4,39 min. Síntese de 4-(3-hidróxi-5-metilciclo-hexil)piridin-3-ilcarbamato de cis (+/-) benzila
[000149] Uma solução de 4-3-(terc-butildimetilsililóxi)-5-metilciclo- hexil)piridin-3-ilcarbamato de cis (+/-) benzila em 1:2:1 6N HCl/THF/MeOH a uma concentração de 0.1 M foi agitada à temperatura ambiente por 6 horas. O pH foi então ajustadp para pH=7 por adição de 6N NaOH e os voláteis foram removidos a vácuo. A camada aquosa foi extraída com EtOAc e o produto orgânico foi lavado com NaCl(sat.), seco sobre MgSO4, filtrado e após remoção dos voláteis a vácuo, 4-(3-hidróxi-5-metilciclo-hexil)piridin-3-ilcarbamato de cis (+/-) benzila foi obtido. O material bruto foi usado como é. LC/MS = 341,2 (M+H), LC = 2,38 min. Síntese de 4-(3-metil-5-oxociclo-hexil)piridin-3-ilcarbamato de cis (+/-)- benzila
[000150] Para uma solução 0 °C de 4-(3-hidróxi-5-metilciclo- hexil)piridin-3-ilcarbamato de cis-(+/-)-benzila em CH2Cl2 úmido a uma concentração de 0,16 M foi adicionado Dess-Martin Periodinano (equiv. 1,5) e a solução foi agitada por 18 horas enquanto aquecia até a temperatura ambiente. A solução foi particionada entre EtOAc e 1:1 Na2S2O3 10 % / NaHCO3(sat.) e separada. Após lavagem adicional com 1:1 Na2S2O3 10% / NaHCO3(sat.) (2x) e NaCl(sat.), secagem sobre MgSO4, filtragem, remoção de solventes e purificação por cromatografia em sílica-gel (75-100% EtOAc/hexanos), 4-(3-metil-5- oxociclo-hexil)piridin-3-ilcarbamato de cis-(+/-)-benzila foi obtido como um sólido branco (53%, 5 etapas). LC/MS = 339,2 (M+H). Síntese de 4-(-3-(benzilamino)-5-metilciclo-hexil)piridin-3-ilcarbamato de cis-(+/-)-benzila
[000151] A solução de 4-(3-metil-5-oxociclo-hexil)piridin-3- ilcarbamato de cis-(+/-)-benzila (equiv. 1,0) e benzilamina (equiv. 3,0) em MeOH, a uma concentração de 0,25 M, foi agitada à temperatura ambiente por 2 horas. Após resfriamento em um banho a -78 oC, LiBH4 (equiv 1,1, 2.0 M em THF) foi adicionado e a solução foi deixada para aquecer até a temperatura ambiente com agitação durante 16 horas. A solução foi particionada entre EtOAc e NaHCO3(sat.), separada, lavada ainda com NaHCO3(sat.) e NaCl(sat.), seca sobre MgSO4, filtrada e após remoção de voláteis a vácuo, 4-(-3-(benzilamino)-5-metilciclo- hexil)piridin-3-ilcarbamato de cis-(+/-)- benzila foi obtido como uma mistura de isômeros 4:1, com todos os cis como predominantes LC/MS = 430,3 (M+H), LC = 0,62 min. Síntese de (-3-(3-aminopiridin-4-il)-5-metilciclo-hexilcarbamato de cis (+/-)-terc-butila
[000152] Para uma solução de 4-(-3-(benzilamino)-5-metilciclo- hexil)piridin-3-ilcarbamato de cis-(+/-)- benzila (equiv. 1,0) em metanol, a uma concentração de 0,07 M, foi adicionado hidróxido de paládio 20% sobre carbono (eq. 2,0). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 14 horas. Neste momento a reação foi purgada com Ar, Boc2O (equiv. 1,0) foi adicionado e a solução foi agitada por 8 horas. Boc2O adicional (equiv. 1,0) foi adicionado e a solução foi agitada por mais 16 horas. Nesse momento a mistura foi filtrada através de uma almofada de celite eluindo com metanol. Após a remoção de voláteis a vácuo, purificação através de cromatografia em sílica-gel (2,5-2,5 MeOH/CH2Cl2 com 0,1% DIEA) e recristalização de EtOAc 10%/hexanos rendeu (-3-(3- aminopiridin-4-il)-5-metilciclo-hexilcarbamato de cis (+/-)-terc-butila (49%). LCMS (m/z): 306,3 (MH+), LC temperatura ambiente = 2,59 min. Enantiômeros puros puderam ser obtidos por cromatografia quiral. Síntese de (+/-)-4-(5-metilciclo-hexa-1,3-dienil)-3-nitropiridina
[000153] Para uma solução de (+/-)-5-metil-3-(3-nitropiridin-4-il)ciclo- hex-2-enol (equiv. 1,0) em dioxano (0.1M) foi adicionado p-TSA (equiv. 1,0), e a reação foi agitada a 100 oC por 3 h. A solução foi resfriada à temperatura ambiente, depois passada através de uma almofada de alumina neutra eluindo com EtOAc para render (+/-)-4-(5-metilciclo- hexa-1,3-dienil)-3-nitropiridina como um óleo amarelo em 68% de rendimento. LC/MS = 217,1 (M+H), LC = 3,908 min. Síntese de (+/-)-2-azido-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-enol OH v N
[000154] Para uma solução de (+/-)-4-(5-metilciclo-hexa-1,3-dienil)-3- nitropiridina (equiv. 1,0) em DCM (0.1 M) at 0 oC foi adicionado m- CPBA (1.1 equiv.) e a reação foi deixada para aquecer à temperatura ambiente. Após 3 horas, a mistura foi extinta com NaHCO3 saturado, extraída com DCM, e a fase orgânica foi seca com sulfato de sódio, filtrada, e concentrada para dar um óleo amarelo. O produto bruto foi dissolvido em etanol e água (3:1, 0,1 M), e azido de sódio (equiv. 2,0) e cloreto de amônio (equiv. 2,0) foram adicionados. A reação foi agitada por 4 horas, depois concentrada a vácuo. Para o produto bruto foram adicionados acetato de etila e água, a fase orgânica foi lavada com salmoura, seca com sulfato de sódio, filtrada, e concentrada. O material bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para render (+/- )-2-azido-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-enol como um óleo em 49% de rendimento. LC/MS = 276,1 (M+H), temperatura ambiente = 0,71 min. Síntese de (+/-)-6-hidróxi-5-metil-3-(3-nitropiridin-4-il)ciclo-hex-2- enilcarbamato de terc-butila
[000155] Para uma solução de (+/-)-2-azido-6-metil-4-(3-nitropiridin- 4-il)ciclo-hex-3-enol (equiv. 1,0) em piridina e hidróxido de amônio (8:1, 0,08 M) foi adicionada trimetilfosfina (equiv. 3,0) e a solução marrom foi agitada à temperatura ambiente por 2 horas. Etanol foi adicionado para a mistura e a solução foi concentrada sob vácuo (2x). A mistura bruta foi então dissolvida em dioxano e NaHCO3 saturado (1:1, 0,08 M) e Boc2O (equiv. 1,0) foi adicionado. A solução foi agitada à temperatura ambiente por 2 horas, depois particionada entre acetato de etila e água. A fase orgânica foi seca com sulfato de magnésio, filtrada, e concentrada a vácuo. O produto bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para render (+/-)-6-hidróxi-5-metil-3-(3-nitropiridin-4- il)ciclo-hex-2-enilcarbamato de terc-butila em 69% de rendimento. LC/MS = 350,1 (M+H), temperatura ambiente = 0,76 min. Síntese de metanossulfonato de (+/-)-2-(terc-butoxicarbonilamino)-6- metil-4-(3-nitropiridin-4-il) ciclo-hex-3-enila
[000156] Para uma solução de (+/-)-6-hidróxi-5-metil-3-(3-nitropiridin- 4-il)ciclo-hex-2-enilcarbamato de terc-butila (equiv. 1,0) em DCM (0.09 M) foi adicionada trietil amina (equiv. 1,5). A mistura de reação foi resfriada a 0 oC e MsCl (equiv. 1,2) foi adicionado para a reação e agitada por 3 horas. Para a solução foi adicionada água, a fase orgânica foi seca com sulfato de sódio, filtrada, e concentrada. O material bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para dar metanossulfonato de (+/-)-2-(terc-butoxicarbonilamino)-6-metil-4-(3- nitropiridin-4-il)ciclo-hex-3-enila como uma espuma branca em 65% de rendimento. LC/MS = 428,2 (M+H), temperatura ambiente = 0,88 min. Síntese de 7-metil-5-(3-nitropiridin-4-il)-2-oxo-3a,6,7,7a-tetra- hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila
[000157] Uma solução de metanossulfonato de (+/-)-2-(terc- butoxicarbonilamino)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-enila (equiv. 1,0) em piridina (0.2 M) em vaso de micro-onda foi aquecida a 110 oC por 10 min. A solução laranja foi então concentrada até secar e manipulada por particionamento entre acetato de etila e água. A fase orgânica foi seca com sulfato de sódio, filtrada e concentrada. O material bruto foi dissolvido em DCM (0.2 M) e trietil amina (equiv. 1,8) foi adicionada para a reação, seguido por Boc2O (1.2 eqiv.). Após agitação à temperatura ambiente por 40 min, a reação foi concentrada a vácuo e purificada através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para dar 7-metil-5-(3- nitropiridin-4-il)-2-oxo-3a,6,7,7a-tetra-hidrobenzo[d]oxazol-3(2H)- carboxilato de (+/-)-terc-butila como uma espuma branca em 66% de rendimento. LC/MS = 376,0 (M+H), temperatura ambiente = 0,87 min. Síntese de 5-(3-aminopiridin-4-il)-7-metil-2- oxohexahidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila
[000158] Para uma solução de 7-metil-5-(3-nitropiridin-4-il)-2-oxo- 3a,6,7,7a-tetra-hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc- butila (equiv. 1,0) em MeOH e acetato de etila (1:1, 0,07 M) foi adicionado Pd/C 10% (equiv. 0,1) e a reação foi agitada à temperatura ambiente sob uma atmosfera de hidrogênio. Após a conclusão da reação, a solução foi filtrada através de uma almofada de Celite, lavada com MeOH e acetato de etila, o filtrado foi concentrado até secar sob vácuo para dar 5-(3-aminopiridin-4-il)-7-metil-2-oxo-hexa- hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila como um mistura de diastereômeros em >99% de rendimento. LC/MS = 348,2 (M+H), temperatura ambiente = 0,50 min. Síntese de (+/-)-6-bromo-5-metil-3-(3-nitropiridin-4-il)ciclo-hex-2-enol
[000159] Para uma solução de 4-(5-metilciclo-hexa-1,3-dienil)-3- nitropiridina (equiv. 1,0) em THF e água (1:1, 0,13 M) foi adicionado NBS (equiv. 1,5) e a reação foi agitada à temperatura ambiente por 30 min. Após a conclusão, acetato de etila e água foram adicionados para uma reação, a fase orgânica foi seca com salmoura, depois sulfato de sódio, filtrada, e concentrada. O material bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para dar (+/-)-6-bromo-5-metil-3-(3-nitropiridin-4-il)ciclo- hex-2-enol como um óleo amarelo em 80% de rendimento. LC/MS = 315,0/313,0 (M+H), LC = 2,966 min. Síntese de (+/-)-2-azido-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-enol
[000160] Para uma solução de (+/-)-6-bromo-5-metil-3-(3-nitropiridin- 4-il)ciclo-hex-2-enol (equiv. 1,0) em THF (0,1 M) foi adicionado terc- butóxido de potássio (equiv. 1,5). A reação tornou-se de laranja para preta quase imediatamente. Por TLC, a formação do produto está completa em 30 min. Extinta através da adição de cloreto de amônio saturado e acetato de etila. A fase orgânica foi seca com salmoura, depois sulfato de sódio, filtrada, e concentrada. O produto bruto foi dissolvido em etanol e água (3:1, 0,1 M), e cloreto de amônio (equiv. 2,0) e azido de sódio (equiv. 2,0) foram adicionados. A reação laranja escura foi agitada à temperatura ambiente durante a noite. A conversão para o produto está completa como indicado por LC/MS. A reação foi concentrada para remover o etanol, acetato de etila e água foram adicionados, e a fase orgânica foi seca com sulfato de sódio, filtrada, e concentrada. O material bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para dar (+/-)-2-azido-6-metil-4-(3-nitropiridin-4-il)ciclo- hex-3-enol em 55% de rendimento. LC/MS = 276,0 (M+H), LC = 2,803 min. Síntese de 6-hidróxi-5-metil-3-(3-nitropiridin-4-il)ciclo-hex-2- enilcarbamato de (+/-)-terc-butila
[000161] Para uma solução de (+/-)-2-azido-6-metil-4-(3-nitropiridin- 4-il)ciclo-hex-3-enol (equiv. 1,0) em piridina e hidróxido de amônio (8:1, 0.08 M) foi adicionada trimetilfosfina (equiv. 3,0) e a solução marrom foi agitada à temperatura ambiente por 2 h. Após a conclusão, EtOH foi adicionado e a solução foi concentrada a vácuo. Mais etanol foi adicionado e a reação foi concentrada de novo. Dioxano e NaHCO3 saturado (1:1, 0,08 M) foram adicionados ao produto bruto, seguido por Boc2O (equiv. 1,0). A mistura de reação foi agitada à temperatura ambiente por 2h, depois adicionados água e acetato de etila. A fase orgânica foi seca com MgSO4, e concentrada. O produto bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para render 6-hidróxi-5-metil-3-(3- nitropiridin-4-il)ciclo-hex-2-enilcarbamato de (+/-)-terc-butila (59%). LC/MS = 350,1 (M+H), Rt: 0,76 min. Síntese de acetato de (+/-)-2-(terc-butoxicarbonilamino)-6-metil-4-(3- nitropiridin-4-il)ciclo-hex-3-enila
[000162] Para uma solução de 6-hidróxi-5-metil-3-(3-nitropiridin-4- il)ciclo-hex-2-enilcarbamato de (+/-)-terc-butila (equiv. 1,0) em piridina (0,1 M) foi adicionado Ac2O (equiv. 2,0) e a reação foi agitada à temperatura ambiente durante a noite. Após a conclusão, a reação foi concentrada até secar, depois estimulada com acetato de etila e água. A fase orgânica foi seca com salmoura, depois sulfato de sódio, filtrada, e concentrada para dar acetato de (+/-)-2-(terc- butoxicarbonilamino)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-enila em 94% de rendimento. LC/MS = 392,2 (M+H), temperatura ambiente = 0,94 min. Síntese de acetato de (+/-)-4-(3-aminopiridin-4-il)-2-(terc- butoxicarbonilamino)-6-metilciclo-hexila
[000163] Para uma solução desgaseificada de acetato de (+/-)-2- (terc-butoxicarbonilamino)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-enila (equiv. 1,0) em MeOH e EtOAc (1:1, 0,1 M) foi adicionado Pd/C 10% (equiv. 0,1) e a reação foi agitada à temperatura ambiente sob um balão de hidrogênio por 3 dias. Após a conclusão, a solução foi filtrada através de uma almofada de Celite, a almofada foi lavada com acetato de etila e o filtrado foi concentrado. O material bruto continha cerca de 10% dos isômeros indesejados. O produto bruto foi dissolvido em acetato de etila (~ 20%) e hexanos e aquecida até todo dissolvido. A solução foi deixada para sentar-se à temperatura ambiente por 2 dias. O precipitado foi depois coletado para dar acetato de (+/-)-4-(3- aminopiridin-4-il)-2-(terc-butoxicarbonilamino)-6-metilciclo-hexila como o produto puro em 59% de rendimento. LC/MS = 364,3 (M+H), temperatura ambiente = 0,63 min. Síntese de metanossulfonato de 2-(terc-butoxicarbonilamino)-6-metil- 4-(3-nitropiridin-4-il)ciclo-hex-3-enila
[000164] Para uma solução de 6-hidróxi-5-metil-3-(3-nitropiridin-4- il)ciclo-hex-2-enilcarbamato de terc-butila (equiv. 1,0) em DCM (0,09 M) foi adicionada trietilamina (equiv. 1,5) e a reação foi resfriada a 0 oC. MsCl (1,2 equiv.) foi adicionado para a reação e agitada por 3 h. Outro equiv. 1,0 de MsCl foi adicionado para a reação e agitado por outras 2 h. Estimulada a reação por adição de água, a fase orgânica foi seca com salmoura, sulfato de sódio, e concentrada. O produto bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com acetato de etila e hexanos (1:1) para render metanossulfonato de 2-(terc-butoxicarbonilamino)-6-metil-4-(3- nitropiridin-4-il)ciclo-hex-3-enila como uma espuma branca em 65% de rendimento. LC/MS = 428,2 (M+H), LC: 3,542 min. Síntese de 7-metil-5-(3-nitropiridin-4-il)-2-oxo-3a,6,7,7a-tetra- hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila
[000165] Uma solução de metanossulfonato de (+/-)-2-(terc- butoxicarbonilamino)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-enila (equiv. 1,0) em piridina (0,2 M) foi aquecida no micro--ondas a 110 oC por 10 min. A reação laranja foi depois concentrada sob vácuo, o produto bruto foi dissolvido em acetato de etila e água, a fase orgânica foi seca com sulfato de sódio e concentrada sob vácuo. O material bruto foi dissolvido em DCM (0,2 M), trietilamina (equiv. 1,8) foi adicionada, seguido por Boc2O (1,2 equiv.). A reação foi agitada por 40 min, depois concentrada até secar. O material bruto foi purificado através de cromatografia em coluna de sílica-gel eluindo com hexano e acetato de etila (1:1) para render 7-metil-5-(3-nitropiridin-4-il)-2-oxo- 3a,6,7,7a-tetra-hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc- butila como uma espuma branca em 66% de rendimento. LC/MS = 376,0 (M+H), LC: 3,424 min. Síntese de 5-(3-aminopiridin-4-il)-7-metil-2-oxo-hexa- hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila
[000166] Para uma solução desgaseificada de 7-metil-5-(3- nitropiridin-4-il)-2-oxo-3a,6,7,7a-tetra-hidrobenzo[d]oxazol-3(2H)- carboxilato de (+/-)-terc-butila (equiv. 1,0) em MeOH e EtOAc (1:1, 0,1 M) foi adicionado Pd/C 10% (equiv. 0,1). A reação foi agitada sob um balão de hidrogênio durante a noite. Após a conclusão, a solução foi filtrada através de uma almofada de Celite e a almofada foi lavada com acetato de etila. O filtrado foi concentrada sob vácuo para dar 5-(3- aminopiridin-4-il)-7-metil-2-oxo-hexa-hidrobenzo[d]oxazol-3(2H)- carboxilato de (+/-)-terc-butila como o produto desejado como um espuma amarela em 93% rendimento. LC/MS = 348,1 (M+H), temperatura ambiente = 0,55 min. Síntese de ((+/-)-(1R,2R,6S)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3- ene-1,2-diol e (+/-)-(1R,2S,6S)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3- eno-1,2-diol)
[000167] Para uma solução de (+/-)-(1S,5S,6S)-6-bromo-5-metil-3-(3- nitropiridin-4-il)ciclo-hex-2-enol (equiv. 1,0) em THF (0,1M) foi adicionado terc-butóxido de potássio (equiv. 1,5) à temperatura ambiente. A mistura de reação foi agitada por 10 min. A mistura de reação foi extinta com solução de NH4Cl e elaborada com EtOAc lavando com água e salmoura. A camada orgânica foi seca sobre sulfato de sódio anídrico, filtrada, e seca a vácuo. O produto bruto foi usado à próxima etapa sem purificação posterior. Rf = 0,5 (50% de EtOAc/Hexanos). LCMS: MH+ 251,2 (como um diol), temperatura ambiente = 0,49 min. Para uma solução de (+/-)-4-((1S,5S)-5-metil-7- oxabiciclo[4.1.0]hept-2-en-3-il)-3-nitropiridina bruta (equiv. 1,0) em 2:1 CH3CN/H2O (0,1 M) foi adicionado ácido acético (equiv. 0,3) à temperatura ambiente. A mistura de reação foi agitada por 16 horas à temperatura ambiente. Após extinta com solução de NaHCO3, a mistura de reação foi concentrada para remover a maioria de CH3CN e o resíduo foi particionado entre EtOAc e água. A camada orgânica combinada foi lavada com água e salmoura, seca sobre sulfato de sódio anídrico, filtrada e concentrada a vácuo. A mistura de dióis ((+/-)- (1R,2R,6S)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-eno-1,2-diol e (+/-)- (1R,2S,6S)-6-metil-4-(3-nitropiridin-4-il)ciclo-hex-3-eno-1,2-diol) foi obtida em 33,1% de rendimento como um sólido branco por cromatografia em coluna rápida. Rf = 0,3 (100% de EtOAc; dióis não eram separáveis sobre TLC). LCMS: MH+ 251,2, temperatura ambiente = 0,49 min. Síntese de (+/-)-4-((3S,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5- metilciclo-hex-1-enil)-3-nitropiridina e (+/-)-4-((3R,4R,5S)-3,4-bis(terc- butildimetilsililóxi)-5-metilciclo-hex-1-enil)-3-nitropiridina
