WO2014184909A1 - 位相差フィルム、偏光板および液晶表示装置 - Google Patents
位相差フィルム、偏光板および液晶表示装置 Download PDFInfo
- Publication number
- WO2014184909A1 WO2014184909A1 PCT/JP2013/063593 JP2013063593W WO2014184909A1 WO 2014184909 A1 WO2014184909 A1 WO 2014184909A1 JP 2013063593 W JP2013063593 W JP 2013063593W WO 2014184909 A1 WO2014184909 A1 WO 2014184909A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- film
- acid
- compound
- retardation
- Prior art date
Links
Images

Classifications
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
- G02B5/3083—Birefringent or phase retarding elements
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/133—Constructional arrangements; Operation of liquid crystal cells; Circuit arrangements
- G02F1/1333—Constructional arrangements; Manufacturing methods
- G02F1/1335—Structural association of cells with optical devices, e.g. polarisers or reflectors
- G02F1/13363—Birefringent elements, e.g. for optical compensation
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/133—Constructional arrangements; Operation of liquid crystal cells; Circuit arrangements
- G02F1/1333—Constructional arrangements; Manufacturing methods
- G02F1/1335—Structural association of cells with optical devices, e.g. polarisers or reflectors
- G02F1/133528—Polarisers
Definitions
- the present invention relates to a retardation film, a polarizing plate, and a liquid crystal display device. More specifically, the present invention relates to a retardation film having a small retardation variation due to humidity fluctuation and a high strength, and a polarizing plate and a liquid crystal display device including the retardation film.
- a liquid crystal display device is composed of a liquid crystal cell in which a transparent electrode, a liquid crystal layer, a color filter, etc. are sandwiched between glass plates, and two polarizing plates provided on both sides thereof. It has a configuration in which a child (also referred to as a polarizing film or a polarizing film) is sandwiched between two films (a protective film for a polarizing plate or a retardation film).
- a polarizing film also referred to as a polarizing film or a polarizing film
- a protective film for a polarizing plate or a retardation film a protective film for a polarizing plate or a retardation film.
- the protective film for polarizing plate a cellulose ester film has been widely used, which has high transparency and can easily ensure adhesion with polyvinyl alcohol used in a polarizer.
- liquid crystal display devices are required to have thinner and lighter polarizing plate members as well as improved contrast of displayed images.
- a technique using a phase difference increasing agent includes, for example, a technique for containing a specific epoxy compound in a cellulose ester resin (Patent Document 1) and a technique for containing a specific epoxy ester compound in a cellulose ester resin. It has been proposed to use a technique such as (Patent Document 2).
- Patent Documents 1 and 2 when the inventors of the present application have confirmed the techniques of Patent Documents 1 and 2 above, it has been found that there is a problem that phase difference variation due to humidity is large and color unevenness occurs when a liquid crystal display device is used. Moreover, when the retardation film described in the above-mentioned Patent Documents 1 and 2 is too thin, tearing strength deteriorates and another problem such as tearing during transportation has occurred.
- the present invention has been made in view of the above circumstances, and an object thereof is to provide a retardation film that can express a desired retardation even in a thin film.
- Another object of the present invention is to provide a retardation film in which the retardation value is less affected by humidity.
- Still another object of the present invention is to provide a retardation film having excellent strength.
- a cellulose ester resin has an epoxy compound or an epoxy ester compound and a compound having at least one 5- or 6-membered aromatic heterocyclic group. It has been found that the above-mentioned problems can be solved by blending and the present invention has been completed.
- R 1 to R 4 each independently represents a hydrogen atom or an alkyl group having 1 to 3 carbon atoms; and R 5 and R 6 each independently represents an alkyl group or glycidyl having a substituent.
- the substituent is at least one selected from the group consisting of a hydroxyl group, an ester group and an aromatic group, At least one compound represented by: The following general formula (2):
- a 1 and A 2 each independently represent an alkyl group, a cycloalkyl group, an aromatic hydrocarbon ring group or an aromatic heterocyclic group
- B represents an aromatic hydrocarbon ring or an aromatic heterocyclic ring
- T 1 and T 2 each independently represent a divalent 1,2,4-triazole ring group
- L 1 , L 2 , L 3 and L 4 each independently represent a single bond Or a divalent linking group
- n represents an integer of 0 to 5
- the present invention it is possible to provide a retardation film capable of expressing a desired retardation even in a thin film.
- the retardation film of the present invention has a small variation in retardation value due to humidity.
- R 1 to R 4 each independently represents a hydrogen atom or an alkyl group having 1 to 3 carbon atoms; and R 5 and R 6 each independently represents an alkyl group or glycidyl having a substituent.
- the substituent is at least one selected from the group consisting of a hydroxyl group, an ester group and an aromatic group, At least one compound represented by: The following general formula (2):
- a 1 and A 2 each independently represent an alkyl group, a cycloalkyl group, an aromatic hydrocarbon ring group or an aromatic heterocyclic group
- B represents an aromatic hydrocarbon ring or an aromatic heterocyclic ring
- T 1 and T 2 each independently represent a divalent 1,2,4-triazole ring group
- L 1 , L 2 , L 3 and L 4 each independently represent a single bond Or a divalent linking group
- n represents an integer of 0 to 5,
- film a retardation film
- the retardation film of the present invention is characterized by containing both the compound of general formula (1) and the compound of general formula (2). With this configuration, the retardation film of the present invention can achieve both good tear strength and retardation humidity fluctuation. The mechanism that produces the effect is unknown, but is presumed as follows.
- the present invention is not limited by the following estimation. That is, by including both the compound of the general formula (1) and the compound of the general formula (2), the interaction with the side chain and the hydrogen atom of the cellulose resin is further strengthened, and the side chain and the hydrogen atom of the cellulose resin are added. Water becomes difficult to coordinate. For this reason, it is estimated that the phase difference humidity fluctuation
- the retardation film of the present invention can achieve both the retardation humidity fluctuation and the tear strength.
- the retardation film of the present invention can express a desired retardation even in the form of a thin film, and the fluctuation of the retardation value due to humidity is small.
- X to Y indicating a range means “X or more and Y or less”, and “weight” and “mass”, “mass%” and “wt%”, “part by weight” and “Part by weight” is treated as a synonym. Unless otherwise specified, measurement of operation and physical properties is performed under conditions of room temperature (20 to 25 ° C.) / Relative humidity 40 to 50%.
- the “retardation film” refers to a film having different refractive indexes in the X-axis direction and the Y-axis direction, and is an optical compensation film that expands the viewing angle.
- the film satisfies the following conditions 1 and 2 as retardation values.
- Such a film has a high retardation value and is suitable for widening the viewing angle of a vertical alignment type liquid crystal display device.
- An in-plane retardation value Ro represented by the following formula (I) measured at a wavelength of 590 nm under an environment of a temperature of 23 ° C. and a relative humidity of 55% is in the range of 20 to 130 nm.
- nx represents the refractive index in the slow axis direction in the plane of the film
- ny represents the refractive index in the direction perpendicular to the slow axis in the plane of the film.
- Nz represents the refractive index in the thickness direction of the film; and
- d represents the thickness (nm) of the film.
- retardation values can be measured using an automatic birefringence meter KOBRA-21ADH (Oji Scientific Instruments).
- the desired retardation value can be adjusted by controlling the stretching ratio at the time of film production, the addition amount of the retardation increasing agent, the type and substitution degree of acyl groups of the cellulose ester, the film thickness, and the like.
- the Rt humidity fluctuation represented by the following formula (1) preferably satisfies 1 to 12%.
- the smaller value of the Rt humidity fluctuation below indicates that it is more stable against the humidity fluctuation. From this viewpoint, the value of the Rt humidity fluctuation is 12% or less.
- the moisture permeability of the retardation film is too small, and water is biased between the protective film and the polarizer, and a load is applied. Therefore, there is a possibility that problems such as peeling off of the protective film and the polarizer, water drainage, and red color of the display device may occur.
- a retardation film satisfying the above range has a small change in the retardation value in the thickness direction of the film due to a change in humidity, and a polarizing plate excellent in moisture and heat resistance, and a liquid crystal display device (hereinafter simply referred to as “display”). Device ").
- the Rt humidity variation is more preferably 1 to 10%, and further preferably 1 to 8%.
- the Rt humidity fluctuation can be controlled within a desired range by adjusting the type and substitution degree of acyl group of cellulose ester, the type and addition amount of retardation increasing agent, the type and addition amount of plasticizer, and the like.
- the film thickness (dry film thickness) of the retardation film or the film thickness is not particularly limited, but is preferably 10 to 40 ⁇ m. With such a film thickness, uniform film formation can be achieved, and even when the area is large, color unevenness can be effectively suppressed and a sufficient phase difference can be achieved. Further, from the viewpoint of uniform film formation and Rt humidity fluctuation, the film thickness (dry film thickness) of the retardation film is more preferably in the range of 20 to 35 ⁇ m, particularly preferably 25 to 30 ⁇ m. The film thickness can be controlled within the desired range by adjusting the thickness of the dope or melt to be cast during film formation and / or the stretching conditions.
- the retardation film according to this embodiment has a thickness variation of 0 to 4 ⁇ m in both the width direction and the longitudinal direction. In such a case, even if the film has a large area, variation in retardation in the film surface is suppressed, and color unevenness can be prevented.
- the film thickness variation is preferably 0 to 2.5 ⁇ m, more preferably 0 to 1.5 ⁇ m.
- the film thickness of the retardation film can be measured using a film thickness meter such as a micrometer. Specifically, the film thickness ( ⁇ m) is measured at 100 or more points at 10 mm intervals in the width direction of the film, and the average value thereof is defined as the film thickness ( ⁇ m) of the film. Further, the difference between the maximum value and the minimum value of the film thickness is defined as film thickness variation ( ⁇ m).
- the film thickness variation can be controlled within a desired range by adjusting the type and degree of substitution of the acyl group of the cellulose ester resin, the type and addition amount of the retardation increasing agent, the type and addition amount of the plasticizer, and the like. Among these, by setting the type and substitution degree of the acyl group of the cellulose ester resin within a predetermined range, the castability and stretchability during film formation can be controlled, and a uniform film thickness can be obtained.
- phase difference of the film there are methods for obtaining the phase difference of the film, such as (1) producing a phase difference with cellulose ester (vinegar cotton); (2) producing a phase difference by adding a phase difference increasing agent.
- Rt varies depending on humidity because cellulose has moisture permeability.
- the latter case is preferable because Rt hardly changes with humidity.
- by adding an elevating agent the retardation development property is improved, so that a thin film can be formed.
- the amount of the raising agent added is too large, the haze may be deteriorated, which is not preferable.
- plasticity is imparted to the film, stress is easily applied to the entire film during stretching, and film thickness variation becomes small (good). Further, since the inside of the film is hydrophobized and does not attract water, the Rt humidity fluctuation is reduced. However, when there is too much plasticizer, haze may deteriorate, which is not preferable.
- the thinner the film thickness the lower the Rt humidity fluctuation because the total water content decreases.
- the film thickness is too thin, it is difficult to form a uniform film, and the film thickness variation increases (deteriorates).
- substitution degree of the acyl group of the cellulose ester is smaller, the retardation development is improved, so that a thin film can be obtained. On the other hand, if the substitution degree of the acyl group is too small, the durability may be deteriorated.
- L * of the water immersion part / L * of the non-immersion part measured by EZ contrast is preferably 1.05 or more and 1.80 or less.
- L * of the water immersion part / L * of the non-immersion part is more preferably 1.05 or more and 1.55 or less, and further preferably 1.05 or more and 1.30 or less, from the viewpoint of further suppressing color unevenness. is there.
- evaluation of L * of a water immersion part / L * of a non-immersion part is performed in the following procedure.
- the retardation film according to the present embodiment has a cellulose ester as a main component, and (a) a retardation increasing agent, (b) a plasticizer, (c) a hydrogen bonding compound, and (d) other optional components as necessary. It further includes other additives such as components.
- the “main component” means a component occupying 50% by weight or more of the entire film, preferably 60% by weight or more, more preferably 70% by weight or more (upper limit: 100% by weight). ).
- Cellulose ester resin (cellulose ester) is formed by acylating some or all of the hydrogen atoms of hydroxyl groups (-OH) at the 2nd, 3rd and 6th positions in the ⁇ -1,4 bonded glucose units constituting cellulose. It means a cellulose acylate resin substituted with a group.
- the cellulose ester resin (cellulose ester) contained in the film of this embodiment is not particularly limited, but is preferably an ester of a linear or branched carboxylic acid having about 2 to 22 carbon atoms.
- the carboxylic acid constituting the ester may be an aliphatic carboxylic acid, may form a ring, or may be an aromatic carboxylic acid.
- the hydrogen atom of the hydroxyl group of cellulose is an acyl group having 2 to 22 carbon atoms such as acetyl group, propionyl group, butyryl group, isobutyryl group, valeryl group, pivaloyl group, hexanoyl group, octanoyl group, lauroyl group, stearoyl and the like.
- Examples include substituted cellulose esters.
- the carboxylic acid (acyl group) constituting the ester may have a substituent.
- the carboxylic acid constituting the ester is particularly preferably a lower fatty acid having 2 to 6 carbon atoms, more preferably a lower fatty acid having 2 to 4 carbon atoms, and a lower fatty acid having 2 or 3 carbon atoms. More preferably it is.
- the acyl group in the cellulose ester may be a single species or a combination of a plurality of acyl groups.
- cellulose esters include cellulose acetate (DAC, TAC), propionate groups in addition to acetyl groups such as cellulose acetate propionate (CAP), cellulose acetate butyrate, and cellulose acetate propionate butyrate.
- CAP cellulose acetate propionate
- CAP cellulose acetate propionate
- bonded the mixed fatty acid ester of the cellulose which the butyrate group couple
- bonded is mentioned.
- Preferred is cellulose acetate, cellulose acetate butyrate or cellulose acetate propionate.
- the butyryl group that can be contained in the cellulose ester may be linear or branched.
- these cellulose esters may use a single kind, and may use it in combination of multiple types.
- the total degree of acyl group substitution (total acyl group substitution degree) of the cellulose ester can be about 1.0 to 3.0.
- the total substitution degree of the acyl group is preferably in the range of 2.0 to 2.95, more preferably 2.1 to 2.5, from the viewpoint of reducing moisture permeability.
- the total substitution degree of the acyl group of the cellulose ester is 2.15 to 2.35. Is preferred.
- the acyl group substitution degree of the cellulose ester can be measured by a method prescribed in ASTM-D817-96.
- the cellulose ester satisfies both the following formulas (a) and (b).
- X represents a substitution degree of an acetyl group
- Y represents a substitution degree of a propionyl group or a butyryl group or a substitution degree thereof.
- the cellulose acetate particularly preferably used is preferably 2.1 ⁇ X ⁇ 2.9, and more preferably 2.2 ⁇ X ⁇ from the viewpoint that the retardation development, Rt humidity fluctuation, and film thickness variation are in the desired ranges.
- cellulose acetate propionate (CAP) or cellulose acetate butyrate particularly preferably used is 0.95 ⁇ X ⁇ 2.25, 0.1 ⁇ Y ⁇ 1.2, 2 .15 ⁇ X + Y ⁇ 2.65.
- cellulose acetate, cellulose acetate propionate, and cellulose acetate butyrate more preferably, cellulose acetate, cellulose acetate propionate
- a retardation film having excellent retardation, mechanical strength, and environmental fluctuation Is obtained.
- the degree of substitution of acyl groups indicates the average number of acyl groups per glucose unit, and how many hydrogen atoms of hydroxyl groups at the 2nd, 3rd and 6th positions of 1 glucose unit are substituted with acyl groups. Show. Therefore, the maximum degree of substitution is 3.0. In this case, it means that the hydrogen atoms of the hydroxyl groups at the 2nd, 3rd and 6th positions are all substituted with acyl groups. These acyl groups may be substituted on the 2nd, 3rd and 6th positions of the glucose unit on average, or may be substituted with a distribution. The degree of substitution is determined by the method prescribed in ASTM-D817-96.
- cellulose acetates having different degrees of substitution may be mixed and used.
- the mixing ratio of different cellulose acetates is not particularly limited.
- the number average molecular weight of the cellulose derivative is preferably in the range of 4 ⁇ 10 4 to 3 ⁇ 10 5 in order to increase the mechanical strength of the obtained film, and is 4.5 ⁇ 10 4 to 2 ⁇ 10 5 .
- the range is more preferable, and the range of 5 ⁇ 10 4 to 7 ⁇ 10 4 is particularly preferable.
- “weight average molecular weight (Mw)” and “number average molecular weight (Mn)” are values measured using gel permeation chromatography (GPC). The measurement conditions are as follows.
- the content of residual sulfuric acid in the cellulose derivative is preferably in the range of 0.1 to 45 ppm by weight in terms of elemental sulfur, and more preferably in the range of 1 to 30 ppm by weight. Sulfuric acid is considered to remain in the film in a salt state. When the content of residual sulfuric acid exceeds 45 ppm by weight, the film tends to break when the film is stretched hot or when slitting is performed after the hot stretch.
- the content of residual sulfuric acid can be measured by the method prescribed in ASTM D817-96.
- the free acid content in the cellulose derivative is preferably in the range of 1 to 500 ppm by weight, more preferably 1 to 100 ppm by weight, and further preferably in the range of 1 to 70 ppm by weight. preferable.
- the content of free acid can be measured by the method prescribed in ASTM D817-96.
- Cellulose derivatives may contain trace amounts of metal components. It is thought that a trace amount metal component originates in the water used in the synthesis process of the cellulose derivative. Like these metal components, the content of components that can become insoluble nuclei is preferably as small as possible.
- metal ions such as iron, calcium, and magnesium may form an insoluble matter by forming a salt with a resin decomposition product or the like that may contain an organic acidic group.
- the calcium (Ca) component easily forms a coordination compound (that is, a complex) with an acidic component such as a carboxylic acid or a sulfonic acid, and many ligands. Insoluble starch, turbidity) may be formed.
- the content of the iron (Fe) component in the cellulose derivative is preferably 1 ppm by weight or less.
- the content of the calcium (Ca) component in the cellulose derivative is preferably 60 ppm by weight or less, more preferably in the range of 0 to 30 ppm by weight.
- the content of the magnesium (Mg) component in the cellulose derivative is preferably in the range of 0 to 70 ppm by weight, particularly preferably in the range of 0 to 20 ppm by weight.
- the content of metal components such as iron (Fe) component, calcium (Ca) component, and magnesium (Mg) component is pretreated by microdigest wet decomposition device (sulfuric acid decomposition) and alkali melting After the measurement, it can be measured using ICP-AES (Inductively Coupled Plasma Atomic Emission Spectrometer).
- microdigest wet decomposition device sulfuric acid decomposition
- alkali melting After the measurement, it can be measured using ICP-AES (Inductively Coupled Plasma Atomic Emission Spectrometer).
- the contents of residual alkaline earth metal, residual sulfuric acid and residual acid can be adjusted by thoroughly washing the cellulose derivative obtained by synthesis.
- Cellulose esters such as cellulose acetate and cellulose acetate propionate can be produced by known methods.
- cellulose is esterified by mixing cellulose as a raw material, a predetermined organic acid (such as acetic acid or propionic acid), an acid anhydride (such as acetic anhydride or propionic anhydride), and a catalyst (such as sulfuric acid). The reaction proceeds until the triester is formed. In the triester, the three hydroxy groups (hydroxyl groups) of the glucose unit are substituted with an acyl acid of an organic acid.
- a mixed ester type cellulose ester such as cellulose acetate propionate or cellulose acetate butyrate can be produced.
- a cellulose ester resin having a desired degree of acyl substitution is synthesized by hydrolyzing the cellulose triester. Thereafter, a cellulose ester resin is completed through steps such as filtration, precipitation, washing with water, dehydration, and drying. Specifically, it can be synthesized with reference to the method described in JP-A-10-45804.
- the cellulose ester may be a commercially available product.
- Commercially available products include Daicel Corporation L20, L30, L40, and L50, Eastman Chemical Co. Ca398-3, Ca398-6, Ca398-10, Ca398-30, Ca394-60S, and the like.
- the retardation film of this invention contains the compound of the following general formula (1).
- the compound of the following general formula (1) acts as a retardation increasing agent (regulator). For this reason, by mix
- the compound of the following general formula (1) has low volatility even under high temperature and high humidity. For this reason, the bleeding resistance of the retardation film, and hence the sharpness of the image can be improved.
- R 1 to R 4 represent a hydrogen atom or an alkyl group having 1 to 3 carbon atoms.
- R 1 to R 4 may be the same or different from each other.
- the alkyl group having 1 to 3 carbon atoms include a methyl group, an ethyl group, a propyl group, and an isopropyl group.
- the retardation value (particularly the retardation value in the thickness direction of the film) is improved.
- a hydrogen atom, a methyl group, and an ethyl group are preferable, and a methyl group is particularly preferable.
- R 5 and R 6 represent an alkyl group or a glycidyl group having a substituent. At this time, R 5 and R 6 may be the same or different.
- the substituent is at least one selected from the group consisting of a hydroxyl group (—OH), an ester group, and an aromatic group.
- the ester group is represented by the formula: —O—C ( ⁇ O) —R or —C ( ⁇ O) —O—R, wherein R is a linear or branched group having 1 to 8 carbon atoms.
- the alkyl group and aromatic group are as defined below.
- R 5 and R 6 may be the same or different from each other.
- the alkyl group as R 5 and R 6 is not particularly limited, but is methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group. And a straight-chain or branched alkyl group having 1 to 8 carbon atoms, such as neopentyl group, hexyl group, heptyl group and octyl group. Among these, an alkyl group having 1 to 5 carbon atoms is preferable, and an alkyl group having 2 to 4 carbon atoms is preferable.
- the aromatic group may be an aryl group having 6 to 24 carbon atoms. More specifically, a phenyl group, p-tolyl group, naphthyl group, biphenyl group, fluorenyl group, anthryl group, pyrenyl group, azulenyl group, acenaphthylenyl group, terphenyl group, phenanthryl group and the like can be mentioned. Of these, a phenyl group and a naphthyl group are preferable, and a phenyl group is more preferable.
- the aromatic group may have a substituent.
- the substituent is not particularly limited, and examples thereof include, for example, an alkyl group having 1 to 3 carbon atoms, an alkoxy group having 1 to 3 carbon atoms, a phenyl group, a methylphenyl group, a phenylphenyl group, and a methylphenylphenyl group. , Cyano group, halogen atom (fluorine atom, chlorine atom, bromine atom, iodine atom), nitro group and the like.
- the said substituent may be one, or may be two or more, and in the latter case, each substituent may be the same or different.
- the aromatic group is a phenyl group, a methylphenyl group, or a methylphenylphenyl group. It is preferable that
- the method for producing the compound of the general formula (1) when R 5 and R 6 are alkyl groups having a substituent is not particularly limited.
- the compound can be obtained by reacting an epoxy compound with an aromatic monocarboxylic acid.
- an epoxy compound the diglycidyl ether type epoxy compound obtained by reaction with biphenols and epichlorohydrin is mentioned.
- this epoxy compound 3,3 ′, 5,5′-tetramethyl-4,4′-diglycidyloxybiphenyl (commercially available product is “jER YX-4000” manufactured by Japan Epoxy Resin Co., Ltd.) Biphenol type epoxy compounds such as epoxy equivalent of 180 to 192)) can be used.
- aromatic monocarboxylic acid examples include benzoic acid, dimethylbenzoic acid, trimethylbenzoic acid, tetramethylbenzoic acid, ethylbenzoic acid, propylbenzoic acid, cumic acid, o-toluic acid, m-toluic acid, p-toluic acid, anisic acid, ethoxybenzoic acid, propoxybenzoic acid, cyanobenzoic acid, fluorobenzoic acid, nitrobenzoic acid, 4-phenylbenzoic acid, 4- (3-methylphenyl) benzoic acid, 4- (4- Methylphenyl) benzoic acid, 4- (3,5-dimethylphenyl) benzoic acid, 2-methyl-4-phenylbenzoic acid, 2,6-dimethyl-4-phenylbenzoic acid, 2,6-dimethyl-4- ( 3,5-dimethylphenyl) benzoic acid, naphthoic acid, nicotinic acid, furoic acid,
- the epoxy group of the epoxy compound and the carboxyl group of the aromatic monocarboxylic acid react to synthesize the compound of the general formula (1).
- the reaction conditions are not particularly limited as long as the reaction proceeds.
- the reaction temperature is 80 to 130 ° C, more preferably 100 ° C to 115 ° C.
- the reaction time is preferably 10 to 25 hours.
- the mixing ratio (preparation ratio) of the epoxy compound and the aromatic monocarboxylic acid is not particularly limited as long as the reaction proceeds.
- the ratio of the number of moles of epoxy groups in the epoxy compound to the number of moles of aromatic monocarboxylic acid is 1 / 0.9 to 1.0. It is preferable that it is the range of these.
- a catalyst may be used as necessary.
- the catalyst include phosphine compounds such as trimethylphosphine, triethylphosphine, tributylphosphine, trioctylphosphine, and triphenylphosphine; 2-methylimidazole, 2-ethylimidazole, 2-isopropylimidazole, 2-ethyl-4-methyl Imidazole compounds such as imidazole and 4-phenyl-2-methylimidazole; triethylamine, tributylamine, trihexylamine, triamylamine, triethanolamine, dimethylaminoethanol, tritylenediamine, dimethylphenylamine, dimethylbenzylamine, 2 -(Dimethylaminomethyl) phenol, amine compounds such as 1,8-diazabicyclo (5,4,0) undecene-7; Such as emission compounds.
- These catalysts are preferably used in an amount of 0.05 to 1
- R 5 and R 6 are preferably an alkyl group having a hydroxyl group and an ester group as a substituent, or a glycidyl group.
- the compounds described in JP 2011-140637 A and JP 2011-116912 A are included in the compound of the general formula (1). More specifically, the following is mentioned as a more preferable example of the compound of General formula (1).
- a compound is prescribed
- the content of the compound of the general formula (1) is not particularly limited.
- the content of the compound of the general formula (1) is, for example, preferably 1 to 30 parts by weight, more preferably 2 to 20 parts by weight, particularly preferably 5 to 10 parts by weight with respect to 100 parts by weight of the cellulose ester resin. is there. With such an amount, the retardation function and tear strength of the retardation film can be improved. In addition, since the volatility under high temperature and high humidity is reduced, the bleeding resistance of the retardation film, and hence the sharpness of the image can be improved.
- the compound represented by the general formula (1) it may be added as a powder to the resin forming the retardation film, and after dissolving in the solvent, the resin forming the retardation film. It may be added.
- the retardation film of the present invention comprises a compound represented by the following general formula (2) having at least one 5-membered or 6-membered aromatic heterocyclic group.
- a 1 and A 2 each independently represents an alkyl group, a cycloalkyl group, an aromatic hydrocarbon ring group, or an aromatic heterocyclic group.
- a 1 and A 2 may be the same or different.
- n is 2 to 5
- each A 2 may be the same or different.
- the alkyl group is not particularly limited, but may be a linear or branched alkyl group having 1 to 24 carbon atoms. More specifically, methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, n-pentyl group, isopentyl group, tert-pentyl group , Neopentyl group, 1,2-dimethylpropyl group, n-hexyl group, isohexyl group, 1,3-dimethylbutyl group, 1-isopropylpropyl group, 1,2-dimethylbutyl group, n-heptyl group, 1,4 -Dimethylpentyl group, 3-ethylpentyl group, 2-methyl-1-isopropylpropyl group, 1-ethyl-3-methylbutyl group, n-octyl
- straight-chain or branched alkyl groups having 1 to 8 carbon atoms methyl group, ethyl group, n-propyl group, isopropyl group, tert-butyl group, n-octyl group, 2-ethylhexyl group, etc. preferable.
- the cycloalkyl group is not particularly limited, but may be a cycloalkyl group having 3 to 24 carbon atoms. More specifically, a cyclopropyl group, a cyclopentyl group, a cyclohexyl group, a 4-n-dodecylcyclohexyl group, and the like can be given.
- the aromatic hydrocarbon ring group is not particularly limited, but may be an aryl group having 6 to 24 carbon atoms. More specifically, a phenyl group, p-tolyl group, naphthyl group, biphenyl group, fluorenyl group, anthryl group, pyrenyl group, azulenyl group, acenaphthylenyl group, terphenyl group, phenanthryl group and the like can be mentioned. Of these, a phenyl group, a p-tolyl group, and a naphthyl group are preferable.
- the aromatic heterocyclic group is not particularly limited, but includes pyridyl group, pyrimidinyl group (for example, 2-pyrimidinyl group), pyrrole group (for example, 2-pyrrole group), pyrazolinone group, pyridinone group, furyl group (for example, 2 -Furyl group), pyrrolyl group, imidazolyl group, benzoimidazolyl group, pyrazolyl group, pyrazinyl group, triazolyl group (for example, 1,2,4-triazol-1-yl group, 1,2,3-triazol-1-yl group) Etc.), oxazolyl group, benzoxazolyl group, thiazolyl group, isoxazolyl group, isothiazolyl group, benzothiazolyl group (for example, 2-benzothiazolyl group), furazanyl group, thienyl group (for example, 2-thienyl group), quinolyl group, benzof
- 2-pyrrole group, 2-furyl group, 2-thienyl group, pyrrole group, imidazolyl group, oxazolyl group, thiazolyl group, benzoimidazolyl group, benzoxazolyl group, 2-benzothiazolyl group, pyrazolinone group, pyridyl group , A pyridinone group and a 2-pyrimidinyl group are preferable.
- the alkyl group, cycloalkyl group, aromatic hydrocarbon ring group and aromatic heterocyclic group represented by A 1 and A 2 may have a substituent.
- substituents include a halogen atom ( Fluorine atom, chlorine atom, bromine atom, iodine atom, etc.), alkyl group (methyl group, ethyl group, n-propyl group, isopropyl group, tert-butyl group, n-octyl group, 2-ethylhexyl group, etc.), cycloalkyl Groups (cyclohexyl group, cyclopentyl group, 4-n-dodecylcyclohexyl group, etc.), alkenyl groups (vinyl group, allyl group, etc.), cycloalkenyl groups (2-cyclopenten-1-yl, 2-cyclohexen-1-yl group, etc.) ), Alkynyl group (ethyny
- a 1 and A 2 are each an aromatic hydrocarbon ring group or an aromatic heterocyclic group, and can easily interact with the cellulose derivative. This is preferable because fluctuation is suppressed.
- B represents an aromatic hydrocarbon ring group or an aromatic heterocyclic group.
- the aromatic hydrocarbon ring represented by B may be a single ring or a condensed ring.
- Preferred examples of the aromatic hydrocarbon ring include benzene ring, naphthalene ring, anthracene ring, phenanthrene ring, perylene ring, tetracene ring, pyrene ring, benzopyrene ring, chrysene ring, triphenylene ring, acenaphthene ring, fluoranthene ring, fluorene ring, etc. More preferably, it is a benzene ring.
- aromatic heterocyclic ring represented by B include a furan ring, a benzofuran ring, a thiophene ring, a benzothiophene ring, a pyrrole ring, a pyrazole ring, an imidazole ring, an oxazole ring, 1, 2, 4-oxadiazole ring, 1,3,4-oxadiazole ring, isoxazole ring, thiazole ring, 1,2,4-thiodiazole ring, 1,3,4-thiodiazole ring, isothiazole ring, indole ring, A carbazole ring, an azacarbazole ring (an azacarbazole ring means one or more carbon atoms constituting the carbazole ring replaced by a nitrogen atom), a 1,2,3-triazole ring, 1,2,4- Triazole ring, pyrroloimidazole ring, pyrrolopyrazole ring, an imidazo
- the aromatic hydrocarbon ring group or aromatic heterocyclic group represented by B may have a substituent, and examples of the substituent include A 1 and A in the general formula (2).
- the same group as the substituent which 2 may have can be mentioned.
- T 1 and T 2 each independently represent a divalent 1,2,4-triazole ring group. Since a compound having an aromatic heterocyclic group has a polarized ⁇ electron, it interacts strongly with the hydrogen atom of the resin and coordinates more strongly with the resin than with the water molecule. It is considered that the fluctuation of the phase difference value is suppressed.
- a compound having a 1,2,4-triazole ring not only strongly coordinates with a resin but also has a strong interaction force with a water molecule, and the resin / 1,2,4-triazole compound / water molecule is stable. This is preferable because it is particularly excellent in suppressing the fluctuation of the retardation value.
- T 1 and T 2 may be the same or different.
- each T 2 may be the same or different.
- the 1,2,4-triazole ring group represented by T 1 and T 2 may be a tautomer. Specifically, any of the following structures represents a 1,2,4-triazole ring group.
- R 5 represents a hydrogen atom or a substituent.
- substituent represented by R 5 include the same groups as the substituent which A 1 in the general formula (2) may have.
- R 5 is preferably a hydrogen atom, an alkyl group or an acyl group.
- T 1 and T 2 may have a substituent, and examples of the substituent include the substituent that A 1 and A 2 in the general formula (2) may have Similar groups can be mentioned.
- L 1 , L 2 , L 3 and L 4 each independently represent a single bond or a divalent linking group.
- L 1 , L 2 , L 3 and L 4 may be the same or different.
- each L 3 and L 4 may be the same or different.
- Specific examples of the divalent linking group include an alkylene group, an alkenylene group, an alkynylene group, O, (C ⁇ O), (C ⁇ O) —O, NR, S, and (O ⁇ S ⁇ O). It represents a divalent linking group selected from the group consisting of these or a combination thereof.
- R represents a hydrogen atom or a substituent.
- Examples of the substituent represented by R include an alkyl group (methyl group, ethyl group, n-propyl group, isopropyl group, tert-butyl group, n-octyl group, 2-ethylhexyl group, etc.), cycloalkyl group ( Cyclohexyl group, cyclopentyl group, 4-n-dodecylcyclohexyl group, etc.), aromatic hydrocarbon ring group (phenyl group, p-tolyl group, naphthyl group, etc.), aromatic heterocyclic group (2-furyl group, 2-thienyl group, etc.) Group, 2-pyrimidinyl group, 2-benzothiazolyl group, 2-pyridyl group, etc.), cyano group and the like.
- the divalent linking group represented by L 1 , L 2 , L 3 and L 4 may have a substituent, and the substituent is not particularly limited. For example, A in the general formula (2)
- n represents an integer of 0 to 5.
- the plurality of A 2 , T 2 , L 3 and L 4 in the general formula (2) may be the same or different.
- a larger n is preferable because the compound represented by the general formula (2) is more likely to interact with the cellulose derivative, so that fluctuations in optical values with respect to changes in environmental humidity are suppressed. The compatibility with the cellulose ester is deteriorated. Therefore, n is preferably an integer of 1 to 5, more preferably an integer of 1 to 4.
- the compound represented by the general formula (2) is preferably a compound represented by the following general formula (5).
- a 1 , A 2 , T 1 , T 2 , L 1 , L 2 , L 3 and L 4 are respectively A 1 , A 2 , T 1 , T 2, L 1, is synonymous with L 2, L 3 and L 4.
- a 3 and T 3 represent the same groups as A 1 and T 1 in the general formula (2), respectively.
- L 5 and L 6 represent the same group as L 1 in the general formula (2).
- Q 1 , Q 2 , Q 3 and Q 4 represent a carbon atom or a nitrogen atom.
- m represents an integer of 0 to 4.
- m is more preferably an integer of 0 to 3 because a smaller m is more compatible with the cellulose ester.
- the compound represented by the general formula (2) may form a hydrate, a solvate or a salt.
- the hydrate may contain an organic solvent
- the solvate may contain water. That is, “hydrate” and “solvate” include mixed solvates containing both water and organic solvents.
- Salts include acid addition salts formed with inorganic or organic acids. Examples of inorganic acids include, but are not limited to, hydrohalic acids (hydrochloric acid, hydrobromic acid, etc.), sulfuric acid, phosphoric acid, and the like.
- organic acids examples include acetic acid, trifluoroacetic acid, propionic acid, butyric acid, oxalic acid, citric acid, benzoic acid, alkylsulfonic acid (methanesulfonic acid, etc.), allylsulfonic acid (benzenesulfonic acid, 4-toluene) Sulfonic acid, 1,5-naphthalenedisulfonic acid, and the like), but are not limited thereto.
- hydrochloride, acetate, propionate and butyrate are preferable.
- salts are those in which the acidic moiety present in the parent compound is a metal ion (eg, an alkali metal salt, such as sodium or potassium salt, an alkaline earth metal salt, such as calcium or magnesium salt, an ammonium salt, an alkali metal ion, alkaline earth And salts formed when substituted with organic bases (ethanolamine, diethanolamine, triethanolamine, morpholine, piperidine, etc.) It is not limited. Of these, sodium salts and potassium salts are preferred.
- a metal ion eg, an alkali metal salt, such as sodium or potassium salt, an alkaline earth metal salt, such as calcium or magnesium salt, an ammonium salt, an alkali metal ion, alkaline earth
- organic bases ethanolamine, diethanolamine, triethanolamine, morpholine, piperidine, etc.
- sodium salts and potassium salts are preferred.
- Examples of the solvent contained in the solvate include any common organic solvent. Specifically, alcohol (eg, methanol, ethanol, 2-propanol, 1-butanol, 1-methoxy-2-propanol, t-butanol), ester (eg, ethyl acetate), hydrocarbon (eg, toluene, hexane) , Heptane), ether (eg, tetrahydrofuran), nitrile (eg, acetonitrile), ketone (acetone) and the like.
- alcohol eg, methanol, ethanol, 2-propanol, 1-butanol, 1-methoxy-2-propanol, t-butanol
- ester eg, ethyl acetate
- hydrocarbon eg, toluene, hexane
- Heptane Heptane
- ether eg, tetrahydrofuran
- nitrile
- solvates of alcohols eg, methanol, ethanol, 2-propanol, 1-butanol, 1-methoxy-2-propanol, t-butanol.
- solvents may be a reaction solvent used at the time of synthesizing the compound, a solvent used at the time of crystallization purification after synthesis, or a mixture thereof.
- two or more kinds of solvents may be included at the same time, or a form containing water and a solvent (for example, water and alcohol (for example, methanol, ethanol, t-butanol, etc.), etc.).
- a solvent for example, water and alcohol (for example, methanol, ethanol, t-butanol, etc.), etc.).
- the molecular weight of the compound represented by the general formula (2) is not particularly limited, but the smaller the value, the better the compatibility with the resin, and the larger the value, the higher the effect of suppressing the fluctuation of the optical value with respect to changes in environmental humidity. Is preferable, 200 to 1500 is more preferable, and 300 to 1000 is more preferable.
- Any raw material may be used for the compound represented by the general formula (2), but a method of reacting a nitrile derivative or imino ether derivative with a hydrazide derivative is preferable.
- the solvent used for the reaction may be any solvent as long as it does not react with the raw material, but may be any ester type (eg, ethyl acetate, methyl acetate), amide type (dimethylformamide, dimethylacetamide, etc.), ether type (Ethylene glycol dimethyl ether, etc.), alcohols (eg, methanol, ethanol, propanol, isopropanol, n-butanol, 2-butanol, ethylene glycol, ethylene glycol monomethyl ether, etc.), aromatic hydrocarbons (eg, toluene, xylene, etc.) ), Water can be mentioned.
- a solvent to be used an alcohol solvent is preferable. These solvents may be used as a mixture.
- the amount of the solvent used is not particularly limited, but is preferably in the range of 0.5 to 30 times, more preferably 1.0 to 25 times the weight of the hydrazide derivative used. Yes, particularly preferably in the range of 3.0 to 20 times the amount.
- a catalyst When reacting a nitrile derivative and a hydrazide derivative, it is not necessary to use a catalyst, but it is preferable to use a catalyst in order to accelerate the reaction.
- a catalyst to be used an acid may be used and a base may be used.
- the acid include hydrochloric acid, sulfuric acid, nitric acid, acetic acid and the like, preferably hydrochloric acid.
- the acid may be added after diluted in water, or may be added by a method of blowing a gas into the system.
- Bases include inorganic bases (potassium carbonate, sodium carbonate, potassium bicarbonate, sodium bicarbonate, potassium hydroxide, sodium hydroxide, etc.) and organic bases (sodium methylate, sodium ethylate, potassium methylate, potassium ethylate, Sodium butyrate, potassium butyrate, diisopropylethylamine, N, N′-dimethylaminopyridine, 1,4-diazabicyclo [2.2.2] octane, N-methylmorpholine, imidazole, N-methylimidazole, pyridine, etc.) Any of them may be used, and the inorganic base is preferably potassium carbonate, and the organic base is preferably sodium ethylate, sodium ethylate or sodium butyrate.
- the inorganic base may be added as a powder or may be added in a state dispersed in a solvent.
- the organic base may be added in a state dissolved in a solvent (for example, a
- the amount of the catalyst used is not particularly limited as long as the reaction proceeds, but it is preferably in the range of 1.0 to 5.0 moles relative to the formed triazole ring, and more preferably 1.05 to 3. A range of 0-fold mole is preferable.
- the target product can be obtained by heating in a solvent.
- the addition method of the raw material, solvent and catalyst used for the reaction is not particularly limited, and the catalyst may be added last, or the solvent may be added last. Also preferred is a method of dispersing or dissolving a nitrile derivative in a solvent, adding a catalyst, and then adding a hydrazide derivative.
- the solution temperature during the reaction may be any temperature as long as the reaction proceeds, but is preferably in the range of 0 to 150 ° C., more preferably in the range of 20 to 140 ° C. Moreover, you may react, removing the water to produce
- any method may be used for treating the reaction solution, but when a base is used as a catalyst, a method of neutralizing the reaction solution by adding an acid is preferable.
- the acid used for neutralization include hydrochloric acid, sulfuric acid, nitric acid, and acetic acid, with acetic acid being particularly preferred.
- the amount of the acid used for neutralization is not particularly limited as long as the pH of the reaction solution is in the range of 4 to 9, but is preferably 0.1 to 3 moles, particularly preferably, relative to the base used. , In the range of 0.2 to 1.5 moles.
- the appropriate organic solvent is a water-insoluble solvent such as ethyl acetate, toluene, dichloromethane, ether, or a mixed solvent of the water-insoluble solvent and tetrahydrofuran or an alcohol solvent, preferably Ethyl acetate.
- compound (2-1) can be synthesized by the following scheme.
- the precipitate collected by filtration was dissolved in 80 ml of methanol, 300 ml of pure water was added, and acetic acid was added dropwise until the pH of the solution reached 7.
- the precipitated crystals were collected by filtration, washed with pure water, and blown dry at 50 ° C. to obtain 38.6 g of compound (2-1).
- the yield was 70% based on benzoylhydrazine.
- the content of the compound of the general formula (2) is not particularly limited, and can be appropriately adjusted to be contained in the retardation film.
- the content of the compound of the general formula (2) is, for example, preferably 1 to 30 parts by weight, more preferably 3 to 20 parts by weight, particularly preferably 5 to 10 parts by weight with respect to 100 parts by weight of the cellulose ester resin. is there. With such an amount, the retardation function and tear strength of the retardation film can be improved. In addition, since the volatility under high temperature and high humidity is reduced, the bleeding resistance of the retardation film, and hence the sharpness of the image can be improved.
- the compound represented by the general formula (2) it may be added as a powder to a resin for forming a retardation film, and after dissolving in a solvent, a resin for forming a retardation film. It may be added.
- the retardation film of the present invention may contain a plasticizer in order to improve the fluidity of the composition during film production and the flexibility and workability of the film.
- plasticizers include sugar ester plasticizers, polyester plasticizers, polyhydric alcohol ester plasticizers, acrylic compounds, polycarboxylic acid ester plasticizers (including phthalate ester plasticizers), glycosates.
- rate plasticizers ester plasticizers (including citrate ester plasticizers, fatty acid ester plasticizers, phosphate ester plasticizers, trimellitic ester plasticizers, etc.), styrene compounds, and the like.
- plasticizers it is effective for moisture permeability to include at least one plasticizer selected from the group consisting of the following sugar ester plasticizers (sugar ester compounds), polyester plasticizers, and acrylic compounds. This is preferable from the viewpoint of achieving both high control and compatibility with the cellulose ester. These may be used alone or in combination of two or more.
- the molecular weight of the plasticizer is preferably 15000 or less, and more preferably 10,000 or less from the viewpoint of achieving both improvement in wet heat resistance and compatibility with the cellulose ester.
- the weight average molecular weight (Mw) is preferably 10,000 or less.
- a preferable molecular weight (Mw) is in the range of 100 to 10,000, and more preferably in the range of 400 to 8,000.
- a sugar ester plasticizer is a compound having 1 to 12 furanose structures or pyranose structures, in which all or part of the hydroxy groups in the compound are esterified.
- the sugar ester plasticizer can be added for the purpose of preventing hydrolysis.
- sugar as a raw material for synthesizing the sugar ester compound according to the present invention examples include the following, but the present invention is not limited to these.
- gentiobiose, gentiotriose, gentiotetraose, xylotriose, galactosyl sucrose, and the like are also included.
- the monocarboxylic acid used for esterifying all or part of the OH group in the pyranose structure or furanose structure is not particularly limited, and is a known aliphatic monocarboxylic acid, alicyclic monocarboxylic acid, aromatic A monocarboxylic acid or the like can be used.
- the carboxylic acid used may be one type or a mixture of two or more types.
- Examples of preferred aliphatic monocarboxylic acids include acetic acid, propionic acid, butyric acid, isobutyric acid, valeric acid, caproic acid, enanthic acid, caprylic acid, pelargonic acid, capric acid, 2-ethyl-hexanecarboxylic acid, undecylic acid, Saturated lauric acid, tridecylic acid, myristic acid, pentadecylic acid, palmitic acid, heptadecylic acid, stearic acid, nonadecanoic acid, arachidic acid, behenic acid, lignoceric acid, serotic acid, heptacosanoic acid, montanic acid, mellicic acid, and laxaric acid
- unsaturated fatty acids such as fatty acids, undecylenic acid, oleic acid, sorbic acid, linoleic acid, linolenic acid, arachidonic acid and oc
- Examples of preferable alicyclic monocarboxylic acids include cyclopentane carboxylic acid, cyclohexane carboxylic acid, cyclooctane carboxylic acid, and derivatives thereof.
- aromatic monocarboxylic acids examples include aromatic monocarboxylic acids having 1 to 5 alkyl groups or alkoxy groups introduced into the benzene ring of benzoic acid such as benzoic acid, phenylacetic acid, toluic acid, cinnamic acid, Examples thereof include aromatic monocarboxylic acids having two or more benzene rings such as benzylic acid, biphenylcarboxylic acid, naphthalenecarboxylic acid, tetralincarboxylic acid, or derivatives thereof, and benzoic acid is particularly preferable.
- sugar esters represented by the following general formula (FA).
- R 1 to R 8 in formula (FA) each independently represent a hydrogen atom, a substituted or unsubstituted alkylcarbonyl group, or a substituted or unsubstituted arylcarbonyl group.
- R 1 to R 8 may be the same as or different from each other.
- the substituted or unsubstituted alkylcarbonyl group is preferably a substituted or unsubstituted alkylcarbonyl group having 2 or more carbon atoms.
- Examples of the substituted or unsubstituted alkylcarbonyl group include a methylcarbonyl group (acetyl group).
- the substituted or unsubstituted arylcarbonyl group is preferably a substituted or unsubstituted arylcarbonyl group having 7 or more carbon atoms.
- the arylcarbonyl group include a phenylcarbonyl group.
- the substituent that the aromatic hydrocarbon ring group has include an alkyl group such as a methyl group, an alkoxyl group such as a methoxy group, and the like.
- the compound represented by the general formula (FA) has an average substitution degree of preferably 3.0 to 7.5, more preferably 3.0 to 6.0, thereby controlling moisture permeability and cellulose ester.
- the compatibility can be highly compatible.
- the degree of substitution of the compound represented by the general formula (FA) represents the number substituted with a substituent other than hydrogen among the eight hydroxyl groups contained in the general formula (FA).
- this represents a number containing a group other than hydrogen. Accordingly, when all of R 1 to R 8 are substituted with a substituent other than hydrogen, the degree of substitution is 8.0, which is the maximum value, and when R 1 to R 8 are all hydrogen atoms, 0.0 It becomes.
- the compound having the structure represented by the general formula (FA) is difficult to synthesize a single kind of compound in which the number of hydroxyl groups and the number of OR groups are fixed. Since it is known that it becomes a compound in which several different components are mixed, it is appropriate to use the average substitution degree as the substitution degree of the general formula (FA) in the present invention.
- the average substitution degree can be measured from the area ratio of the chart showing the substitution degree distribution.
- R 1 to R 8 represent a substituted or unsubstituted alkylcarbonyl group or a substituted or unsubstituted arylcarbonyl group, and R 1 to R 8 may be the same or different.
- R 1 to R 8 are also referred to as acyl groups).
- Specific examples of R 1 to R 8 include acyl groups derived from monocarboxylic acids used during the synthesis of the sugar ester compounds exemplified above.
- sugar ester compound according to the present invention any of R 1 to R 8 may be the same substituent R, and the present invention is not limited thereto.
- polyester compounds are defined by the following symbols.
- sugar ester compounds in which R 1 to R 8 are different groups can be used.
- the sugar ester compound according to the present invention can be produced by reacting the sugar with an acylating agent (also referred to as an esterifying agent, for example, an acid halide such as acetyl chloride, an anhydride such as acetic anhydride).
- an acylating agent also referred to as an esterifying agent, for example, an acid halide such as acetyl chloride, an anhydride such as acetic anhydride.
- the distribution of the degree of substitution is made by adjusting the amount of acylating agent, the timing of addition, and the esterification reaction time, but it is a mixture of sugar ester compounds with different degrees of substitution or purely isolated compounds with different degrees of substitution. Can be used to adjust components having a target average substitution degree and a substitution degree of 4 or less.
- the inside of the Kolben was depressurized to 4 ⁇ 10 2 Pa or less, and after excess pyridine was distilled off at 60 ° C., the inside of the Kolben was depressurized to 1.3 ⁇ 10 Pa or less and the temperature was raised to 120 ° C. Most of the acid and benzoic acid formed were distilled off. Then, 1 L of toluene and 300 g of a 0.5 wt% sodium carbonate aqueous solution were added, and the mixture was stirred for 30 minutes at 50 ° C. and then allowed to stand to separate a toluene layer.
- A-1 was 1.2% by weight
- A-2 was 13.2% by weight
- A-3 was 14.2% by weight
- A-4 was 35.4% by weight
- A-5 and the like were 40.0% by weight.
- the average degree of substitution was 5.2.
- A-5 etc. means a mixture of all components having a substitution degree of 4 or less, that is, compounds having substitution degrees of 4, 3, 2, 1. The average degree of substitution was calculated with A-5 as the degree of substitution of 4.
- the average degree of substitution was adjusted by adding in combination the sugar ester close to the desired degree of average substitution and the isolated A-1 to A-5 etc. by the method prepared here.
- sugar esters examples include compounds described in JP-A Nos. 62-42996 and 10-237084.
- the polyester plasticizer is not particularly limited.
- a polymer in which the terminal hydroxy group of the polyester polyol is sealed with a monocarboxylic acid (end-capped polyester) can be used.
- the ester-forming derivative referred to here is an esterified product of dicarboxylic acid, dicarboxylic acid chloride, or dicarboxylic acid anhydride.
- a polyester plasticizer represented by the following general formula (FB-1) from the viewpoint of high compatibility between moisture permeability control and compatibility with the cellulose ester.
- B represents a linear or branched alkylene group having 2 to 6 carbon atoms or a cycloalkylene group
- A represents an aromatic ring group having 6 to 14 carbon atoms
- n represents 1 or more. Indicates a natural number.
- the compound represented by the above formula is obtained from a dicarboxylic acid having an aromatic ring (also referred to as an aromatic dicarboxylic acid) and a linear or branched alkylene or cycloalkylene diol having 2 to 6 carbon atoms, at both ends. Is not sealed with a monocarboxylic acid.
- aromatic dicarboxylic acids having 6 to 16 carbon atoms include phthalic acid, isophthalic acid, terephthalic acid, 1,5-naphthalenedicarboxylic acid, 1,4-naphthalenedicarboxylic acid, 1,8-naphthalenedicarboxylic acid, 2,3 -Naphthalenedicarboxylic acid, 2,6-naphthalenedicarboxylic acid, 2,8-naphthalenedicarboxylic acid, 2,2'-biphenyldicarboxylic acid, 4,4'-biphenyldicarboxylic acid, and the like.
- 2,6-naphthalenedicarboxylic acid and 4,4'-biphenyldicarboxylic acid are preferable.
- linear or branched alkylene or cycloalkylene diol having 2 to 6 carbon atoms examples include ethylene glycol, 1,2-propanediol, 1,3-propanediol, 1,2-butanediol, and 1,3-butane.
- diol and 1,4-cyclohexanedimethanol examples include diol and 1,4-cyclohexanedimethanol. Of these, ethanediol, 1,2-propanediol, 1,3-propanediol, and 1,3-butanediol are preferable.
- A is a naphthalene ring or a biphenyl ring which may have a substituent.
- the substituent is an alkyl group, alkenyl group, or alkoxyl group having 1 to 6 carbon atoms.
- the hydroxyl value (OH value) of the polyester compound is preferably 100 mgKOH / g or more and 500 mgKOH / g or less, more preferably 170 mgKOH / g to 400 mgKOH / g. When the hydroxyl value is in this range, the compatibility with the cellulose ester and the cellulose ether becomes suitable.
- the hydroxyl value is 400 mgKOH / g or less, the hydrophobicity of the polyester compound does not increase too much, and when the hydroxyl value is 170 mgKOH / g or more, the intermolecular interaction (hydrogen bond, etc.) between the polyester compounds is excessively strong. It is thought that this is because precipitation in the film can be prevented.
- the number average molecular weight (Mn) of the polyester compound can be calculated from the following formula.
- the polyester compound can be obtained by a conventional method such as a hot melt condensation method using a polyesterification reaction or a transesterification reaction between the dicarboxylic acid and a diol, or an interfacial condensation method between an acid chloride of these acids and a glycol. Easy to synthesize.
- a polyester plasticizer represented by the following general formula (FB-2) from the viewpoint of high compatibility between moisture permeability control and compatibility with cellulose ester.
- B represents a hydroxy group or a carboxylic acid residue
- G represents an alkylene glycol residue having 2 to 18 carbon atoms, an aryl glycol residue having 6 to 12 carbon atoms, or 4 carbon atoms.
- A represents an alkylene dicarboxylic acid residue having 4 to 12 carbon atoms or an aryl dicarboxylic acid residue having 6 to 12 carbon atoms
- n represents an integer of 1 or more.
- a hydroxy group or carboxylic acid residue represented by B an alkylene glycol residue, an oxyalkylene glycol residue or an aryl glycol residue represented by G, and an alkylene dicarboxylic acid residue represented by A It is composed of a group or an aryl dicarboxylic acid residue, and can be obtained by a reaction similar to that of a normal ester compound.
- Examples of the carboxylic acid component of the polyester compound represented by the general formula (FB-2) include acetic acid, propionic acid, butyric acid, benzoic acid, p-tert-butylbenzoic acid, orthotoluic acid, metatoluic acid, p-toluic acid, and dimethyl.
- acetic acid propionic acid
- butyric acid butyric acid
- benzoic acid p-tert-butylbenzoic acid
- orthotoluic acid metatoluic acid
- p-toluic acid and dimethyl.
- benzoic acid ethyl benzoic acid
- normal propyl benzoic acid aminobenzoic acid
- acetoxybenzoic acid aliphatic acid and the like
- Examples of the alkylene glycol component having 2 to 18 carbon atoms of the polyester compound represented by the general formula (FB-2) include ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol, and 1,2-butanediol.
- an alkylene glycol having 2 to 12 carbon atoms is particularly preferable because of excellent compatibility with the cellulose ester resin. More preferred are alkylene glycols having 2 to 6 carbon atoms, and still more preferred are alkylene glycols having 2 to 4 carbon atoms.
- Examples of the aryl glycol having 6 to 12 carbon atoms of the polyester plasticizer represented by the general formula (FB-2) include 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, cyclohexanediethanol, 1,4 -There are cyclic glycols such as benzenedimethanol, and these glycols can be used as one kind or a mixture of two or more kinds.
- Examples of the oxyalkylene glycol component having 4 to 12 carbon atoms of the polyester compound represented by the general formula (FB-2) include diethylene glycol, triethylene glycol, tetraethylene glycol, dipropylene glycol, and tripropylene glycol. These glycols can be used as one kind or a mixture of two or more kinds.
- Examples of the alkylene dicarboxylic acid component having 4 to 12 carbon atoms of the polyester compound represented by the general formula (FB-2) include succinic acid, maleic acid, fumaric acid, glutaric acid, adipic acid, azelaic acid, and sebacic acid. , Dodecanedicarboxylic acid and the like, and these are used as one kind or a mixture of two or more kinds, respectively.
- Examples of the aryl dicarboxylic acid component having 6 to 12 carbon atoms of the polyester compound represented by the general formula (FB-2) include phthalic acid, terephthalic acid, isophthalic acid, 1,5-naphthalenedicarboxylic acid, and 1,4-naphthalene. There are dicarboxylic acids and the like.
- the polyester compound represented by the general formula (FB-2) has a weight average molecular weight of preferably 300 to 1500, more preferably 400 to 1,000.
- the acid value is 0.5 mgKOH / g or less
- the hydroxy group (hydroxyl group) value is 25 mgKOH / g or less
- more preferably the acid value is 0.3 mgKOH / g or less
- the hydroxy group (hydroxyl group) value is 15 mgKOH / g or less. Is.
- the weight average molecular weight of the polyester plasticizer is calculated by measurement using gel permeation chromatography (GPC) under the following measurement conditions.
- polyester compounds are defined by the following symbols.
- the viscosity of the polyester plasticizer depends on the molecular structure and molecular weight, but in the case of an adipic acid plasticizer, it has a high compatibility with the cellulose ester and has a high effect of imparting plasticity. It is preferably in the range of s (25 ° C.).
- One type of polyester plasticizer may be used, or two or more types may be used in combination.
- the polyhydric alcohol ester plasticizer is an ester compound (alcohol ester) of a dihydric or higher aliphatic polyhydric alcohol and a monocarboxylic acid, preferably a divalent to 20-valent aliphatic polyhydric alcohol ester.
- the polyhydric alcohol ester compound preferably has an aromatic ring or a cycloalkyl ring in the molecule.
- the acrylic compound is not particularly limited, but at least one selected from the group consisting of (meth) acrylic acid, (meth) acrylic acid ester, (meth) acrylamides, and (meth) acrylonitrile. Examples thereof include a polymer having a repeating unit derived from a certain acrylic monomer. These acrylic compounds can improve the water resistance of the film.
- the acrylic compound is preferably one in which the methyl methacrylate unit is 50 to 99% by weight and the total amount of other monomer units copolymerizable therewith is 1 to 50% by weight.
- Examples of other copolymerizable monomers include alkyl methacrylates having an alkyl group having 2 to 18 carbon atoms; alkyl acrylates having an alkyl group having 1 to 18 carbon atoms; amides such as acryloylmorpholine and N, N-dimethylacrylamide
- the acrylic compound used in the present invention may have a ring structure, specifically, a lactone ring structure, a glutaric anhydride structure, a glutarimide structure, an N-substituted maleimide structure and a maleic anhydride structure. And a pyran ring structure.
- alkyl acrylates having 1 to 18 carbon atoms in the alkyl group examples include alkyl acrylates having 1 to 18 carbon atoms in the alkyl group, amides such as acryloylmorpholine and dimethylacrylamide, from the viewpoint of thermal decomposition resistance and fluidity of the copolymer.
- amides such as acryloylmorpholine and dimethylacrylamide
- Preferred are a vinyl monomer having a group, a methacrylic acid ester or an acrylate ester having an alicyclic hydrocarbon group having 5 to 22 carbon atoms in the ester portion, an N-substituted maleimide structure, a pyran ring structure and the like.
- alkyl acrylate having 1 to 18 carbon atoms in the alkyl group include methyl acrylate, ethyl acrylate, n-propyl acrylate, n-butyl acrylate, s-butyl acrylate, and 2-ethylhexyl acrylate. And methyl acrylate.
- vinyl monomer having an amide group examples include acrylamide, N-methylacrylamide, N-butylacrylamide, N, N-dimethylacrylamide, N, N-diethylacrylamide, acryloylmorpholine, N-hydroxyethylacrylamide, acryloylpyrrolidine, Acryloylpiperidine, methacrylamide, N-methylmethacrylamide, N-butylmethacrylamide, N, N-dimethylmethacrylamide, N, N-diethylmethacrylamide, methacryloylmorpholine, N-hydroxyethylmethacrylamide, methacryloylpyrrolidine, methacryloylpiperidine, N-vinylformamide, N-vinylacetamide, vinylpyrrolidone and the like can be mentioned.
- methacrylic acid ester or acrylate ester having an alicyclic hydrocarbon group having 5 to 22 carbon atoms in the ester moiety include, for example, cyclopentyl acrylate, cyclohexyl acrylate, methyl cyclohexyl acrylate, trimethylcyclohexyl acrylate, Norbornyl acrylate, norbornyl acrylate, cyano norbornyl acrylate, isobornyl acrylate, bornyl acrylate, menthyl acrylate, fentyl acrylate, adamantyl acrylate, dimethyladamantyl acrylate, tricycloacrylate [5.2 .1.0 2,6 ] dec-8-yl, tricyclo [5.2.1.0 2,6 ] dec-4-methyl acrylate, cyclodecyl acrylate, cyclopentyl methacrylate, cyclohexane methacrylate Xylyl, methyl cyclohe
- isobornyl methacrylate dicyclopentanyl methacrylate, dimethyladamantyl methacrylate and the like can be mentioned.
- N-substituted maleimide examples include N-methylmaleimide, N-ethylmaleimide, N-propylmaleimide, Ni-propylmaleimide, N-butylmaleimide, Ni-butylmaleimide, Nt-butylmaleimide, N-laurylmaleimide, N-cyclohexylmaleimide, N-benzylmaleimide, N-phenylmaleimide, N- (2-chlorophenyl) maleimide, N- (4-chlorophenyl) maleimide, N- (4-bromophenyl) phenylmaleimide, N -(2-methylphenyl) maleimide, N- (2-ethylphenylmaleimide), N- (2-methoxyphenyl) maleimide, N- (2,4,6-trimethylphenyl) maleimide, N- (4-benzylphenyl) Maleimide, N- (2,4,6-tribromoph Yl
- the acrylic compound preferably has a weight average molecular weight (Mw) in the range of 15000 or less, more preferably in the range of 10,000 or less, from the viewpoint of achieving both moisture permeability control and compatibility with the cellulose ester. More preferably, it is in the range of 5000 to 10,000.
- Mw weight average molecular weight
- the weight average molecular weight (Mw) of the acrylic compound according to the present invention is calculated by measurement using gel permeation chromatography (GPC) under the following measurement conditions.
- aliphatic polyhydric alcohol examples include ethylene glycol, propylene glycol, trimethylolpropane, pentaerythritol and the like.
- the monocarboxylic acid can be an aliphatic monocarboxylic acid, an alicyclic monocarboxylic acid, an aromatic monocarboxylic acid, or the like.
- One kind of monocarboxylic acid may be used, or a mixture of two or more kinds may be used.
- all of the OH groups contained in the aliphatic polyhydric alcohol may be esterified, or a part of the OH groups may be left as they are.
- the aliphatic monocarboxylic acid is preferably a fatty acid having a straight chain or a side chain having 1 to 32 carbon atoms.
- the number of carbon atoms of the aliphatic monocarboxylic acid is more preferably 1-20, and still more preferably 1-10.
- Examples of such aliphatic monocarboxylic acids include acetic acid, propionic acid, butyric acid, valeric acid, and the like, and acetic acid may be preferable in order to enhance compatibility with the cellulose ester.
- Examples of the alicyclic monocarboxylic acid include cyclopentane carboxylic acid, cyclohexane carboxylic acid, cyclooctane carboxylic acid and the like.
- aromatic monocarboxylic acids examples include benzoic acid; one having 1 to 3 alkyl groups or alkoxy groups (for example, methoxy group or ethoxy group) introduced into the benzene ring of benzoic acid (for example, toluic acid); benzene ring Aromatic monocarboxylic acids having two or more (for example, biphenyl carboxylic acid, naphthalene carboxylic acid, tetralin carboxylic acid, etc.) are included, and benzoic acid is preferred.
- the molecular weight of the polyhydric alcohol ester plasticizer is not particularly limited, but is preferably in the range of 300 to 1500, and more preferably in the range of 350 to 750. In order to make it hard to volatilize, the one where molecular weight is larger is preferable. In order to improve moisture permeability and compatibility with the cellulose ester, a smaller molecular weight is preferable.
- polyhydric alcohol ester plasticizer examples include trimethylolpropane triacetate, trimethylolpropane benzoate, pentaerythritol tetraacetate, and an ester represented by the general formula (I) described in JP-A-2008-88292. Compound (A) and the like are included.
- the polyvalent carboxylic acid ester plasticizer is an ester compound of a divalent or higher, preferably 2 to 20 valent polycarboxylic acid and an alcohol compound.
- the polyvalent carboxylic acid is preferably a 2-20 valent aliphatic polyvalent carboxylic acid, a 3-20 valent aromatic polyvalent carboxylic acid, or a 3-20 valent alicyclic polyvalent carboxylic acid.
- polyvalent carboxylic acids include trivalent or higher aromatic polyvalent carboxylic acids such as trimellitic acid, trimesic acid, pyromellitic acid or derivatives thereof; succinic acid, adipic acid, azelaic acid, sebacic acid, oxalic acid Contains aliphatic polycarboxylic acids such as fumaric acid, maleic acid and tetrahydrophthalic acid; oxypolycarboxylic acids such as tartaric acid, tartronic acid, malic acid and citric acid, etc., and suppresses volatilization from the film. For this, oxypolycarboxylic acids are preferred.
- the alcohol compound examples include an aliphatic saturated alcohol compound having a straight chain or a side chain, an aliphatic unsaturated alcohol compound having a straight chain or a side chain, an alicyclic alcohol compound, or an aromatic alcohol compound.
- the carbon number of the aliphatic saturated alcohol compound or the aliphatic unsaturated alcohol compound is preferably 1 to 32, more preferably 1 to 20, and still more preferably 1 to 10.
- Examples of the alicyclic alcohol compound include cyclopentanol, cyclohexanol and the like.
- the aromatic alcohol compound include phenol, paracresol, dimethylphenol, benzyl alcohol, cinnamyl alcohol and the like.
- the alcohol compound may be one kind or a mixture of two or more kinds.
- the molecular weight of the polyvalent carboxylic acid ester plasticizer is not particularly limited, but is preferably in the range of 300 to 1000, and more preferably in the range of 350 to 750. A larger molecular weight of the polyvalent carboxylic acid ester plasticizer is preferable from the viewpoint of suppressing bleeding out. From the viewpoint of moisture permeability and compatibility with cellulose ester, a smaller one is preferable.
- the acid value of the polyvalent carboxylic acid ester plasticizer is preferably 1 mgKOH / g or less, more preferably 0.2 mgKOH / g or less.
- the acid value refers to the number of milligrams of potassium hydroxide necessary for neutralizing the acid (carboxy group present in the sample) contained in 1 g of the sample. The acid value is measured according to JIS K0070 (1992).
- Examples of the polyvalent carboxylic acid ester plasticizer include an ester compound (B) represented by the general formula (II) described in JP-A-2008-88292.
- the polycarboxylic acid ester plasticizer may be a phthalate ester plasticizer.
- the phthalate ester plasticizer include diethyl phthalate, dimethoxyethyl phthalate, dimethyl phthalate, dioctyl phthalate, dibutyl phthalate, di-2-ethylhexyl phthalate, dicyclohexyl phthalate, dicyclohexyl terephthalate and the like.
- glycolate plasticizers include alkylphthalyl alkyl glycolates.
- alkyl phthalyl alkyl glycolates include methyl phthalyl methyl glycolate, ethyl phthalyl ethyl glycolate, propyl phthalyl propyl glycolate, butyl phthalyl butyl glycolate, octyl phthalyl octyl glycolate and the like. .
- the ester plasticizer includes a fatty acid ester plasticizer, a citrate ester plasticizer, a phosphate ester plasticizer, a trimellitic acid plasticizer, and the like.
- Examples of the fatty acid ester plasticizer include butyl oleate, methylacetyl ricinoleate, dibutyl sebacate and the like.
- Examples of the citrate plasticizer include acetyl trimethyl citrate, acetyl triethyl citrate, acetyl tributyl citrate and the like.
- Examples of the phosphate ester plasticizer include triphenyl phosphate, tricresyl phosphate, cresyl diphenyl phosphate, octyl diphenyl phosphate, diphenyl biphenyl phosphate (BDP), trioctyl phosphate, tributyl phosphate and the like.
- trimellitic acid plasticizers include octyl trimellitic acid, n-octyl trimellitic acid, isodecyl trimellitic acid, and isononyl trimellitic acid.
- the styrene compound may be a homopolymer of a styrene monomer or a copolymer of a styrene monomer and another copolymer monomer.
- the content of the structural unit derived from the styrenic monomer in the styrenic compound may be preferably 30 to 100 mol%, more preferably 50 to 100 mol%, in order for the molecular structure to have a certain bulkiness.
- the styrene monomer is preferably a compound represented by the following formula (A).
- R 101 to R 103 in the formula (A) each independently represent a hydrogen atom, an alkyl group having 1 to 30 carbon atoms, or an aryl group.
- R 104 is a hydrogen atom, an alkyl group having 1 to 30 carbon atoms, a cycloalkyl group, an aryl group, an alkoxy group having 1 to 30 carbon atoms, an aryloxy group, an alkyloxycarbonyl group having 2 to 30 carbon atoms, an aryloxycarbonyl Group, an alkylcarbonyloxy group having 2 to 30 carbon atoms, an arylcarbonyloxy group, a hydroxyl group, a carboxyl group, a cyano group, an amino group, an amide group, and a nitro group.
- Each of these groups may further have a substituent (for example, a hydroxyl group, a halogen atom, an alkyl group, etc.).
- R 104 may be the same as or different from each other, and may be bonded to each other to form a ring.
- styrenic monomers include styrene; alkyl-substituted styrenes such as ⁇ -methylstyrene, ⁇ -methylstyrene, and p-methylstyrene; halogen-substituted styrenes such as 4-chlorostyrene and 4-bromostyrene; p-hydroxy Hydroxystyrenes such as styrene, ⁇ -methyl-p-hydroxystyrene, 2-methyl-4-hydroxystyrene, 3,4-dihydroxystyrene; vinylbenzyl alcohols; p-methoxystyrene, p-tert-butoxystyrene, m Alkoxy substituted styrenes such as tert-butoxystyrene; vinyl benzoic acids such as 3-vinylbenzoic acid and 4-vinylbenzoic acid; 4-vinylbenzyl acetate; 4-acetoxy
- the copolymerizable monomer combined with the styrenic monomer is a (meth) acrylic acid ester compound represented by the following formula (B), maleic anhydride, citraconic anhydride, cis-1-cyclohexene-1,2-dicarboxylic anhydride, Acid anhydrides such as 3-methyl-cis-1-cyclohexene-1,2-dicarboxylic anhydride and 4-methyl-cis-1-cyclohexene-1,2-dicarboxylic anhydride, and nitrile groups such as acrylonitrile and methacrylonitrile -Containing radical polymerizable monomers; amide bond-containing radical polymerizable monomers such as acrylamide, methacrylamide, trifluoromethanesulfonylaminoethyl (meth) acrylate; fatty acid vinyls such as vinyl acetate; chlorine such as vinyl chloride and vinylidene chloride Containing radical polymerizable monomer; 1,
- R 105 to R 107 in the formula (B) each independently represent a hydrogen atom, an alkyl group having 1 to 30 carbon atoms, or an aryl group.
- R 108 represents a hydrogen atom, an alkyl group having 1 to 30 carbon atoms, a cycloalkyl group, or an aryl group.
- Each of these groups may further have a substituent (for example, a hydroxyl group, a halogen atom, an alkyl group, etc.).
- (meth) acrylic acid ester compounds include methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate (i-, n-), butyl (meth) acrylate (n- , I-, s-, tert-), pentyl (meth) acrylate (n-, i-, s-), hexyl (meth) acrylate (n-, i-), heptyl (meth) acrylate (n -, I-), octyl (meth) acrylate (n-, i-), nonyl (meth) acrylate (n-, i-), myristyl (meth) acrylate (n-, i-), (meta ) Acrylic acid (2-ethylhexyl), (meth) acrylic acid ( ⁇ -caprolactone), (meth) acrylic acid (2-hydroxyethyl), acrylic acid (2-hydroxypropyl
- styrene compound examples include styrene / maleic anhydride copolymer, styrene / acrylic ester copolymer, styrene / hydroxystyrene polymer, styrene / acetoxystyrene polymer, and the like. Of these, a styrene / maleic anhydride copolymer is preferable.
- the content of the plasticizer is not particularly limited, but is preferably in the range of 0.1 to 30% by weight, more preferably in the range of 5 to 20% by weight with respect to the cellulose ester resin. . With such an amount, the retardation film hardly causes bleed out.
- the retardation film of the present invention essentially contains a cellulose ester resin, a compound of the general formula (1) and a compound of the general formula (2), and may contain a plasticizer if necessary. Further, the retardation film of the present invention may further contain other additives, if necessary, instead of the plasticizer or in addition to the plasticizer. Examples of such other additives include, but are not limited to, hydrogen bonding compounds, activators, antioxidants, colorants, ultraviolet absorbers, matting agents, acrylic particles, hydrogen bonding solvents, ionic interfaces. An active agent etc. are mentioned.
- the hydrogen bonding compound can reduce the fluctuation of the retardation value Rt with respect to the change in humidity.
- the hydrogen bonding compound preferably has at least a plurality of functional groups selected from a hydroxy group, an amino group, a thiol group, and a carboxylic acid group in one molecule, and a plurality of different functional groups in one molecule. It is more preferable to have a hydroxy group and a carboxylic acid group in one molecule.
- the hydrogen bonding compound preferably contains 1 to 2 aromatic rings as a mother nucleus, and the value obtained by dividing the number of functional groups contained in one molecule by the molecular weight of the compound is 0.00. It is preferably 01 or more.
- the above effect is such that the hydrogen-bonding compound is bonded (hydrogen bond) to a site where the cellulose ester and water molecules interact (hydrogen bonds), thereby suppressing the change in charge distribution due to desorption of water molecules. For the reason.
- the hydrogen bonding compound can be added in the range of 1 to 30 parts by weight with respect to 100 parts by weight of the cellulose ester.
- the antioxidant is not particularly limited, and a commonly known antioxidant can be used.
- lactone, sulfur, phenol, double bond, hindered amine, and phosphorus compounds can be preferably used.
- lactone compound examples include “IrgafosXP40, IrgafosXP60 (trade name)” commercially available from BASF Japan.
- sulfur compound examples include “Sumilizer TPL-R” and “Sumilizer TP-D” commercially available from Sumitomo Chemical Co., Ltd.
- the phenolic compound preferably has a 2,6-dialkylphenol structure. For example, “Irganox 1076”, “Irganox 1010” commercially available from BASF Japan, and “ ADEKA STAB AO-50 ”and the like.
- the above double bond compounds are commercially available from Sumitomo Chemical Co., Ltd. under the trade names “Sumilizer GM” and “Sumilizer GS”.
- the hindered amine compound examples include “Tinvin 144” and “Tinvin 770” commercially available from BASF Japan, and “ADK STAB LA-52” commercially available from ADEKA.
- the phosphorus compounds include “Sumizer GP” commercially available from Sumitomo Chemical Co., Ltd., “ADK STAB PEP-24G”, “ADK STAB PEP-36” and “ADK STAB 3010” commercially available from ADEKA Corporation. “IRGAFOS P-EPQ” commercially available from BASF Japan Ltd. and “GSY-P101” commercially available from Sakai Chemical Industry Ltd.
- the amount of these antioxidants and the like to be added is appropriately determined in accordance with the process for recycling and use, but generally 0.05 to 20 with respect to the resin (cellulose ester) as the main raw material of the film. It is added in the range of wt%, preferably 0.1 to 1 wt%.
- antioxidants can obtain a synergistic effect by using several different types of compounds in combination rather than using only one kind.
- the combined use of lactone, phosphorus, phenol and double bond compounds is preferred.
- the colorant means a dye or a pigment, and in the present invention, a colorant having an effect of making the color tone of a liquid crystal screen blue or adjusting the yellow index and reducing haze.
- Various dyes and pigments can be used as the colorant, but anthraquinone dyes, azo dyes, phthalocyanine pigments and the like are effective.
- the film of the present invention is preferably used on the viewing side or the backlight side of the polarizing plate, it preferably contains an ultraviolet absorber for the purpose of imparting an ultraviolet absorbing function.
- the ultraviolet absorber is not particularly limited, and examples thereof include ultraviolet absorbers such as benzotriazole, 2-hydroxybenzophenone, and salicylic acid phenyl ester.
- ultraviolet absorbers such as benzotriazole, 2-hydroxybenzophenone, and salicylic acid phenyl ester.
- 2- (5-methyl-2-hydroxyphenyl) benzotriazole, 2- [2-hydroxy-3,5-bis ( ⁇ , ⁇ -dimethylbenzyl) phenyl] -2H-benzotriazole, 2- (3 Triazoles such as 5-di-t-butyl-2-hydroxyphenyl) benzotriazole, 2-hydroxy-4-methoxybenzophenone, 2-hydroxy-4-octoxybenzophenone, 2,2′-dihydroxy-4-methoxybenzophenone And benzophenones.
- UV absorbers having a molecular weight of 400 or more are not sublimated or are not easily volatilized at a high boiling point. From the viewpoint of improving weather resistance, it is preferable.
- Examples of the ultraviolet absorber having a molecular weight of 400 or more include 2- [2-hydroxy-3,5-bis ( ⁇ , ⁇ -dimethylbenzyl) phenyl] -2-benzotriazole, 2,2-methylenebis [4- ( Benzotriazoles such as 1,1,3,3-tetrabutyl) -6- (2H-benzotriazol-2-yl) phenol], bis (2,2,6,6-tetramethyl-4-piperidyl) sebacate, Hindered amines such as bis (1,2,2,6,6-pentamethyl-4-piperidyl) sebacate and 2- (3,5-di-t-butyl-4-hydroxybenzyl) -2-n-butyl Bis (1,2,2,6,6-pentamethyl-4-piperidyl) malonate, 1- [2- [3- (3,5-di-t-butyl-4-hydroxyphenyl) propionylo Xyl] ethyl] -4- [3- (3
- 2- [2-hydroxy-3,5-bis ( ⁇ , ⁇ -dimethylbenzyl) phenyl] -2-benzotriazole and 2,2-methylenebis [4- (1,1,3,3- Tetrabutyl) -6- (2H-benzotriazol-2-yl) phenol] is particularly preferred.
- UV absorbers commercially available products may be used, for example, Tinuvin 109, Tinuvin 171, Tinuvin 234, Tinuvin 326, Tinuvin 327, Tinuvin 328, Tinuvin 928, etc.
- 2′-methylenebis [6- (2H-benzotriazol-2-yl) -4- (1,1,3,3-tetramethylbutyl) phenol] (molecular weight 659; examples of commercially available products are manufactured by ADEKA Corporation LA31) can be preferably used.
- the above ultraviolet absorbers can be used alone or in combination of two or more.
- the amount of the UV absorber used is not uniform depending on the type of UV absorber, the operating conditions, etc., but generally 0.05 to 10% by weight, preferably 0.1 to 0.1% by weight based on the resin (cellulose ester). It is added in the range of 5% by weight.
- the matting agent is fine particles imparting slipperiness of the film, and may be either an inorganic compound or an organic compound as long as it does not impair the transparency of the resulting film and has heat resistance during melting. These matting agents can be used alone or in combination of two or more. By using particles having different particle sizes and shapes (for example, acicular and spherical), both transparency and slipperiness can be made highly compatible. Among these, silicon dioxide, which is excellent in transparency (haze), is particularly preferably used because it has a refractive index close to that of the acrylic copolymer or cellulose ester used as a compatible resin.
- silicon dioxide examples include Aerosil 200V, Aerosil R972V, Aerosil R972, R974, R812, 200, 300, R202, OX50, TT600, NAX50 (manufactured by Nippon Aerosil Co., Ltd.), Sea Hoster KEP-10, Sea Hoster KEP- 30, Seahoster KEP-50 (above, manufactured by Nippon Shokubai Co., Ltd.), Silo Hovic 100 (manufactured by Fuji Silysia), Nip Seal E220A (manufactured by Nippon Silica Kogyo), Admafine SO (manufactured by Admatechs), etc. Goods etc. can be preferably used.
- the shape of the particles can be used without particular limitation, such as indefinite shape, needle shape, flat shape, spherical shape, etc. However, the use of spherical particles is preferable because the transparency of the resulting film can be improved.
- the particle size is preferably smaller than the wavelength of visible light, and more preferably 1 ⁇ 2 or less of the wavelength of visible light. . If the size of the particles is too small, the slipperiness may not be improved, so the range of 80 nm to 180 nm is particularly preferable.
- the particle size means the size of the aggregate when the particle is an aggregate of primary particles. Moreover, when a particle is not spherical, it means the diameter of a circle corresponding to the projected area.
- the matting agent is added in an amount of 0.05 to 10% by weight, preferably 0.1 to 5% by weight, based on the resin (cellulose ester).
- the film of the present invention may contain, for example, acrylic particles described in International Publication No. 2010/001668 in an amount within a range where transparency can be maintained.
- the acrylic particles have an action of improving the brittleness of the film.
- acrylic particles examples include, for example, “Metablene W-341” manufactured by Mitsubishi Rayon Co., “Kane Ace” manufactured by Kaneka Corporation, “Paraloid” manufactured by Kureha Co., Ltd., “Roid and Haas Co.” “Acryloid”, “Staffroid” manufactured by Ganz Kasei Kogyo Co., Ltd., Chemisnow MR-2G, MS-300X (above, manufactured by Soken Chemical Co., Ltd.) and “Parapet SA” manufactured by Kuraray Co., Ltd. Or 2 or more types can be used.
- the hydrogen bonding solvent can be added for the purpose of adjusting (reducing) the solution viscosity in a solvent for dissolving the constituent materials of the film when a film is produced by the solution casting method.
- the hydrogen bonding solvent is J.I. N. As described in Israel Ativili, “Intermolecular Forces and Surface Forces” (Takeshi Kondo, Hiroyuki Oshima, Maglow Hill Publishing, 1991) and electrically negative atoms (oxygen, nitrogen, fluorine, chlorine)
- a hydrogen bond is added to the solvent for dissolution.
- a part or all of the solvent may be used.
- An ionic surfactant can be added for the purpose of reducing the peeling force during film formation.
- ionic surfactant examples include a cationic surfactant, an anionic surfactant, and an amphoteric surfactant.
- cationic surfactant examples include aliphatic amine salts, aliphatic quaternary ammonium salts, benzalkonium salts, benzethonium chloride, pyridinium salts, imidazolinium salts, and the like.
- anionic surfactant examples include higher alcohol (C 8 -C 22 ) sulfate salts (for example, sodium salt of lauryl alcohol sulfate, sodium salt of octyl alcohol sulfate, ammonium salt of lauryl alcohol sulfate, “Tepol-81” ( Trade name, manufactured by Shell Chemical Co., Ltd.), secondary sodium alkyl sulfate, etc.), aliphatic alcohol phosphate salts (eg, sodium salt of cetyl alcohol phosphate), alkylaryl sulfonates (eg, dodecylbenzenesulfonic acid) Sodium salt, isopropyl naphthalene sulfonic acid sodium salt, dinaphthalenedisulfonic acid sodium salt, metanitrobenzene sulfonic acid sodium salt), alkylamide sulfonates (eg Examples thereof include C 17 H 33 CON (CH 3 ) CH 2 SO 3 Na) and dibasic fatty acid ester s
- amphoteric surfactants include carboxybetaine type, sulfobetaine type, aminocarboxylate, imidazolinium betaine and the like.
- an anionic surfactant is preferable in the present invention.
- the surfactant is 0.01% by weight or more and 5% by weight or less, preferably 0.05% by weight or more and 3% by weight or less, more preferably 0.2% by weight, based on the total amount of the resin constituting the film. % To 2% by weight is preferable.
- the addition amount is larger than this range, the surfactant is precipitated from the film, or the hygroscopicity of the film is increased, and a quality undesirable for the quality of the retardation film is exhibited. If the addition amount is less than this range, the effect of the present invention using a surfactant may not be obtained.
- a haze value (turbidity) is used as an index for judging the transparency of the retardation film of the present invention.
- the haze value is preferably 1.0% or less, and is 0.5% or less. More preferably.
- the haze value may exceed the above range.
- the film of the present invention preferably has a total light transmittance of 90% or more, more preferably 93% or more. Moreover, as a realistic upper limit, it is about 99%.
- the haze value and transmittance can be measured using a haze meter.
- a film satisfying the above physical properties can be preferably used as a polarizing plate protective film for a large-sized liquid crystal display device or a liquid crystal display device for outdoor use.
- the film forming method is preferably the solution casting film forming method and the melt casting film forming method, and in particular, the solution casting film forming method is a uniform surface. Is more preferable for obtaining.
- the film production method of the present invention includes a cellulose ester resin, a compound of the above general formula (1) and a compound of the above general formula (2), and, if necessary, the plasticizer described above / Or step of dissolving dope in solvent to prepare dope (dissolution step; dope preparation step), step of casting dope onto endless metal support (casting step), casting Step of drying dope as web (solvent evaporation step), step of peeling from metal support (peeling step), step of drying, stretching and width holding (stretching / width holding / drying step), step of winding the finished film (Winding step) is preferably included.
- FIG. 1 is a diagram schematically showing an example of a dope preparation step, a casting step, and a drying step (solvent evaporation step) of a solution casting film forming method preferable for the present invention.
- the main dope is filtered by the main filter 3, and the additive solution is added in-line from 16 to this.
- the main dope may contain about 10 to 50% by weight of recycled material.
- Recycled material is a product obtained by finely pulverizing a retardation film, which is produced when a retardation film is formed.
- a pelletized material (hereinafter also simply referred to as “other compound”) can be preferably used.
- Dissolution process (dope preparation process)
- the cellulose ester, the compound of the above general formula (1) and the compound of the above general formula (2), and optionally other compounds are stirred in a solvent mainly composed of a good solvent for the cellulose ester. It is a step of dissolving the solution while forming a dope, or a step of forming a dope which is a main solution by mixing the cellulose ester solution with another compound solution as the case may be.
- the concentration of cellulose ester in the dope is preferably higher because the drying load after casting on the metal support can be reduced. However, if the concentration of cellulose ester is too high, the load during filtration increases and the filtration accuracy is poor. Become.
- the concentration that achieves both of these is preferably 10 to 35% by weight, and more preferably 15 to 25% by weight.
- Solvents used in the dope may be used alone or in combination of two or more. However, it is preferable to use a mixture of a good solvent and a poor solvent of cellulose ester in terms of production efficiency, and there are many good solvents. This is preferable from the viewpoint of solubility of cellulose acetate.
- the preferable range of the mixing ratio of the good solvent and the poor solvent is 70 to 98% by weight for the good solvent and 2 to 30% by weight for the poor solvent.
- the good solvent used in the present invention is not particularly limited, and examples thereof include organic halogen compounds such as methylene chloride, dioxolanes, acetone, methyl acetate, and methyl acetoacetate. Particularly preferred is methylene chloride or methyl acetate.
- the poor solvent used in the present invention is not particularly limited, but for example, methanol, ethanol, n-butanol, cyclohexane, cyclohexanone and the like are preferably used.
- the dope preferably contains 0.01 to 2% by weight of water.
- the solvent used for dissolving the cellulose ester is used by collecting the solvent removed from the film by drying in the film-forming process and reusing it.
- the recovery solvent may contain trace amounts of additives added to cellulose acetate, such as plasticizers, UV absorbers, polymers, monomer components, etc., but these are preferably reused even if they are included. Can be purified and reused if necessary.
- a general method can be used as a method for dissolving the cellulose ester when preparing the dope described above. Specifically, a method carried out at normal pressure, a method carried out below the boiling point of the main solvent, a method carried out under pressure above the boiling point of the main solvent, JP-A-9-95544, JP-A-9-95557, Various dissolution methods such as a method using a cooling dissolution method as described in Kaihei 9-95538 and a method using a high pressure as described in Japanese Patent Application Laid-Open No. 11-21379 can be used. Among them, a method of performing pressurization at a temperature equal to or higher than the boiling point of the main solvent is preferable.
- a method of stirring and dissolving while heating at a temperature that is equal to or higher than the boiling point of the solvent at normal pressure and does not boil under pressure is preferable in order to prevent the generation of massive undissolved material called gel or mako.
- a method in which cellulose acetate is mixed with a poor solvent and wetted or swollen, and then a good solvent is added and dissolved is also preferably used.
- the pressurization may be performed by a method of injecting an inert gas such as nitrogen gas or a method of increasing the vapor pressure of the solvent by heating. Heating is preferably performed from the outside.
- a jacket type is preferable because temperature control is easy.
- the heating temperature with the addition of the solvent is preferably higher from the viewpoint of the solubility of cellulose acetate, but if the heating temperature is too high, the required pressure increases and the productivity deteriorates.
- the preferred heating temperature is 45 to 120 ° C, more preferably 60 to 110 ° C, and still more preferably 70 ° C to 105 ° C.
- the pressure is adjusted so that the solvent does not boil at the set temperature.
- a cooling dissolution method is also preferably used, whereby the cellulose ester can be dissolved in a solvent such as methyl acetate.
- the cellulose ester solution (doping during or after dissolution) is preferably filtered using a suitable filter medium such as filter paper.
- the absolute filtration accuracy is small in order to remove insoluble matters and the like.
- the absolute filtration accuracy is too small, there is a problem that the filter medium is likely to be clogged.
- a filter medium with an absolute filtration accuracy of 0.008 mm or less is preferable, a filter medium with 0.001 to 0.008 mm is more preferable, and a filter medium with 0.003 to 0.007 mm is more preferable.
- the material of the filter medium there are no particular restrictions on the material of the filter medium, and ordinary filter media can be used. However, plastic filter media such as polypropylene and Teflon (registered trademark), and metal filter media such as stainless steel do not drop off fibers. preferable.
- a bright spot foreign object is placed when two polarizing plates are placed in a crossed Nicol state, a film or the like is placed between them, light is applied from one polarizing plate, and the opposite is observed when observed from the other polarizing plate. It is a point (foreign matter) where light from the side appears to leak, and the number of bright spots having a diameter of 0.01 mm or more is preferably 200 / cm 2 or less. More preferably, it is 100 pieces / cm 2 or less, still more preferably 50 pieces / m 2 or less, still more preferably 0 to 10 pieces / cm 2 . Further, it is preferable that the number of bright spots of 0.01 mm or less is small.
- the dope can be filtered by a normal method, but the method of filtering while heating at a temperature not lower than the boiling point of the solvent at normal pressure and in a range where the solvent does not boil under pressure is the filtration pressure before and after filtration.
- the increase in the difference (referred to as differential pressure) is small and preferable.
- the preferred temperature is 45 to 120 ° C, more preferably 45 to 70 ° C, and still more preferably 45 to 55 ° C.
- the filtration pressure is preferably 1.6 MPa or less, more preferably 1.2 MPa or less, and further preferably 1.0 MPa or less.
- the dope is cast on a metal support. That is, in this step, the dope is fed to the pressurizing die 30 through a liquid feed pump (for example, a pressurized metering gear pump) and transferred indefinitely, for example, an endless metal belt 31 such as a stainless steel belt or a rotating metal drum. The dope is cast from the pressure die slit to the casting position on the metal support.
- a liquid feed pump for example, a pressurized metering gear pump
- ⁇ Pressure dies that can adjust the slit shape of the die base and make the film thickness uniform are preferred.
- the pressure die include a coat hanger die and a T die, and any of them is preferably used.
- the surface of the metal support is preferably a mirror surface.
- two or more pressure dies may be provided on the metal support, and the dope amount may be divided and stacked. Or it is also preferable to obtain the film of a laminated structure by the co-casting method which casts several dope simultaneously.
- the cast width is preferably 1.4 m or more from the viewpoint of productivity. More preferably, it is 1.4 to 4 m. When it exceeds 4 m, there is a risk of streaking in the manufacturing process or lowering of stability in the subsequent transport process. More preferably, it is 2.2 to 3.5 m in terms of transportability and productivity.
- the metal support in the casting process is preferably a mirror-finished surface, and a stainless steel belt or a drum whose surface is plated with a casting is preferably used as the metal support.
- the surface temperature of the metal support in the casting step is ⁇ 50 ° C. to less than the boiling point of the solvent, and a higher temperature is preferable because the web drying rate can be increased. May deteriorate.
- the preferred support temperature is 0 to 55 ° C, more preferably 25 to 50 ° C.
- the method for controlling the temperature of the metal support is not particularly limited, but there are a method of blowing hot air or cold air, and a method of contacting hot water with the back side of the metal support. It is preferable to use warm water because heat transfer is performed efficiently, so that the time until the temperature of the metal support becomes constant is short. When warm air is used, wind at a temperature higher than the target temperature may be used.
- Solvent evaporation step This step is a step of evaporating the solvent by heating the web (the dope is cast on the casting support and the formed dope film is called the web) on the casting support. It is.
- the drying efficiency is good and preferable.
- a method of combining them is also preferably used.
- the web on the support after casting is preferably dried on the support in an atmosphere of 40 to 100 ° C. In order to maintain the atmosphere at 40 to 100 ° C., it is preferable to apply hot air at this temperature to the upper surface of the web or heat by means such as infrared rays.
- peeling step Next, the web is peeled from the metal support. That is, this step is a step of peeling the web where the solvent is evaporated on the metal support at the peeling position. The peeled web is sent to the next process.
- the temperature at the peeling position on the metal support is preferably in the range of ⁇ 50 to 40 ° C., more preferably in the range of 10 to 40 ° C., and most preferably in the range of 15 to 30 ° C.
- the residual solvent amount at the time of peeling of the web on the metal support at the time of peeling is appropriately adjusted depending on the strength of drying conditions, the length of the metal support, and the like.
- the amount of residual solvent when peeling the web from the metal support is preferably 10 to 150% by weight.
- the amount is decided. More preferred is 20 to 40% by weight or 60 to 130% by weight, and particularly preferred is 20 to 30% by weight or 70 to 120% by weight.
- the amount of residual solvent is defined by the following formula.
- M is the weight of a sample collected during or after the production of the web or film
- N is the weight after heating M at 115 ° C. for 1 hour.
- the peeling tension when peeling the metal support and the film is preferably 300 N / m or less. More preferably, it is within the range of 196 to 245 N / m. However, when wrinkles easily occur during peeling, peeling with a tension of 190 N / m or less, preferably 100 to 190 N / m is preferred.
- the web is peeled off from the metal support, and further dried, so that the residual solvent amount is preferably 1% by weight or less, more preferably 0.1% by weight or less, particularly preferably. Is 0 to 0.01% by weight or less.
- a roll drying method (a method in which webs are alternately passed through a plurality of rolls arranged above and below) and a method in which the web is dried while being conveyed by a tenter method are employed.
- a drying device 35 that alternately conveys the web through rollers arranged in the drying device and / or a tenter stretching device 34 that clips and conveys both ends of the web with a clip, dry.
- the means for drying the web is not particularly limited, and can be generally performed with hot air, infrared rays, a heating roll, microwave, or the like, but is preferably performed with hot air in terms of simplicity. Too rapid drying tends to impair the flatness of the finished film. Drying at a high temperature is preferably carried out from about 8% by weight or less of residual solvent. Throughout, the drying is generally carried out in the range of 40-250 ° C. It is particularly preferable to dry within the range of 40 to 200 ° C. The drying temperature is preferably increased stepwise.
- tenter stretching apparatus When using a tenter stretching apparatus, it is preferable to use an apparatus that can independently control the film gripping length (distance from the start of gripping to the end of gripping) left and right by the left and right gripping means of the tenter. In the tenter process, it is also preferable to intentionally create sections having different temperatures in order to improve planarity.
- the web is preferably stretched in at least one direction from the metal support.
- the orientation of molecules in the film can be controlled by the stretching treatment.
- the retardation film has the structure of the present invention, and the refractive index is controlled by controlling the transport tension and stretching. For example, the retardation value can be changed by lowering or increasing the tension in the longitudinal direction.
- two axes are sequentially or simultaneously applied to the longitudinal direction (film forming direction; casting direction; MD direction) of the film and the direction orthogonal to the film plane, that is, the width direction (TD direction).
- Stretching or uniaxial stretching can be performed.
- the biaxially stretched film is biaxially stretched in the casting direction (MD direction) and the width direction (TD direction), but the retardation film according to the present invention may be a uniaxially stretched film. And an unstretched film may be sufficient.
- the stretching operation may be performed in multiple stages. When biaxial stretching is performed, simultaneous biaxial stretching may be performed or may be performed stepwise.
- stepwise means that, for example, stretching in different stretching directions can be sequentially performed, stretching in the same direction is divided into multiple stages, and stretching in different directions is added to any one of the stages.
- stretching steps are possible: -Stretch in the casting direction-> Stretch in the width direction-> Stretch in the casting direction-> Stretch in the casting direction-Stretch in the width direction-> Stretch in the width direction-> Stretch in the casting direction-> Stretch in the casting direction.
- Simultaneous biaxial stretching includes stretching in one direction and contracting the other while relaxing the tension.
- the draw ratios in the biaxial directions perpendicular to each other are preferably in the range of 0.8 to 1.5 times in the casting direction and 1.1 to 2.5 times in the width direction, respectively. It is preferable to carry out in the range of 0.8 to 1.0 times in the extending direction and 1.2 to 2.0 times in the width direction.
- the stretching temperature is usually preferably performed in the temperature range of Tg to Tg + 60 ° C. of the resin constituting the film.
- the stretching temperature is preferably from 120 ° C. to 200 ° C., more preferably from 150 ° C. to 200 ° C., and more preferably from 150 ° C. to 190 ° C. or less.
- the residual solvent in the film at the time of stretching is preferably 20 to 0%, more preferably 15 to 0%.
- the residual solvent is stretched by 11% at 155 ° C., or the residual solvent is stretched by 2% at 155 ° C.
- the residual solvent is stretched at 15% at 160 ° C, or the residual solvent is stretched at less than 1% at 160 ° C.
- the method of stretching the web For example, a method in which a difference in peripheral speed is applied to a plurality of rolls, and the roll peripheral speed difference is used to stretch in the longitudinal direction, the both ends of the web are fixed with clips and pins, and the interval between the clips and pins is increased in the traveling direction. And a method of stretching in the vertical direction, a method of stretching in the horizontal direction and stretching in the horizontal direction, a method of stretching in the vertical and horizontal directions and stretching in both the vertical and horizontal directions, and the like. Of course, these methods may be used in combination. Among them, it is particularly preferable to perform stretching in the width direction (lateral direction) by a tenter method in which both ends of the web are gripped by clips or the like.
- a tenter it may be a pin tenter or a clip tenter.
- the slow axis or the fast axis exists in the film plane, and ⁇ 1 is preferably ⁇ 1 ° or more and + 1 ° or less, assuming that the angle formed with the film forming direction is ⁇ 1. More preferably, it is 5 ° or more and + 0.5 ° or less.
- This ⁇ 1 can be defined as an orientation angle, and ⁇ 1 can be measured using an automatic birefringence meter KOBRA-21ADH (Oji Scientific Instruments).
- KOBRA-21ADH Oji Scientific Instruments
- a phase difference film is obtained by winding the obtained web (finished film). More specifically, it is a step of winding the film as a retardation film by a winder 37 after the residual solvent amount in the web becomes 2% by weight or less, and by setting the residual solvent amount to 0.4% by weight or less.
- a film having good dimensional stability can be obtained. In particular, it is preferable to take up in the range of 0.00 to 0.10% by weight.
- a generally used one may be used, and there are a constant torque method, a constant tension method, a taper tension method, a program tension control method with a constant internal stress, etc., and these may be used properly.
- the end Before winding, the end may be slit and cut to the product width, and knurled (embossed) may be applied to both ends to prevent sticking and scratching during winding.
- the knurling method can process a metal ring having an uneven pattern on its side surface by heating or pressing. Note that the clip holding portions at both ends of the film are usually cut off because the film is deformed and cannot be used as a product. If the material has not deteriorated due to heat, it is reused after recovery.
- the film of the present invention is preferably a long film. Specifically, the film of about 100 m to 10000 m is shown, usually in the form of a roll. Further, the width of the film is preferably 1.4 to 4 m, more preferably 1.4 to 4 m, more preferably 2 to 3 m in order to meet demands for an increase in the size of liquid crystal display devices and production efficiency. More preferably.
- the retardation film of the present invention can also be formed by a melt casting method.
- the melt film-forming method refers to a method in which a composition containing a cellulose ester and the above-mentioned additive is heated and melted to a temperature exhibiting fluidity, and then a melt containing fluid cellulose is cast.
- the molding method for heating and melting can be classified in detail into a melt extrusion molding method, a press molding method, an inflation method, an injection molding method, a blow molding method, a stretch molding method, and the like.
- the melt extrusion method is preferable from the viewpoint of mechanical strength and surface accuracy.
- the plurality of raw materials used in the melt extrusion method are usually preferably kneaded in advance and pelletized.
- Pelletization may be performed by a known method, for example, dry cellulose ester, plasticizer, and other additives are fed to an extruder with a feeder, kneaded using a monoaxial or biaxial extruder, and formed into a strand from a die. It can be carried out by extruding, water cooling or air cooling and cutting.
- Additives may be mixed before being supplied to the extruder, or may be supplied by individual feeders.
- a small amount of additives such as particles and antioxidants are preferably mixed in advance in order to mix uniformly.
- the extruder is preferably processed at as low a temperature as possible so that the shearing force is suppressed and the resin can be pelletized so as not to deteriorate (molecular weight reduction, coloring, gel formation, etc.).
- a twin screw extruder it is preferable to rotate in the same direction using a deep groove type screw. From the uniformity of kneading, the meshing type is preferable.
- the raw material powder can be directly fed to the extruder by a feeder without being pelletized to form a film as it is.
- the melting temperature when extruding the above pellets using a single-screw or twin-screw type extruder is set to a temperature range of 200 to 300 ° C., filtered through a leaf disk type filter, etc.
- a film is cast from the die, the film is nipped by a cooling roller and an elastic touch roller, and solidified on the cooling roller.
- a method for preventing oxidative decomposition or the like under vacuum, reduced pressure, or inert gas atmosphere when introducing from the supply hopper to the extruder is also preferable.
- the extrusion flow rate is preferably carried out stably by introducing a gear pump.
- a stainless fiber sintered filter is preferably used as a filter used for removing foreign substances.
- the stainless steel fiber sintered filter is made by compressing the intricately intertwined state of the stainless steel fiber body and sintering and integrating the contact points. The density changes depending on the thickness of the fiber and the amount of compression, and the filtration accuracy Can be adjusted.
- Additives such as plasticizers and particles may be mixed with the resin in advance, or may be kneaded in the middle of the extruder. In order to add uniformly, it is preferable to use a mixing apparatus such as a static mixer.
- the film temperature on the touch roller side is preferably in the temperature range of Tg to Tg + 110 ° C. of the film.
- a known roller can be used as the roller having an elastic surface used for such a purpose.
- the elastic touch roller is also called a pinching rotary body.
- a commercially available elastic touch roller can also be used.
- the film obtained as described above is stretched by the stretching operation after passing through the step of contacting the cooling roller.
- a known roller stretching machine or tenter can be preferably used. Specific conditions are the same as in the case of the solution pouring method.
- the film obtained as described above is wound to obtain a retardation film.
- the retardation film of the present invention can exhibit a desired retardation even in a thin film. Therefore, the retardation film of this invention can be used conveniently for a polarizing plate and a liquid crystal display device.
- FIG. 2 is a schematic cross-sectional view showing an example of the configuration of a polarizing plate provided with the retardation film of the present invention and a liquid crystal display device provided with the polarizing plate.
- the protective film 102, the active energy ray-curable adhesive 103A, and the polarizer 104 are laminated in this order, and the protective film is disposed. It is a preferable embodiment that the active energy ray-curable adhesive 103B and the retardation film 105 according to the present invention are laminated on the polarizer surface opposite to the surface on which the light is applied. That is, the polarizing plate 101A has a configuration in which the retardation film 105 according to the present invention is bonded to the polarizer 104 with the active energy ray-curable adhesive 103B.
- a functional layer 106 such as an antiglare layer, an antireflection layer, an antifouling layer, and a hard coat layer may be provided on the outer side (outermost surface portion) of the protective film 102 as necessary.
- the retardation film 105 of the polarizing plate 101A is bonded to the liquid crystal cell 107 via an adhesive or an adhesive, and the liquid crystal cell surface (backlight) on the opposite side of the surface where the polarizing plate 101A and the liquid crystal cell 107 are bonded.
- Side: BL is described in the figure
- the retardation film 105 of the present invention of the polarizing plate 101B having the same configuration as the polarizing plate 101A is preferably bonded to constitute the liquid crystal display device 108.
- the retardation film 105 and the polarizer 104 are bonded via an active energy ray-curable adhesive 103B. It is preferable to use an active energy ray-curable adhesive because moisture permeability can be effectively controlled.
- curable adhesives such as urethane adhesives, epoxy adhesives, aqueous polymer-isocyanate adhesives, thermosetting acrylic adhesives, Use moisture-curing urethane adhesive, anaerobic adhesive such as polyether methacrylate type, ester methacrylate type, oxidized polyether methacrylate, cyanoacrylate instant adhesive, acrylate and peroxide two-component instant adhesive be able to.
- the pressure-sensitive adhesive may be a one-component type or a two-component type in which two or more components are mixed before use.
- the adhesive may be a solvent system using an organic solvent as a medium, or an aqueous system such as an emulsion type, a colloidal dispersion liquid type, or an aqueous solution type that is a medium containing water as a main component, or a solvent-free type. It may be a mold.
- concentration of the adhesive solution may be appropriately determined depending on the film thickness after bonding, the coating method, the coating conditions, etc., and is usually 0.1 to 50% by weight.
- an active energy ray-curable adhesive from the viewpoint of effectively controlling moisture permeability. That is, in the polarizing plate of the present invention, it is preferable that the retardation film is bonded to the polarizer with an active energy ray-curable adhesive.
- Preferred examples of the active energy ray-curable adhesive include, for example, ( ⁇ ) cationically polymerizable compound, ( ⁇ ) photocationic polymerization initiator, and ( ⁇ ) as disclosed in JP 2011-028234 A Examples include a photosensitizer that exhibits maximum absorption in light having a wavelength longer than 380 nm, and a photocurable adhesive composition containing ( ⁇ ) a naphthalene-based photosensitization aid.
- active energy ray-curable adhesives may be used.
- a polarizer which is a main component of the polarizing plate, is an element that allows only light having a plane of polarization in a certain direction to pass through.
- a typical polarizer currently known is a polyvinyl alcohol polarizing film.
- the polyvinyl alcohol polarizing film includes those obtained by dyeing iodine on a polyvinyl alcohol film and those obtained by dyeing a dichroic dye.
- polarizer a polarizer obtained by forming a polyvinyl alcohol aqueous solution into a film and dyeing it by uniaxial stretching or dyeing and then uniaxially stretching and then preferably performing a durability treatment with a boron compound may be used.
- the film thickness of the polarizer is preferably in the range of 5 to 30 ⁇ m, particularly preferably in the range of 5 to 15 ⁇ m.
- the ethylene unit content described in JP-A-2003-248123, JP-A-2003-342322, etc. is 1 to 4 mol%
- the degree of polymerization is 2000 to 4000
- the degree of saponification is 99.0 to 99.99 mol. %
- Ethylene-modified polyvinyl alcohol is also preferably used.
- an ethylene-modified polyvinyl alcohol film having a hot water cutting temperature in the range of 66 to 73 ° C. is preferably used.
- a polarizer using this ethylene-modified polyvinyl alcohol film is excellent in polarization performance and durability, and has few color spots, and is particularly preferably used for a large-sized liquid crystal display device.
- an active energy ray-curable adhesive is further provided on the polarizer surface opposite to the surface on which the retardation film of the present invention is disposed, as shown in FIG. It is preferable that the protective film 102 is laminated through the film.
- the protective film can be obtained as a commercial product.
- Konica Minoltak KC4UE, KC8UE, KC8UX, KC5UX, KC8UY, KC4UY, KC4CZ, KC6UA, KC4UA, and KC2UA (above, manufactured by Konica Minolta Co., Ltd.) Is mentioned.
- the protective film disposed on the viewing side is preferably provided with a functional layer such as a hard coat layer, an antistatic layer, an antireflection layer, a slippery layer, an adhesive layer, an antiglare layer, or a barrier layer.
- a functional layer such as a hard coat layer, an antistatic layer, an antireflection layer, a slippery layer, an adhesive layer, an antiglare layer, or a barrier layer.
- the polarizing plate can be produced by laminating the retardation film of the present invention on one surface of the polarizer using an active energy ray-curable adhesive.
- the adhesiveness is different between both surfaces of the retardation film, it is preferable to bond the two on the one having better adhesiveness.
- the polarizing plate includes an adhesive application step of forming an adhesive layer by applying the following active energy ray-curable adhesive to at least one of the adhesive surfaces of the polarizer and the retardation film, and the adhesive layer A bonding step in which the polarizer and the retardation film are bonded and bonded via the adhesive layer, and a curing step in which the adhesive layer is cured in a state where the polarizer and the retardation film are bonded via the adhesive layer; It can manufacture with the manufacturing method containing. Moreover, there may be a pretreatment step in which the surface of the retardation film to which the polarizer is adhered is subjected to an easy adhesion treatment.
- Pretreatment process In the pretreatment step, the surface of the retardation film that adheres to the polarizer is subjected to easy adhesion treatment. When the retardation film and the protective film are adhered to both surfaces of the polarizer, easy adhesion treatment is performed on each of the retardation film and the protective film. In the next adhesive application process, the surface subjected to easy adhesion treatment is treated as a bonding surface with a polarizer, so on both surfaces of the retardation film, on the surface to be bonded with the active energy ray-curable adhesive, Apply easy adhesion treatment. Examples of the easy adhesion treatment include corona treatment and plasma treatment.
- the active energy ray-curable adhesive is applied to at least one of the adhesive surfaces of the polarizer and the retardation film.
- the application method is not particularly limited. For example, various wet coating methods such as a doctor blade, a wire bar, a die coater, a comma coater, and a gravure coater can be used. Also, a method in which an active energy ray-curable adhesive is cast between a polarizer and a retardation film, and then pressed with a roller or the like to uniformly spread the adhesive can also be used.
- Bonding process After apply
- this bonding step for example, when an active energy ray-curable adhesive is applied to the surface of the polarizer in the previous application step, a retardation film is superimposed thereon.
- a polarizer is superimposed thereon.
- an active energy ray-curable adhesive is cast between the polarizer and the retardation film, the polarizer and the retardation film are superposed in that state.
- both sides of the polarizer are respectively connected via an active energy ray-curable adhesive.
- a retardation film and a protective film are overlaid.
- both sides when the retardation film is overlapped on one side of the polarizer, when the retardation film and the protective film are overlapped on the polarizer side and the retardation film side, and on both sides of the polarizer
- rollers or the like As the material of the roller, metal, rubber or the like can be used.
- the rollers arranged on both sides may be made of the same material or different materials.
- an active energy ray curable adhesive is irradiated with active energy rays, and a cationic polymerizable compound (eg, epoxy compound or oxetane compound) or a radical polymerizable compound (eg, acrylate compound, acrylamide type).
- a cationic polymerizable compound eg, epoxy compound or oxetane compound
- a radical polymerizable compound eg, acrylate compound, acrylamide type.
- the active energy ray-curable adhesive containing the compound or the like is cured, and the polarizer and the retardation film, or the polarizer and the retardation film, which are superposed via the active energy ray-curable adhesive, are adhered.
- the active energy ray may be irradiated from either the polarizer side or the retardation film side.
- the active energy is in a state where the retardation film and the protective film are superimposed on both sides of the polarizer via an active energy ray-curable adhesive, respectively. It is advantageous to irradiate the line and simultaneously cure the active energy ray curable adhesive on both sides.
- Visible light, ultraviolet rays, X-rays, electron beams, etc. can be used as the active energy rays applied for curing, but electron beams and ultraviolet rays are generally preferred because they are easy to handle and have a sufficient curing rate. Used.
- the acceleration voltage is preferably in the range of 5 to 300 kV, more preferably in the range of 10 to 250 kV. If the acceleration voltage is less than 5 kV, the electron beam may not reach the adhesive and may be insufficiently cured. If the acceleration voltage exceeds 300 kV, the penetrating force through the sample is too strong and the electron beam rebounds. There is a risk of damaging the polarizer.
- the irradiation dose is in the range of 5 to 100 kGy, more preferably in the range of 10 to 75 kGy.
- the adhesive becomes insufficiently cured, and when it exceeds 100 kGy, the retardation film and the polarizer are damaged, resulting in a decrease in mechanical strength and yellowing to obtain predetermined optical characteristics. I can't.
- Arbitrary appropriate conditions can be employ
- the dose of ultraviolet rays in the range of 50 ⁇ 1500mJ / cm 2 in accumulated light amount, and even more preferably in the range of within the range of 100 ⁇ 500mJ / cm 2.
- the line speed depends on the curing time of the adhesive, but is preferably in the range of 1 to 500 m / min, more preferably 5 to 300 m / min, and still more preferably 10 to 100 m. / Min.
- the line speed is too slow, the productivity is poor or the damage to the retardation film is too great to produce a polarizing plate that can withstand a durability test.
- the line speed is too high, the adhesive is not sufficiently cured, and the target adhesiveness may not be obtained.
- the thickness of the adhesive layer is not particularly limited, but is usually in the range of 0.01 to 10 ⁇ m, and preferably in the range of 0.5 to 5 ⁇ m.
- the polarizing plate of the present invention can be suitably used for a liquid crystal display device. Since the liquid crystal display device using the polarizing plate of the present invention uses a retardation film having a low moisture permeability, color unevenness of the liquid crystal display device due to water content hardly occurs.
- the glass used for the panel of the liquid crystal display device preferably has a thickness in the range of 0.3 to 0.7 mm, and more preferably in the range of 0.3 to 0.5 mm. Since the polarizing plate of the present invention is not easily deformed, it is preferably used particularly when the glass thickness is small.
- Bonding between the surface of the polarizing plate of the present invention on the side of the retardation film and at least one surface of the liquid crystal cell can be performed by a known method. Depending on the case, it may be bonded through an adhesive layer.
- the mode (driving method) of the liquid crystal display device is not particularly limited, and liquid crystal display devices of various drive modes such as STN, TN, OCB, HAN, VA (MVA, PVA), IPS, OCB, and the like can be used.
- the retardation film of the present invention since the retardation film of the present invention has a high retardation value, it is a VA (MVA, PVA) type (vertical alignment type) liquid crystal display device as an optical compensation film (retardation film) that expands the viewing angle.
- VA vertical alignment type liquid crystal display device
- the polarizing plate is provided on at least one surface of a liquid crystal cell.
- the liquid crystal display device By providing the liquid crystal display device with the polarizing plate containing the retardation film of the present invention, the liquid crystal display device is excellent in durability (moisture and heat resistance), and even in a liquid crystal display device having a large screen of 30 type or more, there is variation in retardation. Is suppressed, and a liquid crystal display device with excellent visibility without unevenness of the liquid crystal display device can be obtained.
- Fine particles (matting agent) (Aerosil R812, manufactured by Nippon Aerosil Co., Ltd.) 11 parts by weight Ethanol 89 parts by weight The above was stirred and mixed with a dissolver for 50 minutes, and then dispersed with Manton Gorin.
- Fine particle addition liquid 1 The fine particle dispersion 1 was slowly added to the dissolution tank containing methylene chloride with sufficient stirring. Further, the particles were dispersed by an attritor so that the secondary particles had a predetermined particle size. This was filtered through Finemet NF manufactured by Nippon Seisen Co., Ltd. to prepare a fine particle additive solution 1.
- a main dope solution having the following composition was prepared. First, methylene chloride and ethanol were added to the pressure dissolution tank. Cellulose acetate having an acetyl substitution degree of 2.41 was added to a pressure dissolution tank containing a solvent while stirring. This was heated and stirred to dissolve completely, and this was dissolved in Azumi Filter Paper No. The main dope solution 1 was prepared by filtration using 244.
- the main dope solution 1 was put into a sealed main dissolution vessel and dissolved with stirring to prepare a dope solution.
- the obtained dope solution was uniformly cast on a stainless belt support using an endless belt casting apparatus.
- the solvent was evaporated until the residual solvent amount in the cast (cast) film was 75%, and then peeled off from the stainless steel belt support with a peeling tension of 130 N / m.
- the peeled retardation film was stretched 30% in the width direction using a tenter while applying heat at 160 ° C. At this time, the film was not stretched in the casting direction. Further, the residual solvent at the start of stretching was 15%.
- drying was completed while the drying zone was conveyed by a number of rolls.
- the drying temperature was 130 ° C. and the transport tension was 100 N / m.
- a retardation film 2 having a dry film thickness of 30 ⁇ m and a winding length of 4000 m was obtained.
- a retardation film 8 was produced in the same manner as in the production of the retardation film 2 except that the main dope liquid 1 prepared below was used instead of the main dope liquid 1. .
- sugar ester S has the following structure. This sugar ester is also referred to as “S1” in the table below.
- retardation films 8 to 12 change of film thickness
- the production of the retardation film 1 is the same as the production of the retardation film 1 except that the dope solution is cast on a stainless steel band support so that the film has a thickness described in Table 2 below. Then, retardation films 8 to 12 were produced.
- a retardation film 17 was produced.
- a dehydration condensation reaction was performed for 15 hours, and after the completion of the reaction, unreacted 1,2-propylene glycol was distilled off under reduced pressure at 200 ° C. to obtain a plasticizer S2 (aromatic terminal ester compound).
- the plasticizer S2 had an acid value of 0.10 and a number average molecular weight of 450.
- Plasticizer S3 251 g of 1,2-propylene glycol, 354 g of terephthalic acid, 610 g of benzoic acid, and 0.191 g of tetraisopropyl titanate as an esterification catalyst were charged into a 2 L four-necked flask equipped with a thermometer, a stirrer, and a quick cooling tube. The temperature is gradually raised with stirring until it reaches 230 ° C. in an air stream. A dehydration condensation reaction was carried out for 15 hours, and after the completion of the reaction, unreacted 1,2-propylene glycol was distilled off under reduced pressure at 200 ° C. to obtain a plasticizer S3 (aromatic terminal ester compound). The plasticizer S3 had an acid value of 0.10 and a number average molecular weight of 400.
- Plasticizer S4 Triphenyl phosphate Plasticizer S5: Diphenylbiphenyl phosphate (BDP) Plasticizer S6: Trimethylolpropane benzoate ester.
- the tear load of the Elmendorf method was measured with a light load tear device manufactured by Toyo Seiki Co., Ltd. The tear load was measured under the conditions of 23 ° C. and 55% RH for each of the case where the film was torn in the film conveyance direction (MD direction) and the case where the film was torn in the direction perpendicular to the film conveyance direction (TD direction). went.
- a retardation value Ro represented by the following formula using an automatic birefringence meter KOBRA-21ADH (Oji Scientific Instruments) at a wavelength of 590 nm in an environment of a temperature of 23 ° C. and a relative humidity of 55%. Rt was measured.
- the retardation value Ro in the in-plane direction and the retardation value Rt in the thickness direction were calculated according to the following formulas (I) and (II).
- n x represents a refractive index in the direction x in which the refractive index is maximized in the plane direction of the film.
- n y in-plane direction of the film, the refractive index in the direction y perpendicular to the direction x.
- nz represents the refractive index in the thickness direction z of the film.
- d represents the thickness (nm) of the film.
- the humidity-controlled sample was again conditioned for 5 hours in an environment of 23 ° C. and 55% RH, and measurement was performed to confirm that this change was a reversible change. A smaller value indicates that the value is more stable against humidity fluctuations.
- Rt humidity fluctuation ⁇ Rt / (Rt value measured after leaving the film in a 23% relative humidity 55% environment for 5 hours) ⁇ 100 Rt humidity fluctuation was evaluated based on the following indicators.
- the composition of the retardation film and the results of the above evaluation are summarized in Tables 1 to 11 below.
- the polarizing plates 2-1 to 2-55, 3-1 to 3-55, 4-1 to 4-55, and 5-1 to 5-55 are the same as the polarizing plates 1-1 to 1-55, Similar results were obtained for each.
- a KC6UA film manufactured by Konica Minolta Co., Ltd.
- the prepared active energy ray-curable adhesive solution was microgravure coater (gravure roller: # 300, rotational speed 140%). / Line speed) to form an active energy ray-curable adhesive (103A) by coating to a thickness of 5 ⁇ m.
- the prepared active energy ray-curable adhesive liquid is applied to the prepared retardation film 1 (101) so as to have a thickness of 5 ⁇ m, as described above, and the active energy ray-curable adhesive is thus prepared. (103B) was formed.
- the produced polyvinyl alcohol-iodine polarizer (104) is placed and bonded by a roller machine, and the protective film 1 (102)
- a laminate in which / active energy ray-curable adhesive (103A) / polarizer (104) / active energy ray-curable adhesive (103B) / retardation film 101 (105) was laminated was obtained.
- a polarizing plate 1-1 (polarizing plate 101A) was produced by irradiating an electron beam from both sides of the laminate.
- the line speed was 20 m / min
- the acceleration voltage was 250 kV
- the irradiation dose was 20 kGy.
- Example 1 In Example 1 (paragraphs “0060” to “0066”) of Japanese Patent No. 4962261, the method is the same as that described in Example 1 of Japanese Patent No. 4962261 except that the film thickness of the uniaxially oriented PET film is 80 ⁇ m. Thus, a film H2 was produced.
- ⁇ Active energy ray-curable adhesive Use of radical polymerization type adhesive (in the table, described as radical polymerization type)> [Production of Polarizing Plate 4-1] (Production of polarizer)
- a 70 ⁇ m thick polyvinyl alcohol film was swollen with water at 35 ° C.
- the obtained film was immersed in an aqueous solution composed of 0.075 g of iodine, 5 g of potassium iodide and 100 g of water for 60 seconds, and further immersed in an aqueous solution at 45 ° C. composed of 3 g of potassium iodide, 7.5 g of boric acid and 100 g of water.
- the obtained film was uniaxially stretched under conditions of a stretching temperature of 55 ° C. and a stretching ratio of 5 times. This uniaxially stretched film was washed with water and dried to obtain a polarizer having a thickness of 25 ⁇ m.
- the numerical value in parentheses indicates the number of each component described in FIG.
- the retardation film (105) the above-prepared retardation film 1 (105) was used, and the prepared photocurable adhesive liquid R was applied to a microgravure coater (gravure roll: # 300, rotational speed 140% / line).
- the photocurable resin layer (103B) was formed by coating so as to have a thickness of 5 ⁇ m.
- a Konica Minoltak KC6UA film manufactured by Konica Minolta Co., Ltd.
- the prepared photocurable adhesive liquid R was applied to a thickness of 5 ⁇ m as described above.
- a photocurable resin layer (103A) To form a photocurable resin layer (103A).
- the produced polyvinyl alcohol-iodine polarizer (104) is placed and bonded by a roll machine, and the protective film (102) / photocured.
- a laminate was obtained in which the mold resin layer (103A) / polarizer (104) / photocurable resin layer (103B) / retardation film (105) were laminated. In that case, it bonded by the roll machine so that the slow axis of retardation film (105) and the absorption axis of polarizer (104) might become mutually orthogonal.
- the polarizing plate 4-1 (101A) was produced by irradiating an electron beam from both sides of the laminate under the conditions of a line speed of 20 m / min, an acceleration voltage of 250 kV, and an irradiation dose of 20 kGy.
- the film was uniaxially stretched 6 times in a 50% aqueous solution with a boric acid concentration of 4% under the condition that the tension applied to the film was 700 N / m, and the potassium iodide concentration was 40 g / liter and the boric acid concentration was 40 g / liter. Then, it was immersed in an aqueous solution having a zinc chloride concentration of 10 g / liter and a temperature of 30 ° C. for 5 minutes for fixing.
- the obtained polarizer had an average thickness of 25 ⁇ m, a polarization performance of 43.0% transmittance, a polarization degree of 99.5%, and a dichroic ratio of 40.1.
- the optical film 105 produced as a retardation film and a Konica Minolta KC6UA film (manufactured by Konica Minolta Co., Ltd.) as a protective film were bonded to the polarizer.
- the polarizer was immersed in a storage tank of a polyvinyl alcohol adhesive solution having a solid content of 2% by mass for 1 to 2 seconds.
- KC6UA film as the protective film and the retardation film 1 (105) produced above were subjected to alkali saponification treatment under the following conditions, followed by washing with water, neutralization and washing in this order, and then drying at 100 ° C.
- the excess adhesive adhered to the polarizer immersed in the polyvinyl alcohol adhesive solution in step a was lightly removed, and the KC6UA film and the optical film 105 were sandwiched between the polarizers and laminated. That is, a laminate in which the protective film (102) / polyvinyl alcohol adhesive (103A) / polarizer (104) / polyvinyl alcohol adhesive (103B) / retardation film (105) was laminated was obtained (the polarization shown in FIG. 2). Configuration of plate 101A).
- ⁇ Process d> The sample produced in the above step c was dried in a dryer at a temperature of 80 ° C. for 5 minutes, and a polarizing plate 1202 having the configuration of the polarizing plate 101A in FIG. 2 was produced.
- the prepared liquid crystal display device was laid and placed on a stand or the like, and Bencot (manufactured by Asahi Kasei Fibers Co., Ltd.) was placed on a part of the evaluation polarizing plate to contain water.
- Bencot manufactured by Asahi Kasei Fibers Co., Ltd.
- the bencott was covered with 100 ⁇ m PET so that it would not dry, a black display signal was input to the TV from the PC, and the TV was turned on for 24 hours (the room temperature was set to 23 ° C., and the panel temperature was 38 ° C.). After 24 hours, remove the becot.
- L * of the part where there was a becot was measured by EZ contrast (ELDIM) as L * of the water immersion part.
- the L * of the part without Bencott was measured by EZ contrast as L * of the non-immersed part.
- the measurement with EZ contrast was performed in the color mode with the TV displayed in black.
- the conditions for water immersion were such that the panel was turned on and was allowed to stand for 24 hours in a state where a bencott sufficiently soaked in water was attached.
- L * of the water-immersed part / L * of the non-immersed part was calculated, and color unevenness was evaluated according to the following criteria.
- a liquid crystal display device was produced using the polarizing plates 2-1 to 2-55, and the liquid crystal display device was evaluated in the same manner as described above. As a result, liquid crystals using the polarizing plates 1-1 to 1-55 were used. Similar results were obtained with each of the display devices 1 to 55. Similarly, a liquid crystal display device was produced using the polarizing plates 3-1 to 3-55, and the liquid crystal display device was evaluated in the same manner as described above. As a result, the polarizing plates 1-1 to 1-55 were used. Similar results were obtained with each of the liquid crystal display devices 1 to 55.
- a liquid crystal display device was produced using the polarizing plates 4-1 to 4-55, and the liquid crystal display device was evaluated in the same manner as described above. As a result, the polarizing plates 1-1 to 1-55 were used. Similar results were obtained with each of the liquid crystal display devices 1 to 55.
- a liquid crystal display device was manufactured using the polarizing plates 5-1 to 5-55, and the liquid crystal display device was evaluated in the same manner as described above. As a result, the polarizing plates 1-1 to 1-55 were used. Similar results were obtained with each of the liquid crystal display devices 1 to 55.
- the retardation film of the present invention is superior in tear strength while maintaining a high retardation value as compared with the comparative film. Furthermore, it is also confirmed that the polarizing plate and the liquid crystal display device using the retardation film can suppress the occurrence of color unevenness even when the size is large.
Abstract
本発明は、セルロースエステル樹脂と、下記一般式(1): 式中、R1~R4は、それぞれ独立して、水素原子または炭素数1~3のアルキル基を表し;ならびにR5およびR6は、それぞれ独立して、置換基を有するアルキル基またはグリシジル基を表し、この際、前記置換基は、ヒドロキシル基、エステル基および芳香族基からなる群より選択される少なくとも一種である、 で表わされる化合物の少なくとも1種と、 下記一般式(2): 式中、A1及びA2は、それぞれ独立して、アルキル基、シクロアルキル基、芳香族炭化水素環基または芳香族複素環基を表し;Bは、芳香族炭化水素環または芳香族複素環を表し;T1及びT2は、それぞれ独立して、2価の1,2,4-トリアゾール環基を表し;L1、L2、L3及びL4は、それぞれ独立して、単結合または2価の連結基を表し;ならびにnは、0~5の整数を表す、 で表される化合物の少なくとも1種と、を含有することを特徴とする位相差フィルムを提供する。本発明によると、薄膜でも所望の位相差を発現することができる位相差フィルムが提供できる。
Description
本発明は、位相差フィルム、偏光板および液晶表示装置に関する。さらに詳しくは、湿度変動による位相差バラツキが小さくかつ強度の高い位相差フィルム、ならびにそれを具備してなる偏光板および液晶表示装置に関する。
液晶表示装置は、液晶テレビやパソコンの液晶ディスプレイ等の用途で、需要が拡大している。通常、液晶表示装置は、透明電極、液晶層、カラーフィルタ等をガラス板で挟み込んだ液晶セルと、その両側に設けられた2枚の偏光板で構成されており、それぞれの偏光板は、偏光子(偏光膜、偏光フィルムともいう)を2枚のフィルム(偏光板用保護フィルム、位相差フィルム)で挟んだ構成となっている。このうち、上記偏光板用保護フィルムとしては、高い透明性を有し、偏光子に使用されるポリビニルアルコールとの密着性を容易に確保できる、セルロースエステルフィルムが広く使用されてきた。
一方、液晶表示装置は、表示画像のコントラストの向上と共に、偏光板部材の薄型化や軽量化が求められている。しかし、特に位相差フィルムにおいては、単純に膜厚を薄くすると、位相差値が小さくなり、所望の位相差が達成できないという問題があった。位相差を出すには、位相差上昇剤を使用する技術が広く知られている。位相差上昇剤(レターデーション上昇剤)を使用する技術としては、例えば、特定のエポキシ化合物をセルロースエステル樹脂に含有させる技術(特許文献1)や特定のエポキシエステル化合物をセルロースエステル樹脂に含有させる技術(特許文献2)のような技術を用いることが提案されている。
しかしながら、本願発明者らが上記特許文献1や2の技術を確認したところ、湿度による位相差変動が大きく、液晶表示装置にしたときに色ムラが発生するという問題があることが分かった。また、上記特許文献1や2に記載される位相差フィルムは、膜厚を薄くしすぎると、引き裂き強度が劣化し、搬送時に裂けてしまうなどの別の問題も生じた。
したがって、本発明は、上記事情を鑑みてなされたものであり、薄膜でも所望の位相差を発現することができる位相差フィルムを提供することを目的とする。
本発明の他の目的は、位相差値の湿度による変動が少ない位相差フィルムを提供することである。
本発明のさらなる他の目的は、強度に優れる位相差フィルムを提供することである。
本発明者らは、上記の問題を解決すべく、鋭意研究を行った結果、セルロースエステル樹脂に、エポキシ化合物またはエポキシエステル化合物と5員または6員の芳香族複素環基を少なくとも1種有する化合物とを配合することによって、上記課題を解決できることを見出し、本発明を完成するに至った。
すなわち、上記諸目的は、セルロースエステル樹脂と、下記一般式(1):

式中、R1~R4は、それぞれ独立して、水素原子または炭素数1~3のアルキル基を表し;ならびにR5およびR6は、それぞれ独立して、置換基を有するアルキル基またはグリシジル基を表し、この際、前記置換基は、ヒドロキシル基、エステル基および芳香族基からなる群より選択される少なくとも一種である、
で表わされる化合物の少なくとも1種と、
下記一般式(2):
で表わされる化合物の少なくとも1種と、
下記一般式(2):

式中、A1及びA2は、それぞれ独立して、アルキル基、シクロアルキル基、芳香族炭化水素環基または芳香族複素環基を表し;Bは、芳香族炭化水素環または芳香族複素環を表し;T1及びT2は、それぞれ独立して、2価の1,2,4-トリアゾール環基を表し;L1、L2、L3及びL4は、それぞれ独立して、単結合または2価の連結基を表し;ならびにnは、0~5の整数を表す、
で表される化合物の少なくとも1種と、を含有することを特徴とする位相差フィルムによって達成される。
で表される化合物の少なくとも1種と、を含有することを特徴とする位相差フィルムによって達成される。
本発明によれば、薄膜でも所望の位相差を発現することができる位相差フィルムが提供されうる。また、本発明の位相差フィルムは、位相差値の湿度による変動が少ない。
本発明の一形態によれば、セルロースエステル樹脂と、下記一般式(1):

式中、R1~R4は、それぞれ独立して、水素原子または炭素数1~3のアルキル基を表し;ならびにR5およびR6は、それぞれ独立して、置換基を有するアルキル基またはグリシジル基を表し、この際、前記置換基は、ヒドロキシル基、エステル基および芳香族基からなる群より選択される少なくとも一種である、
で表わされる化合物の少なくとも1種と、
下記一般式(2):
で表わされる化合物の少なくとも1種と、
下記一般式(2):
式中、A1及びA2は、それぞれ独立して、アルキル基、シクロアルキル基、芳香族炭化水素環基または芳香族複素環基を表し;Bは、芳香族炭化水素環または芳香族複素環を表し;T1及びT2は、それぞれ独立して、2価の1,2,4-トリアゾール環基を表し;L1、L2、L3及びL4は、それぞれ独立して、単結合または2価の連結基を表し;ならびにnは、0~5の整数を表す、
で表される化合物の少なくとも1種と、を含有することを特徴とする位相差フィルム(以下、単に「フィルム」ともいう)が提供される。
で表される化合物の少なくとも1種と、を含有することを特徴とする位相差フィルム(以下、単に「フィルム」ともいう)が提供される。
本発明の位相差フィルムは、一般式(1)の化合物及び一般式(2)の化合物双方を含むことを特徴とする。当該構成により、本発明の位相差フィルムは、良好な引き裂き強度及び位相差湿度変動を両立できる。当該効果を奏するメカニズムは、不明であるが、以下のように推測される。なお、本発明は、下記推測によって限定されるものではない。すなわち、一般式(1)の化合物及び一般式(2)の化合物両方を含ませることによって、セルロース樹脂の側鎖や水素原子との相互作用がよりいっそう強まり、セルロース樹脂の側鎖や水素原子に水が配位しにくくなる。このため、得られる位相差フィルムの位相差湿度変動を抑制できると推測される。また、このようによりいっそう相互作用を強めることができるため、フィルムが強靭になり、薄膜にしても、引き裂き強度が劣化されない。ゆえに、本発明の位相差フィルムは、位相差湿度変動と引き裂き強度を両立できると推測される。
したがって、本発明の位相差フィルムは、薄膜形態でも所望の位相差を発現することができ、また、位相差値の湿度による変動が少ない。
以下、本発明の実施の形態を説明する。なお、本発明は、以下の実施の形態のみには限定されない。また、図面の寸法比率は、説明の都合上誇張されており、実際の比率とは異なる場合がある。
また、本明細書において、範囲を示す「X~Y」は「X以上Y以下」を意味し、「重量」と「質量」、「質量%」と「重量%」及び「重量部」と「重量部」は同義語として扱う。また、特記しない限り、操作および物性等の測定は室温(20~25℃)/相対湿度40~50%の条件で測定する。
[位相差フィルム]
本発明において、「位相差フィルム」とはX軸方向とY軸方向とで屈折率が異なるフィルムをいい、視野角を拡大する光学補償フィルムである。好ましくは、レターデーション値として下記条件1および条件2を満たすフィルムである。かかるフィルムは高いレターデーション値の発現性を有し、垂直配向型液晶表示装置の視野角拡大に好適である。
本発明において、「位相差フィルム」とはX軸方向とY軸方向とで屈折率が異なるフィルムをいい、視野角を拡大する光学補償フィルムである。好ましくは、レターデーション値として下記条件1および条件2を満たすフィルムである。かかるフィルムは高いレターデーション値の発現性を有し、垂直配向型液晶表示装置の視野角拡大に好適である。
条件1:温度23℃、相対湿度55%の環境下で、波長590nmで測定した下式(I)で表される面内レターデーション値Roが20~130nmの範囲内である。
条件2:温度23℃、相対湿度55%の環境下で、波長590nmで測定した下式(II)で表される厚さ方向のレターデーション値Rtが、100~300nmの範囲内である。
なお、上記式(I)及び(II)において、nxは、フィルムの面内における遅相軸方向の屈折率を表し;nyは、フィルムの面内における遅相軸に直交する方向の屈折率を表し;nzは、フィルムの厚さ方向の屈折率を表し;およびdは、フィルムの厚み(nm)を表す。
これらのレターデーション値(屈折率)は、自動複屈折計KOBRA-21ADH(王子計測機器)を用いて測定することができる。
所望のレターデーション値は、フィルム作製時の延伸倍率やレターデーション上昇剤の添加量、セルロースエステルのアシル基の種類および置換度、膜厚などを制御することで調整することができる。
本形態の位相差フィルムは、下記式(1)で表されるRt湿度変動が1~12%を満たすことが好ましい。下記Rt湿度変動の値が小さい方が、湿度変動に対して安定であることを示しており、かかる観点からRt湿度変動の値は12%以下である。一方、Rt湿度変動が1%未満である場合には、位相差フィルムの透湿性が小さすぎて、保護フィルムと偏光子とに水が偏り、負荷がかかる。そのため保護フィルムや偏光子の剥がれが生じたり、水抜けが悪くなり表示装置の色味が赤くなったりするなどの問題が生じるおそれがある。

上記範囲を満足する位相差フィルムは、湿度変動によるフィルムの厚さ方向のレターデーション値の変動が小さく、当該フィルムを具備した耐湿熱性に優れた偏光板、および液晶表示装置(以下、単に「表示装置」ともいう)を提供することができる。
フィルムの湿度による位相差変動から起こる色ムラを一層向上させるために、Rt湿度変動は1~10%であることがより好ましく、1~8%であることがさらに好ましい。
上記Rt湿度変動は、セルロースエステルのアシル基の種類および置換度、レターデーション上昇剤の種類および添加量、可塑剤の種類および添加量などを調節することで所望の範囲に制御することができる。
位相差フィルムの膜厚(乾燥膜厚)或は、特に制限されないが、10~40μmであることが好ましい。このような膜厚であれば、均質に製膜でき、大面積とした場合であっても色ムラを有効に抑制でき、十分な位相差を達成できる。また、均質な製膜、Rt湿度変動の観点から、位相差フィルムの膜厚(乾燥膜厚)は、より好ましくは20~35μmの範囲であり、特に好ましくは25~30μmである。上記膜厚は、製膜の際に流延させるドープや溶融物の厚さ、および/または、延伸条件を調整することにより上記所望の範囲に制御することができる。
また、本形態に係る位相差フィルムは幅手方向および長尺方向の膜厚バラツキがいずれも0~4μmである。かかる場合には、大面積のフィルムあってもフィルム面内の位相差バラツキが抑制され、色ムラを防止することができる。位相差バラツキを一層抑制するために、膜厚バラツキは好ましくは0~2.5μmであり、より好ましくは0~1.5μmである。
なお、位相差フィルムの膜厚はマイクロメータなどの膜厚計を用いて測定することができる。具体的には、フィルムの巾方向に、10mm間隔で100箇所以上の点で膜厚(μm)を測定し、これらの平均値をフィルムの膜厚(μm)とする。また、膜厚の最大値と最小値との差を膜厚バラツキ(μm)とする。
上記膜厚バラツキは、セルロースエステル樹脂のアシル基の種類および置換度、レターデーション上昇剤の種類および添加量、可塑剤の種類および添加量などを調節することで所望の範囲に制御することができる。中でも、セルロースエステル樹脂のアシル基の種類および置換度を所定の範囲とすることにより、製膜の際の流延性および延伸性を制御でき、均一な膜厚とすることが可能である。
一般に、フィルムの位相差の出し方は、(1)セルロースエステル(酢綿)で位相差を出す;(2)位相差上昇剤を添加することにより位相差を出す;などの方法がある。前者の場合にはセルロースが透湿性を有するために湿度によりRtが変動する。一方、後者の場合には、湿度によってRtが変動しにくいため好ましい。さらに、上昇剤を添加することにより、位相差発現性が向上するため、薄膜化が可能となる。ただし、上昇剤の添加量が多すぎるとヘイズが悪化するおそれがあり、好ましくない。
可塑剤を添加することにより、フィルムに可塑性が付与され、延伸時にフィルム全体に応力がかかりやすくなって、膜厚バラツキが小さくなる(良好となる)。さらに、フィルム内が疎水化されて水を寄せ付けないためRt湿度変動が低下する。ただし、可塑剤が多すぎるとヘイズが悪化するおそれがあり、好ましくない。
フィルムの膜厚が薄いほど全体の水分量が減少するため、Rt湿度変動が低下する。ただし、フィルムの膜厚が薄すぎると均質な製膜が困難であり、膜厚バラツキが大きくなる(悪化する)。
セルロースエステルのアシル基の置換度が小さいほど位相差発現性が向上するため、薄膜化が可能となる。一方で、アシル基の置換度が小さすぎると、耐久性が悪化するおそれがあり好ましくない。
一方、セルロースエステルのアシル基の置換度が大きいほど位相差が発現しないため、製膜の際に延伸倍率を増加させる必要があるが、高延伸倍率で均一に延伸させることは難しく、このため、膜厚バラツキが大きくなる(悪化する)。また、位相差の湿度変動はセルロースのカルボニル基に水分子が配位することで生じると、推測される。このため、アシル基の置換度が高い、すなわち、セルロース中のカルボニル基が多いほど、Rt湿度変動が悪くなる傾向がある。
本発明では、上記知見に基づき、(a)ドープや溶融物の流延条件(フィルムの膜厚)、(b)延伸条件、(c)セルロースエステルのアシル基の種類および置換度、(d)レターデーション上昇剤の種類および添加量、(e)可塑剤の種類および添加量を制御することにより、(1)膜厚、(2)膜厚バラツキ、(3)Rt湿度変動を所望の範囲とすることができる。
また、本形態の位相差フィルムはEZコントラストで測定した水浸漬部のL*/非浸漬部のL*が1.05以上1.80以下であることが好ましい。かかる場合には、大面積であっても色ムラが抑制された位相差フィルムが得られる。水浸漬部のL*/非浸漬部のL*は、色ムラを一層抑制する点から、より好ましくは1.05以上1.55以下であり、さらに好ましくは1.05以上1.30以下である。なお、水浸漬部のL*/非浸漬部のL*の評価は下記の手順で行う。
(水浸漬部のL*/非浸漬部のL*の評価)
市販のVA型液晶表示装置(SONY製、KDL 40EX720)を用い、液晶セルの表側偏光板を剥離し、本形態の位相差フィルムを用いて構成した評価用偏光板を基材レス両面テープで貼り付ける。液晶表示装置を寝かせて台の上などに置き、ベンコット(旭化成せんい社製)を評価用偏光板の一部に載せて水を含ませる。ベンコットが乾かないよう100μmPETで覆い、テレビにPCから黒表示の信号を入力、テレビの電源ONで24時間放置する(室温は23℃に設定、パネル温度は38℃)。24時間後、ベンコットを取り除く。ベンコットのあった部分のL*を水浸漬部のL*としてEZコントラスト(ELDIM社製)で測定する。ベンコットのない部分のL*を非浸漬部のL*としてEZコントラストで測定する。なお、EZコントラストでの測定はTVを黒表示にしてカラーモードにて行う。水浸漬の条件はパネルの電源をONにし、かつ水を十分に浸み込ませたベンコットを貼り付けた状態で24時間置くものとする。
市販のVA型液晶表示装置(SONY製、KDL 40EX720)を用い、液晶セルの表側偏光板を剥離し、本形態の位相差フィルムを用いて構成した評価用偏光板を基材レス両面テープで貼り付ける。液晶表示装置を寝かせて台の上などに置き、ベンコット(旭化成せんい社製)を評価用偏光板の一部に載せて水を含ませる。ベンコットが乾かないよう100μmPETで覆い、テレビにPCから黒表示の信号を入力、テレビの電源ONで24時間放置する(室温は23℃に設定、パネル温度は38℃)。24時間後、ベンコットを取り除く。ベンコットのあった部分のL*を水浸漬部のL*としてEZコントラスト(ELDIM社製)で測定する。ベンコットのない部分のL*を非浸漬部のL*としてEZコントラストで測定する。なお、EZコントラストでの測定はTVを黒表示にしてカラーモードにて行う。水浸漬の条件はパネルの電源をONにし、かつ水を十分に浸み込ませたベンコットを貼り付けた状態で24時間置くものとする。
本形態に係る位相差フィルムは、セルロースエステルを主成分とし、必要に応じて、(a)レターデーション上昇剤、(b)可塑剤、(c)水素結合性化合物、および(d)他の任意成分等のその他の添加剤をさらに含んで構成される。なお、本発明のフィルムにおいて「主成分」とは、フィルム全体の50重量%以上を占める成分であることをいい、好ましくは60重量%以上、より好ましくは70重量%以上(上限:100重量%)である。
(セルロースエステル樹脂)
セルロースエステル樹脂(セルロースエステル)は、セルロースを構成するβ-1,4結合しているグルコース単位中の2位、3位および6位の水酸基(-OH)の水素原子の一部または全部がアシル基で置換されたセルロースアシレート樹脂を意味する。
セルロースエステル樹脂(セルロースエステル)は、セルロースを構成するβ-1,4結合しているグルコース単位中の2位、3位および6位の水酸基(-OH)の水素原子の一部または全部がアシル基で置換されたセルロースアシレート樹脂を意味する。
本形態のフィルムに含まれるセルロースエステル樹脂(セルロースエステル)は、特に限定されないが、炭素数2~22程度の直鎖または分岐のカルボン酸のエステルであることが好ましい。エステルを構成するカルボン酸は脂肪族カルボン酸でもよいし、環を形成してもよく、芳香族カルボン酸でもよい。例えば、セルロースの水酸基部分の水素原子が、アセチル基、プロピオニル基、ブチリル基、イソブチリル基、バレリル基、ピバロイル基、ヘキサノイル基、オクタノイル基、ラウロイル基、ステアロイル等の炭素数2~22のアシル基で置換されたセルロースエステルが挙げられる。エステルを構成するカルボン酸(アシル基)は、置換基を有してもよい。エステルを構成するカルボン酸は、特に炭素数が2~6の低級脂肪酸であることが好ましく、炭素数が2~4の低級脂肪酸であることがより好ましく、炭素数が2または3の低級脂肪酸であることがさらに好ましい。なお、セルロースエステル中のアシル基は単一種であってもよいし、複数のアシル基の組み合わせであってもよい。
好ましいセルロースエステルの具体例には、セルロースアセテート(DAC、TAC)のほか、セルロースアセテートプロピオネート(CAP)、セルロースアセテートブチレート、セルロースアセテートプロピオネートブチレートのようなアセチル基の他にプロピオネート基またはブチレート基が結合したセルロースの混合脂肪酸エステルが挙げられる。好ましくはセルロースアセテート、セルロースアセテートブチレートまたはセルロースアセテートプロピオネートである。なお、セルロースエステルに含まれうるブチリル基は、直鎖状であっても分岐状であってもよい。また、これらのセルロースエステルは単一種を使用してもよいし、複数種を組み合わせて用いてもよい。
セルロースエステルのアシル基の総置換度(総アシル基置換度)は、1.0~3.0程度としうる。アシル基の総置換度は、透湿性を低くする観点からは、2.0~2.95、より好ましくは2.1~2.5の範囲内であることが好ましい。または、製膜の際の流延性および延伸性を向上させ、膜厚の均一性が一層向上する観点からは、セルロースエステルのアシル基の総置換度は、2.15~2.35であることが好ましい。セルロースエステルのアシル基の置換度は、ASTM-D817-96に規定の方法で測定することができる。
より具体的には、セルロースエステルは、下記式(a)および(b)をともに満足する。式中、Xは、アセチル基の置換度を表し、Yは、プロピオニル基若しくはブチリル基の置換度またはそれらの混合物の置換度を表す。
セルロースエステルは、セルロースアセテート(Y=0)、および、セルロースアセテートプロピオネート(CAP)(Y;プロピオニル基、Y>0)またはセルロースアセテートブチレート(Y;ブチリル基、Y>0)がより好ましく、さらに好ましくは膜厚バラツキを低減させる点からY=0であるセルロースアセテートである。特に好ましく用いられるセルロースアセテートは、位相差発現性、Rt湿度変動、膜厚バラツキを所望の範囲とする点から、好ましくは2.1≦X≦2.9、より好ましくは2.2≦X≦2.5のセルロースジアセテート(DAC)である。また、Y>0の場合には、特に好ましく用いられるセルロースアセテートプロピオネート(CAP)またはセルロースアセテートブチレートは、0.95≦X≦2.25、0.1≦Y≦1.2、2.15≦X+Y≦2.65である。
上述のセルロースアセテート、セルロースアセテートプロピオネート、セルロースアセテートブチレート(より好ましくは、セルロースアセテート、セルロースアセテートプロピオネート)を用いることで、レターデーションに優れ、機械強度、環境変動に優れた位相差フィルムが得られる。
なお、アシル基の置換度は、1グルコース単位あたりのアシル基の平均数を示し、1グルコース単位の2位、3位および6位の水酸基の水素原子のいくつがアシル基に置換されているかを示す。従って、最大の置換度は3.0であり、この場合には2位、3位および6位の水酸基の水素原子がすべてアシル基で置換されていることを意味する。これらアシル基は、グルコース単位の2位、3位、6位に平均的に置換していてもよいし、分布をもって置換していてもよい。置換度は、ASTM-D817-96に規定の方法により求められる。
所望の光学特性を得るために置換度の異なるセルロースアセテートを混合して用いてもよい。異なるセルロースアセテートの混合比は特に限定されない。
セルロース誘導体の数平均分子量は、得られるフィルムの機械的強度を高めるためには、4×104~3×105の範囲であることが好ましく、4.5×104~2×105の範囲であることがより好ましく、5×104~7×104の範囲であることが特に好ましい。本明細書において、「重量平均分子量(Mw)」及び「数平均分子量(Mn)」は、ゲルパーミエーションクロマトグラフィー(GPC)を用いて測定される値である。測定条件は以下のとおりである。

セルロース誘導体中の残留硫酸の含有量は、硫黄元素換算で0.1~45重量ppmの範囲であることが好ましく、1~30重量ppmの範囲がより好ましい。硫酸は、塩の状態でフィルムに残留していると考えられる。残留硫酸の含有量が45重量ppmを超えると、フィルムを熱延伸する際や、熱延伸後にスリッティングする際に破断しやすくなる。残留硫酸の含有量は、ASTM D817-96に規定の方法により測定することができる。
セルロース誘導体中の遊離酸の含有量は、1~500重量ppmの範囲内であることが好ましく、1~100重量ppmであることがより好ましく、1~70重量ppmの範囲内であることがさらに好ましい。遊離酸の含有量が上記範囲であると、前述と同様に、フィルムを熱延伸する際や、熱延伸後にスリッティングする際に破断しにくい。遊離酸の含有量はASTM D817-96に規定の方法により測定することができる。
セルロース誘導体は、微量の金属成分を含有することがある。微量の金属成分は、セルロース誘導体の合成工程で用いられる水に由来すると考えられる。これらの金属成分のように、不溶性の核となりうるような成分の含有量はできるだけ少ないことが好ましい。特に鉄、カルシウム、マグネシウム等の金属イオンは、有機の酸性基を含んでいる可能性のある樹脂分解物等と塩形成して不溶物を形成する場合がある。また、カルシウム(Ca)成分は、カルボン酸やスルホン酸等の酸性成分と、また多くの配位子と配位化合物(すなわち、錯体)を形成しやすく、多くの不溶なカルシウムに由来するスカム(不溶性の澱、濁り)を形成するおそれがある。
具体的には、セルロース誘導体中の鉄(Fe)成分の含有量は、1重量ppm以下であることが好ましい。また、セルロース誘導体中のカルシウム(Ca)成分の含有量は、好ましくは60重量ppm以下であり、より好ましくは0~30重量ppmの範囲内である。セルロース誘導体中のマグネシウム(Mg)成分の含有量は、0~70重量ppmの範囲内であることが好ましく、特に0~20重量ppmの範囲内であることが好ましい。
鉄(Fe)成分、カルシウム(Ca)成分、及びマグネシウム(Mg)成分などの金属成分の含有量は、絶乾したセルロース誘導体をマイクロダイジェスト湿式分解装置(硫硝酸分解)、アルカリ溶融で前処理を行った後、ICP-AES(誘導結合プラズマ発光分光分析装置)を用いて測定することができる。
残留アルカリ土類金属、残留硫酸及び残留酸の含有量は、合成して得られるセルロース誘導体を、十分に洗浄することによって調整することができる。
セルロースアセテート、セルロースアセテートプロピオネートなどのセルロースエステルは、公知の方法により製造することができる。一般的には、原料のセルロースと所定の有機酸(酢酸、プロピオン酸など)と酸無水物(無水酢酸、無水プロピオン酸など)、触媒(硫酸など)と混合して、セルロースをエステル化し、セルロースのトリエステルができるまで反応を進める。トリエステルにおいてはグルコース単位の三個のヒドロキシ基(水酸基)は、有機酸のアシル酸で置換されている。同時に二種類の有機酸を使用すると、混合エステル型のセルロースエステル、例えばセルロースアセテートプロピオネートやセルロースアセテートブチレートを作製することができる。次いで、セルロースのトリエステルを加水分解することで、所望のアシル置換度を有するセルロースエステル樹脂を合成する。その後、濾過、沈殿、水洗、脱水、乾燥などの工程を経て、セルロースエステル樹脂ができあがる。具体的には特開平10-45804号公報に記載の方法を参考にして合成することができる。
セルロースエステルは市販品を使用してもよい。市販品としては、ダイセル社L20、L30、L40、L50、イーストマンケミカル社のCa398-3、Ca398-6、Ca398-10、Ca398-30、Ca394-60S等が挙げられる。
(一般式(1)の化合物)
本発明の位相差フィルムは、下記一般式(1)の化合物を含む。下記一般式(1)の化合物は、レターデーション上昇剤(調整剤)として作用する。このため、セルロースエステル樹脂に配合することにより、レターデーション値(特にフィルムの厚さ方向のレターデーション値)を向上し、透湿性を低減できる。また、下記一般式(1)の化合物は、高温多湿下でも揮発性が低い。このため、位相差フィルムの耐ブリード性、ゆえに映像の鮮明度を向上できる。
本発明の位相差フィルムは、下記一般式(1)の化合物を含む。下記一般式(1)の化合物は、レターデーション上昇剤(調整剤)として作用する。このため、セルロースエステル樹脂に配合することにより、レターデーション値(特にフィルムの厚さ方向のレターデーション値)を向上し、透湿性を低減できる。また、下記一般式(1)の化合物は、高温多湿下でも揮発性が低い。このため、位相差フィルムの耐ブリード性、ゆえに映像の鮮明度を向上できる。

上記一般式(1)において、R1~R4は、水素原子または炭素数1~3のアルキル基を表す。ここで、R1~R4は、それぞれ同じであってもあるいは相互に異なるものであってもよい。上記炭素数1~3のアルキル基としては、メチル基、エチル基、プロピル基及びイソプロピル基があるが、これらのうち、レターデーション値(特にフィルムの厚さ方向のレターデーション値)を向上効果、セルロースエステルとの相溶性などの観点から、水素原子、メチル基、エチル基が好ましく、メチル基が特に好ましい。
R5およびR6は、置換基を有するアルキル基またはグリシジル基を表す。この際、R5およびR6は、同じであってもあるいは異なるものであってもよい。また、前記置換基は、ヒドロキシル基(-OH)、エステル基および芳香族基からなる群より選択される少なくとも一種である。ここで、エステル基は、式:-O-C(=O)-Rまたは-C(=O)-O-Rで表され、ここで、Rは、炭素数1~8の直鎖または分岐鎖のアルキル基または芳香族基である。上記アルキル基及び芳香族基は下記定義と同様である。なお、R5およびR6は、それぞれ同じであってもあるいは相互に異なるものであってもよい。
R5およびR6としてのアルキル基としては、特に制限されないが、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、ヘキシル基、ヘプチル基及びオクチル基等の、炭素数1~8の直鎖または分岐鎖のアルキル基であることが好ましい。これらのうち、炭素数1~5のアルキル基が好ましく、炭素数2~4のアルキル基が好ましい。
芳香族基としては、炭素数6~24のアリール基でありうる。より具体的には、フェニル基、p-トリル基、ナフチル基、ビフェニル基、フルオレニル基、アンスリル基、ピレニル基、アズレニル基、アセナフチレニル基、ターフェニル基、フェナンスリル基などが挙げられる。これらのうち、フェニル基、ナフチル基が好ましく、フェニル基がより好ましい。また、上記芳香族基は、置換基を有していてもよい。ここで、置換基としては、特に制限されないが、例えば、炭素原子数1~3のアルキル基、炭素原子数1~3のアルコキシ基、フェニル基、メチルフェニル基、フェニルフェニル基、メチルフェニルフェニル基、シアノ基、ハロゲン原子(フッ素原子、塩素原子、臭素原子、ヨウ素原子)、ニトロ基等が挙げられる。また、上記置換基は、1個でもあるいは2個以上であってもよく、後者の場合には、各置換基は同じであってもあるいは異なるものであってもよい。これらのうち、レターデーション値(特にフィルムの厚さ方向のレターデーション値)を向上効果、セルロースエステルとの相溶性などの観点から、芳香族基は、フェニル基、メチルフェニル基、メチルフェニルフェニル基であることが好ましい。
R5およびR6が置換基を有するアルキル基である場合の一般式(1)の化合物の製造方法は特に制限されない。具体的には、当該化合物は、エポキシ化合物と芳香族モノカルボン酸とを反応させることにより得ることができる。前記エポキシ化合物としては、ビフェノール類とエピクロルヒドリンとの反応によって得られるジグリシジルエーテル型のエポキシ化合物が挙げられる。このエポキシ化合物の具体的な例として、3,3’,5,5’-テトラメチル-4,4’-ジグリシジルオキシビフェニル(市販品では、ジャパンエポキシレジン株式会社製「jER YX-4000」(エポキシ当量180~192))等のビフェノール型エポキシ化合物を使用できる。
また、前記芳香族モノカルボン酸としては、例えば、安息香酸、ジメチル安息香酸、トリメチル安息香酸、テトラメチル安息香酸、エチル安息香酸、プロピル安息香酸、クミン酸、o-トルイル酸、m-トルイル酸、p-トルイル酸、アニス酸、エトキシ安息香酸、プロポキシ安息香酸、シアノ安息香酸、フルオロ安息香酸、ニトロ安息香酸、4-フェニル安息香酸、4-(3-メチルフェニル)安息香酸、4-(4-メチルフェニル)安息香酸、4-(3,5-ジメチルフェニル)安息香酸、2-メチル-4-フェニル安息香酸、2,6-ジメチル-4-フェニル安息香酸、2,6-ジメチル-4-(3,5-ジメチルフェニル)安息香酸、ナフトエ酸、ニコチン酸、フロ酸、1-ナフタレンカルボン酸、2-ナフタレンカルボン酸等が挙げられる。これらの芳香族モノカルボン酸は、単独で用いることも2種以上併用することもできる。
上記反応において、エポキシ化合物のエポキシ基と芳香族モノカルボン酸のカルボキシル基とが反応して、一般式(1)の化合物が合成される。ここで、上記反応条件は上記反応が進行する条件であれば特に制限されない。例えば、反応温度は、80~130℃、より好ましくは100℃~115℃である。反応時間は、10~25時間であることが好ましい。また、記エポキシ化合物と前記芳香族モノカルボン酸との混合比(仕込み比)は、上記反応が進行する条件であれば特に制限されない。例えば、エポキシ化合物のエポキシ基のモル数と、芳香族モノカルボン酸のモル数の比(エポキシ基モル数)/(芳香族モノカルボン酸のモル数)が、1/0.9~1.0の範囲であることが好ましい。
また、上記反応において、必要に応じて触媒を用いてもよい。この触媒としては、例えば、トリメチルホスフィン、トリエチルホスフィン、トリブチルホスフィン、トリオクチルホスフィン、トリフェニルホスフィン等のホスフィン化合物;2-メチルイミダゾール、2-エチルイミダゾール、2-イソプロピルイミダゾール、2-エチル-4-メチルイミダゾール、4-フェニル-2-メチルイミダゾール等のイミダゾール系化合物;トリエチルアミン、トリブチルアミン、トリヘキシルアミン、トリアミルアミン、トリエタノールアミン、ジメチルアミノエタノール、トリアチレンジアミン、ジメチルフェニルアミン、ジメチルベンジルアミン、2-(ジメチルアミノメチル)フェノール、1,8-ジアザビシクロ(5,4,0)ウンデセン-7等のアミン化合物;ジメチルアミノピリジン等のピリジン化合物などが挙げられる。これらの触媒は、前記エポキシ化合物及び前記芳香族モノカルボン酸の合計100重量部に対して、0.05~1重量部の量で使用されることが好ましい。
これらのうち、R5およびR6としては、置換基としてヒドロキシル基及びエステル基を有するアルキル基、またはグリシジル基であることが好ましい。また、一般式(1)の化合物としては、特開2011-140637号公報及び特開2011-116912号公報に記載の化合物が一般式(1)の化合物に包含される。より具体的には、一般式(1)の化合物のより好ましい例としては、下記が挙げられる。なお、下記番号にて、化合物を規定する。すなわち、下記(1-1)の化合物を「化合物(1-1)」とも称する。

本発明の位相差フィルムにおいて、一般式(1)の化合物の含有量は、特に制限されない。一般式(1)の化合物の含有量は、例えば、セルロースエステル樹脂100重量部に対して、好ましくは1~30重量部、より好ましくは2~20重量部、特に好ましくは5~10重量部である。このような量であれば、位相差フィルムの位相差機能及び引き裂き強度を向上できる。また、高温多湿下での揮発性を低減するため、位相差フィルムの耐ブリード性、ゆえに映像の鮮明度を向上できる。
また、前記一般式(1)で表される化合物の添加方法としては、位相差フィルムを形成する樹脂に粉体で添加しても良く、溶媒に溶解した後、位相差フィルムを形成する樹脂に添加しても良い。
(一般式(2)の化合物)
本発明の位相差フィルムは、5員または6員の芳香族複素環基を少なくとも1種有する下記一般式(2)の化合物を含む。
本発明の位相差フィルムは、5員または6員の芳香族複素環基を少なくとも1種有する下記一般式(2)の化合物を含む。

前記一般式(2)において、A1及びA2は、それぞれ独立して、アルキル基、シクロアルキル基、芳香族炭化水素環基または芳香族複素環基を表す。ここで、A1及びA2は、同じであっても異なるものであってもよい。また、A2が複数存在する(nが2~5である)場合には、各A2は、同じであっても異なるものであってもよい。
アルキル基としては、特に制限されないが、炭素数1~24の直鎖または分岐鎖のアルキル基でありうる。より具体的には、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、イソペンチル基、tert-ペンチル基、ネオペンチル基、1,2-ジメチルプロピル基、n-ヘキシル基、イソヘキシル基、1,3-ジメチルブチル基、1-イソプロピルプロピル基、1,2-ジメチルブチル基、n-ヘプチル基、1,4-ジメチルペンチル基、3-エチルペンチル基、2-メチル-1-イソプロピルプロピル基、1-エチル-3-メチルブチル基、n-オクチル基、2-エチルヘキシル基、3-メチル-1-イソプロピルブチル基、2-メチル-1-イソプロピル基、1-t-ブチル-2-メチルプロピル基、n-ノニル基、3,5,5-トリメチルヘキシル基、n-デシル基、イソデシル基、n-ウンデシル基、1-メチルデシル基、n-ドデシル基、n-トリデシル基、n-テトラデシル基、n-ペンタデシル基、n-ヘキサデシル基、n-ヘプタデシル基、n-オクタデシル基、n-ノナデシル基、n-エイコシル基、n-ヘンエイコシル基、n-ドコシル基、n-トリコシル基、n-テトラコシル基などが挙げられる。これらのうち、炭素数1~8の直鎖もしくは分岐状のアルキル基(メチル基、エチル基、n-プロピル基、イソプロピル基、tert-ブチル基、n-オクチル基、2-エチヘキシル基等)が好ましい。
また、シクロアルキル基としては、特に制限されないが、炭素数3~24のシクロアルキル基でありうる。より具体的には、シクロプロピル基、シクロペンチル基、シクロヘキシル基、4-n-ドデシルシクロヘキシル基などが挙げられる。
芳香族炭化水素環基としては、特に制限されないが、炭素数6~24のアリール基でありうる。より具体的には、フェニル基、p-トリル基、ナフチル基、ビフェニル基、フルオレニル基、アンスリル基、ピレニル基、アズレニル基、アセナフチレニル基、ターフェニル基、フェナンスリル基などが挙げられる。これらのうち、フェニル基、p-トリル基、ナフチル基が好ましい。
芳香族複素環基としては、特に制限されないが、ピリジル基、ピリミジニル基(例えば、2-ピリミジニル基)、ピロール基(例えば、2-ピロール基)、ピラゾリノン基、ピリジノン基、フリル基(例えば、2-フリル基)、ピロリル基、イミダゾリル基、ベンゾイミダゾリル基、ピラゾリル基、ピラジニル基、トリアゾリル基(例えば、1,2,4-トリアゾール-1-イル基、1,2,3-トリアゾール-1-イル基等)、オキサゾリル基、ベンゾオキサゾリル基、チアゾリル基、イソオキサゾリル基、イソチアゾリル基、ベンゾチアゾリル基(例えば、2-ベンゾチアゾリル基)、フラザニル基、チエニル基(例えば、2-チエニル基)、キノリル基、ベンゾフリル基、ジベンゾフリル基、ベンゾチエニル基、ジベンゾチエニル基、インドリル基、カルバゾリル基、カルボリニル基、ジアザカルバゾリル基(カルボリン環を構成する炭素原子の一つが窒素原子で置き換わったものを示す)、キノキサリニル基、ピリダジニル基、トリアジニル基、キナゾリニル基、フタラジニル基などが挙げられる。これらのうち、2-ピロール基、2-フリル基、2-チエニル基、ピロール基、イミダゾリル基、オキサゾリル基、チアゾリル基、ベンゾイミダゾリル基、ベンゾオキサゾリル基、2-ベンゾチアゾリル基、ピラゾリノン基、ピリジル基、ピリジノン基、2-ピリミジニル基が好ましい。
A1及びA2で表されるアルキル基、シクロアルキル基、芳香族炭化水素環基、芳香族複素環基は置換基を有していてもよく、該置換基としては、例えば、ハロゲン原子(フッ素原子、塩素原子、臭素原子、ヨウ素原子等)、アルキル基(メチル基、エチル基、n-プロピル基、イソプロピル基、tert-ブチル基、n-オクチル基、2-エチルヘキシル基等)、シクロアルキル基(シクロヘキシル基、シクロペンチル基、4-n-ドデシルシクロヘキシル基等)、アルケニル基(ビニル基、アリル基等)、シクロアルケニル基(2-シクロペンテン-1-イル、2-シクロヘキセン-1-イル基等)、アルキニル基(エチニル基、プロパルギル基等)、芳香族炭化水素環基(フェニル基、p-トリル基、ナフチル基等)、芳香族複素環基(2-ピロール基、2-フリル基、2-チエニル基、ピロール基、イミダゾリル基、オキサゾリル基、チアゾリル基、ベンゾイミダゾリル基、ベンゾオキサゾリル基、2-ベンゾチアゾリル基、ピラゾリノン基、ピリジル基、ピリジノン基、2-ピリミジニル基、トリアジン基、ピラゾール基、1,2,3-トリアゾール基、1,2,4-トリアゾール基、オキサゾール基、イソオキサゾール基、1,2,4-オキサジアゾール基、1,3,4-オキサジアゾール基、チアゾール基、イソチアゾール基、1,2,4-チオジアゾール基、1,3,4-チアジアゾール基等)、シアノ基、ヒドロキシ基、ニトロ基、カルボキシ基、アルコキシ基(メトキシ基、エトキシ基、イソプロポキシ基、tert-ブトキシ基、n-オクチルオキシ基、2-メトキシエトキシ基等)、アリールオキシ基(フェノキシ基、2-メチルフェノキシ基、4-tert-ブチルフェノキシ基、3-ニトロフェノキシ基、2-テトラデカノイルアミノフェノキシ基等)、アシルオキシ基(ホルミルオキシ基、アセチルオキシ基、ピバロイルオキシ基、ステアロイルオキシ基、ベンゾイルオキシ基、p-メトキシフェニルカルボニルオキシ基等)、アミノ基(アミノ基、メチルアミノ基、ジメチルアミノ基、アニリノ基、N-メチル-アニリノ基、ジフェニルアミノ基等)、アシルアミノ基(ホルミルアミノ基、アセチルアミノ基、ピバロイルアミノ基、ラウロイルアミノ基、ベンゾイルアミノ基等)、アルキル及びアリールスルホニルアミノ基(メチルスルホニルアミノ基、ブチルスルホニルアミノ基、フェニルスルホニルアミノ基、2,3,5-トリクロロフェニルスルホニルアミノ基、p-メチルフェニルスルホニルアミノ基等)、メルカプト基、アルキルチオ基(メチルチオ基、エチルチオ基、n-ヘキサデシルチオ基等)、アリールチオ基(フェニルチオ基、p-クロロフェニルチオ基、m-メトキシフェニルチオ基等)、スルファモイル基(N-エチルスルファモイル基、N-(3-ドデシルオキシプロピル)スルファモイル基、N,N-ジメチルスルファモイル基、N-アセチルスルファモイル基、N-ベンゾイルスルファモイル基、N-(N’フェニルカルバモイル)スルファモイル基等)、スルホ基、アシル基(アセチル基、ピバロイルベンゾイル基等)、カルバモイル基(カルバモイル基、N-メチルカルバモイル基、N,N-ジメチルカルバモイル基、N,N-ジ-n-オクチルカルバモイル基、N-(メチルスルホニル)カルバモイル基等)等の各基が挙げられる。
前記一般式(2)において、A1及びA2は、芳香族炭化水素環基または芳香族複素環基であることが、セルロース誘導体と相互作用しやすくなることで環境湿度の変化に対する光学値の変動が抑制されるため好ましい。
前記一般式(2)において、Bは、芳香族炭化水素環基または芳香族複素環基を表す。
Bで表される芳香族炭化水素環は、単環であっても縮合環であってもよい。芳香族炭化水素環の好ましい例には、ベンゼン環、ナフタレン環、アントラセン環、フェナントレン環、ペリレン環、テトラセン環、ピレン環、ベンゾピレン環、クリセン環、トリフェニレン環、アセナフテン環、フルオランテン環、フルオレン環等が含まれ、より好ましくはベンゼン環である。
Bで表される芳香族複素環基の芳香族複素環の好ましい例には、フラン環、ベンゾフラン環、チオフェン環、ベンゾチオフェン環、ピロール環、ピラゾール環、イミダゾール環、オキサゾール環、1,2,4-オキサジアゾール環、1,3,4-オキサジアゾール環、イソオキサゾール環、チアゾール環、1,2,4-チオジアゾール環、1,3,4-チオジアゾール環、イソチアゾール環、インドール環、カルバゾール環、アザカルバゾール環(アザカルバゾール環とは、カルバゾール環を構成する炭素原子の一つ以上が窒素原子で置き換わったものをいう)、1,2,3-トリアゾール環、1,2,4-トリアゾール環、ピロロイミダゾール環、ピロロピラゾール環、ピロロピロール環、チエノピロール環、チエノチオフェン環、フロピロール環、フロフラン環、チエノフラン環、ベンゾイソオキサゾール環、ベンゾチアゾール環、ベンゾオキサゾール環、ベンゾイソチアゾール環、ベンゾイミダゾール環、フタルイミド環、ピリジン環、ピラジン環、ピリダジン環、ピリミジン環、トリアジン環、キノリン環、イソキノリン環、シノリン環、キノキサリン環、フェナントリジン環、ベンゾイミダゾール環、ペリミジン環、キナゾリン環、キナゾリノン環、アズレン環、シロール環、ジベンゾフラン環、ジベンゾチオフェン環、ジベンゾカルバゾール環、ベンゾジフラン環、ベンゾジチオフェン環、フェナントロリン環、アクリジン環、ベンゾキノリン環、フェナジン環、フェナントリジン環、サイクラジン環、キンドリン環、テペニジン環、キニンドリン環、トリフェノジチアジン環、トリフェノジオキサジン環、フェナントラジン環、アントラジン環、ペリミジン環、ナフトフラン環、ナフトチオフェン環、ナフトジフラン環、ナフトジチオフェン環、アントラフラン環、アントラジフラン環、アントラチオフェン環等が含まれる。なかでも、ピリジン環等がより好ましい。
前記一般式(2)において、Bで表せる芳香族炭化水素環基または芳香族複素環基は置換基を有してもよく、該置換基としては、前記一般式(2)におけるA1及びA2が有してもよい置換基と同様の基を挙げることができる。
前記一般式(2)において、T1およびT2は、それぞれ独立して、2価の1,2,4-トリアゾール環基を表す。芳香族複素環基を有する化合物は、分極したπ電子を有するため、樹脂の水素原子と強く相互作用し、水分子よりも樹脂に強く配位することで、水分子の量が変化しても位相差値の変動が抑制されると考えられる。特に、1,2,4-トリアゾール環を有する化合物は、樹脂と強く配位するだけでなく、水分子との相互作用力も強く、樹脂/1,2,4-トリアゾール系化合物/水分子が安定な構造をとると考えられるため、位相差値の変動の抑制に特に優れるため好ましい。ここで、T1およびT2は、同じであっても異なるものであってもよい。また、T2が複数存在する(nが2~5である)場合には、各T2は、同じであっても異なるものであってもよい。T1及びT2で表される1,2,4-トリアゾール環基は、互変異性体であってもよい。具体的には、下記いずれの構造も、1,2,4-トリアゾール環基を表す。

上記式中、※はL1、L2、L3またはL4との結合位置を表す。R5は水素原子または置換基を表す。R5で表される置換基としては、前記一般式(2)におけるA1が有してもよい置換基と同様の基を挙げることができる。R5は水素原子、アルキル基またはアシル基であることが好ましい。
前記一般式(2)において、T1及びT2は置換基を有してもよく、該置換基としては、前記一般式(2)におけるA1及びA2が有してもよい置換基と同様の基を挙げることができる。
前記一般式(2)において、L1、L2、L3およびL4は、それぞれ独立して、単結合または2価の連結基を表す。ここで、L1、L2、L3およびL4は、同じであっても異なるものであってもよい。また、L3およびL4が複数存在する(nが2~5である)場合には、各L3およびL4は、それぞれ、同じであっても異なるものであってもよい。2価の連結基としては、具体的には、アルキレン基、アルケニレン基、アルキニレン基、O、(C=O)、(C=O)-O、NR、S、(O=S=O)からなる群より選ばれる2価の連結基であるか、それらの組合せかを表す。Rは、水素原子または置換基を表す。Rで表される置換基の例には、アルキル基(メチル基、エチル基、n-プロピル基、イソプロピル基、tert-ブチル基、n-オクチル基、2-エチルヘキシル基等)、シクロアルキル基(シクロヘキシル基、シクロペンチル基、4-n-ドデシルシクロヘキシル基等)、芳香族炭化水素環基(フェニル基、p-トリル基、ナフチル基等)、芳香族複素環基(2-フリル基、2-チエニル基、2-ピリミジニル基、2-ベンゾチアゾリル基、2-ピリジル基等)、シアノ基等が含まれる。L1、L2、L3及びL4で表される2価の連結基は置換基を有してもよく、置換基としては特に制限はないが、例えば、前記一般式(2)におけるA1及びA2が有してもよい置換基と同様の基を挙げることができる。
前記一般式(2)において、L1、L2、L3及びL4は、前記一般式(2)で表される化合物の平面性が高くなることでセルロース誘導体との相互作用が高くなり、環境湿度の変化に応じた位相差値の変動が抑制されるため、単結合、O、(C=O)-O、O-(C=O)、(C=O)-NRまたはNR-(C=O)であることが好ましく、単結合であることがより好ましい。
前記一般式(2)において、nは0~5の整数を表す。nが2以上の整数を表すとき、前記一般式(2)における複数のA2、T2、L3、L4は同じであってもよく、異なっていてもよい。nが大きい程、前記一般式(2)で表される化合物がセルロース誘導体と相互作用しやすくなることで環境湿度の変化に対する光学値の変動が抑制されるため好ましいが、nが大きくなりすぎると、セルロースエステルとの相溶性が悪くなる。このため、nは1~5の整数であることが好ましく、1~4の整数であることがより好ましい。
一般式(2)で表される化合物は、下記一般式(5)で表される化合物であることが好ましい。

上記一般式(5)中、A1、A2、T1、T2、L1、L2、L3及びL4は、それぞれ前記一般式(2)におけるA1、A2、T1、T2、L1、L2、L3及びL4と同義である。A3及びT3は、それぞれ一般式(2)におけるA1及びT1と同様の基を表す。L5及びL6は、前記一般式(2)におけるL1と同様の基を表す。Q1、Q2、Q3及びQ4は炭素原子または窒素原子を表す。mは0~4の整数を表す。
上記一般式(5)において、mが小さい方がセルロースエステルとの相溶性に優れるため、mは0~3の整数であることがより好ましい。
前記一般式(2)で表される化合物は、水和物、溶媒和物若しくは塩を形成してもよい。なお、本発明において、水和物は有機溶媒を含んでいてもよく、また溶媒和物は水を含んでいてもよい。即ち、「水和物」及び「溶媒和物」には、水と有機溶媒のいずれも含む混合溶媒和物が含まれる。塩としては、無機または有機酸で形成された酸付加塩が含まれる。無機酸の例として、ハロゲン化水素酸(塩酸、臭化水素酸など)、硫酸、リン酸などが含まれ、またこれらに限定されない。また、有機酸の例には、酢酸、トリフルオロ酢酸、プロピオン酸、酪酸、シュウ酸、クエン酸、安息香酸、アルキルスルホン酸(メタンスルホン酸など)、アリルスルホン酸(ベンゼンスルホン酸、4-トルエンスルホン酸、1,5-ナフタレンジスルホン酸など)などが挙げられ、またこれらに限定されない。これらのうち好ましくは、塩酸塩、酢酸塩、プロピオン酸塩、酪酸塩である。
塩の例としては、親化合物に存在する酸性部分が、金属イオン(例えばアルカリ金属塩、例えばナトリウムまたはカリウム塩、アルカリ土類金属塩、例えばカルシウムまたはマグネシウム塩、アンモニウム塩アルカリ金属イオン、アルカリ土類金属イオン、またはアルミニウムイオンなど)により置換されるか、あるいは有機塩基(エタノールアミン、ジエタノールアミン、トリエタノールアミン、モルホリン、ピペリジン、など)と調整されたときに形成される塩が挙げられ、またこれらに限定されない。これらのうち好ましくはナトリウム塩、カリウム塩である。
溶媒和物が含む溶媒の例には、一般的な有機溶剤のいずれも含まれる。具体的には、アルコール(例、メタノール、エタノール、2-プロパノール、1-ブタノール、1-メトキシ-2-プロパノール、t-ブタノール)、エステル(例、酢酸エチル)、炭化水素(例、トルエン、ヘキサン、ヘプタン)、エーテル(例、テトラヒドロフラン)、ニトリル(例、アセトニトリル)、ケトン(アセトン)などが挙げられる。好ましくは、アルコール(例、メタノール、エタノール、2-プロパノール、1-ブタノール、1-メトキシ-2-プロパノール、t-ブタノール)の溶媒和物である。これらの溶媒は、前記化合物の合成時に用いられる反応溶媒であっても、合成後の晶析精製の際に用いられる溶媒であってもよく、またはこれらの混合であってもよい。
また、二種類以上の溶媒を同時に含んでもよいし、水と溶媒を含む形(例えば、水とアルコール(例えば、メタノール、エタノール、t-ブタノールなど)など)であってもよい。
なお、前記一般式(2)で表される化合物を、水や溶媒、塩を含まない形態で添加しても、本発明における樹脂組成物または位相差フィルム中において、水和物、溶媒和物または塩を形成してもよい。
前記一般式(2)で表される化合物の分子量は特に制限はないが、小さいほど樹脂との相溶性に優れ、大きいほど環境湿度の変化に対する光学値の変動抑制効果が高いため、150~2000であることが好ましく、200~1500であることがより好ましく、300~1000であることがより好ましい。
以下に、前記一般式(2)で表される化合物の具体例を示すが、本発明で用いることができる前記一般式(2)で表される化合物は、以下の具体例によって何ら限定されることはない。なお、前述のように、以下の具体例は互変異性体であってもよく、水和物、溶媒和物または塩を形成していてもよい。また、下記番号にて、化合物を規定する。すなわち、下記「1」の化合物を「化合物(2-1)」とも称する。













次に、前記一般式(2)で表される化合物の合成方法について説明する。
一般式(2)で表される化合物は、いかなる原料を用いても構わないが、ニトリル誘導体またはイミノエーテル誘導体と、ヒドラジド誘導体を反応させる方法が好ましい。反応に用いる溶媒としては、原料と反応しないと溶媒であれば、いかなる溶媒でも構わないが、エステル系(例えば、酢酸エチル、酢酸メチル等)、アミド系(ジメチルホルムアミド、ジメチルアセトアミド等)、エーテル系(エチレングリコールジメチルエーテル等)、アルコール系(例えば、メタノール、エタノール、プロパノール、イソプロパノール、n-ブタノール、2-ブタノール、エチレングリコール、エチレングリコールモノメチルエーテル等)、芳香族炭化水素系(例えば、トルエン、キシレン等)、水を挙げられることができる。使用する溶媒として、好ましくは、アルコール系溶媒である。また、これらの溶媒は、混合して用いても良い。
溶媒の使用量は、特に制限はないが、使用するヒドラジド誘導体の重量に対して、0.5~30倍量の範囲内であることが好ましく、更に好ましくは、1.0~25倍量であり、特に好ましくは、3.0~20倍量の範囲内である。
ニトリル誘導体とヒドラジド誘導体を反応させる場合、触媒を使用しなくても構わないが、反応を加速させるために触媒を使用する方が好ましい。使用する触媒としては、酸を用いても良く、塩基を用いても良い。酸としては、塩酸、硫酸、硝酸、酢酸等が挙げられ、好ましくは塩酸である。酸は、水に希釈して添加しても良く、ガスを系中に吹き込む方法で添加しても良い。塩基としては、無機塩基(炭酸カリウム、炭酸ナトリウム、炭酸水素カリウム、炭酸水素ナトリウム、水酸化カリウム、水酸化ナトリウム等)及び有機塩基(ナトリウムメチラート、ナトリウムエチラート、カリウムメチラート、カリウムエチラート、ナトリウムブチラート、カリウムブチラート、ジイソプロピルエチルアミン、N,N’-ジメチルアミノピリジン、1,4-ジアザビシクロ[2.2.2]オクタン、N-メチルモルホリン、イミダゾール、N-メチルイミダゾール、ピリジン等)のいずれを用いて良く、無機塩基としては、炭酸カリウムが好ましく、有機塩基としては、ナトリウムエチラート、ナトリウムエチラート、ナトリウムブチラートが好ましい。無機塩基は、粉体のまま添加しても良く、溶媒に分散させた状態で添加しても良い。また、有機塩基は、溶媒に溶解した状態(例えば、ナトリウムメチラートの28%メタノール溶液等)で添加しても良い。
触媒の使用量は、反応が進行する量であれば特に制限はないが、形成されるトリアゾール環に対して1.0~5.0倍モルの範囲内が好ましく、更に1.05~3.0倍モルの範囲内が好ましい。
イミノエーテル誘導体とヒドラジド誘導体を反応させる場合は、触媒を用いる必要がなく、溶媒中で加熱することにより目的物を得ることができる。
反応に用いる原料、溶媒及び触媒の添加方法は、特に制限がなく、触媒を最後に添加しても良く、溶媒を最後に添加しても良い。また、ニトリル誘導体を溶媒に分散若しくは溶解させ、触媒を添加した後、ヒドラジド誘導体を添加する方法も好ましい。
反応中の溶液温度は、反応が進行する温度であればいかなる温度でも構わないが、好ましくは、0~150℃の範囲内であり、更に好ましくは、20~140℃の範囲内である。また、生成する水を除去しながら、反応を行っても良い。
反応溶液の処理方法は、いかなる手段を用いても良いが、塩基を触媒として用いた場合は、反応溶液に酸を加えて中和する方法が好ましい。中和に用いる酸としては、例えば、塩酸、硫酸、硝酸または酢酸等が挙げられるが、特に好ましくは酢酸である。中和に使用する酸の量は、反応溶液のpHが4~9になる範囲であれば特に制限はないが、使用する塩基に対して、0.1~3倍モルが好ましく、特に好ましくは、0.2~1.5倍モルの範囲内である。
反応溶液の処理方法として、適当な有機溶媒を用いて抽出する場合、抽出後に有機溶媒を水で洗浄した後、濃縮する方法が好ましい。ここでいう適当な有機溶媒とは、酢酸エチル、トルエン、ジクロロメタン、エーテル等非水溶性の溶媒、または、前記非水溶性の溶媒とテトラヒドロフランまたはアルコール系溶媒との混合溶媒のことであり、好ましくは酢酸エチルである。
一般式(2)で表される化合物を晶析させる場合、特に制限はないが、中和した反応溶液に水を追加して晶析させる方法、若しくは、一般式(2)で表される化合物が溶解した水溶液を中和して晶析させる方法が好ましい。
例えば、化合物(2-1)は以下のスキームによって合成することができる。

(化合物(2-1)の合成)
n-ブタノール350mlにベンゾニトリル77.3g(75.0mmol)、ベンゾイルヒドラジン34.0g(25.0mmol)、炭酸カリウム107.0g(77.4mmol)を加え、窒素雰囲気下、120℃で24時間撹拌した。反応液を室温まで冷却し、析出物をろ過後、ろ液を減圧下で濃縮した。濃縮物にイソプロパノール20mlを加え、析出物をろ取した。ろ取した析出物をメタノール80mlに溶解し、純水300mlを加え、溶液のpHが7になるまで酢酸を滴下した。析出した結晶をろ取後、純水で洗浄し、50℃で送風乾燥することにより、化合物(2-1)を38.6g得た。収率は、ベンゾイルヒドラジン基準で70%であった。
n-ブタノール350mlにベンゾニトリル77.3g(75.0mmol)、ベンゾイルヒドラジン34.0g(25.0mmol)、炭酸カリウム107.0g(77.4mmol)を加え、窒素雰囲気下、120℃で24時間撹拌した。反応液を室温まで冷却し、析出物をろ過後、ろ液を減圧下で濃縮した。濃縮物にイソプロパノール20mlを加え、析出物をろ取した。ろ取した析出物をメタノール80mlに溶解し、純水300mlを加え、溶液のpHが7になるまで酢酸を滴下した。析出した結晶をろ取後、純水で洗浄し、50℃で送風乾燥することにより、化合物(2-1)を38.6g得た。収率は、ベンゾイルヒドラジン基準で70%であった。
得られた化合物(2-1)の1H-NMRスペクトルは以下のとおりである。
1H-NMR(400MHz、溶媒:重DMSO、基準:テトラメチルシラン)δ(ppm):7.56-7.48(6H、m)、7.62-7.61(4H、m)
例えば、化合物(2-6)は以下のスキームによって合成することができる。
例えば、化合物(2-6)は以下のスキームによって合成することができる。

(化合物(2-6)の合成)
n-ブタノール40mlに1,3-ジシアノベンゼン2.5g(19.5mmol)、ベンゾイルヒドラジン7.9g(58.5mmol)、炭酸カリウム9.0g(68.3mmol)を加え、窒素雰囲気下、120℃で24時間撹拌した。反応液を冷却後、純水40mlを加え、室温で3時間撹拌した後、析出した固体を濾別し、純水で洗浄した。得られた固体に水及び酢酸エチルを加えて分液し、有機層を純水で洗浄した。有機層を硫酸マグネシウムで乾燥し、溶媒を減圧留去した。得られた粗結晶をシリカゲルクロマトグラフィー(酢酸エチル/ヘプタン)で精製し、化合物(2-6)を5.5g得た。収率は、1,3-ジシアノベンゼン基準で77%であった。
n-ブタノール40mlに1,3-ジシアノベンゼン2.5g(19.5mmol)、ベンゾイルヒドラジン7.9g(58.5mmol)、炭酸カリウム9.0g(68.3mmol)を加え、窒素雰囲気下、120℃で24時間撹拌した。反応液を冷却後、純水40mlを加え、室温で3時間撹拌した後、析出した固体を濾別し、純水で洗浄した。得られた固体に水及び酢酸エチルを加えて分液し、有機層を純水で洗浄した。有機層を硫酸マグネシウムで乾燥し、溶媒を減圧留去した。得られた粗結晶をシリカゲルクロマトグラフィー(酢酸エチル/ヘプタン)で精製し、化合物(2-6)を5.5g得た。収率は、1,3-ジシアノベンゼン基準で77%であった。
得られた化合物(2-6)の1H-NMRスペクトルは以下のとおりである。
1H-NMR(400MHz、溶媒:重DMSO、基準:テトラメチルシラン)δ(ppm):8.83(1H、s)、8.16~8.11(6H、m)、7.67-7.54(7H、m)
その他の化合物についても同様の方法によって合成が可能である。
その他の化合物についても同様の方法によって合成が可能である。
本発明の位相差フィルムにおいて、一般式(2)の化合物の含有量は、特に制限されず、適宜量を調整して位相差フィルムに含有することができる。一般式(2)の化合物の含有量は、例えば、セルロースエステル樹脂100重量部に対して、好ましくは1~30重量部、より好ましくは3~20重量部、特に好ましくは5~10重量部である。このような量であれば、位相差フィルムの位相差機能及び引き裂き強度を向上できる。また、高温多湿下での揮発性を低減するため、位相差フィルムの耐ブリード性、ゆえに映像の鮮明度を向上できる。
また、前記一般式(2)で表される化合物の添加方法としては、位相差フィルムを形成する樹脂に粉体で添加しても良く、溶媒に溶解した後、位相差フィルムを形成する樹脂に添加しても良い。
(可塑剤)
本発明の位相差フィルムは、フィルム製造時の組成物の流動性や、フィルムの柔軟性や加工性を向上するために可塑剤を含有していていもよい。可塑剤の例には、糖エステル系可塑剤、ポリエステル系可塑剤、多価アルコールエステル系可塑剤、アクリル系化合物、多価カルボン酸エステル系可塑剤(フタル酸エステル系可塑剤を含む)、グリコレート系可塑剤、エステル系可塑剤(クエン酸エステル系可塑剤、脂肪酸エステル系可塑剤、リン酸エステル系可塑剤、トリメリット酸エステル系可塑剤等を含む)、スチレン系化合物等が含まれる。可塑剤の中でも、下記糖エステル系可塑剤(糖エステル化合物)、ポリエステル系可塑剤、およびアクリル系化合物からなる群から選択される少なくとも1種の可塑剤を含むことが、透湿性の効果的な制御およびセルロースエステルとの相溶性を高度に両立できる観点から好ましい。これらは、単独で用いても、二種類以上を組み合わせて用いてもよい。
本発明の位相差フィルムは、フィルム製造時の組成物の流動性や、フィルムの柔軟性や加工性を向上するために可塑剤を含有していていもよい。可塑剤の例には、糖エステル系可塑剤、ポリエステル系可塑剤、多価アルコールエステル系可塑剤、アクリル系化合物、多価カルボン酸エステル系可塑剤(フタル酸エステル系可塑剤を含む)、グリコレート系可塑剤、エステル系可塑剤(クエン酸エステル系可塑剤、脂肪酸エステル系可塑剤、リン酸エステル系可塑剤、トリメリット酸エステル系可塑剤等を含む)、スチレン系化合物等が含まれる。可塑剤の中でも、下記糖エステル系可塑剤(糖エステル化合物)、ポリエステル系可塑剤、およびアクリル系化合物からなる群から選択される少なくとも1種の可塑剤を含むことが、透湿性の効果的な制御およびセルロースエステルとの相溶性を高度に両立できる観点から好ましい。これらは、単独で用いても、二種類以上を組み合わせて用いてもよい。
当該可塑剤は、分子量が15000以下、さらには10000以下であることが、耐湿熱性の改善とセルロースエステルとの相溶性を両立する観点から好ましい。当該分子量が10000以下である化合物が重合体である場合は、重量平均分子量(Mw)が10000以下であることが好ましい。好ましい分子量(Mw)の範囲は100~10000の範囲内であり、更に好ましくは、400~8000の範囲内である。
糖エステル系可塑剤(糖エステル化合物)は、フラノース構造若しくはピラノース構造を1~12個有する化合物であって、該化合物中のヒドロキシ基の全部または一部がエステル化された化合物をいう。糖エステル系可塑剤は加水分解防止を目的として添加されうる。
本発明に係る糖エステル化合物の合成原料の糖の例としては、例えば以下のようなものを挙げることができるが、本発明はこれらに限定されるものではない。グルコース、ガラクトース、マンノース、フルクトース、キシロース、あるいはアラビノース、ラクトース、スクロース、ニストース、1F-フラクトシルニストース、スタキオース、マルチトール、ラクチトール、ラクチュロース、セロビオース、マルトース、セロトリオース、マルトトリオース、ラフィノースあるいはケストース挙げられる。この他、ゲンチオビオース、ゲンチオトリオース、ゲンチオテトラオース、キシロトリオース、ガラクトシルスクロースなども挙げられる。
ピラノース構造またはフラノース構造中のOH基のすべてもしくは一部をエステル化するのに用いられるモノカルボン酸としては、特に制限はなく、公知の脂肪族モノカルボン酸、脂環族モノカルボン酸、芳香族モノカルボン酸等を用いることができる。用いられるカルボン酸は1種類でもよいし2種以上の混合であってもよい。
好ましい脂肪族モノカルボン酸の例としては、酢酸、プロピオン酸、酪酸、イソ酪酸、吉草酸、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、2-エチル-ヘキサンカルボン酸、ウンデシル酸、ラウリン酸、トリデシル酸、ミリスチン酸、ペンタデシル酸、パルミチン酸、ヘプタデシル酸、ステアリン酸、ノナデカン酸、アラキン酸、ベヘン酸、リグノセリン酸、セロチン酸、ヘプタコサン酸、モンタン酸、メリシン酸、ラクセル酸等の飽和脂肪酸、ウンデシレン酸、オレイン酸、ソルビン酸、リノール酸、リノレン酸、アラキドン酸、オクテン酸等の不飽和脂肪酸等を挙げることができる。
好ましい脂環族モノカルボン酸の例としては、シクロペンタンカルボン酸、シクロヘキサンカルボン酸、シクロオクタンカルボン酸、またはそれらの誘導体を挙げることができる。
好ましい芳香族モノカルボン酸の例としては、安息香酸、フェニル酢酸、トルイル酸等の安息香酸のベンゼン環に1~5個のアルキル基若しくはアルコキシ基を導入した芳香族モノカルボン酸、ケイ皮酸、ベンジル酸、ビフェニルカルボン酸、ナフタリンカルボン酸、テトラリンカルボン酸等のベンゼン環を2個以上有する芳香族モノカルボン酸、またはそれらの誘導体を挙げることができるが、特に安息香酸が好ましい。
そのような糖エステルの好ましい例には、下記一般式(FA)で表されるスクロースエステルが含まれる。
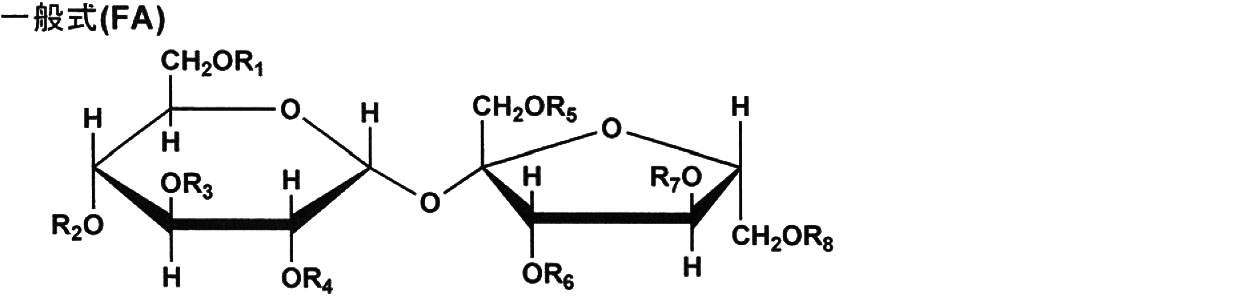
一般式(FA)のR1~R8は、それぞれ独立に、水素原子、置換若しくは無置換のアルキルカルボニル基、または置換若しくは無置換のアリールカルボニル基を表す。R1~R8は、互いに同じであっても、異なってもよい。
置換若しくは無置換のアルキルカルボニル基は、炭素原子数2以上の置換若しくは無置換のアルキルカルボニル基であることが好ましい。置換若しくは無置換のアルキルカルボニル基の例には、メチルカルボニル基(アセチル基)が含まれる。アルキル基が有する置換基の例には、フェニル基等の芳香族炭化水素環基が含まれる。
置換または無置換のアリールカルボニル基は、炭素原子数7以上の置換または無置換のアリールカルボニル基であることが好ましい。アリールカルボニル基の例には、フェニルカルボニル基が含まれる。芳香族炭化水素環基が有する置換基の例には、メチル基等のアルキル基や、メトキシ基等のアルコキシル基等が含まれる。
上記一般式(FA)で表される化合物は、平均置換度が好ましくは3.0~7.5、より好ましくは3.0~6.0であることによって、透湿性の制御とセルロースエステルとの相溶性を高度に両立することができる。
本発明において、一般式(FA)で表される化合物の置換度とは、一般式(FA)に含まれる8つの水酸基のうち、水素以外の置換基で置換されている数を表し、すなわち、一般式(FA)のR1~R8のうち、水素以外の基を含む数を表す。したがって、R1~R8が全て水素以外の置換基により置換された場合に、置換度は最大値の8.0となり、R1~R8が全て水素原子である場合には、0.0となる。
一般式(FA)で表される構造を有する化合物は、水酸基の数、OR基の数が固定された単一種の化合物を合成することは困難であり、式中の水酸基の数、OR基の異なる成分が数種類混合された化合物となることが知られているため、本発明における一般式(FA)の置換度としては、平均置換度を用いることが適当であり、常法により高速液体クロマトグラフィーによって置換度分布を示すチャートの面積比から平均置換度を測定することができる。
一般式(FA)において、R1~R8は、置換または無置換のアルキルカルボニル基、あるいは、置換または無置換のアリールカルボニル基を表し、R1~R8は、同じであっても、異なっていてもよい(以下、R1~R8をアシル基ともいう)。R1~R8としては、具体的には、上記で例示した糖エステル化合物の合成時に用いられるモノカルボン酸由来のアシル基が挙げられる。
以下に、本発明に係る糖エステル化合物の具体例を挙げるが、R1~R8のうちいずれかを同じ置換基Rとした場合であって、本発明はこれに限定されるものではない。また、下記実施例において、ポリエステル化合物を下記記号にて規定する。なお、本発明において、R1~R8はそれぞれ異なる基である糖エステル化合物を使用することができる。



本発明に係る糖エステル化合物は、前記糖に、アシル化剤(エステル化剤ともいう、例えば、アセチルクロライド等の酸ハロゲン化物、無水酢酸等の無水物)を反応させることによって製造することが可能であり、置換度の分布は、アシル化剤の量、添加タイミング、エステル化反応時間の調節によってなされるが、置換度違いの糖エステル化合物の混合、あるいは純粋に単離した置換度違いの化合物を混合することにより、目的の平均置換度、置換度4以下の成分を調整することができる。
(合成例:糖エステル化合物の合成例)

撹拌装置、還流冷却器、温度計および窒素ガス導入管を備えた四頭コルベンに、ショ糖34.2g(0.1モル)、無水安息香酸135.6g(0.6モル)、ピリジン284.8g(3.6モル)を仕込み、撹拌下に窒素ガス導入管から窒素ガスをバブリングさせながら昇温し、70℃で5時間エステル化反応を行った。
次に、コルベン内を4×102Pa以下に減圧し、60℃で過剰のピリジンを留去した後に、コルベン内を1.3×10Pa以下に減圧し、120℃まで昇温させ、無水安息香酸、生成した安息香酸の大部分を留去した。そして、次にトルエン1L、0.5重量%の炭酸ナトリウム水溶液300gを添加し、50℃で30分間撹拌後、静置して、トルエン層を分取した。最後に、分取したトルエン層に水100gを添加し、常温で30分間水洗後、トルエン層を分取し、減圧下(4×102Pa以下)、60℃でトルエンを留去させ、化合物A-1、A-2、A-3、A-4およびA-5等の混合物である糖エステル化合物1を得た。
得られた混合物を高速液体クロマトグラフィー質量分析(HPLC-MS)で解析したところ、A-1が1.2重量%、A-2が13.2重量%、A-3が14.2重量%、A-4が35.4重量%、A-5等が40.0重量%であった。平均置換度は5.2であった。
同様に、無水安息香酸158.2g(0.70モル)、146.9g(0.65モル)、135.6g(0.60モル)、124.3g(0.55モル)と当モルのピリジンとを反応させて、表A記載のような成分の糖エステル化合物を得た。

次いで、得られた混合物の一部を、シリカゲルを用いたカラムクロマトグラフィーにより精製することで、それぞれ純度100%のA-1、A-2、A-3、A-4およびA-5等を得た。
なお、A-5等とは、置換度4以下の全ての成分、つまり置換度4、3、2、1の化合物の混合物であることを意味する。また、平均置換度は、A-5等を置換度4として計算した。
本発明においては、ここで作製した方法により所望の平均置換度に近い糖エステルおよび単離したA-1~A-5等を組み合わせ添加することにより、平均置換度を調整した。

その他の糖エステルの例には、特開昭62-42996号公報及び特開平10-237084号公報に記載の化合物が含まれる。
また、ポリエステル系可塑剤は、特に限定されないが、例えば、ジカルボン酸またはこれらのエステル形成性誘導体とグリコールとの縮合反応により得ることができる末端がヒドロキシ基(水酸基)となる重合体(ポリエステルポリオール)、または、当該ポリエステルポリオールの末端のヒドロキシ基がモノカルボン酸で封止された重合体(末端封止ポリエステル)を用いることができる。ここで言うエステル形成性誘導体とは、ジカルボン酸のエステル化物、ジカルボン酸クロライド、ジカルボン酸の無水物のことである。
好ましくは、下記一般式(FB-1)で表されるポリエステル系可塑剤を用いることが、透湿性の制御とセルロースエステルとの相溶性を高度に両立する観点から好ましい。

上記式中、Bは炭素数が2以上6以下の直鎖もしくは分岐のアルキレン基、もしくはシクロアルキレン基を示し、Aは炭素数が6以上14以下の芳香族環基を、nは1以上の自然数を示す。
上記の式で表される化合物は、芳香環を有するジカルボン酸(芳香族ジカルボン酸ともいう)と、炭素数が2以上6以下の直鎖もしくは分岐のアルキレンもしくはシクロアルキレンジオールから得られ、両末端がモノカルボン酸で封止されていないことが特徴である。
炭素数6以上16以下の芳香族ジカルボン酸としては、フタル酸、イソフタル酸、テレフタル酸、1,5-ナフタレンジカルボン酸、1,4-ナフタレンジカルボン酸、1,8-ナフタレンジカルボン酸、2,3-ナフタレンジカルボン酸、2,6-ナフタレンジカルボン酸、2,8-ナフタレンジカルボン酸、2,2’-ビフェニルジカルボン酸、4,4’-ビフェニルジカルボン酸、等が挙げられる。その中でも好ましくは、2,6-ナフタレンジカルボン酸、4,4’-ビフェニルジカルボン酸である。
炭素数が2以上6以下の直鎖もしくは分岐のアルキレンもしくはシクロアルキレンジオールとしては、エチレングリコール、1,2-プロパンジオール、1,3-プロパンジオール、1,2-ブタンジオール、1,3-ブタンジオール、2-メチル-1,3-プロパンジオール、1,4-ブタンジオール、1,5-ペンタンジオール、3-メチル-1,5-ペンタンジオール、1,6-ヘキサンジオール、1,4-シクロヘキサンジオール、1,4-シクロヘキサンジメタノール等が挙げられる。その中でも、好ましくはエタンジオール、1,2-プロパンジオール、1,3-プロパンジオール、1,3-ブタンジオールである。
中でも、Aが置換基を有していてもよいナフタレン環もしくはビフェニル環であることが本発明の効果を得る上で好ましい。ここで置換基とは、炭素数1以上6以下のアルキル基、アルケニル基、アルコキシル基である。
上記ポリエステル化合物の水酸基価(OH価)としては、100mgKOH/g以上500mgKOH/g以下であることが好ましく、170mgKOH/g~400mgKOH/gであることがさらに好ましい。水酸基価がこの範囲にあると、セルロースエステルおよびセルロースエーテルとの相溶性が好適なものとなる。
水酸基価が400mgKOH/g以下であると、ポリエステル化合物の疎水性が大きくなりすぎず、水酸基価が170mgKOH/g以上であると、ポリエステル化合物同士の分子間相互作用(水素結合等)が過度に強くならず、フィルム中での析出を防止できるためだと考えられる。
また水酸基価の測定は、日本工業規格 JIS K1557-1:2007に記載の無水酢酸法等を適用できる。
上記ポリエステル化合物の数平均分子量(Mn)は、下記式から計算することができる。
上記ポリエステル化合物は、常法により上記ジカルボン酸とジオールとのポリエステル化反応またはエステル交換反応による熱溶融縮合法か、あるいはこれら酸の酸クロライドとグリコール類との界面縮合法のいずれかの方法によっても容易に合成できる。
以下に、上記のポリエステル化合物を例示する。


好ましくは、下記一般式(FB-2)で表されるポリエステル系可塑剤を用いることが、透湿性の制御とセルロースエステルとの相溶性を高度に両立する観点から好ましい。
上記一般式(FB-2)中、Bはヒドロキシ基またはカルボン酸残基を表し、Gは炭素数2~18のアルキレングリコール残基または炭素数6~12のアリールグリコール残基または炭素数が4~12のオキシアルキレングリコール残基を表し、Aは炭素数4~12のアルキレンジカルボン酸残基または炭素数6~12のアリールジカルボン酸残基を表し、nは1以上の整数を表す。
一般式(FB-2)中、Bで示されるヒドロキシ基またはカルボン酸残基と、Gで示されるアルキレングリコール残基またはオキシアルキレングリコール残基またはアリールグリコール残基、Aで示されるアルキレンジカルボン酸残基またはアリールジカルボン酸残基とから構成されるものであり、通常のエステル系化合物と同様の反応により得られる。
一般式(FB-2)で表されるポリエステル系化合物のカルボン酸成分としては、例えば、酢酸、プロピオン酸、酪酸、安息香酸、パラターシャリブチル安息香酸、オルソトルイル酸、メタトルイル酸、パラトルイル酸、ジメチル安息香酸、エチル安息香酸、ノルマルプロピル安息香酸、アミノ安息香酸、アセトキシ安息香酸、脂肪族酸等があり、これらはそれぞれ1種または2種以上の混合物として使用することができる。
一般式(FB-2)で表されるポリエステル系化合物の炭素数2~18のアルキレングリコール成分としては、エチレングリコール、1,2-プロピレングリコール、1,3-プロピレングリコール、1,2-ブタンジオール、1,3-ブタンジオール、1,2-プロパンジオール、2-メチル-1,3-プロパンジオール、1,4-ブタンジオール、1,5-ペンタンジオール、2,2-ジメチル-1,3-プロパンジオール(ネオペンチルグリコール)、2,2-ジエチル-1,3-プロパンジオール(3,3-ジメチロールペンタン)、2-n-ブチル-2-エチル-1,3-プロパンジオール(3,3-ジメチロールヘプタン)、3-メチル-1,5-ペンタンジオール1,6-ヘキサンジオール、2,2,4-トリメチル-1,3-ペンタンジオール、2-エチル-1,3-ヘキサンジオール、2-メチル-1,8-オクタンジオール、1,9-ノナンジオール、1,10-デカンジオール、1,12-オクタデカンジオール等があり、これらのグリコールは、1種または2種以上の混合物として使用される。
特に炭素数2~12のアルキレングリコールがセルロースエステル樹脂との相溶性に優れているため、特に好ましい。より好ましくは炭素数2~6のアルキレングリコールであり、さらに好ましくは炭素数2~4のアルキレングリコールである。
一般式(FB-2)で表されるポリエステル系可塑剤の炭素数6~12のアリールグリコールとしては、例えば、1,4-シクロヘキサンジオール、1,4-シクロヘキサンジメタノール、シクロヘキサンジエタノール、1,4-ベンゼンジメタノール等の環状グリコール類があり、これらのグリコールは、一種または二種以上の混合物として使用できる。
また、上記一般式(FB-2)で表されるポリエステル系化合物の炭素数4~12のオキシアルキレングリコール成分としては、例えば、ジエチレングリコール、トリエチレングリコール、テトラエチレングリコール、ジプロピレングリコール、トリプロピレングリコール等があり、これらのグリコールは、1種または2種以上の混合物として使用できる。
一般式(FB-2)で表されるポリエステル系化合物の炭素数4~12のアルキレンジカルボン酸成分としては、例えば、コハク酸、マレイン酸、フマル酸、グルタル酸、アジピン酸、アゼライン酸、セバシン酸、ドデカンジカルボン酸等があり、これらは、それぞれ1種または2種以上の混合物として使用される。
一般式(FB-2)で表されるポリエステル系化合物の炭素数6~12のアリールジカルボン酸成分としては、フタル酸、テレフタル酸、イソフタル酸、1,5-ナフタレンジカルボン酸、1,4-ナフタレンジカルボン酸等がある。
一般式(FB-2)で表されるポリエステル系化合物は、重量平均分子量が、好ましくは300~1500、より好ましくは400~1000の範囲が好適である。また、その酸価は、0.5mgKOH/g以下、ヒドロキシ基(水酸基)価は25mgKOH/g以下、より好ましくは酸価0.3mgKOH/g以下、ヒドロキシ基(水酸基)価は15mgKOH/g以下のものである。
ポリエステル系可塑剤の重量平均分子量は、下記の測定条件によるゲルパーミエーションクロマトグラフィー(GPC)を用いた測定により算出する。

以下に、本発明に用いることのできる一般式(FB-2)で表されるポリエステル系化合物の具体的化合物を示すが、本発明はこれに限定されない。また、下記実施例において、ポリエステル系化合物を下記記号にて規定する。



ポリエステル系可塑剤の粘度は、分子構造や分子量にもよるが、アジピン酸系可塑剤の場合、セルロースエステルとの相溶性が高く、かつ可塑性を付与する効果が高いこと等から、200~5000mPa・s(25℃)の範囲であることが好ましい。ポリエステル系可塑剤は、一種類であっても、二種類以上を併用してもよい。
多価アルコールエステル系可塑剤は、2価以上の脂肪族多価アルコールと、モノカルボン酸とのエステル化合物(アルコールエステル)であり、好ましくは2~20価の脂肪族多価アルコールエステルである。多価アルコールエステル系化合物は、分子内に芳香環又はシクロアルキル環を有することが好ましい。
アクリル系化合物としては、特に制限されるものではないが、(メタ)アクリル酸、(メタ)アクリル酸エステル、(メタ)アクリルアミド類、および(メタ)アクリロニトリルよりなる群から選択されるいずれか少なくとも1種のアクリル系モノマー由来の繰り返し単位を有する重合体が挙げられる。これらのアクリル系化合物は、フィルムの耐水性を向上できる。
中でも、アクリル系化合物としては、メチルメタクリレート単位が50~99重量%およびこれと共重合可能な他の単量体単位の総量が1~50重量%からなるものが好ましい。
共重合可能な他の単量体としては、アルキル基の炭素数が2~18のアルキルメタクリレート;アルキル基の炭素数が1~18のアルキルアクリレート;アクリロイルモルホリンやN,N-ジメチルアクリルアミドなどのアミド基を有するビニルモノマー;エステル部分に炭素数5~22の脂環式炭化水素基を有するメタクリル酸エステルまたはアクリル酸エステル;アクリル酸、メタクリル酸等のα,β-不飽和カルボン酸;マレイン酸、フマル酸、イタコン酸等の不飽和基含有二価カルボン酸;スチレン、α-メチルスチレン等の芳香族ビニル化合物;アクリロニトリル、メタクリロニトリル等のα、β-不飽和ニトリル;無水マレイン酸、マレイミド、N-置換マレイミド、無水グルタル酸、等が挙げられ、これらは単独で、あるいは2種以上の単量体を併用して用いることができる。
また、本発明に用いられるアクリル系化合物としては、環構造を有してもよく、具体的には、ラクトン環構造、無水グルタル酸構造、グルタルイミド構造、N-置換マレイミド構造および無水マレイン酸構造、ピラン環構造が挙げられる。
これらの中でも、共重合可能な他の単量体は、共重合体の耐熱分解性や流動性の観点から、アルキル基の炭素数が1~18のアルキルアクリレート、アクリロイルモルホリンやジメチルアクリルアミドなどのアミド基を有するビニルモノマー、エステル部分に炭素数5~22の脂環式炭化水素基を有するメタクリル酸エステルまたはアクリル酸エステル、N-置換マレイミド構造、ピラン環構造等が好ましい。
アルキル基の炭素数が1~18のアルキルアクリレートの具体例としては、メチルアクリレート、エチルアクリレート、n-プロピルアクリレート、n-ブチルアクリレート、s-ブチルアクリレート、2-エチルヘキシルアクリレートなどが挙げられ、好ましくは、メチルアクリレートが挙げられる。
アミド基を有するビニルモノマーの具体例としては、アクリルアミド、N-メチルアクリルアミド、N-ブチルアクリルアミド、N,N-ジメチルアクリルアミド、N,N-ジエチルアクリルアミド、アクリロイルモルホリン、N-ヒドロキシエチルアクリルアミド、アクリロイルピロリジン、アクリロイルピペリジン、メタクリルアミド、N-メチルメタクリルアミド、N-ブチルメタクリルアミド、N,N-ジメチルメタクリルアミド、N,N-ジエチルメタクリルアミド、メタクリロイルモルホリン、N-ヒドロキシエチルメタクリルアミド、メタクリロイルピロリジン、メタクリロイルピペリジン、N-ビニルホルムアミド、N-ビニルアセトアミド、ビニルピロリドン等が挙げられる。好ましくは、アクリロイルモルホリン、N,N-ジメチルアクリルアミド、N-ブチルアクリルアミド、ビニルピロリドン、2-ヒドロキシエチルメタクリレートが挙げられる。
エステル部分に炭素数5~22の脂環式炭化水素基を有するメタクリル酸エステルまたはアクリル酸エステルの具体例としては、例えば、アクリル酸シクロペンチル、アクリル酸シクロヘキシル、アクリル酸メチルシクロヘキシル、アクリル酸トリメチルシクロヘキシル、アクリル酸ノルボルニル、アクリル酸ノルボルニルメチル、アクリル酸シアノノルボルニル、アクリル酸イソボルニル、アクリル酸ボルニル、アクリル酸メンチル、アクリル酸フェンチル、アクリル酸アダマンチル、アクリル酸ジメチルアダマンチル、アクリル酸トリシクロ[5.2.1.02,6]デカ-8-イル、アクリル酸トリシクロ[5.2.1.02,6]デカ-4-メチル、アクリル酸シクロデシル、メタクリル酸シクロペンチル、メタクリル酸シクロヘキシル、メタクリル酸メチルシクロヘキシル、メタクリル酸トリメチルシクロヘキシル、メタクリル酸ノルボルニル、メタクリル酸ノルボルニルメチル、メタクリル酸シアノノルボルニル、メタクリル酸フェニルノルボルニル、メタクリル酸イソボルニル、メタクリル酸ボルニル、メタクリル酸メンチル、メタクリル酸フェンチル、メタクリル酸アダマンチル、メタクリル酸ジメチルアダマンチル、メタクリル酸トリシクロ[5.2.1.02,6]デカ-8-イル、メタクリル酸トリシクロ[5.2.1.02,6]デカ-4-メチル、メタクリル酸シクロデシル、メタクリル酸ジシクロペンタニル等が挙げられる。
好ましくは、メタクリル酸イソボルニル、メタクリル酸ジシクロペンタニル、メタクリル酸ジメチルアダマンチルなどが挙げられる。
N-置換マレイミドとしては、例えば、N-メチルマレイミド、N-エチルマレイミド、N-プロピルマレイミド、N-i-プロピルマレイミド、N-ブチルマレイミド、N-i-ブチルマレイミド、N-t-ブチルマレイミド、N-ラウリルマレイミド、N-シクロヘキシルマレイミド、N-ベンジルマレイミド、N-フェニルマレイミド、N-(2-クロロフェニル)マレイミド、N-(4-クロロフェニル)マレイミド、N-(4-ブロモフェニル)フェニルマレイミド、N-(2-メチルフェニル)マレイミド、N-(2-エチルフェニルマレイミド、N-(2-メトキシフェニル)マレイミド、N-(2,4,6-トリメチルフェニル)マレイミド、N-(4-ベンジルフェニル)マレイミド、N-(2,4,6-トリブロモフェニル)マレイミド等が挙げられる。好ましくは、N-メチルマレイミド、N-シクロヘキシルマレイミド、N-フェニルマレイミドなどが挙げられる。
これらのモノマーは市販のものをそのまま使用することができる。
アクリル系化合物は、透湿性の制御とセルロースエステルとの相溶性を両立する観点から、重量平均分子量(Mw)が15000以下の範囲であることが好ましく、10000以下の範囲内であることがより好ましく、さらに好ましくは、5000~10000の範囲内である。なお、本発明に係るアクリル系化合物の重量平均分子量(Mw)は、下記の測定条件によるゲルパーミエーションクロマトグラフィー(GPC)を用いた測定により算出する。

脂肪族多価アルコールの例には、エチレングリコール、プロピレングリコール、トリメチロールプロパン、ペンタエリスリトール等が含まれる。
モノカルボン酸は、脂肪族モノカルボン酸、脂環式モノカルボン酸、芳香族モノカルボン酸等であり得る。モノカルボン酸は、一種類であってもよいし、二種以上の混合物であってもよい。また、脂肪族多価アルコールに含まれるOH基の全部をエステル化してもよいし、一部をOH基のままで残してもよい。
脂肪族モノカルボン酸は、炭素数1~32の直鎖または側鎖を有する脂肪酸であることが好ましい。脂肪族モノカルボン酸の炭素数はより好ましくは1~20であり、さらに好ましくは1~10である。そのような脂肪族モノカルボン酸の例には、酢酸、プロピオン酸、酪酸、吉草酸等が含まれ、セルロースエステルとの相溶性を高めるためには、好ましくは酢酸であり得る。
脂環式モノカルボン酸の例には、シクロペンタンカルボン酸、シクロヘキサンカルボン酸、シクロオクタンカルボン酸等が含まれる。
芳香族モノカルボン酸の例には、安息香酸;安息香酸のベンゼン環にアルキル基またはアルコキシ基(例えばメトキシ基やエトキシ基)を1~3個を導入したもの(例えばトルイル酸等);ベンゼン環を2個以上有する芳香族モノカルボン酸(例えばビフェニルカルボン酸、ナフタリンカルボン酸、テトラリンカルボン酸等)が含まれ、好ましくは安息香酸である。
多価アルコールエステル系可塑剤の分子量は、特に制限されないが、300~1500の範囲内であることが好ましく、350~750の範囲内であることがより好ましい。揮発し難くするためには、分子量が大きい方が好ましい。透湿性を高め、セルロースエステルとの相溶性を高めるためには、分子量が小さい方が好ましい。
多価アルコールエステル系可塑剤の具体例には、トリメチロールプロパントリアセテート、トリメチロールプロパン安息香酸エステル、ペンタエリスリトールテトラアセテート、特開2008-88292号公報に記載の一般式(I)で表されるエステル化合物(A)等が含まれる。
多価カルボン酸エステル系可塑剤は、2価以上、好ましくは2~20価の多価カルボン酸と、アルコール化合物とのエステル化合物である。多価カルボン酸は、2~20価の脂肪族多価カルボン酸、3~20価の芳香族多価カルボン酸または3~20価の脂環式多価カルボン酸であることが好ましい。
多価カルボン酸の例には、トリメリット酸、トリメシン酸、ピロメリット酸のような3価以上の芳香族多価カルボン酸またはその誘導体;コハク酸、アジピン酸、アゼライン酸、セバシン酸、シュウ酸、フマル酸、マレイン酸、テトラヒドロフタル酸のような脂肪族多価カルボン酸;酒石酸、タルトロン酸、リンゴ酸、クエン酸のようなオキシ多価カルボン酸等が含まれ、フィルムからの揮発を抑制するためには、オキシ多価カルボン酸が好ましい。
アルコール化合物の例には、直鎖若しくは側鎖を有する脂肪族飽和アルコール化合物、直鎖若しくは側鎖を有する脂肪族不飽和アルコール化合物、脂環式アルコール化合物または芳香族アルコール化合物等が含まれる。脂肪族飽和アルコール化合物または脂肪族不飽和アルコール化合物の炭素数は、好ましくは1~32であり、より好ましくは1~20であり、さらに好ましくは1~10である。脂環式アルコール化合物の例には、シクロペンタノール、シクロヘキサノール等が含まれる。芳香族アルコール化合物の例には、フェノール、パラクレゾール、ジメチルフェノール、ベンジルアルコール、シンナミルアルコール等が含まれる。アルコール化合物は、一種類でもよいし、二種以上の混合物であってもよい。
多価カルボン酸エステル系可塑剤の分子量は、特に制限はないが、300~1000の範囲内であることが好ましく、350~750の範囲内であることがより好ましい。多価カルボン酸エステル系可塑剤の分子量は、ブリードアウトを抑制する観点では、大きい方が好ましい。透湿性やセルロースエステルとの相溶性の観点では、小さい方が好ましい。
多価カルボン酸エステル系可塑剤の酸価は、1mgKOH/g以下であることが好ましく、0.2mgKOH/g以下であることがさらに好ましい。酸価とは、試料1g中に含まれる酸(試料中に存在するカルボキシ基)を中和するために必要な水酸化カリウムのミリグラム数をいう。酸価は、JIS K0070(1992年)に準拠して測定したものである。
多価カルボン酸エステル系可塑剤の例には、特開2008-88292号公報に記載の一般式(II)で表されるエステル化合物(B)等が含まれる。
多価カルボン酸エステル系可塑剤は、フタル酸エステル系可塑剤であってもよい。フタル酸エステル系可塑剤の例には、ジエチルフタレート、ジメトキシエチルフタレート、ジメチルフタレート、ジオクチルフタレート、ジブチルフタレート、ジ-2-エチルヘキシルフタレート、ジシクロヘキシルフタレート、ジシクロヘキシルテレフタレート等が含まれる。
グリコレート系可塑剤の例には、アルキルフタリルアルキルグリコレート類が含まれる。アルキルフタリルアルキルグリコレート類の例には、メチルフタリルメチルグリコレート、エチルフタリルエチルグリコレート、プロピルフタリルプロピルグリコレート、ブチルフタリルブチルグリコレート、オクチルフタリルオクチルグリコレート等が含まれる。
エステル系可塑剤には、脂肪酸エステル系可塑剤、クエン酸エステル系可塑剤、リン酸エステル系可塑剤、トリメリット酸系可塑剤等が含まれる。
脂肪酸エステル系可塑剤の例には、オレイン酸ブチル、リシノール酸メチルアセチル、セバシン酸ジブチル等が含まれる。クエン酸エステル系可塑剤の例には、クエン酸アセチルトリメチル、クエン酸アセチルトリエチル、クエン酸アセチルトリブチル等が含まれる。リン酸エステル系可塑剤の例には、トリフェニルホスフェート、トリクレジルホスフェート、クレジルジフェニルホスフェート、オクチルジフェニルホスフェート、ジフェニルビフェニルホスフェート(BDP)、トリオクチルホスフェート、トリブチルホスフェート等が含まれる。トリメリット酸系可塑剤の例には、トリメリット酸オクチル、トリメリット酸n-オクチル、トリメリット酸イソデシル、トリメリット酸イソノニル等が含まれる。
スチレン系化合物(スチレン系可塑剤)は、は、スチレン系モノマーの単独重合体であってもよいし、スチレン系モノマーとそれ以外の共重合モノマーとの共重合体であってもよい。スチレン系化合物におけるスチレン系モノマー由来の構成単位の含有割合は、分子構造が一定以上の嵩高さを有するためには、好ましくは30~100モル%、より好ましくは50~100モル%でありうる。
スチレン系モノマーは、下記式(A)で表される化合物であることが好ましい。

式(A)中のR101~R103は、それぞれ独立に、水素原子または炭素数1~30のアルキル基またはアリール基を示す。R104は、水素原子、炭素数1~30のアルキル基、シクロアルキル基、アリール基、炭素数1~30のアルコキシ基、アリールオキシ基、炭素数2~30のアルキルオキシカルボニル基、アリールオキシカルボニル基、炭素数2~30のアルキルカルボニルオキシ基、アリールカルボニルオキシ基、水酸基、カルボキシル基、シアノ基、アミノ基、アミド基、ニトロ基を示す。これらの基は、それぞれ置換基(例えば水酸基、ハロゲン原子、アルキル基など)をさらに有してもよい。R104は、それぞれ同一であっても、異なってもよく、互いに結合して環を形成してもよい。
スチレン系モノマーの例には、スチレン;α-メチルスチレン、β-メチルスチレン、p-メチルスチレンなどのアルキル置換スチレン類;4-クロロスチレン、4-ブロモスチレンなどのハロゲン置換スチレン類;p-ヒドロキシスチレン、α-メチル-p-ヒドロキシスチレン、2-メチル-4-ヒドロキシスチレン、3,4-ジヒドロキシスチレンなどのヒドロキシスチレン類;ビニルベンジルアルコール類;p-メトキシスチレン、p-tert-ブトキシスチレン、m-tert-ブトキシスチレンなどのアルコキシ置換スチレン類;3-ビニル安息香酸、4-ビニル安息香酸などのビニル安息香酸類;4-ビニルベンジルアセテート;4-アセトキシスチレン;2-ブチルアミドスチレン、4-メチルアミドスチレン、p-スルホンアミドスチレンなどのアミドスチレン類;3-アミノスチレン、4-アミノスチレン、2-イソプロペニルアニリン、ビニルベンジルジメチルアミンなどのアミノスチレン類;3-ニトロスチレン、4-ニトロスチレンなどのニトロスチレン類;3-シアノスチレン、4-シアノスチレンなどのシアノスチレン類;ビニルフェニルアセトニトリル;フェニルスチレンなどのアリールスチレン類、インデン類などが含まれる。スチレン系モノマーは、一種類であっても、二種類以上を組み合わせてもよい。
スチレン系モノマーと組み合わされる共重合モノマーは、下記式(B)で表される(メタ)アクリル酸エステル化合物、無水マレイン酸、無水シトラコン酸、シス-1-シクロヘキセン-1,2-無水ジカルボン酸、3-メチル-シス-1-シクロヘキセン-1,2-無水ジカルボン酸、4-メチル-シス-1-シクロヘキセン-1,2-無水ジカルボン酸等の酸無水物、アクリロニトリル、メタクリロニトリルなどのニトリル基含有ラジカル重合性単量体;アクリルアミド、メタクリルアミド、トリフルオロメタンスルホニルアミノエチル(メタ)アクリレートなどのアミド結合含有ラジカル重合性単量体;酢酸ビニルなどの脂肪酸ビニル類;塩化ビニル、塩化ビニリデンなどの塩素含有ラジカル重合性単量体;1,3-ブタジエン、イソプレン、1,4-ジメチルブタジエン等の共役ジオレフィン類などが含まれ、好ましくは下記式(B)で表される(メタ)アクリル酸エステル化合物もしくは無水マレイン酸である。

式(B)中のR105~R107は、それぞれ独立に水素原子または炭素数1~30のアルキル基またはアリール基を示す。R108は、水素原子、炭素数1~30のアルキル基、シクロアルキル基、またはアリール基を示す。これらの基は、それぞれ置換基(例えば水酸基、ハロゲン原子、アルキル基など)をさらに有してもよい。
(メタ)アクリル酸エステル系化合物の例には、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸プロピル(i-、n-)、(メタ)アクリル酸ブチル(n-、i-、s-、tert-)、(メタ)アクリル酸ペンチル(n-、i-、s-)、(メタ)アクリル酸ヘキシル(n-、i-)、(メタ)アクリル酸ヘプチル(n-、i-)、(メタ)アクリル酸オクチル(n-、i-)、(メタ)アクリル酸ノニル(n-、i-)、(メタ)アクリル酸ミリスチル(n-、i-)、(メタ)アクリル酸(2-エチルヘキシル)、(メタ)アクリル酸(ε-カプロラクトン)、(メタ)アクリル酸(2-ヒドロキシエチル)、アクリル酸(2-ヒドロキシプロピル)、(メタ)アクリル酸(3-ヒドロキシプロピル)、(メタ)アクリル酸(4-ヒドロキシブチル)、(メタ)アクリル酸(2-ヒドロキシブチル)、アクリル酸(2-メトキシエチル)、(メタ)アクリル酸(2-エトキシエチル)アクリル酸フェニル、(メタ)メタクリル酸フェニル、(メタ)アクリル酸(2または4-クロロフェニル)、(メタ)アクリル酸(2または3または4-エトキシカルボニルフェニル)、(メタ)アクリル酸(oまたはmまたはp-トリル)、(メタ)アクリル酸ベンジル、(メタ)アクリル酸フェネチル、(メタ)アクリル酸(2-ナフチル)、(メタ)アクリル酸シクロヘキシル、(メタ)アクリル酸(4-メチルシクロヘキシル)、(メタ)アクリル酸(4-エチルシクロヘキシル)等が含まれる。
スチレン系化合物の具体例には、スチレン/無水マレイン酸共重合体、スチレン/アクリル酸エステル共重合体、スチレン/ヒドロキシスチレン重合体、スチレン/アセトキシスチレン重合体などが含まれる。なかでも、スチレン/無水マレイン酸共重合体が好ましい。
上記可塑剤の含有量は、特に制限されないが、セルロースエステル樹脂に対して、0.1~30重量%の範囲内であることが好ましく、5~20重量%の範囲内であることがより好ましい。このような量であれば、位相差フィルムがブリードアウトを生じにくい。
(その他の添加剤)
上述したように、本発明の位相差フィルムは、セルロースエステル樹脂、一般式(1)の化合物及び一般式(2)の化合物を必須に含み、必要であれば、可塑剤を含みうる。また、本発明の位相差フィルムは、上記可塑剤に代えてあるいは可塑剤に加えて、必要であれば、他の添加剤をさらに含みうる。このような他の添加剤としては、特に制限されないが、例えば、水素結合性化合物、活性剤、酸化防止剤、着色剤、紫外線吸収剤、マット剤、アクリル粒子、水素結合性溶媒、イオン性界面活性剤などが挙げられる。
上述したように、本発明の位相差フィルムは、セルロースエステル樹脂、一般式(1)の化合物及び一般式(2)の化合物を必須に含み、必要であれば、可塑剤を含みうる。また、本発明の位相差フィルムは、上記可塑剤に代えてあるいは可塑剤に加えて、必要であれば、他の添加剤をさらに含みうる。このような他の添加剤としては、特に制限されないが、例えば、水素結合性化合物、活性剤、酸化防止剤、着色剤、紫外線吸収剤、マット剤、アクリル粒子、水素結合性溶媒、イオン性界面活性剤などが挙げられる。
上記水素結合性化合物は、湿度の変化に対するレターデーション値Rtの変動を低減できる。ここで、水素結合性化合物としては、一分子中に少なくとも複数のヒドロキシ基、アミノ基、チオール基、カルボン酸基、から選ばれる官能基を有することが好ましく、一分子内に複数の異なる官能基を有することがより好ましく、一分子内にヒドロキシ基とカルボン酸基とを有することが特に好ましい。
当該水素結合性化合物は、母核として、1~2個の芳香族環を含有することが好ましく、一分子中に含有する前記官能基の数を、化合物の分子量で割った値が、0.01以上であることが好ましい。
上記効果は、前記セルロースエステルと水分子とが相互作用(水素結合)する部位に上記水素結合性化合物が結合(水素結合)し、水分子の脱着による電荷分布の変化を抑制するように作用するためと推定している。
具体的な化合物例としては、特開2012-82235号公報に記載の例示化合物E-104が挙げられる。
水素結合性化合物は、セルロースエステル100重量部に対して1~30重量部の範囲で添加することができる。
酸化防止剤は、特に制限されず、通常知られているものを使用することができる。特に、ラクトン系、イオウ系、フェノール系、二重結合系、ヒンダードアミン系、リン系の各化合物を好ましく用いることができる。
上記ラクトン系化合物としては、BASFジャパン株式会社から市販されている「IrgafosXP40、IrgafosXP60(商品名)」等が挙げられる。上記イオウ系化合物は、例えば、住友化学株式会社から市販されている「Sumilizer TPL-R」および「Sumilizer TP-D」を挙げることができる。上記フェノール系化合物としては、2,6-ジアルキルフェノールの構造を有するものが好ましく、例えば、BASFジャパン株式会社から市販されている「Irganox1076」、「Irganox1010」、(株)ADEKAから市販されている「アデカスタブAO-50」等を挙げることができる。上記二重結合系化合物は、住友化学株式会社から「Sumilizer GM」および「Sumilizer GS」という商品名で市販されている。一般には、樹脂に対して、0.05~20重量%、好ましくは0.1~1重量%の範囲で添加される。上記ヒンダードアミン系化合物は、例えば、BASFジャパン株式会社から市販されている「Tinuvin144」および「Tinuvin770」、株式会社ADEKAから市販されている「ADK STAB LA-52」を挙げることができる。上記リン系化合物は、例えば、住友化学株式会社から市販されている「SumilizerGP」、株式会社ADEKAから市販されている「ADK STAB PEP-24G」、「ADK STAB PEP-36」および「ADK STAB 3010」、BASFジャパン株式会社から市販されている「IRGAFOS P-EPQ」、堺化学工業株式会社から市販されている「GSY-P101」を挙げることができる。
さらに、酸捕捉剤として米国特許第4,137,201号明細書に記載されているような、エポキシ基を有する化合物を含有させることも可能である。
これらの酸化防止剤等は、再生使用される際の工程に合わせて適宜添加する量が決められるが、一般には、フィルムの主原料である樹脂(セルロースエステル)に対して、0.05~20重量%、好ましくは0.1~1重量%の範囲で添加される。
これらの酸化防止剤等は、1種のみを用いるよりも数種の異なった系の化合物を併用することで相乗効果を得ることができる。例えば、ラクトン系、リン系、フェノール系および二重結合系化合物の併用は好ましい。
着色剤は、染料や顔料を意味し、本発明では、液晶画面の色調を青色調にする効果またはイエローインデックスの調整、ヘイズの低減を有するものを指す。着色剤としては各種の染料、顔料が使用可能だが、アントラキノン染料、アゾ染料、フタロシアニン顔料などが有効である。
本発明のフィルムは、偏光板の視認側やバックライト側に用いられることが好ましいことから、紫外線吸収機能を付与することを目的として、紫外線吸収剤を含有することが好ましい。
紫外線吸収剤としては、特に限定されないが、例えば、ベンゾトリアゾール系、2-ヒドロキシベンゾフェノン系またはサリチル酸フェニルエステル系等の紫外線吸収剤が挙げられる。例えば、2-(5-メチル-2-ヒドロキシフェニル)ベンゾトリアゾール、2-[2-ヒドロキシ-3,5-ビス(α,α-ジメチルベンジル)フェニル]-2H-ベンゾトリアゾール、2-(3,5-ジ-t-ブチル-2-ヒドロキシフェニル)ベンゾトリアゾール等のトリアゾール類、2-ヒドロキシ-4-メトキシベンゾフェノン、2-ヒドロキシ-4-オクトキシベンゾフェノン、2,2’-ジヒドロキシ-4-メトキシベンゾフェノン等のベンゾフェノン類を例示することができる。
なお、紫外線吸収剤のうちでも、分子量が400以上の紫外線吸収剤は、昇華しにくいか、あるいは高沸点で揮発しにくく、フィルムの高温乾燥時にも飛散しにくいため、比較的少量の添加で効果的に耐候性を改良することができる観点から好ましい。
分子量が400以上の紫外線吸収剤としては、例えば、2-[2-ヒドロキシ-3,5-ビス(α,α-ジメチルベンジル)フェニル]-2-ベンゾトリアゾール、2,2-メチレンビス[4-(1,1,3,3-テトラブチル)-6-(2H-ベンゾトリアゾール-2-イル)フェノール]等のベンゾトリアゾール系、ビス(2,2,6,6-テトラメチル-4-ピペリジル)セバケート、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)セバケート等のヒンダードアミン系、さらには2-(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)-2-n-ブチルマロン酸ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)、1-[2-[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオニルオキシ]エチル]-4-[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオニルオキシ]-2,2,6,6-テトラメチルピペリジン等の分子内にヒンダードフェノールとヒンダードアミンの構造を共に有するハイブリッド系のものが挙げられ、これらは単独で、あるいは2種以上を併用して使用することができる。これらのうちでも、2-[2-ヒドロキシ-3,5-ビス(α,α-ジメチルベンジル)フェニル]-2-ベンゾトリアゾールや2,2-メチレンビス[4-(1,1,3,3-テトラブチル)-6-(2H-ベンゾトリアゾール-2-イル)フェノール]が、特に好ましい。
これら紫外線吸収剤としては、市販品を用いてもよく、例えば、BASFジャパン社製のチヌビン109、チヌビン171、チヌビン234、チヌビン326、チヌビン327、チヌビン328、チヌビン928等のチヌビンシリーズ、あるいは2,2’-メチレンビス[6-(2H-ベンゾトリアゾール-2-イル)-4-(1,1,3,3-テトラメチルブチル)フェノール](分子量659;市販品の例としては、株式会社ADEKA製のLA31)を好ましく使用できる。
上記紫外線吸収剤は、1種単独でまたは2種以上組み合わせて用いることができる。
紫外線吸収剤の使用量は、紫外線吸収剤の種類、使用条件等により一様ではないが、一般には、樹脂(セルロースエステル)に対して、0.05~10重量%、好ましくは0.1~5重量%の範囲で添加される。
マット剤は、フィルムの滑り性を付与する微粒子であり、得られるフィルムの透明性を損なうことがなく、溶融時の耐熱性があれば無機化合物または有機化合物どちらでもよい。これらのマット剤は、単独でも2種以上併用しても使用できる。粒径や形状(例えば針状と球状など)の異なる粒子を併用することで高度に透明性と滑り性を両立させることもできる。これらの中でも、前記アクリル共重合体や相溶させる樹脂として用いるセルロースエステルと屈折率が近いので透明性(ヘイズ)に優れる二酸化珪素が特に好ましく用いられる。
二酸化珪素の具体例としては、アエロジル200V、アエロジルR972V、アエロジルR972、R974、R812、200、300、R202、OX50、TT600、NAX50(以上日本アエロジル(株)製)、シーホスターKEP-10、シーホスターKEP-30、シーホスターKEP-50(以上、株式会社日本触媒製)、サイロホービック100(富士シリシア製)、ニップシールE220A(日本シリカ工業製)、アドマファインSO(アドマテックス製)等の商品名を有する市販品などが好ましく使用できる。
粒子の形状としては、不定形、針状、扁平、球状等特に制限なく使用できるが、特に球状の粒子を用いると得られるフィルムの透明性が良好にできるので好ましい。
粒子の大きさは、可視光の波長に近いと光が散乱し、透明性が悪くなるので、可視光の波長より小さいことが好ましく、さらに可視光の波長の1/2以下であることが好ましい。粒子の大きさが小さすぎると滑り性が改善されない場合があるので、80nmから180nmの範囲であることが特に好ましい。なお、粒子の大きさとは、粒子が1次粒子の凝集体の場合は凝集体の大きさを意味する。また、粒子が球状でない場合は、その投影面積に相当する円の直径を意味する。
マット剤は、樹脂(セルロースエステル)に対して、0.05~10重量%、好ましくは0.1~5重量%の範囲で添加されることが好ましい。
本発明のフィルムは、例えば、国際公開第2010/001668号パンフレットに記載のアクリル粒子を、透明性を維持できる範囲内の量で含有してもよい。該アクリル粒子は、フィルムの脆性を改善する作用がある。
このようなアクリル粒子の市販品の例としては、例えば、三菱レイヨン社製の「メタブレンW-341」、カネカ社製の「カネエース」、クレハ社製の「パラロイド」、ロームアンドハース社製の「アクリロイド」、ガンツ化成工業社製の「スタフィロイド」、ケミスノーMR-2G、MS-300X(以上、綜研化学(株)製)およびクラレ社製の「パラペットSA」などが挙げられ、これらは、単独または2種以上を用いることができる。
水素結合性溶媒は、溶液流延法でフィルムを作製する場合に、フィルムの構成材料を溶解するための溶媒に溶液粘度を調整(低減)する目的で、添加することができる。水素結合性溶媒とは、J.N.イスラエルアチビリ著、「分子間力と表面力」(近藤保、大島広行訳、マグロウヒル出版、1991年)に記載されるように、電気的に陰性な原子(酸素、窒素、フッ素、塩素)と電気的に陰性な原子と共有結合した水素原子間に生ずる、水素原子媒介「結合」を生ずることができるような有機溶媒、すなわち、結合モーメントが大きく、かつ水素を含む結合、例えば、O-H(酸素水素結合)、N-H(窒素水素結合)、F-H(フッ素水素結合)を含むことで近接した分子同士が配列できるような有機溶媒をいう。
これらは、アクリル共重合体やセルロースエステル樹脂、あるいは、相溶化させるための他の樹脂の混合体自身の分子間水素結合よりも、当該樹脂と水素結合性溶媒間との強い水素結合を形成させることで、溶液粘度の変化に期待できる。
本発明で行う溶液流延法においては、用いる該樹脂溶液に対して、溶液粘度を調整することに加えて、製膜時の剥離力を低下させる目的で、溶解のための溶媒に、水素結合性溶媒を一部あるいは全量用いることもできる。
イオン性界面活性剤は、製膜時の剥離力を低下させる目的で添加されうる。
本発明で用いることのできるイオン性界面活性剤としては、例えば、カチオン性界面活性剤、アニオン性界面活性剤、両性界面活性剤等が挙げられる。
カチオン性界面活性剤としては、例えば、脂肪族アミン塩、脂肪族4級アンモニウム塩、ベンザルコニウム塩、塩化ベンゼトニウム、ピリジニウム塩、イミダゾリニウム塩等が挙げられる。
アニオン性界面活性剤としては、高級アルコール(C8~C22)硫酸エステル塩類(例えば、ラウリルアルコールサルフェートのナトリウム塩、オクチルアルコールサルフェートのナトリウム塩、ラウリルアルコールサルフェートのアンモニウム塩、「Teepol-81」(商品名・シェル化学社製)、第二ナトリウムアルキルサルフェート等)、脂肪族アルコールリン酸エステル塩類(例えば、セチルアルコールリン酸エステルのナトリウム塩等)、アルキルアリールスルホン酸塩類(例えば、ドデシルベンゼンスルホン酸のナトリウム塩、イソプロピルナフタレンスルホン酸のナトリウム塩、ジナフタレンジスルホン酸のナトリウム塩、メタニトロベンゼンスルホン酸のナトリウム塩等)、アルキルアミドのスルホン酸塩類(例えば、C17H33CON(CH3)CH2SO3Na等)、二塩基性脂肪酸エステルのスルホン酸塩類(例えば、ナトリウムスルホコハク酸ジオクチルエステル、ナトリウムスルホコハク酸ジヘキシルエステル等)がある。これらの中で特に硫酸塩類やスルホン酸塩類が好適に用いられる。
両性界面活性剤としては、カルボキシベタイン型、スルホベタイン型、アミノカルボン酸塩、イミダゾリニウムベタイン等が挙げられる。
中でも、本発明においては、アニオン性界面活性剤が好ましい。また上記の界面活性剤は、フィルムを構成する樹脂の総量に対して、0.01重量%以上5重量%以下、好ましくは0.05重量%以上3重量%以下、より好ましくは0.2重量%以上2重量%以下で用いることが好ましい。この範囲よりも添加量が多いと、フィルムから界面活性剤が析出したり、フィルムの吸湿性が高くなり、位相差フィルムの品質に好ましくない品質が発現する。またこの範囲よりも添加量が少ないと界面活性剤を用いる本発明の効果が得られなくなったりする。
(フィルムの物性)
以下、本発明における位相差フィルムの物性等についての特徴について説明する。
以下、本発明における位相差フィルムの物性等についての特徴について説明する。
(透明性)
本発明の位相差フィルムの透明性を判断する指標としては、ヘイズ値(濁度)を用いる。特に屋外で用いられる液晶表示装置においては、明るい場所でも十分な輝度や高いコントラストが得られることが求められるため、ヘイズ値は1.0%以下であることが好ましく、0.5%以下であることがさらに好ましい。散乱フィルムとして用いる場合は、ヘイズ値は上記の範囲を超えていてもよい。
本発明の位相差フィルムの透明性を判断する指標としては、ヘイズ値(濁度)を用いる。特に屋外で用いられる液晶表示装置においては、明るい場所でも十分な輝度や高いコントラストが得られることが求められるため、ヘイズ値は1.0%以下であることが好ましく、0.5%以下であることがさらに好ましい。散乱フィルムとして用いる場合は、ヘイズ値は上記の範囲を超えていてもよい。
また、本発明のフィルムは、その全光線透過率が90%以上であることが好ましく、より好ましくは93%以上である。また、現実的な上限としては、99%程度である。
ヘイズ値および透過率はヘイズメーターを用いて測定することができる。
上記のような物性を満たすフィルムは、大型の液晶表示装置や屋外用途の液晶表示装置用の偏光板保護フィルムとしても好ましく用いることができる。
(フィルムの製造方法)
次に、本発明の位相差フィルムの製造方法について説明する。本発明はこれに限定されるものではない。
次に、本発明の位相差フィルムの製造方法について説明する。本発明はこれに限定されるものではない。
本発明のフィルムの製造方法としては、通常のインフレーション法、T-ダイ法、カレンダー法、切削法、流延法、エマルジョン法、ホットプレス法等の製造法が使用できるが、着色抑制、異物欠点の抑制、ダイラインなどの光学欠点の抑制などの観点から製膜方法は、溶液流延製膜法と溶融流延製膜法が好ましく、特に溶液流延製膜法であることが、均一な表面を得るためにより好ましい。
〈溶液流延製膜法〉
溶液流涎法により製膜する場合、本発明のフィルムの製造方法は、セルロースエステル樹脂、上記一般式(1)の化合物および上記一般式(2)の化合物、ならびに必要であれば上述した可塑剤および/または添加剤を溶媒に溶解させてドープを調製する工程(溶解工程;ドープ調製工程)、ドープを無限に移行する無端の金属支持体上に流延する工程(流延工程)、流延したドープをウェブとして乾燥する工程(溶媒蒸発工程)、金属支持体から剥離する工程(剥離工程)、乾燥、延伸、幅保持する工程(延伸・幅保持・乾燥工程)、仕上がったフィルムを巻取る工程(巻き取り工程)を含むことが好ましい。
溶液流涎法により製膜する場合、本発明のフィルムの製造方法は、セルロースエステル樹脂、上記一般式(1)の化合物および上記一般式(2)の化合物、ならびに必要であれば上述した可塑剤および/または添加剤を溶媒に溶解させてドープを調製する工程(溶解工程;ドープ調製工程)、ドープを無限に移行する無端の金属支持体上に流延する工程(流延工程)、流延したドープをウェブとして乾燥する工程(溶媒蒸発工程)、金属支持体から剥離する工程(剥離工程)、乾燥、延伸、幅保持する工程(延伸・幅保持・乾燥工程)、仕上がったフィルムを巻取る工程(巻き取り工程)を含むことが好ましい。
図1は、本発明に好ましい溶液流延製膜法のドープ調製工程、流延工程および乾燥工程(溶媒蒸発工程)の一例を模式的に示した図である。
仕込釜41より濾過器44で大きな凝集物を除去し、ストック釜42へ送液する。その後、ストック釜42より主ドープ溶解釜1へ各種添加液を添加する。
その後主ドープは主濾過器3にて濾過され、これに添加剤添加液が16よりインライン添加される。
多くの場合、主ドープには返材が10~50重量%程度含まれることがある。
返材とは、位相差フィルムを細かく粉砕した物で、位相差フィルムを製膜するときに発生する、フィルムの両サイド部分を切り落とした物や、擦り傷などでスペックアウトしたフィルム原反が使用される。
また、ドープ調製に用いられる樹脂の原料としては、あらかじめセルロースエステル樹脂、上記一般式(1)の化合物および上記一般式(2)の化合物、ならびに必要であれば上述した可塑剤および/または添加剤(以下、単に「その他の化合物」とも称する)などをペレット化したものも、好ましく用いることができる。
以下、各工程について説明する。
1)溶解工程(ドープ調製工程)
本工程は、セルロースエステルに対する良溶媒を主とする溶媒に、溶解釜中で該セルロースエステル、上記一般式(1)の化合物および上記一般式(2)の化合物、場合によって、その他の化合物を攪拌しながら溶解しドープを形成する工程、あるいは該セルロースエステル溶液に、場合によってその他の化合物溶液を混合して主溶解液であるドープを形成する工程である。
本工程は、セルロースエステルに対する良溶媒を主とする溶媒に、溶解釜中で該セルロースエステル、上記一般式(1)の化合物および上記一般式(2)の化合物、場合によって、その他の化合物を攪拌しながら溶解しドープを形成する工程、あるいは該セルロースエステル溶液に、場合によってその他の化合物溶液を混合して主溶解液であるドープを形成する工程である。
ドープ中のセルロースエステルの濃度は、濃い方が金属支持体に流延した後の乾燥負荷が低減できて好ましいが、セルロースエステルの濃度が濃過ぎると濾過時の負荷が増えて、濾過精度が悪くなる。これらを両立する濃度としては、10~35重量%が好ましく、更に好ましくは、15~25重量%である。
ドープで用いられる溶媒は、単独で用いても2種以上を併用してもよいが、セルロースエステルの良溶媒と貧溶媒を混合して使用することが生産効率の点で好ましく、良溶媒が多い方がセルロースアセテートの溶解性の点で好ましい。
良溶媒と貧溶媒の混合比率の好ましい範囲は、良溶媒が70~98重量%であり、貧溶剤が2~30重量%である。良溶媒、貧溶媒とは、使用するセルロースエステルを単独で溶解するものを良溶媒、単独で膨潤するかまたは溶解しないものを貧溶媒と定義している。そのため、セルロースエステルの平均置換度によって良溶媒、貧溶媒が変わる。
本発明に用いられる良溶媒は特に限定されないが、メチレンクロライド等の有機ハロゲン化合物やジオキソラン類、アセトン、酢酸メチル、アセト酢酸メチル等が挙げられる。特に好ましくはメチレンクロライドまたは酢酸メチルが挙げられる。
また、本発明に用いられる貧溶媒は特に限定されないが、例えば、メタノール、エタノール、n-ブタノール、シクロヘキサン、シクロヘキサノン等が好ましく用いられる。また、ドープ中には水が0.01~2重量%含有していることが好ましい。
また、セルロースエステルの溶解に用いられる溶媒は、フィルム製膜工程で乾燥によりフィルムから除去された溶媒を回収し、これを再利用して用いられる。
回収溶媒中に、セルロースアセテートに添加されている添加剤、例えば可塑剤、紫外線吸収剤、ポリマー、モノマー成分などが微量含有されていることもあるが、これらが含まれていても好ましく再利用することができるし、必要であれば精製して再利用することもできる。
上記記載のドープを調製する時の、セルロースエステルの溶解方法としては、一般的な方法を用いることができる。具体的には、常圧で行う方法、主溶媒の沸点以下で行う方法、主溶媒の沸点以上で加圧して行う方法、特開平9-95544号公報、特開平9-95557号公報、または特開平9-95538号公報に記載の如き冷却溶解法で行う方法、特開平11-21379号公報に記載されている高圧で行う方法等種々の溶解方法を用いることができる。中でも、主溶媒の沸点以上で加圧して行う方法が好ましく、加熱と加圧を組み合わせると常圧における沸点以上に加熱できる。
また、溶媒の常圧での沸点以上でかつ加圧下で溶媒が沸騰しない範囲の温度で加熱しながら攪拌溶解する方法も、ゲルやママコと呼ばれる塊状未溶解物の発生を防止するため好ましい。
また、セルロースアセテートを貧溶媒と混合して湿潤あるいは膨潤させた後、更に良溶剤を添加して溶解する方法も好ましく用いられる。
加圧は窒素ガス等の不活性気体を圧入する方法や、加熱によって溶媒の蒸気圧を上昇させる方法によって行ってもよい。加熱は外部から行うことが好ましく、例えばジャケットタイプのものは温度コントロールが容易で好ましい。
溶媒を添加しての加熱温度は、高い方がセルロースアセテートの溶解性の観点から好ましいが、加熱温度が高過ぎると必要とされる圧力が大きくなり生産性が悪くなる。
好ましい加熱温度は45~120℃であり、60~110℃がより好ましく、70℃~105℃が更に好ましい。また、圧力は設定温度で溶媒が沸騰しないように調整される。
または冷却溶解法も好ましく用いられ、これによって酢酸メチルなどの溶媒にセルロースエステルを溶解させることができる。
次に、このセルロースエステル溶液(溶解中または溶解後のドープ)を濾紙等の適当な濾過材を用いて濾過することが好ましい。
濾過材としては、不溶物等を除去するために絶対濾過精度が小さい方が好ましいが、絶対濾過精度が小さ過ぎると濾過材の目詰まりが発生し易いという問題がある。このため絶対濾過精度0.008mm以下の濾材が好ましく、0.001~0.008mmの濾材がより好ましく、0.003~0.007mmの濾材が更に好ましい。
濾材の材質は特に制限はなく、通常の濾材を使用することができるが、ポリプロピレン、テフロン(登録商標)等のプラスチック製の濾材や、ステンレススティール等の金属製の濾材が繊維の脱落等がなく好ましい。
濾過により、原料のセルロースアセテートに含まれていた不純物、特に輝点異物を除去、低減することが好ましい。
輝点異物とは、2枚の偏光板をクロスニコル状態にして配置し、その間にフィルム等を置き、一方の偏光板の側から光を当てて、他方の偏光板の側から観察した時に反対側からの光が漏れて見える点(異物)のことであり、径が0.01mm以上である輝点数が200個/cm2以下であることが好ましい。より好ましくは100個/cm2以下であり、更に好ましくは50個/m2以下であり、更に好ましくは0~10個/cm2以下である。また、0.01mm以下の輝点も少ない方が好ましい。
ドープの濾過は通常の方法で行うことができるが、溶媒の常圧での沸点以上で、かつ加圧下で溶媒が沸騰しない範囲の温度で加熱しながら濾過する方法が、濾過前後の濾圧の差(差圧という)の上昇が小さく、好ましい。
好ましい温度は45~120℃であり、45~70℃がより好ましく、45~55℃であることが更に好ましい。
濾圧は小さい方が好ましい。濾圧は1.6MPa以下であることが好ましく、1.2MPa以下であることがより好ましく、1.0MPa以下であることが更に好ましい。
2)流延工程
続いて、ドープを金属支持体上に流延(キャスト)する。すなわち、本工程は、ドープを、送液ポンプ(例えば、加圧型定量ギヤポンプ)を通して加圧ダイ30に送液し、無限に移送する無端の金属ベルト31、例えばステンレスベルト、あるいは回転する金属ドラム等の金属支持体上の流延位置に、加圧ダイスリットからドープを流延する工程である。
続いて、ドープを金属支持体上に流延(キャスト)する。すなわち、本工程は、ドープを、送液ポンプ(例えば、加圧型定量ギヤポンプ)を通して加圧ダイ30に送液し、無限に移送する無端の金属ベルト31、例えばステンレスベルト、あるいは回転する金属ドラム等の金属支持体上の流延位置に、加圧ダイスリットからドープを流延する工程である。
ダイの口金部分のスリット形状を調整でき、膜厚を均一にし易い加圧ダイが好ましい。加圧ダイには、コートハンガーダイやTダイ等があり、いずれも好ましく用いられる。金属支持体の表面は鏡面となっていることが好ましい。製膜速度を上げるために加圧ダイを金属支持体上に2基以上設け、ドープ量を分割して重層してもよい。あるいは複数のドープを同時に流延する共流延法によって積層構造のフィルムを得ることも好ましい。
キャストの幅は生産性の観点から1.4m以上が好ましい。より好ましくは1.4~4mである。4mを超える場合には、製造工程で縞が入ったり、その後の搬送工程での安定性が低くなったりするおそれがある。さらに好ましくは、搬送性、生産性の点で2.2~3.5mである。
流延(キャスト)工程における金属支持体は、表面を鏡面仕上げしたものが好ましく、金属支持体としては、ステンレススティールベルトもしくは鋳物で表面をメッキ仕上げしたドラムが好ましく用いられる。
流延工程の金属支持体の表面温度は-50℃~溶媒の沸点未満の温度で、温度が高い方がウェブの乾燥速度が速くできるので好ましいが、余り高過ぎるとウェブが発泡したり、平面性が劣化する場合がある。
好ましい支持体温度は0~55℃であり、25~50℃が更に好ましい。あるいは、冷却することによってウェブをゲル化させて残留溶媒を多く含んだ状態でドラムから剥離することも好ましい方法である。
金属支持体の温度を制御する方法は特に制限されないが、温風または冷風を吹きかける方法や、温水を金属支持体の裏側に接触させる方法がある。温水を用いる方が熱の伝達が効率的に行われるため、金属支持体の温度が一定になるまでの時間が短く好ましい。温風を用いる場合は目的の温度よりも高い温度の風を使う場合がある。
3)溶媒蒸発工程
本工程は、ウェブ(流延用支持体上にドープを流延し、形成されたドープ膜をウェブと呼ぶ)を流延用支持体上で加熱し、溶媒を蒸発させる工程である。
本工程は、ウェブ(流延用支持体上にドープを流延し、形成されたドープ膜をウェブと呼ぶ)を流延用支持体上で加熱し、溶媒を蒸発させる工程である。
溶媒を蒸発させるには、ウェブ側から風を吹かせる方法および/または支持体の裏面から液体により伝熱させる方法、輻射熱により表裏から伝熱する方法等があるが、裏面液体伝熱方法が、乾燥効率が良く好ましい。また、それらを組み合わせる方法も好ましく用いられる。流延後の支持体上のウェブを40~100℃の雰囲気下、支持体上で乾燥させることが好ましい。40~100℃の雰囲気下に維持するには、この温度の温風をウェブ上面に当てるか赤外線等の手段により加熱することが好ましい。
面品質、透湿性、剥離性の観点から、30~120秒以内で該ウェブを支持体から剥離することが好ましい。
4)剥離工程
次いで、ウェブを金属支持体から剥離する。すなわち、本工程は金属支持体上で溶媒が蒸発したウェブを、剥離位置で剥離する工程である。剥離されたウェブは次工程に送られる。
次いで、ウェブを金属支持体から剥離する。すなわち、本工程は金属支持体上で溶媒が蒸発したウェブを、剥離位置で剥離する工程である。剥離されたウェブは次工程に送られる。
金属支持体上の剥離位置における温度は-50~40℃の範囲内とするのが好ましく、10~40℃の範囲内がより好ましく、15~30℃の範囲内とするのが最も好ましい。
なお、剥離する時点での金属支持体上でのウェブの剥離時残留溶媒量は、乾燥の条件の強弱、金属支持体の長さ等によって適宜調節される。位相差フィルムが良好な平面性を示すためには、金属支持体からウェブを剥離する際の残留溶媒量は10~150重量%が好ましい。残留溶媒量がより多い時点で剥離する場合、ウェブが柔らか過ぎると剥離時平面性を損ね、剥離張力によるツレや縦スジが発生し易いため、経済速度と品質との兼ね合いで剥離時の残留溶媒量が決められる。更に好ましくは20~40重量%または60~130重量%であり、特に好ましくは、20~30重量%または70~120重量%である。
本発明においては、残留溶媒量は下記式で定義される。
なお、Mはウェブまたはフィルムを製造中または製造後の任意の時点で採取した試料の重量で、NはMを115℃で1時間の加熱後の重量である。
金属支持体とフィルムを剥離する際の剥離張力は、300N/m以下とすることが好ましい。より好ましくは、196~245N/mの範囲内であるが、剥離の際に皺が入り易い場合、190N/m以下、好ましくは100~190N/mの張力で剥離することが好ましい。
5)乾燥・延伸・幅保持工程
(乾燥)
位相差フィルムの乾燥工程においては、ウェブを金属支持体より剥離し、更に乾燥し、残留溶媒量を1重量%以下にすることが好ましく、更に好ましくは0.1重量%以下であり、特に好ましくは0~0.01重量%以下である。
(乾燥)
位相差フィルムの乾燥工程においては、ウェブを金属支持体より剥離し、更に乾燥し、残留溶媒量を1重量%以下にすることが好ましく、更に好ましくは0.1重量%以下であり、特に好ましくは0~0.01重量%以下である。
フィルム乾燥工程では一般にロール乾燥方式(上下に配置した多数のロールにウェブを交互に通し乾燥させる方式)やテンター方式でウェブを搬送させながら乾燥する方式が採られる。例えば、剥離後、ウェブを乾燥装置内に複数配置したローラに交互に通して搬送する乾燥装置35、および/またはクリップでウェブの両端をクリップして搬送するテンター延伸装置34を用いて、ウェブを乾燥する。
ウェブを乾燥させる手段は特に制限なく、一般的に熱風、赤外線、加熱ロール、マイクロ波等で行うことができるが、簡便さの点で熱風で行うことが好ましい。余り急激な乾燥は出来上がりのフィルムの平面性を損ね易い。高温による乾燥は残留溶媒が8重量%以下くらいから行うのがよい。全体を通し、乾燥はおおむね40~250℃の範囲内で行われる。特に40~200℃の範囲内で乾燥させることが好ましい。乾燥温度は、段階的に高くしていくことが好ましい。
テンター延伸装置を用いる場合は、テンターの左右把持手段によってフィルムの把持長(把持開始から把持終了までの距離)を左右で独立に制御できる装置を用いることが好ましい。また、テンター工程において、平面性を改善するため意図的に異なる温度を持つ区画を作ることも好ましい。
また、異なる温度区画の間にそれぞれの区画が干渉を起こさないように、ニュートラルゾーンを設けることも好ましい。
(延伸・幅保持)
続いて、金属支持体よりウェブを少なくとも一方向に延伸処理することが好ましい。延伸処理することでフィルム内の分子の配向を制御することができる。本発明において目標とするレターデーション値Ro、Rtを得るには、位相差フィルムが本発明の構成をとり、更に搬送張力の制御、延伸操作により屈折率制御を行うことが好ましい。例えば、長手方向の張力を低くまたは高くすることでレターデーション値を変動させることが可能となる。
続いて、金属支持体よりウェブを少なくとも一方向に延伸処理することが好ましい。延伸処理することでフィルム内の分子の配向を制御することができる。本発明において目標とするレターデーション値Ro、Rtを得るには、位相差フィルムが本発明の構成をとり、更に搬送張力の制御、延伸操作により屈折率制御を行うことが好ましい。例えば、長手方向の張力を低くまたは高くすることでレターデーション値を変動させることが可能となる。
具体的な延伸方法としては、フィルムの長手方向(製膜方向;流延方向;MD方向)およびフィルム面内で直交する方向、即ち幅手方向(TD方向)に対して、逐次または同時に2軸延伸もしくは1軸延伸することができる。好ましくは、流延方向(MD方向)、幅手方向(TD方向)に二軸延伸を実施した、二軸延伸フィルムであるが、本発明に係る位相差フィルムは一軸延伸フィルムであってもよいし、未延伸フィルムであってもよい。なお、延伸操作は多段階に分割して実施してもよい。また、二軸延伸を行う場合には同時二軸延伸を行ってもよいし、段階的に実施してもよい。この場合、段階的とは、例えば、延伸方向の異なる延伸を順次行うことも可能であるし、同一方向の延伸を多段階に分割し、かつ異なる方向の延伸をそのいずれかの段階に加えることも可能である。即ち、例えば、次のような延伸ステップも可能である:
・流延方向に延伸→幅手方向に延伸→流延方向に延伸→流延方向に延伸
・幅手方向に延伸→幅手方向に延伸→流延方向に延伸→流延方向に延伸
また、同時2軸延伸には、一方向に延伸し、もう一方を、張力を緩和して収縮させる場合も含まれる。
・流延方向に延伸→幅手方向に延伸→流延方向に延伸→流延方向に延伸
・幅手方向に延伸→幅手方向に延伸→流延方向に延伸→流延方向に延伸
また、同時2軸延伸には、一方向に延伸し、もう一方を、張力を緩和して収縮させる場合も含まれる。
互いに直交する2軸方向の延伸倍率は、それぞれ最終的には流延方向に0.8~1.5倍、幅手方向に1.1~2.5倍の範囲とすることが好ましく、流延方向に0.8~1.0倍、幅手方向に1.2~2.0倍に範囲で行うことが好ましい。
延伸温度は、通常、フィルムを構成する樹脂のTg~Tg+60℃の温度範囲で行われることが好ましい。通常、延伸温度は120℃~200℃が好ましく、さらに好ましくは150℃~200℃であり、さらに好ましくは150℃を超えて190℃以下で延伸するのが好ましい。
延伸時におけるフィルム中の残留溶媒は20~0%が好ましく、さらに好ましくは15~0%で延伸するのが好ましい。例えば、155℃で残留溶媒が11%で延伸する、あるいは155℃で残留溶媒が2%で延伸するのが好ましい。もしくは160℃で残留溶媒が15%で延伸するのが好ましく、あるいは160℃で残留溶媒が1%未満で延伸するのが好ましい。
ウェブを延伸する方法には特に限定はない。例えば、複数のロールに周速差をつけ、その間でロール周速差を利用して縦方向に延伸する方法、ウェブの両端をクリップやピンで固定し、クリップやピンの間隔を進行方向に広げて縦方向に延伸する方法、同様に横方向に広げて横方向に延伸する方法、あるいは縦横同時に広げて縦横両方向に延伸する方法などが挙げられる。もちろんこれらの方法は、組み合わせて用いてもよい。中でも、ウェブの両端をクリップ等で把持するテンター方式で幅方向(横方向)に延伸を行うことが特に好ましい。
また、所謂テンター法の場合、リニアドライブ方式でクリップ部分を駆動すると滑らかな延伸を行うことができ、破断等の危険性が減少できるので好ましい。
製膜工程のこれらの幅保持あるいは横方向の延伸はテンターによって行うことが好ましく、ピンテンターでもクリップテンターでもよい。
本発明の位相差フィルムの遅相軸または進相軸がフィルム面内に存在し、製膜方向とのなす角をθ1とするとθ1は-1°以上+1°以下であることが好ましく、-0.5°以上+0.5°以下であることがより好ましい。
このθ1は配向角として定義でき、θ1の測定は、自動複屈折計KOBRA-21ADH(王子計測機器)を用いて行うことができる。θ1が各々上記関係を満たすことは、表示画像において高い輝度を得ること、光漏れを抑制または防止することに寄与でき、カラー液晶表示装置においては忠実な色再現を得ることに寄与できる。
6)巻き取り工程
最後に、得られたウェブ(仕上がったフィルム)を巻取ることにより、位相差フィルムが得られる。より具体的には、ウェブ中の残留溶媒量が2重量%以下となってから位相差フィルムとして巻き取り機37により巻き取る工程であり、残留溶媒量を0.4重量%以下にすることにより寸法安定性の良好なフィルムを得ることができる。特に0.00~0.10重量%の範囲で巻き取ることが好ましい。
最後に、得られたウェブ(仕上がったフィルム)を巻取ることにより、位相差フィルムが得られる。より具体的には、ウェブ中の残留溶媒量が2重量%以下となってから位相差フィルムとして巻き取り機37により巻き取る工程であり、残留溶媒量を0.4重量%以下にすることにより寸法安定性の良好なフィルムを得ることができる。特に0.00~0.10重量%の範囲で巻き取ることが好ましい。
巻き取り方法は、一般に使用されているものを用いればよく、定トルク法、定テンション法、テーパーテンション法、内部応力一定のプログラムテンションコントロール法等があり、それらを使いわければよい。
巻き取る前に、製品となる幅に端部をスリットして裁ち落とし、巻き中の貼り付きや擦り傷防止のために、ナール加工(エンボッシング加工)を両端に施してもよい。ナール加工の方法は凸凹のパターンを側面に有する金属リングを加熱や加圧により加工することができる。なお、フィルム両端部のクリップの把持部分は、通常はフィルムが変形しており、製品として使用できないので切除される。熱による材料の劣化が起こっていない場合は、回収後に再利用される。
本発明のフィルムは、長尺フィルムであることが好ましく、具体的には、100m~10000m程度のものを示し、通常、ロール状で提供される形態のものである。また、フィルムの幅は、液晶表示装置の大型化や生産の効率化の要求に応えるべく、1.4~4mであることが好ましく、1.4~4mであることがより好ましく、2~3mであることがさらに好ましい。
〈溶融流延製膜法〉
また、本発明の位相差フィルムは、溶融流延法により製膜することもできる。
また、本発明の位相差フィルムは、溶融流延法により製膜することもできる。
溶融製膜法とは、セルロースエステルおよび上述した添加剤を含む組成物を、流動性を示す温度まで加熱溶融し、その後、流動性のセルロースを含む溶融物を流延する方法をいう。
加熱溶融する成形方法としては、詳細には、溶融押出成形法、プレス成形法、インフレーション法、射出成形法、ブロー成形法、延伸成形法などに分類できる。これらの成形法の中では、機械的強度および表面精度などの点から、溶融押出し法が好ましい。溶融押出し法に用いる複数の原材料は、通常あらかじめ混錬してペレット化しておくことが好ましい。
ペレット化は、公知の方法でよく、例えば、乾燥セルロースエステルや可塑剤、その他添加剤をフィーダーで押出し機に供給し、1軸や2軸の押出し機を用いて混錬し、ダイからストランド状に押出し、水冷または空冷し、カッティングすることで行うことができる。
添加剤は、押出し機に供給する前に混合しておいてもよいし、それぞれ個別のフィーダーで供給してもよい。
粒子や酸化防止剤等の少量の添加剤は、均一に混合するため、事前に混合しておくことが好ましい。
押出し機は、剪断力を抑え、樹脂が劣化(分子量低下、着色、ゲル生成等)しないようにペレット化可能で、なるべく低温で加工することが好ましい。例えば、2軸押出し機の場合、深溝タイプのスクリューを用いて、同方向に回転させることが好ましい。混錬の均一性から、噛み合いタイプが好ましい。
以上のようにして得られたペレットを用い、フィルム製膜を行う。もちろんペレット化せず、原材料の粉末をそのままフィーダーで押出し機に供給し、そのままフィルム製膜することも可能である。
上記ペレットを、1軸や2軸タイプの押出し機を用いて押出す際の溶融温度は、200~300℃の温度範囲とし、リーフディスクタイプのフィルターなどで濾過し、異物を除去した後、Tダイからフィルム状に流延し、冷却ローラと弾性タッチローラでフィルムをニップし、冷却ローラ上で固化させる。
供給ホッパーから押出し機へ導入する際、真空下または減圧下や不活性ガス雰囲気下にして、酸化分解等を防止する方法も好ましい。
押出し流量は、ギヤポンプを導入するなどして安定に行うことが好ましい。また、異物の除去に用いるフィルターは、ステンレス繊維焼結フィルターが好ましく用いられる。ステンレス繊維焼結フィルターは、ステンレス繊維体の複雑に絡み合った状態を作り出した上で圧縮し、接触箇所を焼結し一体化したもので、その繊維の太さと圧縮量により密度を変え、濾過精度を調整できる。
可塑剤や粒子などの添加剤は、あらかじめ樹脂と混合しておいてもよいし、押出し機の途中で練り込んでもよい。均一に添加するために、スタチックミキサーなどの混合装置を用いることが好ましい。
冷却ローラと弾性タッチローラによりフィルムをニップする際、タッチローラ側のフィルム温度は、フィルムのTg~Tg+110℃の温度範囲にすることが好ましい。このような目的で使用する弾性体表面を有するローラは、公知のローラが使用できる。
弾性タッチローラは、挟圧回転体ともいう。弾性タッチローラとしては、市販されているものを用いることもできる。
冷却ローラからフィルムを剥離する際、張力を制御してフィルムの変形を防止することが好ましい。
また、上記のようにして得られたフィルムは、冷却ローラに接する工程を通過した後、前記延伸操作により延伸することが好ましい。
延伸する方法は、公知のローラ延伸機やテンターなどを好ましく用いることができる。具体的な条件は溶液流涎法の場合と同様である。
最後に、溶液流涎法の場合と同様に、上記のようにして得られたフィルムを巻取ることにより、位相差フィルムが得られる。
<偏光板および液晶表示装置の構成>
本発明の位相差フィルムは、薄膜でも所望の位相差を発現することができる。したがって、本発明の位相差フィルムは、偏光板および液晶表示装置に好適に使用できる。
本発明の位相差フィルムは、薄膜でも所望の位相差を発現することができる。したがって、本発明の位相差フィルムは、偏光板および液晶表示装置に好適に使用できる。
本発明の位相差フィルムが具備される偏光板および液晶表示装置の構成について、図面を参照して説明する。
図2は、本発明の位相差フィルムが具備された偏光板および当該偏光板が具備された液晶表示装置構成の一例を示す概略断面図である。
本発明の一実施形態に係る偏光板101Aは、少なくとも、保護フィルム102と、活性エネルギー線硬化性接着剤103Aと、偏光子104とがこの順序で積層されており、更に、当該保護フィルムが配置されている面とは反対側の偏光子面に、活性エネルギー線硬化性接着剤103Bと、本発明に係る位相差フィルム105を積層した構成であることが好ましい態様である。すなわち、偏光板101Aは、本発明に係る位相差フィルム105が、活性エネルギー線硬化性接着剤103Bで偏光子104と接着されている構成を有する。
また、保護フィルム102の更に外側(最表面部)には、必要に応じて、例えば、防眩層、反射防止層、防汚層、およびハードコート層等の機能性層106を設けても良い。
上記偏光板101Aの位相差フィルム105は液晶セル107と粘着剤または接着剤等を介して貼合され、偏光板101Aと液晶セル107の貼合された面の反対側の液晶セル面(バックライト側:図ではBLと記載。)には、偏光板101Aと同じ構成の偏光板101Bの本発明の位相差フィルム105が貼合されて、液晶表示装置108を構成することが好ましい。
〈接着剤〉
図2に示す形態において、位相差フィルム105と偏光子104とは活性エネルギー線硬化性接着剤103Bを介して接着されている。活性エネルギー線硬化性接着剤を用いることが透湿性を効果的に制御できることから好ましい。ただし、本発明においては、活性エネルギー線硬化性接着剤のみならず、ウレタン系粘着剤、エポキシ系粘着剤、水性高分子-イソシアネート系粘着剤、熱硬化型アクリル粘着剤等の硬化型粘着剤、湿気硬化ウレタン粘着剤、ポリエーテルメタクリレート型、エステル系メタクリレート型、酸化型ポリエーテルメタクリレート等の嫌気性粘着剤、シアノアクリレート系の瞬間粘着剤、アクリレートとペルオキシド系の2液型瞬間粘着剤などを用いることができる。上記粘着剤としては1液型であってもよいし、使用前に2液以上を混合して使用する2液型であってもよい。接着剤は、有機溶媒を媒体とする溶媒系であってもよいし、水を主成分とする媒体であるエマルジョン型、コロイド分散液型、水溶液型等の水系であってもよいし、無溶媒型であってもよい。接着剤液の濃度は、接着後の膜厚、塗布方法、塗布条件等により適宜決定されれば良く、通常は0.1~50重量%である。
図2に示す形態において、位相差フィルム105と偏光子104とは活性エネルギー線硬化性接着剤103Bを介して接着されている。活性エネルギー線硬化性接着剤を用いることが透湿性を効果的に制御できることから好ましい。ただし、本発明においては、活性エネルギー線硬化性接着剤のみならず、ウレタン系粘着剤、エポキシ系粘着剤、水性高分子-イソシアネート系粘着剤、熱硬化型アクリル粘着剤等の硬化型粘着剤、湿気硬化ウレタン粘着剤、ポリエーテルメタクリレート型、エステル系メタクリレート型、酸化型ポリエーテルメタクリレート等の嫌気性粘着剤、シアノアクリレート系の瞬間粘着剤、アクリレートとペルオキシド系の2液型瞬間粘着剤などを用いることができる。上記粘着剤としては1液型であってもよいし、使用前に2液以上を混合して使用する2液型であってもよい。接着剤は、有機溶媒を媒体とする溶媒系であってもよいし、水を主成分とする媒体であるエマルジョン型、コロイド分散液型、水溶液型等の水系であってもよいし、無溶媒型であってもよい。接着剤液の濃度は、接着後の膜厚、塗布方法、塗布条件等により適宜決定されれば良く、通常は0.1~50重量%である。
上述の接着剤のうち、図2にも示されるように、透湿性を効果的に制御できる観点から、活性エネルギー線硬化性接着剤を用いることが好ましい。すなわち、本発明の偏光板は、位相差フィルムが、活性エネルギー線硬化性接着剤で偏光子と接着されてなることが好ましい。
活性エネルギー線硬化性接着剤の好ましい例としては、例えば、特開2011-028234号公報に開示されているような、(α)カチオン重合性化合物、(β)光カチオン重合開始剤、(γ)380nmより長い波長の光に極大吸収を示す光増感剤、および(δ)ナフタレン系光増感助剤の各成分を含有する光硬化性接着剤組成物が挙げられる。ただし、これ以外の活性エネルギー線硬化性接着剤が用いられても、もちろんよい。
〈偏光子〉
偏光板の主たる構成要素である偏光子は、一定方向の偏波面の光だけを通す素子であり、現在知られている代表的な偏光子は、ポリビニルアルコール系偏光フィルムである。ポリビニルアルコール系偏光フィルムには、ポリビニルアルコール系フィルムにヨウ素を染色させたものと、二色性染料を染色させたものとがある。
偏光板の主たる構成要素である偏光子は、一定方向の偏波面の光だけを通す素子であり、現在知られている代表的な偏光子は、ポリビニルアルコール系偏光フィルムである。ポリビニルアルコール系偏光フィルムには、ポリビニルアルコール系フィルムにヨウ素を染色させたものと、二色性染料を染色させたものとがある。
偏光子としては、ポリビニルアルコール水溶液を製膜し、これを一軸延伸させて染色するか、染色した後一軸延伸してから、好ましくはホウ素化合物で耐久性処理を行った偏光子が用いられ得る。偏光子の膜厚は5~30μmの範囲内が好ましく、特に5~15μmの範囲内であることが好ましい。
また、特開2003-248123号公報、および特開2003-342322号公報等に記載のエチレン単位の含有量1~4モル%、重合度2000~4000、ケン化度99.0~99.99モル%のエチレン変性ポリビニルアルコールも好ましく用いられる。なかでも、熱水切断温度が66~73℃の範囲内であるエチレン変性ポリビニルアルコールフィルムが好ましく用いられる。このエチレン変性ポリビニルアルコールフィルムを用いた偏光子は、偏光性能および耐久性能に優れているうえに、色斑が少なく、大型液晶表示装置に特に好ましく用いられる。
〈保護フィルム〉
本発明の偏光板においては、必要に応じて、図2に示すように、本発明の位相差フィルムが配置されている面とは反対側の偏光子面に、更に活性エネルギー線硬化性接着剤を介して保護フィルム102が積層されていることが好ましい。
本発明の偏光板においては、必要に応じて、図2に示すように、本発明の位相差フィルムが配置されている面とは反対側の偏光子面に、更に活性エネルギー線硬化性接着剤を介して保護フィルム102が積層されていることが好ましい。
当該保護フィルムは、市販品として入手することができ、例えば、コニカミノルタタック KC4UE、KC8UE、KC8UX、KC5UX、KC8UY、KC4UY、KC4CZ、KC6UA、KC4UA、およびKC2UA(以上、コニカミノルタ(株)製)等が挙げられる。
特に視認側に配置される保護フィルムには、ハードコート層、帯電防止層、反射防止層、易滑性層、接着層、防眩層、バリアー層等の機能性層を設けることが好ましい。
〈偏光板の製造方法〉
偏光板は、活性エネルギー線硬化性接着剤を用いて、偏光子の一方の面に、本発明の位相差フィルムを貼り合せることにより製造することができる。位相差フィルムの両面で接着性が異なる場合は、接着性の良いほうに貼り合わせるのが好ましい。
偏光板は、活性エネルギー線硬化性接着剤を用いて、偏光子の一方の面に、本発明の位相差フィルムを貼り合せることにより製造することができる。位相差フィルムの両面で接着性が異なる場合は、接着性の良いほうに貼り合わせるのが好ましい。
以下、活性エネルギー線硬化性接着剤を用いた偏光板の製造方法の一例を説明する。
偏光板は、偏光子と位相差フィルムとの接着面のうち、少なくとも一方に、下記の活性エネルギー線硬化性接着剤を塗布して接着剤層を形成する接着剤塗布工程と、当該接着剤層を介して偏光子と位相差フィルムとを接着し、貼り合せる貼合工程と、当該接着剤層を介して偏光子と位相差フィルムとが接着された状態で接着剤層を硬化させる硬化工程とを含む製造方法によって製造することができる。また、位相差フィルムの偏光子を接着する面を易接着処理する前処理工程があってもよい。
(前処理工程)
前処理工程では、偏光子と接着する位相差フィルムの表面が易接着処理される。偏光子の両面にそれぞれ位相差フィルムおよび保護フィルムが接着される場合は、位相差フィルムおよび保護フィルムのそれぞれに対し易接着処理が行われる。次の接着剤塗布工程では、易接着処理された表面が偏光子との貼合面として扱われるので、位相差フィルムの両表面のうち、活性エネルギー線硬化性接着剤と貼合する面に、易接着処理を施す。易接着処理としては、コロナ処理、プラズマ処理等が挙げられる。
前処理工程では、偏光子と接着する位相差フィルムの表面が易接着処理される。偏光子の両面にそれぞれ位相差フィルムおよび保護フィルムが接着される場合は、位相差フィルムおよび保護フィルムのそれぞれに対し易接着処理が行われる。次の接着剤塗布工程では、易接着処理された表面が偏光子との貼合面として扱われるので、位相差フィルムの両表面のうち、活性エネルギー線硬化性接着剤と貼合する面に、易接着処理を施す。易接着処理としては、コロナ処理、プラズマ処理等が挙げられる。
(接着剤塗布工程)
接着剤塗布工程では、偏光子と位相差フィルムとの接着面のうち少なくとも一方に、上記活性エネルギー線硬化性接着剤が塗布される。偏光子または位相差フィルムの表面に直接、活性エネルギー線硬化性接着剤を塗布する場合、その塗布方法に特別な限定はない。例えば、ドクターブレード、ワイヤーバー、ダイコーター、カンマコーター、グラビアコーター等、種々の湿式塗布方式が利用できる。また、偏光子と位相差フィルムの間に、活性エネルギー線硬化性接着剤を流延させたのち、ローラ等で加圧して均一に押し広げる方法も利用できる。
接着剤塗布工程では、偏光子と位相差フィルムとの接着面のうち少なくとも一方に、上記活性エネルギー線硬化性接着剤が塗布される。偏光子または位相差フィルムの表面に直接、活性エネルギー線硬化性接着剤を塗布する場合、その塗布方法に特別な限定はない。例えば、ドクターブレード、ワイヤーバー、ダイコーター、カンマコーター、グラビアコーター等、種々の湿式塗布方式が利用できる。また、偏光子と位相差フィルムの間に、活性エネルギー線硬化性接着剤を流延させたのち、ローラ等で加圧して均一に押し広げる方法も利用できる。
(貼合工程)
上記の方法により活性エネルギー線硬化性接着剤を塗布した後は、貼合工程で処理される。この貼合工程では、例えば、先の塗布工程で偏光子の表面に活性エネルギー線硬化性接着剤を塗布した場合、そこに位相差フィルムが重ね合わされる。先の塗布工程で位相差フィルムの表面に活性エネルギー線硬化性接着剤を塗布した場合は、そこに偏光子が重ね合わされる。また、偏光子と位相差フィルムの間に活性エネルギー線硬化性接着剤を流延させた場合は、その状態で偏光子と位相差フィルムとが重ね合わされる。偏光子の両面に位相差フィルムおよび保護フィルムを接着する場合であって、両面とも活性エネルギー線硬化性接着剤を用いる場合は、偏光子の両面にそれぞれ、活性エネルギー線硬化性接着剤を介して位相差フィルムおよび保護フィルムが重ね合わされる。そして通常は、この状態で両面(偏光子の片面に位相差フィルムを重ね合わせた場合は、偏光子側と位相差フィルム側、また偏光子の両面に位相差フィルムおよび保護フィルムを重ね合わせた場合は、その両面の位相差フィルムおよび保護フィルム側)からローラ等で挟んで加圧することになる。ローラの材質は、金属やゴム等を用いることが可能である。両面に配置されるローラは、同じ材質であってもよいし、異なる材質であってもよい。
上記の方法により活性エネルギー線硬化性接着剤を塗布した後は、貼合工程で処理される。この貼合工程では、例えば、先の塗布工程で偏光子の表面に活性エネルギー線硬化性接着剤を塗布した場合、そこに位相差フィルムが重ね合わされる。先の塗布工程で位相差フィルムの表面に活性エネルギー線硬化性接着剤を塗布した場合は、そこに偏光子が重ね合わされる。また、偏光子と位相差フィルムの間に活性エネルギー線硬化性接着剤を流延させた場合は、その状態で偏光子と位相差フィルムとが重ね合わされる。偏光子の両面に位相差フィルムおよび保護フィルムを接着する場合であって、両面とも活性エネルギー線硬化性接着剤を用いる場合は、偏光子の両面にそれぞれ、活性エネルギー線硬化性接着剤を介して位相差フィルムおよび保護フィルムが重ね合わされる。そして通常は、この状態で両面(偏光子の片面に位相差フィルムを重ね合わせた場合は、偏光子側と位相差フィルム側、また偏光子の両面に位相差フィルムおよび保護フィルムを重ね合わせた場合は、その両面の位相差フィルムおよび保護フィルム側)からローラ等で挟んで加圧することになる。ローラの材質は、金属やゴム等を用いることが可能である。両面に配置されるローラは、同じ材質であってもよいし、異なる材質であってもよい。
(硬化工程)
硬化工程では、未硬化の活性エネルギー線硬化性接着剤に活性エネルギー線を照射して、カチオン重合性化合物(例えば、エポキシ化合物やオキセタン化合物)やラジカル重合性化合物(例えば、アクリレート系化合物、アクリルアミド系化合物等)を含む活性エネルギー線硬化性接着剤を硬化させ、活性エネルギー線硬化性接着剤を介して重ね合わせた偏光子と位相差フィルム、あるいは偏光子と位相差フィルムとを接着させる。偏光子の片面に位相差フィルムを貼合する場合、活性エネルギー線は、偏光子側または位相差フィルム側のいずれから照射してもよい。また、偏光子の両面に位相差フィルムおよび保護フィルムを貼合する場合、偏光子の両面にそれぞれ活性エネルギー線硬化性接着剤を介して位相差フィルムおよび保護フィルムを重ね合わせた状態で、活性エネルギー線を照射し、両面の活性エネルギー線硬化性接着剤を同時に硬化させるのが有利である。
硬化工程では、未硬化の活性エネルギー線硬化性接着剤に活性エネルギー線を照射して、カチオン重合性化合物(例えば、エポキシ化合物やオキセタン化合物)やラジカル重合性化合物(例えば、アクリレート系化合物、アクリルアミド系化合物等)を含む活性エネルギー線硬化性接着剤を硬化させ、活性エネルギー線硬化性接着剤を介して重ね合わせた偏光子と位相差フィルム、あるいは偏光子と位相差フィルムとを接着させる。偏光子の片面に位相差フィルムを貼合する場合、活性エネルギー線は、偏光子側または位相差フィルム側のいずれから照射してもよい。また、偏光子の両面に位相差フィルムおよび保護フィルムを貼合する場合、偏光子の両面にそれぞれ活性エネルギー線硬化性接着剤を介して位相差フィルムおよび保護フィルムを重ね合わせた状態で、活性エネルギー線を照射し、両面の活性エネルギー線硬化性接着剤を同時に硬化させるのが有利である。
硬化に適用される活性エネルギー線としては、可視光線、紫外線、X線、電子線等を用いることができるが、取扱いが容易で硬化速度も十分であることから、一般には電子線や紫外線が好ましく用いられる。
電子線の照射条件は、前記接着剤を硬化しうる条件であれば、任意の適切な条件を採用できる。例えば、電子線照射は、加速電圧が好ましくは5~300kVの範囲内であり、さらに好ましくは10~250kVの範囲内である。加速電圧が5kV未満の場合、電子線が接着剤まで届かず硬化不足となるおそれがあり、加速電圧が300kVを超えると、試料を通る浸透力が強すぎて電子線が跳ね返り、位相差フィルムや偏光子にダメージを与えるおそれがある。照射線量としては、5~100kGyの範囲内、さらに好ましくは10~75kGyの範囲内である。照射線量が5kGy未満の場合は、接着剤が硬化不足となり、100kGyを超えると、位相差フィルムや偏光子にダメージを与え、機械的強度の低下や黄変を生じ、所定の光学特性を得ることができない。
紫外線の照射条件は、前記接着剤を硬化しうる条件であれば、任意の適切な条件を採用できる。紫外線の照射量は積算光量で50~1500mJ/cm2の範囲内であることが好ましく、100~500mJ/cm2の範囲内であるのがさらに好ましい。
前記製造方法を連続ラインで行う場合、ライン速度は、接着剤の硬化時間によるが、好ましくは1~500m/minの範囲内であり、より好ましくは5~300m/min、さらに好ましくは10~100m/minの範囲内である。ライン速度が遅すぎる場合は、生産性が乏しい、または位相差フィルムへのダメージが大きすぎ、耐久性試験などに耐えうる偏光板が作製できない。ライン速度が速やすぎる場合は、接着剤の硬化が不十分となり、目的とする接着性が得られない場合がある。
以上のようにして得られた偏光板において、接着剤層の厚さは、特に限定されないが、通常0.01~10μmの範囲内であり、好ましくは0.5~5μmの範囲内である。
〈液晶表示装置〉
本発明の偏光板は、液晶表示装置に好適に用いることができる。本発明の偏光板が用いられた液晶表示装置は、透湿度の低い位相差フィルムが用いられていることから、含水による液晶表示装置の色ムラが発生しづらい。
本発明の偏光板は、液晶表示装置に好適に用いることができる。本発明の偏光板が用いられた液晶表示装置は、透湿度の低い位相差フィルムが用いられていることから、含水による液晶表示装置の色ムラが発生しづらい。
液晶表示装置のパネルに使用されるガラスは0.3~0.7mmの厚さの範囲が好ましく、さらに、0.3~0.5mmの範囲が好ましい。本発明の偏光板は変形しづらいため、特に、ガラスの厚さが小さいときに、好ましく用いられる。
偏光板の本発明の位相差フィルム側の表面と、液晶セルの少なくとも一方の表面との貼合は、公知の手法により行われ得る。場合によっては、接着層を介して貼合されてもよい。
液晶表示装置のモード(駆動方式)についても特に制限はなく、STN、TN、OCB、HAN、VA(MVA、PVA)、IPS、OCB等の各種駆動モードの液晶表示装置が用いられ得る。
特に、本発明の位相差フィルムは、高いレターデーション値を有することから、視野角を拡大する光学補償フィルム(位相差フィルム)として、VA(MVA,PVA)型(垂直配向型)の液晶表示装置に好適に具備される。すなわち、本発明の一形態によれば、上記偏光板が、少なくとも液晶セルの片面に具備されている、垂直配向型液晶表示装置が提供される。
これらの液晶表示装置に、本発明の位相差フィルムを含む偏光板を具備することで、耐久性(耐湿熱性)に優れるとともに、30型以上の大画面の液晶表示装置であっても位相差バラツキが抑制されて液晶表示装置のムラ等がない視認性に優れた液晶表示装置を得ることができる。
以下、実施例を挙げて本発明を具体的に説明するが、本発明はこれらに限定されるものではない。なお、実施例において「部」あるいは「%」の表示を用いるが、特に断りがない限り「重量部」あるいは「重量%」を表す。また、特に断りがない限り、各操作は、室温(25℃)で行われる。
〔位相差フィルム2の作製〕
〈微粒子分散液1〉
微粒子(マット剤)(アエロジル R812、日本アエロジル(株)製)
11重量部
エタノール 89重量部
以上をディゾルバーで50分間攪拌混合した後、マントンゴーリンで分散を行った。
〈微粒子分散液1〉
微粒子(マット剤)(アエロジル R812、日本アエロジル(株)製)
11重量部
エタノール 89重量部
以上をディゾルバーで50分間攪拌混合した後、マントンゴーリンで分散を行った。
〈微粒子添加液1〉
メチレンクロライドを入れた溶解タンクに十分攪拌しながら、微粒子分散液1をゆっくりと添加した。更に、二次粒子の粒径が所定の大きさとなるようにアトライターにて分散を行った。これを日本精線(株)製のファインメットNFで濾過し、微粒子添加液1を調製した。
メチレンクロライドを入れた溶解タンクに十分攪拌しながら、微粒子分散液1をゆっくりと添加した。更に、二次粒子の粒径が所定の大きさとなるようにアトライターにて分散を行った。これを日本精線(株)製のファインメットNFで濾過し、微粒子添加液1を調製した。
メチレンクロライド 99重量部
微粒子分散液1 5重量部
下記組成の主ドープ液を調製した。まず加圧溶解タンクにメチレンクロライドとエタノールを添加した。溶媒の入った加圧溶解タンクにアセチル置換度2.41のセルロースアセテートを攪拌しながら投入した。これを加熱し、攪拌しながら、完全に溶解し、これを安積濾紙(株)製の安積濾紙No.244を使用して濾過し、主ドープ液1を調製した。
微粒子分散液1 5重量部
下記組成の主ドープ液を調製した。まず加圧溶解タンクにメチレンクロライドとエタノールを添加した。溶媒の入った加圧溶解タンクにアセチル置換度2.41のセルロースアセテートを攪拌しながら投入した。これを加熱し、攪拌しながら、完全に溶解し、これを安積濾紙(株)製の安積濾紙No.244を使用して濾過し、主ドープ液1を調製した。
〈主ドープ液1の組成〉
メチレンクロライド 365重量部
エタノール 40重量部
セルロースアセテート(ジアセチルセルロース:アセチル基置換度
2.41、数平均分子量(Mn)5.6万)(C1) 100重量部
化合物(1-1) 2重量部
化合物(2-1) 3重量部
微粒子添加液1 1重量部
上記主ドープ液1を密閉されている主溶解釜に投入し、攪拌しながら溶解してドープ液を調製した。
メチレンクロライド 365重量部
エタノール 40重量部
セルロースアセテート(ジアセチルセルロース:アセチル基置換度
2.41、数平均分子量(Mn)5.6万)(C1) 100重量部
化合物(1-1) 2重量部
化合物(2-1) 3重量部
微粒子添加液1 1重量部
上記主ドープ液1を密閉されている主溶解釜に投入し、攪拌しながら溶解してドープ液を調製した。
得られたドープ液を、無端ベルト流延装置を用いて、ステンレスベルト支持体上に均一に流延させた。ステンレスベルト支持体上で、流延(キャスト)したフィルム中の残留溶媒量が75%になるまで溶媒を蒸発させ、次いで剥離張力130N/mで、ステンレスベルト支持体上から剥離した。剥離した位相差フィルムを、160℃の熱をかけながらテンターを用いて幅手方向に30%延伸した。この際、流延方向には延伸しなかった。また、延伸開始時の残留溶媒は15%であった。
次いで、乾燥ゾーンを多数のロールで搬送させながら乾燥を終了させた。乾燥温度は130℃で、搬送張力は100N/mとした。以上のようにして、乾燥膜厚30μm、巻長は4000mの位相差フィルム2を得た。
〔位相差フィルム3~7の作製:膜厚変更〕
上記位相差フィルム2の作製において、フィルムが下記表1に記載の膜厚となるようにドープ液をステンレスバンド支持体上に流延したこと以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム3~7を製造した。なお、下記表1において、「C1」は、セルロースアセテート(ジアセチルセルロース:アセチル基置換度2.41、数平均分子量(Mn)5.6万)を示す。
上記位相差フィルム2の作製において、フィルムが下記表1に記載の膜厚となるようにドープ液をステンレスバンド支持体上に流延したこと以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム3~7を製造した。なお、下記表1において、「C1」は、セルロースアセテート(ジアセチルセルロース:アセチル基置換度2.41、数平均分子量(Mn)5.6万)を示す。
〔位相差フィルム1の作製〕
上記位相差フィルム2の作製において、主ドープ液1の代わりに下記で調製された主ドープ液1を使用する以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム8を製造した。
上記位相差フィルム2の作製において、主ドープ液1の代わりに下記で調製された主ドープ液1を使用する以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム8を製造した。
〈主ドープ液2の組成〉
メチレンクロライド 365重量部
エタノール 40重量部
セルロースアセテート(ジアセチルセルロース:アセチル基置換度
2.41、 数平均分子量(Mn)5.6万)(C1) 100重量部
化合物(1-1) 2重量部
化合物(2-1) 3重量部
可塑剤(糖エステルS) 10重量部
微粒子添加液1 1重量部
なお、下記において、糖エステルSは下記構造を有する。また、この糖エステルを、下記表では、「S1」とも称する。
メチレンクロライド 365重量部
エタノール 40重量部
セルロースアセテート(ジアセチルセルロース:アセチル基置換度
2.41、 数平均分子量(Mn)5.6万)(C1) 100重量部
化合物(1-1) 2重量部
化合物(2-1) 3重量部
可塑剤(糖エステルS) 10重量部
微粒子添加液1 1重量部
なお、下記において、糖エステルSは下記構造を有する。また、この糖エステルを、下記表では、「S1」とも称する。

〔位相差フィルム8~12の作製:膜厚の変更〕
上記位相差フィルム1の作製において、フィルムが下記表2に記載の膜厚となるようにドープ液をステンレスバンド支持体上に流延したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム8~12を製造した。
上記位相差フィルム1の作製において、フィルムが下記表2に記載の膜厚となるようにドープ液をステンレスバンド支持体上に流延したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム8~12を製造した。
〔位相差フィルム13~17の作製:化合物(2)の配合量の変更〕
上記位相差フィルム1の作製において、主ドープ液2中の化合物(2-1)の配合量を下記表3に記載される量に変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム13~16を製造した。
上記位相差フィルム1の作製において、主ドープ液2中の化合物(2-1)の配合量を下記表3に記載される量に変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム13~16を製造した。
また、上記位相差フィルム1の作製において、主ドープ液2の調製時に化合物(1-1)及び化合物(2-1)を添加しなかったこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム17を製造した。
〔位相差フィルム18~21の作製:化合物(2)の配合量の変更〕
上記位相差フィルム2の作製において、主ドープ液1中の化合物(2-1)の配合量を下記表3に記載される量に変更したこと以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム18~21を製造した。
上記位相差フィルム2の作製において、主ドープ液1中の化合物(2-1)の配合量を下記表3に記載される量に変更したこと以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム18~21を製造した。
〔位相差フィルム22~25の作製:化合物(1)の配合量の変更〕
上記位相差フィルム1の作製において、主ドープ液2中の化合物(1-1)の配合量を下記表5に記載される量に変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム22~25を製造した。
上記位相差フィルム1の作製において、主ドープ液2中の化合物(1-1)の配合量を下記表5に記載される量に変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム22~25を製造した。
〔位相差フィルム26~29の作製:化合物(1)の配合量の変更〕
上記位相差フィルム2の作製において、主ドープ液1中の化合物(1-1)の配合量を下記表6に記載される量に変更したこと以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム26~29を製造した。
上記位相差フィルム2の作製において、主ドープ液1中の化合物(1-1)の配合量を下記表6に記載される量に変更したこと以外は、上記位相差フィルム2の作製と同様にして、位相差フィルム26~29を製造した。
〔位相差フィルム30~34の作製:セルロースエステル樹脂の変更〕
上記位相差フィルム1の作製において、主ドープ液2中のセルロースアセテート(ジアセチルセルロース:アセチル基置換度2.41、数平均分子量(Mn)5.6万)(C1)を、下記表7に記載されるセルロースエステルに変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム30~34を製造した。なお、下記表7中のセルロースエステルC2~C6は、下記のとおりである:
上記位相差フィルム1の作製において、主ドープ液2中のセルロースアセテート(ジアセチルセルロース:アセチル基置換度2.41、数平均分子量(Mn)5.6万)(C1)を、下記表7に記載されるセルロースエステルに変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム30~34を製造した。なお、下記表7中のセルロースエステルC2~C6は、下記のとおりである:

〔位相差フィルム35~37の作製:可塑剤の配合量の変更〕
上記位相差フィルム1の作製において、主ドープ液2中の可塑剤(糖エステルS)の配合量を、下記表8に記載される量に変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム35~37を製造した。
上記位相差フィルム1の作製において、主ドープ液2中の可塑剤(糖エステルS)の配合量を、下記表8に記載される量に変更したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム35~37を製造した。
〔位相差フィルム38~39の作製:一般式(1)の化合物の変更〕
上記位相差フィルム1の作製において、化合物(1-1)の代わりに化合物(1-2)または化合物(1-5)を使用したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム38~39を製造した。
上記位相差フィルム1の作製において、化合物(1-1)の代わりに化合物(1-2)または化合物(1-5)を使用したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム38~39を製造した。
〔位相差フィルム40~50の作製:一般式(2)の化合物の変更〕
上記位相差フィルム1の作製において、化合物(2-1)の代わりに化合物(2-2)~(2-12)をそれぞれ使用したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム40~50を製造した。
上記位相差フィルム1の作製において、化合物(2-1)の代わりに化合物(2-2)~(2-12)をそれぞれ使用したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム40~50を製造した。
〔位相差フィルム51~55の作製:可塑剤の変更〕
上記位相差フィルム1の作製において、可塑剤(糖エステルS)の代わりに下記表11に示される可塑剤S2~S6をそれぞれ使用したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム51~55を製造した。なお、下記表11中の可塑剤S2~S6は、下記のとおりである:
(可塑剤S2)
1,2-プロピレングリコール251g、無水フタル酸278g、アジピン酸91g、安息香酸610g、エステル化触媒としてテトライソプロピルチタネート0.191gを、温度計、撹拌器、緩急冷却管を備えた2Lの四つ口フラスコに仕込み、窒素気流中230℃になるまで、撹拌しながら徐々に昇温する。15時間脱水縮合反応させ、反応終了後200℃で未反応の1,2-プロピレングリコールを減圧留去することにより、可塑剤S2(芳香族末端エステル化合物)を得た。可塑剤S2の、酸価は0.10、数平均分子量は450であった。
上記位相差フィルム1の作製において、可塑剤(糖エステルS)の代わりに下記表11に示される可塑剤S2~S6をそれぞれ使用したこと以外は、上記位相差フィルム1の作製と同様にして、位相差フィルム51~55を製造した。なお、下記表11中の可塑剤S2~S6は、下記のとおりである:
(可塑剤S2)
1,2-プロピレングリコール251g、無水フタル酸278g、アジピン酸91g、安息香酸610g、エステル化触媒としてテトライソプロピルチタネート0.191gを、温度計、撹拌器、緩急冷却管を備えた2Lの四つ口フラスコに仕込み、窒素気流中230℃になるまで、撹拌しながら徐々に昇温する。15時間脱水縮合反応させ、反応終了後200℃で未反応の1,2-プロピレングリコールを減圧留去することにより、可塑剤S2(芳香族末端エステル化合物)を得た。可塑剤S2の、酸価は0.10、数平均分子量は450であった。
(可塑剤S3)
1,2-プロピレングリコール251g、テレフタル酸354g、安息香酸610g、エステル化触媒としてテトライソプロピルチタネート0.191gを、温度計、撹拌器、緩急冷却管を備えた2Lの四つ口フラスコに仕込み、窒素気流中230℃になるまで、撹拌しながら徐々に昇温する。15時間脱水縮合反応させ、反応終了後200℃で未反応の1,2-プロピレングリコールを減圧留去することにより、可塑剤S3(芳香族末端エステル化合物)を得た。可塑剤S3の、酸価は0.10、数平均分子量は400であった。
1,2-プロピレングリコール251g、テレフタル酸354g、安息香酸610g、エステル化触媒としてテトライソプロピルチタネート0.191gを、温度計、撹拌器、緩急冷却管を備えた2Lの四つ口フラスコに仕込み、窒素気流中230℃になるまで、撹拌しながら徐々に昇温する。15時間脱水縮合反応させ、反応終了後200℃で未反応の1,2-プロピレングリコールを減圧留去することにより、可塑剤S3(芳香族末端エステル化合物)を得た。可塑剤S3の、酸価は0.10、数平均分子量は400であった。
可塑剤S4:トリフェニルホスフェート
可塑剤S5:ジフェニルビフェニルホスフェート(BDP)
可塑剤S6:トリメチロールプロパン安息香酸エステル。
可塑剤S5:ジフェニルビフェニルホスフェート(BDP)
可塑剤S6:トリメチロールプロパン安息香酸エステル。
《位相差フィルムの評価》
上記作製した各位相差フィルムについて、下記の各特性値の測定および評価を行った。
上記作製した各位相差フィルムについて、下記の各特性値の測定および評価を行った。
(引き裂き強度の測定)
JIS K 7128-1991に準拠して、エレメンドルフ法の引き裂き荷重を、東洋精機(株)製の軽荷重引き裂き装置で測定した。引き裂き荷重の測定は、フィルムの搬送方向(MD方向)に引き裂いた場合と、フィルムの搬送方向と直交する方向(TD方向)に引き裂いた場合のそれぞれについて、23℃、55%RHの条件下で行った。
JIS K 7128-1991に準拠して、エレメンドルフ法の引き裂き荷重を、東洋精機(株)製の軽荷重引き裂き装置で測定した。引き裂き荷重の測定は、フィルムの搬送方向(MD方向)に引き裂いた場合と、フィルムの搬送方向と直交する方向(TD方向)に引き裂いた場合のそれぞれについて、23℃、55%RHの条件下で行った。
(レターデーション値の測定)
作製した位相差フィルムについて、温度23℃、相対湿度55%の環境下で、波長590nmで、自動複屈折計KOBRA-21ADH(王子計測機器)を用いて下記式で表されるレターデーション値Ro、Rtを測定した。
作製した位相差フィルムについて、温度23℃、相対湿度55%の環境下で、波長590nmで、自動複屈折計KOBRA-21ADH(王子計測機器)を用いて下記式で表されるレターデーション値Ro、Rtを測定した。
具体的には、位相差フィルムを23℃、55%RHの環境下で、590nmの波長において10カ所で3次元の屈折率測定を行い、屈折率nx、ny、nzの平均値を求めた後、下記式(I)及び(II)に従って面内方向のレターデーション値Ro、厚さ方向のレターデーション値Rtを算出した。
上記式(I)および式(II)において、nxは、フィルムの面内方向において屈折率が最大になる方向xにおける屈折率を表す。nyは、フィルムの面内方向において、前記方向xと直交する方向yにおける屈折率を表す。nzは、フィルムの厚さ方向zにおける屈折率を表す。dは、フィルムの厚さ(nm)を表す。
(湿度変動によるレターデーション値の測定)
位相差フィルムを23℃、20%RHにて5時間調湿した後、同環境で測定したRt値を測定しこれをRt20%(590)とし、同じフィルムを続けて23℃、80%RHにて5時間調湿した後、同環境で測定したRt値を求めこれをRt80%(590)とし、下記の式より変化量ΔRtを求めた。
位相差フィルムを23℃、20%RHにて5時間調湿した後、同環境で測定したRt値を測定しこれをRt20%(590)とし、同じフィルムを続けて23℃、80%RHにて5時間調湿した後、同環境で測定したRt値を求めこれをRt80%(590)とし、下記の式より変化量ΔRtを求めた。
更に調湿後の試料を再度23℃55%RHの環境にて5時間調湿した後測定を行い、この変動が可逆変動であることを確認した。この値が小さい方が、湿度変動に対して安定であることを示す。
上記で求めたΔRtを用いて、下記の式(1)によりRt湿度変動を求めた。
式(1) Rt湿度変動=ΔRt/(23℃相対湿度55%環境下にフィルムを5時間静置した後に測定したRt値)×100
以下の指標に基づいて、Rt湿度変動を評価した。
以下の指標に基づいて、Rt湿度変動を評価した。

位相差フィルムの構成内容および上記評価の結果を、下記表1~11にまとめて示す。なお、上記偏光板2-1~2-55、3-1~3-55、4-1~4-55および5-1~5-55は、上記偏光板1-1~1-55と、それぞれ、同様の結果が得られた。











《偏光板の作製》
<活性エネルギー線硬化性接着剤:カチオン重合型接着剤(表中、カチオン重合型と記載)の使用>
〔偏光板1-1の作製〕
(偏光子の作製)
厚さ30μmのポリビニルアルコールフィルムを、35℃の水で膨潤させた。得られたフィルムを、ヨウ素0.075g、ヨウ化カリウム5gおよび水100gからなる水溶液に60秒間浸漬し、さらにヨウ化カリウム3g、ホウ酸7.5gおよび水100gからなる45℃の水溶液に浸漬した。得られたフィルムを、延伸温度55℃、延伸倍率5倍の条件で一軸延伸した。この一軸延伸フィルムを、水洗した後、乾燥させて、厚さ10μmの偏光子を得た。
<活性エネルギー線硬化性接着剤:カチオン重合型接着剤(表中、カチオン重合型と記載)の使用>
〔偏光板1-1の作製〕
(偏光子の作製)
厚さ30μmのポリビニルアルコールフィルムを、35℃の水で膨潤させた。得られたフィルムを、ヨウ素0.075g、ヨウ化カリウム5gおよび水100gからなる水溶液に60秒間浸漬し、さらにヨウ化カリウム3g、ホウ酸7.5gおよび水100gからなる45℃の水溶液に浸漬した。得られたフィルムを、延伸温度55℃、延伸倍率5倍の条件で一軸延伸した。この一軸延伸フィルムを、水洗した後、乾燥させて、厚さ10μmの偏光子を得た。
(活性エネルギー線硬化性接着剤液の調製:カチオン重合型、表中カチオン重合型と記載)
下記の各成分を混合した後、脱泡して、活性エネルギー線硬化性接着剤液を調製した。なお、トリアリールスルホニウムヘキサフルオロホスフェートは、50%プロピレンカーボネート溶液として配合し、下記にはトリアリールスルホニウムヘキサフルオロホスフェートの固形分量を表示した。
下記の各成分を混合した後、脱泡して、活性エネルギー線硬化性接着剤液を調製した。なお、トリアリールスルホニウムヘキサフルオロホスフェートは、50%プロピレンカーボネート溶液として配合し、下記にはトリアリールスルホニウムヘキサフルオロホスフェートの固形分量を表示した。
3,4-エポキシシクロヘキシルメチル-3,4-エポキシシクロヘキ
サンカルボキシレート 45重量部
エポリードGT-301(ダイセル化学社製の脂環式エポキシ樹脂)
40重量部
1,4-ブタンジオールジグリシジルエーテル 15重量部
トリアリールスルホニウムヘキサフルオロホスフェート 2.3重量部
9,10-ジブトキシアントラセン 0.1重量部
1,4-ジエトキシナフタレン 2.0重量部
(偏光板1-1の作製)
下記の方法に従って、図2の偏光板1Aの構成からなる偏光板1-1を作製した。カッコ内の数値は、図2に記載した各構成要素の番号を示す。
サンカルボキシレート 45重量部
エポリードGT-301(ダイセル化学社製の脂環式エポキシ樹脂)
40重量部
1,4-ブタンジオールジグリシジルエーテル 15重量部
トリアリールスルホニウムヘキサフルオロホスフェート 2.3重量部
9,10-ジブトキシアントラセン 0.1重量部
1,4-ジエトキシナフタレン 2.0重量部
(偏光板1-1の作製)
下記の方法に従って、図2の偏光板1Aの構成からなる偏光板1-1を作製した。カッコ内の数値は、図2に記載した各構成要素の番号を示す。
まず、保護フィルム(102)として、KC6UAフィルム(コニカミノルタ(株)製)を準備し、上記調製した活性エネルギー線硬化性接着剤液を、マイクログラビアコーター(グラビアローラ:#300,回転速度140%/ライン速)を用いて、厚さ5μmになるように塗工して活性エネルギー線硬化性接着剤(103A)を形成した。
次いで、上記作製した位相差フィルム1(101)に、上記調製した活性エネルギー線硬化性接着剤液を、上記と同様に、厚さ5μmとなるように塗工して活性エネルギー線硬化性接着剤(103B)を形成した。
この活性エネルギー線硬化性接着剤(103A)と(103B)の間に、上記作製したポリビニルアルコール-ヨウ素系の偏光子(104)を配置し、ローラ機で貼合し、保護フィルム1(102)/活性エネルギー線硬化性接着剤(103A)/偏光子(104)/活性エネルギー線硬化性接着剤(103B)/位相差フィルム101(105)が積層された積層物を得た。その際に、位相差フィルム(105)の遅相軸と偏光子(104)の吸収軸が互いに直交になるようにローラ機で貼合した。
この積層物の両面側から、電子線を照射して、偏光板1-1(偏光板101A)を作製した。
ライン速度は20m/min、加速電圧は250kV、照射線量は20kGyとした。
〔偏光板1-2~1-55の作製〕
上記偏光板1-1の作製において、位相差フィルム1の代わりに表12に記載の位相差フィルム2~55を使用したこと以外は、上記偏光板1-1の作製と同様にして、偏光板1-2~1-55を作製した。
上記偏光板1-1の作製において、位相差フィルム1の代わりに表12に記載の位相差フィルム2~55を使用したこと以外は、上記偏光板1-1の作製と同様にして、偏光板1-2~1-55を作製した。
〔偏光板2-1~2-55の作製〕
上記偏光板1-1~1-55の作製において、保護フィルム(102)として、KC6UAフィルムの代わりに、下記フィルムH2を使用した以外は、上記偏光板1-1~1-55の作製と同様にして、それぞれ、偏光板2-1~2-55を作製した。
上記偏光板1-1~1-55の作製において、保護フィルム(102)として、KC6UAフィルムの代わりに、下記フィルムH2を使用した以外は、上記偏光板1-1~1-55の作製と同様にして、それぞれ、偏光板2-1~2-55を作製した。
(フィルムH2)
特許第4962261号公報の実施例1(段落「0060」~「0066」)において、一軸配向PETフィルムのフィルム厚みを80μmとする以外は、特許第4962261号の実施例1に記載の方法と同様にして、フィルムH2を作製した。
特許第4962261号公報の実施例1(段落「0060」~「0066」)において、一軸配向PETフィルムのフィルム厚みを80μmとする以外は、特許第4962261号の実施例1に記載の方法と同様にして、フィルムH2を作製した。
〔偏光板3-1~3-55の作製〕
上記偏光板1-1~1-55の作製において、保護フィルム(102)として、KC6UAフィルムの代わりに、下記フィルムH3を使用した以外は、上記偏光板1-1~1-55の作製と同様にして、それぞれ、偏光板3-1~3-55を作製した。
上記偏光板1-1~1-55の作製において、保護フィルム(102)として、KC6UAフィルムの代わりに、下記フィルムH3を使用した以外は、上記偏光板1-1~1-55の作製と同様にして、それぞれ、偏光板3-1~3-55を作製した。
(フィルムH3)
特開2010-055062号公報の偏光子保護フィルムの作製(段落「0096」~「0099」)に記載の方法と同様にして、フィルムH3を作製した。
特開2010-055062号公報の偏光子保護フィルムの作製(段落「0096」~「0099」)に記載の方法と同様にして、フィルムH3を作製した。
<活性エネルギー線硬化性接着剤:ラジカル重合型接着剤(表中、ラジカル重合型と記載)の使用>
〔偏光板4-1の作製〕
(偏光子の作製)
厚さ70μmのポリビニルアルコールフィルムを、35℃の水で膨潤させた。得られたフィルムを、ヨウ素0.075g、ヨウ化カリウム5g及び水100gからなる水溶液に60秒間浸漬し、さらにヨウ化カリウム3g、ホウ酸7.5g及び水100gからなる45℃の水溶液に浸漬した。得られたフィルムを、延伸温度55℃、延伸倍率5倍の条件で一軸延伸した。この一軸延伸フィルムを、水洗した後、乾燥させて、厚さ25μmの偏光子を得た。
〔偏光板4-1の作製〕
(偏光子の作製)
厚さ70μmのポリビニルアルコールフィルムを、35℃の水で膨潤させた。得られたフィルムを、ヨウ素0.075g、ヨウ化カリウム5g及び水100gからなる水溶液に60秒間浸漬し、さらにヨウ化カリウム3g、ホウ酸7.5g及び水100gからなる45℃の水溶液に浸漬した。得られたフィルムを、延伸温度55℃、延伸倍率5倍の条件で一軸延伸した。この一軸延伸フィルムを、水洗した後、乾燥させて、厚さ25μmの偏光子を得た。
(活性エネルギー線硬化性接着剤液の調製:ラジカル重合型、表中ラジカル重合型と記載)
N-ヒドロキシエチルアクリルアミド100質量部に、光重合開始剤(BASFジャパン(株)製;商品名イルガキュア127)3質量部を配合したものを光硬化性接着剤液Rとして用いた。
N-ヒドロキシエチルアクリルアミド100質量部に、光重合開始剤(BASFジャパン(株)製;商品名イルガキュア127)3質量部を配合したものを光硬化性接着剤液Rとして用いた。
(偏光板の作製)
下記の方法に従って、図2の偏光板101Aの構成からなる偏光板4-1を作製した。カッコ内の数値は、図2に記載した各構成要素の番号を示す。
下記の方法に従って、図2の偏光板101Aの構成からなる偏光板4-1を作製した。カッコ内の数値は、図2に記載した各構成要素の番号を示す。
まず、位相差フィルム(105)として、上記作製した位相差フィルム1(105)を用い、上記調製した光硬化性接着剤液Rを、マイクログラビアコーター(グラビアロール:#300回転速度140%/ライン速)を用いて、厚さ5μmになるように塗工して光硬化型樹脂層(103B)を形成した。
次いで、保護フィルム(102)として、コニカミノルタタック KC6UAフィルム(コニカミノルタ(株)製)を用い、上記調製した光硬化性接着剤液Rを、上記と同様に、厚さ5μmとなるように塗工して光硬化型樹脂層(103A)を形成した。
この光硬化型樹脂層(103A)と(103B)の間に、上記作製したポリビニルアルコール-ヨウ素系の偏光子(104)を配置し、ロール機で貼合し、保護フィルム(102)/光硬化型樹脂層(103A)/偏光子(104)/光硬化型樹脂層(103B)/位相差フィルム(105)が積層された積層物を得た。その際に、位相差フィルム(105)の遅相軸と偏光子(104)の吸収軸が互いに直交になるようにロール機で貼合した。 この積層体の両面側から、電子線を、ライン速度は20m/min、加速電圧は250kV、照射線量は20kGyの条件で照射して、偏光板4-1(101A)を作製した。
〔偏光板4-2~4-55の作製〕
上記偏光板4-1の作製において、位相差フィルム1の代わりに表12に記載の位相差フィルム2~55を使用したこと以外は、上記偏光板4-1の作製と同様にして、それぞれ、偏光板4-2~4-55を作製した。
上記偏光板4-1の作製において、位相差フィルム1の代わりに表12に記載の位相差フィルム2~55を使用したこと以外は、上記偏光板4-1の作製と同様にして、それぞれ、偏光板4-2~4-55を作製した。
<ポリビニルアルコール接着剤(表中、ポリビニルアルコールと記載)の使用>
〔偏光板5-1の作製〕
(偏光子の調製)
平均厚さが52μm、水分率が4.4%のポリビニルアルコールフィルムを予備膨潤、染色、湿式法による一軸延伸、固定処理、乾燥、熱処理の順番で、連続的に処理して、偏光子を作製した。すなわち、PVAフィルムを温度30℃の水中に30秒間浸して予備膨潤し、ヨウ素濃度0.4g/リットル、ヨウ化カリウム濃度40g/リットルの温度35℃の水溶液中に3分間浸して膨潤した。続いて、ホウ酸濃度4%の50℃の水溶液中でフィルムにかかる張力が700N/mの条件下で、6倍に一軸延伸を行い、ヨウ化カリウム濃度40g/リットル、ホウ酸濃度40g/リットル、塩化亜鉛濃度10g/リットルの温度30℃の水溶液中に5分間浸漬して固定処理を行った。その後、ポリビニルアルコールフィルムを取り出し、温度40℃で熱風乾燥し、更に温度100℃で5分間熱処理を行った。得られた偏光子は、平均厚さが25μm、偏光性能については透過率が43.0%、偏光度が99.5%、二色性比が40.1であった。
〔偏光板5-1の作製〕
(偏光子の調製)
平均厚さが52μm、水分率が4.4%のポリビニルアルコールフィルムを予備膨潤、染色、湿式法による一軸延伸、固定処理、乾燥、熱処理の順番で、連続的に処理して、偏光子を作製した。すなわち、PVAフィルムを温度30℃の水中に30秒間浸して予備膨潤し、ヨウ素濃度0.4g/リットル、ヨウ化カリウム濃度40g/リットルの温度35℃の水溶液中に3分間浸して膨潤した。続いて、ホウ酸濃度4%の50℃の水溶液中でフィルムにかかる張力が700N/mの条件下で、6倍に一軸延伸を行い、ヨウ化カリウム濃度40g/リットル、ホウ酸濃度40g/リットル、塩化亜鉛濃度10g/リットルの温度30℃の水溶液中に5分間浸漬して固定処理を行った。その後、ポリビニルアルコールフィルムを取り出し、温度40℃で熱風乾燥し、更に温度100℃で5分間熱処理を行った。得られた偏光子は、平均厚さが25μm、偏光性能については透過率が43.0%、偏光度が99.5%、二色性比が40.1であった。
(貼合)
下記工程a~eに従って、偏光子に、位相差フィルムとしての上記で作製した光学フィルム105、および、保護フィルムとしてのコニカミノルタタック KC6UAフィルム(コニカミノルタ(株)製)を貼り合わせた。
下記工程a~eに従って、偏光子に、位相差フィルムとしての上記で作製した光学フィルム105、および、保護フィルムとしてのコニカミノルタタック KC6UAフィルム(コニカミノルタ(株)製)を貼り合わせた。
〈工程a〉
上記偏光子を、固形分2質量%のポリビニルアルコール接着剤溶液の貯留槽中に1~2秒間浸漬した。
上記偏光子を、固形分2質量%のポリビニルアルコール接着剤溶液の貯留槽中に1~2秒間浸漬した。
〈工程b〉
保護フィルムとしてのKC6UAフィルムと上記作製した位相差フィルム1(105)に、下記条件でアルカリ鹸化処理し、水洗、中和、水洗の順に行い、次いで100℃で乾燥した。次いで、工程aでポリビニルアルコール接着剤溶液に浸漬した偏光子に付着した過剰の接着剤を軽く取り除き、この偏光子にKC6UAフィルムと、上記光学フィルム105とを挟み込んで、積層配置した。すなわち、保護フィルム(102)/ポリビニルアルコール接着剤(103A)/偏光子(104)/ポリビニルアルコール接着剤(103B)/位相差フィルム(105)が積層された積層物を得た(図2の偏光板101Aの構成)。
保護フィルムとしてのKC6UAフィルムと上記作製した位相差フィルム1(105)に、下記条件でアルカリ鹸化処理し、水洗、中和、水洗の順に行い、次いで100℃で乾燥した。次いで、工程aでポリビニルアルコール接着剤溶液に浸漬した偏光子に付着した過剰の接着剤を軽く取り除き、この偏光子にKC6UAフィルムと、上記光学フィルム105とを挟み込んで、積層配置した。すなわち、保護フィルム(102)/ポリビニルアルコール接着剤(103A)/偏光子(104)/ポリビニルアルコール接着剤(103B)/位相差フィルム(105)が積層された積層物を得た(図2の偏光板101Aの構成)。
〈アルカリ鹸化処理〉
ケン化工程 1.5モル/L-KOH水溶液 50℃ 45秒
水洗工程 水 30℃ 60秒
中和工程 10質量%HCl水溶液 30℃ 45秒
水洗工程 水 30℃ 60秒。
ケン化工程 1.5モル/L-KOH水溶液 50℃ 45秒
水洗工程 水 30℃ 60秒
中和工程 10質量%HCl水溶液 30℃ 45秒
水洗工程 水 30℃ 60秒。
〈工程c〉
積層物を、2つの回転するローラにて20~30N/cm2の圧力で約2m/minの速度で貼り合わせた。このとき、気泡が入らないように注意して実施した。
積層物を、2つの回転するローラにて20~30N/cm2の圧力で約2m/minの速度で貼り合わせた。このとき、気泡が入らないように注意して実施した。
〈工程d〉
上記工程cで作製した試料を、温度80℃の乾燥機中にて5分間乾燥処理し、図2の偏光板101Aの構成からなる偏光板1202を作製した。
上記工程cで作製した試料を、温度80℃の乾燥機中にて5分間乾燥処理し、図2の偏光板101Aの構成からなる偏光板1202を作製した。
〔偏光板5-2~5-55の作製〕
上記偏光板5-1の作製において、位相差フィルム1の代わりに表12に記載の位相差フィルム2~55を使用したこと以外は、上記偏光板5-1の作製と同様にして、それぞれ、偏光板5-2~5-55を作製した。
上記偏光板5-1の作製において、位相差フィルム1の代わりに表12に記載の位相差フィルム2~55を使用したこと以外は、上記偏光板5-1の作製と同様にして、それぞれ、偏光板5-2~5-55を作製した。
《液晶表示装置の作製》
市販のVA型液晶表示装置(SONY製40型ディスプレイKLV-40J3000)を用い、液晶セルの両面に貼合されていた偏光板を剥離し、上記作製した各偏光板を、図2で示すように液晶セルの両面に貼合して液晶表示装置を作製した。その際、偏光板の吸収軸の向きはあらかじめ貼合されていた偏光板と同じ向きに調整した。
市販のVA型液晶表示装置(SONY製40型ディスプレイKLV-40J3000)を用い、液晶セルの両面に貼合されていた偏光板を剥離し、上記作製した各偏光板を、図2で示すように液晶セルの両面に貼合して液晶表示装置を作製した。その際、偏光板の吸収軸の向きはあらかじめ貼合されていた偏光板と同じ向きに調整した。
《偏光板および液晶表示装置の評価》
上記作製した各液晶表示装置およびその作製に用いた各偏光板について、下記の各評価を行った。
上記作製した各液晶表示装置およびその作製に用いた各偏光板について、下記の各評価を行った。
(耐湿性の評価:含水変動による色ムラの評価)
上記作製した液晶表示装置を寝かせて台の上などに置き、ベンコット(旭化成せんい社製)を評価用偏光板の一部に載せて水を含ませた。ベンコットが乾かないよう100μmPETで覆い、テレビにPCから黒表示の信号を入力、テレビの電源ONで24時間放置した(室温は23℃に設定、パネル温度は38℃)。24時間後、ベンコットを取り除く。ベンコットのあった部分のL*を水浸漬部のL*としてEZコントラスト(ELDIM社製)で測定した。ベンコットのない部分のL*を非浸漬部のL*としてEZコントラストで測定した。なお、EZコントラストでの測定はTVを黒表示にしてカラーモードにて行った。水浸漬の条件はパネルの電源をONにし、かつ水を十分に浸み込ませたベンコットを貼り付けた状態で24時間静置する条件とした。次いで、水浸漬部のL*/非浸漬部のL*を算出し、下記の基準に従って色ムラの評価を行った。
上記作製した液晶表示装置を寝かせて台の上などに置き、ベンコット(旭化成せんい社製)を評価用偏光板の一部に載せて水を含ませた。ベンコットが乾かないよう100μmPETで覆い、テレビにPCから黒表示の信号を入力、テレビの電源ONで24時間放置した(室温は23℃に設定、パネル温度は38℃)。24時間後、ベンコットを取り除く。ベンコットのあった部分のL*を水浸漬部のL*としてEZコントラスト(ELDIM社製)で測定した。ベンコットのない部分のL*を非浸漬部のL*としてEZコントラストで測定した。なお、EZコントラストでの測定はTVを黒表示にしてカラーモードにて行った。水浸漬の条件はパネルの電源をONにし、かつ水を十分に浸み込ませたベンコットを貼り付けた状態で24時間静置する条件とした。次いで、水浸漬部のL*/非浸漬部のL*を算出し、下記の基準に従って色ムラの評価を行った。

位相差フィルムの構成内容および上記評価の結果を、下記表12および表13に、それぞれ、要約する。なお、上記偏光板2-1~2-55を用いて液晶表示装置を作製し、当該液晶表示装置について上記と同様にして評価したところ、上記偏光板1-1~1-55を使用した液晶表示装置1~55と、それぞれ、同様の結果が得られた。同様にして、上記偏光板3-1~3-55を用いて液晶表示装置を作製し、当該液晶表示装置について上記と同様にして評価したところ、上記偏光板1-1~1-55を使用した液晶表示装置1~55と、それぞれ、同様の結果が得られた。同様にして、上記偏光板4-1~4-55を用いて液晶表示装置を作製し、当該液晶表示装置について上記と同様にして評価したところ、上記偏光板1-1~1-55を使用した液晶表示装置1~55と、それぞれ、同様の結果が得られた。同様にして、上記偏光板5-1~5-55を用いて液晶表示装置を作製し、当該液晶表示装置について上記と同様にして評価したところ、上記偏光板1-1~1-55を使用した液晶表示装置1~55と、それぞれ、同様の結果が得られた。




上記表1~13から、本発明の位相差フィルムは、比較例のフィルムに比べて、高いレターデーション値を維持しつつ、引き裂き強度に優れていることが確認される。さらに、当該位相差フィルムを用いた偏光板、液晶表示装置は、大面積サイズとした場合であっても色ムラの発生を抑制できることもまた確認される。
1 溶解釜、
3、6、12、15 濾過器、
4、13 ストックタンク、
5、14 送液ポンプ、
8、16 導管、
10 紫外線吸収剤仕込釜、
20 合流管、
21 混合機、
30 ダイ、
31 金属支持体、
32 ウェブ、
33 剥離位置、
34 テンター装置、
35 ローラ乾燥装置、
41 仕込釜、
42 ストックタンク、
43 ポンプ、
44 濾過器、
101A、101B 偏光板、
102 保護フィルム、
103A、103B 活性エネルギー線硬化性接着剤、
104 偏光子、
105 位相差フィルム、
106 機能性層、
107 液晶セル、
108 液晶表示装置。
3、6、12、15 濾過器、
4、13 ストックタンク、
5、14 送液ポンプ、
8、16 導管、
10 紫外線吸収剤仕込釜、
20 合流管、
21 混合機、
30 ダイ、
31 金属支持体、
32 ウェブ、
33 剥離位置、
34 テンター装置、
35 ローラ乾燥装置、
41 仕込釜、
42 ストックタンク、
43 ポンプ、
44 濾過器、
101A、101B 偏光板、
102 保護フィルム、
103A、103B 活性エネルギー線硬化性接着剤、
104 偏光子、
105 位相差フィルム、
106 機能性層、
107 液晶セル、
108 液晶表示装置。
Claims (6)
- セルロースエステル樹脂と、下記一般式(1):

で表わされる化合物の少なくとも1種と、
下記一般式(2):

で表される化合物の少なくとも1種と、を含有することを特徴とする位相差フィルム。 - 膜厚が10~40μmである、請求項1に記載の位相差フィルム。
- 23℃、相対湿度55%環境下での波長590nmで測定した下記式(I)で定義されるRoが20~130nmであり、かつ23℃、相対湿度55%環境下での波長590nmで測定した下記式(II)で定義されるRtが100~300nmである、請求項1または2に記載の位相差フィルム:

- 前記セルロースエステル樹脂100重量部に対して、可塑剤を0.1~30重量%さらに含有する、請求項1~3のいずれか1項に記載の位相差フィルム。
- 請求項1~4のいずれか1項に記載の位相差フィルムが、活性エネルギー線硬化性接着剤で偏光子と接着されてなる、偏光板。
- 請求項5に記載の偏光板が、少なくとも液晶セルの片面に具備されている、垂直配向型液晶表示装置。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2013/063593 WO2014184909A1 (ja) | 2013-05-15 | 2013-05-15 | 位相差フィルム、偏光板および液晶表示装置 |
| JP2015516822A JP6156492B2 (ja) | 2013-05-15 | 2013-05-15 | 位相差フィルム、偏光板および液晶表示装置 |
| PCT/JP2013/082998 WO2014184981A1 (ja) | 2013-05-15 | 2013-12-09 | 位相差フィルム、偏光板および液晶表示装置 |
| KR1020157032022A KR20150143607A (ko) | 2013-05-15 | 2013-12-09 | 위상차 필름, 편광판 및 액정 표시 장치 |
| CN201380076607.1A CN105209942B (zh) | 2013-05-15 | 2013-12-09 | 相位差膜、偏振片及液晶显示装置 |
| JP2015516881A JP6245260B2 (ja) | 2013-05-15 | 2013-12-09 | 位相差フィルム、偏光板および液晶表示装置 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2013/063593 WO2014184909A1 (ja) | 2013-05-15 | 2013-05-15 | 位相差フィルム、偏光板および液晶表示装置 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014184909A1 true WO2014184909A1 (ja) | 2014-11-20 |
Family
ID=51897922
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/063593 WO2014184909A1 (ja) | 2013-05-15 | 2013-05-15 | 位相差フィルム、偏光板および液晶表示装置 |
| PCT/JP2013/082998 WO2014184981A1 (ja) | 2013-05-15 | 2013-12-09 | 位相差フィルム、偏光板および液晶表示装置 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/082998 WO2014184981A1 (ja) | 2013-05-15 | 2013-12-09 | 位相差フィルム、偏光板および液晶表示装置 |
Country Status (4)
| Country | Link |
|---|---|
| JP (2) | JP6156492B2 (ja) |
| KR (1) | KR20150143607A (ja) |
| CN (1) | CN105209942B (ja) |
| WO (2) | WO2014184909A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015146599A1 (ja) * | 2014-03-25 | 2015-10-01 | コニカミノルタ株式会社 | 位相差フィルムおよび、それを用いた偏光板、表示装置 |
| JP5950174B2 (ja) * | 2013-09-27 | 2016-07-13 | Dic株式会社 | 光学材料用樹脂組成物、光学フィルム及び液晶表示装置 |
| JP2016173467A (ja) * | 2015-03-17 | 2016-09-29 | Dic株式会社 | 光学フィルム及び液晶表示装置 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI494359B (zh) * | 2009-12-07 | 2015-08-01 | Dainippon Ink & Chemicals | A cellulose ester resin composition, an optical film using the same, and a polarizing plate for a liquid crystal display device |
| JP2017021236A (ja) * | 2015-07-13 | 2017-01-26 | コニカミノルタ株式会社 | Va用位相差フィルム、va用位相差フィルムの製造方法、偏光板および垂直配向型液晶表示装置 |
| CN110431455B (zh) * | 2017-03-14 | 2022-07-19 | 柯尼卡美能达株式会社 | λ/4相位差膜、圆偏振片和有机EL显示装置 |
| JP7381357B2 (ja) * | 2020-02-03 | 2023-11-15 | Dicグラフィックス株式会社 | 平版オフセット印刷用活性エネルギー線硬化型インキ、及び印刷物 |
| CN115677994A (zh) * | 2021-07-29 | 2023-02-03 | 武汉科技大学 | 基于联苯二甲酸的聚酯、纳米银导电膜制备及应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010522900A (ja) * | 2007-03-29 | 2010-07-08 | アクロン ポリマー システムズ | 液晶ディスプレイ用の正の複屈折を有する光学補償フィルム |
| JP2011116912A (ja) * | 2009-12-07 | 2011-06-16 | Dic Corp | セルロースエステル樹脂組成物、それを用いた光学フィルム及び液晶表示装置用偏光板 |
| JP2011140637A (ja) * | 2009-12-07 | 2011-07-21 | Dic Corp | セルロースエステル樹脂組成物、それを用いた光学フィルム及び液晶表示装置用偏光板 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63113077A (ja) * | 1986-06-13 | 1988-05-18 | Konica Corp | 分光吸収特性等に優れたジフエニルイミダゾ−ル系染料 |
| JP2864075B2 (ja) * | 1991-12-04 | 1999-03-03 | 富士写真フイルム株式会社 | イミダゾールアゾ色素およびそれを含有する熱転写色素供与材料 |
| JPH07207169A (ja) * | 1994-01-12 | 1995-08-08 | Mitsui Toatsu Chem Inc | 偏光フィルム用アゾ系色素及びこれを用いた偏光フィルム |
| JP2000056123A (ja) * | 1998-08-05 | 2000-02-25 | Toray Ind Inc | カラーフィルター用顔料およびカラーペースト、カラーフィルター、液晶表示素子ならびにそれらの製造方法 |
| JP2003026942A (ja) * | 2001-05-11 | 2003-01-29 | Kanegafuchi Chem Ind Co Ltd | 樹脂組成物 |
| KR101352677B1 (ko) * | 2006-04-25 | 2014-01-16 | 코니카 미놀타 어드밴스드 레이어즈 인코포레이티드 | 위상차 필름, 편광판 및 액정 표시 장치 |
| US8597551B2 (en) * | 2010-12-23 | 2013-12-03 | Sk Innovation Co., Ltd. | Optical film |
| WO2012133169A1 (ja) * | 2011-03-31 | 2012-10-04 | コニカミノルタアドバンストレイヤー株式会社 | 液晶表示装置 |
-
2013
- 2013-05-15 JP JP2015516822A patent/JP6156492B2/ja active Active
- 2013-05-15 WO PCT/JP2013/063593 patent/WO2014184909A1/ja active Application Filing
- 2013-12-09 JP JP2015516881A patent/JP6245260B2/ja active Active
- 2013-12-09 WO PCT/JP2013/082998 patent/WO2014184981A1/ja active Application Filing
- 2013-12-09 CN CN201380076607.1A patent/CN105209942B/zh active Active
- 2013-12-09 KR KR1020157032022A patent/KR20150143607A/ko not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010522900A (ja) * | 2007-03-29 | 2010-07-08 | アクロン ポリマー システムズ | 液晶ディスプレイ用の正の複屈折を有する光学補償フィルム |
| JP2011116912A (ja) * | 2009-12-07 | 2011-06-16 | Dic Corp | セルロースエステル樹脂組成物、それを用いた光学フィルム及び液晶表示装置用偏光板 |
| JP2011140637A (ja) * | 2009-12-07 | 2011-07-21 | Dic Corp | セルロースエステル樹脂組成物、それを用いた光学フィルム及び液晶表示装置用偏光板 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5950174B2 (ja) * | 2013-09-27 | 2016-07-13 | Dic株式会社 | 光学材料用樹脂組成物、光学フィルム及び液晶表示装置 |
| WO2015146599A1 (ja) * | 2014-03-25 | 2015-10-01 | コニカミノルタ株式会社 | 位相差フィルムおよび、それを用いた偏光板、表示装置 |
| JPWO2015146599A1 (ja) * | 2014-03-25 | 2017-04-13 | コニカミノルタ株式会社 | 位相差フィルムおよび、それを用いた偏光板、表示装置 |
| JP2016173467A (ja) * | 2015-03-17 | 2016-09-29 | Dic株式会社 | 光学フィルム及び液晶表示装置 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN105209942B (zh) | 2018-01-30 |
| JP6156492B2 (ja) | 2017-07-05 |
| JPWO2014184981A1 (ja) | 2017-02-23 |
| KR20150143607A (ko) | 2015-12-23 |
| CN105209942A (zh) | 2015-12-30 |
| JPWO2014184909A1 (ja) | 2017-02-23 |
| WO2014184981A1 (ja) | 2014-11-20 |
| JP6245260B2 (ja) | 2017-12-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6156492B2 (ja) | 位相差フィルム、偏光板および液晶表示装置 | |
| JP6292328B2 (ja) | トリアゾール化合物 | |
| TWI569967B (zh) | A polarizing plate and a liquid crystal display device using the same | |
| TWI513994B (zh) | Optical film, circular polarizer and image display device | |
| WO2015001980A1 (ja) | 偏光板およびこれを用いた液晶表示装置 | |
| KR101778477B1 (ko) | 편광판, 그것을 구비한 액정 표시 장치 | |
| WO2014136529A1 (ja) | 光学フィルム、並びにこれを含む偏光板およびva型液晶表示装置 | |
| TWI498371B (zh) | A retardation film, a polarizing plate, and a liquid crystal display device | |
| CN105849599B (zh) | 纤维素酯膜、其制造方法及偏振片 | |
| KR20160060095A (ko) | 위상차 필름, 편광판 및 액정 표시 장치 | |
| JP6627748B2 (ja) | 位相差フィルムおよび、それを用いた偏光板、表示装置 | |
| JP6215869B2 (ja) | 光学フィルム、偏光板、および液晶表示装置 | |
| KR20210006854A (ko) | 편광판의 제조 방법 및 편광판 | |
| WO2016111058A1 (ja) | 垂直配向型液晶表示装置 | |
| JP2014048611A (ja) | 光学フィルム、偏光板及び液晶表示装置 | |
| JP2014153444A (ja) | 位相差フィルム、偏光板及び液晶表示装置 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13884564 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015516822 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13884564 Country of ref document: EP Kind code of ref document: A1 |