WO2014185498A1 - 燃料電池用電極およびその製造方法 - Google Patents
燃料電池用電極およびその製造方法 Download PDFInfo
- Publication number
- WO2014185498A1 WO2014185498A1 PCT/JP2014/062984 JP2014062984W WO2014185498A1 WO 2014185498 A1 WO2014185498 A1 WO 2014185498A1 JP 2014062984 W JP2014062984 W JP 2014062984W WO 2014185498 A1 WO2014185498 A1 WO 2014185498A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water vapor
- carrier
- supported
- catalyst
- fuel cell
- Prior art date
Links
Images

















Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/8647—Inert electrodes with catalytic activity, e.g. for fuel cells consisting of more than one material, e.g. consisting of composites
- H01M4/8657—Inert electrodes with catalytic activity, e.g. for fuel cells consisting of more than one material, e.g. consisting of composites layered
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/90—Selection of catalytic material
- H01M4/9075—Catalytic material supported on carriers, e.g. powder carriers
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/88—Processes of manufacture
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/88—Processes of manufacture
- H01M4/8825—Methods for deposition of the catalytic active composition
- H01M4/8828—Coating with slurry or ink
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/90—Selection of catalytic material
- H01M4/9075—Catalytic material supported on carriers, e.g. powder carriers
- H01M4/9083—Catalytic material supported on carriers, e.g. powder carriers on carbon or graphite
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/86—Inert electrodes with catalytic activity, e.g. for fuel cells
- H01M4/90—Selection of catalytic material
- H01M4/92—Metals of platinum group
- H01M4/925—Metals of platinum group supported on carriers, e.g. powder carriers
- H01M4/926—Metals of platinum group supported on carriers, e.g. powder carriers on carbon or graphite
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M8/1016—Fuel cells with solid electrolytes characterised by the electrolyte material
- H01M8/1018—Polymeric electrolyte materials
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/10—Fuel cells with solid electrolytes
- H01M2008/1095—Fuel cells with polymeric electrolytes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2250/00—Fuel cells for particular applications; Specific features of fuel cell system
- H01M2250/20—Fuel cells in motive systems, e.g. vehicle, ship, plane
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T90/00—Enabling technologies or technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02T90/40—Application of hydrogen technology to transportation, e.g. using fuel cells
Definitions
- the present invention relates to a fuel cell electrode in which an ionomer is coated on a carrier made of carbon on which catalyst particles are supported, and a method for producing the same.
- Solid polymer electrolyte fuel cells are attracting attention as an energy source for transportation instead of conventional internal combustion engines because they have high power density, operate at low temperatures, and emit almost no exhaust gas containing harmful substances. .
- an anode is joined to one surface of a solid polymer electrolyte membrane, and a cathode is joined to the other surface as an electrode.
- Hydrogen as a fuel is supplied to the anode, and oxygen as an oxidant is supplied to the cathode. Then, fuel is oxidized to protons at the anode, and oxygen is reduced to water at the cathode to generate electricity.
- a fuel cell electrode catalyst made of fine powder in which catalyst particles made of noble metal such as Pt are supported on a carrier such as carbon is used for the anode and the cathode.
- the electrode catalyst for use is coated with an ionomer made of a solid polymer electrolyte.
- the utilization rate was increased and the performance was improved when the catalyst particles were in contact with the ionomer, but in recent years, it has been found that the catalyst metal of the catalyst particles is poisoned by the contact of the ionomer.
- the catalyst particles in the support.
- the support usually has low durability. When heat treatment is performed in order to improve durability, it is sometimes difficult to encapsulate the catalyst particles because the pores formed with the carbon black support are collapsed.
- the present invention has been made in view of the above points, and an object of the present invention is to increase the durability of the carrier and to suppress the poisoning of the ionomer to the catalyst particles and the fuel cell electrode. It is in providing the manufacturing method.
- a method for producing an electrode for a fuel cell according to the present invention comprises a heat treatment of a support made of mesoporous carbon having a 002-plane crystallite diameter Lc of 1.5 nm or less at 1700 ° C. or more and less than 2300 ° C. And a step of supporting catalyst particles at least inside the heat-treated carrier, and a step of coating an ionomer on the carrier on which the catalyst particles are supported.
- the pores of mesoporous carbon are not crushed and the durability of mesoporous carbon can be enhanced. That is, by such heat treatment, the G band half-value width of the mesoporous carbon constituting the carrier becomes 70 cm ⁇ 1 or less, and the crystallization of the carrier can be sufficiently enhanced.
- the catalyst particles are supported at least inside the heat-treated carrier (in the mesopores) and the ionomer is coated on the carrier on which the catalyst particles are supported, the pores of the mesoporous carbon (in the mesopores) Since molecules hardly enter, it is possible to prevent the ionomer inside the carrier from coming into contact with the catalyst particles.
- the crystallite diameter Lc of the 002 plane exceeds 1.5 nm
- the pores of the mesoporous carbon are crushed during the heat treatment described above, and the catalyst particles are at least inside (in the mesopores) of the heat-treated carrier. May not be supported.
- the carrier carrying the catalyst particles is coated with an ionomer
- the coverage of the ionomer on the catalyst particles increases, and the catalyst particles are easily poisoned by the ionomer.
- the heat treatment temperature is lower than 1700 ° C.
- the G band half-value width of the mesoporous carbon constituting the support may exceed 70 cm ⁇ 1 , and the support may not be sufficiently crystallized, resulting in a decrease in durability. is there.
- the heat treatment temperature is 2300 ° C. or higher, there may be cases where sufficient pores of mesoporous carbon cannot be secured during the heat treatment.
- the heat treatment is performed so that the mesopore specific surface area of the mesopores having a pore diameter of 2 to 10 nm of the carrier is 400 m 2 / g or more.
- the mesoporous carbon constituting the carrier has a G-band half-value width of 70 cm ⁇ 1 or less, and the catalyst particles can be easily supported at least inside the carrier (in the mesopores).
- the catalyst particles may not be supported at least in the heat-treated support (inside the mesopores). is there. Furthermore, the ionomer can be coated such that the coverage of the ionomer with respect to the total surface area of all the catalyst particles supported on the support is 72% or less. Thereby, it can suppress that the catalyst particle poisons with an ionomer, raising the specific activity of the obtained electrode for fuel cells.
- the method for producing an electrode for a fuel cell comprises a step of oxidizing at least the surface of the mesoporous carbon of the mesoporous carbon of the support after the step of supporting the catalyst particles and before the step of coating the ionomer. Further included.
- the oxidation treatment is performed so that the oxygen content per unit area contained on the surface of the supported catalyst in which the catalyst particles are supported on the carrier is 0.08 mg / m 2 or more.
- the oxygen content satisfies the above-described range, it is possible to suppress a decrease in power generation performance of the fuel cell in a low humidified environment.
- the oxygen content is less than 0.08 mg / m 2 , it cannot be said that the surface of the mesopores is sufficiently hydrophilic, and protons from the ionomer hardly reach the catalyst particles in a low humidified environment.
- the oxygen content is less than 0.08 mg / m 2 , it cannot be said that the surface of the mesopores is sufficiently hydrophilic, and protons from the ionomer hardly reach the catalyst particles in a low humidified environment.
- the supported catalyst subjected to the oxidation treatment is placed in a vacuum environment, and the water vapor is adsorbed on the supported catalyst by increasing the water vapor partial pressure to near the saturated water vapor pressure.
- the water vapor adsorbed on the supported catalyst is desorbed by reducing the water vapor partial pressure from the vicinity of the saturated water vapor pressure in a state where the water vapor is adsorbed, the water vapor partial pressure is increased to 60% of the saturated water vapor pressure.
- the amount of water vapor adsorbed on the supported catalyst when the water vapor partial pressure becomes equal to T1 is T1, and the water vapor partial pressure is decreased so that the water vapor partial pressure becomes 60% of the saturated water vapor pressure.
- the mass of water vapor adsorbed on the catalyst is T2
- the oxidation treatment is performed on the supported catalyst so as to satisfy the relationship of T1 / T2 ⁇ 0.15.
- the amount of water vapor adsorbed on the supported catalyst is increased by increasing the water vapor partial pressure to near the saturated water vapor pressure, and in this state, the water vapor partial pressure is decreased from the vicinity of the saturated water vapor pressure to decrease the supported catalyst.
- the water vapor adsorbed on the desorbed is increased to 60% of the saturated water vapor pressure, and the mass T1 of the water vapor adsorbed on the supported catalyst when the water vapor partial pressure reaches 60% of the saturated water vapor pressure, and the water vapor partial pressure is reduced to 60% of the saturated water vapor pressure.
- the mass T2 of the water vapor adsorbed on the supported catalyst when the water vapor partial pressure becomes% becomes a different value even at the same water vapor partial pressure. That is, the water vapor (water) once entering the mesopores and adsorbed on the surface of the mesopores is difficult to escape from the mesopores, and thus T1 ⁇ T2 described above.
- T1 / T2 is larger (specifically, closer to 1), the drainage of the mesopores is higher.
- the diameter of the mesopores is expanded and the drainage of water adsorbed on the mesopores is enhanced.
- water inside the mesopores is easily discharged, so that oxygen gas easily diffuses inside the mesopores toward the catalyst particles.
- an aqueous nitric acid solution heated to 0.1 mol / L or more and 60 ° C. or more may be brought into contact with at least the surface of the mesoporous carbon of the mesoporous carbon for 1 hour or more.
- an electrode for a fuel cell is also disclosed as an invention.
- An electrode for a fuel cell according to the present invention comprises a carrier made of mesoporous carbon, catalyst particles carried at least inside the carrier, and an ionomer that coats the carrier, and the mesoporous carbon G constituting the carrier.
- the band half width is 70 cm ⁇ 1 or less.
- the mesoporous carbon has a G band half-width of 70 cm ⁇ 1 or less, so that the durability of the mesoporous carbon can be enhanced.
- the G band half-width of the mesoporous carbon constituting the support exceeds 70 cm ⁇ 1 , the support may not be sufficiently crystallized and durability may be lowered.
- the coverage of the ionomer with respect to the total surface area of all catalyst particles supported on the carrier is greater than 0% and 72% or less. According to this aspect, it is possible to suppress the catalyst particles from being poisoned by the ionomer while increasing the specific activity of the fuel cell electrode.
- the oxygen content per unit area contained on the surface of the supported catalyst in which the catalyst particles are supported on the carrier before coating the ionomer on the carrier is 0.08 mg / m 2 or more.
- the oxygen content when the oxygen content satisfies the above-described range, it is possible to suppress a decrease in power generation performance of the fuel cell in a low humidified environment.
- the oxygen content when the oxygen content is less than 0.08 mg / m 2 , it cannot be said that the surface of the mesopores is sufficiently hydrophilic, and protons from the ionomer hardly reach the catalyst particles in a low humidified environment.
- the oxygen content is less than 0.08 mg / m 2 , it cannot be said that the surface of the mesopores is sufficiently hydrophilic, and protons from the ionomer hardly reach the catalyst particles in a low humidified environment.
- the supported catalyst is placed in a vacuum environment before the ionomer is coated on a support, and the supported catalyst is made to adsorb water vapor by increasing the water vapor partial pressure to near the saturated water vapor pressure.
- the water vapor partial pressure is increased to increase the saturated water vapor pressure.
- T1 the mass of water vapor adsorbed on the supported catalyst when the partial pressure of water vapor reaches 60%
- the partial pressure of water vapor is decreased to a partial water vapor pressure of 60% of the saturated water vapor pressure.
- the supported catalyst satisfies the relationship of T1 / T2 ⁇ 0.15.
- the drainage of water (water vapor) adsorbed on the mesopores is enhanced.
- water inside the mesopores is easily discharged, so that oxygen gas easily diffuses inside the mesopores toward the catalyst particles.
- the present invention it is possible to increase the durability of the carrier and suppress the ionomer poisoning of the catalyst particles.
- (A) is a figure for demonstrating the coverage of the ionomer with respect to the catalyst particle of the conventional fuel cell electrode
- (b) is the figure which showed the measurement result of the ionomer coverage with respect to the catalyst particle.
- carrier of the electrode for fuel cells which concerns on this invention.
- (A) is a schematic diagram showing a state where a conventional fuel cell electrode is used in a low humidified state
- (b) is a schematic diagram showing a state where a conventional fuel cell electrode is used in an over humidified state
- (C) is a schematic view showing a state in which the fuel cell electrode shown in FIG.
- FIG. 1 (d) is used in a low humidified state
- (d) is a state in which the fuel cell electrode shown in FIG.
- (A) is a schematic diagram showing the fuel cell electrode shown in FIG. 1 (d)
- (b) is a schematic diagram showing a state where the supported catalyst of the fuel cell electrode shown in (a) is subjected to an oxidation treatment.
- Fig. 3 (c) is a schematic diagram showing a state in which the fuel cell electrode shown in (b) is used in a low humidified state
- Fig. 4 (d) is a diagram showing the fuel cell electrode shown in (b) in an over humidified state. The schematic diagram which showed the state which had been.
- (A) is a figure for demonstrating the change of water vapor
- (b) is the figure which showed the relationship between water vapor
- FIG. The figure which showed the electric power generation characteristic of the fuel cell which concerns on Example 3 and Comparative Example 6.
- FIG. The figure which showed the relationship between the power generation voltage in the low humidification environment at the time of using the electrode for fuel cells which concerns on an Example and a comparative example, and the oxygen content per unit area contained on the surface of a supported catalyst.
- (A) is the figure which showed the relationship between processing time and oxygen content
- (b) is the figure which showed the relationship between processing time and drainage
- (c) is the density
- (d) is the figure which showed the relationship between the density
- FIG. 1 is a schematic diagram for explaining a method for producing a fuel cell electrode according to the present invention, in which (a) is a carrier made of mesoporous carbon, (b) is a carrier after a heat treatment step, and (c).
- FIG. 3 is a view showing a carrier carrying catalyst particles
- FIG. 4D is a diagram showing a fuel cell electrode in which an ionomer is coated on the carrier after carrying catalyst particles.
- a carrier made of mesoporous carbon having a 002-plane crystallite diameter Lc of 1.5 nm or less is prepared (see FIG. 1A).
- mesoporous carbon having a thickness of 1.5 nm or less has a structure in which several layers of graphene sheets are stacked in the wall thickness direction on the pore walls forming mesopores (pores) of mesoporous carbon.
- Graphene is an array of carbon atoms arranged in a hexagonal network, and corresponds to a single layer of graphite.
- the crystallite diameter Lc of the 002 plane is 0.34 nm. Therefore, the crystallite diameter Lc of the 002 plane of the mesoporous carbon constituting the carrier of this embodiment is 0. .34 nm or more is preferable.
- such a carrier preferably has a mesopore specific surface area of 800 m 2 / g or more and a mesopore diameter of 2 to 10 nm.
- the rod-like body or annular body containing carbon has a three-dimensional structure, and the rod-like body or annular body extends three-dimensionally and is connected to each other to form a network.
- a structure having a so-called dendritic shape (dendritic shape) is preferable.
- an acetylene gas is blown into a silver nitrate aqueous ammonia solution while irradiating the solution with ultrasonic waves to form a silver acetylide precipitate in the solution.
- the precipitate is placed in a vacuum electric furnace or a vacuum high-temperature bath and subjected to heat treatment at a temperature of 60 ° C. to 80 ° C. for, for example, 12 hours or more to segregate silver acetylide and encapsulate metal silver particles.
- a nanostructure is formed.
- the carrier has a structure in which carbon particles constituting the carrier, for example, a graphene parcel, are surrounded by a skin made of graphene.
- the crystallite diameter Lc of the 002 plane is 1.5 nm or less” is a value obtained by analysis by a powder X-ray diffraction method using CuK ⁇ rays, and the powdered electrode catalyst body is subjected to powder X-ray diffraction.
- the half-value width ⁇ (radian) of the diffraction peak of each crystal plane is obtained from the obtained diffraction pattern.
- K (shape factor) is 0.89
- ⁇ is the wavelength of X-rays ( ⁇ )
- ⁇ is the diffraction angle (°).
- the peak of the 002 plane appearing near the ⁇ diffraction angle of 26 ° is derived from the stacking direction of the carbon hexagonal network structure, which depends on the wall thickness of the mesopores.
- the support made of mesoporous carbon is heat-treated at 1700 ° C. or more and less than 2300 ° C. Specifically, this makes it possible to obtain a highly crystalline carrier in which the G band half-value width of mesoporous carbon is 70 cm ⁇ 1 or less (see FIG. 1B).
- the time for heating the support is preferably in the range of 30 minutes to 2 hours, and the heat treatment atmosphere is preferably a non-oxidizing atmosphere.
- an inert gas such as argon gas or helium gas is used in the furnace. The inside is filled with heat treatment.
- the heat treatment by setting the heating time and the heating temperature so that the mesopore specific surface area of the mesopores having a pore diameter of 2 to 10 nm of the support is 400 m 2 / g or more.
- the pore diameter of the mesopore exceeds 10 nm, the minimum micelle diameter of the ionomer is about 10 nm, so that the ionomer may enter the mesopore.
- catalyst particles such as platinum particles may not enter the mesopores, and even if some of the catalyst particles enter, the reactants (H + , O 2 ) Diffusion may be slow (diffusion resistance increases), and performance may be degraded.
- the mesoporous carbon As described above, even when heat-treated at 1700 ° C. or more and less than 2300 ° C. by using a support made of mesoporous carbon having a 002-plane crystallite diameter Lc of 1.5 nm or less, the mesoporous carbon The durability of the mesoporous carbon can be increased without crushing the pores (mesopores).
- the pores of the mesoporous carbon are crushed during the heat treatment described above, and the catalyst particles are at least inside (in the mesopores) of the heat-treated carrier. May not be supported.
- the heat treatment temperature is lower than 1700 ° C.
- the G band half-value width of the mesoporous carbon constituting the support may exceed 70 cm ⁇ 1 , and the support may not be sufficiently crystallized, resulting in a decrease in durability. is there.
- the heat treatment temperature is 2300 ° C. or higher, the pores of the mesoporous carbon may be crushed during the heat treatment, and the pores may not be sufficiently secured.
- the mesopore specific surface area of the carrier means the surface area per unit mass of the carrier including the mesoporous carbon of the mesoporous carbon.
- Nitrogen gas is introduced into the carrier as an adsorbed gas, and nitrogen gas is introduced into the carrier. From the adsorption curve at the time of adsorption, the DH method (Dollimore-Heal) is used for analysis, and in the present invention, the pore diameter of 2 to 10 nm is specified, and the surface area formed by these pores (mesopores) is the mesopore specific surface area. It was.
- catalyst particles are supported at least inside the heat-treated carrier (see FIG. 1 (c)).
- the catalyst particles include catalyst metals including platinum, palladium and the like.
- platinum particles when carrying platinum particles, disperse the above-mentioned carrier in pure, add nitric acid to this, add a predetermined amount of dinitrodiamine platinum salt aqueous solution, and then add ethanol further and heat. To reflux (reduction).
- platinum particles can be carried inside mesoporous carbon made of a carrier.
- supported inside a mesoporous carbon can be adjusted by adjusting the time from the addition of dinitrodiamine platinum salt aqueous solution to recirculation
- the ionomer coverage on the catalyst particles (platinum particles) can be adjusted.
- an ionomer is coated on a carrier on which catalyst particles are supported (see FIG. 1 (d)).
- a polymer electrolyte having proton conductivity, and a perfluoro proton exchange resin of a fluoroalkyl copolymer having a fluoroalkyl ether side chain and a perfluoroalkyl main chain is preferably used. Examples include Dufon Nafion (trade name), Asahi Kasei Aciplex (trade name), Asahi Glass Flemion (trade name), Japan Gore-Tex Gore-Select (trade name), etc.
- Examples thereof include a polymer of trifluorostyrene sulfonic acid and a product obtained by introducing a sulfonic acid group into polyvinylidene fluoride.
- styrene-divinylbenzene copolymer which is a hydrocarbon-based proton exchange resin, a polyimide-based resin, and the like are introduced.
- the ionomer can be coated such that the coverage of the ionomer with respect to the total surface area of all the catalyst particles supported on the carrier is greater than 0% and 72% or less. Thereby, it can suppress that the catalyst particle poisons with an ionomer, raising the specific activity of the obtained electrode for fuel cells.
- the ionomer coverage can be calculated by determining (1) the surface area of all the catalyst particles and (2) the surface area of the catalyst particles coated with the ionomer. For example, as shown in FIG. 2 (a), in the case of catalyst particles (platinum particles), if ionomers or water are present on the surface of the catalyst particles, they serve as proton (H + ) paths on the catalyst particles. Proton (H + ) is adsorbed.
- the voltage is set within a predetermined range and speed with respect to the reference electrode (RHE) under the conditions of 100% RH and 20% RH.
- RHE reference electrode
- the area of the hatched portion shown in FIG. 2B becomes the amount of hydrogen adsorption electricity adsorbed on the catalyst particles.
- (Coating ratio of the ionomer with respect to the total surface area of all catalyst particles supported on the support) (Hydrogen adsorption electricity amount at 20% RH) / (hydrogen adsorption electricity amount at 100% RH) ⁇ 100 It can be expressed as.
- the catalyst particles are supported at least inside the heat-treated carrier (in the mesopores), and the carrier on which the catalyst particles are supported (supported catalyst) is coated with the ionomer.
- the ionomer molecules hardly enter the pores (in the mesopores) of the mesoporous carbon, it is possible to prevent the ionomer inside the carrier from contacting the catalyst particles. That is, in the case of this embodiment, the ionomer is coated so that the ionomer coverage is 72% or less with respect to the total surface area of all catalyst particles supported on the support without adjusting the amount of ionomer added. Can do.
- the hydrogen adsorption electricity amount at 20% RH and the hydrogen adsorption electricity amount at 100% RH are almost the same, and as is clear from the examples described later, Compared with the present embodiment, the ionomer coverage on the catalyst particles is high, and it can be said that the catalyst metal by the ionomer is easily poisoned.
- a carrier having a crystallite diameter Lc of 002 plane of 1.5 nm or less (a carrier having the structure described above) is used, even if the carrier is heated under the above-described heating conditions, the meso- The crushing of the holes hardly occurs, and the crystallite diameter Lc of the 002 plane of the mesoporous carbon constituting the carrier after the heat treatment can be maintained at 1.5 nm or less.
- the mesopore specific surface area of the mesopores having a pore diameter of 2 to 10 nm of the support is increased to 400 m 2 / g or more while improving the crystallinity of the support (that is, the G band half-width of the mesoporous carbon is 70 cm ⁇ 1 or less).
- the half band width of G band of mesoporous carbon is preferably 40 cm ⁇ 1 or more, and it is difficult to produce mesoporous carbon having a value less than this value.
- the mesopore specific surface area of mesopores having a pore diameter of 2 to 10 nm is 1100 m 2 / g or less, and mesoporous carbon exceeding this value is difficult to produce.
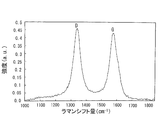
- the peak of the Raman spectroscopic spectrum generally appears at a Raman shift near 1350 cm ⁇ 1 and 1580 cm ⁇ 1. is there.
- this Raman shift is a peak in the vicinity of 1580 cm ⁇ 1 and is a band resulting from the graphite structure.
- the half width of the G band is a width at half the value of the intensity in the G band, and the smaller the G band half width, the higher the carbon crystallinity of the carrier. Become.
- the external particles can be used in either a low humidification environment or a high humidification environment.
- Oxygen gas and protons (H + ) flowing from the ionomer react with each other on the surface of the catalyst particles to generate water.
- the ionomer covers most of the surface of the catalyst particles with the ionomer, the catalyst particles are poisoned, so the activity of the catalyst is not good.
- nitric acid, sulfuric acid, hydrogen peroxide, ozone or the like is brought into contact with the carrier (supported catalyst) on which the catalyst particles shown in FIG. Is oxidized.
- a functional group such as a carbonyl group, a hydroxyl group, or a carboxyl group is imparted to the surface of the mesopores to have hydrophilicity.
- the oxidation treatment is preferably performed so that the oxygen content per unit area contained on the surface of the supported catalyst in which the catalyst particles are supported on the carrier is 0.08 mg / m 2 or more.
- the protons flowing from the ionomer can be reliably reached by the catalyst particles via the water adsorbed on the surface of the mesopores. Can do.
- a decrease in power generation performance of the fuel cell can be suppressed in a low humid environment.
- the oxygen content is less than 0.08 mg / m 2 from the experiment described below, it cannot be said that the surface of the mesopores is sufficiently hydrophilic, and protons from the ionomer reach the catalyst particles in a low humidified environment. It may be difficult.
- the oxygen content is preferably 0.2 mg / m 2 or less. It may be difficult to contain oxygen exceeding this value on the surface of the supported catalyst.
- the water in the mesopores can be drained by expanding the diameter of the mesopores by oxidation treatment.
- This drainage performance depends on the degree of hydrophilicity of the mesopores and the mesopores. Since it also depends on the shape of the mesopore, it cannot be specified simply by the diameter of the mesopores.
- the inventors increase the water vapor partial pressure to near the saturated water vapor pressure, thereby increasing the amount of water vapor adsorbed on the supported catalyst and decreasing the water vapor partial pressure from the vicinity of the saturated water vapor pressure. As a result, water vapor adsorbed on the supported catalyst is desorbed and reduced. At this time, we focused on drawing a hysteresis loop in relation to the partial pressure of water vapor and the mass of adsorbed water (water vapor).
- the mass T1 of water vapor adsorbed on the supported catalyst when the water vapor partial pressure is increased and the mass T2 of water vapor adsorbed on the supported catalyst when the water vapor partial pressure is decreased are the same as the water vapor content. Even pressure is a different value. This is because water vapor (water) once entering the mesopores and adsorbed on the surface of the mesopores is difficult to escape from the mesopores, and even when the water vapor partial pressure is reduced, it is supported by the same water vapor partial pressure.
- the mass of water vapor adsorbed on the catalyst is larger than the mass of water vapor adsorbed on the supported catalyst when the water vapor partial pressure is increased.
- FIG. 7B shows the mass T1 of water vapor adsorbed on the supported catalyst when the water vapor partial pressure is increased to a water vapor partial pressure of 60% of the saturated water vapor pressure, and the water vapor partial pressure Even if the water vapor mass T2 adsorbed on the supported catalyst when the water vapor partial pressure is 60% of the saturated water vapor pressure is reduced, the mass difference between the two water vapor components The value is smaller than the pressure. Therefore, by measuring the mass of water (water vapor) adsorbed on the supported catalyst at a water vapor partial pressure of 60% of the saturated water vapor pressure, it is possible to evaluate the drainage performance of the supported catalyst at the time of excessive humidification.
- FIG. 7B shows the value of T2 / T1 for convenience in order to explain the difference in the amount of water vapor attached at a water vapor partial pressure of 60% of the saturated water vapor pressure. The value is indicated by a value of T1 / T2.
- the water vapor adsorbed on the supported catalyst when the water vapor partial pressure is increased to a water vapor partial pressure of 60% of the saturated water vapor pressure is obtained.
- T1 is the mass of water vapor adsorbed on the supported catalyst when the partial pressure of water vapor is reduced from around the saturated water vapor pressure to a water vapor partial pressure of 60% of the saturated water vapor pressure
- T1 The supported catalyst is oxidized so that /T2 ⁇ 0.15.
- the mesopore diameter is enlarged (pore expansion) as shown in FIG.
- the drainage of the water adsorbed on can be improved.
- the water inside the mesopores is easily discharged even in an overhumidified atmosphere, so that the oxygen gas easily diffuses inside the mesopores toward the catalyst particles. .
- T1 / T2 is preferably 0.3 or less, and it may be difficult to perform the oxidation treatment to exceed this value.
- an aqueous nitric acid solution having a concentration of 0.1 mol / L or more is heated to 60 ° C. or higher (preferably 95 ° C. or higher) and supported on the aqueous nitric acid solution. It is preferable to immerse the catalyst for 1 hour or longer. Thereby, not only the surface of the mesopores is oxidized and hydrophilized, but also the drainage of the supported catalyst (support) can be improved.
- Example 1-1 As shown in Table 1, a mesoporous carbon support was prepared. This carrier has a mesopore diameter of 2 nm or more, and is a carrier produced by the method exemplified in the above-described embodiment. Next, the support was heated under heat treatment conditions at 1700 ° C. for 2 hours in an argon gas environment.
- catalyst particles were supported at least inside the heat-treated carrier.
- the carrier is dispersed in pure, nitric acid is added thereto, a predetermined amount of dinitrodiamine platinum salt aqueous solution is added, ethanol is further added, and reduction is performed by heating.
- platinum particles which are catalyst particles, were supported inside mesoporous carbon made of a carrier. The amount of platinum particles supported is 30% by mass with respect to the supported catalyst supporting platinum particles.
- the obtained catalyst ink was applied onto a substrate using an applicator and vacuum-dried to produce an electrode sheet, and the electrode sheet was thermally transferred to an electrolyte membrane to produce a fuel cell electrode.
- Examples 1-2 and 1-3 A fuel cell electrode was produced in the same manner as Example 1-1. The difference from Example 1-1 is that the carrier was heated under the conditions of 1900 ° C. and 2100 ° C. in an argon gas environment in the heat treatment step.
- Examples 2-1 to 2-3 fuel cell electrodes were fabricated under the conditions corresponding to Examples 1-1 to 1-3, respectively. These differ from Examples 1-1 to 1-3 in that the time from the addition of the dinitrodiamine platinum salt aqueous solution to the reflux is shortened.
- Comparative Examples 1-1 to 1-3 A fuel cell electrode was produced in the same manner as Example 1-1. Comparative Examples 1-1 to 1-3 differ from Example 1-1 in that a commercially available Cabot Corp. product: Vulcan XC-72R (registered trademark) was used as the carrier, and the carrier was heat treated under the heat treatment conditions shown in Table 1. (However, in Comparative Example 1-1, no heat treatment was performed), and the ionomer (Nafion: manufactured by DuPont) and a carrier carrying platinum particles were mixed at the mixing ratio (I / C) shown in Table 1. is there.
- Comparative Examples 2-1 to 2--7 A fuel cell electrode was produced in the same manner as Example 1-1. Comparative Examples 2-1 to 2-7 differ from Example 1-1 in that a commercially available Lion Co., Ltd. product: KetchenEC300J (registered trademark) was used as the carrier, and the carrier was subjected to heat treatment under the heat treatment conditions shown in Table 1. (However, heat treatment was not performed in Comparative Examples 2-1 and 2-2), and the ionomer and the carrier carrying platinum particles were mixed at the mixing ratio (I / C) shown in Table 1 (However, Comparative Example 2- 2 and 2-7 are the same).
- Comparative Examples 3-1 to 3-4 A fuel cell electrode was produced in the same manner as Example 1-1. Comparative Examples 3-1 to 3-4 differ from Example 1-1 in that a commercially available Lion Co., Ltd. product: KetchenEC600JD (registered trademark) was used as the carrier, and the carrier was subjected to heat treatment under the heat treatment conditions shown in Table 1. Application (however, Comparative Example 3-1 was not heat-treated), and the ionomer and the carrier carrying platinum particles were mixed at the mixing ratio (I / C) shown in Table 1.
- the carrier used here has a larger mesopore specific surface area than the carrier used in Comparative Examples 2-1 to 2-7.
- Example 4-3 A fuel cell electrode was produced in the same manner as Example 1-1. The difference from Example 1-1 is that no heat treatment was performed (untreated), and heating was performed under heat treatment conditions of 1300 ° C. and 1500 ° C. in an argon gas environment.
- Example 5-3 A fuel cell electrode was produced in the same manner as in Example 2-1.
- the difference from Example 2-1 is that there was no heat treatment (untreated) and each was heated under heat treatment conditions of 1300 ° C. and 1500 ° C. in an argon gas environment.
- the constant K is a shape factor
- ⁇ is the X-ray wavelength ( ⁇ )
- ⁇ is the diffraction angle (°).
- the peak of the 002 plane appearing near the ⁇ diffraction angle of 26 ° is derived from the stacking direction of the carbon hexagonal network structure and depends on the wall thickness of the mesopores.
- the crystallite diameter Lc of the 002 plane of the carrier before and after the heat treatment was measured, and these values were the same.
- G band half-width of the carrier before platinum particle support (after heat treatment) excluding Comparative Examples 2-2, 2-5, and 2-7 was measured. Specifically, NRS-1000 manufactured by JASCO Corporation is used as a measurement device, excitation light: green laser (wavelength: 532 nm), laser power: 100 mV, and integration is performed twice using a detection device CCD ( ⁇ 60 ° C.). , Exposure time: 30 seconds, dimmer OD2 (dimming rate 1/100). The results are shown in Table 1.
- the measurement conditions are adsorption temperature: 77K, adsorbate: nitrogen, adsorbate cross-sectional area: 0.16 nm 2 , equilibrium waiting time: 500 seconds, and equilibrium waiting time is an adsorption equilibrium state (a pressure change is predetermined during adsorption / desorption). It is the waiting time after reaching the value).
- adsorption temperature 77K
- adsorbate nitrogen
- adsorbate cross-sectional area 0.16 nm 2
- equilibrium waiting time 500 seconds
- equilibrium waiting time is an adsorption equilibrium state (a pressure change is predetermined during adsorption / desorption). It is the waiting time after reaching the value).
- DH method Denso-Heal
- the voltage value at the lower limit of analysis in calculating the amount of hydrogen adsorption electricity was 0.2 V, and the voltage at the upper limit of analysis was set to the maximum current value of 0.4 to 0.6 V. Then, by calculating (hydrogen adsorption electricity amount at 20% RH) / (hydrogen adsorption electricity amount at 100% RH) ⁇ 100, the ionomer coverage with respect to the total surface area of all catalyst particles supported on the carrier is calculated. It was measured. The results are shown in Table 1.
- FIG. 8 shows the relationship between the G band half width of the carrier and the platinum surface area reduction rate
- FIG. 9 shows the mesopore specific surface area of the carrier and the G band half width. Show the relationship.
- FIG. 9 the results of the mesopore specific surface area and the G band half-value width when the support according to Example 1 is heat-treated at 2300 ° C. (see the circles in the figure) are also plotted.
- FIG. 10 shows the relationship between the specific mesopore surface area of the carrier and the crystallite diameter Lc of the 002 plane. Further, FIG. 11 shows the relationship between the specific activity of the fuel cell electrode and the ionomer coverage on the platinum surface. FIG. 12 shows the relationship between the ionomer coverage on the platinum surface of the fuel cell electrode according to the example and the comparative example and the G band half-width of the carrier.
- the platinum surface area reduction rate decreases as the G band half width decreases. That is, this is considered to be caused by an increase in the crystallinity of the carrier when the G band half width is reduced.
- the rate of platinum surface area decrease decreases as the G band half-width of mesoporous carbon decreases from 70 cm ⁇ 1 .
- the specific surface area of the mesopores is decreased by increasing the heat treatment temperature.
- the support was heat treated in the range of 1700 ° C. to 2100 ° C.
- the crystallinity of the support is increased (the G band half width is also reduced), and platinum particles (catalyst particles) can be supported therein.
- Comparative Examples 2-1, 2-3, 2-4, 2-6 and Comparative Examples 3-1 to 3-4 it is considered that the pores of the carrier were crushed by heating.
- the pore walls forming mesoporous carbon mesopores have a structure in which several layers of graphene sheets are laminated in the wall thickness direction. It is considered that the specific surface area of the mesopores did not decrease even after the operation. As shown in FIG. 10, this is achieved by using a support made of mesoporous carbon having a 002-plane crystallite diameter Lc of 1.5 nm or less as in Examples 1-1 to 1-3. Even if heat treatment is performed at a temperature lower than 2300 ° C., the mesoporous carbon pores (mesopores) are not crushed and the durability of the mesoporous carbon can be enhanced.
- the specific activity of Examples 1-1 to 1-3 and Examples 2-1 to 2-3 is higher than the others, and the ionomer coverage on the surface of the platinum particles is It was 72% or less.
- the G band half-width is set to 70 cm by performing heat treatment under the above-described temperature conditions. -1 or less, and the ionomer coverage can be further reduced.
- Comparative Example 1 corresponds to Comparative Examples 1-2 and 1-3
- Comparative Example 2 corresponds to Comparative Examples 2-2 to Corresponds to 2-7.
- the carrier was developed with pores to the inside and formed with only a few layers of laminated structure (that is, mesoporous carbon having a 002-plane crystallite diameter Lc of 1.5 nm or less).
- laminated structure that is, mesoporous carbon having a 002-plane crystallite diameter Lc of 1.5 nm or less.
- Example 3 A fuel cell electrode was produced in the same manner as in Example 1-3. The difference from Example 1-3 is that 30% by mass of catalyst particles are supported inside mesoporous carbon to prepare a supported catalyst, and this is then subjected to an inert gas atmosphere at 700 ° C. before being coated with an ionomer. The heat treatment was carried out at a temperature of 90 ° C. and immersed in a 0.5 mol / L nitric acid aqueous solution for 20 hours for oxidation treatment.
- Comparative Example 6 In the same manner as in Example 3, a fuel cell electrode was produced. The difference from Example 3 is that no oxidation treatment was performed. In addition, the comparative example 6 is an example for comparing with Example 3, and is an Example included in the scope of the present invention.
- FIG. 14 is a diagram showing the influence of power generation characteristics due to oxidation treatment.
- the current value on the horizontal axis is a dimensionless amount obtained by normalizing the maximum current value to 1.
- Example 4-1 to 4-12 A fuel cell electrode was produced in the same manner as in Example 1-3.
- the difference between Examples 4-1 to 4-12 and Example 1-3 is that after the supported catalyst is prepared and before the ionomer is coated, it is heat-treated in an inert gas atmosphere at 700 ° C. And it is the point which immersed in the nitric acid aqueous solution on the conditions shown in Table 2, and performed the oxidation process.
- Examples 4-1 to 4-10 carried 30% by mass of catalyst particles on mesoporous carbon
- Examples 4-11 and 4-12 carried 50% by mass of catalyst particles on mesoporous carbon.
- the heat treatment temperature of the carrier before supporting the catalyst particles is the same as in Example 1-3.
- Comparative Examples 7-1 to 7-5) A fuel cell electrode was prepared in the same manner as in Example 4-1. Comparative Example 7-1 differs from Example 4-1 in that it was immersed in an aqueous nitric acid solution under the conditions shown in Table 2 and subjected to an oxidation treatment. Comparative Example 7-2 to 7-5 is different from Example 4-1 in that no oxidation treatment is performed, and Comparative Examples 7-2 to 7-4 are different from those in which catalyst particles are supported. The heat treatment temperatures of these carriers are different (see Table 2).
- FIG. 15 is a graph showing the relationship between the generated voltage (at a current value of 1.2 A / cm 2 ) in a low humidified environment and the oxygen content per unit area contained on the surface of the supported catalyst.
- the fuel cells according to Examples 4-1 to 4-12 had higher power generation voltages than those of Comparative Examples 7-1 to 7-5. This is because the surface of the mesopores of the supported catalysts according to Examples 4-1 to 4-12 was hydrophilized by containing more oxygen than Comparative Examples 7-1 to 7-5. It is believed that there is. As shown in Examples 4-1 to 4-12, when the oxygen content per unit area contained on the surface of the supported catalyst is 0.08 mg / m 2 or more, the power generation of the fuel cell in a low humidified environment The deterioration of characteristics can be suppressed.
- the fuel cell according to Comparative Example 7-1 has a slightly higher power generation voltage than those of Comparative Examples 7-2 to 7-5, because of the oxidation treatment, compared to Comparative Examples 7-2 to 7-5. This is presumably because the surface of the mesopores of the catalyst is hydrophilized.
- the oxygen content per unit area contained on the surface of the supported catalyst according to Example 4-12 was 0.18 mg / m 2 .
- Examples 5-1, 5-2, 6-1, 6-2 A fuel cell electrode was prepared in the same manner as in Example 4-1.
- Examples 5-1 5-2, 6-1 and 6-2 differ from Example 4-1 in that they were immersed in an aqueous nitric acid solution under the conditions shown in Table 3 and subjected to an oxidation treatment. .
- Examples 5-1 and 5-2 supported 40% by mass of catalyst particles on mesoporous carbon
- Examples 6-1 and 6-2 also supported that 50% by mass of catalyst particles were supported on mesoporous carbon. This differs from that of Example 4-1.
- the heat treatment temperature of the carrier before supporting the catalyst particles is the same as that in Example 1-3.
- Example 7-1 to 7-3 A fuel cell electrode was prepared in the same manner as in Example 5-1.
- Examples 7-1 to 7-3 differ from Example 5-1 in that 30% by mass of catalyst particles are supported on mesoporous carbon and immersed in an aqueous nitric acid solution under the conditions shown in Table 3 for oxidation treatment. It is a point.
- Comparative Examples 8-1, 8-2, 9-1, 9-2) A fuel cell electrode was prepared in the same manner as in Example 5-1.
- the comparative example 8-1 and 9-1 are mainly different from the example 5-1 in that the oxidation treatment is not performed as shown in Table 3, and the comparative examples 8-2 and 9-2 are different.
- the main difference from Example 5-1 is that it was immersed in an aqueous nitric acid solution under the conditions shown in Table 3 and subjected to an oxidation treatment.
- Comparative Examples 9-1 and 9-2 are further different from those of Comparative Example 5-1 in that 50% by mass of catalyst particles are supported on mesoporous carbon.
- Comparative Examples 8-1 and 8-2 are the same as Example 5-1 in that 40% by mass of catalyst particles are supported on mesoporous carbon.
- Comparative Examples 10-1 to 10-3 A fuel cell electrode was prepared in the same manner as in Example 5-1. Comparative Example 10-1 and 10-2 differ from Example 5-1 in that no oxidation treatment is performed. Further, Comparative Example 10-1 is a heat treatment of a carrier before carrying catalyst particles. The temperature is different (see Table 3). The comparative example 10-3 is different from the example 5-1 in that the oxidation treatment was performed by dipping in an aqueous nitric acid solution under the conditions shown in Table 3.
- the measurement conditions were as follows: adsorption temperature: 323.15 K, adsorbate: pure water, saturated water vapor pressure: 12.344 kPa, adsorbate cross-sectional area: 0.12 nm 2 , equilibrium waiting time: 500 seconds, adsorbate molecular weight: 18.020,
- the equilibrium waiting time is a waiting time after reaching an adsorption equilibrium state (a state in which a pressure change becomes a predetermined value or less during adsorption / desorption).