[000168] Para uma solução de uma mistura de dióis (equiv. 1,0) em DMF (0,3 M) foram adicionados TBDMSCI (equiv. 7,0) e imidazol (equiv. 9) à temperatura ambiente. A mistura de reação foi agitada à temperatura ambiente durante a noite. Após extinta com NaHCO3 saturado, a mistura de reação foi extraída com EtOAc. A camada orgânica combinada foi lavada com água e salmoura, seca sobre sulfato de sódio anídrico, filtrada e concentrada a vácuo. A mistura foi purificada por cromatografia em coluna de sílica sequencial automatizada (gradiente eluindo com EtOAc e Hexanos) e fase HPLC reversa preparativa (55% - 95% de acetonitrila em água, depois processado 5% - 95% de acetonitrila em água para render (+/-)-4- ((3S,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5-metilciclo-hex-1-enil)-3- nitropiridina (27,2%) e (+/-)-4-((3R,4R,5S)-3,4-bis(terc- butildimetilsililóxi)-5-metilciclo-hex-1-enil)-3-nitropiridina (50,2%). LCMS: MH+ 479,2, temperatura ambiente = 1,60 e 1,63 min. Síntese de 4-((1S,3S,4S,5R)-3,4-bis(terc-butildimetilsililóxi)-5- metilciclo-hexil)piridin-3-amina e de 4-((1R,3R,4R,5S)-3,4-bis(terc- butildimetilsililóxi)-5-metilciclo-hexil)piridin-3-amina
[000169] Para uma solução de (+/-)-4-((3R,4R,5S)-3,4-bis(terc- butildimetilsililóxi)-5-metilciclo-hex-1-enil)-3-nitropiridina (equiv. 1,0) em etanol/EtOAc, a uma concentração de 0,1 M, foi adicionado paládio sobre carbono 10% (eq. 1,0). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 14 horas. Neste momento a mistura foi filtrada através de uma almofada de celite eluindo com EtOAc. Os voláteis foram removidos a vácuo e o material bruto foi purificado por cromatografia em coluna de sílica automatizada (Rf = 0,2, 40% de EtOAc em Heptano) para render o produto racêmico puro. LCMS: MH+ 451,3, temperatura ambiente = 1,35 min. O composto racêmico foi resolvido por cromatografia quiral (coluna IC, 1 mL/min, 5% de IPA em Heptano) para render 4- ((1S,3S,4S,5R)-3,4-bis(terc-butildimetilsililóxi)-5-metilciclo-hexil)piridin- 3-amina (6,01 min) e 4-((1R,3R,4R,5S)-3,4-bis(terc-butildimetilsililóxi)- 5-metilciclo-hexil)piridin-3-amina (8,34 min). Síntese de 4-((1R,3R,4S,5R)-3,4-bis(terc-butildimetilsililóxi)-5- metilciclo-hexil)piridin-3-amina e de 4-((1S,3S,4R,5S)-3,4-bi(terc- butildimetilsililóxi)-5-metilciclo-hexil)piridin-3-amina
[000170] Para uma solução de (+/-)-4-((3S,4R,5S)-3,4-bis(terc- butildimetilsililóxi)-5-metilciclo-hex-1-enil)-3-nitropiridina (equiv. 1,0) em etanol, a uma concentração de 0.1 M, foi adicionado paládio sobre carbono 10% (eq. 1,0). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 14 horas. Neste momento a mistura foi filtrada através de uma almofada de celite eluindo com etanol. Os voláteis foram removidos a vácuo e o material bruto foi purificado por cromatografia em coluna de sílica automatizada (Rf = 0,2, 40% de EtOAc em Heptano) para render produto racêmico puro (50,4%). LCMS: MH+ 451,3, temperatura ambiente = 1,35 min. O composto racêmico foi resolvido por cromatografia quiral (coluna IC, 1 mL/min, 5% IPA em Heptano) para render 4-((1R,3R,4S,5R)-3,4- bis(terc-butildimetilsililóxi)-5-metilciclo-hexil)piridin-3-amina (6,98 min) e 4-((1S,3S,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5-metilciclo- hexil)piridin-3-amina (8,67 min). Síntese de 6-(metoxicarbonil)-3-oxo-5-(triflúormetil)ciclo-hex-1-enolato de sódio
[000171] Para uma solução recentemente preparada de (eq. 1,0) em t-BuOH de sódio (1 M) foi adicionado acetoacetato de etila (eq. 1,0) em gotas e a mistura agitada em um banho de gelo por um período adicional de 15 min. 4,4,4-triflúorcrotonato de etila (eq. 1,0) foi adicionado em gotas e a mistura agitada à temperatura ambiente por um período adicional de 30 min. Após refluxo por 2 h, a mistura foi resfriada e hexano foi adicionado. O precipitado foi filtrado sem purificação posterior para dar 6-(metoxicarbonil)-3-oxo-5- (triflúormetil)ciclo-hex-1-enolato de sódio (46%). LC/MS (m/z): MH+ = 253,1, temperatura ambiente = 0,70min. Síntese de 5-(triflúormetil)ciclo-hexano-1,3-diona
[000172] 6-(Metoxicarbonil)-3-oxo-5-(triflúormetil)ciclo-hex-1-enolato de sódio (eq. 1,0) foi dissolvido em 1M NaOH (eq. 1,0), e a mistura refluxada por 1 h. Após resfriar à temperatura ambiente, a mistura foi acidificada com 5 M ácido sulfúrico. A mistura foi extraída com EtOAc. Após lavagem com água, a camada orgânica foi seca sobre sulfato de magnésio, o solvente foi removido sob pressão reduzida para dar 5- (triflúormetil)ciclo-hexano-1,3-diona, a qual foi usada para a próxima etapa sem purificação posterior (98%). LC/MS (m/z): MH+ = 181,1, temperatura ambiente = 0,55min. Síntese de triflúormetanossulfonato de 3-oxo-5-(triflúormetil)ciclo-hex- 1-enila
[000173] Para uma suspensão de 5-(triflúormetil)ciclo-hexano-1,3- diona (eq. 1,0) em DCM (0,23 M) foi adicionado TEA (1.2 eq) para dar uma solução clara. A mistura foi resfriada a 0 °C. E depois Tf2O (eq. 1,05) em DCM foi adicionado em gotas. A mistura de reação foi agitada naquela temperatura por 2 horas. A mistura de reação foi diluída com DCM, e lavada com água, NaHCO3 aquoso, salmoura, e foi seca sobre MgSO4, filtrada e concentrada para dar triflúormetanossulfonato de 3-oxo-5-(triflúormetil)ciclo-hex-1-enila, o qual foi usado para a próxima etapa diretamente. LC/MS (m/z): MH+ = 313,0, temperatura ambiente = 1,02 min. Síntese de 3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5- (triflúormetil)ciclo-hex-2-enona
[000174] Todos os reagentes triflúormetanossulfonato de 3-oxo-5- (triflúormetil)ciclo-hex-1-enila (eq. 1,0), NaOAc (eq. 3,0), e bis(pinacolato)diboro (eq. 2,0) foram adicionados a 1,4-dioxano (0,23 M) em um balão de fundo redondo e desgaseificado borbulhando em N2 através de uma mistura por 10 min. Aduto de PdCl2(dppf).CH2Cl2 (eq. 1,0) foi adicionado e a reação aquecida a 80 °C equipado com um condensador de refluxo em um banho de óleo sob N2 por duas horas. A mistura foi resfriada à temperatura ambiente, filtrada através de um funil de frita de vidro grosso, a torta enxaguada com ~10mL de 1,4- dioxano para dar 3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5- (triflúormetil)ciclo-hex-2-enona em 1,4-dioxano, o qual foi usado para a próxima etapa diretamente. LC/MS (m/z): MH+ = 209,1 (ácido borônico), Rt = 0,60 min. Síntese de 3-(3-nitropiridin-4-il)-5-(triflúormetil)ciclo-hex-2-enona
[000175] O éster de boronato de 3-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)-5-(triflúormetil)ciclo-hex-2-enona (eq. 1,0) foi dissolvido em 1,4-dioxano (0.14 M) em um balão de fundo redondo foi desgaseificado por borbulhamento de N2 através de uma solução por 30 minutos. 4-cloro-3-nitropiridina (1.3 eq) e Na2CO3 aquoso (2M, eq. 2,0) foram adicionados e N2 foi borbulhado através por 10 minutos e depois o aduto de PdCl2(dppf).CH2Cl2 (eq. 0,1) foi adicionado. A mistura de reação foi agitada a 100 °C por 2 horas. À mistura foram adicionados EtOAc e salmoura. A mistura resultante foi filtrada através de celite, a torta foi lavada com EtOAc. A camada orgânica foi separada, e lavada com salmoura, seca sobre MgSO4, e filtrada e concentrada. O produto bruto foi purificado por cromatografia em sílica-gel (eluido com EtOAc:Hexanos = 1:10 a 2:1) para dar 3-(3- nitropiridin-4-il)-5-(triflúormetil)ciclo-hex-2-enona (73% por três etapas a partir de dicetona). LC/MS (m/z): MH+ = 287,1, temperatura ambiente = 0,85 min. Síntese de cis-3-(3-nitropiridin-4-il)-5-(triflúormetil)ciclo-hex-2-enol
[000176] 3-(3-Nitropiridin-4-il)-5-(triflúormetil)ciclo-hex-2-enona (eq. 1,0) foi misturada com cloreto de cério (III) hepta-hidrato (eq. 1,0) e etanol absoluto (0,17) foi adicionado. A mistura foi agitada à temperatura ambiente até todos os sólidos dissolvidos. A mistura foi resfriada em um banho de gelo e NaBH4 (eq. 1,2) foi adicionado em porções. A reação foi agitada no banho de gelo por 1h. A mistura foi diluída com EtOAc, lavada com água, seca sobre MgSO4, filtrada e concentrada. O resíduo foi purificado por coluna (1:1 acetato de etila e hexanos) para dar cis-3-(3-nitropiridin-4-il)-5-(triflúormetil)ciclo-hex-2- enol (66%). LC/MS (m/z): MH+ = 289,2, temperatura ambiente = 0,72 min. Síntese de cis-4-(3-azido-5-(triflúormetil)ciclo-hex-1-enil)-3-nitropiridina F3C/,./\ .^3
[000177] Para uma solução de cis-3-(3-nitropiridin-4-il)-5- (triflúormetil)ciclo-hex-2-enol (eq. 1,0) em DCM (0,14 M) foi adicionado TEA (2,5 eq), e seguido por MsCl (eq. 1,8) à temperatura ambiente. A mistura de reação foi agitada à temperatura ambiente por 2 horas. O solvente foi removido. O resíduo foi dissolvido em DMF (0,19 M), e depois à mistura foi adicionado azido de sódio (1,2 eq.). A mistura resultante foi agitada à temperatura ambiente por 1 hora. Outro eq. 1,2 de azido de sódio foi adicionado. A mistura foi agitada à temperatura ambiente durante a noite. A mistura de reação foi diluída com acetato de etila e heptano, e lavada com NaCl saturado. O produto orgânico foi seca sobre MgSO4, filtrada e concentrada. O resíduo foi purificado por coluna (1:1 acetato de etila e hexanos) para dar cis-4-(3-azido-5- (triflúormetil)ciclo-hex-1-enil)-3-nitropiridina (58%). LC/MS (m/z): MH+ = 314,1, Rt = 0,96 min. Síntese de (1R,3R,5S)-3-(3-aminopiridin-4-il)-5-(triflúormetil)ciclo- hexilcarbamato de terc-butila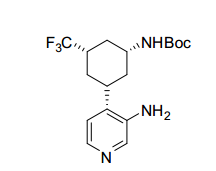
[000178] A solução de cis-4-(3-azido-5-(triflúormetil)ciclo-hex-1-enil)- 3-nitropiridina (eq. 1,0) em etanol (0,13 M) foi borbulhado com N2 por 20 min. depois à mistura de reação foram adicionados Boc-anidrido (eq. 1,5) e Pd/C (eq. 0,2). A mistura de reação foi agitada à temperatura ambiente sob atmosfera de H2 durante a noite. O sólido foi removido por filtração sobre celite e lavado com EtOH. O resíduo foi purificado por coluna (metanol 5% em 1:1 acetato de etila e hexanos) para dar cis-3-(3-aminopiridin-4-il)-5-(triflúormetil) ciclohexilcarbamato racêmico (57%). LC/MS (m/z): MH+ = 360,2 , temperatura ambiente = 0,72 min. O (1R,3R,5S)-3-(3-aminopiridin-4-il)-5-(triflúormetil)ciclo- hexilcarbamato de terc-butila e N-(4-((1S,3S,5R)-3-amino-5- (triflúormetil)ciclohexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5- flúorpicolinamida enantiomericamente puros foram resolvidos por HPLC quiral (para análise Rt = 8,14 min e 10,59 min respectivamente; heptano:isopropanol= 90:10 (v:v), Chiralcel IC 100 x 4,6 mm a 1 mL/min. Para preparação separativa, heptano:isopropanol = 90:10 (v:v), Chiralcel IC 250 x 20 mm a 15 mL/min ). Síntese de (R)-4-benzil-3-((2R,3R)-3-((R)-2,2-dimetil-1,3-dioxolan-4-il)- 3-hidróxi-2-metilpropanoil)oxazolidin-2-ona
[000179] (R)-4-benzil-3-((2R,3R)-3-((R)-2,2-dimetil-1,3-dioxolan-4-il)- 3-hidróxi-2-metilpropanoil)oxazolidin-2-ona foi preparada da maneira reportada (Proc.Nat.Acad.Sciences, 101, 33, 2004, páginas 12042- 12047) pelo composto enantiomérico iniciando com (R)-4-benzil-3- propioniloxazolidin-2-ona e R-gliceraldeído acetonida. Síntese de (R)-4-benzil-3-((2R,3R)-3-(terc-butildimetilsililóxi)-3-((R)-2,2- dimetil-1,3-dioxolan-4-il)-2-metilpropanoil)oxazolidin-2-ona
[000180] (R)-4-benzil-3-((2R,3R)-3-(terc-butildimetilsililóxi)-3-((R)-2,2- dimetil-1,3-dioxolan-4-il)-2-metilpropanoil)oxazolidin-2-ona foi preparada da maneira reportada (Proc.Nat.Acad.Sciences, 101, 33, 2004, páginas 12042-12047) para o composto enantiomérico começando com (R)-4-benzil-3-((2R,3R)-3-((R)-2,2-dimetil-1,3- dioxolan-4-il)-3-hidróxi-2-metilpropanoil)oxazolidin-2-ona. Síntese de (2S,3R)-3-(terc-butildimetilsililóxi)-3-((R)-2,2-dimetil-1,3- dioxolan-4-il)-2-metilpropan-1-ol
[000181] Para uma solução de (R)-4-benzil-3-((2R,3R)-3-(terc- butildimetilsililóxi)-3-((R)-2,2-dimetil-1,3-dioxolan-4-il)-2- metilpropanoil)oxazolidin-2-ona (equiv. 1,0) e etanol (equiv. 3,0) em THF (0,09 M) foi adicionado LiBH4 (equiv. 1,0) a -40oC. A mistura de reação foi deixada para aquecer à temperatura ambiente lentamente e agitada nessa temperatura por 12 horas. A solução foi resfriada de volta a -40 oC e LiBH4 adicional (equiv. 0,3) foi adicionado. Após aquecer novamente à temperatura ambiente e agitar por 2 horas, a solução foi depois diluída com dietil éter e 1N NaOH) foi adicionado. A mistura resultante foi extraída com acetato de etila, a camada orgânica foi separada, lavada com NaCl(sat.), seca sobre sulfato de magnésio, filtrada, e concentrada. O resíduo foi purificado através de cromatografia de coluna de sílica-gel (10-30% de EtOAc/n-heptanos) rendendo (2S,3R)-3-(terc-butildimetilsililóxi)-3-((R)-2,2-dimetil-1,3- dioxolan-4-il)-2-metilpropan-1-ol em (75%). LC/MS = 247,1 (M+H-cetal- H2O), temperatura ambiente = 0,64 min. Síntese de ((1R,2S)-3-azido-1-((R)-2,2-dimetil-1,3-dioxolan-4-il)-2- metilpropóxi)(terc-butil)dimetilsilano
[000182] Para uma solução de (2S,3R)-3-(terc-butildimetilsililóxi)-3- ((R)-2,2-dimetil-1,3-dioxolan-4-il)-2-metilpropan-1-ol (equiv. 1,0), DIAD (equiv. 2,0), e PPh3 (equiv. 2,0) em THF (0.18 M) foi adicionado DPPA (equiv. 1,0, 1M solução em THF). A mistura de reação foi agitada à temperatura ambiente durante a noite. Mediante a remoção dos voláteis sob vácuo, o resíduo foi purificado por cromatografia em coluna de sílica-gel (2-3-5% EtOAc/n-heptanos) rendendo ((1R,2S)-3- azido-1-((R)-2,2-dimetil-1,3-dioxolan-4-il)-2-metilpropóxi)(terc-butil) dimetilsilano (62%). 1H-NMR (400 MHz, CDCl3): δ 4,04 - 4,10 (m, 1H), 3,94 (dd, J=8,0, 6,4, 1H), 3,72 (d, J=7,2, 1H), 3,53 (t, J=8,0, 1H), 3,36 (dd, J=12, 8,0, 1H), 3,19 (dd, J=12,0, 6,7, 1H), 1,52 - 1,60 (m, 1H), 1,41 (s, 3H), 1,34 (s, 3H), 0,92 (d, J=7,2, 3H), 0,90 (s, 9 H), 0,12 (s, 3H), 0,09 (s, 3H). Síntese de(2R,3R,4S)-5-azido-3-(terc-butildimetilsililóxi)-4- metilpentano-1,2-diol
[000183] Para uma solução de ((1R,2S)-3-azido-1-((R)-2,2-dimetil- 1,3-dioxolan-4-il)-2-metilpropóxi)(terc-butil)dimetilsilano (equiv. 1,0) em MeOH (0,1 M) foi adicionado PPTS (equiv. 1,0) e a mistura foi agitada à temperatura ambiente por 14 horas, 50 oC por 2 horas e 80 oC por 1 hora. Os voláteis foram removidos sob vácuo e o resíduo foi purificado através de cromatografia de coluna de sílica-gel (10-25% de EtOAc/n- heptanos) rendendo (2R,3R,4S)-5-azido-3-(terc-butildimetilsililóxi)-4- metilpentano-1,2-diol (40%). Síntese de 4-metilbenzenossulfonato de (2R,3R,4S)-5-azido-3-(terc- butildimetilsililóxi)-2-hidróxi-4-metilpentila OTBS
[000184] Para uma solução de (2R,3R,4S)-5-azido-3-(terc- butildimetilsililóxi)-1-hidróxi-4-metilpentan-2-ilcarbamato de terc-butila (equiv. 1,0) em piridina (0,2 M) foi adicionado pTsCl (equiv. 1,3) a 0 oC. A mistura manteve nessa temperatura por 16 horas. Os voláteis foram removidos a vácuo e o resíduo foi purificado por cromatografia em coluna de sílica-gel (10 - 15 - 20% de EtOAc/n-heptanos) rendendo 4- metilbenzenossulfonato de (2R,3R,4S)-5-azido-3-(terc- butildimetilsililóxi)-2-hidróxi-4-metilpentila. Síntese de 4-metilbenzenossulfonato de (2R,3R,4S)-5-azido-2,3- bis(terc-butildimetilsililóxi)-4-metilpentila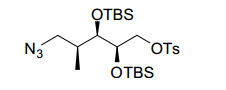
[000185] Para uma solução de 4-metilbenzenossulfonato de (2R,3R,4S)-5-azido-3-(terc-butildimetilsililóxi)-2-hidróxi-4-metilpentila (1,0 equiv.) e 2,6-lutidina (3,4 equiv.) foi adicionado TBDMSOTf (1,7 equiv.) a 0°C. A solução foi agitada por 7 horas conforme foi aquecida à ta. A solução foi diluída com EtOAc, lavada com 10% de CuSO4, H2O, Na2CO3(sat.), NaCl(sat.), secada sobre MgSO4, filtrada e concentrada. O resíduo foi purificado por cromatografia de coluna em sílica-gel (2,5-5-10-20% de EtOAc/n-heptanos) rendendo 4- metilbenzenossulfonato de (2R,3R,4S)-5-azido-2,3-bis(terc- butildimetilsililóxi)-4-metilpentila (75%). Síntese de 4-((3R,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5- metilpiperidin-1-il)-3-nitropiridina
[000186] Uma solução de 4-metilbenzenossulfonato de (2R,3R,4S)- 5-azido-2,3-bis(terc-butildimetilsililóxi)-4-metilpentila em EtOH (0,05 M) foi desgaseificada com argônio. DIEA (1,5 equiv.) foi adicionado, seguido por 10% de Pd/C (0,1 equiv.). A mistura de reação foi agitada sob um balão de hidrogênio por 3 horas. A solução foi desgaseificada e purgada em argônio, ao mesmo tempo que 4-cloro-3-nitropiridina (1,5 equiv.) e DIEA adicional (1,5 equiv.) foram adicionados. Após agitar à ta por 15 horas, a solução foi filtrada para remover o Pd/C, e os voláteis foram removidos em vácuo. O resíduo foi diluído com acetato de etila e lavado com Na2CO3(sat.), NaCl(sat.), secado sobre MgSO4, filtrado e concentrado. O resíduo foi purificado por cromatografia de coluna em sílica-gel (10-15% de EtOAc/n-heptanos) rendendo 4-((3R,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5-metilpiperidin- 1-il)-3-nitropiridina (40%). LC/MS = 482,4 (M+H), Rt = 1,26 min. Síntese de 4-((3R,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5- metilpiperidin-1-il)piridin-3-amina
[000187] Para uma solução de 4-((3R,4R,5S)-3,4-bis(terc- butildimetilsililóxi)-5-metilpiperidin-1-il)-3-nitropiridina (1,0 equiv.) em etanol, a uma concentração de 0,05 M, foram adicionados 10% de paládio em carbono (0,1 eq.). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 14 horas. Neste momento, a mistura foi filtrada através de uma almofada de celite eluindo com etanol. Os voláteis foram removidos em vácuo rendendo 4-((3R,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5-metilpiperidin- 1-il)piridin-3-amina. LC/MS = 452,4 (M+H), Rt = 1,31 min. Síntese de 4-((1R,2R)-3-((R)-4-benzil-2-oxo-oxazolidin-3-il)-1-hidróxi-2- metil-3-oxopropil)-2,2-dimetiloxazolidina-3-carboxilato de (R)-terc-butila
[000188] Para uma solução de (R)-4-benzil-3-propioniloxazolidin-2- ona (1,0 equiv.) em DCM (0,13 M) foi adicionado TiCU (1,0 equiv.) a - 40°C. A mistura foi agitada a -40°C por 10 min (suspensão amarela), depois DIPEA (2,5 equiv.) foi adicionado (solução vermelho-escura) e agitada a 0°C por 20 min. 4-formil-2,2-dimetiloxazolidina-3-carboxilato de (R)-terc-butila (1,0 equiv.) em DCM (0,5 M) foi então adicionado em gotas, e a mistura resultante foi agitada por 1,5 hora. A reação foi extinta pela adição de cloreto de amônio aquoso, e a mistura foi extraída com acetato de etila. A fase orgânica foi separada, lavada com salmoura, secada com sulfato de magnésio, filtrada, e concentrada. O resíduo foi purificado através da cromatografia de coluna eluindo com acetato de etila e hexanos (1:4) para dar 4- ((1R,2R)-3-((R)-4-benzil-2-oxo-oxazolidin-3-il)-1-hidróxi-2-metil-3- oxopropil)-2,2-dimetiloxazolidina-3-carboxilato de (R)-terc-butila como o produto principal (5:2) em 58% de rendimento. LC/MS = 363,3 (M+H- B°C), Rt = 1,09 min. Síntese de 4-((1R,2R)-3-((R)-4-benzil-2-oxo-oxazolidin-3-il)-1-(terc- butildimetilsililóxi)-2-metil-3-oxopropil)-2,2-dimetiloxazolidina-3- carboxilato de (R)-terc-butila
[000189] Para uma solução de 4-((1R,2R)-3-((R)-4-benzil-2-oxo- oxazolidin-3-il)-1-hidróxi-2-metil-3-oxopropil)-2,2-dimetiloxazolidina-3- carboxilato de (R)-terc-butila (1,0 equiv.) e lutidina (1,8 equiv.) em DCM (0,1M) foi adicionado TBSOTf (1,4 equiv.) a -40°C. A mistura de reação foi agitada a -40°C por 2 horas. A solução foi diluída com acetato de etila e lavada com NaHCO3 saturado, NaCl saturado, secada com sulfato de magnésio, filtrada e concentrada. O resíduo foi purificado por cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (1:4) para dar 4-((1R,2R)-3-((R)-4-benzil-2- oxo-oxazolidin-3-il)-1-(terc-butildimetilsililóxi)-2-metil-3-oxopropil)-2,2- dimetiloxazolidina-3-carboxilato de (R)-terc-butila como o produto principal (5:2) em 83% de rendimento. LC/MS = 577,3 (M+H), Rt = 1,33 min (Frac 65%-95% método). Síntese de 4-((1R,2S)-1-(terc-butildimetilsililóxi)-3-hidróxi-2-metilpropil)- 2,2-dimetiloxazolidina-3-carboxilato de (R)-terc-butila
[000190] Para uma solução de 4-((1R,2R)-3-((R)-4-benzil-2-oxo- oxazolidin-3-il)-1-(terc-butildimetilsililóxi)-2-metil-3-oxopropil)-2,2- dimetiloxazolidina-3-carboxilato de (R)-terc-butila (1,0 equiv.) e etanol (3,0 equiv.) em THF (0,09 M) foi adicionado LiBH4 (3,0 equiv.) a -30°C. A mistura de reação foi deixada aquecer a 0°C e agitada àquela temperatura por 3 horas. A solução foi então diluída com éter dietílico, e NaOH a 1N foi adicionado. A mistura resultante foi extraída com acetato de etila, a camada orgânica foi separada, lavada com NaCl saturado, secada sobre sulfato de magnésio, filtrada e concentrada. O resíduo foi purificado através da cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (1:4) para dar 4-((1R,2S)-1- (terc-butildimetilsililóxi)-3-hidróxi-2-metilpropil)-2,2-dimetiloxazolidina-3- carboxilato de (R)-terc-butila como o produto principal (razão 5:2) em 71% de rendimento. LC/MS = 304,3 (M+H-BOC), Rt = 0,95 min (Frac 65%-95% método). Síntese de 4-((1R,2S)-3-azido-1-(terc-butildimetilsililóxi)- 2-metilpropil)-2,2-dimetiloxazolidina-3-carboxilato de (R)-terc-butila
[000191] Para uma solução de 4-((1R,2S)-1-(terc-butildimetilsililóxi)- 3-hidróxi-2-metilpropil)-2,2-dimetiloxazolidina-3-carboxilato de (R)-terc- butila (1,0 equiv.), DIAD (2,0 equiv.), e PPh3 (2,0 equiv.) em THF (0,18 M) foi adicionado DPPA (2,0 equiv., 1M solução em THF). A mistura de reação foi agitada à temperatura ambiente durante a noite. Após a remoção dos voláteis sob vácuo, o resíduo foi purificado por cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (1:6) para dar 4-((1R,2S)-3-azido-1-(terc-butildimetilsililóxi)-2- metilpropil)-2,2-dimetiloxazolidina-3-carboxilato de (R)-terc-butila como o produto principal (5:2) em 86% de rendimento. LC/MS = 329,3 (M+H- BOC), Rt = 1,40 min (Frac 65%-95% método). Síntese de(2R,3R,4S)-5-azido-3-(terc-butildimetilsililóxi)- 1-hidróxi-4-metilpentan-2-ilcarbamato de terc-butila
[000192] Para uma solução de 4-((1R,2S)-3-azido-1-(terc- butildimetilsililóxi)-2-metilpropil)-2,2-dimetiloxazolidina-3-carboxilato de (R)-terc-butila (1,0 equiv.) em EtOH (0,1 M) foi adicionado PPTS (1,3 equiv.), e a mistura foi sumetida a refluxo por 2 dias. Os voláteis foram removidos sob vácuo, o resíduo foi dissolvido em DCM (0,1 M) e DIEA (1,5 equiv.) e BOC2O (1,0 equiv.) foram adicionados à mistura de reação. A solução foi agitada por 3 horas à temperatura ambiente. Os solventes foram removidos sob pressão reduzida, e o resíduo foi diluído com acetato de etila, lavado com água, NaHSO4 aquoso, NaHCO3 aquoso, NaCl saturado, a fase orgânica foi secada com sulfato de magnésio, filtrada e concentrada. O resíduo foi purificado através da cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (1:3) para dar (2R,3R,4S)-5-azido-3-(terc- butildimetilsililóxi)-1-hidróxi-4-metilpentan-2-ilcarbamato de terc-butila como um isômero principal (5:2) em 70% de rendimento. LC/MS = 289,3 (M+H-BOC), Rt = 0,76 min (Frac 65%-95% método). Síntese de metanossulfonato de (2R,3R,4S)-5-azido-2-(terc- butoxicarbonilamino)-3-(terc-butildimetilsililóxi)-4-metilpentila
[000193] Para uma solução de (2R,3R,4S)-5-azido-3-(terc- butildimetilsililóxi)-1-hidróxi-4-metilpentan-2-ilcarbamato de terc-butila (1,0 equiv.) em piridina (0,2 M) foi adicionado MsCl (1,3 equiv.) seguido por DMAP (quantidade catalítica) a 0°C. A mistura foi agitada àquela temperatura por 1 hora. A solução foi diluída com éter e acetato de etila (4:1), lavada com NaHSO4 aquoso, NaHCO3 saturado, salmoura, secada sobre sulfato de magnésio, filtrada e concentrada. O resíduo foi purificado por cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (1:3) para dar metanossulfonato de (2R,3R,4S)-5-azido-2-(terc-butoxicarbonilamino)-3-(terc- butildimetilsililóxi)-4-metilpentila como um isômero principal (5:2) em 90% de rendimento. LC/MS = 367,3 (M+H-BOC), Rt = 0,81 min (Frac 65%-95% método). Síntese de(3R,4R,5S)-4-(terc-butildimetilsililóxi)- 5-metilpiperidin-3-ilcarbamato de terc-butila
[000194] Uma solução de metanossulfonato de (2R,3R,4S)-5-azido- 2-(terc-butoxicarbonilamino)-3-(terc-butildimetilsililóxi)-4-metilpentila em MeOH (0,09 M) foi desgaseificada com nitrogênio por 20 min. DIEA (2,5 equiv.) foi adicionado, seguido por 10% de Pd/C (0,1 equiv.). A mistura de reação foi agitada sob um balão de hidrogênio por 2 horas. A solução foi filtrada, e o filtrado foi concentrado sob vácuo para proporcionar (3R,4R,5S)-4-(terc-butildimetilsililóxi)-5-metilpiperidin-3- ilcarbamato de terc-butila como o isômero principal (5:2) em >99% de rendimento. LC/MS = 345.2 (M+H-Boc), Rt = 0,95 e 0,99 min. Síntese de (3R,4R,5S)-4-(terc-butildimetilsililóxi)-5-metil-1-(3- nitropiridin-4-il)piperidin-3-ilcarbamato de terc-butila
[000195] Para uma solução de (3R,4R,5S)-4-(terc-butildimetilsililóxi)- 5-metilpiperidin-3-ilcarbamato de terc-butila (1,0 equiv.) em i-PrOH (0,09 M) foram adicionados DIEA (2,5 equiv.) e 4-cloro-3-nitropiridina (1,5 equiv.). A mistura de reação foi agitada a 60°C por 2 horas. Os voláteis foram removidos sob vácuo, o resíduo foi diluído com acetato de etila e lavado com NaCl saturado. A fase orgânica foi secada com sulfato de magnésio, filtrada e concentrada. O material bruto foi purificado por cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (1:2) para dar (3R,4R,5S)-4-(terc- butildimetilsililóxi)-5-metil-1-(3-nitropiridin-4-il)piperidin-3-ilcarbamato de terc-butila em 76% de rendimento. LC/MS = 467,3 (M+H), Rt = 1,09 min. Síntese de (3R,4R,5S)-1-(3-aminopiridin-4-il)-4-(terc-butildimetilsililóxi)- 5-metilpiperidin-3-ilcarbamato de terc-butila
[000196] Uma solução de (3R,4R,5S)-4-(terc-butildimetilsililóxi)-5- metil-1-(3-nitropiridin-4-il)piperidin-3-ilcarbamato de terc-butila (1,0 equiv.) em MeOH (0,05 M) foi desgaseificada com nitrogênio por 20 min. 10% de Pd/C (0,2 equiv.) foram adicionados à mistura, e a solução foi agitada sob um balão de hidrogênio por 3 horas. A reação foi filtrada, e o filtrado foi concentrado sob pressão reduzida para dar (3R,4R,5S)-1-(3-aminopiridin-4-il)-4-(terc-butildimetilsililóxi)-5- metilpiperidin-3-ilcarbamato de terc-butila como o produto desejado em 94% de rendimento. LC/MS = 437,4 (M+H), Rt = 1,08 min. 1H-RMN (300 MHz, CDCl3): δ 8.01 (s, 1H), 7.95 (d, J = 6,0 Hz, 1H), 6,76 (d, J = 6,0 Hz, 1H), 4,44 (br s, 1H), 3,74 (br s, 2H), 3,59-3,55 (m, 1H), 3,25- 3,13 (m, 2H), 2,47-2,35 (m, 2H), 1,89 (br s, 2H), 1,44 (s, 9H), 1,04 (d, J = 6,0 , 3H), 0,92 (s, 9H), 0,13 (d, J = 9,0, 6H). Síntese de (2R)-1-(benzilóxi)-3-hidróxi-4-metil-hex-5-en-2-ilcarbamato de terc-butila
[000197] Para uma solução de N-Boc, aldeído de O-benzil-D-Serina (1,0 equiv) em DCM (0,1 M) a -78°C sob uma atmosfera de Ar foi adicionado (Z)-2-(but-2-enil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (1,1 equiv) , e a solução clara agitada por 16 horas conforme foi aquecida à ta. A solução foi adicionada ao EtOAc e foi lavada com H2O (3x), e NaCl(sat.), secada sobre MgSO4 e purificada por cromatografia em sílica-gel (15% de EtOAc/hexanos) para render (2R)-1-(benzilóxi)-3- hidróxi-4-metil-hex-5-en-2-ilcarbamato de terc-butila (54%) como uma mistura 3:1 dos isômeros como julgados por 1H RMN. LCMS (m/z): 236,3 (MH+-Boc); LC Rt = 4,37 e 4,51 min. Síntese de (4R)-4-(benziloximetil)-5-(but-3-en-2-il)oxazolidin-2-ona
[000198] Para uma solução de (2R)-1-(benzilóxi)-3-hidróxi-4-metil- hex-5-en-2- em THF (0,1 M) foram adicionados 60% de hidreto de sódio em óleo mineral (1,5 equiv.). Após agitar por 3 dias, a reação foi extinta pela adição de NH4Cl(sat.), e a solução foi diluída com EtOAc e lavada com NH4Cl(sat.) e NaCl(sat.), secada sobre MgSO4 e purificada por cromatografia em sílica-gel (50% de EtOAc/hexanos) para render (4R)-4-(benziloximetil)-5-(but-3-en-2-il)oxazolidin-2-ona (89%) como uma mistura 3:1. LCMS (m/z): 262,2 (MH+); LC Rt = 3,47 min. Síntese de (4R)-4-(benziloximetil)-5-(1 -hidroxipropan-2-il)oxazolidin-2- ona
[000199] Para uma solução de (4R)-4-(benziloximetil)-5-(but-3-en-2- il)oxazolidin-2-ona (1,0 equiv.) em 2:1 MeOH/ H2O (0,04 M) foram adicionados 4% de tetróxido de ósmio em H2O (0,07 equiv) e periodato de sódio (3,0 equiv.). Após agitar por 3 horas, o precipitado branco foi filtrado e enxaguado com EtOAc. O filtrado combinado foi concentrado em vácuo, e o resíduo foi dissolvido em EtOAc, lavado com NaCl(sat.), secado sobre MgSO4, filtrado e concentrado. O aldeído bruto foi dissolvido em EtOH (0,08 M) e sob resfriamento a 0°C, boridreto de sódio (2,0 equiv.) foi adicionado. Após agitar por 15 horas e chegar à temperatura ambiente, a reação foi extinta pela adição de H2O. Após agitar por 20 minutos, o EtOH foi removido em vácuo, EtOAc foi adicionado, e a solução foi lavada com HCl a 1N, NaHCO3(sat.) e NaCl(sat.), secada sobre MgSO4, filtrada e concentrada rendendo após a purificação por cromatografia em sílica-gel (4R)-4-(benziloximetil)-5- (1-hidroxipropan-2-il)oxazolidin-2-ona como uma mistura 3:1 de isômeros (60%). LCMS (m/z): 266,1 (MH+); LC Rt = 2,28 min. Síntese de (4R)-4-(hidroximetil)-5-(1 -hidroxipropan-2-il)oxazolidin-2- ona
[000200] Para uma solução de (4R)-4-(benziloximetil)-5-(1- hidroxipropan-2-il)oxazolidin-2-ona (1,0 equiv.) em metanol, a uma concentração de 0,1 M, foram adicionados 10% de paládio em carbono (0,1 eq.). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 15 horas. Neste momento, a mistura foi filtrada através de uma almofada de celite eluindo com metanol. Os voláteis foram removidos em vácuo rendendo (4R)-4-(hidroximetil)-5-(1-hidroxipropan-2-il)oxazolidin-2-ona (99%). LCMS (m/z): 176,1 (MH+). Síntese de 4-metilbenzenossulfonato de 2-((4R)-2-oxo-4- (tosiloximetil)oxazolidin-5-il)-propila
[000201] Para uma solução de (4R)-4-(hidroximetil)-5-(1- hidroxipropan-2-il)oxazolidin-2-ona (1,0 equiv.) em piridina (0,15 M) a 0°C foi adicionado cloreto de p-toluenossulfonila (2,1 equiv.). A solução foi deixada aquecer à ta conforme foi agitada por 14 horas, ao mesmo tempo EtOAc foi adicionado, e a solução foi lavada com H2O(3x), CuSO4(sat.) (2x), H2O , Na2CO3(sat.) e NaCl(sat.), secada sobre MgSO4, filtrada, concentrada e purificada por cromatografia em sílica-gel (75% de eluente EtOAc/hexanos) rendendo 4-metilbenzenossulfonato de 2- ((4R)-2-oxo-4-(tosiloximetil)oxazolidin-5-il)propila (68%). LCMS (m/z): 484,1 (MH+); LC Rt = 4,06 min. Síntese de (3aR,7R,7aS)-5-(4-metoxibenzil)-7-metil-hexa-hidro- oxazolo[4,5-c]piridin-2(3H)-ona e (3aR,7S,7aR)-5-(4-metoxibenzil)-7- metil-hexa-hidro-oxazolo[4,5-c]piridin-2(3H)-ona
[000202] Uma solução de 4-metilbenzenossulfonato de 2-((4R)-2- oxo-4-(tosiloximetil)oxazolidin-5-il)propila (1,0 equiv.), di-isopropiletil amina (3,0 equiv.) e para-metoxibenzilamina (1,5 equiv.) em NMP (0,05 M) foi aquecida a 100°C por 14 horas. A solução foi purificada diretamente por RP HPLC. As frações do produto foram dessalgadas pela adição de EtOAc e Na2CO3(s), lavadas adicionalmente com NaCl(sat.), secadas sobre MgSO4 e concentradas rendendo dois isômeros separados de (3aR,7R,7aS)-5-(4-metoxibenzil)-7-metil-hexa- hidro-oxazolo[4,5-c]piridin-2(3H)-ona e (3aR,7S,7aR)-5-(4- metoxibenzil)-7-metil-hexa-hidro-oxazolo[4,5-c]piridin-2(3H)-ona (27% e 8%). LCMS (m/z): 277,2 (MH+) a 0,40 e 0,42 min. Síntese de (3aR,7R,7aS)-7-metil-hexa-hidro-oxazolo[4,5-c]piridin- 2(3H)-ona
[000203] Para uma solução de (3aR,7R,7aS)-5-(4-metoxibenzil)-7- metil-hexa-hidro-oxazolo[4,5-c]piridin-2(3H)-ona (1,0 equiv.) em metanol, a uma concentração de 0,1 M, foram adicionados 20% de hidróxido de paládio em carbono (0,3 eq.). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 2 horas. Neste momento, a mistura foi filtrada através de uma almofada de celite eluindo com metanol. Os voláteis foram removidos em vácuo rendendo (3aR,7R,7aS)-7-metil-hexa-hidro-oxazolo[4,5- c]piridin-2(3H)-ona (99%). LCMS (m/z): 157,1 (MH+) a 0,16 min. Síntese de 7-metil-5-(3-nitropiridin-4-il)-2-oxo-hexa-hidro-oxazolo[4,5- c]piridina-3(2H)-carboxilato de (3aR,7R,7aS)-terc-butila
[000204] Uma solução de 4-cloro-3-nitropiridina (1,3 equiv.) e (3aR,7R,7aS)-7-metil-hexa-hidro-oxazolo[4,5-c]piridin-2(3H)-ona (1,5 equiv.) em CH2CI2, a uma concentração de 0,1 M, foi agitada à ta por 48 horas, a qual piperidina (0,4 equiv) foi adicionada para consumir o excesso de 4-cloro-3-nitropiridina. Após agitar por mais 2 horas, dicarbonato de di-terc-butila (2,0 equiv.) e dimetilaminopiridina (0,1 equiv.) foram adicionados. Após agitar por 4 horas, a solução foi dividida entre EtOAc e NaHCO3 (sat.), foi lavada adicionalmente com NaHCO3 (sat.), e NaCl(sat.), foi secada sobre MgSO4, foi filtrada e purificada por cromatografia em sílica-gel rendendo 7-metil-5-(3- nitropiridin-4-il)-2-oxo-hexa-hidro-oxazolo[4,5-c]piridina- 3(2H)carboxilato de (3aR,7R,7aS)-terc-butila (62%). LCMS (m/z): 379,0 (MH+) a 0,58 min. Síntese de 5-(3-aminopiridin-4-il)-7-metil-2-oxo-hexa-hidro-oxazolo[4,5- c]piridina-3(2H)-carboxilato de (3aR,7R,7aS)-terc-butila
[000205] Para uma solução de 7-metil-5-(3-nitropiridin-4-il)-2-oxo- hexa-hidro-oxazolo[4,5-c]piridina-3(2H)-carboxilato de (3aR,7R,7aS)- terc-butila (1,0 equiv.) em metanol, a uma concentração de 0,1 M, foram adicionados 10% de paládio em carbono (0,1 eq.). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 14 horas. Neste momento, a mistura foi filtrada através de uma almofada de celite eluindo com metanol. Os voláteis foram removidos em vácuo rendendo 5-(3-aminopiridin-4-il)-7-metil-2- oxo-hexa-hidro-oxazolo[4,5-c]piridina-3(2H)-carboxilato de (3aR,7R,7aS)-terc-butila. LCMS (m/z): 349,1 (MH+); LC Rt = 2,06 min. Síntese de (3aR,7S,7aR)-7-metil-hexa-hidro-oxazolo[4,5-c]piridin- 2(3H)-ona
[000206] Para uma solução de (3aR,7S,7aR)-5-(4-metoxibenzil)-7- metil-hexa-hidro-oxazolo[4,5-c]piridin-2(3H)-ona (1,0 equiv.) em metanol, a uma concentração de 0,1 M, foram adicionados 20% de hidróxido de paládio em carbono (0,3 eq.). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 2 horas. Neste momento, a mistura foi filtrada através de uma almofada de celite eluindo com metanol. Os voláteis foram removidos em vácuo rendendo (3aR,7S,7aR)-7-metil-hexa-hidro-oxazolo[4,5- c]piridin-2(3H)-ona (99%). LCMS (m/z): 157,1 (MH+) at 0,17 min. Síntese de 7-metil-5-(3-nitropiridin-4-il)-2-oxo-hexa-hidro-oxazolo[4,5- c]piridina-3(2H)-carboxilato de (3aR,7S,7aR)-terc-butila
[000207] Uma solução de 4-cloro-3-nitropiridina (1,3 equiv.) e (3aR,7S,7aR)-7-metil-hexa-hidro-oxazolo[4,5-c]piridin-2(3H)-ona (1,5 equiv.) em CH2CI2, a uma concentração de 0,1 M, foi agitada à ta por 48 horas, na qual piperidina (0,4 equiv) foi adicionada para consumir o excesso de 4-cloro-3-nitropiridina. Após agitar por mais 2 horas, dicarbonato de di-terc-butila (2,0 equiv.) e dimetilaminopiridina (0,1 equiv.) foram adicionados. Após agitar por 4 horas, a solução foi dividida entre EtOAc e NaHCO3 (sat.), foi lavada adicionalmente com NaHCO3 (sat.), e NaCl(sat.), foi secada sobre MgSO4, foi filtrada e purificada por cromatografia em sílica-gel (75% de eluente EtOAc/hexanos) rendendo 7-metil-5-(3-nitropiridin-4-il)-2-oxo-hexa- hidro-oxazolo[4,5-c]piridina-3(2H)-carboxilato de (3aR,7S,7aR)-terc- butila (35%). LCMS (m/z): 379,0 (MH+). LC Rt = 2,42 min. Síntese de 5-(3-aminopiridin-4-il)-7-metil-2-oxo-hexa-hidro-oxazolo[4,5- c]piridina-3(2H)-carboxilato de (3aR,7R,7aS)-terc-butila
[000208] Para uma solução de 7-metil-5-(3-nitropiridin-4-il)-2-oxo- hexa-hidro-oxazolo[4,5-c]piridina-3(2H)-carboxilato de (3aR,7S,7aR)- terc-butila (1,0 equiv.) em metanol, a uma concentração de 0,1 M, foram adicionados 10% de paládio em carbono (0,1 eq.). A solução heterogênea resultante foi colocada sob uma atmosfera de hidrogênio e foi agitada por 14 horas. Neste momento, a mistura foi filtrada através de uma almofada de celite eluindo com metanol. Os voláteis foram removidos em vácuo rendendo 5-(3-aminopiridin-4-il)-7-metil-2- oxo-hexa-hidro-oxazolo[4,5-c]piridina-3(2H)-carboxilato de (3aR,7S,7aR)-terc-butila. LCMS (m/z): 349,1 (MH+); LC Rt = 2,18 min. Método 1 Síntese de 3-amino-6-(2,6-diflurofenil)picolinato de metila
[000209] Uma solução de 3-amino-6-bromopicolinato de metila (1,0 equiv.), ácido 2,6-di-flúorfenil-borônico (3,0 equiv), e Pd(dppf)Cl2-DCM (0,1 equiv.) em 3:1 DME/ 2M Na2CO3 (0,5 M) foi submetida à irradiação de micro-ondas a 120°C em intervalos de 15 min. A reação foi filtrada e lavada com EtOAc. O orgânico foi dividido com H2O (25mL), foi lavada adicionalmente com NaCl(sat.) (25mL), foi secado sobre MgSO4, e os voláteis foram removidos em vácuo. O resíduo foi diluído em EtOAc e passados através de um plugue de sílica-gel, e os voláteis foram removidos em vácuo rendendo 3-amino-6-(2,6-di- flúorfenil)picolinato de metila (47%). LCMS (m/z): 265,1 (MH+); LC Rt = 2,70 min Síntese de ácido 6-(2,3-di-flúorfenil)-5-flúorpicolínico
