CAMPO TÉCNICO
A presente invenção refere-se, em geral, ao campo de sistemas de liberação controlada e, mais particularmente, aos sistemas de liberação controlada contendo um agente ativo capaz de fornecer um efeito anestésico localizado, onde os sistemas são adequados para uso em conexão com tratamentos cirúrgicos e médicos, e como medicamentos para uso em procedimentos de recuperação pós-operatória.
FUNDAMENTOS DA INVENÇÃO
Sistemas biodegradáveis de liberação controlada para agentes ativos são bastante conhecidos na técnica. Carreadores biodegradáveis para liberação de medicamento são úteis porque eles obviam a necessidade de remoção do dispositivo de depleção de medicamento.
Os materiais carreadores mais comuns usados para os sistemas de liberação controlados são polímeros. O campo dos polímeros biodegradáveis tem se desenvolvido rapidamente desde que a síntese e a biodegradabilidade do ácido polilático foi relatado por Kulkarni et al. (1966). Arch. Surg. 93:839. Exemplos de outros polímeros que foram relatados como úteis como um material de matriz para sistemas de liberação controlados incluem polianidridos, poliésteres, tais como, poliglicolídeos e polilactídeo-co- glicolídeos, poliaminoácidos, tais como, polilisina, polímeros e copolímeros de óxido de polietileno, óxido de polietileno terminado em acrílico, poliamidas, poliuretanos, poliortoésteres, poliacrilonitrilas e polifosfazenos. Vide, por exemplo, Patente U.S. N- 4.891.225 e 4.906.474 (polianidridos); 4.767.628 (polilactídeo, ácido de polilactídeo-co-glicolídeo); 4.530.840 (polilactídeo, poliglicolídeo e copolímeros); e 5.234.520 (polímeros biodegradáveis para liberação controlada no tratamento de doença periodontal).
Os materiais degradáveis de origem biológica são bem conhecidos conhecidos incluindo, por exemplo, gelatina reticulada. Ácido hialurônico foi reticulado e usado como um polímero de intumescimento degradável para aplicações biomédicas (ver, por exemplo, Patente U.S. 4.957.744 e Delia Valle et al. (1991) Polim. Mater. Sci. Eng., 62: 731-735).
Os hidrogéis biodegradáveis também foram desenvolvidos para o uso em sistemas de liberação controlada e servem como carreadores de materiais biologicamente ativos tais como hormônios, enzimas, antibióticos, agentes antineoplásticos e suspensões celulares. Ver, por exemplo, a Patente U.S. N- 5.149.543.
As composições de hidrogel também são habitualmente usadas como substratos para a cultura de célula e tecido, materiais de impressão para protéticos, materiais de pensar ferimento ou como materiais de fase sólida em aplicações na cromatografla por exclusão de tamanho ou afinidade. Por exemplo, composições de hidrogel de agarose não porosas, deformadas e/ou derivadas foram usadas em métodos de cromatografia líquida de alto desempenho e cromatografia de afinidade (Li et al. (1990) Preparative Biochem. 20: 107-121) e pérolas de hidrogel de agarose superporosa foram usadas como um suporte na cromatografia de interação hidrofóbica (Gustaysson et al. (1999) J. Chromatography 830: 275-284).
Muitos sistemas de dispersão também estão correntemente em uso como carreadores de substâncias, particularmente compostos biologicamente ativos. Os sistemas de dispersão usados para as formulações farmacêuticas e cosméticas podem ser categorizados como suspensões ou emulsões. As suspensões são compreendidas de partículas sólidas variando no tamanho de uns poucos nanômetros até centenas de microns, dispersos em um meio líquido usando agentes de suspensão. As partículas sólidas incluem microesferas, microcápsulas e nanoesferas. As emulsões são no geral dispersões de um líquido em um outro estabilizado por uma película interfacial de emulsificadores tais como tensoativos e lipídeos. As formulações de emulsão incluem emulsões de água em óleo e óleo em água, emulsões múltiplas, microemulsões, microgotículas e lipossomas. Microgotículas são vesículas de fosfolipídeo unilamelares que consistem de uma camada lipídica esférica com uma fase oleosa dentro, por exemplo, 5 daquela descrita nas Patentes U.S. Na 4.622.219 e 4.725.442. Os lipossomas são vesículas de fosfolipídeo preparadas misturando-se lipídeos polares insolúveis em água com uma solução aquosa. A entropia desfavorável causada pela mistura do lipídeo insolúvel na água produz uma montagem altamente ordenada de membranas fechadas concêntricas de fosfolipídeo com 10 solução aquosa aprisionada.
Vários sistemas para formar um implante in situforam descritos. Por exemplo, a Patente U.S. N2 4.938.763 descreve um método para formar um implante pela dissolução de um polímero termoplástico não reativo, insolúvel em água em um solvente biocompatível, solúvel em água 15 para formar um líquido, colocando o líquido dentro do corpo e deixando o solvente dissipar para produzir um implante sólido. A solução polimérica pode ser colocada no corpo por intermédio de seringa. O implante pode assumir a forma da sua cavidade circundante. Altemativamente, um implante pode ser formado a partir de polímeros oligoméricos reativos, líquidos que 20 não contenham nenhum solvente e que curem no lugar para formar sólidos, usualmente com a adição de um catalisador de cura.
Vários sistemas de liberação controlada poliméricos para a liberação de anestésicos locais foram descritos na técnica. Embora tais sistemas de liberação poliméricos possam fornecer propriedades de liberação 25 controlada adequadas para o anestésico e ainda superai' as desvantagens associadas com a injeção de anestésicos puros (por exemplo, dispersão para fora do sítio alvo, entrada na comente sanguínea e toxicidades sistêmicas), é difícil superar certas desvantagens associadas com os sistemas poliméricos, tais como insuficiência para evitar arrebentamento de liberação inicial sistêmica do anestésico ou ter que fornecer agentes realçadores de modo a superar a liberação muito pequena do anestésico a partir dos sistemas.
SUMÁRIO DA INVENÇÃO
Os sistemas carreadores de liberação controlada não poliméricos para a administração de um agente anestésico de interesse são fornecidos. E assim um objetivo da presente invenção fornecer um sistema de liberação controlada de ação longa que libere um anestésico em um período prolongado de tempo, suficiente para fornecer um efeito anestésico local em um sítio de administração por pelo menos cerca de 24 horas depois da administração, preferivelmente pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente pelo menos cerca de 48 a 72 horas depois da administração. Também é um objetivo da presente invenção que a liberação do agente anestésico ativo da composição anestésica de ação longa ocorra sem um arrebentamento inicial.
É mais particularmente um objetivo da presente invenção fornecer uma composição contendo um anestésico e um carreador não polimérico farmaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração a um indivíduo sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente pelo menos cerca de 48 a 72 horas depois da administração.
Em um aspecto da invenção, o carreador não polimérico é suficiente para fomecer um perfil de liberação controlada de primeira ordem do anestésico ou um perfil de liberação de ordem pseudo-zero do anestésico. Em certas formas de liberação, o anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster. Em uma forma de liberação preferida, o anestésico é a bupivacaína que pode ser fornecida ainda na forma de base livre. Em outras formas de liberação, a composição é capaz de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml.
Em um outro aspecto da invenção, o carreador não polimérico é substancialmente insolúvel em água ou em um sistema biológico aquoso. Em tais composições, o produto farmacêutico pode conter ainda um solvente que seja dispcrsável, solúvel ou miscível em água ou em um sistema aquoso. O solvente pode ser assim um solvente orgânico que seja capaz de dissipar, difundir ou lixiviar para fora da composição na colocação dentro de um sistema biológico, por meio do qual o carreador pode depois coagular ou precipitar para formar um implante sólido in situ.
Já em um outro aspecto da invenção, o carreador não polimérico é um líquido, preferivelmente um material carreador líquido de alta viscosidade (“HVLCM”) tendo uma viscosidade de pelo menos cerca de 5.000 cP a 37° C e que não cristaliza puro sob condições ambientes ou fisiológicas. Tais materiais carreadores líquidos podem ser combinados com um solvente em que o material carreador seja solúvel. Se um HVLCM é usado, é preferido que o solvente seja suficiente para diminuir a viscosidade do HVLCM. Em certas formas de liberação, o solvente pode ser um segundo agente anestésico tal como álcool benzílico. As composições podem ser fornecidas em qualquer forma adequada, por exemplo, como uma emulsão, uma pasta, um gel, uma lama, um creme, uma película, uma pulverização, um sólido, uma partícula, uma raicropartícula, um pó, um implante ou um líquido. Em certas formas de liberação, a composição ainda inclui um material que seja imiscívél com o carreador não polimérico, por exemplo onde a composição é uma emulsão. Nestas composições, o carreador pode estar presente na fase dispersa ou na contínua da emulsão.
Também é um objetivo da presente invenção fornecer uma composição contendo um anestésico e um carreador não polimérico farmaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração a um indivíduo, onde a composição é capaz ainda de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml.
Em um aspecto da invenção, a composição é capaz de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) por um período de pelo menos cerca de 48 horas. Em um outro aspecto, a composição é caracterizada ainda como não tendo nenhum arrebentamento inicial substancial. Ainda em outros aspectos, o carreador não polimérico é suficiente para fornecer um perfil de liberação controlada de primeira ordem do anestésico ou um perfil de liberação de ordem pseudo-zero do anestésico. Em certas formas de liberação, o anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster. Em um forma de liberação preferida, o anestésico é a bupivacaína que pode ser fornecida ainda na forma de base livre.
Em um outro aspecto da invenção, o carreador não polimérico é substancialmente insolúvel em água ou em um sistema biológico aquoso. Em tais composições, o produto farmacêutico pode conter ainda um solvente que seja dispersável, solúvel ou miscível em água ou em um sistema aquoso. O solvente pode ser assim um solvente orgânico que é capaz de dissipar, difundir ou lixiviar para fora da composição na colocação dentro de um sistema biológico, por meio do qual o carreador pode depois coagular ou precipitar para formai* um implante sólido in situ.
Já em um outro aspecto da invenção, o carreador não polimérico é um líquido, preferivelmente um material carreador líquido de alta viscosidade (“HVLCM”) tendo uma viscosidade de pelo menos cerca de 5.000 cP a 37° C e que não cristaliza puro sob condições ambientes ou fisiológicas. Tais materiais carreadores líquidos podem ser combinados com um solvente em que o material carreador seja solúvel. Se um HVLCM é usado, é preferido que o solvente seja suficiente para diminuir a viscosidade do HVLCM. Em certas formas de liberação, o solvente pode ser um segundo agente anestésico tal como álcool benzílico. As composições podem ser fornecidas em qualquer forma adequada, por exemplo, como uma emulsão, uma pasta, um gel, uma lama, um creme, uma película, uma pulverização, um sólido, uma partícula, uma micropartícula, um pó, um implante ou um líquido. Em certas formas de liberação, a composição ainda inclui um material que é imiscívél com o carreador não polimérico, por exemplo onde a composição é uma emulsão. Nestas composições, o carreador pode estar presente na fase dispersa ou contínua da emulsão.
E um objetivo relacionado da invenção fornecer uma composição contendo um primeiro anestésico, um segundo anestésico e um carreador não polimérico fannaceuticamente aceitável. Na composição, o segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial na administração a um indivíduo. O caneador não polimérico controla a liberação do primeiro anestésico para fornecer um efeito anestésico subsequente caracterizado pela anestesia local prolongada tendo um início dentro de cerca de 2 horas da administração a um indivíduo sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administtação e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração.
Em um aspecto da invenção, o carreador não polimérico é suficiente para fornecer um perfil de liberação controlada de primeira ordem do anestésico ou um perfil de liberação de ordem pseudo-zero do anestésico.
Em outras formas de liberação, a composição é capaz de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml. Em certas outras formas de liberação, o primeiro anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster. Ainda em outras formas de liberação, o segundo anestésico também é um solvente para o carreador não polimérico.
O segundo anestésico pode ser um solvente de álcool, álcool aromático, ácido ou derivado de ácido ou qualquer combinação de tais solventes. Em um forma de liberação preferida, o segundo anestésico é álcool benzílico. Em uma outra forma de liberação preferida, o primeiro anestésico é a bupivacaína que pode ser fornecida ainda na forma de base livre.
Em um outro aspecto da invenção, o carreador não polimérico é substancialmente insolúvel em água ou em um sistema biológico aquoso.
Em tais composições, o produto farmacêutico pode conter ainda um solvente que seja dispersável, solúvel ou miscível em água ou em um sistema aquoso.
O solvente pode ser assim um solvente orgânico que seja capaz de dissipar, difundir ou hxiviar para fora da composição na colocação dentro de um sistema biológico, por meio do qual o carreador pode depois coagular ou precipitar para formar um implante sólido in situ.
Já em um outro aspecto da invenção, o carreador não polimérico é um líquido, preferivelmente um material carreador líquido de alta viscosidade (“HVLCM”) tendo uma viscosidade de pelo menos cerca de 5.000 cP a 37° C e que não cristaliza puro sob condições ambientes ou fisiológicas. Tais materiais carreadores líquidos podem ser combinados com um solvente em que o material carreador seja solúvel. Se um HVLCM é usado, é preferido que o solvente seja suficiente para diminuir a viscosidade do HVLCM. Em certas formas de liberação, o solvente pode ser um segundo agente anestésico tal como álcool benzílico. As composições podem ser fornecidas em qualquer forma adequada, por exemplo, como uma emulsão, uma pasta, um gel, uma lama, um creme, uma película, uma pulverização, um sólido, uma partícula, uma micropartícula, um pó, um implante ou um líquido. Em certas formas de liberação, a composição ainda inclui um material que seja imiscível com o carreador não polimérico, por exemplo onde a composição for uma emulsão. Nestas composições, o carreador pode estar presente na fase dispersa ou contínua da emulsão.
Também é um objetivo relacionado da invenção fornecer uma composição que compreenda um material carreador líquido de alta viscosidade (“HVLCM”) não polimérico, não solúvel em água tendo uma viscosidade de pelo menos 5.000 cP a 37° C que não cristalize puro sob condições ambientes ou fisiológicas, um primeiro anestésico e um segundo anestésico. Aqui mais uma vez o segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial na administração a um indivíduo. O HVLCM controla a liberação do primeiro anestésico para fornecer um efeito anestésico subseqüente caracterizado pela anestesia local prolongada tendo um início dentro de cerca de 2 horas da administração a um indivíduo sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração. Em certas formas de liberação, a composição é capaz de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml.
Em um aspecto da invenção, o primeiro anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster. Em outras formas de liberação, o segundo anestésico também é um solvente para o HVLCM. O segundo anestésico pode ser um solvente de álcool, álcool aromático, ácido ou derivado de ácido ou qualquer combinação de tais solventes. Em uma forma de liberação preferida, o segundo anestésico é álcool benzílico. Em uma outra forma de liberação preferida, o primeiro anestésico é a bupivacaína que pode ser fornecida ainda na forma de base livre. Ainda em outras formas de liberação preferida, o HVLCM é um éster, tal como um éster de açúcar como acetato isobutirato de sacarose. Nestas composições, pode ser útil fornecer um solvente em que o HVVLCM seja solúvel.
E outro objetivo relacionado da invenção fornecer uma composição que compreenda um material carreador líquido de alta viscosidade (“HVLCM”) não polimérico, não solúvel em água tendo uma viscosidade de pelo menos 5.000 cP a 37° C que não cristaliza puro sob condições ambientes ou fisiológicas, um primeiro anestésico e um segundo anestésico. Aqui mais uma vez o segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial na administração a um indivíduo. O HVLCM controla a liberação do primeiro anestésico para fornecer um efeito anestésico subsequente caracterizado pela anestesia local prolongada, onde a composição é capaz ainda de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml.
Em um aspecto da invenção, a composição é capaz de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) por um periodo de pelo menos cerca de 48 horas. Em um outro aspecto, a composição é caracterizada ainda como não tendo nenhum arrebentamento inicial substancial.
Em um outro aspecto da invenção, o primeiro anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster. Em outras formas de liberação, o segundo anestésico também é um solvente para o HVECM. O segundo anestésico pode ser um solvente de álcool, álcool aromático, ácido ou derivado de ácido ou qualquer combinação de tais solventes. Em uma forma de liberação preferida, o segundo anestésico é álcool benzílico. Em uma outra forma de liberação preferida, o primeiro anestésico é a bupivacaína que pode ser fornecida ainda na forma de base livre. Ainda em outras formas de liberação preferida, o HVLCM é um éster, tal como um éster de açúcar como acetato isobutirato de sacarose. Nestas composições, pode ser útil fornecer um solvente em que o HWLCM seja solúvel.
É um outro objetivo da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um indivíduo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um anestésico e um carreador não polimérico farmaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração ao indivíduo sem um arrebentamento inicial e tendo uma duração de pelo menos cerca de 24 horas depois da administração.
Em um aspecto da invenção, o anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster.
É um objetivo relacionado da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um indivíduo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um anestésico e um carreador não polimérico famiaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração ao indivíduo, onde a composição é capaz ainda de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente.
Em um aspecto da invenção, o anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster.
É ainda um outro objetivo da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um indivíduo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um primeiro anestésico, um segundo anestésico e um carreador nào polimérico farmaceuticamente aceitável. O segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial no sítio na administração. O carreador não polimérico controla a liberação do primeiro anestésico para fornecer um efeito anestésico subsequente caracterizado pela anestesia local prolongada no sítio tendo um início dentro de cerca de 2 horas da administração sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração.
Em um aspecto da invenção, o carreador não polimérico é um líquido, preferivelmente um material carreador líquido de alta viscosidade (“HVLCM”) que não seja solúvel em água e tenha uma viscosidade de pelo menos cerca de 5.000 cP a 37° C e ainda que não cristalize puro sob condições ambientes ou fisiológicas. Tais materiais carreadores líquidos podem ser combinados com um solvente em que o material carreador é solúvel. Se um HVLCM é usado, é preferido que o solvente seja suficiente para diminuir a viscosidade do HVLCM. Em certas formas de liberação, o solvente pode ser um segundo agente anestésico tal como álcool benzílico.
Em um outro aspecto da invenção, o primeiro anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster. Em outras formas de liberação, o segundo anestésico também é um solvente para o HVLCM. O segundo anestésico pode ser um solvente de álcool, álcool aromático, ácido ou derivado de ácido ou qualquer combinação de tais solventes. Em uma forma de liberação preferida, o segundo anestésico é álcool benzílico. Em uma outra forma de liberação preferida, o primeiro anestésico é a bupivacaína que pode ser fornecida ainda na forma de base livre. Ainda em outras formas de liberação preferida, o HVLCM é um éster, tal como um éster de açúcar como acetato isobutirato de sacarose. Nestas composições, pode ser útil fornecer um solvente em que o HVVLCM seja solúvel.
Já em um outro aspecto da invenção, a composição é administrada pela administração tópica, administração transdérmica, injeção ou como um implante ao sítio. Em certas formas de liberação, a composição é administrada a um sítio que seja um ferimento cirúrgico e a composição é administrada dentro e/ou adjacente ao ferimento.
E ainda um outro objetivo da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um indivíduo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um primeiro anestésico, um segundo anestésico e um carreador não polimérico farmaceuticamente aceitável. O segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial no sítio na administração. O carreador não polimérico controla a liberação do primeiro anestésico para fornecer um efeito anestésico subseqüente caracterizado pela anestesia local prolongada no sítio e a composição é capaz ainda de fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente.
Em um aspecto da invenção, o carreador não polimérico é um líquido, preferivelmente um material carreador líquido de alta viscosidade (“HVLCM”) que não seja solúvel água e tenha uma viscosidade de pelo menos cerca de 5.000 cP a 37° C e ainda que não cristalize puro sob condições ambientes ou fisiológicas. Tais materiais carreadores líquidos podem ser combinados com um solvente em que o material carreador seja solúvel. Se um HVLCM é usado, é preferido que o solvente seja suficiente para diminuir a viscosidade do HVLCM. Em certas formas de liberação, o solvente pode ser um segundo agente anestésico tal como álcool benzílico.
Em um outro aspecto da invenção, o primeiro anestésico é um anestésico local, por exemplo um anestésico local do tipo amida ou éster. Em outras formas de liberação, o segundo anestésico também é um solvente para o HVLCM. O segundo anestésico pode ser um solvente de álcool, álcool aromático, ácido ou derivado de ácido ou qualquer combinação de tais solventes. Em uma forma de liberação preferida, o segundo anestésico é álcool benzílico. Em uma outra forma de liberação preferida, o primeiro anestésico é a bupivacaína que pode ser fornecida ainda na forma de base livre. Ainda em outras formas de liberação preferida, o HVLCM é um éster, tal como um éster de açúcar como acetato isobutirato de sacarose.
Nestas composições, pode ser útil fornecer um solvente em que o HVVLCM seja solúvel.
Já em um outro aspecto da invenção, a composição é administrada pela administração tópica, administração transdérmica, injeção ou como um implante ao sítio. Em certas formas de liberação, a composição é administrada a um sítio que seja um ferimento cirúrgico e a composição é administrada dentro e/ou adjacente ao ferimento.
É uma vantagem da presente invenção que o material carreador não polimérico seja capaz de controlar a liberação do agente anestésico tanto para evitar um arrebentamento de liberação inicial quanto para fornecer um efeito anestésico prolongado por pelo menos cerca de 24 horas. E uma outra vantagem da invenção que as composições sejam facilmente construídas para fornecer qualquer número de formas farmacêuticas diferentes e ainda para fornecer uma ampla faixa de características de liberação farmacológica diferentes dependendo do sitio pretendido de administração e aplicação médica.
Estes e outros objetivos, aspectos e vantagens da presente invenção facilmente ocorrerão ao técnico habilitado na leitura da presente divulgação e relatório descritivo.
DESCRIÇÃO RESUMIDA DAS FIGURAS
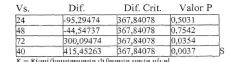
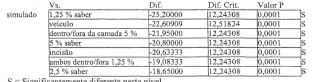
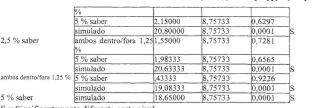
A Figura 1 representa os níveis de bupivacaína plasmática médios em 7 dias (os resultados farmacodinâmicos) do Exemplo 3, em que os dados do Grupo 1 são representados pela curva de fundo e os dados do Grupo 2 são representados pela curva de topo.
A Figura 2 representa os níveis de bupivacaína plasmática média em 0 a 144 horas (os resultados farmacodinâmicos) do Exemplo 4, Grupo 1.
A Figura 3 representa os níveis de bupivacaína plasmática médios em 0 a 12 horas (os resultados farmacodinâmicos) do Exemplo 4, Grupo 1.
A Figura 4 representa os níveis de bupivacaína plasmática médios em 0 a 300 horas (os resultados farmacodinâmicos) do Exemplo 4, Grupo 2, onde os dados do subgrupo 3 são representados pela curva de fundo (0), os dados do subgrupo 2 são representados pela curva intermediária (□) e 5 os dados do subgrupo 1 são representados pela curva de topo (Δ).
A Figura 5 representa os níveis de bupivacaína plasmática médios em 0 a 12 horas (os resultados farmacodinâmicos) do Exemplo 4, Grupo 2, onde os dados do subgrupo 3 são representados pela curva de fundo (0), os dados de subgrupo 2 são representados pela curva intermediária (□) e 10 os dados do subgrupo 1 são representados pela curva de topo (Δ).
A Figura 6 representa as classificações de dor no sítio de incisão “no repouso” médias registradas usando uma escala análoga visual de 0 a 100 mm (VAS) do Exemplo 4, Grupo 2, onde os dados do subgrupo 3 são representados pela curva de topo (Δ), os dados do subgrupo 2 sào 15 representados pela curva intermediária (□) e os dados do subgrupo 1 são representados pela curva de fundo (0).
DESCRIÇÃO DETALHADA DAS FORMAS DE LIBERAÇÃO ESPECÍFICAS
Antes de descrever a presente invenção em detalhes, deve ser 20 entendido que esta invenção não é limitada aos materiais carreadores ou parâmetros de processo particularmente exemplificados visto que tais, naturalmente, podem variar. Também deve ser entendido que a terminologia aqui usada é apenas para o propósito de descrever as formas de liberação particulares da invenção e não é intencionada a ser limitante.
Todas as publicações, patentes e pedidos de patente aqui citadas, seja acima ou abaixo, são por meio deste incorporados por referência em sua totalidade.
Deve ser mencionado que, como usado neste relatório descritivo e nas reivindicações anexas, as formas singulares “um,” “uma” e “o”, “a” incluem referentes plurais a menos que o teor claramente dite de outro modo. Assim, por exemplo, referência a “um carreador não polimérico” inclui uma mistura de dois ou mais de tais carreadores, referência a “um solvente” inclui uma mistura de dois ou mais de tais carreadores, referência a “um anestésico” inclui misturas de dois ou mais de tais agentes e outros.
É um objetivo da presente invenção fornecer um sistema de liberação controlada de ação longa que libera um anestésico em um período prolongado de tempo, suficiente para fornecer um efeito anestésico local em um sítio de administração por pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração. Também é um objetivo da presente invenção que a liberação do agente anestésico ativo da composição anestésica de ação longa ocorre sem um arrebentamento inicial. É um outro objetivo da presente invenção que a composição libere o agente anestésico ativo da composição anestésica de ação longa para fornecer uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml.
Também é um objetivo da presente invenção fornecer uma composição contendo um anestésico e um carreador não polimérico farmaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração a um indivíduo sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração.
Também é um objetivo da presente invenção fornecer uma composição contendo um anestésico e um carreador não polimérico farmaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração a um indivíduo, em que a composição fornece uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml.
E um objetivo relacionado da invenção fornecer uma composição contendo um primeiro anestésico, um segundo anestésico e um carreador não polimérico farmaceuticamente aceitável. Na composição, o segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial na administração a um indivíduo. O carreador não polimérico conttola a liberação do primeiro anestésico para fornecer um efeito anestésico subseqiiente caracterizado pela anestesia local prolongada tendo um início dentro de cerca de 2 horas de administração a um indivíduo sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração.
Também é um objetivo relacionado da invenção fornecer uma composição que compreenda um material carreador líquido de alta viscosidade (“HVLCM”) não polimérico, não solúvel em água tendo uma viscosidade de pelo menos 5.000 cP a 37° C que não cristaliza puro sob condições ambientes ou fisiológicas, um primeiro anestésico e um segundo anestésico. Aqui mais uma vez o segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial na administração a um indivíduo. O HVLCM controla a liberação do primeiro anestésico para fornecer um efeito anestésico subsequente caracterizado pela anestesia local prolongada tendo um início dentro de cerca de 2 horas de administração a um indivíduo sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração.
E um outro objetivo relacionado da invenção fornecer uma composição que compreenda um material carreador líquido de alta viscosidade (“HVLCM”) não polimérico, não solúvel em água tendo uma viscosidade de pelo menos 5.000 cP a 37° C que não cristaliza puro sob condições ambientes ou fisiológicas, um primeiro anestésico e um segundo anestésico. Aqui mais uma vez o segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial na administração a um indivíduo. O HVLCM controla a liberação do primeiro anestésico para fornecer um efeito anestésico subseqüente caracterizado pela anestesia local prolongada e a composição fornece uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente, preferivelmente de pelo menos cerca de 250 ng/ml ou de pelo menos cerca de 300 ng/ml ou de pelo menos cerca de 350 ng/ml.
É um outro objetivo da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um indivíduo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um anestésico e um carreador não polimérico farmaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração ao indivíduo sem um arrebentamento inicial e tendo um duração de pelo menos cerca de 24 horas depois da administração.
É ainda um outro objetivo da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um individuo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um anestésico e um carreador não polimérico farmaceuticamente aceitável. O carreador não polimérico controla a liberação do anestésico para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração ao indivíduo e a composição fornece uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente.
É ainda um outro objetivo da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um indivíduo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um primeiro anestésico, um segundo anestésico e um carreador não polimérico farmaceuticamente aceitável. O segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial no sítio na administração. O carreador não polimérico controla a liberação do primeiro anestésico para fornecer um efeito anestésico subsequente caracterizado pela anestesia local prolongada no sítio tendo um início dentro de cerca de 2 horas de administração sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração.
Também é um objetivo da invenção fornecer um método para fornecer um efeito anestésico a um sítio em um indivíduo. O método compreende administrar uma composição no, próximo, dentro ou adjacente ao sítio, onde a composição inclui um primeiro anestésico, um segundo anestésico e um carreador não polimérico farmaceuticamente aceitável. O segundo anestésico é um solvente para o primeiro anestésico e fornece um efeito anestésico inicial no sítio na administração. O carreador não polimérico controla a liberação do primeiro anestésico para fornecer um efeito anestésico subseqüente caracterizado pela anestesia local prolongada no sítio e a composição fornece uma concentração plasmática de estado estacionário em meio prolongado (Css) do anestésico de pelo menos cerca de 200 ng/ml por um período de pelo menos cerca de 24 horas quando a composição é administrada subcutaneamente.
A frase “sem um arrebentamento inicial,” como aqui usada, pretende que o agente particular que é aludido não libere a composição na administração normal e tome-se farmacologicamente, disponível em uma quantidade apreciável durante um período inicial pré determinado. A presença e nível de um arrebentamento inicial de um agente de uma dada composição podem ser facilmente determinados pelo técnico habilitado utilizando técnicas de teste farmacológicas padrão bem conhecidas no ramo. Os métodos de caracterização de liberação explosiva in vitroadequados incluem o Método Paddle USP II, usando condições de tampão, mistura e aquecimento padrão. As características de liberação explosivas de uma dada composição também podem ser facilmente determinado usando teste in vivo padrão, tal como pela monitoração das concentrações plasmáticas do agente de interesse em um indivíduo animal, em um dado período de tempo. Nas composições da presente invenção, preferivelmente menos do que cerca de 40 a 60 % do agente anestésico é liberado dentro das primeiras 24 horas, mais preferivelmente menos do que cerca de 30 a 50 % e ainda mais preferivelmente menos do que cerca de 20 a 40 % é liberado dentro do seu período de tempo inicial. Em certas outras formas de liberação preferidas, menos do que cerca de 5 a 10 % do agente anestésico é liberado dentro da primeira hora, mais preferivelmente menos do que cerca de 3 a 7 % é liberado dentro deste período de tempo inicial.
Consequentemente, as composições da presente invenção conterão pelo menos um agente anestésico em um sistema de liberação controlada que libera um anestésico em um período prolongado de tempo. Em certas formas de liberação, o anestésico está presente nas composições presentes em uma quantidade de cerca de 95 a cerca de 1 porcento em peso relativo ao peso total da composição (% em peso), em uma quantidade de cerca de 30 a 1 % em peso, em uma quantidade de cerca de 25 a 5 % em peso ou em uma quantidade de cerca de 20 a 10 % em peso, dependendo da identidade do anestésico e do seu uso pretendido.
Como aqui usado, o termo “anestésico” pretende qualquer agente que forneça dormência local reversível, alívio da dor, bloqueios da condução de impulso ao longo dos axions nervosos e outras membranas excitáveis, tais como um bloqueio regional dos caminhos nociceptivos (aferentes e/ou eferentes), analgesia, e/ou anestesia. Ver, por exemplo, Strichartz, G. R. (Ed.) Local Anesthetics, Handbook of Experimental Pharmacology, vol. 81, Springer, Berlin/New York, (1987). O termo também inclui qualquer agente que, quando localmente administrado fornece a inibição localizada (regional) total ou parcial da percepção sensorial e/ou função motora. Os exemplos de agentes habitualmente usados adequados para o uso como anestésicos na prática da invenção incluem, mas nào são limitados à ambucaína, amolanona, amilcaína, benoxinato, álcool benzílico, benzocaína, betoxicaína, bifenamina, bupivacaína, butacaína, butamben, butanilicaína, butetamina, butoxicaína, carticaína, cloroprocaína, cocaetileno, cocaína, ciclometicaína, dibucaína, dimetisoquina, dimetocaína, diperodon, diclonina, ecogonidina, ecogonina, etidocaina, euprocina, fenalcomina, formocaina, hexilcaina, hidroxiteteracaina, isobuanina, p-aminobenzoato de isobutila, leucinocaina, levobupivacaina, levoxadrol, lidocaina, mepivacaina, meprilcaína, metabutoxicaína, cloreto de metila, mirtecaina, naepaina, 5 octacaina, ortocaina, oxetazaina, parentoxicaina, fenacaina, fenol, piperocaina, piridocaina, polidocanol, pramoxina, prilocaina, procaina, propanocafna, proparacaina, propipocaina, propoxicaina, pseudococaina, pirrocaina, ropivacaina, álcool salicilico, tetracaina, tolicaina, trimecaina, xilocaina, zolamina, derivados anestesicamente ativos, análogos e qualquer 10 sal destes farmaceuticamente aceitáveis e qualquer mistura destes.
Os tipos amida e éster de anestésicos locais são preferidos para o uso aqui. Os anestésicos locais do tipo amida são caracterizados por ter uma funcionalidade de amida, enquanto que os anestésicos locais do tipo éster contêm uma funcionalidade éster. Os anestésicos locais do tipo amida 15 preferido incluem lidocaína, bupivacaína, prilocaina, mepivacaina, etidocaina, ropivacaina e dibucaína. Os anestésicos locais do tipo éster preferidos incluem tetracaina, procaina, benzocaína e cloroprocaína. O anestésico local mais preferido é a bupivacaína.
O agente anestésico é fornecido na composição em uma fornia 20 neutra, como uma forma de base livre ou na forma de um sal farmaceuticamente aceitável. O termo “sal farmaceuticamente aceitável,” como aqui usado, pretende aqueles sais que retêm a eficácia biológica e as propriedades de anestésicos neutros e não são de outro modo inaceitáveis para o uso farmacêutico. Os sais farmaceuticamente aceitáveis incluem os sais de 25 grupos ácidos ou básicos, grupos estes que podem estar presentes nos agentes anestésicos. Estes agentes anestésicos que são básicos por natureza são capazes de formar uma ampla variedade de sais com vários ácidos inorgânicos e orgânicos. Os sais de adição de ácido farmaceuticamente aceitáveis de anestésicos básicos adequados para o uso aqui são aqueles que 24 formam sais de adição de ácido não tóxicos, isto é, sais que compreendam ânions farmaceuticamente aceitáveis, tais como os sais de cloridreto, bromidreto, iodidreto, nitrato, sulfato, bissulfato, fosfato, fosfato ácido, isonicotinato, acetato, lactato, salicilato, citrato, tartarato, pantotenato, bitartarato, ascorbato, succinato, maleato, gentisinato, fumarato, gliconato, glicaronato, sacarato, formiato, benzoato, glutamato, metanossulfonato, etanossulfonato, benzeno-sulfonato, p-toluenossulfonato e pamoato (isto é, 1,1 l-metileno-bis-(2-hidróxi-3-naftoato)). Os agentes anestésicos que incluem uma porção amino podem formar sais farmaceuticamente aceitáveis com vários aminoácidos, além dos ácidos mencionados acima. Os sais de base adequados podem ser formados a partir de bases que formam sais não tóxicos, por exemplo, sais de alumínio, cálcio, lítio, magnésio, potássio, sódio, zinco e dietanolamina. Ver, por exemplo, Berge et al. (1977) J. Pharm. Sei. 66: 1-19.
A capacidade de um agente anestésico para fornecer uma condição de anestesia local prolongada refere-se à capacidade do agente objeto para estabelecer um estado avaliável de inibição total ou parcial localizada (regional) de percepção sensorial e/ou função motora. Numerosos métodos e ferramentas para fazer uma tal avaliação facilmente ocorrerão ao técnico habilitado. Com respeito aos indivíduos animais não humanos, estes métodos incluem a medição da locomoção espontânea em ratos de teste (usando, por exemplo, equipamento e software comercialmente disponíveis da Med xVssociates Inc., St. Albans, VT), onde os dados podem ser coletados na distância total percorrida, contagens ambulatoriais, estereotipia, levantamento nas patas traseiras, tempo gasto nos vários movimentos e tempo gasto no repouso para os indivíduos de teste; a visualização da reação à picada de alfinete em ratos; e o modelo da retirada da pata em placa quente de rato, por exemplo, de acordo com o procedimento descrito em detalhes na IACUC N2 95112199.
O teste sensorial em indivíduos humanos também é um modo útil de avaliar o efeito anestésico local. O teste é frequentemente focalizado em três áreas gerais, teste mecânico (picada de alfinete, Pelos de von Frey), térmico (momo, quente, frio) e teste tátil (toque). Tais técnicas de teste são descritas na literatura. Ver, por exemplo, Dahl, et al. (1993) Pain 53: 43-51; Moiniche, et al. (1993) Brit. J. of Anaesthesia 71: 201-205; Moiniche, et al. (1993) Regional Anesthesia 18: 300-303; Pedersen, et al. (1996) Anesthesiology 84(5): 1020-1026; Pedersen, et al. (1996) Brit. J. of Anaesthesia 76(6): 806-810; e Pedersen, et al. (1998) Pain 74: 139-151. Por exemplo, a atividade anestésica local de um agente de teste pode ser examinada com referência ao início, densidade de pico e duração do efeito usando modalidades específicas: 1) teste sensorial mecânico (limiar de detecção da dor mecânica usando pelos de von Frey; 2) teste de supralimiar (mecânico) usando um único pelo de von Frey; 3) teste sensorial térmico (limiar de detecção de calor); 4) limiar de detecção de dor térmica; 5) teste de supralimiar (calor); 6) limiar de detecção de frio; e 7) teste sensorial tátil (limiar de detecção do toque mecânico). Estes dados são indicativos do indivíduo que experiencia alívio da dor local, dormência local e ou bloqueio de nervo local em resposta à administração de um agente anestésico de teste. A resposta à dor pode ser caracterizada usando uma Escala de Classificação Verbal de 0 a 10 (por exemplo, onde 0 = nenhuma dor e 10 — a pior dor imaginável) ou uma Escala Analógica Visual de 0 a 100 mm (por exemplo, onde 0 = nenhuma dor e 100 mm = pior dor imaginável).
Com respeito à seleção de um agente anestésico particular, o técnico habilitado também reconhecerá que as propriedades farmacológicas de cada agente candidato variará, por exemplo, com respeito ao inicio e intensidade, duração e outros do efeito anestésico. Certos agentes podem fornecer um efeito anestésico brando, tendo um início bastante rápido de atividade, mas uma duração curta. Tais agentes podem ser usados com as composições da presente invenção de modo a fornecer um “efeito anestésico inicial,” onde estes são tipicamente unidos com um agente anestésico diferente que forneça uma “anestesia local prolongada,” caracterizado por um início mais gradual de atividade, mas um efeito mais forte e um de duração mais longa. Um exemplo de um anestésico que pode ser usado para fornecer um efeito anestésico inicial é o álcool benzílico. Um exemplo de um anestésico que pode ser usado para fornecer uma anestesia local prolongada é a bupivacaína. Ainda outros agentes que podem ser usados para fornecer um efeito anestésico inicial podem incluir materiais orgânicos habitualmente usados como solventes e/ou agentes de penetração, tais como etanol, sulfóxido de dimetila, N-metilpirrolidona, polietileno glicol e certos ésteres de ácido graxo. Estes e outros agentes similares podem fornecer um efeito anestésico inicial muito brando, por exemplo, quando aplicado estes podem esfriar ou de outro modo dessensibizar/entorpecer um sítio de tecido, inibindo parcialmente deste modo a percepção sensorial naquele sitio. Sempre que um agente é usado na prática da invenção de modo a fornecer um efeito anestésico inicial, o agente é fornecido em uma composição adequada em uma quantidade suficiente para fornecer o efeito objeto e em um tal modo que o agente seja capaz de ser liberado da composição rapidamente de modo a fornecer o efeito intencionado. A montagem de tais composições adequadas (contendo um agente para fornecer um efeito anestésico inicial) está dentro da habilidade da técnica quando tomado em combinação com a orientação e divulgação fornecida pelo presente relatório descritivo.
Em certas formas de liberação da invenção, uma composição é fornecida que inclui dois agentes anestésicos, um primeiro anestésico e um segundo anestésico, em que o segundo agente anestésico é um solvente para o primeiro agente anestésico. Nestas composições particulares, o segundo agente anestésico é tipicamente usado para fornecer um efeito anestésico inicial e o primeiro agente anestésico é usado para fornecer um efeito anestésico subsequente caracterizado pela anestesia local prolongada, tendo um início dentro de cerca de 2 horas de administração a um indivíduo sem um arrebentamento inicial e uma duração de pelo menos cerca de 24 horas depois da administração ou mesmo mais longa. Em certas formas de liberação preferidas, o primeiro agente anestésico fornece a anestesia local prolongada 5 com um início dentro de cerca de 1 a 2 horas da administração e em outras formas de liberação preferidas, o primeiro agente anestésico fornece a anestesia local prolongada com um início dentro de cerca de 30 minutos a 1 hora da administração. Em certas outras formas de liberação, o segundo anestésico também é um solvente para o sistema carreador de liberação 10 controlada.
Um agente anestésico servirá como um solvente para um outro agente anestésico aqui quando um agente é pelo menos parcialmente dissolvido no outro agente solvente na fabricação da composição. Além disso, o solvente anestésico está presente na composição em uma quantidade 15 suficiente para fornecer tanto um efeito anestésico inicial quanto pelo menos parcialmente dissolver o outro agente anestésico. Em certas formas de liberação, o segundo anestésico está assim presente em uma quantidade de cerca de 95 a cerca de 1 porcento em peso relativo ao peso total da composição (% em peso) ou em uma quantidade de cerca de 75 a 10 % em 20 peso ou em uma quantidade de cerca de 50 a 15 % em peso.
Vários agentes anestésicos adequados que também servem como solventes para outros agentes anestésicos podem ser usados na prática da invenção. Os agentes adequados incluem álcoois aromáticos, ácidos e derivados de ácido e combinações destes. Um agente anestésico 25 particularmente preferido que pode ser usado como um solvente por um anestésico adicional é o álcool benzílico.
Os sistemas carreadores de liberação controlada utilizados nas composições da presente invenção são classificados como carreadores não poliméricos. Um carreador não polimérico farmaceuticamente aceitável e tipicamente biocompatível e preferivelmente biodegradável, bioerodível ou bioabsorvível. Uma substância é biocompatível se a mesma e qualquer um de seus produtos de degradação não apresentam nenhum efeito significante, deletério ou indesejável, nem causam irritação de tecido substancial ou 5 necrose quando administrada ao tecido vivo. “Biodegradáveis” ou “bioerodíveis,” usados aqui intercambiavelmente, significam que o material não polimérico objeto degradará ou erodirá in vivo para formar espécies químicas menores, em que tal degradação pode resultar, por exemplo, de processos enzimáticos, químicos e físicos. “Bioabsorvíveis” significa que um 10 dado material não polimérico pode ser quebrado e absorvido dentro de um corpo do indivíduo animal, por exemplo, por uma célula, tecido ou semelhante.
O material carreador não polimérico é usado para controlar a liberação de pelo menos um agente anestésico das composições da presente 15 invenção, em um tal modo como para fornecer uma anestesia local prolongada tendo um início dentro de cerca de 2 horas da administração e uma duração de pelo menos cerca de 24 horas ou mais longa. Em algumas composições da presente invenção, o material carreador não polimérico é suficiente para fornecer um perfil de liberação controlada de primeira ordem 20 do pelo menos um anestésico ou um perfil de liberação de ordem pseudo- zero. Consequentemente, o carreador não polimérico estará presente na composição em uma quantidade de cerca de 99,5 a cerca de 1 porcento em peso relativo ao peso total da composição (% em peso) ou em uma quantidade de cerca de 95 a 10 % em peso ou em uma quantidade de cerca de 75 a 25 % 25 em peso.
A seleção de um carreador não polimérico adequado está dentro da habilidade geral na técnica, usando a divulgação e orientação fornecida pela presente divulgação e relatório descritivo. Por exemplo, numerosos sistemas carreadores não poliméricos farmaceuticamente aceitáveis estão disponíveis ao técnico habilitado para produzir composições farmacêuticas líquidas, em pulverização, creme, loção, unguentos, gel, pasta fluida, óleo, emulsão, microemulsão, sólida, emplastro, película, partícula, micropartícula, pó ou outras formas adequadas. Estes e outros sistemas 5 carreadores são descritos, por exemplo, em Remington's Pharmaceutic Sciences, 16a Edição, 1980 e 17a Edição, 1985, ambas publicadas por Mack Publishing Company, Easton, PA.
As composições da presente invenção podem ainda incluir um ou mais componentes adicionais, por exemplo materiais excipientes 10 farmaceuticamente aceitáveis que possam atuar como agentes dispersantes, agentes de volume, aglutinantes, carreadores, estabilizadores, agentes de deslizamento, antioxidantes, ajustadores de pH, anti-irritantes e outros. O técnico habilitado avaliará que certos materiais excipientes podem servir a diversas das funções aludidas acima em qualquer formulação particular.
Assim, qualquer número de materiais excipientes adequados pode ser misturado com ou incorporado nas composições da presente invenção para fornecer propriedades de volume, alterar as taxas de liberação de agente ativo, aumentar ou impedir a captação de água, controlar o pH, fornecer sustentação estrutural, facilitar os processos de fabricação e outro usos conhecidos por aqueles habilitados na técnica. O termo “excipiente” no geral se refere a um material substancialmente inerte que não é tóxico e não interage com outros componentes da composição em uma maneira deletéria. As proporções em que um excipiente particular pode estar presente na composição depende do propósito para o qual o excipiente é fornecido e da identidade do excipiente.
Por exemplo, excipientes adequados que também podem atuar como estabilizadores para agentes ativos incluem graus farmacêuticos de dextrose, sacarose, lactose, trealose, manitol, sorbitol, inositol, dextrano e outros. Tais estabilizadores podem ser assim um sacarídeo tal como um monossacarídeo, um dissacarídeo, um polissacarídeo ou um álcool de açúcar.
Outros excipientes incluem amido, celulose, fosfatos de sódio ou cálcio, sulfato de cálcio, ácido cítrico, ácido tartárico, glicina e combinações destes. Os exemplos de excipientes hidrofóbicos que podem ser adicionados para diminuir a hidratação e as cinéticas de dissolução incluem ácidos graxos e sais destes farmaceuticamente aceitáveis (por exemplo, estearato de magnésio, ácido esteárico, estearato de zinco, ácido palimítico e palitato de sódio).
Também pode ser útil utilizar um excipiente de lipídeo e/ou detergente carregado nas composições da presente invenção. Os lipídeos carregados adequados incluem, sem limitação, fosfatidilcolinas (lecitina) e outros. Detergentes tipicamente serão um tensoativo não iônico, aniônico, catiônico ou anfotérico. Os exemplos de tensoativos adequados incluem, por exemplo, tensoativos de Tergitol® e Triton® (Union Carbide Chemicals and Plastics); polioxietilenossorbitanos, por exemplo, tensoativos TWEEN® (Atlas Chemical Industries); polissorbatos; éteres de polioxietileno, por exemplo Brij; ésteres de ácido graxo farmaceuticamente aceitáveis, por exemplo, lauril sulfato e sais destes; tensoativos ampifílicos (glicerídeos, etc.); e como materiais.
Outros materiais excipientes podem ser adicionados para alterar a porosidade, por exemplo, materiais como sacarose, dextrose, cloreto de sódio, sorbitol, lactose, polietileno glicol, manitol, frutose, polivinil pirrolidona ou combinações apropriadas destes. Adicionalmente, o agente ou agentes anestésicos podem ser dispersados com óleos (por exemplo, óleo de gergelim, óleo de milho, vegetal) ou um mistura destes com um fosfolipídeo (por exemplo, lecitina) ou triglicerídeos de ácido graxo de cadeia média (por exemplo, Miglyol 812) para fornecer uma suspensão oleosa.
Ainda outros materiais excipientes que podem ser incorporados nas composições da presente invenção incluem diluentes de vários conteúdos de tampão (por exemplo, Tris-HCl, acetato); pH e agentes que alteram a força iônica; aditivos tais como antioxidantes (por exemplo, ácido ascórbico, glutationa, metabissulfito de sódio); preservativos (por exemplo, Timersol, álcool benzílico, metil parabeno, propil parabeno); e agentes dispersantes tais como polissacarídeos solúveis em água (por exemplo, manitol, lactose, glicose, amidos), ácido hialurônico, glicina, fibrina, colágeno e sais inorgânicos (por exemplo, cloreto de sódio).
Em certas formas de liberação da invenção, o carreador não polimérico é substancialmente insolúvel em água ou em um sistema biológico aquoso. Os exemplares de tais materiais carreadores não poliméricos incluem, mas não são limitados a: esteróis tais como colesterol, estigmasterol, β- sitosterol e estradiol; ésteres colesterílico tal como estearato de estearato de colesterila; ácidos graxos C[2-C24 tais como ácido láurico, ácido mirístico, ácido palmítico, ácido esteárico, ácido araquídico, ácido beênico e ácido lignocérico; mono, di e triacilglicerídeos Cig-Cjc tais como monooleato de glicerila, monolinoleato de glicerila, monolaurato de glicerila, monodocosanoato de glicerila, monomiristato de glicerila, monodicenoato de glicerila, dipalmitato de glicerila, didocosanoato de glicerila, dimiristato de glicerila, didecenoato de glicerila, tridocosanoato de glicerila, trimiristato de glicerila, tridecenoato de glicerila, tristearato de glicerol e misturas destes; ésteres de ácido graxo de sacarose tais como diestearato de sacarose e palmitato de sacarose; ésteres de ácido graxo de sorbitano tal como monoestearato de sorbitano, monopalmitato de sorbitano e triestearato de sorbitano; álcoois graxos C16-CiStais como álcool cetílico, álcool miristílico, álcool estearílico e álcool cetoestearílico; ésteres de álcoois graxos e ácidos graxos tais como palmitato de cetila e palmitato de cetearila; anidridos de ácidos graxos tais como anidrido esteárico; fosíòlipídeos incluindo fosfatidilcolina (lecitina), fosfatidilserina, fosfatidiletanolamina, fosfatidilinositol e lisoderivados destes; esfmgosina e derivados desta; espingomielinas tais como estearil, palmitoil e tricosanil espingomielinas; ceramidas tais como estearil e palmitoil ceramidas; glicoesfmgolipídeos; lanolina e álcoois de lanolina; e combinações e misturas destes. Certos carreadores não poliméricos preferidos incluem colesterol, monoestearato de glicerila, triestearato de glicerola, ácido esteárico, anidrido esteárico, monocleato de glicerila, monolinoleato de glicerila e monoglicerídeos acetilados.
Se um dos materiais carreadores não poliméricos mencionados acima é selecionado para o uso em uma composição da presente invenção, este tipicamente será combinado com um solvente orgânico compatível e adequado para o material carreador para formar uma composição tendo uma consistência variando de aquoso a viscoso a uma massa ou pasta espalháveis. A consistência da composição variará de acordo com fatores tais como a solubilidade do carreador não polimérico no solvente, a concentração do carreador não polimérico, a concentração do agente anestésico e/ou a presença de agentes anestésicos adicionais, aditivos e excipientes. A solubilidade de um cameador não polimérico em um solvente particular variará de acordo com fatores tais como a sua cristalimdade, hidrofilicidade, caráter iônico e lipofilicidade. Consequentemente, o caráter iônico e a concentração do carreador não polimérico no solvente pode ser ajustado para se obter a solubilidade desejada. Os materiais carreadores não poliméricos preferidos são aqueles que têm baixa cristalinidade, características não polares e são mais hidrofóbicos.
Os solventes orgânicos adequados para o uso nas composições são no geral aqueles que são biocompatíveis, farmaceuticamente aceitáveis e dissolverão pelo menos parcialmente dissolver o carreador não polimérico. O solvente orgânico terá ainda uma solubilidade em água variando de miscível a solúvel a dispersável. Em certas formas de liberação, o solvente é selecionado tal que é capaz de difundir, dispersar ou lixiviar para fora da composição in situem um sistema aquoso e em fluidos encontrados no sítio administração, formando deste modo um sólido implante. Preferivelmente, o solvente tem uma relação de solubilidade de Hildebrand (HLB) de cerca de 9 a 13 (cal/cm ) Preferivelmente, o grau de polaridade do solvente é eficaz para fornecer pelo menos cerca de 5 % de solubilidade em água.
Os solventes orgânicos adequados assim incluem, mas não são limitados a: compostos heterocíclicos substituídos tais como N-metil-2- pirrolidona (NMP) e 2-pirrolidona (2-pirrol); ésteres de ácido carbônico e álcoois alquílicos tais como carbonato de propileno, carbonato de etileno e carbonato de dimetila; ácidos graxos tais como ácido acético, ácido lático e ácido heptanóico; ésteres alquílicos de ácidos mono, di e tricarboxílicos tais como acetato de 2-etiloxietinila, acetato de etinila, acetato de metinila, lactato de etila, butirato de etila, malonato dietila, glutonato de dietila, citrato de tributila, succinato de dietila, tributirin, miristato de isopropila, adipato de dimetila, succinato de dimetila, oxalato de dimetila, citrato de dimetila, citrato de trietmila, citrato de acetil tributila, triacetato de glicerila; alquil cetonas tais como acetona e metil etil cetona; éter álcoois tais como 2- etoxietanol, éter dimetílico de etileno glicol, glicofiirol e glicerol formal; álcoois tais como etanol e propanol; poliidróxi álcoois tais como propileno glicol, polietileno glicol (PEG), glicerina (glicerol), 1,3-butilenoglicol e isopropilideno glicol (2,2-dimetil-l,3-dioxolona-4-metanol); Solcetal; dialquilamidas tais como dimetilformamida. dimetilacetamida; sulfóxido de dimetila (DMSO) e dimetilsulfona; tetraidrofurano; lactonas tais como oc-caprolactona e butirolactona; alquil amidas cíclicas tais como caprolactama; amidas aromáticas tais como N,N-dimetil-m-toluamida e l-dodecilazacicloeptan-2- ona; e outros; e misturas e combinações destes. Os solventes preferidos incluem N-metil-2-pirrolidona, 2-pirrolidona, sulfóxido de dimetila, lactato de etila, carbonato de propileno, glicofurol, glicerol formal e isopropilideno glicol.
O solvente orgânico será fornecido na composição em uma quantidade de cerca de 99,5 a cerca de 1 porcento em peso relativo ao peso total da composição (% em peso), em uma quantidade de cerca de 95 a 10 % em peso, em uma quantidade de cerca de 75 a 25 % em peso ou em uma quantidade de cerca de 60 a 40 % em peso, dependendo do carreador não polimérico selecionado, solvente orgânico, agente anestésico, aditivo e/ou excipiente que é usado na composição. Em certas formas de liberação, o solvente orgânico difunde ou lixivia para fora da composição em um meio aquoso na colocação dentro de um sistema biológico, por meio do qual o material carreador não polimérico coagula para formar uma matriz sólida. Preferivelmente, o carreador nào polimérico solidifica in situpara formar uma matriz sólida dentro de cerca de 1 a 5 dias depois da administração (implantação), preferivelmente dentro de cerca de 1 a 3 dias, preferivelmente dentro de cerca de 2 horas.
Vários aditivos adequados podem ser incluídos com a composição de modo a comunicar características selecionadas na composição. Por exemplo, o pode incluir uma quantidade menor de um polímero termoplástico biodegradável tal como um polilactídeo, policaprolactona, poliglicolideo ou copolímero deste, de modo a fornecer um implante sólido mais coerente ou uma composição com maior viscosidade de modo a mantê- lo no lugar enquanto o mesmo solidifica. Tais polímeros termoplásticos são divulgados na Patente U.S. Na 4.938.763 a Dunn et al.
Opcionalmente, um agente formador de poro pode ser incluído na composição. O agente formador de poro pode ser qualquer substância orgânica ou inorgânica, farmaceuticamente aceitável que seja substancialmente solúvel em água ou fluido corporal e dissipará do material carreador não polimérico e/ou a matriz sólida de um implante no fluido corporal circundante no sítio de implante. O agente formador de poro pode ser preferivelmente insolúvel no solvente orgânico para formar uma mistura uniforme com o material carreador não polimérico. O agente formador de poro também pode ser uma substância imiscívél em água que rapidamente degrada a uma substância solúvel em água. Em certas composições, o agente formador de poro é combinado com o carreador não polimérico e solvente orgânico em mistura. Os agentes formadores de poro adequados que podem ser usados na composição incluem, por exemplo, açúcares tais como sacarose e dextrose, sais tais como cloreto de sódio e carbonato de sódio, polimeros tais como hidroxilpropilcelulose, carboximetilcelulose, polietileno glicol e polivinilpirrolidona e outros. Os cristais sólidos que fornecerão um tamanho de poro definido, tal como sal ou açúcar, são preferidos.
Em outras formas de liberação da presente invenção, composições são fornecidas em que o carreador não polimérico é um líquido. O líquido carreador não polimérico é preferivelmente um material carreador líquido de alta viscosidade (“HVLCM”) sendo não solúvel em água e tem uma viscosidade de pelo menos 5.000 cP, (e opcionalmente pelo menos 10.000, 15.000; 20.000; 25.000 ou mesmo 50.000 cP) a 37° C que não cristaliza puro sob condições ambientes ou fisiológicas. O termo “não solúvel em água” refere-se a um material que é solúvel em água a um grau de menos do que um porcento em peso sob condições ambientes. O termo “não polimérico” refere-se aos ésteres ou ésteres mistos não tendo essencialmente nenhuma unidade de repetição na porção de ácido do éster, assim como ésteres ou ésteres mistos tendo porções de ácido em que as unidades funcionais na porção ácido são repetidas um número pequeno de vezes (isto é, oligômeros). No geral, materiais tendo mais do que cinco unidades de repetição idênticas e adjacentes ou mers na porção de ácido do éster são excluídos pelo termo “não polimérico” como aqui usado, mas materiais contendo dimeros, trímeros, tetrâmeros ou pentâmeros são incluídos dentro do escopo deste termo. Quando o éster é formado a partir de porções de ácido carboxílico contendo hidróxi que podem ainda esterificar, tais como ácido lático ou ácido glicólico, o número de unidades de repetição é calculado com base no número de porções de lactídeo ou glicolídeo, ao invés do número de porções de ácido lático ou ácido gicólico, onde uma unidade de repetição de lactídeo contém duas porções de ácido lático esterificadas pelas suas respectivas porções de hidróxi e carbóxi e onde uma unidade de repetição de glicolídeo contém duas porções de ácido gicólico esterificadas pelas suas respectivas porções de hidróxi e carbóxi. Os ésteres tendo 1 a cerca de 20 polióis eterifícados na sua porção álcool ou de 1 a cerca de 10 porções de glicerol na sua porção álcool, são considerados não poliméricos como este termo é aqui usado.
Em uma forma de liberação particular, o HVLCM diminui na 10 viscosidade, em alguns casos significantemente, quando misturado com um solvente para formar um carreador de material líquido de viscosidade baixa (“LVLCM”) que pode ser administrado usando dispositivos médicos padrão. A composição de LVLCM é tipicamente mais fácil de colocar no corpo do que uma composição de HVLCM, porque a mesma flui mais facilmente para 15 dentro e fora das seringas ou outros meios de implantação. Também pode ser facilmente formulado como uma emulsão. O LVLCM pode ter qualquer viscosidade desejada, mas a sua viscosidade é no geral mais baixa do que o HVLCM correspondente. Como um exemplo, as faixas de viscosidade para a LVLCM de menos do que aproximadamente 6.000 cP, menos do que aproximadamente 4.000 cP, menos do que aproximadamente 1.000 cP ou menos do que 200 cP, são tipicamente úteis para aplicações in vivo.
O HVLCM particular usado nas composições da invenção podem ser um ou mais de uma variedade de materiais. Os materiais adequados incluem ésteres não poliméricos ou ésteres mistos de um ou mais 25 ácidos carboxíhcos. Em uma forma de liberação particular, o éster é formado a partir de ácidos carboxílicos que são esterificados com um poliol tendo de cerca de 2 a cerca de 20 porções de hidróxi e que podem incluem de 1 a cerca de 20 polióis eterifícados. Particularmente os ácidos carboxílicos adequados para formar a porção de ácido do éster do HVLCM incluem ácidos carboxílicos tendo um ou mais grupos hidróxi, por exemplo, aqueles obtidos pela alcoólise de abertura de anel de lactonas ou carbonatos cíclicos ou pela alcoólise de anidridos de ácido carboxílico. Aminoácidos também são adequados para formar ésteres com o poliol. Em uma forma de liberação particular, o éster ou éster misto contém uma porção de álcool tendo uma ou mais porções de hidróxi terminal que foram esteri ficados com um ou mais ácidos carboxílicos obtidos pela alcoólise de um anidrido do ácido carboxílico, tal como um anidrido cíclico.
Os exemplos não limitantes de ácidos carboxílicos adequados que podem ser esterificados para formar o HVLCM incluem ácido gicólico, ácido lático, ácido s-hidroxicapróico, serina e quaisquer lactonas ou lactamas correspondentes, carbonato de trimetileno e dioxanona. Os ácidos contendo hidróxi podem ser eles próprios esterificados ainda através da reação das suas porções de hidróxi com ácidos carboxílicos adicionais, que podem ser os mesmos ou diferentes de outras porções de ácido carboxílico no material. As lactonas adequadas incluem, mas não são limitados a, glicolídeo, lactídeo, s- caprolactona, butirolactona e valerolactona. Os carbonatos adequados incluem mas não são limitados a carbonato de trimetileno e carbonato de propileno.
A porção de álcool do éster ou éster misto pode ser derivada de um álcool poliidróxi tendo de cerca de 2 a cerca de 20 grupos hidróxi e como indicado acima, pode ser formada pela esteri ficação de 1 a 20 moléculas de poliol. As porções de álcool adequadas incluem aquelas derivadas pela remoção de um ou mais átomos de hidrogênio a partir de: álcoois CrC2Q monofuncionais, álcoois C[-C20 difuncionais, álcoois trifuncionais, ácidos carboxílicos contendo hidróxi, aminoácidos contendo hidróxi, álcoois contendo fosfato, álcoois tetrafuncionais, álcoois de açúcar, monossacarídeos e dissacarídeos, ácidos de açúcar e poliéter polióis. Mais especificamente, as porções de álcool podem incluir um ou mais de: dodecanol, hexanodiol, mais particularmente, 1,6-hexanodiol, glicerol, ácido gicólico, ácido lático, ácido hidroxibutírico, ácido hidroxivalérico, ácido hidroxicapróico, serina, ATP, pentaeritritol, manitol, sorbitol, glicose, frutose, sacarose, ácido glicurônico, éteres de poliglicerol contendo de 1 a cerca de 10 unidades de glicerol, polietileno glicóis contendo de 1 a cerca de 20 unidades de etileno glicol.
Em formas de liberação particulares da invenção, pelo menos uma das porções de ácido carboxílico dos ésteres ou ésteres mistos do HVLCM compreende pelo menos uma porção de óxi. Em uma forma de liberação ainda mais particular, cada uma das porções de ácido carboxílico compreendem pelo menos um porção de óxi.
Em uma outra forma de liberação particular, pelo menos uma das porções de ácido carboxílico dos ésteres ou ésteres mistos da invenção contém de 2 a 4 átomos de carbono. Em uma forma de liberação ainda mais particular, cada uma das porções de ácido carboxílico dos ésteres ou ésteres mistos da invenção contêm de 2 a 4 átomos de carbono.
Em uma outra forma de liberação mais particular da invenção, pelo menos uma das porções de ácido carboxílico do éster ou éster misto da invenção tem de 2 a 4 átomos de carbono e contém pelo menos uma porção óxi. Em uma outra forma de liberação mais particular da invenção, cada uma das porções de ácido carboxílico do éster ou éster misto da invenção tem de 2 a 4 átomos de carbono e contêm pelo menos uma porção óxi.
Em uma forma de liberação particular, o HVLCM pode ser acetato isobutirato de sacarose (SAIB) ou algum outro éster de uma porção de álcool de açúcar com uma ou mais porções de ácido alcanóico.
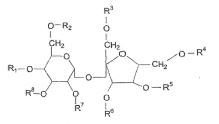
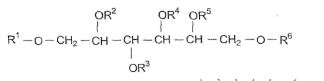
Em uma forma de liberação particular, a invenção inclui composições em que o HVLCM tem uma estrutura selecionada do grupo que consiste de:I:
em que R1, R2, R3, R4, R3, R6 e Rssão independentemente selecionados do grupo que consiste de hidrogênio, alcanoíla, alcanoíla substituído por hidróxi e alcanoíla substituído por acilóxi; em que pelo menos três de R1, R2, R3, R4, R5, R6, R7 e Rs são outros que não hidrogênio; e em que quando R1, R2, R3, R4, R5, R6, R7 e R8 são selecionados • AJl 2 3 do grupo que consiste de acetila e isobutirila, pelo menos três de R , R", R , R4, R5, R6, R7 e R8 são acetila; II
em que R1, R2 e R3 são independenteniente selecionados do grupo que consiste de hidrogênio, alcanoíla, alcanoíla substituído por hidróxi e alcanoíla substituído por acilóxi e em que n está entre 1 e 20; III: R' — O — (CH2), —O— R2 em que n é um número inteiro entre 4 e 8 e R e R" são independentemente selecionados do grupo que consiste de hidrogênio, alcanoíla, alcanoíla substituído por hidróxi e alcanoíla substituído por acilóxi; IV:
V:
em que nas fórmulas IV e V, R1, R2, R3, R4 e R5são independentemente selecionados do grupo que consiste de hidrogênio, alcanoíla, alcanoíla substituído por hidróxi e alcanoíla substituído por acilóxi; VI:
VII:
em que nas fórmulas VI e VII, R1, R2, R4, R4, R5 e Ró são independentemente selecionados do grupo que consiste de hidrogênio, alcanoíla, alcanoíla substituído por hidróxi e alcanoíla substituído por acilóxi;VIII:
em que R1, R2, R’ e R4são independentemente selecionados do grupo que consiste de hidrogênio, alcanoila, alcanoila substituído por hidróxi e alcanoila substituído por acilóxi.
Em cada uma das fórmulas de I até VIII, um ou mais dos grupos alcanoila, alcanoila substituído por hidróxi e alcanoila substituído por acilóxi podem compreender porções alcanoila tendo de 2 a 6 átomos de carbono, incluindo o carbono carbonila. Além disso, em uma outra forma de liberação mais particular da invenção, cada uma das fórmulas I até VIII compreendem pelo menos uma porção alcanoila substituída por hidróxi ou substituída por acilóxi. Em uma forma de liberação ainda mais particular, pelo menos uma destas porções substituídas por hidróxi ou substituídas por acilóxi compreendem porções de alcanoila tendo de 2 a 6 átomos de carbono, incluindo o carbono da carbonila.
Os grupos acila que formam os substituintes acilóxi do HVLCM podem ser qualquer porção derivada de um ácido carboxílico de acordo com a definição habitualmente aceita do termo “acila.” Mais particularmente, os grupos acila das composições da invenção podem ser da forma R9CO-, onde R9 é opcionalmente alquila substituída por óxi de 2 a 6 átomos de carbono. Esta substituição por óxi pode tomar a forma de substituição por hidróxi ou substituição com porções de acila adicionais. Por exemplo R9 pode ser um oligômero de ácidos carboxílicos substituídos por óxi, ligados pela ligação de éster entre o hidróxi de um ácido e o carbóxi de um outro ácido. Em um exemplo mais particular, R J pode compreender de I a 5 unidades de lactídeo ou glicolideo, onde uma unidade de lactídeo contém duas porções de ácido lático esterificadas entre si e uma unidade de glicolídeo contém duas porções de ácido gicólico esterificadas entre si. Altemativamente, R9 pode conter unidades de lactideo e glicolídeo misturadas ou pode conter ácido lático e ácido gicólico misturados, sem a presença de unidades de lactideo ou glicolídeo.
Os materiais de HVLCM particular incluem componentes de acordo com as fórmulas II ou III, em que R , R‘ e R são independentemente lactoíla, polilactoíla, s-caproíla, hidroxiacetila ou poliidroxiacetila, em particular, polilactoíla e s-caproíla ou polilactoíla e poliidroxiacetila.
O uso de cadeia relativamente pequena (2 a 6 átomos de carbono), as porções de ácido carboxílico substituídas por óxi no éster ou éster misto da invenção é vantajoso. Quando estas porções de ácido estão presentes na forma de ésteres oligoméricos (isto é, uma porção de ácido subsequente unida com a porção de ácido anterior através da esterificação do carbóxi subsequente com o óxi anterior), a hidrólise do material é consideravelmente mais fácil do que para os oligômeros fabricados com mais do que 6 átomos de carbono porque o material é mais hidrofilico. No geral, para a liberação de medicamento é desejado que o HVLCM seja insolúvel em água, mas o mesmo pode ser um pouco hidrofílico. No geral, esperar-se-á que os HVLCMs sintetizados com mais unidades hidrofilicas (como determinado por uma relação O:C mais alta) absorverão água mais rapidamente e degradar-se-ão mais rapidamente. Por exemplo, esperar-se-á que um HVLCM fabricado pela ligação covalente de 4 moles de glicolídeo para um mol de glicerol absorverá água mais rapidamente e degradar-se-á mais rapidamente do que um HVLCM fabricado pela ligação covalente de 2 moles de glicolídeo e 2 moles de lactideo a um mol de glicerol. Aumentos similares podem ser esperados para moléculas mais flexíveis e para moléculas mais ramificadas, esféricas com base nos argumentos de volume livre. O uso de moléculas flexíveis e ramificadas também podem ter o beneficio de diminuir a viscosidade do LVLCM. Usando ácidos carboxílicos e/ou polióis de comprimento de cadeia diferentes e usando ácidos carboxílicos tendo substituição óxi permite um controle preciso do grau de hidrofilicidade e da solubilidade do éster resultante. Estes materiais são suficientemente resistentes à dissolução in vivo de modo que eles são capazes de fornecer uma liberação controlada de um agente anestésico carreador no corpo acompanhada ou seguida pela hidrolisação das ligações de óxi in vivo.
Em uma forma de liberação ainda mais particular, o HVLCM exclui os ésteres de acetato e isobutirato da sacarose tendo uma razão de porções ácidas de acetato para isobutirato de 2:6. Entretanto, o éster de acetato isobutirato de sacarose tendo uma razão de porções de acetato para isobutirato de 2:6 está incluído dentro do escopo da invenção para o uso em formulações em aerossol. Este material pode ser fabricado de acordo com os procedimentos descritos na Patente U.S. N2 2.931.802.
No geral, os ésteres de HVLCM adequados podem ser fabricados pela reação de um ou mais álcoois, em particular um ou mais polióis, que formarão a porção de álcool dos ésteres resultantes com um ou mais ácidos carboxílicos, lactonas, lactamas, carbonatos ou anidridos dos ácidos carboxílicos que formarão as porções ácidas dos ésteres resultantes. A reação de esterificaçâo pode ser conduzida simplesmente por aquecimento, embora em alguns casos a adição de um catalisador de esterificaçâo de ácido forte ou base forte pode ser usado. Altemativamente, um catalisador de esterificaçâo tal como 2-etilexanoato estanoso pode ser usado. A mistura de reação aquecida, com ou sem catalisador, é aquecida com agitação depois secada, por exemplo, sob vácuo, para remover quaisquer materiais de partida não reagidos e produzir um produto líquido. Os acetato isobutiratos de sacarose podem ser fabricados seguindo-se os procedimentos descritos na Patente U.S.N2 2.931.802.
A este respeito, o poliol pode ser visualizado como um iniciador de oligomerização, no sentido de que o mesmo fornece um substrato para esterificação de ácidos carboxílicos, em particular, de oligômeros de lactídeo, glicolídeo ou outros ácidos carboxílicos substituídos por hidróxi esterificados.
Em certas formas de liberação, o HVLCM pode ser misturado com um solvente que diminui a viscosidade para formar um material carreador líquido de viscosidade mais baixa (LVLCM), que podem ser depois misturados com o um ou mais agentes anestésicos a serem liberados, antes da administração. Estes solventes podem ser solúveis em água, não solúveis em água ou miscíveis em água e podem incluir, acetona, álcool benzílico, benzoato de benzila, N-(betaidroxietil) lactamidabutileno glicol, caprolactama, caprolactona, óleo de milho, sulfóxido de decilmetila, éter dimetílico, sulfóxido de dimetila, l-dodecilazacicloeptan-2-ona, etanol, acetato de etinila, lactato de etila, oleato de etila, glicerol, glicofurol (tetraglicol), miristato de isopropila, acetato de metinila, metil etil cetona, N- metil-2-pirrolidona, MIGLYOL® (ésteres de ácidos caprílico e/ou cáprico com glicerol ou alquileno glicóis, por exemplo, MIGLYOL® 810 ou 812 (triglicerídeos caprílico/cáprico), MIGLYOL® 818 (triglicerídeo caprílico/cáprico/ linoleico), MIGLYOL® 829 (triglicerídeo caprílico/cáprico/succínico), MIGLYOL® 840 (dicaprilato/caprato de propileno glicol)), ácido oléico, óleo de amendoim, polietileno glicol, carbonato de propileno, 2-pirrolidona, óleo de gergelim, SOLKETAL ([±]- 2,2-dimetil-l,3-dioxolano-4-metanol), tetraidrofurano, TRANSCUTOL49 (éter monoetílico de dietileno glicol, carbitol), triacetina, citrato de trietinila, ftalato de difenila e combinações destes. Adicionalmente, se a composição deva ser aplicada como um aerossol, por exemplo para a aplicação tópica, o solvente pode ser ou pode incluir um ou mais propelentes, tais como propelentes CFC como triclorofluorometano e diclorofluorometano, propelentes não CFC como tetrafluoroetano (R-134a), 1,1,1,2,3,3,3-heptatluoropropano (R-227), éter dimetil i co, propano e butano.
Os solventes e/ou propelentes particularmente adequados incluem benzoato de benzila, álcool benzílico, triacetina, citrato de trietinila, sulfóxido de dimetila, etanol, lactato de etila, glicerol, glicofurol (tetraglicol), N-metil-2-pirrolidona, M1GLYOL® 810, polietileno glicol, carbonato de propileno, 2-pirrolidona e tetrafluoroetano.
Outros solventes possíveis incluem perfluorodecalina, peril uorotributi lamina, metoxiflurano, glicerol formal, álcool tetraidro- furfurílico, diglima e dimetil isossorbida.
Quando a composição é usada como um LVLCM para administrar o agente anestésico, a mesma deve conter um solvente no qual o HVLCM seja solúvel. Em certos casos, o agente anestésico também é solúvel no solvente. Ainda em outros casos, o solvente é um segundo agente anestésico em que o primeiro agente anestésico seja solúvel. O solvente é preferivelmente não tóxico e de outro modo biocompatível.
Em certas formas de liberação, o solvente é pelo menos solúvel em água, de modo que o mesmo se difunda rapidamente nos fluídos corporais ou outro ambiente aquoso na administração, faz com que a composição coagule e/ou se tome mais viscosa. Em uma outra forma de liberação, o solvente não é completamente miscível com água ou fluidos corporais de modo que a difusão do solvente da composição e o aumento correspondente na viscosidade da composição, sejam diminuídos. Os solventes adequados que têm esta propriedade, pelo menos em algum grau, incluem benzoato de benzila, MIGLYOL® 810, álcool benzílico e citrato de trietila. O álcool benzílico pode ser particularmente adequado, visto que o mesmo também é um agente anestésico.
Quando os ésteres de 1,6-hexanodiol ou glicerol são usados como o HVLCM, alguns solventes possíveis são etanol, N-metilpirrolidona, carbonato de propileno e PEG 400.
O solvente é tipicamente adicionado às composições em uma quantidade na faixa de cerca de 99,7 porcento a cerca de 0,5 porcento em peso relativo ao peso total da composição (% em peso), de cerca de 95 porcento a cerca de 1 % em peso, de cerca de 75 a cerca de 10 % em peso ou de cerca de 50 a 15 % em peso. O solvente está tipicamente presente na composição em uma quantidade na faixa de cerca de 55 porcento a 10 % em peso.
Ainda em outras formas de liberação da invenção, a composição inclui um material que não seja miscível com o HVLCM, tal que quando combinado com o HVLCM singularmente ou em combinação com um solvente para o HVLCM, a composição resultante forma uma emulsão. Tais emulsões podem conter o HVLCM na fase dispersa, tal como no caso de misturas de SAIB/M1GLYOL® que são emulsificadas em água ou glicerol ou as mesmas podem conter o HVLCM como um componente da fase contínua, tal como no caso de uma solução aquosa que seja emulsificada no HVLCM ou uma solução do HVLCM em um solvente imiscívél em água.
Qualquer um dos sistemas de liberação controlada não poliméricos descritos acima podem ser formulados como líquido, pulverização, creme, loção, ungüento, gel, pasta fluida, óleo, emulsão, microemulsão, sólido, emplastro, película, partícula, micropartícula, pó ou outras formas adequadas de composições farmacêuticas, adequadas para o uso nos métodos da presente invenção. Em tais composições, o agente anestésico (por exemplo, o primeiro agente anestésico) é incluído em uma quantidade suficiente para liberar ao indivíduo a ser tratado uma quantidade eficaz para alcançar um efeito desejado. A quantidade de agente anestésico incorporada na composição depende da duração e perfil de liberação final desejados e da concentração de anestésico requerida para o efeito pretendido.
A concentração do anestésico na composição também dependerá das taxas de absorção, inativação e excreção daquele agente particular, assim como de outros fatores conhecidos por aqueles de habilidade na técnica. Deve ser mencionado que os valores de dosagem também variarão com a gravidade da condição a ser aliviada. Deve ser ainda entendido que para qualquer indivíduo particular, os regimes de dosagem específicos devem ser ajustados com o tempo de acordo com a necessidade individual e o julgamento profissional da pessoa que administra ou supervisiona a administração das composições e que as faixas de concentração aqui apresentadas são apenas exemplares e não são intencionadas a limitar o escopo ou a prática da composição reivindicada. A composição pode ser administrada em uma dosagem ou pode ser dividida em várias doses menores a serem administradas em intervalos variáveis de tempo, sequencial ou concorrentemente.
O agente ou agentes anestésicos tipicamente estarão presentes na composição na faixa de cerca de 0,1 a cerca de 99,5 porcento em peso relativo ao peso total da composição (% em peso), de cerca de 0,5 a cerca de 70 % em peso ou de cerca de 1 porcento a cerca de 50 % em peso. Entretanto, as faixas tendo pontos finais superiores tão baixos quanto cerca de 40 %, 30 %, 20 % ou 10 % podem ser usadas, como o pode as faixas tendo limites inferiores tão altos quanto cerca de 5 %, 3 % ou 2 %. Para cada um dos agentes anestésicos ativos, as faixas podem ser menores do que 1 % em peso e possivelmente menos do que 0,0001 %.
Os agentes anestésicos tanto solúveis quanto insolúveis podem ser distribuídos usando os materiais carreadores não poliméricos para a liberação controlada. Além disso, as composições podem ser ainda formuladas com excipientes poliméricos para fornecer uma matriz de liberação com propriedades modificadas, por exemplo uma taxa de degradação mais rápida ou mais lenta. A composição resultante pode ser formada em microesferas ou em um implante macroscópico ou outras geometrias e tamanhos de acordo com técnicas conhecidas no ramo. Altemativamente, uma microesfera, implante ou partícula polimérica pré formada com o agente ou agentes anestésicos incorporados nestes podem ser combinados com o carreador não polimérico.
As microesferas podem ser preparadas por vários métodos conhecidos na técnica, assim como métodos descritos nas Patentes U.S. N— 6.291.013 e 6.440.493. A particula polimérica pode ser formada usando a extrusão por fusão, granulação, mistura de solvente, absorção ou como técnicas ou o agente anestésico pode ser absorvido em uma matriz polimérica, tal como uma resina de troca iônica. O material resultante, quando combinado com o material carreador não polimérico adequado pode ser administrado parenteralmente. Em outras formas de liberação, o agente anestésico pode ser combinado com um material não polimérico, tal como fosfato de cálcio ou sacarose, para fornecer propriedades de formação de camada/barreira que prolonguem a degradação. O carreador não polimérico formará então uma barreira secundária para fornecer características de liberação realçadas. A fase de carreador não polimérico pode conter ou não outras substâncias biologicamente ativas, de acordo com a exigência específica da aplicação selecionada. Estes outros agentes biologicamente ativos podem ser qualquer produto farmacêutico terapêutico e/ou profilático adequado, contanto que a substância adicionada seja adequada para a incorporação em microesferas ou implantes de acordo com técnicas conhecidas no ramo.
Como debatido acima, uma variedade de aditivos pode ser opcionalmente adicionada às composições da presente invenção para modificar as suas propriedades e em particular para modificar as propriedades de liberação da composição com respeito aos agentes anestésicos ai contidos. Os aditivos podem estar presentes em qualquer quantidade suficiente para comunicar as propriedades desejadas à composição. A quantidade de aditivo usado no geral será uma função da natureza do aditivo e do efeito a ser obtido e pode ser facilmente determinada pelo encarregado da rotina. Os aditivos adequados são descritos na Patente U.S. N- 5.747.058, os conteúdos inteiros da qual são por meio deste incorporados por referência. Mais particularmente, os aditivos adequados incluem água, polímeros biodegradáveis, polímeros não biodegradáveis, óleos naturais, óleos sintéticos, carboidratos ou derivados de carboidrato, sais inorgânicos, BSA (albumina sérica bovina), tensoativos, 5 compostos orgânicos, tais como açúcares e sais orgânicos, tais como citrato de sódio. No geral, quanto menos solúvel em água, isto é, quanto mais lipofilico, o aditivo, mais diminuirá a taxa de liberação do agente anestésico, comparado com a mesma composição sem o aditivo. Além disso, pode ser desejável incluir aditivos que aumentem as propriedades tais como a 10 concentração ou a porosidade da composição.
A adição de aditivos também pode ser usado para prolongar o tempo de liberação para o agente anestésico, tomando a composição adequada para aplicações médicas que requerem ou responsivas à administração de duração mais longa. Os aditivos adequados a este respeito incluem aquelas 15 divulgadas nas Patentes U.S. N~ 5.747.058 e 5.736.152. Em particular, os aditivos adequados para este propósito incluem aditivos poliméricos, tais como polímeros celulósicos e polímeros biodegradáveis. Os polímeros celulósicos adequados incluem acetatos de celulose, éter de celulose e acetato butiratos de celulose. Os polímeros biodegradáveis adequados incluem 20 polilactonas, polianidridos e poliortoésteres, em particular, ácido polilático, ácido poligicólico, policaprolactona e copolímeros destes.
Quando presente, o aditivo está tipicamente presente nas composições em uma quantidade na faixa de cerca de 0,01 porcento a cerca de 20 porcento em peso, mais particularmente de cerca de 0,1 porcento a 25 cerca de 20 porcento em peso, relativo ao peso total da composição e mais tipicamente, está presente na composição em uma quantidade na faixa de cerca de 1, 2 ou 5 porcento a cerca de 10 porcento em peso. Certos aditivos, tais como tampões, estão apenas presentes em quantidades pequenas na composição.
As seguintes categorias são exemplos não limitantes de classes, de aditivos que podem ser utilizados nas composições da presente invenção.
Uma categoria de aditivos são polímeros e oligômeros biodegradáveis. Os polímeros podem ser usados para alterar o perfil de liberação do agente anestésico a ser liberado, para adicionar a integridade da composição ou de outro modo modificar as propriedades da composição. Os exemplos não limitantes de polímeros e oligômeros biodegradáveis adequados incluem: poli(lactídeo), poli(lactídeo-co- glicolídeo), poli(glicolídeo), poli(caprolactona), poliamidas, poli anidri dos, poliaminoácidos, poliortoésteres, policianoacrilatos, poli(fbsfazinas), poli(fosfoésteres), poliesteramidas, polidioxanonas, poliacetais, policetais, policarbonatos, poliortocarbonatos. poliuretanos degradáveis, poliidroxibutiratos, poliidroxivaleratos, polialquileno oxalatos, polialquileno succinatos, poli(ácido málico), quitina, quitosano e copolímeros, terpolímeros, celulose oxidada ou combinações ou misturas dos materiais acima.
Os exemplos de poli(a-hidróxi ácido)s incluem poli(ácido gicólico), poli(ácido DL-lático) e poli(ácido L-lático) e seus copolímeros. Os exemplos de polilactonas incluem poli(ε-caprolactona), poli(δ-valerolactona) e poli(y-butirolactona).
Embora não se deseje estar ligado por qualquer teoria, acredita-se que quando a composição contém um polímero biodegradável, uma porção do polímero pode precipitar ou coagular na superfície da composição visto que qualquer solvente incluído difunde para fora do material depois da administração ao indivíduo. O polímero pode ser assim adicionado como um agente modificador de liberação para afetar a liberação do agente ou agentes anestésico ou pode ser adicionado como parte de uma composição contendo microesferas, implantes ou partículas poliméricas moídas pré fonnadas. A precipitação ou coagulação do polímero forma uma pele que pelo menos parcialmente circunda o núcleo líquido de ta] composição. Esta pele é porosa e permite que o solvente continue a difundir através dela no tecido circundante. A taxa de liberação do solvente e a extensão da formação da pele, assim como a sua porosidade, podem ser controladas pela quantidade e tipo de solvente e polímero usados na composição.
Outros aditivos para o uso com as presentes composições são polímeros não biodegradáveis. Os exemplos não limitantes de polímeros não erodíveis que podem ser usados como aditivos incluem: poliacrilatos, polímeros de etileno-acetato de vinila, celulose e derivados de celulose, acetatos de celulose substituídos por acila e derivados destes, os poliuretanos não erodíveis, poliestirenos, cloreto de polivinila, fluoreto de polivinila, polivinila (imidazol), poliolefinas cloro-sulfonadas, óxido de polietileno e polietileno.
Os polímeros não biodegradáveis preferidos incluem polivinil pirrolidona, etileno acetato de vinila, polietileno glicol, acetato butirato de celulose (“CAB”) e acetato propionato de celulose (“CAP”).
Uma outra classe de aditivos que podem ser usados nas presentes composições são óleos e gorduras naturais e sintéticos. Os óleos derivados de animais ou de sementes de planta de nozes tipicamente incluem glicerídeos dos ácidos graxos, principalmente oléico, palmítico, esteárico e linoleico. Como uma regra quanto mais hidrogênio a molécula contém mais espesso o óleo se toma.
Os exemplos não limitantes de óleos naturais e sintéticos adequados incluem óleo vegetal, óleo de amendoim, triglicerídeos de cadeia média, óleo de soja, óleo de amêndoa, óleo de oliva, óleo de gergelim, óleo de funcho, óleo de camélia, óleo de milho, óleo de mamona, óleo de semente de algodão e óleo de soja, bruto ou refinado e triglicerídeos de ácido graxo de cadeia média.
As gorduras são tipicamente ésteres de glicerila de ácidos graxos superiores tais como esteáricos e palmíticos. Tais ésteres e suas misturas são sólidos nas temperaturas ambiente e exibem estrutura cristalina. Banha e sebo são exemplos. No geral óleos e gorduras aumentam a hidrofobicidade de um sistema carreador não polimérico, diminuem a degradação e a captação de água.
Todas as composições descritas acima podem ser usadas nos métodos da presente invenção de modo a fornecer anestesia local prolongada em um sítio alvo. Em particular, as composições podem ser formuladas como líquido, pulverização, creme, loção, ungüento, gel, pasta fluida, óleo, emulsão, microemulsão, sólido, emplastro, película, partícula, micropartícula, pó ou qualquer outra forma de composição farmacêutica adequada e depois administrada a um indivíduo por intermédio da liberação tópica, transdérmica, parenteral (por exemplo, injeção, implante, etc.) ou técnicas de liberação semelhantes. As composições, contendo um anestésico e ura carreador não polimérico farmaceuticamente aceitável, são usadas para fornecer um efeito anestésico caracterizado pela anestesia local prolongada depois da administração ao indivíduo sem um arrebentamento inicial e um duração de pelo menos cerca de 24 horas depois da administração, preferivelmente de pelo menos cerca de 36 a 48 horas depois da administração e mais preferivelmente de pelo menos cerca de 48 a 72 horas depois da administração. Em certas formas de liberação, o início da anestesia local ocorre dentro de cerca de 2 horas da administração ao indivíduo, preferivelmente dentro de cerca de 1 hora da administração e em alguns casos dentro de cerca de 30 minutos da administração ao indivíduo.
O termo “indivíduo,” como aqui usado, refere-se a qualquer vertebrado em que é desejado fornecer um estado de anestesia local. O termo assim amplamente refere-se a qualquer animal que seja tratado com as composições da presente invenção, tais como pássaros, peixe e mamíferos incluindo seres humanos. Em certas formas de liberação, os métodos da presente invenção são adequados para fornecer anestesia prolongada na prática veterinária e pecuária, por exemplo, pássaros e mamíferos, sempre que um estado de longa duração de anestesia local for conveniente ou desejável.
Em certos casos, as composições são particularmente adaptadas para o uso com animais de estimação tais como cães ou gatos e adicionalmente podem ser usadas com cavalos. Em formas de liberação preferidas, o termo “indivíduo” pretende um indivíduo humano. Além disso, o termo “indivíduo” não denota uma idade particular e as composições são assim apropriadas para 10 o uso com indivíduos de qualquer idade, tais como indivíduos criança, adolescente, adulto e de idade mais velha.
Em formas de liberação preferidas, as composições da presente invenção são particularmente apropriadas para o uso no tratamento de ferimentos. Os sistemas de carreador não polimérico permitem que o agente 15 ou agentes anestésicos sejam facilmente aplicados ao ferimento, diretamente dentro do ferimento e/ou adjacente ao ferimento, usando técnicas de aplicação muito simples tais como gotejamento sobre, pulverização, pintura, espalhamento, moldagem ou de outro modo manipulação manual de uma composição líquida, pulverização, creme, loção, unguento, gel, pasta fluida, 20 óleo, emulsão, microemulsão, sólido flexível ou emplastro, película, partícula, micropartícula ou pó no ferimento. As composições podem ser assim usadas com qualquer tamanho ou forma de ferimento e fornecerá uma distribuição uniforme do agente ou agentes anestésicos na área inteira do ferimento para melhor retenção e eficácia. Os ferimentos que podem ser tratados usando tais 25 métodos podem variar do mais superficial ao profundo, da superfície para incisional e do cirúrgico (ou de outro modo deliberado) para o acidental. Se a composição deva ser injetada, a mesma pode ser aplicada ao espaço subcutâneo usando uma injeção de arrasto ao longo do ferimento em todos os lados ou fora dos limites. Os métodos de combinação também podem ser utilizados, tais como onde a composição é tanto depositada diretamente dentro do ferimento, por exemplo, antes do fechamento cirúrgico da sonda quanto adicionalmente ao longo do ferimento. Em uma forma de liberação particularmente preferida, os métodos da invenção envolvem o uso das presentes composições como um anestésico local para o tratamento da dor incisional pós operatória. O uso das presentes composições desta maneira pode prevenir ou pelo menos mitigar a necessidade quanto a fornecer terapias suplementares, tais como a administração de analgésicos narcóticos sistêmicos de modo a tratar tal dor pós operatória. Consequentemente, as composições podem ser usadas para tratar a dor pós operatória que acompanha todos os tipos de procedimentos médicos, tais como cirurgias principais (por exemplo toracotomia, reparo aórtico, resseção do intestino), cirurgias intermediárias (por exemplo, corte cesariano, histerectomia e apendectomia) e cirurgias menores (laparoscopia, artroscopia e procedimentos de biópsia), que de outro modo podem ser debilitantes e podem requerer tratamento da dor por 3 a 5 dias depois da cirurgia.
As composições aqui descritas podem ser assim administradas na prática dos presentes métodos usando uma ampla variedade de métodos. Por exemplo, as composições podem ser administradas tópica, sistêmica (por exemplo, mucosicamente (oral, retal, vaginal ou nasalmente), parenteral (intravenosa, subcutânea, intramuscular ou intraperitonealmente) ou semelhante. As composições podem ser aplicadas por intermédio de injeção, irrigação, pulverização, imersão, aerossol ou aplicador de revestimento. Os aerossóis ou névoas da composição podem ser administrados usando um propelente de aerossol, por exemplo, para a administração tópica ou usando um nebulizador adequado, por exemplo, para a administração mucósica nasal ou oral.
Preferivelmente, as composições são administradas como líquidos por intermédio de injeção ou em um aerossol, pasta ou emulsão.
Quando usado em um aerossol, qualquer solvente presente na solução de aerossol tipicamente evaporará na aplicação, deixando a composição assentar como uma película. Altem ativamente, o aerossol ou emulsão podem ser preparados sem um solvente. Nesta situação, o propelente de aerossol também pode funcionar como um solvente. A formação de aerossóis e emulsões podem ser realizadas usando técnicas conhecidas por aqueles habilitados na técnica. Ver, por exemplo, Ansel, H. C. et al.,Pharmaceutic Dosage Forms and Drug Delivery Systems, Sexta Edição (1995).
Além dos usos descritos acima, as presentes composições podem ser administradas através de bombas osmóticas. Em uma forma de liberação, um dispositivo é projetado para ser implantado no tecido do indivíduo e planejado para efetuar a liberação prolongada com o tempo.
Também é possível administrar as composições da invenção usando um tubo poroso ou não poroso, desejavelmente fabricado de polímero biodegradável extrusado. O tubo pode ser preparado com graus variáveis de porosidade dependendo das características da composição e das características de liberação desejadas. A composição da invenção é inserida no tubo e as extremidades do tubo pode ser deixada aberta, possibilitando que o composto biologicamente ativo se difunda das extremidades do tubo ou possa ser bloqueado com polímero poroso ou não poroso adicionais. Tampas porosas e tubos porosos permitem que o composto ativo se difunda através dos poros com o tempo. As tampas não porosas, assim como os tubos não porosos, permitem que os agentes anestésicos que são solúveis no polímero difundam através dele e dentro dos tecidos circundantes. Os materiais não porosos que não são solventes para o anestésico, mas que são biodegradáveis, liberarão o anestésico quando este se degrada suficientemente. As composições da invenção podem ser preparadas e armazenadas como sistemas de componente múltiplo até prontos para a administração. O número de componentes diferentes dependerá, em parte, das características da composição. Antes da administração, os componentes são combinados e misturados, por exemplo, para se obter uma composição homogênea, que pode ser depois administrada ao indivíduo. Os solventes ou aditivos podem ser adicionados a um ou a todos os componentes ou podem formar um componente separado, que também é misturado com os outros antes da administração. A separação da composição em uma mistura de componente múltiplo permite que as condições de armazenagem para cada componente sejam otimizadas e minimiza quaisquer interações nocivas entre os componentes com o tempo. O resultado é a estabilidade na armazenagem aumentada.
EXEMPLOS
Os abaixo são exemplos de formas de liberação especificas para realizar a presente invenção. Os exemplos são oferecidos apenas com propósitos ilustrativos e não são intencionados a limitar o escopo da presente invenção de nenhum modo.
Métodos Gerais
A eficácia in vivo das composições e os métodos da invenção podem ser avaliados no rato usando um modelo de placa quente, por exemplo, de acordo com o procedimento descrito em detalhes na IACUC N° 9511- 2199. Os critérios de eficácia estabelecidos para as composições da invenção 20 são latência média maior do que cerca de 2 segundos, com um corte de 12 segundos (este corte é imposto para impedir qualquer possível dano ao animal). As latências em 2 segundos são demonstrativos de um efeito estatisticamente significante do anestésico local. Preferivelmente, a latência média sob o modelo de placa quente de rato é maior do que 7 segundos.
Preferivelmente, a porcentagem de respondedores é de 50 % ou maior. Preferivelmente, as composições da invenção fornecem uma latência média sob o modelo de placa quente de rato maior do que cerca de 7 segundos a cerca de 12 segundos, com a porcentagem de ratos exibindo o efeito sendo pelo menos cerca de 50 % daquele testado. z metodologia da placa quente de rato é resumida como segue. Ratos Sprague Dawley machos (Harlan Laboratories, Indianápolis, Ind.) com um peso médio de 275 g são usados. O estudo de placa quente consiste de conter suavemente o corpo do animal enquanto a superficie plantar da pata traseira é colocada sobre uma placa quente aquecida a 56° C. A latência de referência é determinada antes da injeção unilateral de composição anestésica em torno do nervo ciático do rato.
O teste sensorial em modelos humanos também é útil no teste das composições da presente invenção. A atividade de anestésico local pode ser examinada com referência ao início, densidade de pico e duração de efeito usando sete modalidades específicas: (a) teste sensonal mecânico (limiar de detecção da dor mecânica usando pelos de von Frey; (b) teste supralimiar (mecânico) usando um pelo de von Frey único; (c) teste sensorial térmico (limiar de detecção térmica); (d) limiar de detecção de dor ao calor; (e) teste supralimiar (térmico); (f) limiar de detecção de frio; e (g) teste sensorial tátil (limiar de detecção de toque mecânico). Os graus ou níveis variáveis ou dos resultados serão indicativos do alívio da dor local que experiencia o indivíduo, dormência e ou bloqueio nervoso local. A atividade anestésica das composições e métodos da invenção pode ser caracterizada ainda com respeito à segurança, por várias medidas de atividade tais como níveis plasmáticos sanguíneos sistêmicos atingidos depois da administração no sítio localizado.
O limiar de detecção da dor mecânica é definido como a força mais baixa ou número de um Pelo de von Frey que produza uma sensação definida de dor ou desconforto e o limiar de detecção de toque mecânico é definido como a força mais baixa ou número de um Pelo de von Frey que produza uma sensação de toque ou pressão. O Limiar de Detecção de Toque Mecânico e os Limiares de Detecção de Dor Mecânica podem ser determinados simultaneamente usando Pelos de von Frey progressivamente rígidos (VFH) (disponíveis da Somedic A/B, Stockholm, Suécia). Foi anteriormente determinado que cada VFH pressionado contra uma balança até que ela levemente curvou-se representa uma força que logaritmicamente aumenta com cada pelo, abrangendo uma faixa total de 3 a 402 miliNewtons (mN) (VFH N2 7=3 mN; VFH N2 8=13 mN; VFH Ns 9=20 mN; VFH N2 10=39 mN; VFH N2 11=59 mN; VFH N2 12=98 mN; VFH N2 13=128 mN; VFH N2 14=133 mN; VFH N2 15=314 mN; VFH N2 16=350 mN; VFH N2 17=402 mN).
Consequentemente, em um indivíduo humano, uma área injetada com uma composição produzida de acordo com a presente invenção pode ser estimulada 8 vezes com cada VFH a uma razão de cerca de 2 estímulos por segundo, partindo com VFH N2 7 e movendo para VFH N2 17. O número de VFH mais baixo que seja sentido como toque ou pressão (Limiar de Detecção de Toque Mecânico) e o número mais baixo do pelo em que metade dos oito estímulos são dolorosos ou desagradáveis (Limiar de Detecção de Dor Mecânica) são registrados. O procedimento é repetido duas vezes mais e a média das três medições é relatada. Se VFH N2 17 não produz a sensação de toque ou pressão um valor de Limiar de Detecção de Toque Mecânico de 18 será designado. Se VFH N217 não produz nenhuma dor ou desconforto um valor de Limiar de Detecção de Dor Mecânica de 18 será designado. A Resposta Mecânica à Dor Supralimiar a um único Pêlo de von Frey é determinado estimulando-se as áreas injetadas cinco vezes com VFH N217 (402 mN). O indivíduo avalia a dor usando uma escala de VRS de 0 a 10, onde zero (0) = nenhuma dor e dez = (10) dor tão intensa quanto imaginável.
Como debatido acima, este teste é conduzido com um único Pêlo de von Frey rígido que seja determinado produzir uma resposta dolorosa nos indivíduos. A resposta é determinada pela estimulação de uma área injetada ou de outro modo tratada 5 vezes com VFH N2 17. O indivíduo avalia a dor na Escala de Classificação Verbal (VRS) de 0 a 10, como acima.
O teste térmico (Resposta de Dor Supralimiar ao Calor) em uma área tratada é determinada por um estímulo de 45° C, durando 5 segundos usando um thermode computadorizado (disponível da Thermostest, Somedic A/B, Stockholm, Suécia) nas áreas tratadas. O indivíduo avalia a dor na Escala de Classificação Verbal (VRS) de 0 a 10.
O Limiar de Detecção de Aquecimento é definido como o aumento mais baixo na temperatura de 32° C percebido, o Limiar de Detecção da Dor Térmica é definido como a temperatura mais baixa percebida como Limiar de Detecção de Dor e Frio é definido como a diminuição mais baixa na temperatura a partir de 32° C percebida. Limiar de Detecção Quente, Limiar de Detecção de Dor Quente e Limiar de Detecção de Frio são determinados com um Thermostest computadorizado (disponível da Somedic A/B, Stockholm, Suécia) nas áreas tratadas. Os indivíduos são instruídos para apertar um botão tão logo a sensação especificada seja atingida. Os limiares térmicos são determinados a partir de uma referência de 32° C e aumentados (Limiar de Detecção Quente e Limiar de Detecção de Dor Térmica) ou diminuída (Limiar de Detecção Fria) a uma taxa de mudança de Io C por segundo. O limite de corte superior é de 52° C para o Limiar de Detecção Quente e Limiar de Detecção de Dor Térmica. O limite de corte inferior é de 25° C para o Limiar de Detecção Fria.
O Limiar de Detecção Quente, Limiar de Detecção de Dor Térmica e Limiar de Detecção Fria são calculados como a média de três medidas, com intervalos de 10 segundos entre cada estímulo. Se o indivíduo não percebeu a quentura ou dor a 52° C, o valor a 53° C é registrado para o Limiar de Detecção Quente; se o indivíduo não percebeu a dor em 52° C, o valor de 53° C é registrado para o Limiar de Detecção de Dor Térmica; e se o indivíduo não percebeu a frieza ou dor a 25° C, o valor de 24° C é registrado para o Limiar de Detecção Frio.
Exemplo 1
Um sistema carreador líquido não polimérico contendo um agente anestésico foi produzido como segue. Acetato isobutirato de sacarose (SAIB) foi combinado com um solvente de N-metilpirrolidona (NMP) para o carreador SAIB para fornecer uma mistura 70:30. A esta mistura 2,5 % (p/v) ou 5 % de bupivacaína (base livre) foram adicionados para fornecer duas composições de teste.
Ratos Sprague Dawley machos, (275 a 300 g) foram divididos em dois grupos de teste de 8 animais cada um. As formulações de teste foram administradas nos quadrúpedes usando agulha e seringa para liberar doses de 25 ou 50 mg da bupivacaína. O teste de resposta à retração da pele foi depois usado para determinar a presença de um efeito anestésico local, em que a retração involuntária no estímulo cutâneo usando um alfinete aplicado a 10 áreas aleatórias dentro de 1 cm da base do sítio de injeção. A inibição percentual da percepção do alfinete foi depois calculada tomando-se a resposta de referência menos a resposta de teste, dividido pela resposta de referência e multiplicados por 100.
Os resultados obtidos indicaram que ambas as composições de teste forneceram efeito anestésico local por até cerca de 60 a 72 horas de duração, com um início de atividade dentro de 1 hora de administração.
Exemplo 2
Indivíduos. Ratos Fisher 344, machos (Charles River Laboratories) (N=96) foram usados para o estudo. Os animais foram mantidos em um ciclo de luz:escuro reverso (escuro das 5:00 às 17:00) em um viveiro de temperatura e umidade controladas. Aos ratos foram dados acesso ad liba alimento e água exceto durante as sessões experimentais. Todos os experimentos foram conduzidos durante a fase escura do ciclo de luz:escuro. Todos os procedimentos foram aprovados pela Institutional Animal Care and Use Committee of Wake Forest University Health Sciences Center.
Procedimento Cirúrgico. A seguir da indução de anestesia com vapor de isoflurano a 5 % em oxigênio, os animais foram raspados no quadrante inferior esquerdo do abdômen. A anestesia foi mantida por todo o procedimento cirúrgico usando vapor de isoflurano de 2,0 a 2,5 % em oxigênio. Uma incisão de 3 cm foi colocada 0,5 cm abaixo e paralela à costela mais baixa no lado esquerdo, penetrando na cavidade peritoneal. As vísceras e musculatura foram vigorosamente manipuladas pela inserção de 5 a 7 cm do dedo indicador dentro da cavidade peritoneal e esticando a musculatura. Aproximadamente 10 cm do intestino delgado foi exteriorizado e suavemente manipulado. O intestino foi colocado dentro da cavidade peritoneal e o ferimento foi suturado em 3 camadas que consistem do revestimento peritoneal, músculo abdominal e pele usando fio crômico 4,0. Curativos foram feitos nos ferimentos exteriores com pó antibiótico (Polisporin®, GlaxoWellcome, Research Triangle Park, NC) e os animais foram ministrados com 75.000 U de penicilina G procaína i.m. os animais tratados com simulado foram anestesiados, raspados, mantidos sob anestesia com isoflurano por 20 min e ministrados com penicilina G procaína i.m.
Administração da composição de bupivacaína de liberação controlada. Os animais de teste foram administrados com veículo (70:30, SAIBJNMP), 1,25 % (p/v), 2,5 % ou 5 % de bupivicaína de acordo com a tabela 1. A composição foi administrada a seguir da sutura da pele usando uma técnica de injeção de arrasto com uma seringa de 1,0 ml e um 1,5 em agulha de 22 ga tal que 0,25 ml da composição foi uniformemente distribuída na área de incisão em aproximadamente 0,5 cm acima do sítio do ferimento.
Uma segunda administração foi dada em uma maneira similar 0,5 cm abaixo do sírio de ferimento. Para os grupos K e L, uma administração adicional de 0,1 ml de bupivicaína a 1,25 % (p/v) ou 5 % foi ministrada no topo do revestimento peritoneal usando a técnica de injeção de arrasto antes da sutura na camada do músculo externo. A composição de bupivicaína foi deixada permanecer sobre o revestimento peritoneal para 1 a 2 min antes de suturar o músculo externo para obter viscosidade suficiente e deste modo impedir a composição de lixiviar através do músculo externo durante o processo de sutura. Tabela 1. Os grupos foram tratados como descrito abaixo. Todos os grupos, com a exceção do grupo B, receberam laparotomia cirúrgica como descrito acima.
Medição da Locomoção Espontânea. O comportamento exploratório foi avaliado começando 24 horas depois da laparotomia usando equipamento e software comercialmente disponível (Med Associates Inc., St. Albans, VT). As câmaras de atividade consistiram de fechamentos de acrílico medindo 17” x 17” que tiveram 15” de altura com um topo aberto. Os bancos de duplicata de 16 transmissores infravermelhos espaçados 1” separadamente foram colocados tanto na direção X quanto na Y, 1” acima da superficie do piso, com detectores infravermelho alinhados nos lados opostos da câmara. Um terceiro banco de transmissores infravermelho e detectores foram localizados na direção X, 7 cm acima da superficie do piso tal que os ratos usados para estes estudos fossem requeridos erguer nas suas patas traseiras de modo a intemomper estes feixes. Cada câmara de atividade foi alojada dentro de um fechamento atenuador de luz e som. As sessões foram conduzidas em 16, 24, 40, 48 e 72 horas no pós operatório e foram de 60 min na duração. As medidas coletadas incluem a distância total viajada, rupturas do feixe totais tanto na direção X quanto Y (contagens de ambulatório), rupturas de feixes repetidos dentro de 3 cm do animal na ausência de locomoção (estereotipia), rupturas de feixe total na direção X superior (levantamento nas patas traseiras), tempo gasto em movimento, tempo gasto na estereotipia, tempo gasto levantando- se nas patas traseiras e tempo gasto no repouso. Todas as medidas foram coletadas em 6 min bins por todo a sessão assim como somado para a totalidade da sessão.
Medição de retomo de anestesia usando o teste de picada de alfinete. Em 96 horas do pós operativo, os animais foram levemente restringidos em uma câmara de contenção de acrílico claro que permite acesso à área abdominal. Uma agulha levemente rombuda de 20 ga foi empurrada dentro de uma região dentro de 0,5 cm do sítio de ferimento abdominal com força suficiente para curvar de modo perceptível o tecido abdominal. A agulha foi mantida no lugar por 10 segundos ou até que o animal reagisse recuando ou puxando fora da agulha. Cada animal foi testado 2 a 3 vezes, no sítio de ferimento. Um sítio pelo menos 10 cm para fora da área de ferimento foi testado em uma maneira similar como uma medida de controle positivo. Análise de dados. Os dados foram analisados usando um ANOVA de 2 fatores para as medições repetidas com índices comportamentais individuais servindo como a medida independente e tanto o grupo de tratamento quanto o tempo pós operatório como as variáveis dependentes. As análises de post-hoc foram realizadas usando o teste t de Bonferroni-Dunn para comparações múltiplas com o grupo de cirurgia simulado servindo como controle e usando LSD Protegido de Fisher para todas as comparações aos pares.
Resultados (distância percorrida). Os efeitos da cirurgia e tratamento peri-operativo com veículo ou bupivicaína são mostrados para cada ponto de tempo nas Figuras 1 a 5. Houve um efeito principal significante de grupo de tratamento e tempo depois da cirurgia na distância percorrida sem nenhuma interação significante entre estas variáveis (Tabela 1). Tabela 2. Resultados de ANOVA de 2 fatores para a distância percorrida (cm).
Dependente: distância.
A análise post-hocusando o teste t de Bonferroni/Dunn para as comparações múltiplas a um grupo de controle revelou que todos os grupos administrados com bupivicaína não foram significantemente diferentes do tratamento simulado, ao passo que os animais de incisão não ministrados com nenhum tratamento ou os animais tratados com veículo permaneceram significantemente diferentes dos controles simulados por todo o estudo (Tabela 2). A análise post-hoc de todas as comparações aos pares usando LSD Protegido de Fisher verificou que apenas os grupos administrados com bupivicaína a 5 % ou bupivicaína a 1,25 % ambas no sítio do revestimento peritoneal e no músculo externo não foram significantemente diferentes do simulado (Tabela 3), Tabela 3. LSD Protegido de Fisher para todas as comparações aos pares,distância percorrida (cm).
S '= Significantemente di ferente neste riivel.
Contagens Ambulativas. Os resultados estatísticos para os dados de contagem ambulativas são qualitativamente similares àqueles descritos para a distância percorrida acima. Os dados para as contagens ambulativas foram avaliados para cada ponto de tempo individual a seguir da cirurgia. Como com a distância percorrida durante a sessão, houve um efeito principal significante tanto do grupo de tratamento quanto tempo depois da cirurgia nas contagens ambulativas, sem nenhuma interação significante entre estas variáveis (Tabela 4).Tabela 4. Resultados de ANOVA de 2 fatores para as contagens ambulativas contagens ambulativas.
Dependente: contagens ambulativas
A análise post-hoccom o teste t de Bonferroni/Dunn usando as contagens ambulativas como a medida independente produziu resultados similares como os dados para a distância percorrida (Tabela 5). Tabela 5. Teste t de Bonferroni/Dunn para as comparações múltiplas,contagens ambulativas.
S = Significantemente diferente neste nível.
A análise de todas as comparações aos pares com LSD Protegido de Fisher verificou que o grupo simulado foi apenas 20 significantemente diferente do grupo de incisão de controle, o grupo tratado com veículo e o grupo tratado com a dose mais baixa de bupivicaína (1,25 %) (Tabela 6). Tabela 6. LSD Protegido de Fisher para todas as comparações aos pares, contagens ambulativas.
S = Significantemente diferente neste nível.
Contagens de Estereotipia. Não houve diferenças significantes encontradas entre os grupos para os movimentos próximos (estereotipia) mas houve um efeito principal significante de tempo depois da cirurgia e uma interação significante entre o grupo de tratamento e o tempo pós cirúrgico (Tabela 7, Figuras 11 a 15). A análise de post-hoc usando o teste t de Bonferroni/Dunn revelou que o ponto de tempo de 40 horas foi [0 significantemente diferente dos outros pontos de tempo para esta medida (Tabela 8). A exclusão do ponto de tempo de 40 horas e a reanálise dos dados remanescentes pelo ANOVA de 2 fatores revelou um efeito marginalmente significante do grupo de tratamento sobre o comportamento estereotípico, um efeito principal significante de tempo pós cirúrgico e uma interação significante entre grupo de tratamento e tempo depois da cirurgia (Tabela 9). A análise de post-hoc pelo Bonferrroni/Dunn não revelou nenhuma diferença significante entre grupos de tratamento, mas o teste t de LSD Protegido de Fisher verificou que todos os grupos foram significantemente diferentes do controle simulado com a exceção da dose mais alta de bupivicaína (5 %) assim como o grupo tratado com bupivicaína a 1,25 % ambos ao nível do revestimento peritoneal e na camada de músculo abdominal (Tabela 10). Tabela 7. ANOVA de 2 Fatores, contagens de estereotipia.

Dependente: contagens de estereotipia
Tabela 8. Bonferroni/Dunn para comparações múltiplas para controle, tempo depois da cirurgia.
S = Significantemente diferente neste nível.
Tabela 9. ANOVA de 2 Fatores, excluindo o ponto de tempo de 40 horas, contagens de estereotipia.
Dependente: contagens de estereotipia
Tabela 10. LSD Protegido de Fisher para todas as comparações aos pares excluindo o ponto de tempo de 40 horas, contagens de estereotipia.
S = Significantemente diferente neste nível.
Contagens Verticais (levantamento nas patas traseiras). Houve um efeito principal significante tanto de grupo de tratamento quanto de tempo depois da cirurgia no levantamento nas patas traseiras, assim como uma interação significante entre estas variáveis (Tabela 11). A análise de post-hoc usando o teste t de Bonferroni/Dunn revelou diferença significante entre o controle simulado e todos os grupos de tratamento (Tabela 12). Os resultados do teste de LSD Protegido de Fisher produziu descobertas compatíveis, sem nenhuma diferença significante encontrada com a exceção do grupo simulado com todos os outros tratamentos (Tabela 13). Tabela 11. ANO VA de 2 fatores, contagens verticais.
Dependente: contagens verticais
Tabela 12. Bonferroni/Dunn para comparações múltiplas para controle, contagens verticais.
S = Significantemente diferente neste nível.
Tabela 13. LSD Protegido de Fisher para todas as comparações aos pares, contagens verticais.
S = Significantemente diferente neste nível.
Estímulo de picada de alfinete. Os animais foram submetidos ao estímulo de picada de alfinete em uma área dentro de 0,5 cm da incisão 96 horas depois da cirurgia. Todos os animais responderam a este estímulo com a exceção de um único animal no grupo HH (bupivicaína a 1,25 % administrada tanto na camada de músculo abdominal quanto no revestimento peritoneal). Este animal respondeu normalmente ao estímulo da picada de alfinete quando este foi administrado em um sítio aproximadamente 3 cm do sítio da incisão. Resultados. O tratamento peri-operatório com o veículo usado para a composição de liberação prolongada (o controle) não teve nenhum efeito significante sobre as medidas de atividade comparadas com a incisão abdominal sem nenhum outro tratamento. Houve um efeito de veículo evidente sobre o comportamento estereotípico em alguns dos pontos de tempo posteriores. Os dados compativelmente sustentam um efeito analgésico significante da bupivicaína a 5 % na composição de liberação prolongada que parece estar presente nos pontos de tempo mais tardios estudados. A composição de liberação controlada de bupivicaína a 5 % compativelmente forneceu analgesia substancial e reverteu o efeito da incisão abdominal sobre todas as medidas de comportamento com a exceção da atividade vertical. Alguns efeitos analgésicos foram descobertos a seguir da administração da bupivicaína a 1,25 % em ambos os níveis do revestimento peritoneal e na camada externa de músculo abdominal, entretanto os efeitos não foram tão compatíveis como aqueles encontrados a seguir da administração da bupivicaína a 5 % apenas na camada de músculo externo. O efeito da bupivicaína a 5 % é mais robusto nos pontos de tempo de 48 e 72 horas. A(s) razão(Ões) para estes animais que demonstram menos atividade nos pontos de tempo mais inicial não é(são) prontamente evidentes visto que estes animais demonstram alimentação e comportamento de arrumar-se normais nas suas gaiolas e não parecem estar em aflição. Estes dados estabelecem que o efeito anestésico local é suficiente para controlar a dor a seguir da cirurgia usando as composições de liberação controlada da invenção contendo um anestésico de bupivicaína.
Exemplo 3
A seguir da escalação de dose, a segurança e avaliação farmacocinética foi realizada em indivíduos voluntários humanos saudáveis de modo a avaliar a segurança/tolerabil idade e desempenho de farmacocinéticas preliminares de composições de bupivacaína de liberação controlada que compreenda um carreador não polimérico de acetato isobutirato de sacarose.
As composições foram formuladas usando base livre de bupivacaína formulada em um carreador não polimérico de acetato isobutirato de sacarose (SAIB) incluindo ainda N-metil-2-pirrolidona (NMP) que atua como um solvente para a bupivacaína e o carreador SAIB. A composição foi preparada combinando-se o carreador SAIB e o solvente NMP (veículo 70:30) com 5 % em peso de bupivacaína, para fornecer dosagens individuais contendo 137,5 mg da bupivacaína em um volume de injeção de 2,5 ml. A composição foi fornecida como um líquido injetável.
Houve dois Grupos no estudo. O Grupo 1 foi compreendido de 6 indivíduos do sexo masculino saudáveis, da idade de 22 a 38. Para o Grupo 1, todos os indivíduos receberam 2,5 ml de injeções de volume total da composição de SAIB/NMP/bupivacaína (contendo 137,5 mg de bupivacaína) em um primeiro sítio de administração, administrado no espaço subcutâneo abdominal como uma injeção de arrasto de 5 cm; e injeções de placebo de 2,5 ml contendo apenas veículo (SAIB/NMP) em um segundo sítio de administração, também administradas no espaço subcutâneo abdominal como uma injeção de arrasto de 5 cm. Depois das administrações, os indivíduos foram avaliados por até oito horas para monitorar as condições de tecido local no sítio de administração e amostras de plasma foram coletadas. Amostras de plasma adicionais foram tiradas nos Dias 1, 2, 3, 4 e 28. Todas as amostras de plasma foram testadas quanto a concentração de bupivacaína no sangue usando métodos padrão.
O Grupo 2 também foi compreendido de 6 indivíduos do sexo masculino saudáveis, de idade de 22 a 38. O Grupo 2 foi dividido em dois subgrupos, o primeiro subgrupo (n = 3) recebeu injeções no volume total de 5 ml da composição de SAIB/NMP/bupivacaína (contendo 275 mg de bupivacaína), administradas no espaço subcutâneo abdominal como duas injeções de arrasto de 5 cm de 2,5 ml cada. O segundo subgrupo (n = 3) recebeu injeções de placebo de volume total de 2,5 ml contendo apenas veículo (SAIB/NMP), também administradas no espaço subcutâneo abdominal como duas injeções de arrasto de 5 cm de 1,25 ml cada. Aqui mais uma vez, depois das administrações, os indivíduos foram avaliados por até oito horas para monitorar as condições de tecido local no sítio de administração e amostras de plasma foram coletadas. As amostras de plasma adicionais foram tiradas nos Dias, 1,2, 3, 4 e 28.
Como um resultado do estudo, foi descoberto que o carreador SA1B e as composições de SAIB/NMP/bupivacaína foram bem toleradas, onde as injeções não resultaram em qualquer vermelhidão, inchaço, coceira, descoloração observáveis ou qualquer outro sintoma adverso nos sítios de injeção ou qualquer reação de tecido inaceitável por toda a duração do estudo. Além disso, as avaliações farmacocinéticas de bupivacaína mostraram liberação lenta adequada (prolongada) do ativo de bupivacaína a partir do carreador SAIB, liberação do ativo de bupivacaína em um período de 3 a 4 dias. Estes resultados farmacocinéticos são apresentados na Figura 1. Como pode ser observado, as concentrações Cmax de bupivacaína plasmática a seguir das injeções do Grupo 1 (84 ng/ml) e Grupo 2 (174 ng/ml) foram bem abaixo das faixas de concentração plasmática tóxica estimada publicadas (cerca de 1 a 4 pg/ml). Além disso, uma comparação entre as curvas do Grupo 1 e Grupo 2 mostram que os níveis plasmáticos de bupivacaína aumentam em uma maneira substancialmente linear com a escalação de dose. Além disso, avaliações da AUC mostram que houve 100 % de biodisponibilidade da bupivacaína.
Exemplo 4
A seguinte avaliação de escalação de dose, farmacocinética, farmacodinâmica (eficácia) é realizada em indivíduos humanos que passam por procedimentos de reparo de hérnia inguinal cirúrgica de modo a avaliar a eficácia e desempenho farmacêutico de composições de bupivacaína de liberação controlada que compreendem um carreador não polimérico de acetato isobutirato de sacarose e preparadas de acordo com a presente invenção. O estudo compara a eficácia das presentes composições de SAIB/bupivacaína administradas subcutaneamente em combinação com um infiltrado de ferimento de solução salina (placebo) ou cloridreto de bupivacaína (Marcain®), em relação a uma solução de bupivacaína comercial mente disponível (Marcain®, Cloridreto de Bupivacaína BP, 5,28 mg/ml, equivalente ao cloridreto de bupivacaína anidro 5 mg/ml) administrada subcutaneamente e como um infiltrado em indivíduos de reparo de hérnia inguinal aberta.
A composição de teste foi/é formulada usando base livre de bupivacaína formulada em um carreador não polimérico de acetato isobutirato de sacarose (SAIB) incluindo ainda álcool benzílico (BA) que atua como um solvente para a bupivacaína e o carreador SA1B. O álcool benzílico também é um agente anestésico. A composição foi/é preparada combinando-se cerca de 66 % em peso do carreador de SAIB, 22 % em peso do solvente de álcool benzil ico/anestésico e 12 % em peso de bupivacaína, para fornecer dosagens individuais contendo 159,5 mg de bupivacaína em um volume de injeção de 1,25 ml (319 mg era um volume total de 2,5 ml). A composição foi/é fornecida como um líquido claro injetável.
O estudo é planejado para incluir 3 Grupos com até 91 indivíduos (6 indivíduos para o Grupo 1; 15 indivíduos para o Grupo 2; e até 70 indivíduos para o Grupo 3). Em particular, o Grupo 1 foi compreendido de 6 indivíduos do sexo masculino saudáveis, de idade de 23 a 52. Para o Grupo 1, todos os indivíduos receberam injeções no volume total de 2,5 ml da composição de SAIB/BA/bupivacaína (contendo 319 mg de bupivacaína), administrada como as injeções subcutâneas de arrasto ao longo de cada lado do ferimento cirúrgico (0,5 ml/cm ao longo de uma incisão de ferimento de comprimento total de 5 cm sugeridos) com 10 ml de solução salina infiltrada no ferimento de incisão (incluindo subfascial) antes do fechamento do ferimento. As injeções de arrasto foram administradas 0,5 a 1,0 cm fora do e paralelo às margens do ferimento de incisão e foram realizadas avançando-se a agulha subcutaneamente, paralelo a e ao longo do comprimento da incisão, injetando continuamente conforme a agulha foi retirada. O efeito anestésico/analgésico foi avaliado usando os testes de Tempo para a Primeira Medicação Analgésica Suplementar e Consumo de Medicação Analgésica Suplementar Total (no curso de 4 dias). A concentração plasmática de bupivacaína foi medida periodicamente por todo o curso do estudo, particularmente através das primeiras 24 horas para avaliar a magnitude da liberação inicial de bupivacaína a partir da composição de liberação controlada de SAIB.
Os resultados do teste de Tempo para a Primeira Medicação Analgésica Suplementar são relatados abaixo na Tabela 14. Tabela 14. Tempo para a Primeira Medicação Analgésica Suplementar.
Os resultados do Consumo de Medicação Analgésica Suplementar Total (no curso de 4 dias) são relatados abaixo na Tabela 15. Tabela 15. Consumo de Medicação Analgésica Suplementar Total.
Como com o estudo do Exemplo 3, foi mais uma vez descoberto que a composição de SAIB/BA/bupivacaína foi bem tolerada, onde as injeções não resultam em qualquer vermelhidão, inchaço, coceira, descoloração observáveis ou qualquer outro sintoma adverso no sítio de injeção ou qualquer reação de tecido inaceitável por toda a duração do estudo.
Além disso, as avaliações farmacocinéticas da bupivacaína mostraram liberação prolongada do ativo de bupivacaína do carreador SA1B, liberando o ativo de bupivacaína em um período de 4 dias. Os resultados farmacêuticos são apresentados nas Figuras 2 e 3. Como pode ser observado, a composição de SAIB/BA/bupivacaína liberou o ativo de bupivacaína rapidamente (dentro de cerca de 1 hora da administração) sem um arrebentamento inicial e mostrou uma liberação constante, estado uniforme substancialmente pelo menos nos primeiros 3 dias de tratamento. A Cmax média observada foi de 277 ng/ml ± 109; o Tmax foi de 23 horas 4 21; e o Css foi 191 ng/ml ±13.
O Grupo 2 foi compreendido de 15 indivíduos do sexo masculino saudáveis, de idade de 26 a 54. O Grupo 2 foi dividido em três subgrupos, o primeiro subgrupo (n = 5) recebeu injeções em volume total de 5,0 ml da composição de SAIB/BA/bupivacaína (contendo 638 mg de bupivacaína), administrada como duas injeções subcutâneas de arrasto ao longo de cada lado do ferimento cirúrgico (0,5 ml/cm ao longo de uma incisão de ferimento de comprimento total de 5 cm sugeridos) com 10 ml de solução salina infiltrados no ferimento da incisão (incluindo subfascial) antes do fechamento de ferimento. O segundo subgrupo (n = 5) receberam injeções de volume total de 5 ml da composição de SAIB/BA/bupivacaína (contendo 638 mg de bupivacaína), administrada como duas injeções subcutâneas de arrasto ao longo de cada lado do ferimento cirúrgico (0,5 ml/cm ao longo uma incisão de ferimento de comprimento total de 5 cm sugeridos) com 10 ml de Marcain® (0,5 % de Bupivacaína-HCl) infiltrados dentro do ferimento da incisão (incluindo subfascial) antes do fechamento do ferimento para produzir um total de 688 mg de bupivacaína administrada por indivíduo. O terceiro subgrupo (n = 5) receberam injeções de volume total de 5 ml da composição de Marcain® (0,5 % de Bupivacaína-HCl) administrados como duas injeções subcutâneas de arrasto ao longo de cada lado do ferimento cirúrgico (0,5 ml/cm ao longo uma incisão de ferimento de comprimento total de 5 cm sugeridos) ao longo com 10 ml de Marcain® infiltrados no ferimento da incisão (incluindo subfascial) antes do fechamento do ferimento para produzir um total de 75 mg de bupivacaína administrada por indivíduo.
O efeito anestésico/analgésico foi avaliado usando os testes de Tempo para a Primeira Medicação Analgésica Suplementar, Contagem de Dor no Sítio de Incisão “no repouso” e Consumo de Medicação Analgésica Suplementar Total (no curso de 4 dias). A concentração plasmática de bupivacaína foi medida periodicamente por todo o curso do estudo, particularmente nas primeiras 24 horas para avaliar a magnitude da liberação inicial de bupivacaína da composição de liberação controlada de SAIB.
Os resultados do teste de Tempo para a Primeira Medicação Analgésica Suplementar e o teste Consumo de Medicação Analgésica Suplementar Total (no curso de 4 dias) para todos os três subgrupos para o Grupo 2 são relatados abaixo na Tabela 16. Tabela 16. Tempo Médio para a Primeira Medicação 5 Analgésica Suplementar e Consumo de Medicação Analgésica Suplementar Total Médio (no curso de 4 dias).
(* Três indivíduos no Subgrupo 1 e dois indivíduos no Subgrupo 2 não tomaram nenhuma dose analgésica suplementar durante o período de 4 dias inteiro).
Mais uma vez, a composição de SAIB/BA/bupivacaína foi 10 bem tolerada (os indivíduos dos subgrupos 1 e 2), onde as injeções não resultaram em qualquer vermelhidão, inchaço, coceira, descoloração observáveis ou qualquer outro sintoma adverso no sítio de injeção ou qualquer reação de tecido inaceitável por toda a duração do estudo. Além disso, as avaliações farmacocinéticas de bupivacaína mostraram liberação 15 prolongada do ativo de bupivacaína a partir do carreador de SAIB, liberando o ativo de bupivacaína em um período de 4 dias. Os resultados farmacocinéticos são apresentados nas Figuras 4 e 5. Como pode ser observado, a composição de SAIB/BA/bupivacaína liberou o ativo de bupivacaína rapidamente (dentro de cerca de 1 hora da administração) sem 20 um arrebentamento inicial e mostrou uma liberação substancialmente constante, de estado uniforme durante pelo menos os primeiros 3 dias de tratamento.
As farmacodinâmicas para todos os três subgrupos do Grupo 2 são relatados abaixo na Tabela 17. Tabela 17. Farmacodinâmicas para o Grupo 2.
Como pode ser observado a partir dos resultados do estudo do Grupo 2, as presentes composições de liberação controlada fornecem efeito anestésico local eficaz durante o curso de pelo menos 4 dias depois da cirurgia, reduzindo enormemente a necessidade quanto a medicações analgésicas suplementares. De fato, 50 % dos indivíduos que receberam as composições de SAIB/BA/Bupivacaína da presente invenção (5 dos 10 indivíduos nos subgrupos 1 e 2) nào requereram nenhuma medicação para dor suplementar durante o período de 4 dias inteiros. Estes indivíduos nos subgrupos 1 e 2 que não requereram medicações analgésicas suplementares foram ainda capazes de esperar as suas primeiras medicações para dor adicionais por cerca de 2 a 3 dias, mostrando o efeito anestésico local eficaz durante o curso de pelo menos 2 dias depois da cirurgia. Além disso, a quantidade de doses de medicações analgésicas suplementares nos subgrupos 1 e 2 foi drasticamente reduzida em relação aos indivíduos de controle (subgrupo 3) que requereram em média 11 doses durante o período de teste de 4 dias como comparado com 2,4 a 2,6 doses durante o mesmo período.
Além disso, uma revisão dos dados farmacocinéticos do Grupo 2 sugere que uma dose subcutânea eficaz de 638 a 688 mg de bupivacaína pode ser reprodutível mente administrada usando as composições de liberação controlada da presente invenção para fornecer uma concentração plasmática de estado uniforme eficaz de bupivacaína de cerca de 300 ng/ml.
Os resultados do teste de Contagens de Dor no Sítio de Incisão “no repouso” para todos os três subgrupos do Grupo 2 são representados na Figura 6. Os dados do subgrupo 3 são representados pela curva de topo (A), os dados do subgrupo 2 são representados pela curva intermediária (□) e os dados do subgrupo 1 são representados pela curva de fundo (0). Por conveniência, o Tempo para a Primeira Medicação Analgésica Suplementar médio é mostrado em cada curva. A intensidade de dor na incisão foi registrado usando uma escala analógica visual de 0 a 100 mm (VAS) com contagens variando de 0 (nenhuma dor) a 100 (pior dor imaginável). Cada contagem de VAS foi registrada como uma linha vertical única na escala. O teste foi administrado como segue. No dia da cirurgia (Dia 0), as contagens de dor na incisão foram registrados inicialmente em 60 minutos depois da administração da composição de teste (como debatido acima, o subgrupo 1 recebeu SAIB/BA/bupivacaína e solução salina; o subgrupo 2 recebeu SAIB/BA/bupivacaína e Marcain®; e o subgrupo 3 recebeu Marcain® e Marcain®). Em seguida, as contagens de dor de incisão foram registrados a cada 30 minutos até o ponto de tempo de 4 horas e depois de hora em hora até o ponto de tempo de avaliação de 8 horas e finalmente no ponto de tempo de avaliação de 12 horas. Nos Dias 1 a 3 na continuidade, as contagens de dor na incisão foram registrados na manhã com base no tempo que a composição de teste foi administrada no Dia 0. Estas medições na continuidade foram tiradas em intervalos de 4 horas até um período avaliação de 12 horas (4 medições). O tempo de uso de qualquer medicação concomitante (suplementar) também foi mencionado durante esta avaliação de 4 dias.
Como pode ser observado pela revisão dos resultados do teste de Contagens de Dor no Sítio de Incisão representado na Figura 6, ambos os subgrupos que receberam as composições de teste de SAIB/BA/bupivacaína (subgrupos 1 e 2) demonstraram contagens VAS médias mais baixos em todos os tempos por todo o teste quando comparado com o grupo que recebeu a composição de teste Marcain® (subgrupo 3). Estes resultados demonstram que as composições da presente invenção fornecem anestesia local prolongada no ferimento do sítio de incisão com uma duração de pelo menos cerca de 36 a 48 horas depois da administração ao indivíduo.
Os indivíduos para o Grupo 3 será dividido em 2 subgrupos de tratamento. O primeiro subgrupo receberá 7 injeções de volume total de 5 ml da composição de SAIB/BA/bupivacaína (contendo 958 mg de bupivacaína), administrada como duas injeções subcutâneas de arrasto ao longo cada lado do ferimento cirúrgico (0,75 ml/cm ao longo de uma incisão de ferimento de comprimento total de 5 cm sugeridos) com 10 ml de Marcain® (0,5 % de Bupivacaína-HCl) infiltrada no ferimento da incisão (incluindo subfascial) antes do fechamento de ferimento para produzir um total de 1,008 mg de bupivacaína administrada por indivíduo. O segundo subgrupo receberá 7 injeções de volume total de 5 ml da composição de Marcain® (0,5 % de Bupivacaína-HCl) administrada como duas injeções subcutâneas de arrasto ao longo de cada lado do ferimento cirúrgico (0,75 ml/cm ao longo de uma incisão de ferimento de comprimento total de 5 cm sugeridos) ao longo com 10 ml de Marcain® infiltrados no ferimento da incisão (incluindo subfascial) antes do fechamento do ferimento para produzir um total de 87,5 mg de bupivacaína administrada por indivíduo.
O efeito anestésico/analgésico será avaliado usando os testes de Tempo para a Primeira Medicação Analgésica Suplementar e Consumo de Medicação Analgésica Suplementar Total (no curso de 4 dias). A concentração plasmática de bupivacaína será medida periodicamente por todo o curso do estudo, particularmente através das primeiras 24 horas para avaliar a magnitude da liberação inicial de bupivacaína a partir da composição de liberação controlada de SAIB. E esperado que a dose mais alta de composições de liberação controlada de SAIB/BA/Bupivacaína preparadas de acordo com a presente invenção fornecerá resultados de eficácia similares ou ainda maiores quando comparada com aquelas dos indivíduos de teste do Grupo 2.
A presente invenção tendo sido assim descrita, suas variações e modificações como estariam evidentes àqueles de habilidade na técnica serão entendidas como estando dentro do escopo das reivindicações anexas.