JP6752935B1 - 樹脂成形体の製造方法 - Google Patents
樹脂成形体の製造方法 Download PDFInfo
- Publication number
- JP6752935B1 JP6752935B1 JP2019099379A JP2019099379A JP6752935B1 JP 6752935 B1 JP6752935 B1 JP 6752935B1 JP 2019099379 A JP2019099379 A JP 2019099379A JP 2019099379 A JP2019099379 A JP 2019099379A JP 6752935 B1 JP6752935 B1 JP 6752935B1
- Authority
- JP
- Japan
- Prior art keywords
- resin
- cellulose nanofibers
- molded product
- mass
- resin molded
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
- C08J3/203—Solid polymers with solid and/or liquid additives
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B17/00—Recovery of plastics or other constituents of waste material containing plastics
- B29B17/0005—Direct recuperation and re-use of scrap material during moulding operation, i.e. feed-back of used material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/002—Methods
- B29B7/007—Methods for continuous mixing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/30—Mixing; Kneading continuous, with mechanical mixing or kneading devices
- B29B7/58—Component parts, details or accessories; Auxiliary operations
- B29B7/60—Component parts, details or accessories; Auxiliary operations for feeding, e.g. end guides for the incoming material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/80—Component parts, details or accessories; Auxiliary operations
- B29B7/88—Adding charges, i.e. additives
- B29B7/90—Fillers or reinforcements, e.g. fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C45/00—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor
- B29C45/0005—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor using fibre reinforcements
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B3/00—Preparation of cellulose esters of organic acids
- C08B3/20—Esterification with maintenance of the fibrous structure of the cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
- C08J3/201—Pre-melted polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/30—Mixing; Kneading continuous, with mechanical mixing or kneading devices
- B29B7/34—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices
- B29B7/38—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary
- B29B7/40—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with single shaft
- B29B7/42—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with single shaft with screw or helix
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/30—Mixing; Kneading continuous, with mechanical mixing or kneading devices
- B29B7/34—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices
- B29B7/38—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary
- B29B7/46—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with more than one shaft
- B29B7/48—Mixing; Kneading continuous, with mechanical mixing or kneading devices with movable mixing or kneading devices rotary with more than one shaft with intermeshing devices, e.g. screws
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/30—Mixing; Kneading continuous, with mechanical mixing or kneading devices
- B29B7/58—Component parts, details or accessories; Auxiliary operations
- B29B7/72—Measuring, controlling or regulating
- B29B7/726—Measuring properties of mixture, e.g. temperature or density
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29B—PREPARATION OR PRETREATMENT OF THE MATERIAL TO BE SHAPED; MAKING GRANULES OR PREFORMS; RECOVERY OF PLASTICS OR OTHER CONSTITUENTS OF WASTE MATERIAL CONTAINING PLASTICS
- B29B7/00—Mixing; Kneading
- B29B7/80—Component parts, details or accessories; Auxiliary operations
- B29B7/82—Heating or cooling
- B29B7/823—Temperature control
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2077/00—Use of PA, i.e. polyamides, e.g. polyesteramides or derivatives thereof, as moulding material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2105/00—Condition, form or state of moulded material or of the material to be shaped
- B29K2105/06—Condition, form or state of moulded material or of the material to be shaped containing reinforcements, fillers or inserts
- B29K2105/16—Fillers
- B29K2105/162—Nanoparticles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2401/00—Use of cellulose, modified cellulose or cellulose derivatives, e.g. viscose, as filler
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/30—Polymeric waste or recycled polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2323/10—Homopolymers or copolymers of propene
- C08J2323/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2377/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2377/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2400/00—Characterised by the use of unspecified polymers
- C08J2400/30—Polymeric waste or recycled polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2401/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2401/02—Cellulose; Modified cellulose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2401/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2401/08—Cellulose derivatives
- C08J2401/10—Esters of organic acids
- C08J2401/12—Cellulose acetate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2423/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2423/02—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers not modified by chemical after treatment
- C08J2423/10—Homopolymers or copolymers of propene
- C08J2423/12—Polypropene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2477/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2477/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/14—Polymer mixtures characterised by other features containing polymeric additives characterised by shape
- C08L2205/16—Fibres; Fibrils
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Materials Engineering (AREA)
- Environmental & Geological Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
Abstract
Description
このように、従来技術によっては、機械強度が良好で、物性の異方性及びそりが少なく、かつ熱による着色を抑制したセルロースナノファイバー含有樹脂組成物は得られていなかった。
すなわち、本発明は以下の態様を包含する。
[1] (A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む樹脂成形体の製造方法であって、前記方法が、
(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む主供給材料と、前記主供給材料の溶融処理生成物である補助供給材料とを準備する工程と、
前記主供給材料と前記補助供給材料とを溶融混合して樹脂組成物を得る樹脂組成物形成工程と、
前記樹脂組成物を成形して樹脂成形体を得る工程と、
を含む、方法。
[2] (A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む樹脂成形体の製造において前記(B)セルロースナノファイバーの解繊性を向上させる方法であって、前記方法が、
(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む主供給材料と、前記主供給材料の溶融処理生成物である補助供給材料とを準備する工程と、
前記主供給材料と前記補助供給材料とを溶融混合することによって、熱履歴が異なる2種以上のセルロースナノファイバーを含む樹脂組成物を得る樹脂組成物形成工程と、
前記樹脂組成物を成形して樹脂成形体を得る工程と、
を含む、方法。
[3] 前記熱履歴が異なる2種以上のセルロースナノファイバーが、互いに異なる繊維長を有する、上記態様2に記載の方法。
[4] 前記樹脂成形体の一部を前記補助供給材料として使用する、上記態様1〜3のいずれかに記載の方法。
[5] 前記主供給材料が、前記(A)熱可塑性樹脂100質量部及び前記(B)セルロースナノファイバー1〜50質量部を含む、上記態様1〜4のいずれかに記載の方法。
[6] 前記主供給材料の構成成分が溶融混合系中で互いに及び補助供給材料と混合される、上記態様1〜5のいずれかに記載の方法。
[7] 前記主供給材料が、前記(A)熱可塑性樹脂100質量部及び前記(B)セルロースナノファイバー1〜50質量部を含む成形体である第1の材料と、前記第1の材料と組成の異なる第2の材料との組合せである、上記態様1〜6のいずれかに記載の方法。
[8] 前記溶融混合を、前記主供給材料と前記補助供給材料との合計100質量%に対する前記補助供給材料の混合比率5〜50質量%にて行う、上記態様1〜7のいずれかに記載の方法。
[9] 前記溶融混合が溶融混練である、上記態様1〜8のいずれかに記載の方法。
[10] 前記樹脂成形体がペレットである、上記態様1〜9のいずれかに記載の方法。
[11] 前記溶融混合が溶融混練であり、前記溶融混練と前記成形とを単一の混練機内で行う、上記態様10に記載の方法。
[12] 前記樹脂成形体の成形収縮率のTD/MD比が1.05〜3.0であり、
前記樹脂成形体の引張強度が90MPa以上である、上記態様1〜11のいずれかに記載の方法。
[13] 前記樹脂成形体において、MD方向の成形収縮率が0.2%〜0.7%であり、TD方向の成形収縮率が0.5%〜1.0%である、上記態様1〜12のいずれかに記載の方法。
[14] 前記方法が、前記樹脂成形体の一部を前記補助供給材料の少なくとも一部として前記樹脂組成物形成工程に戻すことを更に含み、
前記樹脂成形体中のセルロースナノファイバーの総量100質量%に対する、前記樹脂組成物形成工程を2回以上経ているセルロースナノファイバーの比率が、20質量%以下である、上記態様1〜13のいずれかに記載の方法。
[15] 前記樹脂成形体の黄色度(YI)値と前記補助供給材料の黄色度(YI)値との差が10以下である、上記態様1〜14のいずれかに記載の方法。
[16] 前記(A)熱可塑性樹脂がポリアミドである、上記態様1〜15のいずれかに記載の方法。
[17] 前記(B)セルロースナノファイバーが変性セルロースナノファイバーである、上記態様1〜16のいずれかに記載の方法。
[18] 前記変性セルロースナノファイバーの置換度が0.5〜1.5である、上記態様17に記載の方法。
本発明の一態様は、(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む樹脂成形体の製造方法を提供する。一態様において、該方法は、(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む主供給材料と、該主供給材料の溶融処理生成物である補助供給材料とを準備する工程と、該主供給材料と該補助供給材料とを溶融混合して樹脂組成物を得る樹脂組成物形成工程と、該樹脂組成物を成形して樹脂成形体を得る工程と、を含む。
主供給材料は、(A)熱可塑性樹脂と(B)セルロースナノファイバーと任意の追加成分とを含む混合物の形態であってもよいし、主供給材料の構成成分(すなわち(A)熱可塑性樹脂、(B)セルロースナノファイバー、及び任意の追加成分)が個別に準備されたものであってもよい。補助供給材料は、主供給材料の溶融処理生成物である。したがって、補助供給材料は、主供給材料と実質的に同組成である(すなわち構成成分の種類及び量が同じである)が、主供給材料の構成成分の少なくとも一部が溶融処理に起因する変質をしたものであってよい。当該変質は、(B)セルロースナノファイバーの繊維長低下を含む。
(A)熱可塑性樹脂としては、種々の樹脂を使用できる。一態様において、(A)熱可塑性樹脂は数平均分子量5000以上を有する。なお本開示の数平均分子量は、GPC(ゲルパーミエーションクロマトグラフィ)を用い測定したクロマトグラムを、GPC用標準ポリマーで換算した値である。このときのGPC用標準ポリマーとしては、当業者に公知のポリマーを用いることができる。一般的にはポリスチレン、ポリメタクリル酸メチル、ポリエチレングリコール、ポリエチレンオキシド等を例示することができる。どの標準ポリマーを用いるかは、GPC測定時の溶離液の種類により選択される。その一例を挙げると、例えば溶離液が、ヘキサフルオロイソプロパノールの場合、ポリメタクリル酸メチルが使用され、テトラヒドロフラン、クロロホルム、トルエン、1,2,4−トリクロロベンゼンの場合、ポリスチレンが使用され、メタノール、N,N−ジメチルホルムアミド、水系の場合、ポリエチレングリコール 、ポリエチレンオキシドが使用される。
(A)熱可塑性樹脂としては、100℃〜350℃の範囲内に融点を有する結晶性樹脂、又は、100〜250℃の範囲内にガラス転移温度を有する非晶性樹脂が挙げられる。(A)熱可塑性樹脂は、ホモポリマーでもコポリマーでもよい1種又は2種以上のポリマーで構成されてよい。
酸変性されたポリオレフィン系樹脂の酸変性率の下限は、好ましくは0.01質量%であり、より好ましくは0.1質量%であり、更に好ましくは0.3質量%であり、特に好ましくは0.5質量%であり、最も好ましくは0.7質量%である。また上限は、好ましくは10質量%であり、より好ましくは5質量%であり、更に好ましくは3質量%であり、特に好ましくは2質量%であり、最も好ましくは1.5質量%である。セルロースとの界面強度を維持するためには、下限以上が好ましく、酸変性されたポリオレフィンの機械物性を維持するためには上限以下が好ましい。
(B)セルロースナノファイバーは、平均繊維径1000nm以下のセルロースである。(B)セルロースナノファイバーの好適例は、特に限定されないが、例えばセルロースパルプを原料としたセルロースナノファイバー又はこれらセルロースの変性物の1種以上を用いることが出来る。これらの中でも、安定性、性能等の点から、セルロースの変性物の1種以上が好ましく使用可能である。(B)セルロースナノファイバーの平均繊維径は、樹脂成形体の良好な機械的強度(特に引張弾性率)を得る観点から、1000nm以下であり、好ましくは500nm以下、より好ましくは200nm以下である。平均繊維径は小さい方が好ましいが、加工容易性の観点からは、好ましくは10nm以上、より好ましくは20nm以上、更に好ましくは30nm以上であることができる。上記平均繊維径は、レーザー回折/散乱法粒度分布計で、積算体積が50%になるときの粒子の球形換算直径(体積平均粒子径)として求められる値である。
置換度=(Inf)×6/(Inp)
たとえば、修飾基がアセチル基の場合、−CH3に帰属される23ppmのシグナルを用いれば良い。
用いる13C固体NMR測定の条件は、以下が例示できる。
装置 :Bruker Biospin Avance500WB
周波数 :125.77MHz
測定方法 :DD/MAS法
待ち時間 :75sec
NMR試料管 :4mmφ
積算回数 :640回(約14Hr)
MAS :14,500Hz
化学シフト基準:グリシン(外部基準:176.03ppm)
主供給材料は、(A)熱可塑性樹脂及び(B)セルロースナノファイバーに加えて、追加成分を任意に含んでよい。追加成分としては、表面処理剤、酸化防止剤、無機充填剤、潤滑油等が挙げられる。これらの成分は、各々、1種又は2種以上の組み合わせで使用してよい。またこれらの成分は市販の試薬又は製品であってもよい。
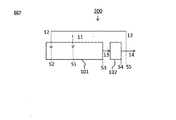
図1〜3は、第1の実施形態に係る樹脂成形体の製造方法の例としてのプロセス100,200,300を説明する図である。図1〜3を参照し、プロセス100,200,300においては、溶融混合部101において、主供給材料11又はその構成成分(すなわち、(A)熱可塑性樹脂11a、(B)セルロースナノファイバー11b、及び任意の追加成分11c)と補助供給材料12とを溶融混合して溶融混合物である樹脂組成物13を生成し、成形部102にて樹脂組成物13を成形して樹脂成形体14を生成する。樹脂成形体14は、ペレット等の形状を有してよい。
主供給材料11は、(A)熱可塑性樹脂11aと(B)セルロースナノファイバー11bと任意の追加成分11cとを含む混合物の形態(図1及び2中の主供給材料11)、又は、主供給材料11の構成成分としての(A)熱可塑性樹脂11a、(B)セルロースナノファイバー11b、及び任意の追加成分11cが個別に準備される形態(図3中の主供給材料11)であってよい。後者の場合には、主供給材料の構成成分が溶融混合系中で互いに及び補助供給材料と混合されることになる。
本工程では、主供給材料11と補助供給材料12とを溶融混合部101に供給してこれらを溶融混合する。一態様において、溶融混合は溶融混練である。溶融混合部101は、例えば単軸押出機、二軸押出機、ロール、バンバリーミキサー等の混合装置における混合部であることができる。上記混合装置の中でも、二軸押出機が好ましく、より具体的には、減圧装置及びサイドフィーダー設備を装備した二軸押出機が挙げられる。二軸押出機のL/Dは、例えば、30〜100、又は35〜75、又は45〜70であってよい。
(1)図1及び2を参照し、(A)熱可塑性樹脂と(B)セルロースナノファイバーと任意の追加成分との混合物としての主供給材料11を溶融混合部101の主供給材料投入部位S1に投入し、補助供給材料12を溶融混合部101の補助供給材料投入部位S2に投入し、両者を溶融混合して樹脂組成物13を生成する。主供給材料投入部位S1と補助供給材料投入部位S2との位置関係は目的に応じて適宜設計できる。例えば、主供給材料投入部位S1の下流側に補助供給材料投入部位S2を配置(図1)してよく、又は、補助供給材料投入部位S2の下流側に主供給材料投入部位S1を配置(図2)してよく、又は、主供給材料11と補助供給材料12とを同時に(例えば、主供給材料11と補助供給材料12とを個別に、又は予め混合して)供給してよい。
本工程では、樹脂組成物13を溶融混合部101の混合終了部位S3から成形部102に送り、成形部102にて目的の形状(例えばペレット、シート、フィルム、三次元的に構造を有する成形体等)に成形して、送出部位S4から目的の樹脂成形体14を取り出す。好ましい態様においては、溶融混合が溶融混練であり、溶融混練と成形とを単一の混練機(例えば、<樹脂組成物形成工程>において例示したもの)内で行う。別の好ましい態様としては、成形が溶融混練とは異なる成形機(例えば、射出成形機)で行う。
図4は、第2の実施形態に係る樹脂成形体の製造方法の例としてのプロセス400を説明する図である。図4を参照し、プロセス400においては、溶融混合部401において、主供給材料41と補助供給材料42とを溶融混合して溶融混合物である樹脂組成物43を生成し、成形部402にて樹脂組成物43を成形して樹脂成形体44を生成する。樹脂成形体44は、更なる加工に供されるための形状(ペレット等)であってもよいし、後述の各種製品形状であってもよい。
主供給材料41は、第1の材料41aと第2の材料41bとを含むことができる。一態様において、第1の材料41aは溶融混合物である。一態様において、第1の材料41aは第1の実施形態で得られる樹脂成形体14である。一態様において、第1の材料41aは(A)熱可塑性樹脂100質量部及び(B)セルロースナノファイバー1〜50質量部を含む成形体である。
本工程では、主供給材料41と補助供給材料42とを溶融混合部401に供給してこれらを溶融混合する。一態様において、溶融混合は溶融混練である。溶融混合部401は、第1の実施形態の溶融混合部101と同様であってよい。すなわち、溶融混合部401は、例えば単軸押出機、二軸押出機、ロール、バンバリーミキサー等の混合装置における混合部であることができる。上記混合装置の中でも、二軸押出機が好ましく、より具体的には、減圧装置及びサイドフィーダー設備を装備した二軸押出機が挙げられる。二軸押出機のL/Dは、例えば、30〜100、又は35〜75、又は45〜70であってよい。
図4を参照し、主供給材料41としての第1の材料41a及び第2の材料41bを、溶融混合部401の複数の主供給材料投入部位S1a,S1bのそれぞれに供給し、補助供給材料42を溶融混合部401の補助供給材料投入部位S2に投入し、両者を溶融混合して樹脂組成物43を生成する。第1の材料41a及び第2の材料41bの供給態様は目的に応じて設計でき、これらを各々個別に又は予め混合した状態で溶融混合部401に供給してよい。また図4においては、溶融混合部401の上流側から順に第1の材料41a、第2の材料41b、補助供給材料42を供給する例を示しているが、供給順はこれに限定されず目的に応じて適宜設定してよい。また主供給材料投入部位S1と補助供給材料投入部位S2との位置関係も適宜設計できる。
本工程では、樹脂組成物43を溶融混合部401の混合終了部位S3から成形部402に送り、成形部402にて目的の形状に成形して、送出部位S4から目的の樹脂成形体44を取り出す。成形部402は、例えば、押出成形、射出成形、真空成形、ブロー成形、射出圧縮成形、加飾成形、他材質成形、ガスアシスト射出成形、発泡射出成形、低圧成形、超薄肉射出成形(超高速射出成形)、金型内複合成形(インサート成形、アウトサート成形)等から選択される成形を行うように構成されている。好ましい態様においては、溶融混合が溶融混練であり、溶融混練と成形とを単一の混練機(例えば、<樹脂組成物形成工程>において例示したもの)内で行う。
本開示の方法で製造される樹脂成形体においては、(B)セルロースナノファイバーの繊維長が特異な態様で制御されており、良好な機械強度と少ない異方性とが両立されている。
得られたペレット状の成形体を、射出成形機を用いて、ISO294−1に準拠し多目的試験片に成形した。得られた多目的試験片について、ISO527−1に準拠し、引張降伏強度を測定した。降伏に至る前に破断した成形片については、その最大強度を代用した。
得られたペレット状の成形体を、射出成形機を用いて、JIS K7152−3に規定された60mm×60mm×2mmの平板に成形した。得られた平板状成形片をISO294−4に準拠し、樹脂流動方向(MD)と流動方向と垂直方向(TD)の寸法を正確に測定し、収縮率を算出した。得られたTD方向の収縮率をMD方向の収縮率で除して、成形収縮比を算出した。
成形収縮測定用に成形した平板を用いて、JIS K7373に準拠し、黄色度を測定した。この時、補助供給材料の黄色度(YI)値に対する、得られた樹脂成形体の黄色度(YI)値の差を黄色度変化率(△YI)として算出した。
得られたペレット状の成形体を、射出成形機を用いて、直径1mmのピンポイントゲートを有する幅50mm、長さ70mm、厚み1mmの平板に成形した。この時、金型温度は25℃に調節した。平滑な面上に、得られた平板状成形片のそり凸部を下にし、ゲート側の一方を面上に押さえつけて、反対側の成形片と平滑面との隙間を測定した。測定は、その隙間を写真で撮影することで測定した。測定は少なくとも5枚の平板について実施し、最大と最小を除いた3点の平均をもってそりの値とした。
得られたペレット状の成形体を、流れに垂直方向にミクロトームで切削し、平滑な面を削りだし、光学顕微鏡(BX53M:オリンパス社製)を用いて写真撮影した。ペレット状成形体の3点について撮影した。得られた写真を、画像解析装置を用いて二値化し、円相当径の直径が5μm以上の個数の合計を算出した。
(A)熱可塑性樹脂
ポリアミド6(以下、単にPA6)
UBEナイロン1013B 宇部興産株式会社製
粘度数:120
カルボキシル末端基比率([COOH]/[全末端基]):0.6
ポリプロピレン(以下、単にPP)
ノバテックPP MA1B(日本ポリプロ株式会社)
MFR(230℃、荷重21.2N)=21g/10分
(B)セルロースナノファイバー(以下、単にCNF)
以下の調製例によって、アセチル化置換していないCNF、及び置換度の異なる3種のCNFを作製した。
(解繊工程)
リンターパルプを裁断し、一軸撹拌機(アイメックス社製 DKV−1 φ125mmディゾルバー)中、ジメチルスルホキサイド(DMSO)中で500rpmにて1時間、常温で攪拌した。続いて、ホースポンプでビーズミル(アイメックス社製 NVM−1.5)にフィードし、120分間循環運転させ、解繊CNFスラリーを得た。
循環運転の際、ビーズミルの回転数は2500rpm、周速12m/sとした。ビーズはジルコニア製、φ2.0mmを用い、充填率は70%とした(このときのビーズミルのスリット隙間は0.6mm)。また、循環運転の際は、摩擦による発熱を吸収するためにチラーによりスラリー温度を40℃に温度管理した。
得られた、解繊CNFの特性を評価したところ、径が65nm、L/Dが約450であった。
解繊工程で得られた解繊CNFスラリー100質量部に対し、酢酸ビニル11質量部、炭酸水素ナトリウム1.63質量部をビーズミル装置内へ加えた後、循環運転を行い、アセチル化CNFスラリーを得た。循環運転の条件は、解繊工程と同一とした。循環運転時間は、30分、60分、120分の3条件で実施し、置換度の異なるアセチル化CNFスラリーを得た。
各条件で得られたCNFの置換度を測定したところ、循環運転時間30分のものは0.50、循環運転時間60分のものは1.02、循環運転時間120分のものは1.49であった。
得られた解繊CNFスラリー、又はアセチル化CNFスラリーの100質量部に対し、純水を192質量部加えて十分に撹拌した後、脱水機に入れて脱水・濃縮し、ウェットケーキを得た。得られたウェットケーキを再度、同量の純水に分散、撹拌、濃縮する洗浄操作を合計5回繰り返し、溶剤置換を実施した。
解繊CNFウェットケーキ、及びそれぞれのアセチル化CNFウェットケーキを、セルロース固形分濃度が10質量%になるように、純水で濃度調整し、CNF100質量部に対し、PEG20000を5質量部添加し、よく撹拌した後、公転・自転方式の攪拌機(EME社製 V−mini300)を用いて約40℃で真空乾燥させることにより、それぞれのCNFの乾燥粉体を得た。
<溶融混合装置>
L/D=4の温度調整バレルを15個有するL/D=60の同方向回転二軸押出機(TEM26SX:東芝機械社製)を用い、押出機の最も上流側バレルであるバレル1に原料供給用のスロート(以下、単にスロートと称す)、バレル4及びバレル7に原料供給用のサイドフィード装置(以下、単にバレル4に設置のサイドフィードをサイド1、バレル7に設置のサイドフィードをサイド2と称す)、バレル14に脱揮用の減圧ポートを設置した。
スクリューデザインは、L/D=0〜18(バレル1〜バレル5の真ん中)の位置に「時計回りスクリュー(以下、単にRS)」を配し、L/D=19〜24(バレル5中央〜バレル6)の位置に、2個の「時計回りニーディングディスク(以下、単にRKD)」、3個の「ニュートラルニーディングディスク(以下、単にNKD)」、1個の「反時計回りニーディングディスク(以下、単にLKD)」をこの順に配し、L/D=24〜32(バレル7〜バレル8)の位置にRSを配し、L/D=32〜36(バレル9)の位置に、1個のRKD、2個のNKD、1個のRKD,2個のNKDをこの順に配し、L/D=36〜40(バレル10)の位置にRSを配し、L/D=40〜44(バレル11)の位置に、1個のRKD、2個のNKD、1個のRKD,3個のNKDをこの順に配し、L/D=44〜48(バレル12)の位置にRSを配し、L/D=48〜52(バレル13)の位置に、2個のNKD、1個のLKD,3個のNKD、1個の「反時計回りスクリュー(以下、単にLS)」をこの順に配し、L/D=52〜56(バレル14)の位置にRSを配し、L/D=56〜60(バレル13)の位置に、1個RS、3個のNKDの順に配した後、残りをすべてRSとするデザインとした。
押出機の先端には、3mm径の紡口を2個有するダイスを設置し、溶融樹脂をストランド状に押し出せるようにした。
溶融混合装置の後工程に設置した水槽で、溶融ストランドを冷却し、その後のペレターザーでペレット状にカットし、ペレット状樹脂成形体を得た。得られたペレット状樹脂成形体の一部を補助供給材料として用いた。(以下、補助供給材料の形状「ペレット」と称す)
更に得られたペレット状樹脂成形体を、射出成形機(ソディックプラステック社製:TR05EH2 型締圧力5トン)を用いて、FPCコネクター(長さ30mm、幅1mm、2個取り、50ピン穴、ピン穴ピッチ:0.5ミリピッチ)金型を用い、射出速度300mm/secで成型し、コネクター状樹脂成形体を得た。本コネクター状成形体を粉砕機を用いて粉砕処理し、メッシュを用いて径が5mm以下となるよう調製し、粉砕補助供給材料を得た。(以下、補助供給材料の形状「粉砕品」と称す)
溶融混合装置のバレル温度の設定を、バレル1〜3を150℃、バレル4〜15及びダイスを250℃に設定し、PA6が60質量%、置換度1.02のCNFが40質量%となるようスロート部より供給し、溶融混合を実施し、ペレット状のPA/CNFの高濃度物(以下、単にPA/CNFMBと称す)を得た。この時の溶融混合装置のスクリュー回転数は、300rpmであり、時間当たりの吐出量は、18kg/hrであった。
溶融混合装置のバレル温度の設定を、バレル1〜3を100℃、バレル4〜15及びダイスを200℃に設定し、PA6をPPに変更した以外は、すべて調製例2と同様に実施し、ペレット状のPP/CNF高濃度物(以下、単にPP/CNF−MBと称す)を得た。
溶融混合装置のバレル温度の設定を、バレル1〜3を150℃、バレル4〜7を260℃、バレル8〜15及びダイスを250℃に設定し主供給材料を表1及び表2に記載の組成になるように主供給材料の添加位置より供給し、主供給材料の溶融混合物である補助供給材料を補助供給材料の添加位置より供給し、それぞれ溶融混合を実施し、ペレット状樹脂成形体を得て、各種特性を評価し、表1及び2に記載した。この時の溶融混合装置のスクリュー回転数は、300rpmであり、時間当たりの吐出量は、25kg/hrであった。
なお、表中に記載した「プロセス」とは、本実施態様で説明に用いたプロセス100、200、300、400の事を指す。
また、実施例6で用いた補助供給材料の粉砕品は、上述で得られたペレット状樹脂成形体を、コネクター状樹脂成形体に成形後、粉砕処理を施して得た。
実施例9の主供給材料は、スロート部よりPA6を供給し、サイド1よりCNFを供給した。分けて供給することで、CNF粉体の供給時の舞い上がりが抑制でき、供給が安定するためか、引張強度の試験片ごとのばらつきが少ないという予想外の効果が得られた。
比較例1、3、及び5は、補助供給材料を添加しなかった例である。比較例2及び4は、主供給材料がない態様、すなわち、補助供給材料のみという態様であり、100%再循環(リサイクル)に相当する。

溶融混合装置のバレル温度の設定を、バレル1〜3を100℃、バレル4〜7を200℃、バレル8〜15及びダイスを190℃に設定し、表3記載の組成・条件に変更した以外は、実施例10と同様に実施し、各種特性を評価した。結果を表3に記載する。
101,401 溶融混合部
102,402 成形部
11,41 主供給材料
11a (A)熱可塑性樹脂
11b (B)セルロースナノファイバー
11c 任意の追加成分
12,42 補助供給材料
13,43 樹脂組成物
14,44 樹脂成形体
41a 第1の材料
41b 第2の材料
S1,S1a,S1b,S1c 主供給材料投入部位
S2 補助供給材料投入部位
S3 混合終了部位
S4 送出部位
S5 分離部位
Claims (19)
- (A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む樹脂成形体の製造方法であって、前記方法が、
(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む主供給材料と、前記主供給材料の溶融処理生成物である補助供給材料とを準備する工程と、
前記主供給材料と前記補助供給材料とを溶融混合して樹脂組成物を得る樹脂組成物形成工程と、
前記樹脂組成物を成形して樹脂成形体を得る工程と、
を含み、
前記溶融混合を、前記主供給材料と前記補助供給材料との合計100質量%に対する前記補助供給材料の混合比率5〜50質量%にて行う、方法。 - (A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む樹脂成形体の製造方法であって、前記方法が、
(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む主供給材料と、前記主供給材料の溶融処理生成物である補助供給材料とを準備する工程と、
前記主供給材料と前記補助供給材料とを溶融混合して樹脂組成物を得る樹脂組成物形成工程と、
前記樹脂組成物を成形して樹脂成形体を得る工程と、
を含み、
前記樹脂成形体の成形収縮率のTD/MD比が1.05〜3.0である、方法。 - (A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む樹脂成形体の製造において前記(B)セルロースナノファイバーの解繊性を向上させる方法であって、前記方法が、
(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む主供給材料と、前記主供給材料の溶融処理生成物である補助供給材料とを準備する工程と、
前記主供給材料と前記補助供給材料とを溶融混合することによって、熱履歴が異なる2種以上のセルロースナノファイバーを含む樹脂組成物を得る樹脂組成物形成工程と、
前記樹脂組成物を成形して樹脂成形体を得る工程と、
を含み、
前記溶融混合を、前記主供給材料と前記補助供給材料との合計100質量%に対する前記補助供給材料の混合比率5〜50質量%にて行う、方法。 - (A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む樹脂成形体の製造において前記(B)セルロースナノファイバーの解繊性を向上させる方法であって、前記方法が、
(A)熱可塑性樹脂及び(B)セルロースナノファイバーを含む主供給材料と、前記主供給材料の溶融処理生成物である補助供給材料とを準備する工程と、
前記主供給材料と前記補助供給材料とを溶融混合することによって、熱履歴が異なる2種以上のセルロースナノファイバーを含む樹脂組成物を得る樹脂組成物形成工程と、
前記樹脂組成物を成形して樹脂成形体を得る工程と、
を含み、
前記樹脂成形体の成形収縮率のTD/MD比が1.05〜3.0である、方法。 - 前記熱履歴が異なる2種以上のセルロースナノファイバーが、互いに異なる繊維長を有する、請求項3又は4に記載の方法。
- 前記樹脂成形体の一部を前記補助供給材料として使用する、請求項1〜5のいずれか一項に記載の方法。
- 前記主供給材料が、前記(A)熱可塑性樹脂100質量部及び前記(B)セルロースナノファイバー1〜50質量部を含む、請求項1〜6のいずれか一項に記載の方法。
- 前記主供給材料の構成成分が溶融混合系中で互いに及び補助供給材料と混合される、請求項1〜7のいずれか一項に記載の方法。
- 前記主供給材料が、前記(A)熱可塑性樹脂100質量部及び前記(B)セルロースナノファイバー1〜50質量部を含む成形体である第1の材料と、前記第1の材料と組成の異なる第2の材料との組合せである、請求項1〜8のいずれか一項に記載の方法。
- 前記溶融混合が溶融混練である、請求項1〜9のいずれか一項に記載の方法。
- 前記樹脂成形体がペレットである、請求項1〜10のいずれか一項に記載の方法。
- 前記溶融混合が溶融混練であり、前記溶融混練と前記成形とを単一の混練機内で行う、請求項11に記載の方法。
- 前記樹脂成形体の引張強度が90MPa以上である、請求項1〜12のいずれか一項に記載の方法。
- 前記樹脂成形体において、MD方向の成形収縮率が0.2%〜0.7%であり、TD方向の成形収縮率が0.5%〜1.0%である、請求項1〜13のいずれか一項に記載の方法。
- 前記方法が、前記樹脂成形体の一部を前記補助供給材料の少なくとも一部として前記樹脂組成物形成工程に戻すことを更に含むことによって、前記樹脂成形体が、前記主供給材料の溶融処理と2回以上の前記樹脂組成物形成工程とを経ているセルロースナノファイバーを含み、
前記樹脂成形体中のセルロースナノファイバーの総量100質量%に対する、前記主供給材料の溶融処理と2回以上の前記樹脂組成物形成工程とを経ているセルロースナノファイバーの比率が、20質量%以下である、請求項1〜14のいずれか一項に記載の方法。 - 前記樹脂成形体の黄色度(YI)値と前記補助供給材料の黄色度(YI)値との差が10以下である、請求項1〜15のいずれか一項に記載の方法。
- 前記(A)熱可塑性樹脂がポリアミドである、請求項1〜16のいずれか一項に記載の方法。
- 前記(B)セルロースナノファイバーが変性セルロースナノファイバーである、請求項1〜17のいずれか一項に記載の方法。
- 前記変性セルロースナノファイバーの置換度が0.5〜1.5である、請求項18に記載の方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019099379A JP6752935B1 (ja) | 2019-05-28 | 2019-05-28 | 樹脂成形体の製造方法 |
| US17/613,198 US11572447B2 (en) | 2019-05-28 | 2020-05-27 | Resin molded body production method |
| PCT/JP2020/020962 WO2020241707A1 (ja) | 2019-05-28 | 2020-05-27 | 樹脂成形体の製造方法 |
| CN202080035183.4A CN113811576A (zh) | 2019-05-28 | 2020-05-27 | 树脂成型体的制造方法 |
| EP20814095.4A EP3978551B1 (en) | 2019-05-28 | 2020-05-27 | Method for producing a resin molded body and method for increasing defibration of cellulose nanofibers during production of the resin molded body |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019099379A JP6752935B1 (ja) | 2019-05-28 | 2019-05-28 | 樹脂成形体の製造方法 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020138788A Division JP2020193345A (ja) | 2020-08-19 | 2020-08-19 | 樹脂成形体の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP6752935B1 true JP6752935B1 (ja) | 2020-09-09 |
| JP2020193266A JP2020193266A (ja) | 2020-12-03 |
Family
ID=72333556
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019099379A Active JP6752935B1 (ja) | 2019-05-28 | 2019-05-28 | 樹脂成形体の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11572447B2 (ja) |
| EP (1) | EP3978551B1 (ja) |
| JP (1) | JP6752935B1 (ja) |
| CN (1) | CN113811576A (ja) |
| WO (1) | WO2020241707A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20220242006A1 (en) * | 2021-01-15 | 2022-08-04 | Innotech Alberta Inc. | Cellulose Particle Mold Release Layer |
| JPWO2023286757A1 (ja) * | 2021-07-14 | 2023-01-19 | ||
| JP7322989B1 (ja) | 2022-02-07 | 2023-08-08 | 大日本印刷株式会社 | 化粧シートおよびその製造方法、ならびに、樹脂層およびその製造方法 |
| CN115417931B (zh) * | 2022-09-22 | 2024-03-22 | 上海同化益生纤生物科技有限公司 | 一种耐温型纤维素的制备方法与应用 |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH07228775A (ja) | 1994-02-17 | 1995-08-29 | Kuraray Co Ltd | 難燃性ポリアミド組成物 |
| JP3156749B2 (ja) | 1995-08-24 | 2001-04-16 | カルソニックカンセイ株式会社 | ポリアミドのリサイクル性評価方法及びポリアミドのリサイクル方法 |
| JP2003055549A (ja) | 2001-06-05 | 2003-02-26 | Kuraray Co Ltd | ポリアミド組成物 |
| JP2004195890A (ja) * | 2002-12-20 | 2004-07-15 | Toray Ind Inc | 繊維強化熱可塑性樹脂成形品 |
| JP5489788B2 (ja) | 2010-03-05 | 2014-05-14 | オリンパス株式会社 | 鏡枠用樹脂構造体 |
| KR101812986B1 (ko) | 2010-04-06 | 2017-12-28 | 유니띠까 가부시키가이샤 | 폴리아미드 수지 조성물 및 폴리아미드 수지 조성물의 제조법 |
| JP5563905B2 (ja) * | 2010-06-25 | 2014-07-30 | 積水化学工業株式会社 | リサイクル樹脂含有ポリエチレン系樹脂組成物を押出成形してなるパイプ成形品、及びポリエチレン系樹脂リサイクル方法 |
| US9114902B2 (en) | 2011-03-22 | 2015-08-25 | Polyone Designed Structures And Solutions Llc | Methods and systems for use in forming an article from a multi-layer sheet structure |
| JP2012201767A (ja) | 2011-03-24 | 2012-10-22 | Nissan Motor Co Ltd | 樹脂組成物 |
| JP2012251039A (ja) * | 2011-06-01 | 2012-12-20 | Asahi Kasei Chemicals Corp | リサイクル樹脂組成物およびその製造方法 |
| KR20160115919A (ko) | 2014-02-03 | 2016-10-06 | 도레이 카부시키가이샤 | 섬유 강화 다층 펠릿, 그것을 성형하여 이루어지는 성형품, 및 섬유 강화 다층 펠릿의 제조 방법 |
| US20160168363A1 (en) | 2014-06-27 | 2016-06-16 | Api Intellectual Property Holdings, Llc | Nanocellulose-polymer composites, and processes for producing them |
| JP6697839B2 (ja) | 2014-07-03 | 2020-05-27 | ユニチカ株式会社 | シート状成形体 |
| SG11201704787QA (en) | 2014-12-22 | 2017-07-28 | Borealis Ag | Composition based on recycled polyethylene from cable waste |
| JP6091589B2 (ja) * | 2015-03-19 | 2017-03-08 | 国立大学法人京都大学 | 化学修飾セルロースナノファイバー及び熱可塑性樹脂を含有する繊維強化樹脂組成物 |
| US11566118B2 (en) | 2016-02-18 | 2023-01-31 | Starlite Co., Ltd. | Nanofiber dispersion, method of producing nanofiber dispersion, powdery nanofibers obtainable from the dispersion, resin composition containing the powdery nanofibers ad molding material for 3D printer using the resin composition |
| CN108779256B (zh) | 2016-02-18 | 2021-12-14 | 日本星光工业株式会社 | 纳米纤维分散体及其粉体以及3d打印机用造型材料 |
| EP3431655A4 (en) | 2016-03-16 | 2020-01-22 | Futamura Kagaku Kabushiki Kaisha | FINE CELLULOSE FIBER AND PRODUCTION METHOD THEREFOR |
| JP6860137B2 (ja) | 2016-07-29 | 2021-04-14 | 日本製紙株式会社 | 繊維性成形品製造用の成形材料およびそれを用いた成形品 |
| JP6469068B2 (ja) | 2016-12-12 | 2019-02-13 | 富士紙管株式会社 | 繊維成分混入合成樹脂組成物及びその製造方法 |
| JP7185215B2 (ja) | 2017-06-22 | 2022-12-07 | 国立大学法人京都大学 | 繊維強化樹脂組成物、繊維強化成形体及びその製造方法 |
| JP6894053B2 (ja) * | 2019-05-13 | 2021-06-23 | 旭化成株式会社 | 低空孔のペレット、及び成形体の製造方法 |
| JP2020193345A (ja) | 2020-08-19 | 2020-12-03 | 旭化成株式会社 | 樹脂成形体の製造方法 |
-
2019
- 2019-05-28 JP JP2019099379A patent/JP6752935B1/ja active Active
-
2020
- 2020-05-27 WO PCT/JP2020/020962 patent/WO2020241707A1/ja unknown
- 2020-05-27 CN CN202080035183.4A patent/CN113811576A/zh active Pending
- 2020-05-27 EP EP20814095.4A patent/EP3978551B1/en active Active
- 2020-05-27 US US17/613,198 patent/US11572447B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US11572447B2 (en) | 2023-02-07 |
| EP3978551A1 (en) | 2022-04-06 |
| EP3978551C0 (en) | 2023-08-16 |
| US20220315714A1 (en) | 2022-10-06 |
| WO2020241707A1 (ja) | 2020-12-03 |
| EP3978551A4 (en) | 2022-07-27 |
| EP3978551B1 (en) | 2023-08-16 |
| JP2020193266A (ja) | 2020-12-03 |
| CN113811576A (zh) | 2021-12-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6752935B1 (ja) | 樹脂成形体の製造方法 | |
| Esmaeili et al. | Poly (lactic acid)/coplasticized thermoplastic starch blend: Effect of plasticizer migration on rheological and mechanical properties | |
| Fowlks et al. | The effect of maleated polylactic acid (PLA) as an interfacial modifier in PLA‐talc composites | |
| JP2024032925A (ja) | 樹脂成形体の製造方法 | |
| Dias et al. | Development of high bio‐content polypropylene composites with different industrial lignins | |
| JP6896946B2 (ja) | 複合粒子及び樹脂組成物 | |
| JP7114376B2 (ja) | セルロース含有樹脂組成物 | |
| JP2020007496A (ja) | セルロース含有樹脂組成物 | |
| WO2019208313A1 (ja) | セルロースナノファイバー含水分散液 | |
| Mucci et al. | Composites made from a soybean oil biopolyurethane and cellulose nanocrystals | |
| JP7561086B2 (ja) | 低空孔のペレット、及び成形体の製造方法 | |
| Abhijit et al. | Melt processing of ethylene‐acrylic acid copolymer composites reinforced with nanocellulose | |
| Simunec et al. | Facilitating the additive manufacture of high-performance polymers through polymer blending: A review | |
| Jubinville et al. | A comparative study of the physico-mechanical properties of material extrusion 3D-printed and injection molded wood-polymeric biocomposites | |
| JP7152558B2 (ja) | ポリアミド樹脂組成物 | |
| VP et al. | Fabrication and characterization of bionanocomposites based on poly (lactic acid), banana fiber and nanoclay | |
| JP7266995B2 (ja) | セルロースナノファイバー組成物 | |
| JP2020176157A (ja) | セルロース組成物 | |
| JP2020007493A (ja) | セルロース含有樹脂組成物 | |
| JP2020015242A (ja) | セルロース含有樹脂組成物 | |
| JP2019183073A (ja) | ポリアミド樹脂組成物 | |
| JP2024070251A (ja) | ポリアセタール樹脂組成物及びその製造方法 | |
| Tuna et al. | Effect of coupling agent on polylactic acid/polypropylene and polylactic acid/polyamide 6 foam composites | |
| Soćko et al. | The Development of Sustainable Polyoxymethylene (POM)-Based Composites by the Introduction of Natural Fillers and Melt Blending with Poly (lactic acid)-PLA | |
| JP2024106126A (ja) | 樹脂組成物及び成形品 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20191108 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20191108 |
|
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20200121 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200128 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20200327 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200527 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200804 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20200819 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6752935 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R157 | Certificate of patent or utility model (correction) |
Free format text: JAPANESE INTERMEDIATE CODE: R157 |