[000210] Para uma solução de ácido 6-bromo-5-flúorpicolínico (1,0 equiv.) em DME e Na2CO3 a 2M (3:1, 0,25 M) foram adicionados ácido 2,3-di-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) em um frasco de micro-ondas. O frasco foi aquecido em micro-ondas a 120°C por 30 minutos. A mistura foi diluída com acetato de etila, e NaOH a1N foi adicionado. A fase orgânica foi separada e extraída mais três vezes com NaOH a 1N e uma vez com NaOH a 6N. As fase aquosas combinadas foram filtradas e acidificadas a pH 1 pela adição de HCl concentrado e extraídas com acetato de etila. A camada orgânica foi secada sobre sulfato de magnésio, filtrada e concentrada para dar ácido 6-(2,3-di-flúorfenil)-5-flúorpicolínico em 78%. LC/MS = 254,1 (M+H), Rt = 0,75 min. Método 2 Síntese de ácido 3-amino-6-(2,6-di-flúorfenil)picolínico
[000211] Para uma solução de 3-amino-6-(2,6-di-flúorfenil)picolinato de metila (1,0 equiv) em THF (0,5 M), foi adicionado 1M LiOH (4,0 equiv). Após agitar por 4 horas a 60°C, HCl a 1N (4,0 equiv.) foi adicionado, e o THF foi removido em vácuo. O sólido resultante foi filtrado e enxaguado com H2O frio (3 x 20mL) para render ácido 3- amino-6-(2,6-di-flúorfenil)picolínico (90%). LCMS (m/z): 251,1 (MH+); LC Rt = 2,1 min. Síntese de ácido 3-amino-6-(2-flúor-5-propoxifenil)picolínico
[000212] Método 1 foi seguido usando ácido 3-amino-6- bromopicolínico (1,0 equiv.) e ácido 2-flúor-5-propoxifenilborônico (1,5 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar ácido 3-amino-6-(2- flúor-5-propoxifenil)picolínico em 75% de rendimento. LC/MS = 291,0 (M+H), Rt = 0,81 min. Síntese de ácido 3-amino-5-flúor-6-(2-flúor-5-propoxifenil)picolínico
[000213] Método 1 foi seguido usando ácido 3-amino-6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido 2-flúor-5-propoxifenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar ácido 3-amino-5- flúor-6-(2-flúor-5-propoxifenil)picolínico em 28% de rendimento. LC/MS = 309,1 (M+H), Rt = 1,00 min. Síntese de 3-amino-5-flúor-6-(2-flúorfenil)picolinato de metila
[000214] Método 1 foi seguido usando 3-amino-6-bromo-5- flúorpicolinato de metila (1,0 equiv.) e ácido 2-flúor-fenilborônico (1,5 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar 3-amino-5-flúor-6-(2- flúorfenil)picolinato de metila em >99% de rendimento. LC/MS = 265,0 (M+H), Rt = 0,77 min. Síntese de ácido 3-amino-5-flúor-6-(2-flúorfenil)picolínico
[000215] Método 2 foi seguido usando 3-amino-5-flúor-6-(2- flúorfenil)picolinato de metila (1,0 equiv.) e LiOH (5,0 equiv.) para dar ácido 3-amino-5-flúor-6-(2-flúorfenil)picolínico em 90% de rendimento. LC/MS = 251,1 (M+H), Rt = 0,80 min. Síntese de 3-amino-6-(2,6-di-flúorfenil)-5-flúorpicolinato de metila
[000216] Método 1 foi seguido usando 3-amino-6-bromo-5- flúorpicolinato de metila (1,0 equiv.) e ácido 2,6-di-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar 3-amino-6-(2,6- di-flúorfenil)-5-flúorpicolinato de metila em 94% de rendimento. LC/MS = 283,0 (M+H), Rt = 0,76 min. Síntese de ácido 3-amino-6-(2,6-di-flúorfenil)-5-flúorpicolínico
[000217] Método 2 foi seguido usando 3-amino-6-(2,6-di-flúorfenil)-5- flúorpicolinato (1,0 equiv.) e LiOH (1,0 equiv.) para dar ácido 3-amino- 6-(2,6-di-flúorfenil)-5-flúorpicolínico em 79% de rendimento. LC/MS = 269,0 (M+H), Rt = 0,79 min. Síntese de ácido 5-flúor-6-(2-flúorfenil)picolínico
[000218] Método 1 foi seguido usando ácido 6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido 2-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar ácido 5-flúor-6-(2- flúorfenil)picolínico em 43% de rendimento. LC/MS = 236,1 (M+H), Rt = 0,72 min. Síntese de ácido 6-(3,4-di-flúorfenil)-5-flúorpicolínico
[000219] Método 1 foi seguido usando ácido 6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido 3,4-di-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar ácido 6-(3,4-di-flúorfenil)-5- flúorpicolínico em 70% de rendimento. LC/MS = 254,1 (M+H), Rt = 0,81 min. Síntese de ácido 6-(2,5-di-flúorfenil)-5-flúorpicolínico
[000220] Método 1 foi seguido usando ácido 6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido 2,5-di-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar ácido 6-(2,5-di-flúorfenil)-5- flúorpicolínico em 80% de rendimento. LC/MS = 254,1 (M+H), Rt = 0,74 min. Síntese de ácido 6-(2,4-di-flúorfenil)-5-flúorpicolínico
[000221] Método flúorpicolínico (1,0 equiv.) e ácido 2,4-di-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar ácido 6-(2,4-di-flúorfenil)-5- flúorpicolínico em 79% de rendimento. LC/MS = 254,1 (M+H), Rt = 0,75 min. Síntese de ácido 5-flúor-6-(2-flúor-5-propoxifenil)picolínico
[000222] Método 1 foi seguido usando ácido 6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido 2-flúor-5-propoxifenilborônico (1,5 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar ácido 5-flúor-6-(2- flúor-5-propoxifenil)picolínico. LC/MS = 294,2 (M+H), Rt = 0,95 min. Síntese de ácido 6-(2-flúorfenil)picolínico
[000223] Método 1 foi seguido usando ácido 6-bromopicolínico (1,0 equiv.) e ácido 2-flúorfenilborônico (1,5 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar o ácido 6-(2-flúorfenil)picolínico em 93% de rendimento. LC/MS = 218,0 (M+H), Rt = 0,66 min. Síntese de ácido 6-(2,6-di-flúorfenil)picolínico
[000224] Método 1 foi seguido usando ácido 6-bromopicolínico (1,0 equiv.) e ácido 2,6-di-flúorfenilborônico (1,5 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar o ácido 6-(2,6-di-flúorfenil)picolínico em 38% de rendimento. LC/MS = 236,0 (M+H), Rt = 0,87 min. Síntese de ácido 6-(2-flúor-5-metoxifenil)picolínico
[000225] Método 1 foi seguido usando 6-bromopicolínico ácido (1,0 equiv.) e ácido 2-flúor-5-metoxifenilborônico (1,3 equiv.) e Pd(dppf)Cl2- DCM (0,15 equiv.) para dar o ácido 6-(2-flúor-5-metoxifenil)picolínico em 95% de rendimento. LC/MS = 248,2 (M+H), Rt = 0,78 min. Síntese de ácido 6-(2-flúor-5-propoxifenil)picolínico
[000226] Método 1 foi seguido usando ácido 6-bromopicolínico (1,0 equiv.) e ácido 2-flúor-5-propoxifenilborônico (1,5 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 6-(2-flúor-5- propoxifenil)picolínico em 20% de rendimento. LC/MS = 276,0 (M+H), Rt = 0,87 min. Síntese de ácido 6-(2,6-di-flúor-4-metoxifenil)picolínico
[000227] Método 1 foi seguido usando ácido 6-bromopicolínico (1,0 equiv.) e ácido 2,6-di-flúor-4-metoxifenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 6-(2,6-di-flúor-4- metoxifenil)picolínico em 42% de rendimento. LC/MS = 266,1 (M+H), Rt = 0,75 min. Síntese de ácido 3-flúor-6-(2-flúorfenil)picolínico
[000228] Método 1 foi seguido usando ácido 6-bromo-3- flúorpicolínico (1,0 equiv.) e ácido 2-flúorfenilborônico (1,5 equiv.) e Pd(dppf)Cl2-DCM (0,05 equiv.) para dar o ácido 3-flúor-6-(2- flúorfenil)picolínico em 81% de rendimento. LC/MS = 236,1 (M+H), Rt = 0,72 min. Síntese de ácido 3-flúor-6-(2-flúor-5-metoxifenil)picolínico
[000229] Método 1 foi seguido usando ácido 6-bromo-3- flúorpicolínico (1,0 equiv.) e ácido 2-flúor-5-metoxifenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 3-flúor-6-(2- flúor-5-metoxifenil)picolínico em 89% de rendimento. LC/MS = 266,1 (M+H), Rt = 0,79 min. Síntese de ácido 5-flúor-6-(2-flúor-5-metoxifenil)picolínico
[000230] Método 1 foi seguido usando ácido 6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido 2-flúor-5-metoxifenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 5-flúor-6-(2- flúor-5-metoxifenil)picolínico em 86% de rendimento. LC/MS = 266,1 (M+H), Rt = 0,79 min. Síntese de ácido 6-(4-(benzilóxi)-2-flúorfenil)-5-flúorpicolínico
[000231] Método 1 foi seguido ácido usando 6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido 4-(benzilóxi)-2-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 6-(4- (benzilóxi)-2-flúorfenil)-5-flúorpicolínico em 28% de rendimento. LC/MS = 342,1 (M+H), Rt = 1,05 min. Síntese de ácido 6-(4-(benzilóxi)-2-flúorfenil)-3-flúorpicolínico
[000232] Método 1 foi seguido usando ácido 6-bromo-3- flúorpicolínico (1,0 equiv.) e ácido 4-(benzilóxi)-2-flúorfenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 6-(4- (benzilóxi)-2-flúorfenil)-3-flúorpicolínico em 41% de rendimento. LC/MS = 342,1 (M+H), Rt = 1,06 min. Síntese de ácido 6-(2,6-di-flúor-4-metoxifenil)-3-flúorpicolínico
[000233] Método 1 foi seguido ácido usando 6-bromo-3- flúorpicolínico (1,0 equiv.) e ácido 2,6-di-flúor-4-metoxifenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 6-(2,6-di- flúor-4-metoxifenil)-3-flúorpicolínico em 9% de rendimento. LC/MS = 284,0 (M+H), Rt = 0,74 min. Síntese de ácido 6-ciclo-hexenil-5-flúorpicolínico
[000234] Método 1 foi seguido usando ácido 6-bromo-5- flúorpicolínico (1,0 equiv.) e ácido ciclo-hexenilborônico (1,3 equiv.) e Pd(dppf)Cl2-DCM (0,15 equiv.) para dar o ácido 6-ciclo-hexenil-5- flúorpicolínico em 61 % de rendimento. LC/MS = 222,0 (M+H), Rt = 0,52 min. Método 3 Síntese de ácido 6-ciclo-hexil-5-flúorpicolínico
[000235] Para uma solução desgaseificada de ácido 6-ciclo-hexenil- 5-flúorpicolínico (1,0 equiv.) em MeOH (0,07M) foram adicionados 10% de Pd/C (0,1 equiv.), e a reação foi agitada sob um balão de hidrogênio durante a noite. A solução foi então filtrada, enxaguada com MeOH, e o filtrado foi concentrado para proporcionar o ácido 6- ciclo-hexil-5-flúorpicolínico em 65% de rendimento. LC/MS = 224,2 (M+H), Rt = 0,95 min. Síntese de ácido 5-flúor-6-(1,4-dioxaspiro[4.5]dec-7-en-8-il)picolínico
[000236] Método 1 foi seguido usando ácido 6-bromo-5- flúorpicolínico (1,0 equiv.) e 4,4,5,5-tetrametil-2-(1,4- dioxaspiro[4.5]dec-7-en-8-il)-1,3,2-dioxaborolano (2,0 equiv.) e Pd(dppf)Cl2-DCM (0,2 equiv.) para dar o ácido 5-flúor-6-(1,4- dioxaspiro[4.5]dec-7-en-8-il)picolínico. LC/MS = 280,2 (M+H), Rt = 0,66 min. Síntese de 5-flúor-6-(1,4-dioxaspiro[4.5]dec-7-en-8-il)picolinato de metila
[000237] Para uma solução de ácido 5-flúor-6-(1,4- dioxaspiro[4.5]dec-7-en-8-il)picolínico (1,0 equiv.) em DCM (0,3 M) foram adicionados EDC-HCl (1,0 equiv.), DMAP (1,0 equiv.), e MeOH (10 equiv.). A mistura de reação foi deixada agitar à temperatura ambiente por 5 dias, depois diluída com acetato de etila, lavada com água, salmoura, secada sobre sulfato de magnésio, filtrada e concentrada. O produto bruto foi purificado por cromatografia de coluna em sílica-gel eluindo com 25-50% de acetato de etila em hexanos para render 5-flúor-6-(1,4-dioxaspiro[4.5]dec-7-en-8- il)picolinato de metila como o produto desejado em 35% de rendimento. LC/MS = 294,2 (M+H), Rt = 0,79 min. Síntese de 5-flúor-6-(1,4-dioxaspiro[4.5]decan-8-il)picolinato de metila
[000238] Para uma solução desgaseificada de 5-flúor-6-(1,4- dioxaspiro[4.5]dec-7-en-8-il)picolinato de metila (1,0 equiv.) em MeOH (0,07M) foram adicionados 10% de Pd/C (0,1 equiv.), e a reação foi agitada sob um balão de hidrogênio durante a noite. A solução foi então filtrada, enxaguada com MeOH, e o filtrado foi concentrado para proporcionar 5-flúor-6-(1,4-dioxaspiro[4.5]decan-8-il)picolinato de metila em 91% de rendimento. LC/MS = 296,2 (M+H), Rt = 0,83 min. Síntese de 5-flúor-6-(4-oxociclo-hexil)picolinato de metila
[000239] Para uma solução de 5-flúor-6-(1,4-dioxaspiro[4.5]decan-8- il)picolinato de metila (1,0 equiv.) em acetona e água (1:1, 0,04 M) foi adicionado deidrato de ácido oxálico (2,0 equiv.), e a mistura de reação foi agitada por 3 dias. A solução foi então neutralizada pela adição de NaHCO3 sólido, a mistura foi adicionada em acetato de etila e salmoura, a fase orgânica foi secada sobre sulfato de magnésio, filtrada, e concentrada. 5-flúor-6-(4-oxociclo-hexil)picolinato de metila foi obtido em 98% de rendimento. LC/MS = 252,1 (M+H), Rt = 0,68 min. Síntese de 5-flúor-6-(4-hidroxiciclo-hexil)picolinato de metila
[000240] Para uma solução a 0°C de 5-flúor-6-(4-oxociclo- hexil)picolinato de metila (1,0 equiv.) com MeOH (0,08 M) foi adicionado NaBH4. A solução foi deixada aquecer à temperatura ambiente durante a noite e depois dividida entre acetato de etila e salmoura, a fase orgânica foi secada sobre sulfato de magnésio, filtrada e concentrada para dar 5-flúor-6-(4-hidroxiciclo-hexil)picolinato de metila como uma mistura de dois isômeros (5:1). LC/MS = 254,2 (M+H), Rt = 0,63 min. Síntese de 6-(4-(terc-butildimetilsililóxi)ciclo-hexil)-5-flúorpicolinato de metila
[000241] Para uma solução de 5-flúor-6-(4-hidroxiciclo- hexil)picolinato de metila (1,0 equiv.) em DMF (0,15 M) foram adicionados imidazol (4,0 equiv.) e TBDMSCl (2,5 equiv.). A mistura de reação foi agitada à temperatura ambiente por 2 dias, depois adicionada em acetato de etila, lavada com água, salmoura, secada sobre sulfato de magnésio, filtrada e concentrada para dar 6-(4-(terc- butildimetilsililóxi)ciclo-hexil)-5-flúorpicolinato de metila em 97% de rendimento como uma mistura de isômeros (3:1). LC/MS = 368,3 (M+H), Rt = 1,4 e 1,42 min. Síntese de ácido 6-(4-(terc-butildimetilsililóxi)ciclo-hexil)-5- flúorpicolínico
[000242] Para hexil)-5-flúorpicolinato de metila (1,0 equiv.) em THF/MeOH (2:1, 0,09 M) foi adicionado LiOH (1,5 equiv.). A mistura de reação foi agitada durante a noite à temperatura ambiente, depois HCl a 1N e acetato de etila foram adicionados, a fase orgânica foi lavada com salmoura, secada sobre sulfato de magnésio, filtrada e concentrada para dar o ácido 6-(4-(terc-butildimetilsililóxi)ciclo-hexil)-5-flúorpicolínico como uma mistura de isômeros (3:1) em 82% de rendimento. LC/MS = 354,2 (M+H), Rt = 1,38 e 1,41 min. Síntese de ácido 6-bromo-5-flúorpicolínico
[000243] Para 2-bromo-3-flúor-6-metilpiridina (1,0 equiv.) em H2O (30 mL) foi adicionado permanganato de potássio (1,0 equiv.). A solução foi aquecida a 100°C por 5 horas, ao mesmo tempo que mais permanganato de potássio (1,0 equiv.) foi adicionado. Após aquecimento por mais 48 horas, o material foi filtrado através de celite (4 cm x 5 cm (2 polegadas)) e enxaguado com H2O (150 mL). O aquoso combinado foi acidificado com HCl a 1N para pH=4, extraído com acetato de etila (200 mL), lavado com NaCl(sat.), secado sobre MgSO4, filtrado e concentrado para render o ácido 6-bromo- 5-flúorpicolínico (17%) como um sólido branco. LCMS (m/z): 221,9 (MH+); LC Rt = 2,05 min. Método 4 Síntese de 2-(2,6-di-flúorfenil)-3-flúor-6-metilpiridina
[000244] Para uma solução de 2-bromo-3-flúor-6-metilpiridina (1,0 equiv.) em THF e Água (10:1, 0,2 M) foram adicionados ácido 2,6-di- flúorfenilborônico (2,0 equiv.) e fluoreto de potássio (3,3 equiv.). A reação foi desgaseificada por 10 minutos, depois Pd2(dba)3 (0,05 equiv.) foi adicionado, seguido por tri-t-butilfosfina (0,1 equiv.). A reação foi agitada a 60°C por 1 hora ponto no qual todo o material de partida foi consumido como indicado por LC/MS. A reação foi deixada resfriar à temperatura ambiente, dividida com acetato de etila e água, a fase orgânica foi secada com sulfato de sódio, filtrada e concentrada. O material bruto foi diluído em EtOH para 0,1 M, e 0,5 equiv. de NaBH4 foi adicionado para reduzir o dba. A reação foi agitada por uma hora à temperatura ambiente, depois foi extinta com água e concentrada sob vácuo para remover o etanol. O produto foi extraído em éter, lavado com salmoura, os orgânicos foram secados sobre sulfato de sódio, filtrados e concentrados. O material bruto foi carregado em sílica-gel e purificado através da cromatografia de coluna (ISCO) eluindo com hexanos e acetato de etila (0%-10% de acetato de etila). As frações puras foram combinadas e concentradas para render 2-(2,6-di- flúorfenil)-3-flúor-6-metilpiridina como um óleo amarelo-claro em 86% de rendimento. LC/MS = 224,0 (M+H), Rt = 0,84 min. Método 5 Síntese de ácido 6-(2,6-di-flúorfenil)-5-flúorpicolínico
[000245] Para uma solução de 2-(2,6-di-flúorfenil)-3-flúor-6- metilpiridina (1,0 equiv.) em água (0,05 M) foi adicionado KMnO4 (2,0 equiv.), e a reação foi aquecida a refluxo durante a noite. Outros 2,0 equiv. de KMnO4 foram adicionados e agitados a refluxos por mais 8 horas. A solução foi resfriada à temperatura ambiente, filtrada através de Celite e lavada com água. O filtrado foi acidificado com HCl a 6N para pH =3, o precipitado branco foi filtrado. O filtrado foi ainda acidificado para pH = 1 e filtrado novamente. O filtrado foi extraído com acetato de etila até que não tenha mais produto na camada aquosa. A fase orgânica foi lavada com salmoura e secada sobre sulfato de magnésio, filtrada, e concentrada. O resíduo foi dissolvido em acetato de etila, lavado com NaOH a 1N, a camada aquosa foi acidificada para pH=1, e os cristais brancos foram filtrados. Os produtos combinados renderam o ácido 6-(2,6-di-flúorfenil)-5-flúorpicolínico em 32% de rendimento como um sólido branco. LC/MS = 254,0 (M+H), Rt = 0,71 min. Síntese de 6-(2,6-di-flúorfenil)-3-flúor-2-metilpiridina
[000246] Para uma solução de 6-bromo-3-flúor-2-metilpiridina (1,0 equiv.) em etanol e tolueno (1:1, 0,2 M) foram adicionados ácido 2,6- di-flúorfenilborônico, DIEA (5 equiv.) e Pd(PPh3)4 (0,2 equiv.). A reação foi aquecida em micro-ondas a 120°C por 30 min. A solução foi filtrada e enxaguada com acetato de etila. Os voláteis foram removidos in vácuo, e o bruto foi purificado através da cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (2,5-20% de acetato de etila). Após concentração das frações puras, 6-(2,6-di-flúorfenil)-3- flúor-2-metilpiridina foi isolada em 88% de rendimento. LC/MS = 224,1 (M+H), Rt = 0,87 min. Síntese de ácido 6-(2,6-di-flúorfenil)-3-flúorpicolínico
[000247] Método 5 foi seguido usando 6-(2,6-di-flúorfenil)-3-flúor-2- metilpiridina (1,0 equiv.) e permanganato de potássio (6,0 equiv.) para dar o ácido 6-(2,6-di-flúorfenil)-3-flúorpicolínico em 30% de rendimento. LC/MS = 254,1 (M+H), Rt = 0,70 min. Síntese de 2-(2,6-di-flúor-3-metoxifenil)-3-flúor-6-metilpiridina