- FIG. 16 is a diagram showing the relationship between the power generation voltage (at a current value of 2.0 A / cm 2 ) in a low humid environment and T1 / T2.
- FIG. 17 is a diagram showing the relationship between the treatment temperature in the oxidation treatment and T1 / T2.
- the fuel cells according to Examples 5-1, 5-2, 6-1, 6-2, 7-1 to 7-3 are comparative examples 8-1, 8-2, 9-.
- the generated voltage was higher than those of 1,9-2,10-1 to 10-3. This is considered to be because the drainage of the mesopores is higher in the example than in the comparative example in an overhumid environment.
- the partial pressure of water vapor is increased to 60% of the saturated water vapor pressure, the mass of water vapor adsorbed on the supported catalyst is T1, and the partial pressure of water vapor is decreased from the vicinity of the saturated water vapor pressure.
- T1 / T2 of Example 6-2 was 0.27.
- Example 8-1 to 8-4 A fuel cell electrode was produced in the same manner as in Example 1-3.
- Examples 8-1 to 8-4 and Examples 9-1 to 9-3 differ from Example 1-3 in that 30% by mass of catalyst particles are supported inside mesoporous carbon, After the production, before the ionomer was coated, it was heat-treated in an inert gas atmosphere at 700 ° C. and subjected to the oxidation treatment shown in Table 4.
- Example 8-1 to 8-4 among the oxidation treatment conditions, the concentration of nitric acid aqueous solution and the treatment temperature were the same (0.5 mol, 90 ° C.), and the treatment time was changed.
- the treatment temperature and the treatment time are the same (90 ° C., 5 hours) among the oxidation treatment conditions, and the concentration of the nitric acid aqueous solution is changed.
- the conditions are the same as in Example 8-2 and Example 9-2.
- FIG. 18A is a diagram showing the relationship between the treatment time and the oxygen content
- FIG. 18B is a diagram showing the relationship between the treatment time and drainage
- FIG. 18C is a nitric acid aqueous solution. It is the figure which showed the relationship between the density
- (d) is the figure which showed the relationship between the density
- the supported catalyst in which the catalyst particles are supported on the support It is considered that the oxygen content per unit area contained on the surface of the film satisfies 0.08 mg / m 2 or more and satisfies T1 / T2 ⁇ 0.15.
Landscapes
- Chemical & Material Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Composite Materials (AREA)
- Life Sciences & Earth Sciences (AREA)
- Sustainable Development (AREA)
- Sustainable Energy (AREA)
- Inert Electrodes (AREA)
- Fuel Cell (AREA)
- Catalysts (AREA)
Abstract
Description
(担体に担持された全触媒粒子の総表面積に対する前記アイオノマーの被覆率)=
(20%RH時の水素吸着電気量)/(100%RH時の水素吸着電気量)×100
と表すことができる。
表1に示すように、メソポーラスカーボンの担体を準備した。この担体は、メソ孔の孔径が2nm以上であり、上述した実施形態において例示した方法で製造された担体である。次に、担体をアルゴンガスの環境下1700℃、2時間の熱処理条件で加熱した。
実施例1-1と同じように、燃料電池用電極を作製した。実施例1-1と相違する点は、それぞれ、熱処理の工程において、担体をアルゴンガスの環境下1900℃、2100℃の条件で加熱した点である。
実施例2-1~2-3では、それぞれ実施例1-1~1-3に対応した条件で、燃料電池用電極を作製した。これらが実施例1-1~1-3と相違する点は、ジニトロジアミン白金塩水溶液の添加から還流までの時間を短くした点である。
実施例1-1と同じように、燃料電池用電極を作製した。比較例1-1~1-3が実施例1-1と相違する点は、担体に、市販のキャボット社製:VulcanXC-72R(登録商標)を用い、表1に示す熱処理条件で担体に熱処理を施し(ただし比較例1-1は熱処理なし)、表1に示す混合比(I/C)で、アイオノマー(ナフィオン:デュポン社製)と、白金粒子が担持された担体とを混合した点である。
実施例1-1と同じように、燃料電池用電極を作製した。比較例2-1~2-7が実施例1-1と相違する点は、担体に、市販のライオン株式会社製:KetchenEC300J(登録商標)を用い、表1に示す熱処理条件で担体に熱処理を施し(ただし比較例2-1、2-2は熱処理なし)、表1に示す混合比(I/C)で、アイオノマーと、白金粒子が担持された担体とを混合した(ただし比較例2-2、2-7は混合比同じ)点である。
実施例1-1と同じように、燃料電池用電極を作製した。比較例3-1~3-4が実施例1-1と相違する点は、担体に、市販のライオン株式会社製:KetchenEC600JD(登録商標)を用い、表1に示す熱処理条件で担体に熱処理を施し(ただし比較例3-1は熱処理なし)、表1に示す混合比(I/C)で、アイオノマーと、白金粒子が担持された担体とを混合した点である。ここで用いた担体は、比較例2-1~2-7で用いた担体よりもメソ孔比表面積が大きい。
実施例1-1と同じように、燃料電池用電極を作製した。実施例1-1と相違する点は、それぞれ、熱処理なし(未処理)、アルゴンガスの環境下1300℃、1500℃の熱処理条件で加熱した点である。
実施例2-1と同じように、燃料電池用電極を作製した。実施例2-1と相違する点は、それぞれ、熱処理なし(未処理)、アルゴンガスの環境下1300℃、1500℃の熱処理条件で加熱した点である。
比較例2-2、2-5、2-7を除く白金粒子担持前(熱処理後)の担体の002面の結晶子径Lcの測定をした。具体的には、CuKα線を用いた粉末X線回折装置を用いて、粉末状の電極触媒体を粉末X線回折法により分析し、得られた回折パターンから、各結晶面(002)の回折ピークの半値幅β(ラジアン)を求めた。そして、シェラーの式:L=Kλ/βcosθにより、担体の結晶子径の平均値L(nm)を算出した。なお、定数Kは、形状因子)、λはX線の波長(Å)、θは回折角(゜)である。ここで、θ回折角26°付近に現れる002面のピークは、炭素6角網目構造の積層方向に由来しており、メソ孔を構成する壁面の肉厚に依存する。なお、実施例1-1~1-3、実施例2-1~2-3は、熱処理前後の担体の002面の結晶子径Lcの測定をしており、これらは同じ値となった。
比較例2-2、2-5、2-7を除く白金粒子担持前(熱処理後)の担体のGバンド半値幅を測定した。具体的には、測定装置として日本分光社製のNRS-1000を用いて励起光:グリーンレーザー(波長532nm)、レーザーパワー:100mVとし、検出装置CCD(-60℃)を用いて積算回数2回、露光時間:30秒、減光器OD2(減光率1/100)とした。この結果を表1に示す。
比較例2-2、2-5、2-7を除く白金粒子担持前(熱処理後)の担体のメソ孔比表面積を測定した。具体的には、担体の2~10nmの孔径を持つメソ孔を含む比表面積を測定した。具体的には、担体を、吸着測定用前処理装置(BELPREP-vacII(日本ベル(株)製)を用いて、150℃で8時間真空脱気した。次に、自動比表面積/細孔分布測定装置(BELPREP-mini(日本ベル(株)製)を用いて、定容法で図7(a)に示したように、窒素による吸着脱離等温線を測定した。
実施例1-1~1-3、比較例1-1~1-3、比較例2-1、比較例3-1、比較例4-1、比較例4-3の燃料電池電極を用いたMEA(電解質膜-電極接合体)に対して、H吸着量から求めた電気化学表面積(ECSA)をそれぞれ求め、その低下率(%/h)を測定した。この結果を表1に示す。具体的には、0.1N過塩素酸(HClO4)水溶液中で、耐久条件、1.5Vvs.RHEで1時間ホールドを10回行って、ECSA低下率(%/h)すなわち、Pt表面積低下速度(%/h)を測定した。ECSA低下率(%/h)が小さいほど、耐久性に優れている。
実施例1-1~1-3、実施例2-1~2-3、比較例1-1、比較例2-1~2-3、2-5~2-7、比較例4-1~4-3、比較例5-1~5-3の燃料電池電極を用いたMEA(電解質膜-電極接合体)に対して質量活性を測定した。この結果を表1に示す。具体的には、温度80℃、湿度100%における電流測定値のターフェルプロットにより算出した。なお、表1に示す規格化比活性は、比較例1に係るMEAの質量活性で正規化した値である。
上述した方法で作成したすべての燃料電池電極における担体に担持された全触媒粒子の総表面積に対する前記アイオノマーの被覆率を測定した。サイクリックボルタンメトリー法を用いて、100%RH(80℃)と、20%RH(44℃)との条件において、アノードに水素ガス、カソードに窒素ガスを流通させ、参照極(RHE)に対して、電圧を0.1-1.0Vの掃引範囲および掃引速度50m/secすることにより、図2(b)に示したごとく、水素吸着電気量を測定した。水素吸着電気量を算出する際の解析下限の電圧値を0.2V、解析上限の電圧を0.4~0.6Vの電流最大値とした。そして、(20%RH時の水素吸着電気量)/(100%RH時の水素吸着電気量)×100を算出することにより、担体に担持された全触媒粒子の総表面積に対するアイオノマーの被覆率を測定した。この結果を表1に示す。

実施例1-3と同じように、燃料電池用電極を作製した。実施例1-3と相違する点は、メソポーラスカーボンの内部に触媒粒子を30質量%担持して、担持触媒を作製した後、アイオノマーを被覆する前に、これを700℃の不活性ガス雰囲気下で熱処理し、90℃に加熱した0.5mol/Lの硝酸水溶液中に20時間浸漬し、酸化処理を行った点である。
実施例3と同じように、燃料電池用電極を作製した。実施例3と相違する点は、酸化処理を行わなかった点である。なお、比較例6は、実施例3と比較するための例であり、本発明の範囲に含まれる実施例である。
過加湿環境下として170%RHの環境下、低加湿環境下として30%RHの環境下において、実施例3および比較例6に係る燃料電池用電極を用いた燃料電池の発電特性(電流電圧特性)を測定した。この結果を、図14に示す。図14は、酸化処理による発電特性の影響を示した図である。なお、図14は、横軸の電流値は、電流の最大値を1に正規化した無次元量である。
(実施例4-1~4-12)
実施例1-3と同じように、燃料電池用電極を作製した。実施例4-1~4-12が実施例1-3と共通して相違する点は、担持触媒を作製した後に、アイオノマーを被覆する前に、これを700℃の不活性ガス雰囲気下で熱処理し、表2に示す条件で硝酸水溶液中に浸漬し、酸化処理を行った点である。また、実施例4-1~4-10は、メソポーラスカーボンに触媒粒子を30質量%担持し、実施例4-11,4-12は、メソポーラスカーボンに触媒粒子を50質量%担持した。なお、実施例4-1~4-12では、触媒粒子を担持する前の担体の熱処理温度等は実施例1-3と同じである。
実施例4-1と同じように、燃料電池用電極を作製した。比較例7-1が実施例4-1と相違する点は、表2に示す条件で硝酸水溶液中に浸漬し、酸化処理を行った点である。比較例7-2~7-5が実施例4-1と相違する点は、酸化処理を行っていない点であり、さらに、比較例7-2~7-4は、触媒粒子を担持する前の担体の熱処理温度が相違する(表2参照)。

実施例4-1~4-12、比較例7-1~7-5において、アイオノマーを担体に被覆する前の、触媒粒子が担持された状態の担体(担持触媒)の表面に含有する単位面積あたりの酸素含有量を測定した。具体的には、各担持触媒を150℃で真空乾燥を8時間行い、不活性ガス中で、酸素窒素分析装置(EMCA-920:(株)堀場製作所製)を用いてインパル加熱・溶融法により担持触媒を溶融し、NDIR検出方式で酸素量を測定した。なお、担持触媒に含有する酸素は、その大半が酸化処理により表面に含まれる酸素であることから、測定した酸素量を上述したBET比表面積で除算し、担持触媒の表面に含有する単位面積あたりの酸素含有量を算出した。
低加湿環境下として30%RHの環境下において、実施例4-1~4-12、比較例7-1~7-5に係る燃料電池用電極を用いた燃料電池の発電特性(電流電圧特性)を測定した。これらの結果を図15に示す。図15は、低加湿環境下における発電電圧(電流値1.2A/cm2時)と、担持触媒の表面に含有する単位面積あたりの酸素含有量との関係を示した図である。
図15に示すように、実施例4-1~4-12に係る燃料電池は、比較例7-1~7-5のものに比べて、発電電圧が高かった。これは、実施例4-1~4-12に係る担持触媒のメソ孔の表面が、比較例7-1~7-5に比べてより多くの酸素を含有することにより、親水化されたからであると考えられる。実施例4-1~4-12に示すように、担持触媒の表面に含有する単位面積あたりの酸素含有量が0.08mg/m2以上であれば、低加湿環境下において、燃料電池の発電特性の低下を抑えることができる。
実施例4-1と同じように、燃料電池用電極を作製した。実施例5-1,5-2,6-1,6-2が実施例4-1と相違する点は、表3に示す条件で硝酸水溶液中に浸漬し、酸化処理を行った点である。さらに、実施例5-1,5-2は、メソポーラスカーボンに触媒粒子を40質量%担持し、実施例6-1,6-2は、メソポーラスカーボンに触媒粒子を50質量%担持した点も、実施例4-1のものと相違する。なお、実施例5-1,5-2,6-1,6-2は、触媒粒子を担持する前の担体の熱処理温度等は実施例1-3と同じである。
実施例5-1と同じように、燃料電池用電極を作製した。実施例7-1~7-3が実施例5-1と相違する点は、メソポーラスカーボンに触媒粒子を30質量%担持し、表3に示す条件で硝酸水溶液中に浸漬し、酸化処理を行った点である。
実施例5-1と同じように、燃料電池用電極を作製した。比較例8-1、9-1が、実施例5-1と主に相違する点は、表3に示すように酸化処理を行っていない点であり、比較例8-2、9-2が、実施例5-1と主に相違する点は、表3に示す条件で硝酸水溶液中に浸漬し、酸化処理を行った点である。比較例9-1,9-2は、メソポーラスカーボンに触媒粒子を50質量%担持した点が、比較例5-1のものとさらに相違する。なお、比較例8-1,8-2は、メソポーラスカーボンに触媒粒子を40質量%担持している点は、実施例5-1と同じである。
実施例5-1と同じように、燃料電池用電極を作製した。比較例10-1,10-2が実施例5-1と相違する点は、酸化処理を行っていない点であり、さらに、比較例10-1は、触媒粒子を担持する前の担体の熱処理温度が相違する(表3参照)。比較例10-3が実施例5-1と相違する点は、表3に示す条件で、硝酸水溶液中に浸漬することで酸化処理を行った点である。

実施例5-1,5-2,6-1,6-2,7-1~7-3、比較例8-1,8-2,9-1,9-2,10-1~10-3にアイオノマーを担持触媒に被覆する前の、担体に触媒粒子が担持された担持触媒を、吸着測定用前処理装置(BELPREP-vacII(日本ベル(株)製)を用いて、150℃で8時間、真空脱気した。次に、高精度蒸気吸着測定装置(BELPREP-aqua3(日本ベル(株)製)を用いて、定容法で図7(a)に示したように、水蒸気による吸着脱離等温線を測定した。
過加湿環境下として170%RHの環境下において、実施例5-1,5-2,6-1,6-2,7-1~7-3、比較例8-1,8-2,9-1,9-2,10-1~10-3に係る燃料電池用電極を用いた燃料電池の発電特性(電流電圧特性)を測定した。これらの結果を図16に示す。図16は、低加湿環境下における発電電圧(電流値2.0A/cm2時)と、T1/T2との関係を示した図である。さらに、図17は、酸化処理における処理温度と、T1/T2との関係を示した図である。
図16に示すように、実施例5-1,5-2,6-1,6-2,7-1~7-3に係る燃料電池は、比較例8-1,8-2,9-1,9-2,10-1~10-3のものに比べて、発電電圧が高かった。これは、過加湿環境下において、実施例のものが比較例のものに比べて、メソ孔の排水性が高いからであると考えられる。そして、水蒸気分圧を増加させて飽和水蒸気圧の60%の水蒸気分圧になったときの担持触媒に吸着している水蒸気の質量をT1とし、飽和水蒸気圧近傍から水蒸気分圧を減少させて飽和水蒸気圧の60%の水蒸気分圧になったときの担持触媒に吸着している水蒸気の質量をT2としたときに、T1/T2≧0.15の関係を満たせば、過加湿環境下であっても、発電特性の低下を抑制することができると考えられる。なお、実施例6-2のT1/T2は、0.27であった。
実施例1-3と同じように、燃料電池用電極を作製した。実施例8-1~8-4,実施例9-1~9-3が、実施例1-3と相違する点は、メソポーラスカーボンの内部に触媒粒子を30質量%担持して、担持触媒を作製した後、アイオノマーを被覆する前に、これを700℃の不活性ガス雰囲気下で熱処理し、表4に示す酸化処理を行った点である。

図18(a),(b)に示すように、酸化処理における処理時間を変化しても、図18(c),(d)に示すように、硝酸水溶液の濃度を変化させても、酸素含有量およびT1/T2の変化は、あまり変わらず、酸化処理条件のうち、処理温度が、酸素含有量およびT1/T2の変化に大きく寄与するものと考えられる。したがって、0.1mol/L以上の濃度で、60℃以上に加熱した硝酸水溶液をメソポーラスカーボンの少なくともメソ孔の表面に1時間以上接触(浸漬)させれば、触媒粒子を担体に担持した担持触媒の表面に含有する単位面積あたりの酸素含有量が、0.08mg/m2以上かつT1/T2≧0.15を満たすと考えられる。
Claims (10)
- 002面の結晶子径Lcが1.5nm以下であるメソポーラスカーボンからなる担体を、1700℃以上かつ2300℃未満で熱処理する工程と、
前記熱処理された担体の少なくとも内部に触媒粒子を担持する工程と、
前記触媒粒子が担持された担体にアイオノマーを被覆する工程と、を少なくとも含むことを特徴とする燃料電池用電極の製造方法。 - 前記担体の2~10nmの孔径を持つメソ孔のメソ孔比表面積が400m2/g以上となるように、前記熱処理を行うことを特徴とする請求項1に記載の燃料電池用電極の製造方法。
- 前記触媒粒子を担持する工程後、前記アイオノマーを被覆する工程の前に、
前記担体のメソポーラスカーボンの少なくともメソ孔の表面を酸化処理する工程をさらに含むことを特徴とする請求項1または2に記載の燃料電池用電極の製造方法。 - 前記触媒粒子を前記担体に担持した担持触媒の表面に含有する単位面積あたりの酸素含有量が、0.08mg/m2以上となるように、前記酸化処理を行うことを特徴とする請求項3に記載の燃料電池用電極の製造方法。
- 前記酸化処理を行った前記担持触媒を真空環境下に配置し、水蒸気分圧を飽和水蒸気圧近傍まで増加させることにより前記担持触媒に水蒸気を吸着させ、前記担持触媒に水蒸気が吸着した状態で、飽和水蒸気圧近傍から水蒸気分圧を減少させることにより、前記担持触媒に吸着した水蒸気を脱離させたときに、
前記水蒸気分圧を増加させて前記飽和水蒸気圧の60%の水蒸気分圧になったときの前記担持触媒に吸着している水蒸気の質量をT1とし、前記水蒸気分圧を減少させて前記飽和水蒸気圧の60%の水蒸気分圧になったときの前記担持触媒に吸着している水蒸気の質量をT2としたときに、T1/T2≧0.15の関係を満たすように、前記担持触媒に対して前記酸化処理を行うことを特徴とする請求項4に記載の燃料電池用電極の製造方法。 - 前記酸化処理において、0.1mol/L以上、60℃以上に加熱した硝酸水溶液を、前記メソポーラスカーボンのメソ孔の少なくとも表面に1時間以上接触させることを特徴とする請求項3に記載の燃料電池用電極の製造方法。
- メソポーラスカーボンからなる担体と、該担体の少なくとも内部に担持された触媒粒子と、該担体を被覆するアイオノマーと、を備え、
前記担体を構成するメソポーラスカーボンのGバンド半値幅が70cm-1以下であることを特徴とする燃料電池用電極。 - 前記担体に担持された全触媒粒子の総表面積に対する前記アイオノマーの被覆率は、0%よりも大きく72%以下であることを特徴とする請求項7に記載の燃料電池用電極。
- 前記アイオノマーを担体に被覆する前の、前記触媒粒子を前記担体に担持した担持触媒の表面に含有する単位面積あたりの酸素含有量が、0.08mg/m2以上であることを特徴とする請求項7または8に記載の燃料電池用電極。
- 前記アイオノマーを担体に被覆する前の、前記担持触媒を真空環境下に配置し、水蒸気分圧を飽和水蒸気圧近傍まで増加させることにより担持触媒に水蒸気を吸着させ、前記担持触媒に水蒸気が吸着した状態で、飽和水蒸気圧近傍から水蒸気分圧を減少させることにより、前記担持触媒に吸着した水蒸気を脱離させたときに、
前記水蒸気分圧を増加させて前記飽和水蒸気圧の60%の水蒸気分圧になったときの前記担持触媒に吸着している水蒸気の質量をT1とし、前記水蒸気分圧を減少させて前記飽和水蒸気圧の60%の水蒸気分圧になったときの前記担持触媒に吸着している水蒸気の質量をT2としたときに、前記担持触媒はT1/T2≧0.15の関係を満たすことを特徴とする請求項9に記載の燃料電池用電極。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015517133A JP6063039B2 (ja) | 2013-05-16 | 2014-05-15 | 燃料電池用電極およびその製造方法 |
| KR1020157031945A KR101759416B1 (ko) | 2013-05-16 | 2014-05-15 | 연료 전지용 전극 및 그 제조 방법 |
| US14/891,167 US9966610B2 (en) | 2013-05-16 | 2014-05-15 | Electrode for fuel cell and method for manufacturing same |
| CN201480026918.1A CN105229834B (zh) | 2013-05-16 | 2014-05-15 | 燃料电池用电极及其制造方法 |
| EP14797181.6A EP2999039B1 (en) | 2013-05-16 | 2014-05-15 | Electrode for fuel cell and method for manufacturing same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-104354 | 2013-05-16 | ||
| JP2013104354 | 2013-05-16 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014185498A1 true WO2014185498A1 (ja) | 2014-11-20 |
Family
ID=51898473
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/062984 WO2014185498A1 (ja) | 2013-05-16 | 2014-05-15 | 燃料電池用電極およびその製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9966610B2 (ja) |
| EP (1) | EP2999039B1 (ja) |
| JP (1) | JP6063039B2 (ja) |
| KR (1) | KR101759416B1 (ja) |
| CN (1) | CN105229834B (ja) |
| WO (1) | WO2014185498A1 (ja) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016100262A (ja) * | 2014-11-25 | 2016-05-30 | 新日鐵住金株式会社 | 固体高分子形燃料電池用触媒 |
| JP2017139141A (ja) * | 2016-02-04 | 2017-08-10 | 新日鐵住金株式会社 | マイクロポア層用炭素材料、マイクロポア層、及び固体高分子形燃料電池 |
| WO2017154359A1 (ja) * | 2016-03-11 | 2017-09-14 | 日産自動車株式会社 | 燃料電池用炭素粉末ならびに当該燃料電池用炭素粉末を用いる触媒、電極触媒層、膜電極接合体および燃料電池 |
| JP2017162660A (ja) * | 2016-03-09 | 2017-09-14 | トヨタ自動車株式会社 | 燃料電池用電極触媒 |
| JP2017188335A (ja) * | 2016-04-06 | 2017-10-12 | トヨタ自動車株式会社 | 膜電極接合体の製造方法 |
| JP2017208224A (ja) * | 2016-05-18 | 2017-11-24 | 新日鐵住金株式会社 | 触媒担体用炭素材料、触媒担体用炭素材料のポリエチレングリコール樹脂吸着量評価試験方法、固体高分子形燃料電池用触媒、固体高分子形燃料電池用触媒層、及び固体高分子形燃料電池 |
| CN107431212A (zh) * | 2015-02-18 | 2017-12-01 | 新日铁住金株式会社 | 催化剂载体用碳材料、固体高分子形燃料电池用催化剂、固体高分子形燃料电池、及催化剂载体用碳材料的制造方法 |
| JP2018098196A (ja) * | 2016-12-09 | 2018-06-21 | トヨタ自動車株式会社 | 燃料電池用電極触媒 |
| EP3392938A1 (en) | 2017-04-17 | 2018-10-24 | Panasonic Intellectual Property Management Co., Ltd. | Electrode catalyst layer of electrochemical device, membrane electrode assembly of electrochemical device, and electrochemical device |
| JP2019185960A (ja) * | 2018-04-05 | 2019-10-24 | ロベルト・ボッシュ・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツングRobert Bosch Gmbh | 燃料電池用触媒およびその製造方法 |
| JP2020024796A (ja) * | 2018-08-06 | 2020-02-13 | トヨタ自動車株式会社 | 燃料電池用触媒の製造方法 |
| EP3641035A1 (en) | 2018-10-17 | 2020-04-22 | Panasonic Intellectual Property Management Co., Ltd. | Electrode catalyst of electrochemical device, electrode catalyst layer of electrochemical device, membrane electrode assembly of electrochemical device, electrochemical device, method for manufacturing electrode catalyst of electrochemical device, and method for manufacturing membrane electrode assembly of electrochemical device |
| WO2021059932A1 (ja) | 2019-09-27 | 2021-04-01 | パナソニックIpマネジメント株式会社 | 触媒、触媒層、膜/電極接合体、電気化学デバイス、触媒の製造方法 |
| US11283093B2 (en) | 2016-12-28 | 2022-03-22 | Kolon Industries, Inc. | Method for manufacturing electrode, electrode manufactured thereby, membrane-electrode assembly comprising same electrode, and fuel cell including same membrane-electrode assembly |
| JPWO2022181261A1 (ja) * | 2021-02-26 | 2022-09-01 | ||
| WO2024202883A1 (ja) * | 2023-03-31 | 2024-10-03 | 日鉄ケミカル&マテリアル株式会社 | 固体高分子型燃料電池の触媒担体用炭素材料、固体高分子型燃料電池用触媒層、燃料電池、及び固体高分子型燃料電池の触媒担体用炭素材料の製造方法 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3147976A4 (en) * | 2014-03-19 | 2017-11-08 | Nippon Steel & Sumitomo Metal Corporation | Supporting carbon material for solid polymer fuel cell and catalyst metal particle-supporting carbon material |
| KR101817771B1 (ko) * | 2016-09-09 | 2018-02-21 | 롯데케미칼 주식회사 | 레독스 흐름 전지의 전극 제조용 슬러리 조성물 및 레독스 흐름 전지의 전극의 제조 방법 |
| CN110537275B (zh) * | 2017-03-31 | 2022-12-02 | 日铁化学材料株式会社 | 固体高分子型燃料电池的催化剂载体用碳材料及其制造方法 |
| CN110915041B (zh) * | 2017-06-29 | 2022-08-30 | 日铁化学材料株式会社 | 固体高分子型燃料电池催化剂载体及制造方法、燃料电池 |
| JP7006497B2 (ja) | 2018-05-11 | 2022-02-10 | トヨタ自動車株式会社 | 燃料電池用触媒層及びその製造方法 |
| EP3796439A4 (en) * | 2018-05-15 | 2022-03-02 | N.E. Chemcat Corporation | ELECTRODE CATALYST, COMPOSITION FOR FORMING A GAS DIFFUSION ELECTRODE, GAS DIFFUSION ELECTRODE, DIAPHRAGM ELECTRODE ASSEMBLY AND FUEL CELL STACK |
| CN115181436B (zh) * | 2018-06-27 | 2024-09-27 | 伊梅科技 | 表面功能化碳质颗粒、其制备方法和应用 |
| KR20200068998A (ko) | 2018-12-06 | 2020-06-16 | 현대자동차주식회사 | 산소반응성이 향상된 연료전지용 전극 및 이의 제조방법 |
| JP7153005B2 (ja) * | 2019-11-29 | 2022-10-13 | 株式会社豊田中央研究所 | メソ多孔カーボン及びその製造方法、並びに、固体高分子形燃料電池 |
| EP4082662A1 (en) * | 2019-12-24 | 2022-11-02 | Agc Inc. | Catalyst layer, membrane electrode assembly for solid polymer type fuel cell, and solid polymer type fuel cell |
| US11715834B2 (en) * | 2019-12-27 | 2023-08-01 | Toyota Motor Engineering And Manufacturing North America, Inc. | Fuel cell cathode catalyst |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002025560A (ja) * | 2000-07-06 | 2002-01-25 | Matsushita Electric Ind Co Ltd | 燃料電池 |
| JP2002226648A (ja) | 2001-01-29 | 2002-08-14 | Sumitomo Rubber Ind Ltd | ダイナミックダンパー用ゴム、ダイナミックダンパー、並びに、ダイナミックダンパーを装着したテニスラケット |
| JP2004071253A (ja) * | 2002-08-02 | 2004-03-04 | Toyota Motor Corp | 燃料電池用電極触媒及び燃料電池 |
| JP2005209615A (ja) * | 2003-11-14 | 2005-08-04 | Nissan Motor Co Ltd | ガス拡散層および固体高分子電解質型燃料電池 |
| JP2008016208A (ja) * | 2006-07-03 | 2008-01-24 | Nissan Motor Co Ltd | 電極触媒及び燃料電池 |
| WO2012053303A1 (ja) * | 2010-10-22 | 2012-04-26 | 日産自動車株式会社 | 固体高分子型燃料電池用電極触媒 |
| WO2013073383A1 (ja) * | 2011-11-17 | 2013-05-23 | 日産自動車株式会社 | 燃料電池用電極触媒層 |
| WO2013129417A1 (ja) * | 2012-02-28 | 2013-09-06 | 日産自動車株式会社 | 燃料電池用カソード電極 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1164651A1 (en) * | 2000-06-12 | 2001-12-19 | Asahi Glass Co., Ltd. | Electrode catalyst for polymer electrolyte fuel cell and method for its production |
| US7169731B2 (en) * | 2003-02-12 | 2007-01-30 | Symyx Technologies, Inc. | Method for the synthesis of a fuel cell electrocatalyst |
| KR100919326B1 (ko) | 2004-04-22 | 2009-09-25 | 신닛뽄세이테쯔 카부시키카이샤 | 연료 전지 및 연료 전지용 가스 확산 전극 |
| JP5151146B2 (ja) * | 2006-03-06 | 2013-02-27 | トヨタ自動車株式会社 | 固体高分子型燃料電池及びそれに用いる固体高分子型燃料電池用meaの製造方法 |
| JP5481748B2 (ja) | 2007-12-12 | 2014-04-23 | 新日鉄住金化学株式会社 | 炭素ナノ構造体、金属内包樹状炭素ナノ構造物の作製方法、及び炭素ナノ構造体の作製方法 |
| JP4880064B1 (ja) | 2010-12-08 | 2012-02-22 | 田中貴金属工業株式会社 | 固体高分子形燃料電池用触媒及びその製造方法 |
| US20150352522A1 (en) * | 2013-02-21 | 2015-12-10 | Nippon Steel & Sumitomo Metal Corporation | Carbon material for catalyst support use |
-
2014
- 2014-05-15 US US14/891,167 patent/US9966610B2/en active Active
- 2014-05-15 KR KR1020157031945A patent/KR101759416B1/ko active IP Right Grant
- 2014-05-15 EP EP14797181.6A patent/EP2999039B1/en active Active
- 2014-05-15 JP JP2015517133A patent/JP6063039B2/ja active Active
- 2014-05-15 CN CN201480026918.1A patent/CN105229834B/zh active Active
- 2014-05-15 WO PCT/JP2014/062984 patent/WO2014185498A1/ja active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002025560A (ja) * | 2000-07-06 | 2002-01-25 | Matsushita Electric Ind Co Ltd | 燃料電池 |
| JP2002226648A (ja) | 2001-01-29 | 2002-08-14 | Sumitomo Rubber Ind Ltd | ダイナミックダンパー用ゴム、ダイナミックダンパー、並びに、ダイナミックダンパーを装着したテニスラケット |
| JP2004071253A (ja) * | 2002-08-02 | 2004-03-04 | Toyota Motor Corp | 燃料電池用電極触媒及び燃料電池 |
| JP2005209615A (ja) * | 2003-11-14 | 2005-08-04 | Nissan Motor Co Ltd | ガス拡散層および固体高分子電解質型燃料電池 |
| JP2008016208A (ja) * | 2006-07-03 | 2008-01-24 | Nissan Motor Co Ltd | 電極触媒及び燃料電池 |
| WO2012053303A1 (ja) * | 2010-10-22 | 2012-04-26 | 日産自動車株式会社 | 固体高分子型燃料電池用電極触媒 |
| WO2013073383A1 (ja) * | 2011-11-17 | 2013-05-23 | 日産自動車株式会社 | 燃料電池用電極触媒層 |
| WO2013129417A1 (ja) * | 2012-02-28 | 2013-09-06 | 日産自動車株式会社 | 燃料電池用カソード電極 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2999039A4 * |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016100262A (ja) * | 2014-11-25 | 2016-05-30 | 新日鐵住金株式会社 | 固体高分子形燃料電池用触媒 |
| CN107431212A (zh) * | 2015-02-18 | 2017-12-01 | 新日铁住金株式会社 | 催化剂载体用碳材料、固体高分子形燃料电池用催化剂、固体高分子形燃料电池、及催化剂载体用碳材料的制造方法 |
| CN107431212B (zh) * | 2015-02-18 | 2020-10-27 | 日铁化学材料株式会社 | 催化剂载体用碳材料、固体高分子形燃料电池用催化剂、固体高分子形燃料电池、及催化剂载体用碳材料的制造方法 |
| JP2017139141A (ja) * | 2016-02-04 | 2017-08-10 | 新日鐵住金株式会社 | マイクロポア層用炭素材料、マイクロポア層、及び固体高分子形燃料電池 |
| JP2017162660A (ja) * | 2016-03-09 | 2017-09-14 | トヨタ自動車株式会社 | 燃料電池用電極触媒 |
| JPWO2017154359A1 (ja) * | 2016-03-11 | 2019-01-31 | 日産自動車株式会社 | 燃料電池用炭素粉末ならびに当該燃料電池用炭素粉末を用いる触媒、電極触媒層、膜電極接合体および燃料電池 |
| US10675611B2 (en) | 2016-03-11 | 2020-06-09 | Nissan Motor Co., Ltd. | Carbon powder for fuel cell and catalyst, electrode catalyst layer, membrane electrode assembly, and fuel cell using the carbon powder for fuel cell |
| WO2017154359A1 (ja) * | 2016-03-11 | 2017-09-14 | 日産自動車株式会社 | 燃料電池用炭素粉末ならびに当該燃料電池用炭素粉末を用いる触媒、電極触媒層、膜電極接合体および燃料電池 |
| JP2017188335A (ja) * | 2016-04-06 | 2017-10-12 | トヨタ自動車株式会社 | 膜電極接合体の製造方法 |
| JP2017208224A (ja) * | 2016-05-18 | 2017-11-24 | 新日鐵住金株式会社 | 触媒担体用炭素材料、触媒担体用炭素材料のポリエチレングリコール樹脂吸着量評価試験方法、固体高分子形燃料電池用触媒、固体高分子形燃料電池用触媒層、及び固体高分子形燃料電池 |
| JP7094075B2 (ja) | 2016-05-18 | 2022-07-01 | 日本製鉄株式会社 | 触媒担体用炭素材料、固体高分子形燃料電池用触媒、固体高分子形燃料電池用触媒層、及び固体高分子形燃料電池 |
| JP2018098196A (ja) * | 2016-12-09 | 2018-06-21 | トヨタ自動車株式会社 | 燃料電池用電極触媒 |
| US11283093B2 (en) | 2016-12-28 | 2022-03-22 | Kolon Industries, Inc. | Method for manufacturing electrode, electrode manufactured thereby, membrane-electrode assembly comprising same electrode, and fuel cell including same membrane-electrode assembly |
| EP3392938A1 (en) | 2017-04-17 | 2018-10-24 | Panasonic Intellectual Property Management Co., Ltd. | Electrode catalyst layer of electrochemical device, membrane electrode assembly of electrochemical device, and electrochemical device |
| US10790526B2 (en) | 2017-04-17 | 2020-09-29 | Panasonic Intellectual Property Management Co., Ltd. | Electrode catalyst layer of electrochemical device, membrane electrode assembly of electrochemical device, and electrochemical device |
| JP7093666B2 (ja) | 2018-04-05 | 2022-06-30 | ロベルト・ボッシュ・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | 燃料電池用触媒およびその製造方法 |
| JP2019185960A (ja) * | 2018-04-05 | 2019-10-24 | ロベルト・ボッシュ・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツングRobert Bosch Gmbh | 燃料電池用触媒およびその製造方法 |
| JP7059861B2 (ja) | 2018-08-06 | 2022-04-26 | トヨタ自動車株式会社 | 燃料電池用触媒の製造方法 |
| JP2020024796A (ja) * | 2018-08-06 | 2020-02-13 | トヨタ自動車株式会社 | 燃料電池用触媒の製造方法 |
| US11367879B2 (en) | 2018-10-17 | 2022-06-21 | Panasonic Intellectual Property Management Co., Ltd. | Electrode catalyst of electrochemical device, electrode catalyst layer of electrochemical device, membrane electrode assembly of electrochemical device, electrochemical device, method for manufacturing electrode catalyst of electrochemical device, and method for manufacturing membrane electrode assembly of electrochemical device |
| EP3641035A1 (en) | 2018-10-17 | 2020-04-22 | Panasonic Intellectual Property Management Co., Ltd. | Electrode catalyst of electrochemical device, electrode catalyst layer of electrochemical device, membrane electrode assembly of electrochemical device, electrochemical device, method for manufacturing electrode catalyst of electrochemical device, and method for manufacturing membrane electrode assembly of electrochemical device |
| WO2021059932A1 (ja) | 2019-09-27 | 2021-04-01 | パナソニックIpマネジメント株式会社 | 触媒、触媒層、膜/電極接合体、電気化学デバイス、触媒の製造方法 |
| US11784320B2 (en) | 2019-09-27 | 2023-10-10 | Panasonic Intellectual Property Management Co., Ltd. | Catalyst, catalyst layer, membrane-electrode assembly, electrochemical device, and method for producing catalyst |
| JPWO2022181261A1 (ja) * | 2021-02-26 | 2022-09-01 | ||
| WO2022181261A1 (ja) * | 2021-02-26 | 2022-09-01 | 三井金属鉱業株式会社 | 電極触媒及びその製造方法並びに燃料電池 |
| JP7418656B2 (ja) | 2021-02-26 | 2024-01-19 | 三井金属鉱業株式会社 | 電極触媒及びその製造方法並びに燃料電池 |
| WO2024202883A1 (ja) * | 2023-03-31 | 2024-10-03 | 日鉄ケミカル&マテリアル株式会社 | 固体高分子型燃料電池の触媒担体用炭素材料、固体高分子型燃料電池用触媒層、燃料電池、及び固体高分子型燃料電池の触媒担体用炭素材料の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20150142007A (ko) | 2015-12-21 |
| EP2999039A4 (en) | 2016-05-25 |
| US9966610B2 (en) | 2018-05-08 |
| JPWO2014185498A1 (ja) | 2017-02-23 |
| CN105229834B (zh) | 2017-09-29 |
| CN105229834A (zh) | 2016-01-06 |
| EP2999039B1 (en) | 2018-11-07 |
| US20160093892A1 (en) | 2016-03-31 |
| KR101759416B1 (ko) | 2017-07-18 |
| JP6063039B2 (ja) | 2017-01-18 |
| EP2999039A1 (en) | 2016-03-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6063039B2 (ja) | 燃料電池用電極およびその製造方法 | |
| KR100696463B1 (ko) | 고농도 탄소 담지 촉매, 그 제조방법, 상기 촉매를 이용한촉매전극 및 이를 이용한 연료전지 | |
| CA2925618C (en) | Carbon powder for catalyst, catalyst, electrode catalyst layer, membrane electrode assembly, and fuel cell using the carbon powder | |
| JP6113836B2 (ja) | 触媒ならびに当該触媒を用いる電極触媒層、膜電極接合体および燃料電池 | |
| KR100868756B1 (ko) | 백금/루테늄 합금 담지 촉매, 그 제조방법 및 이를 이용한연료전지 | |
| CA2910374C (en) | Catalyst and electrode catalyst layer, membrane electrode assembly, and fuel cell using the catalyst | |
| WO2013129417A1 (ja) | 燃料電池用カソード電極 | |
| JP6327681B2 (ja) | 燃料電池用電極触媒、その製造方法、当該触媒を含む燃料電池用電極触媒層ならびに当該触媒または触媒層を用いる燃料電池用膜電極接合体および燃料電池 | |
| EP3032624B1 (en) | Catalyst particles, and electrocatalyst, electrolyte membrane-electrode assembly, and fuel cell using such catalyst particles | |
| EP3214680B1 (en) | Electrode catalyst for fuel cell, electrode catalyst layer for fuel cell, method for producing same, and membrane electrode assembly and fuel cell using catalyst layer | |
| CN111063896A (zh) | 电化学器件的电极催化剂、膜电极接合体、它们的制造方法和电化学器件的电极催化剂层 | |
| WO2016067879A1 (ja) | 燃料電池用電極触媒層、ならびに当該触媒層を用いる燃料電池用膜電極接合体および燃料電池 | |
| WO2014175101A1 (ja) | 触媒の製造方法ならびに当該触媒を用いる電極触媒層、膜電極接合体および燃料電池 | |
| US20060088741A1 (en) | Methanol resistant cathodic catalyst for direct methanol fuel cells | |
| JP2016054065A (ja) | 電極触媒の製造方法 | |
| JP7110842B2 (ja) | 固体高分子形燃料電池触媒担体用炭素材料、固体高分子形燃料電池燃料電池用触媒層、及び固体高分子形燃料電池 | |
| US20170098834A1 (en) | Fuel cell anode catalyst and manufacturing method therefor | |
| JP6846210B2 (ja) | 電極触媒ならびに当該電極触媒を用いる膜電極接合体および燃料電池 | |
| WO2015118922A1 (ja) | 電極触媒およびその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201480026918.1 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14797181 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015517133 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20157031945 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14891167 Country of ref document: US Ref document number: 2014797181 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |