WO2017169399A1 - 形質転換体及びそれを用いるプロトカテク酸又はその塩の製造方法 - Google Patents
形質転換体及びそれを用いるプロトカテク酸又はその塩の製造方法 Download PDFInfo
- Publication number
- WO2017169399A1 WO2017169399A1 PCT/JP2017/007233 JP2017007233W WO2017169399A1 WO 2017169399 A1 WO2017169399 A1 WO 2017169399A1 JP 2017007233 W JP2017007233 W JP 2017007233W WO 2017169399 A1 WO2017169399 A1 WO 2017169399A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- activity
- dna
- seq
- nucleotide sequence
- gene
- Prior art date
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/88—Lyases (4.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
- C12N1/205—Bacterial isolates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0071—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14)
- C12N9/0073—Oxidoreductases (1.) acting on paired donors with incorporation of molecular oxygen (1.14) with NADH or NADPH as one donor, and incorporation of one atom of oxygen 1.14.13
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/40—Preparation of oxygen-containing organic compounds containing a carboxyl group including Peroxycarboxylic acids
- C12P7/42—Hydroxy-carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y114/00—Oxidoreductases acting on paired donors, with incorporation or reduction of molecular oxygen (1.14)
- C12Y114/13—Oxidoreductases acting on paired donors, with incorporation or reduction of molecular oxygen (1.14) with NADH or NADPH as one donor, and incorporation of one atom of oxygen (1.14.13)
- C12Y114/13064—4-Hydroxybenzoate 1-hydroxylase (1.14.13.64)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y401/00—Carbon-carbon lyases (4.1)
- C12Y401/03—Oxo-acid-lyases (4.1.3)
- C12Y401/0304—Chorismate lyase (4.1.3.40)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y402/00—Carbon-oxygen lyases (4.2)
- C12Y402/01—Hydro-lyases (4.2.1)
- C12Y402/01118—3-Dehydroshikimate dehydratase (4.2.1.118)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12R—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES C12C - C12Q, RELATING TO MICROORGANISMS
- C12R2001/00—Microorganisms ; Processes using microorganisms
- C12R2001/01—Bacteria or Actinomycetales ; using bacteria or Actinomycetales
- C12R2001/15—Corynebacterium
Definitions
- the present invention relates to a transformant capable of efficiently producing protocatechuic acid or a salt thereof using a saccharide as a raw material by performing a specific genetic manipulation, and an efficient protocatechu using the transformant.
- the present invention relates to a method for producing an acid.
- Protocatechuic acid is a useful compound used as an antioxidant in addition to being a raw material for pharmaceuticals, agricultural chemicals, fragrances and the like.
- protocatechuic acid is mainly produced by extraction from natural products (agricultural products).
- natural products agricultural products
- Patent Documents 1 and 2 describe that a Escherichia bacterium or Klebsiella bacterium that can convert a carbon source into 3-dehydroshikimate via a common aromatic amino acid biosynthesis pathway, a 3-dehydroshikimate dehydratase gene derived from a Klebsiella bacterium, And a method for producing catechol from saccharides via protocatechuic acid using a transformant introduced with a protocatechuate decarboxylase gene.
- Patent Document 2 further teaches that for the production of catechol via protocatechuic acid, it is preferable to inhibit the conversion of 3-dehydroshikimate to chorismate by inactivating shikimate dehydrogenase. ing.
- Patent Documents 3 and 4 use a transformant in which a 3-dehydroshikimate dehydratase gene, a protocatechuate decarboxylase gene, and a catechol 1,2-dioxygenase gene are introduced into a bacterium of the genus Escherichia or Klebsiella. Teaches how to produce cis, cis-muconic acid or adipic acid from saccharides via protocatechuic acid. Patent Documents 4 and 5 teach that it is preferable to inhibit any enzyme on the metabolic pathway from 3-dehydroshikimic acid to chorismic acid.
- Patent Document 5 discloses protocatechuic acid from saccharides using a transformant in which a 3-dehydroshikimate dehydratase gene and a mutant 4-hydroxybenzoate hydroxylase gene are introduced into a bacterium belonging to the genus Escherichia or Klebsiella. Teaches how to make gallic acid or pyrogallol.
- Patent Documents 1 to 5 do not intend to produce protocatechuic acid, and the problem is that the produced protocatechuic acid is converted into catechol, cis, cis-muconic acid, adipic acid, or gallic acid. is there. In addition, these substances cannot be produced sufficiently efficiently in practice.
- An object of the present invention is to provide a microorganism capable of efficiently producing protocatechuic acid or a salt thereof from a saccharide, and a method for efficiently producing protocatechuic acid or a salt thereof using the microorganism.
- Protocatechuic acid is generally known to be cytotoxic to microorganisms, and the possibility that productivity was limited by the toxicity of the produced protocatechuic acid was considered. Therefore, we compared the effects of protocatechuic acid on the growth of some microorganisms that have been reported to produce aromatic compounds so far. Among erythropolis, Corynebacterium glutamicum was shown to be the most resistant to protocatechuic acid.
- Corynebacterium glutamicum showed high growth ability and sugar consumption ability even in the presence of a high concentration of protocatechuic acid of 500 mM, in which the growth of other microorganisms was completely or significantly suppressed.
- Corynebacterium glutamicum is particularly suitable for the production of protocatechuic acid or a salt thereof because of its extremely high resistance to protocatechuic acid.
- the coryneform bacterial transformant subjected to both (a) and (b) significantly improves the productivity of protocatechuic acid or its salt while blocking the aromatic amino acid biosynthetic pathway. Therefore, there is no requirement for aromatic amino acids such as ⁇ lipptophan, tyrosine, and phenylalanine, and paraaminobenzoic acid, so that it is not necessary to add these compounds to the medium to grow the transformant.
- This transformant has a particularly high production efficiency of protocatechuic acid or a salt thereof when the reaction is carried out under an aerobic and substantially non-proliferating condition.
- the present invention has been completed based on the above findings, and provides the following transformant and a method for producing protocatechuic acid or a salt thereof.
- Item 1 A transformant capable of producing protocatechuic acid, which has been subjected to the following operations (A), (B), and (C).
- a transformant capable of producing protocatechuic acid which has been subjected to the following operations (A), (B), and (C).
- B Enhancement of chorismate pyruvate lyase activity
- C Enhancement of 4-hydroxybenzoate hydroxylase activity 2.
- the genes encoding enzymes with 3-dehydroshikimate dehydratase activity are Corynebacterium glutamicum, Corynebacterium halotolerans, Corynebacterium casei, Corynebacterium efficiens (Corynebacterium Item 3.
- the transformant according to Item 2 or 3, wherein the gene encoding an enzyme having 3-dehydroshikimate dehydratase activity is encoded by the following DNA (a) or (b): (a) DNA consisting of the nucleotide sequence of SEQ ID NO: 7, 134, 135, 145, 147, or 149 (b) a DNA comprising a nucleotide sequence having 90% or more identity with the nucleotide sequence of SEQ ID NO: 7, 134, 135, 145, 147, or 149, comprising a polypeptide having 3-dehydroshikimate dehydratase activity DNA to encode Item 5.
- the enhancement of the chorismate pyruvate lyase activity is caused by the introduction of a gene encoding an enzyme having chorismate pyruvate lyase activity derived from Providencia or Cronobacter bacterium into the host.
- the transformant according to any one of 1 to 4.
- Item 6 Host of genes whose enhanced chorismate pyruvate lyase activity encodes an enzyme with chorismate pyruvate lyase activity from Providencia rustigianii, Providencia stuartii, or Cronobacter sakazakii Item 6.
- the transformant according to Item 5 which is caused by introduction into the cell.
- Item 7. Item 7.
- the enhancement of 4-hydroxybenzoate hydroxylase activity was caused by the introduction of the gene for Corynebacterium glutamicum, which encodes an enzyme having 4-hydroxybenzoate hydroxylase activity, into the host.
- the transformant according to any one of Items 1 to 7. Item 9.
- the enhancement of the DAHP synthase activity was brought about by the introduction of the DNA of the following (g) or (h) into the host, and the enhancement of the 3-dehydroquinate synthase activity was the following (i) or (j) And the enhancement of 3-dehydroquinate dehydratase activity was caused by the introduction of DNA of (k) or (l) below into the host.
- the enhancement of acid dehydrogenase activity was brought about by introduction of the following DNA of (m) or (n) into the host, and the enhancement of shikimate kinase activity was achieved by the following DNA of (o) or (p):
- the enhancement of EPSP synthase activity was caused by introduction of the DNA of the following (q) or (r) into the host, and enhancement of chorismate synthase activity
- the following (s) or (t) DNA may be introduced into the host Item 12.
- Item 13 The transformant according to any one of Items 1 to 12, wherein at least one activity selected from the group consisting of transketolase activity and transaldolase activity is enhanced.
- Item 14 The enhancement of transketolase activity is due to the introduction of the following DNA (u) or (v), and the enhancement of transaldolase activity is due to the introduction of the following DNA (w) or (x): The transformant according to 13.
- Item 17. The transformant according to Item 15 or 16, wherein the host coryneform bacterium is a genus Corynebacterium. Item 18. Item 18. The transformant according to Item 17, wherein the host Corynebacterium bacterium is Corynebacterium glutamicum. Item 19. Item 19. The coryneform bacterium transformant according to Item 18, wherein the host Corynebacterium glutamicum is Corynebacterium glutamicum R (FERM BP-18976), ATCC13032, or ATCC13869. Item 20. Corynebacterium glutamicum PCA4 (Accession number: NITE BP-02217) Item 21. Item 21.
- a method for producing protocatechuic acid or a salt thereof comprising a step of culturing the transformant according to any one of Items 1 to 20 in a reaction solution containing a saccharide to produce protocatechuic acid or a salt thereof.
- Item 22. The method according to Item 21, wherein the transformant is cultured under aerobic conditions where the transformant does not grow.
- the biosynthesis pathway of protocatechuic acid in microorganisms includes (a) a protocatechuic acid production pathway by conversion of 3-dehydroshikimic acid to protocatechuic acid, catalyzed by 3-dehydroshikimate dehydratase, b) Two pathways, chorismate pyruvate lyase and protocatechuate production pathway by conversion of chorismate (the final metabolite of shikimate pathway) to protocatechuate catalyzed by 4-hydroxybenzoate hydroxylase Exists.
- the two metabolic pathways (a) and (b), which branch from 3-dehydroshikimic acid as a base point and lead to the production of protocatechuic acid, are strengthened at the same time.
- Protocatechuic acid production is significantly increased. That is, in coryneform bacteria, (a) 3-dehydroshikimate dehydratase activity, (b) chorismate pyruvate lyase activity, and 4-hydroxybenzoate hydroxylase activity are simultaneously applied (a ) Or only (b), the production amount of protocatechuic acid or a salt thereof from the saccharide is synergistically improved.
- Coryneform bacteria have genes on the chromosome that encode 3-dehydroshikimate dehydratase and 4-hydroxybenzoate hydroxylase among the above three enzymes, but encode chorismate pyruvate lyase. It has no gene to do.
- protocatechuic acid useful as a raw material for pharmaceuticals, fragrances, polymers and the like can be produced at low cost and in large quantities by a fermentation method with a low environmental load.
- the growth of microorganisms is inhibited by the cytotoxicity of aromatic compounds such as protocatechuic acid, it has been difficult to efficiently produce protocatechuic acid using microorganisms.
- coryneform bacteria have a very high resistance to aromatic compounds containing protocatechuic acid, a high concentration of protocatechuic acid or a salt thereof can be efficiently produced using the transformant of the present invention.
- Coryneform bacteria unlike E.
- FIG. 1 schematically shows a protocatechuic acid biosynthesis pathway in a coryneform bacterium transformant.
- a transformant capable of producing protocatechuic acid or a salt thereof Host
- any microorganism having an ability to produce protocatechuic acid can be used as a host.
- Suitable host microorganisms include Corynebacterium bacteria, Escherichia bacteria (especially Escherichia coli), Bacillus bacteria (especially Bacillus subtilis), Pseudomonas bacteria (especially Pseudomonas putida), Brevibacterium bacteria, Streptococcus bacteria , Lactobacillus bacteria, Rhodococcus bacteria (especially Rhodococcus erythropolis, Rhodococcus opacus), Streptomyces bacteria, Saccharomyces yeasts (especially Saccharomyces cerevisiae), Klaveromyces yeasts, Schizosaccharomyces yeasts, Yarrowia Examples include yeast, Trichosporon yeast, Rhodosporidium yeast, Pichia yeast, Candida yeast, Neurospora mold, Aspergillus mold, Trichoderma mold and the like.
- coryneform bacteria are preferably used as the host in terms of production efficiency of protocatechuic acid or a salt thereof.
- Coryneform bacteria are a group of microorganisms defined in Bergey's Manual of Determinative Bacteriology, Vol. 8, 599 (1974), under normal aerobic conditions. If it proliferates, it will not be specifically limited. Specific examples include Corynebacterium, Brevibacterium, Arthrobacter, Mycobacterium, Micrococcus, and the like. Among the coryneform bacteria, the genus Corynebacterium is preferable.
- Corynebacterium glutamicum (Corynebacterium glutamicum), Corynebacterium efficiens, Corynebacterium ammoniagenes, Corynebacterium halotolerance, Corynebacterium alkanoler Examples include riticam (Corynebacterium alkanolyticum). Of these, Corynebacterium glutamicum is preferable because it is safe and has high protocatechuic acid productivity.
- Corynebacterium glutamicum R strain (FERM BP-18976), ATCC13032 strain, ATCC13869 strain, ATCC13058 strain, ATCC13059 strain, ATCC13060 strain, ATCC13232 strain, ATCC13286 strain, ATCC13287 strain, ATCC13655 strain, ATCC13745 Strains, ATCC13746 strain, ATCC13761 strain, ATCC14020 strain, ATCC31831 strain, MJ-233 (FERM BP-1497), MJ-233AB-41 (FERM BP-1498) and the like.
- Corynebacterium glutamicum strains are deposited internationally under the Budapest Treaty and are publicly available. Among these, R strain (FERM BP-18976), ATCC13032 strain, and ATCC13869 strain are preferable.
- corynetypes such as Brevibacterium flavum, Brevibacterium lactofermentum, Brevibacterium divaricatum, Corynebacterium lilium, etc.
- the name of the bacterium is the same as Corynebacterium glutamicum (Liebl, W. et al., Trans Brevibacterium divaricatum DSM 20297T, "Brevibacterium flavum” DSM 20411, "Brevibacterium lactofermentum DSM DSM DSM DSM DSM , And Corynebacterium glutamicum and their distinction by rRNA gene restriction patterns.
- Corynebacterium glutamicum Int J Syst Bacteriol. 41: 255-260. (1991), Komagata Kazuo et al., Coryneform bacteria classification, fermentation and industry, 45: 944-963 (1987) ].
- Examples of the genus Brevibacterium include Brevibacterium ammoniagenes (for example, ATCC6872 strain).
- examples of the genus Arthrobacter include Arthrobacter globiformis (for example, ATCC 8010 strain, ATCC 4336 strain, ATCC 21056 strain, ATCC 31250 strain, ATCC 31738 strain, ATCC 35698 strain) and the like.
- Examples of the genus Mycobacterium include Mycobacterium bovis (for example, ATCC19210 strain, ATCC27289 strain).
- Examples of the genus Micrococcus include Micrococcus freudenreichii (for example, No. 239 strain (FERM P-13221)), Micrococcus leuteus (for example, No.
- Examples include Micrococcus ureae (for example, IAM1010 strain), Micrococcus roseus (for example, IFO3764 strain), and the like. These Brevibacterium, Arthrobacter, Mycobacterium, and Micrococcus strains are internationally deposited under the Budapest Treaty and are publicly available.
- the coryneform bacterium may be a mutant strain or an artificial gene recombinant.
- disrupted strains of genes such as lactate (lactate dehydrogenase: LDH), phosphoenolpyruvate carboxylase, and malate dehydrogenase.
- lactate dehydrogenase lactate dehydrogenase: LDH
- phosphoenolpyruvate carboxylase phosphoenolpyruvate carboxylase
- malate dehydrogenase a disrupted strain of lactate dehydrogenase gene is preferable. In this gene-disrupted strain, the metabolic pathway from pyruvate to lactic acid is blocked because the lactate dehydrogenase gene is disrupted.
- a disrupted strain of Corynebacterium glutamicum particularly a lactate dehydrogenase gene of R (FERM BP-18976) strain is preferable.
- a gene-disrupted strain can be prepared according to a conventional method by genetic engineering techniques.
- WO2005 / 010182A1 describes a lactate dehydrogenase-disrupting strain and a method for producing the same.
- coryneform bacteria are extremely resistant to protocatechuic acid compared to other bacteria. Further, as shown in FIG. 3, the coryneform bacterium exhibited a high sugar consumption ability even in the presence of a high concentration of protocatechuic acid. In these respects, coryneform bacteria are suitable for producing protocatechuic acid or a salt thereof by the method of the present invention.
- Transformants produce better protocatechuic acid transgene present invention efficiently in a host strain, 3-dehydroshikimic acid dehydratase, to strengthen the Collis formate pyruvate lyase, and 4-hydroxybenzoic acid hydroxy each enzyme activity of hydrolase Can be obtained.
- 3-dehydroshikiate dehydratase catalyzes the reaction of producing protocatechuic acid from 3-dehydroshikimate.
- Chorismate pyruvate lyase catalyzes the reaction that produces 4-hydroxybenzoic acid from chorismate.
- 4-hydroxybenzoic acid hydroxylase catalyzes a reaction for producing protocatechuic acid by hydroxylating the 3-position carbon atom of the aromatic ring of 4-hydroxybenzoic acid.
- the activity of these enzymes can be enhanced by introducing a gene encoding these enzymes into a host microorganism.
- the activity enhancement of these enzymes can also be brought about by introducing a mutation into the control sequence of the enzyme gene present on the chromosome of the host microorganism, the gene coding region, or both, or by replacing the base sequence. Of these, it is simple and efficient to enhance the enzyme activity by introducing these enzyme genes into the host microorganism.
- the bacterium When a coryneform bacterium is used as a host, the bacterium has a 3-dehydroshikimate dehydratase gene and a 4-hydroxybenzoate hydroxylase gene on the chromosome, but does not have a chorismate pyruvate lyase gene. Absent.
- the 3-dehydroshikimate dehydratase gene and the 4-hydroxybenzoate hydroxylase gene are also expressed only under specific culture conditions (in the presence of protocatechuic acid or a specific aromatic compound). It is possible that Therefore, the above three genes are preferably introduced into the host coryneform bacterium as a fusion gene placed under the control of an appropriate promoter that provides high expression under the culture conditions used.
- each gene is not particularly limited, for example, the following microorganism genes are mentioned in terms of good production efficiency of protocatechuic acid or a salt thereof.
- the 3-dehydroshikimate dehydratase gene includes bacteria belonging to the genus Corynebacterium (in particular, Corynebacterium glutamicum, Corynebacterium casei), Corynebacterium efficience (Corynebacterium efficience) ), Corynebacterium halotolerans), Rhodococcus bacteria (especially Rhodococcus opacus), Mycobacterium bacteria (especially Mycobacterium smegmatis), Bacillus bacteria ( In particular, Bacillus thuringiensis), Gluconobacter bacterium (especially Gluconobacter oxydans), Rhodopseudomonas bacterium (especially Rhodopseudomonas spp.).
- Corynebacterium in particular, Corynebacterium glutamicum, Corynebacterium casei), Corynebacterium efficience (Corynebacterium efficience) ), Corynebacterium halotolerans),
- genes of Corynebacterium glutamicum, Corynebacterium casei, Corynebacterium efficiens, Corynebacterium halotolerance, Rhodococcus opacus, Methylobacterium exotolquiens, Neurospora crassa, Aspergillus niger, and Aspergillus oryzae are preferable. More preferred are genes of Corynebacterium glutamicum and Corynebacterium halotolerance.
- the 3-dehydroshikimate dehydratase gene of Corynebacterium glutamicum of SEQ ID NO: 7 is called qsuB. Further, it is a DNA that hybridizes under stringent conditions with a DNA comprising a base sequence complementary to any one of SEQ ID NO: 7 and SEQ ID NOs: 134 to 150, and has 3-dehydroshikimate dehydratase activity. DNA encoding a polypeptide having the same can also be used.
- stringent conditions refers to hybridization in a 6 ⁇ SSC salt concentration hybridization solution at a temperature of 50 to 60 ° C. for 16 hours in a 0.1 ⁇ SSC salt concentration solution. This is the condition for cleaning.
- DNA comprising a nucleotide sequence having 90% or more, particularly 95% or more, especially 98% or more identity with any one of SEQ ID NO: 7 and SEQ ID NOs: 134 to 150, and 3-dehydro
- a DNA encoding a polypeptide having shikimate dehydratase activity can also be used.
- the identity of the base sequence is a value calculated by GENETYX® ver. 8 (manufactured by GENETYX® GENETICS).
- the enhancement of 3-dehydroshikimate dehydratase activity of the transformant is confirmed by measuring the 3-dehydroshikimate dehydratase activity in the cell extract of the transformant.
- Collis formate pyruvate lyase gene Collis formate pyruvate but not limited lyase gene derived, in particular, in terms of good production efficiency of protocatechuic acid or a salt thereof, Providencia spp or a gene Chrono Enterobacter bacteria are preferred, among them, The gene of Providencia rustigianii, Providencia stuartii, and Cronobacter sakazakii are more preferred, and the gene of Providencia rustigianii is even more preferred.
- the chorismate pyruvate lyase genes of Providencia rustigianii, Providencia stuartii, and Cronobacter sakazakii consist of the nucleotide sequences shown in SEQ ID NOs: 9, 128, and 129, respectively. Is mentioned.
- the chorismate pyruvate lyase gene of Providencia rustigianii of SEQ ID NO: 9 is called ubiC.
- the encoding DNA can also be used.
- DNA comprising a nucleotide sequence having 90% or more, particularly 95% or more, particularly 98% or more of the nucleotide sequence of any one of SEQ ID NOs: 9, 128 and 129, and has a chorismate pyruvate lyase activity.
- a DNA encoding a polypeptide having can also be used.
- the chorismate pyruvate lyase activity is measured using a method modified from the method described in “Journal of Bacteriology, 174, 5309-5316, 1992“ Materials and Methods ””. That is, by adding a test enzyme solution to a reaction mixture consisting of 50 mM Tris-HCl buffer (pH 7.5), 20 mM NaCl, 0.2 mM NADH, 0.5 mM chorismate, 5 U / ml lactate dehydrogenase at 33 ° C. Beckman DU 800 spectrophotometer The enzyme activity is calculated from the initial reaction rate. The activity at which 1 ⁇ mol of NADH is consumed per minute at 33 ° C.
- the enhancement of the chorismate pyruvate lyase activity of the transformant is confirmed by an increase in the chorismate pyruvate lyase activity in the cell extract of the transformant.
- 4-Hydroxybenzoate hydroxylase gene 4-Hydroxybenzoate hydroxylase is also referred to as phenol monooxygenase.
- the origin of the 4-hydroxybenzoate hydroxylase gene is not particularly limited, but the gene of Corynebacterium genus bacteria, especially the gene of Corynebacterium glutamicum (Corynebacterium glutamicum) is particularly advantageous in terms of good production efficiency of protocatechuic acid or its salts. preferable.
- Examples of the 4-hydroxybenzoate hydroxylase gene of Corynebacterium glutamicum include those consisting of the base sequence shown in SEQ ID NO: 8.
- the Corynebacterium glutamicum 4-hydroxybenzoate hydroxylase gene is called pobA.
- DNA that hybridizes under stringent conditions with DNA consisting of a base sequence complementary to the base sequence of SEQ ID NO: 8 and that encodes a polypeptide having 4-hydroxybenzoate hydroxylase activity can also be used. .
- polypeptide having a 4-hydroxybenzoic acid hydroxylase activity which is a DNA comprising a nucleotide sequence having 90% or more, particularly 95% or more, particularly 98% or more of the nucleotide sequence of SEQ ID NO: 8 DNA can also be used.
- DAHP 3-deoxy-D-arabino-heptulosonate-7-phosphate
- the transformant of the present invention further comprises 3-deoxy-D-arabino-heptulosonate-7-phosphate (DAHP) synthase activity. Is preferably enhanced.
- DAHP synthase is an enzyme that generates DAHP, which is an initial metabolite of an aromatic compound biosynthesis pathway, from erythrose-4-phosphate and phosphoenolpyruvate.
- Enhancement of DAHP synthase activity can be achieved by introducing the DAHP synthase gene into the host microorganism, or introducing a mutation into the DAHP synthase gene (regulatory sequence and / or region, gene coding region, or both) on the chromosome of the host microorganism. Can bring. Of these, it is simple and efficient to enhance DAHP synthase activity by introducing a DAHP synthase gene into a host microorganism.
- the origin of the DAHP synthase gene to be introduced is not particularly limited, but a gene derived from Corynebacterium glutamicum or Escherichia coli is preferable from the viewpoint of good production efficiency of protocatechuic acid or a salt thereof. Of these, genes derived from Escherichia coli are more preferable.
- DNA (aroG S180F ) comprising the nucleotide sequence of SEQ ID NO: 2 is even more preferable.
- This gene is a gene in which the mutation (S180F) that mutates the 180th serine of the amino acid sequence encoded by this gene to phenylalanine is introduced into the aroG gene, which is one of the DAHP synthase genes derived from Escherichia coli.
- the present inventors have found through comparative studies that gene products exhibit resistance to feedback inhibition by aromatic compounds containing aromatic amino acids and high DAHP synthase activity (unpublished).
- a DNA consisting of a nucleotide sequence having 90% or more, particularly 95% or more, especially 98% or more of identity with SEQ ID NO: 2, and a DNA encoding a polypeptide having DAHP synthase activity, or A DNA that hybridizes with a DNA comprising a base sequence complementary to SEQ ID NO: 2 under stringent conditions and that encodes a polypeptide having DAHP synthase activity can also be used.
- DAHP synthase activity as follows. Reaction by adding the test enzyme solution to the reaction mixture consisting of 20 mM bistrispropane buffer (pH 6.8), 500 ⁇ M ⁇ phosphoenolpyruvate (PEP) sodium, 500 ⁇ M erythrose-4-phosphate, 1 mM manganese chloride
- the activity at which 1 ⁇ mol of PEP is consumed per minute at 33 ° C. is defined as 1 ⁇ unit of DAHP synthase activity.
- the enhancement of DAHP synthase activity of the transformant is confirmed by an increase in the DAHP synthase activity value in the cell extract of the transformant.
- the transformant of the present invention preferably further has enhanced transketolase activity, or transketolase activity and transaldolase activity.
- transketolase catalyzes two reactions.
- the first reaction is the conversion of D-xylulose-5-phosphate to glyceraldehyde-3-phosphate and D-ribose-5-phosphate (R5P) in the non-oxidative pentose-phosphate pathway. It is a reaction that catalyzes the conversion of sucrose to sedheptulose-7-phosphate (S7P). These reactions are reversible and conjugated.
- the second reaction consists of the conversion of D-fructose-6-phosphate (F6P) to erythrose-4-phosphate (E4P), and glyceraldehyde-3-phosphate to D-xylulose-5. A reaction that catalyzes the conversion to phosphoric acid. These reactions are reversible and conjugated.
- transaldolase In sugar metabolism, transaldolase is converted from glyceraldehyde-3-phosphate to erythrose-4-phosphate, and from sedoheptulose-7-phosphate to D-fructose-6-phosphate. To catalyze. These reactions are conjugated.
- transketolase and transaldolase play an important role in the production of erythrose-4-phosphate, which is one of the precursors of aromatic compound biosynthesis. Therefore, by enhancing these enzyme activities, the supply of intracellular erythrose-4-phosphate is increased, resulting in an increased metabolic flux into the aromatic compound biosynthetic pathway and an improvement in protocatechuic acid productivity. It is thought to bring.
- the transketolase activity and the enhancement of the transaldolase activity can be achieved by introducing the transketolase gene and the transaldolase gene into the host microorganism or the control sequence of the transketolase gene or transaldolase gene on the chromosome of the host microorganism. , By introducing mutations into the gene coding region or both, and by sequence substitution. Of these, it is simple and efficient to enhance the enzyme activity by introducing the transketolase gene and transaldolase gene into the host microorganism.
- the transketolase gene to be introduced and the origin of the transaldolase gene are not particularly limited, but in terms of good production efficiency of protocatechuic acid or a salt thereof, the transketolase gene of Corynebacterium, particularly Corynebacterium glutamicum, and A transaldolase gene is preferred.
- Examples of the transketolase gene of Corynebacterium glutamicum include DNA (tkt) having the base sequence of SEQ ID NO: 151
- examples of the transaldolase gene of Corynebacterium glutamicum include DNA (tal) having the base sequence of SEQ ID NO: 152 ).
- a DNA comprising a nucleotide sequence having 90% or more, particularly 95% or more, particularly 98% or more of identity with SEQ ID NO: 151 or 152, and transketolase activity or transaldolase activity, respectively. It is also possible to use DNA encoding a polypeptide having Further, in the present invention, DNA that hybridizes under stringent conditions with DNA consisting of a base sequence complementary to SEQ ID NO: 151 or 152, and encodes a polypeptide having transketolase activity or transaldolase activity, respectively. Can also be used.
- the activity that consumes 1 ⁇ mol of NADH per minute at 33 ° C. is defined as 1 unit of transketolase activity, and it is determined that there is transketolase activity when the activity is detected.
- the enhancement of the transketolase activity of the transformant is confirmed by an increase in the transketolase activity value in the cell extract of the transformant.
- the activity at which 1 ⁇ mol of NADH is consumed per minute at 33 ° C. is defined as 1 unit of transaldolase activity. If the activity is detected, it is determined that there is transaldolase activity. In the present invention, the enhancement of the transaldolase activity of the transformant is confirmed by an increase in the transaldolase activity value in the cell extract of the transformant.
- Enhancing the enzyme activities of 3-dehydroquinate synthase, 3-dehydroquinate dehydratase, shikimate dehydrogenase, shikimate kinase, 5-enolpyruvylshikimate 3-phosphate (EPSP) synthase, and chorismate synthase The transformant further comprises a series of enzymes on the shikimate pathway after DAHP synthase: 3-dehydroquinate synthase, 3-dehydroquinate dehydratase, shikimate dehydrogenase, shikimate kinase, 5-enolpyruvylshikimate 3 -Preferably, at least one of the enzyme activities of phosphate (EPSP) synthase and chorismate synthase is enhanced, and more preferably, all of these enzyme activities are enhanced.
- EBP phosphate
- 3-dehydroquinic acid synthase is an enzyme that catalyzes the conversion of DAHP to 3-dehydroquinic acid
- 3-dehydroquinic acid dehydratase is an enzyme that catalyzes the conversion of 3-dehydroquinic acid to 3-dehydroshikimic acid.
- Acid dehydrogenase is an enzyme that catalyzes the conversion of 3-dehydroshikimate to shikimate
- shikimate kinase is an enzyme that catalyzes the conversion of shikimate to shikimate-3-phosphate
- EPSP synthase An enzyme that catalyzes the conversion of acid-3-phosphate to EPSP
- chorismate synthase is an enzyme that catalyzes the conversion of EPSP to chorismate.
- Enhancement of each enzyme activity of 3-dehydroquinate synthase, 3-dehydroquinate dehydratase, shikimate dehydrogenase, shikimate kinase, EPSP synthase, and chorismate synthase can be achieved by introducing a gene encoding each of the enzymes into a host microorganism, or The gene can be brought about by introducing a mutation into the regulatory sequence of the enzyme gene on the chromosome of the host microorganism, the gene coding region, or both, and by replacing the nucleotide sequence. Of these, it is simple and efficient to enhance the enzyme activity encoded by introduction of each enzyme gene into a host microorganism.
- each gene encoding 3-dehydroquinate synthase, 3-dehydroquinate dehydratase, shikimate dehydrogenase, shikimate kinase, EPSP synthase, and chorismate synthase to be introduced is not particularly limited, but the production efficiency of protocatechuic acid and its salts However, it is preferably a gene of Corynebacterium, particularly Corynebacterium glutamicum.
- the 3-dehydroquinate synthase gene includes DNA (aroB) consisting of SEQ ID NO: 153, and the 3-dehydroquinate dehydratase gene is DNA consisting of SEQ ID NO: 5 (aroD
- the shikimate dehydrogenase gene includes DNA (aroE) consisting of SEQ ID NO: 6, the shikimate kinase gene includes DNA (aroK) consisting of SEQ ID NO: 154, and the EPSP synthase gene includes SEQ ID NO: And a chorismate synthase gene includes DNA (aroC) consisting of SEQ ID NO: 156.
- DNA comprising a nucleotide sequence having the identity of SEQ ID NO: 153, 5, 6, 154, 155, or SEQ ID NO: 156 of 90% or more, particularly 95% or more, especially 98% or more,
- DNA encoding a polypeptide having 3-dehydroquinate synthase activity, 3-dehydroquinate dehydratase activity, shikimate dehydrogenase activity, shikimate kinase activity, EPSP synthase activity, or chorismate synthase activity, respectively, can also be used.
- DNA that hybridizes under stringent conditions with DNA consisting of a base sequence complementary to SEQ ID NO: 153, 5, 6, 154, 155, or SEQ ID NO: 156, and each of 3- DNA encoding a polypeptide having dehydroquinate synthase activity, 3-dehydroquinate dehydratase activity, shikimate dehydrogenase activity, shikimate kinase activity, EPSP synthase activity, or chorismate synthase activity can also be used.
- 3-dehydroquinate synthase activity is determined by known methods (Meudi, S. et al., Dehydroquinate synthase from Escherichia coli, and its substrate 3-deoxy-D-arabino-heptulosonic acid 7-phosphate. Methods. Enzymol. 142: 306 -314 (1987)).
- a reaction comprising a crude enzyme solution of 50 mM potassium phosphate buffer (pH 7.0), 0.2 mM DAHP, 0.2 mM NAD + , 1 mM Cobalt (II) chloride ⁇ 6H 2 O, 3-dehydroquinate dehydratase
- the reaction was initiated by adding the test enzyme solution to the mixture, and the absorbance at 234 nm due to 3-dehydroshikimate produced by the coupling reaction of 3-dehydroquinate synthase activity and 3-dehydroquinate dehydratase activity.
- the activity produced by 1 ⁇ mol of 3-dehydroshikimic acid per minute at 33 ° C. is defined as 1 unit of DHQ synthase activity.
- the enhancement of 3-dehydroquinate synthase activity of the transformant is confirmed by an increase in 3-dehydroquinate synthase activity in the cell extract of the transformant.
- 3-Dehydroquinate dehydratase activity is performed according to a known method (Chaudhuri, S. et al., 3-Dehydroquinate dehydratase from Escherichia coli. Methods. Enzymol. 142: 320-324 (1987)). That is, at 33 ° C., the reaction is started by adding the test enzyme solution to a reaction mixture consisting of 50 mM potassium phosphate buffer (pH 7.0) and 0.5 mM 3-dehydroquinic acid, and 3-dehydroshiki-mi produced.
- 1 unit of 3-dehydroquinic acid dehydratase activity is defined as 1 unit of 3-dehydroquinic acid dehydratase activity at 33 ° C per minute, and it is determined that 3-dehydroquinic acid dehydratase activity exists when activity is detected .
- the enhancement of 3-dehydroquinate dehydratase activity of the transformant is confirmed by an increase in 3-dehydroquinate dehydratase activity in the cell extract of the transformant.
- Shikimate dehydrogenase activity is measured according to a known method (Chaudhuri, S. et al., Shikimate dehydrogenase from Escherichia coli. Methods. Enzymol. 142: 315-320 (1987)).
- the reaction was started by adding the test enzyme solution to a reaction mixture consisting of 100 mM Tris-HCl buffer (pH 7.5), 0.2 mM NADPH, 0.5 mM 3-dehydroshikimic acid, and NADPH
- the activity at which 1 ⁇ mol of NADPH is consumed per minute at 33 ° C. is defined as 1 unit of shikimate dehydrogenase activity.
- Shikimate kinase activity is measured according to a known method (Cheng, WC. Et al., Structures of Helicobacter pylori shikimate kinase reveal a selective inhibitor-induced-fit mechanism. PLos One. 7: e33481 (2012)).
- the chorismate synthase activity is measured according to a known method (Kitzing, K. et al., Spectroscopic and Kinetic Characterization of the Bifunctional Chorismate Synthase from Neurospora crassa. J. Biol. Chem. 276: 42658-42666 (2001)).
- the reduction of FMN can be performed by adding 5 mM dithionite or 1 mM NADPH.
- the activity produced by 1 ⁇ mol anthranilic acid per minute at 37 ° C. is defined as 1 unit of the chorismate synthase activity.
- the enhancement of the chorismate synthase activity of the transformant is confirmed by an increase in the chorismate synthase activity in the cell extract of the transformant.
- Protocatechuic acid 3,4-dioxygenase is an enzyme that catalyzes the conversion of protocatechuic acid to ⁇ -carboxy-cis, cis muconic acid by ring opening of protocatechuic acid in the catabolic metabolic pathway of protocatechuic acid.
- Protocatechuate 3,4-dioxygenase activity can be eliminated, inhibited, or decreased by disruption, deletion, or mutation of the protocatechuate 3,4-dioxygenase gene on the chromosome.
- An example of the protocatechuate 3,4-dioxygenase gene of Corynebacterium glutamicum is pcaHG.
- the fact that the protocatechuate 3,4-dioxygenase activity of the transformant is lost, inhibited, or reduced indicates that the protocatechuate 3,4-dioxygenase activity in the cell extract of the transformant is reduced. Measured and confirmed by the decrease or disappearance of the enzyme activity.
- the activity at which 1 ⁇ mol of protocatechuic acid disappears in 1 minute at 33 ° C. is defined as 1 unit of protocatechuic acid 3,4-dioxygenase activity, and when the enzyme activity is detected, protocatechuic acid 3,4-dioxygenase activity Judge that there is.
- sugar Phosphotransferase system is the uptake of sugars such as glucose into the cell and the phosphorus of sugars. It is a sugar transport mechanism existing only in prokaryotes, characterized by performing oxidation in a coupled manner. In Escherichia coli and coryneform bacteria, PTS plays a major role in the uptake of sugar into cells. PTS is a common protein, Enzyme I (PEP Protein kinase), HPr (Histidine-phosphorylatable protein), and a membrane protein involved in the specific transport of various sugars.
- Enzyme I PEP Protein kinase
- HPr Histidine-phosphorylatable protein
- Phosphoenolpyruvic acid composed of glycoprotein II (enzyme II), is used as a phosphate donor and transports sugar into the cell as a phosphorylated form via a phosphate relay between these components System.
- PTS consumes PEP, which is one of the common precursors of aromatic compounds, as a phosphate donating group for producing glucose-6-phosphate as glucose is transported into cells.
- PEP is a key precursor compound in the production of aromatic compounds, and for the high production of aromatic compounds containing protocatechuic acid, the consumption of PEP by competitive metabolic pathways such as PTS is suppressed, and the production route to aromatic compounds is reduced. It is important to increase the availability of PEP.
- the sugar uptake through PTS is inactivated, and at the same time, the PEP is not consumed with the sugar transport, via a sugar transport system (non-PTS sugar transport system) different from PTS. It is preferable that the sugar availability is provided.
- the uptake of sugar into cells via PTS can be eliminated, inhibited or reduced by disruption, deletion or mutation of the gene encoding PTS on the chromosome of coryneform bacteria.
- the gene encoding PTS include ptsI encoding Enzyme I, ptsH encoding Hpr, and ptsG encoding Enzyme II.
- the ptsH gene encoding the Hpr protein, a common component of PTS is disrupted, It is preferably deleted or mutated.
- a deletion type gene is generated by deleting a partial sequence of the gene and modified so as not to produce a normally functioning protein, and a bacterium is transformed with the DNA containing the gene so that the deletion type gene and the chromosome are on the chromosome.
- a bacterium is transformed with the DNA containing the gene so that the deletion type gene and the chromosome are on the chromosome.
- the gene on the chromosome can be replaced with a deletion or destruction type gene. Even if a protein encoded by a deletion-type or disruption-type gene is produced, it has a three-dimensional structure different from that of a wild-type protein, and its function is reduced or lost.
- the ability of the coryneform bacterium transformant to lose, inhibit, or reduce the sugar transport ability via PTS means that sugars (glucose, sucrose, fructose, etc.) transported by PTS in the transformant. ) Is eliminated, inhibited or suppressed, and such phenotype is restored by introduction of a normal pts gene.
- Corynebacterium glutamicum is a sugar transporter different from PTS, and there is a non-PTS glucose transporter that does not consume PEP due to intracellular transport of sugar. To do. Corynebacterium glutamicum whose sugar uptake through PTS is inhibited by disruption of the pts gene, etc.
- glucose uptake into cells and growth of bacteria using glucose as a carbon source are non-PTS glucose transporter activity and glucokinase It is desirable to improve by enhancing the activity. It is considered that this makes it possible to avoid the consumption of PEP accompanying glucose transport and to supply more PEP for biosynthesis of aromatic compounds such as shikimic acid.
- the uptake of glucose into cells by a non-PTS glucose transporter can be achieved by introducing a gene encoding a non-PTS glucose transporter or by a mutation in a non-PTS glucose transporter gene (control sequence or coding region) on the chromosome of a coryneform bacterium.
- the gene expression level can be enhanced by introduction or nucleotide sequence substitution, or the activity of the gene product can be enhanced. Of these, it is simple and efficient to enhance the glucose uptake activity by introducing a non-PTS glucose transporter gene.
- the origin of the non-PTS glucose transporter gene to be introduced is not particularly limited, but it is preferably a gene of Corynebacterium bacteria, particularly Corynebacterium glutamicum, in terms of good shikimic acid production efficiency.
- Any non-PTS glucose transporter may be used as long as it functions in coryneform bacteria, such as inositol transporter (IolT1, IolT2) derived from Corynebacterium glutamicum, galactose permease (GalP) derived from E. coli (Escherichia coli), and zyomonas.
- inositol transporter IolT1, IolT2
- GalP galactose permease
- E. coli Erscherichia coli
- zyomonas examples include glucose facilitator (Glf) derived from Zymomonas mobilis.
- glucose facilitator Glf
- an example of the inositol transporter gene derived from Corynebacterium glutamicum is DNA (iolT1) having the base sequence of SEQ ID NO: 157.
- a DNA comprising a nucleotide sequence having 90% or more, 95% or more, particularly 98% or more identity with SEQ ID NO: 157, and a DNA encoding a polypeptide having inositol transporter activity is also included.
- a DNA that hybridizes with a DNA comprising a base sequence complementary to SEQ ID NO: 159 under stringent conditions and encodes a polypeptide having inositol transporter activity can also be used.
- the protein encoded by DNA is a non-PTS glucose transporter, which means that a host cell that has lost its PTS-dependent glucose transport ability due to disruption of the ptsH gene, etc., and has reduced growth using glucose as a carbon source.
- the growth of the transformant into which the DNA has been introduced and expressed, using glucose as a carbon source, or the rate of glucose consumption being higher than that of the cell before transformation, and the effect of the pts gene disruption It is confirmed as an index that it is not affected by the inhibition of PTS-dependent sugar transport.
- the non-PTS glucose transporter activity of the transformant is enhanced because the growth using glucose as a carbon source or the glucose consumption rate in the transformant deficient in sugar transport by PTS. In the transformant, it is confirmed as an index that it is higher than that before gene introduction.
- Glucokinase is an enzyme that catalyzes the conversion of glucose to glucose-6-phosphate.
- the glucokinase activity is enhanced simultaneously with the enhancement of non-PTS glucose transporter-dependent glucose transport. This is characterized in that glucose uptake into cells and the subsequent glycolysis in the glycolysis and pentose / phosphate pathways are promoted.
- the glucokinase activity is increased by high expression by introduction of the glucokinase gene, or by introduction of mutations into the glucokinase gene (regulatory sequence and gene coding region) on the chromosome, or by substitution of the sequence, It can be enhanced by increasing the activity of the product.
- glucokinase genes cgR_2067 (glk1), cgR_2552 (glk2), and cgR_1739 (ppgK), on the chromosome of Corynebacterium glutamicum R strain.
- cgR_2067 (glk1) and cgR_2552 (glk2) have high homology with glucokinase which uses ATP as a good substrate
- cgR_1739 (ppgK) has high homology with glucokinase which uses polyphosphate as a good substrate.
- one or more of these glucokinase genes are preferably enhanced, and more preferably all three are enhanced.
- the enhancement of glucokinase activity is simple and efficient by introducing a glucokinase gene.
- the origin of the glucokinase gene to be introduced is not particularly limited, it is preferably a gene of Corynebacterium, particularly Corynebacterium glutamicum, in terms of good shikimic acid production efficiency.
- Examples of the glucokinase gene derived from Corynebacterium glutamicum include DNAs having the nucleotide sequences of SEQ ID NOs: 158, 159, and 160 (glk1, glk2, and ppgK, respectively).
- a polypeptide comprising a nucleotide sequence having the identity of SEQ ID NO: 158, 159 or 160 with 90% or more, particularly 95% or more, especially 98% or more, and having a glucokinase activity DNA encoding can also be used.
- a DNA that hybridizes under stringent conditions with a DNA consisting of a base sequence complementary to SEQ ID NO: 158, 159, or 160 and encodes a polypeptide having glucokinase activity. it can.
- Glucokinase activity is a reaction mixture consisting of 100 mM Tris-HCl buffer (pH 7.5), 4 mM magnesium chloride, 1 mM ATP, 0.2 mM NADP + , 20 mM glucose, 1 U glucose-6-phosphate dehydrogenase at 33 ° C.
- the activity at which 1 ⁇ mol of NADPH is produced per minute at 33 ° C. is defined as 1 unit of glucokinase activity.
- GAPDH activity-enhanced glyceraldehyde-3-phosphate dehydrogenase is an enzyme that converts glyceraldehyde 3-phosphate into 1,3-bisphosphoglycerate.
- GAPDH activity is preferably enhanced.
- a coryneform bacterium transformant in which the pts gene is disrupted and the sugar uptake activity via non-PTS glucose transporter and the glucokinase activity are enhanced is a glycolytic metabolic intermediate during culture and reaction.
- Dihydroxyacetone (DHA) which is a metabolite obtained by dephosphorylating dihydroxyacetone phosphate, and glycerol produced by further metabolism of DHA are remarkably accumulated.
- intracellular concentrations of glyceraldehyde-3-phosphate and an upstream glycolytic metabolic intermediate are significantly increased in the transformant.
- reaction step catalyzed by GAPDH is the rate-determining rate of sugar metabolism in the glycolytic system in the transformant, and the high expression of GAPDH in the transformant results in sugar consumption.
- the present inventor has found that the production of the target product is also promoted. Therefore, in the present invention, it is desirable that the GAPDH activity of the transformant is enhanced to release the rate-limiting factor of sugar metabolism to promote sugar consumption and improve protocatechuic acid production ability.
- GAPDH activity is high expression by introduction of the GAPDH gene, increase of gene expression level by introduction of mutation in GAPDH gene (control sequence and gene coding region) on the chromosome, or sequence substitution, or activity of the gene product It can be strengthened by increasing. Among them, the enhancement of GAPDH activity is simple and efficient when carried out by introducing the GAPDH gene.
- the origin of the GAPDH gene to be introduced is not particularly limited, but it is preferably a gene of Corynebacterium, particularly Corynebacterium glutamicum, in terms of good protocatechuic acid production efficiency.
- Examples of the GAPDH gene derived from Corynebacterium glutamicum include DNA (gapA) consisting of the base sequence of SEQ ID NO: 161.
- a DNA comprising a nucleotide sequence having 90% or more, more than 95%, more preferably 98% or more identity with SEQ ID NO: 161, and a DNA encoding a polypeptide having GAPDH activity is also used.
- a DNA that hybridizes with a DNA having a base sequence complementary to SEQ ID NO: 161 under stringent conditions and encodes a polypeptide having GAPDH activity can also be used.
- the fact that the protein encoded by DNA is GAPDH is confirmed by measuring the GAPDH activity of the polypeptide encoded by the DNA.
- GAPDH activity was measured at 33 ° C. using 25 mM phosphate buffer (pH 7.5), 25 mM trisethanolamine (pH 7.5), 0.2 mM EDTA, 5 mM NAD + , 5 mM glyceraldehyde-3-phosphate, The reaction is started by adding an enzyme solution to the reaction mixture consisting of By doing. The activity that 1 ⁇ mol of NADH is produced per minute at 33 ° C. is defined as 1 unit of GAPDH activity.
- the GAPDH activity of the coryneform bacterium transformant is enhanced by measuring the GAPDH activity in the cell extract of the coryneform bacterium transformant.
- DHAP dephosphorylation enzyme catalyzes the conversion of DHAP to dihydroxyacetone (DHA) by dephosphorylation.
- DHA dihydroxyacetone
- the DHAP phosphatase activity is preferably lost, inhibited or decreased.
- coryneform bacteria that take up and use sugar in cells depending on the highly expressed non-PTS glucose transporter and glucokinase produce high amounts of DHA as a by-product. For this reason, it becomes possible to supply more carbon for production of aromatic compounds such as protocatechuic acid by blocking the DHA production pathway.
- Corynebacterium glutamicum has HAD (haloacid dehalogenase) superfamily phosphatase (HdpA) as an enzyme that catalyzes the dephosphorylation of DHAP (Jojima, T. et. Al., Identification of a HAD superfamily phosphatase, HdpA , involved in 1,3-dihydroxyacetone production during sugar catabolism in Corynebacterium glutamicum. FEBS. Lett. 586: 4228-4232 (2012)).
- the DHAP phosphatase activity of Corynebacterium glutamicum can be eliminated, inhibited or reduced by disruption, deletion or mutation of the DHAP phosphatase gene (hdpA) on the chromosome.
- the fact that the DHAP phosphatase activity of the transformant is lost, inhibited, or decreased is determined by measuring the DHAP phosphatase activity in the cell extract of the transformant.
- Inorganic phosphate ions liberated from DHAP according to known methods (Gawronski, JD, et. Al., Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal. Biochem. 327: 114-118 (2004)) Measure by colorimetric determination. When this quantitative value decreases or disappears, it is determined that the dihydroxyacetone phosphate phosphatase activity has disappeared, inhibited, or decreased.
- each protein or DNA encoding the enzyme is integrated into the host chromosome, or They can be cloned into an appropriate vector that can be amplified in the host and introduced into the host.
- the plasmid vector may be any plasmid vector as long as it contains a gene that controls the autonomous replication function in coryneform bacteria. Specific examples thereof include pAM330 derived from Brevibacterium lactofermentum 2256 (Japanese Patent Laid-Open No. 58-67699), [Miwa, K. et al., Cryptic plasmids in glutamic acid-producing bacteria. Agric Biol.
- Preferred promoters include a promoter PgapA of glyceraldehyde 3-phosphate dehydrogenase A gene (gapA) derived from Corynebacterium glutamicum R, a promoter Pmdh of malate dehydrogenase gene (mdh), and a promoter PldhA of lactate dehydrogenase A gene (ldhA) Among them, PgapA is preferable.
- Preferred terminators include the rrnB T1T2 terminator of the E. coli rRNA operon, the trpA terminator of E. coli, the trp terminator of Brevibacterium lactofermentum, and the rrnB T1T2 terminator is preferred.
- Transformation transformation methods can be used without limitation any known methods.
- known methods include calcium chloride / rubidium chloride method, calcium phosphate method, DEAE-dextran mediated transfection, and electric pulse method.
- the electric pulse method is suitable for coryneform bacteria, and the electric pulse method can be performed by a known method (Kurusu, Y. et al., Electroporation-transformation system for Coryneform bacteria by auxotrophic complementation. Agric Biol. Chem. 54: 443-447 (1990)).
- a natural medium or a synthetic medium usually containing a carbon source, a nitrogen source, inorganic salts, and other nutrient substances can be used.
- carbon sources examples include glucose, fructose, sucrose, mannose, maltose, mannitol, xylose, arabinose, galactose, starch, molasses, sorbitol, glycerin and other sugars or sugar alcohols; acetic acid, citric acid, lactic acid, fumaric acid, maleic acid Or organic acids, such as gluconic acid; Alcohol, such as ethanol and a propanol, is mentioned.
- a carbon source can be used individually by 1 type, or may mix 2 or more types. The concentration of these carbon sources in the medium is usually about 0.1 to 10 (w / v%).
- the nitrogen source examples include inorganic or organic ammonium compounds such as ammonium chloride, ammonium sulfate, ammonium nitrate, and ammonium acetate, urea, aqueous ammonia, sodium nitrate, and potassium nitrate. Further, corn steep liquor, meat extract, peptone, NZ-amine, protein hydrolyzate, nitrogen-containing organic compounds such as amino acids, and the like can be used. As the nitrogen source, one kind may be used alone, or two or more kinds may be mixed and used. The nitrogen source concentration in the medium varies depending on the nitrogen compound used, but is usually about 0.1 to 10 (w / v%).
- inorganic salts examples include monopotassium phosphate, dipotassium phosphate, magnesium sulfate, sodium chloride, ferrous nitrate, manganese sulfate, zinc sulfate, cobalt sulfate, and calcium carbonate. These inorganic salts may be used alone or in a combination of two or more. The concentration of inorganic salts in the medium varies depending on the inorganic salt used, but is usually about 0.01 to 1 (w / v%).
- the nutrient substance examples include meat extract, peptone, polypeptone, yeast extract, dry yeast, corn steep liquor, skim milk powder, defatted soy hydrochloride hydrolyzate, or extracts of animal or plant or microbial cells and their degradation products. However, it is usually about 0.1 to 10 (w / v%).
- vitamins can be added as necessary. Examples of vitamins include biotin, thiamine (vitamin B1), pyridoxine (vitamin B6), pantothenic acid, inositol, nicotinic acid and the like.
- the pH of the medium is preferably about 6-8.
- a preferred microorganism culture medium is A medium (Inui, M. et al., Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions.J. Mol. Microbiol. Biotechnol. 7: 182-196 (2004)).
- BT medium [Omumasaba, CA et al., Corynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation. J. Mol. Microbiol. Biotechnol. 8: 91-103 (2004)].
- the culture temperature may be about 15 to 45 ° C.
- the culture time may be about 1 to 7 days.
- Protocatechu is produced by a method comprising a step of producing protocatechuic acid or a salt thereof by culturing or reacting the transformant of the present invention described above in a reaction solution containing a saccharide.
- An acid or a salt thereof can be produced.
- Glucose is preferred as the saccharide, but in addition to monosaccharides such as fructose, mannose, arabinose, xylose and galactose, saccharides capable of producing glucose by metabolism can also be used.
- Such sugars include oligosaccharides or polysaccharides having glucose units, disaccharides such as cellobiose, sucrose (sucrose), lactose, maltose, trehalose, cellobiose, xylobiose; polysaccharides such as dextrin or soluble starch, etc. Is mentioned.
- molasses can also be used as a raw material containing these raw material compounds.
- non-edible agricultural waste such as straw (rice straw, barley straw, wheat straw, rye straw, oat straw), bagasse, corn stover, energy crops such as switchgrass, napiergrass and miscanthus, and wood chips
- a saccharified solution containing a plurality of sugars such as glucose obtained by saccharifying used paper with a saccharifying enzyme or the like can also be used.
- the transformant Prior to the culture in a medium containing microbial growth saccharides, that is, the reaction, the transformant is preferably grown under aerobic conditions at a temperature of about 25 to 38 ° C. for about 12 to 48 hours.
- a natural medium or a synthetic medium containing a carbon source, a nitrogen source, inorganic salts, and other nutrient substances can be used.
- carbon sources sugars (monosaccharides such as glucose, fructose, mannose, xylose, arabinose, galactose; disaccharides such as sucrose, maltose, lactose, cellobiose, xylobiose, trehalose; polysaccharides such as starch; molasses etc.), Sugar alcohols such as mannitol, sorbitol, xylitol, glycerin; organic acids such as acetic acid, citrate fermentation, lactic acid, fumaric acid, maleic acid and gluconic acid; alcohols such as ethanol and propanol; hydrocarbons such as normal paraffin Can also be used.
- a carbon source can be used individually by 1 type or in mixture of 2 or
- inorganic or organic ammonium compounds such as ammonium chloride, ammonium sulfate, ammonium nitrate and ammonium acetate, urea, aqueous ammonia, sodium nitrate, potassium nitrate and the like can be used.
- corn steep liquor, meat extract, peptone, NZ-amine, protein hydrolyzate, nitrogen-containing organic compounds such as amino acids, and the like can also be used.
- a nitrogen source can be used individually by 1 type or in mixture of 2 or more types. The concentration of the nitrogen source in the medium varies depending on the nitrogen compound to be used, but is usually about 0.1 to 10 (w / v%).
- inorganic salts examples include monopotassium phosphate, dipotassium phosphate, magnesium sulfate, sodium chloride, ferrous nitrate, manganese sulfate, zinc sulfate, cobalt sulfate, and calcium carbonate.
- One inorganic salt can be used alone, or two or more inorganic salts can be mixed and used.
- the concentration of inorganic salts in the medium varies depending on the inorganic salt used, but is usually about 0.01 to 1 (w / v%).
- Examples of nutritional substances include meat extract, peptone, polypeptone, yeast extract, dry yeast, corn steep liquor, skim milk powder, defatted soy hydrochloride hydrolyzate, extracts of animals and plants or microbial cells, and degradation products thereof.
- concentration of the nutrient substance in the medium varies depending on the nutrient substance used, but is usually about 0.1 to 10 (w / v%).
- vitamins can be added as necessary. Examples of vitamins include biotin, thiamine (vitamin B1), pyridoxine (vitamin B6), pantothenic acid, inositol, nicotinic acid and the like.
- the pH of the medium is preferably about 6-8.
- a medium (Inui, M. et al., Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J. Mol. Microbiol. Biotechnol. 7: 182- 196 (2004)), BT medium (Omumasaba, CA et al., YneCorynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation. J. Mol. Microbiol. Biotechnol. 8: 91-103 (2004)) Etc.
- the saccharide concentration may be used within the above range.
- a culture solution or a reaction solution a natural reaction solution or a synthetic reaction solution containing a carbon source, a nitrogen source, inorganic salts, and the like can be used.
- the carbon source the saccharide described above or molasses or saccharified solution containing the saccharide may be used.
- a carbon source in addition to sugars, sugar alcohols such as mannitol, sorbitol, xylitol, glycerin; organic acids such as acetic acid, citric acid, lactic acid, fumaric acid, maleic acid, gluconic acid; ethanol, propanol, etc.
- Non-alcoholic; hydrocarbons such as normal paraffin can also be used.
- a carbon source can be used individually by 1 type or in mixture of 2 or more types.
- the concentration of the saccharide as the raw material compound in the reaction solution is preferably about 1 to 20 (w / v%), more preferably about 2 to 10 (w / v%), and about 2 to 5 (w / v%). Is even more preferred. Further, the concentration of the total carbon source including the saccharide as a raw material may be about 2 to 5 (w / v%).
- inorganic or organic ammonium compounds such as ammonium chloride, ammonium sulfate, ammonium nitrate and ammonium acetate, urea, aqueous ammonia, sodium nitrate, potassium nitrate and the like can be used.
- corn steep liquor, meat extract, peptone, NZ-amine, protein hydrolyzate, nitrogen-containing organic compounds such as amino acids, and the like can also be used.
- a nitrogen source can be used individually by 1 type or in mixture of 2 or more types. The concentration of the nitrogen source in the reaction solution varies depending on the nitrogen compound to be used, but is usually about 0.1 to 10 (w / v%).
- inorganic salts include monopotassium phosphate, dipotassium phosphate, magnesium sulfate, sodium chloride, ferrous nitrate, manganese sulfate, zinc sulfate, cobalt sulfate, and calcium carbonate.
- One inorganic salt can be used alone, or two or more inorganic salts can be mixed and used.
- the concentration of the inorganic salt in the reaction solution varies depending on the inorganic salt used, but is usually about 0.01 to 1 (w / v%).
- vitamins can be added as necessary.
- vitamins include biotin, thiamine (vitamin B1), pyridoxine (vitamin B6), pantothenic acid, inositol, nicotinic acid and the like.
- the pH of the reaction solution is preferably about 6-8.
- reaction solution for coryneform bacteria include the BT medium described above.
- saccharide concentration may be used within the above range.
- the culture temperature or reaction temperature that is, the survival temperature of the transformant is preferably about 20 to 50 ° C., more preferably about 25 to 47 ° C. If it is the said temperature range, protocatechuic acid can be manufactured efficiently.
- the culture or reaction time is preferably about 1 to 7 days, more preferably about 1 to 3 days.
- the culture may be any of batch type, fed-batch type, and continuous type. Among these, the batch type is preferable.
- the reaction may be carried out under aerobic conditions or under reducing conditions. The ability of the transformant of the present invention to produce protocatechuic acid or a salt thereof is higher under aerobic conditions.
- Not proliferating in the present invention includes substantially not proliferating or hardly proliferating.
- a reaction solution lacking or limiting one or more of vitamins such as biotin and thiamine, which are essential compounds for the growth of microorganisms, nitrogen sources, or amino acids essential for the growth of auxotrophic transformants By using, the growth of the transformant can be avoided or suppressed.
- the reduction condition is defined by the oxidation-reduction potential of the reaction solution.
- the oxidation-reduction potential of the reaction solution is preferably about ⁇ 200 mV to ⁇ 500 mV, more preferably about ⁇ 150 mV to ⁇ 500 mV.
- the reduction state of the reaction solution can be estimated simply with a resazurin indicator (decolorization from blue to colorless in the reduction state), but using a redox potentiometer (for example, BROADLEY JAMES, ORP Electrodes) Can be measured.
- a known method can be used without limitation.
- an aqueous solution for reaction solution may be used as a liquid medium for the reaction solution instead of distilled water, etc.
- the method for adjusting the aqueous solution for reaction solution is, for example, a culture solution preparation for absolute anaerobic microorganisms such as sulfate-reducing microorganisms.
- an aqueous solution for reaction solution under reducing conditions can be obtained by removing dissolved gas by heat treatment or decompression treatment of distilled water or the like.
- distilled water or the like is treated for about 1 to 60 minutes, preferably about 5 to 40 minutes under reduced pressure of about 10 mmHg or less, preferably about 5 mmHg or less, more preferably about 3 mmHg or less.
- the dissolved gas, particularly dissolved oxygen can be removed to prepare an aqueous solution for reaction solution under reducing conditions.
- an appropriate reducing agent for example, thioglycolic acid, ascorbic acid, cysteine hydrochloride, mercaptoacetic acid, thiolacetic acid, glutathione, sodium sulfide, etc.
- An appropriate combination of these methods is also an effective method for preparing an aqueous solution for reaction solution under reducing conditions.
- the reaction solution is preferably maintained under reducing conditions during the reaction.
- the reaction system is made of an inert gas such as nitrogen gas or carbon dioxide gas.
- the method of enclosing is mentioned.
- a pH maintenance adjusting solution of the reaction system or various nutrient solution in such a case, it is effective to remove oxygen from the added solution in advance.
- protocatechuic acid or a salt thereof is produced in the culture solution or reaction solution.
- the salt of protocatechuic acid varies depending on the medium or components of the reaction solution, and examples thereof include alkali metal salts (sodium salt, potassium salt, etc.) and alkaline earth metal salts (magnesium salt, calcium salt, etc.).
- chromosomal DNA was prepared from the following strains. Corynebacterium glutamicum R (FERM P-18976), Escherichia coli K-12 MG1655, Providencia rustigianii JCM 3953, Corynebacterium casei JCM 12072, Corynebacterium casei JCM 12072 Efficiens (Corynebacterium efficiens NBRC 100395), Pantoea ananatis (Pantoea ananatis LMG 20103), Gluconobacter oxydans ATCC 621H, Pseudomonas putida NBRC 14164, Rhodosdom R , Acinetobacter baylyi ATCC33305, Alteromonas macleodii NBRC 102226, Marinobacter hydrocarbonoclasticus JCM 20777), Methynebacterium glutamicum R (FERM P-18976), Escherichia coli K-12 MG1655, Providencia rustigianii
- Table 2 shows the introduced cloning vectors and the obtained plasmid names. Since tkt and tal (tkt-tal gene; SEQ ID NO: 1), aroC and aroK and aroB (aroCKB; SEQ ID NO: 3) are arranged in the same direction on the chromosome, cloning was performed together. .
- pCRB260, pCRB263, pCRB266, pCRB267, pCRB274 and pCRB279 introduced a restriction enzyme site (unique site) for incorporating a gene into the SSI region by inverse PCR.
- Table 3 shows primer sequences used for isolation and inverse PCR of the SSI region and the resulting vector for chromosome introduction.
- the PgapA promoter fusion enzyme gene fragment was obtained from the PCA production-related gene expression plasmid constructed in Table 2 and introduced into the above-described plasmid for chromosome introduction.
- the obtained plasmids for PCA production-related gene chromosome introduction are shown in Table 4.
- the markerless chromosomal gene transfer vector pCRA725 is a plasmid that cannot replicate in Corynebacterium glutamicum R.
- the double crossover strain shows kanamycin sensitivity due to loss of the kanamycin resistance gene on pCRA725 and growth in a sucrose-containing medium due to loss of the sacR-sacB gene.
- the markerless chromosome gene-introduced strain exhibits kanamycin sensitivity and sucrose-containing medium growth.
- a PCA production-related gene chromosome introduction strain was constructed using the above-mentioned plasmid for PCA production-related gene chromosome introduction and the plasmid for chromosome gene disruption.
- a host strain a xylose cellobiose assimilating coryneform bacterium Corynebacterium glutamicum X5C1 strain [Appl Microbiol Biotechnol. 81 (4): 691-699 (2008)] was used.
- plasmid pCRA728 J Mol Microbiol Biotechnol.
- Corynebacterium glutamicum PCA4 was deposited internationally at the Patent Microorganisms Depositary Center of the National Institute of Technology and Evaluation, 2-5-8 Kazusa Kamashi, Kisarazu, Chiba, Japan (zip code 292-0818) ( Date of international deposit under the Budapest Treaty: March 9, 2016, deposit number: NITE BP-02217). This stock is publicly available.
- Corynebacterium glutamicum which is preferable as a host microorganism in the present invention
- to protocatechuic acid in comparison with other microorganisms, Corynebacterium glutamicum, Escherichia coli, Bacillus subtilis, Pseudomonas putida, Rhodococcus erythropolis, and Saccharomyces cerevisiae The growth inhibitory effect of protocatechuic acid in aerobic culture was investigated.
- Corynebacterium glutamicum R strain grown on the above plate was mixed with A liquid medium containing 4% glucose [(NH 2 ) 2 CO 2 g, (NH 4 ) 2 SO 4 7 g, KH 2 PO 4 0.5 g, K 2 HPO 4 0.5 g, MgSO 4 ⁇ 7H 2 O 0.5 g, 0.06% (w / v) Fe 2 SO 4 ⁇ 7H 2 O + 0.042% (w / v) MnSO 4 ⁇ 2H 2 O 1 ml, 0.02% (w / v) biotin solution 1 ml, 0.01% (w / v) thiamine solution 2 ml, yeast extract 2 g, vitamin assay casamino acid 7 g dissolved in 1 L of distilled water] After inoculation, the cells were cultured under aerobic shaking at 33 ° C for 16 hours.
- Escherichia coli K12, Bacillus subtilis NBRC14144, Pseudomonas putida ATCC700801, and Rhodococcus erythropolis ATCC27854 to LB agar medium [1% polypeptone, 0.5% yeast extract, 0.5% sodium chloride, and 1.5% agar], respectively.
- the Escherichia coli K12 strain and the Bacillus subtilis NBRC14144 strain were cultured at 37 ° C, and the Pseudomonas putida ATCC700801 strain and the Rhodococcus erythropolis ATCC27854 strain were cultured at 30 ° C for 16 hours.
- Each strain grown on the above plate was inoculated into 10 ml of LB liquid medium [1% polypeptone, 0.5% yeast extract, and 0.5% sodium chloride], Escherichia coli K12 strain, and Bacillus subtilis NBRC14144 strain at 37 ° C, Pseudomonas putida ATCC700801 strain and Rhodococcus erythropolis ATCC27854 strain were aerobically cultured at 30 ° C. for 16 hours.
- Escherichia coli K12 strain and Bacillus subtilis NBRC14144 strain were cultured at 37 ° C, and Pseudomonas putida ATCC700801 strain and Rhodococcus erythropolis ATCC27854 strain were aerobically cultured at 30 ° C. Proliferation of cells was carried out by measuring the OD 610.
- Saccharomyces cerevisiae NBRC2376 strain was applied to YPD agar medium [2% polypeptone, 1% yeast extract, 2% glucose, and 1.5% agar] and cultured at 30 ° C. for 16 hours. Saccharomyces cerevisiae NBRC2376 strain grown on the above plate was inoculated into YPD liquid medium [2% polypeptone, 1% yeast extract, and 2% glucose], and aerobically shaken and cultured at 30 ° C. for 16 hours. .
- FIG. 2 shows the results of examining the influence of protocatechuic acid addition to the medium on the aerobic growth of each strain.
- the Escherichia coli K12 strain was significantly inhibited in the presence of 100 mM protocatechuic acid, and the growth was almost completely inhibited at 250 mM.
- Bacillus subtilis NBRC14144 strain was markedly inhibited in the presence of 250 mM protocatechuic acid, and growth was almost completely inhibited at 500 mM.
- Pseudomonas putida ATCC700801 strain was strongly inhibited in the presence of 100 mM protocatechuic acid, and growth was almost completely inhibited at 250 mM.
- Rhodococcus erythropolis ATCC27854 strain was strongly inhibited in the presence of 250 mM protocatechuic acid, and growth was almost completely inhibited at 500 mM.
- Saccharomyces cerevisiae NBRC2376 strain was inhibited from growth in the presence of 250 mM protocatechuic acid and markedly inhibited at 500 mM.
- the Corynebacterium glutamicum R strain was capable of vigorous growth even in the presence of 250-500 mM protocatechuic acid, in which the growth of other strains was markedly or almost completely inhibited.
- Corynebacterium glutamicum has high resistance to protocatechuic acid compared to other microorganisms that have been reported as protocatechuic acid production hosts and representative solvent-resistant bacteria, and therefore has high suitability as a protocatechuic acid production host. It has been shown.
- FIG. 3 shows the glucose consumption of Corynebacterium glutamicum R strain after 24 hours of culture in the presence of various concentrations of protocatechuic acid. As shown in FIG. 3, it can be seen that Corynebacterium glutamicum has a slight decrease in sugar consumption even in the presence of a high concentration of protocatechuic acid.
- Corynebacterium is Corynebacterium glutamicum protocatechuic acid-producing strains of the mixed sugar utilization lines from protocatechuic acid production test Corynebacterium glutamicum R strain by aerobic stationary cell reaction under jar fermentor control of transformants were constructed as a base, PCA1 , PCA2, PCA3, PCA4, PCA5 strains (Example 1 (Table 6)), protocatechuic acid production ability in aerobic stationary cell reaction under the control of jar fermenter (Able Co., Ltd., model: BMJ1L) was confirmed according to the method described below.
- the PCA1 strain was prepared by adding 10 ml of the A liquid medium (phenylalanine, tyrosine, tryptophan 20 ⁇ g / ml each, p-aminobenzoic acid 10 ⁇ g / ml, shikimic acid 3.2 mM, and glucose 4% (each final concentration) ( After inoculation in a test tube), and after inoculation in 10 ml of the A liquid medium (in a test tube) with 10% of glucose added to PCA2, PCA3, PCA4, and PCA5 strains at 33 ° C for 12-16 hours, Aerobic shaking culture was performed.
- the A liquid medium phenylalanine, tyrosine, tryptophan 20 ⁇ g / ml each, p-aminobenzoic acid 10 ⁇ g / ml, shikimic acid 3.2 mM, and glucose 4% (each final concentration)
- Corynebacterium glutamicum for PCA2, PCA3, PCA4, and PCA5 strains glucose 100 g / l, and antifoam (Dishome CB-442) 3 g / l (each final concentration) added, 600 ml
- the A (-UB) liquid medium was inoculated so that OD 610 would be 0.3, and each was cultivated at 33 ° C., pH 7.0 (5 N ammonia water) with a 1000 ml capacity jar fermenter (manufactured by Able Corporation, model: BMJ1L).
- the Corynebacterium glutamicum strain grown under the above conditions is collected by centrifugation (4 ° C, 5000 xg, 10 minutes), and the cells are collected in a BT (-UB) liquid medium [(NH 4 ) 2 SO 4 7 g, KH 2 PO 4 0.5 g, K 2 HPO 4 0.5 g, MgSO 4 ⁇ 7H 2 O 0.5 g, 0.06% (w / v) (Fe 2 SO 4 ⁇ 7H 2 O + 0.042% (w / v) MnSO 4 ⁇ 2H 2 O) 1 ml, 100 ⁇ g / ml thiamine solution 2 ml dissolved in 1 L of distilled water] washed once, then 25 g wet bacteria against the BT (-UB) liquid medium containing 10% glucose / 250 ml (10% of the microbial cells are present in the medium as wet cell weight) and suspended using the 1000 ml jar fermenter at 33 ° C, pH 7.0 (addition of
- the glucose concentration in the reaction solution was measured over time using a glucose sensor (Oji Scientific Instruments, BF-5i), and glucose was added as necessary.
- Aromatic metabolite concentrations in the cell reaction supernatant were determined by high-performance liquid chromatography (Prominence HPLC device (manufactured by Shimadzu Corporation), COSMOSIL Packed column 5C18-AR-II. Acid)].
- Table 8 shows the results of the protocatechuic acid production experiment by the aerobic stationary cell reaction using each strain.
- PCA production amount of each strain 24 hours after the reaction PCA1 strain 273 mM (42.1 g / l), PCA2 strain 153 mM (23.6 g / l), PCA3 strain 515 mM (79.4 g / l), PCA4 The strain was 536 m (82.5 g / l), and the PCA5 strain was 408 m (62.8 g / l).
- the molar yield of sugars for PCA production in each strain was 33.8% for PCA1 strain, 10.0% for PCA2 strain, 34.6% for PCA3 strain, 39.3% for PCA4 strain, and 30.2% for PCA5 strain.
- PCA3 strain into which Corynebacterium glutamicum 3-dehydroshikimate dehydratase gene was introduced and the PCA4 strain into which Corynebacterium halotolerance 3-dehydroshikimate dehydratase gene was introduced showed particularly high PCA productivity.
- these PCA3, PCA4, and PCA5 strains were shown to proliferate vigorously without the addition of supplemental nutrient sources containing aromatic amino acids even when the cells were cultured in a nutrient medium.
- PCA1 strains that depend only on protocatechuic acid production by conversion of 3-dehydroshikimate to protocatechuic acid catalyzed by 3-dehydroshikimate dehydratase also showed a relatively high protocatechuic acid producing ability
- the productivity was inferior to that of PCA3, PCA4, or PCA5 strains that produce PCA from both pathways (a) and (b).
- PCA3 strain shows the requirement for aromatic amino acids and 4-aminobenzoic acid, and this strain grows in nutrient medium. In the case of making them, it was necessary to add those nutrient sources to the medium.
- Example 3 Measurement of enzyme activities of 3-dehydroshikimate dehydratase, chorismate pyruvate lyase, and 4-hydroxybenzoate hydroxylase in protocatechuic acid producing strains PCA1, PCA2, and PCA3
- various enzyme activities of 3-dehydroshikimate dehydratase, chorismate pyruvate lyase, and 4-hydroxybenzoate hydroxylase were measured according to the following methods. Perform the aerobic static cell reaction using jar fermenters of CRZ22 strain, PCA1 strain, PCA2 strain, and PCA3 strain in the same procedure as in Example 1, and collect each strain culture solution 6 hours after the reaction.
- the cells were collected by centrifugation. After washing the cells once with 20 mM Tris-HCl (pH 7.5), 1 ml of a cell disruption buffer (100 mM Tris-HCl (pH 7.5), 20 mM KCl, 20 mM MgCl 2 , 0.1 mM EDTA, and 2 mM DTT)) and crushed using a multi-bead shocker (Yasui Kikai) and glass beads.
- the cell disruption solution was centrifuged at 15000 rpm, 10 min, 4 ° C. to obtain a supernatant fraction as a crude enzyme extract.
- the protein concentration of each crude enzyme extract was quantified using Protein assay kit (Bio-Rad, USA) and BSA (Bovine serum albumin) as a standard. Each enzyme activity in the crude enzyme extract of each strain was measured according to the activity measurement method described above.
- the parental CRZ22 strain did not detect significant activity in any of the three enzyme activities tested. This suggested that these enzymes were weakly expressed or hardly expressed in the strain.
- 3-dehydroshikimate dehydratase (QsuB) activity was detected with the introduction of the 3-dehydroshikimate dehydratase gene (qsuB). Since the aroE gene encoding shikimate dehydrogenase is disrupted in the PCA1 strain, the shikimate pathway is blocked at the enzyme reaction stage. Therefore, it supports that PCA production in the PCA1 strain occurs dependently only on (a) 3-dehydroshikimate dehydratase.
- Example 4 Search for a heterologous gene encoding a competent shikimate dehydratase From the results of Example 2 and Example 3, the productivity of protocatechuic acid by the coryneform bacterial transformant was determined by the enzyme activity of the 3-dehydroshikimate dehydratase gene introduced. It has been shown that the strength is significantly increased by strengthening. On the other hand, in the coryneform bacterial transformants PCA1 and PCA3, the 3-dehydroshikimate dehydratase gene derived from Corynebacterium glutamicum has been introduced, but the more effective 3-dehydroshikimate dehydratase has been introduced. The possibility of being present in microorganisms was also considered.
- 3-dehydroshikimate dehydratase gene qsuB
- shikimate dehydrogenase gene aroE
- Cells grown under the above conditions were glucose 4%, phenylalanine, tyrosine, tryptophan 20 ⁇ g / ml each, p-aminobenzoic acid 10 ⁇ g / ml, shikimic acid 3.2 mM, and kanamycin 50 ⁇ g / ml (each final concentration) was inoculated into 10 ml of the above-mentioned A liquid medium (in a test tube) so that the OD 610 was 0.2, and the culture was shaken aerobically at 33 ° C. for 24 hours.
- protocatechuic acid or a salt thereof can be produced from glucose or the like with practical efficiency using a microorganism.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- Microbiology (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Virology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
Description
従来、プロトカテク酸は主に天然物(農産品)からの抽出法によって製造されている。しかし、このような製造法では天然物原料の生産量が限られることや、天然物からの抽出効率が低いといった問題が存在するため、大量生産が難しい状況にある。
(i) プロトカテク酸は一般的に微生物に対して細胞毒性を示すことが知られており、生産されたプロトカテク酸の毒性により生産性が限定される可能性が考えられた。そこで、これまでに芳香族化合物の生産が報告されている幾つかの微生物について、それらの増殖に及ぼすプロトカテク酸の影響を比較したところ、コリネバクテリウム グルタミカム、エシェリヒア コリ、バチルス サブチリス、シュードモナス プチダ、ロドコッカス エリスロポリスの中では、コリネバクテリウム グルタミカムが最もプロトカテク酸に対する耐性が高いことが示された。具体的には、コリネバクテリウム グルタミカムは、他の微生物の増殖が完全に、もしくは著しく抑制された、500mMという高濃度のプロトカテク酸の存在下においても高い増殖能、及び糖消費能を示した。このように、コリネバクテリウム グルタミカムはプロトカテク酸への耐性が極めて高いため、プロトカテク酸又はその塩の生産に特に好適である。
(ii) コリネ型細菌に、(a)3-デヒドロシキミ酸デヒドラターゼをコードする遺伝子を宿主微生物に導入することによる該酵素活性の強化と、(b)コリスメートピルベートリアーゼをコードする遺伝子、及び4-ヒドロキシ安息香酸ヒドロキシラーゼをコードする遺伝子とを宿主微生物に導入することによる該酵素活性の強化とを組み合わせて行うことにより、(a)のみ行う場合、又は(b)のみ行う場合に比べて、糖類からのプロトカテク酸又はその塩の生産性が相乗的に向上する。
(iii) さらに、(a)と(b)の両者を施したコリネ型細菌形質転換体は、プロトカテク酸又はその塩の生産性が顕著に向上する一方で、芳香族アミノ酸生合成経路は遮断されていないことから、卜リプトファン、チロシン、およびフェニルアラニンといった芳香族アミノ酸やパラアミノ安息香酸の要求性が生じないため、該形質転換体を増殖させるためにこれらの化合物を培地に添加する必要がないという利点を有している。
(iv) この形質転換体は、好気的、かつ実質的に増殖しない条件下で反応を行う場合に、特にプロトカテク酸又はその塩の生産効率が高い。
項1. 下記の(A)、(B)、及び(C)の操作が施されたプロトカテク酸生産能を有する形質転換体。
(A) 3-デヒドロシキミ酸デヒドラターゼ活性の強化
(B) コリスメートピルベートリアーゼ活性の強化
(C) 4-ヒドロキシ安息香酸ヒドロキシラーゼ活性の強化
項2. 3-デヒドロシキミ酸デヒドラターゼ活性の強化が、コリネバクテリウム属、ロドコッカス属、バチルス属、ロドシュードモナス属、アルテロモナス属、マリノバクター属、メチロバクテリウム属、パントエア属、ニューロスポラ属、又はアスペルギルス属に属する微生物由来の3-デヒドロシキミ酸デヒドラターゼ活性を有する酵素をコードする遺伝子の宿主への導入によってもたらされたものである、項1に記載の形質転換体。
項3. 3-デヒドロシキミ酸デヒドラターゼ活性を有する酵素をコードする遺伝子が、コリネバクテリウム グルタミカム (Corynebacterium glutamicum)、コリネバクテリウム ハロトレランス (Corynebacterium halotolerans)、コリネバクテリウム カゼイ (Corynebacterium casei)、コリネバクテリウム エフィシェンス (Corynebacterium efficiens)、アスペルギルス ニガー (Aspergillus niger)、又はアスペルギルス オリゼー (Aspergillus oryzae)の遺伝子である、項2に記載の形質転換体。
項4. 3-デヒドロシキミ酸デヒドラターゼ活性を有する酵素をコードする遺伝子が、下記の(a)または(b)のDNAによってコードされる、項2または3に記載の形質転換体。
(a) 配列番号7、134、135、145、147、又は149の塩基配列からなるDNA
(b) 配列番号7、134、135、145、147、又は149の塩基配列と90%以上の同一性を有する塩基配列からなるDNAであって、3-デヒドロシキミ酸デヒドラターゼ活性を有するポリペプチドをコードするDNA
項5. コリスメートピルベートリアーゼ活性の強化が、プロビデンシア属細菌、又は、クロノバクター属細菌由来のコリスメートピルベートリアーゼ活性を有する酵素をコードする遺伝子の宿主への導入によってもたらされたものである、項1~4のいずれかに記載の形質転換体。
項6. コリスメートピルベートリアーゼ活性の強化が、プロビデンシア ルスティジアニ (Providencia rustigianii)、プロビデンシア スチュアルティ (Providencia stuartii)、又はクロノバクター サカザキ (Cronobacter sakazakii)由来のコリスメートピルベートリアーゼ活性を有する酵素をコードする遺伝子の宿主への導入によってもたらされたものである、項5に記載の形質転換体。
項7. コリスメートピルベートリアーゼ活性の強化が、下記の(c)または(d)のDNAの宿主への導入によってもたらされたものである、項1~6のいずれかに記載の形質転換体。
(c) 配列番号9、128、又は129の塩基配列からなるDNA
(d) 配列番号9、128、又は129の塩基配列と90%以上の同一性を有する塩基配列からなるDNAであって、コリスメートピルベートリアーゼ活性を有するポリペプチドをコードするDNA
項8. 4-ヒドロキシ安息香酸ヒドロキシラーゼ活性の強化が、4-ヒドロキシ安息香酸ヒドロキシラーゼ活性を有する酵素をコードする、コリネバクテリウム グルタミカム (Corynebacterium glutamicum)の遺伝子の宿主への導入によってもたらされたものである、項1~7のいずれかに記載の形質転換体。
項9. 4-ヒドロキシ安息香酸ヒドロキシラーゼ活性の強化が、下記の(e)または(f)のDNAの宿主への導入によってもたらされたものである、項1~8のいずれかに記載の形質転換体。
(e) 配列番号8の塩基配列からなるDNA
(f) 配列番号8の塩基配列と90%以上の同一性を有する塩基配列からなるDNAであって、4-ヒドロキシ安息香酸ヒドロキシラーゼ活性を有するポリペプチドをコードするDNA
項10. プロトカテク酸3,4-ジオキシゲナーゼ活性が消失しているか、阻害されているか、または減少している、項1~9のいずれかに記載のコリネ型細菌形質転換体。
項11. 3-デオキシ-D-アラビノ-ヘプツロソネート-7-リン酸(DAHP)シンターゼ、3-デヒドロキナ酸シンターゼ、3-デヒドロキナ酸デヒドラターゼ、シキミ酸デヒドロゲナーゼ、シキミ酸キナーゼ、5-エノールピルビルシキミ酸3-リン酸(EPSP)シンターゼ、及びコリスミ酸シンターゼからなる酵素群より選ばれる少なくとも一つの酵素活性が強化されている項1~10のいずれかに記載の形質転換体。
項12. DAHPシンターゼ活性の強化が下記の(g)または(h)のDNAの宿主への導入によってもたらされたものであり、3-デヒドロキナ酸シンターゼ活性の強化が下記の(i)または(j)のDNAの宿主への導入によってもたらされたものであり、3-デヒドロキナ酸デヒドラターゼ活性の強化が下記の(k)または(l)のDNAの宿主への導入によってもたらされたものであり、シキミ酸デヒドロゲナーゼ活性の強化が下記の(m)または(n)のDNAの宿主への導入によってもたらされたものであり、シキミ酸キナーゼ活性の強化が下記の(o)または(p)のDNAの宿主への導入によってもたらされたものであり、EPSPシンターゼ活性の強化が下記の(q)または(r)のDNAの宿主への導入によってもたらされたものであり、コリスミ酸シンターゼ活性の強化が下記の(s)または(t)のDNAの宿主への導入によってもたらされたものである、項11に記載の形質転換体。
(g) 配列番号2の塩基配列からなるDNA
(h) 配列番号2と90%以上の同一性を有する塩基配列からなるDNAであって、DAHPシンターゼ活性を有するポリペプチドをコードするDNA
(i) 配列番号153の塩基配列からなるDNA
(j) 配列番号153と90%以上の同一性を有する塩基配列からなるDNAであって、3-デヒドロキナ酸シンターゼ活性を有するポリペプチドをコードするDNA
(k) 配列番号5の塩基配列からなるDNA
(l) 配列番号5と90%以上の同一性を有する塩基配列からなるDNAであって、3-デヒドロキナ酸デヒドラターゼ活性を有するポリペプチドをコードするDNA
(m) 配列番号6の塩基配列からなるDNA
(n) 配列番号6と90%以上の同一性を有する塩基配列からなるDNAであって、シキミ酸デヒドロゲナーゼ活性を有するポリペプチドをコードするDNA
(o) 配列番号154の塩基配列からなるDNA
(p) 配列番号154と90%以上の同一性を有する塩基配列からなるDNAであって、シキミ酸キナーゼ活性を有するポリペプチドをコードするDNA
(q) 配列番号155の塩基配列からなるDNA
(r) 配列番号155と90%以上の同一性を有する塩基配列からなるDNAであって、EPSPシンターゼ活性を有するポリペプチドをコードするDNA
(s) 配列番号156の塩基配列からなるDNA
(t) 配列番号156と90%以上の同一性を有する塩基配列からなるDNAであって、コリスミ酸シンターゼ活性を有するポリペプチドをコードするDNA
項13. トランスケトラーゼ活性、及びトランスアルドラーゼ活性からなる群より選ばれる少なくとも一つの活性が強化されている、項1~12のいずれかに記載の形質転換体。
項14. トランスケトラーゼ活性の強化が下記の(u)又は(v)のDNAの導入によるものであり、トランスアルドラーゼ活性の強化が下記の(w)又は(x)のDNAの導入によるものである、項13に記載の形質転換体。
(u) 配列番号151の塩基配列からなるDNA
(v) 配列番号151と90%以上の同一性を有する塩基配列からなるDNAであって、トランスケトラーゼをコードするDNA
(w) 配列番号152の塩基配列からなるDNA
(x) 配列番号152と90%以上の同一性を有する塩基配列からなるDNAであって、トランスアルドラーゼをコードするDNA
項15. 宿主がコリネ型細菌である、項1~14の何れかに記載の形質転換体。
項16. グルコース、及び、キシロース、アラビノース、及びセロビオースからなる群より選ばれる少なくとも1種の糖の同時利用能を有する、項15に記載の形質転換体。
項17. 宿主のコリネ型細菌が、コリネバクテリウム属細菌である、項15、又は16に記載の形質転換体。
項18. 宿主のコリネバクテリウム属細菌がコリネバクテリウム グルタミカムである、項17に記載の形質転換体。
項19. 宿主のコリネバクテリウム グルタミカムが、コリネバクテリウム グルタミカムR(FERM BP-18976)、ATCC13032、又は、ATCC13869である、項18に記載のコリネ型細菌形質転換体。
項20. コリネバクテリウム グルタミカム PCA4 (受託番号:NITE BP-02217)
項21. 項1~20のいずれかに記載の形質転換体を、糖類を含む反応液中で培養してプロトカテク酸又はその塩を生産させる工程を含むプロトカテク酸又はその塩の製造方法。
項22. 好気的、かつ形質転換体が増殖しない条件下で形質転換体を培養する、項21に記載の方法。
なお、コリネ型細菌は上記3種の酵素のうち、3-デヒドロシキミ酸デヒドラターゼ、及び4-ヒドロキシ安息香酸ヒドロキシラーゼをコードする遺伝子を染色体上に有しているが、コリスメートピルベートリアーゼをコードする遺伝子は有していない。そこで、本発明の実施例においては、高活性なコリスメートピルベートリアーゼをコードすることを我々が見出していた、プロビデンシア ルスティジアニ(Providencia rustigianii)由来の遺伝子を宿主のコリネ型細菌に導入することにより、(b)のプロトカテク酸生合成経路をコリネ型細菌において機能させた。
一般に微生物はプロトカテク酸のような芳香族化合物の細胞毒性により増殖が阻害されるため、微生物を用いて効率的にプロトカテク酸を製造することは困難であった。しかし、コリネ型細菌は、プロトカテク酸を含む芳香族化合物に対する耐性が非常に高いため、本発明の形質転換体を用いれば、高濃度のプロトカテク酸又はその塩を効率よく生産することができる。また、コリネ型細菌は、大腸菌とは異なりエンドトキシンを生成しないため、生産物へのエンドトキシンの残留を懸念する必要がない。また、コリネ型細菌は、培養槽に高細胞密度に充填するとともに、増殖を制限した条件下でも溶菌せずにプロトカテク酸又はその塩の生成反応が進むため、原料の糖が増殖のために消費されずプロトカテク酸又はその塩の収率が高くなる。また増殖を制限した条件下では、微生物の増殖に一般的に求められる芳香族アミノ酸や4-ヒドロキシ安息香酸などを培養液に添加する必要がなく、その分、生産コストを抑えることができる。
本発明の理解を容易にするため、図1に、コリネ型細菌形質転換体におけるプロトカテク酸生合成経路を模式的に図示した。
(1)プロトカテク酸又はその塩の生産能を有する形質転換体
宿主
本発明において、プロトカテク酸を生産する能力を有する微生物であれば、いずれも宿主として用いることができる。
好適な宿主微生物として、コリネバクテリウム属細菌、エシェリヒア属細菌(特に、エシェリヒア コリ)、バチルス属細菌(特にバチルス サブチリス)、シュードモナス属細菌(特に、シュードモナス プチダ)、ブレビバクテリウム属細菌、ストレプトコッカス属細菌、ラクトバチルス属細菌、ロドコッカス属細菌(特にロドコッカス エリスロポリス、ロドコッカス オパカス)、ストレプトマイセス属細菌、サッカロマイセス属酵母(特に、サッカロマイセス セレビシアエ)、クライベロマイセス属酵母、シゾサッカロマイセス属酵母、ヤロウィア属酵母、トリコスポロン属酵母、ロドスポリジウム酵母、ピキア属酵母、キャンディダ属酵母、ノイロスポラ属カビ、アスペルギルス属カビ、トリコデルマ属カビなどが挙げられる。
中でも、プロトカテク酸またはその塩の生産効率の点で、宿主としてコリネ型細菌を用いることが好ましい。
コリネ型細菌とは、バージーズ・マニュアル・デターミネイティブ・バクテリオロジー〔Bergey's Manual of Determinative Bacteriology、Vol. 8、599(1974)〕に定義されている一群の微生物であり、通常の好気的条件で増殖するものならば特に限定されるものではない。具体例を挙げれば、コリネバクテリウム属菌、ブレビバクテリウム属菌、アースロバクター属菌、マイコバクテリウム属菌、マイクロコッカス属菌等が挙げられる。コリネ型細菌の中ではコリネバクテリウム属菌が好ましい。
中でも、安全でかつプロトカテク酸の生産性が高い点で、コリネバクテリウム グルタミカムが好ましい。好適な菌株として、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)R株(FERM BP-18976)、ATCC13032株、ATCC13869株、ATCC13058株、ATCC13059株、ATCC13060株、ATCC13232株、ATCC13286株、ATCC13287株、ATCC13655株、ATCC13745株、ATCC13746株、ATCC13761株、ATCC14020株、ATCC31831株、MJ-233(FERM BP-1497)、MJ-233AB-41(FERM BP-1498)等が挙げられる。これらのコリネバクテリウム グルタミカム株は、ブダペスト条約の下で国際寄託されており、公に利用可能である。
中でも、R株(FERM BP-18976)、ATCC13032株、ATCC13869株が好ましい。
アースロバクター属菌としては、アースロバクター グロビフォルミス(Arthrobacter globiformis)(例えばATCC8010株、ATCC4336株、ATCC21056株、ATCC31250株、ATCC31738株、ATCC35698株)等が挙げられる。
マイコバクテリウム属菌としては、マイコバクテリウム ボビス(Mycobacterium bovis)(例えばATCC19210株、ATCC27289株)等が挙げられる。
マイクロコッカス属菌としては、マイクロコッカス フロイデンライヒ(Micrococcus freudenreichii)(例えばNo. 239株(FERM P-13221))、マイクロコッカス ルテウス(Micrococcus leuteus)(例えばNo. 240株(FERM P-13222))、マイクロコッカス ウレアエ(Micrococcus ureae)(例えばIAM1010株)、マイクロコッカス ロゼウス(Micrococcus roseus)(例えばIFO3764株)等が挙げられる。
これらのブレビバクテリウム属、アースロバクター属、マイコバクテリウム属、及びマイクロコッカス属の菌株は、ブダペスト条約の下で国際寄託されており、公に利用可能である。
このような遺伝子破壊株は、遺伝子工学的手法により常法に従い作製できる。例えば、WO2005/010182A1に、乳酸デヒドロゲナーゼ破壊株、及びその作製方法が記載されている。
本発明のプロトカテク酸を効率よく生成する形質転換体は、宿主菌株において、3-デヒドロシキミ酸デヒドラターゼ、コリスメートピルベートリアーゼ、及び4-ヒドロキシ安息香酸ヒドロキシラーゼの各酵素活性を強化することにより得ることができる。
3-デヒドロシキミ酸デヒドラターゼは、3-デヒドロシキミ酸からプロトカテク酸を生成する反応を触媒する。コリスメートピルベートリアーゼは、コリスミ酸から4-ヒドロキシ安息香酸を生成する反応を触媒する。また、4-ヒドロキシ安息香酸ヒドロキシラーゼは、4-ヒドロキシ安息香酸の芳香環の3位の炭素原子を水酸化することにより、プロトカテク酸を生成する反応を触媒する。
宿主としてコリネ型細菌を用いる場合、本菌は染色体上に、3-デヒドロシキミ酸デヒドラターゼ遺伝子、及び4-ヒドロキシ安息香酸ヒドロキシラーゼ遺伝子を有しているが、コリスメートピルベートリアーゼ遺伝子は有していない。また、3-デヒドロシキミ酸デヒドラターゼ遺伝子、及び4-ヒドロキシ安息香酸ヒドロキシラーゼ遺伝子についても、これらの遺伝子は特定の培養条件下(プロトカテク酸、あるいは特定の芳香族化合物の存在下)でのみ発現が誘導される可能性が考えられる。従って、上記3遺伝子は、用いる培養条件で高発現をもたらす適切なプロモーターの制御下に置いた融合遺伝子として宿主のコリネ型細菌に導入することが好ましい。
3-デヒドロシキミ酸デヒドラターゼ遺伝子としては、コリネバクテリウム属細菌(特に、コリネバクテリウム グルタミカム (Corynebacterium glutamicum)、コリネバクテリウム カゼイ (Corynebacterium casei)、コリネバクテリウム エフィシエンス (Corynebacterium efficience)、コリネバクテリウム ハロトレランス (Corynebacterium halotolerans))、ロドコッカス属細菌 (特に、ロドコッカス オパカス (Rhodococcus opacus))、マイコバクテリウム属細菌 (特に、マイコバクテリウム スメグマティス(Mycobacterium smegmatis))、バチルス属細菌 (特に、バチルス チューリンゲンシス(Bacillus thuringiensis))、グルコノバクター属細菌 (特に、グルコノバクター オキシダンス (Gluconobacter oxydans))、ロドシュードモナス属細菌 (特に、ロドシュードモナス パルストリス (Rhodopseudomonas palustris))、アルテロモナス属細菌 (特に、アルテロモナス マクレオディ (Alteromonas macleodii))、マリノバクター属細菌 (特に、マリノバクター ハイドロカーボノクラスティカス (Marinobacter hydrocarbonoclasticus))、メチロバクテリウム属細菌 (特に、メチロバクテリウム エキソトルキエンス (Methylobacterium extorquens))、シュードモナス属細菌 (特に、シュードモナス プチダ (Pseudomonas putida))、アシネトバクター属細菌 (特に、アシネトバクター ベイリー (Acinetobacter baylyi))、パントエア属細菌 (特に、パントエア アナナティス (Pantoea ananatis))、ニューロスポラ属菌(特に、ニューロスポラ クラッサ (Neurospora crassa))、及び、アスペルギルス属菌(特に、アスペルギルス オリゼー (Aspergillus oryzae)、アスペルギルス ニガー (Aspergillus niger))の遺伝子などが挙げられる。
中でも、コリネバクテリウム グルタミカム、コリネバクテリウム カゼイ、コリネバクテリウム エフィシエンス、コリネバクテリウム ハロトレランス、ロドコッカス オパカス、メチロバクテリウム エキソトルキエンス、ニューロスポラ クラッサ、アスペルギルス ニガー、及びアスペルギルス オリゼーの遺伝子が好ましく、中でも、コリネバクテリウム グルタミカム、及び、コリネバクテリウム ハロトレランスの遺伝子がより好ましい。
配列番号7のコリネバクテリウム グルタミカムの3-デヒドロシキミ酸デヒドラターゼ遺伝子はqsuBと称される。
また、配列番号7、及び、配列番号134~150のいずれかの塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNAであり、かつ3-デヒドロシキミ酸デヒドラターゼ活性を有するポリペプチドをコードするDNAも使用できる。
また、本発明において、形質転換体の3-デヒドロシキミ酸デヒドラターゼ活性が強化されていることは、該形質転換体の細胞抽出液中の3-デヒドロシキミ酸デヒドラターゼ活性を測定することにより確認する。
コリスメートピルベートリアーゼ遺伝子の由来は特に限定されないが、プロトカテク酸又はその塩の生産効率が良い点で、プロビデンシア属細菌、または、クロノバクター属細菌の遺伝子が好ましく、中でも、プロビデンシア ルスティジアニ(Providencia rustigianii)、プロビデンシア ステュアルティ(Providencia stuartii)、クロノバクター サカザキ(Cronobacter sakazakii)の遺伝子がより好ましく、プロビデンシア ルスティジアニ(Providencia rustigianii)の遺伝子がさらにより好ましい。
配列番号9のプロビデンシア ルスティジアニイ(Providencia rustigianii)のコリスメートピルベートリアーゼ遺伝子はubiCと称される。
また、配列番号9、128、及び129のいずれかの塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNAであり、かつコリスメートピルベートリアーゼ活性を有するポリペプチドをコードするDNAも使用できる。
また、本発明において、形質転換体のコリスメートピルベートリアーゼ活性が強化されていることは、該形質転換体の細胞抽出液中のコリスメートピルベートリアーゼ活性の上昇により確認する。
4-ヒドロキシ安息香酸ヒドロキシラーゼは、フェノールモノオキシゲナーゼとも称される。4-ヒドロキシ安息香酸ヒドロキシラーゼ遺伝子の由来は特に限定されないが、プロトカテク酸又はその塩の生産効率が良い点で、コリネバクテリウム属細菌の遺伝子、中でも、コリネバクテリウム グルタミカム(Corynebacterium glutamicum)の遺伝子が好ましい。
また、配列番号8の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNAであり、かつ4-ヒドロキシ安息香酸ヒドロキシラーゼ活性を有するポリペプチドをコードするDNAも使用できる。
また、本発明において、形質転換体の4-ヒドロキシ安息香酸ヒドロキシラーゼ活性が強化されていることは、該形質転換体の細胞抽出液中の4-ヒドロキシ安息香酸ヒドロキシラーゼ活性の上昇により確認する。
本発明の形質転換体は、さらに、3-デオキシ-D-アラビノ-ヘプツロソネート-7-リン酸(DAHP)シンターゼ活性が増強されていることが好ましい。DAHPシンターゼは、エリスロース-4-リン酸、及びホスホエノールピルビン酸とから、芳香族化合物生合成経路の初発代謝産物であるDAHPを生成する酵素である。
DAHPシンターゼ活性の増強は、DAHPシンターゼ遺伝子の宿主微生物への導入、又は、宿主微生物の染色体上のDAHPシンターゼ遺伝子(制御配列ないしは領域、遺伝子コード領域、またはその両者)への変異導入や配列置換によりもたらすことができる。このうち、DAHPシンターゼ遺伝子の宿主微生物への導入によりDAHPシンターゼ活性を増強することが簡便で効率が良い。
エシェリヒア コリ由来のDAHPシンターゼ遺伝子としては、配列番号2の塩基配列からなるDNA(aroG S180F)がさらにより好ましい。この遺伝子は、エシェリヒア コリ由来のDAHPシンターゼ遺伝子の一つであるaroG遺伝子において、この遺伝子がコードするアミノ酸配列の180番目のセリンをフェニルアラニンに変異させる変異(S180F)が導入された遺伝子であり、その遺伝子産物が芳香族アミノ酸を含む芳香族化合物によるフィードバック阻害への耐性、及び、高いDAHPシンターゼ活性を示すことを、本発明者らが比較検討により見出している(未発表)。
本発明の形質転換体は、さらに、トランスケトラーゼ活性、または、トランスケトラーゼ活性とトランスアルドラーゼ活性が増強されていることが好ましい。
このように、トランスケトラーゼ、及びトランスアルドラーゼは芳香族化合物生合成の前駆体の一つであるエリスロース-4-リン酸の生成に重要な役割を果たしている。従って、これらの酵素活性を強化することで細胞内のエリスロース-4-リン酸の供給が増大し、その結果、芳香族化合物生合成経路への代謝フラックスが高まり、プロトカテク酸の生産性向上をもたらすと考えられる。
トランスケトラーゼ活性、及びトランスアルドラーゼ活性の強化は、宿主微生物へのトランスケトラーゼ遺伝子、及びトランスアルドラーゼ遺伝子の導入、又は、宿主微生物の染色体上のトランスケトラーゼ遺伝子、または、トランスアルドラーゼ遺伝子の制御配列、遺伝子コード領域、またはその両者への変異導入や配列置換によりもたらすことができる。このうち、トランスケトラーゼ遺伝子、及びトランスアルドラーゼ遺伝子の宿主微生物への導入により該酵素活性を増強することが簡便で効率が良い。
コリネバクテリウム グルタミカムのトランスケトラーゼ遺伝子としては、配列番号151の塩基配列からなるDNA(tkt)が挙げられ、コリネバクテリウム グルタミカムのトランスアルドラーゼ遺伝子としては、配列番号152の塩基配列からなるDNA(tal)が挙げられる。
また、本発明では、配列番号151又は152と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつそれぞれ、トランスケトラーゼ活性、又はトランスアルドラーゼ活性を有するポリペプチドをコードするDNAも用いることができる。
また、本発明において、形質転換体のトランスケトラーゼ活性が強化されていることは、該形質転換体の細胞抽出液中のトランスケトラーゼ活性値の上昇により確認する。
また、本発明において、形質転換体のトランスアルドラーゼ活性が強化されていることは、該形質転換体の細胞抽出液中のトランスアルドラーゼ活性値の上昇により確認する。
本発明の形質転換体は、さらに、DAHPシンターゼ以降のシキミ酸経路上の一連の酵素群、すなわち、3-デヒドロキナ酸シンターゼ、3-デヒドロキナ酸デヒドラターゼ、シキミ酸デヒドロゲナーゼ、シキミ酸キナーゼ、5-エノールピルビルシキミ酸3-リン酸(EPSP)シンターゼ、及びコリスメートシンターゼの各酵素活性のいずれか一つ以上が強化されていることが好ましく、またこれらの酵素活性のすべてが強化されていることがより好ましい。これらの1以上の酵素活性の強化により、DAHPからコリスミ酸への代謝変換が促進される。
3-デヒドロキナ酸シンターゼは、DAHPから3-デヒドロキナ酸への変換を触媒する酵素であり、3-デヒドロキナ酸デヒドラターゼは3-デヒドロキナ酸から3-デヒドロシキミ酸への変換を触媒する酵素であり、シキミ酸デヒドロゲナーゼは3-デヒドロシキミ酸からシキミ酸への変換を触媒する酵素であり、シキミ酸キナーゼはシキミ酸からシキミ酸-3-リン酸への変換を触媒する酵素であり、EPSPシンターゼは、シキミ酸-3-リン酸からEPSPへの変換を触媒する酵素であり、また、コリスメートシンターゼはEPSPからコリスミ酸への変換を触媒する酵素である。
コリネバクテリウム グルタミカム由来の上記各酵素遺伝子としては、3-デヒドロキナ酸シンターゼ遺伝子は、配列番号153からなるDNA (aroB)が挙げられ、3-デヒドロキナ酸デヒドラターゼ遺伝子は、配列番号5からなるDNA (aroD)が挙げられ、シキミ酸デヒドロゲナーゼ遺伝子は、配列番号6からなるDNA (aroE)が挙げられ、シキミ酸キナーゼ遺伝子は、配列番号154からなるDNA (aroK)が挙げられ、EPSPシンターゼ遺伝子は、配列番号155からなるDNA (aroA)が挙げられ、コリスメートシンターゼ遺伝子は、配列番号156からなるDNA (aroC)が挙げられる。
また、本発明では、配列番号153、5、6、154、155、又は配列番号156と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズするDNAであり、かつ、それぞれ、3-デヒドロキナ酸シンターゼ活性、3-デヒドロキナ酸デヒドラターゼ活性、シキミ酸デヒドロゲナーゼ活性、シキミ酸キナーゼ活性、EPSPシンターゼ活性、又は、コリスメートシンターゼ活性を有するポリペプチドをコードするDNAも使用できる。
また、本発明において、形質転換体の3-デヒドロキナ酸シンターゼ活性が強化されていることは、該形質転換体の細胞抽出液中の3-デヒドロキナ酸シンターゼ活性の上昇により確認する。
また、本発明において、形質転換体の3-デヒドロキナ酸デヒドラターゼ活性が強化されていることは、該形質転換体の細胞抽出液中の3-デヒドロキナ酸デヒドラターゼ活性の上昇により確認する。
また、本発明において、形質転換体のシキミ酸デヒドロゲナーゼ活性が強化されていることは、該形質転換体の細胞抽出液中のシキミ酸デヒドロゲナーゼ活性の上昇により確認する。
また、本発明において、形質転換体のシキミ酸キナーゼ活性が強化されていることは、該形質転換体の細胞抽出液中のシキミ酸キナーゼ活性の上昇により確認する。
また、本発明において、形質転換体のEPSPシンターゼ活性が強化されていることは、該形質転換体の細胞抽出液中のEPSPシンターゼ活性の上昇により確認する。
また、本発明において、形質転換体のコリスメートシンターゼ活性が強化されていることは、該形質転換体の細胞抽出液中のコリスメートシンターゼ活性の上昇により確認する。
本発明の形質転換体は、プロトカテク酸3,4-ジオキシゲナーゼ活性が消失、阻害、又は、減少していることが好ましい。
プロトカテク酸3,4-ジオキシゲナーゼは、プロトカテク酸の異化代謝経路において、プロトカテク酸の開環によるβ-カルボキシ-cis,cisムコン酸への変換を触媒する酵素である。プロトカテク酸3,4-ジオキシゲナーゼ活性は、染色体上のプロトカテク酸3,4-ジオキシゲナーゼ遺伝子の破壊、欠失、又は変異により、消失、阻害、または減少させることができる。
コリネバクテリウム グルタミカムのプロトカテク酸3,4-ジオキシゲナーゼ遺伝子としては、pcaHGが挙げられる。
プロトカテク酸3,4-ジオキシゲナーゼ活性は、33℃において、100 mMトリス塩酸バッファー(pH 7.5)、及び1 mM プロトカテク酸からなる反応用混合液に被験酵素液を添加することで反応を開始し、プロトカテク酸に起因する290 nmの吸光(吸光係数=2800/M・cm)の減少をBeckman DU800 spectrophotometer (ベックマン・コールター社製)によってモニターし、反応初速度から酵素活性を算出する。33℃において、1分間に1μmolのプロトカテク酸が消失する活性を、1 unitのプロトカテク酸3,4-ジオキシゲナーゼ活性とし、該酵素活性が検出される場合に、プロトカテク酸3,4-ジオキシゲナーゼ活性があると判定する。
ホスホエノールピルビン酸:糖 ホスホトランスフェラーゼシステム(PTS)は、グルコースなどの糖の細胞内への取り込みと糖のリン酸化を共役して行うことを特徴とする、原核生物のみに存在する糖輸送機構である。大腸菌やコリネ型細菌において、糖の細胞内への取り込みにはPTSが主要な役割を果たしている。PTSは、共通コンポーネントであるEnzyme I(エンザイムI)(PEP Protein kinase)、HPr (Histidine-phosphorylatable protein;ヒスチジン-ホスフォリラタブルプロテイン)、及び各種糖の特異的な輸送に関わる膜タンパク質である Enzyme II(エンザイムII)によって構成され、解糖系由来のホスホエノールピルビン酸(PEP)をリン酸供与体とし、これら構成因子間のリン酸リレーを介して糖をリン酸化体として細胞内へ輸送するシステムである。一方、PTSはグルコースの細胞内への輸送に伴って、芳香族化合物の共通前駆体の一つであるPEPをグルコース-6-リン酸生成のためのリン酸供与基として消費してしまう。PEPは芳香族化合物生産において鍵となる前駆体化合物であり、プロトカテク酸を含む芳香族化合物高生産のためにはPTSのような競合代謝経路によるPEPの消費を抑え、芳香族化合物生産経路へのPEPの利用能を高めることが重要となる。本発明の形質転換体においては、PTSを介した糖の取り込みが不活性化され、同時に、糖輸送に伴ってPEPを消費しない、PTSとは異なる糖輸送系(非PTS糖輸送系)を介した糖利用能が付与されていることが好ましい。
PTSをコードする遺伝子としては、Enzyme IをコードするptsI、HprをコードするptsH、及び、Enzyme IIをコードするptsG等が挙げられる。PTS依存的なグルコース輸送を阻害するためには、これらの遺伝子の1以上が破壊、欠失、又は変異していればよく、PTSの共通コンポーネントであるHprタンパク質をコードするptsH遺伝子が破壊され、欠失し、又は変異していることが好ましい。
コリネバクテリウム グルタミカムにおいては、PTSとは異なる糖輸送体であり、糖の細胞内輸送に伴ってPEPを消費しない、非PTSグルコース輸送体が存在する。pts遺伝子の破壊等によってPTSを介した糖の取り込みが阻害されたコリネバクテリウム グルタミカムは、グルコースを単一炭素源として増殖できなくなるか、または増殖能が著しく低下するが、その株に対して非PTSグルコース輸送体、及びグルコキナーゼを共に高発現させると、グルコースを単一炭素源とした増殖が回復する(Ikeda, M., et al., Identification and application of a different glucose uptake system that functions as an alternative to the phosphotransferase system in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 90: 1443-1451, Lindner, S. N., et al., Phosphotransferase system-independent glucose utilization in Corynebacterium glutamicum by inositol permeases and glucokinases. Appl. Environ. Microbiol. 77: 3571-3581)。
本発明においては、PTSによる糖輸送が遮断されたコリネバクテリウム グルタミカムにおいて、グルコースの細胞内への取り込み、及び、グルコースを炭素源とする菌の増殖が、非PTSグルコース輸送体活性、及びグルコキナーゼ活性の強化によって改善されていることが望ましい。このことにより、グルコースの輸送に伴うPEPの消費を回避し、より多くのPEPをシキミ酸等の芳香族化合物生合成のために供給することが可能になると考えられる。
これらのうち、非PTSグルコース輸送体遺伝子の導入によりグルコースの取り込み活性を強化することが簡便で効率がよい。
導入する非PTSグルコース輸送体遺伝子の由来は特に限定されないが、シキミ酸生産効率が良い点で、コリネバクテリウム属細菌、中でもコリネバクテリウム グルタミカムの遺伝子であることが好ましい。
また、本発明では、配列番号157と90%以上、中でも95%以上、中でも98%以上の同一性を有する塩基配列からなるDNAであって、イノシトールトランスポーター活性を有するポリペプチドをコードするDNAも用いることができる。
また、本発明では、配列番号159と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつイノシトールトランスポーター活性を有するポリペプチドをコードするDNAも用いることができる。
また、本発明において、形質転換体の非PTSグルコース輸送体活性が強化されていることは、PTSによる糖輸送を欠損した該形質転換体におけるグルコースを炭素源とする生育、または、グルコース消費速度が、該形質転換体において、遺伝子導入前と比べて高まっていることを指標として確認する。
非PTSグルコース輸送体によって細胞内に取り込まれたグルコースが中央代謝系で代謝されるためには、グルコキナーゼによって、グルコース-6-リン酸に変換される必要がある。グルコキナーゼは、グルコースからグルコース-6-リン酸への変換を触媒する酵素である。
本発明においては、非PTSグルコース輸送体依存的なグルコース輸送の強化と同時に、グルコキナーゼ活性が強化されていることが好ましい。このことにより、グルコースの細胞内への取り込みとそれに引き続く解糖系やペントース・リン酸経路での糖代謝が促進されることを特徴としている。
コリネバクテリウム グルタミカム R株の染色体上には、グルコキナーゼ遺伝子として、cgR_2067 (glk1)、cgR_2552 (glk2)、及びcgR_1739 (ppgK)の少なくとも3種が存在する。このうち、cgR_2067 (glk1)、及びcgR_2552(glk2)はATPを良好な基質とするグルコキナーゼと高い相同性を有し、cgR_1739 (ppgK)はポリリン酸を良好な基質とするグルコキナーゼと高い相同性を有している。本発明においては、これらのグルコキナーゼ遺伝子のうちの1種以上が強化されていることが好ましく、3種すべてが強化されていることがより好ましい。
導入するグルコキナーゼ遺伝子の由来は特に限定されないが、シキミ酸生産効率が良い点で、コリネバクテリウム属細菌、中でもコリネバクテリウム グルタミカムの遺伝子であることが好ましい。
コリネバクテリウム グルタミカム由来のグルコキナーゼ遺伝子としては、配列番号158、159、及び、160の塩基配列からなるDNA(それぞれ、glk1、glk2、及びppgK)が挙げられる。
また、本発明では、配列番号158、159、又は、160と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつグルコキナーゼ活性を有するポリペプチドをコードするDNAも用いることができる。
また、本発明において、形質転換体のグルコキナーゼ活性が強化されていることは、該形質転換体の細胞抽出液中のグルコキナーゼ活性を測定することにより確認する。
グリセルアルデヒド-3-リン酸デヒドロゲナーゼ(GAPDH)は、グリセルアルデヒド3-リン酸を1,3-ビスホスホグリセリン酸に変換する酵素である。
本発明の形質転換体においては、GAPDH活性が強化されていることが好ましい。
そこで、本発明においては、形質転換体のGAPDH活性を強化することによって、糖代謝の律速を解除して糖消費を促進させるとともに、プロトカテク酸生産能力を向上させていることが望ましい。
中でも、GAPDH活性の強化は、GAPDH遺伝子の導入により行うことが簡便で効率がよい。
導入するGAPDH遺伝子の由来は特に限定されないが、プロトカテク酸生産効率が良い点で、コリネバクテリウム属細菌、中でもコリネバクテリウム グルタミカムの遺伝子であることが好ましい。
コリネバクテリウム グルタミカム由来のGAPDH遺伝子としては、配列番号161の塩基配列からなるDNA(gapA)が挙げられる。
また、本発明では、配列番号161と相補的な塩基配列からなるDNAとストリンジェントな条件でハイブリダイズし、かつGAPDH活性を有するポリペプチドをコードするDNAも用いることができる。
また、本発明において、コリネ型細菌形質転換体のGAPDH活性が強化されていることは、該コリネ型細菌形質転換体の細胞抽出液中のGAPDH活性を測定することにより確認する。
DHAP脱リン酸化酵素は、DHAPの脱リン酸化によるジヒドロキシアセトン (DHA)への変換を触媒する酵素である。
本発明の形質転換体は、DHAP脱リン酸化酵素活性が消失、阻害、又は、減少していることが好ましい。上述のように、高発現させた非PTSグルコース輸送体及びグルコキナーゼに依存的に糖を細胞内に取り込み利用するコリネ型細菌は、副生物としてDHAを高生成する。このため、DHA生成経路の遮断により、プロトカテク酸等の芳香族化合物生成により多くの炭素を供給することが可能になる。
コリネバクテリウム グルタミカムはDHAPの脱リン酸化を触媒する酵素として、HAD(haloacid dehalogenase)スーパーファミリーホスファターゼ(HdpA)を有している(Jojima, T. et. al., Identification of a HAD superfamily phosphatase, HdpA, involved in 1,3-dihydroxyacetone production during sugar catabolism in Corynebacterium glutamicum. FEBS. Lett. 586: 4228-4232 (2012))。コリネバクテリウム グルタミカムのDHAP脱リン酸化酵素活性は、染色体上のDHAP脱リン酸化酵素遺伝子 (hdpA)の破壊、欠失、又は変異により、消失、阻害、または減少させることができる。
宿主微生物への遺伝子導入により、それがコードするタンパク質ないしは酵素の活性を強化させる場合、各タンパク質ないしは酵素をコードするDNAは、各々宿主の染色体に組み込むか、または、宿主で増幅できる適切なベクターにクローニングして宿主に導入すればよい。
プラスミドベクターは、コリネ型細菌内で自律複製機能を司る遺伝子を含むものであれば良い。その具体例としては、ブレビバクテリウム ラクトファーメンタム(Brevibacterium lactofermentum)2256由来のpAM330〔特開昭58-67699号公報〕、〔Miwa, K. et al., Cryptic plasmids in glutamic acid-producing bacteria. Agric. Biol. Chem. 48:2901-2903(1984)〕及び〔Yamaguchi, R. et al., Determination of the complete nucleotide sequence of the Brevibacterium lactofermentum plasmid pAM330 and the analysis of its genetic information. Nucleic Acids Symp. Ser. 16:265-267(1985)〕、コリネバクテリウム グルタミカム ATCC3058由来のpHM1519 〔Miwa, K. et al., Cryptic plasmids in glutamic acid-producing bacteria. Agric. Biol. Chem. 48:2901-2903(1984)〕及びpCRY30 〔Kurusu, Y. et al., Identification of plasmid partition function in coryneform bacteria. Appl. Environ. Microbiol. 57:759-764 (1991)〕、コリネバクテリウム グルタミカム T250由来のpCG4〔特開昭57-183799号公報〕、〔Katsumata, R. et al., Protoplast transformation of glutamate-producing bacteria with plasmid DNA. J. Bacteriol.、159:306-311 (1984)〕、pAG1、pAG3、pAG14、pAG50〔特開昭62-166890〕、pEK0、pEC5、pEKEx1 〔Eikmanns, B.J. et al., A family of Corynebacterium glutamicum/Escherichia coli shuttle vectors for cloning, controlled gene expression, and promoter probing. Gene, 102:93-98 (1991)〕等が挙げられる。
好ましいプロモーターとしては、コリネバクテリウム グルタミカムR由来のグリセルアルデヒド3-リン酸デヒドロゲナーゼA遺伝子(gapA)のプロモーターPgapA、マレートデヒドロゲナーゼ遺伝子(mdh)のプロモーターPmdh、ラクテートデヒドロゲナーゼA遺伝子(ldhA)のプロモーターPldhA等が挙げられ、中でも、PgapAが好ましい。
好ましいターミネーターとしては、大腸菌rRNAオペロンのrrnB T1T2 ターミネーター、大腸菌のtrpA ターミネーター、ブレビバクテリウム ラクトファーメンタム(Brevibacterium lactofermentum)のtrp ターミネーター等が挙げられ、中でも、rrnB T1T2 ターミネーターが好ましい。
形質転換方法は、公知の方法を制限なく使用できる。このような公知の方法として、例えば、塩化カルシウム・塩化ルビジウム法、リン酸カルシウム法、DEAE-デキストラン介在トランスフェクション、電気パルス法などがあげられる。なかでも、コリネ型細菌には、電気パルス法が好適であり、電気パルス法は公知の方法により行うことができる(Kurusu, Y. et al., Electroporation-transformation system for Coryneform bacteria by auxotrophic complementation. Agric. Biol. Chem. 54: 443-447 (1990))。
炭素源としては、グルコース、フルクトース、スクロース、マンノース、マルトース、マンニトール、キシロース、アラビノース、ガラクトース、でんぷん、糖蜜、ソルビトール、グリセリン等の糖質または糖アルコール;酢酸、クエン酸、乳酸、フマル酸、マレイン酸またはグルコン酸等の有機酸;エタノール、プロパノール等のアルコールが挙げられる。炭素源は、1種を単独で使用でき、または2種以上を混合してもよい。培地中のこれら炭素源の濃度は、通常、約0.1~10(w/v %)とすればよい。
栄養物質としては、例えば肉エキス、ペプトン、ポリペプトン、酵母エキス、乾燥酵母、コーンスティープリカー、脱脂粉乳、脱脂大豆塩酸加水分解物、または動植物もしくは微生物菌体のエキスやそれらの分解物等が挙げられるが、通常、約0.1~10(w/v %)とすればよい。更に、必要に応じて、ビタミン類を添加することもできる。ビタミン類としては、例えばビオチン、チアミン(ビタミンB1)、ピリドキシン(ビタミンB6)、パントテン酸、イノシトール、ニコチン酸等が挙げられる。
培地のpHは約6~8が好ましい。
培養温度は約15~45℃とすればよく、培養時間は約1~7日とすればよい。
上記説明した本発明の形質転換体を、糖類を含有する反応液中で培養又は反応させることによりプロトカテク酸又はその塩を生産させる工程を含む方法によりプロトカテク酸又はその塩を製造することができる。
糖類としては、グルコースが好適であるが、フルクトース、マンノース、アラビノース、キシロース、ガラクトースなどの単糖類の他、代謝によりグルコースを生成し得る糖類も使用できる。このような糖類にはグルコース単位を有するオリゴ糖又は多糖類が含まれ、セロビオース、スクロース(ショ糖)、ラクトース、マルトース、トレハロース、セロビオース、キシロビオースなどの二糖類;デキストリン又は可溶性澱粉などの多糖類などが挙げられる。
また、例えばこれらの原料化合物を含む原料として、糖蜜も用いることができる。また、わら(稲わら、大麦わら、小麦わら、ライ麦わら、オート麦わら等)、バガス、コーンストーバー等の非可食農産廃棄物や、スイッチグラス、ネピアグラス、ミスキャンサス等のエネルギー作物や、木くず、古紙などを糖化酵素などで糖化した、グルコースなどの複数の糖を含む糖化液を用いることもできる。
糖類を含む培地での培養、即ち反応に先立ち、形質転換体を好気条件下で、温度約25~38℃で、約12~48時間培養して増殖させることが好ましい。
反応に先立つ形質転換体の好気的培養に用いる培地は、炭素源、窒素源、無機塩類およびその他の栄養物質等を含有する天然培地または合成培地を用いることができる。
炭素源として、糖類(グルコース、フルクトース、マンノース、キシロース、アラビノース、ガラクトースのような単糖;スクロース、マルトース、ラクトース、セロビオース、キシロビオース、トレハロースのような二糖;澱粉のような多糖;糖蜜等)、マンニトール、ソルビトール、キシリトール、グリセリンのような糖アルコール;酢酸、クエン酵、乳酸、フマル酸、マレイン酸、グルコン酸のような有機酸;エタノール、プロパノールのようなアルコール;ノルマルパラフィンのような炭化水素等も用いることができる。
炭素源は、1種を単独で、又は2種以上を混合して使用できる。
無機塩類としては、リン酸第一カリウム、リン酸第二カリウム、硫酸マグネシウム、塩化ナトリウム、硝酸第一鉄、硫酸マンガン、硫酸亜鉛、硫酸コバルト、炭酸カルシウム等が挙げられる。無機塩は、1種を単独で、又は2種以上を混合して使用できる。無機塩類の培地中の濃度は、使用する無機塩によっても異なるが、通常、約0.01~1(w/v %)とすればよい。
さらに、必要に応じて、ビタミン類を添加することもできる。ビタミン類としては、ビオチン、チアミン(ビタミンB1)、ピリドキシン(ビタミンB6)、パントテン酸、イノシトール、ニコチン酸等が挙げられる。
培地のpHは約6~8が好ましい。
培養液又は反応液としては、炭素源、窒素源、及び無機塩類等を含有する天然反応液または合成反応液を用いることができる。
炭素源としては、上記説明した糖類又はそれを含む糖蜜や糖化液などを用いればよい。また、炭素源として、糖類の他に、マンニトール、ソルビトール、キシリトール、グリセリンのような糖アルコール;酢酸、クエン酸、乳酸、フマル酸、マレイン酸、グルコン酸のような有機酸;エタノール、プロパノールのようなアルコール;ノルマルパラフィンのような炭化水素等も用いることができる。
炭素源は、1種を単独で、又は2種以上を混合して使用できる。
反応液中の原料化合物である糖類の濃度は、約1~20(w/v %)が好ましく、約2~10(w/v %)がより好ましく、約2~5(w/v %)がさらにより好ましい。
また、原料の糖類を含む全炭素源の濃度は、約2~5(w/v %)とすればよい。
無機塩類としては、リン酸第一カリウム、リン酸第二カリウム、硫酸マグネシウム、塩化ナトリウム、硝酸第一鉄、硫酸マンガン、硫酸亜鉛、硫酸コバルト、炭酸カルシウム等が挙げられる。無機塩は、1種を単独で、又は2種以上を混合して使用できる。無機塩類の反応液中の濃度は、使用する無機塩によっても異なるが、通常、約0.01~1(w/v %)とすればよい。
反応液のpHは約6~8が好ましい。
培養温度又は反応温度、即ち形質転換体の生存温度は、約20~50℃が好ましく、約25~47℃がより好ましい。上記温度範囲であれば、効率良くプロトカテク酸を製造できる。
また、培養又は反応時間は、約1~7日間が好ましく、約1~3日間がより好ましい。
培養は、バッチ式、流加式、連続式の何れでもよい。中でも、バッチ式が好ましい。
反応は、好気的条件で行ってもよく、還元条件で行ってもよい。本発明の形質転換体自体のプロトカテク酸又はその塩の生産能力は、好気的条件下の方が高い。しかし、好気的条件下では形質転換体が増殖するため、原料化合物が増殖のために消費され、その分、プロトカテク酸又はその塩の製造効率が低下する。
従って、好気的、かつ形質転換体が増殖しない条件下で反応を行うのが好ましい。本発明で増殖しないことには、実質的に増殖しないこと、又は殆ど増殖しないことが含まれる。例えば、微生物の増殖に必須の化合物であるビオチン、チアミンなどのビタミン類、窒素源、または栄養要求性の形質転換体の増殖に必須のアミノ酸などの1種以上を欠乏、或いは制限させた反応液を用いることにより、形質転換体の増殖を回避または抑制できる。
還元条件は、反応液の酸化還元電位で規定される。反応液の酸化還元電位は、約-200 mV~-500 mVが好ましく、約-150 mV~-500 mVがより好ましい。
反応液の還元状態は簡便にはレサズリン指示薬(還元状態であれば、青色から無色への脱色)で推定できるが、正確には酸化還元電位差計(例えば、BROADLEY JAMES社製、ORP Electrodes)を用いて測定できる。
具体的には、蒸留水などを加熱処理や減圧処理して溶解ガスを除去することにより、還元条件の反応液用水溶液を得ることができる。この場合、約10 mmHg以下、好ましくは約5 mmHg以下、より好ましくは約3 mmHg以下の減圧下で、約1~60分程度、好ましくは約5~40分程度、蒸留水などを処理することにより、溶解ガス、特に溶解酸素を除去して還元条件下の反応液用水溶液を作成することができる。
また、適当な還元剤(例えば、チオグリコール酸、アスコルビン酸、システィン塩酸塩、メルカプト酢酸、チオール酢酸、グルタチオン、硫化ソーダ等)を添加して還元条件の反応液用水溶液を調整することもできる。
これらの方法を適宜組み合わせることも有効な還元条件の反応液用水溶液の調整方法である。
上記のようにして培養することにより、培養液又は反応液中にプロトカテク酸又はその塩が生産される。
プロトカテク酸の塩は、培地又は反応液の成分によっても異なるが、アルカリ金属塩(ナトリウム塩、カリウム塩など)、アルカリ土類金属塩(マグネシウム塩、カルシウム塩など)が挙げられる。
PCA生産株の構築
(1) 染色体DNAの調整
PCA生産関連酵素遺伝子を取得するため、下記菌株から染色体DNAを調整した。
コリネバクテリウム グルタミカム (Corynebacterium glutamicum) R (FERM P-18976)、エシェリヒア コリ(Escherichia coli K-12 MG1655)、プロビデンシア ルスティジアニイ(Providencia rustigianii JCM 3953)、コリネバクテリウム カゼイ(Corynebacterium casei JCM 12072)、コリネバクテリウム エフィシェンス(Corynebacterium efficiens NBRC 100395)、パントエア アナナティス(Pantoea ananatis LMG 20103)、グルコノバクター オキシダンス(Gluconobacter oxydans ATCC 621H)、シュードモナス プチダ(Pseudomonas putida NBRC 14164)、ロドシュードモナス パルストリス(Rhodopseudomonas palustris ATCC BAA-98)、アシネトバクター バイリイ(Acinetobacter baylyi ATCC33305)、アルテロモナス マクレオディイ(Alteromonas macleodii NBRC 102226)、マリノバクター ハイドロカーボノクラスティカス(Marinobacter hydrocarbonoclasticus JCM 20777)、メチロバクテリウム エキストロクエンス(Methylobacterium extorquens JCM 2802)、ニューロスポラ クラッサ(Neurospora crassa ATCC 36373)、アスペルギルス ニガー(Aspergillus niger JCM 22282)、マイコバクテリウム スメグマチス(Mycobacterium smegmatis ATCC 700084)、コリネバクテリウム ハロトレランス(Corynebacterium halotolerans JCM 12676)、ロドコッカス オパクス(Rhodococcus opacus ATCC 51881)、アスペルギルス オリゼ(Aspergillus oryzae JCM 13832)、バチルス チューリンゲンシス(Bacillus thuringiensis NBRC 3951)を菌株入手機関の情報に従って培養した後、DNAゲノム抽出キット(商品名:GenomicPrep Cells and Tissue DNA Isolation Kit、アマシャム社製)を用いて調整した。
目的の酵素遺伝子を単離するために用いたプライマー配列を表1に示す。PCRは、Veritiサーマルサイクラー(アプライド・バイオシステムズ社製)を用い、反応試薬としてPrimeSTAR HS DNA Polymerase(タカラバイオ株式会社製)を用いた。
得られたDNA断片を、PgapAプロモーターを含有するクローニングベクター(pCRB207 [Hasegawa S et al., Improvement of the redox balance increases L-valine production by Corynebacterium glutamicum under oxygen deprivation conditions. Appl Environ Microbiol. 78(3):865-875 (2012)]、pCRB209 [国際公開 WO2012/033112]、pCRB210 [国際公開 WO2012/033112]に導入した。
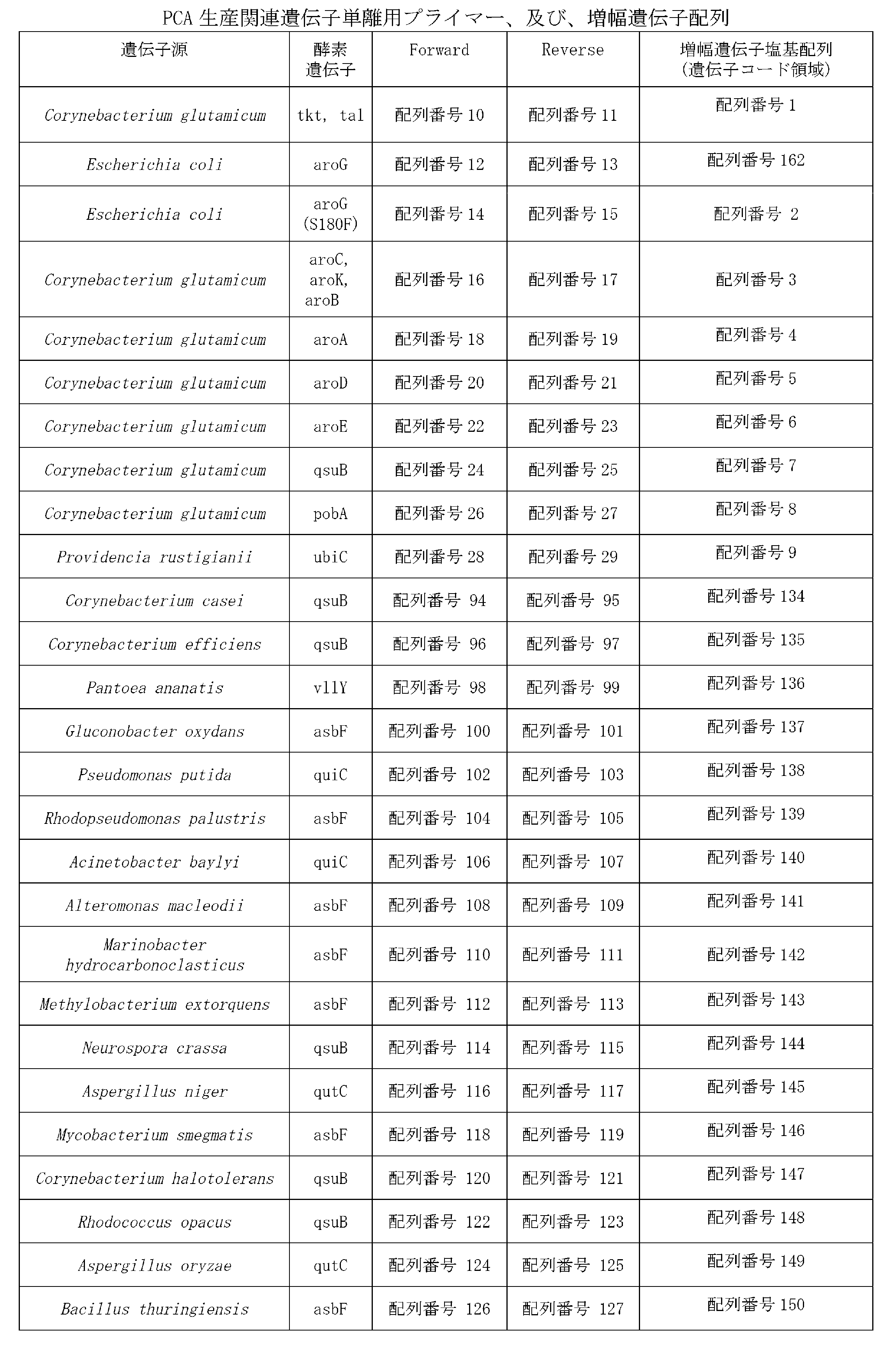
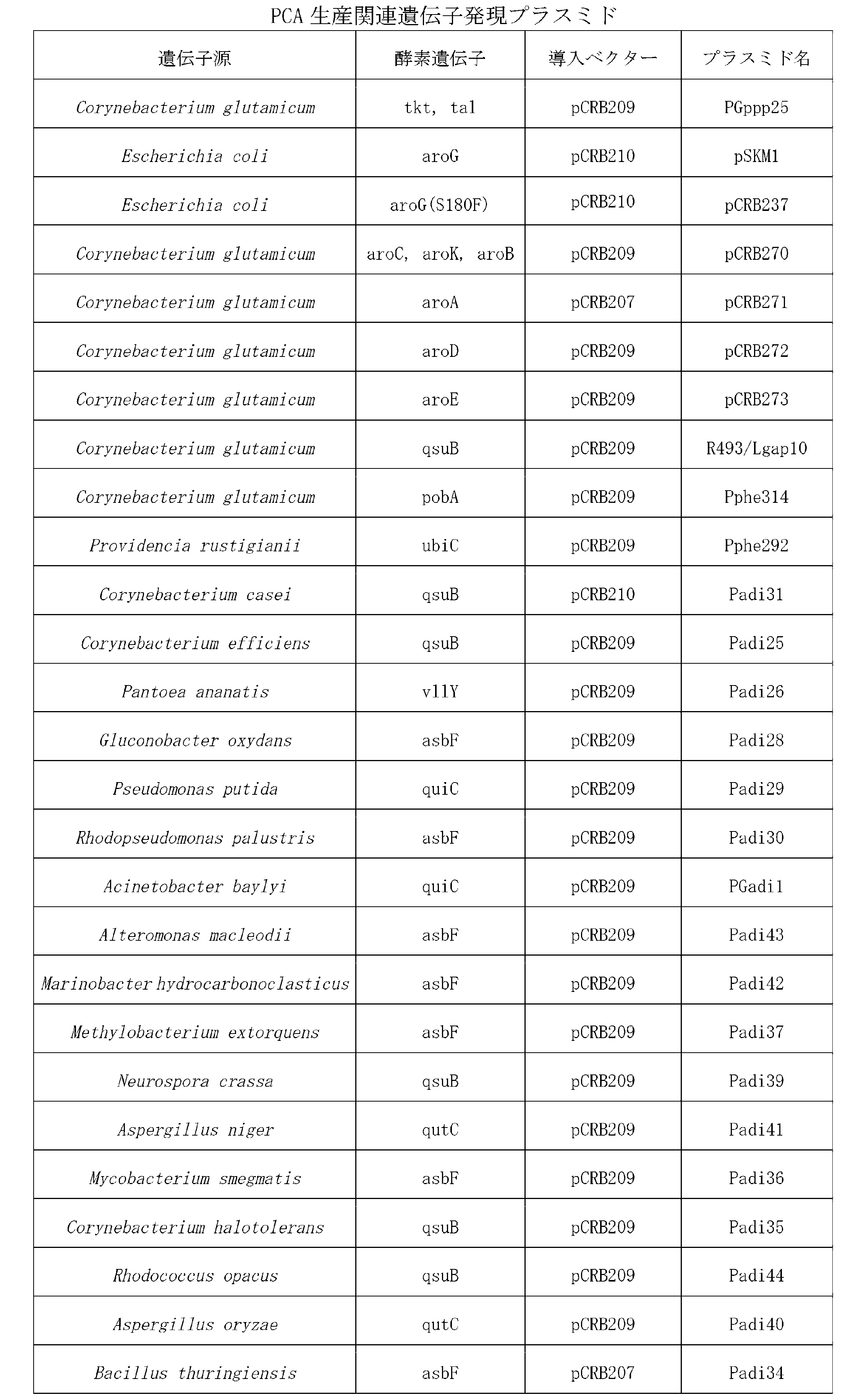
PCA生産関連遺伝子をCorynebacterium glutamicum R株の染色体にマーカーレスで導入するために必要なDNA領域を、Corynebacterium glutamicum R株の生育に必須でないと報告されている配列 [Appl. Environ. Microbiol. 71:3369-3372 (2005)](SSI領域)を基に決定した。このDNA領域PCR法により増幅した。得られたDNA断片をマーカーレス遺伝子導入用プラスミドpCRA725 [J. Mol. Microbiol. Biotechnol. 8:243-254(2004)、(特開2006-124440)] に導入した。なお、pCRB260、pCRB263、pCRB266、pCRB267、pCRB274及びpCRB279は、インバース PCR法によりSSI領域に遺伝子を組み込むための制限酵素部位(ユニークサイト)を導入した。SSI領域の単離及びインバースPCRに用いたプライマー配列および得られた染色体導入用ベクターを表3に示す。

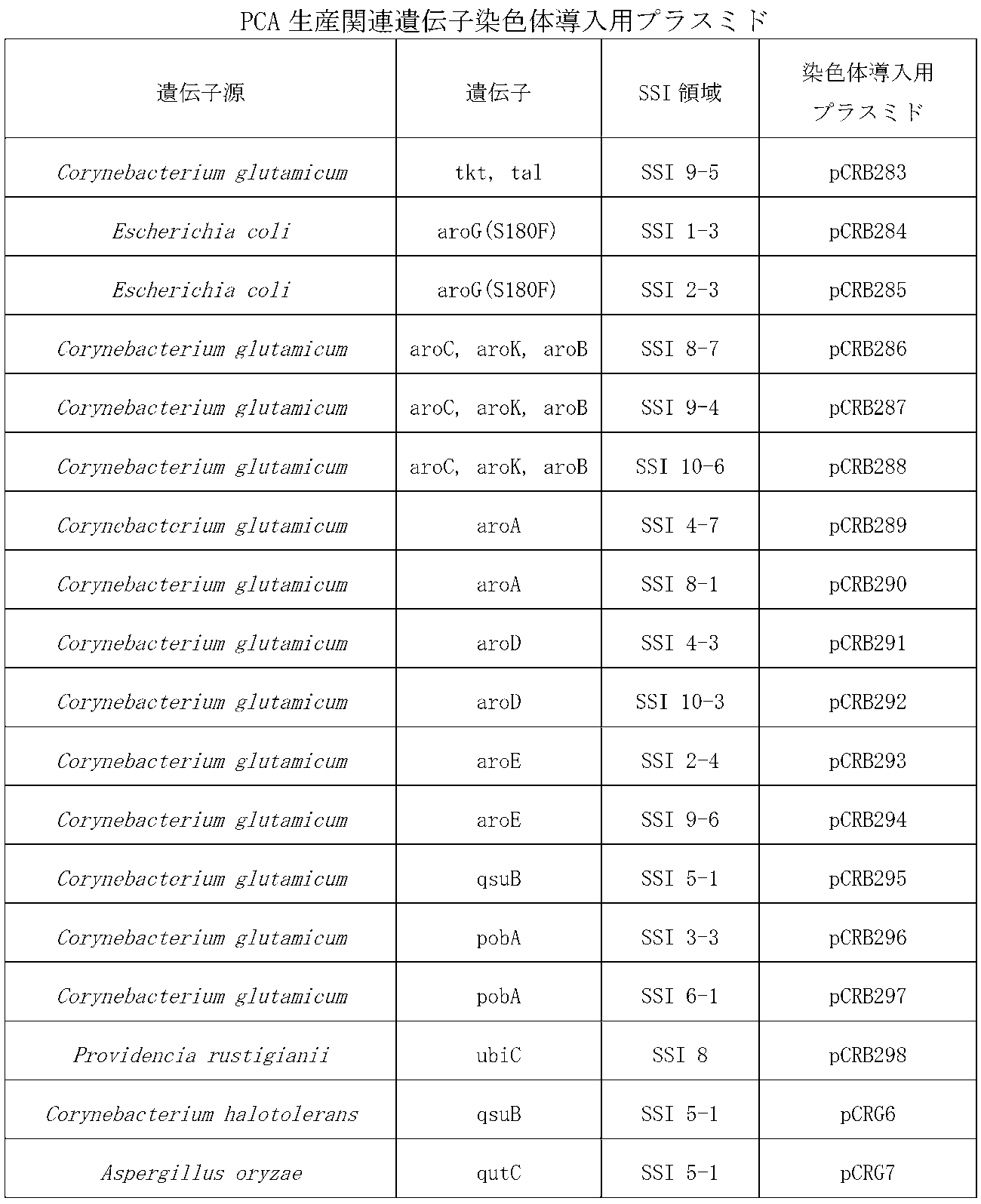
Corynebacterium glutamicum R株の染色体遺伝子をマーカーレスで破壊するために必要なDNA領域をPCR法により増幅した。各PCR断片はオーバーラップ領域により連結可能である。得られたDNA断片をマーカーレス遺伝子破壊用プラスミドpCRA725 [J. Mol. Microbiol. Biotechnol. 8:243-254(2004)、(特開2006-124440)] に導入した。得られた染色体遺伝子破壊用プラスミドを表5に示す。

マーカーレス染色体遺伝子導入用ベクターpCRA725は、コリネバクテリウム グルタミカムR内で複製不能なプラスミドである。プラスミドpCRA725に導入した染色体上の相同領域との一重交叉株の場合、pCRA725上のカナマイシン耐性遺伝子の発現によるカナマイシン耐性と、バチルス サブチリス(Bacillus subtilis)のsacR-sacB遺伝子の発現によるスクロース含有培地での致死性を示すのに対し、二重交叉株の場合、pCRA725上のカナマイシン耐性遺伝子の脱落によるカナマイシン感受性と、sacR-sacB遺伝子の脱落によるスクロース含有培地での生育性を示す。従って、マーカーレス染色体遺伝子導入株は、カナマイシン感受性及びスクロース含有培地生育性を示す。
上記方法により、上述したPCA生産関連遺伝子染色体導入用プラスミド及び染色体遺伝子破壊用プラスミドを用いてPCA生産関連遺伝子染色体導入株を構築した。宿主菌株としてキシロース・セロビオース資化性コリネ型細菌Corynebacterium glutamicum X5C1株 [Appl Microbiol Biotechnol. 81(4):691-699 (2008)]を使用した。また、ldhA遺伝子破壊用プラスミドpCRA728 [J Mol Microbiol Biotechnol. 8(4):243-254 (2004)]、アラビノース資化遺伝子染色体導入用プラスミドpCRD109 [Appl Microbiol Biotechnol. 85(1):105-115 (2009) ]及びアラビノーストランスポーター遺伝子染色体導入用プラスミドpCRD108 [Appl Microbiol Biotechnol. 85(1):105-115 (2009) ]も使用した。尚、本染色体遺伝子組換えの概要は、表6にまとめて示した。

上述の各種微生物由来3-デヒドロシキミ酸デヒドラターゼ遺伝子発現プラスミドを導入したコリネバクテリウム グルタミカム形質転換体を構築した。尚、本プラスミド導入株の概要は、表7にまとめて示した。

他の微生物に比べコリネ型細菌がプロトカテク酸に対して高い耐性を有することの検証
プロトカテク酸のような細胞毒性を有する生産物を微生物を用いて発酵法によって生産させる場合、宿主微生物が、その生産物に対する耐性を持つこと、すなわち、生産物による増殖阻害を受けにくいことが重要である。そこで、本発明における宿主微生物として好ましいCorynebacterium glutamicumのプロトカテク酸に対する耐性度を、他の微生物との比較において検証するため、Corynebacterium glutamicum, Escherichia coli, Bacillus subtilis, Pseudomonas putida, Rhodococcus erythropolis, 及びSaccharomyces cerevisiaeについて、好気培養におけるプロトカテク酸による増殖阻害効果について調べた。
Escherichia coli K12株は、100 mM プロトカテク酸存在下で著しい増殖阻害を受け、250 mMでは、ほぼ完全に増殖が阻害された。
Bacillus subtilis NBRC14144株は250 mM プロトカテク酸存在下で著しい増殖阻害を受け、500 mMでは、ほぼ完全に増殖が阻害された。
Pseudomonas putida ATCC700801株は100 mM プロトカテク酸存在下で強い増殖阻害を受け、250 mMでは、ほぼ完全に増殖が阻害された。
Rhodococcus erythropolis ATCC27854株は250 mM プロトカテク酸存在下で強い増殖阻害を受け、500 mMでは、ほぼ完全に増殖が阻害された。
Saccharomyces cerevisiae NBRC2376株は250 mM プロトカテク酸存在下で増殖阻害を受け、500 mMでは、著しい増殖阻害を受けた。
これに対し、Corynebacterium glutamicum R株は、他の菌株の増殖が著しく、または、ほぼ完全に阻害される250~500 mMのプロトカテク酸存在下においても旺盛な増殖が可能であった。
このように、Corynebacterium glutamicumは、プロトカテク酸生産宿主として報告のある他の微生物や代表的な溶媒耐性菌と比較してプロトカテク酸に対する高い耐性を有することから、プロトカテク酸生産宿主として高い適性を有することが示された。
コリネ型細菌が高濃度のプロトカテク酸存在下において高い糖消費能力を有することの検証
参考例1で示したように、Corynebacterium glutamicumは高濃度のプロトカテク酸存在下でも増殖可能であった。そこでさらに、高濃度のプロトカテク酸存在下におけるCorynebacterium glutamicumのグルコース消費能を以下のように調べた。
Corynebacterium glutamicum R株を4%のグルコースを含んだ前記A寒天培地に塗布し、33℃で16時間培養した。上記プレート上で増殖したCorynebacterium glutamicum R株を4%のグルコースを含んだ前記A液体培地10 mlの入った試験管に一白金耳植菌し、33℃にて16時間、好気的に振とう培養を行った。上記条件で増殖したCorynebacterium glutamicum R株を、4%のグルコースを含んだ前記A液体培地10 mlに初期菌体濃度OD610 = 0.2となるように植菌し、同時にプロトカテク酸が終濃度0, 50, 250, 500 mMとなるように添加し、33℃にて好気的に振とう培養を行った。培養24時間後に培養液を回収し、遠心分離(4℃, 15,000×g、5分)して得られた上清液中のグルコース濃度を、後述する実施例2と同様にしてグルコースセンサーにより測定した。各濃度のプロトカテク酸存在下における培養24時間後のCorynebacterium glutamicum R株のグルコース消費量を図3に示す。
図3に示したように、Corynebacterium glutamicumは高濃度のプロトカテク酸存在下においても糖消費の低下が僅かであることが分かる。
コリネバクテリウム グルタミカム形質転換体によるジャーファーメンター制御下の好気静止菌体反応によるプロトカテク酸生産試験
コリネバクテリウム グルタミカムR株由来の混合糖利用株をベースとして構築したプロトカテク酸生産株である、PCA1、PCA2、PCA3、PCA4、PCA5の各菌株(実施例1(表6))について、ジャーファーメンター(エイブル株式会社製、型式:BMJ1L)制御下の好気的静止菌体反応におけるプロトカテク酸生産能を以下に述べる方法に従って確認した。
PCA1株は、フェニルアラニン、チロシン、トリプトファン各20 μg/ml、p-アミノ安息香酸 10 μg/ml、シキミ酸3.2 mM、及びグルコース4% (各終濃度)を添加した10 mlの前記A液体培地(試験管内)に植菌後、また、PCA2、PCA3、PCA4、及びPCA5株はグルコース4%を添加した10 mlの 前記A液体培地(試験管内)に植菌後、33℃で12-16時間、好気的に振盪培養を行った。
上記条件で増殖したコリネバクテリウム グルタミカム PCA1株については、グルコース 80 g/l、フェニルアラニン、チロシン、トリプトファン各100 μg/ml、p-アミノ安息香酸 50 μg/ml、シキミ酸 16 mM、及び消泡剤(ディスホームCB-442) 3 g/l(各終濃度)を添加した、600 mlの前記A(-UB)液体培地にOD610が0.3となるように植菌し、また、上記条件で増殖したコリネバクテリウム グルタミカム PCA2、PCA3、PCA4、及びPCA5株については、グルコース 100 g/l、及び消泡剤(ディスホームCB-442) 3 g/l(各終濃度)を添加した、600 mlの前記A(-UB)液体培地にOD610が0.3となるように植菌し、各々、1000 ml容量ジャーファーメンター(エイブル株式会社製、型式:BMJ1L)により33℃、pH 7.0 (5 N アンモニア水の添加により一定に制御)、通気量0.6 L/min (air、1 vvm)、溶存酸素濃度(DO) 10%(33℃における大気圧下飽和溶存酸素濃度を100%として)の条件において19-20時間通気撹拌培養を行った。
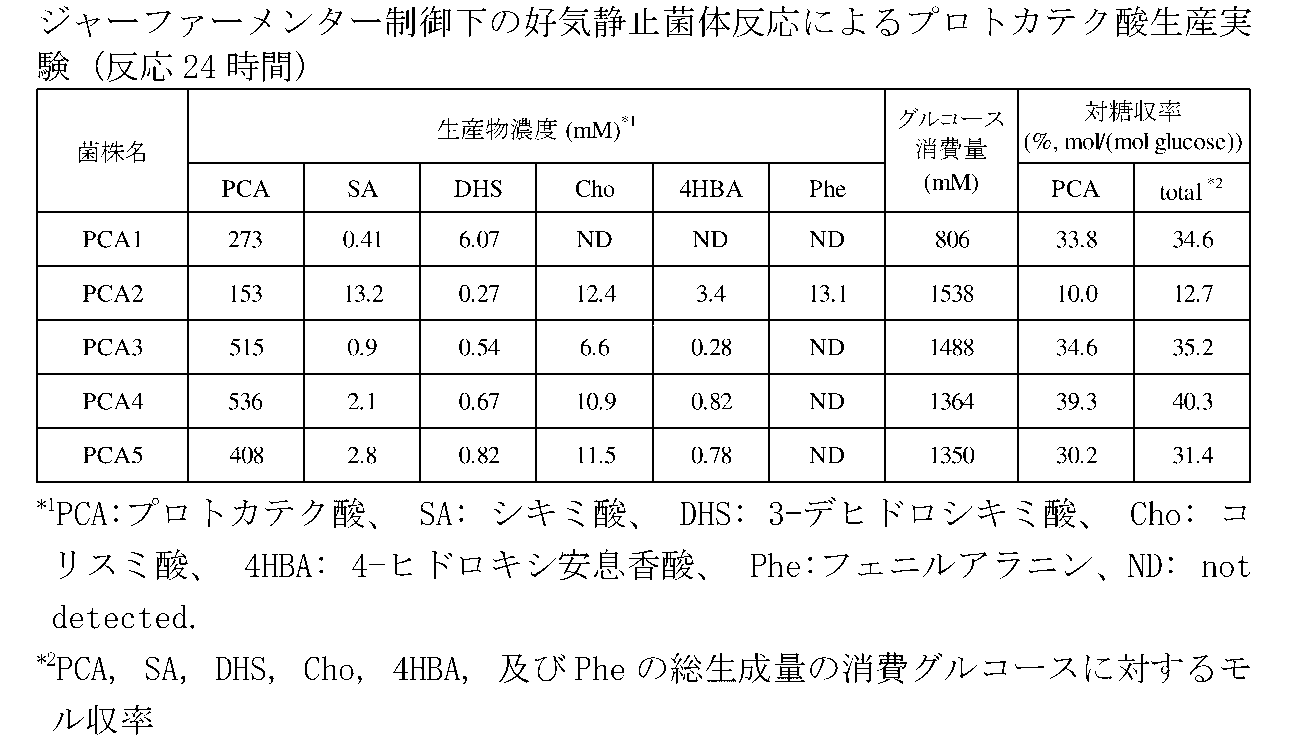
また、(a)3-デヒドロシキミ酸デヒドラターゼによって触媒される、3-デヒドロシキミ酸のプロトカテク酸への変換によるプロトカテク酸生成のみに依存するPCA1株も比較的高いプロトカテク酸生産能を示したが、その生産性は、(a)と(b)の両経路からPCAを生成する、PCA3、PCA4、またはPCA5株と比べ劣っていた。また、PCA1株は芳香族アミノ酸生合成経路がシキミ酸デヒドロゲナーゼ遺伝子(aroE)の破壊により遮断されているため、芳香族アミノ酸及び4-アミノ安息香酸の要求性を示し、本菌株を栄養培地で増殖させる場合には培地にそれら栄養源を添加する必要があった。
一方、(b) コリスメートピルベートリアーゼ、及び4-ヒドロキシ安息香酸ヒドロキシラーゼによって触媒される、コリスミ酸のプロトカテク酸への変換によるプロトカテク酸生成のみに依存してプロトカテク酸を生成するPCA2株のPCA生産性は、上記の他菌株と比べて大幅に低いことが示された。
プロトカテク酸生産株における、3-デヒドロシキミ酸デヒドラターゼ、コリスメートピルベートリアーゼ、及び4-ヒドロキシ安息香酸ヒドロキシラーゼの各酵素活性の測定
PCA1株、PCA2株、PCA3株、及び、これらの菌株の親株であるCRZ22株(表6)について、3-デヒドロシキミ酸デヒドラターゼ、コリスメートピルベートリアーゼ、及び、4-ヒドロキシ安息香酸ヒドロキシラーゼの各種酵素活性を下記の方法に従って測定した。
実施例1と同様の手順で、CRZ22株、PCA1株、PCA2株、及びPCA3株のジャーファーメンターを用いた好気的静止菌体反応を行い、反応6時間後の各菌株培養液を採取し、菌体を遠心分離により回収した。菌体を20 mM Tris-HCl(pH7.5)で1回洗浄した後、1 mlの菌体破砕用バッファー(100 mM Tris-HCl(pH7.5),20 mM KCl, 20 mM MgCl2, 0.1 mM EDTA, and 2 mM DTT))に懸濁し、マルチビーズショッカー(安井器械)及びグラスビーズを用いて破砕した。菌体破砕液を15000 rpm, 10 min, 4℃の条件で遠心分離を行い、上清画分を粗酵素抽出液として得た。各粗酵素抽出液のタンパク質濃度は、Protein assay kit (Bio-Rad, USA)を用い、BSA(Bovine serum albumin)をスタンダードとして定量した。各菌株の粗酵素抽出液中の各酵素活性の測定は、先述の活性測定方法に従って行った。

一方、PCA1株では、3-デヒドロシキミ酸デヒドラターゼ遺伝子(qsuB)の導入に伴って3-デヒドロシキミ酸デヒドラターゼ(QsuB)活性が検出された。PCA1株は、シキミ酸デヒドロゲナーゼをコードするaroE遺伝子が破壊されていることから、該酵素反応段階でシキミ酸経路が遮断される。従って、PCA1株におけるPCA生成が(a) 3-デヒドロシキミ酸デヒドラターゼのみに依存的に起こっていることを支持している。
PCA2株においては、コリスメートピルベートリアーゼ(UbiC)、及び4-ヒドロキシ安息香酸ヒドロキシラーゼ(PobA)遺伝子の導入に対応してそれらの酵素活性が検出されたのに対し、3-デヒドロシキミ酸デヒドラターゼ(QsuB)活性は検出されなかった。このことからPCA2株においては、(a) 3-デヒドロシキミ酸デヒドラターゼ依存的なプロトカテク酸生成は起こっておらず、(b)コリスメートピルベートリアーゼ(UbiC)、及び4-ヒドロキシ安息香酸ヒドロキシラーゼ(PobA)を介した経路によってプロトカテク酸が生成することを支持している。
また、PCA3株においては、上記3種のすべての酵素活性が検出されたことから、本菌株においては、(a)と(b)の双方の経路を介してPCA生成が起こることを支持している。
有能なシキミ酸デヒドラターゼをコードする異種遺伝子の探索
実施例2及び実施例3の結果より、コリネ型細菌形質転換体によるプロトカテク酸の生産性は、3-デヒドロシキミ酸デヒドラターゼ遺伝子導入による該酵素活性の強化により顕著に高まることが示された。一方、コリネ型細菌形質転換体PCA1株、及びPCA3株においては、コリネバクテリウム グルタミカム由来の3-デヒドロシキミ酸デヒドラターゼ遺伝子を導入しているが、より有能な3-デヒドロシキミ酸デヒドラターゼが他の微生物に存在する可能性も考えられた。そこで、有能な異種3-デヒドロシキミ酸デヒドラターゼ遺伝子の探索を行った。
3-デヒドロシキミ酸デヒドラターゼ遺伝子探索用の宿主として、染色体上の3-デヒドロシキミ酸デヒドラターゼ遺伝子(qsuB)、及びシキミ酸デヒドロゲナーゼ遺伝子(aroE)が破壊され、また、シキミ酸経路遺伝子aroGS180F、aroB、及びaroDが導入された、3-デヒドロシキミ酸生産菌株である、DHS1株(表6)を用いた。本菌株に対し、種々の微生物由来の3-デヒドロシキミ酸デヒドラターゼ遺伝子をマルチコピー型発現ベクター(pCRB209、pCRB207、またはpCRB210)を用いて導入した各形質転換株を構築し(表7)、それらについて試験管培養を行い、プロトカテク酸生産能を調べた。
各形質転換体のプロトカテク酸生産能は、以下のようにして測定した。まず、グルコース 4%、フェニルアラニン、チロシン、トリプトファン 各20 μg/ml、p-アミノ安息香酸 10μg/ml、シキミ酸 3.2 mM、及び、カナマイシン 50 μg/ml (各終濃度)を含む10 mlの前記 A液体培地(試験管内)に植菌後、33℃で16-18時間、好気的に振盪培養を行った。

Claims (22)
- 下記の(A)、(B)、及び(C)の操作が施されたプロトカテク酸生産能を有する形質転換体。
(A) 3-デヒドロシキミ酸デヒドラターゼ活性の強化
(B) コリスメートピルベートリアーゼ活性の強化
(C) 4-ヒドロキシ安息香酸ヒドロキシラーゼ活性の強化 - 3-デヒドロシキミ酸デヒドラターゼ活性の強化が、コリネバクテリウム属、ロドコッカス属、バチルス属、ロドシュードモナス属、アルテロモナス属、マリノバクター属、メチロバクテリウム属、パントエア属、ニューロスポラ属、又はアスペルギルス属に属する微生物由来の3-デヒドロシキミ酸デヒドラターゼ活性を有する酵素をコードする遺伝子の宿主への導入によってもたらされたものである、請求項1に記載の形質転換体。
- 3-デヒドロシキミ酸デヒドラターゼ活性を有する酵素をコードする遺伝子が、コリネバクテリウム グルタミカム (Corynebacterium glutamicum)、コリネバクテリウム ハロトレランス(Corynebacterium halotolerans)、コリネバクテリウム カゼイ (Corynebacterium casei)、コリネバクテリウム エフィシェンス (Corynebacterium efficiens)、アスペルギルス ニガー (Aspergillus niger)、又はアスペルギルス オリゼー (Aspergillus oryzae)の遺伝子である、請求項2に記載の形質転換体。
- 3-デヒドロシキミ酸デヒドラターゼ活性を有する酵素をコードする遺伝子が、下記の(a)または(b)のDNAによってコードされる、請求項2または3に記載の形質転換体。
(a) 配列番号7、134、135、145、147、又は149の塩基配列からなるDNA
(b) 配列番号7、134、135、145、147、又は149の塩基配列と90%以上の同一性を有する塩基配列からなるDNAであって、3-デヒドロシキミ酸デヒドラターゼ活性を有するポリペプチドをコードするDNA - コリスメートピルベートリアーゼ活性の強化が、プロビデンシア属細菌、又は、クロノバクター属細菌由来のコリスメートピルベートリアーゼ活性を有する酵素をコードする遺伝子の宿主への導入によってもたらされたものである、請求項1~4のいずれかに記載の形質転換体。
- コリスメートピルベートリアーゼ活性の強化が、プロビデンシア ルスティジアニ (Providencia rustigianii)、プロビデンシア スチュアルティ (Providencia stuartii)、又はクロノバクター サカザキ (Cronobacter sakazakii)由来のコリスメートピルベートリアーゼ活性を有する酵素をコードする遺伝子の宿主への導入によってもたらされたものである、請求項5に記載の形質転換体。
- コリスメートピルベートリアーゼ活性の強化が、下記の(c)または(d)のDNAの宿主への導入によってもたらされたものである、請求項1~6のいずれかに記載の形質転換体。
(c) 配列番号9、128、又は129の塩基配列からなるDNA
(d) 配列番号9、128、又は129の塩基配列と90%以上の同一性を有する塩基配列からなるDNAであって、コリスメートピルベートリアーゼ活性を有するポリペプチドをコードするDNA - 4-ヒドロキシ安息香酸ヒドロキシラーゼ活性の強化が、4-ヒドロキシ安息香酸ヒドロキシラーゼ活性を有する酵素をコードする、コリネバクテリウム グルタミカム (Corynebacterium glutamicum)の遺伝子の宿主への導入によってもたらされたものである、請求項1~7のいずれかに記載の形質転換体。
- 4-ヒドロキシ安息香酸ヒドロキシラーゼ活性の強化が、下記の(e)または(f)のDNAの宿主への導入によってもたらされたものである、請求項1~8のいずれかに記載の形質転換体。
(e) 配列番号8の塩基配列からなるDNA
(f) 配列番号8の塩基配列と90%以上の同一性を有する塩基配列からなるDNAであって、4-ヒドロキシ安息香酸ヒドロキシラーゼ活性を有するポリペプチドをコードするDNA - プロトカテク酸3,4-ジオキシゲナーゼ活性が消失しているか、阻害されているか、または減少している、請求項1~9のいずれかに記載のコリネ型細菌形質転換体。
- 3-デオキシ-D-アラビノ-ヘプツロソネート-7-リン酸(DAHP)シンターゼ、3-デヒドロキナ酸シンターゼ、3-デヒドロキナ酸デヒドラターゼ、シキミ酸デヒドロゲナーゼ、シキミ酸キナーゼ、5-エノールピルビルシキミ酸3-リン酸(EPSP)シンターゼ、及びコリスミ酸シンターゼからなる酵素群より選ばれる少なくとも一つの酵素活性が強化されている請求項1~10のいずれかに記載の形質転換体。
- DAHPシンターゼ活性の強化が下記の(g)または(h)のDNAの宿主への導入によってもたらされたものであり、3-デヒドロキナ酸シンターゼ活性の強化が下記の(i)または(j)のDNAの宿主への導入によってもたらされたものであり、3-デヒドロキナ酸デヒドラターゼ活性の強化が下記の(k)または(l)のDNAの宿主への導入によってもたらされたものであり、シキミ酸デヒドロゲナーゼ活性の強化が下記の(m)または(n)のDNAの宿主への導入によってもたらされたものであり、シキミ酸キナーゼ活性の強化が下記の(o)または(p)のDNAの宿主への導入によってもたらされたものであり、EPSPシンターゼ活性の強化が下記の(q)または(r)のDNAの宿主への導入によってもたらされたものであり、コリスミ酸シンターゼ活性の強化が下記の(s)または(t)のDNAの宿主への導入によってもたらされたものである、請求項11に記載の形質転換体。
(g) 配列番号2の塩基配列からなるDNA
(h) 配列番号2と90%以上の同一性を有する塩基配列からなるDNAであって、DAHPシンターゼ活性を有するポリペプチドをコードするDNA
(i) 配列番号153の塩基配列からなるDNA
(j) 配列番号153と90%以上の同一性を有する塩基配列からなるDNAであって、3-デヒドロキナ酸シンターゼ活性を有するポリペプチドをコードするDNA
(k) 配列番号5の塩基配列からなるDNA
(l) 配列番号5と90%以上の同一性を有する塩基配列からなるDNAであって、3-デヒドロキナ酸デヒドラターゼ活性を有するポリペプチドをコードするDNA
(m) 配列番号6の塩基配列からなるDNA
(n) 配列番号6と90%以上の同一性を有する塩基配列からなるDNAであって、シキミ酸デヒドロゲナーゼ活性を有するポリペプチドをコードするDNA
(o) 配列番号154の塩基配列からなるDNA
(p) 配列番号154と90%以上の同一性を有する塩基配列からなるDNAであって、シキミ酸キナーゼ活性を有するポリペプチドをコードするDNA
(q) 配列番号155の塩基配列からなるDNA
(r) 配列番号155と90%以上の同一性を有する塩基配列からなるDNAであって、EPSPシンターゼ活性を有するポリペプチドをコードするDNA
(s) 配列番号156の塩基配列からなるDNA
(t) 配列番号156と90%以上の同一性を有する塩基配列からなるDNAであって、コリスミ酸シンターゼ活性を有するポリペプチドをコードするDNA - トランスケトラーゼ活性、及びトランスアルドラーゼ活性からなる群より選ばれる少なくとも一つの活性が強化されている、請求項1~12のいずれかに記載の形質転換体。
- トランスケトラーゼ活性の強化が下記の(u)又は(v)のDNAの導入によるものであり、トランスアルドラーゼ活性の強化が下記の(w)又は(x)のDNAの導入によるものである、請求項13に記載の形質転換体。
(u) 配列番号151の塩基配列からなるDNA
(v) 配列番号151と90%以上の同一性を有する塩基配列からなるDNAであって、トランスケトラーゼをコードするDNA
(w) 配列番号152の塩基配列からなるDNA
(x) 配列番号152と90%以上の同一性を有する塩基配列からなるDNAであって、トランスアルドラーゼをコードするDNA - 宿主がコリネ型細菌である、請求項1~14の何れかに記載の形質転換体。
- グルコース、及び、キシロース、アラビノース、及びセロビオースからなる群より選ばれる少なくとも1種の糖の同時利用能を有する、請求項15に記載の形質転換体。
- 宿主のコリネ型細菌がコリネバクテリウム属細菌である、請求項15、又は16に記載の形質転換体。
- 宿主のコリネバクテリウム属細菌がコリネバクテリウム グルタミカムである、請求項17に記載の形質転換体。
- 宿主のコリネバクテリウム グルタミカムが、コリネバクテリウム グルタミカムR(FERM BP-18976)、ATCC13032、又は、ATCC13869である、請求項18に記載のコリネ型細菌形質転換体。
- コリネバクテリウム グルタミカム PCA4 (受託番号:NITE BP-02217)
- 請求項1~20のいずれかに記載の形質転換体を、糖類を含む反応液中で培養してプロトカテク酸又はその塩を生産させる工程を含むプロトカテク酸又はその塩の製造方法。
- 好気的、かつ形質転換体が増殖しない条件下で形質転換体を培養する、請求項21に記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/089,567 US10961526B2 (en) | 2016-03-28 | 2017-02-24 | Transformant, and method for producing protocatechuic acid or salt thereof using same |
| EP17773944.8A EP3438245B1 (en) | 2016-03-28 | 2017-02-24 | Transformant, and method for producing protocatechuic acid or salt thereof using same |
| CN201780021444.5A CN109477066B (zh) | 2016-03-28 | 2017-02-24 | 转化体及使用其的原儿茶酸或其盐的制造方法 |
| JP2018508800A JP6685388B2 (ja) | 2016-03-28 | 2017-02-24 | 形質転換体及びそれを用いるプロトカテク酸又はその塩の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016064516 | 2016-03-28 | ||
| JP2016-064516 | 2016-03-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017169399A1 true WO2017169399A1 (ja) | 2017-10-05 |
Family
ID=59963135
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/007233 WO2017169399A1 (ja) | 2016-03-28 | 2017-02-24 | 形質転換体及びそれを用いるプロトカテク酸又はその塩の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10961526B2 (ja) |
| EP (1) | EP3438245B1 (ja) |
| JP (1) | JP6685388B2 (ja) |
| CN (1) | CN109477066B (ja) |
| WO (1) | WO2017169399A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019211937A1 (ja) | 2018-05-01 | 2019-11-07 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌の形質転換体およびそれを用いる有用化合物の製造方法 |
| WO2020040017A1 (ja) * | 2018-08-23 | 2020-02-27 | 住友ベークライト株式会社 | 医薬品、抗がん剤、医薬中間体および環式カルボン酸化合物またはその誘導体の製造方法 |
| WO2020130095A1 (ja) | 2018-12-20 | 2020-06-25 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌形質転換体およびそれを用いる2-フェニルエタノールの製造方法 |
| WO2020130067A1 (ja) * | 2018-12-20 | 2020-06-25 | 公益財団法人地球環境産業技術研究機構 | カルボニル化合物の製造法 |
| CN111471630A (zh) * | 2020-06-11 | 2020-07-31 | 鲁东大学 | 一株棒杆菌Ytld-phe09及其应用 |
| WO2021241219A1 (ja) * | 2020-05-29 | 2021-12-02 | 花王株式会社 | 没食子酸合成酵素 |
| WO2021241508A1 (ja) * | 2020-05-29 | 2021-12-02 | 花王株式会社 | 没食子酸生産能を有する形質転換体 |
| CN113981014A (zh) * | 2021-08-30 | 2022-01-28 | 黄山科宏生物香料股份有限公司 | 生产原儿茶酸的方法 |
| DE102021000394A1 (de) | 2021-01-27 | 2022-07-28 | Forschungszentrum Jülich GmbH | Herstellung von 3,4-Dihydroxybenzoat aus D-Xylose mit coryneformen Bakterien |
| CN117683759A (zh) * | 2023-12-19 | 2024-03-12 | 江南大学 | 一种3-脱氢莽草酸脱水酶突变体及产原儿茶酸的重组大肠杆菌 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110184288A (zh) * | 2019-05-28 | 2019-08-30 | 南京趣酶生物科技有限公司 | 没食子酸和原儿茶酸的制备方法及其反应催化剂的制备方法 |
| CN114651066A (zh) * | 2019-11-08 | 2022-06-21 | 花王株式会社 | 具有4-氨基苯甲酸羟化活性的多肽及其用途 |
| EP4069252A4 (en) * | 2019-12-02 | 2024-01-10 | Academia Sinica | PDI-A4 INHIBITORS AND THEIR USE TO INHIBIT BETA CELL PATHOGENESIS AND TREAT DIABETES |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009065839A (ja) * | 2007-09-10 | 2009-04-02 | Genaris Inc | 没食子酸の製造法 |
| JP2009082064A (ja) * | 2007-09-28 | 2009-04-23 | Toyota Industries Corp | 組換えプラスミド、形質転換体及び2h−ピラン−2−オン−4,6−ジカルボン酸の製造法 |
| JP2010207094A (ja) * | 2009-03-06 | 2010-09-24 | Genaris Inc | プロトカテク酸の製造法 |
| WO2012128231A1 (ja) * | 2011-03-18 | 2012-09-27 | 三菱化学株式会社 | ポリマーの製造方法、有機酸の製造方法及び有機酸生産菌 |
| WO2015124687A1 (en) * | 2014-02-20 | 2015-08-27 | Bayer Materialscience Ag | Recombinant strain producing o-aminobenzoate and fermentative production of aniline from renewable resources via 2-aminobenzoic acid |
| WO2015174446A1 (ja) * | 2014-05-14 | 2015-11-19 | グリーンフェノール開発株式会社 | 高活性変異型酵素を高発現させたコリネ型細菌形質転換体、及びそれを用いる4-ヒドロキシ安息香酸又はその塩の製造方法 |
| EP2957635A1 (en) * | 2014-06-18 | 2015-12-23 | Rhodia Opérations | Improved selectivity of the production of vanilloids in a recombinant unicellular host |
| WO2016027870A1 (ja) * | 2014-08-21 | 2016-02-25 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌形質転換体、及びそれを用いる有機化合物の製造方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57183799A (en) | 1981-04-17 | 1982-11-12 | Kyowa Hakko Kogyo Co Ltd | Novel plasmid |
| JPS5867699A (ja) | 1981-10-16 | 1983-04-22 | Ajinomoto Co Inc | プラスミド |
| JPS62166890A (ja) | 1986-01-20 | 1987-07-23 | Asahi Chem Ind Co Ltd | イソクエン酸デヒドロゲナ−ゼ産生遺伝子を含むdna断片 |
| JPH07108228B2 (ja) | 1990-10-15 | 1995-11-22 | 味の素株式会社 | 温度感受性プラスミド |
| US5272073A (en) | 1992-06-30 | 1993-12-21 | Purdue Research Foundation | Biocatalytic synthesis of catechol from glucose |
| US5629181A (en) | 1993-09-16 | 1997-05-13 | Purdue Research Foundation | Synthesis of catechol from biomass-derived carbon sources |
| US5487987A (en) | 1993-09-16 | 1996-01-30 | Purdue Research Foundation | Synthesis of adipic acid from biomass-derived carbon sources |
| JP2000262288A (ja) | 1999-03-16 | 2000-09-26 | Ajinomoto Co Inc | コリネ型細菌の温度感受性プラスミド |
| US6472190B1 (en) | 2000-03-16 | 2002-10-29 | Board Of Trustees Operating Michigan State Univerisity | Biocatalytic synthesis of galloid organics |
| ATE462002T1 (de) | 2003-07-29 | 2010-04-15 | Res Inst Innovative Tech Earth | Transformanten eines coryneformen bakteriums und deren verwendung in verfahren zur produktion von dicarbonsäure |
| JP3860189B2 (ja) | 2004-10-27 | 2006-12-20 | 株式会社興人 | 非結晶性ポリエステル樹脂の製造方法 |
| WO2012033112A1 (ja) | 2010-09-08 | 2012-03-15 | グリーンフェノール・高機能フェノール樹脂製造技術研究組合 | コリネ型細菌形質転換体及びそれを用いるフェノールの製造方法 |
| ES2719304T3 (es) * | 2011-08-08 | 2019-07-09 | Int Flavors & Fragrances Inc | Composiciones y métodos para la biosíntesis de vainillina o beta-D-glucósido de vainillina |
| KR102139454B1 (ko) * | 2012-07-03 | 2020-08-03 | 가오 가부시키가이샤 | 유용미생물 및 목적물질의 제조방법 |
| KR102323473B1 (ko) * | 2014-04-08 | 2021-11-08 | 그린 케미칼즈 가부시키가이샤 | 코리네형 세균 형질 전환체 및 이를 이용하는 4-히드록시벤조산 또는 그 염의 제조 방법 |
| WO2016036915A1 (en) * | 2014-09-03 | 2016-03-10 | Coffa Gianguido | Genetically modified microbes for the biological conversion of carbonaceous materials to protocatechuic acid |
-
2017
- 2017-02-24 CN CN201780021444.5A patent/CN109477066B/zh active Active
- 2017-02-24 US US16/089,567 patent/US10961526B2/en active Active
- 2017-02-24 JP JP2018508800A patent/JP6685388B2/ja active Active
- 2017-02-24 WO PCT/JP2017/007233 patent/WO2017169399A1/ja active Application Filing
- 2017-02-24 EP EP17773944.8A patent/EP3438245B1/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009065839A (ja) * | 2007-09-10 | 2009-04-02 | Genaris Inc | 没食子酸の製造法 |
| JP2009082064A (ja) * | 2007-09-28 | 2009-04-23 | Toyota Industries Corp | 組換えプラスミド、形質転換体及び2h−ピラン−2−オン−4,6−ジカルボン酸の製造法 |
| JP2010207094A (ja) * | 2009-03-06 | 2010-09-24 | Genaris Inc | プロトカテク酸の製造法 |
| WO2012128231A1 (ja) * | 2011-03-18 | 2012-09-27 | 三菱化学株式会社 | ポリマーの製造方法、有機酸の製造方法及び有機酸生産菌 |
| WO2015124687A1 (en) * | 2014-02-20 | 2015-08-27 | Bayer Materialscience Ag | Recombinant strain producing o-aminobenzoate and fermentative production of aniline from renewable resources via 2-aminobenzoic acid |
| WO2015174446A1 (ja) * | 2014-05-14 | 2015-11-19 | グリーンフェノール開発株式会社 | 高活性変異型酵素を高発現させたコリネ型細菌形質転換体、及びそれを用いる4-ヒドロキシ安息香酸又はその塩の製造方法 |
| EP2957635A1 (en) * | 2014-06-18 | 2015-12-23 | Rhodia Opérations | Improved selectivity of the production of vanilloids in a recombinant unicellular host |
| WO2016027870A1 (ja) * | 2014-08-21 | 2016-02-25 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌形質転換体、及びそれを用いる有機化合物の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3438245A4 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019211937A1 (ja) | 2018-05-01 | 2019-11-07 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌の形質転換体およびそれを用いる有用化合物の製造方法 |
| US11359217B2 (en) | 2018-05-01 | 2022-06-14 | Research Institute Of Innovative Technology For The Earth | Transformant of coryneform bacterium and production method for useful compound using same |
| EP3789481A4 (en) * | 2018-05-01 | 2022-02-16 | Research Institute Of Innovative Technology For The Earth | TRANSFORMING CORYNEFORM BACTERIA AND METHOD FOR PRODUCING A USEFUL COMPOUND USING THE SAME |
| CN112074599B (zh) * | 2018-05-01 | 2024-09-03 | 公益财团法人地球环境产业技术研究机构 | 棒状细菌的转化体和使用其的有用化合物的制造方法 |
| CN112074599A (zh) * | 2018-05-01 | 2020-12-11 | 公益财团法人地球环境产业技术研究机构 | 棒状细菌的转化体和使用其的有用化合物的制造方法 |
| JPWO2019211937A1 (ja) * | 2018-05-01 | 2021-05-13 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌の形質転換体およびそれを用いる有用化合物の製造方法 |
| JP7317810B2 (ja) | 2018-05-01 | 2023-07-31 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌の形質転換体およびそれを用いる有用化合物の製造方法 |
| WO2020040017A1 (ja) * | 2018-08-23 | 2020-02-27 | 住友ベークライト株式会社 | 医薬品、抗がん剤、医薬中間体および環式カルボン酸化合物またはその誘導体の製造方法 |
| CN112601529A (zh) * | 2018-08-23 | 2021-04-02 | 住友电木株式会社 | 医药品、抗癌剂、药物中间体及环式羧酸化合物或其衍生物的制造方法 |
| WO2020130067A1 (ja) * | 2018-12-20 | 2020-06-25 | 公益財団法人地球環境産業技術研究機構 | カルボニル化合物の製造法 |
| JPWO2020130067A1 (ja) * | 2018-12-20 | 2021-09-27 | 公益財団法人地球環境産業技術研究機構 | カルボニル化合物の製造法 |
| US12006527B2 (en) | 2018-12-20 | 2024-06-11 | Research Institute Of Innovative Technology For The Earth | Coryneform bacterium transformant and method for producing 2-phenylethanol using same |
| CN113260706A (zh) * | 2018-12-20 | 2021-08-13 | 公益财团法人地球环境产业技术研究机构 | 棒状细菌转化体以及使用其的2-苯基乙醇的制造方法 |
| JP7217294B2 (ja) | 2018-12-20 | 2023-02-02 | 公益財団法人地球環境産業技術研究機構 | カルボニル化合物の製造法 |
| WO2020130095A1 (ja) | 2018-12-20 | 2020-06-25 | 公益財団法人地球環境産業技術研究機構 | コリネ型細菌形質転換体およびそれを用いる2-フェニルエタノールの製造方法 |
| WO2021241219A1 (ja) * | 2020-05-29 | 2021-12-02 | 花王株式会社 | 没食子酸合成酵素 |
| WO2021241508A1 (ja) * | 2020-05-29 | 2021-12-02 | 花王株式会社 | 没食子酸生産能を有する形質転換体 |
| CN111471630A (zh) * | 2020-06-11 | 2020-07-31 | 鲁东大学 | 一株棒杆菌Ytld-phe09及其应用 |
| DE102021000394A1 (de) | 2021-01-27 | 2022-07-28 | Forschungszentrum Jülich GmbH | Herstellung von 3,4-Dihydroxybenzoat aus D-Xylose mit coryneformen Bakterien |
| CN113981014A (zh) * | 2021-08-30 | 2022-01-28 | 黄山科宏生物香料股份有限公司 | 生产原儿茶酸的方法 |
| CN117683759A (zh) * | 2023-12-19 | 2024-03-12 | 江南大学 | 一种3-脱氢莽草酸脱水酶突变体及产原儿茶酸的重组大肠杆菌 |
Also Published As
| Publication number | Publication date |
|---|---|
| US10961526B2 (en) | 2021-03-30 |
| EP3438245B1 (en) | 2023-01-11 |
| US20190119664A1 (en) | 2019-04-25 |
| JPWO2017169399A1 (ja) | 2018-09-06 |
| CN109477066B (zh) | 2021-10-01 |
| CN109477066A (zh) | 2019-03-15 |
| EP3438245A1 (en) | 2019-02-06 |
| JP6685388B2 (ja) | 2020-04-22 |
| EP3438245A4 (en) | 2019-10-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6685388B2 (ja) | 形質転換体及びそれを用いるプロトカテク酸又はその塩の製造方法 | |
| JP6302073B2 (ja) | コリネ型細菌形質転換体、及びそれを用いる有機化合物の製造方法 | |
| JP6564929B2 (ja) | コリネ型細菌形質転換体及びそれを用いる4−アミノ安息香酸又はその塩の製造方法 | |
| US10738296B2 (en) | Transformant for producing 4-hydroxybenzoic acid or salt thereof | |
| US9404115B2 (en) | Coryneform bacterium transformant and process for producing phenol using the same | |
| EP2615170B1 (en) | Coryneform bacterium transformant and method for producing phenol using same | |
| KR20160143644A (ko) | 코리네형 세균 형질 전환체 및 이를 이용하는 4-히드록시벤조산 또는 그 염의 제조 방법 | |
| US8846367B2 (en) | Coryneform bacterium transformant and process for producing phenol using the same | |
| US12006527B2 (en) | Coryneform bacterium transformant and method for producing 2-phenylethanol using same | |
| JP7376041B2 (ja) | 形質転換体及びそれを用いる1,3-ブタンジオールの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2018508800 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017773944 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017773944 Country of ref document: EP Effective date: 20181029 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17773944 Country of ref document: EP Kind code of ref document: A1 |