[000248] Método 4 foi seguido usando 2-bromo-3-flúor-6-metilpiridina (1,0 equiv.) e ácido 2,6-di-flúor-3-metoxifenilborônico (2,0 equiv.) para dar 2-(2,6-di-flúor-3-metoxifenil)-3-flúor-6-metilpiridina em 60% de rendimento. LC/MS = 254,1 (M+H), Rt = 0,85 min. Síntese de ácido 6-(2,6-di-flúor-3-metoxifenil)-5-flúorpicolínico
[000249] Método 5 foi seguido usando 2-(2,6-di-flúor-3-metoxifenil)-3- flúor-6-metilpiridina (1,0 equiv.) e permanganato de potássio (4,0 equiv.) para dar o ácido 6-(2,6-di-flúor-3-metoxifenil)-5-flúorpicolínico em 27% de rendimento. LC/MS = 284,1 (M+H), Rt = 0,75 min. Síntese de 3-flúor-6-metil-2-(2,3,5-triflúorfenil)piridina
[000250] Para uma solução de 2-bromo-3-flúor-6-metilpiridina (1,0 equiv.) em dioxano (0,2 M) foi adicionado ácido 2,3,5- triflúorfenilborônico e Pd(dppf)Cl2-DCM (0,1 equiv.). Carbonato de sódio aquoso (solução 2M, 2,0 equiv.) foi adicionado, e a reação foi aquecida em micro-ondas a 120°C por 15 min. A solução foi dividida ente acetato de etila e NaHCO3 saturado, a fase orgânica foi lavada com salmoura, secada com sulfato de magnésio, filtrada e concentrada. O material bruto foi purificado através da cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos (1:3) para dar 3-flúor-6-metil-2-(2,3,5-triflúorfenil)piridina em 87% de rendimento. LC/MS = 242,1 (M+H), Rt = 0,98 min. Síntese de ácido 5-flúor-6-(2,3,5-triflúorfenil)picolínico
[000251] Para uma solução de 3-flúor-6-metil-2-(2,3,5- triflúorfenil)piridina (1,0 equiv.) em água e t-BuOH (2:1, 0,06 M) foi adicionado permanganato de potássio (10 equiv.), e a solução foi aquecida a 90°C por 5 horas. Após resfriamento à temperatura ambiente, a solução foi filtrada, e o filtrado foi concentrada sob pressão reduzida para render o ácido 5-flúor-6-(2,3,5- triflúorfenil)picolínico em 89% de rendimento. LC/MS = 272,0 (M+H), Rt = 0,80 min. Síntese de 6-bromo-5-flúorpicolinato de metila
[000252] Para uma solução de ácido 6-bromo-5-flúorpicolínico (1,0 equiv.) em metanol (0,2 M) foi adicionado H2SO4 (4,2 equiv.), e a reação foi agitada à temperatura ambiente por duas horas. Após a conclusão da reação conforme monitorado por LC/MS, a reação foi diluída com acetato de etila e extinta lentamente com NaHCO3 aquoso saturado. A reação foi derramada em um funil de decantação e extraída com acetato de etila. A fase orgânica foi secada com sulfato de magnésio, filtrada e concentrada in vacuo para fornecer 6-bromo-5- flúorpicolinato de metila como um sólido branco (>99%). LC/MS = 233,9/235,9 (M+H), Rt = 0,69 min. Método 6 Síntese de 6-(3-(benzilóxi)-2,6-di-flúorfenil)-5-flúorpicolinato de metila
[000253] Para uma solução de 6-bromo-5-flúorpicolinato de metila (1,0 equiv.) em THF e água (10:1, 0,1 M) foram adicionados ácido 3- (benzilóxi)-2,6-di-flúorfenilborônico (2,5 equiv.) e fluoreto de potássio (3,3 equiv.). A reação foi desgaseificada com nitrogênio, depois Pd2(dba)3 (0,25 equiv.) e tri-terc-butilfosfina (0,5 equiv.) foram adicionados e a reação foi aquecida a 80°C por uma hora. Análise LC/MS indicou conversão completa do material de partida do produto. A reação foi resfriada à temperatura ambiente, depois concentrada in vacuo e fundida em sílica-gel. O produto bruto foi purificado por cromatografia rápida ISCO eluindo com acetato de etila e hexanos (0% a 30% de acetato de etila) para fornecer 6-(3-(benzilóxi)-2,6-di- flúorfenil)-5-flúorpicolinato de metila como o produto desejado como um óleo amarelo-claro em 96% de rendimento. LC/MS = 374,0 (M+H), Rt = 1,07 min. Síntese de 6-(3-(benzilóxi)-2,6-di-flúorfenil)picolinato de metila
[000254] Método 6 foi seguido usando 6-bromopicolinato de metila (1,0 equiv.) e ácido 3-(benzilóxi)-2,6-di-flúorfenilborônico (2,5 equiv.) para dar 6-(3-(benzilóxi)-2,6-di-flúorfenil)picolinato de metila como um sólido amarelo-claro em 95% de rendimento. LC/MS = 356,2 (M+H), Rt = 1,03 min. Síntese de 6-(2,6-di-flúor-4-metoxifenil)-5-flúorpicolinato de metila
[000255] Método 6 foi seguido usando 6-bromopicolinato de metila (1,0 equiv.) e ácido 2,6-di-flúor-4-metoxifenilborônico (2,5 equiv.) para dar 6-(2,6-di-flúor-4-metoxifenil)-5-flúorpicolinato de metila como um sólido branco em 85% de rendimento. LC/MS = 298,0 (M+H), Rt = 0,89 min. Síntese de ácido 6-(2,6-di-flúor-4-metoxifenil)-5-flúorpicolínico
[000256] Para uma solução de 6-(2,6-di-flúor-4-metoxifenil)-5- flúorpicolinato de metila (1,0 equiv.) em THF/MeOH (2:1, 0,09 M) foi adicionado LiOH (1,5 equiv.), e a reação foi agitada à temperatura ambiente por 1 hora. A solução foi extinta com HCl a 1N, extraída com acetato de etila, lavada com salmoura, secada com sulfato de sódio, filtrada e concentrada para dar o ácido 6-(2,6-di-flúor-4-metoxifenil)-5- flúorpicolínico em 84% de rendimento. LC/MS = 284,1 (M+H), Rt = 0,76 min. Método 7 Síntese de 6-(2,6-di-flúor-3-hidroxifenil)-5-flúorpicolinato de metila
[000257] Para uma solução de 6-(3-(benzilóxi)-2,6-di-flúorfenil)-5- flúorpicolinato de metila (1,0 equiv.) em metanol (0,1 M) foram adicionados 10% de Pd/C (0,1 equiv.) em acetato de etila. A reação foi colocada sob uma atmosfera de hidrogênio e agitada por 2 horas. Após conclusão, a solução foi filtrada sobre uma almofada de Celite, a almofada foi lavada com metanol, o filtrado foi concentrado in vacuo para dar 6-(2,6-di-flúor-3-hidroxifenil)-5-flúorpicolinato de metila como um óleo cinza em 86% de rendimento. LC/MS = 284,0 (M+H), Rt = 0,90 min. Síntese de 6-(2,6-di-flúor-3-hidroxifenil)picolinato de metila
[000258] Método 7 foi seguido usando 6-(3-(benzilóxi)-2,6-di- flúorfenil)picolinato de metila (1,0 equiv.) para render 6-(2,6-di-flúor-3- hidroxifenil)picolinato de metila como um sólido marrom-claro em 96% de rendimento. LC/MS = 266,0 (M+H), Rt = 0,68 min. Síntese de 6-(2-flúor-5-formilfenil)picolinato de metila
[000259] Para uma solução de 6-bromopicolinato de metila (1,0 equiv.) em DME (0,03 M) em um frasco de micro-ondas foi adicionado Pd(dppf)Cl2-DCM (0,05 equiv.), ácido 2-flúor-5-formilfenilborônico (1,5 equiv.) e 2M Na2CO3 (2 equiv.). Os reagentes foram aquecidos a 120°C por 20 min. Uma mistura do produto desejado e o ácido carboxílico correspondente foram detectados por LC/MS, a reação foi diluída com acetato de etila, lavada com HCl (pH=5), a fase ácida foi extraída com acetato de etila, as camadas orgânicas combinadas foram secadas com sulfato de magnésio, filtradas e concentradas in vacuo para fornecer um sólido marrom-claro. O sólido foi dissolvido em MeOH e tratado com 3 equiv. de TMS-diazometano à temperatura ambiente. Após conversão completa do ácido carboxilico ao éster metílico correspondente, a reação foi concentrada in vácuo, e o material bruto foi purificado através da cromatografia de coluna em sílica-gel (ISCO) eluindo com 30% de acetato de etila em hexanos para fornecer 6-(2-flúor-5-formilfenil)picolinato de metila como um sólido amarelo em 58% de rendimento. LC/MS = 260,0 (M+H), Rt = 0,70 min. Síntese de 6-(2-flúor-5-(prop-1-enil)fenil)picolinato de (E)-metila
[000260] Para uma solução de 6-(2-flúor-5-formilfenil)picolinato de metila (1,0 equiv.) em MeOH (0,17 M) foi adicionado brometo de etiltrifenilfosfônio (1,0 equiv.) seguido por metóxido de sódio (1,5 equiv.). A reação foi aquecida a 65°C por 5 horas, depois resfriada à temperatura ambiente e concentrada in vacuo. O material bruto foi purificado através da cromatografia de coluna em sílica-gel (ISCO) eluindo com 50% de acetato de etila em hexanos para fornecer 6-(2- flúor-5-(prop-1-enil)fenil)picolinato de (E)-metila como um sólido branco em 81% de rendimento. LC/MS = 272,0 (M+H), Rt = 0,73 min. Síntese de 6-(2-flúor-5-propilfenil)picolinato de metila
[000261] Para uma solução de 6-(2-flúor-5-(prop-1- enil)fenil)picolinato de (E)-metila (1,0 equiv.) em MeOH (0,04 M) foram adicionados 10% de Pd/C (0,5 equiv.) e a reação foi colocada sob uma atmosfera de hidrogênio e deixada em agitação durante a noite. A mistura foi filtrada sobre uma almofada de Celite e lavada com MeOH. O filtrado foi concentrada in vacuo para fornecer 6-(2-flúor-5- propilfenil)picolinato de metila como um óleo cinza-claro em 97% de rendimento. LC/MS = 274,2 (M+H), Rt = 0,61 min. Síntese de ácido 6-(2-flúor-5-propilfenil)picolínico
[000262] Para uma solução de 6-(2-flúor-5-propilfenil)picolinato de metila (1,0 equiv.) em THF foi adicionado hidróxido de lítio (10 equiv.), e a reação foi agitada à temperatura ambiente por 1 hora. O solvente de THF foi removido in vácuo, e a fase básica restante foi acidificada com HCl concentrado. A camada aquosa foi extraída com acetato de etila (2x), a fase orgânica foi secada com sulfato de sódio, filtrada e concentrada para dar ácido 6-(2-flúor-5-propilfenil)picolínico em 35% de rendimento. LC/MS = 260,2 (M+H), Rt = 0,36 min. Método 8 Síntese de 6-(2,6-di-flúor-3-(triflúormetil-sulfonilóxi)fenil)-5- flúorpicolinato de metila
[000263] Para uma solução de 6-(2,6-di-flúor-3-hidroxifenil)-5- flúorpicolinato de metila (1,0 equiv.) em DCM (0,2 M) foram adicionados DIEA (2,0 equiv.) e 1,1,1-triflúor-N-fenil-N- (triflúormetilsulfonil)metanos-sulfonamida (1,5 equiv.). A reação foi deixada agitar durante a noite à temperatura ambiente. A solução foi extinta com água, a fase orgânica foi secada com sulfato de sódio, e concentrada. O material bruto foi purificado através da cromatografia ISCO eluindo com acetato de etila e hexanos (0-30% de acetato de etila). As frações puras foram concentradas para dar 6-(2,6-di-flúor-3- (triflúormetilsulfonilóxi)fenil)-5-flúorpicolinato de metila como o produto desejado como um óleo claro em 68% de rendimento. LC/MS = 416,1 (M+H), Rt = 1,08 min. Síntese de 6-(2,6-di-flúor-3-(triflúormetilsulfonilóxi)-fenil)picolinato de metila
[000264]Método 8 foi seguido usando 6-(2,6-di-flúor-3- (triflúormetilsulfonilóxi)fenil)-5-flúorpicolinato de metila (1,0 equiv.) para render 6-(2,6-di-flúor-3-(triflúormetilsulfonilóxi)fenil)picolinato de metila como um óleo incolor em >99% de rendimento. LC/MS = 397,9 (M+H), Rt = 1,03 min. Síntese de ácido 6-(2,6-di-flúor-3-metilfenil)-5-flúorpicolínico
[000265] Para uma solução de 6-(2,6-di-flúor-3- (triflúormetilsulfonilóxi) fenil)-5-flúorpicolinato de metila (1,0 equiv.) em dioxano e água (10:1, 0,15 M) foram adicionados ácido metil borônico (3,0 equiv.) e carbonato de potássio (3,0 equiv.). A reação foi desgaseificada com nitrogênio por 10 min, depois Pd(PPh3)4 (0,1 equiv.) foi adicionado à solução e aquecido a 100°C por 3 horas. LC/MS da reação neste ponto indicou conversão completa do produto de ácido carboxílico (M+H = 268). Resfriado à temperatura ambiente e adicionados água e acetato de etila. As duas camadas foram separadas, a fase aquosa foi acidificada com HCl concentrado para pH =1 e extraída com acetato de etila. A fase orgânica foi secada com sulfato de sódio, filtrada e concentrada sob vácuo para dar o ácido 6- (2,6-di-flúor-3-metilfenil)-5-flúorpicolínico como um óleo claro em 97% de rendimento. LC/MS = 268,1 (M+H), Rt = 0,82 min. Síntese de 6-(2,6-di-flúor-3-metilfenil)picolinato de metila
[000266] Para uma solução de 6-(2,6-di-flúor-3- (triflúormetilsulfonilóxi)-fenil)picolinato de metila (1,0 equiv.) em tolueno foi adicionado Pd(dppf)Cl2-DCM (0,1 equiv.) seguido por zinco de dimetila (3 ,0 equiv.). A solução tornou-se de laranja para amarelo brilhante. A reação foi aquecida a 80°C por 2 horas ao mesmo tempo em que a análise de LC/MS indicou conversão completa ao produto. A reação foi resfriada à temperatura ambiente, diluída com acetato de etila e lavada com salmoura. A camada orgânica foi secada com sulfato de magnésio, filtrada e concentrada in vacuo para fornecer 6- (2,6-di-flúor-3-metilfenil)picolinato de metila como um óleo marrom em rendimento quantitativo. LC/MS = 264,0 (M+H), Rt = 0,90 min. Síntese de ácido 6-(2,6-di-flúor-3-metilfenil)picolínico
[000267] Para uma solução de 6-(2,6-di-flúor-3-metilfenil)picolinato de metila (1,0 equiv.) em THF foi adicionado hidróxido de sódio (10 equiv.), e a reação foi agitada por 2 horas. A solução foi diluída com acetato de etila e lavada com NaOH a 1N (2x). As lavagens aquosas básicas combinadas foram combinadas e acidificado com HCl concentrado. A fase aquosa ácida foi extraída com acetato de etila (2x), as camadas orgânicas combinadas foram secadas com sulfato de magnésio, filtradas e concentradas in vacuo para fornecer o ácido 6- (2,6-di-flúor-3-metilfenil)picolínico como um sólido branco em 85% de rendimento. LC/MS = 250,0 (M+H), Rt = 0,76 min. Síntese de ácido 6-(3-etil-2,6-di-flúorfenil)picolínico
[000268] Para uma solução de 6-(2,6-di-flúor-3- (triflúormetilsulfonilóxi)-fenil)picolinato de metila (1,0 equiv.) em tolueno (0,15 M) foi adicionado Pd(dppf)Ch-DCM (0,1 equiv.) seguido por zinco de dietila (3 ,0 equiv.). A solução tornou-se de laranja para amarelo brilhante. A reação foi aquecida a 70°C por 2 horas ao mesmo tempo em que a análise de LC/MS indicou uma mistura de razão 1:3:1 do produto hidrolisado, produto desejado e subproduto desconhecido. A reação foi resfriada à temperatura ambiente, diluída com acetato de etila e lavada com NaOH a 1N (2x). A camada orgânica foi secada sobre sulfato de magnésio, filtrada e concentrada in vacuo para fornecer um óleo marrom. O óleo foi redissolvido em THF e tratado com NaOH a 1N por uma hora. A reação foi então diluída com acetato de etila e lavada com NaOH a 1N (2x). As lavagens básicas foram combinadas, acidificadas com HCl concentrado e extraídas com acetato de etila (3x). A fase orgânica foi secada com sulfato de magnésio, filtrada e concentrada in vacuo para fornecer o ácido 6-(3- etil-2,6-di-flúorfenil)picolínico como um óleo marrom-claro em >99% de rendimento. LC/MS = 264,1 (M+H), Rt = 0,88 min.
[000269] Uma solução homogênea de 1 eq cada amina, ácido carboxílico, HOAT e EDC em DMF, a uma concentração de 0,5 M, foi deixada em espera por 24 horas ao mesmo tempo em que água e acetato de etila foram adicionados. A fase orgânica foi secada com sulfato de sódio e purificada através da cromatografia de coluna em sílica-gel eluindo com acetato de etila e hexanos para dar o produto de amina protegida desejado. Alternativamente, a mistura de reação bruta foi diretamente purificada por HPLC. Após liofilização, o sal de TFA do produto de amina protegida foi obtido. Alternativamente, as frações de HPLC puderam ser adicionadas a EtOAc e Na2CO3 sólido, separadas e lavadas com NaCl(sat.). Após secagem sobre MgSO4, filtragem e remoção dos voláteis in vacuo, o produto de amina protegida foi obtido com uma base livre. Alternativamente, a mistura de reação bruta foi usada para a etapa de desproteção sem purificação adicional.
[000270] Se uma amina protegida N-Boc estiver presente, ela foi removida pelo tratamento com excesso de 4M HCl/ dioxano por 14 horas ou pelo tratamento com 25% de TFA/CH2Cl2 por 2 horas. Após remoção dos voláteis in vacuo, o material foi purificado por RP HPLC rendendo após a liofilização, o produto de amina como o sal de TFA. Alternativamente, as frações de HPLC puderam ser adicionadas a EtOAc e Na2CO3 sólido, separadas e lavadas com NaCl(sat.). Após secagem sobre MgSO4, filtragem e remoção dos voláteis in vacuo, a base livre foi obtida. Após dissolver em MeCN/H2O, adicionar 1 eq. de HCl a 1 N e liofilizar, o sal de HCl do produto de amina foi obtido.
[000271] Se um carbamato cíclico de amino álcool N-Boc1,2 estiver presente, antes da desproteção Boc, o carbamato cíclico poderia ser clivado pelo tratamento com Cs2CO3 (0,5 eq) em etanol a uma concentração de 0,1 M por três horas. Após remoção dos voláteis in vacuo, o grupo amino Boc foi desprotegido como descrito acima.
[000272] Se um grupo N-Boc, OAc estiverem presente, antes da desproteção Boc, o grupo acetato poderia ser clivado pelo tratamento com K2CO3 (2,0 equiv.) em etanol a uma concentração de 0,1 M por 24 horas.
[000273] Se um grupo N-ftalimida estiver presente, a amina foi desprotegida pelo tratamento com hidrazina em MeOH a 65°C por três horas. Após resfriamento e filtragem do precipitado branco, o filtrado foi concentrado e purificado por RP HPLC para render o produto de amino amida.
[000274] Se um éter de TBDMS estiver presente, ele foi desprotegido antes da remoção de Boc pelo tratamento com HCl a 6N, THF, metanol (1:2:1) à temperatura ambiente por 12 h. Após remoção dos voláteis in vacuo, o grupo amino Boc foi desprotegido como descrito acima. Alternativamente, o éter de TBDMS e o grupo Boc puderam ser ambos desprotegidos com HCl a 6N, THF, metanol (1:2:1) se deixados à ta por 24 horas, ou aqeucidos a 60°C por 3 horas.
[000275] Se um grupo OMe estiver presente, ele foi desprotegido pelo tratamento com BBr3 a 1M em DCM (2,0 equiv.) por 24 horas. Água foi adicionada em gotas e os voláteis foram removidos in vacuo. O material foi purificado através da HPLC de fase reversa como descrito acima.
[000276] Se um grupo OBn estiver presente, ele foi desprotegido pelo tratamento com 10% de Pd/C (0,2 equiv.) sob uma atmosfera de hidrogênio em acetato de etila e metanol (1:2). Após conclusão, a reação foi filtrada através de Celite, lavada com metanol, e o filtrado foi concentrado in vacuo. Síntese de(+/-)-3-amino-N-(4-(3-amino-4-hidroxiciclo-hex-1- enil)piridin-3-il)-6-(2,6-di-flúorfenil)picolinamida
[000277] Seguindo o Método 9, butildimetilsililóxi)ciclo-hex-2-enilcarbamato de (+/-)-terc-butila e ácido 3-amino-6-(2,6-di-flúorfenil)picolínico foram acoplados e desprotegidos para render (+/-)-3-amino-N-(4-(3-amino-4-hidroxiciclo-hex-1- enil)piridin-3-il)-6-(2,6-di-flúorfenil)picolinamida como o sal de TFA. LCMS (m/z): 438,2 (MH+), LC Rt = 2,00 min. Síntese de (+/-)-3-amino-N-(4-(3-amino-4-hidroxiciclo-hexil)-piridin-3- il)-6-(2,6-di-flúorfenil)picolinamida
[000278] Seguindo o Método 9, 5-(3-aminopiridin-4-il)-2-(terc- butildimetilsililóxi)ciclo-hexilcarbamato de (+/-)-terc-butila e ácido 3- amino-6-(2,6-di-flúorfenil)picolínico foram acoplados e desprotegidos para render (+/-)-3-amino-N-(4-(3-amino-4-hidroxiciclo-hexil)piridin-3- il)-6-(2,6-di-flúorfenil)picolinamida como o sal de TFA em 18% de rendimento. LCMS (m/z): 440,3 (MH+), LC Rt = 2,04 min.
[000279] Seguindo os procedimentos do Método 9, os seguintes compostos foram preparados:












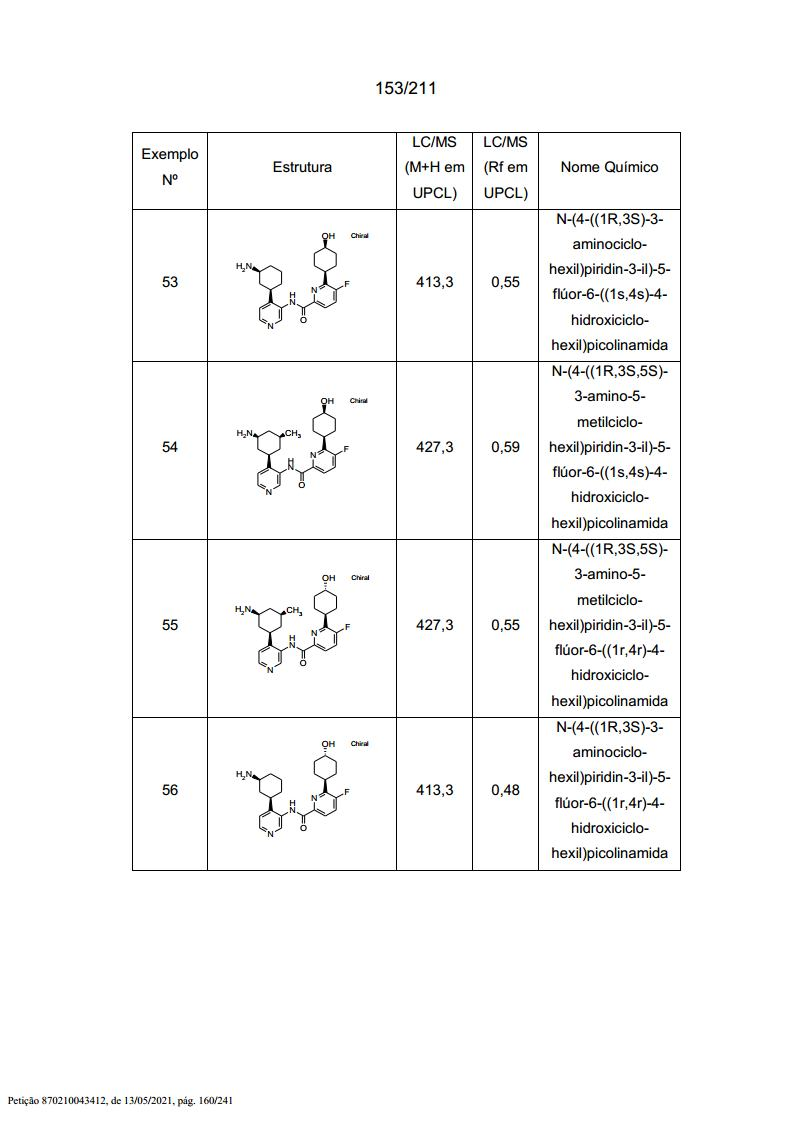












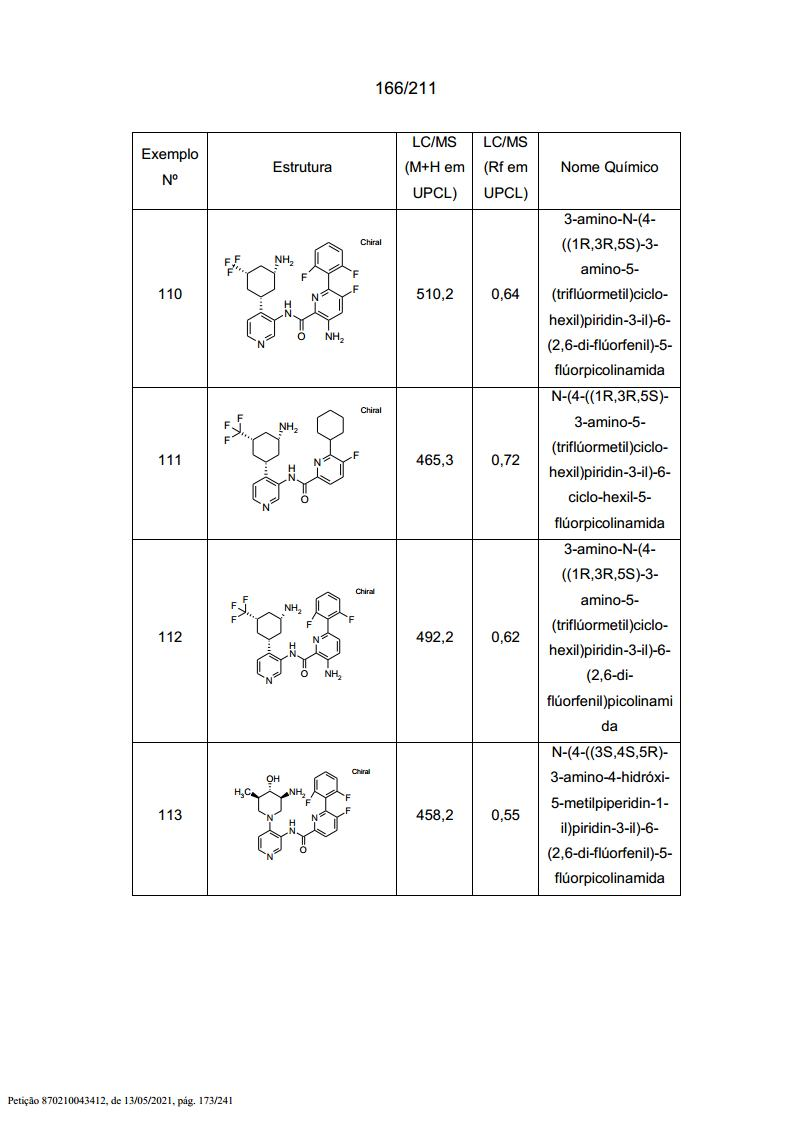

 Síntese de 6-bromo-N-(4-(3-(terc-butildimetilsililóxi)-5-metilciclo-h-hex- 1-enil)piridin-3-il)-5-flúorpicolinamida
Síntese de 6-bromo-N-(4-(3-(terc-butildimetilsililóxi)-5-metilciclo-h-hex- 1-enil)piridin-3-il)-5-flúorpicolinamida
[000280] Seguindo o Método 9, 4-(3-(terc-butildimetilsililóxi)-5- metilciclo-hex-1-enil)piridin-3-amina e ácido 6-bromo-5-flúorpicolínico foram acoplados e seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, 6-bromo-N-(4-(3-(terc- butildimetilsililóxi)-5-metilciclo-hex-1-enil)piridin-3-il)-5-flúorpicolinamida foi obtida. LCMS (m/z): 455,3 (MH+); LC Rt = 2,09 min. Síntese de 6-bromo-N-(4-((1R,3S)-3-(1,3-dioxoisoindolin-2-il)- ciclo-hexil)piridin-3-il)-5-flúorpicolinamida
[000281] Seguindo hexil)isoindolina-1,3-diona e ácido 6-bromo-5-flúorpicolínico foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, 6-bromo-N-(4-((1R,3S)-3-(1,3- dioxoisoindolin-2-il)ciclo-hexil)piridin-3-il)-5-flúorpicolinamida foi obtida. LCMS (m/z): 523,2/525,2 (MH+); LC Rt = 3,31 min. Síntese de 3-amino-6-bromo-N-(4-((1R,3S)-3-(1,3-dioxoisoindolin-2- il)ciclo-hexil)piridin-3-il)-5-flúorpicolinamida
[000282] Seguindo hexil)isoindolina-1,3-diona foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, 3-amino-6-bromo-N-(4-((1R,3S)-3- (1,3-dioxoisoindolin-2-il)ciclo-hexil)piridin-3-il)-5-flúorpicolinamida foi obtida. LCMS (m/z): 538,1/540,1 (MH+); LC Rt = 3,46 min. Síntese de(1S,3R,5S)-3-(3-(6-bromo-5-flúorpicolinamido)- piridin-4-il)-5-metilciclo-hexilcarbamato de terc-butila
[000283] Seguindo o Método 9, (1S,3R,5S)-3-(3-aminopiridin-4-il)-5- metilciclo-hexilcarbamato de terc-butila e ácido 6-bromo-5- flúorpicolínico foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, (1S,3R,5S)-3-(3- (6-bromo-5-flúorpicolinamido)piridin-4-il)-5-metilciclo-hexilcarbamato de terc-butila foi obtido. LCMS (m/z): 507,1/509,1 (MH+), Rt = 0,90 min. Síntese de acetato de (1R,2R,4R,6S)-4-(3-(6-bromo-5- flúorpicolinamido)piridin-4-il)-2-(terc-butoxicarbonilamino)-6-metilciclo- hexila
[000284] Seguindo o Método 9, acetato de (1R,2R,4R,6S)-4-(3- aminopiridin-4-il)-2-(terc-butoxicarbonilamino)-6-metilciclo-hexila e ácido 6-bromo-5-flúorpicolínico foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, acetato de (1R,2R,4R,6S)-4-(3-(6-bromo-5-flúorpicolinamido)piridin-4- il)-2-(terc-butoxicarbonilamino)-6-metilciclo-hexila foi obtido. LCMS (m/z): 567,2 (MH+), Rt = 0,82 min. Síntese de 5-(3-(6-bromo-5-flúorpicolinamido)piridin-4-il)-7-metil-2-oxo- hexa-hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila
[000285] Seguindo o Método 9, 5-(3-aminopiridin-4-il)-7-metil-2-oxo- hexa-hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila e ácido 6-bromo-5-flúorpicolínico foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, 5-(3-(6- bromo-5-flúorpicolinamido)piridin-4-il)-7-metil-2-oxo-hexa- hidrobenzo[d]oxazol-3(2H)-carboxilato de (+/-)-terc-butila foi obtido. LCMS (m/z): 549,2/551,2 (MH+), Rt = 0,78 min. Síntese de 5-(3-(6-bromo-5-flúorpicolinamido)piridin-4-il)-2-oxo-hexa- hidrobenzo[d]oxazol-3(2H)-carboxilato de terc-butila
[000286] Seguindo o Método 9, 5-(3-aminopiridin-4-il)-2-oxo-hexa- hidrobenzo[d]oxazol-3(2H)-carboxilato de terc-butila e ácido 6-bromo- 5-flúorpicolínico foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, 5-(3-(6-bromo-5- flúorpicolinamido)piridin-4-il)-2-oxo-hexa-hidrobenzo[d]oxazol-3(2H)- carboxilato de terc-butila foi obtido. LCMS (m/z): 537,1 (MH+); LCMS Rt = 0,71 min. Síntese de 6-bromo-N-(4-((1R,5R)-5-(1,3-dioxoisoindolin-2-il)-3,3- dimetilciclo-hexil)piridin-3-il)-5-flúorpicolinamida
[000287] Seguindo o Método 9, 2-((1R,5R)-5-(3-aminopiridin-4-il)-3,3- dimetilciclo-hexil)isoindolina-1,3-diona e ácido 6-bromo-5- flúorpicolínico foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, 6-bromo-N-(4- ((1R,5R)-5-(1,3-dioxoisoindolin-2-il)-3,3-dimetilciclo-hexil)piridin-3-il)-5- flúorpicolinamida foi obtido. LCMS (m/z): 551/553 (MH+), Rt = 0,95 min. Síntese de N-(4-((1R,3R,4S,5R)-3,4-bis(terc-butildimetilsililóxi)-5- metilciclo-hexil)piridin-3-il)-6-bromo-5-flúorpicolinamida
[000288] Seguindo o Método 9, 4-((1R,3R,4S,5R)-3,4-bis(terc- butildimetilsililóxi)-5-metilciclo-hexil)piridin-3-amina e ácido 6-bromo- 5-flúorpicolínico foram acoplados e, seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, N-(4- ((1R,3R,4S,5R)-3,4-bis(terc-butildimetilsililóxi)-5-metilciclo-hexil)piridin- 3-il)-6-bromo-5-flúorpicolinamida foi obtido. LCMS (m/z): 652,5, 652,4 (MH+); LC Rt = 5,82 min. Síntese de N-(4-((1S,3S,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5- metilciclo-hexil)piridin-3-il)-6-bromo-5-flúorpicolinamida
[000289] Seguindo o Método 9, butildimetilsililóxi)-5-metilciclo-hexil)piridin-3-amina e ácido 6-bromo- 5-flúorpicolínico foram acoplados e seguindo a adição de EtOAc e lavando com H2O, NaCl(sat.) e secando sobre MgSO4, N-(4- ((1S,3S,4R,5S)-3,4-bis(terc-butildimetilsililóxi)-5-metilciclo-hexil)piridin- 3-il)-6-bromo-5-flúorpicolinamida foi obtido. LCMS (m/z): 652,5, 652,4 (MH+); LC Rt = 5,83 min. Método 10 Síntese de 6-(2,6-di-flúorfenil)-5-flúor-N-(4-(3-hidróxi-5-metilciclo-hex- 1-enil)piridin-3-il)picolinamida
[000290] Uma solução de 6-bromo-N-(4-(3-(terc-butildimetilsililóxi)-5- metilciclo-hex-1-enil)piridin-3-il)-5-flúorpicolinamida (1,0 equiv), ácido 2,6-di-flúorfenil borônico (3,0 equiv.), tetraquistrifenilfosfina (0,2 equiv.) e trietilamina (3,0 equiv.) em 1:1 EtOH/tolueno (0,1 M) foi aquecida a 120 °C com irradiação de micro-ondas por 1200 segundos. Mediante resfriamento, a remoção dos voláteis in vacuo, o produto Suzuki foi diretamente purificado por HPLC de fase reversa. A fração de produto foi liofilizada e o éter de TBDMS resultante foi desprotegido como descrito no Método 9 rendendo, após purificação e liofilização por RP HPLC, 6-(2,6-di-flúorfenil)-5-flúor-N-(4-(3-hidróxi-5-metilciclo-hex-1- enil)piridin-3-il)picolinamida como o sal de TFA. LCMS (m/z): 438,2 (MH+); LC Rt = 2,00 min. Síntese de N-(4-((1R,3S)-3-aminociclo-hexil)piridin-3-il)- 6-(2,6-di-flúorfenil)-5-flúorpicolinamida
[000291] Uma solução de 6-bromo-N-(4-((1R,3S)-3-(1,3- dioxoisoindolin-2-il)ciclo-hexil)piridin-3-il)-5-flúorpicolinamida (1,0 equiv), ácido 2,6-di-flúorfenil borônico (3,0 equiv.), tetraquistrifenilfosfina (0,2 equiv.) e trietilamina (3,0 equiv.) em 1:1 EtOH/tolueno (0,1 M) foi aquecida a 120 °C com irradiação de micro-ondas por 1200 segundos. Mediante resfriamento, a remoção dos voláteis in vacuo, o produto Suzuki foi diretamente purificado por HPLC de fase reversa. A fração de produto foi liofilizada e o grupo ftalimida resultante foi desprotegido como descrito no Método 9 rendendo, após purificação e liofilização por RP HPLC, N-(4-((1R,3S)- 3-aminociclo-hexil)piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida como o sal de TFA. LCMS (m/z): 427,2 (MH+); LC Rt = 2,26 min. Síntese de 3-amino-N-(4-((1R,3S)-3-aminociclo-hexil) piridin-3-il)-6- (2,6-di-flúorfenil)-5-flúorpicolinamida
[000292] Uma solução de 3-amino-6-bromo-N-(4-((1R,3S)-3-(1,3- dioxoisoindolin-2-il)ciclo-hexil)piridin-3-il)-5-flúorpicolinamida (1,0 equiv), ácido 2,6-di-flúorfenil borônico (3,0 equiv.), tetraquistrifenilfosfina (0,2 equiv.) e trietilamina (3,0 equiv.) em 1:1 EtOH/tolueno (0,1 M) foi aquecida a 120 °C com irradiação de micro-ondas por 1200 segundos. Mediante resfriamento, a remoção dos voláteis in vacuo, o produto Suzuki foi diretamente purificado por HPLC de fase reversa. A fração de produto foi liofilizada e o grupo ftalimida resultante foi desprotegido como descrito no Método 9 rendendo, após purificação e liofilização por RP HPLC, 3-amino-N-(4- ((1R,3S)-3-aminociclo-hexil) piridin-3-il)-6-(2,6-di-flúorfenil)-5- flúorpicolinamida como o sal de TFA . LCMS (m/z): 442,2 (MH+); LC Rt = 2,24 min. Síntese de N-(4-(3-amino-4-hidroxiciclo-hexil) piridin-3-il)-6-(2,6-di- flúorfenil)-5-flúorpicolinamida
[000293] Uma solução de 5-(3-(6-bromo-5-flúorpicolinamido) piridin- 4-il)-2-oxo-hexa-hidrobenzo[d]oxazol-3(2H)-carboxilato de terc-butila (1,0 equiv), ácido 2,6-di-flúorfenil borônico (3,0 equiv.), tetraquistrifenilfosfina (0,2 equiv.) e trietilamina (3,0 equiv.) em 1:1 EtOH/tolueno (0,1 M) foi aquecida a 120 °C com irradiação de micro-ondas por 1200 segundos. Mediante resfriamento, a remoção dos voláteis in vacuo, o produto Suzuki foi diretamente purificado por HPLC de fase reversa. A fração de produto foi liofilizada e os grupos carbamato e Boc cíclicos resultantes foram desprotegidos como descrito no método 9 rendendo, após purificação e liofilização por RP HPLC, N-(4-(3-amino-4-hidroxiciclo-hexil) piridin-3-il)-6-(2,6-di- flúorfenil)-5-flúorpicolinamida como o sal de TFA . LCMS (m/z): 443,2 (MH+); LC Rt = 2,11 min. Síntese de 5-amino-N-(4-((1R,3S)-3-aminociclo-hexil) piridin-3-il)-3,3'- diflúor-2,4'-bipiridina-6-carboxamida
[000294] Uma solução de 3-amino-6-bromo-N-(4-((1R,3S)-3-(1,3- dioxoisoindolin-2-il)ciclo-hexil)piridin-3-il)-5-flúorpicolinamida (1,0 equiv), ácido3-flúorpiridin-4-ilborônico (3,0 equiv.), tetraquistrifenilfosfina (0,2 equiv.) e trietilamina (3,0 equiv.) em 1:1 EtOH/tolueno (0,1 M) foi aquecida a 120 °C com irradiação de micro-ondas por 1200 segundos. Mediante resfriamento, a remoção dos voláteis in vacuo, o produto Suzuki foi diretamente purificado por HPLC de fase reversa. A fração de produto foi liofilizada e o grupo ftalimida resultante foi desprotegido como descrito no Método 9 rendendo, após purificação e liofilização por RP HPLC, 5-amino-N-(4- ((1R,3S)-3-aminociclo-hexil) piridin-3-il)-3,3'-diflúor-2,4'-bipiridina-6- carboxamida como o sal de TFA . LCMS (m/z): 425,1 (MH+); LC Rt = 2,08 min. Síntese de N-(4-((1R,3S,5S)-3-amino-5-metilciclo-hexil) piridin-3-il)-6- (2,6-diflúor-3-hidroxifenil)-5-flúorpicolinamida
[000295] A uma solução de (1S,3R,5S)-3-(3-(6-bromo-5- flúorpicolinamido) piridin-4-il)-5-metilciclo-hexilcarbamato de terc-butila (1,0 equiv.) em um frasco de micro-ondas foi adicionado ácido 2,6- diflúor-3-hidroxifenilborônico (5,0 equiv.), KF (5,5 equiv.) e Pd2(dba)3 (0,2 equiv.) seguido por THF e água (10:1, 0,03 M). A esta mistura foi adicionado P(t-Bu)3 (0,4 equiv.) e a reação foi aquecida em microondas a 100 °C por 30 min. A fase orgânica foi a seguir separada, a camada aquosa foi lavada com acetato de etila, e os produtos orgânicos foram combinados e concentrados in vacuo. A mistura bruta foi purificada por meio de prep-HPLC, as frações de produto foram liofilizadas e o grupo BOC resultante foi desprotegido como descrito no Método 9 rendendo, após purificação e liofilização por RP HPLC, N-(4- ((1R,3S,5S)-3-amino-5-metilciclo-hexil)piridin-3-il)-6-(2,6-diflúor-3- hidroxifenil)-5-flúorpicolinamida como o sal de TFA. LCMS (m/z): 457,2 (MH+); LC Rt = 2,17 min.
[000296] Os compostos a seguir foram preparados usando Método 10: TABELA 2


 Exemplo 139 Síntese de N-(4-((1R,3R,4S,5S)-3-amino-4-flúor-5-metilciclo-hexil)- piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida
Exemplo 139 Síntese de N-(4-((1R,3R,4S,5S)-3-amino-4-flúor-5-metilciclo-hexil)- piridin-3-il)-6-(2,6-di-flúorfenil)-5-flúorpicolinamida 
[000297] A uma solução de (1R,2R,3S,5R)-5-(3-(6-(2,6-di-flúorfenil)- 5-flúorpicolinamido)piridin-4-il)-2-hidróxi-3-metilciclo-hexilcarbamato de terc-butila (1,0 equiv.) em DCM (0,04 M) a 0 °C foi adicionado DAST (1,0 equiv.). A reação foi agitada por 1,5 h a 0 °C, a seguir TFA (10 equiv.) foi adicionado à reação. Após 2 h, a reação foi concentrada in vacuo e o resíduo foi purificado por meio de prep-HPLC para render N- (4-((1R,3R,4S,5S)-3-amino-4-flúor-5-metilciclo-hexil)piridin-3-il)-6-(2,6- 'di-flúorfenil)-5-flúorpicolinamida como o sal de TFA. LCMS (m/z): 459,3 (MH+); LC Rt = 2,39 min.
[000298] Em adição à caracterização de LC/MS e LC, compostos representtativos foram analisados por 1H-RMN. A seguir estão os espectros típicos dos compostos da invenção.


[000299] A atividade de PIM1 é medida usando um reagente de detecção de ATP com base em luciferase-luciferina para quantificar a depleção de ATP resultante a partir da transferência de fosforila catalisada por quinase a um substrato de peptídeo. Os compostos a serem testados são dissolvidos em 100% de DMSO e distribuídos diretamente para dentro de placas brancas de 384 poços a 0,5 μl por poço. Para iniciar a reação, 10 μl de Pim1 quinase a 5 nM e peptídeo BAD a 80 μM (RSRHSSIPAGT-OH) em tampão de ensaio (HEPES a 50 mM pH 7,5, MgCl2 a 5 mM, DTT a 1 mM, 0,05% de BSA) são adicionados a cada poço. Após 15 minutos, 10 μl de ATP a 40 μM em tampão de ensaio são adicionados. As concentrações de ensaio finais são PIM1 a 2,5 nM, ATP a 20 μM, peptídeo BAD a 40 μM e 2,5% de DMSO. A reação é realizada até aproximadamente 50% do ATP ser depletado, a seguir parado com a adição de 20 μl de solução de QuinaseGlo Plus (Promega Corporation). A reação parada é incubada por 10 minutos e o ATP restante detectado por meio de luminescência sobre Victor2 (Perkin Elmer). Os compostos dos exemplos anteriores foram testados pelo ensaio de depleção de Pim1 ATP e verificados exibir valores de IC50 como mostrado na Tabela 3, abaixo. IC50, a concentração inibidora máxima média, representa a concentração de um composto de teste que é exibida por 50% de inibição do seu alvo in vitro.
[000300] A atividade de PIM2 é medida usando um reagente de detecção de ATP à base de luciferase-luciferina para quantificar a depleção de ATP resultante da transferência de fosforila catalisada por quinase para um substrato de peptídeo. Os compostos a serem testados são dissolvidos em 100% de DMSO e diretamente distribuídos em placas brancas de 384 poços a 0,5 μl por poço. Para iniciar a reação, 10 μl de Pim2 quinase a 10 nM e peptídeo de BAD a 20 μM (RSRHSSIPAGT-OH) em tampão de ensaio (HEPES a 50 mM pH 7,5, MgCl2 a 5 mM, DTT a 1 mM, 0,05% de BSA) é adicionada a cada poço. Após 15 minutos, 10 μl de ATP a 8 μM em tampão de ensaio são adicionados. As concentrações de ensaio finais são PIM2 a 5 nM, ATP a 4 μM, peptídeo de BAD a 10 μM e 2,5% de DMSO. A reação é realizada até aproximadamente 50% do ATP ser depletada, a seguir parade com a adição de 20 μl de solução de QuinaseGlo Plus (Promega Corporation). A reação parada é incubada por 10 minutos e o ATP restante detectado por meio de luminescência sobre Victor2 (Perkin Elmer). Os compostos dos exemplos anteriores foram testados pelo ensaio de depleção de Pim2 ATP e verificados exibirem valores de IC50 como mostrado na Tabela 3, abaixo.
[000301] A atividade de PIM3 é medida usando um reagente de detecção de ATP à base de luciferase-luciferina para quantificar a depleção de ATP resultante da transferência de fosforila catalisada por quinase a um substrato de peptídeo. Os compostos a serem testados são dissolvidos em 100% de DMSO e diretamente distribuídos em placas brancas de 384 poços a 0,5 μl por poço. Para iniciar a reação, 10 μl de Pim3 quinase a 10 nM e peptídeo de BAD a 200 μM (RSRHSSIPAGT-OH) em tampão de ensaio (HEPES a 50 mM pH 7,5, MgCl2 a 5 mM, DTT a 1 mM, 0,05% de BSA) são adicionados a cada poço. Após 15 minutos, 10 μl de ATP a 80 μM em tampão de ensaio são adicionados. As concentrações de ensaio finais são PIM1 a 5 nM, ATP a 40 μM, peptídeo de BAD a 100 μM e 2,5% de DMSO. A reação é realizada até aproximadamente 50% do ATP ser depletado, a seguir parado pela adição de 20 μl de solução de QuinaseGlo Plus (Promega Corporation). A reação parada é incubada por 10 minutos e o ATP restante detectado por meio de luminescência sobre o Victor2 (Perkin Elmer). Os compostos dos exemplos anteriores foram testados pelo ensaio de depleção de Pim3 ATP e verificados exibirem valores de IC50 como mostrado na Tabela 3, abaixo.
[000302] KMS11s (linhagem celular de mieloma humano) foram cultivados em IMDM suplementado com 10% de FBS, piruvato de sódio e antibiótico. As células foram colocadas em placas no mesmo meio em uma densidade de 2000 células por poço em placas de cultura de tecido de 96 poços, com poços externos vazios, no dia do ensaio. MM1.s (linhagem celular de mieloma humano), foram cultivados em RPMI1640 suplementado com 10% de FBS, piruvato de sódio e antibiótico. As células foram colocadas em placas no mesmo meio em uma densidade de 5000 células por poço em placas de cultura de tecido de 96 poços, poços externos vazios, no dia do ensaio.
[000303] Os compostos de teste supridos em DMSO foram diluídos em DMSO a 500 vezes as concentrações finais desejadas antes da diluição em meios de cultura a 2 vezes as concentrações finais. Os volumes iguais de 2x os compostos foram adicionados às células em placas de 96 poços e incubados a 37 °C por 3 dias.
[000304] Após 3 dias, as placas foram equilibradas até a temperatura ambiente e o volume igual de reagente CellTiter-Glow (Promega) foi adicionado aos poços de cultura. As placas foram agitadas brevemente e o sinal luminescente foi medido com luminômetro. A inibição percentual do sinal vista nas céluals tratadas com DMSO sozinho vs. as células tratadas com composto de controle foi calculada e usada para determinar os valores de EC50 (isto é, a concentração de um composto de teste que é exigida para obter 50% do efeito máximo nas células) para os compostos testados, como mostrado na Tabela 3.
[000305] Usando os procedimentos dos exemplos 140 (ensaio de depleção de Pim1 ATP), 141 (ensaio de depleção de Pim2 ATP), e 142 (ensaio de depleção de Pim3 ATP), a concentração IC50 dos compostos dos exemplos anteriores foi determinada como mostrado na tabela 3 a seguir.
[000306] Usando os procedimentos de Exemplo 143 (ensaio de proliferação de célula ), a concentração EC50 dos compostos dos exemplos foram determinados nas células KMS11 como mostrado na Tabela 3. TABELA 3




















[000307] As células cancerígenas de mieloma múltiplo KMS11-luc, obtidas de Suzanne Trudel (University Health Network, Toronto, Canada), expressam luciferase estável conseguida por transfecção retroviral e foram mantidas em DMEM suplementado com 10% de soro bovino fetal inativado por calor com 1% de glutamina (Invitrogen, Inc.). Os camundongos fêmeas SCID/bg (8-12 semanas de idade, 20-25g, Charles River) foram usados em todos os estudos farmacológicos in vivo. Os camundongos foram alojados e mantidos de acordo com as diretrizes estaduais e federais para o tratamento e cuidado humano de animais de laboratório, e receberam alimento e água ad libitum. As células cancerígenas foram colhidas de culturas de fase mid-log, a contagem de célula viável foi estabelecida com um contador de célula automatizado (Vi-CELL, Beckman-Coulter), e as células foram ressuspensas em partes iguais de HBSS e Matrigel (Invitrogen, Inc.). Dez milhões de células foram injetadas subcutaneamente no flanco direito de cada camundongo. O tratamento com o composto foi iniciado quando o tamanho do tumor atingiu 250-350mm3 para os estudos com PK/PD, e 150-250mm3 para os estudos de eficácia, como volumes de tumor determinados usando o software StudiDirector (StudiLog Systems, Inc.). Todo o tratamento com o composto foi administrado oralmente.
[000308] Para a modulação alvo in vivo em estudos de curso de tempo PK/PD, os camundongos portando o tumor foram administrados uma dose única de veículo ou composto em concentrações diferentes. Em 1, 8 e 24 horas após a dosagem, os tecidos do tumor e as amostras de sangue foram tomados de camundongos individuais. Os tecidos de tumor ressecados foram congelados rapidamente e pulverizados usando um crioalmofariz e pilão resfriados com nitrogênio líquido. Amostras sanguíneas foram tiradas por punctura cardíaca, e plasma foi separado utilizando tubos de centrifugação contendo heparina de lítio e separador de plasma (BD Microtainer). Amostras de tumor congeladas foram lisadas em tampão frio (Meso Scale Discovery) suplementado com inibidor de protease livre de EDTA (Roche), inibidores de fosfatase 1 e 2, e NaF a 1M (Sigma) de acordo com as instruções do fabricante. Após a homogeneízação com um aparelho dounce ou por MagNA Liser (Roche), o sobrenadante claro foi obtido seguindo a centrifugação a 300xg por 30 minutos a 4°C e a concentração de proteína foi determinada por BCA (BioRad). A modulação alvo foi determinada usando o kit de Meso escala fosfo- BadSer112/dúplex total Bad, de acordo com as instruções do fabricante. Brevemente, uma quantidade igual de proteína foi carregada em cada poço de uma placa de 96 poços de Meso escala fosfo-Serina112/dúplex total Bad (Meso Scale Discovery) e as amostras foram incubadas por 30 minutos à temperatura ambiente ou durante a noite a 4°C, agitando. As placas foram lavadas com tampão de lavagem 1x MSD, e anticorpo de detecção Sulfo-Tag foi adicionado aos poços e incubado por 1 hora à temperatura ambiente, agitando. As placas foram lavadas novamente e o analito capturado detectado seguindo a adição de Read Buffer T aos poços. As placas foram lidas em um Instrumento SECTOR Imager 6000 (Meso Scale Discovery). As proporções do sinal de pBad para Bad total foram usadas para corrigir a variabilidade entre as amostras. Os dados mostrados na tabela a seguir expressa o inibição percentual da fosforilação de pBadSer112 em relação à fosforilação de Bad total pelos compostos representativos da invenção, normalizado para o grupo de controle veículo. A extensão de modulação é expressa como o controle percentual relativo ao veículo (n.d., não determinado). Composto de N° do exemplo. 1h 8h 24h
[000309] Para os estudos de eficácia, os camundongos que portam tumor foram randomizados em grupos com variação de volume de tumor equivalente variando de 150-250mm3 utilizando o software StudiDirector (StudiLog Systems, Inc.). Após a randomização, os camundongos foram dosados oralmente diariamente ou duas vezes ao dia em concentrações múltiplas de composto em 200μl de incipiente. O crescimento do tumor e o peso corporal do animal foram medidos pelo menos duas vezes por semana, e observações clínicas diárias foram usadas para monitorar as toxicidades potenciais relacionadas ao tratamento. Os animais foram removidos do estudo se o volume do tumor excedeu 2500mm3, ou se a perda de peso corporal excedeu 20% das medições iniciais.
[000310] A eficácia do composto de Exemplo 99 foi avaliada no modelo de enxerto de KMS11-luc, com camundongos recebendo a administração oral do composto de Exemplo 99 duas vezes ao dia a 50 e 100 mg/kg, e uma vez ao dia a 100 mg/kg por 14 dias. A dosagem foi iniciada quando os tamanhos do tumor alcançaram aproximadamente 250mm3. Como mostrado na figura 1, o composto de Exemplo 99 exibiu efeitos dependentes de dose in vivo, com a inibição do crescimento do tumor observada para 50 mg/kg duas vezes ao dia (92%) e 100 mg/kg duas vezes ao dia (4% de regressão). A administração diária uma só vez de 100 mg/kg foi menos eficaz (65%) do que quando dosado duas vezes ao dia. Estes resultados se correlacionam com a extensão e a magnitude da modulação de pBadSer112, e sugerem que a modulação alvo extensiva e prolongada é necessária para a eficácia máxima.
[000311] A eficácia do composto de Exemplo 70 foi avaliada no modelo de xenoenxerto de KMS11-luc, com camundongos recebendo a administração oral do composto de Exemplo 80 duas vezes ao dia a 25 e 50 mg/kg, e uma vez ao dia a 100 mg/kg por 14 dias. A dosagem foi iniciada quando os tamanhos do tumor alcançaram aproximadamente 225mm3. Como mostrado na figura 2, o composto de Exemplo 70 exibiu efeitos dependentes de dose in vivo, com a inibição do crescimento do tumor observada para 25 mg/kg (65%) e 50 mg/kg (100%). A inibição do crescimento do tumor significante foi também observada para 100 mg/kg uma vez diariamente (84%).
[000312] A eficácia do composto de Exemplo 96 foi avaliada no modelo de xenoenxerto de KMS11-luc, com camundongos recebendo a administração oral do composto de Exemplo 96 duas vezes ao dia a 25 e 50 mg/kg, e uma vez ao dia a 100 mg/kg por 14 dias. A dosagem foi iniciada quando os tamanhos do tumor alcançaram aproximadamente 225mm3. Como mostrado na figura 3, o composto de Exemplo 96 exibiu efeitos dependentes de dose in vivo, com a inibição do crescimento do tumor observada com 25 mg/kg (67%), e 50 mg/kg (96%). A inibição do crescimento do tumor significante foi também observada para 100 mg/kg uma vez diariamente (88%).
[000313] Embora a modalidade preferida da invenção tenha sido ilustrada e descrita, será apreciado que várias mudanças possam ser feitas nela sem se afastar do espírito e escopo da invenção.
Claims (16)
1. Composto, caracterizado pelo fato de que apresenta a Fórmula II, ou um estereoisômero, tautômero, ou sal farmaceuticamente aceitável do mesmo, na qual Y é ciclohexila, substituída com 1 a três substiuintes, os ditos substituintes sendo selecionados dentre hidroxila, amino, C1-4 alquila ou C1-4 haloalquila; R1 é hidrogênio, -NH2 ou halo; R12 são cada, independentemente, hidrogênio ou halo; e R5 é ciclohexila, fenila ou piridila; sendo a dita ciclohexila, a dita fenila e a dita piridila são cada, independentemente, substituída com até três substituintes selecionados dentre halogênio, hidroxila, C14 alquila ou C1-4 alcóxi.
na qual Y é ciclohexila, substituída com 1 a três substiuintes, os ditos substituintes sendo selecionados dentre hidroxila, amino, C1-4 alquila ou C1-4 haloalquila; R1 é hidrogênio, -NH2 ou halo; R12 são cada, independentemente, hidrogênio ou halo; e R5 é ciclohexila, fenila ou piridila; sendo a dita ciclohexila, a dita fenila e a dita piridila são cada, independentemente, substituída com até três substituintes selecionados dentre halogênio, hidroxila, C14 alquila ou C1-4 alcóxi.
2. Composto de Fórmula II, de acordo com a reivindicação 1 , caracterizado pelo fato de que é selecionaod dentre:














3. Composto de Fórmula II, de acordo com a reivindicação 1 caracterizado pelo fato de que é 3-amino-N-(4-((1R,3S)-3- aminociclohexil)piridin-3-il)-6-(2,6-di-flúor-fenil)-5-flúorpicolinamida, apresentando a fórmula: ou um sal farmaceuticamente aceitável do mesmo.
ou um sal farmaceuticamente aceitável do mesmo.
4. Composto de Fórmula II, de acordo com a reivindicação 1, caracterizado pelo fato de que é N-(4-((1R,3R,4R,5S)-3-amino-4- hidróxi-5-metilciclohexil)piridin-3-il)-6-(2,6-di-flúor-fenil)-5- flúorpicolinamida, apresentando a fórmula: ou um sal farmaceuticamente aceitável do mesmo.
ou um sal farmaceuticamente aceitável do mesmo.
5. Composto de Fórmula II, de acordo com a reivindicação 1, caracterizado pelo fato de que é N-(4-((1R,3S,5S)-3-amino-5- metilciclohexil)piridin-3-il)-6-(2,6-di-flúor-fenil)-5-flúorpicolinamida), apresentando a fórmula: ou um sal farmaceuticamente aceitável do mesmo.
ou um sal farmaceuticamente aceitável do mesmo.
6. Composto de Fórmula II, de acordo com a reivindicação 1, caracterizado pelo fato de que é N-(4-((1R,3S)-3- aminociclohexil)piridin-3-il)-6-(2,6-di-flúor-fenil)-5-flúorpicolinamida, apresentando a fórmula: ou um sal farmaceuticamente aceitável do mesmo.
ou um sal farmaceuticamente aceitável do mesmo.
7. Composto de Fórmula II, de acordo com a reivindicação 1, caracterizado pelo fato de que é N-(4-((1R,3R,4S,5S)-3-amino-4- flúor-5-metilciclohexil)piridin-3-il)-6-(2,6-di-flúor-fenil)-5- flúorpicolinamida, apresentando a fórmula: ou um sal farmaceuticamente aceitável do mesmo.
ou um sal farmaceuticamente aceitável do mesmo.
8. Composição farmacêutica, caracterizada pelo fato de que compreende um composto, como definido em qualquer uma das reivindicações 1 a 7.
9. Composição farmacêutica, caracterizada pelo fato de compreendendo um composto, como definido em de qualquer uma das reivindicações 1 a 7, sendo que a referida composição farmacêutica compreende um agente adicional para o tratamento de câncer.
10. Composição farmacêutica, de acordo com a reivindicação 9, caracterizada pelo fato de que o agente adicional é selecionado dentre irinotecano, topotecano, gencitabina, 5-flúoruracil, leucovorina carboplatina, cisplatina, taxanos, tezacitabina, ciclofosfamida, alcalóides vinca, imatinibe (Gleevec), antrumabinas, ruzximab e trituximab.
11. Composição terapêutica, caracterizada pelo fato de que compreende pelo menos um composto, como definido em qualquer uma das reivindicações 1 a 7, em combinação com um ou mais agentes adicionais para o tratamento do câncer.
12. Uso de um composto, como definido em qualquer uma das reivindicações 1 a 7, caracterizado pelo fato de que é para preparação de um medicamento para tratamento de uma condição por modulação da atividade de Integração Provirus de Maloney Quinase (PIM Quinase).
13. Uso, de acordo com a reivindicação 12, caracterizado pelo fato de que a condição é um câncer selecionado de carcinoma dos pulmões, pâncreas, tireóide, ovário, bexiga, mama, próstata ou cólon, melanoma, leucemia mieloide, mieloma múltiplo e eritro leucemia, adenoma viloso do cólon e osteossarcoma.
14. Composição, de acordo com qualquer uma das reivindicações 8 a 11, caracterizada pelo fato de que é para uso no tratamento de uma condição por modulação da atividade de Integração Provirus de Maloney Quinase (PIM Quinase).
15. Composição, de acordo com qualquer uma das reivindicações 8 a 11, caracterizada pelo fato de que a condição é um câncer selecionado de carcinoma dos pulmões, pâncreas, tireóide, ovário, bexiga, mama, próstata ou cólon, melanoma, leucemia mieloide, mieloma múltiplo e eritro leucemia, adenoma viloso do cólon e osteossarcoma.
16. Composição, de acordo com qualquer uma das reivindicações 8 a 11, caracterizada pelo fato de que é para uso no tratamento de distúrbios relacionados com PIM em combinação com pelo menos um agente adicional para o tratamento de câncer.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9366608P | 2008-09-02 | 2008-09-02 | |
| US61/093,666 | 2008-09-02 | ||
| US22566009P | 2009-07-15 | 2009-07-15 | |
| US61/225,660 | 2009-07-15 | ||
| PCT/EP2009/061205 WO2010026124A1 (en) | 2008-09-02 | 2009-08-31 | Picolinamide derivatives as kinase inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| BRPI0918268A2 BRPI0918268A2 (pt) | 2020-11-17 |
| BRPI0918268B1 true BRPI0918268B1 (pt) | 2021-08-03 |
Family
ID=41228825
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0918268-3A BRPI0918268B1 (pt) | 2008-09-02 | 2009-08-31 | Derivados de picolinamida, seu uso, e composição farmacêutica |
Country Status (39)
| Country | Link |
|---|---|
| US (4) | US8329732B2 (pt) |
| EP (1) | EP2344474B1 (pt) |
| JP (2) | JP5412519B2 (pt) |
| KR (1) | KR101345920B1 (pt) |
| CN (3) | CN102203079B (pt) |
| AU (1) | AU2009289319C1 (pt) |
| BR (1) | BRPI0918268B1 (pt) |
| CA (1) | CA2734415C (pt) |
| CL (1) | CL2011000454A1 (pt) |
| CO (1) | CO6351725A2 (pt) |
| CR (1) | CR20110114A (pt) |
| DK (1) | DK2344474T3 (pt) |
| DO (1) | DOP2011000067A (pt) |
| EA (1) | EA020136B1 (pt) |
| EC (1) | ECSP11010859A (pt) |
| ES (1) | ES2551900T3 (pt) |
| GE (1) | GEP20135849B (pt) |
| HK (2) | HK1156627A1 (pt) |
| HN (1) | HN2011000629A (pt) |
| HR (1) | HRP20151410T1 (pt) |
| HU (1) | HUE026381T2 (pt) |
| IL (1) | IL211291A (pt) |
| MA (1) | MA32684B1 (pt) |
| ME (1) | ME01291A (pt) |
| MX (1) | MX2011002365A (pt) |
| MY (1) | MY150136A (pt) |
| NI (1) | NI201100052A (pt) |
| NZ (1) | NZ591449A (pt) |
| PE (1) | PE20110298A1 (pt) |
| PL (1) | PL2344474T3 (pt) |
| PT (1) | PT2344474E (pt) |
| RS (1) | RS54506B1 (pt) |
| SI (1) | SI2344474T1 (pt) |
| SM (1) | SMT201600005B (pt) |
| SV (1) | SV2011003849A (pt) |
| TW (1) | TWI434843B (pt) |
| UY (1) | UY32085A (pt) |
| WO (1) | WO2010026124A1 (pt) |
| ZA (1) | ZA201101118B (pt) |
Families Citing this family (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| UA98449C2 (en) | 2005-12-13 | 2012-05-25 | Инсайт Корпорейшин | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US20140249135A1 (en) * | 2007-03-01 | 2014-09-04 | Novartis Ag | Pim kinase inhibitors and methods of their use |
| RS53245B2 (sr) | 2007-06-13 | 2022-10-31 | Incyte Holdings Corp | Soli inhibitora janus kinaze (r)-3-(4-(7h-pirolo(2,3-d) pirimidin-4-il)-1h-pirazol-1-il)-3-ciklopentilpropan-nitrila |
| EP2342190A1 (en) * | 2008-09-02 | 2011-07-13 | Novartis AG | Bicyclic kinase inhibitors |
| EP2344474B1 (en) * | 2008-09-02 | 2015-09-23 | Novartis AG | Picolinamide derivatives as kinase inhibitors |
| BRPI1012159B1 (pt) | 2009-05-22 | 2022-01-25 | Incyte Holdings Corporation | Compostos derivados de n-(hetero)aril-pirrolidina de pirazol-4-il-pirrolo[2,3-d] pirimidinas e pirrol-3-il-pirrolo[2,3-d] pirimidinas como inibidores de janus cinase, composições farmacêuticas compreendendo os referidos compostos e usos dos mesmos |
| LT2432472T (lt) | 2009-05-22 | 2020-02-10 | Incyte Holdings Corporation | 3-[4-(7h-pirolo[2,3-d]pirimidin-4-il)-1h-pirazol-1-il]oktan- arba heptan-nitrilas, kaip jak inhibitoriai |
| AR078012A1 (es) | 2009-09-01 | 2011-10-05 | Incyte Corp | Derivados heterociclicos de las pirazol-4-il- pirrolo (2,3-d) pirimidinas como inhibidores de la quinasa janus |
| AU2011224484A1 (en) | 2010-03-10 | 2012-09-27 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| EA201290957A1 (ru) * | 2010-04-07 | 2013-04-30 | Ф.Хоффманн-Ля Рош Аг | Пиразол-4-илгетероциклилкарбоксамидные соединения и способы их применения |
| CN103002875B (zh) | 2010-05-21 | 2016-05-04 | 因塞特控股公司 | Jak抑制剂的局部用制剂 |
| US20130109682A1 (en) * | 2010-07-06 | 2013-05-02 | Novartis Ag | Cyclic ether compounds useful as kinase inhibitors |
| ES2536415T3 (es) | 2010-11-19 | 2015-05-25 | Incyte Corporation | Pirrolopiridinas y pirrolopirimidinas sustituidas heterocíclicas como inhibidores de JAK |
| PE20140146A1 (es) | 2010-11-19 | 2014-02-06 | Incyte Corp | Derivados de pirrolopiridina y pirrolopirimidina sustituidos con ciclobutilo como inhibidores de jak |
| WO2012109749A1 (en) * | 2011-02-14 | 2012-08-23 | The Governors Of The University Of Alberta | Boronic acid catalysts and methods of use thereof for activation and transformation of carboxylic acids |
| UY33930A (es) * | 2011-03-04 | 2012-10-31 | Novartis Ag | Inhibidores novedosos de quinasas |
| US20120225061A1 (en) * | 2011-03-04 | 2012-09-06 | Matthew Burger | Tetrasubstituted cyclohexyl compounds as kinase inhibitors |
| MX2013012661A (es) | 2011-04-29 | 2014-03-27 | Amgen Inc | Compuestos de piridazina biciclicos como inhibidores pim. |
| AR086983A1 (es) | 2011-06-20 | 2014-02-05 | Incyte Corp | Derivados de azetidinil fenil, piridil o pirazinil carboxamida como inhibidores de jak |
| WO2013013188A1 (en) | 2011-07-21 | 2013-01-24 | Tolero Pharmaceuticals, Inc. | Heterocyclic protein kinase inhibitors |
| US9458151B2 (en) * | 2011-08-11 | 2016-10-04 | Jikai Biosciences, Inc. | Isothiazole derivatives as PIM kinase inhibitors and preparation methods and use in medicinal manufacture thereof |
| US9453003B2 (en) * | 2011-08-11 | 2016-09-27 | Jikai Biosciences, Inc. | Pyrimidine derivatives as PIM kinase inhibitors and preparation methods and use in medicinal manufacture thereof |
| TW201313721A (zh) | 2011-08-18 | 2013-04-01 | Incyte Corp | 作為jak抑制劑之環己基氮雜環丁烷衍生物 |
| UA111854C2 (uk) | 2011-09-07 | 2016-06-24 | Інсайт Холдінгс Корпорейшн | Способи і проміжні сполуки для отримання інгібіторів jak |
| WO2013173720A1 (en) | 2012-05-18 | 2013-11-21 | Incyte Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as jak inhibitors |
| KR20150013548A (ko) * | 2012-05-21 | 2015-02-05 | 노파르티스 아게 | 키나제 억제제로서의 신규 고리-치환된 n-피리디닐 아미드 |
| WO2014033631A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | N-(3-pyridyl) biarylamides as kinase inhibitors |
| WO2014033630A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | Novel aminothiazole carboxamides as kinase inhibitors |
| WO2014048939A1 (en) | 2012-09-26 | 2014-04-03 | F. Hoffmann-La Roche Ag | Cyclic ether pyrazol-4-yl-heterocyclyl-carboxamide compounds and methods of use |
| CN103724301A (zh) * | 2012-10-10 | 2014-04-16 | 上海特化医药科技有限公司 | (2r)-2-脱氧-2,2-二取代-1,4-核糖内酯及其制备方法和用途 |
| MY191357A (en) | 2012-11-15 | 2022-06-19 | Incyte Holdings Corp | Sustained-release dosage forms of ruxolitinib |
| US20150336960A1 (en) | 2012-12-19 | 2015-11-26 | Novartis Ag | Aryl-substituted fused bicyclic pyridazine compounds |
| WO2014110574A1 (en) * | 2013-01-14 | 2014-07-17 | Incyte Corporation | Bicyclic aromatic carboxamide compounds useful as pim kinase inhibitors |
| ME03780B (me) | 2013-01-15 | 2021-04-20 | Incyte Holdings Corp | Jedinjenja tiazolkarboksamida i piridinkarboksamida korisna kao inhibitori pim kinaze |
| UA120162C2 (uk) | 2013-03-06 | 2019-10-25 | Інсайт Холдінгс Корпорейшн | Способи і проміжні сполуки при отриманні інгібітора jak |
| US9113629B2 (en) * | 2013-03-15 | 2015-08-25 | Dow Agrosciences Llc | 4-amino-6-(4-substituted-phenyl)-picolinates and 6-amino-2-(4-substituted-phenyl)-pyrimidine-4-carboxylates and their use as herbicides |
| PT3030227T (pt) | 2013-08-07 | 2020-06-25 | Incyte Corp | Formas de dosagem de libertação prolongada para um inibidor de jak1 |
| US20160175293A1 (en) * | 2013-08-08 | 2016-06-23 | Novartis Ag | Pim kinase inhibitor combinations |
| PE20160532A1 (es) | 2013-08-23 | 2016-05-21 | Incyte Corp | Compuesto de carboxamida de furo y tienopiridina utiles como inhibidores de cinasas pim |
| EP3757130A1 (en) | 2013-09-26 | 2020-12-30 | Costim Pharmaceuticals Inc. | Methods for treating hematologic cancers |
| EP3074043A1 (en) * | 2013-11-27 | 2016-10-05 | Novartis AG | Combination therapy comprising an inhibitor of jak, cdk and pim |
| JOP20200094A1 (ar) | 2014-01-24 | 2017-06-16 | Dana Farber Cancer Inst Inc | جزيئات جسم مضاد لـ pd-1 واستخداماتها |
| JOP20200096A1 (ar) | 2014-01-31 | 2017-06-16 | Children’S Medical Center Corp | جزيئات جسم مضاد لـ tim-3 واستخداماتها |
| CU24481B1 (es) | 2014-03-14 | 2020-03-04 | Immutep Sas | Moléculas de anticuerpo que se unen a lag-3 |
| KR20160127140A (ko) | 2014-03-18 | 2016-11-02 | 에프. 호프만-라 로슈 아게 | 옥세판-2-일-피라졸-4-일-헤테로사이클릴-카복사마이드 화합물 및 사용 방법 |
| US9498467B2 (en) | 2014-05-30 | 2016-11-22 | Incyte Corporation | Treatment of chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) by inhibitors of JAK1 |
| US9580418B2 (en) | 2014-07-14 | 2017-02-28 | Incyte Corporation | Bicyclic aromatic carboxamide compounds useful as Pim kinase inhibitors |
| WO2016010897A1 (en) | 2014-07-14 | 2016-01-21 | Incyte Corporation | Bicyclic heteroaromatic carboxamide compounds useful as pim kinase inhibitors |
| JO3589B1 (ar) | 2014-08-06 | 2020-07-05 | Novartis Ag | مثبطات كيناز البروتين c وطرق استخداماتها |
| JP6681905B2 (ja) | 2014-09-13 | 2020-04-15 | ノバルティス アーゲー | Alk阻害剤の併用療法 |
| AU2015327868A1 (en) | 2014-10-03 | 2017-04-20 | Novartis Ag | Combination therapies |
| MA41044A (fr) | 2014-10-08 | 2017-08-15 | Novartis Ag | Compositions et procédés d'utilisation pour une réponse immunitaire accrue et traitement contre le cancer |
| CR20170143A (es) | 2014-10-14 | 2017-06-19 | Dana Farber Cancer Inst Inc | Moléculas de anticuerpo que se unen a pd-l1 y usos de las mismas |
| TN2017000375A1 (en) | 2015-03-10 | 2019-01-16 | Aduro Biotech Inc | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
| US9540347B2 (en) | 2015-05-29 | 2017-01-10 | Incyte Corporation | Pyridineamine compounds useful as Pim kinase inhibitors |
| PT3317301T (pt) | 2015-07-29 | 2021-07-09 | Novartis Ag | Terapias de associação compreendendo moléculas de anticorpo contra lag-3 |
| WO2017019896A1 (en) | 2015-07-29 | 2017-02-02 | Novartis Ag | Combination therapies comprising antibody molecules to pd-1 |
| US20180207273A1 (en) | 2015-07-29 | 2018-07-26 | Novartis Ag | Combination therapies comprising antibody molecules to tim-3 |
| TWI734699B (zh) | 2015-09-09 | 2021-08-01 | 美商英塞特公司 | Pim激酶抑制劑之鹽 |
| CN105254624B (zh) * | 2015-09-18 | 2019-08-09 | 上海吉铠医药科技有限公司 | 异噻唑衍生物pim激酶抑制剂及其制备方法与在制药中的应用 |
| CN105130959B (zh) * | 2015-09-18 | 2018-08-03 | 上海吉铠医药科技有限公司 | 嘧啶衍生物pim激酶抑制剂及其制备方法与在制药中的应用 |
| TW201718546A (zh) | 2015-10-02 | 2017-06-01 | 英塞特公司 | 適用作pim激酶抑制劑之雜環化合物 |
| SG11201803520PA (en) | 2015-11-03 | 2018-05-30 | Janssen Biotech Inc | Antibodies specifically binding pd-1 and their uses |
| AU2016369537B2 (en) | 2015-12-17 | 2024-03-14 | Novartis Ag | Antibody molecules to PD-1 and uses thereof |
| CN107522695B (zh) * | 2016-06-21 | 2018-09-14 | 上海方予健康医药科技有限公司 | 一种pim激酶抑制剂的盐酸盐及其制备方法和用途 |
| CN107522696B (zh) * | 2016-06-21 | 2019-02-19 | 上海方予健康医药科技有限公司 | 一种嘧啶类化合物的盐酸盐及其制备方法和用途 |
| CN112043712B (zh) * | 2016-06-21 | 2022-04-29 | 上海方予健康医药科技有限公司 | 嘧啶化合物的盐酸盐在制备用于治疗与flt3相关的疾病或障碍的药物中的应用 |
| WO2018009466A1 (en) | 2016-07-05 | 2018-01-11 | Aduro Biotech, Inc. | Locked nucleic acid cyclic dinucleotide compounds and uses thereof |
| UY37695A (es) | 2017-04-28 | 2018-11-30 | Novartis Ag | Compuesto dinucleótido cíclico bis 2’-5’-rr-(3’f-a)(3’f-a) y usos del mismo |
| WO2018237173A1 (en) | 2017-06-22 | 2018-12-27 | Novartis Ag | ANTIBODY MOLECULES DIRECTED AGAINST CD73 AND CORRESPONDING USES |
| KR102669173B1 (ko) | 2017-06-30 | 2024-05-23 | 브리스톨-마이어스 스큅 컴퍼니 | 치환된 퀴놀리닐시클로헥실프로판아미드 화합물 및 그의 개선된 제조 방법 |
| WO2019113487A1 (en) | 2017-12-08 | 2019-06-13 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| KR20200129099A (ko) | 2018-01-30 | 2020-11-17 | 인사이트 코포레이션 | (1-(3-플루오로-2-(트리플루오로메틸)이소니코티닐)피페리딘-4-온)의 제조 방법 |
| SI3762368T1 (sl) | 2018-03-08 | 2022-06-30 | Incyte Corporation | Aminopirazin diolne spojine kot zaviralci PI3K-y |
| CN112423759A (zh) | 2018-03-30 | 2021-02-26 | 因赛特公司 | 使用jak抑制剂治疗化脓性汗腺炎 |
| MX2020010556A (es) | 2018-04-13 | 2021-03-02 | Sumitomo Pharma Oncology Inc | Inhibidores de cinasa de insercion proviral en linfomas murinos (pim) para el tratamiento de neoplasias mieloproliferativas y fibrosis asociadas con cancer. |
| AR126019A1 (es) | 2018-05-30 | 2023-09-06 | Novartis Ag | Anticuerpos frente a entpd2, terapias de combinación y métodos de uso de los anticuerpos y las terapias de combinación |
| US11046658B2 (en) | 2018-07-02 | 2021-06-29 | Incyte Corporation | Aminopyrazine derivatives as PI3K-γ inhibitors |
| MX2021009371A (es) | 2019-02-12 | 2021-09-10 | Sumitomo Pharma Oncology Inc | Formulaciones que comprenden inhibidores de proteina cinasa heterociclicos. |
| CN110078706B (zh) * | 2019-05-31 | 2022-02-01 | 浙江师范大学 | 一种伊马替尼衍生物及其制备方法和用途 |
| CN110452164B (zh) * | 2019-09-10 | 2022-07-22 | 上海皓鸿生物医药科技有限公司 | Pim447关键中间体的制备方法 |
| EP4031578A1 (en) | 2019-09-18 | 2022-07-27 | Novartis AG | Entpd2 antibodies, combination therapies, and methods of using the antibodies and combination therapies |
| US20230015914A1 (en) * | 2019-11-07 | 2023-01-19 | Crinetics Pharmaceuticals, Inc. | Melanocortin subtype-2 receptor (mc2r) antagonists and uses thereof |
| US11833155B2 (en) | 2020-06-03 | 2023-12-05 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| CN112915086B (zh) * | 2021-01-27 | 2022-04-12 | 广州市力鑫药业有限公司 | 一种含有Akt靶向激酶抑制剂的药物组合物 |
| WO2024097653A1 (en) | 2022-10-31 | 2024-05-10 | Sumitomo Pharma America, Inc. | Pim1 inhibitor for treating myeloproliferative neoplasms |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001055155A1 (fr) | 2000-01-26 | 2001-08-02 | Meiji Seika Kaisha, Ltd. | Nouveaux derives de carbapenem de type sel quaternaire |
| CA2397493A1 (en) | 2000-01-27 | 2001-08-02 | Cytovia, Inc. | Substituted nicotinamides and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| US8618085B2 (en) | 2000-04-28 | 2013-12-31 | Koasn Biosciences Incorporated | Therapeutic formulations of desoxyepothilones |
| MXPA03008560A (es) | 2001-03-22 | 2004-06-30 | Abbot Gmbh & Co Kg | Pirazolopirimidinas como agentes terapeuticos. |
| KR100557093B1 (ko) | 2003-10-07 | 2006-03-03 | 한미약품 주식회사 | 다약제 내성 저해 활성을 갖는 테트라졸 유도체 및 그의제조방법 |
| TW200523252A (en) | 2003-10-31 | 2005-07-16 | Takeda Pharmaceutical | Pyridine compounds |
| AU2004297235A1 (en) | 2003-12-04 | 2005-06-23 | Vertex Pharmaceuticals Incorporated | Quinoxalines useful as inhibitors of protein kinases |
| US7459562B2 (en) | 2004-04-23 | 2008-12-02 | Bristol-Myers Squibb Company | Monocyclic heterocycles as kinase inhibitors |
| US20060004197A1 (en) | 2004-07-02 | 2006-01-05 | Thomas Thrash | Sulfonamide-based compounds as protein tyrosine kinase inhibitors |
| CR9465A (es) | 2005-03-25 | 2008-06-19 | Surface Logix Inc | Compuestos mejorados farmacocineticamente |
| DK1910384T3 (da) | 2005-08-04 | 2012-12-17 | Sirtris Pharmaceuticals Inc | Imidazo [2,1-b] thiazol-derivater som sirtuinmodulerende forbindelser |
| US8053454B2 (en) | 2005-10-06 | 2011-11-08 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PIM-1 and/or PIM-3 |
| GB0520657D0 (en) * | 2005-10-11 | 2005-11-16 | Ludwig Inst Cancer Res | Pharmaceutical compounds |
| HRPK20050957B3 (en) * | 2005-11-11 | 2008-09-30 | Džanko Nikša | Collapsible hanger |
| US9206142B2 (en) | 2006-10-31 | 2015-12-08 | Merck Sharp & Dohme Corp. | Anilinopiperazine derivatives and methods of use thereof |
| US8318735B2 (en) | 2006-10-31 | 2012-11-27 | Merck Sharp & Dohme Corp. | 2-aminothiazole-4-carboxylic amides as protein kinase inhibitors |
| WO2008106692A1 (en) * | 2007-03-01 | 2008-09-04 | Novartis Vaccines And Diagnostics, Inc. | Pim kinase inhibitors and methods of their use |
| BRPI0814441A2 (pt) | 2007-07-19 | 2015-07-14 | Schering Corp | Compostos de amida heterocíclica como inibidores de proteína cinase |
| EP2195286A2 (en) | 2007-09-10 | 2010-06-16 | Cipla Limited | Process for the preparation of a raf kinase inhibitor and intermediates for use in the process |
| UY31679A1 (es) * | 2008-03-03 | 2009-09-30 | Inhibidores de cinasa pim y metodos para su uso | |
| EP2344474B1 (en) | 2008-09-02 | 2015-09-23 | Novartis AG | Picolinamide derivatives as kinase inhibitors |
| WO2011016234A1 (en) | 2009-08-04 | 2011-02-10 | Raqualia Pharma Inc. | Picolinamide derivatives as ttx-s blockers |
| EP2665711A1 (en) | 2011-01-21 | 2013-11-27 | Abbvie Inc. | Picolinamide inhibitors of kinases |
-
2009
- 2009-08-31 EP EP09782396.7A patent/EP2344474B1/en active Active
- 2009-08-31 EA EA201100425A patent/EA020136B1/ru not_active IP Right Cessation
- 2009-08-31 MY MYPI2011000927A patent/MY150136A/en unknown
- 2009-08-31 PE PE2011000444A patent/PE20110298A1/es active IP Right Grant
- 2009-08-31 CN CN200980143187.8A patent/CN102203079B/zh active Active
- 2009-08-31 RS RS20150730A patent/RS54506B1/en unknown
- 2009-08-31 KR KR1020117007556A patent/KR101345920B1/ko active IP Right Grant
- 2009-08-31 MX MX2011002365A patent/MX2011002365A/es active IP Right Grant
- 2009-08-31 NZ NZ591449A patent/NZ591449A/xx not_active IP Right Cessation
- 2009-08-31 GE GEAP2009012163 patent/GEP20135849B/en unknown
- 2009-08-31 WO PCT/EP2009/061205 patent/WO2010026124A1/en active Application Filing
- 2009-08-31 ES ES09782396.7T patent/ES2551900T3/es active Active
- 2009-08-31 ME MEP-2011-37A patent/ME01291A/me unknown
- 2009-08-31 PL PL09782396T patent/PL2344474T3/pl unknown
- 2009-08-31 BR BRPI0918268-3A patent/BRPI0918268B1/pt active IP Right Grant
- 2009-08-31 US US12/584,158 patent/US8329732B2/en active Active
- 2009-08-31 CN CN201410461060.3A patent/CN104311480A/zh active Pending
- 2009-08-31 AU AU2009289319A patent/AU2009289319C1/en active Active
- 2009-08-31 JP JP2011524408A patent/JP5412519B2/ja active Active
- 2009-08-31 DK DK09782396.7T patent/DK2344474T3/en active
- 2009-08-31 PT PT97823967T patent/PT2344474E/pt unknown
- 2009-08-31 CN CN2013102514039A patent/CN103333157A/zh active Pending
- 2009-08-31 HU HUE09782396A patent/HUE026381T2/en unknown
- 2009-08-31 SI SI200931330T patent/SI2344474T1/sl unknown
- 2009-08-31 CA CA2734415A patent/CA2734415C/en active Active
- 2009-09-01 UY UY0001032085A patent/UY32085A/es not_active Application Discontinuation
- 2009-09-01 TW TW098129413A patent/TWI434843B/zh not_active IP Right Cessation
-
2011
- 2011-02-11 ZA ZA2011/01118A patent/ZA201101118B/en unknown
- 2011-02-17 IL IL211291A patent/IL211291A/en active IP Right Grant
- 2011-03-01 NI NI201100052A patent/NI201100052A/es unknown
- 2011-03-01 CL CL2011000454A patent/CL2011000454A1/es unknown
- 2011-03-02 EC EC2011010859A patent/ECSP11010859A/es unknown
- 2011-03-02 DO DO2011000067A patent/DOP2011000067A/es unknown
- 2011-03-02 HN HN2011000629A patent/HN2011000629A/es unknown
- 2011-03-02 CR CR20110114A patent/CR20110114A/es unknown
- 2011-03-03 SV SV2011003849A patent/SV2011003849A/es unknown
- 2011-03-08 CO CO11028419A patent/CO6351725A2/es active IP Right Grant
- 2011-04-01 MA MA33733A patent/MA32684B1/fr unknown
- 2011-10-17 HK HK11111043.1A patent/HK1156627A1/xx not_active IP Right Cessation
- 2011-12-15 US US13/327,358 patent/US8592455B2/en active Active
-
2012
- 2012-03-06 HK HK12102246.4A patent/HK1162022A1/xx not_active IP Right Cessation
-
2013
- 2013-06-25 JP JP2013132978A patent/JP5813701B2/ja active Active
- 2013-11-15 US US14/081,132 patent/US9079889B2/en active Active
-
2015
- 2015-07-13 US US14/797,534 patent/US20150315150A1/en not_active Abandoned
- 2015-12-22 HR HRP20151410TT patent/HRP20151410T1/hr unknown
-
2016
- 2016-01-05 SM SM201600005T patent/SMT201600005B/it unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9079889B2 (en) | Kinase inhibitors and methods of their use | |
| JP5412448B2 (ja) | Pimキナーゼ阻害剤およびその使用方法 | |
| EP2861585B1 (en) | Novel ring-substituted n-pyridinyl amides as kinase inhibitors | |
| US8829193B2 (en) | PIM kinase inhibitors and methods of their use | |
| JP2012501312A (ja) | 二環式キナーゼ阻害剤 | |
| KR20110056399A (ko) | 헤테로시클릭 pim-키나제 억제제 | |
| WO2014033631A1 (en) | N-(3-pyridyl) biarylamides as kinase inhibitors | |
| WO2014033630A1 (en) | Novel aminothiazole carboxamides as kinase inhibitors | |
| AU2011265439B2 (en) | Picolinamide derivatives as kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] |
Free format text: DE ACORDO COM O ARTIGO 229-C DA LEI NO 10196/2001, QUE MODIFICOU A LEI NO 9279/96, A CONCESSAO DA PATENTE ESTA CONDICIONADA A ANUENCIA PREVIA DA ANVISA. CONSIDERANDO A APROVACAO DOS TERMOS DO PARECER NO 337/PGF/EA/2010, BEM COMO A PORTARIA INTERMINISTERIAL NO 1065 DE 24/05/2012, ENCAMINHA-SE O PRESENTE PEDIDO PARA AS PROVIDENCIAS CABIVEIS. |
|
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06U | Preliminary requirement: requests with searches performed by other patent offices: procedure suspended [chapter 6.21 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 31/08/2009, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF, QUE DETERMINA A ALTERACAO DO PRAZO DE CONCESSAO. |