WO2013087004A1 - 咪唑二酮类化合物及其用途 - Google Patents
咪唑二酮类化合物及其用途 Download PDFInfo
- Publication number
- WO2013087004A1 WO2013087004A1 PCT/CN2012/086573 CN2012086573W WO2013087004A1 WO 2013087004 A1 WO2013087004 A1 WO 2013087004A1 CN 2012086573 W CN2012086573 W CN 2012086573W WO 2013087004 A1 WO2013087004 A1 WO 2013087004A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- deuterated
- mmol
- pharmaceutically acceptable
- hydrogen
- Prior art date
Links
- 0 CC(C)(C(N(C1=S)c(cc2)cc(C(F)(F)F)c2C#N)=O)N1c(cc1)cc(F)c1C(N*)=O Chemical compound CC(C)(C(N(C1=S)c(cc2)cc(C(F)(F)F)c2C#N)=O)N1c(cc1)cc(F)c1C(N*)=O 0.000 description 1
- KXNNIMBIYWTKGR-UHFFFAOYSA-N CN(C)C(c(c(F)c1)ccc1N(C1(CCC1)C(N1c(cc2)cc(C(F)(F)F)c2C#N)=O)C1=S)=O Chemical compound CN(C)C(c(c(F)c1)ccc1N(C1(CCC1)C(N1c(cc2)cc(C(F)(F)F)c2C#N)=O)C1=S)=O KXNNIMBIYWTKGR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4166—1,3-Diazoles having oxo groups directly attached to the heterocyclic ring, e.g. phenytoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/08—Antiseborrheics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/28—Antiandrogens
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/86—Oxygen and sulfur atoms, e.g. thiohydantoin
Definitions
- the present invention relates to the field of medicine, and in particular to imidazole diketone compounds and uses thereof, and more particularly to imidazodione compounds and their use as androgen receptor antagonists or for the treatment and prevention of androgen receptors Related diseases. Background technique
- Prostatic cancer Prostatic cancer, PCa in English
- Prostatic cancer is the most common malignant tumor in male reproductive system. The incidence increases with age, and its incidence has obvious regional differences, which is higher in Europe and America. Second only to lung cancer, it is the second death of male cancer. In the past, it was a small disease in China's tumor spectrum and did not receive enough attention. As China's social development progressed, society was aging, population urbanization, Westernization of dietary structure and detection technology progress, the incidence of prostate cancer in China is obvious. Rising momentum.
- a foreign survey on prostate cancer completed by the Tianjin Medical University Second Hospital and the Tianjin Prostate Cancer Collaborative Group in 2011 showed that the incidence of prostate cancer in Tianjin is rising rapidly, and the incidence of prostate cancer has increased four-fold in 20 years. Cancer patients have accounted for 13.4% of hospitalized patients with urinary tumors, and have changed from rare cancers to common tumors. The incidence of prostate cancer in the country has the same trend.
- the androgen receptor is a ligand-dependent trans-regulatory protein with a molecular weight of 110,000 Daltons. Androgen plays a very important role in the pathogenesis of prostate cancer and its deterioration, in male hormone-related diseases such as blue spring pox, male hair loss and the like.
- R 1 and R 2 are dC 4 alkane each independently selected from hydrogen, deuterium, methyl or one or more deuterated or fully deuterated
- R 3 is hydrogen, deuterium, or halogen (such as F, Cl, Br, or I);
- R 4 , R 5 , R 6 , R 9 , R 1Q , R 12 are hydrogen, deuterium, or halogen (such as F, Cl, Br, or I);
- R 7 and R 8 are each independently selected from methyl or one or more deuterated or fully deuterated dC 4 alkyl groups, or C 3 -C 6 (or C 3 - formed by the association of R 7 and R 8 .
- C 8 cycloalkyl
- R 11 is an unsubstituted, one or more deuterated or fully deuterated CC 4 alkyl group, or a partially or fully halogen-substituted CC 4 alkyl group;
- X is S or 0
- the condition is that (1) at least one of Ri, R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 10 R u , R 12 is deuterated or ⁇ , or (2) When both R 1 and R 2 are a methyl group, R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 1Q ,
- R u and R 12 may be deuterated or deuterated, or may be hydrogen or non-deuterated.
- R 1 and R 2 are each independently selected from the group consisting of: hydrogen, deuterated methyl, or deuterated ethyl.
- R 1 when R 1 is hydrogen, R 2 is selected from the group consisting of monomethyl, dimethyl, trimethyl, monoethyl, diethyl, tridecyl , tetradecylethyl, and pentadecylethyl.
- R 1 when R 1 is hydrogen, R 2 is a trimethyl group.
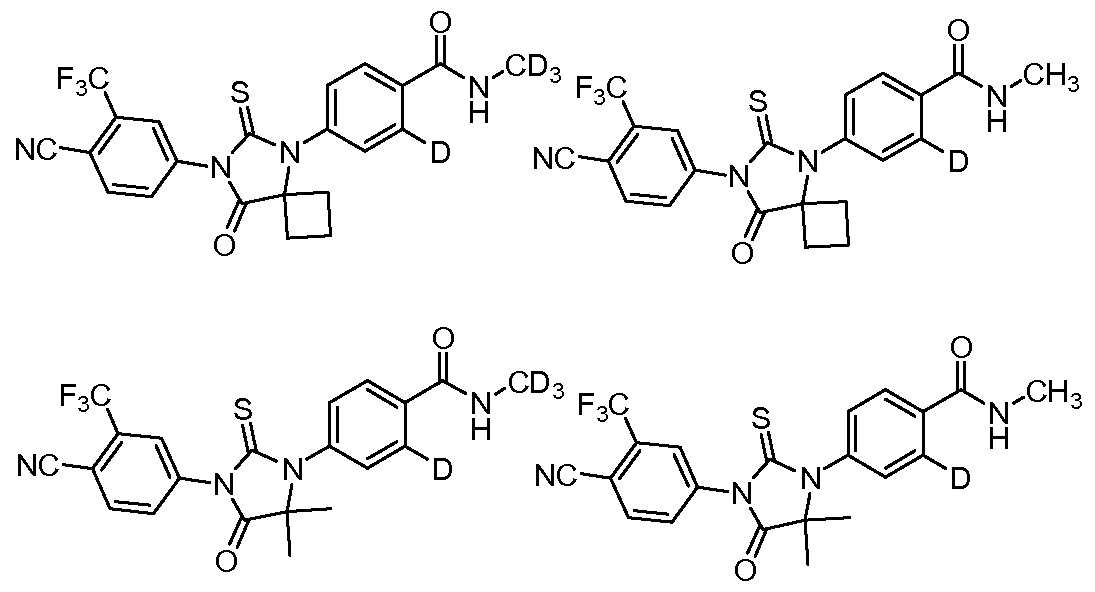
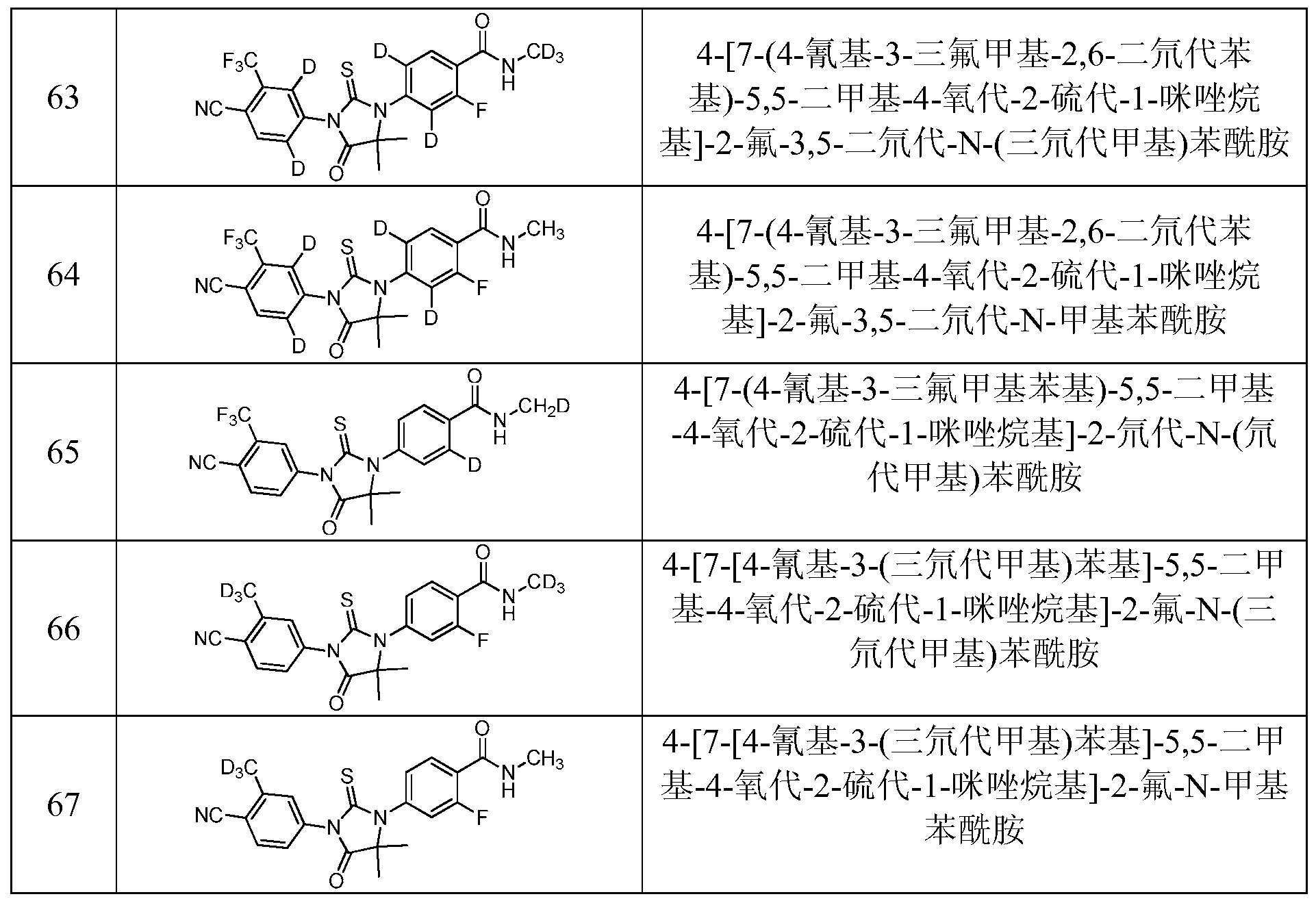
- the compound is selected from the group consisting of
- the compound is selected from the group consisting of
- the compound is selected from the group consisting of
- the compound is selected from the group consisting of
- a second aspect of the present invention provides a process for the preparation of a pharmaceutical composition, which comprises the compound of the first aspect of the invention, or a crystalline form thereof, a pharmaceutically acceptable salt, a hydrate, or a solvate thereof, and a pharmaceutically acceptable Acceptable carriers are mixed to form a pharmaceutical composition.
- a third aspect of the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising (1) the compound of the first aspect of the invention, or a crystalline form thereof, a pharmaceutically acceptable salt, a hydrate or a solvate thereof; and (2) A pharmaceutically acceptable carrier.
- the pharmaceutical composition further comprises an additional therapeutic agent; preferably, the additional therapeutic agent is a medicament for treating hair loss, regenerative hair, acne, acne, or prostate cancer.
- a fourth aspect of the invention provides the use of a compound according to the first aspect of the invention, or a crystalline form, a pharmaceutically acceptable salt, a hydrate or a solvate thereof, which is used as an androgen receptor antagonist, or A medicament for the preparation of a disease for treating and preventing androgen receptor activity.
- the disease is selected from the group consisting of hair loss, regenerative hair, acne, acne, or prostate cancer.
- the pharmaceutical composition is an injection, a sachet, a tablet, a pill, a powder or a granule.
- a fifth aspect of the invention provides a method of treatment comprising the steps of: administering a compound of the first aspect of the invention, or a crystalline form, a pharmaceutically acceptable salt, a hydrate or a solvate thereof, to a subject in need of treatment Or the pharmaceutical composition according to the third invention of the present invention.
- the subject is a human having a disease associated with androgen receptor activity.
- a sixth aspect of the invention provides a process for the preparation of a compound of formula (I) according to the first aspect of the invention, which comprises the steps of: (1) compound 5a and R 7 C in an acidic solvent in the presence of cyanide (0) R 8 reaction, thereby forming H 2 N
- cyanide compound is TMSCN, sodium cyanide or potassium cyanide
- the reaction in the step (2), is carried out in the presence of hydrochloric acid or sulfuric acid.
- step (1) further includes the following steps:
- the reduction is carried out with a reducing agent selected from the group consisting of iron powder, zinc powder, or a combination thereof.
- the acidic solvent is, for example, formic acid, acetic acid, an aqueous solution of hydrochloric acid having a concentration of 1-5% by mass, or an aqueous solution of sulfuric acid having a concentration of 1-5% by mass.
- the step (2) aprotic solvent solvent is dimethylformamide (DMF), dimethyl sulfoxide (DMS0) or CH 3 CN.
- the inert solvent is dichloromethane, ethyl acetate, tetrahydrofuran, chloroform, or acetonitrile.
- the imidazoledione compound of the formula (I) of the present invention or a crystalline form, a pharmaceutically acceptable salt, a hydrate or a solvate thereof, has significantly superior pharmacokinetics. And/or pharmacodynamic properties, and therefore more suitable as an androgen receptor antagonist, and thus more suitable for the preparation of a medicament for treating androgen-related diseases such as cancer.
- the present invention has been completed on this basis. Definition
- halogen refers to F, Cl, Br, and I. More preferably, the halogen atom is selected from the group consisting of F, C1 and Br.
- alkyl includes straight or branched alkyl groups.
- Preferred alkyl groups are dC 4 alkyl groups such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like.
- deuterated refers to the replacement of one or more hydrogens in a compound or group by deuterium. Deuterated can be a substitution, a disubstituted, a polysubstituted or a fully substituted. The terms “one or more deuterated” are used interchangeably with “one or more deuterated”.
- the cerium isotope content of cerium at the cerium substitution site is greater than the natural strontium isotope content (0.015%), more preferably greater than 50%, more preferably greater than 75%, more preferably greater than 95%, more preferably The ground is greater than 97%, more preferably greater than 99%, and even more preferably greater than 99.5%.
- Active ingredient 0.015%, more preferably greater than 50%, more preferably greater than 75%, more preferably greater than 95%, more preferably The ground is greater than 97%, more preferably greater than 99%, and even more preferably greater than 99.5%.
- the term "compound of the invention” refers to a compound of formula (I).
- the term also encompasses various crystalline forms, pharmaceutically acceptable salts, hydrates or solvates of the compounds of formula (I).
- pharmaceutically acceptable salt refers to a salt of the compound of the present invention which is formed with an acid or a base and which is suitable for use as a medicament.
- Pharmaceutically acceptable salts include inorganic and organic salts.
- a preferred class of salts are the salts of the compounds of the invention with acids.
- Suitable acids for forming salts include, but are not limited to, mineral acids such as hydrochloric acid, hydrobromic acid, hydrofluoric acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, Organic acids such as maleic acid, lactic acid, malic acid, tartaric acid, citric acid, picric acid, methanesulfonic acid, benzoic acid, benzenesulfonic acid; and acidic amino acids such as aspartic acid and glutamic acid.
- mineral acids such as hydrochloric acid, hydrobromic acid, hydrofluoric acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid,
- Organic acids such as maleic acid, lactic acid, malic acid, tartaric acid, cit
- the compound of the present invention Since the compound of the present invention has excellent androgen receptor antagonism, the compound of the present invention and various crystal forms thereof, pharmaceutically acceptable inorganic or organic salts, hydrates or solvates, and compounds containing the present invention are mainly active.
- the pharmaceutical composition of the ingredients can be used to treat, prevent, and alleviate the diseases mediated by androgen.
- the compounds of the invention are useful in the treatment of the following disorders: hair loss, regenerative hair, acne, acne, prostate cancer and the like.
- compositions of the present invention comprise a safe or effective amount of a compound of the present invention or a pharmacologically acceptable salt thereof, and a pharmaceutically acceptable excipient or carrier.
- safe and effective amount is meant: The amount of the compound is sufficient to significantly improve the condition without causing serious side effects.
- the pharmaceutical composition contains from 1 to 2000 mg of the compound of the invention per agent, more preferably from 10 to 200 mg of the compound of the invention.
- the "one dose" is a capsule or a tablet.
- “Pharmaceutically acceptable carrier” means: one or more compatible solid or liquid fillers or gel materials which are suitable for human use and which must be of sufficient purity and of sufficiently low toxicity.
- “Compatibility” as used herein refers to the composition The components can be blended with the compounds of the invention and with each other without significantly reducing the potency of the compounds.
- pharmaceutically acceptable carriers are cellulose and its derivatives (such as sodium carboxymethylcellulose, sodium ethylcellulose, cellulose acetate, etc.), gelatin, talc, solid lubricants (such as stearic acid).
- magnesium stearate magnesium stearate
- calcium sulfate vegetable oils (such as soybean oil, sesame oil, peanut oil, olive oil, etc.), polyols (such as propylene glycol, glycerin, mannitol, sorbitol, etc.), emulsifiers (such as Tween®), moist Wet agents (such as sodium lauryl sulfate), colorants, flavoring agents, stabilizers, antioxidants, preservatives, pyrogen-free water, and the like.
- vegetable oils such as soybean oil, sesame oil, peanut oil, olive oil, etc.
- polyols such as propylene glycol, glycerin, mannitol, sorbitol, etc.
- emulsifiers such as Tween®
- moist Wet agents such as sodium lauryl sulfate
- colorants such as sodium lauryl sulfate
- flavoring agents such as pepperminophen®
- the mode of administration of the compound or pharmaceutical composition of the present invention is not particularly limited, and representative modes of administration include, but are not limited to, oral, gastrointestinal (intravenous, intramuscular or subcutaneous), and topical administration.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- the active compound is mixed with at least one conventional inert excipient (or carrier), such as sodium citrate or dicalcium phosphate, or mixed with: (a) a filler or compatibilizer, for example, Starch, lactose, sucrose, glucose, mannitol and silicic acid; (b) binders, for example, hydroxymethylcellulose, alginate, gelatin, polyvinylpyrrolidone, sucrose and acacia; (c) humectants, For example, glycerin; (d) a disintegrant, for example, agar, calcium carbonate, potato starch or tapioca starch, alginic acid, certain complex silicates, and sodium carbonate; (e) a slow solvent such as paraffin; (f) Absorbing accelerators, for example, quaternary amine compounds; (g) wetting agents, such as cetylene glycol
- Solid dosage forms such as tablets, troches, capsules, pills, and granules can be prepared with coatings and shells, such as enteric coatings and other materials known in the art. They may contain opacifying agents and the release of the active compound or compound in such compositions may be released in a portion of the digestive tract in a delayed manner. Examples of embedding components that can be employed are polymeric and waxy materials. If necessary, the active compound may also be in microencapsulated form with one or more of the above-mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups or elixirs.
- the liquid dosage form may contain inert diluents conventionally employed in the art, such as water or other solvents, solubilizers and emulsifiers, for example, ethanol, isopropanol, ethyl carbonate, ethyl acetate, propylene glycol, 1 , 3-butanediol, dimethylformamide and oils, especially cottonseed oil, peanut oil, corn germ oil, olive oil, castor oil and sesame oil or a mixture of these substances.
- inert diluents conventionally employed in the art, such as water or other solvents, solubilizers and emulsifiers, for example, ethanol, isopropanol, ethyl carbonate, ethyl acetate, propylene glycol, 1 , 3-butanediol, dimethyl
- compositions may contain adjuvants such as wetting agents, emulsifying and suspending agents, sweetening agents, flavoring agents, and flavoring agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening agents, flavoring agents, and flavoring agents.
- the suspension may contain a suspending agent, for example, ethoxylated isostearyl alcohol, polyoxyethylene sorbitol and sorbitan ester, microcrystalline cellulose, aluminum methoxide and agar or a mixture of these and the like.
- a suspending agent for example, ethoxylated isostearyl alcohol, polyoxyethylene sorbitol and sorbitan ester, microcrystalline cellulose, aluminum methoxide and agar or a mixture of these and the like.
- compositions for parenteral injection may comprise a physiologically acceptable sterile aqueous or nonaqueous solution, dispersion, suspension or emulsion, and sterile powder for reconstitution into sterile injectable solutions or dispersions.
- Suitable aqueous and nonaqueous vehicles, diluents, solvents or vehicles include water, ethanol, polyols and suitable mixtures thereof.
- Dosage forms for the compounds of the invention for topical administration include ointments, powders, patches, propellants and inhalants.
- the active ingredient is mixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants which may be required if necessary.
- the compounds of the invention may be administered alone or in combination with other pharmaceutically acceptable compounds.
- a pharmaceutical composition e.g., a human
- a safe and effective amount of a compound of the invention is applied to a mammal (e.g., a human) in need of treatment wherein the dosage is a pharmaceutically effective effective dosage, for a 60 kg body weight
- the dose to be administered is usually from 1 to 2000 mg, preferably from 20 to 500 mg.
- specific doses should also consider factors such as the route of administration, the health of the patient, etc., which are within the skill of the skilled physician.
- the compound of the formula (I) of the present invention can be produced by the following synthesis formula.
- each reaction is usually carried out in a solvent at room temperature to reflux temperature (e.g., 0 ° C to 120 ° C, preferably 0 ° C to 80 ° C).
- the reaction time is usually from 0.1 to 60 hours, preferably from 0.5 to 48 hours.
- RR 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , R 1Q , R u , R 12 , and X are as defined in the formula (I).
- Compound la (deuterated or undeuterated aniline) is reacted with thiophosgene (or phosgene) to give compound 2a.
- Compound 4a can be synthesized by amidation of compound 3a.
- Compound 4a can be reduced by a reducing reagent such as zinc powder/acetic acid or iron powder/acetic acid to give compound amine 5a.
- Compound 6a can be obtained by dehydrating aniline and a ketone (e.g., R 7 C(0)R 8 ) in the presence of TMSCN or cyanide (such as sodium cyanide or potassium cyanide).
- TMSCN or cyanide such as sodium cyanide or potassium cyanide
- compound 6a are synthesized by condensation under acidic conditions such as hydrochloric acid or sulfuric acid.
- the preparation of the corresponding deuterated compound can be carried out by the same route using the corresponding deuterated starting compound or the corresponding deuterated reagent as a starting material.
- a starting material such as deuterated methylamine, deuterated acetone.
- deuterated raw materials on the benzene ring can be obtained by the following method (Org Letter, 2008,
- the compound of the present invention is an excellent androgen receptor antagonist which can be used for the preparation of an androgen-related disease, for example, hair loss, regenerative hair, acne, acne, or prostate cancer.
- 35 34 500 mg of Pd/C and 20 ml of heavy water were placed in a 250 ml round bottom flask, and the flask was filled with hydrogen and stirred at room temperature for 3 days.
- Compound 33 (560 mg, 3.0 mmol) in ethyl acetate (10 ml) and triethylamine (303 mg, 3.0 mmol) was then added to the reaction mixture and stirred for 2 h.
- Example 14 4- ⁇ 7-[4-Cyano-3-trifluoromethyl-2,6-didecylphenyl]-5,5-methyl-4-oxo-2-thio- 1-Imidazolium ⁇ -2-fluoro-N-tridemethylbenzamide (Compound 45) It ————— t _ ⁇ .1 , person, -.
- human prostate cancer LNCaP purchased from American ATCC
- 22RV1C purchased from SIBS
- RPMI1640 medium containing 10% charcoal-striped fetal bovine serum (Charcoal-stripped FBS) and cultured for three days.
- the cells were digested with 0.25% trypsin and counted by trypan blue exclusion method.
- the cells were plated, and each well was filled with ⁇ cell suspension containing 5000 cells. To avoid edge effect, add 20 ( ⁇ L medium to the cell plate). Peripheral holes.
- test compound inhibition rate was calculated using the following equation:
- the software can calculate the 50% inhibition rate, IC50
- the in vitro biological activity assay of the androgen receptor antagonist of the compound of the present invention can be tested by the method reported in J. Medcinal Chemistry (2010, pages 2779-2796 and WO201 1/029392).
- Prostate cancer cells LNCaP and 22RV1
- R1881 methyltrienolone, androgen receptor activator
- IC 5 50% inhibitory concentration of the compound on prostate cancer cells (LNCaP and 22RV1) against prostate specific antigen (PSA) was calculated according to the reported method. The results are shown in Table 3 below:
- Example 18 Pharmacokinetic evaluation in mice Healthy Kunming mice, male, weighing 18-20 g. 10 mg/kg AF-484 (Example 6, Compound 21) and AF-486 (Example 4, Compound 17) were administered intragastrically, and the compound was dissolved in DMSO:PEG400:H 2 O 1 :5:14, and administered. The volume is 10 ml/kg. Fasting for 12 h before the test, free drinking water. Eat regularly 2 hours after administration.
- mice After administration, 0.5, 1.0, 2.0, 4.0, 6.0 and B 24 h, 3 mice per time point, 0.3 ml of blood was taken from the posterior venous plexus of the mouse, placed in heparinized tubes, centrifuged at l lOO rpm 5 Min, plasma was separated and frozen in a refrigerator at -20 °C. Pipette 100 ul of serum into a clean plastic centrifuge tube, indicate the name and time of the compound, dilute with acetonitrile (CH 3 CN), and centrifuge. The drug concentration was then analyzed by LC-MS. Serum was stored at -80 °C prior to LC-MS analysis.
- the pharmacokinetic parameters of the deuterated compound (Example 6, Compound 21) and the non-deuterated compound (Example 4, Compound 17) are shown in the following table. According to the experimental results, the compound 21 of the present invention has a significant increase in Cmax and AUC as compared with the corresponding non-deuterated compound 17, wherein Cmax is increased by at least 20%.
Abstract
Description























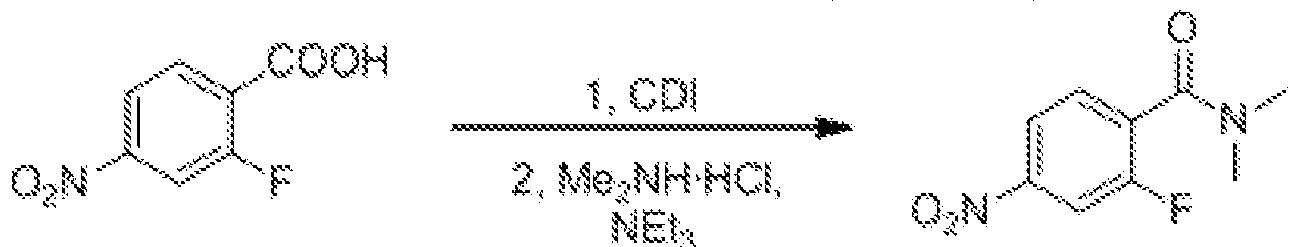


























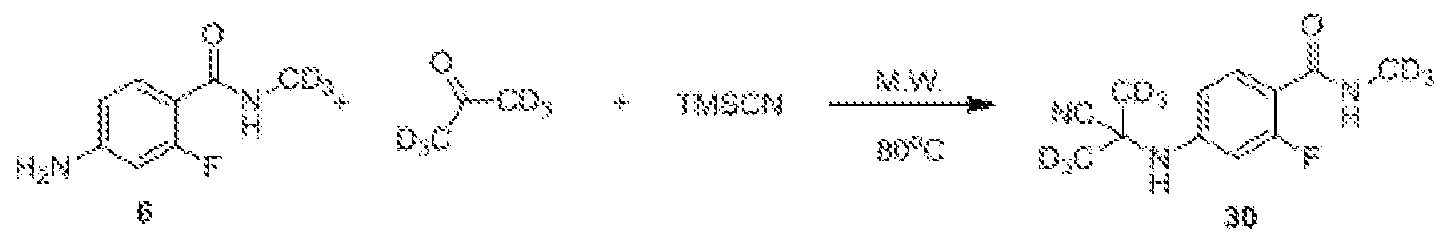























Claims








Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12858174.1A EP2792674B1 (en) | 2011-12-14 | 2012-12-13 | Imidazolidinedione compounds and their uses |
| AU2012350482A AU2012350482B2 (en) | 2011-12-14 | 2012-12-13 | Imidazolidinedione compounds and their uses |
| ES12858174.1T ES2587903T3 (es) | 2011-12-14 | 2012-12-13 | Compuestos de imidazolidinadiona y sus usos |
| JP2014546301A JP5933746B2 (ja) | 2011-12-14 | 2012-12-13 | イミダゾリジンジオン系化合物およびその用途 |
| US14/364,147 US9346764B2 (en) | 2011-12-14 | 2012-12-13 | Imidazolidinedione compounds and their uses |
| CN201280052853.9A CN104024228B (zh) | 2011-12-14 | 2012-12-13 | 咪唑二酮类化合物及其用途 |
| CA2859224A CA2859224C (en) | 2011-12-14 | 2012-12-13 | Imidazolidinedione compounds and their uses |
| HK14111671A HK1198166A1 (zh) | 2011-12-14 | 2014-11-19 | 咪唑二酮類化合物及其用途 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2011104188590A CN103159680A (zh) | 2011-12-14 | 2011-12-14 | 咪唑二酮类化合物及其用途 |
| CN201110418859.0 | 2011-12-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013087004A1 true WO2013087004A1 (zh) | 2013-06-20 |
Family
ID=48583248
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2012/086573 WO2013087004A1 (zh) | 2011-12-14 | 2012-12-13 | 咪唑二酮类化合物及其用途 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US9346764B2 (zh) |
| EP (1) | EP2792674B1 (zh) |
| JP (1) | JP5933746B2 (zh) |
| CN (2) | CN103159680A (zh) |
| AU (1) | AU2012350482B2 (zh) |
| CA (1) | CA2859224C (zh) |
| ES (1) | ES2587903T3 (zh) |
| HK (1) | HK1198166A1 (zh) |
| PL (1) | PL2792674T3 (zh) |
| WO (1) | WO2013087004A1 (zh) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016530208A (ja) * | 2013-09-19 | 2016-09-29 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニーGlaxoSmithKline LLC | 組み合わせ薬物療法 |
| AU2014273618B2 (en) * | 2013-05-29 | 2016-10-13 | Hinova Pharmaceuticals Inc. | Imidazole diketone compound and use thereof |
| WO2017067530A2 (zh) | 2017-02-13 | 2017-04-27 | 康朴生物医药技术(上海)有限公司 | 一种治疗前列腺癌的组合、药物组合物及治疗方法 |
| WO2017071375A1 (zh) * | 2015-10-30 | 2017-05-04 | 成都海创药业有限公司 | 一种氘代咪唑酮化合物的晶型及其制备方法和用途 |
| CN107303278A (zh) * | 2016-04-25 | 2017-10-31 | 成都海创药业有限公司 | 一种hc‑1119固体分散体及其制备方法 |
| WO2019184952A1 (en) * | 2018-03-29 | 2019-10-03 | Hinova Pharmaceuticals Inc. | Deuterated imidazolidinedione compounds and their uses |
| US10626091B2 (en) * | 2014-10-01 | 2020-04-21 | Laurus Labs Limited | Process for the preparation of enzalutamide |
| WO2020095183A1 (en) | 2018-11-05 | 2020-05-14 | Pfizer Inc. | Combination for treating cancer |
| WO2022034504A1 (en) | 2020-08-13 | 2022-02-17 | Pfizer Inc. | Combination therapy |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105188699B (zh) * | 2013-10-14 | 2017-04-26 | 杭州普晒医药科技有限公司 | 恩杂鲁胺的固态形式及其制备方法和用途 |
| CN104803919A (zh) * | 2014-01-26 | 2015-07-29 | 上海医药工业研究院 | 用于制备恩杂鲁胺中间体的方法 |
| CN107954936B (zh) * | 2016-10-17 | 2021-03-19 | 海创药业股份有限公司 | 一种制备氘代咪唑二酮类化合物的方法 |
| WO2019025250A1 (en) | 2017-08-04 | 2019-02-07 | Basf Se | SUBSTITUTED TRIFLUOROMETHYLOXADIAZOLES FOR COMBATING PHYTOPATHOGENIC FUNGI |
| WO2019196945A1 (zh) * | 2018-04-13 | 2019-10-17 | 成都海创药业有限公司 | 一种合成氘代酰胺及氘代磺酰胺的新方法 |
| EP3795153A4 (en) * | 2018-05-14 | 2022-03-09 | Hinova Pharmaceuticals Inc. | FORMULATION BASED ON HC-1119, METHOD FOR PREPARATION AND USE THEREOF |
| CN112442009B (zh) * | 2019-08-30 | 2023-10-03 | 润佳(苏州)医药科技有限公司 | 氘代化合物及其在治疗癌症方面的应用 |
| JP2023517393A (ja) * | 2020-03-17 | 2023-04-25 | メッドシャイン ディスカバリー インコーポレイテッド | タンパク質分解調整剤およびその使用方法 |
| CN114181155A (zh) * | 2021-12-21 | 2022-03-15 | 上海朝晖药业有限公司 | 碘-恩杂鲁胺及其制备方法及其应用 |
| CN115181043A (zh) * | 2022-07-27 | 2022-10-14 | 爱斯特(成都)生物制药股份有限公司 | 一种连续流制备4-异硫氰基-2-(三氟甲基)苯甲腈的方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006124118A1 (en) * | 2005-05-13 | 2006-11-23 | The Regents Of The University Of California | Diarylhydantoin compounds |
| WO2007126765A2 (en) * | 2006-03-27 | 2007-11-08 | The Regents Of The University Of California | Androgen receptor modulator for the treatment of prostate cancer and androgen receptor-associated diseases |
| WO2007127010A2 (en) * | 2006-03-29 | 2007-11-08 | The Regents Of The University Of California | Diarylthiohydantoin compounds |
| CN101817787A (zh) * | 2009-02-26 | 2010-09-01 | 童友之 | 抗前列腺癌的雄性激素受体拮抗剂 |
| WO2011029392A1 (en) | 2009-09-10 | 2011-03-17 | Youzhi Tong | Androgen receptor antagonists and uses thereof |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013067151A1 (en) * | 2011-11-02 | 2013-05-10 | Medivation Prostate Therapeutics, Inc. | Treatment methods using diarylthiohydantoin derivatives |
-
2011
- 2011-12-14 CN CN2011104188590A patent/CN103159680A/zh active Pending
-
2012
- 2012-12-13 EP EP12858174.1A patent/EP2792674B1/en active Active
- 2012-12-13 ES ES12858174.1T patent/ES2587903T3/es active Active
- 2012-12-13 AU AU2012350482A patent/AU2012350482B2/en active Active
- 2012-12-13 CA CA2859224A patent/CA2859224C/en active Active
- 2012-12-13 CN CN201280052853.9A patent/CN104024228B/zh active Active
- 2012-12-13 WO PCT/CN2012/086573 patent/WO2013087004A1/zh active Application Filing
- 2012-12-13 PL PL12858174.1T patent/PL2792674T3/pl unknown
- 2012-12-13 JP JP2014546301A patent/JP5933746B2/ja active Active
- 2012-12-13 US US14/364,147 patent/US9346764B2/en active Active
-
2014
- 2014-11-19 HK HK14111671A patent/HK1198166A1/zh unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006124118A1 (en) * | 2005-05-13 | 2006-11-23 | The Regents Of The University Of California | Diarylhydantoin compounds |
| WO2007126765A2 (en) * | 2006-03-27 | 2007-11-08 | The Regents Of The University Of California | Androgen receptor modulator for the treatment of prostate cancer and androgen receptor-associated diseases |
| WO2007127010A2 (en) * | 2006-03-29 | 2007-11-08 | The Regents Of The University Of California | Diarylthiohydantoin compounds |
| CN101817787A (zh) * | 2009-02-26 | 2010-09-01 | 童友之 | 抗前列腺癌的雄性激素受体拮抗剂 |
| WO2011029392A1 (en) | 2009-09-10 | 2011-03-17 | Youzhi Tong | Androgen receptor antagonists and uses thereof |
Non-Patent Citations (4)
| Title |
|---|
| J. MEDCINAL CHEMISTRY, 2010, pages 2779 - 2796 |
| JIANG, WENFENG ET AL.: "Application of Deuteration in Drug Research", QILU PHARMACEUTICAL AFFAIRS, vol. 29, no. 11, 2010, pages 682 - 684, XP008173943 * |
| ORG LETTER, 2008, pages 4351 - 4353 |
| See also references of EP2792674A4 * |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2014273618B2 (en) * | 2013-05-29 | 2016-10-13 | Hinova Pharmaceuticals Inc. | Imidazole diketone compound and use thereof |
| JP2016530208A (ja) * | 2013-09-19 | 2016-09-29 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニーGlaxoSmithKline LLC | 組み合わせ薬物療法 |
| JP2018158930A (ja) * | 2013-09-19 | 2018-10-11 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニーGlaxoSmithKline LLC | 組み合わせ薬物療法 |
| JP2019196391A (ja) * | 2013-09-19 | 2019-11-14 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニーGlaxoSmithKline LLC | 組み合わせ薬物療法 |
| US10626091B2 (en) * | 2014-10-01 | 2020-04-21 | Laurus Labs Limited | Process for the preparation of enzalutamide |
| WO2017071375A1 (zh) * | 2015-10-30 | 2017-05-04 | 成都海创药业有限公司 | 一种氘代咪唑酮化合物的晶型及其制备方法和用途 |
| CN107303278A (zh) * | 2016-04-25 | 2017-10-31 | 成都海创药业有限公司 | 一种hc‑1119固体分散体及其制备方法 |
| WO2017067530A2 (zh) | 2017-02-13 | 2017-04-27 | 康朴生物医药技术(上海)有限公司 | 一种治疗前列腺癌的组合、药物组合物及治疗方法 |
| WO2017067530A3 (zh) * | 2017-02-13 | 2017-12-21 | 康朴生物医药技术(上海)有限公司 | 一种治疗前列腺癌的组合、药物组合物及治疗方法 |
| WO2019184952A1 (en) * | 2018-03-29 | 2019-10-03 | Hinova Pharmaceuticals Inc. | Deuterated imidazolidinedione compounds and their uses |
| WO2020095183A1 (en) | 2018-11-05 | 2020-05-14 | Pfizer Inc. | Combination for treating cancer |
| WO2022034504A1 (en) | 2020-08-13 | 2022-02-17 | Pfizer Inc. | Combination therapy |
Also Published As
| Publication number | Publication date |
|---|---|
| PL2792674T3 (pl) | 2016-12-30 |
| US9346764B2 (en) | 2016-05-24 |
| CN104024228A (zh) | 2014-09-03 |
| HK1198166A1 (zh) | 2015-03-13 |
| CN103159680A (zh) | 2013-06-19 |
| AU2012350482B2 (en) | 2017-02-16 |
| CA2859224A1 (en) | 2013-06-20 |
| JP2015501820A (ja) | 2015-01-19 |
| US20140371284A1 (en) | 2014-12-18 |
| CA2859224C (en) | 2019-03-19 |
| EP2792674A1 (en) | 2014-10-22 |
| JP5933746B2 (ja) | 2016-06-15 |
| EP2792674A4 (en) | 2015-06-17 |
| EP2792674B1 (en) | 2016-05-25 |
| ES2587903T3 (es) | 2016-10-27 |
| CN104024228B (zh) | 2015-07-08 |
| AU2012350482A1 (en) | 2014-07-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013087004A1 (zh) | 咪唑二酮类化合物及其用途 | |
| AU2014273618B2 (en) | Imidazole diketone compound and use thereof | |
| US8710086B2 (en) | Substituted di-arylhydantoin and di-arylthiohydantoin compounds and methods of use thereof | |
| WO2021129824A1 (zh) | 新型K-Ras G12C抑制剂 | |
| WO2013184698A1 (en) | Solid forms of an antiviral compound | |
| TWI617554B (zh) | 組織胺h受體之苯并咪唑-2-基嘧啶調控劑 | |
| WO2018121689A1 (zh) | 磺酰胺-芳基酰胺类化合物及其治疗乙型肝炎的药物用途 | |
| WO2014183555A1 (zh) | 环烷基甲酸类衍生物、其制备方法及其在医药上的应用 | |
| JPH11292766A (ja) | 哺乳動物の心不全処置法、組成物およびキット | |
| CN110003204B (zh) | 一种bet蛋白抑制剂、其制备方法及用途 | |
| TW479058B (en) | 2,7-substituted octahydro-pyrrolo[1,2-a]pyrazine derivatives | |
| CN106478619B (zh) | 一类黄嘌呤氧化酶抑制剂及其应用 | |
| CN109897036B (zh) | 三唑并吡啶类化合物及其制备方法和用途 | |
| JPWO2002096875A1 (ja) | 鎮痛作用を有する4−ヒドロキシピペリジン誘導体 | |
| WO2020173417A1 (zh) | 含丙烯酰基的核转运调节剂及其用途 | |
| CN111943889A (zh) | 一种双芳香基胺类化合物及其制备方法和应用 | |
| KR20240046891A (ko) | 다환 화합물 및 그의 용도 | |
| JP2022065014A (ja) | 重水素化イミダゾリジンジオン化合物とその用途 | |
| EA008444B1 (ru) | Производные ацилоксипирролидина и их применение в качестве лигандов vили vи vрецепторов | |
| CN104788372B (zh) | 一种氘代卡博替尼衍生物、其制备方法、应用及其中间体 | |
| CN112759546B (zh) | 3-(二甲氨基甲基)哌啶-4-醇衍生物及其制备方法和药物用途 | |
| WO2024027695A1 (zh) | 作为her2抑制剂的化合物 | |
| WO2014029250A1 (zh) | 苯并吡啶氮杂卓类化合物及其应用 | |
| CN115322158A (zh) | 作为krasg12c蛋白抑制剂的取代喹唑啉类化合物 | |
| CN110669049A (zh) | 新型雄激素受体抑制剂及其合成方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12858174 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14364147 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2859224 Country of ref document: CA Ref document number: 2014546301 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012858174 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2012350482 Country of ref document: AU Date of ref document: 20121213 Kind code of ref document: A |