WO2020230827A1 - 低空孔のペレット、及び成形体の製造方法 - Google Patents
低空孔のペレット、及び成形体の製造方法 Download PDFInfo
- Publication number
- WO2020230827A1 WO2020230827A1 PCT/JP2020/019146 JP2020019146W WO2020230827A1 WO 2020230827 A1 WO2020230827 A1 WO 2020230827A1 JP 2020019146 W JP2020019146 W JP 2020019146W WO 2020230827 A1 WO2020230827 A1 WO 2020230827A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- pellet
- pellets
- mass
- resin
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C45/00—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor
- B29C45/0001—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor characterised by the choice of material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/02—Making granules by dividing preformed material
- B29B9/06—Making granules by dividing preformed material in the form of filamentary material, e.g. combined with extrusion
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/002—Methods
- B29B7/007—Methods for continuous mixing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/12—Making granules characterised by structure or composition
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B9/00—Making granules
- B29B9/12—Making granules characterised by structure or composition
- B29B9/14—Making granules characterised by structure or composition fibre-reinforced
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C45/00—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor
- B29C45/0005—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor using fibre reinforcements
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
- C08J3/203—Solid polymers with solid and/or liquid additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/02—Fibres or whiskers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L1/00—Compositions of cellulose, modified cellulose or cellulose derivatives
- C08L1/02—Cellulose; Modified cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L21/00—Compositions of unspecified rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/10—Homopolymers or copolymers of propene
- C08L23/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/30—Mixing; Kneading continuous, with mechanical mixing or kneading devices
- B29B7/34—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices
- B29B7/38—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary
- B29B7/40—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with single shaft
- B29B7/42—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with single shaft with screw or helix
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/30—Mixing; Kneading continuous, with mechanical mixing or kneading devices
- B29B7/34—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices
- B29B7/38—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary
- B29B7/46—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with more than one shaft
- B29B7/48—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with more than one shaft with intermeshing devices, e.g. screws
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/30—Mixing; Kneading continuous, with mechanical mixing or kneading devices
- B29B7/58—Component parts, details or accessories; Auxiliary operations
- B29B7/72—Measuring, controlling or regulating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/001—Combinations of extrusion moulding with other shaping operations
- B29C48/0022—Combinations of extrusion moulding with other shaping operations combined with cutting
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/04—Particle-shaped
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/05—Filamentary, e.g. strands
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/25—Component parts, details or accessories; Auxiliary operations
- B29C48/88—Thermal treatment of the stream of extruded material, e.g. cooling
- B29C48/911—Cooling
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2001/00—Use of cellulose, modified cellulose or cellulose derivatives, e.g. viscose, as moulding material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2105/00—Condition, form or state of moulded material or of the material to be shaped
- B29K2105/06—Condition, form or state of moulded material or of the material to be shaped containing reinforcements, fillers or inserts
- B29K2105/12—Condition, form or state of moulded material or of the material to be shaped containing reinforcements, fillers or inserts of short lengths, e.g. chopped filaments, staple fibres or bristles
- B29K2105/122—Condition, form or state of moulded material or of the material to be shaped containing reinforcements, fillers or inserts of short lengths, e.g. chopped filaments, staple fibres or bristles microfibres or nanofibers
- B29K2105/124—Nanofibers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/10—Homopolymers or copolymers of propene
- C08J2323/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2377/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2377/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
- C08K2201/003—Additives being defined by their diameter
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/002—Physical properties
- C08K2201/004—Additives being defined by their length
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K2201/00—Specific properties of additives
- C08K2201/011—Nanostructured additives
Definitions
- the present invention relates to pellets having low pores and a method for producing a molded product using the pellets.
- thermoplastic resin for the purpose of improving physical properties.
- cellulose derived from a natural product is used as the filler. Is being considered.
- a resin composition containing the thermoplastic resin and cellulose is extruded into a strand in a molten state, cooled, and then pelletized by cutting with a pelletizer, and then this is performed. Generally, the pellets are melted again and formed into a desired molded product.
- Patent Document 1 is a molding material mixture containing at least one base material selected from resins and fibers and cellulose nanofibers, wherein the cellulose nanofibers are ⁇ OH. Described is a molding material mixture characterized in that a hydrophobic polymer is chemically bonded to at least a part of the group, and the substrate and the cellulose nanofibers are pulverized or pelletized.
- Patent Document 1 prevents the generation of hydrogen bonds between cellulose fibers by blocking at least a part of the ⁇ OH groups of the cellulose nanofibers with a hydrophobic polymer, and the pulverized product or pellet remains. It seeks to obtain a stable molding material mixture.
- a resin composition containing a thermoplastic resin and cellulose nanofibers is molded into pellets, vacancies are likely to occur inside the pellets (particularly in the center), and such vacancies. It has been found that when a molded product is produced from pellets having the above, silver streaks (poor appearance) are likely to occur on the surface of the molded product, and the yellowness of the molded product is further increased.
- the present invention solves the above-mentioned problems and enables the production of a pellet containing a thermoplastic resin and cellulose nanofibers, which has a good appearance and suppresses yellowing. And an object of the present invention is to provide a method for producing a molded product using the same.
- the present inventor can produce a molded product in which appearance defects such as silver streaks and occurrence of yellowing are suppressed according to pellets in which pores are appropriately controlled.
- the present invention was completed. That is, the present invention includes the following aspects.
- the pellet according to the above aspect 1 or 2 wherein the number of pore-containing pellets per 100 pellets is 1 or less.
- thermoplastic resin is a polyamide resin and / or a polyolefin resin.
- thermoplastic resin is a polyamide resin and / or a polyolefin resin.
- the pellet according to any one of the above aspects 1 to 9 further comprising an elastomer.
- the pellet according to any one of the above aspects 1 to 10 further comprising cellulose nanocrystals having a diameter of 100 nm or less and less than L / D30, or cellulose microfibers having a fiber diameter of more than 1 ⁇ m to 50 ⁇ m, or a mixture thereof.
- the difference Tcc-Tcp between the temperature-decreasing crystallization peak temperature Tcc of the resin composition measured by the differential scanning calorimeter and the temperature-decreasing crystallization peak temperature Tcp of the thermoplastic resin measured by the differential scanning calorimeter is 5.
- the above embodiment, wherein the ratio Ve / Vc of the total volume (Ve) of the pores of the pellets and the total volume (Vc) of the cellulose nanofibers per 100 pellets is 0% by volume to 4% by volume.
- the resin composition further contains a resin crystallization temperature lowering agent.
- pellets containing a thermoplastic resin and cellulose nanofibers which enable the production of a molded product having a good appearance and suppressed yellowing, and pellets using the same.
- a method for producing a molded product may be provided.
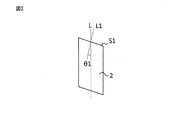
- FIG. 1 is a diagram illustrating a pellet cut surface.
- FIG. 2 is a diagram illustrating a mode in which the pellet cut surface is inclined.
- FIG. 3 is a diagram illustrating a mode in which the pellet cut surface is inclined.
- pellets containing a thermoplastic resin and cellulose nanofibers are provided.
- the number of pore-containing pellets per 100 pellets is 10 or less.
- the term "pore-containing pellet” refers to a pellet in the pellet MD direction (that is, the MD direction (mechanical direction) during pellet formation) and the pellet TD direction (that is, the TD direction (lateral direction) during pellet formation).
- the TD cross section of the pellet obtained by cutting into a TD is observed with a microscope, it means a pellet having at least one visible pore.
- each of 100 randomly selected pellets is cut at the center of the pellet MD direction and further flattened with a microtome to obtain a TD cross section. A morphological image of this TD cross section is photographed with a microscope at a magnification of 20 times and an observation field of view of 10 mm ⁇ 14 mm, and pellets having visible pores are determined to be pore-containing pellets.
- the cause of the pores in the pellet containing the thermoplastic resin and the cellulose nanofibers is that the cellulose nanofibers are mixed with the molten thermoplastic resin to form a strand, and then the cellulose is cooled when the strand is cooled. It is conceivable that nanofibers act as nucleating agents for thermoplastic resins. In particular, when the cooling rate of the strand is high, the crystallization of the thermoplastic resin that causes the strand to solidify proceeds more rapidly near the surface than inside the strand, so that it is considered that vacancies are likely to occur inside the strand. It is also considered that the ability of cellulose nanofibers to exhibit thixotropy in the resin composition is one of the causes of the pores.
- the shearing force applied to the resin composition during melt-mixing is generally applied. Relatively low. Under such a low shear force, it is considered that the increase in viscosity of the resin composition due to the thixotropy of the cellulose nanofibers causes pores.
- a molded product is manufactured using pellets having such pores as a molding material, it is considered that the presence of air during molding causes poor appearance of the molded product and yellowing due to deterioration of the resin composition. Since the pellet according to one aspect of the present invention has few pores, it is useful for producing a molded product having a good appearance and suppressed yellowing.
- the fact that the number of pore-containing pellets is small improves the biteability of the pellets when the pellets are melted to produce a molded product and shortens the molding cycle, thereby improving the productivity and the thermoplastic resin and cellulose nanofibers. It is advantageous in terms of suppressing the decomposition of.
- the number of pore-containing pellets per 100 pellets containing the thermoplastic resin and the cellulose nanofibers is 10 or less, preferably 8 from the viewpoint of obtaining a molded product having less appearance defects and yellowing.
- the number is less than or equal to 5, more preferably 5 or less, further preferably 3 or less, still more preferably 1 or less, and most preferably 0.
- the number of pore-containing pellets is preferably smaller from the viewpoint of obtaining a molded product having less appearance defects and yellowing, but from the viewpoint of ease of pellet production, for example, 1 or more, 3 or more, or 5 or more. Can be.
- the number ratio of the pore-containing pellets needs to be 10% or less, preferably 8% or less, more preferably 5% or less, still more preferably 3% or less, and further. It is preferably 1% by number or less, and most preferably 0% by number.
- the ratio of the pore area to the cross section is preferably 4.0% or less, more preferably 4.0% or less from the viewpoint of obtaining a molded product having less appearance defects and yellowing. Is 2.5% or less, more preferably 1.0% or less.
- the pore area ratio is a value obtained by the following method. First, for 100 randomly selected pellets, the pore-containing pellets are identified by the method of the present disclosure. For the pore-containing pellets, the pore area in the TD cross section was measured with the software attached to the scope, the ratio of the pore area to the total cross-sectional area of the TD cross section was calculated, and the average value for the entire pore-containing pellets.
- the pore area ratio is 0% when no pore-containing pellets are contained in 100 randomly selected pellets.
- the pore area ratio is preferably as low as possible from the viewpoint of obtaining a molded product with few defects and yellowing, and most preferably 0%, but from the viewpoint of ease of pellet production, for example, 0.5% or more, or It can be 1.0% or more, or 2.0% or more.
- the number of pore-containing pellets per 100 pellets is 8 or less, 5 or less, or 3 or less, and the pore area ratio is in any of the above ranges.
- the number of pore-containing pellets per 100 pellets is 0 (therefore, the pore area ratio is 0%).
- the shape of the pellet can be selected according to the purpose, and typically spherical, ellipsoidal, cylindrical, elliptical columnar, etc. can be exemplified.
- the shape of the pellet TD cross section is typically circular (when the pellet is a sphere, cylinder, etc.) or oval (when the pellet is an ellipsoid, ellipsoid, etc.), but polygonal (triangle, square, pentagon, six). It can also be a polygon (such as a polygon) or a variant (a star, a C, an O, etc.).
- the minor axis of the pellet TD cross section is, for example, 1 mm or more, or 2 mm or more, or 2.2 mm or more, or 2.5 mm or more, in that mass production of pellets and molded articles using the pellets is easy. Further, for example, it may be 5 mm or less, 4 mm or less, or 3 mm or less.
