WO2014192903A1 - ジベンジルアミン構造を有するピリミジン化合物の新規形態 - Google Patents
ジベンジルアミン構造を有するピリミジン化合物の新規形態 Download PDFInfo
- Publication number
- WO2014192903A1 WO2014192903A1 PCT/JP2014/064372 JP2014064372W WO2014192903A1 WO 2014192903 A1 WO2014192903 A1 WO 2014192903A1 JP 2014064372 W JP2014064372 W JP 2014064372W WO 2014192903 A1 WO2014192903 A1 WO 2014192903A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrochloride
- pyrimidine compound
- methyl
- ethyl
- phenyl
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/47—One nitrogen atom and one oxygen or sulfur atom, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
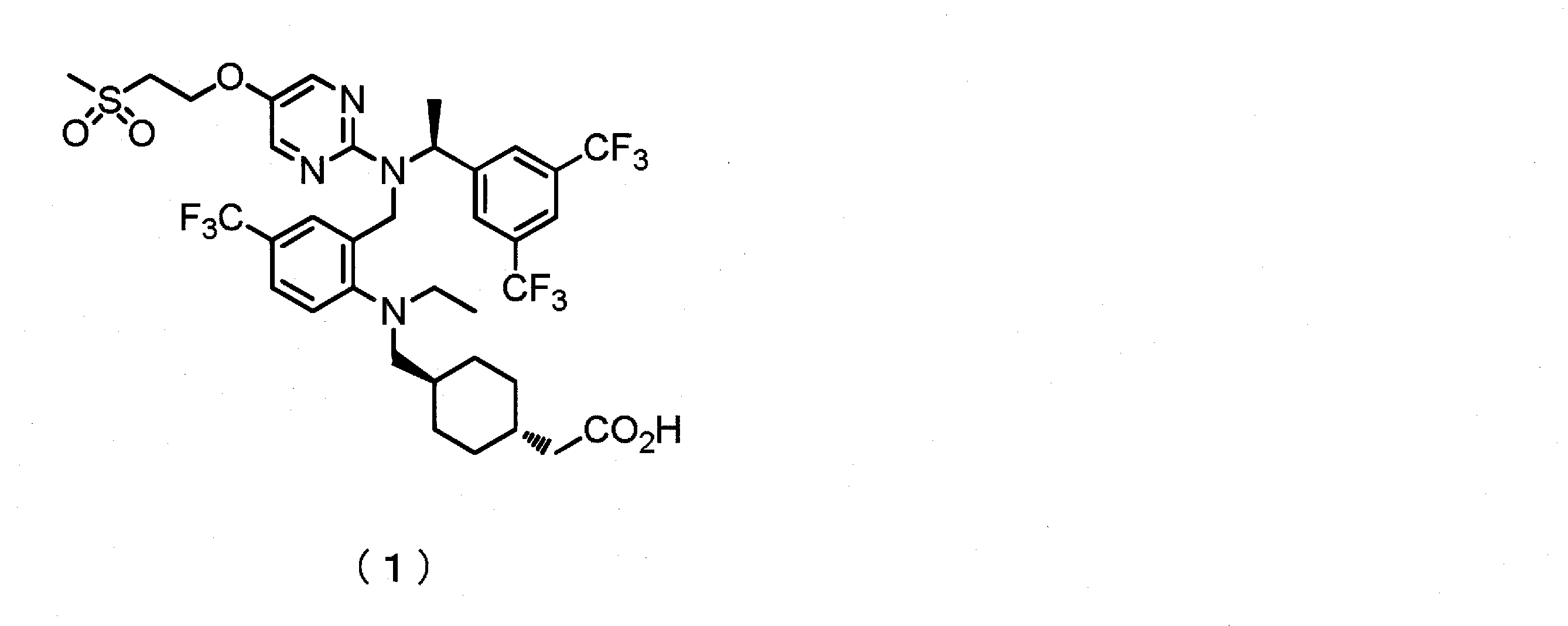
- the present invention is a pyrimidine compound having a dibenzylamine structure useful for the prevention and / or treatment of diseases such as dyslipidemia, (S) -trans- ⁇ 4-[( ⁇ 2-[( ⁇ 1- [ 3,5-bis (trifluoromethyl) phenyl] ethyl ⁇ ⁇ 5- [2- (methylsulfonyl) ethoxy] pyrimidin-2-yl ⁇ amino) methyl] -4- (trifluoromethyl) phenyl ⁇ (ethyl) amino ) Methyl] cyclohexyl ⁇ acetic acid.
- diseases such as dyslipidemia, (S) -trans- ⁇ 4-[( ⁇ 2-[( ⁇ 1- [ 3,5-bis (trifluoromethyl) phenyl] ethyl ⁇ ⁇ 5- [2- (methylsulfonyl) ethoxy] pyrimidin-2-yl ⁇ amino) methyl] -4- (trifluoromethyl) phenyl
- Patent Documents 1, 2, and 3 Prediction of diseases such as abnormalities It is known to be useful for prevention and treatment (Patent Documents 1, 2, and 3).
- Patent Document 1 Example 45 it has been disclosed in Patent Document 1 Example 45 that the racemate has been obtained as a pale yellow oil, and Patent Document 2 Example 1 and Patent Document 3 Production Example 2 have been disclosed so far.
- US Pat. No. 6,057,093 it was disclosed that it was obtained as a white amorphous.
- the crystal of pyrimidine compound (1) has not been reported so far.
- a crystallization technique can be established for a low molecular weight compound that can be used as an active ingredient of a pharmaceutical product, the purity can be improved by recrystallization, so that a high-purity pharmaceutical product can be provided.
- crystals are superior in homogeneity and less susceptible to variations in solubility and the like as compared with forms having poor crystallinity such as amorphous and non-crystalline solids, it is possible to provide homogeneous pharmaceuticals.
- crystals are usually solid and easy to handle, they are advantageous when producing pharmaceutical preparations.
- it is generally desired that a low molecular weight compound that can be used as an active ingredient of a pharmaceutical is in a crystalline form.
- the crystal formation of the compound is extremely unpredictable, and the fact is that the possibility of crystal formation, the conditions for crystal formation, etc. are not understood at all unless actually examined.
- An object of the present invention is to provide a novel form of the pyrimidine compound (1) useful for the prevention and / or treatment of diseases such as dyslipidemia.
- the present inventors first made extensive studies on crystallization of a free form of the pyrimidine compound (1). However, it was difficult to crystallize the pyrimidine compound (1) in a free form, and crystals could not be obtained even when examined under various conditions. Therefore, the inventors of the present invention conducted further crystallization after making pyrimidine compound (1) into various salts, and as a result, sulfate, arginine salt and the like of pyrimidine compound (1), and further hydrohalide salts. In contrast, when the pyrimidine compound (1) is a hydrochloride, which is also a kind of hydrohalide, it cannot be crystallized with the hydrobromide which is a kind of the hydrobromide. An excellent crystal was obtained, and it was found that a stable pharmaceutical composition could be provided by using the crystal, and the present invention was completed.
- the powder X-ray diffraction pattern obtained by irradiation with copper K ⁇ rays shows a value around 14.0 ⁇ 0.2 °, around 18.3 ⁇ 0.2 °, around 20.1 ⁇ 0.2 °, and 20.
- the powder X-ray diffraction pattern obtained by irradiation with copper K ⁇ rays has peaks at diffraction angles (2 ⁇ ) around 18.3 ⁇ 0.2 ° and 20.5 ⁇ 0.2 °, [3] Or crystals according to [4].
- the powder X-ray diffraction pattern obtained by irradiation with copper K ⁇ rays shows a value around 14.0 ⁇ 0.2 °, around 18.3 ⁇ 0.2 °, around 20.1 ⁇ 0.2 °, and 20.
- a pharmaceutical composition comprising the compound according to any one of [1] to [11].
- a pharmaceutical composition comprising the compound according to any one of [1] to [11] above and a pharmaceutically acceptable carrier.
- a method for producing a pharmaceutical composition comprising a step of mixing the compound according to any one of [1] to [11] above with a pharmaceutically acceptable carrier.
- the hydrochloride of the pyrimidine compound (1) according to the present invention can be used as a raw material for crystal formation of the pyrimidine compound (1) which is difficult to crystallize. Moreover, the crystal
- crystallization of pyrimidine compound (1) hydrochloride has favorable heat stability, and is useful for manufacture of a pharmaceutical with favorable quality.
- FIG. 1 is a diagram showing a powder X-ray diffraction pattern of crystals of pyrimidine compound (1) hydrochloride obtained in 1-3 in Example 1.
- FIG. 3 is a diagram showing thermal analysis measurement (TG-DTA measurement) data of pyrimidine compound (1) hydrochloride crystals obtained in 1-3 in Example 1.
- the number of hydrochloric acids is not particularly limited and may be any of monohydrochloride, dihydrochloride, trihydrochloride and tetrahydrochloride, and may be a mixture thereof.
- Monohydrochloride is preferable from the viewpoint of being obtained as a stable acid addition salt.
- the hydrochloride of the pyrimidine compound (1) is represented by the following formula (2):
- the specific crystal form of the pyrimidine compound (1) hydrochloride is not particularly limited, and may be any of different crystal forms, or a mixture thereof. Further, the pyrimidine compound (1) It may be a mixture of amorphous hydrochloride. Whether the pyrimidine compound (1) hydrochloride is a crystal is determined by, for example, X-ray diffraction measurement (specifically, powder X-ray diffraction measurement, etc.), thermal analysis measurement (specifically, differential thermal analysis (DTA)). ), Differential scanning calorimetry (DSC), etc.], polarization confirmation (specifically, observation with a polarizing microscope, etc.), solid NMR measurement, etc., can be confirmed by known methods.
- X-ray diffraction measurement specifically, powder X-ray diffraction measurement, etc.
- thermal analysis measurement specifically, differential thermal analysis (DTA)).
- DSC Differential scanning calorimetry
- polarization confirmation specifically, observation with a polarizing microscope, etc.
- the pyrimidine compound (1) hydrochloride is crystalline. It can be confirmed that there is.
- the method for determining crystallinity can be carried out with reference to descriptions of the Japanese Pharmacopeia, US Pharmacopeia, European Pharmacopeia and the like. These crystals may be confirmed in the presence of other components.
- solid pharmaceutical compositions (tablets, capsules, granules, powders, etc.) containing the hydrochloride of pyrimidine compound (1) and a pharmaceutically acceptable carrier require solid pharmaceutical compositions.
- a pharmaceutically acceptable carrier for example, solid pharmaceutical compositions (tablets, capsules, granules, powders, etc.) containing the hydrochloride of pyrimidine compound (1) and a pharmaceutically acceptable carrier require solid pharmaceutical compositions.
- the crystal of the pyrimidine compound (1) hydrochloride has a powder X-ray diffraction pattern obtained when irradiated with copper K ⁇ rays, at least around 14.0 ⁇ 0.2 °, 18.3 ⁇ 0. Near 2 °, 20.1 ⁇ 0.2 °, 20.5 ⁇ 0.2 °, 21.3 ⁇ 0.2 °, 21.8 ⁇ 0.2 °, 23.3 ⁇ 0.
- a crystal having a peak at one or more diffraction angles (2 ⁇ ) selected from the group consisting of around 2 ° and around 24.0 ⁇ 0.2 ° is preferred, and at least a diffraction angle (2 ⁇ of around 20.5 ⁇ 0.2 °).
- Crystals having peaks at diffraction angles (2 ⁇ ) of at least about 18.3 ⁇ 0.2 ° and 20.5 ⁇ 0.2 ° are more preferable, and at least 14.0 ⁇ Near 0.2 °, 18.3 ⁇ 0.2 °, 20.1 ⁇ 0.2 °, 20.5 ⁇ 0 At diffraction angles (2 ⁇ ) around 2 °, 21.3 ⁇ 0.2 °, 21.8 ⁇ 0.2 °, 23.3 ⁇ 0.2 °, and 24.0 ⁇ 0.2 °. Crystals having peaks are even more preferred, and crystals that are substantially identical to those shown in FIG. 1 are particularly preferred.
- the crystal of the pyrimidine compound (1) hydrochloride is preferably a crystal having an endothermic peak in the vicinity of about 162 ⁇ 5 ° C. in differential thermal analysis (DTA). More preferred are crystals whose thermal analysis (DTA) and thermal mass measurement (TG) results are substantially the same as those shown in FIG.
- hydrochloride of the pyrimidine compound (1) and the crystal thereof according to the present invention may be a solvate such as a hydrate or a non-solvate such as an anhydride. Preferably there is.
- the hydrochloride of the pyrimidine compound (1) and the crystal thereof are, for example, the following steps: (Step 1) Formation of hydrochloride from a free form of pyrimidine compound (1) (Step 2) Production by crystal formation from hydrochloride of pyrimidine compound (1).
- Step 1 Formation of hydrochloride from a free form of pyrimidine compound (1)
- Step 2 Production by crystal formation from hydrochloride of pyrimidine compound (1).
- Step 1 Formation of hydrochloride salt from free form of pyrimidine compound (1)>
- This step is a step of forming a hydrochloride by coexisting the pyrimidine compound (1) and hydrogen chloride in the presence of a solvent.
- this step is a step of forming a salt by dissolving a free form of the pyrimidine compound (1) in a solvent and supplying hydrogen chloride.
- the free form of the pyrimidine compound (1) as a starting material can be produced, for example, according to the method described in Patent Document 2.
- This step is performed in the presence of a solvent.
- the solvent is not particularly limited as long as it does not participate in hydrochloride formation.
- aromatic hydrocarbons such as benzene, toluene, xylene, mesitylene, chlorobenzene, 1,2-dichlorobenzene, nitrobenzene; diethyl ether , Ethers such as diisopropyl ether, tert-butyl methyl ether and 1,4-dioxane; acetates such as ethyl acetate, n-propyl acetate and isopropyl acetate; ketones such as acetone, 2-butanone and 3-pentanone; Aliphatic hydrocarbons such as n-pentane, n-hexane, n-heptane, cyclohexane, n-octane and n-decane; Amides such as N, N-dimethylformamide and N,
- the pyrimidine compound (1) may be dissolved after mixing them, or the remaining solvent after dissolving the pyrimidine compound (1) in one solvent. May be added.
- the solvent is preferably at least one selected from diisopropyl ether, tert-butyl methyl ether, 1,4-dioxane, n-hexane, n-heptane, ethyl acetate, and isopropyl acetate.
- the amount of the solvent is not particularly limited, but the volume ratio of the pyrimidine compound (1) to the free body weight is 1 to 20 times (V / W), preferably 5 to 15 times (V / W). That's fine.
- the supply source of hydrogen chloride is not particularly limited, and hydrogen chloride gas is directly blown into the solution, and easily available concentrated hydrochloric acid, 4M HCl / ethyl acetate solution, 4M HCl / 1,4-dioxane solution, and the like can be used.
- the amount of hydrogen chloride is not particularly limited, but is preferably 1 to 5 molar equivalents and particularly preferably 1 to 4 molar equivalents with respect to the free form of pyrimidine compound (1).
- the salt formation temperature is not particularly limited, but is usually in the range of ⁇ 50 to 150 ° C., preferably ⁇ 20 to 80 ° C., more preferably ⁇ 10 to 40 ° C.
- the time required for salt formation is not particularly limited, but is usually 5 minutes to 48 hours, preferably 30 minutes to 24 hours, more preferably 30 minutes to 3 hours.
- the hydrochloride of the produced pyrimidine compound (1) can be isolated.
- the salt precipitated as a solid may be isolated by a method usually used in the technical field such as filtration, and further dried by a method usually used in the technical field as necessary.
- the drying means is not particularly limited, and examples thereof include heating and / or drying under reduced pressure.
- the drying temperature is preferably 50 ° C. or less, and more preferably 40 to 50 ° C.
- the drying time is preferably 1 to 24 hours, more preferably 6 to 12 hours.
- Step 2 Formation of crystal from hydrochloride of pyrimidine compound (1)>
- This step is a step of crystallizing the hydrochloride (eg, amorphous) of the pyrimidine compound (1) obtained in Step 1 in the presence of a solvent.
- this step is a step of adding the hydrochloride of the pyrimidine compound (1) obtained in Step 1 to a solvent, dissolving it by heating or the like as necessary, and then performing crystallization by cooling or the like. It is.
- This step is performed in the presence of a solvent.
- the solvent include a mixed solution of 2-propanol and heptane, or a mixed solution of methyl ethyl ketone and heptane, and a mixed solution of 2-propanol and heptane is preferable.
- the mixing ratio of the solvent is not particularly limited, but as a volume ratio to the volume of 2-propanol or methyl ethyl ketone, heptane is 0.1 to 2 times (V / V), preferably 0.2 to 1 times (V / V). ) Should be used.
- the hydrochloride of the pyrimidine compound (1) When the hydrochloride of the pyrimidine compound (1) is dissolved in a solvent, the hydrochloride of the pyrimidine compound (1) may be dissolved after mixing the solvent in advance, but the hydrochloride of the pyrimidine compound (1) may be dissolved in 2-propanol or It is preferable to add heptane after dissolving in methyl ethyl ketone.
- the amount of the solvent is not particularly limited, but the total mixed solvent amount is 1 to 20 times (V / W), preferably 5 to 10 times the volume ratio of the pyrimidine compound (1) to the weight of the hydrochloride. V / W) may be used.
- the temperature at which the hydrochloride of the pyrimidine compound (1) is dissolved in the solvent is not particularly limited, but is usually in the range of 40 to 100 ° C., preferably 50 to 80 ° C.
- the crystallization temperature of the hydrochloride of the pyrimidine compound (1) is not particularly limited, but it may be usually in the range of 5 to 40 ° C., preferably 10 to 35 ° C., more preferably 10 to 30 ° C., particularly preferably. Is 15 to 25 ° C. If there is a discrepancy between the temperature at which the hydrochloride of the pyrimidine compound (1) is dissolved in the solvent and the crystallization temperature, it may be cooled slowly in about 1 to 10 hours depending on the temperature difference.
- the time required for crystallization is not particularly limited, but is usually 1 hour or longer, preferably 6 to 24 hours, more preferably 8 to 16 hours.
- the precipitated pyrimidine compound (1) hydrochloride crystals may be isolated by a method commonly used in the technical field such as filtration, and further dried by a method commonly used in the technical field as necessary.
- the drying means is not particularly limited, and examples thereof include heating and / or drying under reduced pressure.
- the drying temperature is preferably 50 ° C. or less, and more preferably 40 to 50 ° C.
- the drying time is preferably 1 to 24 hours, more preferably 6 to 12 hours.
- seed crystal crystallization (seed crystal) of the pyrimidine compound (1) hydrochloride manufactured separately.
- solvent isopropyl acetate may be used instead of the above-described one.
- the amount of seed crystals is not particularly limited, but may be 0.00001 to 0.05 parts by weight, preferably 0.0001 to 0.01 parts by weight, based on the hydrochloride of pyrimidine compound (1).
- the seed crystal is preferably added after the hydrochloride of the pyrimidine compound (1) is dissolved in a solvent.
- hydrochloride of this invention when the crystal
- hydrochloride crystals can be produced from the free form of the pyrimidine compound (1) in the presence of a solvent.
- the solvent is preferably isopropyl acetate. It is also possible to perform step 2 while omitting the time for salt formation in step 1. That is, the free form of the pyrimidine compound (1) can be dissolved in a solvent and hydrogen chloride can be supplied, followed by heating or the like, followed by cooling or the like for crystallization. The operation of other steps is the same as described above.
- the pyrimidine compound (1) has a CETP inhibitory action, a PCSK9 protein amount reducing action, and the like. Therefore, the hydrochloride of the pyrimidine compound (1) and its crystal of the present invention can be used as a pharmaceutical component useful for the prevention and / or treatment of diseases such as dyslipidemia, hyper-LDL and hypoHDL.
- the crystal of the hydrochloride of the pyrimidine compound (1) of the present invention has excellent thermal stability as will be apparent from Test Example 2 described later, and can be particularly suitably used as a component of a stable pharmaceutical composition. Moreover, since the crystal
- a pharmaceutical composition for oral administration include pharmaceutical compositions in dosage forms such as tablets, capsules, granules, powders, oral solutions, syrups, and oral jelly.
- pharmaceutical compositions for parenteral administration include injections, inhalants, eye drops, ear drops, nasal drops, suppositories, external solid preparations, external liquid preparations, sprays, and ointments. And pharmaceutical compositions in dosage forms such as agents, creams, gels and patches.
- compositions can be produced by adding a pharmaceutically acceptable carrier (additive).
- additives include excipients, binders, extenders, disintegrants, surfactants, lubricants, dispersants, buffers, preservatives, flavoring agents, fragrances, coating agents, and diluents. Although it is mentioned, it is not limited to these.
- the dose of the pyrimidine compound (1) varies depending on the patient's weight, age, sex, symptoms, etc., but in the case of a normal adult, about 0.01 to 1000 mg in terms of the free form of the pyrimidine compound (1) is 1
- the administration can be divided into 1 to 4 times a day. Preferably, about 0.1-100 mg can be administered in 1 to 4 divided doses per day.
- Powder X-ray diffraction measurement was performed on the crystals obtained in 1-3. The powder X-ray diffraction was measured by filling the crushed crystal sample in the sample holder portion of the silicon non-reflective sample plate for X-ray diffraction and under the following conditions.
- Powder X-ray diffractometer RINT-UltimaIV-Protectus (manufactured by Rigaku Corporation)
- Sampling width 0.02 °
- Scan speed 2.00 ° / min
- the obtained diffraction pattern is shown in FIG.
- the vertical axis indicates the diffraction intensity (count / second (cps)), and the horizontal axis indicates the diffraction angle 2 ⁇ (°).
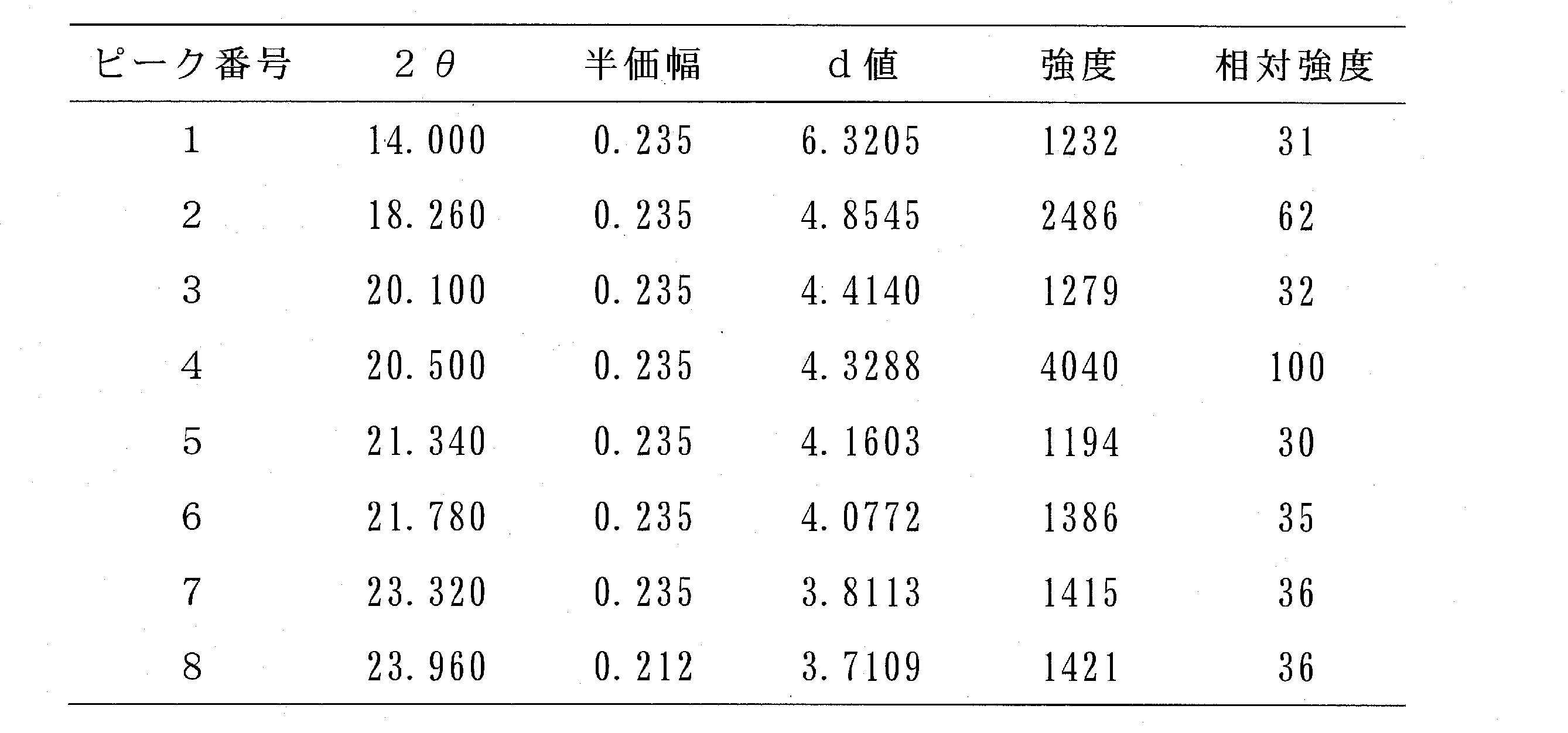
- Table 7 shows the diffraction angle 2 ⁇ , the half width, the d value, the intensity, and the relative intensity for main peaks having a relative intensity of 30 or more. From FIG.
- Example 1-3 The crystal obtained in Example 1-3 was subjected to thermal analysis measurement.
- the thermal analysis measurement about 5 mg of a sample is accurately weighed in an aluminum pan for thermal analysis, Al 2 O 3 is used as a reference material, under a nitrogen atmosphere (150 mL / min), at a heating rate of 10 ° C./min.
- a thermal analyzer Thermo Plus 2 system manufactured by Rigaku Corporation
- DTA differential thermal analysis
- TG thermal mass measurement
- the vertical axis represents the thermoelectromotive force ( ⁇ V) of the thermocouple for the DTA curve
- the mass change (mg) for the TG curve and the horizontal axis represents the temperature (° C.).
- the crystal of pyrimidine compound (1) hydrochloride had an endothermic peak around 162 ⁇ 5 ° C. (specifically, 161.6 ° C.) in differential thermal analysis (DTA).
- DTA differential thermal analysis
- Test Example 2 Thermal stability test The test compound was put in a glass bottle and the residual ratio (%) of the pyrimidine compound (1) in the test compound after being stored for a certain period of time at 80 ° C, 100 ° C or 120 ° C. ) was measured. The residual rate was determined by measuring the ratio of the pyrimidine compound (1) contained in the test compound as a percentage of peak area using high performance liquid chromatography. In the measurement by high performance liquid chromatography, an ODS column is used as a column, two kinds of a 0.1% TFA aqueous solution and a 0.1% TFA acetonitrile solution are mixed and used as a solvent, and a detection wavelength is 242 nm. It was. From the area percentage of the obtained pyrimidine compound (1), the residual ratio was calculated by the following formula.
- test compound used was the crystal (pyrimidine compound (1) hydrochloride crystal) obtained in Example 1-3 and a free form of pyrimidine compound (1).
- the results are shown in Table 8.
- the pyrimidine compound (1) useful for the prevention and / or treatment of diseases such as dyslipidemia can be provided in a form suitable for the production of a pharmaceutical having high purity, homogeneity and excellent quality. It can be used in the pharmaceutical industry.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
ピリミジン化合物(1)については、これまでに特許文献1 実施例45においてそのラセミ体が淡黄色油状物として得られた旨開示されているほか、特許文献2 実施例1や特許文献3 製造例2において白色アモルファスとして得られた旨開示されている。
しかしながら、ピリミジン化合物(1)の結晶については、これまでに報告されていない。
以上のような利点から、一般に、医薬品の有効成分として利用可能な低分子化合物を結晶の形態とすることが望まれる。しかしながら、化合物の結晶形成は予測性が極めて低く、結晶形成の可否・結晶形成の条件等については実際に検討してみなければ全く分からないのが実情である。
そこで、本発明者らは、ピリミジン化合物(1)を種々の塩とした上で結晶化を更に鋭意検討したところ、ピリミジン化合物(1)の硫酸塩やアルギニン塩等、さらにはハロゲン化水素酸塩の一種である臭化水素酸塩では結晶化できなかったのに対し、ピリミジン化合物(1)を、同じくハロゲン化水素酸塩の一種である塩酸塩とした場合に特異的に、熱安定性に優れた結晶が得られ、当該結晶を用いれば安定な医薬組成物が提供できることを見出し、本発明を完成するに至った。
[1](S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸の塩酸塩。
[2]1塩酸塩である、[1]記載の塩酸塩。
[3](S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の結晶。
[4]1塩酸塩である、[3]記載の結晶。
[5]銅Kα線の照射で得られる粉末X線回折パターンが、14.0±0.2°付近、18.3±0.2°付近、20.1±0.2°付近、20.5±0.2°付近、21.3±0.2°付近、21.8±0.2°付近、23.3±0.2°付近及び24.0±0.2°付近よりなる群から選ばれる1以上の回折角(2θ)にピークを有する、[3]又は[4]記載の結晶。
[6]銅Kα線の照射で得られる粉末X線回折パターンが、20.5±0.2°付近の回折角(2θ)にピークを有する、[3]又は[4]記載の結晶。
[7]銅Kα線の照射で得られる粉末X線回折パターンが、18.3±0.2°付近及び20.5±0.2°付近の回折角(2θ)にピークを有する、[3]又は[4]記載の結晶。
[8]銅Kα線の照射で得られる粉末X線回折パターンが、14.0±0.2°付近、18.3±0.2°付近、20.1±0.2°付近、20.5±0.2°付近、21.3±0.2°付近、21.8±0.2°付近、23.3±0.2°付近及び24.0±0.2°付近の回折角(2θ)にピークを有する、[3]又は[4]記載の結晶。
[9]銅Kα線の照射で得られる粉末X線回折パターンが、図1に示されたものと実質的に同一である、[3]又は[4]記載の結晶。
[10]示差熱分析(DTA)において、162±5.0℃付近に吸熱ピークを有する、[3]~[9]のいずれかに記載の結晶。
[11]熱分析測定(示差熱分析(DTA)及び熱質量測定(TG))結果が、図2に示されたものと実質的に同一である、[3]~[9]のいずれかに記載の結晶。
[12]前記[1]~[11]のいずれかに記載の化合物を含有する医薬組成物。
[13]前記[1]~[11]のいずれかに記載の化合物、及び製薬上許容される担体を含有する医薬組成物。
[14]前記[1]~[11]のいずれかに記載の化合物を、製薬上許容される担体と混合する工程を含む、医薬組成物の製造方法。
[15]前記[1]~[11]のいずれかに記載の化合物の、医薬組成物の製造のための使用。
[16]前記[1]~[11]のいずれかに記載の化合物の、医薬組成物の製造原料としての使用。
[17]医薬組成物の製造に使用するための、前記[1]~[11]のいずれかに記載の化合物。
[18]医薬組成物の製造原料として使用するための、前記[1]~[11]のいずれかに記載の化合物。
また、ピリミジン化合物(1) 塩酸塩の結晶は、熱安定性が良好であり、品質の良好な医薬品の製造に有用である。
本発明において、ピリミジン化合物(1)の塩酸塩としては、下記式(2):

ピリミジン化合物(1) 塩酸塩が結晶であるか否かは、例えばX線回折測定(具体的には、粉末X線回折測定等)、熱分析測定(具体的には、示差熱分析法(DTA)、示差走査熱量測定法(DSC)等)、偏光性の確認(具体的には、偏光顕微鏡による観察等)、固体NMR測定など、結晶性を判断する公知の方法により確認できる。例えば、ある固体状のピリミジン化合物(1) 塩酸塩について銅Kα線の照射による粉末X線回折測定を行い、明確なピークが観察される場合には、そのピリミジン化合物(1) 塩酸塩は結晶であると確認できる。なお、結晶性を判断する方法(粉末X線回折測定法、熱分析法など)は、日本薬局方、米国薬局方、欧州薬局方等の記載を参考に実施できる。
また、これらの結晶の確認は他の成分の共存下で行ってもよい。例えば、ピリミジン化合物(1)の塩酸塩、及び製薬上許容される担体を含有する固形状の医薬組成物(錠剤、カプセル剤、顆粒剤、散剤など)においては、固形状の医薬組成物を必要に応じて粉砕し、X線回折測定を行い、ピリミジン化合物(1)の塩酸塩由来のピークが観察される場合には、そのピリミジン化合物(1)の塩酸塩は結晶であると確認できる。
(工程1)ピリミジン化合物(1)のフリー体からの、塩酸塩の形成
(工程2)ピリミジン化合物(1)の塩酸塩からの、結晶の形成
により製造することができる。
以下、各工程に分けて詳述するが、本発明のピリミジン化合物(1)の塩酸塩やその結晶の製造方法は、以下に記載の方法に何ら限定されるものではない。
本工程は、溶媒存在下、ピリミジン化合物(1)と塩化水素を共存させて塩酸塩を形成する工程である。具体的には、本工程は、ピリミジン化合物(1)のフリー体を溶媒に溶解し、塩化水素を供給して塩形成を行う工程である。
溶媒としては、ジイソプロピルエーテル、tert-ブチルメチルエーテル、1,4-ジオキサン、n-ヘキサン、n-ヘプタン、酢酸エチル及び酢酸イソプロピルから選ばれる1種以上が好ましく、ジイソプロピルエーテル、tert-ブチルメチルエーテル、1,4-ジオキサン及び酢酸イソプロピルから選ばれる1種以上がより好ましく、tert-ブチルメチルエーテル又は酢酸イソプロピルが特に好ましい。
溶媒の量は特に制限は無いが、ピリミジン化合物(1)のフリー体の重量に対する容量比として、1~20倍量(V/W)、好ましくは5~15倍量(V/W)を用いればよい。
塩化水素の量は、特に制限は無いが、ピリミジン化合物(1)のフリー体に対して、1~5モル当量が好ましく、1~4モル当量が特に好ましい。
本工程は、溶媒存在下、工程1で得られたピリミジン化合物(1)の塩酸塩(例えば、非晶質のもの)を結晶化する工程である。具体的には、本工程は、工程1で得られたピリミジン化合物(1)の塩酸塩を溶媒に添加し、必要に応じて加熱等して溶解し、その後冷却等して結晶化を行う工程である。
ピリミジン化合物(1)の塩酸塩を溶媒に溶解する場合、あらかじめ溶媒を混合した後にピリミジン化合物(1)の塩酸塩を溶解させてもよいが、ピリミジン化合物(1)の塩酸塩を2-プロパノール又はメチルエチルケトンに溶解させた後、ヘプタンを添加するのが好ましい。
溶媒の量は特に制限は無いが、混合溶媒総量として、ピリミジン化合物(1)の塩酸塩の重量に対する容量比として、1~20倍量(V/W)、好ましくは、5~10倍量(V/W)を用いればよい。
ピリミジン化合物(1)の塩酸塩の結晶化の温度は、特に制限は無いが、通常5~40℃の範囲で行えばよく、好ましくは10~35℃、より好ましくは10~30℃、特に好ましくは15~25℃である。なお、ピリミジン化合物(1)の塩酸塩を溶媒に溶かす際の温度と、結晶化の温度に乖離がある場合は、温度差に応じて、適宜1~10時間程度でゆっくりと冷却すればよい。
結晶化に要する時間は、特に制限は無いが、通常1時間以上、好ましくは6~24時間、より好ましくは8~16時間である。
種晶は、ピリミジン化合物(1)の塩酸塩を溶媒に溶解した後に添加するのが好ましい。
また、工程1において塩形成のための時間を省略して工程2を行うことも可能である。すなわち、ピリミジン化合物(1)のフリー体を溶媒に溶解し、塩化水素を供給した後、加熱等し、その後冷却等して結晶化を行うことも可能である。
その他の各工程の操作等は、前記したものと同様である。
また、本発明のピリミジン化合物(1)の塩酸塩の結晶は優れた熱安定性を有するため、原料として保存する際の安定性も良好であり、医薬組成物の製造原料としても好適に利用できる。斯かる原料としての使用の場合、製造された医薬組成物中において結晶形態が維持されていることは必ずしも要しない。
ピリミジン化合物(1)の投与量は、患者の体重、年齢、性別、症状等によって異なるが、通常成人の場合、ピリミジン化合物(1)のフリー体に換算して、約0.01~1000mgを1日1~4回に分けて投与することができる。好ましくは約0.1~100mgを1日1~4回に分けて投与することができる。
なお、下記実施例中で用いられている略号は下記の意味を示す。
s:シングレット(singlet)
d:ダブレット(doublet)
t:トリプレット(triplet)
q:クアルテット(quartet)
m:マルチプレット(multiplet)
br:ブロード(broad)
J:カップリング定数(coupling constant)
Hz:ヘルツ(Hertz)
DMSO-d6:重ジメチルスルホキシド
1H-NMR:プロトン核磁気共鳴
各種サンプル(ピリミジン化合物(1)のフリー体、及びその塩類(塩酸塩、臭化水素酸塩、硫酸塩、D-(-)-アルギニン塩、シンコニジン塩))について、以下の方法により結晶化条件を検討した。
なお、ピリミジン化合物(1)の塩酸塩は実施例1中、1-1の工程1記載の方法にて得た。また、他の塩類は、メタノールに溶解したピリミジン化合物(1)のフリー体と、水に溶解した酸又は塩基とを、等モルで混合撹拌した後で溶媒を留去することにより得た。
結果を、ピリミジン化合物(1)のフリー体について表1に、塩酸塩について表2に、臭化水素酸塩について表3に、硫酸塩について表4に、D-(-)-アルギニン塩について表5に、シンコニジン塩について表6にそれぞれ示す。





工程1
(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸のフリー体 1.1kg(1.35mol)をアルゴン雰囲気下、tert-ブチルメチルエーテル(15.3kg)に溶解し、0℃に冷却した。次いで、得られた溶液に、16.7%塩酸/1,4-ジオキサン溶液503.9g(塩酸2.31mol)を0~10℃で滴下した後、同温にて1時間攪拌した。析出した固体をろ取した後、冷却したtert-ブチルメチルエーテル(1.85kg)で洗浄し、40~50℃で12時間減圧乾燥し、(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の非晶質1.14kg(収率100%)を得た。
工程1で得られた(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の非晶質(676mg)を2-プロパノール(1.35mL)に50~55℃で加熱溶解させた後、ヘプタン(676μL)を50℃で加え、5~15℃にて14時間密栓静置した。析出した固体をろ取し、40℃にて減圧乾燥し、(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の結晶576mg(収率85%)を得た。
前記1-1 工程1記載の方法によって得た(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の非晶質1.14kg(1.35mol)を酢酸イソプロピル(9.98kg)に懸濁し、65~75℃に加熱し溶解させた。得られた溶液に、同温にて前記1-1 工程2記載の方法によって得た(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の結晶11gを種晶として加え3時間撹拌した。その後、2時間かけて45~55℃に冷却し、さらに3時間かけて15~25℃に冷却し、同温にてさらに16時間撹拌した。析出した結晶をろ取し、酢酸イソプロピル(1720g)で洗浄した後、35~45℃で12時間減圧乾燥し、(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の結晶1.02kg(収率85%)を得た。
なお、元素分析の結果、以下の通り、得られた塩酸塩は、1塩酸塩であることが明らかとなった。
計算値(1塩酸塩として):C 50.91%、H 4.98%、N 6.60%、Cl 4.17%
実測値:C 50.79%、H 4.70%、N 6.40%、Cl 3.94%
(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸のフリー体16.1kg(19.8mol)をアルゴン雰囲気下、酢酸イソプロピル(124kg)に溶解し、40~50℃に加熱した。次いで、得られた溶液に、6.3%塩酸/酢酸イソプロピル溶液15.0kg(塩酸25.98mol)を滴下した後、65~75℃に昇温した。得られた溶液に、同温にて前記1-1 工程2記載の方法によって得た(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の結晶25gを種晶として加え、さらに6.3%塩酸/酢酸イソプロピル溶液7.0kg(塩酸12.08mol)を滴下し、7時間撹拌した。その後、3時間かけて45~55℃に冷却し、さらに4時間かけて15~25℃に冷却し、同温にてさらに16時間撹拌した。析出した結晶をろ取した後、酢酸イソプロピル(32.4kg)で洗浄し、40~50℃で12時間減圧乾燥し、(S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の結晶15.35kg(収率91%)を得た。
なお、元素分析の結果、得られた塩酸塩は、1塩酸塩であることが明らかとなった。
計算値(1塩酸塩として):C 50.91%、H 4.98%、N 6.60%、Cl 4.17%
実測値:C 50.82%、H 4.98%、N 6.56%、Cl 4.15%
前記1-3で得られた結晶について、下記の通り粉末X線回折測定、及び熱分析測定を行った。
前記1-3で得られた結晶について粉末X線回折の測定を行った。粉末X線回折の測定は、粉砕した結晶サンプルをX線回折用シリコン無反射試料板の試料ホルダー部分に充填し、以下の条件で行った。
粉末X線回折測定装置:RINT-UltimaIV-Protectus((株)リガク製)
X線種:銅Kα線(λ=1.54Å)
回折角2θの走査範囲:3.00°~40.00°
サンプリング幅:0.02°
スキャン速度:2.00°/分
また、相対強度が30以上の主要なピークについて、回折角2θ、半価幅、d値、強度及び相対強度を表7に示す。
図1及び表7から、14.0±0.2°付近、18.3±0.2°付近、20.1±0.2°付近、20.5±0.2°付近、21.3±0.2°付近、21.8±0.2°付近、23.3±0.2°付近及び24.0±0.2°付近の回折角(2θ)に主要なピークを有することが明らかとなった。
また、18.3±0.2°付近及び20.5±0.2°付近の回折角(2θ)、特に20.5±0.2°付近の回折角(2θ)に強度の強いピークを有することが明らかとなった。
実施例1-3で得られた結晶について熱分析測定を行った。熱分析測定は、サンプル約5mgを熱分析用アルミパンに精密に秤量し、基準物質としてAl2O3を使用して、窒素雰囲気下(150mL/min)、昇温速度10℃/分とし、熱分析装置Thermo Plus 2 システム(リガク社製)を用いて、示差熱分析法(DTA)及び熱質量測定法(TG)によって行った。
熱分析測定の結果を図2に示す。なお、図2中、縦軸は、DTA曲線に対しては熱電対の熱起電力(μV)を、TG曲線に対しては質量変化(mg)を示し、横軸は温度(℃)を示す。
図2で見られるように、ピリミジン化合物(1) 塩酸塩の結晶は、示差熱分析(DTA)において162±5℃付近(詳細には、161.6℃)に吸熱ピークを有するものであった。以上の熱分析測定結果から、ピリミジン化合物(1) 塩酸塩の結晶は約162±5℃付近に融点を有するものと考えられた。
試験化合物をガラス瓶に入れ、80℃、100℃又は120℃の温度条件下にて一定期間保存した後の試験化合物中のピリミジン化合物(1)の残存率(%)を測定した。
残存率は、試験化合物に含まれるピリミジン化合物(1)の割合を、高速液体クロマトグラフィーを用いてピーク面積百分率として測定した。なお、高速液体クロマトグラフィーによる測定において、カラムとしてはODSカラムを使用し、溶媒としては0.1%TFA水溶液と0.1%TFAアセトニトリル溶液の2種類を混合して使用し、検出波長は242nmとした。
得られたピリミジン化合物(1)の面積百分率より、以下の計算式にて残存率を算出した。

結果を表8に示す。

Claims (11)
- (S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩。
- 塩酸塩が1塩酸塩である、請求項1記載の塩。
- (S)-トランス-{4-[({2-[({1-[3,5-ビス(トリフルオロメチル)フェニル]エチル}{5-[2-(メチルスルホニル)エトキシ]ピリミジン-2-イル}アミノ)メチル]-4-(トリフルオロメチル)フェニル}(エチル)アミノ)メチル]シクロヘキシル}酢酸 塩酸塩の結晶。
- 塩酸塩が1塩酸塩である、請求項3記載の結晶。
- 銅Kα線の照射で得られる粉末X線回折パターンが、14.0±0.2°付近、18.3±0.2°付近、20.1±0.2°付近、20.5±0.2°付近、21.3±0.2°付近、21.8±0.2°付近、23.3±0.2°付近及び24.0±0.2°付近よりなる群から選ばれる1以上の回折角(2θ)にピークを有する、請求項3又は4記載の結晶。
- 請求項1~5のいずれか1項に記載の化合物、及び製薬上許容される担体を含有する医薬組成物。
- 請求項1~5のいずれか1項に記載の化合物を、製薬上許容される担体と混合する工程を含む、医薬組成物の製造方法。
- 請求項1~5のいずれか1項に記載の化合物の、医薬組成物の製造のための使用。
- 請求項1~5のいずれか1項に記載の化合物の、医薬組成物の製造原料としての使用。
- 医薬組成物の製造に使用するための、請求項1~5のいずれか1項に記載の化合物。
- 医薬組成物の製造原料として使用するための、請求項1~5のいずれか1項に記載の化合物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020157033941A KR20160014620A (ko) | 2013-05-31 | 2014-05-30 | 디벤질아민 구조를 갖는 피리미딘 화합물의 신규 형태 |
| EP14804509.9A EP3006430A4 (en) | 2013-05-31 | 2014-05-30 | NOVEL FORM OF A PYRIMIDINE COMPOUND WITH DIBENZYLAMINE STRUCTURE |
| CN201480030617.6A CN105246877A (zh) | 2013-05-31 | 2014-05-30 | 具有二苄胺结构的嘧啶化合物的新形态 |
| US14/784,147 US9682942B2 (en) | 2013-05-31 | 2014-05-30 | Form of pyrimidine compound having dibenzylamine structure |
| US15/595,039 US20170313664A1 (en) | 2013-05-31 | 2017-05-15 | Novel form of pyrimidine compound having dibenzylamine structure |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013115189 | 2013-05-31 | ||
| JP2013-115189 | 2013-05-31 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/784,147 A-371-Of-International US9682942B2 (en) | 2013-05-31 | 2014-05-30 | Form of pyrimidine compound having dibenzylamine structure |
| US15/595,039 Continuation US20170313664A1 (en) | 2013-05-31 | 2017-05-15 | Novel form of pyrimidine compound having dibenzylamine structure |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014192903A1 true WO2014192903A1 (ja) | 2014-12-04 |
Family
ID=51988925
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/064372 WO2014192903A1 (ja) | 2013-05-31 | 2014-05-30 | ジベンジルアミン構造を有するピリミジン化合物の新規形態 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US9682942B2 (ja) |
| EP (1) | EP3006430A4 (ja) |
| JP (2) | JP6109785B2 (ja) |
| KR (1) | KR20160014620A (ja) |
| CN (1) | CN105246877A (ja) |
| TW (1) | TW201512175A (ja) |
| WO (1) | WO2014192903A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015007038A (ja) * | 2013-05-31 | 2015-01-15 | 興和株式会社 | ジベンジルアミン構造を有するピリミジン化合物の新規形態 |
| WO2016084950A1 (ja) * | 2014-11-28 | 2016-06-02 | 興和株式会社 | 医薬組成物 |
| WO2016084949A1 (ja) * | 2014-11-28 | 2016-06-02 | 興和株式会社 | 医薬 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI691331B (zh) * | 2014-09-26 | 2020-04-21 | 日商興和股份有限公司 | 脂質異常症治療劑 |
| US10266558B2 (en) * | 2016-10-07 | 2019-04-23 | Alexandre Vasilievich Ivachtchenko | Macroheterocyclic nucleoside derivatives and their analogues, production and use thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008129951A1 (ja) | 2007-04-13 | 2008-10-30 | Kowa Company, Ltd. | 新規なジベンジルアミン構造を有するピリミジン化合物及びこれを含有する医薬 |
| WO2011152508A1 (ja) | 2010-06-04 | 2011-12-08 | 興和株式会社 | 光学活性ジベンジルアミン誘導体及びその製造方法 |
| WO2012046681A1 (ja) | 2010-10-04 | 2012-04-12 | 興和株式会社 | 脂質代謝関連mRNAの発現抑制剤 |
| WO2013081087A1 (ja) * | 2011-12-02 | 2013-06-06 | 興和株式会社 | 光学活性化合物の製造方法 |
| JP2013136572A (ja) * | 2011-12-02 | 2013-07-11 | Kowa Co | 光学活性ジベンジルアミン誘導体及びその製造方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997009318A1 (fr) | 1995-09-06 | 1997-03-13 | Kowa Co., Ltd. | Derives de la pyrimidine |
| CN1289467C (zh) | 2002-08-30 | 2006-12-13 | 日本烟草产业株式会社 | 二苄胺化合物及其药物用途 |
| JP3630676B2 (ja) | 2002-08-30 | 2005-03-16 | 日本たばこ産業株式会社 | ジベンジルアミン化合物及びその医薬用途 |
| JP5319457B2 (ja) * | 2008-08-25 | 2013-10-16 | 興和株式会社 | 新規なジベンジルアミン構造を有するピリミジン化合物及びこれを含有する医薬 |
| TW201512175A (zh) * | 2013-05-31 | 2015-04-01 | Kowa Co | 具有二苄基胺構造之嘧啶化合物的新穎形態 |
-
2014
- 2014-05-30 TW TW103118969A patent/TW201512175A/zh unknown
- 2014-05-30 KR KR1020157033941A patent/KR20160014620A/ko not_active Application Discontinuation
- 2014-05-30 CN CN201480030617.6A patent/CN105246877A/zh active Pending
- 2014-05-30 WO PCT/JP2014/064372 patent/WO2014192903A1/ja active Application Filing
- 2014-05-30 EP EP14804509.9A patent/EP3006430A4/en not_active Withdrawn
- 2014-05-30 US US14/784,147 patent/US9682942B2/en not_active Expired - Fee Related
- 2014-06-02 JP JP2014113680A patent/JP6109785B2/ja not_active Expired - Fee Related
-
2017
- 2017-03-06 JP JP2017041364A patent/JP2017101077A/ja active Pending
- 2017-05-15 US US15/595,039 patent/US20170313664A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008129951A1 (ja) | 2007-04-13 | 2008-10-30 | Kowa Company, Ltd. | 新規なジベンジルアミン構造を有するピリミジン化合物及びこれを含有する医薬 |
| WO2011152508A1 (ja) | 2010-06-04 | 2011-12-08 | 興和株式会社 | 光学活性ジベンジルアミン誘導体及びその製造方法 |
| WO2012046681A1 (ja) | 2010-10-04 | 2012-04-12 | 興和株式会社 | 脂質代謝関連mRNAの発現抑制剤 |
| WO2013081087A1 (ja) * | 2011-12-02 | 2013-06-06 | 興和株式会社 | 光学活性化合物の製造方法 |
| JP2013136572A (ja) * | 2011-12-02 | 2013-07-11 | Kowa Co | 光学活性ジベンジルアミン誘導体及びその製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| BAVIN, M.: "Polymorphism in Process Development", CHEMISTRY & INDUSTRY, vol. 21, 21 August 1989 (1989-08-21), pages 527 - 529, XP001180136 * |
| BYRN, S. ET AL.: "Pharmaceutical Solids: A Strategic Approach to Regulatory Considerations", PHARMACEUTICAL RESEARCH, vol. 12, no. 7, 1 July 1995 (1995-07-01), pages 945 - 954, XP000996386, DOI: 10.1023/A:1016241927429 * |
| See also references of EP3006430A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015007038A (ja) * | 2013-05-31 | 2015-01-15 | 興和株式会社 | ジベンジルアミン構造を有するピリミジン化合物の新規形態 |
| WO2016084950A1 (ja) * | 2014-11-28 | 2016-06-02 | 興和株式会社 | 医薬組成物 |
| WO2016084949A1 (ja) * | 2014-11-28 | 2016-06-02 | 興和株式会社 | 医薬 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105246877A (zh) | 2016-01-13 |
| JP6109785B2 (ja) | 2017-04-05 |
| JP2017101077A (ja) | 2017-06-08 |
| US9682942B2 (en) | 2017-06-20 |
| US20170313664A1 (en) | 2017-11-02 |
| EP3006430A4 (en) | 2016-11-02 |
| US20160031827A1 (en) | 2016-02-04 |
| KR20160014620A (ko) | 2016-02-11 |
| EP3006430A1 (en) | 2016-04-13 |
| JP2015007038A (ja) | 2015-01-15 |
| TW201512175A (zh) | 2015-04-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2017101077A (ja) | ジベンジルアミン構造を有するピリミジン化合物の新規形態(2) | |
| TWI322805B (ja) | ||
| US9090598B2 (en) | Nilotinib salts and crystalline forms thereof | |
| JP2020183408A (ja) | {[5−(3−クロロフェニル)−3−ヒドロキシピリジン−2−カルボニル]アミノ}酢酸の固体形態、組成物、及びその使用 | |
| EP2547649B1 (en) | Agomelatine hydrochloride hydrate and preparation thereof | |
| JP5337261B2 (ja) | 縮合複素環誘導体の塩及びその結晶 | |
| TW201002323A (en) | Novel piperazine salts as D3/D2 antagonists | |
| JP2022519885A (ja) | Jak2阻害剤の結晶形態 | |
| US10143688B2 (en) | Mesylic acid salt of acylthiourea compound, crystal of the same, and process for producing these | |
| JP2716740B2 (ja) | ブスピロンの薬学的に有用な多形変態 | |
| JP2702519B2 (ja) | 多形結晶形態の転換法 | |
| WO2016084950A1 (ja) | 医薬組成物 | |
| JP2007524569A (ja) | ナテグリニドの結晶形 | |
| JP2022508864A (ja) | チロシンキナーゼ阻害剤のマレイン酸塩の結晶形及びその調製方法 | |
| NO316618B1 (no) | Termodynamisk stabil form av (R)-3-[[(4-fluorfenyl)sulfonyl]amino]-1,2,3,4-tetrahydro-9H-karbazol-9-propansyre (Ramatroban), samt fremgangsmåte og anvendelse derav | |
| TW201238965A (en) | Process for the production of a pemetrexed salt | |
| WO2023125418A1 (zh) | 一种苯磺酸盐结晶及其制备方法 | |
| JP6985137B2 (ja) | スルホンアミド化合物の結晶形 | |
| KR20160035599A (ko) | 아고멜라틴 및 설폰산의 신규한 복합체, 이를 제조하기 위한 방법 및 이를 함유하는 약학적 조성물 | |
| CN109721534A (zh) | 一种马来酸茚达特罗中间体及其制备方法和用途 | |
| TW202016104A (zh) | 新型鹽 | |
| HU231054B1 (hu) | Gyógyászati készítmény előállítására alkalmazható új sók | |
| CZ300352B6 (cs) | Zpusob prípravy fosforecnanu aripiprazolu |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14804509 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14784147 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014804509 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20157033941 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |