WO2013054457A1 - リチウムシリケート系化合物とリチウムイオン二次電池用正極活物質及びこれを用いたリチウムイオン二次電池 - Google Patents
リチウムシリケート系化合物とリチウムイオン二次電池用正極活物質及びこれを用いたリチウムイオン二次電池 Download PDFInfo
- Publication number
- WO2013054457A1 WO2013054457A1 PCT/JP2012/004768 JP2012004768W WO2013054457A1 WO 2013054457 A1 WO2013054457 A1 WO 2013054457A1 JP 2012004768 W JP2012004768 W JP 2012004768W WO 2013054457 A1 WO2013054457 A1 WO 2013054457A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lithium
- positive electrode
- ion secondary
- secondary battery
- silicate compound
- Prior art date
Links
Images






Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01B—CABLES; CONDUCTORS; INSULATORS; SELECTION OF MATERIALS FOR THEIR CONDUCTIVE, INSULATING OR DIELECTRIC PROPERTIES
- H01B1/00—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors
- H01B1/06—Conductors or conductive bodies characterised by the conductive materials; Selection of materials as conductors mainly consisting of other non-metallic substances
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B33/00—Silicon; Compounds thereof
- C01B33/20—Silicates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B33/00—Silicon; Compounds thereof
- C01B33/20—Silicates
- C01B33/32—Alkali metal silicates
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/5825—Oxygenated metallic salts or polyanionic structures, e.g. borates, phosphates, silicates, olivines
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/62—Selection of inactive substances as ingredients for active masses, e.g. binders, fillers
- H01M4/624—Electric conductive fillers
- H01M4/625—Carbon or graphite
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/136—Electrodes based on inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention mainly relates to a lithium silicate compound useful as a positive electrode active material of a lithium ion battery, and a lithium ion secondary battery using the lithium silicate compound as an active material.
- Lithium ion secondary batteries are small and have high energy density, and are widely used as power sources for portable electronic devices.
- a layered compound such as LiCoO 2 is mainly used.
- these compounds have a problem in that the oxygen is easily released at around 150 ° C. in a fully charged state, and this tends to cause an oxidative exothermic reaction of the non-aqueous electrolyte, leading to a decrease in battery safety.
- olivine phosphate compounds LiMPO 4 (LiMnPO 4 , LiFePO 4 , LiCoPO 4, etc.) have been proposed as positive electrode active materials. These compounds improve thermal stability by using a divalent / trivalent redox reaction instead of a trivalent / tetravalent redox reaction using an oxide such as LiCoO 2 as a positive electrode active material. Further, the compound is attracting attention as a compound capable of obtaining a high discharge voltage by disposing a polyanion of a heteroelement having a high electronegativity around a central metal.
- the positive electrode material made of an olivine phosphate compound not only has an average discharge voltage as low as 3.5 V or less, but also has a theoretical capacity limited to about 170 mAh / g due to a large formula weight of phosphate polyanion. Furthermore, since a lithium ion secondary battery using LiCoPO 4 or LiNiPO 4 has an operating voltage exceeding 4.5 V, there is a problem that there is no electrolyte solution that can withstand the charging voltage. Moreover, the lithium ion secondary battery using LiMnPO 4 has a problem that the cycle characteristics are severely deteriorated, and LiFePO 4 has the highest practicality, but has a problem of capacity reduction due to oxidation of divalent iron. .
- Li 2 FeSiO 4 theoretical capacity 331.3mAh / g
- Li 2 MnSiO 4 theoretical capacity 333.2mAh / g
- Lithium silicate compound is a material that is inexpensive, has only abundant resources, has low environmental impact, has a high theoretical charge / discharge capacity of lithium ions, and does not release oxygen at high temperatures. Therefore, it is attracting attention as a positive electrode material for next-generation lithium ion secondary batteries.
- the present invention provides a novel lithium silicate compound having excellent stability due to the presence of iron (Fe) in a trivalent state, and using the lithium silicate compound as a positive electrode active material has a high discharge capacity. It aims at providing the lithium ion secondary battery which expresses.
- the lithium silicate compound of the present invention is characterized by a lithium silicate compound composed of lithium (Li), iron (Fe), silicon (Si), and oxygen (O), and a composition formula: Li 1 + 2 ⁇ FeSiO 4 + ⁇ C ( ⁇ 0.25 ⁇ ⁇ ⁇ 0.25, 0 ⁇ C ⁇ 0.5).
- the lithium silicate compound of the present invention is monoclinic and belongs to the space group P2 1 / n.
- the electronic state of iron (Fe) in the composition formula the trivalence is preferably 70% or more.
- the positive electrode active material for a lithium ion secondary battery may contain carbon (C) and lithium carbonate (Li 2 CO 3 ).
- iron (Fe) When used as a positive electrode of a lithium ion secondary battery, it is preferable that all of iron (Fe) is trivalent after charging, and at least two types of Fe 3+ having different states in 57 Fe Mossbauer spectroscopy are present. .
- Fe 2+ and Fe 3+ when used as a positive electrode of a lithium ion secondary battery, it is preferable that Fe 2+ and Fe 3+ exist in the 57 Fe Mossbauer spectroscopic analysis and 50 to 80% Fe 2+ exists after discharge.
- the present invention is suitable for use as a positive electrode for a lithium secondary battery, a lithium ion secondary battery including a positive electrode for a secondary battery as a constituent element, and a vehicle equipped with a lithium secondary battery.
- lithium silicate compound represented by Li 1 + 2 ⁇ FeSiO 4 + ⁇ C ( ⁇ 0.25 ⁇ ⁇ ⁇ 0.25, 0 ⁇ C ⁇ 0.5)
- Fe iron
- the lithium ion secondary battery using the lithium silicate compound of the present invention as an active material exhibits stable cycle characteristics even after repeated charge and discharge.
- the X-ray-diffraction pattern of the lithium silicate type compound which concerns on an Example is shown.
- the X-ray-diffraction pattern of the lithium silicate type compound which concerns on a comparative example is shown.
- the Rietveld analysis result of the X-ray-diffraction pattern of the lithium silicate type compound which concerns on an Example is shown.
- the 57 Fe Mossbauer spectroscopy measurement result of the lithium silicate type compound which concerns on an Example is shown.
- the 57 Fe Mossbauer spectroscopy measurement result of the lithium silicate type compound which concerns on a comparative example is shown.
- the initial charge / discharge curve of the lithium ion secondary battery which used the lithium silicate type compound which concerns on an Example for the positive electrode active material is shown.
- the lithium ion secondary battery using the lithium silicate-based compound according to Example positive electrode active material shows 57 Fe Mössbauer spectroscopy results of the positive electrode active material after charging.
- the result of 57 Fe Mossbauer spectroscopy measurement of the positive electrode active material after discharge in the lithium ion secondary battery using the lithium silicate compound according to the example as the positive electrode active material is shown.
- the charging / discharging curve of the lithium ion secondary battery using the lithium silicate type compound which concerns on an Example for a positive electrode active material is shown.
- a feature of the lithium silicate compound of the present invention is a lithium silicate compound composed of lithium (Li), iron (Fe), silicon (Si), and oxygen (O), and has a composition formula: Li 1 + 2 ⁇ . FeSiO 4 + ⁇ C ( ⁇ 0.25 ⁇ ⁇ ⁇ 0.25, 0 ⁇ C ⁇ 0.5).
- a lithium silicate compound precursor is formed in advance.
- a method for forming the lithium silicate compound precursor a solid phase reaction method, a hydrothermal method, a sol-gel method, a Pechini method, or the like can be used.
- the case where the molten salt method is used is described in detail.
- lithium silicate represented by Li 2 SiO 3 ;
- An iron source is reacted at 300 ° C. or more and 600 ° C. or less to form a lithium silicate compound precursor.
- an iron source pure iron, iron hydroxide, iron oxalate, iron chloride, iron nitrate, an iron-containing precipitate formed by making an iron-containing aqueous solution alkaline, or the like can be used.
- a lithium silicate compound precursor having a different chemical composition and properties from that of a production method using iron oxalate or the like can be obtained.
- the alkali metal salt include at least one selected from the group consisting of a lithium salt, a potassium salt, a sodium salt, a rubidium salt, and a cesium salt. Of these, lithium salts are desirable.
- alkali metal salt it is desirable to contain at least 1 type of an alkali metal carbonate, an alkali metal chloride, an alkali metal nitrate, and an alkali metal hydroxide.
- the melting temperature is about 700 ° C., but when the molten salt is a mixture of lithium carbonate and other alkali metal salts, the melting temperature can be 600 ° C. or less, 300
- the target lithium silicate compound can be synthesized at a relatively low reaction temperature of ⁇ 600 ° C. As a result, grain growth is suppressed during the synthesis reaction, and a fine lithium silicate compound is formed.
- the molten salt is selected from the above alkali metal salts so that the melting temperature is 600 ° C. or lower, and if the alkali metal salts are mixed and used, the mixing ratio is adjusted so that the melting temperature of the mixture is 600 ° C. or lower. Thus, a mixed molten salt may be obtained. Since the mixing ratio varies depending on the type of salt, it is difficult to define it unconditionally.
- lithium carbonate when the total carbonate mixture is 100 mol%, lithium carbonate is usually 30 mol% or more, and even 30 mol%. It is preferable to contain ⁇ 70 mol%.
- the carbonate mixture include a mixture comprising 30 to 70 mol% lithium carbonate, 0 to 60 mol% sodium carbonate, and 0 to 50 mol% potassium carbonate.
- a mixture comprising 40 to 45 mol% of lithium carbonate, 30 to 35 mol% of sodium carbonate and 20 to 30 mol% of potassium carbonate can be mentioned.
- the melting temperature (melting point) of alkali metal nitrate and alkali metal hydroxide is at most about 450 ° C. (lithium hydroxide), even a molten salt containing one kind of nitrate or hydroxide is low.
- the reaction temperature can be realized.
- the mixing ratio of the lithium silicate represented by Li 2 SiO 3 and the iron source is usually preferably such that the amount of iron (Fe) is 0.9 to 1.2 mol per mol of lithium silicate, It is more preferable that the amount be ⁇ 1.1 mol.
- the raw material mixture In the first stage reaction, it is necessary to react the raw material mixture at 300 to 600 ° C. in a mixed gas atmosphere containing carbon dioxide and a reducing gas in the molten salt.
- the specific reaction method is not particularly limited, but usually, a molten salt raw material containing at least one selected from the alkali metal salts described above, lithium silicate, and an iron source are mixed and uniformly mixed using a ball mill or the like. After mixing, the molten salt raw material may be melted by heating above the melting point of the molten salt raw material. As a result, the reaction of lithium, silicon and iron proceeds in the molten salt, and the target lithium silicate compound precursor can be obtained.
- the mixing ratio of the lithium silicate and iron source and the molten salt raw material is not particularly limited, and may be any amount that can uniformly disperse the raw material in the molten salt.
- the lithium silicate compound and iron The total amount of the molten salt raw material is preferably in the range of 20 to 300 parts by mass, more preferably in the range of 50 to 200 parts by mass, and further in the range of 60 to 80 parts by mass with respect to 100 parts by mass of the total source More preferably, it is an amount.
- the reaction temperature between lithium silicate and the iron source in the molten salt may be 300 to 600 ° C., further 400 to 560 ° C. If it is less than 300 ° C., oxide ions (O 2 ⁇ ) are hardly released into the molten salt, and it takes a long time to synthesize a lithium silicate compound precursor, which is not practical. On the other hand, if it exceeds 600 ° C., the resulting lithium silicate compound precursor particles are likely to be coarsened, which is not preferable.
- the reaction described above is performed in a mixed gas atmosphere containing carbon dioxide and a reducing gas in order to allow iron element to exist stably in the molten salt as a divalent ion during the reaction. Under this atmosphere, even if the oxidation number before reaction is an iron element other than divalent, it can be stably maintained in a divalent state.
- the ratio of carbon dioxide and reducing gas is not particularly limited. However, when a large amount of reducing gas is used, carbon dioxide for controlling the oxidizing atmosphere is reduced, so that decomposition of the molten salt raw material is promoted and the reaction rate is increased. However, if the reducing gas is excessive, the divalent iron element of the lithium silicate compound precursor may be reduced due to too high reducing property, and the reaction product may be destroyed.
- the mixing ratio of the mixed gas is 1 to 40, more preferably 3 to 20, with respect to 100 moles of carbon dioxide in terms of volume ratio.
- the reducing gas for example, hydrogen, carbon monoxide and the like can be used, and hydrogen is particularly preferable.
- the pressure of the mixed gas of carbon dioxide and reducing gas there is no particular limitation on the pressure of the mixed gas of carbon dioxide and reducing gas, and it may be usually atmospheric pressure, but it may be under pressure or under reduced pressure.
- the reaction time between the lithium silicate compound and the iron source is usually 10 minutes to 70 hours, preferably 5 to 25 hours, more preferably 10 to 20 hours.
- the lithium silicate compound precursor is obtained by cooling and removing the alkali metal salt used as the flux.
- the alkali metal salt may be dissolved and removed by washing the product using a solvent capable of dissolving the alkali metal salt solidified by cooling after the reaction.
- a solvent capable of dissolving the alkali metal salt solidified by cooling after the reaction For example, water may be used as the solvent.
- the lithium silicate compound precursor obtained by the above-mentioned first stage reaction is represented by a composition formula of Li 2 FeSiO 4 .
- the lithium silicate compound precursor synthesized at a relatively low temperature has a very large specific surface area because it is in the form of fine particles.
- the specific surface area is preferably 15 m 2 / g or more, 30 m 2 / g or more, and more preferably 35 to 50 m 2 / g.
- the specific surface area can be measured by a nitrogen physical adsorption method using a BET adsorption isotherm.
- the lithium silicate compound precursor produced by the first stage reaction is Li 2 FeSiO 4 , and iron (Fe) is in a divalent state, which causes a problem in stability.
- the lithium silicate compound of the present invention is produced by compounding this lithium silicate compound precursor and carbon (C), and the composition formula: Li 1 + 2 ⁇ FeSiO 4 + ⁇ C ( ⁇ 0.25 ⁇ ⁇ ⁇ 0.25, 0 ⁇ C ⁇ 0.5).
- a method for producing this lithium silicate compound will be described. In this production method, first, a carbon composite is formed by a second-stage reaction, and then a target product is generated by the third-stage storage.
- the lithium silicate compound precursor produced in the first stage and a carbon source are mixed well to form a carbon composite.
- the specific method for forming the carbon composite is not particularly limited, and in addition to a vapor phase method in which heat treatment is performed in an atmosphere containing a carbon-containing gas such as methane gas, ethane gas, and butane gas, A thermal decomposition method in which an organic substance is carbonized by heat treatment after uniformly mixing with a lithium silicate compound precursor is also applicable.
- the lithium silicate compound precursor which is a positive electrode active material, is made amorphous by ball milling, and is uniformly mixed with carbon to increase adhesion. Further, heat treatment further recycles the lithium silicate compound precursor. Simultaneously with crystallization, carbon is uniformly deposited around the lithium silicate compound precursor.
- the half-value width of the diffraction peak derived from the (011) plane of the sample having crystallinity before ball milling is B (011) crystal, ball milling
- the ratio of B (011) crystal / B (011) mill may be in the range of about 0.1 to 0.5.
- acetylene black (AB), ketjen black (KB), graphite or the like can be used as a carbon material.
- the mixing ratio of the lithium silicate compound precursor and the carbon material may be 0.1 to 10 mass ratio with the carbon material as carbon (C) with respect to 1 mass ratio of the lithium silicate compound precursor.
- C carbon
- the surplus carbon material that has not been consumed in the second stage reaction can function as a conductive material in the electrode.
- the ball milling process is performed until the lithium silicate compound precursor becomes amorphous, followed by a heat treatment.
- the heat treatment is performed in a reducing atmosphere in order to keep the iron ions contained in the lithium silicate compound precursor divalent.
- the reducing atmosphere in this case is a mixed gas atmosphere of carbon dioxide and reducing gas in order to suppress the divalent iron ions from being reduced to the metal state, as in the first stage reaction. It is preferable.
- the mixing ratio of carbon dioxide and reducing gas may be the same as in the first stage reaction.
- the heat treatment temperature is preferably 500 to 800 ° C. If the heat treatment temperature is too low, it is difficult to deposit carbon uniformly around the lithium silicate compound precursor. On the other hand, if the heat treatment temperature is too high, decomposition of the lithium silicate compound precursor and lithium deficiency occur. In some cases, the charge / discharge capacity decreases, which is not preferable.
- the heat treatment time is usually 30 minutes to 10 hours.
- ⁇ Third stage reaction> In the carbon composite obtained by the above-mentioned second stage production method, it is considered that a reaction in which carbon absorbs Li occurs, for example, by storing for a long period of time, and Li 2 FeSiO 4 is converted into Li 1 + 2 ⁇ FeSiO 4 + ⁇ C ( -0.25 ⁇ ⁇ ⁇ 0.25, 0 ⁇ C ⁇ 0.5), and Li 2 CO 3 is generated as a by-product.
- This reaction occurs even at room temperature, and a lithium silicate compound is formed by storage for about one year. Thereby, the lithium silicate system active material of the present invention is formed.
- reaction time may be shortened by variously adjusting the reaction conditions such as temperature, pressure, and atmosphere.
- the lithium silicate compound of the present invention can be effectively used as an active material for a positive electrode of a lithium secondary battery.
- a positive electrode using this lithium silicate-based active material can have the same structure as a normal positive electrode for a lithium ion secondary battery.
- the above lithium silicate compounds include acetylene black (AB), ketjen black (KB), vapor grown carbon fiber (Vapor Grown Carbon Fiber (VGCF) and other conductive materials, polyvinylidene fluoride (Poly Vinylidine Fluoride: PVdF), polytetrafluoroethylene (PTFE), binders such as styrene-butadiene rubber (SBR), N-methyl-2-pyrrolidone (A positive electrode can be produced by adding a solvent such as NMP) to form a paste and applying it to a current collector.
- the amount of the conductive material used is not particularly limited. For example, it can be 5 to 20 parts by mass with respect to 100 parts by mass of the lithium silicate active material.
- the amount of the binder used is not particularly limited, but may be 5 to 20 parts by mass with respect to 100 parts by mass of the lithium silicate compound, for example.
- a mixture of the lithium silicate active material, the conductive material and the binder described above is kneaded using a mortar or a press to form a film, and this is crimped to the current collector with a press.
- the positive electrode can be manufactured also by the method to do.
- the current collector is not particularly limited, and materials conventionally used as positive electrodes for lithium ion secondary batteries, such as aluminum foil, aluminum mesh, and stainless steel mesh, can be used. Furthermore, a carbon nonwoven fabric, a carbon woven fabric, etc. can be used as a collector.
- the positive electrode for a lithium ion secondary battery according to the present invention is not particularly limited with respect to the shape, thickness, and the like.
- the positive electrode is filled with an active material and then compressed to have a thickness of 10 to 200 ⁇ m.
- the thickness is preferably 20 to 100 ⁇ m. Therefore, the filling amount of the active material may be appropriately determined so as to have the above-described thickness after compression according to the type and structure of the current collector to be used.
- Lithium silicate active material in charged or discharged state The present inventors investigated the valence of Fe in the carbon composite during the second stage reaction using 57 Fe Mossbauer spectroscopy. In the lithium silicate compound precursor before ball milling, almost all Fe was divalent, but when heat-treated after ball milling, divalent Fe decreased to about 50% and trivalent Fe about 50%. Existed.
- the lithium silicate compound of the present invention formed after the third stage reaction, Fe was all trivalent after the third stage reaction. Moreover, when this lithium silicate type compound was charged and discharged using a positive electrode active material, iron repeated bivalent and trivalent. In the state after discharge, the trivalent Fe is 20 to 80%, preferably 20 to 30%.
- a lithium ion secondary battery using the above-described positive electrode for a lithium ion secondary battery can be produced by a known method. That is, the positive electrode described above is used as the positive electrode material, and as the negative electrode material, carbon-based materials such as known metallic lithium and graphite, silicon-based materials such as silicon thin films, alloy-based materials such as copper-tin and cobalt-tin, An oxide material such as lithium titanate is used, and the electrolyte is a known nonaqueous solvent such as ethylene carbonate, dimethyl carbonate, propylene carbonate, dimethyl carbonate, lithium perchlorate, LiPF 6 , LiBF 4 , LiCF 3 SO 3.
- a lithium ion secondary battery can be assembled according to a conventional method using a solution in which a lithium salt such as 0.5 mol / L to 1.7 mol / L is dissolved, and using other known battery components. That's fine.
- the carbonate mixture consists of lithium carbonate (Kishida Chemical Co., Ltd., purity 99.9%), sodium carbonate (Kishida Chemical Co., Ltd., purity 99.5%), and potassium carbonate (Kishida Chemical Co., Ltd., purity 99.5%). , 0.435 mol: 0.315 mol: 0.25 mol. As for the mixing ratio, the carbonate mixture is 90 parts by mass with respect to 100 parts by mass of the total amount of lithium silicate and iron.
- ⁇ Second stage reaction> The obtained powder and acetylene black (AB) are mixed at a mass ratio of 5: 4, and mechanical milling is performed at 450 rpm for 5 hours in an air atmosphere using a mechanical milling device (manufactured by Fritsch Japan Co., Ltd.). It was. Next, the treated powder was subjected to a heat treatment by heating at 700 ° C. for 2 hours in a mixed gas atmosphere of carbon dioxide and hydrogen in a volume ratio of 100: 3 to obtain a carbon composite.
- a mechanical milling device manufactured by Fritsch Japan Co., Ltd.
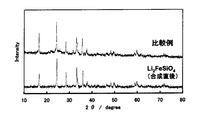
- XRD measurement (radiation source: CuK ⁇ ) was performed on the powders of Examples and Comparative Examples and the powder immediately after being synthesized in the first stage reaction of the Examples, and the results are shown in FIGS.
- the XRD pattern of the powder of the comparative example in FIG. 2 almost coincides with the XRD pattern of the powder immediately after synthesis obtained by the first-stage reaction of the example, whereas the powder of the example of FIG.
- the XRD pattern of the powder was completely different from the XRD pattern of the powder according to the comparative example. This indicates that the composition of the lithium silicate compound contained in the powder of the example and the powder of the comparative example are different. Further, it was found that the powder according to the example shown in FIG. 1 includes XRD patterns corresponding to Li 2 CO 3, Fe 3 O 4 , Li 2 SiO 3 and carbon.
- This Lithium silicate compound is based on a monolith, space group P21 / n, Li 1.5 FeSiO 4.25 model with disordered lithium ions (Li + ) and iron ions (Fe 3+ ), Rietveld Analysis was performed (Figure 3).
- the negative electrode was prepared by pressing a lithium foil (manufactured by Honjo Metal Co., Ltd.) to a copper mesh (# 100 mesh) (size: 20 mm ⁇ 25 mm).
- the positive electrode and the negative electrode were provided with a tab that could be electrically connected to the outside, and a part of this tab extended to the outside of the laminate cell.
- a lithium ion secondary battery in the form of a laminate cell (bipolar pouch cell) was obtained.
- the lithium ion secondary battery was subjected to a charge / discharge test (first time only) at 30 ° C.
- the test conditions are an electrode density of 0.1 C rate (0.05 mA / cm 2 ) and a voltage range of 4.8 to 1.5 V (first charge only for 10 hours at a constant voltage of 4.8 V).
- the obtained charge / discharge curve is shown in FIG. Even if Fe of the lithium silicate-based active material of the positive electrode was all trivalent in the state before charge / discharge, the charge / discharge capacity could be taken out as shown in FIG.
- Test Example 4 The valence of iron in the positive electrode active material after charging (point A in FIG. 6) and discharging (point B in FIG. 6) in Test Example 3 was evaluated by 57 Fe Mossbauer spectroscopy. The respective results are shown in FIG. 7 and FIG.
- Electrolytic solution a solution obtained by dissolving electrolyte LiPF 6 to a concentration of 1.0 mol / L in a mixed solvent in which ethylene carbonate and dimethyl carbonate were mixed at a volume ratio of 1: 1 was used.
- a coin battery was manufactured using the positive electrode and the negative electrode obtained above. Specifically, a separator made of a polypropylene microporous membrane with a thickness of 25 ⁇ m (“Celgard 2400” manufactured by Celgard) and a glass nonwoven fabric filter with a thickness of 500 ⁇ m are sandwiched between a positive electrode and a negative electrode in a dry room. Thus, an electrode body battery was obtained.
- This electrode body battery was accommodated in a battery case (CR2032 type coin battery member) made of a stainless steel container. The electrolyte solution was injected into the battery case. The battery case was sealed with a caulking machine to obtain a lithium ion secondary battery.
- ⁇ Charge / discharge test> The coin battery was subjected to a charge / discharge test at 30 ° C. In other words, it is obtained by charging and discharging 6 cycles at a current density of 0.1 C rate (0.05 mA / cm 2 ) and a voltage range of 4.4 to 0.5 V (only charging for the first time at 4.7 V for 10 hours).
- FIG. 9 shows the charge / discharge curve.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Crystallography & Structural Chemistry (AREA)
- Battery Electrode And Active Subsutance (AREA)
- Silicates, Zeolites, And Molecular Sieves (AREA)
- Silicon Compounds (AREA)
Abstract
リチウムイオン二次電池用正極材料として有用であり、新規なリチウムシリケート系材料を提供する。 Li1.5FeSiO4.25で表されるリチウムシリケート系化合物。リチウム(Li)と、鉄(Fe)と、シリコン(Si)と、酸素(O)とからなるリチウムシリケート系化合物であり、組成式:Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されることを特徴とするリチウムシリケート系化合物。鉄(Fe)が三価に存在できるので、Li2FeSiO4に比べて化学的安定性に優れている。
Description
本発明は、主としてリチウムイオン電池の正極活物質として有用なリチウムシリケート系化合物、及びこのリチウムシリケート系化合物を活物質として用いたリチウムイオン二次電池に関する。
リチウムイオン二次電池は、小型でエネルギー密度が高く、ポータブル電子機器の電源として広く用いられている。その正極活物質としては、主としてLiCoO2などの層状化合物が使われている。しかしながら、これらの化合物は満充電状態において、150℃前後で酸素が脱離しやすく、これが非水電解液の酸化発熱反応を引き起こしやすいという電池の安全性の低下につながる問題点がある。
近年、正極活物質としては、リン酸オリビン系化合物LiMPO4(LiMnPO4、LiFePO4、LiCoPO4など)が提案されている。これらの化合物は、LiCoO2のような酸化物を正極活物質とする三価/四価の酸化還元反応の代わりに、二価/三価の酸化還元反応を用いることにより熱安定性を向上させ、さらに中心金属の周りに電気陰性度の大きいヘテロ元素のポリアニオンを配置することにより高放電電圧の得られる化合物として注目されている。
しかしながら、リン酸オリビン系化合物からなる正極材料は、平均放電電圧が3.5V以下と低いのみならず、リン酸ポリアニオンの大きな式量のため、その理論容量が170mAh/g程度に制限される。さらに、LiCoPO4やLiNiPO4を用いたリチウムイオン二次電池は、動作電圧が4.5Vを超えるため、その充電電圧に耐え得る電解液が無いという問題がある。またLiMnPO4を用いたリチウムイオン二次電池は、サイクル特性の劣化が激しいという問題があり、LiFePO4は最も実用性が高いものの、二価の鉄の酸化による容量の低下という課題を抱えている。
そこで、安価で資源量が多く、環境負荷が低く、高いリチウムイオンの理論充放電容量を有し、かつ高温時に酸素を放出しない正極活物質として、例えば特開2001-266882号公報には、Li2FeSiO4(理論容量331.3mAh/g)、Li2MnSiO4(理論容量333.2mAh/g)等のリチウムシリケート系化合物が記載されている。リチウムシリケート系化合物は、安価で、資源量の豊富な構成金属元素のみからなるために環境負荷が低く、高いリチウムイオンの理論充放電容量を有し、かつ高温時に酸素を放出しない材料であることから、次世代リチウムイオン二次電池正極材料として注目されている。
しかしLi2FeSiO4であっても、鉄(Fe)は二価の状態であるため酸化によって電池容量が低下する場合があるなど、安定性に不安が残る。一方、特開2011-014445公報には、LiFeSi2O6を正極活物質として用いることが記載されている。この化合物では鉄(Fe)は三価の状態であるので、安定性に優れていると考えられる。しかし同公報には、LiFeSi2O6を正極活物質として用いたリチウムイオン二次電池の初回放電容量が102.9mAh/gであったことが記載され、LiFePO4を用いた比較例ほどの容量が得られていない。
本発明は、鉄(Fe)が三価の状態で存在することで安定性に優れた新規なリチウムシリケート系化合物を提供し、そのリチウムシリケート系化合物を正極活物質として用いることで、高い放電容量を発現するリチウムイオン二次電池を提供することを目的とする。
すなわち、本発明のリチウムシリケート系化合物の特徴は、リチウム(Li)と、鉄(Fe)と、シリコン(Si)と、酸素(O)とからなるリチウムシリケート系化合物であり、組成式:Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されることにある。
本発明のリチウムシリケート系化合物は、単斜晶であり、空間群P21/nに帰属する。組成式のδは、δ=0~0.25が好ましい。組成式の鉄(Fe)の電子状態は三価が70%以上が好ましい。リチウムイオン二次電池用正極活物質には炭素(C)と炭酸リチウム(Li2CO3)を含んでもよい。
リチウムイオン二次電池の正極として用いたときに、充電後には鉄(Fe)が全て三価であり57Feメスバウアー分光分析において状態の異なる二種類のFe3+が少なくとも二種類存在するのが好ましい。またはリチウムイオン二次電池の正極として用いたときに、放電後には57Feメスバウアー分光分析においてFe2+とFe3+が存在し、Fe2+が50~80%存在するのが好ましい。
本発明は、リチウム二次電池用正極、二次電池用正極を構成要素として含むリチウムイオン二次電池、リチウム二次電池を搭載した車両の用途として適している。
Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されるリチウムシリケート系化合物では、鉄(Fe)が三価の状態であるため、不安定な二価のFeの場合に比べて安定に存在している。したがって本発明のリチウムシリケート系化合物を活物質として用いたリチウムイオン二次電池は、充放電を繰り返しても安定したサイクル特性を示す。
本発明のリチウムシリケート系化合物の特徴は、リチウム(Li)と、鉄(Fe)と、シリコン(Si)と、酸素(O)とからなるリチウムシリケート系化合物であり、組成式:Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されることにある。
Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されるリチウムシリケート系化合物において、-0.25≦δ≦0.25としたのは、これを活物質として用いたリチウムイオン二次電池の特性の低下を抑制できるためである。
以下、Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されるリチウムシリケート系化合物の製造方法を説明することで、構成の詳細な説明に代える。
<第一段階の反応>
上記リチウムシリケート系化合物を製造するには、事前にリチウムシリケート系化合物前駆体を形成させる。リチウムシリケート系化合物前駆体を形成する方法としては、固相反応法、水熱法、ソルゲル法、Pechini法などを用いることが可能である。以下では溶融塩法を用いた場合について詳細に述べる。
上記リチウムシリケート系化合物を製造するには、事前にリチウムシリケート系化合物前駆体を形成させる。リチウムシリケート系化合物前駆体を形成する方法としては、固相反応法、水熱法、ソルゲル法、Pechini法などを用いることが可能である。以下では溶融塩法を用いた場合について詳細に述べる。
先ず第一段階の反応として、アルカリ金属塩から選ばれた少なくとも一種を含む溶融塩中で、二酸化炭素および還元性ガスを含む混合ガス雰囲気下において、Li2SiO3で表される珪酸リチウムと、鉄源と、を300℃以上600℃以下で反応させ、リチウムシリケート系化合物前駆体を形成する。鉄源としては、純鉄、水酸化鉄、シュウ酸鉄、塩化鉄、硝酸鉄、鉄含有水溶液をアルカリ性にして形成される鉄含有沈殿物などを用いることができる。
鉄含有沈殿物を用いる製造方法によれば、シュウ酸鉄などを用いた製造方法とは化学組成ひいては性質の異なるリチウムシリケート系化合物前駆体が得られる。その結果、特に、電池材料としての特性がより一層優れたリチウムシリケート系化合物前駆体の合成が可能となる。
<溶融塩の組成>
アルカリ金属塩は、リチウム塩、カリウム塩、ナトリウム塩、ルビシウム塩およびセシウム塩からなる群から選ばれる少なくとも一種が挙げられる。なかでも望ましいのは、リチウム塩である。
アルカリ金属塩は、リチウム塩、カリウム塩、ナトリウム塩、ルビシウム塩およびセシウム塩からなる群から選ばれる少なくとも一種が挙げられる。なかでも望ましいのは、リチウム塩である。
また、アルカリ金属塩の種類に特に限定はないが、アルカリ金属炭酸塩、アルカリ金属塩化物、アルカリ金属硝酸塩およびアルカリ金属水酸化物のうちの少なくとも一種を含むことが望ましい。具体的には、炭酸リチウム(Li2CO3)、炭酸カリウム(K2CO3)、炭酸ナトリウム(Na2CO3)、炭酸ルビジウム(Rb2CO3)、炭酸セシウム(Cs2CO3)、塩化リチウム(LiCl)、塩化カリウム(KCl)、塩化ルビシウム(RbCl)、塩化セシウム(CsCl)、硝酸リチウム(LiNO3)、硝酸カリウム(KNO3)、硝酸ナトリウム(NaNO3)、硝酸ルビジウム(RbNO3)、硝酸セシウム(CsNO3)、水酸化リチウム(LiOH)、水酸化カリウム(KOH)、水酸化ナトリウム(NaOH)、水酸化ルビジウム(RbOH)および水酸化セシウム(CsOH)が挙げられ、これらのうちの一種を単独または二種以上を混合して使用するとよい。
たとえば、炭酸リチウム単独では、溶融温度は700℃程度であるが、炭酸リチウムとその他のアルカリ金属塩との混合物の溶融塩とする場合には、溶融温度を600℃以下とすることができ、300~600℃という比較的低い反応温度において、目的とするリチウムシリケート系化合物を合成することが可能となる。その結果、合成反応時に粒成長が抑制されて微細なリチウムシリケート系化合物が形成される。
溶融塩は、溶融温度が600℃以下となるように上記のアルカリ金属塩から選択し、アルカリ金属塩を混合して用いるのであれば混合物の溶融温度が600℃以下となるように混合比を調節して混合溶融塩を得ればよい。混合比は、塩の種類に応じて異なるため、一概に規定することは困難である。
たとえば、炭酸リチウムを必須とし他の炭酸塩を含む炭酸塩混合物を溶融塩として使用するのであれば、通常、炭酸塩混合物全体を100モル%としたとき、炭酸リチウムを30モル%以上さらには30~70モル%含むことが好ましい。炭酸塩混合物の具体例として、炭酸リチウム30~70モル%、炭酸ナトリウム0~60モル%および炭酸カリウム0~50モル%からなる混合物を挙げることができる。このような炭酸塩混合物のさらに好ましい具体例として、炭酸リチウム40~45モル%、炭酸ナトリウム30~35モル%および炭酸カリウム20~30モル%からなる混合物を挙げることができる。
なお、アルカリ金属硝酸塩およびアルカリ金属水酸化物の溶融温度(融点)は高くても450℃程度(水酸化リチウム)であるため、硝酸塩または水酸化物のうち一種を単独で含む溶融塩でも、低い反応温度を実現することができる。
Li2SiO3で表される珪酸リチウムと、鉄源との混合割合については、通常、珪酸リチウム1モルに対して、鉄(Fe)が0.9~1.2モルとなる量とすることが好ましく、0.95~1.1モルとなる量とすることがより好ましい。
第一段階の反応では、上記の溶融塩中で、二酸化炭素および還元性ガスを含む混合ガス雰囲気下において、上記の原料混合物を300~600℃で反応させることが必要である。
具体的な反応方法については特に限定的ではないが、通常は、上記したアルカリ金属塩から選ばれた少なくとも一種を含む溶融塩原料、珪酸リチウム、鉄源を混合し、ボールミル等を用いて均一に混合した後、溶融塩原料の融点以上に加熱して溶融塩原料を溶融させればよい。これにより、溶融塩中において、リチウム、珪素および鉄の反応が進行して、目的とするリチウムシリケート系化合物前駆体を得ることができる。
この際、珪酸リチウム及び鉄源と、溶融塩原料と、の混合割合については特に限定的ではなく、溶融塩中において、原料を均一に分散できる量であればよく、たとえば、珪酸リチウム化合物と鉄源の合計量100質量部に対して、溶融塩原料の合計量が20~300質量部の範囲となる量であることが好ましく、50~200質量部さらには60~80質量部の範囲となる量であることがより好ましい。
溶融塩中における珪酸リチウムと鉄源との反応温度は、300~600℃さらには400~560℃であればよい。300℃未満では、溶融塩中に酸化物イオン(O2-)が放出されにくく、リチウムシリケート系化合物前駆体が合成されるまでに長時間を要するため、実用的ではない。また、600℃を超えると、得られるリチウムシリケート系化合物前駆体の粒子が粗大化し易くなるため好ましくない。
上記した反応は、反応時において、鉄元素を二価イオンとして溶融塩中に安定に存在させるために、二酸化炭素および還元性ガスを含む混合ガス雰囲気下で行う。この雰囲気下では、反応前の酸化数が二価以外の鉄元素であっても二価の状態で安定に維持することが可能となる。二酸化炭素と還元性ガスの比率に特に限定はないが、還元性ガスを多く用いると、酸化雰囲気を制御する二酸化炭素が減少するため、溶融塩原料の分解が促進されて反応速度が速くなる。しかし、還元性ガスが過多では、高過ぎる還元性によりリチウムシリケート系化合物前駆体の二価の鉄元素が還元されて、反応生成物が破壊するおそれがある。そのため、好ましい混合ガスの混合比率は、体積比で、二酸化炭素100モルに対して還元性ガスを1~40さらには3~20とすることが好ましい。還元性ガスとしては、たとえば、水素、一酸化炭素などを用いることができ、水素が特に好ましい。
二酸化炭素と還元性ガスの混合ガスの圧力については、特に限定はなく、通常、大気圧とすればよいが、加圧下、あるいは減圧下のいずれであってもよい。
珪酸リチウム化合物と鉄源との反応時間は、通常、10分~70時間とすればよく、好ましくは5~25時間さらには10~20時間とすればよい。
上記の反応終了後、冷却し、フラックスとして用いたアルカリ金属塩を除去することで、リチウムシリケート系化合物前駆体が得られる。アルカリ金属塩を除去する方法としては、反応後の冷却により固化したアルカリ金属塩を溶解できる溶媒を用いて、生成物を洗浄することによって、アルカリ金属塩を溶解除去すればよい。たとえば、溶媒として、水を用いるとよい。
<リチウムシリケート系化合物前駆体>
上記した第一段階の反応によって得られるリチウムシリケート系化合物前駆体は、Li2FeSiO4の組成式で表される。溶融塩中において、600℃以下という低温で反応を行うことによって、結晶粒の成長が抑制され、平均粒径が数μm以下の微細な粒子となり、さらに、不純物相の量が大きく減少する。
上記した第一段階の反応によって得られるリチウムシリケート系化合物前駆体は、Li2FeSiO4の組成式で表される。溶融塩中において、600℃以下という低温で反応を行うことによって、結晶粒の成長が抑制され、平均粒径が数μm以下の微細な粒子となり、さらに、不純物相の量が大きく減少する。
また、比較的低温で合成されたリチウムシリケート系化合物前駆体は、微粒子状であることから比表面積がきわめて大きい。具体的には、比表面積が15m2/g以上、30m2/g以上さらには35~50m2/gであるのが好ましい。なお比表面積は、BET吸着等温式を用いた窒素物理吸着法により測定することができる。
<第二段階の反応>
しかし上記第一段階の反応によって製造されたリチウムシリケート系化合物前駆体は、Li2FeSiO4であり、鉄(Fe)は二価の状態であるため安定性に問題がある。一方、本発明のリチウムシリケート系化合物は、このリチウムシリケート系化合物前駆体と炭素(C)とを複合化させることにより生成し、組成式:Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表される。以下、このリチウムシリケート系化合物の製造方法を説明する。この製造方法は、先ず第二段階の反応でカーボン複合体を形成し、次いで第三段階の保存に伴う目的物の生成反応を行う。
しかし上記第一段階の反応によって製造されたリチウムシリケート系化合物前駆体は、Li2FeSiO4であり、鉄(Fe)は二価の状態であるため安定性に問題がある。一方、本発明のリチウムシリケート系化合物は、このリチウムシリケート系化合物前駆体と炭素(C)とを複合化させることにより生成し、組成式:Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表される。以下、このリチウムシリケート系化合物の製造方法を説明する。この製造方法は、先ず第二段階の反応でカーボン複合体を形成し、次いで第三段階の保存に伴う目的物の生成反応を行う。
第二段階の反応では、先ず、第一段階で製造されたリチウムシリケート系化合物前駆体と、炭素源と、をよく混合してカーボン複合体を形成する。カーボン複合体を形成する具体的な方法については、特に限定的ではなく、メタンガス、エタンガス、ブタンガスなどのような炭素含有ガスを含む雰囲気において熱処理を行う気相法の他、炭素源となる有機物とリチウムシリケート系化合物前駆体とを均一に混合した後に熱処理によって有機物を炭化させることによる熱分解法も適用可能である。
特に、リチウムシリケート系化合物前駆体にカーボン材料を加え、ボールミルによってリチウムシリケート系化合物前駆体がアモルファス化するまで均一に混合するメカニカルミリング法を適用し、その後熱処理することが好ましい。この方法によれば、ボールミリングによって正極活物質であるリチウムシリケート系化合物前駆体がアモルファス化され、カーボンと均一に混合されて密着性が増加し、さらに熱処理により、リチウムシリケート系化合物前駆体の再結晶化と同時にカーボンがリチウムシリケート系化合物前駆体の周りに均一に析出する。
アモルファス化の程度については、CuKα線を光源とするX線回折測定において、ボールミリング前の結晶性を有する試料についての(011)面由来の回折ピークの半値幅をB(011)crystal、ボールミリングにより得られた試料の同ピークの半値幅をB(011)millとした場合に、B(011)crystal/B(011)millの比が0.1~0.5程度の範囲であればよい。
この第二段階の反応には、カーボン材料として、アセチレンブラック(AB)、ケッチェンブラック(KB)、黒鉛等を用いることができる。リチウムシリケート系化合物前駆体とカーボン材料との混合割合については、リチウムシリケート系化合物前駆体の1質量比に対して、カーボン材料を炭素(C)として0.1~10質量比とすればよい。後述の第三段階の反応を確実に進行させるために、カーボンをリチウムシリケート系化合物前駆体より多くする必要がある。第二段階の反応で消費されなかった余剰のカーボン材料は、電極における導電材として機能させることができる。
リチウムシリケート系化合物前駆体がアモルファス化するまでボールミリング処理を行った後、熱処理を行う。熱処理は、リチウムシリケート系化合物前駆体に含まれる鉄イオンを二価に保持するために、還元性雰囲気下で行う。この場合の還元性雰囲気としては、第一段階の反応と同様に、二価の鉄イオンが金属状態まで還元されることを抑制するために、二酸化炭素と還元性ガスの混合ガス雰囲気中であることが好ましい。二酸化炭素と還元性ガスの混合割合は、第一段階の反応と同様とすればよい。
熱処理温度は、500~800℃とすることが好ましい。熱処理温度が低すぎる場合には、リチウムシリケート系化合物前駆体の周りにカーボンを均一に析出させることが難しく、一方、熱処理温度が高すぎると、リチウムシリケート系化合物前駆体の分解やリチウム欠損が生じることがあり、充放電容量が低下するので好ましくない。また、熱処理時間は、通常、30分~10時間とすればよい。
<第三段階の反応>
上記した第二段階の製造方法によって得られたカーボン複合体では、例えば長期間保存するなどによって、カーボンがLiを吸収する反応が生じると考えられ、Li2FeSiO4がLi1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されるリチウムシリケート系化合物に変化すると共に、副生成物としてLi2CO3が生成する。この反応は室温でも生じ、1年間程度の保存によってリチウムシリケート系化合物が形成される。これにより本発明のリチウムシリケート系活物質が形成される。
上記した第二段階の製造方法によって得られたカーボン複合体では、例えば長期間保存するなどによって、カーボンがLiを吸収する反応が生じると考えられ、Li2FeSiO4がLi1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されるリチウムシリケート系化合物に変化すると共に、副生成物としてLi2CO3が生成する。この反応は室温でも生じ、1年間程度の保存によってリチウムシリケート系化合物が形成される。これにより本発明のリチウムシリケート系活物質が形成される。
なお、室温で保存する以外に、温度条件、圧力条件、雰囲気など、反応条件を種々調整することで、反応時間を短縮できる可能性がある。
<リチウムイオン二次電池用正極>
本発明のリチウムシリケート系化合物はリチウム二次電池正極用活物質として有効に使用できる。このリチウムシリケート系活物質を用いる正極は、通常のリチウムイオン二次電池用正極と同様の構造とすることができる。
本発明のリチウムシリケート系化合物はリチウム二次電池正極用活物質として有効に使用できる。このリチウムシリケート系活物質を用いる正極は、通常のリチウムイオン二次電池用正極と同様の構造とすることができる。
例えば、上記リチウムシリケート系化合物に、アセチレンブラック(AB)、ケッチェンブラック(KB)、気相法炭素繊維(Vapor
Grown Carbon Fiber:VGCF)等の導電材、ポリフッ化ビニリデン(Poly Vinylidine Fluoride:PVdF)、ポリ四フッ化エチレン(PTFE)、スチレン-ブタジエンゴム(SBR)等のバインダー、N-メチル-2-ピロリドン(NMP)等の溶媒を加えてペースト状として、これを集電体に塗布することによって正極を作製することができる。導電材の使用量については、特に限定的ではないが、たとえば、リチウムシリケート系活物質100質量部に対して、5~20質量部とすることができる。また、バインダーの使用量についても、特に限定的ではないが、例えば、リチウムシリケート系化合物100質量部に対して、5~20質量部とすることができる。また、その他の方法として、リチウムシリケート系活物質と、上記の導電材およびバインダーを混合したものを、乳鉢やプレス機を用いて混練してフィルム状とし、これを集電体へプレス機で圧着する方法によっても正極を製造することが出来る。
Grown Carbon Fiber:VGCF)等の導電材、ポリフッ化ビニリデン(Poly Vinylidine Fluoride:PVdF)、ポリ四フッ化エチレン(PTFE)、スチレン-ブタジエンゴム(SBR)等のバインダー、N-メチル-2-ピロリドン(NMP)等の溶媒を加えてペースト状として、これを集電体に塗布することによって正極を作製することができる。導電材の使用量については、特に限定的ではないが、たとえば、リチウムシリケート系活物質100質量部に対して、5~20質量部とすることができる。また、バインダーの使用量についても、特に限定的ではないが、例えば、リチウムシリケート系化合物100質量部に対して、5~20質量部とすることができる。また、その他の方法として、リチウムシリケート系活物質と、上記の導電材およびバインダーを混合したものを、乳鉢やプレス機を用いて混練してフィルム状とし、これを集電体へプレス機で圧着する方法によっても正極を製造することが出来る。
集電体としては、特に限定はなく、従来からリチウムイオン二次電池用正極として使用されている材料、たとえば、アルミ箔、アルミメッシュ、ステンレスメッシュなどを用いることができる。さらに、カーボン不織布、カーボン織布なども集電体として使用できる。
本発明に係るリチウムイオン二次電池用正極は、その形状、厚さなどについては特に限定的ではないが、たとえば、活物質を充填した後、圧縮することによって、厚さを10~200μm、より好ましくは20~100μmとすることが好ましい。従って、使用する集電体の種類、構造等に応じて、圧縮後に上記した厚さとなるように、活物質の充填量を適宜決めればよい。
<充電状態または放電状態のリチウムシリケート系活物質>
本発明者らは、57Feメスバウアー分光法を用いて、第二段階の反応時におけるカーボン複合体のFeの価数を調べた。ボールミリング前におけるリチウムシリケート系化合物前駆体では、Feはほぼ全部が二価であったが、ボールミリング後に熱処理すると、二価のFeは約50%に減少し、三価のFeが約50%存在していた。
本発明者らは、57Feメスバウアー分光法を用いて、第二段階の反応時におけるカーボン複合体のFeの価数を調べた。ボールミリング前におけるリチウムシリケート系化合物前駆体では、Feはほぼ全部が二価であったが、ボールミリング後に熱処理すると、二価のFeは約50%に減少し、三価のFeが約50%存在していた。
それに対し、第三段階の反応後に形成された本発明のリチウムシリケート系化合物では、第三段階の反応後にはFeは全て三価であった。また、このリチウムシリケート系化合物を正極活物質として用いて充放電させると、鉄は二価と三価を繰り返した。放電後の状態では、三価のFeは20~80%、好ましくは20~30%である。
<リチウムイオン二次電池>
上記したリチウムイオン二次電池用正極を用いるリチウムイオン二次電池は、公知の手法により製造することができる。すなわち、正極材料として、上記した正極を使用し、負極材料として、公知の金属リチウム、黒鉛などの炭素系材料、シリコン薄膜などのシリコン系材料、銅-錫やコバルト-錫などの合金系材料、チタン酸リチウムなどの酸化物材料を使用し、電解液として、公知のエチレンカーボネート、ジメチルカーボネート、プロピレンカーボネート、ジメチルカーボネートなどの非水系溶媒に過塩素酸リチウム、LiPF6、LiBF4、LiCF3SO3などのリチウム塩を0.5mol/Lから1.7mol/Lの濃度で溶解させた溶液を使用し、さらにその他の公知の電池構成要素を使用して、常法に従って、リチウムイオン二次電池を組立てればよい。
上記したリチウムイオン二次電池用正極を用いるリチウムイオン二次電池は、公知の手法により製造することができる。すなわち、正極材料として、上記した正極を使用し、負極材料として、公知の金属リチウム、黒鉛などの炭素系材料、シリコン薄膜などのシリコン系材料、銅-錫やコバルト-錫などの合金系材料、チタン酸リチウムなどの酸化物材料を使用し、電解液として、公知のエチレンカーボネート、ジメチルカーボネート、プロピレンカーボネート、ジメチルカーボネートなどの非水系溶媒に過塩素酸リチウム、LiPF6、LiBF4、LiCF3SO3などのリチウム塩を0.5mol/Lから1.7mol/Lの濃度で溶解させた溶液を使用し、さらにその他の公知の電池構成要素を使用して、常法に従って、リチウムイオン二次電池を組立てればよい。
以下に、リチウムシリケート系化合物の製造方法を説明することで、本実施例のリチウムシリケート系化合物の構造の詳細な説明に代える。
<第一段階の反応>
珪酸リチウム(Li2SiO3)(キシダ化学株式会社製、純度99.5%)0.03モルと、鉄(高純度化学株式会社製、純度99.9%)0.03モルとの混合物に、アセトン20mlを加えてジルコニア製ボールミルにて500rpmで60分間混合し、乾燥した。これを炭酸塩混合物と混合した。炭酸塩混合物は、炭酸リチウム(キシダ化学株式会社製、純度99.9%)と、炭酸ナトリウム(キシダ化学株式会社製、純度99.5%)と、炭酸カリウム(キシダ化学株式会社製、純度99.5%)とを、0.435モル:0.315モル:0.25モルのモル比で混合したものである。混合割合は、珪酸リチウムと鉄との合計量100質量部に対して炭酸塩混合物が90質量部である。
珪酸リチウム(Li2SiO3)(キシダ化学株式会社製、純度99.5%)0.03モルと、鉄(高純度化学株式会社製、純度99.9%)0.03モルとの混合物に、アセトン20mlを加えてジルコニア製ボールミルにて500rpmで60分間混合し、乾燥した。これを炭酸塩混合物と混合した。炭酸塩混合物は、炭酸リチウム(キシダ化学株式会社製、純度99.9%)と、炭酸ナトリウム(キシダ化学株式会社製、純度99.5%)と、炭酸カリウム(キシダ化学株式会社製、純度99.5%)とを、0.435モル:0.315モル:0.25モルのモル比で混合したものである。混合割合は、珪酸リチウムと鉄との合計量100質量部に対して炭酸塩混合物が90質量部である。
上記混合物にアセトン20mlを加えてジルコニア製ボールミルにて500rpmで60分間混合し、乾燥した。その後、得られた粉体を金坩堝に入れ、二酸化炭素(流量100ml/min)と水素(流量3ml/min)の混合ガス雰囲気下にて電気炉で500℃に加熱し、炭酸塩混合物が溶融した状態で13時間反応させた。反応後、反応系である炉心全体を電気炉から取り出し、混合ガスを通じた状態で室温まで冷却した。
次いで、得られた反応物に水20mlを加えて乳鉢ですりつぶし、水を用いて洗浄と濾過を繰り返して、塩が除去された粉体を得た。この粉体を100℃の乾燥機に入れて1時間程度乾燥した。
<第二段階の反応>
得られた粉体とアセチレンブラック(AB)とを質量比5:4で混合し、メカニカルミリング装置(フリッチュジャパン(株)製)を用い、大気雰囲気下において450rpmで5時間のメカニカルミリング処理を行った。次いで処理後の粉体を、体積比で二酸化炭素と水素が100:3の混合ガス雰囲気下、700℃で2時間加熱する熱処理を行い、カーボン複合体を得た。
得られた粉体とアセチレンブラック(AB)とを質量比5:4で混合し、メカニカルミリング装置(フリッチュジャパン(株)製)を用い、大気雰囲気下において450rpmで5時間のメカニカルミリング処理を行った。次いで処理後の粉体を、体積比で二酸化炭素と水素が100:3の混合ガス雰囲気下、700℃で2時間加熱する熱処理を行い、カーボン複合体を得た。
<第三段階の反応>
上記熱処理後のカーボン複合体からなる粉体をデシケータに入れ、室温で1年間保存することで、本実施例のリチウムシリケート系化合物を主成分とする粉体を得た。
上記熱処理後のカーボン複合体からなる粉体をデシケータに入れ、室温で1年間保存することで、本実施例のリチウムシリケート系化合物を主成分とする粉体を得た。
[比較例]
実施例において、第一段階の反応と第二段階の反応とを行い、第二段階の反応直後のカーボン複合体からなる粉体を比較例とした。
実施例において、第一段階の反応と第二段階の反応とを行い、第二段階の反応直後のカーボン複合体からなる粉体を比較例とした。
<試験例1>
実施例及び比較例の粉体と、実施例の第一段階の反応で合成された直後の粉体について、XRD測定(線源:CuKα)を行い、結果を図1及び図2に示す。図2の比較例の粉体のXRDパターンは、実施例の第一段階の反応で得られた合成直後の粉体のXRDパターンとほとんど一致しているのに対し、図1の実施例に係る粉体のXRDパターンは、比較例に係る粉体のXRDパターンとは全く異なった。これは実施例の粉体と比較例の粉体とで含まれるリチウムシリケート系化合物の組成が異なることを示している。また図1に示す実施例に係る粉体では、Li2CO3およびFe3O4、Li2SiO3、カーボンに対応するXRDパターンを含んでいることがわかった。
実施例及び比較例の粉体と、実施例の第一段階の反応で合成された直後の粉体について、XRD測定(線源:CuKα)を行い、結果を図1及び図2に示す。図2の比較例の粉体のXRDパターンは、実施例の第一段階の反応で得られた合成直後の粉体のXRDパターンとほとんど一致しているのに対し、図1の実施例に係る粉体のXRDパターンは、比較例に係る粉体のXRDパターンとは全く異なった。これは実施例の粉体と比較例の粉体とで含まれるリチウムシリケート系化合物の組成が異なることを示している。また図1に示す実施例に係る粉体では、Li2CO3およびFe3O4、Li2SiO3、カーボンに対応するXRDパターンを含んでいることがわかった。
このリチウムシリケート系化合物について、単斜晶、空間群P21/n、リチウムイオン(Li+)と鉄イオン(Fe3+)が不規則化したLi1.5FeSiO4.25のモデルを基にリートベルト解析を行った(図3)。実測値は、Li1.5FeSiO4.25の計算値と各相(Li2CO3、Fe3O4、Li2SiO3、カーボン)から得られるパターンでフィッティングすることができた。また解析で得られた信頼度因子(Rwp = 7.58 S = 2.33 RI = 5.13)が小さいことから、Li1.5FeSiO4.25の生成を裏付けることができた。Li1.5FeSiO4.25の格子定数は、 a=8.3888(8)Å, b=5.0278(1) Å, c=8.3546(5) Å, β=103.310(4)度であった。但し、括弧内の数字は標準偏差を表す。この化合物の結晶系は単斜晶、空間群P21/nであった。
<試験例2>
実施例の粉体と比較例の粉体について、57Feメスバウアー分光測定を行った。結果を図4及び図5に示す。測定条件を下記に示す。
実施例の粉体と比較例の粉体について、57Feメスバウアー分光測定を行った。結果を図4及び図5に示す。測定条件を下記に示す。
装置:トポロジックシステムズ製「FGX-100」(γ線源:57CoをRhマトリックスに分散)
測定条件:速度範囲±3mm/s、室温、速度基準はα-Fe
解析:常磁性体の典型的な形状である対称なローレンツ吸収線2本一組をダブレット成分の一成分として解析を実施した。2本の吸収線の中心位置の0速度からのずれを異性体シフト値として算出。鉄の価数はその異性体シフト値に基づき帰属した。成分量は各ダブレット成分の面積割合より算出した。
測定条件:速度範囲±3mm/s、室温、速度基準はα-Fe
解析:常磁性体の典型的な形状である対称なローレンツ吸収線2本一組をダブレット成分の一成分として解析を実施した。2本の吸収線の中心位置の0速度からのずれを異性体シフト値として算出。鉄の価数はその異性体シフト値に基づき帰属した。成分量は各ダブレット成分の面積割合より算出した。
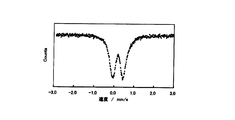
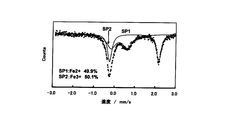
実施例の粉体では、Feは全て三価(異性体シフト値:0.1962(5)mm/s)(図4)であったのに対し、比較例の粉体では二価のFe(異性体シフト値:0.965(1)mm/s)が49.9%、三価のFe(異性体シフト値:0.219(5)mm/s)が50.1%であった(図5)。すなわち実施例の第三段階の反応において、リチウムシリケート系化合物中のFeが全て三価に変化したことが明らかである(図4)。そして実施例の粉体にはLi2CO3が含まれていることから、第三段階の反応では、カーボン複合体中のカーボンがLiを吸収してLi2FeSiO4がLi1.5FeSiO4.25に構造変化したと考えられる。
<試験例3>
実施例で製造された粉体を正極活物質として用いたリチウムイオン二次電池を作製し、その特性を評価した。
実施例で製造された粉体を正極活物質として用いたリチウムイオン二次電池を作製し、その特性を評価した。
<正極の作成>
実施例で製造された粉体:導電助剤(アセチレンブラック(AB))、ポリテトラフルオロエチレン(PTFE)=17.1:4.7:1(質量比)の混合物を混練した後フィルム状にして、アルミニウムメッシュ(#100メッシュ)(サイズ:20mm×25mm)に卓上プレス機で圧着して電極を製作し、140℃で3時間真空乾燥したものを正極として用いた。
実施例で製造された粉体:導電助剤(アセチレンブラック(AB))、ポリテトラフルオロエチレン(PTFE)=17.1:4.7:1(質量比)の混合物を混練した後フィルム状にして、アルミニウムメッシュ(#100メッシュ)(サイズ:20mm×25mm)に卓上プレス機で圧着して電極を製作し、140℃で3時間真空乾燥したものを正極として用いた。
<負極の作成>
負極は、リチウム箔(本城金属(株)製)を銅メッシュ(#100メッシュ)(サイズ:20mm×25mm)に圧着することにより作製した。
負極は、リチウム箔(本城金属(株)製)を銅メッシュ(#100メッシュ)(サイズ:20mm×25mm)に圧着することにより作製した。
<リチウムイオン二次電池の作製>
上記の正極と負極をラミネートフィルムで収容した。この正極および負極の間に、ポリプロピレン微孔質膜からなるセパレータ(「Celgard 2400」Celgard社製)を挟装した。これをラミネートフィルムで覆い、三辺をシールした後、袋状となったラミネートフィルムに上記の電解液を注入した。その後、残りの一辺をシールすることで、四辺が気密にシールされ、極板群および電解液が密閉されたラミネートセルを得た。電解液は、EC(エチレンカーボネート):DMC(ジメチルカーボネート)=1:1(体積比)の混合溶液にLiPF6を1mol/Lとなる濃度で溶解したものを用いた。正極および負極は外部と電気的に接続可能なタブを備え、このタブの一部はラミネートセルの外側に延出した。以上の工程で、ラミネートセル(2極パウチセル)状のリチウムイオン二次電池を得た。
上記の正極と負極をラミネートフィルムで収容した。この正極および負極の間に、ポリプロピレン微孔質膜からなるセパレータ(「Celgard 2400」Celgard社製)を挟装した。これをラミネートフィルムで覆い、三辺をシールした後、袋状となったラミネートフィルムに上記の電解液を注入した。その後、残りの一辺をシールすることで、四辺が気密にシールされ、極板群および電解液が密閉されたラミネートセルを得た。電解液は、EC(エチレンカーボネート):DMC(ジメチルカーボネート)=1:1(体積比)の混合溶液にLiPF6を1mol/Lとなる濃度で溶解したものを用いた。正極および負極は外部と電気的に接続可能なタブを備え、このタブの一部はラミネートセルの外側に延出した。以上の工程で、ラミネートセル(2極パウチセル)状のリチウムイオン二次電池を得た。
このリチウムイオン二次電池について、30℃にて充放電試験(初回のみ)を行った。試験条件は、電極密度0.1Cレート(0.05mA/cm2)、電圧範囲4.8~1.5V(初回充電のみ4.8Vにて10時間定電圧充電)である。得られた充放電曲線を図6に示す。正極のリチウムシリケート系活物質のFeが充放電前の状態において全て三価であっても、図6のように、充放電容量を取り出すことができた。
<試験例4>
試験例3における充電後(図6のA点)及び放電後(図6のB点)の正極活物質中の鉄の価数を57Feメスバウアー分光測定により評価した。それぞれの結果を図7及び図8に示す。
試験例3における充電後(図6のA点)及び放電後(図6のB点)の正極活物質中の鉄の価数を57Feメスバウアー分光測定により評価した。それぞれの結果を図7及び図8に示す。
充電後ではFeは全て三価であり、状態の異なる二種類のFeが47.3%(異性体シフト値:0.45(6)mm/s)と52.7%(異性体シフト値:0.188(5)mm/s)の比率で存在しているのが認められた。放電後には、三価のFeが22.1%(異性体シフト値:0.23(1)mm/s)、二価のFeが77.9%(異性体シフト値:0.964(3)mm/s)であった。
<試験例5>
実施例で製造された粉体を正極活物質として用いたリチウムイオン二次電池を作製し、そのサイクル特性を評価した。
実施例で製造された粉体を正極活物質として用いたリチウムイオン二次電池を作製し、そのサイクル特性を評価した。
<正極の作成>
実施例で製造された粉体:導電助剤(アセチレンブラック(AB)):ポリテトラフルオロエチレン(PTFE)=17.1:4.7:1(質量比)の混合物を混練した後フィルム状にして、φ14mmの円形アルミニウムメッシュ(#100メッシュ)に卓上プレス機で圧着して電極を製作し、140℃で3時間真空乾燥したものを正極として用いた。
実施例で製造された粉体:導電助剤(アセチレンブラック(AB)):ポリテトラフルオロエチレン(PTFE)=17.1:4.7:1(質量比)の混合物を混練した後フィルム状にして、φ14mmの円形アルミニウムメッシュ(#100メッシュ)に卓上プレス機で圧着して電極を製作し、140℃で3時間真空乾燥したものを正極として用いた。
<負極の作成>
黒鉛粉体:導電助剤(ケッチェンブラック(KB)):ポリフッ化ビニリデン(PVdF)=90:2:8 質量%で調合した混合体に、N-メチル-2-ピロリドンを添加し、負極用スラリーを調製した。ドクターブレードを用いてこのスラリーを厚さ18μmの電解銅箔(集電体)に塗布し、負極を作製した。その後、80℃で30分間乾燥し、負極中から有機溶媒を揮発させて除去した。乾燥後、ロールプレス機により、負極の電極密度を調整した後、真空乾燥炉にて170℃で8時間加熱硬化させた。
黒鉛粉体:導電助剤(ケッチェンブラック(KB)):ポリフッ化ビニリデン(PVdF)=90:2:8 質量%で調合した混合体に、N-メチル-2-ピロリドンを添加し、負極用スラリーを調製した。ドクターブレードを用いてこのスラリーを厚さ18μmの電解銅箔(集電体)に塗布し、負極を作製した。その後、80℃で30分間乾燥し、負極中から有機溶媒を揮発させて除去した。乾燥後、ロールプレス機により、負極の電極密度を調整した後、真空乾燥炉にて170℃で8時間加熱硬化させた。
<電解液>
電解液としては、エチレンカーボネートとジメチルカーボネートとを体積比1:1で混合した混合溶媒に、電解質LiPF6を濃度1.0mol/Lとなるように溶解したものを用いた。
電解液としては、エチレンカーボネートとジメチルカーボネートとを体積比1:1で混合した混合溶媒に、電解質LiPF6を濃度1.0mol/Lとなるように溶解したものを用いた。
<リチウムイオン二次電池>
上記で得られた正極および負極を用いて、コイン電池を製作した。詳しくは、ドライルーム内で、厚さ25μmのポリプロピレン微孔質膜からなるセパレータ(「Celgard 2400」Celgard社製)と、厚さ500μmのガラス不織布フィルタと、を正極と負極との間に挟装して、電極体電池とした。この電極体電池を、ステンレス容器からなる電池ケース(CR2032型コイン電池用部材)に収容した。電池ケースには上記電解液を注入した。電池ケースをカシメ機で密閉して、リチウムイオン二次電池を得た。
上記で得られた正極および負極を用いて、コイン電池を製作した。詳しくは、ドライルーム内で、厚さ25μmのポリプロピレン微孔質膜からなるセパレータ(「Celgard 2400」Celgard社製)と、厚さ500μmのガラス不織布フィルタと、を正極と負極との間に挟装して、電極体電池とした。この電極体電池を、ステンレス容器からなる電池ケース(CR2032型コイン電池用部材)に収容した。電池ケースには上記電解液を注入した。電池ケースをカシメ機で密閉して、リチウムイオン二次電池を得た。
<充放電試験>
このコイン電池について30℃にて充放電試験を行った。すなわち、電流密度0.1Cレート(0.05mA/cm2)にて電圧範囲4.4~0.5V(初回充電のみ4.7Vにて10時間定電圧充電は)の条件で6サイクルの充放電を行い、得られた充放電曲線を図9に示す。
このコイン電池について30℃にて充放電試験を行った。すなわち、電流密度0.1Cレート(0.05mA/cm2)にて電圧範囲4.4~0.5V(初回充電のみ4.7Vにて10時間定電圧充電は)の条件で6サイクルの充放電を行い、得られた充放電曲線を図9に示す。
図9から明らかなように、本実施例のリチウムシリケート系活物質を正極に用いることで、初期充電231mAh/g、初期放電147mAh/gの容量を示し、2サイクル目では、充電容量144mAh/g、放電容量138mAh/gを示した。以降も安定した充放電特性を示した。
Claims (11)
- リチウム(Li)と、鉄(Fe)と、シリコン(Si)と、酸素(O)とからなるリチウムシリケート系化合物であり、
組成式:Li1+2δFeSiO4+δ-C(-0.25≦δ≦0.25、0≦C≦0.5)で表されることを特徴とするリチウムシリケート系化合物。 - 単斜晶であり、空間群P21/nに帰属する請求項1に記載のリチウムシリケート系化合物。
- 前記δは、δ=0~0.25である請求項1又は請求項2に記載のリチウムシリケート系化合物。
- 鉄(Fe)の電子状態は三価が50%以上である請求項1~3のいずれかに記載のリチウムシリケート系化合物。
- 請求項1~請求項4のいずれかに記載のリチウムシリケート系化合物を含むことを特徴とするリチウムイオン二次電池用正極活物質。
- 炭素(C)と炭酸リチウム(Li2CO3)を含む請求項5に記載のリチウムイオン二次電池用正極活物質。
- リチウムイオン二次電池の正極として用いたときに、充電後には鉄(Fe)が全て三価であり57Feメスバウアー分光分析において状態の異なる二種類のFe3+が少なくとも二種類存在する請求項5又は請求項6に記載のリチウムイオン二次電池用正極活物質。
- リチウムイオン二次電池の正極として用いたときに、放電後には57Feメスバウアー分光分析においてFe2+とFe3+が存在し、Fe2+が50~80%存在する請求項7に記載のリチウムイオン二次電池用正極活物質。
- 請求項5~8のいずれかに記載のリチウムイオン二次電池用正極活物質を含むリチウム二次電池用正極。
- 請求項9に記載のリチウム二次電池用正極を構成要素として含むリチウムイオン二次電池。
- 請求項10に記載のリチウムイオン二次電池を搭載した車両。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/351,714 US9330805B2 (en) | 2011-10-14 | 2012-07-26 | Lithium silicate-based compound, positive electrode active material for lithium ion secondary battery, and lithium ion secondary battery using the same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-226737 | 2011-10-14 | ||
| JP2011226737A JP5765780B2 (ja) | 2011-10-14 | 2011-10-14 | リチウムシリケート系化合物とリチウムイオン二次電池用正極活物質及びこれを用いたリチウムイオン二次電池 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013054457A1 true WO2013054457A1 (ja) | 2013-04-18 |
Family
ID=48081529
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/004768 WO2013054457A1 (ja) | 2011-10-14 | 2012-07-26 | リチウムシリケート系化合物とリチウムイオン二次電池用正極活物質及びこれを用いたリチウムイオン二次電池 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9330805B2 (ja) |
| JP (1) | JP5765780B2 (ja) |
| WO (1) | WO2013054457A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103746116A (zh) * | 2014-01-10 | 2014-04-23 | 国家纳米科学中心 | 一种碳包覆的硅酸亚铁锂正极材料、制备方法及其用途 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101241810B1 (ko) * | 2009-02-04 | 2013-04-01 | 가부시키가이샤 도요다 지도숏키 | 리튬실리케이트계 화합물의 제조 방법, 이 제조 방법에 의해 얻어진 리튬실리케이트계 화합물로 이루어진 리튬 이온 이차 전지용 정극 활물질, 상기 화합물을 포함하는 리튬 이차 전지용 정극 및 리튬 이차 전지 |
| JP2013258035A (ja) * | 2012-06-12 | 2013-12-26 | Denso Corp | リチウムイオン二次電池用電極活物質及びリチウムイオン二次電池 |
| GB2517460A (en) * | 2013-08-21 | 2015-02-25 | Univ Newcastle | Metal-air batteries |
| CN107210442B (zh) * | 2015-01-28 | 2020-06-16 | 三洋电机株式会社 | 非水电解质二次电池用负极活性物质和非水电解质二次电池 |
| CN111647863B (zh) * | 2020-07-02 | 2022-03-25 | 河北大学 | Li2FexSiO4正极薄膜的制备方法及应用 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2271354C (en) | 1999-05-10 | 2013-07-16 | Hydro-Quebec | Lithium insertion electrode materials based on orthosilicate derivatives |
| CN102037601B (zh) * | 2007-07-12 | 2014-04-23 | A123系统公司 | 用于锂离子电池的多功能混合金属橄榄石 |
| CA2623407A1 (en) * | 2008-02-28 | 2009-08-28 | Hydro-Quebec | Composite electrode material |
| JP5352358B2 (ja) | 2009-07-03 | 2013-11-27 | シャープ株式会社 | 非水電解質二次電池用正極活物質及び非水電解質二次電池 |
| US9318741B2 (en) * | 2010-04-28 | 2016-04-19 | Semiconductor Energy Laboratory Co., Ltd. | Positive electrode active material of power storage device, power storage device, electrically propelled vehicle, and method for manufacturing power storage device |
-
2011
- 2011-10-14 JP JP2011226737A patent/JP5765780B2/ja not_active Expired - Fee Related
-
2012
- 2012-07-26 US US14/351,714 patent/US9330805B2/en not_active Expired - Fee Related
- 2012-07-26 WO PCT/JP2012/004768 patent/WO2013054457A1/ja active Application Filing
Non-Patent Citations (3)
| Title |
|---|
| HUANG,X. ET AL.: "Synthesis and electrochemical performance of Li2FeSi04/C as cathode material for lithium batteries", SOLID STATE IONICS, vol. 181, 2010, pages 1451 - 1455, XP027347167 * |
| LV,D. ET AL.: "A novel Li2FeSi04/C composite: Synthesis, characterization and high storage capacity", JOURNAL OF MATERIALS CHEMISTRY, vol. 21, 14 July 2011 (2011-07-14), pages 9506 - 9512 * |
| MASANOBU NAKAYAMA ET AL.: "Dai Ichi Genri Keisan ni yoru Li2-,FeSiO4 Lithium Ion Denchi Seikyoku Zairyo no Soheiko ni Kansuru Kenkyu", THE ELECTROCHEMICAL SOCIETY OF JAPAN TAIKAI KOEN YOSHISHU, vol. 76, 29 March 2009 (2009-03-29), pages 335 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103746116A (zh) * | 2014-01-10 | 2014-04-23 | 国家纳米科学中心 | 一种碳包覆的硅酸亚铁锂正极材料、制备方法及其用途 |
| CN103746116B (zh) * | 2014-01-10 | 2016-08-17 | 国家纳米科学中心 | 一种碳包覆的硅酸亚铁锂正极材料、制备方法及其用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140231721A1 (en) | 2014-08-21 |
| JP2013086990A (ja) | 2013-05-13 |
| US9330805B2 (en) | 2016-05-03 |
| JP5765780B2 (ja) | 2015-08-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6382649B2 (ja) | 非水電解質二次電池用負極活物質材料、非水電解質電池、電池パック及び車両 | |
| JP5540281B2 (ja) | 非水電解質二次電池用正極の製造方法及びそれを用いた非水電解質二次電池 | |
| WO2010089931A1 (ja) | リチウムシリケート系化合物の製造方法 | |
| TWI445666B (zh) | 矽酸鋰系化合物之製造方法 | |
| EP2936590B1 (en) | Lmfp cathode materials with improved electrochemical performance | |
| EP2872449B1 (en) | Doped nickelate compounds | |
| TWI492443B (zh) | 鋰二次電池用正極活性物質、鋰二次電池用電極以及鋰二次電池 | |
| JP5950389B2 (ja) | リチウムシリケート系化合物、正極活物質、正極活物質の製造方法、非水電解質二次電池およびそれを搭載した車両 | |
| WO2007034823A1 (ja) | 正極活物質の製造方法およびそれを用いた非水電解質電池 | |
| US9590237B2 (en) | Lithium-ion secondary battery and method for producing the same | |
| WO2004001881A2 (en) | Carbon-coated li-containing powders and process for production thereof | |
| WO2015025795A1 (ja) | 異方性構造を有するアルカリ金属チタン酸化物及びチタン酸化物並びにこれらの酸化物を含む電極活物質及び蓄電デバイス | |
| JP4773964B2 (ja) | ホウ素置換されたリチウム挿入化合物、電極の活物質、電池およびエレクトロクロミックデバイス | |
| JP5765780B2 (ja) | リチウムシリケート系化合物とリチウムイオン二次電池用正極活物質及びこれを用いたリチウムイオン二次電池 | |
| CN107408693B (zh) | 二次电池用正极活性物质及其制造方法 | |
| JP5505868B2 (ja) | リチウム二次電池用正極活物質の前駆体とその製造方法 | |
| WO2016143171A1 (ja) | 二次電池用正極活物質及びその製造方法 | |
| WO2012060084A1 (ja) | リチウムボレート系化合物およびその製造方法 | |
| KR20220167538A (ko) | 이차전지용 음극 활물질 및 이의 제조방법 | |
| JP6042512B2 (ja) | 二次電池用正極活物質及びその製造方法 | |
| TW201803803A (zh) | 磷酸釩鋰的製造方法 | |
| JP5769140B2 (ja) | リチウム二次電池用正極活物質の製造方法 | |
| JP2010027604A (ja) | リチウム二次電池用正極活物質及びリチウム二次電池 | |
| JP5598684B2 (ja) | 非水電解質二次電池用正極活物質、正極及び電池 | |
| JP5608856B2 (ja) | リチウムイオン二次電池用正極活物質とその製造方法及びリチウムイオン二次電池用正極とリチウムイオン二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12840221 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14351714 Country of ref document: US |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12840221 Country of ref document: EP Kind code of ref document: A1 |