WO2013094532A1 - 積層フィルム - Google Patents
積層フィルム Download PDFInfo
- Publication number
- WO2013094532A1 WO2013094532A1 PCT/JP2012/082509 JP2012082509W WO2013094532A1 WO 2013094532 A1 WO2013094532 A1 WO 2013094532A1 JP 2012082509 W JP2012082509 W JP 2012082509W WO 2013094532 A1 WO2013094532 A1 WO 2013094532A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- layer
- mass
- poly
- acrylate
- segment
- Prior art date
Links
- 229920005989 resin Polymers 0.000 claims abstract description 83
- 239000011347 resin Substances 0.000 claims abstract description 83
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 claims abstract description 61
- 238000006073 displacement reaction Methods 0.000 claims abstract description 50
- 238000005259 measurement Methods 0.000 claims abstract description 19
- -1 alkylene glycol Chemical compound 0.000 claims description 131
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 claims description 112
- 239000000203 mixture Substances 0.000 claims description 98
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 claims description 78
- 239000004205 dimethyl polysiloxane Substances 0.000 claims description 49
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 claims description 49
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 claims description 48
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 claims description 44
- 239000002537 cosmetic Substances 0.000 claims description 39
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 claims description 27
- 239000012298 atmosphere Substances 0.000 claims description 18
- 238000004090 dissolution Methods 0.000 claims description 16
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 claims description 15
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 claims description 15
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 claims description 15
- 239000005642 Oleic acid Substances 0.000 claims description 15
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 claims description 15
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 claims description 15
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 claims description 15
- 230000003068 static effect Effects 0.000 claims description 5
- 238000000465 moulding Methods 0.000 abstract description 21
- 229920000728 polyester Polymers 0.000 abstract description 12
- 238000011109 contamination Methods 0.000 abstract description 10
- 238000011084 recovery Methods 0.000 abstract description 6
- 239000010410 layer Substances 0.000 description 316
- 239000010408 film Substances 0.000 description 111
- 239000002994 raw material Substances 0.000 description 63
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 54
- 238000000034 method Methods 0.000 description 51
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 30
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 30
- 230000015572 biosynthetic process Effects 0.000 description 28
- 150000001875 compounds Chemical class 0.000 description 27
- 239000000126 substance Substances 0.000 description 27
- 239000003999 initiator Substances 0.000 description 25
- 238000005755 formation reaction Methods 0.000 description 24
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 24
- QNODIIQQMGDSEF-UHFFFAOYSA-N (1-hydroxycyclohexyl)-phenylmethanone Chemical compound C=1C=CC=CC=1C(=O)C1(O)CCCCC1 QNODIIQQMGDSEF-UHFFFAOYSA-N 0.000 description 22
- 239000007787 solid Substances 0.000 description 22
- 238000006243 chemical reaction Methods 0.000 description 21
- 239000000178 monomer Substances 0.000 description 21
- 150000003254 radicals Chemical class 0.000 description 17
- UHESRSKEBRADOO-UHFFFAOYSA-N ethyl carbamate;prop-2-enoic acid Chemical compound OC(=O)C=C.CCOC(N)=O UHESRSKEBRADOO-UHFFFAOYSA-N 0.000 description 16
- 229920001577 copolymer Polymers 0.000 description 15
- 238000010438 heat treatment Methods 0.000 description 15
- 229920001225 polyester resin Polymers 0.000 description 12
- 239000004645 polyester resin Substances 0.000 description 12
- UKLDJPRMSDWDSL-UHFFFAOYSA-L [dibutyl(dodecanoyloxy)stannyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)O[Sn](CCCC)(CCCC)OC(=O)CCCCCCCCCCC UKLDJPRMSDWDSL-UHFFFAOYSA-L 0.000 description 11
- 239000003054 catalyst Substances 0.000 description 11
- 229920000642 polymer Polymers 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 239000005057 Hexamethylene diisocyanate Substances 0.000 description 10
- 230000000052 comparative effect Effects 0.000 description 10
- 239000000463 material Substances 0.000 description 10
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 10
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- RRAMGCGOFNQTLD-UHFFFAOYSA-N hexamethylene diisocyanate Chemical compound O=C=NCCCCCCN=C=O RRAMGCGOFNQTLD-UHFFFAOYSA-N 0.000 description 9
- 239000002245 particle Substances 0.000 description 9
- 229920001223 polyethylene glycol Polymers 0.000 description 9
- 229920001296 polysiloxane Polymers 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- OMIGHNLMNHATMP-UHFFFAOYSA-N 2-hydroxyethyl prop-2-enoate Chemical compound OCCOC(=O)C=C OMIGHNLMNHATMP-UHFFFAOYSA-N 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 8
- 229920002554 vinyl polymer Polymers 0.000 description 8
- RTTZISZSHSCFRH-UHFFFAOYSA-N 1,3-bis(isocyanatomethyl)benzene Chemical compound O=C=NCC1=CC=CC(CN=C=O)=C1 RTTZISZSHSCFRH-UHFFFAOYSA-N 0.000 description 7
- 238000000576 coating method Methods 0.000 description 7
- 150000002009 diols Chemical class 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 239000012948 isocyanate Substances 0.000 description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 238000001035 drying Methods 0.000 description 6
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 6
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- 206010052428 Wound Diseases 0.000 description 5
- 208000027418 Wounds and injury Diseases 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 229920001400 block copolymer Polymers 0.000 description 5
- 239000011248 coating agent Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 150000002513 isocyanates Chemical class 0.000 description 5
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 5
- 229910052753 mercury Inorganic materials 0.000 description 5
- 230000000704 physical effect Effects 0.000 description 5
- 238000006116 polymerization reaction Methods 0.000 description 5
- 229920005992 thermoplastic resin Polymers 0.000 description 5
- SOGAXMICEFXMKE-UHFFFAOYSA-N Butylmethacrylate Chemical compound CCCCOC(=O)C(C)=C SOGAXMICEFXMKE-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 4
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 4
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 4
- 206010047642 Vitiligo Diseases 0.000 description 4
- 230000032683 aging Effects 0.000 description 4
- 239000002216 antistatic agent Substances 0.000 description 4
- 239000001273 butane Substances 0.000 description 4
- 238000010894 electron beam technology Methods 0.000 description 4
- 239000010419 fine particle Substances 0.000 description 4
- 229920000578 graft copolymer Polymers 0.000 description 4
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 4
- 238000007373 indentation Methods 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 229920000139 polyethylene terephthalate Polymers 0.000 description 4
- 239000005020 polyethylene terephthalate Substances 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- 239000006097 ultraviolet radiation absorber Substances 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 229930182556 Polyacetal Natural products 0.000 description 3
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 239000012295 chemical reaction liquid Substances 0.000 description 3
- 238000004132 cross linking Methods 0.000 description 3
- 239000003431 cross linking reagent Substances 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 230000005484 gravity Effects 0.000 description 3
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 239000003505 polymerization initiator Substances 0.000 description 3
- 229920006324 polyoxymethylene Polymers 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 229910000077 silane Inorganic materials 0.000 description 3
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 3
- 229920001187 thermosetting polymer Polymers 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- BQCIDUSAKPWEOX-UHFFFAOYSA-N 1,1-Difluoroethene Chemical compound FC(F)=C BQCIDUSAKPWEOX-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 2
- 229920001342 Bakelite® Polymers 0.000 description 2
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 2
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical class C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- WOBHKFSMXKNTIM-UHFFFAOYSA-N Hydroxyethyl methacrylate Chemical compound CC(=C)C(=O)OCCO WOBHKFSMXKNTIM-UHFFFAOYSA-N 0.000 description 2
- 229920000877 Melamine resin Polymers 0.000 description 2
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical compound ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 2
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- 239000006096 absorbing agent Substances 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 125000002723 alicyclic group Chemical group 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 239000004637 bakelite Substances 0.000 description 2
- 230000005540 biological transmission Effects 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 238000005520 cutting process Methods 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- 229910003460 diamond Inorganic materials 0.000 description 2
- 239000010432 diamond Substances 0.000 description 2
- AYOHIQLKSOJJQH-UHFFFAOYSA-N dibutyltin Chemical compound CCCC[Sn]CCCC AYOHIQLKSOJJQH-UHFFFAOYSA-N 0.000 description 2
- 150000001991 dicarboxylic acids Chemical class 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- 238000002845 discoloration Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- NOPFSRXAKWQILS-UHFFFAOYSA-N docosan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCCCCCO NOPFSRXAKWQILS-UHFFFAOYSA-N 0.000 description 2
- 239000003759 ester based solvent Substances 0.000 description 2
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N ethenoxyethane Chemical compound CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 2
- SUPCQIBBMFXVTL-UHFFFAOYSA-N ethyl 2-methylprop-2-enoate Chemical compound CCOC(=O)C(C)=C SUPCQIBBMFXVTL-UHFFFAOYSA-N 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000007756 gravure coating Methods 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 230000001678 irradiating effect Effects 0.000 description 2
- 239000005453 ketone based solvent Substances 0.000 description 2
- 239000005001 laminate film Substances 0.000 description 2
- 238000003475 lamination Methods 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000012778 molding material Substances 0.000 description 2
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 2
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 2
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 2
- 239000004014 plasticizer Substances 0.000 description 2
- 229920001707 polybutylene terephthalate Polymers 0.000 description 2
- 238000006068 polycondensation reaction Methods 0.000 description 2
- 229920005672 polyolefin resin Polymers 0.000 description 2
- 239000002356 single layer Substances 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- HLZKNKRTKFSKGZ-UHFFFAOYSA-N tetradecan-1-ol Chemical compound CCCCCCCCCCCCCCO HLZKNKRTKFSKGZ-UHFFFAOYSA-N 0.000 description 2
- 238000012719 thermal polymerization Methods 0.000 description 2
- DVKJHBMWWAPEIU-UHFFFAOYSA-N toluene 2,4-diisocyanate Chemical compound CC1=CC=C(N=C=O)C=C1N=C=O DVKJHBMWWAPEIU-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- JHPBZFOKBAGZBL-UHFFFAOYSA-N (3-hydroxy-2,2,4-trimethylpentyl) 2-methylprop-2-enoate Chemical compound CC(C)C(O)C(C)(C)COC(=O)C(C)=C JHPBZFOKBAGZBL-UHFFFAOYSA-N 0.000 description 1
- MIZLGWKEZAPEFJ-UHFFFAOYSA-N 1,1,2-trifluoroethene Chemical group FC=C(F)F MIZLGWKEZAPEFJ-UHFFFAOYSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- YMRMDGSNYHCUCL-UHFFFAOYSA-N 1,2-dichloro-1,1,2-trifluoroethane Chemical compound FC(Cl)C(F)(F)Cl YMRMDGSNYHCUCL-UHFFFAOYSA-N 0.000 description 1
- XSCLFFBWRKTMTE-UHFFFAOYSA-N 1,3-bis(isocyanatomethyl)cyclohexane Chemical compound O=C=NCC1CCCC(CN=C=O)C1 XSCLFFBWRKTMTE-UHFFFAOYSA-N 0.000 description 1
- XFRVVPUIAFSTFO-UHFFFAOYSA-N 1-Tridecanol Chemical compound CCCCCCCCCCCCCO XFRVVPUIAFSTFO-UHFFFAOYSA-N 0.000 description 1
- OVGRCEFMXPHEBL-UHFFFAOYSA-N 1-ethenoxypropane Chemical compound CCCOC=C OVGRCEFMXPHEBL-UHFFFAOYSA-N 0.000 description 1
- 239000012956 1-hydroxycyclohexylphenyl-ketone Substances 0.000 description 1
- STMDPCBYJCIZOD-UHFFFAOYSA-N 2-(2,4-dinitroanilino)-4-methylpentanoic acid Chemical compound CC(C)CC(C(O)=O)NC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O STMDPCBYJCIZOD-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- SJIXRGNQPBQWMK-UHFFFAOYSA-N 2-(diethylamino)ethyl 2-methylprop-2-enoate Chemical compound CCN(CC)CCOC(=O)C(C)=C SJIXRGNQPBQWMK-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- PQJZHMCWDKOPQG-UHFFFAOYSA-N 2-anilino-2-oxoacetic acid Chemical compound OC(=O)C(=O)NC1=CC=CC=C1 PQJZHMCWDKOPQG-UHFFFAOYSA-N 0.000 description 1
- ISPYQTSUDJAMAB-UHFFFAOYSA-N 2-chlorophenol Chemical compound OC1=CC=CC=C1Cl ISPYQTSUDJAMAB-UHFFFAOYSA-N 0.000 description 1
- WDQMWEYDKDCEHT-UHFFFAOYSA-N 2-ethylhexyl 2-methylprop-2-enoate Chemical compound CCCCC(CC)COC(=O)C(C)=C WDQMWEYDKDCEHT-UHFFFAOYSA-N 0.000 description 1
- FBEMBDJJVJHRHZ-UHFFFAOYSA-N 2-hydroxyethyl prop-2-enoate;phthalic acid Chemical compound OCCOC(=O)C=C.OC(=O)C1=CC=CC=C1C(O)=O FBEMBDJJVJHRHZ-UHFFFAOYSA-N 0.000 description 1
- VHSHLMUCYSAUQU-UHFFFAOYSA-N 2-hydroxypropyl methacrylate Chemical compound CC(O)COC(=O)C(C)=C VHSHLMUCYSAUQU-UHFFFAOYSA-N 0.000 description 1
- GWZMWHWAWHPNHN-UHFFFAOYSA-N 2-hydroxypropyl prop-2-enoate Chemical compound CC(O)COC(=O)C=C GWZMWHWAWHPNHN-UHFFFAOYSA-N 0.000 description 1
- LWRBVKNFOYUCNP-UHFFFAOYSA-N 2-methyl-1-(4-methylsulfanylphenyl)-2-morpholin-4-ylpropan-1-one Chemical compound C1=CC(SC)=CC=C1C(=O)C(C)(C)N1CCOCC1 LWRBVKNFOYUCNP-UHFFFAOYSA-N 0.000 description 1
- BSMGLVDZZMBWQB-UHFFFAOYSA-N 2-methyl-1-phenylpropan-1-one Chemical compound CC(C)C(=O)C1=CC=CC=C1 BSMGLVDZZMBWQB-UHFFFAOYSA-N 0.000 description 1
- RUMACXVDVNRZJZ-UHFFFAOYSA-N 2-methylpropyl 2-methylprop-2-enoate Chemical compound CC(C)COC(=O)C(C)=C RUMACXVDVNRZJZ-UHFFFAOYSA-N 0.000 description 1
- CFVWNXQPGQOHRJ-UHFFFAOYSA-N 2-methylpropyl prop-2-enoate Chemical compound CC(C)COC(=O)C=C CFVWNXQPGQOHRJ-UHFFFAOYSA-N 0.000 description 1
- VDHWOHDSOHPGPC-UHFFFAOYSA-N 3,3-dihydroxyoxepan-2-one Chemical compound OC1(O)CCCCOC1=O VDHWOHDSOHPGPC-UHFFFAOYSA-N 0.000 description 1
- LZMNXXQIQIHFGC-UHFFFAOYSA-N 3-[dimethoxy(methyl)silyl]propyl 2-methylprop-2-enoate Chemical compound CO[Si](C)(OC)CCCOC(=O)C(C)=C LZMNXXQIQIHFGC-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- QXBYUPMEYVDXIQ-UHFFFAOYSA-N 4-methyl-3a,4,5,6,7,7a-hexahydro-2-benzofuran-1,3-dione Chemical compound CC1CCCC2C(=O)OC(=O)C12 QXBYUPMEYVDXIQ-UHFFFAOYSA-N 0.000 description 1
- 229920000178 Acrylic resin Polymers 0.000 description 1
- 239000004925 Acrylic resin Substances 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- 229910001369 Brass Inorganic materials 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- MQIUGAXCHLFZKX-UHFFFAOYSA-N Di-n-octyl phthalate Natural products CCCCCCCCOC(=O)C1=CC=CC=C1C(=O)OCCCCCCCC MQIUGAXCHLFZKX-UHFFFAOYSA-N 0.000 description 1
- OWYWGLHRNBIFJP-UHFFFAOYSA-N Ipazine Chemical compound CCN(CC)C1=NC(Cl)=NC(NC(C)C)=N1 OWYWGLHRNBIFJP-UHFFFAOYSA-N 0.000 description 1
- 239000005058 Isophorone diisocyanate Substances 0.000 description 1
- GYCMBHHDWRMZGG-UHFFFAOYSA-N Methylacrylonitrile Chemical compound CC(=C)C#N GYCMBHHDWRMZGG-UHFFFAOYSA-N 0.000 description 1
- CNCOEDDPFOAUMB-UHFFFAOYSA-N N-Methylolacrylamide Chemical compound OCNC(=O)C=C CNCOEDDPFOAUMB-UHFFFAOYSA-N 0.000 description 1
- 229920002292 Nylon 6 Polymers 0.000 description 1
- 229920002302 Nylon 6,6 Polymers 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 239000004734 Polyphenylene sulfide Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 229920006243 acrylic copolymer Polymers 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 229910000410 antimony oxide Inorganic materials 0.000 description 1
- GVFOJDIFWSDNOY-UHFFFAOYSA-N antimony tin Chemical compound [Sn].[Sb] GVFOJDIFWSDNOY-UHFFFAOYSA-N 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000004760 aramid Substances 0.000 description 1
- 229920003235 aromatic polyamide Polymers 0.000 description 1
- 238000007611 bar coating method Methods 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- DVQGYGDSAGBRSZ-UHFFFAOYSA-N bis(1-cyclohexylcyclohexa-2,4-dien-1-yl)methanone Chemical compound C1C=CC=CC1(C1CCCCC1)C(=O)C1(C2CCCCC2)CC=CC=C1 DVQGYGDSAGBRSZ-UHFFFAOYSA-N 0.000 description 1
- BJQHLKABXJIVAM-UHFFFAOYSA-N bis(2-ethylhexyl) phthalate Chemical compound CCCCC(CC)COC(=O)C1=CC=CC=C1C(=O)OCC(CC)CCCC BJQHLKABXJIVAM-UHFFFAOYSA-N 0.000 description 1
- MQDJYUACMFCOFT-UHFFFAOYSA-N bis[2-(1-hydroxycyclohexyl)phenyl]methanone Chemical compound C=1C=CC=C(C(=O)C=2C(=CC=CC=2)C2(O)CCCCC2)C=1C1(O)CCCCC1 MQDJYUACMFCOFT-UHFFFAOYSA-N 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 239000010951 brass Substances 0.000 description 1
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 description 1
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical class [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 229910000420 cerium oxide Inorganic materials 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000008199 coating composition Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 230000008094 contradictory effect Effects 0.000 description 1
- 238000007334 copolymerization reaction Methods 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 239000003484 crystal nucleating agent Substances 0.000 description 1
- KBLWLMPSVYBVDK-UHFFFAOYSA-N cyclohexyl prop-2-enoate Chemical compound C=CC(=O)OC1CCCCC1 KBLWLMPSVYBVDK-UHFFFAOYSA-N 0.000 description 1
- 238000005034 decoration Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000012975 dibutyltin dilaurate Substances 0.000 description 1
- HYXUVVJRDZXCSQ-UHFFFAOYSA-L dibutyltin(2+);sulfite Chemical compound [O-]S([O-])=O.CCCC[Sn+2]CCCC HYXUVVJRDZXCSQ-UHFFFAOYSA-L 0.000 description 1
- KORSJDCBLAPZEQ-UHFFFAOYSA-N dicyclohexylmethane-4,4'-diisocyanate Chemical compound C1CC(N=C=O)CCC1CC1CCC(N=C=O)CC1 KORSJDCBLAPZEQ-UHFFFAOYSA-N 0.000 description 1
- 238000007607 die coating method Methods 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 229960000735 docosanol Drugs 0.000 description 1
- GMSCBRSQMRDRCD-UHFFFAOYSA-N dodecyl 2-methylprop-2-enoate Chemical compound CCCCCCCCCCCCOC(=O)C(C)=C GMSCBRSQMRDRCD-UHFFFAOYSA-N 0.000 description 1
- 239000002019 doping agent Substances 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- XUCNUKMRBVNAPB-UHFFFAOYSA-N fluoroethene Chemical compound FC=C XUCNUKMRBVNAPB-UHFFFAOYSA-N 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 1
- VOZRXNHHFUQHIL-UHFFFAOYSA-N glycidyl methacrylate Chemical compound CC(=C)C(=O)OCC1CO1 VOZRXNHHFUQHIL-UHFFFAOYSA-N 0.000 description 1
- 239000004519 grease Substances 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 239000012760 heat stabilizer Substances 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- RHZWSUVWRRXEJF-UHFFFAOYSA-N indium tin Chemical compound [In].[Sn] RHZWSUVWRRXEJF-UHFFFAOYSA-N 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000001746 injection moulding Methods 0.000 description 1
- 239000010954 inorganic particle Substances 0.000 description 1
- GJRQTCIYDGXPES-UHFFFAOYSA-N iso-butyl acetate Natural products CC(C)COC(C)=O GJRQTCIYDGXPES-UHFFFAOYSA-N 0.000 description 1
- FGKJLKRYENPLQH-UHFFFAOYSA-M isocaproate Chemical compound CC(C)CCC([O-])=O FGKJLKRYENPLQH-UHFFFAOYSA-M 0.000 description 1
- NIMLQBUJDJZYEJ-UHFFFAOYSA-N isophorone diisocyanate Chemical compound CC1(C)CC(N=C=O)CC(C)(CN=C=O)C1 NIMLQBUJDJZYEJ-UHFFFAOYSA-N 0.000 description 1
- OQAGVSWESNCJJT-UHFFFAOYSA-N isovaleric acid methyl ester Natural products COC(=O)CC(C)C OQAGVSWESNCJJT-UHFFFAOYSA-N 0.000 description 1
- 238000010030 laminating Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 238000010550 living polymerization reaction Methods 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- FQPSGWSUVKBHSU-UHFFFAOYSA-N methacrylamide Chemical compound CC(=C)C(N)=O FQPSGWSUVKBHSU-UHFFFAOYSA-N 0.000 description 1
- 125000005395 methacrylic acid group Chemical group 0.000 description 1
- AYLRODJJLADBOB-UHFFFAOYSA-N methyl 2,6-diisocyanatohexanoate Chemical compound COC(=O)C(N=C=O)CCCCN=C=O AYLRODJJLADBOB-UHFFFAOYSA-N 0.000 description 1
- XJRBAMWJDBPFIM-UHFFFAOYSA-N methyl vinyl ether Chemical compound COC=C XJRBAMWJDBPFIM-UHFFFAOYSA-N 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- BFXIKLCIZHOAAZ-UHFFFAOYSA-N methyltrimethoxysilane Chemical compound CO[Si](C)(OC)OC BFXIKLCIZHOAAZ-UHFFFAOYSA-N 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 229940043348 myristyl alcohol Drugs 0.000 description 1
- QOHMWDJIBGVPIF-UHFFFAOYSA-N n',n'-diethylpropane-1,3-diamine Chemical compound CCN(CC)CCCN QOHMWDJIBGVPIF-UHFFFAOYSA-N 0.000 description 1
- 229940088644 n,n-dimethylacrylamide Drugs 0.000 description 1
- YLGYACDQVQQZSW-UHFFFAOYSA-N n,n-dimethylprop-2-enamide Chemical compound CN(C)C(=O)C=C YLGYACDQVQQZSW-UHFFFAOYSA-N 0.000 description 1
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 1
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- HMZGPNHSPWNGEP-UHFFFAOYSA-N octadecyl 2-methylprop-2-enoate Chemical compound CCCCCCCCCCCCCCCCCCOC(=O)C(C)=C HMZGPNHSPWNGEP-UHFFFAOYSA-N 0.000 description 1
- 229940065472 octyl acrylate Drugs 0.000 description 1
- ANISOHQJBAQUQP-UHFFFAOYSA-N octyl prop-2-enoate Chemical compound CCCCCCCCOC(=O)C=C ANISOHQJBAQUQP-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000011146 organic particle Substances 0.000 description 1
- 150000003961 organosilicon compounds Chemical class 0.000 description 1
- BMMGVYCKOGBVEV-UHFFFAOYSA-N oxo(oxoceriooxy)cerium Chemical compound [Ce]=O.O=[Ce]=O BMMGVYCKOGBVEV-UHFFFAOYSA-N 0.000 description 1
- VTRUBDSFZJNXHI-UHFFFAOYSA-N oxoantimony Chemical compound [Sb]=O VTRUBDSFZJNXHI-UHFFFAOYSA-N 0.000 description 1
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 1
- 125000002081 peroxide group Chemical group 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 125000000864 peroxy group Chemical group O(O*)* 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920006122 polyamide resin Polymers 0.000 description 1
- 229920001230 polyarylate Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920005668 polycarbonate resin Polymers 0.000 description 1
- 239000004431 polycarbonate resin Substances 0.000 description 1
- 229920006267 polyester film Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 239000005056 polyisocyanate Substances 0.000 description 1
- 229920001228 polyisocyanate Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000306 polymethylpentene Polymers 0.000 description 1
- 239000011116 polymethylpentene Substances 0.000 description 1
- 229920000069 polyphenylene sulfide Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 238000012805 post-processing Methods 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 229940090181 propyl acetate Drugs 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000007870 radical polymerization initiator Substances 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 238000007348 radical reaction Methods 0.000 description 1
- 229920005604 random copolymer Polymers 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 150000003297 rubidium Chemical class 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 230000003678 scratch resistant effect Effects 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 150000004756 silanes Chemical class 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 229940012831 stearyl alcohol Drugs 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- MUTNCGKQJGXKEM-UHFFFAOYSA-N tamibarotene Chemical compound C=1C=C2C(C)(C)CCC(C)(C)C2=CC=1NC(=O)C1=CC=C(C(O)=O)C=C1 MUTNCGKQJGXKEM-UHFFFAOYSA-N 0.000 description 1
- 238000010345 tape casting Methods 0.000 description 1
- 238000009864 tensile test Methods 0.000 description 1
- 238000004154 testing of material Methods 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
- 238000003856 thermoforming Methods 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 239000012749 thinning agent Substances 0.000 description 1
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 1
- 229910001887 tin oxide Inorganic materials 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 1
- 229940087291 tridecyl alcohol Drugs 0.000 description 1
- PHYFQTYBJUILEZ-IUPFWZBJSA-N triolein Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(OC(=O)CCCCCCC\C=C/CCCCCCCC)COC(=O)CCCCCCC\C=C/CCCCCCCC PHYFQTYBJUILEZ-IUPFWZBJSA-N 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 238000004736 wide-angle X-ray diffraction Methods 0.000 description 1
- 230000037303 wrinkles Effects 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 229910001928 zirconium oxide Inorganic materials 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/043—Improving the adhesiveness of the coatings per se, e.g. forming primers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/46—Polymerisation initiated by wave energy or particle radiation
- C08F2/48—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light
- C08F2/50—Polymerisation initiated by wave energy or particle radiation by ultraviolet or visible light with sensitising agents
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F299/00—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers
- C08F299/02—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers from unsaturated polycondensates
- C08F299/06—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers from unsaturated polycondensates from polyurethanes
- C08F299/065—Macromolecular compounds obtained by interreacting polymers involving only carbon-to-carbon unsaturated bond reactions, in the absence of non-macromolecular monomers from unsaturated polycondensates from polyurethanes from polyurethanes with side or terminal unsaturations
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/0427—Coating with only one layer of a composition containing a polymer binder
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/046—Forming abrasion-resistant coatings; Forming surface-hardening coatings
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J9/00—Adhesives characterised by their physical nature or the effects produced, e.g. glue sticks
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2475/00—Characterised by the use of polyureas or polyurethanes; Derivatives of such polymers
- C08J2475/04—Polyurethanes
- C08J2475/14—Polyurethanes having carbon-to-carbon unsaturated bonds
- C08J2475/16—Polyurethanes having carbon-to-carbon unsaturated bonds having terminal carbon-to-carbon unsaturated bonds
Definitions
- the present invention relates to a laminated film that is excellent in molding followability, scratch resistance, and adhesion to a resin substrate as a molding material, and is advantageous in terms of productivity and cost, and relates to a laminated film that can be suitably used for decorative molding.
- a molding material such as decorative molding is provided with a surface hardness layer to prevent scratches during molding and to prevent scratches in the process of using the article after molding.
- the surface hardened layer lacks the elongation to follow the molding, so cracks occur at the time of molding, and in extreme cases, the film breaks or the surface hardened layer peels off.
- a method of forming a surface hardness layer after molding, or molding in a semi-cured state and then completely curing by heating or actinic radiation is applied.
- the molded article is processed three-dimensionally, it is very difficult to provide a surface hardened layer by post-processing, and when molding in a semi-cured state, depending on the molding conditions, May induce dirt.
- Self-healing materials are capable of self-healing deformation in their own elastic recovery range, and two types are widely known: thermosetting type and active energy ray curable type using ultraviolet rays or electron beams.
- thermosetting materials described in Patent Documents 1 and 2 are excellent in moldability and self-healing property, they have poor stain resistance, so they come into contact with fingers with cosmetics or a resin bag to which a plasticizer is added. It is a problem to cause various problems.
- the stain resistance shown here means that the plasticizer (dioctyl phthalate, etc.) contained in the vinyl chloride sheet, cosmetics, ink components such as oil-based magic, etc. penetrate into the self-healing material and cause coloration and mottle. It is not solved only by wiping the surface.
- An object of the present invention is to provide a laminated film having a self-healing layer excellent in self-healing property and stain resistance.
- the present invention is a laminated film having an A layer on at least one side of a base film,
- the resin contained in the A layer has (1) (poly) caprolactone segment and (2) urethane bond,
- the maximum displacement amount in the thickness direction of the A layer is 1.0 to 3.0 ⁇ m
- the creep displacement amount in the thickness direction of the A layer is 0.2.
- the laminated film has a permanent displacement in the thickness direction of the A layer of 0.3 to 0.65 ⁇ m when the load is released to 0 mN.
- the laminated film of the present invention has a surface flaw repair function (self-healing property) and excellent stain resistance. Therefore, the laminated film of the present invention is particularly useful when a film that is easily damaged by a surface is used as a base film.
- FIG. 3 is a weight-indentation depth diagram when an indentation load / unloading test is performed on the laminated film of the present invention using a regular triangular pyramid.
- the resin constituting the base film may be either a thermoplastic resin or a thermosetting resin, may be a homo resin, or may be a copolymer or a blend of two or more. More preferably, the resin constituting the base film is a thermoplastic resin because of good moldability.
- thermoplastic resins examples include polyolefin resins such as polyethylene, polypropylene, polystyrene, and polymethylpentene, alicyclic polyolefin resins, polyamide resins such as nylon 6 and nylon 66, aramid resins, polyester resins, polycarbonate resins, and polyarylate resins.
- Fluorine resins such as polyacetal resin, polyphenylene sulfide resin, tetrafluoroethylene resin, trifluoroethylene resin, trifluoroethylene chloride resin, tetrafluoroethylene-6 fluoropropylene copolymer, vinylidene fluoride resin, acrylic Resins, methacrylic resins, polyacetal resins, polyglycolic acid resins, polylactic acid resins, and the like can be used.
- the thermoplastic resin is preferably a resin having sufficient stretchability and followability.
- the thermoplastic resin is more preferably a polyester resin from the viewpoint of strength, heat resistance, and transparency.
- the polyester resin in the present invention is a general term for polymers having an ester bond as a main bond chain, and is obtained by polycondensation of an acid component and its ester with a diol component.
- Specific examples include polyethylene terephthalate, polypropylene terephthalate, polyethylene-2,6-naphthalate, polybutylene terephthalate, and the like. These may be copolymerized with other dicarboxylic acids and their esters or diol components as acid components or diol components.
- polyethylene terephthalate and polyethylene-2,6-naphthalate are particularly preferable in terms of transparency, dimensional stability, heat resistance and the like.
- the polyester resin in the present invention is a general term for polymers having an ester bond as a main bond chain, and is obtained by polycondensation of an acid component and its ester with a diol component.
- Specific examples include polyethylene terephthalate, polypropylene terephthalate, polyethylene-2,6-naphthalate, polybutylene terephthalate, and the like. These may be copolymerized with other dicarboxylic acids and their esters or diol components as acid components or diol components.
- polyethylene terephthalate and polyethylene-2,6-naphthalate are particularly preferable in terms of transparency, dimensional stability, heat resistance and the like.
- additives such as an antioxidant, an antistatic agent, a crystal nucleating agent, an inorganic particle, an organic particle, a thinning agent, a heat stabilizer, a lubricant, an infrared absorber, an ultraviolet absorber, A dopant for adjusting the refractive index may be added.
- the base film may be either a single-layer base film or a laminated base film.
- polyester base film when resin which comprises a base film contains 50 mass% or more and 100 mass% or less of polyester resins in 100 mass% of all the components of a base film, a base film is called polyester base film.
- the polyester resin constituting the polyester base film has an intrinsic viscosity (measured in o-chlorophenol at 25 ° C. according to JIS K7367 (2000)) of 0.4 to 1.2 dl / g. 0.5 to 0.8 dl / g is particularly preferable.
- the polyester base film can be any of an unstretched (unoriented) film, a uniaxially stretched (uniaxially oriented) film, and a biaxially stretched (biaxially oriented) film, but is biaxially stretched with excellent dimensional stability and heat resistance. It is preferable to use a film.
- the biaxially stretched film is preferably highly crystallized.
- the biaxial orientation refers to a material that exhibits a biaxial orientation pattern by wide-angle X-ray diffraction.
- the polyester base film may be a polyester film having fine cavities inside.
- the polyester base film may have a single layer structure or a laminated structure.
- polyester resins preferably a layer (C layer) containing 50% by mass to 100% by mass of polyester resin C and 50% by mass to 100% by mass of polyester resin D are included.
- the layer (D layer) to include is laminated
- the different polyester resin means a polyester resin having a different molecular structure or a case where some components of the copolymer polyester resin are different.
- the laminated film of the present invention is a laminated film having an A layer on at least one side of the base film, wherein the resin contained in the A layer is (1) a (poly) caprolactone segment, and (2) a urethane bond,
- the maximum displacement in the thickness direction of the A layer when a 0.5 mN load is applied for 10 seconds in the microhardness measurement is 1.0 to 3.0 ⁇ m
- the creep in the thickness direction of the A layer is The amount of displacement is 0.2 to 0.5 ⁇ m
- the amount of permanent displacement in the thickness direction of the A layer when the load is released to 0 mN is 0.3 to 0.65 ⁇ m.
- the laminated film of the present invention has an excellent self-healing effect by having an A layer on at least one side of the base film.
- the A layer exists only on one side of the base film in consideration of cost, although it depends on the use. In many applications, the A layer exists only on one side of the base film, and the laminated film has sufficient self-healing properties.
- the resin contained in the A layer has (1) (poly) caprolactone segment and (2) urethane bond.
- the layer A can also contain (1) a (poly) caprolactone segment and (2) a resin having a urethane bond, or (1) a resin having a (poly) caprolactone segment and (2) a resin having a urethane bond. It is also possible to contain a plurality of resins.
- the components contained in the A layer will be described.
- the A layer has (1) (poly) caprolactone segments.
- the elastic recovery property self-healing property
- the (poly) caprolactone segment refers to a segment represented by the following chemical formula 1.
- the A layer can have a (poly) caprolactone segment by forming the A layer using a composition containing a resin containing a (poly) caprolactone segment.
- the resin containing a (poly) caprolactone segment preferably has at least one hydroxyl group (hydroxyl group). The hydroxyl group is preferably at the end of the resin containing the (poly) caprolactone segment.
- the layer A can have a self-healing property because the layer A has a component having a (poly) caprolactone segment. That is, even if the surface of the A layer is scratched, the scratch can be extinguished (self-healed) in a short time of several seconds.
- (poly) caprolactone having a bi- to trifunctional hydroxyl group is particularly preferable. Specifically, (poly) caprolactone diol,
- Radical polymerizable caprolactone such as can be used.
- examples of other radical polymerizable caprolactones include (poly) caprolactone-modified hydroxypropyl (meth) acrylate, (poly) caprolactone-modified hydroxybutyl (meth) acrylate, and the like.
- the resin containing the (poly) caprolactone segment may contain (or copolymerize) other segments and monomers in addition to the (poly) caprolactone segment.
- a polydimethylsiloxane segment or a (poly) siloxane segment may be contained (or copolymerized).
- the weight average molecular weight of the (poly) caprolactone segment in the resin containing the (poly) caprolactone segment is preferably 500 to 2,500, more preferably 1,000 to 1. , 500.
- the weight average molecular weight of the (poly) caprolactone segment is from 500 to 2,500, the self-healing effect is further exhibited and the scratch resistance is further improved.
- the (poly) caprolactone segment is copolymerized or added separately, in the 100% by mass of the total components of the composition used to form the A layer, the (poly) caprolactone segment An amount of 5 to 50% by mass is preferable in terms of self-healing properties and stain resistance.
- 100% by mass of all the components of the composition does not include a solvent that does not participate in the reaction.
- the monomer component involved in the reaction is included.
- the resin contained in the A layer has (2) a urethane bond.
- the resin contained in the A layer can have a urethane bond.
- the resin contained in A layer can also have a urethane bond also by making an isocyanate group and a hydroxyl group react and producing
- the resin contained in the A layer preferably has a urethane bond by reacting an isocyanate group with a hydroxyl group to form a urethane bond.
- the toughness of the A layer can be improved and the elastic recovery property (self-healing property) can be improved.
- a resin containing a polysiloxane segment or a resin containing a polydimethylsiloxane segment has a hydroxyl group
- the A layer is formed using a compound containing an isocyanate group, a resin containing a (poly) siloxane segment having a hydroxyl group, or a resin containing a polydimethylsiloxane segment having a hydroxyl group
- the toughness and elastic recovery of the A layer (Self-healing property) and surface slipperiness can be improved, which is preferable.
- the compound containing an isocyanate group means a resin containing an isocyanate group, or a monomer or oligomer containing an isocyanate group.
- the compound containing an isocyanate group include methylene bis-4-cyclohexyl isocyanate, trimethylolpropane adduct of tolylene diisocyanate, trimethylolpropane adduct of hexamethylene diisocyanate, trimethylolpropane adduct of isophorone diisocyanate, and tolylene diisocyanate.
- Polyisocyanates such as isocyanurate bodies, isocyanurate bodies of hexamethylene diisocyanate, burette bodies of hexamethylene isocyanate, and block bodies of the above isocyanates can be mentioned.
- aliphatic isocyanates are preferred because of their high self-healing properties compared to alicyclic and aromatic isocyanates.
- the compound containing an isocyanate group is more preferably hexamethylene diisocyanate.
- the isocyanate group-containing compound is particularly preferably an isocyanate having an isocyanurate ring from the viewpoint of heat resistance, and most preferably an isocyanurate of hexamethylene diisocyanate.
- An isocyanate having an isocyanurate ring forms a layer A having both self-healing properties and heat resistance.
- melamine crosslinking agents such as alkoxymethylol melamine, acid anhydride crosslinking agents such as 3-methyl-hexahydrophthalic anhydride, amine crosslinking agents such as diethylaminopropylamine, etc.
- Other crosslinkers can also be included.
- a crosslinking catalyst such as dibutyltin dilaurate or dibutyltin diethylhexoate may be used to promote the urethane bond formation reaction.
- the A layer preferably has (3) (poly) alkylene glycol segments.
- the (poly) alkylene glycol segment refers to a segment represented by the following chemical formula.
- n is an integer from 2 to 4
- m is an integer from 2 to 11.
- alkylene glycol is a glycol having 2 to 4 carbon atoms. Furthermore, the number of repeating units m of the alkylene glycol is 2 to 11, preferably 3 to 6. When the number of carbons n of the alkylene glycol exceeds 4, or when the number of repeating units m of the alkylene glycol exceeds 11, the molecular chain of the alkylene glycol becomes longer, the crosslink density of the cured product becomes lower, and its hardness becomes lower. As a result, the coating strength, scratch resistance, etc. are reduced. On the other hand, when the number of repeating units m of the alkylene glycol is less than 2, the self-healing property of the cured product is lowered and the workability is further lowered.
- the A layer By forming the A layer using a composition containing a resin containing a (poly) alkylene glycol segment, the A layer can have a (poly) alkylene glycol segment.
- the resin containing a (poly) alkylene glycol segment preferably has at least one hydroxyl group (hydroxyl group).
- the hydroxyl group is preferably at the end of the resin containing the (poly) alkylene glycol segment.
- the resin containing a (poly) alkylene glycol segment is preferably a (poly) alkylene glycol (meth) acrylate having an acrylate group at the end in order to impart elasticity.
- the number of acrylate functional groups (or methacrylate functional groups) of the (poly) alkylene glycol (meth) acrylate is not limited, but is most preferably monofunctional from the viewpoint of the self-healing property of the cured product.
- Examples of the (poly) alkylene glycol (meth) acrylate contained in the composition used for forming the A layer include (poly) ethylene glycol (meth) acrylate, (poly) propylene glycol (meth) acrylate, and (poly) Examples include butylene glycol (meth) acrylate. Each is represented by the following general formula.
- (Poly) ethylene glycol (meth) acrylate: CH 2 CRCOO (C 2 H 4 O) m H
- (Poly) propylene glycol (meth) acrylate: CH 2 CRCOO (CH 2 CH 2 CH 2 O) m H
- the hydroxyl group of the (poly) propylene glycol (meth) acrylate is a primary hydroxyl group, that is, a hydroxyl group bonded to primary carbon.
- polyethylene glycol (meth) acrylate which is ethylene glycol is particularly preferable. Since (poly) ethylene glycol (meth) acrylate has the smallest carbon number n in Chemical Formula 5, it can contribute to both the cosmetic resistance and the scratch resistance of the resulting cured product.
- the above-mentioned compound containing an isocyanate group is reacted with a hydroxyl group of (poly) alkylene glycol (meth) acrylate and used as a urethane (meth) acrylate in the A layer, whereby the A layer is (2 It can have a urethane bond and (3) (poly) alkylene glycol segment, and as a result, the toughness of the A layer can be improved and the self-healing property can be improved.
- hydroxyalkyl (meth) acrylate, long-chain alcohol or the like can be blended.
- hydroxyalkyl (meth) acrylate the hardness of the A layer, which is a cured product, can be increased.
- a long-chain alcohol the surface lubricity of the A layer, which is a cured product, can be increased, and as a result, the scratch resistance can be improved.
- This long-chain alcohol is a compound included in the concept of the long-chain alkyl group-containing compound.
- hydroxyalkyl (meth) acrylate compounded at the same time during the urethanization reaction between the compound containing an isocyanate group and (poly) alkylene glycol (meth) acrylate examples thereof include hydroxybutyl (meth) acrylate.
- Long-chain alcohols to be blended at the same time during the urethanization reaction between the isocyanate group-containing compound and (poly) alkylene glycol (meth) acrylate include tridecanol, myristyl alcohol, cetyl alcohol, stearyl alcohol, behenyl alcohol, polyoxyethylene mono Examples include stearate, polyoxyethylene cetyl ether, polyoxyethylene stearyl ether, and glycerol monostearate.
- Particularly preferred long-chain alcohols include polyether-modified long-chain alcohols such as polyether-modified cetyl alcohol. This is because the use of a polyether-modified long-chain alcohol can impart an antistatic effect to the cured layer A.
- the urethanation reaction between the isocyanate group-containing compound and (poly) alkylene glycol (meth) acrylate is carried out in an organic solvent in the presence of a catalyst, a polymerization inhibitor or the like.
- the reaction temperature in the urethanization reaction is preferably from room temperature to 100 ° C., and the reaction time is preferably from 1 to 10 hours.
- the reaction temperature is lower than normal temperature or when the reaction time is shorter than 1 hour, the progress of the reaction is slow, and the yield of the desired urethane (meth) acrylate is likely to decrease.
- the reaction temperature exceeds 100 ° C. or when the reaction time is longer than 10 hours, a side reaction tends to occur.
- Examples of the organic solvent used in the urethanation reaction between the isocyanate group-containing compound and (poly) alkylene glycol (meth) acrylate are aromatic hydrocarbon solvents such as toluene and xylene; acetone, methyl ethyl ketone, methyl isobutyl ketone, Examples include ketone solvents such as cyclohexanone; ester solvents such as ethyl acetate, propyl acetate, isobutyl acetate, and butyl acetate.
- Examples of the catalyst include dibutyltin laurate, dibutyltin diethylhexoate, dibutyltin sulfite and the like.
- the A layer preferably has (4) a (poly) siloxane segment and / or a polydimethylsiloxane segment.
- the (poly) siloxane segment refers to a segment represented by the following chemical formula.
- R is any one of OH and an alkyl group having 1 to 8 carbon atoms, and each has at least one or more in the formula.
- the composition used for forming the A layer includes a resin containing the (poly) siloxane segment.
- a partial hydrolyzate of a silane compound containing a hydrolyzable silyl group, an organosilica sol or a composition obtained by adding a hydrolyzable silane compound having a radical polymer to the organosilica sol contains a polysiloxane segment. It can be used as a resin.
- Resins containing (poly) siloxane segments are tetraalkoxysilane, methyltrialkoxysilane, dimethyldialkoxysilane, ⁇ -glycidoxypropyltrialkoxysilane, ⁇ -glycidoxypropylalkyldialkoxysilane, ⁇ -methacryloxy Hydrolyzable on the surface of organosilica sol, organosilica sol dispersed in complete or partial hydrolysates of silane compounds with hydrolyzable silyl groups such as propyltrialkoxysilane and ⁇ -methacryloxypropylalkyldialkoxysilane, organic solvents The thing etc. which added the hydrolysis silane compound of the silyl group can be illustrated.
- the resin containing the (poly) siloxane segment may contain (copolymerize) other segments in addition to the (poly) siloxane segment.
- a monomer component having a (poly) caprolactone segment and a polydimethylsiloxane segment may be contained (copolymerized).
- a monomer having a hydroxyl group that reacts with an isocyanate group is copolymerized as a resin containing a (poly) siloxane segment.
- a monomer having a hydroxyl group that reacts with an isocyanate group is copolymerized with a resin containing a (poly) siloxane segment, the toughness of the A layer is improved.
- the resin containing a (poly) siloxane segment is a copolymer having a hydroxyl group
- a composition comprising a resin (copolymer) containing a (poly) siloxane segment having a hydroxyl group and a compound containing an isocyanate group
- the A layer is formed by use, the A layer having (poly) siloxane segments and urethane bonds can be efficiently formed.
- the (poly) siloxane segment is 100% by mass of the total component of the composition used to form the A layer.
- a content of 1 to 20% by mass is preferable in terms of self-healing property, stain resistance, weather resistance, and heat resistance.
- 100% by mass of the total components of the composition does not include a solvent that does not participate in the reaction. The monomer component involved in the reaction is included.
- polydimethylsiloxane segment refers to a segment represented by the following formula.
- the polydimethylsiloxane segment is coordinated to the surface of the A layer.
- the lubricity of the surface of the A layer is improved, and the frictional resistance can be reduced. As a result, scratchability can be suppressed.
- the composition used for forming the A layer can include a resin containing a polydimethylsiloxane segment.
- the resin containing a polydimethylsiloxane segment it is preferable to use a copolymer obtained by copolymerizing a vinyl monomer with a polydimethylsiloxane segment.
- the resin containing a polydimethylsiloxane segment is preferably copolymerized with a monomer having a hydroxyl group that reacts with an isocyanate group.
- a composition containing a resin containing a hydroxyl group-containing polydimethylsiloxane segment (copolymer) and a compound containing an isocyanate group is used.
- the resin containing the polydimethylsiloxane segment When the resin containing the polydimethylsiloxane segment is a copolymer with a vinyl monomer, it may be a block copolymer, a graft copolymer, or a random copolymer. When the resin containing the polydimethylsiloxane segment is a copolymer with a vinyl monomer, this is referred to as a polydimethylsiloxane copolymer.
- Polydimethylsiloxane copolymers can be produced by the living polymerization method, polymer initiator method, polymer chain transfer method, etc., but considering the productivity, the polymer initiator method, polymer chain transfer method can be used. It is preferable to use it.
- a two-stage polymerization is carried out by synthesizing a prepolymer in which a peroxide group is introduced into the side chain by copolymerizing a peroxy monomer and polydimethylsiloxane having an unsaturated group at a low temperature, and then copolymerizing the prepolymer with a vinyl monomer. Can also be done.
- a block copolymer can be synthesized by copolymerization.
- a graft copolymer can be easily obtained by copolymerizing a compound shown in the following, that is, a methacrylic ester of polydimethylsiloxane and a vinyl monomer.
- vinyl monomers used in the copolymer with polydimethylsiloxane include methyl acrylate, ethyl acrylate, n-butyl acrylate, isobutyl acrylate, octyl acrylate, cyclohexyl acrylate, tetrahydrofurfuryl acrylate, methyl methacrylate, ethyl methacrylate, n -Butyl methacrylate, isobutyl methacrylate, 2-ethylhexyl methacrylate, stearyl methacrylate, lauryl methacrylate, methyl vinyl ether, ethyl vinyl ether, n-propyl vinyl ether, styrene, ⁇ -methyl styrene, acrylonitrile, methacrylonitrile, vinyl acetate, vinyl chloride, vinylidene chloride , Vinyl fluoride, vinylidene fluoride, glycidyl accelerator
- Polydimethylsiloxane copolymers include aromatic hydrocarbon solvents such as toluene and xylene, ketone solvents such as methyl ethyl ketone and methyl isobutyl ketone, ester solvents such as ethyl acetate and butyl acetate, ethanol, isopropyl alcohol, etc. It is preferable that the alcoholic solvent is produced by a solution polymerization method alone or in a mixed solvent.
- a polymerization initiator such as benzoyl peroxide or azobisisobutylnitrile is used in combination.
- the polymerization reaction is preferably carried out at 50 to 150 ° C. for 3 to 12 hours.
- the amount of the polydimethylsiloxane segment in the polydimethylsiloxane copolymer in the present invention is 1 to 30 in 100% by mass of all the components of the polydimethylsiloxane copolymer from the viewpoint of lubricity and contamination resistance of the A layer. It is preferable that it is mass%.
- the weight average molecular weight of the polydimethylsiloxane segment is preferably 1,000 to 30,000.
- the dimethylsiloxane segment is 1 to 20 in 100% by mass of the total components of the composition used to form the A layer.
- the mass% is preferable in terms of self-healing property, contamination resistance, weather resistance, and heat resistance. 100% by mass of the total components of the composition does not include a solvent that does not participate in the reaction. The monomer component involved in the reaction is included.
- a resin containing a polydimethylsiloxane segment when used as the composition used to form the A layer, other segments are contained (copolymerized) in addition to the polydimethylsiloxane segment. Also good.
- a (poly) caprolactone segment or a (poly) siloxane segment may be contained (copolymerized).
- the composition used to form the A layer includes a copolymer of (poly) caprolactone segment and polydimethylsiloxane segment, a copolymer of (poly) caprolactone segment and (poly) siloxane segment, (poly) caprolactone segment And a copolymer of a polydimethylsiloxane segment and a (poly) siloxane segment can be used.
- the A layer obtained using such a composition can have a (poly) caprolactone segment, a polydimethylsiloxane segment, and / or a (poly) siloxane segment.
- the reaction can be copolymerized by appropriately adding a (poly) caprolactone segment and a polysiloxane segment during the synthesis of the polydimethylsiloxane copolymer.
- the composition used for forming the A layer preferably contains an initiator, a curing agent, and a catalyst. Initiators and catalysts are used to promote curing of the A layer.
- the initiator those capable of initiating or accelerating polymerization, condensation or cross-linking reaction of the coating composition by anion, cation, radical reaction or the like are preferable.
- initiators, curing agents and catalysts can be used.
- the initiator, the curing agent, and the catalyst may be used alone, or a plurality of initiators, curing agents, and catalysts may be used at the same time.
- acidic catalysts include aqueous hydrochloric acid, formic acid, acetic acid and the like.
- thermal polymerization initiator include peroxides and azo compounds.
- the photopolymerization initiator include alkylphenone compounds, sulfur-containing compounds, acylphosphine oxide compounds, amine compounds, and the like.
- an alkylphenone compound is preferable from the viewpoint of curability.
- the alkylphenone type compound include 1-hydroxy-cyclohexyl-phenyl-ketone, 2.2-dimethoxy-1.2-diphenylethane-1-one, 2-methyl-1- (4-methylthiophenyl)- 2-morpholinopropan-1-one, 2-benzyl-2-dimethylamino-1- (4-phenyl) -1-butane, 2- (dimethylamino) -2-[(4-methylphenyl) methyl]- 1- (4-phenyl) -1-butane, 2-benzyl-2-dimethylamino-1- (4-morpholinophenyl) -1-butane, 2- (dimethylamino) -2-[(4-methylphenyl ) Methyl] -1- [4- (4-morpholinyl) phenyl] -1-butane, 1-cyclohexyl
- a layer can contain a leveling agent, a ultraviolet absorber, a lubricant, an antistatic agent, etc.
- a layer can contain a leveling agent, a ultraviolet absorber, a lubricant, an antistatic agent, etc.
- the leveling agent include an acrylic copolymer or a silicone-based or fluorine-based leveling agent.
- Specific examples of the ultraviolet absorber include benzophenone-based, benzotriazole-based, oxalic anilide-based, triazine-based and hindered amine-based ultraviolet absorbers.
- the antistatic agent include metal salts such as lithium salt, sodium salt, potassium salt, rubidium salt, cesium salt, magnesium salt and calcium salt.
- the A layer is an A layer obtained by curing a composition containing urethane (meth) acrylate B and urethane (meth) acrylate C with active energy rays. It is preferable that acrylate B has at least a (poly) caprolactone segment and the urethane (meth) acrylate C has at least a (poly) alkylene glycol segment. By taking such a structure, it is excellent in self-healing property and stain resistance.
- self-healing materials elastically recover minor scratches by the soft segment component contained acting as a cushion, so increasing the soft segment component can improve self-healing properties. It becomes possible. Specifically, by increasing the (1) (poly) caprolactone segment and the (3) (poly) alkylene glycol segment in the A layer, excellent self-healing properties can be achieved. However, if the method of improving the self-healing property of the A layer by such an improved method is adopted, the ratio of the free volume of the molecules constituting the A layer increases, so that oils and chemicals can easily penetrate between the molecules. The contamination resistance of the A layer deteriorates.
- the maximum displacement amount in the thickness direction of the A layer when a 0.5 mN load is applied for 10 seconds in the microhardness meter measurement is 1.0 to 3.0 ⁇ m. It is important that the creep displacement is 0.2 to 0.5 ⁇ m, and the permanent displacement in the thickness direction of the A layer is 0.3 to 0.65 ⁇ m when the load is released to 0 mN.
- the maximum displacement in the thickness direction of the A layer is more preferably 1.2 to 3.0 ⁇ m.
- the creep displacement amount in the thickness direction of the A layer is more preferably 0.3 to 0.4 ⁇ m.
- the permanent displacement amount in the thickness direction of the A layer is more preferably 0.4 to 0.6 ⁇ m.
- the maximum displacement in the thickness direction of the A layer is 1.0 to 3.0 ⁇ m, the creep displacement in the thickness direction of the A layer is 0.2 to 0.5 ⁇ m, and the permanent displacement in the thickness direction of the A layer When the amount is 0.3 to 0.65 ⁇ m, excellent self-healing properties are exhibited.
- the fracture elongation of the A layer of the laminated film of the present invention is preferably 50% or more.
- the fracture elongation of the A layer is 50% or more, it is excellent in molding followability and can be used for molding decoration.
- the particles used in the layer A include silica fine particles such as dry silica and wet silica, titanium oxide, zirconium oxide, zinc oxide, tin oxide, cerium oxide, antimony oxide, indium tin mixed oxide and antimony tin mixed oxide.
- Metal oxide fine particles such as acryl, styrene, and the like, and various commercial products using an organic solvent as a dispersion medium can also be used.
- the shape of the fine particles is preferably spherical or beaded, but is not particularly limited thereto. In particular, it is preferable to employ silica fine particles because the obtained laminated film has low transparency and has good dispersibility when mixed in a coating agent.
- the cosmetic dissolution concentration of the A layer is preferably 2.9% by mass or less when the cosmetic is applied to the surface of the A layer and kept in a 60 ° C. atmosphere for 6 hours.
- the cosmetic dissolution concentration of the A layer under the above-described conditions is more preferably 0.3% by mass or more and 1.5% by mass or less.
- the cosmetic dissolution concentration of the A layer is 2.9% by mass or less, the stain resistance is good, and when it is 1.5% by mass or less, problems due to the contamination hardly occur.
- the lower the cosmetic dissolution concentration of the A layer the better.
- the A layer contains a (poly) caprolactone segment, it is difficult to make the cosmetic dissolution concentration less than 0.3% by mass. It is considered to be about 3% by mass.
- Contamination resistance refers to the resistance to discoloration of the surface and the occurrence of mottled spots when in contact with creams, oil-based magic, and PVC sheets.
- the mass increase rate of the A layer is preferably 10% by mass or less when oleic acid is applied to the surface of the A layer and kept in a 60 ° C. atmosphere for 1 hour.
- the mass increase rate of the A layer under the above-mentioned conditions is more preferably 0.3% by mass or more and 5.0% by mass or less, and further preferably 0.3% by mass or more and 3.0% by mass or less.
- the rate of mass increase due to oleic acid in the A layer is 10.0% by mass or less, the contamination resistance is good, and when it is 3.0% by mass or less, problems due to the contamination hardly occur.
- Contamination resistance refers to the resistance to discoloration of the surface and the occurrence of mottled spots when in contact with creams, oil-based magic, and PVC sheets.
- the cosmetic dissolution concentration of the A layer is 2.9% by mass or less when the cosmetic is applied to the surface of the A layer and kept in a 60 ° C. atmosphere for 6 hours, or the olein on the surface of the A layer.
- the acid is applied and kept at 60 ° C.
- the increase in mass of the A layer is 10% by mass or less
- the thickness direction of the A layer when a 0.5 mN load is applied for 10 seconds in the microhardness meter measurement A maximum displacement amount of 1.0 to 3.0 ⁇ m, creep displacement amount in the thickness direction of the A layer is 0.05 to 0.5 ⁇ m
- the residual displacement amount in the thickness direction of the A layer when the load is 0 mN Is preferably 0.2 to 0.65 ⁇ m, and it is compatible with cosmetic resistance and self-healing properties.
- This accomplishment method includes the following active energy rays such as ultraviolet rays and electron beams for a composition containing urethane (meth) acrylate B having excellent self-healing properties and urethane (meth) acrylate C having excellent stain resistance. It is important to form the A layer by curing with.
- Urethane (meth) acrylate B having excellent self-healing properties is a layer (hereinafter, referred to as a layer obtained by curing a mixture of urethane (meth) acrylate and a photoinitiator with a high-pressure mercury lamp having an illuminance of 400 mW / cm 2 to a thickness of 30 ⁇ m.
- the physical property of the (B layer) means urethane (meth) acrylate in the following range.
- the cosmetic dissolution concentration of the B layer is 4.0% by mass or less, or oleic acid is applied to the surface of the B layer at 60 ° C.
- the layer B has a mass increase rate of 45% by mass or less when held for 1 hour, and the maximum displacement in the thickness direction of the layer B when a 0.5 mN load is applied for 10 seconds in the microhardness meter measurement is 1. 0 to 3.0 ⁇ m, creep displacement in the thickness direction of the B layer is 0.4 to 0.7 ⁇ m.
- Urethane (meth) acrylate C having excellent cosmetic resistance is a layer obtained by curing a mixture of urethane (meth) acrylate and a photoinitiator to a thickness of 30 ⁇ m with ultraviolet light from a high-pressure mercury lamp having an illuminance of 400 mW / cm 2 (hereinafter, referred to as “urethane (meth) acrylate C”).
- urethane (meth) acrylate C A urethane (meth) acrylate having a physical property of C layer) in the following range is meant.
- the cosmetic dissolution concentration of the A layer is 2.5% by mass or less when a cosmetic product is applied to the surface of the C layer and kept in a 60 ° C. atmosphere for 6 hours, or oleic acid is applied to the C layer surface at 60 ° C.
- the mass increase rate of the C layer is 5.0% by mass or less
- the maximum displacement in the thickness direction of the C layer when a 0.5 mN load is applied for 10 seconds in the microhardness meter measurement is 1.0 to 3.0 ⁇ m
- creep displacement in the thickness direction of the C layer is 0.05 to 0.35 ⁇ m.
- the content ratio of urethane (meth) acrylate B and urethane (meth) acrylate C in the composition used to form the A layer (mass of urethane (meth) acrylate B / mass of urethane (meth) acrylate C) In the range of 70/30 to 30/70, by curing the composition used to form the A layer, the cosmetic dissolution concentration of the obtained A layer is 2.9% by mass or less, or A The oleic acid mass increase rate of the layer is 10% by mass or less, and when the 0.5 mN load is applied for 10 seconds in the micro hardness tester, the maximum displacement amount in the thickness direction of the A layer is 1.0 to 3.0 ⁇ m, A laminate film having a creep displacement amount in the thickness direction of the A layer of 0.2 to 0.5 ⁇ m and a permanent displacement amount of the A layer in the thickness direction of 0.3 to 0.65 ⁇ m can be obtained.
- the content ratio of urethane (meth) acrylate B and urethane (meth) acrylate C is If it deviates from 70/30 to 30/70, it becomes difficult to achieve both self-healing properties and cosmetic resistance properties.
- composition of urethane (meth) acrylate B and urethane (meth) acrylate C is not particularly limited as long as it is a urethane (meth) acrylate containing a (poly) caprolactone segment in one of them.
- the cosmetic dissolution concentration of the A layer when the cosmetic is applied to the surface of the A layer and kept at 60 ° C. for 6 hours is 1.5% by mass or less, or oleic acid is applied to the surface of the A layer.
- the mass increase rate of the A layer when held at 60 ° C. for 1 hour is 3.0% by mass or less, and the maximum displacement in the thickness direction of the A layer when a 0.5 mN load is applied for 10 seconds in the microhardness meter measurement Is 1.2 to 3.0 ⁇ m, the creep displacement amount in the thickness direction of the A layer is 0.3 to 0.4 ⁇ m, and the permanent displacement amount in the thickness direction of the A layer is 0.4 to 0.6 ⁇ m.
- the urethane (meth) acrylate B in the composition used for forming the A layer has at least a (poly) caprolactone segment, and the urethane (meth) acrylate C has at least a (poly) alkylene glycol segment.
- Have Rukoto is required.
- it can achieve by making the physical property of B layer and C layer into the following range. (Physical properties of layer B) When the cosmetic is applied to the surface of the B layer and kept in a 60 ° C. atmosphere for 6 hours, the cosmetic dissolution concentration of the B layer is 2.9% by mass or less, or oleic acid is applied to the surface of the B layer at 60 ° C.
- the mass increase rate of the B layer is 5.0% by mass or less
- the maximum displacement in the thickness direction of the B layer is 1.2 to 3.0 ⁇ m
- the creep displacement in the thickness direction of the B layer The amount is 0.4 to 0.5 ⁇ m.
- the cosmetic dissolution concentration of the C layer is 1.4% by mass or less
- or oleic acid is applied to the surface of the C layer.
- the mass increase rate of the C layer is 3.0% by mass or less
- the maximum displacement in the thickness direction of the C layer is 1.2 to 3.0 ⁇ m
- the thickness direction of the C layer Of creep displacement of 0.1 to 0.25 ⁇ m.
- the above-mentioned laminated film is obtained by curing the composition used for forming the A layer, in which the content ratio of urethane (meth) acrylate B and urethane (meth) acrylate C is mixed as 70/30 to 30 to 70. It has excellent resistance to cosmetics and self-healing. Outside this range, it is not possible to achieve both self-healing properties and cosmetic resistance properties.
- the composition used to form layer A contains urethane (meth) acrylate B and urethane (meth) acrylate C, the urethane (meth) acrylate B has a (poly) caprolactone segment, and the urethane (meth) acrylate
- C has a (poly) alkylene glycol (meth) segment
- an A layer excellent in self-healing property and cosmetic resistance can be obtained. This is because the (poly) alkylene glycol segment with excellent cosmetic resistance is unevenly distributed on the surface during curing due to the difference in surface tension and intermolecular force, and the (poly) caprolactone segment with excellent self-healing properties is unevenly distributed in the inner layer. Is considered to be more prominent.
- the mass m of the (poly) alkylene glycol segment in the resin contained in the A layer and the mass n of the (poly) caprolactone segment in the resin contained in the A layer preferably satisfy 0.3n ⁇ m ⁇ 10n. More preferably, 0.3n ⁇ m ⁇ 5n is satisfied, and more preferably 0.65n ⁇ m ⁇ 1.20n.
- the mass m of the (poly) alkylene glycol segment in the resin contained in the A layer and the mass n of the (poly) caprolactone segment in the resin contained in the A layer satisfy 0.3n ⁇ m ⁇ 10n, the above-described curing is performed.
- the uneven distribution of each segment at the time occurs more remarkably, and it is possible to obtain a layer A further excellent in self-repairing property and cosmetic resistance.
- the mass m of the (poly) alkylene glycol segment in the resin contained in the A layer and the mass n of the (poly) caprolactone segment in the resin contained in the A layer do not satisfy 0.3n ⁇ m ⁇ 10n, The dispersibility of each segment at the time of curing increases, and uneven distribution may be weakened.
- the thickness of the A layer is preferably 10 to 30 ⁇ m in order to obtain a laminated film with good self-restoration and cosmetic resistance.
- the thickness of the A layer is preferably 10 to 30 ⁇ m in order to obtain a laminated film with good self-restoration and cosmetic resistance.
- the dynamic friction coefficient ( ⁇ k) of the A layer is preferably 0.45 or less.
- the dynamic friction coefficient ( ⁇ k) is 0.45 or less, the touch feeling when touched is excellent.
- the coefficient of dynamic friction ( ⁇ k) is 0.12 or more and 0.30 or less, not only the touch feeling is excellent, but also the touch feeling is excellent. What has more excellent scratch resistance can be obtained.
- a resin containing a (poly) siloxane segment or a resin containing a polydimethylsiloxane segment is 100% by mass of all components constituting the A layer. 1 to 5% by mass is preferable.
- the difference ( ⁇ s ⁇ k) between the static friction coefficient ( ⁇ s) and the dynamic friction coefficient ( ⁇ k) of the A layer is preferably 0.05 or less. In the case of minus 0.05 or less, that is, the dynamic friction coefficient is larger than the static friction coefficient. It is preferable that ( ⁇ s ⁇ k) is 0.05 or less because, for example, when this laminated film is applied to the surface of a sheet member that is touched directly with a pen or a finger, characters can be written smoothly.
- ( ⁇ s ⁇ k) is preferably 0.04 or less, and more preferably 0.03 or less.
- the lower limit of ( ⁇ s ⁇ k) is ⁇ 0.15, which is preferable because characters can be written smoothly.
- Examples of a method for adjusting ( ⁇ s ⁇ k) to 0.05 or less include the following methods.
- B) The self-repairing resin layer contains an organosilicon compound and / or a fluorine-based compound.
- the layer A of the laminated film of the present invention can be produced, for example, through a laminating step, a heating step, and an energy ray irradiation step described later in this order.
- Lamination process On at least one side of the base film, (1) a (poly) caprolactone segment and (2) a layer having a urethane bond (A layer) are laminated.
- stacking of the A layer to a base film can mention the method of apply
- a known coating method such as a gravure coating method, a micro gravure coating method, a die coating method, a reverse coating method, a knife coating method, or a bar coating method can be applied.
- the heating method in the heating step is preferably performed with hot air from the viewpoint of heating efficiency, and a known hot air dryer or a hot air furnace capable of continuous conveyance such as roll conveyance or floating can be applied.
- the heating temperature in the heating step is preferably 60 ° C. or higher, more preferably 80 ° C. or higher, whereby the solvent is sufficiently volatilized.
- the heating temperature is preferably 180 ° C. or lower, but in order to improve the adhesion to the base film, the heating is performed at a temperature of 120 ° C. or higher. It is preferable.
- the heating time is 1 minute or longer, preferably 2 minutes or longer, more preferably 3 minutes or longer. From the viewpoint of maintaining productivity, dimensional stability of the base film, and transparency, the heating time is desirably 5 minutes or less.
- the composition used for forming the A layer can be cured.
- Curing with energy rays is preferably electron beams (EB rays) or ultraviolet rays (UV rays) from the viewpoint of versatility.
- EB rays electron beams
- UV rays ultraviolet rays
- Examples of the ultraviolet lamp used when irradiating ultraviolet rays include a discharge lamp method, a flash method, a laser method, and an electrodeless lamp method.
- Case of UV cured using a high pressure mercury lamp is a discharge lamp type, illuminance of ultraviolet rays is 100 mW / cm 2 or more 3,000 mW / cm 2 or less.
- the illuminance of ultraviolet light is 200 mW / cm 2 or more and 1,000 mW / cm 2 or less.
- the ultraviolet illuminance is the irradiation intensity received per unit area, and changes depending on the lamp output, the emission spectral efficiency, the diameter of the light emitting bulb, the design of the reflector, and the light source distance to the irradiated object.
- the illuminance does not change depending on the conveyance speed.
- the UV integrated light amount is irradiation energy received per unit area, and is the total amount of photons reaching the surface.
- the integrated light quantity is inversely proportional to the irradiation speed passing under the light source, and is proportional to the number of irradiations and the number of lamps.
- the preferred use of the laminated film of the present invention is a decorative molding application applied to a housing such as a personal computer or a mobile phone, and a screen protection application such as a touch panel or an antireflection plate.
- the laminated film of the present invention can be formed into a molded body by applying a molding method such as injection molding, pressure molding, vacuum molding, thermoforming, press molding or the like.
- the present invention can be particularly suitably applied to uses that are heated to 80 ° C. to 180 ° C. during molding.
- the characteristic measurement method and effect evaluation method in the present invention are as follows.
- Thickness of A layer A cross section of the laminated film is obtained by freezing and cutting with a diamond knife of a microtome (manufactured by Nippon Microtome, RMS-50). The obtained cross section was magnified with an optical microscope to a magnification range of 100 to 300 times, and the layer thickness of the A layer was measured from an image photograph showing a clear interface between the base film and the A layer. In addition, the measurement was made into the average value of 3 samples.
- a laminated film is cut into 200 mm ⁇ 200 mm, and the mass of this film is defined as A (g).
- a Bakelite plate is fixed, and a 1: 1 mixture (by mass) of ATRIX “Hand Cream A” (NO413) manufactured by Kao Corporation and distilled water is applied to the A layer, and the surface is 95 mm ⁇ 95 mm with a thickness of 3 mm.
- Place a glass plate (at that time, make sure that air does not enter between the laminated film and the glass plate, and wipe off all cosmetics protruding from the glass plate). This was stored for 6 hours in a high temperature and high humidity oven in an atmosphere of 60 ° C. and 95 RH%.
- the amount of displacement in the thickness direction (maximum displacement amount) from when the load is applied to when it is unloaded, and the amount of displacement in the thickness direction (creep displacement when holding for 10 seconds after reaching 0.5 mN) Amount) and a displacement amount (permanent displacement amount) in the thickness direction when the load was unloaded to 0 mN after being held for 10 seconds.
- Each measurement was performed three times, and the average value was adopted.
- Apparatus Dynamic ultra micro hardness tester “DUH-201” (manufactured by Shimadzu Corporation)
- Working indenter Diamond regular triangular pyramid indenter (edge angle 115 °)
- Measurement mode 2
- Maximum load 0.5mN Holding time when 10 mN load is reached: 10 second load speed, unload speed: 0.1422 mN / s.
- Measurement length 100mm
- Output machine Automatic detection by computer
- Load converter capacity 2000g
- Sampling speed 2ms -Static friction coefficient: calculated from the load at the first peak point-Dynamic friction coefficient: calculated from the average load of the last 20 mm in the measured length of 100 mm.
- the samples to be measured were sequentially sampled with the elongation at a rate of 5% lower than the elongation at the time of stopping, and finally, the elongation was such that no cracks were visually observed. Cut out the thin film cross section of the cracked portion of the sample collected, observe the A layer at a magnification such that the thickness of the observed A layer is 30 mm or more on the observation screen of the transmission electron microscope, and the average thickness of the A layer When cracks of 50% or more occur, cracks are present (layer A is broken), and the elongation value of the sample having the lowest elongation among the cracked samples is defined as the fracture elongation. . Then, the same measurement was performed three times in total, and the average value of the breaking elongations was taken as the breaking elongation of the A layer.
- Material 50 parts by mass of toluene, 50 parts by mass of hexamethylene diisocyanate-modified isocyanurate (Takenate D-170N manufactured by Takeda Pharmaceutical Co., Ltd.), 76 parts by mass of (poly) caprolactone-modified hydroxyethyl acrylate (Placcel FA5 manufactured by Daicel Chemical Industries, Ltd.) , 0.02 part by mass of dibutyltin laurate and 0.02 part by mass of hydroquinone monomethyl ether were mixed and held at 70 ° C. for 5 hours. Thereafter, 79 parts by mass of toluene was added to obtain a urethane acrylate mixture having a solid content of 50% by mass.
- (Raw material 2) 50 parts by mass of toluene, 50 parts by mass of hexamethylene diisocyanate biuret-modified (Duranate 24A-90CX, manufactured by Asahi Kasei Corporation, nonvolatile content: 90% by mass, isocyanate content: 21.2% by mass), (poly) caprolactone-modified hydroxy 92 parts by mass of ethyl acrylate (Placcel FA2D manufactured by Daicel Chemical Industries, Ltd.), 0.02 parts by mass of dibutyltin laurate and 0.02 parts by mass of hydroquinone monomethyl ether were mixed and held at 70 ° C. for 5 hours. Thereafter, 82 parts by mass of toluene was added to obtain urethane acrylate having a solid content of 50% by mass. The number of repeating caprolactone units per acrylate monomer residue in this urethane acrylate is 2.
- Raw material 5 100 parts by mass of toluene, 50 parts by mass of methyl-2,6-diisocyanatohexanoate (LDI manufactured by Kyowa Hakko Kogyo Co., Ltd.) and 119 parts by mass of polycarbonate diol (Placcel CD-210HL manufactured by Daicel Chemical Industries, Ltd.) were mixed and heated to 40 ° C. The temperature was raised to and maintained for 8 hours.
- a urethane acrylate (raw material 6) was prepared in the same manner as in the raw material 6 except that the polyethylene glycol monoacrylate was changed to 53 parts by mass of a polyethylene glycol monoacrylate (hydroxyl value: 264 (mgKOH / g)) and the MEK of the reaction liquid was changed to 102 parts by mass.
- Raw material 7) was obtained.
- urethane acrylate (raw material 8) was prepared in the same manner as in raw material 6, except that polyethylene glycol monoacrylate was changed to 68 parts by weight of Bremer AE-200 (hydroxyl value: 205 (mgKOH / g)) and MEK of the reaction liquid was changed to 118 parts by weight.
- urethane acrylate (raw material 9) was the same as raw material 6 except that polyethylene glycol monoacrylate was changed to 142 parts of BLEMMER AE-400 (hydroxyl value: 98 (mg KOH / g)) and MEK of the reaction liquid was changed to 192 parts by weight.
- Example 14 It was obtained by dissolving 50 parts by mass of urethane acrylate (Sin Nakamura Chemical U5201) at 80 ° C. in 50 parts by mass of MEK.
- Example 1 50 parts by mass of raw material 1 being urethane (meth) acrylate B, 50 parts by mass of raw material 2 being urethane (meth) acrylate C, 10 parts by mass of monohydroxyethyl acrylate phthalate (M-5400 manufactured by Toagosei Co., Ltd.), 10 parts by mass of toluene and 3 parts by mass of a photoinitiator (Irgacure 184 manufactured by Ciba Specialty Chemicals) were mixed to prepare a radically polymerizable composition having a solid content of 50% by mass.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. (That is, this composition was coated on a 100 ⁇ m thick polyester base film (“Lumirror” U46, manufactured by Toray Industries, Inc.) with a bar coater so that the thickness of the dried layer was 30 ⁇ m. , Passed through a drying oven at 80 ° C. for 1 minute, and then cured while moving at a conveyor speed of 4 m / min in an ultraviolet (UV) drying oven with a high-pressure mercury lamp with an output of 400 mW / cm 2 to obtain a laminated film .) The results obtained are shown in Table 4.
- UV ultraviolet
- Example 2 50 parts by mass of raw material 3 which is urethane (meth) acrylate B, 50 parts by mass of raw material 4 which is urethane (meth) acrylate C, and 3 parts by mass of a photoinitiator (Irgacure 184 manufactured by Ciba Specialty Chemicals) are mixed to form a solid.
- a radically polymerizable composition having a content of 50% by mass was prepared.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 3 70 parts by mass of raw material 3 which is urethane (meth) acrylate B, 30 parts by mass of raw material 4 which is urethane (meth) acrylate C, and 3 parts by mass of photoinitiator (Irgacure 184 manufactured by Ciba Specialty Chemicals Co., Ltd.)
- a radically polymerizable composition having a content of 50% by mass was prepared.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 4 30 parts by mass of raw material 5 which is urethane (meth) acrylate B, 70 parts by mass of raw material 4 which is urethane (meth) acrylate C, and 3 parts by mass of a photoinitiator (Irgacure 184 manufactured by Ciba Specialty Chemicals) are mixed to form a solid A radically polymerizable composition having a content of 50% by mass was prepared. A layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 5 50 parts by mass of raw material 5 which is urethane (meth) acrylate B, 50 parts by mass of raw material 2 which is urethane (meth) acrylate C, and 3 parts by mass of a photoinitiator (Irgacure 184 manufactured by Ciba Specialty Chemicals) are mixed to form a solid.
- a radically polymerizable composition having a content of 50% by mass was prepared.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- a photopolymerization initiator Irgacure 184 manufactured by Ciba Geigy Co., Ltd.
- Examples 7 to 9 50 parts by mass of raw material 1 that is urethane (meth) acrylate B, 50 parts by mass of each of raw materials 7 to 9 that are urethane (meth) acrylate C, and 3 parts by mass of a photopolymerization initiator (Irgacure 184 manufactured by Ciba Geigy)
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 10 50 parts by mass of raw material 10 which is urethane (meth) acrylate B, 50 parts by mass of raw material 7 which is urethane (meth) acrylate C, and 3 parts by mass of a photopolymerization initiator (Irgacure 184 manufactured by Ciba Geigy Co., Ltd.) 50 mass% of radically polymerizable composition was obtained.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 11 47 parts by mass of raw material 1 which is urethane (meth) acrylate B, 47 parts by mass of raw material 7 which is urethane (meth) acrylate C, MEK dispersion of silica particles (manufactured by JGC Catalysts & Chemicals: Spherica slurry 120, average particle diameter) 5 parts by mass of 120 nm, SiO 2 concentration of 5% by mass) and 3 parts by mass of a photopolymerization initiator (Irgacure 184 manufactured by Ciba Geigy Co., Ltd.) were blended to obtain a radical polymerizable composition having a solid content of 100% by mass.
- a photopolymerization initiator Irgacure 184 manufactured by Ciba Geigy Co., Ltd.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 12 20 parts by mass of raw material 1 that is urethane (meth) acrylate B, 80 parts by mass of raw material 8 that is urethane (meth) acrylate C, and 3 parts by mass of a photopolymerization initiator (Irgacure 184, manufactured by Ciba Geigy Co., Ltd.) 50 mass% of radically polymerizable composition was obtained.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 13 20 parts by mass of raw material 1 which is urethane (meth) acrylate B, 80 parts by mass of raw material 9 which is urethane (meth) acrylate C, and 3 parts by mass of a photopolymerization initiator (Irgacure 184 manufactured by Ciba Geigy Co., Ltd.) 50 mass% of radically polymerizable composition was obtained.
- a photopolymerization initiator Irgacure 184 manufactured by Ciba Geigy Co., Ltd.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Example 14 10 parts by mass of raw material 1 which is urethane (meth) acrylate B, 90 parts by mass of raw material 9 which is urethane (meth) acrylate C, and 3 parts by mass of a photopolymerization initiator (Irgacure 184 manufactured by Ciba Geigy Co., Ltd.) are mixed. 50 mass% of radically polymerizable composition was obtained.
- a photopolymerization initiator Irgacure 184 manufactured by Ciba Geigy Co., Ltd.
- a layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. Table 4 shows the obtained results.
- Comparative Example 2 100 parts by mass of raw material 1 which is urethane (meth) acrylate B and 3 parts by mass of a photopolymerization initiator (Irgacure 184 manufactured by Ciba Geigy Co., Ltd.) were blended to obtain a radical polymerizable composition having a solid content of 50% by mass. A layer A was formed from the obtained composition by the same method as the formation of the B layer or the C layer to obtain a laminated film. The results obtained are shown in Table 5.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Laminated Bodies (AREA)
- Macromonomer-Based Addition Polymer (AREA)
Abstract
Description
A層に含まれる樹脂が、(1)(ポリ)カプロラクトンセグメント、及び、(2)ウレタン結合、を有しており、
微小硬度計測定において0.5mN荷重を10秒間加えたときの、A層の厚み方向の最大変位量が1.0~3.0μmであり、A層の厚み方向のクリープ変位量が0.2~0.5μmであり、荷重を0mNまで解放したときの、A層の厚み方向の永久変位量が0.3~0.65μmである積層フィルムである。
本発明において基材フィルムを構成する樹脂は、熱可塑性樹脂、熱硬化性樹脂のいずれでもよく、ホモ樹脂であってもよく、共重合または2種類以上のブレンドであってもよい。より好ましくは、基材フィルムを構成する樹脂は、成型性が良好であるため、熱可塑性樹脂である。
本発明では、基材フィルムを構成する樹脂が、基材フィルムの全成分100質量%において、ポリエステル樹脂を50質量%以上100質量%以下含む場合、基材フィルムをポリエステル基材フィルムという。
以下、基材フィルムの少なくとも片側に、A層を有する積層フィルムについて説明する。
本発明では、A層が、(1)(ポリ)カプロラクトンセグメントを有する。A層が(ポリ)カプロラクトンセグメントを有することで、A層に弾性回復性(自己治癒性)を賦与することができる。



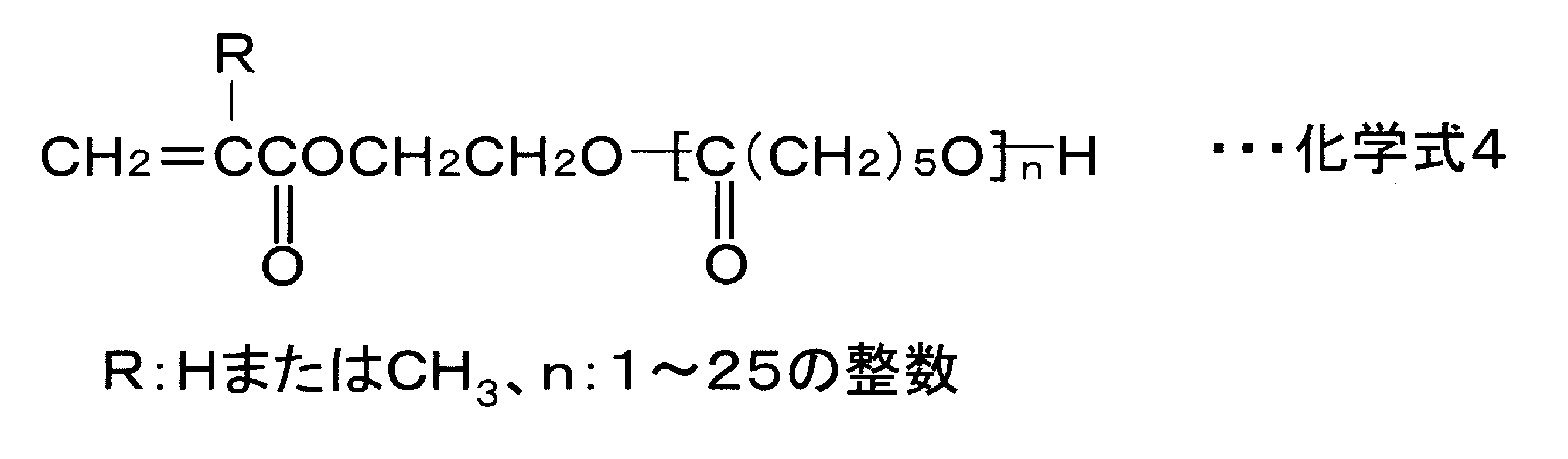
本発明では、A層に含まれる樹脂が(2)ウレタン結合を有する。
本発明では、A層が、(3)(ポリ)アルキレングリコールセグメントを有することが好ましい。本発明において、(ポリ)アルキレングリコールセグメントとは、以下の化学式で示されるセグメントを指す。

CH2=CRCOO(C2H4O)mH
(ポリ)プロピレングリコール(メタ)アクリレート:
CH2=CRCOO(CH2CH2CH2O)mH
この(ポリ)プロピレングリコール(メタ)アクリレートの水酸基は1級水酸基、即ち1級炭素に結合した水酸基である。
CH2=CRCOO(C4H8O)mH
但し、Rは水素(H)又はメチル基(-CH2)、mは2~11となる整数である。
<(ポリ)シロキサンセグメント>
本発明では、A層が(4)(ポリ)シロキサンセグメント及び/又はポリジメチルシロキサンセグメントを有することが好ましい。本発明において、(ポリ)シロキサンセグメントとは、以下の化学式で示されるセグメントを指す。なお、下記式においてRは、OHと炭素数1~8のアルキル基のいずれかであり、式中においてそれぞれを少なくとも1つ以上有する。

本発明において、ポリジメチルシロキサンセグメントとは、下記式で示されるセグメントを指す。



<他の添加剤>
A層を形成するために用いる組成物には、開始剤や硬化剤や触媒を含むことが好ましい。開始剤および触媒は、A層の硬化を促進するために用いられる。開始剤としては、塗料組成物をアニオン、カチオン、ラジカル反応等による重合、縮合または架橋反応を開始あるいは促進できるものが好ましい。
本発明の積層フィルムは、微小硬度計測定において0.5mN荷重を10秒間加えたときのA層の厚み方向の最大変位量が、1.0~3.0μmであり、A層の厚み方向のクリープ変位量が、0.2~0.5μmであり、荷重を0mNまで解放したときの、A層の厚み方向の永久変位量が0.3~0.65μmであることが重要である。
自己修復性の優れたウレタン(メタ)アクリレートBとは、ウレタン(メタ)アクリレートと光開始剤の混合物を、照度400mW/cm2の高圧水銀灯による紫外線で、厚み30μmに硬化させた層(以下、B層という)の物性が、次の範囲にあるウレタン(メタ)アクリレートを意味する。
耐化粧品性の優れたウレタン(メタ)アクリレートCとは、ウレタン(メタ)アクリレートと光開始剤の混合物を、照度400mW/cm2の高圧水銀灯による紫外線で、厚み30μmに硬化させた層(以下、C層という)の物性が、次の範囲にあるウレタン(メタ)アクリレートを意味する。
(B層の物性)
B層表面に化粧品を塗布して60℃雰囲気下に6時間保持したときのB層の化粧品溶解濃度が2.9質量%以下であるか、もしくはB層表面にオレイン酸を塗布して60℃で1時間保持したときのB層の質量増加率が5.0質量%以下であり、B層の厚み方向の最大変位量が、1.2~3.0μm、B層の厚み方向のクリープ変位量が、0.4~0.5μm。
(C層の物性)
C層表面に化粧品を塗布して60℃雰囲気下に6時間保持したときのC層の化粧品溶解濃度が化粧品溶解濃度1.4質量%以下であるか、もしくはC層表面にオレイン酸を塗布して60℃で1時間保持したときのC層の質量増加率が3.0質量%以下であり、C層の厚み方向の最大変位量が、1.2~3.0μm、C層の厚み方向のクリープ変位量が、0.1~0.25μm。
本発明の積層フィルムでは、自己修復性および耐化粧品性のよい積層フィルムを得るため、A層の厚みを、10~30μmとすることが好ましい。A層の厚みを、10~30μmとすることにより、自己修復効果があって、耐汚染性のよい積層フィルムとすることができる。さらに好ましくはA層の厚みを10~20μmとすることにより、A層の化粧品溶解濃度またはA層の質量増加率が少なくなり、耐汚染性が向上する。
本発明の積層フィルムは、A層の動摩擦係数(μk)が、0.45以下であることが好ましい。動摩擦係数(μk)が、0.45以下であると、触ったときの触り心地に優れる。また、動摩擦係数(μk)が0.12以上0.30以下であると、触り心地に優れるだけでなく。より耐傷性に優れたものが得られる。A層の動摩擦係数を低くして、0.45以下にするためには、(ポリ)シロキサンセグメントを含有する樹脂やポリジメチルシロキサンセグメントを含有する樹脂が、A層を構成する全成分100質量%において1~5質量%であるのが好ましい。
イ) 自己修復性樹脂層に、カプロラクトンとイソシアネートの反応によるウレタン結合を有する樹脂を含有させる。
ロ) 自己修復性樹脂層に、有機ケイ素化合物および/またはフッ素系化合物を含有させる。
ハ) 自己修復性樹脂層の自己修復性(傷の消失時間)を調整する。
ニ) 粒子を添加する。
本発明の積層フィルムのA層は、例えば、後述する積層工程、加熱工程、エネルギー線照射工程をこの順に経て、製造することができる。
基材フィルムの少なくとも片側に、(1)(ポリ)カプロラクトンセグメント、及び(2)ウレタン結合を有する層(A層)を積層する。基材フィルムへのA層の積層は、例えば、A層を形成する材料と、必要に応じて溶媒を含む塗液を、基材フィルムの少なくとも片側に、塗布する手法を挙げることができる。また、塗布方法としては、グラビアコート法、マイクログラビアコート法、ダイコート法、リバースコート法、ナイフコート法、バーコート法など公知の塗布方法を適用することができる。
加熱を行うことにより、層中の溶媒が揮発することができる。加熱工程における加熱方法は、加熱効率の点から熱風で行うのが好ましく、公知の熱風乾燥機、または、ロール搬送やフローティングなどの連続搬送が可能な熱風炉などを適用できる。加熱工程における加熱温度は60℃以上であることが好ましく、より好ましくは80℃以上で行うことにより、溶媒の揮発が十分となる。また、基材フィルムの熱収縮によるしわの発生などを考慮すると、加熱温度は180℃以下であることが好ましいが、基材フィルムとの密着性を向上させるためには120℃以上の温度で行うことが好ましい。
エネルギー線を照射する事により、A層を形成するために用いる組成物を硬化させることができる。エネルギー線による硬化は、汎用性の点から電子線(EB線)または紫外線(UV線)が好ましい。また、紫外線を照射する際に用いる紫外線ランプの種類としては、例えば、放電ランプ方式、フラッシュ方式、レーザー方式、無電極ランプ方式等が挙げられる。放電ランプ方式である高圧水銀灯を用いて紫外線硬化させる場合、紫外線の照度は100mW/cm2以上3,000mW/cm2以下である。自己治癒性と耐汚染性を両立させるためには紫外線の照度は200mW/cm2以上1,000mW/cm2以下で行うことが好ましい。ここで、紫外線照度とは、単位面積当たりに受ける照射強度で、ランプ出力、発光スペクトル効率、発光バルブの直径、反射鏡の設計および被照射物との光源距離によって変化する。しかし、搬送スピードによって照度は変化しない。また、紫外線積算光量とは単位面積当たりに受ける照射エネルギーで、その表面に到達するフォトンの総量である。積算光量は、光源下を通過する照射速度に反比例し、照射回数とランプ灯数に比例する。
本発明における特性の測定方法および効果の評価方法は以下のとおりである。
積層フィルムの断面を、凍結させた後にミクロトーム(日本ミクロトーム製、RMS-50)のダイヤモンドナイフにて切削して得る。得られた断面を光学顕微鏡にて、100倍~300倍の倍率の範囲まで拡大し、基材フィルムとA層との明瞭な界面が見られた像写真からA層の層厚みを測定した。なお測定は3サンプルの平均値とした。
積層フィルムを200mm×200mmに切り出し、このフィルムの質量をA(g)とする。ベークライト板に固定し、A層に花王(株)製アトリックス「ハンドクリームA」(NO413)と蒸留水の1:1(質量基準)混合液を塗布し、表面に95mm×95mmで厚み3mmのガラス板を載せる(その際、積層フィルムとガラス板の間には空気が入らないようにし、ガラス板からはみ出た化粧品は全て拭き取っておく)。これを60℃・95RH%の雰囲気下の高温高湿オーブンに6時間保存した。保存後、A層の化粧品をハイゼガーゼで透明になるまで拭き取った後、23℃の雰囲気下で24時間保存した。この後測定したフィルムの質量をB(g)とした。このときの化粧品溶解濃度(質量%)は以下の計算式より求めた。
(B-A)/(400×t×d)×100
t:A層厚み(cm)
d:A層の比重(g/cm3)。
積層フィルムを200mm×200mmに切り出し、この積層フィルムの質量をA(g)とした。ベークライト板に固定し、A層側の100mm×100mm長にオレイン酸を塗布した。塗布する際はプラスチックで囲いを作り、オレイン酸が流れ出ないようにした。これを60℃に加熱したオーブンに1時間保存した。保存後、ハイゼガーゼを用いて積層フィルムが透明になるまで拭き取りを行い、23℃の雰囲気下で24時間保存した。この後測定したフィルムの質量をB(g)とした。このときのオレイン酸による質量増加率は以下の計算式より求めた。測定はそれぞれ3回行い、その平均値を採用した。
(B-A)/(100×t×d)×100
t:A層厚み(cm)
d:A層の比重(g/cm3)。
積層フィルムからA層の切片を片刃ナイフで切り出し、臭化ナトリム水溶液を媒体とした密度勾配管法(JIS K 7112 JISハンドブック(1999年度版))に従い測定した。この時、測定はn数5で行い、その平均値を採用した。
平滑な金属板(ダイス鋼:SKD-11)に、東レ・ダウコーニング社製「ハイバキュームグリース」を1g塗布し、それに積層フィルムの基材フィルム側を塗布部分に貼り付け、表面に濾紙を挟んでハンドプレス機で空気が噛まないようにプレスした。このような方法で得られた静地された試料に対し、正三角錐を用いて押し込み負荷/除荷試験を行い、加重-押し込み深さ線図(図1参照)を取得した。
この線図から、荷重を加えてから除荷するまでの厚み方向の変位量(最大変位量)と、0.5mNに達してから10秒間保持し続けたときの厚み方向の変位量(クリープ変位量)と、10秒間保持してから0mNまで除荷した時の厚み方向の変位量(永久変位量)を求めた。測定はそれぞれ3回行い、その平均値を採用した。
装置:ダイナミック超微小硬度計「DUH-201」(島津製作所製)
使用圧子:ダイヤモンド製正三角錐圧子(稜間角115°)
測定モード:2
最大荷重:0.5mN
10mN荷重に達したときの保持時間:10秒
荷重速度、除荷速度:0.1422mN/s。
5cm角に切り出した試料に花王(株)製アトリックス「ハンドクリームA」(NO413)を0.5g塗布し、温度60℃、相対湿度95%の雰囲気下で6時間放置後、25℃相対湿度65%の雰囲気下で30分間放置し、表面をガーゼできれいに拭き取る。温度25℃、相対湿度65%の雰囲気下で24時間放置後、表面の状態を観察し、下記の基準に則り判定を行った。
○(優):白斑の発生なし。
●(良):白斑の発生がほとんどなし。
△(可):白斑が発生するが、拭き取ればきれいになる。
×(不可):白斑が発生する。拭き取っても温度25℃、相対湿度65%の雰囲気下で24時間放置後に再度発生する。
A層表面を、真鍮ブラシ(TRUSCO製)で、500gの荷重で水平に引っ掻いたときの傷の回復状態を、下記の基準に則り目視で判定を行った。
◎(優):全ての傷が3秒以内に回復する。
○(良): 全ての傷が4~10秒以内に回復する。
●(可):全ての傷が11~30秒以内に回復する。
×(不可):その他(全ての傷の回復が31秒以上かかるか、回復しない傷が存在する)。
23℃、50%(相対湿度)の雰囲気下で、先端形状が半径0.8mmRの半球状のポリアセタール樹脂製タッチペン(スタイラス製)を用いて、荷重200g、速度100mm/secでシート部材表面を移動させたときの動摩擦係数(μk)を、新東科学(株)製の表面性測定機(商品名「トライボギア」、タイプ「14FW」)にて測定した。測定はそれぞれ3回行い、その平均値を採用した。
<測定条件>
・測定長さ;100mm
・出力機;コンピューターに自動検出
・荷重変換器容量;2000g
・サンプリング速度;2ms
・静摩擦係数;最初のピーク点の荷重から計算
・動摩擦係数:測定長さ100mmの中の最後の20mmの平均荷重から計算。
積層フィルムを10mm幅×200mm長に切り出し、長手方向にチャックで把持してインストロン型引っ張り試験機(インストロン社製超精密材料試験機MODEL5848)にて引っ張り速度100mm/分で伸張した。この時の測定雰囲気は23℃・65RH%である。伸張する際に、伸張中のサンプルを観察しておき、目視でクラック(亀裂)が生じたら停止する(停止するときの伸度は5の整数となるように調整する)。次に測定するサンプルは、停止時の伸度より、5%単位で伸張伸度を低くしていったサンプルを順次採取し、最終的に目視にてクラックが入らなくなる伸度まで行った。
採取したサンプルのクラック部分の薄膜断面を切り出し、観察するA層の厚みが、透過型電子顕微鏡の観察画面上において、30mm以上になるような倍率でA層を観察し、A層の平均厚みの50%以上のクラックが発生している場合をクラック有り(A層の破壊有り)として、クラック有りとされたサンプルの中で、最も低い伸度を有するサンプルの伸度値を破壊伸度とした。
そして、同一の測定を計3回行い、それらの破壊伸度の平均値をA層の破壊伸度とした。
ミクロトームを用いて積層フィルムの断面を切り出したサンプルについて、電子顕微鏡観察により求めた。すなわち、透過型電子顕微鏡H-7100FA型((株)日立製作所製)を用い、加速電圧75kVでフィルムの断面を20,000倍に拡大観察し、断面写真を撮影、層構成および各層厚みを測定した。尚、場合によっては、コントラストを高く得るために、公知のRuO4やOsO4などを使用した染色技術を用いても良い。
下記に示す原料1~10、原料12~14の各組成物100質量部に対し、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。この組成物を厚み100μmのポリエステル基材フィルム(東レ(株)製、“ルミラー”U46)上に、乾燥後の層の膜厚が30μmとなるようバーコーターで塗工した。それを、80℃の乾燥炉中に通して1分間乾燥処理し、次いで出力400mW/cm2の高圧水銀灯による紫外線(UV)乾燥炉によりコンベアースピード4m/分で移動させながら硬化させ、積層フィルムを得た。得られた結果を表1および2に示す。
(原料1)
トルエン50質量部、ヘキサメチレンジイソシアネートのイソシアヌレート変性タイプ(武田薬品工業株式会社製タケネートD-170N)50質量部、(ポリ)カプロラクトン変性ヒドロキシエチルアクリレート(ダイセル化学工業株式会社製プラクセルFA5)76質量部、ジブチル錫ラウレート0.02質量部、及びハイドロキノンモノメチルエーテル0.02質量部を混合し、70℃で5時間保持した。その後、トルエン79質量部を加えて固形分50質量%のウレタンアクリレートの混合物を得た。
トルエン50質量部、ヘキサメチレンジイソシアネートのビウレット変性タイプ(旭化成工業株式会社製デュラネート24A-90CX、不揮発分:90質量%、イソシアネート含有量:21.2質量%)50質量部、(ポリ)カプロラクトン変性ヒドロキシエチルアクリレート(ダイセル化学工業株式会社製プラクセルFA2D)92質量部、ジブチル錫ラウレート0.02質量部、及びハイドロキノンモノメチルエーテル0.02質量部を混合し、70℃で5時間保持した。その後、トルエン82質量部を加えて固形分50質量%のウレタンアクリレートを得た。なお、このウレタンアクリレートにおけるアクリレートモノマー残基当たりのカプロラクトン単位の繰り返し数は2である。
トルエン50質量部、ヘキサメチレンジイソシアネートのイソシアヌレート変性タイプ(武田薬品工業株式会社製タケネートD-170N)25質量部、(ポリ)カプロラクトン変性ヒドロキシエチルアクリレート(ダイセル化学工業株式会社製プラクセルFA10)162.8質量部、ジブチル錫ラウレート0.02質量部、及びハイドロキノンモノメチルエーテル0.02質量部を混合し、70℃で5時間保持した。その後、トルエン137.8部を加えて固形分50質量%のウレタンアクリレートを得た。なお、このウレタンアクリレートにおけるアクリレートモノマー残基当たりのカプロラクトン単位の繰り返し数は10である。
トルエン50質量部、ヘキサメチレンジイソシアネートのイソシアヌレート変性タイプ(武田薬品工業株式会社製タケネートD-170N)50質量部、(ポリ)カプロラクトン変性ヒドロキシエチルアクリレート(ダイセル化学工業株式会社製プラクセルFA3)114質量部、ジブチル錫ラウレート0.02質量部、及びハイドロキノンモノメチルエーテル0.02質量部を加え、70℃で3時間保持した。その後、トルエン118.2質量部を加えて固形分50質量%のウレタンアクリレートを得た。なお、このウレタンアクリレートにおけるアクリレートモノマー残基当たりのカプロラクトン単位の繰り返し数は3である。
トルエン100質量部、メチル-2,6-ジイソシアネートヘキサノエート(協和発酵工業社製LDI)50質量部及びポリカーボネートジオール(ダイセル化学工業社製プラクセルCD-210HL)119質量部を混合し、40℃にまで昇温して8時間保持した。それから、2-ヒドロキシエチルアクリレート(共栄社化学社製ライトエステルHOA)28質量部、ジペンタエリストールヘキサアクリレート(東亞合成社製M-400)5部、ハイドロキノンモノメチルエーテル0.02質量部を加えて70℃で30分間保持した後、ジブチル錫ラウレート0.02質量部を加えて80℃で6時間保持した。そして、最後にトルエン97質量部を加えて固形分50質量%のウレタンアクリレートを得た。
ヘキサメチレンジイソシアネートのイソシアヌレート変性体(武田薬品工業製タケネートD-170N、イソシアネート基含有量:20.9質量%)50質量部、ポリエチレングリコールモノアクリレート(日本油脂製ブレンマーAE-90、水酸基価:332(mgKOH/g))42質量部、ジブチルスズラウレート0.02質量部及びハイドロキノンモノメチルエーテル0.02質量部を仕込んだ。そして、70℃で5時間保持して反応を行った。反応終了後、反応液にメチルエチルケトン(MEK)92質量部を加え、固形分50質量%のウレタンアクリレートを得た。
原料6において、ポリエチレングリコールモノアクリレートをブレンマーAE-150(水酸基価:264(mgKOH/g))53質量部、反応液のMEKを102質量部に変更した以外は原料6と同様にしてウレタンアクリレート(原料7)を得た。
また、ポリエチレングリコールモノアクリレートをブレンマーAE-200(水酸基価:205(mgKOH/g))68質量部、反応液のMEKを118質量部に変更した以外は原料6と同様にしてウレタンアクリレート(原料8)を得た。
また、ポリエチレングリコールモノアクリレートをブレンマーAE-400(水酸基価:98(mgKOH/g))142部、反応液のMEKを192質量部に変更した以外は原料6と同様にしてウレタンアクリレート(原料9)を得た。
トルエン50質量部、ヘキサメチレンジイソシアネートのイソシアヌレート変性タイプ(武田薬品工業株式会社製タケネートD-170N)50質量部、(ポリ)カプロラクトン変性ヒドロキシエチルアクリレート(ダイセル化学工業株式会社製プラクセルFA5)70質量部、ポリジメチルシロキサン(信越化学工業社製X-22-160AS)8部ジブチル錫ラウレート0.02質量部、及びハイドロキノンモノメチルエーテル0.02質量部を混合し、70℃で5時間保持した。その後、トルエン79質量部を加えて固形分50質量%のウレタンアクリレートの混合物を得た。
<ポリシロキサン(a)の合成>
攪拌機、温度計、コンデンサ及び窒素ガス導入管を備えた500mlのフラスコにエタノール106質量部、メチルトリメトキシシラン270質量部、γ-メタクリロキシプロピルメチルジメトキシシラン23質量部、脱イオン水100質量部、1質量%塩酸1質量部及びハイドロキノンモノメチルエーテル0.1質量部を仕込み、80℃で3時間反応させ、ポリシロキサン(a)を合成した。これをメチルイソブチルケトンで50質量%に調整した。
ポリシロキサン(a)の合成と同様の装置を用い、トルエン50質量部、およびメチルイソブチルケトン50質量部、ポリジメチルシロキサン系高分子重合開始剤(和光純薬株式会社製、VPS-0501)20質量部、メタクリル酸メチル18質量部、メタクリル酸ブチル38質量部、2-ヒドロキシエチルメタクリレート23質量部、メタクリル酸1重量部および1-チオグリセリン0.5質量部を仕込み、180℃で8時間反応させてポリジメチルシロキサン系ブロック共重合体(a)を得た。得られたブロック共重合体は、固形分50質量%であった。
原料11の組成物は、エージング工程後の層の厚みが30μmとなるようにワイヤーバーを用いて塗布した。厚み100μmのポリエステル基材フィルム(東レ(株)製、“ルミラー”U46)上に、乾燥後の層の膜厚が30μmとなるようバーコーターで塗工した。塗布後、160℃で2分間、熱風乾燥機で加熱した(加熱工程)。その後、40℃で2日間加熱(エージング)を行い(エージング工程)、積層フィルムを得た。得られた結果を表3に示す。
トルエン50質量部、ヘキサメチレンジイソシアネートのイソシアヌレート変性タイプ(武田薬品工業株式会社製タケネートD-170N)34質量部、(ポリ)カプロラクトン変性ヒドロキシエチルアクリレート(ダイセル化学工業株式会社製プラクセルFA10)57質量部、ポリカプロラクトン変性ヒドロキシエチルアクリレート(ダイセル化学工業株式会社製プラクセルFA3)57質量部、ジブチル錫ラウレート0.02質量部、及びハイドロキノンモノメチルエーテル0.02質量部を混合し、70℃で5時間保持した。その後、トルエン137.8質量部を加えて固形分50質量%のウレタンアクリレートを得た。
1,3-ビスイソシアネートメチルシクロヘキサンを50質量部、ヒドロキシアルキルアクリレートを100質量部、ジブチル錫ラウレート0.05質量部、ハイドロキノン2質量部を添加し、70℃で3時間保持した。その後85℃で2時間の熟成を行い、ウレタンアクリレートを得た。
ウレタンアクリレート(新中村化学製 U5201)50質量部を80℃でMEK50質量部に溶解して得た。
「実施例」
(実施例1)
ウレタン(メタ)アクリレートBである原料1を50質量部、ウレタン(メタ)アクリレートCである原料2を50質量部、フタル酸モノヒドロキシエチルアクリレート(東亞合成株式会社製M-5400)10質量部、トルエン10質量部、及び光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。(つまり、この組成物を厚み100μmのポリエステル基材フィルム(東レ(株)製、“ルミラー”U46)上に、乾燥後の層の膜厚が30μmとなるようバーコーターで塗工した。それを、80℃の乾燥炉中に通して1分間乾燥処理し、次いで出力400mW/cm2の高圧水銀灯による紫外線(UV)乾燥炉によりコンベアースピード4m/分で移動させながら硬化させ、積層フィルムを得た。)得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料3を50質量部、ウレタン(メタ)アクリレートCである原料4を50質量部、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料3を70質量部、ウレタン(メタ)アクリレートCである原料4を30質量部、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料5を30質量部、ウレタン(メタ)アクリレートCである原料4を70質量部、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料5を50質量部、ウレタン(メタ)アクリレートCである原料2を50質量部、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料1を50質量部、ウレタン(メタ)アクリレートCである原料6を50質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。
また、このラジカル重合性組成物中の(ポリ)アルキレングリコールセグメントの質量mと(ポリ)カプロラクトンセグメントの質量nの比率はm=0.46nである。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料1を50質量部、ウレタン(メタ)アクリレートCである原料7~9をそれぞれ50質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。
また、このラジカル重合性組成物中の(ポリ)アルキレングリコールセグメントの質量mと(ポリ)カプロラクトンセグメントの質量nの比率は、それぞれm=0.67n、m=0.88n、m=1.6nである。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料10を50質量部、ウレタン(メタ)アクリレートCである原料7を50質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。
また、このラジカル重合性組成物中の(1)(ポリ)アルキレングリコールセグメントの質量mと(3)(ポリ)カプロラクトンセグメントの質量nの比率はm=0.65nである。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料1を47質量部、ウレタン(メタ)アクリレートCである原料7を47質量部、シリカ粒子のMEK分散液(日揮触媒化成製:スフェリカスラリー120、平均粒子径120nm、SiO2濃度5質量%)を5質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分100質量%のラジカル重合性組成物を得た。
また、このラジカル重合性組成物中の(1)(ポリ)アルキレングリコールセグメントの質量mと(3)(ポリ)カプロラクトンセグメントの質量nの比率はm=0.67nである。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料1を20質量部、ウレタン(メタ)アクリレートCである原料8を80質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。
また、このラジカル重合性組成物中の(ポリ)アルキレングリコールセグメントの質量mと(ポリ)カプロラクトンセグメントの質量nの比率はm=3.5nである。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表4に示す。
ウレタン(メタ)アクリレートBである原料1を20質量部、ウレタン(メタ)アクリレートCである原料9を80質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。
ウレタン(メタ)アクリレートBである原料1を10質量部、ウレタン(メタ)アクリレートCである原料9を90質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。
ポリジメチルシロキサン系ブロック共重合体(a)75質量部、ポリシロキサン(a)10質量部および水酸基を有する(ポリ)カプロラクトントリオール(ダイセル化学工業(株)製 プラクセル308、重量平均分子量850)15質量部を配合(混合)し、原料11を得た。原料11を100質量部、ヘキサメチレンジイソシアネートのイソシアヌレート体(武田薬品工業(株)製、タケネートD-170N)を25質量部添加し、さらにメチルエチルケトンを用いて希釈し、固形分40質量%の熱硬化性組成物を得た。得られた組成物を、上記原料11の組成物から形成される層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料1を100質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料3を100質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料12を100質量部、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料3を50質量部、ウレタン(メタ)アクリレートCである原料1を50質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料5を80質量部、ウレタン(メタ)アクリレートCである原料4を20質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分20質量%のラジカル重合性組成物を得た。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料5を50質量部、ウレタン(メタ)アクリレートCである原料3を50質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料13を100質量部、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料14を100質量部、光開始剤(チバスペシャルティケミカルズ社製イルガキュア184)3質量部を混合し、固形分50質量%のラジカル重合性組成物を調製した。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
ウレタン(メタ)アクリレートBである原料5を50質量部、ウレタン(メタ)アクリレートCである原料14を50質量部、光重合開始剤(チバガイギー社製イルガキュア184)3質量部を配合し、固形分50質量%のラジカル重合性組成物を得た。得られた組成物を、上記B層またはC層の形成と同様の方法によってA層を形成し、積層フィルムを得た。得られた結果を表5に示す。
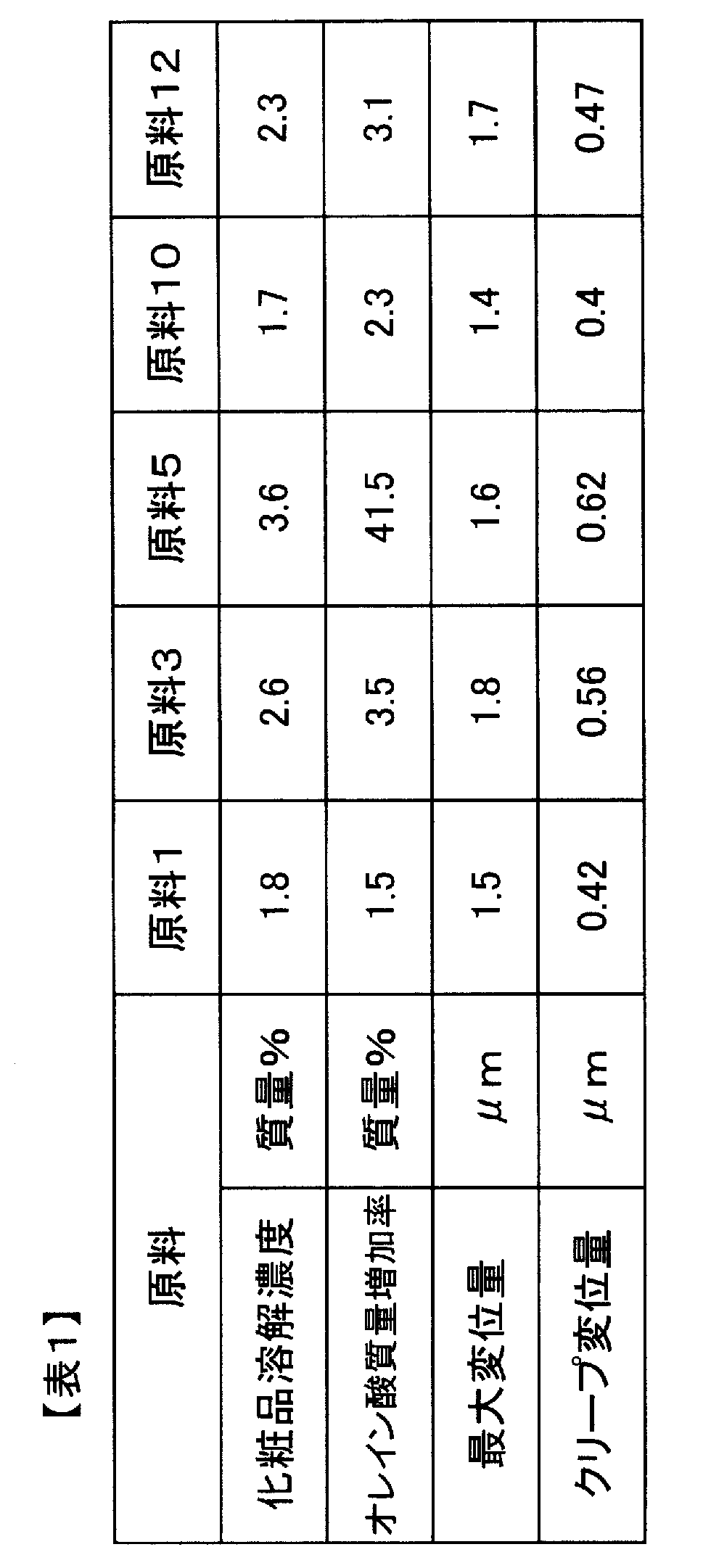
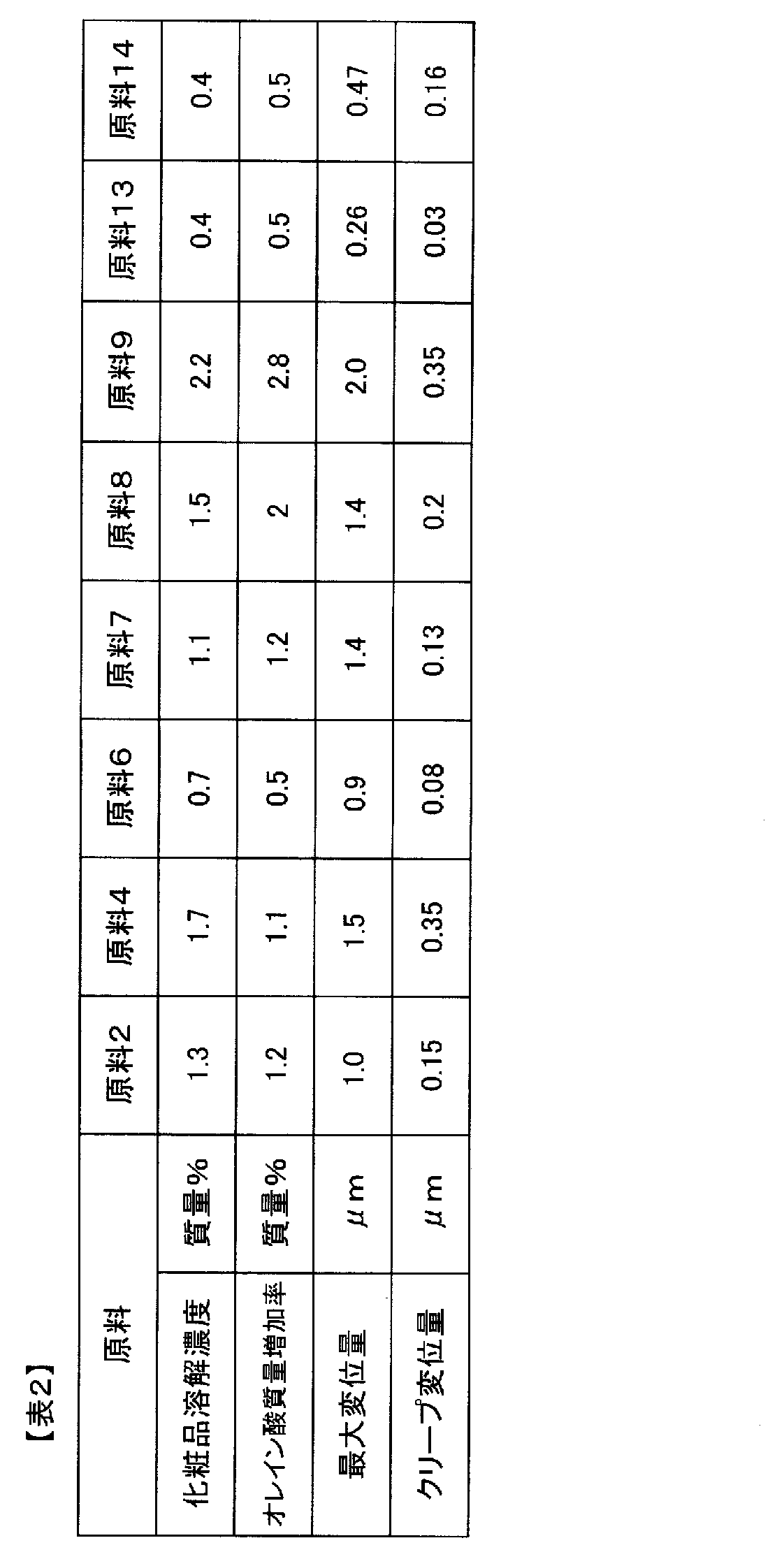
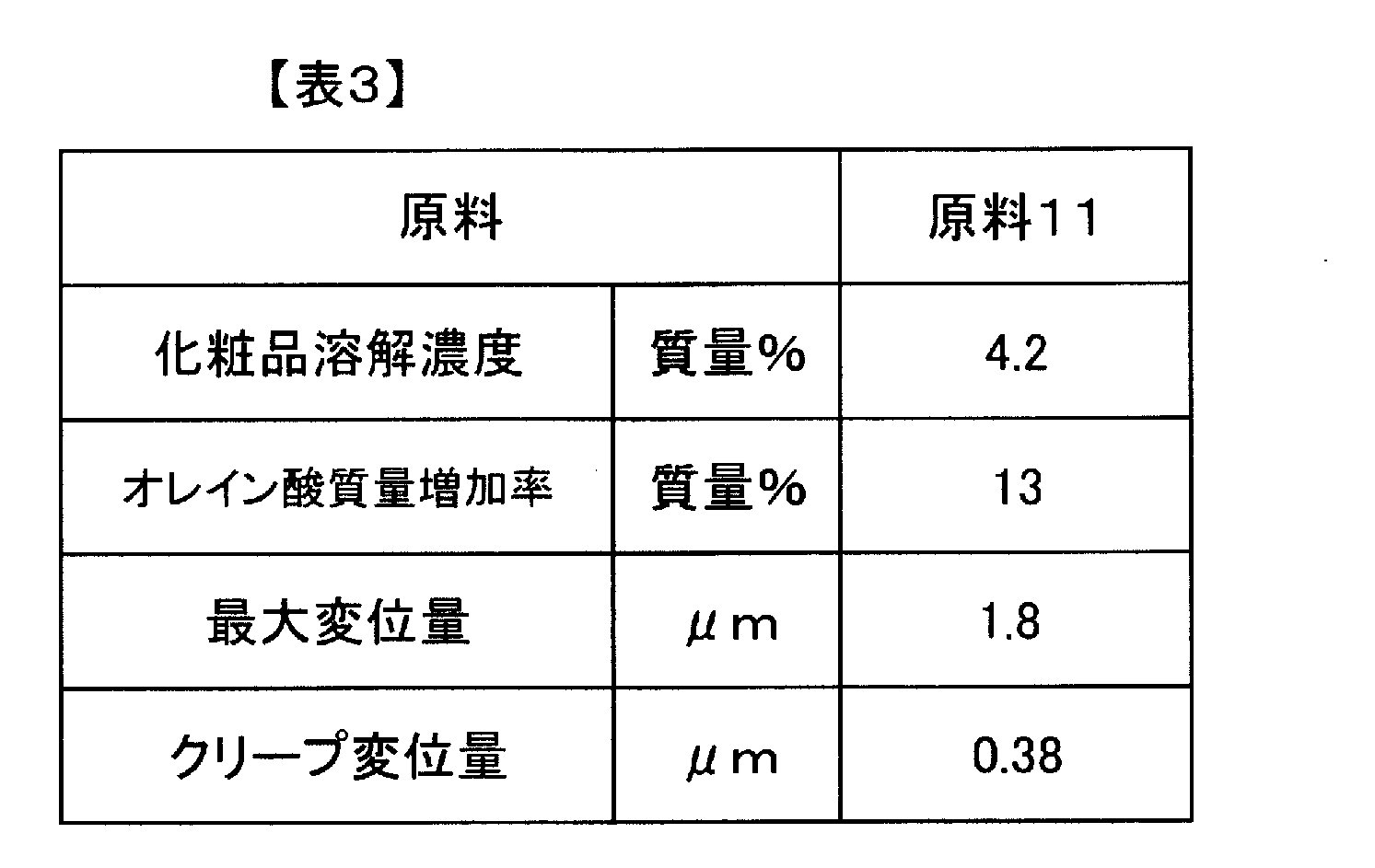
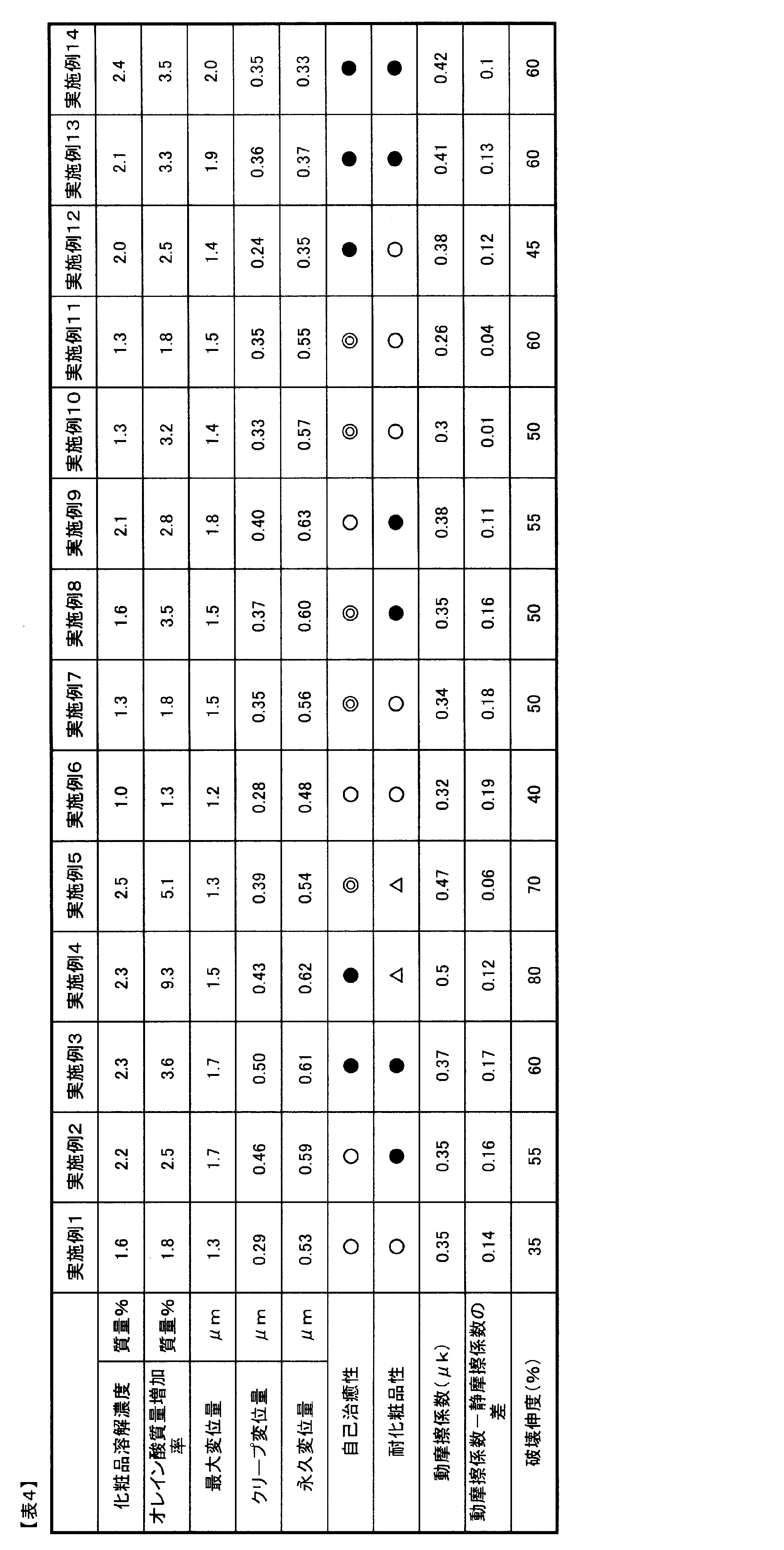

2 保持
3 除荷
a 最大変位量
b クリープ変位量
c 永久変位量
Claims (9)
- 基材フィルムの少なくとも片側に、A層を有する積層フィルムであって、
A層に含まれる樹脂が、(1)(ポリ)カプロラクトンセグメント、及び、(2)ウレタン結合、を有しており、
微小硬度計測定において
0.5mN荷重を10秒間加えたときの、A層の厚み方向の最大変位量が1.0~3.0μmであり、A層の厚み方向のクリープ変位量が0.2~0.5μmであり、
荷重を0mNまで解放したときの、A層の厚み方向の永久変位量が0.3~0.65μmである積層フィルム。 - A層に含まれる樹脂が、(3)(ポリ)アルキレングリコールセグメントを有していることを特徴とする請求項1に記載の積層フィルム。
- A層が、ウレタン(メタ)アクリレートBとウレタン(メタ)アクリレートCとを含んだ組成物を、活性エネルギー線で硬化させて得られるA層であり、
前記ウレタン(メタ)アクリレートBが、少なくとも(ポリ)カプロラクトンセグメントを有しており、
前記ウレタン(メタ)アクリレートCが、少なくとも(ポリ)アルキレングリコールセグメントを有していることを特徴とする、請求項1または2に記載の積層フィルム。 - A層の破壊伸度が50%以上である請求項1から3のいずれかに記載の積層フィルム。
- A層表面に化粧品を塗布して60℃雰囲気下に6時間保持したときのA層の化粧品溶解濃度が2.9質量%以下であることを特徴とする請求項1から4のいずれかに記載の積層フィルム。
- A層表面にオレイン酸を塗布して60℃雰囲気下に1時間保持したときのA層の質量増加率が10質量%以下であることを特徴とする請求項1から5のいずれかに記載の積層フィルム。
- A層に含まれる樹脂が、(4)(ポリ)シロキサンセグメント及び/またはポリジメチルシロキサンセグメントを有していることを特徴とする請求項1から6のいずれかに記載の積層フィルム。
- A層の動摩擦係数(μk)が、0.45以下である、請求項1から7のいずれかに記載の積層フィルム。
- A層の静摩擦係数(μs)と動摩擦係数(μk)の差(μs-μk)が、0.05以下であることを特徴とする請求項1から8のいずれかに記載の積層フィルム。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012556727A JP6236783B2 (ja) | 2011-12-21 | 2012-12-14 | 積層フィルム |
| EP12859078.3A EP2796291B1 (en) | 2011-12-21 | 2012-12-14 | Laminated film |
| KR1020147009403A KR102048801B1 (ko) | 2011-12-21 | 2012-12-14 | 적층 필름 |
| CN201280059267.7A CN103958199B (zh) | 2011-12-21 | 2012-12-14 | 叠层膜 |
| US14/358,768 US9469789B2 (en) | 2011-12-21 | 2012-12-14 | Laminated film |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011279465 | 2011-12-21 | ||
| JP2011-279465 | 2011-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013094532A1 true WO2013094532A1 (ja) | 2013-06-27 |
Family
ID=48668423
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/082509 WO2013094532A1 (ja) | 2011-12-21 | 2012-12-14 | 積層フィルム |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9469789B2 (ja) |
| EP (1) | EP2796291B1 (ja) |
| JP (1) | JP6236783B2 (ja) |
| KR (1) | KR102048801B1 (ja) |
| CN (1) | CN103958199B (ja) |
| TW (1) | TWI673166B (ja) |
| WO (1) | WO2013094532A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016078458A (ja) * | 2014-10-20 | 2016-05-16 | 東レ株式会社 | 積層フィルム |
| JP2016169351A (ja) * | 2015-03-16 | 2016-09-23 | 日油株式会社 | 硬化性樹脂組成物および積層構造体 |
| JPWO2015046049A1 (ja) * | 2013-09-24 | 2017-03-09 | 東レ株式会社 | 積層フィルム |
| JP2017088680A (ja) * | 2015-11-05 | 2017-05-25 | 日油株式会社 | 活性エネルギー線硬化型樹脂組成物 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015115227A1 (ja) * | 2014-01-31 | 2015-08-06 | 東レ株式会社 | 積層フィルム |
| GB201508727D0 (en) | 2015-05-21 | 2015-07-01 | Croda Int Plc | Polyurethane |
| ES2664381T3 (es) | 2015-06-10 | 2018-04-19 | Ube Corporation Europe, S.A.U. | Polímeros de poliuretano autorreparables |
| CN108665810A (zh) * | 2018-05-31 | 2018-10-16 | 昆山国显光电有限公司 | 金属线、显示屏、金属线制作方法和金属线修复方法 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0681822B2 (ja) * | 1986-10-22 | 1994-10-19 | ロード コーポレーション | 放射線硬化被覆組成物 |
| JP2001002744A (ja) | 1999-06-21 | 2001-01-09 | Natoko Kk | 紫外線硬化性組成物及び表面機能材料 |
| JP2003014906A (ja) * | 2001-07-04 | 2003-01-15 | Natoko Kk | 反射防止フィルム |
| JP2004244426A (ja) * | 2002-02-01 | 2004-09-02 | Natoko Kk | 活性エネルギー線硬化性組成物及びその用途 |
| JP2005162908A (ja) * | 2003-12-03 | 2005-06-23 | Natoko Kk | 活性エネルギー線硬化性ウレタン(メタ)アクリレート、それを含有する活性エネルギー線硬化性組成物及びそれらを用いた機能性部材 |
| JP3676260B2 (ja) | 2000-12-28 | 2005-07-27 | ナトコ株式会社 | 活性エネルギー線硬化性ウレタン(メタ)アクリレート及び活性エネルギー線硬化性組成物並びにそれらの用途 |
| JP2006137780A (ja) * | 2004-11-10 | 2006-06-01 | Dainippon Printing Co Ltd | 成形シート |
| JP2007009218A (ja) * | 2006-08-22 | 2007-01-18 | Natoko Kk | 塗料組成物 |
| JP3926461B2 (ja) | 1998-02-13 | 2007-06-06 | ナトコ株式会社 | 塗料組成物 |
| WO2011136042A1 (ja) | 2010-04-27 | 2011-11-03 | 東レ株式会社 | 積層フィルムおよび成型体 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4227979A (en) * | 1977-10-05 | 1980-10-14 | Ppg Industries, Inc. | Radiation-curable coating compositions containing amide acrylate compounds |
| US4762887A (en) * | 1987-01-15 | 1988-08-09 | Wacker Silicones Corporation | Process for preparing acrylate-functional organopolysiloxane-urethane copolymers |
| JP4142330B2 (ja) | 2002-04-12 | 2008-09-03 | ナトコ株式会社 | レンズシート |
| JP2006138879A (ja) * | 2004-11-10 | 2006-06-01 | Dainippon Printing Co Ltd | 反射防止フィルタ及びこれを用いたディスプレイ |
| JP2008106253A (ja) * | 2006-09-29 | 2008-05-08 | Daisan Kogyo Kk | コンベア用潤滑剤 |
| EP2089445A1 (de) * | 2006-10-09 | 2009-08-19 | Basf Se | Strahlungshärtbare verbindungen |
| US20080085947A1 (en) * | 2006-10-10 | 2008-04-10 | Ward Jeanette L | Radiation curable matrix composition |
| CN101970210B (zh) * | 2008-03-14 | 2013-09-04 | 东丽株式会社 | 在表面具有微细的凹凸图案的膜的制造方法和制造装置 |
| CN102076498B (zh) * | 2008-04-30 | 2014-12-17 | 东洋纺织株式会社 | 粘接性改性基材膜和硬涂膜 |
| JP4766122B2 (ja) * | 2009-02-12 | 2011-09-07 | 東洋紡績株式会社 | 成型用ハードコートフィルム |
| JP4623239B1 (ja) * | 2009-09-29 | 2011-02-02 | 東洋紡績株式会社 | 積層フィルム |
| EP2711182B1 (en) * | 2011-05-16 | 2021-06-23 | Toray Industries, Inc. | Laminated film and molded body |
-
2012
- 2012-12-14 CN CN201280059267.7A patent/CN103958199B/zh active Active
- 2012-12-14 US US14/358,768 patent/US9469789B2/en active Active
- 2012-12-14 KR KR1020147009403A patent/KR102048801B1/ko active IP Right Grant
- 2012-12-14 WO PCT/JP2012/082509 patent/WO2013094532A1/ja active Application Filing
- 2012-12-14 JP JP2012556727A patent/JP6236783B2/ja active Active
- 2012-12-14 EP EP12859078.3A patent/EP2796291B1/en active Active
- 2012-12-20 TW TW101148561A patent/TWI673166B/zh active
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0681822B2 (ja) * | 1986-10-22 | 1994-10-19 | ロード コーポレーション | 放射線硬化被覆組成物 |
| JP3926461B2 (ja) | 1998-02-13 | 2007-06-06 | ナトコ株式会社 | 塗料組成物 |
| JP2001002744A (ja) | 1999-06-21 | 2001-01-09 | Natoko Kk | 紫外線硬化性組成物及び表面機能材料 |
| JP3676260B2 (ja) | 2000-12-28 | 2005-07-27 | ナトコ株式会社 | 活性エネルギー線硬化性ウレタン(メタ)アクリレート及び活性エネルギー線硬化性組成物並びにそれらの用途 |
| JP2003014906A (ja) * | 2001-07-04 | 2003-01-15 | Natoko Kk | 反射防止フィルム |
| JP2004244426A (ja) * | 2002-02-01 | 2004-09-02 | Natoko Kk | 活性エネルギー線硬化性組成物及びその用途 |
| JP2005162908A (ja) * | 2003-12-03 | 2005-06-23 | Natoko Kk | 活性エネルギー線硬化性ウレタン(メタ)アクリレート、それを含有する活性エネルギー線硬化性組成物及びそれらを用いた機能性部材 |
| JP2006137780A (ja) * | 2004-11-10 | 2006-06-01 | Dainippon Printing Co Ltd | 成形シート |
| JP2007009218A (ja) * | 2006-08-22 | 2007-01-18 | Natoko Kk | 塗料組成物 |
| WO2011136042A1 (ja) | 2010-04-27 | 2011-11-03 | 東レ株式会社 | 積層フィルムおよび成型体 |
Non-Patent Citations (2)
| Title |
|---|
| "JIS Handbook", 1999, article "JIS K7112" |
| See also references of EP2796291A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2015046049A1 (ja) * | 2013-09-24 | 2017-03-09 | 東レ株式会社 | 積層フィルム |
| EP3050701A4 (en) * | 2013-09-24 | 2017-06-07 | Toray Industries, Inc. | Laminated film |
| JP2016078458A (ja) * | 2014-10-20 | 2016-05-16 | 東レ株式会社 | 積層フィルム |
| JP2016169351A (ja) * | 2015-03-16 | 2016-09-23 | 日油株式会社 | 硬化性樹脂組成物および積層構造体 |
| JP2017088680A (ja) * | 2015-11-05 | 2017-05-25 | 日油株式会社 | 活性エネルギー線硬化型樹脂組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2796291A1 (en) | 2014-10-29 |
| CN103958199B (zh) | 2016-08-17 |
| JPWO2013094532A1 (ja) | 2015-04-27 |
| EP2796291B1 (en) | 2016-10-05 |
| KR20140103901A (ko) | 2014-08-27 |
| KR102048801B1 (ko) | 2019-11-26 |
| TWI673166B (zh) | 2019-10-01 |
| JP6236783B2 (ja) | 2017-11-29 |
| US9469789B2 (en) | 2016-10-18 |
| TW201331035A (zh) | 2013-08-01 |
| US20140303316A1 (en) | 2014-10-09 |
| EP2796291A4 (en) | 2015-08-19 |
| CN103958199A (zh) | 2014-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6236783B2 (ja) | 積層フィルム | |
| KR20170078510A (ko) | 점착제 조성물, 점착필름 및 화상표시장치 | |
| JP6394395B2 (ja) | 積層フィルム | |
| JP5928334B2 (ja) | 積層フィルムおよび成型体 | |
| JP6481607B2 (ja) | 積層フィルム | |
| KR20170077768A (ko) | 점착제 조성물, 점착필름 및 화상표시장치 | |
| KR102242709B1 (ko) | 적층 필름 및 적층 필름의 제조 방법 | |
| JP7468573B2 (ja) | 積層体 | |
| JP2014184610A (ja) | 積層フィルム及びその製造方法 | |
| JP7200562B2 (ja) | 積層体 | |
| JP6531392B2 (ja) | 積層フィルム | |
| JP2023034108A (ja) | 粘着シート、フレキシブル画像表示装置表面保護フィルム用粘着シート及びフレキシブル画像表示装置表面保護フィルム用積層体 | |
| JP2013244650A (ja) | 積層フィルム | |
| JP6582862B2 (ja) | 積層フィルム | |
| WO2023189573A1 (ja) | 粘着シート、離型フィルム付き粘着シート、及びフレキシブル画像表示装置構成部材用粘着シート | |
| JP2019042983A (ja) | 積層フィルム | |
| JP2017064968A (ja) | 積層フィルム | |
| JP2024103132A (ja) | 粘着シート、離型フィルム付き粘着シート、及びフレキシブル画像表示装置構成部材用粘着シート | |
| JP2024141996A (ja) | 積層体、画像表示装置及び、粘着シート |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2012556727 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12859078 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20147009403 Country of ref document: KR Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2012859078 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012859078 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14358768 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |