WO2013104263A1 - 多羟基苯并吡喃酮类化合物的合成及其抗肿瘤作用 - Google Patents
多羟基苯并吡喃酮类化合物的合成及其抗肿瘤作用 Download PDFInfo
- Publication number
- WO2013104263A1 WO2013104263A1 PCT/CN2012/088016 CN2012088016W WO2013104263A1 WO 2013104263 A1 WO2013104263 A1 WO 2013104263A1 CN 2012088016 W CN2012088016 W CN 2012088016W WO 2013104263 A1 WO2013104263 A1 WO 2013104263A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- methyl
- benzopyran
- hydrogen
- compound
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/22—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4
- C07D311/26—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3
- C07D311/28—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3 with aromatic rings attached in position 2 only
- C07D311/30—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring with oxygen or sulfur atoms directly attached in position 4 with aromatic rings attached in position 2 or 3 with aromatic rings attached in position 2 only not hydrogenated in the hetero ring, e.g. flavones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
Definitions
- the present invention relates to polyhydroxybenzopyrone compounds and salts or prodrugs thereof, and to pharmaceutical compositions containing such compounds for use in the manufacture of a medicament for the prevention and/or treatment of neoplastic diseases.
- Estrogen is a group of hormones associated with many key physiological functions in the human body.
- the functions of estrogen include promoting female sexual organ development, preparing for breast and uterus during pregnancy, and breastfeeding after childbirth. Estrogen also plays an important role in maintaining proper cardiovascular function and bone density. It is well known that estrogen can stimulate cell proliferation, which may increase the risk of cancer in women, especially breast cancer and uterine cancer. Estrogen regulates cellular function by binding to estrogen receptors in target cells. Two estrogen receptors (ERs), ER- ⁇ and ER-P have been found in human cells.
- N-terminal domain A/B domain
- C domain mid-DNA binding domain
- D/E/F domain C-terminal ligand binding Domain
- the N-terminal domain has a non-ligand-dependent activation function (AF-1), which interacts with a coactivator and transcriptionally activates a target gene in the absence of a ligand.
- AF-1 non-ligand-dependent activation function
- DNA binding domains play an important role in receptor dimerization and binding to specific DNA sequences.
- the C-terminal ligand binding domain mediates ligand binding and has a ligand-dependent transcriptional activation function (AF-2) that activates gene transcription in the presence of a ligand.
- AF-1 non-ligand-dependent activation function
- AF-2 ligand-dependent transcriptional activation function
- ER- ⁇ The full-length ER- ⁇ is a protein with a molecular weight of 66 kDa and is called ER-a66.
- ER-a66 contains all three functional domains.
- the splice variant of ER-a66 was later discovered and named ER-a46.
- ER-a46 has a molecular weight of approximately 46 kDa, which lacks the N-terminal AF-1 domain of hER-a66.
- ER-a36 a new 36 kDa ER- ⁇ variant, ER-a36, was discovered. It lacks the N-terminal AF-1 domain of ER-a66 and the C-terminal AF-2 domain (see wang et al, Biochem. Biophys. Res. Commun.
- ER-a66 is generally considered Transcription activates its target gene to mediate estrogen-stimulated cell proliferation.
- the binding of estrogen to ER-a66 activates the transcriptional activation domain of ER-a66, thereby stimulating the expression of downstream target genes and ultimately leading to cell proliferation.
- ER-a46 has been shown to mediate rapid NO synthesis initiated by membranes and stimulated by estrogen (see Li et al, Proc. Natl. Acad. Sci. USA 100: 4807-4812, (2003)) and ER-a46 lacking the AF-1 domain was found to inhibit AF-1 activity of ER-a66 (see Flouriot, G., EMBO, 19, 4688-4700, (2000)).
- ER-a36 deletion of AF-1 and The AF-2 transcriptional activation domain which acts as a dominant negative inhibitor, inhibits the AF-1 and AF-2 functions of ER-a and ER- ⁇ .
- ER-a36 is mainly distributed on the cell membrane and mediates Membrance-induced conduction of mitogenic estrogen signaling, which stimulates cell proliferation (see wang et al, Biochem. Biophys. Res. Commun. 336, 1023-1027, (2005); wang et al, Proc. Natl. Acad. Sci. USA 103: 9063-9068, (2006) In-depth studies have shown that estrogen signaling is mediated through traditional nuclear transcriptional activation pathways and non-traditional membrane-primed signaling pathways.
- ER-a66 and ER-a46 appear to act primarily in the nucleus, whereas ER-a36 appears to act primarily through the nucleus. It has also been shown that ER-a36 lacks the helix 8-12 of the ligand binding domain possessed by the original ER-a66, which completely alters the specificity of ER-a36 ligand binding. Therefore, ER-a36 may bind different ligands to ER-a66 and ER- ⁇ . Since diseases associated with estrogen receptors still affect many people, there is an urgent need to find a novel compound and a pharmaceutical composition thereof for preventing and/or treating these related diseases. Summary of the invention
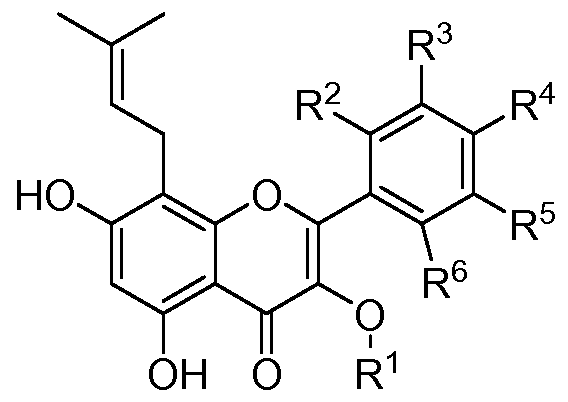
- the present invention provides a class of polyhydroxybenzopyrone compounds and salts or prodrugs thereof which are useful for regulating the function of the novel estrogen receptor ER-a36, and the formula (I):
- R 1 selected from hydrogen, (CC 6) alkyl containing one or more halogen atoms (CC 6) alkyl;
- R 2, R 3, R 4 , R 5 and R 6 may be independently selected from hydrogen, (dC 4) alkyl, containing one or more substituted with halogen atom (dC 4) alkyl, halo, cyano, containing a Or a plurality of halogen atom-substituted (dC 6 ) alkoxy groups; but R 2 , R 3 , R 4 , R 5 and R 6 may not be hydrogen at the same time;
- R 4 may not be a chlorine atom.
- Figure 1 shows the results of Western blotting of ER-a66, ER-a46 and ER-a36 expression in human breast cancer samples.
- Lane 1 normal breast tissue; tract 2: invasive ductal carcinoma; tract 3: invasive ductal carcinoma; tract 4: invasive ductal carcinoma; tract 5: invasive lobular carcinoma; tract 6: invasive lobular carcinoma; Non-invasive ductal carcinoma.
- Figure 2 (top panel) shows the results of immunofluorescence staining of MDA-MB-231 cells.
- MDA-MB-231 cells are ER-negative breast cancer cell lines lacking ER-a66 and ER-a46.
- MDA-MB-231 cells were stained with an antibody that specifically binds to ER-a36 (left panel labeled "ER-a36 Ab” in the figure: positive staining is shown in green). Using 4,6-diindol-2-phenylindole for cells The core is stained. (The middle panel labeled "DAPI'' in the figure: the positive staining is shown in blue.) The combined staining signals are labeled as "merging.” When the antibody is preincubated with the immunogenic polypeptide bound to the antibody, The results were negative (bottom).
- Figure 3 shows the results of Western blotting of ER-0136 expressing different tumor cell lines.
- Lane 1 293 human renal epithelial cell lines transiently overexpressing ER-0136; Human-derived human breast cancer SK-BR-3 cell line; Lane 5-7: Human breast cancer MCF-7 cell line from different laboratory sources; Lane 8-9: Human leukemia HL-60 cells from different laboratory sources Strain 10-11: Human leukemia MV-4-11 cell line from different laboratory sources; Lane 12-13: Human granulocyte leukemia K562 cells from different laboratory sources; Lane 14: Human liver cancer A2780 cells; : Human liver cancer HEL-7402 cells; Lane 16: Human liver cancer HEL-9204 cells; Lane 17: Primary liver cancer Hep-11 cells derived from patients; Lane 18: Primary liver cancer Hep-12 cells derived from patients.
- Figure 8 shows the detection of compound 1 in vitro against gastric cancer BGC-823 cells, lung cancer H460 cells by MTT assay, Growth inhibition of colon cancer LS174T cells, pancreatic cancer PANC-1 and prostate cancer PC-3 cells. The results showed that Compound 1 had significant growth inhibitory effects on these tumor cells in a dose-dependent manner.
- Figure 9 shows the comparison with tamoxifen (0.7 mg/mouse/day), compound 1 (0.7 mg/mouse/day) and negative control blank vehicle (0.2 mL/mouse/day), respectively.
- the average tumor weight (histogram a) of the tumor-bearing human breast cancer BCAP-37 tumor strain after 20 days of continuous administration showed the inhibition of tumor growth of the compound.
- Figure 10 shows that when rituximab was used respectively Positive control, growth curve of tumors administered to nude mice bearing tumor-bearing human B lymphoma Daudi cells for 21 days in a compound 1 (0.7 mg/mouse/day) and a negative control blank vehicle (0.2 mL/mouse/day).
- Figure 11 shows when the positive control medroxyprogesterone acetate (120 mg/kg), and the low (17.5 mg/kg), medium (35 mg/kg), high dose (70 mg/kg) and negative of the compound 1 were respectively negative.
- Control blank vehicle (0.2 mL/mouse/day) on tumor-bearing human endometrial carcinoma Ishikawa tumor strain nude mice Continued administration after 20 days the average tumor weight. Remarkably the growth inhibitory effects of compounds on tumor tumor of mice, and better than the positive control.
- a benzopyrone compound a pharmaceutically acceptable salt or prodrug thereof, and a pharmaceutical composition comprising the same, which are useful in Regulates the function of the novel estrogen receptor ER-a36, and prevents and/or treats diseases mediated by the ER-0136 receptor, such as cancer.
- the invention provides a compound of formula (I):
- R 1 selected from hydrogen, (CC 6) alkyl containing one or more halogen atoms (CC 6) alkyl;
- R 2, R 3, R 4 , R 5 and R 6 may be independently selected from hydrogen, (dC 4) alkyl, containing one or more substituted with halogen atom (dC 4) alkyl,, cyano, containing a Or a plurality of atom-substituted (dC 4 ) alkoxy groups, but R 2 , R 3 , R 4 , R 5 and R 6 may not be hydrogen at the same time; and when R 1 is a methyl group and R 3 and R 5 are hydrogen R 4 may not be a chlorine atom.
- One embodiment of the present invention comprises a group of compounds having the structure of formula (I), referred to as a compound ( ⁇ ), wherein the compound has the formula:
- R 2, R 3, R 4 , R 5 and R 6 are independently selected from hydrogen, (C r C 4) alkyl, containing one or more halogen atoms, substituted with (C r C 4) alkyl, Su, cyano a group having one or more atom-substituted (C r C 4 ) alkoxy groups; but R 2 , R 3 , R 4 , R 5 and R 6 may not be hydrogen at the same time.
- One embodiment of the present invention comprises a group of compounds of the formula (I), referred to as a compound ( ⁇ ), wherein the group of compounds has the formula:
- R 2, R 3, R 4 , R 5 and R 6 may be independently selected from hydrogen, (dC 4) alkyl, containing one or more substituted with halogen atom (dC 4) alkyl,, cyano, containing a Or a plurality of atom-substituted (dC 4 ) alkoxy groups; but R 2 , R 3 , R 4 , R 5 and R 6 may not be hydrogen at the same time.
- R 4 may not be a chlorine atom.
- Particularly preferred compounds of the formula (I) of the present invention include, but are not limited to, the following compounds:
- substitution means that a hydrogen atom in a molecule is replaced by a different atom or molecule.
- An atom or molecule that replaces a hydrogen atom is referred to as a "substituent.”
- the minimum and maximum values of the carbon atom content in the hydrocarbon group are indicated by a prefix, for example, the prefix (C a -C b )alkyl group indicates any alkyl group having from “a” to "b” carbon atoms.
- (dQ alkyl refers to an alkyl group containing from 1 to 6 carbon atoms.
- alkoxy refers to a straight or branched, monovalent, saturated fat bonded to an oxygen atom.
- alkyl refers to a straight chain or a branch. Chained, monovalent, saturated aliphatic chains including, but not limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, pentyl, isopentyl, hexyl, and the like.
- halogen or "halogen atom” means a chlorine, bromine, fluorine and iodine atom or group.
- heteroaryl means that one or more of the carbon atoms have been replaced by a hetero atom such as nitrogen, oxygen or sulfur.
- a hetero atom such as nitrogen, oxygen or sulfur.
- Monocyclic or polycyclic aromatic hydrocarbon groups If a heteroaryl group contains more than one hetero atom, these heteroatoms may be the same or different.
- Heteroaryl groups include, but are not limited to, benzofuranyl, benzothienyl Benzimidazolyl, benzoxazolyl, benzothiazolyl, benzopyranyl, furan , imidazolyl, oxazolyl, fluorenyl, fluorenyl, isobenzofuranyl, isodecyl, isoquinolinyl, isothiazolyl, isoxazolyl, naphthyridinyl, oxadiazolyl, Oxime, oxazolyl, fluorenyl, pteridinyl, fluorenyl, pyranyl, pyridyl, pyrazolyl, fluorenyl, pyrido[3,4-b]decyl, pyridine , pyrimidinyl, pyrrolyl, quinolyl, quinolyl, quinoxalinyl, thiadiazolyl, thiatriazo
- oxo refers to a carbonyl group formed by the combination of a carbon atom and an oxygen atom.
- Prodrugs and solvates of the compounds of the invention are also contemplated.
- prodrug refers to a pre-drug a compound that, after being administered, is chemically converted in the body by metabolic or chemical processes (for example, placed under physiological pH conditions or by the activity of an enzyme) to release the active drug.
- metabolic or chemical processes for example, placed under physiological pH conditions or by the activity of an enzyme
- Prodrugs of the invention may also include metabolic precursors of the compounds of the invention, which may not be active when administered to a subject, but may be in vivo Converted to a compound of formula (I) of the invention or a salt and/or solvate thereof. Prodrugs can also be naturally occurring or chemically synthesized compounds.
- the compounds of formula (I) of the present invention may exist in unsolvated as well as solvated forms of pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is contemplated that the present invention encompasses all solvated and unsolvated forms.
- the solvate of the compound of formula (I) is preferably a hydrate.
- All stereoisomers of the compounds of the invention such as isomers which may exist due to asymmetric carbon atoms on the R substituent of the compound of formula (I), including both enantiomeric and diastereomeric forms, are within the scope of the invention The scope. All stereoisomers of the compounds of formula (I) and mixtures thereof, including racemic mixtures, are also part of the invention. In addition, all geometric isomers and positional isomers are also included. For example, if the compound of the formula (I) contains a double bond, both the cis and trans forms and mixtures thereof are also included in the scope of the present invention.
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physicochemical differences by methods well known to those skilled in the art, such as chromatography and/or fractional crystallization. Separation of enantiomers can be carried out by reaction with an optically active compound, conversion of the enantiomeric mixture to a mixture of diastereomers, followed by separation of the diastereomers, followed by separate diastereoisomers. The isomer is converted (e.g., hydrolyzed) to the corresponding pure enantiomer.
- certain of the compounds of formula (I) may be atropisomers (e.g., substituted biaryls) and are also part of the present invention.
- pharmaceutically acceptable means that a carrier, carrier, diluent, adjuvant, and/or salt formed is generally chemically or physically compatible with the other ingredients that constitute a pharmaceutical dosage form, and is physiologically Compatible with the receptor.
- salt and “pharmaceutically acceptable salt” refer to a compound of formula (I) or a stereoisomer thereof, or a prodrug thereof and an inorganic
- an acid and/or a base salt formed from an organic acid and a base and also a zwitterionic salt (internal salt), and also a quaternary ammonium salt such as an alkylammonium salt.
- These salts can be obtained directly in the final isolation and purification of the compounds. It may also be obtained by mixing a compound represented by the formula (I), or a stereoisomer thereof, or a prodrug thereof with an appropriate amount (e.g., equivalent) of a certain amount of an acid or a base.
- These salts may be precipitated in a solution and collected by filtration, or recovered after evaporation of the solvent, or may be obtained by lyophilization after reaction in an aqueous medium.
- Acid addition salts include, for example, hydrobromide, hydroiodide, hydrochloride, sulfate, hydrogen sulfate, nitrate, acetate
- oxalate alginate, ascorbate, aspartate, butyrate, camphorate, camphorsulfonate, cyclopentane Propionate, digluconate, ethanesulfonate, 2-hydroxyethanesulfonate, 2-naphthalenesulfonate, nicotinate, persulfate, 3-phenylpropionate, picrate, Penic acid salt, propionate, salicylate, besylate, palmitate, stearate, laurate, borate, benzoate, lactate, phosphate, toluene Acid salt, citrate, maleate, fumarate, succinate, tartrate, thiocyanate, naphthate, methanesulfonate, glucoheptonate, lactobionate, dodecyl Sulfonates, adipates, and other similar salts.
- a basic salt for example, a salt formed when the R substituent contains an acidic moiety such as a carboxyl group or a phenolic hydroxyl group
- an ammonium salt for example, an alkali metal salt (for example, a sodium salt, a lithium salt, and a potassium salt), and an alkaline earth metal salt (for example, a calcium salt).
- magnesium salts salts with organic bases (such as organic amines) (eg dibenzylethylenediamine, dicyclohexylamine, sulphamine, N-methyl-D-glucosamine, tert-butylamine), and amino acids Such as arginine, a salt formed by lysine, and the like.
- quaternary ammonium salts with nitrogen-containing alkaline agents including, but not limited to, ammonium, tetramethylammonium, tetraethylammonium, methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, and Other analogues. Further examples are found in the references of this patent, Berge, et al., J. Pharm. Sci., 66, 1-19 (1977). The compounds of formula (I) may also exist as tautomers in equilibrium, and all such forms are embraced within the scope of the invention.
- the invention includes isotopically-labeled compounds of formula (I), said isotopically-labeled compounds being the same as the compounds listed herein, but wherein one or more of the atoms are replaced by another atom
- the atomic mass or mass of the atom is different from the atomic mass or mass number that is common in nature.
- Isotopes which may be introduced into the compounds of formula (I) include hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine and chlorine, SP 2 H, 3 H, 13 C, 14 C, 15 N, 17 0, 18 0, 31 P , 32 P, 35 S, 18 F and 36 C1.
- Compounds of formula (I), and stereoisomers and prodrugs thereof, containing such isotopes and/or other atomic isotopes, as well as pharmaceutically acceptable salts of such compounds, stereoisomers or prodrugs, are intended to be encompassed within the scope of the invention within.
- Certain isotopically-labeled compounds of formula (I), such as those labeled with radioisotopes such as 3 H and 14 C, can be used in tissue distribution analysis of compounds and or substrates. Since the isotope of yttrium (ie) and the isotope of carbon 14 (ie, 14 C) are relatively easy to prepare and detect, we prefer the 3 H isotope and the 14 C isotope.
- isotopes such as deuterated (ie, 2 H)
- the isotopically-labeled compound of formula (I) can generally be prepared by methods known to those of ordinary skill in the art, such as the replacement of non-isotopically labeled reagents with isotopically labeled reagents.
- the compounds of the present invention are modulators of the novel estrogen receptor ER-a36 and can be used to modulate the function of ER-a36 in cells in vitro and in vivo.
- the compounds of formula (I) according to the invention can be used Prevention and/or treatment of related diseases mediated by ER-0136, especially related oncological diseases.
- methods of modulating the function of ER-a36 in a cell comprising administering a compound of formula (I) to a cell expressing ER-0136.
- the cells can endogenously express ER-0136 or exogenously express ER-a36 by genetic engineering, and can simultaneously or not express other estrogen receptors (such as ER-a66, ER-a46 and ER-).
- the cell endogenously expresses ER-a36.
- the The cells are cancer cells that endogenously express ER-a36.
- Cancer cells expressing ER-0136 include, but are not limited to, breast cancer cells, leukemia cells, liver cancer cells, lymphoma cells, lung cancer cells, myeloma cells, prostate cancer cells, ovarian cancer cells, endometrial cancer cells, colon cancer cells. And gastric cancer cells.
- the cell expressing ER-0136 is a breast cancer cell, leukemia cell, liver cancer cell, lymphoma cell, endometrial cancer cell, and ovarian cancer cell endogenously expressing ER-0136.
- Breast cancer cells expressing ER-a36 include, but are not limited to, MCF7, MDA-MB-231, and SKBR-3 cells.
- Leukemia cells expressing ER-a36 include, but are not limited to, K562, MV-4-11, SUM159, HL-60 and Molt-4 cells.
- Endometrial cancer cells that express ER-a36 include, but are not limited to, HeclA cells.
- Hepatoma cells expressing ER-a36 include, but are not limited to, A2780, BEL7402, BEL7404, HEL-9204, Hep2G, Hep3B and primary liver cancer stem cell Hep-12 derived from a patient.
- Lymphoma cells expressing ER-a36 include, but are not limited to, Daudi. Expression of endogenous ER-a36 can be increased or decreased by treatment with one or more agents.
- the invention provides a method of making a cell expressing exogenous ER-a36.
- the cells can be prepared by genetic engineering methods known to those of ordinary skill in the (; See Samb k, etc., Molecular Cloning, A Laboratory Manual ( 2d Ed 1989) (Cold Spring Harbor Laboratory)) 0 Briefly, a first outer prepared The ER-a36 gene is inserted into an expression vector, and the expression vector is transfected into a host cell, and then the host cell is grown in a culture medium suitable for expressing exogenous ER-0136. .
- ER-0136 The gene sequence of human ER-0136 is disclosed in Wang et al., Biochem. Biophys. Res. Commun. 336, 1023-1027 (2005) (GenBank Accession No. BX640939).
- Cells expressing exogenous ER-a36 may or may not express endogenous ER-a36.
- the level of expression of endogenous or exogenous ER-a36 in the cell can be increased or decreased by treatment with one or more other agents. These agents include, for example, serum, ⁇ 2 ⁇ (7 ⁇ -estradiol), tamoxifen, and fulvestrant (ICI 182, 780).
- the compound of the formula (I) according to the present invention can be used for the preparation of a medicament for preventing and/or treating cancer associated with ER-a36, including but not limited to anal cancer, cholangiocarcinoma, bladder cancer, bone cancer, intestinal cancer (colon cancer) , rectal cancer), brain cancer, breast cancer, carcinoid, cervical cancer, endocrine-related cancer, endometrial cancer, eye cancer, gallbladder cancer, head and neck cancer, Kaposi's sarcoma cancer, kidney cancer, laryngeal cancer, leukemia, liver cancer , lung cancer, lymphoma, melanoma, mesothelioma, myeloma, neuroendocrine cancer, esophageal cancer, ovarian cancer, pancreatic cancer, penile cancer, prostate cancer, skin cancer, soft tissue sarcoma cancer, spinal cord cancer, stomach cancer, testicular cancer, Thyroid cancer, vaginal cancer, vulvar cancer or uterine cancer.
- the cancer associated with ER-a36 includes breast cancer, cervical cancer, colon cancer, endometrial cancer, leukemia, liver cancer, lymphoma, lung cancer, myeloma, ovarian cancer, prostate cancer, gastric cancer , pancreatic cancer, kidney cancer, melanoma, thyroid cancer, soft tissue sarcoma cancer, or uterine cancer.
- the cancer associated with ER-a36 includes breast cancer, liver cancer, lymphoma, prostate cancer, gastric cancer, lung cancer, colon cancer, pancreatic cancer, endometrial cancer, ovarian cancer, and leukemia.
- the subject may be a mammal, such as a dog, cat, cow, sheep, horse or human, preferably a human.
- the necessary therapeutic amount of the drug will vary depending on the particular disease and can be readily determined by one of ordinary skill in the art having the benefit of the present disclosure.
- one or more compounds of the invention may be used in combination with one another.
- the compounds of the invention may be used in combination with any other active agent for the preparation of a medicament or pharmaceutical composition that modulates cellular function or treats a disease. If a group of compounds is used, the compounds can be administered to the subject simultaneously, separately or sequentially.
- the compounds of the invention may be used in combination with one or more other anticancer agents.
- Anticancer agents that can be used include, but are not limited to, alkylating agents, nitrogen mustards, folic acid antagonists, guanidine antagonists, pyrimidine antagonists, spindle toxins, topoisomerase inhibitors, apoptosis inducers, blood vessels Production inhibitors, ghost white toxin, nitrosourea, antimetabolites, protein synthesis inhibitors, kinase inhibitors, antiestrogens, cisplatin, carboplatin, interferon, asparaginase, leuprolide, Flutamide, megestrol, mitomycin, bleomycin, doxorubicin, irinotecan and paclitaxel.
- the anticancer agent is an antiestrogen drug, such as tamoxifen and fulvestrant (ICI 182, 780).
- a compound of formula (I), or a stereoisomer or prodrug thereof, or a pharmaceutically acceptable salt of a stereoisomer or prodrug of said compound may be administered as a medicament A form of the composition comprising a pharmaceutically acceptable carrier, carrier or diluent. They are also useful in the preparation of a medicament for the prevention and/or treatment of a disease associated with ER-0136 in a subject.
- the pharmaceutical compositions of the invention are also useful for treating diseases in animals.
- a general veterinarian can administer a compound of the invention, or a veterinary salt thereof, or a veterinary solvent or a prodrug thereof, in a suitable acceptable formulation, based on experience in the art. The veterinarian can determine the most appropriate dosage and route of administration for an animal. If a plurality of active compounds are administered in combination, the active compounds may be administered sequentially, separately or sequentially in a certain order.
- Method of Preparing Compounds The compounds of formula (I) can be prepared by various synthetic methods. A typical preparation process is listed below. Unless otherwise indicated, the definitions of 1 ⁇ , 1 2 , 1 3 , 1 4 , 1 5 , 16 are as previously described. It will be apparent to those skilled in the art that the exact method of preparation of the compounds can vary slightly depending on the chemical structure.
- the compound of the formula (I) can be produced in several steps.
- Compound L is prepared from compound i_ and catalyzed by a Lewis acid by a Houben-Hersch reaction (modified Friedel-Glysylation).
- Lewis acids suitable for the reaction include, for example, anhydrous zinc chloride, anhydrous aluminum trichloride, ferric chloride, titanium tetrachloride, tin chloride, boron trifluoride etherate complex and the like.
- the reaction is usually carried out at a temperature of from about 0 ° C to about 120 ° C for 1 to 20 hours.
- Compound X can be prepared by condensation of compound L with various substituted compounds ⁇ in an inert solvent.
- Suitable inert solvents for this reaction include ethers such as DME, 1,2-diethoxyethane, THF, 1,4-dioxane, DMF, hydrazine, hydrazine-dimethylacetamide, pyridine, hydrazine -Methyl-2-pyrrolidone.
- the reaction can be carried out under basic conditions such as potassium hydroxide, potassium carbonate, cesium carbonate, sodium hydride, sodium methoxide, potassium t-butoxide, DBU 1,8-diazabicyclo-bicyclo (5, 4, 0)-7-undecene), n-butyllithium, LDA (lithium diisopropylamide), LHMDS (lithium hexamethyldisilazide) and the like.
- a stoichiometric or catalytic amount of a phase transfer catalyst such as 18-crown-6, TBAB (tetrabutylammonium bromide), TBAF (tetrabutylammonium fluoride) or the like may be added during the reaction.
- the reaction temperature is usually between about 0 ° C and about 140 ° C, preferably at the reflux temperature of the solvent for 1 to 20 hours.
- the isopentenyl bromide can be reacted with the compound iL under basic conditions to prepare a compound.
- Suitable solvents for the reaction include, for example, methanol, DMF (N,N-dimethylformamide), THF (tetrahydrofuran), water, toluene, DME (1,2-dimethoxyethane), and mixed solvents. Such as methanol-water, DMF-water, tetrahydrofuran-water and the like.
- the solvent used in the reaction is preferably water.
- Bases suitable for the reaction include, for example, potassium hydroxide, potassium carbonate, cesium carbonate, sodium methoxide, sodium hydride, potassium t-butoxide, DBU (1,8-diazabicyclo-bicyclo(5,4,0) -7-undecene), n-butyllithium, LDA (lithium diisopropylamide), LHMDS (lithium hexamethyldisilazide) and the like.
- the reaction temperature is usually between about 0 ° C and about 100 ° C and the reaction time is from 1 to 20 hours.
- Compound ill can be prepared by reacting compound ill with isopentenyl bromide under basic conditions.
- suitable solvents for the reaction include, for example, methanol, DMF (N,N-dimethylformamide), THF (tetrahydrofuran), water, toluene, DME (1,2-dimethoxyethane), and mixed solvents.
- methanol-water, DMF-water, tetrahydrofuran-water and the like The solvent used in the reaction is preferably water.
- Bases suitable for the reaction include, for example, potassium hydroxide, potassium carbonate, cesium carbonate, sodium methoxide, sodium hydride, potassium t-butoxide, DBU (1,8-diazabicyclo-bicyclic (5,4,0) -7-undecene), n-butyllithium, LDA (lithium diisopropylamide), LHMDS (lithium hexamethyldisilazide) and the like.
- the reaction temperature is usually between about 0 ° C and about 100 ° C and the reaction time is from 1 to 20 hours.
- Compound ⁇ can be prepared by condensation of compound IL with various substituted acid chlorides iii in an inert solvent.
- Suitable inert solvents for this reaction include ethers such as DME, 1,2-diethoxyethane, THF, 1,4-dioxane, DMF, hydrazine, hydrazine-dimethylacetamide, pyridine, hydrazine -Methyl-2-pyrrolidone.
- the reaction can be carried out under basic conditions, such as potassium hydroxide, potassium carbonate, cesium carbonate, sodium hydride, sodium methoxide, potassium t-butoxide, DBU 1,8-diazabicyclo-bicyclo (5,4,0 )-7-undecene), n-butyllithium, LDA (lithium diisopropylamide), LHMDS (lithium hexamethyldisilazide) and the like.
- a stoichiometric or catalytic amount of a phase transfer catalyst such as 18-crown-6, TBAB (tetrabutylammonium bromide), TBAF (tetrabutylammonium fluoride), or the like may be added during the reaction.
- the temperature of the reaction is usually between about 0 ° C and about 140 ° C, preferably at the reflux temperature of the solvent for 1 to 20 hours.
- R 1 of the compound of formula (I) is hydrogen, it can be prepared according to the following embodiment
- P is a protecting group for a hydroxyl group.
- the compound is removed by removing the protecting group of the compound.
- the method of removal varies according to the nature of the protection. See mainly "Protective Groups in Organic Synthesis” (Greene T. W et al. John Wiley & Sons, New York, 1991).
- Preferred protecting groups are benzyl, benzoyl, benzyloxycarbonyl, TBDMS (tert-butyldimethylsilyl), THP (tetrahydropyranyl), methyl, MOM (methoxymethyl), PMB (p-methoxybenzyl) and the like.
- the compound siL is reacted with isopentenyl bromide under basic conditions to prepare a compound.
- Suitable solvents for the reaction include, for example, methanol, DMF (N,N-dimethylformamide), THF (tetrahydrofuran), water, toluene, DME (1,2-dimethoxyethane), and mixed solvents.
- methanol-water, DMF-water, tetrahydrofuran-water and the like Such as methanol-water, DMF-water, tetrahydrofuran-water and the like.
- the solvent used in the reaction is preferably water.
- Bases suitable for the reaction include, for example, potassium hydroxide, potassium carbonate, cesium carbonate, sodium methoxide, sodium hydride, potassium t-butoxide, DBU (1,8-diazabicyclo-bicyclo(5,4,0) -7-undecene), n-butyllithium, LDA (lithium diisopropylamide), LHMDS (lithium hexamethyldisilazide) and the like.
- the reaction temperature is usually between about 0 ° C and about 100 ° C and the reaction time is from 1 to 20 hours. detailed description
- room temperature or ambient temperature means in the range of 18-25 ° C
- the solvent is removed using a rotary evaporator under reduced pressure.
- the reaction process was monitored by thin layer chromatography (TLC).
- TLC thin layer chromatography
- the reaction time is for illustrative purposes only.
- the structure and purity of the compound are confirmed by at least one of the following techniques: TLC, mass spectrometry, nuclear magnetic resonance (NMR), high performance liquid chromatography (HPLC).
- the yields are for illustrative purposes only.
- Step 2 Preparation of 2-(4-trifluoromethylphenyl)-3-methoxy-5,7-dihydroxy-4H-benzopyran-4-one 2-methoxy-1-( 2,4,6-Trihydroxyphenyl)ethanone (30 g, 151 mmol) and 4-trifluoromethylbenzoyl chloride (37.5 g, 180 mmol) dissolved in 250 mL of dry pyridine, DBU was added dropwise at room temperature (53.2, 350 mmol). After the dropwise addition was completed, the reaction system was heated to 75 ° C, and the reaction was stirred overnight. The reaction mixture was cooled to room temperature, and most of the solvent was removed under reduced pressure, and the residue was poured into ice-cooled diluted hydrochloric acid.
- reaction system was reacted at room temperature overnight, and the pH was adjusted to 6 with 2N hydrochloric acid. Right, then extracted twice with ethyl acetate. The combined organic phases were washed with a saturated sodium chloride solution and dried over anhydrous sodium sulfate.
- Step 2 Preparation of 2-(3,4-difluorophenyl) -3 -Methoxy-5,7-dihydroxy-8-(3-methyl-2-buten-1-yl)-4H-benzopyran-4-one (Compound 10) under nitrogen protection, 2-methoxy small [2,4,6-trihydroxy-3,5-bis(3-methyl-2-butenyl)phenyl]ethanone (250 mg, 0.94 mmol), anhydrous Potassium powder (779 mg, 5.63 mmol), TBAB (tetrabutylammonium bromide, 454 mg, 1.41 mmol) and 3,4-difluorobenzoyl chloride (331 mg, 1.88 mmol) in toluene (GO mL) , reflux reaction for 6 hours.
- Compound 10 2-methoxy small [2,4,6-trihydroxy-3,5-bis(3-methyl-2-butenyl)phenyl]ethanone (250 mg, 0.94 mmol)
- Example 14 Expression of ER- ⁇ variants in human breast cancer samples
- a film pre-coated with human breast cancer cell tissue was purchased from ProSci Incorporated (Poway, CA).
- the membrane was detected with an anti-ER-a36 antibody and an HRP-conjugated secondary antibody which specifically recognized ER-a36, and was visualized with an enhanced chemical luminescence (ECL) detector (available from Amersham Pharmacia Biotech).
- ECL enhanced chemical luminescence
- the label on the same membrane was eluted, and the anti-estrogen receptor-a antibody H222 (Novocastra Laboratories Ltd, UK), which recognizes all three subtypes of ER- ⁇ , ER-a66, ER-a46 and ER-a36, was used. Provide) for testing.
- Figure 1 shows that three estrogen receptor subtypes of ER-a66, ER-a46 and ER-a36 are not expressed in normal breast tissue (lane 1), but they are in a sample of invasive ductal carcinoma (lane 2). , a sample of invasive lobular carcinoma (lane 5) and non-invasive ductal carcinoma (lane 7) were expressed.
- ER-a36 was expressed in another sample of invasive ductal carcinoma (lane 4) and invasive lobular carcinoma (lane 6).
- Dao 2 and Dao 3 are invasive ductal carcinoma tissues from 2 different patients.
- Lanes 5 and 6 are invasive lobular carcinoma tissues from two different patients, respectively.
- Example 15 Expression of ER-a36 in ER-negative breast cancer cell line MDA-MB-231
- MDA-MB-231 is a well-known cell line lacking ER-a66 and ER-a46 (see Relevance of breast cancer cell lines as models for breast tumours: an update. Marc Lacroix, Guy Leclercq, Brest Cancer Research and Treatment 83: 249-289 (2004)).
- MDA-MB-231 cells are from the American Type Culture Collection (ATCC). Place MDA-MB-231 cells in Dulbecco's Modified Eagle Medium (DMEM) and 10% fetal bovine blood The clear 8-well BIOCOAT slide (supplied by BD Science Discovery Labware) was incubated at 37 ° C and 5% CO 2 atmosphere for 12 hours.
- DMEM Dulbecco's Modified Eagle Medium
- BIOCOAT slide supplied by BD Science Discovery Labware
- the cells were then washed twice with sterile phosphate buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS (pH 7.4) for 30 minutes at room temperature. Thereafter, the cells were washed with PBS and ruptured with 0.5% (v/v) Triton X-100 for 10 minutes. The cells were washed again with PBS, and 3% serum in PBS was added and blocked at room temperature for 1 hour.
- PBS sterile phosphate buffered saline
- PBS 4% paraformaldehyde in PBS
- Figure 2 shows that MDA-MB-231 cells were stained positive by anti-ER-a36 antibody. To further confirm the confidence of the experiment, no specific staining was shown by incubation of the anti-ER-0136 antibody pre-incubated with the immunogenic polypeptide (Fig. 2, lower panel), indicating the specificity of the antibody.
- Example 16 Western blotting detection of ER-0136 expression on different tumor cell lines
- the experimental cells e.g., MDB-MA-231, medium 10%-FBS-DMEM
- the experimental cells were cultured at 37 ° C and 5% CO 2 conditions. After each well of the cells was grown to 60% to 90% fullness, the cells were collected and centrifuged at 4,300 rpm for 5 minutes at 4 °C. After removing the supernatant, an appropriate amount of lysate and Lysis buffer containing 1% ⁇ -40 and 0.7 mM EDTA were added, followed by addition of a protease inhibitor, and cleavage for 30 minutes to 1 hour under ice bath conditions. The cells were again centrifuged at 14,000 rpm for 15 minutes, and the supernatant was taken and protein quantitation was performed.
- Lane 1 293 human renal epithelial cell lines transiently overexpressing ER-0136; lanes 2-4: human breast cancer SK-BR-3 cell lines from different laboratory sources; lanes 5-7: human mammary glands from different laboratory sources Cancer MCF-7 cell line; Lane 8-9: Human leukemia HL-60 cell line from different laboratory sources; Lane 10-11: Human leukemia MV-4-11 cell line from different laboratory sources; Lane 12-13: Different laboratory-derived human chronic myeloid leukemia K562 cells; tract 14: human liver cancer A2780 cells; tract 15: human liver cancer HEL-7402 cells; tract 16: human liver cancer HEL-9204 cells; tract 17: primary cells derived from patients Liver cancer Hep-11 cells; Lane 18: Primary liver cancer Hep-12 cells derived from patients.
- Example 17 Inhibition of compound growth on different breast cancer cells in vitro
- MDA-MB-231 cells were stored at 10% under 37 ° C and 5% CO 2 atmosphere. Fetal bovine serum in DMEM. The cells were seeded at a density of 6 x 10 3 /well in 96-well plates. The test compound was dissolved in DMSO and applied to MDA at a concentration of 0, 0.3 ⁇ , 0.5 ⁇ , 1 ⁇ , 2 ⁇ , 3 ⁇ , 5 ⁇ , 10 ⁇ , 20 ⁇ , 30 ⁇ , 50 ⁇ and 100 ⁇ . MB-231 cells were 72 hours. The CellTiter-Glo® Luminescence Cell Viability Assay Kit (Promega) was then used to detect cells treated with the compound and the luminescence values were recorded using Envision.
- MCF7 cells are breast cancer cell lines that highly express ER-66, ER-46 and ER-36. (Relevance of breast cancer cell lines as models for breast tumours: an update. Marc Lacroix, Guy Leclercq, Breast Cancer Research and Treatment (2004) 83, 249-289; Wang et al., Proc. Natl. Acad. Sci. USA103 :9063-9068 (2006)).
- MCF7 cells were derived from ATCC and maintained at 37 ° C, 5% CO 2 atmosphere and in Dulbecco's Modified Eagle Medium (DMEM) and 10% fetal bovine serum.
- DMEM Dulbecco's Modified Eagle Medium
- the cells were seeded at a density of 6 x 10 3 /well in 96-well plates.
- the test compound was dissolved in DMSO and applied to MCF7 cells at a concentration of 0, 0.3 ⁇ , 0.5 ⁇ , 1 ⁇ , 2 ⁇ , 3 ⁇ , 5 ⁇ , 10 ⁇ , 20 ⁇ , 30 ⁇ , 50 ⁇ and 100 ⁇ . 72 hours.
- the CellTiter-Glo® Luminescence Cell Viability Assay Kit (Promega) was then used to detect cells treated with the compound and the luminescence values were recorded using Envision.
- MDA-MB-231 cells MCF7 cells tamoxifen a) 20.90 ⁇ 1.51 ) 22.55 ⁇ 4.15
- NA stands for no activity and its IC 5Q value is greater than 100 ⁇ .
- Leukemia K562 was obtained from ATCC and maintained at 37 ° C, 5% C0 2 atmosphere and in IMDM and 10% fetal bovine serum. The cells were seeded at a density of 6 x 10 3 /well in 96-well plates. The test compound was dissolved in DMSO and applied to ⁇ 562 cells at a concentration of 0, 0.3 ⁇ , 0.5 ⁇ , 1 ⁇ , 2 ⁇ , 3 ⁇ , 5 ⁇ , 10 ⁇ , 20 ⁇ , 30 ⁇ , 50 ⁇ and 100 ⁇ . 72 hours. The CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega) was then used to detect cells treated with the compound and the luminescence values were recorded using Envision.
- Example 19 Inhibition of Compound Growth on Human B Lymphoma Daudi Cells in Vitro
- Human B lymphoma Daudi cells were obtained from ATCC and maintained at 37 ° C, 5% CO 2 atmosphere and in IMDM and 10% fetal bovine serum. The cells were seeded at a density of 6 x 10 3 /well in 96-well plates. The test compound was dissolved in DMSO and applied to ⁇ 562 cells at a concentration of 0, 0.3 ⁇ , 0.5 ⁇ , 1 ⁇ , 2 ⁇ , 3 ⁇ , 5 ⁇ , 10 ⁇ , 20 ⁇ , 30 ⁇ , 50 ⁇ and 100 ⁇ . 72 hours. The CellTiter-Glo® Luminescence Cell Viability Assay Kit (Promega) was then used to detect cells treated with the compound and the luminescence values were recorded using Envision. The results of in vitro effects of the partial compounds of the present invention on the cell viability of leukemia K562 cells and human B lymphoma Daudi cells are listed in Table 4 below.
- Gleevec and cytarabine are positive control compounds for the K562 cell model and the Daudi cell model, respectively.
- ND stands for no measurement.
- NA stands for no activity and its IC 5Q value is greater than 100 ⁇ .
- Example 20 Inhibition of in vitro growth of compounds on acute leukemia cells
- Acute myeloid leukemia cell HL-60 was obtained from ATCC and maintained at 37 ° C, 5% C0 2 atmosphere and in IMDM and 10% fetal bovine serum. The cells were seeded at a density of 6 x 10 3 /well in 96-well plates. Test compounds dissolved in DMSO, at the concentration of 0, 10_ 4 M, 10_ 5 M, 10_ 6 M, 10 "7 M, 10_ 8 M acting on the HL-60 cells for 72 hours. Then MTT assay was used to detect the OD Value, the percentage of inhibition of cell proliferation was calculated.
- Example 21 Inhibition of growth of compounds on hepatoma cells in vitro
- Hepatoma cell line HL-60 was obtained from ATCC and maintained at 37 ° C, 5% C0 2 atmosphere and in DMEM and 10% 10% NCS and 5 ( ⁇ g/ml KANA. Cells were seeded at a density of 6 x 10 3 /well In a 96-well plate, the test compound was dissolved in DMSO at a concentration of 0, 10" 4 M, 10" 5 M, 10" 6 M, 10" 7 M, 1 (T 8 M was applied to BEL-7402 The cells were subjected to 72 hours. Then, the OD value was measured by the SRB method, and the percentage of inhibition of cell proliferation was calculated. The results of the growth inhibitory effect of some of the compounds of the present invention on liver cancer cells are shown in Table 6.
- the gastric cancer cell BGC-823 was seeded at a density of 3 ⁇ 10 3 /well in a 96-well plate and maintained in a DMEM medium containing 2.5% CS-FBS in phenol red at 37 ° C in a 5% CO 2 atmosphere. 24 hours.
- the test compound was dissolved in DMSO and applied to BGC-823 cells at a concentration of 0, 1, 2, 4, 8, 10, 20 ⁇ for 72 hours. The OD value was then measured by the MTT method to calculate the percentage of inhibition of cell proliferation. The results are shown in Figure 4.
- Example 23 Inhibition of growth of compounds on lung cancer cells in vitro
- Lung cancer cells H460 were seeded at a density of 4.0 ⁇ 10 3 /well in 96-well plates, and cultured in a phenol red-free 1640 medium containing 2.5% CS-FBS at 37 ° C in a 5% CO 2 atmosphere. hour.
- the test compound was dissolved in DMSO and applied to H460 cells at a concentration of 0, 1, 2, 4, 8, 10, 20 ⁇ for 72 hours. Then, the OD value was used to detect the OD value, and the percentage of inhibition of cell proliferation was calculated. The results are shown in Figure 5.
- Example 24 Inhibition of in vitro growth of compounds on colon cancer cells
- Colon cancer cells LS174T were seeded at a density of 4.5 ⁇ 10 3 /well in 96-well plates and maintained in a phenol red-free 1640 medium containing 2.5% CS-FBS at 37 ° C in a 5% CO 2 atmosphere. 24 hours. Test compounds were dissolved in DMSO and applied to LS174T cells for 72 hours at concentrations of 0, 1, 2, 4, 8, 10, 20 ⁇ M. Then, the OD value was used to detect the OD value, and the percentage of inhibition of cell proliferation was calculated. The results are shown in Figure 6.
- Example 25 Inhibition of compound growth on pancreatic cancer cells in vitro
- Prostate cancer cell line PC-3 was seeded in a 96-well plate at a density of 3 ⁇ 10 3 cells/well, and incubated with F12K medium containing 10% fetal calf serum for 24 hours at 37 ° C in a 5% CO 2 atmosphere.
- the test compound was dissolved in DMSO and applied to PC-3 cells at a concentration of 0, 1, 2, 4, 8, 10, 20 ⁇ for 72 hours. Then, the OD value was used to detect the OD value, and the percentage of inhibition of cell proliferation was calculated. The results are shown in Figure 8.
- In Vivo Evaluation Example 27 Growth Inhibition of Compounds on Human Breast Cancer BCAP-37 Cell Transplanted Tumor in Nude Mice
- Test compounds were administered to nude mice bearing breast cancer xenografts to test their effect of inhibiting tumor growth.
- Tumor tissues were extracted from nude mice bearing BCAP-37 cells and cut into small pieces. Several pieces of tumor tissue were implanted into the armpit of the right forelimb of female nude mice. After implantation, the ⁇ 2 ⁇ solution was injected once a day for 6 days in a dose of 7 ⁇ ⁇ per nude mouse to stimulate the growth of the tumor in the mice. From day 7 onwards, the tumor-bearing nude mice were orally administered with a corn oil solution of the test compound at a dose of 35 mg/kg.
- Example 28 Growth inhibition of compound on human B lymphoma Daudi cell xenografts in nude mice
- Test compounds were administered to nude mice bearing human B lymphoma Daudi cell xenografts to test their effect of inhibiting tumor growth.
- Human B lymphoma Daudi cells are derived from ATCC. After 5 passages, lx lO 7 cells were added to 0.2 mL of Matrigel and implanted subcutaneously into the armpit of the right forelimb of male nude mice. When tumors in nude mice reached 150-200 mm 3 , the tumor-bearing nude mice were randomly divided into groups of ten. The mixed oil gavage administration group was used as a negative control, and rituximab was intravenously administered as a positive control group, and the test compound was dissolved in a mixed oil for intragastric administration.
- the administration period was 21 consecutive days, and a mixed oil suspension (35 mg/kg) of the test compound was intragastrically administered to the test group nude mice every day.
- the rituximab was administered to the positive control group twice a week.
- Anti-(20 mg/kg) while daily intragastric administration of negative control group, blank mixed oil solvent Times.
- Tumor volume and body weight of nude mice were measured twice weekly during the dosing cycle. Tumor growth curves were plotted as tumor volume and time of administration ( Figure 10) to assess the inhibitory effect of compounds on tumor growth.
- Example 29 Growth inhibition of compound on human endometrial carcinoma Ishikawa cell xenografts in nude mice
- Test compounds were administered to nude mice bearing endometrial cancer xenografts to test their effect of inhibiting tumor growth.
- Tumor tissues were extracted from nude mice bearing Ishikawa cells and cut into small pieces. Several pieces of tumor tissue were implanted into the armpit of the right forelimb of female nude mice. After implantation, the ⁇ 2 ⁇ solution was injected once a day for 6 days in a dose of 7 ⁇ ⁇ per nude mouse to stimulate the growth of the tumor in the mice. From the 7th day, the tumor-bearing nude mice were orally administered with a mixed oil solution of the test compound at a dose of 35 mg/kg.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyrane Compounds (AREA)
Abstract
Description










Claims




Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/371,866 US9221781B2 (en) | 2012-01-13 | 2012-12-31 | Synthesis of polyhydroxy chromenone compounds and their anti-tumor effects |
| KR1020147022558A KR102030207B1 (ko) | 2012-01-13 | 2012-12-31 | 폴리하이드록시 크로메논 화합물의 합성 및 이의 항종양 효과 |
| BR112014016635A BR112014016635B8 (pt) | 2012-01-13 | 2012-12-31 | composto, composição, e, uso de um composto |
| CA2860999A CA2860999C (en) | 2012-01-13 | 2012-12-31 | Synthesis of polyhydroxy chromenone compounds and their anti-tumor effects |
| EP12864757.5A EP2803665B1 (en) | 2012-01-13 | 2012-12-31 | Synthesis of polyhydroxy benzopyran ketone compound and anti-tumor effect thereof |
| JP2014551506A JP6113751B2 (ja) | 2012-01-13 | 2012-12-31 | ポリヒドロキシベンゾピランケトン化合物の合成およびその抗腫瘍効果 |
| ES12864757.5T ES2627316T3 (es) | 2012-01-13 | 2012-12-31 | Síntesis del compuesto de polihidroxi benzopirano cetona y efecto antitumoral del mismo |
| AU2012365712A AU2012365712B2 (en) | 2012-01-13 | 2012-12-31 | Synthesis of polyhydroxy benzopyran ketone compound and anti-tumor effect thereof |
| MX2014008526A MX364833B (es) | 2012-01-13 | 2012-12-31 | Sintesis de compuestos polihidroxi cromenona y sus efectos anti-tumor. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201210011031.8 | 2012-01-13 | ||
| CN201210011031 | 2012-01-13 | ||
| CN201210573072.6 | 2012-12-25 | ||
| CN201210573072.6A CN103204838B (zh) | 2012-01-13 | 2012-12-25 | 多羟基苯并吡喃酮类化合物的合成及其抗肿瘤作用 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013104263A1 true WO2013104263A1 (zh) | 2013-07-18 |
Family
ID=48752268
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2012/088016 WO2013104263A1 (zh) | 2012-01-13 | 2012-12-31 | 多羟基苯并吡喃酮类化合物的合成及其抗肿瘤作用 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US9221781B2 (zh) |
| EP (1) | EP2803665B1 (zh) |
| JP (1) | JP6113751B2 (zh) |
| KR (1) | KR102030207B1 (zh) |
| CN (1) | CN103204838B (zh) |
| AU (1) | AU2012365712B2 (zh) |
| BR (1) | BR112014016635B8 (zh) |
| CA (1) | CA2860999C (zh) |
| ES (1) | ES2627316T3 (zh) |
| MX (1) | MX364833B (zh) |
| WO (1) | WO2013104263A1 (zh) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101784990B1 (ko) * | 2016-01-13 | 2017-10-12 | 주식회사 파이브지티 | 얼굴인식을 이용한 홈 오토메이션 제어 시스템 및 그 방법 |
| CN107216303B (zh) * | 2016-07-15 | 2020-04-21 | 北京盛诺基医药科技股份有限公司 | 氟可拉定的合成方法 |
| CN107216302B (zh) * | 2016-07-15 | 2020-02-21 | 北京盛诺基医药科技股份有限公司 | 一种氟可拉定的合成方法 |
| AU2017313753B2 (en) | 2016-08-16 | 2022-01-27 | The Trustees Of Columbia University In The City Of New York | GABA(A) receptor modulators and methods to control airway hyperresponsiveness and inflammation in asthma |
| US10266510B2 (en) | 2017-01-18 | 2019-04-23 | King Abdulaziz University | Antimicrobial and cytotoxic compounds and methods for treating cancer, a bacterial infection, and/or a fungal infection |
| CN108947949B (zh) * | 2017-05-19 | 2022-08-19 | 泰州华元医药科技有限公司 | 抗焦虑氘代化合物及其医药用途 |
| CN109020938A (zh) * | 2018-08-17 | 2018-12-18 | 昆明龙津药业股份有限公司 | 一种杨梅素的制备方法 |
| CN110054605A (zh) * | 2019-05-31 | 2019-07-26 | 北京盛诺基医药科技有限公司 | 一种淫羊藿素中间体的制备方法 |
| CN110092770A (zh) * | 2019-05-31 | 2019-08-06 | 北京盛诺基医药科技有限公司 | 一种黄酮化合物中间体的制备方法 |
| CN112569222A (zh) * | 2020-12-31 | 2021-03-30 | 赣南医学院 | 三氟淫羊藿素在制备改善疼痛、肿胀及运动功能的药物中的应用 |
| CN112851615B (zh) * | 2021-01-21 | 2023-04-07 | 厦门大学 | 一种异戊烯基黄酮的制备及其作为治疗宫颈癌药物的应用 |
| CN113717138B (zh) * | 2021-10-21 | 2023-02-24 | 沈阳药科大学 | 一类氮芥类色原酮衍生物与应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101104611A (zh) * | 2007-04-29 | 2008-01-16 | 殷正丰 | 一种3-甲氧基黄酮类化合物,其制备方法和应用 |
| CN102018698A (zh) * | 2008-04-18 | 2011-04-20 | 盛诺基医药科技有限公司 | 治疗雌激素受体相关疾病的化合物及其方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1786864A (zh) | 2004-12-10 | 2006-06-14 | 上海迪比特实业有限公司 | 一种计算机安全认证方法 |
| JP2007332695A (ja) | 2006-06-16 | 2007-12-27 | Advanced Media Inc | 電子錠装置及び音声認証ロッカーシステム |
| WO2008100977A2 (en) | 2007-02-14 | 2008-08-21 | N.V. Organon | Carbamates therapeutic release agents as amidase inhibitors |
| CN102038673B (zh) * | 2009-10-20 | 2012-07-25 | 北京珅奥基医药科技有限公司 | 羟基苯并吡喃酮类化合物在制备治疗白血病药物中的应用 |
| CN102558164B (zh) | 2010-12-31 | 2016-06-15 | 北京盛诺基医药科技有限公司 | 苯并吡喃酮类雌激素受体调节剂 |
-
2012
- 2012-12-25 CN CN201210573072.6A patent/CN103204838B/zh active Active
- 2012-12-31 JP JP2014551506A patent/JP6113751B2/ja active Active
- 2012-12-31 WO PCT/CN2012/088016 patent/WO2013104263A1/zh active Application Filing
- 2012-12-31 AU AU2012365712A patent/AU2012365712B2/en active Active
- 2012-12-31 BR BR112014016635A patent/BR112014016635B8/pt active IP Right Grant
- 2012-12-31 EP EP12864757.5A patent/EP2803665B1/en active Active
- 2012-12-31 ES ES12864757.5T patent/ES2627316T3/es active Active
- 2012-12-31 CA CA2860999A patent/CA2860999C/en active Active
- 2012-12-31 MX MX2014008526A patent/MX364833B/es active IP Right Grant
- 2012-12-31 KR KR1020147022558A patent/KR102030207B1/ko active IP Right Grant
- 2012-12-31 US US14/371,866 patent/US9221781B2/en active Active - Reinstated
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101104611A (zh) * | 2007-04-29 | 2008-01-16 | 殷正丰 | 一种3-甲氧基黄酮类化合物,其制备方法和应用 |
| CN102018698A (zh) * | 2008-04-18 | 2011-04-20 | 盛诺基医药科技有限公司 | 治疗雌激素受体相关疾病的化合物及其方法 |
Non-Patent Citations (16)
| Title |
|---|
| "ACS Symposium Series", vol. 14, article "Produgs as Novel Delivery Systems" |
| "Bioreversible Carriers in Drug Design", 1987, AMERICAN PHARMACEUTICAL ASSOCIATION AND PERGAMON PRESS |
| BERGE ET AL., J. PHARM. SCI., vol. 66, 1977, pages 1 - 19 |
| BIOCHEM. BIOPHYS. RES. COMMUN., vol. 336, 2005, pages 1023 - 1027 |
| FLOURIOT, G., EMBO, vol. 19, 2000, pages 4688 - 4700 |
| GREENE T. W.: "Protective Groups in Organic Synthesis", 1991, JOHN WILEY & SONS |
| GREENE T.W: "protective groups in organic synthesis", 1991, JOHN WILEY & SONS |
| LI ET AL., PROC. NATL. ACAD. SCI., USA, vol. 100, 2003, pages 4807 - 4812 |
| MARC LACROIX; GUY LECLERCQ: "Relevance of breast cancer cell lines as models for breast tumors: an update", BREAST CANCER RESEARCH AND TREATMENT, vol. 83, 2004, pages 249 - 289, XP002502163, DOI: doi:10.1023/B:BREA.0000014042.54925.cc |
| MARC LACROIX; GUY LECLERCQ: "Relevance of breast cancer cell lines as models for breast tumours: an update", BREAST CANCER RESEARCH AND TREATMENT, vol. 83, 2004, pages 249 - 289, XP002502163, DOI: doi:10.1023/B:BREA.0000014042.54925.cc |
| SAMBOOK: "Molecular Cloning, A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY |
| See also references of EP2803665A4 * |
| WANG ET AL., BIOCHEM. BIOPHYS. RES.COMMUN, vol. 336, 2005, pages 1023 - 1027 |
| WANG ET AL., BIOCHEM. BIOPHYS. RES.COMMUN., vol. 336, 2005, pages 1023 - 1027 |
| WANG ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 103, 2006, pages 9063 - 9068 |
| WANG ET AL., PROC.NATL.ACAD.SCI., USA, vol. 103, 2006, pages 9063 - 9068 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2860999A1 (en) | 2013-07-18 |
| US20150011618A1 (en) | 2015-01-08 |
| CN103204838B (zh) | 2017-06-09 |
| JP6113751B2 (ja) | 2017-04-12 |
| JP2015503596A (ja) | 2015-02-02 |
| BR112014016635B1 (pt) | 2021-07-13 |
| BR112014016635B8 (pt) | 2021-08-24 |
| BR112014016635A8 (pt) | 2017-07-04 |
| EP2803665A4 (en) | 2015-07-08 |
| ES2627316T3 (es) | 2017-07-27 |
| KR20140109498A (ko) | 2014-09-15 |
| CA2860999C (en) | 2019-12-31 |
| CN103204838A (zh) | 2013-07-17 |
| US9221781B2 (en) | 2015-12-29 |
| KR102030207B1 (ko) | 2019-10-08 |
| EP2803665B1 (en) | 2017-05-10 |
| MX364833B (es) | 2019-05-08 |
| AU2012365712B2 (en) | 2017-04-20 |
| EP2803665A1 (en) | 2014-11-19 |
| AU2012365712A1 (en) | 2014-07-24 |
| MX2014008526A (es) | 2014-11-25 |
| BR112014016635A2 (pt) | 2017-06-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013104263A1 (zh) | 多羟基苯并吡喃酮类化合物的合成及其抗肿瘤作用 | |
| US9090583B2 (en) | Benzopyrone estrogen receptor regulator | |
| CN102018698B (zh) | 治疗雌激素受体相关疾病的化合物及其方法 | |
| US10398689B2 (en) | Benzopiperidine derivative, preparation method thereof and medical use thereof | |
| CN105008343A (zh) | 作为选择性雌激素受体降解剂的苯并噻吩衍生物及其组合物 | |
| US10315990B2 (en) | Isothiocyanate compounds and isothiocyanate XPO1 protein inhibitor drugs thereof | |
| JP5938400B2 (ja) | ピロリル置換ジヒドロインドール−2−オン誘導体、調製方法及びそれらの用途 | |
| JP6674381B2 (ja) | 癌治療のためのMcl−1調節化合物 | |
| JP2016515585A (ja) | 癌、神経障害及び線維性障害の治療のための、脂質フラン、ピロール及びチオフェン化合物 | |
| CN110240587A (zh) | 一类芳基二氟苄基醚类化合物、制备方法及用途 | |
| WO2014169697A1 (zh) | 长春碱类衍生物及其制备方法和应用 | |
| CN113956236B (zh) | 邻苯二甲酰亚胺类化合物及其用途 | |
| WO2023088257A1 (zh) | 苦鬼臼毒素衍生物、其药物组合物及用途 | |
| CN103755673A (zh) | 治疗雌激素受体相关疾病的化合物及其方法 | |
| WO2023141852A1 (zh) | Cdk2抑制剂及其制备方法和用途 | |
| CN108186630A (zh) | 靛红类似物在制备抗肿瘤药物中的应用 | |
| KR20220104734A (ko) | 올레아난 신남아미드 유도체 및 이의 제조 방법과 용도 | |
| CN108912113A (zh) | 一种化合物及其作为选择性pi3k抑制剂在制备治疗肿瘤的药物中的应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12864757 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012864757 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012864757 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2860999 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14371866 Country of ref document: US Ref document number: MX/A/2014/008526 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2014551506 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2012365712 Country of ref document: AU Date of ref document: 20121231 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20147022558 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014016635 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014016635 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140703 |