- the minor diameter of the pellet TD cross section is the diameter that the inscribed circle can have when the inscribed circle with respect to the outer shape of the pellet TD cross section is subtracted (one or two types depending on the shape of the pellet TD cross section). It means the maximum value of (the above diameters can exist). For example, when the pellet TD cross section is circular, the minor axis is the diameter of the circle, and when the pellet TD cross section is elliptical, the minor axis is the minor axis length of the ellipse.
- the minor axis of the pellet TD cross section is preferably 2 mm to 5 mm.
- the minor axis of the pellet TD cross section is preferably 2 mm to 5 mm, and the pellet MD direction length is preferably 2 mm to 5 mm.
- the above minor diameter and length are preferably not more than the lower limit from the viewpoint of operational stability during extrusion, and preferably not more than the upper limit from the viewpoint of biting into the molding machine in post-processing.
- FIG. 1 is a diagram for explaining a pellet cut surface
- FIGS. 2 and 3 are views for explaining an aspect in which the pellet cut surface is inclined.
- the normal direction L1 of the cut surface S1 is often substantially parallel to the strand MD direction L.
- FIGS. 2 and 3 depending on the manufacturing conditions such as a condition where the strand temperature at the time of pelletizing is high, a condition where a large facility is used (that is, a large amount of composition is discharged), a condition where the line speed is high, and the like.
- the normal directions L1 and L2 may be inclined (that is, not substantially parallel) at angles ⁇ 1 and ⁇ 2 (this angle indicates an acute angle side angle) with respect to the strand MD direction L. ..
- the pellet cut surface may be one surface such as the pellet cut surface S1 shown in FIG. 2, two surfaces such as the pellet cut surfaces S1 and S2 shown in FIG. 3, or three or more surfaces. In a typical embodiment, the pellet cut surface is substantially flat.
- the angles ⁇ 1 and ⁇ 2 formed by the normal directions S1 and S2 of the pellet cutting surfaces S1 and S2 with respect to the pellet MD direction L are large quantities of the pellet and the molded product using the same. It may be 5 ° to 30 ° in terms of ease of production.
- the pellet shape can be controlled relatively uniformly in small-scale production, but it is difficult to control it uniformly in mass production (for example, 100 kg / h scale).
- Pellets having the above angle of 5 ° to 30 ° are preferable because they are easy to mass-produce.
- the angle may be more preferably 8 ° to 20 ° or 10 ° to 15 °.
- the above angle is a value obtained by the following method.
- each of the 10 randomly selected pellets is cut through the central portion in the pellet TD direction and in the direction visually selected so as to maximize the inclination angle of the pellet cutting surface described later, and then flattened by a microtome.
- To obtain an MD cross section A morphological image of this MD cross section is photographed with a microscope at a magnification of 20 times and an observation field of view of 10 mm ⁇ 14 mm.
- the pellet MD direction axis and the normal line for the line segment corresponding to the pellet MD direction end face are defined, and the angle formed by the pellet MD direction axis and the normal line is measured.
- the number average value of 10 pellets is obtained as the inclination angle of the pellet cut surface.
- the pellet contains a thermoplastic resin and cellulose nanofibers, and in one embodiment can further contain any additional components. Hereinafter, each component will be illustrated.
- thermoplastic resin various resins can be used, and examples thereof include a crystalline resin having a melting point in the range of 100 ° C. to 350 ° C. and an amorphous resin having a glass transition temperature in the range of 100 ° C. to 250 ° C. Be done.
- the thermoplastic resin may be composed of one or more polymers, which may be homopolymers or copolymers.
- the melting point of the crystalline resin referred to here is the peak top of the endothermic peak that appears when the temperature is raised from 23 ° C. to 10 ° C./min using a differential scanning calorimetry device (DSC).
- DSC differential scanning calorimetry device
- the enthalpy of the endothermic peak at this time is preferably 10 J / g or more, and more preferably 20 J / g or more.
- the glass transition temperature of the amorphous resin referred to here is when measured at an applied frequency of 10 Hz while raising the temperature from 23 ° C. to 2 ° C./min using a dynamic viscoelasticity measuring device.
- it refers to the peak top temperature at which the storage elastic modulus is greatly reduced and the loss elastic modulus is maximized.
- the measurement frequency be at least once every 20 seconds in order to improve the measurement accuracy.
- the method for preparing the sample for measurement is not particularly limited, but from the viewpoint of eliminating the influence of molding strain, it is desirable to use a cut piece of a hot press molded product, and the size (width and thickness) of the cut piece can be as small as possible. The smaller one is preferable from the viewpoint of heat conduction.
- thermoplastic resin a polyamide resin, a polyester resin, a polyacetal resin, a polycarbonate resin, a polyacrylic resin, and a polyphenylene ether resin (modified polyphenylene modified by blending or graft-polymerizing polyphenylene ether with another resin).
- ether (Including ether), polyarylate resin, polysulfone resin, polyphenylene sulfide resin, polyether sulfone resin, polyketone resin, polyphenylene ether ketone resin, polyimide resin, polyamideimide resin, polyetherimide resin, Examples thereof include polyurethane-based resins, polyolefin-based resins (for example, ⁇ -olefin (co) polymers), polyvinyl-based resins, and various ionomers.
- thermoplastic resin modified with at least one compound selected from unsaturated carboxylic acids, acid anhydrides thereof or derivatives thereof can also be used.
- polyolefin resins polyamide resins, polyester resins, polyacetal resins, polyacrylic resins, polyphenylene ether resins, and polyphenylene sulfide resins
- the thermoplastic resin is a polyamide resin and / or a polyolefin resin
- the advantages of the pellets according to the present embodiment are particularly remarkable and preferable.
- the polyolefin-based resin is a polymer obtained by polymerizing a monomer unit containing olefins (for example, ⁇ -olefins).
- olefins for example, ⁇ -olefins
- Specific examples of the polyolefin-based resin are not particularly limited, but are exemplified by low-density polyethylene (for example, linear low-density polyethylene), high-density polyethylene, ultra-low-density polyethylene, ultra-high-molecular-weight polyethylene, and the like.
- polypropylene, poly1-butene, poly1-pentene, polymethylpentene, ethylene- ⁇ -olefin copolymer or a modified product thereof for example, ethylene-propylene copolymer, ethylene-butene copolymer and the like, and these Modified products
- copolymers of olefins (eg ⁇ -olefins) with other monomer units eg ethylene- (meth) acrylic acid copolymers, ethylene- (meth) methyl acrylate copolymers, ethylene- (eg Meta) Ethyl acrylate copolymer, ethylene-glycidyl methacrylate copolymer, ethylene-propylene-diene ternary copolymer, ethylene-vinyl acetate copolymer, copolymer of non-conjugated olefin and conjugated diene, ethylene and / Or at least the carboxyl groups of the copolymer of
- polyolefins obtained by partially metallizing chlorides, isobutylene halide-paramethylstyrene copolymers, and the like, and modified products thereof.
- the polyolefin-based resin may be a cyclic olefin-based resin.
- polypropylene is mentioned as the most preferable polyolefin resin.
- polypropylene having a melt mass flow rate (MFR) of 3 g / 10 minutes or more and 100 g / 10 minutes or less measured at 230 ° C. and a load of 2.16 kgf in accordance with ISO1133 is preferable.
- the lower limit of MFR is more preferably 5 g / 10 minutes, even more preferably 6 g / 10 minutes, and most preferably 8 g / 10 minutes.
- the upper limit is more preferably 75 g / 10 minutes, even more preferably 60 g / 10 minutes, and most preferably 40 g / 10 minutes. It is desirable that the MFR does not exceed the above upper limit value from the viewpoint of improving the toughness of the composition, and it is desirable not to exceed the above lower limit value from the viewpoint of the fluidity of the composition.
- an acid-modified polyolefin resin can also be preferably used in order to enhance the affinity with cellulose.
- the acid can be appropriately selected from maleic acid, fumaric acid, succinic acid, phthalic acid and anhydrides thereof, polycarboxylic acids such as citric acid and the like. Of these, maleic acid or its anhydride is preferable because of the ease of increasing the denaturation rate.
- the modification method is not particularly limited, but a method of heating the resin to a melting point or higher and melting and kneading it in the presence / absence of a peroxide is common.
- the acid-modified polyolefin resin all of the above-mentioned polyolefin resins can be used, but polypropylene is particularly preferable.
- the acid-modified polyolefin resin may be used alone, but it is more preferable to use the acid-modified polyolefin resin in combination with an unmodified polyolefin resin in order to adjust the modification rate as a composition.
- the ratio of acid-modified polypropylene to total polypropylene is preferably 0.5% by mass to 50% by mass.
- a more preferable lower limit is 1% by mass, still more preferably 2% by mass, even more preferably 3% by mass, particularly preferably 4% by mass, and most preferably 5% by mass.
- the upper limit is more preferably 45% by mass, still more preferably 40% by mass, even more preferably 35% by mass, particularly preferably 30% by mass, and most preferably 20% by mass.
- the lower limit or more is preferable, and in order to maintain the ductility as a resin, the upper limit or less is preferable.
- the lower limit of the acid modification rate of the acid-modified polyolefin resin is preferably 0.01% by mass, more preferably 0.1% by mass, still more preferably 0.3% by mass, and particularly preferably 0.3% by mass. It is 0.5% by mass, most preferably 0.7% by mass.
- the upper limit is preferably 10% by mass, more preferably 5% by mass, further preferably 3% by mass, particularly preferably 2% by mass, and most preferably 1.5% by mass.
- the lower limit or more is preferable, and in order to maintain the mechanical properties of the acid-modified polyolefin, the upper limit or less is preferable.
- Melt mass flow rate (MFR) measured at 230 ° C. and a load of 2.16 kgf in accordance with the preferred ISO 1133 of acid-modified polypropylene should be 50 g / 10 min or more to enhance affinity with the cellulose interface. preferable.
- a more preferable lower limit is 100 g / 10 minutes, even more preferably 150 g / 10 minutes, and most preferably 200 g / 10 minutes.
- the polyamide-based resin preferable as the thermoplastic resin is not particularly limited, but is obtained by a polycondensation reaction of lactams, polyamide 6, polyamide 11, polyamide 12, etc .; 1,6-hexanediamine, 2-methyl-1,5. -Pentanediamine, 1,7-heptanediamine, 2-methyl-1-6-hexanediamine, 1,8-octanediamine, 2-methyl-1,7-heptanediamine, 1,9-nonanediamine, 2-methyl- Diamines such as 1,8-octanediamine, 1,10-decanediamine, 1,11-undecanediamine, 1,12-dodecanediamide, m-xylylene diamine, butane diic acid, pentanic acid, hexane diic acid , Heptane diic acid, octane diacid, nonane diic acid, decane diic acid, benzene-1,2-dicarboxylic acid, benzene-1,3
- polyamide-based resins aliphatic polyamides such as polyamide 6, polyamide 11, polyamide 12, polyamide 6, 6, polyamide 6, 10, polyamide 6, 11, polyamide 6, 12 and polyamide 6, C, polyamide 2 M5, C
- the alicyclic polyamide such as is more preferable.
- the concentration of terminal carboxyl groups in the polyamide resin is not particularly limited, but the lower limit is preferably 20 ⁇ mol / g, more preferably 30 ⁇ mol / g.
- the upper limit of the terminal carboxyl group concentration is preferably 150 ⁇ mol / g, more preferably 100 ⁇ mol / g, and further preferably 80 ⁇ mol / g.
- the ratio of carboxyl terminal groups to total terminal groups is preferably 0.30 to 0.95.
- the lower limit of the carboxyl end group ratio is more preferably 0.35, even more preferably 0.40, and most preferably 0.45.
- the upper limit of the carboxyl end group ratio is more preferably 0.90, even more preferably 0.85, and most preferably 0.80.
- the carboxyl end group ratio is preferably 0.30 or more from the viewpoint of dispersibility in the cellulose pellets, and is preferably 0.95 or less from the viewpoint of the color tone of the obtained resin composition.
- a known method can be used as a method for adjusting the terminal group concentration of the polyamide resin.
- a diamine compound, a monoamine compound, a dicarboxylic acid compound, a monocarboxylic acid compound, an acid anhydride, a monoisocyanate, a monoacid halide, a monoester, a monoalcohol, or the like so as to have a predetermined terminal group concentration during polymerization of polyamide.
- Examples thereof include a method of adding a terminal modifier that reacts with an terminal group to the polymerization solution.
- aliphatic monocarboxylic acids aliphatic monocarboxylic acids
- alicyclic monocarboxylic acids such as cyclohexanecarboxylic acid
- aromatic products such as benzoic acid, toluic acid, ⁇ -naphthalenecarboxylic acid, ⁇ -naphthalenecarboxylic acid, methylnaphthalenecarboxylic acid, phenylacetic acid, etc.
- Carboxylic acids and a plurality of mixtures arbitrarily selected from these.
- acetic acid, propionic acid, butyric acid, valeric acid, caproic acid, caprylic acid, lauric acid, tridecanoic acid, myristic acid, palmitic acid, stearic acid, etc. in terms of reactivity, stability of the sealing end, price, etc.
- One or more terminal modifiers selected from the group consisting of benzoic acid and benzoic acid are preferable, and acetic acid is most preferable.
- Aliphatic compounds such as methylamine, ethylamine, propylamine, butylamine, hexylamine, octylamine, decylamine, stearylamine, dimethylamine, diethylamine, dipropylamine and dibutylamine are examples of the terminal modifier that reacts with the terminal carboxyl group.
- one or more terminals selected from the group consisting of butylamine, hexylamine, octylamine, decylamine, stearylamine, cyclohexylamine and aniline from the viewpoints of reactivity, boiling point, stability of sealing terminal, price and the like. Regulators are preferred.
- the concentrations of these amino-terminal groups and carboxyl-terminal groups are preferably obtained by 1 H-NMR from the integrated value of the characteristic signal corresponding to each terminal group in terms of accuracy and simplicity.
- a method for determining the concentration of those terminal groups specifically, the method described in JP-A-7-228775 is recommended. When this method is used, heavy trifluoroacetic acid is useful as a measurement solvent.
- the number of integrations of 1 1 H-NMR requires at least 300 scans even when measured with an instrument having sufficient resolution.
- the concentration of the terminal group can also be measured by a measurement method by titration as described in JP-A-2003-055549. However, in order to reduce the influence of mixed additives, lubricants, etc. as much as possible, 1 H-NMR quantification is more preferable.
- the viscosity number [VN] of the polyamide resin measured under the condition of 30 ° C. in concentrated sulfuric acid is preferably 60 to 200 mL / g, more preferably 70 to 180 mL / g, and 70 to 140 mL / g. It is more preferably g, and particularly preferably 70 to 120 mL / g.
- a polyamide-based resin having an intrinsic viscosity in the above range is advantageous in that it improves the fluidity in the mold when the molded product is manufactured by injection molding of the resin composition and improves the appearance of the molded product.
- the "viscosity number” is an index of viscosity measured with sulfuric acid having a concentration of 96% in accordance with ISO307.
- the polyester-based resin preferable as the thermoplastic resin is not particularly limited, but is not particularly limited, but is polyethylene terephthalate (PET), polybutylene terephthalate (PBT), polyethylene naphthalate (PEN), polybutylene succinate (PBS), polybutylene succinate adipate ( Selected from PBSA), polybutylene terephthalate (PBAT), polyallylate (PAR), polyhydroxyalkanoic acid (PHA) (polyester resin composed of 3-hydroxyalkanoic acid), polylactic acid (PLA), polycarbonate (PC), etc.
- PET polyethylene terephthalate
- PBT polybutylene terephthalate
- PEN polyethylene naphthalate
- PBS polybutylene succinate
- PBS polybutylene succinate adipate
- PAR polyallylate
- PHA polyhydroxyalkanoic acid
- PDA polylactic acid
- PC polycarbonate
- more preferable polyester-based resins include PET, PBS,
- the terminal group can be freely changed depending on the monomer ratio at the time of polymerization and the presence / absence and amount of the terminal stabilizer added, but the carboxyl terminal group ratio to the total terminal group of the polyester resin.
- ([COOH] / [all terminal groups]) is preferably 0.30 to 0.95.
- the lower limit of the carboxyl end group ratio is more preferably 0.35, further preferably 0.40, and most preferably 0.45.
- the upper limit of the carboxyl end group ratio is more preferably 0.90, further preferably 0.85, and most preferably 0.80.
- the carboxyl end group ratio is preferably 0.30 or more from the viewpoint of dispersibility in the cellulose pellets, and is preferably 0.95 or less from the viewpoint of the color tone of the obtained composition.
- Preferable polyacetal resins as thermoplastic resins are generally homopolyacetal made from formaldehyde and copolyacetal containing trioxane as a main monomer and, for example, 1,3-dioxolane as a comonomer component, and both can be used.
- copolyacetal can be preferably used.
- the amount of the comonomer component (for example, 1,3-dioxolane) is preferably in the range of 0.01 to 4 mol%.
- a more preferable lower limit amount of the comonomer component amount is 0.05 mol%, more preferably 0.1 mol%, and particularly preferably 0.2 mol%.
- the more preferable upper limit amount is 3.5 mol%, more preferably 3 mol%, particularly preferably 2.5 mol%, and most preferably 2.3 mol%. From the viewpoint of thermal stability during extrusion and molding, it is desirable that the lower limit is within the above range, and from the viewpoint of mechanical strength, the upper limit is preferably within the above range.
- PV-based resins as thermoplastic resins include vinyl aliphatic (co) copolymers (polyvinyl chloride, vinyl chloride-ethylene copolymers and ethylene-vinyl acetate copolymers (these are also olefinic resins)) and These saponates, etc.), vinyl aromatic (co) copolymers (polystyrene, conjugated diene and vinyl aromatic hydrocarbon block copolymers, conjugated diene and vinyl aromatic hydrocarbon block copolymer hydrides) Etc.), acrylic (co) polymer (poly (meth) acrylic acid ester, etc.), acrylonitrile-based (co) polymer (acrylonitrile-butanediene-styrene (ABS) resin, acrylonitrile-styrene (AS) resin, etc.), etc. Can be mentioned.
- vinyl aliphatic (co) copolymers polyvinyl chloride, vinyl chloride-ethylene copolymers and ethylene-vinyl a
- the fiber diameter of the cellulose nanofibers is preferably 50 nm or more, 60 nm or more, or 70 nm or more from the viewpoint of improving the dispersibility of the cellulose nanofibers in the pellet or the molded body, and the cellulose nanofibers are the molded body. In one embodiment, it is 1000 nm or less, preferably 600 nm or less, 400 nm or less, or 200 nm or less from the viewpoint of satisfactorily obtaining the effect of improving the physical properties of the product.
- the fiber diameter is a value obtained as a spherical equivalent diameter (volume average particle diameter) of particles when the integrated volume becomes 50% by a laser diffraction / scattering method particle size counter.
- the fiber diameter can be measured by the following method. With the solid content of cellulose nanofibers as 40% by mass, knead in a planetary mixer (for example, 5DM-03-R manufactured by Shinagawa Kogyo Co., Ltd., stirring blade is hook type) at 126 rpm for 30 minutes at room temperature and normal pressure. Then, a pure water suspension was prepared at a concentration of 0.5% by mass, and a high shear homogenizer (for example, manufactured by Nippon Seiki Co., Ltd., trade name "Excel Auto Homogenizer ED-7" treatment condition) was used, and the rotation speed was 15,000 rpm.
- a high shear homogenizer for example, manufactured by Nippon Seiki Co., Ltd., trade name "Excel Auto Homogenizer ED-7" treatment condition
- centrifuge for example, manufactured by Kubota Shoji Co., Ltd., trade name “6800 type centrifuge”, rotor type RA-400 type
- processing conditions centrifugal force 39200 m 2 / s 10
- the supernatant is collected by centrifuging for a minute, and the supernatant is further centrifuged at 116000 m 2 / s for 45 minutes, and the supernatant after centrifugation is collected.
- a laser diffraction / scattering particle size counter for example, manufactured by Horiba Seisakusho Co., Ltd., trade name "LA-910” or trade name "LA-950", ultrasonic treatment 1 minute, refractive index 1.
- the integrated 50% particle diameter in the volume frequency particle size distribution obtained in 20 (that is, the spherical equivalent diameter of the particles when the integrated volume becomes 50% with respect to the total volume of the particles) is defined as the volume average particle diameter. ..
- the L / D ratio of cellulose nanofibers is 30 or more, 40 or more, 50 or more, or 100 or more in one embodiment.
- the upper limit is not particularly limited, but is 10,000 or less in one aspect from the viewpoint of handleability.
- it is desirable that the L / D ratio of the cellulose nanofibers is within the above range.
- the lengths (L) and diameters (D) of cellulose nanofibers, and cellulose nanocrystals and cellulose microfibers are calculated from these.
- L / D ratio an aqueous dispersion of cellulose components was used with a high shear homogenizer (for example, manufactured by Nippon Seiki Co., Ltd., trade name "Excel Auto Homogenizer ED-7"), and treatment conditions: 15,000 rpm x 5
- the aqueous dispersion dispersed in 1 minute is diluted with pure water to 0.1 to 0.5% by mass, cast on mica, and air-dried as a measurement sample.
- An optical microscope or a high-resolution scanning microscope It is determined by measuring with SEM) or an interatomic force microscope (AFM). Specifically, the length (L) and diameter (D) of 100 randomly selected cellulose components are measured in an observation field in which the magnification is adjusted so that at least 100 cellulose components are observed. Then, the (L / D) ratio is calculated. The number average value of the above 100 celluloses is defined as the length (L), diameter (D) and (L / D) ratio.
- Cellulose nanofibers are crushed by a pulverization method using a high-pressure homogenizer, microfluidizer, ball mill, disc mill, etc. after treating pulp or the like with hot water or the like at 100 ° C. or higher to hydrolyze and weaken the hemicellulose portion. It may be delicate cellulose.
- the cellulose nanofibers may be modified products (ie, modified cellulose nanofibers).
- modified cellulose nanofibers are those in which cellulose is modified by one or more modifiers selected from esterifying agents, silylating agents, isocyanate compounds, halogenating alkylating agents, alkylene oxides and / or glycidyl compounds. Can be mentioned.
- the cellulose nanofibers are free of unmodified or oxo acid-modifying groups (ie, sites where the hydroxyl groups of cellulose are converted with oxo acids (eg, carboxylic acids) or salts thereof (eg, carboxylates)).
- the esterifying agent as a modifier includes an organic compound having at least one functional group capable of reacting with a hydroxyl group on the surface of cellulose nanofibers to esterify the hydroxyl group. Esterification can also be carried out by the method described in paragraph [0108] of WO 2017/159823.
- the esterifying agent may be a commercially available reagent or product.
- esterifying agent are not particularly limited, but for example, acetic acid, propionic acid, butyric acid, valeric acid, caproic acid, capricic acid, lauric acid, tridecanoic acid, myristic acid, palmitic acid, stearic acid, pivalic acid, iso.
- Alipid monocarboxylic acid such as butyric acid; alicyclic monocarboxylic acid such as cyclohexanecarboxylic acid; aromatics such as benzoic acid, toluic acid, ⁇ -naphthalenecarboxylic acid, ⁇ -naphthalenecarboxylic acid, methylnaphthalenecarboxylic acid, and phenylacetic acid.
- Monocarboxylic acids and a plurality of mixtures arbitrarily selected from these, and esters of these acids and vinyl alcohols (eg, vinyl acetate, vinyl propionate, vinyl butyrate, vinyl valerate, vinyl caproate, vinyl caprylate). , Vinyl laurate, etc.), and symmetric anhydrides (eg, acetic anhydride, maleic anhydride, cyclohexane-carboxylic acid anhydride, benzene-sulfonic acid anhydride), mixed acid anhydride, which are arbitrarily selected from the above-mentioned acids.
- vinyl alcohols eg, vinyl acetate, vinyl propionate, vinyl butyrate, vinyl valerate, vinyl caproate, vinyl caprylate.
- vinyl laurate, etc. and symmetric anhydrides (eg, acetic anhydride, maleic anhydride, cyclohexane-carboxylic acid anhydride, benzene-sulfonic acid anhydride), mixed
- butyric acid-valeric acid anhydride for example, butyric acid-valeric acid anhydride
- cyclic anhydride for example, succinic anhydride, phthalic anhydride, naphthalene-1,8: 4,5-tetracarboxylic hydride, cyclohexane-1,2 , 3,4-Tetracarboxylic acid 3,4-anhydride
- esteric anhydride for example, 3- (ethoxycarbonyl) propanoic anhydride, benzoylethyl carbonate
- acetic acid, propionic acid, butyric acid, valeric acid, caproic acid, caprylic acid, lauric acid, tridecanoic acid, myristic acid, palmitic acid, stearic acid, and these acids are used in terms of reactivity, stability, price, etc.
- Esterized products of vinyl alcohol, benzoic acid, acetic acid anhydride, maleic anhydride, succinic anhydride, and phthalic anhydride can be preferably used.
- the silylating agent as a modifier includes a Si-containing compound having at least one reactive group capable of reacting with the hydroxyl group on the surface of cellulose or the group after hydrolysis thereof.
- the silylating agent may be a commercially available reagent or product.
- silylating agent are not particularly limited, but are chlorodimethylisopropylsilane, chlorodimethylbutylsilane, chlorodimethyloctylsilane, chlorodimethyldodecylsilane, chlorodimethyloctadecylsilane, chlorodimethylphenylsilane, and chloro (1-hexenyl).
- hexamethyldisilazane, octadecyldimethylmethoxysilane, dimethyloctylmethoxysilane, and trimethylethoxysilane can be preferably used from the viewpoints of reactivity, stability, price, and the like.
- the halogenated alkylating agent as a modifier includes an organic compound having at least one functional group capable of reacting with a hydroxyl group on the surface of cellulose to alkylate the halogen.
- the halogenated alkylating agent may be a commercially available reagent or product.
- halogenated alkylating agent are not particularly limited, but chloropropane, chlorobutane, bromopropane, bromohexane, bromoheptane, iodomethane, iodoethane, iodooctane, iodooctadecane, iodobenzene and the like can be used.
- bromohexane and iodooctane can be preferably used from the viewpoints of reactivity, stability, price and the like.
- the isocyanate compound as a modifier includes an organic compound having at least one isocyanate group capable of reacting with a hydroxyl group on the surface of cellulose.
- the isocyanate compound may be a blocked isocyanate compound capable of regenerating an isocyanate group by desorbing a blocking group at a specific temperature, or a dimer or trimeric of polyisocyanate, or a buretted isocyanate.
- the modified product such as Polymethylenepolyphenylpolyisocyanate (Polymeric MDI) may be used. These may be commercially available reagents or products.
- isocyanate compound examples include, but are not limited to, aliphatic polyisocyanate, alicyclic polyisocyanate, aromatic polyisocyanate, aromatic aliphatic polyisocyanate, blocked isocyanate compound, polyisocyanate and the like.
- tetramethylene diisocyanate dodecamethylene diisocyanate, hexamethylene diisocyanate, 2,2,4-trimethylhexamethylene diisocyanate, 2,4,4-trimethylhexamethylene diisocyanate, lysine diisocyanate, 2-methylpentane-1,5-diisocyanate, 3-Methylpentane-1,5-diisocyanate, isophorone diisocyanate, hydrogenated xylylene diisocyanate, 4,4'-dicyclohexylmethane diisocyanate, 1,4-cyclohexane diisocyanate, methylcyclohexylene diisocyanate, 1,3-bis (isocyanate methyl) Cyclohexane), tolylene diisocyanate (TDI), 2,2'-diphenylmethane diisocyanate, 2,4'-diphenylmethane diisocyanate, 4,4''-dip
- TDI, MDI, hexamethylene diisocyanate, and blocked isocyanate made from a hexamethylene diisocyanate modified product and hexamethylene diisocyanate as raw materials can be preferably used from the viewpoints of reactivity, stability, price, and the like.
- the dissociation temperature of the block group of the blocked isocyanate compound has an upper limit of preferably 210 ° C, more preferably 190 ° C, and further preferably 150 ° C.
- the lower limit is preferably 70 ° C., more preferably 80 ° C., and even more preferably 110 ° C.
- the blocking agent in which the dissociation temperature of the blocking group falls within this range include methylethylketone oxime, ortho-secondary butylphenol, caprolactam, sodium bisulfite, 3,5-dimethylpyrazole, 2-methylimidazole and the like.
- the alkylene oxide and / or glycidyl compound as a modifier includes an organic compound having at least one alkylene oxide group, a glycidyl group and / or an epoxy group capable of reacting with a hydroxyl group on the surface of cellulose.
- the alkylene oxide and / or glycidyl compound may be a commercially available reagent or product.
- alkylene oxide and / or the glycidyl compound are not particularly limited, but for example, methyl glycidyl ether, ethyl glycidyl ether, butyl glycidyl ether, 2-ethylhexyl glycidyl ether, 2-methyloctyl glycidyl ether, phenyl glycidyl ether, p.
- Glycidyl ester ethylene glycol diglycidyl ether, propylene glycol diglycidyl ether, 1,4-butanediol diglycidyl ether, hexamethylene glycol diglycidyl ether, resorcinol diglycidyl ether, bisphenol A diglycidyl ether, polyethylene glycol diglycidyl ether, polypropylene Glycol diglycidyl ether, polybutylene glycol diglycidyl ether, glycerol triglycidyl ether, trimethylol propantriglycidyl ether, pentaerythritol tetraglycidyl ether, sorbitol polyglycidyl ether, sorbitan polyglycidyl ether, polyglycerol polyglycidyl ether, diglycerol polyglycidyl Examples thereof include polyhydric alcohol glycidyl ether such as ether.
- 2-methyloctyl glycidyl ether hexamethylene glycol diglycidyl ether, and pentaerythritol tetraglycidyl ether can be preferably used from the viewpoints of reactivity, stability, price and the like.
- the cellulose nanofibers are hydrophobic cellulose nanofibers.
- the denaturing agents exemplified above can be suitably used for producing hydrophobic cellulose nanofibers.
- acetylated cellulose nanofibers are preferable as the hydrophobic cellulose fibers.
- the content of the cellulose nanofibers in the pellets is preferably 0.1% by mass or more, more preferably 1% by mass or more, still more preferably 3 from the viewpoint of obtaining a good effect of improving the physical properties of the cellulose nanofibers on the molded product.
- the pellets may optionally contain additional components in addition to the thermoplastic resin and cellulose nanofibers. Additional ingredients include cellulose nanocrystals, cellulose microfibers, elastomers, surface treatment agents, compatibilizers, plasticizers, colorants, pigments, flow modifiers, antioxidants, UV absorbers, UV dispersants, inorganic fillers. , Lubricating oil and the like. Each of these components may be used alone or in combination of two or more. Further, these components may be commercially available reagents or products.
- the additional component comprises cellulose nanocrystals having a diameter of 100 nm or less and less than L / D 30, or cellulose microfibers having a fiber diameter of more than 1 ⁇ m to 50 ⁇ m, or a mixture thereof.
- the diameter of the cellulose nanocrystal is 100 nm or less, 80 nm or less, or 70 nm or less in one embodiment, and 3 nm or more, 5 nm or more, or 10 nm or more in one embodiment.
- the L / D of cellulose nanocrystals is less than 30, 20 or less, or 15 or less, or 10 or less in one embodiment, and 1 or more, 2 or more, or 4 or more, or 5 or more in one embodiment. ..
- the fiber diameter of the cellulose microfiber is more than 1 ⁇ m, 2 ⁇ m or more, 5 ⁇ m or more, or 10 ⁇ m or more in one embodiment, and 50 ⁇ m or less, 45 ⁇ m or less, 40 ⁇ m or less, or 30 ⁇ m or less, or 20 ⁇ m or less in one embodiment. Or it is 15 ⁇ m or less.
- the L / D ratio of the cellulose microfiber is 30 or more, 50 or more, or 70 or more in one embodiment, and 2000 or less, 1000 or less, or 500 or less in one aspect.
- cellulose nanocrystals and / or cellulose microfibers may be modified products.
- Examples of denaturation may be similar to those described above for cellulose nanofibers.
- an elastomer means a substance (specifically, a natural or synthetic polymer substance) that is an elastic substance at room temperature (23 ° C.).
- the elastomer include natural rubber, conjugated diene compound polymer, aromatic compound-conjugated diene copolymer, hydrogenated additive of aromatic compound-conjugated diene copolymer, polyolefin-based elastomer, polyester-based elastomer, and polyurethane-based elastomer. Examples thereof include elastomers, polyamide-based elastomers, and elastomers having a core-shell structure.
- the elastomer is a polymer that is different from the thermoplastic resin.
- a polymer that is different from the thermoplastic resin for example, from the viewpoint of easiness of modification reaction for obtaining an elastomer having an acidic functional group described later, an aromatic compound-conjugated diene copolymer, a hydrogenated additive of an aromatic compound-conjugated diene copolymer, and the like.
- Polyolefin-based elastomers and elastomers having a core-shell structure are preferable.
- both aromatic compound-conjugated diene block copolymer and aromatic compound-conjugated diene block are used.
- Hydrogenated polymers are more preferable, and among polyolefin-based elastomers, a copolymer of ethylene and ⁇ -olefin is more preferable.
- the aromatic compound-conjugated diene block copolymer referred to here is a block composed of a polymer block (A) mainly composed of an aromatic vinyl compound and a polymer block (B) mainly composed of a conjugated diene compound. It is a polymer.
- a block copolymer in which the binding form of each block is any of AB type, ABA type, and ABAB type is preferable from the viewpoint of developing impact strength, and more preferably ABBA type or ABAB type.
- the mass ratio of the aromatic vinyl compound and the conjugated diene compound in the block copolymer is 10/90 to 70/30. More preferably, it is 15/85 to 55/45, and most preferably 20/80 to 45/55. Further, two or more kinds of these having different mass ratios of the aromatic vinyl compound and the conjugated diene compound may be blended.
- the aromatic vinyl compound include styrene, ⁇ -methylstyrene, vinyltoluene and the like, and one or more compounds selected from these are used, and styrene is particularly preferable.
- the conjugated diene compound examples include butadiene, isoprene, piperylene, 1,3-pentadiene and the like, and one or more compounds selected from these are used. Among them, butadiene, isoprene and a combination thereof are preferable. ..
- the microstructure of the polybutadiene block portion may be 1,2-vinyl content or 1,2-vinyl content from the viewpoint of suppressing crystallization of soft segments.
- the total amount with the 3,4-vinyl content is preferably 5 to 80%, more preferably 10 to 50%, and most preferably 15 to 40% on a molar basis.
- the hydrogenated additive of the block copolymer of the aromatic vinyl compound and the conjugated diene compound is mainly composed of the diene compound by hydrogenating the block copolymer of the above-mentioned aromatic vinyl compound and the conjugated diene compound.
- This refers to a compound in which the aliphatic double bond of the polymer block is controlled in the range of more than 0% to 100%.
- the hydrogenation rate of the hydrogenated product of the block copolymer is preferably 50% or more, more preferably 80% or more, and most preferably 98% or more from the viewpoint of suppressing thermal deterioration during processing.
- the number average molecular weight (Mn) is 10,000 to 10,000 or more from the viewpoint of achieving both impact strength and fluidity. 500,000 is preferable, and 40,000 to 250,000 is most preferable.
- the number average molecular weight referred to here is a value measured by a GPC apparatus using chloroform as an elution solvent at a measurement temperature of 40 ° C. and converted with a polystyrene standard polymer.
- the block copolymers of these aromatic vinyl compounds-conjugated diene compounds have different bonding forms, different molecular weights, different aromatic vinyl compound species, different conjugated diene compound species, and 1,2-vinyl content.
- two or more kinds of compounds having different total amounts of 1,2-vinyl content and 3,4-vinyl content, different aromatic vinyl compound component contents, different hydrogen addition rates, etc. are used. It doesn't matter.
- an ethylene- ⁇ -olefin copolymer can be preferably used from the viewpoint of developing impact resistance.
- Monomers that can be copolymerized with ethylene units include propylene, butene-1, pentene-1, 4-methylpentene-1, hexene-1, heptene-1, octene-1, nonen-1, decene-1, and undecene-1.
- Dodecene-1 tridecene-1, tetradecene-1, pentadecene-1, hexadecene-1, heptadecene-1, octadecene-1, nonadecene-1, or aliphatic substituted vinyl monomers such as eicosen-1, isobutylene, and styrene.
- Aromatic vinyl monomers such as substituted styrene, vinyl acetate, acrylic acid ester, methacrylic acid ester, glycidyl acrylate ester, glycidyl methacrylic acid ester, acrylate vinyl monomer such as hydroxyethyl methacrylic acid ester, acrylamide, allylamine , Nitrogen-containing vinyl monomers such as vinyl-p-aminobenzene and acrylonitrile, and diene such as butadiene, cyclopentadiene, 1,4-hexadiene and isoprene.
- It is preferably a copolymer of ethylene and one or more ⁇ -olefins having 3 to 20 carbon atoms, more preferably a copolymer of ethylene and one or more ⁇ -olefins having 3 to 16 carbon atoms, and most preferably ethylene. It is a copolymer with one or more ⁇ -olefins having 3 to 12 carbon atoms.
- the molecular weight of the ethylene- ⁇ -olefin copolymer was measured with a gel permeation chromatography measuring device at 140 ° C. using polystyrene standard using 1,2,4-trichlorobenzene as a solvent from the viewpoint of developing impact resistance.
- the number average molecular weight (Mn) obtained is preferably 10,000 or more, more preferably 10,000 to 100,000, and further preferably 20,000 to 60,000.
- the molecular weight distribution (weight average molecular weight / number average molecular weight: Mw / Mn) is preferably 3 or less, and more preferably 1.8 to 2.7, from the viewpoint of achieving both fluidity and impact resistance.
- the content of the ethylene unit of the ethylene- ⁇ -olefin copolymer is preferably 30 to 95% by mass with respect to the total amount of the ethylene- ⁇ -olefin copolymer from the viewpoint of handleability during processing.
- ethylene- ⁇ -olefin copolymers are, for example, JP-A-4-12833, JP-A-60-35006, JP-A-60-35007, JP-A-60-3508, and JP-A. It can be produced by the production method described in Kaihei 5-155930, JP-A-3-163088, US Pat. No. 5,272,236 and the like.
- the elastomer having a core-shell structure is a core-shell type impact-resistant modification having a core which is a particulate rubber and a shell which is a glassy graft layer formed on the outside of the core.
- Agents can be mentioned.
- the component of the rubber as the core butadiene rubber, acrylic rubber, silicone / acrylic composite rubber and the like can be preferably used.
- a glassy polymer such as a styrene resin, an acrylonitrile-styrene copolymer, or an acrylic resin can be preferably used for the shell.
- the thermoplastic resin is a polyamide resin
- an elastomer having a core-shell structure having a butadiene rubber core and an acrylic resin shell can be preferably used from the viewpoint of compatibility with the polyamide.
- the elastomer has at least a part of the elastomer having an acidic functional group.
- the fact that the elastomer has an acidic functional group means that the acidic functional group is added to the molecular skeleton of the elastomer via a chemical bond.
- the acidic functional group means a functional group capable of reacting with a basic functional group or the like, and specific examples thereof include a hydroxyl group, a carboxyl group, a carboxylate group, a sulfo group, an acid anhydride group and the like. Can be mentioned.
- the affinity between the elastomer and the cellulose nanofibers is high, which is preferable.
- the amount of the acidic functional group added to the elastomer is preferably 0.01% by mass or more, more preferably 0.01% by mass or more, based on 100% by mass of the elastomer from the viewpoint of affinity with cellulose nanofibers. It is 0.1% by mass or more, more preferably 0.2% by mass or more, preferably 5% by mass or less, more preferably 3% by mass or less, still more preferably 2% by mass or less.
- the number of acidic functional groups is determined based on the calibration curve prepared by measuring a calibration curve sample mixed with an acidic substance in advance with an infrared absorption spectrum measuring device and using the characteristic absorption band of the acid. It is a value obtained by measuring a sample.
- the elastomer having an acidic functional group examples include an elastomer having a core-shell structure having a layer formed by using acrylic acid or the like as a copolymerization component as a shell, an ethylene- ⁇ olefin copolymer containing acrylic acid or the like as a monomer, a polyolefin, and an aroma.
- Acrylic acid, ⁇ -unsaturated dicarboxylic acid or a derivative thereof is grafted onto a hydrogenated additive of a group compound-conjugated diene copolymer or an aromatic compound-conjugated diene copolymer in the presence or absence of a peroxide.
- examples thereof include an elastomer which is a modified product.
- the elastomer is an acid anhydride-modified elastomer.
- ⁇ , ⁇ in the hydrogenated product of a polyolefin-based elastomer, an aromatic compound-conjugated diene copolymer, or an aromatic compound-conjugated diene copolymer in the presence or absence of a peroxide is particularly preferable, and a peroxide is particularly added to a hydrogenated product of an ethylene- ⁇ -olefin copolymer or an aromatic compound-conjugated diene block copolymer.
- a modified product obtained by grafting an ⁇ , ⁇ -unsaturated dicarboxylic acid and a derivative thereof in the presence or absence of is particularly preferable.
- ⁇ , ⁇ -unsaturated dicarboxylic acid and its derivatives include maleic acid, fumaric acid, maleic anhydride, and fumaric anhydride, and maleic anhydride is particularly preferable among these.
- the elastomer When the elastomer has an acidic functional group, the elastomer may be a mixture of an elastomer having an acidic functional group and an elastomer having no acidic functional group.
- the mixing ratio of the elastomer having an acidic functional group and the elastomer not having an acidic functional group is 100% by mass in total, the elastomer having an acidic functional group has high toughness and physical property stability of the resin composition. From the viewpoint of maintaining a good condition, the content is preferably 10% by mass or more, more preferably 20% by mass or more, still more preferably 30% by mass or more, and most preferably 40% by mass or more.
- substantially all elastomers may be elastomers having an acidic functional group, but 80% by mass or less is desirable from the viewpoint of not causing a problem in fluidity.
- the amount of elastomer with respect to 100 parts by mass of the thermoplastic resin is preferably in the range of 1 to 50 parts by mass.
- the upper limit is more preferably 40 parts by mass, more preferably 35 parts by mass, still more preferably 30 parts by mass, and most preferably 25 parts by mass.
- the content is not more than the above upper limit.
- the lower limit is more preferably 2 parts by mass, further preferably 3 parts by mass, still more preferably 4 parts by mass, and most preferably 5 parts by mass. In order to enhance the toughness and physical property stability of the resin composition, it is preferably at least the above lower limit.
- the dispersed particle diameter is preferably 3 ⁇ m or less, more preferably 2 ⁇ m or less, and most preferably 1 ⁇ m as the number average particle diameter. It is as follows.
- the lower limit is not particularly limited, but is, for example, 0.1 ⁇ m. From the viewpoint of high toughness and physical property stability, it is preferably within the above range.
- the elastomer has high uniformity of dispersed particle size. From this viewpoint, it is preferable that the volume ratio of the dispersed particles having a particle diameter of 1 ⁇ m or more to the entire dispersed particles of the elastomer is 30% by volume or less. The upper limit is more preferably 25% by volume, even more preferably 20% by volume, even more preferably 15% by volume, and most preferably 10% by volume. In the distributed particle size distribution on a volume basis, even if the number is very small, if coarse particles are present, the volume ratio of the dispersed particles having a particle size of 1 ⁇ m or more is expressed in a large volume at once.
- the volume ratio When the volume ratio is within the above range, the uniformity of the dispersed particle size is high, which is preferable. From the viewpoint of ease of production of the resin composition, the volume ratio may be, for example, 2% by volume or more, or 5% by volume or more.
- a resin composition is produced by extrusion-kneading the compounding components of the resin composition, and the screw rotation speed during extrusion-kneading is increased to increase the shear strain in the compounding components.
- a method of finely dispersing the elastomer by giving a method, for example, a method of arranging screw parts such as a seal ring in which a narrow clearance is uniformly present to give a stretch flow strain to a compounding component, a method of passing a special narrow slit through a molten polymer, Examples thereof include a method of applying stretch flow strain at the slit portion, and any of these methods may be used.
- the method of applying high shear the polymer temperature rises remarkably during processing, so a method using stretch flow strain is used. Is more preferable.
- a resin composition in the form of a molded product, pellets or the like is cut as an ultrathin section, the thermoplastic resin phase is stained with phosphotoxyic acid or the like, and then observed with a transmission electron microscope.
- Method A method in which the surface of a resin composition in the form of a molded product, pellet, etc. is uniformly surfaced, then immersed in a solvent that selectively dissolves only the elastomer, the elastomer is extracted, and the resin composition is observed with a scanning electron microscope. And so on.
- the obtained image is binarized by an image analyzer, the diameters of the dispersed particles in the dispersed phase (at least 500 randomly selected) are calculated as the equivalent circle diameters, and the respective particle diameters are counted to obtain the dispersed particles. It is possible to calculate the number average particle diameter and the volume ratio of particles having a predetermined particle diameter (for example, the above particle diameter of 1 ⁇ m or more).
- the surface treatment agent include a compound having a hydrophilic segment and a hydrophobic segment in the molecule, and more specifically, a compound giving a hydrophilic segment (for example, polyethylene glycol) and a hydrophobic segment.
- a copolymer for example, a block copolymer of propylene oxide and ethylene oxide, tetrahydrofuran obtained by using one or more of each of the given compounds (for example, polypropylene glycol, poly (tetramethylene ether) glycol (PTMEG), polybutadiene diol, etc.) Block copolymer of ethylene oxide) and the like.
- the preferable content of the surface treatment agent in the pellet is 0.1% by mass or more, 0.2% by mass or more, or 0.5% by mass from the viewpoint of enhancing the dispersibility of the cellulose nanofibers in the pellet.
- % Or more preferably 50% by mass or less, 30% by mass or less, or 20% by mass or less, or 18% by mass or less, or 15 from the viewpoint of suppressing plasticization of the molded product and maintaining good strength. It is 0% by mass or less, 10% by mass or less, or 5% by mass or less.
- the preferable amount of the surface treatment agent with respect to 100 parts by mass of the cellulose nanofibers is preferably 0.1 part by mass or more, 0.5 parts by mass or more, or 1 from the viewpoint of enhancing the dispersibility of the cellulose nanofibers in the pellet. From the viewpoint of suppressing plasticization of the molded product and maintaining good strength, it is preferably 100 parts by mass or less, 99 parts by mass or less, 90 parts by mass or less, or 80 parts by mass or less, or It is 70 parts by mass or less, 50 parts by mass or less, or 40 parts by mass or less.
- antioxidants hindered phenolic antioxidants, sulfur-based antioxidants, and phosphorus-based antioxidants are preferable from the viewpoint of the effect of preventing deterioration due to heat, and phosphorus-based antioxidants and hindered phenol-based oxidations are preferable.
- Inhibitors are more preferred, and phosphorus-based antioxidants and / or hindered phenol-based antioxidants in combination with hindered amine-based light stabilizers (HALS) are even more preferred.
- HALS hindered amine-based light stabilizers
- the preferable amount of the antioxidant is 0.01% by mass or more, 0.02% by mass or more, or 0.03% by mass or more, or 0.05% by mass or more, preferably 0.01% by mass or more, based on the whole pellet. Is 5% by mass or less, or 4% by mass or less, or 3% by mass or less, or 2% by mass or less, or 1% by mass or less.
- the inorganic filler examples include fibrous particles, plate-like particles, and inorganic pigments.
- the fibrous particles and the plate-like particles may have an average aspect ratio of 5 or more.
- the amount of the inorganic filler in the pellet is preferably 0.002 part by mass to 50 part by mass with respect to 100 parts by mass of the thermoplastic resin from the viewpoint of improving the handleability when molding the pellet into the molded product.
- lubricating oil examples include natural oils (engine oils, cylinder oils, etc.), synthetic hydrocarbons (paraffin oils, naphthenic oils, aroma oils, etc.), silicone oils, and the like.
- the molecular weight of the lubricating oil may be, for example, 100 or more, 400 or more, or 500 or more, and may be, for example, 5 million or less, 2 million or less, or 1 million or less.
- the melting point of the lubricating oil may be, for example, -50 ° C or higher, -30 ° C or higher, or -20 ° C or higher, and may be, for example, 50 ° C or lower, 30 ° C or lower, or 20 ° C or lower.
- the melting point is 2.5 ° C. lower than the pour point of the lubricating oil, and the pour point can be measured in accordance with JIS K2269.
- the content of the lubricating oil with respect to 100 parts by mass of the thermoplastic resin is preferably 0.1 part by mass or more, 0.2 parts by mass or more, or 0.3 parts by mass or more. From the viewpoint of avoiding undesired softening of the molded product, it is preferably 5.0 parts by mass or less, 4.5 parts by mass or less, or 4.2 parts by mass or less.
- the total amount of additional components in the pellet may be, for example, 0.01% by mass or more, 0.1% by mass or more, or 1% by mass or more, for example, 20% by mass or less, or 10% by mass or less, or. It may be 5% by mass or less.
- the pellet comprises 50% to 99% by weight of the thermoplastic resin, 1% to 30% by weight of the cellulose nanofibers, and 0.01% to 20% by weight of the additional component.
- the pellets are thermoplastic resin 70% to 99% by weight, cellulose nanofibers 1% to 10% by weight, and additional components 0.01% to 30% by weight, or thermoplastic resin. It contains 75% by mass to 99% by mass, 1% by mass to 5% by mass of cellulose nanofibers, and 0.01% by mass to 20% by mass of additional components.
- the crystallization temperature lowering agent of the present disclosure is a crystallization temperature lowering agent that lowers the temperature-decreasing crystallization peak temperature of the resin composition of the present disclosure, which is measured by a differential scanning calorimetry (DSC), by 5 to 30 ° C.
- the resin composition containing the above compound has a temperature-decreasing crystallization peak temperature that is 5 to 30 ° C. lower than that of the same resin composition except that it does not contain the crystallization temperature lowering agent).
- the lowering of the temperature-decreasing crystallization peak temperature by the crystallization temperature lowering agent is preferably 5 ° C. to 25 ° C., or 10 ° C. to 20 ° C.
- the temperature-decreasing crystallization peak temperature of the present disclosure is a temperature rise rate of 10 ° C./min using DSC, a melting point of + 30 ° C. when the thermoplastic resin is a crystalline resin, and glass when the thermoplastic resin is an amorphous resin. This is the peak top temperature of the crystallization peak when the temperature is raised to the transition point + 30 ° C. and then lowered at 10 ° C./min.
- a crystallization temperature lowering agent when the thermoplastic resin and the cellulose nanofibers are melt-mixed and then cooled to produce pellets of the resin composition, crystallization by cooling of the resin composition proceeds slowly. The voids in the pellet can be reduced.
- crystallization temperature lowering agent examples include pentaerythritol, dipentaerythritol, trimethylolethane, niglocin and the like, and pentaerythritol is particularly preferable.
- the mass ratio of the crystallization temperature lowering agent to 100% by mass of the resin composition is preferably 0.01% by mass to 10% by mass, or 0.05% by mass to 5% by mass, or 0.8% by mass to 3% by mass. %.
- thermoplastic resins having different temperature-decreasing crystallization peak temperatures Tcc and the temperature-decreasing crystallization peak temperature (temperature-decreasing crystallization peak temperature) of the thermoplastic resin contained in the resin composition.
- the difference Tcc-Tcp from the peak temperature on the highest temperature side) Tcp is preferably 5 ° C to 30 ° C, or 5 ° C to 25 ° C, or 10 ° C to 20 ° C.
- the ratio of the pores (Ve) of the pellets to the total volume (Vc) of the cellulose nanofibers per 100 pellets Ve / Vc is the formation of voids relative to the amount of cellulose nanofibers used. From the viewpoint of obtaining pellets with a small amount of cellulose, it is preferably 0% by volume to 4% by volume, 0% by volume to 3% by volume, or 0% by volume to 1% by volume.
- Ve / Vc is calculated by the following formula.
- Ve / Vc (pore area ratio x pore-containing pellet ratio) / (cellulose volume ratio when no pores exist x (100-pore area ratio x pore-containing pellet ratio / 100)) x 100
- the cellulose volume ratio in the absence of pores is calculated from the addition amount and density of each component of the resin composition.
- the pellets of the present disclosure are obtained by melt-kneading a thermoplastic resin, cellulose nanofibers, and any additional components using, for example, a single-screw or twin-screw extruder, extruding them into strands, cooling and solidifying them in a water bath, and pelletizing them.
- a single-screw or twin-screw extruder extruding them into strands, cooling and solidifying them in a water bath, and pelletizing them.
- Examples of the method for melt-kneading the thermoplastic resin, the cellulose nanofibers, and any additional component include the following methods. (1) A method of batch melting and kneading a thermoplastic resin, cellulose nanofibers, and any additional component.
- thermoplastic resin and an optional additional component are melt-kneaded, and then cellulose nanofibers and an optional additional component are added and further melt-kneaded.
- a thermoplastic resin, cellulose nanofibers, and any additional component are melt-kneaded, and then cellulose nanofiber, water, and if necessary, any additional component are mixed and then melt-kneaded all at once.
- a thermoplastic resin and, if necessary, an arbitrary additional component are melt-kneaded, and then a thermoplastic resin and cellulose nanofibers mixed in a desired ratio and an arbitrary additional component are added and further melt-kneaded.
- a method in which the above (1) to (4) are added separately by Top and Side at an arbitrary ratio using a single-screw or twin-screw extruder, and melt-kneaded.
- the air cooling distance between the extruder die and the water surface of the strand bath , Strand bath temperature, Strand bath immersion length, Strand take-up speed, etc. can be controlled to change the properties of the pellets. From the viewpoint of reducing the pore-containing pellets, it is useful to control the melt-kneading temperature, the resin discharge amount, the number of die holes, the air-cooling distance, the strand bath temperature, and the strand immersion length.
- the design conditions for the extruder include screw diameter, number of die holes, and die hole diameter. Further, as the cooling conditions of the strands discharged from the extruder, the air-cooling distance between the extruder die and the water surface of the strand bath, the strand bath temperature, the strand bath immersion length, and the range of the strand take-up speed can be mentioned.
- the melt-kneading temperature can be adjusted according to the type of thermoplastic resin. Further, the cooling rate of the strand can be reduced by increasing the air cooling distance between the extruder die and the water surface of the strand bath, increasing the water temperature in the strand bath, and the like. For example, by setting the melting temperature to the melting point of the crystalline thermoplastic resin or the glass transition point of the amorphous thermoplastic resin of about +20 to 100 ° C. and controlling the cooling rate of the strands, the temperature of the resin composition during strand cooling It is possible to increase the time required for the temperature to drop from the melting temperature to less than the melting point of the crystalline thermoplastic resin or the glass transition point of the amorphous thermoplastic resin.
- the melt-kneading temperature is 180 ° C. to 300 ° C.
- the strand cooling condition is an air cooling distance of 150 to 300 mm
- the strand bath temperature is 40 ° C.
- the immersion length is set. It may be set to 300 to 500 mm.
- the melt-kneading temperature is 180 ° C to 250 ° C
- the strand cooling conditions are an air cooling distance of 150 to 300 mm
- the strand bath temperature is 40 ° C
- the immersion length is 500 to 500. It can be set to 2000 mm.
- the cooled strands are cut into pellets.
- the pellet shape may differ depending on the cutting method at the time of extrusion processing. For example, pellets cut by a cutting method called underwater cut are often spherical, and pellets cut by a cutting method called hot cut are spherical. Alternatively, it often has an ellipsoidal shape, and pellets cut by a cutting method called strand cutting often have a columnar shape.
- the pellet of this embodiment is vacuum molded by extrusion molding (cold runner method, hot runner method), injection molding, injection compression molding, gas assisted injection molding, foam injection molding, ultra-thin injection molding (ultra-high-speed injection molding), etc. , Blow molding, decorative molding, molding of other materials, low pressure molding, composite molding in a mold (insert molding, outsert molding) and the like can be used to manufacture a molded product.
- extrusion molding cold runner method, hot runner method
- injection molding injection compression molding
- gas assisted injection molding foam injection molding
- ultra-thin injection molding ultra-thin injection molding (ultra-high-speed injection molding)
- Blow molding decorative molding, molding of other materials, low pressure molding, composite molding in a mold (insert molding, outsert molding) and the like can be used to manufacture a molded product.
- injection molding the advantages of using the pellets of the present disclosure are particularly remarkable.
- One aspect of the present invention is a step of preparing the pellets of the present disclosure, and a step of injecting the pellets in a mold to obtain a molded product.
- the injection molding machine suitable as a drive system is a hydraulic, electric, or hydraulic electric hybrid injection molding machine.
- a plunger type, a pre-plastic type, and a screw type injection molding machine can be exemplified.
- suitable molding conditions are that the melting point is the melting point of the crystalline thermoplastic resin or the glass transition point of the amorphous thermoplastic resin + 20 to 100 ° C., and the mold temperature is the melting temperature of the crystalline thermoplastic resin. Examples thereof include a melting point or a glass transition point of an amorphous thermoplastic resin of ⁇ 50 to 200 ° C.
- the molded product may be provided in various shapes such as a sheet, a film, a fiber, a solid or hollow molded product, and the like.
- various uses of the molded body can be exemplified, for example, industrial machine parts (for example, electromagnetic equipment chassis, roll material, transport arm, medical equipment member, etc.), general machine parts, automobile / railway / vehicle parts (for example). Skin panels, chassis, aerodynamic members, seats, friction materials inside transmissions, etc.), ship members (eg hulls, seats, etc.), aviation-related parts (eg fuselage, main wings, tail wings, moving wings, fairings, cowls, doors, etc.
- the molded product when provided as a resin composite film, it is suitable for reinforcing a laminated plate in a printed wiring board.
- Case crossbar, insulation shaft, fan blade, mechanical parts, transparent resin substrate, speaker diaphragm, eta diaphragm, TV screen, fluorescent lamp cover, antenna for communication equipment / aerospace, horn cover, radome, case, Mechanical parts, wiring boards, aircraft, rockets, electronic equipment parts for artificial satellites, railway parts, marine parts, bathtubs, septic tanks, corrosion resistant equipment, chairs, safety caps, pipes, tank lorries, cooling towers, floating breakers, underground It can also be applied to applications such as buried tanks and containers.
- automobile members and electronic product members that require resin molding are preferable in that they can exert superiority due to the excellent heat resistance of the molded product containing cellulose nanofibers.
- Ve / Vc (pore area ratio x pore-containing pellet ratio) / (cellulose volume ratio when no pores exist x (100-pore area ratio x pore-containing pellet ratio / 100)) x 100
- the pore area ratio and the pore-containing pellet ratio were measured by the method described later.
- the cellulose volume ratio in the absence of pores was calculated from the addition amount and density of each component of the resin composition.
- density of polyamide 6 (PA6) 1.14g / cm 3 , polypropylene (PP) 0.90g / cm 3, maleic anhydride-modified ethylene - octene copolymer (MEOR) 0.87g / cm 3, cellulose nano Calculated as crystal (CNC) and cellulose nanofibers (CNF) 1.50 g / cm 3 , hydrophobicized cellulose nanofibers (hydrophobicized CNF) 1.43 g / cm 3 , and pentaerythritol (PET) 1.40 g / cm 3 .
- PA6 polyamide 6
- PP polypropylene
- MEOR maleic anhydride-modified ethylene - octene copolymer
- CNF cellulose nano Calculated as crystal
- CNF cellulose nanofibers
- hydrophobicized cellulose nanofibers hydrophobicized CNF
- PET pentaerythritol
- ⁇ Ratio of pore area to cross-sectional area of pellet TD cross section in pore-containing pellets 100 pellets were selected and the TD cross-section was observed in the same manner as in the evaluation of ⁇ the number of pore-containing pellets per 100 pellets>.
- the pore area in the TD cross section of the pore-containing pellet was measured with the software attached to the scope, the ratio of the pore area to the total cross-sectional area of 100% of the TD cross section was calculated, and averaged for the entire pore-containing pellet. The value was taken as the pore area ratio. When no pore-containing pellet is contained in 100 pellets, the ratio of the pore area is 0%.
- the pellet MD direction axis and the normal to the line segment corresponding to the pellet MD direction end face were defined, and the angle formed by the pellet MD direction axis and the normal was measured.
- the average value of the number of 10 pellets was obtained as the inclination angle of the pellet cut surface.
- ⁇ Molding cycle> The pellet was injection molded into the shape of an ISO 294-3 multipurpose test piece. The average value of the molding cycle (seconds) when 30 pieces were produced was defined as the molding cycle.
- YI ⁇ Yellowness
- ⁇ Cellulose> Cellulose nanocrystal (hereinafter referred to as CNC)
- CNC Commercially available DP pulp (average degree of polymerization 1600) was cut and hydrolyzed in a 10 mass% hydrochloric acid aqueous solution at 105 ° C. for 30 minutes.
- the obtained acid-insoluble residue was filtered, washed, and adjusted in pH to prepare a crystalline cellulose dispersion having a solid content concentration of 14% by mass and a pH of 6.5.
- the crystalline cellulose dispersion was spray-dried to obtain a dried product of crystalline cellulose.
- the supply amount was 10 kg / hr
- the dried product obtained above was supplied to an airflow type crusher (STJ-400 type, manufactured by Seishin Enterprise Co., Ltd.) and pulverized to obtain CNC as crystalline cellulose fine powder.
- the diameter was 30 nm and the L / D was 8.
- CNF Cellulose nanofibers
- defibrated cellulose was obtained by defibrating with a high-pressure homogenizer (operating pressure: treated 10 times at 85 MPa) at the same concentration.
- a disc refiner is used, and after processing with a beating blade having a high cutting function (hereinafter referred to as a cutting blade) for 4 hours, a beating blade having a high defibration function (hereinafter referred to as a defibrating blade) is used. An additional 1.5 hours of beating was performed.
- the diameter was 90 nm and the L / D was 30 or more (about 300).
- hydrophobicized CNF Hydrophobicized CNF (hereinafter referred to as hydrophobicized CNF) (Defibration process)
- DMSO dimethyl sulfoxide
- IMEX uniaxial stirrer
- the mixture was fed to a bead mill (NVM-1.5 manufactured by Imex) with a hose pump and circulated for 120 minutes using only DMSO to obtain a defibrated slurry.
- the rotation speed of the bead mill was 2500 rpm and the peripheral speed was 12 m / s.
- the beads were made of zirconia, ⁇ 2.0 mm, and the filling rate was 70% (the slit gap of the bead mill at this time was 0.6 mm).
- the slurry temperature was controlled to 40 ° C. by a chiller in order to absorb heat generated by friction.
- the diameter was 65 nm and the L / D was 30 or more (about 450).
- Example 1 90 parts by mass of PA6 and 10 parts by mass of CNF obtained in Preparation Example 2 are mixed and melt-kneaded with a twin-screw extruder (TEM-37SS manufactured by Toshiba Machine Co., Ltd., screw diameter 37 mm) at a kneading temperature of 250 ° C. Extruded into strands.
- the resin discharge rate of the twin-screw extruder was 20 kg / h, and the number of holes in the die was three.
- the distance from the die hole to the water surface of the strand bath was adjusted so that the air-cooling distance until the strand that came out of the die landed on the water was 150 mm.
- After cooling with a strand bath whose water temperature was adjusted to 40 ° C. with an immersion length of 400 mm it was cut with a strand cutter to obtain pellets.
- Example 2 Pellets were obtained in the same manner as in Example 1 except that the pellet production conditions were changed as shown in Table 1.
- Example 8 Pellets were prepared in the same manner as in Example 3 except that the pellet production conditions were changed as shown in Table 1, the take-up speed of the strand cutter was adjusted in the low speed direction, and the minor axis of the pellet TD cross section was set to the value shown in Table 1. Obtained.
- Example 10 70 parts by mass of PA6, 20 parts by mass of MEOR, and 10 parts by mass of CNF were mixed and melt-kneaded to obtain pellets in the same manner as in Example 3 except that the pellet production conditions were changed as shown in Table 1. It was.
- Example 11 Except that 90 parts by mass of PA6, 5 parts by mass of CNC obtained in Preparation Example 1 and 5 parts by mass of CNF obtained in Preparation Example 2 were mixed and melt-kneaded, and the pellet production conditions were changed as shown in Table 1. Pellets were obtained in the same manner as in Example 3.
- Example 12 and 13 90 parts by mass of PA6 and 10 parts by mass of the hydrophobized CNF obtained in Preparation Example 3 were mixed and melt-kneaded, and pellets were prepared in the same manner as in Example 3 except that the pellet production conditions were changed as shown in Table 1. Obtained.
- Example 14 89 parts by mass of PA6, 1 part by mass of PET, and 10 parts by mass of the hydrophobic CNF obtained in Preparation Example 3 were mixed and melt-kneaded, and the pellet production conditions were changed as shown in Table 1, except that the pellet production conditions were changed to those of Example 3. Pellets were obtained in the same manner.
- Example 4 Same as Example 3 except that 90 parts by mass of PP and 10 parts by mass of CNF obtained in Preparation Example 2 were mixed, melt-kneaded at a kneading temperature of 200 ° C., and the pellet production conditions were changed as shown in Table 1. To obtain pellets.
- Example 1 From the results shown in Table 1, in Examples 1 to 15, although the pellets contained cellulose nanofibers, there was little yellowing of the molded product and the appearance was good.
- Example 2, 6, 7, 11 and 12 in which the water temperature was relatively high and the solidification rate of the thermoplastic resin was slowed down, the pore-containing pellet ratio was 0%, and the generation of pores was not cellulose nanofibers. It was reduced to a level comparable to that of Reference Example 1 in use, and showed a good appearance comparable to that of Reference Example 1 in which cellulose nanofibers were not used.
- Example 8 in which the pellet size was increased, and in Examples 10 and 15 in which the type of the thermoplastic resin was changed, molded articles having less yellowing and a good appearance were obtained.
- Example 12 and 13 using the hydrophobized CNF the yellowing of the molded product was particularly small.
- Example 14 in which the resin crystallization temperature lowering agent was added, a molded product having a shorter molding cycle and less yellowing was obtained.
- the pellet according to the present invention is useful for producing a molded product having a good appearance and suppressed yellowing.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Injection Moulding Of Plastics Or The Like (AREA)
- Processing And Handling Of Plastics And Other Materials For Molding In General (AREA)
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202311549950.5A CN117417635A (zh) | 2019-05-13 | 2020-05-13 | 低孔隙的粒料以及成型体的制造方法 |
| EP20806009.5A EP3970934A4 (en) | 2019-05-13 | 2020-05-13 | Low-pore pellets and method for producing molded body |
| JP2020543123A JP6894053B2 (ja) | 2019-05-13 | 2020-05-13 | 低空孔のペレット、及び成形体の製造方法 |
| US17/610,886 US12186947B2 (en) | 2019-05-13 | 2020-05-13 | Low-pore pellets and method for producing molded body |
| CN202080033561.5A CN113795549B (zh) | 2019-05-13 | 2020-05-13 | 低孔隙的粒料以及成型体的制造方法 |
| US18/961,880 US20250091263A1 (en) | 2019-05-13 | 2024-11-27 | Low-Pore Pellets and Methods for Producing Molded Body |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019090819 | 2019-05-13 | ||
| JP2019-090819 | 2019-05-13 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US17/610,886 A-371-Of-International US12186947B2 (en) | 2019-05-13 | 2020-05-13 | Low-pore pellets and method for producing molded body |
| US18/961,880 Continuation US20250091263A1 (en) | 2019-05-13 | 2024-11-27 | Low-Pore Pellets and Methods for Producing Molded Body |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020230827A1 true WO2020230827A1 (ja) | 2020-11-19 |
Family
ID=73288883
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/019146 Ceased WO2020230827A1 (ja) | 2019-05-13 | 2020-05-13 | 低空孔のペレット、及び成形体の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US12186947B2 (https=) |
| EP (1) | EP3970934A4 (https=) |
| JP (2) | JP6894053B2 (https=) |
| CN (2) | CN113795549B (https=) |
| WO (1) | WO2020230827A1 (https=) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022209138A1 (https=) * | 2021-03-29 | 2022-10-06 | ||
| JP2023019943A (ja) * | 2021-07-30 | 2023-02-09 | 公立大学法人 富山県立大学 | バイオマスナノファイバー添加剤及び樹脂組成物 |
| JP2023043785A (ja) * | 2021-09-16 | 2023-03-29 | 旭化成株式会社 | 発泡用熱可塑性樹脂ペレット群及びその製造方法 |
| SE2330258A1 (en) * | 2023-06-02 | 2024-12-03 | Woodcomposite Sweden Ab | Cellulosic composite |
| JP2025505659A (ja) * | 2022-02-24 | 2025-02-28 | リグナム インコーポレーティッド | プラスチック添加用耐スクラッチ性バイオ添加剤の製造方法、及びこれにより製造されたプラスチック添加用耐スクラッチ性バイオ添加剤 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6752935B1 (ja) * | 2019-05-28 | 2020-09-09 | 旭化成株式会社 | 樹脂成形体の製造方法 |
| JP2023152719A (ja) * | 2022-03-30 | 2023-10-17 | 旭化成株式会社 | ペレット及びペレットの製造方法 |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6035007A (ja) | 1983-06-06 | 1985-02-22 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | ポリオレフインの密度及び分子量を調節するための方法と触媒 |
| JPS6035006A (ja) | 1983-06-06 | 1985-02-22 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | 反応器ブレンドポリオレフインの製造方法及びその触媒 |
| JPS6035008A (ja) | 1983-06-06 | 1985-02-22 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | 幅広い分子量分布を有するポリエチレンの製造方法及びその触媒 |
| JPH03163088A (ja) | 1989-08-31 | 1991-07-15 | Dow Chem Co:The | 第4族金属配位錯体 |
| JPH0412283B2 (https=) | 1981-07-09 | 1992-03-04 | Hoechst Ag | |
| JPH05155930A (ja) | 1991-05-31 | 1993-06-22 | Mitsui Petrochem Ind Ltd | オレフィン重合用触媒およびオレフィンの重合方法 |
| US5272236A (en) | 1991-10-15 | 1993-12-21 | The Dow Chemical Company | Elastic substantially linear olefin polymers |
| JPH07228775A (ja) | 1994-02-17 | 1995-08-29 | Kuraray Co Ltd | 難燃性ポリアミド組成物 |
| JP2003055549A (ja) | 2001-06-05 | 2003-02-26 | Kuraray Co Ltd | ポリアミド組成物 |
| WO2007034805A1 (ja) * | 2005-09-21 | 2007-03-29 | Kureha Corporation | ポリグリコール酸樹脂組成物の製造方法 |
| JP2007290384A (ja) * | 2006-03-29 | 2007-11-08 | Toray Ind Inc | 熱可塑性樹脂組成物溶融混練装置および熱可塑性樹脂組成物の製造方法 |
| JP2010221622A (ja) * | 2009-03-25 | 2010-10-07 | Konica Minolta Holdings Inc | 繊維複合材料の製造方法 |
| JP2012117066A (ja) * | 2010-12-02 | 2012-06-21 | Ems-Patent Ag | 透明成形物を製造するための、透明コポリアミド及び脂肪族のホモポリアミドから作製された混合物をベースとするポリアミド成形組成物 |
| WO2017159823A1 (ja) | 2016-03-16 | 2017-09-21 | 株式会社Kri | セルロース微細繊維およびその製造方法 |
| JP2018016896A (ja) | 2016-07-25 | 2018-02-01 | トヨタ車体株式会社 | 成形材料混合物及びその製造方法 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0170245B1 (en) * | 1981-01-21 | 1999-04-28 | Kawasaki Chemical Holding Co., Inc. | Pellets of fibre-reinforced compositions and methods for producing such pellets |
| JP2854949B2 (ja) | 1990-09-13 | 1999-02-10 | 三井化学株式会社 | ポリイミドペレットの製造方法 |
| US5288772A (en) | 1992-06-23 | 1994-02-22 | Clemson University | Pre-treated cellulosic materials for producing molded composite articles therefrom and process |
| JP2000015631A (ja) * | 1998-07-03 | 2000-01-18 | Kanegafuchi Chem Ind Co Ltd | 成形加工性に優れる熱可塑性樹脂ペレット |
| US20080146701A1 (en) | 2003-10-22 | 2008-06-19 | Sain Mohini M | Manufacturing process of cellulose nanofibers from renewable feed stocks |
| US9371616B2 (en) | 2009-01-05 | 2016-06-21 | Konica Minolta Holdings, Inc. | Laminate and production method thereof |
| JP5675066B2 (ja) * | 2009-06-26 | 2015-02-25 | 株式会社ダイセル | 微小セルロース系繊維含有樹脂組成物及びその製造方法 |
| JP5371683B2 (ja) | 2009-10-20 | 2013-12-18 | 三菱エンジニアリングプラスチックス株式会社 | ポリアミド樹脂組成物ペレットの製造方法 |
| JP5838105B2 (ja) * | 2012-03-05 | 2015-12-24 | 住化カラー株式会社 | ストランド切断方法ならびにペレット製造方法および製造装置 |
| DE102015211632A1 (de) * | 2015-06-23 | 2016-12-29 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Polymerzusammensetzung mit verzögertem Kristallisationsverhalten, das Kristallisationsverhalten beeinflussende Additivzusammensetzung, Verfahren zur Herabsetzung des Kristallisationspunktes sowie Verwendung einer Additivzusammensetzung |
| KR102070374B1 (ko) * | 2016-12-28 | 2020-01-29 | 아사히 가세이 가부시키가이샤 | 셀룰로오스 함유 수지 조성물 및 셀룰로오스 제제 |
| WO2018123150A1 (ja) * | 2016-12-28 | 2018-07-05 | 旭化成株式会社 | セルロース含有樹脂組成物及びセルロース製剤 |
| JP7108375B2 (ja) | 2017-01-18 | 2022-07-28 | パナソニックホールディングス株式会社 | 複合樹脂組成物 |
| JP6360954B1 (ja) * | 2017-09-22 | 2018-07-18 | 株式会社吉川国工業所 | 繊維強化複合材料 |
-
2020
- 2020-05-13 CN CN202080033561.5A patent/CN113795549B/zh active Active
- 2020-05-13 US US17/610,886 patent/US12186947B2/en active Active
- 2020-05-13 WO PCT/JP2020/019146 patent/WO2020230827A1/ja not_active Ceased
- 2020-05-13 JP JP2020543123A patent/JP6894053B2/ja active Active
- 2020-05-13 CN CN202311549950.5A patent/CN117417635A/zh active Pending
- 2020-05-13 EP EP20806009.5A patent/EP3970934A4/en active Pending
-
2021
- 2021-06-02 JP JP2021093204A patent/JP7561086B2/ja active Active
-
2024
- 2024-11-27 US US18/961,880 patent/US20250091263A1/en active Pending
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0412283B2 (https=) | 1981-07-09 | 1992-03-04 | Hoechst Ag | |
| JPS6035007A (ja) | 1983-06-06 | 1985-02-22 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | ポリオレフインの密度及び分子量を調節するための方法と触媒 |
| JPS6035006A (ja) | 1983-06-06 | 1985-02-22 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | 反応器ブレンドポリオレフインの製造方法及びその触媒 |
| JPS6035008A (ja) | 1983-06-06 | 1985-02-22 | エクソン・リサ−チ・アンド・エンジニアリング・カンパニ− | 幅広い分子量分布を有するポリエチレンの製造方法及びその触媒 |
| JPH03163088A (ja) | 1989-08-31 | 1991-07-15 | Dow Chem Co:The | 第4族金属配位錯体 |
| JPH05155930A (ja) | 1991-05-31 | 1993-06-22 | Mitsui Petrochem Ind Ltd | オレフィン重合用触媒およびオレフィンの重合方法 |
| US5272236A (en) | 1991-10-15 | 1993-12-21 | The Dow Chemical Company | Elastic substantially linear olefin polymers |
| JPH07228775A (ja) | 1994-02-17 | 1995-08-29 | Kuraray Co Ltd | 難燃性ポリアミド組成物 |
| JP2003055549A (ja) | 2001-06-05 | 2003-02-26 | Kuraray Co Ltd | ポリアミド組成物 |
| WO2007034805A1 (ja) * | 2005-09-21 | 2007-03-29 | Kureha Corporation | ポリグリコール酸樹脂組成物の製造方法 |
| JP2007290384A (ja) * | 2006-03-29 | 2007-11-08 | Toray Ind Inc | 熱可塑性樹脂組成物溶融混練装置および熱可塑性樹脂組成物の製造方法 |
| JP2010221622A (ja) * | 2009-03-25 | 2010-10-07 | Konica Minolta Holdings Inc | 繊維複合材料の製造方法 |
| JP2012117066A (ja) * | 2010-12-02 | 2012-06-21 | Ems-Patent Ag | 透明成形物を製造するための、透明コポリアミド及び脂肪族のホモポリアミドから作製された混合物をベースとするポリアミド成形組成物 |
| WO2017159823A1 (ja) | 2016-03-16 | 2017-09-21 | 株式会社Kri | セルロース微細繊維およびその製造方法 |
| JP2018016896A (ja) | 2016-07-25 | 2018-02-01 | トヨタ車体株式会社 | 成形材料混合物及びその製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3970934A4 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2022209138A1 (https=) * | 2021-03-29 | 2022-10-06 | ||
| WO2022209138A1 (ja) * | 2021-03-29 | 2022-10-06 | 三井化学株式会社 | グラフト変性エチレン系重合体、グラフト変性エチレン系重合体を含むポリアミド組成物およびその用途 |
| JP7639121B2 (ja) | 2021-03-29 | 2025-03-04 | 三井化学株式会社 | グラフト変性エチレン系重合体、グラフト変性エチレン系重合体を含むポリアミド組成物およびその用途 |
| JP2023019943A (ja) * | 2021-07-30 | 2023-02-09 | 公立大学法人 富山県立大学 | バイオマスナノファイバー添加剤及び樹脂組成物 |
| JP7738834B2 (ja) | 2021-07-30 | 2025-09-16 | 公立大学法人 富山県立大学 | バイオマスナノファイバー添加剤及び樹脂組成物 |
| JP2023043785A (ja) * | 2021-09-16 | 2023-03-29 | 旭化成株式会社 | 発泡用熱可塑性樹脂ペレット群及びその製造方法 |
| JP2025505659A (ja) * | 2022-02-24 | 2025-02-28 | リグナム インコーポレーティッド | プラスチック添加用耐スクラッチ性バイオ添加剤の製造方法、及びこれにより製造されたプラスチック添加用耐スクラッチ性バイオ添加剤 |
| SE2330258A1 (en) * | 2023-06-02 | 2024-12-03 | Woodcomposite Sweden Ab | Cellulosic composite |
| SE547316C2 (en) * | 2023-06-02 | 2025-07-01 | Woodcomposite Sweden Ab | Cellulosic composite and method for producing the cellulosic composition |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2021138965A (ja) | 2021-09-16 |
| US20250091263A1 (en) | 2025-03-20 |
| JP6894053B2 (ja) | 2021-06-23 |
| JPWO2020230827A1 (ja) | 2021-05-20 |
| CN117417635A (zh) | 2024-01-19 |
| CN113795549B (zh) | 2023-12-12 |
| JP7561086B2 (ja) | 2024-10-03 |
| EP3970934A1 (en) | 2022-03-23 |
| CN113795549A (zh) | 2021-12-14 |
| US12186947B2 (en) | 2025-01-07 |
| EP3970934A4 (en) | 2022-06-29 |
| US20220203585A1 (en) | 2022-06-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6894053B2 (ja) | 低空孔のペレット、及び成形体の製造方法 | |
| Petchwattana et al. | Mechanical properties, thermal degradation and natural weathering of high density polyethylene/rice hull composites compatibilized with maleic anhydride grafted polyethylene | |
| Fowlks et al. | The effect of maleated polylactic acid (PLA) as an interfacial modifier in PLA‐talc composites | |
| JP6896946B2 (ja) | 複合粒子及び樹脂組成物 | |
| JP6752935B1 (ja) | 樹脂成形体の製造方法 | |
| CN108884272A (zh) | 含纤维素的树脂组合物和纤维素制剂 | |
| CN113652029B (zh) | 一种微发泡聚丙烯组合物及其制备方法和应用 | |
| JP7117181B2 (ja) | セルロース含有樹脂組成物 | |
| JP2021066789A (ja) | セルロース樹脂組成物 | |
| JP2022136237A (ja) | セルロース含有樹脂組成物 | |
| JP6889681B2 (ja) | ポリアミド樹脂組成物 | |
| JP2020204112A (ja) | 化学修飾セルロース繊維 | |
| JP6915107B2 (ja) | 微細セルロース含有樹脂組成物 | |
| JP2021084925A (ja) | セルロース組成物 | |
| Jagtap et al. | Influence of ethylene-methacrylic acid copolymer on thermo-mechanical, morphological and rheological properties of recycled PET/SEBS blend | |
| JP2024032925A (ja) | 樹脂成形体の製造方法 | |
| JP2020176157A (ja) | セルロース組成物 | |
| JP2020015242A (ja) | セルロース含有樹脂組成物 | |
| EP4725989A1 (en) | Resin composition, molded body using same, and method for producing resin composition | |
| Xia et al. | Overcoming the trade-off between toughness and stiffness of fully polymer-based alloys by elastomeric salami particles through reactive blending | |
| JP4826238B2 (ja) | 多層容器及び多層配管 | |
| WO2021079908A1 (ja) | セルロース樹脂組成物 | |
| JP2021169623A (ja) | 微細セルロース含有樹脂組成物 | |
| Yuan et al. | Synergistic Dynamic Vulcanization and Reactive Compatibilization for MVQ/HDPE Thermoplastic Vulcanizate with Excellent Low-Temperature Toughness |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2020543123 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20806009 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020806009 Country of ref document: EP Effective date: 20211213 |